Область техники, к которой относится изобретение
Настоящее изобретение относится к способу превращения соединения или полимера, включающего первичный спирт и амидогруппу, в амидокарбоновую кислоту. В особенности, первичный амидоспирт превращают в карбоновую кислоту с неожиданно высокими выходами в том случае, когда выбирают правильный растворитель (например, воду). Кроме того, когда в качестве окислителей используют хлор или хлорированные молекулы, конкретные параметры обработки (технологического процесса), используемые в одном варианте осуществления изобретения, гарантированно обеспечивают то, что хлоринованный амидный азот не образуется.
Предпосылки создания изобретения
Амидокарбоновые кислоты являются желательными поверхностно-активными соединениями по той причине, что они имеют хорошую растворимость в воде, хорошие моющую способность и вспенивающие свойства и имеют мягкое воздействие на кожу и волосы. Один способ для получения такого поверхностно-активного соединения представляет собой окисление спирта, содержащего амидогруппу (например, кокомоноэтаноламид или CMEA).
Проблема, однако, заключается в том, что очень трудно проводить оксиление спирта до карбоновой кислоты эффективно. Реакция будет часто останавливаться на альдегидной стадии, и выходы карбоновой кислоты в качестве конечных продуктов являются довольно низкими.
Японский выложенный патент: Japanese Patent Laid-Open No. 05/194334 (Sandoz), раскрывает процесс, в котором гидроксилсодержащее соединение (которое может представлять собой, например, алкиламид-полиоксиалканол) подвергают реакции с, по меньшей мере, эквимолярным количеством неорганического или органического галогенсодержащего окислителя, например NaOCl, в присутствии слабого основания и каталитического количества пространственно затрудненного нитроксида, примером которого служит 2,2,6,6-тетраметилпиперидин-1-оксил, в дальнейшем в этом документе сокращенно называемый TEMPO, и его химические производные. В этом патенте не приводят информацию ни о выходе, ни о степени чистоты. Раскрытый процесс ограничен спиртами, которые имеют полиэтиленгликолевое или полипропиленгликолевое замещение, или полиглюкозидами, в качестве исходных реагентов. Такие соединения являются водорастворимыми или диспергируемыми в воде, что дает возможность использовать воду в качестве растворителя. Патент не обучает способу с использованием гидрофобных первичных спиртов (то есть амидоспиртов) изобретения в качестве исходного реагента.
Японский выложенный патент: Japanese Patent Laid-Open No. 04/283537 (Shell), раскрывает процесс c использованием окислителя, такого как гипохлорит натрия, в присутствии TEMPO. Впрочем, процесс относится к получению алкоксиалкановой кислоты из соответствующего алкоксиалканола, а не к получению амидокарбоновой кислоты из спирта, имеющего амидогруппу.
Японский выложенный патент: Japanese Patent Laid-Open No. 10/087554 (Lion Corporation), раскрывает процесс для получения амидокарбоновой кислоты из спирта, имеющего амидогруппу, с использованием окислителя хлорного типа (например, NaOCl) в присутствии нитроксидной радикальной группы (например, TEMPO) и дополнительно в присутствии галогенида щелочного металла или галогенида щелочноземельного металла (например, хлорида калия). В Примерах 3 и 5, например, спирт, содержащий амидогруппу, нитроксидную радикальную группу и 10%-ный раствор хлорида щелочного металла (бромид калия или натрия) в воде, дополнительную воду, и ацетонитрил (растворитель) загружают в химический стакан и перемешивают. В этих условиях ацетонитрил и воду смешивают вместе с образованием одной жидкой фазы. В каждом примере степень чистоты карбоновой кислоты вычисляют из кислотного числа, но в отношении выхода ничего не указано. Кислотное число не является селективным в отношении желательной карбоновой кислоты, но может включать все присутствующие кислотные компоненты.
Неожиданно авторы заявки как раз обнаружили, что тип растворителя или растворителей, используемого(ых) во время реакции окисления, является решающим для выхода продукта (карбоновая кислота). Не желая быть связанными теорией, авторы заявки полагают, что исходный амидоспирт не должен находиться в той же самой фазе, что и окислитель. Авторы заявки обнаружили, что такое разделение окислителя и спирта может быть осуществлено, по меньшей мере, двумя различными путями. В соответствии с отдельной заявкой, которую авторы подали с той же датой подачи заявки, что и представленную на рассмотрение заявку, конечный продукт (например, амидокарбоновая кислота) является распределенным в органический растворитель (то есть с использованием растворителя, который будет образовывать как гидрофобную жидкую фазу, так и водную жидкую фазу, а не с образованием одной в значительной степени водной фазы). Таким образом, подвергаемая воздействию амидогруппа в амидоспирте является защищенной от расщепления (например, отбеливающее вещество, которое оказывается распределенным в основном в водную фазу, не будет воздействовать разрушающе на амидоспирт, находящийся в отдельной фазе), и в результате получают намного больший выход амидокарбоновой кислоты. То есть важно то, что в присутствии окислителя образуются как богатый растворителем слой (в значительной степени свободный от окислителя), так и водный слой (в основном содержащий окислитель).
Во втором варианте осуществления, который является заявленным в представленной на рассмотрение заявке, окислитель и спирт также поддерживают в отдельных фазах. Здесь, однако, две фазы представляют собой водную фазу и твердую фазу, и в качестве растворителя используют одну воду. То есть амидоспирт, который является гидрофобным, не растворяется или не диспергируется в водной фазе (скорее он остается в твердой, неводной фазе), наряду с тем, что NaOCl остается в непрерывной (диффузионной) водной фазе. В предпочтительной особенности этого варианта осуществления (вода в качестве растворителя), когда в качестве окислителя используют хлор или хлорированную молекулу (например, NaOCl), должны быть использованы особые технологические стадии (например, нагревание) для гарантированного обеспечения того, чтобы амидный азот конечной карбоновой кислоты не был хлорированным, поскольку это было бы нежелательным побочным продуктом.
Как указано, представленная на рассмотрение заявка относится к способам, где в качестве растворителя применяют воду, и при добавлении окислителя окислитель распределяется в растворитель, в то время как амидоспирт и/или амидокарбоновая кислота остаются в твердой фазе.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение обеспечивает способ для превращения первичного спирта, имеющего амидную группу, в амидокарбоновую кислоту с высоким выходом (например, с выходом ≥75%, предпочтительно ≥80%, более предпочтительно ≥85%, более предпочтительно ≥90%), где способ включает реакционное взаимодействие первичного спирта, имеющего амидогруппу (амидоспирт), с окислителем, предпочтительно с хлорсодержащим оксидантом, подобным NaOCl, в присутствии нитроксидной радикальной группы и необязательно в присутствии галогенида щелочного металла или галогенида щелочноземельного металла. В этом способе растворитель, в котором протекает реакция, выбирают из условия, чтобы в присутствии окислителя первичный амидоспирт распределялся или оставался (после добавления отбеливающего вещества или другого окислителя) в твердой органической фазе, в то время как отбеливающее вещество или окислитель распределялся в основном в жидкую водную фазу. Такое распределение гарантированно обеспечивает высокие выходы, отмеченные выше (например, амидная связь не является доступной для разрыва посредством окислителя, так как окислитель распределен в жидкую водную фазу). Иными словами, амидоспирт остается в твердой фазе, а окислитель достаточно быстро распределяется в водную фазу с избежанием образования нежелательного побочного продукта. Совершенно неожиданным является то, что вода, выбранная в качестве единственного растворителя, могла дать такую решающую разницу.
Также важной особенностью изобретения является то, что катализатор, используемый в этой реакции, представляет собой пространственно затрудненную нитроксидную радикальную группу. Также может быть использован необязательный сокатализатор на основе галогенида щелочного металла или галогенида щелочноземельного металла или сокатализатор может представлять собой, например, тетраборат натрия.
Конкретно, в одном варианте осуществления изобретения, в реационную смесь добавляют основание (например, гидроксид натрия), достаточное для гарантированного обеспечения того, чтобы реакция протекала при рН выше 6, предпочтительно 7-10, более предпочтительно 7,5-9, еще более предпочтительно 8-9. Добавление основания используют для компенсирования расходования окислителя (например, гипохлорита натрия) во время образования амидокарбоновой кислоты. Основание может быть добавлено в раствор окислителя до добавления окислителя в реакционную смесь или оно может быть добавлено в ходе реакции (например, для поддерживания постоянного рН).
Эти и другие особенности, отличительные признаки и преимущества станут очевидны средним специалистам в данной области после прочтения следующего подробного описания и прилагаемой формулы изобретения. Во избежание сомнений любой отличительный признак одной особенности настоящего изобретения может быть применен в любой другой особенности данного изобретения. Отмечают, что примеры, приведенные в описании ниже, предназначены для прояснения изобретения и не предназначены для ограничения изобретения по сути непосредственно теми примерами. За исключением экспериментальных примеров или тех случаев, где указано иное, все числа, выражающие количества ингредиентов или реакционные условия, используемые в этом документе, следует понимать как модифицированные во всех случаях посредством термина «приблизительно». Подобным образом, все процентные содержания представляют собой процентные содержания по массе относительно массы всей композиции, если не оговорено иное. Полагают, что численные диапазоны, выраженные в формате «от х до y», включают х и y. Когда для конкретного отличительного признака в формате «от х до y» описывают многочисленные предпочтительные диапазоны, то подразумевают, что все диапазоны, объединяющие различные конечные точки, также являются рассматриваемыми. В тех случаях, где в описании или в формуле изобретения используют термин «включающий/содержащий», он не предназначен для исключения каких-либо терминов, стадий или отличительных признаков, особым образом не изложенных. Все температуры представлены в градусах Цельсия (°С), если не определено иное. Все измерения представлены в единицах системы СИ, если не определено иное. Все перечисленные документы - в релевантном разделе - включены в данный документ путем ссылки.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
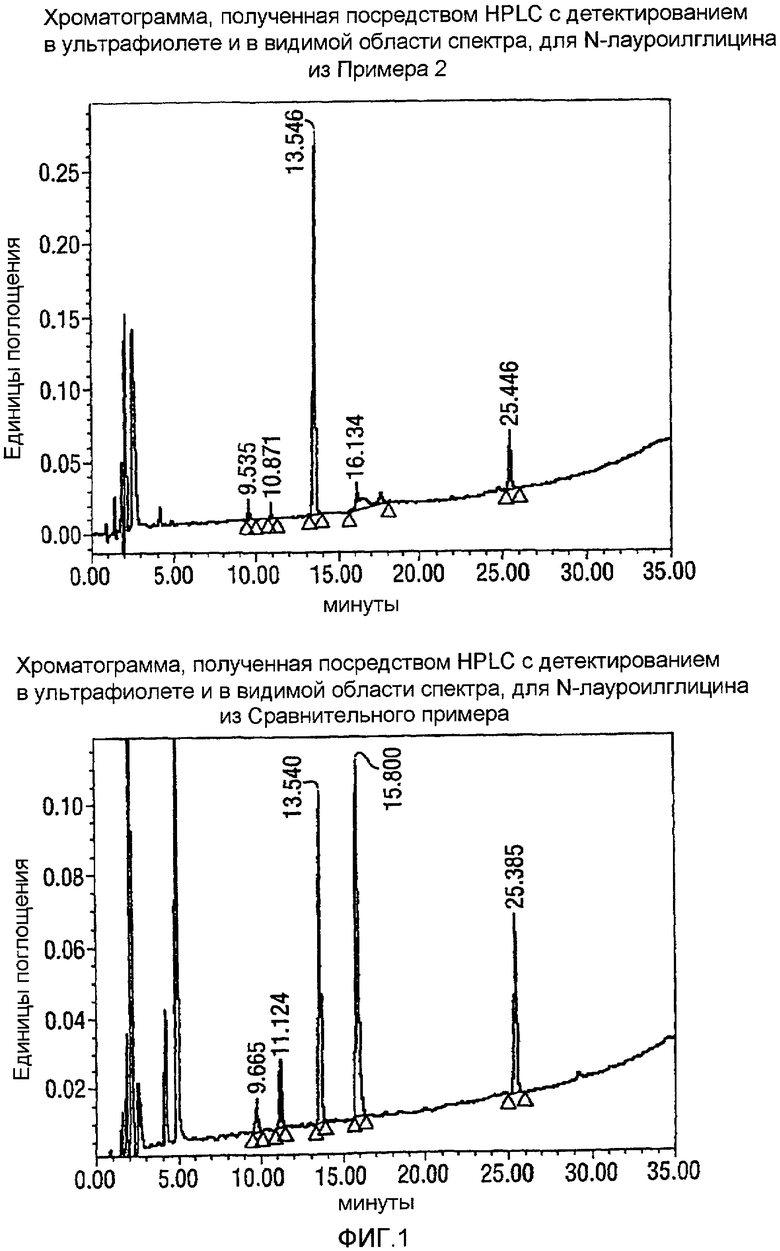
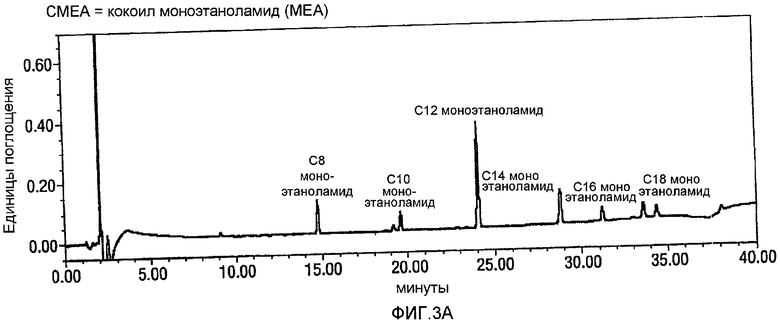
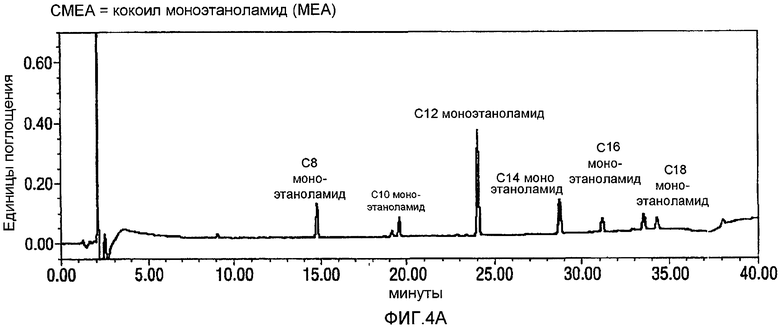
Фигура 1 представляет собой профиль хроматограмм, сделанных на жидкостном хроматографе (HPLC-UV-Vis = высокоэффективная жидкостная хроматография-детектирование в ультрафиолете-детектирование в видимой области спектра), для продуктов, образованных, когда реакцию проводят в растворителе CH3CN/вода, и когда была образована только одна жидкая фаза (нижняя фигура, соответствующая примеру из ссылки на японский патент JP 10/087554 (Lion Corporation)), в сравнении с тем, когда растворитель представлял собой смесь THF/вода, и когда образовывались две жидкие фазы (сверху). На фигурах 1-4 AU относится к Единицам Поглощения.
Как видно, растворитель, который разделяется на две жидкие фазы, приводит к высоким выходам N-лауроилглицина (LG) из N-лауроилмоноэтаноламидного (LMEA) исходного вещества. В противоположность тому, когда органический растворитель образовывал одну жидкую фазу с водой, выход и степень чистоты LG (глицинат) были более низкими. Так, например, когда образовывались две жидкие фазы, то за 13,54 минуты был преимущественно образован чистый продукт LG, но когда образовывалась только одна фаза, то через 15,80 секунд было огромное количество примесей.
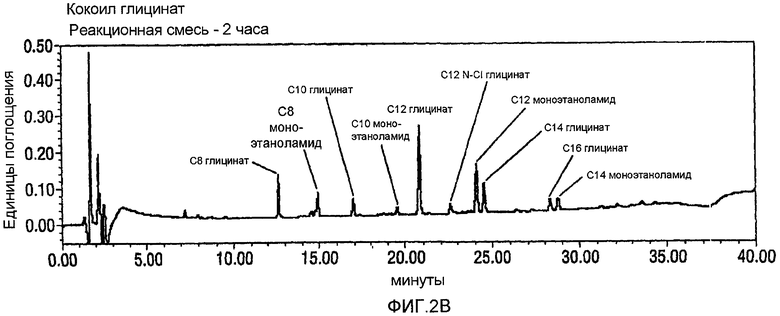
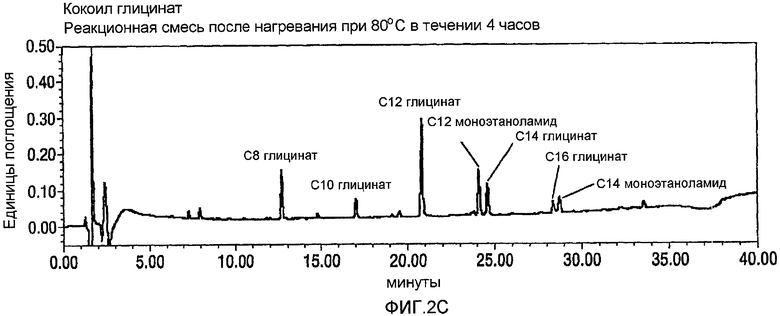
Фигура 2 представляет собой график HPLC-UV-Vis-анализа окисления кокоил-моноэтаноламида (CMEA) до карбоновой кислоты с использованием 1,6-2,5 эквивалентов NaOCl. Верхняя панель А представляет собой реагент CMEA до добавления NaOCl; средняя панель В представляет собой анализ продукта с использованием воды, подвергаемой анализу через два часа после добавления NaOCl и до нагревания; и нижняя панель С представляет собой анализ продукта с использованием способа с участием воды в качестве растворителя через 24 часа после добавления NaOCl и с дополнительным нагреванием при 80°С в течение 4 часов. Как видно, когда не используют стадию нагревания (Панель В), образуется С12 N-Cl глицинатное промежуточное соединение (то есть между 22 и 23 минутой), тогда как, когда используют стадию нагревания, такое хлорированное промежуточное соединение не образуется.
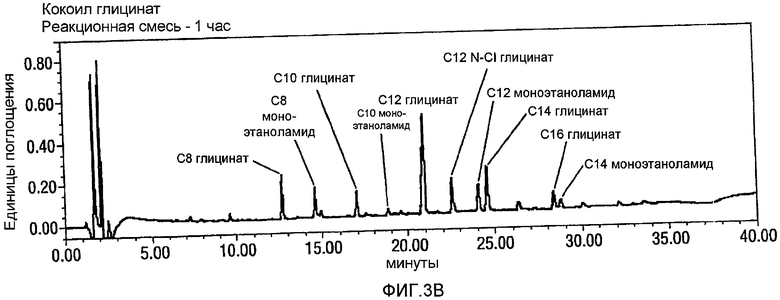
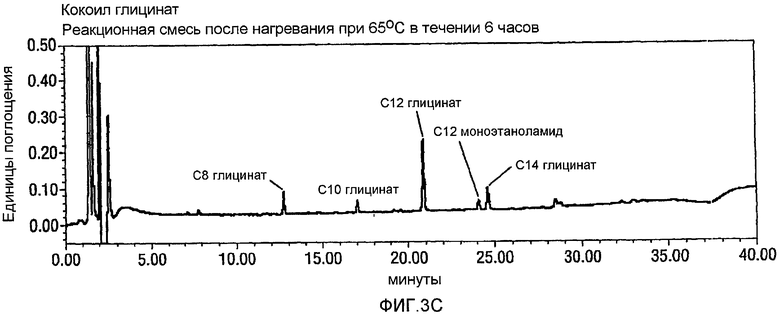
Фигура 3 представляет собой график, аналогичный графику фигуры 2, но при использовании 2,3-3,2 эквивалентов NaOCl для окисления CMEA до кокоил-глицината. Панель А опять показывает CMEA до добавления NaOCl, Панель В представляет собой реакционную смесь через 1 час после добавления NaOCl, и Панель С представляет собой реакционную смесь через 24 часа после добавления NaOCl и дополнительного нагревания при 65°С в течение 6 часов. Опять, видно, что без стадии нагревания образуется С12 N-Cl глицинатное промежуточное соединение, и в том случае, когда используют стадию нагревания, такое промежуточное соединение не образуется.
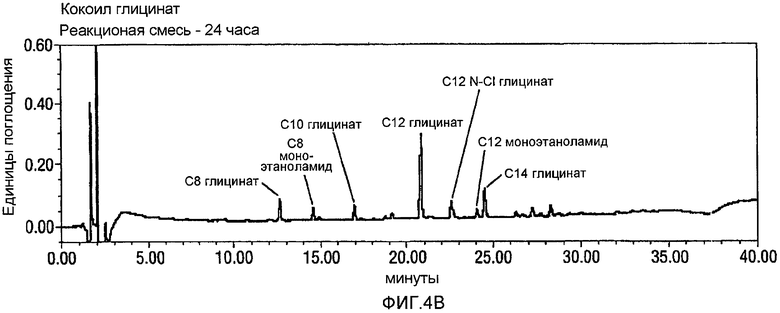
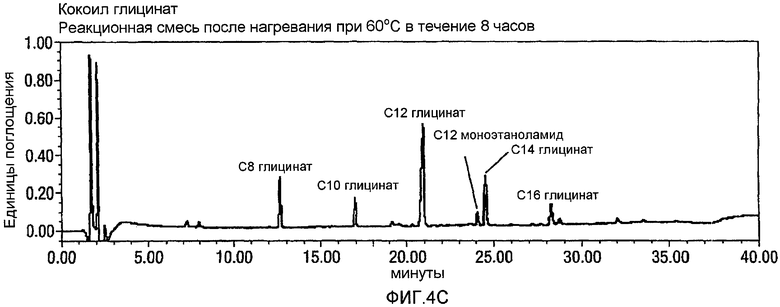
Фигура 4 представляет собой реакционный профиль окисления CMEA до кокоил-глицината с использованием 3,2-4,0 эквивалентов NaOCl. Панель А представляет собой CMEA до добавления NaOCl, Панель В представляет реакционную смесь через 24 часа после добавления NaOCl, и Панель С отражает реакционную смесь через 24 часа после добавления NaOCl и дополнительного нагревания при 60°С в течение 8 часов. Вновь, в отсутствии стадии нагревания, будет образовываться С12 N-Cl глицинат.
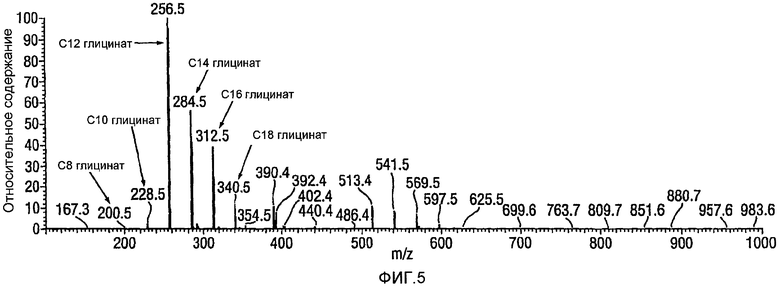
Фигура 5 представляет собой типичный спектр с учетом общего числа ионов для введенного образца кокоил-глицината, выработанного с использованием методики, описанной в Примере 10. Этот спектр демонстрирует отсутствие кокоил N-Cl глицинатного промежуточного соединения.
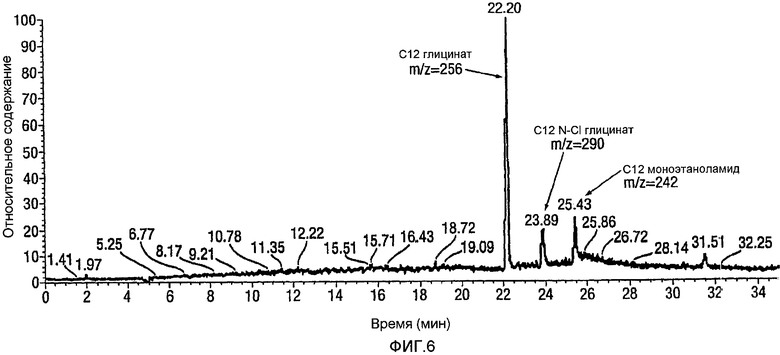
Для анализа методом жидкостной хроматографии в сочетании с масс-спектрометрией образец глицината с концентрацией 1 мг/мл, полученный в подвижной фазе, был разделен методом высокоэффективной жидкостной хроматографии (HPLC) и проанализирован посредством детектирования в ультрафиолете (UV) и методом масс-спектрометрии (MS). На фигуре 6 показана типичная HPLC-хроматограмма с профилем масс-детектирования общего числа ионов для аликвоты реакционной смеси после окисления лауроил-моноэтаноламида (С12 MEA) до лауроил-глицината (С12 глицинат).
Подробное описание изобретения
Настоящее изобретение относится к новому и улучшенному способу для превращения первичного спирта, содержащего амидную группу (например, С8-С22 алкоилмоноалканоламида, такого как лауроил-моноэтаноламид), в соответствующую амидокарбоновую кислоту (например, смесь N-лауроил-глицина и N-лауроил-глицината щелочного металла), и где способ обеспечивает очень высокие выходы продукта (например, выход ≥75%, предпочтительно ≥80%, более предпочтительно ≥85%). Конкретнее, способ включает реакционное взаимодействие первичного спирта, содержащего такую амидную группу, с окислителем в присутствии нитроксидной радикальной группы и необязательного катализатора (например, галогенида щелочного металла), где растворитель, в котором протекает реакция, выбирают таким образом, что образуются две фазы, отделяющие амидоспирт от окислителя. Иными словами, амидоспирт остается или является распределенным в органическую фазу двухфазной системы, а окислитель остается преимущественно в водной фазе. Это защищает амидную группу в амидоспирте от дальнейшего расщепления и обеспечивает высокие выходы, которые указаны выше. В особом варианте осуществления заявленного изобретения, амидоспирт отделяют от окислителя с использованием только воды в качестве растворителя по той причине, что окислитель распределяется в жидкую водную фазу, в то время как амидоспирт остается в нерастворенном твердом состоянии (гетерогенная система твердое вещество-жидкость). В предпочтительном варианте осуществления этого способа с участием воды в качестве растворителя, после добавления окислителя (например, NaOCl) реакции дают пройти до завершения. Это, как правило, занимает 30 минут-24 часа, типично 1-10 часов. Затем реакционную смесь нагревают до температуры, по меньшей мере, 40°С - вплоть до 100°С в течение от 1 до 24 часов. Стадия нагревания не является обязательной, если в способе с участием воды в качестве растворителя не используют хлорсодержащую молекулу, в таком случае хлорированный амидный азот не образуется.
Конкретнее, исходный реагент представленного на рассмотрение изобретения представляет собой спирт, имеющий амидную группу, который может быть определен следующим образом:
R 1 -CONR 2 (CH 2 ) m OH
где R1 представляет собой линейную или разветвленную алкильную или алкенильную группу, имеющую 7-22 атома углерода; R2 представляет собой Н, алкильную или гидроксиалкильную группу с 1-6 атомом(ами) углерода; и m представляет собой целое число от 1 до 6.
Примерами соединений, которые могут быть охвачены такой структурой, являются N-алканоилмоноэтаноламины, такие как N-лауроил-моноэтаноламид (LMEA) или N-кокоил-моноэтаноламид (CMEA).
Исходный продукт может представлять собой смесь моноалканоламидов (например, моноэтаноламин), включающую смеси моноалканоламидов, полученные из смесей жирных кислот, имеющих природное происхождение. N-кокоил-моноэтаноламин, например, может включать смесь С8, С10 и С12 жирных кислот в качестве основного компонента, смешанных с С14, С16 и С18 жирными кислотами.
Окислитель, используемый для окисления исходного спирта, может представлять собой любой окислитель, который будет давать возможность окислять спиртовую группу до карбоновой кислоты. Обычно такие окислители включают окислители хлорного типа. Они могут включать хлор, гипохлорит (например, гипохлорит щелочного металла), трихлоризоциануровую кислоту и дихлоризоциановую кислоту. Предпочтительные окислители включают гипохлорит натрия (например, отбеливатель технического сорта содержит 5-13% гипохлорита натрия), гипохлорит кальция, хлор сам по себе и органические хлорсодержащие соединения, например трихлоризоциануровую кислоту. Могут быть использованы окислители, не содержащие хлор, например оксон (2KHSO5•KHSO4•K2SO4), NaOBr, N-бромсукцинимид, или трибромизоциануровая кислота. Также могут быть использованы окислители, не содержащие галоген, в качестве примера которых могут быть приведены H2O2 необязательно в присутствии катализатора на основе дигидрата вольфрамата натрия.
Количество окислителя может варьироваться, но используют обычно эквимолярное - 8-молярное количество, предпочтительно 1-7 эквивалентов, более предпочтительно 2-, 6-молярное количество.
Исходный спирт изобретения окисляют посредством окислителя (как указано выше) в присутствии катализатора на основе пространственно затрудненной радикальной пиперидинилоксигруппы (нитроксид) и необязательно в присутствии сокатализатора, который будет описан ниже.
Нитроксидную каталитическую радикальную группу, используемую в изобретении (например, пространственно затрудненный нитроксид), получают окислением циклического или ациклического вторичного амина, не содержащего α-водороды (то есть отсутствуют водороды на углероде, смежном с N), посредством пероксида или окислением соответствующего гидроксиламина. Примеры стабильных нитроксидных радикальных групп, подходящих для использования в этом изобретении, указаны в следующих документах. Они включают линейные, циклические, дициклические или макромолекулярные соединения, к которым присоединена одна или более нитроксильных радикальных групп.
Chem. Review, 78, 37 (1979):
G. Rozantsev, «Free Nitroxyl Radicals», Plenum Publishing Corporation, New York, 1970; и
E. G. Rozantsev, V. D., Scholle, Synthesis, 1971, 190.
Предпочтительные примеры нитроксидной радикальной группы представляют собой следующее.
2,2,6,6-Тетраметил-пиперидин-1-оксил (TEMPO).
2,2,5,5-Тетраметил-пирролидин-1-оксил.
1-Аза-2,2,7,7-тетраметил-циклогептан-1-оксил.
TEMPO и его химические производные являются предпочтительными, примеры которых следуют далее.
4-Гидроксил-2,2,6,6-тетраметил-пиперидин-1-оксил.
4-Метокси-2,2,6,6-тетраметил-пиперидин-1-оксил.
4-Этокси-2,2,6,6-тетраметил-пиперидин-1-оксил.
4-Ацетиламидо-2,2,6,6-тетраметил-пиперидин-1-оксил.
4-Карбамоил-2,2,6,6-тетраметил-пиперидин-1-оксил.
4-Бензоиламино-2,2,6,6-тетраметил-пиперидин-1-оксил.
4-Оксо-2,2,6,6-тетраметил-пиперидин-1-оксил.
2,2,6,6-Тетраметил-пиперидин-1-оксил 4-сульфат.
2,2,6,6-Тетраметил-пиперидин-1-оксил 4-фосфат.
3-Карбамоил-2,2,6,6-тетраметил-пирролидин-1-оксил.
Вещества, задерживающие процесс старения под воздействием УФ-излучения, содержащие 2,2,6,6-тетраметилпиперидиновую функциональность (вещества, задерживающие процесс старения под воздействием света, на основе пространственно затрудненных аминов, сокращенно называемые как HALS), как мономерные, так и олигомерные, также могут служить в качестве предшественников стабильных нитроксильных радикальных групп при использовании окисления.
Также является возможным, когда используют амин или гидроксиламин, который является предшественником нитроксидной радикальной группы, и, в действительности, его окисляют и затем используют. Количество нитроксидной радикальной группы, используемое на 1 эквивалент исходного спиртового вещества, обычно составляет от 0,01 до 10 мольных % или предпочтительно от 0,1 до 5 мольных %, исходя из амидоспирта.
Необязательный сокатализатор часто используют вместе с основным катализатором на основе нитроксидной радикальной группы. Сокатализатор, если используют, может представлять собой, например, галогенид щелочного металла или галогенид щелочноземельного металла. Они могут включать бромид щелочного металла, например бромид натрия, и хлорид щелочного металла, например хлорид натрия и хлорид калия, бромид щелочноземельного металла, например бромид кальция и бромид магния, хлорид щелочноземельного металла, например хлорид кальция и хлорид магния.
Обычно сокатализатор используют в количестве от 0,01 до 10 мольных %, предпочтительно 0,1-5 мольных % эквивалента, исходя из амидоспирта. Тетраборат натрия может быть использован вместо бромида или хлорида.
Растворитель
Ключ к изобретению находится в выборе правильного растворителя, то есть растворителя, который будет разделяться на органическую фазу и водную фазу при соединении окислителя и амидоспирта в растворителе.
Хотя идеальные растворители являются, по меньшей мере, частично смешивающимися с водой (например, тетрагидрофуран), ключевой момент состоит в том, что в присутствии окислителя (например, водного гипохлорита натрия) будут образовываться, по меньшей мере, два несмешивающихся слоя (например, богатый растворителем слой, обычно верхний слой; и богатый водой слой, обычно нижний слой).
Не желая быть связанными теорией, авторы изобретения полагают, что важно то, что амидоспирт не находится в той же фазе, что и окислитель при их соединении. Авторы изобретения обнаружили, что это может быть осуществлено двумя различными путями. В соответствии с формулами изобретения одной из двух заявок, находящихся одновременно на рассмотрении патентного ведомства, это может быть выполнено путем распределения конечного продукта (амидокарбоновой кислоты) в жидкий органический растворитель (то есть с использованием растворителя, который будет образовывать две фазы, а не с образованием одной в значительной степени водной фазы). Подвергаемая воздействию амидная группа в молекуле спирта, содержащего эту амидную группу, таким образом, является защищенной от расщепления (например, посредством разрушающего воздействия отбеливателя, который в основном распределился в отдельную жидкую водную фазу), и в результате получают намного больший выход карбоновой кислоты. То есть важно то, что в присутствии окислителя образуются как богатый растворителем слой (в значительной степени свободный от окислителя), так и водный слой (состоящий в основном из окислителя). Следует отметить, что то, насколько быстро происходит разделение двух фаз, зависит, как правило, от объема реакционной смеси. Обычно разделение фаз будет происходить за 1 час или менее и может происходить относительно мгновенно.
Второй путь (как заявлено в представленном на рассмотрение изобретении) поддерживать окислитель и спирт в отдельных фазах, в этом случае в жидкой водной фазе и в твердой фазе, представляет собой использование воды как таковой в качестве растворителя. Амидоспирт является гидрофобным и не растворяется или не диспергируется в водной фазе (он остается в твердой фазе), в то время как NaOCl остается в непрерывной (диффузионной) жидкой водной фазе.
Реакция, в которой растворитель будет образовывать только одну жидкую фазу (например, растворитель на основе смеси CH3CN/вода, используемый в японском патенте JP 10/087554), таким образом, не является подходящей и будет давать продукт с более низкими выходом и степенью чистоты.
Неожиданно, для реакции могут быть подходящими растворители с противоположными максимально отдаленными значениями по шкале полярности. Подходящие полярные растворители могут включать окисленные (кислородосодержащие) углеводороды, конкретнее циклические и ациклические простые эфиры и простые полиэфиры. Подходящие неполярные растворители могут включать циклические и ациклические алифатические растворители и ароматические растворители.
Конкретные примеры циклических кислородосодежащих растворителей (например, полярных растворителей), которые могут быть использованы, включают тетрагидрофуран (THF) и диоксолан. Примеры ациклических кислородосодержащих растворителей включают 1,2-диметоксиэтан, диметоксиметан, диэтоксиметан и 2-метоксиэтиловый эфир.
Предпочтительно, что растворители не содержат антиокислители (например, бутилированный гидроксилтолуол, сокращенно называемый как ВНТ), поскольку эти антиокислители могут помешать реакции окисления. Такие антиокислители часто обнаруживают в циклических и ациклических простых эфирах и простых полиэфирах. Таким образом, предпочтительно растворители изобретения являются в основном свободными от антиокислителей.
Конкретные примеры циклических алифатических растворителей (например, неполярных растворителей) включают циклогексан; примеры ациклических алифатических растворителей включают гептаны и гексаны; и примеры ароматических растворителей включают толуол, о-, м- или л-ксилол и смеси ксилолов.
В предпочтительной реакции, вследствие расходования окислителя (например, гипохлорита натрия) и образования карбоновой кислоты в результате реакции, в реакции следует использовать основание, достаточное для поддерживания рН выше 6, более предпочтительно выше 7, предпочтительно 8-9. Примером основания, которое может быть использовано, является гидроксид щелочного металла (например, NaOH).
Основание может быть добавлено к окислителю до введения окислителя в реакцию или, альтернативно, основание может быть добавлено, например, по каплям в ходе реакции по мере необходимости для поддерживания рН постоянным.
Реакция сама по себе обычно протекает при комнатной температуре, но является экзотермической. Повышения температуры вплоть до приблизительно 35°С происходят без охлаждения. Для снижения выделяемой теплоты экзотермичеческой реакции может быть использована охлаждающая баня.
Типичный пример окисления моноэтаноламида (N-лауроил-моноэтаноламид, или MEA) до N-лауроилглицина (LG), а также реакционные условия, методология выделения и степень химического превращения в N-лауроилглицин изложены ниже:
Реакционные условия:
Следует отметить, что в зависимости от рН при выделении может быть получена смесь N-лауроилглицина и N-лаурилглицината натрия (например, солевая форма), и, следовательно, выходы могут быть рассчитаны отдельно для каждого соединения.
В случае, когда в способе с участием воды в качестве растворителя используют хлор или хлорсодержащую молекулу, обычно после введения катализатора и окислителя реакции дают протекать 30 минут-24 часа и затем раствор нагревают в течение 1-24 часов при температуре, по меньшей мере, 40°С - вплоть до приблизительно 100°С. После нагревания рН «подкисляют» и твердый продукт отфильтровывают из раствора.
ПРИМЕРЫ
Протокол
Метод экстракции для выделения карбоновой кислоты (например, N-лаурилглицина)
При доведении реакции оксиления до завершения реакционную смесь подкисляют до рН приблизительно 3,0 (например, посредством добавления HCl) и слои разделяют. Нижний водный слой экстрагируют тетрагидрофураном и объединенные тетрагидрофурановые слои концентрируют на роторном испарителе и сушат в вакууме с получением карбоновой кислоты (например, N-лауроилглицин) в виде твердого вещества белого цвета.
Метод экстракции для выделения соли карбоновой кислоты с щелочным металлом или щелочноземельным металлом (например, N-лауроилглицината натрия)
Реакция здесь аналогична реакции, описанной выше, за исключением того, что тетрагидрофурановый слой отделяют без подкисления. Водный слой должен иметь рН в диапазоне 6-10, предпочтительно 6-8. Водный слой экстрагируют (предпочтительно дважды) посредством тетрагидрофурана. Объединенные тетрагидрофурановые слои концентрируют на роторном испарителе и сушат в вакууме с получением соли (например, N-лауроилглицината).
Альтернативный метод экстракции для карбоновой кислоты
Кроме экстракции тетрагидрофураном карбоновая кислота может быть выделена посредством методики «погружения» и фильтрации. В этой методике реакционную смесь подкисляют до рН приблизительно 2-3 и добавляют к избытку воды (приблизительно 3-4 объема по сравнению с объемом реакционной смеси) при энергичном перемешивании с использованием перемешивающей лопасти. Осадок собирают фильтрацией, промывают водой и сушат в вакууме с получением карбоновой кислоты (например, N-алканоил-глицина).
Мониторинг реакции окисления кокоил-моноэтиламида (CMEA) до кокоил-глицината (CG) посредством высокоэффективной жидкостной хроматографии (HPLC)
Прибор: модуль разделений Waters 2695, оснащенный матричным фотодиодным детектором Waters 2996
Программное обеспечение: Empower Pro (версия 5,00, Waters Corp.)
Колонка: Restec Pinnacle DB C18 5мкм, 4,6×150 мм, поддерживаемая при 30°С
Скорость потока: 1 мл/мин
Проба: 1-2 мг/мл в смеси вода:ацетонитрил (W:AcN) 1:1, содержащей 0,04% уксусной кислоты (AcOH)
Вводимый объем: 15 мкл
Подвижная фаза: А=2 мМ ацетата аммония, 0,04% AcOH
B=2 мМ ацетата аммония, 0,04% AcOH в 90% водном AcN
Градиент: А:В 95:5 - 100% В (градиент, 35 мин) с последующим 100% В (изократическая хроматография, 5 мин)
Детектирование: 205 нм
Анализ кокоил-глицината натрия с использованием жидкостной хроматографии в сочетании с масс-спектрометрией (LC-MS)
Прибор: Finnigan Mat LCQ
Колонка: Restec Pinnacle DB C18 5 мкм, 4,6×150 мм, поддерживаемая при 30°С
Скорость потока: 1 мл/мин
Проба: 1-2 мг/мл для LC-MS и 50 миллионных долей (ppm) раствора для инфузии в смесь вода:ацетонитрил (W:AcN) 1:1, содержащей 0,04% уксусной кислоты (AcOH) и 2 мМ ацетата аммония (АА)
Подвижная фаза: А=2 мМ ацетата аммония, 0,04% AcOH
B=2 мМ ацетата аммония, 0,04% AcOH, AcN:W (90:10)
Градиент: А:В 95:5 - 100% В (градиент, 35 мин) с последующим 100% В (изократическая хроматография, 5 мин)
Детектирование: УФ - 205 нм: масс-спектрометрия - метод ионизации электрораспылением (-)
Анализ: для анализа общего числа ионов в качестве способа введения пробы используют инфузию. Образец кокоил-глицината (кислотная форма, 1 мг) растворяют в тетрагидрофуране (1 мл) и разбавляют путем взятия аликвоты 50 мкл и разбавления посредством 95 мкл тетрагидрофурана. Разбавленный раствор инфундируют в масс-спектрометр и регистрируют общее число ионов. Характерный спектр с учетом общего числа ионов для инфундированной пробы кокоил-глицината, приготовленной с использованием методики, описанной в Примере 10, показан на фигуре 5 ниже. Для анализа методом жидкостной хроматографии в сочетании с масс-спектрометрией (LC-MS) пробу глицината с концентрацией 1 мг/мл, полученную в подвижной фазе, разделяют посредством высокоэффективной жидкостной хроматографии (HPLC) и анализируют детектированием в ультрафиолете (UV) и методом масс-спектрометрии (MS). Характерный спектр с учетом общего числа ионов для аликвоты реакционной смеси при окислении лауроил-моноэтаноламида (С12 MEA) до лауроил-глицината (С12 Глицинат) показан на фигуре 6 ниже.
Пример 1. Окисление N-лауроилэтаноламида (LMEA) в THF посредством 6,5 экв. NaOCl и кислотная обработка. 33 мг (4,5 мольных %) KBr (сокатализатор) растворяют в 6 мл воды. Тетрагидрофурановый растворитель (THF) (31 мл), катализатор AA-TEMPO (25 мг, 2,5 мольных %) и 1,5 г N-лауроилэтаноламида (LMEA) добавляют при перемешивании, что дает гомогенный бесцветный как вода раствор. Смешивают окислитель, гипохлорит натрия (22 мл 11,5%-ного водного раствора, 6,5 эквивалентов) и 2,3 мл 2н. NaOH (для поддержания рН приблизительно 7). Объединенный раствор добавляют по каплям к раствору LMEA и катализатора в течение периода времени 1,5 часа. При добавлении раствора гипохлорита натрия мгновенно образуется отдельный водный слой. рН водного слоя составляет 12,7 после добавления первых 3,5 мл. Температуру поддерживают ниже 32°С посредством водно-ледяной бани. Реакционную смесь перемешивают в течение более 0,5 часа до полного превращения LMEA в LG, что подтверждено обращеннофазной жидкостной хроматографией высокого давления, сокращенно называемой как HPLC. рН в конце реакции составляет 7,6.
При завершении реакции смесь подкисляют до рН 3,0 (для получения очищенной карбоновой кислоты) путем добавления 8,5 мл 1н. HCl и слои разделяют. Нижний водный слой экстрагируют посредством 30 мл THF и объединенные тетрагидрофурановые слои концентрируют на роторном испарителе и сушат в вакууме с получением N-лауроилглицина со 116%-ным выходом (присутствует остаточная вода).
Пример 2. Окисление LMEA посредством 3,25 экв. NaOCl и кислотная обработка. Следуют методике Примера 1, за исключением того, что количество гипохлорита натрия снижают до 3,25 эквивалентов. Выход продукта после выделения составляет 103% (включает остаточную воду), что показывает, что окисление происходит с более низким количеством гипохлорита натрия.
Пример 3. Окисление LMEA посредством 3,25 экв. NaOCl, выделение натриевой соли. В этом примере N-лауроилглицинат натрия получают с использованием небольшой модификации методики выделения. Следуют методике Примера 2. рН составляет 7,8 по завершении реакции. В этом случае тетрагидрофурановый слой отделяют без подкисления. Водный слой экстрагируют дважды посредством 30 мл тетрагидрофурана. После экстракции рН водного слоя составляет 8,3. Объединенные тетрагидрофурановые слои концентрируют на роторном испарителе и сушат в вакууме с получением N-лауроилглицината натрия с 99%-ным выходом. В отличие от N-лауроилглицина, N-лауроилглицинат натрия растворяется в воде и дает пену при взбалтывании.
Пример 4. Окисление LMEA в THF посредством 6,5 экв. NaOCl, методика «Погружения» при рН=2,6. Следуют методике Примера 1, за исключением стадии выделения. После завершения реакции смесь подкисляют до рН=2,6 посредством 9,25 мл 1н. HCl. Всю реакционную смесь (как тетрагидрофурановую, так и водную фазы) выливают в 240 мл воды при энергичном перемешивании. Осажденный продукт выделяют посредством безнапорной фильтрации и промывают посредством 200 мл воды. После сушки воздухом и дополнительной сушки в вакууме получают продукт с 77%-ным выходом. Выход может быть улучшен посредством более совершенного/подходящего метода фильтрации, например посредством фильтрации под давлением через фильтр с порами 0,45 мкм или менее.
Пример 5. Окисление LMEA в THF посредством 3,25 экв. NaOCl, методика «Погружения», влияние рН на выход. Следуют методике Примера 2, за исключением стадии выделения. После завершения реакции смесь подкисляют лишь частично (до рН=5,2 посредством 3,6 мл 1н. HCl). Всю реакционную смесь (как тетрагидрофурановую, так и водную фазы) выливают в 240 мл воды при энергичном перемешивании. Осажденный продукт выделяют посредством безнапорной фильтрации и промывают посредством 200 мл воды. После сушки в вакууме в течение ночи получают продукт с 59%-ным выходом (исходя из свободной карбоновой кислоты) и с 55%-ным выходом (исходя из карбоксилата натрия). Более низкий выход по сравнению с Примером 4 относят к большей доле водорастворимого карбоксилата натрия при более высоком рН.
Пример 6. Окисление LMEA посредством 3,25 экв. NaOCl в воде, кислотная обработка. KBr (33 мг, 4,5 мольных %) и АА-TEMPO (25 мг, 2 мольных %) растворяют в 50 мл воды. Добавляют 1,50 г LMEA и смесь перемешивают 1,5 часа с образованием гомогенной суспензии. Разбавленный гипохлорит натрия (5%) добавляют порциями по 2,0 мл в течение 1,3 часа. После каждого добавления вводят 0,1н. HCl, если необходимо поддерживать рН 8-9.
Подробности добавления приведены в таблице ниже.
После перемешивания в течение 20,5 часов рН понижают до 5,9. Смесь представляет собой полупрозрачную эмульсию белого цвета. Ее доводят до состояния с рН=3,0 путем добавления 3,5 мл 1н. HCl и экстрагируют дважды посредством 75 мл тетрагидрофурана. Объединенные тетрагидрофурановые слои концентрируют на роторном испарителе и дополнительно сушат в вакууме с получением 1,81 г N-лауроилглицина с 114%-ным выходом (включает остаточную воду).
Пример 7. Окисление N-кокоилмоноэтаноламида (CMEA) посредством 3,25 экв. NaOCl с использованием толуола в качестве растворителя, кислотная обработка. Реакция окисления также происходит со смесями моноэтаноламидов, включая смеси моноэтаноламидов, полученных из смесей жирных кислот, встречающихся в природе. В этом примере, N-кокоилмоноэтаноламин (смесь моноэтаноламидов С-8, С-10, С-12 (основной компонент), С-14, С-16 и С-18) окисляют в аналогичных условиях с получением смеси соответствующих N-кокоилглицинов. В 6 мл воды растворяют KBr (33 мг, 4,5 мольных %) и АА-ТЕМРО (25 мг, 2 мольных %). CMEA (1,50 г, 6,16 ммоль при условии 100% LG) растворяют в 60 мл толуола при 32°С и раствор добавляют к KBr и AA-TEMPO. Перемешиваемую смесь поддерживают при 31-37°С в ходе добавления и в период выдержки. Раствор гипохлорита натрия (11 мл 11,5%-ного водного раствора, 3,25 экв.) и 1,15 мл 2н. NaOH добавляют порциями по 2,0 мл в течение периода времени 50 минут. В конце добавления рН составляет 6,9. Через 40 минут при 33°С рН корректируют с доведением до 8,6 посредством 0,5 мл 1н. NaOH. Перемешивание продолжают в течение дополнительных 3,5 часов и затем раствору дают остыть до комнатной температуры.
Гелеобразную реакционную смесь разбавляют посредством 70 мл THF и доводят до состояния с рН=2,2 посредством 5,5 мл 1н. HCl. Слои разделяют и водный слой экстрагируют посредством дополнительных 20 мл THF. Объединенные тетрагидрофурановые слои концентрируют на роторном испарителе и сушат в вакууме с получением N-кокоилглицина с 84%-ным выходом.
Пример 8. Окисление N-кокоилмоноэтаноламида (CMEA) посредством 3,25 экв. NaOCl c использованием циклогексана в качестве растворителя, кислотная обработка. Следуют методике Примера 4, за исключением того, что CMEA растворяют в 120 мл циклогексана при 43°С вместо толуола. По завершении реакции получают полупрозрачную эмульсию (рН=6,2). Эмульсию доводят до состояния с рН=3,1 посредством 3,0 мл 1н. HCl и экстрагируют дважды посредством 100 мл THF. Объединенные тетрагидрофурановые слои концентрируют на роторном испарителе и сушат в вакууме с получением N-кокоилглицина с 73%-ным выходом.
Сравнительный Пример
Для того чтобы показать необходимость правильного растворителя, авторы изобретения проводят эксперимент, в котором сравнивают реакцию получения N-лауроил-моноэтаноламина, изложенную в Примере 5 в японской заявке на патент: Japanese Patent Application 10/087554 (Приписываемой компании Lion Corporation), с реакцией своего изобретения. Результаты изложены ниже.
NaOCl
Краткое изложение Примеров окисления
В таблице ниже изложено краткое изложение Примеров окисления LMEA до LG.
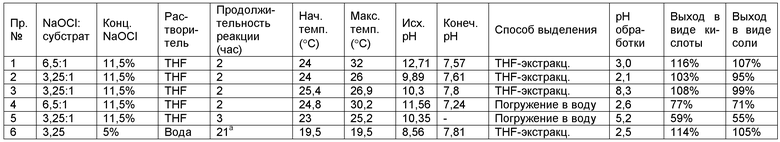
а) Истинная продолжительность реакции может быть меньше.
В таблице ниже изложено краткое изложение Примеров окисления N-кокоилметаноламида до N-кокоилглицина.

а) Истинная продолжительность реакции может быть меньше.
b) Полагают, что CMEA и CG представляют собой, каждый, соответствующие 100% С-12 соединения.
Пример 9. Окисление CMEA посредством 1,6-2,5 экв. NaOCl в воде.
CMEA (6 г) суспендируют в воде (150 мл) и перемешивают в течение 30 минут при высоких скоростях с использованием механической мешалки. АА-TEMPO (0,101 г, 0,02 экв.) и KBr (0,113 г, 0,04 экв.) добавляют в реакционную смесь с последующим введением по каплям NaOCl (10-14%, 1,6~2,5 экв.) при энергичном перемешивании с поддерживанием температуры между 22 и 33°С. Между добавлениями рН поддерживают около 8-9 путем введения 1н. NaOH. Протекание реакции внимательно контролируют посредством HPLC, и оно продолжается в течение 24 часов до тех пор, пока не прекратится расходование CMEA. На этой стадии реакционную смесь нагревают в течение 6 часов при 65°С для гарантированного обеспечения полного превращения в кокоилглицинатный продукт. Смесь подкисляют до рН 3 посредством 1н. HCl и отфильтровывают твердый продукт белого цвета и сушат в высоком вакууме с получением 5,45 г. Типичный реакционный профиль, полученный посредством HPLC, демонстрирующий превращение CMEA в кокоилглицинат, показан на фигуре 2 ниже.
Пример 10. Окисление CMEA посредством 2,3-3,2 экв. NaOCl в воде.
CMEA (6 г) суспендируют в воде (150 мл) и перемешивают в течение 30 минут при высоких скоростях с использованием механической мешалки. АА-TEMPO (0,101 г, 0,02 экв.) и KBr (0,113 г, 0,04 экв.) добавляют к реакционной смеси с последующим введением по каплям NaOCl (10-14%, 2,3~3,2 экв.) при энергичном перемешивании с поддерживанием температуры между 22 и 33°С. Между добавлениями рН поддерживают около 8-9 путем добавления 1н. NaOH. Протекание реакции внимательно контролируют посредством HPLC, и оно продолжается в течение 24 часов до тех пор, пока не прекратится расходование CMEA. На этой стадии реакционную смесь нагревают в течение 8 часов при 60°С для гарантированного обеспечения полного превращения в кокоилглицинатный продукт. Смесь подкисляют до рН 3 посредством 1н. HCl, отфильтровывают твердый продукт белого цвета и сушат в высоком вакууме с получением 5,75 г. Типичный реакционный профиль, полученный посредством HPLC, демонстрирующий превращение CMEA в кокоилглицинат, показан на фигуре 3 ниже.
Пример 11. Окисление CMEA посредством 3,2-4,0 экв. NaOCl в воде.
CMEA (6 г) суспендируют в воде (150 мл) и перемешивают в течение 30 минут при высоких скоростях с использованием механической мешалки. АА-TEMPO (0,101 г, 0,02 экв.) и KBr (0,113 г, 0,04 экв.) добавляют к реакционной смеси с последующим введением по каплям NaOCl (10-14%, 3,2~4,0 экв.) при энергичном перемешивании с поддерживанием температуры между 22 и 26°С. Между добавлениями рН поддерживают около 8-9 путем добавления 1н. NaOH. Протекание реакции внимательно контролируют посредством HPLC, и оно продолжается в течение 24 часов до тех пор, пока не прекратится расходование CMEA. На этой стадии реакционную смесь нагревают в течение 8 часов при 60°С для гарантированного обеспечения полного превращения в кокоилглицинатный продукт. Смесь подкисляют до рН 3 посредством 1н. HCl, отфильтровывают твердый продукт белого цвета и сушат в высоком вакууме с получением 5,50 г. Типичный реакционный профиль, полученный посредством HPLC, демонстрирующий превращение CMEA в кокоилглицинат, показан на фигуре 4 ниже.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОЛУЧЕНИЯ ФТОРИРОВАННЫХ КАРБОНОВЫХ КИСЛОТ И ИХ СОЛЕЙ | 2010 |
|
RU2545172C2 |
| ПРЕВРАЩЕНИЕ СПИРТОВ В ЛИНЕЙНЫЕ И РАЗВЕТВЛЕННЫЕ ФУНКЦИОНАЛИЗИРОВАННЫЕ АЛКАНЫ | 2017 |
|
RU2732124C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОКИСЛЕННОГО ПРОИЗВОДНОГО ГИАЛУРОНОВОЙ КИСЛОТЫ И СПОСОБ ЕГО МОДИФИКАЦИИ | 2010 |
|
RU2559447C2 |
| ПРОИЗВОДНОЕ ГИАЛУРОНОВОЙ КИСЛОТЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ЕГО МОДИФИКАЦИИ | 2010 |
|
RU2550602C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОСНОВАННЫХ НА N-АЦИЛАМИНОКИСЛОТЕ ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ С ПРИМЕНЕНИЕМ ОСНОВАННЫХ НА N-АЦИЛАМИНОКИСЛОТЕ ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ ИЛИ СООТВЕТСТВУЮЩИХ АНГИДРИДОВ В КАЧЕСТВЕ КАТАЛИЗАТОРОВ | 2012 |
|
RU2624026C2 |
| ПРОИЗВОДНЫЕ СУЛЬФАТИРОВАННЫХ ПОЛИСАХАРИДОВ, ИХ СПОСОБ ПОЛУЧЕНИЯ, МОДИФИКАЦИЯ И ПРИМЕНЕНИЕ | 2016 |
|
RU2708327C2 |
| СПОСОБ СЕЛЕКТИВНОГО ОКИСЛЕНИЯ 5-ГИДРОКСИМЕТИЛФУРФУРОЛА | 2015 |
|
RU2703511C2 |
| НЕНАСЫЩЕННЫЕ ПРОИЗВОДНЫЕ ПОЛИСАХАРИДОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2017 |
|
RU2725500C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЦИКЛИДЕНОВОГО АЦЕТАМИДНОГО ПРОИЗВОДНОГО | 2020 |
|
RU2813203C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОКИСЛЕННЫХ ДРЕВЕСНЫХ ПРОДУКТОВ | 2020 |
|
RU2809043C2 |
Изобретение относится к усовершенствованному способу для окисления первичного амидоспирта до соответствующей амидокарбоновой кислоты с высоким выходом. Способ превращения спирта, содержащего амидную группу, в соответствующую карбоновую кислоту заключается в реакционном взаимодействии первичного амидоспирта формулы: R'-CONR2(CH2)mOH, где R1 представляет собой линейную или разветвленную алкильную или алкенильную группу, имеющую 7-22 атома углерода; R2 представляет собой Н, алкильную или гидроксиалкильную группу с 1-6 атомом(ами) углерода; и m представляет собой целое число от 1 до 6, с окислителем в присутствии пространственно затрудненной радикальной группы и сокатализатора, где растворитель представляет собой только воду, и в присутствии окислителя первичный амидоспирт остается в твердой органической фазе, в то время как окислитель распределяется в основном в жидкую фазу, причем к реакционной смеси дополнительно добавляют основание. 9 з.п. ф-лы, 12 ил., 11 пр.
1. Способ превращения спирта, содержащего амидную группу, в соответствующую карбоновую кислоту, включающий реакционное взаимодействие первичного амидоспирта формулы:
R1-CONR2(CH2)mOH,
где R1 представляет собой линейную или разветвленную алкильную или алкенильную группу, имеющую 7-22 атома углерода; R2 представляет собой Н, алкильную или гидроксиалкильную группу с 1-6 атомом(ами) углерода; и m представляет собой целое число от 1 до 6, с окислителем в присутствии пространственно затрудненной радикальной группы и сокатализатора, где растворитель представляет собой только воду, и в присутствии окислителя первичный амидоспирт остается в твердой органической фазе, в то время как окислитель распределяется в основном в жидкую фазу, причем к реакционной смеси дополнительно добавляют основание.
2. Способ по п.1, где спирт, содержащий амидную группу, представляет собой алканоилмоноалканоламин.
3. Способ по п.2, где алканоилмоноалканоламин представляет собой лауроил-моноалканоламид или кокомоноэтаноламид.
4. Способ по п.1, где окислитель выбирают из группы, состоящей из хлора, гипохлорита, хлоризоциануровой кислоты и их смесей.
5. Способ по п.1, где окислитель выбирают из группы, состоящей из NaOBr, бромсукцинимида, бромизоциануровой кислоты, перкислот, оксона, Н2O2 и их смесей.
6. Способ по п.1, где окислитель присутствует в количестве от эквимолярного до 8-молярного.
7. Способ по п.1, где нитроксидный катализатор представляет собой 4-ацетамидо-ТЕМРО.
8. Способ по п.1, где сокатализатор представляет собой галогенид щелочного металла или галогенид щелочноземельного металла.
9. Способ по п.1, где добавляют основание, достаточное для поддержания рН выше 6.
10. Способ по п.1, где окислитель представляет собой хлор или хлорсодержащую молекулу, где окислитель добавляют, и реакции дают 30 мин-24 ч для достижения завершения, и, где получающийся в результате раствор нагревают в течение 1-24 ч при температуре, по меньшей мере, 40-100°С.
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| US 4016287 А, 05.04.1977 | |||
| NOOY DE А Е J ЕТ AL: "ON THE USE OF STABLE ORGANIC NITROXYL RADICALS FOR THE OXIDATION OF PRIMARY AND SECONDARY ALCOHOLS" SYNTHESIS, №10, 1996, p.1153-1174 | |||
| СПОСОБ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНЫХ ЭФИРОВ ЭРИТРО-3-АМИНО-2-ОКСИМАСЛЯНЫХ КИСЛОТ И СООТВЕТСТВУЮЩИХ ИСХОДНЫХ КИСЛОТ | 1997 |
|
RU2169138C2 |
| US 5506224 A, 09.04.1996. | |||

