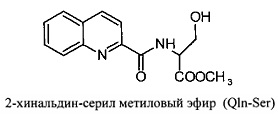
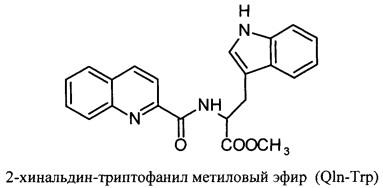
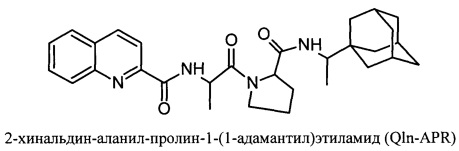
Изобретение относится к вирусологии и медицине, в нем представлены новые синтетические соединения, а именно: 2-хинальдин-серил метиловый эфир (Qln-Ser) и 2-хинальдин-триптофанил метиловый эфир (Qln-Trp) и 2-хинальдин-аланил-пролин-1(1-адамантил)этиламид (Qln-APR), которые могут быть использованы для создания новых противовирусных препаратов в виде индивидуального лекарства и/или в виде композиции.
Серьезную опасность для человечества представляют нетипичные для человека вирусы гриппа птиц: A(H5N1), A(H7N9), A(H7N7), A(H7N3), A(H9N2). В настоящее время большую опасность представляют A(H5N1), A(H7N9) и A(H3N2)v, которые характеризуются высокой летальностью для человека. При определенных условиях вирусы гриппа A(H5N1), A(H7N9) могут приобрести способность передаваться от человека к человеку и вызвать чрезвычайную пандемическую ситуацию.
Существует три основных серотипа вируса гриппа: А, В и С. Мембрана вириона вируса гриппа А содержит большое число равномерно расположенных гликопротеинов: гемагглютинина (НА) и нейраминидазы (NA), в соотношении приблизительно четыре к одному, соответственно [1]. Мембрана также содержит матричный белок 2 (М2 или АМ2 для гриппа А), хотя его значительно меньше чем гемагглютинина (1:10-100 М2:НА) [3]. Вирусный нуклеокапсид состоит из восьми отдельных сегментов одноцепочечной (-)РНК замкнутых в кольца. Шесть из этих сегментов РНК (1-6) кодируют по одному вирусному белку, а два сегмента (7 и 8) кодируют по два белка [2]. Белок М2 кодируется седьмым сегментом РНК вместе с матричным белком 1 (M1). Рибонуклеопротеидная (РНП) и липидная оболочки предположительно связаны между собой через взаимодействие с белком M1. В отличие от вириона вируса гриппа А, вирион вируса гриппа В имеет четыре мембранных белка: НА, NA, ВМ2 (белок М2, вируса гриппа В), и NB, а вирион вируса гриппа С содержит только два мембранных «шипа»: СМ2 (белок М2, вируса гриппа С) и гликопротеин gp88, который имеет комбинированную функцию: НА и NA (нейраминат-О-ацетил-эстеразы (гликопептид HEF)) [2]. Соответственно вирус гриппа С распознается другим клеточным рецептором - мукопептидом, содержащим ацетил-9-O-ацетилнейраминовую кислоту. Это обстоятельство обусловливает отсутствие конкуренции на стадии адсорбции между вирусом типа С и вирусами других типов.
Белок М2 образует достаточно специфичные ионные каналы, специализирующиеся на транспорте ионов водорода (протонов). Наиболее важную роль в транспорте протонов через канал в М2, играют аминокислотные остатки гистидина (Н37) и триптофана (W41) в трансмембранном (ТМ) домене. Ранее для канала АМ2 показано, что Н37 выполняет важную роль в протонной избирательности и рН-регуляции [3, 4].
Канал М2 представляет собой мишень, на которую направлено действие известных препаратов против гриппа, таких как Амантадин (1-аминоадамантан гидрохлорид) и Ремантадин (1-(1-адамантил)-этиламин гидрохлорид). В последние годы они в значительной мере потеряли свою противогриппозную активность, что явилось следствием изменения канала М2 на молекулярном уровне, и это делает вирус устойчивым к действию обоих препаратов. В результате секвенирования трансмембранной области белка М2 вируса гриппа А было установлено, что штаммы, резистентные к препаратам адамантана, содержат аминокислотную замену в основном в положении 31 серина на аспарагин (S31N). В настоящее время среди адамантан-резистентных вирусов гриппа А, изолированных в минувшее десятилетие, 98% содержат эту замену. Она стала одним из сигналов к тому, что штамм устойчив к действию амантадина и ремантадина [5].
Один из способов восстановления противовирусных свойств соединений адамантана - это обеспечение их дополнительными функционально активными группами, которые в процессе взаимодействия с трансмембранным доменом были бы способны нарушать процесс транспорта протонов через мембрану вируса. Источником таких функционально активных групп могут являться аминокислотные остатки, а также ряд других физиологически активных соединений, введенных в адамантановый карбоцикл методами пептидного синтеза [6].
Ингибиторы функции белка М2, как правило, состоят из гидрофобной части молекулы, соединенной с полярной функциональной группой, которая, как правило, положительно заряжена. В амантадине или ремантадине гидрофобная часть представлена адамантаном, а заместитель представлен амино- или этил аминогруппой. Адамантильный остаток может быть заменен на другие гидрофобные группы, в том числе сопряженные и спиро-сопряженные мультициклические алканы, разветвленные ациклические алканы и силаны [7, 8]. Эти соединения показали высокую активность в отношении дикого типа, а некоторые были весьма активны в отношении мутантов V27A и L26F [8, 9]. Однако ни одно из этих производных не показало противовирусной активности в отношении мутанта S31N, превосходящей активность амантадина.
Хинолиновые молекулы находят применение в медицине, сельском хозяйстве и фармацевтике. Известно производное изохинолина с аминокислотой аспарагин, которое проявляет ингибирующий эффект на вирусные ферменты. Они ингибируют протеазы вирусного начала, и их можно использовать для профилактики или лечения вирусных инфекций, в частности заболеваний, вызываемых вирусом ВИЧ и ретроидными вирусами [10]. Известно пептидное производное аспарагил-пролина с хинолинкарбоновой кислотой, которое также обладает ингибирующим действием в отношении протез производимых ВИЧ [11].
В качестве гидрофобного компонента предлагаемых соединений была выбрана 2-хинальдинкарбоновая кислота. Эта конденсированная ароматическая система помимо гидрофобных свойств обладает повышенной электронной плотностью, что может быть важно при образовании нековалентных взаимодействий с белками вируса. Использование именно 2-хинольдинкарбоновой кислоты, отчасти обусловлено ее синтетической и экономической доступностью в условиях современного производства, что немаловажно для создания фармацевтической композиции в будущем.
В качестве полярной функциональной группы в конструкцию молекул предлагаемых соединений были выбраны эфиры аминокистот: метиловый эфир серина и метиловый эфир триптофана, а также амид пептида. В структуре дипептидного соединения сложноэфирная группировка заменена на амид 1-адамантилэтиламина.
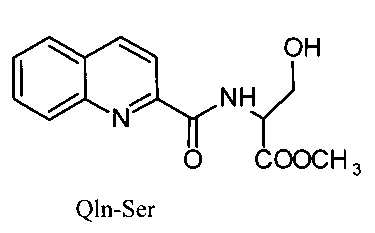
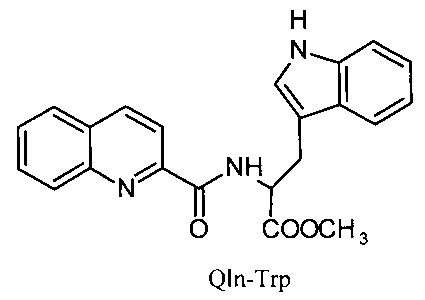
Сущность изобретения заключается в создании новых синтетических соединений, являющихся производными 2-хинальдинкарбоновой кислоты, а именно: 2-хинальдин-серил метилового эфира (Qln-Ser) и 2-хинальдин-триптофанил метилового эфира (Qln-Trp) и 2-хинальдин-аланил-пролин-1(1-адамантил)этиламида (Qln-APR), которые ингибируют репродукцию патогенных штаммов вируса гриппа A/H1N1pdm2009 и A/H5N1, резистентных к действию ремантадина, а также обладают вирулицидным действием по отношению к штамму вируса гриппа A/H5N1. Более того, предлагаемые соединения обладают меньшим токсическим эффектом на монослой клеток Madin Darby Canine Kidney (MDCK) и Vero-E6, чем римантадина гидрохлорид. Соединения Qln-Ser, Qln-Trp и Qln-APR имеют следующие структурные формулы:
Технический результат - получены новые соединения: аминокислотные производные 2-хинальдинкарбоновой кислоты, малотоксичные, обладающие противогриппозной, в том числе вирулицидной, активностью и действующие на штаммы вируса гриппа А, резистентные к действию препаратов ремантадина и амантадина.
Краткое описание чертежей
Для более ясного понимания сути заявленного изобретения, которое отражено в формуле изобретения, а также для демонстрации ее особенностей и преимуществ далее приводится подробное описание со ссылками на чертежи.
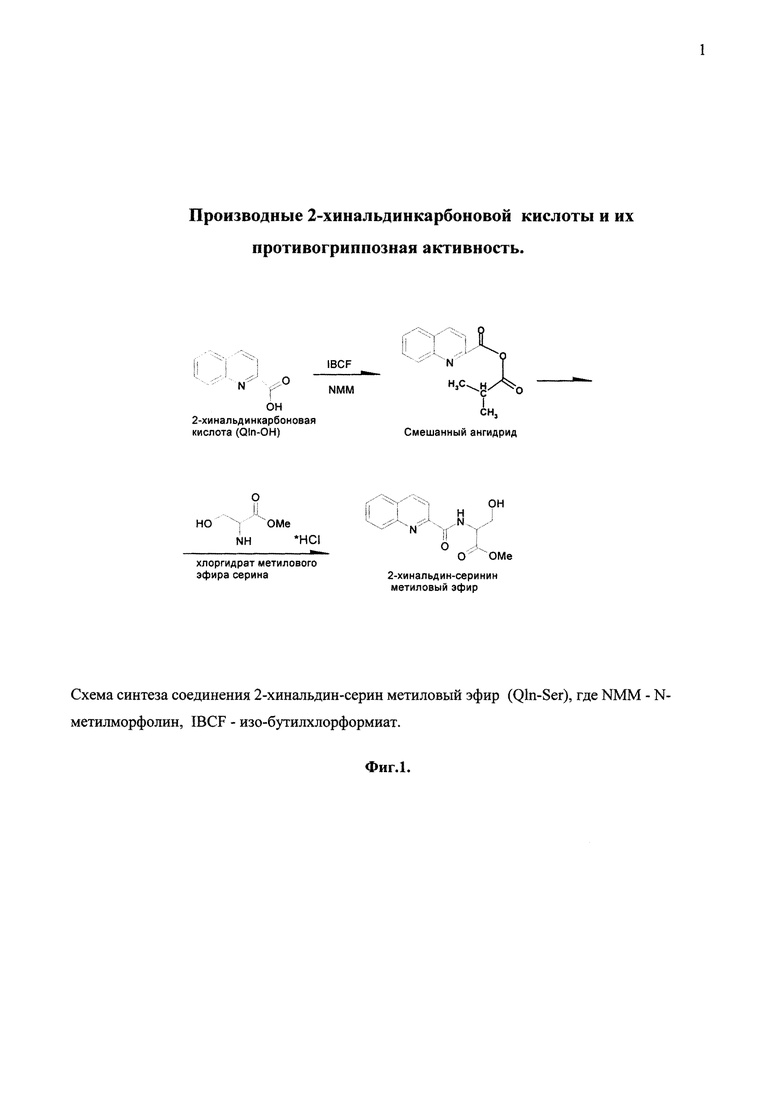
На фиг. 1 представлена схема синтеза соединения Qln-Ser методом смешанных ангидридов.
На фиг. 2а представлены спектральные данные соединения Qln-Ser
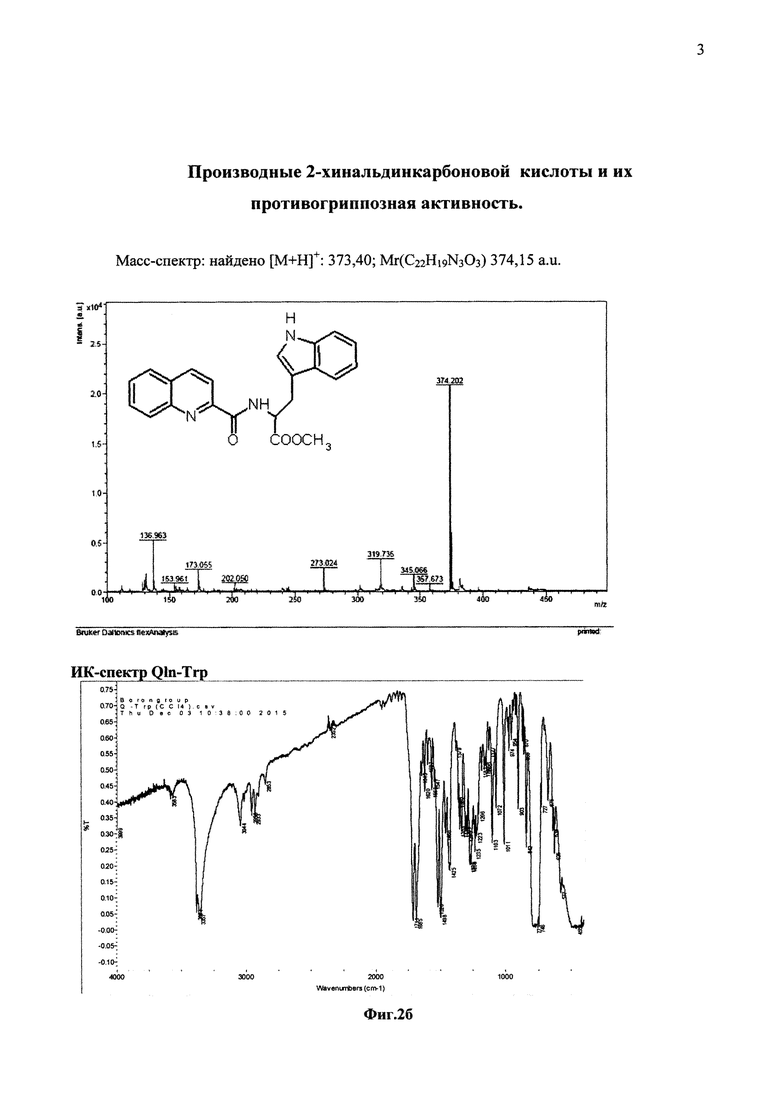
На фиг. 2б представлены спектральные данные соединения Qln-Trp
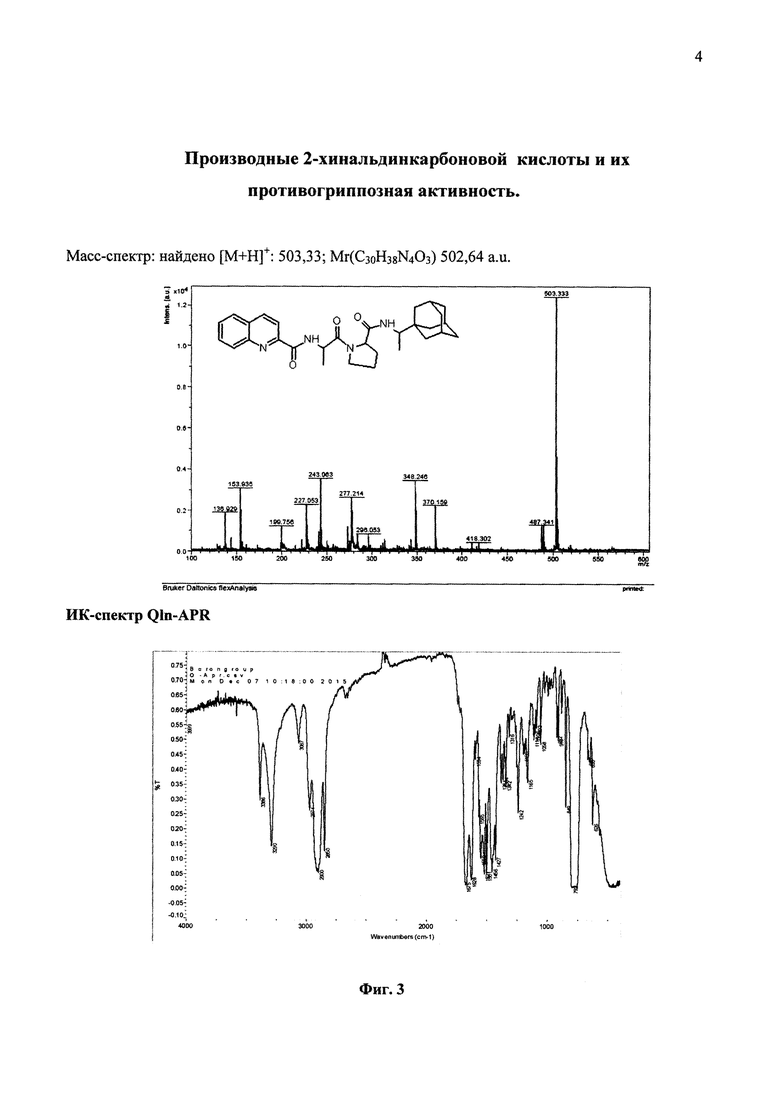
На фиг. 3 представлены спектральные данные соединения Qln-APR
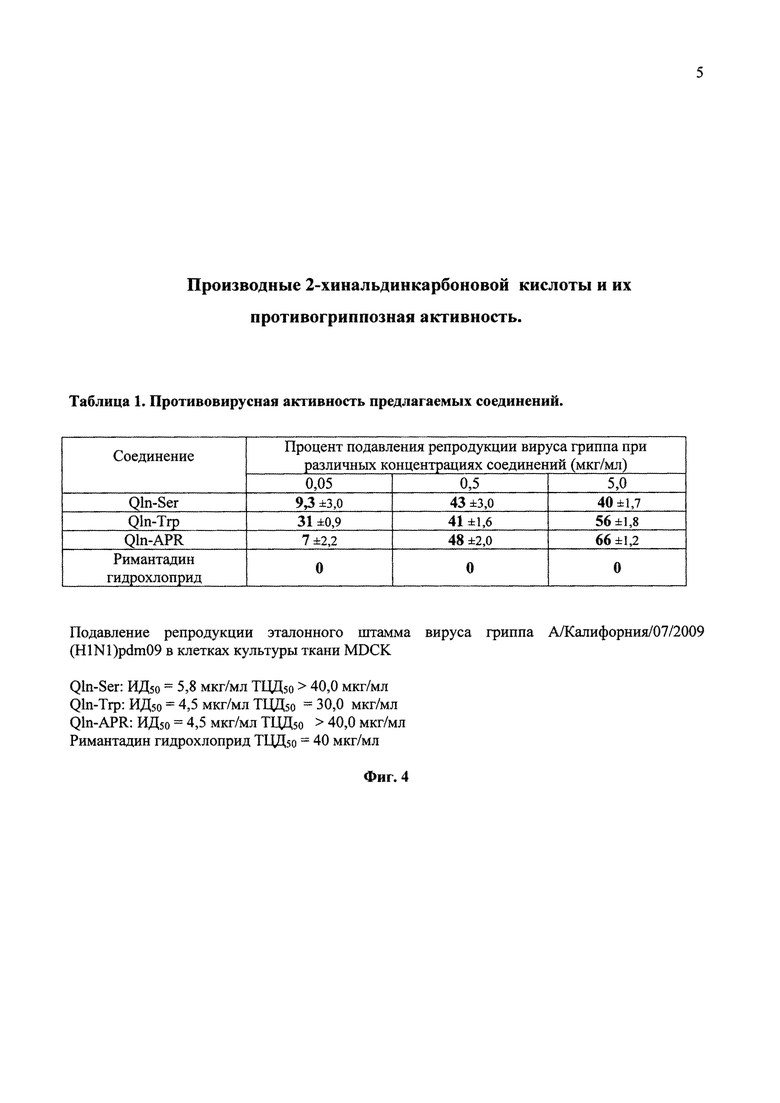
На фиг. 4 в виде таблицы 1 представлены данные влияния различных концентраций соединений Qln-Ser, Qln-Trp и Qln-APR на репродукцию пандемического штамма вируса гриппа А/Калифорния/07/2009(H1N1)рdm09 в культуре клеток Madin Darby Canine Kidney (MDCK) при добавлении вещества одномоментно с вирусом. Испытания противовирусной активности соединений Qln-Ser, Qln-Trp и Qln-APR проведены в сравнении с ремантадином.
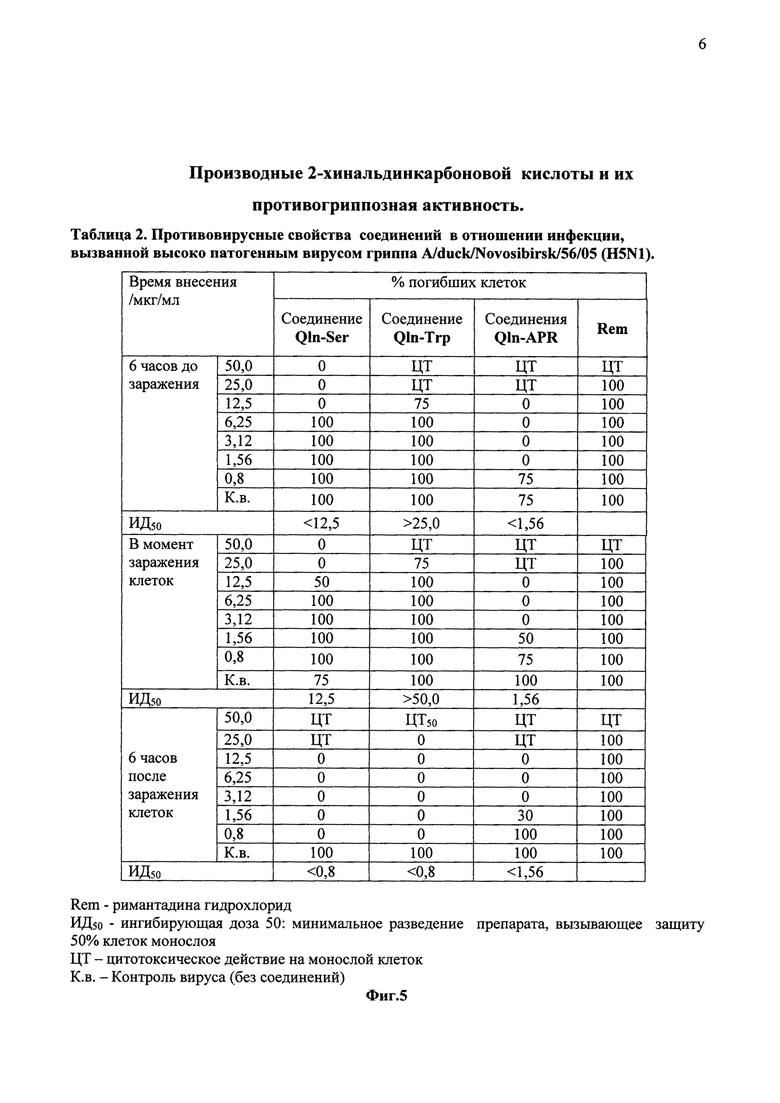
На фиг. 5 в виде таблицы 2 представлены данные противовирусных свойств соединений Qln-Ser, Qln-Trp и Qln-APR в отношении инфекции, вызванной высоко патогенным вирусом гриппа A/duck/Novosibirsk/56/05 (H5N1)
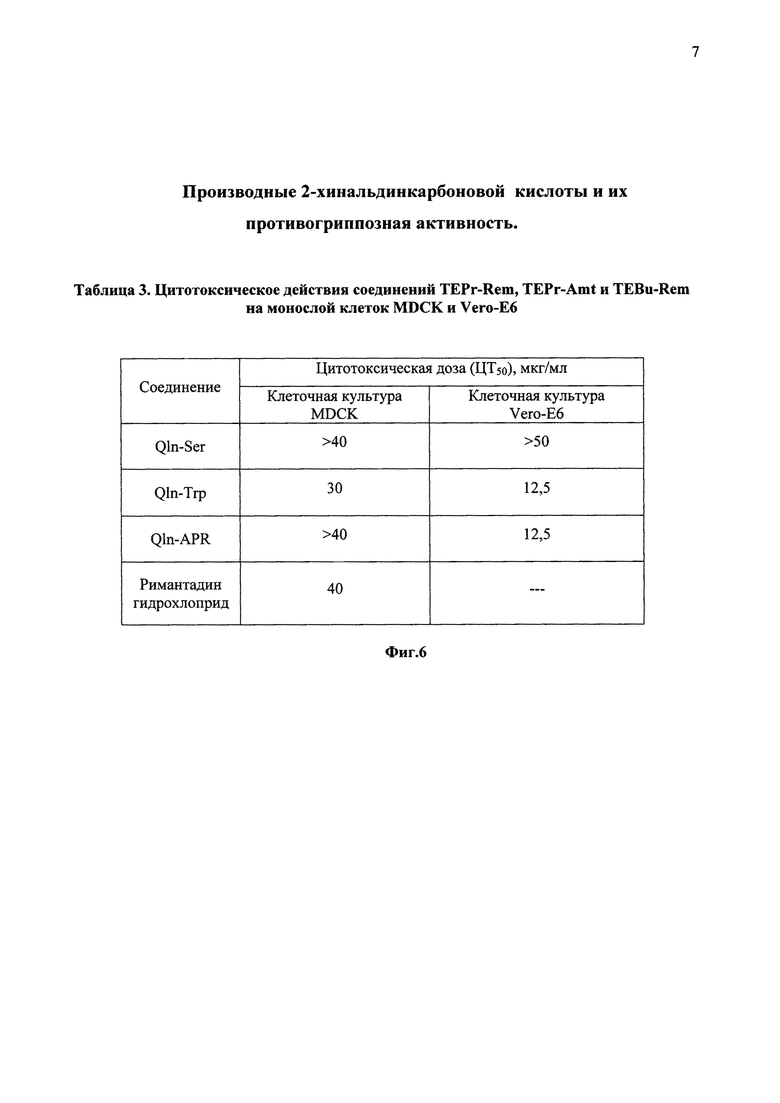
На фиг. 6 в виде таблицы 3 представлены данные цитотоксического действия на культуры клеток MDCK и Vero-E6.
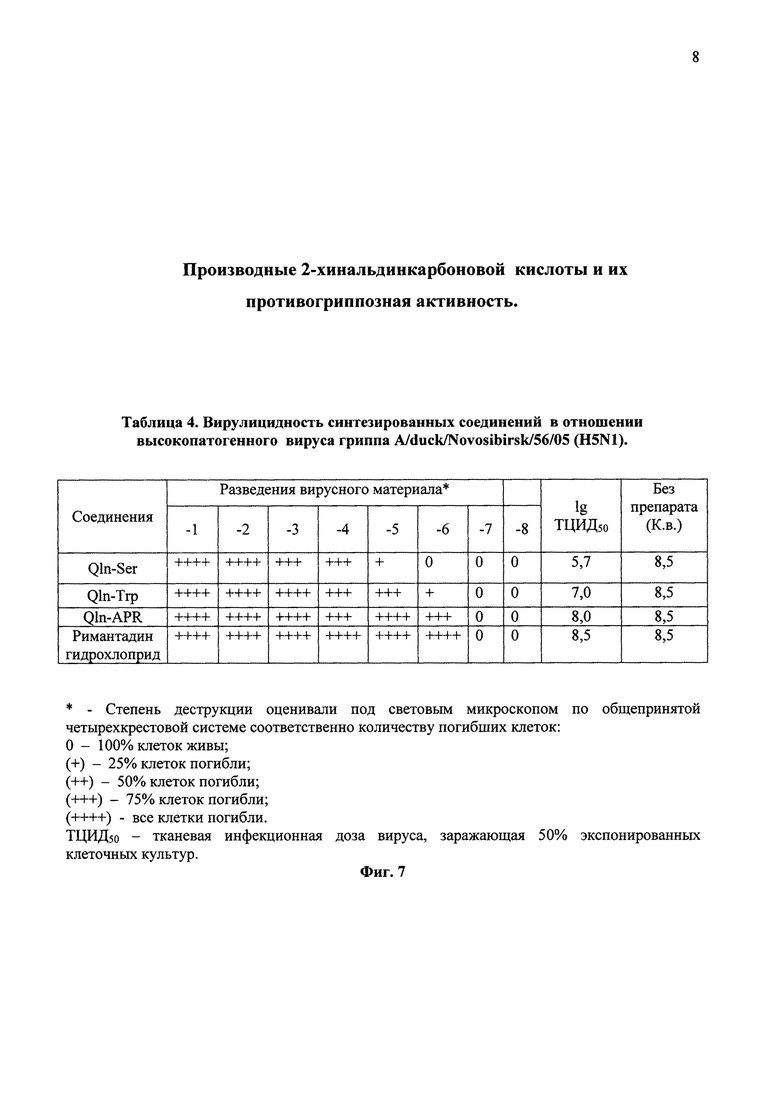
На фиг. 7 в виде таблицы 4 представлены данные вирулицидной активности синтезированных соединений в отношении высокопатогенного вируса гриппа A/duck/Novosibirsk/56/05 (H5N1).
Для получения соединений Qln-Ser, Qln-Trp и Qln-APR могут быть использованы различные подходы. Один из способов получения соединений Qln-Ser, Qln-Trp и Qln-APR - метод смешанных ангидридов (Фиг. 1). Однако предложенный метод синтеза не должен рассматриваться как некое ограничение объема настоящего изобретения во всех отношениях.
Образование пептидной связи между 2-хинольдинкарбоновой кислотой и аминокислотами проводили в одну стадию в эквимолярном соотношении.
При синтезе соединений использовали 2-хинальдинкарбоновую кислоту фирмы Sigma-Aldrich (США), L-аминокислоты фирм Sigma-Aldrich (США) и Nova Biochem (США), рацемический римантадин гидрохлорид фирмы Zhejiang Kangyu Pharmaceutical Со (Китай). Использовали изо-бутилхлорформиат (IBCF) фирмы «Fluka» (Швейцария), N-метилморфолин (NMM) фирмы Sigma-Aldrich (США). Все используемые для конденсации и удаления защитных групп растворители предварительно абсолютировали и перегоняли по стандартным методикам. Идентификация полученных соединений осуществлялась с помощью тонкослойной хроматографии (ТСХ) на пластинах Silufol (Чехия) в системах: метанол-хлороформ, 13:60 (А), втор-бутанол - 3%-ный аммиак, 100:44 (В), н-бутанол - уксусная кислота - вода - пиридин, 30:3:12:10 (С), позволяющих констатировать полное отсутствие в испытуемых образцах следов римантадина гидрохлорида и 2-хинальдинкарбоновой кислоты. Молекулярный вес был установлен на MALDI-TOF-времяпролетном масс-спектрометре Bruker UltraFlex II с программным обеспечением для сбора и обработки масс-спектров flexControl 1.1. и flexAnalys 2.2. Инфракрасные спектры получены на ИК Фурье спектрометре ИнфраЛЮМ ФТ-10. Температуру плавления полученных соединений измеряли на цифровом приборе для определения точки плавления SMP20 Stuart Scientific.
Настоящее изобретение проиллюстрировано нижеследующими примерами. Однако эти примеры не должны рассматриваться как некое ограничение объема настоящего изобретения во всех отношениях.
Пример 1. Получение Qln-Ser (2-хинальдин-серил метиловый эфир).
Схема синтеза соединения 2-хинальдин-серин метилового эфира (Qln-Ser) представлена на фиг. 1.
Qln-OH (2-хиналъдинкарбоповая кислота) была получена от фирмы Sigma-Aldrich (США). Чистота 98.0%, температура плавления 156-158°С. Коммерческая Qln-OH была использована в синтезе конечного соединения.
В трехгорлую колбу, снабженную механической мешалкой, термометром и хлоркальцевой трубкой, вносят 1,5 г (8,66 мМ) Qln-OH в смеси 7,0 мл СНCH3 с 15,0 мл тетрагидрофурана и прибавляют 1,0 мл (8,66 мМ) N-метилморфолина (NMM). Охлаждают до -25°С и при перемешивании в реакционную массу добавляют 1,3 мл (8,66 мМ) изо-бутилхлорформиата (IBCF). Перемешивают 10 мин. Затем добавляют заранее приготовленный и охлажденный до -20°С раствор 1,35 г (8,66 мМ) хлоргидрата метилового эфира серина в 10 мл СНCl3 с 1,0 мл (8,66 мМ) NMM. Перемешивают 30 мин при -15°С, затем еще 1 час при 0°С и 10 часов при комнатной температуре.
Растворители удаляют на роторном испарителе при 45°С и 15 мм рт. ст. Остаток растворяют в 35,0 мл этилацетата и 10,0 мл Н2О. Раствор переносят в делительную воронку и последовательно промывают 0,5 н серной кислотой (4,0 мл × 1), 0,5 н. KНСО3 (10,0 мл × 2), затем подкисляют 1н. H2SO4 и промывают Н2О (5,0 мл × 1). Органический слой отделяют и сушат безводным Na2SO4. Этилацетат удаляют в вакууме, получают вспененное масло, которое через некоторое время твердеет.
Выход: сероватые кристаллы, 2,07 г (78%), твердый, аморфный, [α]D20 = +112°, Rf = 0,84(А); Rf = 0,86(B); Rf = 0,75 (С).
Данные ИК-спектра продукта реакции. Образец приготовлен в виде суспензии в CCl4 v(OH) кислоты ~3300 см-1; v(NH) 3364 см-1; v(CH) СН3-группы 2970, 2955, 2890 см-1; v(C=O) 1739, 1653 см-1; v(циклов) 1620-700 см-1.
Об образовании серил метилового эфира исходной хинальдинкарбоновой кислоты свидетельствует наличие в спектре интенсивной узкой полосы валентных колебаний v(NH) при 3364 см-1 на фоне уширенной полосы средней интенсивности v(OH) гидроксогруппы около ~3300 см-1. Наличие эфирной СН3-группы подтверждается проявлением в спектре полос валентных колебаний v(CH) при 2970, 2955, 2890 см-1. В спектре также присутствуют две полосы v(C=O), соответствующие двум типам карбоксилатных групп, при 1739, 1653 см-1.
Масс-спектр: найдено [М+Н]+: 275,10; Mr(C14H14N2O4) 274,27 atomic unit (a.u.).
Соединение Qln-Trp (2-хинальдин-триптофанил метиловый эфир) было получено аналогичным образом с использованием в синтезе метилового эфира триптофана вместо гидрохлорида метилового эфира серина.
Выход: аморфные хлопья, 2,02 г (98%), [α]D20 = -20°, Rf = 0.95(A); Rf = 0,93(B); Rf = 0,92 (С).
Данные ИК-спектра продукта реакции. Образец приготовлен в виде суспензии в CCl4 v(NH) 3381, 3357 см-1; v(CH) СН3-группы 2958, 2933, 2853 см-1; v(C=O)1711, 1685 см-1; v(циклов) 1620-700 см-1.
Наличию двух типов NH-групп в спектре соответствуют две полосы валентных колебаний v(NH) при 3381, 3357 см-1. Присутствие полос валентных колебаний v(CH) свидетельствует о наличии эфирной СН3-группы. Двум типам карбонильных групп соответствуют в спектре две полосы валентных колебаний v(C=O) при 1711, 1685 см-1.
Масс-спектр: найдено [М+Н]+: 373,40; Mr(C22H19N3O3) 374,15 a.u.
Пример 2. Получение Qln-APR (2-хинальдин-аланил-пролин-1-(1-адамантил)этиламида)
Хлоргидрат этилового эфира дипептида аланил-пролина (НCl* H-Ala-Pro-OEt) был получен классическим методом пептидного синтеза с использованием стратегии трет-бутилоксикарбонильной защиты аминогруппы.
В трехгорлую колбу, снабженную механической мешалкой, термометром и хлоркальцевой трубкой, вносят 0,69 г (3,98 мМ) Qln-OH в смеси 7,0 мл СНСl3 с 15,0 мл тетрагидрофурана и прибавляют 0,44 мл (3,98 мМ) NMM. Охлаждают до -25°С и при перемешивании в реакционную массу добавляют 0,52 мл (3,98 мМ) IBCF. Перемешивают 10 мин. Затем добавляют, заранее приготовленный и охлажденный до -20°С раствор 1,0 г (3,98 мМ) НCl* H-Ala-Pro-OEt в 10,0 мл СНСl3 с 0,44 мл (3,98 мМ) NMM. Перемешивают 30 мин при -15°С, затем еще 1 час при 0°С и 10 часов при комнатной температуре.
Растворители удаляют на роторном испарителе при 50°С и 15 мм рт. ст. Остаток растворяют в 30,0 мл этилацетата и 10,0 мл Н2О. Раствор переносят в делительную воронку и последовательно промывают, 0,5н. серной кислотой (7,0 мл × 1), 0,5н. KНСО3 (10,0 мл × 2), затем подкисляют 1н. H2SO4 и промывают Н2О (5,0 мл × 1). Органический слой отделяют и сушат безводным Na2SO4. Этилацетат удаляют в вакууме, получают вязкое масло.
Выход: прозрачное желтое масло, 1,4 г (~95%), [α]D20 = +54°, Rf 0,94(А); Rf 0,89(B); Rf 0,86(С).
Полученный таким образом эфир N-хинальдин-дипептида (Qln-Ala-Pro-OEt) впоследствии подвергался омылению в смеси 0,2н. NaOH - Ацетон (1:1).
К раствору 1,4 г (3,79 мМ) Qln-Ala-Pro-OEt в 12,0 мл ацетона прибавляют 16,0 мл (4,0 мМ) 0,25н. NaOH. Выдерживают реакционную смесь при 20°С в течение 45 мин. Ацетон удаляют в вакууме, водную фракцию подкисляют 10% лимонной кислотой до рН 4, выпадает маслянистый осадок. Переносят в делительную воронку и вносят 20,0 мл этилацетата, перемешивают до образования двух прозрачных слоев, водный слой отделяют и промывают водой (2×4 мл). Водный слой вновь отделяют, органический сушат безводным Na2SO4. Этилацетат удаляют в вакууме, выпадают белые кристаллы, которые многократно промывают водой.
Выход: белые кристаллы, 0,52 г (40%), Тпл.=126-128°С, [α]D20 = +23°, Rf = 0,59(А); Rf = 0,47(B); Rf = 0,59(C).
Омыленный продукт Qln-Ala-Pro-OH участвовал в конденсации с хлоргидратом 1-(1-адамантил)этиламином (НСl *Rem) в условиях реакции смешанных ангидридов.
В трехгорлую колбу, снабженную механической мешалкой, термометром и хлоркальцевой трубкой, вносят 0,5 г (1,47 мМ) Qln-OH в 15,0 мл СНСl3 и прибавляют 0,16 мл (1,47 мМ) NMM. Охлаждают до -25°С и при перемешивании в реакционную массу добавляют 0,19 мл (1,47 мМ) IBCF. Перемешивают 10 мин. Затем добавляют заранее приготовленный и охлажденный до -18°С раствор 0,32 г (1,47 мМ) НСl *Rem в 10,0 мл СНСl3 с 0,16 мл (1,47 мМ) NMM. Перемешивают 30 мин при -15°С, затем еще 1 час при 0°С и 10 часов при комнатной температуре.
Растворители удаляют на роторном испарителе при 50°С и 15 мм рт. ст. Остаток растворяют в 25,0 мл этилацетата и 10,0 мл Н2O. Раствор переносят в делительную воронку и последовательно промывают, 0,5н. серной кислотой (10,0 мл × 1), 0,5н. KНСО3 (10,0 мл × 2), затем промывают Н2O (5,0 мл × 1). Органический слой отделяют и сушат безводным Na2SO4. Этилацетат удаляют в вакууме, получают белые кристаллы
Выход: кристаллический, 0,66 г (90,4%), Тпл.=145-147°С, [α]D20 = +16°, Rf = 0,81(А); Rf = 0,92(B); Rf = 0,88(С).
Данные ИК-спектра продукта реакции (Qln-APR). Образец приготовлен в виде суспензии в ССl4. v(NH) 3386, 3290 см-1; v(CH) СН3-группы 2974, 2900, 2850 см-1; v(C=O)1675, 1628 см-1; v(циклов) 1620-700 см-1.
В спектре присутствуют две полосы v(NH) при 3386, 3290 см-1, что соответствует наличию двух имино-групп в составе сложного эфира. О замещении ОН-групп исходной кислоты на радикал замещенного амида свидетельствует наличие в спектре полос валентных колебаний v(CH) с максимумами при 2974, 2900, 2850 см-1. Наличию двух типов карбонильных групп соответствует присутствие в спектре двух полос v(C=O) при 1675, 1628 см-1.
Масс-спектр: найдено [М+Н]+: 503,33; Mr(С30Н38N4О3) 502,64 a.u.
Пример 3. Определение противовирусной активности предлагаемых соединений в отношении вируса гриппа А/-Калифорния/07/2009(H1N1)pdm09
Изучение противовирусной активности синтезированных соединений проводили на 96-луночных панелях со сформировавшимся монослоем клеток культуры ткани MDCK.
Одномоментно с инфицированием в монослой клеток вносили ремантадин и изучаемые синтетические соединения в концентрациях 0,05, 0,5 и 5,0 мкг/мл. Панели инкубировали 24 часа при 37°С, а затем останавливали реакцию фиксированием клеток 80% ацетоном на фосфатном буфере. Постановку метода клеточного иммуноферментного анализа (ИФА) проводили согласно методике, описанной ранее [12, 13]. Процент ингибирования вирусной активности соединениями определяли как отношение оптической плотности опытной лунки (с веществом) при 492 н.м. к оптической плотности клеточного контроля.
На фиг. 4 представлены средние значения результатов испытания противовирусной активности синтезированных соединений из параллельных опытов, проводимых в аналогичных условиях.
Результаты эксперимента показывают, что синтезированные соединения Qln-Ser, Qln-Trp и Qln-APR защищают клетки монослоя MDCK от цитопатического действия вируса. В условиях in vitro отмечено значительное ингибирование репродукции штамма вируса гриппа А/-Калифорния/07/2009(Н1N1)рdm09, устойчивого к действию ремантадина. Ингибирующая доза (ИД50) для соединения Qln-Ser составила 5,8 мкг/мл, а для Qln-Trp ИД50 составила 4,5 мкг/мл. Хинальдиновое производное дипептида (Qln-APR) не уступает Qln-Trp, ИД50 для Qln-APR составила также 4,5 мкг/мл. Отсутствие ингибирующего эффекта римантадина гидрохлорида косвенно свидетельствует о резистентности данного штамма к препаратам ремантадин и амантадин.
Пример 4. Определение противовирусной активности синтетических соединений в отношении вируса гриппа A/duck/Novosibirsk/56/05 (H5N1)
Изучение противовирусной активности синтезированных соединений проводили на панелях со сформировавшимся монослоем клеток Vero-Е6 (перевиваемая линия почки африканской зеленой мартышки). Противовирусную активность проверяли в трех схемах введения соединений в культуру клеток: за 6 ч до заражения клеток, в момент заражения и через 6 ч после заражения культур клеток. Соединения вносили в концентрациях 50, 25, 12,5, 6,25, 3,15, 1,56, и 0,8 мкг/мл. В качестве контроля (К.в.) использовали инфицированную культуру клеток без добавления соединений. Оценку цитотоксического действия определяли колориметрическим методом после инкубации клеток с синтезированными соединениями (Qln-Ser, Qln-Trp и Qln-APR) в течение 72 ч при 37°С.
Противовирусный эффект соединения оценивается по проценту жизнеспособных инфицированных клеток, путем сравнения интенсивности окрашивания раствора в контрольных и опытных лунках при добавлении нейтрального красного на автоматическом спектрофотометре при длине волны 450 нм (Фиг. 5).
Высоковирулентный штамм вируса гриппа A/duck/Novosibirsk/56/05 (H5N1) в большей или меньшей степени был чувствителен ко всем трем соединениям. Из соединений 2-хинальдинкарбоновой кислоты с эфирами аминокислот (Qln-Ser и Qln-Trp) несколько большим противовирусным эффектом обладает производное с остатком серина (Qln-Ser). Из данных таблицы 2 (Фиг. 5) видно, что аминокислотные производные хинальдиновой кислоты (Qln-Ser и Qln-Trp) имеют очень низкий уровень противовирусной активности в условиях профилактической схемы внесения, для соединения Qln-Ser ИД50 составило 12,5 мкг/мл и более, а для Qln-Trp ИД50 составляют еще большие значения. Однако оба соединения (Qln-Ser и Qln-Trp) проявляют значительный противовирусный эффект при внесении после заражения клеток, ИД50<0,08 мкг/мл. Производное 2-хинальдинкарбоновой кислоты с дипептид амидом (Qln-APR) эффективно во всех схемах внесения соединения: ИД50 составило 1,56 мкг/мл для одномоментного введения вещества с вирусом и менее 1,56 мкг/мл для схем до инфицирования и через 6 ч после инфицирования.
Пример 5. Исследование цитотоксического действия соединений Qln-Ser, Qln-Trp и Qln-APR на монослой клеток Vero-Е6 и MDCK
Оценку цитотоксического действия синтезированных соединений для клеток Vero-Е6 проводили по стандартной методике. Клеточную суспензию в ростовой среде (концентрация клеток - 4*105 клеток в 1 мл) вносили в 96-луночные пластиковые панели и через 24 ч после инкубации при 37°С монослой клеток дважды отмывали раствором Хенкса. Добавляли по 20,0 мкл соединений в различных концентрациях в среду поддержки, при этом общий объем жидкости в лунке составлял 200,0 мкл. Обработанные и необработанные таким образом культуры клеток инкубировали в течение 72 ч, после чего клетки, окрашенные метиленовым синим, подсчитывали, используя слайдный цитометр (Cauntess) фирмы Invitrogen (США). Рассчитывали цитотоксическую дозу 50 (ЦД50), которая соответствовала минимальной концентрации соединений, приводящей к гибели 50% клеток монослоя к 72 часам. Оценку цитотоксического действия синтезированных соединений для клеток MDCK проводили по методике, описанной нами ранее [13].
Для соединения Qln-Ser ЦД50 составила более 50 мкг/мл в культуре клеток Vero-E6 более 40 мкг/мл для клеточной культуры MDCK. Таким образом соединение Qln-Ser оказалось менее токсичным для обеих клеточных линий, чем римантадин гидрохлорид. Для соединений Qln-Trp и Qln-APR в культуре клеток Vero-Е6 ЦД50 составила 12,5 мкг/мл, что значительно меньше, чем для клеточной культуры MDCK, для которой этот показатель составил 30 и более 40 мкг/мл соответственно (фиг. 6).
Пример 6. Исследование вирулицидной активности соединений в отношении вируса гриппа A/duck/Novosibirsk/56/05 (H5N1)
Вирулицидная активность соединения связана с прямым инактивирующим действием на вирионы в составе вирусной популяции, в результате чего частично или полностью утрачивается инфекционная активность вируса. Чтобы проверить вирулицидные свойства соединения достаточно провести инкубацию смеси вируса и соединения в течение определенного времени, после чего проверить инфекционные свойства вируса без исследуемого соединения и вируса в смеси с веществом методом титрования в культурах клеток. Достоверное снижение инфекционной активности вируса на 1,0 и более логарифмов (lg) или ее полная утрата по сравнению с вирусом без вещества свидетельствует о проявлении вирулицидной активности исследуемого соединения. В опыте использованы концентрации соединений 10,0 мг/мл, которые смешивали с вирусом следующим образом: 200,0 мкл раствора соединения с добавлением 100,0 мкл вируссодержащего материала в исходной концентрации. Экспозиция с вируссодержащим материалом проведена при комнатной температуре в течение 20 мин, после чего титровали инфекционную активность вируса в каждом варианте опыта в культурах клеток Vero-Е6 при различных разведениях смеси соединений с вирусом. В качестве контроля (К.в.) использовали инфицированную культуру клеток без добавления соединений. По разнице титров вируса в контрольных и опытных экспериментах судили о вирулицидной активности соединения - его способности подавлять инфекционную активность вируса гриппа A/duck/Novosibirsk/56/05 (H5N1). Вирулицидная активность синтетических соединений проиллюстрирована в таблице 4 (Фиг. 7).
В результате была обнаружена умеренная вирулицидная активность для соединения Qln-Ser. Снижение инфекционного титра составило 2,8 логарифма (100-1000 раз) по отношению к вирусному контролю (1gТЦИД50/0,2=8,5). Соединение Qln-Trp практически не проявляло вирулицидных свойств, снижение инфекционного титра составило лишь 1,5 логарифма (10-100 раз) по отношению к вирусному контролю (lgТЦИД50/0,2=8,5). Вирулицидных свойств для соединения Qln-APR обнаружить не удалось, снижение инфекционного титра составило менее одного логарифма. Из полученных данных таблицы 4 (Фиг.7) можно сделать вывод, что дипептидное производное (Qln-APR) действует на вирусную частицу несколько иначе, чем аминокислотные производные (Qln-Ser и Qln-Trp) 2-хинальдинкарбоновой кислоты в силу стерических особенностей более крупной молекулы Qln-APR.
Предложенные соединения ингибируют репродукцию патогенных штаммов вируса гриппа A/H1N1pdm2009 и A/H5N1. Предлагаемые соединения также обладают вирулицидным действием по отношению к вирусным частицам гриппа A/H5N1, что доказывает прямое действие соединений на вирус. Предлагаемые соединения могут быть применены для создания новых противовирусных препаратов, ингибиторов функции протонселективного канала М2 вируса гриппа А с использованием как в виде индивидуального лекарства, так и в составе комплексной терапии.
ЛИТЕРАТУРА
1. Bouvier N.M., Palese P., The biology of influenza viruses, Vaccine 26 (Suppl 4), (2008) D49-D53.
2. Palese P., Shaw M.L., Orthomyxoviridae: the viruses and their replication, in: D.M. Knipe, P.M. Howley (Eds.), Fields virology, Williams & Wilkins, 2007.
3. Wang C., Lamb R.A., Pinto L.H., Activation of the M2 ion channel of influenza virus: a role for the transmembrane domain histidine residue, Biophys. J. 1995, v. 69, p. 1363-1371.
4. Tang Y., Zaitseva F., Lamb R.A., Pinto L.H., The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue, J. Biol. Chem. 2002, v. 277 p. 39880-39886.
5. Chuang G.Y., Kozakov D., Brenke R., Beglov D., Guarnieri F., Vajda S. Binding hot spots and amantadine orientation in the influenza A virus M2 proton channel. Biophys. J. 2009. v. 97(10), p. 2846-2853.
6. Shibnev V.A., Garaev T.M., Finogenova M.P., Shevchenko E.S.; Burtseva E.I. New adamantane derivatives capable of overcoming the resistance of influenza A (H1N1) pdm2009 and A (H3N2) for "rimantadine" Bull. Exp. Biol. Med. 2012, v. 153(2), p. 233-235
7. Hu, W.; Zeng, S.; Li, C; Jie, Y.; Li, Z.; Chen, L. Identification of hits as matrix-2 protein inhibitors through the focused screening of a small primary amine library. J. Med. Chem. 2010, v. 53, p. 3831-3834.
8. Wang, J.; Ma, C; Balannik, V.; Pinto, L.H.; Lamb, R.A.; Degrado, W.F. Exploring the Requirements for the Hydrophobic Scaffold and Polar Amine in inhibitors of M2 from Influenza A Virus. ACS Med. Chem. Lett. 2011, v. 2, p. 307-312.
9. Wang, J.; Ma, C; Fiorin, G.; Carnevale, V.; Wang, Т.; Hu, F.; Lamb, R.A.; Pinto, L.H.; Hong, M.; Klein, M.L.; DeGrado, W.F. Molecular dynamics simulation directed rational design of inhibitors targeting drug-resistant mutants of influenza A virus M2. J. Am. Chem. Soc. 2011, v. 133, p. 12834-12841.
10. Joseph Armstrong Martin, Sally Redshaw. «Amino acid derivatives)) EP 0432695 B1.
11. Susumu Higashida, Mitsuya Sakurai, Yuichiro Yabe, Takashi Nishihgaki, Tomoaki Komai, Limited Handa, С «Peptides capable of inhibiting the activity of HIV protease, their preparation and their therapeutic use» EP 0587311 A1.
12. Бурцева Е.И., Шевченко Е.С., Ленева И.А. и др. Чувствительность к ремантадину и арбидолу вирусов гриппа, вызвавших эпидемические подъемы заболеваемости в России в сезоне 2004-2005 гг. Вопр. вирусол., 2007, №2, с. 24-29.
13. Ленева И.А., Фадеева Н.И., Федякина И.Т. и др. Применение иммуноферментной индикации вирусспецифических антигенов в изучении нового противовирусного препарата. Хим.-фарм. журнал, 1994, №9, с. 4-15.
| название | год | авторы | номер документа |
|---|---|---|---|
| АДАМАНТИЛАМИДЫ СЕРОСОДЕРЖАЩИХ КИСЛОТ И ИХ ПРОТИВОГРИППОЗНАЯ АКТИВНОСТЬ | 2015 |
|
RU2617850C1 |
| ПРОИЗВОДНЫЕ 1-(1-АДАМАНТИЛ)ЭТИЛАМИН-N-АЦИЛАМИНОКИСЛОТ И ИХ ПРОТИВОГРИППОЗНАЯ АКТИВНОСТЬ | 2014 |
|
RU2572102C1 |
| Аминокислотные производные 2-норборнануксусной кислоты и их противогриппозная активность | 2017 |
|
RU2676699C1 |
| ПРОТИВОВИРУСНОЕ СРЕДСТВО НА ОСНОВЕ СУХОГО ЭКСТРАКТА ЛИШАЙНИКА Cetraria islandica | 2015 |
|
RU2580305C1 |
| ПРОИЗВОДНЫЕ 1-(1-АДАМАНТИЛ)ЭТИЛАМИНА И ИХ ПРОТИВОВИРУСНАЯ АКТИВНОСТЬ | 2011 |
|
RU2461544C1 |
| ЦИКЛОГЕКСАНОКСИКАРБОНИЛ-ДИПЕПТИД И ЕГО ПРОТИВОВИРУСНАЯ АКТИВНОСТЬ В ОТНОШЕНИИ ВИРУСА ГЕПАТИТА С | 2017 |
|
RU2641297C1 |
| ПРОИЗВОДНОЕ 1,3-АДАМАНТАНДИУКСУСНОЙ КИСЛОТЫ И ЕГО ПРОТИВОВИРУСНАЯ АКТИВНОСТЬ | 2013 |
|
RU2553991C1 |
| ПЕПТИДНЫЕ ПРОИЗВОДНЫЕ 1-(1-АДАМАНТИЛ)ЭТИЛАМИНА И ИХ ПРОТИВОВИРУСНОЕ ДЕЙСТВИЕ | 2013 |
|
RU2524216C1 |
| ПРОТИВОВИРУСНОЕ СРЕДСТВО НА ОСНОВЕ СУХОГО ЭКСТРАКТА ПЛОДОВОГО ТЕЛА КСИЛОТРОФНОГО БАЗИДИОМИЦЕТА Bjerkandera adusta | 2015 |
|
RU2580296C1 |
| ГИДРОХЛОРИДЫ (3-R-1-АДАМАНТИЛ)-1-ЭТИЛАМИНОВ, ОБЛАДАЮЩИЕ ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2001 |
|
RU2247714C2 |

Изобретение относится к противогриппозному средству, обладающему противовирусной активностью в отношении вирусов гриппа А и действующему на штаммы, резистентные к действию ремантадина и амантадина. Средство представляет собой аминокислотные производные 2-хинальдинкарбоновой кислоты: 2-хинальдин-серил метиловый эфир (Qln-Ser), 2-хинальдин-триптофанил метиловый эфир (Qln-Trp) и 2-хинальдин-аланил-пролин-1(1-адамантил)этиламид (Qln-APR), и может найти применение при создании новых противовирусных препаратов. Изобретение относится также к новому соединению - 2-хинальдин-аланил-пролин-1(1-адамантил)этиламиду (Qln-APR). 2 н. и 1 з.п. ф-лы, 7 ил., 4 табл., 6 пр.
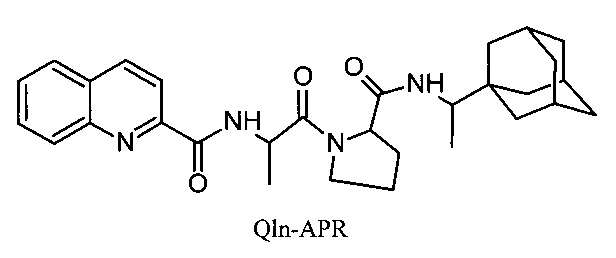
1. Противогриппозное средство, обладающее противовирусной активностью в отношении вирусов гриппа А и действующее на штаммы, резистентные к действию ремантадина и амантадина, представляющее собой аминокислотные производные 2-хинальдинкарбоновой кислоты:
2-хинальдин-серил метиловый эфир (Qln-Ser)
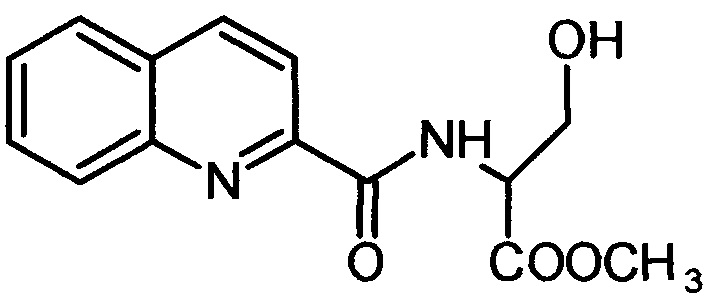 ,
,
2-хинальдин-триптофанил метиловый эфир (Qln-Trp)
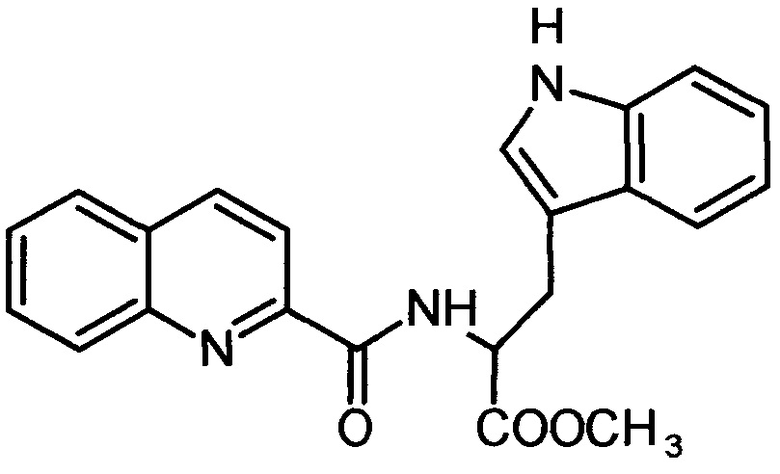 ,
,
2-хинальдин-аланил-пролин-1(1-адамантил)этиламид (Qln-APR)
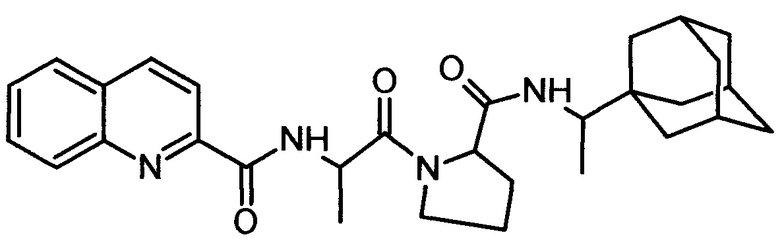 .
.
2. Средство по п.1, представляющее собой Qln-Ser и Qln-Trp, обладающее вирулицидной активностью в отношении вирусов гриппа А.
3. Соединение 2-хинальдин-аланил-пролин-1(1-адамантил)этиламид (Qln-APR)
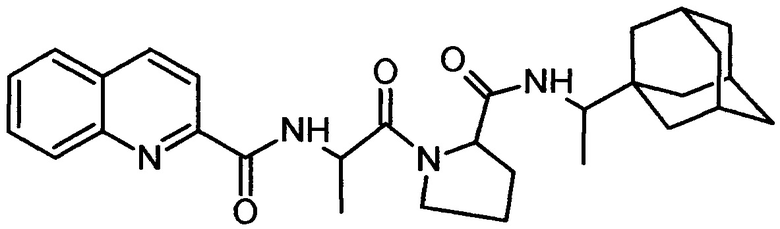 .
.
| M.A | |||
| CIUFOLINI ET AL., Synthesis of a model depsipeptide segment of luzopeptins (BBM 928), potent antitumor and antiretroviral antibiotics, TETRAHEDRON LETTERS, 1989, 30(23), pp.3027-3028 | |||
| C.W | |||
| HOLZAPFEL ET AL., Application of aminocarbonylation in the synthesis of a lavendamycin synthon, J | |||
| CHEM | |||
| RES | |||
| (S), 2002, pp.22-24 | |||
| ХИНОЛИНОВЫЕ КАРБОКСАМИДЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2170730C2 |

