Настоящее изобретение относится к мономерам, способные к разъединению по требованию (РпТ) и их использованию в качестве мономерных компонентов в стоматологических материалах, в частности, для подготовки адгезивов и цементов.
Адгезивные соединения, которые могут быть сняты, имеют все большее значение в различных областях техники. Примеры включают компоненты конструкций автоматизированных производственных процессов, ремонт сложных компонентов с адгезивно связанными компонентами или упрощение отделения материалов при утилизации таких компонентов в конце срока использования продукта. Раскрепление адгезивных соединений может быть достигнуто через значительное снижение прочности слоя адгезивного соединения, например, нагреванием.
Например, в DE 198 32 629 A1 описана адгезивная система для образования обратимых адгезивных соединений на основе полиуретанов, полимочевин или эпоксидных смол, в которой дополнительный компонент может быть активирован при применении энергии так, что происходит разрушение адгезивных компонентов. Например, органические основания или кислоты, которые вызывают разрушение адгезивного связующего, могут выделяться из блокированных предшественников при применении тепла или облучения.
В WO 2010/128042 A1 описаны промышленные адгезивные композиции для разъемных адгезивных соединений для воздушного судна или транспортного средства, состоящие из обычной связующей матрицы и порошкового расширяющего материала, такого как азодикарбонамид. Компоненты разъединяются нагреванием адгезивного соединения до по меньшей мере температуры расширения расширяющего материала.
В стоматологии раскрепление адгезивных связей является важным, кроме прочего, в ортодонтии, где скобки, которые приклеивают к поверхности зуба для исправления неправильного прикуса, после успешной коррекции должны быть удалены без ущерба для зубной эмали. Кроме того, в случае ремонта или полной замены высокопрочных керамических пломб или коронок, которые тяжело снимать механически, были бы желательны цементные соединения, которые могут быть легко размягчены или отделены.
Для применения в ортодонтии, в US 2007/0142498 A1 описаны стоматологические композиции, содержащие термически контролируемые добавки, такие как, например, термопластичные полимеры.
В US 2007/0142497 A1 описаны стоматологические композиции на основе диметакрилатов с кислотонеустойчивыми третичными карбонатными группами и фотокислотами, такими как триарилсульфониевые соли. Эти композиции могут быть фотохимически отверждены с применением подходящих инициаторов, таких как, например, оксид бисацилфосфина Irgacure 819, светом в видимом диапазоне (фотофотоскреплены) и снова размягчены при облучении УФ светом при повышенной температуре (фототермическое раскрепление).
Целью данного изобретения является получение адгезивных стоматологических пломбировочных материалов, которые являются полимеризуемыми, показывают хорошую адгезию к субстрату, в частности, к структуре зуба, и/или стоматологической керамике, и способны открепляться от субстрата при нагревании, и которые, таким образом, прежде всего, подходят для производства адгезивов или композитных цементов, способных к разъединению по требованию.
Эта цель достигается в соответствии с данным изобретением посредством стоматологических пломбировочных материалов на основе термолабильного или фотолабильного полимеризуемого соединения формулы I:

в котором
Т является термолабильной или фотолабильной группой,
Z1 и Z2 каждый независимо представляет собой полимеризуемую группу, выбранную из винильных групп, CH2=CR1-CO-O- и CH2=CR1-CO-NR2-, или связывающую группу, выбранную из -Si(OR)3, -СООН, -O-PO(OH)2, -PO(OH)2, -SO2OH и -SH, где по меньшей мере один Z1 или Z2 представляет собой полимеризуемую группу,
Q1 в каждом случае независимо отсутствует или представляет собой (m+1)-валентный линейный или разветвленный алифатический C1-С20-радикал, который может прерываться -O-, -S-, -CO-O-, -O-CO-, -CO-NR3-, -NR3-CO-, -O-CO-NR3-, -NR3-CO-O- или -NR3-CO-NR3-,
Q2 в каждом случае независимо отсутствует или представляет собой (n+1)-валентный линейный или разветвленный алифатический C1-C20-радикал, который может прерываться -O-, -S-, -CO-O-, -O-CO-, -CO-NR3-, -NR3-CO-, -O-CO-NR3-, -NR3-CO-O- или -NR3-CO-NR3-,
X и Y каждый независимо друг от друга отсутствуют или представляет собой -O-, -S-, -CO-O, -O-CO, -CO-NR3-, -NR3-CO, -O-CO-NR3-, NR3-CO-O- или -NR3-CO-NR3-,
R, R1, R2 и R3 каждый независимо представляет собой Н или С1-С7-алкильный радикал, и
k, l, m и n каждый независимо равен 1, 2 или 3.
В одном варианте по меньшей мере один из Z1 или Z2 является полимеризуемой группой, и по меньшей мере один из Z1 или Z2 является связывающей группой. В этом контексте, соединения формулы I, в которых один из Z1 и Z2 представляет собой полимеризуемую группу, а другой из Z1 и Z2 представляет собой связывающую группу, являются предпочтительными. В другом варианте, Z1 и Z2 оба представляет собой полимеризуемую группу.
Примечание о том, что радикал может быть прерван группой, такой как -O-, понимается как то, что группа вставляется в углеродную цепь радикала, т.е. ограничена с двух сторон атомами углерода. Число этих групп, следовательно по меньшей мере на 1 меньше, чем число атомов углерода, и группа не может быть концевой. Согласно изобретению, радикалы, которые не прерываются указанными группами, являются предпочтительными.
В соответствии с изобретением рассматриваются только те соединения, которые совместимы с теорией химической валентности.
Особенно предпочтительными являются такие соединения формулы I, в которых, в каждом случае, независимо
один из Z1 и Z2 в каждом случае независимо представляет собой полимеризуемую группу, выбранную из CH2=CR1-CO-O- и CH2=CR1-CO-NR2-, другой Z1 и Z2 в каждом случае независимо представляет собой полимеризуемую группу, выбранную из CH2=CR1-CO-O- и CH2=CR1-CO-NR2-, или предпочтительно связывающую группу, выбранную из -Si(OR)3, -СООН, -O-PO(OH)2, -PO(OH)2, -SO2OH и -SH,
Q1 в каждом случае независимо отсутствует или представляет собой (m+1)-валентный линейный или разветвленный алифатический C1-C10 радикал, который может прерываться -O-, -CO-O-, -O-CO-, -CO-NR3- или -NR3-CO-,
Q2 в каждом случае независимо отсутствует или представляет собой (n+1)-валентный линейный или разветвленный алифатический C1-C10 радикал, который может прерываться -O-, -CO-O-, -O-CO-,-CO-NR3- или -NR3-CO-,
X и Y в каждом случае независимо отсутствует или представляет собой -O-, -CO-O-, -O-CO, -CO-NR3- или -NR3-CO-,
R в каждом случае независимо представляет собой Н, СН3 или C2H5,
R1 в каждом случае независимо представляет собой Н или СН3,
R2 в каждом случае независимо представляет собой Н, СН3 или С2Н5,
R3 в каждом случае независимо представляет собой Н, СН3 или C2H5, и/или
k, l, m и n в каждом случае независимо равны 1 или 2.
Особенно предпочтительными являются соединения, в которых все переменные имеют один из определенных выше предпочтительных значений.
В предпочтительном варианте T является термолабильной группой. В этом контексте предпочтительными являются такие соединения формулы I, в которых
Z1 и Z2 каждый независимо представляет собой полимеризуемую группу, выбранную из винильных групп, CH2=CR1-CO-O- и CH2=CR1-CO-NR2- или кислотную группу, выбранную из -O-PO(OH)2, -PO(OH)2 и -SO2OH, где по меньшей мере один из Z1 и Z2 является полимеризуемой группой, и по меньшей мере один из Z1 и Z2 является кислотной группой,
Q1 в каждом случае независимо отсутствует или представляет собой (m+1)-валентный линейный или разветвленный алифатический C1-C10 радикал, который может прерываться -O-, -S-, -CO-O-, -O-CO-, -CO-NR3- или -NR3-CO-,
Q2 в каждом случае независимо отсутствует или представляет собой (n+1)-валентный линейный или разветвленный алифатический C1-C10 радикал, который может прерываться -O-, -S-, -CO-O-, -O-CO-, -CO-NR3- или -NR3-CO-,
X и Y в каждом случае независимо отсутствует или представляет собой -O-, -S-, -CO-O-, -O-CO, -CO-NR3- или -NR3-CO-,
R1, R2 и R3 в каждом случае независимо представляет собой Н или С1-С7 алкильный радикал, и
k, l, m и n в каждом случае независимо равны 1, 2 или 3.
В этом контексте предпочтительны такие соединения формулы I, в которых один из Z1 и Z2 представляет собой полимеризуемую группу, и другой из Z1 и Z2 представляет собой кислотную группу.
В этом контексте особенно предпочтительными являются такие соединения формулы I, в которых в каждом случае независимо:
один из Z1 и Z2 в каждом случае независимо представляет собой полимеризуемую группу, выбранную из CH2=CR1-CO-O- и CH2=CR1-CO-NR2-, и другой Z1 и Z2 в каждом случае независимо представляет собой кислотную группу, выбранную из -O-PO(OH)2 и -PO(OH)2 и -SO2OH,
Q1 в каждом случае независимо отсутствует или представляет собой (m+1)-валентный линейный или разветвленный алифатический C1-C10 радикал, который может прерываться -O-, -CO-O-, -O-CO-, -CO-NR3- или -NR3-CO-,
Q2 в каждом случае независимо отсутствует или представляет собой (n+1)-валентный линейный или разветвленный алифатический C1-C10 радикал, который может прерываться -O-, -CO-O-, -O-CO-,-CO-NR3- или -NR3-CO-,
X и Y в каждом случае независимо отсутствует или представляет собой -O-, -CO-O-, -O-CO, -CO-NR3- или -NR3-CO-,
R1 в каждом случае независимо представляет собой Н или СН3,
R2 в каждом случае независимо представляет собой Н, СН3 или С2Н5,
R3 в каждом случае независимо представляет собой Н, СН3 или С2Н5, и/или
k, l, m и n в каждом случае независимо равны 1 или 2.
Особенно предпочтительными являются соединения, в которых все переменные имеют одно из определенных выше предпочтительных значений.
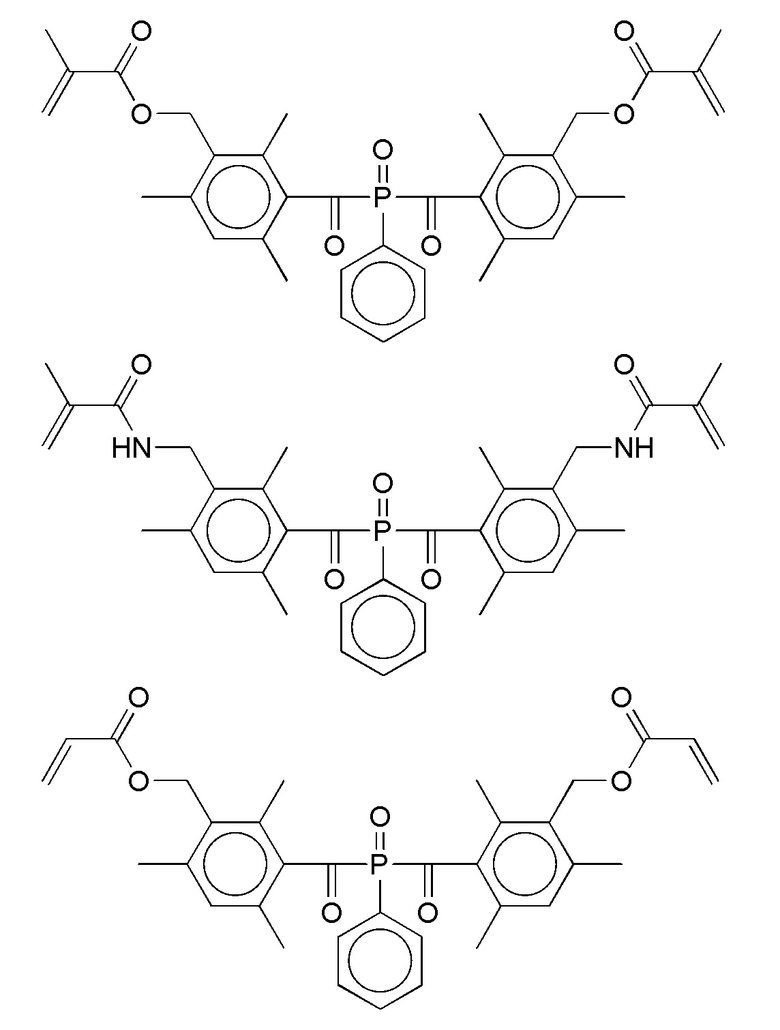
Подходящие термолабильные группы известны. Они характеризуются в соответствии с изобретением, тем, что они содержат одну или более термолабильных ковалентных связей. Предпочтительные термолабильные группы с термолабильными ковалентными связями включают термолабильные аддукты циклоприсоединения, такие как аддукты Дильса-Альдера, гетеро-аддукты Дильса-Альдера и термолабильный алкоксиамин, оксим-сложный эфир, оксим-уретан или азогруппы. Примеры термолабильных групп также описаны в RJ Wojtecki et al., Nature Materials 2011, 10, 14-27.
В частности, предпочтительны такие соединения формулы I, в которых Т является термолабильной группой, которую выбирают из группы, включающей:
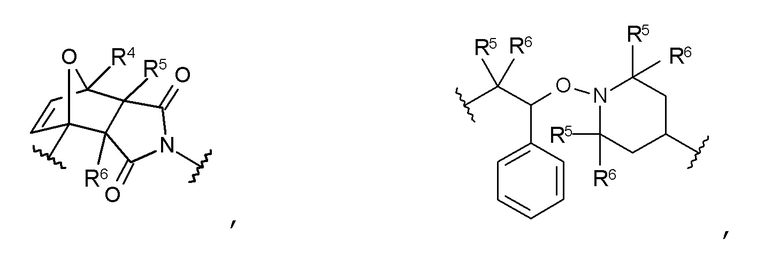
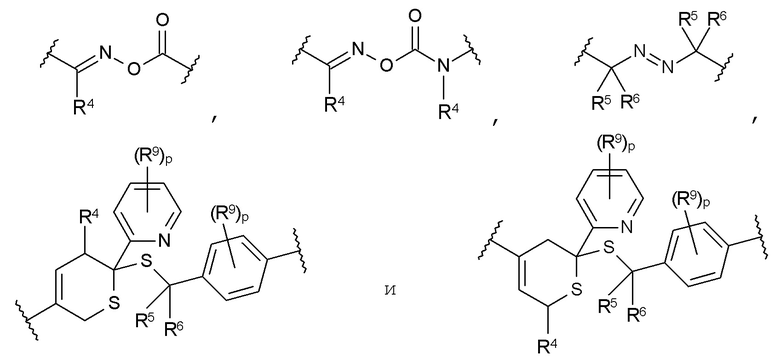
где
R4 представляет собой Н или C1-C10 алкильный радикал;
R5 представляет собой Н, C1-C5 алкильный радикал, F или CN,
R6 представляет собой Н, C1-C5 алкильный радикал, F или CN,
R9 в каждом случае независимо представляет собой CH3, C2H5, ОСН3 или OC2H5, и
р в каждом случае независимо равен 0, 1, 2 или 3.
В соответствии с изобретением, особенно предпочтительными являются стоматологические материалы, в которых термолабильным полимеризуемым соединением формулы I является аддукт Дильса-Альдера формулы II:
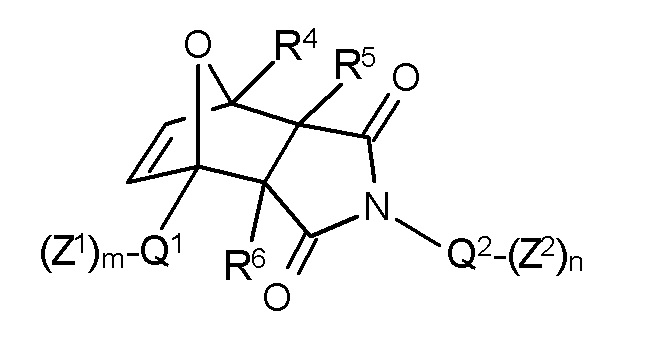
Формула II,
где указанные переменные имеют значения, определенные выше. В соответствии с данным изобретением, формула II включает чистые экзо продукты или чистые эндо продукты и смеси экзо и эндо продуктов.
В этом контексте, предпочтительными являются такие соединения формулы II, в которых в каждом случае независимо
один из Z1 и Z2 в каждом случае независимо представляет собой полимеризуемую группу, выбранную из CH2=CR1-CO-O- и CH2=CR1-CO-NR2-, и другой из Z1 и Z2 в каждом случае независимо представляет собой полимеризуемую группу, выбранную из CH2=CR1-CO-O- и CH2=CR1-CO-NR2- или предпочтительно связывающую группу, выбранную из Si(OR)3, -СООН, -O-PO(OH)2, -PO(OH)2, -SO2OH и -SH,
Q1 в каждом случае независимо отсутствует или представляет собой (m+1)-валентный линейный или разветвленный алифатический C1-C15 радикал, предпочтительно C1-C10 радикал, предпочтительно C1-C8 радикал, в частности C2-C6 радикал, и особенно предпочтительно C1-C2 радикал, который может прерываться -O-, -CO-O-, -O-CO-, -CO-NR3-, -NR3-CO-, -O-CO-NR3-, -NR3-CO-O- или -NR3-CO-NR3-,
Q2 в каждом случае независимо отсутствует или представляет собой (n+1)-валентный линейный или разветвленный алифатический C1-C15 радикал, предпочтительно C1-C10 радикал, предпочтительно C1-C8 радикал, в частности C2-C6 радикал, и особенно предпочтительно C2-C3 радикал, который может прерываться -O-, -CO-O-, -O-CO-, -CO-NR3-, -NR3-CO-, -O-CO-NR3-, -NR3-CO-O- или -NR3-CO-NR3-,
R в каждом случае независимо является СН3 или C2H5,
R1 в каждом случае независимо является Н или СН3,
R2 в каждом случае независимо является Н, СН3 или С2Н5,
R3 в каждом случае независимо является Н, СН3 или C2H5,
R4 является Н, СН3 или С2Н5;
R5 является Н, F или CN, и, в частности, Н,
R6 является Н, F или CN, и, в частности, Н, и/или
m и n в каждом случае независимо равны 1 или 2.
Особенно предпочтительными являются соединения формулы II, в которых в каждом случае независимо
один из Z1 и Z2 в каждом случае независимо представляет собой полимеризуемую группу, выбранную из CH2=CR1-CO-O- и CH2=CR1-CO-NR2-, и другой из Z1 и Z2 в каждом случае независимо представляет собой полимеризуемую группу, выбранную из CH2=CR1-CO-O- и CH2=CR1-CO-NR2- или предпочтительно связывающую группу, выбранную из Si(OR)3, -O-PO(OH)2, -PO(OH)2 и -SH,
Q1 представляет собой метиленовый или этиленовый радикал,
Q2 представляет собой этиленовый или пропиленовый радикал,
R в каждом случае независимо представляет собой СН3 или C2H5,
R1 в каждом случае независимо представляет собой Н или СН3,
R2 в каждом случае независимо представляет собой Н, СН3 или С2Н5,
R3 в каждом случае независимо представляет собой Н, СН3 или C2H5,
R4 представляет собой Н, СН3 или С2Н5;
R5 представляет собой Н, F или CN, и, в частности, Н,
R6 представляет собой Н, F или CN, и, в частности, Н, и/или
m и n каждый равен 1.
Особенно предпочтительными являются соединения, в которых все переменные имеют одно из представленных выше предпочтительных значений.
В другом варианте T является фотолабильной группой. Подходящие фотолабильные группы известны. Они обычно характеризуются тем, что содержат одну или несколько фотолабильных ковалентных связей. Предпочтительные фотолабильные группы с фотолабильными ковалентными связями включают простые эфиры бензоина, алкиалкилфенилацетофеноны, диалкилоксиацетофеноны, оксиды бензоилдифенилфосфина, оксиды дибензоилфенилфосфина, диалкил-бензоил и производные диалкилдибензоилгермания.
В частности, предпочтительными являются такие соединения формулы I, где Т представляет собой фотолабильную группу, выбранную из группы, включающей:
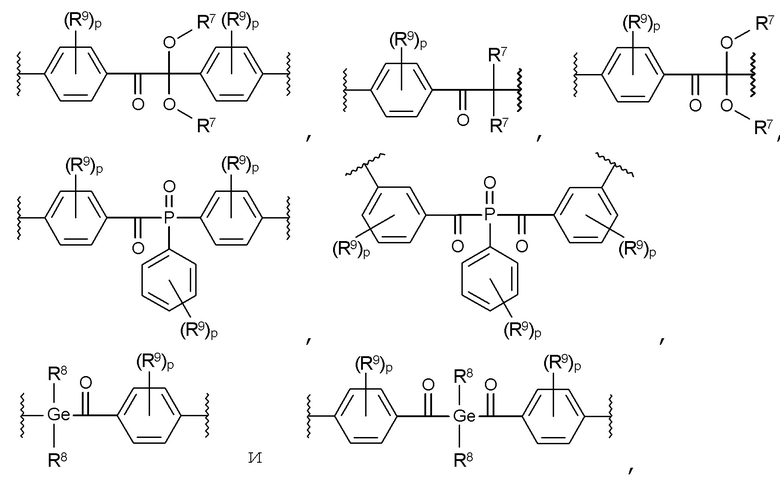
где:
R7 в каждом случае независимо представляет собой C1-C10 алкильный радикал,
R8 в каждом случае независимо представляет собой C1-C7 алкильный радикал,
R9 в каждом случае независимо представляет собой CH3, C2H5, ОСН3 или OC2H5, и
р в каждом случае независимо равен 0, 1, 2 или 3.
В соответствии с данным изобретением, особенно предпочтительны стоматологические материалы, в которых фотолабильным полимеризуемым соединением формулы I является оксид дибензоилфенилфосфина формулы III или производное диалкилдибензоилгермания формулы IV:
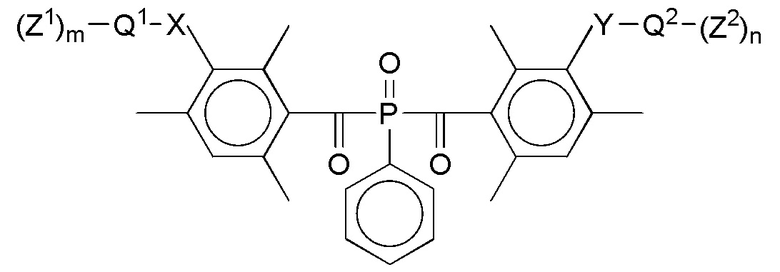
Формула III
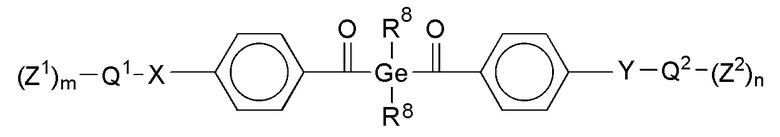
Формула IV,
где указанные переменные имеют определенные выше значения.
В этом контексте предпочтительными являются такие соединения формул III и IV, в которых, в каждом случае, независимо
один из Z1 и Z2 в каждом случае независимо представляет собой полимеризуемую группу, выбранную из CH2=CR1-CO-O- и CH2=CR1-CO-NR2-, и другой из Z1 и Z2 в каждом случае независимо представляет собой полимеризуемую группу, выбранную из CH2=CR1-CO-O- и CH2=CR1-CO-NR2- или, предпочтительно связывающую группу, выбранную из Si(OR)3, -O-PO(OH)2, -PO(OH)2 и -SH,
Q1 в каждом случае независимо отсутствует или представляет собой (m+1)-валентный линейный или разветвленный алифатический C1-C15 радикал, предпочтительно C1-C10 радикал, предпочтительно C1-C8 радикал, в частности C2-C6 радикал, и особенно предпочтительно C1-C2 радикал, который может прерываться -O-, -CO-O-, -O-CO-, -CO-NR3-, -NR3-CO-, -O-CO-NR3-, -NR3-CO-O- или -NR3-CO-NR3-,
Q2 в каждом случае независимо отсутствует или представляет собой (n+1)-валентный линейный или разветвленный алифатический C1-C15 радикал, предпочтительно C1-C10 радикал, предпочтительно C1-C8 радикал, в частности C2-C6 радикал, и особенно предпочтительно C2-C3 радикал, который может прерываться -O-, -CO-O-, -O-CO-, -CO-NR3-, -NR3-CO-, -O-CO-NR3-, -NR3-CO-O- или -NR3-CO-NR3-,
X и Y в каждом случае независимо отсутствует или представляет собой -O-, -CO-O-, -O-CO, -CO-NR3-, -NR3-CO-, -O-CO-NR3-, -NR3-CO-O- или -NR3-CO-NR3-,
R в каждом случае независимо представляет собой СН3 или C2H5,
R8 в каждом случае независимо представляет собой C1-C5 алкильный радикал, и
m и n в каждом случае равны 1 или 2.
Особенно предпочтительными являются соединения формул III и IV, в которых в каждом случае независимо
один из Z1 и Z2 в каждом случае независимо представляет собой полимеризуемую группу, выбранную из CH2=CR1-CO-O- и CH2=CR1-CO-NR2-, и другой из Z1 и Z2 в каждом случае независимо представляет собой полимеризуемую группу, выбранную из CH2=CR1-CO-O- и CH2=CR1-CO-NR2- или предпочтительно связывающую группу, выбранной из Si(OR)3, -O-PO(OH)2, -PO(OH)2 и -SH,
Q1 в каждом случае независимо отсутствует или представляет собой (m+1)-валентный линейный или разветвленный алифатический C1-C8 радикал, в частности C2-C6 радикал, и особенно предпочтительно C1-C2 радикал, который может прерываться -O-, -CO-O- или -O-CO-,
Q2 в каждом случае независимо отсутствует или представляет собой (n+1)-валентный линейный или разветвленный алифатический C1-C8 радикал, в частности C2-C6 радикал, и особенно предпочтительно C2-C3 радикал, который может прерываться -O-, -CO-O- или -O-CO-,
X и Y в каждом случае независимо отсутствует или представляет собой -O-, -CO-O- или -O-CO,
R в каждом случае независимо является СН3 или C2H5,
R8 в каждом случае независимо является C1-C4 алкильным радикалом, и
m и n в каждом случае равны 1.
Особенно предпочтительными являются соединения, в которых все переменные имеют одно из представленных выше предпочтительных значений.
Неожиданно было обнаружено, что после полимеризации стоматологические пломбировочные материалы в соответствии с данным изобретением, которые содержат по меньшей мере одно термолабильное и/или фотолабильное полимеризуемое соединение формулы I и предпочтительно по меньшей мере одно термолабильное полимеризуемое соединение формулы II и/или фотолабильное полимеризуемое соединение формулы III или IV, с одной стороны, демонстрируют отличные механические свойства и отличное сцепление с тканями зуба и стоматологической керамикой, и с другой стороны, могут быть легко отцеплены от субстрата под воздействием тепла (термолабильные соединения) или облучения УФ светом или видимым светом (фотолабильные соединения).
Полимеризуемые аддукты Дильса-Альдера формулы II могут быть легко получены. Например, подходящим образом функционализированные производные фурана могут быть подвергнуты взаимодействию с подходящими N-функционализированными малеимидамми в условиях реакции, обычных для реакции Дильса-Альдера, и, в частности, при 80-120°С, например, в ароматических растворителях, при необязательном добавлении подходящего катализатора (например, кислот Бренстеда или Льюиса), а также ингибитора полимеризации (см. коллектив авторов, Organikum, Wiley-VCH 21-е издание, Weinheim etc. 2001, 330 и далее) с получением соответствующего полимеризуемого аддукта Дильса-Альдера:
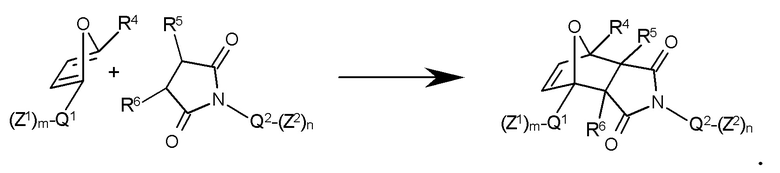
Конкретный пример: реакция Дильса-Альдера метакрилата фурфурила (Z1 = CH2=CR1-CO-O, Q1 = -CH2-, R1 = CH3, R4 = Н и m = 1) и N-[3-(дигидроксифосфорил)пропил]малеинимида (Z2 = -PO(OH)2, Q2 = -(CH2)3-, R5 и R6 = Н и n = 1):
Подходящие исходные материалы для синтеза производных фурана, функционализированных полимеризуемыми или сильно кислотными группами, являются коммерчески доступными, например, фурфурол, фурфуриловый спирт или пирослизевая кислота (см. Ullmann’s Encyclopedia of Industrial Chemistry, 5th ed., Vol. 12, VCH, Weinheim etc. 1989, стр. 119 и далее). Замещенные производные фурана могут быть получены, например, синтезом Пааля-Кнорра путем нагревания соответствующих 1,4-дикетосоединений (см. W. Walter, W. Francke, Beyer-Walter Lehrbuch der Organischen Chemie, S. Hirzel Verlag, Stuttgart und Leipzig 2004, 24-е издание, стр. 769). Малеинимиды, функционализированные полимеризуемыми или сильно кислотными группами, могут быть получены наиболее просто взаимодействием малеинового ангидрида с соответствующим образом функционализированными аминами.
Методика синтеза аддуктов Дильса-Альдера формулы II также может быть постадийной, так что сначала соответствующей аддукт Дильса-Альдера получают из подходящим образом функционализированного малеинимида и производного фурана, и только затем вводят полимеризуемые или сильно кислотные группы, синтез необязательно проводят с применением защитных групп. Например, аддукта Дильса-Альдера из приведенного выше примера также может быть получен с полимеризуемой метакрилатной группой и сильной кислой группой фосфоновой кислоты так, что сначала фурфуриловый спирт, который защищен, например, тетрагидропиранильной (ТГП) группой, превращают в аддукт Дильса-Альдера с N-(3-бромпропил)малеинимидом. После введения группы фосфоновой кислоты, например, в реакции Михаэлиса-Арбузова при взаимодействии аддукта Дильса-Альдера, например, с триэтилфосфитом (P(OC2H5)3), защитная группа ТГФ может быть отщеплена, полученная группа OH может быть метакрилирована, например, ангидридом метакриловой кислоты (АМАК) и, наконец, группа фосфоновой кислоты может быть гидролитически выделена:
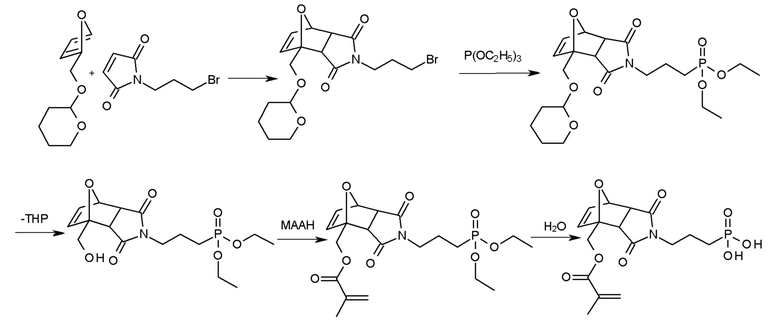
Примеры термолабильных аддуктов Дильса-Альдера формулы II в соответствии с данным изобретением включают:
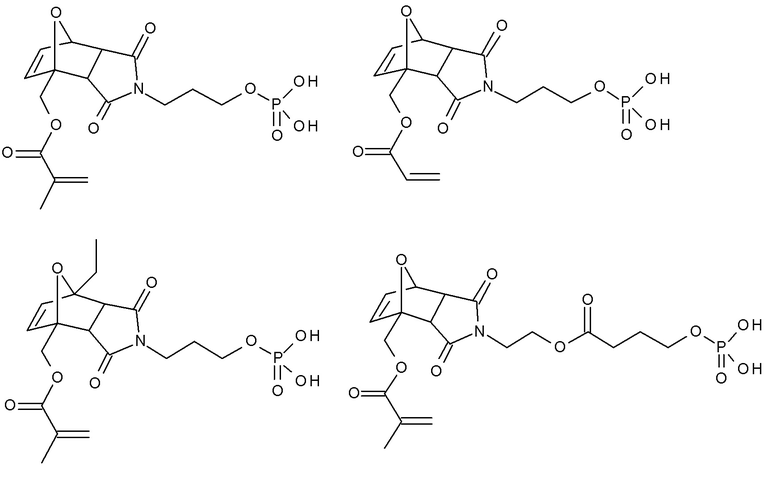
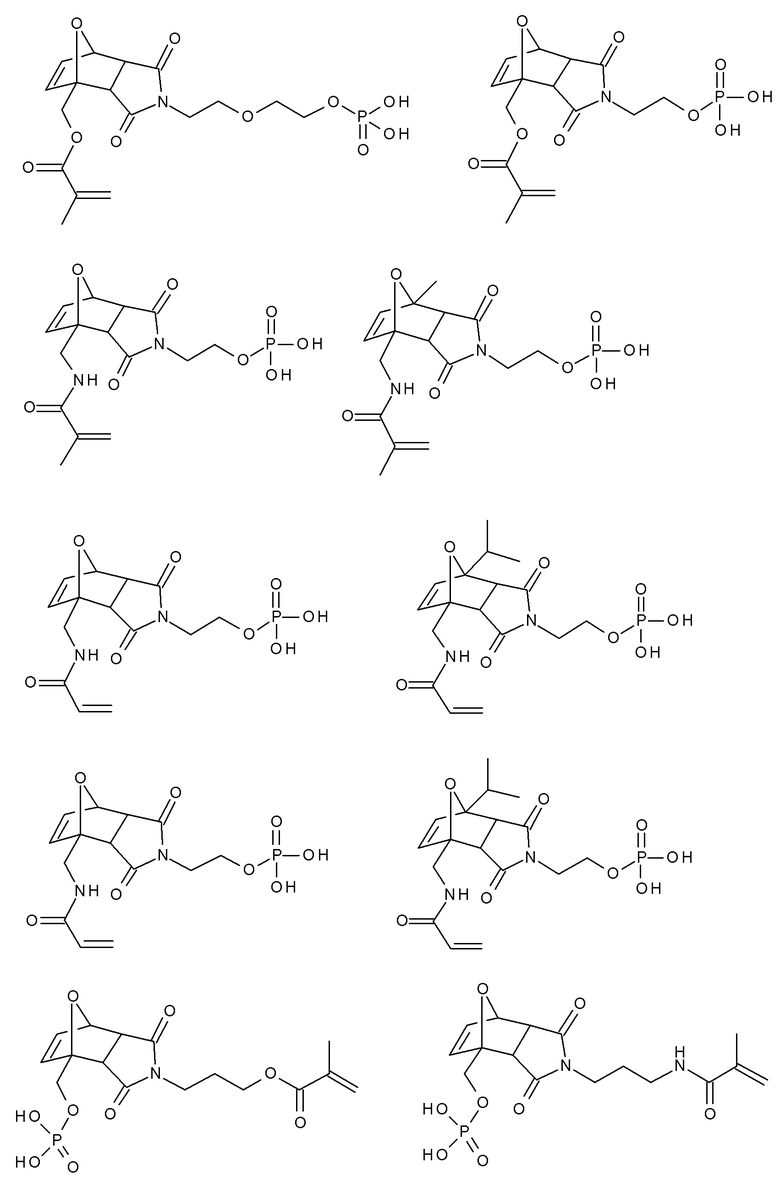
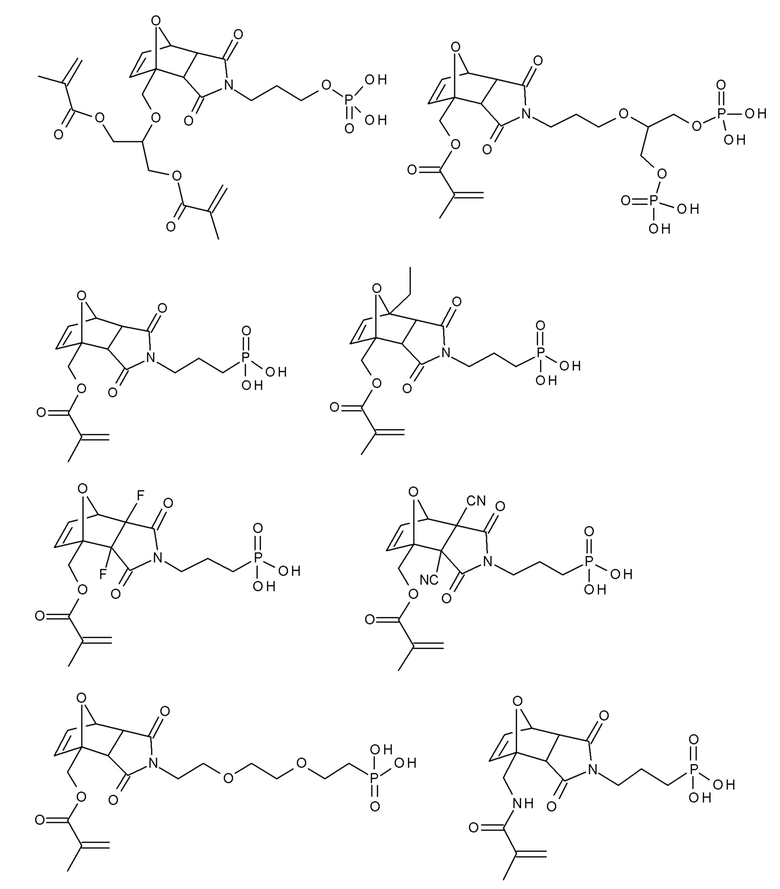
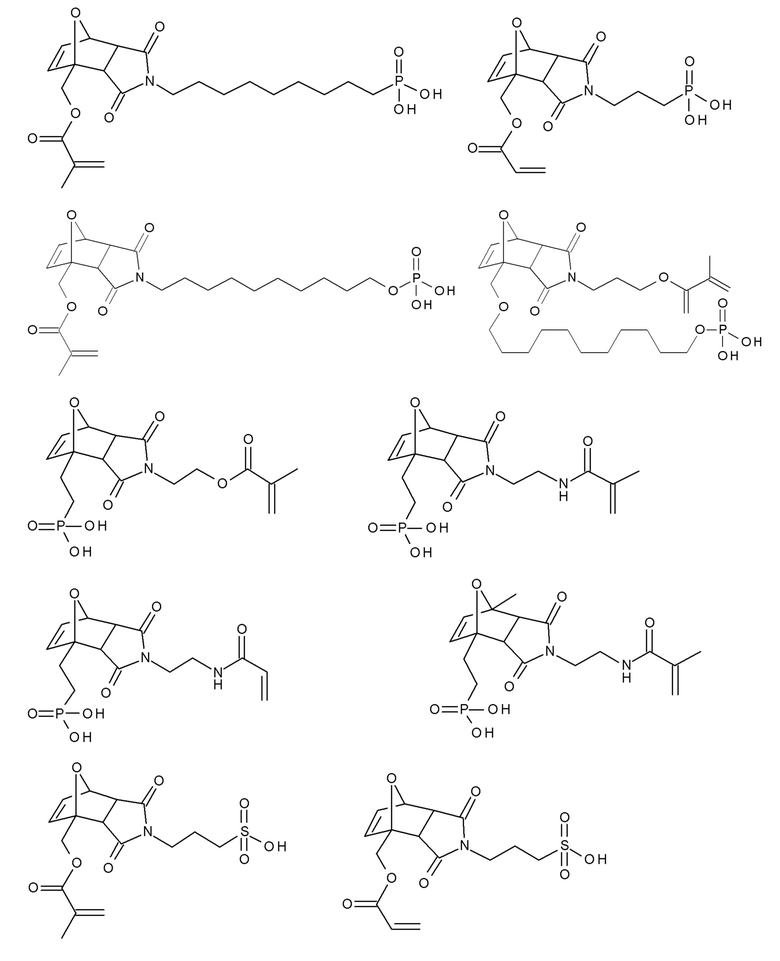
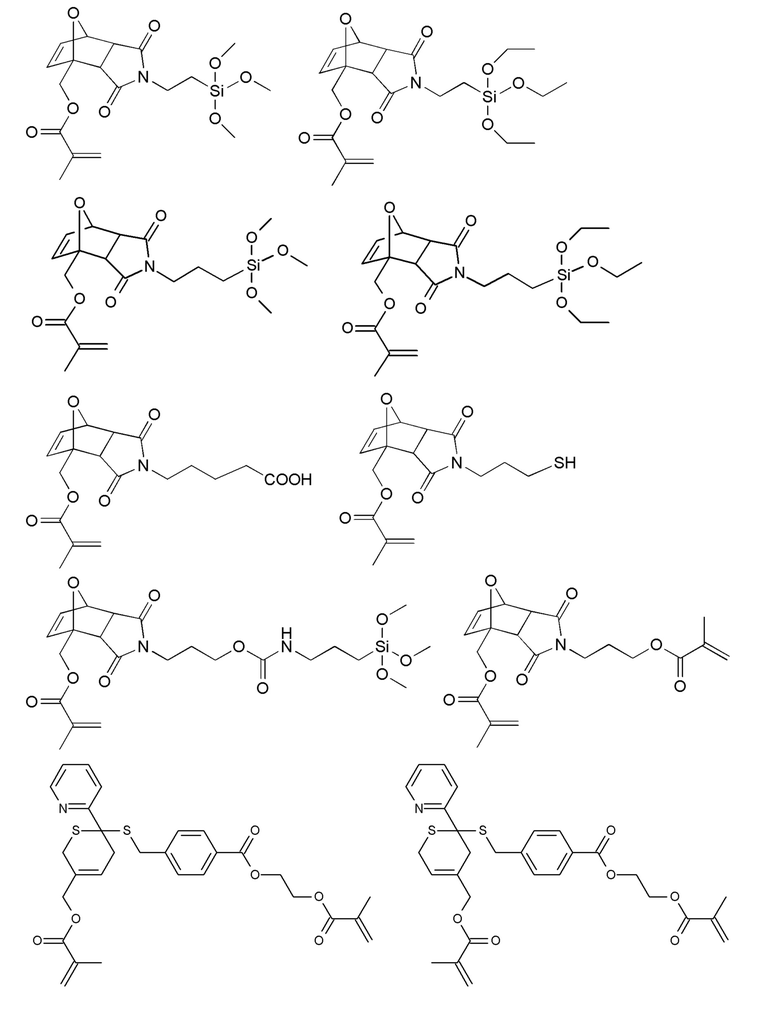
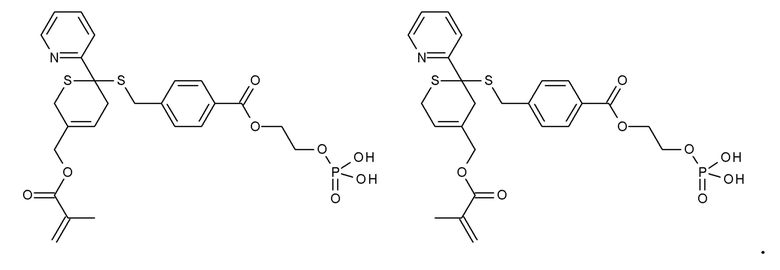
Примеры фотолабильных оксидов дибензоилфенилфосфина формулы III в соответствии с данным изобретением включают:
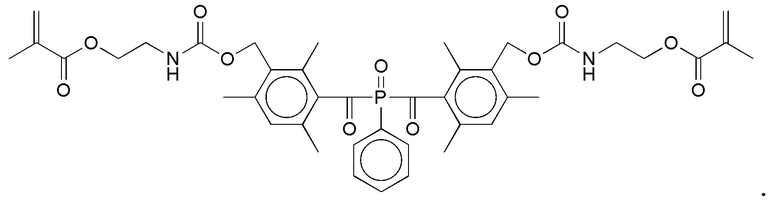
Примеры фотолабильных производных диалкилдибензоилгермания формулы IV в соответствии с данным изобретением включают:
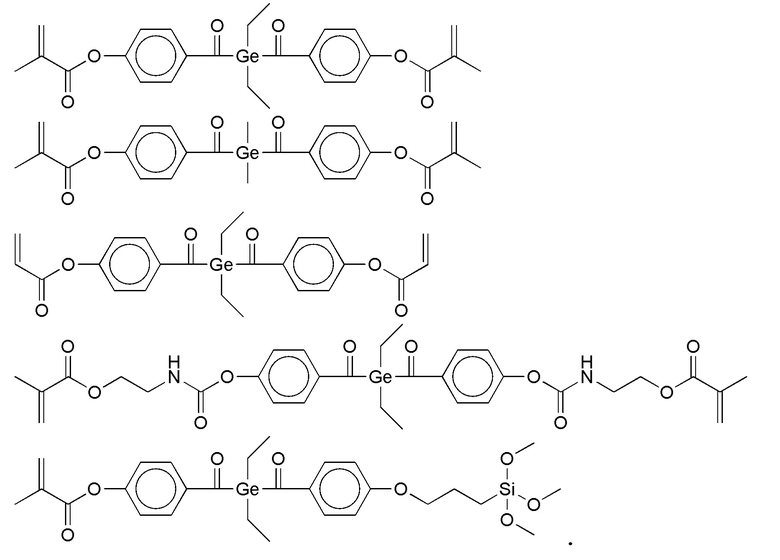
Стоматологические материалы в соответствии с изобретением предпочтительно содержат, в дополнение к термолабильному или фотолабильному полимеризуемому соединению формулы I, один или более дополнительных радикально полимеризуемых мономеров (сомономеров), в частности, моно- или полифункциональных производных (мет)акриловой кислоты. Монофункциональные производные (мет)акриловой кислоты означают соединения с одной группой (мет)акриловой кислоты, полифункциональные производные (мет)акриловой кислоты означают соединения с двумя или более, предпочтительно от 2 до 4, группами (мет)акриловой кислоты. Полифункциональные мономеры обладают поперечно-сшивающим действием.
Предпочтительные моно- или полифункциональные производные (мет)акриловой кислоты в соответствии с изобретением включают метил, этил, гидроксиэтил, бутил, бензил, тетрагидрофурфурил или изоборнил(мет)акрилат, бисфенол-А-ди(мет)акрилат, бис-GMA (продукт присоединения метакриловой кислоты и бисфенола-A-диглицидилового эфира), UDMA (продукт присоединения 2-гидроксиэтилметакрилата (ГЭМА) и 2,2,4-триметилгексаметилендиизоцианата), ди(мет)акрилат ди-, три- или тетра-этиленгликоля, три(мет)акрилат триметилолпропана, тетра(мет)акрилат пентаэритритола, ди(мет)акрилат глицерина, ди(мет)акрилат 1,4-бутандиола, ди(мет)акрилат 1,10-декандиола и ди(мет)акрилат 1,12-додекандиола.
Особенно предпочтительными моно- или полифункциональными производными (мет)акриловой кислоты являются N-моно- или дизамещенные акриламиды, такие как N-этилакриламид, N,N-диметакриламид, N-(2-гидроксиэтил)акриламид или N-метил-N-(2-гидроксиэтил)акриламид , N-монозамещенные метакриламиды, такие как N-этилметакриламид и N-(2-гидроксиэтил)метакриламид, а также N-винилпирролидон и аллиловый эфир. Эти мономеры характеризуется высокой устойчивостью к гидролизу и, в силу их относительно низкой вязкости, особенно предпочтительны в качестве разбавляющих мономеров.
Предпочтительные полифункциональные производные (мет)акриловой кислоты с высокой устойчивостью к гидролизу включают поперечно-сшивающие пирролидоны, такие как 1,6-бис(3-винил-2-пирролидонил)гексан, бисакриламиды, такие как бисакриламид метилена или этилена, и бис(мет)акриламиды, такие как N,N'-диэтил-1,3-бис(акриламидо)пропан, 1,3-бис(метакриламидо)пропан, 1,4-бис(акриламидо)бутан или 1,4-бис(акрилоил)пиперазин, которые могут быть синтезированы взаимодействием соответствующих диаминов с хлоридом (мет)акриловой кислоты.
Термолабильные поперечно-сшивающие мономеры также особенно подходят в соответствии с данным изобретением в качестве сомономеров. Термолабильные поперечно-сшивающие мономеры имеют по меньшей мере одну термолабильную группу между двумя полимеризуемыми группами. Примеры включают полифункциональные (мет)акрилаты или (мет)акриламиды с по меньшей мере одной термолабильной группой между двумя (мет)акрильными группами. В принципе, в качестве термолабильных групп во внимание принимаются те же группы, которые определены выше для соединений формулы I, и, в частности, термолабильные аддукты циклоприсоединения, такие как аддукты Дильса-Альдера, гетероаддукты Дильса-Альдера и термолабильные алкоксиамины, а также оксим-сложный эфир, оксим-уретан или азогруппы. Примеры включают аддукты Дильса-Альдера, такие как аддукт Дильса-Альдера из фурфурилметакрилата и N-(3-(метакрилоилокси)пропил)малеинимида, продукты реакции N-гидрокси(мет)акриламида с ди- или триизоцианатами, такие как гексаметилен-1,6-диизоцианат (ГДИ), 2,2,4-триметилгексаметилен-1,6-диизоцианат или тример ГДИ, также как продукты, полученные стехиометрической реакцией ди- или триизоцианатов с 1-гидроксиметилакриловыми эфирами, такими как 1-гидроксиметилэтилакрилат, или с (мет)акрилатами β-кетоэфира, такими как 2-ацетоацетоксиэтилметакрилат. Особенно подходят выделяющие газ термолабильные поперечно-сшивающие мономеры. Примеры включают продукты этерификации азобис(4-циановалериановой кислоты) с гидроксиалкил(мет)акрилатами, такими как гидроксиэтил(мет)акрилат или гидроксипропил(мет)акрилат, или с N-(гидроксиалкил)(мет)акриламидами, такими как N-(5-гидроксипентил)метакриламид или N-метил-N-(2-гидроксиэтил)акриламид.
Кроме термолабильного или фотолабильного полимеризуемого соединения формулы I и, необязательно, указанных выше мономеров, стоматологические материалы в соответствии с данным изобретением могут предпочтительно также содержать радикально полимеризуемые, содержащие кислотные группы мономеры (связывающие мономеры). Предпочтительные кислотные группы включают группы карбоновой кислоты, группы фосфоновой кислоты, группы фосфорной кислоты и группы сульфоновой кислоты.
Предпочтительными мономерами с полимеризуемыми карбоновыми кислотами являются малеиновая кислота, акриловая кислота, метакриловая кислота, 2-(гидроксиметил)акриловая кислота, 4-(мет)акрилоилоксиэтилтримеллитовый ангидрид, 10-метакрилоилоксидецилмалоновая кислота, N-(2-гидрокси-3-метакрилоилоксипропил)-N-фенилглицин и 4-винилбензойная кислота.
Предпочтительными мономерами с полимеризуемыми группами фосфоновой кислоты являются винилфосфоновая кислота, 4-винилфенилфосфоновая кислота, 4-винилбензилфосфоновая кислота, 2-метакрилоилоксиэтилфосфоновая кислота, 2-метакриламидоэтилфосфоновая кислота, 4-метакриламидо-4-метилпентилфосфоновая кислота, 2-[4-(дигидроксифосфорил)-2-оксабутил]акриловая кислота, этиловый и 2,4,6-триметилфениловый эфир 2-[4-(дигидроксифосфорил)-2-оксабутил]акриловой кислоты.
Предпочтительными мономерами с полимеризуемыми группами фосфорной кислоты являются моно- или дигидрофосфат 2-метакрилоилоксипропила, моно- или дигидрофосфат 2-метакрилоилоксиэтила, гидрофосфат 2-метакрилоилоксиэтилфенила, дипентаэритритолпентаметакрилоилоксифосфат, дигидрофосфат 10-метакрилоилоксидецила, моно-(1-акрилоилпиперидин-4-иловый) эфир фосфорной кислоты, дигидрофосфат 6-(метакриламидо)гексила и дигидрофосфат 1,3-бис-(N-акрилоил-N-пропиламино)пропан-2-ила.
Предпочтительными мономерами с полимеризуемыми группами сульфоновой кислоты являются винилсульфоновая кислота, 4-винилфенилсульфоновая кислота и 3-(метакриламидо)пропилсульфоновая кислота.
Предпочтительно применяют смеси указанных выше мономеров. На основе общей массы смеси мономеров, предпочтительные мономерные смеси содержат:
от 1 до 90% масс., предпочтительно от 5 до 80% масс., особенно предпочтительно от 5 до 70 соединения формулы I, и, в частности, формулы II, III и/или IV,
от 0 до 70% масс., предпочтительно от 1 до 60% масс., особенно предпочтительно от 5 до 50, и наиболее предпочтительно от 10 до 30% масс. сомономера, в частности, моно- и/или полифункциональных (мет)акрилатов,
от 0 до 70% масс., предпочтительно от 1 до 60% масс. и особенно предпочтительно от 5 до 50% масс. термолабильного поперечно-сшивающего мономера и
от 0 до 40% масс., предпочтительно от 1 до 30% масс. и особенно предпочтительно от 5 до 20% масс. клеевого мономера.
Особенно предпочтительные мономерные смеси (в каждом случае на основе общей массы мономерной смеси) приведены в следующей таблице:
Кроме того, стоматологические пломбировочные материалы в соответствии с данным изобретением предпочтительно также содержат инициатор радикальной полимеризации.
Предпочтительно бензофенон, бензоин и их производные или α-дикетоны или их производные, такие как 9,10-фенантренхинон, 1-фенилпропан-1,2-дион, диацетил или 4,4'-дихлорбензил применяют для инициирования радикальной фотополимеризации, в частности, для термолабильных соединений формулы I. Предпочтительно применяют камфорхинон и 2,2-диметокси-2-фенилацетофенон, и особенно предпочтительно α-дикетоны в сочетании с аминами, такими как 4-(диметиламино)бензоат, N,N-диметиламиноэтилметакрилат, N,N-диметил-сим-ксилидин или триэтаноламин в качестве восстанавливающих агентов. Также особенно подходят фотоинициаторы Норриша, тип I, в частности ацил- или бисацилфосфиноксиды, соединения моноацилтриалкил- или диацилдиалкилгермания, такие как бензоилтриметилгерманий, дибензоилдиэтилгерманий или бис-(4-метоксибензоил)диэтилгерманий. Также могут быть использованы смеси различных фотоинициаторов, такие как, например, дибензоилдиэтилгерманий в сочетании с камфорохиноном и этиловым эфиром 4-диметиламинобензойной кислоты.
Предпочтительно, сочетания редокс-инициаторов, таких как, например, сочетания перекиси бензоила с N,N-диметил-сим.-ксилидином или N,N-диметил-п-толуидином, применяют в качестве инициаторов для полимеризации, проводимой при комнатной температуре, в частности для фотолабильных соединений формулы I. Кроме того, окислительно-восстановительные системы, состоящие из перекисей и восстановительных агентов, таких как, например, аскорбиновая кислота, барбитураты или сульфиновые кислоты, также являются особенно подходящими.
Предлагаемые в изобретении стоматологические пломбировочные материалы также могут содержать выделяющую газ при нагревании добавку. Подходящие выделяющие газ добавки включают, например, азосоединения, такие как азодикарбонамид, 2,2'-азобисизобутиронитрил или 2,2'-азобис(4-цианопентановую кислоту), N-нитрозосоединения, гидразиды, такие как гидразид бензолсульфонила, перекиси, такие как перекись дикумила или ацетондикарбоновой кислоты. Примеры таких соединений описаны, например, в St. Quinn Plastics, Additives & Compounding 2001, 3, 16-21. Температуру разложения, например, для азосоединения, можно регулировать известным методом по схеме замещения (см. D. Brown, R. Jakobi, Monatshefte Chemie 1982, 113, 1403-1414).
Кроме того, стоматологические пломбировочные материалы в соответствии с изобретением могут содержать добавку, которая может превращать испускаемое электромагнитное излучение в тепло. Такие так называемые радиационно-тепловые преобразователи включают органические, неорганические или металлорганические вещества или гибридные компоненты, которые способны превращать УФ, БИК или ИК излучение, видимый свет, микроволны или радиоволны в тепло для отщепления термолабильных групп. Примерами включают красители и пигменты, которые поглощают УФ, БИК или ИК излучение. Примеры красителей, которые поглощают в ИК диапазоне, включают азо, метиновые, антрахиноновые или порфириновые красители. Примеры пигментов, которые поглощают БИК излучение, включают сурьму и оксид индия и олова, фталоцианиновые пигменты, сажу, комплексы дитиолена и Ni и Pt. Примеры соединений, которые поглощают в УФ диапазоне, включают бензотриазолы, триазины, бензофеноны, цианоакрилаты, производные салициловой кислоты и затрудненные аминовые светостабилизаторы (ЗАСС). Примеры добавок, которые поглощают в диапазоне частот микроволн (от 1 до 300 ГГц) или радиоволн (от 10 кГц до 1 ГГц) включают ферромагнитные керамические вещества, так называемые ферриты, которые состоят из оксидов железа гематита (Fe2O3) или магнетита (Fe2O4) и другие оксиды, например, металлов Zn, Mn или Ni, и являются коммерчески доступными в виде порошков.
Кроме того, стоматологические пломбировочные материалы в соответствии с данным изобретением также включают органический или неорганический наполнитель для улучшения механических свойств или для регулирования вязкости. Предпочтительные неорганические порошковые наполнители включают аморфные сферические материалы на основе оксидов, таких как ZrO2 и TiO2, или смешанных оксидов SiO2, ZrO2 и/или TiO2 со средним размером частиц от 0,005 до 2 мкм, предпочтительно от 0,1 до 1 мкм, в форме наночастиц или мелкодисперсные наполнители, такие как пирогенная кремниевая кислота или осажденная двуокись кремния со средним размером частиц от 5 до 200 нм, предпочтительно от 10 до 100 нм, мининаполнители, такие как кварц, стеклокерамика или стеклянный порошок со средним размером частиц от 0,01 до 10 мкм, предпочтительно от 0,1 до 1 мкм, и не прозрачные для рентгеновских лучей наполнители, такие как трифторид иттербия или наночастицы оксида тантала (V) или сульфата бария со средним размером частиц от 10 до 1000 нм, предпочтительно от 100 до 300 нм.
Кроме того, стоматологические пломбировочные материалы в соответствии с данным изобретением могут содержать дополнительные добавки, в частности растворители, такие как вода или этанол, или соответствующие смеси растворителей, а также, например, стабилизаторы, вкусовые добавки, красители, микробиоцидные активные ингредиенты, фторидные выделяющие ионы добавки, оптические отбеливатели или пластификаторы.
Особенно предпочтительно стоматологические пломбировочные материалы на основе термолабильного или фотолабильного полимеризуемого соединения формулы I, и, в частности, формулы II, III и/или IV, которые включают следующие компоненты:
а) от 0,1 до 50% масс., в частности от 1 до 40% масс., предпочтительно от 2 до 30% масс. и особенно предпочтительно от 5 до 30% масс. соединения формулы I и, в частности, формулы II, III и/или IV,
b) от 0,01 до 10% масс., предпочтительно от 0,1 до 3,0% масс. и более предпочтительно от 0,2 до 2% масс. инициатора,
c) от 0 до 80% масс., предпочтительно от 1 до 60% масс. и особенно предпочтительно от 5 до 50% масс. сомономера,
d) от 0 до 30% масс., предпочтительно от 0,5 до 15% масс. и особенно предпочтительно от 1 до 5% масс. клеевого мономера,
е) от 0 до 80% масс. наполнителя,
f) от 0 до 70% масс. растворителя.
Предпочтительное содержание наполнителя зависит от желаемого применения. Адгезивы предпочтительно содержат от 0 до 20% масс. и цементы и композиты предпочтительно содержат от 20 до 80% масс. наполнителя.
То же самое относится и к содержанию растворителя. Адгезивы предпочтительно содержат от 0 до 60% масс. и особенно предпочтительно от 1 до 50% масс. растворителя. Предпочтительны стоматологические материалы, содержащие воду в качестве растворителя. Особенно предпочтительны стоматологические материалы, которые содержат от 0 до 20% масс., и, в частности, от 1 до 10% масс. воды.
Свойства открепления стоматологических пломбировочных материалов в соответствии с данным изобретением могут корректироваться намеренно через состав материалов. Корректировка композиции, подходящей для определенной цели, находится в пределах общих знаний и квалификации специалиста в данной области. Таким образом, способность открепляться по требованию при нагревании увеличивается в зависимости от используемой концентрации термолабильных или фотолабильных компонентов, т.е., в частности, термолабильного или фотолабильного полимеризуемого соединения формулы I, а также, необязательно, термолабильных поперечно-сшивающих мономеров и выделяющих газ добавок. Кроме того, свойства открепления также могут изменяться выбором сомономеров, где плотность поперечного сшивания и, следовательно, прочность и модуль упругости, можно изменять в зависимости от доли поперечно-сшивающих мономеров или добавлением монофункциональных мономеров.
Стоматологические материалы в соответствии с данным изобретением на основе термолабильного или фотолабильного полимеризуемого соединения формулы I, и предпочтительно формулы II, III и/или IV, могут применяться, в частности, для обратимого присоединения, например, брекетов, коронок или виниров. Предпочтительно связь изначально формируется отверждением материалов (адгезивных или цементных) на основе термолабильного или фотолабильного полимеризуемого соединения формулы I. Для открепления приклеенные части необходимо быстро нагреть до температуры, превышающей температуру, при которой происходит отщепление термолабильных связей, или облучить светом с подходящей длиной волны. Целевое применение энергии может осуществляться, например, с помощью источника ИК излучения или лазера. Кроме того, индукционное нагревание может быть достигнуто под воздействием переменного магнитного поля, если ферромагнитные частицы, такие как, например, ферромагнитные наночастицы, введены в стоматологические материалы в соответствии с данным изобретением.
Целью данного изобретения также является использование термолабильного или фотолабильного полимеризуемого соединения формулы I, и, в частности, формулы II, III и/или IV, для получения стоматологических пломбировочных материалов предпочтительно адгезивов или цементов, более предпочтительно самопротравливающих адгезивов или цементов.
Ниже данное изобретение поясняется более подробно с помощью примеров.
ПРИМЕРЫ
Пример 1
Синтез 3,5-диоксо-4-(3-фосфонооксипропил)-10-окса-4-аза-трицикло[5.2.1.02,6]дец-8-ен-1-илметилового эфира метакриловой кислоты (МАТФК)
Стадия 1: 4,10-Диоксатрицикло[5.2.1.02,6]дец-8-ен-3,5-дион
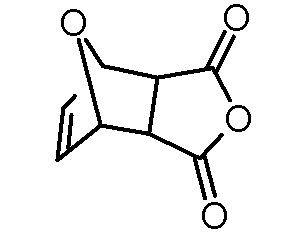
Раствор малеинового ангидрида (98,06 г, 1,0 моль) и фурана (102,12 г, 1,5 моль) в ацетонитриле (200 мл) перемешивали при комнатной температуре в течение 96 ч. Выпавший осадок отфильтровывали, промывали ацетонитрилом (100 мл) и сушили в вакуумной печи (125 мбар, 50°C). Получали 123,30 г (740 ммоль, выход 74%) белого твердого вещества.
1H-ЯМР (ДМСО-d6, 400 МГц): δ = 3,31 (с, 2H), 5,35 (с, 2H), 6,58 (с, 2H).
13C-ЯМР (ДМСО-d6, 100 МГц): δ = 49,0, 81,6, 136,8, 171,5.
Стадия 2: 4-(3-гидроксипропил)-10-окса-4-аза-трицикло[5.2.1.02,6]дец-8-ен-3,5-дион
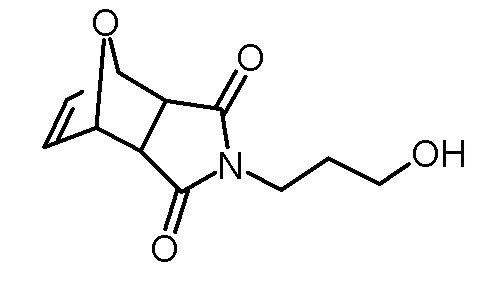
Раствор 3-амино-1-пропанола (15,02 г, 200 ммоль) в метаноле (30 мл) добавляли по каплям к суспензии 4,10-диоксатрицикло[5.2.1.02,6]дец-8-ен-3,5-диона (33,23 г, 200 ммоль) в метаноле (70 мл). Реакционную смесь затем кипятили с обратным холодильником. Через 24 ч раствор концентрировали на роторном испарителе. Желтоватое твердое вещество растворяли в воде (100 мл) и экстрагировали дихлорметаном (3×200 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали, концентрировали на роторном испарителе и сушили в высоком вакууме. Получали 25,40 г (114 ммоль, выход 57%) белого твердого вещества.
1H-ЯМР (ДМСО-d6, 400 МГц): δ = 1,59 (м, 2H), 2,91 (с, 2H), 3,38 (м, 4H), 4,45 (ш с, 1H), 5,12 (с, 2H), 6,55 (с, 2H).
13C-ЯМР (ДМСО-d6, 100 МГц): δ = 30,5, 35,5, 47,0, 58,3, 80,3, 136,4, 176,4.
Стадия 3: 1-(3-гидроксипропил)пиррол-2,5-дион
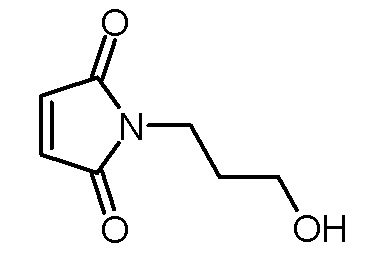
Раствор 4-(3-гидроксипропил)-10-окса-4-аза-трицикло[5.2.1.02,6]дец-8-ен-3,5-диона (17,80 г, 79,8 ммоль) в толуоле (300 мл) кипятили с обратным холодильником в течение 16 ч. Раствор концентрировали на роторном испарителе и остаток сушили в высоком вакууме. Получали 11,92 г (76,8 ммоль, выход 96%) белого твердого вещества.
1H-ЯМР (ДМСО-d6, 400 МГц): δ = 1,65 (м, 2H), 3,40 (т, 2H; J=6,2 Гц), 3.47 (т, 2H; J=7.,4 Гц), 4,48 (ш с, 1H), 6,99 (с, 2H).
13C-ЯМР (ДМСО-d6, 100 МГц): δ = 31,2, 34,7, 58,4, 134,4, 171,0.
Стадия 4: 4-(3-гидроксипропил)-3,5-диоксо-10-окса-4-аза-трицикло[5.2.1.02,6]дец-8-ен-1-илметиловый эфир метакриловой кислоты
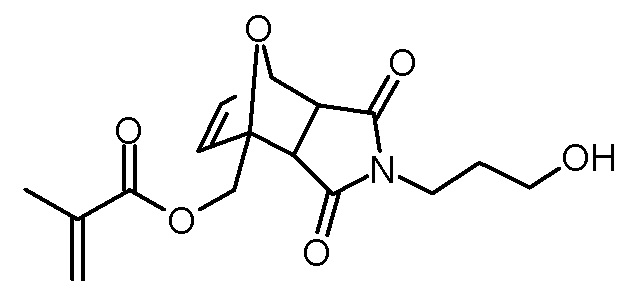
1-(3-гидроксипропил)пиррол-2,5-дион (5,17 г, 33,3 ммоль), метакрилат фурфурила (5,65 г, 34,0 ммоль) и BHT (10 мг) растворяли в бензоле (60 мл). Раствор нагревали при кипячении с обратным холодильником при одновременном введении потока разреженного воздуха. Растворитель отгоняли через 20 ч. Коричневатое масло очищали хроматографией на колонке (SiO2, этилацетат). Получали 2,84 г (8,8 ммоль, 27% выход, смесь экзо- и эндоизомеров) желтоватого масла.
1H-ЯМР (ДМСО-d6, 400 МГц): δ (эндо изомер) = 1,55–1,64 (м, 2H), 1,88 (с, 3H), 3,01–3,05 (д, 1H; J=6,4 Гц), 3,09 (д, 1H; J=6,4 Гц), 3,32–3,49 (м, 4H), 4,41 (д, 1H; J=12,8 Hz), 4,45–4,48 (м, 1H), 4,78–4,84 (м, 1H), 5,15 (д, 1H; J=1,5 Hz), 5,68–5,70 (м, 1H), 6,00–6,03 (м, 1H), 6,47–6,52 (м, 1H), 6,58–6,64 (м, 1H).
13C-ЯМР (ДМСО-d6, 100 МГц): δ (эндо изомер) = 17,8, 30,5, 35,6, 48,1, 49,6, 58,2, 61,7, 80,5, 88,8, 126,2, 135,4, 136,7, 137,3, 166,1, 174,7, 176,0.
Стадия 5: 3,5-диоксо-4-(3-фосфонооксипропил)-10-окса-4-аза-трицикло[5.2.1.02,6]дец-8-ен-1-илметиловый эфир метакриловой кислоты (МАТФК)
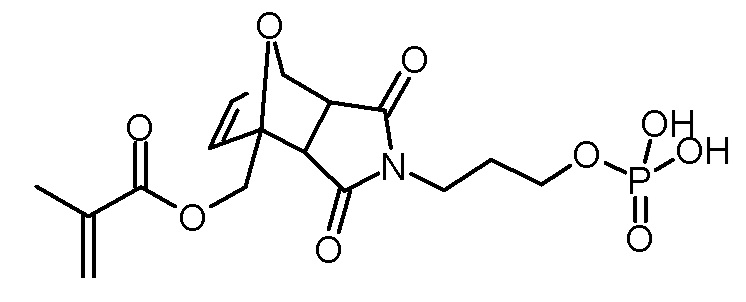
Раствор 4-(3-гидроксипропил)-3,5-диоксо-10-окса-4-аза-трицикло[5.2.1.02,6]дец-8-ен-1-илметилового эфира метакриловой кислоты (2,6 г, 8,2 ммоль), ВНТ (10 мг) и триэтиламина (910 мг, 9,0 ммоль) в тетрагидрофуране (20 мл) добавляли по каплям к раствору оксихлорида фосфора (1,39 г, 9,0 ммоль) в тетрагидрофуране (30 мл) при -5°С. После завершения добавления суспензию перемешивали в течение 3 ч при -5°С и затем по каплям добавляли воду (2 мл). Суспензию перемешивали в течение еще 30 мин при -5°С, и затем осадок отфильтровывали холодным. Желтоватый фильтрат промывали насыщенным водным раствором NaCl (3×30 мл). Объединенные водные фазы повторно экстрагировали тетрагидрофураном (2×30 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали на роторном испарителе. В коричневатое масло добавляли ацетонитрил (2×50 мл) для удаления воды, и затем концентрировали на роторном испарителе. К остатку добавляли диэтиловый эфир (50 мл) и перемешивали при комнатной температуре. Через 1 ч растворитель декантировали. Коричневое масло сушили на роторном испарителе и в высоком вакууме. Получали 2,46 г (6,1 ммоль, 75% выход, смесь экзо и эндо изомеров) коричневатой смолы.
1H-NMR (ДМСО-d6, 400 МГц): δ (эндо изомер) = 1,73–1,82 (м, 2H), 1,87 (с, 3H), 3,03 (д, 1H; J=6,5 Гц), 3,10 (д, 1H; J=6,5 Гц), 3,42–3,48 (м, 2H), 3,76–3,84 (м, 2H), 4,41 (д, 1H; J=12,5 Гц), 4,84 (д, 1H; J=12,5 Гц), 5,15 (с, 1H), 5,69 (с, 1H), 6,01 (с, 1H), 6,50 (д, 1H; J=5,7 Гц), 6,59–6,63 (м, 1H), 6,94 (ш, 2H).
13C-NMR (ДМСО-d6, 100 МГц): δ (эндо изомер) = 17,8, 28,2 (д, J = 7 Гц), 35,2, 48,2, 49,7, 61,6, 63,0 (д, J=5 Гц), 80,4, 88,8, 126,2, 135,4, 136,7, 137,3, 166,1, 174,7, 176,0.
31P-ЯМР (ДМСО-d6, 162 МГц): δ = -1,3.
Пример 2
Синтез 3-(3,5-диоксо-1-фосфонооксиметил-10-окса-4-аза-трицикло[5.2.1.02,6]дец-8-ен-4-ил)пропилового эфира метакриловой кислоты
Стадия 1: 3-(2,5-диоксо-2,5-дигидропиррол-1-ил)пропиловый эфир метакриловой кислоты
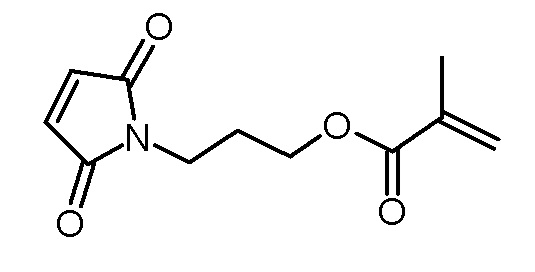
1-(3-гидроксипропил)пиррол-2,5-дион (5,36 г, 34,5 ммоль), триэтиламин (3,85 г, 38,0 ммоль) и N,N-диметиламинопиридин (120 мг, 1,0 ммоль) растворяли в дихлорметане (80 мл). Раствор метакрилового ангидрида (5,86 г, 38,0 ммоль) и ВНТ (10 мг) в дихлорметане (20 мл) добавляли по каплям при 0°C, и реакционную смесь затем перемешивали в течение 2 ч при 0°C и 22 ч при комнатной температуре. Реакционный раствор промывали водой (3×50 мл). Объединенные водные фазы повторно экстрагировали дихлорметаном (50 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали на роторном испарителе. Неочищенный продукт очищали хроматографией на колонке (SiO2, н-гексан/этилацетат 1:1). Получали 2,81 г (12,5 ммоль, 35% выход) желтоватого масла.
1H-NMR (CDCl3, 400 МГц): δ = 1,95 (2, 3H), 1,97–2,04 (м, 2H), 3,66 (т, 2H; J=6,9 Гц), 4,15 (т, 2H; J=6,2 Гц), 5,57 (м, 1H), 6,12 (с, 1H), 6,72 (с, 2H).
13C-ЯМР (CDCl3, 100 МГц): δ = 18,3, 27,6, 34,9, 61,8, 125,6, 134,2, 126,2, 167,2, 170,6.
Стадия 2: 3-(1-гидроксиметил-3,5-диоксо-4-10-окса-4-азатрицикло[5.2.1.02,6]дец-8-ен-4-ил)пропиловый эфир метакриловой кислоты
3-(2,5-диоксо-2,5-дигидро-пиррол-1-ил)пропиловый эфир метакриловой кислоты (2,71 г, 12,1 ммоль), фурфуриловый спирт (1,28 г, 13,0 ммоль) и ВНТ (10 мг) растворяли в бензоле (40 мл). Раствор нагревали при кипячении с обратным холодильником при введении потока разреженного воздуха. Растворитель отгоняли через 20 ч. Коричневатое масло, полученное в виде неочищенного продукта, очищали хроматографией на колонке (SiO2, этилацетат). Получали 3,10 г (9,6 ммоль, 80% выход, смесь экзо- и эндо-изомеров) желтого масла.
1H-ЯМР (CDCl3, 400 МГц): δ = 1,78–1,89 (м, 0,4H; экзо), 1,93–2,02 (м, 5,8H; экзо/эндо), 2,94–3,02 (м, 3H; эндо), 3,43–3,48 (м, 0,6H; экзо), 3,55–3,69 (м, 2,2H; экзо/эндо), 4,05–4,14 (м, 4,4H; экзо/эндо), 4,15–4,22 (м, 0,2H; экзо), 4,25–4,31 (м, 0,2H; экзо), 5,25 (м, 1H; эндо), 5,28–5,32 (м, 0,2H; экзо), 5,57–5,60 (м, 1,2H; экзо/эндо), 6,10–6,12 (м, 0,2H; эндо), 6,12–6,14 (м, 1H; эндо), 6,35–6,38 (м, 0,2H; экзо), 6,46–6,49 (м, 0,2H; экзо), 6,52–6,56 (м, 1H; эндо), 6,59–6,62 (м, 1H; эндо).
13C-ЯМР (CDCl3, 100 МГц): δ = 18,3, 26,6 (эндо), 26,7 (экзо), 35,5 (экзо), 35,8 (эндо), 46,0 (экзо), 48,1 (эндо), 49,9, 60,7 (эндо), 61,3 (экзо), 61,4 (эндо), 61,6 (экзо), 79,5 (экзо), 80,9 (эндо), 91,5 (эндо), 92,1 (экзо), 125,7 (эндо), 125,8 (экзо), 134,9 (экзо), 135,7 (экзо), 136,1 (экзо), 136,2 (эндо), 137,0 (эндо), 138,3 (эндо), 167,2 (экзо), 167,3 (эндо), 174,7 (экзо), 175,1 (экзо), 175,8 (эндо), 175,9 (эндо).
Стадия 3: 3-(3,5-диоксо-1-фосфонооксиметил-10-окса-4-аза-трицикло[5.2.1.02,6]дец-8-ен-4-ил)пропиловый эфир метакриловой кислоты
Раствор 3-(1-гидроксиметил-3,5-диоксо-10-окса-4-азатрицикло[5.2.1.02,6]дец-8-ен-4-ил)пропилового эфира метакриловой кислоты (3,00 г, 9,3 ммоль), ВНТ (10 мг) и триэтиламина (1,04 г, 10,3 ммоль) в тетрагидрофуране (20 мл) по каплям добавляли к раствору хлорокиси фосфора (1,57 г, 10,3 ммоль) в тетрагидрофуране (30 мл) при -5°С. После завершения добавления суспензию перемешивали в течение 3 ч при температуре от -5°С, затем по каплям добавляли воду (2 мл). Суспензию перемешивали при -5°С в течение еще 30 мин, и затем осадок отфильтровывали холодным. Желтоватый фильтрат промывали насыщенным водным раствором NaCl (3×30 мл). Объединенные водные фазы повторно экстрагировали тетрагидрофураном (2×30 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали на роторном испарителе. В коричневатое масло добавляли ацетонитрил (2×50 мл) для удаления воды, и смесь концентрировали на роторном испарителе. Остаток обрабатывали диэтиловым эфиром (2×50 мл) и перемешивали при комнатной температуре. Через 1 ч растворитель декантировали. Коричневое масло сушили в роторном испарителе и в высоком вакууме. Получали 3,19 г (7,9 ммоль, выход 85%, смесь экзо и эндо изомеров) гигроскопической белой пены.
1H-ЯМР (ДМСО-d6, 400 МГц): δ = 1,65–1,75 (м, 0,4H; экзо), 1,75–1,85 (м, 2H; эндо), 1,87 (с, 3,6H; экзо/эндо), 3,02 (дд, 2H; J = 28,2 Гц, 6,4 Гц; эндо), 3,29 (т, 0,4H; J = 7,0 Гц; экзо), 3,41–3,49 (м, 2,2H; экзо/эндо), 3,66 (дд, 0,2H; J = 7,8 Гц, 5,6 Гц; экзо), 3,94–4,08 (м, 3,4H; экзо/эндо), 4,32 (дд, 0,2H; J = 12,2 Гц, 5,2 Гц; экзо), 4,42 (дд, 0,2H; J = 12,2 Гц, 5,8 Гц; экзо), 4,54 (кв, 1H; J = 6,1 Гц; эндо), 5,08–5,11 (м, 1H; эндо), 5,25–5,29 (м, 0,2H; экзо), 5,64–5,68 (м, 1,2H; экзо/эндо), 6,00–6,05 (м, 1,2H; экзо/эндо), 6,36 (д, 0,2H, J = 5,5 Гц; экзо), 6,44–6,50 (м, 1,2H; экзо/эндо), 6,54–6,58 (м, 1H; эндо), 6,66 (ш с, 2,4H; экзо/эндо).
13C-ЯМР (ДМСО-d6, 100 МГц): δ = 17,9, 26,1 (экзо), 26,2 (эндо), 34,6 (экзо), 34,8 (эндо), 45,6 (экзо), 47,3 (экзо), 48,0 (эндо), 49,8 (эндо), 61,5 (эндо), 61,6 (экзо), 62,9 (д, J = 5 Гц; эндо), 63,3 (д, J = 5 Гц; экзо), 78,7 (экзо), 80,4 (эндо), 89,7 (д, J = 10 Гц; эндо), 90,0 (д, J = 10 Гц; экзо), 125,6, 134,5 (экзо), 135,4 (экзо), 135,8 (эндо), 136,8 (эндо), 137,0 (эндо), 166,4, 174,7 (экзо), 174,7 (эндо), 174,8 (экзо), 176,1 (эндо).
Пример 3
Радикальная фотополимеризация метакрилата фосфорной кислоты МАТФК из примера 1
Получали смесь 2,97 г метакрилата фосфорной кислоты МАТФК из примера 1, 6,95 г поперечно-сшивающего агента N,N'-диэтил-1,3-бис(акриламидо)пропана, 0,03 г фотоинициатора камфорохинона и 0,05 г аминового ускорителя 4-(диметиламино)бензоата. Каплю смеси помещали на стеклянную пластину, покрывали пленкой ПЭТ и облучали полимеризационной лампой Bluephase (Ivoclar Vivadent AG, интенсивность света 1000 мВт/см-2) в течение 20 с. Облученный слой затем отверждали. Затем смесь исследовали с помощью фото-ДСК (дифференциальной сканирующей калориметрии, Perkin Elmer DSC 7) и измеряли теплу полимеризации 273 Дж/г.
Пример 4
Получение светоотверждаемого клея на основе метакрилата фосфорной кислоты МАТФК из примера 1
Клей получали из 1,09 г метакрилата фосфорной кислоты МАТФК из примера 1, 1,49 г монофункционального сомономера 2-гидроксиэтилметакрилата, 3,25 г поперечно-сшивающего агента бис-GMA, 0,99 г поперечно-сшивающего агента UDMA, 1,01 г поперечно-сшивающего агента диметакрилата глицерина, 0,02 г фотоинициатора камфорохинона, 0,05 г аминового ускорителя 4-(диметиламино)бензоата, 0,10 г фотоинициатора на основе оксида ацилфосфина Lucirin TPO и 2,00 г растворителя этанола. Клей может отверждаться с использованием полимеризационной лампы Bluephase (Ivoclar Vivadent AG, интенсивность света 1000 мВт/см-2).
Пример 5
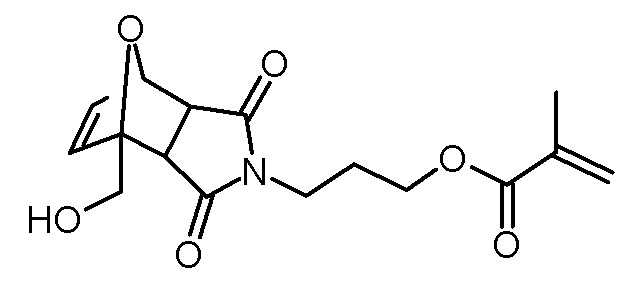
Синтез 3-[3,5-диоксо-1-(11-фосфонооксиундецилоксиметил)-10-окса-4-аза-трицикло[5.2.1.02,6]дец-8-ен-4-ил]пропилового эфира метакриловой кислоты
Стадия 1: 2-(11-бромундецилокси)тетрагидропиран
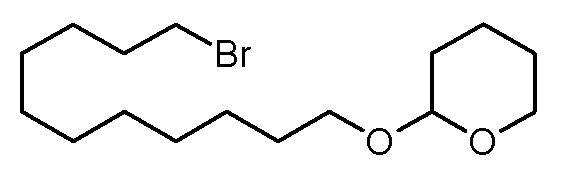
3,4-дигидро-2H-пиран (21,87 г, 260 ммоль) по каплям добавляли к раствору 11-бромундеканола (50,24 г, 200 ммоль) и моногидрата толуол-4-сульфоновой кислоты (80 мг, 0,4 ммоль) в дихлорметане (100 мл). Реакционную смесь перемешивали при комнатной температуре. Через 24 ч коричневый раствор фильтровали через тонкий слой силикагеля. Фильтрат концентрировали на роторном испарителе и сушили в высоком вакууме. Получали 65,85 г (196 ммоль, выход 98%) светло-желтоватого масла.
1H-ЯМР (CDCl3, 400 МГц): δ = 1,23–1,37 (м, 12H), 1,37–1,47 (м, 2H), 1,48–1,64 (м, 6H), 1,65–1,76 (м, 1H), 1,77–1,90 (м, 3H), 3,35–3,42 (м, 3H), 3,46–3,53 (м, 1H), 3,70–3,75 (м, 1H), 3,84–3,90 (м, 1H), 4,56–4,58 (м, 1H).
13C-ЯМР (CDCl3, 100,6 МГц): δ = 19,7, 25,4, 26,2, 28,2, 28,8, 29,4, 29,5, 29,6, 29,8, 30,8, 32,9, 33,9, 62,3, 67,7, 98,8.
Стадия 2: 2-[11-(фуран-2-илметокси)ундецилокси]тетрагидропиран
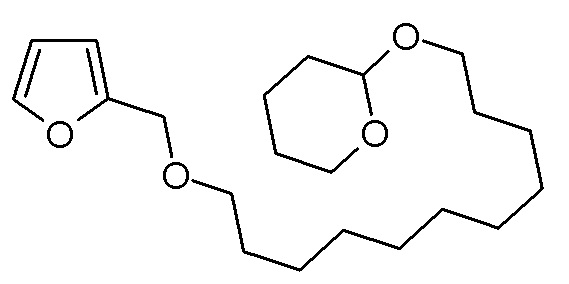
Фурфуриловый спирт (9,81 г, 100 ммоль) добавляли по каплям к суспензии гидрида натрия (2,40 г, 100 ммоль) в ТГФ (100 мл). Суспензию перемешивали при комнатной температуре в течение 1 ч, затем по каплям добавляли раствор 2-(11-бромундецилокси)тетрагидропирана (33,53 г, 100 ммоль) в ТГФ (100 мл). Реакционную смесь нагревали в течение 16 ч при кипячении с обратным холодильником. После охлаждения смесь гасили насыщенным водным раствором NH4Cl (100 мл). Двухфазную смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали, концентрировали на роторном испарителе и сушили в высоком вакууме. Неочищенный продукт очищали хроматографией на колонке (SiO2, дихлорметан). Получали 20,58 г (58,4 ммоль, выход 58%) желтоватого масла.
1H-ЯМР (CDCl3, 400 МГц): δ = 1,25–1,40 (м, 16H), 1,48–1,62 (м, 6H), 1,66–1,74 (м, 1H), 1,78–1,87 (м, 1H), 3,34–3,40 (м, 1H), 3,45 (т, 2H; J = 6,8 Hz), 3,47–3,52 (м, 1H), 3,70–3,75 (м, 1H), 3,84–3,90 (м, 1H), 4,42 (с, 2H), 4,56–4,58 (м, 1H), 6,28–6,30 (м, 1H), 6,32–6,33 (с, 1H), 7,38–7,39 (м, 1H).
13C-ЯМР (CDCl3, 100,6 МГц): δ = 19,7, 25,5, 26,1, 26,3, 29,1, 29,4, 29,5, 29,5, 29,5, 29,6, 29,7, 29,8, 30,8, 62,3, 64,7, 67,7, 70,4, 98,8, 108,9, 110,2, 142,6, 152,2.
Стадия 3: 11-(фуран-2-илметокси)ундекан-1-ол
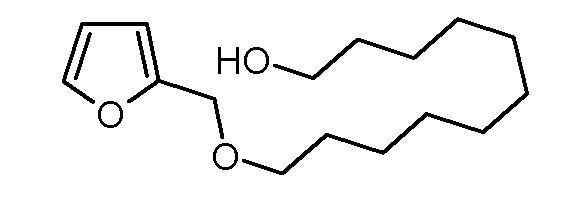
Раствор 2-[11-(фуран-2-илметокси)ундецилокси]тетрагидропирана (20,48 г, 58,1 ммоль) и моногидрата толуол-4-сульфоновой кислоты (480 мг, 2,4 ммоль) в метаноле (100 мл) перемешивали в течение 20 ч при комнатной температуре. Реакционную смесь концентрировали на роторном испарителе и неочищенный продукт очищали хроматографией на колонке (SiO2, этилацетат). Получали 7,18 г (26,8 ммоль, выход 46%) желтоватого твердого вещества.
1H-ЯМР (CDCl3, 400 МГц): δ = 1,25–1,37 (м, 14H), 1,51–1,62 (м, 4H), 1,84 (с, 1H), 3,45 (т, 2H; J = 6,8 Гц), 3,61 (т, 2H; J = 6,8 Гц), 4,43 (с, 2H), 6,29–6,30 (м, 1H), 6,32–6,34 (м, 1H), 7,39–7,40 (м, 1H).
13C-ЯМР (CDCl3, 100,6 МГц): δ = 25,8, 26,1, 29,4, 29,5, 29,5, 29,6, 29,6, 32,8, 62,9, 64,7, 70,4, 108,9, 110,2, 142,6, 152,1.
Стадия 4: 3-[3,5-диоксо-1-(11-гидроксиундецилоксиметил)-10-окса-4-азатрицикло[5.2.1.02,6]дец-8-ен-4-ил]пропиловый эфир метакриловой кислоты
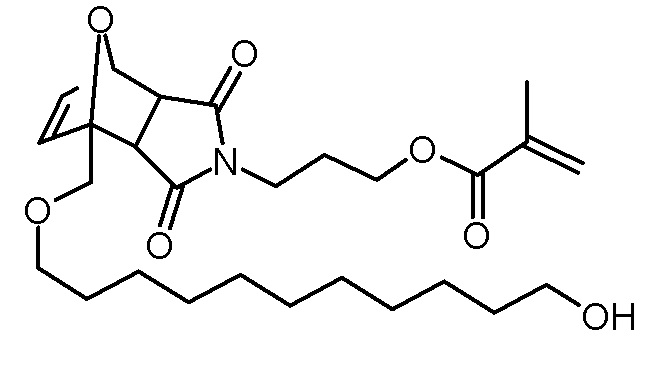
Раствор 11-(фуран-2-илметокси)ундекан-1-ола (7,00 г, 26,1 ммоль), 3-(2,5-диоксо-2,5-дигидропиррол-1-ил)пропилового эфира метакриловой кислоты (5,82 г, 26,1 ммоль) и BHT (10 мг) в толуоле (100 мл) нагревали до 80°С с введением потока разреженного воздуха. Через 20 ч раствор концентрировали на роторном испарителе и неочищенный продукт очищали хроматографией на колонке (SiO2, этилацетат). Получали 6,16 г (12,5 ммоль, выход 48%, смесь экзо и эндо изомеров) в виде желтоватого масла.
1H-ЯМР (CDCl3, 400 МГц): δ = 1,25-1,36 (м, 22,4H; экзо/эндо), 1,51–1,63 (м, 6,6H; экзо/эндо), 1,76 (с, 1,6H; экзо/эндо), 1,81–1,88 (м, 1,2H; экзо), 1,92–1,98 (м, 6,4H; экзо/эндо), 2,91 (дд, 2H; J = 40,2 Гц, 6,4 Гц; эндо), 3,43–3,51 (м, 2,4H; экзо), 3,52-3,65 (м, 8,4H), 3,81 (д, 1H; J = 11,6 Гц; эндо), 4,01 (д, 0,6H; J = 11,6 Гц; эндо), 4,06–4,15 (м, 5H; экзо/эндо), 5,23–5,24 (м, 1H; эндо), 5,28–5,30 (м, 0,6H; экзо), 5,57–5,58 (м, 1,6H; экзо/эндо), 6,11–6,14 (м, 1,6H; экзо/эндо), 6,30–6,32 (д, 0,6H; J = 5,8 Гц; экзо), 6,44–6,46 (м, 0,6H; экзо), 6,51–6,54 (м, 2H; эндо).
13C-ЯМР (CDCl3, 100,6 МГц): δ = 18,3, 18,3, 25,7, 26,0, 26,7, 29,4, 29,4, 29,4, 29,5, 29,5, 29,5, 29,6, 32,8, 35,4, 35,7, 45,7, 47,8, 48,3, 49,9, 61,5, 61,6, 62,9, 67,9, 68,4, 72,1, 72,2, 79,6, 81,0, 90,7, 91,4, 125,6, 125,7, 135,1, 135,3, 136,1, 136,2, 136,6, 138,1, 167,2, 174,5, 174,8, 175,0, 176,0.
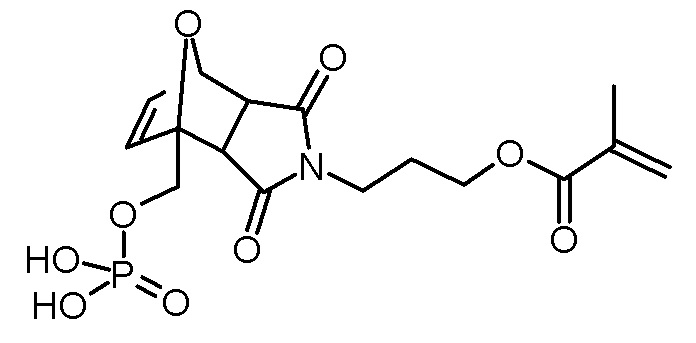
Стадия 5: 3-[3,5-диоксо-1-(11-фосфонооксиундецилокси-метил)-10-окса-4-азатрицикло[5.2.1.02,6]дец-8-ен-4-ил]пропиловый эфир метакриловой кислоты
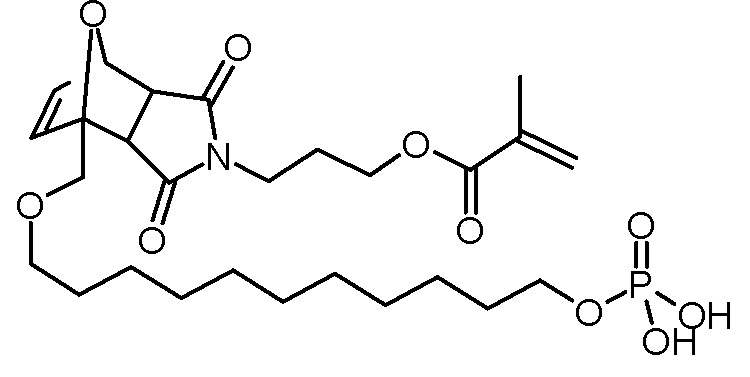
Раствор 3-[3,5-диоксо-1-(11-гидроксиундецилоксиметил)-10-окса-4-аза-трицикло[5.2.1.02,6]дец-8-ен-4-ил]пропилового эфира метакриловой кислоты (6,06 г, 12,3 ммоль), ВНТ (10 мг) и триэтиламина (1,37 г, 13,6 ммоль) в ТГФ (30 мл) добавляли по каплям при -5°С к раствору хлорокиси фосфора (2,08 г, 13,6 ммоль) в ТГФ (50 мл). После завершения добавления суспензию перемешивали в течение 3 ч при -5°С, затем по каплям добавляли воду (2 мл). Суспензию перемешивали в течение еще 30 мин на бане со льдом и осадок отфильтровывали холодным. Желтоватый фильтрат промывали насыщенным водным раствором NaCl (3×50 мл). Объединенные водные фазы повторно экстрагировали ТГФ (2×30 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали на роторном испарителе. К коричневатому маслу добавляли ацетонитрил (2×50 мл) для удаления воды и концентрировали на роторном испарителе. Коричневое масло обрабатывали диэтиловым эфиром (4×100 мл) и перемешивали при комнатной температуре в течение 10 мин. Темно-коричневое масло выпадало в осадок. Растворитель декантировали. Объединенный эфирный раствор концентрировали на роторном испарителе и сушили в высоком вакууме. Получали 3,87 г (6,8 ммоль, выход 55%, смесь экзо и эндо изомеров) коричневатого масла.
1H-ЯМР (ДМСО-d6, 400 МГц): δ = 1,22-1,32 (м, 22,4H; экзо/эндо), 1,43-1,60 (м, 6,6H; экзо/эндо), 1,69–1,76 (м, 1,2H; экзо), 1,80–1,85 (м, 2H; эндо), 1,87–1,91 (м, 5,8H; экзо/эндо), 2,96 (дд, 2H; J = 55,6 Гц, 6,4 Гц; эндо), 3,30 (т, 1H; J = 6,4 Гц; эндо), 3,36–3,54 (м, 6H; эндо), 3,59–3,66 (м, 1,6H; экзо/эндо), 3,90–4,09 (м, 7,2H; экзо/эндо), 5,08–5,09 (м, 1H; эндо), 5,24–5,26 (м, 0,6H; экзо), 5,67–5,69 (м, 1,6H; экзо/эндо), 6,00–6,06 (м, 1,6H; экзо/эндо), 6,32–6,34 (м, 0,6H; экзо), 6,43–6,47 (м, 1,6H; экзо/эндо), 6,52–6,55 (м, 1H; эндо), 8,68 (ш с, 3,2H; экзо/эндо).
13C-ЯМР (ДМСО-d6, 100,6 МГц): δ = 17,9, 25,1, 25,5, 26,1, 28,6, 28,8, 29,0, 29,0, 29,0, 29,8, 29,9, 30,4, 34,7, 45,5, 47,3, 48,1, 49,6, 61,3, 61,6, 65,2 (д, J = 5 Гц), 67,6, 68,1, 70,9, 71,0, 78,7, 80,3, 90,3, 90,8, 125,6, 134,5, 135,9, 135,8, 136,4, 137,6, 166,3, 166,4, 174,7, 174,9, 175,0, 176,2.
31P-ЯМР (ДМСО-d6, 162 MHz): δ = -1.1.
Пример 6
Синтез 3,5-диоксо-4-(10-фосфонооксидецил)-10-окса-4-азатрицикло[5.2.1.02,6]дец-8-ен-1-илметиловый эфир метакриловой кислоты
Стадия 1: 4-(10-гидроксидецил)-4-10-окса-4-азатрицикло[5.2.1.02,6]дец-8-ен-3,5-дион
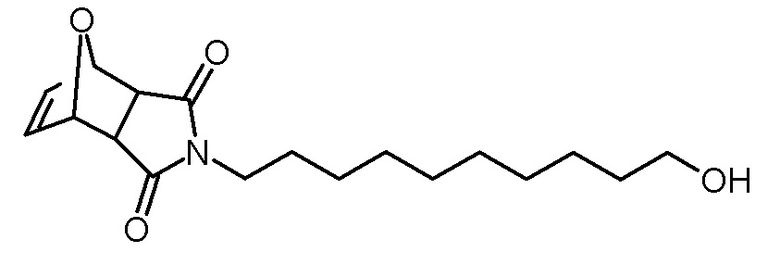
Раствор 10-амино-1-деканола (5,36 г, 30,9 ммоль) в метаноле (20 мл) добавляли по каплям к суспензии 4,10-диоксатрицикло[5.2.1.02,6]дец-8-ен-3,5-диона (5,13 г, 30,9 ммоль) в метаноле (30 мл). Реакционную смесь нагревали при кипячении с обратным холодильником в течение 24 ч и затем концентрировали на роторном испарителе. Сырой продукт очищали хроматографией на колонке (SiO2, этилацетат). Получали 1,52 г (4,7 ммоль, выход 15%) желтоватого твердого вещества.
1H-ЯМР (CDCl3, 400 Мгц): δ = 1,22–1,37 (м, 12H), 1,51–1,59 (м, 4H), 2,60 (ш с, 1H), 2,84 (с, 2H), 3,46 (т, 2H; J = 7,4 Гц), 3,62 (т, 2H; J = 6,5 Hz), 5,26 (с, 2H), 6,51 (с, 2H).
13C-ЯМР (CDCl3, 100,6 МГц): δ = 25,7, 26,6, 27,5, 29,0, 29,3, 29,3, 29,4, 32,7, 39,0, 47,4, 62,9, 80,9, 136,6, 176,4.
Стадия 2: 1-(10-гидроксидецил)пиррол-2,5-дион
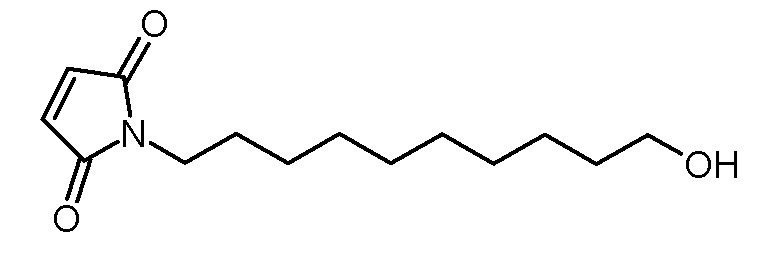
Суспензию 4-(10-гидроксидецил)-10-окса-4-азатрицикло[5.2.1.02,6]дец-8-ен-3,5-диона (1,52 г, 4, 7 ммоль) в толуоле (50 мл) нагревали при кипячении с обратным холодильником в течение 16 ч. Раствор декантировали из нерастворенного остатка, концентрировали на роторном испарителе и сушили в высоком вакууме. Получали 1,15 г (4,5 ммоль, 97% выход) белого твердого вещества.
1H-ЯМР (CDCl3, 400 МГц): δ = 1,25–1,36 (м, 12H), 1,52–1,61 (м, 4H), 2,63 (ш с, 1H), 3,50 (т, 2H; J = 7,2 Hz), 3,63 (т, 2H; J = 6,8 Hz), 6,69 (с, 2H).
13C-ЯМР (CDCl3, 100,6 МГц): δ = 25,7, 26,7, 28,5, 29,0, 29,3, 29,4, 32,7, 37,9, 63,0, 134,0, 170,9.
Стадия 3: 4-(10-гидроксидецил)-3,5-диоксо-10-окса-4-азатрицикло[5.2.1.02,6]дец-8-ен-1-илметиловый эфир метакриловой кислоты
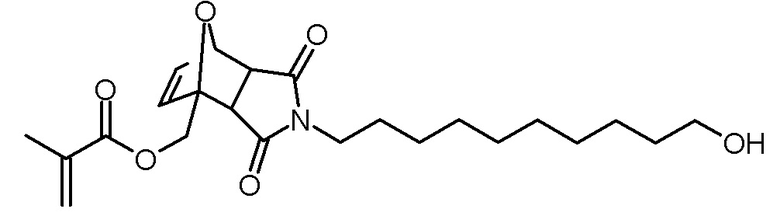
Раствор фурфурилметакрилата (750 мг, 4,5 ммоль), 1-(10-гидроксидецил)пиррол-2,5-диона (1,05 г, 4,1 ммоль) и BHT (5 мг) в толуоле (30 мл) нагревали до 80°С с введением потока разреженного воздуха. Через 20 ч реакционный раствор концентрировали на роторном испарителе и сушили в высоком вакууме. Неочищенный продукт очищали хроматографией на колонке (SiO2, н-гексан/этилацетат 1:1). Получали 560 мг (1,3 ммоль, выход 33%) эндо-изомера в виде белого твердого вещества и 430 мг (1,0 ммоль, 25% выход) экзо-изомера в виде желтоватого масла.
эндо-изомер:
1H-ЯМР (CDCl3, 400 МГц): δ = 1,19–1,36 (м, 12H), 1,37–1,46 (м, 2H), 1,52–1,61 (м, 3H), 1,97 (с, 3H), 3,31 (т, 2H; J = 7,6 Гц), 3,38 (д, 1H; J = 7,6 Гц), 3,61–3,66 (м, 3H), 4,69 (д, 1H; J = 12,8 Гц), 4,91 (д, 1H; J = 12,8 Гц), 5,31 (дд, 1H; J = 5,3 Гц, 1,7 Гц), 5,61–5,61 (м, 1H), 6,17 (с, 1H), 6,36 (д, 1H; J = 5,8 Гц), 6,45 (дд, 1H; J = 5,7 Гц, 1,6 Гц).
13C-ЯМР (CDCl3, 100,6 МГц): δ = 18,3, 25,7, 26,8, 27,4, 29,0, 29,3, 29,4, 32,8, 38,7, 46,8, 47,7, 62,2, 62,9, 79,6, 89,8, 126,5, 134,4, 135,6, 135,7, 166,8, 174,4, 174,6.
экзо-изомер:
1H-ЯМР (CDCl3, 400 МГц): δ = 1,25–1,35 (м, 12H), 1,52–1,58 (м, 4H), 1,95 (с, 3H), 1,99 (ш с, 1H), 2,95 (дд, 2H; J = 28,2 Гц, 6,5 Гц), 3,46 (т, 2H; J = 7,5 Гц), 3,61 (т, 2H; J = 6,8 Гц), 4,52 (д, 1H; J = 12,8 Гц), 4,98 (д, 1H; J = 12,8 Гц), 5,27 (д, 1H; J = 1,6 Гц), 5,59–5,61 (м, 1H), 6,13 (с, 1H), 6,45 (д, 1H; J = 5,8 Гц), 6,56 (дд, 1H; J = 5,6 Гц, 1,6 Гц),
13C-ЯМР (CDCl3, 100,6 МГц): δ = 18,3, 25,8, 26,6, 27,5, 29,0, 29,3, 29,3, 29,4, 32,8, 39,0, 48,3, 49,9, 61,6, 62,9, 81,1, 89,6, 126,3, 135,8, 137,1, 137,4, 166,8, 174,3, 175,8.
Стадия 4: 3,5-диоксо-4-(10-фосфонооксидецил)-10-4-азатрицикло[5.2.1.02,6]дец-8-ен-1-илметиловый эфир метакриловой кислоты
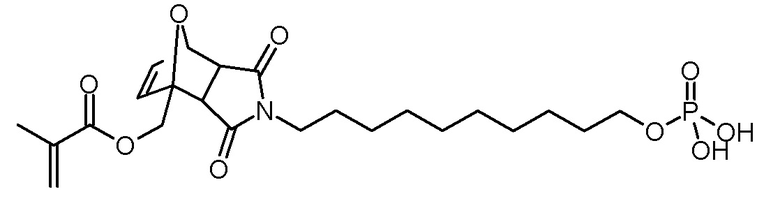
Раствор эндо/экзо 4-(10-гидроксидецил)-3,5-диоксо-10-окса-4-азатрицикло[5.2.1.02,6]дец-8-ен-1-илметилового эфира метакриловой кислоты (890 мг, 2,1 ммоль), ВНТ (5 мг) и триэтиламина (240 мг, 2,3 ммоль) в ТГФ (30 мл) по каплям добавляли к раствору хлорокиси фосфора (360 мг, 2,3 ммоль) в ТГФ (20 мл) при -5°С. После завершения добавления суспензию перемешивали в течение 3 ч при -5°С, затем по каплям добавляли воду (2 мл). Суспензию перемешивали в течение еще 30 мин на бане со льдом, осадок отфильтровывали холодным. Желтоватый фильтрат промывали насыщенным водным раствором NaCl (3×50 мл). Объединенные водные фазы повторно экстрагировали ТГФ (2×30 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали на роторном испарителе. К коричневатому маслу добавляли ацетонитрил для удаления воды (2×50 мл) и концентрировали на роторном испарителе. Остаток сушили в высоком вакууме. Получали 1,01 г (2,0 ммоль, выход 95%, смесь экзо- и эндо-изомеров) бесцветного масла.
Пример 7
Подготовка ОпТ адгезивов на основе фосфатов метакрилата из примеров 5 и 6
3 грунтовочных раствора готовили для исследования прочности сцепления при сдвиге между ZrO2 керамикой и композитным цементом в соответствии с данным изобретением. Растворы содержали 1% масс. каждого из фосфата метакрилата из примера 5 (грунтовка А), фосфата метакрилата из примера 6 (грунтовка В) или фосфата 10-метакрилоилоксидецила (грунтовка С, сравнение) в этаноле, соответственно.
Для определения прочности сцепления соответствующие грунтовочные растворы наносили на ZrO2 керамические тестеры (IPS e.max ZirCAD, Ivoclar Vivadent, стабилизированный иттрием оксид циркония) и растворитель выдували. Затем на слой грунтовки наносили композитный цемент Multilink Automix (Ivoclar Vivadent) и отверждали в течение 20 сек светодиодной лампой Bluephase C8 (Ivoclar Vivadent), а затем в течение 3 мин в световой печи Spectramat (Ivoclar Vivadent). Тестеры затем хранили в воде в течение 24 ч при 37°С и прочность сцепления при сдвиге измеряли аналогично инструкции ISO "ISO 1994-ISO TR 11405: Dental Materials Guidance on Testing of Adhesion to Tooth Structure". Во втором опыте тестеры дополнительно хранили в сушильной печи при 130°С течение 60 мин после хранения в воде и только потом определяли прочность сцепления при сдвиге после быстрого охлаждения тестеров. Результаты представлены в таблице 1, и они показывают, что при температурной нагрузке на сцепление керамического композита с грунтовками с адгезивными мономерами в соответствии с данным изобретением возникает значительное большее снижение прочности сцепления, и поэтому такое сцепление может быть легче расцеплено.
Прочность сцепления при сдвиге (ПСС, в МПа) сцепления между ZrO2 керамикой и композитным цементом
b)ХВ+ТО = хранение в воде и тепловая обработка
Пример 8
Синтез 2-(метакрилоилокси)этилового эфира 4-[4(5)-(метакрилоилоксиметил)-2-пиридин-2-ил-3,6-дигидро-2Н-тиопиран-2-илсульфанилметил]бензойной кислоты
Стадия 1: 2-Бензолсульфонилметилпиридин
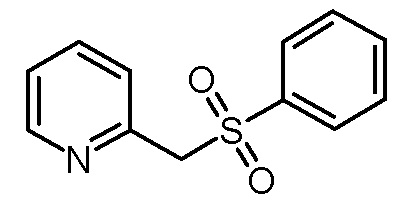
Суспензию гидрохлорида 2-(хлорметил)пиридина (32,81 г, 200 ммоль) в ацетонитриле (200 мл) обрабатывали фенилсульфонатом натрия (49,24 г, 300 ммоль), бромидом тетрапропиламмония (10,64 г, 40 ммоль) и 1,8-диазабицикло[5.4.0]ундец-7-еном (30,44 г, 200 ммоль). Реакционную смесь нагревали при кипячении с обратным холодильником в течение 16 ч и затем концентрировали на роторном испарителе. Остаток помещали в дихлорметан (200 мл), промывали насыщенным водным раствором NaCl (3×100 мл), сушили над Na2SO4, фильтровали и концентрировали до приблизительно половины объема на роторном испарителе. Коричневый раствор фильтровали через слой силикагеля. Получали 41,55 г (178 ммоль, 89% выход) желтоватого твердого вещества.
1H-ЯМР (CDCl3, 400 МГц): δ = 4,56 (с, 2H), 7,21–7,24 (м, 1H), 7,42–7,48 (м, 3H), 7,58–7,62 (м, 1H), 7,66–7,70 (м, 3H), 8,41–8,42 (м, 1H).
13C-ЯМР (CDCl3, 100,6 МГц): δ = 64,6, 123,4, 125,7, 128,4, 129,0, 133,8, 136,7, 138,2, 148,8, 149,7.
Стадия 2: 4-(пиридин-2-карботиоилсульфанилметил)бензойная кислота
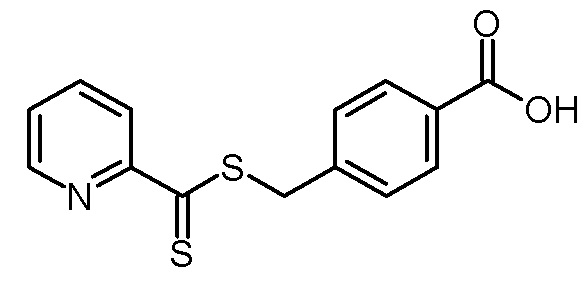
Раствор 1,8-диазабицикло[5.4.0]ундец-7-ена (79,56 г, 523 ммоль) в ацетонитриле (100 мл) добавляли по каплям к суспензии 2-бензолсульфонилметилпиридина (40,64 г, 174 ммоль) и серы (16,76 г, 523 ммоль) в ацетонитриле (500 мл) при охлаждении льдом. После завершения добавления темно-красный раствор перемешивали в течение 22 ч при комнатной температуре. Затем порциями добавляли 4-(бромметил)бензойную кислоту (37,46 г, 174 ммоль). Реакционную смесь перемешивали в течение еще 4 ч при комнатной температуре и затем добавляли 2н хлористоводородную кислоту (200 мл) (рН = 1). Из красного раствора выпадал красный осадок. Суспензию фильтровали и остаток промывали ацетонитрилом (100 мл). Фильтрат обрабатывали трет-бутилметиловым эфиром (200 мл) и насыщенным водным раствором NaCl (100 мл). Фазы разделяли и органическую фазу промывали насыщенным водным раствором NaCl (2×100 мл). Затем объединенные водные фазы повторно экстрагировали трет-бутилметиловым эфиром (100 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали на роторном испарителе. Остаток объединяли с полученным ранее остатком фильтрации, добавляли ацетонитрил (200 мл) и перемешивали в течение 4 ч при комнатной температуре. Полученную суспензию фильтровали. Остаток промывали ацетонитрилом (50 мл) и сушили в вакуумной печи (50°С, 125 мбар). Получали 44,95 г (155 ммоль, выход 89%) красного твердого вещества.
1H-ЯМР (ДМСО-d6, 400 МГц): δ = 4,66 (с, 2H), 7,56–7,58 (м, 2H), 7,70–7,73 (м, 1H), 7,94–7,96 (м, 2H), 7,00–8,03 (м, 1H), 8,26–8,28 (м, 1H), 8,66–8,68 (м, 1H), 13,03 (с, 1H).
13C-ЯМР (ДМСО-d6, 100,6 МГц): δ = 39,6, 121,9, 127,8, 129,4, 129,5, 129,9, 137,7, 140,6, 148,3, 155,3, 166,9, 226,0.
Стадия 3: 2-(метакрилоилокси)этиловый эфир 4-(пиридин-2-карботиоилсульфанилметил)бензойной кислоты
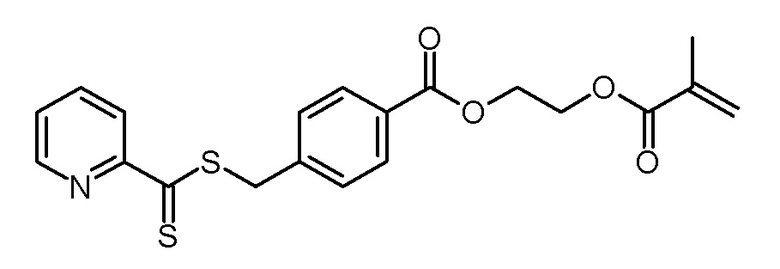
Суспензию 4-(пиридин-2-карботиоилсульфанилметил)бензойной кислоты (10,39 г, 35,9 ммоль), 2-гидроксиэтилметакрилата (4,67 г, 35,9 ммоль), N,N-диметиламинопиридина (600 мг, 5,0 ммоль) и ВНТ (10 мг) в дихлорметане (100 мл) охлаждали до 0°C. Добавляли гидрохлорид 3-(этилиминометилиденамино)-N,N-диметилпропан-1-амина (8,26 г, 43,1 ммоль) и реакционную смесь перемешивали в течение 1 ч при 0°C и в течение 16 ч при комнатной температуре. Красный реакционный раствор фильтровали через слой силикагеля (SiO2, дихлорметан) и фильтрат концентрировали на роторном испарителе. Маслянистое красное твердое вещество обрабатывали н-гексаном (100 мл), перемешивали в течение 20 ч при комнатной температуре и фильтровали. Остаток промывали н-гексаном (50 мл) и сушили в вакуумной печи (50°C, 125 мбар). Получали 11,22 г (27,9 ммоль, 78% выход) светло-красного твердого вещества.
1H-ЯМР (CDCl3, 400 МГц): δ = 1,95 (с, 3H), 4,47–4,50 (м, 2H), 4,54–4,58 (м, 4H), 5,58–5,59 (м, 1H), 6,13–6,14 (м, 1H), 7,46–7,49 (м, 3H), 7,7–7,81 (м, 1H), 7,98–8,00 (м, 2H), 8,31–8,33 (м, 1H), 8,59–8,61 (м, 1H).
13C-ЯМР (CDCl3, 100,6 МГц): δ = 18,3, 40,8, 62,4, 62,7, 122,3, 126,2, 127,0, 129,0, 129,5, 130,0, 135,9, 137,0, 141,0, 148,0, 156,1, 166,0, 167,1, 225,4.
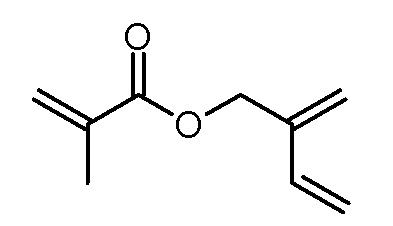
Стадия 4: 2-метиленбут-3-ениловый эфир метакриловой кислоты
Диизопропиламид лития (30% масс. в парафиновом масле, 53,58 г, 150 моль) обрабатывали диэтиловым эфиром (150 мл) и охлаждали до -5°С. Затем по каплям добавляли моноокись изопрена (11,78 г, 140 ммоль). Реакционную смесь перемешивали в течение 3 ч при -5°С и затем в течение 1 ч при комнатной температуре. При охлаждении льдом добавляли 2н хлористоводородную кислоту (100 мл) и фазы разделяли. Органическую фазу промывали водным раствором NaHCO3 (5% масс., 100 мл) и объединенные водные фазы повторно экстрагировали диэтиловым эфиром (5×50 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали на роторном испарителе (40°С, 750-90 мбар). Полученное желтоватое масло экстрагировали ацетонитрилом (3×100 мл) и объединенные растворы ацетонитрила концентрировали на роторном испарителе (40°С, 150-90 мбар). Полученную желтую жидкость растворяли в дихлорметане (70 мл). Добавляли триэтиламин (5,06 г, 50,0 ммоль) и N,N-диметиламинопиридин (600 мг, 5,0 ммоль) и раствор охлаждали до -5°С. Раствор метакрилового ангидрида (7,71 г, 50,0 ммоль) и BHT (10 мг) в дихлорметане (30 мл) добавляли по каплям. Реакционную смесь перемешивали в течение 2 ч при -5°C и в течение 22 ч при комнатной температуре, промывали водой (3×100 мл) и сушили над Na2SO4, фильтровали и концентрировали на роторном испарителе. Желто-коричневое масло растворяли в дихлорметане (50 мл) и фильтровали через слой силикагеля. Фильтрат концентрировали на роторном испарителе и сушили в высоком вакууме. Получили 3,85 г (25,3 ммоль, выход 51%) желтой жидкости.
1H-NMR (CDCl3, 400 MHz): δ = 1,97 (с, 3H), 4,84 (с, 2H), 5,14 (д, J = 11,2 Гц; 1H), 5,22–5,29 (м, 3H), 5,58 (с, 1H), 6,14 (с, 1H), 6,39 (дд, J = 11,0 Гц, 17,8 Гц; 1H).
13C-NMR (CDCl3, 100,6 MHz): δ = 18,3, 63,7, 114,6, 117,8, 125,7, 136,2, 136,2, 140,7, 167,0.
Стадия 5: 2-(метакрилоилокси)этиловый эфир 4-[4(5)-(метакрилоилоксиметил)-2-пиридин-2-ил-3,6-дигидро-2H-тиопиран-2-илсульфанилметил]бензойной кислоты
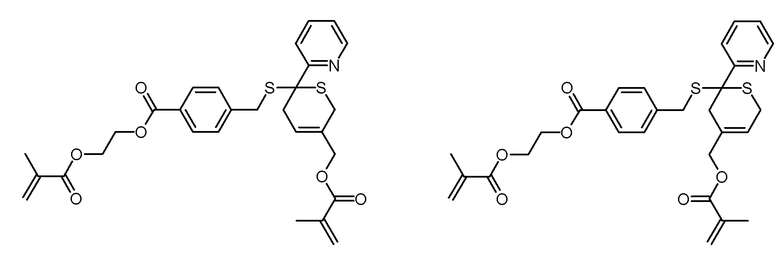
Этиловый эфир 4-(пиридин-2-карботиоилсульфанилметил)бензойной кислоты (6,59 г, 16,4 ммоль) и трифторуксусную кислоту (1,87 г, 16,4 ммоль) растворяли в хлороформе (70 мл). По каплям добавляли раствор 2-метиленбут-3-енилового эфира метакриловой кислоты (3,75 г, 24,6 ммоль) в хлороформе (30 мл). Изначально красный раствор перемешивали в течение 24 ч при комнатной температуре, при этом происходит усиливающееся обесцвечивание раствора. Затем раствор промывали водным раствором NaHCO3 (5% масс.;. 3×50 мл), сушили над Na2SO4, фильтровали и концентрировали на роторном испарителе. Слегка красный неочищенный продукт очищали хроматографией на колонке (SiO2, н-гексан/этилацетат 4:1). Получали 7,22 г (13,0 ммоль, 80% выход) неразделяемой изомерной смеси в виде желтоватого масла.
1H-ЯМР (CDCl3, 400 МГц): δ = 1,94 (с, 3H), 1,95 (с, 3H), 2,84–2,95 (м, 1H), 3,07–3,20 (м, 2H), 3,44–3,56 (м, 2H), 3,78 (d, J = 12,8 Hz; 1H), 4,46–4,48 (м, 2H), 4,53–4,55 (м, 2H), 4,57–4,63 (м, 2H), 5,56–5,59 (м, 2H), 5,88–5,89 (м, 0,7H), 5,97–5,98 (м, 0,3H), 6,12 (с, 1H), 6,13 (с, 1H), 7,13–7,16 (м, 3H), 7,60–7,65 (м, 1H), 7,69–7,74 (м, 1H), 7,82–7,85 (м, 2H), 8,51–8,54 (м, 1H).
13C-NMR (CDCl3, 100,6 МГц): δ = 18,3, 18,3, 26,1, 26,4, 35,1, 38,9, 39,4, 61,1, 62,4, 62,6, 68,1, 68,7, 121,5, 121,6, 122,5, 122,6, 124,9, 125,8, 126,1, 128,2, 128,2, 129,0, 129,0, 129,6, 129,7, 129,8, 132,2, 135,9, 136,1, 136,7, 136,7, 143,2, 143,4, 148,1, 148,2, 161,2, 161,4, 166,0, 167,0, 167,1.
Пример 9
Синтез 4-[3-(метакрилоилокси)пропил]-3,5-диоксо-10-окса-4-азатрицикло[5.2.1.02,6]дец-8-ен-1-илметилового эфира метакриловой кислоты
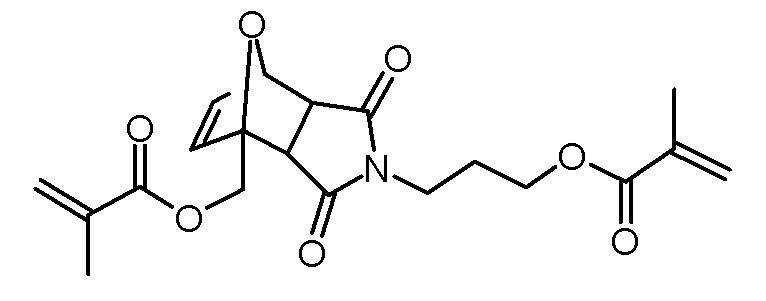
1-гидроксиметил-4-(3-гидроксипропил)-10-окса-4-азатрицикло[5.2.1.02,6]дец-8-ен-3,5-дион (18,29 г, 72,2 ммоль), триэтиламин (16,08 г, 159 ммоль), N,N-диметиламинопиридин (600 мг, 5,0 ммоль) и BHT (10 мг) растворяли в дихлорметане (100 мл). Раствор метакрилового ангидрида (24,49 г, 159 ммоль) в дихлорметане (50 мл) по каплям добавляли при 0°C. Прозрачный желтый раствор перемешивали в течение 2 ч при -0°C, затем ледяную баню удаляли и перемешивание продолжали при комнатной температуре. Через 22 ч реакционный раствор промывали водой (3×100 мл). Объединенные органические фазы повторно экстрагировали дихлорметаном (100 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали и концентрировали на роторном испарителе. Коричневатое масло очищали хроматографией на колонке (SiO2, н-гексан/этилацетат 2:1). Получали 15,04 г (38,6 ммоль, 53% выход, смесь экзо- и эндо-изомеров) желтого масла.
1H-ЯМР (ДМСО-d6, 400 МГц): δ = 1,81-1,90 (м, 9,6H; экзо/эндо), 3,04 (д, 1H; J = 6,5 Гц; эндо), 3,10 (д, 1H; J = 6,5 Гц; эндо), 3,31–3,35 (м, 0,4H; экзо), 3,47–3,51 (м, 2,2H; экзо/эндо), 3,70–3,73 (м, 0,2H; экзо), 3,99–4,05 (м, 2,4H; экзо/эндо), 4,41 (д, 1H; J = 12,6 Гц; эндо), 4,63 (д, 0,2H; J = 12,8 Гц; экзо), 4,79 (д, 0,2H; J = 12,8 Гц; экзо), 4,84 (д, 1H; J = 12,6 Гц; эндо), 5,15 (д, 1H; J = 1,6 Гц; эндо), 5,32 (dд, 0,2H; J = 5,6 Гц, 1,5 Гц; экзо), 5,67–5,69 (м, 2,2H; экзо/эндо), 5,71-5,73 (м, 0,2H; экзо), 6,01–6,08 (м, 2,4H; экзо/эндо), 6,42 (д, 0,2H; J = 6,1 Гц; экзо), 6,49–6,53 (м, 1,2H; экзо/эндо), 6,60–6,62 (м, 1H; эндо).
13C-ЯМР (ДМСО-d6, 100,6 МГц): δ = 17,8 (эндо), (17,8; экзо), 17,9, (26,2), 26,2, (34,6), 34,9, (46,4), (47,3), 48,2, 49,7, 61,4, (61,5), 61,6, (62,0), (78,8), 80,5, 88,8 (89,1), 125,5, 126,1, (126,3), (134,3), (135,4), 135,5, (135,7), 135,8, 136,6, 137,2, 166,0, 166,4, (174,5), (174,6), 174,7, 176,0.
Пример 10
Синтез 3,5-диоксо-4-(3-триэтоксисилилпропил)-10-окса-4-азатрицикло[5.2.1.02,6]дец-8-ен-1-илметиловый эфир метакриловой кислоты
Стадия 1: N-(3-триэтоксисилилпропил)малеимид (SI126)
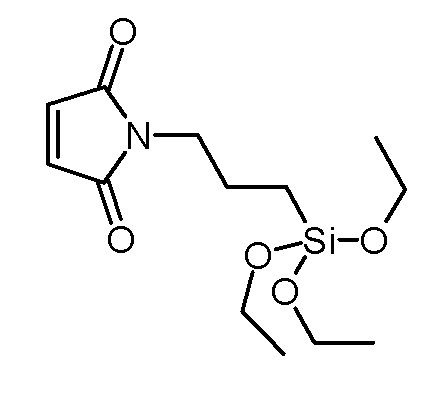
Малеиновый ангидрид (29,42 г, 300 ммоль) суспендировали в толуоле (100 мл). По каплям добавляли раствор 3-аминопропилтриэтоксисилана (66,41 г, 300 ммоль) в толуоле (75 мл) и реакционную смесь перемешивали при комнатной температуре. Через 2 ч сначала добавляли хлорид цинка (13,63 г, 100 ммоль), затем по каплям добавляли раствор гексаметилдисилазана (60,52 г, 375 ммоль) в толуоле (75 мл). Суспензию нагревали при кипячении с обратным холодильником в течение 24 ч и фильтровали над Celatom после охлаждения до комнатной температуры. Фильтрат концентрировали на роторном испарителе и сушили в высоком вакууме. Неочищенный продукт очищали вакуумной дистилляцией (т. кип.: 125°C/0,03 мбар). Получали 15,14 г (50,3 ммоль, выход 17%) бесцветной жидкости.
1H-NMR (CDCl3, 400 MHz): δ = 0,41–0,49 (м, 2H), 1,08 (т, 9H; J = 7,1 Гц), 1,49–1,60 (м, 2H), 3,37 (т, 2H; J = 7,3 Гц), 3,60–3,70 (м, 6H), 6,59 (с, 2H).
13C-ЯМР (CDCl3, 100,6 МГц): δ = 6,1, 16,7, 20,5, 38,7, 56,8, 132,5, 169,2.
29Si-ЯМР (CDCl3, 79,5 МГц): δ = -46,4.
Стадия 2: 3,5-диоксо-4-(3-триэтоксисилилпропил)-10-окса-4-азатрицикло[5.2.1.02,6]дец-8-ен-1-илметиловый эфир метакриловой кислоты
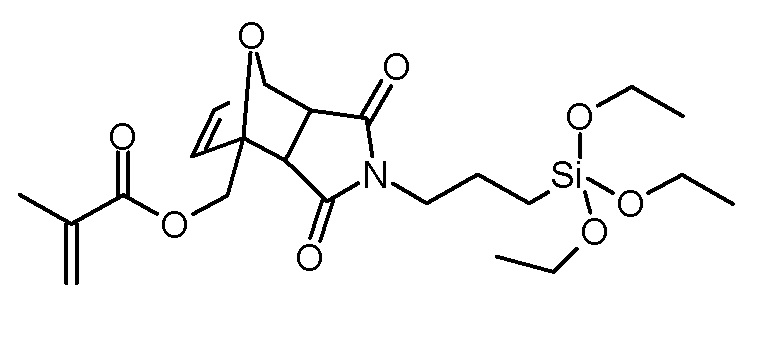
Раствор фурфурилметакрилата (8,24 г, 49,6 ммоль), N-(3-триэтоксисилилпропил)малеимида (14,94 г, 49,6 ммоль) и BHT (10 мг) в толуоле (150 мл) нагревали до 80°С с введением потока разреженного воздуха. Через 20 ч растворитель концентрировали на роторном испарителе и неочищенный продукт очищали хроматографией на колонке (SiO2, н-гексан/этилацетат 2:1). Получали 5,67 г (12,1 ммоль, выход 24%, смесь экзо и эндо изомеров) в виде бесцветного масла.
1H-ЯМР (CDCl3, 400 МГц): δ = 0,43–0,47 (м, 2,2H; экзо/эндо), 1,06–1,10 (м, 9,9H; экзо/эндо), 1,37–1,46 (м, 0,2H; экзо), 1,50–1,58 (м, 2H, эндо), 1,82 (с, 3H; эндо), 1,84 (с, 0,3H; экзо), 2,85 (дд, 2H, J = 28,2 Гц, 6,4 Гц; эндо), 3,19 (т, 0,2H; J = 7,3 Гц; экзо), 3,28 (д, 0,2H; J = 7,3 Гц; экзо), 3,35 (т, 2H; J = 7,3 Гц; эндо), 3,63–3,71 (м, 6,6H; экзо/эндо), 4,38 (д, 1H, J = 12,7 Гц; эндо), 4,56 (д, 0,1H, J = 12,7 Гц; экзо), 4,78 (д, 0,1H, J = 12,7 Гц; экзо), 4,87 (д, 1H, J = 12,7 Гц; эндо), 5,15 (с, 1H; эндо), 5,18–5,20 (м, 0,1H; экзо), 5,48 (с, 1H; эндо), 5,50 (с, 0,1H; экзо), 6,00 (с, 1H; эндо), 6,05 (с, 0,1H; экзо), 6,22 (д, 0,1H, J = 5,4 Гц; экзо), 6,34 (д, 1,1H, J = 5,4 Гц; эндо/экзо), 6,45–6,47 (м, 1H; эндо).
13C-ЯМР (CDCl3, 100,6 МГц): δ = 5,9 (эндо), 6,3 (экзо), 16,6, 19,5, 45,2 (экзо), 46,1 (экзо), 46,7 (эндо), 48,3 (эндо), 56,7, 60,0 (эндо), 60,6 (экзо), 77,9 (экзо), 79,4 (эндо), 87,9 (эндо), 78,1 (экзо), 124,6 (эндо), 124,8 (экзо), 132,8 (экзо), 134,0 (экзо), 134,1 (экзо), 134,1 (эндо), 135,5 (эндо), 135,8 (эндо), 165,1, 172,7 (эндо), 172,8 (экзо), 172,9 (экзо), 174,2 (эндо).
29Si-ЯМР (CDCl3, 79,5 МГц): δ = -46,6 (экзо), -46,3 (эндо).
Пример 11
Синтез бис-(4-метакрилоилоксибензоил)диэтилгермания
Стадия 1: 2-(4-метоксифенил)-1,3-дитиан
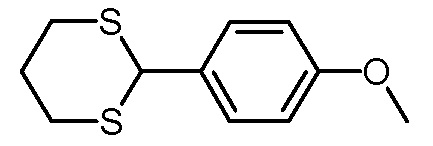
Раствор п-анисового альдегида (136,2 г, 1,0 моль) в хлороформе (500 мл) обрабатывали 1,3-пропандитиолом (108,2 г, 1,0 моль) и охлаждали до -10°С. Поток газа HCl пропускали через суспензию в течение 45 минут. Затем смесь дополнительно перемешивали в течение 30 мин при 0°С, затем охлаждающую баню удаляли и реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Растворитель удаляли на роторном испарителе и к остатку добавляли метанол (300 мл). Суспензию перемешивали в течение 24 ч при комнатной температуре и фильтровали. Остаток промывали метанолом (50 мл) и сушили в вакуумной печи (125 мбар, 50°C). Получали 219,9 г (970 ммоль, выход 97%) белого твердого вещества (т.пл.: 117-119°C).
1H-ЯМР (CDCl3, 400 МГц): δ = 1,87-1,97 (м, 1H, -CH2-), 2,13-2,20 (м, 1H, -CH2-), 2,88-2,93 (м, 2H, S-CH2-), 3,02-3,09 (м, 2H, S-CH2-), 3,79 (с, 3H, O-CH3), 5,13 (с, 1H, S-CH-S), 6,85–6,87 (м, 2H, Ar-H3,5), 7,38–7,40 (м, 2H, Ar-H2,6).
13C-ЯМР (CDCl3, 100,6 МГц): δ = 25,1 (-CH2-), 32,2 (-CH2-), 50,1 (S-C-S), 55,3 (O-CH3), 114,1 (Ar-C3,5), 128,9 (Ar-C2,6), 131,3 (Ar-C1), 159,6 (Ar-C4).
Стадия 2: Бис-[2-(4-метоксифенил)-1,3-дитиан-2-ил]диэтилгерманий
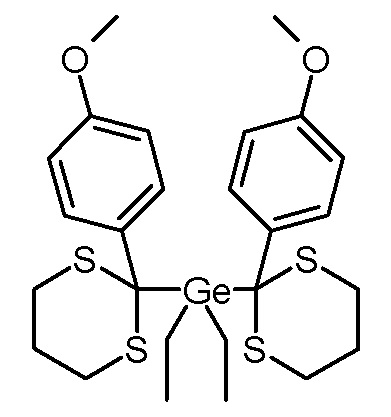
2-(4-метоксифенил)-1,3-дитиан (113,2 г, 500 ммоль) растворяли в абсолютном ТГФ (500 мл) под защитным газом и охлаждали до -5°С. По каплям добавляли 2,5 М раствор бутиллития в н-гексане (200 мл, 500 ммоль). Коричневый раствор затем перемешивали в течение 3 часов при -5°С, затем по каплям добавляли раствор дихлорида диэтилгермания (42,0 г, 208 ммоль) в абсолютном ТГФ (100 мл). Реакционную смесь перемешивали в течение ночи на тающей ледяной бане, затем добавляли воду (200 мл) и этилацетат (400 мл) и фазы разделяли. Органическую фазу промывали водой (2×125 мл) и объединенные водные фазы повторно экстрагировали этилацетатом (150 мл). Объединенные органические фазы промывали насыщенным водным раствором NaCl (150 мл), сушили над Na2SO4, фильтровали, концентрировали на роторном испарителе и сушили в высоком вакууме. Остаток обрабатывали этилацетатом (100 мл) и суспензию перемешивали при комнатной температуре. Через 20 часов добавляли метанол (100 мл) и через 24 часа суспензию фильтровали. Остаток промывали этилацетатом (20 мл) и сушили в вакуумной печи (125 мбар, 50°C). Получали 88,8 г (153 моль, выход 73%) белого твердого вещества (т.пл.: 115-116°C).
1H-ЯМР (CDCl3, 400 МГц): δ = 1,15 (м, 6H, -CH3), 1,28 (м, 4H, Ge-CH2-), 1,77–1,81 (м, 2H, -CH2-), 1,94–2,05 (м, 2H, -CH2-), 2,24–2,29 (м, 4H, S-CH2-), 2,70–2,77 (м, 4H, S-CH2-), 3,82 (с, 6H, O-CH3), 6,80–6,82 (м, 4H, Ar-H3,5), 7,78–7,80 (м, 4H, Ar-H2,6).
13C-ЯМР (CDCl3, 100,6 МГц): δ = 4,5 (-CH3), 10,2 (Ge-CH2-), 25,2 (-CH2-), 25,6 (S-CH2-), 51,2 (Ge-C-S), 55,2 (O-CH3), 113,3 (Ar-C3,5), 131,4 (Ar-C2,6), 132,5 (Ar-C1), 157,5 (Ar-C4).
Стадия 3: бис-(4-метоксибензоил)диэтилгерманий
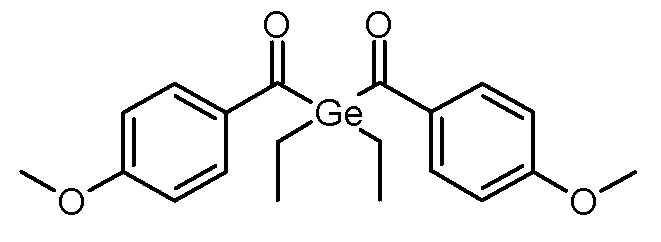
Раствор бис-[2-(4-метоксифенил)-1,3-дитиан-2-ил]диэтилгермания (87,21 г, 150 моль) в ТГФ (900 мл) обрабатывали водой (220 мл). Карбонат кальция (180,2 г, 1,80 моль) и йод (456,9 г, 1,80 моль) разделяли на восемь равных частей. Затем с интервалами 30 мин одну порцию каждого из CaCO3 и йода добавляли в реакционную смесь при периодическом охлаждении льдом. После завершения добавления реакционную смесь перемешивали в течение 24 ч при комнатной температуре и затем фильтровали через тонкий слой силикагеля. К красно-коричневому фильтрату добавляли насыщенный водный раствор дитионита натрия (1600 мл) при интенсивном перемешивании до полного изменения цвета на желтый. Суспензию фильтровали и фильтрат промывали этилацетатом (400 мл). Фильтрат разбавляли этилацетатом (800 мл) и фазы разделяли. Органическую фазу промывали водой (2×250 мл) и объединенные водные фазы повторно экстрагировали этилацетатом (2×200 мл). Объединенные органические фазы промывали насыщенным водным раствором NaCl (200 мл), сушили над Na2SO4, фильтровали, концентрировали на роторном испарителе и сушили в высоком вакууме. Неочищенный продукт очищали хроматографией на колонке (SiO2, н-гексан/этилацетат 9:1). Получали 38,4 г (95,9 ммоль, выход 64%) желтого твердого вещества (т.пл.: 47-50°С).
1H-ЯМР (CDCl3, 400 МГц): δ = 1,12 (т, 6H; J = 7,9 Гц, -CH3), 1,47 (кв, 4H; J = 7,9 Гц, Ge-CH2-), 3,80 (с, 6H, O-CH3), 6,87–6,91 (м, 4H, Ar-H3,5), 7,71–7,75 (м, 4H, Ar-H2,6).
13C-ЯМР (CDCl3, 100,6 МГц): δ = 6,4 (-CH3), 9,0 (Ge-CH2-), 55,5 (O-CH3), 114,1 (Ar-C3,5), 130,5 (Ar-C2,6), 135,0 (Ar-C1), 163,8 (Ar-C4), 227,4 (C=O).
Стадия 4: бис-(4-гидроксибензоил)диэтилгерманий
Бис-(4-метоксибензоил)диэтилгерманий (8,0 г, 20,0 ммоль) растворяли в безводном толуоле (200 мл) в атмосфере защитного газа и добавляли Celatom (10 г) и хлорид алюминия (9,6 г, 72,0 ммоль). Реакционную смесь нагревали в течение 2 ч при кипячении с обратным холодильником. После охлаждения добавляли воду (10 мл) и суспензию перемешивали в течение 10 мин при комнатной температуре. Растворитель удаляли на роторном испарителе. Остаток обрабатывали этилацетатом (300 мл). Суспензию перемешивали в течение 16 ч при комнатной температуре и фильтровали через тонкий слой силикагеля. Фильтрат концентрировали на роторном испарителе. К маслянистому коричневому остатку добавляли хлороформ (200 мл). Суспензию перемешивали в течение 16 ч при комнатной температуре и фильтровали. Остаток промывали хлороформом (80 мл) и сушили в вакуумной печи (50°С, 125 мбар). Получали 4,23 г (11,3 ммоль, выход 57%) светло-желтого твердого вещества (т.пл.: 167-168°C).
1H-ЯМР (ДМСО-d6, 400 МГц): δ = 1,04 (т, 6H; J = 7,9 Гц, -CH3), 1,38 (кв, 4H; J = 7,9 Гц, Ge-CH2-), 6,88 (д, 4H; J = 8,5 Гц, Ar-H3,5), 7,58 (д, 4H; J = 8,5 Гц, Ar-H2,6), 10,53 (с, 2H, OH).
13C-ЯМР (ДМСО-d6, 100,6 МГц): δ = 6,1 (-CH3), 8,9 (Ge-CH2-), 115,7 (Ar-C3,5), 130,3 (Ar-C2,6), 133,3 (Ar-C1), 162,7 (Ar-C4), 225,5 (C=O).
Стадия 5: Бис-(4-метакрилоилоксибензоил)диэтилгерманий
Раствор метакрилового ангидрида (5,89 г, 38,2 ммоль) и BHT (10 мг) в дихлорметане (50 мл) по каплям добавляли к раствору бис-(4-гидроксибензоил)диэтилгермания (6,78 г, 18,2 ммоль), триэтиламина (3,86 г, 38,2 ммоль) и N,N-диметиламинопиридина (240 мг, 2,0 ммоль) в дихлорметане (100 мл) при -5°C. После завершения добавления раствор перемешивали в течение 1 ч при -5°C и в течение 20 ч при комнатной температуре. Реакционный раствор промывали водой (3×100 мл). Объединенные водные фазы повторно экстрагировали дихлорметаном (2×50 мл). Объединенные органические фазы сушили над Na2SO4, фильтровали, концентрировали на роторном испарителе и сушили в высоком вакууме. Неочищенный продукт очищали хроматографией на колонке (SiO2, н-гексан/этилацетат 4:1). Получали 4,40 г (8,6 ммоль, выход 47%) желтого твердого вещества.
1H-ЯМР (CDCl3, 400 МГц): δ = 1,13 (т, 6H; J = 7,9 Гц, -CH3), 1,51 (кв, 4H; J = 7,9 Гц, Ge-CH2-), 2,04 (с, 6H, CH3), 5,79–5,77 (м, 2H, C=CH), 6,34 (с, 2H, C=CH), 7,23–7,20 (м, 4H, Ar-H3,5), 7,80–7,77 (м, 4H, Ar-H2,6).
13C-ЯМР (CDCl3, 100,6 МГц): δ = 6,5 (Ge-CH2-), 9,0 (Ge-CH2-CH3), 18,3 (CH3), 122,3 (Ar C3,5), 128,0 (C=CH2), 129,6 (Ar-C2,6), 135,4 (C=C), 138,6 (Ar-C1), 155,0 (Ar-C4), 165,1 (C=O), 228,3 (Ge-C=O).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 2-(ИМИНОМЕТИЛ)АМИНОФЕНИЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ ВЕЩЕСТВА | 1998 |
|
RU2183211C2 |
| ИНГИБИТОРЫ ГЛЮКОЗИЛЦЕРАМИД-СИНТАЗЫ | 2012 |
|
RU2645675C2 |
| ПРОИЗВОДНЫЕ ТАКРИНА В КАЧЕСТВЕ ИНГИБИТОРОВ АЦЕТИЛХОЛИНЭСТЕРАЗЫ | 2004 |
|
RU2402545C2 |
| ИНГИБИТОРЫ ДВУХ САЙТОВ СВЯЗЫВАНИЯ АЦЕТИЛХОЛИНЭСТЕРАЗЫ ДЛЯ ЛЕЧЕНИЯ БОЛЕЗНИ АЛЬЦГЕЙМЕРА | 2003 |
|
RU2325379C2 |
| ИНГИБИТОРЫ АРОМАТАЗЫ | 2010 |
|
RU2572244C2 |
| Имидазопиридиновые соединения в качестве ингибиторов PAD | 2018 |
|
RU2782743C2 |
| ПРОИЗВОДНЫЕ БЕНЗОПИПЕРИДИНА, БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1992 |
|
RU2142945C1 |
| ПРОИЗВОДНЫЕ 2-АМИНО-2-ФЕНИЛАЛКАНОЛА, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2009 |
|
RU2486174C2 |
| ДИАРИЛЬНЫЕ СОЕДИНЕНИЯ С МОСТИКОВОЙ СВЯЗЬЮ | 2002 |
|
RU2297409C2 |
| ИНГИБИТОРЫ ФОСФАТАЗ Сdc25 | 2005 |
|
RU2395510C2 |
Группа изобретений относится к области стоматологических материалов. Предлагается стоматологический пломбировочный материал, содержащий термолабильное полимеризуемое соединение формулы II:
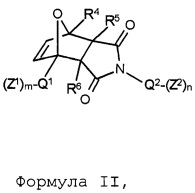
в которой Z1 и Z2 каждый независимо представляет собой полимеризуемую группу, выбранную из винильных групп, CH2=CR1-CO-O- и CH2=CR1-CO-NR2-, или связывающую группу, выбранную из -Si(OR)3, -СООН, -O-РО(ОН)2, -РО(ОН)2, -SO2OH и -SH, где по меньшей мере один Z1 или Z2 является полимеризуемой группой. Q1 в каждом случае независимо отсутствует или представляет собой (m+1)-валентный линейный или разветвленный алифатический C1-С20-радикал, который может прерываться -О-, -S-, -CO-O-, -O-CO-, -CO-NR3 -, -NR3-CO-, -O-CO-NR3-, -NR3-CO-O- или -NR3-CO-NR3-. Q2 в каждом случае независимо отсутствует или представляет собой (n+1)-валентный линейный или разветвленный алифатический C1-С20-радикал, который может прерываться -О-, -S-, -СО-O-, -O-CO-, -CO-NR3-, -NR3-CO-, -O-CO-NR3-, -NR3-CO-O- или -NR3-CO-NR3-. R, R1, R2 и R3 каждый независимо друг от друга представляет собой Н или С1-С7-алкильный радикал; R4 - Н или C1-С10 алкильный радикал; R5 - Н, С1-С5 алкильный радикал, F или CN; R6 - Н, С1-С5 алкильный радикал, F или CN. m и n каждый независимо равен 1, 2 или 3. Предлагается также применение вышеуказанного соединения формулы II для приготовления стоматологического пломбировочного материала. Использование группы изобретений обеспечивает получение адгезивных стоматологических пломбировочных материалов, которые являются полимеризуемыми, показывают хорошую адгезию к субстрату, в частности к структуре зуба и/или стоматологической керамике, и способны открепляться от субстрата при нагревании, и которые, таким образом, прежде всего подходят для производства адгезивов или композитных цементов, способных к «разъединению по требованию». 2 н. и 15 з.п. ф-лы, 11 пр., 2 табл.
1. Стоматологический пломбировочный материал, содержащий термолабильное полимеризуемое соединение формулы II:
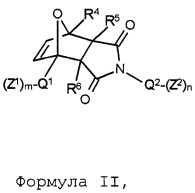
в которой
Z1 и Z2 каждый независимо представляет собой полимеризуемую группу, выбранную из винильных групп, CH2=CR1-CO-O- и CH2=CR1-CO-NR2-, или связывающую группу, выбранную из -Si(OR)3, -СООН, -O-РО(ОН)2, -РО(ОН)2, -SO2OH и -SH, где по меньшей мере один Z1 или Z2 является полимеризуемой группой,
Q1 в каждом случае независимо отсутствует или представляет собой (m+1)-валентный линейный или разветвленный алифатический C1-С20-радикал, который может прерываться -О-, -S-, -CO-O-, -O-CO-, -CO-NR3 -, -NR3-CO-, -O-CO-NR3-, -NR3-CO-O- или -NR3-CO-NR3-,
Q2 в каждом случае независимо отсутствует или представляет собой (n+1)-валентный линейный или разветвленный алифатический C1-С20-радикал, который может прерываться -О-, -S-, -СО-O-, -O-CO-, -CO-NR3-, -NR3-CO-, -O-CO-NR3-, -NR3-CO-O- или -NR3-CO-NR3-,
R, R1, R2 и R3 каждый независимо друг от друга представляет собой Н или С1-С7-алкильный радикал,
R4 представляет собой Н или C1-С10 алкильный радикал;
R5 представляет собой Н, С1-С5 алкильный радикал, F или CN,
R6 представляет собой Н, С1-С5 алкильный радикал, F или CN,
и
m и n каждый независимо равен 1, 2 или 3.
2. Стоматологический пломбировочный материал по п. 1, в котором
Z1 и Z2 каждый независимо представляет собой полимеризуемую группу, выбранную из винильных групп, CH2=CR1-CO-O- и CH2=CR1-CO-NR2-, или кислотную группу, выбранную из -O-РО(ОН)2, -РО(ОН)2 и -SO2OH, где по меньшей мере один Z1 или Z2 является полимеризуемой группой и по меньшей мере один Z1 или Z2 является кислотной группой,
Q1 в каждом случае независимо отсутствует или представляет собой (m+1)-валентный линейный или разветвленный алифатический C1-С10 радикал, который может прерываться -О-, -S-, -CO-O-, -O-CO-, -CO-NR3- или -NR3-CO-,
Q2 в каждом случае независимо отсутствует или представляет собой (n+1)-валентный линейный или разветвленный алифатический C1-С10 радикал, который может прерываться -O-, -S-, -CO-O-, -O-CO-, -CO-NR3- или -NR3-CO-,
R1, R2 и R3 в каждом случае независимо представляют собой Н или С1-С7 алкильный радикал, и
m и n в каждом независимо равны 1, 2 или 3.
3. Стоматологический пломбировочный материал по п. 1, в котором в каждом случае независимо
один из Z1 и Z2 в каждом случае независимо представляет собой полимеризуемую группу, выбранную из CH2=CR1-CO-O- и CH2=CR1-CO-NR2-, и другой из Z1 и Z2 в каждом случае независимо представляет собой полимеризуемую группу, выбранную из CH2=CR1-CO-O- и CH2=CR1-CO-NR2- или предпочтительно связывающую группу, выбранную из -Si(OR)3, -СООН, -O-РО(ОН)2, -РО(ОН)2, -SO2OH и -SH,
Q1 в каждом случае независимо отсутствует или представляет собой (m+1)-валентный линейный или разветвленный алифатический C1-С15 радикал, предпочтительно C1-С10 радикал, предпочтительно C1-C8 радикал, в частности С2-С6 радикал, и особенно предпочтительно C1-С2 радикал, который может прерываться -O-, -CO-O-, -O-CO-, -CO-NR3-, -NR3-CO-, -O-CO-NR3-, -NR3-CO-O- или -NR3-CO-NR3-,
Q2 в каждом случае независимо отсутствует или представляет собой (n+1)-валентный линейный или разветвленный алифатический C1-С15 радикал, предпочтительно C1-С10 радикал, предпочтительно C1-C8 радикал, в частности С2-С6 радикал, и особенно предпочтительно С2-С3 радикал, который может прерываться -О-, -СО-O-, -O-CO-, -CO-NR3-, -NR3-CO-, -O-CO-NR3-, -NR3-CO-O- или -NR3-CO-NR3-,
R в каждом случае независимо представляет собой СН3 или C2H5,
R1 в каждом случае независимо представляет собой Н или СН3,
R2 в каждом случае независимо представляет собой Н, СН3 или С2Н5,
R3 в каждом случае независимо представляет собой Н, СН3 или С2Н5,
R4 представляет собой Н, СН3 или С2Н5;
R5 представляет собой Н, F или CN и, в частности, Н,
R6 представляет собой Н, F или CN и, в частности, Н, и/или
m и n в каждом случае независимо равны 1 или 2.
4. Стоматологический пломбировочный материал по одному из пп. 1-3, содержащий один или более дополнительных радикально полимеризуемых мономеров.
5. Стоматологический пломбировочный материал по п. 4, который содержит
метил-, этил-, гидроксиэтил-, бутил-, бензил-, тетрагидрофурфурил- или изоборнил(мет)акрилат, бисфенол-А-ди(мет)акрилат, бис-GMA, UDMA, ди-, три- или тетраэтиленгликоль ди(мет)акрилат, триметилолпропантри(мет)акрилат, пентаэритритол тетра(мет)акрилат, глицеринди(мет)акрилат, 1,4-бутандиол ди(мет)акрилат, 1,10-декандиолди(мет)акрилат, 1,12-додекандиол ди(мет)акрилат,
и/или
один или более N-моно- или дизамещенных акриламидов, N-этилакриламид, N,N-диметакриламид, N-(2-гидроксиэтил)акриламид, N-метил-N-(2-гидроксиэтил)акриламид, один или более N-монозамещенных метакриламидов, N-этилметакриламид, N-(2-гидроксиэтил)метакриламид, N-винилпирролидон, один или более поперечно-сшивающих аллиловых эфиров,
и/или
один или более поперечно-сшивающих пирролидонов, 1,6-бис(3-винил-2-пирролидонил)гексан, один или более поперечно-сшивающих бисакриламидов, метилен- или этиленбисакриламид, один или более поперечно-сшивающих бис(мет)акриламидов, N,N'-диэтил-1,3-бис(акриламидо)пропан, 1,3-бис(метакриламидо)пропан, 1,4-бис(акриламидо)бутан, 1,4-бис(акрилоил)пиперазин,
и/или
один или более термолабильных поперечно-сшивающих мономеров или их смесь.
6. Стоматологический пломбировочный материал по одному из пп. 1-3, содержащий один или более термолабильных поперечно-сшивающих мономеров.
7. Стоматологический пломбировочный материал по одному из пп. 1-3, содержащий один или более радикально полимеризуемых мономеров, содержащих кислотную группу.
8. Стоматологический пломбировочный материал по п. 7, который содержит
малеиновую кислоту, акриловую кислоту, метакриловую кислоту, 2-(гидроксиметил)акриловую кислоту, 4-(мет)акрилоилоксиэтилтримеллитовый ангидрид, 10-метакрилоилоксидецилмалоновую кислоту, N-(2-гидрокси-3-метакрилоилоксипропил)-N-фенилглицин, 4-винилбензойную кислоту,
и/или
винилфосфоновую кислоту, 4-винилфенилфосфоновую кислоту, 4-винилбензилфосфоновую кислоту, 2-метакрилоилоксиэтилфосфоновую кислоту, 2-метакриламидоэтилфосфоновую кислоту, 4-метакриламидо-4-метилпентилфосфоновую кислоту, 2-[4-(дигидроксифосфорил)-2-оксабутил]акриловую кислоту, этиловый или 2,4,6-триметилфениловый эфиры 2-[4-(дигидроксифосфорил)-2-оксабутил]акриловой кислоты,
и/или
моно- или дигидрофосфат 2-метакрилоилоксипропила, гидрофосфат 2-метакрилоилоксиэтилфенила, дипентаэритритолпентаметакрилоилоксифосфат, дигидрофосфат 10-метакрилоилоксидецила, моно-(1-акрилоилпиперидин-4-иловый)эфир фосфорной кислоты, дигидрофосфат 6-(метакриламидо)гексила, дигидрофосфат 1,3-бис-(N-акрилоил-N-пропиламино)пропан-2-ила,
и/или
винилсульфоновую кислоту, 4-винилфенилсульфоновую кислоту, 3-(метакриламидо)пропилсульфоновую кислоту,
или их смесь.
9. Стоматологический пломбировочный материал по одному из пп. 1-3, содержащий инициатор радикальной полимеризации.
10. Стоматологический пломбировочный материал по одному из пп. 1-3, содержащий добавку, выделяющую газ при нагревании.
11. Стоматологический пломбировочный материал по одному из пп. 1-3, содержащий добавку, которая может превращать излучаемое электромагнитное излучение в тепло.
12. Стоматологический пломбировочный материал по одному из пп. 1-3, включающий органический и/или неорганический наполнитель.
13. Стоматологический пломбировочный материал по одному из пп. 1-3, содержащий
a) от 0,1 до 50% масс., в частности от 1 до 40% масс., предпочтительно от 2 до 30% масс. и особенно предпочтительно от 5 до 30% масс. соединения формулы формулы II,
b) от 0,01 до 10% масс., предпочтительно от 0,1 до 3,0% масс. и более предпочтительно от 0,2 до 2% масс. инициатора,
c) от 0 до 80% масс., предпочтительно от 1 до 60% масс. и особенно предпочтительно от 5 до 50% масс. сомономера,
d) от 0 до 30% масс., предпочтительно от 0,5 до 15% масс. и особенно предпочтительно от 1 до 5% масс. склеивающего мономера,
e) от 0 до 80% масс. наполнителя,
f) от 0 до 70% масс. растворителя.
14. Стоматологический пломбировочный материал по п. 12 для применения в качестве клея, содержащего от 0 до 20% масс. наполнителя.
15. Стоматологический пломбировочный материал по п. 12 для применения в качестве композита, содержащего от 20 до 80% масс. наполнителя.
16. Применение соединения формулы II, определенного в любом из пп. 1-3, для приготовления стоматологического пломбировочного материала.
17. Применение по пп. 16 для приготовления адгезива или цемента и, в частности, самопротравливающего адгезива или цемента.
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| АЛКИЛФЕНИЛБИСАЦИЛФОСФИНОКСИДЫ, ИХ СМЕСИ, ФОТОПОЛИМЕРИЗУЕМАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ИХ, СПОСОБ ФОТОПОЛИМЕРИЗАЦИИ И СУБСТРАТ, ПОКРЫТЫЙ ЭТОЙ КОМПОЗИЦИЕЙ | 1997 |
|
RU2180667C2 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |

