ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННУЮ ЗАЯВКУ
Настоящая заявка на патент испрашивает приоритет заявки на патент Кореи № 10-2013-0043100, поданной 18 апреля 2013, и №10-2014-0045343, поданной 16 апреля 2014, содержание которых включено в настоящее описание посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
1. Область техники, к которой относится изобретение
Настоящее изобретение относится к новому производному 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты, способу его получения и к фармацевтической композиции для профилактики и лечения метаболического заболевания, включающая его в качестве активного ингредиента.
2. Описание связанной области техники
Диабет представляет собой серьезное заболевание, которое постоянно угрожает нашему здоровью, и которым страдают по меньшей мере сто миллионов человек по миру. Диабет может быть классифицирован на две категории клинических симптомов, которые составляют диабет типа I и диабет типа II. Диабет типа I, называемый инсулинзависимым сахарным диабетом (IDDM), вызывается аутоиммунной деструкцией бета-клеток поджелудочной железы, которые продуцируют инсулин, так, что при этом состоянии требуется регулярное введение экзогенного инсулина. Диабет типа II, называемый инсулиннезависимым сахарным диабетом (NIDDM), развивается в результате нарушения регуляции сахара в крови. Так, люди, которые имеют диабет типа II, характеристически демонстрируют нарушение секреции инсулина или инсулинорезистентность, что позволяет предположить, что они с трудом секретируют инсулин in vivo или не могут использовать инсулин эффективно.
Диабет характеризуется высокой концентрацией глюкозы в крови и моче, в результате чего это заболевание является причиной полиурии, жажды, голода и других связанных с метаболизмом липидов и белков проблем. Диабет может вызывать такие угрожающие жизни осложнения как потеря зрения, отказ почек и болезнь сердца. Диабет также является причиной повреждений сетчатки и увеличивает риск катаракты и глаукомы. Диабет также понижает реакцию на боль, связанную с повреждением нерва в ногах и ступнях и может быть причиной обширной инфекции.
Лекарственными средствами для лечения диабета, разработанными в последнее время, являются инсулин, средство, усиливающее секрецию инсулина, глюкозопонижающий эффектор, активатор активируемого пролифератором пероксисомы рецептора и т.д. Однако разработанные в последнее время способы лечения имеют проблемы, связанные со стимуляцией низкого уровня сахара в крови, увеличением массы тела, уменьшением ответа на лекарственное средство в течение времени, возникновение проблем желудочно-кишечного тракта и отеков и т.д. Поэтому были предприняты исследования для поиска более эффективного способа лечения. Одна из тех попыток состоит в том, чтобы использовать рецептор, связанный с G-белком (GPCR).
GPR40 был недавно идентифицирован как один из рецепторов, связанных с G-белком (GPCR). Он известен как рецептор свободной жирной кислоты I, который суперэкспрессируется в β-клетках в поджелудочной железе. Внутриклеточная концентрация кальция повышается таким соединением, которое активирует GPR40 (FFAR1), и соответственно промотируется стимулируемая глюкозой секреция инсулина (GSIS) (Current Drug Targets, 2008, 9, 899-910). Когда активатор GPR40 вводили нормальной мыши или трансгенной мыши, пригодным для наблюдения в тесте диабетом и переносимости глюкозы, он показал увеличенную переносимость глюкозы. Подвергнутая лечению мышь продемонстрировала кратковременное увеличение инсулина в плазме крови. В исследовании функций GPR40 было подтверждено, что жирная кислота в свободном состоянии, которая является лигандом GPR40, действовала в панкреатических β-клетках, и в результате β-клетки секретировали инсулин зависимым от концентрации глюкозы образом. В результате анализа с использованием мыши с нокаутом по GPR было подтверждено, что GPR40 участвует в развитии ожирения и диабета (Can J Diabetes 2012, 36, 275-280). Поэтому GPR40 расценивается как новая мишень исследований диабета.
В ходе исследования активатора GPR40 авторы настоящего изобретения подтвердили, что новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты, его фармацевтически приемлемая соль или оптический изомер имеет родственную GPR40 активность, что привело к подтверждению превосходных in vivo эффектов, таких как увеличение внутриклеточной концентрации кальция и эффект снижения глюкозы крови, в результате чего было осуществлено настоящее изобретение.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является получение нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты, его оптического изомера или фармацевтически приемлемой соли.
Другой целью настоящего изобретения является разработка способа получения указанного производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты.
Также целью настоящего изобретения является получение фармацевтической композиции, включающей указанное производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты в качестве активного ингредиента для профилактики или лечения метаболического заболевания.
Для достижения вышеупомянутых целей, настоящее изобретение предоставляет соединение, представленное ниже формулой 1, его оптический изомер или фармацевтически приемлемую соль.
[Формула 1]
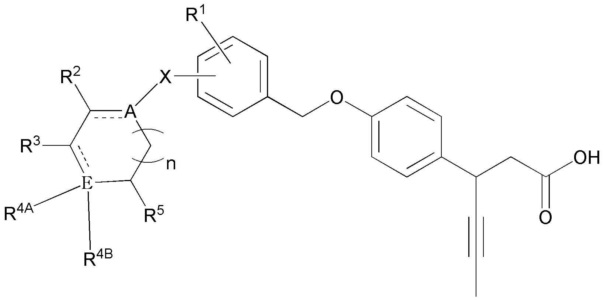
(В формуле 1,
 обозначает простую связь или двойную связь;
обозначает простую связь или двойную связь;
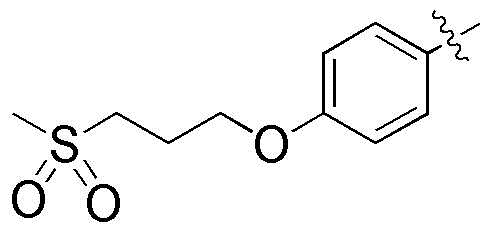
A и E независимо обозначают C, N или O;
n означает целое число 0-5;
X обозначает простую связь или C1-10 прямой или разветвленный алкилен;
R1 обозначает -H, -ОН, галоген, C1-10 прямой или разветвленный алкил, C1-10 прямой или разветвленный алкокси, C5-10 циклоалкил или C5-10 циклоалкенил;
R2, R3 и R5 независимо обозначают -H, -ОН, галоген, C1-10 прямой или разветвленный алкил или C1-10 прямой или разветвленный алкокси;
причем R2 и R3 могут образовывать C5-10 циклоалкил, C6-10 арил, 5-10-членный гетероциклоалкил или 5-10-членный гетероарил вместе с атомами, которые к ним присоединены. 5-10-членный гетероциклоалкил может содержать один или более гетероатомов, выбранных из группы, состоящей из N, O и S, и 5-10-членный гетероарил может содержать один или более гетероатомов, выбранных из группы, состоящей из N, O и S;
R4A обозначает -H, -ОН, =O, незамещенный или замещенный C6-10 арил или незамещенный или замещенный 5-10-членный гетероарил, содержащий один или более гетероатомов, выбранных из группы, состоящей из N, O и S,
В упомянутом замещенном C6-10 ариле и замещенном 5-10-членном гетероариле один или более заместителей, выбраных из группы, состоящей из -ОН, галогена, нитрила, незамещенного или замещенного C1-5 прямого или разветвленного алкила, в котором один или более галогенов замещены, незамещенного или замещенного C1-5 прямого или разветвленного алкокси, в котором один или более галогенов замещены, C1-10 прямого или разветвленного алкилсульфонила,  и
и  могут быть замещены. Причем, m и q независимо означают целые числа 1-10,
могут быть замещены. Причем, m и q независимо означают целые числа 1-10,
В упомянутом незамещенном или замещенном 5-10-членном гетероариле фенил может быть конденсирован;
причем, R3 и R4A могут образовывать C5-10 циклоалкил, C6-10 арил, 5-10-членный гетероциклоалкил или 5-10-членный гетероарил вместе с атомами, которые к нему присоединены. 5-10-членный гетероциклоалкил может содержать один или более гетероатомов, выбранных из группы, состоящей из N, O и S, и 5-10-членный гетероарил может содержать один или более гетероатомов, выбранных из группы, состоящей из N, O и S;
В упомянутом C5-10 циклоалкиле, C6-10 ариле, 5-10-членном гетероциклоалкиле и 5-10-членном гетероариле, C1-5 прямой или разветвленный алкокси может быть замещен;
R4B отсутствует или может образовывать 5-10-членный гетероцикл, содержащий один или более гетероатомов, выбранных из группы, состоящей из N, O и S, вместе с атомами, которые к нему присоединены, и R4A).
Настоящее изобретение также относится к способу получения соединения, представленного формулой 1, включающему следующие стадии, как показано в приведенной ниже схеме реакции 1:
получение соединения, представленного формулой 4, реакцией конденсации соединения, представленного формулой 2, и соединения, представленного формулой 3 (стадия 1); и
получение соединения, представленного формулой 1, реакцией восстановления соединения, представленного формулой 4, полученного на стадии 1) (стадия 2).
[Схема реакции 1]
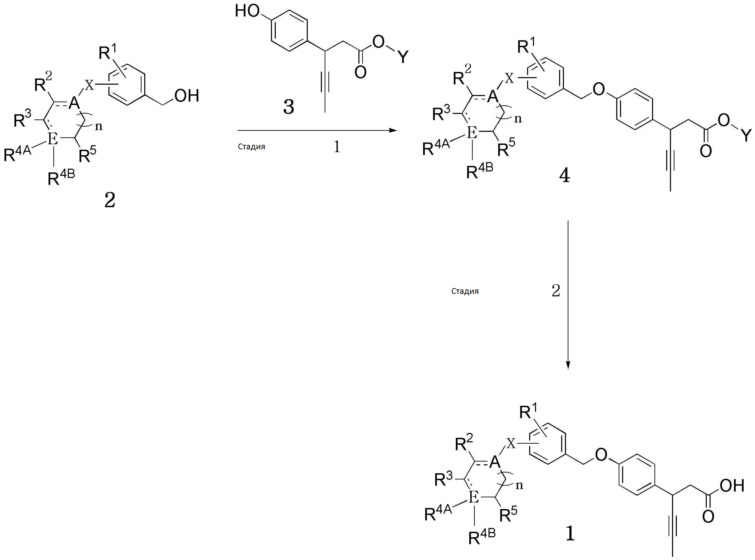
(В схеме реакции 1,
R1, R2, R3, R4A, R4B, R5, A, E, n и X имеют значения, определенные в формуле 1; и Y обозначает C1-10 прямой или разветвленный алкил).
Далее, настоящее изобретение относится к способу получения соединения, представленного формулой 1 по п.1 формулы изобретения, включающему следующие стадии как показано в представленной ниже схеме реакции 3;
получение соединения, представленного формулой 6, реакцией сочетания соединения, представленного формулой 5, и соединения, представленного формулой 3 (стадия 1);
получение соединения, представленного формулой 7, мезилатной реакцией соединения, представленного формулой 6, полученного на стадии 1) (стадия 2);
получение соединения, представленного формулой 4, заменой мезилатной части соединения, представленного формулой 7, полученного на стадии 2), соединением, представленным формулой 13 (стадия 3); и
получение соединения, представленного формулой 1, реакцией восстановления соединения, представленного формулой 4, полученного на стадии 3) (стадия 4).
[Схема реакции 3]
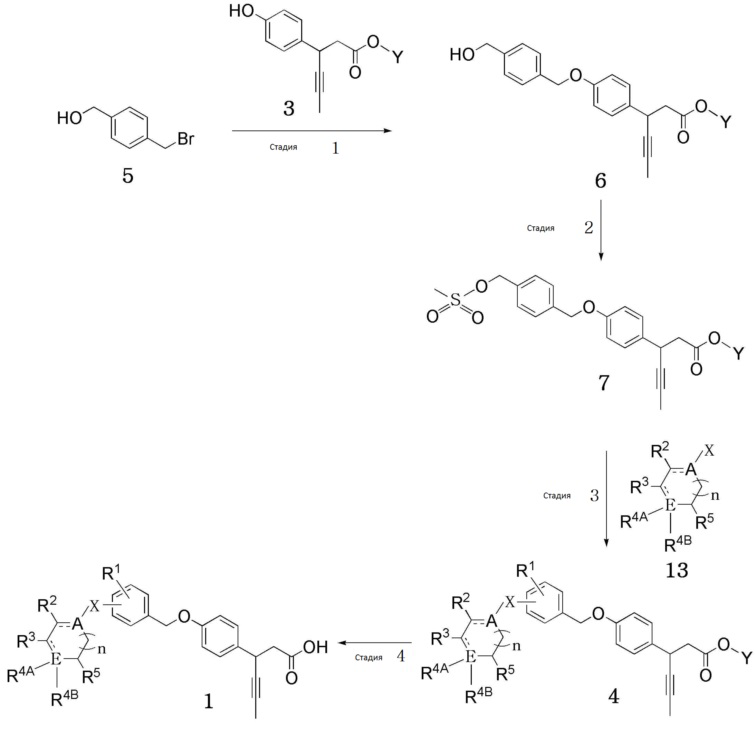
(В схеме реакции 3,
R1, R2, R3, R4A, R4B, R5, A, E, n и X имеют значения, определенные в формуле 1; и Y обозначает C1-10 прямой или разветвленный алкил).
Настоящее изобретение также относится к способу получения соединения, представленного формулой 1, включающему стадию получения соединения, представленного формулой 1b, реакцией размыкания кольца соединения, представленного формулой 1a (стадия 1) как показано в приведенной ниже схеме реакции 4.
[Схема реакции 4]
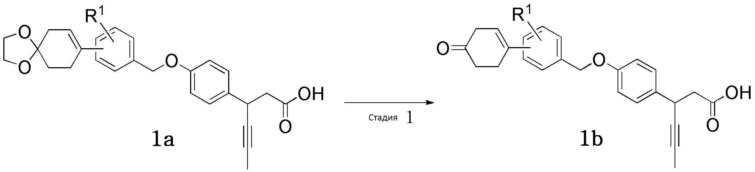
(В схеме реакции 4,
R1 имеет значения, определенные в формуле 1; и соединения, представленные формулой 1a и формулой 1b, включены в соединение, представленное формулой 1).
Настоящее изобретение также относится к способу получения соединения, представленного формулой 1, включающему стадию получения соединения, представленного формулой 1c, реакцией восстановления соединения, представленного формулой 1b (стадия 1), как показано в приведенной ниже схеме реакции 5.
[Схема реакции 5]
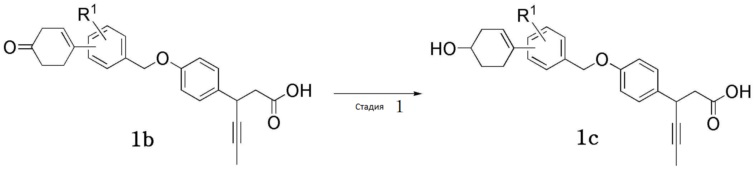
(В схеме реакции 5,
R1 имеет значения, определенные в формуле 1; и соединения, представленные формулой 1b и формулой 1c, включены в соединение, представленное формулой 1).
Кроме того, настоящее изобретение относится к фармацевтической композиции, включающей соединение, представленное формулой 1, его оптический изомер или его фармацевтически приемлемую соль в качестве активного ингредиента.
ВЫГОДНЫЙ ЭФФЕКТ
Новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты, его оптический изомер или его фармацевтически приемлемая соль согласно настоящему изобретению имеют превосходные активности активации белка GPR40 и промотирования секреции инсулина, соответственно, но не имеют никакой токсичности при совместном введении с другими лекарственными средствами. Таким образом, новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты, его оптический изомер или его фармацевтически приемлемую соль согласно настоящему изобретению может быть введено совместно с другими лекарственными средствами и может существенно промотировать активацию белка GPR40, так, чтобы композиция, включающая его в качестве активного ингредиента, могла эффективно использоваться как фармацевтическая композиция для профилактики и лечения метаболического заболевания, такого как ожирение, диабет типа I, диабет типа II, несовместимая переносимость глюкозы, инсулинорезистентность, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром X, и т.д.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Осуществление предпочтительных вариантов осуществления настоящего изобретения лучше всего может быть понято с опорой на сопутствующие чертежи, на которых:
Фиг. 1 представляет собой график, иллюстрирующий структуру активации GPR40 согласно концентрации соединений Примера 9, Сравнительного Примера 1 и Сравнительного Примера 3.
Фиг. 2 представляет собой график, иллюстрирующий содержание GLP-1 в крови крысы SD (крыса Sprague Dawley) согласно пероральному введению соединений Примера 9 и Сравнительного Примера 1.
ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
Далее настоящее изобретение описано подробно.
Настоящее изобретение относится к соединению, представленному приведенной ниже формулой 1, его оптическому изомеру или его фармацевтически приемлемой соли.
[Формула 1]
(В формуле 1,
Обозначает простую связь или двойную связь;
A и E независимо обозначают C, N или O;
n означает целое число 0-5;
X обозначает простую связь или C1-10 прямой или разветвленный алкилен;
R1 обозначает -H, -OH, галоген, C1-10 прямой или разветвленный алкил, C1-10 прямой или разветвленный алкокси, C5-10 циклоалкил или C5-10 циклоалкенил;
R2, R3 и R5 независимо обозначают -H, -OH, галоген, C1-10 прямой или разветвлены алкил или C1-10 прямой или разветвленный алкокси;
причем R2 и R3 могут образовывать C5-10 циклоалкил, C6-10 арил, 5-10-членный гетероциклоалкил или 5-10-членный гетероарил вместе с атомами, которые к ним присоединены. 5-10-членный гетероциклоалкил может содержать один или более гетероатомов, выбранных из группы, состоящей из N, O и S, и 5-10-членный гетероарил может содержать один или более гетероатомов, выбранных из группы, состоящей из N, O и S;
R4A обозначает -H, -OH, =O, незамещенный или замещенный C6-10 арил или незамещенный или замещенный 5-10-членный гетероарил, содержащий один или более гетероатомов, выбранных из группы, состоящей из N, O и S,
В упомянутом замещенном C6-10 ариле и замещенном 5-10-членном гетероариле один или более заместителей, выбранных из группы, состоящей из -OH, галогена, нитрила, незамещенного или замещенного C1-5 прямого или разветвленного алкила, в котором один или более галогенов замещены, незамещенного или замещенного C1-5 прямого или разветвленного алкокси, в котором один или более галогенов замещены, C1-10 прямого или разветвленного алкилсульфонила, и могут быть замещены. Причем m и q независимо означают целые числа 1-10,
В упомянутом незамещенном или замещенном 5-10-членном гетероариле фенил может быть конденсирован;
причем R3 и R4A могут образовывать C5-10 циклоалкил, C6-10 арил, 5-10-членный гетероциклоалкил или 5-10-членный гетероарил вместе с атомами, которые к ним присоединены. 5-10-членный гетероциклоалкил может содержать один или более гетероатомов, выбранных из группы, состоящей из N, O и S, и 5-10-членный гетероарил может содержать один или более гетероатомов, выбранных из группы, состоящей из N, O и S;
В упомянутом C5-10 циклоалкиле, C6-10 ариле, 5-10-членном гетероциклоалкиле и 5-10-членном гетероариле C1-5 прямой или разветвленный алкокси может быть замещен;
R4B отсутствует или может образовывать 5-10-членный гетероцикл, содержащий один или более гетероатомов, выбранных из группы, состоящей из N, O и S, вместе с атомами, которые к нему присоединены, и R4A).
Предпочтительно,
обозначает простую связь или двойную связь;
A и E независимо обозначают C, N или O;
n означает целое число 0-3;
X обозначает простую связь или C1-5 прямой или разветвленный алкилен;
R1 обозначает -H, -OH, галоген, C1-5 прямой или разветвленный алкил, C1-5 прямой или разветвленный алкокси, C5-8 циклоалкил или C5-8 циклоалкенил;
R2, R3 и R5 независимо обозначают -H, -OH, галоген, C1-5 прямой или разветвлены алкил или C1-5 прямой или разветвленный алкокси;
причем R2 и R3 могут образовывать C5-8 циклоалкил, C6-8 арил, 5-8-членный гетероциклоалкил или 5-8-членный гетероарил вместе с атомами, которые к ним присоединены. 5-8-членный гетероциклоалкил может содержать один или более гетероатомов, выбранных из группы, состоящей из N, O и S, и 5-8-членный гетероарил может содержать один или более гетероатомов, выбранных из группы, состоящей из N, O и S;
R4A обозначает -H, -OH, =O, незамещенный или замещенный C6-8 арил, или незамещенный или замещенный 5-8-членный гетероарил, содержащий один или более гетероатомов, выбранных из группы, состоящей из N, O и S,
В упомянутом замещенном C6-8 ариле и замещенном 5-8-членном гетероариле один или более заместителей, выбранных из группы, состоящей из -OH, галогена, нитрила, незамещенного или замещенного C1-5 прямого или разветвленного алкила, в котором один или более галогенов замещены, незамещенного или замещенного C1-5 прямого или разветвленного алкокси, в котором один или более галогенов замещены, C1-8 прямого или разветвленного алкилсульфонила, и могут быть замещены. Причем m и q независимо обозначают целые числа 1-5,
В упомянутом незамещенном или замещенном 5-8-членном гетероариле фенил может быть конденсирован;
причем R3 и R4A могут образовывать C5-8 циклоалкил, C6-8 арил, 5-8-членный гетероциклоалкил или 5-8-членный гетероарил вместе с атомами, которые к ним присоединены. 5-8-членный гетероциклоалкил может содержать один или более гетероатомов, выбранных из группы, состоящей из N, O и S, и 5-8-членный гетероарил может содержать один или более гетероатомов, выбранных из группы, состоящей из N, O и S;
В упомянутом C5-8 циклоалкиле, C6-8 ариле, 5-8-членном гетероциклоалкиле и 5-8-членном гетероариле C1-5 прямой или разветвленный алкокси может быть замещен;
R4B отсутствует или может образовывать 5-8-членный гетероцикл, содержащий один или более гетероатомов, выбранных из группы, состоящей из N, O и S, наряду с атомами, которые к нему присоединены, и R4A.
Более предпочтительно,
обозначает простую связь или двойную связь;
A и E независимо обозначают C или N;
n означает целое число 0-1;
X обозначает простую связь или C1-3 прямой или разветвленный алкилен;
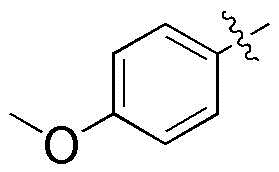
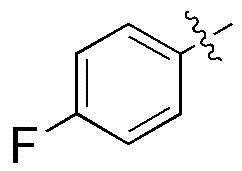
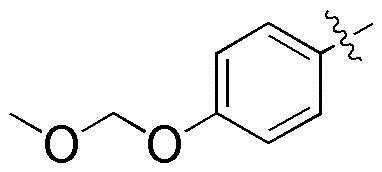
R1 обозначает -H или 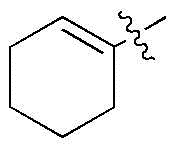 ;
;
R2, R3 и R5 независимо обозначают -H,
причем R2 и R3 могут образовывать фенил;
R4A обозначает -H, -OH, =O,
 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  или
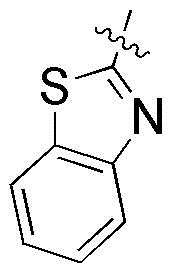
или 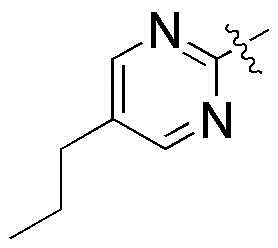 ,
,
причем R3 и R4A могут образовывать фенил вместе с атомами, которые к ним присоединены. В упомянутом фениле, метокси может быть замещен;
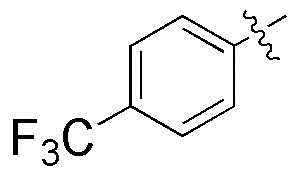
R4B отсутствует или может образовывать 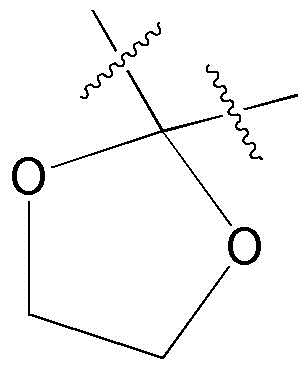 вместе с атомами, которые к нему присоединены, и R4A.
вместе с атомами, которые к нему присоединены, и R4A.
Соединение, представленное формулой 1, может иллюстрироваться следующими соединениями.
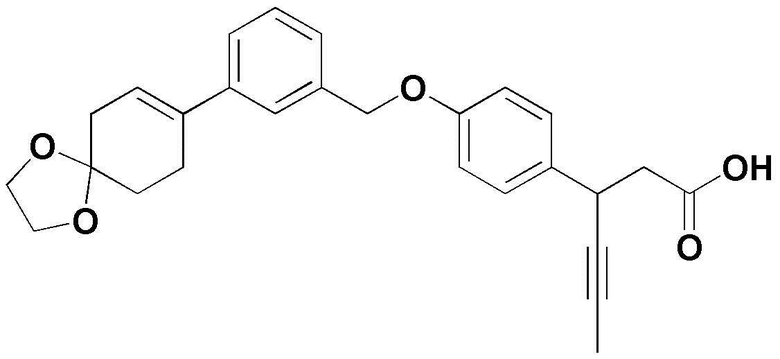
(1) 3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновая кислота;
(2) L-лизин 3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат;
(3) 4-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновая кислота;
(4) 3-(4-(3-(4-оксоциклогекс-1-енил)бензилокси)фенил)гекс-4-иновая кислота;
(5) 3-(4-(3-(4-гидроксициклогекс-1-енил)бензилокси)фенил)гекс-4-иновая кислота;
(6) L-лизин 3-(4-(3-(4-гидроксициклогекс-1-енил)бензилокси)фенил)гекс-4-иноат;
(7) (3S)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновая кислота;
(8) (3R)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновая кислота;
(9) L-лизин (3S)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат;
(10) L-лизин (3R)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат;
(11) (3S)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат натрия;
(12) 3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(13) 3-(4-(3-циклогексенил-4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(14) 3-(4-(4-((4-фенил-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(15) 3-(4-(4-((4-фенилпиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(16) 3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(17) 3-(4-(4-((4-фенилпиперидин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(18) 3-(4-(4-((4-(4-фторфенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(19) 3-(4-(4-((4-(4-(трифторметил)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(20) 3-(4-(4-((4-(4-(3-(метилсульфонил)пропокси)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(21) (S)-3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(22) (S)-3-(4-(4-((4-(4-(трифторметил)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(23) (S)-3-(4-(4-((4-(4-фторфенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(24) (S)-3-(4-(4-((4-(4-(трифторметил)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иноат калия;
(25) (S)-3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(26) (S)-3-(4-(4-((4-фенилпиперидин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(27) (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иновая кислота;
(28) (S)-3-(4-(4-((4-фенил-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(29) (S)-3-(4-(4-((4-(4-(метоксиметокси)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
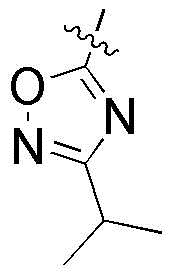
(30) (S)-3-(4-(4-((4-(5-изопропил-1,2,4-оксадиазол-3-ил)пиперидин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(31) (S)-3-(4-(4-((4-(5-изопропил-1,2,4-оксадиазол-3-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(32) (S)-3-(4-(4-((4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(33) (S)-3-(4-(4-((4-(4-(3-(метилсульфонил)пропокси)фенил)-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(34) (3S)-3-(4-(4-(1-(3,4-дигидроизохинолин-2(1H)-ил)этил)бензилокси)фенил)гекс-4-иновая кислота;
(35) (S)-3-(4-(4-((4-(4-гидроксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(36) (S)-3-(4-(4-((4-(4-(3-(метилсульфонил)пропокси)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(37) натрий (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иноат;
(38) L-лизин (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иноат;
(39) (S)-3-(4-(4-((4-(4-фторфенил)-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(40) (S)-3-(4-(4-((4-(4-метоксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(41) натрий (S)-3-(4-(4-((3,4-дигидрохинолин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иноат;
(42) (S)-3-(4-(4-((3,4-дигидрохинолин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иноат калия;
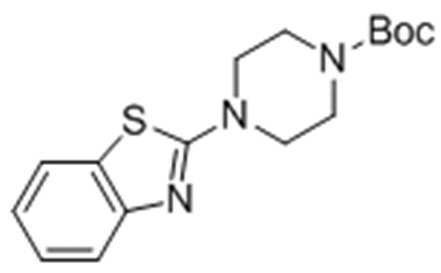
(43) (S)-3-(4-(4-((4-(бензо[d]тиазол-2-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
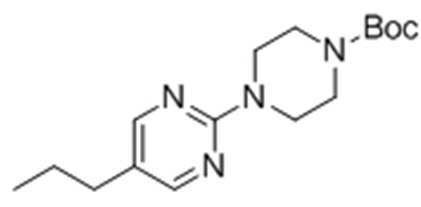
(44) (S)-3-(4-(4-((4-(5-пропилпиримидин-2-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(45) (S)-3-(4-(4-((4-(5-цианопиридин-2-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(46) (3S)-3-(4-(4-((3-фенилпирролидин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(47) натрий (S)-3-(4-(4-((4-(4-метоксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иноат;
(48) (S)-3-(4-(4-(2-(6-метокси-3,4-дигидроизохинолин-2(1H)-ил)этил)бензилокси)фенил)гекс-4-иновая кислота;
(49) (S)-3-(4-(4-(2-(изоиндолин-2-ил)этил)бензилокси)фенил)гекс-4-иновая кислота;
(50) (S)-3-(4-(4-(2-(3,4-дигидроизохинолин-2(1H)-ил)этил)бензилокси)фенил)гекс-4-иновая кислота; и
(51) (S)-3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноат натрия.
Соединение, представленное формулой 1 согласно настоящему изобретению, может использоваться в форме фармацевтически приемлемой соли, причем соль является предпочтительно солью присоединения с кислотой, образованной фармацевтически приемлемыми свободными кислотами. Соль присоединения с кислотой по изобретению может быть получена из неорганических кислот, таких как соляная кислота, азотная кислота, фосфорная кислота, серная кислота, бромистоводородная кислота, йодистоводородная кислота, азотистая кислота и фосфористая кислота; нетоксичных органических кислот, таких как алифатический моно/дикарбоксилат, замещенный фенилом алканоат, гидрокси алканоат, алкандиоат, кислоты ароматического ряда, и алифатические/ароматические сульфоновые кислоты; или органических кислот, таких как уксусная кислота, бензойная кислота, лимонная кислота, молочная кислота, малеиновая кислота, глюконовая кислота, метансульфоновая кислота, 4-толуолсульфоновая кислота, винная кислота и фумаровая кислота. Фармацевтически нетоксичные соли иллюстрируются сульфатом, пиросульфатом, бисульфатом, сульфитом, бисульфитом, нитратом, фосфатом, моногидрофосфатом, дигидрофосфатом, метафосфатом, пирофосфатом, хлоридом, бромидом, йодидом, фторидом, ацетатом, пропионатом, деканоатом, каприлатом, акрилатом, формиатом, изобутилатом, капратом, гептаноатом, пропиолатом, оксалатом, малонатом, сукцинатом, субератом, себакатом, фумаратом, малиатом, бутин-1,4-диоатом, гексан-1,6-диоатом, бензоатом, хлорбензоатом, метилбензоатом, динитробензоатом, гидроксибензоатом, метоксибензоатом, фталатом, терефталатом, бензолсульфонатом, толуолсульфонатом, хлорбензолсульфонатом, ксилолсульфонатом, фенилацетатом, фенилпропионатом, фенилбутилатом, цитратом, лактатом, гидроксибутилатом, гликолятом, малатом, тартратом, метансульфонатом, пропансульфонатом, нафталин-1-сульфонатом, нафталин-2-сульфонатом и манделатом.
Соль присоединения с кислотой в этом изобретении может быть получена обычным способом, известным специалисту в данной области техники. Например, соединение, представленное формулой 1, растворяют в органическом растворителе, таком как метанол, этанол, ацетон, метиленхлорид или ацетонитрил, к которому добавляют органическую кислоту или неорганическую кислоту, чтобы вызвать осаждение. Затем осадок отфильтровывают и высушивают, получая соль. Или растворитель и избыток кислоты перегоняют при пониженном давлении и высушивают, получая соль. Или осадок кристаллизуют в органическом растворителе, получая соль.
Соль фармацевтически приемлемого металла может быть получена при использовании основания. Соль щелочного металла или щелочно-земельного металла получают следующими способами: растворение соединения в избытке раствора гидроксида щелочного металла или гидроксида щелочно-земельного металла; фильтрование нерастворимой соли соединения; упаривание оставшегося раствора и высушивание остатка. В это время соль металла предпочтительно получают в фармацевтически приемлемой форме соли натрия, калия или кальция. И соответствующую соль серебра получают реакцией соли щелочного металла или щелочно-земельного металла с соответствующей солью серебра (например, нитратом серебра).
Фармацевтически приемлемая соль может также быть получена при использовании аминокислоты, в которой аминогруппа присоединена на органическую кислоту, и в это время соль аминокислоты предпочтительно получают как природные аминокислоты, такие как глицин, аланин, фенилаланин, валин, лизин и глутаминовая кислота, и более предпочтительно, L-лизин.
Настоящее изобретение включает не только соединение, представленное формулой 1, но также и его фармацевтически приемлемую соль и сольват, оптический изомер или гидрат, которые могут быть получены из него.
Кроме того, настоящее изобретение относится к способу получения соединения, представленного формулой 1.
Способ получения 1
Соединение, представленное формулой 1 согласно настоящему изобретению, может быть получено способом, включающим следующие стадии, как показано в приведенной ниже схеме реакции 1:
получение соединения, представленного формулой 4 реакцией конденсации соединения, представленного формулой 2, и соединения, представленного формулой 3 (стадия 1); и
получение соединения, представленного формулой 1, реакцией восстановления соединения, представленного формулой 4, полученного на стадии 1) (стадия 2).
[Схема реакции 1]
(В схеме реакции 1,
R1, R2, R3, R4A, R4B, R5, A, E, n и X имеют значения, определенные в формуле 1; и Y обозначает C1-10 прямой или разветвленный алкил).
Далее способ получения соединения, представленного формулой 1 согласно настоящему изобретению, проиллюстрирован более подробно, постадийно.
В способе получения соединения, представленного формулой 1 согласно настоящему изобретению, стадия 1) представляет собой получение соединения, представленного формулой 4, путем индукции реакции сочетания между соединением, представленным формулой 2, и соединением, представленным формулой 3. Более точно, соединение, представленное формулой 2, соединение, представленное формулой 3, и трифенилфосфин смешивают с получением смешанного раствора. Азокарбоксилатный реагент медленно добавляют к смешанному раствору при температуре от -5°C ~ 10°C, с последующей индукцией реакции Mitsunobu, получая соединение, представленное формулой 4.
На этой стадии азодикарбоксилатный реактив может быть выбран из группы, состоящей из диэтил азодикарбоксилата (DEAD) и диизопропил азодикарбоксилата (DIAD), и предпочтительно выбирают диизопропил азодикарбоксилат (DIAD).
Растворитель реакции здесь может быть выбран из группы, состоящей из тетрагидрофурана (THF), дихлорметана (DCM), толуола и ацетонитрила, и предпочтительно выбирают тетрагидрофуран.
Температура реакции предпочтительно составляет от 0°C до ~ температуры кипения растворителя, и время реакции не ограничено, но предпочтительна реакция в течение 0,5~10 часов.
В способе получения соединения, представленного формулой 1 согласно настоящему изобретению, стадия 2) представляет собой получение соединения, представленного формулой 1, путем индукции реакции восстановления соединения, представленного формулой 4, полученного на стадии 1) в присутствии основания. Более точно, соединение, представленное формулой 4, полученное на стадии 1), вводят в реакцию с основанием при комнатной температуре, в результате чего сложноэфирная группа, входящая в состав соединения, представленного формулой 4, восстанавливается до карбоксильной группы, приводя к получению соединения, представленного формулой 1.
На этой стадии основание может быть выбрано из группы, состоящей из гидроксида калия (KOH), гидроксида натрия (NaOH) и гидроксида лития (LiOH), и предпочтительно выбирают гидроксид калия (KOH).
Растворитель реакции здесь может быть выбран из группы, состоящей из тетрагидрофурана (THF), дихлорметана (DCM), толуола и ацетонитрила, и предпочтительно выбирают тетрагидрофуран.
Температура реакции предпочтительно составляет от 0°C до ~ температуры кипения растворителя, и время реакции не ограничено, но предпочтительна реакция в течение 0,5~10 часов.
Получение исходного материала (соединение, представленное формулой 2)
В схеме реакции 1 согласно настоящему изобретению, соединение, представленное формулой 2, может быть получено способом, включающим следующие стадии, как показано в приведенной ниже схеме реакции 2:
получение соединения, представленного формулой 10, путем введения в реакцию соединения, представленного формулой 8, и соединения, представленного формулой 9 (стадия 1);
получение соединения, представленного формулой 12, путем введения в реакцию соединения, представленного формулой 10, полученного на стадии 1), и соединения, представленного формулой 11 (стадия 2); и
получение соединения, представленного формулой 2, реакцией восстановления соединения, представленного формулой 12, полученного на стадии 2) (стадия 3).
[Схема реакции 2]
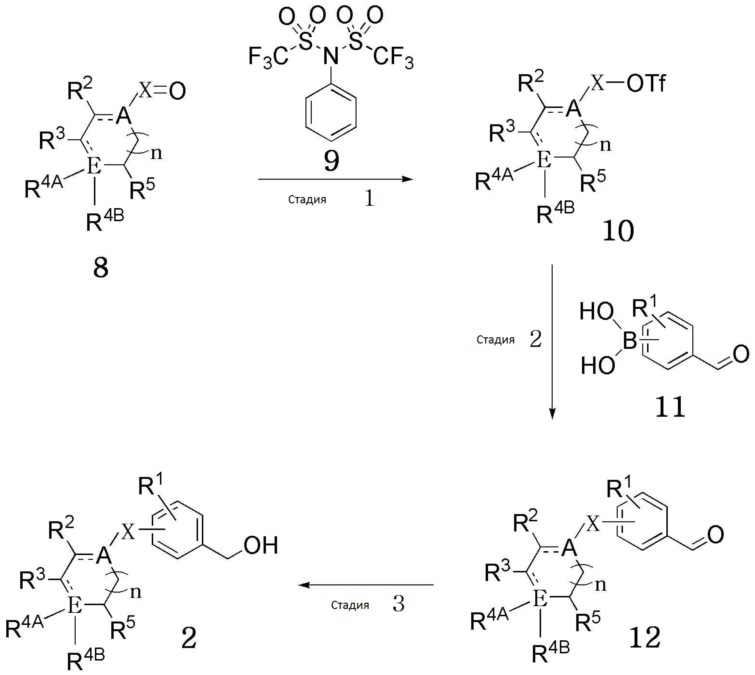
(В схеме реакции 2,
R1, R2, R3, R4A, R4B, R5, A, E, n и X имеют значения, определенные в формуле 1; и -OTf обозначает трифторметансульфонат).
Далее способ получения соединения, представленного формулой 2 согласно настоящему изобретению, проиллюстрирован более подробно, постадийно.
В способе получения соединения, представленного формулой 2 согласно настоящему изобретению, стадия 1) представляет собой получение соединение, представленного формулой 10, путем введения в реакцию соединения, представленного формулой 8, и соединения, представленного формулой 9. Более точно, соединение, представленное формулой 8, и соединение, представленное формулой 9, растворяют в органическом растворителе при температуре от -80°C до ~ -70°C, медленно добавляют комплекс металла и бис(триметилсилил)амида, с последующим перемешиванием с подъемом температуры, получая соединение, представленное формулой 10.
На этой комплекс металла и бис(триметилсилил)амида может быть выбран из группы, состоящей из бис(триметилсилил)амида калия, бис(триметилсилил)амида лития и бис(триметилсилил)амида натрия, и предпочтительно выбирают бис(триметилсилил)амид калия.
Органический растворитель здесь может быть выбран из группы, состоящей из тетрагидрофурана (THF), простого диэтилового эфира, простого дифенилового эфира, простого диизопропилового эфира (DIPE), диметилформамида (DMF), диметилацетамида (DMA), диметилсульфоксида (ДМСО), дихлорметана (DCM), хлорбензола, толуола и бензола.
Температура реакции предпочтительно составляет от -80°C до ~ температуры кипения растворителя, и время реакции не ограничено, но предпочтительна реакция в течение 0,5~10 часов.
В способе получения соединения, представленного формулой 2 согласно настоящему изобретению, стадия 2) представляет собой получение соединение, представленного формулой 12, путем введения в реакцию соединения, представленного формулой 10, полученного на стадии 1), и соединения, представленного формулой 11. Более точно, соединение, представленное формулой 12, получают, индуцирую реакцию сочетания Suzuki между соединением, представленным формулой 10, полученным на стадии 1), и боронатным соединением, представленным формулой 11.
На этой стадии катализатор на основе палладия может представлять собой тетракис(трифенилфосфин) (Pd(PPh3)4), бис(трифенилфосфин)палладий (Ⅱ) дихлорид (PdCl2(PPh3)2), дихлорид палладия (PdCl2) или ацетат палладия (Pd(OCOCH3)2), и тетракис(трифенилфосфин) (Pd(PPh3)4) является более предпочтительным.
Органический растворитель здесь выбирают из группы, состоящей из тетрагидрофурана (THF), простого диэтилового эфира, простого дифенилового эфира, простого диизопропилового эфира (DIPE), диметилформамида (DMF), диметилацетамида (DMA), диметилсульфоксида (ДМСО), дихлорметана (DCM), хлорбензола, толуола и бензола, и предпочтительно выбирают толуол.
Температура реакции предпочтительно составляет от 0°C до ~ температуры кипения растворителя, и время реакции не ограничено, но предпочтительна реакция в течение 0,5~10 часов.
В способе получения соединения, представленного формулой 2 согласно настоящему изобретению, стадия 3) представляет собой получение соединение, представленного формулой 2, путем индукции реакции восстановления соединения, представленного формулой 12, полученного на стадии 2) в присутствии основания. Более точно, соединение, представленное формулой 12, полученное на стадии 2), растворяют в органическом растворителе, к которому добавляют основание. При этом альдегидная группа, входящая в соединение, представленное формулой 12, восстанавливается до гидроксильной группы, что приводит к соединению, представленному формулой 2.
На этой стадии органический растворитель может представлять собой метанол, этанол, этилацетат, тетрагидрофуран, простой диэтиловый эфир или смешанный раствор, включающим два или более этих растворителей, но предпочтительно здесь используется смешанный растворитель тетрагидрофуран:метанол (4:1).
Основание может представлять собой боргидрид натрия (NaBH3) или литий-алюминийгидрид (LiAlH4), и боргидрид натрия (NaBH3) является более предпочтительным.
Температура реакции предпочтительно составляет от 0°C до ~ температуры кипения растворителя, и время реакции не ограничено, но предпочтительна реакция в течение 0,5~10 часов.
Способ получения 2
Соединение, представленное формулой 1 согласно настоящему изобретению, может быть получено способом, включающим следующие стадии, как показано в приведенной ниже схеме реакции 3:
получение соединения, представленного формулой 6, реакцией сочетания соединения, представленного формулой 5, и соединения, представленного формулой 3 (стадия 1);
получение соединения, представленного формулой 7, мезилатной реакцией соединения, представленного формулой 6, полученного на стадии 1) (стадия 2);
получение соединения, представленного формулой 4, заменой мезилатной части соединения, представленного формулой 7, полученного на стадии 2), соединением, представленным формулой 13 (стадия 3); и
получение соединения, представленного формулой 1, реакцией восстановления соединения, представленного формулой 4, полученного на стадии 3) (стадия 4).
[Схема реакции 3]
(В схеме реакции 3,
R1, R2, R3, R4A, R4B, R5, A, E, n и X имеют значения, определенные в формуле 1; и Y обозначает C1-10 прямой или разветвленный алкил).
Далее способ получения соединения, представленного формулой 1 согласно настоящему изобретению, проиллюстрирован более подробно, постадийно.
В способе получения соединения, представленного формулой 1 согласно настоящему изобретению, стадия 1) представляет собой получение соединения, представленного формулой 6, путем индукции реакции сочетания между соединением, представленным формулой 5, и соединением, представленным формулой 3.
Органический растворитель здесь выбран из группы, состоящей из тетрагидрофурана (THF), простого диэтилового эфира, простого дифенилового эфира, простого диизопропилового эфира (DIPE), диметилформамида (DMF), диметилацетамида (DMA), диметилсульфоксида (ДМСО), дихлорметана (DCM), хлорбензола, толуола и бензола, и предпочтительно выбирают диметилформамид (DMF).
Основание здесь может представлять собой карбонат цезия (Cs2CO3), боргидрид натрия (NaBH3) или литий-алюминийгидрид (LiAlH4), и карбонат цезия (Cs2CO3) является более предпочтительным.
Температура реакции предпочтительно составляет от 0°C до ~ температуры кипения растворителя, и время реакции не ограничено, но предпочтительна реакция в течение 0,5~10 часов.
В способе получения соединения, представленного формулой 1 согласно настоящему изобретению, стадия 2) представляет собой получение соединение, представленного формулой 7, путем индукции мезилатной реакции соединения, представленного формулой 6, полученного на стадии 1), в растворителе.
На этой стадии образец, используемый для мезилатной реакции, может быть метан сульфонил хлоридом (MsCl).
Органический растворитель здесь выбирают из группы, состоящей из триэтиламина (ТЕА), тетрагидрофурана (THF), простого диэтилового эфира, простого дифенилового эфира, простого диизопропилового эфира (DIPE), диметилформамида (DMF), диметилацетамида (DMA), диметилсульфоксида (ДМСО), дихлорметана (DCM), хлорбензола, толуола и бензола, и предпочтительно выбирают триэтиламин (ТЕА).
Температура реакции предпочтительно составляет от 0°C до ~ температуры кипения растворителя, и время реакции не ограничено, но предпочтительна реакция в течение 0,5~10 часов.
В способе получения соединения, представленного формулой 1 согласно настоящему изобретению, стадия 3) представляет собой получение соединение, представленного формулой 4, заменой мезилатной части соединения, представленного формулой 7, полученного на стадии 2), соединением, представленным формулой 13.
На этой стадии органический растворитель выбирают из группы, состоящей из тетрагидрофурана (THF), простого диэтилового эфира, простого дифенилового эфира, простого диизопропилового эфира (DIPE), диметилформамида (DMF), диметилацетамида (DMA), диметилсульфоксида (ДМСО), дихлорметана (DCM), хлорбензола, толуола и бензола, и предпочтительно выбирают дихлорметан (DCM).
Основание здесь может представлять собой карбонат цезия (Cs2CO3), боргидрид натрия (NaBH3) или литий-алюминийгидрид (LiAlH4), и карбонат цезия (Cs2CO3) является более предпочтительным.
Температура реакции предпочтительно составляет от 0°C до ~ температуры кипения растворителя, и время реакции не ограничено, но предпочтительна реакция в течение 0,5~10 часов.
В способе получения соединения, представленного формулой 1 согласно настоящему изобретению, стадия 4) представляет собой получение соединения, представленного формулой 1, путем индукции реакции восстановления соединения, представленного формулой 4, полученного на стадии 3), в присутствии основания. Более точно, соединение, представленное формулой 4, полученное на стадии 3), вводят в реакцию с основанием при комнатной температуре для восстановления сложноэфирной группы, входящей в соединение, представленное формулой 4, до карбоксильной группы, что приводит к получению соединения, представленного формулой 1.
На этой стадии основание может представлять собой гидроксид калия (KOH), гидроксид натрия (NaOH) или гидроксид лития (LiOH), и гидроксид калия (KOH) является более предпочтительныма.
Растворитель реакции может представлять собой тетрагидрофуран (THF), дихлорметан (DCM), толуол или ацетонитрил, и тетрагидрофуран (THF) является более предпочтительным.
Температура реакции предпочтительно составляет от 0°C до ~ температуры кипения растворителя, и время реакции не ограничено, но предпочтительна реакция в течение 0,5~10 часов.
Способ получения 3
Соединение, представленное формулой 1 согласно настоящему изобретению, может быть получено способом, включающим стадию получения соединения, представленного формулой 1b, реакцией размыкания кольца соединения, представленного формулой 1a (стадия 1), как показано в приведенной ниже схеме реакции 4.
[Схема реакции 4]
(В схеме реакции 4,
R1 имеет значения, определенные в формуле 1; и соединения, представленные формулой 1a и формулой 1b, включены в соединение, представленное формулой 1).
Далее способ получения согласно настоящему изобретению описан более подробно, постадийно.
В этом способе получения стадия 1) представляет собой получение соединение, представленного формулой 1b, путем индукции реакции размыкания кольца соединения, представленного формулой 1a, в присутствии кислоты. Более точно, соединение, представленное формулой 1a, включенное в соединение, представленное формулой 1, подвергают реакции размыкания кольца в присутствии кислоты. В результате гетероцикл соединения, представленного формулой 1a, раскрывается, образуя соединение, представленное формулой 1b, содержащее карбонил.
На этой стадии кислота может быть соляной кислотой, серной кислотой или фосфорной кислотой, и соляная кислота является более предпочтительной.
Растворитель реакции может представлять собой тетрагидрофуран (THF), дихлорметан (DCM), толуол или ацетонитрил, и тетрагидрофуран (THF) является более предпочтительным.
Температура реакции предпочтительно составляет от 0°C до ~ температуры кипения растворителя, и время реакции не ограничено, но предпочтительна реакция в течение 0,5~10 часов.
Способ получения 4
Соединение, представленное формулой 1 согласно настоящему изобретению, может быть получено способом, включающим стадию получения соединения, представленного формулой 1c, реакцией восстановления соединения, представленного формулой 1b (стадия 1), как показано в приведенной ниже схеме реакции 5.
[Схема реакции 5]
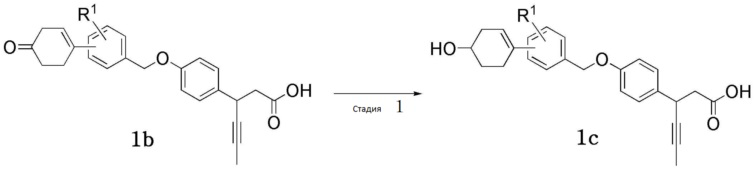
(В схеме реакции 5,
R1 имеет значения, определенные в формуле 1; и соединения, представленные формулой 1b и формулой 1c, включены в соединение, представленное формулой 1).
Далее способ получения согласно настоящему изобретению описан более подробно, постадийно.
В этом способе получения стадия 1) представляет собой получение соединение, представленного формулой 1c, путем индукции реакции восстановления соединения, представленного формулой 1b, в присутствии основания. Более точно, соединение, представленное формулой 1b, одно из соединений, представленных формулой 1, восстанавливают в присутствии основания. Таким образом, карбонильная группа соединения, представленного формулой 1b, восстанавливается до гидроксильной группы, приводя к соединению, представленному формулой 1c.
На этой стадии основание здесь может представлять собой боргидрид натрия (NaBH3) или литий-алюминийгидрид (LiAlH4), и боргидрид натрия (NaBH3) является более предпочтительным.
Растворитель реакции может представлять собой тетрагидрофуран (THF), дихлорметан (DCM), толуол или ацетонитрил, и тетрагидрофуран (THF) является более предпочтительным.
Температура реакции предпочтительно составляет от 0°C до ~ температуры кипения растворителя, и время реакции не ограничено, но предпочтительна реакция в течение 0,5~10 часов.
Настоящее изобретение также относится к фармацевтической композиции, включающей соединение, представленное формулой 1, его оптический изомер или его фармацевтически приемлемую соль в качестве активного ингредиента для профилактики или лечения метаболического заболевания.
Фармацевтическая композиция характерно функционирует, активируя фермент GPR40.
GPR40 представляет собой сцепленный с G-белком рецептор (GPCR), главным образом экспрессирующийся в инсулинсекретирующих клетках в поджелудочной железе. Профиль экспрессии GPR40 имеет потенциальную пригодность для лечения различных метаболических заболеваний, включая ожирение и диабет.
Поэтому авторы изобретения исследовали структуру активации рецептора GPR40 согласно соединению, представленному формулой 1, его оптическому изомеру или его фармацевтически приемлемой соли согласно настоящему изобретению. В результате все экспериментальные соединения согласно настоящему изобретению могли активировать рецептор GPR40 на 50% (EC50) в низкой концентрации, что позволяет предположить, что активирующий эффект соединений согласно настоящему изобретению был превосходным (см. Экспериментальные Примеры 1 и 2 и фигуру 1).
Относительно метаболизма лекарственного средства соединения, представленного формулой 1, его оптического изомера или его фармацевтически приемлемой соли согласно настоящему изобретению, авторы изобретения оценили его степень ингибирования фермента CYP. В результате было подтверждено, что все экспериментальные соединения не вызывают токсичности при совместном введении с другими лекарственными средствами, независимо от концентрации, что позволяет предположить, что их можно вводить совместно с другими лекарственными средствами, когда требуется лечение осложнений (см. Экспериментальный Пример 3).
Авторы настоящего изобретения также провели тест на пероральную переносимость глюкозы с соединением, представленным формулой 1, его оптическим изомером или его фармацевтически приемлемой солью по изобретению. В результате все экспериментальные соединения по изобретению продемонстрировали подобный или более сильный эффект в отношении снижения глюкозы крови, чем обычный активатор GPR40, что позволяет предположить, что они все превосходно активируют GPR40 in vivo (см. Экспериментальные Примеры 4, 5 и 6).
Авторы настоящего изобретения также исследовали степень повышения GLP-1 крови при пероральном введении соединения, представленного формулой 1, его оптического изомера или его фармацевтически приемлемой соли по изобретению. В результате соединение Сравнительного Примера 1 не показывало эффекта повышения GLP-1 крови после введения по сравнению с группой, получавшей глюкозу (носитель), в то время как соединение Примера 9 по настоящему изобретению увеличило GLP-1 крови при введении крысам SD (см. Экспериментальный Пример 7 и Фигуру 2).
Поэтому соединение, представленное формулой 1 согласно настоящему изобретению, не только превосходно активирует белок GPR40 и таким образом промотирует секрецию инсулина, но также и может быть использовано совместно с другими лекарственными средствами, так, чтобы композиция, включающая соединение формулы 1, которое является превосходным в активации белка GPR40 in vivo, в качестве активного ингредиента, могла эффективно использоваться как фармацевтическая композиция для профилактики или лечения метаболического заболевания, такого как ожирение, диабет типа I, диабет типа II, несовместимая переносимость глюкозы, инсулинорезистентность, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром X и т.д.
Соединение, представленное формулой 1 согласно настоящему изобретению, может вводиться перорально или парентерально и может быть использовано в обычных формах фармацевтического состава. Таким образом, соединение согласно настоящему изобретению может быть получено для перорального или парентерального введения путем смешивания с обычно используемыми разбавителями или эксципиентами, такими как наполнители, экстендеры, связующие, смачивающие вещества, разрыхлители и поверхностно-активные вещества.
Твердые составы для перорального ввдеения представляют собой таблетки, пилюли, порошки, гранулы, капсулы и пастилки и т.д. Эти твердые составы получают, смешивая соединение по изобретению с одним или более подходящими эксципиентами, такими как крахмал, карбонат кальция, сахароза или лактоза и желатин и т.д. Помимо простых эксципиентов, могут использоваться лубриканты, например, стеарат магния, тальк и т.д. Жидкие составы для перорального введения представляют собой суспензии, растворы, эмульсии и сиропы, и вышеупомянутые составы могут содержать различные эксципиенты, такие как смачивающие вещества, подсластители, ароматизаторы и консерванты в дополнение к обычно используемым простым разбавителям, таким как вода и жидкий парафин.
Составы для парентерального введения представляют собой стерилизованные водные растворы, водонерастворимые эксципиенты, суспензии, эмульсии, лиофилизированные препараты и суппозитории. Водонерастворимые эксципиенты и суспензии могут содержать, в дополнение к активному соединению или соединениям, пропиленгликоль, полиэтиленгликоль, растительное масло, такое как оливковое масло, инъецируемый сложный эфир, такой как этилолеат, и т.д. Суппозитории могут содержать, в дополнение к активному соединению или соединениям, witepsol, macrogol, tween 61, масло какао, лауриновое масло, глицерин, желатин и т.д.
Эффективная дозировка соединения согласно настоящему изобретению может быть отрегулирована в зависимости от возраста, массы тела и пола пациента, пути введения, состояния здоровья, серьезности заболевания и т.д. В целом, дозировка составляет 0,001~100 мг/кг/сутки, и предпочтительно 0,01~35 мг/кг/сутки. Соединение согласно настоящему изобретению может вводиться в количестве 0,07~7000 мг/сутки для взрослого пациента, который весит 70 кг, и более предпочтительно в количестве 0,7~2500 мг/сутки, причем это количество может быть введено от 1 до ~ нескольких раз в сутки с регулярными интервалами на усмотрение врача или фармаколога.
Практические и предпочтительные варианты осуществления настоящего изобретения проиллюстрированы в следующих Примерах.
Однако следует понимать, что специалист в данной области техники на основании этого раскрытия могут сделать модификации и улучшения в рамках духа и объема настоящего изобретения.
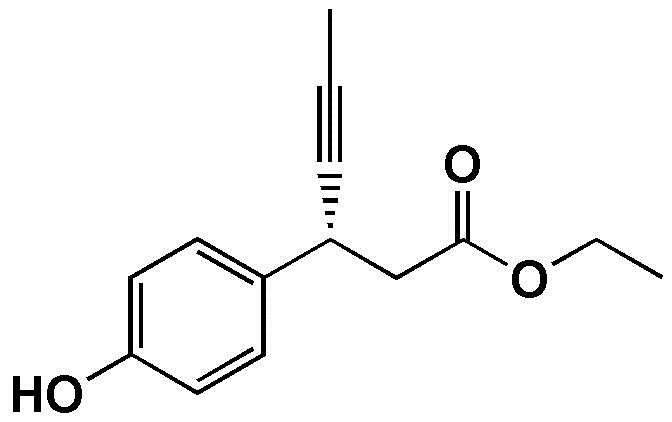
Пример получения 1: Получение этил-3-(4-гидроксифенил)гекс-4-иноата
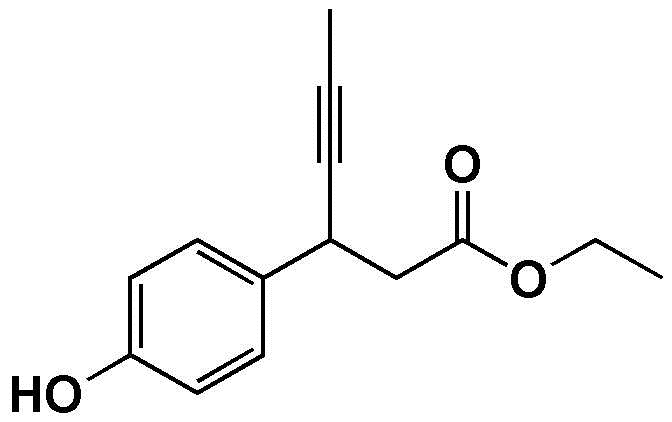
3-(4-гидроксифенил)-гекс-4-иновую кислоту (20,0 г) и этанол (200 мл) загружали в колбу на 250 мл в атмосфере азота с последующим перемешиванием для того, чтобы растворить их. Серную кислоту (9,6 мл) медленно добавляли при комнатной температуре. Смесь перемешивали при нагревании с обратным холодильником в течение по меньшей мере 6 часов. После завершения реакции медленно добавляли дистиллированную воду (150 мл), с последующей экстракцией с использованием этилацетата (200 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение (19,5 г, 85,7%).
1H ЯМР (400 МГц, CDCl3): δ 7,25 (2H, д), 6,78 (2H, д), 4,95 (1H, с), 4,14 (2H, м), 4,04 (1H, м), 2,68 (2H, м), 1,84 (3H, д), 1,29 (3H, т).
Пример получения 2: Получение (S)-этил-3-(4-гидроксифенил)гекс-4-иноата
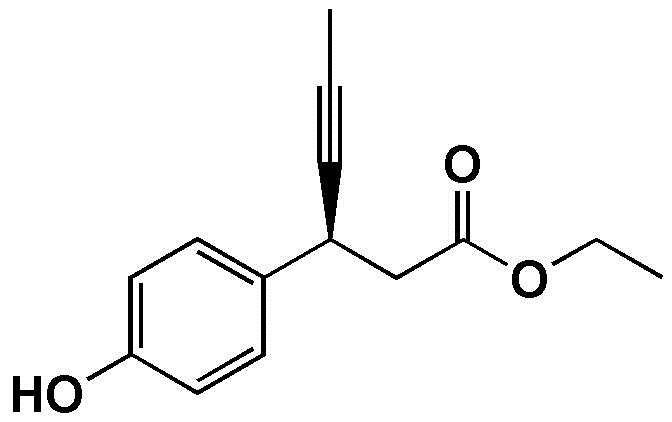
(S)-3-(4-гидроксифенил)-гекс-4-иновую кислоту (20,0 г) и этанол (200 мл) загружали в колбу на 250 мл в атмосфере азота, с последующим перемешиванием для того, чтобы растворить их. Серную кислоту (9,6 мл) медленно добавляли при комнатной температуре. Смесь перемешивали при нагревании с обратным холодильником в течение по меньшей мере 6 часов. После завершения реакции медленно добавляли дистиллированную воду (150 мл), с последующей экстракцией с использованием этилацетата (200 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение (21,2 г, 93,2%).
1H ЯМР (400 МГц, CDCl3): δ 7,25 (2H, д), 6,78 (2H, д), 4,95 (1H, с), 4,14 (2H, м), 4,04 (1H, м), 2,68 (2H, м), 1,84 (3H, д), 1,29 (3H, т).
Пример получения 3: Получение (R)-этил-3-(4-гидроксифенил)гекс-4-иноата
(R)-3-(4-гидроксифенил)-гекс-4-иновую кислоту (20,0 г) и этанол (200 мл) загружали в колбу на 250 мл в атмосфере азота, с последующим перемешиванием для того, чтобы растворить их. Серную кислоту (9,6 мл) медленно добавляли при комнатной температуре. Смесь перемешивали при нагревании с обратным холодильником в течение по меньшей мере 6 часов. После завершения реакции медленно добавляли дистиллированную воду (150 мл), с последующей экстракцией с использованием этилацетата (200 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение (20,6 г, 90,6%).
1H ЯМР (400 МГц, CDCl3): δ 7,25 (2H, д), 6,78 (2H, д), 4,95 (1H, с), 4,14 (2H, м), 4,04 (1H, м), 2,68 (2H, м), 1,84 (3H, д), 1,29 (3H, т).
Пример получения 4: Получение (3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)фенил)метанола
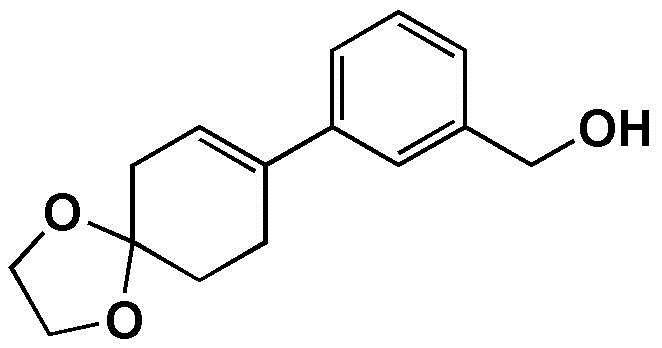
Стадия 1: Получение 1,4-диоксаспиро[4,5]дец-7-ен-8-ил трифторметансульфоната
1,4-диоксаспиро[4,5]декан-8-он (30,0 г) и толуол (300 мл) загружали в колбу на 1000 мл в атмосфере азота, с последующим перемешиванием для их растворения. Затем N-фенилбис(трифторметансульфонимид) (64,3 г) добавляли. 0,7 М раствор бис(триметилсилил)амида калия (257 мл) медленно добавляли при использовании капельной воронки при -78°C, с последующим перемешиванием в течение по меньшей мере 4 часов с подъемом температуры до комнатной температуры. После завершения реакции медленно добавляли дистиллированную воду (200 мл) с последующей экстракцией с использованием этилацетата (300 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение (54,7 г, 98,8%).
1H ЯМР (400 МГц, CDCl3): δ 5,68 (1H, т), 4,01 (4H, с), 2,55 (2H, т), 2,42 (2H, д), 1,92 (2H, т).
Стадия 2: Получение 3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензальдегида
1,4-диоксаспиро[4,5]дец-7-ен-8-ил трифторметансульфонат (54,70 г) и толуол (300 мл) загружали в колбу на 1000 мл в атмосфере азота с последующим перемешиванием для их растворения. Добавляли 3-формилфенилбороновую кислоту (28,7 г) и карбонат цезия (156 г). Смесь охлаждали до 0°C, затем медленно добавляли тетракис(трифенилфосфин)палладий (11,09 г). Смесь перемешивали в течение по меньшей мере 3 часов с подъемом температуры до комнатной температуры. После завершения реакции медленно добавляли дистиллированную воду (200 мл) с последующей экстракцией с использованием этилацетата (300 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение (45,9 г, 99%).
1H ЯМР (400 МГц, CDCl3): δ 10,03 (1H, с), 7,92 (1H, с), 7,76 (1H, д), 7,67 (1H, д), 7,47 (1H, т), 6,11 (1H, с), 4,05 (4H, с), 2,71 (2H, т), 2,51 (2H, с), 1,97 (2H, т).
Стадия 3: Получение (3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)фенил)метанола
3-(1,4-диоксаспиро[4.5]дец-7-ен-8-ил)бензальдегид (46,9 г), тетрагидрофуран (160 мл) и метанол (40 мл) загружали в колбу на 500 мл в атмосфере азота с последующим перемешиванием для их растворения. Смесь охлаждали до 0°C. Затем медленно добавляли боргидрид натрия (10,9 г) с последующим перемешиванием в течение по меньшей мере 3 часов с подъемом температуры до комнатной температуры. После завершения реакции медленно добавляли дистиллированную воду (150 мл) с последующей экстракцией с использованием этилацетата (150 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение (37,8 г, 81,7%).
1H ЯМР (400 МГц, CDCl3): δ 7,34 (1H, с), 7,25 (3H, м), 6,01 (1H, м), 4,69 (2H, д), 4,04 (4H, с), 2,68 (2H, м), 2,48 (2H, с), 1,94 (2H, т), 1,80 (1H, т).
Пример получения 5: Получение (4-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)фенил)метанола
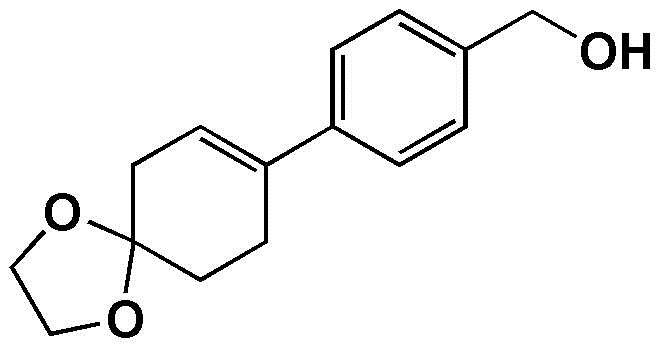
Стадия 1: Получение 4-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензальдегида
1.4-диоксаспиро[4.5]дец-7-ен-8-ил трифторметансульфонат (3,0 г) и толуол (50 мл) загружали в колбу на 250 мл в атмосфере азота с последующим перемешиванием для их растворения. Добавляли 3-формилфенил бороновую кислоту (1,8 г) и карбонат цезия (8,47 г). Смесь охлаждали до 0°C, затем медленно добавляли тетракис(трифенилфосфин)палладий (601 мг). Смесь перемешивали в течение по меньшей мере 3 часов с подъемом температуры до комнатной температуры. После завершения реакции медленно добавляли дистиллированную воду (500 мл) с последующей экстракцией с использованием этилацетата (100 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение (2,0 г, 78,7%).
1H ЯМР (400 МГц, CDCl3): δ 10,00 (1H, с), 7,84 (2H, д), 7,57 (2H, д), 6,19 (1H, с), 4,06 (4H, с), 2,71 (2H, т), 2,53 (2H, с), 1,97 (2H, т).
Стадия 2: Получение (4-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)фенил)метанола
4-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензальдегид (2,0 г), тетрагидрофуран (40 мл) и метанол (10 мл) загружали в колбу на 250 мл в атмосфере азота с последующим перемешиванием для их растворения. Смесь охлаждали до 0°C. Затем медленно добавляли боргидрид натрия (619 мг) с последующим перемешиванием в течение по меньшей мере 3 часов с подъемом температуры до комнатной температуры. После завершения реакции медленно добавляли дистиллированную воду (50 мл) с последующей экстракцией с использованием этилацетата (100 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение (1,6 г, 52,9%).
1H ЯМР (400 МГц, CDCl3): δ 7,40 (2H, д), 7,32 (2H, д), 6,01 (1H, м), 4,70 (2H, д), 4,13 (4H, с), 2,68 (2H, т), 2,49 (2H, с), 1,93 (2H, т), 1,60 (1H, т).
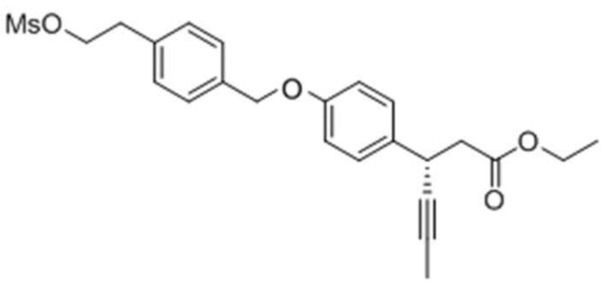
Пример получения 6: Получение этил-3-(4-(4-((метилсульфонилокси)метил)бензилокси)фенил)гекс-4-иноата
Стадия 1: Получение (4-(бромметил)фенил)метанола
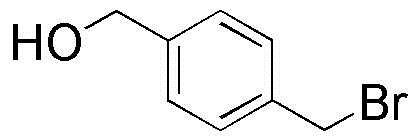
Метил-4-(бромметил)бензоат (5,0 г) и МС (20 мл) загружали в 1 л колбу в атмосфере азота с последующим перемешиванием для их растворения. Затем медленно добавляли 70 мл DIBAL-Н при -78°C, с последующим перемешиванием в течение 5 часов. После завершения реакции, смесь охлаждали до 0°C, и медленно добавляли дистиллированну. воду с последующей экстракцией с использованием МС. Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,42 (2H, д), 7,38 (2H, д), 4,73 (2H, с), 4,52 (2H, м).
Стадия 2: Получение этил-3-(4-(4-(гидроксиметил)бензилокси)фенил)гекс-4-иноата

4,0 г этил-3-(4-гидроксифенил)гекс-4-иноат, полученный в Примере получения 1, и 5,0 г (4-(бромметил)фенил)метанола, полученного на стадии 1), загружали в колбу на 500 мл, содержащую 50 мл DMF в атмосфере азота, с последующим перемешиванием для их растворения. Затем загружали 9,0 г Cs2CO3, с последующим перемешиванием при комнатной температуре в течение 12 часов. После завершения реакции, медленно добавляли дистиллированную воду с последующей экстракцией с использованием этилацетата. Экстракт промывали солевым раствором, высушивали над безводным MgSO4 и концентрировали. Затем проводили хроматографию на колонках с силикагелем, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,42 (2H, д), 7,38 (2H, д), 7,29 (2H, д), 6,93 (2H, д), 5,06 (2H, с), 4,73 (2H, д), 4,15 (2H, м), 4,06 (1H, м), 2,68 (2H, м), 1,84 (3H, с), 1,69 (1H, м), 1,24 (3H, м).
Стадия 3: Получение этил-3-(4-(4-((метилсульфонилокси)метил)бензилокси)фенил)гекс-4-иноата
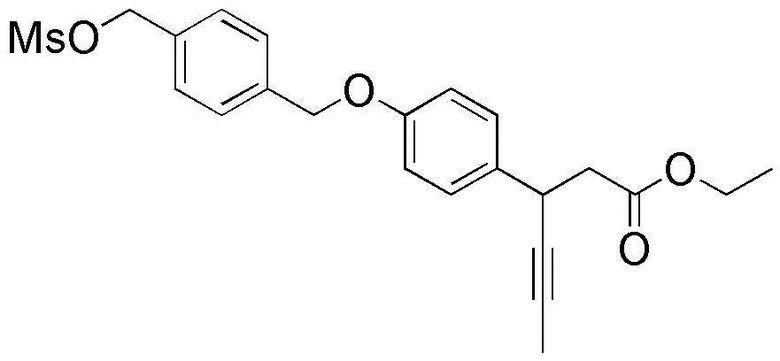
3,0 г этил-3-(4-(4-(гидроксиметил)бензилокси)фенил)гекс-4-иноата, полученного на стадии 2), загружали в колбу на 500 мл, содержащую 30 мл МС в атмосфере азота, с последующим перемешиванием для их растворения. Затем 4,0 мл ТЕА загружали при 0°C. 30 минут спустя медленно добавляли 2,1 мл MsCl. Один час спустя, когда реакция была закончена, медленно добавляли дистиллированную воду с последующей экстракцией с использованием МС. Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,49 (4H, м), 7,29 (2H, д), 6,93 (2H, д), 5,27 (2H, с), 5,08 (2H, с), 4,15 (2H, м), 4,06 (1H, м), 2,95 (3H, с), 2,68 (2H, м), 1,84 (3H, с), 1,69 (1H, м), 1,24 (3H, м).
Пример получения 7: Получение (S)-этил-3-(4-(4-((метилсульфонилокси)метил)бензилокси)фенил)гекс-4-иноата
Стадия 1: Получение (S)-этил-3-(4-(4-(гидроксиметил)бензилокси)фенил)гекс-4-иноата
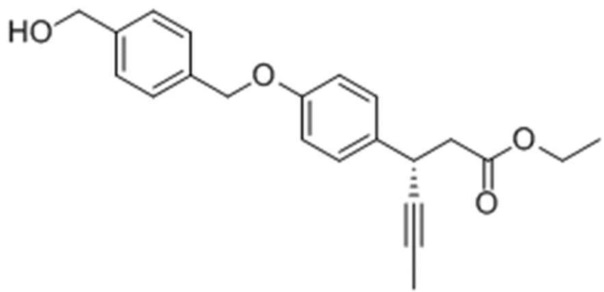
Целевое соединение получали тем же самым образом, как описано на стадии 2) Примера получения 6 за исключением того, что (S)-этил 3-(4-гидроксифенил)гекс-4-иноат использовали вместо этил-3-(4-гидроксифенил)гекс-4-иноата.
1H ЯМР (400 МГц, CDCl3): δ 7,42 (2H, д), 7,38 (2H, д), 7,29 (2H, д), 6,93 (2H, д), 5,06 (2H, с), 4,73 (2H, д), 4,15 (2H, м), 4,06 (1H, м), 2,68 (2H, м), 1,84 (3H, с), 1,69 (1H, м), 1,24 (3H, м).
Стадия 2: Получение (S)-этил-3-(4-(4-((метилсульфонилокси)метил)бензилокси)фенил)гекс-4-иноата
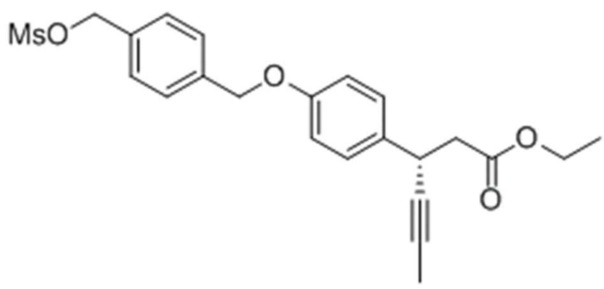
Целевое соединение получали тем же самым образом, как описано на стадии 3) Примера получения 6, за исключением того, что (S)-этил 3-(4-(4-(гидроксиметил)бензилов)фенил)гекс-4-иноат, полученный на стадии 1), использовали вместо этил-3-(4-(4-(гидроксиметил)бензилокси)фенил)гекс-4-иноата.
1H ЯМР (400 МГц, CDCl3): δ 7,49 (4H, м), 7,29 (2H, д), 6,93 (2H, д), 5,27 (2H, с), 5,08 (2H, с), 4,15 (2H, м), 4,06 (1H, м), 2,95 (3H, с), 2,68 (2H, м), 1,84 (3H, с), 1,69 (1H, м), 1,24 (3H, м).
Пример получения 8: Получение 6-метокси-1,2,3,4-тетрагидроизохинолина
Стадия 1: Получение этил-3-метоксифенэтилкарбамата
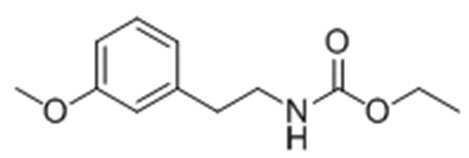
25 г 2-(3-метоксифенил)этанамина загружали в колбу, содержащую 300 мл МС в атмосфере азота, с последующим перемешиванием для их растворения. Затем 24,2 мл ТЕА загружали при 0°C. 30 минут спустя медленно добавляли 16,6 мл этил хлорформиата. Один час спустя, когда реакция была закончена, медленно добавляли дистиллированную воду с последующей экстракцией с использованием МС. Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,25 (1H, м), 6,79 (3H, м), 4,70 (1H, с), 4,13 (2H, м), 3,81 (3H, с), 3,46 (2H, м), 2,80 (2H, м), 1,25 (3H, м).
Стадия 2: Получение 6-метокси-3,4-дигидроизохинолин-1(2H)-она
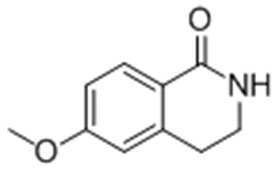
36 г этила-3-метоксифенэтилкарбамата, полученного на стадии 1), и 120 г полифосфорной кислоты загружали в колбу на 500 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем смесь нагревали с обратным холодильником в течение по меньшей мере 3 часов. Смесь охлаждали до комнатной температуры. Медленно добавляли этилацетат и дистиллированную воду с последующей экстракцией по меньшей мере три раза. Экстрагированный органический слой промывали солевым раствором, высушивали над безводным MgSO4 и концентрировали. Затем проводили хроматографию на колонках с силикагелем, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 8,03 (1H, д), 6,87 (1H, д), 6,72 (1H, с), 6,44 (1H, с), 3,86 (3H, с), 3,57 (2H, м), 2,98 (2H, м).
Стадия 3: Получение 6-метокси-1,2,3,4-тетрагидроизохинолина
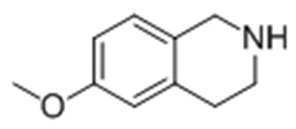
10 г 6-метокси-3,4-дигидроизохинолин-1(2H)-она загружали в колбу, содержащую 150 мл THF в атмосфере азота, с последующим перемешиванием для их растворения. Затем 4,3 г LAH медленно добавляли при 0°C. После индукции нагревания с обратным холодильником в течение по меньшей мере 5 часов, когда реакция была закончена, медленно добавляли дистиллированную воду с последующей экстракцией с использованием этилацетата. Экстракт промывали солевым раствором, высушивали над безводным MgSO4 и концентрировали. Затем осуществляли отверждение, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 6,94 (1H, д), 6,73 (1H, д), 6,65 (1H, с), 4,14 (2H, с), 3,80 (3H, с), 3,13 (2H, м), 2,79 (2H, м).
Пример получения 9: Получение 4-(4-(метилсульфонил)фенил)-1,2,3,6-тетрагидропиридин гидрохлорида
Стадия 1: Получение трет-бутил-4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилата
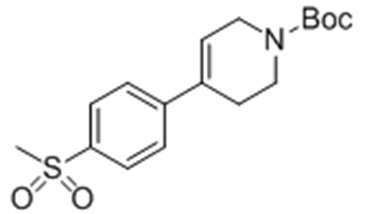
3,31 г трет-бутил-4-(трифторметилсульфонилокси)-5,6-дигидропиридин-1(2H)-карбоксилата и 50 мл толуола загружали в колбу на 1000 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем добавляли 2,0 г 4-(метилсульфонил)фенилбороновую кислоту и 6,6 г карбонат цезия. Смесь охлаждали до 0°C, затем медленно добавляли 1,16 г тетракис(трифенилфосфин)палладия (11,09 г). Смесь перемешивали в течение по меньшей мере 3 часов с подъемом температуры до комнатной температуры. После завершения реакции медленно добавляли дистиллированную воду с последующей экстракцией с использованием этилацетата. Экстрагированный органический слой высушивали при пониженном давлении. Затем проводили хроматографию на колонках с силикагелем, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,92 (2H. д), 7,56 (2H, д), 6,21 (1H, с), 4,14 (2H, д), 3,68 (2H, м), 3,07 (3H, с), 2,56 (2H, с), 1,49 (9H, c).
Стадия 2: Получение 4-(4-(метилсульфонил)фенил)-1,2,3,6-тетрагидропиридин гидрохлорида
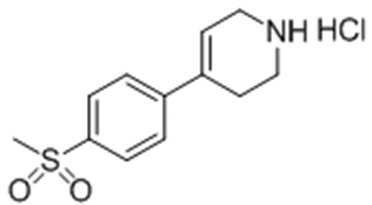
1,4 г трет-бутил-4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилата, полученного на стадии 1), растворяли в 20 мл МС, затем добавляли 10,4 мл 4н. HCl. 5 часов спустя, когда реакция была закончена, добавляли простой диэтиловый эфир. Затем осуществляли отверждение, получая целевое соединение.
1H ЯМР (400 МГц, D2O): δ 7,92 (2H, д), 7,56 (2H, д), 6,21 (1H, с), 4,14 (2H, д), 3,68 (2H, м), 3,07 (3H, с), 2,56 (2H, с).
Пример получения 10: Получение 4-(1,2,3,6-тетрагидропиридин-4-ил)фенол гидрохлорида
Стадия 1: Получение трет-бутил-4-(4-гидроксифенил)-5,6-дигидропиридин-1(2H)-карбоксилата
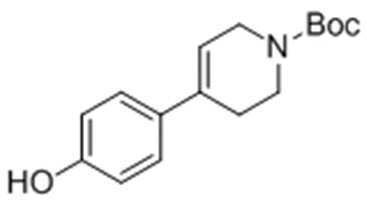
Целевое соединение получали тем же самым образом, как описано на стадии 1) Примера получения 9, за исключением того, что 4-гидроксифенилбороновую кислоту использовали вместо 4-(метилсульфонил) фенилбороновой кислоты.
1H ЯМР (400 МГц, CDCl3): δ 7,26 (2H, д), 6,83 (2H, д), 5,93 (1H, с), 5,47 (1H, с), 4,07 (2H, с), 3,66 (2H, м), 2,50 (2H, с), 1,52 (9H, с).
Стадия 2: Получение 4-(1,2,3,6-тетрагидропиридин-4-ил)фенол гидрохлорида
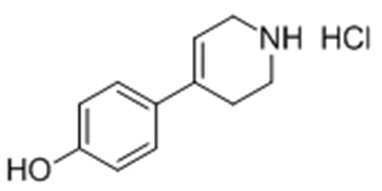
Целевое соединение получали тем же самым образом, как описано на стадии 2) Примера получения 9, за исключением того, что трет-бутил-4-(4-гидроксифенил)-5,6-дигидропиридин-1(2H)-карбоксилат, полученный на стадии 1), использовали вместо трет-бутил-4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилата.
1H ЯМР (400 МГц, D2O): δ 7,26 (2H, д), 6,83 (2H, д), 5,93 (1H, с), 5,47 (1H, с), 4,07 (2H, с), 3,66 (2H, м), 2,50 (2H, с).
Пример получения 11: Получение 4-(4-(3-(метилсульфонил)пропокси)фенил)-1,2,3,6-тетрагидропиридин гидрохлорида
Стадия 1: Получение 3-(метилтио)пропил-4-метилбензолсульфоната

25,4 г 3-(метилтио)пропан-1-ола загружали в колбу на 500 мл, содержащую 500 мл МС в атмосфере азота, с последующим перемешиванием для их растворения. Затем 44 мл ТЕА добавляли при 0°C. 30 минут спустя медленно добавляли 46 г TsCl. Один час спустя, когда реакция была закончена, медленно добавляли дистиллированную воду с последующей экстракцией с использованием МС. Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,81 (2H, д), 7,38 (2H, д), 4,16 (2H, м), 2,53 (2H, м), 2,47 (3H, с), 2,05 (3H, с), 1,94 (2H, м).
Стадия 2: Получение 3-(метилсульфонил)пропил-4-метилбензолсульфоната

62 г 3-(метилтио)пропил-4-метилбензолсульфоната, полученного на стадии 1), загружали в смесь THF/дистиллированная вода (150/100 мл) в колбе в атмосфере азота с последующим перемешиванием для их растворения. Затем 310 г добавляли оксон. Смесь перемешивали в течение 12 часов при комнатной температуре. После завершения реакции, медленно добавляли дистиллированную воду с последующей экстракцией с использованием этилацетата. Экстракт промывали солевым раствором, высушивали над безводным MgSO4, и концентрировали, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,81 (2H, д), 7,38 (2H, д), 4,20 (2H, м), 3,13 (2H, м), 2,93 (3H, с), 2,48 (3H, с), 2,23 (2H, м).
Стадия 3: Получение трет-бутил-4-(4-(3-(метилсульфонил)пропокси)фенил)-5,6-дигидропиридин-1(2H)-карбоксилата
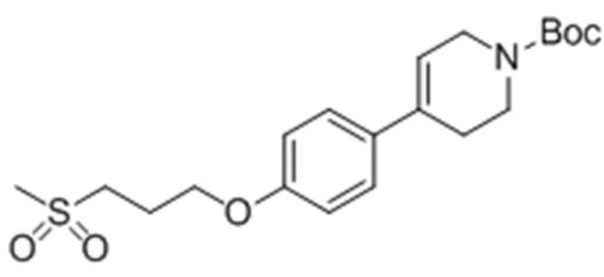
Целевое соединение получали тем же самым образом, как описано на стадии 2) Примера получения 6, за исключением того, что использовали трет-бутил-4-(4-гидроксифенил)-5,6-дигидропиридин-1(2H)-карбоксилат, полученный на стадии 1) Примера получения 10, и 3-(метилсульфонил)пропил-4-метилбензолсульфонат, полученный на стадии 2) Примера получения 10.
1H ЯМР (400 МГц, CDCl3): δ 7,34 (2H, д), 6,85 (2H, д), 6,00 (1H, с), 4,12 (2H, с), 3,28 (2H, м), 3,18 (2H, с), 2,97 (3H, с), 2,72 (2H, м), 2,56 (2H, м), 2,36 (2H, м), 1,52 (9H, с).
Стадия 4: Получение 4-(4-(3-(метилсульфонил)пропокси)фенил)-1,2,3,6-тетрагидропиридин гидрохлорида
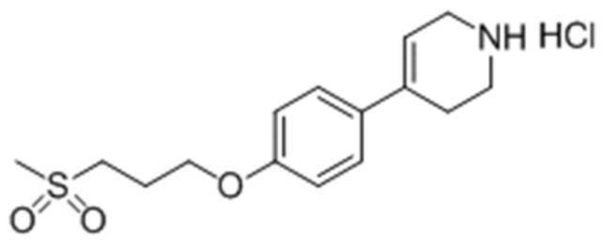
Целевое соединение получали тем же самым образом, как описано на стадии 2) Примера получения 9, за исключением того, что трет-бутил-4-(4-(3-(метилсульфонил)пропокси)фенил)-5,6-дигидропиридин-1(2H)-карбоксилат, полученный на стадии 3), использовали вместо трет-бутил-4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилата.
1H ЯМР (400 МГц, D2O): δ 7,34 (2H, д), 6,85 (2H, д), 6,00 (1H, с), 4,12 (2H, с), 3,28 (2H, м), 3,18 (2H, с), 2,97 (3H, с), 2,72 (2H, м), 2,56 (2H, м), 2,36 (2H, м).
Пример получения 12: Получение (3S)-этил-3-(4-(4-(1-бромэтил)бензилокси)фенил)гекс-4-иноата
Стадия 1: Получение 1-(4-(бромметил)фенил)этанона
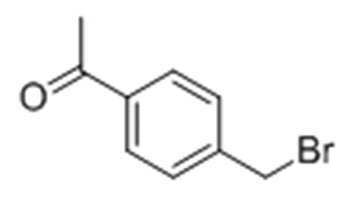
5,0 г 1-п-толилэтана растворяли в 100 мл CCl4 в колбе в атмосфере азота при перемешивании, затем 14,6 г NBS и 6,7 г AIBN добавляли при 0°C. Затем смесь нагревали с обратным холодильником в течение по меньшей мере 5 часов. После завершения реакции, медленно добавляли дистиллированную воду с последующей экстракцией с использованием МС. Экстрагированный органический слой промывали солевым раствором, высушивали над безводным MgSO4 и концентрировали. Затем проводили хроматографию на колонках с силикагелем, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,95 (2H, д), 7,50 (2H, д), 4,52 (2H, с), 2,62 (3H, с).
Стадия 2: Получение (S)-этил-3-(4-(4-ацетилбензилокси)фенил)гекс-4-иноата
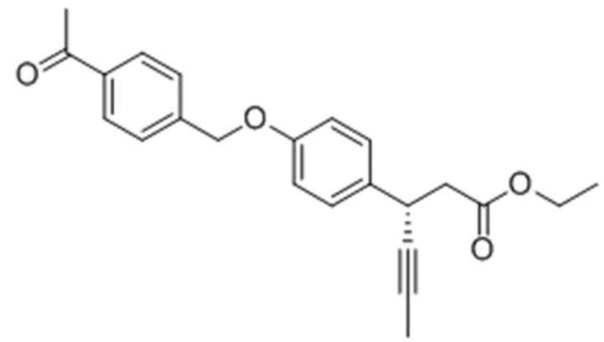
Целевое соединение получали тем же самым образом, как описано на стадии 2) Примера получения 6, за исключением того, что использовали (S)-этил-3-(4-гидроксифенил)гекс-4-иноат, полученный в Примере получения 2, и 1-(4-(бромметил)фенил)этанон, полученный на стадии 1).
1H ЯМР (400 МГц, CDCl3): δ 7,99 (2H, д), 7,53 (2H, д) 7,31 (2H, д), 6,92 (2H, д), 5,13 (2H, с), 4,15 (2H, м), 4,09 (1H, м), 2,75 (2H, м), 2,64 (3H, с), 1,84 (3H, д), 1,24 (3H, м).
Стадия 3: Получение (3S)-этил-3-(4-(4-(1 оксиэтил)бензилокси)фенил)гекс-4-иноата
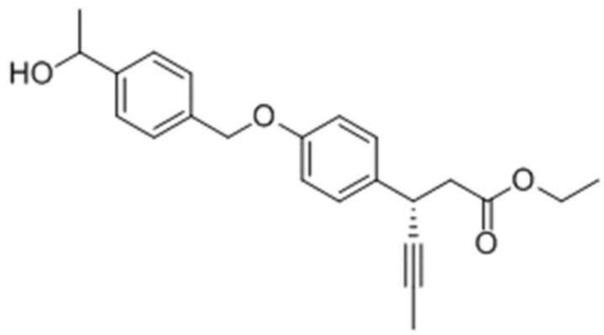
1,0 г (S)-этил-3-(4-(4-ацетилбензилокси)фенил)гекс-4-иноата, полученного на стадии 2), растворяли в 50 мл THF в колбе при перемешивании в атмосфере азота, затем 0,16 г NaBH4 добавляли при 0°C. После перемешивания смеси при комнатной температуре в течение по меньшей мере 2 часов, когда реакция была закончена, медленно добавляли дистиллированную воду с последующей экстракцией с использованием ЕА. Экстрагированный органический слой промывали солевым раствором, высушивали над безводным MgSO4 и концентрировали, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 8,02 (2H, д), 7,57 (2H, д) 7,36 (2H, д), 6,99 (2H, д), 5,21 (2H, с), 4,23 (2H, м), 4,17 (1H, м), 3,81 (1H, с), 2,75 (2H, м), 2,64 (3H, с), 1,84 (3H, д), 1,24 (3H, м).
Стадия 4: Получение (3S)-этил-3-(4-(4-(1-бромэтил)бензилокси)фенил)гекс-4-иноата
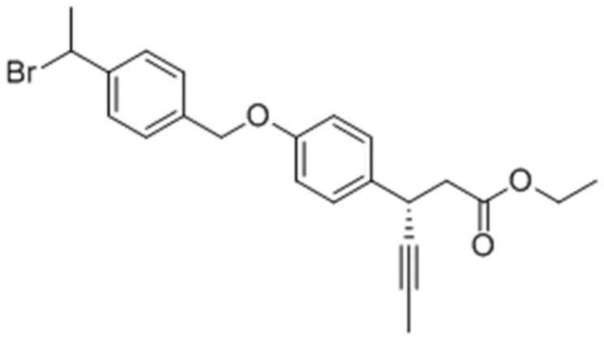
0,76 г (3S)-этил-3-(4-(4-(1 оксиэтил)бензилокси)фенил)гекс-4-иноата, полученного на стадии 3), растворяли в 50 мл МС в колбе при перемешивании в атмосфере азота, затем 0,6 г трифенилфосфина и 0,75 г CBr4 добавляли при 0°C. После перемешивания смеси при комнатной температуре в течение по меньшей мере 2 часов, когда реакция была закончена, медленно добавляли дистиллированную воду с последующей экстракцией с использованием ЕА. Экстрагированный органический слой промывали солевым раствором, высушивали над безводным MgSO4 и концентрировали, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 8,02 (2H, д), 7,57 (2H, д) 7,36 (2H, д), 6,99 (2H, д), 5,21 (2H, с), 4,23 (2H, м), 4,17 (1H, м), 3,92 (1H, с), 2,85 (2H, м), 2,44 (3H, с), 1,86 (3H, д), 1,27 (3H, м).
Пример получения 13: Получение 2-(пиперазин-1-ил)бензо[d]тиазол гидрохлорида
Стадия 1: Получение трет-бутил-4-(бензо[d]тиазол-2-ил)пиперазин-1-карбоксилата
2,0 г трет-бутилпиперазин-1-карбоксилата растворяли в смеси AN/дистиллированная вода (100/50 мл) в колбе при перемешивании в атмосфере азота, затем 2,1 мл DIPEA добавляли при 0°C. Добавляли 0,9 г 2-хлорбензо[d]тиазола с последующим нагреванием с обратным холодильником в течение по меньшей мере 2 часов. После завершения реакции медленно добавляли дистиллированную воду с последующей экстракцией с использованием ЕА. Экстрагированный органический слой промывали солевым раствором, высушивали над безводным MgSO4 и концентрировали, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,61 (1H, д), 7,60 (1H, д), 7,29 (1H, м), 7,09 (1H, м), 3,77 (4H, м), 2,62 (4H, м), 1,52 (9H, с).
Стадия 2: Получение 2-(пиперазин-1-ил)бензо[d]тиазол гидрохлорида
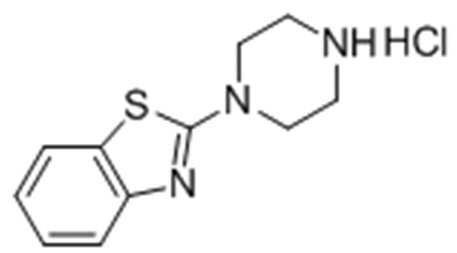
Целевое соединение получали тем же самым образом, как описано на стадии 2) Примера получения 9, за исключением того, что трет-бутил-4-(бензо[d]тиазол-2-ил)пиперазин-1-карбоксилат, полученный на стадии 1), использовали вместо трет-бутил-4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилата.
1H ЯМР (400 МГц, D2O): δ 7,61 (1H, д), 7,60 (1H, д), 7,29 (1H, м), 7,09 (1H, м), 3,77 (4H, м), 2,62 (4H, м).
Пример получения 14: Получение 2-(пиперазин-1-ил)-5-пропилпиримидин гидрохлорида
Стадия 1: Получение трет-бутил-4-(5-пропилпиримидин-2-ил)пиперазин-1-карбоксилата
Целевое соединение получали тем же самым образом, как описано на стадии 1) Примера получения 13, за исключением того, что 2-хлор-5-пропилпиримидин использовали вместо 2-хлорбензо[d]тиазола.
1H ЯМР (400 МГц, CDCl3): δ 8,19 (2H, с), 3,77 (4H, м), 2,62 (4H, м), 2,41 (2H, м), 1,61 (2H, м), 1,52 (9H, с), 0,96 (3H, м).
Стадия 2: Получение 2-(пиперазин-1-ил)-5-пропилпиримидин гидрохлорида
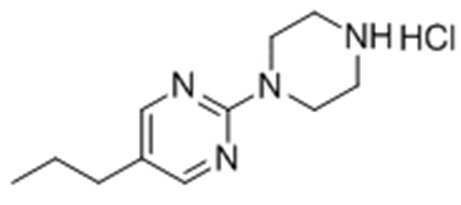
Целевое соединение получали тем же самым образом, как описано на стадии 2) Примера получения 9, за исключением того, что трет-бутил-4-(5-пропилпиримидин-2-ил)пиперазин-1-карбоксилат, полученный на стадии 1), использовали вместо трет-бутил-4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилата.
1H ЯМР (400 МГц, D2O): δ 8,19 (2H, с), 3,77 (4H, м), 2,62 (4H, м), 2,41 (2H, м), 1,61 (2H, м), 0,96 (3H, м).
Пример получения 15: Получение 6-(пиперазин-1-ил)никотинонитрил гидрохлорида
Стадия 1: Получение трет-бутил-4-(5-цианопиридин-2-ил)пиперазин-1-карбоксилата
Целевое соединение получали тем же самым образом, как описано на стадии 1) Примера получения 13, за исключением того, что 6-хлорникотинонитрил использовали вместо 2-хлорбензо[d]тиазола.
1H ЯМР (400 МГц, CDCl3): δ 8,41 (1H, с), 7,61 (1H, д), 6,59 (1H, д), 3,77 (4H, м), 2,62 (4H, м), 1,52 (9H, с).
Стадия 2: Получение 6-(пиперазин-1-ил)никотинонитрил гидрохлорида
Целевое соединение получали тем же самым образом, как описано на стадии 2) Примера получения 9, за исключением того, что трет-бутил-4-(5-цианопиридин-2-ил)пиперазин-1-карбоксилат, полученный на стадии 1), использовали вместо трет-бутил-4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-карбоксилата.
1H ЯМР (400 МГц, D2O): δ 8,41 (1H, с) 7,61 (1H, д), 6,59 (1H, д), 3,77 (4H, м), 2,62 (4H, м).
Пример получения 16: Получение (S)-этил-3-(4-(4-(2-(метилсульфонилокси)этил)бензилокси)фенил)гекс-4-иноата
Стадия 1: Получение 2-(4-(бромметил)фенил)этанола
5 г 2-(4-(бромметил)фенил)уксусной кислоты растворяли в 100 мл THF в колбе при перемешивании в атмосфере азота, затем 70 мл раствора гидрид бора-THF медленно добавляли при 0°C. После перемешивания смеси в течение 2 часов, когда реакция была закончена, температуру понижали до 0°C и медленно добавляли дистиллированную воду с последующей экстракцией с использованием ЕА. Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 37 (2H, д), 7,24 (2H, д), 4,51 (2H, с), 3,89 (2H, м), 2,89 (2H, м).
Стадия 2: Получение (S)-этил-3-(4-(4-(2-гидроксиэтил)бензилокси)фенил)гекс-4-иноата
Целевое соединение получали тем же самым образом, как описано на стадии 2) Примера получения 6, за исключением того, что 2-(4-(бромметил)фенил)этанол, полученный на стадии 1), использовали вместо (4-(бромметил)фенил)метанола.
1H ЯМР (400 МГц, CDCl3): δ 7,40 (2H, д), 7,30 (2H, д), 7,27 (2H, д), 6,95 (2H, д), 5,04 (2H, с), 4,18 (2H, м), 4,11 (1H, м), 3,89 (2H, м), 2,91 (2H, м), 2,71 (2H, м), 1,84 (3H, с), 1,38 (1H, м), 1,25 (3H, м).
Стадия 3: Получение (S)-этил-3-(4-(4-(2-(метилсульфонилокси)этил)бензилокси)фенил)гекс-4-иноата
Целевое соединение получали тем же самым образом, как описано на стадии 3) Примера получения 6, за исключением того, что (S)-этил-3-(4-(4-(2-гидроксиэтил)бензилокси)фенил)гекс-4-иноат, полученный на стадии 2), использовали вместо этил-3-(4-(4-(гидроксиметил)бензилокси)фенил)гекс-4-иноата.
1H ЯМР (400 МГц, CDCl3): δ 7,40 (2H, д), 7,30 (2H, д), 7,27 (2H, д), 6,95 (2H, д), 5,04 (2H, с), 4,18 (2H, м), 4,11 (1H, м), 3,99 (2H, м), 2,95 (3H, с), 2,93 (2H, м), 2,71 (2H, м), 1,84 (3H, с), 1,25 (3H, м).
Пример 1: Получение 3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновой кислоты
Стадия 1: Получение этил-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноата
(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)фенил)метанол (19,54 г), полученный в Примере получения 4, и тетрагидрофуран (80 мл) загружали в колбу на 500 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем медленно добавляли этил-3-(4-гидроксифенил)гекс-4-иноат (18,42 г), полученный в Примере получения 1, и трифенил фосфин (31,21 г). Диизопропил азодикарбоксилат (23,4 мл) медленно добавляли при использовании капельной воронки при 0°C с последующим перемешиванием в течение по меньшей мере 4 часов с подъемом температуры до комнатной температуры. После завершения реакции медленно добавляли дистиллированную воду (200 мл) с последующей экстракцией с использованием этилацетата (300 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение (32,1 г, 87,9%).
1H ЯМР (400 МГц, CDCl3): δ 7,46 (1H, с), 7,31 (5H, м), 6,93 (2H, д), 6,02 (1H, м), 5,04 (2H, с), 4,13 (2H, м), 4,08 (1H, м), 4,04 (4H, с), 2,69 (4H, м), 2,49 (2H, с), 1,94 (2H, т), 1,84 (3H, д), 1,31 (3H, т).
Стадия 2: Получение 3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновой кислоты
Этил-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат (32,1 г), полученный на стадии 1), метанол (50 мл) и дистиллированную воду (50 мл) загружали в колбу на 500 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем гидроксид калия (19,5 г) медленно добавляли при комнатной температуре с последующим перемешиванием по меньшей мере 1 час. После завершения реакции, смесь подкисляли (рН: 2~3) при использовании 1М водного раствора HCl с последующей экстракцией с использованием этилацетата (300 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение (24,1 г, 79,9%).
1H ЯМР (400 МГц, CDCl3): δ 7,44 (1H, с), 7,34 (5H, м), 6,91 (2H, д), 6,00 (1H, т), 5,02 (2H, с), 4,08 (1H, м), 4,04 (4H, с), 2,73 (4H, м), 2,48 (2H, с), 1,92 (2H, т), 1,82 (3H, с).
Пример 2: Получение L-лизин 3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноата
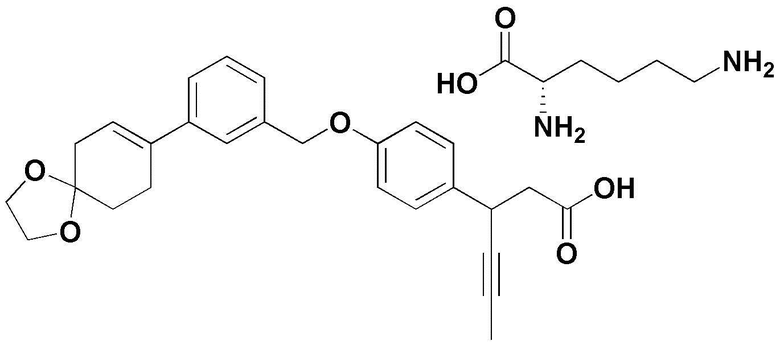
3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновую кислоту (24,1 г), полученную в Примере 1, и этанол (170 мл) загружали в колбу на 500 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем добавляли L-лизин (7,33 г). Температуру реакции повышали до 50°C, и смесь перемешивали в течение 30 минут при 50°C. Смесь охлаждали до комнатной температуры с последующим перемешиванием в течение 30 минут. После завершения реакции, полученное твердое вещество отфильтровывали, получая целевое соединение (31,5 г, 73,3%).
1H ЯМР (400 МГц, D2O): δ 7,11 (3H, м), 6,99 (3H, м), 6,64 (2H, д), 5,65 (1H, с), 4,59 (2H, с), 3,79 (5H, с), 3,60 (1H, т), 2,88 (2H, т), 2,35 (2H, д), 2,23 (2H, с), 2,14 (2H, с), 1,75 (2H, м), 1,59 (7H, м), 1,38 (2H, м).
Пример 3: Получение 4-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновой кислоты
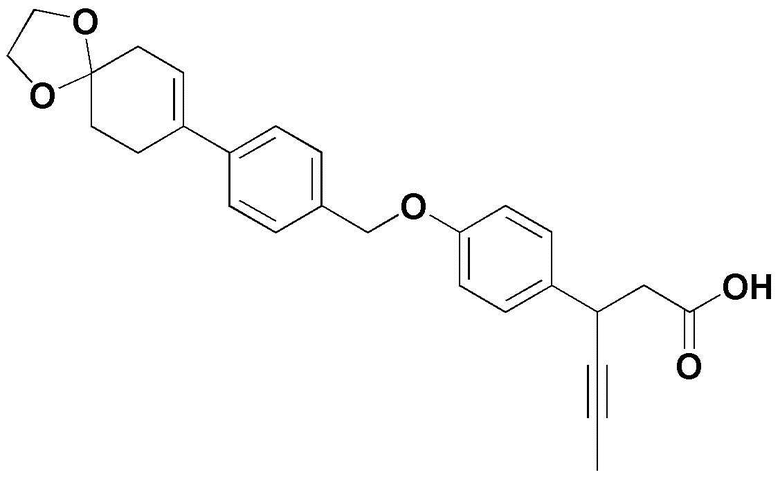
Стадия 1: Получение этил-4-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноата
(4-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)фенил)метанол (1,5 г), полученный в Примере получения 5, и тетрагидрофуран (20 мл) загружали в колбу на 100 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем медленно добавляли этил-3-(4-гидроксифенил)гекс-4-иноат (1,41 г), полученный в Примере получения 1, и трифенил фосфин (2,39 г). Диизопропил азодикарбоксилат (9,38 мл) медленно добавляли при использовании капельной воронки при 0°C с последующим перемешиванием в течение по меньшей мере 4 часов с подъемом температуры до комнатной температуры. После завершения реакции медленно добавляли дистиллированную воду (50 мл) с последующей экстракцией с использованием этилацетата (100 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение (1,38 г, 49,2%).
1H ЯМР (400 МГц, CDCl3): δ 7,42 (2H, д), 7,37 (2H, д), 7,30 (2H, д), 6,92 (2H, д), 6,01 (1H, с), 5,01 (2H, с), 4,14 (2H, м), 4,06 (5H, м), 2,70 (4H, м), 2,49 (2H, с), 1,94 (2H, т), 1,84 (3H, д), 1,24 (3H, т).
Стадия 2: Получение 4-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновой кислоты
Этил-4-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат (1,38 г), полученный на стадии 1), метанол (10 мл) и дистиллированную воду (10 мл) загружали в колбу на 500 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем гидроксид калия (1,25 г) медленно добавляли при комнатной температуре с последующим перемешиванием в течение по меньшей мере 1 часа. После завершения реакции, смесь подкисляли (рН: 2~3) при использовании 1М водного раствора HCl с последующей экстракцией с использованием этилацетата (50 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение (0,98 г, 75,6%).
1H ЯМР (400 МГц, CDCl3): δ 7,41 (2H, д), 7,36 (2H, д), 7,29 (2H, д), 6,92 (2H, д), 6,01 (1H, с), 5,01 (2H, с), 4,04 (5H, м), 2,77 (4H, м), 2,49 (2H, с), 1,96 (2H, т), 1,83 (3H, д).
Пример 4: Получение 3-(4-(3-(4-оксоциклогекс-1-енил)бензилокси)фенил)гекс-4-иновой кислоты
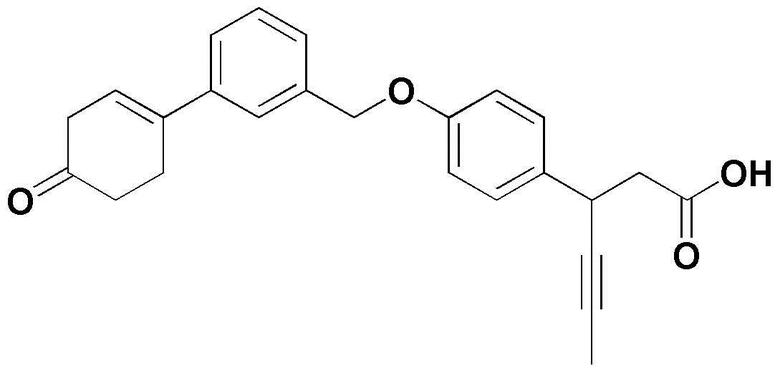
3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновую кислоту (1 г), полученную в Примере 1, и тетрагидрофуран (5 мл) загружали в колбу в атмосфере азота с последующим перемешиванием для их растворения. Добавляли 6н. водный раствор HCl (5 мл) с последующим перемешиванием при комнатной температуре в течение по меньшей мере 1 часа. После завершения реакции медленно добавляли дистиллированную воду (50 мл) с последующей экстракцией с использованием этилацетата (50 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение (0,76 г, 84,6%).
1H ЯМР (400 МГц, CDCl3): δ 7,48 (1H, с), 7,40 (5H, м), 6,94 (2H, д), 6,13 (1H, с), 5,07 (2H, с), 4,05 (1H, м), 3,10 (1,5H, т), 2,93 (1,5H, т), 2,82 (2H, м), 2,67 (2H, т), 1,85 (3H, с).
Пример 5: Получение 3-(4-(3-(4-гидроксициклогекс-1-енил)бензилокси)фенил)гекс-4-иновой кислоты
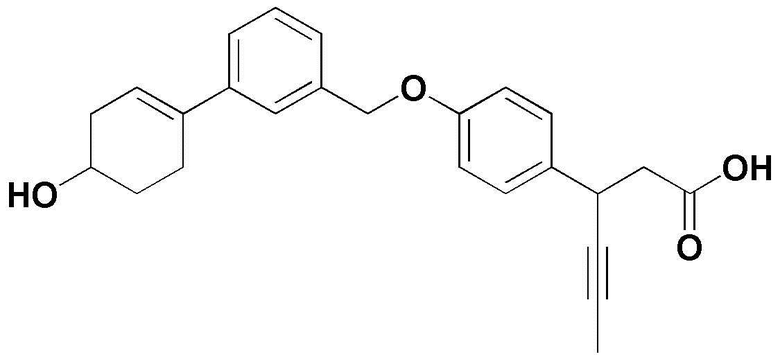
3-(4-(3-(4-оксоциклогекс-1-енил)бензилокси)фенил)гекс-4-иновую кислоту (1 г), полученную в Примере 4, и этанол (10 мл) загружали в колбу на 100 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем боргидрид натрия (0,3 г) добавляли при комнатной температуре с последующим перемешиванием в течение по меньшей мере 3 часов. После завершения реакции, смесь подкисляли (рН: 4~5) при использовании 1н. водного раствора HCl с последующей экстракцией с использованием этилацетата (100 мл) и дистиллированной воды (100 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение (0,81 г, 80,6%).
1H ЯМР (400 МГц, CDCl3): δ 7,44 (1H, с), 7,33 (5H, м), 6,93 (2H, д), 6,02 (1H, с), 5,03 (2H, с), 4,08 (2H, с), 2,78 (2H, м), 2,55 (2,5H, м), 2,22 (1H, м), 2,04 (1H, м), 1,85 (3H, с).
Пример 6: Получение L-лизин 3-(4-(3-(4-гидроксициклогекс-1-енил)бензилокси)фенил)гекс-4-иноата

3-(4-(3-(4-гидроксициклогекс-1-енил)бензилокси)фенил)гекс-4-иновую кислоту (1 г), полученную в Примере 5, и этанол (170 мл) загружали в колбу на 100 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем добавляли L-лизин (0,7 г). Температуру реакции повышали до 50°C, и смесь перемешивали в течение 30 минут при 50°C. Смесь охлаждали до комнатной температуры с последующим перемешиванием в течение 30 минут. После завершения реакции, полученное твердое вещество отфильтровывали, получая целевое соединение (0,95 г, 69,1%).
1H ЯМР (400 МГц, D2O): δ 7,11 (3H, м), 6,99 (3H, м), 6,64 (2H, д), 5,65 (1H, с), 4,59 (2H, с), 3,79 (1H, с), 3,60 (1H, т), 2,88 (2H, т), 2,35 (2H, д), 2,23 (2H, с), 2,14 (2H, с), 1,75 (2H, м), 1,59 (7H, м), 1,38 (2H, м).
Пример 7: Получение (3S)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновой кислоты
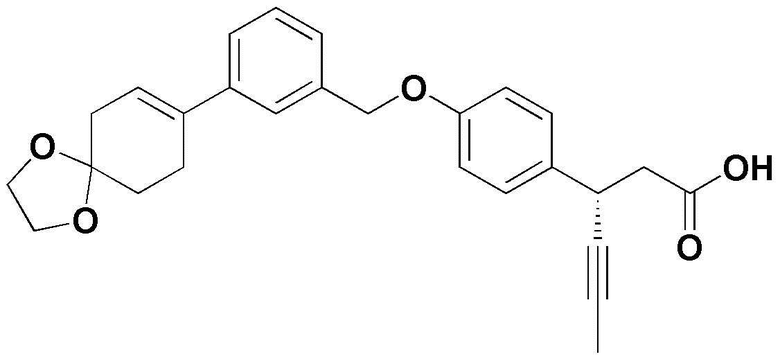
Стадия 1: Получение этил-(3S)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноата
(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)фенил)метанол (19,54 г), полученный в Примере получения 4, и тетрагидрофуран (80 мл) загружали в колбу на 500 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем медленно добавляли (S)-этил-3-(4-гидроксифенил)гекс-4-иноат (18,42 г), полученный в Примере получения 2, и трифенил фосфин (31,21 г). Диизопропил азодикарбоксилат (23,4 мл) медленно добавляли при использовании капельной воронки при 0°C с последующим перемешиванием в течение по меньшей мере 4 часов с подъемом температуры до комнатной температуры. После завершения реакции медленно добавляли дистиллированную воду (200 мл) с последующей экстракцией с использованием этилацетата (300 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,46 (1H, с), 7,31 (5H, м), 6,93 (2H, д), 6,02 (1H, м), 5,04 (2H, с), 4,13 (2H, м), 4,08 (1H, м), 4,04 (4H, с), 2,69 (4H, м), 2,49 (2H, с), 1,94 (2H, т), 1,84 (3H, д), 1,31 (3H, т).
Стадия 2: Получение (3S)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновой кислоты
Этил-(3S)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат (32,1 г), полученный на стадии 1), метанол (50 мл) и дистиллированную воду (50 мл) загружали в колбу на 500 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем гидроксид калия (19,5 г) медленно добавляли при комнатной температуре с последующим перемешиванием в течение по меньшей мере 1 часа. После завершения реакции, смесь подкисляли (рН: 2~3) при использовании 1М водного раствора HCl с последующей экстракцией с использованием этилацетата (300 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение (24,1 г, 66,2%).
1H ЯМР (400 МГц, CDCl3): δ 7,44 (1H, с), 7,34 (5H, м), 6,91 (2H, д), 6,00 (1H, т), 5,02 (2H, с), 4,08 (1H, м), 4,04 (4H, с), 2,73 (4H, м), 2,48 (2H, с), 1,92 (2H, т), 1,82 (3H, с).
Пример 8: Получение (3R)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновой кислоты
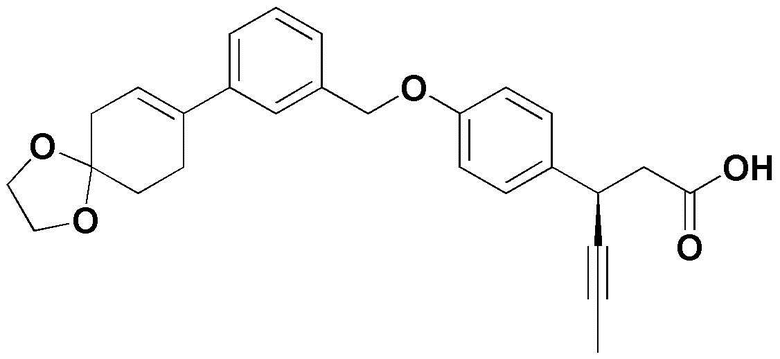
Стадия 1: Получение этил-(3R)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноата
(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)фенил)метанол (19,54 г), полученный в Примере получения 4, и тетрагидрофуран (80 мл) загружали в колбу на 500 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем медленно добавляли (R)-этил-3-(4-гидроксифенил)гекс-4-иноат (18,42 г), полученный в Примере получения 3, и трифенил фосфин (31,21 г). Диизопропил азодикарбоксилат (23,4 мл) медленно добавляли при использовании капельной воронки при 0°C с последующим перемешиванием в течение по меньшей мере 4 часов с подъемом температуры до комнатной температуры. После завершения реакции медленно добавляли дистиллированную воду (200 мл) с последующей экстракцией с использованием этилацетата (300 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,46 (1H, с), 7,31 (5H, м), 6,93 (2H, д), 6,02 (1H, м), 5,04 (2H, с), 4,13 (2H, м), 4,08 (1H, м), 4,04 (4H, с), 2,69 (4H, м), 2,49 (2H, с), 1,94 (2H, т), 1,84 (3H, д), 1,31 (3H, т).
Стадия 2: Получение (3R)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновой кислоты
Этил-(3R)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат (32,1 г), полученный на стадии 1), метанол (50 мл) и дистиллированную воду (50 мл) загружали в колбу на 500 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем гидроксид калий (19,5 г) медленно добавляли при комнатной температуре с последующим перемешиванием в течение по меньшей мере 1 часа. После завершения реакции, смесь подкисляли (рН: 2~3) при использовании 1М водного раствора HCl с последующей экстракцией с использованием этилацетата (300 мл). Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение (17,3 г, 47,5%).
1H ЯМР (400 МГц, CDCl3): δ 7,44 (1H, с), 7,34 (5H, м), 6,91 (2H, д), 6,00 (1H, т), 5,02 (2H, с), 4,08 (1H, м), 4,04 (4H, с), 2,73 (4H, м), 2,48 (2H, с), 1,92 (2H, т), 1,82 (3H, с).
Пример 9: Получение L-лизин (3S)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноата
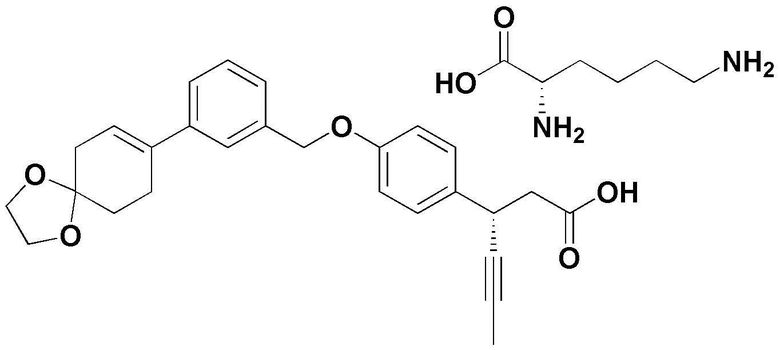
(3S)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновую кислоту (24,1 г), полученную в Примере 7, и этанол (170 мл) загружали в колбу на 500 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем добавляли L-лизин (7,33 г). Температуру реакции повышали до 50°C, и смесь перемешивали в течение 30 минут при 50°C. Смесь охлаждали до комнатной температуры с последующим перемешиванием в течение 30 минут. После завершения реакции, полученное твердое вещество отфильтровывали, получая целевое соединение (22,5 г, 69,8%).
1H ЯМР (400 МГц, D2O): δ 7,11 (3H, м), 6,99 (3H, м), 6,64 (2H, д), 5,65 (1H, с), 4,59 (2H, с), 3,79 (5H, с), 3,60 (1H, т), 2,88 (2H, т), 2,35 (2H, д), 2,23 (2H, с), 2,14 (2H, с), 1,75 (2H, м), 1,59 (7H, м), 1,38 (2H, м).
Пример 10: Получение L-лизин (3R)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноата
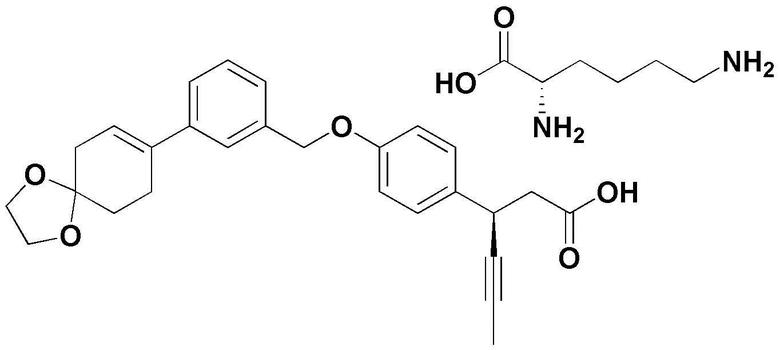
(3R)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновую кислоту (24,1 г), полученную в Примере 8, и этанол (170 мл) загружали в колбу на 500 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем добавляли L-лизин (7,33 г). Температуру реакции повышали до 50°C, и смесь перемешивали в течение 30 минут при 50°C. Смесь охлаждали до комнатной температуры с последующим перемешиванием в течение 30 минут. После завершения реакции, полученное твердое вещество отфильтровывали, получая целевое соединение (16,2 г, 71,4%).
1H ЯМР (400 МГц, D2O): δ 7,11 (3H, м), 6,99 (3H, м), 6,64 (2H, д), 5,65 (1H, с), 4,59 (2H, с), 3,79 (5H, с), 3,60 (1H, т), 2,88 (2H, т), 2,35 (2H, д), 2,23 (2H, с), 2,14 (2H, с), 1,75 (2H, м), 1,59 (7H, м), 1,38 (2H, м).
Пример 11: Получение (3S)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноата натрия
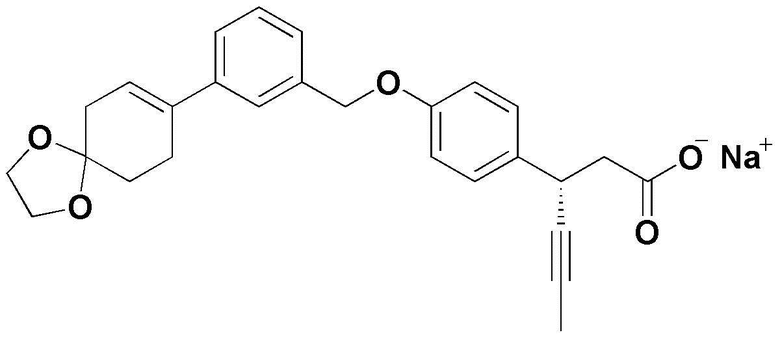
(3S)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновую кислоту (1 г), полученную в Примере 7, и этанол (170 мл) загружали в колбу на 500 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем добавляли 3н. водный раствор гидрокисда натрия (0,77 мл) с последующим перемешиванием при комнатной температуре. После завершения реакции, реакционную смесь концентрировали при пониженном давлении. Затем добавляли изопропиловый спирт (10 мл), и полученное твердое вещество отфильтровывали, получая целевое соединение (0,73 г, 69,2%).
1H ЯМР (400, CDCl3): δ 7,44 (1H, с), 7,34 (5H, м), 6,91 (2H, д), 6,00 (1H, т), 5,02 (2H, с), 4,08 (1H, м), 4,04 (4H, с), 2,73 (4H, м), 2,48 (2H, с), 1,92 (2H, т), 1,82 (3H, с)
Пример 12: Получение 3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
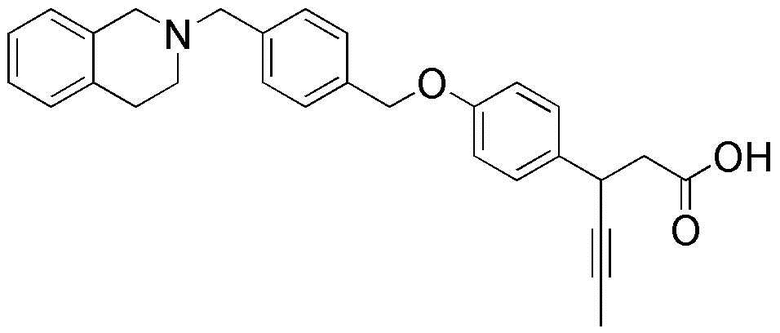
Стадия 1: Получение этил-3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноата
0,5 г 1,2,3,4-тетрагидроизохинолина загружали в 20 мл DMF в колбе в атмосфере азота с последующим перемешиванием. 1,2 г карбоната цезия добавляли при комнатной температуре. 30 минут спустя добавляли 1,0 г этил-3-(4-(4-((метилсульфонилокси)метил)бензилокси)фенил)гекс-4-иноата, полученного в Примере получения 6, с последующим перемешиванием при комнатной температуре в течение 12 часов. После завершения реакции медленно добавляли дистиллированную воду с последующей экстракцией с использованием этилацетата. Экстракт промывали солевым раствором, высушивали над безводным MgSO4 и концентрировали. Затем проводили хроматографию на колонках с силикагелем, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,38 (2H, д), 7,31 (2H, д), 7,22 (2H, д), 7,16 (3H, м), 6,97 (3H, м), 4,98 (2H, с), 4,14 (2H, м), 4,09 (1H, с), 3,91 (1H, д), 3,70 (3H, м), 2,92 (4H, с), 2,73 (2H, м), 1,83 (3H, с), 1,29 (3H, м).
Стадия 2: Получение 3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
0,7 г этил-3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноата, полученного на стадии 1), THF и дистиллированную воду загружали в колбу в атмосфере азота с последующим перемешиванием для их растворения. Затем гидроксид лития (0,7 г) медленно добавляли при комнатной температуре с последующим перемешиванием в течение по меньшей мере 1 часа. После завершения реакции, смесь подкисляли (рН: 2~3) при использовании 1М водного раствора HCl с последующей экстракцией с использованием этилацетата. Экстракт высушивали при пониженном давлении, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,38 (2H, д), 7,31 (2H, д), 7,22 (2H, д), 7,16 (3H, м), 6,97 (3H, м), 4,98 (2H, с), 4,09 (1H, с), 3,91 (1H, д), 3,70 (3H, м), 2,92 (4H, с), 2,73 (2H, м), 1,83 (3H, с).
Пример 13: Получение 3-(4-(3-циклогексенил-4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
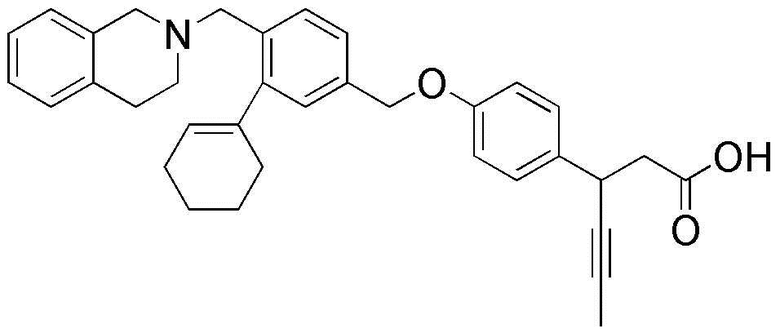
Стадия 1: Получение этил-3-(4-(3-циклогексенил-4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноата
1,0 г (3-циклогексенил-4-((3,4-дигидроизохинолин-2(1H)-ил)метил)фенил)метанола и 30 мл тетрагидрофурана загружали в колбу в атмосфере азота с последующим перемешиванием для их растворения. Затем медленно добавляли 0,8 г этил-3-(4-гидроксифенил)гекс-4-иноата, полученного в Примере получения 1, и 0,6 г трифенил фосфина. 0,5 мл диизопропила азодикарбоксилат медленно добавляли при использовании капельной воронки при 0°C с последующим перемешиванием в течение по меньшей мере 4 часов с подъемом температуры до комнатной температуры. После завершения реакции медленно добавляли дистиллированную воду с последующей экстракцией с использованием этилацетата. Экстрагированный органический слой высушивали при пониженном давлении, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 12,56 (1H, с), 8,26 (1H, д), 7,43 (2H, д), 7,25 (6H, м), 7,21 (1H, д), 7,02 (1H, д), 6,89 (2H, д), 5,46 (1H, с), 5,03 (2H, с), 4,14 (2H, м), 4,05 (1H, с), 3,92 (1H, с), 3,70 (1H, с), 3,35 (1H, с), 3,27 (1H, с), 3,03 (1H, с), 2,83 (2H, м), 2,01 (4H, м), 1,84 (3H, д), 1,51 (4H, м), 1,29 (3H, м).
Стадия 2: Получение 3-(4-(3-циклогексенил-4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
Целевое соединение получали тем же самым образом, как описано на стадии 2) Примера 12, за исключением того, что этил-3-(4-(3-циклогексенил-4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноат использовали вместо этил-3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноата.
1H ЯМР (400 МГц, CDCl3): δ 12,56 (1H, с), 8,26 (1H, д), 7,43 (2H, д), 7,25 (6H, м), 7,21 (1H, д), 7,02 (1H, д), 6,89 (2H, д), 5,46 (1H, с), 5,03 (2H, с), 4,05 (1H, с), 3,92 (1H, с), 3,70 (1H, с), 3,35 (1H, с), 3,27 (1H, с), 3,03 (1H, с), 2,83 (2H, м), 2,01 (4H, м), 1,84 (3H, д), 1,51 (4H, м).
Пример 14: Получение 3-(4-(4-((4-фенил-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
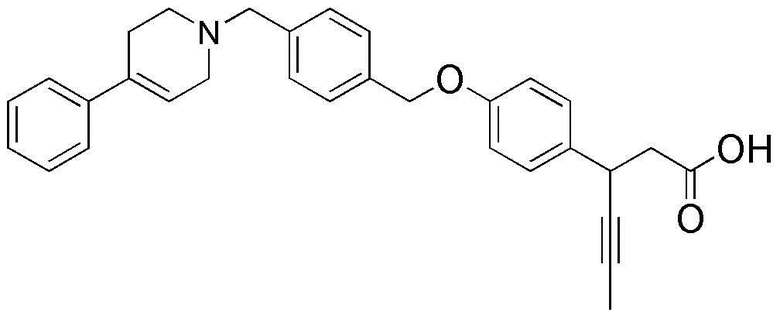
Целевое соединение получали тем же самым образом, как описано в Примере 12, за исключением того, что 4-фенил-1,2,3,6-тетрагидропиридин гидрохлорид использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,25 (2H, д), 6,78 (2H, д), 4,95 (1H, с), 4,14 (2H, м), 4,04 (1H, м), 2,68 (2H, м), 1,84 (3H, д), 1,29 (3H, т).
Пример 15: Получение 3-(4-(4-((4-фенилпиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
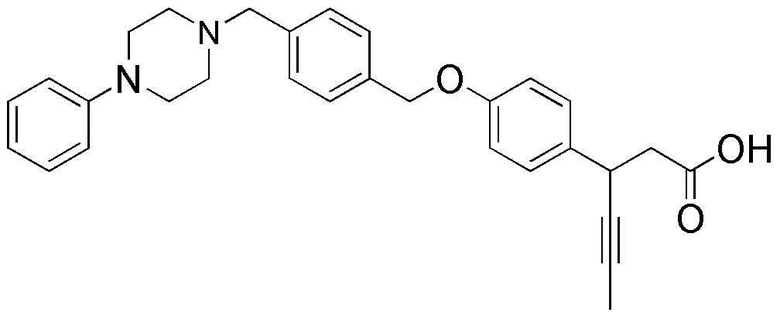
Целевое соединение получали тем же самым образом, как описано в Примере 12, за исключением того, что 1-фенилпиперазин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,37 (2H, д), 7,29 (4H, м), 7,11 (2H, д), 6,93 (5H, м), 4,96 (2H, с), 4,13 (1H, с), 3,66 (2H, м), 3,23 (4H, с), 2,83 (2H, м), 2,66 (2H, с), 1,82 (3H, с).
Пример 16: Получение 3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
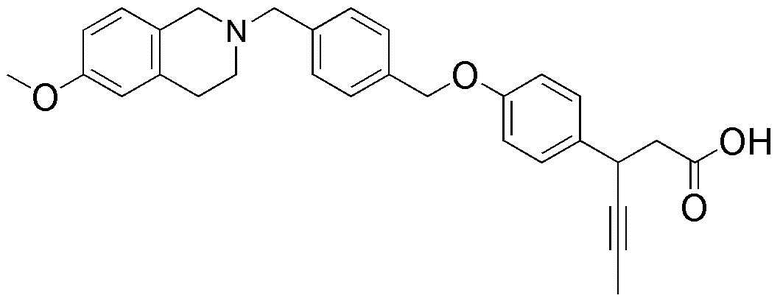
Целевое соединение получали тем же самым образом, как описано в Примере 12, за исключением того, что 6-метокси-1,2,3,4-тетрагидроизохинолин, полученный в Примере получения 8, использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,40 (4H, кв.), 7,26 (2H, д), 6,92 (3H, кв.), 6,66 (2H, д), 5,06 (2H, с), 3,94 (1H, с), 3,73 (3H, с), 3,63 (2H, с), 3,35 (3H, с), 2,78 (2H, т), 2,62 (2H, т), 2,58 (2H, с), 1,77 (3H, с).
Пример 17: Получение 3-(4-(4-((4-фенилпиперидин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
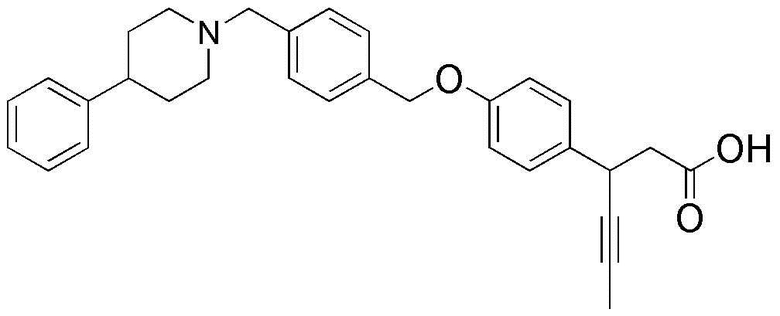
Целевое соединение получали тем же самым образом, как описано в Примере 12, за исключением того, что 4-фенилпиперидин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,44 (2H, д), 7,32 (2H, д), 7,23 (5H, т), 7,13 (2H, д), 6,96 (2H, д), 4,92 (2H, с), 4,16 (1H, с), 3,85 (2H, кв.), 3,33 (2H, т), 2,90 (1H, д), 2,78 (1H, м), 2,58 (1H, т), 2,38 (2H, т), 2,02 (2H, м), 1,83 (5H, м).
Пример 18: Получение 3-(4-(4-((4-(4-фторфенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
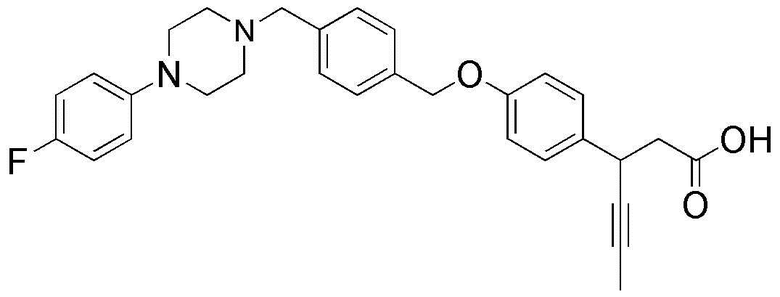
Целевое соединение получали тем же самым образом, как описано в Примере 12, за исключением того, что 1-(4-фторфенил)пиперазин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,60 (2H, д), 7,46 (2H, д), 7,30 (3H, д), 6,97 (2H, т), 6,86 (4H, м), 5,01 (2H, с), 4,21 (2H, с), 4,04 (1H, т), 3,50 (4H, д), 3,25 (4H, с), 2,78 (2H, м), 1,80 (3H, д).
Пример 19: Получение 3-(4-(4-((4-(4-(трифторметил)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
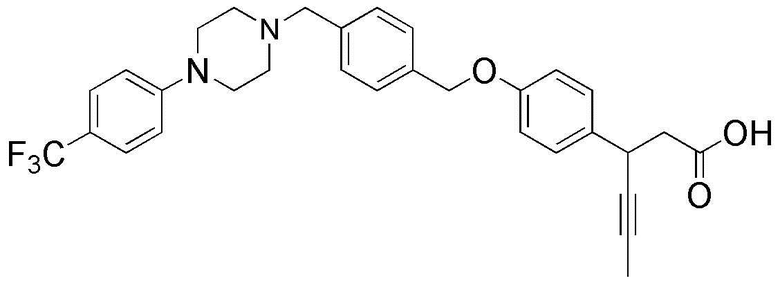
Целевое соединение получали тем же самым образом, как описано в Примере 12, за исключением того, что 1-(4-(трифторметил)фенил)пиперазин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,63 (2H, д), 7,51 (4H, д), 7,21 (2H, д), 6,93 (2H, д), 6,74 (2H, с), 5,03 (2H, с), 4,13 (2H, м), 4,01 (1H, т), 3,73 (4H, с), 2,96 (4H, с), 2,71 (2H, м), 1,78 (3H, с).
Пример 20: Получение 3-(4-(4-((4-(4-(3-(метилсульфонил)пропокси)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Целевое соединение получали тем же самым образом, как описано в Примере 12, за исключением того, что 1-(4-(3-(метилсульфонил)пропокси)фенил)пиперазин гидрохлорид использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,65 (2H, д), 7,49 (2H, д), 7,30 (2H, д), 6,87 (6H, м), 5,07 (2H, с), 4,20 (2H, д), 4,08 (2H, т), 4,01 (1H, т), 6,63 (2H, с), 3,49 (4H, м), 3,26 (2H, т), 3,01 (2H, с), 2,97 (3H, с), 2,71 (2H, м), 2,34 (2H, м), 1,83 (2H, д).
Пример 21: Получение (S)-3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
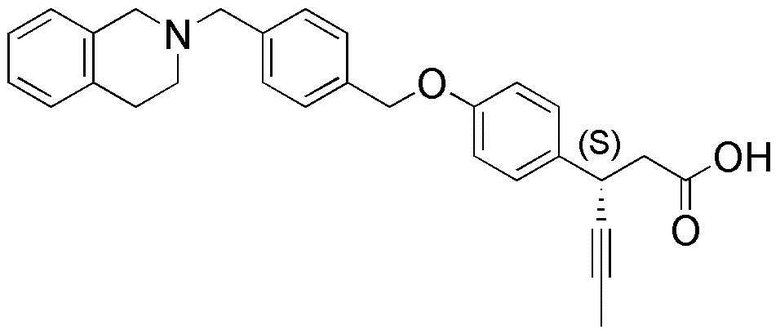
Стадия 1: Получение этил-(S)-3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноата
0,5 г 1,2,3,4-тетрагидроизохинолина загружали в 20 мл DMF в колбе в атмосфере азота с последующим перемешиванием. 1,1 г карбонат цезия добавляли при комнатной температуре. 30 минут спустя добавляли 1,0 г (S)-этил-3-(4-(4-((метилсульфонилокси)метил)бензилокси)фенил)гекс-4-иноата, полученного в Примере получения 7, с последующим перемешиванием при комнатной температуре в течение 12 часов. После завершения реакции медленно добавляли дистиллированную воду с последующей экстракцией с использованием этилацетата. Экстракт промывали солевым раствором, высушивали над безводным MgSO4 и концентрировали. Затем проводили хроматографию на колонках с силикагелем, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,38 (2H, д), 7,31 (2H, д), 7,22 (2H, д), 7,16 (3H, м), 6,97 (3H, м), 4,98 (2H, с), 4,14 (2H, м), 4,09 (1H, с), 3,91 (1H, д), 3,70 (3H, м), 2,92 (4H, с), 2,73 (2H, м), 1,83 (3H, с), 1,29 (3H, м).
Стадия 2: Получение (S)-3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
0,5 г (S)-3-(4-(4-((3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноата, полученного на стадии 1), THF, метанол и дистиллированную воду загружали в колбу в атмосфере азота с последующим перемешиванием для их растворения. Затем 0,5 г гидроксида лития медленно добавляли при комнатной температуре с последующим перемешиванием в течение по меньшей мере 1 часа. После завершения реакции, смесь подкисляли (рН: 2~3) при использовании 1М водного раствора HCl с последующей экстракцией с использованием этилацетата. Экстракт высушивали при пониженном давлении, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,38 (2H, д), 7,31 (2H, д), 7,22 (2H, д), 7,16 (3H, м), 6,97 (3H, м), 4,98 (2H, с), 4,09 (1H, с), 3,91 (1H, д), 3,70 (3H, м), 2,92 (4H, с), 2,73 (2H, м), 1,83 (3H, с).
Пример 22: Получение (S)-3-(4-(4-((4-(4-(трифторметил)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
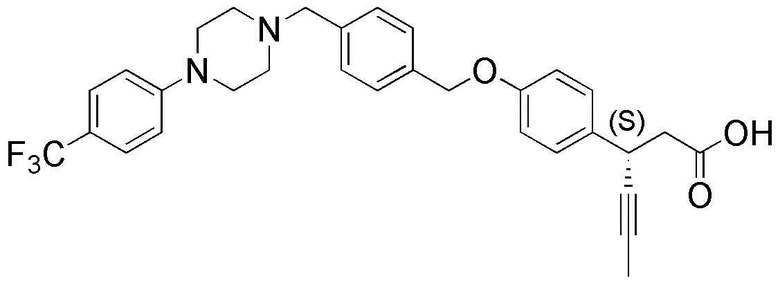
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 1-(4-(трифторметил)фенил)пиперазин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,65 (2H, д), 7,51 (4H, м), 7,30 (2H, д), 6,61 (2H, д), 6,85 (2H, д), 5,05 (2H, с), 4,21 (2H, с), 4,03 (1H, т), 3,68 (4H, с), 3,49 (2H, с), 2,84 (2H, с), 2,70 (2H, м), 1,82 (3H, с).
Пример 23: Получение (S)-3-(4-(4-((4-(4-фторфенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 1-(4-фторфенил)пиперазин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,39 (2H, д), 7,30 (2H, д), 7,19 (2H, д), 6,96 (4H, м), 6,87 (2H, м), 4,97 (2H, с), 4,10 (2H, с), 3,81 (1H, д), 3,51 (1H, д), 3,15 (4H, с), 2,80 (6H, м), 1,82 (3H, с).
Пример 24: Получение (S)-3-(4-(4-((4-(4-(трифторметил)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иноата калия
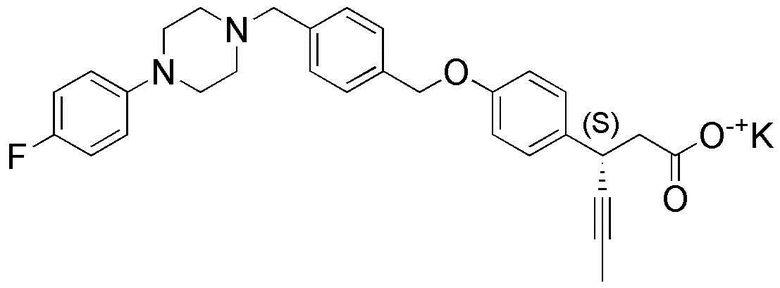
0,4 г (S)-3-(4-(4-((4-(4-фторфенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты, полученной в Примере 23, и 10 мл этанола загружали в колбу в атмосфере азота с последующим перемешиванием для их растворения. Затем добавляли 0,3 мл 3н. водного раствора гидроксида калия с последующим перемешиванием при комнатной температуре. После завершения реакции, реакционную смесь концентрировали при пониженном давлении. Затем добавляли изопропиловый спирт, и полученное твердое вещество отфильтровывали, получая целевое соединение.
1H ЯМР (400 МГц, D2O): δ 7,10 (4H, м), 6,98 (2H, д), 6,57 (4H, д), 6,38 (2H, с), 4,55 (2H, с), 3,82 (1H, с), 3,07 (2H, с), 2,59 (4H, с), 2,36 (2H, с), 2,13 (4H, с), 1,51 (3H, с).
Пример 25: Получение (S)-3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
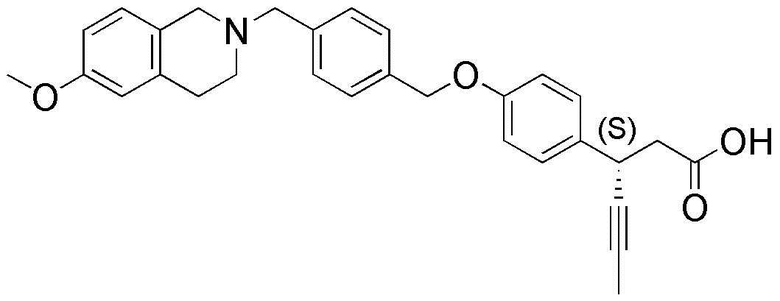
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 6-метокси-1,2,3,4-тетрагидроизохинолин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, ДМСО): δ 7,40 (4H, кв.), 7,26 (2H, д), 6,94 (3H, м), 6,68 (2H, м), 5,06 (2H, с), 3,95 (1H, т), 3,70 (3H, с), 3,51 (2H, с), 3,43 (2H, с), 2,77 (2H, т), 2,66 (2H, т), 2,57 (2H, д), 1,75 (3H, д).
Пример 26: Получение (S)-3-(4-(4-((4-фенилпиперидин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 4-фенилпиперидин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,66 (2H, д), 7,49 (2H, д), 7,30 (7H, м), 6,87 (2H, д), 5,04 (2H, с), 4,19 (2H, с), 4,06 (1H, т), 3,59 (2H, д), 2,73 (7H, м), 2,00 (2H, д), 1,82 (3H, с).
Пример 27: Получение (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иновой кислоты
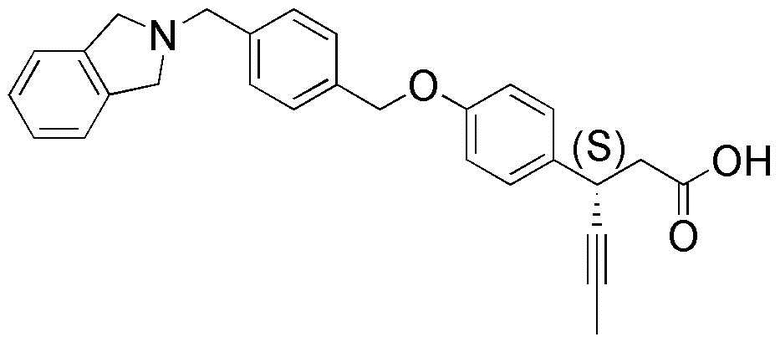
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что изоиндолин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,68 (2H, д), 7,47 (2H, д), 7,38 (2H, м), 7,30 (4H, м), 6,87 (2H, д), 5,06 (2H, с), 4,90 (2H, с), 4,32 (4H, м), 4,05 (1H, т), 2,81 (2H, м), 1,83 (3H, с).
Пример 28: Получение (S)-3-(4-(4-((4-фенил-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 4-фенил-1,2,3,6-тетрагидропиридин гидрохлорид использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,47 (2H, д), 7,36 (9H, м), 6,88 (2H, д), 5,99 (1H, с), 4,99 (2H, с), 4,18 (1H, м), 4,06 (2H, м), 3,53 (2H, с), 3,22 (2H, с), 2,82 (4H, м), 1,82 (3H, с).
Пример 29: Получение (S)-3-(4-(4-((4-(4-(метоксиметокси)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
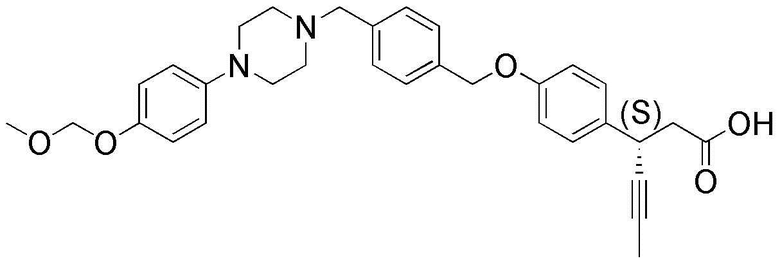
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 1-(4-(метоксиметокси)фенил)пиперазин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,57 (2H, д), 7,46 (2H, д), 7,26 (2H, д), 6,97 (2H, д), 6,87 (2H, д), 6,80 (2H, д), 5,13 (2H, с), 5,01 (2H, с), 4,13 (2H, с), 4,02 (1H, т), 3,51 (11H, м), 2,72 (2H, м), 1,79 (3H, с).
Пример 30: Получение (S)-3-(4-(4-((4-(5-изопропил-1,2,4-оксадиазол-3-ил)пиперидин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
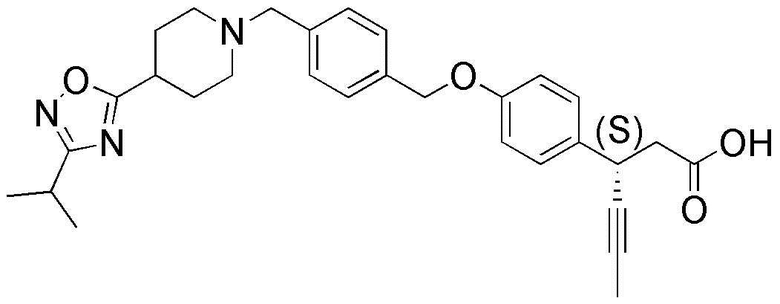
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 3-изопропил-5-(пиперидин-4-ил)-1,2,4-оксадиазол использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,63 (2H, д), 7,46 (2H, д), 7,30 (2H, д), 6,86 (2H, д), 5,05 (2H, д), 4,13 (2H, м), 4,03 (1H, т), 3,61 (1H, с), 3,43 (2H, с), 3,10 (1H, м), 2,92 (4H, м), 2,73 (2H, м), 2,30 (2H, м), 1,83 (3H, с), 1,32 (6H, д).
Пример 31: Получение (S)-3-(4-(4-((4-(5-изопропил-1,2,4-оксадиазол-3-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
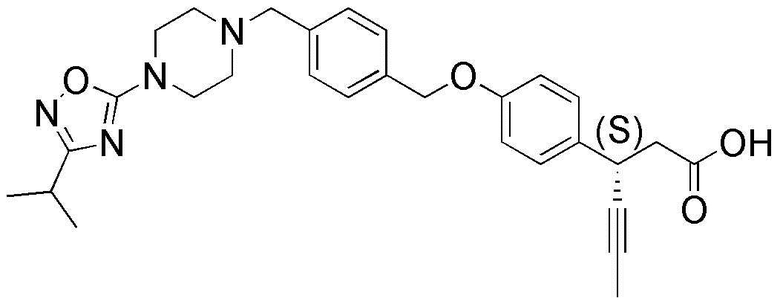
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 3-изопропил-5-(пиперазин-1-ил)-1,2,4-оксадиазол использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,61 (2H, д), 7,49 (2H, д), 7,30 (2H, д), 6,87 (2H, д), 5,05 (2H, с), 4,15 (4H, м), 4,02 (1H, т), 3,49 (3H, м), 2,81 (3H, м), 1,83 (3H, с), 1,24 (6H, д).
Пример 32: Получение (S)-3-(4-(4-((4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
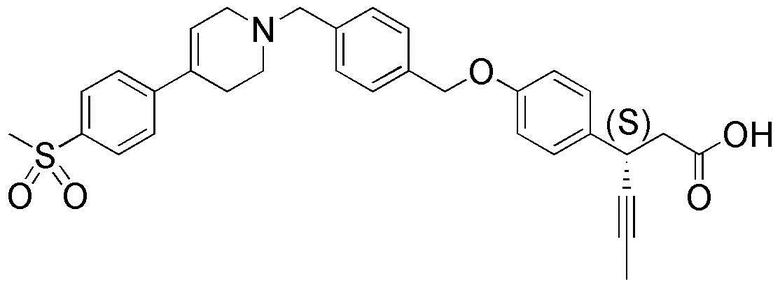
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 4-(4-(метилсульфонил)фенил)-1,2,3,6-тетрагидропиридин гидрохлорид, полученный в Примере получения 9, использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, ДМСО): δ 7,95 (2H, д), 7,75 (2H, д), 7,63 (2H, д), 7,44 (2H, д), 7,30 (2H, д), 6,98 (2H, д), 6,37 (1H, с), 5,14 (2H, с), 4,45 (2H, т), 6,97 (1H, с), 6,82 (4H, м), 3,27 (4H, с), 2,84 (2H, с), 2,59 (2H, д), 1,77 (3H, с).
Пример 33: Получение (S)-3-(4-(4-((4-(4-(3-(метилсульфонил)пропокси)фенил)-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты

Целевое соединение получали тем же самым образом, как описано в Примере 21 за исключением того, что 4-(4-(3-(метилсульфонил)пропокси)фенил)-1,2,3,6-тетрагидропиридин гидрохлорид, полученный в Примере получения 11, использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,66 (2H, д), 7,49 (2H, д), 7,32 (2H, д), 7,15 (2H, д), 6,90 (2H, д), 6,82 (2H, д), 5,06 (2H, с), 4,18 (2H, с), 4,09 (3H, м), 3,58 (2H, с), 3,26 (2H, м), 2,97 (3H, с), 2,81 (5H, м), 2,62 (3H, с), 2,32 (2H, м), 1,96 (2H, д), 1,83 (3H, с).
Пример 34: Получение (3S)-3-(4-(4-(1-(3,4-дигидроизохинолин-2(1H)-ил)этил)бензилокси)фенил)гекс-4-иновой кислоты
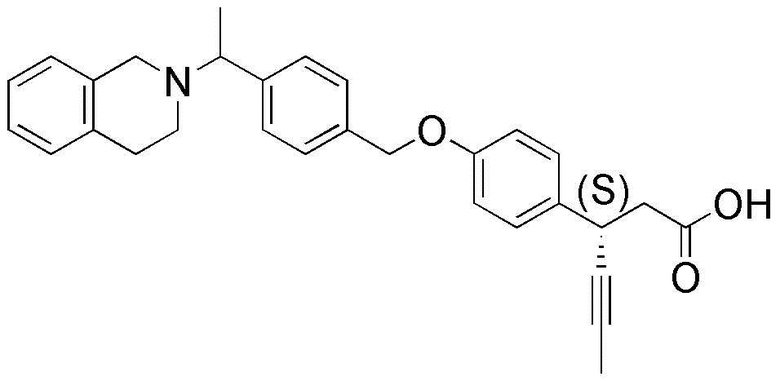
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что (3S)-этил-3-(4-(4-(1-бромэтил)бензилокси)фенил)гекс-4-иноат, полученный в Примере получения 12, использовали вместо (S)-этил-3-(4-(4-((метилсульфонилокси)метил)бензилокси)фенил)гекс-4-иноата.
1H ЯМР (400 МГц, CDCl3): δ 12,98 (1H, с), 7,61 (6H, м), 7,30 (4H, м), 6,92 (2H, т), 5,08 (2H, с), 4,29 (2H, с), 4,06 (1H, с), 3,81 (1H, с), 3,51 (2H, с), 3,21 (2H, м), 2,75 (2H, м), 1,95 (2H, д), 1,84 (3H, с).
Пример 35: Получение (S)-3-(4-(4-((4-(4-гидроксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
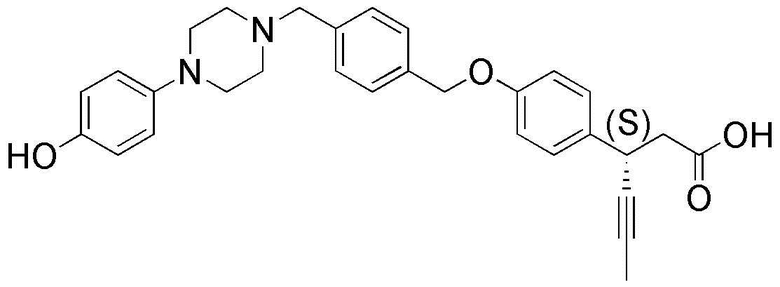
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 4-(1,2,3,6-тетрагидропиридин-4-ил)фенол гидрохлорид, полученный в Примере получения 10, использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 8,80 (1H, с), 7,41 (2H, д), 735 (2H, д), 7,28 (2H, д), 6,94 (2H, д), 6,74 (2H, д), 6,63 (2H, д), 5,06 (2H, с), 3,94 (1H, т), 3,62 (3H, с), 2,95 (4H, с), 2,61 (2H, д), 1,77 (3H, с).
Пример 36: Получение (S)-3-(4-(4-((4-(4-(3-(метилсульфонил)пропокси)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
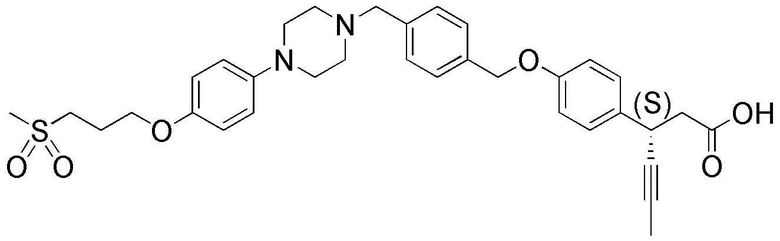
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 1-(4-(3-(метилсульфонил)пропокси)фенил)пиперазин гидрохлорид использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 12,32 (1H, с), 7,42 (4H, м), 7,29 (2H, д), 6,96 (2H, д), 6,83 (4H, кв.), 5,06 (2H, с), 4,02 (2H, т), 3,92 (1H, т), 3,52 (2H, с), 3,25 (2H, т), 3,01 (7H, м), 2,61 (2H, д), 2,09 (2H, м), 1,77 (3H, д).
Пример 37: Получение (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иноата натрия
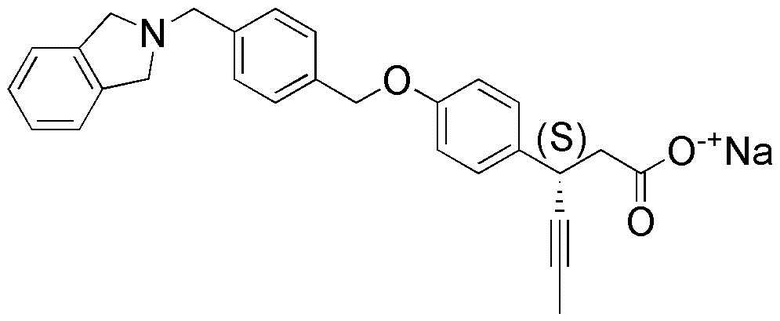
0,4 г (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иновой кислоты, полученной в Примере 27, и этанол загружали в колбу на 500 мл в атмосфере азота с последующим перемешиванием для их растворения. Затем добавляли 0,3 мл 3н. водного раствора гидроксида натрия с последующим перемешиванием при комнатной температуре. После завершения реакции, реакционную смесь концентрировали при пониженном давлении, затем добавляли изопропиловый спирт. Затем полученное твердое вещество отфильтровывали, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,09 (2H, д), 7,03 (2H, д), 6,97 (2H, д), 6,85 (2H, м), 6,75 (2H, м), 6,57 (2H, д), 4,54 (2H, с), 3,81 (1H, т), 3,36 (4H, с), 3,31 (2H, с), 2,33 (2H, д), 1,54 (3H, д).
Пример 38: Получение L-лизин (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иноата
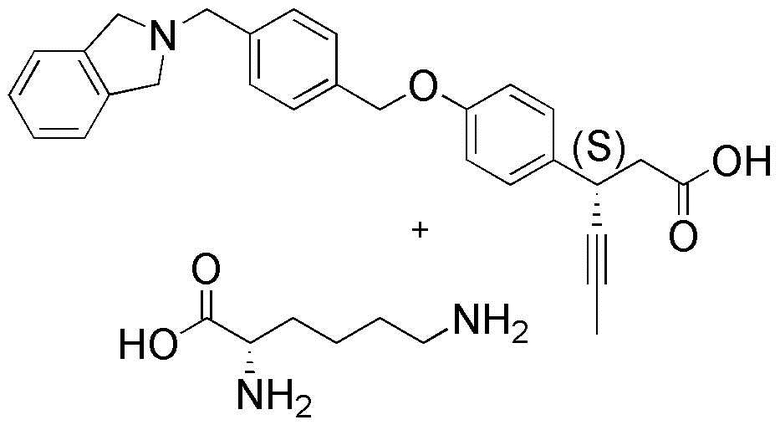
0,4 г (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иновой кислоты, полученной в Примере 27, и этанол загружали в колбу в атмосфере азота с последующим перемешиванием для их растворения. Затем добавляли 0,12 г L-лизина. Температуру реакции повышали до 50°C, и смесь перемешивали в течение 30 минут при 50°C. Смесь охлаждали до комнатной температуры с последующим перемешиванием в течение 30 минут. После завершения реакции, полученное твердое вещество отфильтровывали, получая целевое соединение.
1H ЯМР (400 МГц, D2O): δ 7,03 (6H, с), 6,83 (2H, с), 6,74 (2H, с), 6,54 (2H, с), 4,53 (2H, с), 3,77 (1H, с), 3,54 (5H, м), 2,88 (2H, т), 2,28 (2H, с), 1,74 (2H, м), 1,62 (3H, м), 1,42 (3H, с), 1,35 (3H, м).
Пример 39: Получение (S)-3-(4-(4-((4-(4-фторфенил)-5,6-дигидропиридин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
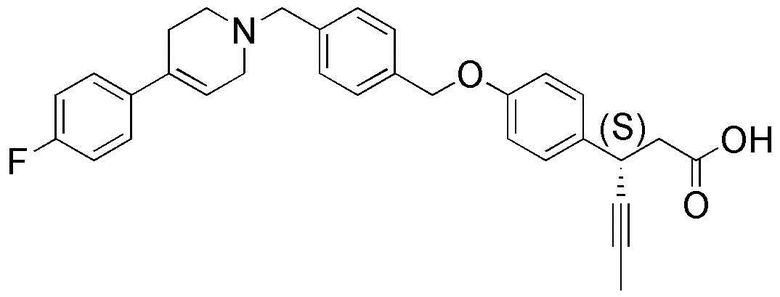
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что (4-фторфенил)-1,2,3,6-тетрагидропиридин гидрохлорид использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,69 (2H, д), 7,48 (2H, д), 7,32 (4H, м), 7,04 (2H, т), 6,86 (2H, д), 5,90 (1H, с), 5,03 (2H, с), 4,30 (2H, с), 4,02 (1H, т), 3,71 (2H, с), 3,54 (2H, с), 3,31 (2H, с), 2,73 (2H, м), 1,81 (3H, д).
Пример 40: Получение (S)-3-(4-(4-((4-(4-метоксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
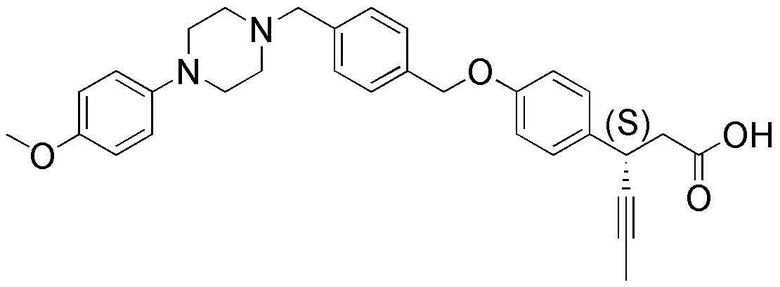
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 4-(4-метоксифенил)-1,2,3,6-тетрагидропиридин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,64 (2H, д), 7,48 (2H, д), 7,31 (2H, д), 6,94 (2H, с), 6,86 (4H, т), 5,04 (2H, с), 4,21 (2H, с), 4,03 (1H, т), 3,78 (3H, с), 3,60 (2H, с), 3,47 (2H, с), 3,05 (2H, с), 2,73 (2H, м), 1,82 (3H, с).
Пример 41: Получение (S)-3-(4-(4-((3,4-дигидрохинолин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иноата натрия
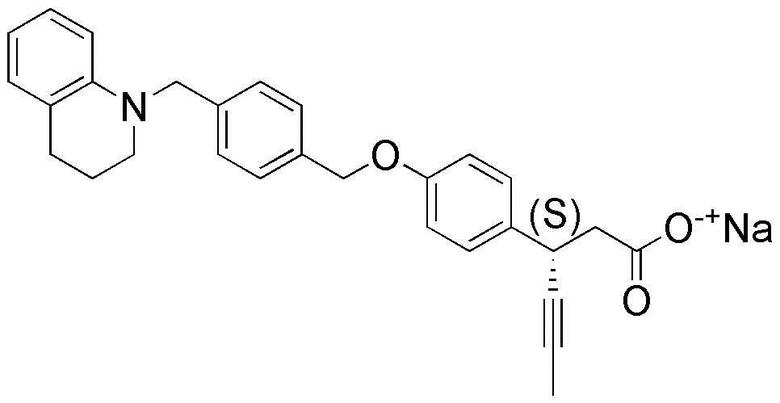
Стадия 1: Получение (S)-3-(4-(4-((3,4-дигидрохинолин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 1,2,3,4-тетрагидрохинолин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 7,02 (2H, д), 6,76 (2H, д), 6,69 (2H, д), 6,43 (4H, м), 6,21 (1H, с), 6,02 (1H, с), 4,24 (2H, с), 3,84 (3H, с), 2,68 (2H, с), 2,37 (2H, д), 2,14 (2H, с), 1,47 (3H, с), 1,35 (2H, с).
Стадия 2: Получение (S)-3-(4-(4-((3,4-дигидрохинолин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иноата натрия
Целевое соединение получали тем же самым образом, как описано в Примере 37, за исключением того, что (S)-3-(4-(4-((3,4-дигидрохинолин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновую кислоту, полученную на стадии 1), использовали вместо (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иновой кислоты.
1H ЯМР (400 МГц, D2O): δ 7,01 (2H, д), 6,74 (2H, д), 6,68 (2H, д), 6,42 (4H, м), 6,15 (1H, с), 6,02 (1H, с), 4,25 (2H, с), 3,79 (3H, с), 2,62 (2H, с), 2,34 (2H, д), 2,12 (2H, с), 1,45 (3H, с), 1,32 (2H, с).
Пример 42: Получение (S)-3-(4-(4-((3,4-дигидрохинолин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иноата калия

Целевое соединение получали тем же самым образом, как описано в Примере 25, за исключением того, что (S)-3-(4-(4-((3,4-дигидрохинолин-1(2H)-ил)метил)бензилокси)фенил)гекс-4-иновую кислоту, полученную на стадии 1) Примера 41, использовали вместо (S)-3-(4-(4-((4-(4-фторфенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты.
1H ЯМР (400 МГц, D2O): δ 6,97 (2H, д), 6,71 (2H, д), 6,63 (2H, д), 6,45 (2H,с), 6,38 (2H, д), 6,13 (1H, с), 5,98 (1H, с), 4,20 (2H, с), 3,71 (3H, м), 2,58 (2H, с), 2,32 (2H, с), 2,15 (2H, с), 1,43 (3H, с), 1,29 (2H, с).
Пример 43: Получение (S)-3-(4-(4-((4-(бензо[d]тиазол-2-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
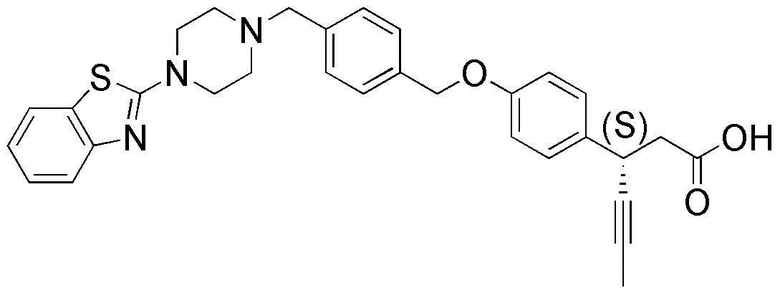
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 2-(пиперазин-1-ил)бензо[d]тиазол гидрохлорид, полученный в Примере получения 13, использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, ДМСО): δ 10,87 (1H, с), 7,85 (1H, д), 7,55 (5H, м), 7,31 (3H, м), 7,14 (2H, т), 6,96 (2H, д), 5,13 (2H, с), 4,40 (2H, с), 4,17 (2H, д), 3,95 (1H, т), 3,57 (3H, т), 3,22 (3H, с), 2,57 (2H, д), 1,78 (3H, д).
Пример 44: Получение (S)-3-(4-(4-((4-(5-пропилпиримидин-2-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
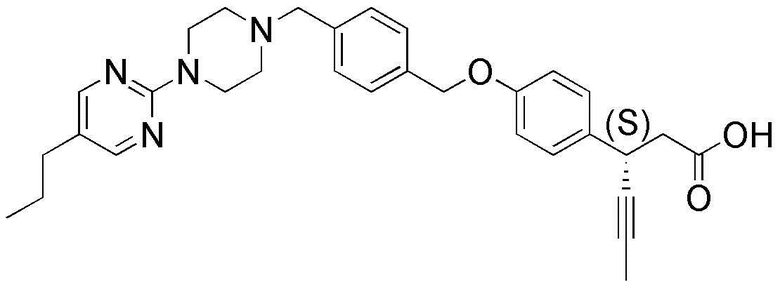
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 2-(пиперазин-1-ил)-5-пропилпиримидин гидрохлорид, полученный в Примере получения 14, использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 8,20 (2H, с), 7,62 (2H, д), 7,47 (2H, д), 7,30 (2H, д), 6,85 (2H, д), 5,08 (2H, с), 4,80 (2H, д), 4,17 (2H, с), 4,03 (1H, т), 3,84 (1H, т), 3,43 (2H, с), 2,74 (4H, м), 2,43 (2H, т), 1,83 (3H, д), 1,59 (2H, кв.), 0,94 (3H, т).
Пример 45: Получение (S)-3-(4-(4-((4-(5-цианопиридин-2-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
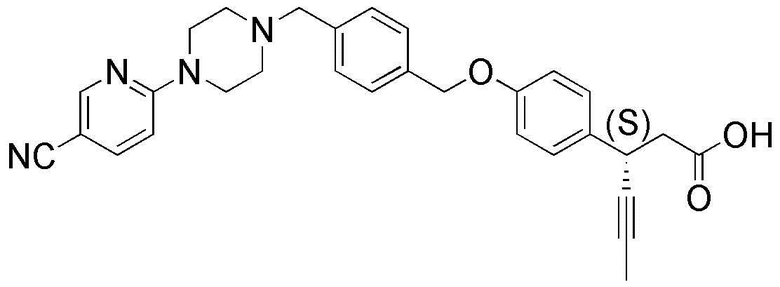
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 6-(пиперазин-1-ил)никотинонитрил гидрохлорид, полученный в Примере получения 15, использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, ДМСО): δ 11,20 (1H, с), 8,56 (1H, с), 7,99 (1H, д), 7,63 (1H, д), 7,55 (1H, д), 7,27 (2H, д), 7,04 (1H, д), 6,95 (2H, д), 5,12 (2H, с), 4,57 (2H, д), 4,35 (2H, с), 3,95 (1H, т), 3,39 (5H, м), 2,90 (2H, м), 2,59 (2H, д), 1,77 (3H, д).
Пример 46: Получение (3S)-3-(4-(4-((3-фенилпирролидин-1-ил)метил)бензилокси)фенил)гекс-4-иновой кислоты
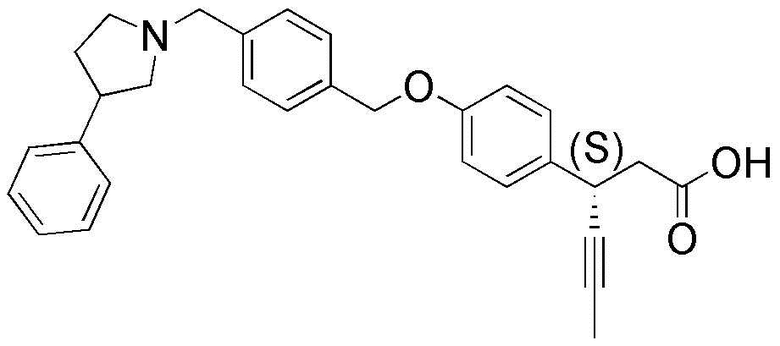
Целевое соединение получали тем же самым образом, как описано в Примере 21, за исключением того, что 3-фенилпирролидин использовали вместо 1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 12,64 (1H, с), 7,66 (2H, с), 7,46 (2H, д), 7,32 (7H, м), 6,86 (2H, д), 5,02 (2H, с), 4,28 (2H, м), 4,04 (1H, т), 3,87 (2H, с), 3,73 (1H, с), 3,18 (1H, с), 2,89 (1H, м), 2,84 (3H, м), 2,61 (1H, с), 2,41 (1H, с), 2,19 (1H, с), 1,81 (3H, д).
Пример 47: Получение (S)-3-(4-(4-((4-(4-метоксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иноата
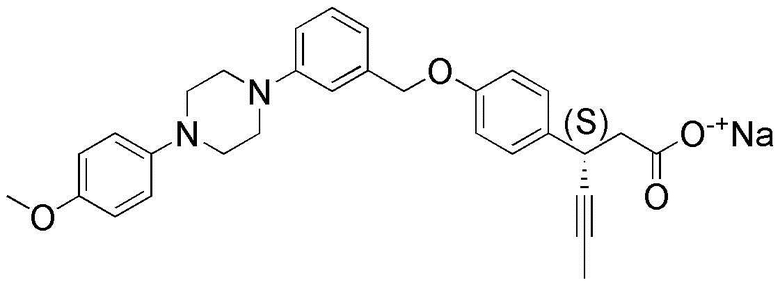
Целевое соединение получали тем же самым образом, как описано в Примере 37, за исключением того, что (S)-3-(4-(4-((4-(4-метоксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновую кислоту, полученную в Примере 40, использовали вместо (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иновой кислоты.
1H ЯМР (400 МГц, MEOC): δ 7,33 (2H, д), 7,26 (1H, д), 7,11 (1H, с), 6,96 (8H, м), 5,04 (2H, с), 4,04 (1H, т), 3,76 (3H, с), 3,32 (4H, м), 3,21 (4H, м), 2,52 (2H, м), 1,80 (3H, с).
Пример 48: Получение (S)-3-(4-(4-(2-(6-метокси-3,4-дигидроизохинолин-2(1H)-ил)этил)бензилокси)фенил)гекс-4-иновой кислоты
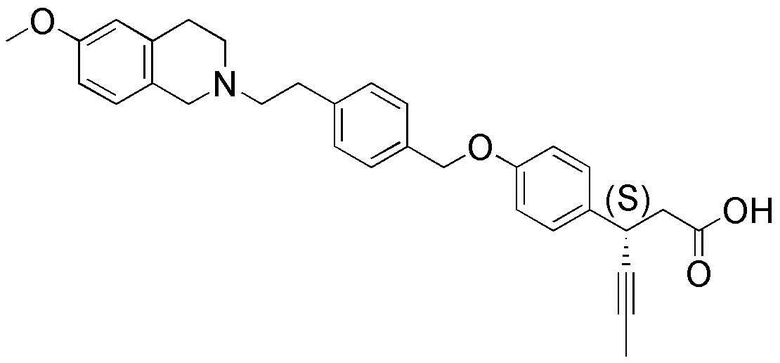
Стадия 1: Получение этил-(S)-3-(4-(4-(2-(6-метокси-3,4-дигидроизохинолин-2(1H)-ил)этил)бензилокси)фенил)гекс-4-иноата
0,5 г 6-метокси-1,2,3,4-тетрагидроизохинолина загружали в 20 мл DMF в колбе в атмосфере азота с последующим перемешиванием. 1,1 г карбоната цезия добавляли при комнатной температуре. 30 минут спустя добавляли 1,0 г (S)-этил-3-(4-(4-(2-(метилсульфонилокси)этил)бензилокси)фенил)гекс-4-иноата, полученного в Примере получения 16, с последующим перемешиванием при комнатной температуре в течение 12 часов. После завершения реакции медленно добавляли дистиллированную воду с последующей экстракцией с использованием этилацетата. Экстракт промывали солевым раствором, высушивали над безводным MgSO4 и концентрировали. Затем проводили хроматографию на колонках с силикагелем, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,35 (2H, д), 7,30 (2H, д), 7,23 (2H, д), 7,00 (1H, д), 6,85 (2H, д), 6,80 (1H, д), 6,70 (1H, д), 5,00 (2H, с), 4,30 (2H, м), 4,13 (2H, м) 4,03 (1H, т), 3,80 (3H, с), 3,58 (6H, м), 3,30 (2H, с), 2,78 (2H, м), 1,86 (3H, д), 1,28 (3H, м).
Стадия 2: Получение (S)-3-(4-(4-(2-(6-метокси-3,4-дигидроизохинолин-2(1H)-ил)этил)бензилокси)фенил)гекс-4-иновой кислоты
0,5 г этил-(S)-3-(4-(4-(2-(6-метокси-3,4-дигидроизохинолин-2(1H)-ил)этил)бензилокси)фенил)гекс-4-иноата, полученного на стадии 1), THF, метанол и дистиллированную воду загружали в колбу в атмосфере азота с последующим перемешиванием для их растворения. Затем 0,5 г гидроксида лития медленно добавляли при комнатной температуре с последующим перемешиванием в течение по меньшей мере 1 часа. После завершения реакции, смесь подкисляли (рН: 4~5) при использовании 1М водного раствора HCl с последующей экстракцией с использованием этилацетата. Экстракт высушивали при пониженном давлении, получая целевое соединение.
1H ЯМР (400 МГц, CDCl3): δ 7,35 (2H, д), 7,30 (2H, д), 7,23 (2H, д), 7,00 (1H, д), 6,85 (2H, д), 6,80 (1H, д), 6,70 (1H, д), 5,00 (2H, с), 4,30 (2H, м), 4,03 (1H, т), 3,80 (3H, с), 3,58 (6H, м), 3,30 (2H, с), 2,78 (2H, м), 1,86 (3H, д).
Пример 49: Получение (S)-3-(4-(4-(2-(изоиндолин-2-ил)этил)бензилокси)фенил)гекс-4-иновой кислоты
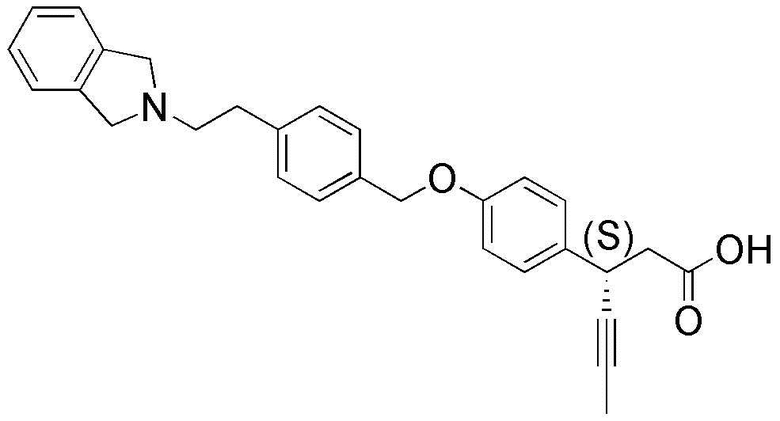
Целевое соединение получали тем же самым образом, как описано в Примере 48, за исключением того, что изоиндолин использовали вместо 6-метокси-1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, CDCl3): δ 13,57 (1H, с), 7,38 (3H, м), 7,29 (7H, м), 6,90 (2H, д), 5,03 (4H, м), 4,28 (2H, с), 4,08 (1H, т), 3,48 (2H, м), 3,34 (2H, м), 2,80 (2H, м), 1,83 (3H, д).
Пример 50: Получение (S)-3-(4-(4-(2-(3,4-дигидроизохинолин-2(1H)-ил)этил)бензилокси)фенил)гекс-4-иновой кислоты
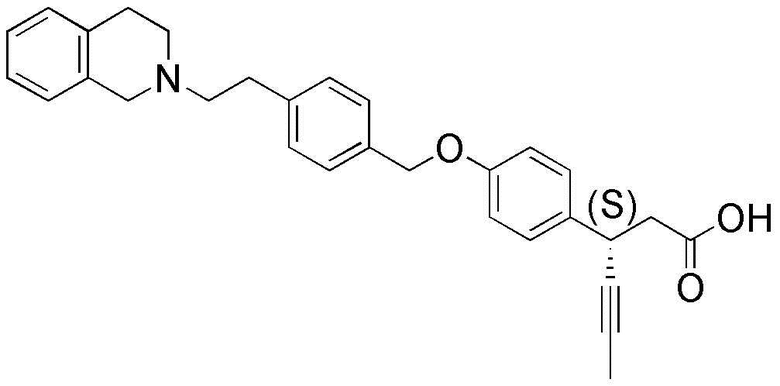
Целевое соединение получали тем же самым образом, как описано в Примере 48, за исключением того, что 1,2,3,4-тетрагидроизохинолин использовали вместо 6-метокси-1,2,3,4-тетрагидроизохинолина.
1H ЯМР (400 МГц, ДМСО): δ 7,44 (2H, д), 7,38 (2H, д), 7,27 (5H, м), 7,22 (1H, д), 6,94 (2H, д), 5,07 (2H, с), 4,64 (1H, д), 4,38 (1H, с), 3,95 (1H, т), 3,77 (1H, с), 3,39 (2H, с), 3,16 (4H, м), 2,26 (2H, д), 1,77 (3H, д), 1,84 (3H, д), 1,29 (3H, т).
Пример 51: Получение (S)-3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иноата натрия
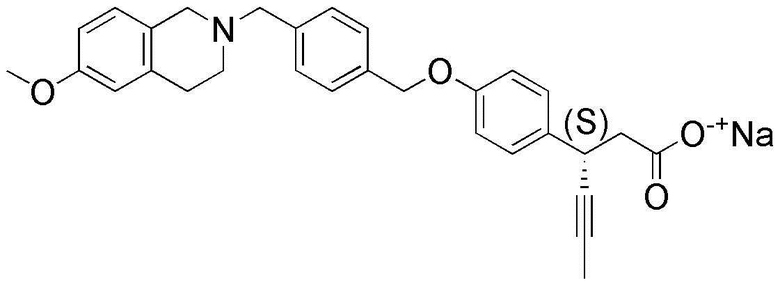
Целевое соединение получали тем же самым образом, как описано в Примере 37, за исключением того, что (S)-3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1H)-ил)метил)бензилокси)фенил)гекс-4-иновую кислоту, полученную в Примере 25, использовали вместо (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иновой кислоты.
1H ЯМР (400 МГц, D32O): δ 7,10 (2H, д), 7,02 (2H, д), 6,95 (2H, д), 6,55 (2H, д), 6,40 (1H, д), 6,34 (2H, с), 4,53 (2H, с), 3,83 (1H, т), 3,39 (3H, с), 3,17 (2H, с), 3,05 (2H, с), 2,37 (4H, м), 2,20 (2H, с), 1,57 (3H, с).
Сравнительный Пример 1: Получение [(3S)-6-({(2',6’-диметил-4’-[3-(метансульфонил)пропокси]-[1,1’-дифенил]-3-ил)}метокси)-2,3-дигидро-1-бензофуран-3-ил]уксусной кислоты
[(3S)-6-({(2',6’-диметил-4’-[3-(метансульфонил)пропокси]-[1,1’-дифенил]-3-ил)}метокси)-2,3-дигидро-1-бензофуран-3-ил]уксусная кислота была получена способом, описанным в международной патентной публикации No. 2008/001931.
Сравнительный Пример 2: Получение (3S)-3-(4-{[4-(1'H-спиро[инден-1,4’-пиперидин]-1’-илметил)бензил]окси}фенил)гекс-4-иновой кислоты
(3S)-3-(4-{[4-(1'H-спиро[инден-1,4’-пиперидин]-1’-илметил)бензил]окси}фенил)гекс-4-иновая кислота была получена способом, описанным в международной патентной публикации No. WO2011/046851.
Сравнительный Пример 3: Получение 4-(3-феноксибензиламино)фенилпропионовой кислоты
4-(3-феноксибензиламино)фенилпропионовая кислота была получена обычным способом.
Химические формулы соединений, полученных в Примерах 1~51, представлены в Таблице 1.
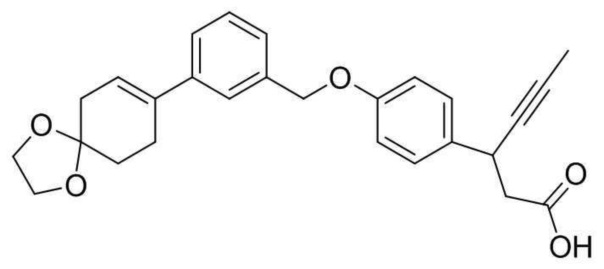
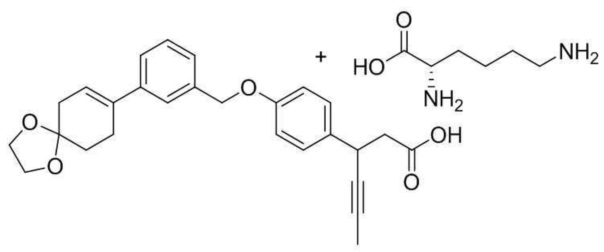
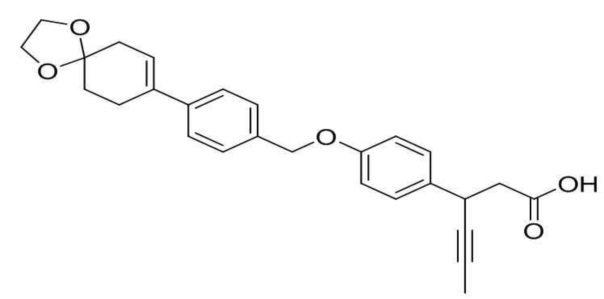
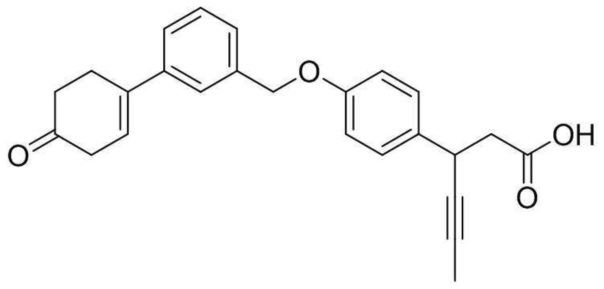
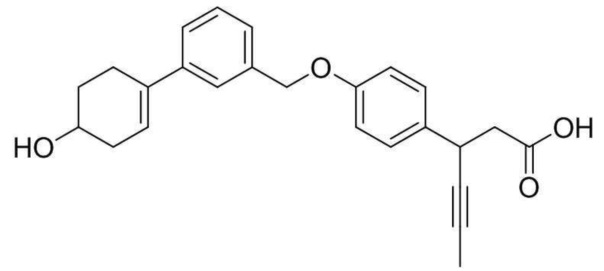
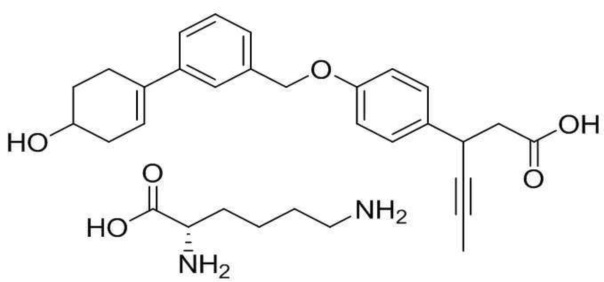
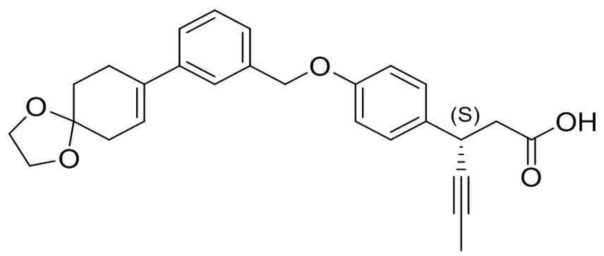
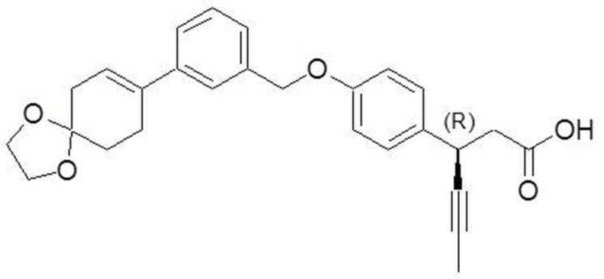


Экспериментальный Пример 1: Оценка производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты в отношении активности белка GPR40
Для оценки активности нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты согласно настоящему изобретению в отношении GPR40 проводили следующий эксперимент.
Активность нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты согласно настоящему изобретению в отношении белка GPR40 измеряли, исследуя изменения внутриклеточной концентрации кальция, на которую влияет активация GRP40. Сначала клетки HEK-293 трансфицировали человеческой ДНК GPR40 (Origene, RC218370) при использовании Fugene HD (Promega, E2311). Трансфицированные клетки HEK-293 распределяли в черном планшете с 96 лунками с прозрачным дном (Costar, 3603), затем культивировали. 24 часа спустя среду культуры клеток заменяли модифицированной по Дульбекко средой Игла (DMEM, 50 мкл/лунка), дополненной 1% зародышевой телячьей сывороткой (FBS). Для измерения концентрации кальция 50 мкл реагента Fluo-4 (Invitrogen, F10471) добавляли в каждую лунку, затем культивировали в инкубаторе при 37°C в течение 2 часов. В ходе культивирования, соединения из Примеров и соединения Сравнительных примеров 1 и 2 разбавляли 1 × HBSS (Hank's Buffered Salt Solution), содержащим 20 мМ 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновой кислоты (HEPES) в качестве буфера, получая образцы для обработки клеток. Спустя 2 часа после начала культивирования, эти полученные образцы автоматически впрыскивали к клеткам при использовании Flexstation 3 (Molecular Devices). Затем внутриклеточную концентрацию кальция измеряли в течение 120 секунд при использовании программного обеспечения SoftMax®Pro. На этой стадии диметилсульфоксид (ДМСО) впрыскивали к клеткам необработанной группы, затем измеряли концентрацию кальция. Активность белка GPR40 вычисляли с помощью приведенной ниже математической формулы 1 с результатами измерения концентрации кальция. Результаты показаны в Таблице 2.
Математическая формула 1
Активность GPR40 = (внутриклеточная концентрация кальция, увеличенная образцом)/(внутриклеточная концентрация кальция необработанной группы) × 100
В Таблице 2,
A: менее 0,20 мкМ;
B: 0,20-0,30 мкМ; и
C: более 0,30 мкМ.
Как показано в Таблице 2, было подтверждено, что соединения из Примеров настоящего изобретения превосходно промотируют активацию белков GPR 40 в низкой концентрации. В частности, соединения Примеров 7, 9, 11, 12, 14, 27, 28, 37 и 38 были в состоянии промотировать активацию белка GPR40 на 50% в очень низкой концентрации (менее 0,20 мкМ), что позволяет предположить, что их способность увеличивать внутриклеточную концентрацию Ca2+ является превосходной, по сравнению с таковой соединения Сравнительного Примера 1 (B, 0,28 мкМ).
Поэтому было подтверждено, что новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты согласно настоящему изобретению превосходно усиливают активацию белка GPR40. Эта активность по меньшей мере подобна таковой или превосходит активность обычного антидиабетического средства (Сравнительный Пример 1), известного как промотирующее секрецию инсулина путем индукции активации белка GPR40. Таким образом, композиция, включающая соединение по изобретению в качестве активного ингредиента, может эффективно использоваться как фармацевтическая композиция для профилактики и лечения метаболического заболевания, такого как ожирение, диабет типа I, диабет типа II, несовместимая переносимость глюкозы, инсулинорезистентность, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром X, и т.д.
Экспериментальный Пример 2: Анализ потока кальция
Поток кальция согласно активации GPR40 новым производным 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты согласно настоящему изобретению оценивался Millipore, учреждением, специализирующимся на тесте GPCR.
Соединения Примеров по изобретению, растворенные в ДМСО (диметилсульфоксид), PBS (фосфатный буферный солевой раствор) и DW (дистиллированная вода) и т.д, разбавляли три раза профильным буфером теста GPCR EMD Millipore. Аналогично, получали необработанную группу (носитель) и положительные контрольные группы (Сравнительные Примеры 1 и 3), чтобы увеличить точность анализа. Каждую лунку заполняли профильным буфером теста GPCR EMD Millipore. Упомянутым профильным буфером теста GPCR EMD Millipore был HBSS (Сбалансированный солевой раствор Хэнкса), содержащий 20 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота) и 2,5 мМ Probenecid (4-(дипропилсульфамоил)бензойнай кислота), чей рН составил 7,4.
Соединения Примеров дублировали в каждой концентрации. Получали положительную контрольную группу (Сравнительный Пример 1 или 3) для каждого сцепленного с G-белком рецептора (GPCR) и необработанную группу (носитель). Положительная контрольная группа (Сравнительный Пример 1 или 3) для каждого GPCR была включена в Emax в такой концентрации, которая показывала самую высокую активность. Тест на агонизм осуществляли при использовании FLIPRTETRA. Измеряли базовые флюоресценцию и люминесценцию. Соединения Примеров, необработанную группу (носитель) и положительную контрольную группу (Сравнительный Пример 1 или 3) включали в тестовый планшет. Чтобы измерить активность соединений Примеров, тест активности GPCR проводили в течение 180 секунд.
Значения флюоресценции, исключая базовое, сравнивали с Emax положительных контролей (Сравнительные Примеры 1 и 3) и необработанной группы, и активность выражали как %. Полученные данные показывают, что степень ингибирования (%) является результатом сравнения EC50 с необработанной группой, и качество каждого планшета оценивали статистическими числами, представляющими % активности от повторных данных. Когда данные теста не были удовлетворительными, проводили дополнительный эксперимент.
Все зависимые от концентрации графики получали при использовании GraphPad Prism. График модифицировали Сигмовидной кривой доза-ответ. Минимальное значение было зафиксировано как 0, и максимальное значение было зафиксировано как 100 для прогноза значения лучшего эффекта.
Результаты показаны на фигуре 1 и в Таблице 3.
Фигура 1 представляет собой график, иллюстрирующий структуру активации GPR40 согласно концентрации соединений Примера 9, Сравнительного Примера 1 и Сравнительного Примера 3.
Как показано на Фигуре 1, соединение Примера требовало намного более низкой концентрации, чем соединения Сравнительных Примеров 1 и 3, чтобы поднять активность GPR40 до 50% (еще ниже чем 1 нМ, самая низкая детектируемая концентрация). Особенно, как показано в Таблице 3, соединение Примера 9 согласно настоящему изобретению могло индуцировать активацию GPR40 в более низкой концентрации, чем соединения Сравнительного Примера 1 (14 нМ) и Сравнительный Пример 3 (27 нМ).
Поэтому было подтверждено, что новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты согласно настоящему изобретению превосходно промотирует активацию белка GPR40, гораздо лучше, чем обычные антидиабетические средства (соединения Сравнительных Примеров 1 и 3), известные как увеличивающие секрецию инсулина путем активации белка GPR40. Таким образом, композиция, включающая соединение по изобретению в качестве активного ингредиента, может эффективно использоваться как фармацевтическая композиция для профилактики и лечения метаболического заболевания такое как ожирение, диабет типа I, диабет типа II, несовместимая переносимость глюкозы, инсулинорезистентность, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром X, и т.д.
Экспериментальный Пример 3: Анализ ингибирования CYP
Для оценки взаимодействия между новым производным 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты согласно настоящему изобретению и другими лекарственными средствами проводили следующий эксперимент.
CYP представляет собой фермент, участвующий в метаболизме лекарственных средств. Так, ингибирование этого фермента может изменить прогноз дозы лекарственного средства и токсичности, вызываемой комбинированным лечением с другими лекарственными средствами. Поэтому авторы изобретения измерили ингибирующее действие соединений из Примеров изобретения на эндогенный CYP3A4, CYP2C9, CYP1A2, CYP2D6 и CYP2C19. В этом исследовании Invitrogen (P2862) использовали в качестве набора для ингибирования CYP2D6, и BD GENTEST (459100, 459300, 459400, 459500) использовали в качестве набора для ингибирования CYP1A2, CYP2C9, CYP2C19 и CYP3A4. Для получения набора Invitrogen, тестируемый образец разбавляли в дистиллированной воде в 2,5× от конечной экспериментальной концентрации.
Реактив P450 BACULOSOMES® и дубликатор (100x), входящие в набор Invitrogen, разбавляли в буфере реакции Vivid® CYP450 (2×) в концентрации, которая соответствовала целевому CYP450. Полученный 2,5× образец (80 мкл) и разбавленную смесь реактива P450 BACULOSOMES® (100 мкл) смешивали в каждой лунке 96-луночного планшета с U-дном, затем предварительно культивировали в течение 20 минут. Субстрат Vivid® CYP450 и NADP+ (100×) разбавляли в реакционном буфере Vivid® CYP450 (2×) в концентрации, которая соответствовала целевому CYP450 и субстрату. После завершения предварительного культивирования, добавляли смешанный раствор (20 мкл) субстрат-NADP (никотинамид аденин динуклеотид фосфат), с последующей реакцией в течение 1 часа. После завершения реакции, реагент переносили на белый планшет, и затем флюоресценцию измеряли с помощью устройтсва для считывания микропланшетов (CYP 2D6 длина волны возбуждения: 400 нм, длина волны поглощения: 502 нм).
Тестируемый образец для набора BD GENTEST разбавляли в ацетонитриле в 50× от конечной экспериментальной концентрации. Смесь NADPH-кофермент получали, разбавляя кофермент, G6PDH, и регулирующий белок, входящие в набор, дистиллированной водой в концентрации, указанной во входящей в набор инструкции. Полученный 50× образец (4 мкл) и смесь NADPH-кофермент (96 мкл) смешивали в каждой лунке 96-луночного планшета с U-дном, затем предварительно культивировали в течение 10 минут в термостате при 37°C. Смешанный раствор фермент/субстрат получали, разбавляя буфер (0,5 М фосфата калия, рН 7,4), смешанный раствор каждый фермент/субстрат CYP450, входящий в набор, дистиллированной водой в концентрации, указанной в инструкции. После завершения предварительного культивирования, 100 мкл смешанного раствора фермент/субстрат добавляли в каждую лунку планшета, затем культивировали в термостате при 37°C в течение 15 минут (CYP 1A2), 30 минут (CYP 3A4 и CYP 2C19) или 1,5 часов (CYP 2C9). После завершения реакции, реагент переносили на белый планшет, и затем флюоресценцию измеряли с помощью устройства для считывания микропланшетов (CYP 1A2 и CYP 2C19 длина волны возбуждения: 410 нм, длина волны поглощения: 460 нм; CYP 2C9 и CYP 3A4 длина волны возбуждения: 409 нм, длина волны поглощения: 530 нм). Значения, полученные выше, конвертировали в % как степень ингибирования образца от значения необработанной группы. Результаты показаны в Таблице 4.
(10 мкМ)
Как показано в Таблице 4, соединения из Примеров настоящего изобретения показывают низкую активность ингибирования CYP450, что позволяет предположить, что риск возникновения побочных эффектов вследствие взаимодействия с различными лекарственными средствами является очень низким. Более точно, почти все соединения по изобретению показали степень ингибирования 50% в лучшем случае по отношению к ферментам CYP 1A2, CYP 2C9, CYP 2C19, CYP 2D6 и CYP 3A4. В частности, по сравнению с соединением Сравнительного Примера 1 (81,2%), которое использовали как обычное антидиабетическое средство, которое может промотировать секрецию инсулина путем активации белка GPR40, соединения из Примеров изобретения демонстрировали значительно более низкую ингибирующую фермент активность особенно против CYP 2C9. По сравнению с соединением Сравнительного Примера 2 (63,2%), соединения из Примеров изобретения демонстрировали сравнительно более низкую ингибирующую фермент активность особенно против CYP 2D6.
Поскольку новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты согласно настоящему изобретению имеет существенно низкое ингибирующее действие в отношении фермента CYP, фармацевтическая композиция, включающая его в качестве активного ингредиента, может быть использована для лечения совместно с другими лекарственными средствами, и таким образом, может эффективно использоваться для лечения осложнений, включая нарушение обмена веществ, такое как ожирение, диабет типа I, диабет типа II, несовместимая переносимость глюкозы, инсулинорезистентность, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром X, и т.д.
Экспериментальный Пример 4: Тест на толерантность к пероральной глюкозе (OGTT) 1
Чтобы исследовать гипогликемический эффект нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты согласно настоящему изобретению in vivo, проводили следующий эксперимент.
Самцов крыс Sprague Dawley в возрасте 8~10 недель с индуцированной диетой моделью ожирения адаптировали по меньшей мере в течение 7 дней. Затем выбирали только здоровых животных и проводили OGTT. После ограничения пищи в течение 16~18 часов, рандомизированно выбирали по 5 крыс на группу и перорально вводили соединения, полученные в Примерах 2, 3, 4, 6, 9, 12, 14, 16, 25, 29, 36, 37, 41, 43 и 44 в дозе 10 мг/кг каждое. В это время 5%-ый полиэтиленгликоль (ПЭГ) вводили перорально в той же самой дозе крысам в необработанной группе (носитель). Спустя 30 минут после введения образца, глюкозу (4 г/кг) вводили перорально в дозе 5 мл/кг. Затем глюкозу крови измеряли при использовании активной полоски Accu-chek (Roche diagnostic Co.). Время для измерения было установлено в 30 минут перед введением глюкозы (-30), 0 минут, 20 минут, 40 минут, 60 минут и 120 минут после введения глюкозы, и глюкозу крови измеряли через пункцию хвостовой вены. Результаты показаны в Таблице 5.
Как показано в Таблице 5, соединения по изобретению показали гипогликемический эффект 21,9% от необработанной группы, что позволяет предположить, что они имели превосходное глюкозопонижающее действие in vivo. Более точно, было подтверждено, что соединение из Сравнительного Примера 1, известное как обычный активатор белка GPR40, имеет гипогликемический эффект 16,2%, в то время как соединения из Примеров по изобретению демонстрировали более высокий гипогликемический эффект. В частности, гипогликемические эффекты соединений Примеров 9, 12, 14, 29 и 37 составили, соответственно, 24,7%, 31,0%, 27,7%, 27,1% и 28,5%, что показывает, что их активность в отношении снижения глюкозы крови была превосходной по сравнению с активностью соединения из Сравнительного Примера 1.
Поэтому было подтверждено, что новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты согласно настоящему изобретению имеет превосходный эффект в отношении активации белка GPR 40, и соответственно, имеет значительный эффект в отношении снижения глюкозы крови, промотируя секрецию инсулина. Таким образом, композиция, включающая это соединение в качестве активного ингредиента, может эффективно использоваться как фармацевтическая композиция для лечения метаболического заболевания, такого как ожирение, диабет типа I, диабет типа II, несовместимая переносимость глюкозы, инсулинорезистентность, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром X, и т.д.
Экспериментальный Пример 5: Тест на толерантность к пероральной глюкозе (OGTT) 2
Чтобы исследовать гипогликемический эффект нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты согласно настоящему изобретению in vivo, проводили следующий эксперимент.
Самцов крыс Goto-Kakizaki (GK) в возрасте 22~23 недели, животная модель диабета типа II (не ожирение), адаптировали по меньшей мере в течение 7 дней. Затем выбирали только здоровых животных и проводили OGTT. После ограничения пищи в течение 16~18 часов, рандомизированно выбирали по 5 крыс на группу и перорально вводили соединения, полученные в Примерах 5, 9, 14, 28, 37 и 47 в дозе 0,3~10 мг/кг. В это время 5%-ый полиэтиленгликоль (ПЭГ) вводили перорально в той же самой дозе крысам в необработанной группе. Спустя 60 минут после введения образца, глюкозу (4 г/кг) вводили перорально в дозе 5 мл/кг. Затем глюкозу крови измеряли при использовании активной полоски Accu-chek (Roche diagnostic Co.). Время для измерения было установлено в 60 минут перед введением глюкозы (-60), 0 минут, 20 минут, 40 минут, 60 минут и 120 минут после введения глюкозы, и глюкозу крови измеряли через пункцию хвостовой вены. Результаты показаны в Таблице 6.
В Таблице 6,
A: более чем 35,0%;
B: 25,0–35,0%; и
C: менее чем 25,0%.
Как показано в Таблице 6, соединения из Примеров по изобретению демонстрировали по меньшей мере средний 30,0% гипогликемический эффект по сравнению с необработанной группой в той же самой дозе соединения из Сравнительного Примера 1 (10 мг/кг). Более точно, соединение из Сравнительного Примера 1 показало гипогликемический эффект 25,3% (B) в дозе 10 мг/кг, в то время как соединения из Примеров 5, 9, 14, 28, 37 и 47 демонстрировали в дозе 3 мг/кг гипогликемический эффект, подобный таковому соединения из Сравнительного Примера 1. В частности соединения Примеров 9 и 37 показали гипогликемический эффект больше чем 35,0% в дозе 10 мг/кг, который был значительно более высоким по сравнению с таковым соединения из Сравнительного Примера 1.
Поэтому было подтверждено, что новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты согласно настоящему изобретению имеет превосходный эффект в отношении активации белка GPR 40, и соответственно, имеет значительный эффект в отношении снижения глюкозы крови, промотируя секрецию инсулина. Таким образом, композиция, включающая это соединение в качестве активного ингредиента, может эффективно использоваться как фармацевтическая композиция для лечения метаболического заболевания, такого как ожирение, диабет типа I, диабет типа II, несовместимая переносимость глюкозы, инсулинорезистентность, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром X, и т.д.
Экспериментальный Пример 6: Тест на толерантность к пероральной глюкозе (OGTT) 3
Чтобы исследовать гипогликемический эффект нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты согласно настоящему изобретению in vivo, проводили следующий эксперимент.
Самцов крыс OLETF (Otsuka Long-Evans Tokushima fatty) в возрасте 29~30 недель, животная модель диабета типа II (ожирение), адаптировали по меньшей мере в течение 7 дней. Затем выбирали только здоровых животных и проводили OGTT. После ограничения пищи в течение 16~18 часов, рандомизированно выбирали по 5 крыс на группу и перорально вводили соединения, полученные в Примерах 5, 9, 14, 28, 37 и 47 в дозе 1~10 мг/кг. В это время 5%-ый полиэтиленгликоль (ПЭГ) вводили перорально в той же самой дозе крысам в необработанной группе. Спустя 60 минут после введения образца, глюкозу (4 г/кг) вводили перорально в дозе 5 мл/кг. Затем глюкозу крови измеряли при использовании активной полоски Accu-chek (Roche diagnostic Co.). Время для измерения было установлено в 60 минут перед введением глюкозы (-60), 0 минут, 20 минут, 40 минут, 60 минут и 120 минут после введения глюкозы, и глюкозу крови измеряли через пункцию хвостовой вены. Результаты показаны в Таблице 7.
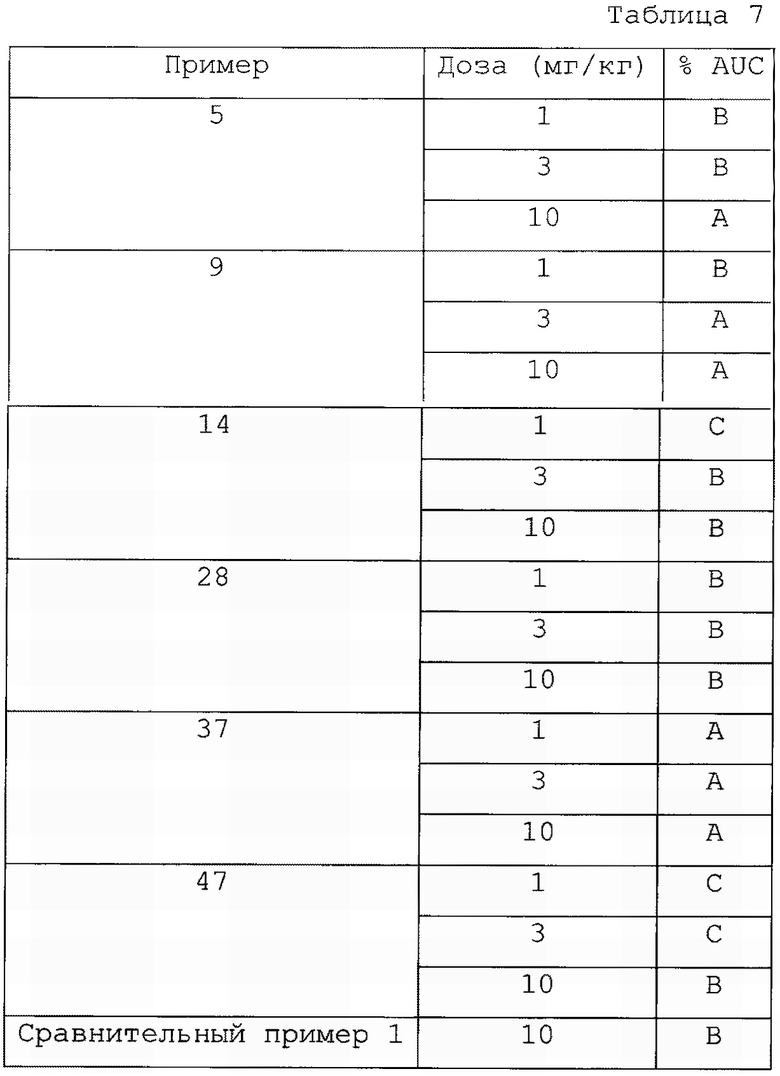
В Таблице 7,
A: более чем 35,0%;
B: 25,0-35,0%; и
C: менее чем 25,0%.
Соединения из Примеров по изобретению демонстрировали по меньшей мере средний 35,0% гипогликемический эффект по сравнению с необработанной группой в той же самой дозе соединения из Сравнительного Примера 1 (10 мг/кг). Более точно, соединение из Сравнительного Примера 1 показало гипогликемический эффект 31,6% (B) в дозе 10 мг/кг, в то время как соединения из Примеров 9 и 37 демонстрировали более высокий гипогликемический эффект в дозе 1 мг/кг, чем соединение из Сравнительного Примера 1. В частности, соединения Примеров 9 и 37 показали гипогликемический эффект больше чем 35,0% в дозе 10 мг/кг, который был значительно более высоким по сравнению с таковым соединения из Сравнительного Примера 1.
Поэтому было подтверждено, что новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты согласно настоящему изобретению имеет превосходный эффект в отношении активации белка GPR 40, и соответственно, имеет значительный эффект в отношении снижения глюкозы крови, промотируя секрецию инсулина. Таким образом, композиция, включающая это соединение в качестве активного ингредиента, может эффективно использоваться как фармацевтическая композиция для лечения метаболического заболевания, такого как ожирение, диабет типа I, диабет типа II, несовместимая переносимость глюкозы, инсулинорезистентность, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром X, и т.д.
Экспериментальный Пример 7: Измерение GLP-1 (глюкагоноподобный пептид-1) крови после перорального введения
Чтобы исследовать повышение GLP-1 крови после перорального введения нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты по изобретению, проводили следующий эксперимент.
Самцов крыс Sprague Dawley в возрасте 10~12 недель с индуцированной диетой моделью ожирения адаптировали по меньшей мере в течение 7 дней. Только здоровые животные были выбраны после адаптации для следующего эксперимента. После ограничения пищи в течение 16~18 часов, рандомизированно выбирали по 5 крыс на группу и перорально вводили соединение, полученное в Примере 9 в дозе 10~100 мг/кг (объем растворителя для введения: 5 мл/кг). В это время 5%-ый полиэтиленгликоль (ПЭГ) вводили перорально в той же самой дозе крысам в необработанной группе. Спустя 60 минут после введения образца, глюкозу вводили перорально в дозе 2 г/кг. 20 минут спустя кровь забирали из сердца (0,5 мл цельной крови). Пробу крови немедленно загружали в пробирку для проб, обрабатывали ингибитором DPP-4 (дипептидилпептидазы-4) и EDTA (этилендиаминтетрауксусная кислота), затем помещали в сосуд со льдом для охлаждения. Пробу крови центрифугировали при 3600 об/мин в течение 10 минут, чтобы отделить плазму крови. Затем содержание GLP-1 в отделенной плазме крови измеряли при использовании набора ELISA для измерения GLP-1 (Millipore, США). Результаты показаны на Фигуре 2.
Фигура 2 представляет собой график, иллюстрирующий содержание GLP-1 в крови крысы SD (крыса Sprague Dawley) при пероральном введении соединений из Примера 9 и Сравнительного Примера 1.
Как показано на Фигуре 2, соединение из Сравнительного Примера 1 не показывало никакого увеличения GLP-1, который может промотировать секрецию инсулина, после введения по сравнению с группой, обработанной глюкозой (носитель). Однако было подтверждено, что соединение из Примера 9 увеличивает GLP-1 в крови крыс SD.
Поэтому было подтверждено, что новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты согласно настоящему изобретению имеют превосходную активность промотирования секреции гормона GLP-1 по сравнению с соединением из Сравнительного Примера 1, и особенно этот эффект был превосходным в животных моделях диабета. Также на основании такой активности по промотированию секреции GLP-1 нового производного 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты согласно настоящему изобретению ожидается, что оно может предотвращать функциональный дефект бета-клеток и увеличение массы тела. Таким образом, композиция, включающая новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты согласно настоящему изобретению в качестве активного ингредиента может эффективно использоваться как фармацевтическая композиция для лечения метаболического заболевания, такого как ожирение, диабет типа I, диабет типа II, несовместимая переносимость глюкозы, инсулинорезистентность, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром X, и т.д.
В то же время, соединение, представленное формулой 1 согласно настоящему изобретению, может быть составлено в различных формах в зависимости от цели использования. Ниже приведены примеры способов составления с использованием соединения, представленного формулой 1 согласно настоящему изобретению, в качестве активного ингредиента, но настоящее изобретение не ограничено этими примерами.
Препаративный Пример 1: Получение фармацевтических составов
<1-1> Получение порошков
Порошки получали, смешивая все вышеупомянутые компоненты, которые заполняли в воздухонепроницаемые упаковки согласно обычному способу получения порошков.
<1-2> получение таблеток
Таблетки получали, смешивая все вышеупомянутые компоненты обычным способом получения таблеток.
<1-3> Получение капсул
Капсулы получали, смешивая все вышеупомянутые компоненты, которые заполняли в желатиновые капсулы согласно обычному способу получения капсул.
<1-4> Получение инъецируемых растворов
Инъецируемые растворы получали, смешивая все вышеупомянутые компоненты, помещая смесь в ампулы на 2 мл и стерилизуя обычным способом получения инъецируемых растворов.
<1-5> Получение жидких составов
Все вышеупомянутые компоненты растворяли в очищенной воде. После добавления лимонного ароматизатора, полный объем доводили до 100 мл добавлением очищенной воды. Жидкие составы получали, помещая смесь в бутыли из коричневого стекла и стерилизуя обычным способом получения жидких составов.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты, его оптический изомер или фармацевтически приемлемая соль согласно настоящему изобретению имеют превосходную активность в отношении активации белка GPR40 и промотирования секреции инсулина, соответственно, но не имеют никакой токсичности при совместном введении с другими лекарственными средствами. Таким образом, новое производное 3-(4-(бензилокси)фенил)гекс-4-иновой кислоты, его оптический изомер или фармацевтически приемлемую соль согласно настоящему изобретению можно вводить совместно с другими лекарственными средствами, и оно может значительно промотировать активацию белка GPR40, так, что композиция, включающая его в качестве активного ингредиента, может эффективно использоваться как фармацевтическая композиция для профилактики и лечения метаболического заболевания, такого как ожирение, диабет типа I, диабет типа II, несовместимая переносимость глюкозы, инсулинорезистентность, гипергликемия, гиперлипидемия, гипертриглицеридемия, гиперхолестеринемия, дислипидемия и синдром X, и т.д.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМБИНАЦИЯ, СОДЕРЖАЩАЯ НОВОЕ ПРОИЗВОДНОЕ 3-(4-(БЕНЗИЛОКСИ)ФЕНИЛ)ГЕКС-4-ИНОВОЙ КИСЛОТЫ И ДРУГОЙ АКТИВНЫЙ ИНГРЕДИЕНТ, ДЛЯ АКТИВИРОВАНИЯ ФЕРМЕНТА РЕЦЕПТОРА G-БЕЛКА 40 | 2015 |
|
RU2680248C1 |
| НОВЫЕ СОЕДИНЕНИЯ, ИХ ИЗОМЕР ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ В КАЧЕСТВЕ АНТАГОНИСТА ВАНИЛОИДНОГО РЕЦЕПТОРА И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2007 |
|
RU2448108C2 |
| СПОСОБ ОЦЕНКИ КАЧЕСТВА (3S)-3-(4-(3-(1,4-ДИОКСАСПИРО[4,5]ДЕЦ-7-ЕН-8-ИЛ)БЕНЗИЛОКСИ)ФЕНИЛ)ГЕКС-4-ИНОВОЙ КИСЛОТЫ | 2021 |
|
RU2804883C1 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА ДЛЯ ТОРМОЖЕНИЯ РОСТА РАКОВЫХ КЛЕТОК И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2005 |
|
RU2362773C2 |
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ИНГИБИРОВАНИЯ ТИРОЗИНКИНАЗНОЙ АКТИВНОСТИ | 2011 |
|
RU2585177C2 |
| АГОНИСТ РЕЦЕПТОРА GPR40, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЕГО В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2013 |
|
RU2650506C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА И ИНДАЗОЛА, ОБЛАДАЮЩИЕ КОНСЕРВИРУЮЩИМ ДЕЙСТВИЕМ ПО ОТНОШЕНИЮ К КЛЕТКАМ, ТКАНЯМ И ОРГАНАМ | 2009 |
|
RU2460525C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2004 |
|
RU2366659C2 |
| СОЕДИНЕНИЯ ИНДОЛА И ИНДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА НЕКРОЗА КЛЕТКИ | 2008 |
|
RU2437883C1 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2020 |
|
RU2833354C1 |
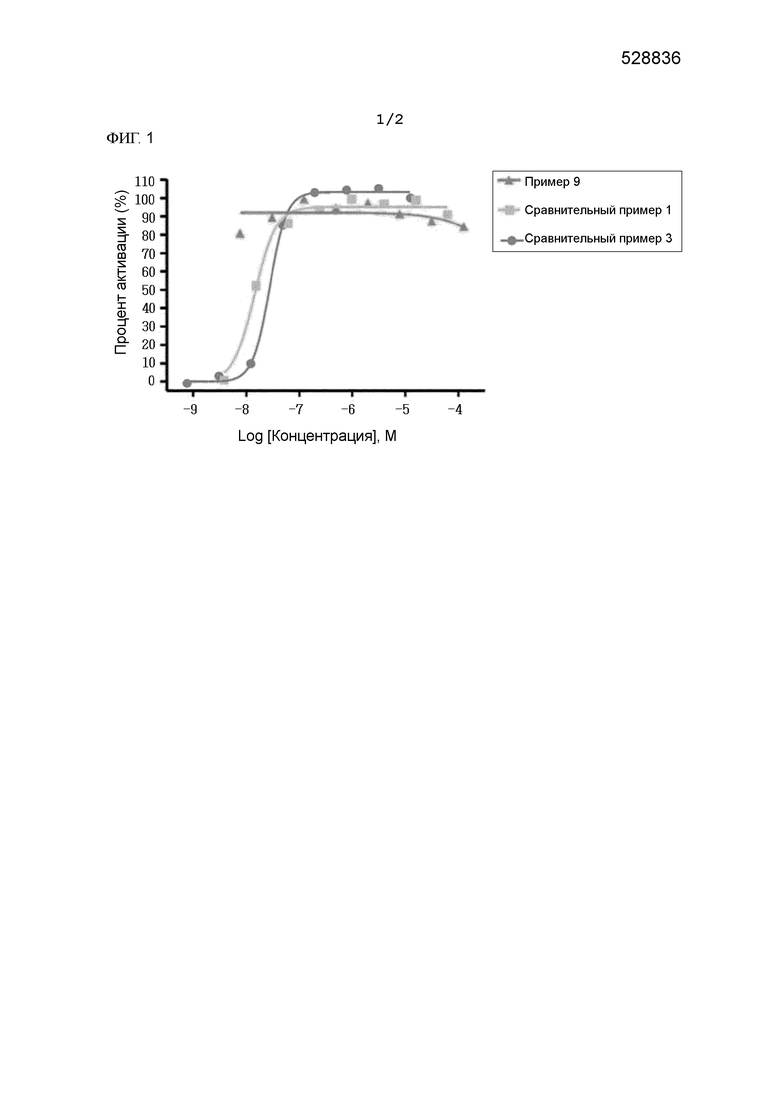
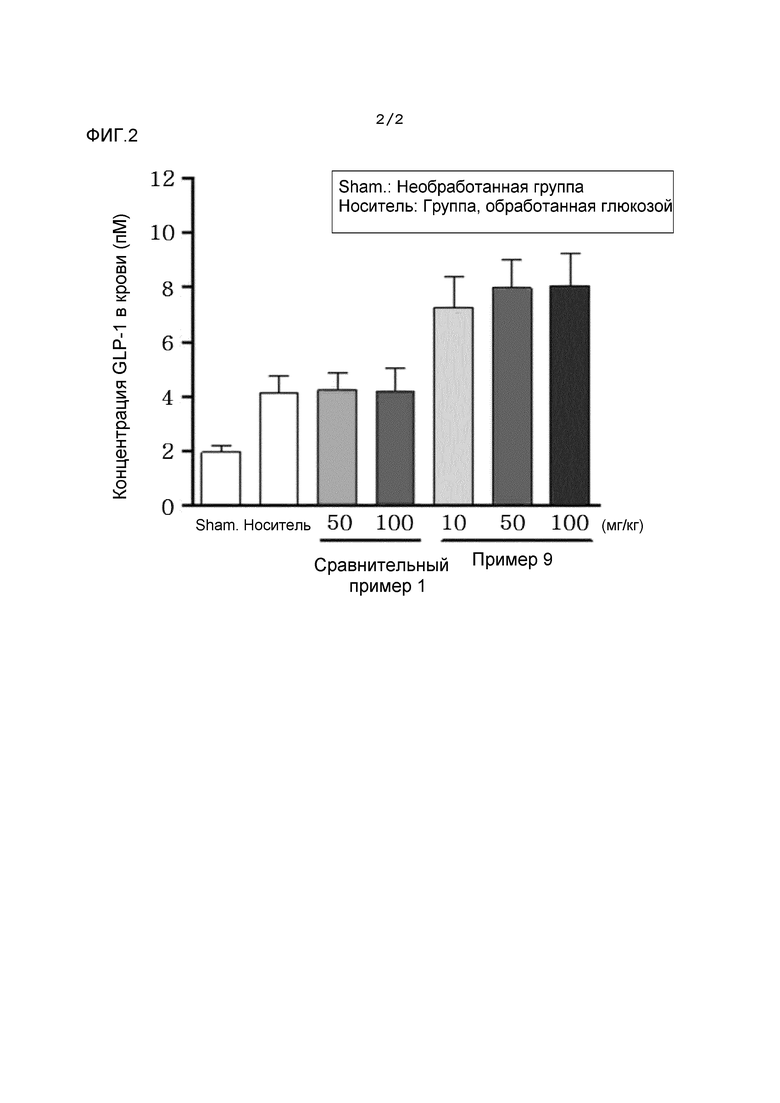
Изобретение относится к соединению, представленному формулой 1, его оптическому изомеру или его фармацевтически приемлемой соли:
[Формула 1]
 , а также к способам его получения и фармацевтической композиции на его основе. Технический результат: получены новые соединения, обладающие способностью активировать фермент GPR40, пригодные для применения для профилактики или лечения метаболического заболевания. 7 н. и 3 з.п. ф-лы, 7 табл., 77 пр., 2 ил.
, а также к способам его получения и фармацевтической композиции на его основе. Технический результат: получены новые соединения, обладающие способностью активировать фермент GPR40, пригодные для применения для профилактики или лечения метаболического заболевания. 7 н. и 3 з.п. ф-лы, 7 табл., 77 пр., 2 ил.
1. Соединение, представленное приведенной ниже формулой 1, его оптический изомер или его фармацевтически приемлемая соль:
[Формула 1]
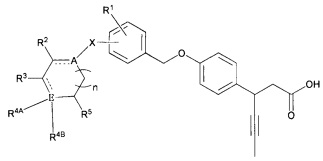
(в формуле 1
 обозначает простую связь или двойную связь;
обозначает простую связь или двойную связь;
А и Е независимо обозначают С или N;
n означает целое число 0-1;
X обозначает простую связь или C1-3 прямой или разветвленный алкилен;
R1 обозначает -Н или  ;
;
R2 обозначает -Н или может образовывать фенил с R3;
R3 обозначает -Н или может образовывать фенил с R2 или может образовывать фенил с R4A вместе с атомами, которые к ним присоединены; в упомянутом фениле, образованном R3 и R4A, метокси может быть замещен;
R4A обозначает -Н, -ОН, =O, 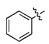 ,
, 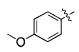 ,
,  ,
,  ,
, 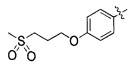 ,
, 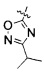 ,
, 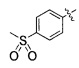 ,
, 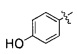 ,
, 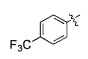 ,
, 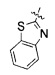 ,
, 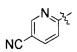 ,
, 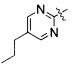 или может образовывать фенил с R3 вместе с атомами, которые к ним присоединены; в упомянутом фениле, образованном R3 и R4A, метокси может быть замещен;
или может образовывать фенил с R3 вместе с атомами, которые к ним присоединены; в упомянутом фениле, образованном R3 и R4A, метокси может быть замещен;
R4B отсутствует или может образовывать 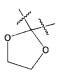 вместе с атомами, которые к нему присоединены, и R4A; и
вместе с атомами, которые к нему присоединены, и R4A; и
R5 обозначает -Н.
2. Соединение, представленное формулой 1, его оптический изомер или его фармацевтически приемлемая соль по п. 1, причем соединение, представленное формулой 1, выбрано из группы, состоящей из следующих соединений:
(1) 3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновая кислота;
(2) L-лизин 3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат;
(3) 4-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновая кислота;
(4) 3-(4-(3-(4-оксоциклогекс-1-енил)бензилокси)фенил)гекс-4-иновая кислота;
(5) 3-(4-(3-(4-гидроксициклогекс-1-енил)бензилокси)фенил)гекс-4-иновая кислота;
(6) L-лизин 3-(4-(3-(4-гидроксициклогекс-1-енил)бензилокси)фенил)гекс-4-иноат;
(7) (3S)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновая кислота;
(8) (3R)-3-(4-(3-(l,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иновая кислота;
(9) L-лизин (3S)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат;
(10) L-лизин (3R)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат;
(11) (3S)-3-(4-(3-(1,4-диоксаспиро[4,5]дец-7-ен-8-ил)бензилокси)фенил)гекс-4-иноат натрия;
(12) 3-(4-(4-((3,4-дигидроизохинолин-2(1Н)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(13) 3-(4-(3-циклогексенил-4-((3,4-дигидроизохинолин-2(1Н)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(14) 3-(4-(4-((4-фенил-5,6-дигидропиридин-1(2Н)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(15) 3-(4-(4-((4-фенилпиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(16) 3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1Н)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(17) 3-(4-(4-((4-фенилпиперидин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(18) 3-(4-(4-((4-(4-фторфенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(19) 3-(4-(4-((4-(4-(трифторметил)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(20) 3-(4-(4-((4-(4-(3-(метилсульфонил)пропокси)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(21) (S)-3-(4-(4-((3,4-дигидроизохинолин-2(1Н)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(22) (S)-3-(4-(4-((4-(4-(трифторметил)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(23) (S)-3-(4-(4-((4-(4-фторфенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(24) (S)-3-(4-(4-((4-(4-(трифторметил)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иноат калия;
(25) (S)-3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1Н)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(26) (S)-3-(4-(4-((4-фенилпиперидин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(27) (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иновая кислота;
(28) (S)-3-(4-(4-((4-фенил-5,6-дигидропиридин-1(2Н)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(29) (S)-3-(4-(4-((4-(4-(метоксиметокси)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(30) (S)-3-(4-(4-((4-(5-изопропил-1,2,4-оксадиазол-3-ил)пиперидин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(31) (S)-3-(4-(4-((4-(5-изопропил-1,2,4-оксадиазол-3-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(32) (S)-3-(4-(4-((4-(4-(метилсульфонил)фенил)-5,6-дигидропиридин-1(2Н)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(33) (S)-3-(4-(4-((4-(4-(3-(метилсульфонил)пропокси)фенил)-5,6-дигидропиридин-1(2Н)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(34) (3S)-3-(4-(4-(1-(3,4-дигидроизохинолин-2(1Н)-ил)этил)бензилокси)фенил)гекс-4-иновая кислота;
(35) (S)-3-(4-(4-((4-(4-гидроксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(36) (S)-3-(4-(4-((4-(4-(3-(метилсульфонил)пропокси)фенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(37) (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иноат натрия;
(38) L-лизин (S)-3-(4-(4-(изоиндолин-2-илметил)бензилокси)фенил)гекс-4-иноат;
(39) (S)-3-(4-(4-((4-(4-фторфенил)-5,6-дигидропиридин-1(2Н)-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(40) (S)-3-(4-(4-((4-(4-метоксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(41) (S)-3-(4-(4-((3,4-дигидрохинолин-1(2Н)-ил)метил)бензилокси)фенил)гекс-4-иноат натрия;
(42) (S)-3-(4-(4-((3,4-дигидрохинолин-1(2Н)-ил)метил)бензилокси)фенил)гекс-4-иноат калия;
(43) (S)-3-(4-(4-((4-(бензо[d]тиазол-2-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(44) (S)-3-(4-(4-((4-(5-пропилпиримидин-2-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(45) (S)-3-(4-(4-((4-(5-цианопиридин-2-ил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(46) (3S)-3-(4-(4-((3-фенилпирролидин-1-ил)метил)бензилокси)фенил)гекс-4-иновая кислота;
(47) (S)-3-(4-(4-((4-(4-метоксифенил)пиперазин-1-ил)метил)бензилокси)фенил)гекс-4-иноат натрия;
(48) (S)-3-(4-(4-(2-(6-метокси-3,4-дигидроизохинолин-2(1Н)-ил)этил)бензилокси)фенил)гекс-4-иновая кислота;
(49) (S)-3-(4-(4-(2-(изоиндолин-2-ил)этил)бензилокси)фенил)гекс-4-иновая кислота;
(50) (S)-3-(4-(4-(2-(3,4-дигидроизохинолин-2(1Н)-ил)этил)бензилокси)фенил)гекс-4-иновая кислота; и
(51) (S)-3-(4-(4-((6-метокси-3,4-дигидроизохинолин-2(1Н)-ил)метил)бензилокси)фенил)гекс-4-иноат натрия.
3. Способ получения соединения, представленного формулой 1 по п. 1, включающий следующие стадии, как показано в приведенной ниже схеме реакции 1:
получение соединения, представленного формулой 4, реакцией конденсации соединения, представленного формулой 2, и соединения, представленного формулой 3 (стадия 1); и
получение соединения, представленного формулой 1, реакцией восстановления соединения, представленного формулой 4, полученного на стадии 1 (стадия 2).
[Схема Реакции 1]
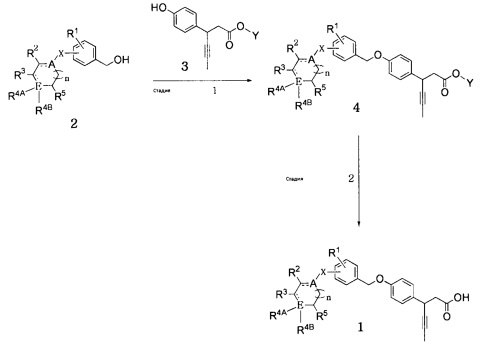
(в схеме реакции 1
R1, R2, R3, R4A, R4B, R5, А, Е, n, и X имеют значения, определенные в формуле 1; и Y обозначает C1-10 прямой или разветвленный алкил).
4. Способ получения соединения, представленного формулой 1 по п. 1, в котором соединение, представленное формулой 2, получают способом, включающим следующие стадии, как показано в приведенной ниже схеме реакции 2:
получение соединения, представленного формулой 10, путем реакции соединения, представленного формулой 8, и соединения, представленного формулой 9 (стадия 1);
получение соединения, представленного формулой 12, реакцией соединения, представленного формулой 10, полученного на стадии 1, и соединения, представленного формулой 11 (стадия 2); и
получение соединения, представленного формулой 2, реакцией восстановления соединения, представленного формулой 12, полученного на стадии 2 (стадия 3).
[Схема реакции 2]
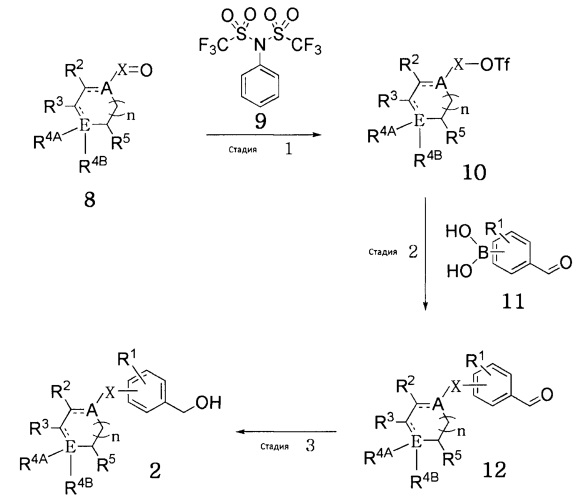
(В схеме реакции 2
R1, R2, R3, R4A, R4B, R5, А, Е, n, и X имеют значения, определенные в формуле 1; и -OTf обозначает трифторметансульфонат).
5. Способ получения соединения, представленного формулой 1 по п. 1, включающий следующие стадии, как показано в приведенной ниже схеме реакции 3:
получение соединения, представленного формулой 6, реакцией сочетания соединения, представленного формулой 5, и соединения, представленного формулой 3 (стадия 1);
получение соединения, представленного формулой 7, мезилатной реакцией соединения, представленного формулой 6, полученного на стадии 1 (стадия 2);
получение соединения, представленного формулой 4, путем замены мезилатной части соединения, представленного формулой 7, полученного на стадии 2, соединением, представленным формулой 13 (стадия 3); и
получение соединения, представленного формулой 1, реакцией восстановления соединения, представленного формулой 4, полученного на стадии 3 (стадия 4).
[Схема реакции 3]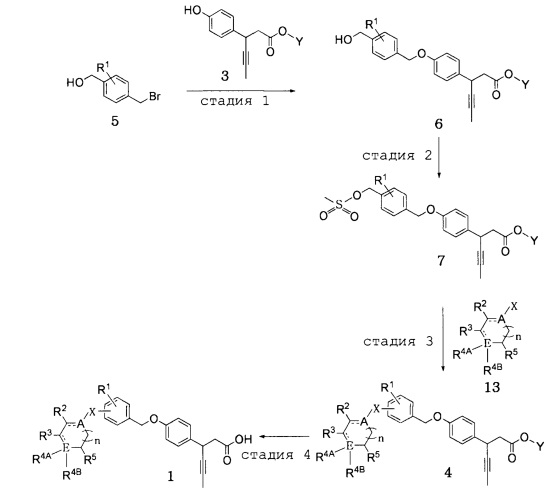
(В схеме реакции 3 R1, R2, R3, R4A, R4B, R5, A, E, n, и X имеют значения, определенные в формуле 1; и Y обозначает C1-10 прямой или разветвленный алкил).
6. Способ получения соединения, представленного формулой 1 по п. 1, включающий стадию получения соединения, представленного формулой 1b, реакцией размыкания кольца соединения, представленного формулой 1а (стадия 1), как показано в приведенной ниже схеме реакции 4:
[Схема реакции 4]

(В схеме реакции 4
R1 имеет значения, определенные в формуле 1; и соединения, представленные формулой 1а и формулой 1b, включены в соединение, представленное формулой 1).
7. Способ получения соединения, представленного формулой 1 по п. 1, включающий стадию получения соединения, представленного формулой 1с, реакцией восстановления соединения, представленного формулой 1b (стадия 1), как показано в приведенной ниже схеме реакции 5:
[Схема реакции 5]
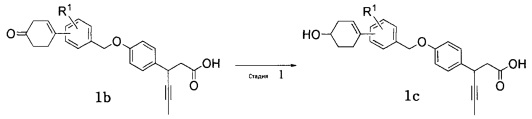
(В схеме реакции 5
R1 имеет значения, определенные в формуле 1; и соединения, представленные формулой 1b и формулой 1с, включены в соединение, представленное формулой 1).
8. Фармацевтическая композиция, включающая эффективное количество соединения, представленного формулой 1 по п. 1, его оптического изомера или его фармацевтически приемлемой соли в качестве активного ингредиента для профилактики или лечения метаболического заболевания.
9. Фармацевтическая композиция для профилактики или лечения метаболического заболевания по п. 8, причем соединение характеристически способно активировать фермент GPR40.
10. Фармацевтическая композиция для профилактики или лечения метаболического заболевания по п. 8, причем метаболическое заболевание выбрано из группы, состоящей из ожирения, диабета типа 1, диабета типа II, несовместимой переносимости глюкозы, инсулинорезистентности, гипергликемии, гиперлипидемии, гипертриглицеридемии, гиперхолестеринемии и дислипидемии.
| EA201270560 A1, 28.09.2012 | |||
| WO2008030618 A1, 13.03.2008 | |||
| WO2005086661 A2, 22.09.2005 | |||
| RU2010121160 A, 10.12.2011 | |||
| Shawn D Walker; Christopher J Borths; Evan Divirgilio; Liang Huang; Pingli Liu; Henry Morrison; Kiyoshi Sugi; Masahide Tanaka; Jacqueline C S Woo; Margaret M Faul | |||
| Приспособление с иглой для прочистки кухонь типа "Примус" | 1923 |
|
SU40A1 |
| ORGANIC PROCESS RESEARCH AND DEVELOPMENT, 15, No:3, стр | |||
| Секретный замок | 1923 |
|
SU570A1 |

