Область техники, к которой относится настоящее изобретение
Настоящая заявка относится к зародышеобразователям для термопластичных полимеров, композициям на основе термопластичных полимеров, содержащим такие зародышеобразователи, изделиям, полученным из таких композиций на основе термопластичных полимеров, и способам получения и формования таких композиций на основе термопластичных полимеров.
Предшествующий уровень техники настоящего изобретения
Различные зародышеобразователи для термопластичных полимеров известны в данной области техники. Эти зародышеобразователи обычно действуют путем образования зародышей кристаллизации или обеспечения мест для образования и/или роста кристаллов в термопластичном полимере, когда он отверждается из расплавленного состояния. Зародыши кристаллизации или места, обеспеченные зародышеобразователем, делают возможным образование кристаллов в охлаждающемся полимере при более высокой температуре и/или с большей скоростью, чем кристаллы будут образовываться в чистом термопластичном полимере без зародышеобразователя. Эти явления могут затем обеспечивать переработку композиции на основе термопластичного полимера с зародышеобразователем при более коротких длительностях производственного цикла, чем для чистого термопластичного полимера без зародышеобразователя.
Хотя полимерные зародышеобразователи могут действовать аналогичным образом, не все зародышеобразователи разрабатываются одинаковыми. Например, определенный зародышеобразователь может быть очень эффективным для повышения пиковой температуры перекристаллизации термопластичного полимера, но большая скорость кристаллизации, вызванная таким зародышеобразователем, может вызывать неравномерную усадку отформованной детали, полученной из композиции на основе термопластичного полимера, содержащей зародышеобразователь. Такой зародышеобразователь может также быть неэффективным для повышения жесткости отформованной детали до требуемого уровня. Также, хотя зародышеобразователи для полиэтиленовых полимеров известны в данной области техники, относительно небольшое число этих зародышеобразователей, как оказалось, улучшает физические свойства полиэтиленового полимера до какого-либо коммерчески значительного уровня.
Учитывая сложную взаимосвязь этих свойств и того факта, что многие зародышеобразователи проявляют характер хуже оптимального по меньшей мере в одном отношении, потребность остается в зародышеобразователях, которые способны давать композиции на основе термопластичных полимеров, характеризующиеся более желательной комбинацией высокой пиковой температуры перекристаллизации полимера, регулируемой усадки и высокой жесткости. Заявители считают, что зародышеобразователи и композиции на основе термопластичных полимеров, раскрытые в настоящей заявке, удовлетворяют данную потребность.
Краткое раскрытие настоящего изобретения
Как указано выше, настоящая заявка в общем относится к зародышеобразователям, композициям на основе термопластичных полимеров, содержащим такие зародышеобразователи, изделиям (например, формованным изделиям), полученным из таких композиций на основе термопластичных полимеров, и способам получения и формования таких композиций на основе термопластичных полимеров. Зародышеобразователи и композиции на основе термопластичных полимеров согласно настоящему изобретению, как считается, являются особенно подходящими для получения изделий из термопластичных полимеров (например, формованных изделий из термопластичных полимеров), характеризующихся желательной комбинацией физических свойств. В частности, изделия, полученные при помощи зародышеобразователей и композиций на основе термопластичных полимеров настоящего изобретения, как считается, характеризуются желательной комбинацией более высокой пиковой температуры перекристаллизации полимера и улучшенных физических свойств (например, прочности на разрыв) по сравнению с изделиями, полученными из термопластичного полимера без зародышеобразователя. Заявители считают, что данная комбинация физических свойств показывает, что зародышеобразователи и композиции на основе термопластичных полимеров согласно настоящему изобретению хорошо подходят для использования при производстве изделий из термопластичных полимеров.
Согласно первому варианту осуществления настоящее изобретение обеспечивает композицию на основе термопластичного полимера, содержащую:
(a) полиолефиновый полимер и
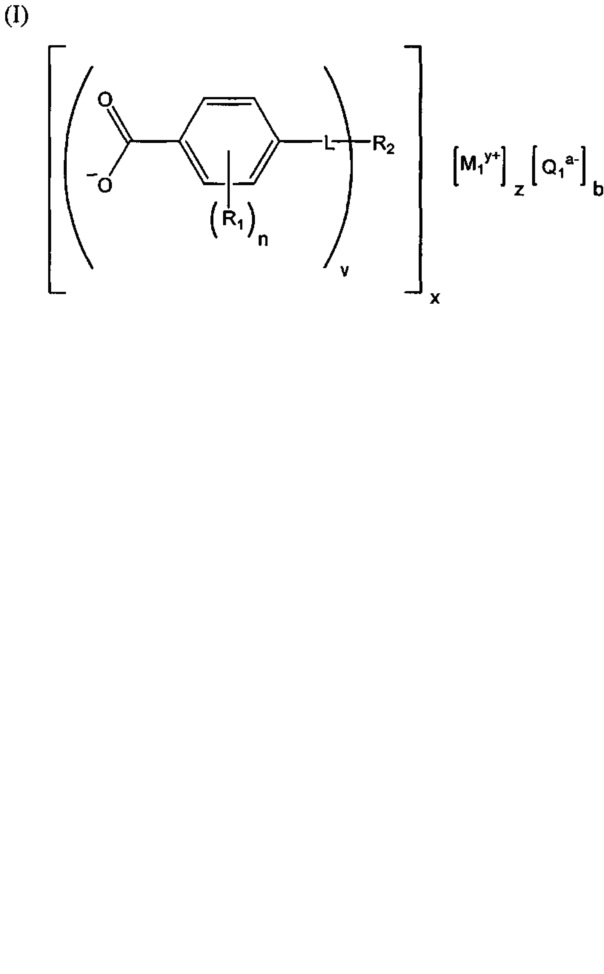
(b) зародышеобразователь, причем зародышеобразователь содержит соединение, соответствующее структуре формулы (I)
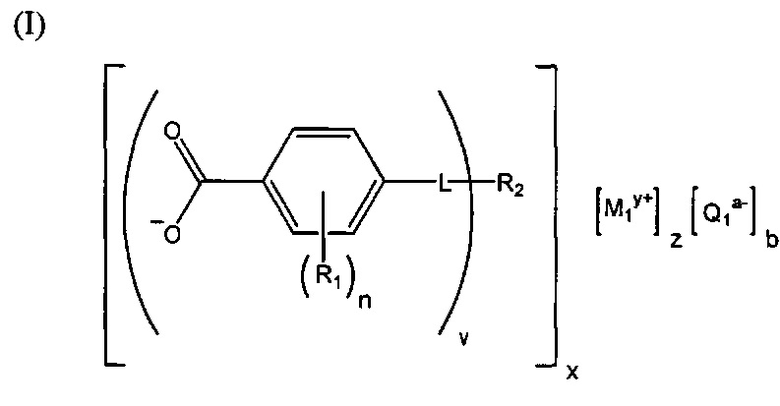 ,
,
где R1 выбрана из группы, состоящей из гидроксигруппы, галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп; n равняется нулю или целому положительному числу от 1 до 4; L представляет собой связующую группу, содержащую два или более атомов и по меньшей мере одну двойную связь между двумя атомами в связующей группе; v представляет собой целое положительное число от 1 до 3; R2: (i) выбрана из группы, состоящей из алкильных групп, замещенных алкильных групп, циклоалкильных групп, замещенных циклоалкильных групп, арильных групп, замещенных арильных групп, гетероарильных групп и замещенных гетероарильных групп, если L представляет собой двухвалентную связующую группу, а v равняется 1, (ii) выбрана из группы, состоящей из алкандиильных групп, замещенных алкандиильных групп, циклоалкандиильных групп, замещенных циклоалкандиильных групп, арендиильных групп, замещенных арендиильных групп, гетероарендиильных групп и замещенных гетероарендиильных групп, если L представляет собой трехвалентную связующую группу, а v равняется 1, (iii) выбрана из группы, состоящей из алкандиильных групп, замещенных алкандиильных групп, циклоалкандиильных групп, замещенных циклоалкандиильных групп, арендиильных групп, замещенных арендиильных групп, гетероарендиильных групп и замещенных гетероарендиильных групп, если L представляет собой двухвалентную связующую группу, а v равняется 2, и (iv) выбрана из группы, состоящей из алкантриильных групп, замещенных алкантриильных групп, циклоалкантриильных групп, замещенных циклоалкантриильных групп, арентриильных групп, замещенных арентриильных групп, гетероарентриильных групп и замещенных гетероарентриильных групп, если L представляет собой двухвалентную связующую группу, а v равняется 3; x представляет собой целое положительное число; каждый M1 представляет собой катион металла; y представляет собой валентность катиона; z представляет собой целое положительное число; b равняется нулю или целому положительному числу; если b представляет собой целое положительное число, каждый Q1 представляет собой отрицательно заряженный противоион, и а представляет собой валентность отрицательно заряженного противоиона; а значения v, x, y, z, a и b удовлетворяют уравнению (vx)+(ab)=yz; при этом циклическая часть циклоалкильной группы или замещенной циклоалкильной группы содержит не более двух кольцевых структур, сконденсированных вместе, если L представляет собой двухвалентную связующую группу, v равняется 1, а R2 представляет собой циклоалкильную группу или замещенную циклоалкильную группу.
Согласно второму варианту осуществления настоящее изобретение обеспечивает соединение, соответствующее структуре формулы (С)
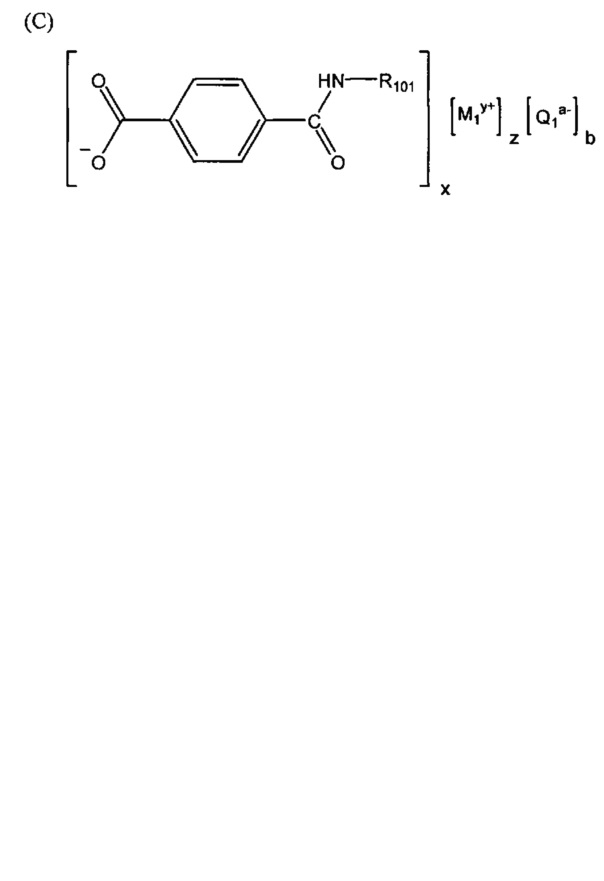 ,
,
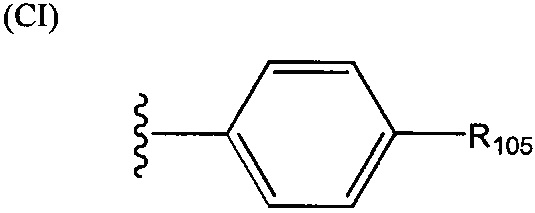
где R101 выбрана из группы, состоящей из циклопентильной группы и фрагментов, соответствующих структуре формулы (CI); формула (CI) представляет собой
 ,
,
R105 выбрана из группы, состоящей из водорода и галогенов; x представляет собой целое положительное число; каждый M1 представляет собой катион металла; y представляет собой валентность катиона; z представляет собой целое положительное число; b равняется нулю или целому положительному числу; если b представляет собой целое положительное число, каждый Q1 представляет собой отрицательно заряженный противоион, и а представляет собой валентность отрицательно заряженного противоиона; а значения x, y, z, а и b удовлетворяют уравнению х+(ab)=yz.
Согласно третьему варианту осуществления настоящее изобретение обеспечивает соединение, соответствующее структуре формулы (СХ)
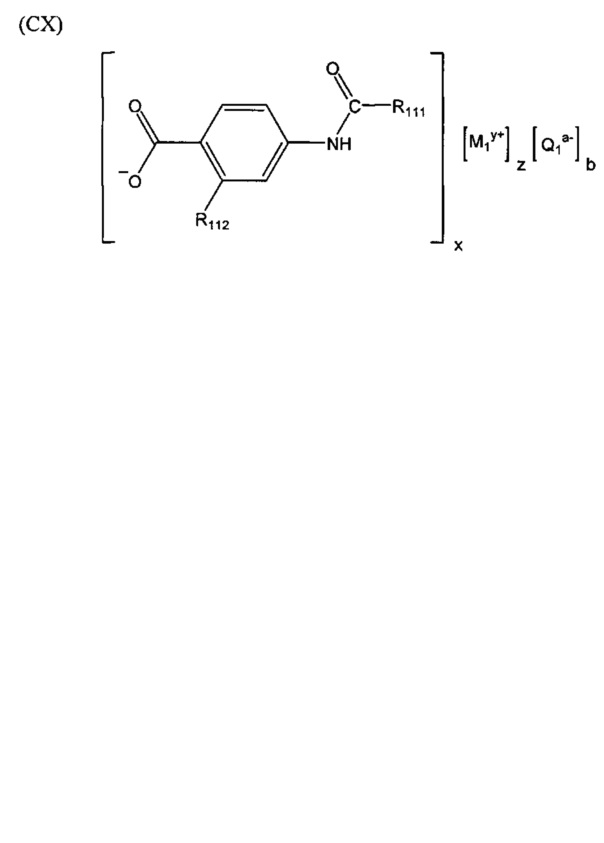 ,
,
где R111 выбрана из группы, состоящей из циклопентильной группы и фрагментов, соответствующих структуре формулы (CXI); R112 выбрана из группы, состоящей из водорода и гидроксигруппы, при этом формула (CXI) представляет собой
 ,
,
R115 выбрана из группы, состоящей из водорода, галогена, метоксигруппы и фенила; x представляет собой целое положительное число; каждый M1 представляет собой катион металла; y представляет собой валентность катиона; z представляет собой целое положительное число; b равняется нулю или целому положительному числу; если b представляет собой целое положительное число, каждый Q1 представляет собой отрицательно заряженный противоион, и a представляет собой валентность отрицательно заряженного противоиона; а значения x, y, z, а и b удовлетворяют уравнению х+(ab)=yz; при условии, что, если R112 представляет собой водород, тогда R115 представляет собой водород, x равняется 1, M1 представляет собой катион лития, y равняется 1, z равняется 1, а b равняется нулю; и при условии, что, если представляет собой метоксигруппу, тогда R112 представляет собой гидроксигруппу.
Согласно четвертому варианту осуществления настоящее изобретение обеспечивает соединение, соответствующее структуре формулы (СХХ)
 ,
,
где x представляет собой целое положительное число; каждый M1 представляет собой катион металла, выбранного из группы, состоящей из щелочных металлов, щелочноземельных металлов и цинка; y представляет собой валентность катиона; z представляет собой целое положительное число; b равняется нулю или целому положительному числу; если b представляет собой целое положительное число, каждый Q1 представляет собой отрицательно заряженный противоион, и a представляет собой валентность отрицательно заряженного противоиона; а значения x, y, z, а и b удовлетворяют уравнению х+(ab)=yz.
Подробное раскрытие настоящего изобретения
Следующие определения представлены для обозначения некоторых выражений, используемых во всей настоящей заявке.
При использовании в настоящем документе выражение "замещенные алкильные группы" относится к одновалентным функциональным группам, полученным из замещенных алканов путем отщепления атома водорода от атома углерода алкана. В данном определении выражение "замещенные алканы" относится к соединениям, полученным из ациклических неразветвленных и разветвленных углеводородов, в которых (1) один или несколько атомов водорода углеводорода замещены не являющимся водородом атомом (например, атомом галогена) или неалкильной функциональной группой (например, гидроксигруппой, арильной группой или гетероарильной группой), и/или (2) углерод-углеродная цепочка углеводорода прерывается атомом кислорода (как в эфире), атомом азота (как в амине) или атомом серы (как в сульфиде).
При использовании в настоящем документе выражение "замещенные циклоалкильные группы" относится к одновалентным функциональным группам, полученным из замещенных циклоалканов путем отщепления атома водорода от атома углерода циклоалкана. В данном определении выражение "замещенные циклоалканы" относится к соединениям, полученным из насыщенных моноциклических и полициклических углеводородов (с боковыми цепочками или без них), в которых (1)один или несколько атомов водорода углеводорода замещены не являющимся водородом атомом (например, атомом галогена) или неалкильной функциональной группой (например, гидроксигруппой, арильной группой или гетероарильной группой), и/или (2) углерод-углеродная цепочка углеводорода прерывается атомом кислорода, атомом азота или атомом серы.
При использовании в настоящем документе выражение "замещенные алкоксигруппы" относится к одновалентным функциональным группам, полученным из замещенных гидроксиалканов путем отщепления атома водорода от гидроксигруппы. В данном определении выражение "замещенные гидроксиалканы" относится к соединениям с одной или несколькими гидроксигруппами, присоединенными к замещенному алкану, а выражение "замещенный алкан" определено как указано выше в определении замещенных алкильных групп.
При использовании в настоящем документе выражение "замещенные арильные группы" относится к одновалентным функциональным группам, полученным из замещенных аренов путем отщепления атома водорода от атома углерода кольца. В данном определении выражение "замещенные арены" относится к соединениям, полученным из моноциклических и полициклических ароматических углеводородов, в которых один или несколько атомов водорода углеводорода замещены не являющимся водородом атомом (например, атомом галогена) или неалкильной функциональной группой (например, гидроксигруппой).
При использовании в настоящем документе выражение "замещенные гетероарильные группы" относится к одновалентным функциональным группам, полученным из замещенных гетероаренов путем отщепления атома водорода от атома кольца. В данном определении выражение "замещенные гетероарены" относится к соединениям, полученным из моноциклических и полициклических ароматических углеводородов, в которых (1) один или несколько атомов водорода углеводорода замещены не являющимся водородом атомом (например, атомом галогена) или неалкильной функциональной группой (например, гидроксигруппой), и (2) по меньшей мере одна метановая группа (-C=) углеводорода замещена трехвалентным гетероатомом, и/или по меньшей мере одна винилиденовая группа (-CH=CH-) углеводорода замещена двухвалентным гетероатомом.
При использовании в настоящем документе выражение "алкандиильные группы" относится к двухвалентным функциональным группам, полученным из алканов путем отщепления двух атомов водорода от алкана. Эти атомы водорода можно отщеплять от одного и того же атома углерода в алкане (как в этан-1,1-дииле) или от различных атомов углерода (как в этан-1,2-дииле).
При использовании в настоящем документе выражение "замещенные алкандиильные группы" относится к двухвалентным функциональным группам, полученным из замещенных алканов путем отщепления двух атомов водорода от алкана. Эти атомы водорода можно отщеплять от одного и того же атома углерода в замещенном алкане (как в 2-фторэтан-1,1-дииле) или от различных атомов углерода (как в 1-фторэтан-1,2-дииле). В данном определении выражение "замещенные алканы" имеет такое же значение, как указано выше в определении замещенных алкильных групп.
При использовании в настоящем документе выражение "циклоалкандиильные группы" относится к двухвалентным функциональным группам, полученным из циклоалканов путем отщепления двух атомов водорода от циклоалкана. Эти атомы водорода можно отщеплять от одного и того же атома углерода в циклоалкане или от различных атомов углерода.
При использовании в настоящем документе выражение "замещенные циклоалкандиильные группы" относится к двухвалентным функциональным группам, полученным из замещенных циклоалканов путем отщепления двух атомов водорода от алкана. В данном определении выражение "замещенные циклоалканы" имеет такое же значение, как указано выше в определении замещенных циклоалкильных групп.
При использовании в настоящем документе выражение "арендиильные группы" относится к двухвалентным функциональным группам, полученным из аренов (моноциклических и полициклических ароматических углеводородов) путем отщепления двух атомов водорода от атомов углерода кольца.
При использовании в настоящем документе выражение "замещенные арендиильные группы" относится к двухвалентным функциональным группам, полученным из замещенных аренов путем отщепления двух атомов водорода от атомов углерода кольца. В данном определении выражение "замещенные арены" относится к соединениям, полученным из моноциклических и полициклических ароматических углеводородов, в которых один или несколько атомов водорода углеводорода замещены не являющимся водородом атомом (например, атомом галогена) или неалкильной функциональной группой (например, гидроксигруппой).
При использовании в настоящем документе выражение "гетероарендиильные группы" относится к двухвалентным функциональным группам, полученным из гетероаренов путем отщепления двух атомов водорода от атомов кольца. В данном определении выражение "гетероарены" относится к соединениям, полученным из моноциклических и полициклических ароматических углеводородов, в которых по меньшей мере одна метановая группа (-C=) углеводорода замещена трехвалентным гетероатомом, и/или по меньшей мере одна винилиденовая группа (-CH=CH-) углеводорода замещена двухвалентным гетероатомом.
При использовании в настоящем документе выражение "замещенные гетероарендиильные группы" относится к двухвалентным функциональным группам, полученным из замещенных гетероаренов путем отщепления двух атомов водорода от атомов кольца. В данном определении выражение "замещенные гетероарены" имеет такое же значение, как указано выше в определении замещенных гетероарильных групп.
При использовании в настоящем документе выражение "алкантриильные группы" относится к трехвалентным функциональным группам, полученным из алканов путем отщепления трех атомов водорода от алкана. Эти атомы водорода можно отщеплять от одного и того же атома углерода в алкане или от различных атомов углерода.
При использовании в настоящем документе выражение "замещенные алкантриильные группы" относится к трехвалентным функциональным группам, полученным из замещенных алканов путем отщепления трех атомов водорода от алкана. Эти атомы водорода можно отщеплять от одного и того же атома углерода в замещенном алкане или от различных атомов углерода. В данном определении выражение "замещенные алканы" имеет такое же значение, как указано выше в определении замещенных алкильных групп.
При использовании в настоящем документе выражение "циклоалкантриильные группы" относится к трехвалентным функциональным группам, полученным из циклоалканов путем отщепления трех атомов водорода от циклоалкана.
При использовании в настоящем документе выражение "замещенные циклоалкантриильные группы" относится к трехвалентным функциональным группам, полученным из замещенных циклоалканов путем отщепления трех атомов водорода от алкана. В данном определении выражение "замещенные циклоалканы" имеет такое же значение, как указано выше в определении замещенных циклоалкильных групп.
При использовании в настоящем документе выражение "арентриильные группы" относится к трехвалентным функциональным группам, полученным из аренов (моноциклических и полициклических ароматических углеводородов) путем отщепления трех атомов водорода от атомов углерода кольца.
При использовании в настоящем документе выражение "замещенные арентриильные группы" относится к трехвалентным функциональным группам, полученным из замещенных аренов путем отщепления трех атомов водорода от атомов углерода кольца. В данном определении выражение "замещенные арены" имеет такое же значение, как указано выше в определении замещенных арендиильных групп.
При использовании в настоящем документе выражение "гетероарентриильные группы" относится к трехвалентным функциональным группам, полученным из гетероаренов путем отщепления трех атомов водорода от атомов кольца. В данном определении выражение "гетероарены" имеет такое же значение, как указано выше в определении гетероарендиильных групп.
При использовании в настоящем документе выражение "замещенные гетероарентриильные группы" относится к трехвалентным функциональным группам, полученным из замещенных гетероаренов путем отщепления трех атомов водорода от атомов кольца. В данном определении выражение "замещенные гетероарены" имеет такое же значение, как указано выше в определении замещенных гетероарильных групп.
Согласно первому варианту осуществления настоящее изобретение обеспечивает композицию на основе термопластичного полимера, содержащую термопластичный полимер и зародышеобразователь. Термопластичный полимер композиции на основе термопластичного полимера может представлять собой любой подходящий термопластичный полимер. При использовании в настоящем документе выражение "термопластичный полимер" используют для обозначения полимерного материала, который будет плавиться под воздействием достаточного тепла с образованием текучей жидкости и будет возвращаться в затвердевшее состояние при достаточном охлаждении. В своем затвердевшем состоянии такие термопластичные полимеры характеризуются или кристаллической, или частично кристаллической морфологией. Подходящие термопластичные полимеры включают, помимо прочего, полиолефины (например, полиэтилены, полипропилены, полибутилены и любые их комбинации), полиамиды (например, нейлон), полиуретаны, сложные полиэфиры (например, полиэтилентерефталат) и подобное, а также любые их комбинации. Эти термопластичные полимеры могут находиться в виде порошка, пыли, крошки, гранул или дробинок, полученных из свежеприготовленного полимера, повторно измельченного полимера, отходов после использования продуктов и изделий или промышленных отходов.
Согласно некоторым вариантам осуществления термопластичный полимер может представлять собой полиолефин, такой как полипропилен, полиэтилен, полибутилен, поли(4-метил-1-пентен) и поли(винилциклогексан). Согласно предпочтительному варианту осуществления термопластичный полимер представляет собой полиолефин, выбранный из группы, состоящей из гомополимеров полипропилена (например, атактического полипропилена, изотактического полипропилена и синдиотактического полипропилена), сополимеров полипропилена (например, статистических сополимеров полипропилена), ударопрочных сополимеров полипропилена, полиэтилена, сополимеров полиэтилена, полибутилена, поли(4-метил-1-пентена) и их смесей. Подходящие сополимеры полипропилена включают, помимо прочего, статистические сополимеры, полученные полимеризацией пропилена в присутствии сомономера, выбранного из группы, состоящей из этилена, бут-1-ена (т.е. 1-бутена) и гекс-1-ена (т.е. 1-гексена). В таких статистических сополимерах полипропилена сомономер может присутствовать в любом подходящем количестве, но обычно присутствует в количестве менее чем приблизительно 10 масс. % (например, от приблизительно 1 до приблизительно 7 масс. %). Подходящие ударопрочные сополимеры полипропилена включают, помимо прочего, полученные добавлением сополимера, выбранного из группы, состоящей из этиленпропиленового каучука (EPR), этиленпропилендиенового мономера (EPDM), полиэтилена и пластомеров, в гомополимер полипропилена или статистический сополимер полипропилена. В таких ударопрочных сополимерах полипропилена сомономер может присутствовать в любом подходящем количестве, но обычно присутствует в количестве от приблизительно 5 до приблизительно 25 масс. %.
Согласно другому предпочтительному варианту осуществления термопластичный полимер может представлять собой полиэтилен. Подходящие полиэтилены включают, помимо прочего, полиэтилен низкой плотности, линейный полиэтилен низкой плотности, полиэтилен средней плотности, полиэтилен высокой плотности и их комбинации. Согласно некоторым предпочтительным вариантам осуществления термопластичный полимер выбран из группы, состоящей из линейного полиэтилена низкой плотности, полиэтилена высокой плотности и их смесей. Согласно другому предпочтительному варианту осуществления термопластичный полимер представляет собой полиэтилен высокой плотности.
Полиэтиленовые полимеры высокой плотности, подходящие для использования в настоящем изобретении, обычно характеризуются плотностью больше чем приблизительно 0,940 г/см3. Нет верхнего предела для подходящей плотности полимера, но полиэтиленовые полимеры высокой плотности обычно характеризуются плотностью, которая меньше чем приблизительно 0,980 г/см3 (например, меньше чем приблизительно 0,975 г/см3).
Полиэтиленовые полимеры высокой плотности, подходящие для использования в настоящем изобретении, могут представлять собой или гомополимеры, или сополимеры этилена с одним или несколькими α-олефинами. Подходящие α-олефины включают, помимо прочего, 1-бутен, 1-гексен, 1-октен, 1-децен и 4-метил-1-пентен. Сомономер может присутствовать в сополимере в любом подходящем количестве, например, в количестве приблизительно 5 масс. % или менее (например, приблизительно 3 мольн. % или менее). Специалистам в данной области техники будет понятно, что количество сомономера, подходящее для сополимера, в значительной степени обусловлено конечным использованием сополимера и требуемыми или желаемыми свойствами полимера, обусловленными таким конечным использованием.
Полиэтиленовые полимеры высокой плотности, подходящие для использования в настоящем изобретении, можно получать любым подходящим способом. Например, полимеры можно получать свободнорадикальным способом при помощи очень высоких давлений, как описано, например, в патенте США №2816883 (Larchar и соавт.), но обычно полимеры получают каталитическим способом под "низким давлением". В данном контексте выражение "низкое давление" используют для обозначения способов, проводимых под давлениями менее 6,9 МПа (например, 1000 фунтов на кв. дюйм), таких как 1,4-6,9 МПа (200-1000 фунтов на кв. дюйм). Примеры подходящих каталитических процессов под низким давлением включают, помимо прочего, процессы полимеризации в растворе (т.е. процессы, в которых полимеризацию проводят при помощи растворителя для полимера), процессы суспензионной полимеризации (т.е. процессы, в которых полимеризацию проводят при помощи углеводородной жидкости, в которой полимер не растворяется или не набухает), процессы газофазной полимеризации (например, процессы, в которых полимеризацию проводят без использования жидкой среды или разбавителя) или процессы полимеризации с многоступенчатыми реакторами. Подходящие процессы газофазной полимеризации также включают так называемые процессы "в конденсированном режиме" или "сверхконденсированном режиме", в которых жидкий углеводород вводят в псевдоожиженный слой для повышения поглощения тепла, образующегося в процессе полимеризации. В этих процессах в конденсированном режиме и сверхконденсированном режиме жидкий углеводород обычно конденсируется в рециркуляционном потоке и повторно используется в реакторе. В процессах с многоступенчатыми реакторами можно использовать комбинацию реакторов для суспензионного процесса (корпусных или петлевых реакторов), которые соединены последовательно, параллельно или комбинацией последовательного или параллельного соединения так, что катализатор (например, хромовый катализатор) подвергается действию не более одного набора реакционных условий. Процессы с многоступенчатыми реакторами можно также проводить путем последовательного соединения двух петлевых реакторов, последовательного соединения одного или нескольких корпусных и петлевых реакторов, путем использования множества последовательно соединенных газофазных реакторов или при помощи конфигурации петлевой-газофазный реактор. Из-за возможности подвергать катализатор в них различным наборам реакционных условий процессы с многоступенчатыми реакторами часто используют для получения полимодальных полимеров, таких как обсуждаемые ниже. Подходящие процессы также включают такие, в которых проводят стадию предварительной полимеризации. На этой стадии предварительной полимеризации катализатор обычно подвергают действию сокатализатора и этилена при мягких условиях в меньшем отдельном реакторе, и реакции полимеризации позволяют продолжаться до тех пор, пока катализатор не будет содержать относительно небольшое количество (например, от приблизительно 5% до приблизительно 30% общей массы) полученной композиции. Этот предварительно полимеризованный катализатор затем вводят в промышленный реактор, в котором должна проводиться полимеризация.
Полиэтиленовые полимеры высокой плотности, подходящие для использования в настоящем изобретении, можно получать при помощи любого подходящего катализатора или комбинации катализаторов. Подходящие катализаторы включают катализаторы на основе переходных металлов, такие как восстановленный оксид молибдена на подложке, молибдат кобальта на оксиде алюминия, оксид хрома и галогениды переходных металлов. Хромоксидные катализаторы обычно получают пропиткой соединением хрома пористого оксидного носителя с высокой удельной поверхностью, такого как диоксид кремния, а затем прокаливанием его в сухом воздухе при 500-900°C. Это превращает хром в шестивалентный поверхностный сложный эфир хромата или сложный эфир дихромата. Хромоксидные катализаторы можно использовать совместно с сокатализаторами на основе алкильных соединений металлов, такими как алкилбор, алкилалюминий, алкилцинк и алкиллитий. Носители для оксида хрома включают диоксид кремния, диоксид кремния-диоксид титана, диоксид кремния-оксид алюминия, оксид алюминия и алюмофосфаты. Дополнительные примеры хромоксидных катализаторов включают катализаторы, получаемые осаждением низковалентного хроморганического соединения, такого как бис(арен)Cr0, аллил CR2+ и Cr3+, бета-стабилизированные алкилы CR2+ и Cr4+ и бис(циклопентадиенил)CR2+, на хромоксидный катализатор, такой как описанные выше. Подходящие катализаторы на основе переходных металлов также включают хромовые катализаторы на носителе, такие как катализаторы на основе хромоцена или силилхромата (например, би(трисфенилсилил)хромат). Эти хромовые катализаторы можно наносить на любой подходящий носитель с высокой удельной поверхностью, как, например, описанные выше для хромоксидных катализаторов, при этом обычно используют диоксид кремния. Хромовые катализаторы на носителе можно также использовать совместно с сокатализаторами, такими как сокатализаторы на основе алкильных соединений металлов, перечисленные выше для хромоксидных катализаторов. Подходящие катализаторы на основе галогенидов переходных металлов включают галогениды титана (III) (например, хлорид титана (III)), галогениды титана (IV) (например, хлорид титана (IV)), галогениды ванадия, галогениды циркония и их комбинации. Эти галогениды переходных металлов часто наносят на твердые вещества с высокой удельной поверхностью, такие как хлорид магния. Катализаторы на основе галогенидов переходных металлов обычно используют совместно с сокатализатором на основе алкильного соединения алюминия, такого как триметилалюминий (т.е. Al(CH3)3) или триэтилалюминий (т.е. Al(С2Н5)3). Эти галогениды переходных металлов можно также использовать в процессах с многоступенчатыми реакторами. Подходящие катализаторы также включают металлоценовые катализаторы, такие как галогениды циклопентадиенилтитана (например, хлориды циклопентадиенилтитана), галогениды циклопентадиенилциркония (например, хлориды циклопентадиенилциркония), галогениды циклопентадиенилгафния (например, хлориды циклопентадиенилгафния) и их комбинации. Металлоценовые катализаторы на основе переходных металлов, образующих комплексы с инденильными или флуоренильными лигандами, также известны и могут использоваться для получения полиэтиленовых полимеров высокой плотности, подходящих для использования в настоящем изобретении. Катализаторы обычно содержат множество лигандов, и лиганды могут быть замещены различными группами (например, н-бутильной группой) или связаны при помощи мостиковых групп, таких как -CH2CH2- или >SiPh2. Металлоценовые катализаторы обычно используют совместно с сокатализатором, таким как метилалюмоксан (т.е. (Al(CH3)xOy)n). Другие сокатализаторы включают сокатализаторы, описанные в патенте США №5919983 (Rosen и соавт.), патенте США №6107230 (McDaniel и соавт.), патенте США №6632894 (McDaniel и соавт.) и патенте США №6300271 (McDaniel и соавт). Другие "одноцентровые" катализаторы, подходящие для использования при получении полиэтилена высокой плотности, включают дииминные комплексы, такие как описанные в патенте США №5891963 (Brookhart и соавт.).
Полиэтиленовые полимеры высокой плотности, подходящие для использования в настоящем изобретении, могут характеризоваться любой подходящей молекулярной массой (например, среднемассовой молекулярной массой). Например, среднемассовая молекулярная масса полиэтилена высокой плотности может составлять от 20000 г/моль до приблизительно 1000000 г/моль или более. Как будет понятно специалистам в данной области техники, подходящая среднемассовая молекулярная масса полиэтилена высокой плотности будет зависеть, по меньшей мере, частично от конкретного применения или конечного использования, для которого полимер предназначен. Например, полиэтиленовый полимер высокой плотности, предназначенный для применений в выдувном формовании, может характеризоваться среднемассовой молекулярной массой от приблизительно 100000 г/моль до приблизительно 1000000 г/моль. Полиэтиленовый полимер высокой плотности, предназначенный для применений для труб или применений для пленки, может характеризоваться среднемассовой молекулярной массой от приблизительно 100000 г/моль до приблизительно 500000 г/моль. Полиэтиленовый полимер высокой плотности, предназначенный для применений в литье под давлением, может характеризоваться среднемассовой молекулярной массой от приблизительно 20000 г/моль до приблизительно 80000 г/моль. Полиэтиленовый полимер высокой плотности, предназначенный для применений для изоляции проводов, применений для изоляции кабелей, применений для лент или применений для нитей, может характеризоваться среднемассовой молекулярной массой от приблизительно 80000 г/моль до приблизительно 400000 г/моль. Полиэтиленовый полимер высокой плотности, предназначенный для применений в центробежном формовании, может характеризоваться среднемассовой молекулярной массой от приблизительно 50000 г/моль до приблизительно 150000 г/моль.
Полиэтиленовые полимеры высокой плотности, подходящие для использования в настоящем изобретении, могут также характеризоваться любой подходящей полидисперсностью, которую определяют как значение, полученное путем деления среднемассовой молекулярной массы полимера на среднечисленную молекулярную массу полимера. Например, полиэтиленовый полимер высокой плотности может характеризоваться полидисперсностью от более 2 до приблизительно 100. Как понимается специалистами в данной области техники, на полидисперсность полимера сильно влияет каталитическая система, используемая для получения полимера, причем металлоцен или другие "одноцентровые" катализаторы обычно дают полимеры с относительно низкой полидисперсностью и узкими распределениями молекулярных масс, а другие катализаторы на основе переходных металлов (например, хромовые катализаторы) дают полимеры с более высокой полидисперсностью и более широким распределением молекулярных масс. Полиэтиленовые полимеры высокой плотности, подходящие для использования в настоящем изобретении, могут также характеризоваться полимодальным (например, бимодальным) распределением молекулярной массы. Например, полимер может иметь первую фракцию, характеризующуюся относительно низкой молекулярной массой, и вторую фракцию, характеризующуюся относительно высокой молекулярной массой. Разница между среднемассовой молекулярной массой фракций в полимере может представлять собой любое подходящее значение. На самом деле не обязательно, чтобы разница между среднемассовыми молекулярными массами была настолько большой, чтобы две отдельные молекулярно-весовые фракции можно было разделять при помощи гельпроникающей хроматографии (ГПХ). Однако, в некоторых полимодальных полимерах разница между среднемассовыми молекулярными массами фракций может быть достаточно большой, чтобы два или более отдельных пиков можно было выделить на кривой ГПХ для полимера. В данном случае выражение "отдельный" не обязательно означает, что части кривой ГПХ, соответствующие каждой фракции, не перекрываются, а подразумевает только индикацию того, что отдельный пик для каждой фракции можно выделить на кривой ГПХ для полимера. Полимодальные полимеры, подходящие для использования в настоящем изобретении, можно получать при помощи любого подходящего способа. Как указано выше, полимодальные полимеры можно получать при помощи процессов с многоступенчатыми реакторами. Одним подходящим примером будет многоступенчатый процесс в среде растворителя, включающий ряд смесителей. Альтернативно, полимодальные полимеры можно получать в одном реакторе при помощи комбинации катализаторов, каждый из которых разработан для получения полимера, характеризующегося различной среднемассовой молекулярной массой.
Полиэтиленовые полимеры высокой плотности, подходящие для использования в настоящем изобретении, могут характеризоваться любым подходящим индексом расплава. Например, полиэтиленовый полимер высокой плотности может характеризоваться индексом расплава от приблизительно 0,01 дг/мин до приблизительно 40 дг/мин. Как и в случае среднемассовой молекулярной массы, специалистам в данной области техники будет понятно, что подходящий индекс расплава полиэтиленового полимера высокой плотности будет зависеть, по меньшей мере, частично от конкретного применения или конечного использования, для которого полимер предназначен. Таким образом, например, полиэтиленовый полимер высокой плотности, предназначенный для применений в выдувном формовании, может характеризоваться индексом расплава от приблизительно 0,01 дг/мин до приблизительно 1 дг/мин. Полиэтиленовый полимер высокой плотности, предназначенный для применений для пленки, получаемой экструзией с раздувом, может характеризоваться индексом расплава от приблизительно 0,5 дг/мин до приблизительно 3 дг/мин. Полиэтиленовый полимер высокой плотности, предназначенный для применений для получаемой поливом пленки, может характеризоваться индексом расплава от приблизительно 2 дг/мин до приблизительно 10 дг/мин. Полиэтиленовый полимер высокой плотности, предназначенный для применений для труб, может характеризоваться индексом расплава от приблизительно 2 дг/мин до приблизительно 40 дг/мин. Полиэтиленовый полимер высокой плотности, предназначенный для применений для литья под давлением, может характеризоваться индексом расплава от приблизительно 2 дг/мин до приблизительно 80 дг/мин. Полиэтиленовый полимер высокой плотности, предназначенный для применений для центробежного формования, может характеризоваться индексом расплава от приблизительно 0,5 дг/мин до приблизительно 10 дг/мин. Полиэтиленовый полимер высокой плотности, предназначенный для применений для лент, может характеризоваться индексом расплава от приблизительно 0,2 дг/мин до приблизительно 4 дг/мин. Полиэтиленовый полимер высокой плотности, предназначенный для применений для нитей, может характеризоваться индексом расплава от приблизительно 1 дг/мин до приблизительно 20 дг/мин. Индекс расплава полимера измеряют при помощи стандарта ASTM D1238-04c.
Полиэтиленовые полимеры высокой плотности, подходящие для использования в настоящем изобретении, обычно не содержат высокие степени длинноцепочечной разветвленности. Выражение "длинноцепочечная разветвленность" используют для обозначения ответвлений, которые присоединены к полимерной цепи и имеют достаточную длину для влияния на реологию полимера (например, ответвления длиной приблизительно 130 углеродов или более). Если необходимо для применения, в котором полимер будут использовать, полиэтиленовый полимер высокой плотности может содержать небольшие степени длинноцепочечной разветвленности. Однако, полиэтиленовые полимеры высокой плотности, подходящие для использования в настоящем изобретении, обычно содержат очень небольшую степень длинноцепочечной разветвленности (например, менее чем приблизительно 1 длинноцепочечное ответвление на 10000 углеродов, менее чем приблизительно 0,5 длинноцепочечного ответвления на 10000 углеродов, менее чем приблизительно 0,1 длинноцепочечного ответвления на 10000 углеродов или менее чем приблизительно 0,01 длинноцепочечного ответвления на 10000 углеродов).
Полиэтиленовые полимеры средней плотности, подходящие для использования в настоящем изобретении, обычно характеризуются плотностью от приблизительно 0,926 г/см3 до приблизительно 0,940 г/см3. Выражение "полиэтилен средней плотности" используют для обозначения полимеров этилена, которые характеризуются плотностью между плотностью полиэтилена высокой плотности и плотностью линейного полиэтилена низкой плотности и содержат относительно короткие ответвления, по меньшей мере, по сравнению с длинными ответвлениями, находящимися в полиэтиленовых полимерах низкой плотности, полученных свободнорадикальной полимеризацией этилена под высокими давлениями.
Полиэтиленовые полимеры средней плотности, подходящие для использования в настоящем изобретении, обычно представляют собой сополимеры этилена и по меньшей мере одного α-олефина, такого как 1 бутен, 1-гексен, 1-октен, 1-децен и 4-метил-1-пентен. α-Олефиновый сомономер может присутствовать в любом подходящем количестве, но обычно присутствует в количестве менее чем приблизительно 8 масс. % (например, менее чем приблизительно 5 мольн. %). Специалистам в данной области техники будет понятно, что количество сомономера, подходящее для сополимера, в значительной степени обусловлено конечным использованием сополимера и требуемыми или желаемыми свойствами полимера, обусловленными таким конечным использованием.
Полиэтиленовые полимеры средней плотности, подходящие для использования в настоящем изобретении, можно получать любым подходящим способом. Аналогично полиэтиленовым полимерам высокой плотности, полиэтиленовые полимеры средней плотности обычно получают каталитическими процессами под "низким давлением", такими как любые из процессов, описанных выше касательно полиэтиленовых полимеров высокой плотности, подходящих для использования в настоящем изобретении. Примеры подходящих процессов включают, помимо прочего, процессы газофазной полимеризации, процессы полимеризации в растворе, процессы суспензионной полимеризации и процессы с многоступенчатыми реакторами. Подходящие процессы с многоступенчатыми реакторами могут включать любую подходящую комбинацию процессов газофазной полимеризации, полимеризации в растворе и суспензионной полимеризации, описанных выше. Как и в случае полиэтиленовых полимеров высокой плотности, процессы с многоступенчатыми реакторами часто используют для получения полимодальных полимеров.
Полиэтиленовые полимеры средней плотности, подходящие для использования в настоящем изобретении, можно получать при помощи любого подходящего катализатора или комбинации катализаторов. Например, полимеры можно получать при помощи катализаторов Циглера, таких как галогениды или сложные эфиры переходных металлов (например, титана), используемые в комбинации с алюмоорганическими соединениями (например, триэтилалюминием). Эти катализаторы Циглера могут быть нанесены, например, на хлорид магния, диоксид кремния, оксид алюминия или оксид магния. Полиэтиленовые полимеры средней плотности, подходящие для использования в настоящем изобретении, можно также получать при помощи так называемых "двойных катализаторов Циглера", которые содержат один вид катализатора для димеризации этилена в 1-бутен (например, комбинацию сложного эфира титана и триэтилалюминия) и другой катализатор для сополимеризации этилена и образовавшегося 1-бутена (например, хлорид титана на носителе из хлорида магния). Полиэтиленовые полимеры средней плотности, подходящие для использования в настоящем изобретении, можно также получать при помощи хромоксидных катализаторов, таких как катализаторы, получаемые осаждением соединения хрома на носитель из диоксида кремния-диоксида титана, окислением полученного катализатора в смеси кислорода и воздуха, а затем восстановлением катализатора монооксидом углерода. Эти хромоксидные катализаторы обычно используют совместно с сокатализаторами, такими как соединения триалкилбора или триалкилалюминия. Хромоксидные катализаторы можно также использовать совместно с катализатором Циглера, таким как катализатор на основе галогенида титана или сложного эфира титана. Полиэтиленовые полимеры средней плотности, подходящие для использования в настоящем изобретении, можно также получать при помощи хромовых катализаторов на носителе, таких как описанные выше при обсуждении катализаторов, подходящих для получения полиэтилена высокой плотности. Полиэтиленовые полимеры средней плотности, подходящие для использования в настоящем изобретении, можно также получать при помощи металлоценовых катализаторов. Несколько различных типов металлоценовых катализаторов можно использовать. Например, металлоценовый катализатор может содержать бис(металлоценовый) комплекс циркония, титана или гафния с двумя циклопентадиенильными кольцами и метилалюмоксаном. Как и в катализаторах, используемых при получении полиэтилена высокой плотности, лиганды могут быть замещены различными группами (например, н-бутильной группой) или присоединены при помощи мостиковых групп. Другой класс металлоценовых катализаторов, который можно использовать, состоит из бис(металлоценовых) комплексов циркония или титана и анионов перфторированных борароматических соединений. Третий класс металлоценовых катализаторов, который можно использовать, называется катализаторами со стесненной геометрией и содержит моноциклопентадиенильные производные титана или циркония, в которых один из атомов углерода в циклопентадиенильном кольце связан с атомом металла при помощи мостиковой группы. Эти комплексы активируют путем их реакции с метилалюмоксаном или путем образования ионных комплексов с некоординационными анионами, такими как B(C6F5)4- или B(C6F5)3CH3-. Четвертый класс металлоценовых катализаторов, который можно использовать, представляет собой комплексы на основе металлоценов переходного металла, такого как титан, содержащие один циклопентадиенильный лиганд в комбинации с другим лигандом, таким как фосфинимин или -O-SiR3. Этот класс металлоценовых катализаторов также активируют метилалюмоксаном или соединением бора. Другие катализаторы, подходящие для использования в получении полиэтилена средней плотности, подходящего для использования в настоящем изобретении, включают, помимо прочего, катализаторы, раскрытые в патенте США №6649558.
Полиэтиленовые полимеры средней плотности, подходящие для использования в настоящем изобретении, могут характеризоваться любой подходящей композиционной однородностью, что является выражением, используемым для описания однородности разветвленности в молекулах сополимера полимера. Многие коммерчески доступные полиэтиленовые полимеры средней плотности характеризуются относительно низкой композиционной однородностью, в случае чего высокомолекулярная фракция полимера содержит относительно небольшое количество α-олефинового сомономера и характеризуется относительно низкой степенью разветвленности, тогда как низкомолекулярная фракция полимера содержит относительно высокое количество α-олефинового сомономера и характеризуется относительно высокой степенью разветвленности. Альтернативно, другой ряд полиэтиленовых полимеров средней плотности характеризуется относительно низкой композиционной однородностью, в случае чего высокомолекулярная фракция полимера содержит относительно большое количество α-олефинового сомономера, тогда как низкомолекулярная фракция полимера содержит относительно небольшое количество α-олефинового сомономера. Композиционную однородность полимера можно измерить при помощи любого подходящего способа, такого как фракционирование посредством элюирования при повышении температуры.
Полиэтиленовые полимеры средней плотности, подходящие для использования в настоящем изобретении, могут характеризоваться любой подходящей молекулярной массой. Например, полимер может характеризоваться среднемассовой молекулярной массой от приблизительно 50000 г/моль до приблизительно 200000 г/моль. Как будет понятно специалистам в данной области техники, подходящая среднемассовая молекулярная масса полиэтилена средней плотности будет зависеть, по меньшей мере, частично от конкретного применения или конечного использования, для которого полимер предназначен.
Полиэтиленовые полимеры средней плотности, подходящие для использования в настоящем изобретении, могут также характеризоваться любой подходящей полидисперсностью. Многие коммерчески доступные полиэтиленовые полимеры средней плотности характеризуются полидисперсностью от приблизительно 2 до приблизительно 30. Полиэтиленовые полимеры средней плотности, подходящие для использования в настоящем изобретении, могут также характеризоваться полимодальным (например, бимодальным) распределением молекулярной массы. Например, полимер может иметь первую фракцию, характеризующуюся относительно низкой молекулярной массой, и вторую фракцию, характеризующуюся относительно высокой молекулярной массой. Как и в случае полиэтиленовых полимеров высокой плотности, подходящих для использования в настоящем изобретении, разница между среднемассовой молекулярной массой фракций в полимодальном полиэтиленовом полимере средней плотности может представлять собой любую подходящую величину. На самом деле не обязательно, чтобы разница между среднемассовыми молекулярными массами была настолько большой, чтобы две отдельные молекулярно-весовые фракции можно было разделять при помощи гельпроникающей хроматографии (ГПХ). Однако, в некоторых полимодальных полимерах разница между среднемассовыми молекулярными массами фракций может быть достаточно большой, чтобы два или более отдельных пиков можно было выделить на кривой ГПХ для полимера. В данном случае выражение "отдельный" не обязательно означает, что части кривой ГПХ, соответствующие каждой фракции, не перекрываются, а подразумевает только индикацию того, что отдельный пик для каждой фракции можно выделить на кривой ГПХ для полимера. Полимодальные полимеры, подходящие для использования в настоящем изобретении, можно получать при помощи любого подходящего способа. Как указано выше, полимодальные полимеры можно получать при помощи процессов с многоступенчатыми реакторами. Одним подходящим примером будет многоступенчатый процесс в среде растворителя, включающий ряд смесителей. Альтернативно, полимодальные полимеры можно получать в одном реакторе при помощи комбинации катализаторов, каждый из которых разработан для получения полимера, характеризующегося различной среднемассовой молекулярной массой.
Полиэтиленовые полимеры средней плотности, подходящие для использования в настоящем изобретении, могут характеризоваться любым подходящим индексом расплава. Например, полиэтиленовый полимер средней плотности может характеризоваться индексом расплава от приблизительно 0,01 дг/мин до приблизительно 200 дг/мин. Как и в случае среднемассовой молекулярной массы, специалистам в данной области техники будет понятно, что подходящий индекс расплава полиэтиленового полимера средней плотности будет зависеть, по меньшей мере, частично от конкретного применения или конечного использования, для которого полимер предназначен. Таким образом, например, полиэтиленовый полимер средней плотности, предназначенный для применений в выдувном формовании или применений для труб, может характеризоваться индексом расплава от приблизительно 0,01 дг/мин до приблизительно 1 дг/мин. Полиэтиленовый полимер средней плотности, предназначенный для применений для получаемой экструзией с раздувом пленки, может характеризоваться индексом расплава от приблизительно 0,5 дг/мин до приблизительно 3 дг/мин. Полиэтиленовый полимер средней плотности, предназначенный для применений для получаемой поливом пленки, может характеризоваться индексом расплава от приблизительно 2 дг/мин до приблизительно 10 дг/мин. Полиэтиленовый полимер средней плотности, предназначенный для применений для литья под давлением, может характеризоваться индексом расплава от приблизительно 6 дг/мин до приблизительно 200 дг/мин. Полиэтиленовый полимер средней плотности, предназначенный для применений для центробежного формования, может характеризоваться индексом расплава от приблизительно 4 дг/мин до приблизительно 7 дг/мин. Полиэтиленовый полимер средней плотности, предназначенный для применений для изоляции проводов и кабелей, может характеризоваться индексом расплава от приблизительно 0,5 дг/мин до приблизительно 3 дг/мин. Индекс расплава полимера измеряют при помощи стандарта ASTM D1238-04c.
Полиэтиленовые полимеры средней плотности, подходящие для использования в настоящем изобретении, обычно не содержат значительную степень длинноцепочечной разветвленности. Например, полиэтиленовые полимеры средней плотности, подходящие для использования в настоящем изобретении, обычно содержат менее чем приблизительно 0,1 длинноцепочечного ответвления на 10000 атомов углерода (например, менее чем приблизительно 0,002 длинноцепочечного ответвления на 100 этиленовых звеньев) или менее чем приблизительно 0,01 длинноцепочечного ответвления на 10000 атомов углерода.
Линейные полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, обычно характеризуются плотностью 0,925 г/см3 или менее (например, от приблизительно 0,910 г/см3 до приблизительно 0,925 г/см3). Выражение "линейный полиэтилен низкой плотности" используют для обозначения полимеров этилена низкой плотности, которые содержат относительно короткие ответвления, по меньшей мере, по сравнению с длинными ответвлениями, находящимися в полиэтиленовых полимерах низкой плотности, полученных свободнорадикальной полимеризацией этилена под высокими давлениями.
Линейные полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, обычно представляют собой сополимеры этилена и по меньшей мере одного α-олефина, такого как 1 бутен, 1-гексен, 1-октен, 1-децен и 4-метил-1-пентен. α-Олефиновый сомономер может присутствовать в любом подходящем количестве, но обычно присутствует в количестве менее чем приблизительно 6 мольн. % (например, от прибилизтельно 2 мольн. % до приблизительно 5 мольн. %). Специалистам в данной области техники будет понятно, что количество сомономера, подходящее для сополимера, в значительной степени обусловлено конечным использованием сополимера и требуемыми или желаемыми свойствами полимера, обусловленными таким конечным использованием.
Линейные полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, можно получать любым подходящим способом. Аналогично полиэтиленовым полимерам высокой плотности, линейные полиэтиленовые полимеры низкой плотности обычно получают каталитическими процессами под "низким давлением", такими как любые из процессов, описанных выше касательно полиэтиленовых полимеров высокой плотности, подходящих для использования в настоящем изобретении. Подходящие процессы включают, помимо прочего, процессы газофазной полимеризации, процессы полимеризации в растворе, процессы суспензионной полимеризации и процессы с многоступенчатыми реакторами. Подходящие процессы с многоступенчатыми реакторами могут включать любую подходящую комбинацию процессов газофазной полимеризации, полимеризации в растворе и суспензионной полимеризации, описанных выше. Как и в случае полиэтиленовых полимеров высокой плотности, процессы с многоступенчатыми реакторами часто используют для получения полимодальных полимеров.
Линейные полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, можно получать при помощи любого подходящего катализатора или комбинации катализаторов. Например, полимеры можно получать при помощи катализаторов Циглера, таких как галогениды или сложные эфиры переходных металлов (например, титана), используемые в комбинации с алюмоорганическими соединениями (например, триэтилалюминием). Эти катализаторы Циглера могут быть нанесены, например, на хлорид магния, диоксид кремния, оксид алюминия или оксид магния. Линейные полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, можно также получать при помощи так называемых "двойных катализаторов Циглера", которые содержат один вид катализатора для димеризации этилена в 1-бутен (например, комбинацию сложного эфира титана и триэтилалюминия) и другой катализатор для сополимеризации этилена и образованного 1-бутена (например, хлорид титана на носителе из хлорида магния). Линейные полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, можно также получать при помощи хромоксидных катализаторов, таких как катализаторы, получаемые осаждением соединения хрома на носитель из диоксида кремния-диоксида титана, окислением полученного катализатора в смеси кислорода и воздуха, а затем восстановлением катализатора монооксидом углерода. Эти хромоксидные катализаторы обычно используют совместно с сокатализаторами, такими как соединения триалкилбора или триалкилалюминия. Хромоксидные катализаторы можно также использовать совместно с катализатором Циглера, таким как катализатор на основе галогенида титана или сложного эфира титана. Линейные полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, можно также получать при помощи хромовых катализаторов на носителе, таких как описанные выше при обсуждении катализаторов, подходящих для получения полиэтилена высокой плотности. Линейный полиэтилен низкой плотности, подходящий для использования в настоящем изобретении, можно также получать при помощи металлоценовых катализаторов. Несколько различных типов металлоценовых катализаторов можно использовать. Например, металлоценовый катализатор может содержать бис(металлоценовый) комплекс циркония, титана или гафния с двумя циклопентадиенильными кольцами и метилалюмоксаном. Как и в катализаторах, используемых при получении полиэтилена высокой плотности, лиганды могут быть замещены различными группами (например, н-бутильной группой) или присоединены при помощи мостиковых групп. Другой класс металлоценовых катализаторов, который можно использовать, состоит из бис(металлоценовых) комплексов циркония или титана и анионов перфторированных борароматических соединений. Третий класс металлоценовых катализаторов, который можно использовать, называется катализаторами со стесненной геометрией и содержит моноциклопентадиенильные производные титана или циркония, в которых один из атомов углерода в циклопентадиенильном кольце связан с атомом металла при помощи мостиковой группы. Эти комплексы активируют путем их реакции с метилалюмоксаном или путем образования ионных комплексов с некоординационными анионами, такими как B(C6F5)4- или B(C6F5)3CH3-. Четвертый класс металлоценовых катализаторов, который можно использовать, представляет собой комплексы на основе металлоценов переходного металла, такого как титан, содержащие один циклопентадиенильный лиганд в комбинации с другим лигандом, таким как фосфинимин или -O-SiR3. Этот класс металлоценовых катализаторов также активируют метилалюмоксаном или соединением бора. Другие катализаторы, подходящие для использования в получении линейного полиэтилена низкой плотности, подходящего для использования в настоящем изобретении, включают, помимо прочего, катализаторы, раскрытые в патенте США №6649558.
Линейные полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, могут характеризоваться любой подходящей композиционной однородностью, что является выражением, используемым для описания однородности разветвленности в молекулах сополимера полимера. Многие коммерчески доступные линейные полиэтиленовые полимеры низкой плотности характеризуются относительно низкой композиционной однородностью, в случае чего высокомолекулярная фракция полимера содержит относительно небольшое количество α-олефинового сомономера и характеризуется относительно низкой степенью разветвленности, тогда как низкомолекулярная фракция полимера содержит относительно высокое количество α-олефинового сомономера и характеризуется относительно высокой степенью разветвленности. Альтернативно, другой ряд линейных полиэтиленовых полимеров низкой плотности характеризуется относительно низкой композиционной однородностью, в случае чего высокомолекулярная фракция полимера содержит относительно большое количество α-олефинового сомономера, тогда как низкомолекулярная фракция полимера содержит относительно небольшое количество α-олефинового сомономера. Композиционную однородность полимера можно измерить при помощи любого подходящего способа, такого как фракционирование посредством элюирования при повышении температуры.
Линейные полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, могут характеризоваться любой подходящей молекулярной массой. Например, полимер может характеризоваться среднемассовой молекулярной массой от приблизительно 20000 г/моль до приблизительно 250000 г/моль. Как будет понятно специалистам в данной области техники, подходящая среднемассовая молекулярная масса линейного полиэтилена низкой плотности будет зависеть, по меньшей мере, частично от конкретного применения или конечного использования, для которого полимер предназначен.
Линейные полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, могут также характеризоваться любой подходящей поли дисперсностью. Многие коммерчески доступные линейные полиэтиленовые полимеры низкой плотности характеризуются относительно узким распределением молекулярной массы и, таким образом, относительно низкой полидисперсностью, например, от приблизительно 2 до приблизительно 5 (например, от приблизительно 2,5 до приблизительно 4,5 или от приблизительно 3,5 до приблизительно 4,5). Линейные полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, могут также характеризоваться полимодальным (например, бимодальным) распределением молекулярной массы. Например, полимер может иметь первую фракцию, характеризующуюся относительно низкой молекулярной массой, и вторую фракцию, характеризующуюся относительно высокой молекулярной массой. Как и в случае полиэтиленовых полимеров высокой плотности, подходящих для использования в настоящем изобретении, разница между среднемассовой молекулярной массой фракций в полимодальном линейном полиэтиленовом полимере низкой плотности может представлять собой любую подходящую величину. В действительности не обязательно, чтобы разница между среднемассовыми молекулярными массами была настолько большой, чтобы две отдельные молекулярно-весовые фракции можно было разделять при помощи гельпроникающей хроматографии (ГПХ). Однако, в некоторых полимодальных полимерах разница между среднемассовыми молекулярными массами фракций может быть достаточно большой, чтобы два или более отдельных пиков можно было выделить на кривой ГПХ для полимера. В данном случае выражение "отдельный" не обязательно означает, что части кривой ГПХ, соответствующие каждой фракции, не перекрываются, а подразумевает только индикацию того, что отдельный пик для каждой фракции можно выделить на кривой ГПХ для полимера. Полимодальные полимеры, подходящие для использования в настоящем изобретении, можно получать при помощи любого подходящего способа. Как указано выше, полимодальные полимеры можно получать при помощи процессов с многоступенчатыми реакторами. Одним подходящим примером будет многоступенчатый процесс в среде растворителя, включающий ряд смесителей. Альтернативно, полимодальные полимеры можно получать в одном реакторе при помощи комбинации катализаторов, каждый из которых разработан для получения полимера, характеризующегося различной среднемассовой молекулярной массой.
Линейные полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, могут характеризоваться любым подходящим индексом расплава. Например, линейный полиэтиленовый полимер низкой плотности может характеризоваться индексом расплава от приблизительно 0,01 дг/мин до приблизительно 200 дг/мин. Как и в случае среднемассовой молекулярной массы, специалистам в данной области техники будет понятно, что подходящий индекс расплава линейного полиэтиленового полимера низкой плотности будет зависеть, по меньшей мере, частично от конкретного применения или конечного использования, для которого полимер предназначен. Таким образом, например, линейный полиэтиленовый полимер низкой плотности, предназначенный для применений в выдувном формовании или применений для труб, может характеризоваться индексом расплава от приблизительно 0,01 дг/мин до приблизительно 1 дг/мин. Линейный полиэтиленовый полимер низкой плотности, предназначенный для применений для получаемой экструзией с раздувом пленки, может характеризоваться индексом расплава от приблизительно 0,5 дг/мин до приблизительно 3 дг/мин. Линейный полиэтиленовый полимер низкой плотности, предназначенный для применений для получаемой поливом пленки, может характеризоваться индексом расплава от приблизительно 2 дг/мин до приблизительно 10 дг/мин. Линейный полиэтиленовый полимер низкой плотности, предназначенный для применений для литья под давлением, может характеризоваться индексом расплава от приблизительно 6 дг/мин до приблизительно 200 дг/мин. Линейный полиэтиленовый полимер низкой плотности, предназначенный для применений для центробежного формования, может характеризоваться индексом расплава от приблизительно 4 дг/мин до приблизительно 7 дг/мин. Линейный полиэтиленовый полимер низкой плотности, предназначенный для применений для изоляции проводов и кабелей, может характеризоваться индексом расплава от приблизительно 0,5 дг/мин до приблизительно 3 дг/мин. Индекс расплава полимера измеряют при помощи стандарта ASTM D1238-04c.
Линейные полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, обычно не содержат высокие степени длинноцепочечной разветвленности. Например, линейные полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, обычно содержат менее чем приблизительно 0,1 длинноцепочечного ответвления на 10000 атомов углерода (например, менее чем приблизительно 0,002 длинноцепочечного ответвления на 100 этиленовых звеньев) или менее чем приблизительно 0,01 длинноцепочечного ответвления на 10000 атомов углерода.
Полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, обычно характеризуются плотностью менее 0,935 г/см3 и в отличие от полиэтилена высокой плотности, полиэтилена средней плотности и линейного полиэтилена низкой плотности характеризуются относительно высокой степенью длинноцепочечной разветвленности в полимере.
Полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, могут представлять собой или гомополимеры этилена, или сополимеры этилена и полярного сомономера. Подходящие полярные сомономеры включают, помимо прочего, винилацетат, метилакрилат, этилакрилат и акриловую кислоту. Эти сомономеры могут присутствовать в любом подходящем количестве, причем содержание сомономера до 20 масс. % используется для некоторых применений. Специалистам в данной области техники будет понятно, что количество сомономера, подходящее для полимера, в значительной степени обусловлено конечным использованием полимера и требуемыми или желаемыми свойствами полимера, обусловленными таким конечным использованием.
Полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, можно получать при помощи любого подходящего процесса, но обычно полимеры получают путем инициированной свободными радикалами полимеризации этилена под высоким давлением (например, от приблизительно 81 до приблизительно 276 МПа) и при высокой температуре (например, от приблизительно 130 до приблизительно 330°C). Любой подходящий инициатор свободнорадикальной полимеризации можно использовать в таких процессах, причем пероксиды и кислород являются наиболее распространенными. Механизм свободнорадикальной полимеризации является причиной короткоцепочечной разветвленности в полимере, а также относительно высокой степени длинноцепочечной разветвленности, которая отличает полиэтилен низкой плотности от других полимеров этилена (например, полиэтилена высокой плотности и линейного полиэтилена низкой плотности). Реакцию полимеризации обычно проводят в реакторе-автоклаве (например, реакторе-автоклаве с мешалкой), трубчатом реакторе или комбинации таких реакторов, расположенных последовательно.
Полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, могут характеризоваться любой подходящей молекулярной массой. Например, полимер может характеризоваться среднемассовой молекулярной массой от приблизительно 30000 г/моль до приблизительно 500000 г/моль. Как будет понятно специалистам в данной области техники, подходящая среднемассовая молекулярная масса полиэтилена низкой плотности будет зависеть, по меньшей мере, частично от конкретного применения или конечного использования, для которого полимер предназначен. Например, полиэтиленовый полимер низкой плотности, предназначенный для применений в выдувном формовании, может характеризоваться среднемассовой молекулярной массой от приблизительно 80000 г/моль до приблизительно 200000 г/моль. Полиэтиленовый полимер низкой плотности, предназначенный для применений для труб, может характеризоваться среднемассовой молекулярной массой от приблизительно 80000 г/моль до приблизительно 200000 г/моль. Полиэтиленовый полимер низкой плотности, предназначенный для применений в литье под давлением, может характеризоваться среднемассовой молекулярной массой от приблизительно 30000 г/моль до приблизительно 80000 г/моль. Полиэтиленовый полимер низкой плотности, предназначенный для применений для пленки, может характеризоваться среднемассовой молекулярной массой от приблизительно 60000 г/моль до приблизительно 500000 г/моль.
Полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, могут характеризоваться любым подходящим индексом расплава. Например, полиэтиленовый полимер низкой плотности может характеризоваться индексом расплава от приблизительно 0,2 до приблизительно 100 дг/мин. Как указано выше, индекс расплава полимера измеряют при помощи стандарта ASTM D1238-04c.
Как указано выше, одним из основных различий между полиэтиленом низкой плотности и другими полимерами этилена является относительно высокая степень длинноцепочечной разветвленности в полимере. Полиэтиленовые полимеры низкой плотности, подходящие для использования в настоящем изобретении, могут характеризоваться любой подходящей степенью длинноцепочечной разветвленности, такой как приблизительно 0,01 или более длинноцепочечных ответвлений на 10000 атомов углерода, приблизительно 0,1 или более длинноцепочечных ответвлений на 10000 атомов углерода, приблизительно 0,5 или более длинноцепочечных ответвлений на 10000 атомов углерода, приблизительно 1 или более длинноцепочечных ответвлений на 10000 атомов углерода или приблизительно 4 или более длинноцепочечных ответвлений на 10000 атомов углерода. Хотя нет строгого предела в отношении максимального уровня длинноцепочечной разветвленности, которая может присутствовать в полиэтиленовых полимерах низкой плотности, подходящих для использования в настоящем изобретении, длинноцепочечная разветвленность во многих полиэтиленовых полимерах низкой плотности составляет менее чем приблизительно 100 длинноцепочечных ответвлений на 10000 атомов углерода.
Композиция на основе термопластичного полимера также содержит зародышеобразователь. При использовании в настоящем документе выражение "зародышеобразователь" используют для обозначения соединений или добавок, которые образуют зародыши кристаллизации или обеспечивают места для образования и/или роста кристаллов в полимере, когда он отверждается из расплавленного состояния. Согласно первому варианту осуществления зародышеобразователь содержит соединение, соответствующее структуре формулы (I)
 .
.
В структуре формулы (I) R1 выбрана из группы, состоящей из гидроксигрупп, галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп. Переменная n равняется нулю или целому положительному числу от 1 до 4. L представляет собой связующую группу, содержащую два или более атомов и по меньшей мере одну двойную связь между двумя атомами в связующей группе. Переменная v представляет собой целое положительное число от 1 до 3. R2: (i) выбрана из группы, состоящей из алкильных групп, замещенных алкильных групп, циклоалкильных групп, замещенных циклоалкильных групп, арильных групп, замещенных арильных групп, гетероарильных групп и замещенных гетероарильных групп, если L представляет собой двухвалентную связующую группу, а v равняется 1, (ii) выбрана из группы, состоящей из алкандиильных групп, замещенных алкандиильных групп, циклоалкандиильных групп, замещенных циклоалкандиильных групп, арендиильных групп, замещенных арендиильных групп, гетероарендиильных групп и замещенных гетероарендиильных групп, если L представляет собой трехвалентную связующую группу, а v равняется 1, (iii) выбрана из группы, состоящей из алкандиильных групп, замещенных алкандиильных групп, циклоалкандиильных групп, замещенных циклоалкандиильных групп, арендиильных групп, замещенных арендиильных групп, гетероарендиильных групп и замещенных гетероарендиильных групп, если L представляет собой двухвалентную связующую группу, а v равняется 2, и (iv) выбрана из группы, состоящей из алкантриильных групп, замещенных алкантриильных групп, циклоалкантриильных групп, замещенных циклоалкантриильных групп, арентриильных групп, замещенных арентриильных групп, гетероарентриильных групп и замещенных гетероарентриильных групп, если L представляет собой двухвалентную связующую группу, а v равняется 3. Переменная x представляет собой целое положительное число. Каждый M1 представляет собой катион металла; переменная y представляет собой валентность катиона; а переменная z представляет собой целое положительное число. Переменная b равняется нулю или целому положительному числу. Если b представляет собой целое положительное число, каждый Q1 представляет собой отрицательно заряженный противоион, а a представляет собой валентность отрицательно заряженного противоиона. Значения v, x, y, z, а и b удовлетворяют уравнению (vx)+(ab)=yz. В структуре формулы (I) циклическая часть циклоалкильной группы или замещенной циклоалкильной группы содержит не более двух кольцевых структур, сконденсированных вместе, если L представляет собой двухвалентную связующую группу, v равняется 1, а R2 представляет собой циклоалкильную группу или замещенную циклоалкильную группу.
Согласно предпочтительному варианту осуществления R1 представляет собой галоген или гидроксигруппу, причем n=1 является особенно предпочтительным.
Согласно более конкретному варианту осуществления n может равняться 1, R1 может представлять собой гидроксигруппу и присоединяться к арильному кольцу в орто-положении относительно карбоксилатной группы. Согласно другому предпочтительному варианту осуществления n равняется 0, подразумевая, что замещенное карбоксилатом арильное кольцо не замещено R1-группами.
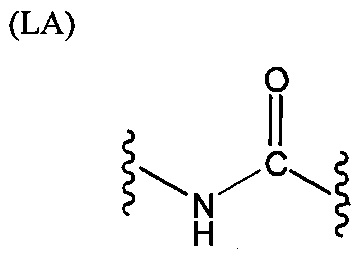
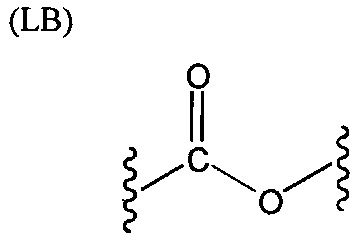
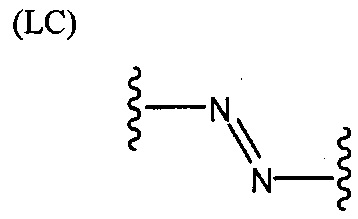
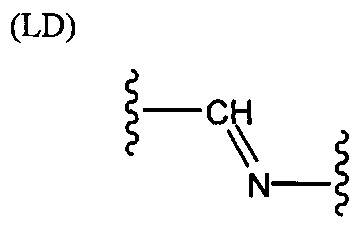
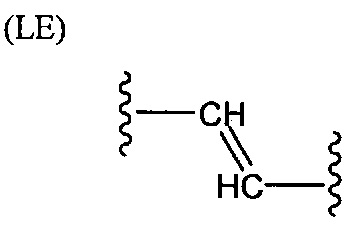
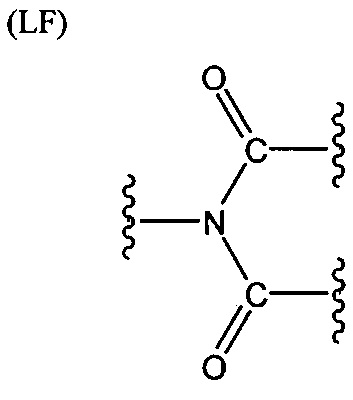
L представляет собой связующую группу, содержащую два или более атомов и по меньшей мере одну двойную связь между двумя атомами в связующей группе. При наличии по меньшей мере одной двойной связи между двумя атомами в связующей группе, два атома в связующей группе sp2-гибридизованы, а сумма валентных углов вокруг по меньшей мере одного из этих атомов составляет приблизительно 360 градусов. Присутствие двойной связи в связующей группе ограничивает вращение молекулы вокруг двойной связи и, как полагают без ограничения какой-либо теорией, поддерживает соединение в конфигурации, которая является более подходящей для образования зародышей кристаллизации в полимере. Согласно ряду предпочтительных вариантов осуществления L выбрана из группы, состоящей из фрагментов, соответствующих структуре одной из формул (LA)-(LF) ниже
 ,
,
 ,
,
 ,
,
 ,
,
 ,
,
 .
.
Как видно из этих структур, подходящие связующие группы содержат по меньшей мере два атома и двойную связь между двумя атомами в связующей группе. В случае каждой из этих L-групп любой подходящий конец связующей группы может присоединяться к замещенному карбоксилатом арильному кольцу, а другой конец(концы) может присоединяться к группе R2. Согласно предпочтительному варианту осуществления L представляет собой фрагмент, выбранный из группы, состоящей из фрагментов, соответствующих структуре формул (LA) и (LD). Согласно особенно предпочтительному варианту осуществления L представляет собой фрагмент, соответствующий структуре формулы (LA). Согласно такому варианту осуществления фрагмент может иметь атом азота, присоединенный к замещенному карбоксилатом арильному кольцу или группе R2.
Группа R2 может представлять собой одновалентный, двухвалентный или трехвалентный фрагмент. Валентность R2 зависит от валентности связующей группы L и числа замещенных карбоксилатом арильных колец в соединении. Таким образом, если L представляет собой двухвалентную связующую группу, v равняется 1, то R2 можно выбирать из группы, состоящей из фрагментов, соответствующих структуре одной из формул (АА)-(AG) ниже. Структура формулы (АА) представляет собой
 .
.
В структуре формулы (АА) переменная d равняется нулю или целому положительному числу от 1 до 5, а каждая R10 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп. Структура формулы (AB) представляет собой
 .
.
В структуре формулы (АВ) переменная h равняется нулю или целому положительному числу от 1 до 10, а каждая R13 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп. Структура формулы (AC) представляет собой
 .
.
В структуре формулы (АС) переменная е равняется нулю или целому положительному числу от 1 до 8, а каждая R15 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп. Структура формулы (AD) представляет собой
 .
.
В структуре формулы (AD) переменная g равняется нулю или целому положительному числу от 1 до 6, а каждая R20 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп. Структура формулы (AE) представляет собой
 .
.
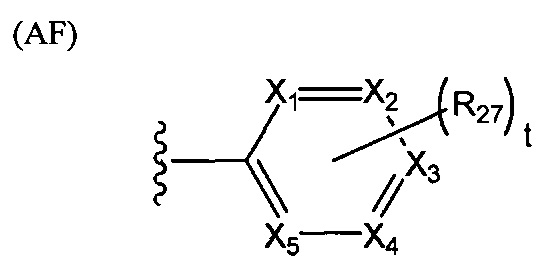
В структуре формулы (АЕ) переменная j равняется нулю или целому положительному числу от 1 до 4, а каждая R25 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп. Структура формулы (AF) представляет собой
 .
.
В структуре формулы (AF) переменные X1, X2, Х3, X4 и Х5 независимо выбраны из группы, состоящей из атома углерода и атома азота, при условии, что по меньшей мере один и не более трех из X1, X2, Х3, X4 и Х5 представляют собой атомы азота; t равняется нулю или целому положительному числу, равному 5-Х, где X представляет собой число атомов азота; а каждая R27 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп. Структура формулы (AG) представляет собой
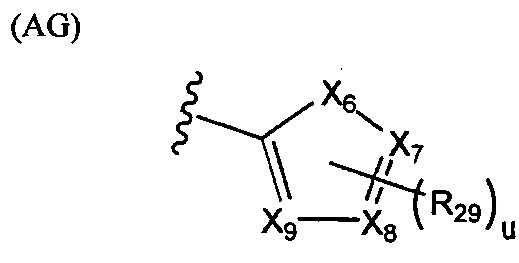 .
.
В структуре формулы (AG) переменная X6 выбрана из группы, состоящей из атома углерода, атома кислорода, атома серы и вторичной аминогруппы, X7, X8 и X9 независимо выбраны из группы, состоящей из атома углерода и атома азота, по меньшей мере один и не более трех из X6, X7, X8 и X9 представляют собой атомы, не являющиеся углеродом; и равняется нулю или целому положительному числу, равному 4-Y, где Y представляет собой число атомов, не являющихся углеродом, в кольцевой структуре; а каждая R29 независимо выбрана из группы, состоящей из галогенов, цианогрупп, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп.
Если L представляет собой трехвалентную связующую группу, v равняется 1, то R2 можно выбирать из группы, состоящей из фрагментов, соответствующих структуре одной из формул (АН)-(AJ) ниже. Структура формулы (АН) представляет собой
 .
.
В структуре формулы (АН) переменная к равняется нулю или целому положительному числу от 1 до 8, а каждая R30 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп. Структура формулы (AI) представляет собой
 .
.
В структуре формулы (AI) переменная m равняется нулю или целому положительному числу от 1 до 4, а каждая R35 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп. Структура формулы (AJ) представляет собой
 .
.
В структуре формулы (AJ) переменная p равняется нулю или целому положительному числу от 1 до 3, p' равняется нулю или целому положительному числу от 1 до 3, а каждая из R40 и R45 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп.
Если L представляет собой двухвалентную связующую группу, v равняется 2, то R2 можно выбирать из группы, состоящей из фрагментов, соответствующих структуре формулы (ВА) ниже
 .
.
В структуре формулы (ВА) переменная q равняется нулю или целому положительному числу от 1 до 4, r равняется нулю или целому положительному числу от 1 до 4, а каждая из R50 и R55 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп.
Если L представляет собой двухвалентную связующую группу, v равняется 3, то R2 можно выбирать из группы, состоящей из фрагментов, соответствующих структуре формулы (СА) ниже
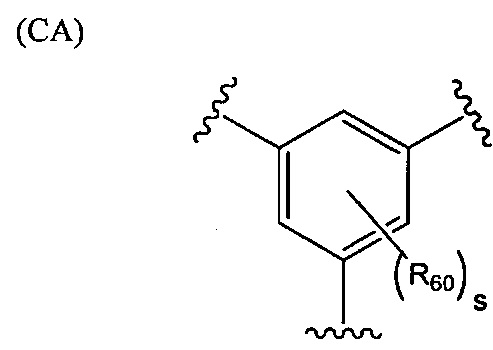 .
.
В структуре формулы (СА) переменная s равняется нулю или целому положительному числу от 1 до 3, а каждая R60 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп.
Согласно ряду предпочтительных вариантов осуществления L представляет собой двухвалентную связующую группу, v равняется 1, а R2 представляет собой фрагмент, соответствующий структуре формулы (АА). В этом ряду предпочтительных вариантов осуществления переменная d предпочтительно равняется нулю или 1. Если d равняется 1, группа R10 предпочтительно присоединена к арильному кольцу в пара-положении относительно связи со связующей группой L. Кроме того, если d равняется 1, группа R10 предпочтительно представляет собой галоген (например, бром), алкоксигруппу (например, метоксигруппу) или арильную группу (например, фенильную группу).
Согласно ряду предпочтительных вариантов осуществления L представляет собой двухвалентную связующую группу, v равняется 1, а R2 представляет собой фрагмент, соответствующий структуре формулы (АС). В этом ряду предпочтительных вариантов осуществления переменная d предпочтительно равняется нулю или 1, причем значение ноль является особенно предпочтительным.
Как указано выше, M1 представляет собой катион металла. Подходящие катионы металлов включают, помимо прочего, катионы щелочных металлов (например, натрия), катионы щелочноземельных металлов (например, кальция), катионы переходных металлов (например, цинка) и катионы металлов 13 группы (например, алюминия). При использовании в настоящем документе выражение "переходный металл" используют для обозначения таких элементов в d-блоке Периодической таблицы элементов, которые соответствуют 3-12 группам Периодической таблицы элементов. Согласно предпочтительному варианту осуществления M1 представляет собой катион металла, выбранный из группы, состоящей из лития, натрия, магния, алюминия, калия, кальция и цинка. Согласно другому предпочтительному варианту осуществления M1 представляет собой катион лития. Согласно тем вариантам осуществления, в которых соединение содержит более одного катиона металла M1, каждый M1 может быть одинаковым или отличным.
Согласно ряду предпочтительных вариантов осуществления зародышеобразователь может содержать соединение, соответствующее структуре одной из формул (IA)-(IM) ниже
 ,
,
 ,
,
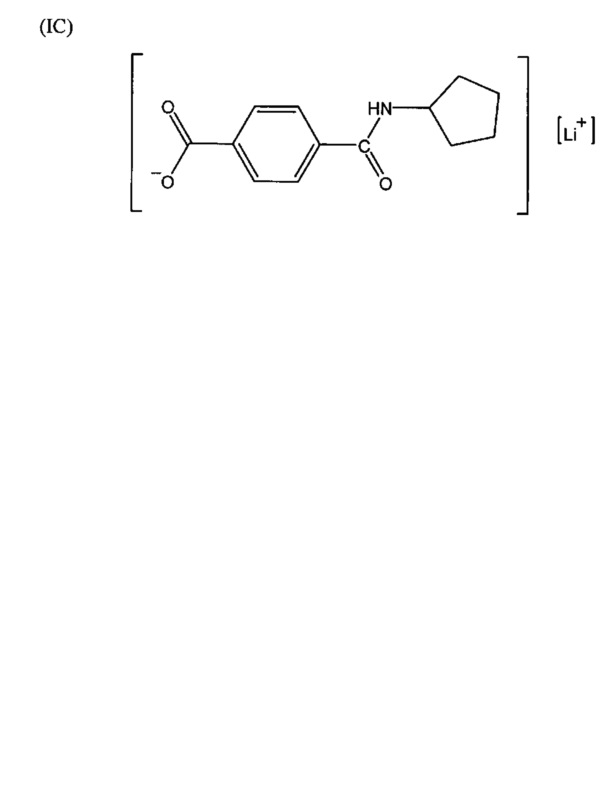 ,
,
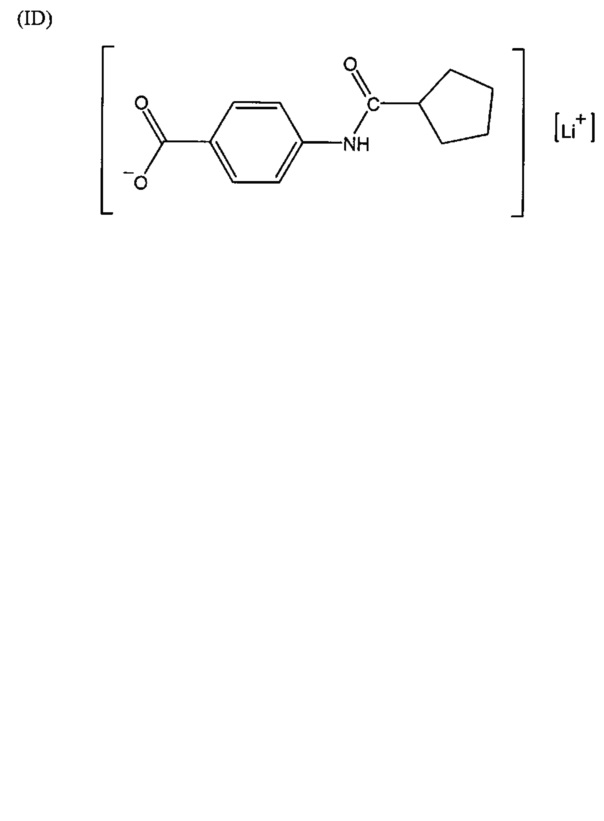 ,
,
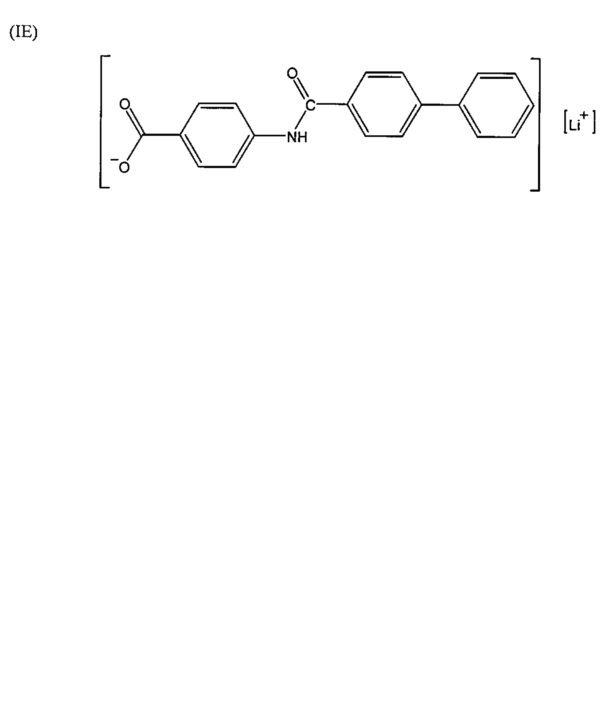 ,
,
 ,
,
 ,
,
 ,
,
 ,
,
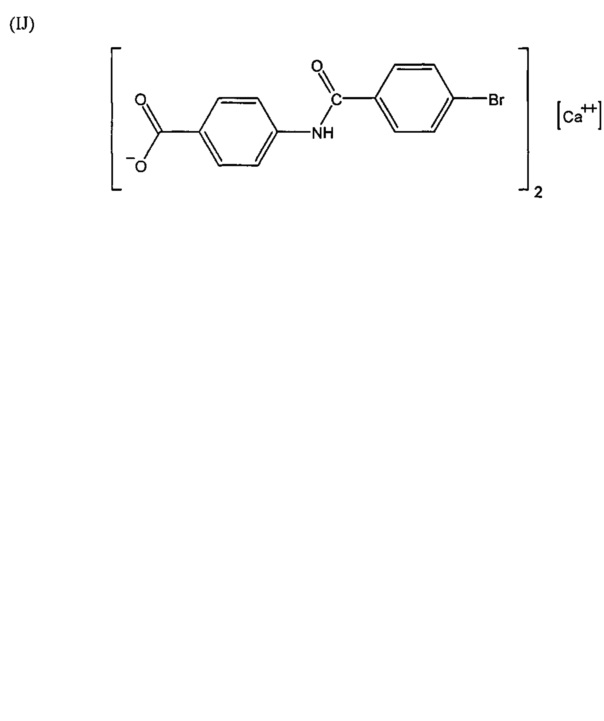 ,
,
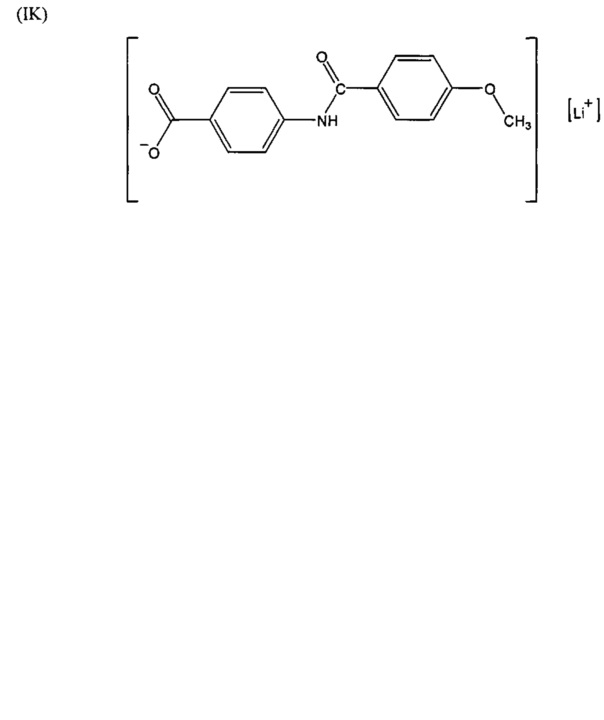 ,
,
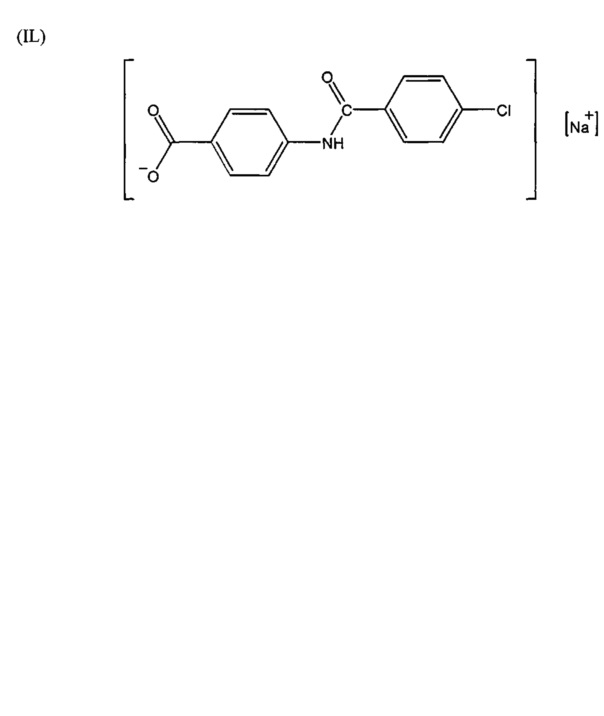 ,
,
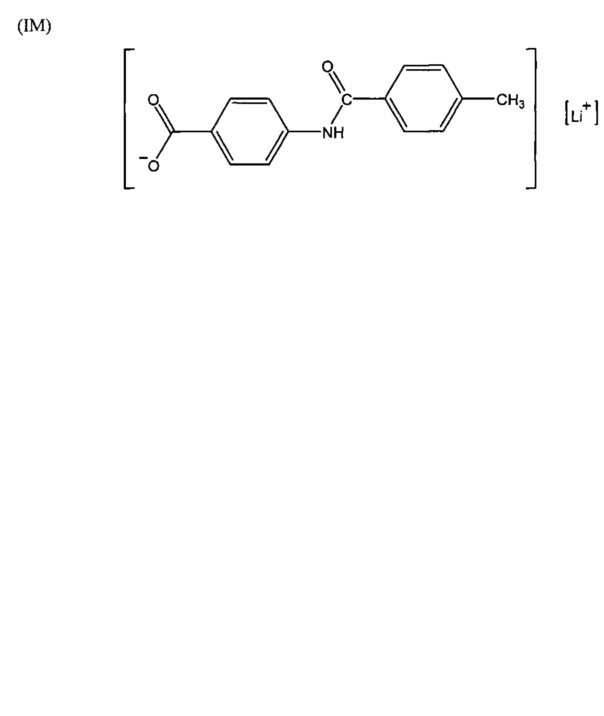 .
.
Композиция может содержать одно или несколько соединений солей металлов, соответствующих структуре формулы (I). Например, композиция может содержать любую подходящую комбинацию соединений, соответствующих структурам (IA)-(IM), изображенным выше. Более конкретно, композиция может содержать соединение, соответствующее структуре формулы (IA), и соединение, соответствующее структуре формулы (IL). Согласно другому конкретному варианту осуществления композиция может содержать соединение, соответствующее структуре формулы (IB), и соединение, соответствующее структуре формулы (IL). Согласно еще одному конкретному варианту осуществления композиция может содержать соединение, соответствующее структуре формулы (IC), и соединение, соответствующее структуре формулы (IL). Смеси этих соединений можно использовать для получения композиций, которые характеризуются желательной комбинацией свойств, причем одно соединение обеспечивает одно преимущество, а другое соединение обеспечивает дополнительное преимущество.
Соединения солей металлов формулы (I) и более конкретные структуры, охваченные формулой (I), можно синтезировать при помощи любой подходящей техники, многие из которых будут абсолютно очевидны для специалистов в данной области техники. Например, если кислота, используемая при получении соединения, коммерчески доступна, соединение можно получать путем реакции кислоты с подходящим основанием (например, основанием, содержащим желаемый катион металла, и основанием Брэнстеда) в подходящей среде (например, водной среде). Если кислота, используемая для получения соединения соли металла, не является коммерчески доступной, кислоту можно синтезировать, например, при помощи любой из техник, показанных ниже в примерах. Как только получают желаемую кислоту, соединение можно получать, как описано выше (например, реакцией кислоты с подходящим основанием в соответствующей среде).
Соединения солей металлов формулы (I) и более конкретные структуры, охваченные формулой (I), можно получать с различными формами и размерами частиц. В общем, эти соединения солей образуют слоистые кристаллические структуры, в которых ионы металлов находятся в каналах, которые размещаются между чередующимися слоями органических поверхностей. В результате часто получаются плоские частицы в виде пластинок, при этом образующие зародыши кристаллизации поверхности находятся сверху и снизу частиц, а не на кромках. Отношение размеров этих частиц в виде пластинок обычно определяют как диаметр или ширину относительно толщины. Продолговатые пластинки или "плосковытянутые" кристаллы представляют собой другую морфологию частиц, возможную в случае этих соединений солей металлов. В этих продолговатых структурах отношение размеров обычно определяют как отношение длины к ширине. Возможны отношения размеров от 2:1 вплоть до 50:1. Частицы с такими отношениями размеров могут выстраиваться в полях течения расплавленного полимера так, что плоские поверхности параллельны машинному направлению или направлению потока и параллельны поперечному или перпендикулярному направлению. В результате образующие зародыши кристаллизации поверхности открыты только в нормальном направлении полимерного расплава во время частичной переработки (исключения будут случаться, когда пластинчатые частицы обладают отношением размеров, несоответствующим плоскому регистру, и происходит переворачивание в направлении течения полимера). Предпочтительные ориентации частиц или "регистры" совместно со специфическими кристаллографическими взаимодействиями с полиэтиленом во время события образования зародышей кристаллизации могут образовывать направленный рост ламелей, что может приводить к уникальным и полезным ориентациям кристаллов полиэтилена в получаемых изделиях.
Частицы зародышеобразователя, обсуждаемого выше, могут характеризоваться любым подходящим размером. Предпочтительно частицы зародышеобразователя являются достаточно мелкими, чтобы их не было видно в готовом изделии, полученном из композиции термопластичного полимера. Таким образом, согласно предпочтительному варианту осуществления частицы зародышеобразователя предпочтительно характеризуются диаметром менее 25 микрон, более предпочтительно диаметром менее 20 микрон и наиболее предпочтительно диаметром менее 15 микрон.
Зародышеобразователь может присутствовать в композиции на основе термопластичного полимера в любом подходящем количестве. Зародышеобразователь может присутствовать в композиции термопластичного полимера в количестве приблизительно 50 ppm (частей на миллион) или более, приблизительно 100 частей на миллион или более, приблизительно 250 частей на миллион или более или приблизительно 500 частей на миллион или более на основании общей массы композиции на основе термопластичного полимера. Зародышеобразователь обычно присутствует в композиции термопластичного полимера в количестве приблизительно 10000 частей на миллион или менее, приблизительно 7500 частей на миллион или менее, приблизительно 5000 частей на миллион или менее, приблизительно 4000 частей на миллион или менее или приблизительно 3000 частей на миллион или менее на основании общей массы композиции термопластичного полимера. Таким образом, согласно некоторым вариантам осуществления композиции термопластичного полимера зародышеобразователь присутствует в композиции термопластичного полимера в количестве от приблизительно 50 до приблизительно 10000 частей на миллион, от приблизительно 100 до приблизительно 7500 частей на миллион (например, от приблизительно 100 до приблизительно 5000 частей на миллион), от приблизительно 250 до приблизительно 5000 частей на миллион (например, от приблизительно 250 до приблизительно 4000 частей на миллион или от приблизительно 250 до приблизительно 3000 частей на миллион) или от приблизительно 500 до приблизительно 5000 частей на миллион (например, от приблизительно 500 до приблизительно 4000 частей на миллион или от приблизительно 500 до приблизительно 3000 частей на миллион) на основании общей массы композиции термопластичного полимера.
Композицию термопластичного полимера настоящего изобретения можно также обеспечивать в виде композиции мастербатча, разработанной для добавления или введения в чистый термопластичный полимер. Согласно такому варианту осуществления композиция на основе термопластичного полимера будет обычно содержать большее количество зародышеобразователя по сравнению с композицией на основе термопластичного полимера, предназначенной для использования в образовании готового изделия, и вводится без дополнительного разбавления в чистый термопластичный полимер. Например, зародышеобразователь может присутствовать в такой композиции на основе термопластичного полимера в количестве от приблизительно 1 масс. % до приблизительно 10 масс. % (например, от приблизительно 1 масс. % до приблизительно 5 масс. % или от приблизительно 2 масс. % до приблизительно 4 масс. %) на основании общей массы композиции на основе термопластичного полимера.
Композиция на основе термопластичного полимера настоящего изобретения может содержать другие полимерные добавки в дополнение к вышеуказанному зародышеобразователю. Подходящие дополнительные полимерные добавки включают, помимо прочего, антиоксиданты (например, фенольные антиоксиданты, фосфитные антиоксиданты и их комбинации), ангиадгезивы (например, аморфный диоксид кремния и диатомитовую землю), пигменты (например, органические пигменты и неорганические пигменты) и другие окрашивающие вещества (например, красители и полимерные окрашивающие вещества), наполнители и армирующие средства (например, стекло, стекловолокна, тальк, карбонат кальция и нити оксисульфата магния), зародышеобразователи, осветляющие средства, поглотители кислот (например, металлические соли жирных кислот, такие как металлические соли стеариновой кислоты и дигидроталькит), полимерные технологические добавки (например, фторполимерные полимерные технологические добавки), полимерные сшивающие средства, понижающие трение средства (например, соединения жирной кислоты и амида, полученные реакцией жирной кислоты и аммиака или содержащего амин соединения), соединения сложных эфиров жирных кислот (например, соединения сложных эфиров жирных кислот, полученные реакцией жирной кислоты и гидроксисодержащего соединения, такого как глицерин, диглицерин и их комбинации) и комбинации вышеперечисленного.
Как указано выше, композиция на основе термопластичного полимера настоящего изобретения может содержать другие зародышеобразователи в дополнение к соединениям, которые соответствуют структуре формулы (I). Подходящие зародышеобразователи включают, помимо прочего, 2,2'-метилен-бис-(4,6-ди-трет-бутилфенил)фосфатные соли (например, 2,2'-метилен-бис-(4,6-ди-трет-бутилфенил)фосфат натрия или 2,2'-метилен-бис-(4,6-ди-трет-бутилфенил)фосфат алюминия), бицикло[2.2.1]гептан-2,3-дикарбоксилатные соли (например, бицикло[2.2.1]гептан-2,3-дикарбоксилат динатрия или бицикло[2.2.1]гептан-2,3-дикарбоксилат кальция), циклогексан-1,2-дикарбоксилатные соли (например, циклогексан-1,2-дикарбоксилат кальция, циклогексан-1,2-дикарбоксилат одноосновный алюминия, циклогексан-1,2-дикарбоксилат дилития или циклогексан-1,2-дикарбоксилат стронция), глицеролатные соли (например, глицеролат цинка), фталатные соли (например, фталат кальция), соли фенилфосфоновых кислот (например, фенилфосфонат кальция) и их комбинации. В случае бицикло[2.2.1]гептан-2,3-дикарбоксилатных солей и циклогексан-1,2-дикарбоксилатных солей карбоксилатные фрагменты могут располагаться как в цис-, так и транс-конфигурации, причем цис-конфигурация предпочтительна.
Как указано выше, композиция на основе термопластичного полимера настоящего изобретения может также содержать осветляющее средство. Подходящие осветляющие средства включают, помимо прочего, трисамиды и ацетальные соединения, которые представляют собой продукт конденсации многоатомного спирта и ароматического альдегида. Подходящие трисамидные осветляющие средства включают, помимо прочего, амидные производные бензол-1,3,5-трикарбоновой кислоты, производные N-(3,5-бис-формиламинофенил)-формамида (например, N-[3,5-бис-(2,2-диметилпропиониламино)-фенил]-2,2-диметилпропионамид), производные 2-карбамоилмалонамида (например, N,N'-бис-(2-метилциклогексил)-2-(2-метилциклогексилкарбамоил)-малонамида) и их комбинации. Как указано выше, осветляющее средство может представлять собой ацетальное соединение, которое представляет собой продукт конденсации многоатомного спирта и ароматического альдегида. Подходящие многоатомные спирты включают ациклические полиолы, такие как ксилит и сорбит, а также ациклические дезоксиполиолы (например, 1,2,3-тридезоксинонит или 1,2,3-тридезоксинон-1-енит). Подходящие ароматические альдегиды обычно содержат одну альдегидную группу, причем оставшиеся положения в ароматическом кольце являются или незамещенными, или замещенными. Следовательно, подходящие ароматические альдегиды включают бензальдегид и замещенные бензальдегиды (например, 3,4-диметилбензальдегид или 4-пропилбензальдегид). Ацетальное соединение, полученное вышеуказанной реакцией, может представлять собой моноацетельное, диацетальное или триацетальное соединение (т.е. соединение, содержащее одну, две или три ацетальных группы, соответственно), причем диацетальные соединения предпочтительны. Подходящие осветляющие средства на основе ацеталей включают, помимо прочего, осветляющие средства, раскрытые в патентах США №5049605; №7157510 и №7262236.
Композицию на основе термопластичного полимера настоящего изобретения можно получать любым подходящим способом или процессом. Например, композицию на основе термопластичного полимера можно получать путем простого перемешивания отдельных компонентов композиции на основе термопластичного полимера (например, термопластичного полимера, зародышеобразователя и других добавок при необходимости). Композицию на основе термопластичного полимера можно также получать путем смешивания отдельных компонентов при условиях перемешивания с большими сдвиговыми усилиями или с высокой интенсивностью. Композицию на основе термопластичного полимера настоящего изобретения можно обеспечивать в любой форме, подходящей для использования для дальнейшей переработки с получением готового изделия из композиции на основе термопластичного полимера. Например, композиции на основе термопластичного полимера можно обеспечивать в виде порошка (например, свободнотекучего порошка), крошек, дробинок, гранул, пластинок, агломератов и подобного.
Композиция на основе термопластичного полимера настоящего изобретения, как считается, пригодна для получения готовых изделий из термопластичного полимера. Композицию на основе термопластичного полимера настоящего изобретения можно формовать в желаемое готовое изделие из термопластичного полимера при помощи любой подходящей техники, такой как литье под давлением (например, тонкостенное литье под давлением, многокомпонентное формование, напрессовка или 2-компонентное формование), формование раздувом (например, формование экструзией с раздувом, литье под давлением с раздувом или литье под давлением с раздувом и ориентированием), экструзия (например, экструзия волокон, экструзия лент (например, щелевая экструзия лент), изготовление листов экструзией, экструзия пленки, экструзия поливной пленки, экструзия труб, нанесение покрытия экструзией или экструзия пенопластов), термоформование, центробежное формование, получение пленки экструзией с раздувом (пленка, получаемая экструзией с раздувом), получение пленки поливом (поливная пленка), компрессионное формование, экструзионно-компрессионное формование, экструзионно-компрессионное формование с раздувом и подобное. Изделия из термопластичного полимера, полученные при помощи композиции на основе термопластичного полимера настоящего изобретения, могут состоять из множества слоев (например, многослойные получаемые экструзией с раздувом или поливом пленки или многослойные полученным литьем под давлением изделия), причем один или любое подходящие количество из множества слоев содержат композицию на основе термопластичного полимера настоящего изобретения.
Композицию на основе термопластичного полимера настоящего изобретения можно использовать для получения любого подходящего готового изделия. Подходящие готовые изделия включают, помимо прочего, медицинские устройства (например, предварительно заполненные шприцы для реторт-упаковок, контейнеры для внутривенного введения веществ и устройство забора крови), упаковку для пищевых продуктов, контейнеры для жидкости (например, контейнеры для напитков, лекарственных препаратов, составов для личной гигиены, шампуней и подобного), детали одежды, изделия, которые можно подвергать воздействию микроволнового излучения, полки, дверцы шкафов, механические детали, автомобильные детали, листы, трубы, трубопроводы, полученные ротационным формованием детали, полученные литьем с раздувом детали, пленки, волокна и подобное.
Введение гетерогенных зародышеобразователей, описанных выше, как было последовательно показано, способствует образованию зародышей кристаллизации в термопластичном полимере (например, полиолефине, таком как полиэтилен), что отмечали, например, вследствие увеличения пиковой температуры перекристаллизации полимера. Кроме того, введение зародышеобразователя, как наблюдалось, подходящим образом улучшает некоторые физические свойства термопластичного полимера, такие как мутность, прочность на разрыв (или абсолютная прочность на разрыв, или равновесие между прочностью на разрыв в машинном и поперечном направлениях), жесткость и барьерные свойства. Когда композицию на основе термопластичного полимера используют для получения изделия, влияние зародышеобразователя на физические свойства полимера можно увеличивать путем управления характеристическим временем процесса (T) и/или путем выбора полимера, характеризующегося соответствующим средним временем релаксации (λ). В этом случае характеристическое время процесса (T) представляет собой время, в течение которого расплавленный полимер подвергают деформации, которая приводит к напряжению (например, растягивающему напряжению в расплаве) в полимерном расплаве. Среднее время релаксации (λ) является характеристикой полимера и представляет собой количество времени, которое необходимо полимерному расплаву для снятия напряжения. Среднее время релаксации (λ) зависит, помимо прочего, от молекулярной массы полимера, распределения молекулярной массы полимера и степени разветвленности в полимере. Например, известно, что λ пропорционально молекулярной массе полимера, причем более высокие молекулярные массы приводят к более длительному времени релаксации. Кроме того, большинство коммерческих полиолефинов являются в той или иной степени полидисперсными, причем степень полидисперсности обычно выражают как Mw/Mn, определенных при помощи ГПХ. Эта полидисперсность по существу дает ряд зависящих от молекулярной массы времен релаксации, хотя многие техники могут измерять только одно среднее время релаксации для таких полидисперсных систем. Полидисперсность полимера и ряд зависящих от молекулярной массы времен релаксации и/или среднее время релаксации можно специально дополнительно увеличивать или регулировать путем получения бимодальных смесей, как описано выше.
Многие термопластичные полимеры, такие как полиэтилен, кристаллизуются путем складывания цепи, давая кристаллические ламели, чередующиеся с аморфной фазой. В процессах, в которых расплавленный полимер подвергают относительно небольшой деформации, полимерные цепочки в полимерном расплаве плохо выравниваются, и полимерный расплав (например, полиэтиленовый расплав) охлаждается до тех пор, пока не произойдет достаточное выравнивание цепочек, чтобы вызвать самопроизвольный рост кристаллических ламелей. Когда происходит этот самопроизвольный рост ламелей, плотность образования зародышей кристаллизации относительно низкая, и растущие ламели перемещаются перед соударением друг с другом. Это позволяет ламелям начинать изменение своего направления или расширяться, причем крайняя степень расширения представляла собой образование полных сферолитов. Поскольку при этих условиях самообразование зародышей кристаллизации занимает относительно длительное время, зародышеобразователь (такой как описанный в настоящей заявке), добавленный в полимерный расплав, будет иметь возможность регулировать значительную часть роста ламелей. И при условии, что большая часть ламелей образуется зародышеобразователем, зародышеобразователь будет эффективно влиять на физические свойства полимера и изделия.
Некоторые процессы, такие как получение пленки экструзией с раздувом, могут придавать значительную пространственную деформацию полимерному расплаву в машинном направлении (т.е. направлении, в котором расплавленный полимер выходит из экструзионной головки). Конечное напряжение вызывает развертывание полимерных цепочек из их энтропийной статистической спирали, приводя в результате к выравниванию вытянутых полимерных цепочек в машинном направлении. Если эта ориентация сохраняется при охлаждении полимерного расплава, некоторые из этих выровненных, вытянутых сегментов цепи могут кристаллизоваться из расплава с образованием относительно длинных фибрилл. Фибриллы очень эффективны для образования зародышей роста ламелей путем складывания цепи. Ламели образуются и начинают расти перпендикулярно оси фибриллы и практически радиально вокруг фибрилл. Поскольку плотность образования зародышей кристаллизации высокая, растущие ламели могут сталкиваться друг с другом перед тем, как начнется значительное слияние. Этот процесс называется в настоящем документе "обусловленным напряжением самообразованием зародышей кристаллизации на фибриллах". При некоторых условиях, как описано ниже, это обусловленное напряжением самообразование зародышей кристаллизации на фибриллах может становиться главным в полимере (например, полиэтиленовом полимере). Таким образом, любой гетерогенный зародышеобразователь должен конкурировать с этим обусловленным напряжением самообразованием зародышей кристаллизации на фибриллах, что делает зародышеобразователь менее эффективным для соответствующего влияния на физические свойства полимера и изделия. Влияния λ и T на обусловленное напряжением самообразование зародышей кристаллизации на фибриллах и эффективность зародышеобразователей описаны ниже.
При условии, что T постоянно, более короткое λ означает, что происходит большая релаксация напряжений, и ориентация меньшего количества цепей полимера (например, ориентация цепей полимера, вызванная растягивающей деформацией полимерного расплава) сохраняется в конце T. При таких условиях обусловленное напряжением самообразование зародышей кристаллизации на фибриллах будет менее заметным в полимере, и зародышеобразователь будет более эффективным для регулирования роста ламелей и воздействия на физические свойства полимера и изделия. При таком же T более длительное λ означает, что происходит меньшая релаксация напряжений, и сохраняется ориентация большего количества цепей полимера в конце Т. При этом наборе условий обусловленное напряжением самообразование зародышей кристаллизации на фибриллах будет более заметным в полимере, и зародышеобразователь будет менее эффективным для регулирования роста ламелей и воздействия на физические свойства полимера и изделия.
При определении влияния λ и Т на обусловленное напряжением самообразование зародышей кристаллизации на фибриллах и эффективность гетерогенного зародышеобразователя (такого как описанные в настоящем документе), например, в процессах получения пленки экструзией с раздувом, может быть полезно рассмотреть отношение 1 к Т (А/Т), которое будет называться далее в настоящем документе "коэффициент времени переработки" (FTR). FTR имеет какую же форму и приблизительно соответствует числу Деборы (De). Как показано в вышеуказанном обсуждении, меньший FTR означает, что меньшее обусловленное напряжением самообразование зародышей кристаллизации на фибриллах будет происходить в полимере, что делает зародышеобразователь более эффективным для влияния на физические свойства. И больший FTR означает, что большее обусловленное напряжением самообразование зародышей кристаллизации на фибриллах будет происходить в полимере, что делает зародышеобразователь менее эффективным для влияния на физические свойства. Поскольку технологическое время большинства коммерческих процессов может изменяться только в относительно узком интервале, более осуществимый вариант изменения FTR для улучшения или оптимизации влияния зародышеобразователя состоит в изменении λ, что осуществляют путем изменения свойств полимера. В частности, для данного процесса влияние зародышеобразователя можно оптимизировать для достижения желаемого результата путем изменения свойств полимера и λ так, чтобы они лучше соответствовали времени процесса Т.
Таким образом, если невозможно достичь желаемого уровня полезных эффектов от образования зародышей кристаллизации (например, улучшенных свойств непроницаемости или увеличенной прочности на разрыв) при помощи заданного зародышеобразователя и полимера в процессе, можно улучшить результаты путем выбора другого полимера с более коротким λ. Например, можно выбирать бимодальный полимер, содержащий первую фракцию с относительно низким индексом расплава (что обычно указывает на более высокую молекулярную массу и, таким образом, более длительное λ) и вторую фракцию с относительно высоким индексом расплава (что обычно указывает на более низкую молекулярную массу и, таким образом, более короткое λ). В данной системе фракция с большим индексом расплава может обеспечивать λ, для всего полимера, которое дает меньшее обусловленное напряжением самообразование зародышей кристаллизации на фибриллах и улучшенную реакцию на гетерогенный зародышеобразователь. Альтернативно, зародышеобразователь может образовывать зародыши только во фракции с высоким индексом расплава (вследствие короткого λ, проявляемого фракцией), предоставляя фракции с низким индексом расплава подвергаться обусловленному напряжением самообразованию зародышей кристаллизации на фибриллах по существу таким же образом, как если бы никакого зародышеобразователя не присутствовало. Независимо от действующего механизма, конечный результат состоит в том, что зародышеобразователь регулирует рост большего числа ламелей в полимере и имеет повышенное влияние на физические свойства полимера. Хотя вышеуказанный пример описывает использование бимодальных полимеров, такие же эффекты можно получить, используя полимодальные полимеры и физические смеси отдельных полимеров, поскольку каждый из этих альтернативных вариантов также обеспечивает средства снижения λ. Кроме того, аналогичных улучшений можно достичь путем выбора полимера с более узким распределением молекулярной массы (что показано меньшим индексом течения расплава). Более узкое распределение молекулярной массы обычно указывает на отсутствие высокомолекулярного "хвоста" или фракции в полимере, которая может увеличивать λ для полимера. Также, аналогичных улучшений можно достичь путем выбора полимера с меньшей степенью длинноцепочечной разветвленности, поскольку длинноцепочечная разветвленность может приводить к переплетению цепей молекул в расплаве, что может увеличивать λ.
Согласно второму варианту осуществления настоящее изобретение обеспечивает соединение, соответствующее структуре формулы (C)
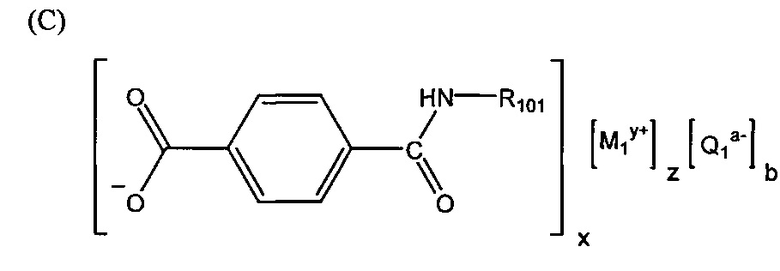 .
.
В структуре формулы (С) R101 выбрана из группы, состоящей из циклопентильной группы и фрагментов, соответствующих структуре формулы (CI). Структура формулы (CI) представляет собой
 .
.
В структуре (CI) R105 выбрана из группы, состоящей из водорода и галогенов. Переменная x представляет собой целое положительное число, каждый M1 представляет собой катион металла, y представляет собой валентность катиона; а z представляет собой целое положительное число. Переменная b равняется нулю или целому положительному числу. Если b представляет собой целое положительное число, каждый Q1 представляет собой отрицательно заряженный противоион, и а представляет собой валентность отрицательно заряженного противоиона. Значения x, y, z, а и b удовлетворяют уравнению х+(ab)=yz.
M1 может представлять собой любой из катионов, описанных выше как подходящие для соединения, соответствующего структуре формулы (I), включая катионы, указанные как предпочтительные для структуры формулы (I). Согласно предпочтительному варианту осуществления M1 представляет собой катион металла, выбранного из группы, состоящей из щелочных металлов и щелочноземельных металлов. Согласно другому предпочтительному варианту осуществления M1 представляет собой катион металла, выбранного из группы, состоящей из щелочных металлов. Согласно предпочтительному варианту осуществления M1 представляет собой катион лития. Q1, если присутствует, может представлять собой любой из анионов, описанных выше как подходящие для соединения, соответствующего структуре формулы (I), включая анионы, указанные как предпочтительные для структуры формулы (I).
Согласно предпочтительному варианту осуществления R101 представляет собой циклопентильную группу. Циклопентильная группа может быть незамещенной или замещенной. Замещенная циклопентильная группа может соответствовать структуре формулы (АС) выше. Предпочтительно циклопентильная группа является незамещенной. Согласно более конкретному варианту осуществления R101 представляет собой циклопентильную группу, переменная x равняется 1, M1 представляет собой катион лития, y равняется 1, z равняется 1, а b равняется нулю.
Согласно другому предпочтительному варианту осуществления R101 представляет собой фрагмент, соответствующий структуре формулы (CI). Согласно более конкретному варианту осуществления R101 представляет собой фрагмент, соответствующий структуре формулы (CI), а R105 представляет собой водород. Согласно другому конкретному варианту осуществления R101 представляет собой фрагмент, соответствующий структуре формулы (CI), R105 представляет собой водород, x равняется 1, M1 представляет собой катион лития, y равняется 1, z равняется 1, а b равняется нулю. Согласно другому конкретному варианту осуществления R101 представляет собой фрагмент, соответствующий структуре формулы (CI), а R105 представляет собой галоген, предпочтительно бром. Согласно более конкретному варианту осуществления R101 представляет собой фрагмент, соответствующий структуре формулы (CI), R105 представляет собой бром, x равняется 1, M1 представляет собой катион лития, y равняется 1, z равняется 1, а b равняется нулю.
В ряду дополнительных вариантов осуществления соединение данного второго варианта осуществления можно использовать в качестве зародышеобразователя для термопластичного полимера, как описано выше в первом варианте осуществления настоящего изобретения. В частности, эти дополнительные варианты осуществления включают композиции на основе термопластичного полимера, содержащие термопластичный полимер, предпочтительно полиолефиновый полимер (например, полиэтиленовый полимер), и одно или несколько конкретных соединений, описанных в предыдущих параграфах.
Согласно третьему варианту осуществления настоящее изобретение обеспечивает соединение, соответствующее структуре формулы (CX)
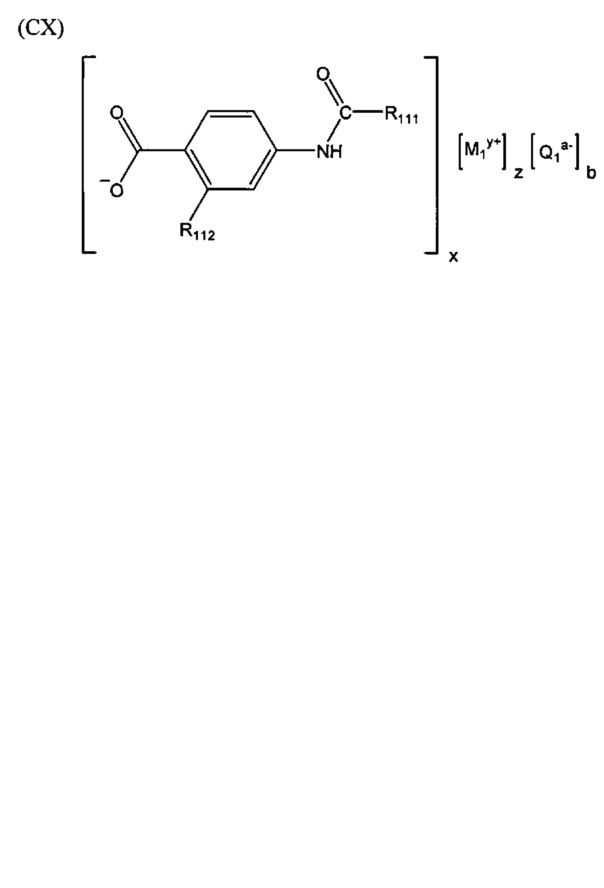 .
.
В структуре (СХ) R111 выбрана из группы, состоящей из циклопентильной группы и фрагментов, соответствующих структуре формулы (CXI); а R112 выбрана из группы, состоящей из водорода и гидроксигруппы. Структура формулы (CXI) представляет собой
 .
.
В структуре (CXI) R115 выбрана из группы, состоящей из водорода, галогена, метоксигруппы и фенила. Переменная x представляет собой целое положительное число, каждый M1 представляет собой катион металла, y представляет собой валентность катиона; а z представляет собой целое положительное число. Переменная b равняется нулю или целому положительному числу. Если b представляет собой целое положительное число, каждый Q1 представляет собой отрицательно заряженный противоион, и а представляет собой валентность отрицательно заряженного противоиона. Значения x, y, z, а и b удовлетворяют уравнению х+(ab)=yz. Кроме того, если R115 представляет собой водород, тогда R112 представляет собой водород, x равняется 1, M1 представляет собой катион лития, y равняется 1, z равняется 1, а b равняется нулю. Также, если R115 представляет собой метоксигруппу, тогда R112 представляет собой гидроксигруппу.
M1 может представлять собой любой из катионов, описанных выше как подходящие для соединения, соответствующего структуре формулы (I), включая катионы, указанные как предпочтительные для структуры формулы (I). Согласно предпочтительному варианту осуществления M1 представляет собой катион металла, выбранного из группы, состоящей из щелочных металлов и щелочноземельных металлов. Согласно другому предпочтительному варианту осуществления M1 представляет собой катион металла, выбранного из группы, состоящей из щелочных металлов. Согласно предпочтительному варианту осуществления M1 представляет собой катион лития. Q1, если присутствует, может представлять собой любой из анионов, описанных выше как подходящие для соединения, соответствующего структуре формулы (I), включая анионы, указанные как предпочтительные для структуры формулы (I).
Согласно предпочтительному варианту осуществления R111 представляет собой циклопентильную группу. Циклопентильная группа может быть незамещенной или замещенной. Замещенная циклопентильная группа может соответствовать структуре формулы (АС) выше. Предпочтительно циклопентильная группа является незамещенной. Согласно более конкретному варианту осуществления R111 представляет собой циклопентильную группу, переменная x равняется 1, M1 представляет собой катион лития, y равняется 1, z равняется 1, а b равняется нулю.
Согласно другому предпочтительному варианту осуществления R111 представляет собой фрагмент, соответствующий структуре формулы (CXI). Согласно более конкретному варианту осуществления R111 представляет собой фрагмент, соответствующий структуре формулы (CXI), а R115 представляет собой водород. Согласно другому более конкретному варианту осуществления R111 представляет собой фрагмент, соответствующий структуре формулы (CXI), а R115 представляет собой метоксигруппу. Согласно еще одному конкретному варианту осуществления R111 представляет собой фрагмент, соответствующий структуре формулы (CXI), R115 представляет собой метоксигруппу, x равняется 1, M1 представляет собой катион лития, y равняется 1, z равняется 1, а b равняется нулю. Согласно другому более конкретному варианту осуществления R111 представляет собой фрагмент, соответствующий структуре формулы (CXI), а R115 представляет собой галоген, предпочтительно хлор. Согласно еще более конкретному варианту осуществления R111 представляет собой фрагмент, соответствующий структуре формулы (CXI), а R115 представляет собой галоген, предпочтительно хлор, и R112 представляет собой водород. Согласно другому более конкретному варианту осуществления R111 представляет собой фрагмент, соответствующий структуре формулы (CXI), R115 представляет собой хлор, R112 представляет собой водород, а M1 представляет собой катион металла, выбранного из группы, состоящей из щелочных металлов, предпочтительно натрий. Согласно более конкретному варианту осуществления R111 представляет собой фрагмент, соответствующий структуре формулы (CXI), R115 представляет собой хлор, R112 представляет собой водород, x равняется 1, M1 представляет собой катион натрия, y равняется 1, z равняется 1, а b равняется нулю.
В ряду дополнительных вариантов осуществления соединение данного третьего варианта осуществления можно использовать в качестве зародышеобразователя для термопластичного полимера, как описано выше в первом варианте осуществления настоящего изобретения. В частности, эти дополнительные варианты осуществления включают композиции на основе термопластичного полимера, содержащие термопластичный полимер, предпочтительно полиолефиновый полимер (например, полиэтиленовый полимер), и одно или несколько конкретных соединений, описанных в предыдущих параграфах.
Согласно четвертому варианту осуществления настоящее изобретение обеспечивает соединение, соответствующее структуре формулы (СХХ)
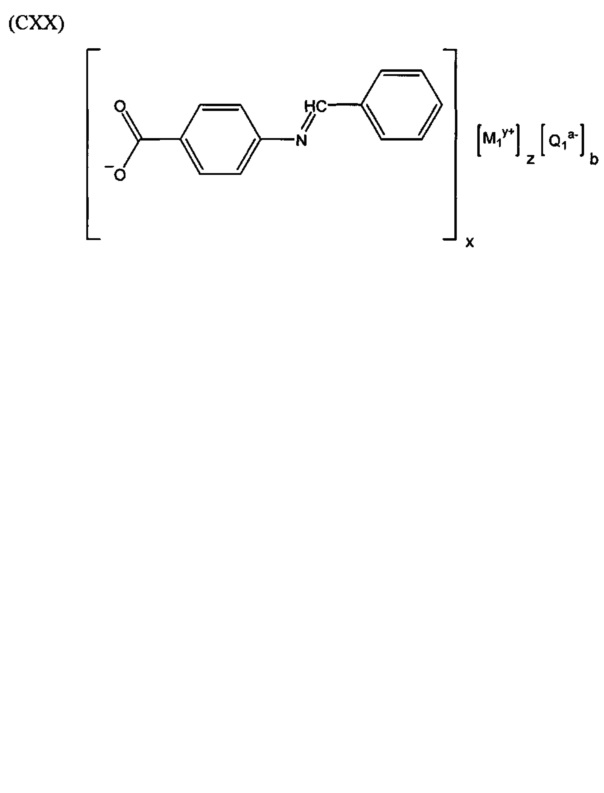 .
.
В структуре (СХХ) переменная x представляет собой целое положительное число. Каждый M1 представляет собой катион металла, выбранного из группы, состоящей из щелочных металлов, щелочноземельных металлов и цинка; y представляет собой валентность катиона; а z представляет собой целое положительное число. Переменная b равняется нулю или целому положительному числу. Если b представляет собой целое положительное число, каждый Q1 представляет собой отрицательно заряженный противоион, и а представляет собой валентность отрицательно заряженного противоиона. Значения x, y, z, а и b удовлетворяют уравнению х+(ab)=yz.
Согласно предпочтительному варианту осуществления M1 представляет собой катион металла, выбранного из группы, состоящей из щелочных металлов и щелочноземельных металлов. Согласно другому предпочтительному варианту осуществления M1 представляет собой катион металла, выбранного из группы, состоящей из щелочных металлов. Согласно более конкретному варианту осуществления M1 представляет собой катион лития. Согласно другому конкретному варианту осуществления x равняется 1, M1 представляет собой катион лития, y равняется 1, z равняется 1, а b равняется нулю.
В ряду дополнительных вариантов осуществления соединение данного четвертого варианта осуществления можно использовать в качестве зародышеобразователя для термопластичного полимера, как описано выше в первом варианте осуществления настоящего изобретения. В частности, эти дополнительные варианты осуществления включают композиции на основе термопластичного полимера, содержащие термопластичный полимер, предпочтительно полиолефиновый полимер (например, полиэтиленовый полимер), и одно или несколько конкретных соединений, описанных в предыдущих параграфах.
Согласно другому варианту осуществления настоящее изобретение обеспечивает композицию добавки, содержащую зародышеобразователь, описанный выше, и соединение поглотителя кислот. Зародышеобразователь, присутствующий в композиции, может быть любым одним или несколькими из соединений зародышеобразователей, описанных выше, таких как соединение, соответствующее структуре формулы (I), соединение, соответствующее структуре формулы (С), соединение, соответствующее структуре формулы (СХ), соединение, соответствующее структуре формулы (СХХ), или любой подходящей смесью таких соединений. Предпочтительно зародышеобразователь в композиции добавки выбран из группы, состоящей из соединений, соответствующих структуре формулы (СХ). Более предпочтительно зародышеобразователь представляет собой соединение, соответствующее структуре формулы (СХ), в которой R111 представляет собой водород, R112 представляет собой фрагмент, соответствующий структуре формулы (CXI), а R115 представляет собой галоген. Согласно более конкретному предпочтительному варианту осуществления зародышеобразователь представляет собой соединение, соответствующее структуре формулы (СХ), в которой R112 представляет собой водород, R111 представляет собой фрагмент, соответствующий структуре формулы (CXI), R115 представляет собой хлор, M1 представляет собой катион натрия, x равняется 1, y равняется 1, z равняется 1, а b равняется 0.
Предпочтительно поглотитель кислот выбран из группы, состоящей из металлических солей жирных кислот и синтетических гидроталькитных соединений. Подходящие металлические соли жирных кислот включают, помимо прочего, металлические соли С12-С22 жирных кислот, таких как стеариновая кислота. Согласно предпочтительному варианту осуществления поглотитель кислот выбран из группы, состоящей из цинковых, калиевых и лантановых солей стеариновой кислоты. Подходящие синтетические гидроталькитные соединения включают, помимо прочего, поглотитель кислот DHT-4A, продаваемый Kyowa Chemical Industry Co., Ltd.
Зародышеобразователь и поглотитель кислот могут присутствовать в композиции добавки в любых подходящих количествах. Например, зародышеобразователь и поглотитель кислот могут присутствовать в композиции добавки в отношении (зародышеобразователь к поглотителю кислот) от приблизительно 10:1 до приблизительно 1:10 на основании массы зародышеобразователя и поглотителя кислот в композиции. Более предпочтительно зародышеобразователь и поглотитель кислот присутствуют в композиции добавки в отношении (зародышеобразователь к поглотителю кислот) от приблизительно 4:1 до приблизительно 1:4, от приблизительно 3:1 до приблизительно 1:3, от приблизительно 1:1 до приблизительно 1:4 или от приблизительно 1:1 до приблизительно 1:3 на основании массы зародышеобразователя и поглотителя кислот в композиции добавки.
Неожиданно обнаружили, что зародышеобразователь и поглотитель кислот синергично взаимодействуют, когда композицию добавки, описанную выше, вводят в термопластичный полимер. В частности, обнаружили, что введение поглотителя кислот может улучшать работу зародышеобразователя. Например, введение как зародышеобразователя, так и поглотителя кислот может улучшать физические свойства полимера относительно тех, которые получают, если зародышеобразователь используют сам по себе. Кроме того, введение поглотителя кислот может обеспечивать достижение желаемого уровня улучшений физических свойств для полимера, используя меньшее количество зародышеобразователя, чем потребовалось бы, если бы зародышеобразователь добавляли сам по себе. Этот синергизм является особенно неожиданным, учитывая, что не наблюдалось того, что поглотитель кислот образует зародыши кристаллизации в полимере сам по себе. Например, введение поглотителя кислот самого по себе не имеет ощутимого влияния на физические свойства полимера.
Композиция добавки, описанная выше, предназначена для введения в термопластичный полимер, такой как полиэтиленовые и полипропиленовые полимеры, описанные ранее в настоящей заявке. В частности, считается, что композиция добавки является особенно эффективной при использовании в полиэтиленовом полимере высокой плотности. В этих полимерах введение композиции добавки, как наблюдали, значительно снижает усадку в машинном направлении, что указывает на улучшенную ориентацию в машинном направлении кристаллических ламелей, и значительно увеличивает прочность и деформационную теплостойкость полимера.
Следующие примеры также иллюстрируют объект, описанный выше, но, конечно, не должны рассматриваться как ограничивающие его объем каким-либо образом.
Пример получения EX1
Данный пример показывает получение сложного метилового эфира 4-хлоркарбонилбензойной кислоты, имеющего следующую структуру
 .
.
В 4 л бак с механической мешалкой, обратным холодильником, капельной воронкой, термометром, водяной баней и нагревательной плиткой вводили 438 г диметилтерефталата (DMT) и 2700 мл толуола. Бак нагревали до приблизительно 65°C для растворения всего DMT. После растворения раствор гидроксида калия (144,54 г в 700 мл метанола) добавляли по каплям в течение 45 минут. Реакционную смесь перемешивали при 65°C в течение трех часов, а затем реакционную смесь охлаждали до комнатной температуры в течение ночи. Твердое вещество выделяли после фильтрования и промывки при помощи 3750 мл толуола при 80°C. Продукт снова отфильтровывали и сушили в печи при 110°C. Выход составлял 465,9 г (95,3%).
В 2 л трехгорлую круглодонную колбу с механической мешалкой, капельной воронкой, водяной баней, термометром, продувкой азотом и нагревательной плиткой вводили 130,31 г продукта, полученного на предыдущей стадии, и 1000 мл толуола. Затем 48 мл тионилхлорида добавляли по каплям. После окончания добавления смесь нагревали до 67°C в течение трех часов. Реакционную смесь охлаждали до комнатной температуры и перемешивали в течение ночи. Содержимое отфильтровывали для сбора фильтрата. Избыточный растворитель удаляли под вакуумом и получали 86,52 г продукта (выход 73%).
Пример получения EX2
Данный пример показывает синтез N-циклопентилтерефталевой кислоты, имеющей следующую структуру
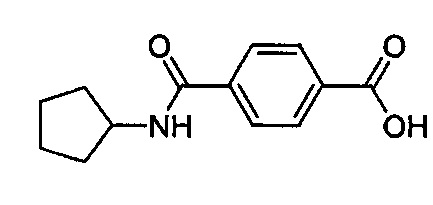 .
.
В 2 л кругло донную колбу загружали 15,44 г бикарбоната натрия, 15,75 г циклопентиламина, 0,5 г триэтиламина и 200 мл тетрагидрофурана (THF). Колбу охлаждали на ледяной бане, а затем раствор сложного метилового эфира 4-хлоркарбонилбензойной кислоты (36,78 г в приблизительно 100 мл THF) добавляли по каплям в колбу. После добавления смесь нагревали с обратным холодильником. Реакцию контролировали при помощи инфракрасной спектроскопии (ИК) до тех пор, пока пик на 1780 см-1 не исчезал. Затем смесь выливали в приблизительно 2 л воды и перемешивали в течение приблизительно 20 минут. Твердый продукт выделяли после фильтрования и сушки в печи при 100°C.
В 2 л трехгорлую кругло донную колбу вводили 21 г продукта, полученного на предыдущей стадии, и 150 мл метанола. Смесь нагревали с обратным холодильником и добавляли гидроксид калия (4,76 г, предварительно растворенный в метаноле). Реакцию контролировали при помощи ИК-спектроскопии до тех пор, пока пик на 1720 см-1 не исчезал. Затем добавляли 400 мл воды и отфильтровывали любые нерастворимые примеси. pH фильтрата доводили приблизительно до 2, и образовывался осадок. Твердый продукт отфильтровывали и сушили в печи при 100°C.
Пример получения EX3
Данный пример показывает получение калиевой соли N-циклопентилтерефталевой кислоты, имеющей следующую структуру
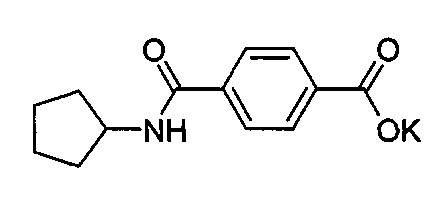 .
.
В лабораторном стакане 10 г N-циклопентилтерефталевой кислоты вводили в 50 мл H2O. Затем 2,41 г гидроксида калия растворяли в отдельном лабораторном стакане в приблизительно 20 мл H2O. Раствор гидроксида калия добавляли в суспензию N-циклопентилтерефталевой кислоты, и большая часть твердого вещества растворялась. Для удаления какого-либо нерастворенного материала смесь отфильтровывали. Собирали фильтрат и выпаривали воду с получением продукта. Продукт сушили в течение ночи в печи при 110°C.
Пример получения EX4
Данный пример показывает получение N-фенилтерефталевой кислоты, имеющей следующую структуру
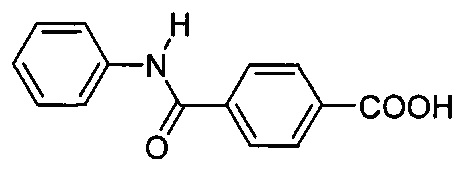 .
.
В 1 л трехгорлую круглодонную колбу с магнитной мешалкой, капельной воронкой, ледяной баней, продувкой азотом, газоочистителем и нагревательной плиткой вводили 93,13 г анилина, 42,30 г бикарбоната натрия, 0,5 г триэтиламина и 300 мл тетрагидрофурана (THF). Смесь охлаждали ниже 10°C, а затем добавляли по каплям раствор 100 г сложного метилового эфира 4-хлоркарбонилбензойной кислоты в 100 мл тетрагидрофурана. Температуру поддерживали приблизительно на 10°C при добавлении. После добавления смесь нагревали с обратным холодильником и контролировали завершение реакции при помощи ИК-спектроскопии (исчезновение пика при 1780 см-1). После завершения реакции реакционную смесь разбавляли до 2 л холодной деионизированной (DI) водой и перемешивали в течение приблизительно 20 минут. Твердый продукт отфильтровывали и сушили в печи при 110°C. После сушки получали 105,6 г продукта (выход 82,2%).
В 1 л колбу Эрленмейера с магнитной мешалкой и пластиной для смешивания вводили 15,94 г продукта, полученного на предыдущей стадии, и 200 мл метанола. Затем вводили гидроксид калия (3,87 г), который предварительно растворяли в метаноле. Реакцию контролировали при помощи ИК-спектроскопии (исчезновение пика приблизительно при 1720 см-1). После завершения реакции реакционную смесь разбавляли 400 мл воды. Твердые примеси удаляли фильтрованием и pH фильтрата доводили приблизительно до 2. Продукт осаждался на этой стадии, и его выделяли фильтрованием. Продукт промывали дополнительным количеством DI-воды до нейтрального состояния, и сушили его в печи при 100°C. После сушки получали 14,47 г продукта (выход 95%).
Пример получения EX5
Данный пример показывает получение литиевой соли N-фенилтерефталевой кислоты, имеющей следующую структуру
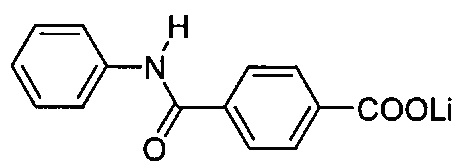 .
.
В 500 мл колбу Эрленмейера с магнитной мешалкой и пластиной для смешивания вводили 13,3 г N-фенилтерефталевой кислоты и 200 мл воды. Смесь нагревали практически до кипения, а затем вводили водный раствор гидроксида лития (содержащий 1,49 г безводного гидроксида лития). Реакцию контролировали при помощи ИК-спектроскопии (исчезновение пика приблизительно при 1677 см-1). После завершения реакции реакционную смесь охлаждали и отфильтровывали для выделения продукта. Продукт сушили в печи при 110°C и получали 11,56 г продукта.
Пример получения EX6
Данный пример показывает получение 4-(4-бромбензоиламино)бензойной кислоты, имеющей следующую структуру
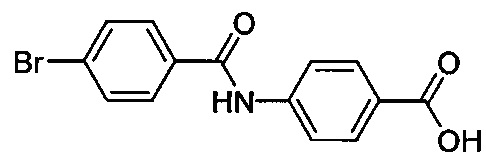 .
.
В 1 л трехгорлую круглодонную колбу вводили 40 г 4-аминобензойной кислоты и 400 мл диоксана. Смесь перемешивали до растворения кислоты. Затем раствор 4-бромбензоилхлорида (32,04 г в 100 мл диоксана) добавляли по каплям в реакционную смесь. После добавления реакционную смесь перемешивали в течение ночи, а затем отфильтровывали для выделения твердого вещества. Твердое вещество промывали кипящей водой, а затем холодной DI-водой, пока pH не становился нейтральным. После сушки продукт получали с выходом 99,6%.
Пример получения ЕХ7
Данный пример показывает получение калиевой соли 4-(4-бромбензоиламино)бензойной кислоты, имеющей следующую структуру
Пример получения EX9
Данный пример показывает получение кальциевой соли 4-(4-бромбензоиламино)бензойной кислоты, имеющей следующую структуру
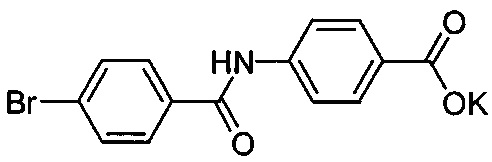 .
.
В лабораторный стакан вводили 25 г 4-(4-бромбензоиламино)бензойной кислоты и 200 мл DI-воды. Смесь перемешивали до тех пор, пока она не образовывала однородную суспензию. Затем вводили раствор гидроксида калия (4,4 г в 100 мл воды). Реакционную смесь перемешивали в течение ночи, и значение pH падало до 10,6. Твердый продукт отфильтровывали и сушили в печи при 110°C.
Пример получения ЕХ8
Данный пример показывает получение литиевой соли 4-(4-бромбензоиламино)бензойной кислоты, имеющей следующую структуру
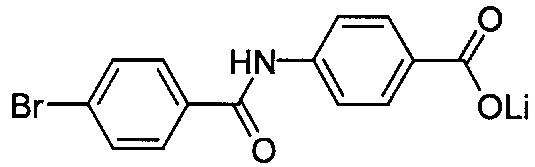 .
.
В лабораторном стакане 3 г 4-(4-бромбензоиламино)бензойной кислоты диспергировали в приблизительно 50 мл воды при перемешивании. Затем в суспензию добавляли раствор моногидрата гидроксида лития (0,39 г в 50 мл H2O). Реакционную смесь перемешивали в течение ночи, а затем твердое вещество выделяли фильтрованием. Фильтрат промывали DI-водой, а затем сушили в печи при 110°C.
Пример получения ЕХ9
Данный пример показывает получение литиевой соли 4-(4-бромбензоиламино)бензойной кислоты, имеющей следующую структуру
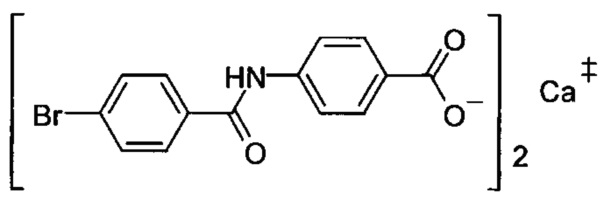 .
.
В лабораторном стакане 3 г 4-(4-бромбензоиламино)бензойной кислоты диспергировали в приблизительно 50 мл воды при перемешивании. Затем в суспензию вводили раствор гидроксида кальция (0,35 г в 50 мл воды). Реакционную смесь перемешивали в течение выходных, а затем отфильтровывали для выделения конечного твердого вещества. Фильтрат промывали DI-водой, а затем сушили в печи при 110°C.
Пример получения ЕХ10
Данный пример показывает получение 4-(циклопропанкарбониламино)-бензойной кислоты, имеющей следующую структуру
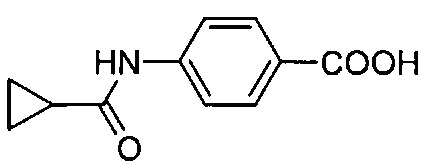 .
.
В трехгорлой колбе 20,3 г карбоната натрия диспергировали в 80 мл THF в атмосфере N2. При перемешивании 13,1 г дисперсии 4-аминобензойной кислоты (в 15 мл THF) и 10,0 г раствора циклопропанкарбонилхлорида (в 15 мл THF) отдельно добавляли по каплям. Реакционную смесь перемешивали в течение ночи. Затем добавляли 10,15 г карбоната натрия и смесь перемешивали в течение дополнительных 3 часов. Затем THF выпаривали и реакционную смесь переносили в 1 л лабораторный стакан и разбавляли 600 мл воды. pH доводили приблизительно до 2 при помощи соляной кислоты с получением продукта в виде осадка. Смесь отфильтровывали для выделения осадка и осадок сушили в вакуумной печи.
Пример получения EX11
Данный пример показывает получение литиевой соли 4-(циклопропанкарбониламино)бензойной кислоты, имеющей следующую структуру
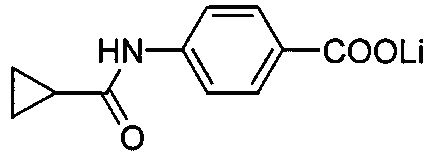 .
.
В лабораторном стакане 5 г 4-(циклопропанкарбониламино)бензойной кислоты диспергировали в 20 мл воды, а затем добавляли 1,13 г моногидрата гидроксида лития. После 20 минут перемешивания реакционную смесь концентрировали под вакуумом с получением продукта. Продукт сушили, и выход составлял приблизительно 2,59 г.
Пример получения ЕХ12
Данный пример показывает получение натриевой соли 4-(циклопропанкарбониламино)бензойной кислоты, имеющей следующую структуру
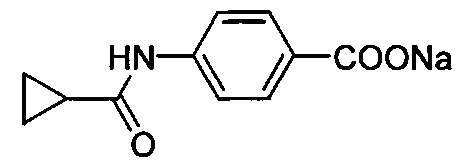 .
.
В лабораторном стакане 20 г 4-(циклопропанкарбониламино)бензойной кислоты перемешивали в 80 мл воды, а затем добавляли 8,58 г раствора гидроксида натрия (50% в воде). После 20 минут перемешивания реакционную смесь концентрировали под вакуумом с получением продукта.
Пример получения ЕХ13
Данный пример показывает получение литиевой соли 4-стильбенкарбоновой кислоты, имеющей следующую структуру
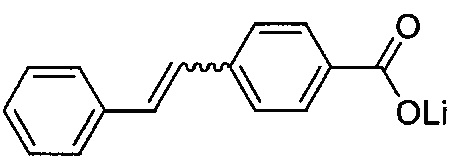 .
.
В лабораторном стакане 1 г 4-стильбенкарбоновой кислоты (смеси транс- и цис-изомеров) диспергировали в 25 мл воды. Затем 0,19 г моногидрата гидроксида лития растворяли в 25 мл воды, а затем добавляли в суспензию кислоты. Реакционную смесь перемешивали в течение ночи. Твердый продукт выделяли фильтрованием, промывали три раза водой, а затем сушили в печи при 110°C.
Пример получения EX14
Данный пример показывает получение 4-(1,3-диоксооктагидроизоиндол-2-ил)-2-гидроксибензойной кислоты, имеющей следующую структуру
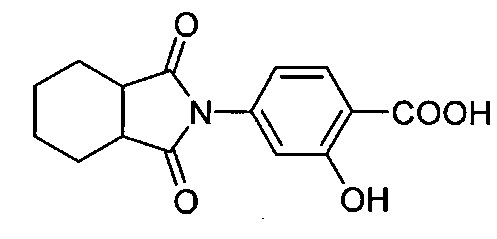 .
.
В 500 мл четырехгорлую круглодонную колбу, оборудованную датчиком температуры, колбонагревателем, мешалкой и конденсатором, загружали 21,20 г гексагидрофталевого ангидрида и 50 мл уксусной кислоты. При 70°C смесь перемешивали до однородности, а затем загружали 21,7 г 4-аминосалициловой кислоты и 100 мл уксусной кислоты. После нагревания с обратным холодильником в течение 6 часов содержимое выливали в ледяную DI H2O и отфильтровывали под вакуумом для выделения твердого вещества. После промывки при помощи DI H2O и сушки получали 33,07 г продукта.
Пример получения ЕХ15
Данный пример показывает получение цинковой соли 4-(1,3-диоксооктагидроизоиндол-2-ил)-2-гидроксибензойной кислоты, имеющей следующую структуру
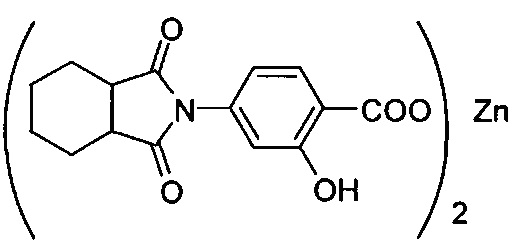 .
.
В лабораторном стакане суспендировали 4-(1,3-диоксооктагидроизоиндол-2-ил)-2-гидроксибензойную кислоту в приблизительно 100-150 мл воды при помощи магнитной мешалки. Затем 25% раствор гидроксида натрия медленно добавляли, пока pH не устанавливался на 12,5, и раствор не становился прозрачным. Затем один эквивалент хлорида цинка добавляли (использовали 1 экв. вместо 0,5, поскольку ионы металла также могут образовывать комплекс с мета-гидроксигруппой). Продукты осаждались, и смесь отфильтровывали для выделения продукта.
Пример получения EX16
Данный пример показывает получение 4-(2,2-диметилпропиониламино)-бензойной кислоты, имеющей следующую структуру
 .
.
В трехгорлую круглодонную колбу с закрепленной сверху мешалкой, датчиком температуры, баней с сухим льдом и обратным холодильником вводили 25 г 4-аминобензойной кислоты, 15,12 г кальцинированной соды и 200 мл THF. При перемешивании 21,98 г пивалоилхлорида добавляли по каплям в течение 1-1,5 часов. Затем добавляли 22,68 г кальцинированный соды и смесь нагревали до 40°C, чтобы довести реакцию до конца. Полученную смесь разбавляли 2 л DI H2O. pH смеси доводили до 2,37 концентрированной соляной кислотой, а затем смесь отфильтровывали для выделения продукта.
Пример получения EX17
Данный пример показывает получение калиевой соли 4-(2,2-диметилпропиониламино)бензойной кислоты, имеющей следующую структуру
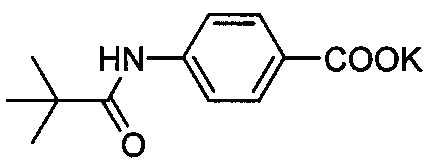 .
.
В лабораторном стакане 4-(2,2-диметилпропиониламино)бензойную кислоту суспендировали в приблизительно 100-150 мл воды при помощи магнитной мешалки. Затем 25% раствор гидроксида калия медленно добавляли, пока pH не устанавливался на 12,5, и раствор не становился прозрачным. Воду отгоняли и получали продукт.
Пример получения EX18
Данный пример показывает получение кальциевой соли 4-(2,2-диметилпропиониламино)бензойной кислоты, имеющей следующую структуру
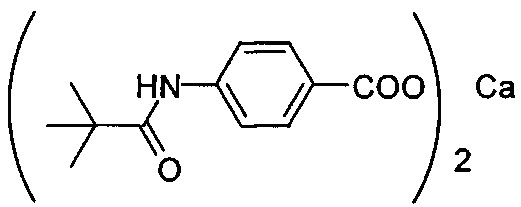 .
.
В лабораторном стакане 4-(2,2-диметилпропиониламино)бензойную кислоту суспендировали в приблизительно 100-150 мл воды при помощи магнитной мешалки. Затем 25% раствор гидроксида калия медленно добавляли, пока pH не устанавливался на 12,5, и раствор не становился прозрачным. Затем добавляли один эквивалент хлорида кальция. Продукт осаждался, и смесь отфильтровывали для выделения продукта.
Пример получения ЕХ19
Данный пример показывает получение N-4-метоксибензоиламиносалициловой кислоты, имеющей следующую структуру
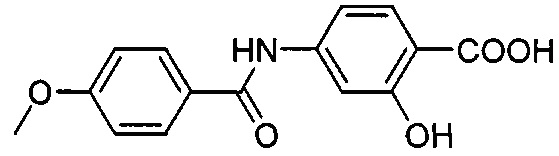 .
.
Трехгорлую круглодонную колбу оборудовали закрепленной сверху мешалкой, датчиком температуры, баней с сухим льдом и обратным холодильником. Затем в колбу загружали 12,36 г 4-аминосалициловой кислоты, 16,75 г кальцинированной соды и 500 мл тетрагидрофурана. Смесь охлаждали ниже 10°C, а затем 14,85 г 4-метоксибензоилхлорида добавляли по каплям в течение 1-1,5 часов. Полученную смесь разбавляли 2 л воды и отфильтровывали для выделения продукта.
Пример получения ЕХ20
Данный пример показывает получение N-4-метоксибензоиламиносалициловой кислоты, имеющей следующую структуру
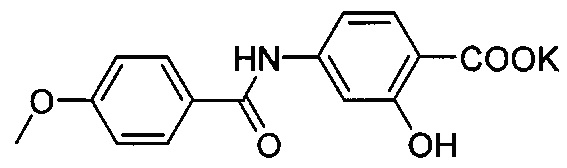 .
.
В лабораторном стакане N-4-метоксибензоиламиносалициловую кислоту суспендировали в приблизительно 100-150 мл воды при помощи магнитной мешалки. Затем 25% раствор гидроксида калия медленно добавляли, пока pH не устанавливался на 12,5, и раствор не становился прозрачным. Воду отгоняли и получали продукт.
Пример получения EX21
Данный пример показывает получение литиевой соли N-4-метоксибензоиламиносалициловой кислоты, имеющей следующую структуру
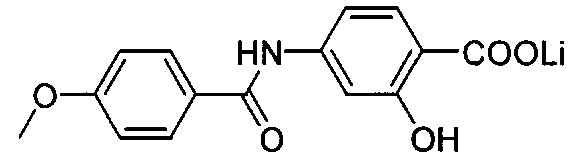 .
.
В лабораторном стакане N-4-метоксибензоиламиносалициловую кислоту суспендировали в приблизительно 100-150 мл воды при помощи магнитной мешалки. Затем медленно добавляли 25% раствор гидроксида лития, пока pH не устанавливался на 12,5, и раствор не становился прозрачным. Воду отгоняли и получали продукт.
Пример получения ЕХ22
Данный пример показывает получение натриевой соли N-4-метоксибензоиламиносалициловой кислоты, имеющей следующую структуру
 .
.
В лабораторном стакане N-4-метоксибензоиламиносалициловую кислоту суспендировали в приблизительно 100-150 мл воды при помощи магнитной мешалки.
Затем медленно добавляли 25% раствор гидроксида натрия, пока pH не устанавливался на 12,5, и раствор не становился прозрачным. Воду отгоняли и получали продукт.
Пример получения ЕХ23
Данный пример показывает получение 4-(циклобутанкарбониламино)бензойной кислоты, имеющей следующую структуру
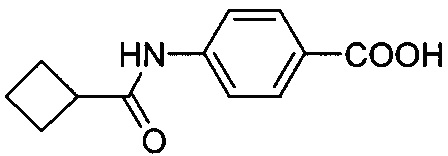 .
.
В колбу вводили 20,3 г карбоната натрия, 6,3 г 4-аминобензойной кислоты и 80 мл THF. Затем вводили 5 г циклобутанкарбонилхлорида (разбавленного в 15 мл THF). Реакционную смесь перемешивали в атмосфере азота в течение выходных и выпаривали THF. Смесь переносили в 1 л лабораторный стакан и разбавляли 400 мл воды. Раствор подкисляли соляной кислотой, пока pH не становился равным приблизительно 2, и продукт не осаждался. Продукт выделяли фильтрованием, затем промывали водой и сушили.
Пример получения ЕХ24
Данный пример показывает получение калиевой соли 4-(циклобутанкарбониламино)бензойной кислоты, имеющей следующую структуру
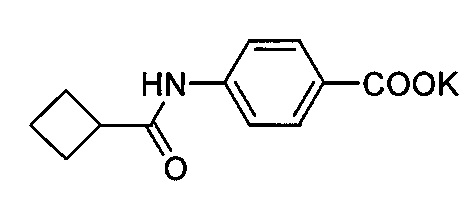 .
.
В лабораторном стакане 4-(циклобутанкарбониламино)бензойную кислоту суспендировали в приблизительно 100-150 мл воды при помощи магнитной мешалки. Затем добавляли 25% раствор гидроксида натрия для повышения pH раствора приблизительно до 12,5. Получали прозрачный раствор и затем воду отгоняли для выделения продукта в виде порошка.
Пример получения ЕХ25
Данный пример показывает получение 4-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)бензойной кислоты, имеющей следующую структуру
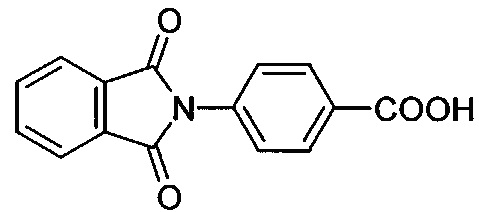 .
.
В 500 мл четырехгорлую кругло донную колбу, оборудованную датчиком температуры, колбонагревателем, мешалкой и конденсатором, загружали 25,03 г фталевого ангидрида и 87 мл уксусной кислоты. При 70°C реакционную смесь перемешивали, пока не получали прозрачный раствор. Затем 24,37 г 4-аминобензойной кислоты загружали и смесь нагревали с обратным холодильником в течение 2 часов. Затем добавляли еще 50 мл уксусной кислоты. Содержимое выливали в DI H2O. Продукт выделяли фильтрованием, а затем промывали DI H2O. После сушки получали 43,345 г продукта (выход 96%).
Пример получения ЕХ26
Данный пример показывает получение литиевой соли 4-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)бензойной кислоты, имеющей следующую структуру
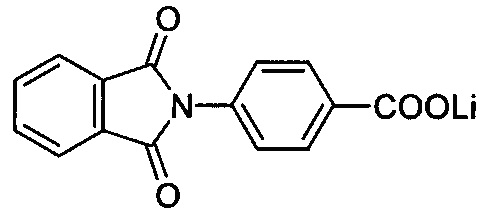 .
.
В 1000 мл лабораторный стакан, оборудованный мешалкой, загружали 5,03 г 4-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-бензойной кислоты и 100 мл DI H2O. Гидроксид лития загружали в лабораторный стакан и смесь перемешивали до полного растворения кислоты. Если кислота полностью не растворялась, добавляли гидроксид лития с шагом 0,1 г, пока кислота полностью не растворялась. Выпаривание в роторном испарителе использовали для извлечения продукта.
Пример получения ЕХ27
Данный пример показывает получение натриевой соли 4-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)бензойной кислоты, имеющей следующую структуру
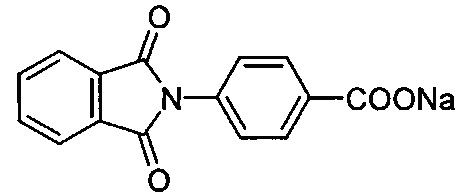 .
.
В 1000 мл лабораторный стакан, оборудованный размещенной сверху мешалкой, вводили 4-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)бензойную кислоту и 100 мл DI H2O. Раствор перемешивали и 25% раствор гидроксида натрия медленно добавляли до полного растворения кислоты. Воду удаляли путем выпаривания в роторном испарителе для извлечения продукта.
Пример получения ЕХ28
Данный пример показывает получение сложного метилового эфира N-циклобутилтерефталевой кислоты, имеющего следующую структуру
 .
.
В трехгорлую круглодонную колбу вводили 14,8 г карбоната натрия и 50 мл тетрагидрофурана. Затем добавляли 5 г циклобутиламина. Затем 11,59 г метилбензоата 4-карбонилхлорида (разбавленного в 30 мл тетрагидрофурана) добавляли по каплям. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь затем переносили в лабораторный стакан и перемешивали с 200 мл воды. Смесь подкисляли 1М соляной кислотой. Затем смесь переносили в разделительную воронку и экстрагировали этилацетатом три раза (80 мл каждый). Органическую фазу концентрировали для выделения продукта.
Продукт, полученный на предыдущей стадии, перемешивали с 200 мл воды, а затем нагревали до 80°C. 50% раствор гидроксид натрия добавляли во время нагревания для поддержания pH выше 12. Через 4 часа реакционную смесь подкисляли до pH приблизительно 2, и продукт осаждался. Продукт выделяли фильтрованием.
Пример получения ЕХ29
Данный пример показывает получение литиевой соли N-циклобутилтерефталевой кислоты, имеющей следующую структуру
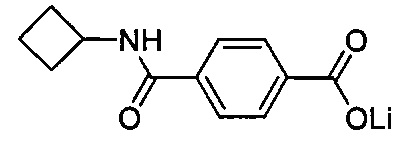 .
.
В лабораторном стакане 300 мг N-циклобутилтерефталевой кислоты диспергировали в приблизительно 40 мл воды. Гидроксид лития медленно добавляли, пока pH не становился равным приблизительно 12. Затем раствор концентрировали для получения желаемого продукта.
Пример получения ЕХ30
Данный пример показывает получение N-циклопропилтерефталевой кислоты, имеющей следующую структуру
 .
.
В колбу вводили 9,3 г карбоната натрия, 5 г циклопропиламина и 80 мл тетрагидрофурана. Затем 16,43 г сложного метилового эфира 4-хлоркарбонилбензойной кислоты разбавляли в 30 мл THF, а затем добавляли по каплям в реакционную смесь. Реакционную смесь перемешивали в течение ночи. Продукт извлекали из смеси при помощи 400 мл воды. Продукт выделяли и сушили, получилось приблизительно 18 г.
В колбе 18 г продукта, полученного на предыдущей стадии, перемешивали с 200 мл воды, а затем нагревали до 80°C. 50% раствор гидроксид натрия добавляли во время нагревания для поддержания pH выше 12. Через 4 часа реакционную смесь подкисляли до pH приблизительно 2, и продукт осаждался. Раствор отфильтровывали для получения продукта.
Пример получения EX31
Данный пример показывает получение литиевой соли N-циклопропилтерефталевой кислоты, имеющей следующую структуру
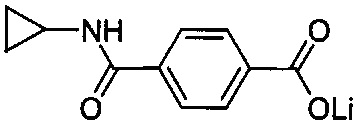 .
.
В лабораторном стакане 2,46 г влажной N-циклопропилтерефталевой кислоты перемешивали с 100 мл воды, а затем моногидрат гидроксида лития добавляли, пока pH не составил 12. Реакционную смесь перемешивали в течение 20 минут и концентрировали для получения продукта.
Пример получения EX32
Данный пример показывает получение кальциевой соли N-циклопропилтерефталевой кислоты, имеющей следующую структуру
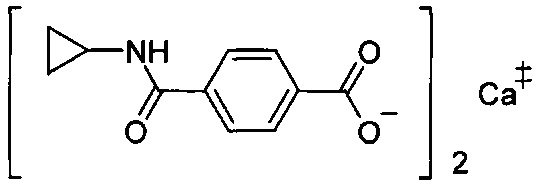 .
.
В лабораторном стакане 2,51 г N-циклопропилтерефталевой кислоты смешивали с 50 мл воды. Затем 50% раствор гидроксида натрия добавляли, пока pH не составил 12. Реакционную смесь перемешивали в течение 20 минут. Затем 3,52 г дигидрата хлорида кальция добавляли в раствор для получения продукта. Продукт выделяли фильтрованием и сушили в печи.
Пример получения EX33
Данный пример показывает получение цинковой соли N-циклопроилтерефталевой кислоты, имеющей следующую структуру
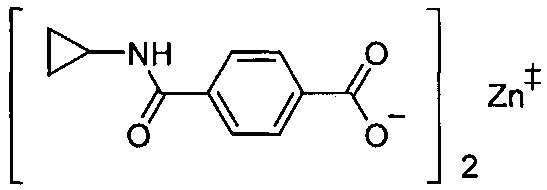 .
.
В лабораторном стакане 2,51 г N-циклопропилтерефталевой кислоты смешивали с 50 мл воды. Затем 50% раствор гидроксида натрия добавляли, пока pH не составил 12. Реакционную смесь перемешивали в течение 20 минут. Затем 3,27 г хлорида цинка добавляли в раствор для получения продукта. Продукт выделяли фильтрованием и сушили в печи.
Пример получения ЕХ34
Данный пример показывает получение 4-(4-метоксибензоиламино)бензойной кислоты, имеющей следующую структуру
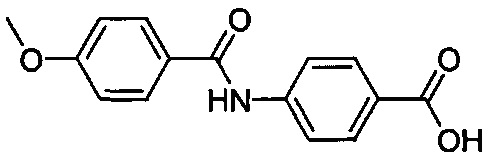 .
.
В 1 л трехгорлую колбу, оборудованную закрепленной сверху мешалкой, датчиком температуры, баней с сухим льдом и обратным холодильником, вводили 25 г 4-аминобензойной кислоты, 45,39 г кальцинированной соды и 200 мл тетрагидрофурана. При перемешивании 31,10 г 4-метоксибензоилхлорида добавляли по каплям в течение периода 1-1,5 часа. Температуру поддерживали приблизительно на 10°C при добавлении. После завершения реакции смесь разбавляли 2 л воды. pH снижали приблизительно до 2 при помощи соляной кислоты для осаждения продукта. Продукт выделяли фильтрованием и сушили в печи.
Пример получения ЕХ35
Данный пример показывает получение натриевой соли 4-(4-метоксибензоиламино)бензойной кислоты, имеющей следующую структуру
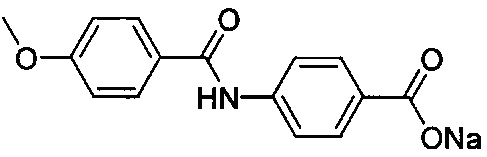 .
.
В лабораторном стакане 24 г 4-(4-метоксибензоиламино)бензойной кислоты смешивали с 200 мл воды. Затем 50% раствор гидроксида натрия медленно добавляли, пока не получали постоянное значение pH 12. Раствор концентрировали под вакуумом для обеспечения натриевой соли 4-(4-метоксибензоиламино)бензойной кислоты.
Пример получения ЕХ36
Данный пример показывает получение литиевой соли 4-(4-метоксибензоиламино)бензойной кислоты, имеющей следующую структуру
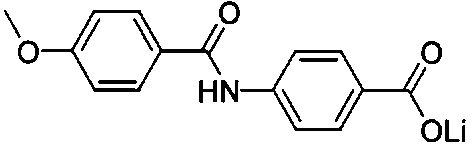 .
.
В лабораторном стакане 6 г 4-(4-меоксибензоиламино)бензойной кислоты перемешивали с 100 мл воды, и моногидрат гидроксида лития медленно добавляли, пока pH не устанавливался на 12. Реакционную смесь перемешивали в течение 20 минут, а затем концентрировали под вакуумом для получения продукта.
Пример получения ЕХ37
Данный пример показывает получение N-циклогептилтерефталевой кислоты, имеющей следующую структуру
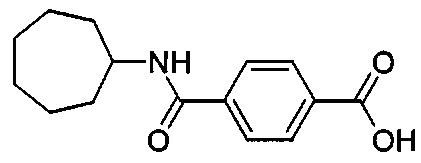 .
.
В 1 л круглодонную колбу загружали 9,3 г бикарбоната натрия, 5 г циклогептиламина и 80 мл тетрагидрофурана (THF). Колбу охлаждали на ледяной бане. Затем раствор сложного метилового эфира 4-хлоркарбонилбензойной кислоты (8,32 г в приблизительно 30 мл THF) добавляли по каплям в колбу. После добавления реакционную смесь нагревали с обратным холодильником. Реакцию контролировали при помощи ИК-спектроскопии до тех пор, пока пик на 1780 см-1 не исчезал. Затем смесь выливали в приблизительно 400 л воды и перемешивали в течение приблизительно 20 минут. Продукт выделяли фильтрованием и сушили в печи при 100°C.
В колбе 9,1 г продукта с предыдущей стадии смешивали с 200 мл воды. 50% раствор NaOH добавляли, пока pH не становился равным приблизительно 12. Реакционную смесь нагревали до 80°C, перемешивали в течение 4 часов и pH поддерживали на 12 во время реакции. После того как тонкослойная хроматография показала завершение реакции, pH доводили до 2 для осаждения продукта. Продукт отфильтровывали и промывали.
Пример получения ЕХ38
Данный пример показывает получение натриевой соли N-циклогептилтерефталевой кислоты, имеющей следующую структуру
 .
.
В колбе 8,8 г N-циклогептилтерефталевой кислоты смешивали с 200 мл воды и 50% раствор гидроксида натрия медленно добавляли, пока pH не устанавливался на 12. Еще 20 минут раствор перемешивали, а затем концентрировали под вакуумом для получения продукта.
Пример получения ЕХ39
Данный пример показывает получение 4-(1,3-диоксо-1Н,3H-бензо[де]изохинолин-2-ил)-2-гидроксибензойной кислоты, имеющей следующую структуру
 .
.
В 500 мл четырехгорлую круглодонную колбу, оборудованную датчиком температуры, колбонагревателем, мешалкой и конденсатором, загружали 17,95 г нафтойного ангидрида и 87 мл уксусной кислоты. Смесь нагревали до 70°C и перемешивали, пока не получали прозрачный раствор. Раствор имел светло-желтый цвет. Затем 14,58 г 4-аминосалициловой кислоты добавляли в раствор. После нагревания с обратным холодильником в течение 6 часов реакционную смесь выливали в воду. Продукт выделяли фильтрованием, а затем промывали водой. После сушки 22,18 г продукта получали в виде коричневого порошка.
Пример получения ЕХ40
Данный пример показывает получение натриевой соли 4-(1,3-диоксо-1Н,3H-бензо[де]изохинолин-2-ил)-2-гидроксибензойной кислоты, имеющей следующую структуру
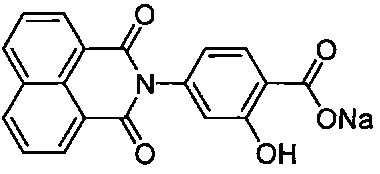 .
.
В лабораторном стакане 4-(1,3-диоксо-1Н,3H-бензо[де]изохинолин-2-ил)-2-гидроксибензойную кислоту смешивали с 200 мл воды. Затем 50% раствор гидроксида натрия медленно добавляли, пока не получали постоянное значение pH 12. Раствор концентрировали под вакуумом для получения натриевой соли 4-(1,3-диоксо-1Н,3H-бензо[де]изохинолин-2-ил)-2-гидроксибензойной кислоты.
Пример получения ЕХ41
Данный пример показывает получение калиевой соли 4-(1,3-диоксо-1Н,3H-бензо[де]изохинолин-2-ил)-2-гидроксибензойной кислоты, имеющей следующую структуру
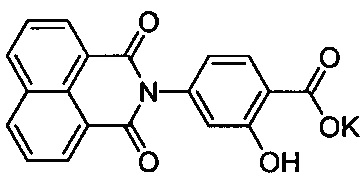 .
.
В лабораторном стакане 4-(1,3-диоксо-1Н,3H-бензо[де]изохинолин-2-ил)-2-гидроксибензойную кислоту смешивали с 200 мл воды. Затем гидроксид калия медленно добавляли, пока не получали постоянное значение pH 12. Раствор концентрировали под вакуумом для получения калиевой соли 4-(1,3-диоксо-1Н,3H-бензо[де]изохинолин-2-ил)-2-гидроксибензойной кислоты.
Пример получения ЕХ42
Данный пример показывает получение N-(3,4-диметилфенил)терефталевой кислоты, имеющей следующую структуру
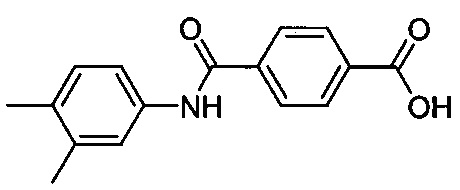 .
.
Продукт получали таким же способом, как использованный в примере получения ЕХ37, используя 3,4-диметиланилин вместо циклогептиламина.
Пример получения ЕХ43
Данный пример показывает получение калиевой соли N-(3,4-диметилфенил)терефталевой кислоты, имеющей следующую структуру
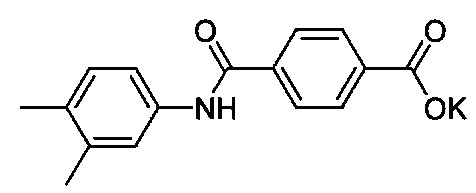 .
.
В лабораторном стакане N-(3,4-диметилфенил)терефталевую кислоту смешивали с 200 мл воды. Затем гидроксид калия медленно добавляли, пока не получали постоянное значение pH 12 и прозрачный раствор. Раствор концентрировали под вакуумом, получая желаемый продукт.
Пример получения EX44
Данный пример показывает получение литиевой соли N-(3,4-диметилфенил)терефталевой кислоты, имеющей следующую структуру
 .
.
В лабораторном стакане N-(3,4-диметилфенил)терефталевую кислоту смешивали с 200 мл воды. Затем моногидрат гидроксида лития медленно добавляли, пока не получали постоянное значение pH 12. Раствор концентрировали под вакуумом, получая желаемый продукт.
Пример получения ЕХ45
Данный пример показывает получение 4-бензоиламинобензойной кислоты, имеющей следующую структуру
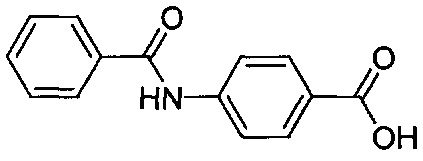 .
.
В 1 л лабораторном стакане с механической мешалкой перемешивали 27,4 г 4-аминобензойной кислоты (0,2 моль) в 300 мл DI H2O. Затем 21,2 г (0,2 моль) карбоната натрия добавляли, пока значение pH не составило 9,1, и вся 4-аминобензойная кислота не растворялась в воде.
Затем 56,24 г (0,4 моль) бензоилхлорида добавляли по каплям в лабораторный стакан при комнатной температуре. Реакционную смесь перемешивали в течение ночи. Твердое вещество образовалось во время реакции, и pH устанавливался на 4,0. pH дополнительно снижали приблизительно до 2 при помощи соляной кислоты. Продукт выделяли фильтрованием и промывали горячей водой для удаления избытка бензойной кислоты. Твердый продукт сушили в печи при 110°C и получали 44,21 г продукта (выход 96,7%).
Пример получения EX46
Данный пример показывает получение литиевой соли 4-бензоиламинобензойной кислоты, имеющей следующую структуру
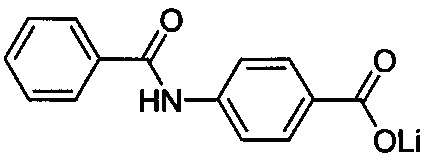 .
.
В 500 мл лабораторном стакане 44,21 г 4-бензоамидобензойной кислоты смешивали с приблизительно 250 мл воды. Затем вводили 7,69 г моногидрата гидроксида лития (растворенного в приблизительно 100 мл воды). Реакционную смесь перемешивали в течение ночи, и значение pH становилось нейтральным. Твердый продукт выделяли фильтрованием и сушили в печи при 110°C, получали 39,7 г материала (выход 88%).
Пример получения ЕХ47
Данный пример показывает получение магниевой соли 4-бензоиламинобензойной кислоты, имеющей следующую структуру
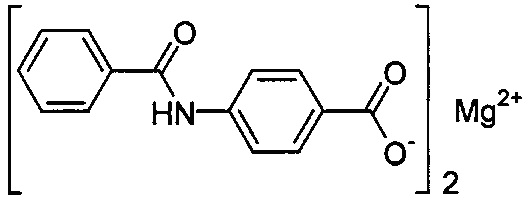 .
.
В 500 мл лабораторном стакане 30 г 4-бензоамидобензойной кислоты смешивали с приблизительно 250 мл воды. Затем вводили 6,98 г гидроксида калия (растворенного в приблизительно 50 мл воды). Полученную смесь перемешивали в течение ночи. Все твердые вещества растворялись, а значение pH становилось нейтральным. Затем вводили 25,3 г гексагидрата хлорида магния в приблизительно 100 мл воды. Продукт сразу же осаждался. Смесь перемешивали в течение еще одного часа после добавления и затем отфильтровывали для выделения продукта. Продукт промывали DI-водой и сушили в печи при 110°C.
Пример получения EX48
Данный пример показывает получение 4-N-циклогексиламидобензойной кислоты, имеющей следующую структуру
Пример получения ЕХ49
Данный пример показывает получение калиевой соли 4-N-циклогексиламидобензойной кислоты, имеющей следующую структуру
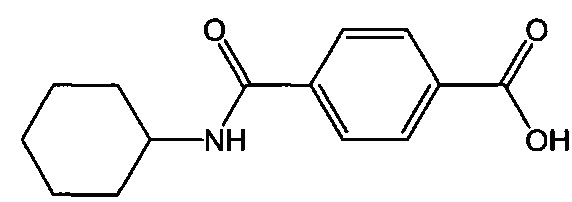 .
.
В 2 л круглодонную колбу, оборудованную ледяной баней, вводили 3,83 г бикарбоната натрия, 4,53 г циклогексиламина, 0,5 г триэтиламина и 200 мл тетрагидрофурана. Затем 9,06 г 4-карбометоксибензоилхлорида (растворенного в 9,70 г тетрагидрофурана) добавляли по каплям в течение часа в колбу. После добавления реакционную смесь медленно нагревали с обратным холодильником. При помощи ИК-спектроскопии контролировали завершение реакции (исчезновение пика при 1780 см-1). После завершения реакции реакционную смесь разбавляли 2 л H2O, перемешивали 20-30 минут, а затем отфильтровывали для выделения твердого вещества в качестве продукта. Продукт сушили в печи при 110°C, получали 11,31 г продукта.
В 2 л трехгорлую круглодонную колбу вводили 11,31 г продукта, полученного на предыдущей стадии, и 150 мл метанола. Затем 2,72 г гидроксида калия (растворенного в метаноле) добавляли по каплям в колбу. После завершения добавления реакционную смесь нагревали с обратным холодильником. При помощи ИК-спектроскопии контролировали завершение реакции (исчезновение пика при 1720 см-1). После завершения реакции добавляли 750 мл воды и фильтровали для удаления нерастворимых примесей. pH фильтрата доводили приблизительно до 2 при помощи соляной кислоты для осаждения продукта. Смесь отфильтровывали для выделения продукта и продукт промывали DI-водой. Продукт сушили в печи при 110°C.
Пример получения ЕХ 49
Данный пример показывает получение калиевой соли 4-N-циклогексиламидобензойной кислоты, имеющей следующую структуру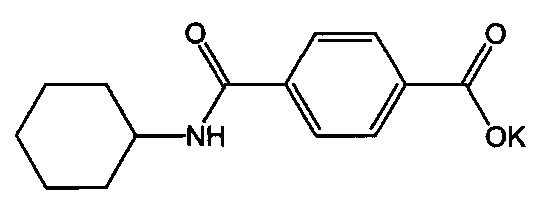 .
.
В лабораторном стакане 6 г N-циклогексиламидобензойной кислоты диспергировали в 50 мл H2O. Затем 1,36 г гидроксида калия растворяли в другом лабораторном стакане в приблизительно 20 мл H2O, а затем добавляли в суспензию. Большая часть материала растворялась, и оставшееся нерастворимое твердое вещество удаляли фильтрованием. H2O отгоняли из фильтрата для выделения продукта. Продукт сушили в печи в течение ночи при 110°C.
Пример получения ЕХ50
Данный пример показывает получение алюминиевой соли 4-N-циклогексиламидобензойной кислоты, имеющей следующую структуру
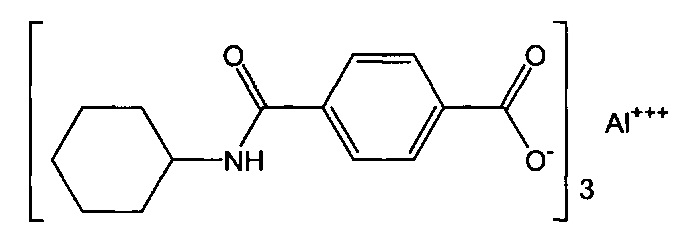 .
.
1 г калиевой соли 4-N-циклогексиламидобензойной кислоты растворяли в лабораторном стакане в приблизительно 25 мл H2O. В другом лабораторном стакане 0,78 г октадекагидрата сульфата алюминия растворяли в 15 мл H2O. Два раствора смешивали, и осадок образовывался сразу же. Твердое вещество выделяли вакуум-фильтрованием и сушили в печи в течение ночи при 110°C.
Пример получения ЕХ51
Данный пример показывает получение 4-(1,3-диоксо-1Н,3H-бензо[де]изохинолин-2-ил)бензойной кислоты, имеющей следующую структуру
 .
.
В 1 л четырехгорлую круглодонную колбу, оборудованную датчиком температуры, колбонагревателем, мешалкой и конденсатором, загружали 25 г нафтойного ангидрида и 80 мл уксусной кислоты. После образования раствора с цветом от темно-красного до оранжевого вводили 17,31 г 4-аминобензойной кислоты и реакционную смесь нагревали с обратным холодильником в течение ночи. Реакционную смесь выливали в избыточное количество DI-воды для осаждения продукта. Продукт выделяли фильтрованием, промывали дополнительным количеством DI-воды, а затем сушили в печи.
Пример получения ЕХ52
Данный пример показывает получение литиевой соли 4-(1,3-диоксо-1Н,3H-бензо[де]изохинолин-2-ил)бензойной кислоты, имеющей следующую структуру
 .
.
В 1000 мл лабораторный стакан, оборудованный закрепленной сверху мешалкой, загружали 5,09 г 4-(1,3-диоксо-1Н,3H-бензо[де]изохинолин-2-ил)бензойной кислоты и 100 мл DI H2O. Реакционную смесь перемешивали и pH регулировали гидроксидом лития, пока вся кислота не растворялась. Воду удаляли роторным испарителем. Получали 5,231 г продукта.
Пример получения ЕХ53
Данный пример показывает получение литиевой соли N-бензилтерефталевой кислоты, имеющей следующую структуру
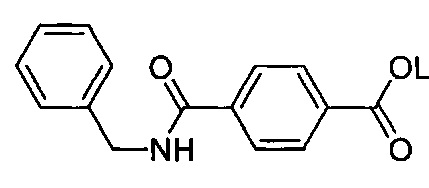 .
.
В трехгорлую круглодонную колбу, оборудованную конденсатором и капельной воронкой, вводили 6,345 г бикарбоната натрия, 8,09 г бензиламина, 0,5 г триэтиламина и 350 мл тетрагидрофурана. Температуру смеси снижали ниже 10°C. Затем 15 г карбометоксибензоилхлорида (растворенного в приблизительно 150 мл тетрагидрофурана) добавляли по каплям в течение одного часа. После добавления смесь медленно нагревали с обратным холодильником. Завершение реакции контролировали при помощи ИК-спектроскопии (исчезновение пика при 1780 см-1). После завершения реакции смесь разбавляли, приблизительно 2 л DI-воды и перемешивали в течение 20-30 минут. Продукт выделяли фильтрованием и сушили в печи при 100°C. Получали 18,68 г материала (выход 91,84%).
В 2 л лабораторный стакан вводили 2,12 г продукта с предыдущей стадии и 300 мл DI-воды. Затем 1,89 г 10% раствора гидроксида лития добавляли и перемешивали до завершения реакции (исчезновение пика при 1720 см-1 в ИК-спектроскопии). Затем всю воду удаляли роторным испарителем для выделения продукта, получали 1,94 г продукта (выход 94,37%).
Пример получения ЕХ54
Данный пример показывает получение литиевой соли 1Я-пиридин-2-илтерефталевой кислоты, имеющей следующую структуру
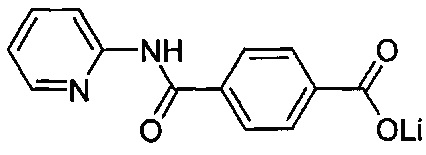 .
.
В 250 мл трехгорлую круглодонную колбу, оборудованную закрепленной сверху мешалкой, датчиком температуры, ледяной баней и обратным холодильником, вводили 4,71 г 3-аминопиридина, 4,2 г бикарбоната натрия, приблизительно 0,1 г триэтиламина и 50 мл тетрагидрофурана. Температуру снижали ниже 10°C, а затем 9,9 г карбометоксибензоилхлорида (раствор в 20 мл тетрагидрофурана) добавляли по каплям в течение 1-1,5 часов. Реакционную смесь перемешивали в течение ночи, а затем нагревали с обратным холодильником в течение приблизительно 2 часов. Затем 500 мл DI-воды использовали для разбавления реакционной смеси и полученную смесь перемешивали в течение 20-30 минут. Твердый продукт выделяли фильтрованием и сушили в печи при 110°C.
В 250 мл лабораторный стакан, оборудованный магнитной мешалкой, вводили 2,56 г продукта, полученного на предыдущей стадии, 0,42 г моногидрата гидроксида лития и 50 мл DI-воды. Лабораторный стакан нагревали до 90°C, пока pH не становился ниже 10. Твердый продукт выделяли фильтрованием и всю воду удаляли выпариванием.
Пример получения ЕХ55
Данный пример показывает получение N-(2-хлорфенил)терефталевой кислоты, имеющей следующую структуру
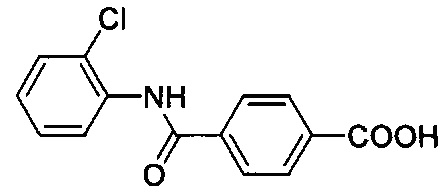 .
.
В 2 л круглодонную колбу вводили 6,34 г бикарбоната натрия, 9,63 г 2-хлоранилина, 0,5 г триэтиламина и 200 мл тетрагидрофурана. После охлаждения реакционной смеси при помощи ледяной бани 15 г карбометоксибензоилхлорида (растворенного в приблизительно 100 мл THF) добавляли по каплям в колбу. После добавления реакционную смесь нагревали с обратным холодильником. При помощи ИК-спектроскопии контролировали завершение реакции (исчезновение пика при 1780 см-1). После завершения реакции раствор разбавляли 2 л DI H2O и перемешивали 20-30 минут. Твердый продукт выделяли фильтрованием и сушили в печи при 110°C.
В 2 л трехгорлую круглодонную колбу вводили 20,12 г продукта с предыдущей стадии и 150 мл метанола. Реакционную смесь нагревали с обратным холодильником. Когда начинали нагревание, 3,90 г гидроксида калия (растворенного в метаноле) добавляли по каплям в реакционную смесь. При помощи ИК-спектроскопии контролировали завершение реакции (исчезновение пика при 1720 см-1). После завершения реакции раствор разбавляли избытком H2O. Фильтрование использовали для удаления любого остаточного твердого вещества, а затем HCl добавляли в фильтрат, пока значение pH не составило приблизительно 2. Продукт, который осаждался на этой стадии, выделяли фильтрованием, а затем сушили в печи при 110°C.
Пример получения ЕХ56
Данный пример показывает получение литиевой соли N-(2-хлорфенил)терефталевой кислоты, имеющей следующую структуру
 .
.
В лабораторном стакане 1 г N-(2-хлорфенил)терефталевой кислоты суспендировали в приблизительно 20 мл воды, а затем добавляли 0,1527 г моногидрата гидроксида лития. Реакционную смесь перемешивали, пока значение pH не падало ниже 10. Твердый продукт выделяли фильтрованием.
Пример получения ЕХ57
Данный пример показывает получение калиевой соли N-(2-хлорфенил)терефталевой кислоты, имеющей следующую структуру
 .
.
В лабораторном стакане 12 г N-(2-хлорфенил)терефталевой кислоты суспендировали в 200 мл воды, а затем добавляли 2,448 г гидроксида калия. Реакционную смесь перемешивали, пока значение pH не падало ниже 10. Продукт собирали после выпаривания в роторном испарителе для удаления избытка воды.
Пример получения ЕХ58
Данный пример показывает получение N-(3,5-дициано-4-метилтиофен-2-ил)терефталевой кислоты, имеющей следующую структуру
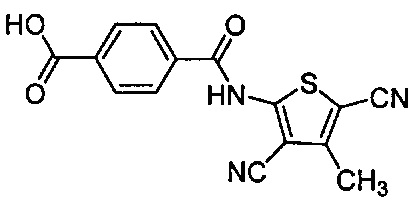 .
.
В трехгорлую круглодонную колбу, оборудованную закрепленной сверху мешалкой, датчиком температуры, баней с сухим льдом и обратным холодильником, вводили 12,32 г 5-амино-3-метилтиофен-2,4-дикарбонитрила, 6,27 г кальцинированной соды и 200 мл тетрагидрофурана. Температуру снижали ниже 10°C, а затем 30 г раствора карбометоксибензоилхлорида (50% раствор в тетрагидрофуране) добавляли по каплям в течение 1-1,5 часов. После добавления смесь нагревали приблизительно до 40°C до завершения реакции (контролировали при помощи ИК-спектроскопии, пик при 1780 см-1 исчезал). Затем реакционную смесь разбавляли 2 л DI-воды и отфильтровывали для выделения продукта.
В емкости на 32 унции, оборудованной магнитной мешалкой, 17,4 г продукта, полученного на предыдущей стадии, растворяли в 300 мл метанола. А затем добавляли 30,05 г раствора гидроксида калия (10% в метаноле). Реакцию контролировали при помощи ИК-спектроскопии. После завершения реакции смесь разбавляли 1 л воды. Смесь отфильтровывали для удаления любых нерастворимых примесей, а фильтрат подкисляли при помощи соляной кислоты, пока pH не составил приблизительно 2. Продукт осаждался на этой стадии. Смесь отфильтровывали для выделения продукта. Продукт промывали DI-водой и сушили.
Пример получения ЕХ59
Данный пример показывает получение натриевой соли N-(3,5-дициано-4-метилтиофен-2-ил)терефталевой кислоты, имеющей следующую структуру
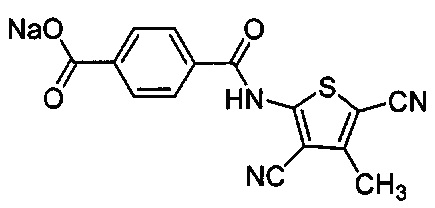 .
.
В лабораторном стакане Н-(3,5-дициано-4-метилтиофен-2-ил)терефталевую кислоту смешивали с 200 мл воды. Затем 25% раствор гидроксида натрия медленно добавляли, пока не получали постоянное значение pH 12. Раствор концентрировали под вакуумом, получая желаемый продукт.
Пример получения ЕХ60
Данный пример показывает получение литиевой соли N-(3,5-дициано-4-метилтиофен-2-ил)терефталевой кислоты, имеющей следующую структуру
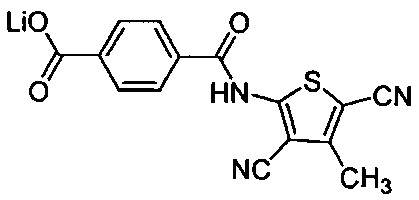 .
.
В лабораторном стакане N-(3,5-дициано-4-метилтиофен-2-ил)терефталевую кислоту смешивали с 200 мл воды. Затем моногидрат гидроксида лития медленно добавляли, пока не получали постоянное значение pH 12. Раствор концентрировали под вакуумом, получая желаемый продукт.
Пример получения ЕХ61
Данный пример показывает получение цинковой соли N-(3,5-дициано-4-метилтиофен-2-ил)терефталевой кислоты, имеющей следующую структуру
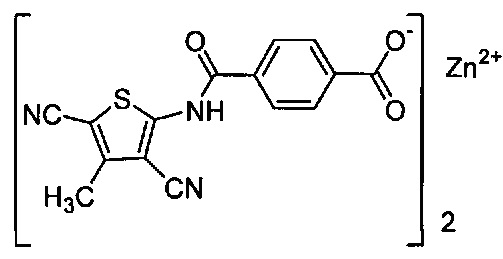 .
.
В лабораторном стакане N-(3,5-дициано-4-метилтиофен-2-ил)терефталевую кислоту смешивали с 200 мл воды. Затем 25% раствор гидроксида калия медленно добавляли, пока не получали постоянное значение pH 12. Один эквивалент хлорида цинка (растворенного в воде) затем добавляли в раствор, и продукт осаждался. Продукт выделяли фильтрованием и промывали DI-водой.
Пример получения EX62
Данный пример показывает получение литиевой соли N-пиридин-3-илтерефталевой кислоты, имеющей следующую структуру
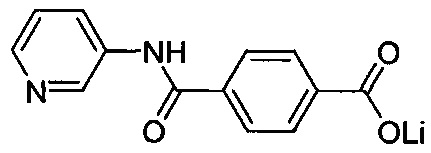 .
.
В 250 мл трехгорлую колбу, оборудованную закрепленной сверху мешалкой, датчиком температуры, ледяной баней и обратным холодильником, вводили 4,71 г 3-аминопиридина, 4,2 г бикарбоната натрия, приблизительно 0,1 г триэтиламина и 50 мл тетрагидрофурана. Температуру снижали ниже 10°C, а затем 9,9 г карбометоксибензоилхлорида (раствор в 20 мл тетрагидрофурана) добавляли по каплям в течение 1-1,5 часов. Реакционную смесь перемешивали в течение ночи, а затем нагревали с обратным холодильником в течение приблизительно 2 часов. Затем реакционную смесь разбавляли приблизительно 500 мл DI-воды и перемешивали в течение приблизительно 20-30 минут. Твердый продукт выделяли фильтрованием и сушили в печи при 110°C.
В 250 мл лабораторный стакан, оборудованный магнитной мешалкой, вводили 2,56 г продукта, полученного на предыдущей стадии, 0,42 г моногидрата гидроксида лития и 75 мл DI-воды. Лабораторный стакан нагревали до 90°C, пока pH не составил менее 10. Любые твердые вещества удаляли посредством фильтрования и выделяли фильтрат. Продукт собирали после удаления избытка воды посредством выпаривания.
Пример получения ЕХ63
Данный пример показывает получение N-(2-метоксифенил)терефталевой кислоты, имеющей следующую структуру
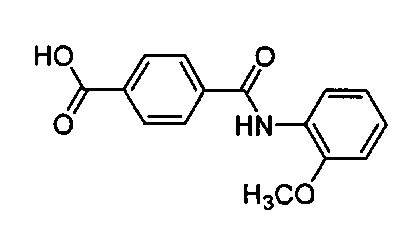 .
.
В трехгорлую колбу, оборудованную закрепленной сверху мешалкой, датчиком температуры, баней с сухим льдом и обратным холодильником, вводили 7,44 г o-анизидина, 5,01 г кальцинированной соды и 200 мл тетрагидрофурана. Температуру снижали ниже 10°C, а затем 25 г раствора карбометоксибензоилхлорида (48% в тетрагидрофуране) добавляли по каплям в течение 1-1,5 часов. После добавления реакционную смесь нагревали приблизительно до 40°C до завершения реакции (контролировали при помощи ИК-спектроскопии, пик при 1780 см-1 исчезал). Затем смесь разбавляли 2 л DI-воды и отфильтровывали для выделения продукта.
В емкости на 32 унции, оборудованной магнитной мешалкой, 13,95 г продукта, полученного на предыдущей стадии, растворяли в 300 мл метанола. Затем добавляли 27,5 г раствора гидроксида калия (10% в метаноле). Реакцию контролировали при помощи ИК-спектроскопии. После завершения реакции смесь разбавляли приблизительно 1 л воды. Нерастворимые примеси удаляли фильтрованием. Фильтрат подкисляли соляной кислотой, пока pH не становился равным приблизительно 2. Продукт осаждался на этой стадии. Продукт выделяли фильтрованием, промывали DI-водой и сушили.
Пример получения ЕХ64
Данный пример показывает получение калиевой соли N-(2-метоксифенил)терефталевой кислоты, имеющей следующую структуру
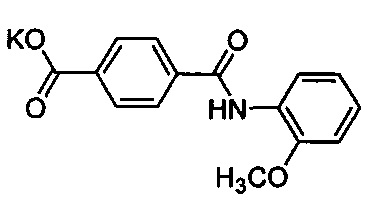 .
.
В лабораторном стакане N-(2-метоксифенил)терефталевую кислоту смешивали с 200 мл воды. Затем 25% раствор гидроксида калия медленно добавляли, пока не получали постоянное значение pH 12. Раствор концентрировали под вакуумом с получением желаемого продукта.
Пример получения ЕХ65
Данный пример показывает получение магниевой соли 4-N-фенилтерефталевой кислоты, имеющей следующую структуру
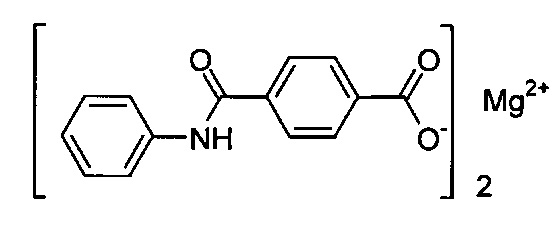 .
.
В 250 л лабораторный стакан, оборудованный магнитной мешалкой и пластиной для смешивания, вводили 10 г 4-N-фениламидобензойной кислоты и 50 мл воды. Реакционную смесь нагревали практически до кипения и добавляли 1,5 г оксида магния. ИК-спектроскопию использовали для контроля завершения реакции, а продукт выделяли фильтрованием.
Пример получения ЕХ66
Данный пример показывает получение литиевой соли 4-N-(3,4-дихлорфенил)амидобензойной кислоты, имеющей следующую структуру
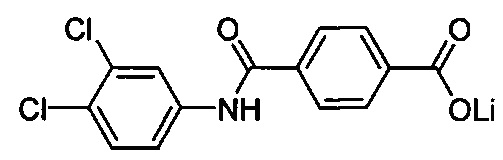 .
.
В 1 л трехгорлую круглодонную колбу, оборудованную закрепленной сверху мешалкой, капельной воронкой, ледяной баней, впуском азота и нагревательной плиткой, вводили 12,48 г 3,4-дихлоранилина, 6,34 г бикарбоната натрия, 0,5 г триэтиламина и 200 мл тетрагидрофурана. Температуру снижали ниже 10°C, а затем 15 г карбометоксибензоилхлорида (раствор в 100 мл тетрагидрофурана) добавляли по каплям в течение 1-1,5 часов. После добавления реакционную смесь нагревали приблизительно до 40°C до завершения реакции (контролировали при помощи ИК-спектроскопии, пик при 1780 см-1 исчезал). Затем реакционную смесь разбавляли 2 л DI-воды и отфильтровывали для выделения продукта. Получали 23,87 г продукта (выход: 97,5%).
В 250 мл лабораторном стакане 3 г продукта с предыдущей стадии смешивали с 50 мл воды. Смесь нагревали практически до кипения и вводили 2,22 г 10% раствора гидроксида лития. Завершение реакции контролировали при помощи ИК-спектроскопии. Реакционную смесь выпаривали практически досуха и продукт выделяли фильтрованием. Получали 1,92 г продукта (выход: 65,6%).
Пример получения EX67
Данный пример показывает получение кальциевой соли 4-N-(2,6-диизопропилфенил)амидобензойной кислоты, имеющей следующую структуру
Пример получения ЕХ68
Данный пример показывает получение 4-бензоиламино-2-гидроксибензойной кислоты, имеющей следующую структуру
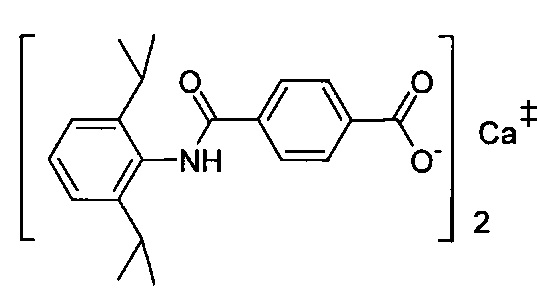 .
.
В 1 л трехгорлую круглодонную колбу, оборудованную магнитной мешалкой, капельной воронкой, ледяной баней, продувом азотом, газоочистителем и нагревательной плиткой, вводили 14,73 г 2,6-диизопропиланилина, 6,34 г бикарбоната натрия, 0,5 г триэтиламина и 200 мл тетрагидрофурана. Смесь охлаждали ниже 10°C, а затем 15 г карбометоксибензоилхлорида (растворенного в 100 мл тетрагидрофурана) добавляли по каплям в течение 1-1,5 часов. После добавления реакционную смесь медленно нагревали с обратным холодильником. После завершения реакции (исчезновения пика при 1780 см-1 в ИК), реакционную смесь разбавляли холодной DI-водой и перемешивали в течение 20-30 минут. Продукт выделяли фильтрованием и сушили в печи при 110°C.
В 600 мл лабораторном стакане 13,79 г продукта с предыдущей стадии смешивали с 200 мл воды. Смесь нагревали практически до кипения и вводили 22,8 г 10% раствора гидроксида калия. После того, как завершение реакции контролировали при помощи ИК-спектроскопии, вводили 6,72 г дигидрата хлорида кальция (растворенного с образованием 10% раствора). Продукт осаждался, и его выделяли фильтрованием.
Пример получения ЕХ68
Данный пример показывает получение 4-бензоиламино-2-гидроксибензойной кислоты, имеющей следующую структуру
 .
.
В трехгорлую колбу, оборудованную закрепленной сверху мешалкой, датчиком температуры, баней с сухим льдом и обратным холодильником, вводили 25 г 4-аминосалициловой кислоты, 5,01 г кальцинированной соды и 200 мл тетрагидрофурана. Температуру снижали ниже 10°C и 7,32 г бензоилхлорида добавляли по каплям в течение 1-1,5 часов. После добавления колбу медленно нагревали до 40°C. После завершения реакции (контролировали при помощи ИК-спектроскопии, пока пик при 1780 см-1 не исчезал) реакционную смесь разбавляли 300 мл воды. Органический слой отделяли. После сушки от растворителя получали приблизительно 16 г продукта.
Пример получения ЕХ69
Данный пример показывает получение литиевой соли 4-бензоиламино-2-гидроксибензойной кислоты, имеющей следующую структуру
 .
.
В лабораторном стакане 3 г 4-бензоиламино-2-гидроксибензойной кислоты смешивали с 20 мл воды. Затем добавляли 1,05 г моногидрата гидроксида лития. Смесь перемешивали в течение 20 минут, а затем реакционную смесь концентрировали под вакуумом для получения желаемой литиевой соли.
Пример получения ЕХ70
Данный пример показывает получение кальциевой соли 4-бензоиламино-2-гидроксибензойной кислоты, имеющей следующую структуру
 .
.
В лабораторном стакане 3 г 4-бензоиламино-2-гидроксибензойной кислоты смешивали с 20 мл воды. Затем добавляли 2,51 г 50% раствора гидроксида натрия. После того как раствор становился прозрачным, добавляли раствор, содержащий 3,53 г дегидрата хлорида кальция. Продукт осаждался, и его выделяли фильтрованием.
Пример получения ЕХ71
Данный пример показывает получение 4-[(бифенил-4-карбонил)-амино]-бензойной кислоты, имеющей следующую структуру
 .
.
В 5 л трехгорлой круглодонной колбе 316,5 г 4-аминобензойной кислоты растворяли в приблизительно 3 л диоксана. Затем 250 г бифенил-4-карбонилхлорида (растворенного в приблизительно 150 мл диоксана) добавляли по каплям в течение 1 часа. Реакционную смесь перемешивали в течение ночи и отфильтровывали для выделения твердого вещества. Твердое вещество промывали кипящей DI-водой, а затем холодной DI-водой, пока pH не становился приблизительно нейтральным. Промытое твердое вещество затем сушили в вакуумной печи.
Пример получения ЕХ72
Данный пример показывает получение литиевой соли 4-[(бифенил-4-карбонил)-амино]-бензойной кислоты, имеющей следующую структуру
 .
.
В лабораторном стакане 364,62 г 4-[(бифенил-4-карбонил)-амино]-бензойной кислоты смешивали с 3 л воды. Затем раствор моногидрата гидроксида лития (41,96 г в приблизительно 500 мл воды) вводили в суспензию. Реакционную смесь перемешивали в течение ночи, и значение pH становилось 7,5. Твердый продукт выделяли фильтрованием, промывали водой и сушили в печи при 110°C. Получали 334,7 г продукта (выход 90%).
Пример получения ЕХ73
Данный пример показывает получение 4-(бензилиденамино)бензойной кислоты, имеющей следующую структуру
 .
.
В 500 мл трехгорлую круглодонную колбу, оборудованную конденсатором, колбонагревателем, магнитной мешалкой и двумя пробками, вводили 10 г 4-аминобензойной кислоты, 7,75 г бензальдегида и 200 мл этанола. Реакционную смесь нагревали с обратным холодильником в течение 6 часов. Продукт кристаллизовался из раствора после того, как раствор охлаждали до комнатной температуры. Продукт выделяли фильтрованием. Дополнительное количество продукта извлекали концентрированием фильтрата. Получали 15,41 г продукта (выход: 94%).
Пример получения ЕХ74
Данный пример показывает получение литиевой соли 4-(бензилиденамино)бензойной кислоты, имеющей следующую структуру
 .
.
В 2 л лабораторном стакане 15,41 г 4-(бензилиденамино)бензойной кислоты растворяли в 200 мл воды. Смесь медленно нагревали и перемешивали на нагревательной плитке до получения прозрачного раствора. Затем медленно добавляли 2,85 г моногидрата гидроксида лития. Раствор становился немного мутным. После завершения реакции реакционную смесь охлаждали и воду выпаривали. Выделяли желтое твердое вещество. Продукт промывали ацетоном, а затем сушили в печи при 110°C.
Пример получения ЕХ75
Данный пример показывает получение 4-хлорфениламидобензойной кислоты, имеющей следующую структуру
 .
.
В 5 л колбу вводили 274,3 г 4-аминобензойной кислоты (2 моль) и 2800 мл ацетона. Реакционную смесь перемешивали до получения однородной суспензии. Затем 175 г 4-хлорбензоилхлорида добавляли по каплям в 5 л колбу, в то же время перемешивая содержимое. Реакционную смесь перемешивали в течение ночи, а затем отфильтровывали для выделения твердого вещества. Продукт промывали приблизительно 500 мл ацетона, а затем три раза водой (500 мл каждый раз). После промывки твердое вещество переносили в 4 л лабораторный стакан и суспендировали приблизительно в 2 л кипящей воды в течение часа. Твердый продукт выделяли фильтрованием и промывали дополнительным количеством кипящей водой, пока вода не становилась бесцветной.
Пример получения ЕХ76
Данный пример показывает получение натриевой соли 4-хлорфениламидобензойной кислоты, имеющей следующую структуру
 .
.
В 2 л лабораторный стакан, оборудованный механической мешалкой, вводили 400 мл воды и 27,5 г 4-хлорфениламидобензойной кислоты. В другом лабораторном стакане 8,4 г NaOH (50% раствор) разбавляли в 100 мл воды. Раствор NaOH добавляли в суспензию 4-хлорфениламидобензойной кислоты и смесь перемешивали в течение ночи. Продукт выделяли фильтрованием. Продукт промывали DI-водой, пока pH воды не становился менее 10, а продукт затем сушили в печи при 110°C.
Пример получения ЕХ77
Данный пример показывает получение 4-(4-фторбензоиламино)бензойной кислоты, имеющей следующую структуру
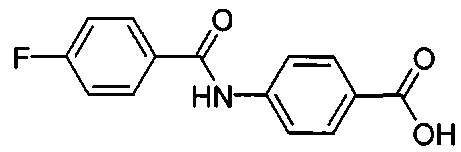 .
.
В 4 л лабораторный стакан вводили 21,27 г 4-аминобензойной кислоты и 1 л DI H2O. Затем вводили 33,38 г карбоната натрия. Затем 100 г 4-фторбензоилхлорида добавляли по каплям в колбу (в течение приблизительно 45 минут - 1 часа) и реакционную смесь перемешивали в течение ночи. Твердый продукт выделяли вакуумным фильтрованием и промывали кипящей водой для удаления избытка 4-фторбензойной кислоты. Продукт сушили в течение ночи в вакуумной печи. Получали 59,08 г продукта.
Пример получения ЕХ78
Данный пример показывает получение литиевой соли 4-(4-фторбензоиламино)бензойной кислоты, имеющей следующую структуру
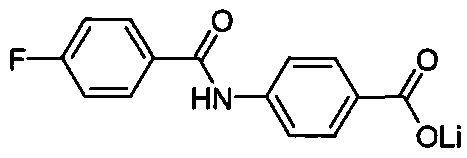 .
.
В лабораторном стакане 10 г 4-(4-фторбензоиламино)бензойной кислоты суспендировали в 100 мл DI-воды. Затем 1,62 г моногидрата гидроксида лития сначала растворяли в 25 мл DI-воды, а затем добавляли в суспензию кислоты. Реакционную смесь перемешивали в течение ночи и продукт выделяли выпариванием воды.
Пример получения EX79
Данный пример показывает получение 4-бензоиламино-2,3,4,5-тетрафторбензойной кислоты, имеющей следующую структуру
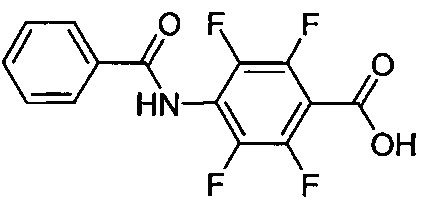 .
.
В 250 л колбу, оборудованную мешалкой, вводили 3,37 г 4-амино-2,3,4,5-тетрафторбензойной кислоты, 1,06 г карбоната натрия и 20 мл воды. Затем 6,8 г 4-бензоилхлорида добавляли по каплям в колбу (в течение приблизительно 45 минут - 1 часа). pH отмечали как составляющий менее 1 на следующее утро. Твердый продукт выделяли фильтрованием, промывали DI-водой 5 раз, а затем сушили в печи при 110°C.
Пример получения ЕХ80
Данный пример показывает получение литиевой соли 4-(4-фторбензоиламино)бензойной кислоты, имеющей следующую структуру
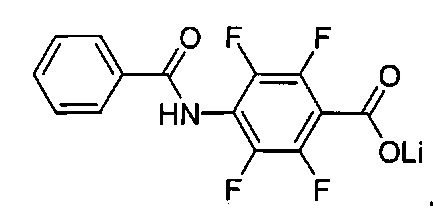
В 250 мл лабораторный стакан вводили 2 г 4-бензоиламино-2,3,4,5-тетрафторбензойной кислоты и 20 мл DI-воды. Затем 0,27 г моногидрата гидроксида лития сначала растворяли в 10 мл DI-воды, а затем добавляли в суспензию кислоты. Реакционную смесь перемешивали в течение ночи и продукт выделяли выпариванием воды.
Пример получения ЕХ81
Данный пример показывает получение трис-(4-карбоксибензол)амида бензол-1,3,5-трикарбоновой кислоты, имеющего следующую структуру
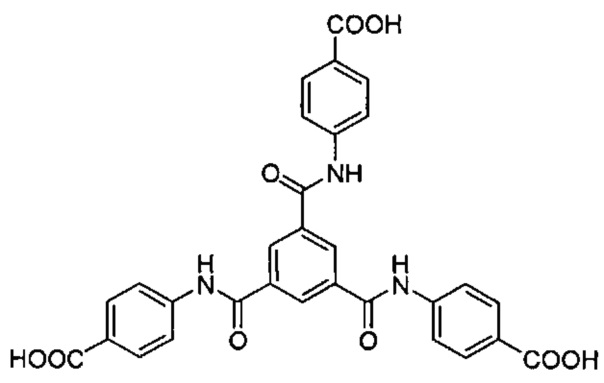 .
.
В трехгорлую круглодонную колбу вводили 21,2 г карбоната натрия и 100 мл тетрагидрофурана. Затем 13,75 г 4-аминобензойной кислоты и 8 г 1,3,5-бензолтрикарбонилтрихлорида отдельно разбавляли в 15 мл THF и добавляли в реакционную смесь одновременно через две капельные воронки. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Приблизительно 80 мл тетрагидрофурана добавляли для компенсации испарения во время реакций и добавляли дополнительные 10,6 г карбоната натрия. Через три часа реакционную смесь переносили в 1 л лабораторный стакан с 600 мл воды. pH доводили приблизительно до 2 при помощи соляной кислоты. Продукт осаждался, и его выделяли фильтрованием. Продукт затем частично сушили в вакуумной печи при 40°C. Получали приблизительно 35 г влажного продукта.
Пример получения ЕХ82
Данный пример показывает получение натриевой соли трис-(4-карбоксибензол)амида бензол-1,3,5-трикарбоновой кислоты, имеющей следующую структуру
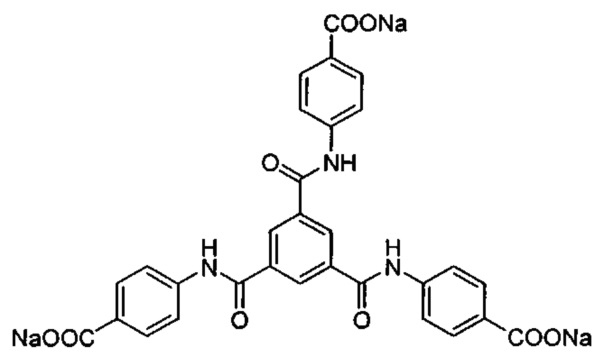 .
.
В лабораторном стакане 32 г трис-(4-карбоксибензол)амида бензол-1,3,5-трикарбоновой кислоты смешивали с 200 мл воды. Затем 50% раствор гидроксида натрия медленно добавляли в смесь, пока pH не становился равным 12. Смесь перемешивали в течение 20 минут, а затем концентрировали под вакуумом для получения продукта.
Пример получения ЕХ83
Данный пример показывает получение бис-(4-карбоксибензол)амида бифенил-4,4'-дикарбоновой кислоты, имеющего следующую структуру
 .
.
В трехгорлую круглодонную колбу вводили 5,67 г карбоната натрия и приблизительно 40 мл тетрагидрофурана. Затем 4,9 г 4-аминобензойной кислоты и 5,0 г 4,4'-бифенилдикарбонилхлорида отдельно разбавляли в 15 мл THF, а затем добавляли в реакционную смесь одновременно через две капельные воронки. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем реакционную смесь переносили в 1 л лабораторный стакан с 600 мл воды. pH доводили приблизительно до 2 при помощи соляной кислоты. Продукт осаждался, и его выделяли фильтрованием. Продукт сушили в вакуумной печи при 50°C.
Пример получения ЕХ84
Данный пример показывает получение натриевой соли бис-(4-карбоксибензол)амида бифенил-4,4'-дикарбоновой кислоты, имеющей следующую структуру
 .
.
В лабораторном стакане 12 г бис-(4-карбоксибензол)амида бифенил-4,4'-дикарбоновой кислоты смешивали с 100 мл воды. Затем 50% раствор гидроксида натрия медленно добавляли в смесь, пока pH не становился равным 12,5. Смесь перемешивали в течение 20 минут, а затем концентрировали под вакуумом для получения продукта.
Пример получения EX85
Данный пример показывает получение 4-(4-метилбензоиламино)бензойной кислоты, имеющей следующую структуру
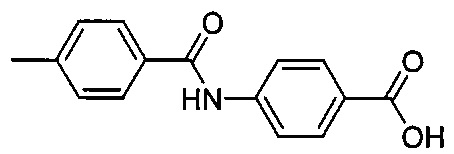 .
.
В 5 л трехгорлую круглодонную колбу вводили 274 г 4-аминобензойной кислоты и 3000 мл ацетона. Смесь перемешивали с образованием прозрачного раствора. Затем 154,5 г 4-метилбензоилхлорида добавляли по каплям в реакционную смесь. После добавления реакционную смесь перемешивали в течение ночи, а затем отфильтровывали для выделения твердого вещества. Твердое вещество промывали кипящей водой, а затем холодной DI-водой, пока pH не становился нейтральным. Продукт сушили при 110°C.
Пример получения ЕХ86
Данный пример показывает получение литиевой соли 4-(4-метилбензоиламино)бензойной кислоты, имеющей следующую структуру
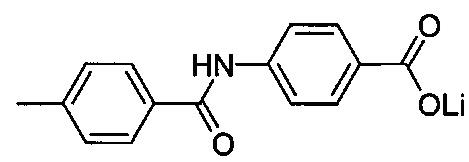 .
.
В лабораторный стакан вводили 25,5 г 4-(4-метилбензоиламино)бензойной кислоты и 200 мл DI-воды. Смесь перемешивали до тех пор, пока она не образовывала однородную суспензию. Затем добавляли 4,2 г моногидрата гидроксида лития. Реакционную смесь перемешивали в течение ночи, и значение pH падало до 10. Твердый продукт отфильтровывали, а затем сушили в печи при 110°C.
Пример получения ЕХ87
Данный пример показывает получение литиевой соли N-циклопентилтерефталевой кислоты, имеющей следующую структуру
 .
.
В лабораторном стакане 23,3 г N-циклопентилтерефталевой кислоты добавляли в 100 мл H2O. Затем 4,2 г моногидрата гидроксида лития растворяли в отдельном лабораторном стакане в приблизительно 50 мл H2O. Раствор гидроксида лития добавляли в суспензию N-циклопентилтерефталевой кислоты и перемешивали, пока значение pH не становилось приблизительно нейтральным. Продукт был частично растворимым в воде. Воду удаляли выпариванием для получения продукта. Продукт сушили в течение ночи в печи при 110°C.
Пример получения ЕХ88
Данный пример показывает получение литиевой соли 4-(циклопентанкарбониламино)бензойной кислоты, имеющей следующую структуру
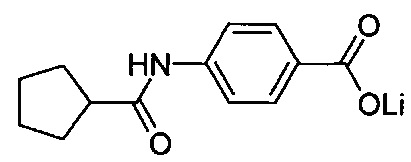 .
.
В 1 л 2-горлой круглодонной колбе 40 г 4-аминобензойной кислоты растворяли приблизительно в 400 мл диоксана. Затем 19,35 г циклопентанкарбонилхлорида добавляли по каплям в раствор. Промежуточный продукт реакции, 4-(циклопентанкарбониламино)бензойная кислота, образовывался в виде белого твердого вещества на данной стадии, и его выделяли фильтрованием. После промывки продукта приблизительно 200 мл диоксана, а затем приблизительно 1 л кипящей воды, промежуточный продукт реакции сушили в печи при 110°C. Выход на этой стадии составлял приблизительно 27,7 г (81%).
27,7 г 4-(циклопентанкарбониламино)бензойной кислоты суспендировали в приблизительно 277 мл воды. Затем добавляли 5 г моногидрата гидроксида лития. Смесь перемешивали в течение ночи, и значение pH становилось равным приблизительно 7. После выпаривания избытка воды готовый продукт (литиевую соль 4-(циклопентанкарбониламино)бензойной кислоты) выделяли в виде белого твердого вещества и сушили в печи при 110°C.
Пример T1
Различные добавки из вышеописанных примеров получения отдельно измельчали и смешивали с полиэтиленовым полимером высокой плотности, характеризующимся плотностью приблизительно 0,952 г/см3 и индексом текучести расплава приблизительно 19 дг/минуту (ExxonMobil™ HDPE HD 6719). Затем смесь или формовали под давлением в полосы, или формовали поливом в тонкие пленки. Пиковую температуру перекристаллизации полимера (Tc) для каждой композиции на основе термопластичного полимера измеряли при помощи дифференциального сканирующего калориметра (дифференциального сканирующего калориметра Mettler-Toledo DSC822). В частности, образец отбирали из заданной части и нагревали со скоростью 20°C/минуту от температуры 60°C до 220°C, выдерживали при 220°C в течение двух минут и охлаждали со скоростью приблизительно 10°C/минуту до температуры 60°C. Температура, при которой возникало пиковое преобразование кристаллов полимера (что соответствует пиковой температуре перекристаллизации полимера) записывали для каждого образца и указывали в таблице 1 ниже.
Сравнительный пример CTCEX1 представляет собой полиэтиленовый полимер высокой плотности, характеризующийся плотностью приблизительно 0,952 г/см3 и индексом текучести расплава приблизительно 19 дг/минуту (ExxonMobil™ HDPE HD 6719), который формовали литьем под давлением в эталонные полосы. Сравнительные примеры CTCEX2 и CTCEX3 представляют собой такой же полиэтиленовый полимер высокой плотности, содержащий 1000 частей на миллион бензоата натрия и алюминий-бис[4-1(1,1-диметилэтил)бензоат] гидроксид (Al-pTBBA), соответственно. Сравнительный пример CTCEX4 представляет собой такой же полиэтиленовый полимер высокой плотности, отлитый в пленку. Примеры TCEX1-TCEX56 представляют собой полученную поливом пленку из полиэтиленового полимера высокой плотности, содержащую 1500 частей на миллион соединений примеров получения, раскрытых в настоящей заявке.

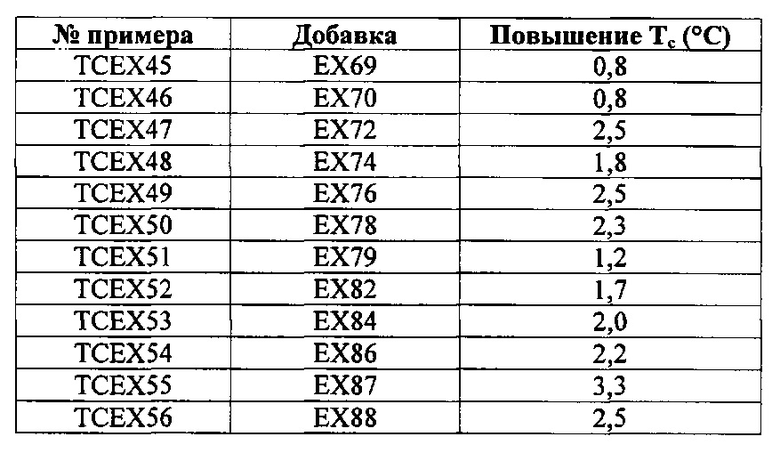
Из таблицы 1 видно, что все соединения солей металлов настоящего изобретения могут повышать температуру перекристаллизации (Tc) полиэтилена в некоторой степени. Хотя Tc является не единственным важным фактором при выборе подходящего инициатора образования зародышей для частично кристаллического термопластичного полимера, повышение Tc является очень желательным, поскольку оно увеличивает скорость кристаллизации при обработке, уменьшает производственный цикл и улучшает эффективность производства.
Производство получаемых экструзией с раздувом пленок с зародышеобразователем кристаллизации
Для всех примеров получаемых экструзией с раздувом пленок используемые полиэтиленовые смолы сначала измельчали в порошок приблизительно 35 меш. Затем 1000 частей на миллион Irganox 1010, 800 частей на миллион Irgafos 168, 1000 частей на миллион DHT4-A и зародышеобразователь согласно изобретению вводили в смолу и перемешивали в высокоэффективной мешалке Henschel в течение приблизительно 2 минут при скорости лопасти мешалки приблизительно 2100 об/мин. Образцы затем компаундировали в расплаве в одношнековом экструдере МРМ с диаметром шнека 38 мм. Температуру цилиндра экструдера быстро увеличивали от 160 до 190°C. Экструдат в виде нитей охлаждали в водяной бане и затем далее гранулировали.
Пленки получали на опытной линии для получения пленок экструзией с раздувом при помощи 4-дюймовой головки для получения монослойной пленки, используя щель головки экструдера 2 мм. Линия включала кольцевой зазор с двумя кромками для подачи воздуха от Future Design с холодным воздухом. Экструдер имел барьерный шнек с диаметром 55 мм и отношением длины к ширине 24:1. Температуру цилиндра экструдера быстро увеличивали от 190 до 220°C.
Испытание получаемых экструзией с раздувом пленок с зародышеобразователем кристаллизации
% мутности деталей измеряли при помощи мутномера BYK Gardner согласно ASTM D1023. Прозрачность деталей измеряли при помощи мутномера BYK Gardner. Проницаемость, измеренную как скорость проникания водяного пара, измеряли при помощи анализатора проницания водяного пара от Illinois Instruments серии 7000 согласно ASTM Е398. Прочность на разрыв измеряли при помощи прибора для испытания на разрыв ProTear согласно ASTM D1922. Испытание на прокол проводили при помощи прибора для испытания полимеров на прокол Dynisco Model D2085AB-P согласно ASTM D1709. Испытание пленки на разрыв проводили при помощи устройства MTS Q-Test-5 согласно ASTM D882.
Пиковую температуру перекристаллизации полимера (Tc) для композиций на основе термопластичного полимера измеряли при помощи дифференциального сканирующего калориметра (дифференциального сканирующего калориметра Mettler-Toledo DSC822). В частности, полученную компрессионным формованием пластинку получали из гранул, а образец отбирали из пластинки и нагревали со скоростью 20°C/минуту от температуры 60°C до 220°C, выдерживали при 220°C в течение двух минут и охлаждали со скоростью приблизительно 10°C/минуту до температуры 60°C. Температура, при которой возникало пиковое преобразование кристаллов полимера (что соответствует пиковой температуре перекристаллизации полимера) записывали для каждого образца.
Пример F1
Данный пример показывает некоторые физические свойства, проявляемые полиэтиленовым полимером высокой плотности, в который вводили зародышеобразователь кристаллизации согласно настоящему изобретению. Полимерные композиции получали примешиванием (как описано выше) 2000 частей на миллион ЕХ5 в коммерчески доступный полиэтиленовый полимер высокой плотности (Sclair® 19G от Nova Chemicals), характеризующийся плотностью приблизительно 0,962 г/см3 и индексом текучести расплава приблизительно 1,2 дг/минуту. Образованные гранулы из полимерной композиции затем использовали для получения пленок экструзией с раздувом (толщиной 3 мил) при помощи следующих установок: головка экструдера 101,6 мм (4 дюйма), щель головки экструдера 2,0 мм, BUR 2,3, DDR 11,4 и производительность 30 кг/ч. Пиковую температуру перекристаллизации полимера, проницаемость, прочность на разрыв, стойкость к проколу, секущий модуль при 1% деформации и оптические свойства полученных пленок измеряли и указывали в таблицах F1-F4.
Пример F2
Пример F2 получали таким же образом, как пример F1, за исключением того, что ЕХ46 использовали вместо ЕХ5.
Пример F3
Пример F3 получали таким же образом, как пример F1, за исключением того, что ЕХ76 использовали вместо ЕХ5.
Сравнительный пример CF1
Сравнительный пример CF1 получали таким же образом, как пример F1, за исключением того, что не использовали зародышеобразователь.
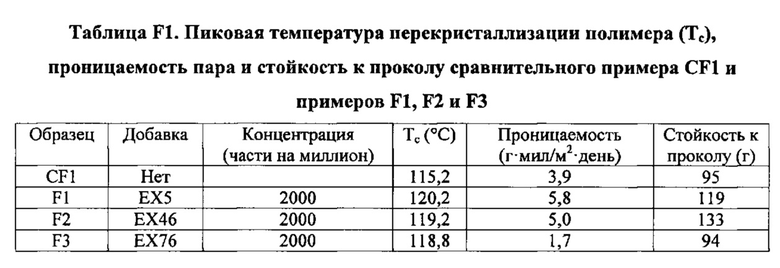
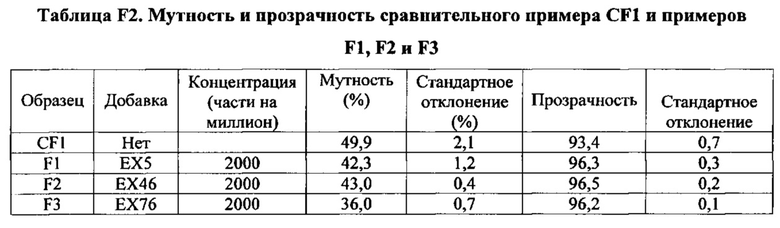
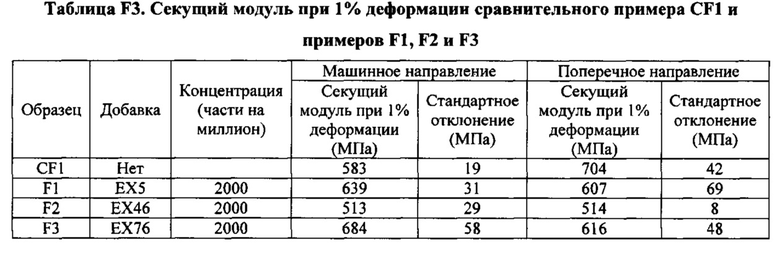

Из данных в таблицах F1-F4 видно, что все добавки, ЕХ5, ЕХ46 и ЕХ76, повышают пиковую температуру перекристаллизации полимера, снижают мутность и повышают прозрачность. Кроме того, ЕХ5 и ЕХ46 повышают прочность на разрыв в машинном направлении, стойкость к проколу и проницаемость пара. ЕХ76 также повышает прочность на разрыв в машинном направлении. Более того, ЕХ76 дает равномерную прочность на разрыв в машинном и поперечном направлениях, улучшает барьерное свойство (подтвержденное низким числом проницаемости) и повышенную жесткость в машинном направлении (секущий модуль при 1% деформации).
Пример F4
Данный пример показывает некоторые физические свойства, проявляемые линейным полиэтиленовым полимером низкой плотности, в который вводили зародышеобразователь кристаллизации согласно настоящему изобретению. Полимерные композиции получали примешиванием (как описано выше) 2000 частей на миллион ЕХ5 в коммерчески доступный линейный полиэтиленовый полимер низкой плотности (ExxonMobil™ LLDPE LL 1001.32), характеризующийся плотностью приблизительно 0,918 г/см3 и индексом текучести расплава приблизительно 1,0 дг/минуту. Образованные гранулы из полимерной композиции затем использовали для получения пленок экструзией с раздувом (толщиной 2 мил) при помощи следующих установок: головка экструдера 101,6 мм (4 дюйма), щель головки экструдера 2,0 мм, BUR 2,35, DDR 17 и производительность 30 кг/ч. Пиковую температуру перекристаллизации полимера, проницаемость, стойкость к проколу, секущий модуль при 1% деформации и прочность на разрыв измеряли и указывали в таблицах F5 и F6.
Пример F5
Пример F5 получали таким же образом, как пример F4, за исключением того, что ЕХ46 использовали вместо ЕХ5.
Пример F6
Пример F6 получали таким же образом, как пример F4, за исключением того, что ЕХ76 использовали вместо ЕХ5.
Сравнительный пример CF2
Сравнительный пример CF2 получали таким же образом, как пример F4, за исключением того, что не использовали зародышеобразователь.

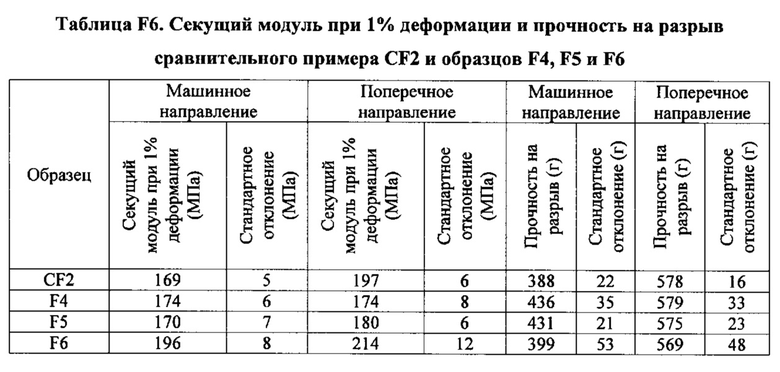
Из данных в таблицах F5 и F6 видно, что все добавки, ЕХ5, ЕХ46 и ЕХ76, повышают пиковую температуру перекристаллизации полимера. ЕХ5 и ЕХ46 повышают прочность на разрыв в машинном направлении, а ЕХ76 значительно повышает модуль в машинном направлении. Все три зародышеобразователя настоящего изобретения незначительно повышают стойкость к проколу.
Пример F7
Данный пример показывает некоторые физические свойства, проявляемые линейным полиэтиленовым полимером низкой плотности, в который вводили зародышеобразователь кристаллизации согласно настоящему изобретению. Полимерные композиции получали примешиванием (как описано выше) 2000 частей на миллион ЕХ46 в коммерчески доступный линейный полиэтиленовый полимер низкой плотности (Dowlex™ 2056G), характеризующийся плотностью приблизительно 0,922 г/см и индексом текучести расплава приблизительно 1,0 дг/минуту. Образованные гранулы из полимерной композиции затем использовали для получения пленок экструзией с раздувом (толщиной 1 мил) при помощи следующих установок: головка экструдера 101,6 мм (4 дюйма), щель головки экструдера 2,0 мм, BUR 2,38, DDR 33 и производительность 22 кг/ч. Пиковую температуру перекристаллизации полимера, проницаемость, стойкость к проколу, секущий модуль при 1% деформации и прочность на разрыв измеряли и указывали в таблицах F7 и F8.
Сравнительный пример CF3
Сравнительный пример CF3 получали таким же образом, как пример F7, за исключением того, что не использовали зародышеобразователь.

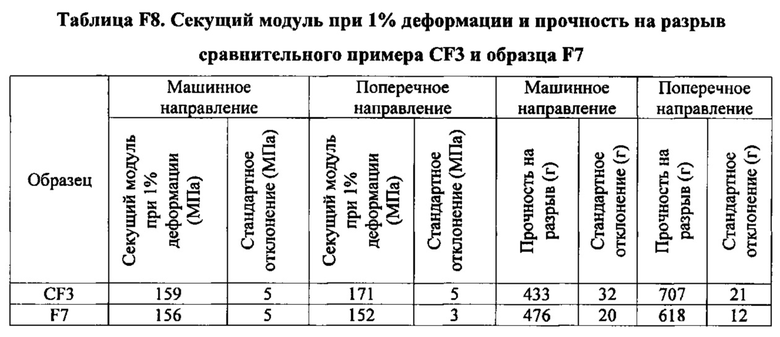
Из данных в таблицах F7 и F8 видно, что добавка ЕХ46 повышает пиковую температуру перекристаллизации полимера, повышает прочность на разрыв в машинном направлении и стойкость к проколу.
Пример F8
Данный пример показывает некоторые физические свойства, проявляемые линейным полиэтиленовым полимером низкой плотности, в который вводили зародышеобразователь кристаллизации согласно настоящему изобретению. Полимерные композиции получали примешиванием (как описано выше) 2000 частей на миллион ЕХ76 в коммерчески доступный линейный полиэтиленовый полимер низкой плотности (Dowlex™ 2056G), характеризующийся плотностью приблизительно 0,922 г/см3 и индексом текучести расплава приблизительно 1,0 дг/минуту. Образованные гранулы из полимерной композиции затем использовали для получения пленок экструзией с раздувом (толщиной 3 мил) при помощи следующих установок: головка экструдера 101,6 мм (4 дюйма), щель головки экструдера 2,0 мм, BUR 2,38, DDR 11 и производительность 23 кг/ч. Пиковую температуру перекристаллизации полимера, проницаемость, стойкость к проколу, секущий модуль при 1% деформации и прочность на разрыв измеряли и указывали в таблицах F9 и F10.
Сравнительный пример CF4
Сравнительный пример CF4 получали таким же образом, как пример F8, за исключением того, что не использовали зародышеобразователь.

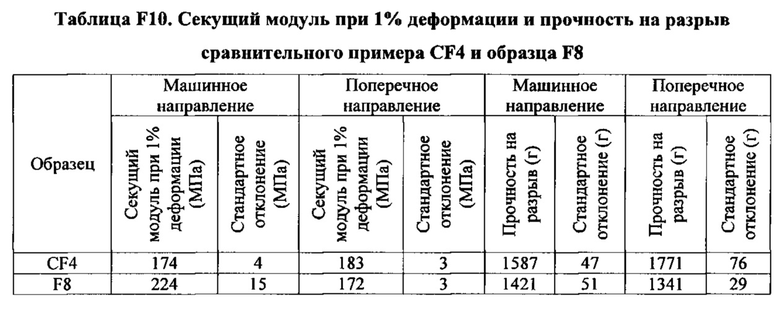
Из данных в таблицах F9 и F10 видно, что добавка ЕХ76 повышала пиковую температуру кристаллизации, стойкость к проколу и секущий модуль при 1% деформации в MD. Также она обеспечивала равномерную прочность на разрыв в машинном и поперечном направлениях.
Пример F9
Данный пример показывает некоторые физические свойства, проявляемые линейным полиэтиленовым полимером низкой плотности, в который вводили зародышеобразователь кристаллизации согласно настоящему изобретению. Полимерные композиции получали примешиванием (как описано выше) 2000 частей на миллион ЕХ5 в коммерчески доступный линейный полиэтиленовый полимер низкой плотности (Dow Elite™ 5100), характеризующийся плотностью приблизительно 0,922 г/см3 и индексом текучести расплава приблизительно 0,85 дг/минуту. Образованные гранулы из полимерной композиции затем использовали для получения пленок экструзией с раздувом (толщиной 2 и 3 мил) при помощи следующих установок: головка 101,6 мм (4 дюйма), щель головки экструдера 2,0 мм, BUR 2,38, DDR 16,5 и 11, соответственно для пленок 2 мил и 3 мил, и производительность 30 кг/ч. Пиковую температуру перекристаллизации полимера, проницаемость, секущий модуль при 1% деформации и прочность на разрыв измеряли и указывали в таблицах F11 и F12.
Пример F10
Пример F10 получали таким же образом, как пример F9, за исключением того, что ЕХ46 использовали вместо ЕХ5.
Пример F11
Пример F11 получали таким же образом, как пример F9, за исключением того, что ЕХ76 использовали вместо ЕХ5.
Сравнительный пример CF5
Сравнительный пример CF1 получали таким же образом, как пример F9, за исключением того, что не использовали зародышеобразователь.
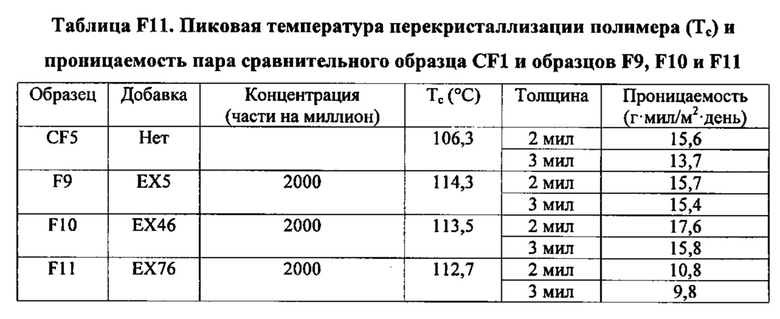
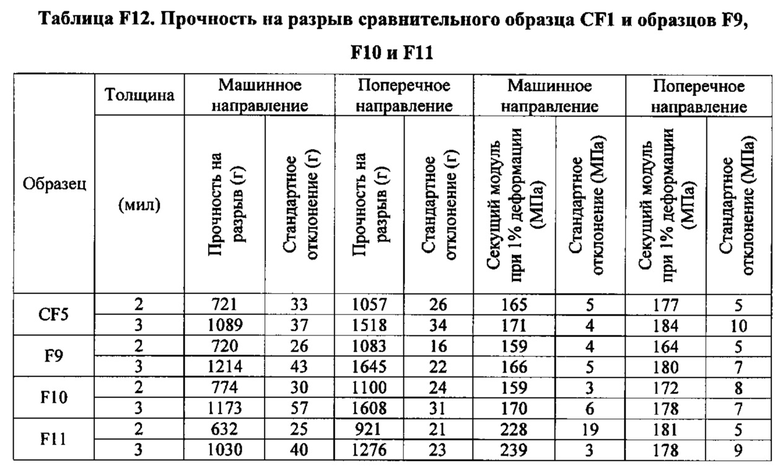
Из данных в таблицах F11-F12 видно, что все добавки, ЕХ5, ЕХ46 и ЕХ76, повышали пиковую температуру перекристаллизации полимера. Кроме того, ЕХ5 и ЕХ46 повышали прочность на разрыв в машинном направлении, в частности, когда толщина пленки составляла 3 мил. ЕХ76 повышала модуль упругости при растяжении в машинном направлении и давала более равномерную прочность на разрыв в машинном и поперечном направлениях. ЕХ5 и ЕХ46 повышали проницаемость, в то же время ЕХ76 снижала проницаемость.
Переработка полиэтилена с зародышеобразователем кристаллизации литьем под давлением
В следующих примерах для литья под давлением используемые полиэтиленовые смолы сначала измельчали в порошок приблизительно 35 меш. Затем вводили зародышеобразователь настоящего изобретения в смолу и перемешивали в высокоэффективной мешалке Henschel в течение приблизительно 2 минут при скорости лопасти мешалки приблизительно 2100 об/мин. Образцы затем компаундировали в расплаве в одношнековом экструдере DeltaPlast со шнеком диаметром 25 мм и отношением длины к диаметру 30:1. Температуру цилиндра быстро повышали от 160 до 190°C и скорость шнека устанавливали на приблизительно 130 об/мин. Экструдат в виде нити охлаждали на водяной бане, а затем далее гранулировали.
Пластинки и полосы получали посредством литья под давлением на машине для литья под давлением Arburg 40 ton со шнеком диаметром 25,4 мм. Температура цилиндра машины для литья под давлением составляла 230°C, если иное специально не указано, а температуру формы контролировали на 25°C.
Если иное не указано, скорость впрыска для пластинок составляла 2,4 см3/с, а их размеры представляли собой длину приблизительно 60 мм, ширину 60 мм и толщину 2 мм. Эти пластинки использовали для измерения температуры перекристаллизации, жесткости в двух направлениях и сопротивления многоосному напряженному состоянию при ударных нагрузках.
Если иное не указано, скорость впрыска для полос составляла 15 см3/с, а их размеры представляли собой длину приблизительно 127 мм, ширину 12,7 мм и толщину 3,3 мм. Эти полосы использовали для измерения секущего модуля при 1% деформации, HDT и ударопрочности по Изоду.
Испытание полиэтилена с зародышеобразователем кристаллизации
Исследование свойств при изгибе (указанных как модуль упругости в двух направлениях) проводили на вышеуказанных пластинках, используя установку MTS Q-Test-5 с шириной захвата 32 мм, фиксированной скоростью деформации 8,53 мм/минуту и номинальной шириной образца 50,8 мм. Образцы получали вырезанием квадратных секций (приблизительно 50 мм × 50 мм) из центра пластинок для получения образцов с одинаковыми параметрами во всех направлениях. Кроме испытания образцов поперек машинного направления/направления потока, как принято (обозначенного как "поперечное направление" в таблице результатов), образцы также испытывали путем сгибания поперек поперечного направления относительно потока, чтобы также измерить жесткость в этом направлении (обозначенном как "машинное направление" в таблице результатов) для анализа жесткости пластинок в двух направлениях.
Исследование многоосного напряженного состояния при ударных нагрузках проводили на вышеуказанных пластинках при помощи тестера Instron Ceast 9350 согласно стандарту ISO 6603, используя скорость 2,2 м/с и температуру в камере -30°C. Испытание на модуль упругости при изгибе (указанный как секущий модуль при 1% деформации) проводили на вышеуказанных полосах при помощи установки MTS Qtest/5 согласно процедуре B из ASTM D790. Испытание на деформационную теплостойкость проводили на вышеуказанных полосах при помощи прибора Ceast HDT 3 VICAT согласно способу B из ASTM D648-07. Испытание на ударную вязкость по Изоду проводили на вышеуказанных полосах при помощи прибора Tinius-Olsen 892Т согласно ASTM D256, способу А.
Пиковую температуру перекристаллизации полимера (Тс) для композиций на основе термопластичного полимера измеряли при помощи дифференциального сканирующего калориметра (дифференциального сканирующего калориметра Mettler-Toledo DSC822). В частности, образец отбирали из заданной части и нагревали со скоростью 20°C/минуту от температуры 60°C до 220°C, выдерживали при 220°C в течение двух минут и охлаждали со скоростью приблизительно 10°C/минуту до температуры 60°C. Температура, при которой возникало пиковое преобразование кристаллов полимера (что соответствует пиковой температуре перекристаллизации полимера) записывали для каждого образца.
Пример I1-I3
Данные примеры показывают некоторые физические свойства, проявляемые полиэтиленовым полимером высокой плотности, в который вводили зародышеобразователь кристаллизации согласно настоящему изобретению. Полимерные композиции получали примешиванием (как описано выше) соединения примера получения ЕХ5 и различных поглотителей кислот в коммерчески доступный полиэтилен высокой плотности (Dowlex™ IP 40), характеризующийся плотностью приблизительно 0,954 г/см3 и индексом текучести расплава приблизительно 40 дг/минуту. Смолу сначала измельчали, перемешивали с добавками, а затем компаундировали и экструдировали с образованием гранул. Образованные гранулы из полимерной композиции затем отливали под давлением в тестовые пластинки и полосы.
Информация о составах для примеров I1-I3 и сравнительного примера CI1 указана в таблице I1. Пиковая температура перекристаллизации полимера (Tc), многоосное напряженное состояние при ударных нагрузках при температуре -30°C и модуль упругости в двух направлениях (измеренные на пластинках) и секущий модуль при 1% деформации и деформационная теплостойкость (измеренные на полосах) указаны в таблицах I2 и I3 ниже.

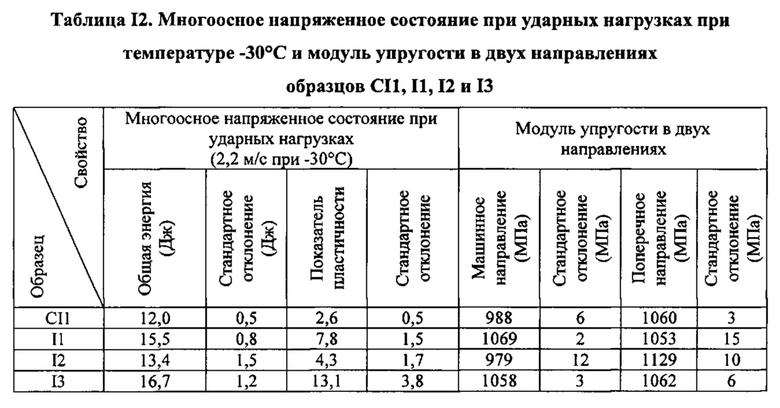

Пример I4-I6
Данные примеры показывают некоторые физические свойства, проявляемые полиэтиленовым полимером высокой плотности, в который вводили зародышеобразователь кристаллизации согласно настоящему изобретению. Полимерные композиции получали примешиванием (как описано выше) соединения примера получения ЕХ46 и различных поглотителей кислот в коммерчески доступный полиэтилен высокой плотности (Dowlex™ IP 40), характеризующийся плотностью приблизительно 0,954 г/см3 и индексом текучести расплава приблизительно 40 дг/минуту. Смолу сначала измельчали, перемешивали с добавками, а затем компаундировали и экструдировали с образованием гранул. Образованные гранулы из полимерной композиции затем отливали под давлением в тестовые пластинки и полосы. Информация о составах для примеров I4-I6 и сравнительного примера CI2 указана в таблице I4. Пиковая температура перекристаллизации полимера (Тс), многоосное напряженное состояние при ударных нагрузках при температуре -30°C и модуль упругости в двух направлениях (измеренные на пластинках) и секущий модуль при 1% деформации и деформационная теплостойкость (измеренные на полосах) указаны в таблицах 15 и I6 ниже.
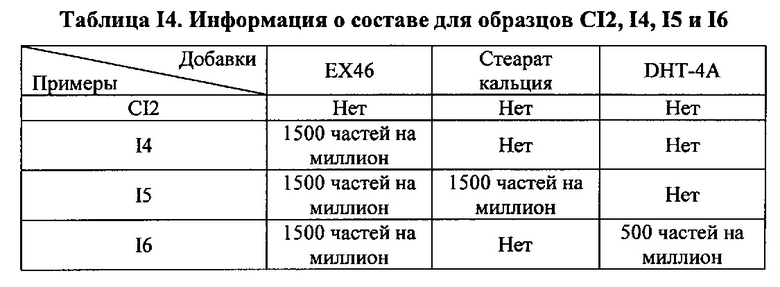
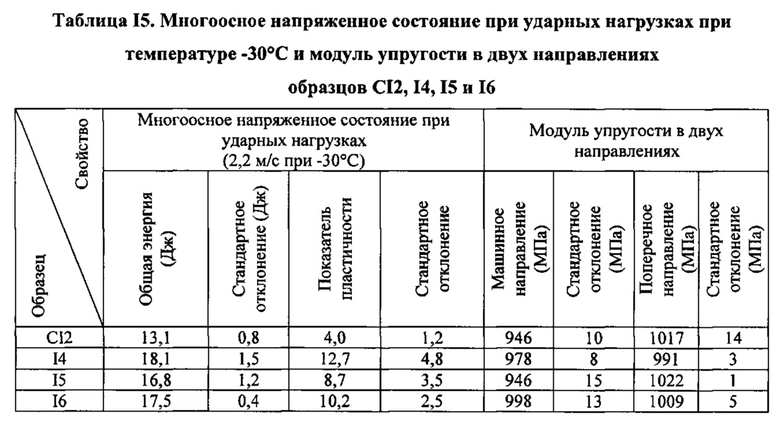
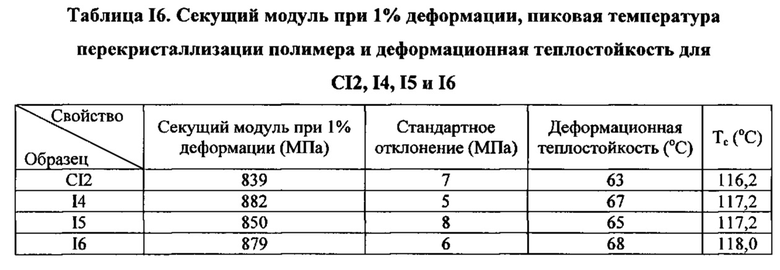
Пример I7
Данный пример показывает некоторые физические свойства, проявляемые полиэтиленовым полимером высокой плотности, в который вводили зародышеобразователи кристаллизации согласно настоящему изобретению. Полимерные композиции получали примешиванием (как описано выше) соединения примера получения ЕХ76 в коммерчески доступный полиэтилен высокой плотности (Dowlex™ IP 40), характеризующийся плотностью приблизительно 0,954 г/см3 и индексом текучести расплава приблизительно 40 дг/минуту. Смолу сначала измельчали, перемешивали с добавками, а затем компаундировали и экструдировали с образованием гранул. Образованные гранулы из полимерной композиции затем отливали под давлением в тестовые пластинки и полосы. Информация о составах для примера I7 и сравнительного примера CI3 указана в таблице I7. Пиковая температура перекристаллизации полимера (Тс), многоосное напряженное состояние при ударных нагрузках при температуре -30°C и модуль упругости в двух направлениях (измеренные на пластинках) и секущий модуль при 1% деформации и деформационная теплостойкость (измеренные на полосах) измерены и указаны в таблицах I8 и I9 ниже.



Пример I8-I10
Данные примеры показывают некоторые физические свойства, проявляемые полиэтиленовым полимером высокой плотности, в который вводили зародышеобразователи кристаллизации согласно настоящему изобретению. Полимерные композиции получали примешиванием (как описано выше) соединения примера получения ЕХ5 и различных поглотителей кислот в коммерчески доступный полиэтилен высокой плотности (ExxonMobil™ HDPE HD 6719), характеризующийся плотностью приблизительно 0,952 г/см3 и индексом текучести расплава приблизительно 19 дг/минуту. Смолу сначала измельчали, перемешивали с добавками, а затем компаундировали и экструдировали с образованием гранул. Образованные гранулы из полимерной композиции затем отливали под давлением в тестовые пластинки и полосы. Информация о составах для примеров I8-I10 и сравнительного примера CI4 указана в таблице I10. Пиковая температура перекристаллизации полимера, многоосное напряженное состояние при ударных нагрузках при температуре -30°C и модуль упругости в двух направлениях (измеренные на пластинках) и секущий модуль при 1% деформации и деформационная теплостойкость (измеренные на полосах) измерены и указаны в таблицах I11 и I12 ниже.

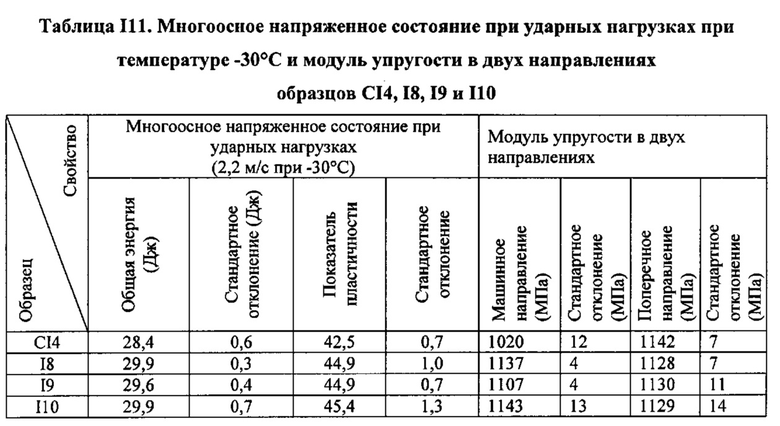
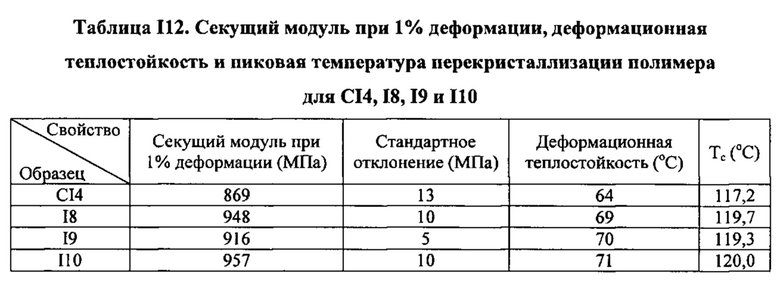
Пример I11-I12
Данные примеры показывают некоторые физические свойства, проявляемые полиэтиленовым полимером высокой плотности, в который вводили зародышеобразователи кристаллизации согласно настоящему изобретению. Полимерные композиции получали примешиванием (как описано выше) соединений примеров получения ЕХ46 и ЕХ76 в коммерчески доступный полиэтилен высокой плотности (ExxonMobil™ HDPE HD 6719), характеризующийся плотностью приблизительно 0,952 г/см3 и индексом текучести расплава приблизительно 19 дг/минуту. Смолу сначала измельчали, перемешивали с добавками, а затем компаундировали и экструдировали с образованием гранул. Образованные гранулы из полимерной композиции затем отливали под давлением в тестовые пластинки и полосы. В данном примере пластинки формовали при 15 см3/с, а полосы при 40 см3/с, поддерживая другие условия переработки такими, как описано выше. Информация о составах для примеров I11 и I12 и сравнительного примера CI5 указана в таблице I13. Пиковая температура перекристаллизации полимера, многоосное напряженное состояние при ударных нагрузках при температуре -30°C и модуль упругости в двух направлениях (измеренные на пластинках) и секущий модуль при 1% деформации, ударная вязкость по Изоду и деформационная теплостойкость (измеренные на полосах) измерены и указаны в таблицах I14 и I15 ниже.
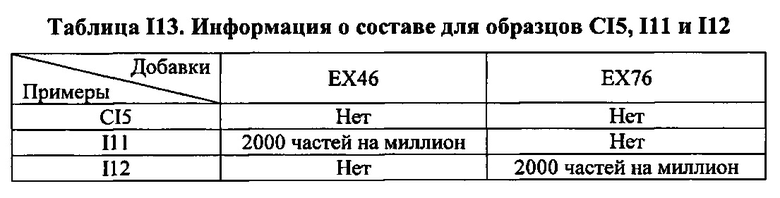
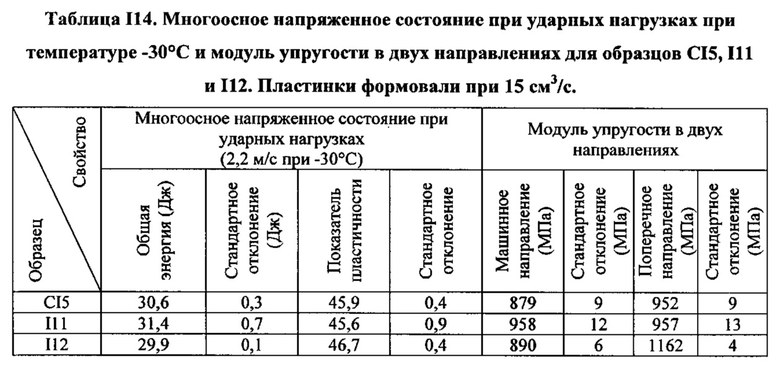

Пример I13-I15
Данные примеры показывают некоторые физические свойства, проявляемые полиэтиленовым полимером высокой плотности, в который вводили зародышеобразователи кристаллизации согласно настоящему изобретению. Полимерные композиции получали примешиванием соединений примеров получения ЕХ5, ЕХ46 и ЕХ76 в коммерчески доступный полиэтилен высокой плотности (LyondellBasell Hostalen® АСР 6541A UV), характеризующийся плотностью приблизительно 0,954 г/см3 и индексом текучести расплава приблизительно 1,5 дг/минуту. Смолу сначала измельчали, перемешивали с добавками, а затем компаундировали и экструдировали с образованием гранул. Образованные гранулы из полимерной композиции затем отливали под давлением в тестовые пластинки и полосы. В данном примере пластинки формовали при 220°C и 20 см3/с, а полосы при 220°C и 40 см3/с, поддерживая другие условия переработки такими, как описано выше. Информация о составах для примеров I13, I14 и I15 и сравнительного примера CI6 указана в таблице I16. Пиковая температура перекристаллизации полимера, многоосное напряженное состояние при ударных нагрузках при температуре -30°C и модуль упругости в двух направлениях (измеренные на пластинках) и секущий модуль при 1% деформации, ударная вязкость по Изоду и деформационная теплостойкость (измеренные на полосах) измерены и указаны в таблицах I17 и I18 ниже.



Производство полученных тонкостенным литьем под давлением с зародышеобразователем кристаллизации одноразовых стаканчиков
Используемые полиэтиленовые смолы сначала измельчали до порошка в 35 меш. Вводили зародышеобразователь настоящего изобретения в смолу и перемешивали в высокоэффективной мешалке Henschel в течение приблизительно 2 минут при скорости лопасти мешалки приблизительно 2100 об/мин. Образцы затем компаундировали в расплаве в одношнековом экструдере МРМ с диаметром шнека 38 мм. Температуру цилиндра экструдера быстро увеличивали от 160 до 190°C. Экструдат в виде нитей охлаждали в водяной бане и затем далее гранулировали. Одноразовые стаканчики емкостью 16 унций получали на машине для литья под давлением Husky S-90 RS40/32 с усилием смыкания 90 тонн и оснащенным аккумулятором/высокоскоростным узлом впрыска, используя одногнездную форму. Машина для литья под давлением имела шнек с возвратно-поступательным движением с диаметром 32 мм и отношением длины к диаметру 25:1. Температура цилиндра экструдера составляла от 190 до 210°C в зависимости от индекса расплава смолы, при этом температуры обогреваемых литников также устанавливали приблизительно на 210°C. Температуру формы устанавливали приблизительно на 12°C. Размеры одноразовых стаканчиков представляли собой диаметр приблизительно 117 мм и высоту 76 мм.
Испытание одноразовых стаканчиков из полиэтилена с зародышеобразователем кристаллизации
% мутности изделий измеряли на боковой стенке при помощи мутномера BYK Gardner согласно ASTM D1023. Прозрачность изделий измеряли на боковой стенке при помощи мутномера BYK Gardner. Верхний предел нагрузки на изделия измеряли при помощи установки MTS Q-Test-5 согласно ASTM D 2659. Пиковую температуру перекристаллизации полимера (Тс) для композиций на основе термопластичного полимера измеряли при помощи дифференциального сканирующего калориметра (дифференциального сканирующего калориметра Mettler-Toledo DSC822). В частности, полученную компрессионным формованием пластинку получали из гранул, а образец отбирали из пластинки и нагревали со скоростью 20°C/минуту от температуры 60°C до 220°C, выдерживали при 220°C в течение двух минут и охлаждали со скоростью приблизительно 10°C/минуту до температуры 60°C. Температура, при которой возникало пиковое преобразование кристаллов полимера (что соответствует пиковой температуре перекристаллизации полимера) записывали для каждого образца.
Пример I16-I18
Данные примеры показывают некоторые физические свойства, проявляемые изделиями (одноразовыми стаканчиками) из полиэтиленового полимера высокой плотности, которые получали с использованием композиции, содержащей зародышеобразователь согласно настоящему изобретению. Полиэтиленовые изделия получали примешиванием (как описано выше) соединений примеров получения ЕХ5, ЕХ46 и ЕХ76 в коммерчески доступный полиэтилен высокой плотности (Dowlex IP 40), характеризующийся плотностью приблизительно 0,954 г/см3 и индексом текучести расплава приблизительно 40 дг/минуту. Смолу сначала измельчали, перемешивали с добавками, а затем компаундировали и экструдировали с образованием гранул, как описано выше. Образованные гранулы из полимерной композиции затем перерабатывали путем тонкостенного литья под давлением (TWIM) с получением полиэтиленовых изделий. В данном примере одноразовые стаканчики получали, используя время заполнения 0,21 с. Информация о составе для примеров I16, I17, I18 и сравнительного примера CI7 указана в таблице I19. Пиковое время перекристаллизации (измеренное в полученной компрессионным формованием пластинке, полученной из гранул), прозрачность, мутность и верхний предел нагрузки на одноразовые стаканчики измеряли и указывали в таблице I20 ниже.
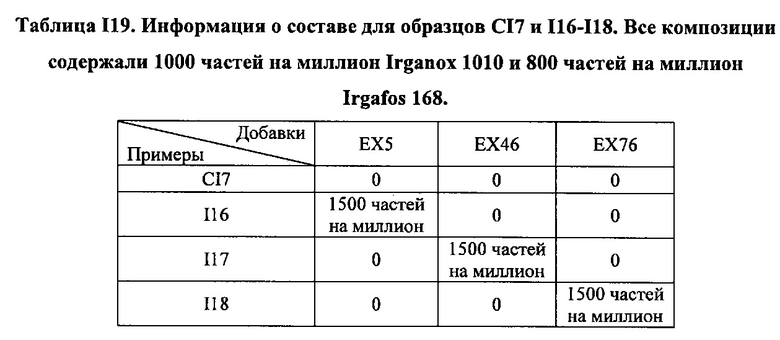
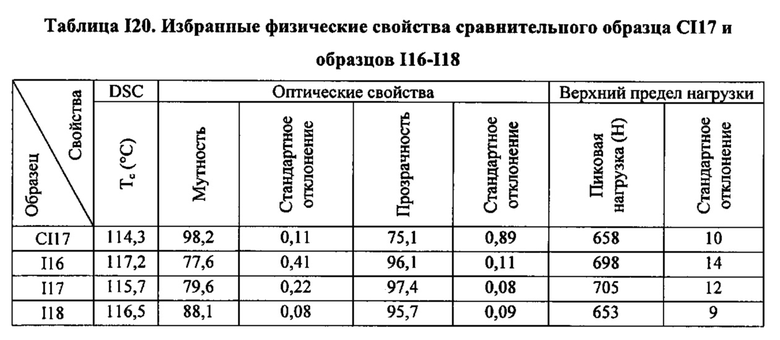
Производство полученного литьем под давлением с зародышеобразователем кристаллизации контейнера для хранения продуктов питания
Используемые полиэтиленовые смолы сначала измельчали до порошка в 35 меш. Вводили зародышеобразователи настоящего изобретения в смолу и перемешивали в высокоэффективной мешалке Henschel в течение приблизительно 2 минут при скорости лопасти мешалки приблизительно 2100 об/мин. Образцы затем компаундировали в расплаве в одношнековом экструдере МРМ с диаметром шнека 38 мм. Температуру цилиндра экструдера быстро увеличивали от 160 до 190°C. Экструдат в виде нитей охлаждали в водяной бане и затем далее гранулировали. Многоразовые контейнеры для хранения продуктов питания с приблизительной массой 62 г получали на машине для литья под давлением Husky S-90 RS40/32 с усилием смыкания 90 тонн и оснащенным аккумулятором/высокоскоростным узлом впрыска, используя одногнездную форму. Машина для литья под давлением имела шнек с возвратно-поступательным движением с диаметром 32 мм и отношением длины к диаметру 25:1. Температура цилиндра экструдера составляла от 190 до 220°C в зависимости от индекса расплава смолы, при этом температуры обогреваемых литников также устанавливали приблизительно на 220°C. Температуру формы устанавливали приблизительно на 12°C. Размеры контейнеров для хранения продуктов питания составляли 190,5 мм X 98,4 мм X 76,2 мм, а толщина стенки составляла приблизительно 1 мм.
Испытание контейнеров для хранения продуктов питания из полиэтилена с зародышеобразователем кристаллизации
% мутности изделий измеряли на боковой стенке при помощи мутномера BYK Gardner согласно ASTM D1023. Прозрачность изделий измеряли на боковой стенке при помощи мутномера BYK Gardner. Верхний предел нагрузки на изделия измеряли при помощи установки MTS Q-Test-5 согласно ASTM D 2659. Пиковую температуру перекристаллизации полимера (Тс) для композиций на основе термопластичного полимера измеряли при помощи дифференциального сканирующего калориметра (дифференциального сканирующего калориметра Mettler-Toledo DSC822). В частности, полученную компрессионным формованием пластинку получали из гранул, а образец отбирали из пластинки и нагревали со скоростью 20°C/минуту от температуры 60°C до 220°C, выдерживали при 220°C в течение двух минут и охлаждали со скоростью приблизительно 10°C/минуту до температуры 60°C. Температура, при которой возникало пиковое преобразование кристаллов полимера (что соответствует пиковой температуре перекристаллизации полимера) записывали для каждого образца.
Пример H1-H3
Данные примеры показывают некоторые физические свойства, проявляемые изделием (контейнером для хранения продуктов питания) из полиэтиленового полимера высокой плотности, в который вводили зародышеобразователи кристаллизации согласно настоящему изобретению. Полиэтиленовые изделия получали примешиванием соединений примеров получения ЕХ5, ЕХ46 и ЕХ76 в коммерчески доступный полиэтиленовый полимер высокой плотности (ExxonMobil™ HDPE HD 6719), характеризующийся плотностью приблизительно 0,952 г/см3 и индексом текучести расплава приблизительно 19 дг/минуту. Смолу сначала измельчали, перемешивали с добавками, а затем компаундировали и экструдировали с образованием гранул, как описано выше. Образованные гранулы из полимерной композиции затем перерабатывали путем литья под давлением (IM) с получением полиэтиленовых изделий. В данном примере посуду получали, используя время заполнения 2,8 с. Информация о составе для примеров H1, H2, H3 и сравнительного примера CH1 указана в таблице TH1. Пиковое время перекристаллизации (измеренное в полученной компрессионным формованием пластинке, полученной из гранул), прозрачность, мутность и верхний предел нагрузки измеряли и указывали в таблице TH2 ниже.

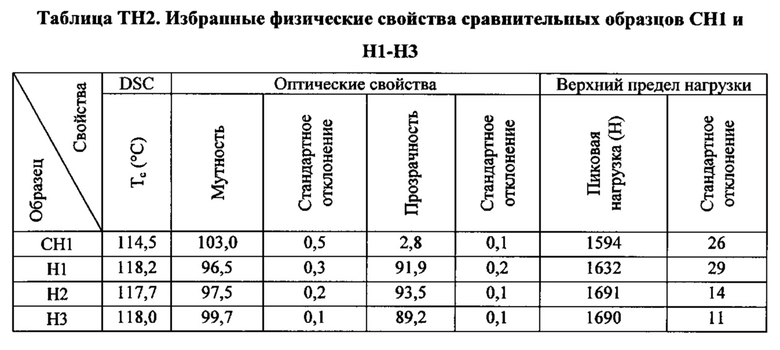
Пример Н4-Н6
Данные примеры показывают некоторые физические свойства, проявляемые изделием (контейнером для хранения продуктов питания) из полиэтиленового полимера высокой плотности, которое получали из смолы с зародышеобразователем кристаллизации согласно настоящему изобретению. Полиэтиленовые изделия получали примешиванием соединений примеров получения ЕХ5, ЕХ46 и ЕХ76 в коммерчески доступный полиэтилен высокой плотности (Dow™ HDPE DMDA-8965 NT 7), характеризующийся плотностью приблизительно 0,954 г/см3 и индексом текучести расплава приблизительно 66 дг/минуту. Смолу сначала измельчали, перемешивали с добавками, а затем компаундировали и экструдировали с образованием гранул, как описано выше. Образованные гранулы из полимерной композиции затем перерабатывали путем литья под давлением (IM) с получением полиэтиленовых изделий. В данном примере посуду получали, используя время заполнения 3,0 с. Информация о составе для примеров H4, H5, H6 и сравнительного примера СН2 указана в таблице TH3. Пиковое время перекристаллизации (измеренное в полученной компрессионным формованием пластинке, полученной из гранул), прозрачность, мутность и верхний предел нагрузки измеряли и указывали в таблице TH4 ниже.
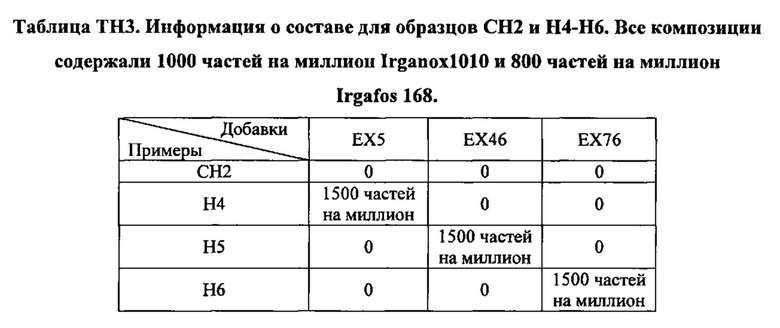
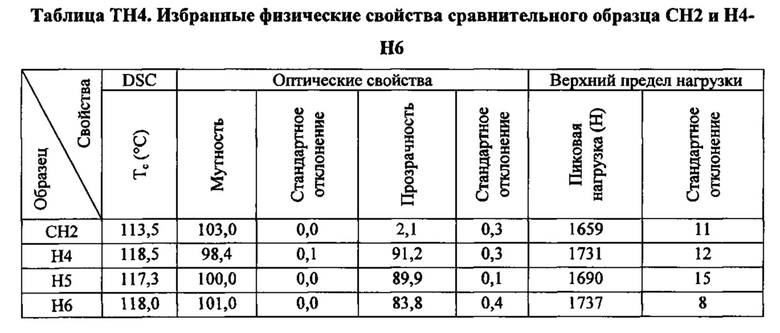
Пример Н7-Н9
Данные примеры показывают некоторые физические свойства, проявляемые изделием (контейнером для хранения продуктов питания) из полиэтиленового полимера высокой плотности, которое получали, используя смолу с зародышеобразователем кристаллизации согласно настоящему изобретению. Полиэтиленовые изделия получали примешиванием соединений примеров получения ЕХ5, ЕХ46 и ЕХ76 в коммерчески доступный линейный полиэтиленовый полимер низкой плотности (ExxonMobil™ LLDPE LL 6100.17), характеризующийся плотностью приблизительно 0,925 г/см3 и индексом текучести расплава приблизительно 20 дг/минуту. Смолу сначала измельчали, перемешивали с добавками, а затем компаундировали и экструдировали с образованием гранул, как описано выше. Образованные гранулы из полимерной композиции затем перерабатывали путем литья под давлением (IM) с получением полиэтиленовых изделий. В данном примере посуду получали, используя время заполнения 2,7 с. Информация о составе для примеров H7, H8, H9 и сравнительного примера CH3 указана в таблице TH5. Пиковое время перекристаллизации (измеренное в полученной компрессионным формованием пластинке, полученной из гранул), прозрачность, мутность и верхний предел нагрузки измеряли и указывали в таблице TH6 ниже.
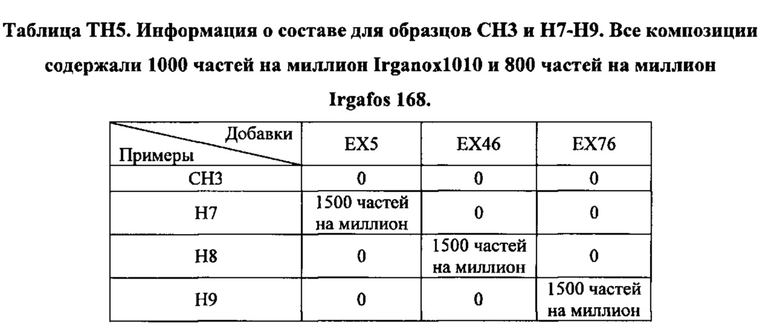
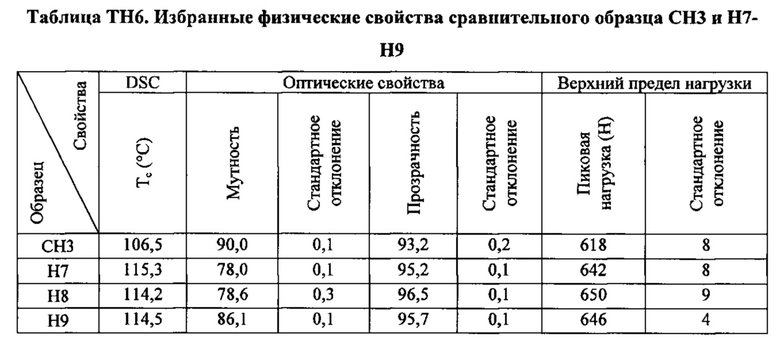
Пример Н10-Н12
Данные примеры показывают некоторые физические свойства, проявляемые изделием (контейнером для хранения продуктов питания) из полиэтиленового полимера высокой плотности, которое получали из смолы с зародышеобразователем кристаллизации согласно настоящему изобретению. Полиэтиленовые изделия получали примешиванием соединений примеров получения ЕХ5, ЕХ46 и ЕХ76 в коммерчески доступный линейный полиэтиленовый полимер низкой плотности (Dowlex™ 2517), характеризующийся плотностью приблизительно 0,919 г/см3 и индексом текучести расплава приблизительно 25 дг/минуту. Смолу сначала измельчали, перемешивали с добавками, а затем компаундировали и экструдировали с образованием гранул, как описано выше. Образованные гранулы из полимерной композиции затем перерабатывали путем литья под давлением (IM) с получением полиэтиленовых изделий. В данном примере посуду получали, используя время заполнения 2,5 с. Информация о составе для примеров H10, H11, H12 и сравнительного примера СН4 указана в таблице ТН7. Пиковое время перекристаллизации (измеренное на полученной компрессионным формованием пластинке, полученной из гранул), прозрачность, мутность и верхний предел нагрузки измеряли и указывали в таблице ТН8 ниже.
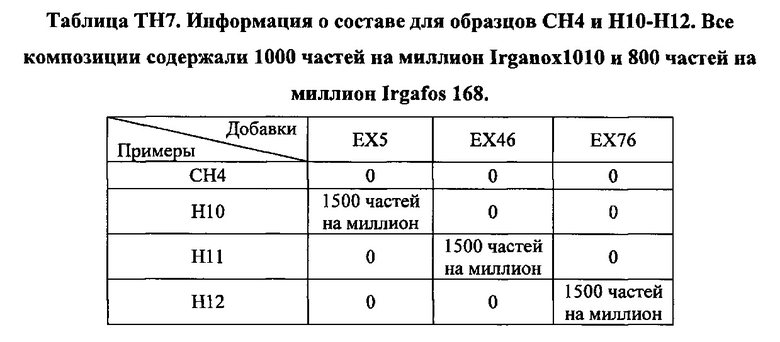

Образование полипропилена с зародышеобразователем кристаллизации
Вводили различные добавки в смолу на основе полипропилена и перемешивали в высокоэффективной мешалке Henschel в течение приблизительно 2 минут при скорости лопасти мешалки приблизительно 2100 об/мин. Образцы затем компаундировали в расплаве в одношнековом экструдере DeltaPlast со шнеком диаметром 25 мм и отношением длины к диаметру 30:1. Температуру цилиндра быстро повышали от 190 до 230°C и скорость шнека устанавливали на приблизительно 130 об/мин. Экструдат в виде нити охлаждали на водяной бане, а затем далее гранулировали.
Пластинки и полосы получали посредством литья под давлением на машине для литья под давлением Arburg 40 ton со шнеком диаметром 25,4 мм. Температура цилиндра машины для литья под давлением составляла 230°C, а температуру формы контролировали на 25°C. Скорость впрыска для пластинок составляла 2,4 см3/с, а их размеры представляли собой длину приблизительно 60 мм, ширину 60 мм и толщину 2 мм. Эти пластинки использовали для измерения температуры перекристаллизации, жесткости в двух направлениях. Скорость впрыска для полос составляла 15 см3/с, а их размеры представляли собой длину приблизительно 127 мм, ширину 12,7 мм и толщину 3,3 мм. Эти полосы использовали для измерения секущего модуля при 1% деформации, HDT и ударопрочности по Изоду.
Испытание полипропилена с зародышеобразователем кристаллизации
Исследование свойств при изгибе (указанных как модуль упругости в двух направлениях) проводили на вышеуказанных пластинках, используя установку MTS Q-Test-5 с шириной захвата 32 мм, фиксированной скоростью деформации 8,53 мм/минуту и номинальной шириной образца 50,8 мм. Образцы получали вырезанием квадратных секций (приблизительно 50 мм × 50 мм) из центра пластинок для получения образцов с одинаковыми параметрами во всех направлениях. Кроме испытания образцов поперек машинного направления/направления потока, как принято (обозначенного как "поперечное направление" в таблице результатов), образцы также испытывали путем сгибания поперек поперечного направления относительно потока, чтобы также измерить жесткость в этом направлении (обозначенном как "машинное направление" в таблице результатов) для анализа жесткости в двух направлениях пластинок.
Испытание на модуль упругости при изгибе (указанного как секущий модуль при 1% деформации) проводили на вышеуказанных полосах, используя установку MTS Qtest/5 согласно процедуре B из ASTM D790. Испытание на деформационную теплостойкость проводили на вышеуказанных полосах, используя установку Ceast HDT 3 VICAT согласно способу B из ASTM D648-07. Испытание на ударную вязкость по Изоду проводили на вышеуказанных полосах, используя установку Tinius-Olsen 892Т согласно ASTM D256, способу А. Пиковую температуру перекристаллизации полимера (Tc) для композиций на основе термопластичного полимера измеряли при помощи дифференциального сканирующего калориметра (дифференциального сканирующего калориметра Mettler-Toledo DSC822). Это выполняли путем нагревания приблизительно 5 миллиграммового образца, полученного из целевых пластинок, при 20°C/минуту от 50°C до 220°C, выдерживания при 220°C в течение 2 минут, охлаждения пластинок со скоростью приблизительно 20°C/минуту назад до 50°C и записи температуры, при которой происходило пиковое преобразование кристаллов полимера (Tc).
Пример Р1-Р6
Данные примеры показывают некоторые физические свойства, проявляемые полипропиленовым полимером, в который вводили зародышеобразователь кристаллизации согласно настоящему изобретению. Полимерные композиции получали примешиванием соединений примеров получения ЕХ5, ЕХ46, ЕХ9, ЕХ8, ЕХ36 и ЕХ76 в коммерчески доступный гомополимер полипропилена (LyondellBasell Pro-fax™ 6301), характеризующийся индексом текучести расплава приблизительно 12 дг/минуту. Смолу сначала смешивали с зародышеобразователем настоящего изобретения с антиоксидантом и поглотителями кислот, затем смесь компаундировали и экструдировали с получением гранул. Образованные гранулы отливали под давлением в тестовые пластинки и полосы, как описано выше. Информация о составах для примеров Р1-Р6 и сравнительного примера CP1 указана в таблице Р1. Пиковую температуру перекристаллизации полимера, модуль упругости в двух направлениях, ударную вязкость по Изоду и деформационную теплостойкость измеряли и указывали в таблицах P2 и P3 ниже.
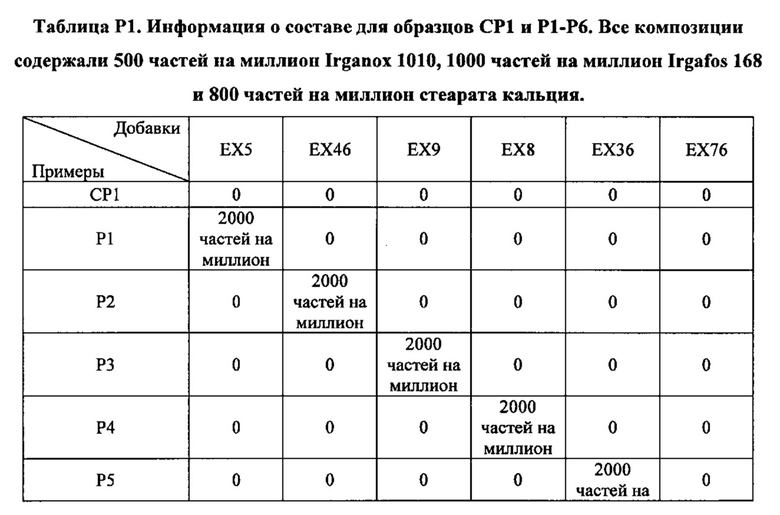
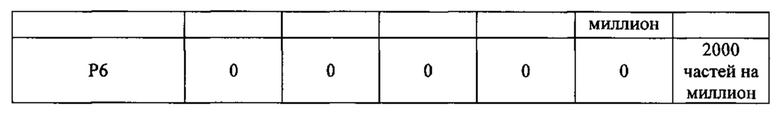
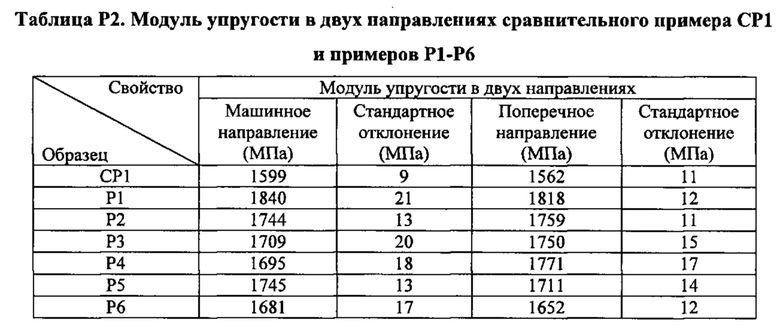
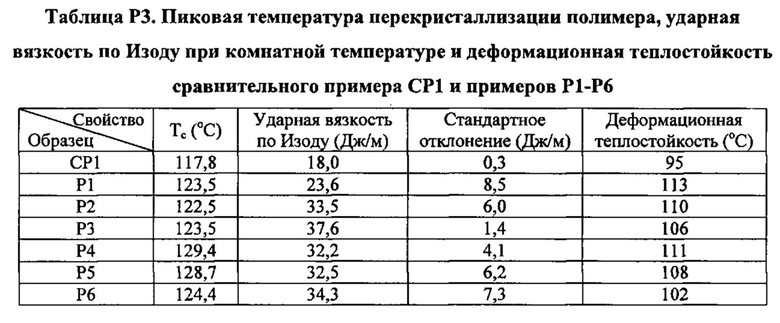
Пример Р7-Р12
Данные примеры показывают некоторые физические свойства, проявляемые полипропиленовым полимером, в который вводили зародышеобразователь кристаллизации согласно настоящему изобретению. Полимерные композиции получали примешиванием соединений примеров получения ЕХ5, ЕХ46, EX9, ЕХ8, ЕХ36 и ЕХ76 в коммерчески доступный гомополимер полипропилена (LyondellBasell Pro-fax™ 6301), характеризующийся индексом текучести расплава приблизительно 12 дг/минуту. Смолу сначала смешивали с зародышеобразователями настоящего изобретения с антиоксидантом и поглотителями кислот, затем смесь компаундировали и экструдировали с получением гранул. Образованные гранулы отливали под давлением в тестовые пластинки и полосы, как описано выше. Информация о составах для примеров Р7-Р12 и сравнительного примера CP2 указана в таблице P4. Пиковую температуру перекристаллизации полимера, модуль упругости в двух направлениях, ударную вязкость по Изоду и деформационную теплостойкость измеряли и указывали в таблицах Р5 и P6 ниже.
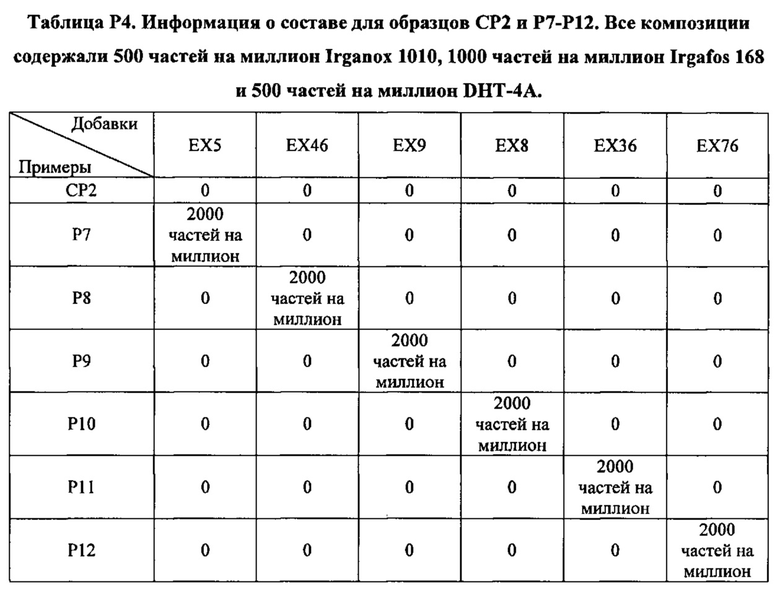


Пример Р13-Р18
Данные примеры показывают некоторые физические свойства, проявляемые полипропиленовым полимером, в который вводили зародышеобразователь кристаллизации согласно настоящему изобретению. Полимерные композиции получали примешиванием соединений примеров получения ЕХ5, ЕХ46, EX9, ЕХ8, ЕХ36 и ЕХ76 в коммерчески доступный ударопрочный сополимер полипропилена (LyondellBasell Pro-fax™ SD375S), характеризующийся индексом текучести расплава приблизительно 18 дг/минуту.
Смолу сначала смешивали с зародышеобразователями настоящего изобретения с антиоксидантом и поглотителями кислот, затем смесь компаундировали и экструдировали с получением гранул. Образованные гранулы отливали под давлением в тестовые пластинки и полосы, как описано выше. Информация о составах для примеров Р13-Р18 и сравнительного примера CP3 указана в таблице Р7. Пиковую температуру перекристаллизации полимера, модуль упругости в двух направлениях, ударную вязкость по Изоду и деформационную теплостойкость измеряли и указывали в таблицах P8 и P9 ниже.

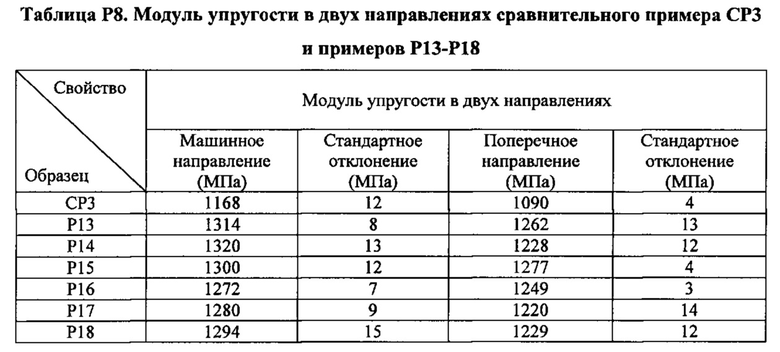

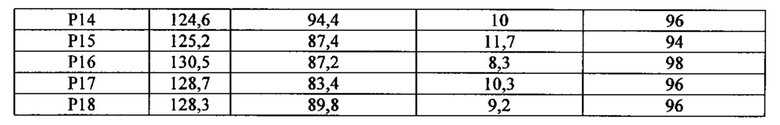
Пример P19-Р24
Данные примеры показывают некоторые физические свойства, проявляемые полипропиленовым полимером, в который вводили зародышеобразователь кристаллизации согласно настоящему изобретению. Полимерные композиции получали примешиванием соединений примеров получения ЕХ5, ЕХ46, EX9, ЕХ8, ЕХ36 и ЕХ76 в коммерчески доступный ударопрочный сополимер полипропилена (LyondellBasell Pro-fax™ SD375S), характеризующийся индексом текучести расплава приблизительно 18 дг/минуту.
Смолу сначала смешивали с зародышеобразователями настоящего изобретения с антиоксидантом и поглотителями кислот, затем смесь компаундировали и экструдировали с получением гранул. Образованные гранулы отливали под давлением в тестовые пластинки и полосы, как описано выше. Информация о составах для примеров Р19-Р24 и сравнительного примера CP4 указана в таблице Р10. Пиковую температуру перекристаллизации полимера, модуль упругости в двух направлениях, ударную вязкость по Изоду и деформационную теплостойкость измеряли и указывали в таблицах P11 и Р12 ниже.
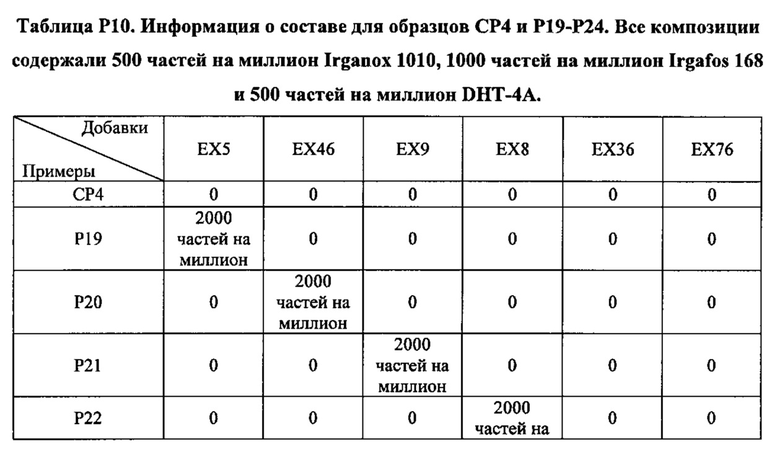
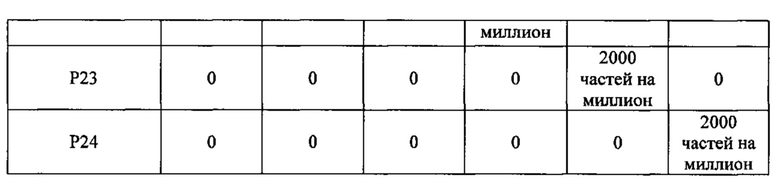
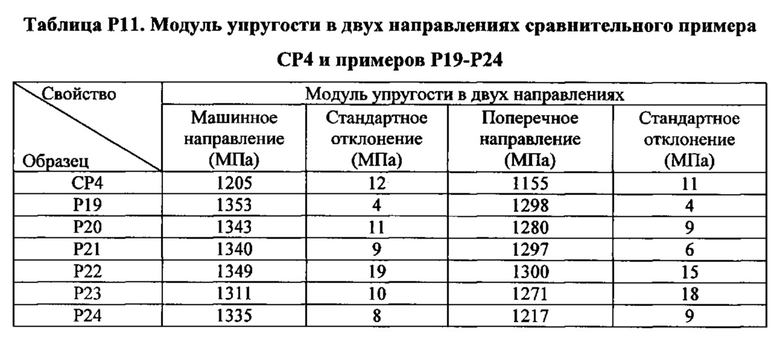
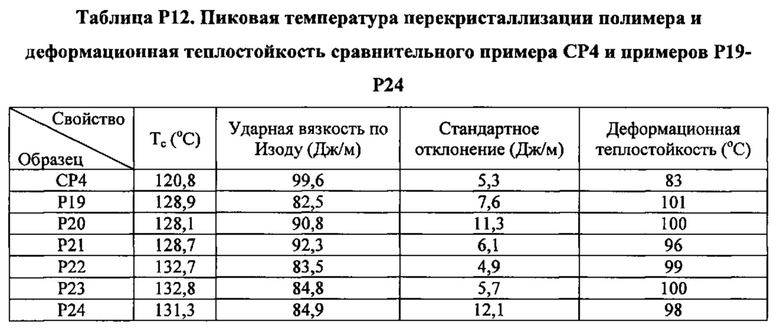
Переработка полиэтилена с зародышеобразователем кристаллизации литьем под давлением
В следующих примерах для литья под давлением полиэтиленовые смолы получали, как описано выше в отношении предыдущих примеров для литья под давлением. Пластинки и полосы получали посредством литья под давлением на машине для литья под давлением Arburg 40 ton со шнеком диаметром 25,4 мм. Температура цилиндра машины для литья под давлением составляла от 190 до 230°C в зависимости от индекса расплава смолы, а температуру формы контролировали на 25°C.
Если иное не указано, скорость впрыска для пластинок составляла 15 см3/с, а их размеры представляли собой длину приблизительно 60 мм, ширину 60 мм и толщину 2 мм. Эти пластинки использовали для измерения усадки в двух направлениях, температуры перекристаллизации и жесткости в двух направлениях.
Если иное не указано, скорость впрыска для полос составляла 40 см3/с, а их размеры представляли собой длину приблизительно 127 мм, ширину 12,7 мм и толщину 3,3 мм. Эти полосы использовали для измерения секущего модуля при 1% деформации и HDT.
Испытание полиэтилена с зародышеобразователем кристаллизации Усадку измеряли на пластинках как в машинном направлении (MD), так и в поперечном направлении (TD) после 48 часов состаривания при условиях окружающей среды согласно ASTM D955. Усадку в процентах для каждого направления рассчитывали при помощи следующего уравнения:
Исследование свойств при изгибе (указанных как модуль упругости в двух направлениях) проводили на вышеуказанных пластинках, используя установку MTS Q-Test-5 с шириной захвата 32 мм, фиксированной скоростью деформации 8,53 мм/минуту и номинальной шириной образца 50,8 мм. Образцы получали вырезанием квадратных секций (приблизительно 50 мм × 50 мм) из центра пластинок для получения образцов с одинаковыми параметрами во всех направлениях. Кроме испытания образцов поперек машинного направления/направления потока, как принято (обозначенного как "поперечное направление" в таблице результатов), образцы также испытывали путем сгибания поперек направления, перпендикулярного направлению потока для измерения жесткости в этом направлении (обозначенного как "машинное направление" в таблице результатов) для анализа жесткости в двух направлениях пластинок.
Пиковую температуру перекристаллизации полимера (Tc) для композиций на основе термопластичного полимера измеряли при помощи дифференциального сканирующего калориметра (дифференциального сканирующего калориметра Mettler-Toledo DSC822). В частности, образец отбирали из заданной части и нагревали со скоростью 20°C/минуту от температуры 60°C до 220°C, выдерживали при 220°C в течение двух минут и охлаждали со скоростью приблизительно 10°C/минуту до температуры 60°C. Температура, при которой возникало пиковое преобразование кристаллов полимера (что соответствует пиковой температуре перекристаллизации полимера) записывали для каждого образца.
Примеры Q1-Q12
Эти примеры показывают некоторые физические свойства, проявляемые полиэтиленовым полимером высокой плотности, в который вводили зародышеобразователь кристаллизации в виде смеси ЕХ76 и поглотителя кислот, в частности стеарата цинка (ZnSt) или синтетического дигидроталькитного соединения (DHT-4A). Полимерные композиции получали примешиванием (как описано выше) соединения примера получения ЕХ76 и различных поглотителей кислот в коммерчески доступный полиэтилен высокой плотности (Nova Sclair® 19G), характеризующийся плотностью приблизительно 0,960 г/см3 и индексом текучести расплава приблизительно 1,2 дг/минуту. Смолу сначала измельчали, перемешивали с добавками, а затем компаундировали и экструдировали с образованием гранул. Образованные гранулы из полимерной композиции затем отливали под давлением в тестовые пластинки и полосы.
Информация о составах для примеров Q1-Q12 и сравнительного примера CQ1 указана в таблице Q1. Пиковая температура перекристаллизации полимера (Tc), модуль упругости в двух направлениях (измеренные на пластинках) и секущий модуль при 1% деформации и деформационная теплостойкость (измеренные на полосах) указаны в таблицах Q2 и Q3 ниже.


ЕХ76 без ZnSt или DHT-4A влияет в некоторой мере на ориентацию роста кристаллов в машинном направлении (MD) о чем свидетельствует снижение усадки в MD. Когда DHT-4A использовали в качестве поглотителя кислот при отношении 3:1 ЕХ76 к DHT-4A, более сильная ориентация в MD (меньшая усадка в MD, чем в TD) появлялась при концентрациях смеси 1500 частей на миллион. Когда ZnSt использовали в качестве поглотителя кислот, сильная ориентация в MD была очевидна при концентрациях смеси всего лишь 500 частей на миллион. Это очевидно из более низкой усадки в MD, большей жесткости в MD и снижения жесткости в TD.
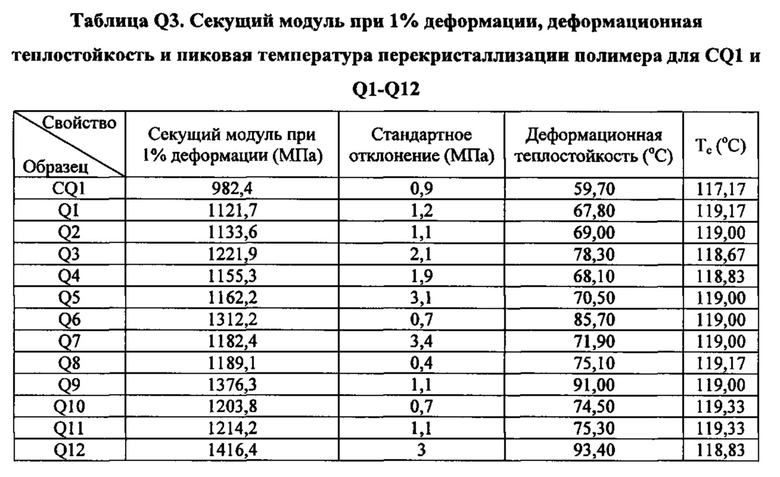
Как можно видеть из таблицы Q3, смеси ЕХ76 с поглотителем кислот имеют такой же эффект на Tc, как ЕХ76 сама по себе. Измерения жесткости и HDT на гибких полосах подтверждали, что использование ЕХ76 вместе с ZnSt или DHT-4A значительно улучшает работу ЕХ76 по сравнению с ЕХ76 самой по себе. Когда использовали смесь зародышеобразователя и поглотителя кислот, более низкие концентрации зародышеобразователя (ЕХ76) способны придавать аналогичные или лучшие свойства, чем более высокие концентрации ЕХ76 самой по себе.
Примеры R1-R9
Данные примеры показывают некоторые физические свойства, проявляемые полиэтиленовым полимером высокой плотности, в который вводили смеси ЕХ76 и ZnSt в различных соотношениях. Полимерные композиции получали примешиванием (как описано выше) соединения примера получения ЕХ76 и ZnSt в коммерчески доступный полиэтилен полимер высокой плотности (Nova Sclair® 19G), характеризующийся плотностью приблизительно 0,960 г/см3 и индексом текучести расплава приблизительно 1,2 дг/минуту. Смолу сначала измельчали, перемешивали с добавками, а затем компаундировали и экструдировали с образованием гранул. Образованные гранулы из полимерной композиции затем отливали под давлением в тестовые пластинки и полосы.
Информация о составах для примеров R1-R9 и сравнительного примера CR1 указана в таблице R1. Пиковая температура перекристаллизации полимера (Tc), модуль упругости в двух направлениях (измеренные на пластинках) и секущий модуль при 1% деформации и деформационная теплостойкость (измеренные на полосах) указаны в таблицах R2 и R3 ниже.
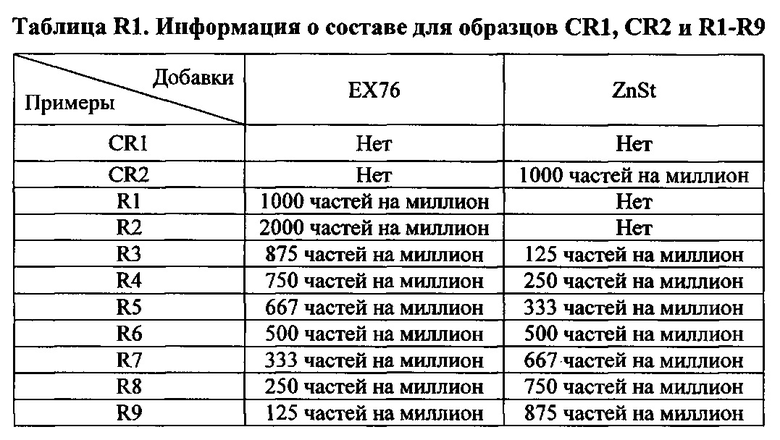
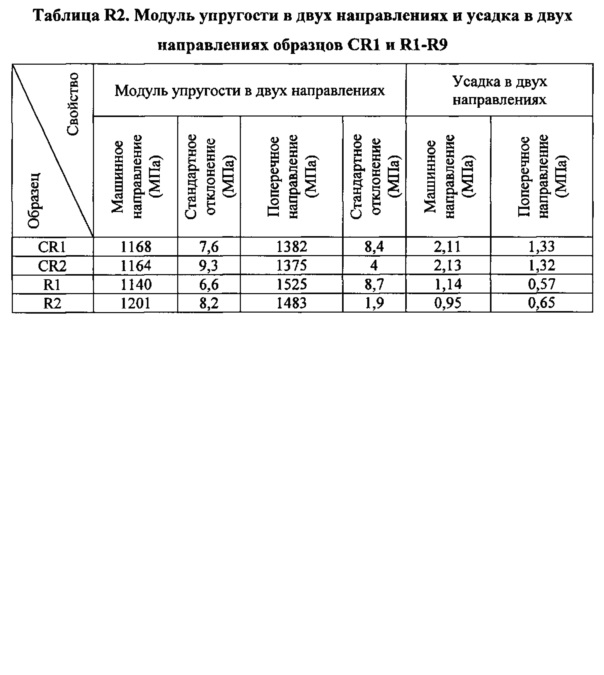
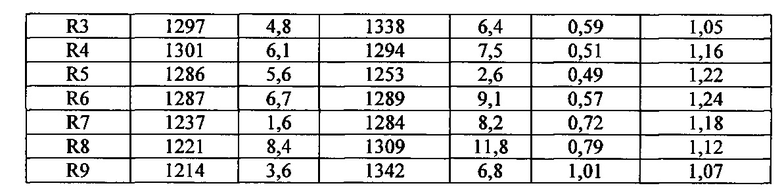
Данные для образца CR2 показывали, что введение ZnSt самого по себе не имело значительного влияния на жесткость в двух направлениях или усадку данной смолы. Это свидетельствует о том, что ZnSt не образует зародыши кристаллизации в смоле. Образцы R1 и R2 показали, что ЕХ76 без ZnSt придает ориентацию росту кристаллов в MD (снижая усадку в MD по сравнению с CR1 и CR2). Когда ZnSt использовали вместе с ЕХ76, наблюдали более сильную ориентацию в MD (очень низкую усадку в MD). Это верно даже для смеси с отношением 1:4, где ЕХ76 присутствует только в количестве 125 частей на миллион в смоле.
Когда ЕХ76 и ZnSt использовали вместе, наблюдали очень высокую жесткость в MD и снижение жесткости в TD, что указывает на очень сильную ориентацию в MD. Жесткость в MD, придаваемая всеми смесями, выше, чем жесткость в MD ЕХ76 самой по себе как при 1000 частях на миллион, так и 2000 частях на миллион. Это неожиданно, поскольку смолы, которые компаундировали со смесями, содержали меньше ЕХ76. Наибольшую жесткость в MD получали со смесями ЕХ76 и ZnSt с отношениями в диапазоне 4:1, 3:1, 2:1 и 1:1. Но даже при отношениях 1:3 и 1:4 (которые соответствуют концентрациям ЕХ76 в 250 частей на миллион и 125 частей на миллион, с ZnSt в количестве 750 частей на миллион и 875 частей на миллион, соответственно) жестокость в MD аналогична или незначительно выше, чем с ЕХ76 самой по себе при 2000 частях на миллион.
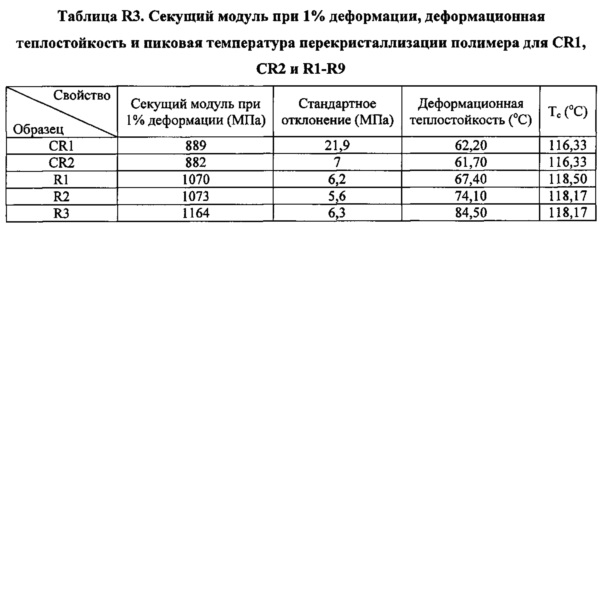

Как можно увидеть из данных в таблице R3, ЕХ76 повышала Tc смолы. Различные смеси ЕХ76 с ZnSt не повышали Tc свыше той, которую наблюдали с ЕХ76 самой по себе. Конечно, Tc незначительно снижалась, когда количество ЕХ76 повышалось.
Измерения жесткости и HDT на гибких полосах подтверждали синергический эффект ЕХ76 и ZnSt. Смеси с отношениями 4:1, 3:1, 2:1, 1:1 и 1:2 (EX76:ZnSt) придавали намного более высокую жесткость и HDT, чем ЕХ76 сама по себе. А смеси ЕХ76 с ZnSt при отношениях 1:3 и 1:4 придавали значения жесткости и HDT, аналогичные таким для ЕХ76 самой по себе. Это означает, что можно использовать смолу, содержащую смесь ЕХ76 в количестве 125 частей на миллион и ZnSt в количестве 875 частей на миллион, и все еще получать характеристики, аналогичные таким для смолы, содержащей ЕХ76 саму по себе в количестве 2000 частей на миллион.
Примеры S1-S5
Данные примеры показывают некоторые физические свойства, проявляемые полиэтиленовым полимером высокой плотности, в который вводили смеси ЕХ76 и ZnSt в различных соотношениях. Полимерные композиции получали примешиванием (как описано выше) соединения примера получения ЕХ76 и различных поглотителей кислот в коммерчески доступный полиэтилен высокой плотности (Dow HDPE DMDA-8007 NT7), характеризующийся плотностью приблизительно 0,967 г/см3 и индексом текучести расплава приблизительно 8,3 дг/минуту. Смолу сначала измельчали, перемешивали с добавками, а затем компаундировали и экструдировали с образованием гранул. Образованные гранулы из полимерной композиции затем отливали под давлением в тестовые пластинки и полосы.
Информация о составах для примеров S1-S5 и сравнительных примеров CS1 и CS2 указана в таблице S1. Пиковая температура перекристаллизации полимера (Tc), модуль упругости в двух направлениях (измеренные на пластинках) и секущий модуль при 1% деформации и деформационная теплостойкость (измеренные на полосах) указаны в таблицах S2 и S3 ниже.
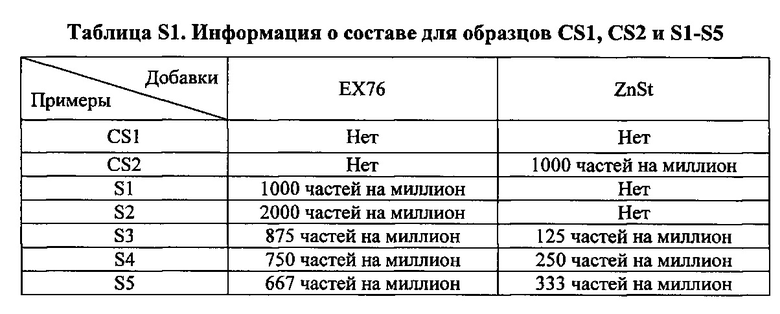
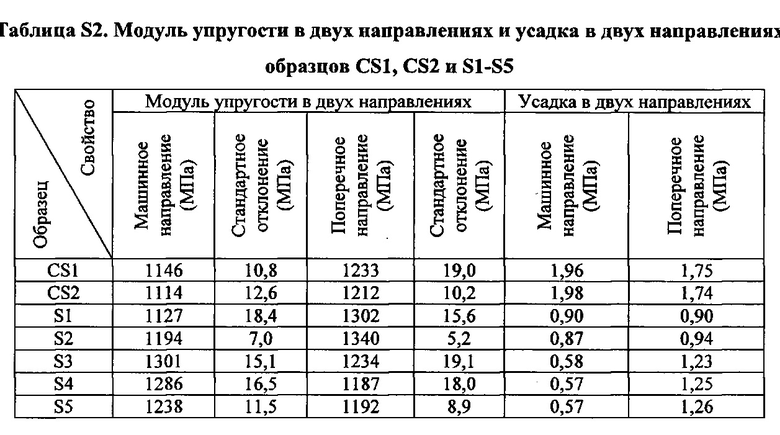
Данные для образца CS2 показывали, что введение ZnSt самого по себе не имело значительного влияния на жесткость в двух направлениях или усадку данной смолы. Эти наблюдения подтверждают, что ZnSt не образует зародыши кристаллизации в смоле. Данные для образцов S1 и S2 показали, что ЕХ76 сама по себе придает ориентацию росту кристаллов в MD (снижая усадку в MD по сравнению с CS1 и CS2). Когда ZnSt использовали вместе с ЕХ76, наблюдали намного более сильную ориентацию в MD (очень низкую усадку в MD). Это справедливо для всех протестированных отношений компонентов в смеси.
Когда использовали смесь ЕХ76 и ZnSt, наблюдали очень высокую жесткость в MD и снижение жесткости в TD, что указывает на очень сильную ориентацию в MD. Жесткость в MD, придаваемая любой из смесей при общей концентрации 1000 частей на миллион, выше, чем такая придаваемая ЕХ76 самой по себе, даже при концентрации 2000 частей на миллион. Эти результаты соответствуют наблюдаемым с полиэтиленовыми смолами с более низким индексом текучести расплава.
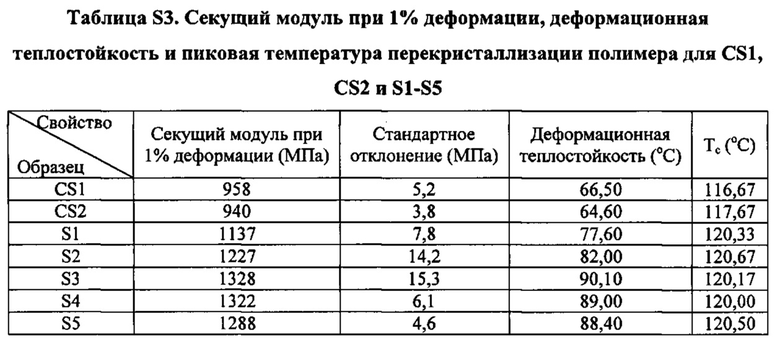
Как можно увидеть из данных в таблице S3, ЕХ76 повышала Tc смолы. Различные смеси ЕХ76 с ZnSt не повышали Tc свыше той, которую наблюдали с ЕХ76 самой по себе. В действительности, Tc незначительно снижалась, когда количество ЕХ76 повышалось.
Измерения жесткости и HDT на гибких полосах подтверждали синергический эффект ЕХ76 и ZnSt. Смеси с отношениями 3:1, 2:1 и 1:1 (EX76:ZnSt) придавали намного более высокие значения жесткости и HDT, чем ЕХ76 сама по себе.
Примеры Т1-Т5
Данные примеры показывают некоторые физические свойства, проявляемые полиэтиленовым полимером высокой плотности, в который вводили смеси ЕХ76 и ZnSt в различных соотношениях. Полимерные композиции получали примешиванием (как описано выше) соединения примера получения ЕХ76 и различных поглотителей кислот в коммерчески доступный полиэтилен высокой плотности (Dowlex™ IP40), характеризующийся плотностью приблизительно 0,952 г/см3 и индексом текучести расплава приблизительно 40 дг/минуту. Смолу сначала измельчали, перемешивали с добавками, а затем компаундировали и экструдировали с образованием гранул. Образованные гранулы из полимерной композиции затем отливали под давлением в тестовые пластинки и полосы.
Информация о составах для примеров Т1-Т5 и сравнительных примеров CT1 и CT2 указана в таблице T1. Пиковая температура перекристаллизации полимера (Tc), модуль упругости в двух направлениях (измеренные на пластинках) и секущий модуль при 1% деформации и деформационная теплостойкость (измеренные на полосах) указаны в таблицах T2 и T3 ниже.
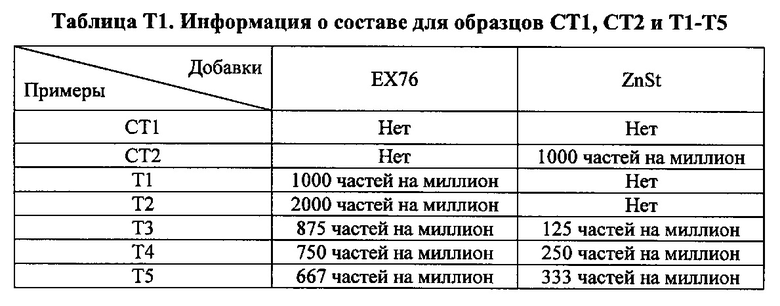
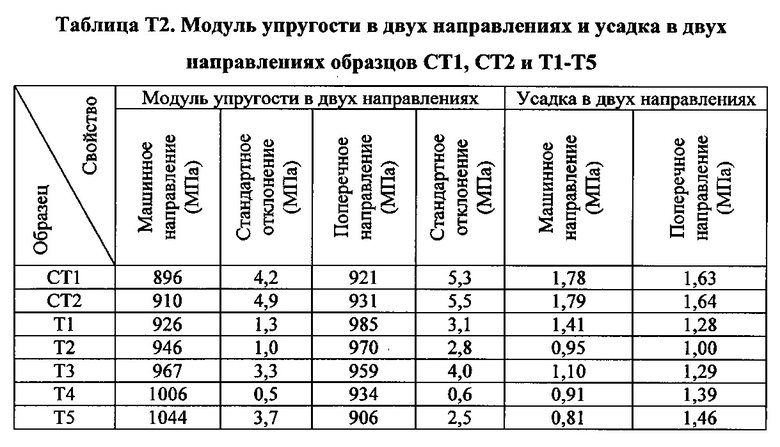
Данные для образца CT2 показывали, что введение ZnSt самого по себе не имело значительного влияния на жесткость в двух направлениях или усадку данной смолы. Это свидетельствует о том, что ZnSt не образует зародыши кристаллизации в смоле. Образцы T1 и T2 показали, что ЕХ76 сама по себе придает ориентацию росту кристаллов в MD (снижая усадку в MD по сравнению с CT1 и CT2). Когда ZnSt и ЕХ76 использовали вместе, присутствовала намного более сильная ориентация в MD (очень низкая усадка в MD). Это справедливо для всех различных протестированных отношений компонентов в смеси.
Когда ЕХ76 и ZnSt использовали вместе, наблюдали очень высокую жесткость в MD и снижение жесткости в TD, что указывает на очень сильную ориентацию в MD. Жесткость в MD, придаваемая всеми смесями, выше, чем жесткость в MD для ЕХ76 самой по себе при концентрациях 1000 частей на миллион и 2000 частей на миллион. Эти результаты соответствуют наблюдаемым с другими HDPE смолами.
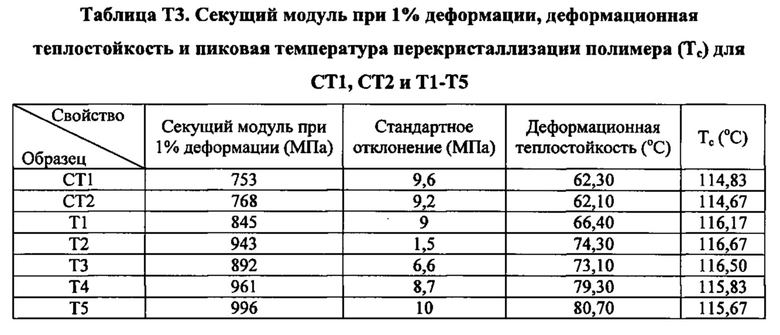
Как можно увидеть из данных в таблице ТЗ, ЕХ76 повышала Tc смолы. Различные смеси ЕХ76 с ZnSt не повышали Tc свыше той, которую наблюдали с ЕХ76 самой по себе.
Измерение жесткости и HDT на гибких полосах подтверждало синергический эффект ЕХ76 и ZnSt. Смеси с отношениями 3:1, 2:1 и 1:1 придавали намного более высокие значения жесткости и HDT, чем ЕХ76 сама по себе.
Все ссылки, включая публикации, патентные заявки и патенты, цитируемые в настоящем документе, таким образом включены ссылкой в том же объеме, как если бы каждая ссылка была отдельно и конкретно указана как включенная ссылкой и изложена во всей своей полноте в настоящем документе.
Использование выражений в форме единственного числа в контексте описания объекта настоящей заявки (в частности в контексте следующей формулы изобретения) следует толковать как охватывающие формы и единственного, и множественного числа, если иное не указано в настоящем документе или явно не противоречит контексту. Выражения "содержащий", "характеризующийся", "включающий" и "содержащий в себе" следует толковать как открытые выражения (т.е. означающие "включающий, помимо прочего"), если иное не указано. Перечисление диапазонов значений в настоящем документе предназначено для использования только в качестве краткого способа для обозначения отдельной ссылки на каждое отдельное значение, попадающее в пределы диапазона, если иное не указано в настоящем документе, и каждое отдельное значение включено в описание, как если бы его отдельно указывали в настоящем документе. Все способы, описанные в настоящем документе, можно осуществлять в любом подходящем порядке, если иное не указано в настоящем документе или иным образом явно не противоречит контексту. Использование любого или всех примеров или типичного выражения (например, "такой как"), представленных в настоящем документе, предназначено только для лучшего освещения объекта данной заявки, а не накладывает ограничения на объем объекта, если иное не заявлено. Никакие выражения в описании не следует рассматривать как указывающие какой-либо незаявленный элемент как необходимый для осуществления на практике объекта, описанного в настоящем документе.
Предпочтительные варианты осуществления объекта настоящей заявки описаны в настоящем документе, включая наилучший способ, известный авторам настоящего изобретения, для выполнения заявленного объекта. Вариации этих предпочтительных вариантов осуществления станут очевидными специалистам в данной области техники при прочтении вышеуказанного описания. Авторы настоящего изобретения предполагают, что специалисты в данной области используют такие варианты при необходимости, и авторы настоящего изобретения подразумевают, что объект, описанный в настоящем документе, можно осуществлять на практике отличным образом от конкретно описанного в настоящем документе. Следовательно, настоящее раскрытие включает все модификации и эквиваленты объекта, изложенного в формуле изобретения, приложенной к нему, в соответствии с действующим законодательством. Кроме того, любая комбинация вышеописанных элементов во всех их возможных вариациях охвачена настоящим раскрытием, если иное не указано в настоящем документе или иным образом явно не противоречит контексту.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛИЭТИЛЕНОВЫЕ ИЗДЕЛИЯ | 2014 |
|
RU2643145C2 |
| ПОЛИЭТИЛЕНОВЫЕ ПОЛИМЕРНЫЕ КОМПОЗИЦИИ И ИЗГОТОВЛЕННЫЕ ИЗ НИХ ИЗДЕЛИЯ | 2022 |
|
RU2835555C1 |
| ТЕРМОПЛАСТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2801939C2 |
| ПОЛИЭТИЛЕНОВЫЕ ПОЛИМЕРНЫЕ КОМПОЗИЦИИ И ИЗГОТОВЛЕННЫЕ ИЗ НИХ ИЗДЕЛИЯ | 2022 |
|
RU2835554C1 |
| ПОЛИМЕР НА ПРОПИЛЕНОВОЙ ОСНОВЕ, ИЗДЕЛИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2009 |
|
RU2527036C2 |
| СТАБИЛИЗИРОВАННЫЙ ПОЛИЭТИЛЕН ВЫСОКОЙ ПЛОТНОСТИ С УЛУЧШЕННОЙ УСТОЙЧИВОСТЬЮ К ПОВРЕЖДЕНИЯМ И СТАБИЛИЗИРУЮЩЕЙ СИСТЕМОЙ | 2013 |
|
RU2615747C2 |
| ПОЛИОЛЕФИНОВЫЕ КОМПОЗИЦИИ, УСТОЙЧИВЫЕ К ДИНАМИЧЕСКИМ НАГРУЗКАМ | 2004 |
|
RU2371457C2 |
| ПОЛИЭТИЛЕН ВЫСОКОЙ ПЛОТНОСТИ | 2006 |
|
RU2419640C2 |
| Электретные полотна с добавками, способствующими накоплению заряда | 2016 |
|
RU2673299C1 |
| ПОЛИЭТИЛЕНОВЫЕ КОМПОЗИЦИИ С УЛУЧШЕННЫМИ РАЗРЫВНЫМИ ХАРАКТЕРИСТИКАМИ | 2004 |
|
RU2349613C2 |
Изобретение относится к композициям на основе термопластичных полимеров, подходящих для получения готовых изделий, например формованных изделий, Композиция содержит полиолефиновый полимер и зародышеобразователь, содержащий соединение, соответствующее структуре формулы (I). Изделия, полученные из композиций на основе термопластичных полимеров настоящего изобретения, характеризуются желательной комбинацией более высокой пиковой температуры перекристаллизации полимера и улучшенных таких физических свойств, как мутность, прочность на разрыв (или абсолютная прочность на разрыв, или равновесие между прочностью на разрыв в машинном и поперечном направлениях), жесткость и барьерные свойства. 19 з.п. ф-лы, 65 табл.
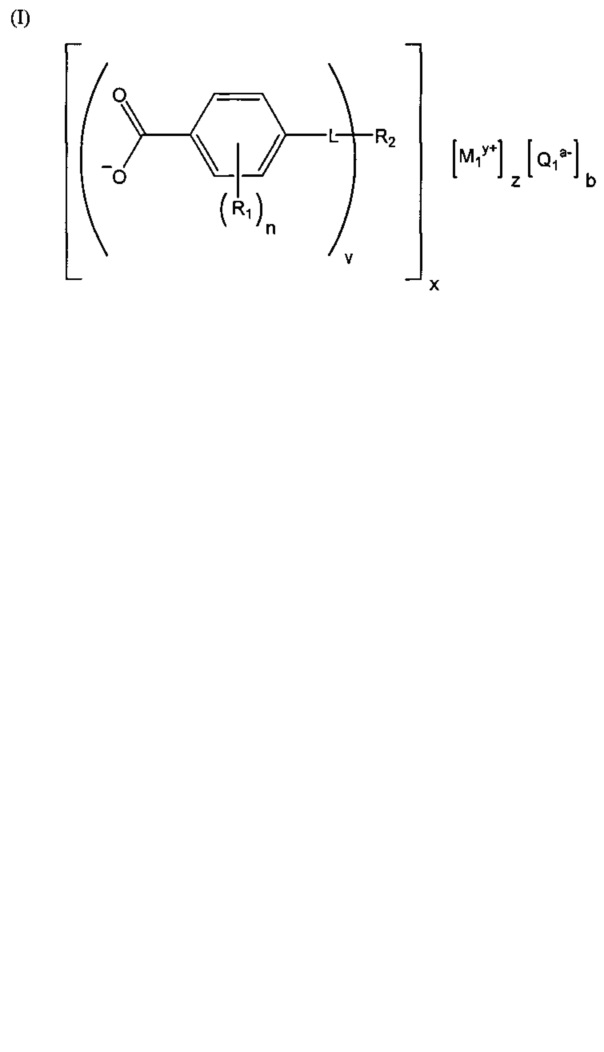 .
.
1. Композиция на основе термопластичного полимера, содержащая:
(a) полиолефиновый полимер и
(b) зародышеобразователь, причем зародышеобразователь содержит соединение, соответствующее структуре формулы (I)
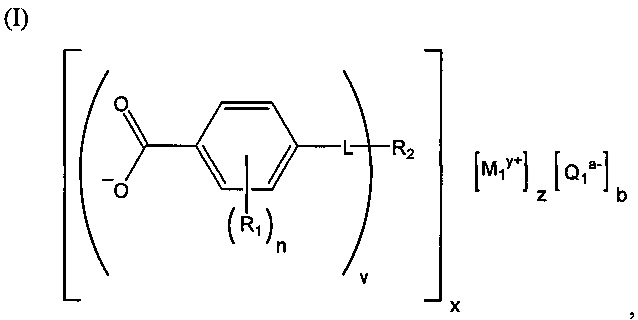
где R1 выбрана из группы, состоящей из гидроксигруппы, галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп; n равняется нулю или целому положительному числу от 1 до 4; L представляет собой связующую группу, содержащую два или более атома и по меньшей мере одну двойную связь между двумя атомами в связующей группе; v представляет собой целое положительное число от 1 до 3; R2: (i) выбрана из группы, состоящей из алкильных групп, замещенных алкильных групп, циклоалкильных групп, замещенных циклоалкильных групп, арильных групп, замещенных арильных групп, гетероарильных групп и замещенных гетероарильных групп, если L представляет собой двухвалентную связующую группу, a v равняется 1, (ii) выбрана из группы, состоящей из алкандиильных групп, замещенных алкандиильных групп, циклоалкандиильных групп, замещенных циклоалкандиильных групп, арендиильных групп, замещенных арендиильных групп, гетероарендиильных групп и замещенных гетероарендиильных групп, если L представляет собой трехвалентную связующую группу, a v равняется 1, (iii) выбрана из группы, состоящей из алкандиильных групп, замещенных алкандиильных групп, циклоалкандиильных групп, замещенных циклоалкандиильных групп, арендиильных групп, замещенных арендиильных групп, гетероарендиильных групп и замещенных гетероарендиильных групп, если L представляет собой двухвалентную связующую группу, a v равняется 2, и (iv) выбрана из группы, состоящей из алкантриильных групп, замещенных алкантриильных групп, циклоалкантриильных групп, замещенных циклоалкантриильных групп, арентриильных групп, замещенных арентриильных групп, гетероарентриильных групп и замещенных гетероарентриильных групп, если L представляет собой двухвалентную связующую группу, a v равняется 3; x представляет собой целое положительное число; каждый M1 представляет собой катион металла; y представляет собой валентность катиона; z представляет собой целое положительное число; b равняется нулю или целому положительному числу; если b представляет собой целое положительное число, каждый Q1 представляет собой отрицательно заряженный противоион, и а представляет собой валентность отрицательно заряженного противоиона; а значения v, x, y, z, а и b удовлетворяют уравнению (vx)+(ab)=yz; при этом, если L представляет собой двухвалентную связующую группу, v равняется 1, a R2 представляет собой циклоалкильную группу или замещенную циклоалкильную группу, циклическая часть циклоалкильной группы или замещенной циклоалкильной группы содержит не более двух кольцевых структур, сконденсированных вместе.
2. Композиция на основе термопластичного полимера по п. 1, в которой L выбрана из группы, состоящей из фрагментов, соответствующих структуре одной из формул (LA)-(LF) ниже
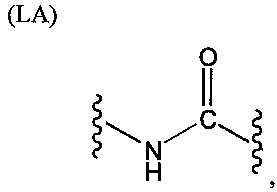
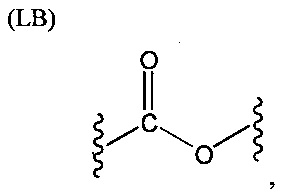

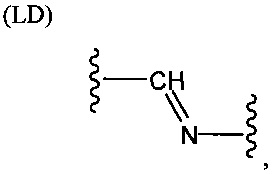
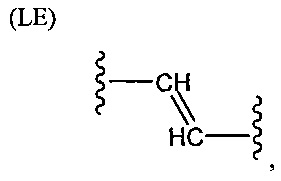
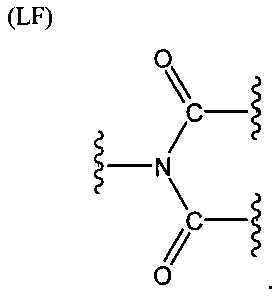
3. Композиция на основе термопластичного полимера по п. 1 или 2, в которой L представляет собой двухвалентную связующую группу, v равняется 1, a R2 выбрана из группы, состоящей из фрагментов, соответствующих структуре одной из формул (АА)-(AG) ниже
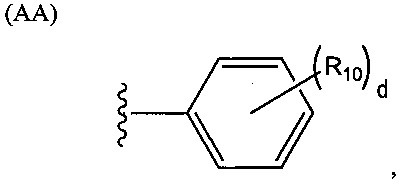
где d равняется нулю или целому положительному числу от 1 до 5, а каждая R10 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп;

где h равняется нулю или целому положительному числу от 1 до 10, а каждая R13 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп;
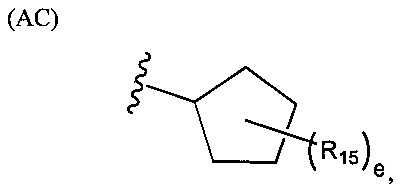
где е равняется нулю или целому положительному числу от 1 до 8, а каждая R15 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп;

где g равняется нулю или целому положительному числу от 1 до 6, а каждая R20 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп;

где j равняется нулю или целому положительному числу от 1 до 4, а каждая R25 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп;
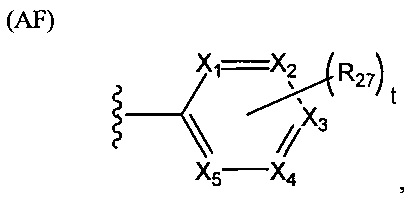
где X1, X2, X3, X4 и X5 независимо выбраны из группы, состоящей из атома углерода и атома азота, при условии, что по меньшей мере один и не более трех из X1, X2, X3, X4 и X5 представляют собой атомы азота; t равняется нулю или целому положительному числу, равному 5-Х, где X представляет собой число атомов азота; а каждая R27 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп;

где X6 выбран из группы, состоящей из атома углерода, атома кислорода, атома серы и вторичной аминогруппы, X7, X8 и X9 независимо выбраны из группы, состоящей из атома углерода и атома азота, по меньшей мере один и не более трех из X6, X7, X8 и X9 представляют собой атомы, не являющиеся углеродом; u равняется нулю или целому положительному числу, равному 4-Y, где Y представляет собой число атомов, не являющихся углеродом, в кольцевой структуре; а каждая R29 независимо выбрана из группы, состоящей из галогенов, цианогрупп, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп.
4. Композиция на основе термопластичного полимера по п. 1 или 2, в которой L представляет собой трехвалентную связующую группу, v равняется 1, a R2 выбрана из группы, состоящей из фрагментов, соответствующих структуре одной из формул (АН)-(AJ) ниже

где k равняется нулю или целому положительному числу от 1 до 8, а каждая R30 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп;

где m равняется нулю или целому положительному числу от 1 до 4, а каждая R35 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп;
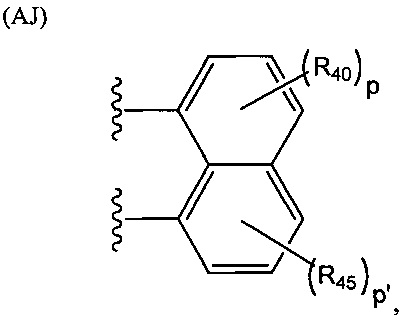
где p равняется нулю или целому положительному числу от 1 до 3, p' равняется нулю или целому положительному числу от 1 до 3, а каждая из R40 и R45 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп.
5. Композиция на основе термопластичного полимера по п. 1 или 2, в которой L представляет собой двухвалентную связующую группу, v равняется 2, a R2 выбрана из группы, состоящей из фрагментов, соответствующих структуре формулы (ВА) ниже
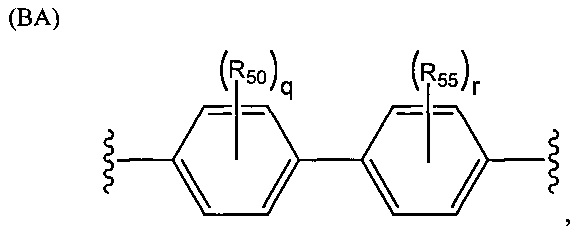
где q равняется нулю или целому положительному числу от 1 до 4, r равняется нулю или целому положительному числу от 1 до 4, а каждая из R50 и R55 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп.
6. Композиция на основе термопластичного полимера по п. 1 или 2, в которой L представляет собой двухвалентную связующую группу, v равняется 3, a R2 выбрана из группы, состоящей из фрагментов, соответствующих структуре формулы (СА) ниже

где s равняется нулю или целому положительному числу от 1 до 3, а каждая R60 независимо выбрана из группы, состоящей из галогенов, алкильных групп, замещенных алкильных групп, алкоксигрупп, замещенных алкоксигрупп, арильных групп и замещенных арильных групп.
7. Композиция на основе термопластичного полимера по п. 1 или 2, в которой M1 выбран из группы, состоящей из катионов щелочных металлов, катионов щелочноземельных металлов и катионов переходных металлов.
8. Композиция на основе термопластичного полимера по п. 1 или 2, в которой M1 представляет собой катион элемента, выбранного из группы, состоящей из лития, натрия, магния, алюминия, калия, кальция и цинка.
9. Композиция на основе термопластичного полимера по п. 8, в которой M1 представляет собой катион лития.
10. Композиция на основе термопластичного полимера по п. 9, в которой зародышеобразователь содержит соединение, соответствующее структуре одной из формул (IA)-(IH) ниже
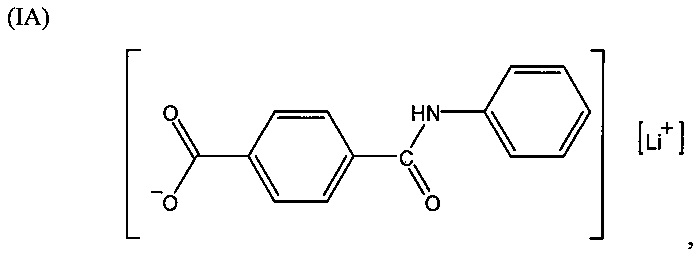
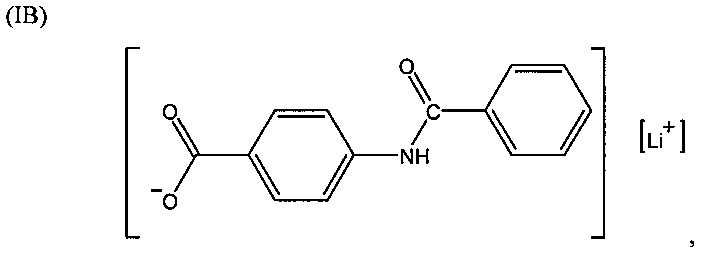
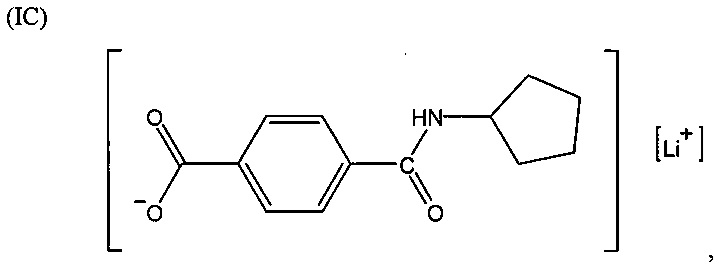
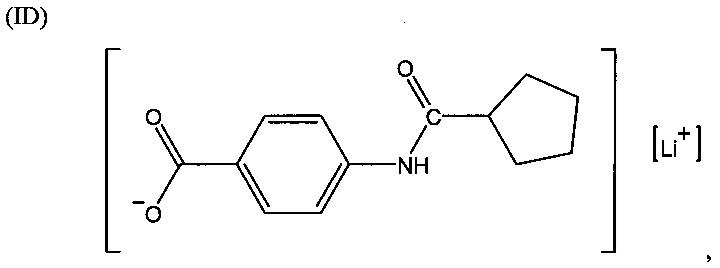

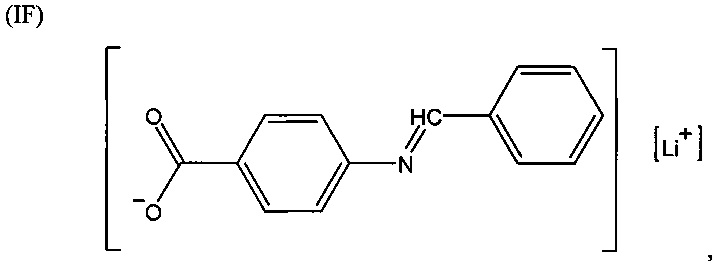

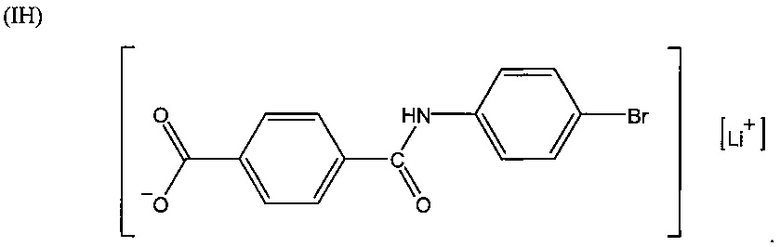
11. Композиция на основе термопластичного полимера по п. 1 или 2, в которой термопластичный полимер выбран из группы, состоящей из полипропиленов, полиэтиленов и их смесей.
12. Композиция на основе термопластичного полимера по п. 11, в которой термопластичный полимер представляет собой полиэтилен.
13. Композиция на основе термопластичного полимера по п. 12, в которой термопластичный полимер представляет собой линейный полиэтилен низкой плотности.
14. Композиция на основе термопластичного полимера по п. 12, в которой термопластичный полимер представляет собой полиэтилен высокой плотности.
15. Композиция на основе термопластичного полимера по п. 1 или 2, в которой зародышеобразователь находится в композиции на основе термопластичного полимера в количестве от приблизительно 100 до приблизительно 5000 частей на миллион (ppm) на основании общей массы композиции на основе термопластичного полимера.
16. Композиция на основе термопластичного полимера по п. 15, в которой зародышеобразователь находится в композиции на основе термопластичного полимера в количестве от приблизительно 250 до приблизительно 3000 частей на миллион (ppm) на основании общей массы композиции на основе термопластичного полимера.
17. Композиция на основе термопластичного полимера по п. 1, причем полимерная композиция дополнительно содержит поглотитель кислот, выбранный из группы, состоящей из синтетических гидроталькитных соединений и металлических солей С12-С22-жирных кислот.
18. Композиция на основе термопластичного полимера по п. 17, в которой поглотитель кислот выбран из группы, состоящей из цинковых, калиевых и лантановых солей стеариновой кислоты.
19. Композиция на основе термопластичного полимера по п. 17, в которой зародышеобразователь и поглотитель кислот находятся в композиции в соотношении от приблизительно 4:1 до приблизительно 1:4 на основании массы зародышеобразователя и поглотителя кислот в композиции.
20. Композиция на основе термопластичного полимера по п. 18, в которой зародышеобразователь и поглотитель кислот находятся в композиции в соотношении от приблизительно 4:1 до приблизительно 1:4 на основании массы зародышеобразователя и поглотителя кислот в композиции.
| Клапанный регулятор для паровозов | 1919 |
|
SU103A1 |
| ЧЕТЫРЕХТАКТНЫЙ РОТОРНО-ПОРШНЕВОЙ ДВИГАТЕЛЬ ВНУТРЕННЕГО СГОРАНИЯ | 2008 |
|
RU2392458C2 |
| EP 0557721 A2, 01.09.1993 | |||
| ДИАЦЕТАЛЕВАЯ КОМПОЗИЦИЯ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ, ЗАРОДЫШЕОБРАЗОВАТЕЛЬ ДЛЯ ПОЛИОЛЕФИНОВ, СОДЕРЖАЩИЙ УКАЗАННУЮ КОМПОЗИЦИЮ, КОМПОЗИЦИИ ПОЛИОЛЕФИНОВОЙ СМОЛЫ И ФОРМОВАННЫЕ ИЗДЕЛИЯ | 1998 |
|
RU2177479C2 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| WO 00/17265 A1, 30.03.2000 | |||
| QIAO, WEN-QIANG ET AL: "Synthesis of a novel nucleating agent with thermotropic crystalline liquid behaviorand its influence on the crystallizationof polyethylene - (I) | |||
| Synthesis and characterization of a novel nucleating agent with thermotropic liquid crystalline behavior", https://books.google.com/books/about/Nucleating_Agents.html?hl=ru&id=K6gWHXVE37YC, с.40, реферат. | |||

