Область техники
Настоящее изобретение относится к области фармацевтики. В частности, настоящее изобретение относится к новому соединению дейтерированного диаминопиримидина и фармацевтическим композициям, содержащим указанное соединение.
Предшествующий уровень техники
Киназа анапластической лимфомы (ALK) представляет собой член суперсемейства инсулиновых тирозинкиназных рецепторов, слитый белок ALK, мутация и сверхэкспрессия которого связаны с разными заболеваниями. ALK впервые была открыта в линиях клеток анапластической крупноклеточной лимфомы (ALCL), причем ген слитого белка, образованный транслокацией t2;5, содержит 3'-концевой участок гена ALK (включая внутриклеточный домен и протеинкиназный домен) и ген ядрышкового фосфопротеина (ген нуклеофозмина, NPM). Открыли более двадцати типов слитых белков ALK, образованных в результате разных хромосомных перестроек. Они участвуют в патогенезе заболеваний, включающих анапластическую крупноклеточную лимфому, диффузную B-крупноклеточную лимфому, воспалительную миофибробластическую опухоль, нейробластому и т.д. Слитый белок EML4-ALK и четыре других слитых белка ALK играют фундаментальную роль в развитии примерно 5% случаев немелкоклеточного рака легкого. Последующие сигнальные пути, включающие слитые белки ALK, включают модуль пролиферации Ras/Raf/MEK/ERK1/2 и путь клеточного выживания JAK/STAT, и данная внутренняя сеть сигнальной трансдукции влияет на клеточную пролиферацию, дифференциацию и апоптоз. Ингибитор киназы ALK можно использовать для лечения рака, аутоиммунных заболеваний и тому подобное. В августе 2011 селективный двойной ингибитор ALK и c-Met кризотиниб компании Pfizer (торговая марка Ксалкори) был одобрен Администрацией США по пищевым продуктам и лекарственным веществам для лечения мелкоклеточного рака легкого на поздних стадиях. Во время клинического применения Кризотиниба возникает устойчивость к лекарственному средству, стимулируя, таким образом, разработку лекарственных средств - ингибиторов киназ ALK второго поколения для лечения немелкоклеточного рака легкого и других заболеваний.
Соединения диаминопиримидинов и их производных представляют собой класс ингибиторов для протеинкиназ, таких как киназа AKL. Ряд диаминопиримидиновых производных, имеющих двойные 2,4-заместители на пиримидиновом кольце, были описаны в WO 2008073687 и WO 2012106540. В этих документах соединение LDK378 (Церитиниб), химическим названием которого является 5-хлор-N2-(2-изопропокси-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин, представляет собой селективный ингибитор киназы ALK, и его можно использовать в лечении рака и заболеваний, связанных с нарушением пролиферации клеток, и других родственных заболеваний. В настоящее время соединение находится на Стадии II клинических испытаний лечения заболеваний, связанных с нарушением пролиферации клеток (таких как немелкоклеточный рак легкого).
Несмотря на то, что направленное ингибирование различных протеинкиназ является благоприятным для лечения разных заболеваний, связанных с киназами, открытие новых соединений, которые специфично ингибируют какую-либо протеинкиназу и обладают хорошей доступностью лекарственного средства, такой как пероральная биодоступность, все еще является актуальным. Кроме того, у некоторых доступных в настоящее время ингибиторов протеинкиназ существуют некоторые побочные эффекты и проблемы устойчивости к лекарственным средствам.
Таким образом, в данной области все еще существует потребность в разработке соединений, обладающих ингибиторной активностью в отношении киназ (например, киназ ALK) или лучшими фармакодинамическими/фармакокинетическими свойствами.
Краткое описание изобретения
Целью настоящего изобретения является предложение типа новых соединений, обладающих ингибирующей активностью в отношении киназ ALK и/или лучшими фармакодинамическими/фармакокинетическими свойствами, и способов их применения.
В первом аспекте настоящего изобретения предложено соединение дейтерированного диаминопиримидина формулы (I) или его кристаллическая форма, фармацевтически приемлемая соль, гидрат или сольват:
 ,
,
где R1a, R1b, R1c, R2a, R2b, R2c, R3, R4, R5, R6, R7, R9, R10, R11, R12, R13a, R13b, R13c, R14a, R14b, R14c, R15a, R15b, R15c, R16, R17a, R17b, R18a, R18b, R19a, R19b, R20a и R20b независимо представляют собой водород, дейтерий или галоген;
R8 представляет собой водород, дейтерий, галоген, циано, недейтерированный C1-C6 алкил или C1-C6 алкокси, однократно или многократно дейтерированный или пердейтерированный C1-C6 алкил или C1-C6 алкокси или однократно или многократно галогенированный или пергалогенированный C1-C6 алкил или C1-C6 алкокси;
при условии, что по меньшей мере один из R1a, R1b, R1c, R2a, R2b, R2c, R3, R4, R5, R6, R7, R9, R10, R11, R12, R13a, R13b, R13c, R14a, R14b, R14c, R15a, R15b, R15c, R16, R17a, R17b, R18a, R18b, R19a, R19b, R20a или R20b является дейтерированным или дейтерием.
В другом предпочтительным воплощении содержание изотопа дейтерий в замещенном дейтерием положении по меньшей мере выше, чем природное содержание изотопа дейтерий (примерно 0,015%), предпочтительно выше чем 30%, более предпочтительно выше чем 50%, более предпочтительно выше чем 75%, более предпочтительно выше чем 95%, более предпочтительно выше чем 99%.
В другом предпочтительном воплощении соединение формулы (I) содержит по меньшей мере один атом дейтерия, более предпочтительно два атома дейтерия, более предпочтительно четыре атома дейтерия, более предпочтительно 6 атомов дейтерия.
В другом предпочтительном воплощении R1a, R1b, R1c, R2a, R2b, R2c и R3 независимо представляют собой водород или дейтерий.
В другом предпочтительном воплощении R12, R13a, R13b, R13c, R14a, R14b и R14c независимо представляют собой водород или дейтерий.
В другом предпочтительном воплощении R15a, R15b и R15c независимо представляют собой водород или дейтерий.
В другом предпочтительном воплощении R16, R17a, R17b, R18a, R18b, R19a, R19b, R20a и R20b независимо представляют собой водород или дейтерий.
В другом предпочтительном воплощении каждый R8 независимо выбран из: галогена, циано, однократно или многократно дейтерированного или пердейтерированного метила или метоксила, или трифторметила.
В другом предпочтительном воплощении R3 представляет собой дейтерий;
В другом предпочтительном воплощении R8 представляет собой хлор;
В другом предпочтительном воплощении R12 представляет собой дейтерий;
В другом предпочтительном воплощении R15a, R15b и R15c представляют собой дейтерий;
В другом предпочтительном воплощении R19a, R19b, R20a и R20b представляют собой дейтерий;
В другом предпочтительном воплощении соединение представляет собой одно из следующих соединений или его фармацевтически приемлемую соль:
5-хлор-N2-(2-изопропокси-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамин; 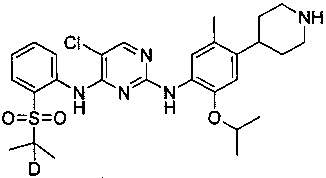
5-хлор-N2-(2-изопропокси-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-((d7-изопропил)сульфонил)фенил)пиримидин-2,4-диамин; 
5-хлор-N2-((2-d-проп-2-илокси)-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин; 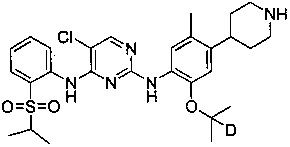
5-хлор-N2-(2-(d7-изопропокси)-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин; 
5-хлор-N2-(2-изопропокси-5-(d3-метил)-4-(пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин; 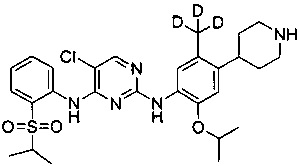
5-хлор-N2-(2-изопропокси-5-метил-4-(4-d-пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин; 
5-хлор-N2-(2-изопропокси-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин;

5-хлор-N2-(2-изопропокси-5-метил-4-(2,2,4,6,6-d5-пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин;

5-хлор-N2-((2-d-проп-2-илокси)-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамин;

5-хлор-N2-(2-(d7-изопропокси)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-((d7-изопропил)сульфонил)фенил)пиримидин-2,4-диамин;

5-хлор-N2-((2-d-проп-2-илокси)-5-метил-4-(4-d-пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамин;

5-хлор-N2-((2-d-проп-2-илокси)-5-(d3-метил)-4-(4-d-пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамин;

5-хлор-N2-(2-(2-d-проп-2-илокси)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамин;

5-хлор-N2-(2-(2-d-проп-2-илокси)-5-метил-4-(2,2,4,6,6-d5-пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамин;
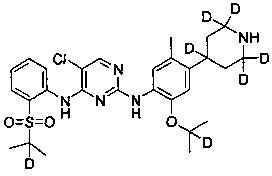
5-хлор-N2-(2-(2-d-проп-2-илокси)-5-(d3-метил)-4-(2,2,4,6,6-d5-пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамин;

5-хлор-N2-(2-(2-d-проп-2-илокси)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин;
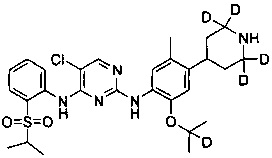
5-хлор-N2-(2-(2-d-проп-2-илокси)-5-(d3-метил)-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин;
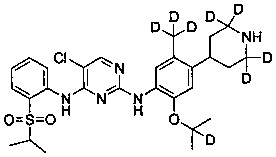
5-хлор-N2-(2-(2-d-проп-2-илокси)-5-(d3-метил)-4-(2,2,4,6,6-d5-пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин;
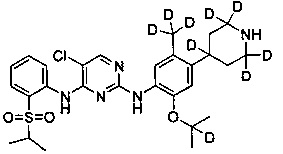
5-хлор-N2-(2-(2-d-проп-2-илокси)-5-(d3-метил)-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамин;

5-хлор-N2-(2-(d7-изопропокси)-5-(d3-метил)-4-(2,2,4,6,6-d5-пиперидин-4-ил)фенил)-N4-(2-((d7-изопропил)сульфонил)фенил)пиримидин-2,4-диамин;

5-хлор-N2-(2-изопропокси-5-метил-4-(2,2,4,6,6-d5-пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамин;
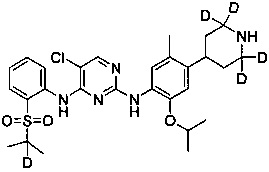
5-хлор-N2-(2-изопропокси-5-метил-4-(2,2,4,6,6-d5-пиперидин-4-ил)фенил)-N4-(4-фтор-2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин;

В другом предпочтительном воплощении соединение представляет собой  ; которое имеет следующие характеристики:
; которое имеет следующие характеристики:
МС (масс-спектр) расчетный: 561; МС обнаруженный: 562 (M+H)+, 584 (M+Na)+.
В другом предпочтительном воплощении соединение представляет собой 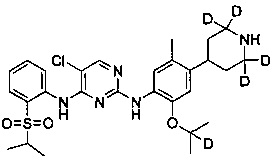 ; которое имеет следующие характеристики:
; которое имеет следующие характеристики:
МС расчетный: 562; МС обнаруженный: 563 (M+H)+, 585 (M+Na)+.
В другом предпочтительном воплощении соединение представляет собой  ; которое имеет следующие характеристики:
; которое имеет следующие характеристики:
МС расчетный: 563; МС обнаруженный: 564 (M+H)+, 586 (M+Na)+.
В другом предпочтительном воплощении соединение представляет собой  ; которое имеет следующие характеристики:
; которое имеет следующие характеристики:
МС расчетный: 566; МС обнаруженный: 567 (M+H)+, 589 (M+Na)+.
В другом предпочтительном воплощении соединение представляет собой  ; которое имеет следующие характеристики:
; которое имеет следующие характеристики:
МС расчетный: 562; МС обнаруженный: 563 (M+H)+, 585 (M+Na)+.
В другом предпочтительном воплощении недейтерированные соединения не включены в соединение.
В другом предпочтительном воплощении недейтерированное соединение представляет собой 5-хлор-N2-(2-изопропокси-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин.
В другом предпочтительном воплощении соединение получают способом, описанным в примерах 1-16.
Во втором аспекте настоящего изобретения предложен способ получения фармацевтической композиции, который включает следующую стадию: смешивание соединений согласно первому аспекту настоящего изобретения или их кристаллической формы, фармацевтически приемлемой соли, гидрата или сольвата с фармацевтически приемлемым носителем с получением фармацевтической композиции.
В третьем аспекте настоящего изобретения предложена фармацевтическая композиция, которая содержит фармацевтически приемлемый носитель и соединение согласно первому аспекту настоящего изобретения или его кристаллическую форму, фармацевтически приемлемую соль, гидрат или сольват.
В другом предпочтительном воплощении фармацевтическая композиция представляет собой инъекцию, капсулу, таблетку, пилюлю, порошок или гранулу.
В другом предпочтительном воплощении фармацевтическая композиция содержит другие терапевтические средства, и другие терапевтические средства представляют собой терапевтические средства для лечения онкологических заболеваний, нарушений пролиферации клеток, сердечнососудистых заболеваний, воспалений, инфекций, аутоиммунных заболеваний, вирусных заболеваний или нарушений метаболизма.
Более предпочтительно, другое терапевтическое средство включает: 5-фторурацил, фолфокс, Avastin™ (авастин, бевацизумаб), бексаротен, бортезомиб, кальцитриол, канертиниб, капецитабин, гемцитабин, карбоплатин, целекоксиб, цетуксимаб, цисплатин, дазатиниб, дигоксин, энзастаурин, эрлотиниб, этопозид, эверолимус, фулвестрант, гефинитиб, генистеин, иматиниб, иринотекан, лапатиниб, леналидомид, летрозол, лейковорин, матузумаб, оксалиплатин, Таксол (паклитаксел), доцетаксел, панитумумаб, пегилированный гранулоцитарный колониестимулирующий фактор (пэгфилграстин), пегилированный альфа-интерферон, пеметрексед, Полифенон®Е, сатраплатин, сиролимус, сунитиниб (сутент, сунитиниб), сулиндаковая кислота (сулиндак), Таксотер (таксотер), темозоломид (темодар, темозомоломид), Торизел, темсиролимус, типифарниб, транстузумаб, вальпроевая кислота, винфлунин, Волоциксимаб, Вориностат, сорафениб, Кризотиниб, Лкотиниб, лапатиниб, Тофацитиниб, PD-0332991 (Палбоциклиб), амбризентан, CD40 и/или CD154-специфичные антитела, слитые белки, ингибиторы NF-kB (ядерного фактора «каппа-би»), нестероидное противовоспалительное лекарственное средство, ингибиторы фактора свертывания крови FXa (например, ривароксабан и т.д.), антитела против ФНО (фактора некроза опухоли), антибиотики, такие как калихимицин, актиномицин, Адриамицин (доксорубицин) и другие терапевтические средства на основе простагландинов или монтелукаст (но не ограничивается ими).
В четвертом аспекте настоящего изобретения предложено применение соединения первого аспекта настоящего изобретения или его кристаллической формы, фармацевтически приемлемой соли, гидрата или сольвата в получении фармацевтических композиций, которые ингибируют протеинкиназы (например, киназы ALK).
В другом предпочтительном воплощении фармацевтическую композицию согласно изобретению можно применять для лечения следующих заболеваний: онкологические заболевания, нарушения пролиферации клеток, воспаления, инфекции, аутоиммунные соединения, трансплантации органов, вирусные заболевания, сердечнососудистые заболевания или нарушения метаболизма.
В другом предпочтительном воплощении онкологические заболевания включают: рак легкого, рак головы и шеи, рак молочной железы, рак предстательной железы, рак пищевода, рак толстой и прямой кишки, рак толстой кишки, рак носоглотки, рак матки, рак поджелудочной железы, лимфому, лейкоз, остеосаркому, меланому, рак почки, рак желудка, рак печени, рак мочевого пузыря, рак щитовидной железы или рак толстой кишки (но не ограничиваются ими).
В другом предпочтительном воплощении иммунные заболевания или воспаление включают: ревматоидный артрит, остеоартрит, ревматоидный спондилит, подагру, астму, бронхит, ринит, хроническую обструктивную болезнь легких, заболевание муковисцидоз (но не ограничиваются ими).
В другом предпочтительном воплощении нарушения пролиферации клеток включают: рак легкого, рак головы и шеи, рак молочной железы, рак предстательной железы, рак пищевода, рак толстой и прямой кишки, рак толстой кишки, рак носоглотки, рак матки, рак поджелудочной железы, лимфому, лейкоз, остеосаркому, меланому, рак почки, рак желудка, рак печени, рак мочевого пузыря, рак щитовидной железы или рак толстой кишки (но не ограничиваются ими).
В другом предпочтительном воплощении онкологическое заболевание представляет собой немелкоклеточный рак легкого.
В пятом аспекте настоящего изобретения предложен способ ингибирования протеинкиназы (например, киназ ALK) или способ лечения заболеваний (таких как рак, нарушения пролиферации клеток, воспаление, инфекция, иммунопатологические заболевания, трансплантация органов, вирусное заболевание, сердечнососудистые заболевания или метаболическое заболевание), включающий следующие стадии: введение соединения первого аспекта настоящего изобретения или его кристаллической формы, фармацевтически приемлемой соли, гидрата или сольвата, или введение фармацевтической композиции третьего аспекта настоящего изобретения нуждающемуся в этом субъекту.
Следует понимать, что в настоящем изобретении каждый из технических признаков, более конкретно описанных выше и ниже (например, технические признаки в разделе Примеры), можно объединять один с другим, образуя, таким образом, новые или предпочтительные технические решения, которые нет необходимости снова подробно описывать в данном документе.
Описание графических материалов:
Фиг. 1 представляет собой кривую время - концентрация соединения в плазме у самцов крыс, которым, соответственно, вводили 5 мг/кг контрольного соединения Церитиниб и соединения согласно Примеру 1 посредством зондового кормления.
Фиг. 2 представляет собой кривую время - концентрация соединения в плазме у самцов крыс, которым, соответственно, вводили 5 мг/кг контрольного соединения Церитиниб и соединения согласно Примеру 2 посредством зондового кормления.
Фиг. 3 представляет собой кривую время - концентрация соединения в плазме у самцов крыс, которым, соответственно, вводили 5 мг/кг контрольного соединения Церитиниб и соединения согласно Примеру 3 посредством зондового кормления.
Фиг. 4 представляет собой кривую время - концентрация соединения в плазме у самцов крыс, которым, соответственно, вводили 5 мг/кг контрольного соединения Церитиниб и соединения согласно Примеру 13 посредством зондового кормления.
Воплощения для осуществления изобретения
При проведении исследования автор изобретения неожиданно обнаружил, что соединение дейтерированного диаминопиримидина или его фармацевтически приемлемые соли явно превосходят недейтерированное соединение по фармакокинетическим и/или фармакодинамическим свойствам, которые, вследствие этого, больше подходят для применения в качестве соединений ингибиторов киназ ALK и более подходят для применения в получении лекарственных средств для лечения рака и заболеваний, связанных с киназами ALK. Настоящее изобретение осуществлено, исходя из этого.
Определения
В том виде, как он используется в данном документе, термин «галоген» относится к F, Cl, Br и I. Более предпочтительно, галоген выбран из F, Cl и Br.
В том виде, как он используется в данном документе, термин «C1-C6 алкил» относится к прямому или разветвленному алкилу, который содержит от 1 до 6 атомов углерода, такому как метил, этил, пропил, изопропил, бутил, изобутил, mpem-бутил и тому подобное.
В том виде, как он используется в данном документе, термин «C1-C6 алкокси» относится к прямому из разветвленному алкокси, который содержит от 1 до 6 атомов углерода, такому как метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, трет-бутокси и тому подобное.
В том виде, как он используется в данном документе, термин «дейтерированный» означает, что один или более чем один водород в соединении или группе замещен дейтерием. «Дейтерированный» может быть однозамещенным, двузамещенным, многократно замещенным или полностью замещенным. Термин «однократно или многократно дейтерированный» и «дейтерированный один или более чем один раз» можно использовать взаимозаменяемо.
В том виде, как он используется в данном документе, «недейтерированное соединение» относится к соединению, доля атомов дейтерия которого составляет не больше чем природное содержание изотопа дейтерий (0,015%).
В другом предпочтительном воплощении содержание изотопа дейтерий в замещенном дейтерием положении выше чем природное содержание изотопа дейтерий (0,015%), более предпочтительно выше чем 50%, более предпочтительно выше чем 75%, более предпочтительно выше чем 95%, более предпочтительно выше чем 97%, более предпочтительно выше чем 99%, более предпочтительно выше чем 99,5%.
В другом предпочтительном воплощении соединение формулы (I) содержит по меньшей мере два атома дейтерия, более предпочтительно четыре атома дейтерия, более предпочтительно шесть атомов дейтерия, более предпочтительно восемь атомов дейтерия.
В соединении формулы (I) N может представлять собой 14N и/или 15N; O может представлять собой 16O и/или 18O.
Предпочтительно, в соединении формулы (I) N представляет собой 14N и/или O представляет собой 16O.
В другом предпочтительном воплощении в соединении содержание изотопа 14N в положении атома азота составляет ≥95%, предпочтительно ≥99%.
В другом предпочтительном воплощении в соединении содержание изотопа 16O в положении атома кислорода составляет ≥95%, предпочтительно ≥99%.
Активные ингредиенты
В том виде, как он используется в данном документе, термин «соединение согласно настоящему изобретению» относится к соединению формулы (I). Термин также включает кристаллические формы, фармацевтически приемлемые соли, гидраты или сольваты соединения формулы (I).
Среди которых термин «фармацевтически приемлемая соль» относится к соли, образованной соединением согласно настоящему изобретению и кислотой или основанием, которая подходит для лекарственного средства. Фармацевтически приемлемые соли включают неорганические и органические соли. Предпочтительным типом солей являются соли, образованные соединениями согласно настоящему изобретению и кислотой. Подходящие солеобразующие кислоты включают: неорганические кислоты, такие как соляная кислота, бромоводородная кислота, фтороводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное; органические кислоты, такие как муравьиная кислота, уксусная кислота, трифторуксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, пикриновая кислота, бензойная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, бензолсульфоновая кислота, нафталинсульфоновая кислота и тому подобное; и аминокислоты, такие как пролин, фенилаланин, аспарагиновая кислота, глутаминовая кислота и тому подобное, но не ограничиваются ими. Другим предпочтительным типом солей являются соли, образованные соединениями согласно настоящему изобретению и основаниями, например, соли щелочных металлов (например, соли натрия или калия), соли щелочно-земельных металлов (например, соли кальция или магния), аммонийные соли (например, аммонийные соли низших алканолов или другие фармацевтически приемлемые соли амина), например, соль метиламина, соль этиламина, соль пропиламина, соль диметиламина, соли триметиламина, соли диэтиламина, соли триэтиламина, соли трет-бутиламина, соли этилендиамина, соли гидроксиэтиламина, соли дигидроксиэтиламина, соли тригидроксиэтиламина и соли амина, образованные морфолином, пиперазином и лизином.
Термин «сольват» относится к комплексу определенного соотношения, образованному в результате образования координационных связей между соединением согласно настоящему изобретению и молекулами растворителя. Термин «гидрат» относится к комплексу, образованному в результате образования координационных связей между соединением согласно настоящему изобретению и водой.
Кроме того, соединения согласно настоящему изобретению дополнительно содержат пролекарства соединений диаминопиримидина формулы (I). Термин «пролекарство» включает тип соединений, которые обладают биологической активностью или неактивностью, и будут превращаться в соединение формулы (I) в результате метаболизма или химических реакций в организме человека при введении соответствующим способом или соль или сольват, образованные соединением формулы (I). Пролекарство включает сложный эфир карбоновой кислоты, сложный эфир угольной кислоты, фосфат, нитрат, сульфат, сложный эфир сульфоновой кислоты, сложные эфиры сульфоксида, аминосоединения, карбаматы, азосоединения, фосфорамиды, глюкозид, простой эфир, ацеталь соединения и т.д. (но не ограничивается ими).
Способ получения
Ниже в данном документе будет подробно описано получение соединений формулы (I), но такие конкретные способы не накладывают какого-либо ограничения настоящего изобретения. Соединения согласно изобретению можно также легко получать, возможно, путем комбинирования разных способов синтеза, описанных в данном описании или известных в данной области, такую комбинацию может легко осуществить средний специалист в данной области, к которой принадлежит настоящее изобретение.
Известны способы, используемые в настоящем изобретении для получения соединений недейтерированных диаминопиримидинов и их физиологически совместимых солей. Получение соответствующих соединений дейтерированных диаминопиримидинов можно проводить с использованием соответствующего исходного дейтерированного соединения посредством того же пути синтеза. Например, соединение формулы (I) согласно настоящему изобретению можно получать способом, описанным в WO 2008073687, за исключением того, что вместо недейтерированных веществ используют дейтерированные вещества.
Как правило, в способе получения каждую реакцию обычно проводят в инертном растворителе, при температуре от комнатной до температуры кипения (например, 0°C~80°C, предпочтительно от 0°C~50°C). Время реакции обычно составляет от 0,1 часа до 60 часов, предпочтительно от 0,5 до 48 часов.
В синтезе соединений формулы (I) согласно настоящему изобретению можно использовать следующий общий путь получения.


где X, X1, X2 выбраны из F, Cl, Br, I, OTs, OMs, OTf; и R1a, R1b, R1c, R2a, R2b, R2c, R3, R4, R5, R6, R7, R9, R10, R11, R12, R13a, R13b, R13c, R14a, R14b, R14c, R15a, R15b, R15c, R16, R17a, R17b, R18a, R18b, R19a, R19b, R20a или R20b представляют собой такие, как определено выше.
Как показано в Схеме синтеза I, соединение II нитробензол взаимодействует с алканольным соединением III в щелочных условиях с получением соединения IV. 4-Хлор-пиперидиновое соединение V защищают группой Вос(трет-бутоксикарбонил) с получением соединения VI и в результате реакции с бораном получают соединение бора VII. Соединение IV и соединение бора VII сочетают с получением соединения VIII в результате сочетания Сузуки, и затем путем восстановления получают анилиновое соединение IX. В основных условиях посредством реакции замещения из тиофенольного соединения X и алкилгалидного соединения XI получают бензолсульфидное соединение XII, затем соединение XII и 2,4-дигалогенированное соединение пиримидина XIV подвергают реакции замещения в основных условиях с получением 2-галозамещенного соединения пиримидина XIII, и затем посредством реакции окисления получают соединение XV. Соединение XV и анилиновое соединение IX используют для получения соединения XVI посредством сочетания; и соединение I согласно настоящему изобретению получают из соединения XVI посредством реакции снятия защитной группы Вое. Приведенные выше реакции проводят в инертном растворителе, таком как дихлорметан, дихлорэтан, ацетонитрил, н-гексан, толуол, тетрагидрофуран, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, изопропанол, н-бутанол, трет-бутанол, диоксан и т.д, при температуре 0-200°C. Основание выбрано из карбоната калия, карбоната цезия, гидроксида натрия, гидроксида калия, н-бутиллития, трет-бутиллития, изо-бутиллития, диизопропиламинолития, фосфата калия, бис(триметилсилил)аминолития, бис(триметилсилил)аминокалия, трет-бутоксида калия, трет-бутоксида натрия, этоксида натрия, 4-диметиламинопиридина, пиридина, триэтиламина, диизопропилэтиламина и тому подобное.

где X, X1, X2 выбраны из F, Cl, Br, I, OTs, OMs, OTf; и R1a, R1b, R1c, R2a, R2b, R2c, R3, R4, R5, R6, R7, R9, R10, R11, R12, R13a, R13b, R13c, R14a, R14b, R14c, R15a, R15b, R15c, R16, R17a, R17b, R18a, R18b, R19a, R19b, R20a или R20b представляют собой такие, как определено выше. PG представляет собой защитную группу аминогруппы, такую как трет-бутоксикарбонильная группа, бензильная группа, бензилоксикарбонильная группа, п-метоксибензильная группа и тому подобное.
Как показано в Схеме синтеза II, соединение XVII о-нитрофенол взаимодействует с соединением A1 в основных условиях с образованием соединения IV. В щелочных условиях соединение A2 4-пиперидон взаимодействует с N-фенил-бис(трифторметилсульфонил)имидом с получением соединения A3, затем оно взаимодействует с бис(пинаколато)дибораном с получением соединения A4. Соединение A5 получают из соединения IV и соединения A4 посредством реакции сочетания Сузуки; соединение гидрохлорид анилина A6 получают из соединения A5 путем восстановления водородом или газом дейтерия, снятия защиты и солеобразования с использованием гидрохлорида; и соединение A6 и соединение XV конденсируют с получением соединения I согласно настоящему изобретению. Приведенные выше реакции проводят в инертном растворителе, таком как дихлорметан, дихлорэтан, ацетонитрил, н-гексан, толуол, тетрагидрофуран, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид, изопропанол, н-бутанол, трет-бутанол, диоксан и т.д, при температуре 0-200°C. Основание выбрано из карбоната калия, карбоната цезия, гидроксида натрия, гидроксида калия, н-бутиллития, трет-бутиллития, изо-бутиллития, диизопропиламинолития, фосфата калия, бис(триметилсилил)аминолития, бис(триметилсилил)аминокалия, трет-бутоксида калия, трет-бутоксида натрия, этоксида натрия, 4-диметиламинопиридина, пиридина, триэтиламина, диизопропилэтиламина и тому подобное.
Дейтерированное соединение A2 может быть получено следующими путями:

Ссылаясь на журнал Labelled Compounds and Radiopharmaceuticals 2007, 50, 131-137, бензиламина трифторацетат 16 взаимодействует с аллил-триметилсилиловым эфиром и дейтерированным формальдегидом с получением соединения 17, и затем соединение 18 получают путем окисления по методу Десс-Мартина. Соединение 18 подвергают обмену водорода на дейтерий с помощью дейтерированного хлороформа/тяжелой воды в условиях 1,5,7-триазабицикло(4.4.0)дец-5-ена с получением соединения 19.

Соединение 20 подвергают замене водорода на дейтерий с помощью дейтерированного хлороформа/тяжелой воды в условиях 1,5,7-триазабицикло(4.4.0)дец-5-ена с получением соединения 21.

Соединение 22 защищают этиленгликолем с получением кетального соединения 23, которое затем восстанавливают LiAlD4 с получением соединения 24; соединение 24 защищают мезилом с образованием соединения 25, и соединение 25 взаимодействует с бензиламином с получением соединения 26, с которого снимают защиту соляной кислотой с получением соединения 18.
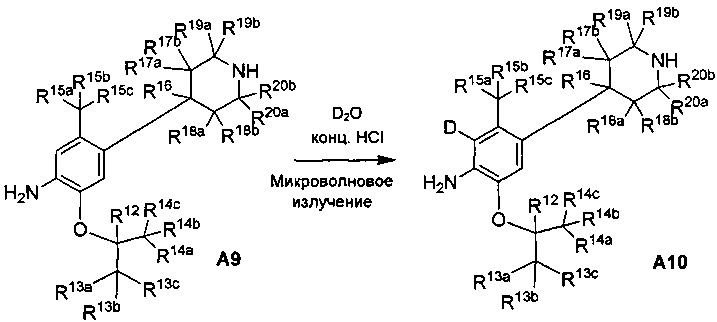
Дейтерированное соединение A8 и A10 можно получать следующими путями:
 ,
,
где R1a, R1b, R1c, R2a, R2b, R2c, R3 представляют собой такие, как определено выше.
Ссылаясь на Organic Letters, 2008, pp. 4351-4353, соединение A7 нагревают микроволновым излучением в концентрированной соляной кислоте и тяжелой воде с получением соединения A8.
 ,
,
где R12, R13a, R13b, R13c, R14a, R14b, R14c, R15a, R15b, R15c, R16, R17a, R17b, R18a, R18b, R19a, R19b, R20a или R20b представляют собой такие, как определено выше.
Ссылаясь на Organic Letters, 2008, pp. 4351-4353, соединение A9 нагревают микроволновым облучением в концентрированной соляной кислоте и тяжелой воде с получением соединения A10.
Фармацевтическая композиция и ее введение
Соединения согласно настоящему изобретению обладают выдающейся активностью ингибирования протеинкиназы, такой как киназы ALK. Вследствие этого соединение согласно настоящему изобретению и его кристаллические формы, фармацевтически приемлемые неорганические или органические соли, гидраты или сольваты, и фармацевтическую композицию, содержащую соединение согласно настоящему изобретению в качестве основного активного ингредиента, можно использовать для лечения, предупреждения и облегчения заболеваний, опосредованных протеинкиназой (например, киназы ALK). На основе предшествующего уровня техники соединения согласно изобретению можно использовать для лечения следующих заболеваний: онкологические заболевания, нарушения пролиферации клеток, сердечнососудистые соединения, воспаления, инфекции, аутоиммунные соединения, трансплантации органов, вирусные заболевания, сердечнососудистые заболевания или метаболические заболевания.
Фармацевтическая композиция согласно изобретению содержит соединение согласно настоящему изобретению или его фармацевтически приемлемые соли в диапазоне безопасных и эффективных доз и фармацевтически приемлемые вспомогательные вещества или носители. При этом, термин «безопасная и эффективная доза» относится к количеству соединения, которого достаточно для улучшения состояния пациента без какого-либо серьезного побочного эффекта. Как правило, фармацевтическая композиция содержит 1-2000 мг соединения согласно изобретению на дозу, предпочтительно 10-1000 мг соединений согласно изобретению на дозу. Предпочтительно, «на дозу» означает одну капсулу или таблетку.
«Фармацевтически приемлемый носитель» означает один или более чем один совместимый твердый или жидкий наполнитель или гелевое вещество, которые подходят для человека и должны иметь достаточную чистоту и достаточно низкую токсичность. «Совместимость» в данном документе означает, что компоненты композиции можно смешивать с соединениями согласно изобретению или друг с другом, и они не будут значительно снижать эффективность соединений. Некоторые примеры фармацевтически приемлемых носителей включают целлюлозу и ее производные (такие как натрий-карбоксиметилцеллюлоза, натрий-этилцеллюлоза, ацетат целлюлозы и т.д.), желатин, тальк, твердые смазывающие вещества (такие как стеариновая кислота, стеарат магния), сульфат кальция, растительные масла (такие как соевое масло, кунжутное масло, арахисовое масло, оливковое масло и т.д.), полиолы (такие как пропиленгликоль, глицерин, маннит, сорбит и т.д.), эмульгаторы (такие как Tween®), увлажнитель (такой как додецилсульфат натрия), красители, корригенты, стабилизаторы, антиоксиданты, консерванты, апирогенную воду и т.д.
Не существует какого-либо специального ограничения по способу введения в отношении соединения или фармацевтических композиций согласно настоящему изобретению, и типичный способ введения включает: пероральное, внутриопухолевое, ректальное, парентеральное (внутривенное, внутримышечное или подкожное) и местное введение (но не ограничивается ими).
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В данных твердых лекарственных формах активные соединения смешивают по меньшей мере с одним традиционным инертным вспомогательным веществом (или носителем), таким как цитрат натрия или CaHPO4, или смешивают с любым из следующих компонентов: (а) наполнители или агент, улучшающий совместимость, например, крахмал, лактоза, сахароза, глюкоза, маннит или салициловая кислота: (б) связующие вещества, например, гидроксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и гуммиарабик; (в) увлажнитель, такой как глицерин; (г) разрыхлители, такие как агар, карбонат кальция, картофельный крахмал или тапиоковый крахмал, альгиновая кислота, определенные смешанные силикаты и карбонат натрия; (д) средства, замедляющие растворение, такие как парафин; (е) ускорители абсорбции, например, четвертичные аммониевые соединения; (ж) пластификаторы, такие как цетиловый спирт и глицерилмоностеарат; (з) адсорбенты, например, каолин; и (и) смазывающие вещества, такие как тальк, стеарин кальция, стеарат магния, твердый полиэтиленгликоль, лаурилсульфат натрия или их смеси; В капсулах, таблетках и пилюлях лекарственные формы также содержат буферные вещества.
Твердые лекарственные формы, такие как таблетки, капсулы, пилюли и гранулы можно получать с использованием материалов покрытия и оболочки, таких как растворимые в кишечнике покрытия и любые другие материалы, известные в данной области. Они могут содержать непрозрачный агент. Высвобождение активных соединений или соединений в композициях может быть замедленным в определенной части пищеварительного тракта. Примеры капсулирующих компонентов включают полимеры и воска. При необходимости активные соединения и одно или более чем одно приведенное выше вспомогательное вещество могут образовывать микрокапсулы.
Жидкие лекарственные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы или настойки. Помимо активных соединений жидкие лекарственные формы могут содержать любые традиционные инертные разбавители, известные в данной области, такие как вода или другие растворители, солюбилизаторы и эмульгаторы, например, этанол, изопропанол, этилкарбонат, этилацетат, пропиленгликоль, 1,3-бутандиол, диметилформамид, а также масло, в частности, хлопковое масло, арахисовое масло, масло из зародышей кукурузы, оливковое масло, касторовое масло и кунжутное масло или их сочетание.
Помимо данных инертных разбавителей композиция может также содержать добавки, такие как увлажнители, эмульгаторы и суспендирующие агенты, подсластители, корригенты и отдушки.
Помимо активных соединений суспензия может содержать суспендирующий агент, например, этоксилированный изооктадеканол, полиоксиэтиленсорбит и сложные эфиры сорбитана, микрокристаллическую целлюлозу, метанол алюминий и агар или их сочетание.
Композиции для парентеральной инъекции могут содержать физиологически приемлемые стерильные водные или безводные растворы, дисперсии, суспензии или эмульсии и стерильные порошки, которые могут быть повторно растворены в стерильных инъецируемых растворах или дисперсиях. Подходящие водные и неводные носители, разбавители, растворители или эксципиенты включают воду, этанол, полиолы и их любые подходящие смеси.
Лекарственные формы для местного введения соединений согласно изобретению включают мази, порошки, пластыри, аэрозоль и средства для ингаляции. Активные ингредиенты при необходимости смешивают в стерильных условиях с физиологически приемлемыми носителями и любыми консервантами, буферами или пропеллентом.
Соединения согласно настоящему изобретению можно вводить отдельно или в комбинации с любыми другими фармацевтически приемлемыми соединениями.
При использовании фармацевтических композиций безопасное и эффективное количество соединения согласно настоящему изобретению применяют для нуждающегося в этом млекопитающего (такого как человек), где доза введения представляет собой фармацевтически эффективную дозу. Для человека с массой 60 кг суточная доза обычно составляет 1-2000 мг, предпочтительно 50-1000 мг. Конечно, конкретная доза будет также зависеть от разных факторов, таких как путь введения, состояние здоровья пациента, которые хорошо известны специалистам - опытным терапевтам.
По сравнению с недейтерированными соединениями, известными в предшествующем уровне техники, соединения согласно настоящему изобретению обладают рядом преимуществ. Основными преимуществами настоящего изобретения являются следующие:
(1) Соединения согласно настоящему изобретению обладают хорошей ингибирующей активностью в отношении протеинкиназы (такой как киназа ALK).
(2) Технология дейтерирования меняет метаболизм дейтерированных соединений в организме, придавая, таким образом, соединению лучшие фармакокинетические характерные параметры. В данном случае доза может варьировать, и для улучшения применимости можно получать композицию длительного действия.
(3) Концентрацию лекарственного соединения у животных можно увеличивать посредством замены водорода дейтерием в соединении благодаря эффекту изотопа дейтерия, улучшая, таким образом, эффективность лекарственного средства.
(4) Безопасность соединения можно улучшать путем замены в соединении водорода на дейтерий, так как некоторые метаболиты подавляются.
Настоящее изобретение будет дополнительно проиллюстрировано ниже со ссылкой на конкретные примеры. Следует понимать, что данные примеры даны только для иллюстрации изобретения, а не для ограничения объема изобретения. Экспериментальные способы без конкретных условий, описанные в следующих примерах, обычно осуществляют в традиционных условиях или в соответствии с инструкциями изготовителя. Если не указано иное, доли и процентное содержание рассчитаны по массе.
Дейтерированное исходное вещество, используемое в следующих воплощениях (например, 1-бром-5-фтор-2-(d3-метил)-4-нитробензол), может представлять собой дейтерированные соединения, полученные из недейтерированных соединений (таких как 1-бром-5-фтор-2-метил-4-нитрофенил) посредством реакции дейтерирования традиционным способом.
Пример 1: Получение 5-хлор-N2-(2-изопропокси-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамина (соединение 13)

1. Получение трет-бутилового эфира 4-бром-2,2,6,6-d4-пиперидин-1-карбоновой кислоты (соединение 2)
Соединение 4-бром-2,2,6,6-d4-пипередин (0,84 г, 5 ммоль) и дихлорметан (10 мл) последовательно добавляли в колбу. При 0°C по каплям добавляли N,N-диизопропилэтиламин (0,65 г, 5 ммоль). После перемешивания в течение 0,5 часа по каплям добавляли ди-трет-бутилдикарбонат (1,64 г, 7,5 ммоль) в дихлорметане (10 мл). Сразу после добавления реакционный раствор перемешивали при комнатной температуре всю ночь. Реакционный раствор соответственно промывали водным раствором хлороводорода (1 н., 2×10 мл) и насыщенным солевым раствором (10 мл). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали посредством роторного испарителя в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (элюент: н-гексан / этилацетат = 5/1, об./об.) с получением желаемого продукта в виде бесцветного масла (1,23 г, выход 92%). 1H-ЯМР (ядерный магнитный резонанс) (400 МГц, CDCl3) δ 4,34 (тт, J=7,69, 3,8 Гц, 1H), 2,16-2,05 (м, 2H), 1,95-1,82 (м, 2H), 1,48 (с, 9H).
2. Получение трет-бутилового эфира 4-(4,4,5,5-тетрабутил-1,3,2-диокса боролан-2-ил)-2,2,6,6-d4-пиперидин-1-карбоновой кислоты (соединение 3)
Йодид меди (48 мг, 0,25 ммоль), трифенилфосфин (86 мг, 0,33 ммоль), метоксид лития (0,2 г, 5 ммоль) и бис(пинаколато)дибор (0,965 г, 3,8 ммоль) последовательно добавляли в реакционную пробирку Шленка. В системе заменяли аргон 3-4 раза. В условиях аргона добавляли трет-бутиловый эфир 4-бром-2,2,6,6-d4-пиперидин-1-карбоновой кислоты (0,67 г, 2,5 ммоль) в N,N-диметилформамиде (5 мл). При 40°C реакцию перемешивали в течение 24 часов. Реакционную жидкость охлаждали до комнатной температуры. Этилацетат добавляли для разбавления реакционной жидкости, фильтровали через целит и осадок на фильтре промывали этилацетатом. Фильтрат объединяли и концентрировали с помощью роторного испарителя в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле с получением желаемого продукта в виде землисто-желтого масла (0,47 г, выход 60%). ГХ (газовая хроматография)-МС расчетный: 315; ГХ-МС обнаруженный: 315; 1H-ЯМР (CDCl3, 400 МГц) δ 1,50-1,58 (м, 2H), 1,40-1,46 (м, 2H), 1,38 (с, 9H), 1,16 (с, 12H), 1,00-1,08 (м, 1H).
3. Получение 1-бром-5-изопропокси-2-метил-4-нитробензола (соединение 5)
Соединение 1-бром-5-фтор-2-метил-4-нитробензол (4,68 г, 20 ммоль) и водный N,N-диметилформамид (15 мл) последовательно добавляли в колбу. Под защитой азота последовательно добавляли карбонат калия (8,29 г, 60 ммоль) и изопропанол (2,40 г, 40 ммоль). Температуру поднимали до 50°C и реакционную смесь перемешивали всю ночь. После охлаждения до комнатной температуры для разбавления реакционной жидкости добавляли воду (20 мл) и этилацетат (30 мл); реакционную жидкость перемешивали в течение 15 мин и разделяли на слои. Затем водный слой дважды экстрагировали этилацетатом (30 мл). Органические слои объединяли и последовательно промывали водой и солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали с помощью роторного испарителя в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (элюент: петролейный эфир / этилацетат = 10/1, об./об.) с получением желаемого продукта в виде желтого твердого вещества (4,44 г, выход 81%).
4. Получение трет-бутилового эфира 4-(5-изопропокси-2-метил-4-нитрофенил)-2,2,6,6-d4-пиперидин-1-карбоновой кислоты (соединение 6)
Соединение 1-бром-5-изопропокси-2-метил-4-нитробензол (1,37 г, 5,0 ммоль), водный карбонат натрия (2 М, 8,35 мл, 16,7 ммоль) и 1,2-метоксиэтан (50 мл) последовательно добавляли в колбу. При перемешивании добавляли трет-бутиловый эфир 4-(4,4,5,5-тетрабутил-1,3,2-диоксаборолан-2-ил)-2,2,6,6-d4-пиперидин-1-карбоновой кислоты (1,50 г, 4,76 ммоль). Описанную выше суспензию дегазировали в атмосфере аргона в течение 5 мин, к которой добавляли трифенилфосфин (0,26 г, 1 ммоль) и ацетат палладия (0,11 г, 0,5 ммоль), нагревали с обратным холодильником в течение 24 ч при 85°C. Ее охлаждали до комнатной температуры, добавляли этилацетат (50 мл) и воду (40 мл) и разделяли на слои. Затем водный слой дважды экстрагировали этилацетатом. Органические слои объединяли и последовательно промывали водой и солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали с помощью роторного испарителя в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением желаемого продукта в виде белого твердого вещества (1,37 г, выход 75%). МС расчетный: 382; МС обнаруженный: 383 (M+H)+, 405 (M+Na)+.
5. Получение трет-бутилового эфира 4-(4-амино-5-изопропокси-2-метилфенил)-2,2,6,6-d4-пиперидин-1-карбоновой кислоты (соединение 7)
Трет-бутиловый эфир 4-(5-изопропокси-2-метил-4-нитрофенил)-2,2,6,6-d4-пиперидин-1-карбоновой кислоты (1,30 г, 3,40 ммоль), этанол (10 мл) и воду (1 мл) последовательно добавляли в реакционную колбу для гидрогенизации. 10% палладий на угле (0,13 г) добавляли в среде азота. После замены в системе водорода 3-4 раза ее перемешивали при 40°C в течение 5 часов при давлении водорода 1 атм. Реакцию детектировали с осуществлением ВЭЖХ (высокоэффективная жидкостная хроматография). Реакционный раствор охлаждали до комнатной температуры, фильтровали через Целит и осадок на фильтре промывали этанолом. Фильтрат объединяли и концентрировали с помощью роторного испарителя в вакууме с получением желаемого продукта, не совсем белого твердого вещества (1,16 г, выход: 97%). МС расчетный: 352; МС обнаруженный: 353 (M+H)+.
6. Получение 2-(изопропилтио)анилина (Соединение 9)
Под защитой азота соединение 2-амино-тиофенол (12,52 г, 0,1 мол), 2-йодпропан (18,71 г, 0,11 моль) и эталон (130 мл) последовательно добавляли в колбу. При перемешивании при 0°C медленно добавляли трет-бутоксид калия (14,61 г, 0,13 моль). Реакцию нагревали до комнатной температуры, и проводили в течение 4 часов, и реакцию контролировали по существу в виде осуществления ВЭЖХ. Реакционную смесь фильтровали через Целит, осадок на фильтре промывали этанолом и объединенный фильтрат концентрировали в вакууме с помощью роторного испарителя. Осадок растворяли в этилацетате (200 мл), последовательно промывали водой и солевым раствором. Его сушили над безводным сульфатом натрия, фильтровали и концентрировали с помощью роторного испарителя в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением желаемого продукта в виде желтоватого масла. МС расчетный: 167; МС обнаруженный: 168 (M+H)+.
7. Получение 2,5-дихлор-N-(2-(изопропилтио)фенил)пиримидин-4-амина (Соединение 10)
Под защитой азота в колбу последовательно добавляли 2-(изопропилтио)анилин (0,50 г, 3,0 ммоль), 2,4,5-трихлорпиримидин (0,37 г, 2,0 ммоль) и безводный N,N-димитилформамид (12 мл). При перемешивании медленно добавляли трет-бутоксид калия (0,67 г, 6,0 моль) при 0°C. Реакцию нагревали до комнатной температуры в течение 2 часов. Реакционную смесь добавляли в чистую воду (100 мл) для остановки реакции. Затем ее три раза экстрагировали этилацетатом. Органические слои объединяли и последовательно промывали водой и солевым раствором. Осуществляли сушку над безводным сульфатом натрия, фильтровали и концентрировали посредством роторного испарителя в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле с получением желаемого продукта в виде белого твердого вещества. МС расчетный: 314; МС обнаруженный: 315 (M+H)+, 337 (M+Na)+.
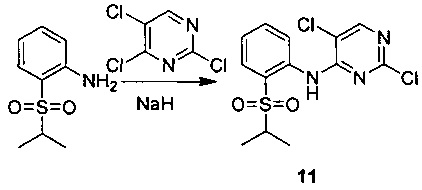
8. Получение 2,5-дихлор-N-(2-(изопропилсульфонил)фенил)пиримидин-4-амина (Соединение 11)
Под защитой азота в колбу последовательно добавляли 2,5-дихлор-N-(2-(изопропилтио)фенил)пиримидин-4-амин (0,50 г, 1,59 ммоль) и дихлорметан (50 мл). 3-хлорпербензойную кислоту (85%, 0,41 г, 2,0 ммоль) добавляли при перемешивании. Реакцию выдерживали при комнатной температуре в течение 5 часов. Добавляли дихлорметан (20 мл) и последовательно осуществляли промывание водой, насыщенным раствором бикарбоната натрия и солевым раствором. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали посредством роторного испарителя в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле с получением желаемого продукта (0,33 г, выход: 60%). МС расчетный: 345; МС обнаруженный: 346 (M+H)+, 368 (M+Na)+. 1H-ЯМР (CDCl3, 400 МГц) δ 10,10 (с, 1H), 8,65 (д, 1H), 8,32 (с, 1H), 7,92 (м, 1H), 7,71-7,76 (м, 1H), 7,29-7,32 (м, 1H), 3,18-3,25 (м, 1H), 1,36 (с, 6H).
9. Получение трет-бутилового эфира 4-(4-(5-хлор-4-(2-(изопропилсульфонил)фенил)амино)пиримидин-2-ил-амино)-5-изопропокси-2-метилфенил)-2,2,6,6-d4-пиперидин-1-карбоновой кислоты (соединение 12)
Под защитой азота трет-бутиловый эфира 4-(4-амино-5-изопропокси-2-метилфенил)-2,2,6,6-d4-пиперидин-1-карбоновой кислоты (353 мг, 1 ммоль), 2,5-дихлор-N-(2-(изопропилсульфонил)фенил)пиримидин-4-амин (346 мг, 1 ммоль), 4,5-бисдифенилфосфин-9,9-диметилксантен (ксантфос, 58 мг, 0,1 ммоль), ацетат палладия (18 мг, 0,08 ммоль), карбонат цезия (978 мг, 3 ммоль) и тетрагидрофуран (10 мл) последовательно добавляли в колбу. При перемешивании смесь последовательно барботировали в азоте в течение 10 мин. Смесь загружали в пробирку, пробирку плотно закрывали и реакцию проводили с использованием микроволнового облучения при 150°C в течение 30 мин. Затем смесь охлаждали и разбавляли тетрагидрофураном, фильтровали и концентрировали посредством роторного испарителя в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии с получением желаемого продукта (0,20 г, выход: 30%). МС расчетный: 661; МС обнаруженный: 662 (M+H)+, 684 (M+Na)+.
10. Получение 5-хлор-N2-(2-изопропокси-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамина (соединение 13)
Под защитой азота в колбу последовательно добавляли трет-бутиловый эфир 4-(4-(5-хлор-4-(2-(изопропилсульфонил)фениламино)пиримидин-2-ил-амино)-5-изопропокси-2-метилфенил)-2,2,6,6-d4-пиперидин-1-карбоновой кислоты (0,18 г, 0,27 ммоль) и дихлорметан (5 мл). При перемешивании по каплям добавляли трифторуксусную кислоту (3 мл). Реакцию выдерживали при комнатной температуре в течение 1,5 часов. Добавляли насыщенный водный карбонат натрия, реакционный раствор концентрировали посредством роторного испарителя в вакууме и три раза экстрагировали этилацетатом. Органические слои объединяли и последовательно промывали водой и солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали посредством роторного испарителя в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ЖХ-МС (жидкостная хроматография с масс-спектрометрией) с получением желаемого продукта в виде белого твердого вещества (0,12 г, выход: 80%). МС расчетный: 561; МС обнаруженный: 562 (M+H)+ 584 (M+Na)+.
Другой способ получения соединения 13:

Под защитой азота в колбу добавляли соединение дигидрохлорид 2-изопропокси-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)анилина (0,50 г, 1,54 ммоль), 2,5-дихлор-N-(2-(изопропилсульфонил)фенил)пиримидин-4-амин (0,58 г, 1,69 ммоль) и изопропанол (6 мл), реакцию нагревали до 85°C, и проводили всю ночь, и реакцию контролировали по существу в виде осуществления ВЭЖХ. Реакционную смесь охлаждали до комнатной температуры и перемешивали в течение 3 часов для осаждения твердого вещества. Смесь фильтровали и осадок на фильтре с измельчением промывали холодным изопропанолом с получением гидрохлорида желаемого продукта. Гидрохлорид растворяли в чистой воде, водный раствор карбоната натрия добавляли по каплям для нейтрализации pH до 8.5 и затем три раза экстрагировали этилацетатом. Объединенную органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением желательного продукта в виде белого твердого вещества (0,68 г, выход: 78%). МС расчетный: 561; МС обнаруженный: 562 (M+H)+, 584 (M+Na)+. 1H-ЯМР (ДМСО (диметилсульфоксид)-d6+D2O, 400 МГц) δ 8,47 (д, J=7,6 Гц, 1H), 8,24 (с, 1H), 7,85-7,83 (дд, J=7,6, 2,0 Гц, 1H), 7,62 (т, J=7,6 Гц, 1H), 7,52 (с, 1H), 7,36 (т, J=7,6 Гц, 1H), 6,79 (с, 1H), 4,53-4,50 (м, 1H), 3,45-3,38 (м, 1H), 2,94-2,92 (м, 1H), 2,33 (с, 3H), 1,79-1,64 (м, 4H), 1,21 (д, 6H), 1,15 (д, 6H).
Пример 2: Получение 5-хлор-N2-(2-(2-d-проп-2-илокси)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамина

1. Получение 1-бром-5-(2-d-проп-2-илоксил)-2-метил-4-нитробензола (соединение 14)

В колбу последовательно добавляли соединение 1-бром-5-фтор-2-метил-4-нитробензол (468 мг, 2 ммоль) и безводный N,N-диметилформамид (5 мл). Под защитой азота последовательно добавляли карбонат цезия (1,95 г, 6 ммоль) и 2-d-изопропанол (240 мг, 4 ммоль). Температуру повышали до 50°C и реакционную смесь перемешивали всю ночь. После охлаждения до комнатной температуры для разбавления реакционной жидкости добавляли воду и этилацетат; ее перемешивали в течение 15 мин и разделяли на слои. Затем водный слой дважды экстрагировали этилацетатом. Органические слои объединяли, последовательно промывали водой и солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали посредством роторного испарителя в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной хроматографии на пластинах (ТСХ) (элюент: петролейный эфир / этилацетат = 10/1, об./об.) с получением желаемого продукта в виде желтого твердого вещества (468 мг, выход 85%).
Соединение 14 можно также получать следующими путями:

Под защитой азота соединение 5-бром-4-метил-2-нитрофенол (194 мг, 0,84 ммоль), N,N-диметилформамид (5 мл), 2-d-изопропил п-толуолсульфонат (150 мг, 0,70 ммоль), карбонат цезия (454 мг, 1,40 ммоль) добавляли в колбу, нагревали до 60°C всю ночь. После охлаждения до комнатной температуры реакцию останавливали путем добавления воды, и экстрагировали этилацетатом, выдерживали для разделения, и органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом магния, и фильтровали, и фильтрат концентрировали в вакууме с помощью роторного испарителя с получением неочищенного продукта, и очищали посредством препаративной хроматографии на пластинах ТСХ (проявляющий растворитель: петролейный эфир / этилацетат = 10/1, об./об.) с получением желаемого продукта в виде желтого твердого вещества (140 мг, выход 73%). 1H-ЯМР (ДМСО-d6, 400 МГц) δ 8,08 (с, 1H), 7,46 (с, 1H), 2,41 (с, 3H), 1,28 (с, 6H).
2. Получение диэтил-2,2'-(1,3-диоксолан-2,2-диил)диацетата
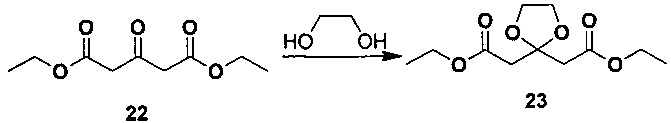
Под защитой азота соединение диэтил-1,3-ацетон-дикарбоксилат (8 г, 40 ммоль), дихлорметан (80 мл) и этиленгликоль (8,9 мл, 160 ммоль) добавляли в колбу, охлаждали до 0°C и по каплям добавляли диэтилэфират трехфтористого бора (7,6 мл, 60 ммоль). Сразу после добавления реакционную смесь перемешивали в течение 1 часа при 0°C и затем в естественных условиях нагревали до комнатной температуры и перемешивали всю ночь. После охлаждения до 0°C по каплям добавляли воду (40 мл) и водную фазу разделяли и экстрагировали дихлорметаном. Объединенную органическую фазу промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали. Фильтрат концентрировали посредством роторного испарителя в вакууме с получением желаемого продукта в виде желтой жидкости (9 г, выход 92%). 1H-ЯМР (CDCl3, 400 МГц) δ 4,20-4,15 (кв, 4H), 4,03 (с, 4H), 2,96 (с, 4H), 1,30-1,27 (т, 6H).
3. Получение 2,2'-(1,3-диоксолан-2,2-диил)-1,1,1',1'-d4-диэтанола
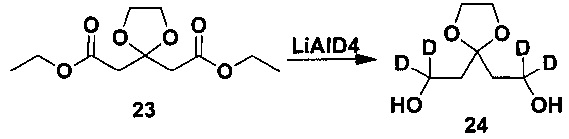
Под защитой азота дейтерированный литийалюминийгидрид (510 мг, 12 мг) и тетрагидрофуран (40 мл) последовательно добавляли в колбу и охлаждали до 0°C. Соединение диэтил-2,2'-(1,3-диоксолан-2,2-диил)диацетат (1,5 г, 6 ммоль) растворяли в тетрагидрофуране (10 мл) и раствор по каплям добавляли в описанный выше реакционный раствор. Сразу после добавления реакционную смесь перемешивали в течение 1 ч при 0°C и затем в естественных условиях нагревали до комнатной температуры и перемешивали всю ночь. Реакционную жидкость охлаждали до 0°C на ледяной бане, водный раствор 2 н. NaOH добавляли по каплям, фильтровали, и осадок на фильтре по каплям промывали этилацетатом, и фильтрат концентрировали посредством роторного испарителя в вакууме с получением желаемого продукта в виде бесцветной жидкости (800 мг, выход 79%). 1H-ЯМР (CDCl3, 400 МГц) δ 4,08 (с, 4H), 3,75 (с, 2H), 1,99 (с, 4H).
4. Получение (1,3-диоксолан-2,2-диил)бис(1,1-d2-этил-2,1-диил)диметансульфоната

Под защитой азота 2,2'-1,3-диоксолан-2,2-диил)-1,1,1',1'-d4-диэтанол (800 мг, 4,81 ммоль), триэтиламин (1,15 г, 11,38 ммоль) и тетрагидрофуран (16 мл) последовательно добавляли в колбу и охлаждали до -30°C. Метансульфонилхлорид (1,38 г, 12,03 ммоль) растворяли в тетрагидрофуране (2 мл) и раствор по каплям добавляли в описанный выше реакционный раствор. Сразу после добавления баню с сухим льдом удаляли, реакционную жидкость нагревали в естественных условиях до комнатной температуры и перемешивали всю ночь. В реакционную жидкость последовательно добавляли метиленхлорид и насыщенный раствор хлорида аммония, и органический слой отделяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом магния и фильтровали, и фильтрат концентрировали в вакууме посредством роторного испарителя с получением неочищенного продукта и очищали посредством препаративной хроматографии на пластинах ТСХ (проявляющий растворитель: петролейный эфир / этилацетат = 1/50, об./об.) с получением желаемого продукта в виде белого твердого вещества (900 мг, выход 58%). 1H-ЯМР (CDCl3, 400 МГц) δ 4,00 (с, 4H), 3,05 (с, 6H), 2,15 (с, 4H).
5. Получение 1-бензил-4-(2,2,6,6-d4-пиперидин)-этиленкеталя
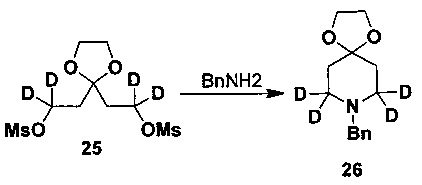
(1,3-Диоксолан-2,2-диил)бис(1,1-d2-этил-2,1-диил)диметансульфонат (300 мг, 0,89 ммоль) и чистый этанол (10 мл) добавляли в колбу и при перемешивании добавляли бензиламин (973 мг, 8,90 ммоль). Реакционную жидкость перемешивали и нагревали с обратным холодильником всю ночь. Реакционную жидкость охлаждали до комнатной температуры и для остановки реакции добавляли воду. Экстрагированную дихлорметаном, объединенную органическую фазу последовательно промывали насыщенным раствором хлорида аммония, водой и солевым раствором и сушили над безводным сульфатом натрия. Объединенную органическую фазу концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт отделяли и очищали посредством препаративной ТСХ (проявляющий растворитель: метанол / дихлорметан = 5/95, об./об.) с получением желаемого продукта (145 мг, выход 66%). 1H-ЯМР (CDCl3, 400 МГц) δ 7,39-7,31 (м, 5H), 3,97 (с, 4H), 3,62 (с, 2H), 1,80 (с, 4H).
6. Получение 1-бензил-2,2,6,6-d4-пиперидин-4-она

В колбу последовательно добавляли соединение 1-бензил-4-(2,2,6,6-d4-пиперидин)-этиленкеталь (135 мг, 0,57 ммоль) и чистый метанол (5 мл). При перемешивании добавляли 2 М соляную кислоту (3 мл, 5,7 ммоль) и нагревали с обратным холодильником в течение 6 ч. Растворитель удаляли посредством концентрирования при пониженном давлении и для нейтрализации добавляли насыщенный раствор бикарбоната натрия и экстрагировали дихлорметаном. Масляную фазу сушили над безводным сульфатом натрия и концентрировали, неочищенный продукт очищали посредством препаративной хроматографии на пластинах ТСХ (проявляющий растворитель: метанол / метиленхлорид = 4/96, об./об.) с получением желаемого продукта в виде желтого масла (83 мг, выход 75%).
7. Получение 1-бензил-1,2,3,6-(2,2,6,6-d4-тетрагидропиридин)-4-ил-трифторметансульфоната

Соединение 1-бензил-2,2,6,6-d4-пиперидин-4-он (80 мг, 0,41 ммоль) и безводный тетрагидрофуран (5 мл) последовательно добавляли в колбу и охлаждали до -78°C. Под защитой азота добавляли LiHMDS (гексаметилдисилазан лития) в тетрагидрофуране (0,5 мл, 0,49 ммоль, 1М) и выдерживали, перемешивая в течение 30 мин. После этого добавляли PhN(Tf)2 (175 мг, 0,49 ммоль), перемешивали при комнатной температуре в течение одного часа, затем перемешивали с обратным холодильником в течение 2 часов. Для остановки реакции добавляли воду. Смесь экстрагировали этилацетатом, и масляные фазы отдельно промывали насыщенным раствором бикарбоната натрия, сушили над безводным сульфатом натрия и концентрировали, и неочищенный продукт очищали посредством препаративной хроматографии на пластинах ТСХ (проявляющий растворитель: этилацетат / петролейный эфир = 15/85, об./об.) с получением желаемого продукта в виде желтого масла (85 мг, выход 63%). 1H-ЯМР (CDCl3, 400 МГц) δ 7,37-7,30 (м, 5H), 5,75 (с, 1H), 3,65 (с, 2H), 2,46 (с, 2H).
8. Получение 1-бензил-2,2,6,6-d4-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,6-тетрагидропиридина

В колбу последовательно добавляли соединение 1-бензил-1,2,3,6-(2,2,6,6-d4-тетрагидропиридин)-4-ил-трифторметансульфонат (85 мг, 0,26 ммоль) и диоксан (3 мл). Под защитой азота при перемешивании добавляли бис(пинаколато)дибор (73 мг, 0,29 ммоль), Pd2(dba)3 (10 мг), PCy3 (8 мг) и ацетат калия (38 мг, 0,39 ммоль). Реакционную смесь нагревали до 80°C и перемешивали всю ночь. Воду и дихлорметан добавляли для экстракции; масляную фазу сушили над безводным сульфатом натрия и концентрировали. Неочищенный продукт очищали посредством препаративной хроматографии на пластинах ТСХ с получением желаемого продукта в виде не совсем белого твердого вещества (56 мг, выход 75%).
9. Получение 1-бензил-2,2,6,6-d4-4-(5-(2-d-проп-2-ил)оксил-2-метил-4-нитрофенил)-1,2,3,6-тетрагидропиридина

Под защитой азота в колбу добавляли соединение 1-бром-5-(2-d-проп-2-ил)окси-2-метил-4-нитробензол (0,24 г, 0,87 ммоль), 1-бензил-2,2,6,6-d4-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,6-тетрагидропиридин (0,35 г, 1,13 ммоль), ацетат палладия (18 мг, 0,078 ммоль), 4,5-бисдифенилфосфин-9,9-диметил-ксантен (35 мг, 0,061 ммоль), фосфат калия тригидрат (0,59 г, 2,62 ммоль) и тетрагидрофуран (8 мл). Сразу после добавления реакционную смесь нагревали до 85°C и выдерживали всю ночь. В реакционную смесь добавляли воду и три раза экстрагировали этилацетатом. Объединенную органическую фазу последовательно промывали водой и солевым раствором, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ТСХ с получением желаемого продукта (0,24 г, выход: 75%). МС расчетный: 371; МС обнаруженный: 372 (M+H)+.
10. Получение дигидрохлорида 2-((2-d-проп-2-ил)оксил)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)анилина

Соединение 1-бензил-2,2,6,6-d4-4-(5-(2-d-проп-2-ил)окси-2-метил-4-нитрофенил)-1,2,3,6-тетрагидропиридин (0,22 г, 0,59 ммоль), палладий на угле (0,022 г, содержание палладия 50%) и метанол (8 мл) добавляли в колбу и три раза заменяли водород. Реакционную смесь нагревали до 45°C и выдерживали в течение 40 часов. Палладий на угле удаляли посредством фильтрации; фильтрат концентрировали с получением серого твердого вещества. Полученное серое вещество и раствор соляной кислоты в изопропаноле (2 М, 2 мл) последовательно добавляли в колбу и перемешивали в течение 2 ч. Реакционный раствор концентрировали при пониженном давлении с получением желаемого продукта в виде белого твердого вещества (0,18 г, выход: 95%).
11. Получение 2,5-дихлор-N-(2-(изопропилсульфонил)фенил)пиримидин-4-амина
Под защитой азота 2-(изопропилсульфонил)анилин (0,65 г, 3,29 ммоль) и безводный N,N-диметилформамид (6 мл) последовательно добавляли в колбу, охлаждали до 0°C и порциями добавляли гидрид натрия (0,20 г, 60%, 8,24 ммоль). Сразу после добавления реакционную смесь перемешивали в течение 1 часа. 2,4,5-трихлорпиримидин (1,21 г, 6,59 ммоль) в N,N-диметилформамиде (3 мл) добавляли по каплям, и затем нагревали до комнатной температуры, и перемешивали всю ночь. Реакционную смесь добавляли в чистую воду для остановки реакции. Затем ее три раза экстрагировали этилацетатом. Органические слои объединяли и последовательно промывали водой и солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали посредством роторного испарителя в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии с получением желаемого продукта в виде белого твердого вещества (0,70 г, 63%). МС расчетный: 345; МС обнаруженный: 346 (M+H)+, 368 (M+Na)+. 1H-ЯМР (CDCl3, 400 МГц) δ 10,08 (с, 1H), 8,65 (д, J=8,4 Гц, 1H), 8,32 (с, 1H), 7,94 (д, J=8,0 Гц, 1H), 7,75 (т, J=8,4 Гц, 1H), 7,33 (т, J=8,0 Гц, 1H), 3,25-3,12 (м, 1H), 1,34 (д, 6H).
12. Получение 5-хлор-N2-(2-(2-d-проп-2-илоксил)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамина
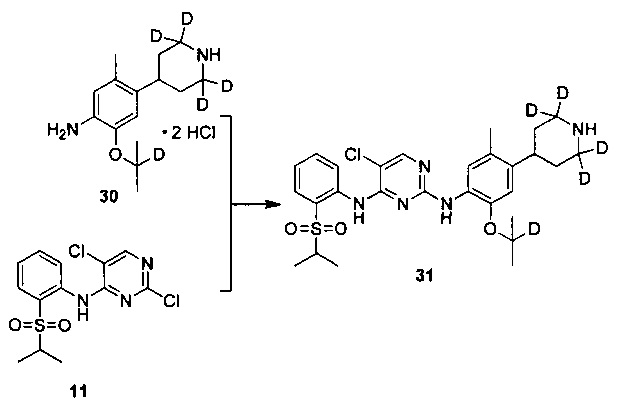
Под защитой азота соединение дигидрохлорид 2-((2-d-проп-2-ил)оксил)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)анилина (0,180 г, 0,52 ммоль), 2,5-дихлор-N-(2-(изопропилсульфонил)фенил)пиримидин-4-амин (0,198 г, 0,57 ммоль) и изопропанол (4 мл) добавляли в колбу, нагревали до 85°C и выдерживали всю ночь. Реакцию контролировали по существу в виде осуществления ВЭЖХ, охлаждали до комнатной температуры и перемешивали в течение 3 часов для осаждения твердого вещества. Твердое вещество фильтровали и осадок на фильтре с измельчением промывали холодным изопропанолом с получением гидрохлорида желаемого продукта. Гидрохлорид растворяли в чистой воде, водный раствор карбоната натрия медленно добавляли по каплям для нейтрализации pH до 8,5 и затем экстрагировали этилацетатом. Объединенную органическую фазу сушили над безводным сульфатом натрия и концентрировали с получением желаемого продукта в виде белого твердого вещества (0,176 г, выход: 60%). МС расчетный: 562; МС обнаруженный: 563 (M+H)+, 585 (M+Na)+. 1H-ЯМР (ДМСО-d6+D2O, 400 МГц) δ 8,49 (д, J=7,6 Гц, 1H), 8,23 (с, 1H), 7,80-7,78 (дд, J=7,6, 2,0 Гц, 1H), 7,46 (т, J=7,6 Гц, 1H), 7,30 (с, 1H), 7,26 (т, J=7,6 Гц, 1H), 6,88 (с, 1H), 3,45-3,38 (м, 1H), 3,02-2,98 (м, 1H), 2,32 (с, 3H), 1,77-1,63 (м, 4H), 1,18 (д, 6H), 1,12 (с, 6H).
Пример 3: Получение 5-хлор-N2-(2-(2-d-проп-2-илоксил)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамина

1. Получение 2-((2-d-проп-2-ил)тио)анилина (Соединение 15)

Под защитой азота соединение 2-аминотиофенол (0,5 г, 4 ммоль), 2-йод-2-d-пропан (0,75 г, 4,4 ммоль) и этанол (5 мл) последовательно добавляли в колбу. При перемешивании медленно добавляли трет-бутоксид калия (0,59 г, 5,2 ммоль) при 0°C. Осуществляли нагревание до комнатной температуры и выдерживали в течение 4 часов. Реакцию контролировали по существу в виде осуществления ВЭЖХ. Реакционную смесь фильтровали через целит, осадок на фильтре промывали этанолом и объединенный фильтрат концентрировали в вакууме посредством роторного испарителя. Остаток растворяли в этилацетате (20 мл), последовательно промывали водой и солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали посредством роторного испарителя в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле с получением желаемого продукта в виде желтоватого масла. МС расчетный: 168; МС обнаруженный: 169 (M+H)+.
Соединение 15 можно также получать следующими путями:

Под защитой азота соединение 2-амино-тиофенол (0,105 г, 0,84 ммоль), п-толуолсульфо-2-d-изопропиловый эфир (150 мг, 0,70 ммоль), карбонат цезия (0,681 г, 2,09 ммоль) и N,N-диметилформамид (5 мл) последовательно добавляли в колбу, нагревали до 60°C и выдерживали в течение 20 часов. Реакцию контролировали по существу в виде осуществления ВЭЖХ. В реакционную смесь добавляли чистую воду для остановки реакции. Затем реакционную смесь три раза экстрагировали этилацетатом. Органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали посредством роторного испарителя в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством хроматографии ТСХ с получением желаемого продукта в виде слегка желтоватого масла. 1H-ЯМР (CDCl3, 400 МГц) δ 7,37-7,35 (дд, J=7,6, 2,0 Гц, 1H), 7,12 (т, J=7,6 Гц, 1H), 6,74-6,72 (дд, J=7,6, 2,0 Гц, 1H), 6,67 (т, J=7,6 Гц, 1H), 4,42 (широкий, 2H), 1,23 (с, 6H).
2. Получение 2,5-дихлор-N-(2-((2-d-проп-2-ил)тио)фенил)пиримидин-4-амина
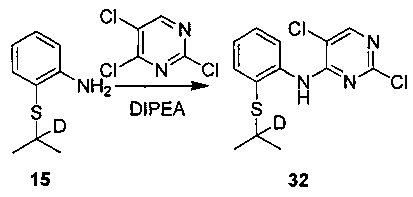
Соединение 2-((2-d-проп-2-ил)тио)анилин (137 мг, 0,81 ммоль), 2,4,5-трихлорпиримидин (299 мг, 1,63 ммоль), диизопропилэтиламин (157 мг, 1,22 ммоль) и н-бутанол (4 мл) последовательно добавляли в колбу, нагревали до 100°C и выдерживали в течение 20 часов. Реакционную смесь концентрировали и затем добавляли чистую воду. Полученную смесь три раза экстрагировали этилацетатом. Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали посредством роторного испарителя в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством ТСХ с получением желаемого продукта в виде бледно-желтого твердого вещества (128 мг, выход 50%). 1H-ЯМР (CDCl3, 400 МГц) δ 9,31 (s, 1H), 8,69-8,67 (дд, J=8,4, 1,2 Гц, 1H), 8,26 (с, 1H), 7,62-7,60 (дд, J=7,6, 1,6 Гц, 1H), 7,48 (т, J=8,8 Гц, 1H), 7,15-7,11 (т, J=8,8 Гц, 1H), 1,29 (с, 6H).
3. Получение 2,5-дихлор-N-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-4-амина

Соединение 2,5-дихлор-N-(2-((2-d-проп-2-ил)тио)фенил)пиримидин-4-амин (290 мг, 0,92 ммоль), 3-хлор-пероксибензойную кислоту (476 мг, 85%, 2,76 ммоль) и дихлорметан (6 мл) последовательно добавляли в колбу, нагревали до 40°C и выдерживали в течение 4 часов. Реакцию контролировали по существу в виде осуществления ВЭЖХ. В реакционную смесь добавляли 5% раствор бикарбоната натрия и затем добавляли чистую воду. Полученную смесь три раза экстрагировали дихлорметаном. Объединенный органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали посредством роторного испарителя в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством ТСХ с получением желаемого продукта в виде белого твердого вещества (307 мг, выход 96%). 1H-ЯМР (CDCl3, 400 МГц) δ 10,08 (с, 1H), 8,65 (д, J=7,6 Гц, 1H), 8,32 (с, 1H), 7,95-7,93 (дд, J=8,0, 1,6 Гц, 1H), 7,75 (т, J=8,4 Гц, 1H), 7,34 (т, J=8,0 Гц, 1H), 1,33 (с, 6H).
4. Получение 5-хлор-N2-(2-(2-d-проп-2-илокси)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамина
Под защитой азота соединение дигидрохлорид 2-((2-d-проп-2-ил)оксил)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)анилина (0,124 г, 0,43 ммоль), 2,5-дихлор-N-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-4-амин (0,164 г, 0,473 ммоль) и изопропанол (3 мл) добавляли в колбу, нагревали до 85°C и выдерживали в течение 20 ч. После охлаждения до комнатной температуры, в реакционную смесь добавляли чистую воду и водный раствор карбоната натрия добавляли для нейтрализации pH до 8,5. Полученную смесь три раза экстрагировали этилацетатом. Органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ТСХ (проявляющий растворитель: метанол / дихлорметан = 1/9, об./об.) с получением желаемого продукта в виде белого твердого вещества (0,070 г, выход: 30%). МС расчетный: 563; МС обнаруженный: 564 (M+H)+, 586 (M+Na)+. 1H-ЯМР (ДМСО-d6+D2O, 400 МГц) δ 8,50 (д, J=7,6 Гц, 1H), 8,22 (с, 1H), 7,80-7,78 (дд, J=7,6, 2,0 Гц, 1H), 7,46 (т, J=7,6 Гц, 1H), 7,30 (с, 1H), 7,26 (т, J=7,6 Гц, 1H), 6,89 (с, 1H), 3,00-2,98 (м, 1H), 2,34 (с, 3H), 1,77-1,63 (м, 4H), 1,23 (с, 6H), 1,12 (с, 6H).
Пример 4: Получение 5-хлор-N2-(2-изопропокси-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамина

1. Получение 1-бензил-2,2,6,6-d4-4-(5-изопропокси-2-метил-4-нитрофенил)-1,2,3,6-тетрагидропиридина
Под защитой азота соединение 1-бром-5-изопропокси-2-метил-4-нитробензол (0,30 г, 1,09 ммоль), 1-бензил-2,2,6,6-d4-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,6-тетрагидропиридин (0,43 г, 1,42 ммоль), ацетат палладия (22 мг, 0,098 ммоль), 4,5-бис-дифенилфосфин-9,9-диметил-ксантен (45 мг, 0,076 ммоль), фосфат калия тригидрат (0,74 г, 3,27 ммоль) и тетрагидрофуран (6 мл) добавляли в колбу, и затем нагревали до 85°C, и выдерживали всю ночь. В реакционную смесь добавляли чистую воду для остановки реакции и полученную смесь экстрагировали этилацетатом. Объединенную органическую фазу последовательно промывали водой и солевым раствором, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ТСХ с получением желаемого продукта (0,30 г, выход: 75%). МС расчетный: 370; МС обнаруженный: 371 (M+H)+.
2. Получение дигидрохлорида 2-изопропокси-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)анилина

В колбу добавляли соединение 1-бензил-2,2,6,6-d4-4-(5-изопропокси-2-метил-4-нитрофенил)-1,2,3,6-тетрагидропиридин (0,30 г, 0,81 ммоль), палладий на угле (0,03 г, содержание палладия 50%) и метанол (10 мл). В системе три раза заменяли водород, нагревали до 45°C и выдерживали в течение 40 часов. Палладий на угле удаляли путем фильтрации; фильтрат концентрировали с получением серого твердого вещества. Полученное серое твердое вещество и раствор соляной кислоты в изопропаноле (2 М, 2 мл) последовательно добавляли в колбу и перемешивали в течение 2 ч. Реакционный раствор концентрировали при пониженном давлении с получением желаемого продукта в виде белого твердого вещества (0,26 г, выход: 94%).
3. Получение 5-хлор-N2-(2-изопропокси-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамина

Под защитой азота соединение дигидрохлорид 2-изопропокси-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)анилина (0,17 г, 0,52 ммоль), 2,5-дихлор-N-(2-((2-d-проп-2-ил))сульфонил)фенил)пиримидин-4-амин (0,20 г, 0,57 ммоль) и изопропанол (5 мл) добавляли в колбу, нагревали до 85°C и выдерживали всю ночь. После охлаждения до комнатной температуры в реакционную смесь добавляли воду для остановки реакции и водный раствор карбоната натрия добавляли для нейтрализации pH до 8,5. Полученную смесь экстрагировали этилацетатом. Органический слой объединяли, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством ТСХ (проявляющий растворитель: метанол / дихлорметан = 1/9, об./об.) с получением желаемого продукта в виде белого твердого вещества (0,10 г, выход: 35%). МС расчетный: 562; МС обнаруженный: 563 (M+H)+, 585 (M+Na)+. 1H-ЯМР (ДМСО-d6+D2O, 400 МГц) δ 8,50 (д, J=7,6 Гц, 1H), 8,22 (с, 1H), 7,80-7,78 (дд, J=7,6, 2,0 Гц, 1H), 7,46 (т, J=7,6 Гц, 1H), 7,30 (с, 1H), 7,26 (т, J=7,6 Гц, 1H), 6,88 (с, 1H), 4,52-4,48 (м, 1H), 3,00-2,97 (м, 1H), 2,32 (с, 3H), 1,77-1,63 (м, 4H), 1,22 (с, 6H), 1,12 (д, 6H).
Пример 5: Получение 5-хлор-N2-(2-(2-d-проп-2-илокси)-5-(d3-метил)-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамина

Эксперимент проводили согласно способу примера 1, за исключением того, что для замещения 1-бром-5-изопропокси-2-метил-4-нитробензола (соединение 5) и 2-(изопропилтио)анилина (соединение 9) соответственно использовали 1-бром-5-(2-d-проп-2-илоксил)-2-метил-4-нитробензол (соединение 14) и 2-((2-d-проп-2-ил)тио)анилин (соединение 15) и 1-бром-5-фтор-2-(d3-метил)-4-нитробензол использовали вместо 1-бром-5-фтор-2-метил-4-нитробензола (соединение 4) с получением целевого соединения.
Пример 6: Получение 5-хлор-N2-(2-изопропокси-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамина

1. Получение трет-бутил-4-(5-изопропокси-2-метил-4-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилата
Под защитой азота соединение 1-хлор-5-изопропокси-2-метил-4-нитробензол (0,245 г, 1,07 ммоль), пинаколовый эфир 3,6-дигидро-2H-пиридин-1-трет-бутоксикарбонил-бороновой кислоты (0,495 г, 1,60 ммоль), ацетат палладия (22 мг, 0,096 ммоль), 4,5-бисдифенилфосфин-9,9-диметил-ксантен (43 мг, 0,075 ммоль), фосфат калия тригидрат (0,73 г, 3,20 ммоль) и тетрагидрофуран (10 мл) добавляли в колбу и затем нагревали до 85°C и выдерживали всю ночь. В реакционную смесь добавляли чистую воду для остановки реакции и ее три раза экстрагировали этилацетатом. Объединенную органическую фазу последовательно промывали водой и солевым раствором, сушили над водным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством ТСХ с получением желаемого продукта в виде бесцветной прозрачной жидкости (0,29 г, выход 73%). 1H-ЯМР (CDCl3, 400 МГц) δ 7,63 (с, 1H), 6,80 (с, 1H), 5,62 (м, 1H), 4,65-4,62 (м, 1H), 4,08 (м, 2H), 3,65 (т, 2H), 2,34 (м, 2H), 2,25 (с, 3H), 1,52 (с, 9H), 1,39 (м, 6H).
2. Получение дигидрохлорида 2-изопропокси-5-метил-4-(пиперидин-4-ил)анилина

В колбу добавляли соединение трет-бутил 4-(5-изопропокси-2-метил-4-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилат (0,29 г, 0,77 ммоль), палладий на угле (0,03 г, содержание палладия 50%) и метанол (10 мл). В реакционной системе три раза замещали водород, ее нагревали до 45°C и выдерживали всю ночь. Палладий на угле удаляли посредством фильтрации; и фильтрат концентрировали с получением розового твердого вещества. 1H-ЯМР (CD3OD, 400 МГц) δ 6,66 (с, 1H), 6,61 (с, 1H), 4,51-4,48 (м, 1H), 4,23-4,20 (м, 2H), 2,87-2,79 (м, 3H), 2,22 (с, 3H), 1,73-1,70 (м, 2H), 1,54-1,47 (м, 11H), 1,32 (д, 6H).
Розовое твердое вещество и раствор гидрохлорида в изопропаноле (2 М, 2 мл) последовательно добавляли в колбу, нагревали до 55°C и выдерживали в течение 2 ч. Реакцию контролировали в виде осуществления ВЭЖХ. Реакционный раствор концентрировали при пониженном давлении с получением желаемого продукта в виде белого твердого вещества (0,23 г, выход: 94%).
3. Получение 5-хлор-N2-(2-изопропокси-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамина

Под защитой азота соединение дигидрохлорид 2-изопропокси-5-метил-4-(пиперидин-4-ил)анилина (0,10 г, 0,31 ммоль), 2,5-дихлор-N-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-4-амин (0,12 г, 0,34 ммоль) и изопропанол (3 мл) добавляли в колбу, нагревали до 85°C и выдерживали всю ночь. После охлаждения до комнатной температуры в реакционную смесь добавляли чистую воду для остановки реакции и водный раствор карбоната натрия добавляли для нейтрализации pH to 8,5. Полученную смесь экстрагировали этилацетатом. Органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством ТСХ (проявляющий растворитель: метанол / дихлорметан = 1/9, об./об.) с получением желаемого продукта в виде белого твердого вещества (0,054 г, выход: 31%). МС расчетный: 558; МС обнаруженный: 559 (M+H)+, 581 (M+Na)+. 1H-ЯМР (ДМСО-d6+D2O, 400 МГц) δ 8,48 (д, J=7,6 Гц, 1H), 8,20 (с, 1H), 7,79-7,77 (дд, J=7,6, 2,0 Гц, 1H), 7,46 (т, J=7,6 Гц, 1H), 7,29 (с, 1H), 7,26 (т, J=7,6 Гц, 1H), 6,86 (с, 1H), 4,52-4,48 (м, 1H), 3,30-3,28 (м, 2H), 3,00-2,91 (м, 3H), 2,30 (с, 3H), 1,77-1,63 (м, 4H), 1,22 (с, 6H), 1,12 (д, 6H).
Пример 7: 5-хлор-N2-(2-изопропокси-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-((d7-изопропил)сульфонил)фенил)пиримидин-2,4-диамин

1. Получение d7-изопропил-4-метилбензолсульфоната

Соединение d8-изопропанол (1,0 г, 27,74 ммоль), триэтиламин (8,4 г, 83,22 ммоль) и дихлорметан (150 мл) добавляли в колбу, нагревали до 40°C и выдерживали в течение двух суток. В реакционную жидкость добавляли воду и три раза экстрагировали дихлорметаном. Объединенную органическую фазу последовательно промывали водным карбонатом натрия и водой, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством колоночной хроматографии с получением желаемого продукта в виде белого твердого вещества.
2. Получение 2-(d7-изопропилтио)анилина

Синтез проводили в соответствии со стадией 1 примера 3 и слегка желтоватый масляный целевой продукт получали путем замещения п-толуолсульфо-2-d-изопропила п-толуолсульфо-d7-изопропилом.
3. Получение 2,5-дихлор-N-(2-(d7-изопропилтио)фенил)пиримидин-4-амина

Синтез проводили в соответствии со стадией 2 примера 3 и слегка желтоватый масляный целевой продукт получали путем замещения 2-((2-d-проп-2-ил)тио)анилина 2-(d7-изопропилтио)анилином.
4. Получение 2,5-дихлор-N-(2-(d7-изопропилсульфонил)фенил)пиримидин-4-амина

Соединение 2,5-дихлор-N-(2-(d7-изопропилтио)фенил)пиримидин-4-амин (200 мг, 0,62 ммоль), 3-хлор-пероксибензойную кислоту (379 мг, 85%, 1,87 ммоль) и метиленхлорид (4 мл) последовательно добавляли в колбу, нагревали до 40°C и выдерживали в течение 6 часов. Реакцию контролировали по существу в виде проведения ВЭЖХ. В реакционную смесь добавляли 5% раствор карбонат натрия для нейтрализации pH до нейтрального pH и затем добавляли чистую воду. Полученную смесь экстрагировали метиленхлоридом. Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали посредством роторного испарителя в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ТСХ с получением желаемого продукта в виде белого твердого вещества (197 мг, выход 90%). 1H-ЯМР (CDCl3, 400 МГц) δ 10,05 (с, 1H), 8,65-8,62 (дд, J=8,4, 0,8 Гц, 1H), 8,30 (с, 1H), 7,96-7,93 (дд, J=8,0, 1,6 Гц, 1H), 7,73 (т, J=8,4 Гц, 1H), 7,34 (т, J=8,4 Гц, 1H).
5. 5-хлор-N2-(2-изопропокси-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-((d7-изопропилсульфонил)фенил)пиримидин-2,4-диамин

Под защитой азота соединение дигидрохлорид 2-изопропокси-5-метил-4-(пиперидин-4-ил)анилина (0,10 г, 0,31 ммоль), 2,5-дихлор-N-(2-(d7-изопропилсульфонил)фенил)пиримидин-4-амин (0,12 г, 0,34 ммоль) и изопропанол (3 мл) добавляли в колбу, нагревали до 85°C и выдерживали всю ночь. После охлаждения до комнатной температуры для остановки реакции в реакционную смесь добавляли чистую воду и для нейтрализации pH до 8,5 водный добавляли раствор карбоната натрия. Полученную смесь экстрагировали этилацетатом. Органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством ТСХ (проявляющий растворитель: метанол / дихлорметан = 1/9, об./об.) с получением желаемого продукта в виде белого твердого вещества (0,063 г, выход: 36%). МС расчетный: 564; МС обнаруженный: 565 (M+H)+, 587 (M+Na)+.
8. 5-хлор-N2-((2-d-проп-2-илокси)-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин

1. Получение трет-бутил 4-(5-(2-d-проп-2-ил)окси-2-метил-4-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилата

Под защитой азота соединение 1-бром-5-(2-d-проп-2-ил)окси-2-метил-4-нитробензол (0,30 г, 1,09 ммоль), пинаколовый эфир 3,6-дигидро-2H-пиридин-1-трет-бутоксикарбонил-бороновой кислоты (0,51 г, 1,64 ммоль), ацетат палладия (22 мг, 0,098 ммоль), 4,5-бисдифенилфосфин-9,9-диметилксантен (44 мг, 0,076 ммоль), фосфат калия тригидрат (0,74 г, 3,27 ммоль) и тетрагидрофуран (10 мл) добавляли в колбу и затем нагревали до 85°C и выдерживали в течение 20 ч. В реакционную смесь добавляли воду и три раза экстрагировали этилацетатом. Объединенную органическую фазу последовательно промывали водой и солевым раствором, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали ТСХ с получением желаемого продукта в виде бесцветной прозрачной жидкости (0,30 г, выход 73%). 1H-ЯМР (CDCl3, 400 МГц) δ, 7,60 (с, 1H), 6,88 (с, 1H), 5,62 (м, 1H), 4,05 (м, 2H), 3,63 (т, 2H), 2,33 (м, 5H), 1,52 (с, 9H), 1,41 (с, 6H).
2. Получение дигидрохлорида 2-((2-d-проп-2-ил)оксил)-5-метил-4-(пиперидин-4-ил)анилина
В колбу добавляли соединение трет-бутил 4-(5-(2-d-проп-2-ил)окси-2-метил-4-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилат (0,130 г, 0,34 ммоль), палладий на угле (0,013 г, содержание палладия 50%) и метанол (10 мл). В реакционной системе три раза заменяли водород, ее нагревали до 45°C и выдерживали в течение 20 ч. Палладий на угле удаляли путем фильтрации; фильтрат концентрировали с получением розового твердого вещества.
Розовый твердый осадок и раствор гидрохлорида в изопропаноле (2 М, 2 мл) последовательно добавляли в колбу, нагревали до 55°C и выдерживали в течение 2 ч. Реакцию контролировали по существу в виде проведения ВЭЖХ. Реакционный раствор концентрировали при пониженном давлении с получением желаемого осадка в виде белого твердого вещества (0,106 г, выход: 96%). 1H-ЯМР (ДМСО-d6, 400 МГц) δ 9,75 (широкий, 3H), 8,99 (широкий, 2H), 7,20 (с, 1H), 7,03 (с, 1H), 3,36-3,33 (м, 2H), 3,03-2,98 (м, 3H), 2,32 (с, 3H), 1,80-1,70 (м, 4H), 1,30 (с, 6H).
3. 5-хлор-N2-((2-d-проп-2-илокси)-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин

Под защитой азота соединение дигидрохлорид 2-((2-d-проп-2-ил)оксил)-5-метил-4-(пиперидин-4-ил)анилина (0,107 г, 0,43 ммоль), 2,5-дихлор-N-(2-(изопропилсульфонил)фенил)пиримидин-4-амин (0,164 г, 0,473 ммоль) и изопропанол (2,5 мл) добавляли в колбу, нагревали до 85°C и выдерживали 20 ч. После охлаждения до комнатной температуры в реакционную смесь добавляли воду и водный раствор карбоната натрия добавляли для нейтрализации pH до 8,5. Полученную смесь три раза экстрагировали этилацетатом. Органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством ТСХ (проявляющий растворитель: метанол / дихлорметан = 1/9, об./об.) с получением желаемого продукта в виде белого твердого вещества (0,07 г, выход: 30%). МС расчетный: 558; МС обнаруженный: 559 (M+H)+, 581 (M+Na)+. 1H-ЯМР (ДМСО-d6+D2O, 400 МГц) δ 8,47 (д, J=7,6 Гц, 1H), 8,19 (с, 1H), 7,79-7,77 (дд, J=7,6, 2,0 Гц, 1H), 7,45 (т, J=7,6 Гц, 1H), 7,29 (с, 1H), 7,27 (т, J=7,6 Гц, 1H), 6,89 (с, 1H), 3,45-3,38 (м, 1H), 3,31-3,29 (м, 2H), 3,02-2,92 (м, 3H), 2,33 (с, 3H), 1,79-1,64 (м, 4H), 1,16 (д, 6H), 1,12 (с, 6H).
Пример 9: 5-хлор-N2-(2-(d7-изопропокси)-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин

1. Получение 1-бром-5-(d7-изопропокси)-2-метил-4-нитробензол

Синтез проводили в соответствии со стадией 1 примера 2 и посредством замещения п-толуолсульфо-2-d-изопропила п-толуолсульфо-d7-изопропилом получали слегка желтоватый масляный целевой продукт.
2. Получение трет-бутил-4-(5-(d7-изопропокси)-2-метил-4-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилата
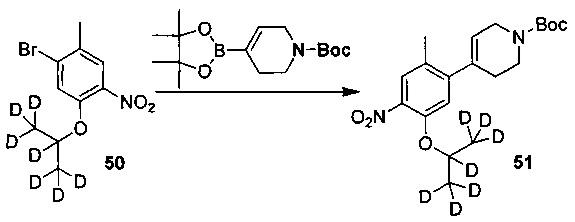
Под защитой азота соединение 1-бром-5-(d7-изопропокси)-2-метил-4-нитробензол (0,30 г, 1,07 ммоль), пинаколовый эфир 3,6-дигидро-2H-пиридин-1-трет-бутоксикарбонил-бороновой кислоты (0,495 г, 1,60 ммоль), ацетат палладия (22 мг, 0,096 ммоль), 4,5-бисдифенилфосфин-9,9-диметил-ксантен (43 мг, 0,075 ммоль), фосфат калия тригидрат (0,73 г, 3,20 ммоль) и тетрагидрофуран (10 мл) добавляли в колбу, и затем нагревали до 85°C, и выдерживали всю ночь. В реакционную смесь для остановки добавляли чистую воду и три раза экстрагировали этилацетатом. Объединенную органическую фазу последовательно промывали водой и солевым раствором, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт отделяли и очищали посредством ТСХ с получением желаемого продукта в виде бесцветной прозрачной жидкости (0,29 г, выход 70%).
3. Получение дигидрохлорида 2-(d7-изопропокси)-5-метил-4-(пиперидин-4-ил)анилина

Соединение трет-бутил 4-(5-(d7-изопропоксил)-2-метил-4-нитрофенил)-5,6-дигидропиридин-1(2H)-карбоксилат (0,28 г, 0,73 ммоль), палладий на угле (0,028 г, содержание палладия 50%) и метанол (10 мл) добавляли в колбу, и в системе три раза заменяли водород, нагревали до 45°C, и выдерживали всю ночь. Палладий на угле удаляли путем фильтрации; и фильтрат концентрировали с получением розового твердого вещества.
Розовое твердое вещество и раствор гидрохлорида в изопропаноле (2М, 2 мл) последовательно добавляли в колбу, нагревали до 55°C и выдерживали в течение 2 ч. Реакцию контролировали в виде осуществления ВЭЖХ. Реакционный раствор концентрировали при пониженном давлении с получением желаемого продукта в виде белого твердого вещества (0,24 г, выход: 98%).
4. 5-хлор-N2-(2-(d7-изопропокси)-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин
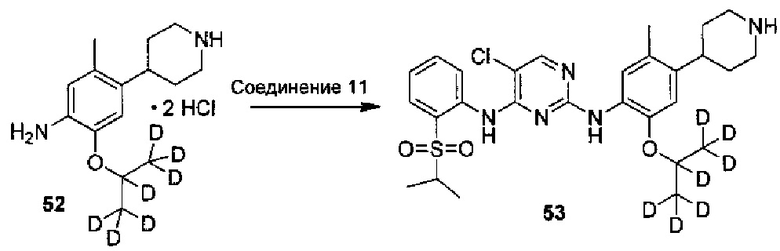
Под защитой азота соединение дигидрохлорид 2-(d7-изопропоксил)-5-метил-4-(пиперидин-4-ил)анилина (0,12 г, 0,37 ммоль), 2,5-дихлор-N-(2-(изопропилсульфонил)фенил)пиримидин-4-амин (0,14 г, 0,40 ммоль) и изопропанол (3 мл) добавляли в колбу, нагревали до 85°C и выдерживали всю ночь. После охлаждения до комнатной температуры в реакционную смесь добавляли воду и для нейтрализации pH до 8,5 добавляли водный раствор карбоната натрия. Полученную смесь экстрагировали этилацетатом. Органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ТСХ (проявляющий раствор: метанол / дихлорметан = 1/9, об./об.) с получением желаемого продукта в виде белого твердого вещества. МС расчетный: 564; МС обнаруженный: 565 (M+H)+, 587 (M+Na)+.
Пример 10: Получение 5-хлор-N2-((2-d-проп-2-илокси)-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамина

Под защитой азота соединение дигидрохлорид 2-((2-d-проп-2-ил)оксил)-5-метил-4-(пиперидин-4-ил)анилина (0,055 г, 0,17 ммоль), 2,5-дихлор-N-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-4-амин (0,066 г, 0,19 ммоль) и изопропанол (3 мл) добавляли в колбу, нагревали до 85°C и выдерживали всю ночь. После охлаждения до комнатной температуры в реакционную смесь добавляли воду и для нейтрализации pH до 8,5 добавляли водный раствор карбоната натрия. Полученную смесь экстрагировали этилацетатом. Органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ТСХ (проявляющий растворитель: метанол / дихлорметан = 1/9, об./об.) с получением желаемого продукта в виде белого твердого вещества (39 мг, выход: 40%). МС расчетный: 559; МС обнаруженный: 560 (M+H)+, 582 (M+Na)+. 1H-ЯМР (ДМСО-d6+D2O, 400 МГц) δ 8,49 (д, J=8,0 Гц, 1H), 8,19 (с, 1H), 7,79-7,77 (дд, J=8,0, 1,6 Гц, 1H), 7,45 (м, 1H), 7,29-7,25 (м, 2H), 6,89 (с, 1H), 3,32-3,29 (м, 2H), 3,03-2,93 (м, 3H), 2,33 (с, 3H), 1,79-1,65 (м, 4H), 1,16 (с, 6H), 1,12 (с, 6H).
Пример 11: 5-хлор-N2-(d7-изопропилокси-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-((d7-изопропил)сульфонил)фенил)пиримидин-2,4-диамин
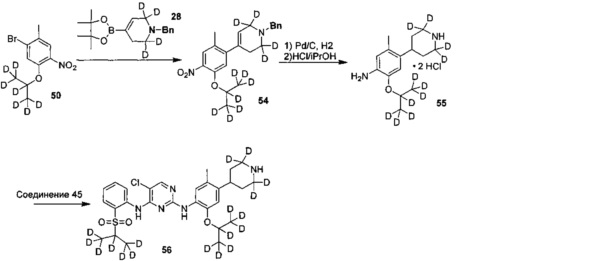
1. Получение 1-бензил-2,2,6,6-d4-4-(5-(d7-изопропоксил)-2-метил-4-нитрофенил)-1,2,3,6-тетрагидропиридина

Под защитой азота соединение 1-бром-5-(d7-изопропокси)-2-метил-4-нитробензол (0,24 г, 0,71 ммоль), 1-бензил-2,2,6,6-d4-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,6-тетрагидропиридин (0,28 г, 0,93 ммоль), ацетат палладия (15 мг, 0,064 ммоль), 4,5-бис-дифенилфосфин-9,9-диметил-ксантен (29 мг, 0,050 ммоль), фосфат калия тригидрат (0,48 г, 2,13 ммоль) и тетрагидрофуран (8 мл) добавляли в колбу, и затем нагревали до 85°C, и выдерживали всю ночь. Для остановки реакции в реакционную смесь добавляли чистую воду и экстрагировали этилацетатом. Объединенную органическую фазу последовательно промывали водой и солевым раствором, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ТСХ (тонкослойная хроматография) с получением желаемого продукта (0,20 г, выход: 75%). МС расчетный: 377; МС обнаруженный: 378 (M+H)+.
2. Получение дигидрохлорида 2-(d7-изопропоксил)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)анилина
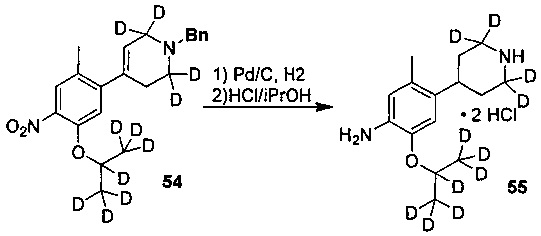
В колбу добавляли соединение 1-бензил-2,2,6,6-d4-4-(5-(d7-изопропоксил)-2-метил-4-нитрофенил)-1,2,3,6-тетрагидропиридин (0,38 г, 1,01 ммоль), палладий на угле (0,038 г, содержание палладия 50%) и метанол (10 мл). В системе три раза заменяли водород, нагревали до 45°C и выдерживали в течение 40 часов. Палладий на угле удаляли путем фильтрования и фильтрат концентрировали с получением серого твердого вещества. Полученное серое твердое вещество и раствор соляной кислоты в изопропаноле (2 М, 2 мл) последовательно добавляли в колбу и перемешивали в течение 2 ч. Реакционный раствор концентрировали при пониженном давлении с получением желаемого продукта в виде белого твердого вещества (0,32 г, выход: 95%).
3. 5-хлор-N2-(d7-изопропилокси)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)фенил)-N4-(2-((d7-изопропил)сульфонил)фенил)пиримидин-2,4-диамин;
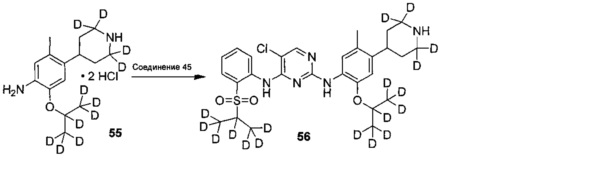
Под защитой азота соединение дигидрохлорид 2-(d7-изопропоксил)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)анилина (0,09 г, 0,27 ммоль), 2,5-дихлор-N-(2-(d7-изопропилсульфонил)фенил)пиримидин-4-амин (0,11 г, 0,30 ммоль) и изопропанол (3 мл) добавляли в колбу, нагревали до 85°C и выдерживали всю ночь. После охлаждения до комнатной температуры в реакционную смесь добавляли воду и водный раствор карбоната натрия добавляли для нейтрализации pH 8,5. Полученную смесь экстрагировали этилацетатом. Органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ТСХ (проявляющий растворитель: метанол / дихлорметан = 1/9, об./об.) с получением желаемого продукта в виде белого твердого вещества (54 мг, выход: 35%).
Пример 12: 5-хлор-N2-((2-d-проп-2-илокси)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)-6-d-фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамин

1. Получение дигидрохлорида 2-((2-d-проп-2-ил)оксил)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)-6-d-анилина
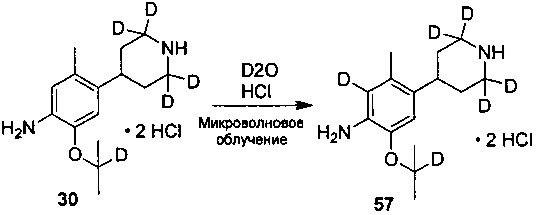
Дигидрохлорид 2-((2-d-проп-2-ил)оксил)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)анилина (0,42 г, 1,29 ммоль) и тяжелую воду (2 мл) последовательно добавляли в пробирку для реакции, проводимой в условиях микроволнового облучения, и реакционную пробирку плотно закрывали и нагревали в условиях микроволнового облучения до 180°C в течение 40 минут. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт добавляли к раствору хлороводорода в изопропаноле для превращения в массу так, чтобы получить желаемый продукт в виде белого твердого вещества (0,28 г, 67%).
2. 5-хлор-N2-((2-d-проп-2-илокси)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)-6-d-фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамин

Под защитой азота соединение дигидрохлорид 2-((2-d-проп-2-ил)оксил)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)-6-d-анилина (0,087 г, 0,30 ммоль), 2,5-дихлор-N-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-4-амин (0,12 г, 0,33 ммоль) и изопропанол (3 мл) добавляли в колбу, нагревали до 85°C и выдерживали всю ночь. После охлаждения до комнатной температуры в реакционную смесь добавляли чистую воду и для нейтрализации pH до 8,5 добавляли водный раствор карбоната натрия. Полученную смесь три раза экстрагировали этилацетатом. Органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ТСХ (проявляющий растворитель: метанол / дихлорметан = 1/9, об./об.) с получением желаемого продукта в виде белого твердого вещества (0,05 г, выход: 30%). МС расчетный: 564; МС обнаруженный: 565 (M+H)+, 587 (M+Na)+. 1H-ЯМР (ДМСО-d6+D2O, 400 МГц) δ 8,50 (д, J=7,6 Гц, 1H), 8,22 (с, 1H), 7,80-7,78 (дд, J=7,6, 2,0 Гц, 1H), 7,46 (т, J=7,6 Гц, 1H), 7,30 (с, 1H), 7,26 (т, J=7,6 Гц, 1H), 3,00-2,98 (м, 1H), 2,34 (с, 3H), 1,77-1,65 (м, 4H), 1,23 (с, 6H), 1,12 (с, 6H).
Пример 13: 5-хлор-N2-((2-d-проп-2-илоксил)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)-6-d-фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин

Под защитой азота соединение дигидрохлорид 2-((2-d-проп-2-ил)оксил)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)-6-d-анилина (0,087 г, 0,30 ммоль), 2,5-дихлор-N-(2-(изопропилсульфонил)фенил)пиримидин-4-амин (0,12 г, 0,33 ммоль) и изопропанол (3 мл) добавляли в колбу, нагревали до 85°C и выдерживали всю ночь. После охлаждения до комнатной температуры в реакционную смесь добавляли чистую воду и для нейтрализации pH 8,5 добавляли водный раствор карбоната натрия. Полученную смесь три раза экстрагировали этилацетатом. Органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ТСХ (проявляющий растворитель: метанол / метиленхлорид = 1/9, об./об.) с получением желаемого продукта в виде белого твердого вещества (0,06 г, выход: 35%). МС расчетный: 563; МС обнаруженный: 564 (M+H)+, 586 (M+Na)+.
Пример 14: 5-хлор-N2-(2-изопропокси-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)-6-d-фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамин
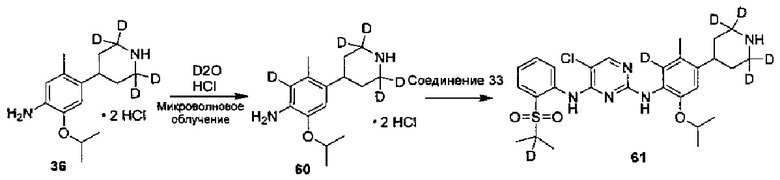
1. Получение дигидрохлорида 2-изопропокси-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)-6-d-анилина

Дигидрохлорид 2-изопропокси-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)анилина (0,21 г, 0,65 ммоль) и тяжелую воду (2 мл) последовательно добавляли в пробирку для реакции, проводимой в условиях микроволнового облучения, и реакционную пробирку плотно закрывали и нагревали в условиях микроволнового облучения до 180°C в течение 40 минут. Реакционную смесь концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт добавляли к раствору хлороводорода в изопропаноле для превращения в массу так, чтобы получить желаемый продукт в виде белого твердого вещества (0,15 г, 70%).
2. Получение 5-хлор-N2-(2-изопропокси)-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)-6-d-фенил)-N4-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-2,4-диамина
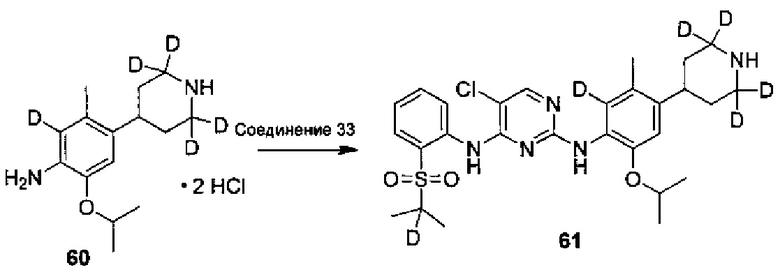
Под защитой азота соединение дигидрохлорид 2-изопропокси-5-метил-4-(2,2,6,6-d4-пиперидин-4-ил)-6-d-анилина (0,070 г, 0,21 ммоль), 2,5-дихлор-N-(2-((2-d-проп-2-ил)сульфонил)фенил)пиримидин-4-амин (0,083 г, 0,24 ммоль) и изопропанол (3 мл) добавляли в колбу, нагревали до 85°C и выдерживали всю ночь. После охлаждения до комнатной температуры в реакционную смесь добавляли воду и водный раствор карбоната натрия добавляли для нейтрализации pH до 8,5. Полученную смесь экстрагировали этилацетатом. Органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ТСХ (проявляющий растворитель: метанол / метиленхлорид = 1/9, об./об.) с получением желаемого продукта в виде белого твердого вещества (0,045 г, выход: 38%). МС расчетный: 563; МС обнаруженный: 564 (M+H)+, 586 (M+Na)+.
Пример 15: 5-хлор-N2-((2-d-проп-2-илокси)-5-метил-4-(d9-пиперидин-4-ил)-фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин
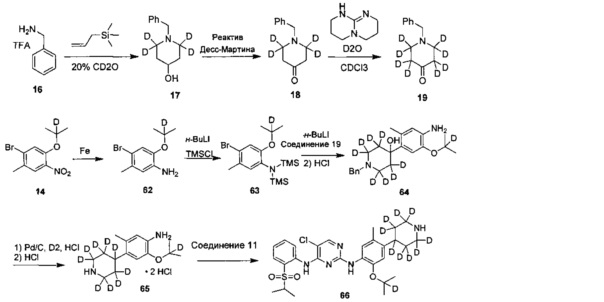
1. Получение 4-бром-2-((2-d-проп-2-ил)оксил)-5-метиланилин-1-бензил-2,2,6,6-d4-пиперидин-4-ола

Желаемый продукт получали согласно Journal of Labelled Compounds and Radiopharmaceuticals 2007, 50, 131-137.
2. Получение 1-бензил-2,2,6,6-d4-пиперидин-4-она
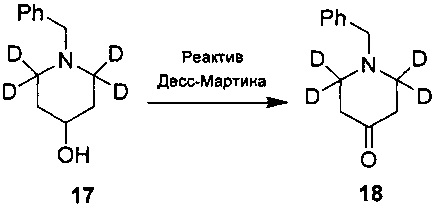
4-бром-2-((2-d-проп-2-ил)оксил)-5-метиланилин-1-бензил-2,2,6,6-d4-пиперидин-4-ол (1,0 г, 5,12 ммоль) и метиленхлорид (10 мл) добавляли в реакционную пробирку и охлаждали до 5°C. Порциями добавляли окислитель Десс-Мартина (2,61 г, 6,14 ммоль). Сразу после добавления полученную смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. Реакцию контролировали по существу в виде выполнения ВЭЖХ. Реакцию останавливали тиосульфатом натрия. Затем ее экстрагировали этилацетатом. Органические слои объединяли и последовательно промывали водой и солевым раствором и сушили над безводным сульфатом натрия. Неочищенный продукт получали путем концентрирования при пониженном давлении и очищали посредством препаративной ТСХ с получением желаемого продукта (614 мг, выход: 62%).
3. Получение 1-бензил-2,2,3,3,5,5,6,6-d8-пиперидин-4-она
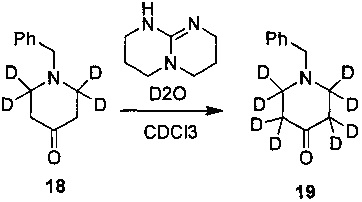
1-Бензил-2,2,6,6-d4-пиперидин-4-он (0,26 г) растворяли в дейтерохлороформе (CDCl3, 5 мл 99,9% D) и тяжелой воде (5 мл, 99,9% D) и добавляли 1,5,7-триазабицикло[4.4.0]дец-5-ен. Полученную смесь плотно закрывали и перемешивали при комнатной температуре всю ночь. Реакционную смесь контролировали с помощью H-ЯМР до тех пор, пока пик водорода в орто-положении к карбонилу не становился невидимым. Для остановки реакции добавляли разбавленную соляную кислоту. Полученную смесь экстрагировали этилацетатом и сушили над безводным сульфатом натрия. Неочищенный продукт получали в результате концентрирования при пониженном давлении и очищали посредством препаративной ТСХ с получением желаемого продукта. 1H-ЯМР (CDCl3, 400 МГц) δ 7,39-7,31 (m, 5H), 3,61 (s, 2H).
4. Получение 4-бром-2-((2-d-проп-2-ил)оксил)-5-митиланилина

Под защитой азота соединения 1-бром-5-(2-d-проп-2-илоксил)-2-метил-4-нитробензол (100 мг, 0,36 ммоль), опилки восстановленного железа (306 мг, 5,5 ммоль) и уксусную кислоту (2 мл) добавляли в колбу и перемешивали всю ночь при комнатной температуре. Воду и этилацетат последовательно добавляли в реакционную смесь, водную фазу отделяли и экстрагировали этилацетатом, объединенную органическую фазу промывали раствором бикарбоната натрия, сушили над безводным сульфатом магния, фильтровали и фильтрат концентрировали в вакууме посредством роторного испарителя с получением неочищенного продукта и очищали посредством препаративной хроматографии на пластинах ТСХ (проявляющий растворитель: петролейный эфир / этилацетат = 1/10, об./об.) с получением желаемого продукта в виде коричневого твердого вещества (70 мг, выход 78%). 1H-ЯМР (ДМСО-d6, 400 МГц) δ 6,82 (с, 1H), 6,78 (с, 1H), 4,70 (с, 2H), 2,17 (с, 3H), 1,26 (с, 6H).
5. Получение 4-(4-амино-5-((2-d-проп-2-ил)оксил)-2-метилфенил)-1-бензил-(d8-пиперидин)-4-ола
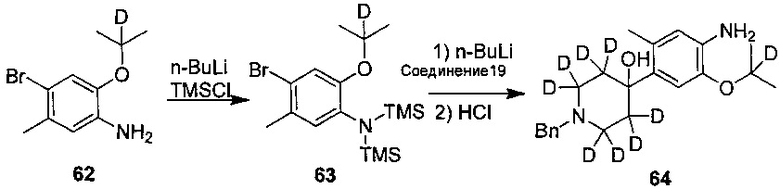
Под защитой азота соединение 4-бром-2-((2-d-проп-2-ил)оксил)-5-метиланилин (0,37 г, 1,5 ммоль) и тетрагидрофуран (5 мл) добавляли в колбу и охлаждали до температуры ниже -78°C.Затем добавляли н-бутиллитий в н-гексане (1,6 М, 0,94 мл, 1,5 ммоль) и перемешивали в течение 5 минут. Температуру держали на уровне ниже -70°C, и по каплям добавляли триметилхлорсилан (0,16 г, 1,5 ммоль), и перемешивали в течение 20 минут. Температуру поддерживали на уровне ниже -70°C и добавляли н-бутиллитий в н-гексане (0,94 М, 3,12 мл, 1,5 ммоль) и триметилхлорсилане (0,18 г, 1,7 ммоль). Сразу после добавления температуру постепенно повышали до -5°C. Добавляли н-гексан и водный бикарбонат натрия и водный слой отделяли и экстрагировали н-гексаном. Органические слои объединяли, последовательно промывали водой и солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного соединения 63, которое использовали на следующей стадии реакции без очистки.
Под защитой азота соединение 63 в виде неочищенного продукта растворяли в безводном тетрагидрофуране (3 мл) и охлаждали до температуры ниже -78°C. Добавляли н-бутиллитий в н-гексане (1,6 М, 0,94 мл, 1,5 ммоль) и перемешивали в течение 60 минут. Температуру поддерживали на уровне ниже -70°C и по каплям добавляли 1-бензил-2,2,3,3,5,5,6,6-d8-пиперидин-4-он (0,25 г, 1,3 ммоль) в тетрагидрофуране (1 мл). Сразу после добавления полученную смесь медленно нагревали до комнатной температуры и перемешивали всю ночь. Добавляли концентрированную соляную кислоту (0,5 мл) и перемешивали всю ночь. Полученную смесь нейтрализовали насыщенным раствором бикарбоната натрия до нейтральной реакции и затем экстрагировали этилацетатом. Органические слои объединяли, последовательно промывали бикарбонатом натрия и водой, сушили над безводным сульфатом натрия и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ТСХ с получением желаемого продукта.
6. Получение дигидрохлорида 2-((2-d-проп-2-ил)оксил)-5-метил-4-(d9-пиперидин-4-ил)анилина
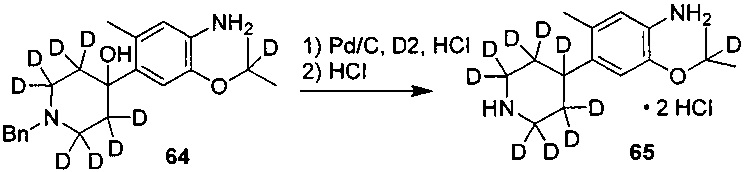
Соединение 4-(4-амино-5-((2-d-проп-2-ил)окси)-2-метилфенил)-1-бензил-(d8-пиперидин)-4-ол (0,125 г, 0,34 ммоль), палладий на угле (15 мг, 50% Pd), концентрированную соляную кислоту и дейтерированный метанол (CD3OD, 8 мл) добавляли в колбу и в системе три раза заменяли дейтерий, ее нагревали до 40°C в течение 8 часов при 50 фунтах/кв. дюйм (344,738 кПа). Палладий на угле удаляли путем фильтрации; и фильтрат концентрировали с получением серого твердого вещества. Полученное серое твердое вещество и раствор соляной кислоты в изопропаноле (2 М, 2 мл) последовательно добавляли в колбу и перемешивали в течение 2 ч. Реакционный раствор концентрировали при пониженном давлении с получением желаемого продукта в виде белого твердого вещества.
7. Получение 5-хлор-N2-((2-d-проп-2-илокси)-5-метил-4-(d9-пиперидин-4-ил)-фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамина
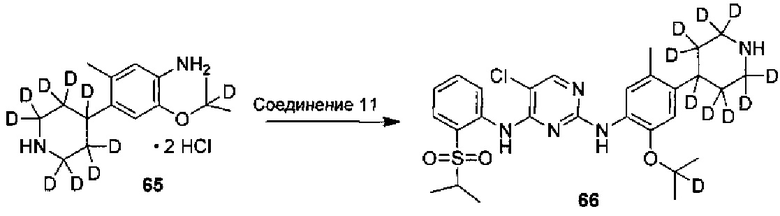
Под защитой азота соединение дигидрохлорид 2-((2-d-проп-2-ил)окси)-5-метил-4-(d9-пиперидин-4-ил)анилина (0,050 г, 0,15 ммоль), 2,5-дихлор-N-(2-(изопропилсульфонил)фенил)пиримидин-4-амин (0,063 г, 0,18 ммоль) и изопропанол (2 мл) добавляли в колбу, нагревали до 85°C и выдерживали всю ночь. После охлаждения до комнатной температуры для остановки реакции в реакционную смесь добавляли чистую воду и для нейтрализации pH до 8,5 добавляли водный раствор карбоната натрия. Полученную смесь экстрагировали этилацетатом. Органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт отделяли посредством препаративной ТСХ (проявляющий растворитель: метанол / метиленхлорид = 1/9, об./об.) с получением желаемого продукта в виде белого твердого вещества. МС расчетный: 567; МС обнаруженный: 568 (M+H)+, 590 (M+Na)+.
Пример 16: 5-хлор-N2-((2-d-проп-2-илокси)-5-метил-4-(2,2,3,3,5,5,6,6-d8-пиперидин-4-ил)-фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин

1. Получение дигидрохлорида 2-((2-d-проп-2-ил)оксил)-5-метил-4-(2,2,3,3,5,5,6,6-d8-пиперидин-4-ил)анилина

Соединение 4-(4-амино-5-((2-d-проп-2-ил)окси)-2-метилфенил)-1-бензил-(d8-пиперидин)-4-ол (0,125 г, 0,34 ммоль), палладий на угле (15 мг, 50% Pd), концентрированную соляную кислоту и метанол (8 мл) добавляли в колбу и в системе три раза заменяли водород, нагревали до 40°C в течение 8 часов при 50 фунтах/кв. дюйм (344,738 кПа). Палладий на угле удаляли путем фильтрации и фильтрат концентрировали с получением серого твердого вещества. Полученное серое твердое вещество и раствор соляной кислоты в изопропаноле (2 М, 2 мл) последовательно добавляли в колбу и перемешивали в течение 2 ч. Реакционный раствор концентрировали при пониженном давлении с получением желаемого продукта в виде белого твердого вещества.
2. Получение 5-хлор-N2-((2-d-проп-2-илокси)-5-метил-4-(2,2,3,3,5,5,6,6-d8-пиперидин-4-ил)-фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамина
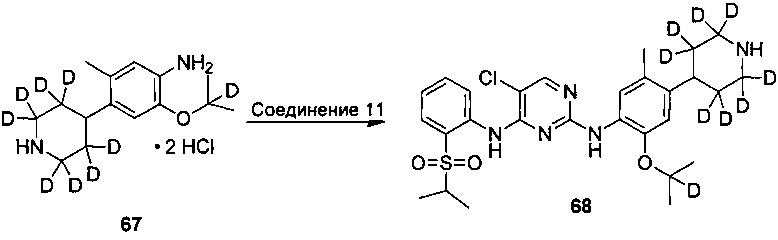
Под защитой азота соединение дигидрохлорид 2-((2-d-проп-2-ил)окси)-5-метил-4-(2,2,3,3,5,5,6,6-d8-пиперидин-4-ил)анилина (0,045 г, 0,14 ммоль), 2,5-дихлор-N-(2-(изопропилсульфонил)фенил)пиримидин-4-амин (0,058 г, 0,17 ммоль) и изопропанол (2 мл) добавляли в реакционную пробирку, нагревали до 85°C и выдерживали всю ночь. После охлаждения до комнатной температуры для остановки реакции в реакционную смесь добавляли чистую воду и для нейтрализации pH до 8,5 добавляли водный раствор карбоната натрия. Полученную смесь экстрагировали этилацетатом. Органические слои объединяли, сушили над безводным сульфатом натрия и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали посредством препаративной ТСХ (проявляющий растворитель: метанол / дихлорметан = 1/9, об./об.) с получением желаемого продукта в виде белого твердого вещества. МС расчетный: 566; МС обнаруженный: 567 (M+H)+, 589 (M+Na)+.
Пример 17: Фармакокинетическая оценка у крыс
8 самцов крыс Спрег-Доули (возраст 7-8 недель, масса тела приблизительно 200 г) делили на две группы, по четыре в каждой группе. Дозу 5 мг/кг (а) контрольного соединения 5-хлор-N2-(2-изопропокси-5-метил-4-(пиперидин-4-ил)фенил)-N4-(2-(изопропилсульфонил)фенил)пиримидин-2,4-диамин (Церитиниб) и (б) тестируемого соединения - соединений, полученных в примере 1-16 - каждый раз вводили перорально и у двух групп сравнивали различие в фармакокинетике.
Крыс кормили стандартным кормом и им неограниченно давали воду, и они начинали голодать за 16 часов до испытания. Лекарственное средство растворяли в ПЭГ 400 и диметилсульфоксиде. Отбор крови из глазничной области проводили через 0,083 часа, 0,25 часа, 0,5 часа, 1 час, 2 часа, 4 часа, 6 часов, 8 часов, 12 часов и 24 часа после введения.
Крысам быстро давали наркоз посредством вдыхания простого эфира; в пробирку отбирали 300 мкл крови из глазничной области. В пробирке находилось 30 мкл 1% солевых растворов гепарина. Перед применением пробирки сушили всю ночь при 60°C. После того, как образец крови был отобран в последующие моменты времени, крысам давали наркоз с использованием простого эфира и подвергали эвтаназии.
После того, как был отобран образец крови, пробирки сразу же осторожно переворачивали по меньшей мере 5 раз для обеспечения правильного смешивания и помещали на лед. Образцы крови центрифугировали при 4°C, при 5000 об/мин в течение 5 минут для разделения плазмы и эритроцитов. 100 мкл плазмы отбирали пипеткой в чистую пластиковую центрифужную пробирку и указывали название соединений и момент времени. Перед проведением анализа плазму хранили при - 80°C. Концентрацию соединения согласно изобретению в плазме определяли с помощью ЖХ-МС/МС. Фармакокинетические параметры рассчитывали на основе концентрации соединения в плазме у каждого животного в разные моменты времени.
Из результатов может быть видно, что, по сравнению с контрольным соединением, соединения согласно настоящему изобретению имеют лучшую фармакокинетику у животных (более высокие концентрации в плазме (например, Фиг. 1-4), больший период полураспада и более низкую скорость клиренса), что имеет лучшие фармакодинамические и терапевтические эффекты.
Пример 18: Оценка фармакодинамики in vitro соединений согласно изобретению в отношении киназ ALK
Определение IC50 (концентрация полумаксимального ингибирования) ингибирующего эффекта в отношении киназы ALK
96-луночные планшеты покрывали слоем покрывающего буфера при 37°C (125 мкл/лунка) на ночь, и покрывающий буфер представлял собой полипептидный субстрат [Поли (4: 1 Glu, Tyr) пептид, имеющийся у SignalChem], содержащий 2,5 мкг/лунку киназы ALK в ФСБ (фосфатно-солевой буферный раствор). Затем, каждую лунку промывали 200 мкл буфера (ФСБ, содержащий 0,05% Tween 80) и помещали при 37°C по меньшей мере на 2 часа до сухого состояния.
Тестируемые соединения серийного разведения в разных концентрациях (соединения, полученные в любом из Примеров 1-16, растворенные в ДМСО) добавляли в каждую лунку в объеме 5 мкл/лунку с последующим добавлением киназного буфера (25 мМ Hepes, pH 7,5, 5 мМ MnCl2, 5 мМ MgCl2), 0,3 мМ АТФ и 100 нг/лунку рекомбинантной AKL человека (Abnova Corporation) до общего объема 100 мкл в каждой лунке. После выдерживания при 30°C в течение 15 минут реакционную смесь удаляли и 5 раз промывали 200 мкл промывочного буфера (ФСБ, содержащий 0,05% Tween 80).
100 мкл/лунку мышиного моноклонального антитела против фосфотирозина (клон 4G10, приобретенный у EMD Millipore Corporation) использовали в выявлении фосфорилированного пептидного субстрата. Моноклональное антитело разводили в соотношении 1:500 PBS, содержащим 4% бычий сывороточный альбумин. После инкубирования при комнатной температуре в течение 30 минут раствор с антителами удаляли и каждую лунку 5 раз промывали 200 мкл промывочного буфера (ФСБ, содержащий 0,05% Tween 80).
Добавляли 100 мкл/лунку вторичного антитела (против мышиного IgG (иммуноглобулин G)). Вторичное антитело разводили в соотношении 1:1000 PBS, содержащем 4% бычий сывороточный альбумин, инкубировали в течение 30 минут при комнатной температуре; и лунки снова промывали, как указано выше.
100 мкл/лунку раствора субстрата ТМБ (тетраметилбензидин) использовали для развития окраски и такой же объем 0,18 М H2SO4 добавляли для остановки развития окраски. Наконец, считывали поглощение при 450 нм или 490 нм и рассчитывали IC50.
Результаты показаны в таблице 1. Видно, что соединения согласно настоящему изобретению имеют превосходную ингибирующую активность и селективность в отношении киназы ALK.

Пример 19: Фармацевтическая композиция
Приведенные выше вещества смешивали традиционными способами, затем ими наполняли обычные желатиновые капсулы с получением 100 капсул.
Все литературные источники, упомянутые в настоящей заявке, включены в данный документ путем ссылки, как если бы каждый литературный источник был отдельно включен путем ссылки. Кроме того, следует понимать, что после прочтения описанных выше идей, специалисты в данной области могут вносить разные изменения или модификации настоящего изобретения. Данные эквиваленты также попадают в объем, определенный прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ ПИРИМИДИН-4,6-ДИАМИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2022 |
|
RU2834353C2 |
| ХИМИЧЕСКИЙ СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРИМИДИНА И ИХ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2015 |
|
RU2703300C2 |
| Регулятор на основе азотсодержащего гетероциклического производного, способ его получения и его применение | 2020 |
|
RU2829553C2 |
| ПРОИЗВОДНОЕ ПИРИМИДИНА И ПЯТИЧЛЕННОГО АЗОТСОДЕРЖАЩЕГО ГЕТЕРОЦИКЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКИЕ ПРИМЕНЕНИЯ | 2019 |
|
RU2775229C1 |
| Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака | 2015 |
|
RU2607371C1 |
| ПРОИЗВОДНЫЕ 2,4-ДИЗАМЕЩЕННОГО ФЕНИЛЕН-1,5-ДИАМИНА И ИХ ПРИМЕНЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КОМПОЗИЦИИ, ПОЛУЧЕННЫЕ ИЗ НИХ | 2015 |
|
RU2649001C1 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ 5-ХЛОР-N2-(2-ИЗОПРОПОКСИ-5-МЕТИЛ-4-ПИПЕРИДИН-4-ИЛ-ФЕНИЛ)-N4-[2-(ПРОПАН-2-СУЛЬФОНИЛ)-ФЕНИЛ]-ПИРИМИДИН-2,4-ДИАМИНА | 2011 |
|
RU2746159C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| СОЕДИНЕНИЯ-ИНГИБИТОРЫ EGFR | 2017 |
|
RU2751341C2 |
| ИМИДАЗОПИРАЗИНЫ | 2012 |
|
RU2600327C2 |


Настоящее изобретение относится к новым дейтерированным диаминопиримидинам общей формулы (I) и их фармацевтически приемлемым солям. Соединения обладают свойствами ингибирования ALK протеинкиназ и могут быть использованы для лечения и/или предупреждения онкологических заболеваний, нарушения пролиферации клеток, сердечнососудистых заболеваний, воспаления, инфекции, аутоиммунных заболеваний, трансплантации органов, вирусных заболеваний, сердечнососудистых заболеваний или метаболических заболеваний. В общей формуле (I)

R1a, R1b, R1c, R2a, R2b, R2c, R3, R4, R6, R7, R9, R10, R11, R12, R13a, R13b, R13c, R14a, R14b, R14c, R15a, R15b, R15c, R16, R17a, R17b, R18a, R18b, R19a, R19b, R20a и R20b независимо представляют собой водород или дейтерий; R5 представляет собой водород, дейтерий или галоген, R8 представляет собой галоген; при условии, что по меньшей мере четыре из R1a, R1b, R1c, R2a, R2b, R2c, R3, R12, R13a, R13b, R13c, R14a, R14b, R14c, R19a, R19b, R20a или R20b являются дейтерием. Изобретение также относится к способу получения соединений общей формулы (I) путем взаимодействия соединения А6 с соединением XV и к промежуточным соединениям А6 и XV.

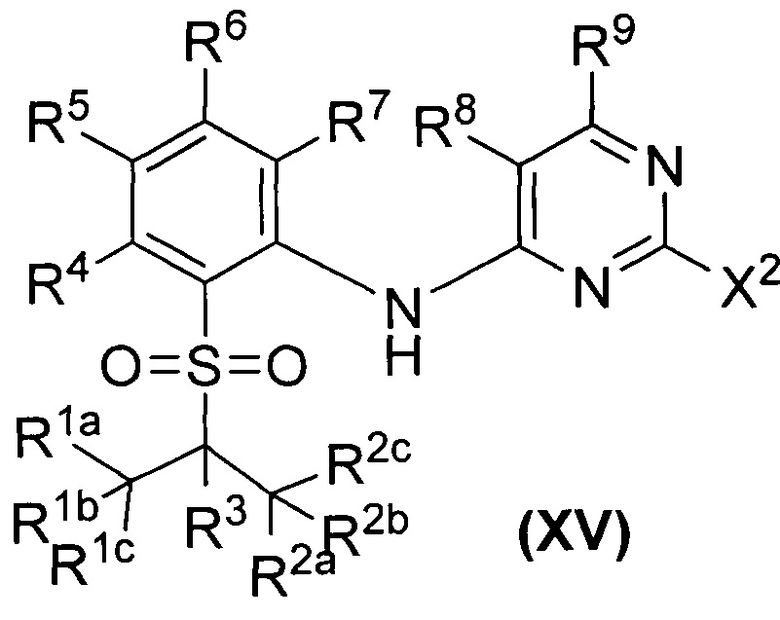 .
.
При этом в соединении А6 R10, R11, R12, R13a, R13b, R13c, R14a, R14b, R14c, R15a, R15b, R15c, R16, R17a, R17b, R18a, R18b, R19a, R19b, R20a или R20b независимо выбраны из дейтерия или водорода и по меньшей мере один из них представляет собой дейтерий; в соединении XV R1a, R1b, R1c, R2a, R2b, R2c, R3, R4, R6, R7, R9 независимо выбраны из водорода или дейтерия и по меньшей мере один из них представляет собой дейтерий; и R5 представляет собой водород, дейтерий или галоген; и R8 представляет собой галоген; X2 выбран из F, Cl, Br, I. 7 н. и 11 з.п. ф-лы, 4 ил., 1 табл.
1. Соединение дейтерированного диаминопиримидина или его фармацевтически приемлемая соль:
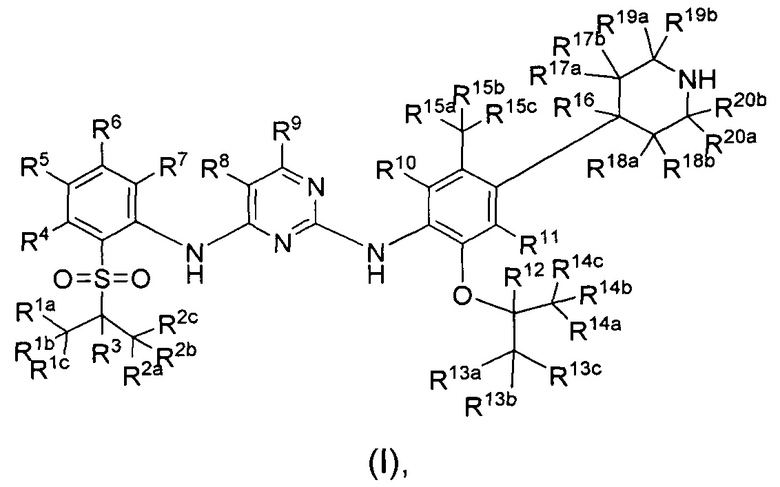
где
R1a, R1b, R1c, R2a, R2b, R2c, R3, R4, R6, R7, R9, R10, R11, R12, R13a, R13b, R13c, R14a, R14b, R14c, R15a, R15b, R15c, R16, R17a, R17b, R18a, R18b, R19a, R19b, R20a и R20b независимо представляют собой водород или дейтерий;
R5 представляет собой водород, дейтерий или галоген,
R8 представляет собой галоген;
при условии, что по меньшей мере четыре из R1a, R1b, R1c, R2a, R2b, R2c, R3, R12, R13a, R13b, R13c, R14a, R14b, R14c, R19a, R19b, R20a или R20b являются дейтерием.
2. Соединение по п. 1, где R1a, R1b, R1c, R2a, R2b, R2c и R3 независимо представляют собой дейтерий или водород.
3. Соединение по п. 1, где R12, R13a, R13b, R13c, R14a, R14b и R14c независимо представляют собой дейтерий или водород.
4. Соединение по п. 1, где R15a, R15b и R15c независимо представляют собой дейтерий или водород.
5. Соединение по п. 1, где R16, R17a, R17b, R18a, R18b, R19a, R19b, R20a и R20b независимо представляют собой дейтерий или водород.
6. Соединение по любому из пп. 1-4, где R8 представляет собой хлор.
7. Соединение по любому из пп. 1-4, где R12 представляет собой дейтерий.
8. Соединение по любому из пп. 1-4, где R19a, R19b, R20a и R20b представляют собой дейтерий.
9. Соединение по п. 1, где соединение представляет собой одно из следующих соединений или его фармацевтически приемлемую соль:


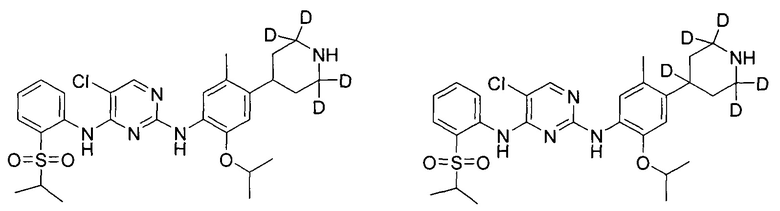

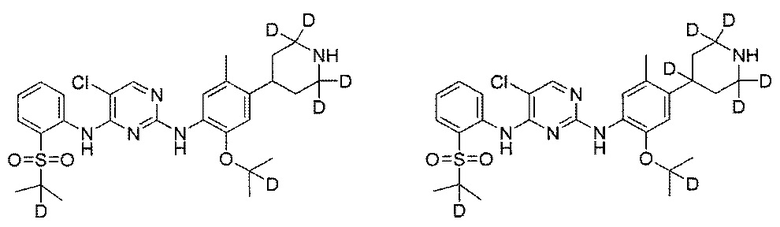

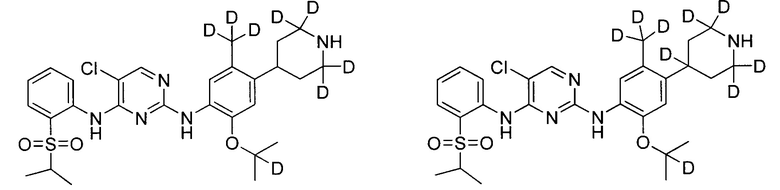
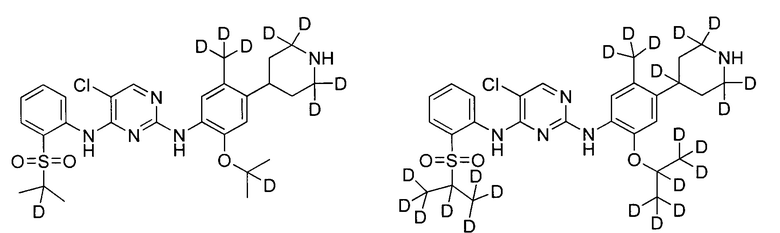
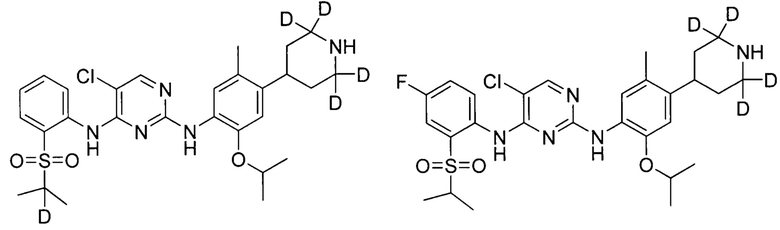





10. Способ получения фармацевтической композиции, обладающей свойствами ингибитора протеинкиназы ALK, включающий смешивание соединения по п. 1 или его фармацевтически приемлемой соли с фармацевтически приемлемым носителем с получением таким образом фармацевтической композиции.
11. Фармацевтическая композиция, обладающая свойствами ингибитора протеинкиназы ALK, содержащая фармацевтически приемлемый носитель и соединение по п. 1 или его фармацевтически приемлемую соль в эффективном количестве.
12. Применение соединения по п. 1 или его фармацевтически приемлемой соли для получения фармацевтической композиции, обладающей свойствами ингибирования ALK протеинкиназ.
13. Применение по п. 12, где фармацевтическую композицию применяют для лечения или предупреждения любого из следующих заболеваний: онкологические заболевания, нарушения пролиферации клеток, сердечнососудистые заболевания, воспаления, инфекции, аутоиммунные заболевания, трансплантации органов, вирусные заболевания, сердечнососудистые заболевания или метаболические заболевания.
14. Способ получения соединения по п. 1 или его фармацевтически приемлемой соли, включающий реакцию соединения А6 с соединением XV с получением таким образом соединения I в форме гидрохлорида или свободного основания

где X2 выбран из F, Cl, Br, I; и R1a, R1b, R1c, R2a, R2b, R2c, R3, R4, R5, R6, R7, R8, R9, R10, R11, R12, R13a R13b, R13c, R14a, R14b, R14c, R15a, R15b, R15c, R16, R17a, R17b, R18a, R18b, R19a, R19b, R20a или R20b определены выше.
15. Способ получения по п. 14, отличающийся тем, что соединение А6 получают посредством следующих стадий:
(а) реакция соединения IV с соединением А4 с получением таким образом соединения А5;
(б) проведение реакции восстановления и реакции снятия защиты в отношении соединения А5, получая таким образом соединение А6;

где X выбран из F, Cl, Br, I; OTf; R10, R11, R12, R13a, R13b, R13c, R14a, R14b, R14c, R15a, R15b, R15c, R16, R17a, R17b, R18a, R18b, R19a, R19b, R20a или R20b определены выше; и PG представляет собой защитную группу аминогруппы.
16. Способ получения по п. 14, отличающийся тем, что соединение XV получают посредством следующих стадий:
(а) реакция соединения X с соединением XI с получением таким образом соединения XII;
(б) реакция соединения XII с соединением XIV с получением таким образом соединения XIII;
(в) проведение реакции окисления в отношении соединения XIII с получением таким образом соединения XV;
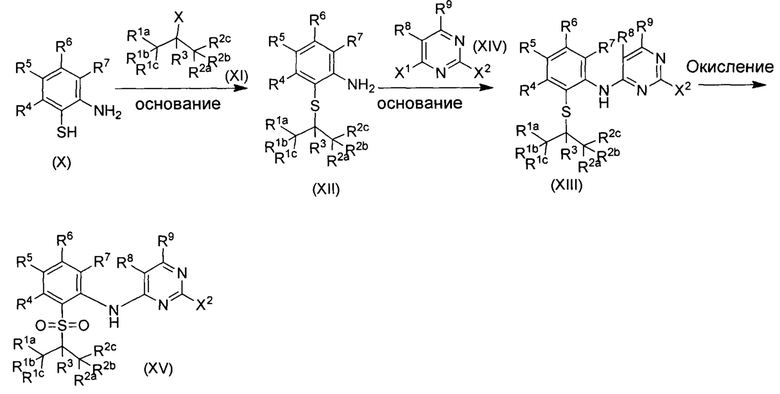
где X, X1, X2 выбраны из F, Cl, Br, I, OTs, OMs, OTf; и R1a, R1b, R1c, R2a, R2b, R2c, R3, R4, R5, R6, R7, R8, R9 определены выше.
17. Промежуточное соединение формулы А6 или его свободное основание

где R10, R11, R12, R13a, R13b, R13c, R14a, R14b, R14c, R15a, R15b, R15c, R16, R17a, R17b, R18a, R18b, R19a, R19b, R20a или R20b независимо выбраны из дейтерия или водорода и по меньшей мере один из них представляет собой дейтерий.
18. Промежуточное соединение формулы XV
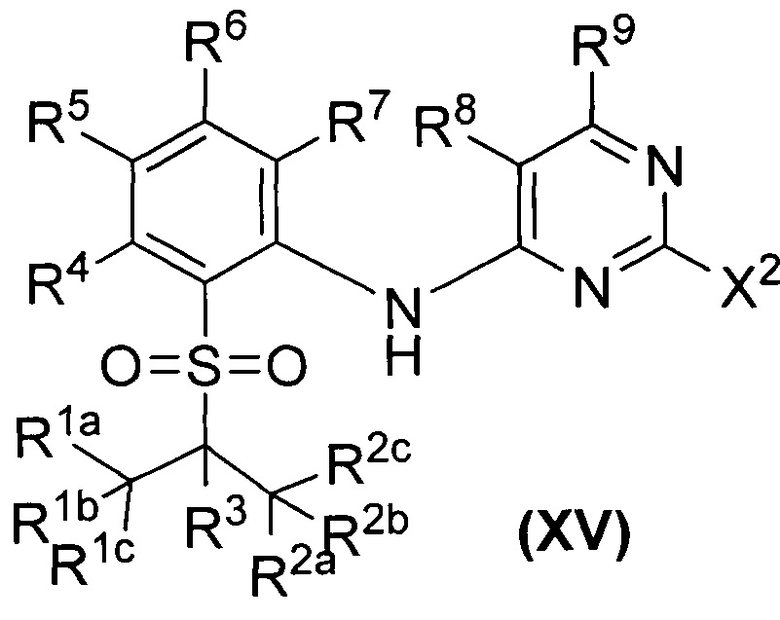 ,
,
где R1a, R1b, R1c, R2a, R2b, R2c, R3, R4, R6, R7, R9 независимо выбраны из водорода или дейтерия и по меньшей мере один из них представляет собой дейтерий; и R5 представляет собой водород, дейтерий или галоген; и R8 представляет собой галоген; X2 выбран из F, Cl, Br, I.
| US 8039479 B2, 18.10.2011 & CN 101616895 A, 30.12.2009 & EA 20110688, 30.12.2011 & EA 20110687, 30.12.2011 | |||
| US 2010291025 A1, 18.11.2010 | |||
| CN 102131788 A, 20.07.2011 & EA 018282 B1, 28.06.2013 | |||
| CN 103458881 A, 18.12.2013 & EA 023404 B1, 31.05.2016 | |||
| Опорно-поворотное устройство | 1975 |
|
SU751926A1 |
| US 2013040957 A1, 14.02.2013 | |||
| ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ | 2005 |
|
RU2401260C2 |

