Область техники
Настоящее изобретение относится к области медицинской техники, в частности к производным 2,4-дизамещенного бензол-1,5-диамина и их применениям в качестве ингибиторов тирозинкиназы рецептора эпидермального фактора роста (EGFR), а также к полученным из них фармацевтическим композициям и лекарственным композициям.
Предшествующий уровень техники
Рак легкого является самым распространенным заболеванием в мире. В Китае заболеваемость раком легких занимает первое место среди всех раковых заболеваний, и заболеваемость и смертность от рака также являются самыми высокими в Китае. В Китае 30% пациентов с раком легких имеют мутации EGFR, более 90% которых представляют собой мутацию L858R и делецию в 19-м экзоне, и эти пациенты являются более чувствительными к ингибиторам EGFR. Имеющееся в продаже первое поколение ингибиторов EGFR, таких как эрлотиниб, гефитиниб, оказывает положительное воздействие на этих пациентов, опухоль у более чем 60% пациентов уменьшается в размере, значительно продлевая выживаемость без прогрессирования заболевания. Однако у большинства пациентов формируется резистентность в течение 6-12 месяцев, первое поколение ингибиторов EGFR более не оказывает воздействия, и в настоящее время не существует никаких лекарств для таких пациентов. У 50% пациентов, демонстрирующих резистентность к первому поколению ингибиторов EGFR, клинически обнаруживается мутация EGFR Т790М. В клеточной линии Н1975, несущей мутацию Т790М, активность первого поколения ингибиторов EGFR, таких как гефитиниб и эрлотиниб, была больше чем 3 мкм, что означает практически отсутствие активности.
У пациентов, страдающих раком легких, с мутациями EGFR терапевтический эффект второго поколения необратимых ингибиторов пан-EGFR (Афатиниб (BIBW2992)), запущенных в производство в настоящее время, был значительно лучше, чем терапевтический эффект первого поколения ингибиторов EGFR. Однако второе поколение ингибиторов EGFR обладает также сильной ингибирующей активностью в отношении EGFR дикого типа, причем ингибирующая активность в отношении EGFR дикого типа была значительно выше, чем ингибирующая активность в отношении резистентной мутации Т790М, и у пациентов наблюдались серьезные побочные эффекты, такие как сыпь на коже, и воздействие второго поколения ингибиторов на пациентов с лекарственной резистентностью было слабым, только небольшая часть пациентов с резистентностью к первому поколению ингибиторов EGFR отвечала на лечение этими лекарствами.
Для повышения ингибирующей активности в отношении резистентной мутации Т790М с одновременным снижением ингибирующей активности в отношении EGFR дикого типа большое значение имеет разработка третьего поколения селективных ингибиторов для мутантов EGFR с более высокой активностью, лучшей селективностью и более низкой токсичностью.
Краткое описание изобретения
Целью настоящего изобретения является обеспечение производных 2,4-дизамещенного бензол-1,5-диамина или их фармацевтически приемлемой соли, стереоизомера, сольвата или пролекарства; а также фармацевтической композиции, включающей вышеупомянутое производное.
Второй целью настоящего изобретения является обеспечение применения вышеуказанного производного или композиции для получения лекарственного средства для регулирования активности тирозинкиназы EGFR или лечения EGFR-зависимых заболеваний.
В первом аспекте настоящего изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль, стереоизомер, сольват или пролекарство:

где А является любым, выбранным из:
 или
или  ;
;
где Z2 представляет собой CR5 или N; R5 представляет собой водород, галоген, трифторметил или С1-10 алкокси; Z21 представляет собой N или СН; каждый из R4, R42, R43 независимо представляет собой водород или С1-10 алкил, С3-10 циклоалкил, С3-10 гетероциклоалкил (например пиперидинил) или С1-10 галогеналкил; R41 представляет собой водород, галоген, трифторметил, С1-10 алкил или С1-10 алкокси; (предпочтительно, каждый из R4, R42, R43 независимо представляет собой водород, метил или этил; R41 представляет собой водород, хлор, фтор, трифторметил, метил или метокси); каждый из R1', R2' и R5' независимо представляет собой водород, С1-10 алкил; (предпочтительно, каждый из R1', R2' и R5' независимо представляет собой водород, метил или этил); каждый из R3', R4' независимо представляет собой водород, С1-10, или R3' и R4' образуют мостиковую кольцевую структуру; (предпочтительно, каждый из R3', R4' независимо представляет собой водород, метил или этил; или R3' и R4' образуют -СН2-); Y представляет собой О, СН2, NRa'; где Ra' представляет собой водород, метил, трет-бутокси карбон ил, ацетил, метилсульфонил; и n равно 0 или 1;
R0 представляет собой водород, галоген, С1-10 алкил, -NRaRb или -CO-NRaRb, где каждый из Ra, Rb независимо представляет собой С1-10 алкил;
Z1 представляет собой CR6 или N; где R6 представляет собой водород, галоген, трифторметил, метокси или -CO2C1-10 алкил;
R1 представляет собой водород, или является любым, выбранным из группы, состоящей из:
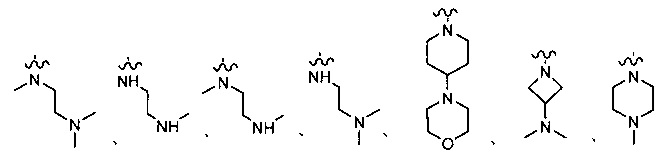 ,
,
 или
или 
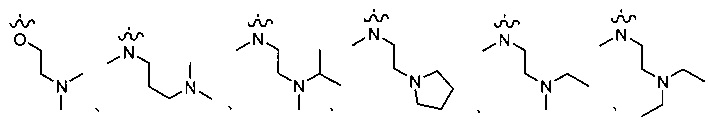 ;
;
каждый или R2 и R3 независимо представляет собой водород или -CH2NR7R8; где (1) каждый из R7, R8 независимо представляет собой водород или метил; или (2) R7 и R8 вместе с атомом азота, к которому они присоединены, образуют 5-6-членное азотсодержащее насыщенное гетероциклическое кольцо;
R12 представляет собой водород, галоген, С1-10 алкил или С1-10 алкокси (предпочтительно метокси).
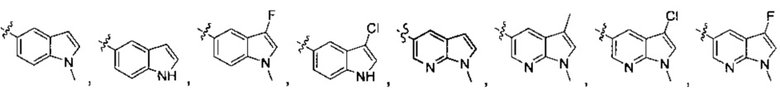
В другом предпочтительном варианте осуществления А формулы (I) является любым, выбранным из группы, состоящей из:
 ;
;
где n, R4, R41, R42, R43, R1', R2', R3', R4', R5', Ra' являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления R4 формулы (I) представляет собой водород, метил, этил, изопропил, циклопропил, дифторметил или пиперидинил; R41 представляет собой водород, хлор, фтор, трифторметил, метил или метокси; R42 представляет собой водород, метил или этил; R43 представляет собой водород, метил или этил.
В другом предпочтительном варианте осуществления каждый из R2 и R3 формулы (I) независимо представляет собой водород; и R12 представляет собой метокси.
В другом предпочтительном варианте осуществления Z1 формулы (I) представляет собой CR6 or N; где R6 представляет собой водород, фтор, хлор, трифторметил, метокси или -СО2С1-10 алкил;
В другом предпочтительном варианте осуществления Z1 формулы (I) представляет собой CR6, где R6 представляет собой водород, фтор, хлор, трифторметил, метокси или -СО2СН3,
В другом предпочтительном варианте осуществления R0 формулы (I) представляет собой водород.
В другом предпочтительном варианте осуществления А формулы (I) является любым, выбранным из группы, состоящей из:
 ,
,
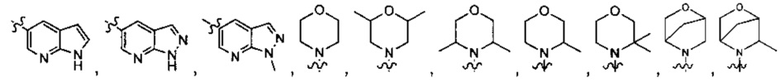 ,
,
 и
и
 ; при этом
; при этом 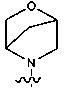 и
и  могут быть
могут быть  и
и  соответственно.
соответственно.
В другом предпочтительном варианте осуществления каждый из R2 and R3 формулы (I) независимо представляет собой водород.
В другом предпочтительном варианте осуществления каждый из R2 and R3 формулы (I) независимо представляет собой водород или -CH2NR7R8, и каждый из R7 и R8 независимо представляет собой водород или метил.
В другом предпочтительном варианте осуществления каждый из R2 и R3 формулы (I) независимо представляет собой водород, или -CH2NR7R8, и R7 и R8 вместе с атомом азота, к которому они присоединены, образуют 5-6-членное насыщенное азотсодержащее гетероциклическое кольцо.
В другом предпочтительном варианте осуществления каждый из R2 и R3 формулы (I) независимо представляет собой водород или -CH2NR7R8, и R7 и R8 вместе с соседним атомом азота образуют 5-6-членное азотсодержащее насыщенное гетероциклическое кольцо, и структура 5-6-членного азотсодержащего насыщенного гетероциклического кольца представлена формулой (III):
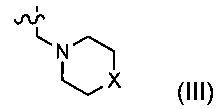
где X представляет собой О, S, NR9 или CR10R11; где каждый из R9, R10, R11 независимо представляет собой водород, С1-10 алкокси или -СО-С1-10 алкил.
В другом предпочтительном варианте осуществления соединение формулы (I) представляет собой соединение формулы (IV-1):

где A, R0, R1, R2, R3 и R6 являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления соединение формулы (I) представляет собой соединение формулы (IV-2):

где A, R0, R1, R2 и R3 являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления соединение формулы (I) представляет собой соединение формулы (V-1):

где R1, R2, R3, R4, R41 и R6 являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления соединение формулы (I) представляет собой соединение формулы (V-2):

где R1, R2, R3, R4 и R41 являются такими как определено в формуле (I).
В другом предпочтительном варианте осуществления соединение формулы (I) представляет собой соединение формулы (VI-1):

где R1, R2, R3, R4, R41 и R6 являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления соединение формулы (I) представляет собой соединение формулы (VI-2):

где R1, R2, R3, R4 и R41 являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления соединение формулы (I) представляет собой соединение формулы (VII-1):

где R1, R2, R3 и R6 являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления соединение формулы (I) показано в формуле (VII-2):

где R1, R2 и R3 являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления соединение формулы (I) представляет собой соединение, представленное формулой (VIII-1):

где R1', R2', R3', R4', R5', R1, R2, R3, R6 и Y являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления соединение формулы (I) представляет собой соединение, представленное формулой (VIII-2):

где R1', R2', R3', R4', R5', R1, R2, R3 и Y являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления соединение формулы (I) представляет собой соединение формулы (IX-1):

где R1', R2', R4', R5', R1, R2, R3 и R6 являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления соединение формулы (I) представляет собой соединение формулы (IX-2):

где R1', R2', R4', R5', R1, R2 и R3 являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления соединение формулы (I) представляет собой соединение формулы (Х-1):
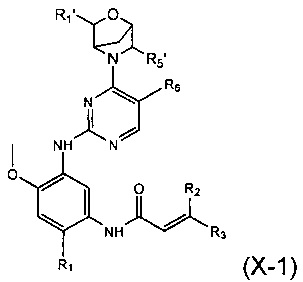
где R1', R5', R1, R2, R3 и R6 являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления соединение формулы (I) представляет собой соединение формулы (Х-2):

где R1', R5', R1, R2 и R3 являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления соединение формулы (I) представляет собой соединение формулы (XI-1):

где R1, R2, R3, R4, R42, R43 и R6 являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления соединение формулы (I) представляет собой соединение формулы (XI-2):

где R1, R2, R3, R4, R42 и R43 являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления, соединение формулы (I) представляет собой соединение формулы (XII-1):
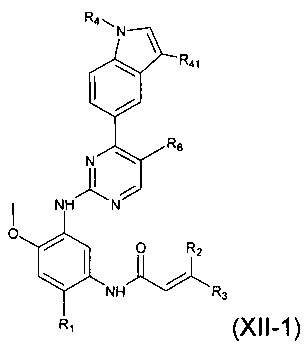
где R1, R2, R3, R4, R41 и R6 являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления соединение формулы (I) представляет собой соединение формулы (XII-2):

где R1, R2, R3, R4 и R41 являются такими, как определено в формуле (I).
В другом предпочтительном варианте осуществления в соединении формулы (XI-1) R6 представляет собой водород, фтор, хлор или трифторметил.
В другом предпочтительном варианте осуществления в соединении формулы (XI-1) или формулы (XI-2) каждый из R2 и R3 независимо представляет собой водород.
В другом предпочтительном варианте осуществления в соединении формулы (XI-1) или формулы (XI-2) R1 выбирают из:
 или
или 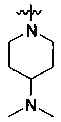 .
.
В другом предпочтительном варианте осуществления в соединении формулы (XI-1) или формулы (XI-2) каждый из R4, R42, R43 независимо представляет собой водород, С1-10 алкил, С1-10 галогеналкил или пиперидинил;
В другом предпочтительном варианте осуществления в соединении формулы (XI-1) или формулы (XI-2) каждый из R4, R42 и R43 независимо представляет собой водород, метил, этил, дифторметил или пиперидинил.
В другом предпочтительном варианте осуществления в соединении формулы (XI-1) или формулы (XI-2) R4 представляет собой водород, метил, дифторметил или пиперидинил и каждый из R42, R43 независимо представляет собой водород или метил.
В другом предпочтительном варианте осуществления в соединении формулы (V-1), формулы (V-2), формулы (VI-1), формулы (VI-2), формулы (XII-1) или формулы (XII-2) R4 представляет собой водород, С1-10 алкил, С3-10 циклоалкил или С1-10 галогеналкил.
В другом предпочтительном варианте осуществления в соединении формулы (V-1), формулы (V-2), формулы (VI-1), формулы (VI-2), формулы (XII-1) или формулы (XII-2) R4 представляет собой водород, метил, этил, изопропил, циклопропил или дифторметил.
В другом предпочтительном варианте осуществления в соединении формулы (V-1), формулы (V-2), формулы (VI-1), формулы (VI-2), формулы (XII-1) или формулы (XII-2) R41 представляет собой водород, хлор, фтор, трифторметил, метил или метокси.
В другом предпочтительном варианте осуществления в соединении формулы (V-1), формулы (V-2), формулы (VI-1), формулы (VI-2), формулы (XII-1) или формулы (XII-2) каждый из R2 и R3 независимо представляет собой водород.
В другом предпочтительном варианте осуществления в соединении формулы (V-1), формулы (VI-1) или формулы (XI1-1) R6 представляет собой водород, фтор, хлор, трифторметил, метокси или -CO2CH3,
В другом предпочтительном варианте осуществления в соединении формулы (V-1), формулы (V-2), формулы (VI-1), формулы (VI-2), формулы (XII-1) или формулы (XII-2) R1 выбирают из:
 ,
,
 или
или 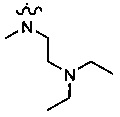 .
.
В другом предпочтительном варианте осуществления соединение выбирают из:
 ,
,
 ,
,
 ,
,
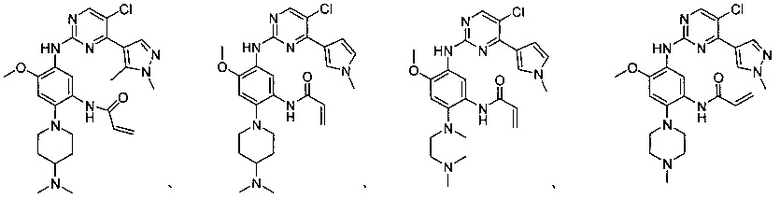 ,
,
 ,
,
 ,
,
 ,
,
 ,
,
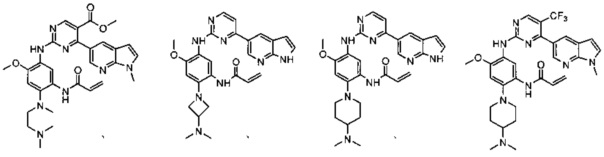 ,
,
 ,
,
 ,
,
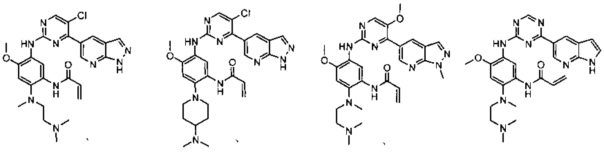 ,
,
 ,
,
 ,
,
 ,
,
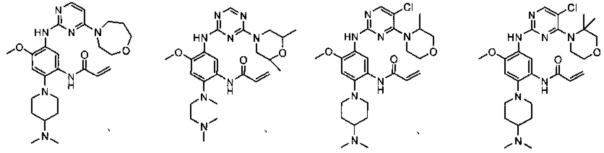 ,
,
 ,
,
 ,
,
 ,
,
 ,
,
 или
или  ,
,
 ,
,
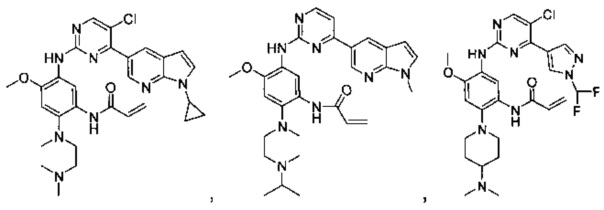 или
или  .
.
Во втором аспекте настоящего изобретения предложена фармацевтическая композиция, включающая соединение по первому аспекту настоящего изобретения или его фармацевтически приемлемую соль, стереоизомер, сольват или пролекарство, и фармацевтически приемлемый носитель.
В другом предпочтительном варианте осуществления фармацевтическая композиция включает терапевтически эффективное количество соединения по первому аспекту настоящего изобретения, такого как соединения формулы (I), соединения формулы (IV-1), соединения формулы (IV-2), соединения формулы (V-1), соединения формулы (V-2), соединения формулы (VI-1), соединения формулы (VI-2), соединения формулы (VII-1), соединения формулы (VII-2), соединения формулы (VIII-1), соединение формулы (VIII-2), соединения формулы (IX-1), соединения формулы (IX-2), соединения формулы (Х-1), соединения формулы (Х-2), соединения формулы (XI-1), соединения формулы (XI-2) или указанных выше иллюстративных соединений, или их фармацевтически приемлемых солей, стереоизомеров, сольватов или пролекарств, и фармацевтически приемлемые носители.
Как правило, соединение по настоящему изобретению или его фармацевтически приемлемая соль может образовывать подходящую лекарственную форму для введения с одним или несколькими фармацевтически приемлемыми носителями. Эти лекарственные формы пригодны для перорального, ректального, местного, интраорального введения и другого парентерального введения (например подкожного, внутримышечного, внутривенного введения и т.д.). Например, лекарственные формы, пригодные для перорального введения, включают капсулы, таблетки, гранулы и сиропы. Соединения по настоящему изобретению, содержащиеся в этих лекарственных формах, могут представлять собой твердые порошки или гранулы; водные или неводные жидкие растворы или суспензии; эмульсии вода-в-масле или масло-в-воде. Такие лекарственные формы могут быть получены с активными соединениями и одним или несколькими носителями или эксципиентами с помощью обычных фармацевтических способов. Вышеупомянутые носители должны быть совместимы с активными веществами или другими наполнителями. В твердых лекарственных формах обычные нетоксичные носители включают в себя, но не ограничиваются ими, маннит, лактозу, крахмал, стеарат магния, целлюлозу, глюкозу, сахарозу и тому подобное. Носители, используемые для жидких препаратов, включают воду, физиологический раствор, водную декстрозу, этиленгликоль и полиэтиленгликоль. Активные соединения могут образовывать раствор или суспензию с вышеупомянутыми носителями.
Композиции по настоящему изобретению составляют, количественно измеряют и вводят способом в соответствии с медицинской практикой. "Эффективное количество" вводимого соединения зависит от таких факторов, как конкретное подлежащее лечению заболевание, индивидуум, который подвергается лечению, причина заболевания, мишень лекарственного средства и способ введения и т.д.
В третьем аспекте настоящего изобретения предложено применение соединения по первому аспекту настоящего изобретения или его фармацевтически приемлемой соли, стереоизомера, сольвата или пролекарства для получения лекарственного средства для регулирования активности тирозинкиназы EGFR или лечения EGFR-зависимых заболеваний.
Предпочтительно, EGFR-зависимое заболевание представляет собой рак, диабет, заболевания иммунной системы, нейродегенеративные заболевания или сердечно-сосудистые заболевания.
Предпочтительно, рак представляет собой немелкоклеточный рак легкого, рак головы и шеи, рак молочной железы, рак почки, рак поджелудочной железы, рак шейки матки, рак пищевода, рак поджелудочной железы, рак предстательной железы, рак мочевого пузыря, рак толстой и прямой кишки, рак яичника, рак желудка, злокачественные опухоли мозга, в том числе глиобластому и т.д., или любую их комбинацию.
Предпочтительно, немелкоклеточный рак легкого обусловлен мутациями EGFR, в том числе чувствительными мутациями (такими как мутация L858R или делеции в 19-м экзоне) и резистентными мутациями (такими как мутация EGFR Т790М).
Предпочтительно, соединение или его фармацевтически приемлемая соль, стереоизомер, сольват или пролекарство могут также быть применены для получения лекарственного средства для лечения заболевания с аномальной экспрессией EGFR, или заболевания, которое приобрело резистентность в ходе лечения с использованием модуляторов EGFR.
Предпочтительно, приобретенная резистентность представляет собой резистентность, обусловленную мутациями Т790, кодированными 20-м экзоном EGFR, или содержанием мутаций Т790, кодированных 20-м экзоном EGFR, таких как Т790М.
В настоящем изобретении термин модулятор EGFR относится к низкомолекулярному ингибитору тирозинкиназы, нацеленному на EGFR, такому как гефитиниб, эрлотиниб, икотиниб, лапатиниб или афатиниб.
В четвертом аспекте настоящего изобретения предложена фармацевтическая композиция, включающая терапевтически эффективное количество соединения по первому аспекту настоящего изобретения или его фармацевтически приемлемой соли, стереоизомера, сольвата или пролекарства, а также одно или более других лекарственных средств, выбранных из группы, состоящей из гефитиниба, эрлотиниба, икотиниба, лапатиниба, XL647, NVP-AEE-788, ARRY-334543, EKB-569, BIBW2992, HKI272, ВМС-690514, CI-1033, вандетаниба, PF00299804, WZ4002, цетуксимаба, трастузумаба, панитумумаба, матузумаба, нимотузумаба, залутумумаба, пертузумаба, MDX-214, CDX-110, IMC-11F8, Zemab, вакцины Her2 РХ 1041, ингибиторов HSP90, CNF2024, танеспимицина, алвеспимицина, IPI-504, SNX-5422, NVP-AUY922 или их комбинаций.
В дополнение к соединению по настоящему изобретению или его фармацевтически приемлемой соли, стереоизомеру, сольвату или пролекарству, другие лекарственные средства в фармацевтической композиции, описанной выше, представляют собой противоопухолевые средства, хорошо известные специалистам в данной области техники.
В настоящей заявке термин "терапевтически эффективное количество" относится к количеству, обеспечивающему функцию или активность в отношении людей и/или животных, и переносимому людьми и/или животными.
Терапевтически эффективное количество соединения по настоящему изобретению или его фармацевтически приемлемой соли, стереоизомера, сольвата или пролекарства, содержащееся в фармацевтической композиции или лекарственной композиции по настоящему изобретению, составляет предпочтительно от 0,1 мг - 5 г/кг (масса).
Фармацевтические композиции по настоящему изобретению могут быть использованы для лечения EGFR-зависимых заболеваний, таких как рак, диабет, нарушения иммунной системы, нейродегенеративные заболевания или сердечно-сосудистые заболевания, или для лечения заболевания с приобретенной резистентностью в ходе лечения с помощью модулятора EGFR.
Заболевание с приобретенной резистентностью обусловлено мутациями Т790, кодированными 20-м экзоном EGFR, или содержанием мутаций Т790, кодированных 20-м экзоном EGFR.
В другом предпочтительном варианте осуществления мутация Т790, кодированная 20-м экзоном EGFR, представляет собой Т790М.
При определенных заболеваниях для достижения желаемого терапевтического эффекта соединения по настоящему изобретению могут быть использованы в комбинации с другими лекарствами. Примером такого заболевания является немелкоклеточный рак легких (NSCLC). Например, терапевтически эффективное количество соединения формулы I по настоящему изобретению используют в комбинации с ингибиторами mTOR (мишени рапамицина млекопитающих) (например рапамицином); или ингибиторами Met (включая Met антитела MetMAb (моноклональные антитела) и низкомолекулярные ингибиторы Met PF02341066) MS; или ингибиторами IGF1R (рецептора инсулиноподобного фактора роста 1) (например OSI-906); или ингибиторами белков теплового шока и тому подобное.
Следует понимать, что каждый из указанных выше технических признаков изобретения и каждый технический признак, в частности описанный ниже (например в Примерах), может быть скомбинирован с другим в пределах объема настоящего изобретения для создавать новых или предпочтительных технических решений.
Подробное описание изобретения
В ходе долгого и интенсивного исследования изобретатели неожиданно обнаружили класс селективных ингибиторов мутаций EGFR. Анализы in vitro показали, что селективные ингибиторы в наномолярных концентрациях могут ингибировать двойной мутантный фермент EGFR T790M/L858R и пролиферацию клеточной линии Н1975, а также демонстрируют высокую ингибирующую активность в отношении EGFR чувствительной мутантной клеточной линии НСС827 (делеция в 19-м экзоне), но имеют относительно слабую ингибирующую активность в отношении фермента EGFR дикого типа и клеточной линии А431, Таким образом, соединения с такими структурами могут быть использованы не только для лечения EGFR чувствительного мутантного рака, но и для лечения случаев вторичной резистентности, генерируемых в ходе проводимой в настоящее время терапии EGFR-TKI (EGFR-ингибитор тирозинкиназы); в то же время, побочные эффекты, вызванные ингибированием EGFR дикого типа, значительно снижаются вследствие мутантной селективности соединений. Кроме того, эти соединения проявляют низкую цитотоксичность в нормальных клетках (таких как клетки 3Т3), тем самым значительно уменьшая неспецифические побочные эффекты, и являются идеальными заменителями второго поколения EGFR-TKI.
Значения терминов
В настоящей заявке термин "С1-10 алкил" относится к прямой или разветвленной насыщенной алифатической углеводородной группе, имеющей от 1 до 10 атомов углерода, например метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, гексил, гептил, октил, нонил, децил и тому подобное.
В настоящей заявке термин "С1-10 алкокси" относится к С1-10 алкил -О-, например метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси и тому подобное.
В настоящей заявке термин "галоген" обозначает фтор, хлор, бром или йод.
В настоящей заявке термин "С3-10 циклоалкил" относится к насыщенному или частично ненасыщенному моноциклическому или полициклическому циклическому углеводородному заместителю, содержащему от 3 до 10 атомов углерода, предпочтительно циклоалкилу, содержащему от 3 до 8 атомов углерода (С3-8 циклоалкил), наиболее предпочтительно к циклоалкилу, содержащему от 3 до 6 атомов углерода (С3-6 циклоалкил). Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил и циклооктил, предпочтительно циклопропил, циклопентил, циклогексенил. Полициклический циклоалкил включает спиро-кольцо, конденсированное кольцо и мостиковое кольцо.
В настоящей заявке термин "С3-10 гетероциклоалкил" относится к насыщенному или частично ненасыщенному моноциклическому или полициклическому циклическому углеводородному заместителю, содержащему от 3 до 10 кольцевых атомов, где один или несколько кольцевых атомов представляют собой гетероатомы, выбранные из азота, кислорода или S(O)t (где t представляет собой целое число от 0 до 2), и другие кольцевые атомы представляют собой углерод, но С3-10 гетероциклоалкил не включает в себя кольцевой фрагмент, такой, как -О-О-, -O-S- или -S-S-. Предпочтительно, гетероциклоалкил включает от 3 до 8 кольцевых атомов, где от 1 до 3 кольцевых атомов представляют собой гетероатомы, предпочтительно, гетероциклоалкил, содержащий от 3 до 6 кольцевых атомов, и, более предпочтительно, гетероциклоалкил, включающий от 5 до 6 кольцевых атомов. Неограничивающие примеры моноциклических гетероциклических групп включают пирролидинил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил, пиранил, тетрагидрофуранил и тому подобное. Полициклический гетероциклоалкил включает в себя спиро-кольцо, конденсированное кольцо и мостиковое кольцо.
В настоящей заявке термин "С1-10 галогеналкил" относится к алкилу, замещенному 1, 2, 3, 4 или 5 атомами галогена, где алкил является таким, как определено выше, предпочтительно представляет собой С1-10 галогеналкил, более предпочтительно С1-6 галогеналкил и наиболее предпочтительно С1-3 галогеналкил, в том числе (но не ограничиваясь этим) хлорэтил, дихлорметил, 1,2-дихлорэтил, бромэтил, фторэтил, фторметил, дифторметил, трифторметил и тому подобное.
Активные ингредиенты
Термин "активный материал по настоящему изобретению" или "активное соединение по настоящему изобретению" относится к соединению формулы (I) по настоящему изобретению или его фармацевтически приемлемой соли, или его сольвату, или его стереоизомеру, или его пролекарству, обладающему значительной ингибирующей активностью в отношении резистентности к мутации EGFR Т790М (в частности двойной мутации EGFR T790M/L858R).
В настоящей заявке термин "фармацевтически приемлемая(ые) соль(и)" включает в себя фармацевтически приемлемую(ые) соль(и) присоединения кислоты и соль(и) присоединения основания. "Фармацевтически приемлемые соли присоединения кислоты" относятся к солям, которые способны сохранять биологическую эффективность свободного основания без других побочных эффектов и образованы с неорганическими или органическими кислотами. Соли неорганических кислот включают, но не ограничиваются ими, гидрохлорид, гидробромид, сульфат, фосфат и тому подобное; соли органических кислот включают в себя, но не ограничиваются ими, формиат, ацетат, пропионат, гликолят, глюконат, лактат, оксалат, малеат, сукцинат, фумарат, тартрат, цитрат, глутамат, аспартат, бензоат, метансульфонат, п-толуолсульфонат, салицилат и т.п. Эти соли могут быть получены способами, известными в данной области техники. "Фармацевтически приемлемые соли присоединения основания" включают в себя, но не ограничиваются ими, соли неорганических оснований, такие как соли натрия, калия, кальция и магния, или включают в себя, но не ограничиваются ими, соли органических оснований, таких как соль аммония, соль триэтиламина, соль лизина, соль аргинина и тому подобное. Эти соли могут быть получены способами, известными в данной области техники.
В настоящей заявке соединения формулы (I) могут быть получены в одной или нескольких кристаллических формах, активные соединения по настоящему изобретению включают различные полиморфы и их смеси.
В настоящей заявке термин "сольват" относится к комплексу, образованному соединением по настоящему изобретению с растворителем. Сольват может быть образован либо путем реакции в растворителе, или осажден, или кристаллизован из растворителя. Например, комплекс, образованный с водой, называется "гидрат". Сольваты соединений формулы (I) находятся в пределах объема настоящего изобретения.
Соединения формулы (I) по настоящему изобретению могут содержать один или несколько хиральных центров и могут существовать в различных оптически активных формах. Когда соединение содержит хиральный центр, соединение включает энантиомеры. Настоящее изобретение включает в себя оба изомера и их смесь, такие как рацемические смеси. Энантиомеры могут быть разделены с использованием способов, известных в данной области техники, таких как кристаллизация и хиральная хроматография и тому подобное. Когда соединение формулы (I) содержит больше одного хирального центра, соединения могут включать диастереомеры. Настоящее изобретение включает конкретные изомеры, разделенные на оптически чистые изомеры, а также смеси диастереомерных изомеров. Диастереомерные изомеры могут быть разделены с использованием способов, известных в данной области техники, таких как кристаллизация и препаративная хроматография.
Настоящее изобретение включает пролекарства вышеуказанных соединений. Пролекарства включают известные аминозащитные группы и карбоксилзащитные группы, которые гидролизуются в физиологических условиях или высвобождаются в ходе ферментативной реакции с получением исходных соединений. Конкретные способы получения пролекарств можно найти в (Saulnier, MG; Frennesson, DB; Deshpande, MC; Hansel, SB and Vysa, DMBioorg. Med. Chem Lett. 1994, 4, 1985-1990; and Greenwald, RB; Choe, YH; Conover, CD; Shum, K.; Wu, D.; Royzen, M.J. Med. Chem. 2000, 43, 475).
Способ получения
Настоящее изобретение предлагает способ получения соединений формулы (I), соединения по настоящему изобретению могут быть легко получены с помощью различных синтетических операций, эти операции известны специалистам в данной области техники. Примеры получения этих соединений могут включать в себя (но не ограничиваются ими) способы, описанные ниже.
Предпочтительно, соединения формулы (I) могут быть получены с помощью следующих схем и иллюстративных способов, описанных в варианте осуществления, а также связанных публикациях, доступных для специалистов в данной области техники.
На практике, процедуры способа могут быть по желанию расширены или скомбинированы.
Схема 1:

Стадия 1: Реакция по хлору в 4 положении пиримидина с боратом или борной кислотой, или с соответствующим амином может быть проведена в присутствии подходящего катализатора и подходящего растворителя при определенной температуре. Палладиевым катализатором, используемым в реакции Сузуки, может быть, но не ограничиваясь им, PdCl2(dppf) ([1,1'-бис(дифенилфосфино)ферроцен]палладий(II) дихлорид), и используемым основанием может быть, но не ограничиваясь им, карбонат натрия.
Стадия 2: Замена хлора во 2 положении пиримидина амином может происходить в присутствии подходящего катализатора и подходящего растворителя при определенной температуре. В качестве катализатора может быть использована кислота, которой может являться, но не ограничиваясь ею, трифторуксусная кислота (TFA) или пара-толуолсульфонат. Может быть использовано аминирование Бухвальд-Хартвига, используемым палладиевым катализатором может быть, но не ограничивается им, Pd2(dba)3 (трис(дибензилиденацетон)дипалладий(0)), используемым лигандом может быть, но не ограничивается им, Ксантфос (4,5-бис(дифенилфосфино)-9,9-диметилксантен), используемым основанием может быть, но не ограничивается им, карбонат цезия.
Стадия 3: Замена фтора в 4 положении бензола амином может происходить в присутствии подходящего основания и подходящего растворителя при определенной температуре. Используемым основанием может быть, но не ограничивается им, карбонат калия.
Стадия 4: Нитросоединение может быть превращено в соответствующее аминосоединение в кислотлных условиях с использованием металла (металлами могут быть, но не ограничиваются ими, порошок железа, порошок цинка) или двухлористого олова путем восстановления; или путем гидрирования в присутствии катализатора палладия на угле.
Стадия 5: Для получения амида в основных условиях аминосоединение можно конденсировать с соответствующим ацилхлоридом, или для получения амида в присутствии агента конденсации аминосоединение можно конденсировать с соответствующей карбоновой кислотой.
Реакции, проводимые на каждой из этих стадий, представляют собой обычные реакции, знакомые специалистам в данной области техники.
Соединения формулы (I), способы их получения, фармацевтические композиции и стратегии лечения, раскрытые в настоящем изобретении, могут быть воспроизведены специалистом в данной области техники с помощью ссылки на эту статью и соответствующих улучшений параметров процесса.
Следует особо отметить, что все подобные модификации и изменения очевидны для специалиста в данной области техники и они считаются включенными в настоящее изобретение. Предпочтительные варианты продуктов, способов и применений настоящего изобретения описаны, и соответствующие специалисты, очевидно, могут модифицировать или изменять и комбинировать способы и применения настоящего изобретения, не отступая от содержания, сути и объема настоящего изобретения, для реализации и применения настоящей технологии.
По сравнению с предшествующим уровнем техники основные преимущества настоящего изобретения заключаются в следующем:
(1) Соединения по настоящему изобретению проявляют высокую ингибирующую активность по отношению к EGFR Т790М мутантным (в частности с двойной мутацией EGFR T790M/L858R) фермантам и клеткам, но низкую ингибирующую активность по отношению к ферментам и клеткам с EGFR дикого типа (EGFR WT) и, следовательно, обладают высокой селективной ингибирующей активностью.
(2) Соединения по настоящему изобретению не только демонстрируют высокоселективное ингибирование в отношении ферментов и клеток с двойной мутацией EGFR, но также обладают низкой неспецифической цитотоксичностью.
(3) Соединения по настоящему изобретению также обладают улучшенными физическими свойствами (например высокой растворимостью в воде), благоприятными характеристиками токсичности (например низкой склонностью к блокированию hERG (ген человека, кодирующий калиевые каналы)), и преимущественными метаболическими характеристиками (например лучшими фармакокинетическими характеристиками, такими как биологическая доступность) по сравнению с другими известными EGFR-мутантными ингибиторами.
Для того, чтобы специалисты в данной области техники могли лучше понять техническое решение по изобретению, настоящее изобретение будет дополнительно подробно описано в сочетании со следующими конкретными вариантами осуществления.
Реагенты и инструменты
В настоящем изобретении структуру и чистоту соединений определяют методом ядерного магнитного резонанса (1Н-ЯМР) и/или методом жидкостной хроматографии с масс-спектрометрией (ЖХ-МС). 1Н-ЯМР: спектрометр Bruker AVANCE-400 NMR, внутренний стандарт - тетраметилсилан (TMS). ЖХ-МС: Agilent 1200 ВЭЖХ система/6140 МС жидкостный хромато-масс спектрометр (от компании Agilent), колонка WatersX-Bridge, 150×4,6 мм, 3,5 мкм. Препаративная высокоэффективная жидкостная хроматография (пре-ВЭЖХ): Waters PHW007, колонка XBridge С18, 4,6*150 мм, 3,5 мкм.
Автоматизированная элюирующая система Combiflash-Rf75 или Rf200 от компании ISCO, одноразовая колонка с силикагелем Agela 4 г, 12 г, 20 г, 40 г, 80 г, 120 г.
Тонкослойные пластины с силикагелем представляют собой пластины с силикагелем Yantai Huanghai HSGF254 или Qingdao GF254, тонкослойная хроматография (ТСХ), пластины с силикагелем, используемые для детектирования реакции, имеют размер 0,15 мм - 0,2 мм, пластины с силикагелем для очистки продукта с помощью тонкослойной хроматографии ТСХ имеют размер 0,4 мм - 0,5 мм. В качестве носителя, как правило, используют силикагель Yantai Huanghai 200-300 меш. В качестве носителя в колонке из щелочного оксида алюминия обычно используют FCP 200-300 меш щелочной оксид алюминия.
Известные исходные материалы согласно данному изобретению могут быть синтезированы с использованием способов, известных в данной области техники, или могут быть приобретены у компаний ABCR GmbH & Co.KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc и Darui Chemical Company и т.п.
Все примеры проводят в атмосфере азота или в атмосфере аргона и раствор представляет собой водный раствор, если не указано иное.
В настоящей заявке термин DMF обозначает диметилформамид, термин DMCO обозначает диметилсульфоксид, термин THF обозначает тетрагидрофуран, термин DIEA относится к N,N-диизопропилэтиламину, термин ЕА обозначает этилацетат, термин РЕ относится к петролейному эфиру, термин BINAP относится к (2R,3S)-2,2'-бис-дифенилфосфино-1,1'-бинафтилу, термин NBS относится к N-бромсукцинимиду, термин NCS относится к N-хлорсукцинимиду, термин Pd2(dba)3 Accela ChemBio относится к трис(дибензилиденацетон)дипалладию, Pd(dppf)Cl2 обозначает [1,1'-бис(дифенилфосфино)ферроцен]палладия дихлорид.
В настоящей заявке комнатная температура означает температуру приблизительно 25°С.
Получение соединения 1а

Стадия а: в реакционную колбу емкостью 500 мл добавляют соединение 1а1 (10,6 г, 58 ммоль) и смешанный раствор THF/вода (100 мл/60 мл) добавляют для растворения соединения. Последовательно при перемешивании при комнатной температуре добавляют хлорид аммония (15,5 г, 292 ммоль) и порошок восстановленного железа (26 г, 467 ммоль) и затем реакционную систему нагревают до 65°С и перемешивают непрерывно в течение 3 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции избыток порошка железа удаляют фильтрованием и осадок на фильтре промывают три раза с помощью ЕА. Фильтрат трижды экстрагируют системой ЕА/вода, отделяют органический слой, промывают водой, насыщенным солевым раствором, сушат над безводным Na2SO4 и концентрируют при пониженном давлении, получая соединение 1а2 (8,0 г), которое используют непосредственно в следующей реакции. Выход: 93%; чистота: 90%; МС m/z (с электрораспылительной ионизацией (ЭРИ)): 142,0 [М+Н]+.
Стадия b: В реакционную колбу емкостью 500 мл добавляют соединение 1а2 (8,0 г, 43 ммоль), для растворения промежуточного соединения добавляют при постоянном перемешивании концентрированную серную кислоту (100 мл) при -20°С. Концентрированную азотную кислоту (6,15 мл, 48 ммоль) медленно, по каплям добавляют при перемешивании и реакционную смесь перемешивают в течение 5 мин при этой температуре. Ход реакции контролируют с помощью ТСХ. После завершения реакции смесь выливают в ледяную воду. Раствор гидроксид натрия/вода (150 мл/300 мл) медленно добавляют к реакционной системе при температуре -20°С на ледяной бане и значение pH полученной смеси доводят до 8-9, Реакционный раствор трижды экстрагируют с помощью системы ЕА/вода, органический слой отделяют, промывают водой, насыщенным солевым раствором, сушат над безводным Na2SO4 и концентрируют при пониженном давлении, получая соединение 1а (8,7 г), которое используют непосредственно в следующей реакции. Выход: 80%; чистота: 100%; МС m/z (ЭРИ): 187,0 [М+Н]+; 1Н-ЯМР (400 МГц, DMCO-d6): δ 7,34 (d, J=7,8 Гц, 1Н), 7,04 (d, J=13,4 Гц, 1Н), 5,25 (brs, 2Н), 3,90 (s, 3Н).
Получение промежуточного соединения 2а

Стадия 1: Соединение 1а (11,16 г, 60 ммоль) растворяют в 150 мл дихлорметана, ди-трет-бутилдикарбонат (15,60 г, 72 ммоль), триэтиламин (12,24 г, 120 ммоль) и 4-диметиламинопиридин (0,74 г, 6 ммоль) добавляют и смесь перемешивают при комнатной температуре в течение 18 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции реакционный раствор концентрируют при пониженном давлении, изолируют и подвергают очистке с помощью колоночной хроматографии [РЕ:ЕА=80:20], с получением целевого соединения 2а2 (12,56 г, 73%). МС m/z (ЭРИ): 285 [М-Н]+.
Стадия 2: Соединение 2а2 (11,46 г, 40 ммоль) растворяют в 60 мл N,N-диметилацетамида и добавляют N,N,N'-триметилэтилендиамин (4,90 г, 48 ммоль), N,N-диизопропилэтиламин (7,74 г, 60 ммоль) и нагревают до 90°С и перемешивают в течение 6 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции реакционный раствор охлаждают до комнатной температуры, выливают в воду со льдом, экстрагируют этилацетатом, промывают водой и насыщенным солевым раствором, сушат над безводным Na2SO4 и концентрируют при пониженном давлении с получением целевого соединения 2а3 (12,51 г, 85%). Полученное соединение используют непосредственно в следующей реакции. МС m/z (ЭРИ): 369 [М+Н]+.
Стадия 3: Соединение 2а3 (12 г, 32,6 ммоль) растворяют в 200 мл метанола и добавляют 1,0 г 10% Pd/C. После замены воздуха водородом реакционный раствор перемешивают в течение 1 ч в атмосфере водорода, заполняющего баллон, при комнатной температуре. Ход реакции контролируют с помощью ТСХ. После завершения реакции реакционную систему фильтруют с помощью воронки Бюхнера, осадок на фильтре промывают небольшим количеством метанола и фильтрат концентрируют, получая целевое соединение 2а4 (10,70 г, 97%). Полученное соединение непосредственно используют в следующей реакции. МС m/z (ЭРИ): 339 [M+H]+.
Стадия 4: Соединение 2а4 (10,1 г, 30 ммоль) и триэтиламин (6,12 г, 60 ммоль) растворяют в 200 мл дихлорметана и охлаждают до 0°С. Добавляют акрилоил хлорид (3,24 г, 36 ммоль) и перемешивают в атмосфере азота при комнатной температуре в течение 3 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции реакционный раствор промывают последовательно насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором, сушат над безводным Na2SO4, фильтруют и концентрируют при пониженном давлении с получением целевого соединения 2а5 (9,64 г, 82%). Полученное соединение непосредственно используют в следующей реакции. МС m/z (ЭРИ): 393 [М+Н]+.
Стадия 5: Соединение 2а5 (9,41 г, 24 ммоль) растворяют в 100 мл дихлорметана, охлаждают до 0°С, добавляют 20 мл трифторуксусной кислоты, и реакционную смесь перемешивают в атмосфере азота при комнатной температуре в течение 18 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции реакционный раствор концентрируют при пониженном давлении. Остаток растворяют в 300 мл дихлорметана, промывают насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют при пониженном давлении, получая неочищенный продукт, который очищают с помощью колоночной хроматографии [DCM (дихлорметан) : МеОН = 10:1] с получением целевого соединения 2а (3,26 г, 46,5%). МС m/z (ЭРИ): 293 [M+H]+.
Получение промежуточного соединения 3а

Получение промежуточного соединения 4а

Пример 1: N-(5-(5-хлор-4-(1-метил-1H-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-амино)-2-((2-(диметиламино)этил)метиламино)-4-метоксифенил)акриламид (соединение Z-1)

Стадия 1: 5-бром-1-метил-1Н-пирроло[2,3-b]пиридин (соединение 1-2)
При температуре 0°С раствор соединения 5-бром-1Н-пирроло[2,3-b]пиридина (5 г, 25,4 ммоль, коммерчески доступный) в DMF добавляют к раствору гидрида натрия (4,32 г, 30,5 ммоль) в 40 мл DMF и энергично перемешивают при 0°С в течение 0,5 ч, затем добавляют йодистый метил (1,22 г, 30,5 ммоль) и энергично перемешивают при комнатной температуре в течение 3 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции реакционный раствор выливают в воду со льдом. После полного осаждения твердой фазы ее отфильтровывают и растворяют в дихлорметане и метаноле (10:1), трижды промывают водой, и сушат. Органический слой отделяют и концентрируют при пониженном давлении, получая соединение 1-2 (5,4 г, 99%). Продукт используют непосредственно на следующей стадии. МС m/z (ЭРИ): 211,0 [М+Н]+.
Стадия 2: 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пирроло[2,3-b]пиридин (соединение 1-3)
Бис(пинаколато)борат (11,26 г, 43,6 ммоль), PdCl2(dppf) (814 мг, 1,11 ммоль) и ацетат калия (6,53 г, 32,7 ммоль) добавляют к раствору соединения 1-2 (4,6 г, 21,8 ммоль) в 50 мл 1,4-диоксана и энергично перемешивают в атмосфере N2 при температуре 90°С в течение 8 часов. После завершения реакции смесь фильтруют и фильтрат концентрируют с получением неочищенного соединения 1-3 (13,9 г), которое используют непосредственно на следующей стадии. МС m/z (ЭРИ): 259,1 [М+Н]+.
Стадия 3: 5-(2,5-дихлорпиримидин-4-ил)-1-метил-1Н-пирроло[2,3-b]пиридин (соединение 1-4)
2,4,5-трихлорпиримидин (4,93 г, 26,2 ммоль), PdCl2(dppf) (1,66 г, 2,26 ммоль, коммерчески доступен) и 23 мл 2,0 моль/л раствора карбоната натрия добавляют к раствору соединения 1-3 (13,9 г, 21,8 ммоль) в 50 мл ацетонитрила и интенсивно перемешивают в атмосфере N2 при 85°С в течение 4 часов. После завершения реакции реакционный раствор разбавляют водой, экстрагируют системой ЕА/вода, трижды промывают водой и сушат и органический слой концентрируют при пониженном давлении с получением неочищенного соединения, которое очищают с помощью колоночной комби-флэш хроматографии [РЕ:ЕА=100:0-20:80] с получением соединения 1-4 (2,16 г, 36%). МС m/z (ЭРИ): 279,0 [М+Н]+.
Стадия 4: 5-хлоро-N-(4-фторо-2-метокси-5-нитрофенил)-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-амин (соединение 1-5)
Соединение 4-фтор-2-метокси-5-нитроанилин (1,13 г, 6,09 ммоль), Pd2(dba)3 (558 мг, 0,61 ммоль), Ксантфос (705 мг, 1,22 ммоль) и карбонат цезия (3,97 г, 12,2 ммоль) добавляют к раствору соединения 1-4 (1,70 г, 6,09 ммоль) в 20 мл 1,4-диоксана и энергично перемешивают в атмосфере N2 при температуре 120°С в течение 5 ч. После завершения реакции фильтруют и фильтрат концентрируют при пониженном давлении с получением неочищенного продукта, очищают колоночной комби-флэш хроматографией [РЕ:ЕА=100:0-0:100] с получением соединения 1-5 (420 мг, 16%). МС m/z (ЭРИ): 429,0 [М+Н]+.
Стадия 5: N1-(5-хлор-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамин (соединение 1-6)
N,N,N'-триметилэтилендиамин (72 мг, 0,70 ммоль) и карбонат калия (193 мг, 1,40 ммоль) добавляют к раствору соединения 1-5 (200 мг, 0,47 ммоль) в 4 мл DMF, и энергично перемешивают при температуре 100°С в течение 2 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции добавляют 10 мл воды и трижды экстрагируют системой ЕА/вода. Органический слой отделяют и концентрируют при пониженном давлении с получением соединения 1-6 (240 мг, 90%). МС m/z (ЭРИ): 511,3 [М+Н]+.
Стадия 6: N4-(5-хлор-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-ил)-N1-(2-(диметиламино)этил)-5-метокси-N1-метилбензол-1,2,4-триамин (соединение 1-7)
Pd/C (20 мг) добавляют к раствору соединения 1-6 (100 мг, 0,20 ммоль) в 20 мл метанола и энергично перемешивают в атмосфере Н2 при комнатной температуре в течение 4 часов. После завершения реакции реакционный раствор фильтруют и фильтрат концентрируют, получая соединение 1-7 (80 мг, 83%), которое непосредственно используют на следующей стадии. МС m/z (ЭРИ): 481,2 [М+Н]+.
Стадия 7: N-(5-(5-хлор-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-амино)-2-((2-(диметиламино)этил)метиламино)-4-метоксифенил)акриламид (соединение Z-1)
Акрилоил хлорид (17 мг, 0,17 ммоль) и триэтиламин (28 мг, 0,25 ммоль) добавляют к раствору соединения 1-7 (80 мг, 0,17 ммоль) в 2 мл дихлорметана при температуре 0°С, и энергично перемешивают при 0°С в течение 2 ч. После завершения реакции смесь разбавляют водой и трижды экстрагируют системой дихлорметан/вода и органический слой концентрируют при пониженном давлении с получением неочищенного продукта, который отделяют и очищают с помощью препаративной жидкостной хроматографии с получением соединения Z-1 (2,39 мг, 6%). МС m/z (ЭРИ): 534,9 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6) δ 10,11 (s, 1Н), 8,87-8,75 (m, 2Н), 8,57 (d, J=17,2 Гц, 3Н), 7,62 (d, J=3,5 Гц, 1Н), 7,00 (s, 1Н), 6,57 (d, J=3,4 Гц, 1Н), 6,44-6,35 (m, 1Н), 6,29 (d, J=16,7 Гц, 1Н), 5,77 (d, J=12,0 Гц, 1Н), 3,87 (s, 3Н), 3,82 (s, 3Н), 2,85 (s, 2H), 2,70 (s, 3Н), 2,29 (d, J=5,7 Гц, 2H), 2,19 (s, 6H).
Пример 2: N-(5-(5-хлор-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-амино)-2-(4-(диметиламино)пиперидин-1-ил)-4-метоксифенил)акриламид (соединение Z-2)

Стадия 1: 5-хлор-N-(4-(4-(диметиламино)пиперидин-1-ил)-2-метокси-5-нитрофенил)-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-амин (соединение 2-6)
Соединение 1-5 (260 мг, 0,607 ммоль) и карбонат калия (251 мг, 1,822 ммоль) добавляют в реакционную колбу емкостью 25 мл, для частичного растворения промежуточного соединения добавляют DMF (10 мл), а затем добавляют N,N-диметиламинопиперидин (85,4 мг, 0,67 ммоль) и реакционную систему поддерживают при 70°С в течение 3 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции реакционный раствор трижды экстрагируют системой ЕА/вода, органический слой отделяют, промывают водой, насыщенным раствором соли, сушат над безводным Na2SO4 и концентрируют при пониженном давлении с получением неочищенного промежуточного соединения 2-6, 220 мг, выход 68%. МС M/Z (ЭРИ) 537,2 [M+H]+.
Стадия 2: N1-(5-хлор-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-ил)-4-(4-(диметиламино)пиперидин-1-ил)-6-метоксибензол-1,3-диамин (соединение 2-7)
Промежуточное соединение реакции 2-6 (110 мг, 0,205 ммоль) добавляют в реакционную колбу емкостью 25 мл и добавляют этанол (6 мл). Двухлористое олово (138 мг, 0,615 ммоль) добавляют в реакционную колбу при перемешивании при комнатной температуре и реакционную систему поддерживают при 60°С в течение 2 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции, реакционный раствор концентрируют путем выпаривания и трижды экстрагируют системой ЕА/вода и органический слой отделяют и промывают водой, насыщенным раствором соли и сушат над безводным Na2SO4, Фильтрат концентрируют при пониженном давлении с получением неочищенного продукта 2-7 (80 мг, 76,9%), который используют непосредственно в следующей реакции.
Стадия 3: N-(5-(5-хлор-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-амино)-2-(4-(диметиламино)пиперидин-1-ил)-4-метоксифенил)акриламид (соединение Z-2)
Акрилоил хлорид (17 мг, 0,188 ммоль) и DIEA(60,8 мг, 0,471 ммоль) добавляют к раствору соединения 2-7 (80 мг, 0,157 ммоль) в 3 мл THF при 0°С и энергично перемешивают при 0°С в течение 2 ч. После завершения реакции реакционный раствор разбавляют водой и трижды экстрагируют системой дихлорметан/вода, а органический слой концентрируют при пониженном давлении с получением неочищенного продукта, который отделяют и очищают с помощью препаративной жидкостной хроматографии с получением указанного соединения Z-2 (9,0 мг, 10%). МС m/z (ЭРИ): 560,8 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6) δ 8,98 (s, 1Н), 8,75-8,74 (d, J=4,0 Гц, 1Н), 8,48-8,53 (m, 4Н), 7,62-7,61 (d, J=4,0 Гц, 1Н), 6,84 (s, 1Н), 6,71-6,57 (m, 2Н), 6,29-6,25 (d, J=16,0 Гц, 1Н), 5,77-5,74 (d, J=12,0 Гц, 1Н), 3,87 (s, 3Н), 3,82 (s, 3Н), 3,05-3,02 (d, J=12,0 Гц, 2Н), 2,69-2,63 (m, 2Н), 2,30-2,19 (m, 7Н), 1,91-1,83 (m, 2Н), 1,70-1,68 (m, 2Н).
Пример 3: N-(5-(5-хлор-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-амино)-4-метокси-2-(4-метилпиперазин-1-ил)фенил)акриламид(соединение Z-3)
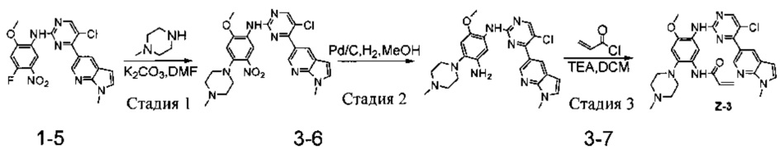
Стадия 1: 5-хлор-N-(2-метокси-4-(4-метилпиперазин-1-ил)-5-нитрофенил)-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-амин (соединение 3-6)
N-метилпиперазин (126 мг, 1,26 ммоль) и карбонат калия (261 мг, 1,9 ммоль) добавляют к раствору соединения 1-5 (200 мг, 0,47 ммоль) в 4 мл DMFA и энергично перемешивают при 100°С в течение 2 ч, ход реакции контролируют с помощью ТСХ. После завершения реакции добавляют 10 мл воды, экстрагируют системой ЕА/вода, органический слой отделяют и концентрируют при пониженном давлении с получением соединения 3-6 (200 мг, 84%). МС m/z (ЭРИ): 509,2 [М+Н]+.
Стадия 2: N1-(5-хлор-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-ил)-6-метокси-4-(4-метилпиперазин-1-ил)бензол-1,3-диамин (соединение 3-7)
С использованием соединения 3-6 (200 мг, 0,39 ммоль) в качестве исходного материала соединение 3-7 (140 мг, 75%) синтезируют согласно стадии 6 Примера 1 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 479,1 [М+Н]+.
Стадия 3: N-(5-(5-хлор-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-амино)-4-метокси-2-(4-метилпиперазин-1-ил)фенил)акриламид (соединение Z-3)
Используя соединение 3-7 (140 мг, 0,29 ммоль) в качестве исходного материала соединение Z-3 (1,60 мг, 1%) синтезируют согласно стадии 7 Примера 1, МС m/z (ЭРИ): 533,2 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6) δ 8,98 (s, 1Н), 8,76 (s, 1Н), 8,51 (d, J=32,1 Гц, 3Н), 7,63 (s, 1Н), 6,86 (s, 1Н), 6,63 (d, J=28,9 Гц, 2Н), 6,27 (d, J=16,2 Гц, 1Н), 5,76 (d, J=9,1 Гц, 1Н), 5,33 (s, 1Н), 3,88 (s, 3Н), 3,84 (s, 3Н), 2,86 (s, 4Н), 2,55 (s, 2Н), 2,26 (s, 3Н), 1,99 (s, 2Н).
Пример 4: N-(2-(4-(диметиламино)пиперидин-1-ил)-4-метокси-5-(4-(1-метил-1Н-пиразол[3,4-b]пиридин-5-ил)пиримидин-2-иламино)фенил)акриламид (соединение Z-4)

Стадия 1: 5-бром-1-метил-1Н-пиразол[3,4-b]пиридин (соединение 4-2)
Гидрид натрия (720 мг, 30 ммоль) добавляют в реакционную колбу емкостью 100 мл, и безводный THF (40 мл) добавляют в реакционную колбу. После перемешивания при 0°С в течение 5 мин промежуточное соединение реакции 5-бром-1Н-пиразол[3,4-b]пиридина (4,0 г, 20 ммоль, коммерчески доступный) растворяют в THF (20 мл) и полученный раствор медленно по каплям добавляют в реакционную колбу через воронку постоянного давления и перемешивают в течение 30 мин. Затем йодистый метил (1,6 мл, 26 ммоль) добавляют по каплям к реакционной системе. После завершения добавления реакционный раствор медленно нагревают до комнатной температуры и перемешивают в течение ночи. Ход реакции контролируют с помощью ТСХ. После завершения реакции 10 мл ледяной воды добавляют к реакционной системе для подавления реакции, THF удаляют концентрированием при пониженном давлении и остаток экстрагируют дихлорметаном (60 мл) и водой (20 мл × 3), Органический слой сушат над безводным Na2SO4 и концентрируют при пониженном давлении с получением 4,1 г твердого вещества коричневого цвета, которое отделяют и очищают с помощью колоночной комби-флэш хроматографии [РЕ:ЕА=10:90-40:60] с получением целевого соединения 4-2 (3,1 г, 73%). МС m/z (ЭРИ): 211,9 [М+Н]+.
Стадия 2: 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразоло[3,4-b]пиридин (соединение 4-3)
Соединение 4-2 (3,1 г, 14,6 ммоль) и бис(пинаколато)борат (11,1 г, 43,8 ммоль) добавляют в реакционную колбу емкостью 250 мл, и добавляют DMF (150 мл) и ацетат калия (4,3 г, 43,8 ммоль). После того, как воздух реакционной системы трижды заменяют аргоном, добавляют Pd(dppf)Cl2 (0,55 г, 0,73 ммоль), а затем воздух заменяют азотом еще три раза. Реакционную смесь нагревают до 90°С, и перемешивают в течение 8 часов. Протекание реакции контролируют с помощью ЖХ-МС. После завершения реакции реакционный раствор фильтруют, осадок на фильтре промывают ЕА (30 мл × 3) и фильтрат концентрируют при пониженном давлении для удаления DMF и получают соединение 4-3 (13,5 г) в виде твердого вещества коричневого цвета, которое непосредственно используют в следующей реакции. МС m/z (ЭРИ): 260,2 [М+Н]+.
Стадия 3: 5-(2-хлорпиримидин-4-ил)-1-метил-1Н-пиразол[3,4-b]пиридин (соединение 4-4)
Промежуточное соединение 4-3 (3,8 г, 14,6 ммоль) и 2,4-дихлорпиримидин (2,6 г, 17,5 ммоль) добавляют в реакционную колбу емкостью 250 мл и добавляют ацетонитрил (130 мл) и раствор карбоната натрия (22 мл, 2М). После того, как воздух реакционной системы заменяют аргоном три раза, добавляют Pd(dppf)Cl2 (0,55 г, 0,73 ммоль), а затем воздух заменяют азотом еще три раза. Реакционную смесь нагревают до 85°С, и непрерывно перемешивают в течение 3 часов. Протекание реакции контролируют с помощью ЖХ-МС. После завершения реакции реакционную смесь охлаждают до комнатной температуры и осаждают большое количество твердой фазы и фильтруют, осадок на фильтре промывают ЕА (10 мл × 3) и сушат до постоянной массы с получением 2,2 г твердого вещества желтого цвета, фильтрат экстрагируют ЕА и водой, сушат, концентрируют, а затем очищают с помощью комби-флэш хроматографии [РЕ:ЕА=10:90-70:30] с получением 0,6 г желтого твердого вещества. Твердые вещества объединяют с получением целевого соединения 4-4 (2,8 г, 78%). МС m/z (ЭРИ): 246,1 [М+Н]+.
Стадия 4: N-(4-фтор-2-метокси-5-нитрофенил)-4-(1-метил-1Н-пиразоло[3,4-b]пиридин-5-ил)пиримидин-2-амин (соединение 4-5)
Промежуточное соединение реакции 4-4 (420 мг, 1,7 ммоль) и 4-фтор-2-метокси-5-нитроанилин (316 мг, 1,7 ммоль) помещают в 100 мл герметично закрытую пробирку и добавляют изобутанол (40 мл) для частичного растворения промежуточного соединения. Затем добавляют пара-толуолсульфоновую кислоту (807 мг, 4,25 ммоль), реакционную систему герметизируют и нагревают при 130°С в течение 16 часов. После завершения реакции реакционную смесь охлаждают до комнатной температуры и с осаждением из раствора большого количество твердой фазы. Твердые вещества отфильтровывают с помощью воронки Бюхнера для получения осадка на фильтре, ресуспендируют в этаноле при нагревании с обратным холодильником с получением целевого соединения 4-5 (524 мг, 78%), которое используют непосредственно в следующей реакции. Чистота: 47%. МС m/z (ЭРИ): 396,2 [М+Н]+
Стадия 5: N-(4-(4-(диметиламино)пиперидин-1-ил)-2-метокси-5-нитрофенил)-4-(1-метил-1Н-пиразоло[3,4-b]пиридин-5-ил)пиримидин-2-амин (соединение 4-6)
Промежуточное соединение 4-5 (146 мг, 0,25 ммоль) и карбонат калия (104 мг, 0,75 ммоль) добавляют в реакционную колбу емкостью 25 мл, DMF (10 мл) добавляют до полного растворения промежуточного соединения. Затем добавляют 4-диметиламинопиперидин (42 мг, 0,325 ммоль) и реакционную систему нагревают при 100°С в течение 2 ч. Ход реакции контролируют с помощью ТСХ. После завершения реакции, реакционный раствор трижды экстрагируют системой ЕА/вода, органический слой отделяют, промывают водой, насыщенным раствором соли, сушат над безводным Na2SO4 и концентрируют при пониженном давлении с получением целевого соединения 4-6 (120 мг, 98%). МС m/z (ЭРИ): 504,2 [М+Н]+.
Стадия 6: 4-(4-(диметиламино)пиперидин-1-ил)-6-метокси-N1-(4-(1-метил-1Н-пиразоло[3,4-b]пиридин-5-ил)пиримидин-2-ил)бензол-1,3-диамин (соединение 4-7)
С использованием соединения 4-6 (120 мг, 0,24 ммоль) в качестве исходного материала соединение 4-7 (113 мг) синтезируют согласно стадии 6 Примера 1 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 474,3 [М+Н]+.
Стадия 7: N-(2-(4-(диметиламино)пиперидин-1-ил)-4-метокси-5-(4-(1-метил-1Н-пиразоло[3,4-b]пиридин-5-ил)пиримидин-2-иламино)фенил)акриламид (соединение Z-4).
С использованием соединения 4-7 (113 мг, 0,24 ммоль) в качестве исходного материала полученный неочищенный продукт синтезируют согласно стадии 7 Примера 1 и очищают с помощью препаративной ТСХ-хроматографии [DCM:MeOH:NH4OH=9:1:0,1] с получением целевого соединения Z-4 (6 мг, 5%). МС m/z (ЭРИ): 528,3 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6) δ 9,33 (s, 1Н), 9,22 (s, 1Н), 9,04 (s, 1Н), 8,80 (s, 1H), 8,51-8,50 (d, J=5,6 Гц, 2Н), 8,13 (s, 1Н), 7,53-7,51 (d, J=5,6 Гц, 1Н), 6,86 (s, 1H), 6,74-6,67 (m, 1Н), 6,32-6,28 (d, J=17,0 Гц, 2Н), 5,79-5,67 (d, J=13,4 Гц, 1Н), 4,24 (s, 3Н), 3,86 (s, 3Н), 3,33 (s, 1Н), 3,05-3,03 (d, J=10,7 Гц, 2Н), 2,70-2,64 (t, J=11,7 Гц, 3Н), 2,22 (s, 6Н), 1,85-1,82 (d, J=11,3 Гц, 2Н), 1,70-1,67 (d, J=12,1 Гц, 2Н).
Пример 5: N-(4-метокси-5-(4-(1-метил-1Н-пиразол[3,4-b]пиридин-5-ил)пиримидин-2-иламино)-2-(4-метилпиперазин-1-ил)фенил)акриламид (соединение Z-5)

Стадия 1: N-(2-метокси-4-(4-метилпиперазин-1-ил)-5-нитрофенил)-4-(1-метил-1Н-пиразоло[3,4-b]пиридин-5-ил)пиримидин-2-амин (соединение 5-6)
Соединение 4-5 (146 мг, 0,25 ммоль) и карбонат калия (104 мг, 0,75 ммоль) добавляют в реакционную колбу емкостью 25 мл и добавляют DMF (10 мл) до полного растворения промежуточного соединения. Затем добавляют N-метилпиперазин (32 мг, 0,325 ммоль) и реакционную систему поддерживают при 100°С в течение 2 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции реакционный раствор троекратно экстрагируют системой ЕА/вода и органический слой отделяют, промывают водой, насыщенным раствором соли, сушат над безводным Na2SO4 и концентрируют при пониженном давлении с получением целевого соединения 5-6, которое непосредственно используют на следующей стадии. Чистота: 88%. МС m/z (ЭРИ): 476,2 [М+Н]+.
Стадия 2: 6-метокси-N1-(4-(1-метил-1Н-пиразоло[3,4-b]пиридин-5-ил)пиримидин-2-ил)-4-(4-метилпиперазин-1-ил)бензол-1,3-диамин (соединение 5-7)
Используя соединение 5-6 (119 мг, 0,25 ммоль) в качестве исходного материала, соединение 5-7 синтезируют согласно стадии 6 Примера 1 и используют непосредственно в следующей реакции. Чистота: 59%. МС m/z (ЭРИ): 446,2 [М+Н]+.
Стадия 3: N-(4-метокси-5-(4-(1-метил-1Н-пиразоло[3,4-b]пиридин-5-ил)пиримидин-2-иламино)-2-(4-метилпиперазин-1-ил)фенил)акриламид (соединение Z-5)
Используя соединение 5-7 (111 мг, 0,25 ммоль) в качестве исходного материала, полученный неочищенный продукт синтезируют согласно стадии 7 Примера 1 и очищают с помощью препаративной ТСХ [DCM:MeOH:NH4OH=9:1:0,1] с получением целевого соединения Z-5 (8 мг, 6%). Чистота: 96,5%. МС m/z (ЭРИ): 500,2 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6) δ 9,33 (s, 1Н), 9,21 (s, 1Н), 9,03 (s, 1Н), 8,79 (s, 1H), 8,51-8,50 (d, J=5,2 Гц, 2Н), 8,12 (s, 1Н), 7,52-7,51 (d, J=5,2 Гц, 1Н), 6,88 (s, 1H), 6,68-6,61 (m, 1Н), 6,31-6,27 (d, J=16,9 Гц, 2H), 5,78-5,76 (d, J=9,9 Гц, 1H), 4,24 (s, 3Н), 3,87 (s, 3Н), 2,86 (s, 4Н), 2,53-2,49 (d, J=13,6 Гц, 4H), 2,25 (s, 3Н).
Пример 6: N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-(4-(1-метил-1Н-пиразол[3,4-b]пиридин-5-ил)пиримидин-2-иламино)фенил)акриламид (соединение Z-6)
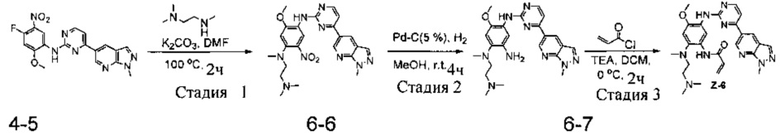
Стадия 1: N1-(2-(диметиламино)этил)-5-метокси-N1-метил-N4-(4-(1-метил-1Н-пиразоло[3,4-b]пиридин-5-ил)пиримидин-2-ил)-2-нитробензол-1,4-диамин (соединение 6-6)
Соединение 4-5 (146 мг, 0,25 ммоль) и карбонат калия (104 мг, 0,75 ммоль) добавляют в реакционную колбу емкостью 25 мл и добавляют DMF (10 мл) до полного растворения промежуточного соединения. Затем добавляют N,N,N-триметилэтилендиамин (34 мг, 0,325 ммоль) и реакционную систему поддерживают при 100°С в течение 2 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции реакционный раствор трижды экстрагируют системой ЕА/вода и органический слой отделяют, промывают водой, насыщенным раствором соли, сушат над безводным Na2SO4 и концентрируют при пониженном давлении с получением целевого соединения 6-6, которое непосредственно используют на следующей стадии. Чистота: 94%. МС m/z (ЭРИ): 478,2 [М+Н]+.
Стадия 2: N1-(2-(диметиламино)этил)-5-метокси-N1-метил-N4-(4-(1-метил-1Н-пиразоло[3,4-b]пиридин-5-ил)пиримидин-2-ил)бензол-1,2,4-триамин (соединение 6-7)
Используя соединение 6-6 (119 мг, 0,25 ммоль) в качестве исходного материала, соединение 6-7 синтезируют согласно стадии 6 Примера 1 и используют непосредственно в следующей реакции. Чистота: 83%. МС m/z (ЭРИ): 448,2 [М+Н]+.
Стадия 3: N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-(4-(1-метил-1Н-пиразоло[3,4-b]пиридин-5-ил)пиримидин-2-иламино)фенил)акриламид (соединение Z-6)
С использованием соединения 6-7 (111 мг, 0,25 ммоль) в качестве исходного материала, полученный неочищенный продукт синтезируют согласно стадии 7 Примера 1 и очищают с помощью преп-ВЭЖХ с получением целевого соединения Z-6 (1 мг, 1%). Чистота: 96,5%. МС m/z (ЭРИ): 502,2 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6) δ 10,13 (s, 1Н), 9,35 (s, 1Н), 9,27 (s, 1Н), 9,12 (s, 1H), 8,52-8,51 (d, J=5,3 Гц, 1H), 8,50 (s, 2Н), 8,20 (s, 1Н), 8,14 (s, 1H), 7,54-7,53 (d, J=5,2 Гц, 1H), 7,03 (s, 1H), 6,50-6,44 (d, J=10,0 Гц, 1H), 6,34-6,29 (d, J=17,3 Гц, 1H), 5,80-5,78 (d, J=10,6 Гц, 1H), 4,24 (s, 3Н), 3,87 (s, 3Н), 2,92-2,90 (d, J=5,4 Гц, 2H), 2,69 (s, 3Н), 2,39-2,36 (t, J=5,4 Гц, 2H), 2,25 (s, 6H).
Пример 7: N-(5-(5-хлор-4-морфолинопиримидин-2-иламино)-2-(4-(диметиламино)пиперидин-1-ил)-4-метоксифенил)акриламид (соединение Z-7) формиат
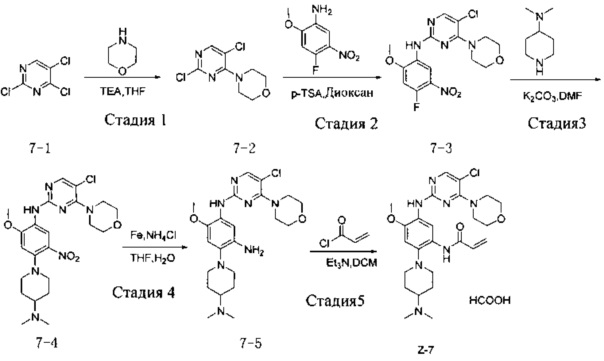
Стадия 1: 4-(2,5-дихлор-4-ил)морфолин (соединение 7-2)
При температуре 25°С соединение 2,4,5-трихлорпиримидин (5 г, 27,0 ммоль) добавляют к 25 мл THF, добавляют триэтиламин (5,5 г, 54,0 ммоль) и добавляют по каплям морфолин (2,37 г, 27,0 ммоль) в реакционную смесь и перемешивают при комнатной температуре в течение 1 ч. Ход реакции контролируют с помощью ТСХ. После завершения реакции добавляют воду и ЕА и органический слой отделяют, промывают насыщенным водным раствором хлорида натрия, сушат и концентрируют при пониженном давлении с получением соединения 7-2 (5,2 г), которое используют непосредственно на следующей стадии. МС m/z (ЭРИ): 234,0 [М+Н]+.
Стадия 2: 5-хлор-N-(4-фтор-2-метокси-5-нитрофенил)-4-морфолинопиримидин-2-амин (соединение 7-3)
Соединение 7-2 (1,0 г, 4,2 ммоль), 4-фтор-2-метокси-5-нитроанилин (0,83 г, 4,4 ммоль) и пара-толуолсульфоновую кислоту (2,57 г, 15,0 ммоль) добавляют к 10 мл 1,4 диоксана и энергично перемешивают в атмосфере N2 при температуре 110°С в течение 8 часов. После завершения реакции реакционный раствор фильтруют и осадок на фильтре промывают ЕА и сушат при температуре 50°С с получением неочищенного соединения 7-3 (1,16 г), которое используют непосредственно на следующей стадии. МС m/z (ЭРИ): 284,1 [М+Н]+.
Стадия 3: 5-хлоро-N-(4-(4-(диметиламино)пиперидин-1-ил)-2-метокси-5-нитрофенил)-4-морфолинопиримидин-2-амин (соединение 7-4)
Соединение 7-3 (0,1 г, 0,26 ммоль), 4-диметиламинопиперидин (0,033 г, 0,26 ммоль) и карбонат калия (0,072 г, 0,52 ммоль) добавляют к DMF и интенсивно перемешивают в атмосфере N2 при 100°С в течение 2 ч. После завершения реакции реакционную смесь разбавляют водой и трижды экстрагируют с помощью системы ЕА/вода, сушат, органический слой концентрируют при пониженном давлении с получением неочищенного соединения 7-4 (0,21 г), которое непосредственно используют на следующей стадии. МС m/z (ЭРИ): 492,2 [M+H]+.
Стадия 4: N-(5-хлоро-4-морфолинопиримидин-2-ил)-4-(4-(диметиламино)пиперидин-1-ил)-6-метоксибензол-1,3-диамин (соединение 7-5)
Соединение 7-4 (0,2 г, 0,41 ммоль), порошок железа (0,51 г, 8,1 ммоль) и хлорид аммония (0,11 г, 2,0 ммоль) добавляют к 16 мл смешанного раствора воды и THF и интенсивно перемешивают в атмосфере N2 при 65°С в течение 3 часов. После завершения реакции реакционную смесь фильтруют и фильтрат экстрагируют с помощью ЕА, промывают водой, сушат и концентрируют при пониженном давлении с получением соединения 7-5 (0,19 г), которое используют непосредственно на следующей стадии. МС m/z (ЭРИ): 462,2 [М+Н]+.
Стадия 5: N-(5-(5-хлоро-4-морфолинопиримидин-2-ил)амино)-2-(4-(диметиламино)пиперидин-1-ил)-4-метоксифенил)акриламид (соединение Z-7) формиат
При температуре 0°С акрилоилхлорид (40 мг, 0,43 ммоль) и триэтиламин (50 мг, 0,49 ммоль) добавляют к раствору соединения 7-5 (190 мг, 0,41 ммоль) в 5 мл дихлорметана и энергично перемешивают при 0°С в течение 2 ч. После завершения реакции реакционную смесь разбавляют водой и трижды экстрагируют системой дихлорметан/вода, органический слой концентрируют при пониженном давлении с получением неочищенного продукта, который очищают с помощью препаративной жидкостной хроматографии с получением формиата соединения Z-7 (4,27 мг, выход трех стадий 3,1%). МС m/z (ЭРИ): 562,2 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6) δ 8,98 (s, 1Н), 8,57 (s, 1Н), 8,28 (s, 1Н), 8,06 (s, 1Н), 7,83 (s, 1Н), 6,80 (s, 1Н), 6,65 (dd, J=17,0, 10,1 Гц, 1Н), 6,21 (d, J=17,0 Гц, 1Н), 5,72 (d, J=10,3 Гц, 1Н), 3,84 (s, 3Н), 3,65 (d, J=7,6 Гц, 8Н), 3,02 (d, J=11,0 Гц, 2Н), 2,64 (t, J=11,0 Гц, 2Н), 2,27 (перекрывание, 7Н), 1,84 (d, J=10,7 Гц, 2Н), 1,68 (d, J=9,6 Гц, 2Н).
Пример 8: N-(5-(5-хлор-4-морфолинопиримидин-2-иламино)-4-метокси-2-(4-метилпиперазин-1-ил)фенил)акриламид (соединение Z-8)

Стадия 1: 5-хлоро-N-(2-метокси-4-(4-метилпиперазин-1-ил)-5-нитрофенил)-4-м орфолинопиримидин-2-амин (соединение 8-4)
Соединение 7-3 (0,5 г, 1,3 ммоль), N-метилпиперазин (0,13 г, 1,3 ммоль) и карбонат калия (0,36 г, 2,6 ммоль) добавляют к DMF и перемешивают в атмосфере N2 при температуре 100°С в течение 2 часов. После завершения реакции реакционную смесь разбавляют водой и экстрагируют системой ЕА/вода, трижды промывают водой и сушат и органический слой концентрируют при пониженном давлении с получением неочищенного соединения 8-4 (0,6 г), которое используют непосредственно на следующей стадии. МС m/z (ЭРИ): 464,1 [М+Н]+.
Стадия 2: N-(5-хлор-4-морфолинопиримидин-2-ил)-6-метокси-4-(4-метилпипер азин-1-ил)бензол-1,3-диамин (соединение 8-5)
Используя соединение 8-4 (0,6 г, 1,3 ммоль) в качестве исходного материала, соединение 8-5 (0,56 г) синтезируют согласно стадии 4 Примера 7 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 434,2 [М+Н]+.
Стадия 3: N-(5-(5-хлоро-4-морфолинопиримидин-2-иламино)-4-метокси-2-(4-метилпиперазин-1-ил)фенил)акриламид (соединение Z-8)
Используя соединение 8-5 (0,56 г, 1,3 ммоль) в качестве исходного материала, синтезируют неочищенный продукт согласно стадии 7 Примера 1 и очищают с помощью препаративной жидкостной хроматографии с получением соединения Z-8 (44,51 мг, выход трех стадий 7,0%). МС m/z (ЭРИ): 488,2 [М+Н]+; 1Н-ЯМР (400 МГц, CDCl3) δ 9,26 (s, 1Н), 8,46 (s, 1Н), 7,94 (s, 1Н), 7,43 (s, 1Н), 6,69 (s, 1Н), 6,29 (d, J=16,8 Гц, 1Н), 6,18 (dd, J=16,8, 10,0 Гц, 1Н), 5,66 (dd, J=10,0, 1,3 Гц, 1Н), 3,78 (s, 3Н), 3,76 (s, 8Н), 2,82 (d, J=4,2 Гц, 4Н), 2,55 (d, J=13,4 Гц, 4Н), 2,32 (s, 3Н).
Пример 9: N-(5-(5-хлор-4-морфолинопиримидин-2-иламино)-2-((2-(диметиламино)этил)метиламино)-4-метоксифенил)акриламид (соединение Z-9)

Стадия 1: N-(5-хлоро-4-морфолинопиримидин-2-ил)-N-(2-(диметиламино)этил)-2-метокси-N-метил-5-нитробензол-1,4-диамин (соединение 9-4)
Соединение 7-3 (0,5 г, 1,3 ммоль), N,N,N'-триметил этилендиамин (0,26 г, 2,6 ммоль) и карбонат калия (0,36 г, 2,6 ммоль) добавляют к DMF и перемешивают в атмосфере N2 при 100°С в течение 2 часов. После завершения реакции, реакционную смесь разбавляют водой и экстрагируют системой ЕА/вода, трижды промывают водой, сушат, органический слой концентрируют при пониженном давлении с получением неочищенного соединения 9-4 (0,6 г), которое непосредственно используют на следующей стадии. МС m/z (ЭРИ): 466,1 [М+Н]+.
Стадия 2: N-(5-хлоро-4-морфолинопиримидин-2-ил)-N-(2-(диметиламино)этил)-5-метокси-N-метилбензол-1,2,4-триамин (соединение 9-5)
Используя соединение 9-4 (0,6 г, 1,3 ммоль) в качестве исходного материала, соединение 9-5 (0,56 г) синтезируют согласно стадии 4 Примера 7 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 436,2 [М+Н]+.
Стадия 3: N-(5-((5-хлоро-4-морфолинопиримидин-2-ил)амино)-2-((2-(диметиламино)этил)метиламино)-4-метоксифенил)акриламид (соединение Z-9)
С использованием соединения 9-5 (0,50 г, 1,2 ммоль) в качестве исходного материала, синтезируют неочищенный продукт согласно стадии 7 Примера 1 и очищают с помощью препаративной жидкостной хроматографии с получением соединения Z-9 (77,19 мг, выход трех стадий 12,1%). МС m/z (ЭРИ): 490,2 [М+Н]+; 1Н-ЯМР (400 МГц, CDCl3) δ 10,00 (s, 1Н), 9,29 (s, 1Н), 7,94 (s, 1Н), 7,44 (s, 1Н), 6,69 (s, 1Н), 6,31 (dd, J=16,9, 1,8 Гц, 1Н), 6,21 (d, J=9,6 Гц, 1Н), 5,59 (dd, J=10,0, 1,8 Гц, 1Н), 3,78 (d, J=7,3 Гц, 9Н), 2,79 (s, 2Н), 2,62 (s, 3Н), 2,18 (s, 6Н), 1,53 (s, 4Н).
Пример 10: N-2-(3-(диметиламино)азетидин-1-ил)-4-метокси-5-((4-(хинолин-3-ил)пиримидин-2-ил)амино)фенил)акриламид (соединение Z-10)
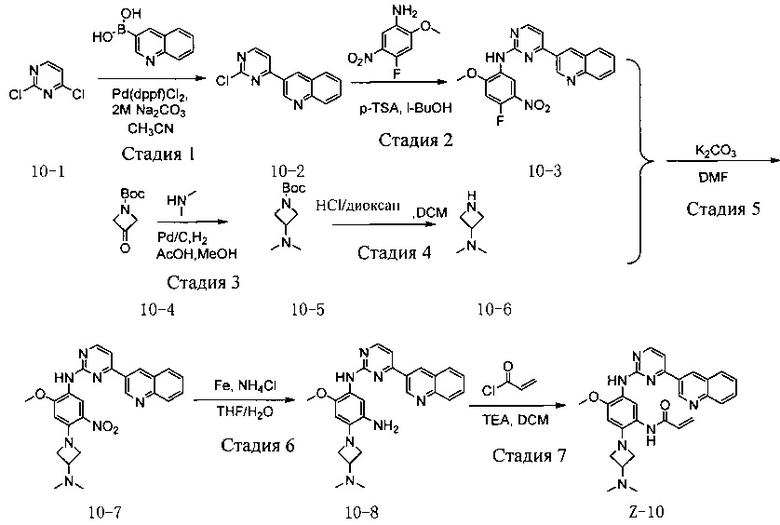
Стадия 1: 3-(2-хлорпиримидин-4-ил)хинолин (соединение 10-2)
Соединение 2,4-дихлорпиримидин (5,0 г, 34 ммоль, коммерчески доступен) и хинолин-3-борную кислоту (5,8 г, 34 ммоль, коммерчески доступна) добавляют к раствору ацетонитрила (120 мл) и карбоната натрия (50 мл, 2М). После троекратной замены воздуха реакционной системы аргоном добавляют Pd(dppf)Cl2 (494 мг, 0,68 ммоль), а затем воздух заменяют аргоном еще три раза. Реакционную смесь нагревают до 80°С и непрерывно перемешивают в течение 6 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции реакционную смесь разбавляют водой, осажденные серое твердое вещество отфильтровывают с помощью воронки Бюхнера и осадок на фильтре промывают водой и сушат до постоянного веса с получением соединения 10-2 (4,6 г, 57%) которое непосредственно используют на следующей стадии. МС m/z (ЭРИ): 242,7 [М+Н]+.
Стадия 2: N-(4-фтор-2-метокси-5-нитрофенил)-4-(хинолин-3-ил)пиримидин-2-амин (соединение 10-3)
4-фтор-2-метокси-5-нитроанилин (2,2 г, 12 ммоль) и соединение 10-2 (2,9 г, 12 ммоль) добавляют к раствору пара-толуолсульфоновой кислоты (p-TSA) (2,1 г, 12 ммоль) в 40 мл н-бутанол и реакционную смесь нагревают при 130°С в течение 6 ч в герметично закрытой пробирке. После завершения реакции реакционную смесь охлаждают до комнатной температуры с осаждением большого количества твердой фазы из раствора. Твердую фазу отфильтровывают с помощью воронки Бюхнера с получением осадка на фильтре, который ресуспендируют в этаноле при нагревании с обратным холодильником с получением соединения 10-3 (4,3 г, 64%), которое используют непосредственно в следующей реакции. МС m/z (ЭРИ): 392,0 [M+H]+.
Стадия 3: трет-бутил-3-(диметиламино)азетидин-1-карбоксилат (соединение 10-5)
Соединение трет-бутил-3-оксоазетидин-1-карбоновую кислоту (2,0 г, 11,7 ммоль, коммерчески доступна) и гидрохлорид диметиламина (1,3 г, 17 ммоль) добавляют к 100 мл раствора уксусной кислоты (1 мл) в метаноле и добавляют палладий на угле (1,4 г). После троекратной замены воздуха водородом реакционную смесь перемешивают в атмосфере водорода в течение 5 ч. После завершения реакции оставшийся палладий на угле отфильтровывают, а фильтрат концентрируют при пониженном давлении с получением соединения 10-5 (2,2 г, 80%), которое используют непосредственно в следующей реакции. МС m/z (ЭРИ): 200,1 [M+H]+
Стадия 4: 3-(диметиламино)азетидина (соединение 10-6) гидрохлорид
Раствор соляной кислоты в 1,4-диоксане (10 мл) добавляют к 15 мл соединения 10-5 (2,2 г, 11 ммоль) и перемешивают при комнатной температуре в течение 3 часов. После завершения реакции реакционную смесь концентрируют при пониженном давлении с получением соединения 10-6 (1,3 г, 90%), которое используют непосредственно в следующей реакции. МС m/z (ЭРИ): 100,1 [M+H]+.
Стадия 5: N-(4-(3-(диметиламино)азетидин-1-ил)-2-метокси-5-нитрофенил)-4-(хинолин-3-ил)пиримидин-2-амин(соединение 10-7)
Соединение 10-6 (160 мг, 1,16 ммоль) и карбонат калия (350 мг, 2,3 ммоль) добавляют к раствору соединения 10-3 (300 мг, 0,58 ммоль) в 4 мл диметилформамида, перемешивают при 100°С в течение 2 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции добавляют 10 мл воды, реакционную смесь троекратно экстрагируют с помощью системы ЕА/вода и органический слой отделяют и концентрируют при пониженном давлении, с получением соединения 10-7 (130 мг, 50%), которое используют непосредственно в следующей реакции. МС m/z (ЭРИ): 472,2 [M+H]+.
Стадия 6: 4-(3-(диметиламино)азетидин-1-ил)-6-метокси-N1-(4-(хинолин-3-ил)пиримидин-2-ил)фенил-1,3-диамин (соединение 10-8)
Используя соединение 10-7 (130 мг, 0,27 ммоль) в качестве исходного материала, соединение 10-8 (80 мг, 70%) синтезируют согласно стадии 4 Примера 7 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 441,2 [М+Н]+.
Стадия 7: N-(2-(3-(диметиламино)азетидин-1-ил)-4-метокси-5-((4-(хинолин-3-ил)пиримидин-2-ил)амино)фенил)акриламид (соединение Z-10)
Используя соединение 10-8 (80 мг, 0,17 ммоль) в качестве исходного материала, полученный неочищенный продукт синтезируют согласно стадии 7 Примера 1 и очищают с помощью препаративной жидкостной хроматографии с получением соединения Z-10 (30,69 мг, 30%). МС m/z (ЭРИ): 496,2 [М+Н]+, 1Н-ЯМР (400 МГц, CDCl3) δ 9,40 (d, J=2,0 Гц, 1Н), 9,32 (s, 1Н), 9,16 (s, 1Н), 8,45 (d, J=5,1 Гц, 1Н), 8,25 (s, 1Н), 8,06 (d, J=8,5 Гц, 1Н), 7,68 (dd, J=18,2, 10,0 Гц, 2Н), 7,54-7,43 (m, 2Н), 7,22 (d, J=5,2 Гц, 1Н), 6,49-6,37 (m, 2Н), 6,33 (d, J=10,1 Гц, 1Н), 5,73 (d, J=10,2 Гц, 1Н), 3,92-3,78 (m, 5Н), 3,58 (t, J=6,5 Гц, 2Н), 3,07 (d, J=6,3 Гц, 1Н), 2,13 (d, J=17,8 Гц, 6Н).
Пример 11: (R)-N-(2-(3-(диметиламино)пирролидин-1-ил)-4-метокси-5-((4-(хинолин-3-ил)пиримидин-2-ил)амино)фенил)акриламид (соединение Z-11)

Стадия 1: (R)-N-(4-(3-(диметиламино)пирролидин-1-ил)-2-метокси-5-нитрофенил)-4-(хинолин-3-ил)пиримидин-2-амин (соединение 11-7)
(R)-N,N-диметилпирролидин-3-амин (80 мг, 0,70 ммоль, коммерчески доступу) и карбонат калия (193 мг, 1,40 ммоль) добавляют к раствору соединения 10-3 (200 мг, 0,47 ммоль) в 4 мл DMF и энергично перемешивают при 100°С в течение 2 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции добавляют 10 мл воды, трижды экстрагируют системой ЕА/вода, органический слой отделяют и концентрируют при пониженном давлении с получением соединения 11-7 (180 мг, 79%). МС m/z (ЭРИ): 486,2 [М+Н]+.
Стадия 2: (R)-4-(3-(диметиламино)пирролидин-1-ил)-6-метокси-N1-(4-хинолин-3-ил)пиримидин-2-ил)бензол-1,3-диамин (соединение 11-8)
Используя соединение 11-7 (180 мг, 0,37 ммоль) в качестве исходного материала, синтезируют согласно стадии 6 Примера 1 соединение 11-8 (150 мг, 89%), которое используют непосредственно в следующей стадии. МС m/z (ЭРИ): 456,2 [М+Н]+.
Стадия 3: (R)-N-(2-(3-(диметиламино)пирролидин-1-ил)-4-метокси-5-((4-(хинолин-3-ил)пиримидин-2-ил)амино)фенил)акриламид (соединение Z-11)
Используя соединение 11-8 (150 мг, 0,3 ммоль) в качестве исходного материала, синтезируют согласно стадии 7 Примера 1 неочищенный продукт и очищают с помощью препаративной жидкостной хроматографии с получением соединения Z-11 (23,58 мг, 9,87%). МС m/z (ЭРИ): 509,9 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6): δ 9,67 (1s, 1Н), 9,40 (1s, 1Н), 8,59 (d, J=4 Гц, 1Н), 8,39 (s, 1Н), 8,31 (s, 1Н), 8,07 (d, J=8 Гц, 1Н), 7,70 (m, 2Н), 7,50 (m, 1Н), 7,27 (m, 1Н), 6,74 (s, 1Н), 6,35 (m, 2Н), 5,73 (d, J=8 Гц, 1Н), 3,83 (s, 3Н), 3,05 (m, 4Н), 1,16 (m, 1Н), 2,24 (s, 6Н), 2,05 (m, 2Н).
Пример 12: N-(4-метокси-2-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)-5-((4-(хинолин-3-ил)пиримидин-2-ил)амино)фенил)акриламид (соединение Z-12)

Стадия 1: 3-(2-хлорпиримидин-4-ил)хинолин (соединение 10-2, согласно стадии 1 Примера 10)
Стадия 2: 4-бром-2-метокси-1-нитробензол (соединение 12-4)
Соединение 12-3 (10,0 г, 46 ммоль, коммерчески доступно) растворяют в 150 мл безводного метанола при комнатной температуре, добавляют метоксид натрия (4,37 г, 81 ммоль) и реакционную смесь нагревают с обратным холодильником и энергично перемешивают в течение 8 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции реакционную смесь разбавляют водой, экстрагируют системой ЕА/вода, трижды промывают водой, сушат, органический слой концентрируют при пониженном давлении с получением неочищенного соединения 12-4 (9,5 г), которое используют непосредственно на следующей стадии. МС m/z (ЭРИ): 231,0 [М+Н]+.
Стадия 3: 4-бром-2-метоксианилин (соединение 12-5)
При комнатной температуре восстановленный железный порошок (4,8 г, 80 ммоль) и хлорид аммония (2,65 г, 50 ммоль) добавляют к смешанному раствору соединения 12-4 (2,3 г, 10,0 ммоль) в 60 мл THF/вода (объемное соотношение THF и воды 2:1), энергично перемешивают при 65°С в течение 4 часов. После завершения реакции реакционную смесь фильтруют, фильтрат разбавляют водой, экстрагируют системой ЕА/вода, трижды промывают водой, сушат, органический слой концентрируют при пониженном давлении с получением неочищенного соединения 12-5 (2, г), которое используют непосредственно на следующей стадии. МС m/z (ЭРИ): 201,9 [М+Н]+.
Стадия 4: 4-бром-2-метокси-5-нитроанилин (соединение 12-6)
Нитрат калия (217 мг, 2,25 ммоль) добавляют к раствору соединения 12-5 (400 мг, 2,0 ммоль) в 3,5 мл концентрированной серной кислоты при температуре -20°С и энергично перемешивают при -20°С в течение 5 мин. После завершения реакции реакционную смесь разбавляют водой, экстрагируют системой ЕА/вода, промывают водой трижды и сушат и органический слой концентрируют при пониженном давлении с получением неочищенного соединения 12-6 (300 мг), которое используют непосредственно на следующей стадии. МС m/z (ЭРИ): 246,9 [М+Н]+.
Стадия 5: трет-бутил-4-(4-амино-5-метокси-2-нитрофенил)-5,6-дигидропиридин-1(2Н)-карбоксилат (соединение 12-7)
Соединение 12-6 (100 мг, 0,40 ммоль), Pd(PPh3)4 (24 мг, 0,02 ммоль) и 0,5 мл 2,0 моль/л раствора карбоната натрия добавляют к раствору соединения трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2Н)-карбоксилата (125 мг, 0,40 ммоль) в 5 мл 1,4-диоксана и энергично перемешивают в атмосфере N2 при температуре 100°С в течение 4 часов. После завершения реакции реакционную смесь фильтруют и фильтрат концентрируют при пониженном давлении с получением неочищенного продукта, который очищают с помощью колоночной комби-флэш хроматографии [РЕ:ЕА=100:0-0:100] с получением соединения 12-7 (50 мг, 35%). МС m/z (ЭРИ): 350,1 [М+Н]+.
Стадия 6: трет-бутил-4-(5-метокси-2-нитро-4-((4-(хинолин-3-ил)пиримидин-2-ил)амино)фенил)-5,6-дигидропиридин-1(2Н)-карбоксилат (соединение 12-8)
Соединение 12-7 (500 мг, 1,43 ммоль), Pd2(dba)3 (131 мг, 0,143 ммоль), Ксантофос (165 мг, 0,286 ммоль) и карбонат цезия (925 мг, 2,86 ммоль) добавляют к раствору соединения 10-2 (345 мг, 1,43 ммоль) в 30 мл 1,4-диоксана, и энергично перемешивают в атмосфере N2 при температуре 100°С в течение 5 ч. После завершения реакции реакционную смесь фильтруют и фильтрат концентрируют при пониженном давлении с получением неочищенного продукта, который очищают с помощью колоночной комби-флэш хроматографии [DCM:MeOH=100:0-0:100] с получением соединения 12-8 (600 мг, 76%). МС m/z (ЭРИ): 545,0 [М+Н]+.
Стадия 7: N-(2-метокси-5-нитро-4-(1,2,3,6-тетрагидропиридин-4-ил)фенил)-4-(хинолин-3-ил)пиримидин-2-амин (соединение 12-9)
1 мл трифторуксусной кислоты добавляют по каплям к раствору соединения 12-8 (150 мг, 0,27 ммоль) в дихлорметане (10 мл) при комнатной температуре и затем перемешивают при комнатной температуре в течение 3 часов. Реакционный раствор нейтрализуют насыщенным водным раствором бикарбоната натрия и отделяют органическую фазу и водную фазу экстрагируют дихлорметаном (2*10 мл), объединенные органические фазы промывают насыщенным раствором соли (10 мл), сушат над безводным Na2SO4, фильтруют и концентрируют при пониженном давлении с получением соединения 12-9 (115 мг, 93%), которое используют непосредственно в следующей реакции. МС m/z (ЭРИ): 455,2 [М+Н]+.
Стадия 8: N-(2-метокси-4-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)-5-нитрофенил)-4-(хинолин-3-ил)пиримидин-2-амин (соединение 12-10)
Параформальдегид (45 мг, 0,5 ммоль) и триацетоксиборгидрид натрия (106 мг, 0,5 ммоль) добавляют к раствору соединения 12-9 (115 мг, 0,25 ммоль) в метаноле (5 мл) при комнатной температуре и после перемешивания при комнатной температуре в течение 3 часов добавляют 20 мл дихлорметана. Органическую фазу последовательно промывают водой (10 мл), насыщенным раствором соли (10 мл), сушат над безводным Na2SO4, фильтруют и концентрируют с получением соединения 12-10 (95 мг, 81%), которое используют непосредственно в следующей реакции. МС m/z (ЭРИ): 469,2 [M+H]+.
Стадия 9: 6-метокси-4-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)-N1-(4-(хинолин-3-ил)пиримидин-2-ил)бензол-1,3-диамин (соединение 12-11)
С использованием соединения 12-10 (95 мг, 0,2 ммоль) в качестве исходного материала, соединение 12-11 (74 мг, 84%) синтезируют согласно стадии 4 Примера 7 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 439,2 [М+Н]+.
Стадия 10: N-(4-метокси-2-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)-5-((4-(хинолин-3-ил)пиримидин-2-ил)амино)фенил)акриламид (соединение Z-12)
С использованием соединения 12-11 (74 мг, 0,17 ммоль) в качестве исходного материала, полученный неочищенный продукт синтезируют согласно стадии 7 Примера 1 и очищают с помощью препаративной жидкостной хроматографии с получением соединения Z-12 (14 мг, 17%). МС m/z (ЭРИ): 493,2 [М+Н]+.
Пример 13: N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-(5-метокси-4-(хинолин-3-ил)пиримидин-2-иламино)фенил)акриламид (Z-13)

Стадия 1: 3-(2-хлор-5-метоксипиримидин-4-ил)хинолин (соединение 13-2)
Хинолин-3-борную кислоту (2,93 г, 16,9 ммоль), PdCl2(dppf) (250 мг, 0,34 ммоль) и 50 мл 2,0 моль/л раствора карбоната натрия добавляют к раствору соединения 13-1 (3 г, 16,9 ммоль, коммерчески доступно) в 80 мл ацетонитрила и интенсивно перемешивают в атмосфере N2 при температуре 95°С в течение 8 часов. После завершения реакции реакционную смесь выливают в большое количество холодной воды до полного осаждения твердой фазы, которую отфильтровывают, тщательно промывают водой и сушат с получением соединения 13-2 (4 г, 100%). МС m/z (ЭРИ): 272,7 [М+Н]+.
Стадия 2: N-(4-фтор-2-метокси-5-нитрофенил)-5-метокси-4-(хинолин-3-ил)пиримидин-2-амин (соединение 13-3)
Соединение 4-нитро-2-метокси-5-нитроанилин (343 мг, 1,85 ммоль), Pd2(dba)3 (169 мг, 0,18 ммоль), Ксантфос (214 мг, 0,37 ммоль) и карбонат цезия (1,2 г, 3,69 ммоль) добавляют к раствору соединения 13-2 (500 мг, 1,85 ммоль) в 9 мл 1,4-диоксана при интенсивном перемешивании в атмосфере N2 при температуре 120°С в течение 5 ч.
После завершения реакции реакционную смесь фильтруют и фильтрат концентрируют при пониженном давлении с получением неочищенного продукта, который очищают с помощью колоночной комби-флэш хроматографии [РЕ:ЕА=100:0-0:100] с получением соединения 13-3 (180 мг, 65%). МС m/z (ЭРИ): 422,1 [М+Н]+.
Стадия 3: N1-(2-(диметиламино)этил)-5-метокси-N4-(5-метокси-4-(хинолин-3-ил)пиримидин-2-ил)-N1-метил-2-нитробензол-1,4-диамин (соединение 13-4)
N,N,N'-триметилэтилендиамин (65 мг, 0,64 ммоль) и карбонат калия (193 мг, 1,29 ммоль) добавляют к раствору соединения 13-3 (180 мг, 0,43 ммоль) в 3 мл DMF, и энергично перемешивают при 100°С в течение 2 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции добавляют 10 мл воды, полученную смесь трижды экстрагируют с помощью системы ЕА/вода и органический слой отделяют и концентрируют при пониженном давлении с получением соединения 13-4 (200 мг, 91%). МС m/z (ЭРИ): 504,3 [М+Н]+.
Стадия 4: N1-(2-(диметиламино)этил)-5-метокси-N4-(5-метокси-4-(хинолин-3-ил)пиримидин-2-ил)-N1-метилбензол-1,2,4-триамин (соединение 13-5)
Используя соединение 13-4 (200 мг, 0,40 ммоль) в качестве исходного материала, соединение 13-5 (100 мг, 92%) синтезируют согласно стадии 6 Примера 1 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 474,1 [М+Н]+.
Стадия 5: N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-(5-метокси-4-(хинолин-3-ил)пиримидин-2-иламино)фенил)акриламид (соединение Z-13)
Используя соединение 13-5 (100 мг, 0,21 ммоль) в качестве исходного материала, полученный неочищенный продукт синтезируют согласно стадии 7 Примера 1 и очищают с помощью препаративной жидкостной хроматографии с получением соединения Z-13 (4,55 мг, 4%). МС m/z (ЭРИ): 527,8 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6) δ 9,57 (s, 1Н), 9,21 (d, J=16,7 Гц, 2Н), 8,57 (s, 1Н), 8,28-8,19 (m, 2Н), 8,07 (d, J=8,2 Гц, 1Н), 7,96 (s, 1Н), 7,85 (t, J=7,4 Гц, 1Н), 7,65 (t, J=7,3 Гц, 1Н), 7,02 (s, 1Н), 6,61-6,47 (m, 1Н), 6,32 (d, J=17,1 Гц, 1Н), 5,80 (d, J=10,8 Гц, 1Н), 3,97 (s, 3Н), 3,88 (s, 3Н), 2,94 (s, 2Н), 2,68 (s, 3Н), 2,45 (s, 2Н), 2,30 (s, 6Н).
Пример 14: N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-(4-(хинолин-3-ил)-1,3,5-триазин-2-ил)фенил)акриламид (Z-14)


Стадия 1: 4-хлор-N-(4-фтор-2-метокси-5-нитрофенил)-1,3,5-триазин-2-амин (соединение 14-2)
Соединение 1а (200 мг, 1,1 ммоль, коммерчески доступно) добавляют в реакционную колбу объемом 250 мл и добавляют THF (10 мл) для растворения промежуточного соединения. Раствор 4-фтор-2-метокси-5-нитроанилина (200 мг, 1,1 ммоль) в THF (30 мл) по каплям добавляют в реакционную колбу емкостью 50 мл при перемешивании при 80°С. После того, как добавление завершено, реакцию продолжают в течение 10 мин. Ход реакции контролируют с помощью ТСХ. После завершения реакции реакционный раствор концентрируют при пониженном давлении с получением неочищенного продукта, который очищают с помощью колоночной комби-флэш хроматографии с получением целевого соединения 14-2 (110 мг, 34%). МС m/z (ЭРИ): 300,0 [М+Н]+.
Стадия 2: N-(4-фтор-2-метокси-5-нитрофенил)-4-(хинолин-3-ил)-1,3,5-триазин-2-амин (соединение 14-3)
Промежуточное соединение 14-2 (1,2 г, 4 ммоль), 3-хинолинилбороновую кислоту (5,8 г, 34 ммоль), карбонат калия (1,11 г, 8 ммоль) добавляют в реакционную колбу емкостью 250 мл и добавляют диметиловый эфир этиленгликоля (20 мл) и воду (5 мл). После того, как воздух реакционной системы трижды заменяют аргоном, добавляют Pd(dppf)Cl2 (300 мг, 0,4 ммоль), а затем воздух опять трижды заменяют азотом. Реакционную смесь нагревают до 100°С и непрерывно перемешивают в течение 4 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции реакционный раствор концентрируют при пониженном давлении с получением неочищенного продукта, который очищают с помощью колоночной комби-флэш хроматографии с получением целевого соединения 14-3 (700 мг, 45%). МС m/z (ЭРИ): 393,1 [М+Н]+.
Стадия 3: N1-(2-(диметиламино)этил)-5-метокси-N1-метил-2-нитро-N4-(4-(хинолин-3-ил)-1,3,5-триазин-2-ил)бензол-1,4-диамин (соединение 14-4)
Промежуточное соединение 14-3 (400 мг, 1 ммоль), N,N,N'-триметилэтилендиамин (156 мг, 1,5 ммоль) и карбонат цезия (670 мг, 2 ммоль) добавляют в реакционную колбу емкостью 50 мл и добавляют диметиламино формамид (6 мл). После того, как воздух реакционной системы трижды заменяют азотом, реакционную смесь нагревают до 80°С и непрерывно перемешивают в течение 4 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции, реакционный раствор разбавляют водой и фильтруют с получением неочищенного продукта, который очищают с помощью колоночной комби-флэш хроматографии с получением целевого соединения 14-4 (200 мг, 42%). МС m/z (ЭРИ): 475,2 [М+Н]+.
Стадия 4: N1-(2-(диметиламино)этил)-5-метокси-N-метил-N4-(4-(хинолин-3-ил)-1,3,5-триазин-2-ил)бензол-1,2,4-триамин (соединение 14-5)
Используя соединение 14-4 (190 мг, 0,4 ммоль) в качестве исходного материала, синтезируют согласно стадии 6 Примера 1 соединение 14-5 (180 мг), которое используют непосредственно на следующей стадии. МС m/z (ЭРИ): 445,2 [М+Н]+.
Стадия 5: N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-(4-(хинолин-3-ил)-1,3,5-триазин-2-ил)фенил)акриламид (соединение Z-14)
Используя соединение 14-5 (180 мг, 0,41 ммоль) в качестве исходного материала, синтезируют неочищенный продукт согласно стадии 7 Примера 1 и очищают с помощью препаративной тонкослойной хроматографии с получением целевого соединения Z-14 (2,01 мг, 1%). Чистота: 94,99%. МС m/z (ЭРИ): 499,3 [M+H]+.
Пример 15: N-(2-(4-(диметиламино)пиперидин-1-ил)-4-метокси-5-(4-(хинолин-3-ил)-1,3,5-триазин-2-иламино)фенил)акриламид (Z-15)

Стадия 1: N-(4-(4-(диметиламино)пиперидин-1-ил)-2-метокси-5-нитрофенил)-4-(хинолин-3-ил)-1,3,5-триазин-2-амин (соединение 15-4)
Соединение 14-3 (100 мг, 0,25 ммоль), 4-диметиламинопиперидин (66 мг, 0,51 ммоль) и карбонат калия (140 мг, 1,02 ммоль) добавляют в реакционную колбу емкостью 20 мл и добавляют диметилформамид (4 мл). После того, как воздух реакционной системы трижды заменяют азотом, реакционную смесь нагревают до 100°С и непрерывно перемешивают в течение 4 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции, реакционный раствор трижды экстрагируют с помощью системы ЕА/вода, органический слой отделяют, промывают водой, насыщенным солевым раствором, сушат над безводным Na2SO4 и концентрируют при пониженном давлении с получением неочищенного продукта, который очищают с помощью колоночной комби-флэш хроматографии с получением целевого соединения 15-4 (110 мг, 43%). МС m/z (ЭРИ): 501,2 [М+Н]+.
Стадия 2: 4-(4-(диметиламино)пиперидин-1-ил)-6-метокси-N1-(4-(хинолин-3-ил)-1,3,5-триазин-2-ил)бензол-1,3-диамин (соединение 15-5)
Используя соединение 15-4 (100 мг, 0,2 ммоль) в качестве исходного материала, синтезируют согласно стадии 6 Примера 1 соединение 15-5 (90 мг) и используют непосредственно в следующей реакции. МС m/z (ЭРИ): 471,3 [М+Н]+
Стадия 3: N-(2-(4-(диметиламино)пиперидин-1-ил)-4-метокси-5-(4-(хинолин-3-ил)-1,3,5-триазин-2-иламино)фенил)акриламид (соединение Z-15)
С использованием соединения 15-5 (90 мг, 0,2 ммоль) в качестве исходного материала, синтезируют согласно стадии 7 Примера 1 неочищенный продукт и очищают с помощью препаративной ВЭЖХ с получением целевого соединения Z-15 (2,09 мг, 2%, FA соль). Чистота: 100,0%. МС m/z (ЭРИ): 525,3 [М+Н]+
Пример 16: N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-ил)амино)фенил)акриламид (соединение Z-16)

Стадия 1: 5-(2-хлорпиримидин-4-ил)-1-метил-1Н-пирроло[2,3-b]пиридин (соединение 16-2)
Соединение 16-1 (2,0 г, 13,5 ммоль), PdCl2(dppf) (1,04 г, 1,35 ммоль) и 13,5 мл 2,0 моль/л раствора карбоната натрия добавляют к раствору соединения 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пирроло[2,3-b]пиридин (8,6 г, 13,5 ммоль) в 50 мл ацетонитрила и интенсивно перемешивают в атмосфере N2 при 80°С в течение 4 часов. После завершения реакции реакционную смесь разбавляют водой и экстрагируют системой ЕА/вода, трижды промывают водой, сушат и органический слой концентрируют при пониженном давлении с получением неочищенного продукта, который очищают с помощью комби-флэш колоночной хроматографии [РЕ:ЭА=100:0-20:80] с получением соединения 16-2 (2,1 г, 36%). МС m/z (ЭРИ): 245,0 [М+Н]+.
Стадия 2: N-(4-фтор-2-метокси-5-нитрофенил)-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-амин (соединение 16-3)
4-фтор-2-метокси-5-нитроанилин (1,6 г, 8,6 ммоль) и соединение 16-2 (2,1 г, 8,6 ммоль) добавляют к раствору пара-толуолсульфоновой кислоты (3,7 г, 21,5 ммоль) в 20 мл изобутанола и реакционную смесь нагревают при 130°С в течение 6 часов в герметически закрытой пробирке. После завершения реакции реакционную смесь охлаждают до комнатной температуры с осаждением большого количества твердой фазы из раствора. Твердые вещества отфильтровывают с помощью воронки Бюхнера с получением осадка на фильтре, который ресуспендируют в этаноле при нагревании с обратным холодильником с получением соединения 16-3 (1,2 г, 44%), которое используют непосредственно в следующей реакции. МС m/z (ЭРИ): 429,0 [М+Н]+.
Стадия 3: N1-(2-(диметиламино)этил)-5-метокси-N-метил-N4-(4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-ил)-2-нитробензол-1,4-диамин (соединение 16-4)
Используя соединение 16-3 (370 мг, 0,62 ммоль) в качестве исходного материала, соединение 16-4 (300 мг, 90%) синтезируют согласно стадии 5 Примера 1, МС m/z (ЭРИ): 477,2 [М+Н]+.
Стадия 4: N1-(2-(диметиламино)этил)-5-метокси-N-метил-N4-(4-(1-метил-1H-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-ил)бензол-1,2,4-триамин (соединение 16-5)
Используя соединение 16-4 (300 мг, 0,6 ммоль) в качестве исходного материала, синтезируют согласно стадии 4 Примера 7 соединение 16-5 (200 мг), которое непосредственно используют на следующей стадии. МС m/z (ЭРИ): 447,2 [М+Н]+.
Стадия 5: N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-ил)амино)фенил)акриламид (соединение Z-16)
Используя соединение 16-5 (200 мг, 0,44 ммоль) в качестве исходного материала, полученный неочищенный продукт синтезируют согласно стадии 7 Примера 1 и очищают с помощью препаративной жидкостной хроматографии, с получением соединения Z-16 (23 мг, 10%). МС m/z (ЭРИ): 501,3 [М+Н]+; 11Н-ЯМР (400 МГц, CDCl3) δ 10,04 (s, 1Н), 9,78 (s, 1Н), 9,20 (s, 1Н), 8,90 (d, J=2,0 Гц, 1Н), 8,41 (d, J=5,2 Гц, 1Н), 7,70 (s, 1Н), 7,15 (dd, J=6,9, 4,4 Гц, 2Н), 6,73 (s, 1Н), 6,59 (d, J=3,4 Гц, 1Н), 6,49-6,42 (m, 1Н), 6,30 (d, J=10,1 Гц, 1Н), 5,65 (d, J=10,1 Гц, 1Н), 3,84 (d, J=13,7 Гц, 6Н), 2,86-2,77 (m, 2Н), 2,64 (s, 3Н), 2,22 (d, J=5,8 Гц, 2Н), 2,19 (s, 6Н).
Пример 17: N-(2-(4-(диметиламино)пиперидин-1-ил)-4-метокси-5-((4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-ил)амино)фенил)акриламид (Z-17)
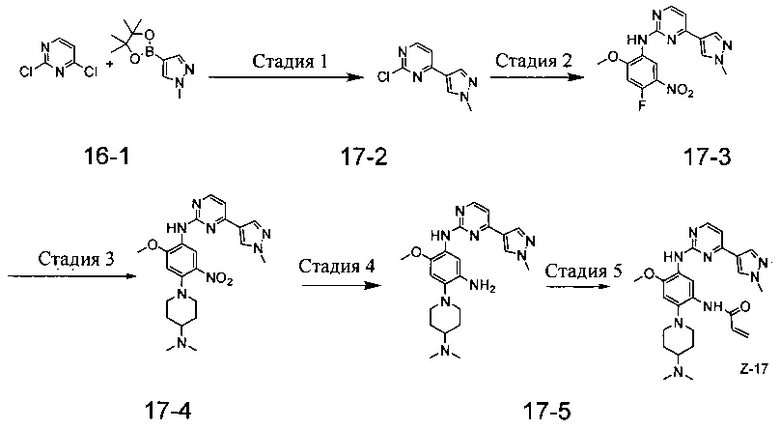
Стадия 1: 2-хлор-4-(1-метил-1Н-пиразол-4-ил) пиримидин (соединение 17-2)
Соединение 16-1 (745 мг, 5 ммоль) и пинаколовый эфир метил-4-пиразолборной кислоты (1,092 г, 5,25 ммоль, коммерчески доступный) добавляют в 100 мл реакционную колбу и добавляют ацетонитрил (30 мл) и раствор карбоната натрия (5 мл, 2М). После того, как воздух реакционной системы трижды заменяют азотом, добавляют Pd(dppf)Cl2 (109,7 мг, 0,15 ммоль), а затем воздух опять трижды заменяют азотом. Реакционную смесь нагревают до 110°С и непрерывно перемешивают в течение 6 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции, реакционный раствор разбавляют водой, трижды экстрагируют системой ЕА/вода и органический слой отделяют, промывают водой, насыщенным раствором соли, сушат над безводным Na2SO4 и концентрируют при пониженном давлении с получением 1,1 г неочищенного продукта, который очищают с помощью комби-флэш колоночной хроматографии [ЕА:РЕ=1:5-1:2] с получением целевого соединения 17-2 (600 мг, 93%). МС m/z (ЭРИ): 195,1 [М+Н]+.
Стадия 2: N-(4-фтор-2-метокси-5-нитрофенил)-4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-амин (соединение 17-3)
Соединение 17-2 (500 мг, 2,577 ммоль) и 4-фтор-2-метокси-5-нитроанилин (479 г, 2,577 ммоль) добавляют в запаянную пробирку объемом 50 мл и добавляют н-бутанол (12 мл) для частичного растворения промежуточного соединения. Затем добавляют пара-толуолсульфоновую кислоту (1,108 г, 6,44 ммоль) и реакционную смесь нагревают при 130°С в течение 6 часов в герметически закрытой пробирке. После завершения реакции реакционную смесь охлаждают до комнатной температуры с осаждением большого количества твердой фазы из раствора. Твердые вещества отфильтровывают с помощью воронки Бюхнера с получением осадка на фильтре, который ресуспендируют в этаноле с обратным холодильником с получением целевого соединения 17-3 (600 мг, 67,8%), которое используют непосредственно в следующей реакции.
Стадия 3: N-(4-(4-(диметиламино)пиперидин-1-ил)-2-метокси-5-нитрофенил)-4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-амин (соединение 17-4)
Соединение 17-3 (70 мг, 0,203 ммоль) и карбонат калия (84 мг, 0,609 ммоль) добавляют в реакционную колбу емкостью 25 мл и добавляют DMF (5 мл) для частичного растворения промежуточного соединения. Затем добавляют N,N-диметиламино пиперидин (31,2 мг, 0,244 ммоль) и реакционную систему поддерживают при 70°С в течение 2 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции реакционный раствор трижды экстрагируют с помощью системы ЕА/вода и органический слой отделяют, промывают водой, насыщенным раствором соли, сушат над безводным Na2SO4 и концентрируют при пониженном давлении с получением соединения 17-4 (70 мг, 76%). МС m/z (ЭРИ): 453,2 [М+Н]+.
Стадия 4: 4-(4-(диметиламино)пиперидин-1-ил)-6-метокси-N1-(4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-ил)бензол-1,3-диамин (соединение 17-5)
При использовании соединения 17-4 (70 мг, 0,155 ммоль) в качестве исходного материала, неочищенное соединение 17-5 (60 мг, 92%) синтезируют согласно стадии 6 Примера 1 и используют непосредственно на следующей стадии.
Стадия 5: N-(2-(4-(диметиламино)пиперидин-1-ил)-4-метокси-5-((4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-ил)амино)фенил)акриламид (Z-17)
С использованием соединения 17-5 (50 мг, 0,118 ммоль) в качестве исходного материала, полученный неочищенный продукт синтезируют согласно стадии 7 Примера 1 и очищают с помощью препаративной пластины [DCM:MeOH=10:1] с получением соединения Z-17 (30 мг, 44%). МС m/z (ЭРИ): 477,2 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6): δ 9,14 (s, 1Н), 9,05 (s, 1Н), 8,81 (s, 1Н), 8,36-8,35 (d, J=4,0 Гц, 1Н), 8,21 (s, 1Н), 7,76 (s, 1Н), 7,10-7,09 (d, J=4,0 Гц, 1Н), 6,86 (s, 1Н), 6,76-6,69 (m, 1Н), 6,33-6,29 (d, J=16,0 Гц, 1Н), 5,80-5,77 (d, J=12,0 Гц, 1Н), 3,92 (s, 3Н), 3,88 (s, 3Н), 3,03-3,00 (m, 2Н), 2,69-2,64 (m, 2Н), 2,27-2,15 (m, 7Н), 1,85-1,82 (m, 2Н), 1,73-1,68 (m, 2Н).
Пример 18: N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-ил)амино)фенил)акриламид (Z-18)

Стадия 1: N1-(2-(диметиламино)этил)-5-метокси-N-метил-N4-(4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-ил)-2-нитробензол-1,4-диамин (соединение 18-4)
Соединение 17-3 (130 мг, 0,378 ммоль, полученное согласно стадии 2 Примера 17) и карбонат калия (156 мг, 1,133 ммоль) добавляют в реакционную колбу объемом 50 мл, добавляют DMF (5 мл) для частичного растворения промежуточного соединения. Затем добавляют N,N,N'-триметил этилендиамин (57,8 мг, 0,567 ммоль) и реакционную систему поддерживают при 70°С в течение 3 часов. Ход реакции контролируют с помощью ТСХ. После завершения реакции, реакционный раствор трижды экстрагируют системой ЕА/вода, органический слой отделяют, промывают водой, насыщенным раствором соли, сушат над безводным Na2SO4 и концентрируют при пониженном давлении с получением неочищенного соединения 18-4 (110 мг, 68%). МС m/z (ЭРИ): 427,2 [М+Н]+.
Стадия 2: N1-(2-(диметиламино)этил)-5-метокси-N-метил-N4-(4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-ил)бензол-1,2,4-триамин (соединение 18-5)
Используя соединение 18-4 (110 мг, 0,258 ммоль) в качестве исходного материала, синтезируют неочищенное соединение 18-5 (110 мг, 100%) согласно стадии 6 Примера 1 и соединение используют непосредственно на следующей стадии,
Стадия 3: N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-ил)амино)фенил)акриламид (соединение Z-18)
Используя соединение 18-5 (110 мг, 0,278 ммоль) в качестве исходного материала, полученный неочищенный продукт синтезируют согласно стадии 7 Примера 1 и очищают с помощью препаративной пластинки [DCM:MeOH=10:1] с получением целевого соединения продукта Z-18 (10,0 мг, 8,2%). МС m/z (ЭРИ): 451,1 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6): δ 10,18 (s, 1Н), 9,42 (s, 1Н), 8,90 (s, 1Н), 8,38-8,37 (d, J=4,0 Гц, 1Н), 8,23 (s, 1Н), 7,79 (s, 1Н), 7,12-7,11 (d, J=4,0 Гц, 1Н), 7,03 (s, 1Н), 6,46-6,32 (m, 2Н), 5,82-5,76 (m, 1Н), 3,93 (s, 3Н), 3,86 (s, 3Н), 2,97-2,93 (m, 2Н), 2,68 (s, 3Н), 2,08-2,47 (m, 8Н).
Пример 19: N-(5-((5-хлор-4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (Z-19)

Стадия 1: 2,5-дихлор-4-(1-метил-1Н-пиразол-4-ил)пиримидин (соединение 19-2)
Используя 2,4,5-трихлорпиримидин (1,32 г, 7,21 ммоль) и пинаколовый эфир метил-4-пиразолборной кислоты (1,5 г, 7,21 ммоль) в качестве исходных веществ, синтезируют неочищенный продукт согласно стадии 1 Примера 17 и очищают с помощью колоночной комби-флэш хроматографии (ЕА:РЕ=1:5) с получением соединения 19-2 (1,4 г, 84,8%). МС m/z (ЭРИ): 229,3 [М+Н]+.
Стадия 2: 5-хлор-N-(4-фтор-2-метокси-5-нитрофенил)-4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-амин (соединение 19-3)
Соединение 19-2 (1,1 г, 4,8 ммоль), Pd2(dba)3 (439 мг, 0,48 ммоль), Ксантфос (277 мг, 0,48 ммоль) и карбонат цезия (4,68 г, 14,4 ммоль) добавляют к раствору 4 фтор-2-метокси-5-нитроанилина (892 мг, 4,8 ммоль) в 30 мл 1,4-диоксана и энергично перемешивают в атмосфере N2 при температуре 100°С в течение 5 ч. После завершения реакции реакционную смесь фильтруют и фильтрат концентрируют при пониженном давлении с получением неочищенного продукта, который очищают с помощью колоночной комби-флэш хроматографии [РЕ:ЕА=100:0-0:100] с получением соединения 19-3 (600 мг, 33,1%). МС m/z (ЭРИ): 379,3 [М+Н]+.
Стадия 3: N1-(5-хлор-4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N-4-метил-5-нитробензол-1,4-диамин (соединение 19-4)
Используя соединение 19-3 (600 мг, 1,587 ммоль) в качестве исходного материала, синтезируют неочищенное соединение 19-4 (400 мг, 57,7%) согласно стадии 5 Примера 1 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 461,1 [М+Н]+.
Стадия 4: N4-(5-хлор-4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-ил)-N1-(2-(диметиламино)этил)-5-метокси-N-метилбензол-1,2,4-триамин (соединение 19-5)
Используя соединение 19-4 (200 мг, 0,435 ммоль) в качестве исходного материала, неочищенное соединение 19-5 (200 мг) синтезируют согласно стадии 2 Примера 2 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 431,3 [М+Н]+.
Стадия 5: N-(5-((5-хлор-4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (соединение Z-19)
Используя соединение 19-5 (200 мг, 0,465 ммоль) в качестве исходного материала, синтезируют неочищенный продукт согласно стадии 7 Примера 1, очищают с помощью препаративной ВЭЖХ с получением соединения Z-19 (23 мг, 10%). МС m/z (ЭРИ): 485,2 1Н-ЯМР (400 МГц, DMSO-d6) δ 10,21 (s, 1Н), 9,11 (s, 1Н), 8,99 (s, 1Н), 8,46 (s, 1Н), 8,37 (s, 1Н), 8,07 (s, 1Н), 7,04 (s, 1Н), 6,29-6,47 (m, 2Н), 5,79-5,81 (m, 1Н), 3,97 (s, 3Н), 3,87 (s, 3Н), 2,89 (m, 2Н), 2,70 (s, 3Н), 2,15-2,39 (m, 8Н).
Пример 20: N-(2-((2-(диметиламино)этил)(метил)амино)-5-((5-фтор-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (Z-20)

Стадия 1: 5-(2-хлор-5-фторпиримидин-4-ил)-1-метил-1Н-пирроло[2,3-b]пиридин (соединение 20-2)
2,4-дихлор-5-фторпиримидин (2,0 г, 7,8 ммоль), PdCl2(dppf)2 (0,57 г, 0,78 ммоль) и карбонат натрия (1,64 г, 15,6 ммоль) добавляют к смешанному раствору соединения 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пирроло[2,3-b]пиридин (2,0 г, 7,8 ммоль, коммерчески доступно) в 20 мл ацетонитрила и 4 мл воды и интенсивно перемешивают в атмосфере N2 при 80°С в течение 2 часов. После завершения реакции реакционную смесь фильтруют и фильтрат концентрируют при пониженном давлении с получением неочищенного продукта, который очищают с помощью колоночной комби-флэш хроматографии [РЕ:ЕА=100:0-60:40] с получением соединения 20-2 (1,12 г, 55%). МС m/z (ЭРИ): 263,1 [М+Н]+.
Стадия 2: 5-фтор-N-(4-фтор-2-метокси-5-нитрофенил)-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-амин (соединение 20-3)
Используя соединение 20-2 (1,5 г, 5,46 ммоль) в качестве исходного материала, полученный неочищенный продукт синтезируют согласно стадии 2 Примера 19 и очищают с помощью комби-флэш колоночной хроматографии (РЕ:ЕА=50:50) с получением соединения 20-3 (0,8 г, 45%), за исключением того, что на стадии 2 Примера 20 реакционную смесь энергично перемешивают в атмосфере N2 при 120°С в течение 21 ч. МС m/z (ЭРИ): 413,1 [М+Н]+.
Стадия 3: N1-(2-(диметиламино)этил)-N4-(5-фтор-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-ил)-5-метокси-N-метил-2-нитробензол-1,4-диамин (соединение 20-4)
Используя соединение 20-3 (300 мг, 1,18 ммоль) в качестве исходного материала, соединение 20-4 (330 мг, 92%) синтезируют согласно стадии 5 Примера 1 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 495,2 [М+Н]+.
Стадия 4: N1-(2-(диметиламино)этил)-N4-(5-фтор-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-ил)-5-метокси-N1-метилбензол-1,2,4-триамин (соединение 20-5)
Используя соединение 20-4 (330 мг, 0,67 ммоль) в качестве исходного материала, соединение 20-5 (317 мг, 100%) синтезируют согласно стадии 6 Примера 1 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 465,2 [М+Н]+.
Стадия 5: N-(2-((2-(диметиламино)этил)(метил)амино)-5-((5-фтор-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-ил)амино)-4-метоксифенил)акриламид (соединение Z-20)
Используя соединение 20-5 (317 мг, 0,68 ммоль) в качестве исходного материала, синтезируют неочищенный продукт согласно стадии 7 Примера 1 и очищают с помощью препаративной пластины [DCM:MeOH:NH4OH=90:10:1] с получением соединения Z-20 (11,9 мг, 3/4%). МС m/z (ЭРИ): 519,2 [М+Н]+; 1Н-ЯМР (400 МГц, CDCl3) = 9,92 (br. s., 1 Н), 9,64 (s, 1 Н), 9,07 (d, J=9,2 Гц, 2 H), 8,31 (d, J=2,8 Гц, 1 H), 7,66 (s, 1 Н), 7,14 (d, J=1,6 Гц, 1 H), 6,70 (s, 1 Н), 6,59 (d, J=1,6 Гц, 1 H), 6,42 (br. s., 2 H), 5,66-5,63 (m, 1 H), 3,86 (s, 3 H), 3,81 (s, 3 H), 2,87 (br. s., 2 H), 2,63 (s, 3 H), 2,25 (br. s., 8 H).
Пример 21: N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((5-метокси-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-ил)амино)фенил)акриламид (соединение Z-21)

Стадия 1: 5-(2-хлор-5-метоксипиримидин-4-ил)-1-метил-1Н-пирроло[2,3-b]пиридин (соединение 21-2)
2,4-дихлор-5-метоксипиримидин (1,25 г, 7,0 ммоль), PdCl2(dppf)2 (0,5 г, 0,7 ммоль) и карбонат натрия (1,5 г, 14,0 ммоль) добавляют к смешанному раствору соединения 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пирроло[2,3-b]пиридин (1,8 г, 7,0 ммоль, коммерчески доступно) в 20 мл ацетонитрила и 4 мл воды и интенсивно перемешивают в атмосфере N2 при 80°С в течение 2 часов. После завершения реакции реакционную смесь фильтруют и фильтрат концентрируют при пониженном давлении с получением неочищенного продукта, который очищают с помощью колоночной комби-флэш хроматографии [РЕ:ЕА=100:0-60:40] с получением соединения 21-2 (1,54 г, 80%). МС m/z (ЭРИ): 275,1 [М+Н]+.
Стадия 2: N-(4-фтор-2-метокси-5-нитрофенил)-5-метокси-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-амин (соединение 21-3)
Используя соединение 21-2 (1,5 г, 5,46 ммоль) в качестве исходного материала, полученный неочищенный продукт синтезируют согласно стадии 2 Примера 19 и очищают с помощью колоночной комби-флэш хроматографии [РЕ:ЕА=50:50] с получением соединения 21-3 (1,2 г, 52%), за исключением того, что на стадии 2 Примера 21 реакционную смесь энергично перемешивают в атмосфере N2 при 120°С в течение 21 ч. МС m/z (ЭРИ): 425,2 [М+Н]+.
Стадия 3: N1-(2-(диметиламино)этил)-5-метокси-N4-(5-метокси-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-ил)-N1-метил-2-нитробензол-1,4-диамин (соединение 21-4)
Используя соединение 21-3 (500 мг, 1,18 ммоль) в качестве исходного материала, соединение 21-4 (620 мг, 100%) синтезируют согласно стадии 5 Примера 1 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 507,3 [М+Н]+.
Стадия 4: N1-(2-(диметиламино)этил)-5-метокси-N4-(5-метокси-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-ил)-N1-метилбензол-1,2,4-триамин (соединение 21-5)
Используя соединение 21-4 (500 мг, 0,99 ммоль) в качестве исходного материала, соединение 21-5 (332 мг, 70,3%) синтезируют согласно стадии 6 Примера 1 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 477,2 [М+Н]+.
Стадия 5: N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((5-метокси-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)пиримидин-2-ил)амино)фенил)акриламид (соединение Z-21)
Используя соединение 21-5 (332 мг, 0,7 ммоль) в качестве исходного материала полученный неочищенный продукт синтезируют согласно стадии 7 Примера 1 и очищают с помощью препаративной пластины [DCM:MeOH:NH4OH=90:10:1] с получением соединения Z-21 (59 мг, 15,6%). МС m/z (ЭРИ): 531,2 [М+Н]+; 1Н-ЯМР (400 МГц, CDCl3): 9,83 (br. s., 1 Н), 9,63 (s, 1 Н), 9,13 (d, J=1,6 Гц, 1 H), 8,93 (s, 1 Н), 8,23 (s, 1 Н), 7,52 (s, 1 Н), 7,11 (d, J=3,6 Гц, 1 H), 6,67 (s, 1 Н), 6,53 (d, J=3,2 Гц, 1 H), 6,48-6,34 (m, 2 Н), 5,63 (d, J=11,6 Гц, 1 H), 3,84 (s, 3 Н), 3,81 (s, 3 Н), 3,80 (s, 3 Н), 2,87 (br. s., 2 H), 2,61 (s, 3 H), 2,37 (br. s., 2 H), 2,28 (br. s., 6 H).
Пример 22: N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламид (соединение Z-22)

Стадия 1: 5-(2-хлор-5-(трифторметил)пиримидин-4-ил)-1-метил-1Н-пирроло[2,3-b]пиридин (соединение 22-2)
2,4-дихлор-5-трифторметилпиримидин (1,0 г, 6,5 ммоль), PdCl2(dppf) (0,5 г, 0,65 ммоль) и 6,5 мл 2,0 моль/л раствора карбоната натрия добавляют к раствору соединения 1-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пирроло[2,3-b]пиридина (4,3 г, 6,5 ммоль) в 25 мл ацетонитрила и энергично перемешивают в атмосфере N2 при 80°С в течение 4 часов. После завершения реакции реакционную смесь разбавляют водой и экстрагируют системой ЕА/вода, трижды промывают водой и сушат и органический слой концентрируют при пониженном давлении с получением неочищенного соединения, которое очищают с помощью колоночной комби-флэш хроматографии [РЕ:ЕА=100:0-20:80] с получением соединения 22-2 (0,6 г, 36%). МС m/z (ЭРИ): 313,0 [М+Н]+.
Стадия 2: N-(4-фтор-2-метокси-5-нитрофенил)-4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)-5-(трифторметил)пиримидин-2-амин (соединение 22-3)
4-фтор-2-метокси-5-нитроанилин (760 мг, 4,1 ммоль) и соединение 22-2 (1,0 г, 4,1 ммоль) добавляют к раствору пара-толуолсульфоновой кислоты (p-TSA) (1,7 г, 10,5 ммоль) в 20 мл изобутанола и реакционную смесь нагревают при 130°С в течение 6 часов в герметично закрытой пробирке. После завершения реакции реакционную смесь охлаждают до комнатной температуры и из раствора осаждается большое количество твердой фазы. Твердую фазу отфильтровывают с помощью воронки Бюхнера с получением осадка на фильтре, который ресуспендируют в этаноле при нагревании с обратным холодильником с получением соединения 22-3 (740 мг, 44%), которое используют непосредственно в следующей реакции. МС m/z (ЭРИ): 463,1 [М+Н]+.
Стадия 3: N1-(2-(диметиламино)этил)-5-метокси-N-метил-N4-(4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)-5-(трифторметил)пиримидин-2-ил)-2-нитробензол-1,4-диамин (соединение 22-4)
Используя соединение 22-3 (370 мг, 0,62 ммоль) в качестве исходного материала, соединение 22-4 (300 мг, 90%) синтезируют согласно стадии 5 Примера 1, МС m/z (ЭРИ): 544,2 [М+Н]+.
Стадия 4: N1-(2-(диметиламино)этил)-5-метокси-N-метил-N4-(4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)-5-(трифторметил)пиримидин-2-ил)бензол-1,2,4-триамин (соединение 22-5)
Используя соединение 22-4 (300 мг, 0,6 ммоль) в качестве исходного материала, соединение 22-5 (200 мг) синтезируют согласно стадии 4 Примера 7 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 514,2 [М+Н]+.
Стадия 5: N-(2-((2-(диметиламино)этил)(метил)амино)-4-метокси-5-((4-(1-метил-1Н-пирроло[2,3-b]пиридин-5-ил)-5-(трифторметил)пиримидин-2-ил)амино)фенил)акриламид (соединение Z-22)
Используя соединение 22-5 (200 мг, 0,44 ммоль) в качестве исходного материала, полученный неочищенный продукт синтезируют согласно стадии 7 Примера 1 и очищают с помощью препаративной жидкостной хроматографии с получением соединения Z-22 (44,9 мг, 20%). МС m/z (ЭРИ): 568,8 [М+Н]+; 1Н-ЯМР (400 МГц, CDCl3) δ 10,03 (s, 1Н), 9,60 (s, 1Н), 8,76 (s, 1Н), 8,56 (s, 1Н), 7,85 (s, 1Н), 7,16 (s, 2Н), 6,72 (s, 1Н), 6,52 (s, 1Н), 6,42 (d, J=16,8 Гц, 1Н), 6,24 (dd, J=17,0, 10,1 Гц, 1Н), 5,64 (d, J=10,2 Гц, 1Н), 3,83 (d, J=24,8 Гц, 6Н), 2,79 (s, 2Н), 2,63 (s, 3Н), 2,20 (d, J=12,4 Гц, 8Н).
Пример 23: N-(5-((5-хлор-4-(1,4-оксазепан-4-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (Z-23)

Стадия 1: 4-(2,5-дихлорпиримидин-4-ил)-1,4-оксазепан (соединение 23-2)
Соединение 2,4,5-трихлорпиримидина (0,3 г, 1,65 ммоль) добавляют к 10 мл THF при 0°С, добавляют триэтиламин (0,49 г, 4,9 ммоль), гомоморфолин (0,22 г, 1,65 ммоль, коммерчески доступен) в реакционный раствор, перемешивают при комнатной температуре в течение 1 ч. Ход реакции контролируют с помощью ТСХ. После завершения реакции, добавляют воду и ЕА, органический слой отделяют, промывают насыщенным водным раствором хлорида натрия, сушат и концентрируют при пониженном давлении, получая соединение 23-2 (0,5 г), которое используют непосредственно на следующей стадии. МС m/z (ЭРИ): 248,0 [M+H]+.
Стадия 2: 5-хлор-N-(4-фтор-2-метокси-5-нитрофенил)-4-(1,4-оксазепан-4-ил)пиримидин-2-амин (соединение 23-3)
Соединение 23-2 (0,5 г, 2,0 ммоль), 4-фтор-2-метокси-5-нитроанилин (0,37 г, 2,0 ммоль) и пара-толуолсульфоновую кислоту (0,87 г, 5,0 ммоль) добавляют к 10 мл 1,4-диоксана и перемешивают в атмосфере N2 при температуре 110°С в течение 8 часов. После завершения реакции реакционную смесь фильтруют, осадок на фильтре промывают ЕА, сушат при 50°C с получением неочищенного соединения 23-3 (0,45 г), которое используют непосредственно на следующей стадии. МС m/z (ЭРИ): 398,1 [М+Н]+.
Стадия 3: N1-(5-хлор-4-(1,4-оксазепан-4-ил)пиримидин-2-ил)-N4-(2-(диметиламино)этил)-2-метокси-N4-метил-5-нитробензол-1,4-диамин (соединение 23-4)
Используя соединение 23-3 (0,45 г, 1,1 ммоль) в качестве исходного материала, соединение 23-4 (0,4 г) синтезируют согласно стадии 5 Примера 1 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 480,2 [М+Н]+.
Стадия 4: N4-(5-хлор-4-(1,4-оксазепан-4-ил)пиримидин-2-ил)-N1-(2-(диметиламино)этил)-5-метокси-N-метилбензол-1,2,4-триамин (соединение 23-5)
Используя соединение 23-4 (0,4 г, 0,83 ммоль) в качестве исходного материала, соединение 23-5 (0,25 г) синтезируют согласно стадии 4 Примера 7 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 450,2 [М+Н]+.
Стадия 5: N-(5-((5-хлор-4-(1,4-оксазепан-4-ил)пиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (Z-23)
Используя соединение 23-5 (0,25 г, 0,55 ммоль) в качестве исходного материала, неочищенный продукт синтезируют согласно стадии 7 Примера 1, очищают с помощью препаративной жидкостной хроматографии с получением соединения Z-23 (69,8 мг, 8,4%). МС m/z (ЭРИ): 504,2 [М+Н]+; 1Н-ЯМР (400 МГц, CDCl3) δ 9,99 (s, 1Н), 9,18 (s, 1Н), 7,89 (s, 1Н), 7,32 (s, 1Н), 6,68 (s, 1Н), 6,30 (dd, J=17,0, 1,9 Гц, 1Н), 6,20 (d, J=9,9 Hz, 1Н), 5,58 (dd, J=10,0, 1,9 Гц, 1H), 4,03-3,98 (m, 2Н), 3,96 (t, J=6,2 Гц, 2H), 3,83-3,79 (m, 2Н), 3,78 (s, 3Н), 3,72-3,66 (m, 2Н), 2,84-2,75 (m, 2Н), 2,61 (s, 3Н), 2,19 (s, 2Н), 2,17 (s, 6Н), 1,98 (dd, J=11,9, 5,9 Гц, 2H).
Пример 24: N-(5-((4-((1S,4S)-2-окса-5α-азабицикло[2,2,1]гептан-5-ил)-5-хлорпиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (Z-24)

Стадия 1: (1S,4S)-5-(2,5-дихлор-пиримидин-4-ил)-2-окса-5-азабицикло[2,2,1]гептан (соединение 24-2)
Соединение 2,4,5-трихлорпиримидин (0,3 г, 1,65 ммоль) добавляют к 10 мл THF при 0°С, добавляют триэтиламин (0,5 г, 4,9 ммоль), (1S,4S)-2-окса-5-аза-бицикло[2,2,1]гептан (0,22 г, 1,65 ммоль, коммерчески доступен) добавляют к реакционному раствору и перемешивают при комнатной температуре в течение 1 ч. Ход реакции контролируют с помощью ТСХ. После завершения реакции добавляют воду и ЕА и органический слой отделяют, промывают насыщенным водным раствором хлорида натрия, сушат и концентрируют при пониженном давлении с получением соединения 24-2 (0,36 г), которое используют непосредственно на следующей стадии. МС m/z (ЭРИ): 246,0 [М+Н]+.
Стадия 2: 4-((1S,4S)-2-окса-5-азабицикло[2,2,1]гепт-5-ил)-5-хлор-N-(4-фтор-2-метокси-5-нитрофенил)пиримидин-2-амин (соединение 24-3)
Используя соединение 24-2 (0,36 г, 1,4 ммоль) в качестве исходного материала, соединение 24-3 (0,32 г) синтезируют согласно стадии 2 Примера 23, и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 396,1 [М+Н]+.
Стадия 3: N1-(4-((1S,4S)-2-окса-5-азабицикло[2,2,1]гепт-5-ил)-5-хлорпиримидин-2-ил)-N4-2-(диметиламино)этил)ил-2-метокси-N-4-метил-5-нитрофенил-1,4-диамин (соединение 24-4)
Используя соединение 24-3 (0,32 г, 0,81 ммоль) в качестве исходного материала, соединение 24-4 (0,31 г) синтезируют согласно стадии 5 Примера 1 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 478,1 [М+Н]+.
Стадия 4: N4-(4-((1S,4S)-2-окса-5-азабицикло[2,2,1]гепт-5-ил)-5-хлор-пиримидин-2-ил)-N1-(2-(диметиламино)этил)ил-5-метокси-N1-метилфенил-1,2,4-триамин (соединение 24-5)
Используя соединение 24-4 (0,31, 0,65 ммоль) в качестве исходного материала, соединение 24-5 (0,22 г) синтезируют согласно стадии 4 Примера 7 и используют непосредственно на следующей стадии. МС m/z (ЭРИ): 448,2 [М+Н]+.
Стадия 5: N-(5-(4-((1S,4S)-2-окса-5α-азабицикло[2,2,1]гептан-5-ил)-5-хлорпиримидин-2-ил)амино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламид (Z-24)
Используя соединение 24-5 (0,2 г, 0,44 ммоль) в качестве исходного материала, неочищенный продукт синтезируют согласно стадии 7 Примера 1 и очищают с помощью препаративной жидкостной хроматографии с получением соединения Z-24 (60,1 мг, 7,3%). МС m/z (ЭРИ): 502,2 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6) δ 10,09 (s, 1H), 8,85 (s, 1H), 7,96 (s, 1H), 7,67 (s, 1H), 6,96 (s, 1H), 6,37 (dd, J=16,9, 10,0 Гц, 1H), 6,22 (dd, J=16,9, 2,1 Гц, 1H), 5,73 (dd, J=10,0, 2,1 Гц, 1H), 5,32 (s, 1H), 4,61 (s, 1H), 3,88 (d, J=10,5 Гц, 1H), 3,85 (s, 3Н), 3,81 (d, J=7,6 Гц, 1H), 3,73 (d, J=7,5 Гц, 1H), 3,59 (d, J=10,5 Гц, 1H), 2,85 (t, J=5,2 Гц, 2H), 2,68 (s, 3Н), 2,25 (t, J=5,9 Гц, 2H), 2,18 (s, 6H), 1,90 (d, J=9,9 Гц, 1H), 1,78 (d, J=9,7 Гц, 1H).
Пример 25-78, 87, 88:

Целевые соединения Примеров 25-78, 87 и 88 показаны в виде формулы (IV-1), где заместители R0, R2 и R3 представляют собой водород, а другие заместители А, R1, R6 являются такими, как показано в следующей таблице.
Общий способ: Способы получения соединений Z-25-Z-56, Z-87, Z-88 аналогичны способу Примера 1, с использованием боронатного эфира или борной кислоты, замещенной различными заместителями, и с использованием 5-замещенного 2,4-дихлорпиримидина в качестве исходных веществ.
Используя различные амины и 5-замещенный 2,4-дихлорпиримидин в качестве исходных веществ, соединения Z-57 до Z-78 получают согласно способу, аналогичному способу Примера 7.






Примеры 79-86

Целевые соединения Примеров 79-86 представлены в виде формулы (IV-2), где заместители R0, R2 и R3 представляют собой водород, а другие заместители А, R1 показаны в виде следующей таблицы.
Общий Способ: Способы получения соединений Z-79-Z-82 аналогичны способу Примера 23, с использованием различных аминов и 2,4-дихлор-1,3,5-триазина в качестве исходных веществ.
Используя боронатный эфир или борную кислоту, замещенную различными заместителями, и 2,4-дихлор-1,3,5-триазин в качестве исходных веществ, соединения Z-83 до Z-86 получают согласно способу, аналогичному описанному в Примере 14.


Пример 89: Получение N-(5-(5-хлор-4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-иламино)-2-(4-(диметиламино)пиперидин-1-ил)-4-метоксифенил)акриламида (Z-89)
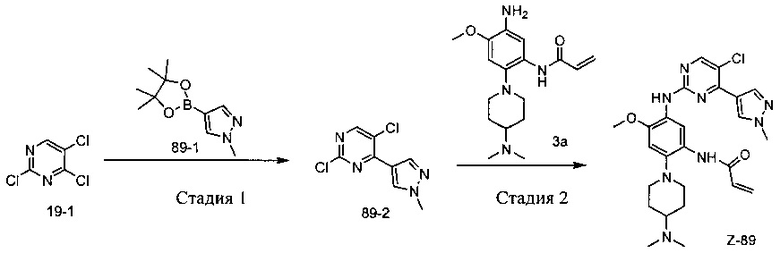
Стадия 1: Смешанный раствор из 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразола (1,1 г, 5,25 ммоль), соединение 19-1 (745 мг, 4 ммоль), Pd(dppf)Cl2 (109,7 мг, 0,15 ммоль) и раствор карбоната натрия (5 мл, 2М) в ацетонитриле (30 мл) перемешивают в атмосфере азота при 85°С в течение 6 часов. После завершения реакции реакцию гасят водой, добавляют ЕА (150 мл) к реакционной смеси, слои разделяют, водную фазу дважды экстрагируют ЕА (50 мл × 2) и объединенную органическую фазу сушат над Na2SO4 и концентрируют с получением неочищенного продукта, который очищают с помощью комби-флэш колоночной хроматографии [РЕ:ЕА=5:1-2:1] с получением 0,6 г целевого соединения 89-2, МС m/z (ЭРИ): 229 [М+Н]+.
Стадия 2: Карбонат цезия (487,5 мг, 0,5 ммоль) добавляют к раствору соединения 3а (159 мг, 0,5 ммоль), соединение 89-2 (114 мг, 0,5 ммоль), Pd2(dba)3 (45,75 мг, 0,05 ммоль) и Ксантфос (28,4 мг, 0,05 ммоль) в 1,4-диоксане (4 мл), реакционную смесь перемешивают при 100°С в течение 3 часов, ЕА (50 мл × 2) и воду (10 мл) добавляют к реакционной смеси, органическую фазу промывают водой (10 мл × 3), сушат над Na2SO4 и концентрируют с получением неочищенного продукта, полученный неочищенный продукт отделяют и очищают с помощью препаративной жидкостной хроматографии с получением целевого соединения Z-89 (19 мг, 4%), МС m/z (ЭРИ): 511,2 [М+Н]+; 1Н-ЯМР (400 МГц, CDCl3) δ 9,50 (s, 1Н), 9,19 (s, 1Н), 8,63 (s, 1Н), 8,40 (s, 1Н), 8,24 (s, 1Н), 7,66 (s, 1Н), 6,69 (s, 1Н), 6,28 (ddd, J=26,8, 16,9, 5,7 Гц, 2Н), 5,69 (dd, J=9,9, 1,6 Гц, 1Н), 4,00 (s, 3Н), 3,81 (s, 3Н), 2,96 (d, J=12,1 Гц, 2Н), 2,66 (t, J=10,9 Гц, 2Н), 2,15 (dd, J=9,3, 5,5 Гц, 1Н), 2,04-1,91 (m, 2Н), 1,62 (s, 2Н).
Пример 90: Получение N-(2-(4-(диметиламино)пиперидин-1-ил)-5-(5-фтор-4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-иламино)-4-метоксифенил)акриламида (Z-90)

Способ получения целевого соединения Z-90 (42 мг, выход 9%) аналогичен способу Примера 89, за исключением того, что соединение 19-1 на стадии 1 Примера 89 заменяют соединением 90-1, МС m/z (ЭРИ): 495,0 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6) δ 9,05 (1s, 1Н), 9,01 (1s, 1Н), 8,80 (s, 1Н), 8,48 (d, J=4 Гц, 1Н), 8,18 (s, 1Н), 7,89 (s, 1Н), 6,87 (s, 1Н), 6,73 (m, 1Н), 6,32 (m, 1Н), 5,78 (t, 1Н), 3,97 (s, 3Н), 3,88 (s, 3Н), 3,01 (d, J=12 Гц, 2Н), 2,66 (t, 2Н), 2,19 (s, 6Н), 2,16 (m, 1Н), 1,84 (m, 2Н), 1,71 (m, 2Н).
Пример 91: Получение N-(5-(5-хлор-4-(1,3,5-триметил-1Н-пиразол-4-ил)пиримидин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида (Z-91)
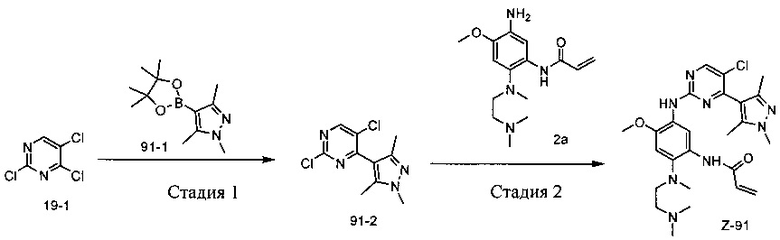
Способ получения целевого соединения Z-91 (22 мг, выход 11%) аналогичен способу Примера 89, за исключением того, что соединение 89-1 на стадии 1 Примера 89 заменяют соединением 91-1 и соединение 3а на стадии 2 заменяют соединением 2а. Соединение Z-91 представляет собой твердое вещество желтого цвета. МС m/z (ЭРИ): 513,2 [М+Н]+; 1Н-ЯМР (400 МГц, CDCl3) δ 9,32 (br. s., 1Н), 8,43 (s, 1Н), 7,54 (s, 1Н), 6,66 (s, 1Н), 6,35 (d, J=16,0 Hz, 1H), 5,62 (d, J=12,0 Hz, 1H), 3,78 (s, 3H), 3,70 (s, 3H), 2,92 (br. s., 2H), 2,63 (s, 3H), 2,34 (br. s., 6H), 2,21 (s, 3H), 2,19 (s, 3H), 1,60 (br. s., 2H).
Пример 92: Получение N-(5-(5-хлор-4-(1,3,5-триметил-1Н-пиразол-4-ил)пиримидин-2-иламино)-2-(4-(диметиламино)пиперидин-1-ил)-4-метоксифенил)акриламида (Z-92)

Пример 93: Получение N-(2-((2-(диметиламино)этил)(метил)амино)-5-(5-фтор-4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-иламино)-4-метоксифенил)акриламид (Z-93)

Пример 94: Получение N-(5-(5-хлор-4-(1,3-диметил-1Н-пиразол-4-ил)пиримидин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида (Z-94)

Способ получения целевого соединения Z-94 (13 мг, 2,6%) аналогичен способу Примера 91, за исключением того, что соединение 91-1 на стадии 1 Примера 91 заменяют соединением 94-1, МС m/z (ЭРИ): 499,2 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6) δ 10,13 (s, 1Н), 8,46 (d, J=3,1 Гц, 3Н), 8,40 (s, 1Н), 7,00 (s, 1Н), 6,41 (dd, J=16,7, 10,0 Гц, 1Н), 6,22 (dd, J=16,9, 2,0 Гц, 1Н), 5,75 (dd, J=10,1, 1,9 Гц, 1Н), 3,81 (d, J=17,7 Гц, 6Н), 2,90 (s, 2Н), 2,72 (s, 3Н), 2,33 (s, 2Н), 2,21 (d, J=24,6 Гц, 9Н).
Пример 95: Получение N-(5-(5-хлор-4-(1,3-диметил-1Н-пиразол-4-ил)пиримидин-2-иламино)-2-(4-(диметиламино)пиперидин-1-ил)-4-метоксифенил)акриламида (Z-95)
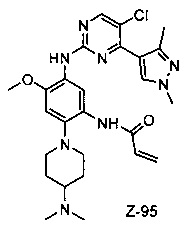
Пример 96: Получение N-(5-(5-хлор-4-(1-(пиперидин-4-ил)-1Н-пиразол-4-ил)пиримидин-2-иламино)-2-(4-(диметиламино)пиперидин-1-ил)-4-метоксифенил)акриламид (Z-96)

Стадия 1: Триэтиламин (2,0 г, 20 ммоль) добавляют к раствору трет-бутил-4-гидроксипиперидин-1-карбоксилата (2,0 г, 10 ммоль) в дихлорметане (20 мл) при 0°С и добавляют раствор метансульфонилхлорида (1,73 г, 15 ммоль) в дихлорметане (5 мл) по каплям. После перемешивания в течение 1 ч реакционную смесь выливают в лед, экстрагируют дихлорметаном и органическую фазу промывают насыщенным раствором соли, сушат над безводным Na2SO4 и концентрируют с получением 2,5 г неочищенного продукта 96-2, который используют без очистки на следующей стадии; МС m/z (ЭРИ): 223 [М-56]+.
Стадия 2: Соединение 96-2 (134 мг, 0,5 ммоль) и карбонат цезия (245 мг, 0,75 ммоль) добавляют к раствору 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразола (97 мг, 0,5 ммоль) в DMF, реакционную смесь перемешивают при 90°С в течение 12-16 ч и реакционный раствор охлаждают до комнатной температуры, разбавляют водой, экстрагируют с помощью ЕА и объединенную органическую фазу промывают насыщенным раствором соли, сушат над безводным Na2SO4 и концентрируют с получением неочищенного продукта, который очищают с помощью комби-флэш колоночной хроматографии [РЕ:ЕА=100:0-50:50] с получением целевого соединения 96-3 (2 г, выход 44%), МС m/z (ЭРИ): 378,2 [М+1]+.
Стадия 3: Используя соединение 19-1 (549 мг, 3,0 ммоль) и соединение 96-3 (754 мг, 2,0 ммоль) в качестве исходных веществ, целевое соединение 96-4 получают согласно стадии 1 Примера 89 (580 мг, выход 73%). МС m/z (ЭРИ): 398 [М+Н]+.
Стадия 4: Pd2(dba)3 (46 мг, 0,05 ммоль) добавляют к раствору соединения 96-4 (298 мг, 0,75 ммоль), соединения 2а (159 мг, 0,5 ммоль), карбоната цезия (326 мг, 1,0 ммоль) и Ксантфоса (58 мг, 0,1 ммоль) в 1,4-диоксане (2 мл), реакционный раствор трижды продувают азотом. После проведения микроволновой реакции при температуре 160°С в течение 0,5 ч реакционную смесь фильтруют и концентрируют с получением неочищенного продукта. Неочищенный продукт очищают с помощью комби-флэш колоночной хроматографии [РЕ:ЕА=100:0-0:100] с получением целевого соединения 96-5 (250 мг) в виде желтого твердого вещества. МС m/z (ЭРИ): 680,3 [М+Н]+.
Стадия 5: Смешанный раствор соляной кислоты/1,4-диоксана добавляют к раствору соединения 96-5 (240 мг, 0,35 ммоль) в дихлорметане (10 мл), реакционную смесь перемешивают при комнатной температуре в течение 2 ч, с помощью Na2CO3 доводят pH до 9, а затем добавляют дихлорметан. Органическую фазу промывают соляной кислотой и концентрируют с получением неочищенного продукта. Полученный неочищенный продукт очищают с помощью препаративной жидкостной хроматографии с получением 20 мг целевого соединения Z-96 в виде желтого твердого вещества. МС m/z (ЭРИ): 580 [М+Н]+.
Пример 97: Получение N-(5-(5-хлор-4-(1,5-диметил-1Н-пиразол-4-ил)пиримидин-2-иламино)-2-(4-(диметиламино)пиперидин-1-ил)-4-метоксифенил)акриламида (Z-97)

Способ получения целевого соединения Z-97 (56,5 мг, выход 26%) аналогичен способу Примера 89, за исключением того, что соединение 89-1 на стадии 1 Примера 89 заменяют соединением 97-1. Соединение Z-97 представляет собой желтое твердое вещество. МС m/z (ЭРИ): 525,2 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6) δ 8,99 (s, 1 Н), 8,44 (s, 1 Н), 8,39 (s, 1 Н), 8,20 (s, 1,5 Н), 7,99 (s, 1 Н), 6,81 (s, 1 Н), 6,66 (dd, J=16,8, 10,0 Гц, 1 Н), 6,22 (dd, J=16,8 Гц, 2,0 Гц, 1 Н), 5,73 (d, J=10,4 Гц, 1 Н), 3,79 (s, 3 Н), 3,76 (s, 3 Н), 3,07 (d, J=11,0 Гц, 2 Н), 2,68 (t, J=11,0 Гц, 2 Н), 2,46 (br. s., 1 Н), 2,37 (s, 6 Н), 2,34 (s, 3 Н), 1,89 (d, J=10,5 Гц, 2 Н), 1,75 (t, J=11,0 Гц, 2 Н).
Пример 98: Получение N-(5-(5-хлор-4-(1-метил-1Н-пиррол-3-ил)пиримидин-2-иламино)-2-(4-(диметиламино)пиперидин-1-ил)-4-метоксифенил)акриламида (Z-98)
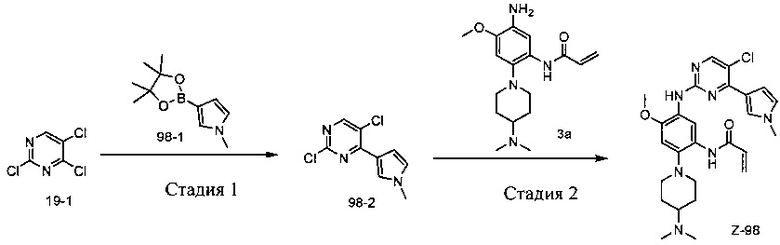
Стадия 1: Используя соединение 19-1 (400 мг, 2,2 ммоль) и 1-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиррол (250 мг, 2,0 ммоль) в качестве исходных веществ, неочищенный продукт получают согласно стадии 1 Примера 89, очищают с помощью комби-флэш колоночной хроматографии [РЕ:ЕА=3:1] с получением целевого соединения 98-2 (80 мг, выход 17,6%). МС m/z (ЭРИ): 228 [М+Н]+.
Стадия 2: Соединение 3а (140 мг, 0,4 ммоль), Pd2(dba)3 (41 мг, 0,044 ммоль), BINAP (55 мг, 0,088 ммоль) и карбонат цезия (286 мг, 0,88 ммоль) добавляют к раствору соединения 98-2 (100 мг, 0,44 ммоль) в 1,4-диоксане (6 мл). Реакционную смесь подвергают воздействию в микроволнового излучения при 130°С в течение 30 минут. Реакционную смесь фильтруют с получением неочищенного продукта, который очищают с помощью препаративной жидкостной хроматографии с получением 94,47 мг целевого соединения Z-98 в виде желтого порошка. МС m/z (ЭРИ): 510,3 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6) δ 9,06 (s, 1Н), 8,80 (s, 1Н), 8,34 (s, 1Н), 8,28 (s, 1Н), 8,06 (s, 1Н), 7,92 (s, 1Н), 6,98 (s, 1Н), 6,84 (dd, J=5,6, 3,2 Гц, 2Н), 6,71 (dd, J=16,9, 10,2 Гц, 1Н), 6,29 (dd, J=17,0, 1,6 Гц, 1Н), 5,77 (d, J=11,5 Гц, 1Н), 3,87 (s, 3Н), 3,74 (s, 3Н), 3,05 (d, J=11,4 Гц, 2Н), 2,68 (t, J=11,0 Гц, 2Н), 2,42 (t, J=11,1 Гц, 1Н), 2,34 (s, 6Н), 1,89 (d, J=10,6 Гц, 2Н), 1,79-1,67 (m, 2Н).
Пример 99: Получение N-(5-(5-хлор-4-(1-метил-1Н-пиррол-3-ил)пиримидин-2-иламино)-2-((2-(диметиламино)этил)(метил)амино)-4-метоксифенил)акриламида (Z-99)

Пример 100: Получение N-(5-(5-хлор-4-(1-метил-1Н-пиразол-4-ил)пиримидин-2-иламино)-4-метокси-2-(4-метилпиперазин-1-ил)фенил)акриламида (Z-100)

Используя соединение 4а (100 мг, 0,34 ммоль) и соединение 89-2 (80 мг, 0,34 ммоль) в качестве исходного материала, целевое соединение Z-100 (200 мг, выход 70%) синтезируют согласно стадии 2 Примера 89. МС m/z (ЭРИ): 483 [М+Н]+; 1Н-ЯМР (400 МГц, CDCl3) δ 9,51 (s, 1Н), 9,10 (s, 1Н), 8,41 (s, 2Н), 8,26 (s, 1Н), 8,20 (s, 1Н), 7,70 (s, 1Н), 6,73 (s, 1Н), 6,43-6,32 (m, 1Н), 6,26 (d, J=10,0 Гц, 1Н), 5,82-5,65 (m, 1Н), 3,99 (s, 3Н), 3,83 (s, 3Н), 3,03 (s, 8Н), 2,62 (s, 4Н).
Пример 101: Получение N-(5-(5-хлор-4-(1-(дифторметил)-1Н-пиразол-4-ил)пиримидин-2-иламино)-2-(4-(диметиламино)пиперидин-1-ил)-4-метоксифенил)акриламида (Z-101)

Стадия 1: К ацетонитрилу (100 мл) добавляют соединение 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол 101-1 (4,0 г, 0,21 моль), хлордифторацетат натрия (3,77 г 0,25 моль), 15-краун-5 (103,8 г, 0,65 моль) и реакционную смесь нагревают с обратным холодильником в атмосфере азота и перемешивают в течение 24 ч, охлаждают до комнатной температуры, реакционную смесь выливают в воду (50 мл) для гашения реакции и экстрагируют ЕА (100 мл × 3), промывают насыщенным раствором соли (20 мл), сушат над Na2SO4, фильтруют и фильтрат выпаривают с получением соединения 101-2 (4,1 г, выход 80%) в виде белого твердого вещества. МС m/z (ЭРИ): 245,1 [М+Н]+.
Стадия 2: В реакционную колбу, содержащую тетракис(трифенилфосфин) палладий (0,264 г, 2,58×10-4 моль), добавляют раствор соединения 101-2 (2,1 г, 8,9×10-3 моль) в диметоксиэтане (20 мл), раствор соединения 19-1 (1,4 г, 8,0×10-3 моль) в диметоксиэтане (20 мл) и раствор карбоната натрия (1,8 г, 1,72×10-2 моль) в воде (11 мл) и реакционную смесь нагревают до 100°С в атмосфере азота и перемешивают в течение ночи. Реакционный раствор охлаждают до комнатной температуры, используют воду (50 мл) для гашения реакции и полученную смесь экстрагируют ЕА (3×100 мл), промывают насыщенным раствором соли (30 мл), сушат над Na2SO4, фильтруют и концентрируют с получением масла, которое очищают с помощью колоночной хроматографии (силикагель; 300-400 меш), петролейный эфир : ЕА = 30:1 с получением соединения 101-3 (870 мг, выход 30%) в виде белого твердого вещества. МС m/z (ЭРИ): 265,0 [М+Н]+, чистота = 96,08% (UV214);1Н-ЯМР δ (400 МГц, DMSO) δ: 9,21 (s, 1Н), 8,95 (s, 1Н), 8,56 (s, 1Н), 7,94 (t, J=58 Гц, 1Н).
Стадия 3: Используя соединение 101-3 и соединение 3а в качестве исходных материалов, согласно стадии 2 Примера 89 синтезируют неочищенный продукт и очищают с помощью препаративной жидкостной хроматографии с получением целевого соединения Z-101 в виде желтого твердого вещества.
МС m/z (ЭРИ): 547 [М+Н]+; 1Н-ЯМР (400 МГц, DMSO-d6) δ 9,28 (s, 1Н), 9,06 (s, 1Н), 8,76 (s, 1Н), 8,65 (s, 1Н), 8,55 (s, 1Н), 8,28 (d, J=3,6 Гц, 2Н), 7,92 (t, J=58,6 Гц, 1Н), 6,87 (s, 1Н), 6,70 (dd, J=17,0, 10,2 Гц, 1Н), 6,29 (dd, J=16,9, 1,7 Гц, 1Н), 5,87-5,65 (m, 1Н), 3,86 (s, 3Н), 3,04 (d, J=11,6 Гц, 2Н), 2,68 (t, J=10,9 Гц, 2Н), 2,26 (s, 7Н), 1,85 (d, J=10,5 Гц, 2Н), 1,71 (d, J=8,9 Гц, 2Н).
Пример 102: Получение N-(5-(5-хлор-4-(1Н-пиразол-4-ил)пиримидин-2-иламино)-2-(4-(диметиламино)пиперидин-1-ил)-4-метоксифенил)акриламида (Z-102)

Стадия 1: DTSA (463 мг, 2,6 ммоль) добавляют к раствору соединения 101-1 (5,0 г, 25,8 ммоль) и 2,3-дигидропирану (4,33 г, 51,5 ммоль) в дихлорметане (100 мл) при перемешивании и реакционную смесь перемешивают при 40°С в течение 2 ч. После завершения реакции реакционный раствор промывают водой, экстрагируют ЕА и концентрируют при пониженном давлении с получением неочищенного продукта 102-2 (7,0 г, 60%), который используют на следующей стадии без дополнительной очистки. МС m/z (ЭРИ): 279 [М+Н]+.
Стадия 2: Используя соединение 19-1 (4,5 г, 25 ммоль) и соединение 102-2 (7,0 г, 25 ммоль) в качестве исходных веществ, согласно стадии 1 Примера 89 синтезируют неочищенный продукт и очищают с помощью комби-флэш колоночной хроматографии с получением целевого соединения 102-3 (850 мг, выход 10%). МС m/z (ЭРИ): 299 [М+Н]+.
Стадия 3: Используя соединение 102-3 (300 мг, 1 ммоль) и соединение 3а (320 мг, 1 ммоль) в качестве исходных веществ, согласно стадии 2 Примера 89 получают неочищенный продукт 102-4 (60 мг, выход 70%), который используют непосредственно на следующей стадии. МС m/z (ЭРИ): 581 [М+Н]+.
Стадия 4: К раствору соединения 102-4 (700 мг, 1 ммоль) в дихлорметане (5 мл) добавляют раствор соляной кислоты/1,4-диоксана (2 мл) при перемешивании, и реакционную смесь перемешивают при комнатной температуре в течение 4 часов, получая неочищенный продукт, который очищают с помощью препаративной колоночной хроматографии с получением целевого соединения Z-102 (80 мг, выход 60%) в виде желтого твердого вещества. МС m/z (ЭРИ): 497 [М+Н]+; 1Н-ЯМР (400 МГц, CDCl3) δ 9,55 (s, 1Н), 8,92 (s, 2Н), 8,70 (s, 1Н), 8,35 (s, 1Н), 7,72 (s, 1Н), 6,76 (s, 1Н), 6,32 (dt, J=17,0, 12,9 Гц, 2H), 5,76 (d, J=9,9 Гц, 1H), 3,89 (s, 3Н), 3,04 (d, J=11,9 Гц, 2H), 2,73 (t, J=11,0 Гц, 2H), 2,36 (s, 6H), 2,24 (d, J=7,4 Гц, 1H), 2,05 (d, J=11,5 Гц, 2H), 1,70 (s, 2H).
Примеры 103-127
Целевые соединения примеров 103-127 показаны в виде формулы (IV-1), где заместители R0, R2 и R3 представляют собой водород, а другие заместители A, R1, R6 являются такими, как показано в следующей таблице.
Общий способ: Способы получения соединений Z-103-Z-127 аналогичны способу Примера 1, с использованием боронатного эфира или борной кислоты, замещенной различными заместителями, и с использованием 5-замещенного 2,4-дихлорпиримидина в качестве исходных веществ.

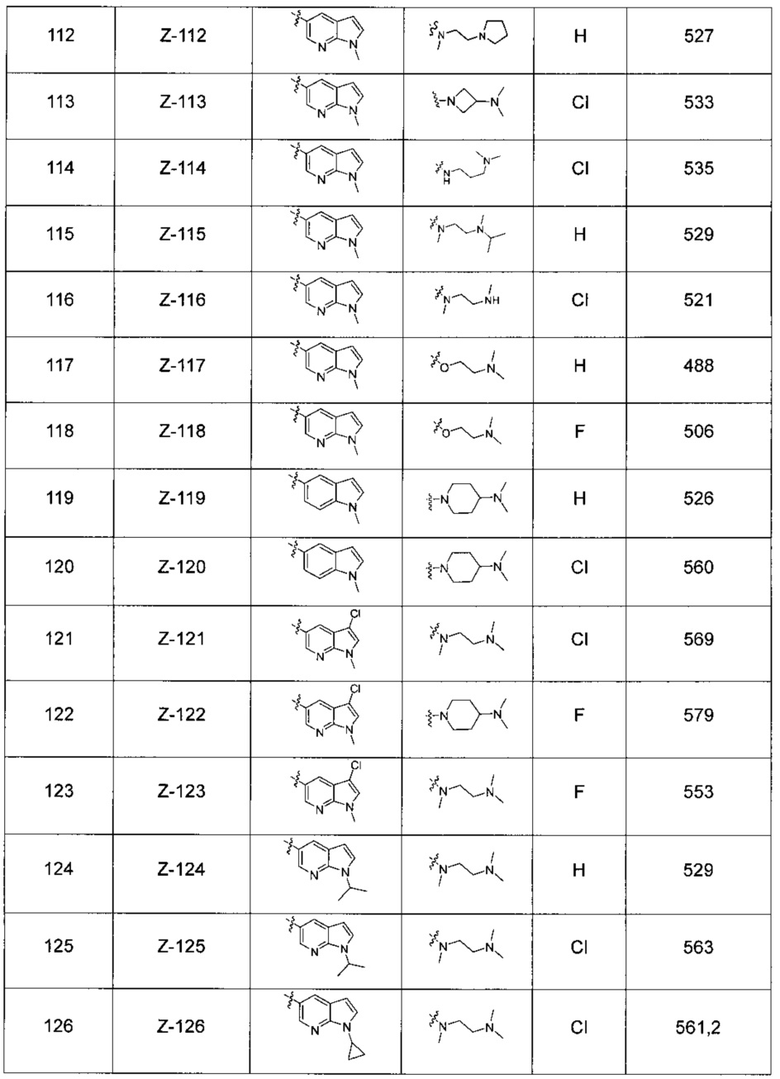




Сравнительный пример
Используя соответствующий сложный эфир бора и незамещенный или 5-замещенный 2,4-дихлорпиримидин в качестве исходных материалов, согласно Примеру 1 получают сравнительные соединения 2 и 3.

Исследование 1: Исследование ингибирования киназы EGFR дикого типа и мутантного EGFR.
Все последующие реагенты, используемые в тесте z-lyte, являются коммерчески доступными от фирмы Invitrogen.
Ингибирующие действия тестируемых соединений на киназу EGFR с двойной мутацией (киназа EGFR T790M/L858R) (Invitrogen, PV4879) и на киназу EGFR дикого типа (EGFR WT) (Invitrogen, PV3872) измеряют с помощью методов z-lyte.
Рабочая концентрация каждого компонента в 10 мкл реакционной системы киназы T790M/L858R составляет: 25 мкм АТФ, 0,08 (или 0,1) нг/мкл киназы EGFR T790M/L858R, 2 мкМ субстрата Tyr04 (Invitrogen, PV3193, в дальнейшем - аналогично). После добавления соединений (т.е. тестируемых соединений), полученных в приведенных выше в этом документе примерах, концентрация DMSO составляет 2%.
Методы исследований:
10 мМ маточные растворы исследуемых соединений, растворенные при комнатной температуре, постепенно разводят 4% DMSO в воде до конечной концентрации 10-0,005 мкМ. В каждую лунку добавляют 2,5 мкл раствора тестируемых соединений и 5 мкл смеси киназы EGFR T790M/L858R (или киназы EGFR WT) и субстрата Tyr04, разведенной реакционным буфером, а затем добавляют 2,5 мкл АТФ для инициирования реакции. В лунки С1 вместо АТФ добавляют реакционный буфер, в лунки С2 не добавляют никаких лекарственных средств, в лунки С3 добавляют фосфорилированные субстраты в соответствии с инструкцией.
Реакцию проводят на вибростенде при комнатной температуре в течение 60 мин. Затем добавляют 5 мкл Проявляющего Реагента В (Invitrogen) и проводят реакцию на вибростенде при комнатной температуре в течение 60 мин. Планшеты считывают на микропланшетном ридере VictorX5 (PerkinElmer), для измерения оптической плотности при длине волны возбуждения 405 нм и длинах волн эмиссии 450 нм и 520 нм. (Например, С3520 нм представляет показание при 520 нм для лунки С3).
Показатель ингибирования рассчитывают следующим образом:
1. ER = Эмиссия кумарина (450 нм) / эмиссия флуоресцеина (520 нм)
2. Показатель фосфорилирования = (1-((ER×C3520 нм - C3450 нм)/((C1450 нм - C3450 нм)+ER×(С3520 нм - С1520 нм))))×100%
3. Показатель ингибирования (IR) = (1 - показатель фосфорилирования тестируемого соединения) / (показатель фосфорилирования С2))×100%
IC50 (концентрацию полумаксимального ингибирования) определяют путем вычисления подбором с помощью XLfit 5.0 (компания IDBS, Великобритания). Результаты ингибирующей активности и селективной ингибирую щей активности в отношении фермента показаны в Таблице 1-Таблице 3.






Как видно из Таблицы 1, Таблицы 2 и Таблицы 3 иллюстративные соединения по настоящему изобретению демонстрируют сильные ингибирующие активности в отношении мутантного фермента EGFR (T790M/L858R и L858R), но слабую ингибирующую активность в отношении фермента EGFR дикого типа (Т790М WT), соединения по настоящему изобретению обладают значительной селективной ингибирующей активностью в отношении мутантного фермента EGFR по сравнению с положительным контролем BIBW2992 (афатиниб), и селективная ингибирующая активность иллюстративных соединений по изобретению в отношении мутантного фермента EGFR была выше, чем у сравнительного соединения 1 (конкретная структура показана ниже и может быть найдена в WO 2013014448 A1). Самая высокая селективность в 18 раз превышала селективность сравнительного соединения 1.
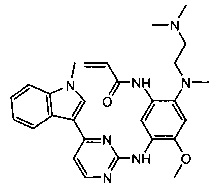
Исследование 2: Проверка цитостатической активности методом МТТ (на основе 3-(4,5-диметилтиазол-2)-2,5-дифенилтетразолия бромида)
МТТ-тест хорошо известен специалистам в данной области техники, и реагенты, используемые в этом методе, коммерчески доступны.
2.1 Метод исследования:
Во-первых, среду удаляют и добавляют 0,25% трипсин/EDTA (этилендиаминтетрауксусная кислота) (Gibco, 25200-056). После однократного промывания добавляют 1,5 мл трипсин/EDTA для переваривания прилипающих клеток до полного их открепления, а затем останавливают трипсинизацию путем добавления 3,5 мл среды. Суспензию переваренных клеток переносят в пробирку Falcon объемом 15 мл и центрифугируют при 1300 об/мин. в течение 3 минут, супернатант удаляют и клетки суспендируют в свежей среде. Затем клетки подсчитывают и разбавляют до следующих концентраций: 2,78×104 клеток/мл (А431 и Н1975), 3,33×104 клеток/мл (NIH3T3). Клетки высевают на 96-луночные планшеты (BD 3072), 90 мкл/лунка, и инкубируют в течение ночи.
Среда для культивирования клеток А431: 10% FBS (фетальная бычья сыворотка) (Gibco, 10099-141) DMEM (среда Игла, модифицированная по способу Дульбекко) (Hyclone SH30243,01B);
Среда для культивирования клеток NIH3T3: 10% FBS (Gibco, 10099-141) DMEM (Hyclone SH30243,01B);
Среда для культивирования клеток Н1975: 10% FBS (Gibco, 10099-141) RPMI-1640 (Hyclone SH30809,01В);
Берут 20 мкл 10 мМ тестируемого соединения и разбавляют согласно следующему градиенту концентраций: (2000, 666,67, 222,22, 74,07, 24,69, 8,23, 2,74, 0,91 мкМ), затем добавляют бессывороточную среду (конечная концентрация: 10, 3,333, 1,111, 0,370, 0,123, 0,041, 0,014, 0,005 мкМ) и 10 мкл тестриуемого соединения из каждой лунки планшета добавляют в культуральные чашки, конечная концентрация DMSO составляет 0,5%.
После добавления тестируемого соединения клетки помещают в инкубатор и инкубируют в течение 72 ч, 10 мкл 5 мг/мл раствора МТТ (Sigma, М5655) добавляют в каждую лунку планшета и инкубируют 96-луночные планшеты при 37°С и 5% СО2 в инкубаторе в течение 4 ч.
Планшеты центрифугируют при 2000 об/мин в течение 5 минут. После удаления супернатанта в каждую лунку добавляют 150 мкл DMSO и планшеты встряхивают на шейкере до полного растворения фиолетового кристалла (приблизительно 10-20 мин). И, наконец, измеряют оптическую плотность при 492 нм с использованием микропланшетного ридера и вычисляют IC50 с помощью программного обеспечения XLfit 5.0 (компания UK IDBS). Ингибирующая активность и селективная ингибирующая активность иллюстративных соединений в отношении клеток приведены в Таблице 4 - Таблице 7.

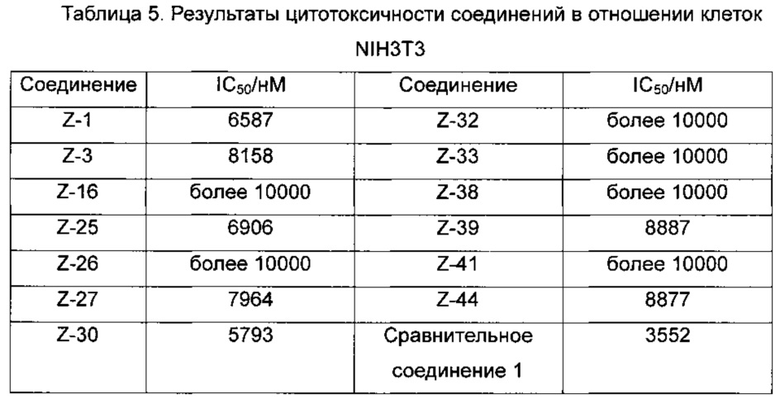



Как видно из Таблицы 4 и Таблицы 6 иллюстративные соединения по изобретению демонстрируют высокие ингибирующие активности в отношении EGFR мутантных клеток (клетки Н1975), но слабые ингибирующие активности в отношении клеток EGFR дикого типа (клетки А431), а также соединения по настоящему изобретению обладают значительными селективными ингибирующими активностями в отношении роста EGFR мутантных клеток по сравнению с положительным контролем BIBW2992 (афатиниб). Селективные ингибирующие активности большинства иллюстративных соединений в отношении роста EGFR мутантных клеток выше, чем у сравнительных соединений 1, 2 и 3, Наивысшая селективность была почти в четыре раза выше, чем у сравнительного соединения 1. Исследование показало, что активности в отношении клеток Н1975 и селективные ингибирующие активности в отношении роста клеток значительно снижаются после того, как кольцо А соединения заменяют пиридином или хинолином.
Как видно из Таблицы 5 и Таблицы 7 иллюстративные соединения по изобретению обладают более высокими значениями IC50 в отношении клеток NIH3T3 и демонстрируют меньшую токсичность.
Исследование 3: Изучение клеточной активности ингибитора EGFR Т790М методом ELISA (иммуно-ферментный анализ)
Реагенты, способы приготовления раствора, процедуры обработки клеток и получения лизата, а также стадии ELISA в следующем методе проводят в соответствии с инструкциями для R&D DYC3570, R&D DYC1095E и R&D DYC1095BE.
1. Реагенты и растворы
Лизирующий буфер: 1% NP-40, 20 мМ Трис (pH 8,0), 137 мМ NaCl, 10% глицерин, 1 мМ NaVO3, 2 мМ EDTA.
Клеточные лизаты: лизирующий буфер + 10 мкг/мл апротинина (Sigma), 10 мкг/мл лейпептина (Sigma). Все реагенты готовят непосредственно перед использованием.
1×PBS (фосфатно-солевой) буфер: NaCl: 0,137 М, KCl: 0,0027 М, Na2PO4-12H2O: 0,01 М, KH2PO4: 0,0015М, pH 7,4.
Отмывочный буфер: 0,05% Tween-20 в PBS.
IC Разбавитель: 20 мМ Трис, 137 мМ NaCl, 0,05% Tween-20, 0,1% BSA (альбумин бычьей сыворотки), pH 7,2-7,4.
Блокирующий буфер: 1% BSA в PBS.
Наборы для ELISA: R&D DYC3570, R&D DYC1095E и R&D DYC1095BE.
2. Клетки Н1975
2.1 Обработка клеток Н1975 и получение лизатов
(1) клетки Н1975 высевают в 96-луночный планшет с плотностью 1×104 клеток/лунка с 90 мкл с 10% FBS, 1640 среды на каждую лунку, и инкубируют при 37°С в атмосфере 5% CO2 в течение ночи.
(2) Тестируемые соединения разводят в соответствии с методом МТТ и 10 мкл разведенного соединения или DMSO добавляют в соответствующую лунку планшета и инкубируют при 37°С в атмосфере 5% CO2 в течение 1 ч. Конечная концентрация DMSO составляет 0,5%. В качестве контроля клеток используют клеточную культуру, обработанную только DMSO.
(3) 100 мкл клеточного лизата добавляют после отсасывания среды, герметизируют и помещают в холодильник при температуре -80°С в течение ночи. В качестве пустого контроля используют лизирующий буфер.
2.2 Процедура ELISA анализа
Анализ проводят в соответствии с инструкциями для R&D DYC1095E или R&D DYC1095BE.
(1) R&D иммобилизированное антитело ((DYC1095BE или DYC1095E)) разводят в PBS в соотношении 1:180 и планшет для ELISA (Corning Costar 42592) покрывают разведенным антителом в количестве 100 мкл/лунка, и инкубируют при 25°С в течение ночи при встряхивании;
(2) отмывают три раза, используя 360 мкл отмывочного буфера;
(3) добавляют 300 мкл блокирующего буфера, инкубируют при 25°С в течение 2 часов при встряхивании;
(4) отмывают три раза, используя 360 мкл отмывочного буфера;
(5) добавляют 40 мкл лизирующего буфера и 60 мкл клеточных лизатов и инкубируют при 25°С в течение 2 ч при встряхивании;
(6) отмывают три раза, используя 360 мкл отмывочного буфера;
(7) детекторные антитела разводят в соответствии с заранее определенным соотношением, как это предусмотрено в инструкции к набору с использованием IC разбавителя, и 100 мкл разведенного детекторного антитела добавляют в каждую лунку и инкубируют при 25°С в течение 1 ч при встряхивании в темноте;
(8) отмывают три раза, используя 360 мкл отмывочного буфера;
(9) реагент А и реагент В субстрата ТМВ (3,3',5,5'-тетраметилбензидин) (R&D DY999) смешивают в соотношении 1:1 и 100 мкл субстрата добавляют в каждую лунку и инкубируют при 25°С в течение 20 минут при встряхивании в темноте;
(10) 50 мкл 2N H2SO4 добавляют в каждую лунку;
(11) Значения OD450 и OD570 (опитческая плотность) клеточного контроля, пустого контроля и после обработки соединением измеряют соответственно с помощью микропланшетного ридера (Thermo Multiskan К3), и соответствующие значения OD570 вычитают из значений OD450 для одних и тех же лунок с получением ODклетки, ОDпустого контроля и ODпосле обработки соединением, соответственно.
2.3 Анализ данных
Показатель ингибирования (%) = 100%×(ODклетки - ODпосле обработки соединением)/(ODклетки - ODпустого контроля)
2.4 Величины IC50 рассчитывают с помощью программного обеспечения XLfit 5.0 от расчитанного показателя ингибирования и они представлены в Таблице 8,
3. Клетки А431
3.1 Обработка клеток А431 и процедура исследования
(1) Клетки А431 высевают на 96-луночный планшет в количестве 1×104 клеток/лунка с 90 мкл 10% FBS, 1640 среды на каждую лунку и инкубируют при 37°C с 5% CO2 в течение ночи.
(2) Среду клеток А431 заменяют на 90 мкл среды DMEM без FBS и инкубируют в течение ночи.
(3) Тестируемые соединения разводят в соответствии с методом МТТ, 10 мкл разведенного соединения или DMSO добавляют в соответствующую лунку планшета и инкубируют при 37°C с 5% CO2 в течение 1 ч. Конечная концентрация DMSO составляет 0,5%. затем добавляют в каждую лунку, за исключением лунки с клеточным контролем, 10 мкл 2 мкг/л EGF (эпидермальный фактор роста), затем добавляют в лунку 10 мкл среды DMEM без FBS и инкубируют в течение 45 минут; в качестве клеточного контроля используют клетки без EGF и необработанные соединением, в качестве контроля EGF используют клетки необработанные соединением, но обработанные EGF.
(4) 100 мкл клеточного лизата добавляют после удаления среды, герметезируют и помещают в холодильник при температуре -80°С на ночь.
3.2 Процедура анализа ELISA
Анализ проводят в соответствии с инструкциями для R&D DYC3570E.
(1) Иммобилизированные антитела R&D (DYC3570E) разводят в PBS в соотношении 1:180 и разведенные антитела добавляют в планшет для ELISA (Corning Costar 42592) 100 мкл/лунка и инкубируют при 25°С в течение ночи при встряхивании;
(2) отмывают три раза, используя 360 мкл отмывочного буфера;
(3) добавляют 200 мкл блокирующего буфера, инкубируют при 25°С в течение 2-х часов при встряхивании;
(4) отмывают три раза, используя 360 мкл отмывочного буфера;
(5) добавляют 40 мкл лизирующего буфера и 60 мкл клеточных лизатов и инкубируют при 25°С в течение 2 ч при встряхивании;
(6) отмывают три раза, используя 360 мкл отмывочного буфера;
(7) детекторные антитела разводят в соответствии с заранее определенным соотношением в соответствии с инструкцией к набору, используя IC разведение, и 100 мкл разведенных детекторных антител добавляют в каждую лунку и инкубируют при 25°С в течение 1 ч при встряхивании в темноте;
(8) отмывают три раза, используя 360 мкл отмывочного буфера;
(9) реагент А и реагент В субстрата ТМВ (R&D DY999) смешивают в соотношении 1:1 и 100 мкл субстрата добавляют в каждую лунку и инкубируют при 25°С в течение 20 минут при встряхивании в темноте;
(10) в каждую лунку добавляют 50 мкл 2N H2SO4;
(11) Значения OD450 и OD570 клеточного контроля, пустого контроля и после обработки соединением измеряют соответственно с помощью микропланшетного ридера (Thermo Multiskan K3) и соответствующие значения OD570 вычитают из значений OD450 для одних и тех же лунок с получением ODклетки, ODпустого контроля и ODпосле обработки соединением соответственно.
3.3 Анализ данных
Показатель ингибирования (%) = 100%×(ODEGF - ODсоединения)/(ODEGF - ODклетки)
3.4 Значения IC50 рассчитывают с помощью программного обеспечения XLFIT 5.0 из вычисленного показателя ингибирования, они показаны в Таблице 8.
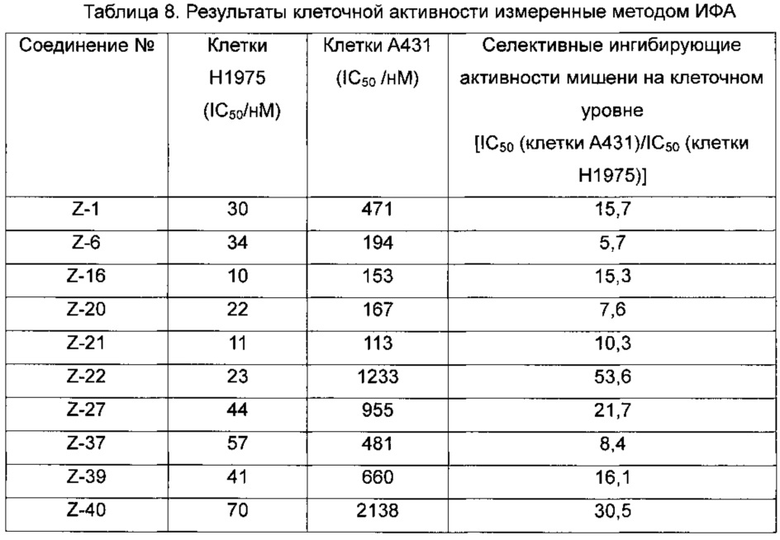


Как видно из Таблицы 8, иллюстративные соединения по изобретению демонстрируют очевидную селективную ингибирующую активность в отношении мишени на клеточном уровне по сравнению с положительным контролем BIBW2992. Наибольшая селективная ингибирующая активность по отношению к мишени на клеточном уровне увеличилась до 90 раз по сравнению с таковой сравнительных соединений 1, 2 и 3. Исследование показало, что активность в отношении клеток Н1975 и селективная ингибирующая активность в отношении мишени на клеточном уровне существенно ниже или даже отсутствует после того, как кольцо А соединения заменяют пиридином или хинолином.
Исследования in vitro ингибирования ферментов и роста клеток показывают, что соединения по изобретению проявляют сильную ингибирующую активность в отношении EGFR мутантных ферментов и клеток, но демонстрируют слабую ингибирующую активность в отношении ферментов и клеток EGFR дикого типа и, таким образом, обладают более высокой селективностью; кроме того, соединения оказывают очень слабое ингибирующее действие на клетки NIH-3T3 в исследованиях цитотоксичности, и, таким образом, демонстрируют более низкую токсичность. Следовательно, такие соединения обладают более высокой селективной ингибирующей активностью в отношении мутаций Т790М и более низкой цитотоксичностью.
Исследование 4: Исследование in vivo на крысах или мышах
Для определения концентрации лекарства в плазме в различные моменты времени после того, как иллюстративные соединения перорально и внутривенно вводили крысам или мышам с целью изучения фармакокинетического поведения соединений по изобретению in vivo у крыс или мышей и оценки их фармакокинетических характеристик применяют метод ЖХ/МС/МС (жидкостной хроматографии с тандемной масс-спектрометрией).
Протокол:
Подопытные животные: здоровые взрослые самцы крыс SD (вес 200-300 г, 6, без пищи) или самцы мышей CD1 (вес 20-30 г, 18, свободный доступ к воде и пище), предоставлены компанией SLAC;
Введение и дозировка: (1 мг/кг, 5 мл/кг, 5% DMAC (диметилацетамид), 5% Solutol HS 15 (полиэтиленгликоль-15 гидроксистеарат) и 90% физиологического раствора вводят внутривенно через дорсальную вену стопы и (20 мг/кг, 10 мл/кг, 0,5% раствор CMC-Na (натрий-карбоксиметилцеллюлоза)) вводят через желудочный зонд крысам SD; (1 мг/кг, 5 мл/кг, 5% DMAC, 5% Solutol HS 15 и 90% солевой раствор) вводят внутривенно через хвостовую вену и (5 мг/кг, 10 мл/кг, 0,5% водного раствора CMC-Na) вводят через желудочный зонд мышам CD1.
Отбор крови: во-первых, животных, удовлетворяющих требованиям эксперимента перед введением, взвешивают. Крысы или мышей связывают перед отбором крови, у каждой крысы после введения отбирают кровь в заранее определенные моменты времени (внутривенное введение: кровь отбирают в 0,083, 0,25, 0,5, 1, 2, 4, 8, 24 часа до и после введения, соответственно, суммарно 9 временных точек; через желудочный зонд: кровь отбирают в 0, 083, 0,25, 0,5, 1, 2, 4, 8, 24 часа до и после введения, суммарно 9 временных точек), около 150 мкл крови отбирают из хвостовой вены или сердца. У каждой мыши после введения отбирают кровь в заранее определенные моменты времени (через хвостовую вену: кровь отбирают в 0,083, 0,25, 0,5, 1, 2, 4, 8, 24 часа, суммарно 8 временных точек; через желудочный зонд: кровь отбирают в 0,25, 0,5, 1, 2, 4, 8, 24 часа, суммарно 7 временных точек), около 150 мкл крови отбирают из вены глаза или сердца. Кровь переносят в 1,5 мкл пробирку, в которую заранее добавляют K2EDTA. Отобранные образцы крови помещают на лед и центрифугируют с получением образца плазмы (2000 г, 5 мин при 4°С) в течение 15 минут. Все образцы плазмы хранят при приблизительно -70°С до проведения анализа.
Для определения концентрации лекарства был применен метод ЖХ/МС/МС. В Таблице 9 показаны фармакокинетические параметры некоторых иллюстративных соединений по изобретению при той же дозе и способе введения у крыс и мышей:


Как видно из Таблицы 9, иллюстративные соединения по настоящему изобретению хорошо абсорбируются и одновременно демонстрируют очевидный абсорбционный эффект и высокую биодоступность.
Все публикации, упомянутые в настоящем описании, включены в качестве ссылки, как если бы каждый отдельный документ был приведен в качестве ссылки, как в настоящей заявке. Следует также понимать, что после прочтения вышеизложенных идей настоящего изобретения специалисты в данной области техники могут вносить различные изменения или модификации, эквиваленты которых входят в объем формулы изобретения, как это определено в прилагаемой формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОР EGFR И ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2702631C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИМИДОНА ИЛИ ПИРРОЛОТРИАЗОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2014 |
|
RU2674977C2 |
| Соединения триазоло-пиримидина и их применение | 2019 |
|
RU2802866C2 |
| ЗАМЕЩЕННЫЙ 2-АМИНОПИРИДИН В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗЫ | 2014 |
|
RU2671212C2 |
| Ингибиторы ErbB/BTK | 2019 |
|
RU2764069C1 |
| ПРОИЗВОДНОЕ ПИРИМИДИН-4,6-ДИАМИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2022 |
|
RU2834353C2 |
| ПРОИЗВОДНЫЕ АНИЛИНПИРИМИДИНА И ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2734849C2 |
| НОВОЕ ХИНОЛИН-ЗАМЕЩЕННОЕ СОЕДИНЕНИЕ | 2014 |
|
RU2689158C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРОВ ERBB | 2019 |
|
RU2810215C2 |
| ПРОИЗВОДНЫЕ ПИРИДИЛКЕТОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2014 |
|
RU2667892C2 |
Изобретение относится к новому производному 2,4-замещенного фенилен-1,5-диамина формулы (I) или его фармацевтически приемлемой соли, обладающим активностью ингибитора в отношении тирозинкиназы EGFR. Соединения могут найти применение для изготовления лекарственного средства для регулирования активности тирозинкиназы EGFR (рецептора эпидермального фактора роста) или лечения EGFR-зависимых заболеваний. Таким заболеванием являются рак, диабет, заболевания иммунной системы, нейродегенеративные заболевания, сердечно-сосудистые заболевания или заболевание с приобретенной резистентностью в ходе лечения с использованием модуляторов EGFR. При этом заболевание с приобретенной резистентностью обусловлено мутациями Т790, кодируемыми 20-м экзоном EGFR, или содержанием мутаций Т790, кодируемых 20-м экзоном EGFR. В соединении формулы (I)
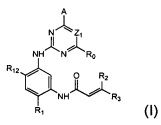
А является любым, выбранным из  или
или 
где Z2 представляет собой CR5 или N; R5 представляет собой водород; Z21 представляет собой N; каждый из R4, R42, R43 независимо представляет собой водород или C1-6 алкил, 6-членный гетероциклоалкил, содержащий группу -NH в кольце или C1-6 галогеналкил; R41 представляет собой водород или галоген; R0 представляет собой водород; Z1 представляет собой CR6, где R6 представляет собой водород, галоген, трифторметил, метокси или -СО2С1-10 алкил; R1 представляет собой водород или является любым, выбранным из группы, состоящей из:



каждый из R2 и R3 независимо представляет собой водород; R12 представляет собой С1-6 алкокси. 3 н. и 6 з.п. ф-лы, 9 табл., 127 пр.
1. Соединение формулы (I) или его фармацевтически приемлемая соль:

где А является любым, выбранным из  или
или 
где Z2 представляет собой CR5 или N; R5 представляет собой водород;
Z21 представляет собой N;
каждый из R4, R42, R43 независимо представляет собой водород или C1-6 алкил, 6-членный гетероциклоалкил, содержащий группу -NH в кольце или C1-6 галогеналкил;
R41 представляет собой водород или галоген;
R0 представляет собой водород;
Z1 представляет собой CR6, где R6 представляет собой водород, галоген, трифторметил, метокси или -СО2С1-10 алкил;
R1 представляет собой водород или является любым, выбранным из группы, состоящей из:



каждый из R2 и R3 независимо представляет собой водород;
R12 представляет собой С1-6 алкокси.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где А является любым, выбранным из группы, состоящей из

где R4, R41, R42, R43 являются такими, как определено в п. 1.
3. Соединение по п. 1 или его фармацевтически приемлемая соль, где соединение формулы (I) является соединением, представленным формулой (IV-1):

в формуле (IV-1) A, R0, R1, R2, R3 и R6 являются такими, как определено в п. 1.
4. Соединение по п. 1 или его фармацевтически приемлемая соль, где соединение формулы (I) является соединением, представленным формулой (V-1), (V-2), (VI-1), (VI-2), (XI-1) или (XI-2):
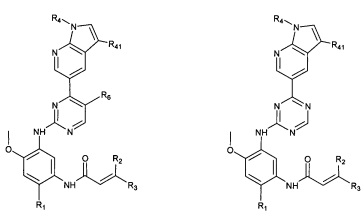

в формуле (V-1) R1, R2, R3, R4, R41 и R6 являются такими, как определено в п. 1;
в формуле (V-2) R1, R2, R3, R4 и R41 являются такими, как определено в п. 1;

в формуле (VI-1) R1, R2, R3, R4, R41 и R6 являются такими, как определено в п. 1;
в формуле (VI-2) R1, R2, R3, R4 и R41 являются такими, как определено в п. 1;

в формуле (XI-1) R1, R2, R3, R4, R42, R43 и R6 являются такими, как определено в п. 1;
в формуле (XI-2) R1, R2, R3, R4, R42 и R43 являются такими, как определено в п. 1.
5. Соединение по п. 1 или его фармацевтически приемлемая соль, где соединение формулы (I) выбрано из:





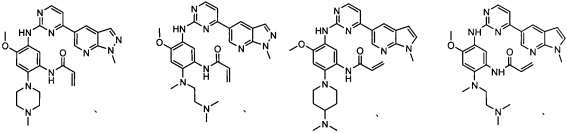





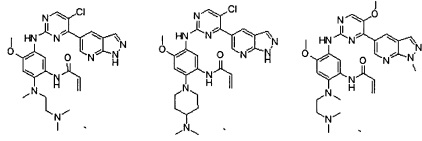


 или
или 
6. Фармацевтическая композиция, обладающая активностью ингибитора в отношении тирозинкиназы EGFR, включающая эффективное количество соединения по любому из пп. 1-5 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
7. Применение соединения по любому из пп. 1-5 или его фармацевтически приемлемой соли для изготовления лекарственного средства для регулирования активности тирозинкиназы EGFR (рецептора эпидермального фактора роста) или лечения EGFR-зависимых заболеваний.
8. Применение по п. 7, где EGFR-зависимым заболеванием являются рак, диабет, заболевания иммунной системы, нейродегенеративные заболевания, сердечно-сосудистые заболевания или заболевание с приобретенной резистентностью в ходе лечения с использованием модуляторов EGFR.
9. Применение по п. 8, где заболевание с приобретенной резистентностью обусловлено мутациями Т790, кодируемыми 20-м экзоном EGFR, или содержанием мутаций Т790, кодируемых 20-м экзоном EGFR.
| CN 101910152 A, 08.12.2010 & US 8309718 B2, & EA 020777 B1, 30.01.2015 | |||
| CN 102740847 A, 17.10 | |||
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| CN 103501612 A, 08.01.2014 & EA 201391626 A1, 31.03.2014 | |||
| WO 2006021458 A2, 02.03.2006 | |||
| КОМБИНАЦИЯ, ВКЛЮЧАЮЩАЯ КОМПЛЕКСОН ЖЕЛЕЗА И ПРОТИВООПУХОЛЕВЫЙ АГЕНТ, И ЕЕ ПРИМЕНЕНИЕ | 2007 |
|
RU2469721C2 |
| WO 2013014448 A1,31.01.2013 | |||
| Finlay, M | |||
| Raymond V | |||
| et al | |||
| Рельсовая подкладка | 1928 |
|
SU9291A1 |

