ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение принадлежит к области медицины и относится к производному пиримидо-пятичленного азотсодержащего гетероцикла, к способу его получения и к его применению в медицине. В частности, настоящее изобретение относится к производному пиримидо-пятичленного азотсодержащего гетероцикла формулы (I), способу его получения, содержащей его фармацевтической композиции, его применению в качестве ингибитора SHP2 и к его применению в получении лекарственного средства для профилактики и/или лечения опухоли или рака.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Тирозинфосфатаза-2, содержащая домен 2 гомологии Src (SHP2), представляет собой эволюционно консервативный белок тирозинкиназу (PTP), кодируемый геном PTPN11. SHP2 в основном состоит из двух доменов SH2 (N-SH2 и C-SH2) и одного каталитического домена PTP. SHP2 широко экспрессируется в различных тканях человека и играет важную роль в поддержании развития тканей, клеточном гомеостазе и т.п. SHP2 связан с сигналами посредством активируемой митогеном Ras протеинкиназы JAK-STAT или пути протеинкиназы В, представляющей собой фосфоинозитид-3-киназу. Мутации в гене PTPN11 и последующие мутации SHP2 идентифицированы при ряде заболеваний человека, таких как синдром Нунан, синдром Leopard, ювенильный миеломоноцитарный лейкоз, нейробластома, меланома, острый миелоидный лейкоз, рак молочной железы, рак легкого и рак ободочной кишки (как в п. 19 формулы изобретения). Таким образом, SHP2 представляет собой в высшей степени привлекательную мишень для разработки новых видов терапии для лечения различных заболеваний.
Опубликованные заявки на патенты, относящиеся к мишени SHP2, включают WO2018136264A, WO2015003094A, WO2018160731A, WO2018130928A1, WO2018136265A, WO2018172984A, WO2018081091, WO2016203405, WO2017211303A, WO2018013597A и т.п. В настоящее время оба ингибитора SHP2 - TNO155, разработанный компанией Novartis, и JAB-3068, разработанный компанией JACOBIO, - проходят клиническое исследование I фазы, поэтому в продаже препараты, нацеленные на эту мишень, отсутствуют. Таким образом, все еще существует необходимость в продолжении разработки новых ингибиторов SHP2 с высокой эффективностью в целях обеспечения пациентов новыми и эффективными противораковыми лекарственными препаратами.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложено соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер, атропизомер или их смесь, или его фармацевтически приемлемая соль
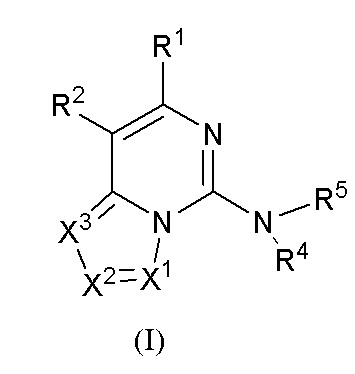 ,
,
где:
R1 выбран из группы, состоящей из атома водорода, атома дейтерия, гидрокси, циано, нитро, галогена, карбокси, алкила, алкокси, галогеналкила, галогеналкокси, амино, алкенила и гидроксиалкила;
R2 представляет собой 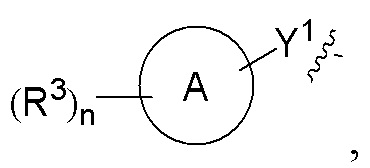
Y1 выбран из группы, состоящей из -S-, -NH-, -S(O)2-, -S(O)2-NH-, -C(=CH2)-, -S(O)- и химической связи;
кольцо A выбрано из группы, состоящей из циклоалкила, гетероциклила, арила и гетероарила, где циклоалкил, гетероциклил, арил и гетероарил, каждый независимо, представляет собой 5-12-членный моноцикл или полицикл;
каждый R3 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, алкила, алкокси, циано, амино, нитро, карбокси, гидрокси, гидроксиалкила, C3-8 циклоалкила, 3-12-членного гетероциклила, арила, гетероарила, C2-6 алкенила, C4-8 циклоалкенила, C2-6 алкинила, -CHRaRb, -NRaRb, -алкенил-NRaRb, -алкенил-O-Ra, -алкенил-C(O)2Ra, -алкенил-Ra, -алкенил-CO-NRaRb, -алкенил-NRa-CO-NRaRb, -алкенил-NRa-C(O)Rb, -C(O)NRaRb, -C(O)Ra, -CO-алкенил-NRaRb, -NRa C(O)Rb, -C(O)2Ra, -O-алкенил-CO-ORa, -O-алкенил-CO-NRaRb, -O-алкенил-NRaRb, -ORa, -SRa, -NRa-CO-NRaRb, -NRa-алкенил-NRaRb, -NRa-алкенил-Rb, -NRaS(O)2Rb, -NRaS(O)Rb, -NRaS(O)2NRaRb, -NRaS(O)NRaRb, -S(O)2NRaRb, -S(O)NRaRb, -S(O)Ra, -S(O)2Ra, -P(O)RaRb, -N(S(O)RaRb) и -S(O)(NRa)Rb, где алкил, алкокси, арил и гетероарил, каждый независимо необязательно дополнительно замещен одним или более заместителем, выбранным из группы, состоящей из галогена, атома водорода, атома дейтерия, циано, амино, нитро, карбокси, гидрокси, гидроксиалкила, алкила, алкокси, галогеналкила и галогеналкокси;
n выбрано из группы, состоящей из 0, 1, 2, 3, 4 и 5;
X1, X2 и X3, каждый независимо выбран из группы, состоящей из CRc и N, где по меньшей мере один из них представляет собой N, и предпочтительно X1 представляет собой CRc;
Rc выбран из группы, состоящей из атома водорода, атома дейтерия, C1-6 алкила, C1-6 алкокси, C1-6 алкилтио, амино, нитро, гидрокси, карбонила, карбокси, галогена и циано;
R4 выбран из группы, состоящей из водорода, C1-6 алкила, 3-12-членного моноциклического гетероциклила или полициклического гетероциклила и C3-8 циклоалкила, где алкил, гетероциклил и циклоалкил, каждый независимо необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, C1-3 алкила, амино, алкиламино, гидроксиалкила и алкокси;
R5 выбран из группы, состоящей из атома водорода, гидрокси, C1-6 алкила и C3-8 циклоалкила, где алкил или циклоалкил необязательно замещен одним или более амино; или
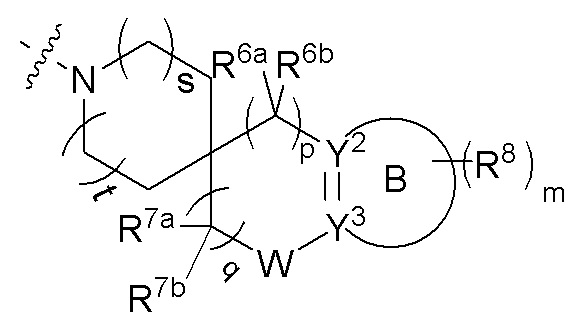
R4 и R5 вместе с атомом азота, к которому они присоединены, образуют 3-12-членный моноциклический гетероцикл или полициклический гетероцикл, где моноциклический гетероцикл или полициклический гетероцикл необязательно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, галоген-замещенного или незамещенного C1-6 алкила, амино, алкокси, гидроксиалкила, арила, гетероарила, гетероциклила, алкиламино, галоген-замещенного или незамещенного алкокси и -NRaS(O)NRaRb, и полициклический гетероцикл включает без ограничений мостиковый гетероцикл и спиро-гетероцикл.
Примеры колец, образованных R4 и R5 вместе с атомом азота, к которому они присоединены, включают без ограничений:
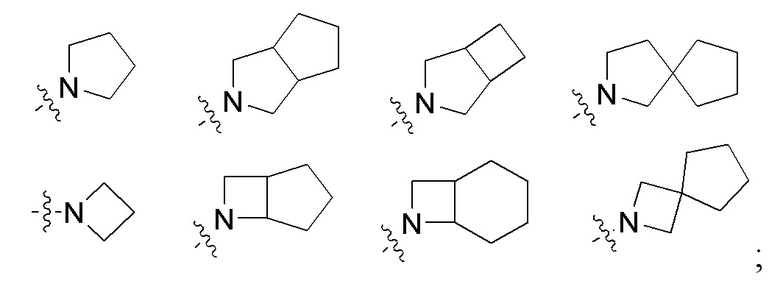
или R4 и R5 вместе с атомом азота, к которому они присоединены, образуют структуру
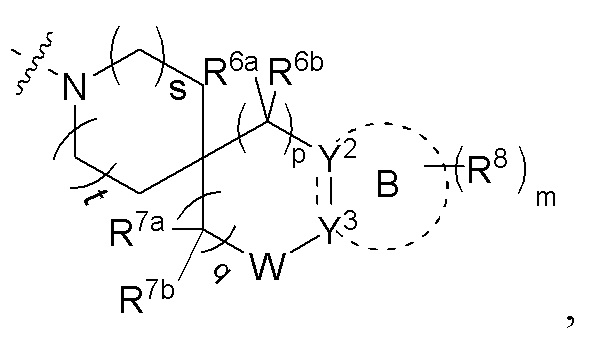
где s и t, каждый независимо, выбран из группы, состоящей из 0 и 1;
R6a и R6b, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, атома фтора, амино, гидрокси, циано, нитро, карбокси, фторзамещенного или незамещенного алкила и фторзамещенного или незамещенного алкокси; или R6a и R6b вместе с атомом углерода, к которому они присоединены, образуют CO, C=NH, C=N-OH, 3-12-членный гетероциклил или C3-8 циклоалкил;
p выбрано из группы, состоящей из 0, 1, 2, 3 и 4;
R7a и R7b, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, атома фтора, амино, гидрокси, циано, нитро, карбокси, фторзамещенного или незамещенного алкила, фторзамещенного или незамещенного алкокси и -NRaS(O)NRaRb;
или R7a и R7b вместе с атомом углерода, к которому они присоединены, образуют 3-12-членный гетероциклил, 5-10-членный гетероарил, C3-8 циклоалкил и C=NR7c, где кольца необязательно замещены;
R7c выбран из группы, состоящей из атома водорода, атома дейтерия и C1-6 алкила;
q выбрано из группы, состоящей из 0, 1, 2, 3 и 4;
W отсутствует или выбран из группы, состоящей из -O-, -S- и -NRw-;
Rw выбран из группы, состоящей из атома водорода, галогена, амино, гидрокси, циано, нитро, карбокси, -C(O)C1-6 алкила, -C(O)2C1-6 алкила, C1-6 алкилэфира, галоген-замещенного или незамещенного C1-6 алкила и галоген-замещенного или незамещенного C1-6 алкокси;
кольцо B отсутствует или представляет собой 3-10-членное кольцо;
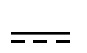 представляет собой простую связь или двойную связь;
представляет собой простую связь или двойную связь;
когда кольцо B отсутствует, тогда Y2 представляет собой CR2aR2b, NR2a или O, Y3 представляет собой CR3aR3b, NR3a или O;
когда кольцо B представляет собой 3-10-членное кольцо, тогда
1) Y2 представляет собой CR2a или N, Y3 представляет собой CR3a или N, представляет собой простую связь; или
2) Y2 представляет собой C, и Y3 представляет собой C, представляет собой двойную связь;
R2a, R2b, R3a и R3b, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, циано, амино, нитро, карбокси, гидрокси, гидроксиалкила, C3-8 циклоалкила, 3-12-членного гетероциклила, арила, гетероарила, C2-6 алкенила, C4-8 циклоалкенила, C2-6 алкинила, -NRaRb, -алкенил-NRaRb, -алкенил-O-Ra, -алкенил-C(O)2Ra, -алкенил-Ra, -алкенил-CO-NRaRb, -алкенил-NRa-CO-NRaRb, -алкенил-NRa-C(O)Rb, -C(O)NRaRb, -C(O)Ra, -CO-алкенил-NRaRb, -NRaC(O)Rb, -C(O)2Ra, -O-алкенил-CO-ORa, -O-алкенил-CO-NRaRb, -O-алкенил-NRaRb, -ORa, -SRa, -NRa-CO-NRaRb, -NRa-алкенил-NRaRb, -NRa-алкенил-Rb, -NRaS(O)2Rb, -NRaS(O)Rb, -NRaS(O)2NRaRb, -NRaS(O)NRaRb, -S(O)2NRaRb, -S(O)NRaRb, -S(O)Ra, -S(O)2Ra, -P(O)RaRb, -N(S(O)RaRb) и -S(O)(NRa)Rb, где арил и гетероарил, каждый независимо необязательно дополнительно замещен одним или более заместителей, выбранных из группы, состоящей из галогена, атома водорода, атома дейтерия, циано, амино, нитро, карбокси, гидрокси, гидроксиалкила, алкила, алкокси, галогеналкила и галогеналкокси;
Ra и Rb, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, амино, гидрокси, циано, нитро, карбокси, алкила, алкокси, галогеналкила, галогеналкокси, C3-8 циклоалкила, 5-10-членного гетероарила и арила, где арил и гетероарил, каждый независимо необязательно дополнительно замещен одним или более заместителей, выбранных из группы, состоящей из галогена, атома водорода, атома дейтерия, циано, амино, нитро, карбокси, гидрокси, гидроксиалкила, алкила, алкокси, галогеналкила и галогеналкокси;
m выбрано из группы, состоящей из 0, 1, 2, 3 и 4; и
каждый R8 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, амино, гидрокси, циано, нитро, карбокси, C1-6 алкила и C1-6 алкокси;
или R8 соединены вместе с образованием фенила, 5-членного гетероарила, 6-членного гетероарила или 3-6-членного гетероциклила, где каждое кольцо необязательно замещено одним или более заместителей, выбранных из группы, состоящей из галогена, амино, гидрокси, циано, нитро и C1-6 алкила.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, атропизомере или их смеси, или его фармацевтически приемлемой соли R4 и R5 вместе с атомом азота, к которому они присоединены, образуют структуру
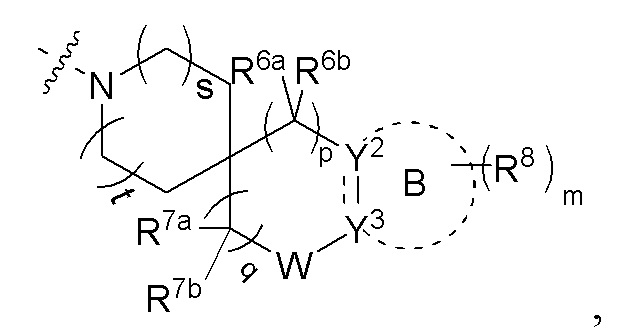
где s и t, каждый независимо выбран из группы, состоящей из 0 и 1;
R6a и R6b, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, C1-6 алкила и C1-6 алкокси; или R6a и R6b вместе с атомом углерода, к которому они присоединены, образуют 3-12-членный гетероциклил или C3-8 циклоалкил;
p выбран из группы, состоящей из 0, 1 и 2;
R7a и R7b, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, амино, C1-6 алкила и -NRaS(O)NRaRb, где Ra и Rb являются такими, как определено в формуле (I) выше;
q равно 1 или 2;
W отсутствует;
кольцо B отсутствует или представляет собой 3-10-членное кольцо;
представляет собой простую связь или двойную связь;
когда кольцо B отсутствует, тогда Y2 представляет собой CR2aR2b или O, Y3 представляет собой CR3aR3b; или
когда кольцо B представляет собой 3-10-членное кольцо, тогда
Y2 представляет собой CR2a или N, Y3 представляет собой CR3a или N, представляет собой простую связь; или
Y2 представляет собой C, и Y3 представляет собой C, представляет собой двойную связь;
R2a, R2b и R3a, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия и C1-6 алкила;
m выбрано из группы, состоящей из 0, 1, 2, 3 и 4; и
каждый R8 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, амино, гидрокси, циано, нитро, карбокси, C1-6 алкила и C1-6 алкокси;
или два R8 соединены вместе с образованием фенила, 5-членного гетероарила, 6-членного гетероарила или 3-6-членного гетероциклила, где каждое кольцо необязательно замещено одним или более заместителей, выбранных из группы, состоящей из галогена, амино, гидрокси, циано, нитро и C1-6 алкила.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, атропизомере или их смеси, или его фармацевтически приемлемой соли R4 и R5 вместе с атомом азота, к которому они присоединены, образуют структуру
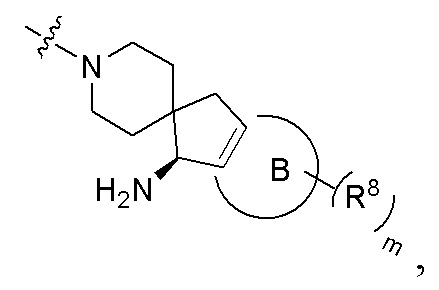
где:
кольцо B выбрано из группы, состоящей из бензольного кольца, 5-членного гетероароматического кольца и 6-членного гетероароматического кольца, предпочтительно бензольного кольца или пиридинового кольца;
каждый R8 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, циано, C1-6 алкила и C1-6 алкокси; и
m выбрано из группы, состоящей из 0, 1, 2, 3 и 4.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, атропизомере или их смеси, или его фармацевтически приемлемой соли R4 и R5 вместе с атомом азота, к которому они присоединены, образуют структуру 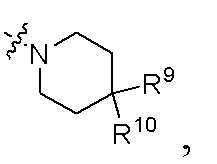
где R9 и R10, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, гидрокси, C1-6 алкила, C1-6 алкокси, галогена, C1-6 гидроксиалкила, арила, гетероарила, гетероциклила, амино, C1-6 алкиламино и -NRaS(O)NRaRb, предпочтительно выбраны из группы, состоящей из атома водорода, атома дейтерия, C1-6 алкила, амино и -NRaS(O)NRaRb; или
Ra и Rb являются такими, как определено в формуле (I) выше.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, атропизомере или их смеси, или его фармацевтически приемлемой соли
Y1 представляет собой -S- или связь;
кольцо A представляет собой арил или гетероарил;
каждый R3 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, C1-6 алкила, галоген-C1-6 алкила, галоген-C1-6 алкокси, C1-6 алкокси, циано, амино, нитро, карбокси, гидрокси и фенила, где фенил необязательно дополнительно замещен одним или более заместителем, выбранным из группы, состоящей из галогена, атома водорода, атома дейтерия, циано, амино, нитро, карбокси, гидрокси, гидроксиалкила, алкила, алкокси, галогеналкила и галогеналкокси; каждый R3 предпочтительно выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, галоген-C1-6 алкила, C1-6 алкила, C1-6 алкокси, галоген-C1-6 алкокси и фенила, где фенил необязательно дополнительно замещен одним или более заместителем, выбранным из группы, состоящей из галогена, атома водорода, атома дейтерия, циано, амино, нитро, карбокси, гидрокси, гидроксиалкила, алкила, алкокси, галогеналкила и галогеналкокси; и
n выбрано из группы, состоящей из 0, 1, 2, 3, 4 и 5.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, атропизомере или их смеси, или его фармацевтически приемлемой соли X1, X2 и X3, каждый независимо выбран из группы, состоящей из CRc и N, где по меньшей мере один из них представляет собой N, предпочтительно X1 представляет собой CRc, и Rc представляет собой атом водорода.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, атропизомере или их смеси, или его фармацевтически приемлемой соли X1 и X2 оба представляют собой CRc, и X3 представляет собой N, или X1 представляет собой CRc, и X2 и X3 оба представляют собой N, и Rc представляет собой атом водорода.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, атропизомере или их смеси, или его фармацевтически приемлемой соли R1 выбран из группы, состоящей из атома водорода, атома дейтерия, C1-6 алкила, C1-6 алкокси, амино и гидрокси.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, атропизомере или их смеси, или его фармацевтически приемлемой соли
R1 выбран из группы, состоящей из атома водорода, атома дейтерия, C1-6 алкила и амино;
Y1 представляет собой -S- или связь;
кольцо A представляет собой арил или гетероарил;
каждый R3 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, галоген-C1-6 алкила, C1-6 алкила, C1-6 алкокси, галоген-C1-6 алкокси и замещенного фенила;
n выбрано из группы, состоящей из 0, 1, 2, 3, 4 и 5;
X1, X2 и X3, каждый независимо выбран из группы, состоящей из CRc и N, где по меньшей мере один из них представляет собой N, предпочтительно X1 представляет собой CRc, и Rc представляет собой атом водорода;
R4 и R5 вместе с атомом азота, к которому они присоединены, образуют структуру и
R9 и R10, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, C1-6 алкила, амино и -NRaS(O)NRaRb, где Ra и Rb являются такими, как определено в формуле (I) выше.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, атропизомере или их смеси, или его фармацевтически приемлемой соли
R1 выбран из группы, состоящей из атома водорода, атома дейтерия, C1-6 алкила и амино;
Y1 представляет собой -S- или связь;
кольцо A представляет собой арил или гетероарил;
каждый R3 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, галоген-C1-6 алкила, C1-6 алкила, C1-6 алкокси, галоген-C1-6 алкокси и замещенного фенила;
n выбрано из группы, состоящей из 0, 1, 2, 3, 4 и 5;
X1, X2 и X3, каждый независимо выбран из группы, состоящей из CRc и N, где по меньшей мере один из них представляет собой N, предпочтительно X1 представляет собой CRc, и Rc представляет собой атом водорода;
R6a и R6b, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, C1-6 алкила и C1-6 алкокси; или R6a и R6b вместе с атомом углерода, к которому они присоединены, образуют 3-12-членный гетероциклил или C3-8 циклоалкил;
p равно 1 или 2;
R7a и R7b, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, амино, C1-6 алкила и -NRaS(O)NRaRb, где Ra и Rb являются такими, как определено в формуле (I) выше;
q равно 1 или 2;
W отсутствует;
кольцо B отсутствует, Y2 представляет собой CR2aR2b или O, Y3 представляет собой CR3aR3b; и
R2a, R2b, R3a и R3b, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия и C1-6 алкила.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, атропизомере или их смеси, или его фармацевтически приемлемой соли
R1 выбран из группы, состоящей из атома водорода, атома дейтерия, C1-6 алкила и амино;
Y1 представляет собой -S- или связь;
кольцо A представляет собой арил или гетероарил;
каждый R3 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, галоген-C1-6 алкила, C1-6 алкила, C1-6 алкокси, галоген-C1-6 алкокси и замещенного фенила;
n выбрано из группы, состоящей из 0, 1, 2, 3, 4 и 5;
X1, X2 и X3, каждый независимо выбран из группы, состоящей из CRc и N, где по меньшей мере один из них представляет собой N, предпочтительно X1 представляет собой CRc, и Rc представляет собой атом водорода;
R6a и R6b, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, C1-6 алкила и C1-6 алкокси;
p равно 1 или 2;
R7a и R7b, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, амино, C1-6 алкила и -NRaS(O)NRaRb, где Ra и Rb являются такими, как определено в формуле (I) выше;
q равно 1 или 2;
W отсутствует;
кольцо B выбрано из группы, состоящей из фенила, 5-членного гетероарила и 6-членного гетероарила;
Y2 представляет собой C, и Y3 представляет собой C, 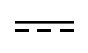 представляет собой двойную связь;
представляет собой двойную связь;
каждый R8 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, амино, гидрокси, циано, нитро, карбокси, C1-6 алкила и C1-6 алкокси; и
m выбрано из группы, состоящей из 0, 1, 2, 3 и 4.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере, атропизомере или их смеси, или его фармацевтически приемлемой соли R4 и R5 вместе с атомом азота, к которому они присоединены, образуют структуру
R1 выбран из группы, состоящей из атома водорода, C1-6 алкила и амино;
Y1 представляет собой -S- или связь;
кольцо A представляет собой арил или гетероарил; предпочтительно фенил или пиридил;
каждый R3 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, циано, амино, галоген-C1-6 алкила, C1-6 алкила, C1-6 алкокси, галоген-C1-6 алкокси, C1-6 алкиламино, галоген-C1-6 алкиламино, C3-8 циклоалкила, 3-12-членного гетероциклила, -ORa, -CHRaRb и -NRaR;
Ra и Rb, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, гидрокси, C1-6 алкила и C3-8 циклоалкила, где алкил, гетероциклил и циклоалкил, каждый независимо необязательно дополнительно замещены одним или более заместителей, выбранных из группы, состоящей из галогена, атома дейтерия, циано, амино и гидрокси;
n выбрано из группы, состоящей из 0, 1, 2, 3, 4 и 5;
X3 представляет собой N, X1 и X2, каждый независимо представляет собой CRc, и Rc представляет собой атом водорода;
s и t, каждый независимо выбран из группы, состоящей из 0 и 1;
R6a и R6b, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, C1-6 алкила и C1-6 алкокси;
p равно 1;
R7a и R7b, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, амино и C1-6 алкила;
q равно 1;
W отсутствует;
кольцо B выбрано из группы, состоящей из бензольного кольца, 5-членного гетероароматического кольца и 6-членного гетероароматического кольца, предпочтительно бензольного кольца или пиридинового кольца;
Y2 представляет собой C, и Y3 представляет собой C;
каждый R8 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, амино, гидрокси, циано, нитро, карбокси, C1-6 алкила и C1-6 алкокси; и
m выбрано из группы, состоящей из 0, 1, 2, 3 и 4.
В предпочтительном воплощении настоящего изобретения соединение формулы (I), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, атропизомер или их смесь, или его фармацевтически приемлемая соль, представляет собой соединение формулы (II), или его таутомер, мезомер, рацемат, энантиомер, диастереомер, атропизомер или их смесь, или его фармацевтически приемлемую соль
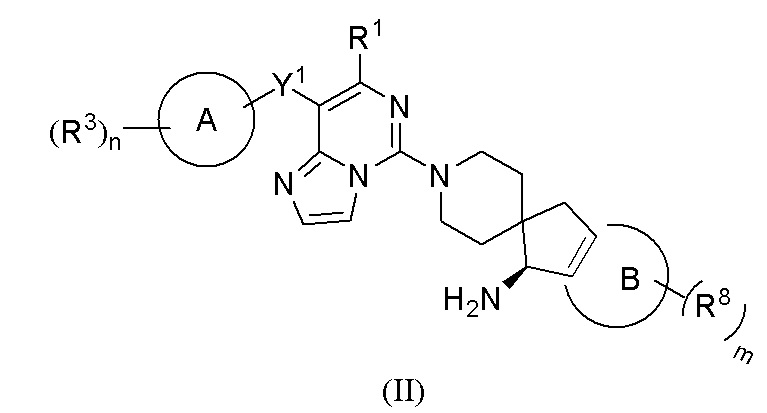 ,
,
где:
R1 выбран из группы, состоящей из атома водорода, C1-6 алкила, галогеналкила и амино;
Y1 представляет собой -S- или связь;
кольцо A представляет собой арил или гетероарил; предпочтительно фенил или пиридил;
каждый R3 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, циано, амино, C1-6 алкила, C1-6 алкокси, галоген-C1-6 алкила, галоген-C1-6 алкокси, C3-8 циклоалкила, 3-12-членного гетероциклила, -ORa, -CHRaRb и -NRaRb;
Ra и Rb, каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, гидрокси, C1-6 алкила и C3-8 циклоалкила, где алкил, гетероциклил и циклоалкил, каждый независимо необязательно дополнительно замещены одним или более заместителем, выбранным из группы, состоящей из галогена, атома дейтерия, циано, амино и гидрокси;
кольцо B выбрано из группы, состоящей из бензольного кольца, 5-членного гетероароматического кольца и 6-членного гетероароматического кольца, предпочтительно бензольного кольца или пиридинового кольца;
каждый R8 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, циано, C1-6 алкила и C1-6 алкокси;
m выбрано из группы, состоящей из 0, 1, 2, 3 и 4; и
n выбрано из группы, состоящей из 1, 2, 3 и 4.
В настоящем изобретении, когда Y1 представляет собой химическую связь, то соединение, предложенное настоящим изобретением, может существовать в виде смеси атропизомеров в связи с ограничением вращения вокруг связи, и его энантиомерный избыток составляет от 0 до 98%. Когда соединение представляет собой чистый атропизомер, стереохимия каждого хирального центра может быть указана как aR или aS. Эти термины можно также использовать для смеси, обогащенной одним атропизомером. Атропизомеры aR и aS можно разделить методом хиральной хроматографии.
Дополнительное описание атропизомерии и осевой хиральности можно найти в Eliel, E.L. & Wilen, S. H. 'Stereochemistry of Organic compounds' John Wiley and Sons, Inc. 1994.
Типовые соединения формулы (I) настоящего изобретения включают без ограничений:
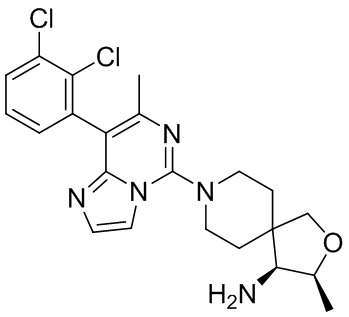
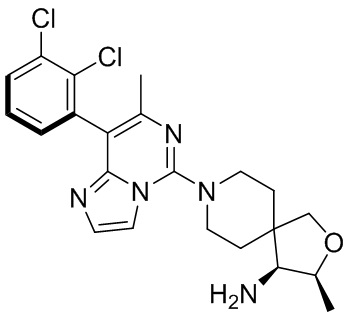
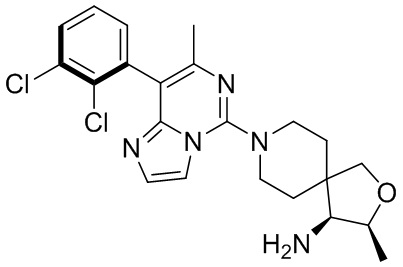
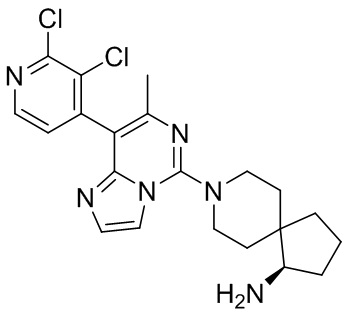
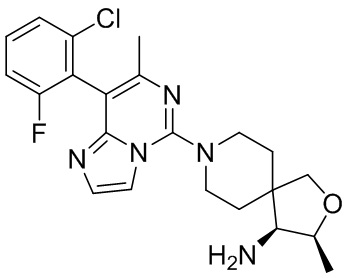
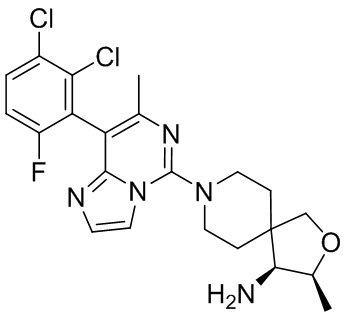
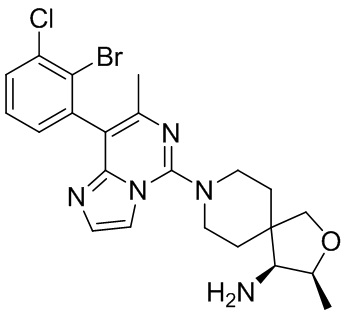
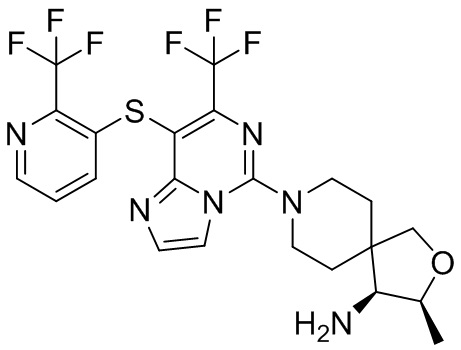
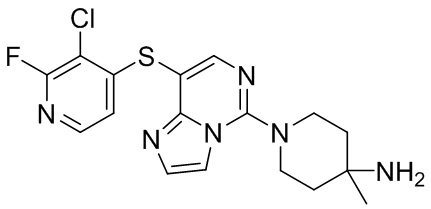
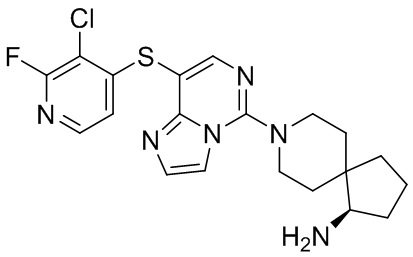
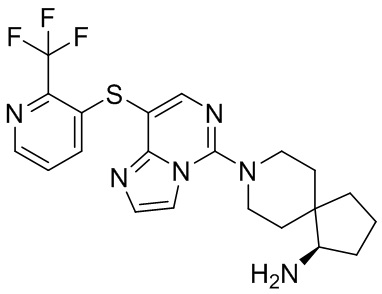
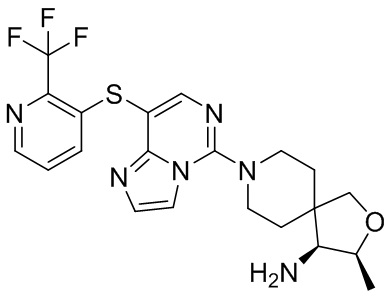
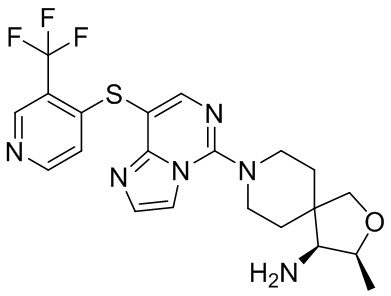
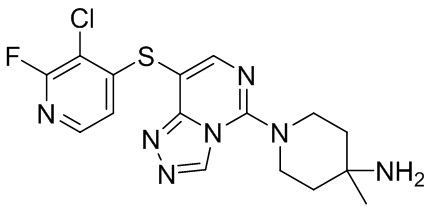
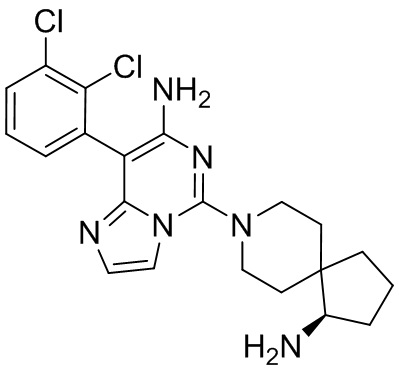
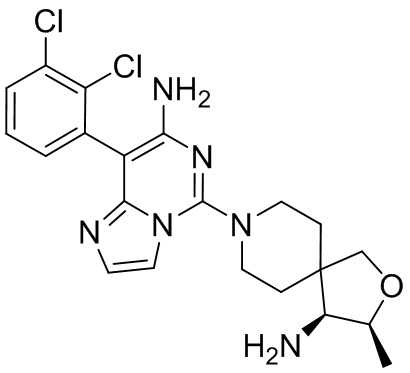
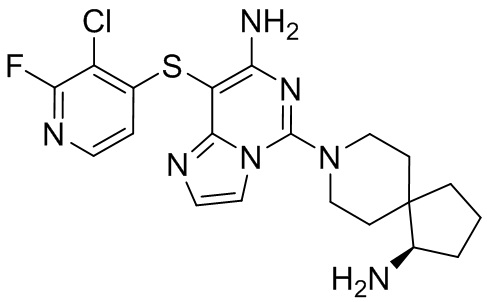
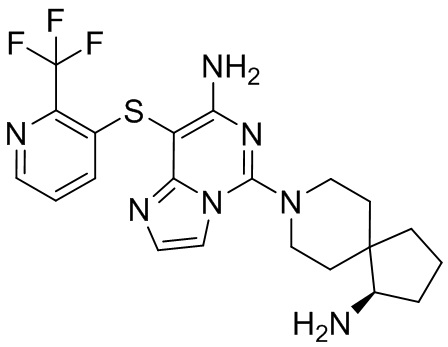
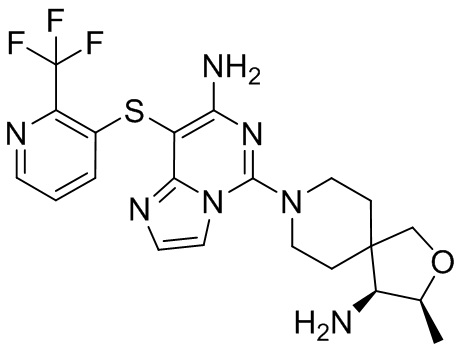
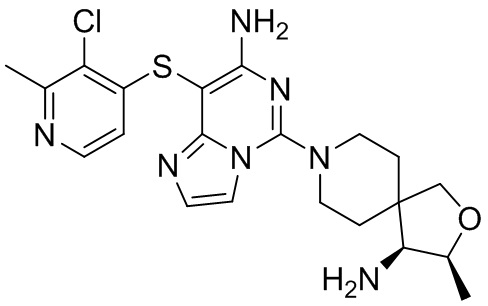
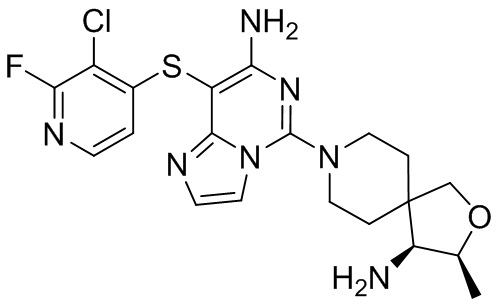
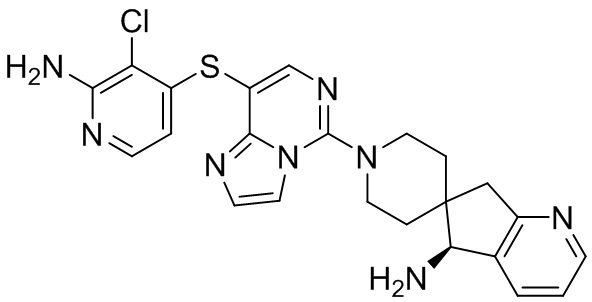
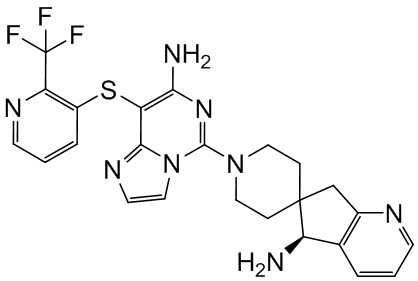
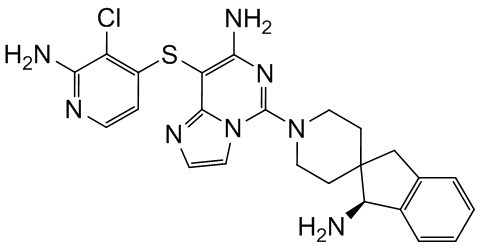
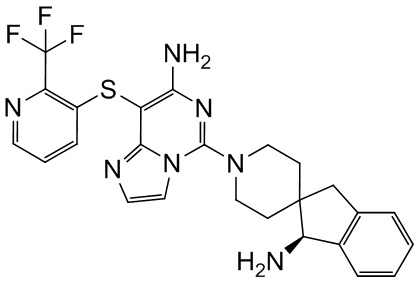
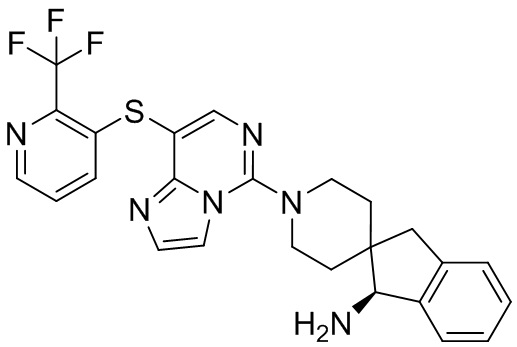
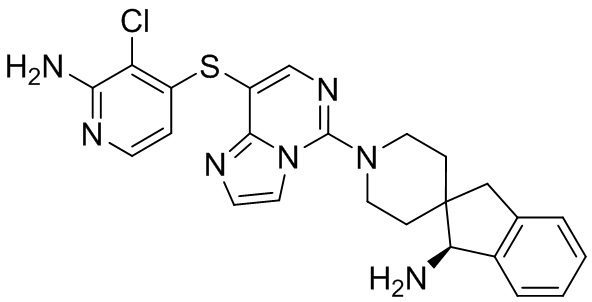
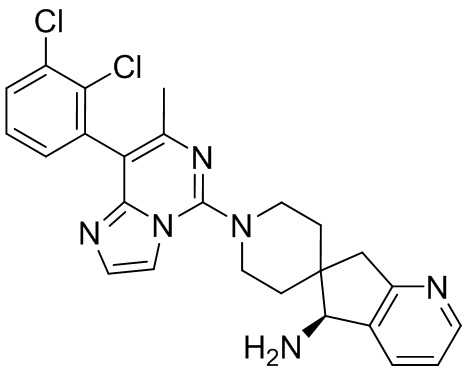
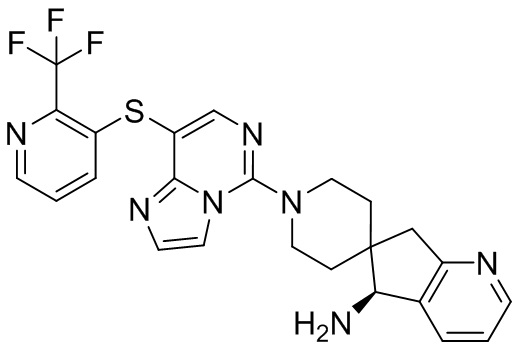
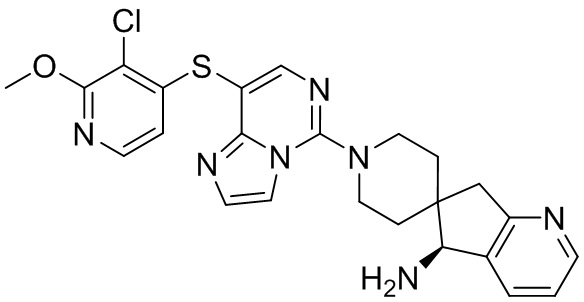
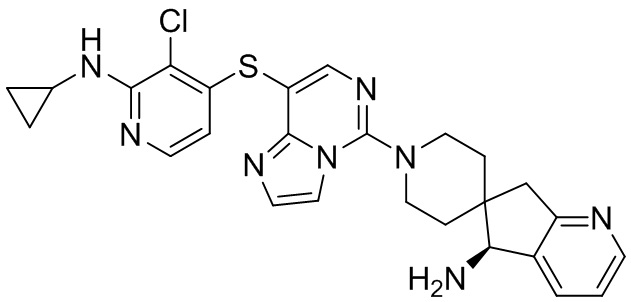
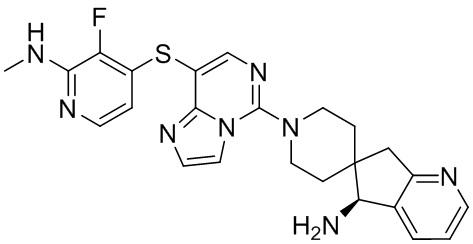
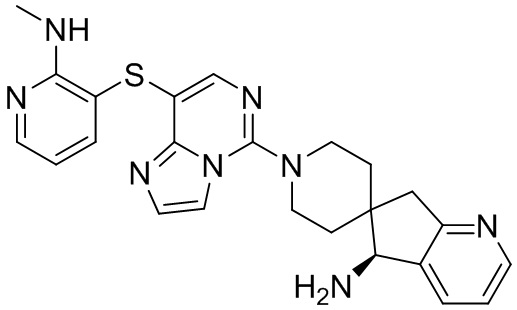
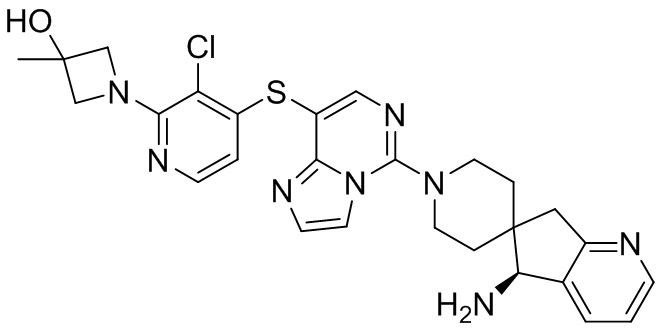
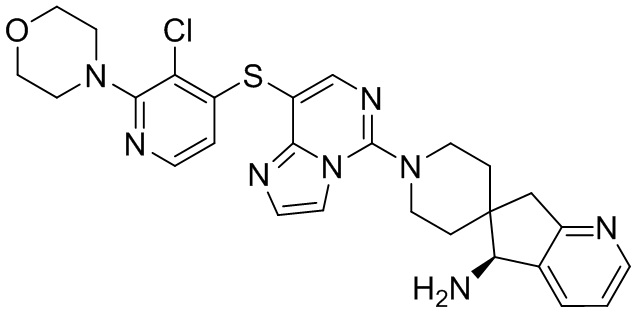
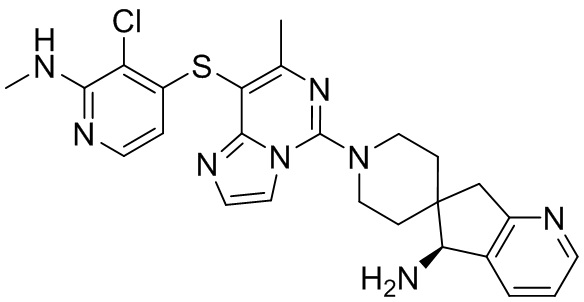
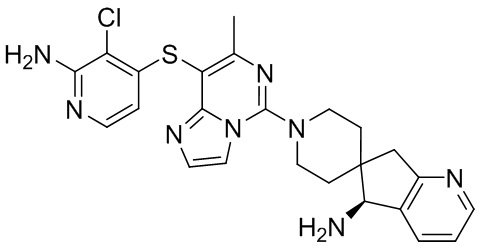
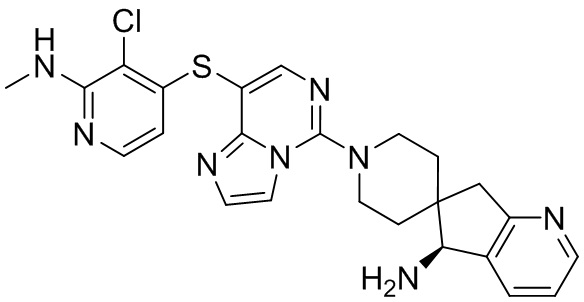
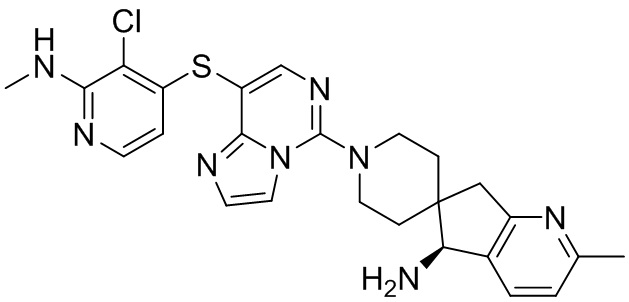
или их таутомер, мезомер, рацемат, энантиомер, диастереомер, атропизомер или их смесь, или их фармацевтически приемлемую соль.
В настоящем изобретении предложен способ получения соединения формулы (I), в котором соединение формулы (I) представляет собой соединение формулы (I-A) или соединение формулы (I-B), отличающийся тем, что он включает стадии
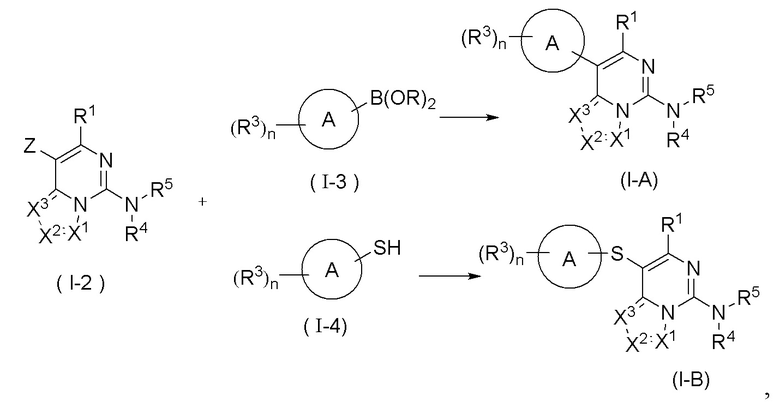
на которых соединение формулы (I-2) и соединение формулы (I-3) подвергают реакции сочетания Сузуки в щелочных условиях в присутствии катализатора с получением соединения формулы (I-A), причем катализатор выбран из группы, состоящей из палладия на углероде, никеля Ренея, тетракис(трифенилфосфин)палладия, дихлорида палладия, ацетата палладия, [1,1'-бис(дифенилфосфино)ферроцен]палладия (II) дихлорида, 1,1'-бис(дибензилфосфино)дихлорферроценпалладия (II), трис(дибензилиденацетон)дипалладия и 2-дициклогексилфосфино-2',6'-диметоксидифенила, и предпочтительно [1,1'-бис(дифенилфосфино)ферроцен]палладия (II) дихлорида и 2-дициклогексилфосфино-2',6'-диметоксидифенила; или
подвергают соединение формулы (I-2) и соединение формулы (I-4) реакции C-S сочетания в щелочных условиях с получением соединения формулы (I-B);
где реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания; органическое основание выбрано из группы, состоящей из триэтиламина, N,N-диизопропилэтиламина, н-бутиллития, диизопропиламида лития, бистриметилсилиламида лития, ацетата калия, трет-бутилата натрия и трет-бутилата калия; неорганическое основание выбрано из группы, состоящей из гидрида натрия, фосфата калия, карбоната натрия, карбоната калия, ацетата калия, карбоната цезия, гидроксида натрия и гидроксида лития;
B(OR)2 представляет собой борат или борную кислоту, которые включают без ограничений 4,4,5,5-тетраметил-1,3,2-диоксаборолан, 4,4,4',4',5,5,5',5'-октаметил-2,2'-бис(1,3,2-диоксаборолан), бис(неопентилгликолято)дибор, B(OBu-n)3 и B(OPr-i)3;
Z выбран из группы, состоящей из галогена и сульфонила; и
R1, X1, X2, X3, R3, R4 и R5 являются такими, как определено в формуле (I) выше.
Для достижения цели настоящего изобретения в настоящем изобретении можно применять следующую схему синтеза:
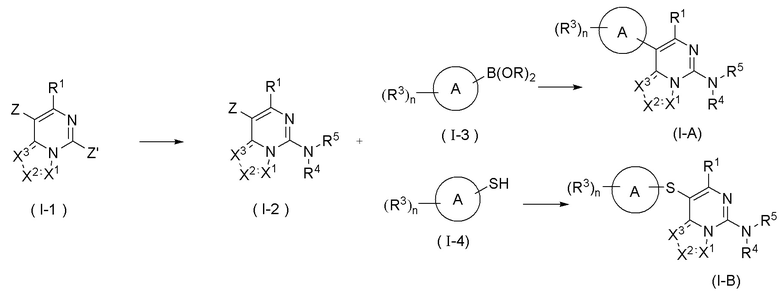
введение аммиака в соединение формулы (I-1) с получением соединения формулы (I-2), в которой Z и Z', каждый независимо выбран из группы, состоящей из галогена и сульфонила, другие заместители являются такими, как определено в предыдущем воплощении, растворитель реакции в схеме синтеза по настоящему изобретению включает без ограничений уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N,N-диметилформамид и их смеси.
В настоящем изобретении предложен способ получения соединения формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, атропизомера или их смеси, или его фармацевтически приемлемой соли, где соединение формулы (II) представляет собой соединение формулы (II-A) или соединение формулы (II-B), включающий следующие стадии, на которых:
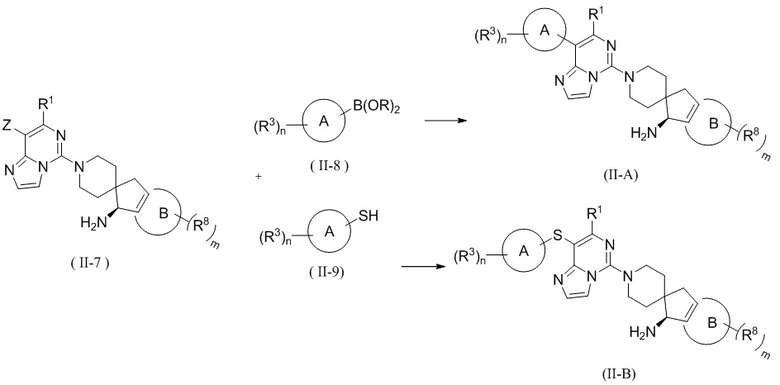
подвергают соединение формулы (II-7) и соединение формулы (II-8) реакции сочетания Сузуки в щелочных условиях в присутствии катализатора с получением соединения формулы (II-A);
или подвергают соединение формулы (II-7) и соединение формулы (II-9) реакции C-S сочетания в щелочных условиях с получением соединения формулы (II-B);
где катализатор выбран из группы, состоящей из палладия на углероде, никеля Ренея, тетракис(трифенилфосфин)палладия, дихлорида палладия, ацетата палладия, [1,1'-бис(дифенилфосфино)ферроцен]палладия (II) дихлорида, 1,1'-бис(дибензилфосфино)дихлорферроценпалладия (II), трис(дибензилиденацетон)дипалладия и 2-дициклогексилфосфино-2',6'-диметоксидифенила, и предпочтительно [1,1'-бис(дифенилфосфино)ферроцен]палладия (II) дихлорида и 2-дициклогексилфосфино-2',6'-диметоксидифенила;
реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания; органическое основание выбрано из группы, состоящей из триэтиламина, N,N-диизопропилэтиламина, н-бутиллития, диизопропиламида лития, бистриметилсилиламида лития, ацетата калия, трет-бутилата натрия и трет-бутилата калия; неорганическое основание выбрано из группы, состоящей из гидрида натрия, фосфата калия, карбоната натрия, карбоната калия, ацетата калия, карбоната цезия, гидроксида натрия и гидроксида лития;
B(OR)2 представляет собой борат или борную кислоту, которые включают без ограничений 4,4,5,5-тетраметил-1,3,2-диоксаборолан, 4,4,4',4',5,5,5',5'-октаметил-2,2'-бис(1,3,2-диоксаборолан), бис(неопентилгликолято)диборон, B(OBu-n)3 и B(OPr-i)3;
Z выбран из группы, состоящей из галогена, сульфонила и сульфинила; и
кольцо A, кольцо B, R1, R3, R8, B, m и n являются такими, как определено в формуле (II) выше.
Способ получения соединения формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, атропизомера или их смеси, или его фармацевтически приемлемой соли, предложенный в настоящем изобретении, дополнительно включает стадию взаимодействия соединения формулы (II-5) с соединением формулы (II-6) в щелочных условиях с получением соединения формулы (II-7),
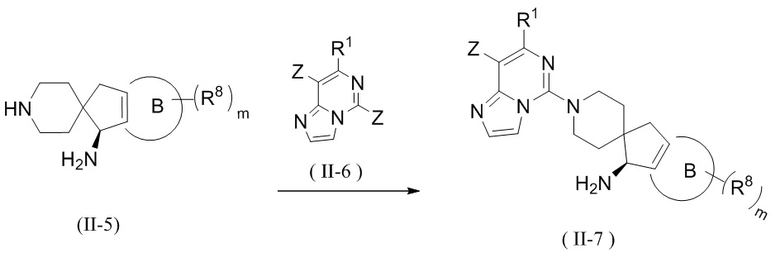
где реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания; органическое основание выбрано из группы, состоящей из триэтиламина, N,N-диизопропилэтиламина, н-бутиллития, диизопропиламида лития, бистриметилсилиламида лития, ацетата калия, трет-бутилата натрия и трет-бутилата калия; неорганическое основание выбрано из группы, состоящей из гидрида натрия, фосфата калия, карбоната натрия, карбоната калия, ацетата калия, карбоната цезия, гидроксида натрия и гидроксида лития;
Z, R1, R8, кольцо B и m являются такими, как определено в формуле (II).
Необязательно способ получения соединения формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, атропизомера или их смеси, или его фармацевтически приемлемой соли, предложенный в настоящем изобретении, дополнительно включает следующие стадии, на которых:
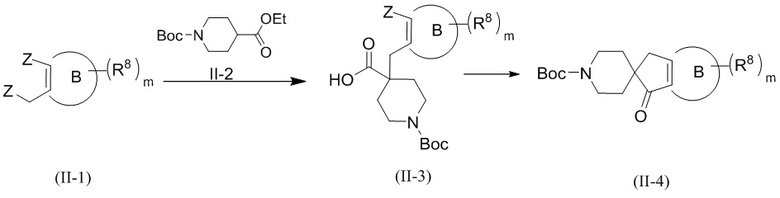
1) подвергают взаимодействию соединение формулы (II-1) с соединением формулы (II-2) в щелочных условиях с получением соединения формулы (II-3); 2) подвергают соединение формулы (II-3) реакции внутримолекулярного замыкания цикла в присутствии н-бутиллития с получением соединения формулы (II-4); подвергают взаимодействию соединение формулы (II-4) хиральному селективному восстановительному аминированию с последующим удалением защитной группы амино с получением соединения формулы (II-5); при этом реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания; органическое основание выбрано из группы, состоящей из триэтиламина, N,N-диизопропилэтиламина, н-бутиллития, диизопропиламида лития, бистриметилсилиламида лития, ацетата калия, трет-бутилата натрия и трет-бутилата калия; неорганическое основание выбрано из группы, состоящей из гидрида натрия, фосфата калия, карбоната натрия, карбоната калия, ацетата калия, карбоната цезия, гидроксида натрия и гидроксида лития; и
Z, R8, кольцо B и m являются такими, как определено в формуле (II).
В настоящем изобретении предложено соединение формулы (I-2) или его фармацевтически приемлемая соль
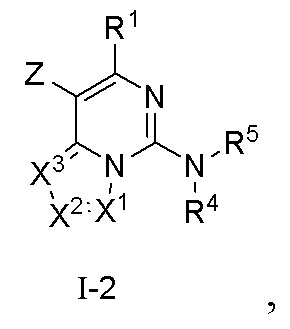
где R1, X1, X2, X3, R4 и R5 являются такими, как определено в формуле (I);
Z выбран из группы, состоящей из галогена и сульфонила.
В настоящем изобретении предложено соединение формулы (I-1) или его фармацевтически приемлемая соль
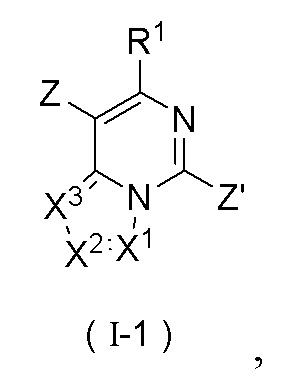
где R1, X1, X2 и X3 являются такими, как определено в формуле (I);
Z и Z', каждый независимо выбран из группы, состоящей из галогена и сульфонила.
В настоящем изобретении предложен способ получения соединения формулы (I) из соединения формулы (I-2) или его фармацевтически приемлемой соли или из соединения формулы (I-1) или его фармацевтически приемлемой соли.
В другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы (I) или формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, атропизомера или их смеси, или его фармацевтически приемлемой соли, и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов, и терапевтически эффективное количество по настоящему изобретению может составлять от 0,1 до 2000 мг. Настоящее изобретение также относится к способу получения фармацевтической композиции, включающий стадию смешивания соединения формулы (I) или формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, атропизомера или их смеси, или его фармацевтически приемлемой соли, либо соединения формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, атропизомера или их смеси, или его фармацевтически приемлемой соли с фармацевтически приемлемым носителем, разбавителем или эксципиентом.
Настоящее изобретение дополнительно относится к применению соединения формулы (I) или формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, атропизомера или их смеси, или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции в получении ингибитора SHP2.
Настоящее изобретение дополнительно относится к применению соединения формулы (I) или формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, атропизомера или их смеси, или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции в получении лекарственного средства для лечения заболевания или состояния, опосредованного активностью SHP2.
Настоящее изобретение дополнительно относится к применению соединения формулы (I) или формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, атропизомера или их смеси, или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции в качестве ингибитора SHP2 в получении лекарственного средства для профилактики и/или лечения опухоли или рака.
Настоящее изобретение дополнительно относится к применению соединения формулы (I) или формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, атропизомера или их смеси, или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции в получении лекарственного средства для профилактики или лечения синдрома Нунан, синдрома Leopard, ювенильного миеломоноцитарного лейкоза, нейробластомы, меланомы, острого миелогенного лейкоза, рака молочной железы, рака пищевода, рака легкого, рака ободочной кишки, рака головы, рака поджелудочной железы, плоскоклеточной карциномы головы и шеи, рака желудка, рака печени, анапластической крупноклеточной лимфомы или глиобластомы.
Настоящее изобретение дополнительно относится к соединению формулы (I) или формулы (II) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, атропизомеру или их смеси, или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для применения в качестве лекарственного средства.
Настоящее изобретение дополнительно относится к соединению формулы (I) или формулы (II) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, атропизомеру или их смеси, или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для применения в качестве ингибитора SHP2.
Настоящее изобретение также относится к соединению формулы (I) или формулы (II) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру, атропизомеру или их смеси, или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для применения в качестве ингибитора SHP2 в профилактике и/или лечении опухоли или злокачественного новообразования.
Настоящее изобретение также относится к соединению формулы (I) или формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, атропизомера или их смеси, или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для применения в профилактике или лечении синдрома Нунан, синдрома Leopard, ювенильного миеломоноцитарного лейкоза, нейробластомы, меланомы, острого миелогенного лейкоза, рака молочной железы, рака пищевода, рака легкого, рака ободочной кишки, рака головы, рака поджелудочной железы, плоскоклеточной карциномы головы и шеи, рака желудка, рака печени, анапластической крупноклеточной лимфомы или глиобластомы.
Настоящее изобретение также относится к способу профилактики и/или лечения опухоли или рака, включающему стадию введения нуждающемуся в этом пациенту терапевтически эффективной дозы соединения формулы (I) или формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, атропизомера или их смеси, или его фармацевтически приемлемой соли в качестве ингибитора SHP2.
Настоящее изобретение также относится к a способу профилактики и/или лечения синдрома Нунан, синдрома Leopard, ювенильного миеломоноцитарного лейкоза, нейробластомы, меланомы, острого миелогенного лейкоза, рака молочной железы, рака пищевода, рака легкого, рака ободочной кишки, рака головы, рака поджелудочной железы, плоскоклеточной карциномы головы и шеи, рака желудка, рака печени, анапластической крупноклеточной лимфомы или глиобластомы, включающему стадию введения нуждающемуся в этом пациенту терапевтически эффективной дозы соединения формулы (I) или формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера, атропизомера или их смеси, или его фармацевтически приемлемой соли в качестве ингибитора SHP2.
Фармацевтическая композиция, содержащая действующее вещество, может быть в форме, пригодной для перорального введения, например, таблетки, троше, пастилки для рассасывания, водной или масляной суспензии, диспергируемого порошка или гранул, эмульсии, твердой или мягкой капсулы, сиропа или эликсира. Композицию для перорального применения можно готовить в соответствии с любым известным в данной области техники способом приготовления фармацевтической композиции. Такая композиция может содержать один или более ингредиентов, выбранных из группы, состоящей из подсластителей, вкусоароматических добавок, красителей и консервантов, чтобы обеспечить фармацевтический состав приятного внешнего вида и вкуса. Таблетка содержит действующее вещество в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, подходящими для производства таблеток. Эти эксципиенты могут представлять собой инертные эксципиенты, гранулирующие агенты, вещества для улучшения распадаемости таблеток, связующие и смазывающие вещества. Таблетка может быть непокрытой или покрытой оболочкой с помощью известного метода для маскировки вкуса лекарственного средства или замедления распада и всасывания действующего вещества в желудочно-кишечном тракте, в результате чего обеспечивается пролонгированное высвобождение в течение длительного периода времени.
Лекарственная форма для перорального применения может быть также представлена в виде мягких желатиновых капсул, в которых либо действующее вещество смешано с инертным твердым разбавителем, либо действующее вещество смешано с растворимым в воде носителем или масляной средой.
Водная суспензия содержит действующее вещество в смеси с эксципиентами, подходящими для производства водной суспензии. Такие эксципиенты представляют собой суспендирующие агенты, диспергирующие или смачивающие агенты. Водная суспензия может также содержать один или более консервантов, один или более красителей, одну или более вкусоароматическтих добавок и один или более подсластителей.
Масляную суспензию можно готовить путем суспендирования действующего вещества в растительном масле или минеральном масле. Масляная суспензия может содержать загуститель. Вышеупомянутые подсластители и вкусоароматические добавки можно добавлять, чтобы обеспечить лекарственную форму с приятным вкусом. Эти композиции можно консервировать путем добавления антиоксиданта.
Фармацевтическая композиция по настоящему изобретению может также принимать форму эмульсии масло-в-воде. Масляная фаза может представлять собой растительное масло, или минеральное масло, или их смесь. Подходящие эмульгирующие агенты могут представлять собой природные фосфолипиды. Эмульсия может также содержать подсластитель, вкусоароматическую добавку, консервант и антиоксидант. Такая лекарственная форма может также содержать мягчительное средство, консервант, краситель и антиоксидант.
Фармацевтическая композиция по настоящему изобретению может также принимать форму стерильного водного раствора для инъекций. Приемлемыми несущими средами и растворителеями, которые можно применять, являются вода, раствор Рингера или изотонический раствор хлорида натрия. Стерильная лекарственная форма для инъекций может представлять собой стерильную микроэмульсию масло-в-воде для инъекций, в которой действующее вещество растворено в масляной фазе. Раствор или микроэмульсию для инъекций можно вводить в кровоток пациента посредством локальной болюсной инъекции. Альтернативно раствор или микроэмульсию предпочтительно вводят таким образом, чтобы поддерживать постоянную концентрацию соединения по настоящему изобретению в кровообращении. Чтобы поддерживать такую постоянную концентрацию, можно использовать устройство для непрерывной внутривенной доставки. Примером такого устройства является помпа Deltec CADD-PLUS. TM. 5400 для внутривенных инъекций.
Фармацевтическая композиция по настоящему изобретению может принимать форму стерильной водной или масляной суспензии для внутримышечного и подкожного введения. Такую суспензию можно готовить с подходящими диспергирующими или смачивающими агентами и суспендирующими агентами, как описано выше, в соответствии с известными методами. Стерильная лекарственная форма для инъекций может также представлять собой стерильный раствор или суспензию для инъекций, которые готовят в нетоксичном парентерально приемлемом разбавителе или растворителе. Кроме того, в качестве растворителя или суспендирующей среды можно использовать стерильные нелетучие масла. Для этой цели можно использовать любое смешанное нелетучее масло. В дополнение к этому для приготовления инъекций можно также использовать жирные кислоты.
Соединение по настоящему изобретению можно вводить в форме суппозитория для ректального введения. Эти фармацевтические композиции можно готовить путем смешивания лекарственного средства с подходящим нераздражающим эксципиентом, который представляет собой твердое вещество при комнатной температуре, но становится жидким в прямой кишке и в результате плавления в прямой кишке высвобождает лекарственное средство.
Специалистам в данной области техники хорошо известно, что дозировка лекарственного средства зависит от ряда факторов, включающих без ограничений следующие факторы: активность конкретного соединения, возраст пациента, масса тела пациента, общее состояние здоровья пациента, поведение пациента, рацион питания пациента, время введения, путь введения, скорость выведения, комбинация лекарственных средств и т. п. В дополнение к этому оптимальное лечение, такое как режим лечения, суточная доза соединения формулы (I) или тип его фармацевтически приемлемой соли, можно верифицировать с помощью традиционных схем лечения.
ОПРЕДЕЛЕНИЯ
Если не указано иное, термины, используемые в описании и формуле изобретения, имеют описанные ниже значения.
Термин «алкил» относится к насыщенной алифатической углеводородной группе, которая представляет собой группу с нормальной или разветвленной цепью, содержащую от 1 до 20 атомов углерода, предпочтительно алкил, имеющий от 1 до 12 атомов углерода и более предпочтительно алкил, имеющий от 1 до 6 атомов углерода. Неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их различные разветвленные изомеры. Более предпочтительно алкильная группа представляет собой низший алкил, имеющий от 1 до 6 атомов углерода, не ограничивающие примеры которого включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т. п. Алкильная группа может быть замещенной или незамещенной. При замещении группа(-ы) заместителя(-ей) могут быть присоединены в любой доступной точке. Группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси и алкоксикарбонила.
Термин «циклоалкил» относится к насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе заместителя, имеющей от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода и более предпочтительно от 3 до 6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т. п. Полициклический циклоалкил включает циклоалкил, имеющий спиро-кольцо, конденсированное кольцо или кольцо с внутренним мостиком.
Термин «спиро-циклоалкил» относится к 5-20-членной полициклической группе с отдельными кольцами, соединенными посредством одного общего атома углерода (называемого спиро-атомом), причем кольца могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной π-электронной системы. Спиро-циклоалкил предпочтительно представляет собой 6-14-членный спиро-циклоалкил и более предпочтительно 7-10-членный спиро-циклоалкил. В зависимости от числа общих между кольцами спиро-атомов спиро-циклоалкил можно разделить на моно-спиро-циклоалкил, ди-спиро-циклоалкил или поли-спиро-циклоалкил, и спиро-циклоалкил предпочтительно представляет собой моно-спиро-циклоалкил или ди-спиро-циклоалкил и более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моно-спиро-циклоалкил. Неограничивающие примеры спиро-циклоалкила включают:
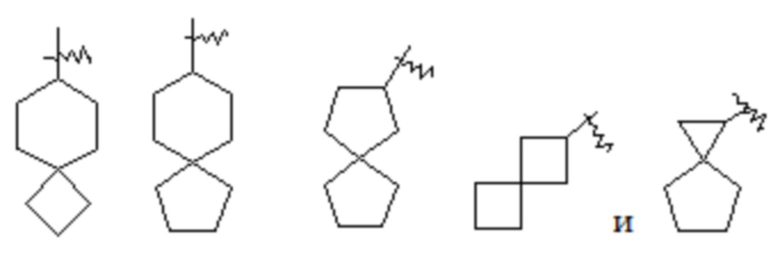 .
.
Термин «гетероциклил» относится к 3-20-членной насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе заместителя, где один или более кольцевых атомов представляют собой гетероатомы, выбранные из группы, состоящей из N, O и S(O)m (где m представляет собой целое число от 0 до 2), но за исключением -O-O-, -O-S- или -S-S- в кольце, при этом остальные кольцевые атомы представляют собой атомы углерода. Предпочтительно гетероциклил имеет от 3 до 12 кольцевых атомов, причем от 1 до 4 атомов представляют собой гетероатомы; наиболее предпочтительно от 3 до 8 кольцевых атомов, причем от 1 до 3 атомов представляют собой гетероатомы; и наиболее предпочтительно от 3 до 6 кольцевых атомов, причем от 1 до 2 атомов представляют собой гетероатомы. Неограничивающие примеры моноциклического гетероциклила включают азетидинил, пирролидинил, имидазолидинил, тетрагидрофуранил, тетрагиротиенил, дигидроимидазолил, дигидрофуранил, дигидропиразолил, дигидропирролил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил, пиранил и т.п., и предпочтительно азетидинил, пиперидинил, пиперазинил или морфолинил. Полициклический гетероциклил включает гетероциклил, имеющий спиро-кольцо, конденсированное кольцо или кольцо с внутренним мостиком.
Гетероциклильное кольцо может быть конденсировано с кольцом арила, гетероарила или циклоалкила, причем связанным с исходной структурой кольцом является гетероциклил. Его неограничивающие примеры включают:
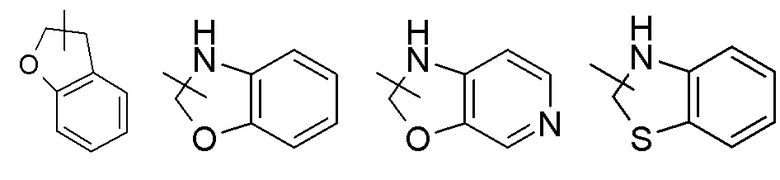 и т.п.
и т.п.
Термин «арил» относится к 6-14-членному полностью углеродному моноциклическому кольцу или полициклическому конденсированному кольцу (т.е. такое кольцо в системе имеет общую пару соседних атомов углерода с другим кольцом в системе), имеющему конъюгированную π-электронную систему, предпочтительно к 6-10-членному арилу, например фенилу и нафтилу, и более предпочтительно к фенилу. Арильное кольцо может быть конденсировано с кольцом гетероарила, гетероциклила или циклоалкила, причем связанным с исходной структурой кольцом является арильное кольцо. Его неограничивающие примеры включают:
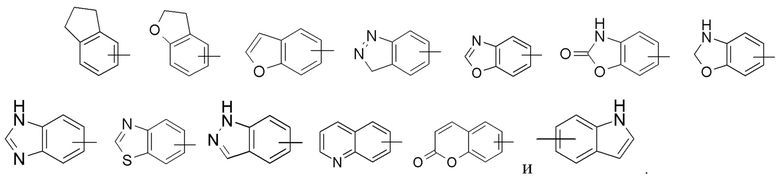
Арил может быть замещенным или незамещенным. При замещении группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси и алкоксикарбонила.
Термин «гетероарил» относится к 5-14-членной гетероароматической системе, имеющей от 1 до 4 гетероатомов, выбранных из группы, состоящей из O, S и N. Гетероарил предпочтительно представляет собой 5-10-членный гетероарил, имеющий от 1 до 3 гетероатомов, более предпочтительно 5- или 6-членный гетероарил, имеющий 1-2 гетероатома; предпочтительно, например, имидазолил, фурил, тиенил, тиазолил, пиразолил, оксазолил, пирролил, тетразолил, пиридил, пиримидинил, тиадиазолил, пиразинил и т.п., предпочтительно имидазолил, тетразолил, пиридил, тиенил, пиразолил, пиримидинил, тиазолил, и более предпочтительно пиридил. Гетероарильное кольцо может быть конденсировано с кольцом арила, гетероциклила или циклоалкила, причем связанным с исходной структурой кольцом является гетероарильное кольцо. Его неограничивающие примеры включают:
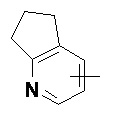
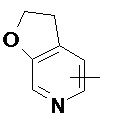
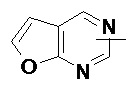
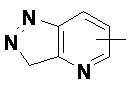
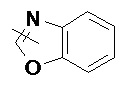
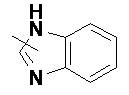
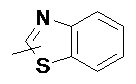
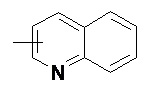 и
и 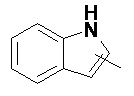 .
.
Гетероарил может быть необязательно замещенным или незамещенным. При замещении группа(-ы) заместителя(-ей) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси и алкоксикарбонила.
Термин «галоген» относится к атому фтора, хлора, брома или йода.
Термин «галогеналкил» относится к алкильной группе, замещенной одним или более атомов галогена, где алкил является таким, как определено выше.
Термин «галогеналкокси» относится к алкоксигруппе, замещенной одним или более атомов галогена, где алкил является таким, как определено выше.
Термин «гидроксиалкил» относится к алкильной группе, замещенной гидроксигруппой (-ами), где алкил является таким, как определено выше.
Термин «алкиламино» относится к аминогруппе, замещенной одним или двумя алкилами, где алкил является таким, как определено выше.
Термин «гидрокси» относится к группе -OH.
Термин «галоген» относится к атому фтора, хлора, брома или йода.
Термин «амино» относится к группе -NH2.
Термин «циано» относится к группе -CN.
Термин «нитро» относится к группе -NO2.
Термин «оксо» относится к группе =O.
Термин «карбонил» относится к группе C=O.
Термин «карбокси» относится к группе -C(O)OH.
Термин «тио» относится к группе -S-.
Термин «тиол» относится к группе -SH.
«Необязательный» или «необязательно» означает, что описанное далее событие или обстоятельство может произойти, но не обязательно происходит, и такое описание включает ситуацию, в которой это событие или обстоятельство либо происходит, либо не происходит. Например, «гетероциклил, необязательно замещенный алкилом» означает, что алкильная группа может присутствовать, но не обязательно присутствует, и такое описание включает ситуацию, где гетероциклил замещен алкилом и где гетероциклил не замещен алкилом.
«Замещенный» относится к одному или более атомов водорода в группе, предпочтительно в количестве до 5 и более предпочтительно от 1 до 3 атомов водорода, независимо замещенных соответствующим количеством заместителей. Без слов понятно, что заместители могут существовать только в их возможном химическом положении. Специалист в данной области техники способен экспериментальным или теоретическим путем без лишних усилий определить, возможно или невозможно замещение. Например, объединение амино- или гидроксигруппы, имеющей свободный атом водорода и атомы углерода, имеющие нестабильные связи (такие как олефиновые), может быть нестабильным.
«Фармацевтическая композиция» относится к смеси одного или более соединений в соответствии с настоящим изобретением или их физиологически/фармацевтически приемлемых солей или пролекарств с другими химическими компонентами и другими компонентами, такими как физиологически/фармацевтически приемлемые носители и эксципиенты. Цель фармацевтической композиции состоит в том, чтобы способствовать введению соединения в организм, что создает условия для всасывания действующего вещества, позволяющего ему проявить биологическую активность.
«Фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, которая безопасна и эффективна для млекопитающих и обладает необходимой биологической активностью.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение будет дополнительно описано со ссылкой на следующие ниже примеры, но эти примеры не следует истолковывать как ограничивающие объем настоящего изобретения.
ПРИМЕРЫ
Структуры соединений идентифицировали методом ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС). Сдвиги на ЯМР (δ) представлены в 10-6 (ppm (млн-1)). Спектры ЯМР определяли на устройстве Bruker AVANCE-400. Растворители для определения представляли собой дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD), и внутренний стандарт представлял собой тетраметилсилан (TMS).
МС определяли на масс-спектрометре Shimadzu 2010 или на спектрометре Agilent 6110A MSD.
Высокоэффективную жидкостную хроматографию (ВЭЖХ) проводили на системах серии Shimadzu LC-20A, Shimadzu LC-2010HT или на жидкостном хроматографе высокого давления Agilent 1200 LC (колонка Ultimate XB-C18 3,0×150 мм или Xtimate C18 2,1×30 мм).
Хиральную ВЭЖХ проводили на колонке Chiralpak IC-3 100×4,6 мм (внутр. диаметр, I.D.), 3 мкм, Chiralpak AD-3 150×4,6 мм I.D., 3 мкм, Chiralpak AD-3 50×4,6 мм I.D., 3 мкм, Chiralpak AS-3 150×4,6 мм I.D., 3 мкм, Chiralpak AS-3 100×4,6 мм I.D., 3 мкм, ChiralCel OD-3 150×4,6 мм I.D., 3 мкм, Chiralcel OD-3 100×4,6 мм I.D., 3 мкм, ChiralCel OJ-H 150×4,6 мм I.D., 5 мкм, Chiralcel OJ-3 150×4,6 мм I.D., 3 мкм.
Для тонкослойной хроматографии (ТСХ) использовали пластину силикагеля Yantai Huanghai HSGF254 или Qingdao GF254. Размер пластин силикагеля, использованных в ТСХ, составлял от 0,15 мм до 0,2 мм, и размер пластин силикагеля, использованных в очистке продукта тонкослойной хроматографией, составлял от 0,4 мм до 0,5 мм.
В качестве носителя для колоночной хроматографии использовали силикагель Yantai Huanghai от 100 до 200 меш, от 200 до 300 меш или от 300 до 400 меш.
Для хирального получения использовали колонку DAICEL CHIRALPAK IC (250×30 мм, 10 мкм) или Phenomenex-Amylose-1 (250 мм×30 мм, 5 мкм).
Для быстрого получения CombiFlash использовали прибор Combiflash Rf150 (TELEDYNE ISCO).
Средние значения коэффициентов ингибирования киназы и значения IC50 определяли методом иммуносорбентного ферментного анализа (ИФА) NovoStar (BMG Co., Германия).
Известные исходные материалы по настоящему изобретению могут быть получены известными в данной области техники способами или приобретены у компаний ABCR GmbH & Co. KG, Acros Organnics, Aldrich Chemical Company, Accela ChemBio Inc., Dari chemical Company и т.д.
Если не указано иное, реакции проводили в атмосфере аргона или в атмосфере азота.
«Атмосфера аргона» или «атмосфера азота» означает, что реакционную колбу оснащают баллоном с аргоном или азотом (около 1 л).
«Атмосфера водорода» означает, что реакционную колбу оснащают баллоном с водородом (около 1 л).
Реакции гидрогенизации под давлением выполняли в гидрогенизаторе Parr 3916EKX с генератором водорода Qinglan QL-500 или гидрогенизаторе HC2-SS.
В реакциях гидрогенизации, как правило, из системы откачивали воздух и заполняли водородом, повторяя вышеуказанную операцию три раза.
В микроволновых реакциях использовали микроволновый реактор типа CEM Discover-S 908860.
Если не указано иное, «раствор» относится к водному раствору.
Если не указано иное, реакции проводили при комнатной температуре от 20°C до 30°C.
Процесс реакции в примерах контролировали методом тонкослойной хроматографии (ТСХ). Используемые в реакции растворитель для проявления, система элюентов колоночной хроматографии и система растворителей для проявления в тонкослойной хроматографии для очистки соединений включали: A: Система B дихлорметан/метанол: Система н-гексан/этилацетат, C: петролейный эфир/этилацетат и D: петролейный эфир/этилацетат/метанол. Долю объема растворителя регулировали в зависимости от полярности соединений, для чего можно было также добавить небольшое количество щелочного реагента, такого как триэтиламин, или кислого реагента, такого как уксусная кислота.
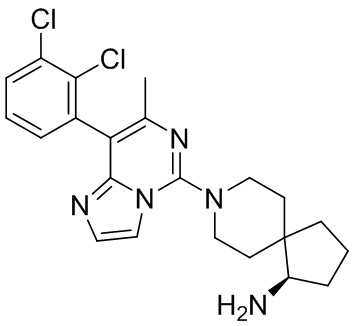
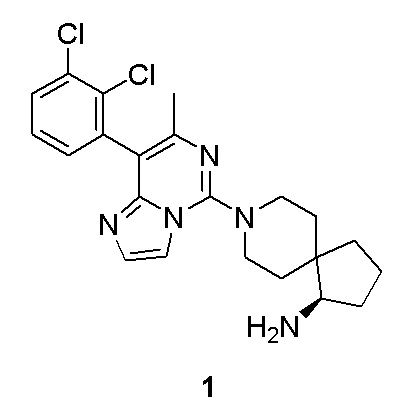
Пример 1
(R)-8-(8-(2,3-дихлорфенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4,5]декан-1-амин
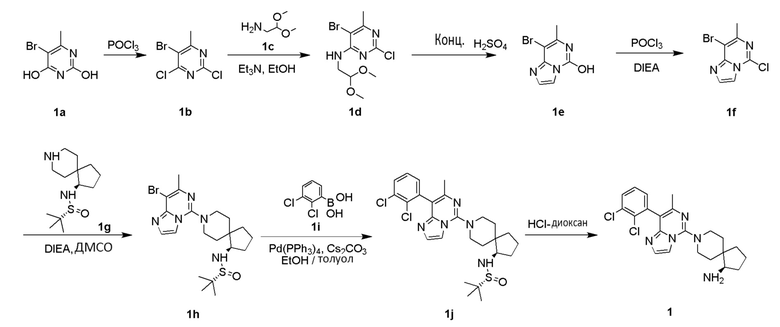
DIEA - диизопропилэтиламин
Стадия 1
5-бром-2,4-дихлор-6-метилпиримидин 1b
5-Бром-6-метилпиримидин-2,4-диол 1a (1,5 г, 7,32 ммоль) растворяли в 8 мл оксихлорида фосфора. Добавляли 0,3 мл N,N-диметилформамида, подогревали реакционный раствор до 115°C и перемешивали в течение 4 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении и добавляли к 20 мл смеси льда и воды. Реакционный раствор экстрагировали этилацетатом (3 раза по 10 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (5 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении с получением указанного в заголовке соединения 1b (950 мг, выход: 54%) в виде желтого твердого вещества.
МС, ионизация электрораспылением (ИЭР) m/z 242,8 [M+H]+
1H ЯМР (400 МГц, CDCl3) δ 2,72 (s, 3H).
Стадия 2
5-Бром-2-хлор-N-(2,2-диметоксиэтил)-6-метилпиримидин-4-амин 1d
Соединение 1b (940 мг, 3,89 ммоль) и 2,2-диметоксиэтиламин 1c (817 мг, 7,77 ммоль) растворяли в 15 мл этанола. Добавляли 1,1 мл триэтиламина (785 мг, 7,77 ммоль) при 0°C, и реакционный раствор перемешивали при комнатной температуре в течение 12 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении. Добавляли воду и экстрагировали реакционный раствор этилацетатом (3 раза по 10 мл). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с петролейным эфиром и этилацетатом в качестве элюента с получением указанного в заголовке соединения 1d (790 мг, выход: 65%) в виде белого твердого вещества.
МС (ИЭР) m/z 311,8 [M+H]+
1H ЯМР (400 МГц, CDCl3) δ 5.82 (s, 1H), 4.49 (t, J = 4.8 Гц, 1H), 3.66 (t, J = 5.6 Гц, 2H), 3.45 (s, 6H), 2.48 (s, 3H).
Стадия 3
8-Бром-7-имидазо[1,2-c]пиримидин-5-ол 1e
Соединение 1d (780 мг, 2,51 ммоль) растворяли в 8 мл концентрированной серной кислоты и подвергали взаимодействию при 65°C в течение 2 часов. После завершения реакции добавляли 100 мл смеси льда и воды и добавляли насыщенный раствор гидроксида натрия до pH=6. Реакционный раствор экстрагировали смешанным растворителем дихлорметана и изопропанола (3 раза по 50 мл, объемное соотношение: 3:1). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (150 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении с получением указанного в заголовке соединения 1e (560 мг, выход: 92%) в виде желтого твердого вещества.
МС (ИЭР) m/z 227,9, 229,9 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ 7.81 (d, J = 1,6 Гц, 1H), 7.36 (d, J = 1,6 Гц, 1H), 2.43 (s, 3H).
Стадия 4
8-Бром-5-хлор-7-метилимидазо[1,2-c]пиримидин 1f
N,N-Диизопропилэтиламин (5,6 мл, 5,57 ммоль) добавляли к суспензии соединения 1e (300 мг, 1,32 ммоль) и оксихлорида фосфора (7,58 г). Реакционный раствор подвергали взаимодействию при 110°C в течение 3 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении. Добавляли 50 мл насыщенного раствора бикарбоната натрия и экстрагировали реакционный раствор этилацетатом (3 раза по 50 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (200 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении с получением указанного в заголовке соединения 1f (210 мг, выход: 36%) в виде желтого твердого вещества.
МС (ИЭР) m/z 247,9 [M+H] +
1H ЯМР (400 МГц, ДМСО-d6) δ 8.10 (s, 1H), 7.75 (s, 1H), 2.56 (s, 3H).
Стадия 5
(R)-N-(R)-8-(8-Бром-7-метилимидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4.5]декан-1-ил)-2-метилпропан-2-сульфинамид 1h
Соединение 1f (180 мг, 0,73 ммоль), (R)-2-метил-N-((R)-8-азаспиро[4.5]декан-1-ил)пропан-2-сульфинамид 1g (268 мг, 0,73 ммоль, получен способом, раскрытым в заявке на патент WO2016203406 A1) и N,N-диизопропилэтиламин (0,36 мл, 2,19 ммоль) растворяли в 5 мл диметилсульфоксида. Реакционный раствор подвергали взаимодействию при 90°C в течение 30 минут. После завершения реакции добавляли 20 мл этилацетата и 40 мл воды и экстрагировали реакционный раствор этилацетатом (3 раза по 20 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с дихлорметаном и метанолом в качестве элюента с получением указанного в заголовке соединения 1h (200 мг, выход: 56%) в виде белого твердого вещества.
МС (ИЭР) m/z 468,0, 470,0 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ 7.77 (d, J = 1.6 Гц, 1H), 7.54 (d, J = 1.6 Гц, 1H), 5.02 (d, J = 8.4 Гц, 1H), 3.84-3.77 (m, 2H), 3.37-3.33 (m, 1H), 3.17-3.03 (m, 2H), 2.55 (s, 3H), 2.21-2.15 (m, 1H), 2.09-1.89 (m, 3H), 1.81-1.64 (m, 3H), 1.62-1.54 (m, 1H), 1.53-1.39 (m, 2H), 1.26 (s, 9H).
Стадия 6
(R)-N-((R)-8-(8-(2,3-Дихлорфенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4.5]декан-1-ил)-2-метилпропан-2-сульфинамид 1j
Соединение 1h (170 мг, 0,36 ммоль), 2,3-дихлорфенилбороновую кислоту 1i (138 мг, 0,73 ммоль), карбонат цезия (355 мг, 1,09 ммоль) и тетракис(трифенилфосфин)палладий (42 мг, 0,036 ммоль) последовательно растворяли в смешанном растворе 2 мл толуола и 2 мл этанола в атмосфере азота. Реакционный раствор подвергали взаимодействию при 120°C в течение 1 часа. Реакционный раствор охлаждали до комнатной температуры. Добавляли 10 мл этилацетата и 10 мл воды, и реакционный раствор экстрагировали этилацетатом (3 раза по 10 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с дихлорметаном и метанолом в качестве элюента с получением указанного в заголовке соединения 1j (37 мг, выход: 19%) в виде желтого масла.
МС (ИЭР) m/z 534,4 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ 7.83-7.25 (m, 5H), 3.92-3.75 (m, 2H), 3.22-2.95 (m, 2H), 2.18 (s, 3H), 2.11-1.18 (m, 20H).
Стадия 7
(R)-8-(8-(2,3-Дихлорфенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4.5]декан-1-амин 1
Соединение 1j (25 мг, 0,047 ммоль) растворяли в 2 мл 1,4-диоксана. Добавляли 1 мл 4 М раствора хлорида водорода в 1,4-диоксана при 0°C, и реакционный раствор перемешивали при комнатной температуре в течение 30 часов. Добавляли 10 мл этилацетата, затем реакционный раствор фильтровали и промывали этилацетатом (3 раза по 10 мл). Полученное в результате твердое вещество растворяли в 10 мл воды и добавляли насыщенный раствор бикарбоната натрия до pH 9. Смесь экстрагировали хлороформом (2 раза по 20 мл). Органическую фазу промывали насыщенным раствором бикарбоната натрия (10 мл) и насыщенным раствором хлорида натрия (2 раза по 20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении с получением указанного в заголовке соединения 1 (9,3 мг, выход: 42%).
МС (ИЭР) m/z 430,1 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ 7.70-7.62 (m, 2H), 7.47-7.39 (m, 2H), 7.31 (d, J = 1.6 Гц, 1H), 3.97-3.81 (m, 2H), 3.29-3.18 (m, 2H), 3.11-3.04 (m, 1H), 2.19 (s, 3H), 2.17-2.10 (m, 1H), 2.06-1.90 (m, 3H), 1.88-1.82 (m, 1H), 1.81-1.70 (m, 2H), 1.66-1.50 (m, 3H).
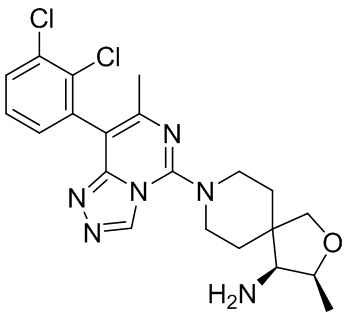
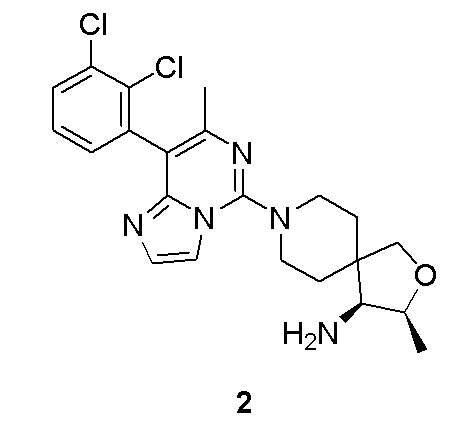
Пример 2
(3S,4S)-8-(8-(2,3-Дихлорфенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин
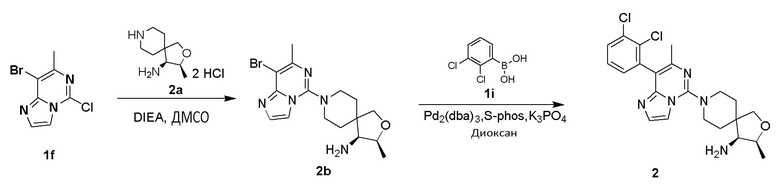
Стадия 1
(3S,4S)-8-(8-Бром-7-метилимидазо[1,2-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин 2b
Соединение 1f (150 мг, 0,53 ммоль), (3S,4S)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин 2а (154 мг, 0,63 ммоль, получен способом, раскрытым в заявке на патент WO2015107495 A1) и N,N-диизопропилэтиламин (31 мг, 1,06 ммоль) растворяли в 3 мл диметилсульфоксида. Реакционный раствор подвергали взаимодействию при 90°C в течение 1 часа. После завершения реакции добавляли 15 мл этилацетата и 30 мл воды и экстрагировали реакционный раствор этилацетатом (3 раза по 10 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (3 раза по 10 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с дихлорметаном и метанолом в качестве элюента с получением указанного в заголовке соединения 2b (150 мг, выход: 75%) в виде желтого твердого вещества.
МС (ИЭР) m/z 380,1, 382,1 [M+H] +
1H ЯМР (400 МГц, CDCl3) δ 7.59 (s, 1H), 7.42 (s, 1H), 4.23-4.16 (m, 1H), 3.83 (d, J = 8.8 Гц, 1H), 3.72 (d, J = 8.8 Гц, 1H), 3.64-3.55 (m, 2H), 3.30-3.22 (m, 1H), 3.20-3.14 (m, 1H), 3.04 (d, J = 4.4 Гц, 1H), 2.57 (s, 3H), 2.03-1.98 (m, 1H), 1.93-1.86 (m, 1H), 1.84-1.74 (m, 2H), 1.26 (d, J = 6.4 Гц, 3H).
Стадия 2
(3S,4S)-8-(8-(2,3-Дихлорфенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин 2
Соединение 2b (50 мг, 0,131 ммоль), 2,3-дихлорфенилбороновой кислоты 1i (30 мг, 0,158 ммоль), фосфат калия (55,6 мг, 0,262 ммоль), трис(дибензилиденацетон)дипалладий (5,95 мг, 0,007 ммоль) и 2-дициклогексилфосфино-2',6'-диметоксидифенил последовательно суспендировали в 1 мл 1,4-диоксана в атмосфере азота. Реакционный раствор подвергали взаимодействию при 100°C в течение 1 часа. Реакционный раствор охлаждали до комнатной температуры. Добавляли 5 мл этилацетата и 4 мл воды, и реакционный раствор экстрагировали этилацетатом (3 раза по 8 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (8 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с этилацетатом, метанолом и аммиаком в качестве элюента с получением указанного в заголовке соединения 2 (9,7 мг, выход: 16%).
МС (ИЭР) m/z 446,1 [M+H] +
1H ЯМР (400 МГц, CDCl3) δ 7.57-7.50 (m, 2H), 7.41 (d, J = 1.2 Гц, 1H), 7.32 (t, J = 7.6 Гц, 1H), 7.28 (d, J = 1.6 Гц, 1H), 4.25-4.19 (m, 1H), 3.86 (d, J = 8.8 Гц, 1H), 3.76 (d, J = 8.8 Гц, 1H), 3.72-3.63 (m, 2H), 3.40-3.18 (m, 2H), 3.07 (d, J = 4.8 Гц, 1H), 2.23 (s, 3H), 2.10-2.01 (m, 1H), 1.98-1.77 (m, 3H), 1.27 (d, J = 6.4 Гц, 3H).
Пример 3
a(R)-(3S,4S)-8-(8-(2,3-Дихлорфенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амина атропизомер 1
Пример 4
a(S)-(3S,4S)-8-(8-(2,3-Дихлорфенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амина атропизомер 2
Атропизомер 2
Атропизомер 1
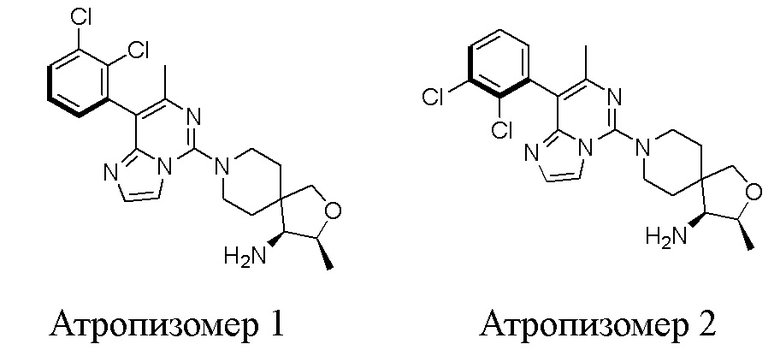
(3S,4S)-8-(8-(2,3-Дихлорфенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин (17 мг), полученный в примере 2, восстанавливали с помощью сверхкритической жидкостной хроматографии (подвижная фаза: 45% EtOH + 0,1% NH3H2O /55% scCO2, скорость потока: 80 мл/мин) на хиральной колонке (DAICEL CHIRALPAK IC (250 мм×30 мм, 10 мкм)). 5,2 мг атропизомера (диастереомерный избыток (д.и.) 98,91%) был получен из первого пика элюирования. 1H ЯМР (400МГц, CDCl3) δ 7.72-7.60 (m, 2H), 7.47-7.39 (m, 2H), 7.34-7.25 (m, 1H), 4.95-4.91 (m, 1H), 4.42-4.23 (m, 1H), 3.96-3.88 (m, 1H), 3.80-3.68 (m, 2H), 3.55-3.42 (m, 1H), 3.30-3.07 (m, 2H), 2.18 (s, 3H), 2.05-1.95 (m, 2H), 1.89-1.72 (m, 2H), 1.5 (d, J = 6.8 Гц, 3H). Метод хирального анализа: Chiralpak IC-3 100 7.47-7.39 (m, 2H), подвижная фаза: A - сверхкритический диоксид углерода, B - EtOH + 0,05% ДЭА, скорость потока: 2,8 мл/мин, изократическое элюирование 40% B. Время удерживания (RT): 1,495 минут.
4,4 мг атропизомера (диастереомерный избыток (д.и.) 99,33%) был получен из второго пика элюирования. 1H ЯМР (400 МГц, CDCl3) δ 7.72-7.60 (m, 2H), 7.46-7.39 (m, 2H), 7.32-7.28 (m, 1H), 5.01-4.90 (m, 1H), 4.45-4.23 (m, 1H), 3.97-3.87 (m, 1H), 3.80-3.65 (m, 2H), 3.55-3.44 (m, 1H), 3.27-3.07 (m, 2H), 2.18 (s, 3H), 2.05-1.94 (m, 2H), 1.87-1.70 (m, 2H), 1.25 (d, J = 6.8 Гц, 3H). Метод хирального анализа: Chiralpak IC-3 100 7.46-7.39 (m, 2H), подвижная фаза: A - сверхкритический диоксид углерода, B - EtOH и 0,05% ДЭА, скорость потока: 2,8 мл/мин, изократическое элюирование 40% B. Время удерживания (RT): 2,716 минут.
Пример 5
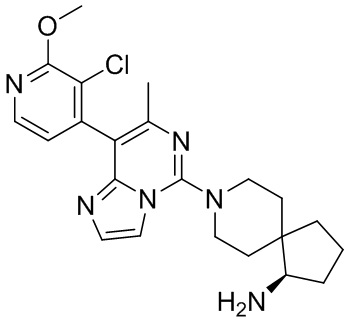
(R)-8-(8-(2,3-Дихлорпиридин-4-ил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4.5]декан-1-амин
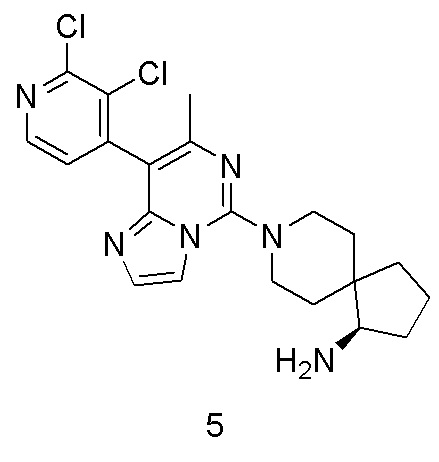
В соответствии со стадиями синтеза примера 1 соединение 1i заменяли соединением (2,3-дихлорпиридин-4-ил)бороновой кислоты, соответственно, получили соединение примера 5.
МС (ИЭР) m/z 431,0 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ 8.42 (d, J= 4.8 Гц, 1H), 7.72 (d, J=2.0 Гц, 1H), 7.46 (d, J=1.6 Гц, 1H), 7.42 (d, J=4.8 Гц, 1H), 3.97-3.84 (m, 2H), 3.29-3.15 (m, 2H), 2.89 (t, J=7.2 Гц, 1H), 2.22 (s, 3H), 2.13-2.04 (m, 1H), 2.01-1.87 (m, 3H), 1.83-1.74 (m, 1H),1.73-1.67 (m, 1H), 1.66-1.58 (m, 1H), 1.55-1.41 (m, 3H).
Пример 6
(R)-8-(8-(3-Хлор-2-метоксипиридин-4-ил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4.5]декан-1-амин
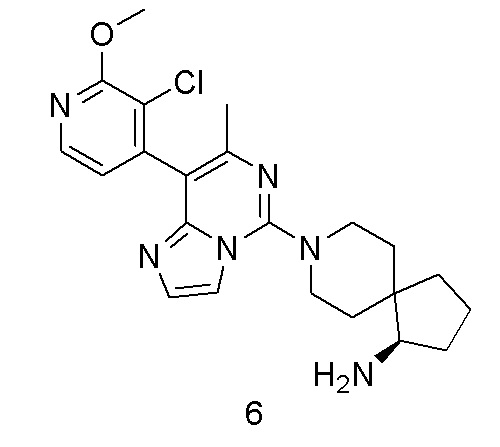
В соответствии со стадиями синтеза примера 1 соединение 1i заменяли соединением (3-хлор-2-метоксипиридин-4-ил)бороновой кислоты, соответственно, получили соединение примера 6.
МС (ИЭР) m/z 427,2 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ 8.16 (d, J=5.2 Гц, 1H), 7.69 (s, 1H), 7.45 (s, 1H), 6.95 (d, J=5.2 Гц, 1H), 4.06 (s, 3H), 3.94-3.82 (m, 2H), 3.27-3.16 (m, 2H), 3.01 (t, J=7.2 Гц, 1H), 2.21 (s, 3H), 2.16-2.08 (m, 1H), 2.00-1.91 (m, 2H), 1.88-1.77 (m, 2H), 1.74-1.64 (m, 2H), 1.62-1.45 (m, 3H).
Пример 7
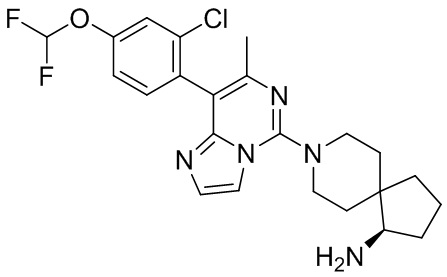
(R)-8-(8-(2-Хлор-4-(дифторметокси)фенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4.5]декан-1-амин
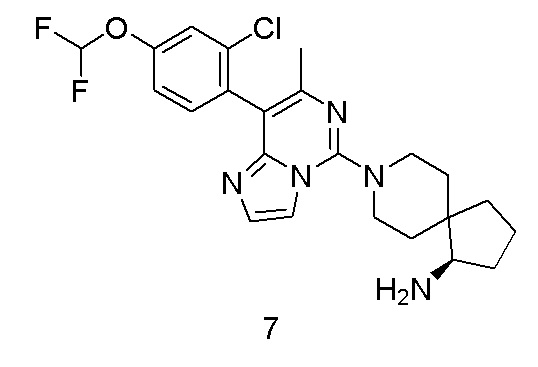
В соответствии со стадиями синтеза примера 1 соединение 1i заменяли соединением (2-хлор-4-(дифторметокси)фенил)бороновой кислоты, соответственно, получили соединение примера 7.
МС (ИЭР) m/z 462,2 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ 7.68 (d, J = 1.2 Гц, 1H), 7.44 (d, J = 1.2 Гц, 1H), 7.24 (dd, J = 2.0, 8.4 Гц, 1H), 6.97 (t, J = 73.6 Гц, 1H), 3.90-3.80 (m, 2H), 3.26-3.13 (m, 2H), 2.92-2.88 (m, 1H), 2.19 (s, 3H), 2.10-1.95 (m, 2H), 1.95-1.87 (m, 2H), 1.84-1.75 (m, 1H), 1.74-1.68 (m, 1H), 1.66-1.58 (m, 1H), 1.56-1.41 (m, 3H).
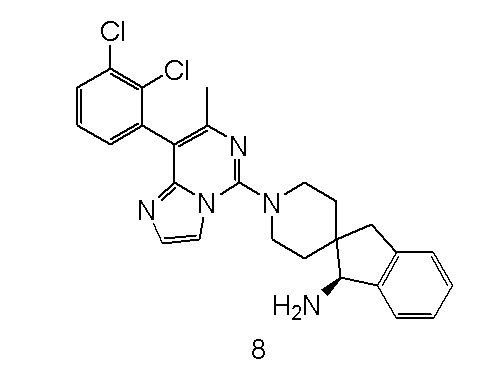
Пример 8
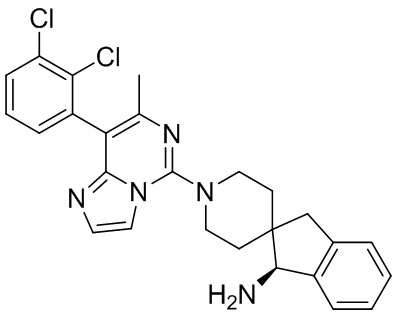
(S)-1'-(8-(2,3-Дихлорфенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
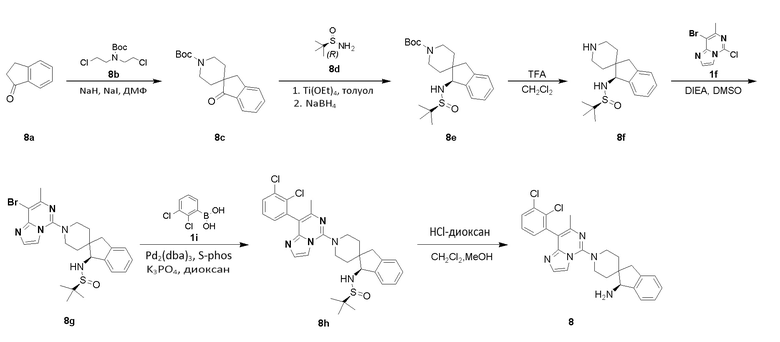
Стадия 1
Трет-бутил-1-оксо-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат 8c
Соединение 8a (10 г, 75,66 ммоль) растворяли в ДМФ (300 мл) в атмосфере азота с последующим добавлением гидрида натрия (60%, смесь с керосином, 9,1 г, 226,98 ммоль) при 10°C. Реакционную систему перемешивали при комнатной температуре в течение 30 минут. Добавляли соединение трет-бутил-бис(2-хлорэтил)карбамат 8b (18,3 г, 75,66 ммоль) и гидрид натрия (22,6 г, 151,32 ммоль). Реакционный раствор подвергали взаимодействию при комнатной температуре в течение 1 часа и нагревали до 50°C в течение 12 часов. После завершения реакции добавляли насыщенный водный раствор хлорида аммония (50 мл) и экстрагировали реакционный раствор этилацетатом (200 мл). Органические фазы объединяли, промывали водой (2 раза по 80 мл) и насыщенным раствором хлорида натрия (2 раза по 80 мл), высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный в результате неочищенный продукт очищали хроматографией на силикагеле с петролейным эфиром и этилацетатом в качестве элюента с получением указанного в заголовке соединения 8c (1,9 г, выход: 8,34%) в виде коричневого твердого вещества.
МС (ИЭР) m/z 246,0 [M+H-56] +
1H ЯМР: (400 МГц, CDCl3) δ 7.78 (d, J = 8.0 Гц, 1H), 7.62 (t, J = 7.2 Гц, 1H), 7. 48 (d, J = 7.2 Гц, 1H), 7.40 (t, J = 7.2 Гц, 1H), 4.15-4.13 (m, 2H), 3.08 (s, 2H), 3.05-2.99 (m, 2H), 1.92 (dt, J = 4.4 Гц, J = 13.2 Гц, 2H), 1.49 (s, 9H), 1.40-1.35 (m, 2H).
Стадия 2
(S)-1-((S)-трет-бутилсульфиниламино)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1'-карбоксилат 8e
Соединение 8c (1,60 г, 5,31 ммоль) растворяли в безводном толуоле (20 мл) с последующим добавлением тетраэтоксида титана (2,42 г, 10,62 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 20 минут. Добавляли соединение (R)-2-метилпропан-2-сульфинамид 8d (965 мг, 7,96 ммоль) и подвергали реакционную систему взаимодействию при 90°C в течение 15 часов. После охлаждения до 0°C добавляли боргидрид лития (139 мг, 6,37 ммоль) и подвергали взаимодействию реакционный раствор в течение 30 минут. После завершения реакции добавляли по каплям метанол (8 мл) при 0°C. Добавляли воду (20 мл) и этилацетат (30 мл) и перемешивали реакционный раствор в течение 5 минут. Суспендированное вещество отфильтровывали через диатомовую землю и промывали этилацетатом (50 мл). Реакционный раствор экстрагировали этилацетатом (2 раза по 70 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (2 раза по 30 мл), высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный в результате неочищенный продукт очищали хроматографией на силикагеле с петролейным эфиром и этилацетатом в качестве элюента с получением соединения 8e (530 мг, выход: 24,5%) в виде желтого твердого вещества.
МС (ИЭР) m/z 307,2 [M+H-Boc] +
1H ЯМР (400 МГц, MeOH-d4) δ 7.31 (d, J = 5.6 Гц, 1H), 7.22-7.18 (m, 3H), 5.56 (d, J = 8.0 Гц, 1H), 4.48 (d, J = 10.4 Гц, 1H), 4.02-3.97 (m, 1H), 3.13 (d, J = 15.6 Гц, 1H), 3.08-2.95 (m, 2H), 2.73 (d, J = 16.0 Гц, 1H), 2.05-1.96 (m, 1H), 1.73-1.72 (m, 1H), 1.54-1.52 (m, 1H), 1.46 (s, 9H), 1.31 (s, 9H).
Стадия 3
(S)-N-((S)-1,3-Дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид 8f
Соединение 8e (460 мг, 1,13 ммоль) растворяли в дихлорметане (5 мл) с последующим добавлением трифторуксусной кислоты (1 мл) при 0°C. Реакционный раствор перемешивали при комнатной температуре в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного продукта и добавляли насыщенный раствор бикарбоната натрия до pH 7-8. Реакционный раствор экстрагировали дихлорметаном (3 раза по 10 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (8 мл), высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 8f (230 мг, выход: 66%) в виде желтого масла.
МС, ионизация электрораспылением (ИЭР) m/z 307,2 [M+H]+
Стадия 4
(S)-N-((S)-1'-(8-Бром-7-метилимидазо[1,2-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид 8g
Соединение 8f (123 мг, 0,50 ммоль) и соединение 1f (230 мг, 0,75 ммоль) растворяли в диметилсульфоксиде (3 мл) в атмосфере азота с последующим добавлением диизопропилэтиламина (129 мг, 1,0 ммоль). Реакционный раствор перемешивали при 90°C в течение 1 часа. Добавляли этилацетат (20 мл) и воду (10 мл), и реакционный раствор экстрагировали этилацетатом (2 раза по 10 мл). Органические фазы объединяли, промывали водой (2 раза по 8 мл) и насыщенным раствором хлорида натрия (2 раза по 8 мл), высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный в результате неочищенный продукт очищали хроматографией на силикагеле с петролейным эфиром и этилацетатом в качестве элюента с получением соединения 8g (250 мг, выход: 97%) в виде белого твердого вещества.
МС (ИЭР) m/z 516,1, 518,1 [M+H] +
Стадия 5
(S)-N-((S)-1'-(8-(2,3-Дихлорфенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-ил)-2-метилпропан-2-сульфинамид 8h
Соединение 8g (100 мг, 0,19 ммоль) и соединение 1i (55,3 мг, 0,29 ммоль) растворяли в 1,4-диоксане (2 мл) в атмосфере азота с последующим добавлением карбоната калия (161 мг, 0,76 ммоль) при комнатной температуре. Добавляли трис(дибензилиденацетон)дипалладий (8,7 мг, 0,0095 ммоль) и 2-дициклогексилфосфино-2',6'-диметоксидифенил (7,8 мг, 0,019 ммоль), нагревали реакционный раствор до 100°C и перемешивали в течение 1 часа. Добавляли этилацетат (10 мл) и воду (8 мл), и реакционный раствор экстрагировали этилацетатом (2 раза по 8 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (5 мл), высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный в результате неочищенный продукт очищали хроматографией на силикагеле с петролейным эфиром и этилацетатом в качестве элюента с получением соединения 8h (15 мг, выход: 13,5%) в виде желтого твердого вещества.
МС (ИЭР) m/z 582,3 [M+H] +
Стадия 6
(S)-1'-(8-(2,3-Дихлорфенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин 8
Соединение 8h (15 мг, 0,026 ммоль) растворяли в дихлорметане (2 мл) и метаноле (0,2 мл) с последующим добавлением раствора хлорида водорода в 1,4-диоксане (0,5 мл, 4 н.) при 0°C. Реакционный раствор подвергали взаимодействию при комнатной температуре в течение 20 минут. Реакционный раствор концентрировали при пониженном давлении и добавляли насыщенный раствор бикарбоната натрия до pH 7-8. Реакционный раствор экстрагировали дихлорметаном (3 раза по 8 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (5 мл), высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 8 (6,3 мг, выход: 50,7%).
МС (ИЭР) m/z 478,1 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ 7.72 (s, 1H), 7.65 (d, J = 8 Гц, 1H), 7.45-7.41 (m, 3H), 7.32-7.24 (m, 4H), 4.97-4.95 (m, 1H), 4.86-4.85 (m, 1H), 4.13 (s, 1H), 3.91-3.88 (m, 2H), 3.20 (d, J = 8 Гц, 1H), 2.93 (d, J = 8 Гц, 1H), 2.20 (s, 3H), 2.09-2.06 (m, 2H), 1.72 (d, J = 13.6 Гц, 1H), 1.62 (d, J = 13.6 Гц, 1H).
Пример 9
(3S,4S)-8-(8-(2,3-Дихлорфенил)-7-метил-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин
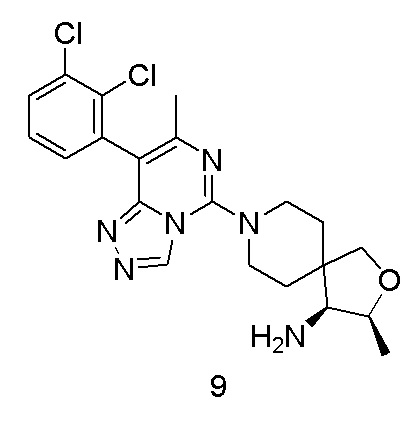
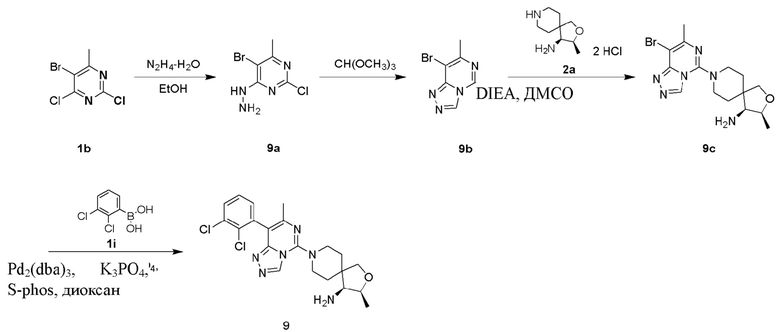
Стадия 1
5-Бром-2-хлор-4-гидразинил-6-метилпиримидин 9a
Соединение 1b (3,0 г, 12,40 ммоль) растворяли в этаноле (30 мл) с последующим добавлением гидрата гидразина (1,86 г, 37,21 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 4 минут, и затем фильтровали. Фильтрационный осадок промывали этанолом (3 раза по 10 мл). Полученное в результате твердое вещество высушивали в вакууме с получением соединения 9a (2,8 г, выход: 95%) в виде желтого твердого вещества.
МС (ИЭР) m/z 236,7, 238,7 [M+H]+
1H ЯМР (400 МГц, MeOH-d4) δ 2,43 (s, 3H).
Стадия 2
8-Бром-5-хлор-7-метил-[1,2,4]триазоло[4,3-c]пиримидин 9b
Соединение 9a (1,4 г, 5,90 ммоль) растворяли в триметилортоформиате (20 мл) и подвергали взаимодействию реакционный раствор при 100°C в течение 2 часов. После завершения реакции реакционный раствор концентрировали в вакууме, и полученный в результате неочищенный продукт (1,4 г) очищали хроматографией на силикагеле с петролейным эфиром и этилацетатом в качестве элюента с получением соединения 9b (330 мг, выход: 23%) в виде белого твердого вещества.
МС (ИЭР) m/z 246,4, 248,4 [M+H] +
Стадия 3
(3S,4S)-8-(8-Бром-7-метил-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин 9c
Соединение 9b (120 мг, 0,48 ммоль) и диизопропилэтиламин (ДИЭА) (310 мг, 2,40 ммоль) растворяли в ДМСО (диметилсульфоксид) (4,0 мл) с последующим добавлением соединения 2a (140 мг, 0,58 ммоль). Реакционный раствор подвергали взаимодействию при 90°C в течение 1 часа. После завершения реакции добавляли воду (50 мл) и экстрагировали реакционный раствор этилацетатом (3 раза по 25 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (3 раза по 20 мл), высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный в результате остаток очищали хроматографией на силикагеле с дихлорметаном и метанолом в качестве элюента с получением указанного в заголовке соединения 9c (150 мг, выход: 60%) в виде желтого твердого вещества.
МС (ИЭР) m/z 381,1, 383,1 [M+H] +
1H ЯМР (400 МГц, CDCl3) δ 8.69 (s, 1H), 4.21-4.15 (m, 1H), 3.82 (d, J = 8.8 Гц, 1H), 3.75-3.62 (m, 3H), 3.44-3.25 (m, 2H), 3.04 (d, J = 4.4 Гц, 1H), 2.53 (s, 3H), 2.10-1.98 (m, 1H), 1.95-1.74 (m, 3H), 1.24 (d, J = 6.0 Гц, 3H).
Стадия 4
(3S,4S)-8-(8-(2,3-Дихлорфенил)-7-метил-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин 9
Соединение 9c (50 мг, 0,13 ммоль), соединение 1i (30 мг, 0,16 ммоль) и фосфат калия (55,2 мг, 0,26 ммоль) суспендировали в 1,4-диоксане (2 мл) в атмосфере азота. Добавляли 2-дициклогексилфосфино-2',6'-диметоксидифенил (S-Phos, 5,3 мг, 0,013 ммоль) и трис(дибензилиденацетон)дипалладий (Pd2(dba)3, 5,5 мг, 0,006 ммоль). Реакционный раствор подвергали взаимодействию при 100,06°C в течение 1 часа. Реакционный раствор фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с этилацетатом, метанолом и аммиаком в качестве элюента с получением указанного в заголовке соединения 9 (12,1 мг, выход: 20,6%).
МС (ИЭР) m/z 447,1 [M+H] +
1H ЯМР (400 МГц, CDCl3) δ 8.70 (s, 1H), 7.57 (dd, J = 7.6 Гц, J = 1.6 Гц, 1H), 7.36-7.27 (m, 2H), 4.26-4.18 (m, 1H), 3.89-3.70 (m, 4H), 3.52-3.31 (m, 2H), 3.08 (br s, 1H), 2.23 (s, 3H), 2.11-2.01 (m, 1H), 1.98-1.77 (m, 3H), 1.27 (d, J = 6.4 Гц, 3H).
Пример 10
(3S,4S)-8-(8-(2-Хлор-6-фторфенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин
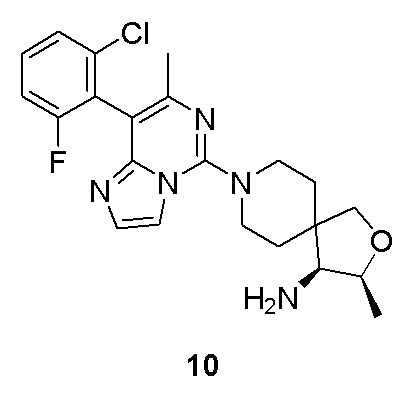
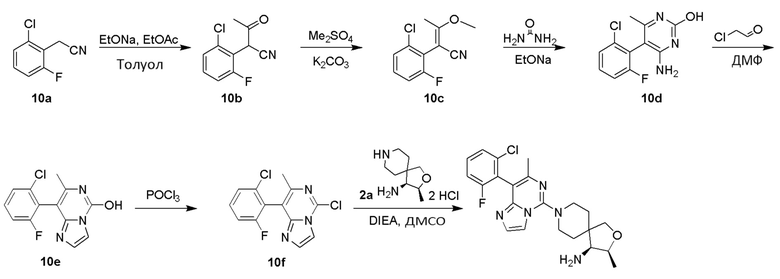
Стадия 1
2-(2-Хлор-6-фторфенил)-3-оксобутаннитрил 10b
2-(2-Хлор-6-фторфенил)ацетонитрил 10a (2,0 г, 11,8 ммоль) растворяли в 3,6 мл этилацетата с последующим добавлением этилата натрия (6,8 г, 11,8 ммоль). Реакционный раствор перемешивали при 85°C в течение 5 часов. После завершения реакции к реакционному раствору добавляли 30 мл воды и насыщенный водный раствор лимонной кислоты до доведения pH до 4-5. Реакционный раствор экстрагировали этилацетатом (3 раза по 30 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (40 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении с получением соединения 10b (2,2 г, выход: 88%) в виде желтого твердого вещества.
1H ЯМР: (400 МГц, CDCl3) δ 7.42-7.36 (m, 1H), 7.34-7.30 (m, 1H), 7.17-7.11 (m, 1H), 5.18 (s, 1H), 2.43 (s, 3H).
Стадия 2
(Z)-2-(2-Хлор-6-фторфенил)-3-метоксибут-2-еннитрил 10c
Соединение 10b (2,0 г, 9,45 ммоль) растворяли в 30 мл тетрагидрофурана в атмосфере азота с последующим добавлением карбоната калия (2,61 г, 18,9 ммоль) и диметилсульфат (1,79 мл, 18,9 ммоль). Реакционный раствор подвергали взаимодействию при комнатной температуре в течение 10 часов. После завершения реакции реакционный раствор концентрировали. Добавляли 30 мл воды и экстрагировали реакционный раствор этилацетатом (3 раза по 30 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (2 раза по 40 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с петролейным эфиром и этилацетатом в качестве элюента с получением указанного в заголовке соединения 10c (выход 2,6 г: 97%) в виде желтого масла.
МС (ИЭР) m/z = 225,8 [M+H]+
1H ЯМР: (400 МГц, MeOH-d4) δ 7.34-7.28 (m, 1H), 7.22-7.15 (m, 1H), 7.14-7.08 (m, 1H), 3.81 (s, 3H), 2.46 (s, 3H).
Стадия 3
4-Амино-5-(2-хлор-6-фторфенил)-6-метилпиримидин-2-ол 10d
К раствору соединения 10c (1,0 г, 4,4 ммоль) добавляли этилат натрия (0,90 г, 13,3 ммоль) и мочевину (0,40 г, 6,6 ммоль) в этаноле (5 мл). Реакционный раствор подвергали взаимодействию при 80°C в течение 12 часов. После завершения реакции реакционный раствор фильтровали. К фильтрату добавляли 2 M соляную кислоту до доведения pH 6 и экстрагировали реакционный раствор дихлорметаном и изопропанолом (3 раза по 20 мл, об./об. = 3:1). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с дихлорметаном и метанолом в качестве элюента с получением соединения 10d (0,42 г, выход: 37%) в виде желтого твердого вещества.
МС (ИЭР) m/z = 254,1 [M+H]+
1H ЯМР: (400 МГц, MeOH-d4) δ 7.46-7.54 (m, 2H), 7.24-7.28 (m, 1H), 1.92 (s, 3H).
Стадия 4
8-(2-Хлор-6-фторфенил)-7-метилимидазо[1,2-c]пиримидин-5-ол 10e
К раствору соединения 10d (0,32 г, 1,3 ммоль) в ДМФ (10 мл) добавляли 2-хлорацетальдегид (0,40 мл, 2,52 ммоль). Реакционный раствор подвергали взаимодействию при 80°C в течение 2 часов. После завершения реакции добавляли 50 мл воды и экстрагировали реакционный раствор этилацетатом (50 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с петролейным эфиром и этилацетатом в качестве элюента с получением указанного в заголовке соединения 10e (30 мг, выход: 8%).
МС (ИЭР) m/z 277,9 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 7.77 (d, J = 1.6 Гц, 1H), 7.58-7.47 (m, 2H), 7.39-7.32 (m, 1H), 7.26 (d, J = 1.6 Гц, 1H), 6.96 (s, 1H), 2.01 (s, 3H).
Стадия 5
5-Хлор-8-(2-Хлор-6-фторфенил)-7-метилимидазо[1,2-c]пиримидин 10f
К соединению 10e (30 мг, 0,11 ммоль) добавляли оксихлорид фосфора (1 мл) и DIEA (диизопропилэтиламин) (0,2 мл, 1,2 ммоль) . Реакционный раствор подвергали взаимодействию при 110°C в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении, добавляли дихлорметан (50 мл) и ледяную воду (100 мл) и насыщенный раствор бикарбоната натрия до доведения pH 8. Реакционный раствор экстрагировали дихлорметаном (50 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (150 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении с получением указанного в заголовке соединения 10f (24 мг, выход: 75%) в виде желтого твердого вещества.
МС (ИЭР) m/z 295,8 [M+H]+
1H ЯМР: (400 МГц, ДМСО_d6) δ 8.12 (d, J = 0.8 Гц, 1H), 7.71 (d, J = 0.8 Гц, 1H), 7.66-7.60 (m, 1H), 7.58-7.54 (m, 1H), 7.44 (t, J = 8.8 Гц, 1H), 2.25 (s, 3H).
Стадия 6
(3S,4S)-8-(8-(2-Хлор-6-фторфенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин 10
Соединение 10f (60 мг, 0,20 моль) растворяли в ДМСО (1,5 мл) с последующим добавлением соединения 2a (49 мг, 0,20 ммоль) и DIEA (0,1 мл, 0,61 ммоль). Реакционный раствор подвергали взаимодействию при 90°C в течение 30 минут. После завершения реакции добавляли воду (40 мл) и экстрагировали реакционный раствор этилацетатом (3 раза по 30 мл). Органическую фазу промывали насыщенным раствором хлорида натрия (2 раза по 50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с дихлорметаном и метанолом (содержащим 0,1% аммиак) в качестве элюента с получением указанного в заголовке соединения 10 (32 мг, выход: 36,4%).
МС (ИЭР) m/z 430,2 [M+H]+
1H ЯМР: (400МГц, MeOD_d4) δ 7.69 (d, J = 1.2 Гц, 1H), 7.52-7.46 (m, 1H), 7.45 (d, J = 1.2 Гц, 1H), 7.44-7.41 (m, 1H), 7.26-7.20 (m, 1H), 4.31-4.22 (m, 1H), 3.91 (d, J = 8.8 Гц, 1H), 3.83-3.72 (m, 3H), 3.37-3.31 (m, 1H), 3.28-3.20 (m, 1H), 3.11 (d, J = 5.2 Гц, 1H), 2.19 (s, 3H), 2.11-1.95 (m, 2H), 1.87-1.76 (m, 2H), 1.25 (d, J = 6.4 Гц, 3H).
Пример 11
(3S,4S)-8-(8-(2,3-Дихлор-6-фторфенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин
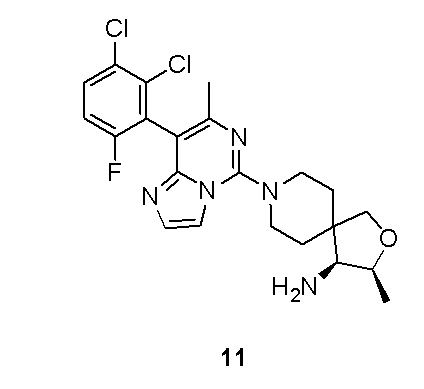
В соответствии со стадиями синтеза примера 10 соединение 10a заменяли соединением 2-(2,3-дихлор-6-фторфенил)ацетонитрил, соответственно, получили соединение примера 11.
МС (ИЭР) m/z 464,1 [M+H] +
1H ЯМР: (400 МГц, CD3OD) δ 7.78-7.65 (m, 2H), 7.46 (d, J = 1.2 Гц, 1H), 7.28 (t, J = 8.4 Гц, 1H), 4.35-4.19 (m, 1H), 3.91 (d, J = 8.8 Гц, 1H), 3.87-3.73 (m, 3H), 3.42-3.33 (m, 1H), 3.30-3.19 (m, 1H), 3.12 (d, J = 4.8 Гц, 1H), 2.20 (s, 3H), 2.11-1.97 (m, 2H), 1.92-1.76 (m, 2H), 1.25 (d, J = 6.4 Гц, 3H).
Пример 12
(3S,4S)-8-(8-(2-Бром-3-хлорфенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин
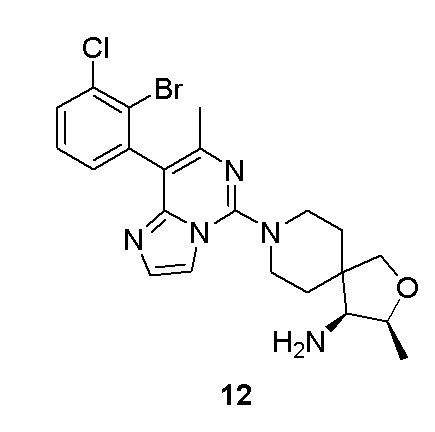
В соответствии со стадиями синтеза примера 10 соединение 10a заменяли соединением 2-(2-бром-3-дихлорфенил)ацетонитрил, соответственно, получили соединение примера 12.
МС (ИЭР) m/z 492,3 [M+H] +
1H ЯМР: (400МГц, CD3OD) δ 7.70 (d, J = 1.6 Гц, 1H), 7.63 (dd, J = 1.6 Гц, 8.0 Гц, 1H), 7.49-7.43 (m, 2H), 7.27 (dd, J = 1.2 Гц, 7.6 Гц, 1H), 4.31-4.22 (m, 1H), 3.90 (d, J = 8.8 Гц, 1H), 3.80-3.71 (m, 3H), 3.37-3.32 (m, 1H), 3.27-3.19 (m, 1H), 3.10 (d, J = 4.8 Гц, 1H), 2.17 (s, 3H), 2.07-1.96 (m, 2H), 1.88-1.76 (m, 2H), 1.25 (d, J = 7.2 Гц, 3H).
Пример 13
(3S,4S)-3-Метил-8-(7-(трифторметил)-8-((2-(трифторметил)пиридин-3-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-2-окса-8-азаспиро[4.5]декан-4-амин
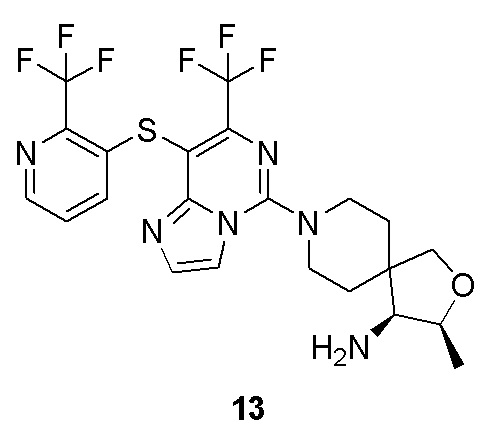
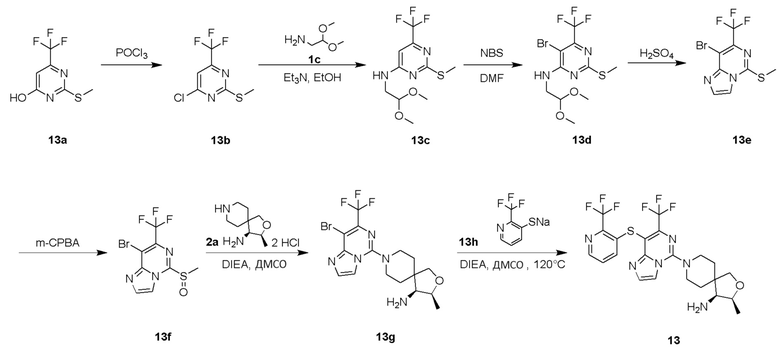
NBS - N-бромсукцинимид
Стадия 1
4-Хлор-2-(метилтио)-6-(трифторметил)пиримидин 13b
2-(Метилтио)-6-(трифторметил)пиримидин-4-ол 13a (100 мг, 0,21 ммоль) растворяли в оксихлориде фосфора (20 мл). Реакционный раствор подвергали взаимодействию в атмосфере азота при 120°C в течение 5 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении с получением указанного в заголовке соединения 13b (4 г, неочищенный продукт) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2
N-(2,2-Диметоксиэтил)-2-(метилтио)-6-(трифторметил)пиримидин-4-амин 13c
Соединение 13b (4 г, 17,50 ммоль) растворяли в N,N-диметилформамиде (10 мл) с последующим добавлением соединения 1c (2,84 мл, 26,24 ммоль) и триэтиламина (5,31 г, 52,49 ммоль). Реакционный раствор подвергали взаимодействию при 6-11°C в течение 12 часов. После завершения реакции реакционный раствор наливали в 40 мл воды. Добавляли насыщенный водный раствор гидроксида натрия для доведения pH до 10 и экстрагировали реакционный раствор этилацетатом (3 раза по 25 мл). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (2 раза по 25 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали полученный в результате неочищенный продукт хроматографией на силикагеле с этилацетатом и петролейным эфиром в качестве элюента с получением указанного в заголовке соединения 13c (1,12 г, выход за две стадии: 33%) в виде желтого твердого вещества.
МС (ИЭР) m/z 298,2 [M+H]+
1H ЯМР: (400 МГц, CD3OD) δ 6.52 (s, 1H), 4.54 (t, J = 5.2 Гц, 1H), 3.59 (d, J = 5.2 Гц, 2H), 3.40 (s, 6H), 2.50 (s, 3H).
Стадия 3
5-Бром-N-(2,2-диметоксиэтил)-2-(метилтио)-6-(трифторметил)пиримидин-4-амин 13d
Соединение 13c (1.02 г, 3.43 ммоль) растворяли в N,N-диметилформамиде (15 мл) с последующим добавлением N-бромсукцинимида (0,67 г, 3,77 ммоль). Реакционный раствор подвергали взаимодействию в атмосфере азота при 70°C в течение 1 часа. После завершения реакции реакционный раствор наливали в 100 мл воды и экстрагировали этилацетатом (3 раза по 40 мл). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (2 раза по 50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали полученный в результате неочищенный продукт хроматографией на силикагеле с этилацетатом и петролейным эфиром в качестве элюента с получением указанного в заголовке соединения 13d (1,12 г, выход: 85%) в виде желтого твердого вещества.
МС (ИЭР) m/z 377,8 [M+H+2]+
1H ЯМР: (400 МГц, CD3OD) δ 4.62 (t, J = 5.2 Гц, 1H), 3.65 (d, J = 5.6 Гц, 2H), 3.41 (s, 6H), 2.51 (s, 3H).
Стадия 4
8-Бром-5-(метилтио)-7-(трифторметил)имидазо[1,2-c]пиримидин 13e
Соединение 13d (1,05 г, 2,79 ммоль) растворяли в концентрированной серной кислоте (15 мл) и подвергали взаимодействию при 65°C в течение 12 часов. После завершения реакции реакционный раствор разбавляли 25 мл этилацетата. Реакционный раствор наливали в 100 мл ледяной воды и добавляли насыщенный раствор гидроксида натрия до доведения pH до 11-13. Водную фазу отделяли и экстрагировали этилацетатом (3 раза по 35 мл). Фазы этилацетата объединяли, промывали насыщенным водным раствором хлорида натрия (2 раза по 25 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 13e (642 мг, выход: 74%) в виде желтого твердого вещества.
МС (ИЭР) m/z 313,8 [M+H+2]+
1H ЯМР: (400 МГц, CD3OD) δ 8.01 (s, 1H), 7.87 (d, J = 1.6 Гц, 1H), 2.85 (s, 3H).
Стадия 5
8-Бром-5-(метилсульфинил<сульфенил>)-7-(трифторметил)имидазо[1,2-c]пиримидин 13f
Соединение 13e (290 мг, 0,93 ммоль) растворяли в дихлорметане (8 мл) с последующим добавлением м-хлорпероксибензойной кислоты (481 мг, 2,79 ммоль) при 0-3°C. Реакционный раствор подвергали взаимодействию при 0-3°C в течение 2 часов. После завершения реакции реакционную смесь гасили 25 мл насыщенным раствором бисульфита натрия при 0-3°C. Реакционный раствор перемешивали при 0-3°C в течение 10 минут и экстрагировали дихлорметаном (3 раза по 25 мл). Органические фазы объединяли, последовательно промывали насыщенным раствором бикарбоната натрия (2 раза по 30 мл) и насыщенным водным раствором хлорида натрия (30 мл) , высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 13f (225 мг, выход: 74%) в виде желтого твердого вещества.
МС (ИЭР) m/z 327,7 [M+H+2]+
Стадия 6
(3S,4S)-8-(8-Бром-7-(трифторметил)имидазо[1,2-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин 13g
Соединение 13f (163 мг, 0,67 ммоль) растворяли в диметилсульфоксиде (5 мл) с последующим добавлением (3S,4S)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амина дигидрохлорида 2a (163 мг, 0,67 ммоль) и N,N-диизопропилэтиламина (260 мг, 2,01 ммоль). Реакционный раствор подвергали взаимодействию при 60°C в течение 2 часов. После завершения реакции реакционный раствор разбавляли 30 мл воды, и затем экстрагировали смешанным растворителем из хлороформа и изопропанола (3/1, 3 раза по 30 мл). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали полученный в результате неочищенный продукт хроматографией на силикагеле с метанолом и дихлорметаном в качестве элюента с получением указанного в заголовке соединения 13g (115 мг, выход: 39%) в виде желтого твердого вещества.
МС (ИЭР) m/z 435,7 [M+H+2]+
1H ЯМР: (400 МГц, CD3OD) δ 7.78 (d, J = 1.2 Гц, 1H), 7.57 (d, J = 0.8 Гц, 1H), 4.23-4.17 (m, 1H), 3.84 (d, J = 9.2 Гц, 1H), 3.74-3.66 (m, 3H), 3.42-3.35 (m, 1H), 3.33-3.27 (m, 1H), 3.06 (d, J = 4.4 Гц, 1H), 2.09-2.00 (m, 1H), 1.95-1.88 (m, 1H), 1.86-1.78 (m, 2H), 1.26 (d, J = 6.8 Гц, 3H).
Стадия 7
(3S,4S)-3-метил-8-(7-(трифторметил)-8-((2-(трифторметил)пиридин-3-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-2-окса-8-азаспиро[4.5]декан-4-амин 13
Соединение 13g (100 мг, 0,23 ммоль) и соединение 13h (139 мг, 0,69 ммоль; получено способом, раскрытым в заявке на патент WO2016203405 A1) растворяли в диметилсульфоксиде (5 мл), с последующим добавлением N,N-диизопропилэтиламина (238 мг, 1,84 ммоль). Реакционный раствор подвергали взаимодействию при 120°C в течение 4 часов. После завершения реакции реакционный раствор разбавляли 30 мл этилацетата, промывали насыщенным водным раствором хлорида натрия (2 раза по 20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали препаративной высокоэффективной жидкостной хроматографией (Welch Xtimate C18 150×30 мм×5 мкм; условия: 37-67% B (A: вода (содержащая 0,05% аммиака), B: ацетонитрил); скорость потока: 25 мл/мин) с получением указанного в заголовке соединения 13 (8,9 мг, выход: 7%).
МС (ИЭР) m/z 533,1 [M+H]+
1H ЯМР: (400 МГц, CD3OD) δ 8.44-8.30 (m, 1H), 7.95 (d, J = 1.2 Гц, 1H), 7.65 (d, J = 1.2 Гц, 1H), 7.34-7.25 (m, 2H), 4.32-4.19 (m, 1H), 4.07-3.95 (m, 2H), 3.91 (d, J = 8.8 Гц, 1H), 3.76 (d, J = 8.8 Гц, 1H), 3.62-3.52 (m, 1H), 3.51-3.42 (m, 1H), 3.09 (d, J = 4.8 Гц, 1H), 2.10-1.93 (m, 2H), 1.89-1.75 (m, 2H), 1.24 (d, J = 6.4 Гц, 3H).
Пример 14
1-(8-((3-Хлор-2-фторпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-4-метилпиперидин-4-амин
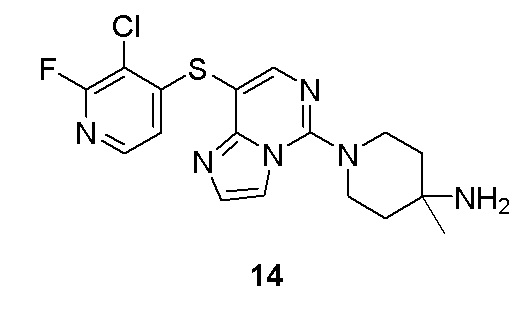
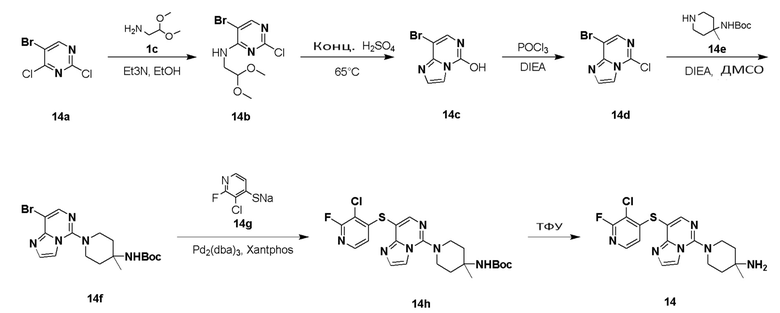
Стадия 1
5-Бром-2-Хлор-N-(2,2-диметоксиэтил)пиримидин-4-амин 14b
5-Бром-2,4-дихлорпиримидин 14a (600 мг, 2,63 ммоль) растворяли в 15 мл этанола с последующим добавлением 2,2-диметоксиэтиламина 1c (554 мг, 5,27 ммоль) и триэтиламина (0,73 мл, 5,27 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 12 часов. После завершения реакции реакционный раствор концентрировали. Добавляли 20 мл смеси льда и воды и экстрагировали реакционный раствор этилацетатом (3 раза по 10 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (5 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с петролейным эфиром и этилацетатом в качестве элюента с получением указанного в заголовке соединения 14b (710 мг, выход: 91%) в виде желтого твердого вещества.
МС (ИЭР) m/z = 295,9, 297,9 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 8.14 (s, 1H), 5.77 (br s, 1H), 4.50 (t, J = 5.2 Гц, 1H), 3.67 (t, J = 5.2 Гц, 2H), 3.45 (s, 6H).
Стадия 2
8-Бром-7-имидазо[1,2-c]пиримидин-5-ол 14c
Соединение 14b (700 мг, 2,36 ммоль) растворяли в 7 мл концентрированной серной кислоты и подвергали реакционный раствор взаимодействию при 65°C в течение 2 часов. После завершения реакции добавляли 100 мл смеси льда и воды и добавляли насыщенный раствор гидроксида натрия до pH=6. Реакционный раствор экстрагировали смешанным растворителем дихлорметана и изопропанола (по 50 мл, объемное соотношение: 3:1). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (150 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении с получением указанного в заголовке соединения 14c (330 мг, выход: 65,3%) в виде желтого твердого вещества.
МС (ИЭР) m/z = 213,7, 215,7 [M+H]+
1H ЯМР: (400 МГц, MeOH-d4) δ 7.88 (d, J = 1.6 Гц, 1H), 7.51 (s, 1H), 7.43 (d, J = 1.6 Гц, 1H).
Стадия 3
8-Бром-5-хлор-имидазо[1,2-c]пиримидин 14d
N,N-диизопропилэтиламин (6,0 мл, 36,45 ммоль) добавляли к суспензии соединения 14c (300 мг, 1,4 ммоль) и оксихлорида фосфора (19,25 г). Реакционный раствор подвергали взаимодействию при 110°C в течение 3 часов. После завершения реакции реакционный раствор концентрировали при пониженном давлении. Добавляли 50 мл насыщенного раствора бикарбоната натрия и экстрагировали реакционный раствор этилацетатом (50 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (80 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении с получением указанного в заголовке соединения 14d (230 мг, выход: 55%) в виде желтого твердого вещества.
МС (ИЭР) m/z = 231,8, 233,8 [M+H]+
Стадия 4
Трет-бутил(1-(8-бромимидазо[1,2-c]пиримидин-5-ил)-4-метилпиперидин-4-ил)карбамат 14f
Соединение 14d (220 мг, 0,95 ммоль), трет-бутил (4-метилпиперидин-4-ил)карбамат 14e (203 мг, 0,95 ммоль) и N,N-диизопропилэтиламин (0,47 мл, 2,84 ммоль) растворяли в 5 мл диметилсульфоксида. Реакционный раствор подвергали взаимодействию при 90°C в течение 1,5 часов. После завершения реакции добавляли 20 мл этилацетата и 40 мл воды и экстрагировали реакционный раствор этилацетатом (3 раза по 20 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с дихлорметаном и метанолом в качестве элюента с получением указанного в заголовке соединения 14f (220 мг, выход: 54,3%) в виде желтого твердого вещества.
МС (ИЭР) m/z 409,9 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 7.88 (s, 1H), 7.66 (d, J = 1.6 Гц, 1H), 7.49 (d, J = 1.6 Гц, 1H), 4.45 (br s, 1H), 3.57-3.49 (m, 2H), 3.37-3.27 (m, 2H), 2.26-2.20 (m, 2H), 1.85-1.77 (m, 2H), 1.45 (s, 12H).
Стадия 5
Трет-бутил(1-(8-((3-хлор-2-фторпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-4-метилпиперидин-4-ил)карбамат 14h
Соединение 14f (210 мг, 0,51 ммоль), соединение 14g (237 мг, 0,77 ммоль), N,N-диизопропилэтиламин (0,17 мл, 1,02 ммоль), трис(дибензилиденацетон)дипалладий (23,4 мг, 0,026 ммоль) и 4,5-бис(дифенилфосфино)-9,9-диметилксантин (29,6 мг, 0,051 ммоль) последовательно растворяли в 6 мл 1,4-диоксана в атмосфере азота. Реакционный раствор подвергали взаимодействию при 100°C в течение 15 часа и нагревали до 120°C в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры. Добавляли 10 мл этилацетата и 10 мл воды, и реакционный раствор экстрагировали этилацетатом (10 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с петролейным эфиром и этилацетатом в качестве элюента с получением указанного в заголовке соединения 14h (25 мг, выход: 9,9%) в виде желтого твердого вещества.
МС (ИЭР) m/z 493,1 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 8.03 (s, 1H), 7.72 (d, J = 5.2 Гц, 1H), 7.63 (d, J = 1.2 Гц, 1H), 7.52 (d, J = 1.2 Гц, 1H), 6.41 (d, J = 5.2 Гц, 1H), 4.48 (br s, 1H), 3.78-3.70 (m, 2H), 3.53-3.44 (m, 2H), 2.32-2.25 (m, 2H), 1.88-1.79 (m, 2H), 1.48-1.44 (m, 12H).
Стадия 6
1-(8-((3-Хлор-2-фторпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-4-метилпиперидин-4-амин 14
Соединение 14h (25 мг, 0,051 моль) растворяли в 3 мл дихлорметана с последующим добавлением 1 мл трифторуксусной кислоты при 0°C. Реакционный раствор подвергали взаимодействию при 25°C в течение 2 часов. Добавляли насыщенный раствор бикарбоната натрия до pH 9 и экстрагировали реакционный раствор хлороформом (2 раза по 20 мл). Органическую фазу промывали насыщенным раствором хлорида натрия (2 раза по 20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат собирали и концентрировали при пониженном давлении с получением указанного в заголовке соединения 14 (3,7 мг, выход: 18,6%).
МС (ИЭР) m/z 393,1 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 8.04 (s, 1H), 7.72 (d, J = 5.2 Гц, 1H), 7.62 (d, J = 1.6 Гц, 1H), 7.51 (d, J = 1.6 Гц, 1H), 6.43 (d, J = 5.2 Гц, 1H), 3.71-3.64 (m, 4H), 1.87-1.77 (m, 2H), 1.73-1.66 (m, 2H), 1.30 (s, 3H).
Пример 15
(R)-8-(8-((3-Хлор-2-фторпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4.5]декан-1-амин
В соответствии со стадиями синтеза примера 14 соединение 14e заменяли соединением 1g, соответственно, получили соединение примера 15.
МС (ИЭР) m/z 433,1 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 8.03 (s, 1H), 7.72 (d, J= 5.2 Гц, 1H), 7.62 (s, 1H), 7.53 (s, 1H), 6.43 (d, J = 4.4 Гц, 1H), 4.02-3.91 (m, 2H), 3.33-3.19 (m, 2H),2.94 (t, J = 7.2 Гц, 1H), 2.13-2.00 (m, 1H), 1.92-1.84 (m, 3H),1.80-1.73 (m, 1H), 1.70-1.65 (m, 1H), 1.51-1.46 (m, 2H), 1.45-1.37 (s, 2H).
Пример 16
(R)-8-(8-((2-(Трифторметил)пиридин-3-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4.5]декан-1-амин
В соответствии со стадиями синтеза примера 14 соединение 14e заменяли соединением 1g, и соединение 14g заменяли соединением 13h, соответственно, получили соединение примера 16.
МС (ИЭР) m/z 449,1 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 8.44 (s, 1H), 7.98 (s, 1H), 7.62 (s, 1H), 7.57-7.47 (m, 2H), 7.25-7.18 (m, 1H), 3.96-3.84 (m, 2H), 3.22 (q, J= 8.0Hz, 2H), 2.93 (t, J= 7.2Hz, 1H), 2.12-2.00 (m, 1H), 1.92-1.83 (m, 3H),1.79-1.74 (m, 1H), 1.66-1.61 (m, 1H), 1.54-1.48 (m, 2H), 1.44-1.38 (m, 2H).
Пример 17
(3S,4S)-3-Метил-8-(8-((2-(трифторметил)пиридин-3-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-2-окса-8-азаспиро[4.5]декан-4-амин
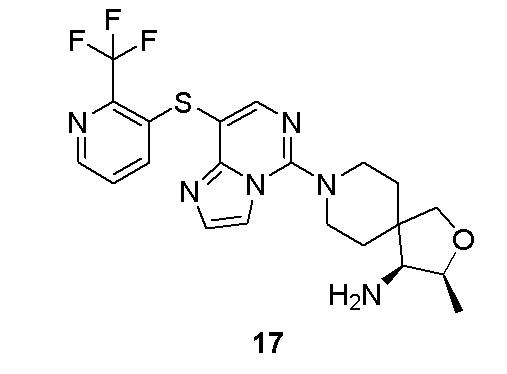
В соответствии со стадиями синтеза примера 14 соединение 14e заменяли соединением 2a, и соединение 14g заменяли соединением 13h, соответственно, получили соединение примера 17.
МС (ИЭР) m/z 479,0 [M+H] +
1H ЯМР: (400 МГц, CD3OD) δ 8.36 (d, J = 4.4 Гц, 1H), 7.75 (d, J = 1.2 Гц, 1H), 7.49 (d, J = 1.6 Гц, 1H), 7.31 (dd, J = 4.4, 8.4 Гц, 1H), 7.24 (d, J = 8.4 Гц, 1H), 4.30-4.22 (m, 1H), 3.94-3.83 (m, 3H), 3.76 (d, J = 8.8 Гц, 1H), 3.46-3.38 (m, 1H), 3.37-3.32 (m, 1H), 3.09 (d, J = 4.8 Гц, 1H), 2.53 (s, 3H), 2.07-1.95 (m, 2H), 1.86-1.76 (m, 2H), 1.24 (d, J = 6.4 Гц, 3H).
Пример 18
(3S,4S)-3-Метил-8-(8-((3-(трифторметил)пиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-2-окса-8-азаспиро[4.5]декан-4-амин
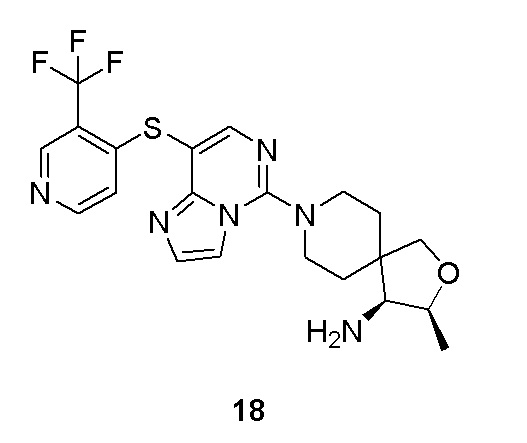
В соответствии со стадиями синтеза примера 14 соединение 13e заменяли соединением 2a, и соединение 14g заменяли соединением 3-(трифторметил)пиридин-4-тиолат натрия, соответственно, получили соединение примера 18.
МС (ИЭР) m/z 465,1 [M+H] +
1H ЯМР: (400 МГц, CD3OD) δ 8.77-8.63 (m, 1H), 8.36-8.24 (m, 1H), 8.14-8.04 (m, 1H), 7.91-7.80 (m, 1H), 7.61-7.51 (m, 1H), 6.94-6.83 (m, 1H), 4.33-4.20 (m, 1H), 4.00-3.84 (m, 3H), 3.80-3.70 (m, 1H), 3.53-3.35 (m, 2H), 3.14-3.05 (m, 1H), 2.11-1.93 (m, 2H), 1.90-1.74 (m, 2H), 1.24 (t, J = 6.0 Гц, 3H).
Пример 19
1-(8-((3-Хлор-2-фторпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-4-метилпиперидин-4-амин
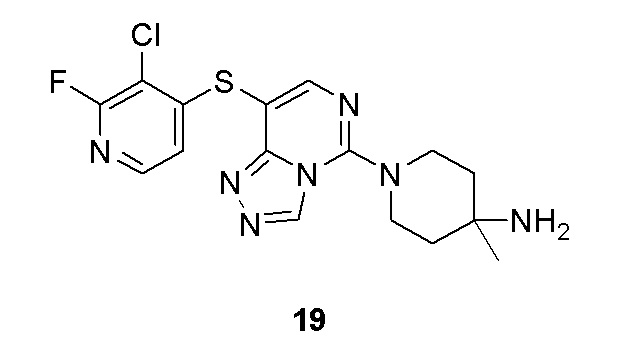
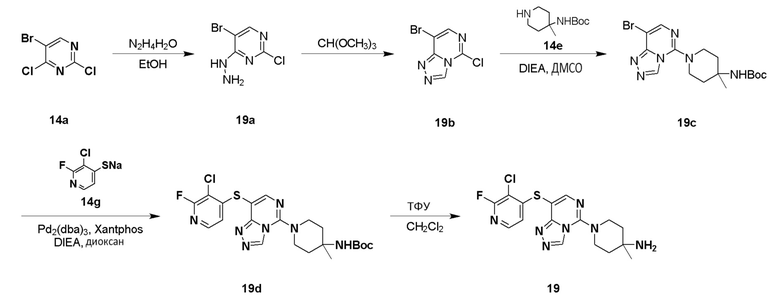
Стадия 1
5-Бром-2-Хлор-4-гидразинилпиримидин 19a
Соединение 14a (5,0 г, 22,0 ммоль) растворяли в этаноле (55 мл) с последующим добавлением гидрата гидразина (1,4 г, 26,4 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 4 часов, и затем фильтровали. Фильтрационный кек промывали н-гексаном (30 мл). Полученное в результате твердое вещество высушивали в вакууме с получением соединения 19a (3,4 г, выход: 69%) в виде желтого твердого вещества.
МС (ИЭР) m/z 222,9, 224,9 [M+H]+
1H ЯМР (400 МГц, ДМСО-d6) δ 9.03 (br s, 1H), 8.15 (s, 1H), 4.62 (br s, 2H).
Стадия 2
8-Бром-5-хлор-[1,2,4]триазоло[4,3-c]пиримидин 19b
Соединение 19a (3,2 г, 14,35 ммоль) растворяли в триметилортоформиате (32 мл) и подвергали взаимодействию реакционный раствор при 90°C в течение 2 часов. После фильтрования реакционного раствора концентрировали в вакууме, и полученный в результате неочищенный продукт очищали хроматографией на силикагеле с петролейным эфиром и этилацетатом в качестве элюента с получением указанного в заголовке соединения (840 мг, выход: 25%) в виде желтого твердого вещества.
МС (ИЭР) m/z 234,7 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 9.02 (s, 1H), 8.04 (s, 1H).
Стадия 3
Трет-бутил(1-(8-бром-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-4-метилпиперидин-4-ил)карбамат 19c
Соединение 19b (200 мг, 0,856 ммоль), соединение 14e (220 мг, 1,03 ммоль) и DIEA (221 мг, 1,71 ммоль) растворяли в ДМСО (4,0 мл). Реакционный раствор подвергали взаимодействию при 28°C в течение 2 часов. Медленно добавляли по каплям воду (150 мл) при 0°C и фильтровали реакционный раствор. Фильтрационный кек высушивали в вакууме с получением соединения 19c (400 мг, выход: 90%) в виде желтого твердого вещества.
МС (ИЭР) m/z 410,8, 412,8 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 8.76 (s, 1H), 7.82 (s, 1H), 4.46 (br s, 1H), 3.69-3.60 (m, 2H), 3.51-3.39 (m, 2H), 2.32-2.22 (m, 2H), 1.84-1.76 (m, 2H), 1.45 (s, 12H).
Стадия 4
Трет-бутил(1-(8-((3-хлор-2-фторпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-4-метилпиперидин-4-ил)карбамат 19d
Соединение 19c (200 мг, 0,486 ммоль), соединение 14g (108 мг, 0,583 ммоль) и DIEA (188 мг, 1,46 ммоль) добавляли к 1,4-диоксану (4 мл) в атмосфере азота. Добавляли трис(дибензилиденацетон)дипалладий (44 мг, 0,048 ммоль) и 4,5-бис(дифенилфосфино)-9,9-диметилксантин (56 мг, 0,097 ммоль). Реакционный раствор подвергали взаимодействию при 100°C в течение 16 часов. После завершения реакции добавляли воду (30 мл) и экстрагировали реакционный раствор этилацетатом (3 раза по 30 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (3 раза по 30 мл) и высушивали над безводным сульфатом натрия. Продукт очищали хроматографией на силикагеле с петролейным эфиром и этилацетатом в качестве элюента с получением указанного в заголовке соединения 19d (129 мг, выход: 54%) в виде желтого твердого вещества.
МС (ИЭР) m/z 494,0 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 8.82 (s, 1H), 7.99 (s, 1H), 7.75 (d, J = 5.6 Гц, 1H), 6.49 (d, J = 5.6 Гц, 1H), 4.48 (s, 1H), 3.94-3.85 (m, 2H), 3.68-3.59 (m, 2H), 2.38-2.27 (m, 2H), 1.87-1.77 (m, 2H), 1.46 (s, 12H).
Стадия 5
1-(8-((3-Хлор-2-фторпиридин-4-ил)тио)-[1,2,4]триазоло[4,3-c]пиримидин-5-ил)-4-метилпиперидин-4-амин 19
Соединение 19d (129 мг, 0,26 ммоль) растворяли в дихлорметане (3,0 мл) с последующим добавлением трифторуксусной кислоты (1,0 мл). Реакционный раствор подвергали взаимодействию при комнатной температуре в течение 1 часа. После завершения реакции добавляли 1 н. водный раствор NaOH до доведения pH до 10. Реакционный раствор экстрагировали хлороформом (3 раза по 15 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (3 раза по 15 мл), высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 19 (34,5 мг, выход: 33%).
МС (ИЭР) m/z 393,9 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 8.81 (s, 1H), 7.97 (s, 1H), 7.75 (d, J = 5.6 Гц, 1H), 6.49 (d, J = 5.6 Гц, 1H), 3.87-3.78 (m, 4H), 1.84-1.77 (m, 2H), 1.69-1.60 (m, 2H), 1.30 (s, 3H).
Пример 20
(R)-8-(7-амино-8-(2,3-Дихлорфенил)имидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4.5]декан-1-амин
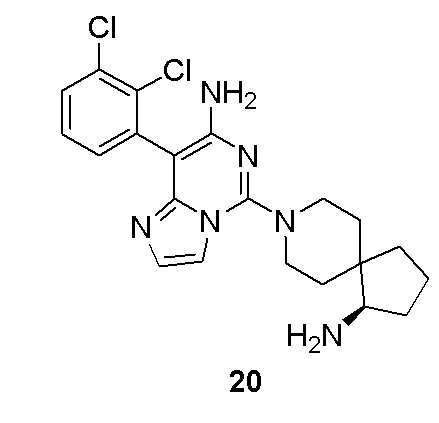
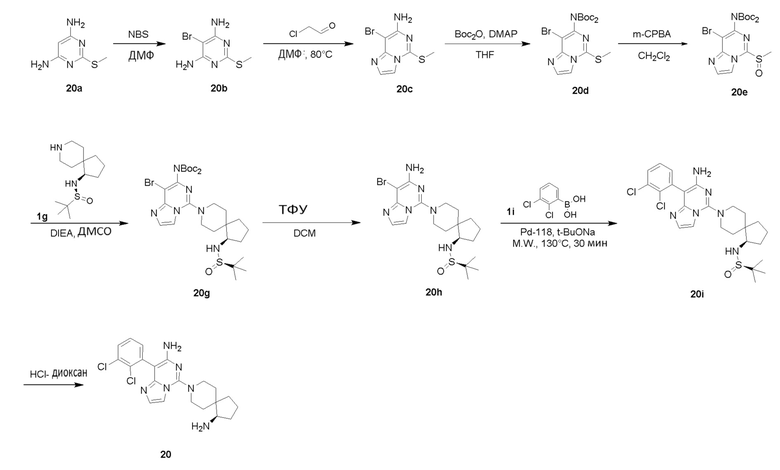
DMAP - 5-диметиламинофенол
Стадия 1
5-Бром-2-(метилтио)пиримидин-4,6-диамин 20a
Соединение 20a (10 г, 64 ммоль) растворяли в ДМФ (100 мл) с последующим добавлением N-бромсукцинимида (13,7 г, 76,8 ммоль). Реакционный раствор подвергали взаимодействию при 3-7°C в течение 12 часов. После завершения реакции добавляли 500 мл воды и 12 мл насыщенного раствора гидроксида натрия до доведения pH до 10. Реакционный раствор экстрагировали этилацетатом (5 раз по 200 мл). Органические фазы объединяли и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с метанолом и дихлорэтаном в качестве элюента с получением указанного в заголовке соединения 20b (12 г, выход: 80%) в виде желтого твердого вещества.
МС (ИЭР) m/z 236,7 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 5.12 (br s, 4H), 2.45 (s, 3H).
Стадия 2
8-Бром-5-(метилтио)имидазо[1,2-c]пиримидин-7-амин 20c
Соединение 20b (5,7 г, 24 ммоль) растворяли в ДМФ (70 мл) с последующим добавлением 2-хлорацетальдегидом (7,1 г, 36 ммоль). Реакционный раствор подвергали взаимодействию при 80°C в течение 2 часов. После завершения реакции добавляли 500 мл воды и насыщенный раствор гидроксида натрия до доведения pH до 10. Реакционный раствор экстрагировали этилацетатом (5 раз по 200 мл). Органические фазы объединяли и концентрировали при пониженном давлении. Неочищенный остаток очищали хроматографией на силикагеле с петролейным эфиром и этилацетатом в качестве элюента с получением указанного в заголовке соединения 20c (3,6 г, выход: 57%) в виде желтого твердого вещества.
МС (ИЭР) m/z 260,8 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 7.45 (d, J = 1.2 Гц, 1H), 7.35 (d, J = 1.6 Гц, 1H), 4.81 (br s, 2H), 2.71 (s, 3H).
Стадия 3
Ди-трет-бутил(8-бром-5-(метилтио)имидазо[1,2-c]пиримидин-7-ил)аминодикарбоксилат 20d
Соединение 20c (3,8 g, 15 ммоль) растворяли в ТГФ (40 мл) с последующим добавлением ди-трет-бутилдикарбоната (9,6 г, 44 ммоль) и DMAP (0,34 г, 2,9 ммоль). Реакционный раствор подвергали взаимодействию при 20°C в течение 12 часов. После завершения реакции добавляли 100 мл воды и экстрагировали реакционный раствор этилацетатом (3 раза по 70 мл). Органические фазы объединяли и концентрировали при пониженном давлении. Неочищенный остаток очищали хроматографией на силикагеле с петролейным эфиром и этилацетатом в качестве элюента с получением указанного в заголовке соединения 20d (3,2 г, выход: 48%) в виде желтого твердого вещества.
МС (ИЭР) m/z 460,9 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 7.74 (d, J = 1.6 Гц, 1H), 7.60 (d, J = 1.6 Гц, 1H), 2.75 (s, 3H), 1.45 (s, 18H).
Стадия 4
Ди-трет-бутил(8-бром-5-(метилсульфинил)имидазо[1,2-c]пиримидин-7-ил)аминодикарбоксилат 20e
Соединение 20d (0,40 г, 0,87 ммоль) растворяли в ДХМ (дихлорметан) (10 мл) с последующим добавлением м-хлорпероксибензойной кислоты (0,53 г, 2,6 ммоль). Реакционный раствор подвергали взаимодействию при 0°C в течение 1 часа. После завершения реакции добавляли 15 мл насыщенного водного раствора бисульфита натрия, и реакционный раствор перемешивали в течение 15 минут. Добавляли 30 мл воды и экстрагировали реакционный раствор дихлорметаном (3 раза по 25 мл). Органические фазы объединяли, последовательно промывали насыщенным водным раствором бикарбоната натрия (20 мл) и насыщенным водным раствором хлорида натрия (20 мл) и концентрировали при пониженном давлении с получением указанного в заголовке соединения 20e (0,4 г, выход: 97%) в виде желтого твердого вещества.
МС (ИЭР) m/z 476,7 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 8.77 (d, J = 1.2 Гц, 1H), 7.88 (d, J = 1.2 Гц, 1H), 3.18 (s, 3H), 1.45 (s, 18H).
Стадия 5
Ди-трет-бутил(8-бром-5-((R)-1-(((R)-трет-бутилсульфинил)амино)-8-азаспиро[4.5]декан-8-ил)имидазо[1,2-c]пиримидин-7-ил)аминодикарбоксилат 20g
Соединение 20e (1,9 г, 4,0 ммоль) и соединение 1g (0,58 г, 2,3 ммоль) растворяли в ДМСО (20 мл), с последующим добавлением DIEA (0,87 г, 6,8 ммоль). Реакционный раствор подвергали взаимодействию при 50°C в течение 2 часов. После завершения реакции добавляли 50 мл воды и экстрагировали реакционный раствор этилацетатом (3 раза по 50 мл). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (3 раза по 50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный в результате неочищенный остаток очищали хроматографией на C-18 с аммиаком (10 мМ) и метанолом в качестве элюента с получением указанного в заголовке соединения 20g (1,0 г, выход: 37%) в виде желтого твердого вещества.
МС (ИЭР) m/z 669,3 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 7.65 (s, 1H), 7.55 - 7.50 (m, 1H), 3.83 - 3.64 (m, 2H), 3.41 (q, J=6.8 Гц, 1H), 3.25 (d, J=6.0 Гц, 1H), 3.16 - 2.97 (m, 2H), 2.19 - 2.04 (m, 2H), 1.92 - 1.82 (m, 2H), 1.80 - 1.64 (m, 4H), 1.63 - 1.53 (m, 2H), 1.45 (s, 18H), 1.25 (s, 9H).
Стадия 6
(R)-N-((R)-8-(7-Амино-8-Бромимидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4.5]декан-1-ил)-2-метилпропан-2-сульфинамид 20h
Соединение 20g (100 мг, 0,15 ммоль) растворяли в ДХМ (3 мл) с последующим добавлением TFA (трифторуксусная кислота) (1 мл). Реакционный раствор подвергали взаимодействию при 5-8°C в течение 1 часа. После завершения реакции реакционный раствор концентрировали при пониженном давлении. Добавляли 20 мл воды и добавляли насыщенный раствор гидроксида натрия до доведения до pH 13. Реакционный раствор последовательно экстрагировали этилацетатом (3 раза по 10 мл) и дихлорметаном (3 раза по 20 мл). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 20h (37 мг, выход: 38%) в виде желтого твердого вещества.
МС (ИЭР) m/z 469,0 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 7.42 - 7.36 (m, 1H), 7.29 - 7.27 (m, 1H), 4.68 (s, 2H), 3.79 - 3.73 (m, 1H), 3.45 - 3.33 (m, 1H), 3.24 (d, J=5.2 Гц, 1H), 3.10 - 2.97 (m, 2H), 2.19 - 2.06 (m, 1H), 2.05 - 1.96 (m, 1H), 1.90 - 1.65 (m, 6H), 1.48 - 1.42 (m, 2H), 1.24 (s, 9H).
Стадия 7
(R)-N-((R)-8-(7-Амино-8-(2,3-дихлорфенил)имидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4.5]декан-1-ил)-2-метилпропан-2-сульфинамид 20i
Соединение 20h (100 мг, 0,21 ммоль) и 2,3-дихлорфенилбороновую кислоту 1i (203 мг, 1,07 ммоль) растворяли в диоксане (4 мл) в атмосфере азота с последующим добавлением Pd-118 (42 мг, 0,064 ммоль) и трет-бутилата натрия (82 мг, 0,85 ммоль). Реакционный раствор подвергали взаимодействию при 130°C в течение 30 минут. После завершения реакции реакционный раствор концентрировали при пониженном давлении и очищали полученный в результате остаток очищали хроматографией на силикагеле с метанолом и дихлорэтаном в качестве элюента с получением указанного в заголовке соединения 20i (85 мг, выход: 43%).
МС (ИЭР) m/z 535,0 [M+H]+
Стадия 8
(R)-8-(7-Амино-8-(2,3-дихлорфенил)имидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4.5]декан-1-амин 20
Соединение 20i (80 мг, 0,085 ммоль) растворяли в диоксане (3 мл) с последующим добавлением раствора (1 мл, 4 моль/л) хлорида водорода в диоксане. Реакционный раствор перемешивали при 0-7°C в течение 30 минут. После завершения реакции добавляли воду (25 мл) и промывали реакционный раствор этилацетатом (3 раза по 25 мл). Добавляли насыщенный раствор гидроксида натрия до доведения pH до 8-9 и экстрагировали водную фазу дихлорметаном (3 раза по 25 мл). Органические фазы объединяли, промывали насыщенным водным раствором хлорида натрия (30 мл), высушивали над сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате неочищенный продукт очищали препаративной высокоэффективной жидкостной хроматографией (Phenomenex Gemini-NX 150×30 мм×5 мкм; условия: 33-63% B (A: вода (содержащая 0,04% аммиак и 10 мМ бикарбонат аммония), B: ацетонитрил); скорость потока: 30 мл/мин) с получением указанного в заголовке соединения 20 (19,3 мг, выход: 53%).
МС (ИЭР) m/z 431,1 [M+H]+
1H ЯМР: (400 МГц, CD3OD) δ 7.59 (dd, J = 1.6, 8.0 Гц, 1H), 7.42-7.37 (m, 2H), 7.34 (dd, J = 1.6, 8.0 Гц, 1H), 7.18 (d, J = 2.0 Гц, 1H), 3.92-3.75 (m, 2H), 3.24-3.07 (m, 2H), 2.95 (t, I = 7.2 Гц, 1H), 2.14-2.03 (m, 1H), 1.97-1.88 (m, 2H), 1.86-1.77 (m, 2H), 1.73-1.63 (m, 2H), 1.55-1.43 (m, 3H).
Пример 21
(3S,4S)-8-(7-Амино-8-(2,3-дихлорфенил)имидазо[1,2-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин
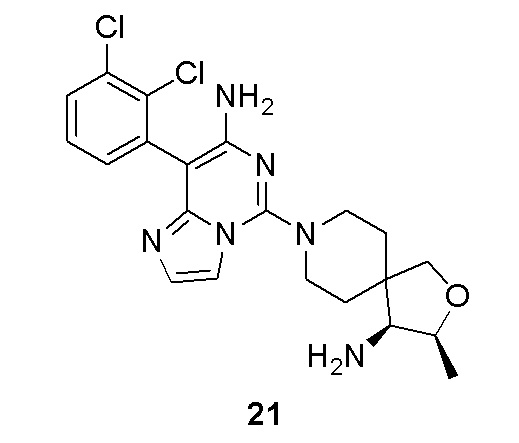
В соответствии со стадиями синтеза примера 20 соединение 1g заменяли соединением 2a, соответственно, получили соединение примера 21.
МС (ИЭР) m/z 477,2 [M+H] +
1H ЯМР: (400 МГц, CD3OD) δ 7.60 (dd, J = 2.0 Гц, 8.0 Гц, 1H), 7.43 (d, J = 2.0 Гц, 1H), 7.40 (t, J = 7.6 Гц, 1H), 7.34 (dd, J = 2.0 Гц, 7.6 Гц, 1H), 7.19 (d, J = 1.6 Гц, 1H), 4.30-4.23 (m, 1H), 3.90 (d, J = 8.8 Гц, 1H), 3.79-3.70 (m, 3H), 3.28-3.22 (m, 1H), 3.21-3.15 (m, 1H), 3.14-3.10 (m, 1H), 2.09-1.93 (m, 2H), 1.88-1.72 (m, 2H), 1.25 (d, J = 6.4 Гц, 3H).
Пример 22
(R)-8-(7-Амино-8-((3-хлор-2-фторпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4.5]декан-1-амин
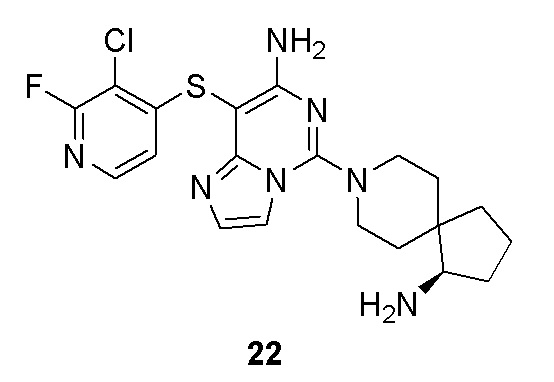
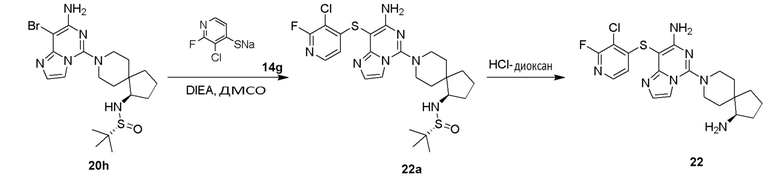
Стадия 1
(R)-N-((R)-8-(7-Амино-8-((3-хлор-2-фторпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4.5]декан-1-ил)-2-метилпропан-2-сульфинамид 22a
Соединение 20h (50 мг, 0,11 ммоль) и 3-хлор-2-фторпиридин-4-тиолата натрия 14g (49 мг, 0,27 ммоль) растворяли в диметилсульфоксиде (4,0 мл), с последующим добавлением N,N-диизопропилэтиламина (76 мг, 0,59 ммоль). Реакционный раствор подвергали взаимодействию при 90°C в течение 20 часов. После завершения реакции добавляли этилацетат (20 мл). Реакционный раствор три раза промывали 15 мл насыщенного раствора хлорида натрия, высушивали над сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали полученный в результате неочищенный продукт хроматографией на силикагеле с дихлорметаном и метанолом в качестве элюента с получением соединения 22a (36 мг, выход: 54%) в виде желтого твердого вещества.
МС (ИЭР) m/z 552,0 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 7.74 (d, J = 5.6 Гц, 1H), 7.29 (s, 1H), 7.27 (s, 1H), 6.52 (d, J = 5.2 Гц, 1H), 5.10 (br s, 2H), 4.05-3.85 (m, 2H), 3.40 (q, J = 6.8 Гц, 1H), 3.31-3.24 (m, 1H), 3.23-3.07 (m, 2H), 2.14-2.03 (m, 2H), 1.91-1.84 (m, 2H), 1.78-1.56 (m, 4H), 1.52-1.43 (m, 2H), 1.23 (s, 9H).
Стадия 2
(R)-8-(7-Амино-8-((3-хлор-2-фторпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4.5]декан-1-амин 22
Соединение 22a (33 мг, 0,052 ммоль) растворяли в диоксане (3,0 мл) с последующим добавлением соляной кислоты в метаноле (0,5 мл). Реакционный раствор подвергали взаимодействию при 6-9°C в течение 0,5 часа. После завершения реакции добавляли 25 мл воды и промывали реакционный раствор 20 мл этилацетата. К водной фазе добавляли насыщенный раствор бикарбоната натрия до доведения pH до 8-9 и экстрагировали водную фазу дихлорметаном (20 мл). Реакционный раствор три раза промывали 20 мл насыщенного водного раствора хлорида натрия, высушивали над сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате неочищенный продукт очищали препаративной высокоэффективной жидкостной хроматографией (Phenomenex Gemini-NX 150*30 мм*5 мкм; условия: 31-61% B (A: вода (содержащая 0,04% аммиак и 10 мМ бикарбонат аммония), B: ацетонитрил); скорость потока: 30 мл/мин) с получением указанного в заголовке соединения 22 (10 мг, выход: 38%).
МС (ИЭР) m/z 448,1 [M+H]+
1H ЯМР: (400 МГц, CDCl3) δ 7.75 (d, J = 5 .2 Гц, 1H), 7.33 (s, 1H), 7.29-7.27 (m, 1H), 6.54 (d, J = 5.6 Гц, 1H), 5.06 (br s, 2H), 4.06-3.83 (m, 2H), 3.28-3.12 (m, 2H), 2.94 (t, J = 7.2 Гц, 1H), 2.11-1.99 (m, 1H), 1.90-1.82 (m, 3H), 1.79-1.75 (m, 1H), 1.73-1.63 (m, 2H), 1.58-1.52 (m, 1H), 1.49-1.42 (m, 2H).
Пример 23
(R)-8-(7-Амино-8-((2-(трифторметил)пиридин-3-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-8-азаспиро[4.5]декан-1-амин
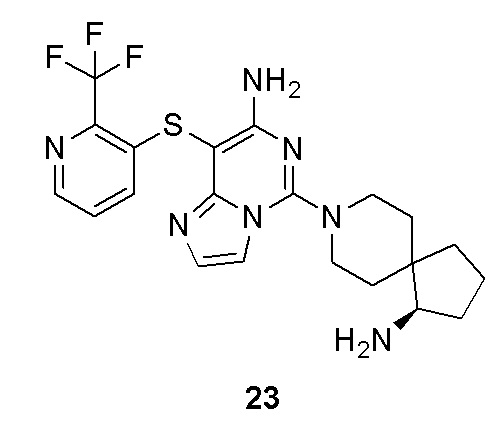
В соответствии со стадиями синтеза примера 22 соединение 14g заменяли соединением 13h, соответственно, получили соединение примера 23.
МС (ИЭР) m/z 464,2 [M+H] +
1H ЯМР: (400 МГц, CD3OD) δ 8.39-8.29 (m, 1H), 7.47 (s, 1H), 7.34 (d, J = 2.4 Гц, 2H), 7.19 (s, 1H), 3.97 (t, J = 13.2 Гц, 2H), 3.29-3.16 (m, 2H), 2.94 (t, J = 7.2 Гц, 1H), 2.15-2.04 (m, 1H), 1.97-1.87 (m, 2H), 1.86-1.75 (m, 2H), 1.75-1.61 (m, 2H), 1.59-1.48 (m, 2H), 1.47-1.39 (m, 1H).
Пример 24
(3S,4S)-8-(7-Амино-8-((2-(трифторметил)пиридин-3-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин
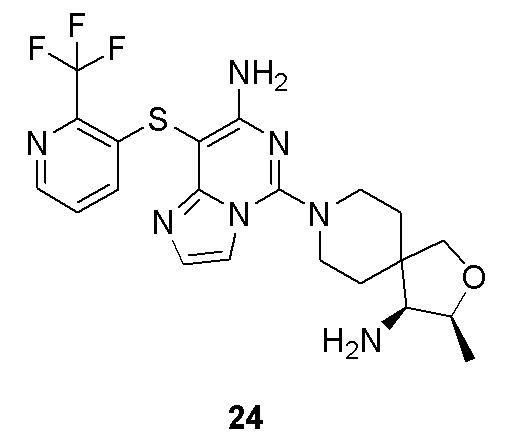
В соответствии со стадиями синтеза примера 22 соединение 1g заменяли соединением 2a, и соединение 14g заменяли соединением 13h, соответственно, получили соединение примера 24.
МС (ИЭР) m/z 480,2 [M+H] +
1H ЯМР: (400 МГц, CD3OD) δ 8.34 (t, J = 2.8 Гц, 1H), 7.48 (d, J = 1.6 Гц, 1H), 7.38-7.31 (m, 2H), 7.19 (d, J = 1.6 Гц, 1H), 4.30-4.19 (m, 1H), 3.88 (d, J = 8.8 Гц, 1H), 3.87-3.78 (m, 2H), 3.74 (d, J = 8.8 Гц, 1H), 3.41-3.33 (m, 1H), 3.30-3.23 (m, 1H), 3.07 (d, J = 5.2 Гц, 1H), 2.04-1.88 (m, 2H), 1.84-1.72 (m, 2H), 1.23 (d, J = 6.4 Гц, 3H).
Пример 25
(3S,4S)-8-(7-Амино-8-((3-хлор-2-метилпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин
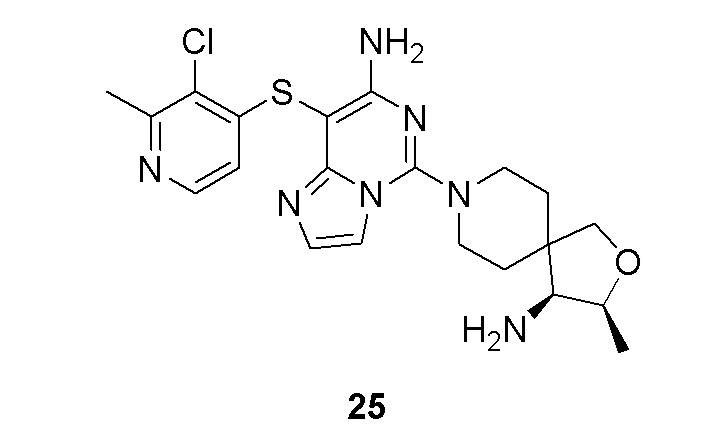
В соответствии со стадиями синтеза примера 22 соединение 1g заменяли соединением 2a, и соединение 14g заменяли соединением 3-хлор-2-метилпиридин-4-тиолат натрия, соответственно, получили соединение примера 25.
МС (ИЭР) m/z 460,1 [M+H] +
1H ЯМР: (400 МГц, CD3OD) δ 7.95 (d, J = 5.6 Гц, 1H), 7.48 (s, 1H), 7.18 (d, J = 1.6 Гц, 1H), 6.56 (d, J = 5.2 Гц, 1H), 4.28-4.22 (m, 1H), 3.91-3.81 (m, 3H), 3.78-3.73 (m, 1H), 3.41-3.33 (m, 1H), 3.27-3.11 (m, 1H), 3.10-3.06 (m, 1H), 2.03-1.94 (m, 2H), 1.83-1.74 (m, 2H), 1.24 (d, J = 6.4 Гц, 3H).
Пример 26
(3S,4S)-8-(7-Амино-8-((3-хлор-2-фторпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-3-метил-2-окса-8-азаспиро[4.5]декан-4-амин
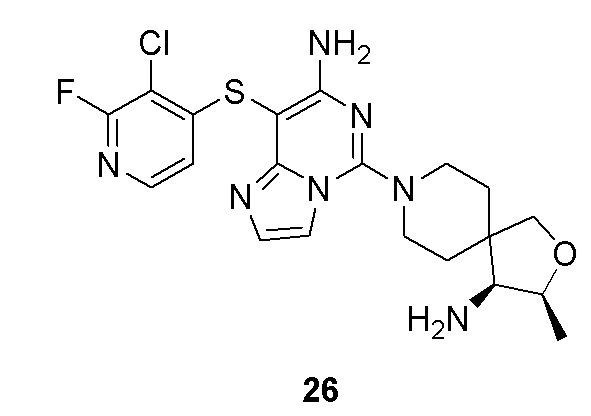
В соответствии со стадиями синтеза примера 22 соединение 1g заменяли соединением 2a, соответственно, получили соединение примера 26.
МС (ИЭР) m/z 464,2 [M+H] +
1H ЯМР: (400 МГц, CD3OD) δ 7.74 (d, J = 5.6 Гц, 1H), 7.49 (d, J = 2.0 Гц, 1H), 7.18 (d, J = 1.2 Гц, 1H), 6.60 (d, J = 5.2 Гц, 1H), 4.29-4.21 (m, 1H), 3.91-3.81 (m, 3H), 3.75 (d, J = 8.8 Гц, 1H), 3.43-3.34 (m, 1H), 3.29-3.24 (m, 1H), 3.07 (d, J = 4.8 Гц, 1H), 2.03-1.91 (m, 2H), 1.85-1.72 (m, 2H), 1.24 (d, J = 6.4 Гц, 3H).
Пример 27
(S)-1'-(8-((2-Амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амин
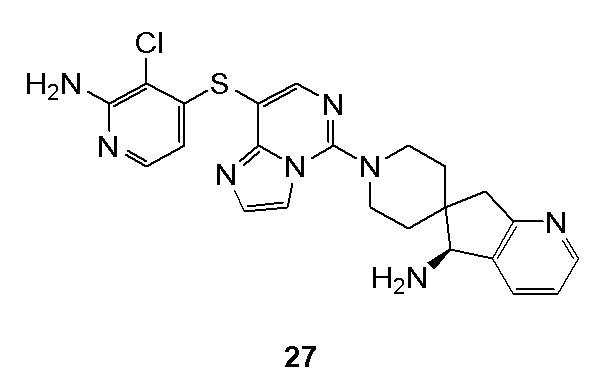
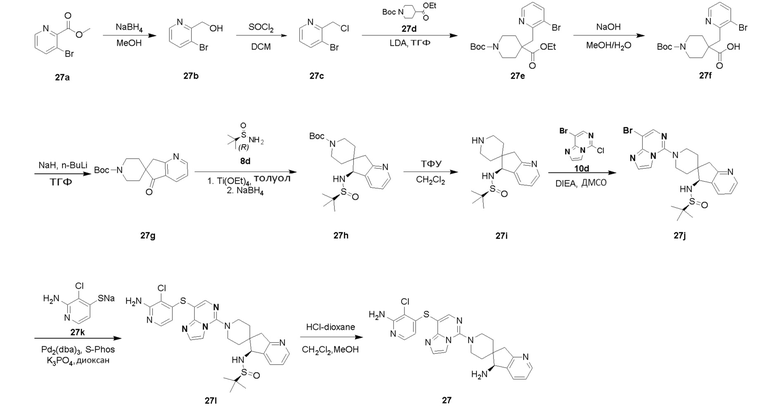
Стадия 1
(3-Бромпиридин-2-ил)метанол 27b
Соединение 27a (17,2 г, 79,6 ммоль) растворяли в метаноле (50 мл) с последующим добавлением боргидрида натрия (15,1 г, 398 ммоль) при 0°C. Реакционный раствор перемешивали при комнатной температуре в течение 12 часов. После завершения реакции добавляли насыщенный водный раствор хлорида аммония (600 мл) и экстрагировали реакционный раствор этилацетатом (3 раза по 200 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (2 раза по 200 мл), высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 27b (9,7 г, выход: 64,8%) в виде белого твердого вещества.
МС (ИЭР) m/z 187,8 [M+H] +
1H ЯМР: (400 МГц, MeOD-d4) δ = 8.52 (d, J = 4.8 Гц, 1H), 8.01 (dd, J = 1.2, 8.0 Гц, 1H), 7.26 (dd, J = 4.4, 6.4 Гц, 1H), 4.77 (s, 2H).
Стадия 2
3-Бром-2-(хлорметил)пиридин 27c
Соединение 27b (9,70 г, 51,6 ммоль) растворяли в дихлорметане (20 мл) с последующим добавлением тионилхлорида (7,48 мл, 103 ммоль) при комнатной температуре. Реакционный раствор перемешивали при комнатной температуре в течение 3 часов. После завершения реакции добавляли насыщенный водный раствор бикарбоната натрия (300 мл) и экстрагировали реакционный раствор этилацетатом (3 раза по 80 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл), высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 27c (10,3 г, выход: 96,9%) в виде розового масла.
МС (ИЭР) m/z 207,7 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ = 8.55-8.45 (m, 1H), 8.12-7.99 (m, 1H), 7.37-7.21 (m, 1H), 4.84-4.80 (m, 2H).
Стадия 3
1-(Трет-бутил)-4-этил-4-((3-бромпиридин-2-ил)метил)пиперидин-1,4-дикарбоксилат 27e
Соединение 27c (9,97 г, 38,7 ммоль) растворяли в тетрагидрофуране (80 мл) в атмосфере азота и добавляли по каплям LDA (13,5 мл, 2 M раствор в тетрагидрофуране и н-гексане) при -78°C. После завершения добавления реакционный раствор перемешивали при -78°C в течение 1 часа. Соединение 27d (8,8 г, 35,07 ммоль) добавляли по каплям при -78°C и перемешивали реакционный раствор при -78°C в течение 9 часов. После завершения реакции добавляли насыщенный водный раствор хлорида аммония (400 мл) и экстрагировали реакционный раствор этилацетатом (3 раза по 100 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (2 раза по 100 мл) и высушивали над безводным сульфатом натрия. Органическую фазу концентрировали, и полученный в результате неочищенный продукт очищали хроматографией на силикагеле с петролейным эфиром и этилацетатом в качестве элюента с получением соединения 27e (14,8 г, выход: 89,4%) в виде желтого масла.
МС (ИЭР) m/z 429,0 [M+H]+
Стадия 4
4-((3-Бромпиридин-2-ил)метил)-1-(трет-бутоксикарбонил)пиперидин-4-карбоновая кислота 27f
Соединение 27e (14,8 г, 34,6 ммоль) растворяли в метаноле (3 мл) с последующим добавлением водного раствора гидроксида натрия (13,8 г, 346 ммоль, растворенный в 40 мл воды) при 0°C. Реакционный раствор перемешивали при 80°C в течение 12 часов. После завершения реакции реакционный раствор концентрировали и добавляли к нему этилацетат (300 мл) и воду (300 мл). Добавляли насыщенный водный раствор гидроксида натрия (10 мл) до доведения pH до 12. Водную фазу отделяли и промывали этилацетатом (2 раза по 80 мл). Добавляли 2 н. раствор соляной кислоты (25 мл) до доведения pH до 3 и экстрагировали водную фазу этилацетатом (3 раза по 100 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (150 мл), высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 27f (11,4 г, выход: 82,4%) в виде белого твердого вещества.
МС (ИЭР) m/z 344,0 [M-56+H] +
Стадия 5
Трет-бутил-5-оксо-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-1'-карбоксилат 27g
К раствору соединения 27f (11,0 г, 27,6 ммоль) в тетрагидрофуране (100 мл) добавляли гидрид натрия (60% смесь с керосином, 1,32 г, 33,1 ммоль) в атмосфере азота при -15°C. Реакционный раствор перемешивали при -15°C в течение 1 часа. Реакционный раствор охлаждали до -78°C и добавляли по каплям 2,5 M раствор (16,5 мл, 41,3 ммоль) н-бутиллития в н-гексане. Реакционный раствор перемешивали при -78°C в течение 1 часа. После завершения реакции добавляли насыщенный водный раствор хлорида аммония (400 мл) при 0°C и экстрагировали реакционный раствор этилацетатом (3 раза по 100 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (2 раза по 100 мл), высушивали над безводным сульфатом натрия и концентрировали в вакууме. Полученный в результате неочищенный продукт очищали хроматографией на силикагеле с дихлорметаном и метанолом в качестве элюента с получением соединения 27g (4,60 г, выход: 55,2%) в виде белого твердого вещества.
МС (ИЭР) m/z 246,9 [M-56+H]+
1H ЯМР (400 МГц, MeOH-d4) δ = 8.82 (dd, J = 1.6, 4.8 Гц, 1H), 8.12 (dd, J = 1.6, 7.6 Гц, 1H), 7.50 (dd, J = 4.8, 7.6 Гц, 1H), 4.08 (td, J = 3.6, 13.6 Гц, 2H), 3.25 (s, 2H), 3.12 (br s, 2H), 1.88-1.77 (m, 2H), 1.51 (br s, 2H), 1.49 (s, 9H).
Стадия 6
Трет-бутил(S)-5-((S)-трет-бутилсульфиниламино)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-1'-карбоксилат 27h
Тетраэтилтитанат (9,4 мл, 44,6 ммоль) добавляли к раствору соединения 27g (4,50 г, 14,9 ммоль) в безводном толуоле (80 мл) в атмосфере азота. Реакционный раствор перемешивали при комнатной температуре в течение 10 минут. Добавляли соединение 8d (5,4 г, 44,6 ммоль), и реакционный раствор подвергали взаимодействию при 120°C в течение 5 часов. После охлаждения до 0°C добавляли боргидрид лития (1,58 г, 89,2 ммоль) и подвергали взаимодействию реакционный раствор в течение 30 минут. Реакционный раствор подогревали до комнатной температуры и перемешивали в течение 1 часа. После завершения реакции добавляли по каплям метанол (20 мл) при 0°C. Добавляли воду (100 мл) и этилацетат (100 мл) и перемешивали реакционный раствор в течение 5 минут. Суспендированное вещество отфильтровывали через диатомовую землю и промывали этилацетатом (300 мл) и водой (300 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (500 мл), высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный в результате неочищенный продукт очищали хроматографией на силикагеле с петролейным эфиром и этилацетатом в качестве элюента с получением соединения 27h (4,40 г, выход: 72,6%) в виде желтого твердого вещества.
МС (ИЭР) m/z 408,1 [M+H] +
Стадия 7
(S)-N-((S)-5,7-Дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамид 27i
Соединение 27g (4,40 г, 10,8 ммоль) растворяли в дихлорметане (15 мл) с последующим добавлением трифторуксусной кислоты (5 мл) при 0°C. Реакционный раствор перемешивали при 0°C в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного продукта и добавляли 4 М водный раствор гидроксида натрия до pH 11. Реакционный раствор экстрагировали хлороформом и изопропанолом (объемное соотношение: 3:1). Органические фазы объединяли, высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении с получением конечного продукта 27i (3,32 г, выход: 100%) в виде желтого масла.
МС (ИЭР) m/z 307,9 [M+H]+
Стадия 8
(S)-N-((S)-1'-(8-Бромимидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамид 27j
Соединение 27i (3,30 мг, 10.7 ммоль) и соединение 10d (2,50 г, 10,7 ммоль) растворяли в диметилсульфоксиде (40 мл) в атмосфере азота с последующим добавлением диизопропилэтиламина (7,7 г, 59,8 ммоль). Реакционный раствор перемешивали при 90°C в течение 2 часов. Добавляли этилацетат (50 мл) и воду (100 мл), и реакционный раствор экстрагировали этилацетатом (2 раза по 50 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (3 раза по 50 мл), высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный в результате неочищенный продукт очищали хроматографией на силикагеле с дихлорметаном и метанолом в качестве элюента с получением соединения 27j (2,96 г, выход: 54,6%).
МС (ИЭР) m/z 503,1 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ = 8.41 (d, J=4.8 Гц, 1H), 7.97 (s, 1H), 7.92 (d, J=1.5 Гц, 1H), 7.81 (d, J=7.5 Гц, 1H), 7.66 (d, J=1.5 Гц, 1H), 7.32 (dd, J=5.0, 7.5 Гц, 1H), 4.61 (br s, 2H), 3.95 - 3.83 (m, 2H), 3.30 - 3.21 (m, 2H), 2.99 (d, J=16.6 Гц, 1H), 2.40 (dt, J=4.0, 12.7 Гц, 1H), 2.14 (dt, J=3.6, 12.4 Гц, 1H), 1.82 (br d, J=13.3 Гц, 1H), 1.54 (br d, J=12.3 Гц, 1H), 1.36 (s, 9H).
Стадия 9
(S)-N-((S)-1'-(8-((2-Амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамид 27l
Соединение 27j (70 мг, 0,14 ммоль) и соединение 27k (33 мг, 0,21 ммоль, получено способом, раскрытым в заявке на патент WO2016203405 A1) растворяли в 1,4-диоксане (1 мл) в атмосфере азота с последующим добавлением диизопропилэтиламина (54 мг, 0,42 ммоль) при комнатной температуре. Добавляли трис(дибензилиденацетон)дипалладий (13 мг, 0,014 ммоль) и 2-дициклогексилфосфино-2',6'-диметоксидифенил (14 мг, 0,028 ммоль). Реакционный раствор нагревали до 110°C и перемешивали в течение 12 часов. После завершения реакции реакционный раствор фильтровали. Фильтрат концентрировали и очищали остаток обращенно-фазовой хроматографией на C-18 с водой и метанолом в качестве элюента с получением соединения 22l (45 мг, выход: 55,1%) в виде коричневого масла.
МС (ИЭР) m/z 583,1 [M+H]+
Стадия 10
(S)-1'-(8-((2-Амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амин 27
Соединение 27l (25 мг, 0,035 ммоль) растворяли в 1,4-диоксане с последующим добавлением раствора хлорида водорода в 1,4-диоксане (0,2 мл, 4 н.) при 0°C. Реакционный раствор подвергали взаимодействию при 2-7°C в течение 1 часа. После завершения реакции добавляли воду (30 мл) и экстрагировали реакционный раствор этилацетатом (2 раза по 15 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали обращенно-фазовой хроматографией на C-18 с получением соединения 27 (3,9 мг, выход: 19,0%).
МС (ИЭР) m/z 479,1 [M+H]+
1H ЯМР: (400 МГц, MeOD-d4) δ = 8.38 (d, J= 4.8 Гц, 1H), 8.06 (s, 1H), 7.90-7.84 (m, 2H), 7.57 (s, 1H), 7.50 (d, J = 5.2 Гц, 1H), 7.30 (dd, J = 5.6, 7.6 Гц, 1H), 5.90 (d, J = 6.0 Гц, 1H), 4.16 (s, 1H), 4.06 (br d, J = 13.6 Гц, 2H), 3.48-3.36 (m, 2H), 3.30-3.24(m, 1H), 3.01(br d, J = 16.4 Гц, 1H), 2.20-2.01 (m, 2H), 1.80-1.71 (m, 1H), 1.61-1.53 (m, 1H).
Пример 28
(S)-1'-(7-Амино-8-((2-(трифторметил)пиридин-3-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амин
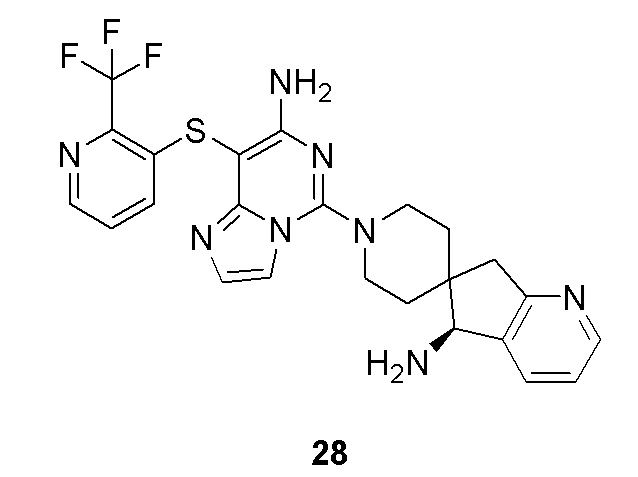
В соответствии со стадиями синтеза примера 22 соединение 1g заменяли соединением 27i, и соединение 14g заменяли соединением 13h, соответственно, получили соединение примера 28.
МС (ИЭР) m/z 513,2 [M+H] +
1H ЯМР: (400 МГц, MeOD-d4) δ 8.39-8.31 (m, 2H), 7.85 (d, J = 7.6 Гц, 1H), 7.51 (d, J = 1.6 Гц, 1H), 7.40-7.31 (m, 2H), 7.28 (dd, J = 5.2, 7.6 Гц, 1H), 7.20 (d, J = 1.2 Гц, 1H), 4.09 (s, 1H), 4.07-3.94 (m, 2H), 3.42-3.32 (m, 2H), 3.25 (d, J = 16.4 Гц, 1H), 2.95 (d, J = 16.8 Гц, 1H), 2.15-2.00 (m, 2H), 1.76-1.66 (m, 1H), 1.59-1.43 (m, 1H).
Пример 29
(S)-1'-(7-Амино-8-((2-амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
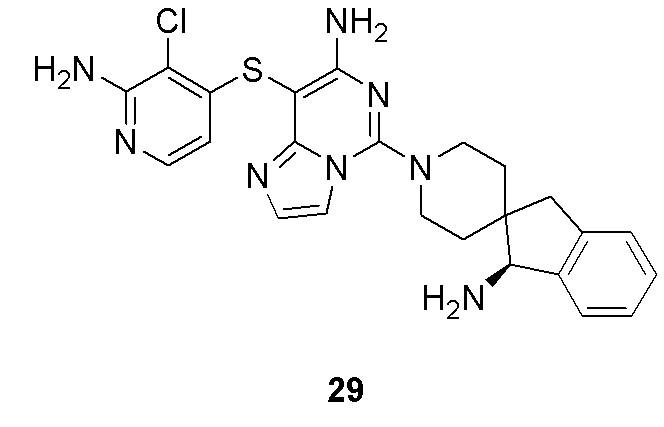
В соответствии со стадиями синтеза примера 22 соединение 1g заменяли соединением 8f, и соединение 14g заменяли соединением 27k, соответственно, получили соединение примера 29.
МС (ИЭР) m/z 493,1 [M+H] +
1H ЯМР: (400 МГц, CD3OD) δ 7.71-7.13 (m, 7H), 5.98 (br s, 1H), 4.16-3.88 (m, 3H), 3.52-3.36 (m, 2H), 3.22-3.09 (m, 1H), 2.95-2.78 (m, 1H), 2.22-1.90 (m, 2H), 1.82-1.40 (m, 2H).
Пример 30
(S)-1'-(7-Амино-8-((2-(трифторметил)пиридин-3-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
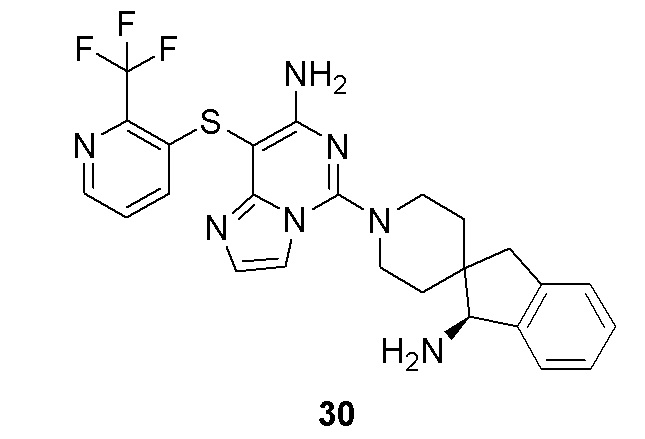
В соответствии со стадиями синтеза примера 22 соединение 1g заменяли соединением 8f, и соединение 14g заменяли соединением 13h, соответственно, получили соединение примера 30.
МС (ИЭР) m/z 512,2 [M+H] +
1H ЯМР: (400 МГц, CD3OD) δ 8.34 (dd, J = 1.6, 4.0 Гц, 1H), 7.49 (d, J = 2.0 Гц, 1H), 7.41-7.37 (m, 1H), 7.36-7.31 (m, 2H), 7.25-7.17 (m, 4H), 4.04-3.92 (m, 3H), 3.39-3.33 (m, 1H), 3.31-3.28 (m, 1H), 3.16 (d, J = 15.6 Гц, 1H), 2.81 (d, J = 15.6 Гц, 1H), 2.12-1.92 (m, 2H), 1.66 (d, J = 13.2 Гц, 1H), 1.49 (d, J = 13.2 Гц, 1H).
Пример 31
(S)-1'-(8-((2-(Трифторметил)пиридин-3-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
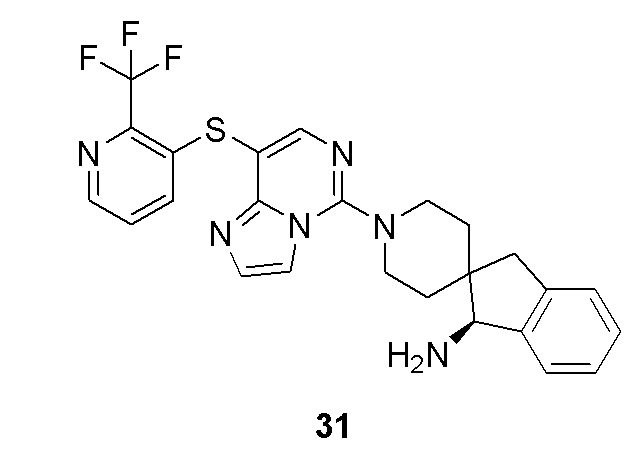
В соответствии со стадиями синтеза примера 27 соединение 27i заменяли соединением 8f, и соединение 27k заменяли соединением 13h, соответственно, получили соединение примера 31.
МС (ИЭР) m/z 497,1 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ 8.39 (s, 1H), 8.05 (d, J= 5.6 Гц, 1H), 7.86 (d, J = 5.2 Гц, 1H), 7.56 (d, J = 5.2 Гц, 1H), 7.47-7.30 (m, 3H), 7.28-7.16 (m, 3H), 4.12-3.95 (m, 3H), 3.51-3.37 (m, 2H), 3.19 (dd, J = 5.2, 15.2 Гц, 1H), 2.86 (dd, J = 5.2, 15.2 Гц, 1H), 2.16-1.96 (m, 2H),1.77-1.66 (m, 1H), 1.62-1.49 (m, 1H).
Пример 32
(S)-1'-(8-((2-Амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-1,3-дигидроспиро[инден-2,4'-пиперидин]-1-амин
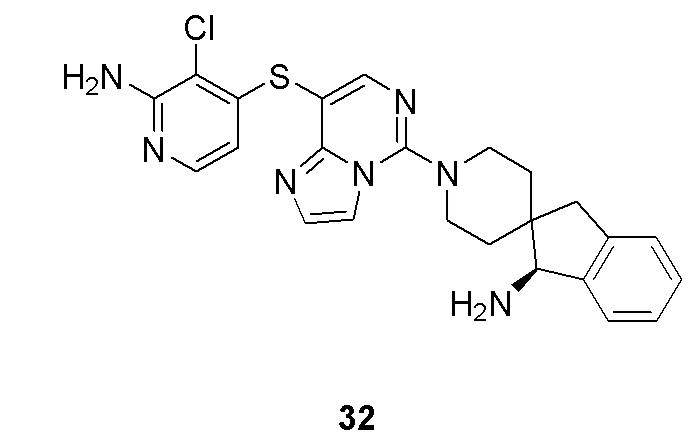
В соответствии со стадиями синтеза примера 27 соединение 27i заменяли соединением 8f, соответственно, получили соединение примера 32.
МС (ИЭР) m/z 478,1 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ 8.06 (s, 1H), 7.85 (d, J = 1.2 Гц, 1H), 7.56 (d, J = 1.6 Гц, 1H), 7.50 (d, J = 5.2 Гц, 1H), 7.44-7.40 (m, 1H), 7.28-7.23 (m, 3H), 5.89 (d, J = 5.6 Гц, 1H), 4.11 (s, 1H), 4.06-4.00 (m, 2H), 3.45-3.38 (m, 2H), 3.20 (d, J = 16.0 Гц, 1H), 2.92 (d, J = 16.0 Гц, 1H), 2.08-2.01 (m, 2H), 1.75-1.68 (m, 1H), 1.63-1.57 (m, 1H).
Пример 33
(S)-1'-(8-(2,3-Дихлорфенил)-7-метилимидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амин
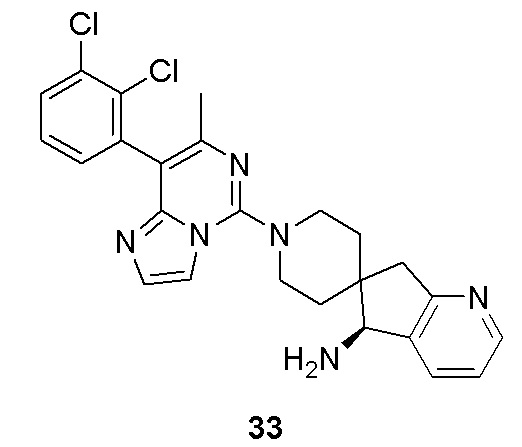
В соответствии со стадиями синтеза примера 8 соединение 8f заменяли соединением 27i, соответственно, получили соединение примера 33.
МС (ИЭР) m/z 479,1 [M+H] +
1H ЯМР: (400 МГц, CD3OD) δ 8.35 (d, J = 4.8 Гц, 1H), 7.86 (d, J = 7.6 Гц, 1H), 7.72 (s, 1H), 7.64 (d, J = 8.0 Гц, 1H), 7.47-7.39 (m, 2H), 7.35-7.25 (m, 2H), 4.11 (s, 1H), 3.91 (d, J = 11.2 Гц, 2H), 3.39-3.31 (m, 1H), 3.29-3.23 (m, 1H), 2.95 (d, J = 16.4 Гц, 1H), 2.19 (s, 3H), 2.16-2.02 (m, 2H), 1.74 (d, J = 13.2 Гц, 1H), 1.53 (d, J = 13.2 Гц, 1H).
Пример 34
(S)-1'-(8-((2-(Трифторметил)пиридин-3-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амин
В соответствии со стадиями синтеза примера 27 соединение 27k заменяли соединением 13h, соответственно, получили соединение примера 34.
МС (ИЭР) m/z 498,1 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ 8.39 (d, J = 4.8 Гц, 1H), 8.36 (d, J = 4.0 Гц, 1H), 8.05 (s, 1H), 7.88-7.83 (m, 1H), 7.56 (d, J = 1.6 Гц, 1H), 7.44 (d, J = 8.0 Гц, 1H), 7.36-7.26 (m, 2H), 4.11 (s, 1H), 4.08-4.01 (m, 2H), 3.47-3.36 (m, 2H), 3.27 (d, J = 16.4 Гц, 1H), 2.97 (d, J = 16.4 Гц, 1H), 2.18-2.02 (m, 2H), 1.75 (d, J = 12.8 Гц, 1H), 1.53 (d, J = 14.0 Гц, 1H)
Пример 35
(S)-1'-(8-((3-Хлор-2-метоксипиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амин
В соответствии со стадиями синтеза примера 27 соединение 27k заменяли соединением 3-хлор-2-метоксипиридин-4-тиолат натрия, соответственно, получили соединение примера 35.
МС (ИЭР) m/z 494,1 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ 8.38 (d, J=4.4 Гц, 1H), 8.06 (s, 1H), 7.90 - 7.83 (m, 2H), 7.67 (d, J=5.6 Гц, 1H), 7.55 (d, J=1.2 Гц, 1H), 7.30 (dd, J=5.2, 7.6 Гц, 1H), 6.21 (d, J=5.2 Гц, 1H), 4.17 (s, 1H), 4.07 (br d, J=13.2 Гц, 2H), 3.96 (s, 3H), 3.49-3.38 (m, 2H), 3.26 (d, J=16.8 Гц, 1H), 3.01 (d, J=16.8 Гц, 1H), 2.17 - 2.04 (m, 2H), 1.75 (d, J=13.6 Гц, 1H), 1.58 (d, J=13.6 Гц, 1H).
Пример 36
(S)-1'-(8-((3-Хлор-2-(метиламино)пиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амин
В соответствии со стадиями синтеза примера 27 соединение 27k заменяли соединением 3-хлор-2-(циклопропиламино)пиридин-4-тиолат натрия, соответственно, получили соединение примера 36.
МС (ИЭР) m/z 519,3 [M+H] +
1H ЯМР: (400 МГц, MeOD_d4) δ 8.36 (d, J = 4.4 Гц, 1H), 8.04 (s, 1H), 7.88-7.83 (m, 2H), 7.62 (d, J = 5.6 Гц, 1H), 7.56 (d, J = 1.6 Гц, 1H), 7.29 (dd, J = 5.2, 7.6 Гц, 1H), 5.91 (d, J = 5.6 Гц, 1H), 4.13-4.00 (m, 3H), 3.50-3.37 (m, 2H), 3.27-3.19 (m, 1H), 2.70-2.63 (m, 1H), 2.18-2.03 (m, 2H), 1.75 (br d, J = 14.0 Гц, 1H), 1.54 (br d, J = 13.2 Гц, 1H), 0.82-0.76 (m, 2H), 0.58-0.52 (m, 2H).
Пример 37
(S)-1'-(8-((3-Фтор-2-(метиламино)пиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амин
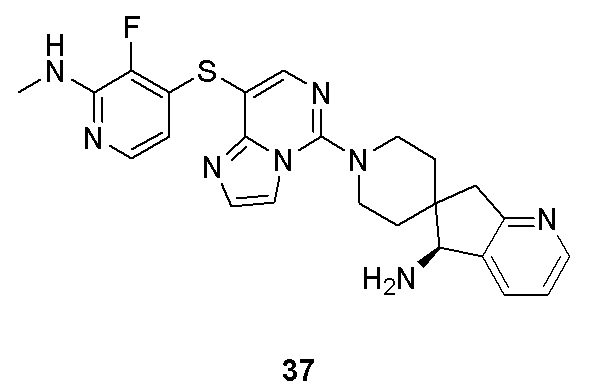
В соответствии со стадиями синтеза примера 27 соединение 27k заменяли соединением 3-фтор-2-(метиламино)пиридин-4-тиолат натрия, соответственно, получили соединение примера 37.
МС (ИЭР) m/z 477,3 [M+H] +
1H ЯМР: (400 МГц, MeOD_d4) δ 8.37 (d, J = 4.8 Гц, 1H), 8.04 (s, 1H), 7.90-7.82 (m, 2H), 7.57 (d, J = 1.6 Гц, 1H), 7.47 (d, J = 5.6 Гц, 1H), 7.30 (dd, J = 4.8, 7.2 Гц, 1H), 5.90 (t, J = 5.2 Гц, 1H), 4.14 (s, 1H), 4.03 (br d, J = 13.6 Гц, 2H), 3.47-3.35 (m, 2H), 3.27 (d, J = 16.8 Гц, 1H), 2.99 (d, J = 16.8 Гц, 1H), 2.92 (s, 3H), 2.14-2.01 (m, 2H), 1.74 (br d, J = 13.2 Гц 1H), 1.55 (br d, J = 14.0 Гц, 1H).
Пример 38
(S)-1'-(8-((2-(Метиламино)пиридин-3-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амин
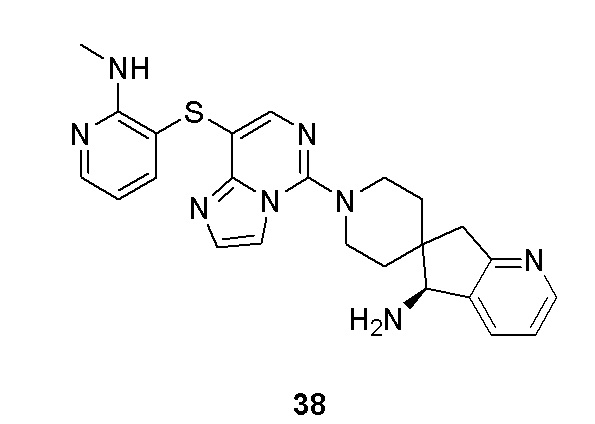
В соответствии со стадиями синтеза примера 27 соединение 27k заменяли соединением 2-(метиламино)пиридин-3-тиолат натрия, соответственно, получили соединение примера 38.
МС (ИЭР) m/z 459,2 [M+H] +
1H ЯМР: (400 МГц, MeOD_d4) δ 8.34 (d, J = 4.0 Гц, 1H), 8.04 (dd, J = 1.6, 5.2 Гц, 1H), 7.85-7.74 (m, 3H), 7.64 (d, J = 1.6 Гц, 1H), 7.54 (s, 1H), 7.27 (dd, J = 5.2, 7.6 Гц, 1H), 6.58 (dd, J = 4.8, 7.6 Гц, 1H), 4.08 (s, 1H), 3.84 (br d, J = 13.2 Гц, 2H), 3.30-3.18 (m, 3H), 2.95-2.89 (m, 4H), 2.13-1.98 (m, 2H), 1.69 (br d, J = 13.6 Гц, 1H), 1.48 (br d, J = 14.0 Гц, 1H)
Пример 39
(S)-1-(4-((5-(5-Амино-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-1'-ил)имидазо[1,2-c]пиримидин-8-ил)тио)-3-хлорпиридин-2-ил)-3-метилазетидин-3-ол
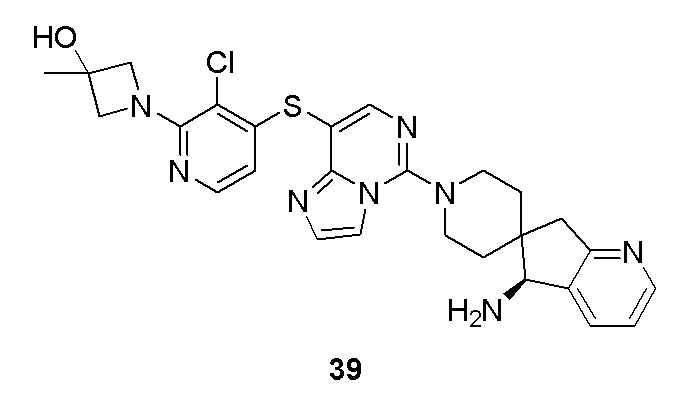
В соответствии со стадиями синтеза примера 27 соединение 27k заменяли соединением 1-(3-хлор-4-тиолпиридин-2-ил)-3-метилазетидин-3-ол, соответственно, получили соединение примера 39.
МС (ИЭР) m/z 549,1 [M+H] +
1H ЯМР: (400 МГц, MeOD_d4) δ 8.36 (d, J = 4.0 Гц, 1H), 8.05 (s, 1H), 7.88-7.82 (m, 2H), 7.62 (d, J = 5.2 Гц, 1H), 7.56 (d, J = 1.6 Гц, 1H), 7.29 (dd, J = 5.2, 7.6 Гц, 1H), 5.98 (d, J = 5.6 Гц, 1H), 4.16-4.12 (m, 2H), 4.12-4.02 (m, 5H), 3.50-3.36 (m, 2H), 3.27 (br d, J = 16.8 Гц, 1H), 2.97 (d, J = 16.4 Гц, 1H), 2.18-2.02 (m, 2H), 1.75 (br d, J = 13.2 Гц, 1H), 1.55 (s, 4H).
Пример 40
(S)-1-(4-((5-(5-Амино-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-1'-ил)имидазо[1,2-c]пиримидин-8-ил)тио)-3-хлорпиридин-2-ил)-3-метилазетидин-3-ол
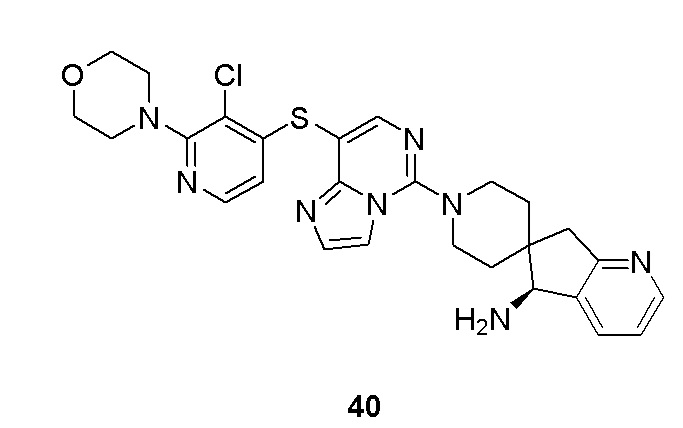
В соответствии со стадиями синтеза примера 27 соединение 27k заменяли соединением 3-хлор-2-морфолинопиридин-4-тиолат натрия, соответственно, получили соединение примера 40.
МС (ИЭР) m/z 549,2 [M+H] +
1H ЯМР: (400 МГц, MeOD_d4) δ 8.36 (d, J =5.2 Гц, 1H), 8.07 (s, 1H), 7.83-7.89 (m, 2H), 7.79 (d, J =5.2 Гц, 1H), 7.56 (s, 1H), 7.29 (dd, J =5.2, 7.6 Гц, 1H), 6.27 (d, J = 5.6 Гц, 1H), 4.02-4.14 (m, 3H), 3.80-3.88 (m, 4H), 3.32-3.50 (m, 4H), 3.24-3.29 (m, 3H), 2.97 (br d, J = 16.0 Гц, 1H), 2.04-2.18 (m, 2H), 1.75 (br d, J = 12.8 Гц, 1H), 1.54 (br d, J = 12.8 Гц, 1H).
Пример 41
(S)-1'-(8-((3-Хлор-2-(метиламино)пиридин-4-ил)тио)-7-метилимидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амин
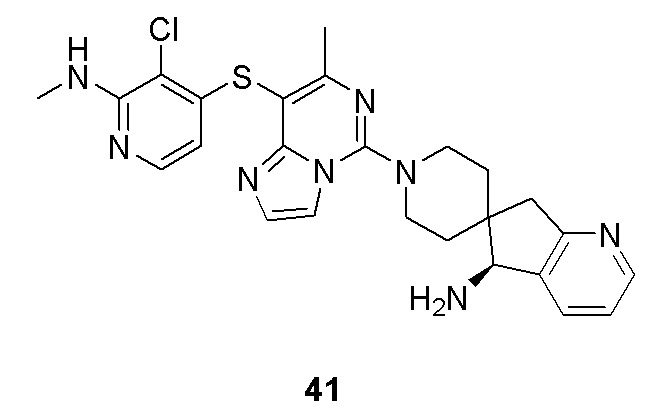
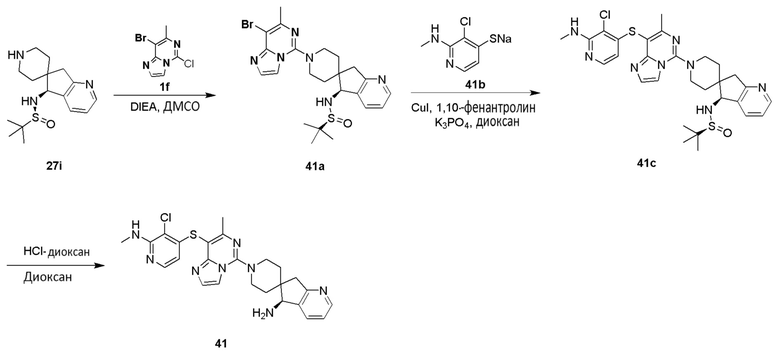
Стадия 1
(S)-N-((S)-1'-(8-Бром-7-метилимидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамид 41a
Соединение 27i (260 мг, 0,85 ммоль) и соединение 1f (271 мг, 1,10 ммоль) растворяли в диметилсульфоксиде (3 мл), с последующим добавлением диизопропилэтиламина (547 мг, 4,23 ммоль). Реакционный раствор перемешивали при 90°C в течение 1 часа. Добавляли воду (30 мл) и экстрагировали реакционный раствор этилацетатом (3 раза по 30 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (2 раза по 50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали полученный в результате неочищенный продукт хроматографией на силикагеле с метанолом и дихлорметаном в качестве элюента с получением соединения 41a (370 мг, выход: 84,5%) в виде желтого твердого вещества.
МС (ИЭР) m/z 518,8 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ 8.40 (d, J = 4.8 Гц, 1H), 7.87 (d, J = 1.2 Гц, 1H), 7.82 (d, J = 7.6 Гц, 1H), 7.63 (d, J = 2.0 Гц, 1H), 7.33 (dd, J = 5.2, 7.6 Гц, 1H), 4.72-4.66 (m, 1H), 3.95-3.84 (m, 2H), 3.31-3.19 (m, 3H), 3.01-2.94 (m, 1H), 2.58 (s, 3H), 2.37 (dt, J = 4.0, 12.8 Гц, 1H), 2.13 (dt, J = 4.0, 12.8 Гц, 1H), 1.80 (d, J = 12.8 Гц, 1H), 1.52 (d, J = 14.0 Гц, 1H), 1.34 (s, 9H).
Стадия 2
(S)-N-((S)-1'-(8-((3-Хлор-2-(метиламино)пиридин-4-ил)тио)-7-метилимидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-ил)-2-метилпропан-2-сульфинамид 41c
Соединение 41a (50 мг, 0,10 ммоль), соединение 41b (77 мг, 0,39 ммоль, полученное способом, раскрытым в заявке на патент WO2018013597 A1) и фосфат калия (41 мг, 0,19 ммоль) растворяли в 1,4-диоксане (1 мл). Реакционный раствор три раза продували азотом при перемешивании. Быстро добавляли 1,10-фенантролин (3,5 мг, 0,02 ммоль) и йодид меди (1,8 мг, 0,01 ммоль) в атмосфере азота. Реакционный раствор три раза продували азотом, нагревали до 130°C и перемешивали в течение 10 часов. Добавляли воду (50 мл) и экстрагировали реакционный раствор этилацетатом (3 раза по 40 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (2 раза по 70 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали полученный в результате неочищенный продукт хроматографией на силикагеле с дихлорметаном и метанолом в качестве элюента с получением соединения 41c (36 мг, выход: 58,5%) в виде белого твердого вещества.
МС (ИЭР) m/z 611,1 [M+H] +
Стадия 3
(S)-1'-(8-((3-Хлор-2-(метиламино)пиридин-4-ил)тио)-7-метилимидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амин 41
Соединение 41c (36 мг, 0,059 ммоль) растворяли в безводном диоксане (1 мл) с последующим добавлением по каплям раствора (1 мл, 4 н.) хлорида водорода в 1,4-диоксане при 10°C. Реакционный раствор подвергали взаимодействию при 10°C в течение 15 минут. К мутному реакционному раствору добавляли воду (30 мл), и затем экстрагировали этилацетатом (3 раза по 30 мл). К водной фазе добавляли насыщенный раствор бикарбоната натрия до доведения до pH 8 и экстрагировали водную фазу хлороформом (4 раза по 40 мл). Все органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, и полученный в результате неочищенный продукт очищали высокоэффективной жидкостной хроматографией с получением соединения 41 (2,3 мг, выход: 7,7%).
МС (ИЭР) m/z 507,3 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ = 8.35 (d, J = 4.4 Гц, 1H), 7.85 (d, J = 7.6 Гц, 1H), 7.76 (d, J = 1.6 Гц, 1H), 7.58 (d, J = 5.6 Гц, 1H), 7.48 (d, J = 1.6 Гц, 1H), 7.29 (dd, J = 5.2 Гц, 7.6 Гц, 1H), 5.75 (d, J = 6.0 Гц, 1H), 4.12-4.00 (m, 3H), 3.46-3.34 (m, 2H), 3.29-3.23 (m, 1H), 3.01-2.92 (m, 4H), 2.55 (s, 3H), 2.17-2.01 (m, 2H), 1.74 (d, J = 13.6 Гц, 1H), 1.53 (d, J = 13.6 Гц, 1H).
Пример 42
(S)-1'-(8-((2-Амино-3-хлорпиридин-4-ил)тио)-7-метилимидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амин
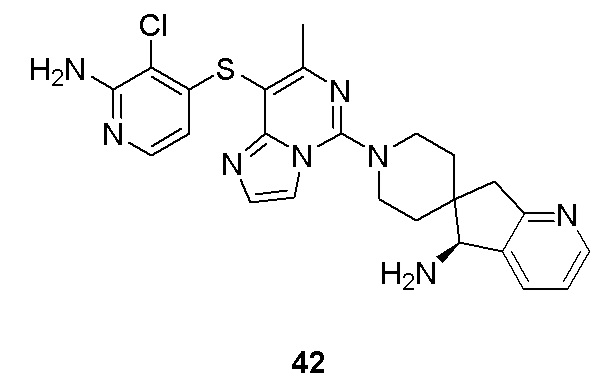
В соответствии со стадиями синтеза примера 41 соединение 41b заменяли соединением 27k, соответственно, получили соединение примера 42.
МС (ИЭР) m/z 493,1 [M+H] +
1H ЯМР: (400 МГц, CDCl3) δ 8.45 (d, J=4.4 Гц, 1H), 7.67 (d, J=7.6 Гц, 1H), 7.61 (d, J=5.2 Гц, 1H), 7.56 (d, J=1.2 Гц, 1H), 7.44 (d, J=1.2 Гц, 1H), 7.18 (dd, J=5.2, 7.6 Гц, 1H), 5.86 (d, J=5.2 Гц, 1H), 4.89 (s, 2H), 4.11 (s, 1H), 4.05 - 3.93 (m, 2H), 3.39 - 3.28 (m, 2H), 3.25 (d, J=16.4 Гц, 1H), 2.93 (d, J=16.4 Гц, 1H), 2.58 (s, 3H), 2.15 - 2.06 (m, 1H), 2.05 - 1.98 (m, 1H), 1.82 - 1.73 (m, 1H), 1.54 - 1.45 (m, 1H).
Пример 43
(S)-1'-(8-((2-Амино-3-хлорпиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амин
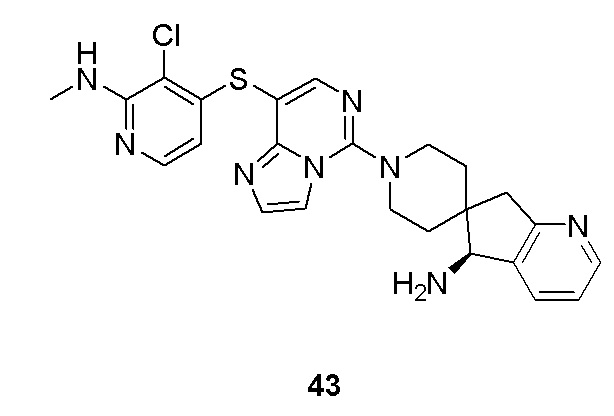
В соответствии со стадиями синтеза примера 27 соединение 27k заменяли соединением 41b, соответственно, получили соединение примера 43.
МС (ИЭР) m/z 493,1 [M+H] +
1H ЯМР (400 МГц, MeOH-d4) δ 8.36 (d, J=4.8 Гц, 1H), 8.04 (s, 1H), 7.88 - 7.81 (m, 2H), 7.60 - 7.52 (m, 2H), 7.29 (dd, J=5.2, 7.2 Гц, 1H), 5.83 (d, J=5.2 Гц, 1H), 4.12 (s, 1H), 4.05 (d, J=13.6 Гц, 2H), 3.48-3.36 (m, 2H), 3.29-3.23 (m, 1H), 3.01-2.95 (m, 1H), 2.94 (s, 3H), 2.18 - 2.03 (m, 2H), 1.74 (br d, J=13.2 Гц, 1H), 1.54 (br d, J=13.2 Гц, 1H).
Пример 44
(S)-1'-(8-((3-Хлор-2-(метиламино)пиридин-4-ил)тио)имидазо[1,2-c]пиримидин-5-ил)-2-метил-5,7-дигидроспиро[циклопента[b]пиридин-6,4'-пиперидин]-5-амин
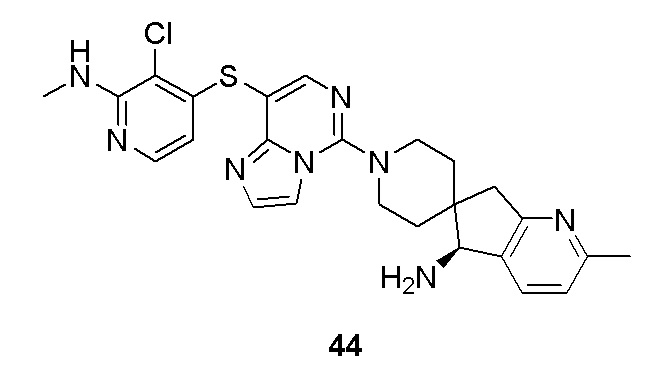
В соответствии со стадиями синтеза примера 27 соединение 27a заменяли соединением метил-3-бром-6-метилпиколинат, и соединение 27k заменяли соединение 41b, соответственно, получили соединение примера 44.
МС (ИЭР) m/z 507,2 [M+H] +
1H ЯМР: (400 МГц, CD3OD) δ 8.04 (s, 1H), 7.85 (d, J = 1.2 Гц, 1H), 7.76 (d, J = 7.6 Гц, 1H), 7.59-7.55 (m, 2H), 7.18 (d, J = 8.0 Гц, 1H), 5.83 (d, J = 5.6 Гц, 1H), 4.15 (s, 1H), 4.12-3.96 (m, 2H), 3.48-3.37 (m, 2H), 3.23 (d, J = 16.8 Гц, 1H), 2.99 (d, J = 16.8 Гц, 1H), 2.94 (s, 3H), 2.53 (s, 3H), 2.14-2.04 (m, 2H), 1.73 (br d, J = 13.2 Гц, 1H), 1.62 (br d, J = 13.2 Гц, 1H).
Биологический анализ
Настоящее изобретение будет дополнительно описано со ссылкой на следующие ниже тестовые примеры, но эти примеры не следует истолковывать как ограничивающие объем настоящего изобретения.
Тестовый пример 1. Определение активности соединений настоящего изобретения in vitro в отношении фосфатазы SHP2 дикого типа
1. Экспериментальные материалы и приборы
2. Экспериментальные методики
0,2 нМ рекомбинантно экспрессированного полноразмерного SHP2 (aa 1-593), 0,5 нМ активированного полипептида IRS1 с двойными сайтами фосфорилирования (последовательность: H2N-LN(pY)IDLDLY(dPEG8)LST(pY)ASINFQK-амид) и добавляли серию концентраций исследуемого соединения (конечные концентрации составляли 1 мкМ, 0,3 мкМ, 0,1 мкМ, 0,03 мкМ, 0,01 мкМ, 0,003 мкМ, 0,001 мкМ, 0,0003 мкМ, 0,0001 мкМ, 0,00003 мкМ) к раствору для фосфатазной реакции (60 мМ ГЭПЭС (4-(2-гидроксиэтил)-1-пиперазин этансульфоновоя кислота), pH 7,5 0,005% Brij-35, 75 мМ NaCl, 75 мМ KCl, 1 мМ ЭДТА, 5 мМ ДТТ (дитиотреитол)). Реакционный раствор перемешивали (350 об/мин) при комнатной температуре в течение 30 минут. Субстрат реакции DiFMUP с конечной концентрацией 30 мкМ и проводили реакцию в растворе при комнатной температуре в течение 30 минут. Фосфатазную реакцию останавливали добавлением 5 мкл стоп-раствора (60 мМ ГЭПЭС, pH 7,5, 0,2% додецилсульфата натрия (ДСН)). Значение флуоресценции Ex358nm/Em455 считывали на ридере для микропланшетов для считывания флуоресценции MD SpectraMax.
Значение IC50 соединения рассчитывали с помощью четырехпараметрического логит-метода. В приведенной ниже формуле x представляет собой логарифмическую форму концентрации соединения, и F(x) представляет собой величину эффекта (показатель ингибирования клеточной пролиферации в условиях данной концентрации): F(x) = ((A-D)/(1+ ((x/C) ^B))) + D. A, B, C и D представляют собой четыре параметра. Различные концентрации соответствуют различным показателям ингибирования, и на их основании строили кривую обратной зависимости, по которой рассчитывали IC50 ингибитора. IC50 соединения рассчитывали с помощью программы Primer premier 6.0.
Активность соединений настоящего изобретения in vitro в отношении SHP2 определяли с помощью описанного выше теста. В качестве лекарственных препаратов для положительного контроля были выбраны ингибиторы SHP2, обладающие активностью при пероральном применении: SHP099 и RMC4550. Структура соединения SHP099 опубликована в литературе J. Med. Chem. 2016, 59, 7773-7782, и соединение приобретали у Shanghai Haoyuan Chemexpress Co., Ltd. Структура соединения RMC4550 опубликована в литературе Nature Cell Biology, 2018, 20, 1064-1073, и соединение приобретали у Shanghai AppTec Co., Ltd.
Полученные в результате значения IC50 приведены в таблице 1.
Таблица 1. IC50 соединений по настоящему изобретению в отношении фосфатазы SHP2
минут в примерах 3 и 4
Тестовый пример 2. Определение активности соединений по настоящему изобретению in vitro в отношении мутантных фосфатаз SHP2 E67K и E69K
1. Экспериментальные материалы и приборы: описаны выше для определения активности in vitro в отношении фосфатазы дикого типа
2. Экспериментальные методики
Поскольку мутантные белки SHP2E69K и E76K сами обладают фоновой ферментативной активностью, не зависящей от активации фосфорилированного полипептида, ингибирование соединением ферментативной активности мутанта определяли в присутствии или в отсутствие активированного полипептида.
0,2 нМ рекомбинантно экспрессированного полноразмерного SHP2 (aa 1-593) с мутациями E69K и E76K (производства компании Novoprotein Scientific Inc.), 0,5 нМ активированного полипептида IRS1 с двойными сайтами фосфорилирования (последовательность: H2N-LN(pY)IDLDLY(dPEG8)LST(pY)ASINFQK-амид) (добавляли или не добавляли) и серию концентраций исследуемого соединения (конечные концентрации составляли 1 мкМ, 0,3 мкМ, 0,1 мкМ, 0,03 мкМ, 0,01 мкМ, 0,003 мкМ, 0,001 мкМ, 0,0003 мкМ, 0,0001 мкМ, 0,00003 мкМ) к раствору для фосфатазной реакции (60 мМ ГЭПЭС, pH 7,5 0,005% Brij-35, 75 мМ NaCl, 75 мМ KCl, 1 мМ ЭДТА, 5 мМ ДТТ). Реакционный раствор перемешивали (350 об./мин) при комнатной температуре в течение 30 минут. Субстрат реакции DiFMUP с конечной концентрацией 30 мкМ и проводили реакцию в растворе при комнатной температуре в течение 30 минут. Фосфатазную реакцию останавливали добавлением 5 мкл стоп-раствора (60 мМ ГЭПЭС, pH 7,5, 0,2% додецилсульфата натрия (ДСН)). Значение флуоресценции Ex358nm/Em455 считывали на ридере для микропланшетов для считывания флуоресценции MD SpectraMax. Значение IC50 соединения при ингибировании ферментативной активности мутантов рассчитывали с помощью непараметрического логит-метода со ссылкой на тестовый пример 1.
Таблица 2. IC50 соединений по настоящему изобретению на мутантных фосфатазах SHP2 E67K и E69K
активированный полипептид добавляли
IC50 (нМ)
активированный полипептид не добавляли
IC50 (нМ)
активированный полипептид добавляли
IC50 (нМ)
активированный полипептид не добавляли IC50 (нМ)
Тестовый пример 3. Определение p-ERK в клетках KYSE-520
1. Экспериментальные материалы и приборы
2. Экспериментальные методики
Клетки KYSE-520 (Nanjing Cobioer Biosciences CO., Ltd.) в логарифмической фазе роста инокулировали (30000 клеток/лунка) в среде 1640, содержащей 10% фетальную бычью сыворотку (ФБС), оставляли на ночь в 96-луночном планшете для прикрепления (5% CO2, 37°C). Добавляли серию концентраций исследуемого соединения (10 мкМ, 3 мкМ, 1 мкМ, 0,3 мкМ, 0,1 мкМ, 0,03 мкМ, 0,01 мкМ, 0,003 мкМ, 0,001 мкМ) и проводили реакцию в растворе при 37°C, 5% CO2 в течение 2 часов. Рост культуры останавливали путем лизиса клеток. Уровень фосфорилированной ERK в клетках KYSE-520 определяли способом, основанным на однородной времяпролетной флуоресценции HTRF (Cisbio, 64ERKPEG). Значения флуоресценции (Ex337 нм/Em625/665 нм) считывали на совместимом ридере HTRF (MD SpectraMax). Значение IC50 соединений по ингибированию фосфорилирования внутриклеточной фосфорилированной ERK рассчитывали с помощью четырехпараметрического логит-метода. В приведенной ниже формуле x представляет собой логарифмическую форму концентрации соединения, и F(x) представляет собой величину эффекта (показатель ингибирования клеточной пролиферации в условиях данной концентрации): F(x) = ((A-D)/(1+ ((x/C) ^B))) + D. A, B, C и D представляют собой четыре параметра. Различные концентрации соответствуют дифференциальным показателям ингибирования, на основании которых строили кривую обратной зависимости и рассчитывали IC50 ингибитора по этой кривой. IC50 соединения рассчитывали с помощью программы Primer premier 6.0.
Таблица 3. IC50 соединения по настоящему изобретению на p-ERK в клетках KYSE-520
в примерах 3 и 4
Тестовый пример 4. Эксперимент по пролиферации клеток KYSE-520
1. Экспериментальные материалы и приборы
2. Экспериментальные методики
Клетки KYSE-520 в логарифмической фазе роста прикрепляли (600 клеток/лунка) в среде 1640, содержащей 10% ФБС на 96-луночном планшете в течение ночи, и затем обрабатывали серией концентраций исследуемого соединения (10 мкМ, 3 мкМ, 1 мкМ, 0,3 мкМ, 0,1 мкМ, 0,03 мкМ, 0,01 мкМ, 0,003 мкМ, 0,001 мкМ). Планшет с обработанными клетками инкубировали при 37°C, 5% CO2 в течение 7 дней. Для определения количества жизнеспособных клеток использовали CellTiter-Glo (Promega, G7573). В каждую лунку добавляли по100 мкл детектирующего реагента и инкубировали планшет при комнатной температуре в течение 10 минут. Затем измеряли сигнал флуоресценции в каждой лунке с помощью ридера Envision (Perkin Elmer). Значение IC50 KYSE-520 рассчитывали с помощью четырехпараметрического логит-метода. В приведенной ниже формуле x представляет собой логарифмическую форму концентрации соединения, и F(x) представляет собой величину эффекта (показатель ингибирования клеточной пролиферации в условиях данной концентрации): F(x) = ((A-D)/(1+ ((x/C) ^B))) + D. A, B, C и D представляют собой четыре параметра. Дополнительно значение IC50 рассчитывали как концентрацию соединения, требующуюся для 50% ингибирования пролиферации на кривой наилучшей аппроксимации в программе Primer premier 6.0.
Таблица 4. IC50 соединения по настоящему изобретению для пролиферации клеток KYSE-520
Тестовый пример 5. Эксперимент по ингибированию тока hERG
1. Экспериментальные материалы и приборы
2. Экспериментальные методики
В этом эксперименте выполняли регистрацию цельноклеточного тока с помощью ручной системы пэтч-клэмп (систему амплификации сигнала HEKA EPC-10 и цифрового преобразования приобретали в HEKA Electronic, Германия). Округлое покровное стекло, на котором были выращены клетки CHO hERG (предоставленные Sophion Bioscience Inc., Дания, номе поколения клеток P21), помещали в щель электрофизиологической регистрации под инвертированным микроскопом. Щель для регистрации непрерывно перфузировали внеклеточной жидкостью (приблизительно 1 мл в минуту). В эксперименте использовали общепринятый метод регистрации цельноклеточного тока пэтч-клэмп. Эксперименты выполняли при нормальной комнатной температуре (~25°C). Клетки подвергали режиму фиксации потенциала при исходном потенциале -80 мВ. Для активации калиевого канала hERG напряжение пэтч-клэмп клеток деполяризовали до +20 мВ, и через 5 секунд до -50 мВ, чтобы устранить инактивацию и создать следовые токи. Максимум следового тика использовали в качестве значения тока hERG. Если калиевый ток hERG, зарегистрированный на вышеописанных стадиях, достигал равновесия, при непрерывной перфузии внеклеточной жидкости в щели для регистрации, при перфузии можно было добавлять исследуемый препарат до тех пор, пока ингибирующее действие препарата на ток hERG не достигал равновесного состояния. Как правило, перекрывание трех самых последних последовательных линий регистрации тока использовали в качестве критерия для определения стабильности состояния. После достижения равновесного состояния щель для регистрации перфузировали внеклеточной жидкостью до тех пор, пока ток hERG не восстанавливался до значения, предшествующего добавлению препарата. Данные теста анализировали с помощью программы HEKA Patchmaster (V2x73.2), Microsoft Excel, и программного обеспечения анализа данных, предложенного в Graphpad Prism 5.0.
Таблица 5. IC50 соединения по настоящему изобретению для пролиферации клеток CHO hERG
Оценка фармакокинетики
Тестовый пример 6. Фармакокинетический анализ соединения по настоящему изобретению
1. Аннотация
В качестве испытуемых животных использовали крыс. Концентрацию лекарственного средства в плазме крови в различные моменты времени определяли методом жидкостной хроматографии с тандемной масс-спектрометрией (ЖХ/МС/МС) после внутривенного введения или внутрижелудочного введения крысам соединения по настоящему изобретению. У крыс изучали и оценивали фармакокинетические свойства соединения по настоящему изобретению.
2. Протокол испытания
2.1 Исследуемые соединения
Атропизомер с RT 1,495 минут в примерах 3 и 4, соединения примера 17, примера 41 и примера 43.
2.2 Испытуемые животные
Здоровые самцы крыс линии SD (возраст 6-8 недель), 3 крысы на группу.
2.3 Приготовление исследуемых соединений
Внутривенное введение: Готовили навеску некоторого количества исследуемого соединения, к которой добавляли 10% N,N-диметилацетамида по объему, 33% триэтиленгликоля по объему, 57% воды по объему с получением 1 мг/мл бесцветного, чистого и прозрачного раствора;
Внутрижелудочное введение: Готовили навеску некоторого количества исследуемого соединения, к которой добавляли 0,5% гипромеллозы по массе, 0,1% Твин 80 по объему и 99,4% воды по объему с получением 1 мг/мл белой суспензии.
2.4 Введение
После ночного голодания крысам SD вводили исследуемое соединение в дозе 1 мг/кг внутривенно или в дозе 5 мг/кг внутрижелудочно.
3. Способ
Крыса v внутривенно вводили соединение по настоящему изобретению. Брали 0,2 мл крови из яремной вены через 0,083, 0,25, 0,5, 1, 2, 4, 8 и 24 часов после введения. Образцы помещали в пробирки, содержащие ЭДТА-K2, и центрифугировали при 4000 об/мин и 4°C в течение 5 минут для отделения плазмы крови. Образцы плазмы крови хранили при -75°C.
Или крысам вводили соединение по настоящему изобретению внутрижелудочно. Брали 0,2 мл крови из яремной вены через 0,25, 0,5, 1, 2, 4, 8 и 24 часа после введения. Образцы помещали в пробирки, содержащие ЭДТА-K2, и центрифугировали при 4000 об./мин и 4°C в течение 5 минут для отделения плазмы крови. Образцы плазмы крови хранили при -75°C.
Определяли содержание исследуемого соединения в образцах плазмы крови крыс после внутрижелудочного введения исследуемого соединения в различных концентрациях: в каждый момент времени после введения брали по 50 мкл плазмы крови и добавляли 200 мкл раствора (50 нг/мл) дексаметазона в ацетонитриле в качестве внутреннего стандарта. Плазму крови перемешивали на вихревой мешалке в течение 30 секунд и центрифугировали при 4700 об./мин и 4°C в течение 15 минут. Из образцов плазмы крови отбирали супернатант и разводили в три раза добавлением воды. Для анализа ЖХ/МС/МС использовали 2,0 мкл супернатанта.
4. Результаты определения фармакокинетических показателей
Фармакокинетические показатели соединений по настоящему изобретению показаны ниже:
(нг/мл)
(ч)
(мл/мин/кг)
(мл/кг)
(%)
1 мг/кг
5 мг/кг
1 мг/кг
5 мг/кг
1 мг/кг
5 мг/кг
1 мг/кг
5 мг/кг
Вывод: соединения по настоящему изобретению хорошо всасываются и обладают фармакокинетическим преимуществом.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ПРОТЕИНТИРОЗИНФОСФАТАЗЫ | 2020 |
|
RU2799449C2 |
| ИНГИБИТОРЫ МУТАЦИИ HER2 | 2022 |
|
RU2834124C2 |
| ПРОИЗВОДНЫЕ N2-(2-ФЕНИЛ)-ПИРИДО[3,4-d]ПИРИМИДИН-2,8-ДИАМИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ MPS1 ИНГИБИТОРА | 2015 |
|
RU2693460C2 |
| СОЕДИНЕНИЯ СО СПИРО- И АРОМАТИЧЕСКИМИ КОЛЬЦАМИ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2781100C1 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ ПИРИМИДИН-4(3H)-ОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2839367C1 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| СОЕДИНЕНИЯ ДЛЯ ИНГИБИРОВАНИЯ КИНАЗЫ EGFR, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2020 |
|
RU2832925C2 |
| ЗАМЕЩЕННЫЕ 1,2-ДИГИДРО-3Н-ПИРАЗОЛО[3,4-D]ПИРИМИДИН-3-ОНЫ | 2019 |
|
RU2812726C2 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АЛЛОСТЕРИЧЕСКИХ ИНГИБИТОРОВ SHP2 | 2018 |
|
RU2776846C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКОГО ЦИАНОЕНОНА КАК МОДУЛЯТОРЫ KEAP1 | 2021 |
|
RU2822828C1 |
Изобретение относится к пиримидину и производному пятичленного азотсодержащего гетероцикла общей формулы (I), способу его получения, содержащей производное фармацевтической композиции, его применению в качестве ингибитора SHP2 для применения в профилактике и/или лечении опухоли или рака. 6 н. и 6 з. п. ф-лы, 50 пр., 5 табл.
1. Соединение формулы (I)
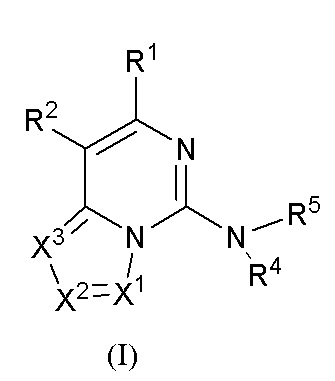
или его фармацевтически приемлемая соль,
где:
R1 выбран из группы, состоящей из атома водорода, атома дейтерия, C1-6 алкила и амино;
R2 представляет собой 
Y1 выбран из группы, состоящей из –S–, –S(O)2–, –S(O)– и связи;
кольцо A выбрано из группы, состоящей из бензольного и пиридинового колец;
каждый R3 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, галоген-C1-6 алкила, C1-6 алкила, C1-6 алкокси, галоген-C1-6 алкокси и -NRaRb;
n выбрано из группы, состоящей из 0, 1, 2, 3, 4 и 5;
где X1 и X2 оба представляют собой CRc, и X3 представляет собой N, и Rc представляет собой атом водорода;
R4 и R5 вместе с атомом азота, к которому они присоединены, образуют структуру
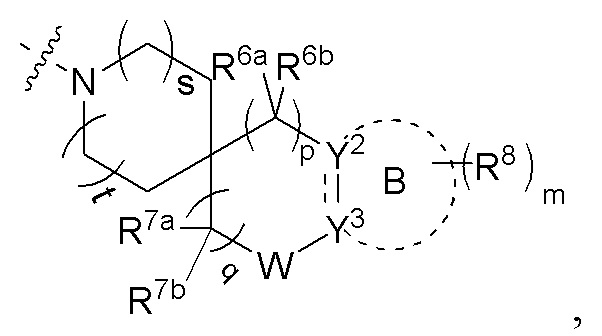
где s и t, каждый независимо, выбраны из группы, состоящей из 0 и 1;
R6a и R6b, каждый независимо, выбраны из группы, состоящей из атома водорода, атома дейтерия, С1-6 алкила и С1-6 алкокси;
p выбрано из группы, состоящей из 0, 1, 2, 3 и 4;
R7a и R7b, каждый независимо, выбраны из группы, состоящей из атома водорода, атома дейтерия, амино и С1-6 алкила;
q выбрано из группы, состоящей из 0, 1, 2, 3 и 4;
W отсутствует;
кольцо B отсутствует или представляет собой 3–10-членное кольцо;
 представляет собой простую связь или двойную связь;
представляет собой простую связь или двойную связь;
когда кольцо B отсутствует, тогда Y2 представляет собой CR2aR2b, NR2a или O, Y3 представляет собой CR3aR3b, NR3a или O;
когда кольцо B представляет собой 3–10-членное кольцо, тогда
3) Y2 представляет собой CR2a или N, Y3 представляет собой CR3a или N, представляет собой простую связь; или
4) Y2 представляет собой C, и Y3 представляет собой C, представляет собой двойную связь;
R2a, R2b, R3a и R3b, каждый независимо, выбраны из группы, состоящей из атома водорода, атома дейтерия и C1-6 алкила;
Ra и Rb, каждый независимо, выбраны из группы, состоящей из атома водорода, атома дейтерия и C1-6 алкила;
m выбрано из группы, состоящей из 0, 1, 2, 3 и 4; и
каждый R8 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, амино, гидрокси, циано, нитро, карбокси, C1-6 алкила и C1-6 алкокси.
2. Соединение по п. 1, где R4 и R5 вместе с атомом азота, к которому они присоединены, образуют структуру
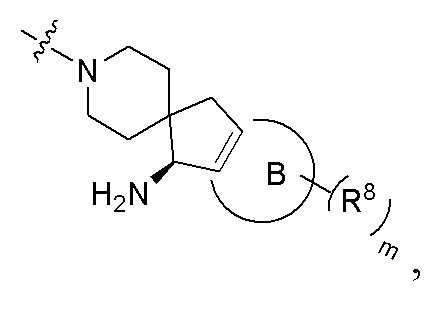
где:
кольцо B выбрано из группы, состоящей из бензольного кольца и пиридинового кольца;
каждый R8 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, циано, C1-6 алкила и C1-6 алкокси; и
m выбрано из группы, состоящей из 0, 1, 2, 3 и 4.
3. Соединение по п. 1, где
Y1 представляет собой –S– или связь;
p равно 1 или 2;
q равно 1 или 2;
W отсутствует;
кольцо B отсутствует, Y2 представляет собой CR2aR2b или O, Y3 представляет собой CR3aR3b; и
R2a, R2b, R3a и R3b, каждый независимо, выбраны из группы, состоящей из атома водорода, атома дейтерия и C1-6 алкила.
4. Соединение по п. 1, где
Y1 представляет собой –S– или связь;
p равно 1 или 2;
q равно 1 или 2;
кольцо B выбрано из группы, состоящей из фенила, 5-членного гетероарила и 6-членного гетероарила, где указанный гетероарил содержит от 1 до 2 гетероатомов, выбранных из группы, состоящей из O, S и N;
Y2 представляет собой C, и Y3 представляет собой C, представляет собой двойную связь.
5. Соединение по п. 1, где R4 и R5 вместе с атомом азота, к которому они присоединены, образуют структуру
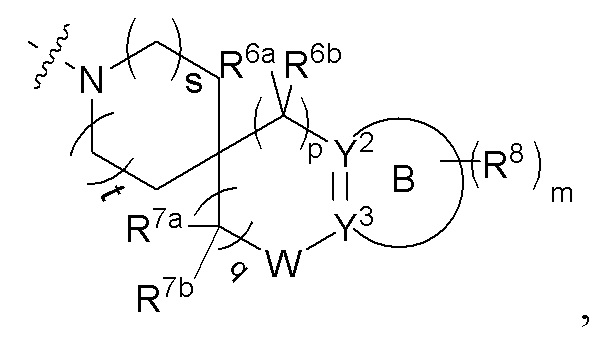
R1 выбран из группы, состоящей из атома водорода и C1-6 алкила;
Y1 представляет собой –S– или связь;
p равно 1;
q равно 1;
кольцо B выбрано из группы, состоящей из бензольного кольца и пиридинового кольца;
Y2 представляет собой C, и Y3 представляет собой C;
кольцо А, R3, n, m, s, t, W, X1, X2, X3, Ra, Rb, R6a, R6b, R7a, R7b и R8 являются такими, как определено в п. 1.
6. Соединение по п. 1, представляющее собой соединение формулы (II):
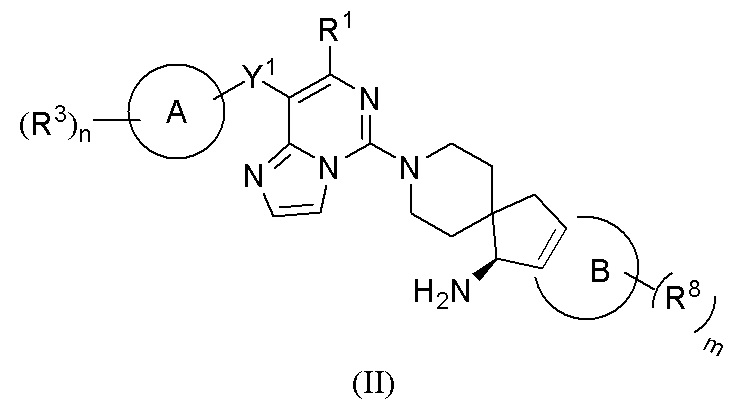
или его фармацевтически приемлемая соль,
где:
R1 представляет собой C1-6 алкил;
Y1 представляет собой –S–;
кольцо A представляет собой пиридил;
каждый R3 независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена и –NRaRb;
Ra и Rb, каждый независимо, выбраны из группы, состоящей из водорода, атома дейтерия и C1-6 алкила;
кольцо B представляет собой пиридиновое кольцо;
m представляет собой 0; и
n выбрано из группы, состоящей из 1, 2, 3 и 4.
7. Соединение, выбранное из группы, состоящей из
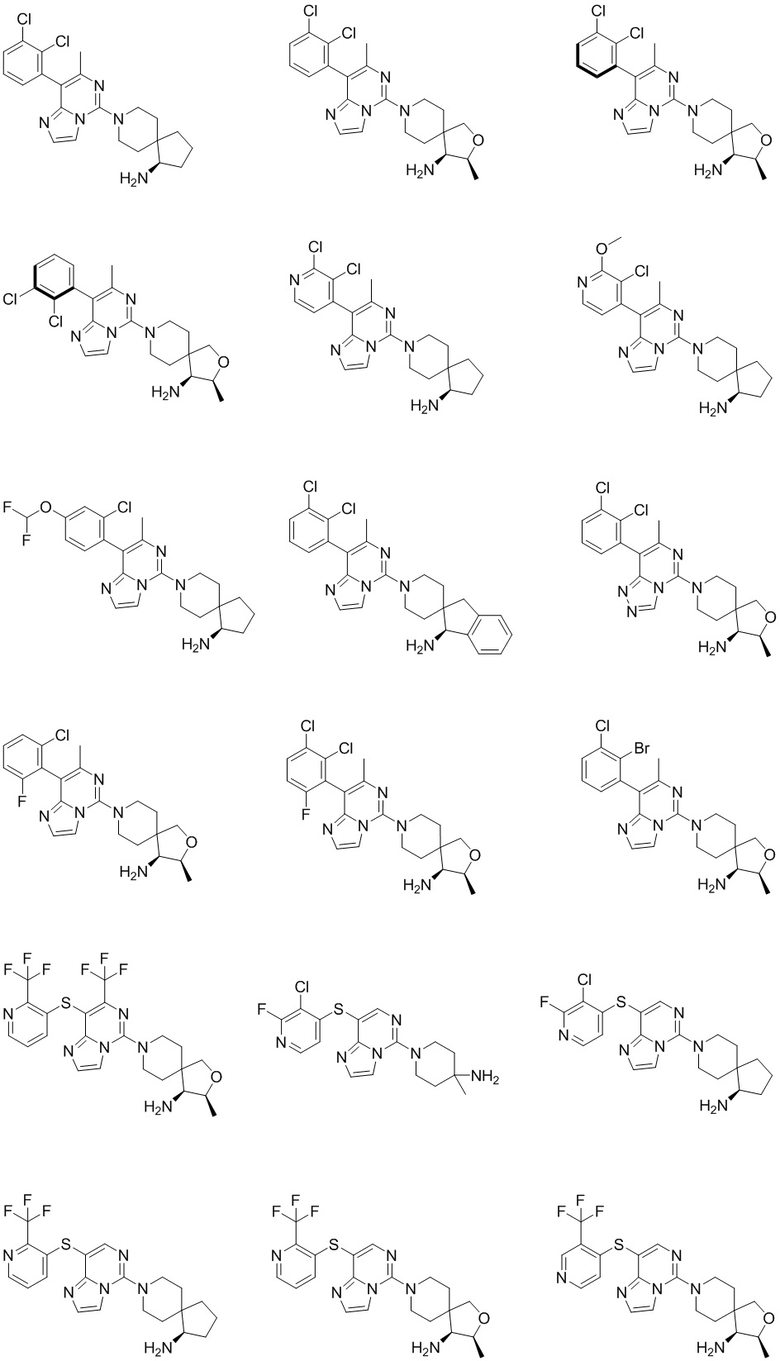
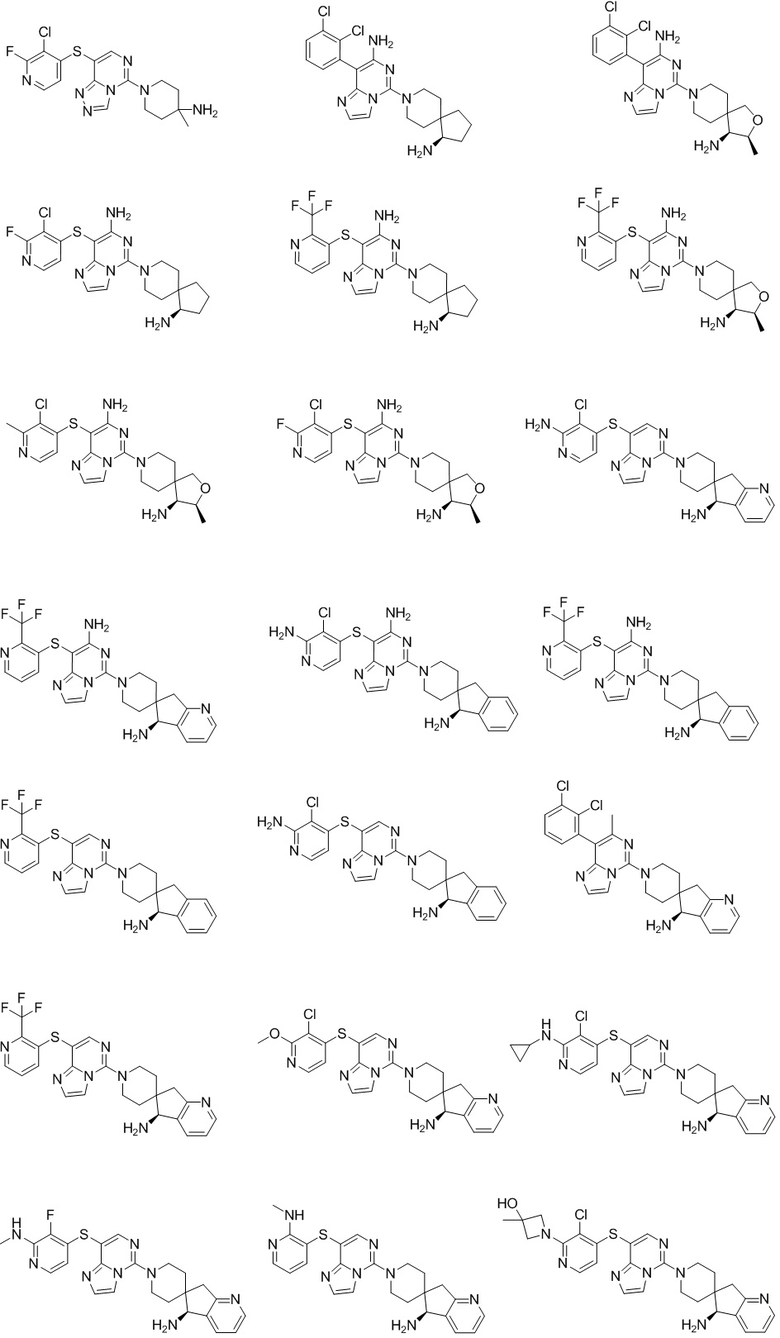
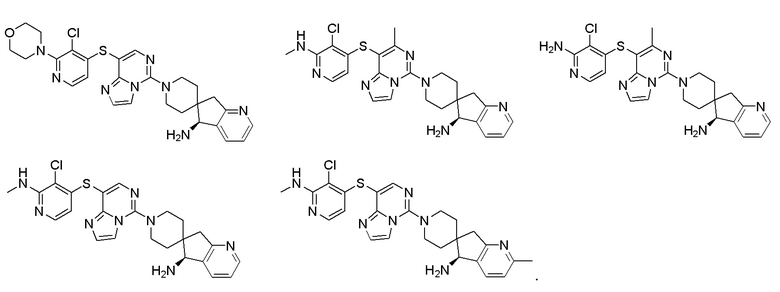
8. Способ получения соединения по любому из пп. 1–5 и 7, где соединение формулы (I) представляет собой соединение формулы (I-B), в котором
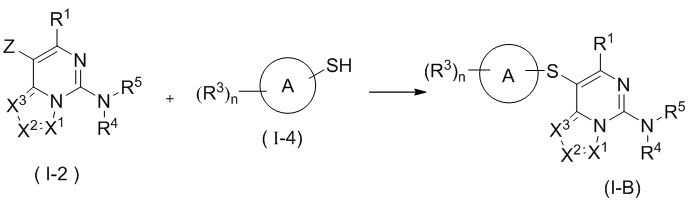
соединение формулы (I-2) и соединение формулы (I-4) подвергают реакции C–S сочетания в щелочных условиях с получением соединения формулы (I-B);
где реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания; органическое основание выбрано из группы, состоящей из триэтиламина, N,N-диизопропилэтиламина, н-бутиллития, диизопропиламида лития, бистриметилсилиламида лития, ацетата калия, трет-бутилата натрия и трет-бутилата калия; неорганическое основание выбрано из группы, состоящей из гидрида натрия, фосфата калия, карбоната натрия, карбоната калия, ацетата калия, карбоната цезия, гидроксида натрия и гидроксида лития;
Z представляет собой галоген или сульфонил; и
кольцо A, R1, X1, X2, X3, R3, R4, R5 и n являются такими, как определено в п. 1.
9. Способ получения соединения по п. 6, где соединение формулы (II) представляет собой соединение формулы (II-B), включающий следующие стадии, на которых:
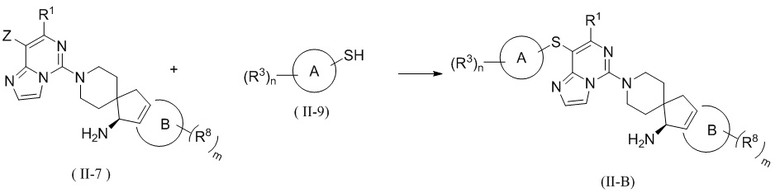
соединение формулы (II-7) и соединение формулы (II-9) подвергают реакции C–S сочетания в щелочных условиях с получением соединения формулы (II-B);
реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания; органическое основание выбрано из группы, состоящей из триэтиламина, N,N-диизопропилэтиламина, н-бутиллития, диизопропиламида лития, бистриметилсилиламида лития, ацетата калия, трет-бутилата натрия и трет-бутилата калия; неорганическое основание выбрано из группы, состоящей из гидрида натрия, фосфата калия, карбоната натрия, карбоната калия, ацетата калия, карбоната цезия, гидроксида натрия и гидроксида лития;
Z выбран из группы, состоящей из галогена, сульфонила и сульфинила;
кольцо A, кольцо B, R1, R3, R8, B, m и n являются такими, как определено в п. 6.
10. Способ по п. 9, дополнительно включающий стадию взаимодействия соединения формулы (II-5) с соединением формулы (II-6) в щелочных условиях с получением соединения формулы (II-7)
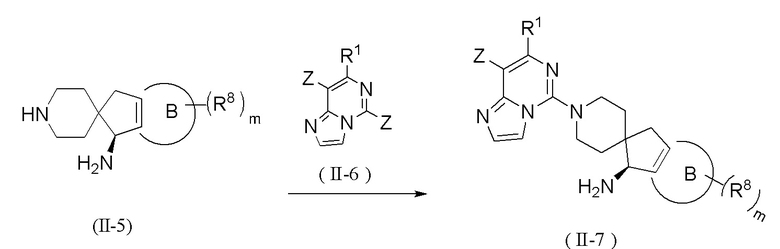
где реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания; органическое основание выбрано из группы, состоящей из триэтиламина, N,N-диизопропилэтиламина, н-бутиллития, диизопропиламида лития, бистриметилсилиламида лития, ацетата калия, трет-бутилата натрия и трет-бутилата калия; неорганическое основание выбрано из группы, состоящей из гидрида натрия, фосфата калия, карбоната натрия, карбоната калия, ацетата калия, карбоната цезия, гидроксида натрия и гидроксида лития.
11. Фармацевтическая композиция для профилактики или лечения заболевания или состояния, опосредованного активностью SHP2, где заболевание или состояние выбрано из синдрома Нунан, синдрома Leopard, ювенильного миеломоноцитарного лейкоза, нейробластомы, меланомы, острого миелогенного лейкоза, рака молочной железы, рака пищевода, рака легкого, рака ободочной кишки, рака головы, рака поджелудочной железы, плоскоклеточной карциномы головы и шеи, рака желудка, рака печени, анапластической крупноклеточной лимфомы или глиобластомы, содержащая соединение или его фармацевтически приемлемую соль по любому из пп. 1–7 и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
12. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1–7 или фармацевтической композиции по п. 11 в получении лекарственного средства для профилактики или лечения заболевания или состояния, опосредованного активностью SHP2, где заболевание или состояние выбрано из синдрома Нунан, синдрома Leopard, ювенильного миеломоноцитарного лейкоза, нейробластомы, меланомы, острого миелогенного лейкоза, рака молочной железы, рака пищевода, рака легкого, рака ободочной кишки, рака головы, рака поджелудочной железы, плоскоклеточной карциномы головы и шеи, рака желудка, рака печени, анапластической крупноклеточной лимфомы или глиобластомы.
| WO 2018184590 A1, 11.10.2018 | |||
| АНТЕННА УСТРОЙСТВА ДЛЯ КОНТРОЛЯ И ДИАГНОСТИКИ ЛИНИИ ЭНЕРГОСНАБЖЕНИЯ | 2012 |
|
RU2545058C1 |
| УСТРОЙСТВО ДЛЯ ПРЕССОВАНИЯ ИЗДЕЛИЙ ИЗ ПОРОШКА | 1997 |
|
RU2131790C1 |
| Sergio Lopez et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Устройство для выпрямления многофазного тока | 1923 |
|
SU50A1 |
| ПУСКОВОЕ ПРИСПОСОБЛЕНИЕ ДЛЯ ДВИГАТЕЛЕЙ ВНУТРЕННЕГО ГОРЕНИЯ | 1926 |
|
SU6022A1 |
| REGISTRY [STN online], RN: 2173116-56-6, 08.02.2018 | |||
| Kikkeri N | |||

