ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001] Настоящее изобретение касается способа получения производного бензилового эфира 2-аминоникотиновой кислоты. В частности, настоящее изобретение касается способа получения с высоким выходом и высокой степенью чистоты производного бензилового эфира 2-аминоникотиновой кислоты, которое представляет собой соединение, применимое в качестве активного компонента сельскохозяйственного фунгицида.
УРОВЕНЬ ТЕХНИКИ
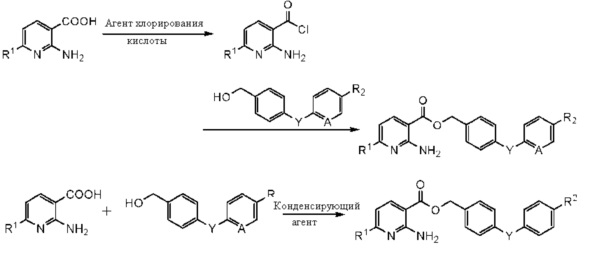
[0002] Примеры способа получения производного эфира 2-аминоникотиновой кислоты охватывают способ, включающий хлорирование производного 2-аминоникотиновой кислоты агентом галогенирования и взаимодействие хлорированного продукта с производным бензилового спирта в присутствии основания в органическом растворителе, и способ, включающий взаимодействие производного 2-аминоникотиновой кислоты и производного бензилового спирта с использованием конденсирующего агента в органическом растворителе.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0003] В обычных методиках выход является низким, и требуется очистка, например, методом колоночной хроматографии на силикагеле для удаления остающегося непрореагировавшего сырого материала, производного 2-аминоникотиновой кислоты. Таким образом, требуется разработать способ промышленного получения целевого продукта с высоким выходом и высокой степенью чистоты.
[0004] Заявители настоящего изобретения провели тщательное исследование с целью решения упоминаемых выше проблем и в результате обнаружили, что хлорирование производного 2-аминоникотиновой кислоты изменяет цвет реакционного раствора до темно-коричневого, препятствует развитию указанного взаимодействия и увеличивает количество побочных продуктов. Кроме того, обнаружено, что взаимодействие не завершается, даже если используют конденсирующий агент. Таким образом, получение производного бензилового эфира 2-аминоникотиновой кислоты по обычным методикам нуждается в способе очистки и имеет низкий выход.
Заявители настоящего изобретения также обнаружили, что можно получить производное бензилового эфира 2-аминоникотиновой кислоты с высоким выходом и высокой степенью чистоты взаимодействием производного бензилгалогенида с производным 2-аминоникотиновой кислоты в присутствии заданного основания в полярном растворителе и выполнить настоящее изобретение.
[0005] То есть аспект настоящего изобретения касается способа получения производного бензилового эфира 2-аминоникотиновой кислоты, представленного следующей формулой [I]:
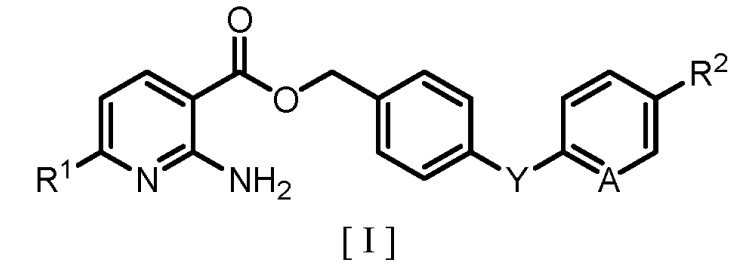
где R1 обозначает атом водорода или C1-C4 алкильную группу; R2 обозначает атом водорода, атом галогена, C1-C4 алкильную группу или C1-C4 алкоксигруппу; A обозначает атом азота или метиновую группу (CH); и Y обозначает атом кислорода, метиленовую группу (CH2) или метиленоксигруппу (OCH2), причем способ включает:
(a) взаимодействие производного 2-аминоникотиновой кислоты, представленного следующей формулой [II]
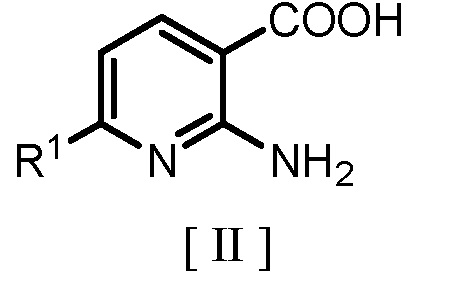
где R1 является таким же, как заместитель, определенный для формулы [I], с гидридом щелочного металла или карбонатом щелочного металла с получением соединения, представленного следующей формулой [III]:
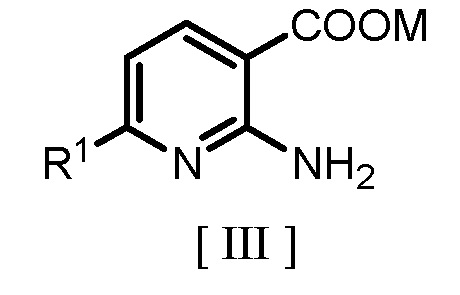
где R1 является таким же, как заместитель, определенный для формулы [I], и M обозначает щелочной металл; и
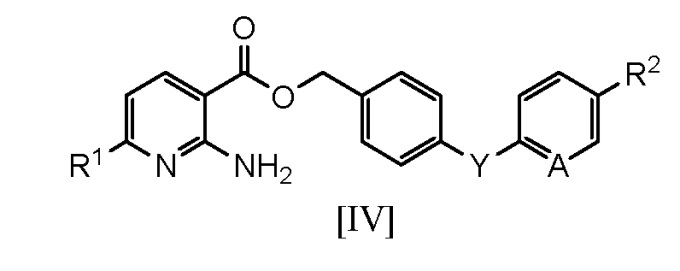
(b) взаимодействие соединения, полученного на стадии (a) и представленного формулой [III], с производным бензилгалогенида, представленным следующей формулой [IV]:
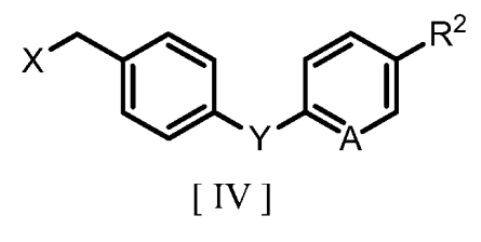
где R2, A и Y являются такими же, как группы, определенные для формулы [I], и X обозначает атом галогена, в полярном растворителе с получением производного бензилового эфира 2-аминоникотиновой кислоты, представленного формулой [I].
[0006] Другой аспект настоящего изобретения касается способа получения производного бензилового эфира 2-аминоникотиновой кислоты, представленного следующей формулой [I]:
где R1 обозначает атом водорода или C1-C4 алкильную группу; R2 обозначает атом водорода, атом галогена, C1-C4 алкильную группу или C1-C4 алкоксигруппу; A обозначает атом азота или метиновую группу (CH); Y обозначает атом кислорода, метиленовую группу (CH2) или метиленоксигруппу (OCH2), причем способ включает:
(a) взаимодействие производного 2-аминоникотиновой кислоты, представленного следующей формулой [II]:
где R1 является таким же, как заместитель, определенный для формулы [I], с гидридом щелочного металла или карбонатом щелочного металла с получением соединения, представленного следующей формулой [III]:
где R1 является таким же, как заместитель, определенный для формулы [I], и M обозначает щелочной металл;
(b) взаимодействие производного бензилового спирта, представленного следующей формулой [V]:
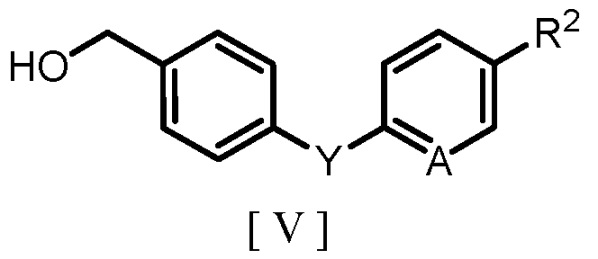
где R2, A и Y являются такими же, как группы, определенные для формулы [I], с агентом галогенирования с получением производного бензилгалогенида, представленного следующей формулой [IV]:
где R2, A и Y являются такими же, как группы, определенные для формулы [I], и X обозначает атом галогена; и
(c) взаимодействие производного бензилгалогенида, полученного на стадии (b) и представленного формулой [IV], без выделения с соединением, полученным на стадии (a) и представленным формулой [III], в полярном растворителе с получением производного бензилового эфира 2-аминоникотиновой кислоты, представленного формулой [I].
[0007] Способами получения по настоящему изобретению можно получить производное бензилового эфира 2-аминоникотиновой кислоты более высокой степени чистоты и с большим выходом по сравнению с общепринятыми способами.
Описание вариантов осуществления.
[0008] Теперь настоящее изобретение будет описано подробно.
В формулах [I], [II], [III], [IV] и [V] примеры C1-C4 алкильной группы, представленной R1 или R2, включают метильную, этильную, н-пропильную, изопропильную, н-бутильную, изобутильную, втор-бутильную и трет-бутильную группы; примеры атома галогена, представленного R2, включают атомы фтора, хлора, брома и йода; примеры C1-C4 алкоксигруппы, представленной R2, включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси и трет-бутоксигруппы; и примеры щелочного металла, представленного M, включают литий, натрий, калий и цезий.
[0009] В вариантах осуществления настоящего изобретения способ получения может включать следующие стадии: добавление соединения формулы [II] к полярному растворителю, такому как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, 1,3-диметил-2-имидазолидон, ацетонитрил или диметилсульфоксид, затем добавление основания, такого как карбонат натрия, карбонат калия или карбонат цезия, к полярному растворителю в (1,0-3,0)-кратном мольном количестве, предпочтительно (1,5-2,0)-кратном мольном количестве, относительно количества соединения формулы [II] и перемешивание смеси при температуре от 0 до 60°C в течение периода от 5 мин до 2 ч, предпочтительно при температуре от 30 до 50°C в течение 10-30 мин с получением суспензии (здесь далее обозначаемой как суспензия 1) соединения формулы [III]. Способ также может включать следующие стадии: добавление соединения формулы [V] к полярному растворителю, такому как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, 1,3-диметил-2-имидазолидон, ацетонитрил или диметилсульфоксид, затем добавление агента галогенирования, такого как тионилхлорид, тионилбромид или оксихлорид фосфора, к полярному растворителю в (1,0-1,5)-кратном мольном количестве, предпочтительно в (1,0-1,1)-кратном мольном количестве относительно количества соединения формулы [V] и перемешивание смеси при температуре от -5 до 30°C в течение периода от 5 мин до 1 ч, предпочтительно при температуре от 0 до 10°C в течение 20-40 мин с получением раствора соединения формулы [IV]. Способ также может включать стадию добавления полученного раствора по капле к суспензии 1 и перемешивание смеси с нагреванием при температуре от 0 до 100°C в течение 1-20 ч, предпочтительно при температуре от 60 до 80°C в течение 2-16 ч для проведения взаимодействия с получением соединения формулы [I]. После взаимодействия реакционный раствор можно перегнать при пониженном давлении для удаления от 50 до 95% органического растворителя, можно влить в раствор воду со льдом и перемешивать смесь в течение 5-30 мин, предпочтительно 10-20 мин для осаждения кристаллов и последующего сбора кристаллов фильтрованием. Собранные кристаллы можно промыть водой и затем высушить. Таким образом, можно достаточно легко получить целевое соединение формулы [I] с высоким выходом и высокой степенью чистоты.
[0010] В другом варианте осуществления способ получения по настоящему изобретению может включать следующие стадии: добавление соединения формулы [II] к полярному растворителю, такому как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, 1,3-диметил-2-имидазолидон, ацетонитрил или диметилсульфоксид, затем добавление основания, такого как карбонат натрия, карбонат калия или карбонат цезия, к полярному растворителю в (1,0-3,0)-кратном мольном количестве, предпочтительно (1,5-2,0)-кратном мольном количестве относительно количества соединения формулы [II], перемешивание смеси при температуре от 0 до 60°C в течение периода от 5 мин до 2 ч, предпочтительно при температуре от 30 до 50°C в течение 10-30 мин с получением суспензии и затем добавление по капле соединения формулы [IV] к суспензии и перемешивание смеси с нагреванием при температуре от 0 до 100°C в течение 1-20 ч, предпочтительно при температуре от 60 до 80°C в течение 2-16 ч для проведения взаимодействия с получением соединения формулы [I]. После взаимодействия реакционный раствор можно перегнать при пониженном давлении для удаления от 50 до 95% органического растворителя, можно влить в реакционный раствор воду со льдом и перемешивать смесь в течение 5-30 мин, предпочтительно 10-20 мин для осаждения кристаллов с последующим сбором кристаллов фильтрованием. Собранные кристаллы можно промыть водой и затем высушить. Таким образом, можно достаточно легко получить целевое соединение формулы [I] с высоким выходом и высокой степенью чистоты.
[0011] В способе получения по настоящему изобретению гидрид щелочного металла, который взаимодействует с соединением формулы [II], не имеет особых ограничений и включает гидрид лития, гидрид натрия, гидрид калия и гидрид цезия.
В способе получения по настоящему изобретению карбонат щелочного металла, который взаимодействует с соединением формулы [II], не имеет особых ограничений и включает карбонат натрия, карбонат калия и карбонат цезия.
В способе получения по настоящему изобретению количество гидрида щелочного металла или карбоната щелочного металла, который взаимодействует с соединением формулы [II], не имеет особых ограничений, но составляет, например, (1,0-3,0)-кратное мольное количество, предпочтительно (1,5-2,0)-кратное мольное количество относительно количества соединения формулы [II].
В способе получения по настоящему изобретению взаимодействие соединения формулы [II] с гидридом щелочного металла или карбонатом щелочного металла не имеет особых ограничений и происходит, например, при перемешивании реагентов в полярном растворителе, таком как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, 1,3-диметил-2-имидазолидон, ацетонитрил или диметилсульфоксид, например, при температуре от 0 до 60°C в течение периода от 5 мин до 2 ч, предпочтительно при температуре от 30 до 50°C в течение 10-30 мин.
[0012] В способе получения по настоящему изобретению взаимодействие между соединением формулы [III] и соединением формулы [IV] не имеет особых ограничений и происходит, например, при добавлении по капле раствора соединения формулы [IV] к суспензии соединения формулы [III] и перемешивании смеси при нагревании, например, при температуре от 0 до 100°C в течение 1-20 ч, предпочтительно при температуре от 60 до 80°C в течение 2-16 ч.
В способе получения по настоящему изобретению агент галогенирования, который взаимодействует с соединением формулы [V], не имеет особых ограничений и включает тионилхлорид, тионилбромид или оксихлорид фосфора. Агент галогенирования применяют, например, в (1,0-1,5)-кратном мольном количестве, предпочтительно в (1,0-1,1)-кратном мольном количестве, относительно количества соединения формулы [V].
В способе получения по настоящему изобретению взаимодействие между соединением формулы [V] и агентом галогенирования не имеет особых ограничений и происходит, например, в полярном растворителе, таком как N,N-диметилформамид, N,N-диметилацетамид, N-метилпирролидон, 1,3-диметил-2-имидазолидон, ацетонитрил или диметилсульфоксид, например, при температуре от -5 до 30°C в течение периода от 5 мин до 1 ч, предпочтительно при температуре от 0 до 10°C в течение 20-40 мин.
В способе получения по настоящему изобретению соединение формулы [IV], полученное при взаимодействии соединения формулы [V] и агента галогенирования, может взаимодействовать с соединением формулы [III] без выделения.
В способе получения по настоящему изобретению полярный растворитель, который используют при взаимодействии между соединением формулы [II] и гидридом щелочного металла или карбонатом щелочного металла, может быть таким же или отличаться от полярного растворителя, который используют при взаимодействии между соединением формулы [V] и агентом галогенирования, предпочтительно, они являются одинаковыми.
[0013] Производное никотиновой кислоты, представленное формулой [II] и предполагаемое для использования в способе получения по настоящему изобретению, можно синтезировать непосредственно из известных соединений, например, способом, описанным в JP-A-2010-083861.
Производное спирта, представленное формулой [V] и предполагаемое для использования в способе получения по настоящему изобретению, можно синтезировать непосредственно из известных соединений, например, способом, описанным в Journal of Medicinal Chemistry, Vol. 43, p. 1826 (2000).
Соединение формулы [I], получаемое способом получения по настоящему изобретению, применимо в качестве сельскохозяйственного фунгицида.
Примеры
[0014] Далее настоящее изобретение будет описано с помощью примеров, но область настоящего изобретения не ограничена следующими примерами.
[0015] Пример 1
Синтез 4-феноксибензилового эфира 2-амино-6-метилникотиновой кислоты.
4-Феноксибензиловый спирт (4,00 г) растворяют в N,N-диметилформамиде (10 мл) и раствор охлаждают до 5°C. К этому раствору добавляют по капле тионилхлорид (1,45 мл) и затем перемешивают смесь в течение 30 мин, получая раствор (I).
2-Амино-6-метилникотиновую кислоту (3,04 г) суспендируют в N,N-диметилформамиде (60 мл). Добавляют к суспензии карбонат калия (5,53 г) с последующим перемешиванием при 40°C в течение 30 мин. Раствор (I) добавляют по капле к полученной суспензии и далее перемешивают с нагреванием при 80°C в течение 2 ч. Реакционный раствор охлаждают до комнатной температуры и удаляют N,N-диметилформамид (40 мл, 57%) перегонкой при пониженном давлении. Добавляют к остатку воду со льдом (100 мл) и далее перемешивают при комнатной температуре в течение 10 мин. Выпавшие в осадок кристаллы собирают фильтрованием и сушат, получая 6,41 г (выход: 96%) целевого продукта (соединение 2, показанное в таблице 1). Согласно анализу чистоты методом жидкостной хроматографии, чистота получаемого таким образом продукта является высокой, 99,6%. Температура плавления продукта составляет от 122 до 124°C.
1H-ЯМР (CDCl3) δ, м.д.: 2,38 (3H, с), 5,25 (2H, с), 6,06-6,72 (2H, ш.с), 6,44 (1H, д), 6,99-7,04 (4H, м), 7,12 (1H, т), 7,31-7,41 (4H, м), 8,04 (1H, д).
[0016] Пример 2
Синтез 4-феноксибензилового эфира 2-амино-6-метилникотиновой кислоты.
4-Феноксибензиловый спирт (4,00 г) растворяют в ацетонитриле (10 мл) и раствор охлаждают до 5°C. К этому раствору (1,45 мл) добавляют по капле тионилхлорид и затем перемешивают смесь в течение 30 мин, получая раствор (I).
2-Амино-6-метилникотиновую кислоту (3,04 г) суспендируют в ацетонитриле (50 мл) и добавляют к суспензии карбонат калия (5,53 г) с последующим перемешиванием при 40°C в течение 30 мин. Раствор (I) добавляют по капле к полученной суспензии, затем кипятят смесь с обратным холодильником в течение 16 час. Реакционный раствор охлаждают до комнатной температуры и концентрируют при пониженном давлении. Добавляют к остатку воду со льдом (200 мл) и затем перемешивают при комнатной температуре в течение 10 мин. Выпавшие в осадок кристаллы собирают фильтрованием и сушат, получая 6,05 г (выход: 91%) целевого продукта (соединение 2, показанное в таблице 1). Согласно анализу чистоты методом жидкостной хроматографии, чистота получаемого таким образом продукта является высокой, 98,9%. Температура плавления продукта составляет от 122 до 124°C.
1H-ЯМР (CDCl3) δ, м.д.: 2,38 (3H, с), 5,25 (2H, с), 6,06-6,72 (2H, ш.с), 6,44 (1H, д), 6,99-7,04 (4H, м), 7,12 (1H, т), 7,31-7,41 (4H, м), 8,04 (1H, д).
[0017] Пример 3
Синтез 4-феноксибензилового эфира 2-амино-6-метилникотиновой кислоты.
2-Амино-6-метилникотиновую кислоту (3,04 г) суспендируют в N,N-диметилформамиде (60 мл) и добавляют к суспензии карбонат калия (5,53 г) с последующим перемешиванием при 40°C в течение 30 мин. К полученной суспензии добавляют по капле 4-феноксибензилхлорид (4,37 г), далее перемешивают смесь с нагреванием при 80°C в течение 2 час. Реакционный раствор охлаждают до комнатной температуры и удаляют N,N-диметилформамид (35 мл, 58%) перегонкой при пониженном давлении. Добавляют к остатку воду со льдом (100 мл) и затем перемешивают смесь при комнатной температуре в течение 10 мин. Выпавшие в осадок кристаллы собирают фильтрованием и сушат, получая 6,42 г (выход: 96%) целевого продукта (соединение 2, показанное в таблице 1). Согласно анализу чистоты методом жидкостной хроматографии, чистота получаемого таким образом продукта является высокой, 99,7%. Температура плавления продукта составляет от 122 до 124°C.
1H-ЯМР (CDCl3) δ, м.д.: 2,38 (3H, с), 5,25 (2H, с), 6,06-6,72 (2H, ш.с), 6,44 (1H, д), 6,99-7,04 (4H, м), 7,12 (1H, т), 7,31-7,41 (4H, м), 8,04 (1H, д).
[0018] Пример сравнения 1.
4-Феноксибензилхлорид, используемый в примере 3, синтезируют следующим способом.
Синтез 4-феноксибензилхлорида.
4-Феноксибензиловый спирт (20,0 г) растворяют в толуоле (100 мл) и добавляют к раствору тионилхлорид (13,1 г) за 30 мин при комнатной температуре. Через 2 ч реакционный раствор концентрируют при пониженном давлении. Остаток отгоняют, получая 16,7 г (выход: 76%) целевого продукта. Температура кипения продукта составляет 137°C/3 мм рт.ст.
[0019] Пример 4
Синтез 4-(4-метилфенокси)бензилового эфира 2-амино-6-метилникотиновой кислоты.
4-(4-Метилфенокси)бензиловый спирт (4,26 г) растворяют в N,N-диметилформамиде (10 мл) и раствор охлаждают до 5°C. К этому раствору добавляют по капле тионилхлорид (1,45 мл) и затем перемешивают смесь в течение 30 мин, получая раствор (I).
2-Амино-6-метилникотиновую кислоту (3,04 г) суспендируют в N,N-диметилформамиде (60 мл) и добавляют к суспензии карбонат калия (5,53 г) с последующим перемешиванием при 40°C в течение 30 мин. Раствор (I) добавляют по капле к полученной суспензии и далее перемешивают смесь с нагреванием при 80°C в течение 2 ч. Реакционный раствор охлаждают до комнатной температуры и удаляют N,N-диметилформамид (45 мл, 64%) перегонкой при пониженном давлении. Добавляют к остатку воду со льдом (100 мл) с последующим перемешиванием при комнатной температуре в течение 10 мин. Выпавшие в осадок кристаллы собирают фильтрованием и сушат, получая 6,42 г (выход: 92%) целевого продукта (соединение 4 показанное в таблице 1). Согласно анализу чистоты методом жидкостной хроматографии, чистота получаемого таким образом продукта является высокой, 99,1%. Температура плавления продукта составляет от 94 до 96°C.
1H-ЯМР (CDCl3) δ, м.д.: 2,33 (3H, с), 2,40 (3H, с), 5,27 (2H, с), 6,08-6,82 (2H, ш.с), 6,44 (1H, д), 6,90-7,00 (5H, м), 7,14 (2H, д), 7,37 (2H, д), 8,02 (1H, д).
[0020] Пример 5
Синтез 4-фенилметилбензилового эфира 2-амино-6-метилникотиновой кислоты.
4-Фенилметилбензиловый спирт (3,96 г) растворяют в N,N-диметилформамиде (10 мл) и раствор охлаждают до 5°C. Добавляют к этому раствору по капле тионилхлорид (1,45 мл) и затем перемешивают смесь в течение 30 мин, получая раствор (I).
2-Амино-6-метилникотиновую кислоту (3,04 г) суспендируют в N,N-диметилформамиде (60 мл) и добавляют к суспензии карбонат калия (5,53 г) с последующим перемешиванием при 40°C в течение 30 мин. Раствор (I) добавляют по капле к полученной суспензии и далее перемешивают смесь с нагреванием при 80°C в течение 2 ч. Реакционный раствор охлаждают до комнатной температуры и удаляют N,N-диметилформамид (40 мл, 57%) перегонкой при пониженном давлении. Добавляют к остатку воду со льдом (100 мл) с последующим перемешиванием при комнатной температуре в течение 10 мин. Выпавшие в осадок кристаллы собирают фильтрованием и сушат, получая 6,08 г (выход: 91%) целевого продукта (соединение 8, показанное в таблице 1). Согласно анализу чистоты методом жидкостной хроматографии, чистота получаемого таким образом продукта является высокой, 98,3%. Температура плавления продукта составляет от 106 до 108°C.
1H-ЯМР (CDCl3) δ, м.д.: 2,40 (3H, с), 3,99 (2H, с), 5,25 (2H, с), 6,10-6,74 (2H, ш.с), 6,43 (1H, д), 7,16-7,22 (4H, м), 7,24-7,34 (5H, м), 8,02 (1H, д).
[0021] Пример 6
Синтез 4-(2-пиридилокси)бензилового эфира 2-амино-6-метилникотиновой кислоты.
4-(2-Пиридилокси)бензиловый спирт (4,02 г) растворяют в N,N-диметилформамиде (10 мл) и раствор охлаждают до 5°C. Добавляют к этому раствору по капле тионилхлорид (1,45 мл) и затем перемешивают смесь в течение 30 мин, получая раствор (I). 2-Амино-6-метилникотиновую кислоту (3,04 г) суспендируют в N,N-диметилформамиде (60 мл) и добавляют к суспензии карбонат калия (5,53 г) с последующим перемешиванием при 40°C в течение 30 мин. Раствор (I) добавляют по капле к полученной суспензии и затем перемешивают смесь с нагреванием при 80°C в течение 2 ч. Реакционный раствор охлаждают до комнатной температуры и удаляют N,N-диметилформамид (50 мл, 71%) перегонкой при пониженном давлении. Добавляют к остатку воду со льдом (100 мл) и далее перемешивают смесь при комнатной температуре в течение 10 мин. Выпавшие в осадок кристаллы собирают фильтрованием и сушат, получая 6,13 г (выход: 91%) целевого продукта (соединение 6, показанное в таблице 1). Согласно анализу чистоты методом жидкостной хроматографии, чистота получаемого таким образом продукта является высокой, 98,5%. Температура плавления продукта составляет от 120°C до 121°C.
1H-ЯМР (CDCl3) δ, м.д.: 2,40 (3H, с), 5,31 (2H, с), 6,10-6,91 (2H, ш.с), 6,46 (1H, д), 6,89 (1H, д), 7,00 (1H, т), 7,16 (2H, д), 7,43 (2H, д), 7,67-7,72 (1H, т), 8,03 (1H, д), 8,20 (1H, д).
[0022] Пример сравнения 1
Синтез 4-феноксибензилового эфира 2-амино-6-метилникотиновой кислоты.
Смешивают 2-амино-6-метилникотиновую кислоту (3,04 г) и тионилхлорид (15 мл) и перемешивают с нагреванием при 85°C в течение 1 час. Целевой хлорангидрид кислоты частично разлагается и окрашивает реакционный раствор в коричневый цвет. Избыточное количество тионилхлорида удаляют из реакционного раствора перегонкой. Остаток охлаждают до комнатной температуры и добавляют к остатку тетрагидрофуран (30 мл), 4-феноксибензиловый спирт (4,00 г) и триэтиламин (6,06 г). Смесь перемешивают при комнатной температуре в течение 1 ч и добавляют в нее воду (80 мл). Нерастворимые вещества удаляют фильтрованием и слои фильтрата разделяют. Органический слой промывают насыщенным солевым раствором, сушат безводным сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают методом колоночной хроматографии на силикагеле (гексан:этилацетат=2:1), получая 1,23 г (выход: 18%) целевого продукта. Температура плавления продукта составляет от 122 до 124°C.
[0023] Пример сравнения 2
Синтез 4-феноксибензилового эфира 2-амино-6-метилникотиновой кислоты.
2-Амино-6-метилникотиновую кислоту (3,04 г) суспендируют в метиленхлориде (100 мл) и суспензию охлаждают до 5°C. Добавляют к этой суспензии оксалилхлорид (2,58 мл) и N,N-диметилформамид (несколько капель) и далее перемешивают смесь в течение 2 ч, получая раствор (I). 4-Феноксибензиловый спирт (4,00 г) растворяют в метиленхлориде (100 мл) и добавляют к раствору 4-диметиламинопиридин (3,36 г). Полученный раствор охлаждают до 5°C и добавляют к нему по капле раствор (I) с последующим перемешиванием в течение 1 ч. Добавляют к реакционному раствору воду (200 мл), затем разделяют слои жидкости. Органический слой промывают насыщенным солевым раствором, сушат безводным сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают методом колоночной хроматографии на силикагеле (гексан:этилацетат=2:1), получая 2,47 г (выход: 37%) целевого продукта. Температура плавления продукта составляет от 122 до 124°C.
[0024] Пример сравнения 3
Синтез 4-феноксибензилового эфира 2-амино-6-метилникотиновой кислоты.
2-Амино-6-метилникотиновую кислоту (3,04 г) суспендируют в 1,2-дихлорэтане (50 мл). Добавляют к суспензии 4-феноксибензиловый спирт (4,00 г), 1-этил-3-(3-диметиламинопропил)карбодиимид (4,60 г) и 4-диметиламинопиридин (2,92 г) и далее перемешивают с нагреванием при 70°C в течение 12 ч. Реакционный раствор охлаждают до комнатной температуры и проводят разделение слоев, используя 1,2-дихлорэтан (100 мл) и воду (150 мл). Органический слой промывают насыщенным солевым раствором, сушат безводным сульфатом натрия и концентрируют при пониженном давлении. Остаток очищают методом колоночной хроматографии на силикагеле (градиент смеси гексан/этилацетат), получая 3,87 г (выход: 58%) целевого продукта. Температура плавления продукта составляет от 121 до 123°C.
[0025] Ниже показаны соединения по настоящему изобретению, которые получают способами, аналогичными способу примера 1.
[0026]
[Таблица 1]
[0027] Как описано выше, способ получения по настоящему изобретению представляет собой промышленно важный способ получения сельскохозяйственного фунгицида, то есть производного бензилового эфира 2-аминоникотиновой кислоты.
Изобретение относится к способу получения производного бензилового эфира 2-аминоникотиновой кислоты формулы
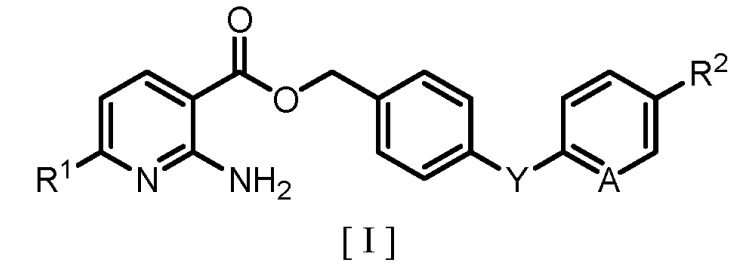
с высоким выходом и высокой степенью чистоты. Способ заключается во взаимодействии производного бензилгалогенида с производным 2-аминоникотиновой кислоты в полярном растворителе в присутствии основания. 2 н.п. ф-лы, 1 табл., 6 пр.
1. Способ получения производного бензилового эфира 2-аминоникотиновой кислоты, представленного формулой [I]
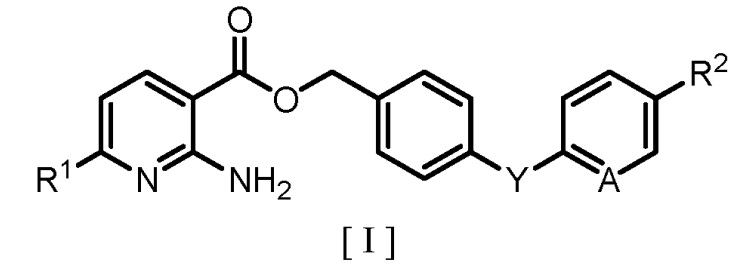
где R1 обозначает атом водорода или C1-C4 алкильную группу; R2 обозначает атом водорода, атом галогена, C1-C4 алкильную группу или C1-C4 алкоксигруппу; A обозначает атом азота или метиновую группу (CH); и Y обозначает атом кислорода, метиленовую группу (CH2) или метиленоксигруппу (OCH2), причем способ включает:
(a) взаимодействие производного 2-аминоникотиновой кислоты, представленного формулой [II]
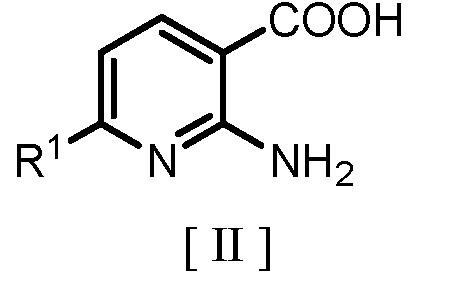
где R1 является таким же, как определено для формулы [I], с гидридом щелочного металла или карбонатом щелочного металла с получением соединения, представленного формулой [III]
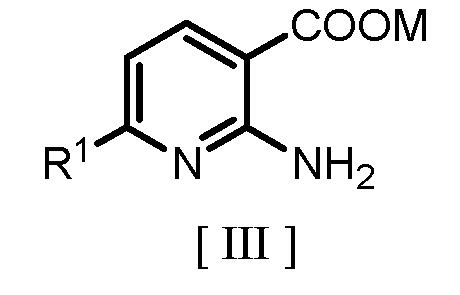
где R1 является таким же, как определено для формулы [I], и M обозначает щелочной металл; и
(b) взаимодействие соединения, полученного на стадии (a) и представленного формулой [III], с производным бензилгалогенида, представленным формулой [IV]
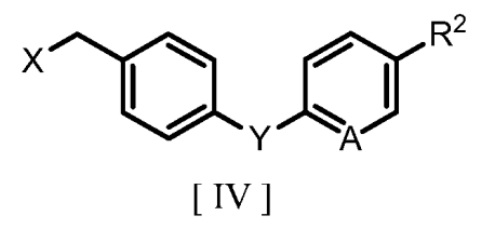
где R2, A и Y являются такими же, как определено для формулы [I], и X обозначает атом галогена, в полярном растворителе с получением производного бензилового эфира 2-аминоникотиновой кислоты, представленного формулой [I].
2. Способ получения производного бензилового эфира 2-аминоникотиновой кислоты, представленного формулой [I]
где R1 обозначает атом водорода или C1-C4 алкильную группу; R2 обозначает атом водорода, атом галогена, C1-C4 алкильную группу или C1-C4 алкоксигруппу; A обозначает атом азота или метиновую группу (CH); Y обозначает атом кислорода, метиленовую группу (CH2) или метиленоксигруппу (OCH2), причем способ включает:
(a) взаимодействие производного 2-аминоникотиновой кислоты, представленного формулой [II]
где R1 является таким же, как определено для формулы [I],
с гидридом щелочного металла или карбонатом щелочного металла с получением соединения, представленного формулой [III]
где R1 является таким же, как определено для формулы [I], и M обозначает щелочной металл;
(b) взаимодействие производного бензилового спирта, представленного формулой [V]
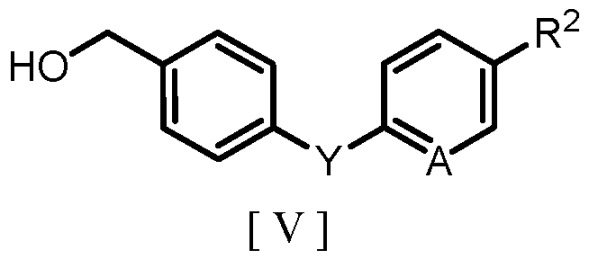
где R2, A и Y являются такими же, как группы, определенные для формулы [I],
с агентом галогенирования с получением производного бензилгалогенида, представленного формулой [IV]:
где R2, A и Y являются такими же, как определено для формулы [I], и X обозначает атом галогена; и
(c) взаимодействие производного бензилгалогенида, представленного формулой [IV] и полученного на стадии (b), без выделения с соединением, представленным формулой [III] и полученным на стадии (a), в полярном растворителе с получением производного бензилового эфира 2-аминоникотиновой кислоты, представленного формулой [I].
| WO 2014006945 A1, 09.01.2014 | |||
| JP 2010083861 A, 15.04.2010 | |||
| 0 |
|
SU217301A1 | |

