Область техники, к которой относится изобретение
[0001]
Настоящее изобретение относится к способу получения производного бензилового эфира 2-аминоникотиновой кислоты. Точнее, настоящее изобретение относится к промышленному способу получения производного бензилового эфира 2-аминоникотиновой кислоты, которое является полезным соединением в качестве эффективного компонента сельскохозяйственного фунгицида, с высоким выходом при высокой чистоте с высоким объемным коэффициентом полезного действия и слабым воздействием на окружающую среду.
Уровень техники
[0002]
В качестве общих способов получения производного сложного эфира известен способ, включающий хлорирование производного карбоновой кислоты с использованием галогенирующего реагента и взаимодействия полученного продукта с производным спирта в органическом растворителе в присутствии основания, способ включает взаимодействие производного карбоновой кислоты с производным спирта в органическом растворителе с использованием конденсирующего реагента, и аналогичные способы.
[0003]
В качестве способа получения производного эфира 2-аминоникотиновой кислоты, в Патентной литературе 1 раскрыт способ получения производного эфира 2-аминоникотиновой кислоты, описывающийся следующим уравнением реакции:
в которой M означает щелочной металл и X означает атом галогена.
Хотя указанный выше способ, описанный в Патентной литературе 1, может дать производное эфира 2-аминоникотиновой кислоты с высоким выходом при высокой чистоте, вязкость раствора реакционной смеси склонна быть высокой и при производстве затруднительно обеспечить объемный коэффициент полезного действия, равный 10% или более. Кроме того, поскольку используется растворимый в воде полярный растворитель, невозможно исключить затруднения, связанные с воздействием на окружающую среду при рециркуляции растворителя и обработкой жидких отходов после проведения реакции.
Список литературы
Патентная литература
[0004]
Патентная литература 1: WO2015/097850
Сущность изобретения
Задача, решаемая изобретением
[0005]
В Патентной литературе 1 указано, что при обычном подходе общи способ получения производного сложного эфира не является предпочтительным для получения производного эфира 2-аминоникотиновой кислоты. В частности, когда производное эфира 2-аминоникотиновой кислоты хлорируют, раствор реакционной смеси становится коричневым. В этом случае целевая реакция замедляется и увеличивается количество побочных продуктов. Кроме того, реакция не завершается даже при использовании конденсирующего реагента. Кроме того, способ, описанный в Патентной литературе 1, не является необходимым оптимальным способом получения вследствие низкого объемного коэффициента полезного действия и воздействия на окружающую среду. Поэтому необходима разработка промышленного способа производства для получения целевого соединения с высоким выходом высокой чистоты и с обеспечением высокого объемного коэффициента полезного действия и слабым воздействием на окружающую среду.
Средства решения задачи
[0006]
Авторы настоящего изобретения тщательно исследовали решение задач, описанных выше, и в результате разработали способ, включающий взаимодействие производного 2-аминоникотиновой кислоты с производным бензила при высокой концентрации с использованием ароматического углеводородного растворителя в присутствии межфазного катализатора или каталитического количества третичного амина, и завершили настоящее изобретение.
[0007]
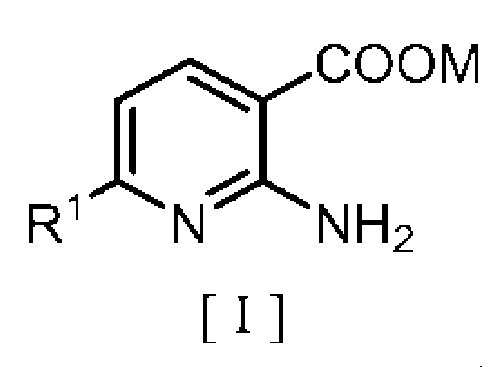
Точнее, настоящее изобретение относится к способу получения производного бензилового эфира 2-аминоникотиновой кислоты, включающему:
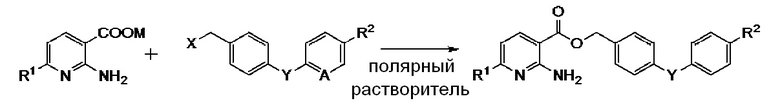
взаимодействие производного 2-аминоникотиновой кислоты, описывающегося следующей формулой [I]:
в которой R1 означает атом водорода или C1-C4 алкильную группу и M означает атом водорода или щелочной металл, с
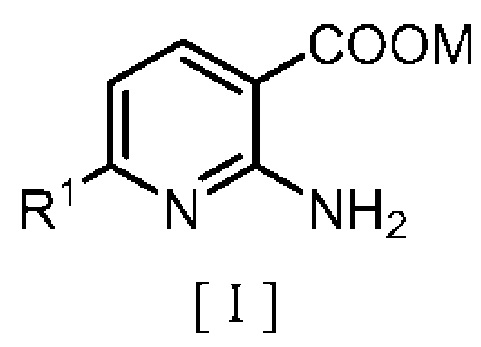
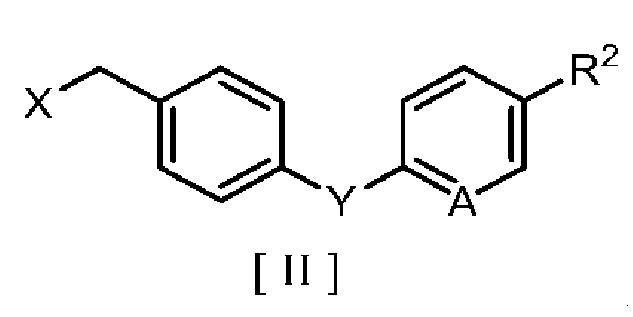
производным бензила, описывающимся следующей формулой [II]:
в которой R2 означает атом водорода, атом галогена, C1-C4 алкильную группу или C1-C4 алкоксигруппу; A означает атом азота или метиновую группу (CH); X означает гидроксигруппу или атом галогена; и Y означает атом кислорода, метиленовую группу (CH2) или метиленоксигруппу (OCH2),
a) с использованием основания, где M означает атом водорода в формуле [I], и
b) с использованием галогенирующего реагента, где X означает гидроксигруппу в формуле [II], и
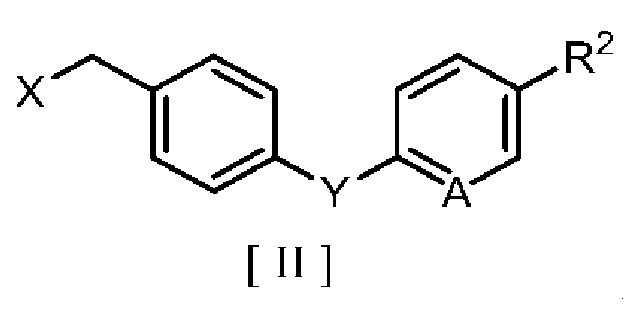
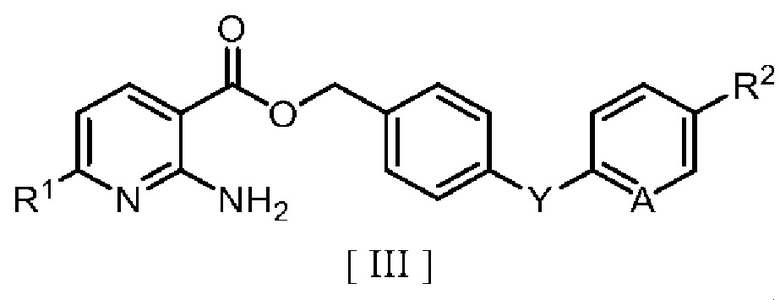
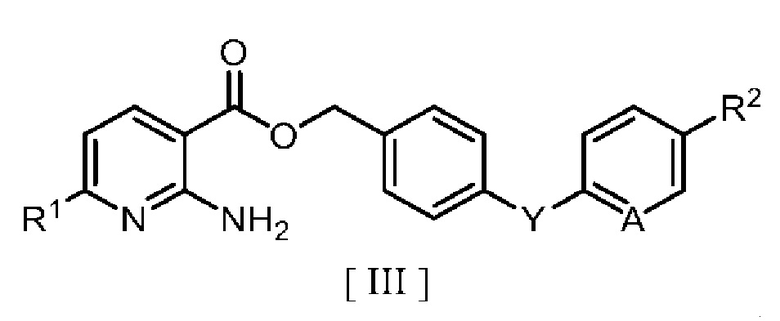
затем взаимодействие производного 2-аминоникотиновой кислоты с производным бензила в ароматическом углеводородном растворителе в присутствии межфазного катализатора или третичного амина с получением производного бензилового эфира 2-аминоникотиновой кислоты, описывающегося следующей формулой [III]:
в которой R1, R2, A и Y являются такими, как определено для формул [I] и [II].
Эффект изобретения
[0008]
В соответствии с предложенным способом получения предполагается, что производное бензилового эфира 2-аминоникотиновой кислоты получают с высоким выходом, с более высоким объемным коэффициентом полезного действия и меньшим воздействием на окружающую среду, чем в обычных способах. Точнее, использование ароматического углеводородного растворителя в качестве плохого растворителя может подавить повышение вязкости даже для взвеси высокой концентрации и обеспечить перемешиваемость, лучшую, чем при использовании полярного растворителя. Это повышает производительность при использовании такой же производственной аппаратуры. Кроме того, поскольку ароматический углеводородный растворитель можно легко использовать повторно, настоящее изобретение эффективно для уменьшения воздействия на окружающую среду.
Описание варианта осуществления изобретения
[0009]
Настоящее изобретение подробно описано ниже.
В формулах [I], [II] и [III], описанных выше, C1-C4 алкильная группа, описывающаяся с помощью R1 и R2, включает метильную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу и т. п.; атом галогена, описывающийся с помощью R2 и X, включает атом фтора, атом хлора, атом брома и атом йода; C1-C4 алкоксигруппа, описывающаяся с помощью R2, включает метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, н-бутоксигруппу, изобутоксигруппу, втор-бутоксигруппу и трет-бутоксигруппу; и щелочной металл, описывающийся с помощью M включает натрий и калий.
[0010]
Способ получения, предлагаемый в настоящем изобретении, может включать стадии: если M в приведенной выше формуле [I] означает водород, добавление основания, такого как гидроксид щелочного металла, например, гидроксид натрия или гидроксид калия, карбонат щелочного металла, такой как карбонат натрия или карбонат калия и т. п., в количестве, равном, например, от 1 до 10 молей на 1 моль, предпочтительно от 1 до 5 молей на 1 моль в пересчете на соединение приведенной выше формулы [I], в ароматическом углеводородном растворителе, таком как бензол, толуол или ксилол, с последующим перемешиванием, например, в течение от 1 мин до 1 ч при температуре от 0 до 100°C, предпочтительно в течение от 5 до 30 мин при температуре от 20 до 60°C; и если X в приведенной выше формуле [II] означает гидроксигруппу, добавление галогенирующего реагента, такого как тионилхлорид, тионилбромид или оксихлорид фосфора в количестве, равном, например, от 1,0 до 3,0 молей на 1 моль, предпочтительно от 1,0 до 1,5 молей на 1 моль в пересчете на соединение приведенной выше формулы [II], в ароматическом углеводородном растворителе, таком как бензол, толуол или ксилол, с последующим перемешиванием, например, в течение от 5 мин до 1 ч при температуре от -5 до 30°C, предпочтительно в течение от 10 до 40 мин при температуре от 0 до 25°C. После проведения указанной выше обработки так, как это необходимо, соединения приведенной выше формул [I] и [II] смешивают в ароматическом углеводородном растворителе, таком как бензол, толуол или ксилол, и затем межфазный катализатор, такой как тетрабутиламмонийхлорид, тетрабутиламмонийбромид, тетрабутиламмониййодид, бензилтриэтиламмонийбромид, 15-краун-5-эфир или 18-краун-6-эфир или третичный амин, такой как триметиламин, триэтиламин, диметилизопропиламин, тетраметилэтилендиамин или 1,4-диазабицикло[2.2.2]октан добавляют в количестве, равном, например, от 0,001 до 0,1 молей на 1 моль, предпочтительно от 0,005 до 0,05 молей на 1 моль в пересчете на соединение приведенной выше формулы [I], с последующим перемешиванием в течение от 5 мин до 10 ч при температуре от 0 до 130°C, предпочтительно в течение от 2 до 6 ч при температуре от 70 до 120°C. После проведения реакции раствор реакционной смеси перегоняют при пониженном давлении для выпаривания, например, от 10 до 100% органического растворителя. Воду со льдом выливают в раствор реакционной смеси, с последующим перемешиванием, например, в течение от 5 до 30 мин, предпочтительно в течение от 10 до 20 мин, и осадившиеся кристаллы собирают фильтрованием и сушат. Таким путем соединение приведенной выше формулы [III] в качестве целевого соединения можно очень легко получить с высоким выходом с высокой чистотой. Кроме того достаточно использовать ароматический углеводородный растворитель только в количеств, например, от 3 до 5 раз превышающем максимальное теоретическое значение для получаемого целевого соединения, и объемный коэффициент полезного действия достигает примерно от 20 до 35%. Кроме того, ароматический углеводородный растворитель можно легко отделить от воды путем перегонки раствора реакционной смеси или фильтрата при пониженном давлении и затем использовать повторно для уменьшения воздействия на окружающую среду.
[0011]
В способе получения, предлагаемом в настоящем изобретении,, ароматический углеводородный растворитель, использующийся в реакции между гидроксидом щелочного металла и соединением приведенной выше формулы [I], где M в формуле [I] означает атом водорода и ароматический углеводородный растворитель, использующийся в реакции между галогенирующим реагентом и соединением приведенной выше формулы [II], могут быть одинаковыми или разными, но предпочтительно если они являются одинаковыми.
[0012]
Производное никотиновой кислоты, описывающееся указанной выше формулой [I], которое используется в способе получения, предлагаемом в настоящем изобретении, можно легко синтезировать из общеизвестного соединения, например, по методике, описанной в JP-A-2010-083861.
Производное спирта, описывающееся указанной выше формулой [II], в которой X означает гидроксигруппу, которое используется в способе получения, предлагаемом в настоящем изобретении, можно легко синтезировать из общеизвестного соединения, например, по методике, описанной в Journal of Medicinal Chemistry, vol. 43, p. 1826 (2000).
Соединение приведенной выше формулы [III], которое получают способом получения, предлагаемым в настоящем изобретении, применимо в качестве сельскохозяйственного фунгицида.
Примеры
[0013]
Настоящее изобретение дополнительно описано ниже со ссылкой на примеры, но объем настоящего изобретения ни в коей мере не ограничивается приведенными ниже примерами.
[0014]
Пример 1
Синтез 4-феноксибензилового эфира 2-амино-6-метилникотиновой кислоты
Сначала 2-амино-6-метилникотинат калия (0,95 г) суспендировали в толуоле (5 мл) и к нему добавляли (1-хлорметил)-4-феноксибензол (1,10 г) и 18-краун-6-эфир (40 мг), затем перемешивали в течение 6 ч с нагреванием при 105°C. Раствор реакционной смеси охлаждали до комнатной температуры и примерно 80% толуола выпаривали при пониженном давлении. К остатку добавляли воду со льдом, затем перемешивали в течение 20 мин при комнатной температуре. Осадившиеся кристаллы собирали фильтрованием и сушили и получали 1,52 г (выход 91,6%, объемный коэффициент полезного действия равен 33,4%) искомого соединения (соединение 2, описанное в таблице 1).
1H-ЯМР (CDCl3) δ част./млн.: 2,38 (3H,s), 5,25 (2H,s), 6,06-6,72 (2H.br), 6,44 (1H,d), 6,99-7,04 (4H,m), 7,12 (1H,t), 7,31-7,41 (4H,m), 8,04 (1H,d)
[0015]
Пример 2
Синтез 4-феноксибензилового эфира 2-амино-6-метилникотиновой кислоты
Сначала 2-амино-6-метилникотинат калия (0,95 г) суспендировали в м-ксилол (5 мл) и к нему добавляли (1-хлорметил)-4-феноксибензол (1,10 г) и тетраметилэтилендиамин (19 мг), затем перемешивали в течение 6 ч с нагреванием при 110°C. Раствор реакционной смеси охлаждали до комнатной температуры и примерно 80% м-ксилола выпаривали при пониженном давлении. К остатку добавляли воду со льдом, затем перемешивали в течение 20 мин при комнатной температуре. Осадившиеся кристаллы собирали фильтрованием и сушили и получали 1,41 г (выход 84,4%, объемный коэффициент полезного действия равен 33,4%) искомого соединения (соединение 2, описанное в таблице 1).
[0016]
Пример 3
Синтез 4-феноксибензилового эфира 2-амино-6-метилникотиновой кислоты
Сначала 2-амино-6-метилникотинат калия (0,95 г) суспендировали в м-ксилол (5 мл) и к нему добавляли (1-хлорметил)-4-феноксибензол (1,10 г) и тетрабутиламмонийбромид (49 мг), затем перемешивали в течение 6 ч с нагреванием при 110°C. Раствор реакционной смеси охлаждали до комнатной температуры и примерно 90% м-ксилола выпаривали при пониженном давлении. К остатку добавляли воду со льдом, затем перемешивали в течение 20 мин при комнатной температуре. Осадившиеся кристаллы собирали фильтрованием и сушили и получали 1,55 г (выход 92,8%, объемный коэффициент полезного действия равен 33,4%) искомого соединения (соединение 2, описанное в таблице 1).
[0017]
Пример 4
Синтез 4-феноксибензилового эфира 2-амино-6-метилникотиновой кислоты
Сначала 2-амино-6-метилникотиновую кислоту (2,28 г) суспендировали в м-ксилол (15 мл) и к нему добавляли гидроксид натрия (1,20 г), затем перемешивали в течение 30 мин при 40°C и получали раствор 1. С другой стороны, (4-феноксифенил)метанол (3,00 г) растворяли в м-ксилоле (5 мл), при охлаждении льдом по каплям добавляли тионилхлорид (1,1 мл), затем перемешивали в течение 30 мин, давая нагреться до комнатной температуры. Затем к этому реакционному раствору добавляли раствор 1, к нему добавляли тетрабутиламмонийбромид (145 мг), затем перемешивали в течение 6 ч с нагреванием при 110°C. Раствор реакционной смеси охлаждали до комнатной температуры и примерно 90% м-ксилола выпаривали при пониженном давлении. К остатку добавляли воду со льдом, затем перемешивали в течение 30 мин при комнатной температуре. Осадившиеся кристаллы собирали фильтрованием и сушили и получали 4,17 г (выход 83,2%, объемный коэффициент полезного действия равен 20%) искомого соединения (соединение 2, описанное в таблице 1).
[0018]
Пример 5
Синтез 4-феноксибензилового эфира 2-амино-6-метилникотиновой кислоты
Сначала 2-амино-6-метилникотиновую кислоту (3,04 г) суспендировали в м-ксилол (20 мл), к нему добавляли карбонат калия (4,15 г), затем перемешивали в течение 30 мин при 40°C и получали раствор 1. С другой стороны, (4-феноксифенил)метанол (4,00 г) растворяли в м-ксилоле (7 мл), при охлаждении льдом по каплям добавляли тионилхлорид (1,46 мл), затем перемешивали в течение 30 мин, давая нагреться до комнатной температуры. Затем, хлорид водорода, диоксид серы и тионилхлорид выпаривали из системы, добавляли раствор 1, добавляли тетрабутиламмонийбромид (194 мг), затем перемешивали в течение 4 ч с нагреванием при 110°C. Раствор реакционной смеси охлаждали до комнатной температуры и примерно 95% м-ксилола выпаривали при пониженном давлении. К остатку добавляли воду со льдом, затем перемешивали в течение 30 мин при комнатной температуре. Осадившиеся кристаллы собирали фильтрованием и сушили и получали 6,39 г (выход 95,7%, объемный коэффициент полезного действия равен 25%) искомого соединения (соединение 2, описанное в таблице 1).
[0019]
В представленной ниже таблице 1 приведены данные не только для соединения 2, предлагаемого в настоящем изобретении, но и для соединений 1 и 3-8, предлагаемых в настоящем изобретении, которые получены таким же способом, как в примере 1.
[0020]
Таблица 1
[0021]
Как показано выше, предложенный способ получения является способом, обладающим большой промышленной ценностью и дает производное бензилового эфира 2-аминоникотиновой кислоты в качестве сельскохозяйственного фунгицида.
Настоящее изобретение относится к способу получения производного бензилового эфира 2-аминоникотиновой кислоты. Способ получения производного бензилового эфира 2-аминоникотиновой кислоты, включает взаимодействие производного 2-аминоникотиновой кислоты, описывающегося следующей формулой [I], в которой R1 означает атом водорода или C1-C4 алкильную группу и M означает атом водорода или щелочной металл, с производным бензила, описывающимся следующей формулой [II], в которой R2 означает атом водорода, атом галогена, C1-C4 алкильную группу или C1-C4 алкоксигруппу, A означает атом азота или метиновую группу (CH), X означает гидроксигруппу или атом галогена, и Y означает атом кислорода, метиленовую группу (CH2) или метиленоксигруппу (OCH2), (a) с использованием основания, где M означает атом водорода в формуле [I], и (b) с использованием галогенирующего реагента, где X означает гидроксигруппу в формуле [II], и затем взаимодействие производного 2-аминоникотиновой кислоты с производным бензила в ароматическом углеводородном растворителе в присутствии межфазного катализатора или третичного амина с получением производного бензилового эфира 2-аминоникотиновой кислоты, описывающегося следующей формулой [III], в которой R1, R2, A и Y являются такими, как определено для формул [I] и [II]. Также предложены варианты способа получения производного бензилового эфира 2-аминоникотиновой кислоты. Технический результат – получение производного бензилового эфира 2-аминоникотиновой кислоты с высоким выходом при высокой чистоте, с высоким объемным коэффициентом полезного действия. 3 н.п. ф-лы, 1 табл., 5 пр.
 ,
,  ,
,
1. Способ получения производного бензилового эфира 2-аминоникотиновой кислоты, включающий:
взаимодействие производного 2-аминоникотиновой кислоты, описывающегося следующей формулой [I]:
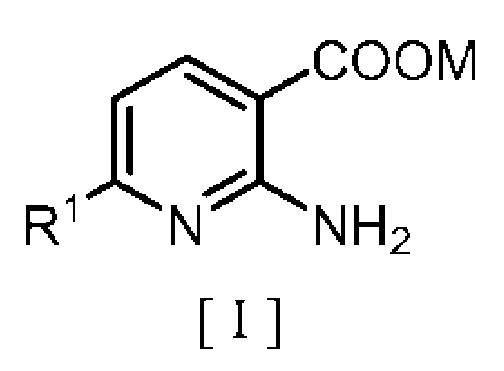
в которой R1 означает атом водорода или C1-C4 алкильную группу и M означает атом водорода или щелочной металл, с
производным бензила, описывающимся следующей формулой [II]:
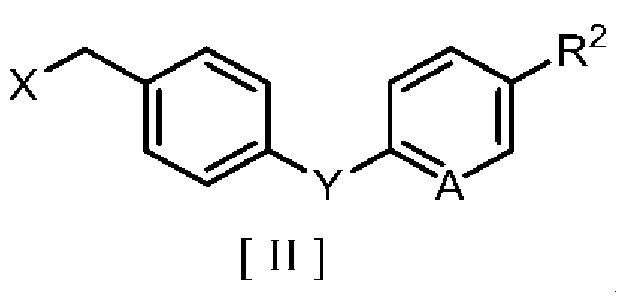
в которой R2 означает атом водорода, атом галогена, C1-C4 алкильную группу или C1-C4 алкоксигруппу, A означает атом азота или метиновую группу (CH), X означает гидроксигруппу или атом галогена, и Y означает атом кислорода, метиленовую группу (CH2) или метиленоксигруппу (OCH2),
a) с использованием основания, где M означает атом водорода в формуле [I], и
b) с использованием галогенирующего реагента, где X означает гидроксигруппу в формуле [II], и
затем взаимодействие производного 2-аминоникотиновой кислоты с производным бензила в ароматическом углеводородном растворителе в присутствии межфазного катализатора или третичного амина с получением производного бензилового эфира 2-аминоникотиновой кислоты, описывающегося следующей формулой [III]:
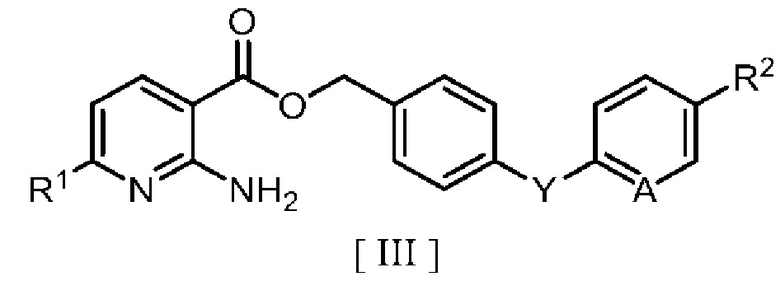
в которой R1, R2, A и Y являются такими, как определено для формул [I] и [II].
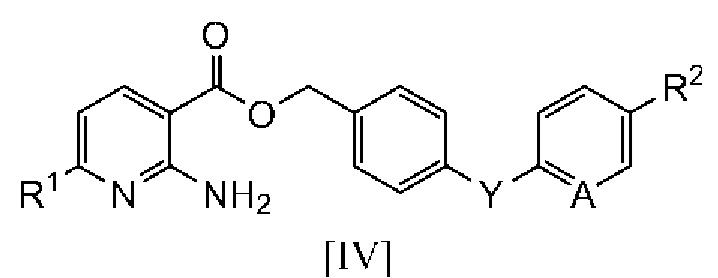
2. Способ получения производного бензилового эфира 2-аминоникотиновой кислоты, включающий:
взаимодействие производного неорганической соли 2-аминоникотиновой кислоты, описывающегося следующей формулой [IV]:
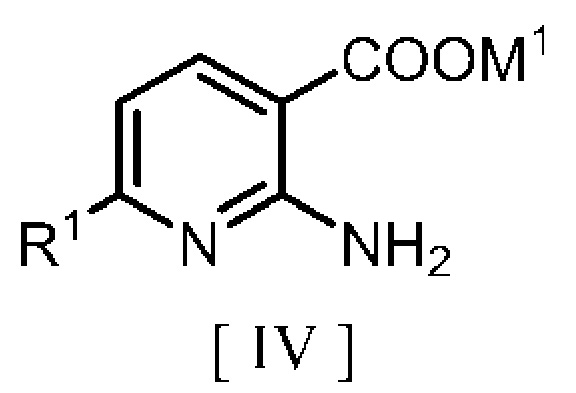
в которой R1 означает атом водорода или C1-C4 алкильную группу и M1 означает щелочной металл, с
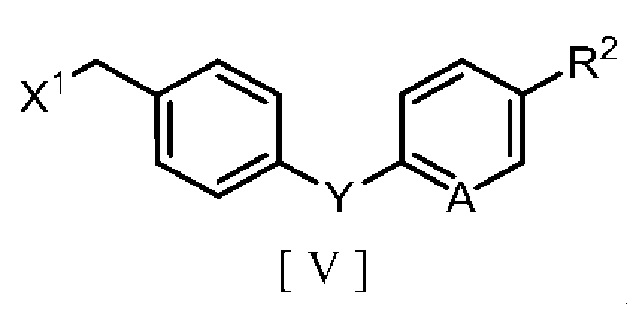
производным бензилгалогенида, описывающимся следующей формулой [V]:
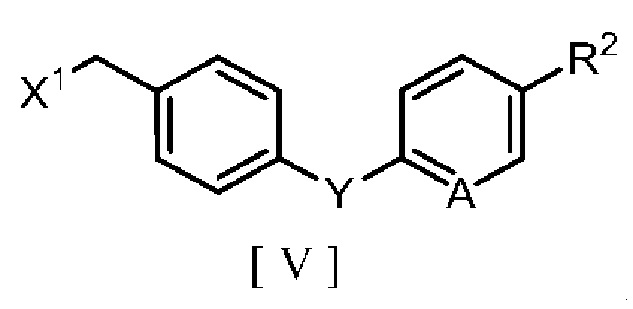
в которой R2 означает атом водорода, атом галогена, C1-C4 алкильную группу или C1-C4 алкоксигруппу, A означает атом азота или метиновую группу (CH), X1 означает атом галогена, и Y означает атом кислорода, метиленовую группу (CH2) или метиленоксигруппу (OCH2), в ароматическом углеводородном растворителе в присутствии межфазного катализатора или третичного амина с получением производного бензилового эфира 2-аминоникотиновой кислоты, описывающегося следующей формулой [III]:
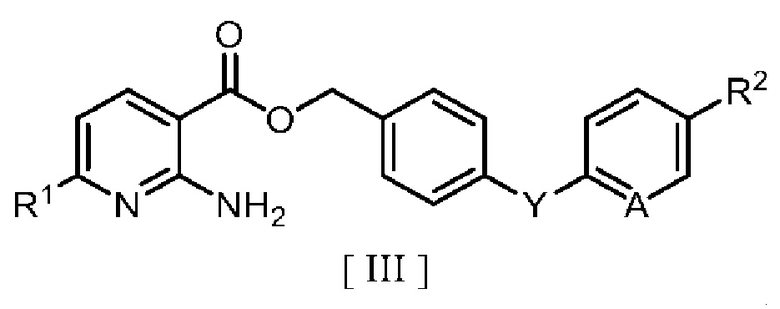
в которой R1, R2, A и Y являются такими, как определено для формул [IV] и [V].
3. Способ получения производного бензилового эфира 2-аминоникотиновой кислоты, включающий:
взаимодействие производного 2-аминоникотиновой кислоты, описывающегося следующей формулой [VI]:
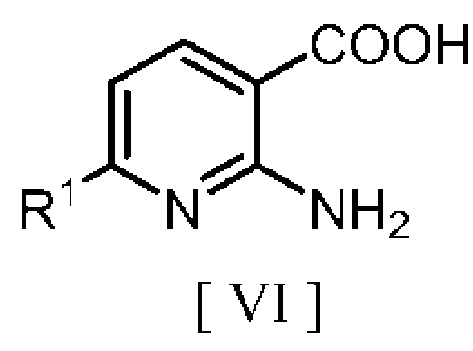
в которой R1 означает атом водорода или C1-C4 алкильную группу, с
соединением, полученным взаимодействием с галогенирующим реагентом производного бензилового спирта, описывающегося следующей формулой [VII]:
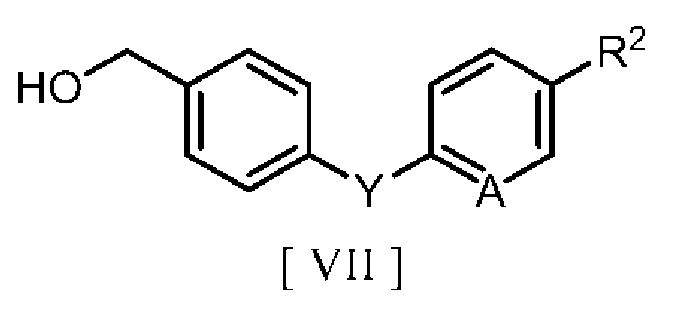
в которой R2 означает атом водорода, атом галогена, C1-C4 алкильную группу или C1-C4 алкоксигруппу, A означает атом азота или метиновую группу (CH), и Y означает атом кислорода, метиленовую группу (CH2) или метиленоксигруппу (OCH2), т.е. производного бензилгалогенида, описывающегося следующей формулой [II]:
в которой R2, A и Y являются такими, как определено для формулы [VII], и X1 означает атом галогена, в ароматическом углеводородном растворителе в присутствии основания и межфазного катализатора или третичного амина с получением производного бензилового эфира 2-аминоникотиновой кислоты, описывающегося следующей формулой [III]:
в которой R1, R2, A и Y являются такими, как определено для формул [VI] и [VII].
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| US 6200999 B1, 13.03.2001 | |||
| Цепной ветряный двигатель | 1923 |
|
SU1625A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |

