Область техники, к которой относится изобретение
Настоящее изобретение относится к новым аналогам гликосфинголипидов, являющимся промежуточными продуктами при их получении. Более конкретно, изобретение относится к новым способам получения гликосфинголипидов. В частности, изобретение относится к способам синтеза и применению новых альфа-связанных гликосфинголипидных соединений.
Уровень техники
Исследования показывают, что естественные киллерные Т-клетки (NKT-клетки), уникальная субпопуляция лимфоцитов, характеризуются коэкспрессией инвариантного антигенного рецептора и рецепторов естественных киллеров. Естественные киллерные Т-клетки человека (Vα24-Jα18) активируются специфическим гликолипидным антигеном, CD1d-зависимым способом. Молекулы CD1d представляют собой гетеродимеры, состоящие из тяжелой полипептидной цепи, нековалентно связанной с 2-микроглобулином, и обладают существенным структурным сходством с протеинами главного комплекса гистосовместимости (МНС) класса I. После активации естественные киллерные Т-клетки проявляют МНС-независимую противоопухолевую активность в отношении опухолевых клеток in vitro и in vivo по нескольким механизмам. Активированные Vα24 NKT-клетки продуцируют значительное количество цитокинов, таких как интерферон-гамма (IFN-γ), тем самым объединяя врожденный и приобретенный иммунитет активацией других эффекторных клеток, включая дендритные клетки (DC), естественные киллерные клетки (NK-клетки) и CD8+ Т-клетки. NKT-клетки играют регуляторную роль в иммунной системе, следовательно, они являются перспективными целями в иммунотерапии.
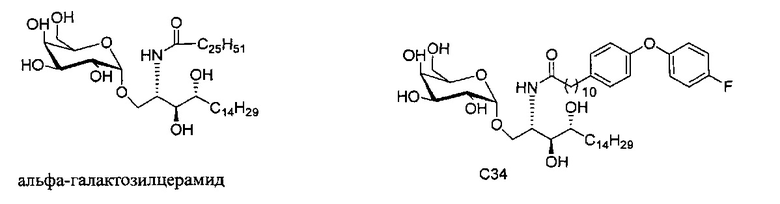
В настоящее время альфа-галактозилцерамид (αGalCer, KRN-7000) является наиболее полно изученным CD1d презентируемым антигеном. Первоначально он вызвал интерес, когда были продемонстрированы новые противоопухолевые свойства экстрактов из морской губки, Agelas mauritianus. Фармацевтическая компания Kirin Beer (патент США №5849716). Позже эта выраженная активность была отмечена у группы альфа-связанных гликосфинголипидов (GSLs), от которых структурной оптимизацией был получен αGalCer. Гликосфинголипиды состоят из сахаридной группы альфа-связанной с церамидом, образованным амидной связью жирной кислоты с длинноцепочечным основанием.
При активации αGalCer естественные киллерные Т-клетки высвобождают провоспалительный (Th1) и иммуномодулирующий (Th2) цитокин. Считается, что выработка ТН1 цитокинов коррелирует с противоопухолевым, противовирусным/антибактериальным и адъювантным действием, тогда как выработка ТН2 цитокина считается подавляющей аутоиммунные заболевания. αGalCer был объектом клинических испытаний в связи с его противораковым потенциалом, но они были прекращены на этапе I. Неспецифическая природа профиля цитокина, индуцированного αGalCer , а также обоих Th1 и Th2, делает его менее эффективным в плане терапевтического лечения. Это объяснение побудило многих исследователей сосредоточить свое внимание на поиске соединений которые повышают селективность в отношении ответной реакции на TH1 или на TH2 цитокины. Уонг и коллеги (Wong et al.) синтезировали ряд гликолипидов, несущих ароматические группы в ацильной цепи и установили, что эти молекулы смещают профиль высвобождения цитокина в сторону ответной реакции на TH1 (J. Am. Chem. Soc. 2006, 128, 9022-9023. US 2007/0238871). В эксперименте in vivo на мышах с агрессивными раковыми опухолями легкого (клеточная линия ТС1) и раковыми опухолями молочной железы (клеточная линия 4Т1) было показано, что у мышей с раком легкого, которых лечили новыми гликолипидами, значительно увеличилось время жизни по сравнению с мышами, которых лечили αGalCer. У мышей, пораженных раком молочной железы, лечение новыми гликолипидами замедляло темп роста опухоли на 75% относительно группы, не получавшей лечения, по сравнению с 50% замедлением у мышей, которых лечили α-GalCer (Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 10299-10304).
Следовательно, существует потребность в эффективном средстве для синтеза альфа-гликосфинголипидов, таких как соединения αGalCer, а также потребность в синтезе новых альфа-гликосфинголипидных соединений, обладающих иммуномодулирующим эффектом.
Раскрытие изобретения
Настоящее изобретение предоставляет новые способы синтеза галактозилсфинголипидов, включая новые соединения, относящиеся к альфа-галактозилцерамидам и их активным аналогам, таким как С34.
По одному из вариантов осуществления изобретение предоставляет соединение структурной формулы (1):
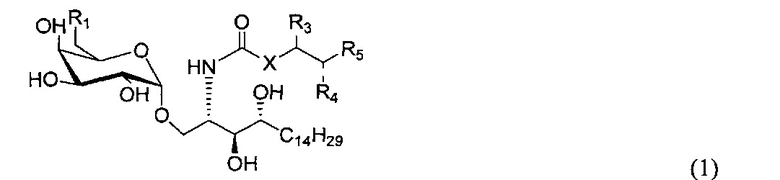
где R1=OH, NH2, NHCOR2; R2=Н или алкил, алкенил или алкил с концевой арильной группой, замещенный арил, гетероарил, или замещенный гетероарил; X = алкильная группа, алкенил; R3, R4=H, ОН; R5 = арил, замещенный арил, гетероарил или замещенный гетероарил.
По одному из вариантов осуществления изобретения соединение формулы 1 может быть получено способом, включающим в числе прочих этап удаления бензилиденовой защитной группы и гидрогенизации соединения структурной формулы (2):
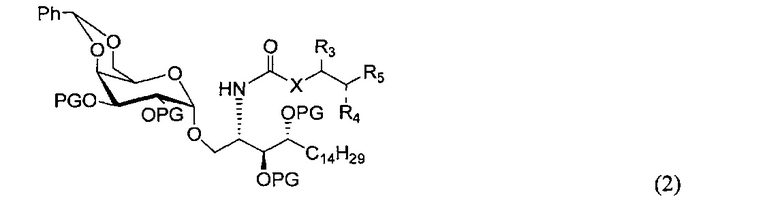
где PG является защитной группой гидроксила.
По другому варианту осуществления изобретения защитной группой гидроксила может быть бензил.
По одному из вариантов осуществления изобретения соединение формулы 2, где X=(CH2)8, R3=H, R4=H, R5=4-F-фенокси-фенил, может быть получено способом, включающим в числе прочих этап образования амидной связи между соединениями формулы (3) и формулы (4).
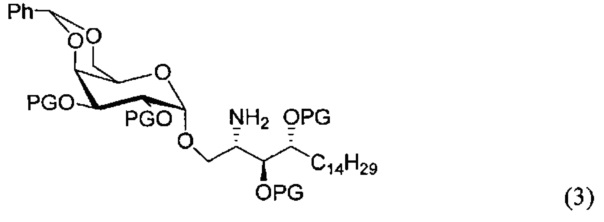
где по одному из вариантов осуществления PG является бензилом

где по одному из вариантов осуществления, X = алкильная группа, алкенил, R3=H, ОН, R4=Н, OH, R5 = арил, замещенный арил, гетероарил, или замещенный гетероарил.
По одному из вариантов осуществления изобретения соединение формулы (3), где R является бензилом, может быть получено способом, включающим в числе прочих этап восстановления азидной группы соединения структурной формулы (5):
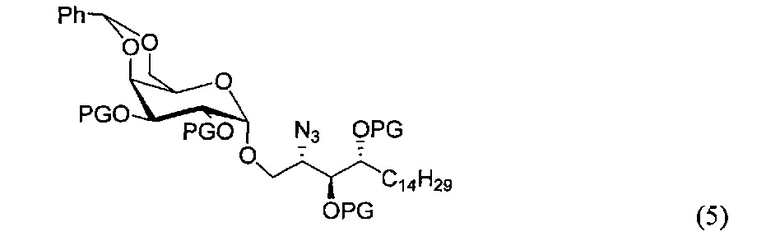
По одному из вариантов осуществления изобретения соединение формулы 5, где PG является бензилом, может быть получено способом, включающим в числе прочих этап реакции соединения структурной формулы (6)

где PG является защитной группой гидроксила и LG является уходящей группой, с соединением структурной формулы (7)

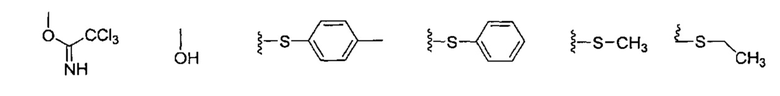
где PG является защитной группой гидроксила, с образованием альфа-гликозидной связи, с получением, таким образом, соединения формулы (5). По другому варианту осуществления изобретения замещаемой группой из формулы (6) может быть любая группа из числа:
По одному из вариантов осуществления изобретения соединение формулы 1 может быть получено способом, включающим в числе прочих этап удаления бензилиденовой защитной группы и гидрогенизации соединения структурной формулы (8):
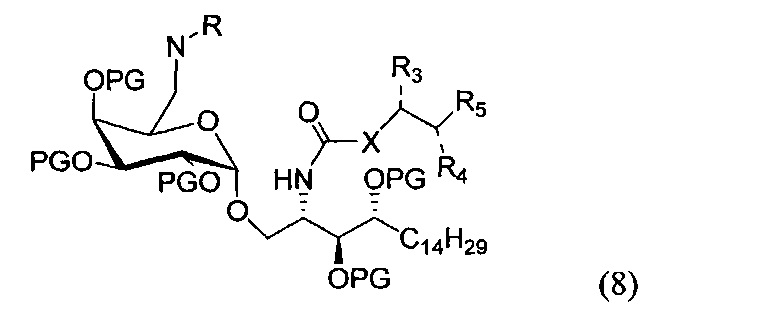
где PG является защитной группой гидроксила. По другому варианту осуществления изобретения PG может быть в числе прочих бензилом.
По одному из вариантов осуществления изобретения соединение формулы (8) может быть получено способом, включающим в числе прочих этап реакции соединения структурной формулы (9) с соединением, представленным алкановой кислотой, арильной кислотой, арил-алкановой кислотой, замещенной арил-алкановой кислотой и гетероциклической кислотой.
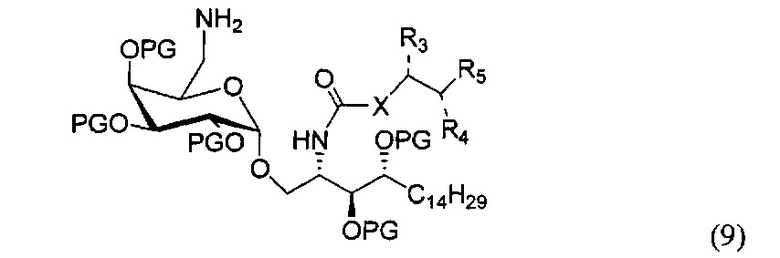
где PG является защитной группой гидроксила.
По одному из вариантов осуществления изобретения соединение (9) может быть получено способом, включающим в числе прочих этап восстановления азида структурной формулы (10)
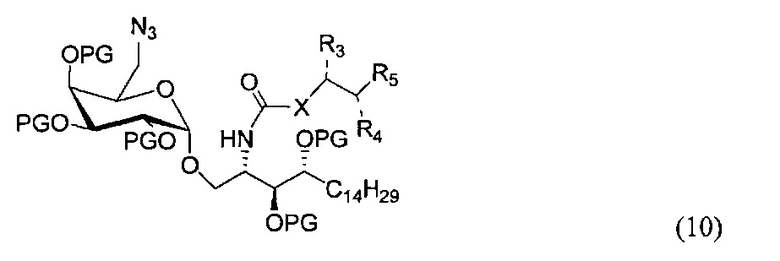
где PG является защитной группой гидроксила.
По одному из вариантов осуществления изобретения соединение (10) может быть получено способом, включающим в числе прочих этап замещения соединения структурной формулы (11)
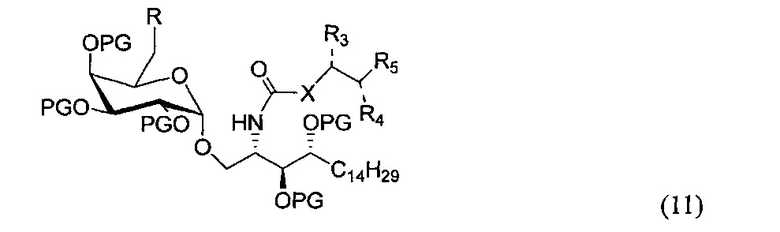
где R является уходящей группой, с получением, таким образом, соединения формулы (11).
По другому варианту осуществления изобретения R может быть в числе прочих метансульфонилом или толуолсульфонилом. По одному из вариантов осуществления изобретения соединение формулы (11) может быть получено способом, включающим в числе прочих этап проведения замещения гидроксильной группы соединения структурной формулы (12)
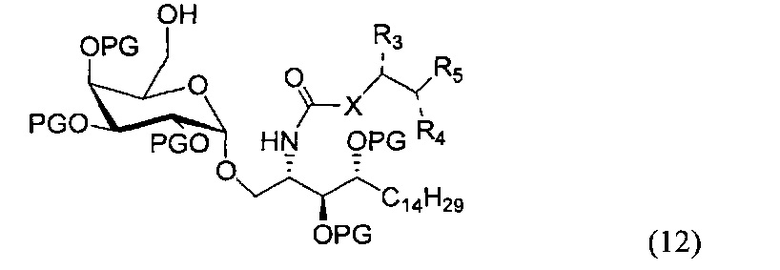
По другому варианту осуществления изобретения замещение может проводиться в присутствии основания и метансульфонил хлорида или толуолсульфонил хлорида.
По одному из вариантов осуществления изобретения соединение (12) может быть получено способом, включающим в числе прочих этап гидролиза соединения структурной формулы (13)
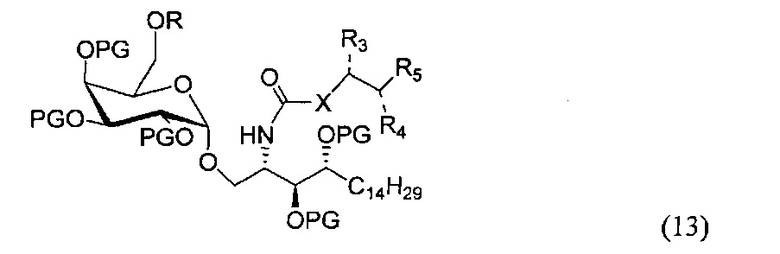
где R является защитной группой гидроксила, по одному из вариантов осуществления изобретения R является сложным эфиром алкила. По другому варианту осуществления изобретения R является ацетатом.
По одному из вариантов осуществления изобретения соединение (13) может быть получено способом, включающим в числе прочих этап образования амидной связи между соединением структурной формулы (14),
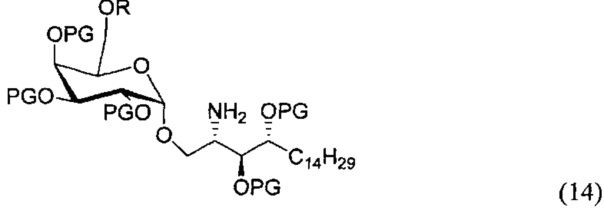
и соединением структурной формулы (4).
По одному из вариантов осуществления изобретения соединение (14) может быть получено способом, включающим в числе прочих этап восстановления азида структурной формулы (15).
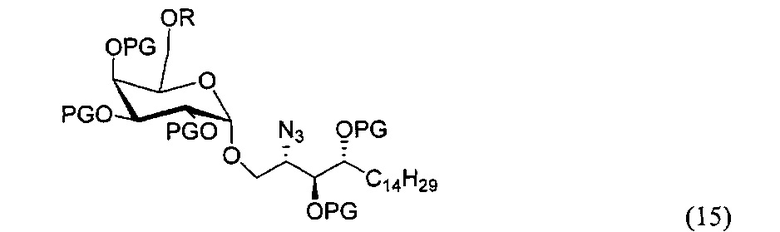
По одному из вариантов осуществления изобретения соединение (15) может быть получено способом, включающим в числе прочих этап реакции соединения структурной формулы (16)

где PG является защитной группой гидроксила, LG является уходящей группой и R является сложным эфиром, с соединением структурной формулы (7), где PG является защитной группой гидроксила с образованием альфа-гликозидной связи, с получением, таким образом, соединения формулы (15).
По другому варианту осуществления изобретения уходящей группой может быть любая группа из числа:
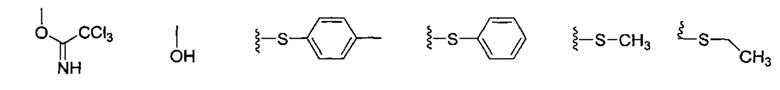
По одному из вариантов осуществления изобретения соединение формулы (17) может быть получено способом, включающим в числе прочих этап удаления защитной группы гидроксила PG1, с получением, таким образом, соединения формулы (17), при этом PG1 может быть в числе прочих тритилом.
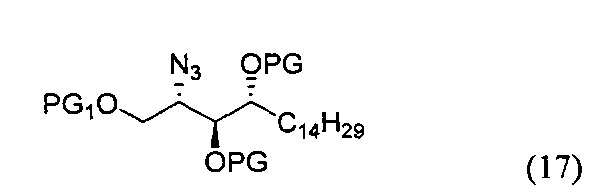
Эти и другие аспекты станут очевидными из следующего описания предпочтительных вариантов осуществления в сочетании с приведенными ниже чертежами, причем очевидно, что могут быть осуществлены другие реализации и модификации в рамках изобретения без отступления от существа и объема новых концепций раскрытия.
Краткое описание чертежей
Следующие чертежи являются частью настоящего описания, они были включены для дополнительной демонстрации некоторых аспектов настоящего раскрытия, изобретения в рамках которого будут более понятными при наличии ссылок на один или несколько этих чертежей в сочетании с подробным описанием конкретных вариантов осуществления, представленных в настоящем документе.
Фиг.1 является схематической иллюстрацией, описывающей синтез С34, А15-21. Схема 1. Реактивы и условия: (a) TfN3, K2CO3, CuSO4, дихлорметан, МеОН, H2O; (b) тритил хлорид, триэтиламин, толуол; (c) бензилхлорид (BnCl), NaH, диметилформамид, толуол; (d) HCl, толуол, МеОН; (е) А5, Me2S2-Tf2O, тетрагидрофуран, 4 Å MS; (f) LiAlH4, тетрагидрофуран; (g) RCO2H, HBTU, N-метилморфолин, дихлорметан; (h) Pd(OH)2, H2, МеОН, дихлорметан.
Фиг.2 является схематической иллюстрацией, описывающей синтез соединения А23-А25. Схема 2. Реактивы и условия: (a) Pd(OH)2, Н2 (80 фунтов/кв. дюйм), МеОН, дихлорметан, АсОН; (b) RCO2H, HBTU, N-метилморфолин, дихлорметан МеОН.
Фиг.3 является схематической иллюстрацией, описывающей синтез соединения формулы (4). Схема 3.
Фиг.4 является схематической иллюстрацией, описывающей синтез соединений C5-C7. Схема 4. Реактивы и условия: (a) PPh3, тетрагидрофуран, H2O; (b) Ph(CH2)nCO2H, HBTU, N-метилморфолин, дихлорметан; (c) Pd(OH)2, Н2, дихлорметан, МеОН.
Фиг.5 является схематической иллюстрацией, описывающей синтез соединений C20-C31. Схема 5. Реактивы и условия: (a) TsCl, пиридин; (b) NaN3, диметилформамид; (c) PPh3, тетрагидрофуран, H2O; (d) RCH2CO2H, HBTU, N-метилморфолин, МеОН, дихлорметан.
Фиг.6 показывает индуцированную гликосфинголипидами секрецию IL-2 в клеточной системе A20CD1d и mNK1,2. Данные приведены как среднее ± среднеквадратическое отклонение; "-" указывает на отсутствие соединения.
Фиг.7 показывает секрецию INF-гамма цитокина спленоцитами самки мыши C57BL/6.
Фиг.8 показывает секрецию IL-4 цитокина спленоцитами самки мыши C57BL/6.
Фиг.9 показывает соотношения секреции цитокина, полученные при сравнении фиг.7 и 8.
Осуществление изобретения
Термины, использованные в этом описании, как правило, имеют свое обычное значение в данной области в контексте изобретения и в конкретном контексте, в котором использован каждый термин. Некоторые термины, которые были использованы для описания изобретения, обсуждаются ниже или в других местах описания для предоставления дополнительных указаний практикам применительно к описанию изобретения. Для удобства некоторые термины могут быть выделены, например, курсивом и/или кавычками. Использование выделения не влияет на объем и значение термина; объем и значение термина остаются теми же в том же контексте, независимо от того, был он выделен или нет. Следует понимать, что одно и то же может быть выражено несколькими способами. Следовательно, для любого или нескольких обсуждаемых здесь терминов могут быть использованы альтернативные выражения и синонимы, независимо от того, развивается или обсуждается термин в настоящем документе или нет, он не принимает никакого специального значения. Для некоторых терминов приведены синонимы. Указание одного или нескольких синонимов не исключает использование других синонимов. Использование примеров в любой части этого описания, включая примеры любых обсуждаемых здесь терминов, приведено только для иллюстрации, и ни в коей мере не ограничивает объем и значение изобретения или любого термина, представленного в качестве примера. Аналогично, изобретение не ограничивается различными вариантами осуществления, приведенными в этом описании.
Если не определено иное, все технические и научные термины, использованные в настоящем документе, имеют такое значение, в котором их обычно понимает специалист в области, к которой относится изобретение. При возникновении споров, преимущественной силой будет обладать настоящий документ, включая определения.
Новые способы синтеза соединений формулы (1) в целом включают связывание церамида с сахаридом, но также существует возможность сначала провести связывание с фитосфингозином, а затем преобразовать аминогруппу в амидогруппу для формирования соединения формулы 1.
Примером такого синтеза может служить синтез соединения формулы (1), в котором галактоза C6' является гидроксильной группой во время последовательных этапов (см. Фиг.1-3).
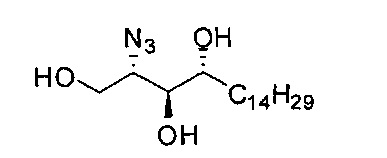
Исходным материалом является фитосфингозин гидрохлорид ((2S,3S,4R)-2-амино-1,3,4-октадекантриол), хотя существует ряд способов синтеза фитосфингозина согласно описанию в Curr. Org. Chem. 2002, 6, 365-391. Промышленно выпускаемый источник - фитосфингозин гидрохлорид готовят из соответствующего дрожжевого ферментативного бульона, который можно получать в больших количествах по разумной цене (Evonik Degussa Taiwan Ltd.). Изомерные сфингозины, конфигурация которых отличается от таковой природного сфингозина, можно приготовить в соответствии с методами, описанными в Helvetica Chimica Acta 1957, 470, 1145; или в Chem. Commun. 1991, 820.
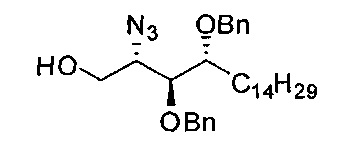
На первом этапе аминогруппа фитосфингозина была преобразована в азидогруппу реакцией диазопереноса свежеприготовленным TfN3 с получением А1. С приготовлением TfN3 можно ознакомиться в Tetrahedron Lett 1996, 37, 6029-6032. Защита тритилом первичного спирта А1 дала неочищенный А2, который был напрямую подвергнут воздействию условий бензилирования NaH и BnCl с получением соединения A3. В данном методе, когда бензильная группа применяется как защитная группа гидроксильной группы, можно также применять другие соответствующие группы, такие как бензоат, (p-метокси)-бензил или изопропилиден. Для синтеза соединения с А1 по A3 в качестве растворителя был выбран толуол. Реакцию бензилирования вторичных спиртов не удавалось провести в толуоле, если он был единственным растворителем. Для решения проблемы низкой химической активности реакции бензилирования в толуоле была использована система сорастворителей из 10% диметилформамида в толуоле с целью повышения растворимости NaH и промежуточного алкоксида. После промывания водой был получен неочищенный A3 в виде раствора в толуоле, и было проведено удаление защитной группы кислотой с получением акцептора гликозила А4. Для удаления защитной тритиловой группы можно использовать разные кислоты, такие как соляная кислота, серная кислота, бромистый водород, трифторуксусная кислота, BF3.OEt, азотная кислота, смоляная кислота (например, амберлит (Amberlite IR120®)) и тому подобные.
Раньше при синтезе гликолипидов применяли различные способы гликозилирования, включая использование гликозил фторида, гликозил трихлорацетоимидата, гликозил бромида и гликозил иодида20-23. Tetrahedron 1998, 54, 3141-3150; J Org Chem 2005, 70, 10260-10270; J Org Chem 2002, 67, 4559-4564; Chem Commun 2007, 2336-2338. Первоначально в нашем синтезе применяли гликозил имидат, который обеспечивал отличный выход (89%) и селективность по отношению к аномерам (α/β=9/1). В связи с тем, что имидат легко подвергается гидролизу, и обычно необходимо применять свежеприготовленный раствор, при применении этой замещаемой группы для гликозилирования при проведении синтеза в большом масштабе могут возникать проблемы с хранением и очисткой. В качестве альтернативы можно в качестве донора получить тиогликозид А5 с использованием кислоты Льюиса, такой как TMSOTf, Tf2O, BF3⋅OEt2, TfOH, Me2S2-Tf2O, в качестве катализаторов и с применением молекулярных сит для дегидратации. Соединение А5 содержит азидо-группу, которая способствует гликозилированию, тогда как аминогруппа фитосфингозина защищена амидом или карбаматом (t-бутил карбаматом) как показано в US 5849716; US 2007/0238871; J. Am. Chem. Soc. 2004, 126, 13602-13603; J. Org. Chem. 2005, 70, 10260-10270; Tetrahedron 1998, 54, 3141-3150; Synthesis 2004, 847-850; Bioorg. Med. Chem. Lett. 2006, 16, 2195-2199. 2-NH и 1-OH могут образовывать внутримолекулярную водородную связь, которая мешает ему как нуклеофилу вступать в реакцию с активированным гликозидом, результатом этого является низкий выход реакции гликозилирования. После гликозилирования между А4 и А5 очистка в колонне может дать А6 в чистой α-форме и в чистой β-форме.
Соединение А6, которое содержит азидо-группу, можно восстановить с помощью любого из: алюмогидрида лития, борогидрида натрия, боранового комплекса, фосфинового комплекса, ферментного восстановления, гидрогенизации или трансферной гидрогенизации. Вместо использования фосфинового комплекса, образующего побочный продукт - оксид фосфина, который трудно удалить, восстановление азида алюмогидридом лития (LiAlH4) давало высокочистый амин А7. Соединение А7 сочетали с различными полученными карбоновыми кислотами (способы получения, см. Фиг. 3) с образованием соответствующих амидных соединений. В целом, снятие защитных групп у этих соединений достигалось в условиях гидрогенолиза в присутствии катализатора гидрогенизации, выбранного из Pd/C, Pd(OH)2 или ренеевского никелевого катализатора, и Н2 в смешанных растворителях МеОН и CH2CI2 с получением аналогов С34, соединений А15-21.
Восстановительное дегалогенирование арил галидов на ацильной цепи проводится реакцией гидрогенизации хлоро- и бромо-ацил содержащих соединений. Поэтому для предупреждения реакции дегалогенирования с А6 удаляли защитные группы и восстанавливали гидрогенизацией в присутствии каталитического Pd(OH)2 и Н2. После этого образовавшийся амин А22 сочетали с соответствующими кислотами (способы приготовления, см. Фиг. 3) в условиях сочетания с получением аналогов А23-25 (Фиг. 2).
Для этого процесса известен ряд способов проведения реакций, в частности для амидирования. Можно также использовать ацил хлорид и ангидрид кислоты или карбоновую кислоту. Карбоновая кислота используется в реакции конденсации в присутствии соответствующего конденсирующего реагента. Соответствующие конденсирующие реагенты, используемые в этой реакции, включают дициклогексилкарбодиимид (DCC), 1-этил-3-(3'диметиламинопропил)карбодиимин (EDC), а также 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурониум гексафторфосфат (HBTU), гидроксибензотриазол (HOBt) или тому подобные. Для обеспечения быстрого протекания реакции добавляют органическое основание, такое как триэтиламин, пиридин, N-метилморфолин, диметиланилин, 4-диметиламинопиридин, N-метилпиперидин, N-метилпирролидин. Растворителем может быть любой инертный растворитель, который не будет вовлечен в реакцию, такой как тетрагидрофуран, простой этиловый эфир, толуол, хлороформ, метилен хлорид, этилацетат, ацетон или тому подобные.
Как можно видеть на Фиг.3 для получения различных замещенных фенилалкановых кислот применяется реакция Виттига. В этом способе ω-бромо-алкановые кислоты смешивают с трифенил фосфином в присутствии растворителя. Реакцию обычно проводят с соответствующим растворителем, но если реакция протекает вяло, ее можно усилить проведением реакции в отсутствие растворителя. Растворителем может быть любой инертный растворитель, который не будет вовлечен в реакцию, например, толуол, бензол, диглим, диметилсульфид или тому подобные.
В качестве еще одного примера синтеза, можно также синтезировать соединение формулы (1) проведением различных замещений в позиции галактозы С6', выполнив следующие этапы (см. Фиг.4-5).
Синтез модифицированных аналогов С6 был начат с соединения С1 согласно описанию в Org Lett 2002, 4, 1267-1270. Восстановление азида, затем амидирование с различными выпускаемыми ароматическими кислотами и полное снятие защитных групп дало С5-С6 (Фиг.4, схема 4). Кроме того, для предотвращения трудоемкого взаимного преобразования защитных групп для приготовления из соединения C1 при синтезе С6'' и би-модифицированных соединений с ацильной цепью (Фиг.5) была применена иная стратегия. Гидроксильную группу C6'' А15 можно тозилировать или мезилировать толуол сульфонил хлоридом или метан сульфонил хлоридом в присутствии основания, такого как пиридин, диметилпиридин, триэтиламин, диэтилпропиламин, DBU или тому подобных, и затем соответствующую группу тозилата или мезилата можно заместить азидом реакцией с азидом натрия с получением С18. Восстановление азида по Штаудингеру, амидирование с различными кислотами дает C20-31.
Применение соединения по настоящему изобретению
Соединения формулы (1), обладающие следующей физиологической активностью, можно применять в качестве иммунотерапевтического средства против рака или в качестве иммуностимулирующего средства против других болезней.
Активность по активации АРС: секрецию IL-2 можно измерить в системе клеток A20CD1d и mNK1,2 как АРС и эффекторных клеток, как показано в примере 2.
Иммуностимулирующая активность: секрецию цитокина IFN-γ и Il-4 можно измерить, как показано в примере 2, в котором самка мыши C57BL/6 (16w4d) была забита, и селезенка использована для анализа.
Противоопухолевое средство: соединение по настоящему изобретению обладает смещенным в сторону Th1 профилем секреции цитокина и может быть использовано как вещество, обладающее противоопухолевой активностью и иммуностимулирующим действием.
Наряду с тем, что соединения в настоящем изобретении могут применяться как иммунотерапевтическое средство против опухолей, их можно применять отдельно или в сочетании с химиотерапией или радиотерапией.
Соединения по настоящему изобретению в качестве противоопухолевого средства или иммуностимулирующего средства можно вводить любыми соответствующими способами введения дозы. Соединение обычно превращают в препарат, в растворенной форме или сформованный с фармацевтически приемлемым носителем (липосома или мицелла). При применении соединения по настоящему изобретению, его можно вводить перорально или парентерально человеку или млекопитающему. Например, соединения по настоящему изобретению при применении в форме инъекции, можно вводить внутривенно, внутримышечно, подкожно или в виде ингаляций в форме раствора, суспензии или эмульсии с соответствующим растворителем. В этом случае при необходимости можно добавить в качестве солюбилизирующего вещества полисорбаты или макрогол, холестерол. При пероральном приеме соединений по настоящему изобретению они могут быть в форме таблетки, порошка, гранулы, или сухого сиропа в соответствующем разбавителе.
ПРИМЕРЫ
Без намерения ограничить объем изобретения ниже приведены типовые приборы, аппарат, способы и связанные с ними результаты, соответствующие вариантам осуществления настоящего изобретения. Следует иметь в виду, что для удобства читателя в примерах могут быть использованы заглавия или подзаголовки, которые ни в коей мере не будут ограничивать объем изобретения. Кроме того, в документе предложены и раскрыты некоторые теории; однако, независимо от того, являются ли они правильными или нет, они ни в коем случае не будут ограничивать объем изобретения, поскольку изобретение воплощается на практике по изобретению без учета какой бы то ни было конкретной теории или плана действий.
Пример 1
Ниже описаны методы синтеза и физико-химические свойства соединений по настоящему изобретению (номера соединений, участвующих в процессе синтеза, приведены на схемах реакций, которые показаны на Фиг.1-5).
На приведенных в настоящем документе схемах использованы следующие сокращения:
Другие сокращения имеют те же значения, как и сокращения на схемах, приведенные выше.
Схема синтеза 1 (Фиг.1)
Пути конкретно показывают способ получения соединения С34, А13, А14, А15, А16, А18, А19, А20, А21, этим способом можно также синтезировать соединения по настоящему изобретению.
Синтез (2S,3S,4R)-2-азидо-D-рибо-октадекан-1,3,4-триола (А1)
К раствору азида натрия (64,3 г, 989 мМоль) в 250 мл воды добавили дихлорметан (350 мл). Двухфазную смесь охладили до 5°C в ледяной бане и в течение 20 мин добавляли трифторметансульфоновый ангидрид (47,5 мл, 283 мМоль), поддерживая температуру на уровне ниже 10°C. После перемешивания в течение 2,5 часов в ледяной бане реакцию в реакционной смеси остановили добавлением 70 мл насыщенного раствора K2CO3. Органическую фазу отделили, а водную фазу экстрагировали CH2Cl2 (200 мл). Органические фазы соединили и получили раствор азида трифторметансульфоновой кислоты в дихлорметане. [Осторожно! Азид трифторметансульфоновой кислоты является взрывчатым веществом, его необходимо хранить с растворителем.]
К раствору сульфата меди (0,35 г, 1,4 мМоль) в воде (150 мл) добавили фитосфингозин гидрохлорид (фитосфингозин гидрохлорид высокой степени очистки [(2S,3S,4R)-2-амино-1,3,4-октадекантриол] из соответствующего дрожжевого ферментативного бульона, который производят для продажи в больших количествах по разумной цене, 50,0 г, 141 мМоль), карбонат калия (29,28 г, 211,9 мМоль) и метанол (1,0 л). Суспензию охладили до 0-5°C в льдосоляной бане и в течение 10 мин добавляли раствор азида трифторметансульфоновой кислоты (в 550 мл CH2Cl2). После перемешивания реакционной смеси в течение 12 часов при комнатной температуре, смесь концентрировали. Остаток развели в воде (1,0 л) и перемешивали при комнатной температуре в течение 12 часов. Осадок отфильтровали и промыли водой (500 мл×2). Осадок высушили азеотропной дистилляцией (70-80°C, 200-250 мм рт.ст.) с толуолом (1,5 л) и получили (2S,3S,4R)-2-азидо-D-рибо-октадекан-1,3,4-триол (А1) (47,0 г, 137 мМоль, 97%) в виде белого твердого вещества с сероватым или желтоватым оттенком, точка плавления: 87°C. 1H-NMR (CD3OD, 400 мГц) δ 3,85 (dd, J=11,6, 3,3 Гц, 1Н), 3,69 (dd, J=11,6, 7,9 Гц, 1H), 3,50-3,55 (m, 1Н), 3,43-3,47 (m, 2Н), 1,22-1,60 (m, 26Н), 0,83 (t, J=6,4 Гц, 3Н). 13C-NMR (CD3OD, 100 МГц) δ 74,6, 71,5, 65,3, 61,2, 32,5, 31,7, 29,4, 29,4, 29,1, 25,4, 22,4, 13,1. HRMS (ESI) в пересчете на O18H37N3O3Na [M+Na]+: 366,2733, установлено: 366,2729.
Синтез (2S,3S,4R)-2-азидо-3,4-ди-O-бензил-D-рибо-октадекан-1,3,4-триола (А4)
К смеси (2S,3S,4R)-2-азидо-D-рибо-октадекан-1,3,4-триола (47,0 г, 137 мМоль) в толуоле (1,0 л) добавили триэтиламин (46 мл, 332 мМоль) и тритил хлорид (42,0 г, 151 мМоль). После перемешивания при 50-55°C в течение 6 часов добавили триэтиламин (4,6 мл, 33 мМоль) и трифенилметил хлорид (4,20 г, 15,1 мМоль), затем смесь перемешивали еще 15 часов. К смеси добавили воду (1,0 л) и перемешали в течение 3 мин. Органическую фазу вымыли водой (1,0 л, 500 мл) провели концентрацию и получили неочищенный (2S,3S,4R)-2-азидо-1-тритил-D-рибо-октадекан-1,3,4-триол (А2). Очистку образца для анализа методом NMR провели колоночной хроматографией. 1Н-NMR (CDCl3, 400 МГц) δ 7,22-7,47 (m, 15Н), 3,62 (dd, J=3,7, 10,1 Гц, 2Н), 3,53 (m, 2Н), 3,40 (dd, J=6, 10,1 Гц, 1H), 2,35 (brs, 1H), 1,83 (brs, 1H), 1,20-1,52 (m, 26Н), 0,87 (t, J=6,8 Гц, 3Н). 13C-NMR (CDCl3, 100 МГц) δ 143,35, 128,54, 128,01, 127,27, 87,78, 74,19, 72,18, 63,48, 62,31, 31,90, 31,74, 29,67, 29,64, 29,60, 29,55, 29,34, 25,59, 22,67, 14,10.
К раствору неочищенного (2S,3S,4R)-2-азидо-1-тритил-D-рибо-октадекан-1,3,4-триола (А2) в толуоле (750 мл) и диметилформамида (75 мл) добавили гидрид натрия (60% в минеральном масле, 21,9 г, 548 мМоль) тремя порциями в течение 10 мин. Смесь перемешивали в течение 30 мин., после чего в реакционную смесь добавили бензилхлорид (50,5 мл, 0,438 мМоль). Смесь нагрели до 50-60°C и перемешивали в течение 18 часов. Затем смесь охладили до 0°C и по каплям добавили воду (50 мл). Органическую фазу промыли насыщенным раствором хлорида аммония (500 мл×2) и водой (500 мл×2). Органическую фазу профильтровали через целитовую подушку, и провели концентрацию фильтрата для получения неочищенного (2S,3S,4R)-2-азидо-2,3-ди-бензил-1-тритил-D-рибо-октадекан-1,3,4-триола (A3).
К раствору A3 в смеси толуол/метанол (600 мл, 1/1) добавили водный раствор HCl (33%, 6,0 мл). Смесь нагрели до 50-60°C и перемешивали в течение 20 часов. Реакцию в реакционной смеси остановили добавлением 1,0 N NaOH (55 мл) и провели концентрацию. Остаток разделили толуолом (500 мл) и водой (500 мл). Провели концентрацию органической фазы, и очистку остатка провели колоночной хроматографией (неочищенная фаза 100 г, силикагель 500 г, этилацетат/n-гексан = 1/10) с получением (2S,3S,4R)-2-азидо-2,3-ди-бензил-D-рибо-октадекан-1,3,4-триола (А4) (27,5 г, 52,5 мМоль, 38% за 4 этапа) в форме желтого жидкого масла. 1H-NMR (CDCl3, 200 МГц) δ 7,26-7,39 (m, 10Н), 4,69 (d, J=1,4 Гц, 2Н), 4,59 (d, J=4,3 Гц, 2Н), 3,59-3,94 (m, 5Н), 1,26-1,61 (m, 26Н), 0,88 (t, J=6,4 Гц, 3Н). 13C-NMR (CDCl3, 50 МГц) δ 137,9, 137,6, 128,5, 128,4, 128,1, 127,97, 127,81, 127,10, 80,38, 78,96, 73,59, 72,49, 63,03, 62,20, 21,90, 30,16, 29,66, 29,35, 25,43, 22,67, 14,11. HRMS (ESI) в пересчете на C32H49N3O3Na [M+Na]+: 546,3672, установлено: 546,3689.
Синтез 4-метилфенил 2,3-O-дибензил-4,6-O-бензилиден 1-тиол-D-галактопиранозида (А5)
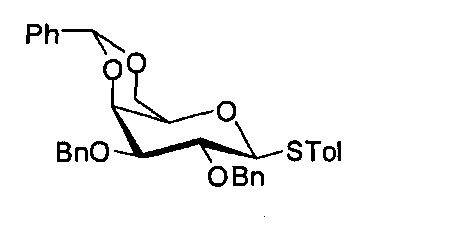
Соединение А5 может быть получено методом, описанным в Plettenburg, О. et al. J. Org. chem. 2002, 67, 4559-4564.
Данные о соединении А5: 1Н-NMR (CDCl3, 400 МГц) δ 7,58 (d, J=8,1 Гц, 1Н), 7,50 (m, 2Н), 7,23-7,42 (m, 15Н), 6,98 (d, J=8,0 Гц, 1H), 5,46 (s, 1Н), 4,69 (m, 4Н), 3,54 (d, J=9,5 Гц, 1Н), 4,35 (dd, J=12,3, 1,5 Гц, 1H), 4,12 (d, J=3,2 Гц, 1Н), 4,03 (dd, J=9,8, 3,6 Гц, 1Н), 3,82 (t, J=9,3 Гц, 1H), 3,60 (dd, J=9,2, 3,4 Гц, 1H), 3,39 (s, 1Н), 2,28 (s, 3Н).
Синтез 2-азидо-3,4-ди-O-бензил-1-O-(2,3-ди-O-бензил-4,6-O-бензилиден-α-D-галактопиранозил)-D-рибо-октадекан-1,3,4-триола (А6)
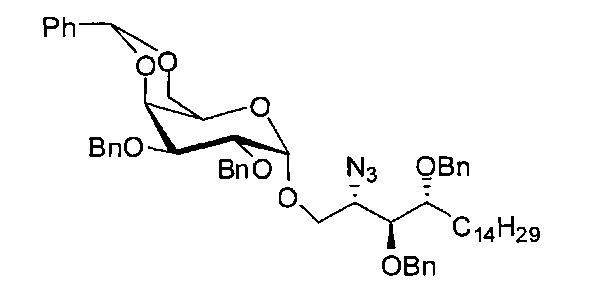
К раствору диметил дисульфида (10,0 мл, 113 мМоль) в дихлорметане (75 мл) добавили трифторметансульфоновый ангидрид (17 мл, 100 мМоль) при 0-5°C в льдосоляной бане. После перемешивания реакционной смеси в льдосоляной бане в течение 30 мин получили Me2S2-Tf2O в виде 1,0 М раствора в дихлорметане, его можно хранить в ледяной бане в течение 3 часов.
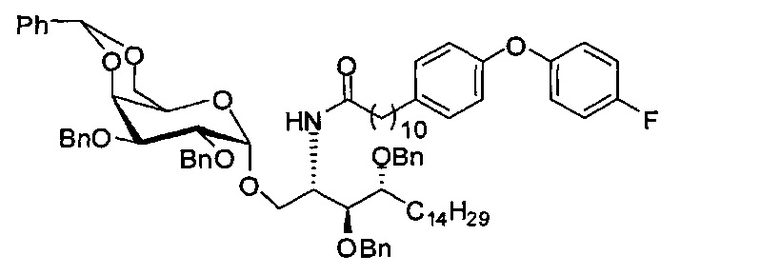
Соединение А4 (27,3 г, 52,2 мМоль), А5 (34,7 г, 62,6 мМоль) и молекулярное сито 4 Å (33 г) смешали и высушили под вакуумом в течение 1 часа, затем к смеси добавили тетрагидрофуран (520 мл). Смесь охладили до -10°C в льдосоляной бане перед добавлением Me2S2Tf2O (1,0 М раствор в CH2Cl2, 94 мл, 94 мМоль). После перемешивания в течение 20 мин реакцию в реакционной смеси остановили добавлением триэтиламина (22 мл), затем разбавили дихлорметаном (200 мл). Смесь профильтровали через целитовую подушку и промыли дихлорметаном (50 мл). Провели концентрацию объединенного фильтрата и разделили его дихлорметаном (500 мл) и водой (500 мл). Провели концентрацию органической фазы, и остаток очистили колоночной хроматографией (вес неочищенной фазы = 153 г, 600 г силикагеля, этилацетат : n-гексан = 1:15 до 1:12 до 1:10) и получили 2-азидо-3,4-ди-O-бензил-1-O-(2,3-ди-O-бензил-4,6-O-бензилиден-α-D-галактопиранозил)-D-рибо-октадекан-1,3,4-триол (А6) (32,07 г, 33,60 мМоль, 64%) в виде белого воска с сероватым или желтоватым оттенком, точка плавления: 59°C. 1H-NMR (CDCl3, 400 МГц) δ 7,22-7,50 (m, 25Н), 5,44 (s, 1H), 4,96 (d, J=3,4 Гц, 1Н), 4,85 (d, J=11,9 Гц, 1Н), 4,79 (d, J=12,3 Гц, 1H), 4,73 (d, J=12,3 Гц, 1H), 4,66-4,69 (m, 2Н), 4,59 (d, J=8,5 Гц, 1H), 4,56 (d, J=12,8 Гц, 1H), 4,48 (d, J=11,5 Гц, 1Н), 4,15 (d, J=2,8 Гц, 1H), 4,06-4,12 (m, 1H), 3,98-4,04 (m, 3Н), 3,86 (dd, J=12,5, 1,5 Гц, 1H), 3,68-3,73 (m, 3H), 3,60-3,62 (m, 1H), 3,55 (s, 1H), 1,25-1,55 (m, 26H), 0,87 (t, J=7,1 Гц, 3H). 13C-NMR (CDCl3, 100 МГц) δ 138,75, 138,36, 138,01, 137,82, 128,85, 128,37, 128,34, 128,26, 128,22, 128,09, 127,90, 127,79, 127,75, 127,70, 127,66, 127,61, 127,50, 127,45, 126,33, 101,05, 99,13, 79,41, 78,95, 76,68, 75,80, 75,44, 74,66, 73,77, 73,49, 72,06, 72,03, 69,31, 68,43, 62,97, 61,80, 31,91, 30,01, 29,75, 29,69, 29,67, 29,65, 29,63, 29,60, 29,35, 25,44, 22,68, 14,10. 
Синтез 3,4-ди-O-бензил-1-O-(2,3-ди-O-бензил-4,6-O-бензилиден-α-D-галактопиранозил)-2-(11-(4-(4-фторфенокси)фенил)ундеканоил)амино-D-рибо-октадекан-1,3,4-триола (А8)
К раствору соединения А7 (32,07 г, 33,61 мМоль) в тетрагидрофуране (340 мл), охлажденному в ледяной бане, за два приема добавили алюмогидрид лития (1,910 г, 50,33 мМоль). Смесь была возвращена к комнатной температуре, и ее перемешивали в течение 70 мин. Смесь охладили до 0°C перед тем, как последовательно остановить реакцию добавлением воды (1,9 мл), 1,0 N NaOH (3,8 мл) и воды (1,9 мл). Смесь профильтровали через целитовую подушку и промыли дихлорметаном (100 мл). Фильтрат концентрировали и разделили дихлорметаном (350 мл) и водой (350 мл).
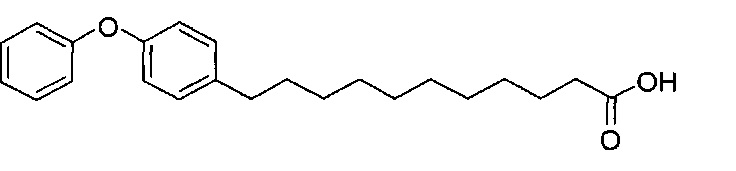
К выделенной органической фазе добавили 11-(4-(4-фторфенокси)фенил)) ундекановую кислоту (В4) (11,27 г, 30,26 мМоль), затем N-метилморфолин (9,2 мл, 84 мМоль) и HBTU (19,12 г, 50,41 мМоль). После перемешивания при комнатной температуре в течение 12 часов смесь профильтровали и промыли 50 мл CH2Cl2. Объединенный фильтрат промыли насыщенным раствором хлорида аммония (400 мл) и водой (400 мл). Провели концентрацию органической фазы и очистку колоночной хроматографией (вес неочищенной фазы = 46 г, 350 г силикагеля, этилацетат/n-гексан = 1/6 до 1/5 до 1/4) с получением А8 (36,06 г, 28,11 мМоль, 84%) в виде белого воска с сероватым или желтоватым оттенком. 1H-NMR (CDCl3, 400 МГц) δ 6,86-7,51 (m, 33Н), 5,84 (d, J=8,0 Гц, 1Н), 5,44 (s, 1Н), 4,94 (d, J=3,5 Гц, 1Н), 4,83 (d, J=11,6 Гц, 1H), 4,69-1,74 (m, 3Н), 4,63 (d, J=11,6 Гц, 1Н), 4,57 (d, J=11,6 Гц, 1Н), 4,50 (d, J=3,9 Гц, 1H), 4,47 (d, J=3,9 Гц, 1Н), 4,24-А31 (m, 1Н), 4,16 (d, J=3,1 Гц, 1Н) 4,03-4,12 (m, 3Н), 3,92 (dd, J=10,3, 3,6 Гц, 2Н), 3,73-3,79 (m, 2Н), 3,56 (s, 1Н), 3,51-3,53 (m, 1Н), 2,55 (t, J=7,6 Гц, 2Н), 1,87-1,91 (m, 2Н), 1,19-1,69 (m, 42H), 0,87 (t, J=6,6 Гц, 3H). 13C-NMR (CDCl3, 100 МГц) δ 172,89, 159,76, 157,36, 155,36, 153,36, 153,33, 138,63, 138,52, 138,50, 138,38, 137,81, 137,79, 129,53, 128,84, 128,64, 128,42, 128,35, 128,31, 128,29, 128,08, 127,88, 127,80, 127,69, 127,59, 127,57, 127,55, 126,29, 120,08, 120,00, 118,31, 116,24, 116,01, 100,99, 99,59, 79,81, 79,48, 76,68, 76,14, 75,68, 74,33, 73,81, 73,28, 71,88, 71,69, 69,39, 68,13, 62,91, 60,36, 50,32, 36,69, 35,17, 31,90, 31,61, 30,24, 29,78, 29,69, 29,67, 29,65, 29,59, 29,56, 29,51, 29,42, 29,35, 29,29, 25,81, 25,68, 22,66, 14,17, 14,10. HRMS (ESI) в пересчете на C82H105FNO10 [M+H]+: 1282,7723, установлено: 1282,7731.
Синтез 1-O-(α-D-галактопиранозил)-2-(11-(4-(4-фторфенокси)фенил)ундеканоил)амино-D-рибо-октадекан-1,3,4-триола (С34)
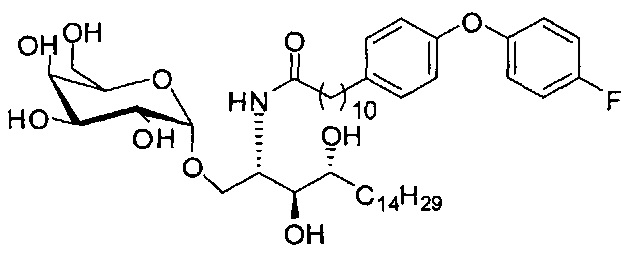
К раствору А8 (36,06 г, 28,11 мМоль) в смеси дихлорметан/метанол (200 мл, дихлорметан/метанол = 1/1) добавили гидроксид палладия (1,8 г). Смесь перемешивали в атмосфере водорода (5 бар) при комнатной температуре в течение 10 часов. Реакционную смесь профильтровали через целитовую подушку и промыли смесью дихлорметан/метанол (100 мл, дихлорметан/метанол = 1/1). Провели концентрацию объединенного фильтрата и очистку колоночной хроматографией (300 г силикагеля, смесь дихлорметан/метанол = 15/1 до 10/1) с получением неочищенного С34 (17,46 г, 20,93 мМоль, чистота = 95,72% по площади методом ВЭЖХ) в виде белого твердого вещества с сероватым или желтоватым оттенком, выход 75%. К неочищенному С34 добавили этанол (87,5 мл) и нагрели до 50°C, затем добавили ацетон (87,5 мл). Раствор охладили до комнатной температуры в течение 3 часов, затем охлаждали в ледяной бане. Осадок профильтровали и промыли ацетоном (200 мл) с получением С34 (16,02 г, 19,21 мМоль, 68%, чистота = 97,15%) по площади методом ВЭЖХ) в виде белого вещества, выход 92%, точка плавления: 163°C. 1H-NMR (CDCl3/CD3OD=1/1, 400 МГц) δ 7,26 (d, J=8,5 Гц, 2Н), 7,07-7,17 (m, 4Н), 7,00 (dd, J=6,6, 2,0 Гц, 2Н), 5,03 (d, J=3,7 Гц, 1H), 4,33 (q, J=4,7 Гц, 1Н), 4,05 (d, J=2,7 Гц, 1Н), 4,01 (dd, J=4,6, 10,8 Гц, 1H), 3,79-3,97 (m, 6Н), 3,64-3,72 (m, 2Н), 2,71 (t, J=7,4 Гц, 2Н), 2,34 (t, J=7,5 Гц, 2Н), 1,29-1,43 (m, 42Н), 1,01 (t, J=6,6 Гц, 3Н). 13C-NMR (CDCl3/CD3OD=1/1, 100 МГц) δ 173,99, 154,82, 152,97, 137,34, 128,95, 119,39, 119,31, 117,71, 115,52, 115,29, 99,21, 73,92, 71,28, 70,41, 69,69, 69,17, 68,37, 66,63, 61,12, 49,92, 35,74, 34,50, 31,66, 31,27, 31,02, 29,10, 29,06, 28,99, 28,95, 28,88, 28,85, 28,77, 28,70, 28,60, 25,28, 25,23, 21,99, 13,10.
Синтез 3,4-ди-O-бензил-1-O-(2,3-ди-O-бензил-4,6-O-бензилиден-α-D-галактопиранозил)-2-(11-(4-феноксифенил)ундеканоил)амино-D-рибо-октадекан-1,3,4-триола (А9)
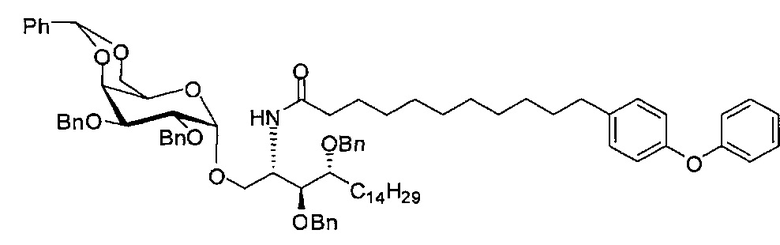
По тому же способу синтеза, что и способ синтеза А8 соединение А6 (100 мг, 0,105 мМоль) и соединение В12 (33 мг, 0,093 мМоль) являются исходными материалами для получения соединения А9 (45 мг, 0,036 мМоль, 38%) в виде белого воска. 1H-NMR (CDCl3, 400 МГц) δ 7,48-7,52 (m, 2Н), 7,20-7,41 (m, 25Н), 7,12 (d, J=8,8 Гц, 2Н), 7,06 (t, J=1,3 Гц, 1H), 6,96-7,00 (m, 2Н), 6,92 (d, J=10,4 Гц, 2Н), 5,83 (d, J=8,4 Гц, 1H), 5,45 (s, 1Н), 4,94 (d, J=3,6, 1Н), 4,84 (d, J=11,6 Гц, 1H), 4,69-4,75 (m, 3Н), 4,63 (d, J=11,6 Гц, 1H), 4,57 (d, J=11,6 Гц, 1H), 4,49 (d, J=11,6 Гц, 1H), 4,48 (d, J=11,6 Гц, 1H), 4,24-4,31 (m, 1H), 4,17 (d, J=3,2 Гц, 1H) 4,03-4,12 (m, 3H), 3,87-3,96 (m, 3H), 3,74-3,80 (m, 2H), 3,56 (s, 1H), 3,50-3,56 (m, 1H), 2,57 (t, J=7,6 Гц, 2H), 1,82-1,95 (m, 2H), 1,15-1,65 (m, 42H), 0,88 (t, J=6,6 Гц, 3Н). 13C-NMR (CDCl3, 100 МГц) δ 172,89, 159,69, 154,86, 138,62, 138,51, 138,50, 138,38, 137,89, 137,79, 129,60, 129,49, 128,83, 128,41, 128,34, 128,31, 128,29, 128,07, 127,87, 127,82, 127,80, 127,68, 127,57, 127,55, 126,29, 122,79, 118,92, 118,41, 100,98, 99,60, 79,82, 79,48, 76,14, 75,68, 74,33, 73,80, 73,27, 71,88, 71,69, 69,38, 68,14, 62,91, 50,32, 36,70, 35,21, 31,90, 31,61, 30,24, 29,78, 29,69, 29,67, 29,65, 29,59, 29,56, 29,51, 29,41, 29,34, 29,29, 25,80, 25,68, 22,66, 14,10. HRMS (ESI) в пересчете на C82H106NO10 [М+Н]+: 1264,7817, установлено: 1264,7834.
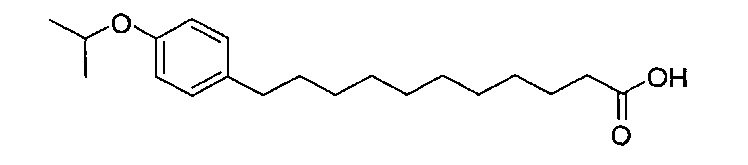
Синтез 3,4-ди-O-бензил-1-O-(2,3-ди-O-бензил-4,6-O-бензилиден-α-D-галактопиранозил)-2-(11-(4-изопропоксифенил)ундеканоил)амино-D-рибо-октадекан-1,3,4-триола (А10)
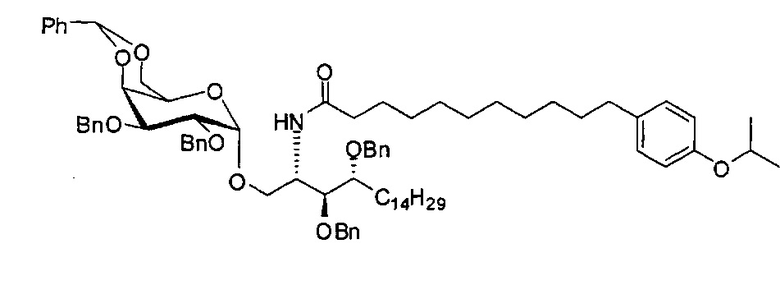
По тому же способу синтеза, что и способ синтеза А8, соединение А6 (100 мг, 0,105 мМоль) и В13 (30 мг, 0,094 мМоль) использовали в качестве исходных материалов для получения А10 (72,0 мг, 0,059 мМоль, 63%) в виде белого воска. 1H-NMR (CDCl3, 400 МГц) δ 7,21-7,55 (m, 25Н), 7,06 (d, J=8,4 Гц, 2Н), 7,78-7,84 (m, 2Н), 5,86 (d, J=8,4 Гц, 1Н), 5,46 (s, 1Н), 4,95 (d, J=3,2, 1H), 4,85 (d, J=11,6 Гц, 1H), 4,71-4,80 (m, 3Н), 4,63 (d, J=11,6 Гц, 1H), 4,58 (d, J=11,6 Гц, 1H), 4,46-4,54 (m, 3H), 4,18 (d, J=3,2 Гц, 1H), 4,05-4,14 (m, 3H), 3,88-3,97 (m, 3H), 3,75-3,82 (m, 2H), 3,58 (s, 1H), 3,51-3,57 (m, 1H), 2,53 (t, J=7,6 Гц, 2H), 1,83-1,96 (m, 2H), 1,15-1,71 (m, 48H), 0,89 (t, J=6,6 Гц, 3Н). 13C-NMR (CDCl3, 100 МГц) δ 172,84, 139,68, 138,60, 138,49, 138,46, 138,36, 137,77, 128,81, 128,39, 128,32, 128,28, 128,26, 128,05, 127,86, 127,79, 127,78, 127,66, 127,54, 126,26, 124,00, 116,89, 116,72, 100,96, 99,54, 79,75, 79,46, 76,10, 75,66, 74,29, 73,79, 73,25, 71,84, 71,65, 69,35, 68,08, 62,88, 50,28, 36,66, 35,04, 31,87, 31,20, 30,19, 29,75, 29,67, 29,64, 29,50, 29,48, 29,38, 29,37, 29,31, 29,04, 25,87, 25,64, 22,64, 14,07. HRMS (ESI) в пересчете на C79H108NO10 [M+H]+: 1230,7973, установлено: 1230,7968.
Синтез 3,4-ди-O-бензил-1-O-(2,3-ди-O-бензил-4,6-O-бензилиден-α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амино-D-рибо-октадекан-1,3,4-триола (АН)
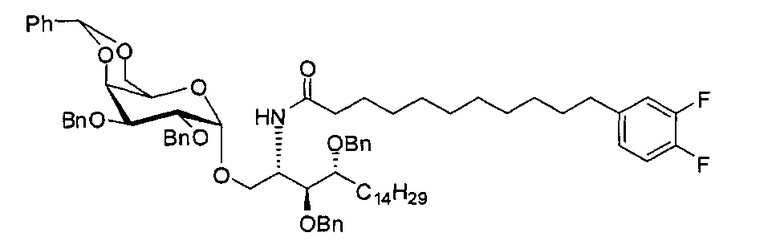
По тому же способу синтеза, что и способ синтеза А8, соединение А7 (100 мг, 0,105 мМоль) и В14 (28 мг, 0,093 мМоль) использовали в качестве исходных материалов для получения А11 (63 мг, 0,052 мМоль, 56%), точка плавления: 98°C. 1Н-NMR (CDCl3, 400 МГц) δ 7,21-7,54 (m, 25Н), 6,92-7,06 (m, 2Н), 6,81-6,87 (m, 1H), 5,87 (d, J=8,4 Гц, 1Н), 5,45 (s, 1H), 4,95 (d, J=3,2, 1Н), 4,84 (d, J=11,6 Гц, 1Н), 4,69-4,79 (m, 3Н), 4,63 (d, J=11,6 Гц, 1Н), 4,58 (d, J=11,6 Гц, 1Н), 4,50 (d, J=11,6 Гц, 1H), 4,49 (d, J=11,6 Гц, 1H), 4,25-4,33 (m, 1H), 4,17 (d, J=2,8 Гц, 1H) 4,04-4,13 (m, 2Н), 3,88-3,97 (m, 3Н), 3,74-3,81 (m, 2Н), 3,57 (s, 1H), 3,51-3,56 (m, 1H), 2,54 (t, J=7,6 Гц, 2Н), 1,82-1,96 (m, 2Н), 1,15-1,69 (m, 42Н), 0,89 (t, J=6,8 Гц, 3Н). 13C-NMR (CDCl3, 100 МГц) δ 172,84, 150,02 (dd, J=245, 13 Гц), 148,55 (dd, J=244, 13 Гц), 139,68, 138,60, 138,49, 138,46, 138,36, 137,77, 128,81, 128,39, 128,32, 128,28, 128,26, 128,05, 127,86, 127,79, 127,78, 127,66, 127,54, 126,26, 124,00, 116,89, 116,72, 100,96, 99,54, 79,75, 79,46, 76,01, 75,66, 74,29, 73,79, 73,25, 71,84, 71,65, 69,35, 68,08, 62,88, 50,28, 36,66, 35,04, 31,87, 31,20, 30,19, 29,75, 29,67, 29,64, 29,50, 29,48, 29,38, 29,37, 29,31, 29,04, 25,78, 25,64, 22,64, 14,07. HRMS (ESI) в пересчете на C76H100F2NO9 [М+Н]+: 1208,7366, установлено: 1208,7398.
Синтез 3,4-ди-O-бензил-1-O-(2,3-ди-O-бензил-4,6-O-бензилиден-α-D-галактопиранозил)-2-(11-(2,4-дифторфенил)ундеканоил)амино-D-рибо-октадекан-1,3,4-триола (А12)
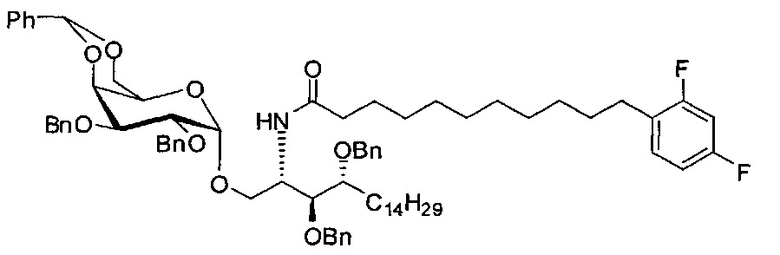
По тому же способу синтеза, что и способ синтеза А8, соединение А6 (100 мг, 0,105 мМоль) и В15 (28 мг, 0,093 мМоль) использовали в качестве исходных материалов для получения А12 (70 мг, 0,058 мМоль, 62%) в виде белого воска. 1H-NMR (CDCl3, 400 МГц) δ 7,20-7,55 (m, 25Н), 7,04-7,14 (m, 1Н), 6,69-6,81 (m, 2Н), 5,89 (d, J=8,3 Гц, 1H), 5,45 (s, 1H), 4,95 (d, J=3,2 Гц, 1H), 4,85 (d, J=11,6 Гц, 1H), 4,70-4,79 (m, 3Н), 4,64 (d, J=11,6 Гц, 1H), 4,58 (d, J=11,6 Гц, 1H), 4,50 (d, J=11,6 Гц, 1H), 4,49 (d, J=11,6 Гц, 1Н), 4,25-4,33 (m, 1Н), 4,17 (d, J=2,8 Гц, 1Н) 4,04-4,13 (m, 2Н), 3,88-3,97 (m, 3Н), 3,74-3,80 (m, 2Н), 3,57 (s, 1H), 3,51-3,56 (m, 1Н), 2,57 (t, J=7,6 Гц, 2Н), 1,82-1,96 (m, 2Н), 1,15-1,69 (m, 42Н), 0,88 (t, J=6,6 Гц, 3Н). 13C-NMR (CDCl3, 100 МГц) δ 172,90, 162,30, 138,59, 138,50, 138,45, 138,36, 137,78, 130,89, 128,82, 128,40, 128,33, 128,30, 128,27, 128,06, 127,87, 127,79, 127,68, 127,55, 126,27, 110,07, 103,41, 99,53, 79,75, 79,47, 76,11, 75,67, 74,29, 73,81, 73,27, 71,85, 71,65, 69,36, 68,05, 62,89, 50,29, 36,67, 31,88, 30,19, 30,15, 29,76, 29,67, 29,65, 29,51, 29,37, 29,32, 29,17, 28,38, 25,78, 25,66, 22,65, 14,08. HRMS (ESI) в пересчете на C76H100F2NO9 [М+Н]+: 1208,7366, установлено: 1208,7377.
Синтез 1-O-(α-O-галактопиранозил)-2-(11-(4-феноксифенил)ундеканоил)амино-D-рибо-1,3,4-октадекантриола (А15)
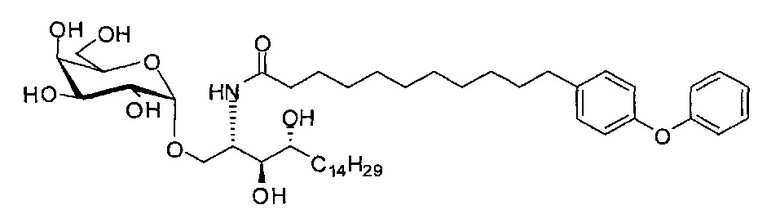
Способом, аналогичным получению С34, был получен А15 (21 мг, 0,026 мМоль, 72%) из А9 (45 мг, 0,036 мМоль), в виде белого твердого вещества с сероватым или желтоватым оттенком, точка плавления: 131°C. 1H-NMR (CD3OD/CDCl3=1/1, 400 МГц) δ 7,45 (t, J=8,3 Гц, 2Н), 7,28 (d, J=1,1 Гц, 2Н), 7,20 (t, J=1,3 Гц, 1H), 7,10 (d, J=8,1 Гц, 2Н), 7,04 (d, J=8,1 Гц, 2H), 5,04 (d, J=3,3 Гц, 1H), 4,26 (q, J=7,1 Гц, 1H), 3,79-4,13 (m, 10Н), 2,73 (t, J=1,1 Гц, 2H), 2,36 (t, J=7,5 Гц, 2H), 1,65-1,82 (m, 4H), 1,41 (brs, 38H), 0,89 (t, J=7,5 Гц, 3H). 13C-NMR (CD3OD/CDCl3=1/1, 100 МГц) δ 174,09, 157,18, 154,36, 137,40, 128,98, 128,92, 122,22, 118,25, 117,71, 99,18, 73,88, 71,30, 70,41, 69,68, 69,17, 68,37, 66,63, 61,10, 60,01, 35,80, 34,56, 31,61, 31,29, 31,04, 29,08, 29,02, 28,91, 28,80, 28,72, 28,64, 25,30, 22,01, 19,90, 13,15.
Синтез 1-O-(α-D-галактопиранозил)-2-(11-(4-изопропокси)фенил)ундеканоил)амино-D-рибо-1,3,4-октадекантриола (А 16)
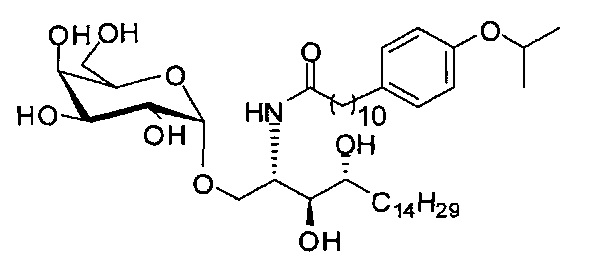
Способом, аналогичным получению С34, было получено соединение А16 (34 мг, 0,044 мМоль, 74%) из А10 (72 мг, 0,059 мМоль). Данные о соединении А16: точка плавления: 120°C. 1H-NMR (CD3OD/CDCl3=1/1, 400 МГц) δ 7,16 (d, J=8,5 Гц, 2Н), 6,90 (d, J=8,5 Гц, 2Н), 5,01 (d, J=3,7 Гц, 1H), 4,58-4,66 (m, 1H), 4,32 (m, 1Н), 4,03 (d, J=2,6 Гц, 1Н), 3,99 (dd, J=10,6, 4,8, 1Н), 3,88-3,96 (m, 2Н), 3,78-3,88 (m, 4Н), 3,62-3,73 (m, 2Н), 2,64 (t, J=7,6 Гц, 2Н), 2,32 (t, J=7,4 Гц, 2Н), 1,61-1,81 (m, 4Н), 1,32-1,52 (m, 44Н), 0,99 (t, J=6,8 Гц, 3Н). 13C-NMR (CD3OD/CDCl3=1/1, 100 МГц) δ 174,06, 155,10, 134,52, 128,54, 115,33, 99,16, 73,81, 71,25, 70,40, 69,65, 69,62, 69,12, 68,33, 66,60, 61,04, 35,75, 35,70, 34,35, 31,53, 31,25, 31,08, 29,09, 29,03, 28,97, 28,94, 28,87, 28,85, 28,76, 28,68, 28,59, 25,27, 25,22, 21,96, 21,08, 20,88, 13,08.
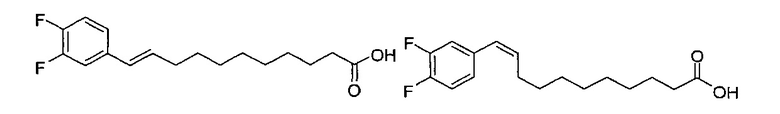
Синтез 1-O-(α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амкко-D-рибо-1,3,4-октадекантриола (А17)
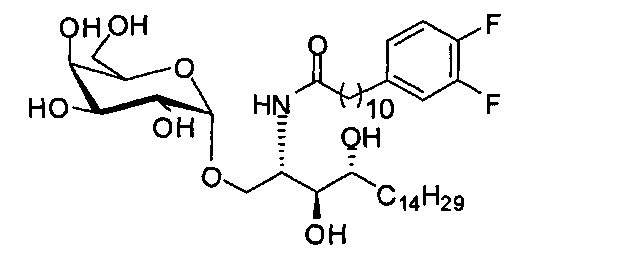
Способом, аналогичным получению С34, было получено соединение А17 (37 мг, 0,049 мМоль, 94%) из А11 (63 мг, 0,052 мМоль). Данные о соединении А18: точка плавления: 140°C. 1H-NMR (CD3OD/CDCl3=1/1, 400 МГц) δ 7,06-7,22 (m, 2Н), 6,98-7,04 (m, 1Н), 5,03 (d, J=3,3 Гц, 1H), 4,29-4,36 (m, 1Н), 4,05 (d, J=2,5 Гц, 1Н), 4,00 (dd, J=10,5, 4,8, 1H), 3,89-3,96 (m, 2Н), 3,78-3,89 (m, 4Н), 3,64-3,73 (m, 2Н), 2,69 (t, J=7,5 Гц, 2H), 2,34 (t, J=7,6 Гц, 2H), 1,63-1,83 (m, 4Н), 1,33-1,48 (m, 38Н), 1,00 (t, J=6,5 Гц, 3Н). 13C-NMR (CD3OD/CDCl3=1/1, 100 МГц) δ 174,07, 149,33 (d, J=247, 13 Гц), 147,95 (d, J=244, 13 Гц), 139,47, 123,56, 116,22, 116,06, 99,18, 73,86, 71,82, 70,40, 69,67, 69,15, 68,34, 66,63, 61,08, 45,00, 35,78, 35,73, 34,40, 31,58, 31,28, 30,66, 29,12, 29,06, 29,00, 28,91, 28,85, 28,77, 28,71, 28,44, 25,28, 21,99, 13,12.
Синтез 1-O-(α-D-галактопиранозил)-2-(11-(2,4-дифторфенил)ундеканоил)амино-O-рибо-1,3,4-октадекантриола (А18).
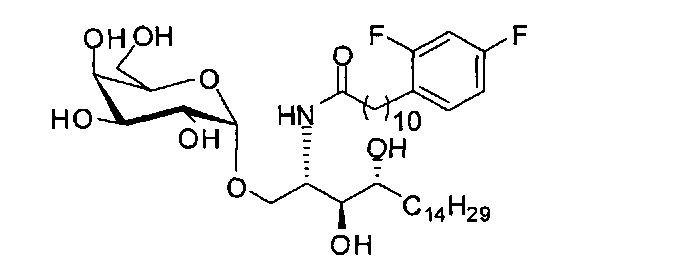
Способом, аналогичным получению С34, было получено соединение А18 (39 мг, 0,051 мМоль, 88%) из соединения А12 (70 мг, 0,058 мМоль). Данные о соединении А18: точка плавления: 149°C. 1H-NMR (CD3OD/CDCl3=1/1, 400 МГц) δ 7,29 (q, J=8,1 Гц, 1Н), 6,87-6,97 (m, 2Н), 5,05 (d, J=3,7 Гц, 1H), 4,30-4,38 (m, 1Н), 4,07 (d, J=2,9 Гц, 1H), 4,03 (dd, J=10,6, 4,4 Гц, 1Н), 3,91-3,98 (m, 2Н), 3,66-3,75 (m, 4Н), 3,64-3,73 (m, 2Н), 2,73 (t, J=7,5 Гц, 2Н), 2,36 (t, J=7,6 Гц, 2Н), 1,65-1,86 (m, 4Н), 1,24-1,60 (m, 38Н), 1,02 (t, J=6,6 Гц, 3Н). 13C-NMR (CD3OD/CDCl3=1/1, 100 МГц) δ 174,09, 161,65, 159,20, 130,48, 124,73, 110,06, 102,62, 99,19, 73,90, 71,31, 70,40, 69,69, 69,17, 68,36, 66,66, 61,11, 50,12, 35,81, 35,76, 31,63, 31,30, 29,59, 29,16, 29,14, 29,09, 29,02, 28,92, 28,87, 28,78, 28,76, 28,73, 28,56, 27,72, 25,30, 22,01, 13,16.
Синтез 1-O-(α-D-галактопиранозил)-2-((10R,11S)-11-(3,4-дифторфенил)-10,11-дигидроксиундеканоил)амино-D-рибо-1,3,4-октадекантриола и 1-O-(α-D-галактопиранозил)-2-((10S,11R)-11-(3,4-дифторфенил)-10,11-дигидроксиундеканоил)амино-D-рибо-1,3,4-октадекантриола (А19) в форме смеси антиизомеров диола.
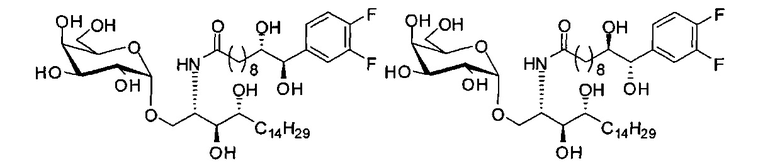
К раствору А7 (101 мг, 0,109 мМоль) в дихлорметане (3 мл) добавили анти-В21 (32 мг, 0,097 мМоль), HBTU (62 мг, 0,16 мМоль) и N-метилморфолин (24 мкл, 0,23 мМоль). После перемешивания при комнатной температуре в течение 12 часов провели концентрацию смеси и очистку остатка колоночной хроматографией (этилацетат/n-гексан=1/4 до 1/2 до 1/1). Полученный белый воск растворили в смеси дихлорметан/метанол (1/1, 10 мл) и затем добавили Pd(OH)2 (10 мг). После перемешивания при комнатной температуре в атмосфере водорода в течение 15 часов, смесь профильтровали через целитовую подушку и промыли смесью дихлорметан/метанол (1/1). Провели концентрацию фильтрата, и остаток очистили колоночной хроматографией (дихлорметан/метанол=10/1, до 8/1) с получением А19 (34 мг, 0,043 мМоль, 44%) в виде белого твердого вещества с сероватым или желтоватым оттенком, точка плавления: 105°C. 1H-NMR (CDCl3/CD3OD=1/1, 400 МГц) δ 7,17-7,40 (m, 3Н), 5,01 (d, J=3,6 Гц, 1H), 4,62 (d, J=4,8 Гц, 0,75Н), 4,50 (d, J=6,0 Гц, 0,25Н), 4,28-4,36 (m, 1Н), 4,03 (d, J=2,8 Гц, 1Н), 3,99 (dd, J=4,8, 10,8 Гц, 1H), 3,87-3,95 (m, 2Н), 3,76-3,87 (m, 4Н), 3,61-3,71 (m, 2Н), 2,31 (t, J=7,4 Гц, 2Н), 1,31-1,83 (m, 40Н), 0,99 (t, J=6,9 Гц, 3Н). 13C-NMR (CDCl3/CD3OD=1/1, 100 МГц) δ 173,97, 149,42 (dd, J=245, 13 Гц), 148,86 (dd, J=245, 13 Гц), 136,65, 122,46, 116,09, 115,77, 115,27, 115,10, 99,16, 75,70, 75,35, 74,74, 74,21, 73,94, 71,25, 70,40, 69,65, 69,10, 68,31, 66,52, 61,03, 49,91, 35,63, 31,94, 31,69, 31,23, 31,18, 29,09, 29,04, 29,00, 28,95, 28,83, 28,76, 28,68, 28,65, 28,53, 25,17, 25,08, 24,96, 21,94, 13,04.
Синтез 1-O-(α-D-галактопиранозил)-2-((10S,11S)-11-(3,4-дифторфенил)-10,11-дигидроксиундеканоил)амино-D-рибо-1,3,4-октадекантриола и 1-О-(α-D-галактопиранозил)-2-((10R,11R)-11-(3,4-дифторфенил)-10,11-дигидроксиундеканоил)амино-D-рибо-1,3,4-октадекантриола, (А20) в форме смеси син-изомеров диола.
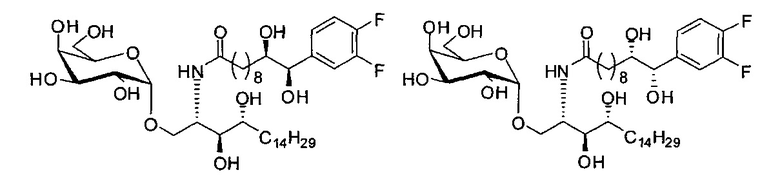
К раствору А7 (101 мг, 0,109 мМоль) в дихлорметане (3 мл) добавили син-А21 (14 мг, 0,042 мМоль), HBTU (62 мг, 0,16 мМоль) и N-метилморфолин (24 мкл, 0,22 мМоль). После перемешивания при комнатной температуре в течение 12 часов провели концентрацию смеси и очистку остатка колоночной хроматографией (этилацетат/n-гексан=1/4 до 1/2 до 1/1). Полученный белый воск растворили в смеси дихлорметан/метанол (1/1, 10 мл) и затем добавили Pd(OH)2 (10 мг). После перемешивания при комнатной температуре в атмосфере водорода в течение 15 часов смесь профильтровали через целитовую подушку и промыли смесью дихлорметан/метанол (1/1). Провели концентрацию фильтрата, и остаток очистили колоночной хроматографией (дихлорметан/метанол=10/1, до 8/1) с получением А20 (20 мг, 0,025 мМоль, 60%) в виде твердого белого вещества, точка плавления: 80°C. 1H-NMR (CDCl3/CD3OD=1/1, 400 МГц) δ 7,19-7,49 (m, 3Н), 5,02 (d, J=3,6 Гц, 1Н), 4,52 (d, J=6 Гц, 1H), 4,30-4,37 (m, 1Н), 4,04 (d, J=2,8 Гц, 1H), 4,01 (dd, J=4,4, 10,8 Гц, 1Н), 3,89-3,96 (m, 2Н), 3,78-3,88 (m, 4Н), 3,63-3,73 (m, 3Н), 2,33 (t, J=7,6 Гц, 2Н), 1,26-1,84 (m, 40Н), 1,01 (t, J=6,8 Гц, 3Н). 13C-NMR (CDCl3/CD3OD=1/1, 100 МГц) δ 174,00, 149,58 (dd, J=245, 12 Гц), 149,07 (dd, J=245, 12 Гц), 138,90, 122,46, 116,13, 115,14, 99,21, 75,47, 74,79, 74,01, 71,28, 70,41, 69,69, 69,15, 68,34, 66,57, 61,09, 49,90, 35,67, 31,98, 31,77, 31,27, 29,12, 29,03, 28,98, 28,80, 28,68, 28,56, 25,20, 25,00, 21,98, 13,08.
Синтез 1-O-(α-D-галактопиранозил)-2-(11-(4-бромфенил)-10,11-дигидроксиундеканоил)амино-D-рибо-1,3,4-октадекантриола (А21)
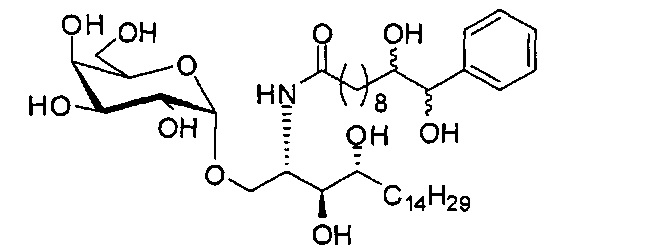
Способом, аналогичным получению А19, было получено соединение А21 (18 мг, 0,024 мМоль, 28%) из А7 (90 мг, 0,094 мМоль) и В19 (32 мг, 0,086 мМоль). Данные о соединении А21: белый воск с сероватым или желтоватым оттенком. 1H-NMR (CD3OD/CDCl3=1/1, 400 МГц) δ 7,26-7,51 (m, 5Н), 5,02-5,10 (m, 1H), 5,02 (d, J=4,0 Гц, 1H), 4,23-4,29 (m, 1H), 4,06-4,15 (m, 1H), 3,97-4,03 (m, 2H), 3,83-3,96 (m, 6H), 3,68 (t, J=10,2 Гц, 1H), 2,84-2,89 (m, 1H), 2,52 (t, J=7,4 Гц, 2H), 1,27-1,89 (m, 40H), 1,02 (t, J=6,8 Гц, 3Н). 13C-NMR (CD3OD/CDCl3=1/1, 150 МГц) δ 174,08, 173,54, 138,50, 128,72, 127,55, 125,39, 99,10, 72,40, 71,92, 70,84, 70,16, 69,92, 69,57, 69,24, 68,23, 63,43, 61,13, 52,30, 43,34, 36,69, 36,39, 35,91, 35,69, 35,26, 33,69, 32,83, 32,03, 31,67, 31,25, 30,90, 30,59, 29,81, 29,30, 29,01, 28,97, 28,86, 28,77, 28,67, 28,54, 28,46, 25,22, 25,00, 24,36, 24,27, 21,97, 13,09.
Схема 2: синтез соединения А23-25
Синтез 1-O-(α-D-галактопиранозил)-2-амино-D-рибо-1,3,4-октадекантриола (А22)
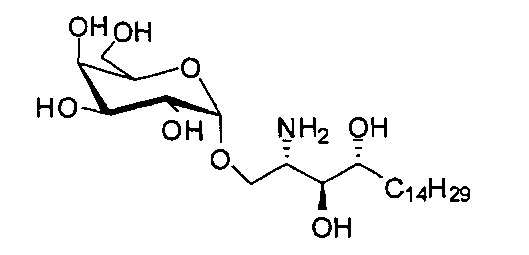
К раствору А7 (520 мг, 0,545 мМоль) в смеси дихлорметан/метанол (1/1, 20 мл) добавили Pd(OH)2 (220 мг) и три капли уксусной кислоты. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода при давлении 80 фунтов на кв. дюйм в течение 16 часов. Смесь профильтровали через целитовую подушку, и осадок на фильтре промыли метанолом. Провели концентрацию фильтрата и высушили его в вакууме с получением неочищенного А22 (302 мг, оценивали по количеству) в виде твердого белого вещества. HRMS (ESI) в пересчете на C24H49NO8H [М+Н]+: 480,3536, установлено: 480,3515.
Синтез 1-O-(α-D-галактопиранозил)-2-(11-(3,4-дихлорфенил)ундеканоил)амино-D-рибо-1,3,4-октадекантриола (А23)
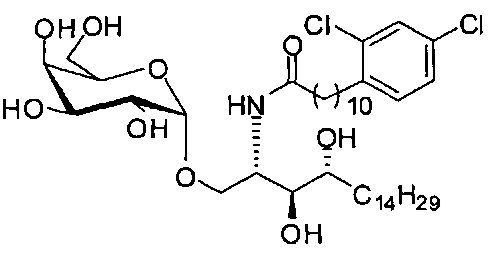
раствору А22 (50 мг, 0,10 мМоль) в смеси дихлорметан/метанол (1/1, 3 мл) добавили В16 (34 мг, 0,10 мМоль), HBTU (59 мг, 0,16 мМоль) и N-метилморфолин (23 мкл, 0,21 мМоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Провели концентрацию смеси, и остаток очистили колоночной хроматографией (дихлорметан/метанол = 15/1 до 12/1 до 9/1) с получением А23 (16 мг, 0,020 мМоль, 20%) в виде белого твердого вещества с сероватым или желтоватым оттенком, точка плавления: 147°C. 1H-NMR (CD3OD/CDCl3=1/1, 400 МГц) δ 7,51 (s, 1Н), 7,35 (s, 2Н), 5,08 (d, J=3,3 Гц, 1H), 4,33-4,43 (m, 1Н), 4,08-4,12 (m, 1Н), 4,06 (dd, J=10,7, 4,0 Гц, 1H), 3,82-4,02 (m, 6Н), 3,67-3,77 (m, 2Н), 2,86 (t, J=7,6 Гц, 2Н), 2,39 (t, J=7,5 Гц, 2Н), 1,66-1,88 (m, 4Н), 1,34-1,60 (m, 38Н), 1,06 (t, J=6,4 Гц, 3Н). 13C-NMR (CD3OD/CDCl3=1/1, 100 МГц) δ 174,05, 138,47, 133,89, 131,36, 130,64, 128,41, 126,35, 99,25, 73,99, 71,36, 70,42, 69,74, 69,22, 68,42, 66,71, 61,19, 49,97, 35,82, 32,41, 31,76, 31,34, 29,20, 29,17, 29,12, 29,06, 28,95, 28,91, 28,81, 28,76, 28,69, 25,33, 25,29, 22,06, 13,21.
Синтез 1-O-(α-D-галактопиранозил)-2-(11-(4-хлорфенил)ундеканоил)амино-D-рибо-1,3,4-октадекантриола (А24)
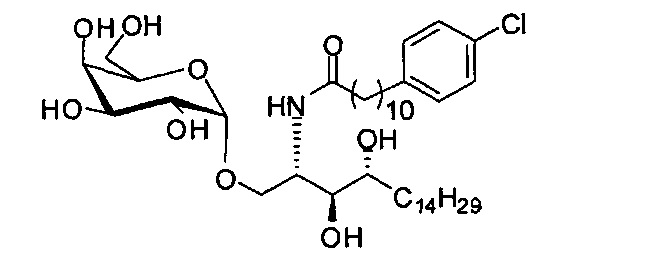
Способом, аналогичным получению А23, было получено соединение А24 (18 мг, 0,024 мМоль, 22%) из А22 (52 мг, 0,11 мМоль) и В17 (32 мг, 0,11 мМоль). Данные о соединении А24: белое твердое вещество с сероватым или желтоватым оттенком, точка плавления: 136°C. 1H-NMR (CD3OD/CDCl3=1/1, 400 МГц) δ 7,28 (d, J=8,4 Гц, 2Н), 7,17 (d, J=8,4 Гц, 2Н), 4,96 (d, J=3,6 Гц, 1Н), 4,21-4,29 (m, 1Н), 3,98 (d, J=2,5 Гц, 1H), 3,95 (dd, J=10,6, 4,4, 1Н), 3,83-3,90 (m, 2H), 3,72-3,82 (m, 4H), 3,58-3,68 (m, 2H), 2,64 (t, J=7,6 Гц, 2H), 2,28 (t, J=7,8 Гц, 2H), 1,57-1,77 (m, 4H), 1,20-1,51 (m, 38H), 0,95 (t, J=6,7 Гц, 3Н). 13C-NMR (CD3OD/CDCl3=1/1, 100 МГц) δ 173,99, 140,73, 130,53, 129,08, 127,51, 99,15, 73,79, 71,23, 70,34, 69,62, 69,17, 68,32, 66,58, 61,06, 49,87, 35,67, 34,52, 31,53, 31,23, 30,73, 29,01, 28,95, 28,87, 28,81, 28,75, 28,65, 28,46, 25,24, 21,94, 13,03.
Синтез 1-O-(α-D-галактопиранозил)-2-(11-(4-бромфенил)ундеканоил)амино-D-рибо-1,3,4-октадекантриола (А25)
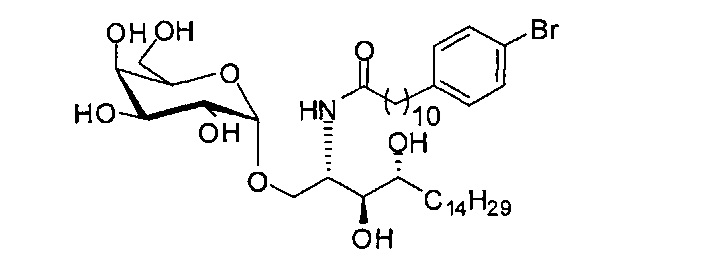
Способом, аналогичным получению А23, было получено соединение А25 (22 мг, 0,027 мМоль, 25%) из А22 (52 мг, 0,11 мМоль) и В28 (56 мг, 0,16 мМоль). Данные о соединении А25: белый воск с сероватым или желтоватым оттенком. 1H-NMR (CD3OD/CDCl3=1/1, 400 МГц) δ 7,28 (d, J=8,4 Гц, 2Н), 7,20-7,47 (m, 2Н), 5,00 (d, J=3,6 Гц, 1Н), 4,28-4,33 (m, 1H), 4,02 (d, J=2,8 Гц, 1Н), 3,98 (dd, J=10,6, 4,6 Гц, 1Н), 3,863-3,94 (m, 2Н), 3,77-3,87 (m, 4Н), 3,62-3,70 (m, 2Н), 2,69 (t, J=7,6 Гц, 2Н), 2,31 (t, J=7,6 Гц, 2Н), 1,29-1,81 (m, 42Н), 0,98 (t, J=6,6 Гц, 3Н). 13C-NMR (CD3OD/CDCl3=1/1, 100 МГц) δ 171,21, 141,18, 130,44, 129,44, 118,39, 99,41, 74,54, 70,41, 70,13, 69,58, 69,50, 69,07, 68,41, 67,22, 60,95, 49,68, 35,58, 34,51, 31,36, 31,18, 30,62, 28,95, 28,90, 28,81, 28,76, 28,68, 28,61, 28,40, 27,00, 25,21, 24,88, 21,89, 19,90, 19,74, 19,57, 12,96.
(3) Схема синтеза 3: Синтез арил-алкановой кислоты
Синтез (9-карбоксинонил)трифенилфосфониум бромида (В2)
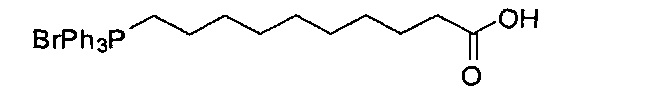
Смешали 10-бромдекановую кислоту (19,65 г, 78,24 мМоль) и трифенилфосфин (21,45 г, 81,78 мМоль) и перемешивали при 150°C в течение 24 часов. Был получен реактив Виттига В2 в виде светло-желтого сиропа с выходом 100%, его использовали без дополнительной очистки.
Синтез 11-(4-(4-фторфенокси)фенил))ундекановой кислоты (В4)
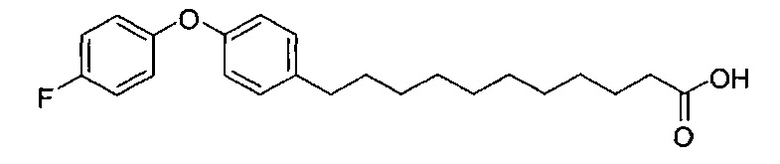
К смеси B2 (19,89 г, 38,81 мМоль) и тетрагидрофурана (150 мл) добавили t-бутоксид калия (10,40 г, 92,68 мМоль) при 0°C. Реакционная смесь превратилась в раствор красного цвета. Реакционную смесь подогрели до комнатной температуры и перемешивали в течение 1 часа. К реакционной смеси при 0°C добавили 4-(4-фторфенокси)бензальдегид (B1) (7,50 г, 34,7 мМоль), и смесь перемешивали дополнительно в течение 30 мин при комнатной температуре. Реакционную смесь нейтрализовали добавлением 1,0 N HCl и провели концентрацию. Остаток разделили этилацетатом (200 мл), водой (200 мл) (показатель pH довели до 5 добавлением 1,0 N HCl) и соляным раствором (200 мл). Органическую фазу отделили и провели концентрацию при пониженном давлении. Остаток рекристаллизовали со смесью этанол/вода (1/1, 80 мл) и промыли водой, при этом получили В3 (9,99 г, 27,0 мМоль, 78%) в виде белого твердого вещества. В3 растворили в смеси этанол/этилацетат (1/1, 80 мл) и затем добавили Pd/C (10%, 1,08 г). Смесь перемешивали при комнатной температуре в атмосфере водорода в течение 12 часов. Реакционную смесь профильтровали через целитовую подушку и промыли этилацетатом. Фильтрат концентрировали и рекристаллизовали со смесью метанол/вода (5/1, 12 мл), профильтровали и промыли водой и получили 11-(4-{4-фторфенокси)фенил))ундекановую кислоту (В4) (9,080 г, 24,38 мМоль, 90%) в виде белого твердого вещества, точка плавления: 73°C. 1Н-NMR (CDCl3, 400 МГц) δ 6,85-7,13 (m, 8Н), 2,56 (t, J=7,6 Гц, 2Н), 2,33 (t, J=7,4 Гц, 2Н), 1,22-1,65 (m, 16Н). 13C-NMR (CDCl3, 100 МГц) δ 179,69, 159,78, 157,38, 155,36, 153,39, 137,88, 129,56, 120,09, 120,01, 118,33, 116,25, 116,02, 35,18, 33,97, 31,59, 29,69, 29,48, 29,44, 29,38, 29,24, 29,20, 29,03, 24,65. HRMS (ESI) в пересчете на C23H29FO3Na [M+Na]+: 395,1998, установлено: 395,2003.
Синтез 11-(4-фенокси)фенилундекановой кислоты (В12)
Аналогично способу, описанному для соединения B4, было синтезировано соединение В12 (1,34 г, 3,78 мМоль, 97%) из B2 (3,47 г, 6,76 мМоль) и 4-феноксибензальдегида (1,03 г, 5,20 мМоль). Данные о соединении В12: белое твердое вещество с сероватым или желтоватым оттенком, точка плавления: 55°C. 1H-NMR (CDCl3, 400 МГц) δ 6,93-7,35 (m, 9Н), 2,60 (t, J=7,4 Гц, 2Н), 2,37 (t, J=7,3 Гц, 2Н), 1,65 (m, 4Н), 1,33 (m, 12Н). 13C-NMR (CDCl3, 100 МГц) δ 180,35, 157,71, 154,83, 137,87, 130,13, 129,51, 122,73, 118,97, 118,32, 35,18, 34,10, 31,55, 29,45, 29,42, 29,36, 29,22, 29,18, 29,00, 24,63. HRMS (ESI) в пересчете на C23H30O3Na [M+Na]+: 377,2093, установлено: 377,2053.
Синтез 11-(4-изопропокси)фенилундекановой кислоты (В13)
По тому же способу синтеза, что и способ синтеза B4, соединение В2 (2,25 г, 4,38 мМоль) и 4-изопропоксибензальдегид (479 мг, 2,92 мМоль) использовали в качестве исходных материалов для получения соединения В13 (562 мг, 1,75 мМоль, 60%) в виде белого твердого вещества, точка плавления: 46°C. 1H-NMR (CDCl3, 400 МГц) δ 7,04 (d, J=8,5 Гц, 2Н), 6,78 (d, J=8,6 Гц, 2Н), 4,48 (m, 1Н), 2,50 (t, J=7,5 Гц, 2Н), 2,32 (t, J=7,5 Гц, 2Н), 1,50-1,61 (m, 4Н), 1,25-1,35 (m, 18Н). 13C-NMR (CDCl3, 100 МГц) δ 179,73, 155,82, 134,93, 129,20, 115,77, 69,91, 35,04, 34,11, 31,69, 29,50, 29,47, 29,40, 29,26, 29,22, 29,12, 29,06, 24,71, 22,11, 21,88. HRMS (ESI) в пересчете на C20H32O3Na [M+Na]+: 343,2249, установлено: 343,2227.
Синтез (10E или 10Z)-11-(3,4-дифторфенил)ундек-10-еноевой кислоты ((E)-В7, и (Z)-B7)
По тому же способу, что и способ синтеза В3, В2 (12,93 г, 25,18 мМоль) и тетрагидрофуран (80 мл) и 3,4-дифторбензальдегид (2,35 г, 16,5 мМоль) были использованы как исходные материалы для получения соединения В7 (3,77 г, 12,7 мМоль, 77%). Продукты в Z-форме и в E-форме разделяли рекристаллизацией с n-гексаном. (E)-В7 был получен в виде белого твердого вещества, точка плавления: 66°C. 1H-NMR (CDCl3, 400 МГц) δ 6,98-7,14 (m, 3Н), 6,25 (d, J=15,8 Гц, 1H), 6,12 (m, 1H), 2,33 (t, J=7,5 Гц, 2Н), 2,16 (q, J=7,0 Гц, 2Н), 1,61 (m, 2Н), 1,43 (m, 2Н), 1,30 (s, 8Н). 13C-NMR (CDCl3, 100 МГц) δ 180,57, 150,49 (dd, J=246, 12 Гц), 149,31 (dd, J=241,13 Гц), 135,29, 132,35, 127,88, 121,99, 117,13, 114,18, 34,15, 32,91, 29,31, 29,22, 29,18, 29,08, 24,70. HRMS (ESI) в пересчете на C17H22F2O2Na [M+Na]+: 319,1486, установлено: 319,1485. (Z)-B7 был получен в виде бесцветного масла с 25% неотделимого (E)-B7.
Синтез (3,4-дифторфенил)ундекановой кислоты (В14)
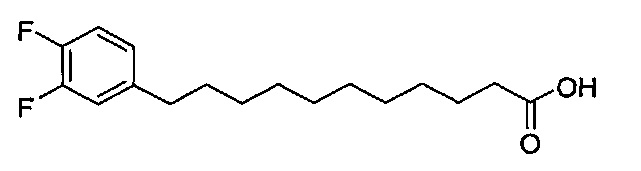
Соединение В7 (1,61 г, 5,43 мМоль) растворили в смеси этанол/этилацетат (1/1, 30 мл) и к раствору добавили Pd/C (10%, 160 мг). Смесь перемешивали при комнатной температуре в атмосфере водорода в течение 12 часов. Смесь профильтровали через целитовую подушку и промыли этилацетатом. Провели концентрацию фильтрата и высушили его под вакуумом. Соединение В14 было получено в виде белого твердого вещества (1,61 г, 5,40 мМоль, 99%), точка плавления: 51°C. 1H-NMR (CDCl3, 400 МГц) δ 6,90-7,03 (m, 2Н), 6,84 (m, 1H), 2,53 (t, J=7,7 Гц, 2Н), 2,33 (t, J=7,5 Гц, 2Н), 1,52-1,64 (m, 4Н), 1,26 (m, 12Н). 13C-NMR (CDCl3, 100 МГц) δ 180,24, 150,13 (dd, J=13, 247 Гц), 148,49 (dd, J=13, 246 Гц), 147,40, 139,77, 124,05, 116,85, 35,09, 34,01, 31,23, 29,43, 29,36, 29,35, 29,18, 29,05, 29,01, 24,64. HRMS (ESI) в пересчете на C14H24F2O2Na [M+Na]+: 321,1642, установлено: 321,1594.
Синтез 11-(2,4-дифторфенил)ундекановой кислоты (В15)
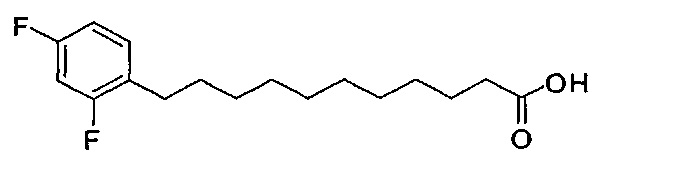
По тому же способу синтеза, что и способ синтеза В4, соединение В2 (2,76 г, 5,38 мМоль) и 2,4-дифторбензальдегид (588 мг, 4,14 мМоль) использовали в качестве исходных материалов для получения соединения В15 (431 мг, 1,45 мМоль, 35%) в виде белого твердого вещества с сероватым или желтоватым оттенком, точка плавления: 56°C. 1H-NMR (CDCl3, 400 МГц) δ 7,05-7,13 (m, 1Н), 6,70-6,79 (m, 2Н), 2,66 (t, J=7,6 Гц, 2Н), 2,31 (t, J=7,4 Гц, 2Н), 1,50-1,62 (m, 4Н), 1,26 (m, 12Н). 13C-NMR (CDCl3, 100 МГц) δ 180,00, 162,19, 159,80, 130,90, 125,31, 110,69, 103,44, 34,11, 30,15, 29,44, 29,36, 29,33, 29,19, 29,16, 29,03, 28,41, 24,69. HRMS (ESI) в пересчете на C17H24F2O2Na [M+Na]+: 321,1642, установлено: 321,1637.
Синтез 11-(2,4-дихлорфенил)ундекановой кислоты (В16)
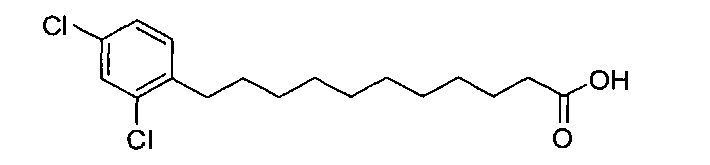
По тому же способу синтеза, что и способ синтеза В7, В2 (2,25 г, 4,38 мМоль) и 2,4-дихлорбензальдегид (500 мг, 2,86 мМоль) использовали в качестве исходных материалов для получения (10E или 10Z))-11-(2,4-дихлорфенил)ундек-10-еноевой кислоты (В9) (576 мг, 1,75 мМоль, 61%). Затем это соединение (210 мг, 0,638 мМоль) растворяли в этилацетате (10 мл) и после этого добавляли Pd/BaSO4 (21 мг). Затем смесь перемешивали при комнатной температуре в атмосфере водорода в течение 12 часов. Смесь профильтровали через целитовую подушку и промыли этилацетатом. Провели концентрацию объединенного фильтрата и высушили его в вакууме с получением соединения В16 (210 мг, 0,634 мМоль, 99%) в виде белого твердого вещества, точка плавления: 78°C. 1H-NMR (CDCl3, 400 МГц) δ 7,32 (d, J=1,9 Гц, 1Н), 7,09-7,15 (m, 2Н), 2,65 (t, J=7,8 Гц, 2Н), 2,33 (t, J=7,5 Гц, 2Н), 1,53-1,63 (m, 4Н), 1,26 (m, 12Н). 13C-NMR (CDCl3, 100 МГц) δ 180,00, 162,19, 159,80, 130,90, 125,31, 110,69, 103,44, 34,11, 30,15, 29,44, 29,36, 29,33, 29,19, 29,16, 29,03, 28,41, 24,69. HRMS (ESI) в пересчете на C17H24Cl2O2Na [M+Na]+: 353,1051, установлено: 353,1046.
Синтез 11-(4-хлорфенил)ундекановой кислоты (В17)
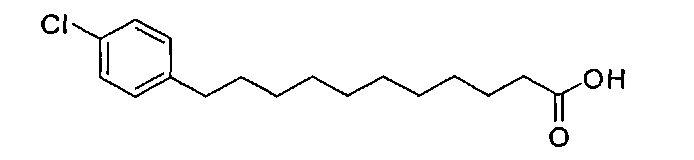
По тому же способу синтеза, что и способ синтеза В10, соединение В2 (2,20 г, 4,28 мМоль) и 4-хлорбензальдегид (401 мг, 2,85 мМоль) использовали в качестве исходных материалов для получения В17 (526 мг, 1,77 мМоль, 62%), точка плавления: 93°C. 1H-NMR (CDCl3, 400 МГц) δ 7,21 (d, J=8,4 Гц, 1H), 7,08 (d, J=8,4 Гц, 2Н), 2,54 (t, J=7,5 Гц, 2Н), 2,33 (t, J=7,5 Гц, 2Н), 1,53-1,63 (m, 4Н), 1,26 (m, 12Н). 13C-NMR (CDCl3, 100 МГц) δ 179,90, 141,28, 131,21, 129,71, 128,27, 35,26, 34,00, 31,35, 29,69, 29,44, 29,39, 29,36, 29,19, 29,13, 29,01, 24,64. HRMS (ESI) в пересчете на C17H25ClO2Na [M+Na]+: 319,1441, установлено: 319,1435.
Синтез 11-(4-бромфенил)ундекановой кислоты (В18)
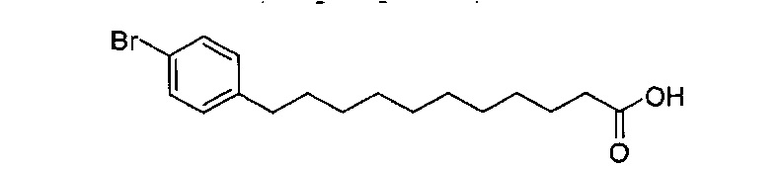
По тому же способу синтеза, что и способ синтеза В10, соединение В2 (330 мг, 0,643 мМоль) и 4-бромобензальдегид (91,5 мг, 0,495 мМоль) использовали в качестве исходных материалов для получения В18 (98,0 мг, 0,287 мМоль, 58%) в виде белого твердого вещества с сероватым или желтоватым оттенком, точка плавления: 91°C. 1H-NMR (CDCl3, 400 МГц) δ 7,37 (d, J=8,3 Гц, 1Н), 7,02 (d, J=8,3 Гц, 2Н), 2,53 (t, J=7,6 Гц, 2Н), 2,34 (t, J=1,5 Гц, 2Н), 1,52-1,63 (m, 4Н), 1,20-1,37 (m, 12Н). 13C-NMR (CDCl3, 100 МГц) δ 179,94, 141,79, 141,79, 131,22, 130,14, 119,22, 35,31, 34,11, 31,27, 29,44, 29,38, 29,36, 29,19, 29,12, 29,03, 24,68. HRMS (ESI) в пересчете на C17H24BrO2 [М+Н]+: 341,1116, установлено: 341,1111.
11-(4-бромфенил)-10,11-дигидроксиундекановая кислота (В20)
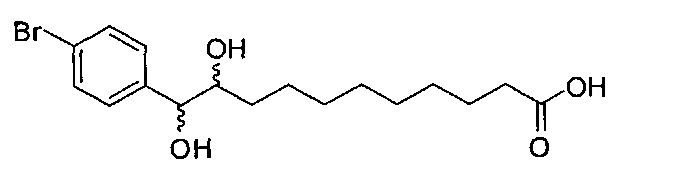
К раствору В11 (389 мг, 1,15 мМоль) в смеси t-бутанол/вода (4/3, 35 мл) добавили NMO (462 мг, 3,94 мМоль) и тетраоксид осмия (2,5 масс.% в t-BuOH, 170 мкл, 0,167 мМоль). После перемешивания при комнатной температуре в течение 15 часов, реакцию в реакционной смеси остановили добавлением насыщенного Na2S2O3 и провели концентрацию. Остаток разделили дихлорметаном (50 мл) и насыщенным Na2S2O3 (50 мл). Органическую фазу промыли соляным раствором (50 мл), провели концентрацию и очистили колоночной хроматографией (этилацетат/n-гексан = 1/2, затем 1/1) с получением В20 (256 мг, 0,686 мМоль, 60%). 1H-NMR (CD3OD/CDCl3=1/1, 400 МГц) δ 7,49-7,56 (m, 2Н), 7,28-7,37 (m, 2Н), 4,59 (d, J=5,1 Гц, 0,7Н), 4,44 (d, J=6,6 Гц, 0,3Н), 3,71-3,77 (m, 0,7Н), 3,62-3,66 (m, 0,3Н), 2,45 (t, J=1,2 Гц, 2Н), 1,27-1,78 (m, 14Н). 13C-NMR (CD3OD/CDCl3=1/1, 100 МГц) δ 169,05, 140,40, 140,16, 130,60, 130,30, 128,18, 120,64, 120,27, 76,44, 75,80, 74,81, 74,27, 31,88, 30,94, 28,92, 28,78, 28,67, 28,63, 28,56, 28,48, 28,45, 28,38, 25,10, 24,88, 24,28. HRMS (ESI) в пересчете на C17H25BrO4Na [M+Na]+: 395,0834, установлено: 395,0813.
Синтез L-(-)-ментил(10E)-11-(3,4-дифторфенил)-ундек-10-еноата(ментил (Е)-В7)
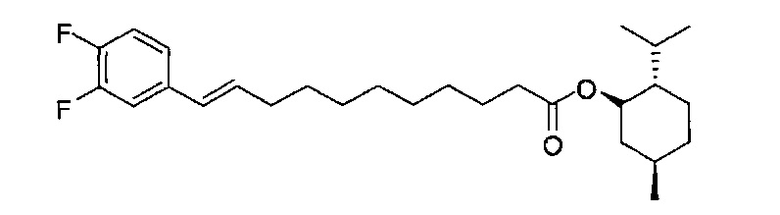
К раствору (E)-В1 (298 мг, 1,01 мМоль) в дихлорметане (3 мл) добавили L-(-)-ментол (314 мг, 2,01 мМоль), EDC⋅HCl (347 мг, 1,81 мМоль) и DMAP (1,2 мг, 0,010 мМоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Смесь разбавили дихлорметаном (20 мл), промыли водой (20 мл) и провели концентрацию. Очистку остатка провели колоночной хроматографией (этилацетат/n-гексан=1/50) с получением ментил (Е)-В7 (112 мг, 0,258 мМоль, 26%) в виде бесцветного масла. 1H-NMR (CDCl3, 400 МГц) δ 6,95-7,13 (m, 3Н), 6,24 (d, J=15,6 Гц, 1H), 6,15-6,24 (m, 1H), 4,11-4,20 (m, 1Н), 2,27 (t, J=7,6 Гц, 2Н), 2,15 (q, J=6,8 Гц, 2Н), 1,92-1,99 (m, 1H), 1,80-1,89 (m, 1H), 1,55-1,68 (m, 4Н), 1,38-1,50 (m, 3Н), 1,22-1,35 (m, 10Н), 0,77-1,09 (m, 9Н), 0,73 (d, J=7,0 Гц, 3Н). 13C-NMR (CDCl3, 100 МГц) δ 173,34, 150,39 (dd, 7=245, 13 Гц), 149,23 (dd, 7=246, 13 Гц), 135,15, 132,24, 127,77, 121,87, 117,00, 114,08, 73,82, 47,00, 40,93, 34,67, 34,25, 32,82, 31,33, 29,67, 29,26, 29,14, 29,08, 29,06, 26,22, 25,06, 23,39, 21,97, 20,70, 16,25. HRMS (ESI) в пересчете на C27H40F2O2Na [M+Na]+: 457,2894, установлено: 457,2863.
Синтез (10S,11S)-11-(3,4-дифторфенил)-10,11-дигидроксиундекановой кислоты и (10R,11R)-11-(3,4-дифторфенил)-10,11-дигидроксиундекановой кислоты, син-(В21) в форме смеси син-изомеров диола
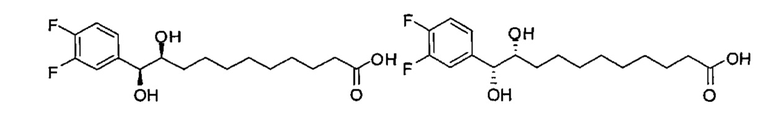
К раствору ментил (Е)-В7 (110 мг, 0,253 мМоль) в смеси t-бутанол/вода (2/1, 6 мл) добавили NMO (103 мг, 0,879 мМоль) и тетраоксид осмия (2,5 масс.% в t-BuOH, 38 мкл, 0,0037 мМоль). После перемешивания при комнатной температуре в течение 20 часов, реакцию в реакционной смеси остановили добавлением насыщенного Na2S2O3 (10 мл). Смесь концентрировали, и остаток растворили в этилацетате (20 мл), промыли насыщенным Na2S2O3 (20 мл) и соляным раствором (20 мл), высушили над MgSO4, профильтровали и провели концентрацию. Очистку остатка провели колоночной хроматографией (этилацетат/n-гексан=1/4, затем 1/3). Полученное в результате бесцветное масло растворили в метаноле (5 мл) и затем добавили 1,0 N NaOH (5 мл). После перемешивания при комнатной температуре в течение 12 часов реакционную смесь нейтрализовали добавлением 1,0 N HCl (5 мл) и провели концентрацию. Очистку остатка провели колоночной хроматографией (этилацетат/n-гексан=1/2, затем 1/1) с получением син-В21 (35 мг, 0,11 мМоль, 43%) в виде белого твердого вещества, точка плавления: 104°C. 1H-NMR (CD3OD, 400 МГц) δ 6,99-7,25 (m, 3Н), 4,36 (d, J=5,6 Гц, 1Н), 3,45-3,52 (m, 1Н), 2,18 (t, J=7,4 Гц, 2Н), 1,11-1,54 (m, 14Н). 13C-NMR (CD3OD, 100 МГц) δ 177,70, 151,29 (dd, J=244, 12 Гц), 150,76 (dd, J=244, 13 Гц), 141,48, 124,36, 117,64, 116,87, 77,23, 76,39, 34,39, 33,72, 30,51, 30,41, 30,27, 30,15, 26,80, 26,03. HRMS (ESI) в пересчете на C17H24F2O4Na [M+Na]+: 353,1540, установлено: 353,1550.
Синтез (10R,11S)- и (10S,11R)-11-(3,4-дифторфенил)-(10,11)-дигидроксиундекановой кислоты, анти-(В21) в виде смеси анти- изомеров диола
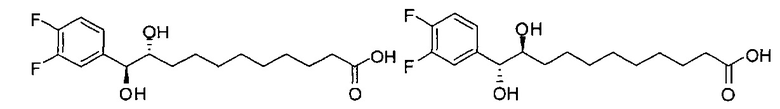
К раствору (Z)-B7 (361 мг, 1,22 мМоль) в смеси t-бутанол/вода (2/1, 6 мл) добавили NMO (494 мг, 4,22 мМоль) и тетраоксид осмия (2,5 масс.% в t-BuOH, 186 мкл, 0,0183 мМоль). После перемешивания при комнатной температуре в течение 20 часов реакцию в реакционной смеси остановили добавлением насыщенного Na2S2O3 (15 мл). Провели концентрацию смеси, остаток растворили в этилацетате (50 мл), промыли насыщенным Na2S2O3 (50 мл) и соляным раствором (50 мл), высушили над MgSO4, профильтровали и провели концентрацию. Очистку остатка провели колоночной хроматографией (этилацетат/n-гексан=1/2, затем 1/1) с получением анти-В21 (246 мг, 0,745 мМоль, 61%, смесь анти-изомеров диола) в виде бесцветного масла. 1H-NMR (CD3OD, 400 МГц) δ 7,03-7,25 (m, 3Н), 4,36 (d, J=5,5 Гц, 1H), 3,47-3,57 (m, 1Н), 2,16-2,22 (m, 2Н), 1,18-1,56 (m, 14Н). 13C-NMR (CD3OD, 100 МГц) δ 177,73, 151,19 (dd, J=245, 13 Гц), 150,72 (dd, J=245, 13 Гц), 141,46, 124,57, 117,49, 117,08, 77,34, 77,25, 76,42, 76,06, 34,95, 33,75, 33,50, 30,63, 30,51, 30,44, 30,34, 30,20, 26,83, 26,07. HRMS (ESI) в пересчете на C17H24F2O4Na [M+Na]+: 353,1540, установлено: 353,1566.
(4) Схема синтеза 4: синтез Соединения С5-С7
Синтез 3,4-ди-O-бензил-1-O-(6-азидо-2,3,4-три-O-бензил-6-дезокси-α-D-галактопиранозил)-2-гексакозаноиламино-D-рибо-октадекан-1,3,4-триола (С1)
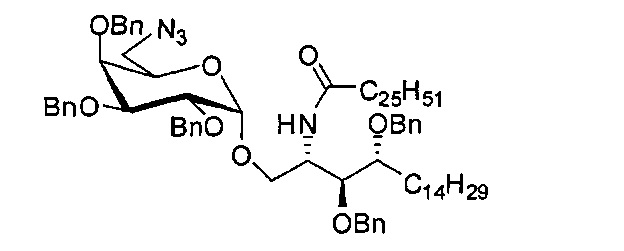
Соединение C1 можно синтезировать по Zhou, X.T. et al. Org Lett 2002, 4, 1267-1270. Данные по Cl: 1Н-NMR (CDCl3, 400 Гц) δ 7,35-7,21 (m, 25Н), 5,94 (d, J=6,0 Гц, 1H), 4,95 (d, J=11,4 Гц, 1H), 4,82 (d, J=3,2 Гц, 1H), 4,79-4,91 (m, 4H), 4,63-4,55 (m, 3H), 4,37-4,48 (m, 2H), 4,18-4,30 (m, 2H), 4,00 (dd, J=3,6, 10,1 Гц, 1H), 3,87-3,81 (m, 7H), 3,52-3,50 (m, 1H), 1,78-1,79 (m, 74H), 0,86 (t, J=7,0 Гц, 6H).
Синтез 1-O-(6-фенилацетамидо-6-дезокси-α-D-галактопиранозил)-2-гексакозаноиламино-D-рибо-октадекан-1,3,4-триола (С5)
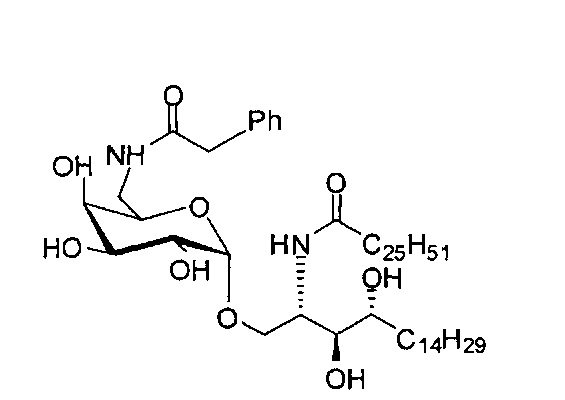
К раствору C1 (24 мг, 0,018 мМоль) в смеси тетрагидрофуран/вода (10/1, 5 мл) добавили трифенилфосфин (10 мг, 0,038 мМоль). После перемешивания при комнатной температуре в течение 2 дней провели концентрацию смеси и высушивание в вакууме. Остаток растворили в хлороформе (3 мл). К этому раствору добавили фенилуксусную кислоту (3 мг, 0,02 мМоль), N-метилморфолин (5 мкл, 0,05 мМоль) и HBTU (10 мг, 0,026 мМоль). После перемешивания при комнатной температуре в течение 12 часов провели концентрацию смеси и очистку колоночной хроматографией (этилацетат/n-гексан=1/8, до 1/6, до 1/4) с получением соединения С2. Полученное в результате промежуточное соединение C2 растворили в смеси дихлорметан/метанол (1/1, 5 мл) и затем добавили Pd(OH)2 (5,0 мг). После перемешивания при комнатной температуре в атмосфере водорода в течение 15 часов смесь профильтровали через целитовую подушку и промыли смесью дихлорметан/метанол (1/1). Провели концентрацию фильтрата и очистку колоночной хроматографией (дихлорметан/метанол=1/1) с получением С5 (4,0 мг, 0,004 мМоль, 22%) в виде белого воска. 1H-NMR (пиридин-d5, 400 Гц) δ 7,24-7,43 (m, 5Н), 5,51 (d, J=3,8 Гц, 1H), 4,50-4,67 (m, 2Н), 4,09-4,51 (m, 8Н), 3,89 (s, 2Н), 3,82-4,01 (m, 1Н), 2,21-2,57 (m, 4Н), 1,08-1,96 (m, 74Н), 0,88 (t, J=6,1 Гц, 6Н). 13C-NMR (пиридин-d6, 150 Гц) δ 173,75, 173,46, 142,53, 129,24, 129,20, 126,75, 101,71, 77,08, 72,95, 71,64, 71,34, 70,91, 70,50, 68,82, 51,89, 41,50, 38,77, 37,20, 34,81, 32,67, 32,51, 30,78, 30,55, 30,42, 30,39, 30,32, 30,20, 29,99, 26,90, 26,79, 23,32, 14,66.
Синтез 1-O-(6-(3-фенилпропиламидо)-6-дезокси-α-D-галактопиранозил)-2-гексакозаноиламино-D-рибо-октадекан-1,3,4-триола (С6)
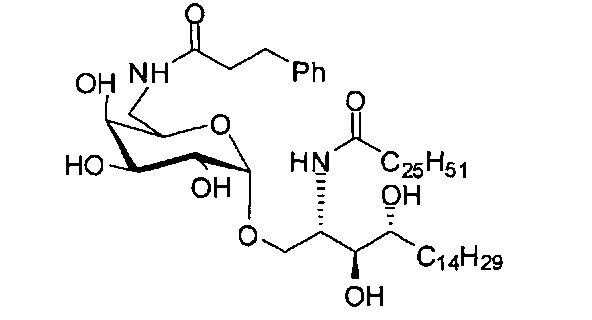
По тому же способу синтеза, что и способ синтеза С5, соединение C1 (24 мг, 0,018 мМоль) и 3-фенилпропионовую кислоту (2,2 мг, 0,018 мМоль) использовали в качестве исходных материалов для получения С6 (10 мг, 0,010 мМоль, 55%). 1H-NMR (пиридин-d5, 400 Гц) δ 7,29-7,30 (m, 5Н), 5,50 (d, J=3,7 Гц, 1H), 5,20-5,30 (m, 1H), 4,56-4,65 (m, 2Н), 4,48 (t, J=6,6 Гц, 1Н), 4,25-4,38 (m, 5Н), 4,15-4,23 (m, 1Н), 3,86-3,95 (m, 1H), 3,17-3,26 (m, 2Н), 2,74-2,89 (m, 2Н), 2,41-2,55 (m, 2Н), 1,08-2,04 (m, 74Н), 0,89 (t, J=6,7 Гц, 3Н). 13C-NMR (пиридин-d5, 400 Гц) δ 173,81, 173,52, 149,78, 142,60, 129,32, 129,27, 126,83, 101,78, 77,15, 73,00, 71,70, 71,41, 70,98, 70,56, 68,88, 51,94, 38,84, 37,26, 34,88, 32,74, 32,58, 30,86, 30,63, 20,50, 30,47, 30,39, 26,97, 26,87, 23,40, 14,74.
Синтез 1-O-(6-(4-фенилбутиламидо)-6-дезокси-α-D-галактопиранозил)-2-гексакозаноиламино-D-рибо-октадекан-1,3,4-триола (С7)
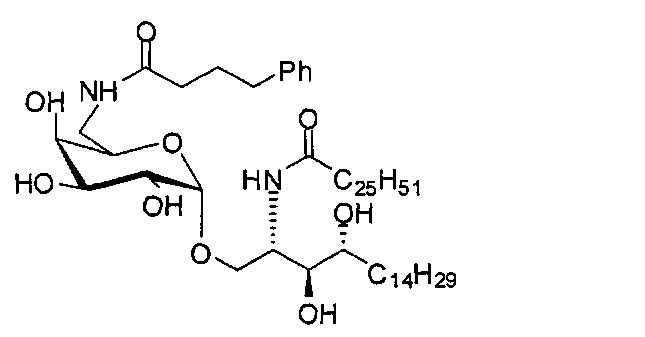
По тому же способу синтеза, что и способ синтеза С5 соединение С1 (24 мг, 0,018 мМоль) и 4-фенилбутановую кислоту (2,0 мг, 0,018 мМоль) использовали в качестве исходных материалов для получения С7 (9,0 мг, 0,090 мМоль, 50%). 1H-NMR (пиридин-d5, 400 Гц) δ 7,27-7,40 (m, 5Н), 5,50 (d, J=3,7 Гц, 1Н), 5,21-5,27 (m, 1H), 4,55-4,66 (m, 2Н), 4,48 (t, J=6,6 Гц, 1H), 4,25-4,37 (m, 6Н), 4,15-4,23 (m, 1Н), 3,87-3,95 (m, 1H), 3,16-3,25 (m, 2Н), 2,74-2,89 (m, 2Н), 2,41-2,55 (m, 2Н), 1,02-1,98 (m, 74Н), 0,88 (t, J=7,2 Гц, 6Н). 13C-NMR (пиридин-d5, 400 Гц) δ 173,30, 173,02, 142,07, 128,82, 128,70, 126,33, 101,28, 76,66, 72,50, 71,19, 70,90, 70,48, 70,05, 68,39, 51,44, 41,06, 38,34, 36,77, 34,38, 32,25, 32,10, 30,37, 30,13, 30,01, 29,98, 29,91, 29,79, 29,76, 29,59, 29,58, 26,47, 26,37, 22,91, 14,25.
(5) Схема синтеза 5: Синтез соединения С20-С31
Синтез 1-O-(6-(9-толуолсульфонил-α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амино-D-рибо-1,3,4-октадекантриола (С17)
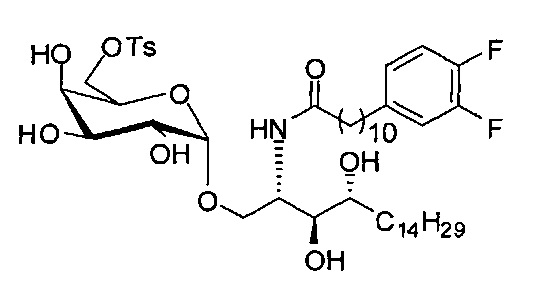
К раствору А15 (2,58 г, 3,40 мМоль) в пиридине (30 мл) добавили раствор p-толуолсульфонил хлорида (712 мг, 3,74 мМоль) в пиридине (20 мл) в ледяной бане. Реакционную смесь довели до комнатной температуры. После перемешивания в течение 16 часов, растворитель испарился, и очистку остатка провели колоночной хроматографией (дихлорметан/метанол = 100/1, до 50/1, до 20/1, до 15/1) с получением С17 в виде желтого воска (782 мг, 0,855 мМоль, 25%, 100% на базе извлеченного исходного материала, при извлечении исходного материала 2,01 г). 1H-NMR (CD3OD/CDCl3=1/1, 400 МГц) δ 7,96 (d, J=8,3 Гц, 2Н), 7,56 (d, J=8,1 Гц, 2Н), 7,10-7,27 (m, 2Н), 7,02-7,09 (m, 1Н), 5,01 (d, J=3,2 Гц, 1Н), 4,29-4,42 (m, 3Н), 4,21 (t, J=5,9 Гц, 1H), 3,96-4,04 (m, 2Н), 3,85-3,95 (m, 2Н), 3,81 (dd, J=4,0, 10,6 Гц, 1H), 3,70-3,76 (m, 2Н), 2,74 (t, J=7,6 Гц, 2Н), 2,62 (s, 3Н), 2,35-2,43 (m, 2Н), 1,67-1,88 (m, 4Н), 1,36-1,60 (m, 38Н), 1,05 (t, J=6,7 Гц, 3Н). 13C-NMR (CD3OD/CDCl3=1/1, 100 МГц) δ 173,84, 149,47 (dd, J=246, 13 Гц), 148,00 (dd, J=244, 13 Гц), 144,74, 139,43, 132,04, 129,32, 127,44, 123,60, 116,22, 114,55, 99,09, 73,87, 71,39, 69,27, 68,98, 68,51, 68,15, 68,07, 66,95, 35,85, 34,46, 31,71, 31,34, 30,72, 29,21, 29,17, 29,12, 29,06, 28,96, 28,91, 28,82, 28,77, 28,50, 25,30, 22,06, 20,67, 13,22. HRMS (ESI) в пересчете на C48H78F2NO11S [М+Н]+: 914,5264, установлено: 914,5228.
Синтез 1-O-(6-азидо-6-дезокси-α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амино-D-рибо-1,3,4-октадекантриола (С18)
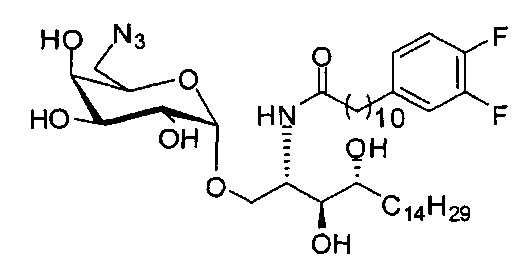
К раствору С17 (1,63 г, 1,78 мМоль) в диметилформамиде (15 мл) добавили азид натрия (322 мг, 4,95 мМоль). Реакционную смесь перемешивали при 100°C в течение 2 дней. Провели концентрацию смеси, и остаток очистили колоночной хроматографией (дихлорметан/метанол = 20/1 до 15/1) с получением неочищенного С18 (1,15 г, 1,47 мМоль, 82%) в виде твердого вещества желтого цвета, точка плавления: 101°C. 1H-NMR (CD3OD/CDCl3=1/1, 400 МГц) δ 7,25-7,51 (m, 3Н), 5,36 (d, J=3,2 Гц, 1Н), 4,57-4,67 (m, 1H), 4,12-4,42 (m, 6H), 3,94-4,06 (m, 3H), 3,74 (dd, J=12,8, 4,8 Гц, 1H), 3,00 (t, J=7,7 Гц, 2H), 2,60-2,67 (m, 2H), 1,62-2,15(m, 42H), 1,32 (t, J=6,8 Гц, 3Н). HRMS (ESI) в пересчете на C41H71F2N4O8 [М+Н]+: 785,5240, установлено: 785,5267.
Синтез 1-O-(6-амино-6-дезокси-α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амино-D-рибо-1,3,4-октадекантриола (С19)
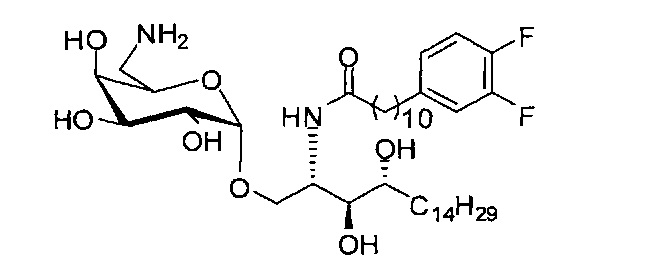
К раствору С18 (1,11 г, 1,41 мМоль) в смеси тетрагидрофуран/вода (10/1, 33 мл) добавили трифенилфосфин (0,74 г, 2,8 мМоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 дней. Провели концентрацию смеси, и остаток очистили колоночной хроматографией (1% триэтиламин, дихлорметан/метанол=6/1, до 4/1, до 2/1) с получением С19 (566 мг, 0,746 мМоль, 53%) в виде белого твердого вещества, точка плавления: 161°C. 1H-NMR (CD3OD, 400 МГц) δ 6,95-7,09 (m, 2Н), 6,86-6,88 (m, 1Н), 4,82 (d, J=3,2 Гц, 1H), 4,13-4,19 (m, 1H), 3,83 (dd, J=10,4, 4,4 Гц, 1H), 3,75 (d, J=2,0, 1H), 3,63-3,73 (m, 3Н), 3,59 (dd, J=10,8, 5,6 Гц, 1H), 3,44-3,54 (m, 2Н), 2,87 (dd, J=13,2, 7,6 Гц, 1H), 2,72 (dd, J=13,2, 4,4 Гц, 1H), 2,51 (t, J=7,6 Гц, 2H), 2,14 (t, J=7,5 Гц, 2H), 1,50-1,63 (m, 4H), 1,16-1,32 (m, 38H), 0,82 (t, J=6,8 Гц, 3Н). 13C-NMR (CD3OD, 100 МГц) δ 176,07, 151,5 (d, J=246, 13 Гц), 150,8 (d, 243, 12 Гц), 141,74, 125,79, 118,16, 118,02, 101,43, 75,90, 73,12, 72,46, 72,02, 71,63, 70,33, 68,35, 52,18, 43,44, 37,46, 36,20, 33,37, 33,26, 32,68, 31,06, 31,02, 30,96, 30,89, 30,81, 30,73, 30,67, 30,62, 30,39, 27,30, 27,21, 23,29. HRMS (ESI) пересчете на C41H73F2N2O8 [M+H]+: 759,5335, установлено: 759,5319.
Синтез 1-O-(6-(4-нитрофенилацетамидо)-6-дезокси-α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амино-D-рибо-1,3,4-октадекан триола (С20)
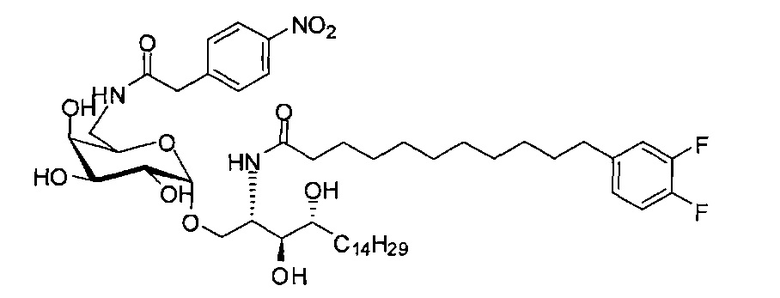
К раствору С19 (34 мг, 0,045 мМоль) в смеси дихлорметан/метанол (1/1, 3 мл) добавили 4-нитрофенилуксусную кислоту (8,1 мг, 0,045 мМоль), HBTU (34 мг, 0,090 мМоль) и N-метилморфолин (20 мкл, 0,18 мМоль). После перемешивания при комнатной температуре в течение 16 часов провели концентрацию смеси и очистку колоночной хроматографией (дихлорметан/метанол = 30/1 до 20/1). Неочищенный продукт растворили в смеси дихлорметан/метанол (1/1, 3 мл) и к раствору добавили Si-карбонатный силикагель (поглотитель НОВТ, 100 мг). После перемешивания при комнатной температуре в течение 1 часа смесь профильтровали и промыли смесью дихлорметан/метанол (1/1). Провели концентрацию фильтрата и высушили его в вакууме с получением С20 (13 мг, 0,014 мМоль, 31%) в виде светло-желтого воска. 1H-NMR (CDCl3/CD3OD=1/1, 200 МГц) δ 8,25 (d, J=8,7 Гц, 2Н), 7,68 (d, J=8,7 Гц, 2Н), 6,90-7,21 (m, 3Н), 4,94 (d, J=3,2 Гц, 1Н), 4,20-4,26 (m, 1H), 3,77-3,92 (m, 5Н), 3,58-3,74 (m, 6Н), 2,63 (t, J=7,4 Гц, 2Н), 2,28 (t, J=7,6 Гц, 2Н), 1,30-1,81 (m, 42Н), 0,98 (t, J=6,8 Гц, 3Н). 13C-NMR (CDCl3/CD3OD=1/1, 50 МГц) δ 173,93, 170,81, 149,17 (dd, J=246, 13 Гц), 148,05 (dd, J=244, 13 Гц), 146,43, 142,53, 139,35, 129,58, 123,59, 122,95, 116,28, 116,11, 98,99, 73,65, 71,21, 69,35, 69,18, 68,41, 68,14, 66,16, 49,81, 41,69, 39,69, 36,34, 35,69, 34,36, 31,61, 31,25, 30,66, 29,04, 28,74, 28,41, 25,27, 21,97, 13,07.
Синтез 1-O-(6-(2,4-динитрофенилацетамидо)-6-дезокси-α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амино-D-рибо-1,3,4-октадекантриола (С21)
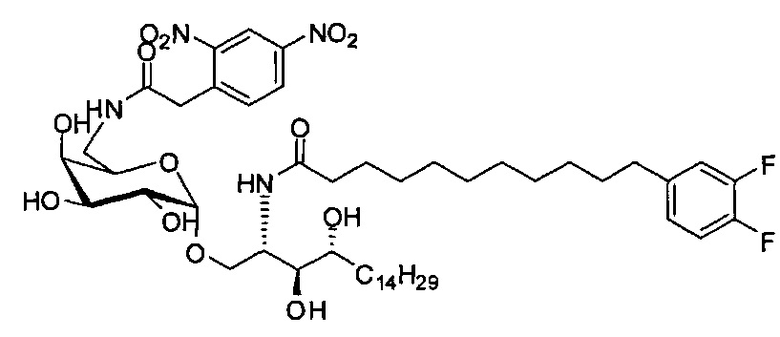
По тому же способу синтеза, что и способ синтеза С20, соединение С19 (34 мг, 0,045 мМоль) и 2,4-динитрофенилуксусную кислоту (10 мг, 0,045 мМоль) использовали в качестве исходных материалов для получения С21 (4,0 мг с неотделимыми примесями, 0,0041 мМоль, 9%). 1H-NMR (CDCl3/CD3OD=1/1, 600 МГц) δ 7,39-7,48 (m, 3Н), 6,97-7,17 (m, 3Н), 4,98 (d, J=3,2 Гц, 1Н), 4,24-4,31 (m, 1H), 3,59-3,93 (m, 10Н), 2,65 (t, J=7,4 Гц, 2Н), 2,26-2,31 (m, 2Н), 1,14-1,87 (m, 42Н), 0,90-1,02 (m, 3Н). 13C-NMR (CDCl3/CD3OD=1/1, 150 МГц) δ 173,95, 169,65, 148,64, 147,80, 139,33, 134,76, 132,72, 129,90, 129,13, 123,53, 123,15, 116,92, 116,57, 116,13, 110,69, 99,01, 73,64, 73,42, 71,16, 69,32, 69,08, 68,12, 66,07, 49,80, 39,45, 39,60, 38,18, 38,13, 36,30, 35,66, 34,33, 34,30, 33,63, 32,01, 31,60, 31,43, 30,67, 30,58, 29,73, 29,23, 28,39, 28,35, 28,23, 26,30, 25,23, 25,17, 23,06, 22,25, 22,20, 21,78, 12,98.
1-O-(6-(4-трет-бутилфенилацетамидо)-6-дезокси-α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амино-D-рибо-1,3,4-октадекантриол (С22)
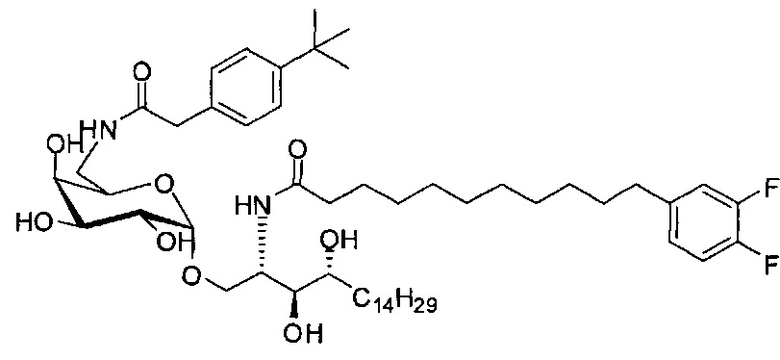
По тому же способу синтеза, что и способ синтеза С20, соединение С19 (34 мг, 0,045 мМоль) и 4-трет-бутилфенилуксусную кислоту (8,6 мг, 0,044 мМоль) использовали в качестве исходных материалов для получения С22 (12 мг, 0,013 мМоль, 29%), точка плавления: 170°C. 1H-NMR (CDCl3/CD3OD=1/1, 400 МГц) δ 7,47 (d, J=8,0 Гц, 2Н), 7,32 (d, J=8,0 Гц, 2Н), 6,98-7,22 (m, 3Н), 4,97 (d, J=3,2 Гц, 1H), 4,29-4,34 (m, 1H), 3,80-3,95 (m, 5Н), 3,76 (dd, J=10,8, 4,4 Гц, 1H), 3,61-3,71 (m, 5Н), 3,36 (dd, J=7,8, 13,8 Гц, 1H), 2,68 (t, J=7,6 Гц, 2Н), 2,33 (t, J=7,6 Гц, 2Н), 1,63-1,82 (m, 4Н), 1,31-1,53 (m, 47Н), 1,00 (t, J=6,5 Гц, 3Н). 13C-NMR (CDCl3/CD3OD=1/1, 100 МГц) δ 173,91, 172,88, 149,40 (dd,7=246, 12 Гц), 148,02 (dd,243, 13 Гц), 139,36, 131,28, 124,96, 123,57, 116,21, 116,05, 99,06, 73,79, 71,30, 69,35, 68,97, 68,40, 68,20, 66,42, 49,84, 41,82, 39,39, 35,72, 34,37, 33,66, 31,66, 31,25, 30,65, 30,38, 29,14, 29,09, 29,05, 29,03, 28,97, 28,89, 28,83, 28,75, 28,71, 28,68, 28,42, 25,28, 25,24, 21,96, 13,06.
Синтез 1-O-(6-(4-бромфенилацетамидо)-6-дезокси-α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амино-D-рибо-1,3,4-октадекан триола (С23)
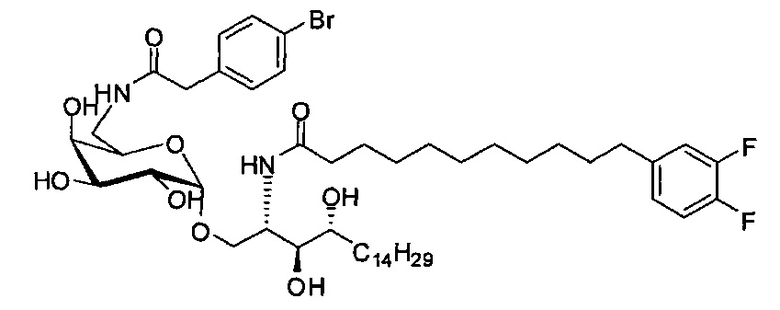
По тому же способу синтеза, что и способ синтеза С20, соединение С19 (34 мг, 0,045 мМоль) и 4-бромфенилуксусную кислоту (9,7 мг, 0,044 мМоль) использовали в качестве исходных материалов для получения С23 (15 мг, 0,016 мМоль, 35%) в виде белого твердого вещества с сероватым или желтоватым оттенком, точка плавления = 177°C. 1H-NMR (CDCl3/CD3OD=1/1, 400 МГц) δ 7,54 (d, J=8,4 Гц, 2Н), 7,29 (d, J=8,0 Гц, 2Н), 6,97-7,20 (m, 3Н), 4,97 (d, J=3,6 Гц, 1H), 4,28-4,33 (m, 1H), 3,78-3,92 (m, 5H), 3,59-3,74 (m, 6H), 3,37 (dd, J=7,6, 13,6 Гц, 1H), 2,66 (t, J=7,6 Гц, 2H), 2,31 (t, J=7,6 Гц, 2H), 1,30-1,81 (m, 42H), 0,98 (t, J=6,8 Гц, 3Н). 13C-NMR (CDCl3/CD3OD=1/1, 100 МГц) δ 173,90, 171,91, 149,53 (dd, J=247, 13 Гц), 147,86 (dd, J=245, 12 Гц), 139,35, 133,70, 130,98, 130,29, 123,58, 120,22, 120,22, 116,18, 116,02, 99,03, 73,77, 71,26, 69,34, 69,08, 68,38, 68,15, 66,32, 49,81, 41,48, 39,51, 35,68, 34,35, 31,63, 31,23, 30,62, 29,11, 29,08, 29,01, 28,95, 28,87, 28,81, 28,73, 28,68, 28,66, 28,40, 25,25, 25,22, 21,94, 13,03.
Синтез 1-O-(6-(4-метоксифенилацетамидо)-6-дезокси-α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амино-D-рибо-1,3,4-октадекан триола (С24)
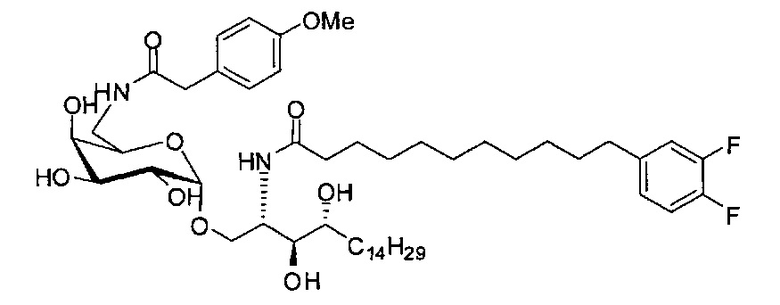
По тому же способу синтеза, что и способ синтеза С20, соединение С19 (34 мг, 0,045 мМоль) и 4-метоксифенилуксусную кислоту (7,5 мг, 0,045 мМоль) использовали в качестве исходных материалов для получения С24 (15 мг, 0,017 мМоль, 38%) в виде белого твердого вещества с сероватым или желтоватым оттенком, точка плавления: 172°C. 1H-NMR (CDCl3/CD3OD=1/1, 400 МГц) δ 7,36 (d, J=8,8 Гц, 2Н), 7,12-7,26 (m, 2Н), 7,01-7,09 (m, 3Н), 5,02 (d, J=3,6 Гц, 1Н), 4,32-4,38 (m, 1H), 3,97 (s, 3Н), 3,72-3,96 (m, 5Н), 3,69-3,78 (m, 4Н), 3,65 (s, 2Н), 3,40 (dd, J=7,8, 14,0 Гц, 1H), 2,74 (t, J=7,6 Гц, 2Н), 2,38 (t, J=7,6 Гц, 2Н), 1,38-1,89 (m, 42Н), 1,05 (t, J=6,8 Гц, 3Н). 13C-NMR (CDCl3/CD3OD=1/1, 100 МГц) δ 173,91, 173,06, 149,59 (dd, J=247, 13 Гц), 148,08 (dd, J=244, 13 Гц), 158,27, 129,40, 129,66, 126,43, 123,61, 116,29, 116,12, 114,55, 113,62, 99,08, 73,92, 71,39, 61,41, 69,03, 68,41, 68,24, 66,43, 54,47, 49,78, 41,57, 39,46, 35,82, 34,46, 31,80, 31,33, 30,71, 29,22, 29,17, 29,12, 29,11, 29,05, 28,96, 28,90, 28,82, 28,78, 28,75, 28,49, 25,34, 25,31, 22,04, 13,19.
Синтез 1-O-(6-(3,4-ди(трифторометил)фенил-ацетамидо)-6-дезокси-α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амино-D-рибо-1,3,4-октадекантриола (С25)
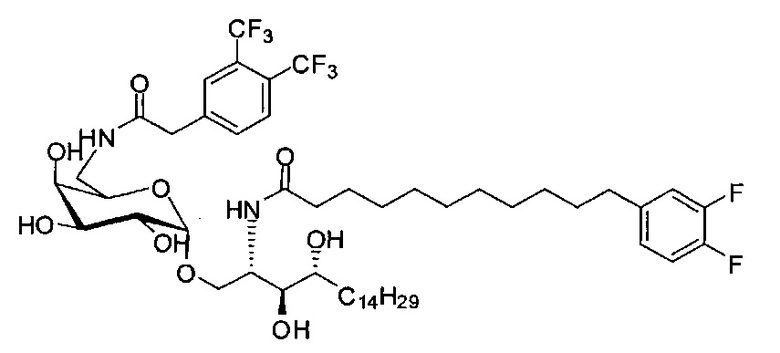
По тому же способу синтеза, что и способ синтеза С20, соединение С19 (34 мг, 0,045 мМоль) и 3,4-ди(трифторометанил)фенилуксусную кислоту (12 мг, 0,045 мМоль) использовали в качестве исходных материалов для получения С25 (11 мг, 0,011 мМоль, 24%) в виде белого твердого вещества с сероватым или желтоватым оттенком, точка плавления: 180°C 1H-NMR (CDCl3/CD3OD=1/1, 400 МГц) δ 7,98 (s, 2Н), 7,91 (s, 1H), 769-7,22 (m, 3Н), 5,01 (d, J=3,6 Гц, 1H), 4,28-4,33 (m, 1Н), 3,84-3,99 (m, 5Н), 3,83 (s, 2Н), 3,78 (dd, J=4,2, 10,8 Гц, 1Н), 3,22-3,32 (m, 3Н), 3,42-3,51 (m, 1H), 2,69 (t, J=7,6 Гц, 2Н), 2,33 (t, J=7,4 Гц, 2Н), 1,39-1,82 (m, 42Н), 1,00 (t, J=6,8 Гц, 3Н). (13C-NMR CDCl3/CD3OD=1/1, 100 МГц) δ 174,04, 170,56, 143,17, 139,37, 137,65, 131,13, 130,80, 129,10, 124,18, 123,56, 121,47, 120,09, 116,22, 116,06, 114,50, 99,07, 73,77, 71,32, 69,40, 69,27, 68,42, 68,20, 66,30, 49,99, 41,14, 39,78, 35,75, 34,39, 31,73, 31,27, 30,66, 29,14, 29,09, 29,04, 28,99, 28,88, 28,75, 28,71, 28,69, 28,42, 25,29, 25,24, 21,98, 13,08.
Синтез 1-O-(6-(3,4-дифторфенилацетамидо)-6-дезокси-α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амино-D-рибо-1,3,4-октадекантриола (С26)
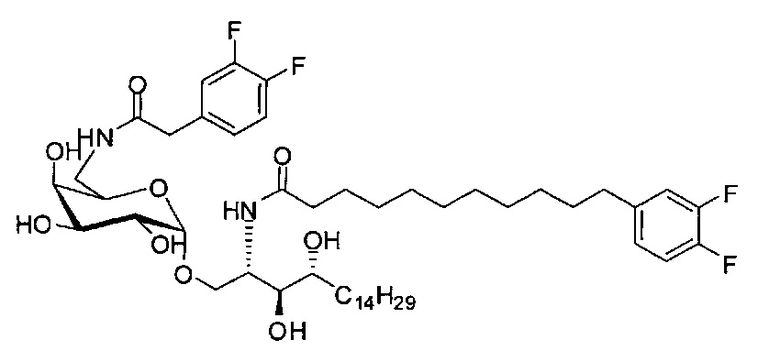
По тому же способу синтеза, что и способ синтеза С20, соединение С19 (34 мг, 0,045 мМоль) и 3,4-дифторфенилуксусную кислоту (7,7 мг, 0,045 мМоль) использовали в качестве исходных материалов для получения С26 (17 мг, 0,019 мМоль, 42%) в виде белого твердого вещества с сероватым или желтоватым оттенком, точка плавления: 182°C 1H-NMR (CDCl3/CD3OD=1/1, 400 МГц) δ 6,97-7,43 (m, 6Н), 4,97 (d, J=3,6 Гц, 1H), 4,28 (q, J=4,4, 9,6 Гц, 1Н), 3,83-4,01 (m, 4Н), 3,80 (dd, J=3,2, 10,0 Гц, 1H), 3,71 (dd, J=4,4, 10,6 Гц, 1H), 3,60-3,68 (m, 3Н), 3,59 (s, 2Н), 3,38 (dd, J=8,0, 13,6 Гц, 1Н), 2,66 (t, J=7,6 Гц, 2H), 2,30 (t, J=7,6 Гц, 2H), 1,32-1,81 (m, 42H), 0,97 (t, J=6,8 Гц, 3Н). 13C-NMR CDCl3/CD3OD=1/1, 100 МГц) δ 173,92, 171,63, 150,67, 150,03, 149,13, 148,25, 147,65, 146,72, 139,34, 131,81, 124,72, 123,54, 117,52, 117,34, 116,97, 116,59, 116,42, 116,25, 116,08, 115,93, 99,02, 73,75, 71,23, 69,34, 69,15, 68,38, 68,14, 66,24, 49,82, 41,06, 39,57, 35,66, 34,33, 31,63, 31,23, 30,61, 29,10, 29,07, 29,00, 28,94, 28,85, 28,79, 28,71, 28,65, 28,38, 25,24, 25,20, 21,93, 13,01.
Синтез 1-O-(6-(3-трифторметилфенил-ацетамидо)-6-дезокси-α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амино-α-D-рибо-1,3,4-октадекан триола (С27)
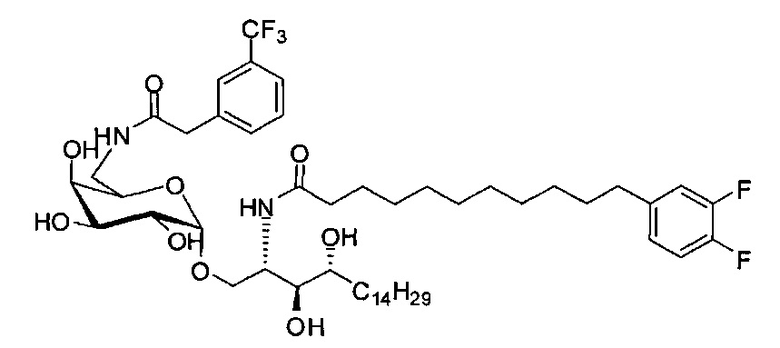
По тому же способу синтеза, что и способ синтеза С20, соединение С19 (34 мг, 0,045 мМоль) и 3-трифторометилфенилуксусную кислоту (9,2 мг, 0,045 мМоль) использовали в качестве исходных материалов для получения С27 (12 мг, 0,013 мМоль, 29%) в виде белого твердого вещества с сероватым или желтоватым оттенком, точка плавления: 157°C. 1H-NMR (CDCl3/CD3OD=1/1, 400 МГц) δ 6,96-7,82 (m, 7Н), 4,96 (d, J=3,6 Гц, 1H), 4,25-41,30 (m, 1H), 4,03-41,10 (m, 1Н), 3,77-3,91 (m, 5Н), 3,53-3,74 (m, 5Н), 3,36-3,44 (m, 1H), 2,65 (t, J=7,6 Гц, 2Н), 2,29 (t, J=7,6 Гц, 2Н), 1,27-1,76 (m, 42Н), 0,97 (t, J=6,8 Гц, 3Н). 13C-NMR (CDCl3/CD3OD=1/1, 100 МГц) δ 174,52, 172,19, 150,04 (dd, 247, 13 Гц), 148,52 (dd, J=244, 13 Гц), 139,90, 136,43, 132,64, 129,02, 127,67, 125,80, 124,17, 116,76, 116,60, 111,27, 99,64, 74,33, 71,87, 69,93, 69,73, 68,98, 68,76, 66,90, 60,57, 50,44, 42,27, 40,16, 36,25, 34,92, 32,20, 31,82, 31,21, 29,68, 29,65, 29,59, 29,53, 29,44, 29,38, 29,30, 29,24, 28,97, 25,83, 25,80, 22,52, 13,60.
Синтез 1-O-(6-(4-метилфенилацетамидо)-6-дезокси-α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амино-D-рибо-1,3,4-октадекан триола (С28)
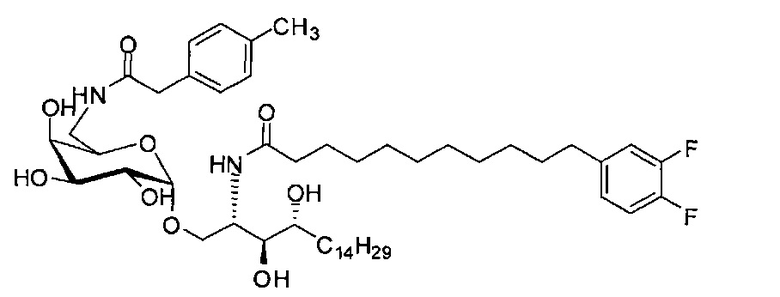
По тому же способу синтеза, что и способ синтеза С20, соединение С19 (34 мг, 0,045 мМоль) и 4-метилфенилуксусную кислоту (6,8 мг, 0,045 мМоль) использовали в качестве исходных материалов для получения С28 (11 мг, 0,012 мМоль, 27%) в виде белого твердого вещества с сероватым или желтоватым оттенком, точка плавления: 171°C. 1H-NMR (CDCl3/CD3OD=1/1, 400 МГц) δ 7,28-44 (m, 4Н), 7,12-7,26 (m, 2Н), 7,03-7,08 (m, 1Н), 5,01 (d, J=3,6 Гц, 1Н), 4,32-4,41 (m, 1Н), 3,83-3,95 (m, 5Н), 3,67-3,79 (m, 3Н), 3,66 (s, 2Н), 3,40 (dd, J=7,6, 13,6 Гц, 1Н), 2,73 (t, J=7,6 Гц, 2Н), 2,37 (t, J=7,6 Гц, 2Н), 1,32-1,87 (m, 42Н), 1,05 (t, J=6,8 Гц, 3Н). 13C-NMR (CDCl3/CD3OD=1/1, 100 МГц) δ 173,94, 172,96, 149,83 (dd, J=247, 13 Гц), 148,02 (dd, J=244, 13 Гц), 139,42, 136,24, 131,26, 128,86, 128,47, 123,42, 116,31, 116,15, 99,10, 73,89, 71,43, 69,40, 68,98, 68,41, 68,25, 66,49, 49,82, 42,07, 39,42, 35,83, 34,47, 31,78, 31,34, 30,73, 29,23, 29,19, 29,12, 29,06, 28,97, 28,92, 28,84, 28,80, 28,77, 28,51, 25,35, 25,32, 22,06, 20,10, 13,22.
Синтез 1-O-(6-(3-метилфенилацетамидо)-6-дезокси-α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амино-D-рибо-1,3,4-октадекан триола (С29)
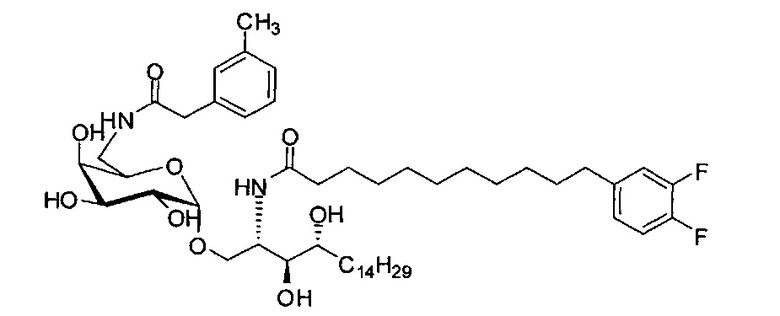
По тому же способу синтеза, что и способ синтеза С20, соединение С19 (34 мг, 0,045 мМоль) и 3-метилфенилуксусную кислоту (6,8 мг, 0,045 мМоль) использовали в качестве исходных материалов для получения С29 (14 мг, 0,016 мМоль, 35%) в виде белого твердого вещества с сероватым или желтоватым оттенком, точка плавления: 167°C. 1H-NMR (CDCl3/CD3OD=1/1, 400 МГц) δ 7,18-7,51 (m, 7Н), 5,08 (d, J=4,0 Гц, 1H), 4,36-4,43 (m, 1H), 3,91-4,02 (m, 5Н), 3,23-3,35 (m, 4Н), 3,73 (s, 2Н), 3,47 (dd, J=7,8, 13,8 Гц, 1H), 2,79 (t, J=7,8 Гц, 2H), 2,57 (s, 3Н), 2,43 (t, J=7,6 Гц, 2H), 1,46-1,94 (m, 42H), 1,11 (t, J=6,8 Гц, 3Н). 13C-NMR (CDCl3/CD3OD=1/1, 100 МГц) δ 173,96, 172,85, 149,52 (dd, J=245, 13 Гц), 148,13 (dd, J=243, 13 Гц), 139,43, 137,90, 134,27, 129,41, 128,13, 127,37, 125,61, 123,67, 116,35, 116,19, 99,15, 73,99, 71,49, 69,44, 69,00, 68,41, 68,30, 66,53, 49,88, 42,50, 39,46, 35,89, 34,52, 31,91, 31,39, 30,76, 29,28, 29,23, 29,11, 29,01, 28,96, 28,88, 28,84, 28,81, 28,55, 25,38, 25,36, 22,11, 13,29.
Синтез 1-O-(6-(2-метилфенилацетамидо)-6-дезокси-α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амино-D-рибо-1,3,4-октадекан триола (С30)
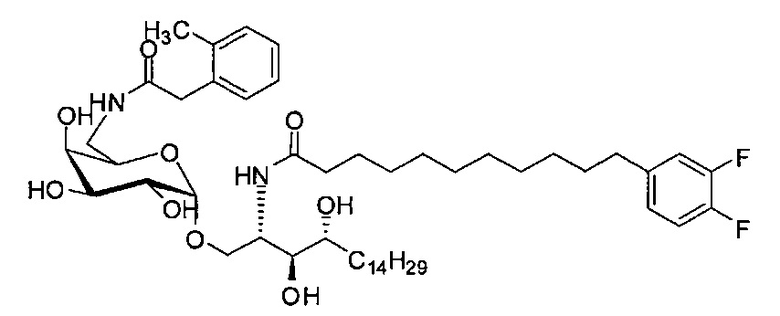
По тому же способу синтеза, что и способ синтеза С20, соединение С19 (34 мг, 0,045 мМоль) и 2-метилфенилускусную кислоту (6,8 мг, 0,045 мМоль) использовали в качестве исходных материалов для получения C30 (16 мг, 0,018 мМоль, 40%) в виде белого твердого вещества с сероватым или желтоватым оттенком, точка плавления: 182°C. 1H-NMR (CDCl3/CD3OD=1/1, 400 МГц) δ 7,31-7,37 (m, 4Н), 7,01-7,26 (m, 3Н), 4,99 (d, J=3,6 Гц, 1Н), 4,30-4,35 (m, 1Н), 3,83-3,94 (m, 5Н), 3,68-3,75 (m, 6Н), 3,40 (dd, J=8,0, 13,8 Гц, 1Н), 2,72 (t, J=7,8 Гц, 2Н), 2,45 (s, 3Н), 2,36 (t, J=7,6 Гц, 2Н), 1,32-1,87 (m, 42Н), 1,04 (t, J=6,8 Гц, 3Н). 13C-NMR (CDCl3/CD3OD=1/1, 100 МГц) δ 173,86, 172,42, 149,48 (dd, J=246, 13 Гц), 147,94 (dd, J=243, 13 Гц), 139,41, 136,38, 132,67, 129,91, 129,65, 126,99, 125,79, 123,63, 116,28, 116,11, 99,15, 73,91, 71,38, 69,38, 69,11, 68,35, 68,20, 66,56, 49,74, 40,27, 39,49, 35,80, 34,45, 31,78, 31,32, 30,70, 29,21, 29,16, 29,10, 29,04, 28,95, 28,90, 28,81, 28,77, 28,74, 28,48, 25,33, 25,29, 22,03, 18,65, 13,17.
Синтез 1-O-(6-(2-нафтилацетамидо)-6-дезокси-α-D-галактопиранозил)-2-(11-(3,4-дифторфенил)ундеканоил)амино-D-рибо-1,3,4-октадекантриола (С31)
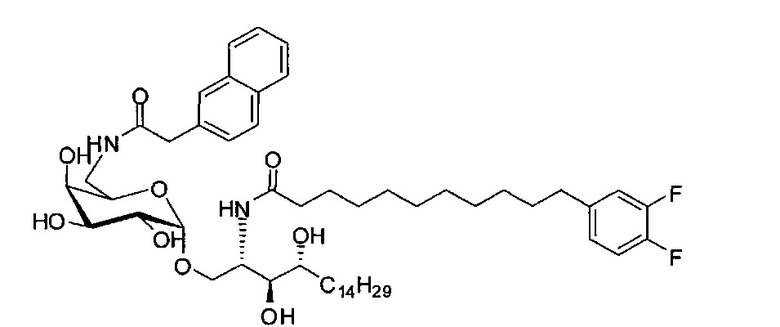
По тому же способу синтеза, что и способ синтеза С20, соединение С19 (34 мг, 0,045 мМоль) и 2-нафтилуксусную кислоту (8,4 мг, 0,045 мМоль) использовали в качестве исходных материалов для получения С31 (12 мг, 0,013 мМоль, 29%) в виде твердого белого вещества, точка плавления: 178°C. 1H-NMR (CDCl3/CD3OD=1/1, 400 МГц) δ 7,85-8,04 (m, 3Н), 7,40-7,69 (m, 4Н), 7,05-7,29 (m, 3Н), 5,03 (d, J=3,6 Гц, 1H), 4,31-4,40 (m, 1H), 3,87-4,01 (m, 6Н), 3,72-3,81 (m, 4Н), 3,46 (dd, J=7,8, 14,0 Гц, 1H), 2,76 (t, J=7,6 Гц, 2Н), 2,38 (t, J=8,0 Гц, 2Н), 1,32-1,90 (m, 42Н), 1,08 (t, J=6,6 Гц, 3Н). 13C-NMR (CDCl3/CD3OD=1/1, 150 МГц) δ 173,92, 172,58, 149,54 (dd, J=244, 12 Гц), 147,93 (dd, J=240, 13 Гц), 142,73, 139,39, 133,06, 131,96, 127,78, 127,34, 127,01, 126,97, 126,48, 125,61, 125,20, 123,60, 123,30, 123,14, 117,04, 116,24, 116,09, 110,83, 99,07, 73,85, 71,38, 69,38, 69,05, 68,42, 68,21, 66,45, 49,81, 42,35, 39,52, 35,75, 34,41, 31,74, 31,29, 30,67, 29,72, 29,18, 29,13, 29,07, 29,01, 28,92, 28,68, 28,78, 28,72, 28,45, 25,30, 25,28, 22,00, 13,12.
Синтез соединений формулы (1)
Был синтезирован ряд гликосфинголипидов и протестирован на предмет активации NKT клеток. Структура соединений соответствовала формуле 1.
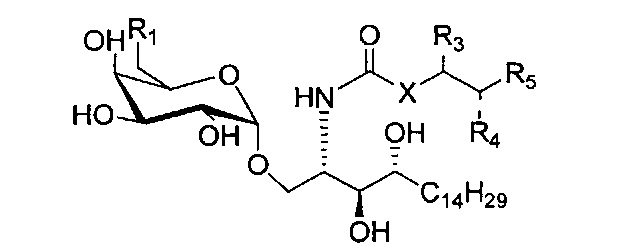
где для получения соответствующих соединений соединение №(R) выбрано из таблицы 1.
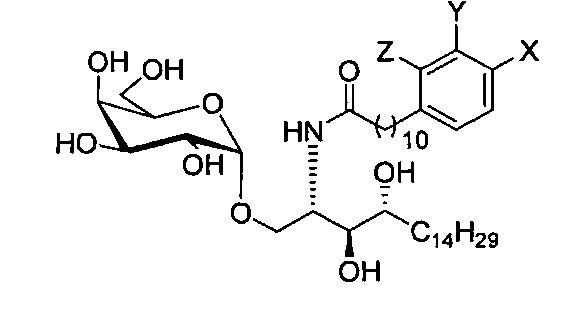
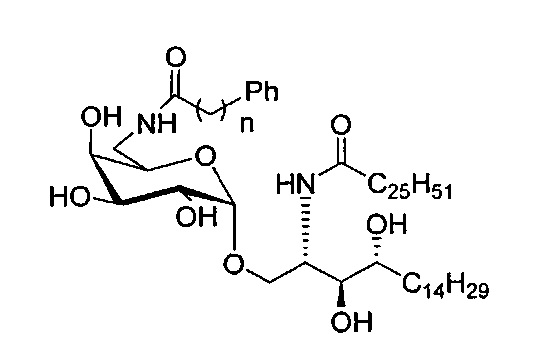
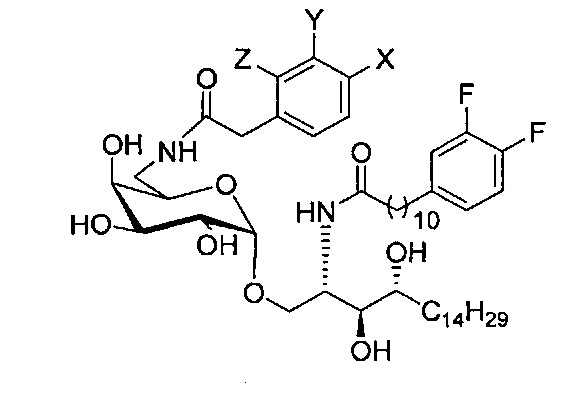
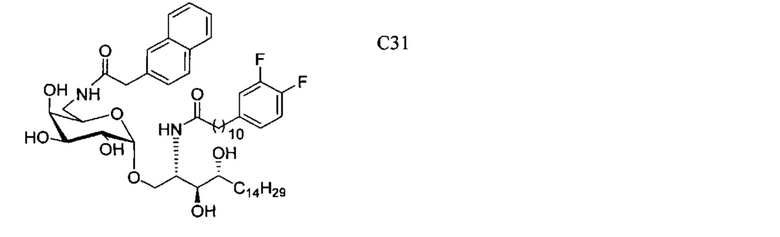
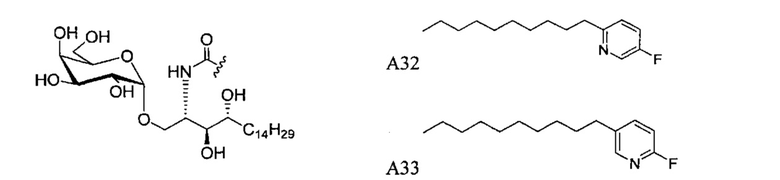
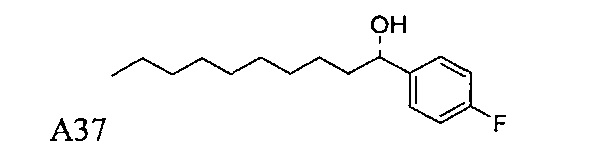
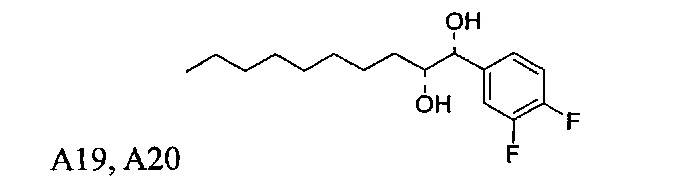
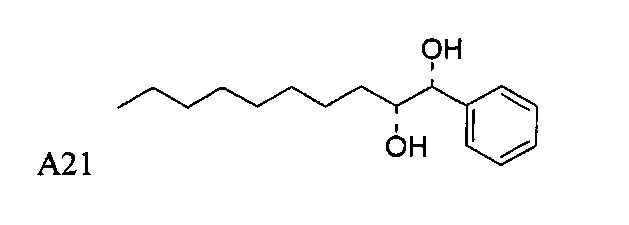
Пример 2
Активация антиген-презентирующих клеток (АРС)
Клетки A20CD1d и клетки mNK1,2 использовали в качестве АРС и эффекторных клеток, соответственно. Для определения жизнеспособности клеток и числа жизнеспособных клеток с помощью системы Guava EasyCyte Plus использовали реагент Guava ViaCount. Для обнаружения выработки IL-2 использовали исследовательскую систему Mouse IL-2 DuoSet ELISA. Клетки и гликолипиды ко-культивировали при 37°С, надосадочную жидкость собирали после 24 часов культивирования. После двух дней культивирования собирали клетки для определения жизнеспособности; результаты показывают, что эти гликосфинголипиды не являются токсичными. Как показано на фиг. 6, в этой связи все испытанные соединения проявляют свойство активировать АРС.
Секреция IFN-γ и Il-4 цитокина
Самку мыши C57BL/6 (16w4d) забили и для опыта взяли селезенку. Клетки и гликолипиды ко-культивировали при 37°С в течение 3 дней, и надосадочную жидкость собирали на 3-й день (~60 часов) культивирования. Затем добавили alarma Blue (5%/200 мкл), и клетки культивировали в течение 7 часов для определения пролиферации клеток. Для определения выработки цитокина использовали исследовательскую систему Mouse IL-4 и IFN-γ Duo Set ELISA. В этом исследовании негативным контролем был диметисульфоксид, и KRN-7000 был позитивным контролем. Как показано на фиг. 7-9, соединения обладали профилем секреции цитокина, смещенным в сторону Th1, что указывает на возможность их применения для обеспечения противоопухолевого, противовирусного/антибактериального действия и в качестве адъюванта.
Все публикации и патентные заявки, процитированные в этом описании, включены в настоящую заявку посредством ссылки, как если бы каждая отдельная публикация или патентная заявка были конкретно и индивидуально указаны, чтобы быть включенными посредством ссылки.
Хотя представленное выше изобретение описано в некоторых деталях в качестве иллюстрации и с примерами для ясности понимания, рядовому специалисту данной области техники не составит труда понять, в свете принципов настоящего изобретения, что могут быть осуществлены определенные его изменения и модификации, без отступления от сущности или объема притязаний приложенной формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ СПОСОБ СИНТЕЗА АГОМЕЛАТИНА | 2014 |
|
RU2683279C1 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| ПРОИЗВОДНОЕ НА ОСНОВЕ ДИГИДРОПИРИМИДО-КОЛЬЦА В КАЧЕСТВЕ ИНГИБИТОРА HBV | 2015 |
|
RU2693897C2 |
| СОЕДИНЕНИЯ ДИГИДРОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В ФАРМАЦЕВТИЧЕСКИХ ПРЕПАРАТАХ | 2013 |
|
RU2655914C9 |
| Новые производные имидазо[4,5-с]хинолина в качестве ингибиторов LRRK2 | 2018 |
|
RU2773516C2 |
| ГИДРОКСАМОВЫЕ КИСЛОТЫ, СОДЕРЖАЩИЕ ЯДРА 1,2,4- И 1,3,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ПОТЕНЦИАТОРОВ ДЕЙСТВИЯ АНТИБИОТИКОВ | 2023 |
|
RU2830361C1 |
| ТРИАЗОЛЫ ДЛЯ РЕГУЛЯЦИИ ГОМЕОСТАЗА ВНУТРИКЛЕТОЧНОГО КАЛЬЦИЯ | 2017 |
|
RU2753509C2 |
| НОВЫЕ N-МОДИФИЦИРОВАННЫЕ 5-МЕТИЛ-2'-ДЕЗОКСИЦИТИДИНЫ, ПРОЯВЛЯЮЩИЕ АНТИМИКОЗНУЮ АКТИВНОСТЬ | 2021 |
|
RU2766333C1 |
| ПРОИЗВОДНОЕ ЦИКЛОПЕНТИЛАКРИЛАМИДА | 2009 |
|
RU2565070C2 |
| 2,4,5-ТРИ(МЕТОКСИФЕНИЛ) ЦИС-ИМИДАЗОЛИН И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2019 |
|
RU2730497C1 |
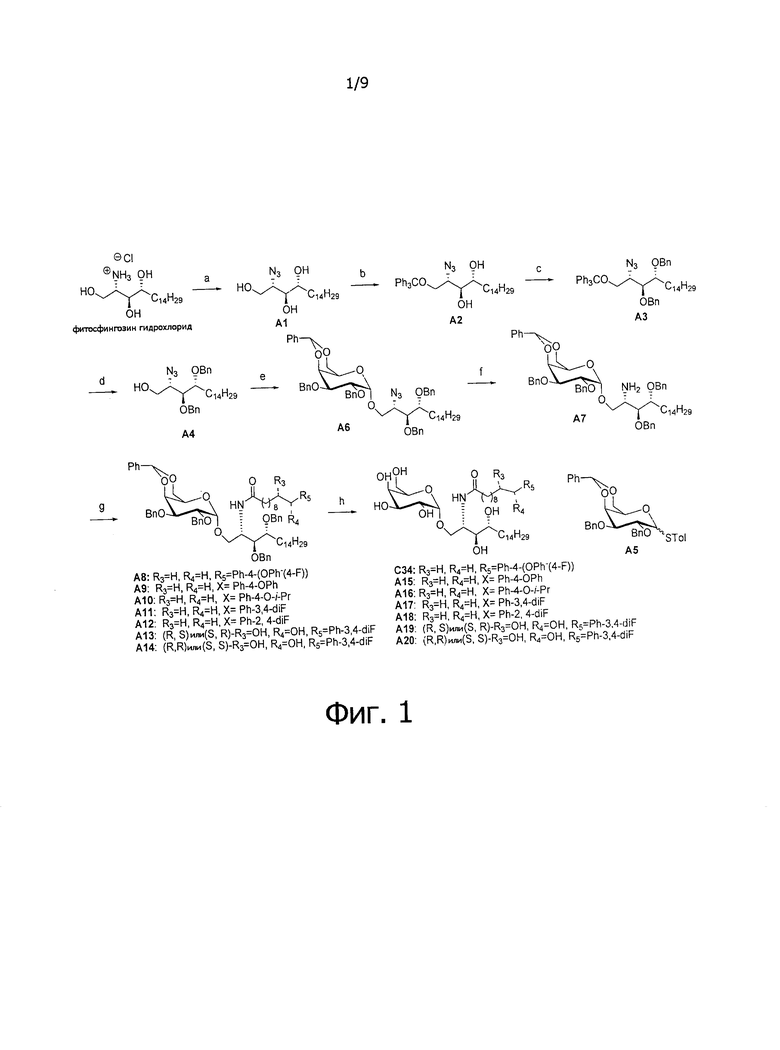
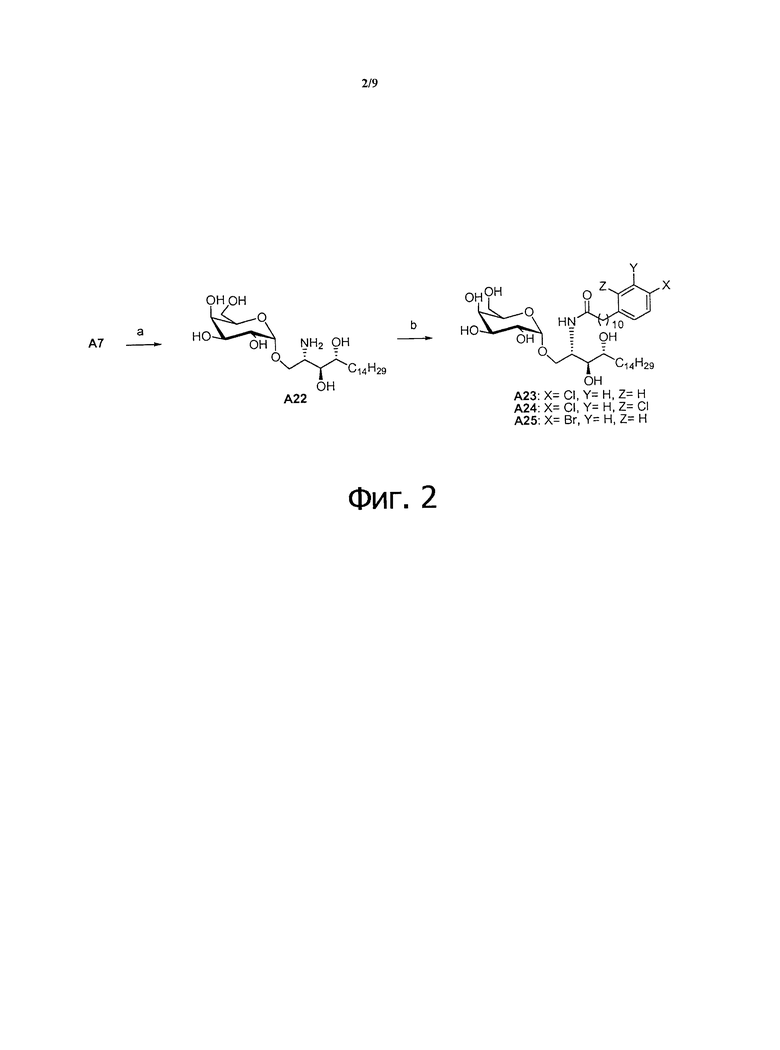
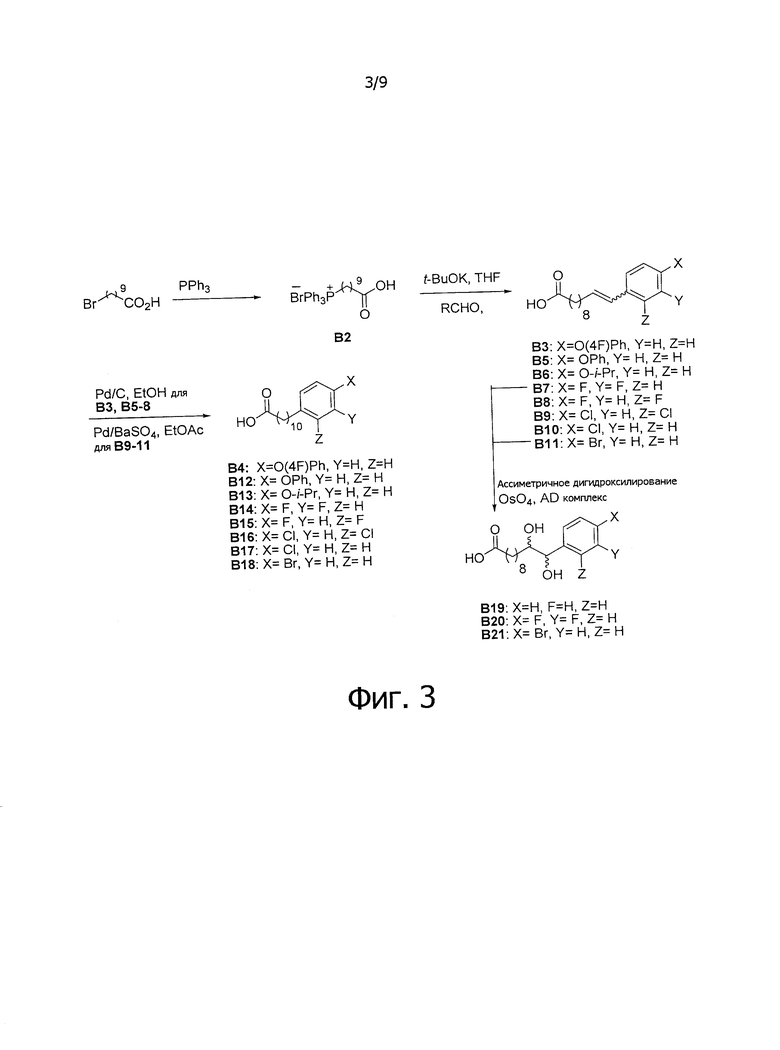
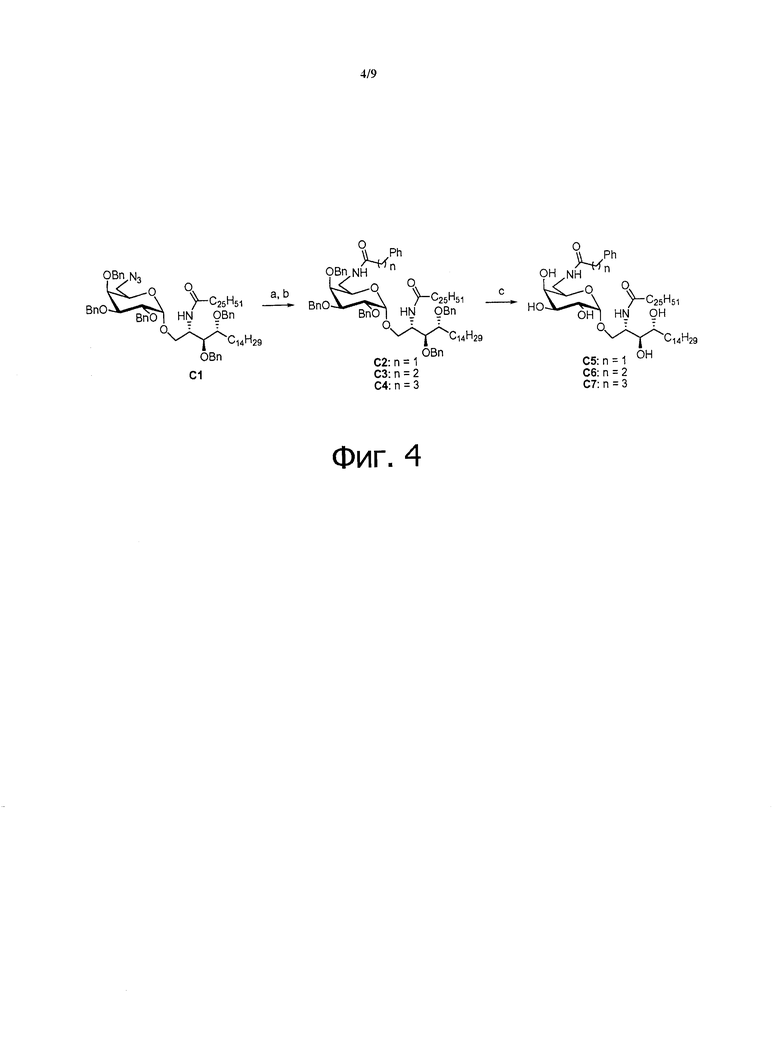
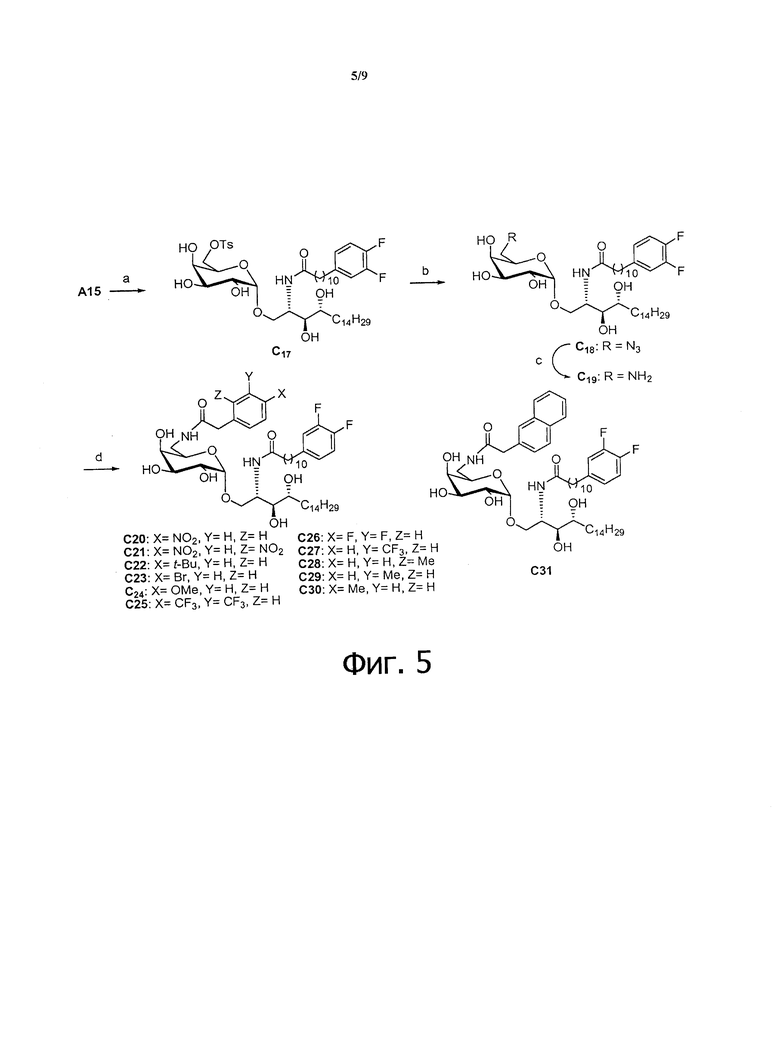
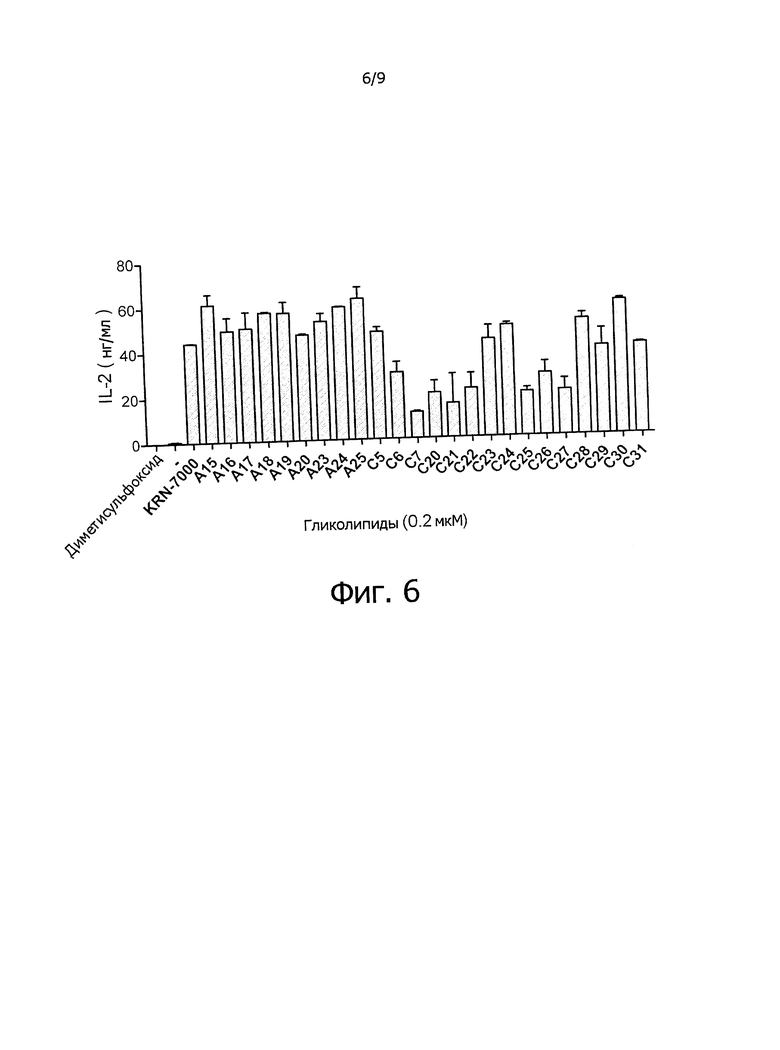
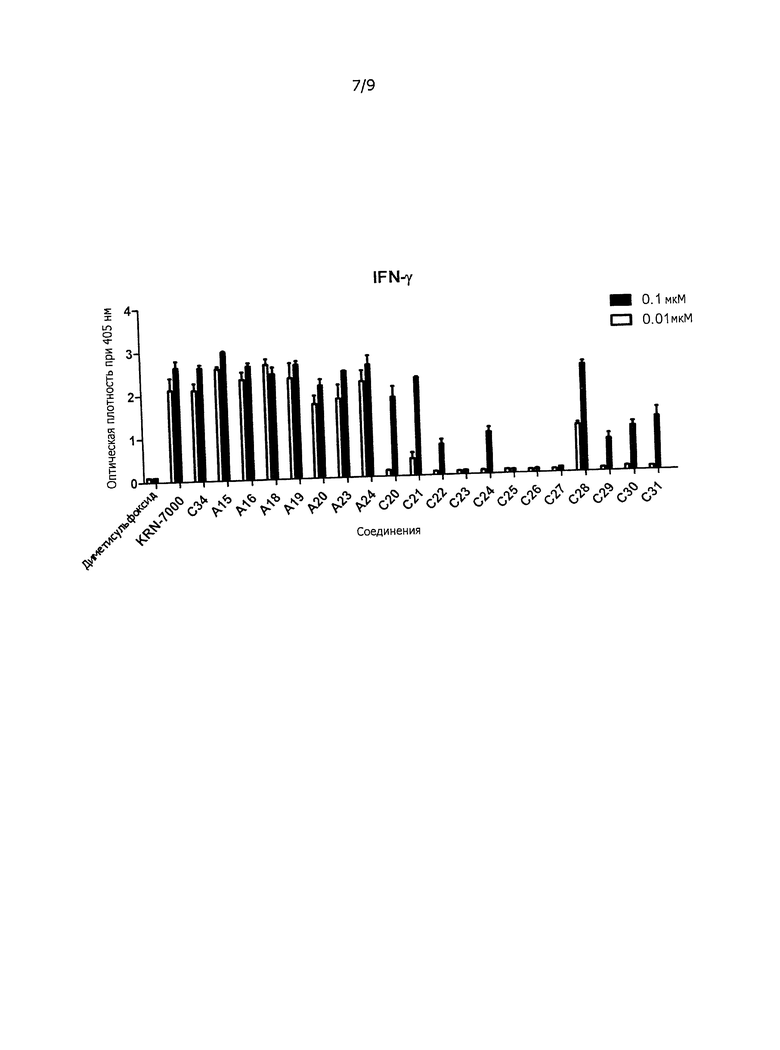
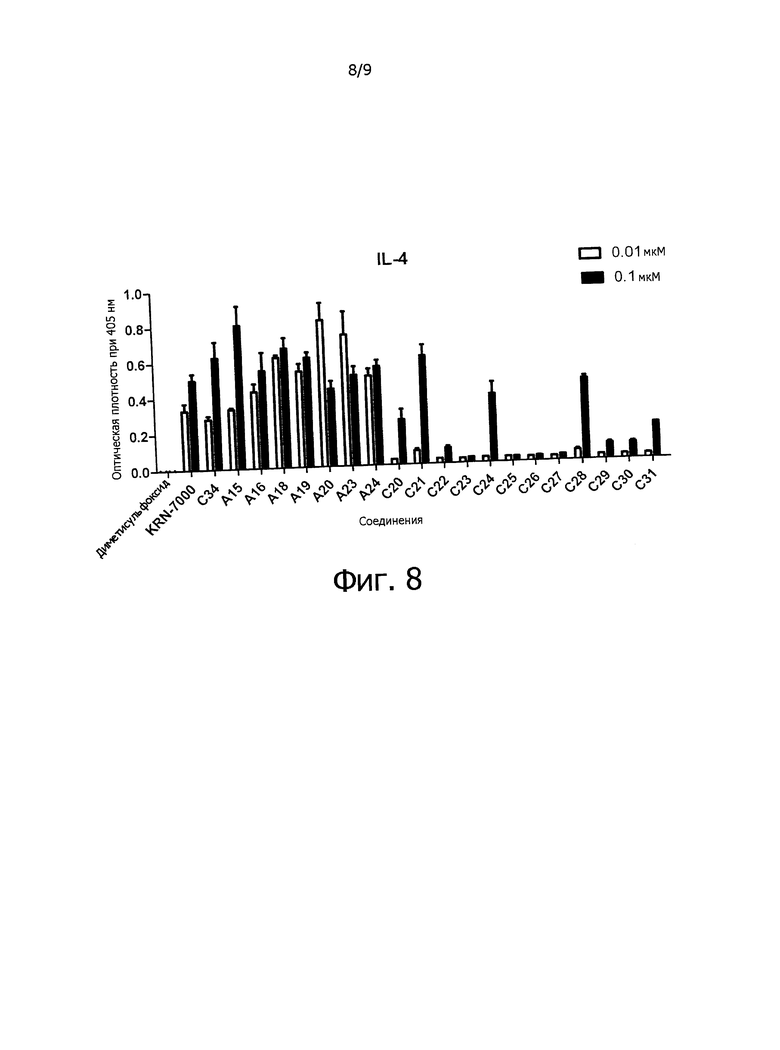
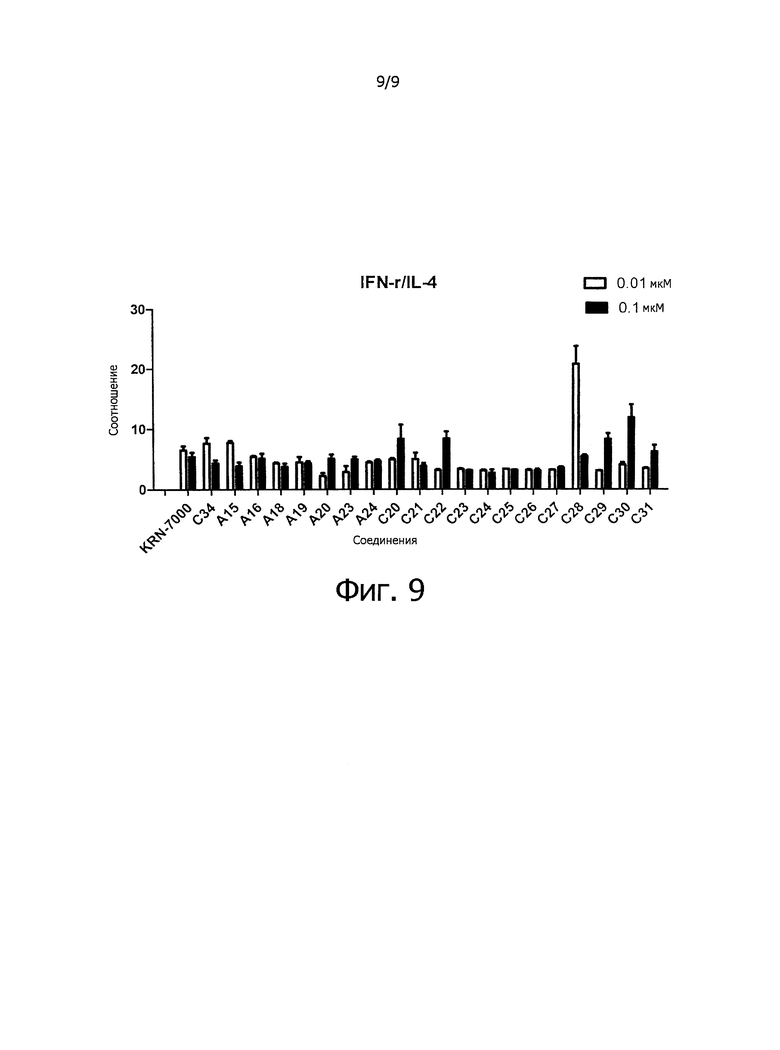
Изобретение относится к способам получения альфа-гликосфинголипидов и новым альфа-гликосфинголипидам, фармацевтическим композициям на их основе, применимым в фармацевтической промышленности. Способ получения соединения формулы (1)
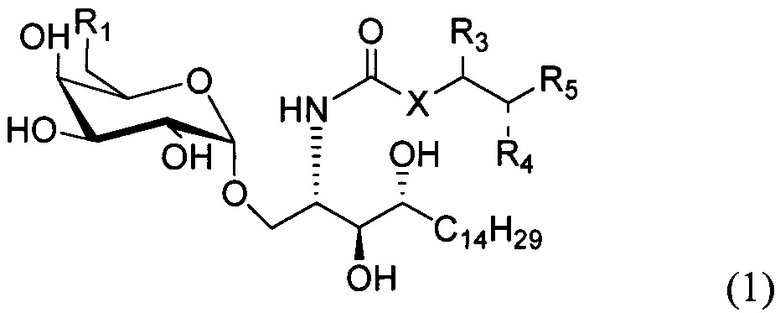
где R1 представляет собой ОН, X представляет собой С8 алкилен, R3 представляет собой ОН или Н, R4 представляет собой ОН или Н, R5 представляет собой арил, замещенный арил, гетероарил или замещенный гетероарил, при условии, что когда R3 представляет собой Н и R4 представляет собой Н, то R5 представляет собой фенил, замещенный одной или двумя группами, независимо выбранными из -F, -Cl, -Br, -ОСН3, -ОСН(СН3)2, -OPh, -OPh(4-F), 2-(5-F)-пиридинил, -CF3, -NO2, -N(CH3)2 и 3-(6-F)-O-пиридинил; или R5 представляет собой пиридинил, замещенный группой, независимо выбранной из -F- или -OPh(4-F); когда R3 представляет собой Н и R4 представляет собой ОН, то R5 представляет собой фенил, замещенный группой, независимо выбранной из -F- или -ОСН3, или R5 представляет собой фенил, замещенный двумя -F; когда R3 представляет собой ОН и R4 представляет собой ОН, то R5 представляет собой фенил, замещенный одной или двумя группами, независимо выбранными из -F или -ОСН3; включает:
(а) реакцию соединения формулы (6) с соединением формулы (7) в присутствии кислоты Льюиса с образованием соединения формулы (5):
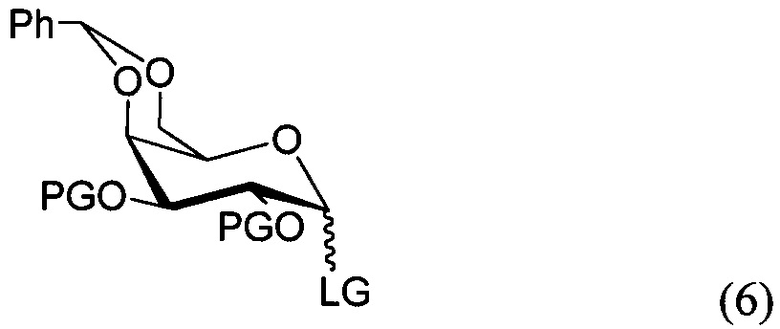
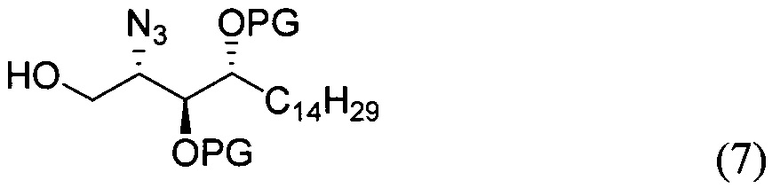
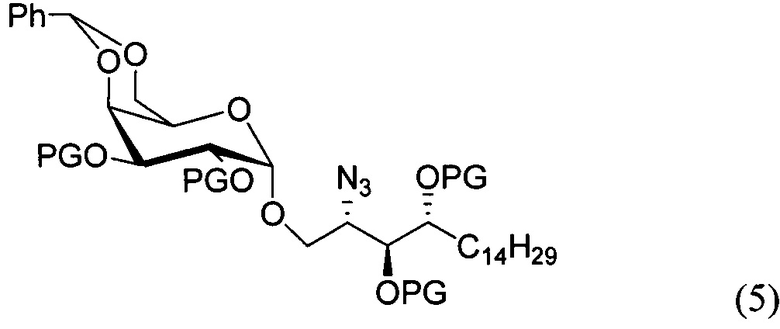 ,
,
где PG является защитной группой гидроксила и LG является тиолсодержащей уходящей группой формулы:
 ,
, 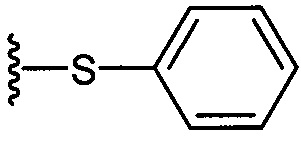 ,
,  или
или  ;
;
(b) восстановление соединения формулы (5) с получением соединения формулы (3):
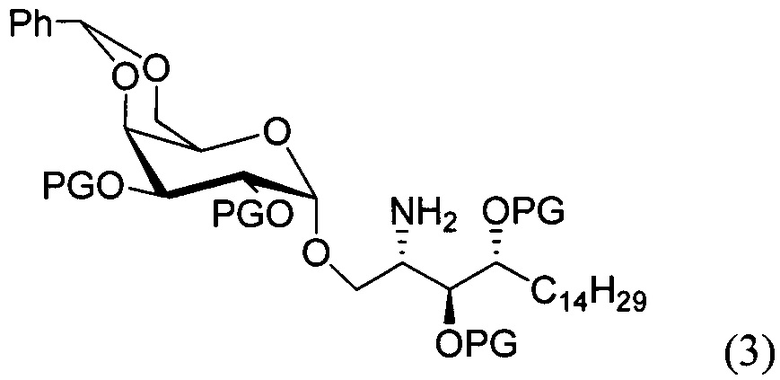 ;
;
(c) сочетание соединения формулы (3) с соединением формулы (4):
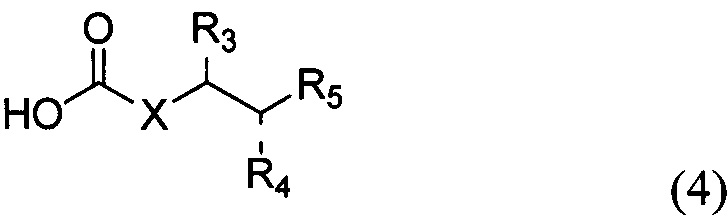 ,
,
где X означает C8 алкилен или C8 алкенилен, с получением соединения формулы (2):
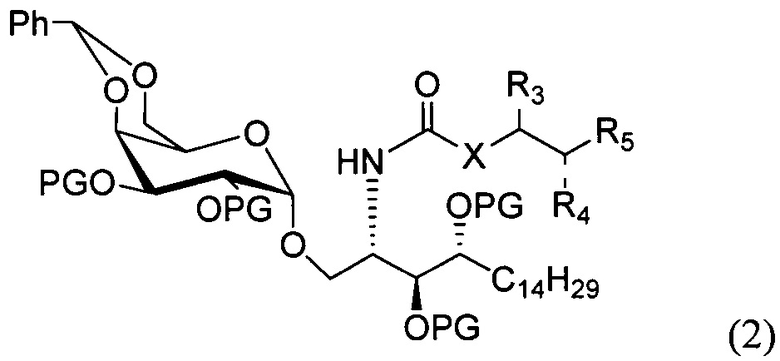
и удаление защитной группы с соединения формулы (2):
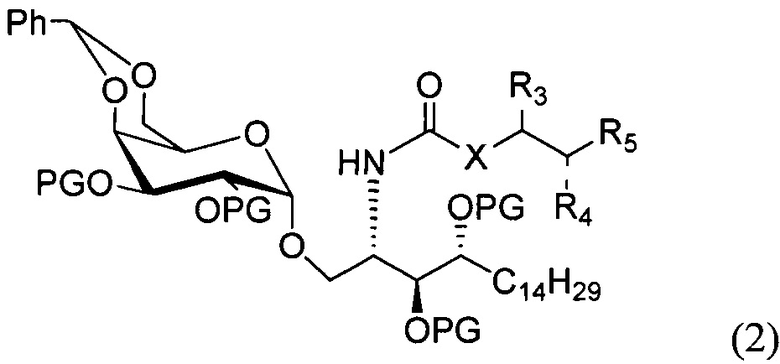 ,
,
где PG является защитной группой гидроксила, водородом в условиях гидрогенизационного катализа с получением соединения формулы (1). Предложены новые соединения, эффективные в качестве лигандов для естественных киллерных Т-клеток, новые эффективные способы их получения и новые антивирусные, антибактериальные, противоопухолевые средства на их основе. 7 н. и 16 з.п. ф-лы, 39 пр., 1 табл., 9 ил.
1. Способ получения хирального соединения, включающего R-форму или S-форму гликосфинголипида формулы (1)
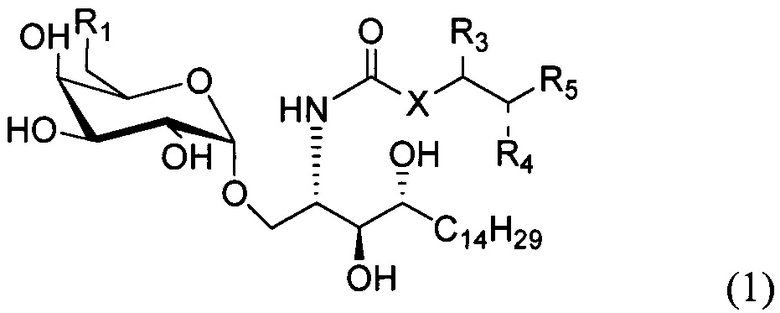
где:
R1 представляет собой ОН,
X представляет собой С8 алкилен,
R3 представляет собой ОН или Н,
R4 представляет собой ОН или Н,
R5 представляет собой арил, замещенный арил, гетероарил или замещенный гетероарил,
при том условии, что:
когда R3 представляет собой Н и R4 представляет собой Н, то R5 представляет собой фенил, замещенный одной или двумя группами, независимо выбранными из -F, -Cl, -Br, -ОСН3, -ОСН(СН3)2, -OPh, -OPh(4-F), 2-(5-F)-пиридинил, -CF3, -NO2, -N(CH3)2 и 3-(6-F)-O-пиридинил; или R5 представляет собой пиридинил, замещенный группой, независимо выбранной из -F- или -OPh(4-F);
когда R3 представляет собой Н и R4 представляет собой ОН, то R5 представляет собой фенил, замещенный группой, независимо выбранной из -F- или -ОСН3, или R5 представляет собой фенил, замещенный двумя -F;
когда R3 представляет собой ОН и R4 представляет собой ОН, то R5 представляет собой фенил, замещенный одной или двумя группами, независимо выбранными из -F или -ОСН3;
и способ включает:
(а) реакцию соединения формулы (6):
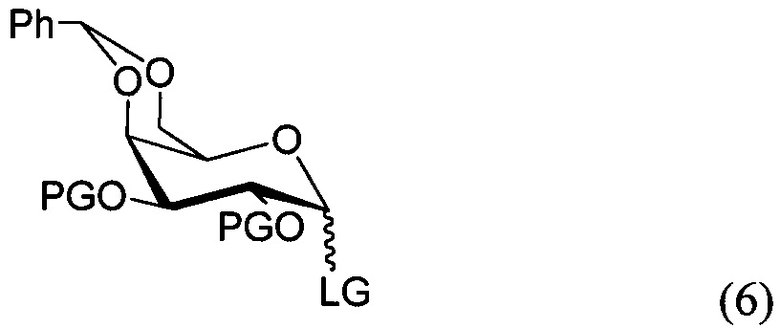
с соединением формулы (7):
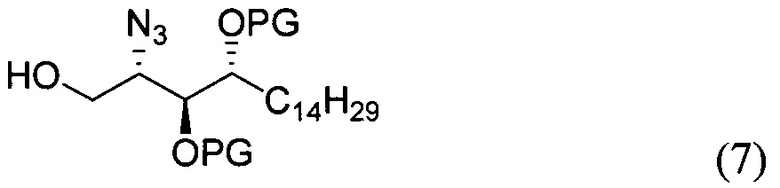
в присутствии кислоты Льюиса с образованием соединения формулы (5):
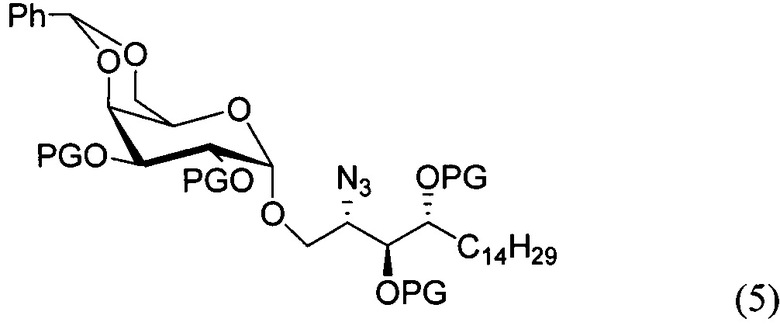 ,
,
где PG является защитной группой гидроксила и LG является тиолсодержащей уходящей группой формулы:
 ,
, 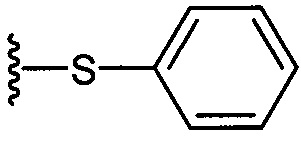 ,
, 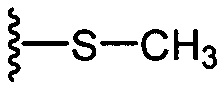 или
или 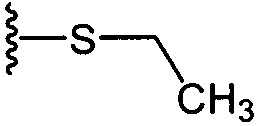 ;
;
(b) восстановление соединения формулы (5) с получением соединения формулы (3):
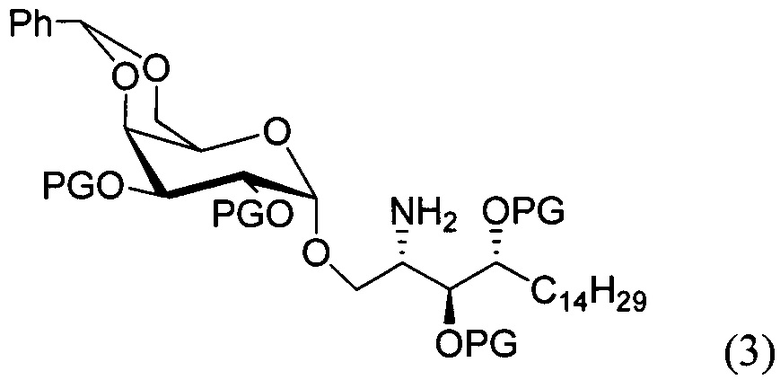
(c) сочетание соединения формулы (3) с соединением формулы (4):
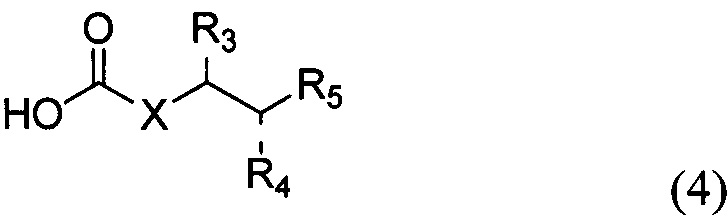 ,
,
где X означает C8 алкилен или C8 алкенилен, с получением соединения формулы (2):
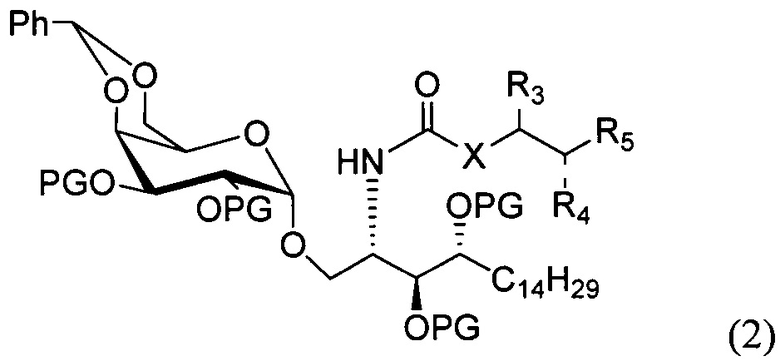
и
удаление защитной группы с соединения формулы (2):
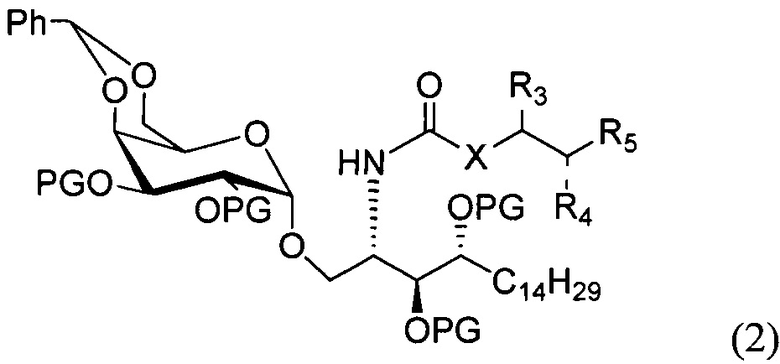 ,
,
где PG является защитной группой гидроксила,
водородом в условиях гидрогенизационного катализа с получением соединения формулы (1).
2. Способ по п. 1, где катализатор гидрогенизации выбирают из Pd/C, Pd(OH)2 или ренеевского никелевого катализатора.
3. Способ по п. 1, где стадию восстановления (b) осуществляют с применением алюмогидрида лития, борогидрида натрия, боранового комплекса, фосфинового комплекса, ферментного восстановления, гидрогенизации или трансферной гидрогенизации.
4. Способ по п. 1, где кислота Льюиса представляет собой TMSOTf, Tf2O, BF3⋅OEt2, TfOH, или Me2S2-Tf2O.
5. Способ по п. 1, где молекулярные сита используют на реакционной стадии (а).
6. Способ получения хирального соединения, включающего R-форму или S-форму гликосфинголипида формулы (1)
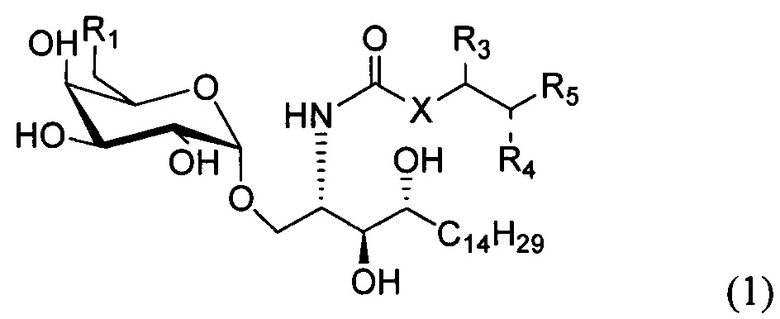
где:
R1 представляет собой NH2 или NHCOR2,
R2 представляет собой Н или C1-3алкил, C1-3алкенил или C1-3алкил с концевой арильной группой, выбранной из нафтильной группы или фенильной группы, необязательно замещенной одной или двумя группами, независимо выбранными из -F, -Br, -ОСН3, -СН3, -CF3, -С(СН3)3 и -NO2,
X представляет собой C8 алкилен или C8 алкенилен;
R3 представляет собой Н;
R4 представляет собой Н;
R5 представляет собой фенил, замещенный двумя -F,
и способ включает:
(a) реакцию соединения формулы (16):
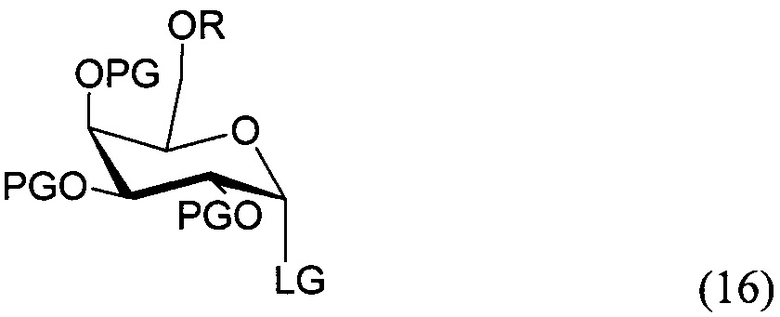
с соединением формулы (7):
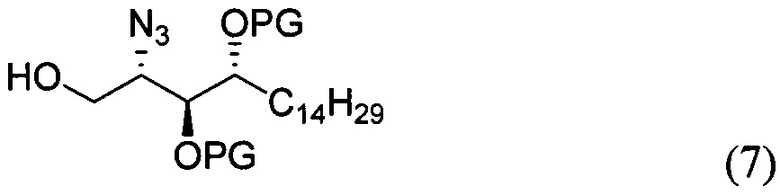
в присутствии кислоты Льюиса с образованием соединения формулы (15):
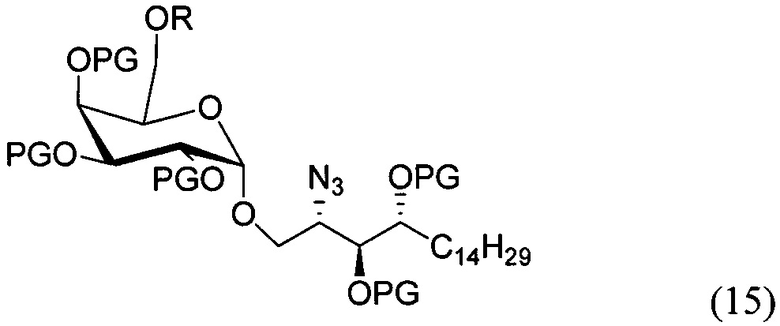 ,
,
где каждый PG независимо является защитной группой гидроксила, a R является сложноэфирной защитной группой гидроксила и LG является тиолсодержащей уходящей группой формулы:
 ,
, 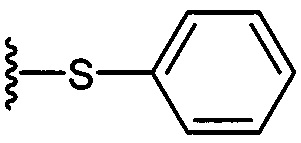 ,
,  или
или  ;
;
(b) восстановление соединения формулы (15) с получением соединения формулы (14):
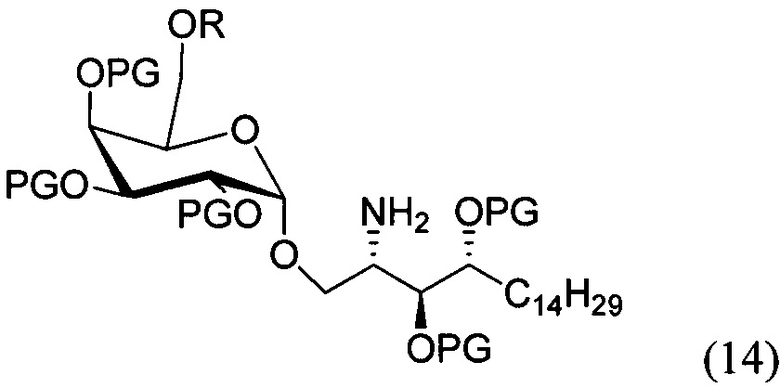 ;
;
(c) сочетание соединения формулы (14) с соединением формулы (4):
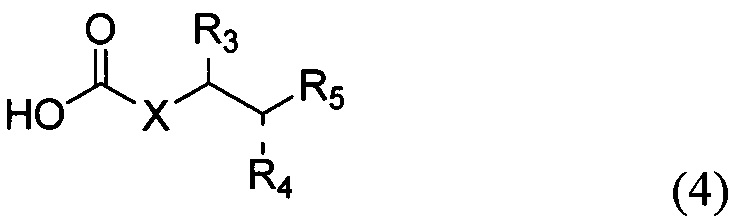 ,
,
где X означает C8 алкилен или C8 алкенилен, с получением соединения формулы (13):
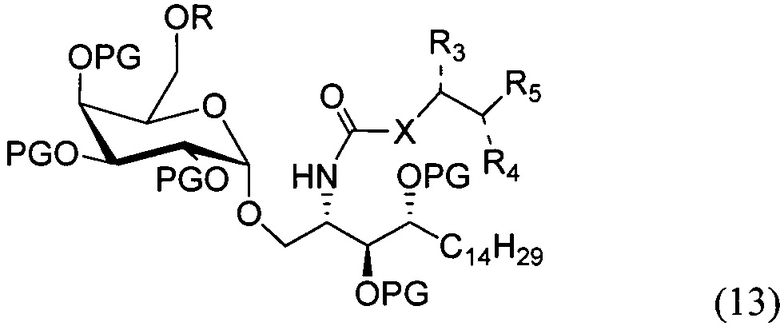 ;
;
(d) гидролиз соединения формулы (13) с получением соединения формулы (12):
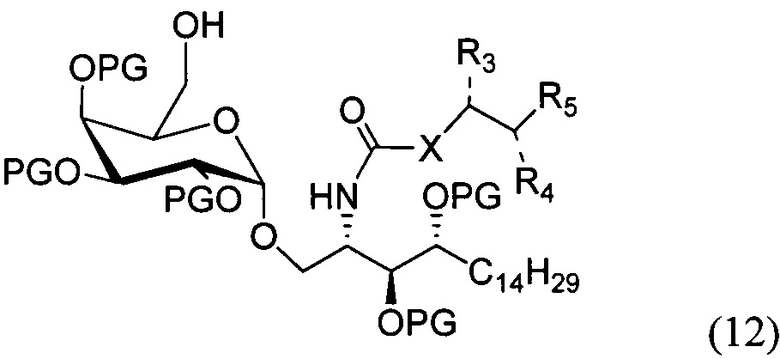 ;
;
(e) замещение соединения формулы (12) с получением соединения формулы (11): 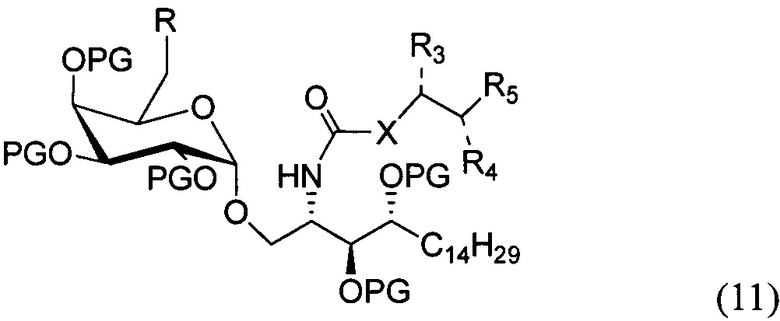 ,
,
где R является уходящей группой;
(f) проведение реакции соединения формулы (11) с азидом натрия с получением соединения формулы (10):
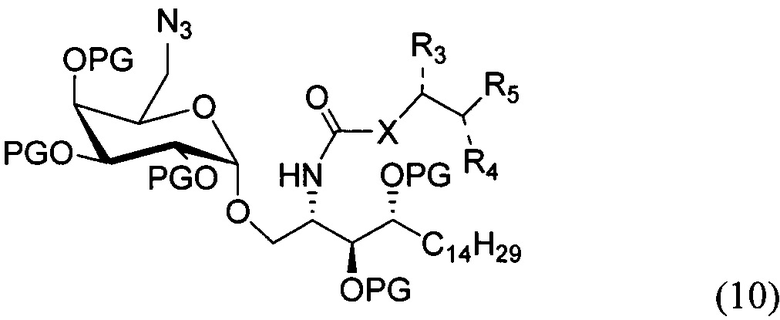 ;
;
(g) восстановление соединения формулы (10) с получением соединения формулы (9):
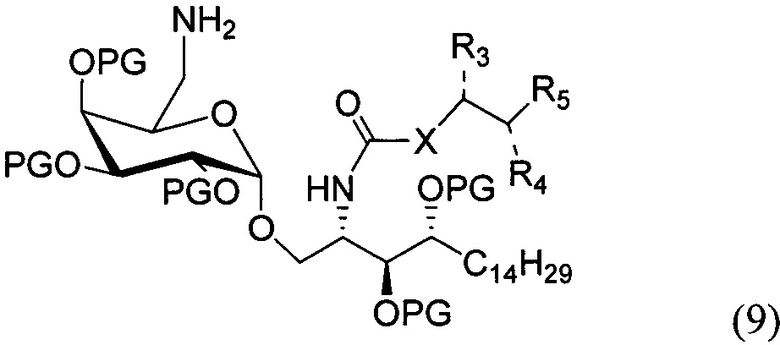 ;
;
(h) необязательно, сочетание соединения формулы (9) с кислотой формулы HOCOR2 с получением соединения формулы (8):
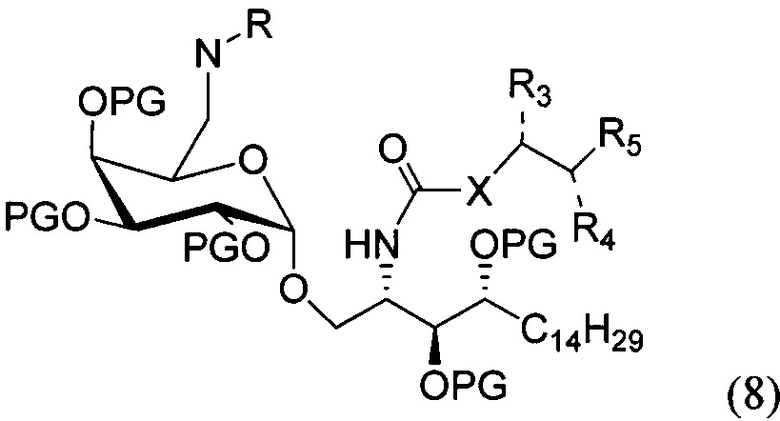 ,
,
где R означает -COR2; и
(i) удаление защитных групп с соединения, полученного на стадии (g) или (h), водородом в условиях гидрогенизационного катализа с получением соединения формулы (1).
7. Способ по п. 1, характеризующийся тем, что соединение формулы 1 является 1-O-(α-D-галактопиранозил)-2-(11-(4-(4-фторфенокси)фенил)ундеканоил) амино-D-рибо-октадекан-1,3,4-триолом (С34), имеющим формулу:
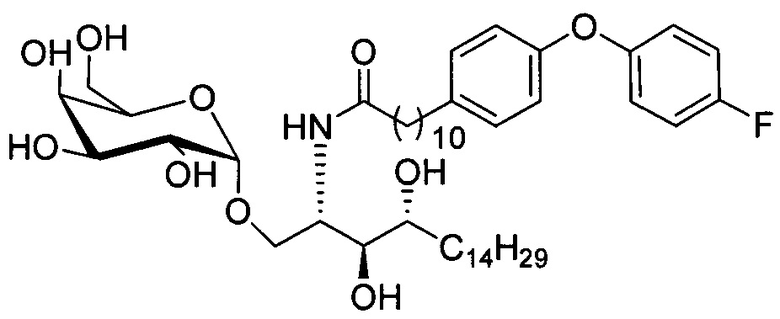
8. Способ по п. 1, где соединение формулы (3) представляет собой:
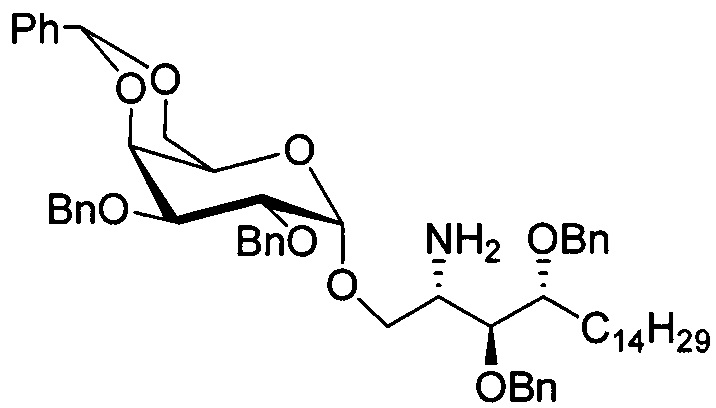 .
.
9. Способ по п. 1, где соединение формулы (5) представляет собой:
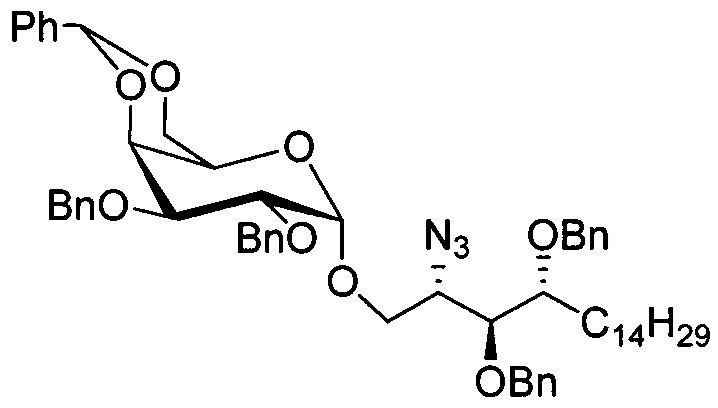 .
.
10. Способ по п. 1, где соединение формулы (4) представляет собой соединение, выбранное из группы, включающей:
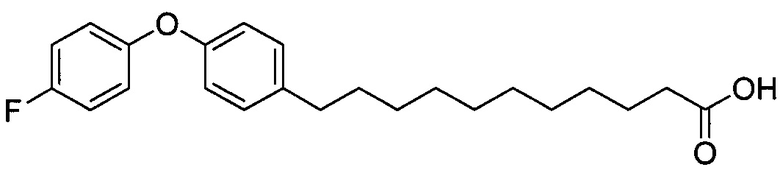 ;
;
 ;
;
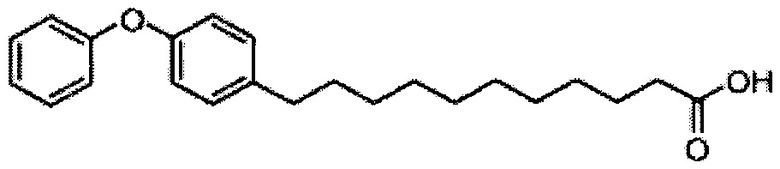 ;
;
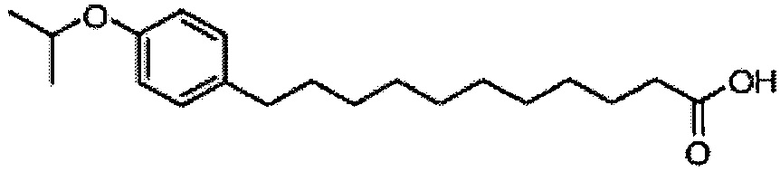 ;
;
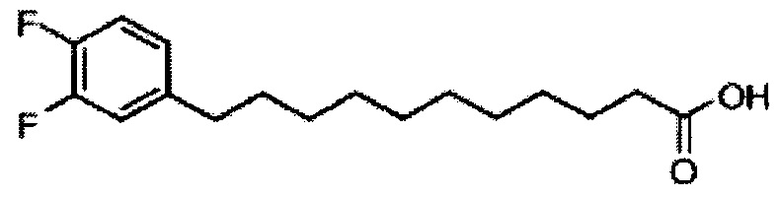 ;
;
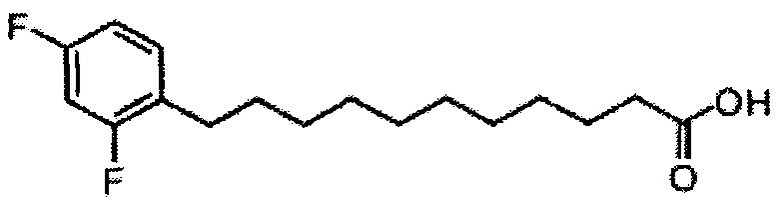 ;
;
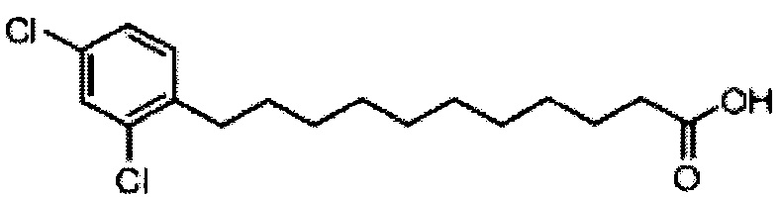 ;
;
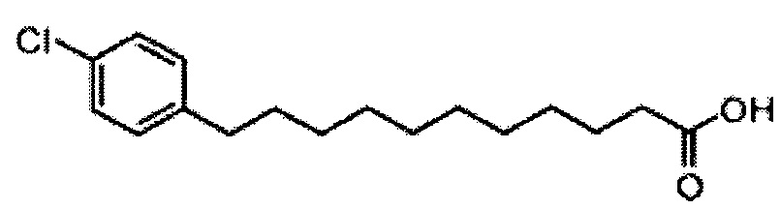 ;
;
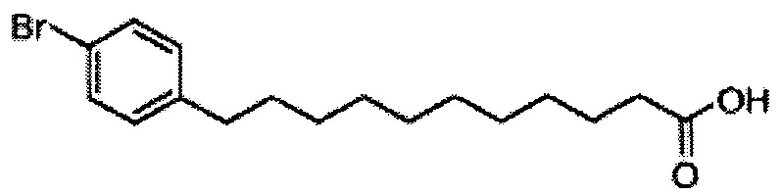 ;
;
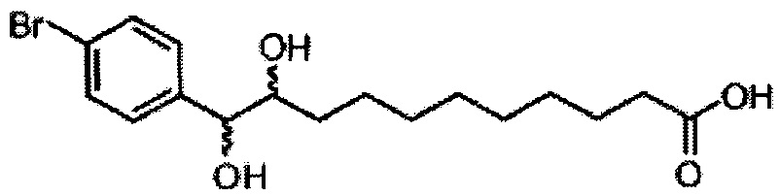 ;
;
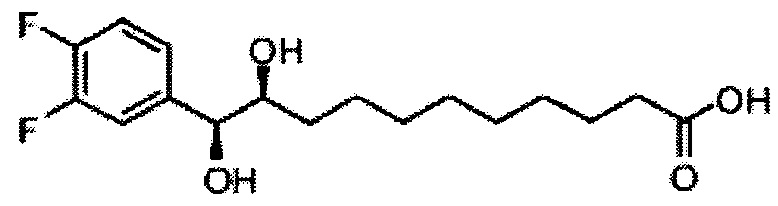 ;
;
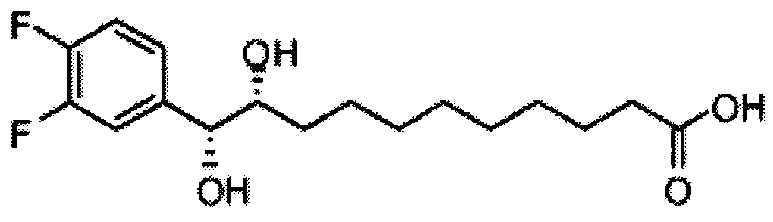 ;
;
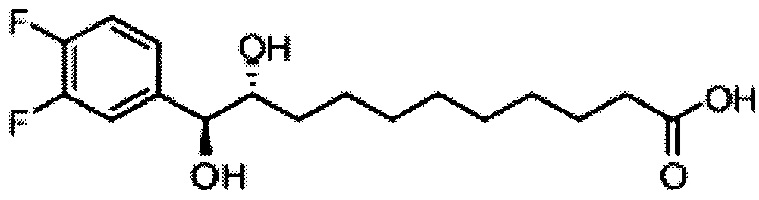 ;
;
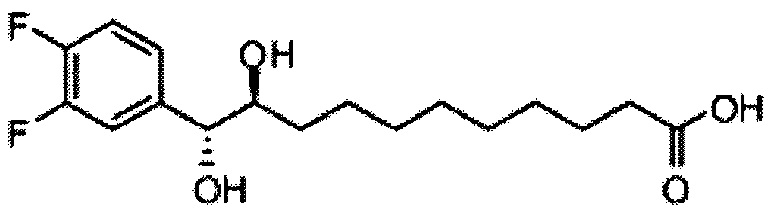 ;
;
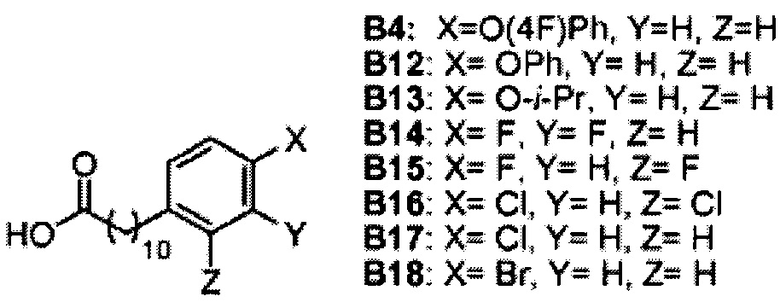 ;
;
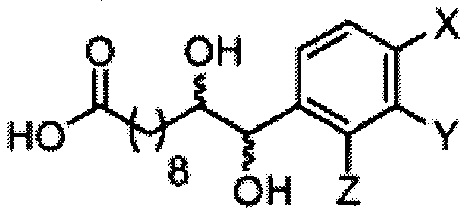
и
В19: Х=Н, Y=H, Z=H
B21:X=F,Y=F,Z=H
B20: X=Br, Y=H, Z=H.
11. Способ по п. 1, где соединение формулы (1) представляет собой соединение, выбранное из группы, включающей:
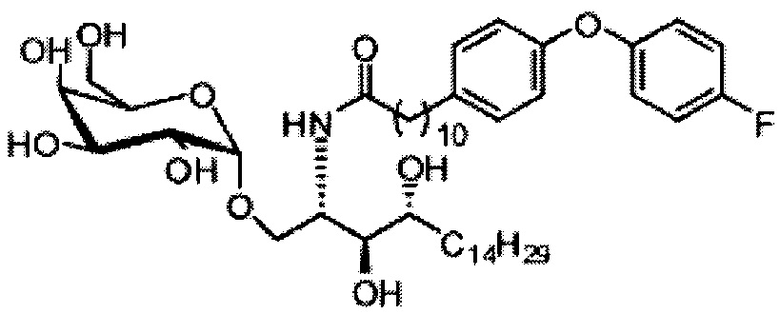 ;
;
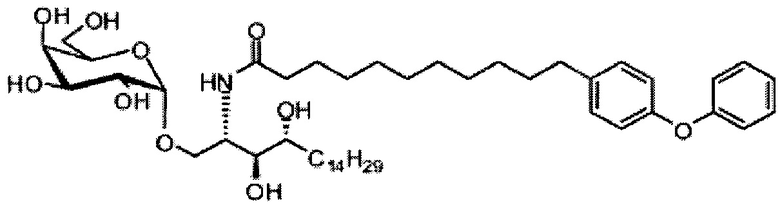 ;
;
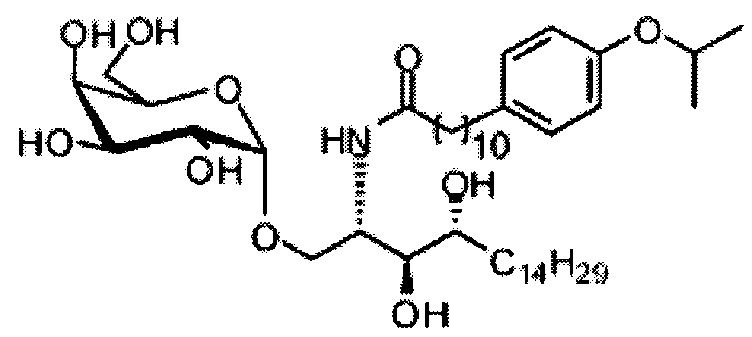 ;
;
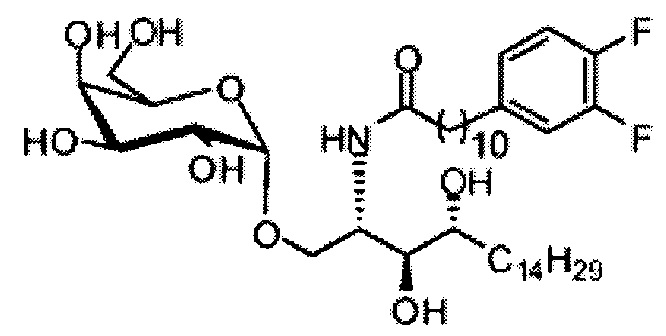 ;
;
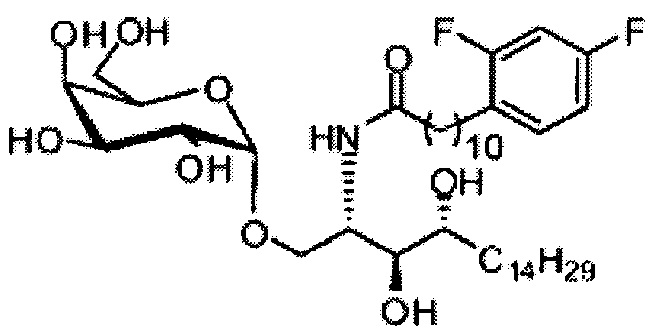 ;
;
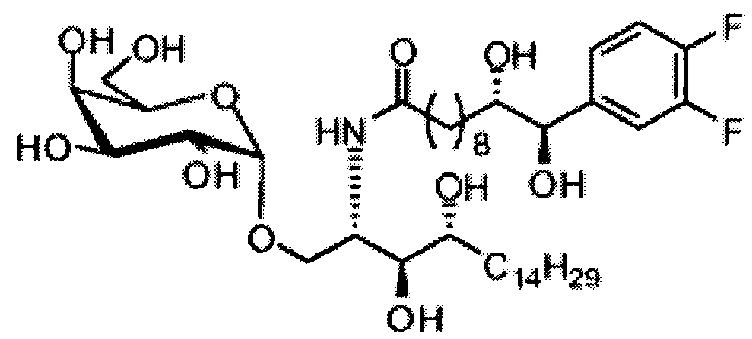 ;
;
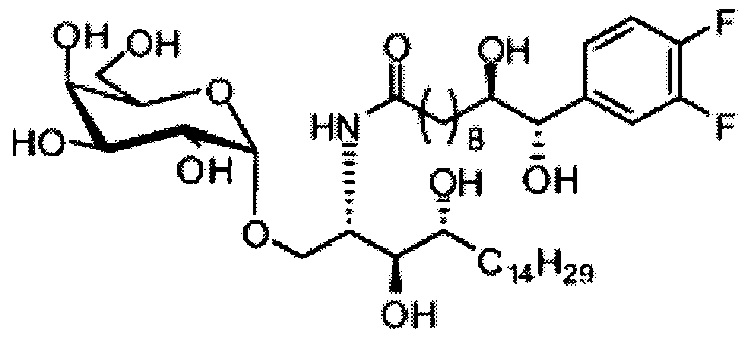 ;
;
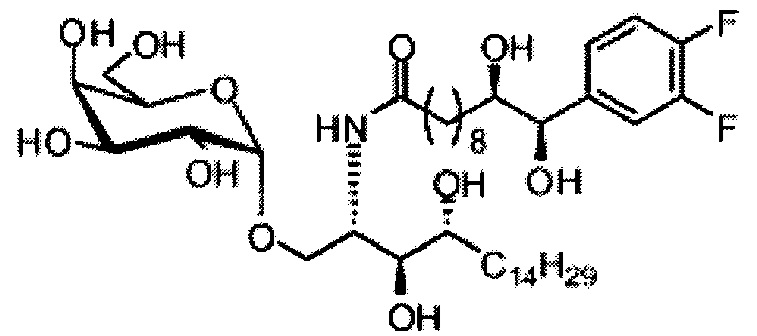 ;
;
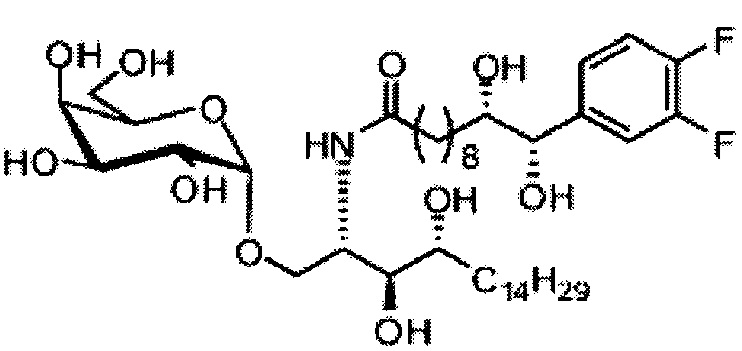 ;
;
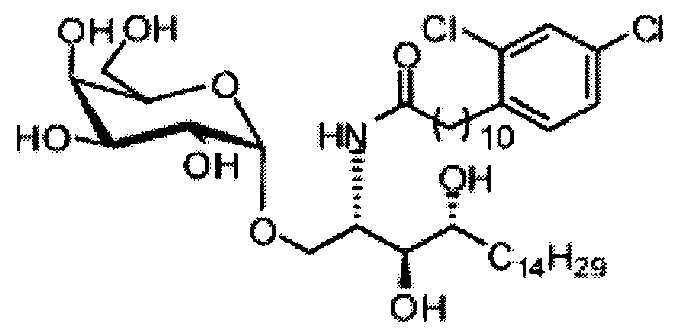 ;
;
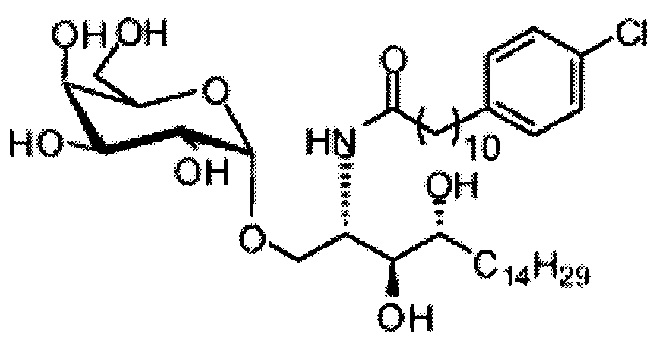 ;
;
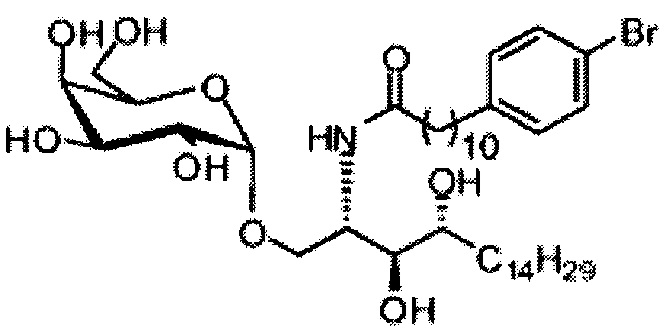 ;
;
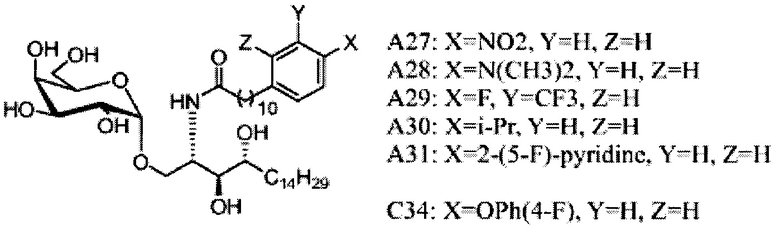 ;
;
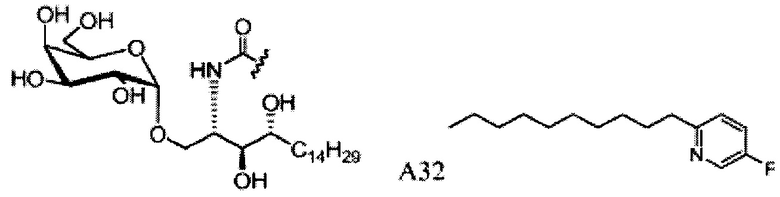 ;
;
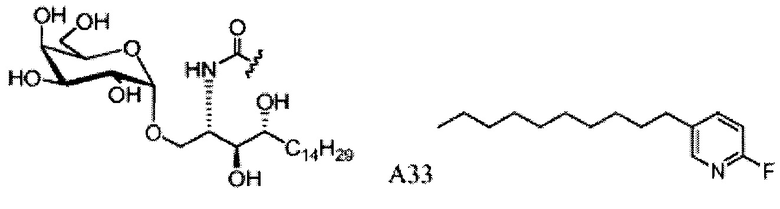 ;
;
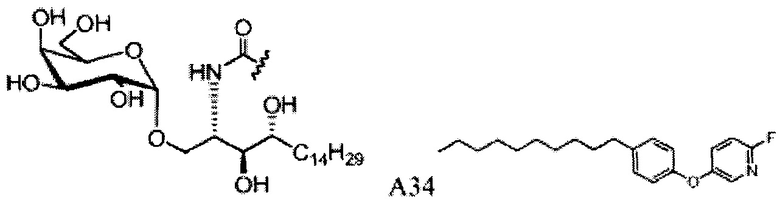 ;
;
 ;
;
 ;
;
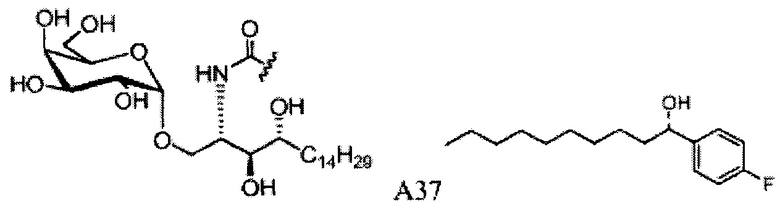 ;
;
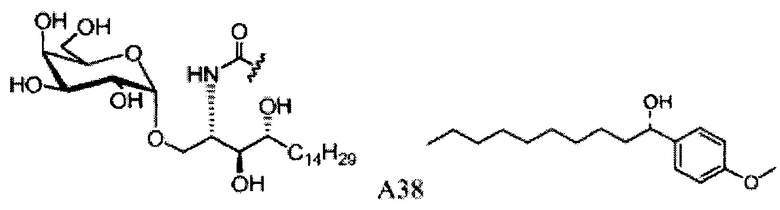 ;
;
 ;
;
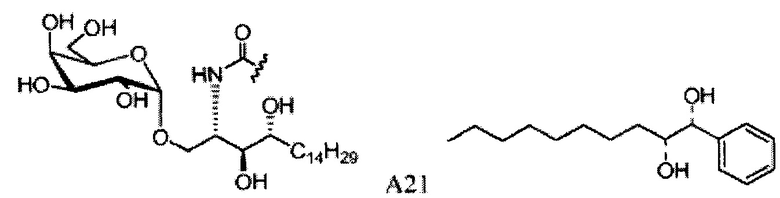 ;
;
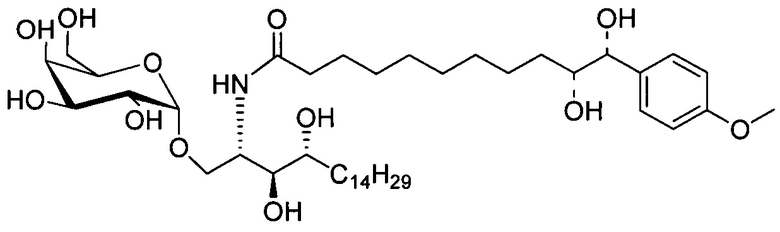 ; и
; и
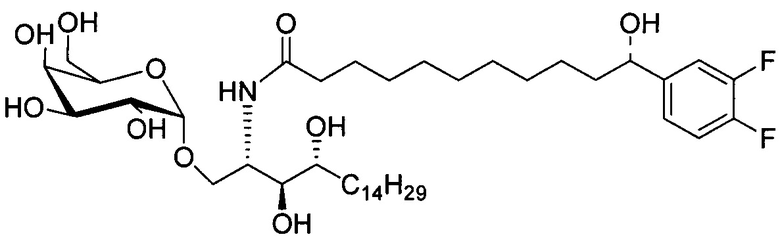 .
.
12. Способ по п. 6, где соединение формулы (1) представляет собой соединение, выбранное из группы, включающей:
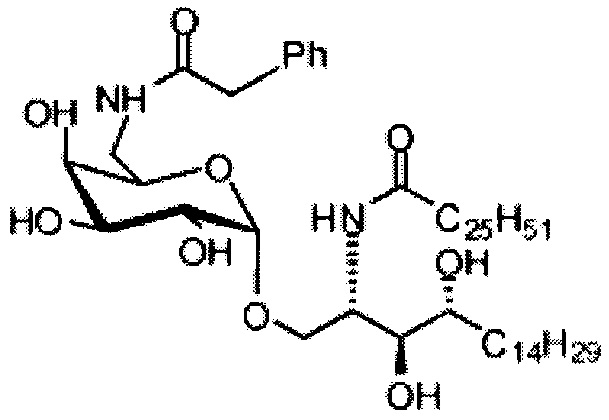 ;
;
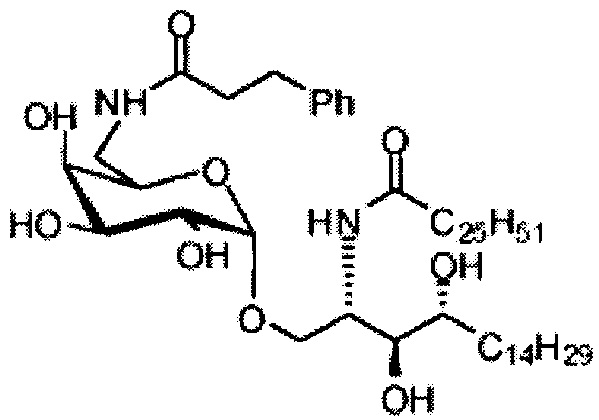 ;
;
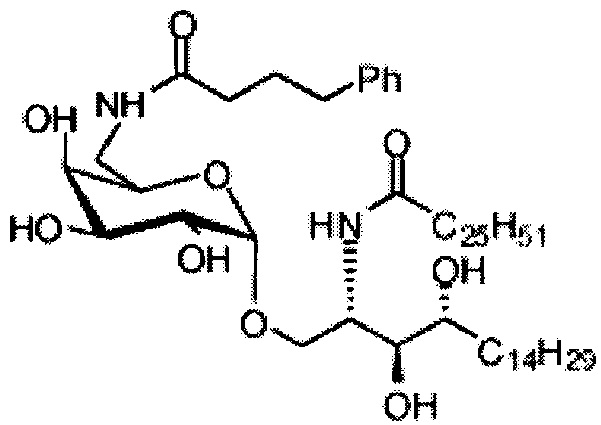 ;
;
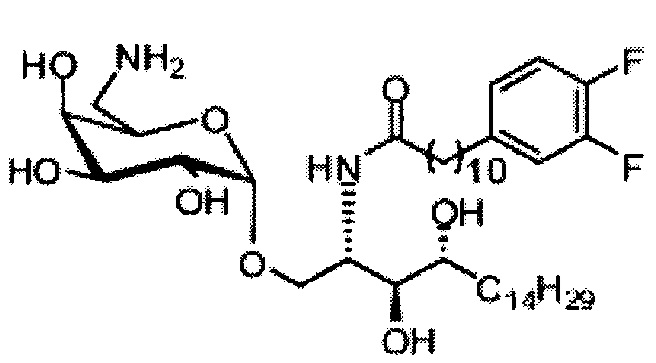 ;
;
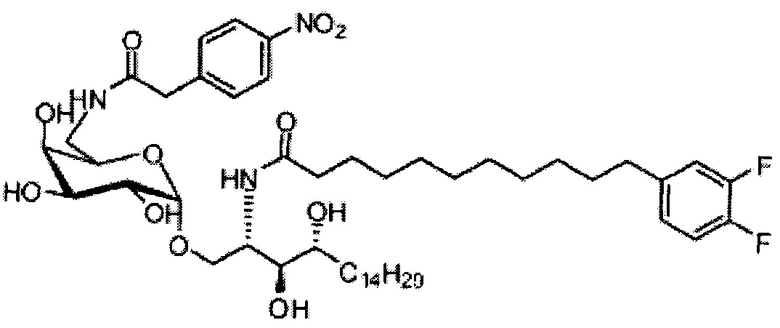 ;
;
 ;
;
 ;
;
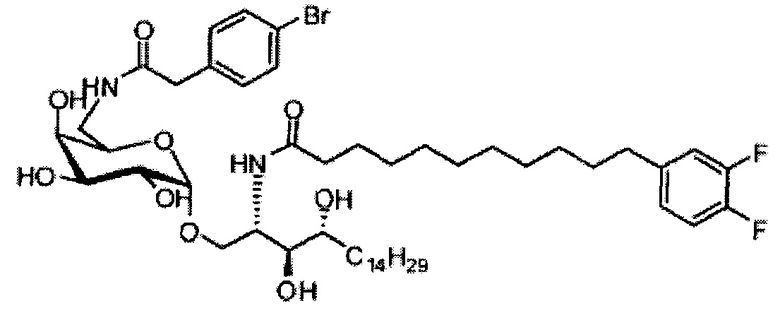 ;
;
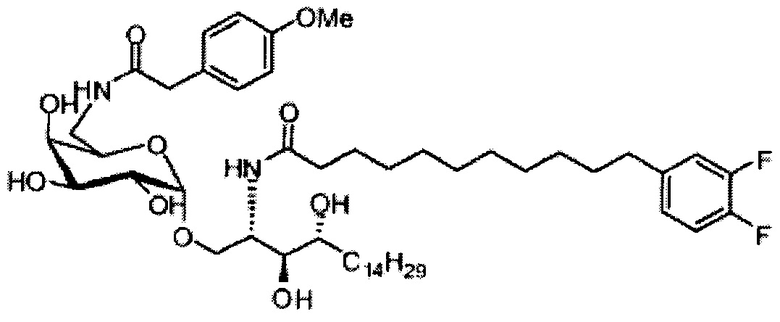 ;
;
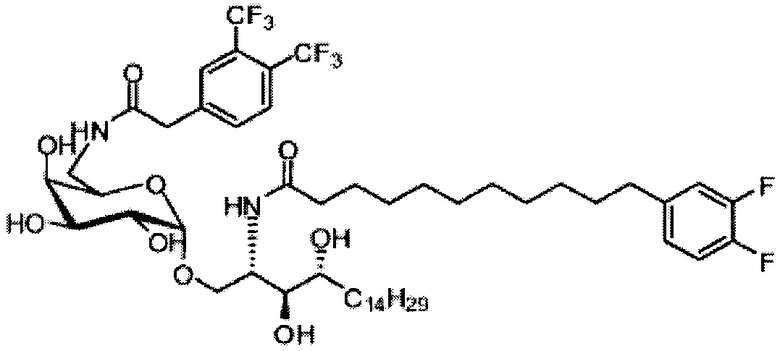 ;
;
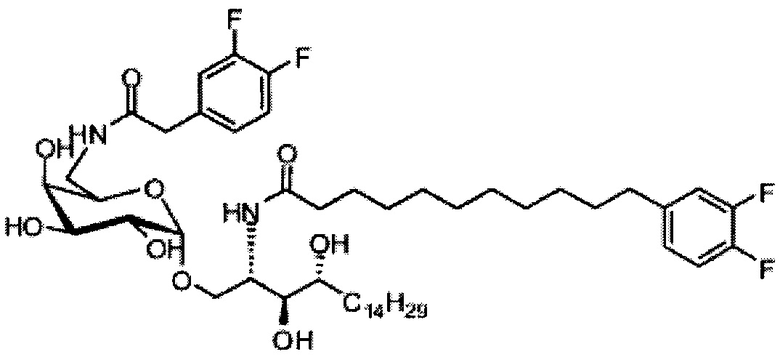 ;
;
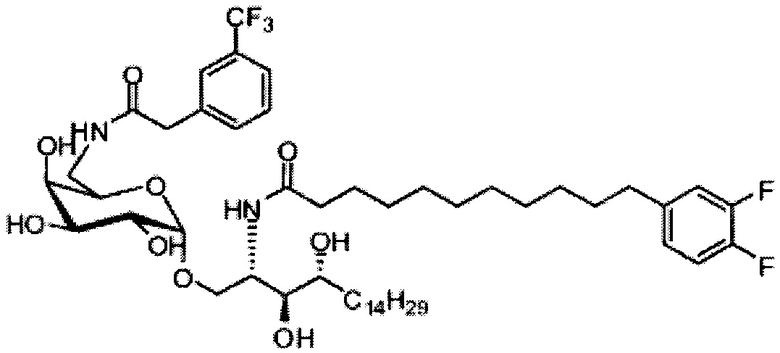 ;
;
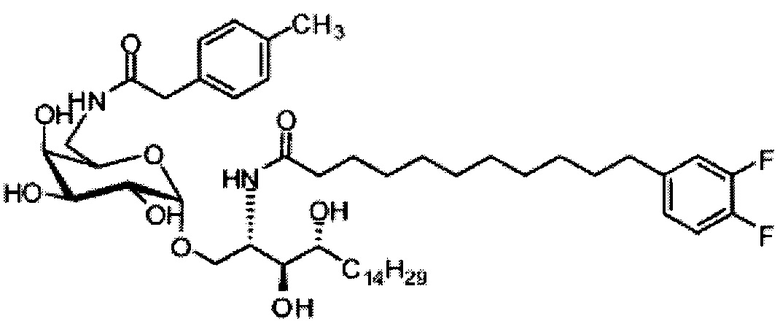 ;
;
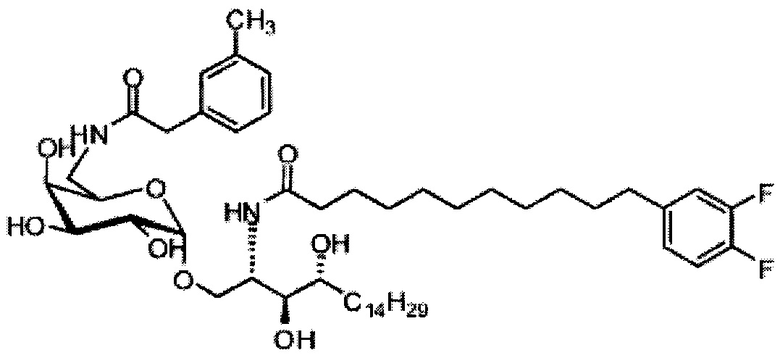 ;
;
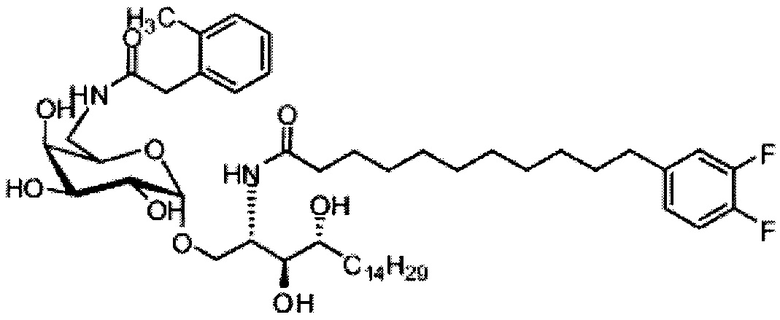 ; и
; и
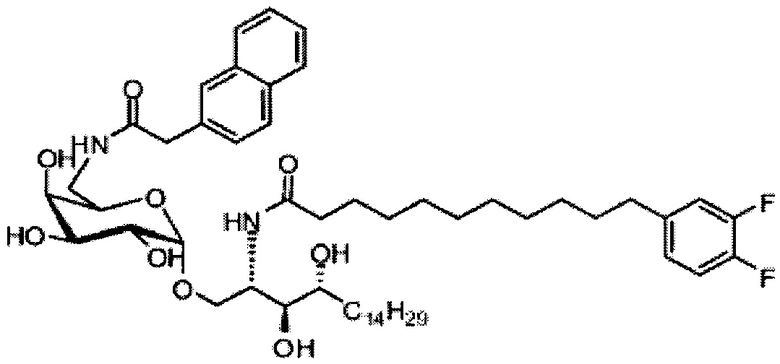 .
.
13. Соединение, выбранное из группы, включающей:
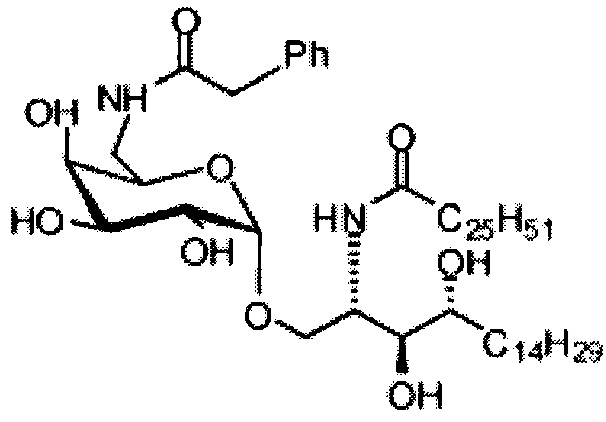 ;
;
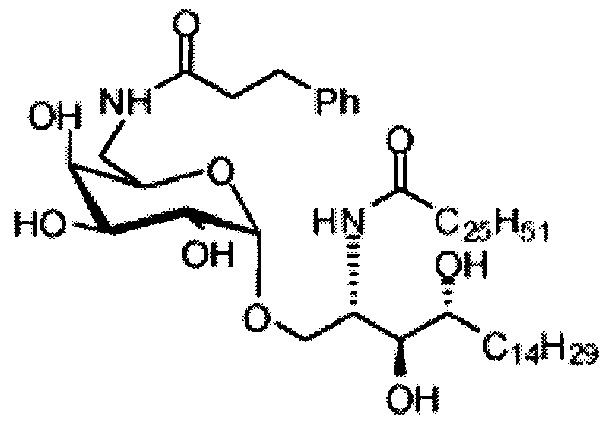 ;
;
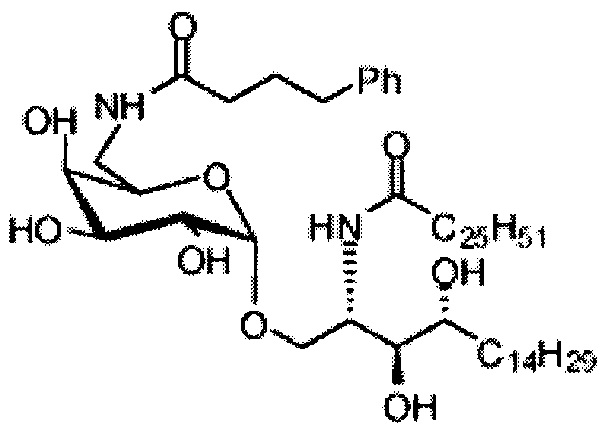 ;
;
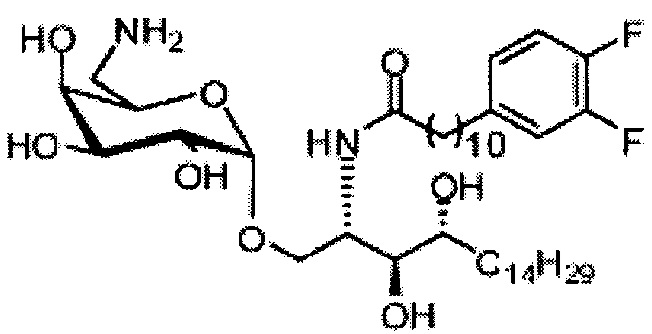 ;
;
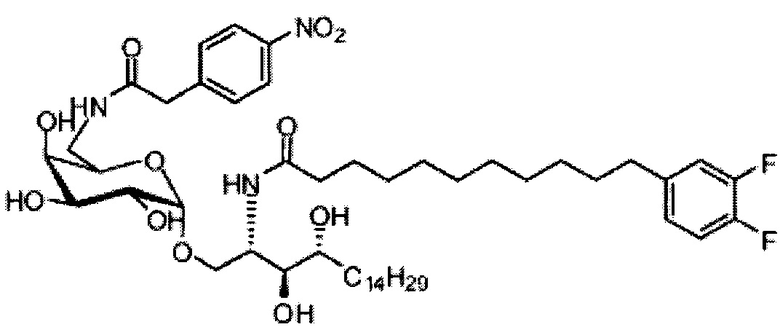 ;
;
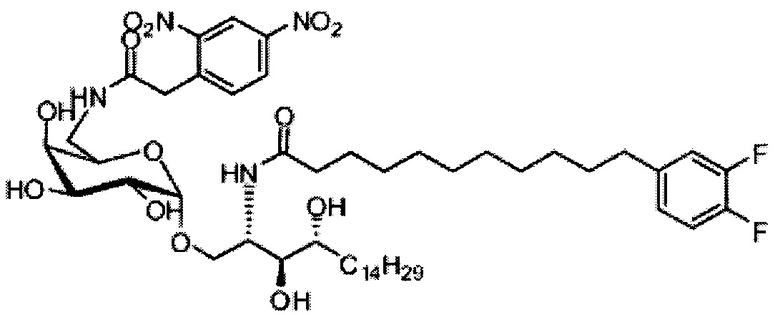 ;
;
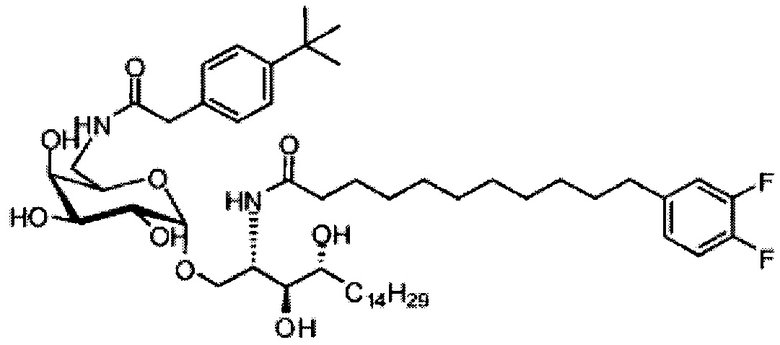 ;
;
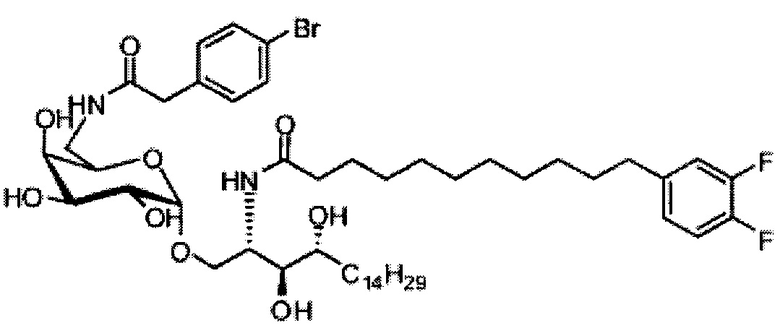 ;
;
 ;
;
 ;
;
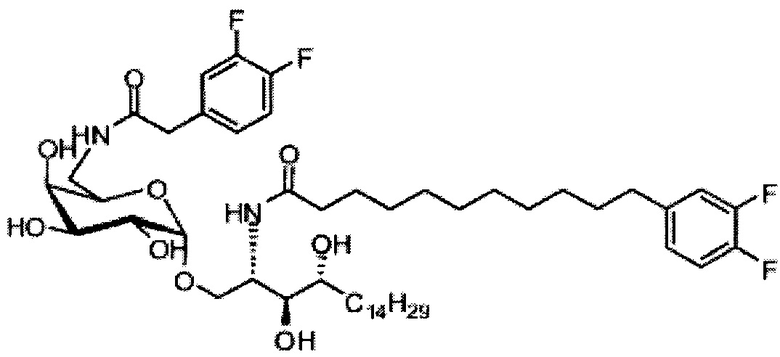 ;
;
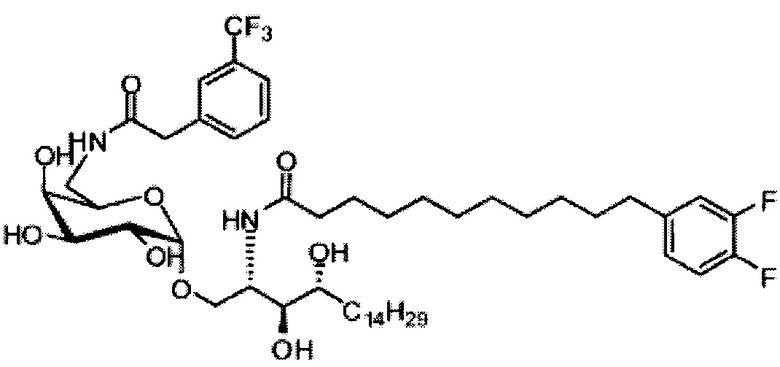 ;
;
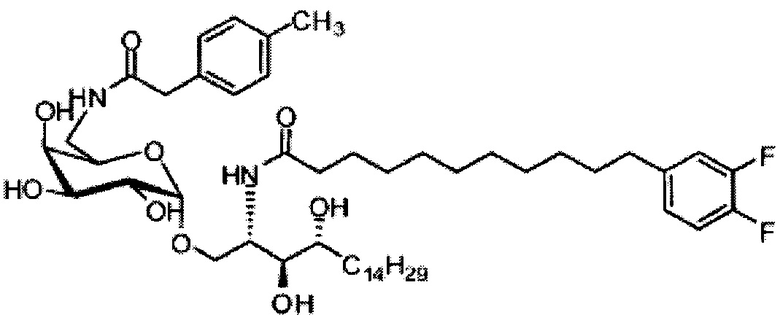 ;
;
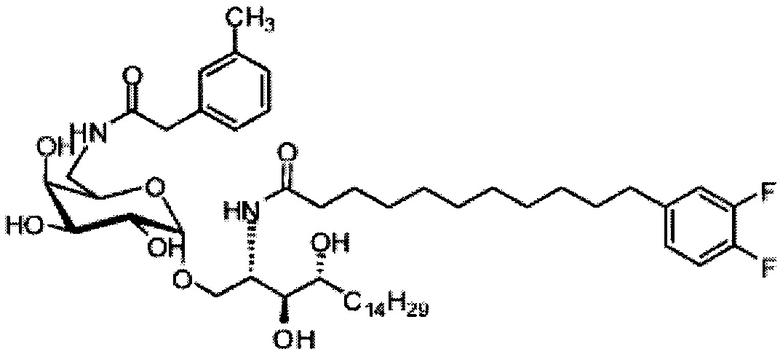 ;
;
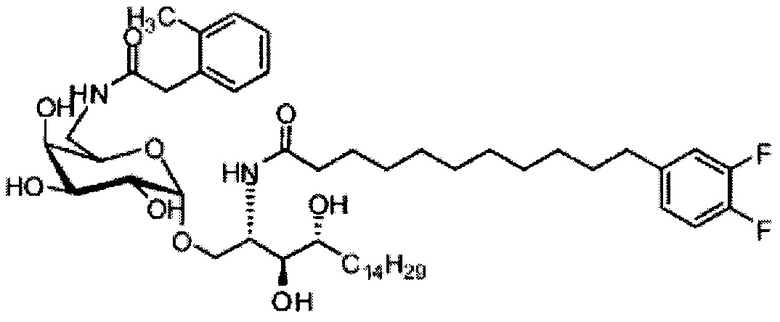 ; и
; и
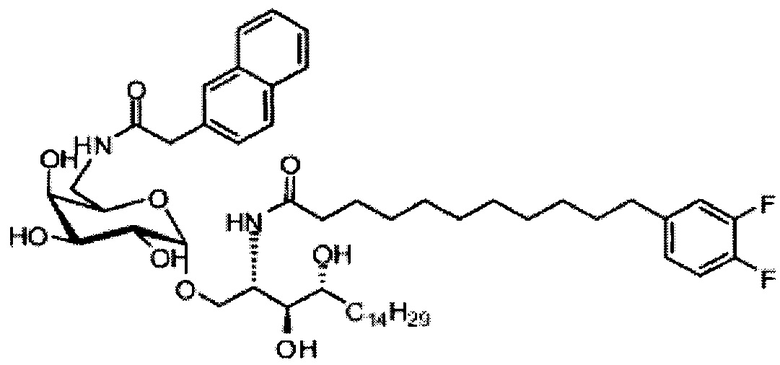 .
.
14. Фармацевтическая композиция для применения в качестве противоопухолевого, противовирусного, антибактериального средства или адъюванта, включающая соединение по п. 13, обладающее профилем секреции цитокина, сдвинутым в сторону ТН-1, и фармацевтически приемлемый носитель или эксципиент.
15. Применение фармацевтической композиции по п. 14 для изготовления лекарственного средства для повышения секреции цитокина, сдвинутой в сторону ТН-1, у субъекта, где:
композицию вводят субъекту в количестве, достаточном для повышения секреции цитокина, сдвинутой в сторону ТН-1.
16. Применение по п. 15, где цитокин выбирают из IL-2, IFN-гамма и IL-4.
17. Применение по п. 15, где результатом повышения секреции цитокина, сдвинутой в сторону ТН-1, является противоопухолевое, противовирусное, антибактериальное или адъювантное действие.
18. Соединение формулы (6):
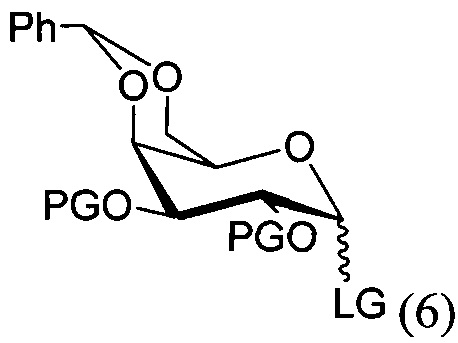 ,
,
где PG является бензильной защитной группой гидроксила и LG является тиолсодержащей уходящей группой формулы:
 ,
,  ,
, 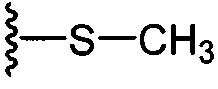 или
или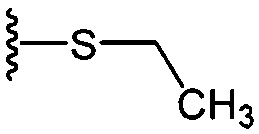 .
.
19. Способ получения соединения формулы (5):
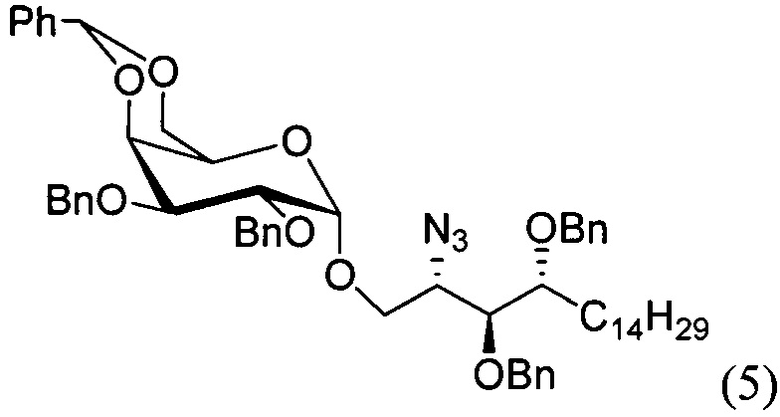 ,
,
который включает взаимодействие соединения формулы (6):
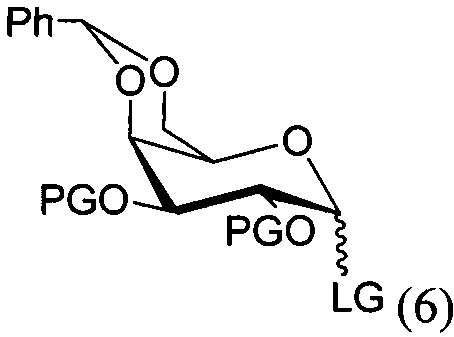 ,
,
где PG является бензильной защитной группой гидроксила и LG является тиолсодержащей уходящей группой формулы:
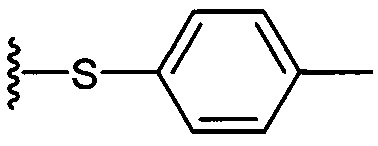 ,
, 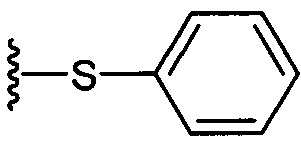 ,
,  или
или  ;
;
с соединением формулы (7):
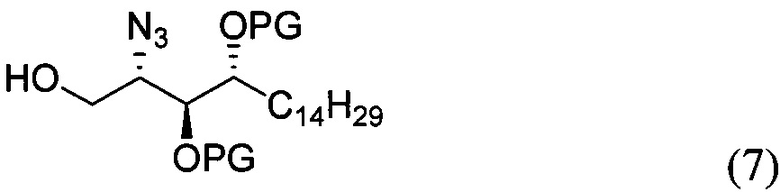 ,
,
и проведение альфа-гликозилирования в присутствии кислоты Льюиса с образованием соединения формулы (5).
20. Способ по п. 19, где кислота Льюиса представляет собой TMSOTf, Tf2O, BF3⋅OEt2, TfOH или Me2S2-Tf2O.
21. Способ по п. 19, пригодный для дальнейшего превращения соединения формулы (5):
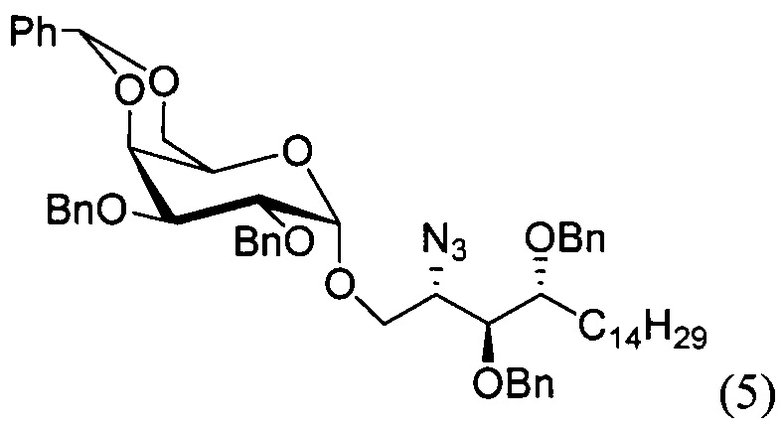
путем восстановления с получением соединения формулы (3):
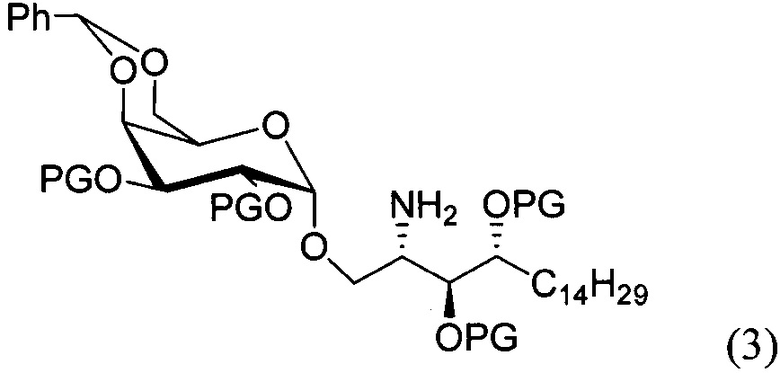 ,
,
где PG является бензильной защитной группой гидроксила.
22. Способ по п. 21, пригодный для дальнейшего превращения соединения формулы (3):
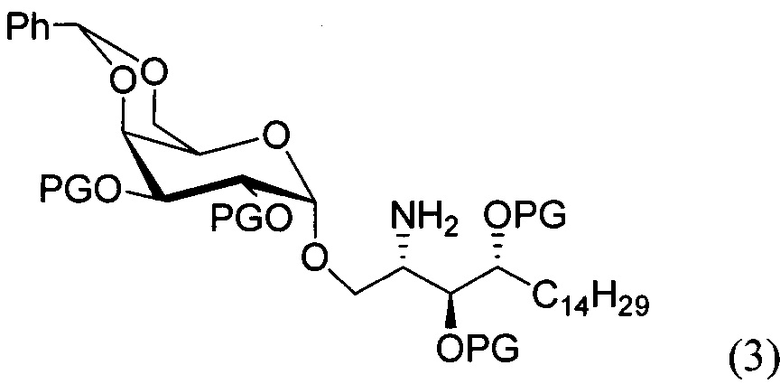 ,
,
где PG является бензильной защитной группой гидроксила, с соединением формулы (4):
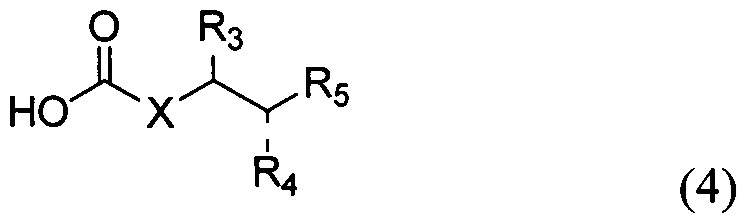 ,
,
где:
X, R3, R4 и R5 имеют значения, как определено в п. 1, с получением соединения формулы (2):
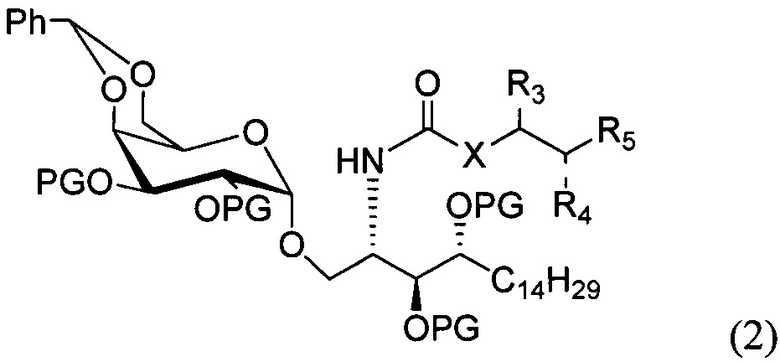 .
.
23. Способ по п. 22, пригодный для дальнейшего превращения соединения формулы (2):
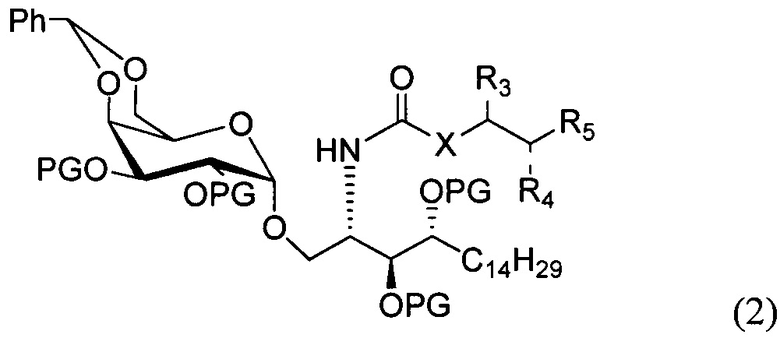 ,
,
где PG является бензильной защитной группой гидроксила,
путем удаления защитных групп под водородом в условиях гидрогенизационного катализа с получением соединения формулы (1)
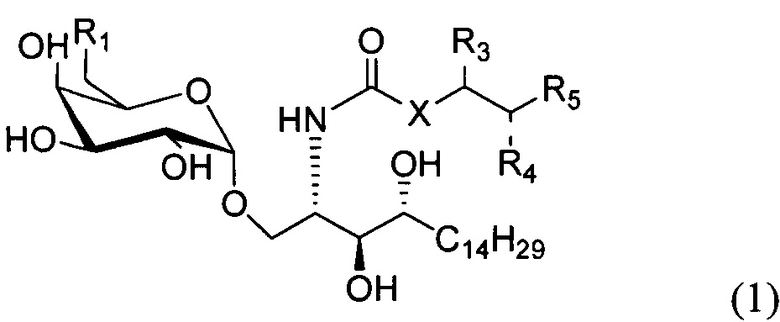 ,
,
где X, R1, R3, R4 и R5 имеют значения, как определено в п. 1.
| Kun-Hsien Lin et al | |||
| Antimicrobial Agents an Chemotherapy, 2010, 4129-4136 | |||
| WO 2008128062 A1, 23.10.2008 | |||
| Matthias Trappeniers et al | |||
| ChemMedChem, 2008, 3, 1061-1070 | |||
| НОВЫЕ АРОМАТИЧЕСКИЕ ФТОРГЛИКОЗИДНЫЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ ЛЕКАРСТВЕННЫЕ СРЕДСТВА, И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2340622C2 |

