Изобретение относится к области органической химии, медицинской химии, к малым молекулам, эффективным в потенцировании действия антибиотиков в отношении грамотрицательных бактерий. Представленные малые молекулы - функциональные производные гидроксамовых кислот, содержащие ядра арилзамещенных 1,2,4- и 1,3,4-оксадиазолов представляют собой ингибиторы фермента УДФ-3-О-(R-3-гидроксимиристоил)-N-ацетилглюкозамин деацетилазы (LpxC, UDP-3-О-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase), который является одним из важнейших ферментов, участвующих в биосинтезе липополисахарида (ЛПС) бактерий.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Человеческие инфекции, вызванные патогенными бактериями, ежегодно убивают миллионы людей, многие из которых вызваны диарейными заболеваниями, бактериальным туберкулезом и инфекциями нижних дыхательных путей [1-3]. Ежегодно от тяжелого сепсиса, вызванного бактериальными инфекциями, в мире страдают до 18 миллионов человек, при этом уровень смертности составляет от 28% до 50% [1, 4]. Бактериальные инфекции являются серьезной проблемой здравоохранения в мире, а лечение этих инфекционных заболеваний становится все более затруднительным из-за развития устойчивости к антибиотикам [5-10]. Развивающаяся антибиотикорезистентность усугубляется бесконтрольным применением антибиотиков в медицине и сельском хозяйстве [6]. Устойчивые штаммы не только встречаются в окружающей среде (вода, почва и воздух), но и могут размножаться и распространяться при несоблюдении мер профилактики и контроля инфекции.
Политика, рекомендованная Всемирной Организацией Здравоохранения (ВОЗ), включает в себя разработку новых антибиотиков, однако, со времен даптомицина и линезолида в 1980-х годах не было обнаружено ни одного нового класса антибиотиков [7]. Устойчивость клинически важных патогенных бактерий к доступным антибиотикам в настоящее время является глобальной проблемой из-за постоянно растущего числа штаммов, устойчивых уже к нескольким классам антибиотиков. Это означает, что прогресс современной медицины, опирающейся на наличие эффективных антибактериальных препаратов, находится под угрозой. Таким образом, актуальна необходимость разработки не только новых антибиотиков, систем доставки и бактериальной диагностики, но и разработки «антибиотических адъювантов/потенциаторов», действующих в сочетании с известными антибиотиками [11].
Большинство патогенных микроорганизмов, которые могут вызывать различные серьезные заболевания, представляют собой грамотрицательные бактерии. В частности, грамотрицательные бактерии с множественной лекарственной устойчивостью стали серьезной угрозой для госпитализированных пациентов, уровень смертности которых колеблется от 30% до 70% [12]. Например, Е. coli является основной причиной инфекций мочевыводящих путей, P. aeruginosa вызывает внутрибольничную пневмонию и инфекции кровотока, N gonorrhoeae является источником гонореи, а K. pneumoniae вызывает инфекции мочевыводящих путей, а также пневмонию [1, 13]. Кроме того, вызывает тревогу тот факт, что множественная лекарственная устойчивость также распространена среди наиболее смертоносных и патогенных для человека видов бактерий, таких как возбудитель чумы Y. pestis [14].
Липополисахарид (ЛПС) представляет собой поверхностный гликолипид бактерий, продуцируемый грамотрицательными бактериями и присутствующий в их внешней клеточной стенке. ЛПС состоит из трех доменов: липида А, основного олигосахарида и антигена О. В качестве основных биологических функций ЛПС можно отметить структурную и защитную - выступать барьером против токсических для бактерий агентов, например антибиотиков [15]. Липид А, являющийся гидрофобным якорем липополисахарида (ЛПС), является привлекательной мишенью для разработки как новых антибиотиков, так и потенциаторов действия уже известных антибиотиков, поскольку бактерии, лишенные липида А, как правило, нежизнеспособны, а мутанты со сниженным биосинтезом липида А имеют медленный рост и высокую чувствительность к широкому спектру антибиотиков [1].
Цинк-зависимая металлоамидаза, УДФ-3-O-(R-3-тидроксимирштоия)-N-ацетилглюкозаминдеацетилаза (LpxC), считается многообещающей мишенью, что обусловлено ключевой ролью LpxC в биосинтезе липида А и отсутствие гомологии с белками млекопитающих, для стадии детерминации, участвующей в катализе биосинтеза липида А [16]. Деацетилаза LpxC - один из наиболее изученных ферментов, участвующих в биосинтезе липида А, ингибиторы которого потенциально могут выступать как антибиотики, так и потенциаторы действия уже известных антибиотиков.
В настоящее время в литературе описано антибактериальное и потенцирующее действие множества ингибиторов фермента LpxC в отношении различных грамотрицательных микроорганизмов, показавшие перспективность при лечении заболеваний, вызванных соответствующими бактериальными инфекциями (P. aeruginosa, Е. coli, N. gonorrhoeae, K. pneumoniae, В. pseudomallei, В. pertussis).
Целью настоящего изобретения является разработка потенциальных ингибиторов фермента LpxC, относящихся к классу функциональных производных гидроксамовых кислот, содержащих ядра арилзамещенных 1,2,4- и 1,3,4-оксадиазола, которые могут быть использованы в качестве потенциаторов действия антибиотиков в отношении грамотрицательных бактерий.
УРОВЕНЬ ТЕХНИКИ
В настоящее время существует неудовлетворенная медицинская потребность, связанная с развитием антибиотикорезистентности, в частности, у патогенных грамотрицательных бактерий, в связи с чем существующие антибиотики не обладают достаточной эффективностью в отношении данных микроорганизмов.
В этой связи было исследовано несколько подходов, проведен поиск и анализ источников информации из общедоступного уровня техники до даты испрашиваемого приоритета.
Первые синтетические ингибиторы деацетилазы LpxC появились в 1996 году и были разработаны учеными Merck. На сегодняшний день до фазы клинических испытаний дошли только два ингибитора - ACHN-975 (NCT01597947, NCT01870245, данные из https://clinicaltrials.gov/) и RC-01 (NCT03832517). К сожалению, оба эти ингибитора (Таблица 1) были исключены из клинических испытаний I фазы из соображений безопасности. Исходя из представленных данных, основными проблемами потенциальных ингибиторов LpxC являются растворимость, проницаемость и токсичность. Данные проблемы необходимо решать за счет химических модификаций уже известных ингибиторов, либо синтезом ингибиторов новых хемотипов [1].
Большинство ингибиторов LpxC в настоящее время относятся к структурному классу гидроксамовых кислот, которые катализируют хелатирование ионов Zn2+ в активном сайте фермента, увеличивая сродство и ингибирующую активность. Однако, в это же время гидроксамовые кислоты способны метаболизироваться в организме до токсичного гидроксиламина. В настоящее время установлено, что ингибиторы LpxC, не относящиеся к классу гидроксамовых кислот, не демонстрируют превосходного профиля антимикробной активности, по сравнению с гидроксамовыми кислотами [1]. Таким образом, становится актуальным вопрос снижения профиля токсичности с одновременным сохранением противомикробной активности ингибиторов LpxC гидроксаматной структуры, наряду с поиском новых потенциальных ингибиторов LpxC, обладающих уже улучшенными показателями ADMET свойств.
Предложенные в изобретении малые молекулы, потенциальные ингибиторы LpxC представляют собой новый хемотип, потенцирующая активность которого в присутствии различных антибиотиков в отношении грамотрицательных бактерий ранее не исследовалась.
Проанализировав патентные источники, содержащие данные по известным на настоящий момент ингибиторам LpxC, наиболее близкой структуры к заявляемым в настоящем изобретении соединениям формулы (I) можно отметить нижеследующие.
Известен патент на изобретение US 8748466 (В2) от 10.06.2014 «Isoxazole derivatives useful as antibacterial agents» [19], в котором в качестве формул Map куша I и II заявлено:
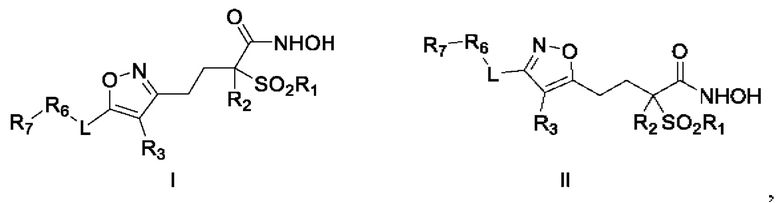
Где R1 представляет собой алкильную цепь C1-C3, R2 представляет собой атом водорода или алкильную цепь C1-C3, R3 представляет собой атом водорода, алкокси-цепь C1-C3, алкильную цепь C1-C3, цианогруппу, галогензамещенную алкокси-цепь C1-C3, галогензамещенную алкильную цепь C1-C3, атом галогена или гидрокси-группу. L - представляет собой связь С-С или линкер (CH2)n, (CH2)nO(CH2)p, (CH2)nS(CH2)p, (CH2)nNR4(CH2)p, (CH2)nSO2NR4(CH2)p, (CH2)nNR4SO2(CH2)p, (CH2)nCONR4(CH2)p, (CH2)nNR4CO(CH2)p. Заместители R4 и R5 представляют собой атом водорода, алкильную группу C1-C6, алкилкарбонильную группу C1-C6, циклоалкил С3-С8, (С3-С8)циклоалкилзамещенная алкильная группа C1-C6, формильную группу.
n=0, 1, 2, 3, 4,
p=0, 1, 2, 3, 4.
R6 представляет собой (C1-С6)алкоксизамещенный алкил C1-С6, алкоксикарбонильную группу C1-С6, алкилкарбонильную группу C1-С6, (C1-С6)алкил-NR4-(C1-С6)алкил, (C1-С6)алкилтио(C1-С6)алкил, (C1-С6)алкилтиокарбонил, (С6-С12)арил, (С6-С12)арилокси, (С6-С12)арилтио, (С6-С12)apnn-NR4, (С3-С8)циклоалкил, (С3-С8)циклоалкилокси, (С3-С8)циклоалкилтио, (С3-С8)никлоалкил-NR4, (С5-С12)гетероарил, (С5-С12)гетероарил, (С5-С12)гетероарилокси, (С5-С12)гетероарилтио, (С5-С12)гетероарил-NR4, (С3-С13)гетероарил, (С3-С13)-гетероарилокси, (С3-С13)-гетероарилтио, (С3-С13)гетероарил-NR4, гидроксиалкил CR, меркаптоалкил C1-С10, (NR4R5)алкил или (NR4R5)карбонил.
R7 представляет собой (С6-С12)арил, (С6-С12)арил(C1-С6)алкил, (С3-С8)циклоалкил, (С3-С8)циклоалкил(C1-С6)алкил, (С5-С12)гетероарил, (С5-С12)гетероарил(C1-С6)алкил, (С3-С13)гетероцикл или (С3-С13)гетероцикл(C1-С6)алкил.
Известен источник информации - патентная заявка WO 9742179 (А1) от 13.11.1997 «Antibacterial agents» [20], в котором в качестве формулы Маркуша Ia заявлено:
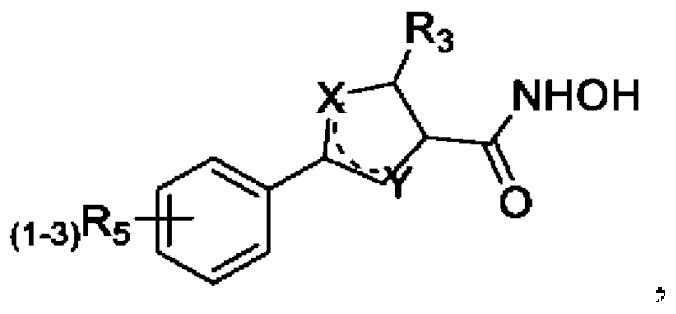
где один из пары X или Y представляет собой N(R2)0-1, а второй - О, S или N(R2)0-1. Заместитель R2 представляет собой водород или алкильную цепь C1-С6, пунктирная линия представляет собой дополнительную (кратную связь), R3 представляет собой водород или алкильную цепь C1-C6, которая дополнительно может содержать OR2, CO2R2, N(R2)2 группу. Заместитель R5 представляет собой алкильную цепь C1-C6, алкокси-цепь C1-C6, атом галогена, трифторметильную группу, N(R2)2, фенокси, CO2R2, гидрокси, R2SO2, CON(R2)2, бензилокси. В случае, когда заместитель R5 представляет собой фенокси или бензилокси-группу, то они могут быть дополнительно замещены алкильными группами C1-С6, алкокси группами C1-С6, атомами галогенов, трифторметильными группами или гидрокси-группами. Техническое решение, описанное в источнике WO 9742179 (А1) от 13.11.1997, является наиболее близким аналогом предлагаемого изобретения и выбрано за прототип.
К недостаткам известных ингибиторов LpxC (оксазолиновым, ацетиленовым и бутадииновым, а также содержащих сульфоновый заместитель [1]) можно отнести:
Неудовлетворительные ADMET-свойства кандидатов (плохая растворимость, проницаемость и высокая токсичность).
- В большинстве случаев - многостадийный синтез целевых ингибиторов, что значительно сказывается на технологичности получения данных кандидатов. В случае использования в качестве исходных соединений для синтеза целевых молекул «N - 2», «N - 3» интермедиатов - остро встает вопрос экономического характера, в связи с высокой стоимостью исходных материалов.
- Наличие стереоцентра(-ов) в известных на настоящее время ингибиторах, с одной стороны, может являться важным параметром для проявления нужной фармакологической активности, но, с другой стороны, значительно усложняет синтез, очистку и выделение целевых молекул.
Структуры известных ингибиторов LpxC различного структурного типа представлены в Таблице 2.
Целью настоящего технического решения является разработка потенциальных ингибиторов LpxC, обладающих потенцирующей действие антибиотиков активностью в отношении грамотрицательных бактерий, нового структурного типа (гидроксамовые кислоты, содержащие ядра 1,2,4- и 1,3,4-оксадиазола), характеризующихся сравнительно коротким синтезом, кристалличностью полупродуктов синтеза, их повышенной универсальностью с точки зрения создания широкого химического пространства структурных аналогов, потенциально большими возможностями по повышению технологичности процесса производства полупродуктов и целевых субстанций, удовлетворяющих целевому профилю для данного класса фармацевтических препаратов.
В результате проведенного анализа уровня техники можно сделать вывод о том, что предлагаемое изобретение может быть признано соответствующим критериям патентоспособности «новизна» и «изобретательский уровень» до даты испрашиваемого приоритета.
РАСКРЫТИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Заявителем предложен новый хемотип ингибиторов биосинтеза липополисахарида бактерий, обладающих потенцирующей активностью в сочетании с антибиотиками в отношении грамотрицательных бактерий, охарактеризованный общей формулой (I). Представлен способ синтеза предлагаемых соединений, а также данные по исследованиям их ADME-свойств, цитотоксичности, антибактериальной и потенцирующей активности в отношении клеточных линий Escherichia coli MG1655, Pseudomonas aeruginosa АТСС 27853.
Техническими результатами от реализации заявленного изобретения являются:
- синтез новых химических соединений, являющимися ингибиторами фермента LpxC, обладающих фармакологической активностью,
- уменьшение цитотоксичности синтезированных химических соединений, являющимися ингибиторами фермента LpxC,
- улучшение ADME-свойств синтезированных химических соединений, являющимися ингибиторами фермента LpxC.
Техническая проблема может заключаться в необходимости расширения арсенала ингибиторов биосинтеза ЛПС бактерий.
Чтобы устранить недостатки известных ингибиторов фермента LpxC, катализирующего биосинтез липополисахарида бактерий [1], используя принцип биоизостерических замен, авторы настоящего изобретения предложили новый хемотип потенциальных ингибиторов LpxC -функциональные производные гидроксамовых кислота, содержащие ядра 1,2,4- и 1,3,4-оксадиазола, сопряженные с функциональной группой кислоты напрямую или через алифатический/ароматический линкер. Пять полученных соединений общей формулы I обладают значительной эффективностью в потенцировании действия налидиксовой кислоты, рифампицина и канамицина в отношении клеточных линий Escherichia coli MG1655, Pseudomonas aeruginosa АТСС 27853. Полученные активные в отношении микроорганизмов гидроксамовые кислоты обладают хорошими ADME-свойствами и удовлетворительными показателями цитотоксической концентрации. Это позволяет утверждать, что предложенное авторами решение технической проблемы обладает ценностью, а также представляет собой перспективное направление для дальнейших, более детальных исследований потенцирующего действия представителей данного хемотипа и установления более глубоких взаимосвязей структура-активность с целью выявления пригодного кандидата для дальнейших клинических испытаний.
Основная гипотеза настоящего изобретения заключается в том, что
- препараты на основе малых молекул нового предложенного хемотипа, имея большое сродство с целевой мишенью LpxC in vitro, будут замедлять биосинтез липополисахарида бактерий, что облегчит проникновение антибиотиков через клеточную стенку грамотрицательных бактерий, что позволит организму вновь эффективно бороться с инфекциями, вызванными этими бактериями;
- ингибиторы предложенного хемотипа будут обладать антибактериальной активностью, и/или при их совместном с различными антибиотиками использовании будет наблюдаться синергетический эффект взаимного усиления антибактериальной активности;
- ингибиторы предложенного хемотипа будут обладать улучшенными показателями ADMET-свойств, по сравнению с известными эффективными ингибиторами LpxC (в частности, бутадииновыми, ацетиленовыми).
КРАТКОЕ ОПИСАНИЕ ФИГУР, ТАБЛИЦ, ИНЫХ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
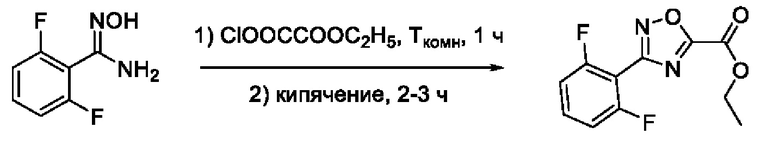
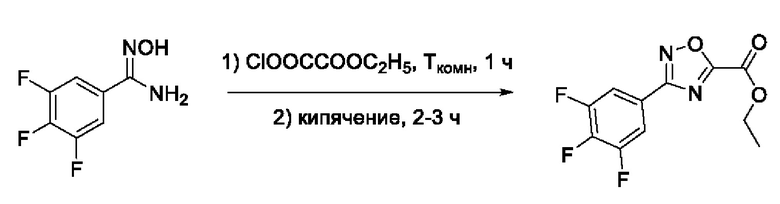
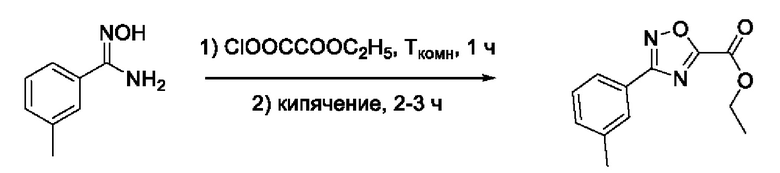
На фигуре 1 представлен синтез гидроксамовых кислот, функциональная группа которых сопряжена с положением 5 ядра 1,2,4-оксадиазола, содержащего ароматический заместитель в положении 3 гетероцикла (соединения I(1-6)).
На фигуре 2 представлен синтез гидроксамовых кислот, функциональная группа которых связана с положением 5 ядра 1,2,4-оксадиазола, содержащего ароматический заместитель в положении 3 гетероцикла, через метиленовый линкер (соединения I(7-10)).
На фигуре 3 представлен синтез гидроксамовой кислоты, функциональная группа которой сопряжена с положением 3 ядра 1,2,4-оксадиазола, содержащего ароматический заместитель в положении 5 гетероцикла, через мета-замещеикьт фениленовый линкер (соединение I(11)).
На фигуре 4 представлен синтез гидроксамовых кислот, функциональная группа которых сопряжена с положением 5 ядра 1,2,4-оксадиазола, содержащего ароматический заместитель в положении 3 гетероцикла, через мета-замещетпт фениленовый линкер (соединения I(12-14)).
На фигуре 5 представлен синтез гидроксамовых кислот, функциональная группа которых сопряжена с положением 5 ядра 1,2,4-оксадиазола, содержащего ароматический заместитель в положении 3 гетероцикла, через пара-замещенный фениленовый линкер (соединения I(15-16)).
На фигуре 6 представлен синтез гидроксамовых кислот, функциональная группа которых связана с положением 3 ядра 1,2,4-оксадиазола, содержащего ароматический заместитель в положении 5 гетероцикла, через метиленовый линкер (соединения I(17-20)).
На фигуре 7 представлен синтез гидроксамовых кислот, функциональная группа которых связана с положением 5 ядра 1,2,4-оксадиазола, содержащего ароматический заместитель в положении 3 гетероцикла, через этиленовый линкер (соединения I(21-24)).
На фигуре 8 представлен синтез гидроксамовых кислот, функциональная группа которых сопряжена с положением 2 ядра 1,3,4-оксадиазола, содержащего ароматический заместитель в положении 5 гетероцикла (соединения I(25-30)).
На фигуре 9 представлены структуры соединений-лидеров, обладающих эффективностью в потенцировании действия налидиксовой кислоты в отношении клеточной линии Е. coli MG1655.
На фигуре 10 представлены репрезентативные кривые роста Е. coli MG1655 в присутствии налидиксовой кислоты (NaI, 4 мкг/мл), без или с ингибиторами LpxC в концентрации 50-100 мкМ.
На фигуре 11 представлены репрезентативные кривые роста Е. coli MG1655 в присутствии рифампипина (5 мкг/мл) и канамицина (0,3 мкг/мл) без или с ингибиторами LpxC в концентрации 50 или 100 мкМ.
На фигуре 12 представлены структуры соединений-лидеров, показавших наилучшую эффективность в потенцировании действия трех антибиотиков в отношении клеточной линии Е. coli MG1655.
На фигуре 13 представлены репрезентативные кривые роста P. aeruginosa АТСС 27853 в присутствии налидиксовой кислоты (слева) и рифампицина (справа) в присутствии гидроксамовых кислот I(9) и I(10) или без них.
В таблице 1 представлены первые ингибиторы LpxC, дошедшие до клинических испытаний.
В таблице 2 представлены известные гидроксаматные ингибиторы LpxC, относящиеся к различным структурным типам: бутадииновые, ацетиленовые, «сульфоновые», оксазолиновые.
В таблице 3 представлены структурные формулы и названия по номенклатуре ИЮПАК соединений общей формулы I(1-30) по примерам в соответствии с настоящим изобретением.
В таблице 4 представлены данные исследований цитотоксичности гидроксамовых кислот общей формулы I(1-30) по примерам в соответствии с настоящим изобретением.
В таблице 5 представлены данные исследований растворимости соединений-лидеров.
В таблице 6 представлены данные исследований стабильности соединений-лидеров в микросомах печени крыс.
В таблице 7 представлены данные исследований проницаемости соединений-лидеров на клетках линии Сасо-2.
В таблице 8 представлены данные исследований стабильности соединений-лидеров в плазме крови человека.
В таблице 9 представлены данные исследований стабильности соединений-лидеров в искусственном кишечном соке (SIF) и искусственном желудочном соке (SGF).
В таблице 10 представлены данные исследований доли соединений-лидеров, связавшихся с белками плазмы крови человека.
В таблице 11 представлены данные исследований коэффициентов распределения logD7.4 соединений-лидеров между н-октанолом и 0,1 М фосфатным буферным раствором.
ОБЪЕКТ ИЗОБРЕТЕНИЯ
Указанный технический результат достигается совокупностью существенных признаков, представленной в формуле изобретения, где заявлены молекулы - ингибиторы нового хемотипа (содержащие ядра арилзамещенных 1,2,4- и 1,3,4-оксадиазола) биосинтеза липополисахарида, охарактеризованные общей формулой (I), как указано ниже.
Предлагаемые соединения могут быть использованы в качестве потенциаторов действия антибиотиков (в частности, налидиксовой кислоты, рифампицина и канамицина) в отношении грамотрицательных бактерий и применены для лечения человеческих инфекций, вызванных патогенными бактериями (например, отита, псевдомонадный фолликулит, гангренозной эктимы, глазных инфекций, инфекций мягких тканей, пневмонии, инфекции мочевыводящих путей, инфекций кровотока, костей и суставов, возбудителем которых является P. aeruginosa).
В одной неограничивающей реализации изобретения предложено соединение формулы (I):
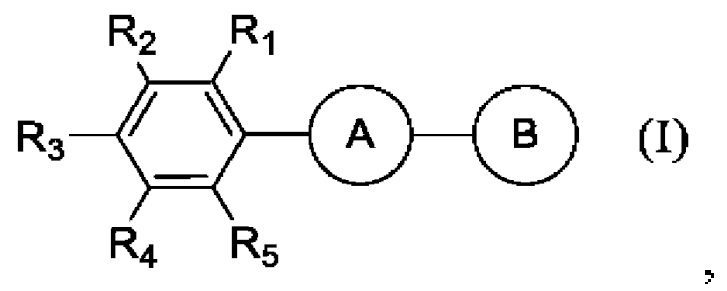
и его фармацевтически приемлемые соли,
где А представляет собой 3,5-дизамещенное ядро 1,2,4-оксадиазола (в виде двух региоизомеров) или 2,5-дизамещенное ядро 1,3,4-оксадиазола:
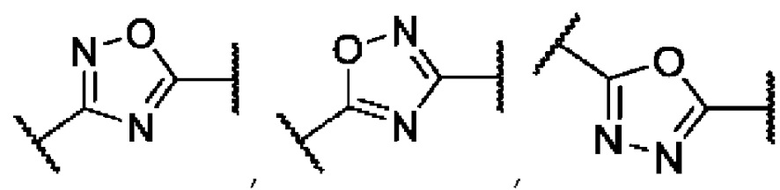
В представляет собой функциональную группу гидроксамовой кислоты (C(O)NHOH), свободную или сопряженную с алифатическим или ароматическим линкером:
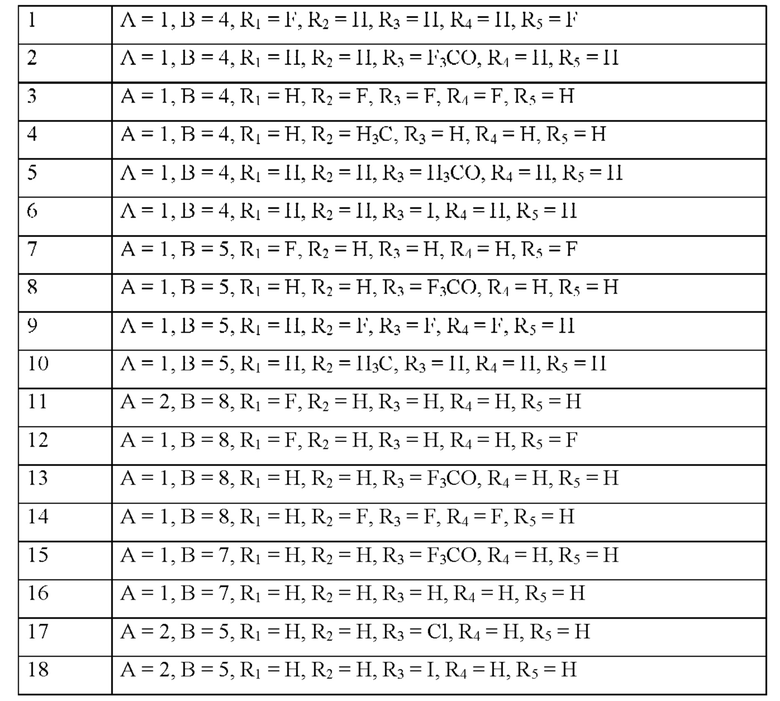
R1 представляет собой: Н, F, Н3СО, Н3С,
R2 представляет собой: Н, F, Cl, Br, I, Н3СО, Н3С,
R3 представляет собой: Н, F, Cl, Br, I, Н3СО, F3CO, Н3С,
R4 представляет собой: Н, F, CI, Br, I,
R5 представляет собой: Н, F,
где I(1- 30) представляют собой:

В одном примере реализации представлено соединение по формуле (I), имеющее формулу:
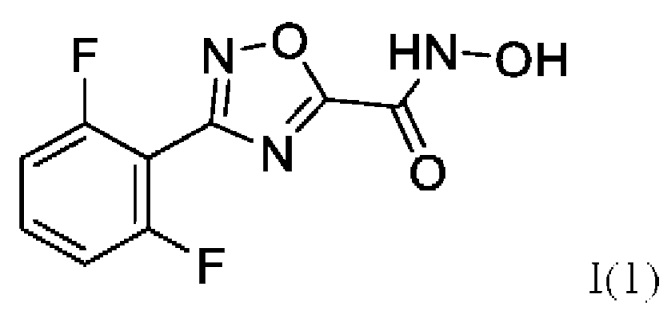
3-(2',6'-Дифторфенил)-N-гидрокси-1,2,4-оксадиазол-5-карбоксамид (1) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
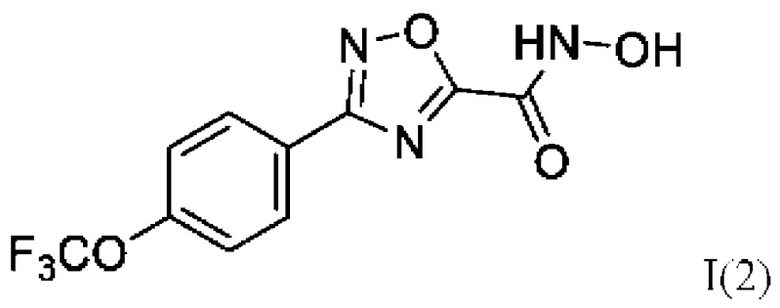
N-Гидрокси-3-(4'-(трифторметокси)фенил)-1,2,4-оксадиазол-5-карбоксамид (2) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
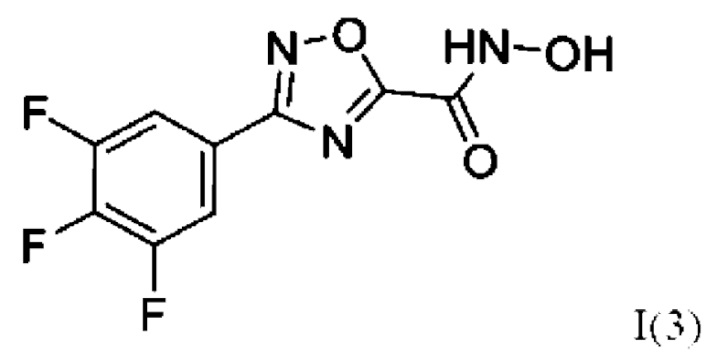
N-Гидрокси-3-(3',4',5'-трифторфенил)-1,2,4-оксадиазол-5-карбоксамид (3) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
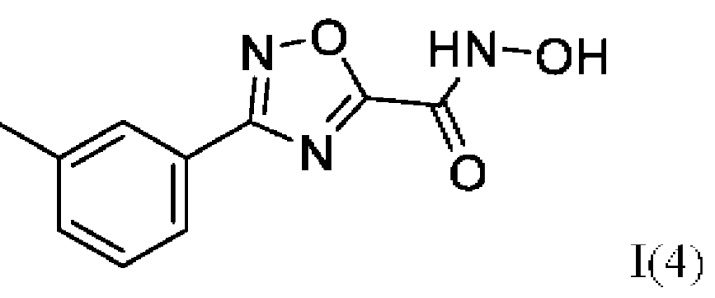
N-Гидрокси-3-(3'-метилфенил)-1,2,4-оксадиазол-5-карбоксамид (4) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
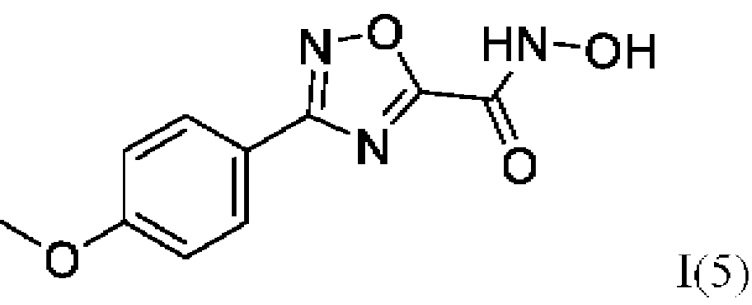
N-Гидрокси-3-(4'-метоксифенил)-1,2,4-оксадиазол-5-карбоксамид (5) или его фармацевтически приемлемая соль.
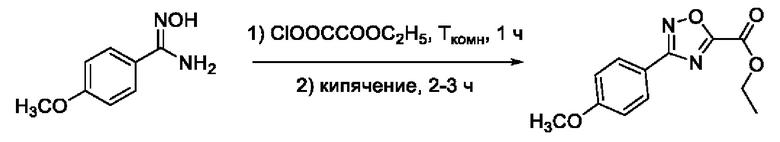
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
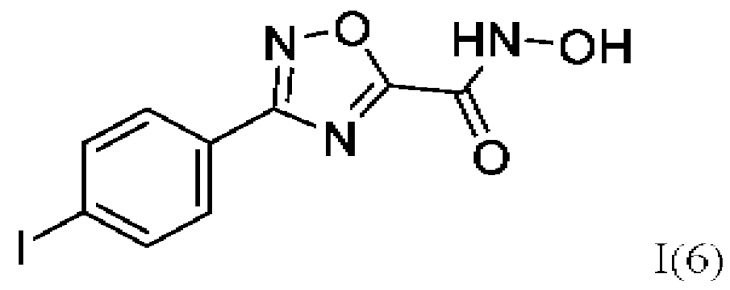
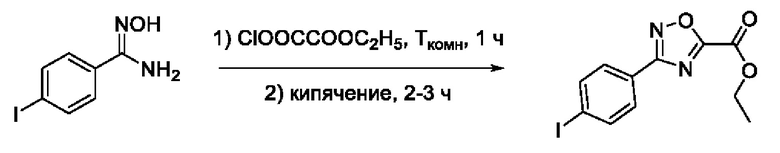
N-Гидрокси-3-(4,-йодфенил)-1,2,4-оксадиазол-5-карбоксамид (6) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
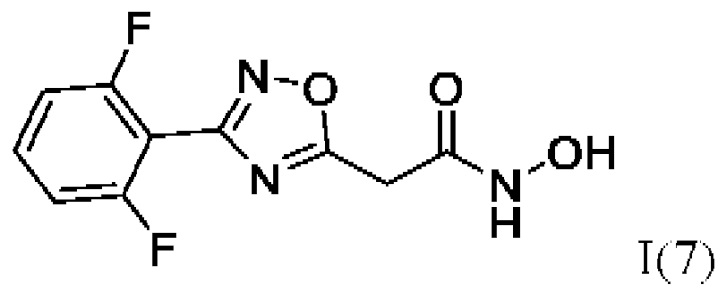
2-(3'-(2'',6''-Дифторфенил)-1',2',4'-оксадиазол-5'-ил)-N-гидроксиацетамид (7) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
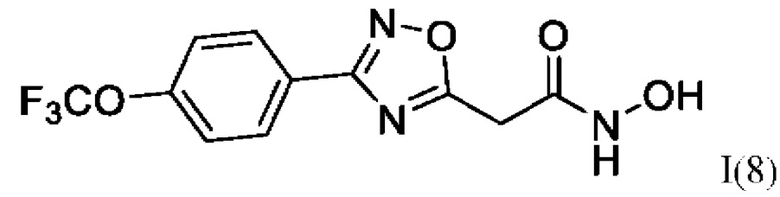
N-Гидрокси-2-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5,-ил) ацетамид (8) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
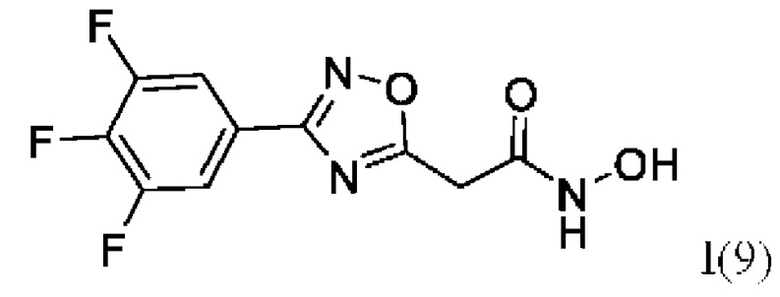
N-Гидрокси-2-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)ацетамид (9) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
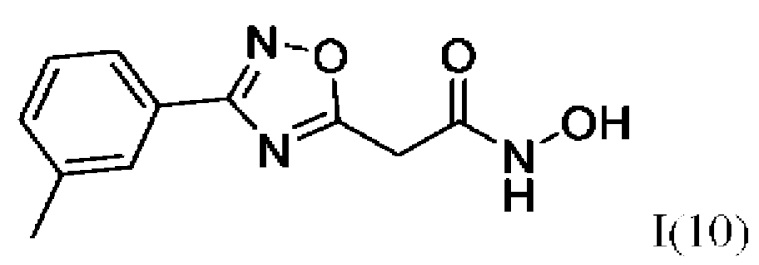
N-Гидрокси-2-(3''-метилфенил)-1',2',4'-оксадиазол-5'-ил)ацетамид (10) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
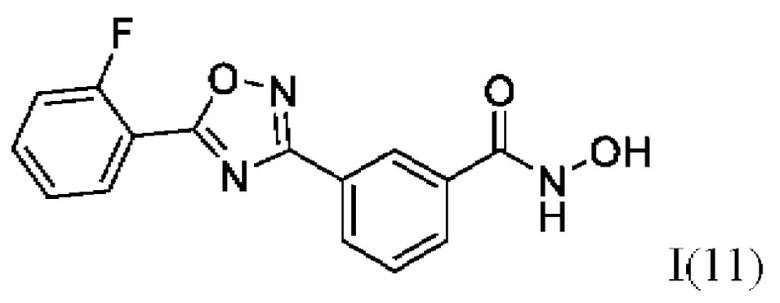
3-(5'-(2''-Фторфенил)-1',2',4'-оксадиазол-3,-ил)-N-гидроксибензамид (11) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
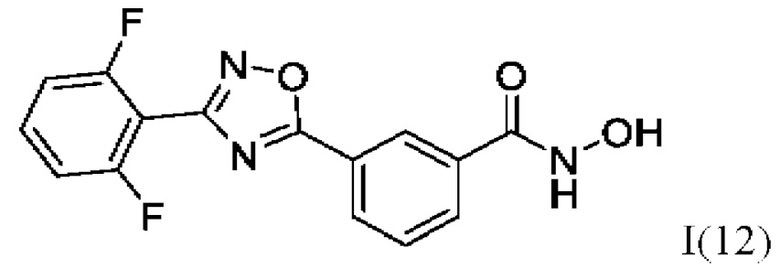
3-(3-(2'',6''-Дифторфенил)-1',2';4'-оксадиазол-5'-ил)-N-гидроксибензамидд (12) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
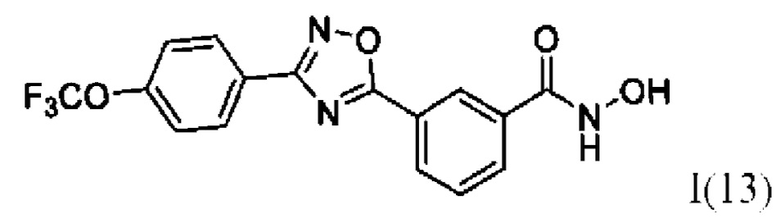
N-Гидрокси-3-(3'-(4''-(трифторметокси)фенил)-1',2,,4'-оксадиазол-5,-ил) бензамид (13) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
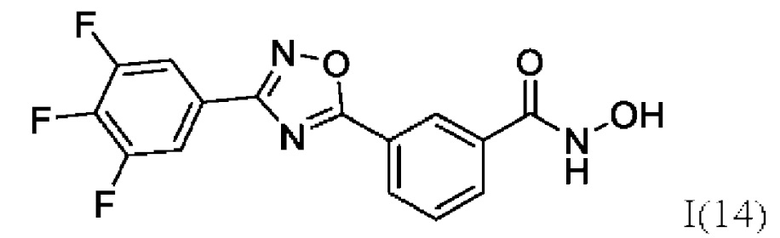
N-Гидрокси-3-(3,-(3',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)бензамид (14) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
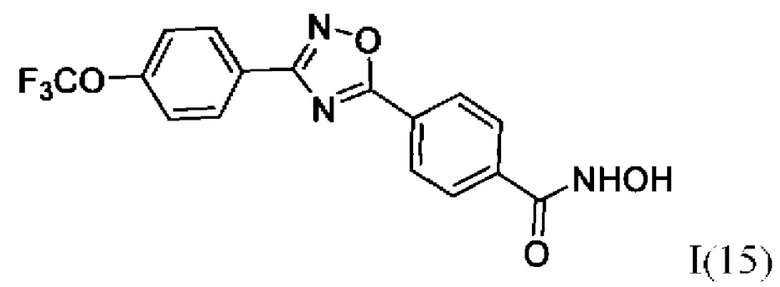
N-Гидрокси-4-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5,-ил) бензамид (15) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
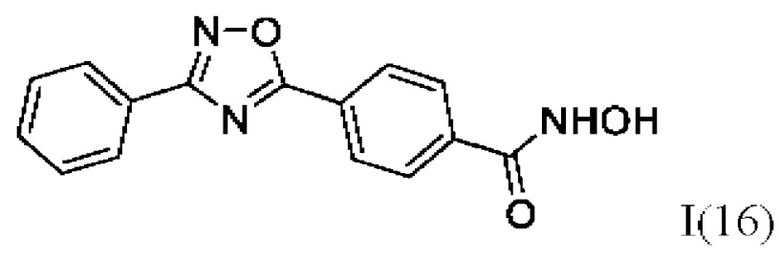
N-Гидрокси-4-(3'-фенил-1',2',4'-оксадиазол-5'-ил)бензамид (16) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
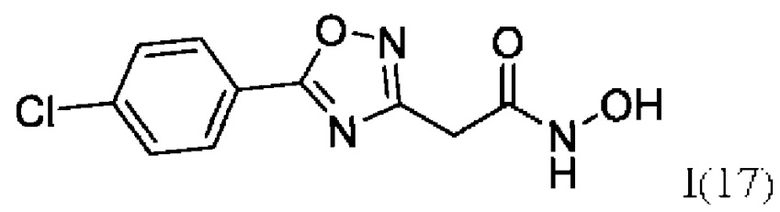
2-(5'-(4''-Хлорфенил)-1',2',4'-оксадиазол-3'-ил)-N-гидроксиацетамид (17) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
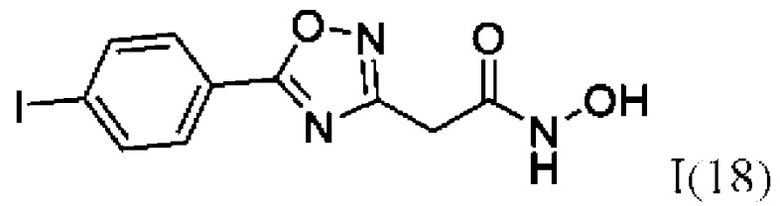
2-(5'-(4''-Йодфенил)-1',2',4'-оксадиазол-3'-ил)-N-гидроксиацетамид (18) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
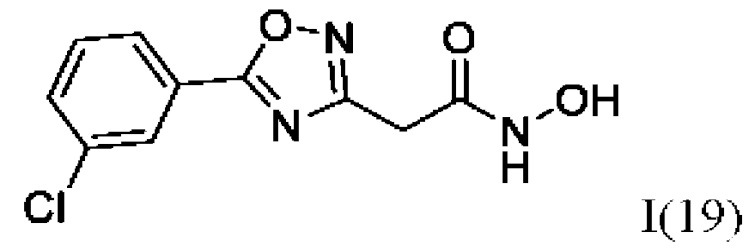
2-(5'-(3''-Хлорфенил)-1',2',4'-оксадиазол-3'-ил)-N-гидроксиацетамид (19) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
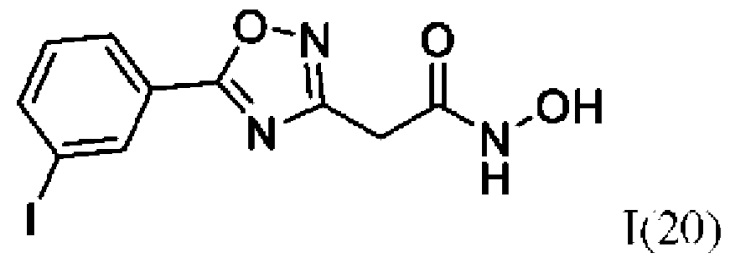
2-(5'-(3''-Йодфенил)-1',2',4'-оксадиазол-3'-ил)-N-гидроксиацетамид (20) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
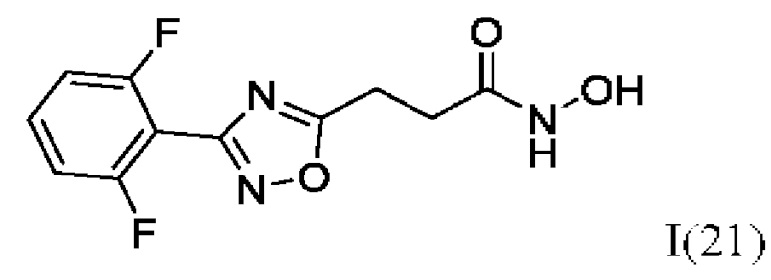
3-(3'-(2'',6''-Дифторфенил)-1',2',4'-оксадиазол-5'-ил)-N-гидроксипропанамид (21) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
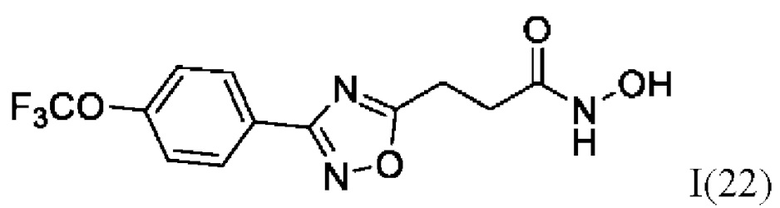
N-Гидрокси-3-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5'-ил)пропанамид (22) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
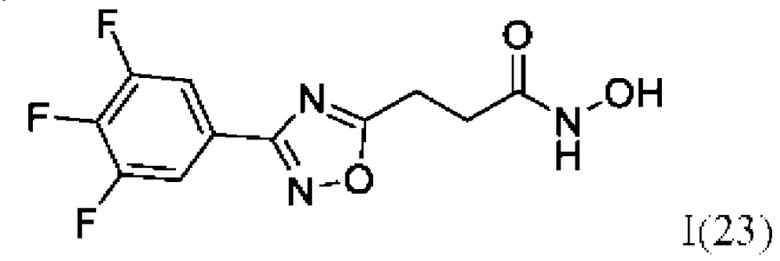
N-Гидрокси-3-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)пропанамид (23) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
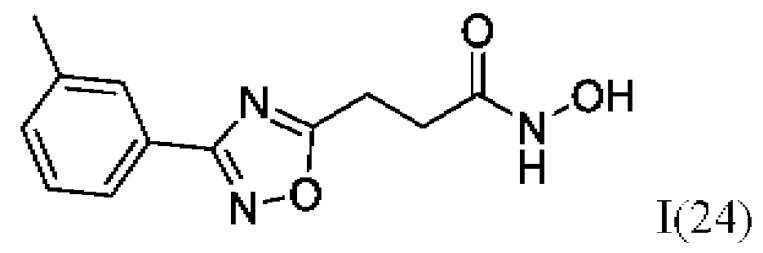
N-Гидрокси-3-(3'-(3''-метилфенил)-1',2',4'-оксадиазол-5,-ил)пропанамид (24) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
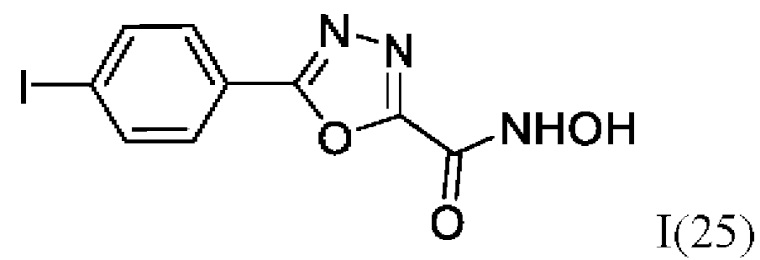
N-Гидрокси-5-(4-йодфенил)-1,3,4-оксадиазол-2-карбоксамид (25) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
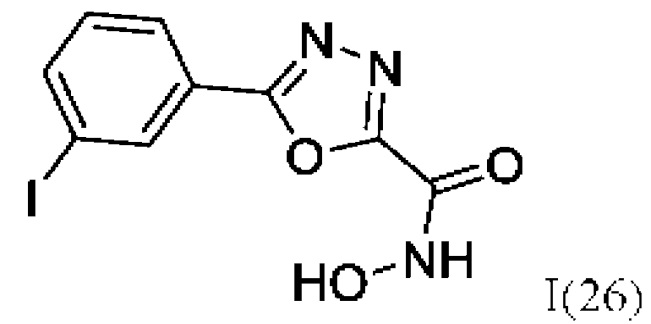
N-Гидрокси-5-(3-йодфенил)-1,3,4-оксадиазол-2-карбоксамид (26) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
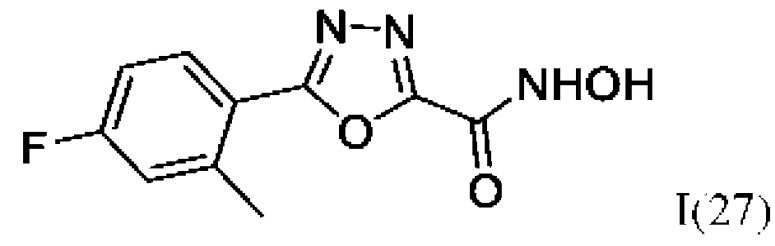
5-(4-Фтор-2-Метилфенил)-N-гидрокси-1,3,4-оксадиазол-2-карбоксамид (27) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
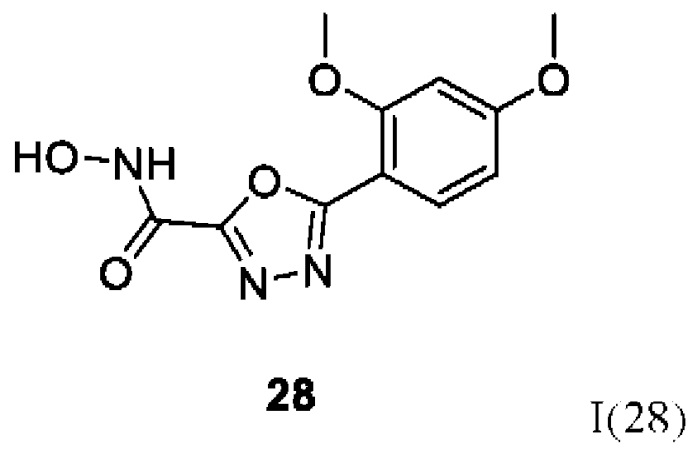
5-(2,4-Диметоксифенил)-N-гидрокси-1,3,4-оксадиазол-2-карбоксамид (28) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
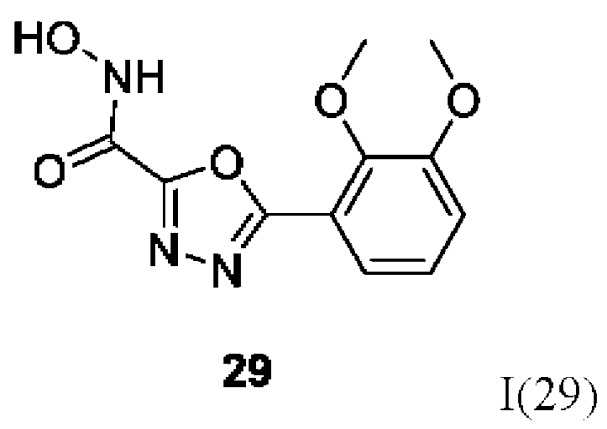
5-(2,3-Диметоксифенил)-N-гидрокси-1,3,4-оксадиазол-2-карбоксамид (29) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), имеющее формулу:
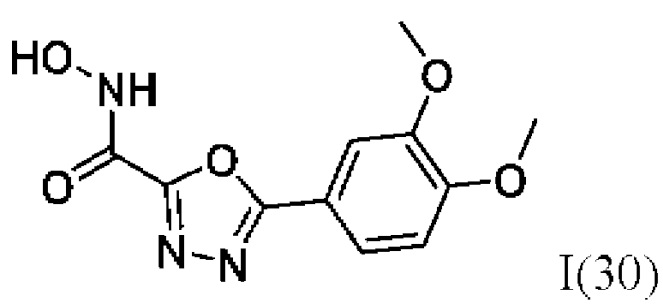
5-(3,4-Диметоксифенил)-N-гидрокси-1,3,4-оксадиазол-2-карбоксамид (30) или его фармацевтически приемлемая соль.
Еще в одном примере реализации представлено соединение по формуле (I), представляющее собой функциональное производное гидроксамовой кислоты, содержащей ядро дизамещенного 1,2,4-оксадиазола или ядро дизамещенного 1,3,4-оксадиазола, являющееся ингибитором фермента уридиндифосфат-3-O-(гидроксимиристоил)-N-ацетилглюкозаминдеацетилаза (LpxC).
Еще в одном примере реализации представлено соединение по формуле (I), представляющее собой ингибитор биосинтеза структурного липида А, компонента липополисахарида, являющегося основным компонентом клеточной стенки грамотрицательных бактерий.
Еще в одном примере реализации представлено соединение по формуле (I), обладающее эффективностью в потенцировании действия в отношении грамотрицательных бактерий антибиотиков, выбираемых из группы: налидиксовая кислота, рифампицин, канамицин.
Еще в одном примере реализации представлено соединение по формуле (I), обладающее антибактериальной активностью и применяемое для лечения заболеваний, вызываемых антибиотикорезистентными грамотрицательными бактериями, выбираемыми из группы: Escherichia coli MG1655, Pseudomonas aeruginosa АТСС 27853.
Множество соединений по примерам в соответствии с настоящим изобретением представлены в таблице 3.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Определения и общая терминология.
Далее будут приведены ссылки на определенные варианты осуществления изобретения, примеры которых проиллюстрированы прилагаемыми структурами и формулами. Подразумевается, что изобретение охватывает все альтернативы, модификации и эквиваленты, которые могут быть включены в объем настоящего изобретения, как определено в формуле изобретения. Специалисту в данной области известны многие способы и материалы, аналогичные или эквивалентные описанным в данном документе, которые могут быть использованы при практическом осуществлении настоящего изобретения. Настоящее изобретение не ограничивается изложенными в данном документе способами и материалами. В случае, если один или несколько из включенных литературных ссылок, патентов и аналогичных материалов отличается от или противоречит данной заявке, включая, в частности, определенные термины, использование терминов, описанные приемы и т.п., преимущественную силу имеет настоящая заявка.
Далее следует принять во внимание, что определенные признаки изобретения, которые для ясности описаны в контексте отдельных вариантов осуществления изобретения, могут быть представлены также совместно в одном варианте. И наоборот, разнообразные признаки изобретения, которые для краткости описаны в контексте одного варианта осуществления изобретения, могут быть представлены отдельно или в любой подходящей подкомбинации.
Для описания настоящего изобретения используются следующие термины.
Все технические и научные термины, использованные в данном документе, имеют такое значение, какое обычно понимается специалистами в области, к которой относится изобретение, если не указано иное. Все патенты и публикации, на которые имеются ссылки в данном документе, полностью включены в изобретение посредством ссылки.
В контексте данного документа будут применяться следующие определения, если не указано иное. В целях предложенного изобретения химические элементы обозначены в соответствии с Периодической таблицей элементов, версия CAS, и Справочником по химии и физике, 75-й выпуск, 1994 и т.д.
В тех случаях, когда термин конкретно не определен, этот термин имеет принятое специалистами в данной области значение и означает, что этот термин в данном контексте используется для описания настоящего изобретения.
Термин «соединение», как он используется в настоящем документе, если не указано иное, относится к любому конкретному химическому соединению, раскрытому в данном документе, включая таутомеры, где это применимо в контексте. Согласно его использованию в контексте, термин «соединение» обычно относится к единственному соединению.
Отмечается, что при описании настоящих соединений, среди прочих, описано множество заместителей и связанных с ними вариантов. Специалисту в данной области понятно, что молекулы, которые описаны в настоящем документе, представляют собой стабильные соединения, как это обычно предусматривается далее.
Термин «соединение-лидер», как он используется в настоящем документе, если не указано иное, относится к соединению, которое проявило необходимую фармакологическую активность в отношении определенной мишени (например, потенцирующую или антибактериальную), а также, вероятно, обладает другими благоприятными свойствами, например, низкой цитотоксичностью или ADME-свойствами, характерными для кандидата в лекарственный препарат.
Термин «пациент» используется в описании для обозначения животного, предпочтительно человека, или одомашненного животного, которому проводят лечение, включая профилактическое лечение, с помощью композиций в соответствии с настоящим изобретением. При лечении таких инфекций, состояний или заболеваний, которые являются специфическими для конкретного животного, такого как пациент-человек, термин «пациент» относится к конкретным животным, включая домашних животных, таких как собаки или кошки, грызуны, или сельскохозяйственных животных, таких как лошадь, корова, овца и т.д. В основном, в настоящем изобретении термин «пациент» относится к пациенту-человеку, если другое не указано или не подразумевается из контекста использования термина.
Термин «эффективный» используется для описания количества соединения, композиции или компонента, которое, при использовании в контексте его предназначения, приводит в предполагаемому результату. Термин «эффективный» охватывает все другие термины, относящееся к эффективному количеству или эффективным концентрациям, которые иным образом описаны или использованы в настоящей заявке.
Термин «заболевание» используется для описания какого-либо заболевания или состояния, где происходит дисрегуляция белка (то есть, количество белка, экспрессированного в организме пациента, повышается) и где деградация одного или более белков в организме пациента может обеспечить успешное лечение или облегчение симптомов пациенту, нуждающемуся в этом. В некоторых случаях заболевание или состояние может быть вылечено.
Термин «фармацевтически приемлемые соли» относится к органическим или неорганическим солям предложенного в настоящем изобретении соединения. Термин «фармацевтически приемлемая соль» используется в описании для обозначения, где это применимо, солевой формы одного или нескольких соединений, описанных в настоящем документе, которые предложены для увеличения растворимости соединения в желудочных соках желудочно-кишечного тракта пациента для способствования растворения и биодоступности соединений. Фармацевтически приемлемые соли включают полученные с фармацевтически приемлемыми неорганическими или органическими основаниями и кислотами, где это применимо. Подходящие соли включают соли, полученные с щелочными металлами, такими как калий и натрий, щелочноземельными металлами, такими как кальций, магний, и соли аммония, среди многочисленных других кислот и оснований, хорошо известных в фармацевтической области. Соли натрия и калия являются особенно предпочтительными в качестве солей нейтрализации фосфатов в соответствии с настоящим изобретением. Фармацевтически приемлемые соли хорошо известны специалистам. Например, S.M. Berge et al., подробно описывает фармацевтически приемлемые соли в публикации в журнале J. Pharmaceutical Sciences, 1977, 66: 1-19.
Таким образом, как описано в изобретении, предложенные соединения являются конкретными химическими соединениями, раскрытыми в данном документе, включая таутомерные формы.
Термин «таутомер» или «таутомерная форма» относится к структурным изомерам с различной энергией, которые являются взаимопревращаемыми с низким энергетическим барьером. В тех случаях, когда возможна таутомеризация (например, в растворе), может достигаться химическое равновесие таутомеров. Например, протонные таутомеры (называемые также прототропными таутомерами) включают взаимопревращения посредством миграции протона, такие как кето-енольная и имин-энаминная изомеризация. Таутомеры валентности включают взаимные превращения посредством реорганизации некоторых связывающих электронов. Все таутомерные формы соединений по настоящему изобретению входят в объем изобретения, если не указано иное.
Термин «арил» будет обозначать в своем контексте циклический и ароматический углеводородный радикал, предпочтительно Се, который может содержать заместители при углеродных атомах. Примерами арильных групп являются, среди прочих, фенил, нафтил, 3-метилфенил, 4-хлорфенил, 2-фторфенил и т.д.
Термин «алкил» будет обозначать в своем контексте циклический или ациклический углеводородный радикал, содержащий неисчислимое количество углеродных атомов. Примерами алкильных и циклоалкильных групп являются, среди прочих, метил и циклогексил, соответственно.
Термин «заместитель» подразумевает собой атом или группу атомов, связанных с упоминаемым ароматическим, алифатическим или гетероароматическим остовом посредством связи углерод-углерод или углерод-гетероатом. Примерами заместителей, среди прочих, являются фтор, фенил, 3,4,5-трифторфенил, циклогексил, 1,2,4-оксадиазолил и т.д.
Термин «гетероцикл» будет обозначать в своем контексте пятичленный или шестичленный циклический ароматический или неароматический остов, содержащий один или более гетероатомов (атомов отличных от углерода и водорода, например, азот и кислород). В контексте настоящего документа данный термин в основном относится к ароматическим кольцам 1,2,4- или 1,3,4-оксадиазола, содержащих заместители в положениях 3,5 или 2,5, соответственно. В то же время «ядром» гетероцикла будет обозначаться гетероциклический остов, как основа, не содержащая заместителей при атомах углерода или гетероатомах, например, 1,2,4-оксадиазол, 1,3,4-оксадиазол.
Термин «незамещенный» будет обозначать углеродный остов, замещенный только атомами водорода.
Термин «замещённый» (каждый заместитель является независимым от любого другого заместителя) также будет обозначать в своём контексте использование заместителей при углеродных атомах, отличных от атома водорода.
Термины «лечение», «лечить» или «подвергать лечению» и т.д., как используется в настоящем документе, относятся к какому-либо действию, обеспечивающему благоприятное воздействие на пациента, которому могут быть введены настоящие соединения. Заболевания или состояния, включая злокачественное новообразование, которые можно лечить с использованием соединений в соответствии с настоящим изобретением, перечислены выше.
Термин «гидроксамовая кислота» будет обозначать соединение из числа отображенных в изобретении, содержащее функциональную группу -C(O)NHOH, связанную с sp3 или sp2 гибридным атомом углерода.
Термин «антибиотикорезистентность» подразумевает собой частный случай устойчивости к противомикробным препаратам, когда бактерии становятся устойчивыми к антибиотикам.
Термин «потенциатор действия» или «потенциатор действия антибиотиков» подразумевает под собой химическое соединение из числа отображенных в изобретении, добавление которых совместно с антибиотиком приводит к уменьшению минимальной ингибирующей концентрации данного антибиотика в отношении грамотрицательных микроорганизмов. В то же время термин «потенцирование» означает увеличение активности соединения (например, антибиотика, в настоящем документе) в присутствии другого соединения (потенциатора, например, одного из заявляемых соединений).
Термин «ингибитор» подразумевает под собой химическое вещество, которое замедляет тот или иной химический или биохимический процесс за счет связывания с мишенью, в частности, в настоящем документе потенциальными «ингибиторами» являются заявляемые молекулы, которые, вероятно, связываются с ферментом LpxC, катализирующего биосинтез липополисахарида бактерий.
Термин «химическая модификация» подразумевает под собой направленное изменение химического состава указанной молекулы посредством химических превращений с целью синтеза новых соединений.
Термин «интермедиат» обозначает в контексте данного документа промежуточное вещество (не являющееся целевой, конечной молекулой) из которого далее синтезируют целевую молекулу в одну или несколько стадий.
Термин «сопряженный» обозначает в контексте данного документа связанный, посредством сопряжения - образования в структуре молекулы единого делокализованного электронного облака в результате перекрывания негибридизованных p-орбиталей.
Термин «функциональная группа» подразумевает собой структурный фрагмент (атом или группу атомов) указанной органической молекулы, определяющий ее химические свойства. В качестве функциональных групп, упоминаемых в контексте настоящего документа можно отметить функциональную группу гидроксамовой кислоты (-C(O)NHOH), наличие которой в молекуле определяет ее кислотные свойства, а также определенные химические свойства, например, способность к хелатированию ионов металла в активном сайте фермента.
Термин «линкер» обозначает в контексте настоящего документа атом или группу атомов, соединяющих два указанных фрагмента в единую молекулу. В частности, из числа упоминаемых в документе можно отметить «функциональная группа гидроксамовой кислоты, связанная через метиленовый линкер с положением 5 гетероцикла», обозначающее, что два фрагмента - гетероцикл и -C(O)NHOH связаны друг с другом посредством СН2-группы.
Для описания настоящего изобретения используются следующие обозначения.
ADMET - «Absorption, Distribution, Metabolism, Excretion, and Toxicity)) (Абсорбция, Распределение, Метаболизм, Экскреция и Токсичность)
CAS - chemical abstracts service (химическая реферативная служба)
СС50 - 50%-cytotoxicity concentration (цитотоксическая концентрация)
CDI - N,N-carbonyldiimidazole (N,N'-карбонилдиимидазол)
CLSI - Clinical and Laboratory Standards Institute (институт клинических и лабораторных стандартов)
DIPEA - diisopropylethylamine (диизопропилэтиламин)
DMAP - 4-Dimethylaminopyridine (4-диметиламинопиридин)
DMEM (ДМЕМ) - Dulbecco's Modified Eagle Medium
ESI - electrospray ionization (ионизация электрораспылением)
Et3N - triethylamine (триэтиламин)
EtOAc - ethyl acetate (этилацетат)
HEK - Human Embryonic Kidney (эмбриональная почка человека)
HRMS - high-resolution mass spectra (масс-спектрометрия высокого разрешения)
Km - kanamycin (канамицин)
LpxC -UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (УДФ-3-O-(R-3-гидроксимиристоил)-N-ацетилглюкозамин деацетилаза)
NADPH - Nicotinamide adenine dinucleotide (Никотинамидадениндинуклеотид)
NaI - nalidixic acid (налидиксовая кислота)
OD - optical density (оптическая плотность)
Rif- rifampicin (рифампицин)
SGF - simulated gastric fluid (искусственный желудочный сок)
SIF - Simulated Intestinal Fluid (искусственный кишечный сок)
TBAF - tetrabutylammonium fluoride (тетрабутиламмоний фторид)
TsCl - Tosyl chloride (пара-толуолсульфонилхлорид)
UDP - Uridine diphosphate (уридин дифосфат)
UDPGA - Uridine Diphosphate Glucuronic Acid (уридиндифосфатглюкуроновая кислота)
ВОЗ - всемирная организация здравоохранения
ВЭЖХ - высокоэффективная жидкостная хроматография
ДМСО - диметилсульфоксид
ДМФА - диметилформамид
ИЮПАК (IUPAC) - International Union of Pure and Applied Chemistry (Международный союз теоретической и прикладной химии)
КОЕ - Колониеобразующая единица
ЛПС - липополисахарид
МИК - минимальная ингибирующая концентрация
ТГФ - тетрагидрофуран
ТСХ - тонкослойная хроматография
УДФ - уридин дифосфат
ЭДТА - Этилендиаминтетрауксусная кислота
ЯМР - ядерно-магнитный резонанс
ОБЩИЕ СИНТЕТИЧЕСКИЕ ПОДХОДЫ
Далее описана общая схема синтеза заявленных соединений. Немаловажным является то, что синтез предлагаемых ингибиторов LpxC характеризуется значительным упрощением, принимая во внимание количество синтетических стадий, отсутствие стереоцентров и коммерческую доступность исходных материалов, по сравнению с известными ингибиторами LpxC.
Вкратце, соединения по настоящему изобретению синтезируют в соответствии с общими схемами, как представлено ниже.
Общим подходом к синтезу заявленных соединений 1(1-30) является - сборка ядер 1,2,4- или 1,3,4-оксадиазола из амидоксимов или гидразидов, соответственно, с образованием соответствующих оксадиазолил-замещенных сложных эфиров и их последующим взаимодействием с избытком гидроксиламина напрямую, либо через стадию получения соответствующих карбоновых кислот.
Общий подход к синтезу предлагаемых ингибиторов LpxC, содержащих функциональную группу гидроксамовой кислоты, сопряженную с положением 5 ядра 1,2,4-оксалиазола, сводится к нуклеофильному присоединению гидроксиламина к замещенным бензонитрилам с последующей гетероциклизацией полученных амидоксимов в 1,2,4-оксадиазолы действием этил оксалилхлорида и превращением в целевые кислоты действием избытка гидроксиламина по сложноэфирной группе (Фигура 1).
Общий подход к синтезу гидроксамовых кислот, связанных с положением 5 ядра 1,2,4-оксадиазола, заключается во взаимодействии монометилового эфира малоновой кислоты с рядом амидоксимов с образованием О-ациламидоксимов, с последующей их гетероциклизацией в 1,2,4-оксадиазолы и реакцией последних с избытком гидроксиламина (Фигура 2).
Общий подход к синтезу гидроксамовых кислот, связанных с ядром 1,2,4-оксадиазола ароматическим линкером заключался в той же синтетической последовательности, что и на фигуре 2, за исключением стартового соединения - монометилового эфира изофталевой кислоты. В данном случае, полученные сложные эфиры, содержащие ядро 1,2,4-оксадиазола, подвергались гидролизу до соответствующих карбоновых кислот, которые далее переводили в целевые гидроксамовые кислоты общей формулы I (Фигуры 3-5).
Общий подход к синтезу гидроксамовых кислот, связанных с положением 3 ядра 1,2,4-оксадиазола через метиленовый линкер, заключался во взаимодействии продукта нуклеофильного присоединения гидроксиламина к цианоуксусному эфиру с рядом замещенных бензойных кислот с образованием О-ациламидоксимов и их последующей гетероциклизацией в сложные эфиры, содержащие ядро 1,2,4-оксадиазола. На заключительной стадии, аналогично предыдущим примерам, сложные эфиры превращали в гидроксамовые кислоты действием избытка гидроксиламина (Фигура 6).
Общим подходом к синтезу целевых гидроксамовых кислот, содержащих функциональную группу гидроксамовой кислоты, связанную с ядром 1,2,4-оксадиазола через этиленовый линкер, являлось взаимодействие ангидрида яблочной кислоты с рядом амидоксимов в условиях без растворителя, с образованием пропионовых кислот, содержащих ядро 1,2,4-оксадиазола, которые далее последовательно переводили в соответствующие метиловые эфиры и целевые гидроксамовые кислоты (Фигура 7).
Общим подходом к получению гидроксамовых кислот, функциональная группа которых сопряжена с ядром 1,3,4-оксадиазола, являлось взаимодействие сложных эфиров замещенных бензойных кислот с гидразином, в результате которого получали ряд гидразидов, которые последовательно гетероциклизовали под действием этил оксалилхлорида и переводили в целевые гидроксамовые кислоты аналогично вышеописанным подходам (Фигура 8).
Все полученные вышеописанными подходами соединения общей формулы I (1-30) исследовались далее в качестве потенциаторов действия антибиотиков в отношении Escherichia coli MG1655. Обнаружено, что пять соединений общей формулы I обладают высокой эффективностью в потенцировании действия налидиксовой кислоты, рифампицина и канамицина на рост клеточной культуры Escherichia coli MG1655 (Фигуры 10-11). Дополнительно были исследованы цитотоксические свойства соединений 1-30, результаты представлены в таблице 4. Для выделенных пяти соединений-лидеров (Фигура 9) были исследованы ADME-свойства (Таблицы 5-11). На основании полученных данных по цитотоксичности соединений общей формулы I, а также их потенцирующего действия были выбраны два соединения-лидера для дальнейших исследований (Фигура 12), обладающие наивысшей эффективностью в потенцировании антибиотиков в сочетании с низкой токсичностью и наилучшими ADME-свойствами. Выбранные соединения-лидеры исследовались в качестве потенциаторов действия налидиксовой кислоты и рифампицина в отношении клеточной линии Pseudomonas aeruginosa АТСС 27853. Данные соединения значительно снижали рост P. aeruginosa АТСС 27853 и снижали МИК тестируемых антибиотиков при совместном использовании в сочетании с исследуемыми соединениями (Фигура 13). Значение МИК налидиксовой кислоты в присутствии 100 мкМ исследуемых соединения снижалось примерно в 8 раз с 4 мкг/мл до 0,5 мкг/мл. Комбинация 50 мкМ соединения-лидера 9 (Фигура 12) с 0,5 мкг налидиксовой кислоты полностью ингибировала рост Р aeruginosa в течение 14 часов, тогда как добавление 50 мкМ соединения-лидера 10 к такому же количеству налидиксовой кислоты подавляло рост культуры только в течение 8 часов (Фигура 13). Отличные результаты были показаны при исследовании эффективности потенцирования рифампицина - соединения 9 и 10 одинаково эффективно снижали МИК рифампицина в 64 раза с 4 мкг/мл до 0,0625 мкг/мл при обеих концентрациях 100 мкМ и 50 мкМ (Фигура 13).
Таким образом, соединения общей формулы I представляют собой новый хемотип потенциальных ингибиторов биосинтеза ЛПС бактерий, обладающих эффективностью в потенцировании действия различных антибиотиков в отношении грамотрицательных бактерий, которая подтверждена экспериментами in vitro. Заявленные соединения общей формулы I обладают хорошими ADME-свойствами и показателями цитотоксичности, и, во многом, по данным свойствам превосходят уже известные ингибиторы LpxC, что делает перспективным дальнейшие исследования данного хемотипа, включающие в себя структурно-ориентированный дизайн новых потенциальных ингибиторов с еще большей эффективностью в потенцировании действия антибиотиков с одновременным сохранением цитотоксических и ADME-свойств. Представленные в настоящем изобретении соединения обладают значительным потенциалом в разработке новых эффективных антибактериальных препаратов, в том числе в разработке потенциаторов уже известных антибиотиков, и, таким образом, они обладают ценностью для научного сообщества в борьбе с антибиотикорезистентностью.
Все задействованные реактивы были получены из коммерческих источников и использовались без дополнительной очистки. Растворители были подготовлены к использованию согласно распространенным методикам (перегонка над подходящим осушителем).
Спектры ядерно-магнитного резонанса ЯМР 1Н и 13С зарегистрированы на спектрометрах 300 МГц (300,1, 75,5 МГц соответственно) и 400 МГц (400,1, 100,6 МГц соответственно) (Bruker Bio Spin GmbH и Bruker Avance Neo, соответственно) в растворах CDCl3, ДМСО-d6 и CD3OD с использованием 0,05% (H3C)4Si в качестве внешнего или внутреннего стандарта. Химические сдвиги 5 выражены в миллионных долях (ppm); кратность: с, синглет; д, дублет; т, триплет; к, квартет; дд, дублет дублетов; тт, триплет триплетов; дт, дублет триплетов; тд, триплет дублетов; ддд, дублет дублетов дублетов; м, мультиплет; уш.с., уширенный синглет; константы спин-спинового взаимодействия J указаны в герцах [Гц]. Структура продуктов определена путем анализа спектров ЯМР 1Н и 13С с привлечением двумерных {1Н-13С} корреляционных ЯМР спектров (HSQC, НМВС). ВЭЖХ-МС анализ проводили на хроматографе «Vanquish Flex» (Thermo Scientific, США) с диодно-матричным детектором «DAD FG» (Thermo Scientific, США) и одноквадрупольным масс-спектрометрическим детектором «ISQ ЕМ» (Thermo Scientific, США). Разделение определяемых соединений проводили на колонке Agilent Poroshell 120 ЕС-С18 (100 мм-2,0 мм, размер частиц 1,9 мкм) в следующих хроматографических условиях: с 0 по 7 мин -элюирование от 5% до 95% подвижной фазы Б, с 7 по 13 мин - элюирование в 95% подвижной фазы Б, с 13 по 17 мин - уравновешивание хроматографической колонки в 95% подвижной фазы А. Подвижная фаза А - 0,1%) муравьиная кислота в воде, подвижная фаза Б - 0,1%) муравьиная кислота в ацетонитриле. Температура колонки - 40°С. Объем вводимой пробы - 1 мкл, скорость потока - 0,3 мл/мин. Для ионизации образцов использовали источник ионизации электрораспылением. Детектировали ионы положительной полярности в режиме регистрации полного ионного тока, диапазон регистрируемых масс 50-700 m/z. Регистрацию спектров поглощения проводили на диодно-матричном детекторе на 4 длинах волн: 210 нм, 240 нм, 280 нм, 300 нм. Масс-спектры высокого разрешения (HRMS) регистрировали с использованием масс-спектрометра Bruker maXis II 4G ETD и хроматографа UltiMate 3000, оснащенного колонкой Acclaim RSLC 120 С18 (2,2 мкм, 2,1-100 мм), Режим регистрации спектра: ионизация электрораспылением (ESI) с полным сканированием в диапазоне m/z от 100 до 1500, тандемная регистрация масс-спектра (МС/МС) с выбором трех наиболее интенсивных молекулярных ионов; диссоциация, индуцированная столкновениями (CID) при 10-40 эВ; и азот в качестве газа столкновения. Температуры плавления определяли на приборе Melting Point Apparatus SMP50. Для лиофилизации веществ использовали компактную настольную лиофильную сушку LABCONCO FreeZone 2,5L (для этого образец предварительно замораживали в морозильной камере на -80°С в течение 1 ч, сублимация проводилась 12 ч, остаточное давление 0,003 мбар). Для ТСХ анализа применяли хроматографические пластины Merck TLC Silica gel 60 F254, детектирование веществ производили при помощи УФ-лампы на длине волны 254 нм (SPECTROLINE, СМ-10А, USA), стандартными растворами для проявления (нингидрина, перманганата калия и фосфорномолибденовой кислоты).
Колоночную хроматографию осуществляли на силикагеле (60-200 mesh, Sisco).
Каждый способ представлен со ссылкой на конкретное соединение или группу соединений, подробные детали синтеза которого представлены выше, далее и на фигурах 1-8. Все пронумерованные соединения 1-30 в рамках настоящего изобретения (см. таблицу 3), входящие в предлагаемую структуру Маркуша, обозначенную формулой (I), а также интермедиаты их синтеза могут быть синтезированы относительно легко, используя напрямую способы, которые были изложены. В некоторых случаях для некоторых предпочтительных вариантов осуществления представлено больше подробностей синтеза, чтобы предоставить такие сведения, которые могут служить в качестве модели для синтеза ряда других соединений, описанных в данном документе.
Следуя общим способам синтеза, представленным далее и как описано выше, следующие соединения были синтезированы по аналогии в соответствии с установленным общим способом, очищены стандартными хроматографическими способами и охарактеризованы данными 1Н и 13С ЯМР, ВЭЖХ-МС, HRMS, показателями Тпл. Примеры соединений I(1-30) представлены в таблице 3.
На фигуре 9 представлены соединения-лидеры, обладающие эффективностью в потенцировании действия антибиотиков в отношение клеточной линии Е. coli MG1655, из представленных в таблице 3 в соответствии с настоящим изобретением. На фигурах 10-11 представлены репрезентативные кривые роста Е. coli MG1655 в присутствии антибиотиков без или с добавлением соединений-лидеров данного изобретения. На фигуре 12 представлены соединения-лидеры, обладающие наивысшей эффективностью в потенцировании действия антибиотиков в отношении Е. coli MG1655 и наименьшей цитотоксичностью (данные представлены в таблице 4). На фигуре 13 представлены репрезентативные кривые роста клеточной линии P. aeruginosa АТСС 27853 в присутствии антибиотиков без или с добавлением соединений-лидеров данного изобретения, изображенных на фигуре 12
Притязания по формуле изобретения подтверждаются приведенными примерами реализации заявляемого изобретения, но не ограничиваются ими.
ПРИМЕРЫ
Пример 1. Получение 2,6-дифторо-N'-гидроксибензимидамида
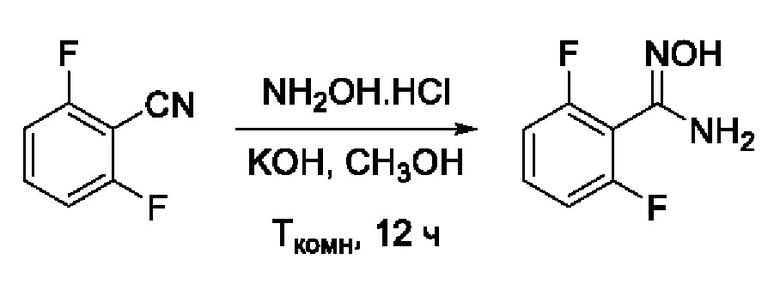
Навеску NH2OH⋅HCl (375 мг, 5,4 ммоль, 2 экв) растворяли в 5 мл СН3ОН в круглодонной колбе. К образовавшемуся раствору добавляли раствор KOH (378 мг, 6,8 ммоль, 2,5 экв) в 5 мл СН3ОН. Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 10 мин, после чего добавляли раствор 2,6-дифторбензонитрила (375 мг, 2,7 ммоль, 1 экв) в 5 мл СН3ОН. Образовавшуюся реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 12 ч. После окончания реакции осадок отфильтровывали, растворитель удаляли на роторном испарителе, твердый остаток растворяли в 40 мл EtOAc. Полученный раствор последовательно обрабатывали деионизированной водой (2⋅15 мл) и насыщенным раствором хлорида натрия (1⋅15 мл). Органический слой сушили над безводным Na2SO4 при интенсивном перемешивании, после чего осадок отфильтровывали, а фильтрат упаривали досуха с получением 2,6-дифторо-N'-гидроксибензимидамида (460 мг, 90%). Желтоватый порошок. ВЭЖХ-МС (ESI+): найдено m/z 173,1 [М+Н]+; вычислено C7H7F2N2O+ 173,0.
Пример 2. Получение N'-гидрокси-4-(трифторметокси)бензимидамида
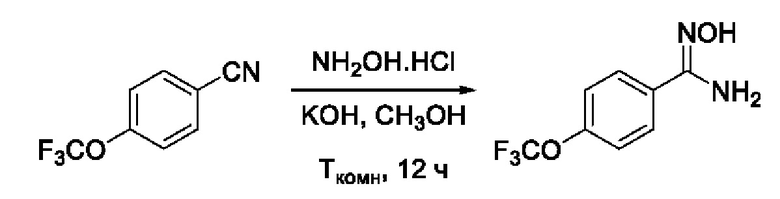
Получен способом, описанном в Примере 1 (690 мг, 95%). ВЭЖХ-МС (ESI+): найдено m/z 221,1 [М+Н]+; вычислено C8H8F3N2O2+ 221,1.
Пример 3. Получение 3,4,5-трифтор-N'-гидроксибензимидамида
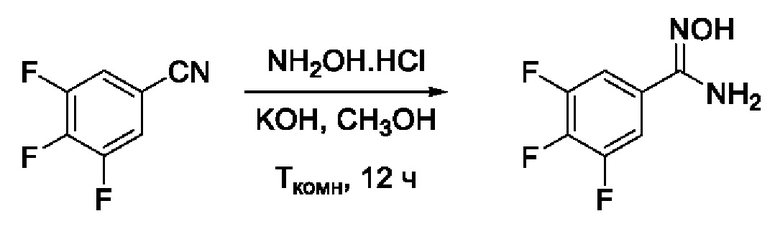
Получен способом, описанном в Примере 1 (320 мг, 90%). ВЭЖХ-МС (ESI+): найдено m/z 191,1 [М+Н]+; вычислено C7H6F3N2O+ 191,0.
Пример 4. Получение N'-гидрокси-3-метилбензимидамида
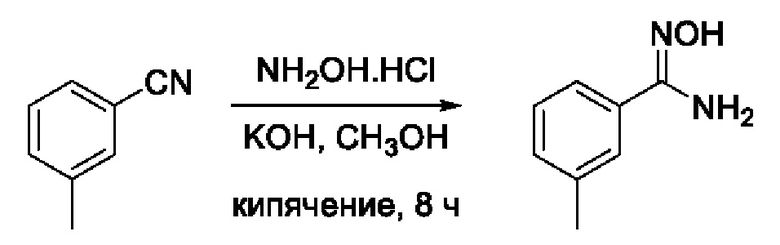
Навеску NH2OH⋅HCl (278 мг, 4 ммоль, 4 экв) растворяли в 10 мл СН3ОН в круглодонной колбе. К образовавшемуся раствору добавляли раствор KOH (280 г, 5 ммоль, 5 экв) в 10 мл СН3ОН. Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 10 мин, после чего добавляли раствор 3,4,5-трифторбензонитрила (117 мг, 1 ммоль, 1 экв) в 5 мл СН3ОН. Образовавшуюся реакционную смесь кипятили с обратным холодильником в течение 8 ч, ход реакции контролировали ТСХ (элюент CH2Cl2:изо-С3Н7ОН=19:1). После окончания реакции осадок отфильтровывали, растворитель удаляли на роторном испарителе, твердый остаток растворяли в 40 мл EtOAc. Полученный раствор последовательно обрабатывали деионизированной водой (2-15 мл) и насыщенным раствором хлорида натрия (1-15 мл). Органический слой сушили над безводным Na2SO4 при интенсивном перемешивании, после чего осадок отфильтровывали, а фильтрат упаривали досуха с получением 3,4,5-трифторо-N'-гидроксибензимидамида (150 мг, 90%). Желтоватый порошок. ВЭЖХ-МС (ESI+): найдено m/z 151,1 [М+Н]+; вычислено C8H11N2O+ 151,1.
Пример 5. Получение N'-гидрокси-4-метоксибензимидамида
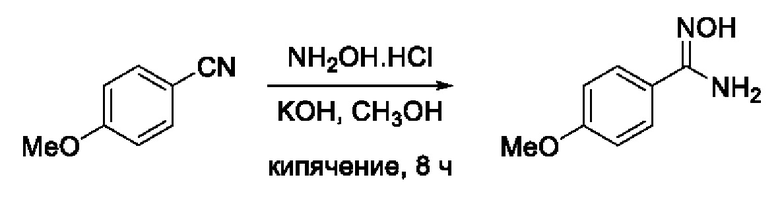
Получен способом, описанном в Примере 4 (620 мг, 90%). ВЭЖХ-МС (ESI+): найдено m/z 167,1 [М+Н]+; вычислено C8H11N2O2+ 167,1.
Пример 6. Получение N'-гидрокси-4-йодобензимидамида
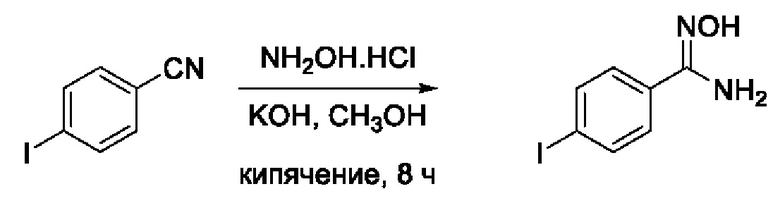
Получен способом, описанном в Примере 4 (260 мг, 80%). ВЭЖХ-МС (ESI+): найдено m/z 263,0 [М+Н]+; вычислено C7H8IN2O+ 263,0.
Пример 7. Получение этил 3-амино-3-(гидроксиимино)пропаноата
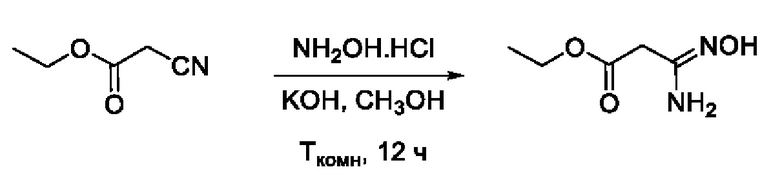
Навеску NH2OH⋅HCl (2 г, 28,8 ммоль, 1,2 экв) растворяли в 10 мл СН3ОН в кругло донной колбе. К полученному раствору добавляли раствор KOH (1,6 г, 28,8 ммоль, 1,2 экв) в 10 мл СН3ОН. Реакционную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 10 мин. После завершения реакции осадок отфильтровывали и к фильтрату добавляли раствор этил 2-цианоацетата (2,55 мл, 24 ммоль, 1 экв) в 5 мл СН3ОН. Реакционную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 12 ч. После завершения реакции раствор упаривали досуха с получением этил 3-амино-3-(гидроксиимино)пропаноата в виде коричневого масла, который использовали на следующей стадии без очистки.
Пример 8. Получение монометилового эфира мало новой кислоты
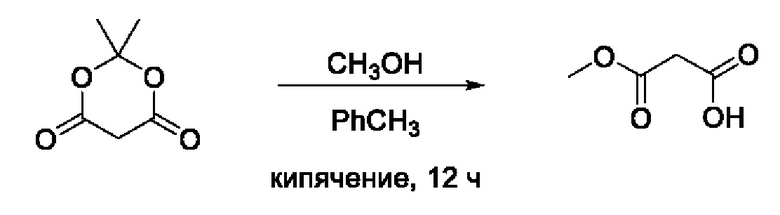
Кислоту Мельдрума (1,44 г, 10 ммоль, 1 экв) растворяли в 30 мл толуола в кругло донной колбе. Затем добавляли СН3ОН (4 мл, 100 ммоль, 10 экв) и полученную смесь кипятили с обратным холодильником в течение 12 ч, ход реакции контролировали методом ТСХ (элюент CH2Cl2:изо-С3Н7ОН:HCOOH=9:1:0,01) с использованием кислоты Мельдрума (Rƒ=0,9) и мало новая кислота (Rƒ=0,1) в качестве свидетелей. После завершения реакции раствор упаривали досуха, получая 3-метокси-3-оксопропановую кислоту (Rƒ=0,7) в виде бесцветной жидкости, 1,12 г (95%). Полученный эфир в дальнейшем использовали на следующем этапе без очистки и характеристики физико-химическими методами анализа.
Пример 9. Получение метил 3-(((амино(2',6'-дифторфенил)метилен)амино)окси)-3-оксопропаноата
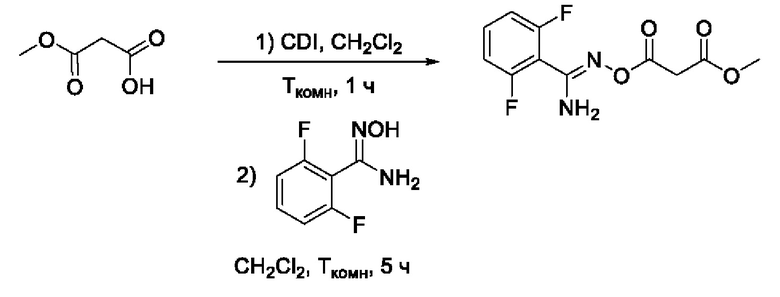
Смесь монометилового эфира малоновой кислоты (1,0 экв) и N,N'-карбонилдиимидазола (CDI) (1,2 экв) растворяли в 10 мл CH2Cl2 и раствор перемешивали при комнатной температуре в атмосфере воздуха в течение 1 ч. Затем добавляли раствор соответствующего 2,6-дифтор-N-гидроксибензимидамида (1,2 экв) в 10 мл CH2Cl2; полученный раствор перемешивали при комнатной температуре в атмосфере воздуха в течение 5 ч, ход реакции контролировали методом ТСХ (элюент CH2Cl2:изо-С3Н7ОН = 19:1). После завершения реакции раствор упаривали досуха и остаток очищали колоночной хроматографией на силикагеле (элюент CH2Cl2:iso-C3H7OH с градиентом от 100:0 до 95:5). Объединенные органические фракции, содержащие целевой метил 3-(((амино(2',6'-дифторфенил)метилен)амино)окси)-3-оксопропаноат, собирали, раствор упаривали досуха с получением соответствующего О-ациламидоксима (245 мг, 70%). Белый порошок, 1Н ЯМР (300 МГц, CDCl3, δ): 7,47-7,35 (1H, м, 4'-СН), 7,02-6,93 (2Н, м, 3'-СН, 5'-СН), 5,43 (2Н, уш.с., 5-NH2), 3,78 (3Н, с, 6-СН3), 3,60 (2Н, с, 2-СН2). 13С ЯМР (75 МГц, CDCl3, δ): 167,13 (С-1), 164,10 (С-3), 160,78 (С-2', С-6', dd, JF=254.1, 6.1 Гц), 149,37 (С-4), 132,53-131,96 (С-4', т), 112,28-111,75 (С-31, С-5', м), 109,10 (С-1'), 52,66 (С-6), 40,38 (С-2). ВЭЖХ-МС (ESI+): найдено m/z 273,1 [М+Н]+; вычислено C11H11F2N2O4+ 273,1.
Пример 10. Получение метил 3-(((амино(4'-(трифторметокси)фенил) метилен)амино)окси)-3-оксопропаноата
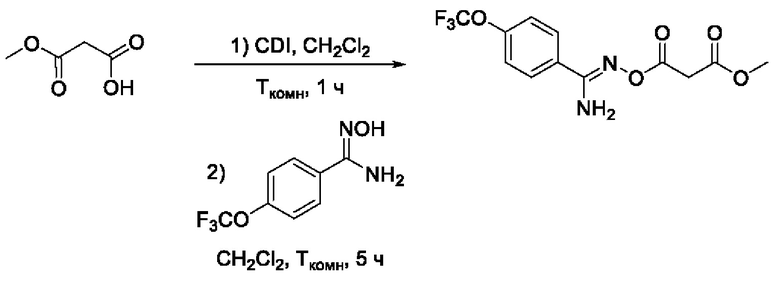
Получен способом, описанном в Примере 9 из монометилового эфира малоновой кислоты и 4-трифторметокси-N-гидроксибензимидамида в виде желтого порошка с выходом 250 мг, 80%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 95:5). 1Н ЯМР (400 МГц, CDCl3, δ): 7,68-7,64 (2Н, м, 2''-СН, 6''-СН), 7,18-7,17 (2Н, м, 3''-СН, 5''-СН), 5,30 (2Н, уш.с., 5-NH2), 3,78 (3Н, с, 6-СН3), 3,60 (2Н, с, 2-СН2). 13С ЯМР (101 МГц, CDCl3, δ): 167,35 (С-1), 164,45 (С-3), 156,48 (С-4), 151,28 (С-4'), 129,29 (С-1'), 128,53 (С-2'', С-6''), 120,97 (С-3'', С-5''), 119,02 (С-7), 52,71 (С-6), 40,50 (С-2). ВЭЖХ-МС (ESI+): найдено m/z 321,0 [М+Н]+; вычислено C12H12F3N2O5+ 321,1.
Пример 11. Получение метил 3-(((амино(3',4',5'-трифторфенил)метилен)амино)окси)-3-оксопропаноата
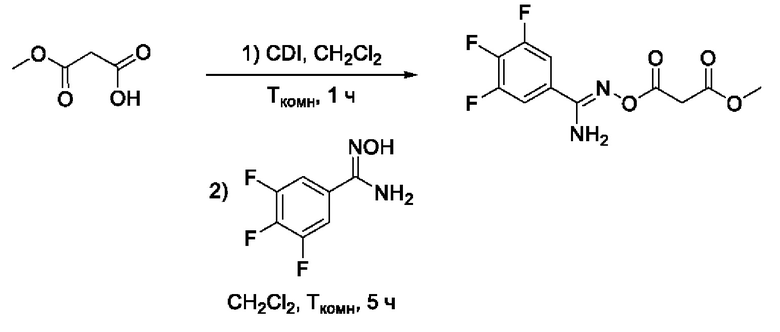
Получен способом, описанном в Примере 9 из монометилового эфира малоновой кислоты и 3,4,5-трифтор-N-гидроксибензимидамида в виде желтоватого порошка с выходом 320 мг, 80%. Элюент для очистки колоночной хроматографией-CH2Cl2:изо-C3H7OH (градиент от 100:0 до 95:5). 1Н ЯМР (300 МГц, CDCl3, δ): 7,45-7,34 (2Н, м, 2'-CH, 6'-СН), 5,44 (2H, уш.с., 5-NH2), 3,80 (3Н, с, 6-СН3), 3,61 (2Н, с, 2-СН2). 13С ЯМР (75 МГц, CDCl3, δ): 167,24 (С-1), 164,17 (С-3), 155,08 (С-4), 151,25 (С-3', С-5', ддд, JF=251.6, 10.1, 3.8 Гц), 141,59 (С-4', дт, JF=257.1, 15.2 Гц), 111,69-111,38 (С-2', С-6', м), 110,39-110,08 (С-1', м), 52,75 (С-6), 40,40 (С-2). ВЭЖХ-МС (ESI+): найдено m/z 291,0 [М+Н]+; вычислено C11H10F3N2O4+ 291,1.
Пример 12. Получение метил 3-(((амино(3'-метилфенил)метилен)амино)окси)-3-оксопропаноата
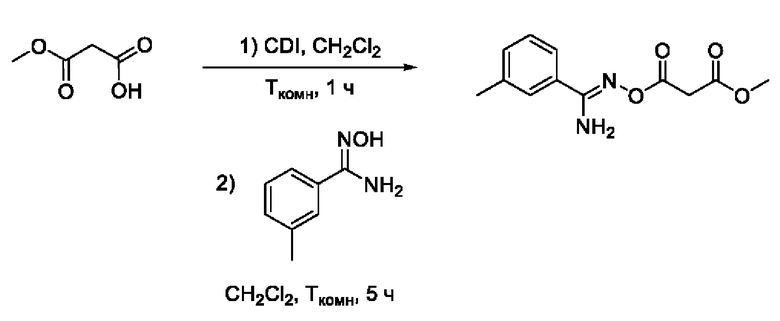
Получен способом, описанном в Примере 9 из монометилового эфира малоновой кислоты и 3-метил-N-гидроксибензимидамида в виде белого порошка с выходом 190 мг, 70%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 95:5). 1Н ЯМР (400 МГц, CDCl3, δ): 7,53 (1H, уш.с., 2'-СН), 7,48-7,44 (1H, м, 5'-СН), 7,32-7,30 (2Н, м, 4'-СН, 6'-СН), 5,31 (2H,уш.с., 5-NH2), 3,79 (3Н, с, 6-СН3), 3,62 (2Н, с, 2-СН2), 2,39 (3Н, с, 7-СН3). 13С ЯМР (101 МГц, CDCl3, δ): 167,38 (С-1), 164,62 (С-3), 157,70 (С-4), 138,65 (С-3'), 131,97 (С-4'), 130,61 (С-1'), 128,65 (С-6'), 127,40 (С-2'), 123,66 (С-5'), 52,66 (С-6), 40,59 (С-2), 21,30 (С-7). ВЭЖХ-МС (ESI+): найдено m/z 251,1 [М+Н]+; вычислено C12H15N2O4+ 251,1.
Пример 13. Получение метил 3-((((амино(2',6'-дифторфенил)метилен)амино)окси)карбонил)бензоата
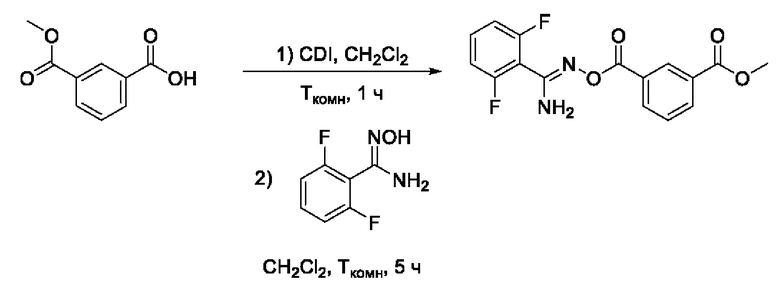
Получен способом, описанном в Примере 9 из монометилового эфира изофталевой кислоты и 2,6-дифтор-N-гидроксибензимидамида в виде желтоватого порошка с выходом 200 мг, 60%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 98:2). 1H ЯМР (300 МГц, ДМСО-d6, δ): 8,62-8,61 (1H, м, 2-СН), 8,55-8,51 (1Н, м, 4-СН), 8,25-8,21 (1H, м, 6-СН), 7,75-7,69 (1H, м, 5-СН), 7,65-7,58 (1H, м, 4'-СН), 7,35 (2Н, уш.с., 11-NH2), 7,29-7,21 (2H, м, 3'-СН, 5'-СН), 3,92 (3Н, с, 8-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 166,03 (С-7), 163,01 (С-9), 160,60 (С-2', С-6', дд, JF=250.0, 6.7 Гц), 150,02 (С-10), 134,54 (С-4), 133,88 (С-6), 132,96 (С-4', т, JF=10.2 Гц), 130,63 (С-1), 130,47 (С-2), 130,31 (С-3), 129,76 (С-5), 112,51-112,11 (С-3', С-5', м), 110,75 (С-1', т, JF=20.7 Гц), 52,90 (С-8). ВЭЖХ-МС (ESI+): найдено m/z 335,1 [М+Н]+; вычислено C16H13F2N2O4+ 335,1
Пример 14. Получение метил 3-((((амино(4'-(трифторметокси)фенил)метилен)амино)окси)карбонил)бензоата
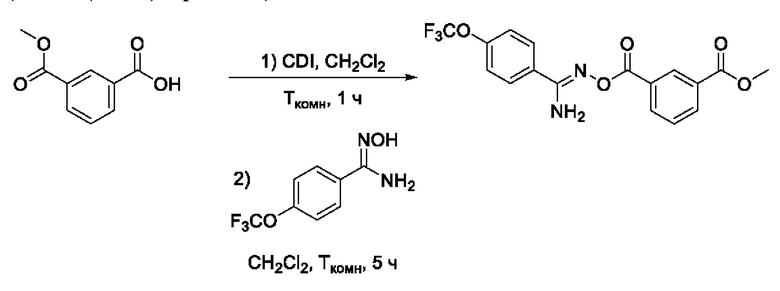
Получен способом, описанном в Примере 9 из монометилового эфира изофталевой кислоты и 4-трифторметокси-N-гидроксибензимидамида в виде белого порошка с выходом 420 мг, 85%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 98:2). 1Н ЯМР (400 МГц, CDCl3, δ): 8,64-8,63 (1Н, м, 2-СН), 8,22-8,17 (2Н, м, 4-СН, 6-СН), 7,75-7,72 (2Н, м, 2'-СН, 6'-СН), 7,50 (1H, т, J=7,8 Гц, 5-СН), 7,21-7,19 (2Н, м, 3'-СН, 5'-СН), 5,25 (2Н, уш.с., 11-NH2), 3,89 (3Н, с, 8-СН3). 13С ЯМР (101 МГц, CDCl3, δ): 166,17 (С-7), 163,13 (С-9), 156,27 (С-10), 151,25 (С-4'), 133,96 (С-6), 133,85 (С-4), 130,71 (С-1), 130,39 (С-2), 129,91 (С-3), 129,60 (С-1'), 128,87 (С-5), 128,67 (С-2', С-6'), 120,99 (С-3', С-5'), 119,04 (С-12), 52,45 (С-8). ВЭЖХ-МС (ESI+): найдено m/z 383,1 [М+Н]+; вычислено C17H14F3N2O5+ 383,1.
Пример 15. Получение метил 3-((((амино(3',4',5'-трифторфенил) метилен)амино)окси)карбонил)бензоата
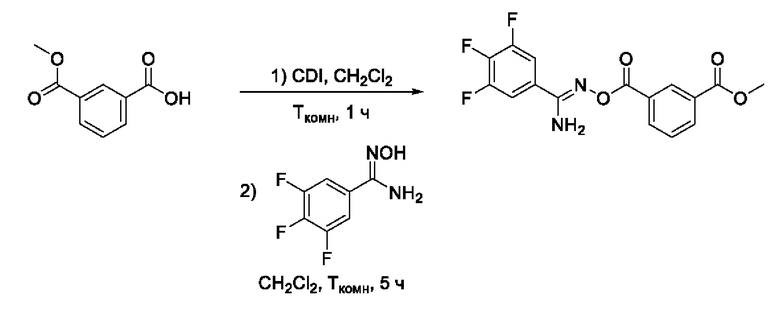
Получен способом, описанном в Примере 9 из монометилового эфира изофталевой кислоты и 3,4,5-трифтор-N-гидроксибензимидамида в виде белого порошка с выходом 175 мг, 85%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 98:2). 1Н ЯМР (300 МГц, CDCl3, δ): 8,87-8,86 (1H, м, 2-СН), 8,39 (1H, ддд, J=7.8, 1.8, 1.2 Гц, 4-СН), 8,32 (1H, ддд, J=7.9, 1.7, 1.2 Гц, 6-СН), 7,92-7,81 (2Н, м, 2'-СН, 6'-СН), 7,69 (1H, тд, J=7.8, 0.6 Гц, 5-СН), 4,02 (3Н, с, 8-СН3). 13С ЯМР (75 МГц, CDCl3, δ): 175,49 (С-9), 166,92 (С-10), 165,73 (С-7), 151,55 (С-3', С-5', ддд, JF=251.4, 10.3, 3.9 Гц), 141,80 (С-4', дм, JF=257.1, 15.2 Гц), 133,90 (С-6), 132,10 (С-4), 131,49 (С-1), 129,48 (С-5), 129,30 (С-2), 124,21 (С-3), 122,94-122,61 (С-1', м), 112,24-111,93 (С-2', С-6', м), 52,57 (С-8). ВЭЖХ-МС (ESI+): найдено m/z 353,1 [М+Н]+; вычислено C16H12F3N2O4+ 353,1.
Пример 16. Получение метил 4-((((амино(4'-(трифторметокси)фенил)метилен)амино)окси)карбонил)бензоата
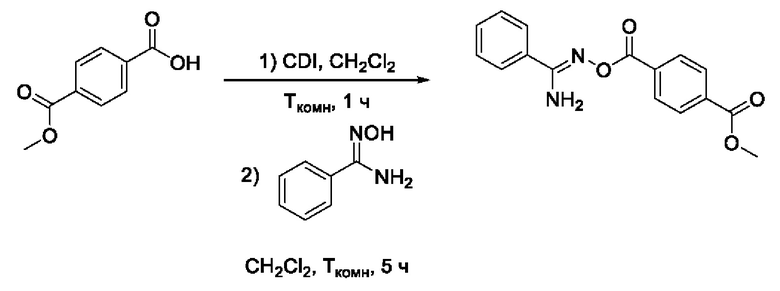
Получен способом, описанном в Примере 9 из монометилового эфира терефталевой кислоты и 4-трифторметокси-N-гидроксибензимидамида в виде белого порошка с выходом 100 мг, 85%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 98:2). ВЭЖХ-МС (ESI+): найдено m/z 299,1 [М+Н]+; вычислено C16H15N2O4+ 299,1. Целевой метил 4-((((амино(4'-(трифторметокси)фенил)метилен)амино)окси)карбонил)бензоат включали в следующую стадию синтеза без регистрации спектров ЯМР.
Пример 17. Получение метил 4-((((амино(фенил)метилен)амино)окси)карбонил)бензоата
Получен способом, описанном в Примере 9 из монометилового эфира терефталевой кислоты и N-гидроксибензимидамида в виде белого порошка с выходом 95 мг, 80%. Элюент для очистки колоночной хроматографией -CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 9:1). ВЭЖХ-МС (ESI+): найдено m/z 299,1 [М+Н]+; вычислено C16H15N2O4+ 299,1. Целевой метил 4-((((амино(фенил)метилен)амино)окси)карбонил)бензоат включали в следующую стадию синтеза без регистрации спектров ЯМР.
Пример 18. Получение этил 3-амино-3-(((4'-хлорбензоил)окси)имино)пропаноата
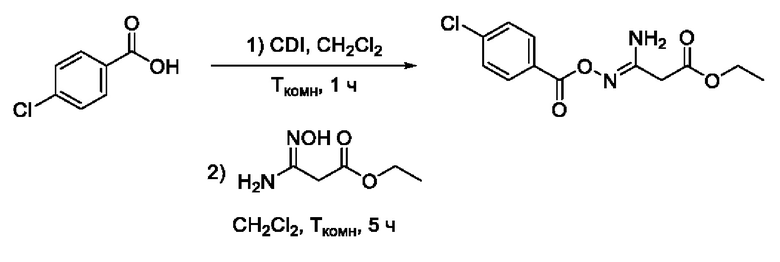
Получен способом, описанном в Примере 9 из 4-хлорбензойной кислоты и этил 3-амино-3-(гидроксиимино)пропаноата в виде светло-желтого порошка с выходом 720 мг, 80%. Элюент для очистки колоночной хроматографией -CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 95:5). 1H ЯМР (400 МГц, CDCl3, δ): 8,00-7,97 (2H,м, 2'-СН, 6'-СН), 7,46-7,43 (2H, м, 3'-СН, 5'-СН), 5,51 (2Н,уш.с., 4-NH2), 4,23 (2Н, к, J=7.1 Гц, 6-СН2), 3,41 (2Н, с, 2-СН2), 1,31 (3Н, т, J=7.1 Гц, 7-СН3). 13С ЯМР (101 МГц, CDCl3, δ): 168,85 (С-1), 163,06 (С-5), 153,73 (С-3), 139,59 (С-1'), 130,81 (С-2', С-6'), 128,88 (С-3', С-5'), 127,78 (С-4'), 61,88 (С-6), 35,85 (С-2), 14,05 (С-7). ВЭЖХ-МС (ESI-): найдено m/z 283,1 [М-H]-; вычислено C12H12ClN2O4- 283,1.
Пример 19. Получение этил 3-амино-3-(((4'-йодбензоил)окси)имино)пропаноата
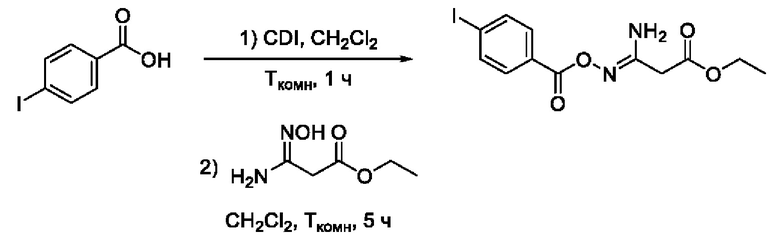
Получен способом, описанном в Примере 9 из 4-йодбензойной кислоты и этил 3-амино-3-(гидроксиимино)пропаноата в виде белого порошка с выходом 140 мг, 65%. Элюент для очистки колоночной хроматографией -CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 95:5). 1Н ЯМР (400 МГц, CDCl3, δ): 7,76-7,74 (2Н, м, 3'-СН, 5'-СН), 7,68-7,66 (2Н, м, 2'-СН, 6'-СН), 5,39 (2H, уш.с., 4-NH2), 4,15 (2Н, к, J=7.1 Гц, 6-СН2), 3,33 (2Н, с, 2-СН2), 1,23 (3Н, т, J=7.1 Гц, 7-СН3). 13С ЯМР (101 МГц, CDCl3, δ): 168,87 (С-1), 163,39 (С-5), 153,73 (С-3), 137,88 (С-3', С-5'), 130,82 (С-2', С-6'), 128,82 (С-1'), 100,84 (С-4'), 61.89 (С-6), 35,81 (С-2), 14,06 (С-7). ВЭЖХ-МС (ESI+): найдено m/z 377,0 [М+Н]+; вычислено C12H14N2O4+ 377,0.
Пример 20. Получение этил 3-амино-3-(((3'-хлорбензоил)окси)имино)пропаноата
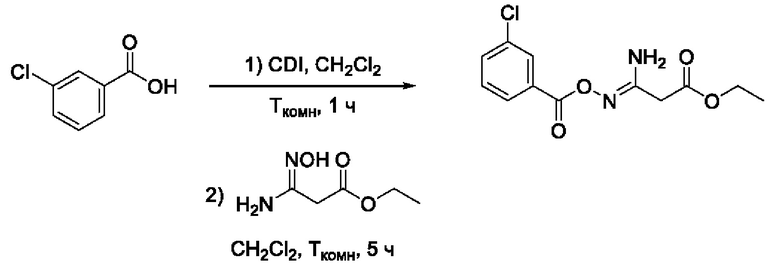
Получен способом, описанном в Примере 9 из 3-хлорбензойной кислоты и этил 3-амино-3-(гидроксиимино)пропаноата в виде светло-желтого порошка с выходом 740 мг, 80%. Элюент для очистки колоночной хроматографией -CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 95:5). 1Н ЯМР (300 МГц, CDCl3, δ): 8,03-8,01 (1H, м, 2'-СН), 7,96-7,92 (1Н, м, 6'-СН), 7,58-7,54 (1H, м, 4'-СН), 7,44-7,39 (1H, м, 5'-СН), 5,52 (2H, уш.с., 4-NH2), 4,24 (2Н, к, J=7.1 Гц, 6-СН2), 3,41 (2Н, с, 2-СН2), 1,31 (3Н, m, J=1.2 Гц, 7-СН3). 13С ЯМР (75 МГц, CDCl3, δ): 168,81 (С-1), 162,69 (С-5), 153,80 (С-3), 134,67 (С-3'), 133,12 (С-4'), 131,11 (С-1'), 129,85 (С-2'), 129,42 (С-5'), 127,60 (С-6'), 61,88 (С-6), 35,82 (С-2), 14,03 (С-7). ВЭЖХ-МС (ESI+): найдено m/z 285,1 [М+Н]+; вычислено C12H14ClN2O4+ 285,1.
Пример 21. Получение этил 3-амино-3-(((3'-йодбензоил)окси)имино)пропаноата
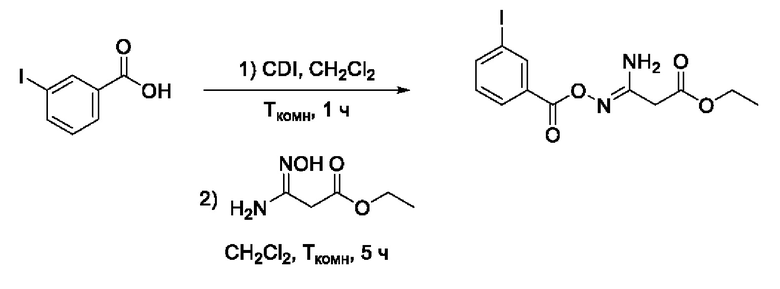
Получен способом, описанном в Примере 9 из 3-йодбензойной кислоты и этил 3-амино-3-(гидроксиимино)пропаноата в виде белого порошка с выходом 480 мг, 90%. Элюент для очистки колоночной хроматографией -CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 95:5). 1H ЯМР (300 МГц, CDCl3, δ): 8,37 (1H, т, J=1.7 Гц, 2'-СН), 8,02 (1H, дт, J=7.8, 1.4 Гц, 6'-СН), 7,92 (1H, ддд, J=7.9, 1.8, 1.1 Гц, 4'-СН), 7,22 (1H, т, J=7.9 Гц, 5'-СН), 5,49 (2H, уш.с., 4-NH2), 4,24 (2Н, к, J=7.2 Гц, 6-СН2), 3,42 (2Н, с, 2-СН2), 1,32 (3Н, т, J=7.1 Гц, 7-СН3). 13С ЯМР (75 МГц, CDCl3, δ): 168,84 (С-1), 162,37 (С-7), 153,78 (С-3), 141,93 (С-4'), 138,17 (С-2'), 131,30 (С-1'), 130,17 (С-5'), 128,62 (С-6'), 93,89 (С-3'), 61,89 (С-6), 35,80 (С-2), 14,05 (С-7). ВЭЖХ-МС (ESI+): найдено m/z 377,0 [М+Н]+; вычислено C12H14IN2O4+ 377,0.
Пример 22. Получение этил 3-(2',6'-дифторфенил)-1,2,4-оксадиазол-5-карбоксилата
Навеску 2,6-дифтор-N-гидроксибензимидамида (360 мг, 2,1 ммоль, 1 экв) растворяли в 5 мл ТГФ при охлаждении и интенсивном перемешивании. К полученному раствору по каплям при охлаждении последовательно добавляли раствор этил оксалилхлорида (345 мкл, 3,1 ммоль, 1,5 экв) в 15 мл ТГФ и диизопропилэтиламин DIPEA (750 мкл, 4,2 ммоль, 2 экв). Образовавшуюся реакционную смесь перемешивали 0,5 ч при охлаждении и 1 ч при комнатной температуре. Далее, реакционную смесь кипятили с обратным холодильником при интенсивном перемешивании, ход реакции контролировали ТСХ (элюент - гексан:EtOAc = 3:1). После окончания реакции, раствор упаривали досуха на роторном испарителе, твердый остаток растворяли в 40 мл EtOAc, последовательно промывали деионизированной водой (3-15 мл) и насыщенным водным раствором хлорида натрия (3-15 мл). Объединенную органическую фазу сушили безводным Na2SO4 при интенсивном перемешивании, после чего осадок отфильтровывали, а фильтрат упаривали досуха. Полученный твердый остаток очищали колоночной хроматографии на силикагеле, элюент - CH2Cl2:изо-C3H7OH (градиент от 96:4 до 85:15). Объединенные органические фракции, содержащие целевой продукт, объединяли, растворитель упаривал досуха с образованием целевого этил 3-(2',6'-дифторфенил)-1,2,4-оксадиазол-5-карбоксилата с выходом 320 мг (60%). Светло-желтый порошок, 1H ЯМР (300 МГц, CDCl3, δ): 7,59-7,49 (1Н, м, 4'-СН), 7,13-7,06 (2Н, м, 3'-СН, 5'-СН), 4,59 (2Н, к, J=7.1 Гц, 7-СН2), 1,50 (3Н, m,J=7.1 Гц, 8-СН3). 13С ЯМР (75 МГц, CDCl3, δ): 166,77 (С-3), 162,07 (С-5), 160,93 (С-2', С-6', дд, JF=257.6, 5.5 Гц), 153,83 (С-6), 133,38 (С-4, т, JF=10.3 Гц), 112,16 (С-3', С-5', дд, JF=21.7, 3.2 Гц), 104,65 (С-1'), 64,08 (С-7), 14,01 (С-8). ВЭЖХ-МС (ESI+): найдено m/z 255,1 [М+Н]+; вычислено C11H9F2N2O3+ 255,1.
Пример 23. Получение этил 3-(4'-(трифторметокси)фенил)-1,2,4-оксадиазол-5-карбоксилата
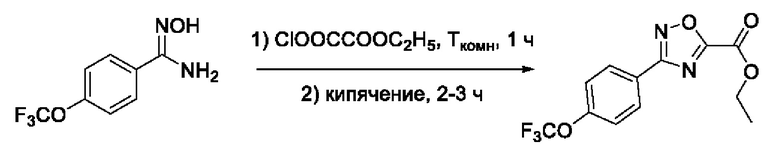
Получен способом, описанном в Примере 22 из 4-трифторметокси-N-гидроксибензимидамида и этил оксалилхлорида в виде светло-желтого порошка с выходом 350 мг, 80%). Элюент для очистки колоночной хроматографией - гексан:EtOAc (градиент от 9:1 до 5:1). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 8,22-8,18 (2Н, м, 2'-СН, 6'-СН), 7,62-7,58 (2Н, м, 3'-СН, 5'-СН), 4,48 (2Н, к, J=7.1 Гц, 7-СН2), 1,39 (3Н, т, J=7.1 Гц, 8-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 168,00 (С-3), 167,62 (С-5), 154,03 (С-6), 151,24 (С-4'), 129,98 (С-2', С-6'), 124,96 (С-9), 122,20 (С-3', С-5'), 118,70 (С-1'), 63,85 (С-7), 14,24 (С-8). ВЭЖХ-МС (ESI+): найдено m/z 303,1 [М+Н]+; вычислено C12H10F3N2O4+ 303,1.
Пример 24. Получение этил 3-(3',4',5',-трифторфенил)-1,2,4-оксадиазол-5-карбоксилата
Получен способом, описанном в Примере 22 из 3,4,5-трифтор-N-гидроксибензимидамида и этил оксалилхлорида в виде светло-желтого порошка с выходом 200 мг, 60%. Элюент для очистки колоночной хроматографией - гексан:EtOAc = 3:1. 1Н ЯМР (300 МГц, ДМСО-d6, δ): 8,04-7,93 (2Н, м, 2'-СН, 6'-СН), 4,48 (2Н, к, J=7.1 Гц, 7-СН2), 1,38 (3Н, т, J=7.1 Гц, 8-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 167,90 (С-3), 166,94 (С-5), 153,87 (С-6), 151,31 (С-3', С-5', ддд, JF=249.7, 10.2, 3.9 Гц), 141,75 (С-4', дт, JF=254.8, 15.3 Гц), 122,52-122,21 (С-1', м), 113,06-112,75 (С-2', С-6', м), 63,94 (С-7), 14,24 (С-8). ВЭЖХ-МС (ESI+): найдено m/z 273,0 [М+Н]+; вычислено C11H8F3N2O3+ 273,0.
Пример 25. Получение этил 3-(3'-метилфенил)-1,2,4-оксадиазол-5-карбоксилата
Получен способом, описанном в Примере 22 из 3-метил-N-гидроксибензимидамида и этил оксалилхлорида в виде светло-желтого порошка с выходом 260 мг, 65%. Элюент для очистки колоночной хроматографией - гексан:EtOAc = 19:1. 1Н ЯМР (300 МГц, CDCl3, δ): 8,00-7,96 (2H,м, 2'-СН, 6'-СН), 7,44-7,35 (2H,м, 4'-СН, 5'-СН), 4,59 (2Н, к, J=7.1 Гц, 7-СН2), 2,45 (3Н, с, 9-СН3), 1,51 (3Н, т, J=7.1 Гц, 8-СН3). 13С ЯМР (75 МГц, CDCl3, δ): 169,56 (С-3), 166,53 (С-5), 154,21 (С-6), 138,87 (С-3'), 132,61 (С-4'), 128,88 (С-5'), 128,23 (С-2'), 125,51 (С-1'), 124,79 (С-6'), 63,92 (С-7), 21,26 (С-9), 14,04 (С-8). ВЭЖХ-МС (ESI+): найдено m/z 233,1 [М+Н]+; вычислено C12H13N2O3+ 233,1.
Пример 26. Получение этил 3-(4'-метоксифенил)-1,2,4-оксадиазол-5-карбоксилата
Получен способом, описанном в Примере 22 из 4-метокси-N-гидроксибензимидамида и этил оксалилхлорида в виде светло-желтого порошка с выходом 400 мг, 75%. Элюент для очистки колоночной хроматографией - гексан:EtOAc (градиент от 99:1 до 0:100). 1Н ЯМР (300 МГц, CDCl3, δ): 8,13-8,08 (2Н, м, 3'-СН, 5'-СН), 7,04-6,99 (2Н, м, 2'-СН, 6'-СН), 4,58 (2Н, к, J=7.1 Гц, 7-СН2), 3,89 (3Н, с, 9-СН3), 1,50 (3Н, т, J=7.1 Гц, 8-СН3). 13С ЯМР (75 МГц, CDCl3, δ): 169,16 (С-3), 166,34 (С-5), 162,44 (С-4'), 154,27 (С-6), 129,36 (С-3', С-5'), 118,06 (С-1'), 114,39 (С-2', С-6'), 63,86 (С-7), 55,41 (С-9), 14,04 (С-8). ВЭЖХ-МС (ECI+): найдено m/z 249,1 [М+Н]+; вычислено C12H13N2O4+ 249,1.
Пример 27. Получение этил 3-(4'-йодфенил)-1,2,4-оксадиазол-5-карбоксилата
Получен способом, описанном в Примере 22 из 4-йод-N-гидроксибензимидамида и этил оксалилхлорида в виде светло-желтого порошка с выходом 400 мг, 60%. Элюент для очистки колоночной хроматографией - гексан:EtOAc (градиент от 98:2 до 95:5). 1Н ЯМР (300 МГц, CDCl3, δ): 7,92-7,86 (4Н, м, 2'-СН, 3'-СН, 5'-СН, 6'-СН), 4,59 (2Н, к, J=7.1 Гц, 7-СН2), 1,51 (3Н, т, J=7.1 Гц, 8-СН3). 13С ЯМР (75 МГц, CDCl3, δ): 168,93 (С-3), 166,76 (С-5), 154,02 (С-6), 138,29 (С-2', С-6'), 129,09 (С-3', С-5'), 125,14 (С-1'), 98,80 (С-4'), 64,04 (С-7), 14,04 (С-8). ВЭЖХ-МС (ESI+): найдено m/z 345,0 [М+Н]+; вычислено C11H10IN2O3+ 345,0.
Пример 28. Получение метил 2-(3'-(2'',6''-дифторфенил)-1',2',4'-оксадиазол-5'-ил)ацетата

К раствору метил 3-(((амино(2',6'-дифторфенил)метилен)амино)окси)-3-оксопропаноата (232 мг, 0,85 ммоль, 1 экв) в 20 мл ТГФ добавили раствор тетрабутиламмоний фторида (TBAF, 1М раствор в ТГФ, 0,43 ммоль, 0,5 экв). Реакционную смесь кипятили с обратным холодильником при интенсивном перемешивании, ход реакции контролировали ТСХ (элюент - CH2Cl2:изо-С3Н7ОН=19:1). После окончания реакции, раствор упаривали досуха на роторном испарителе, твердый остаток растворяли в 30 мл EtOAc, последовательно промывали деионизированной водой (2⋅15 мл) и насыщенным водным раствором NaCl (1⋅15 мл). Органический слой сушили безводным Na2SO4 при интенсивном перемешивании, осадок отфильтровывали, фильтрат упаривали досуха. Образовавшийся твердый остаток очищали колоночной хроматографией на силикагеле (элюент -CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 99:1). Объединенные органические фракции собирали, растворитель упаривали на роторном испарителе досуха с образованием метил 2-(3'-(2'',6''-дифторфенил)-1',2',4'-оксадиазол-5'-ил)ацетата с выходом 130 мг (60%). Желтоватое масло, 1Н ЯМР (300 МГц, CDCl3, δ): 7,55-7,46 (1H, м, 4''-СН), 7,12-7,04 (2H,м, 3''-СН, 5''-СН), 4,13 (2Н, с, 2-СН2), 3,82 (3Н, с, 3-СН3). 13С ЯМР (75 МГц, CDCl3, δ): 172,76 (С-5'), 165,88 (С-1), 161,34 (С-3'), 160,98 (С-2'', С-6'', дд, JF=257.1, 5.7 Гц), 132,81 (С-4'', т, JF=10.4 Гц), 112,30-111,96 (С-3'', С-5'', м), 105,33 (С-1''), 53,06 (С-3), 32,90 (С-2). ВЭЖХ-MS (ESI+): найдено m/z 255,1 [М+Н]+; вычислено C11H9F2N2O3+ 255,1.
Пример 29. Получение метил 2-(3'-(4''-трифторметокси)фенил)-1',2',4'-оксадиазол-5'-ил)ацетата

Получен способом, описанном в Примере 28 из метил 3-(((амино(4'-(трифторметокси)фенил)метилен)амино)окси)-3-оксопропаноата в виде светло-желтого масла с выходом 170 мг, 75%. Элюент для очистки колоночной хроматографией -CH2Cl2:изо-C3H7OH (градиент от 100:0 до 99:1). 1H ЯМР (300 МГц, CDCl3, δ): 8,18-8,13 (2Н, м, 2''-СН, 6''-СН), 7,36-7,33 (2Н, м, 3''-СН, 5''-СН), 4,08 (2Н, с, 2-СН2), 3,82 (3Н, с, 3-СН3). 13С ЯМР (75 МГц, CDCl3, δ): 172,84 (С-5'), 167,69 (С-3'), 166,03 (С-1), 151,39 (С-4''), 129,22 (С-2'', С-6''), 125,07 (С-4), 121,07 (С-3'', С-5''), 118,64 (С-1''), 53,05 (С-3), 32,92 (С-2). ВЭЖХ-МС (ESI+): найдено m/z 302,9 [М+Н]+; вычислено C12H10F3N2O4+ 303,1.
Пример 30. Получение метил 2-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)ацетата
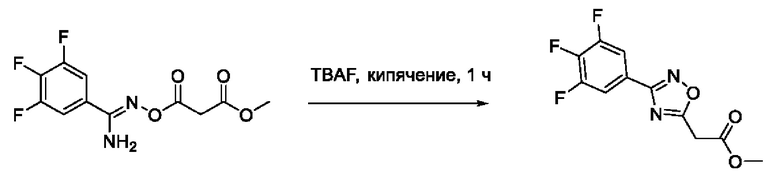
Получен способом, описанном в Примере 28 из метил 3-(((амино(3',4',5'-трифторфенил)метилен)амино)окси)-3-оксопропаноата в виде светло-желтого масла с выходом 180 мг, 60%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 99:1). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 7,95-7,85 (2Н, м, 2''-СН, 6''-СН), 4,42 (2Н, с, 2-СН2), 3,72 (3Н, с, 3-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 175,14 (С-5'), 167,08 (С-1), 166,31 (С-3'), 151,25 (С-3'', С-5'', ддд, JF=249.0, 10.1, 3.8 Гц), 141,48 (С-4'', дт, JF=254.3,15.4 Гц), 123,05-122,74 (C-1'', м), 112,79-112,48 (C-2'', С-6'', м), 53,16 (С-3), 32,91 (С-2). ВЭЖХ-МС (ESI-): найдено m/z 271,0 [М-Н]-; вычислено C11H6F3N2O3- 271,0.
Пример 31. Получение 2-(3'-(3''-метилфенил)-1',2',4'-оксадиазол-5'-ил)ацетата

Получен способом, описанном в Примере 28 из метил 3-(((амино(3'-метилфенил)метилен)амино)окси)-3-оксопропаноата в виде светло-желтого масла с выходом 120 мг, 70%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 99:1). 1Н ЯМР (400 МГц, CDCl3, δ): 7,93-7,89 (2Н, м, 2''-СН, 6''-СН), 7,41-7,33 (2Н, м, 5''-СН, 4''-СН), 4,08 (2Н, с, 2-СН2), 3,82 (3Н, с, 3-СН2), 2,44 (ЗН, с, 4-СН3). 13С ЯМР (101 МГц, CDCl3, δ): 172,43 (С-5'), 168,80 (С-3'), 166,16 (С-1), 138,69 (С-3''), 132,11 (С-4''), 128,78 (С-5''), 128,03 (С-2''), 126,29 (С-1''), 124,59 (С-6''), 53,03 (С-3), 32,97 (С-2), 21,31 (С-4). ВЭЖХ-МС (ESI+): найдено m/z 233,0 [М+Н]+; вычислено C12H13N2O3+ 233,1.
Пример 32. Получение метил 3-(3'-(2'',6''-дифторфенил)-1',2',4'-оксадиазол-5'-ил)бензоата

Получен способом, описанном в Примере 28 из метил 3-((((амино(2',6'-дифторфенил)метилен)амино)окси)карбонил)бензоата в виде белого порошка с выходом 130 мг, 70%. Элюент для очистки колоночной хроматографией - CH2Cl2:изоС3Н7ОН (градиент от 100:0 до 9:1). 1H ЯМР (400 МГц, CDCl3, δ): 8,81-8,80 (1H,м, 2-СН), 8,33 (1H, ддд, J=7.8, 1.8, 1.2 Гц, 4-СН), 8,23 (1Н, ддд, J=7.9, 1.7, 1.2 Гц, 6-СН), 7,59 (1H, тд, J=7.8, 0.6 Гц, 5-СН), 7,44 (1H, мм, J=8.5, 6.2 Гц, 4''-СН), 7,05-6,99 (2Н, м, 3''-СН, 5''-СН), 3,91 (3Н, с, 8-СН3). 13С ЯМР (101 МГц, CDCl3, δ): 175,14 (С-5'), 165,80 (С-3'), 161,72 (С-7), 161,06 (С-2'', С-6'', дд, JF=256.8, 5.8 Гц), 133,82 (С-6), 132,79 (С-4'', м, JF=10.3 Гц), 132,23 (С-4), 131,45 (С-1), 129,45 (С-5), 129,34 (С-2), 124,34 (С-3), 112,26-112,01 (С-3'', С-5'', м), 104,97-104,00 (С-1'', м), 52,52 (С-8). ВЭЖХ-МС (ESI+): найдено m/z 317,1 [М+Н]+; вычислено C16H11F2N2O3+ 317,1.
Пример 33. Получение метил 3-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5'-ил)бензоата
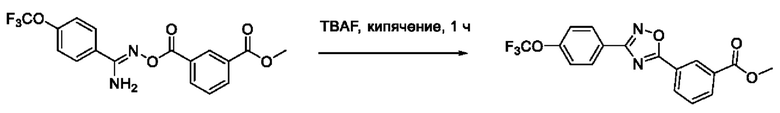
Получен способом, описанном в Примере 28 из 4-((((амино(4'-(трифторметокси)фенил)метилен)амино)окси)карбонил)бензоа та в виде белого порошка с выходом 400 мг, 65%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 9:1). 1Н ЯМР (300 МГц, CDCl3, δ): 8,90-8,89 (1H, м, 2-СН), 8,43-8,40 (1H, м, 4-СН), 8,33-8,29 (1H, м, 6-СН), 8,28-8,23 (2Н, м, 2''-СН, 6''-СН), 7,68 (1H, м, J=7.8 Гц, 5-СН), 7,40-7,37 (2Н, м, 3''-СН, 5''-СН), 4,02 (3Н, с, 8-СН3). 13С ЯМР (75 МГц, CDCl3, δ): 175,11 (С-5'), 168,08 (С-3'), 165,82 (С-7), 151,39 (С-4''), 133,67 (С-6), 132,11 (С-4), 131,42 (С-1), 129,40 (С-5), 129,30 (С-2'', С-6'', С-2), 125,37 (С-9), 124,54 (С-3), 121,09 (С-3'', С-5''), 118,67 (С-1''), 52,53 (С-8). ВЭЖХ-МС (ESI+): найдено m/z 365,1 [М+Н]+; вычислено C17H12F3N2O4+ 365,1.
Пример 34. Получение метил 3-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)бензоата

Получен способом, описанном в Примере 28 из метил 3-((((амино(3',4',5'-трифторфенил)метилен)амино)окси)карбонил)бензоата в виде белого порошка с выходом 100 мг, 60%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 9:1). ВЭЖХ-МС (ESI+): найдено m/z 335,1 [М+Н]+; вычислено C16H10F3N2O3+ 335,1. Целевой метил 3-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)бензоат включали в следующую стадию синтеза без регистрации спектров ЯМР.
Пример 35. Получение метил 4-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5'-ил)бензоата

Получен способом, описанном в Примере 28 из метил 4-((((амино(4'-(трифторметокси)фенил)метилен)амино)окси)карбонил)бензоата в виде белого порошка с выходом 67 мг, 70%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 9:1). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 8,35-8,31 (2H,м, 2''-СН, 6''-СН), 8,25-8,19 (4Н, м, 3-СН, 5-СН, 2-СН, 6-СН), 7,61-7,56 (2Н, м, 3''-СН, 5''-СН), 3,94 (3Н, s, 8-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 175,40 (С-5'), 168,07 (С-7), 165,81 (С-3'), 151,19 (С-4''), 134,19 (С-1), 130,59 (С-2, С-6), 129,91 (С-3, С-5), 128,81 (С-2'', С-6''), 127,55 (С-9), 125,68 (С-4), 121,98 (С-3'', С-5''), 52,93 (С-8). ВЭЖХ-МС (ESI+): найдено m/z 365,1 [М+Н]+; вычислено C17H12F3N2O4+ 365,1.
Пример 36. Получение метил 4-(3'-фенил-1',2',4'-оксадиазол-5'-ил)бензоата
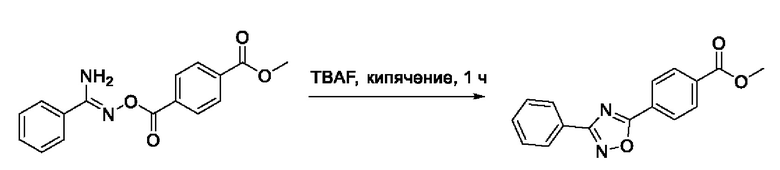
Получен способом, описанном в Примере 28 из метил 4-((((амино(фенил)метилен)амино)окси)карбонил)бензоата в виде белого порошка с выходом 185 мг, 75%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-C3H7OH (градиент от 100:0 до 9:1). 1Н ЯМР (400 МГц, ДМСО-d6, δ): 8,32-8,30 (2Н, м, 2-СН, 6-СН), 8,20-8,18 (2Н, м, 3-СН, 5-СН), 8,11-8,09 (2H, м, 2''-СН, 6''-СН), 7,63-7,61 (3Н, м, 3''-СН, 4''-СН, 5''-СН), 3,91 (3Н, с, 8-СН3). 13С ЯМР (101 МГц, ДМСО-d6, δ): 175,03 (С-5'), 168,91 (С-7), 165,78 (С-3'), 133,85 (С-1), 132,25 (С-4''), 130,61 (С-3, С-5), 129,77 (С-3'', С-5''), 128,79 (С-2, С-6), 127,81 (С-4), 127,59 (С-2'', С-6''), 126,41 (С-1''), 53,05 (С-8). ВЭЖХ-МС (ESI+): найдено m/z 281,1 [М+Н]+; вычислено C16H13N2O3+ 281,1.
Пример 37. Получение этил 2-(5'-(4''-хлорфенил)-1',2',4'-оксадиазол-3'-ил)ацетата
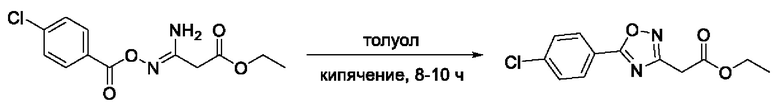
Этил 3-амино-3-(((4'-хлорбензоил)окси)имино)пропаноата растворяли в 20 мл толуола в круглодонной колбе и кипятили с обратным холодильником с использованием насадки Дина-Старка в течение 8-10 ч, ход реакции контролировали ТСХ (элюент - CH2Cl2:изо-С3Н7ОН=19:1). После окончания реакции раствор упаривали досуха на роторном испарителе, твердый остаток очищали колоночной хроматографией на силикагеле, элюент - CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 97:3). Объединенные органические фракции собирали, растворитель упаривали на роторном испарителе досуха с образованием этил 2-(5'-(4''-хлорфенил)-1',2',4'-оксадиазол-3'-ил)ацетата с выходом 440 мг (65%). Белый порошок, 1Н ЯМР (300 МГц, CDCl3, δ): 8,12-8,07 (2Н, м, 2''-СН, 6''-СН), 7,55-7,50 (2Н, м, 3''-СН, 5''-СН), 4,26 (2Н, к, J=7.1 Гц, 3-СН2), 3,89 (2Н, с, 2-СН2), 1,31 (3Н, м, J=7.1 Гц, 4-СН3). 13С ЯМР (75 МГц, CDCl3, δ): 175,16 (C-5'), 167,55 (C-1), 165,35 (C-3'), 139,34 (С-1''), 129,51 (C-3'', C-5''), 129,43 (С-2'', C-6''), 122,47 (C-4''), 61,75 (C-3), 32,38 (C-2), 14,09 (C-4). ВЭЖХ-МС (ESI+): найдено m/z 267,1 [M+H]+; вычислено C12H12ClN2O3+ 267,1.
Пример 38. Получение этил 2-(5'-(4''-йодфенил)-1',2',4'-оксадиазол-3'-ил)ацетата
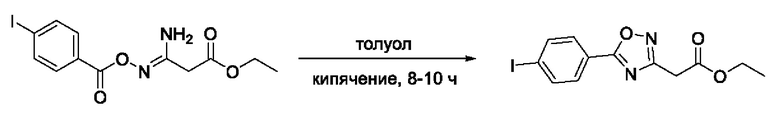
Получен способом, описанном в Примере 37 из этил 3-амино-3-(((4'-йодбензоил)окси)имино)пропаноата в виде светло-желтого порошка с выходом 80 мг, 60%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 99:1). 1Н ЯМР (400 МГц, CDCl3, δ): 7,83-7,76 (4Н, м, 2''-СН, 6''-СН, 3''-СН, 5''-СН), 4,17 (2Н, к, J=7.1 Гц, 3-СН2), 3,81 (2Н, с, 2-СН2), 1,22 (3Н, т, J=7.1 Гц, 4-СН3). 13С ЯМР (101 МГц, CDCl3, δ): 175,46 (С-5'), 167,56 (С-1), 165,36 (С-3'), 138,46 (С-3'', С-5''), 129,40 (С-2'', С-6''), 123,41 (С-1''), 100,31 (С-4''), 61,78 (С-3), 32,39 (С-2), 14,10 (С-4). ВЭЖХ-МС (ESI+): найдено m/z 359,0 [М+Н]+; вычислено C12H12IN2O3+ 359,0.
Пример 39. Получение этил 2-(5'-(3''-хлорфенил)-1',2',4'-оксадиазол-3'-ил)ацетата
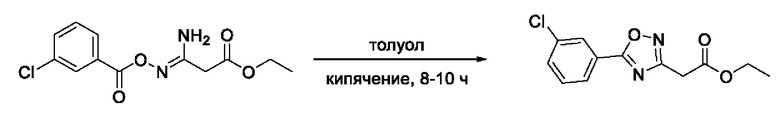
Получен способом, описанном в Примере 37 из этил 3-амино-3-(((3'-хлорбензоил)окси)имино)пропаноата в виде светло-желтого порошка с выходом 350 мг, 50%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 97:3). 1Н ЯМР (300 МГц, CDCl3, δ): 8,16-8,15 (1H, м, 2''-СН), 8,04 (1H, дт, J=7.7, 1.4 Гц, 6''-СН), 7,59 (1H, ддд, J=8.1, 2.1, 1.2 Гц, 4''-СН), 7,51-7,46 (1H, м, 5''-СН), 4,27 (2Н, к, J=7.1 Гц, 3-СН2), 3,90 (2Н, с, 2-СН2), 1,32 (3Н, т, J=7.1 Гц, 4-СН3). 13С ЯМР (75 МГц, CDCl3, δ): 174,84 (С-5'), 167,50 (С-1), 165,39 (С-3'), 135,31 (С-1''), 132,88 (С-4''), 130,44 (С-5''), 128,18 (С-2''), 126,18 (С-6''), 125,58 (С-3''), 61,78 (С-3), 32,38 (С-2), 14,09 (С-4). ВЭЖХ-МС (ESI+): найдено m/z 267,1 [М+Н]+; вычислено C12H12ClN2O3+ 267,1.
Пример 40. Получение этил 2-(5'-(3''-йодфенил)-1',2',4'-оксадиазол-3'-ил)ацетата
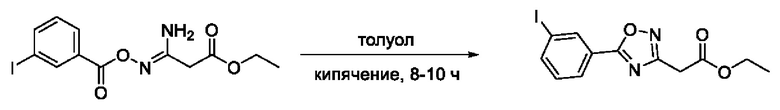
Получен способом, описанном в Примере 37 из этил 3-амино-3-(((3'-йодбензоил)окси)имино)пропаноата в виде желтого порошка с выходом 200 мг, 45%. Элюент для очистки колоночной хроматографией - гексан:EtOAc = 8:1. 1H ЯМР (300 МГц, CDCl3, δ): 8,53-8,51 (1Н, м, 2''-СН), 8,12 (1H, ддд, J=7.9, 1.7, 1.1 Гц, 6''-СН), 7,95 (1H, ддд, J=7.9, 1.8, 1.1 Гц, 4''-СН), 7,31-7,26 (1H, м, 5''-СН), 4,27 (2Н, к, J=7.1 Гц, 3-СН2), 3,90 (2Н, с, 2-СН2), 1,32 (3Н, м, J=7.1 Гц, 4-СН3). 13С ЯМР (75 МГц, CDCl3, δ): 174,52 (С-5'), 167,51 (С-1), 165,35 (С-3'), 141,71 (С-4''), 136,84 (С-2''), 130,66 (С-5''), 127,16 (С-6''), 125,77 (С-1''), 94,31 (С-3''), 61,79 (С-3), 32,38 (С-2), 14,10 (С-4). ВЭЖХ-МС (ESI+): найдено m/z 359,0 [М+Н]+; вычислено C12H12IN2O3+ 359,0.
Пример 41. Получение 3-(5-(2-фторфенил)-1,2,4-оксадиазол-3-ил) бензойной кислоты
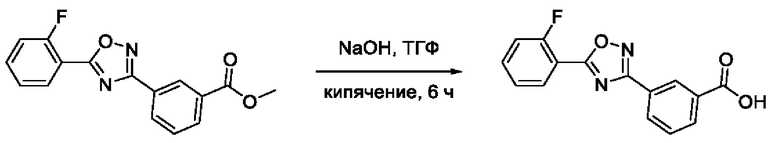
Метиловый эфир 3-(5-(2-фторфенил)-1,2,4-оксадиазол-3-ил)бензойной кислоты (350 мг, 1,17 ммоль, 1 экв) растворяли в 5 мл ТГФ в круглодонной колбе. К полученному раствору добавляли водный раствор гидроксида натрия (1,5 М, 10 мл). Реакционную смесь кипятили с обратным холодильником при интенсивном перемешивании в течение 6 ч, ход реакции контролировали ТСХ (элюент - CH2Cl2:изо-С3Н7ОН = 9:1). После окончания реакции, водный слой реакционной массы отделяли, органический дополнительно промывали деионизированной водой. Водную фазу объединяли и подкисляли 5% водным раствором серной кислоты до рН 3-4. Образовавшийся осадок отфильтровывали на водоструйном насосе и сушили на воздухе с получением 3-(5-(2-фторфенил)-1,2,4-оксадиазол-3-ил)бензойной кислоты с выходом 300 мг (90%). Белый порошок, ВЭЖХ-МС (ESI-): найдено m/z 283,0 [М-Н]-; вычислено C15H8FN2O3- 283,1. Целевую 3-(5-(2-фторфенил)-1,2,4-оксадиазол-3-ил)бензойной кислоту включали в следующую стадию синтеза без регистрации спектров ЯМР.
Пример 42. Получение 3-(3'-(2'',6''-дифторфенил)-1',2',4'-оксадиазол-5'-ил)бензойной кислоты
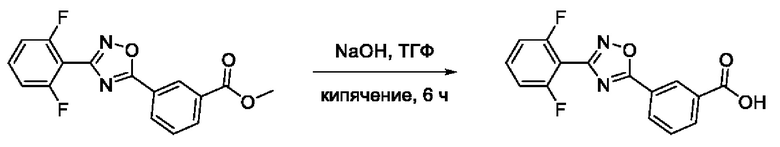
Получен способом, описанном в Примере 41 из метилового эфира 3-(3'-(2'',6''-дифторфенил)-1',2',4'-оксадиазол-5'-ил)бензойной кислоты в виде белого порошка с выходом 110 мг, 95%. 1Н ЯМР (400 МГц, ДМСО-d6, δ): 8,66-8,65 (1Н, м, 2-СН), 8,43-8,40 (1H, м, 4-СН), 8,29-8,26 (1H, м, 6-СН), 7,82 (1H, т, J=7.8 Гц, 5-СН), 7,79-7,73 (1H, м, 4''-СН), 7,42-7,37 (2Н, м, 3''-СН, 5''-СН). 13С ЯМР (101 МГц, ДМСО-d6, δ): 175,42 (С-5'), 166,61 (С-3'), 161,51 (С-7), 160,54 (С-2'', С-6'', дд, JF=254.7, 5.8 Гц), 134,85 (С-4'', т, JF=10.6 Гц), 134,39 (С-6), 132,57 (С-1), 132,43 (С-4), 130,78 (С-5), 128,95 (С-2), 123,89 (С-3), 113,29-113,05 (С-3'', С-5'', м), 104,97-104,00 (С-1'', м). ВЭЖХ-МС (ESI-): найдено m/z 301,1 [М-Н]-; вычислено C15H7F2N2O3- 301,1.
Пример 43. Получение 3-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5'-ил)бензойной кислоты
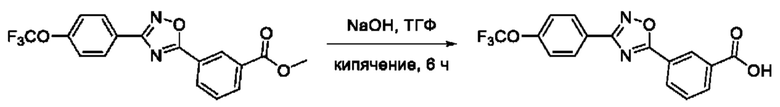
Получен способом, описанном в Примере 41 из метилового эфира 3-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5'-ил)бензойной кислоты в виде белого порошка с выходом 90 мг, 95%. 1Н ЯМР (300 МГц, ДМСО-d6, δ): 13,35 (1H, уш.с, 9-ОН), 8,67-8,66 (1H, м, 2-СН), 8,40-8,36 (1H, м, 4-СН), 8,27-8,26 (1H, м, 6-СН), 8,25-8,20 (2Н, м, 2''-СН, 6''-СН), 7,79 (1H, m, J=7.8 Гц, 5-СН), 7,59-7,56 (2Н, м, 3''-СН, 5''-СН). 13С ЯМР (75 МГц, ДМСО-d6, δ): 175,46 (С-5'), 167,80 (С-3'), 166,68 (С-7), 150,99 (С-4''), 134,20 (С-6), 132,90 (С-4), 132,16 (С-1), 130,57 (С-5), 129,88 (С-2'', С-6'') 128,95 (С-2), 125,61 (С-8), 124,07 (С-3), 122,05 (С-3'', С-5''), 118,72 (С-1''). ВЭЖХ-МС (ESI-): найдено m/z 349,1 [М-H]-; вычислено C16H8F3N2O4– 349,1.
Пример 44. Получение 3-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)бензойной кислоты
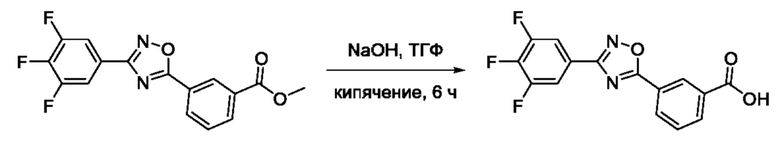
Получен способом, описанном в Примере 41 из метилового эфира 3-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)бензойной кислоты в виде белого порошка с выходом 85 мг, 90%. 1Н ЯМР (300 МГц, ДМСО-d6, δ): 13,46 (1H, уш.с, 8-ОН), 8,63-8,61 (1H, м, 2-СН), 8,38-8,34 (1H, м, 4-СН), 8,26-8,23 (1H, м, 6-СН), 8,00-7,90 (2Н, м, 2''-СН, 6''-СН), 7,79 (1H, m, J=7.8 Гц, 5-СН). 13С ЯМР (75 МГц, ДМСО-d6, δ): 175,71 (С-5'), 166,73 (С-3'), 166,54 (С-7), 151,21 (С-3'', С-5'', ∂∂∂, JF=249.3, 10.1, 3.8 Гц), 141,46 (С-4'', ∂m, JF=254.5, 15.4 Гц), 134,33 (С-6), 132,54 (С-4), 132,29 (С-1), 130,62 (С-5), 128,95 (С-2), 123,83 (С-3), 123,10-122,86 (C-1'', м), 112,84-112,53 (С-2'', С-6'', м). ВЭЖХ-МС (ESI-): найдено m/z 319,0 [М-Н]-; вычислено C15H6F3N2O3- 319,0.
Пример 45. Получение 4-(3'-(3'',4'',5''-трифторфенил)-1',2,,4'-оксадиазол-5'-ил)бензойной кислоты
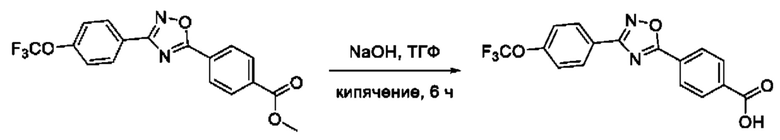
Получен способом, описанном в Примере 41 из метилового эфира 4-(3'-(4''-(трифторметокси)фенил)-1',2',4,-оксадиазол-5,-ил)бензойной кислоты в виде белого порошка с выходом 61 мг, 95%. ВЭЖХ-МС (ESI-): найдено m/z 349,1 [М-Н]-; вычислено C16H8F3N2O4- 349,1. Целевую 4-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)бензойной кислоту включали в следующую стадию синтеза без регистрации спектров ЯМР.
Пример 46. Получение 4-(3'-фенил-1',2',4'-оксадиазол-5'-ил)бензойной кислоты
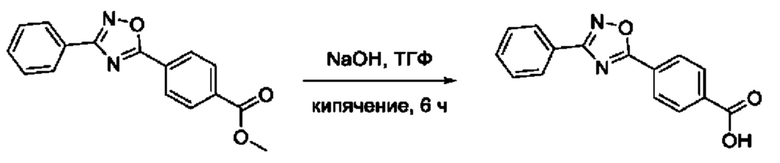
Получен способом, описанном в Примере 41 из метилового эфира 4-(3'-фенил-1',2',4'-оксадиазол-5'-ил)бензойной кислоты в виде белого порошка с выходом 60 мг, 95%. ВЭЖХ-МС (ESI-): найдено m/z 265,1 [М-Н]-; вычислено C15H9N2O3- 265,1. Целевую 4-(3'-фенил-1',2',4'-оксадиазол-5'-ил)бензойной кислоту включали в следующую стадию синтеза без регистрации спектров ЯМР.
Пример 47. Получение 3-(3'-(2'',6''-дифторфенил)-1',2',4'-оксадиазол-5'-ил)пропановой кислоты
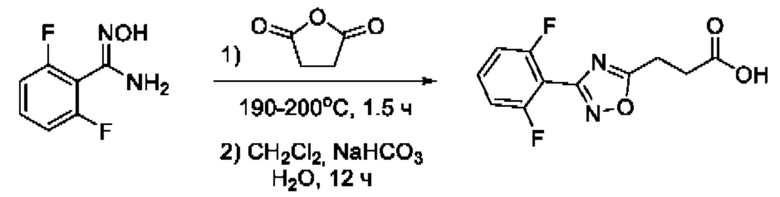
Навеску 2,6-дифторо-N'-гидроксибензимидамида смешивали в круглодонной колбе с янтарным ангидридом. Полученную смесь нагревали с обратным холодильником без растворителя до 190-200°С (температура нагревателя) до образования однородной смеси бурого цвета. Реакционную смесь выдерживали при заданной температуре в течение 1,5 ч. После этого смесь охлаждали до комнатной температуры и растворяли в 10 мл CH2Cl2. В полученный раствор добавляли 15 мл насыщенного водного раствора NaHCO3. Полученную смесь перемешивали в течение 12 ч, после чего отделяли водную фазу от органической, органическую фазу дополнительно промывали деионизированной водой. Водные фазы объединяли и подкисляли 5% водным раствором серной кислоты до рН=3. Целевую карбоновую кислоты экстрагировали этилацетатом (4⋅20 мл). Объединенную органическую фазу сушили безводным Na2SO4 при интенсивном перемешивании, после чего осушитель отфильтровывали на водоструйном насосе, а фильтрат упаривали досуха с образованием 3-(3'-(2'',6''-дифторфенил)-1',2',4'-оксадиазол-5'-ил)пропановой кислоты с выходом 330 мг (75%). Оранжевая аморфная субстанция, 1Н ЯМР (300 МГц, ДМСО-d6, δ): 12,32 (1H, уш.с, 4-ОН), 7,78-7,68 (1H, м, 4''-СН), 7,38-7,31 (2Н, м, 3''-СН, 5''-СН), 3,25 (2Н, m, J=6.9 Гц, 3-СН2), 2,84 (2Н, m, J=6.9 Гц, 2-СН2). 13С ЯМР (75 МГц, ДМСО-d6, δ): 180,41 (С-5'), 174,01 (С-3'), 173,13 (С-1), 160,46 (С-2'', С-6'', ∂∂, JF=254.0, 5.9 Гц), 134,50 (С-4'', m, JF=10.5 Гц), 113,18-112,86 (С-3'', С-5'', м), 105,22 (С-1'', m, J=18.0 Гц), 30,24 (С-2), 22,13 (С-3). ВЭЖХ-МС (ESI+): найдено m/z 255,1 [М+Н]+; вычислено C11H9F2N2O3+ 255,1.
Пример 48. Получение 3-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5'-ил)пропановой кислоты
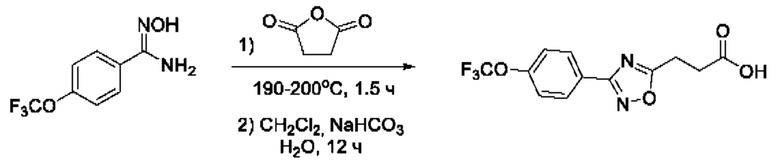
Получен способом, описанном в Примере 47 из 4-трифторметокси-N'-гидроксибензимидамида в виде белого порошка с выходом 380 мг, 70%. 1Н ЯМР (300 МГц, ДМСО-d6, δ): 12,34 (1H, уш.с, 5-ОН), 8,15-8,10 (2Н, м, 2''-СН, 6''-СН), 7,59-7,53 (2Н, м, 3''-СН, 5''-СН), 3,22 (2Н, m, J=6.9 Гц, 3-СН2), 2,85 (2Н, m, J=6.9 Гц, 2-СН2). 13С ЯМР (75 МГц, ДМСО-d6, δ): 180,52 (С-5'), 174,00 (С-3'), 173,18 (С-1), 150,82 (С-4''), 129,67 (С-2'', С-6''), 125,84 (С-1''), 122,08 (С-3'', С-5''), 118,72 (С-4), 30,34 (С-2), 22,19 (С-3). ВЭЖХ-МС (ESI+): найдено m/z 303,0 [М+Н]+; вычислено for C12H10F3N2O4+ 303,1.
Пример 49. Получение 3-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)пропановой кислоты
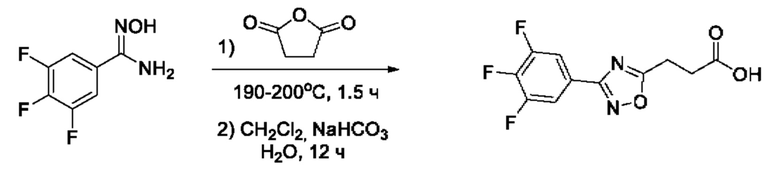
Получен способом, описанном в Примере 47 из 3,4,5-трифтор-N'-гидроксибензимидамида в виде белого порошка с выходом 340 мг, 70%. 1Н ЯМР (300 МГц, ДМСО-d6, δ): 12,32 (1Н, уш.с, 4-ОН), 7,92-7,82 (2Н, м, 2''-СН, 6''-СН), 3,22 (2Н, m, J=6.9 Гц, 3-СН2), 2,85 (2Н, m, J=6.9 Гц, 2-СН2). 13С ЯМР (75 МГц, ДМСО-d6, δ): 180,99 (С-5'), 174,00 (С-3'), 173,13 (С-1), 151,22 (С-3'', С-5'', ∂∂∂, JF=249.3, 10.4, 3.9 Гц), 141,34 (С-4'', ∂m, JF=253.6, 15.4 Гц), 123,41-123,11 (С-1'', м), 112,65-112,35 (С-2'', С-6'', м), 30,30 (С-2), 22,21 (С-3). ВЭЖХ-МС (ESI+): найдено m/z 273,0 [М+Н]+; вычислено C11H8F3N2O3+ 273,0.
Пример 50. Получение 3-(3'-(3''-метилфенил)-1',2',4'-оксадиазол-5'-ил)пропановой кислоты
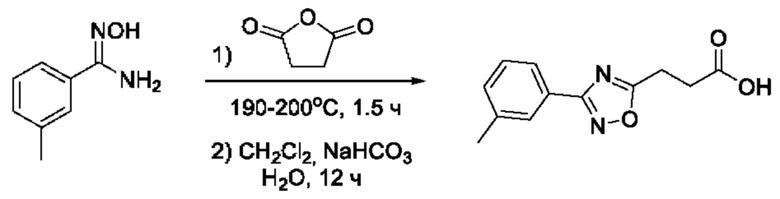
Получен способом, описанном в Примере 47 из 3-метил-N'-гидроксибензимидамида в виде белого порошка с выходом 370 мг, 65%. 1Н ЯМР (300 МГц, ДМСО-d6, δ): 12,46 (1H, уш.с, 5-ОН), 7,82-7,77 (2Н, м, 2''-СН, 6''-СН), 7,47-7,38 (2Н, м, 4''-СН, 5''-СН), 3,20 (2Н, m, J=6.9 Гц, 3-СН2), 2,85 (2Н, m, J=6.9 Гц, 2-СН2), 2,40 (3Н, с, 4-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 180,04 (С-5'), 173,21 (С-1), 167,96 (С-3'), 139,07 (С-3''), 132,56 (С-4''), 129,58 (С-5''), 127,80 (С-2''), 126,65 (С-1''), 124,58 (С-6''), 30,41 (С-2), 22,19 (С-3), 21,33 (С-4). ВЭЖХ-МС (ESI+): найдено m/z 233,2 [М+Н]+; вычислено C12H13N2O3+233,1.
Пример 51. Получение метил 3-(3'-(2'',6''-дифторфенил)-1',2',4'-оксадиазол-5'-ил)пропаноата
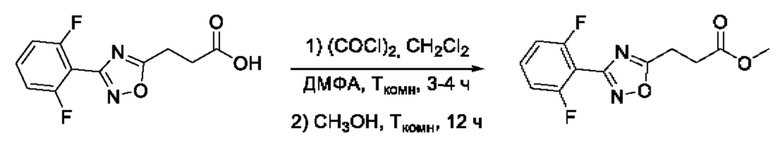
В круглодонной колбе растворяли 3-(3'-(2'',6''-дифторфенил)-1',2',4'-оксадиазол-5'-ил)пропановую кислоту (316 мг, 1,24 ммоль, 1 экв) в 10 мл CH2Cl2, к полученному раствору добавляли две капли ДМФА. Реакционную смесь охлаждали в водяной бане со льдом, после чего в раствор добавляли оксалилхлорид (130 мкл, 1,5 ммоль, 1,2 экв) при интенсивном перемешивании. Далее реакционную смесь перемешивали при комнатной температуре в течение 3-4 ч, ход реакции контролировали ТСХ (элюент -CH2Cl2:изо-С3Н7ОН=9:1). После окончания реакции раствор упаривали досуха на роторном испарителе, твердый остаток сушили в глубоком вакууме, после чего растворяли его в 5 мл метанола. Полученный раствор перемешивали в течение 12 ч, после чего раствор упаривали досуха, а полученный твердый остаток очищали колоночной хроматографией на силикагеле, элюент - гексан:EtOAc = 3:1. Органические фракции, содержащие целевой эфир объединяли, растворитель упаривали, остаток сушили в глубоком вакууме с образованием метил 3-(3'-(2'',6''-дифторфенил)-1',2',4'-оксадиазол-5'-ил)пропаноата с выходом 200 мг (60%). Светло-желтая аморфная субстанция, 1Н ЯМР (400 МГц, CDCl3, δ): 7,40 (1Н, mm, J=8.5, 6.2 Гц, 4''-СН), 7,00-6,94 (2H, м, 3''-СН, 5''-СН), 3,66 (3Н, с, 4-СН3), 3,25 (2Н, m, J=7.4 Гц, 3-СН2), 2,88 (2Н, m, J=7.4 Гц, 2-СН2). 13С ЯМР (101 МГц, CDCl3, δ): 178,52 (С-5'), 171,59 (С-1), 160,98 (С-2'', С-6'', ∂∂, JF=256.6, 5.9 Гц), 160,93 (С-3'), 132,62 (С-4'', m, JF=10.4 Гц), 112,20-111,95 (С-3'', С-5'', м), 105,70 (С-1''), 52,11 (С-4), 30,24 (С-2), 22,08 (С-3). ВЭЖХ-МС (ESI+): найдено m/z 269,0 [М+Н]+; вычислено C12H11F2N2O3+ 269,1.
Пример 52. Получение метил 3-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5'-ил)пропаноата
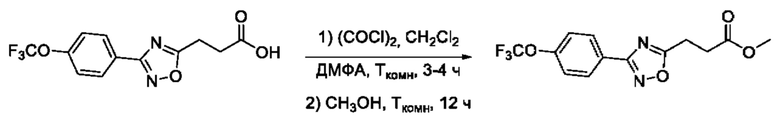
Получен способом, описанном в Примере 51 из 3-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5'-ил)пропановой кислоты в виде светло-желтой аморфной субстанции с выходом 320 мг, 80%. 1Н ЯМР (300 МГц, CDCl3, δ): 8,15-8,11 (2Н, м, 2''-СН, 6''-СН), 7,36-7,31 (2Н, м, 3''-СН, 5''-СН), 3,76 (3Н, с, 5-СН3), 3,29 (2Н, m, J=7.3 Гц, 3-СН2), 2,97 (2Н, m, J=7.3 Гц, 2-СН2). 13С ЯМР (75 МГц, CDCl3, δ): 178,61 (С-5'), 171,65 (С-1), 167,29 (С-3'), 151,25 (С-4''), 129,16 (С-2'', С-6''), 125,38 (С-1''), 121,04 (С-3'', С-5''), 118,65 (С-4), 52,10 (С-5), 30,25 (С-2), 22,05 (С-3). ВЭЖХ-МС (ESI+): найдено m/z 317,0 [М+Н]+; вычислено C13H12F3N2O4+ 317,1.
Пример 53. Получение метил 3-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)пропаноата
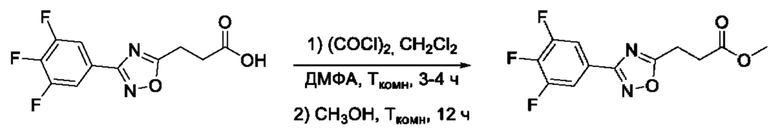
Получен способом, описанном в Примере 51 из 3-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)пропановой кислоты в виде светло-желтой аморфной субстанции с выходом 285 мг, 80%. 1Н ЯМР (400 МГц, CDCl3, δ): 7,69-7,61 (2H, м, 2''-СН, 6''-СН), 3,67 (3Н, с, 4-СН3), 3,19 (2Н, m, J=7.2 Гц, 3-СН2), 2,87 (2Н, m, J=7.2 Гц, 2-СН2). 13С ЯМР (101 МГц, CDCl3, δ): 179,12 (С-5'), 171,59 (С-1), 166,20 (С-3'), 151,50 (С-3'', С-5'', ∂∂∂, JF=251.4, 10.4, 3.9 Гц), 141,68 (С-4'', ∂m, JF=257.1, 15.2 Гц), 122,88-122,71 (С-1'', м), 112,07-111,84 (С-2'', С-6'', м), 52,15 (С-4), 30,14 (С-2), 22,02 (С-3). ВЭЖХ-МС (ESI+): найдено m/z 287,1 [М+Н]+; вычислено C12H10F3N2O3+ 287,1.
Пример 54. Получение метил 3-(3'-(3''-метилфенил)-1',2',4,-оксадиазол-5'-ил)пропаноата
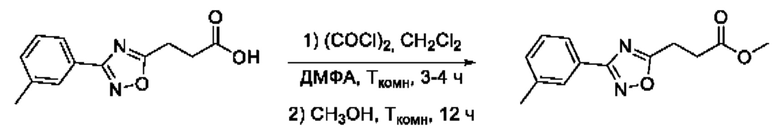
Получен способом, описанном в Примере 51 из 3-(3'-(3''-метилфенил)-1',2',4'-оксадиазол-5'-ил)пропановой кислоты в виде светло-желтой аморфной субстанции с выходом 310 мг, 80%. ВЭЖХ-МС (ESI+): найдено m/z 247,1 [М+Н]+; вычислено C13H15N2O3+ 247,1. Целевой метил 3-(3'-(3''-метилфенил)-1',2',4'-оксадиазол-5'-ил)пропаноат включали в следующую стадию синтеза без регистрации спектров ЯМР.
Пример 55. Получение 3-(2',6'-дифторфенил)-N-гидрокси-1,2,4-оксадиазол-5-карбоксамида 1(1)
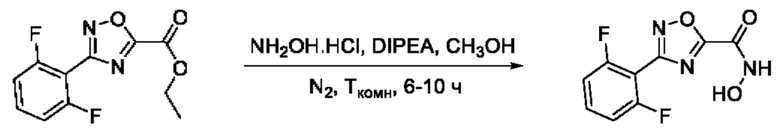
Навеску гидроксиламина солянокислого (346 мг, 5 ммоль, 4 экв) растворяли в 15 мл метанола в круглодонной колбе, далее добавляли диизопропилэтиламин (1,7 мл, 10 ммоль, 8 экв), и полученный раствор перемешивали в атмосфере N2 в течение 10 мин. К полученному раствору добавляли раствор этил 3-(2',6'-дифторфенил)-1,2,4-оксадиазол-5-карбоксилата (316 мг, 1,24 ммоль, 1 экв) в 10 мл метанола. Реакционную смесь выдерживали при комнатной температуре и интенсивном перемешивании в атмосфере азота в течение 6-10 ч, ход реакции контролировали ТСХ (элюент - CH2Cl2:изо-С3Н7ОН:НСООН=19:1:0,1). После окончания реакции к раствору добавляли 5% водный раствор серной кислоты до рН=2. Целевую гидроксамовую кислоту экстрагировали EtOAc (2-30 мл). Органическую фазу собирали и промывали насыщенным водным раствором NaCl, после чего сушили безводным Na2SO4 при интенсивном перемешивании, осушитель отфильтровывали, фильтрат упаривали досуха. Твердый остаток очищали колоночной хроматографией на силикагеле, элюент - CH2Cl2:изо-С3Н7ОН (градиент от 99:1 до 9:1). Фракции, содержащие целевую кислоту, собирали, растворитель упаривали на роторном испарителе, остаток сушили в глубоком вакууме и лиофилизировали с образованием 3-(2',6'-дифторфенил)-N-гидрокси-1,2,4-оксадиазол-5-карбоксамида с выходом 270 мг (90%). Светло-желтый порошок, Тпл 117,4°С (д). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 10,53 (1Н, уш.с, 8-ОН, 7-NH), 7,82-7,72 (1Н, м, 4'-СН), 7,47-7,32 (2H, м, 3'-СН, 5'-СН). 13С ЯМР (75 МГц, ДМСО-d6, δ): 169,86 (С-3), 160,92 (С-5), 160,45 (С-2', С-6', ∂∂, JF=254.9, 5.7 Гц), 150,89 (С-6), 135,03 (С-4', m, JF=10.6 Гц), 113,14 (С-3', С-5', ∂∂, JF=21.5, 2.8 Гц), 104,49 (С-1', m, JF=17.7 Гц). HRMS (ESI-): найдено m/z 240,0221 [М-Н]-; вычислено C9H4F2N3O3- 240,0226. ВЭЖХ-МС (ESI-): найдено m/z 240,1 [М-Н]-; вычислено C9H4F2N3O3- 240,0.
Пример 56. Получение N-гидрокси-3-(4'-(трифторметокси)фенил)-1,2,4-оксадиазол-5-карбоксамида I(2)
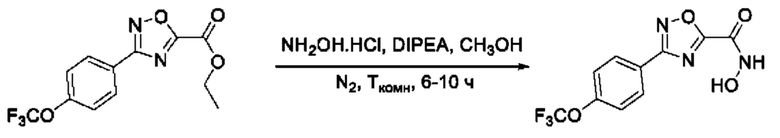
Получен способом, описанном в Примере 55 из этил 3-(4'-(трифторметокси)фенил)-1,2,4-оксадиазол-5-карбоксилата в виде белого порошка с выходом 250 мг, 75%. Элюент для очистки колоночной хроматографией - CH2Cl2-изо-С3Н7ОН (градиент от 99:1 до 9:1). Tпл 108,6°С (д). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 10,45 (1H, уш.с, 7-NH, 8-ОН), 8,20-8,16 (2Н, м, 2'-СН, 6'-СН), 7,64-7,59 (2Н, м, 3'-СН, 5'-СН). 13С ЯМР (75 МГц, ДМСО-d6, δ): 169,55 (С-3), 167,43 (С-5), 151,14 (С-6), 150,87 (С-4'), 129,96 (С-2', С-6'), 125,19 (С-9), 122,18 (С-3', С-5'), 118,71 (С-1'). HRMS (ESI-): найдено m/z 288,0233 [М-Н]-; вычислено C10H5F3N3O4- 288,0238. ВЭЖХ-МС (ESI-): найдено m/z 288,1 [М-Н]-; вычислено C10H5F3N3O4- 288,0.
Пример 57. Получение N-гидрокси-3-(3',4',5'-трифторфенил)-1,2,4-оксадиазол-5-карбоксамида I(3)
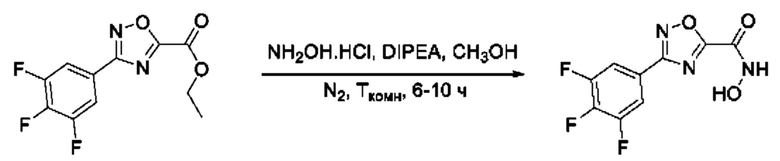
Получен способом, описанном в Примере 55 из этил 3-(3',4',5'-трифторфенил)-1,2,4-оксадиазол-5-карбоксилата в виде светло-желтого порошка с выходом 170 мг, 90%. Аналитически чистый продукт был получен без очистки колоночной хроматографией. Tпл 133,6°С (д). 1H ЯМР (300 МГц, ДМСО-d6, δ): 12,26 (1H, уш.с, 8-ОН), 10,04 (1H, уш.с, 7-NH), 7,99-7,89 (2H, м, 2'-СН, 6'-СН). 13С ЯМР (75 МГц, ДМСО-d6, δ): 169,48 (С-3), 166,40 (С-5), 151,27 (С-3', С-5', ∂∂∂, JF=249.2, 10.1, 3.8 Гц), 150,38 (С-6), 141,66 (С-4', ∂m, JF=254.7,15.4 Гц), 122,69-122,38 (С-1', м), 113,02-112,71 (С-2', С-6', м). HRMS (ESI-): найдено m/z 258,0136 [М-Н]-; вычислено C9H3F3N3O3- 258,0132. ВЭЖХ-МС (ESI-): найдено m/z 258,0 [М-Н]-; вычислено C9H3F3N3O3- 258,0.
Пример 58. Получение N-гидрокси-3-(3'-метилфенил)-1,2,4-оксадиазол-5-карбоксамида I(4)
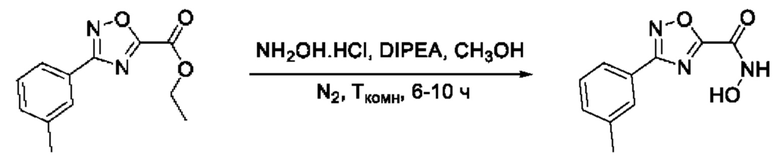
Получен способом, описанном в Примере 55 из этил 3-(3'-метилфенил)-1,2,4-оксадиазол-5-карбоксилата в виде белого порошка с выходом 240 мг, 98%. Аналитически чистый продукт был получен без очистки колоночной хроматографией. Тпл 106,0°С (д). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 12,25 (1Н, уш.с, 8-ОН), 9,92 (1H, уш.с, 7-NH), 7,89-7,83 (2Н, м, 2'-СН, 6'-СН), 7,52-7,43 (2Н, м, 4'-СН, 5'-СН), 2,42 (3Н, с, 9-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 168,93 (С-3), 168,50 (С-5), 150,84 (С-6), 139,27 (С-3'), 133,10 (С-4'), 129,74 (С-5'), 128,05 (С-2'), 125,86 (С-1'), 124,82 (С-6'), 21,33 (С-9). HRMS (ESI-): найдено m/z 218,0565 [М-Н]-; вычислено C10H8N3O3- 218,0571. ВЭЖХ-МС (ESI-): найдено m/z 218,1 [М-Н]-; вычислено C10H8N3O3- 218,1.
Пример 59. Получение N-гидрокси-3-(4'-метоксифенил)-1,2,4-оксадиазол-5-карбоксамида I(5)
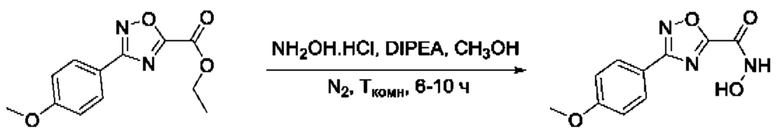
Получен способом, описанном в Примере 55 из этил 3-(4'-метоксифенил)-1,2,4-оксадиазол-5-карбоксилата в виде светло-желтого порошка с выходом 340 мг, 90%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 99:1 до 9:1). Tпл 144,0°С (д). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 11,54-10,29 (2Н, уш.с, 8-ОН, 7-NH), 8,01-7,97 (2Н, м, 3'-СН, 5'-СН), 7,17-7,13 (2Н, м, 2'-СН, 6'-СН), 3,85 (3Н, с, 9-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 169,05 (С-3), 168,12 (С-5), 162,48 (С-4'), 151,11 (С-6), 129,37 (С-3', С-5'), 118,22 (С-1'), 115,25 (С-2', С-6'), 55,93 (С-9). HRMS (ESI-): найдено m/z 234,0515 [М-Н]-; вычислено C10H8N3O4- 234,0520. ВЭЖХ-МС (ESI-): найдено m/z 234,1 [М-Н]-; вычислено C10H8N3O4- 234,1.
Пример 60. Получение N-гидрокси-3-(4'-йодфенил)-1,2,4-оксадиазол-5-карбоксамида I(6)
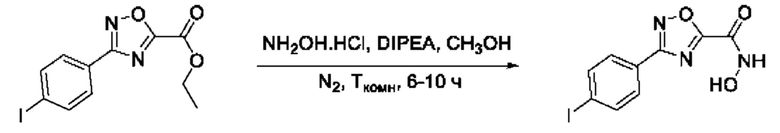
Получен способом, описанном в Примере 55 из этил 3-(4'-йодфенил)-1,2,4-оксадиазол-5-карбоксилата в виде белого порошка с выходом 380 мг, 98%. Аналитически чистый продукт был получен без очистки колоночной хроматографией. Тпл 181,3°С (д). 1H ЯМР (300 МГц, ДМСО-d6, δ): 12,27 (1H, уш.с, 8-ОН), 9,95 (1H, уш.с, 7-NH), 8,02-7,98 (2Н, м, 3'-СН, 5'-СН), 7,84-7,80 (2Н, м, 2'-СН, 6'-СН). 13С ЯМР (75 МГц, ДМСО-d6, δ): 169,11 (С-3), 168,01 (С-5), 150,68 (С-6), 138,78 (С-2', С-6'), 129,37 (С-3', С-5'), 125,42 (С-1'), 100,02 (С-4'). HRMS (ESI-): найдено m/z 329,9372 [М-Н]-; вычислено C9H5IN3O3- 329,9381. ВЭЖХ-МС (ESI+): найдено m/z 332,0 [М+Н]+; вычислено C9H7IN3O3+ 332,0.
Пример 61. Получение 2-(3'-(2'',6''-дифторфенил)-1',2',4'-оксадиазол-5'-ил)-N-гидроксиацетамида I(7)
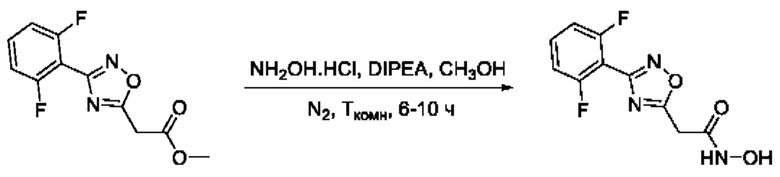
Получен способом, описанном в Примере 55 из метил 2-(3'-(2'',6''-дифторфенил)-1',2',4,-оксадиазол-5'-ил)ацетата в виде светло-желтого порошка с выходом 40 мг, 35%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-C3H7OH (градиент от 99:1 до 9:1). Тпл 149,7°С (д). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 10,94 (1H, уш.с., 4-ОН), 9,17 (1H, c, 3-NH), 7,79-7,69 (1H, м, 4''-СН), 7,40-7,32 (2Н, м, 3''-СН, 5''-СН), 3,96 (2Н, с, 2-СН2). 13С ЯМР (75 МГц, ДМСО-d6, δ): 175,85 (С-5'), 161,90 (С-1), 160,78 (С-3'), 160,47 (С-2'', С-6'', ∂∂, JF=254.3, 6.0 Гц), 134,63 (С-4'', m, JF=10.5 Гц), 113,24-112,92 (С-3'', С-5'', м), 105,00 (С-1'', m, JF=17.8 Гц), 31,88 (С-2). HRMS (ESI-): найдено m/z 254,0380 [М-Н]-; вычислено C10H6F2N3O3- 254,0383. ВЭЖХ-МС (ESI-): найдено m/z 254,0 [М-Н]-; вычислено C10H6F2N3O3- 254,0.
Пример 62. Получение N-гидрокси-2-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5'-ил)ацетамида I(8)
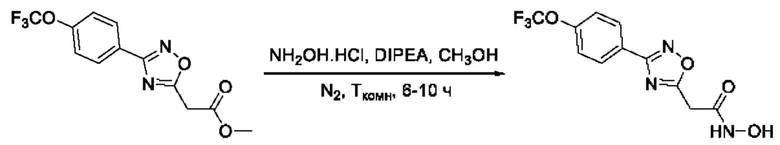
Получен способом, описанном в Примере 55 из метил 2-(3'-(4''-трифторметокси)фенил)-1',2',4,-оксадиазол-5'-ил)ацетата в виде белого порошка с выходом 140 мг, 85%). Очистка осуществлялась перекристаллизацией из CH2Cl2. Тпл 177,0°С (д). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 10,93 (1Н, уш.с, 4-ОН), 9,17 (1H, уш.с, 3-NH), 8,16-8,11 (2Н, м, 2''-СН, 6''-СН), 7,60-7,55 (2Н, м, 3''-СН, 5''-СН), 3,93 (2Н, с, 2-СН2). 13С ЯМР (75 МГц, ДМСО-d6, δ): 175,96 (С-5'), 167,26 (С-3'), 161,99 (С-1), 150,90 (С-4''), 129,70 (С-2'', С-6''), 125,67 (С-4), 122,10 (С-3'', С-5''), 118,72 (С-1''), 115,31 (С-5), 53,05 (С-3), 32,92 (С-2). HRMS (ESI-): найдено m/z 302,0393 [М-Н]-; вычислено C11H7F3N3O4- 302,0394. ВЭЖХ-МС (ESI-): найдено m/z 301,9 [М-Н]-; вычислено C11H7F3N3O4- 302,0.
Пример 63. Получение N-гидрокси-2-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)ацетамида I(9)
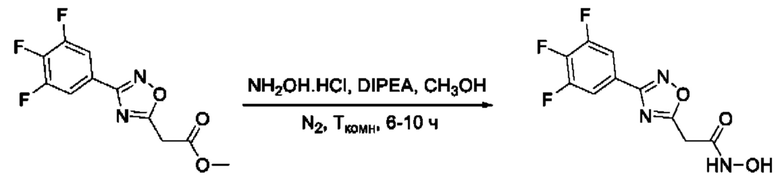
Получен способом, описанном в Примере 55 из метил 2-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)ацетата в виде светло-желтого порошка с выходом 120 мг, 70%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 99:1 до 9:1). Tпл 159,2°С (д). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 10,93 (1H, уш.с, 4-ОН), 9,17 (1H, c, 3-NH), 7,94-7,83 (2Н, м, 2''-СН, 6''-СН), 3,94 (2Н, с, 2-СН2). 13С ЯМР (75 МГц, ДМСО-d6, δ): 176,38 (С-5'), 166,21 (С-3'), 161,85 (С-1), 151,24 (С-3'', С-5'', ∂∂∂, JF=249.0, 9.8, 3.8 Гц), 141,42 (С-4'', ∂m, JF=254.4, 15.5 Гц), 123,22-122,92 (С-1'', м), 112,71-112,40 (С-2'', С-6'', м), 31,93 (С-2). HRMS (ESI-): найдено m/z 272,0289 [М-Н]-; вычислено C10H5F3N3O3- 272,0288. ВЭЖХ-МС (ESI-): найдено m/z 272,0 [М-Н]-; вычислено C10H5F3N3O3- 272,0.
Пример 64. Получение N-гидрокси-2-(3''-метилфенил)-1',2',4'-оксадиазол-5'-ил)ацетамида I(10)
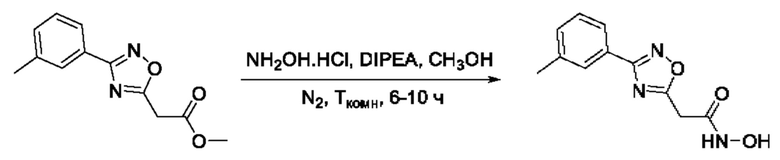
Получен способом, описанном в Примере 55 из метил 2-(3''-метил фенил)-1',2',4',-оксадиазол-5'-ил)ацетата в виде белого порошка с выходом 70 мг, 65%. Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 99:1 до 9:1). Тпл 165,4°С (д). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 10,94 (1Н, уш.с, 4-ОН), 9,17 (1H, уш.с, 3-NH), 7,84-7,82 (1H, м, 2''-СН), 7,82-7,78 (1H, м, 6''-СН), 7,49-7,42 (1H, м, 5''-СН), 7,42-7,39 (1H, м, 4''-СН), 3,90 (2Н, с, 2-СН2), 2,40 (3Н, с, 5-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 175,51 (С-5'), 168,27 (С-3'), 162,08 (С-1), 139,14 (С-3''), 132,68 (С-4''), 129,64 (С-5''), 127,83 (С-2''), 126,46 (С-1''), 124,57 (С-6''), 31,94 (С-2), 21,33 (С-5). HRMS (ESI-): найдено m/z 232,0724 [М-Н]-; вычислено C11H10N3O3- 232,0728. ВЭЖХ-МС (ESI-): найдено m/z 232,0 [М-Н]-; вычислено C11H10N3O3- 232,1.
Пример 65. Получение 3-(5'-(2''-фторфенил)-1',2',4',-оксадиазол-3'-ил)-N-гидроксибензамида I(11)
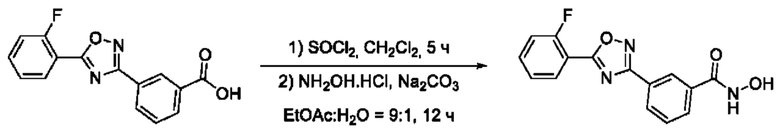
Навеску 3-(5-(2-фторфенил)-1,2,4-оксадиазол-3-ил) бензойной кислоты (295 мг, 1,04 ммоль, 1 экв) растворяли в 10 мл CH2Cl2 в круглодонной колбе. К полученному раствору добавляли SOCl2 (750 мкл, 10,4 ммоль, 10 экв), реакционную смесь перемешивали при комнатной температуре в течение 5 ч, ход реакции контролировали ТСХ (элюент - CH2O2:изо-C3H7OH=9:1). После окончания реакции раствор упаривали досуха, твердый остаток сушили в глубоком вакууме, после чего растворяли в 20 мл смеси EtOAc:H2O=9:1 (по объему), содержащей NH2OH⋅HCl (145 мг, 2,08 ммоль, 2 экв) и Na2CO3 (275 мг, 2,6 ммоль, 2.5 экв). Реакционную смесь перемешивали при комнатной температуре в течение 12 ч, после чего образовавшийся осадок соответствующей гидроксамовой кислоты отфильтровывали на водоструйном насосе, промывали смесью EtOAc:H2O=4:1 (по объему) и сушили на воздухе 2 ч с образованием 3-(5'-(2''-фторфенил)-1',2',4'-оксадиазол-3'-ил)-N-гидроксибензамида с выходом 280 мг (90%). Красный порошок, Тпл 199,3°С (д). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 11,48 (1H, уш.с, 9-ОН), 9,18 (1H, уш.с, 8-NH), 8,50 (1H, с, 2-СН), 8,28-8,22 (2H, м, 6''-СН, 6-СН), 8,00 (1H, ∂, J=7.3 Гц, 5-СН), 7,84-7,78 (1H, м, 4''-СН), 7,70 (1H, m, J=7.5 Гц, 4-СН), 7,59-7,48 (2Н, м, 3''-СН, 5''-СН). 13С ЯМР (75 МГц, ДМСО-d6, δ): 173,16 (С-5', ∂, JF=4.0 Гц), 168,07 (С-3'), 163,77 (С-7), 160,45 (С-2'', ∂, JF=257.8 Гц), 136,25 (С-4'', ∂, JF=8.8 Гц), 134,28 (С-1), 131,38 (С-6), 130,33 (С-6''), 130,07 (С-5), 130,00 (С-4), 126,70 (С-3), 126,30 (С-2), 125,97 (С-5'', ∂, JF=3.3 Гц), 117,78 (С-3'', ∂, JF=20.6 Гц), 112,19 (С-1'', ∂, JF=11.0 Гц). HRMS (ESI-): найдено m/z 298,0622 [М-Н]-; вычислено C15H9FN3O3- 298,0633. ВЭЖХ-МС (ESI-): найдено m/z 298,0 [М-H]-; вычислено C15H9FN3O3- 298,1.
Пример 66. Получение 3-(3-(2'',6''-дифторфенил)-1',2',4'-оксадиазол-5'-ил)-N-гидроксибензамида I(12)
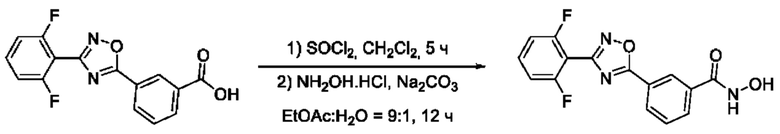
Получен способом, описанном в Примере 65 из 3-(3'-(2'',6''-дифторфенил)-1',2',4,-оксадиазол-5'-ил)бензойной кислоты в виде белого порошка с выходом 100 мг, 90%. Тпл 204,2°С (д). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 11,47 (1Н, уш.с, 9-ОН), 9,35 (1H, уш.с, 8-NH), 8,57-8,56 (1Н, м, 2-СН), 8,34-8,30 (1Н, м, 4-СН), 8,14-8,10 (1Н, м, 6-СН), 7,83-7,73 (2Н, м, 4''-СН, 5-СН), 7,44-7,36 (2Н, м, 3''-СН, 5''-СН). 13С ЯМР (75 МГц, ДМСО-d6, δ): 175,62 (С-5'), 163,05 (С-3'), 161,49 (С-7), 160,55 (С-2'', С-6'', ∂∂, JF=254.5, 5.8 Гц), 134,81 (С-4'', m, JF=10.5 Гц), 134,50 (С-1), 132,15 (С-6), 130,86 (С-5), 130,42 (С-4), 126,88 (С-2), 123,67 (С-3), 113,14 (С-3'', С-5'', ∂∂ JF=21.6, 3.1 Гц), 104,97 (С-1'', m, JF=17.7 Гц). HRMS (ESI-): найдено m/z 316,0534 [М-Н]-; вычислено C15H8F2N3O3- 316,0539. ВЭЖХ-МС (ESI-): найдено m/z 316,0 [М-Н]-; вычислено C15H8F2N3O3- 316,1.
Пример 67. Получение N-гидрокси-3-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5'-ил)бензамида I(13)
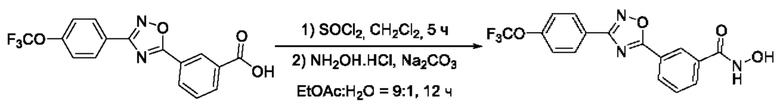
Получен способом, описанном в Примере 65 из 3-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5'-ил)бензойной кислоты в виде белого порошка с выходом 80 мг, 90%. Тпл 191,0°С (д). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 11,54 (1H, уш.с, 10-ОН), 9,23 (1H, уш.с, 9-NH), 8,58-8,57 (1Н, м, 2-СН), 8,34-8,31 (1H, м, 4-СН), 8,27-8,22 (2H, м, 2''-СН, 6''-СН), 8,12-8,08 (1H, м, 6-СН), 7,77 (1H, m, J=7.8 Гц, 5-СН), 7,63-7,58 (2Н, м, 3''-СН, 5''-СН). 13С ЯМР (75 МГц, ДМСО-d6, δ): 175,64 (С-5'), 167,84 (С-3'), 163,26 (С-7), 151,02 (С-4''), 134,42 (С-1), 131,97 (С-6), 130,86 (С-4), 130,35 (С-5), 129,89 (С-2'', С-6''), 126,93 (С-2), 125,66 (С-8), 123,95 (С-3), 122,11 (С-3'', С-5''), 118,73 (С-1''). HRMS (ESI-): найдено m/z 364,0544 [М-Н]-; вычислено C16H9F3N3O4-364,0551. ВЭЖХ-МС (ESI-): найдено m/z 364,0 [М-Н]-; вычислено C16H9F3N3O4- 364,1.
Пример 68. Получение N-гидрокси-3-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)бензамида I(14)
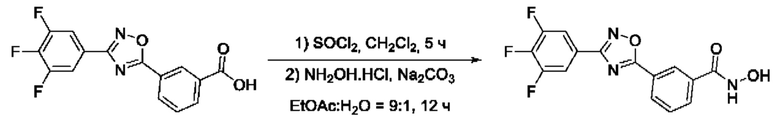
Получен способом, описанном в Примере 65 из 3-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)бензойной кислоты в виде белого порошка с выходом 70 мг, 80%. Тпп 188,5°С (д). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 11,49 (1H, уш.с, 9-ОН), 9,25 (1Н, уш.с, 8-NH), 8,57-7,77 (6H, м, 2-СН, 4-СН, 6-СН, 2''-СН, 6''-СН, 5-СН). 13С ЯМР (75 МГц, ДМСО-d6, δ): 175,96 (С-5'), 166,78 (С-3'), 163,03 (С-7), 151,27-149,57 (С-3'', С-5'', м), 143,39-139,72 (С-4'', м), 134,42 (С-6), 132,07 (С-4), 130,86 (С-1), 130,37 (С-5), 126,98 (С-2), 123,72 (С-3), 123,30-122,90 (С-1'', м), 112,88-112,62 (С-2'', С-6'', м). HRMS (ESI-): найдено m/z 334,0441 [М-Н]-; вычислено C15H7F3N3O3- 334,0445. ВЭЖХ-МС (ESI-): найдено m/z 334,0 [М-Н]-; вычислено C15H7F3N3O3- 334,0.
Пример 69. Получение N-гидрокси-4-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5'-ил)бензамида I(15)
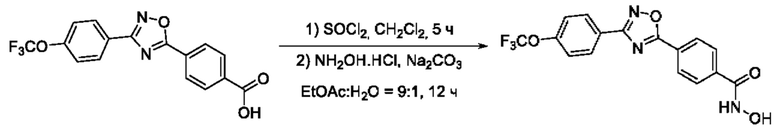
Получен способом, описанном в Примере 65 из 4-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5'-ил)бензойной кислоты в виде белого порошка с выходом 50 мг, 80%. Тпл 236,0°С (д). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 11,35 (1H, уш.с, 10-ОН), 9,32 (1H, уш.с, 9-NH), 8,29-8,22 (4H, м, 2''-СН, 6''-СН, 2-СН, 6-СН), 8,02 (2Н, ∂, J=8.1 Гц, 3-СН, 5-СН), 7,64-7,61 (2Н, м, 3''-СН, 5''-СН). 13С ЯМР (75 МГц, ДМСО-d6, δ): 175,59 (С-5'), 167,89 (С-3'), 163,25 (С-7), 151,03 (С-4''), 137,53 (С-1), 129,91 (CAr), 128,57 (CAr), 128,47 (CAr), 125,69 (CAr), 122,16 (CAr). HRMS (ESI-): найдено m/z 364,0544 [М-Н]-; вычислено C16H9F3N3O4- 364,0551. ВЭЖХ-МС (ESI-): найдено m/z 364,0 [М-H]-; вычислено C16H9F3N3O4- 364,1.
Пример 70. Получение N-гидрокси-4-(3'-фенил-1',2',4'-оксадиазол-5'-ил)бензамида I(16)
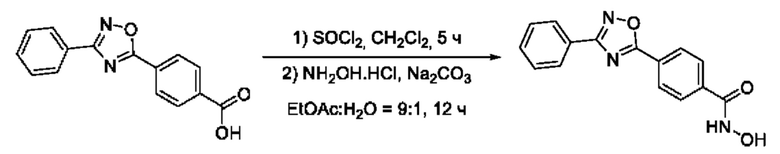
Получен способом, описанном в Примере 65 из 4-(3'-фенил-1',2',4'-оксадиазол-5'-ил)бензойной кислоты в виде белого порошка с выходом 50 мг, 80%. Тпл 265,4°С (д). 1H ЯМР (300 МГц, ДМСО-d6, δ): 11,73 (1H, уш.с, 9-ОН), 10,01 (1H, уш.с, 8-NH), 8,10-8,07 (4Н, м, 2-СН, 6-СН, 3-СН, 5-СН), 7,71-7,62 (5Н, м, 2''-СН, 6''-СН, 3''-СН, 4''-СН, 5''-СН). 13С ЯМР (75 МГц, ДМСО-d6, δ): 175,31 (С-5'), 168,87 (С-7), 163,47 (С-3'), 137,35 (С-1), 132,24 (С-4''), 129,79 (С-3'', С-5''), 128,53 (С-2, С-3, С-5, С-6), 127,61 (С-2'', С-6''), 126,51 (С-4), 125,92 (С-1''). HRMS (ESI-): найдено m/z 280,0709 [М-Н]-; вычислено C15H10N3O3- 280,0728. ВЭЖХ-МС (ESI-): найдено m/z 280,1 [М-Н]-; вычислено C15H10N3O3- 280,1.
Пример 71. Получение 2-(5'-(4''-хлорфенил)-1',2',4'-оксадиазол-3'-ил)-N-гидроксиацетамида I(17)
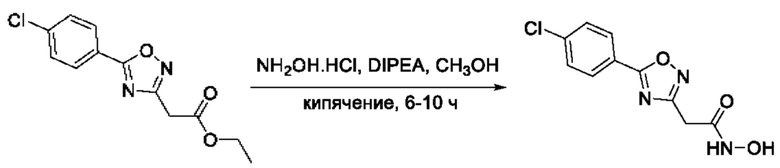
Получен способом, описанном в Примере 55 (за исключением условия кипячения реакционной смеси в данном случае) из этил 2-(5'-(4''-хлорфенил)-1',2',4'-оксадиазол-3'-ил)ацетата в виде белого порошка с выходом 250 мг, 60%. Тпл 174,0°С (д). Целевая гидроксамовая кислота очищалась путем осаждения гексаном из насыщенного раствора твердого остатка (после упаривания метанола) в CH2Cl2. 1Н ЯМР (300 МГц, ДМСО-d6, δ): 10,86 (1H, уш.с, 4-ОН), 9,06 (1H, уш.с, 3-NH), 8,13-8,08 (2H, м, 2''-СН, 6''-СН), 7,73-7,68 (1H, м, 3''-СН, 5''-СН), 3,62 (2Н, с, 2-СН2). 13С ЯМР (75 МГц, ДМСО-d6, δ): 174,61 (С-5'), 166,94 (С-3'), 163,46 (С-1), 138,59 (С-4''), 130,23 (С-3'', С-5''), 130,03 (С-2'', С-6''), 122,62 (С-1''), 30,95 (С-2). HRMS (ESI-): найдено m/z 252,0178 [М-Н]-; вычислено C10H7ClN3O3- 252,0181. ВЭЖХ-МС (ESI+): найдено m/z 254,0 [М+Н]+; вычислено C10H9ClN3O3+ 254,0.
Пример 72. Получение 2-(5'-(4''-йодфенил)-1',2',4'-оксадиазол-3'-ил)-N-гидроксиацетамида I(18)
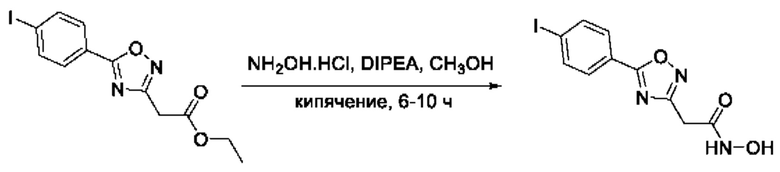
Получен способом, описанном в Примере 55 (за исключением условия кипячения реакционной смеси в данном случае) из этил 2-(5'-(4''-йодфенил)-1',2',4'-оксадиазол-3'-ил)ацетата в виде белого порошка с выходом 50 мг, 70%. Тпл 211,6°С (д). Целевая гидроксамовая кислота очищалась путем осаждения гексаном из насыщенного раствора твердого остатка (после упаривания метанола) в CH2Cl2. 1Н ЯМР (300 МГц, ДМСО-d6, δ): 11,03 (1H, уш.с, 4-ОН), 9,08 (1H, уш.с, 3-NH), 8,05-8,01 (2H, м, 3''-СН, 5''-СН), 7,87-7,83 (2H, м, 2''-СН, 6''-СН), 3,64 (2Н, с, 2-СН2). 13С ЯМР (75 МГц, ДМСО-d6, δ): 175,00 (С-5'), 166,99 (С-3'), 163,44 (С-1), 138,98 (С-3'', С-5''), 129,76 (С-2'', С-6''), 123,14 (С-1''), 102,11 (С-4''), 30,93 (С-2). HRMS (ESI-): найдено m/z 343,9530 [М-Н]-; вычислено C10H7IN3O3- 343,9538. ВЭЖХ-МС (ESI+): найдено m/z 345,8 [М+Н]+; вычислено C10H9IN3O3+ 346,0.
Пример 73. Получение 2-(5'-(3''-хлорфенил)-1',2',4,-оксадиазол-3'-ил)-N-гидроксиацетамида I(19)
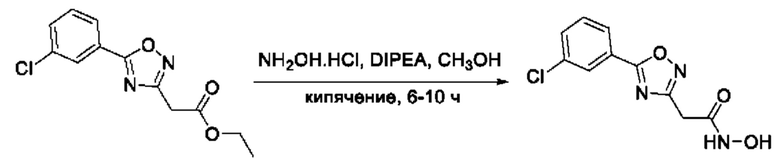
Получен способом, описанном в Примере 55 (за исключением условия кипячения реакционной смеси в данном случае) из этил 2-(5'-(3''-хлорфенил)-1',2',4'-оксадиазол-3'-ил)ацетата в виде белого порошка с выходом 230 мг, 70%. Тпл 146,0°С (д). Целевая гидроксамовая кислота очищалась путем осаждения гексаном из насыщенного раствора твердого остатка (после упаривания метанола) в CH2Cl2. 1H ЯМР (300 МГц, ДМСО-d6, δ): 10,91 (1H, уш.с, 4-ОН), 10,33 (1Н, уш.с., 3-NH), 8,09-8,04 (2Н, м, 2''-СН, 6''-СН), 7,81-7,77 (1H, м, 4''-CH), 7,70-7,65 (1H, м, 5''-СН), 3,64 (2Н, с, 2-СН2). 13С ЯМР (75 МГц, ДМСО-d6, δ): 174,23 (С-5'), 166,99 (С-3'), 163,40 (С-1), 134,64 (С-3''), 133,55 (С-4''), 132,10 (С-5''), 127,67 (С-2''), 126,93 (С-6''), 125,62 (С-1''), 30,94 (С-2). HRMS (ESI-): найдено m/z 252,0176 [М-Н]-; вычислено C10H7ClN3O3- 252,0181. ВЭЖХ-МС (ESI+): найдено m/z 254,0 [М+Н]+; вычислено C10H9ClN3O3+ 254,0.
Пример 74. Получение 2-(5'-(3''-йодфенил)-1',2',4'-оксадиазол-3'-ил)-N-гидроксиацетамида I(20)
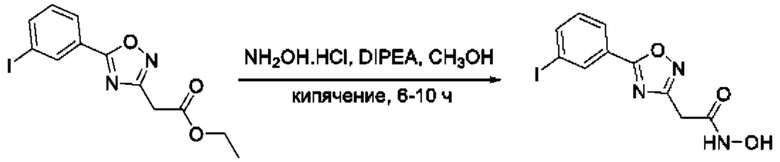
Получен способом, описанном в Примере 55 (за исключением условия кипячения реакционной смеси в данном случае) из этил 2-(5'-(3''-йодфенил)-1',2',4'-оксадиазол-3'-ил)ацетата в виде белого порошка с выходом 100 мг, 55%. Тпл 156,3°С (д). Целевая гидроксамовая кислота очищалась путем осаждения гексаном из насыщенного раствора твердого остатка (после упаривания метанола) в CH2Cl2. 1Н ЯМР (300 МГц, ДМСО-d6, δ): 10,83 (1Н, уш.с, 4-ОН), 9,05 (1H, уш.с, 3-NH), 8,39-8,37 (1H, м, 6''-СН), 8,12-8,06 (1H, м, 2''-СН, 4''-СН), 7,44 (1H, m, J=7.9 Гц, 5''-СН), 3,62 (2Н, с, 2-СН2). 13С ЯМР (75 МГц, ДМСО-d6, δ): 174,05 (С-5'), 166,91 (С-3'), 163,42 (С-1), 142,20 (С-2''), 136,20 (С-6''), 132,07 (С-5''), 127,48 (С-4''), 125,66 (С-1''), 95,94 (С-3''), 30,96 (С-2). HRMS (ESI-): найдено m/z 343,9530 [М-Н]-; вычислено C10H7IN3O3- 343,9538. ВЭЖХ-МС (ESI+): найдено m/z 345,9 [М+Н]+; вычислено C10H9IN3O3+ 346,0.
Пример 75. Получение 3-(3'-(2'',6''-дифторфенил)-1',2',4'-оксадиазол-5'-ил)-N-гидроксипропанамида I(21)
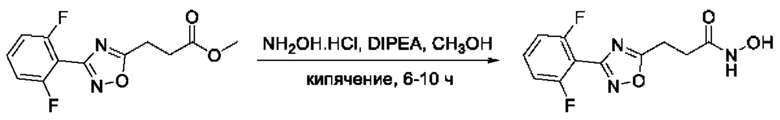
Получен способом, описанном в Примере 55 (за исключением условия кипячения реакционной смеси в данном случае) из метил 3-(3'-(2'',6''-дифторфенил)-1',2',4'-оксадиазол-5'-ил)пропаноата в виде оранжевого порошка с выходом 85 мг, 45%. Tпл 99,0°С (д). Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 75:25). 1Н ЯМР (400 МГц, ДМСО-d6, δ): 10,53 (1H, с, 5-ОН), 8,81 (1H, с, 4-NH), 7,73 (1H, mm, J=8.6, 6.5 Гц, 4''-СН), 7,38-7,32 (2Н, м, 3''-СН, 5''-СН), 3,26 (2Н, m, J=7.1 Гц, 3-СН2), 2,58 (2Н, m, J=7.1 Гц, 2-СН2). 13С ЯМР (101 МГц, ДМСО-d6, δ): 180,45 (С-5'), 167,41 (С-1), 160,47 (С-3'), 160,46 (С-2'', С-6'', ∂∂, JF=254.0, 5.9 Гц), 134,51 (С-4'', m, JF=10.4 Гц), 113,17-112,92 (С-3'', С-5'', м), 105,43 (С-1''), 28,62 (С-2), 22,23 (С-3). HRMS (ESI-): найдено m/z 268,0530 [М-Н]-; вычислено C11H8F2N3O3- 268,0539. ВЭЖХ-МС (ESI+): найдено m/z 270,1 [М+Н]+; вычислено C11H10F2N3O3+ 270,1.
Пример 76. Получение N-гидрокси-3-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5'-ил)пропанамида I(22)

Получен способом, описанном в Примере 55 (за исключением условия кипячения реакционной смеси в данном случае) из метил 3-(3'-(4''-(трифторметокси)фенил)-1',2',4'-оксадиазол-5'-ил)пропаноата в виде светло-желтого порошка с выходом 160 мг, 50%. Тпл 120,0°С (д). Целевая гидроксамовая кислота очищалась путем осаждения гексаном из насыщенного раствора твердого остатка (после упаривания метанола) в CH2Cl2. 1Н ЯМР (400 МГц, ДМСО-d6, δ): 10,66 (1H, с, 5-ОН), 8,83 (1H, с, 4-NH), 8,14-8,10 (2Н, м, 2''-СН, 6''-СН), 7,58-7,56 (2Н, м, 3''-СН, 5''-СН), 3,22 (2Н, m, J=7.21 Гц, 3-СН2), 2,59 (2Н, m, J=7.2 Гц, 2-СН2). 13С ЯМР (101 МГц, ДМСО-d6, δ): 180,59 (С-5'), 167,45 (С-1), 166,94 (С-3'), 150,81 (С-4''), 129,69 (С-2'', С-6''), 125,91 (С-1''), 122,09 (С-3'', С-5''), 121,71 (С-6), 28,80 (С-2), 22,34 (С-3). HRMS (ESI-): найдено m/z 316,0543 [М-Н]-; вычислено C12H9F3N3O4- 316,0551. ВЭЖХ-МС (ESI+): найдено m/z 318,0 [М+Н]+; вычислено C12H11F3N3O4+ 318,1.
Пример 77. Получение N-гидрокси-3-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)пропанамида I(23)
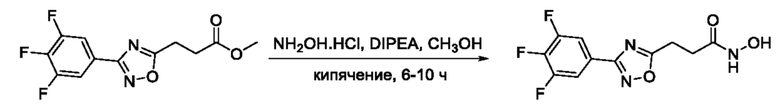
Получен способом, описанном в Примере 55 (за исключением условия кипячения реакционной смеси в данном случае) из метил 3-(3'-(3'',4'',5''-трифторфенил)-1',2',4'-оксадиазол-5'-ил)пропаноата в виде бежевого порошка с выходом 140 мг, 50%. Тпл 122,0°С (д). Элюент для очистки колоночной хроматографией - CH2Cl2:изо-С3Н7ОН (градиент от 100:0 до 75:25). 1H ЯМР (400 МГц, ДМСО-d6, δ): 10,55 (1Н, с, 5-ОН), 8,81 (1Н, с, 4-NH), 7,92-7,84 (2Н, м, 2''-СН, 6''-СН), 3,23 (2Н, m, J=7.1 Гц, 3-СН2), 2,58 (2Н, m, J=7.1 Гц, 2-СН2). 13С ЯМР (101 МГц, ДМСО-d6, δ): 181,04 (С-5'), 167,44 (С-1), 165,90 (С-3'), 151,23 (С-3'', С-5'', ∂∂∂, JF=248.9, 10.2, 3.9 Гц), 141,34 (С-4'', ∂m, JF=254.0, 15.3 Гц), 123,37-123,25 (С-1'', м), 112,63-112,40 (С-2'', С-6'', м), 28,73 (С-2), 22,34 (С-3). HRMS (ESI-): найдено m/z 286,0438 [М-Н]-; вычислено C11H7F3N3O3- 286,0445. ВЭЖХ-МС (ESI+): найдено m/z 288,0 [М+Н]+; вычислено C11H9F3N3O3+ 288,0.
Пример 78. Получение N-гидрокси-3-(3'-(3''-метилфенил)-1',2',4'-оксадиазол-5'-ил)пропанамида I(24)
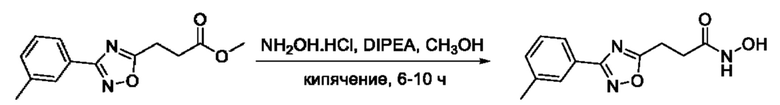
Получен способом, описанном в Примере 55 (за исключением условия кипячения реакционной смеси в данном случае) из метил 3-(3'-(3''-метил фенил)-1',2',4'-оксадиазол-5'-ил)пропаноата в виде бежевого порошка с выходом 125 мг, 40%. Тпл 115,8°С (д). Целевая гидроксамовая кислота очищалась путем осаждения гексаном из насыщенного раствора твердого остатка (после упаривания метанола) в CH2Cl2. 1Н ЯМР (300 МГц, ДМСО-d6, δ): 10,54 (1H, уш.с, 5-ОН), 8,81 (1H, уш.с, 4-NH), 7,83-7,80 (1H, м, 2''-СН), 7,79-7,77 (1H, м, 6''-СН), 7,45 (1H, m, J=7.3 Гц, 5''-СН), 7,43-7,38 (1H, м, 4''-СН), 3,21 (2Н, m, J=7.2 Гц, 3-СН2), 2,58 (2Н, m, J=7.2 Гц, 2-СН2), 2,40 (3Н, с, 6-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 180,07 (С-5'), 167,96 (С-3'), 167,51 (С-1), 139,06 (С-3''), 132,55 (С-4''), 129,58 (С-5''), 127,82 (С-2''), 126,69 (С-1''), 124,58 (С-6''), 28,81 (С-2), 22,29 (С-3), 21,34 (С-6). HRMS (ESI-): найдено m/z 246,0875 [М-Н]-; вычислено C12H12N3O3- 246,0884. ВЭЖХ-МС (ESI+): найдено m/z 248,1 [М+Н]+; вычислено C12H14N3O3+ 248,1.
Пример 79. Получение этил 5-(4-йод фенил)-1,3,4-оксадиазол-2-карбоксилата
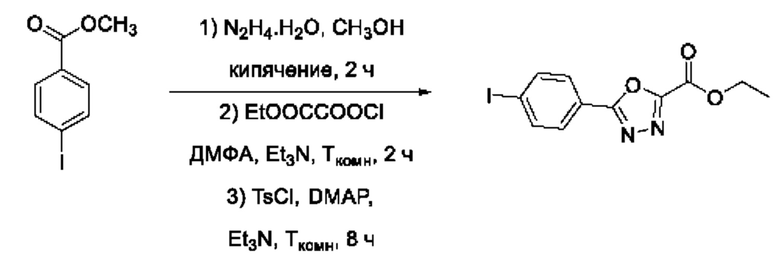
К раствору метил 4-йодбензоата (248 мг, 1,01 ммоль, 1 экв) в 5 мл метанола добавляли 60%) водный раствор N2H4⋅H2O (100 мкл, 10,1 ммоль, 10 экв). Реакционную смесь кипятили с обратным холодильником в течение 2 ч, ход реакции контролировали ТСХ (элюент - гексан:EtOAc = 3:1). После окончания реакции смесь охлаждали до комнатной температуры, раствор упаривали досуха на роторном испарителе, сушили с толуолом (3⋅10 мл) и лиофилизировали.
Полученный на первом этапе гидразид 4-йодбензойной кислоты растворяли в 10 мл ДМФА, добавляли Et3N (420 мкл, 3,03 ммоль, 3 экв), полученный раствор перемешивали при комнатной температуре 10 мин. Затем раствор охлаждали до +5°С и добавляли раствор этил оксалилхлорида (ПО мкл, 1,01 ммоль, 1 экв) в 10 мл ДМФА. Полученный после добавления раствор перемешивали при комнатной температуре в течение 2 ч, ход реакции контролировали ТСХ (элюент - гексан:EtOAc = 1:1).
После окончания ацилирования гидразида 4-йодбензойной кислоты, в реакционную смесь добавляли последовательно Et3N (140 мкл, 1,01 ммоль, 1 экв), DMAP (123 мг, 1,01 ммоль, 1 экв), раствор TsCl (190 мг, 1,01 ммоль, 1 экв) в 5 мл ДМФА. Реакционная смесь перемешивалась при комнатной температуре в течение 8 ч, после чего раствор обрабатывали деионизированной водой, целевой этил 5-(4-йодфенил)-1,3,4-оксадиазол-2-карбоксилат экстрагировали EtOAc (3⋅20 мл). Объединенную органическую фазу дополнительно промывали насыщенным водным раствором NaCl (2⋅10 мл) и сушили безводным Na2SO4 при интенсивном перемешивании, осушитель отфильтровывали, фильтрат упаривали досуха на роторном испарителе. Полученный твердый остаток очищали колоночной хроматографией на силикагеле, элюент - гексан:EtOAc = 3:1. Органические фракции, содержащие целевой эфир собирали, растворитель упаривали досуха на роторном испарителе с образованием этил 5-(4-йодфенил)-1,3,4-оксадиазол-2-карбоксилат с выходом 136 мг (55%). Розоватый порошок, 1Н ЯМР (300 МГц, ДМСО-d6, δ): 8,06-8,00 (2Н, м, 3-СН, 5-СН), 7,85-7,80 (2Н, м, 2-СН, 6-СН), 4,47 (2Н, к, J=7.1 Гц, 8-СН2), 1,38 (3Н, m, J=7.1 Гц, 9-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 165,44 (С-7), 157,00 (С-2' или С-5'), 154,42 (С-5' или С-2'), 138,99 (С-3, С-5), 129,16 (С-2, С-6), 122,46 (С-4), 101,29 (С-1), 63,45 (С-8), 14,34 (С-9). ВЭЖХ-МС (ESI+): найдено m/z 345,1 [М+Н]+; вычислено C11H10IN2O3 345,1.
Пример 80. Получение этил 5-(3-йод фенил)-1,3,4-оксадиазол-2-карбоксилата
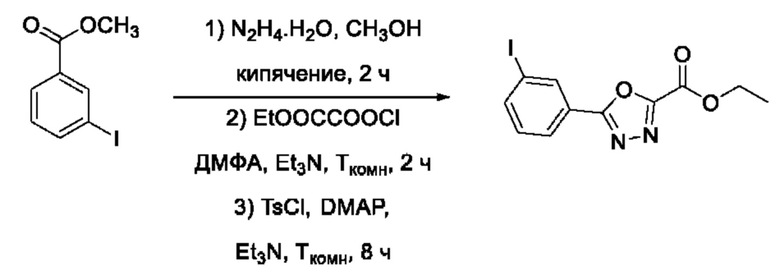
Получен способом, описанном в Примере 79 из метил 3-йодбензоата в виде белого порошка с выходом 385 мг, 60%. 1Н ЯМР (400 МГц, ДМСО-d6, δ): 8,33 (1H, m, J=1.7 Гц, 2-СН), 8,09-8,05 (2Н, м, 4-СН, 6-СН), 7,45 (1H, m, J=7.9 Гц, 5-СН), 4,47 (2Н, к, J=7.1 Гц, 8-СН2), 1,38 (3Н, m, J=7.1 Гц, 9-СН3). 13С ЯМР (101 МГц, ДМСО-d6, δ): 164,44 (С-7), 157,10 (С-2' или С-5'), 154,40 (С-5' или С-2'), 141,82 (С-4), 135,50 (С-2), 132,10 (С-5), 126,93 (С-6), 125,02 (С-1), 95,94 (С-3), 63,50 (С-8), 14,35 (С-9). ВЭЖХ-МС (ESI+): найдено m/z 345,0 [М+Н]+; вычислено C11H10IN2O3+ 345,0
Пример 81. Получение этил 5-(4-фтор-2-метилфенил)-1,3,4-оксадиазол-2-карбоксилата
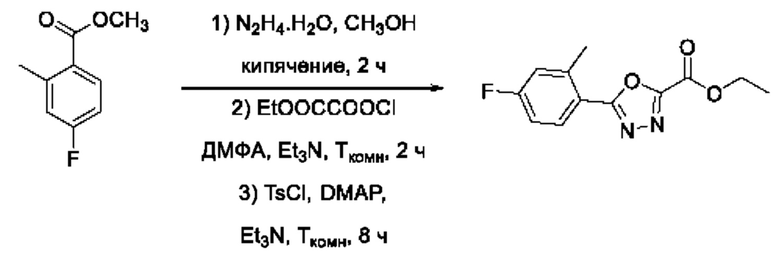
Получен способом, описанном в Примере 79 из метилового эфира 4-фтор-2-бензойной кислоты в виде белого порошка с выходом 175 мг, 45%. 1Н ЯМР (300 МГц, ДМСО-d6, δ): 8,00 (1Н, ∂∂, J=8.7, 5.9 Гц, 6-СН), 7,41-7,36 (1Н, м, 3-СН), 7,34-7,27 (1H, м, 5-СН), 4,47 (2Н, к, J=7.1 Гц, 9-СН2), 2,66 (3Н, с, 7-СН3), 1,38 (3Н, m, J=7.1 Гц, 10-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 165,99 (С-8), 164,32 (С-4, ∂, JF=251.2 Гц), 156,54 (С-2' или С-5'), 154,55 (С-5' или С-2'), 142,46 (С-1, ∂, JF=93 Гц), 142,40 (С-2), 132,43 (С-6, ∂, JF=9.8 Гц), 119,06 (С-3, ∂, JF=21.9 Гц), 114,38 (С-5, ∂, JF=22.2 Гц), 63,40 (С-9), 21,69 (С-7), 14,34 (С-10). ВЭЖХ-МС (ESI+): найдено m/z 251,1 [М+Н]+; вычислено for C12H12FN2O3+ 251,1.
Пример 82. Получение этил 5-(2,4-диметоксифенил)-1,3,4-оксадиазол-2-карбоксилата
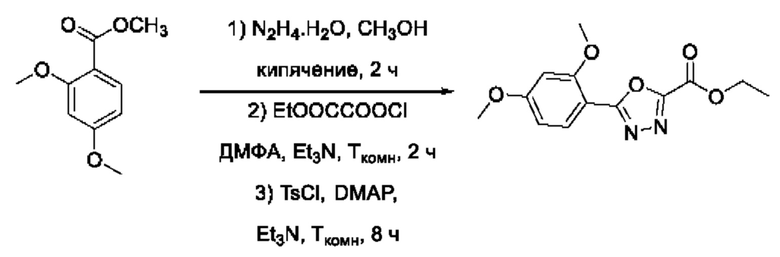
Получен способом, описанном в Примере 79 из метилового эфира 2,4-диметоксибензойной кислоты в виде белого порошка с выходом 385 мг, 40%. 1H ЯМР (300 МГц, ДМСО-d6, δ): 7,81-7,75 (1H, м, 6-СН), 6,67-6,60 (2Н, м, 5-СН, 3-СН), 4,30 (2Н, к, J=7.1 Гц, 10-СН2), 3,92 (3Н, с, 7-СН3 или 8-СН3), 3,85 (3Н, с, 8-СН3 или 7-СН3), 1,31 (3Н, m, J=7.1 Гц, 11-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 163,95 (С-9), 163,93 (С-2' или С-5'), 160,43 (С-2 или С-4), 159,34 (С-5' или С-2'), 156,64 (С-4 или С-2), 133,05 (С-6), 113,34 (С-1), 106,36 (С-5), 98,91 (С-3), 62,67 (С-10), 56,47 (С-8), 56,05 (С-7), 14,28 (С-11). ВЭЖХ-МС (ESI+): найдено m/z 279,1 [М+Н]+; вычислено C13H15N2O5+ 279,1.
Пример 83. Получение этил 5-(2,3-диметоксифенил)-1,3,4-оксадиазол-2-карбоксилата
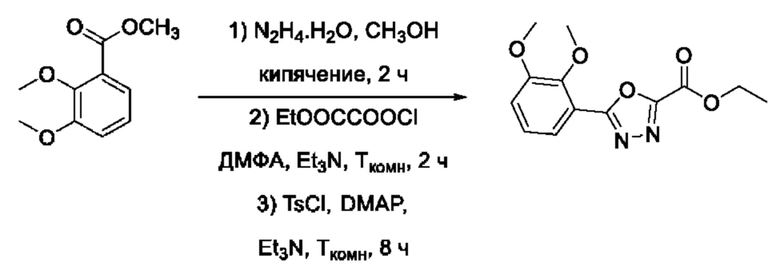
Получен способом, описанном в Примере 79 из метилового эфира 2,3-диметоксибензойной кислоты в виде белого порошка с выходом 135 мг, 65%. 1H ЯМР (300 МГц, ДМСО-d6, δ): 7,49 (1H, ∂∂, J=7.7, 1.7 Гц, 6-СН), 7,39 (1H, ∂∂, J=8.3, 1.7 Гц, 4-СН), 7,31 (1H, ∂∂, J=8.2, 7.7 Гц, 5-СН), 4,46 (2Н, к, J=7.1 Гц, 10-СН2), 3,90 (3Н, с, 7-СН3 или 8-СН3), 3,86 (3Н, с, 8-СН3 или 7-СН3), 1,38 (3H, m, J=7.1 Гц, 11-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 164,35 (С-9), 157,03 (С-2 или С-3), 154,52 (С-3 или С-2), 153,91 (С-2', С-5'), 148,12 (С-1), 125,52 (С-5), 121,51 (С-6), 117,59 (С-4), 63,3 5 (С-10), 61,43 (С-7 или С-8), 56,56 (С-8 или С-7), 14,32 (С-11). ВЭЖХ-МС (ESI+): найдено m/z 279,1 [М+Н]+; вычислено C13H15N2O5+ 279,1.
Пример 84. Получение этил 5-(3,4-диметоксифенил)-1,3,4-оксадиазол-2-карбоксилата
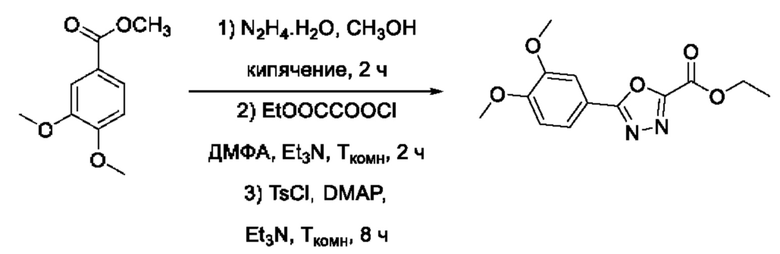
Получен способом, описанном в Примере 79 из метилового эфира 3,4-диметоксибензойной кислоты в виде белого порошка с выходом 96 мг, 50%. 1H ЯМР (400 МГц, ДМСО-d6, δ): 7,65 (1H, ∂∂, J=8.4, 2.1 Гц, 5-СН), 7,53 (1H, ∂, J=2.0 Гц, 2-СН), 7,20 (1H, ∂, J=8.5 Гц, 6-СН), 4,46 (2Н, к, J=7.1 Гц, 10-СН2), 3,88 (3Н, с, 7-СН3 или 8-СН3), 3,88 (3Н, с, 8-СН3 или 7-СН3), 1,38 (3Н, m, J=7.1 Гц, 11-СН3). 13С ЯМР (101 МГц, ДМСО-d6, δ): 165,93 (С-9), 156,59 (С-2' или С-5'), 154,55 (С-5' или С-2'), 153,13 (С-3), 149,68 (С-4), 121,46 (С-5), 115,10 (С-1), 112,64 (С-6), 109,99 (С-2), 63,34 (С-10), 56,29 (С-7 или С-8), 56,22 (С-8 или С-7), 14,36 (С-11). ВЭЖХ-МС (ESI+): найдено m/z 279,1 [М+Н]+; вычислено C13H15N2O5+ 279,1.
Пример 85. Получение N-гидрокси-5-(4-йодфенил)-1,3,4-оксадиазол-2-карбоксамида I(25)
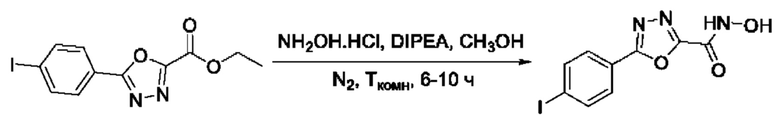
Получен способом, описанном в Примере 55 (за исключением очистки - целевая гидроксамовая кислота осаждалась при подкислении реакционной смеси 5% раствором серной кислоты) из этил 5-(4-йодфенил)-1,3,4-оксадиазол-2-карбоксилата. Белый порошок, 110 мг (85%). Тпл 184,4°С (д). 1H ЯМР (300 МГц, ДМСО-d6, δ): 12,15 (1H, уш.с, 9-ОН), 9,76 (1H, уш.с, 8-NH), 8,05-8,01 (2H, м, 3-СН, 5-СН), 7,87-7,82 (2H, м, 2-СН, 6-СН). 13С ЯМР (75 МГц, ДМСО-d6, δ): 164,82 (С-7), 157,96 (С-5'), 151,06 (С-2'), 138,89 (С-3, С-5), 129,10 (С-2, С-6), 122,70 (С-1), 100,88 (С-4). HRMS (ESI-): найдено m/z 329,9367 [М-Н]-; вычислено C9H5IN3O3- 329,9381. ВЭЖХ-МС (ESI+): найдено m/z 332,0 [М+Н]+; вычислено C9H7IN3O2+ 332,0.
Пример 86. Получение N-гидрокси-5-(3-йодфенил)-1,3,4-оксадиазол-2-карбоксамида I(26)
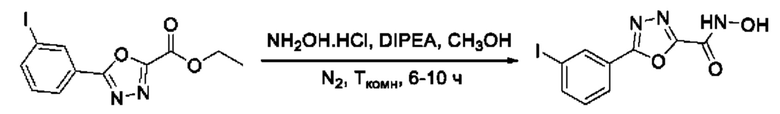
Получен способом, описанном в Примере 55 (за исключением очистки - целевая гидроксамовая кислота осаждалась при подкислении реакционной смеси 5% раствором серной кислоты) из этил 5-(3-йодфенил)-1,3,4-оксадиазол-2-карбоксилата. Белый порошок, 110 мг (85%). Тпл 274,9°С (д). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 8,37 (1H, с, 8-NH), 8,27 (1H, m, J=1.7 Гц, 2-СН), 8,01-7,97 (2Н, м, 4-СН, 6-СН), 7,41 (1H, m, J=7.9 Гц, 5-СН). 13С ЯМР (75 МГц, ДМСО-d6, δ): 163,81 (С-7), 161,78 (С-2'), 154,30 (С-5'), 140,56 (С-4), 134,86 (С-2), 131,95 (С-5), 126,23 (С-6), 126,10 (С-1), 95,80 (С-3). HRMS (ESI-): найдено m/z 329,9363 [М-Н]-; вычислено C9H5IN3O3- 329,9381. ВЭЖХ-МС (ESI+): найдено m/z 332,0 [М+Н]+; вычислено C9H7IN3O3+ 332,0.
Пример 87. Получение N-гидрокси-5-(4-фтор-2-метилфенил)-1,3,4-оксадиазол-2-карбоксамида I(27)
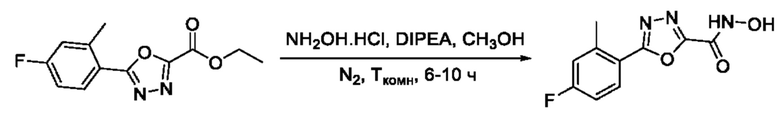
Получен способом, описанном в Примере 55 (за исключением очистки - целевая гидроксамовая кислота осаждалась при подкислении реакционной смеси 5% раствором серной кислоты) из этил 5-(4-фтор-2-метилфенил)-1,3,4-оксадиазол-2-карбоксилата. Розоватый порошок, 90 мг (60%). Тпл 259,6°С (д). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 8,50 (1H, уш.с, 8-NH), 7,93 (1Н, ∂∂, J=8.7, 6.0 Гц, 6-СН), 7,34 (1H, ∂∂, J=10.1, 2.7 Гц, 3-СН), 7,26 (1H, m∂, J=8.5, 2.8 Гц, 5-СН), 2,64 (3Н, с, 10-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 163,69 (С-4, ∂, JF=249.3 Гц), 162,84 (С-7), 162,59 (С-2'), 154,12 (С-5'), 141,54 (С-2, ∂, JF=8.8 Гц), 131,63 (С-6, ∂, JF=9.5 Гц), 120,09 (С-1, ∂, JF=2.7 Гц), 118,79 (С-3, ∂, JF=22.0 Гц), 114,07 (С-5, ∂, JF=22.0 Гц), 21,81 (С-10). HRMS (ESI-): найдено m/z 236,0463 [М-Н]-; вычислено C10H7FN3O3-236,0477. ВЭЖХ-МС (ESI+): найдено m/z 238,1 [М+Н]+; вычислено C10H9FN3O3+ 238,1.
Пример 88. Получение N-гидрокси-5-(2,4-диметоксифенил)-1,3,4-оксадиазол-2-карбоксамида I(28)
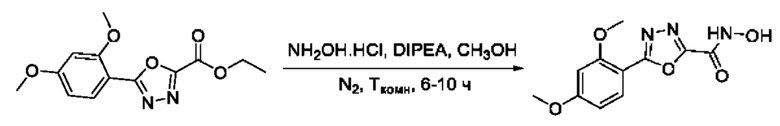
Получен способом, описанном в Примере 55 (за исключением очистки - целевая гидроксамовая кислота осаждалась при подкислении реакционной смеси 5% раствором серной кислоты) из этил 5-(2,4-диметоксифенил)-1,3,4-оксадиазол-2-карбоксилата. Белый порошок, 185 мг (65%). Тпл 197,3°С (д). 1Н ЯМР (300 МГц, ДМСО-d6, δ): 12,05 (1H, уш.с, 9-ОН), 9,68 (1H, с, 8-NH), 7,86 (1H, ∂, J=8.6 Гц, 6-СН), 6,79 (1H, ∂, J=2.2 Гц, 3-СН), 6,75 (1Н, ∂∂, J=8.7, 2.3 Гц, 5-СН), 3,92 (3Н, с, 10-СН3 или 11-СН3), 3,88 (3Н, с, 11-СН3 или 10-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 164,50 (С-7), 164,14 (С-2' или С-5'), 159,79 (С-2, С-4), 157,32 (С-5' или С-2'), 132,20 (С-6), 106,88 (С-5), 104,61 (С-1), 99,55 (С-3), 56,59 (С-10 или С-11), 56,20 (С-11 или С-10). HRMS (ESI-): найдено m/z 264,0615 [М-Н]-; вычислено C11H10N3O5- 264,0626. ВЭЖХ-МС (ESI-): найдено m/z 264,0 [М-Н]-; вычислено C11H10N3O5- 264,1.
Пример 89. Получение N-гидрокси-5-(2,3-диметоксифенил)-1,3,4-оксадиазол-2-карбоксамида I(29)
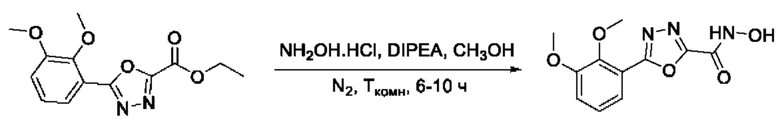
Получен способом, описанном в Примере 55 (за исключением очистки - целевая гидроксамовая кислота осаждалась при подкислении реакционной смеси 5% раствором серной кислоты) из этил 5-(2,3-диметоксифенил)-1,3,4-оксадиазол-2-карбоксилата. Розоватый порошок, 48 мг (85%). Тпл 166,7°С (д). 1H ЯМР (300 МГц, ДМСО-d6, δ): 12,11 (1Н, уш.с, 11-ОН), 9,74 (1H, уш.с, 10-NH), 7,49 (1Н, ∂∂, J=7.6, 1.7 Гц, 6-СН), 7,39-7,27 (2Н, м, 4-СН, 5-СН), 3,90 (3Н, с, 7-СН3 или 8-СН3), 3,85 (3Н, с, 8-СН3 или 7-СН3). 13С ЯМР (75 МГц, ДМСО-d6, δ): 163,65 (С-9), 157,98 (С-2' или С-5'), 153,85 (С-5' или С-2'), 151,29 (С-2 или С-3), 148,04 (С-3 или С-2), 125,41 (С-5), 121,58 (С-6), 117,80 (С-1), 117,31 (С-4), 61,42 (С-7 или С-8), 56,55 (С-8 или С-7). HRMS (ESI-): найдено m/z 264,0614 [М-Н]-; вычислено C11H10N3O5- 264,0626. ВЭЖХ-МС (ESI-): найдено m/z 264,1 [М-Н]-; вычислено C11H10N3O5- 264,1.
Пример 90. Получение N-гидрокси-5-(3,4-диметоксифенил)-1,3,4-оксадиазол-2-карбоксамида I(30)
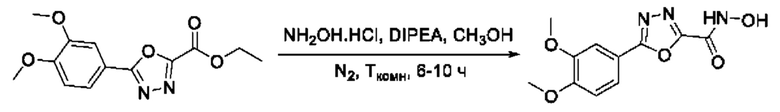
Получен способом, описанном в Примере 55 (за исключением очистки - целевая гидроксамовая кислота осаждалась при подкислении реакционной смеси 5% раствором серной кислоты) из этил 5-(3,4-диметоксифенил)-1,3,4-оксадиазол-2-карбоксилата. Белый порошок, 183 мг (65%). Тпл 130,7°С (д). 1Н ЯМР (400 МГц, ДМСО-d6, δ): 11,81 (1H, уш.с, 11-ОН), 9,85 (1H, уш.с, 10-NH), 7,68 (1Н, ∂∂, J=8.4, 2.0 Гц, 6-СН), 7,56 (1Н, ∂, J=2.0 Гц, 2-СН), 7,20 (1H, ∂, J=8.5 Гц, 5-СН), 3,88 (3Н, с, 7-СН3 или 8-СН3), 3,87 (3Н, с, 8-СН3 или 7-СН3). 13С ЯМР (101 МГц, ДМСО-d6, δ): 165,22 (С-9), 157,51 (С-2' или С-5'), 152,85 (С-5' или С-2'), 151,23 (С-4), 149,60 (С-3), 121,26 (С-6), 115,32 (С-1), 112,58 (С-5), 110,01 (С-2), 56,26 (С-7 или С-8), 56,20 (С-8 или С-7). HRMS (ESI-): найдено m/z 264,0610 [М-Н]-; вычислено C11H10N3O5- 264,0626. ВЭЖХ-МС (ESI-): найдено m/z 264,0 [М-Н]-; вычислено C11H10N3O5- 264,1.
Пример 91. Исследование цитотоксических свойств соединений I(1-30)
Общую цитотоксичность определяли в тесте на клеточной линии почки эмбриона человека HEK293 (1-1-609, Институт Цитологии Российской Академии Наук, Россия). Клетки культивировали в среде ДМЕМ (ПанЭко, Россия) с добавлением 10%) фетальной бычьей сыворотки (HyClone, Cytiva, Австрия), 2 мМ L-глутамина и 1% пенициллин/стрептомицин (100Х, Capricorn Scientific, Германия) не менее 1-2 пассажей после разморозки. Клетки трипсинизировали 0,05% раствором Тринсин-ЭДТА и повторно высевали в 96-луночные планшеты (Corning, США) в концентрации 1⋅104 клеток/100 мкл на лунку. Инкубировали во влажной атмосфере в СО2 инкубаторе при 5% СО2 и 37°С в течении 24 часов. Исследуемые соединения титровали последовательно в ДМСО и в клеточной среде, переносили полученные после титрования растворы в 96-луночные планшеты с клетками и оставляли на инкубацию в течении 72 часов в CO2 инкубаторе при 5% CO2 и 37°С. После инкубации проводили детекцию при помощи витального красителя AlamarBlue (Invitrogen, США), в лунки клеточного планшета вносили по 10 мкл красителя, и оставляли планшет на 3 часа инкубации в СО2 инкубаторе при 5% СО2 и 37°С. Флуоресцентный сигнал детектировали на спектрофотометре (BMG Labtech, Германия) при длине волны возбуждения 545 нм и испускания 600 нм. Для определения соответствующих значений 50% клеточной цитотоксической концентрации (СС50) к полученным данным использовали четырехпараметрическую модель кривой при помощи программного обеспечения GraphPad Prism 8. Полученные результаты представлены в таблице 4. В качестве контрольного соединения с высокой токсичностью использовали Доксорубицин (Sigma-Aldrich, США).
Пример 92. Исследования антибактериальной и потенцирующей активности соединений I(1-30)
Штамм Escherichia coli MG1655 и штамм Pseudomonas aeruginosa АТСС 27853 были получены из Американской коллекции типовых культур (АТСС; Роквилл, Мэриленд, США). Субкультивирование и подготовку инокулята готовили в соответствии с рекомендованным Институтом клинических и лабораторных стандартов (CLSI) методом М7-А9 [17] с использованием агара Мюллера-Хинтона и бульона Мюллера-Хинтона, которые были приобретены у компании «HiMedia» (Индия). Рифампицин и налидиксовая кислота приобретены у компании «Sigma Aldrich» (США). Кривые роста получали на автоматизированной системе анализа роста Bioscreen С. Культуры Е. Coli MG1655 выращивали в течение ночи при 37°С, разбавляли свежей средой в соотношении 1:100, инокулировали в лунки в трех повторностях и выращивали при 37°С при максимальном встряхивании на платформе прибора Bioscreen С. Когда культуры достигали значения оптической плотности (OD600) равного 0,2, клетки обрабатывали налидиксовой кислотой (4 мг/мл), рифампицином (5 мг/мл) или канамицином (0,3 мг/мл) в присутствии или в отсутствии потенциальных ингибиторов LpxC 1-30 в концентрации 0,01-0,1 мМ и инкубировали при 37°С в течение 19 часов. Значения OD600 записывали автоматически в заданное время и наносили на график средние значения трехкратных культур. Структурные формулы соединений общей формулы I, показавших эффект потенцирования действия вышеупомянутых антибиотиков, представлены на фигуре 9. Репрезентативные кривые роста E. Coli MG1655 в присутствии или отсутствии потенциальных ингибиторов представлены на фигурах 10-11. Минимальные ингибирующие концентрации (МИК) для рифампицина и налидиксовой кислоты в отношении P. aeruginosa АТСС 27853 определяли с помощью модифицированного метода микроразведения в бульоне в трех экземплярах в соответствии с рекомендациями CLSI-М7-А9 [17] и М100-А30 [18]. Антибиотики серийно разводили в два раза в 100 мкл бульона Мюллера-Хинтона. Инокулят бактерий составлял 100 мкл разведения 1,0⋅106 КОЕ/мл в бульоне Мюллера-Хинтона. МИК для антибиотиков в присутствии соединений 9 и 10, изображенных на фигуре 12, определяли тем же методом. Рифампицин или налидиксовая кислота находились в диапазоне от 4⋅МИК до 1/128⋅МИК и включали нулевую концентрацию для каждого антибиотика в сочетании с соединениями 9 или 10 (100 мкМ, 50 мкМ и 25 мкМ) в равном объеме 50 мкл. Исходные растворы соединений 9 и 10 готовили в ДМСО, общая концентрация ДМСО в лунке не превышала 1%. Конечные значения МИК определяли после 24 часов инкубации с помощью считывателя поглощения микропланшетов iMark (Bio-Rad, США). Кривые роста P. aeruginosa АТСС 27853, изображенные на фигуре 13, при добавлении налидиксовой кислоты или рифампицина в присутствии гидроксамовых кислот 9 и 10 или без них получали с помощью многофункционального микропланшетного считывателя SuperMax 3100 (ShanghaiFlash, Китай). Клетки выращивали в трех повторностях при 37°С с аэрацией.
Пример 93. Определение кинетической растворимости соединений-лидеров общей формулы I
Исходные растворы анализируемых соединений готовили в ДМСО с концентрацией 10 мМ. В полипропиленовую пробирку объемом 1,5 мл вносили 990 мкл 0,1 М фосфатного буферного раствора (рН=7,4) и добавляли 10 мкл исходного раствора анализируемого соединения. Эксперимент проводили в трех повторах. Полученные растворы помещали в термошейкер и перемешивали в течении 2 часов при 300 об/мин при 25°С, далее фильтровали с использованием шприцевых фильтров (PVDF, 0,45 мкм). В виалу вместимостью 2 мл переносили 320 мкл фильтрата и добавляли 80 мкл ацетонитрила, перемешивали и проводили ВЭЖХ-УФ анализ на жидкостном хроматографе Vanquish Flex (Thermo Scientific, США) с диодно-матричным детектором DAD FG (Thermo Scientific, США). Разделение определяемых соединений проводили на колонке Agilent Poroshell 120 ЕС CI8 (100 мм⋅2,0 мм, размер частиц 1,9 мкм) в следующих хроматографических условиях: с 0 по 3,5 мин - элюирование от 5% до 95% подвижной фазы Б, с 3,5 по 4,5 мин - элюирование в 95% подвижной фазы Б, с 4,5 по 8 мин - уравновешивание хроматографической колонки в 95% подвижной фазы А. Подвижная фаза А - 0,1% муравьиная кислота в воде, подвижная фаза Б - 0,1% муравьиная кислота в ацетонитриле. Температура колонки - 40°С. Объем вводимой пробы - 5 мкл, скорость потока - 0,4 мл/мин. Концентрацию анализируемого соединения в фильтратах (растворимость) определяли по калибровочной кривой построенной в диапазоне от 1 мкМ до 100 мкМ. В качестве контрольных соединений использовали диклофенак натрия (высокая растворимость) и никардипин (низкая растворимость).
На основе полученных данных оценивали растворимость 5 соединений-лидеров. Все соединения обладали высокой растворимостью (>100 мкМ), результаты представлены в таблице 5.
Пример 94. Определение стабильности соединений лидеров общей формулы I в микросомах печени
Раствор анализируемого соединения в концентрации 1 мкМ инкубировали с микросомальной фракцией печени крыс (Gibco, США) при 37°С в присутствии NADPH (кофактор ферментов фазы I метаболизма) или UDPGA (кофактор ферментов фазы II метаболизма). В качестве буферного раствора использовали 0,1 М фосфатный буферный раствор (рН=7,4). Объем инкубационной смеси 600 мкл. Во временные точки 0, 5, 15, 30, 45 мин отбирали в полипропиленовую пробирку объемом 0,5 мл 50 мкл инкубационной смеси и останавливали реакцию путем добавления 100 мкл ацетонитрила. Центрифугировали пробирки при 10000⋅g в течение 10 мин при температуре 4°С. Супернатант объемом 100 мкл переносили в виалу вместимостью 2 мл со стеклянной вставкой и проводили ВЭЖХ-МС/МС анализ на жидкостном хроматографе UltiMate 3000 UHPLC (Thermo, США) с масс-спектрометрическим детектором EVOQ Elite (Bruker, США) с ионизацией электрораспылением. По кинетике убыли тестируемого соединения в процессе инкубации рассчитывали период полувыведения (t1/2) и клиренс (CLint). С помощью программы MS Workstation software (version 8.2.1; Bruker, Германия) рассчитывали площадь пика анализируемого соединения. Далее строили график в координатах «натуральный логарифм площади пика» - «время инкубации».
Период полувыведения рассчитывали по формуле: t1/2=-0,693/slope, где t1/2 - период полувыведения, мин; Slope - угол наклона графика зависимости натурального логарифма площади пика от времени инкубации. Клиренс рассчитывали по формуле: CLint=(0,693/ti/2)-(incubation volume/microsomal protein), где CLint - клиренс, мкл/мин/мг; t1/2 - период полувыведения, мин; Incubation volume - объем инкубационной смеси, мкл; Microsomal protein - содержание микросомального белка в инкубационной смеси, мг.
Эксперимент проводили в двух повторах. В качестве контрольных соединений использовали верапамил (субстрат цитохрома Р450) и 7-гидроксикумарин (субстрат УДФ-глюкуронилтрансферазы). Результаты экспериментов показали, что 5 соединений-лидеров общей формулы I обладают высокой стабильностью к действию ферментов I фазы метаболизма (изоферменты цитохрома Р450) и ферментов II фазы метаболизма (УДФ-глюкуронилтрансферазы), результаты представлены в таблице 6.
Пример 95. Определение проницаемости соединений-лидеров на клетках линии Сасо-2
На полупроницаемую мембрану (ПЭТ-мембрана, размер пор: 0,4 мкм, площадь поверхности: 0,33 см2, Corning, США) планшета, состоящего из двух камер, высеивали клетки колоректальной аденокарциномы Сасо-2 и культивировали их в течение 21 дня до образования монослоя. Целостность монослоя оценивали с использованием красителя Lucifer yellow. Для оценки транспорта в прямом направлении А-В (от апикальной к базолатеральной мембране) в апикальную камеру (камера-донор) добавляли 300 мкл раствора анализируемого соединения (10 мкМ в растворе Хенкса). В базолатеральную камеру (камера-реципиент) добавляли 800 мкл раствора Хенкса и инкубировали планшет при 37°С в атмосфере CO2 (5%) в течение 2 часов. Для оценки транспорта в направлении В-А (от базолатеральной к апикальной мембране) в апикальную камеру (камера-реципиент) добавляли 300 мкл раствора Хенкса. В базолатеральную камеру (камера-донор) добавляли 800 мкл раствора анализируемого соединения (10 мкМ в растворе Хенкса) и инкубировали планшет при 37°С в атмосфере СО2 (5%) в течение 2 часов. В полипропиленовые пробирки объемом 0,5 мл отбирали аликвоты (100 мкл) из базолатеральной и апикальной камер, добавляли 300 мкл ацетонитрила и перемешивали. Центрифугировали пробирки при 10000в течение 10 мин при температуре 4°С. Супернатант объемом 100 мкл переносили в виалу вместимостью 2 мл со стеклянной вставкой и проводили ВЭЖХ-МС/МС анализ на жидкостном хроматографе UltiMate 3000 UHPLC (Thermo, США) с масс-спектрометрическим детектором EVOQ Elite (Braker, США) с ионизацией электрораспылением. Коэффициент кажущейся проницаемости рассчитывали по формуле: Рарр=(V⋅ΔC)/(Δt⋅A⋅C0), где Рарр - коэффициент кажущейся проницаемости, см/сек; V - объем раствора Хенкса в камере-реципиенте (в тесте А-В - 0,8 см3; в тесте В-А - 0,3 см3), см3; Δt - время инкубации (7200 сек), сек; А - площадь поверхности мембраны, на которой культивируются клетки (0,33 см2), см2; С0 - начальная концентрация анализируемого соединения в камере-доноре (10 мкМ), мкМ; ΔС - изменение концентрации анализируемого соединения после инкубации (в тесте А-В -концентрация в образце из базолатеральной камеры; в тесте В-А -концентрация в образце из апикальной камеры), мкМ. Эксперимент проводили в двух повторах. В качестве контрольных соединений использовали Атенолол (соединение с низкой проницаемостью) и пропранолол (соединение с высокой проницаемостью). На основе полученных данных оценивали проницаемость 5 соединений-лидеров. Обнаружено, что все соединения-лидеры обладают высокой проницаемостью, результаты представлены в таблице 7. Отношение коэффициентов кажущейся проницаемости Рарр(В-А) к Рарр(А-В) меньше 2, что свидетельствует о том, что все лидерные соединения не являются субстратом транспортных белков гликопротеина-Р и белка резистентности рака молочной железы (BCRP).
Пример 96. Определение стабильности соединений-лидеров общей формулы I в плазме крови человека
В полипропиленовую пробирку объемом 1,5 мл вносили 98 мкл пулированной плазмы крови человека, добавляли 2 мкл растворов соответствующих анализируемых соединений-лидеров с концентрацией 500 мкМ. Пробирки помещали в термошейкер на 4 часа при 37°С, 400 rpm. Пробирки вынимали из термошейкера, вносили 400 мкл ацетонитрила и перемешивали, после чего центрифугировали при 10000⋅g в течение 10 мин при температуре 4°С. Супернатант объемом 100 мкл переносили в виалу вместимостью 2 мл со стеклянной вставкой и проводили ВЭЖХ-МС/МС анализ на жидкостном хроматографе UltiMate 3000 UHPLC (Thermo, США) с масс-спектрометрическим детектором EVOQ Elite (Bruker, США) с ионизацией электрораспылением. Процент оставшегося после инкубации соединения рассчитывали по следующей формуле: % remaining plasma=(Sпосле инкубации, плазма /S0, плазма)⋅100, где % remaining plasma - процент оставшегося соединения после инкубации с плазмой крови в течение 4 часов при 37°С, %; Sпосле инкубации, плазма - площадь хроматографического пика, измеренная в образце после инкубации с плазмой крови в течение 4 часов при 37°С; S0, плазма - площадь хроматографического пика, измеренная в образце плазмы крови без проведения инкубации (нулевая точка). Эксперимент проводили в трех повторах. В качестве контрольных соединений использовали верапамил (стабильное в плазме крови соединение) и пропантелин бромид (нестабильное в плазме крови соединение). Обнаружено, что все соединения-лидеры являются стабильными в плазме крови человека, результаты представлены в таблице 8.
Пример 97. Определение стабильности соединений-лидеров общей формулы I в искусственном кишечном соке и искусственном желудочном соке
Искусственный кишечный сок (SIF) готовили путем добавления панкреатина к искусственному кишечному соку без фермента (Ricca, США, кат. номер 7109.75-16). Концентрация панкреатина в искусственном кишечном соке составляла 10 мг/мл. Искусственный желудочный сок (SGF) готовили путем добавления пепсина к искусственному желудочному соку без фермента (Ricca, США, кат. номер 7108-16). Концентрация пепсина в искусственном желудочном соке составляли 1,6 мг/мл. В полипропиленовую пробирку объемом 1,5 мл вносили 98 мкл искусственного кишечного сока или искусственного желудочного сока, добавляли 2 мкл раствора анализируемого соединения с концентрацией 500 мкМ. Помещали пробирки в термошейкер на 2 часа при 37°С, 400 rpm. После, вынимали пробирки из термошейкера, вносили 400 мкл ацетонитрила и перемешивали. Центрифугировали при 10000 -g в течение 10 мин при температуре 4°С. Супернатант объемом 100 мкл переносили в виалу вместимостью 2 мл со стеклянной вставкой и проводили ВЭЖХ-МС/МС анализ на жидкостном хроматографе UltiMate 3000 UHPLC (Thermo, США) с масс-спектрометрическим детектором EVOQ Elite (Bruker, США) с ионизацией электрораспылением. Процент оставшегося после инкубации соединения рассчитывали по следующей формуле: % remaining SIF/SGF=(Sпосле инкубации, SIF/SGF / S0, SIF/SGF)⋅100, где % remaimng SGF/SIF - процент оставшегося соединения после инкубации в искусственном кишечном соке или искусственном желудочном соке в течение 2 часов при 37°С, %; Sпосле инкубации, SIF/SGF - площадь хроматографического пика, измеренная в образце после инкубации в искусственном кишечном соке или искусственном желудочном соке в течение 2 часов при 37°С; S0, SIF/SGF - площадь хроматографического пика, измеренная в образце искусственного кишечного сока или искусственного желудочного сока без проведения инкубации (нулевая точка). Эксперимент проводили в трех повторах. В качестве контрольных соединений использовали верапамил (стабильное соединение) и омепразол (нестабильное в кислой среде соединение). Обнаружено, что все соединения-лидеры являются стабильными в искусственном кишечном соке и искусственном желудочном соке, результаты приведены в таблице 9.
Пример 98. Определение свободной и связанной фракции соединения в плазме крови методом равновесного диализа
В плашку для проведения равновесного диализа RED Device Reusable Base Plate (Thermo, США) помещали вставки RED Device Inserts (Thermo, США). В правую камеру вставки приливали 500 мкл фосфатного буферного солевого раствора. В левую камеру (красного цвета) вставки приливали 300 мкл пулированной плазмы крови человека с концентрацией исследуемого соединения 5 мкМ. Плашку инкубировали при 37°С в атмосфере CO2 (5%) в течение 6 часов. Вынимали плашку из инкубатора, отбирали в новую полипропиленовую пробирку объемом 1,5 мл 100 мкл плазмы из левой камеры, приливали 100 мкл фосфатного буферного солевого раствора и 600 мкл ацетонитрила, перемешивали. Из правой камеры отбирали в новую полипропиленовую пробирку объемом 1,5 мл 100 мкл фосфатного буферного солевого раствора, приливали 100 мкл пулированной плазмы крови человека и 600 мкл ацетонитрила, перемешивали. Центрифугировали пробирки при 10000⋅g в течение 10 мин при температуре 4°С. Супернатант объемом 100 мкл переносили в виалу вместимостью 2 мл со стеклянной вставкой и проводили ВЭЖХ-МС/МС анализ на жидкостном хроматографе UltiMate 3000 UHPLC (Thermo, США) с масс-спектрометрическим детектором EVOQ Elite (Bruker, США) с ионизацией электрораспылением. Долю соединения, связавшегося с белками плазмы рассчитывали по следующей формуле: fb (%)=[1-(Cb/Ср)]⋅100, где fb (%) - доля соединения, связавшегося с белками плазмы; Cb - концентрация соединения в камере с буферным раствором (правая камера); Ср - концентрация соединения в камере с плазмой (левая камера).
Эксперимент проводили в двух повторах. В качестве контрольных соединений использовали пропранолол (высокая степень связывания с белками плазмы крови) и атенолол (низкая степень связывания с белками плазмы крови).
Соединения общей формулы I(17, 19, 26) обладают очень высокой степенью связывания с белками плазмы крови человека (>97%), в то время как соединения I(9, 10) - обладают высокой степенью связывания с белками плазмы крови человека (85-90%), результаты представлены в таблице 10.
Пример 99. Определение коэффициента распределения logD7.4 для соединений-лидеров общей формулы I
В полипропиленовую пробирку объемом 1,5 мл вносили 245 мкл н-октанола, насыщенного 0,1 М фосфатным буферным раствором. Далее добавляли 5 мкл раствора анализируемого соединения (10 мМ в ДМСО) и 500 мкл 0,1 М фосфатного буферного раствора, насыщенного н-октанолом. Полученную смесь интенсивно перемешивали на вортексе в течении 3 часов при комнатной температуре. Центрифугировали пробирки при 1500⋅g в течение 5 мин при комнатной температуре и оставляли их на 3 часа при комнатной температуре для полного разделения слоев. Отбирали 50 мкл верхней фазы (н-октанол) и переносили в полипропиленовую пробирку объемом 0,5 мл, добавляли 100 мкл н-октанола, насыщенного 0,1 М фосфатным буферным раствором и перемешивали. Отбирали 50 мкл нижней фазы (0,1 М фосфатный буферный раствор) и переносили в полипропиленовую пробирку объемом 0,5 мл, добавляли 100 мкл 0,1 М фосфатного буферного раствора, насыщенного н-октанолом и перемешивали. Далее 100 мкл каждого из растворов переносили в виалы вместимостью 2 мл со стеклянной вставкой и проводили ВЭЖХ-МС/МС анализ на жидкостном хроматографе UltiMate 3000 UHPLC (Thermo, США) с масс-спектрометрическим детектором EVOQ Elite (Bruker, США) с ионизацией электрораспылением.
Коэффициент распределения между н-октанолом и 0,1 М фосфатным буферным раствором рассчитывали по следующей формуле: logD7.4=log10 (Soctanol/Sbuffer), где logD7.4 - коэффициент распределения анализируемого соединения между н-октанолом и 0,1 М фосфатным буферным раствором; Soctanol - площадь хроматографического пика, измеренная в образце н-октанола (верхний слой); Sbuffer - площадь хроматографического пика, измеренная в образце 0,1 М фосфатного буферного раствора (нижний слой). Эксперимент проводили в трех повторах. В качестве контрольных соединений использовали парацетамол (logD7.4<1) и кетоконазол (logD7.4>3). С помощью программы ACD/Labs Percepta Platform (ACD/Labs Release 2021.2.2, Advanced Chemistry Development, Канада, https://www.acdlabs.com) определяли расчетные значения logD7.4 и сравнивали их с экспериментальными. Измеренные значения logD7.4 для соединений-лидеров общей формулы I находятся в диапазоне от 0,5 до 1,5, результаты представлены в таблице 11.
Заявителем предприняты меры, направленные на повышение достоверности данных исследования, минимизацию/исключение субъективности оценки. Молекулярная структура и чистота синтезированных веществ подтверждены с помощью комплекса физико-химических методов анализа, что сводит вероятность ошибки и необъективной интерпретации результатов к минимуму. Все конечные результаты настоящего исследования являются объективными, поскольку получены с использованием инструментальных методов анализа, в которых возможность влияния на результаты измерений сведена к минимуму. Меры, направленные на повышение достоверности данных исследования, включают в себя использование подходящих вспомогательных и расходных материалов высокого качества, однозначное установление структуры полупродуктов и конечных веществ с помощью методов ЯМР и ВЭЖХ-МС, использование подходящих методов во время их выделения и очистки.
Соединения, которые являются эффективными в потенцировании действия антибиотиков в отношении грамотрицательных бактерий, будут затем образовывать основу для следующего этапа разработки потенциальных лекарственных средств. В первую очередь соединения-лидеры, обладающие эффективностью в потенцировании действия антибиотиков в отношении клеточных линий Е. coli MG1655, P. aeruginosa АТСС 27853, а также показавшие наилучшие ADME-свойства и низкую цитотоксичность, далее будут тестироваться на расширенной панели грамотрицательных бактерий, а также с расширенным рядом антибиотиков различного строения для установления необходимых закономерностей. В то же время структура соединений-лидеров (в частности, (9 и 10) будет подвергаться химическим модификациям, а параллельно с этим будет осуществляться синтез структурно близких к соединениям-лидерам данного изобретения гидроксамовых кислот с целью получения ингибиторов, обладающих сравнимой/улучшенной эффективностью в потенцировании в совокупности с улучшенным профилем ADMET-свойств. Вместе с тем, вероятно, будет привлечена процедура молекулярного моделирования с целью поиска наиболее оптимальных структур потенциальных ингибиторов, которые далее будут синтезироваться и исследоваться в аналогичных экспериментах.
Предлагаемые малые молекулы-ингибиторы биосинтеза ЛПС бактерий, отвечающие целевому профилю, могут быть востребованы фармацевтическими компаниями для создания на их основе терапевтического препарата для лечения различных заболеваний, вызванных грамотрицательными микроорганизмами.
Эквиваленты.
Специалистам в данной области будут очевидны, или они смогут достоверно установить при помощи обычных экспериментальных методик, многочисленные эквиваленты конкретных вариантов реализации, описанные в настоящем документе. Предполагается, что такие эквиваленты входят в объем охраны следующей формулы изобретения.
Резюмируя вышесказанное, предлагаемое изобретение может быть признано соответствующей критерию патентоспособности «Промышленная применимость» во всех частных формах воплощения признаков формулы изобретения до даты испрашиваемого приоритета.
Источники информации
1. Niu, Z.; Lei, P.; Wang, Y.; Wang, J.; Yang, J.; Zhang, J. Small Molecule LpxC Inhibitors against Gram-Negative Bacteria: Advances and Future Perspectives. Eur. J. Med. Chem. 2023, 253, 115326.
2. Kalinin, D. V.;Holl, R. LpxC Inhibitors: A Patent Review (2010-2016). Expert Opin. Ther. Pat. 2017, 27, 1227-1250.
3. Frackenpohl, J.; Schneider, L.; Decker, L.J.B.; Dittgen, J.; Fenkl, F.; Fischer, C; Franke, J.; Freigang, J.; Getachew, R.; Gonzalez Fernandez-Nino, S.M.; et al. Identifying New Lead Structures to Enhance Tolerance towards Drought Stress via High-Throughput Screening Giving Crops a Quantum of Solace. Bioorg. Med. Chem. 2019, 27, 115142.
4. Perman, S.M.; Goyal, M.; Gaieski, D.F. Initial Emergency Department Diagnosis and Management of Adult Patients with Severe Sepsis and Septic Shock. Scand. J. Trauma. Resusc. Emerg. Med. 2012, 20, 1-11.
5. Negash; Norris; Hodgkinson Siderophore-Antibiotic Conjugate Design: New Drugs for Bad Bugs? Molecules 2019, 24, 3314.
6. Chernyshov, V.V.; Kuzovlev, A.S.; Cherepanova, N.D.; Kasatkina, M.A.; Ivanov, R.A. Siderophore-antibiotic Conjugates: Structural Diversity and Antibacterial Activity. Clin. Microbiol. Antimicrob. Chemother. 2022,24, 314-344.
7. Durand, G.A.; Raoult, D.; Dubourg, G. Antibiotic Discovery: History, Methods and Perspectives. Int. J. Antimicrob. Agents 2019, 53, 371-382.
8. Baym, M.; Stone, L.K.; Kishony, R. Multidrug Evolutionary Strategies to Reverse Antibiotic Resistance. Science 2016, 351.
9. Cal, P.M.S.D.; Matos, M.J.; Bernardes, G.J.L. Trends in Therapeutic Drag Conjugates for Bacterial Diseases: A Patent Review. Expert Opin. Ther. Pat. 2017, 27, 179-189.
10. Skwarczynski, M.; Bashiri, S.; Yuan, Y.; Ziora, Z.M.; Nabil, O.; Masuda, K.; Khongkow, M.; Rimsueb, N.; Cabral, H.; Ruktanonchai, U.; et al. Antimicrobial Activity Enhancers: Towards Smart Delivery of Antimicrobial Agents. Antibiotics 2022, 11, 412.
11. Chawla, M.; Verma, J.; Gupta, R.; Das, B. Antibiotic Potentiators Against Multidrug-Resistant Bacteria: Discovery, Development, and Clinical Relevance. Front. Microbiol. 2022, 13, 1-19.
12. Tamma, P.D.; Cosgrove, S.E.; Maragakis, L.L. Combination Therapy for Treatment of Infections with Gram-Negative Bacteria. Clin. Microbiol. Rev. 2012, 25, 450-470.
13. Barb, A.W.; Jiang, L.; Raetz, C.R.H.; Zhou, P. Structure of the Deacetylase LpxC Bound to the Antibiotic CHIR-090: Time-Dependent Inhibition and Specificity in Ligand Binding. Proc. Natl. Acad. Sci. 2007,104, 18433-18438.
14. Lemaitre, N.; Liang, X.; Najeeb, J.; Lee, C.-J.; Titecat, M.; Leteurtre, E.; Simonet, M.; Toone, E.J.; Zhou, P.; Sebbane, F. Curative Treatment of Severe Gram-Negative Bacterial Infections by a New Class of Antibiotics Targeting LpxC. MBio 2017, 8, e00674-17.
15. Candelli, M.; Franza, L.; Pignataro, G.; Ojetti, V.; Covino, M.; Piccioni, A.; Gasbarrini, A.; Franceschi, F. Interaction between Lipopolysaccharide and Gut Microbiota in Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2021, 22, 6242.
16. Pirrung, M.C.; Tumey, L.N.; McClerren, A.L.; Raetz, C.R.H. High-Throughput Catch-and-Release Synthesis of Oxazoline Hydroxamates. Structure-Activity Relationships in Novel Inhibitors of Escherichia Coli LpxC: In Vitro Enzyme Inhibition and Antibacterial Properties. J. Am. Chem. Soc. 2003,125,1575-1586.
17. Clinical and Laboratory Standards Institute. 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 9th ed. CLSI document M7-A9. Clinical and Laboratory Standards Institute, Wayne PA.
18. Clinical and Laboratory Standards Institute. 2020. Performance Standards for Antimicrobial Susceptibility Testing, 30th ed. CLSI document M100-A30. Clinical and Laboratory Standards Institute, Wayne PA.
19. Патент на изобретение US 8748466 (В2) от 10.06.2014 (ABRAMITE JOSEPH ALAN [US]; BROWN MATTHEW FRANK [US]; CHEN JINSHAN MICHAEL [US]; MELNICK MICHAEL JOSEPH [US]; MONTGOMERY JUSTIN IAN [US]; REILLY USA [US]; PFIZER [US]) «Isoxazole derivatives useful as antibacterial agents».
20. Патентная заявка WO 9742179 (A1) от 13.11.1997 (MERCK & CO INC [US]; PATCHETT ARTHUR A [US]; NARGUND RAVI [US]; CHEN MENG HSIN [US]; ONI SHI H RUSSELL [US]), «Antibacterial agents» (прототип).
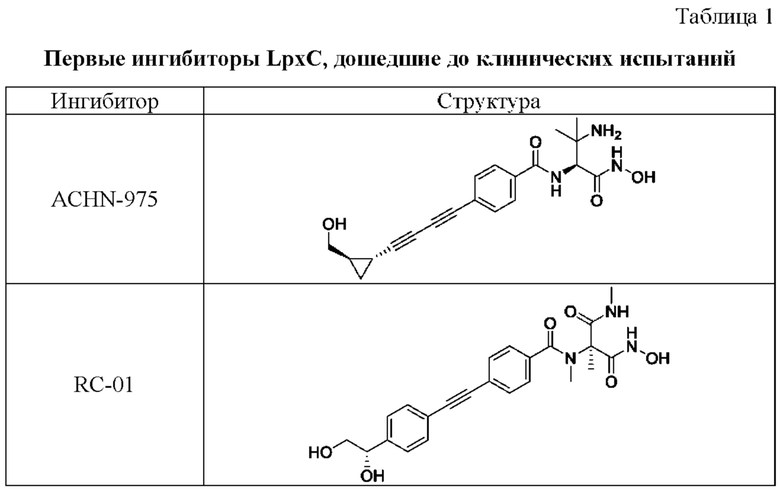
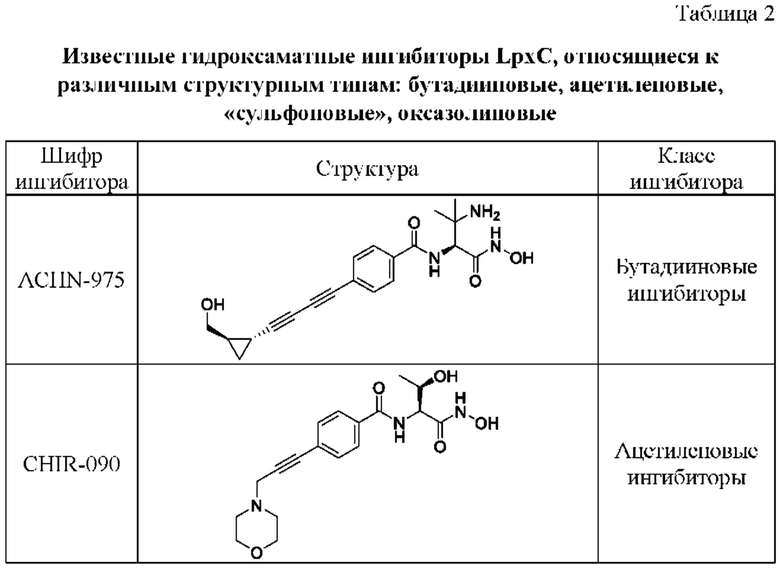
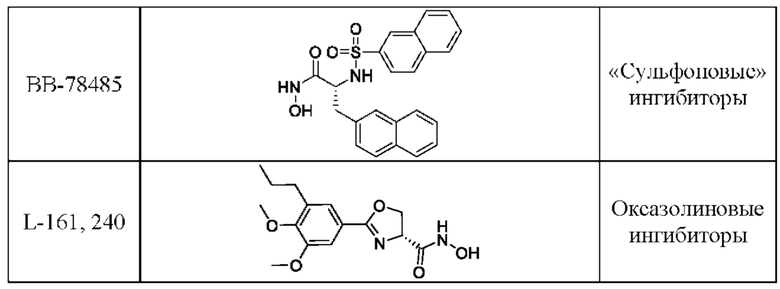
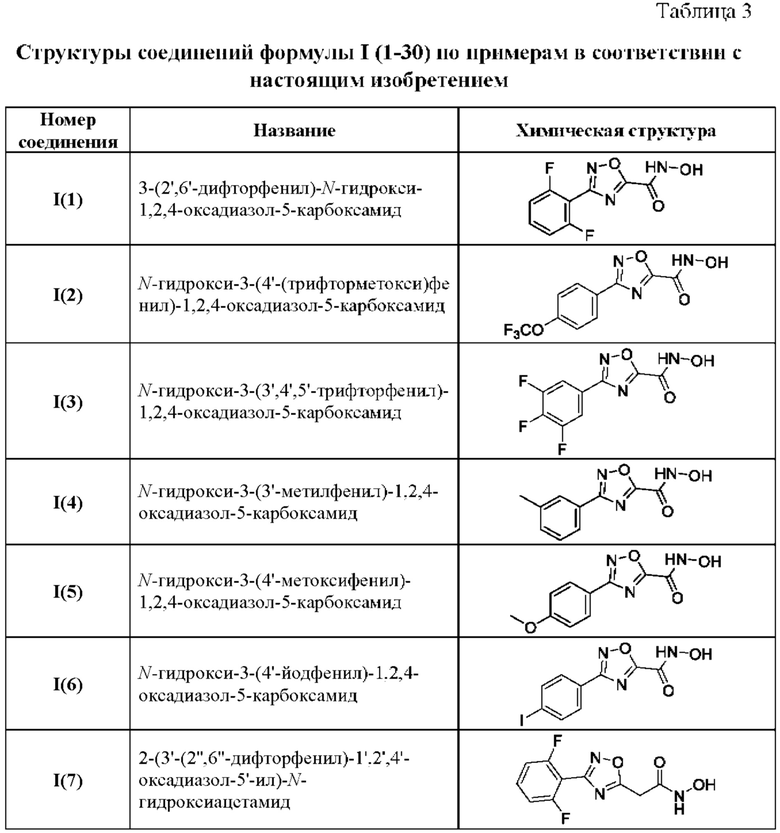
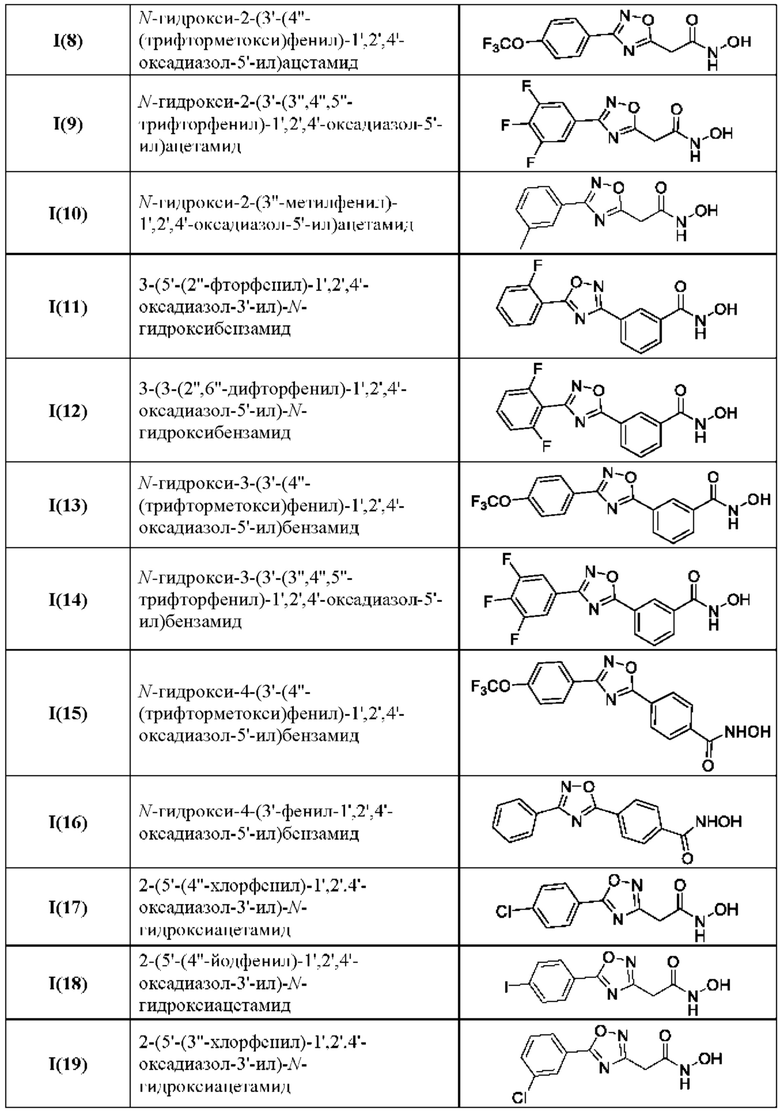
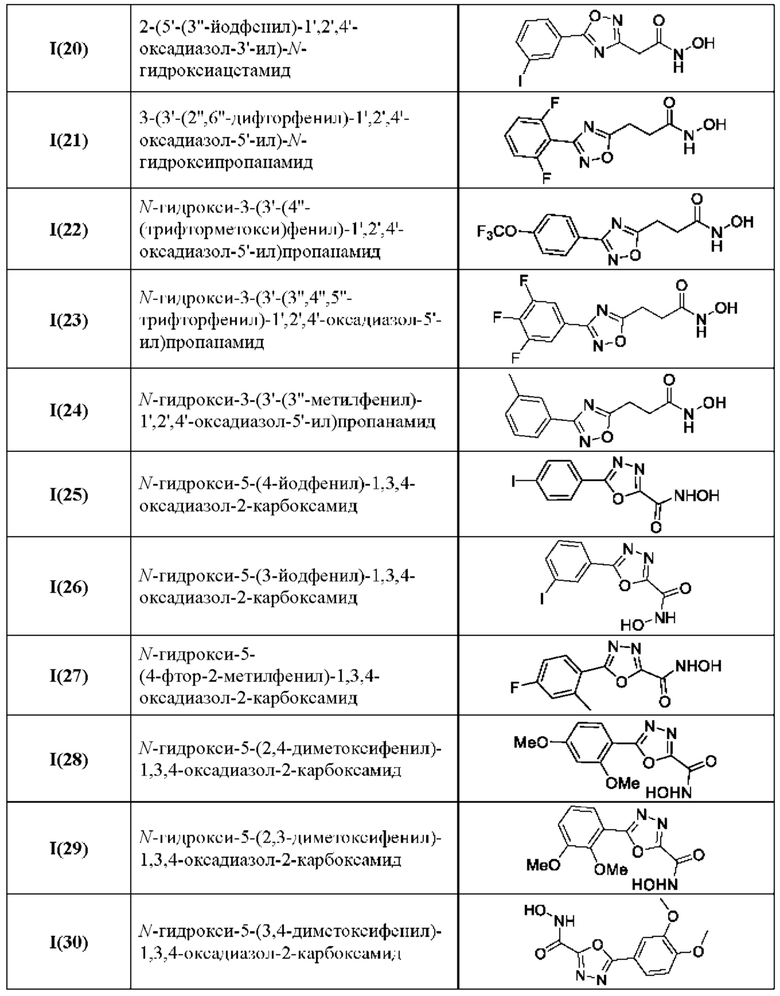
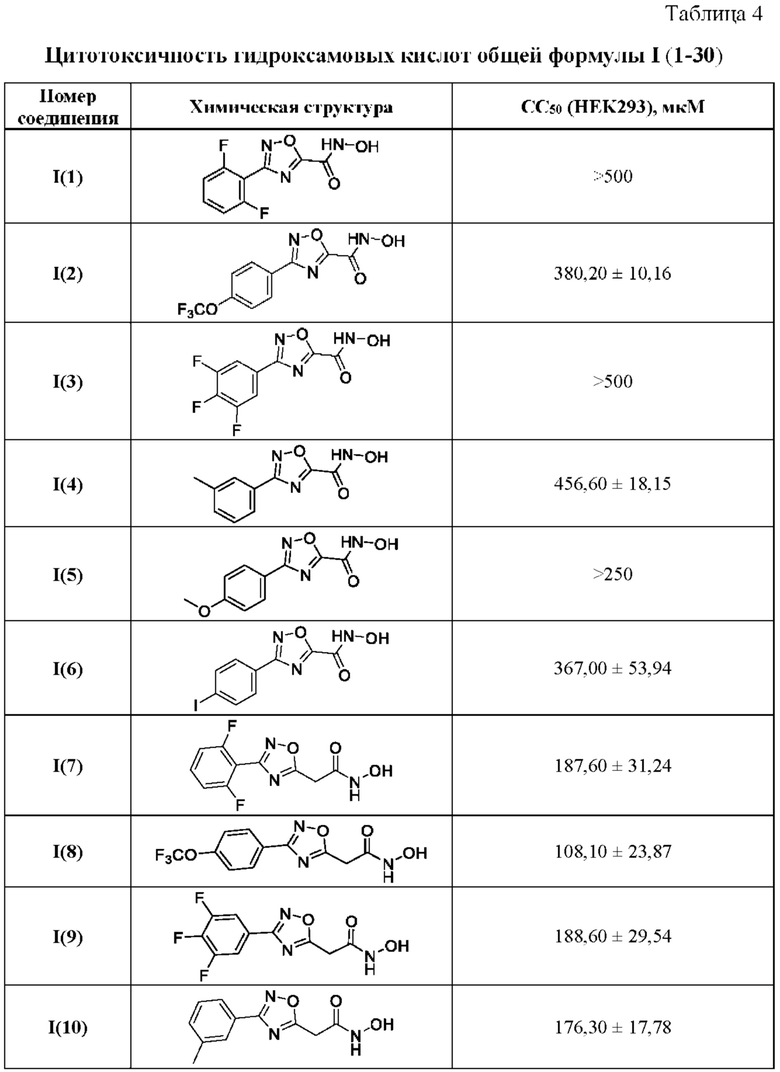
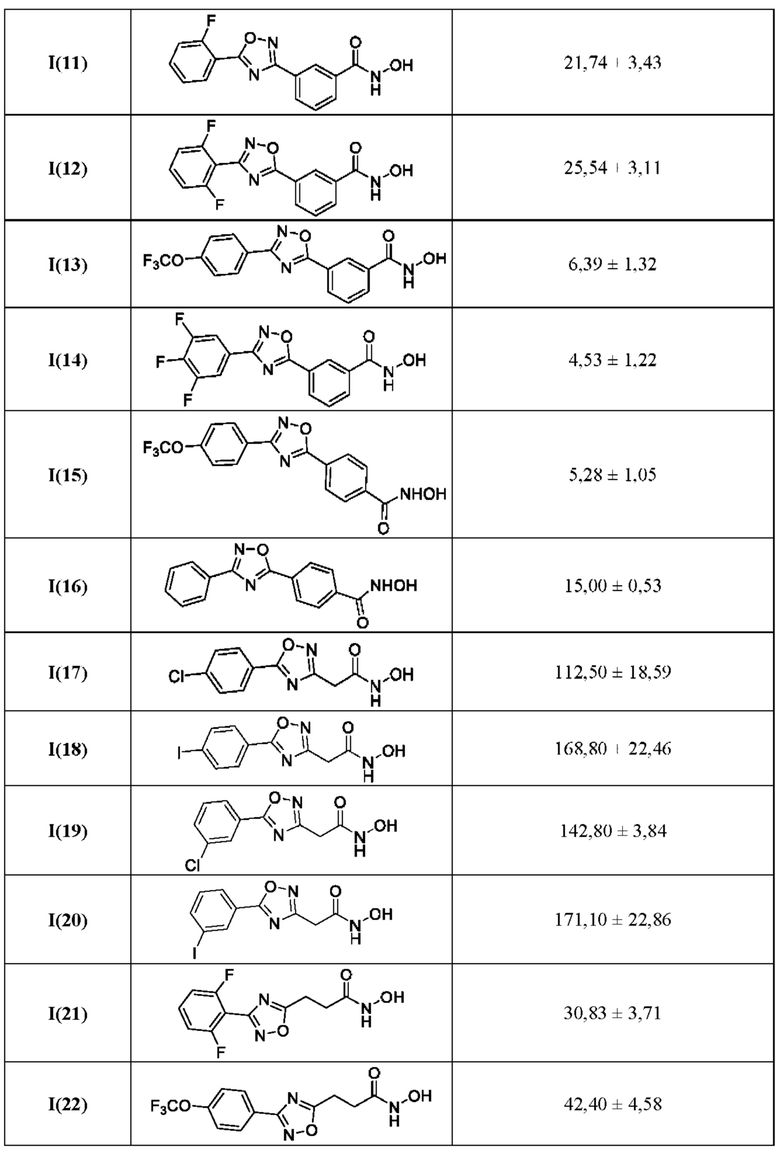
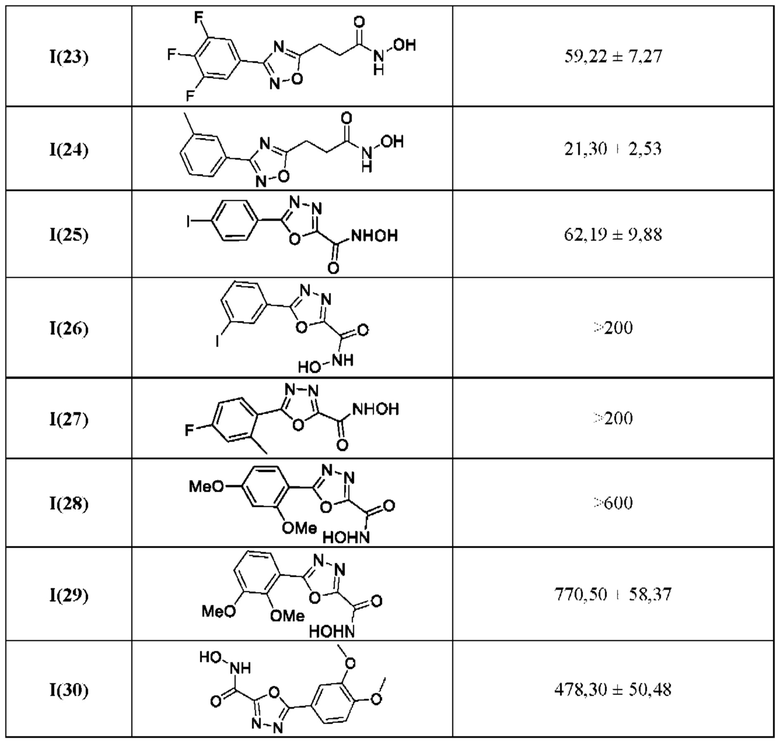
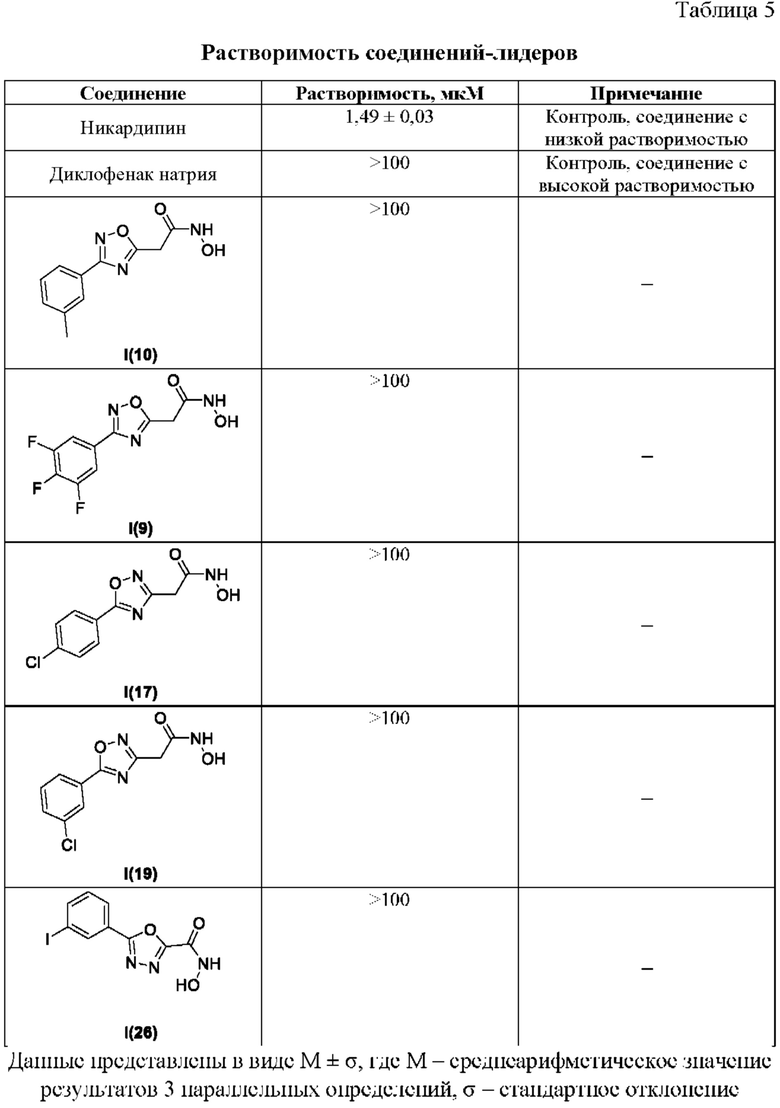
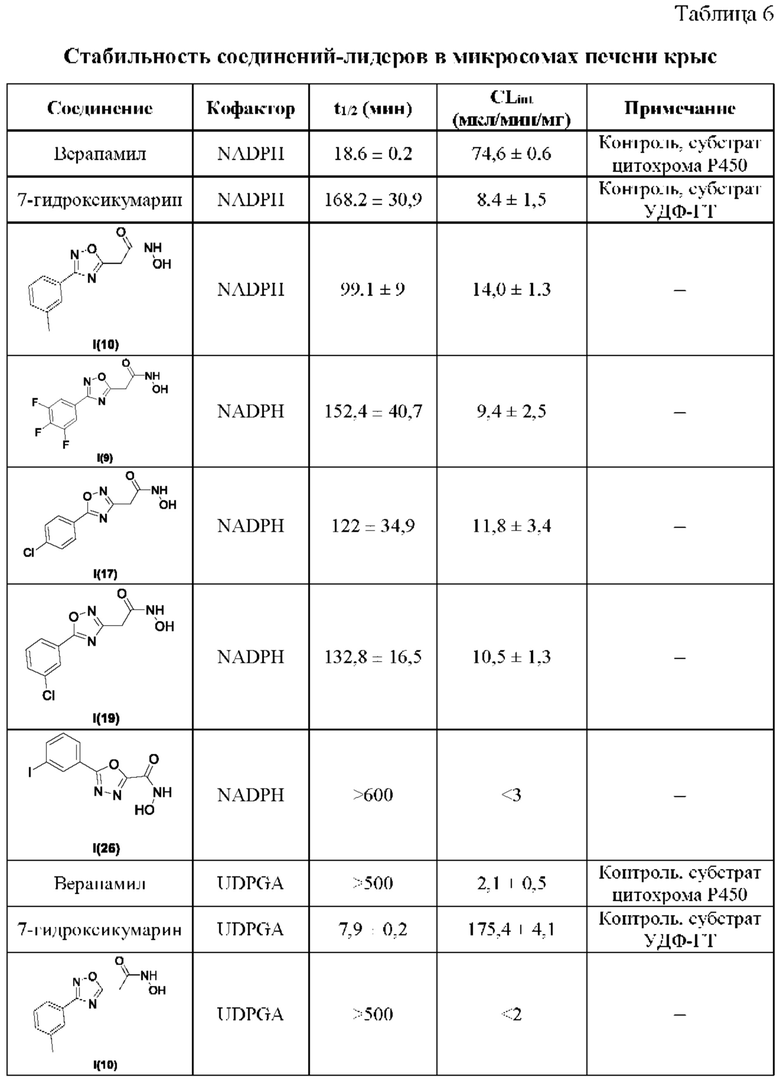
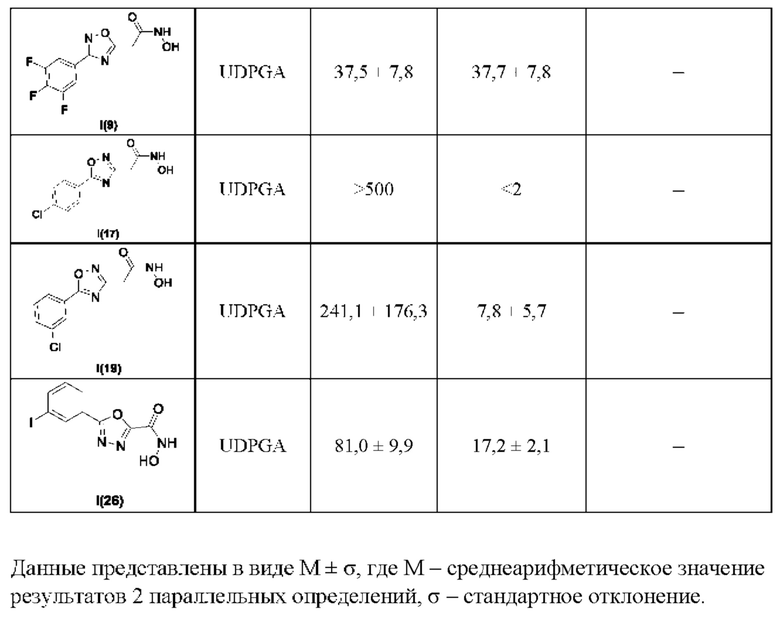
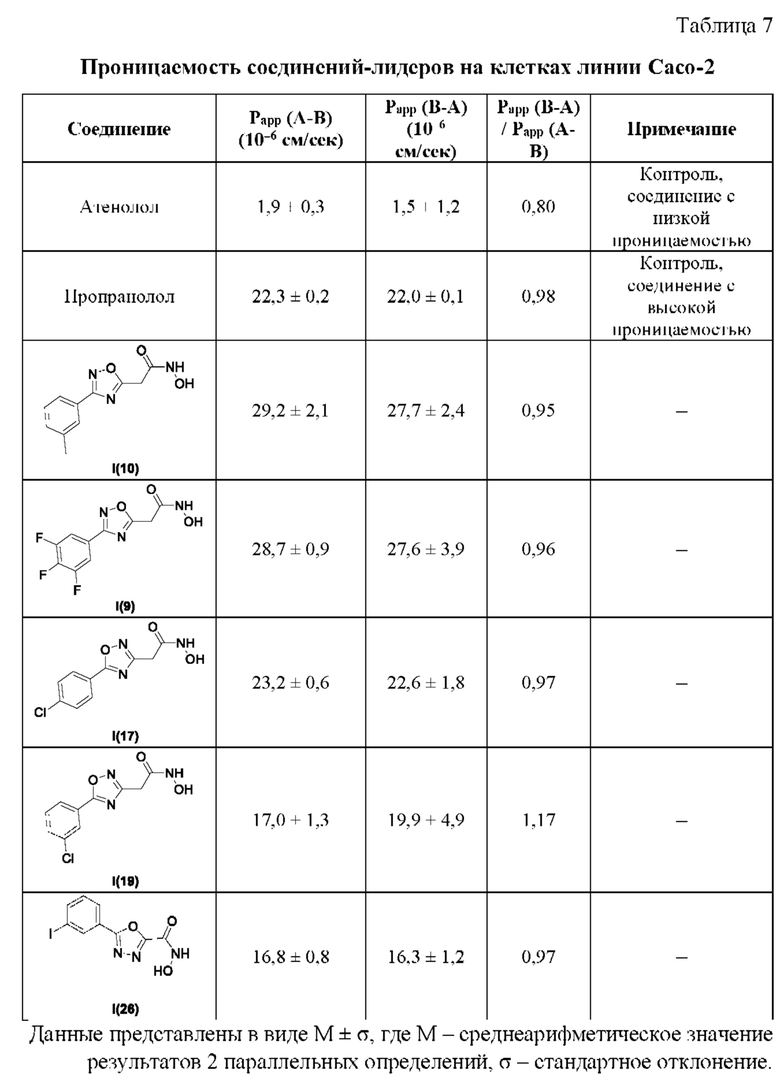
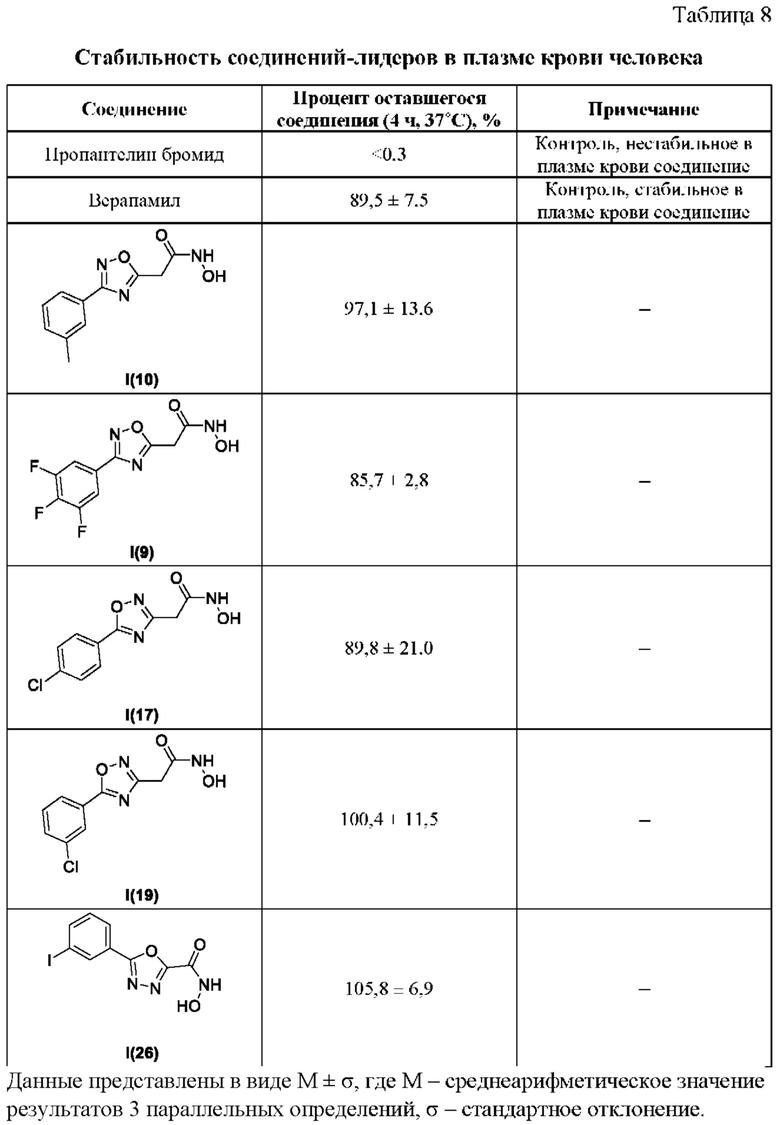
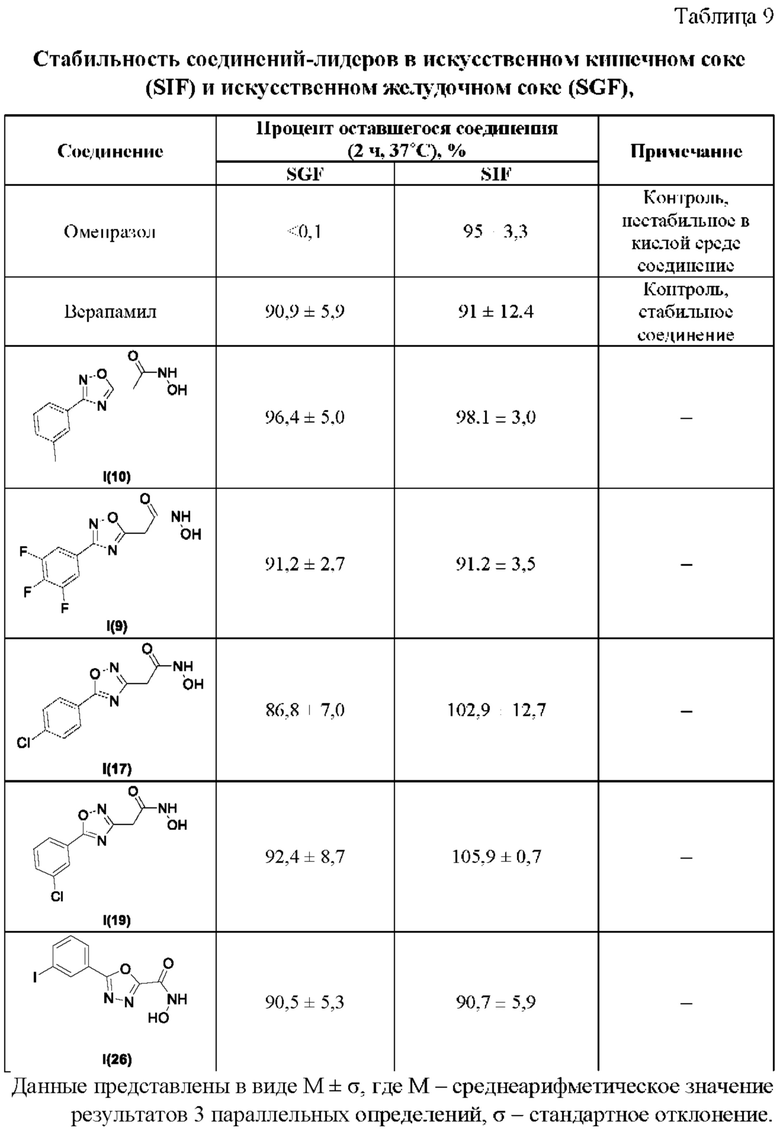
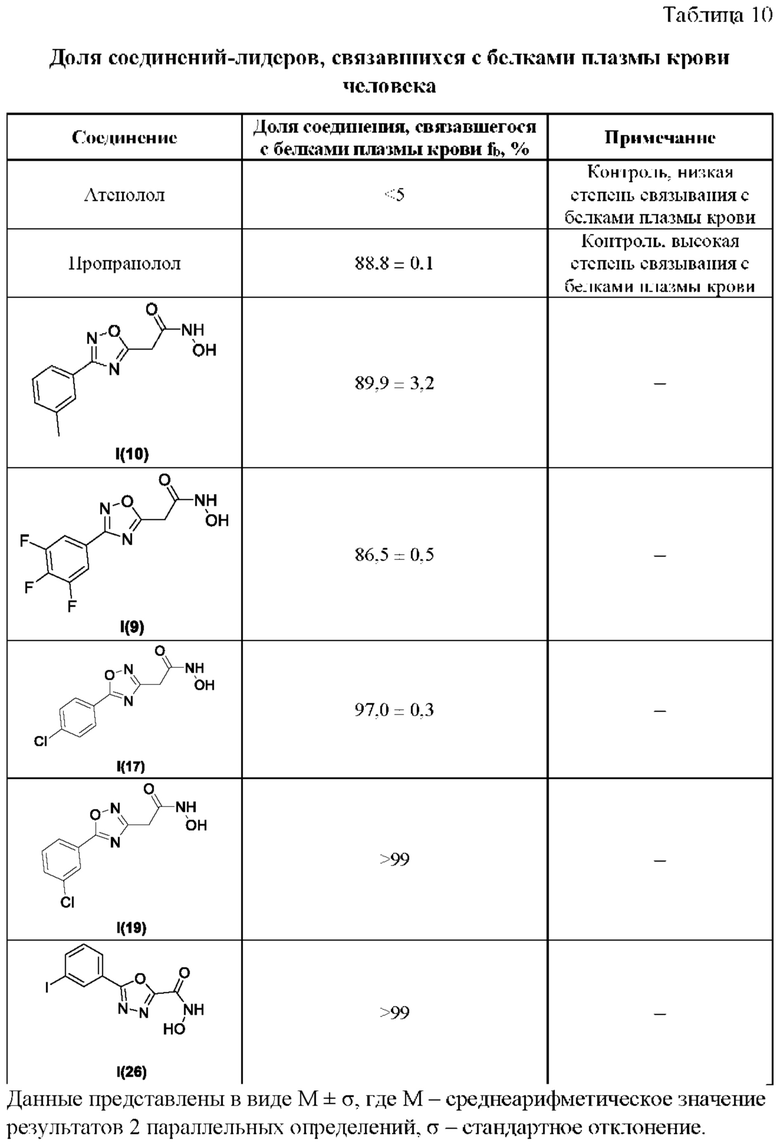
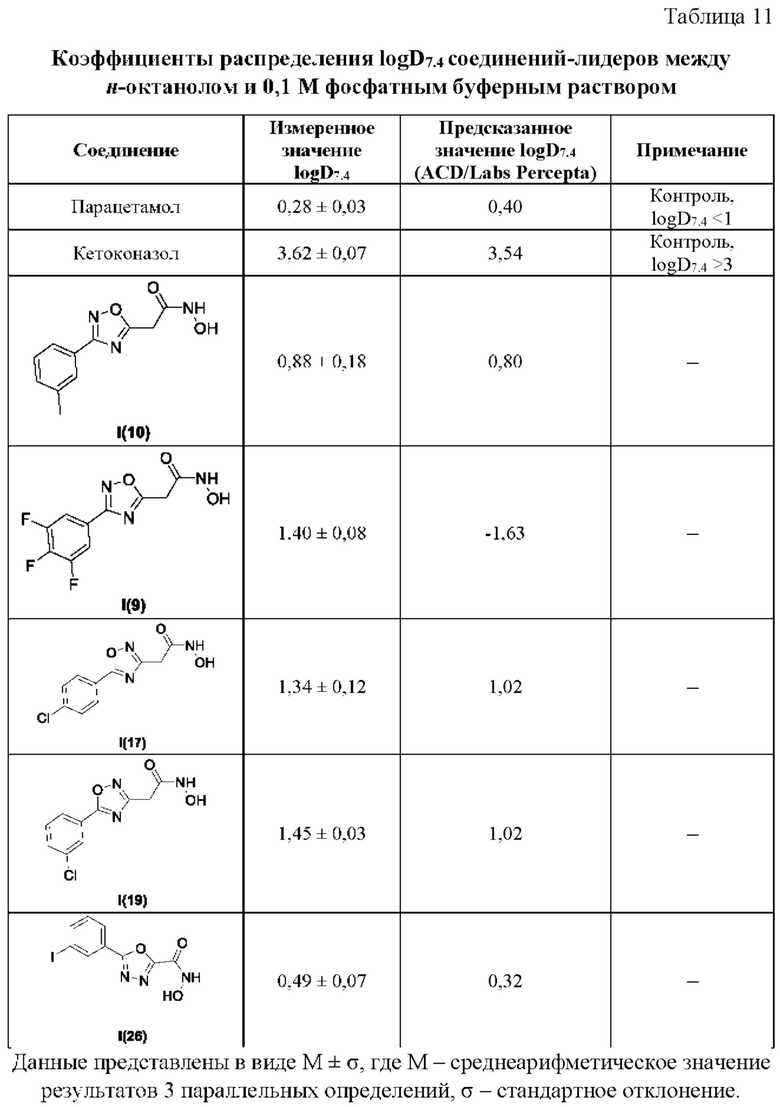
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ИНГИБИРОВАНИЯ ОПУХОЛЕВЫХ КЛЕТОК НОВЫМИ ПРОИЗВОДНЫМИ 3-ТРИФТОРМЕТИЛХИНОКСАЛИН 1,4-ДИОКСИДА | 2020 |
|
RU2746395C1 |
| Производные 1,4-диоксида хиноксалин-2-карбонитрила, ингибирующие рост опухолевых клеток | 2016 |
|
RU2640304C1 |
| Четвертичные аммониевые соли на основе производных витамина В6 | 2015 |
|
RU2607522C1 |
| ПРОИЗВОДНЫЕ 2-((4-(4-(БЕНЗИЛОКСИ)ПИРИМИДИН-2-ИЛ)ПИПЕРАЗИН-1-ИЛ)МЕТИЛ)-1Н-БЕНЗО[d]ИМИДАЗОЛА В КАЧЕСТВЕ НИЗКОМОЛЕКУЛЯРНЫХ АГОНИСТОВ РЕЦЕПТОРА ГЛЮКАГОНОПОДОБНОГО ПЕПТИДА-1 | 2024 |
|
RU2838323C1 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2015 |
|
RU2701188C2 |
| Ингибиторы образования конечных продуктов гликирования на основе азопроизводных фенилсульфокислот | 2016 |
|
RU2634594C1 |
| 8,10-Динитро-дегидро-1,5-5Н,11Н-[1,2,3,4]тетразино[5',6':4,5][1,2,3]триазоло-[2,1-a][1,2,3]бензотриазол-1,3-диоксид и способ его получения | 2017 |
|
RU2643363C1 |
| Азопроизводные аминофенолов, обладающие способностью ингибировать образование конечных продуктов гликирования | 2024 |
|
RU2839138C1 |
| ЗАМЕЩЕННЫЕ ФЕНИЛНАФТАЛИНЫ В КАЧЕСТВЕ ЭСТРОГЕННЫХ АГЕНТОВ | 2002 |
|
RU2314283C2 |
| Соединения на основе пиридоксина, обладающие способностью активировать фермент глюкокиназу | 2017 |
|
RU2644355C1 |
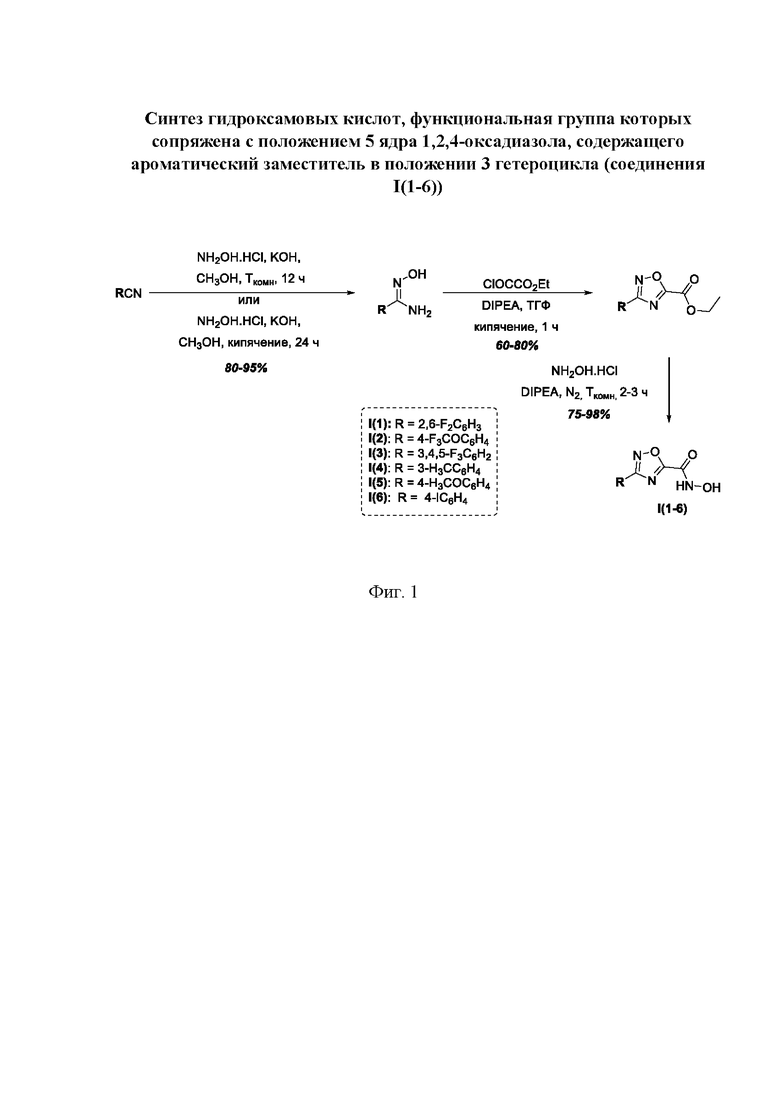
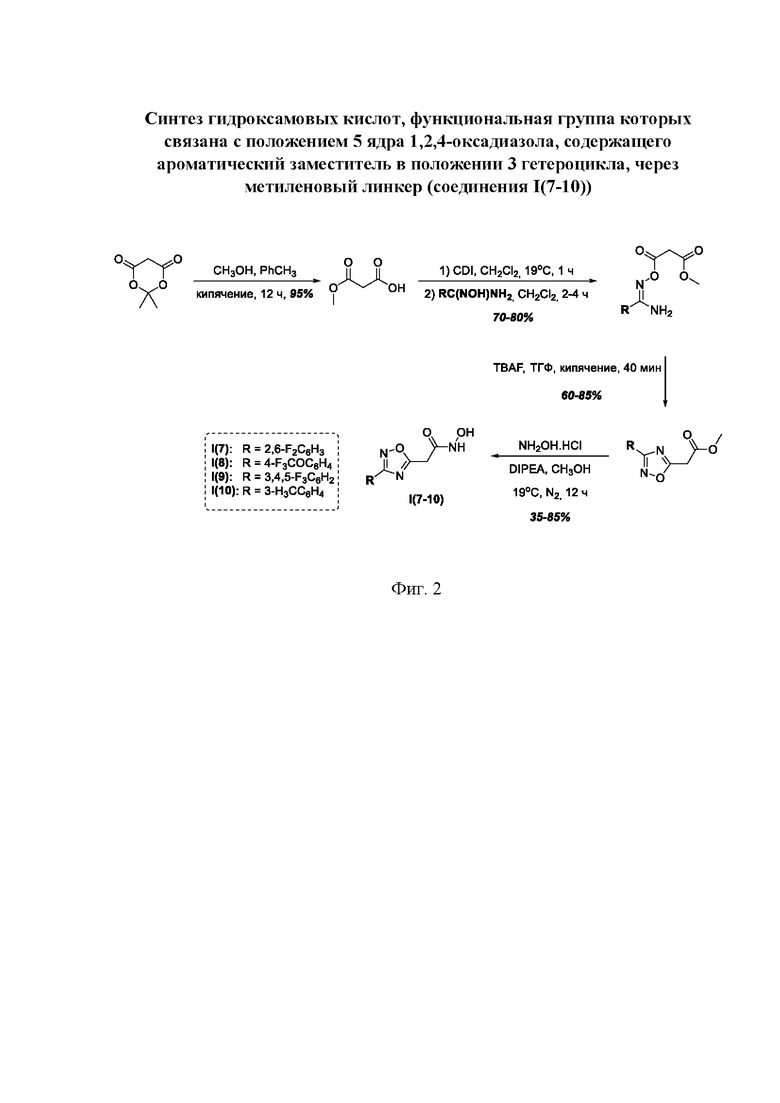
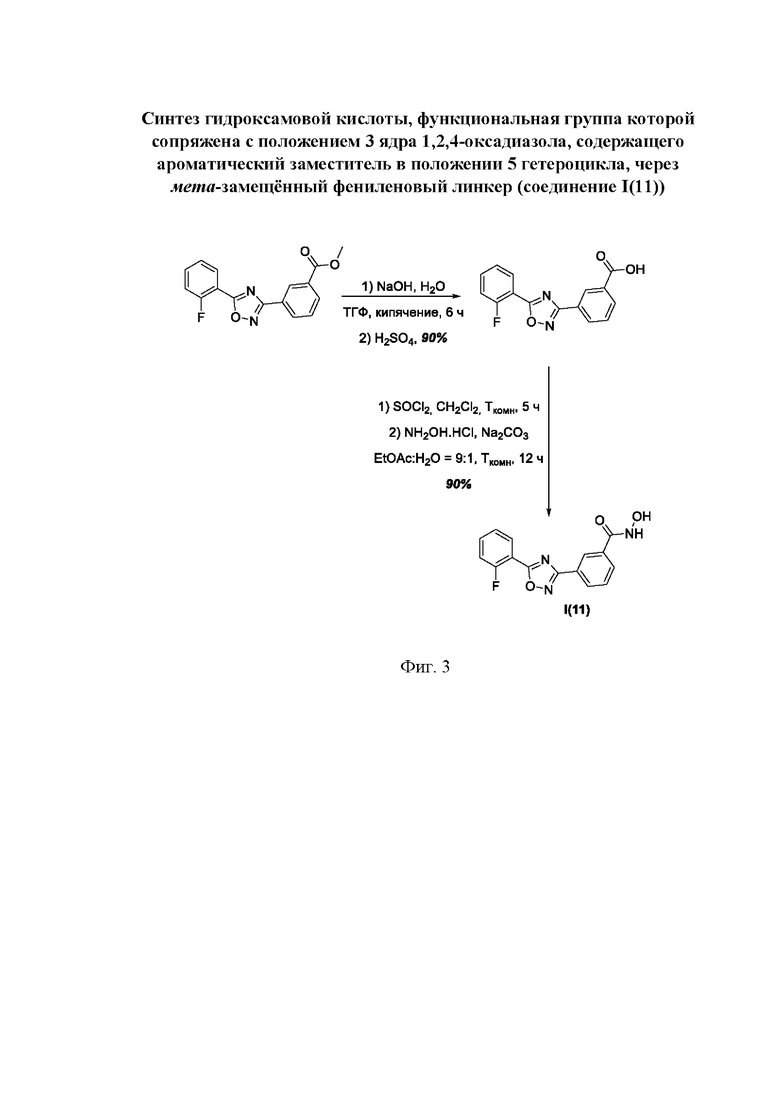
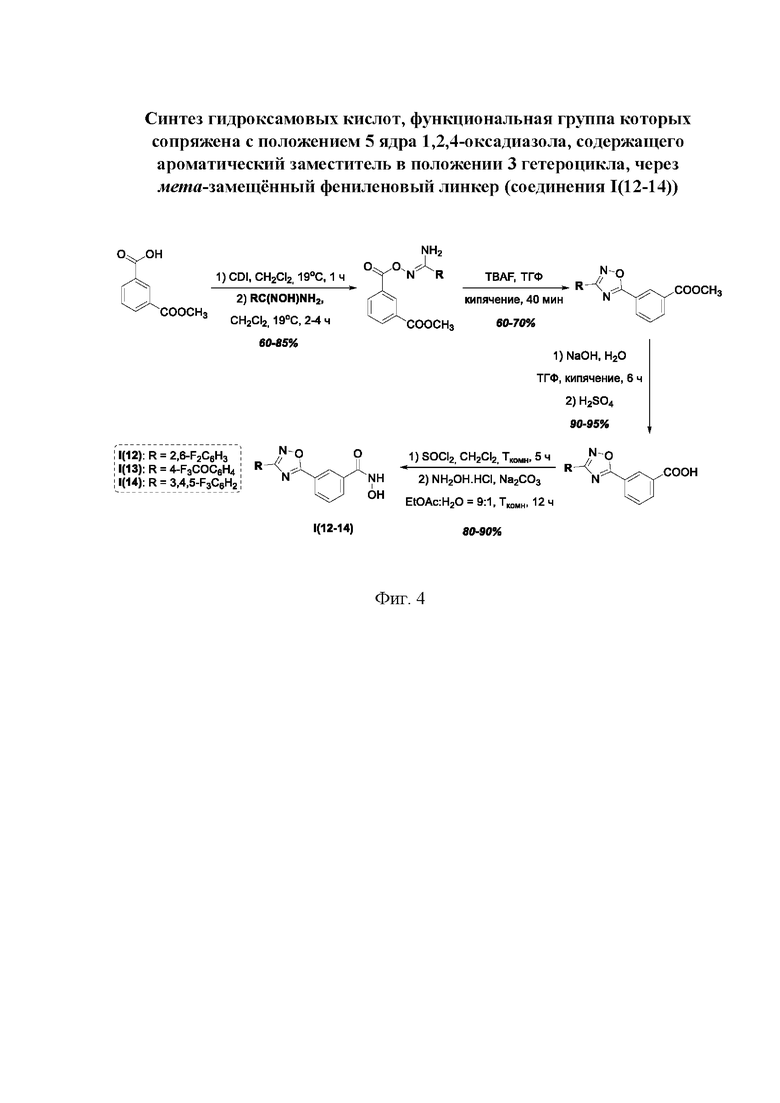
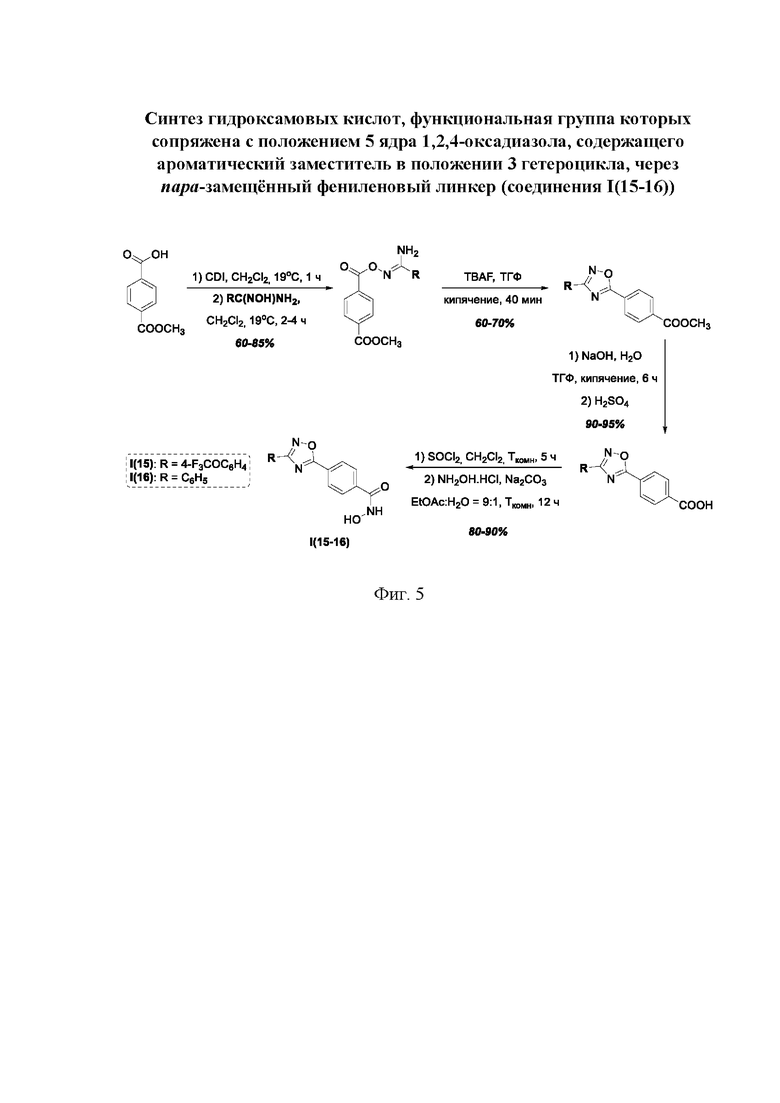
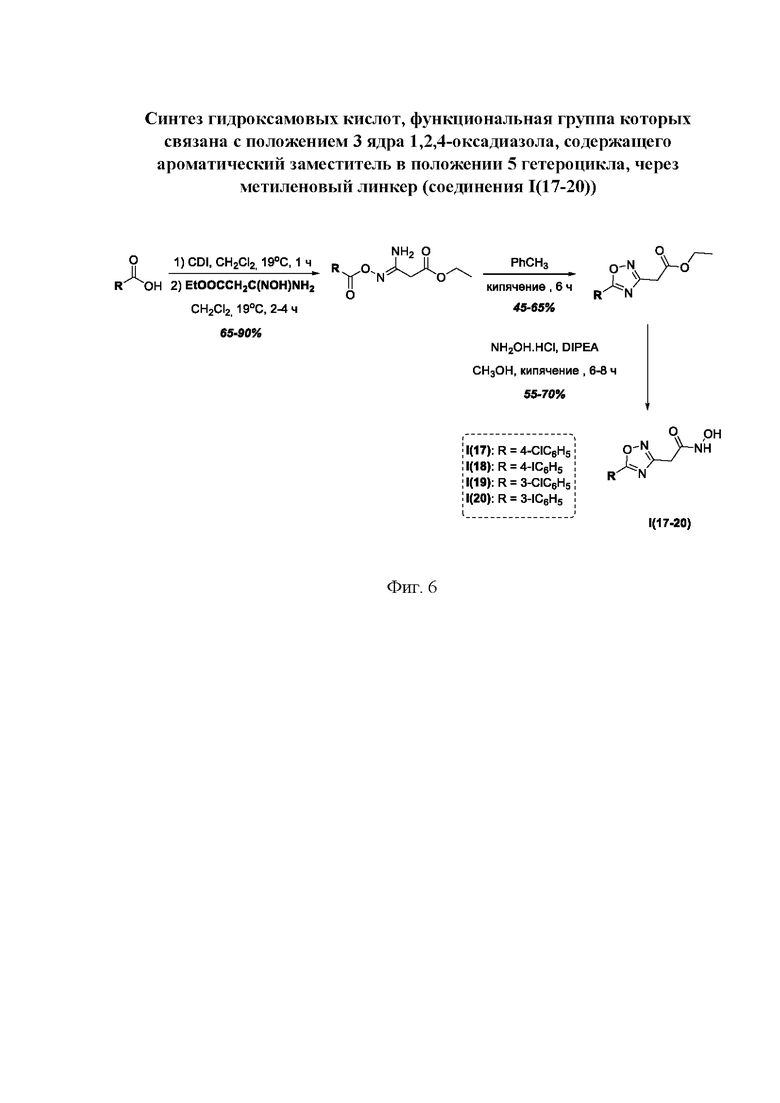
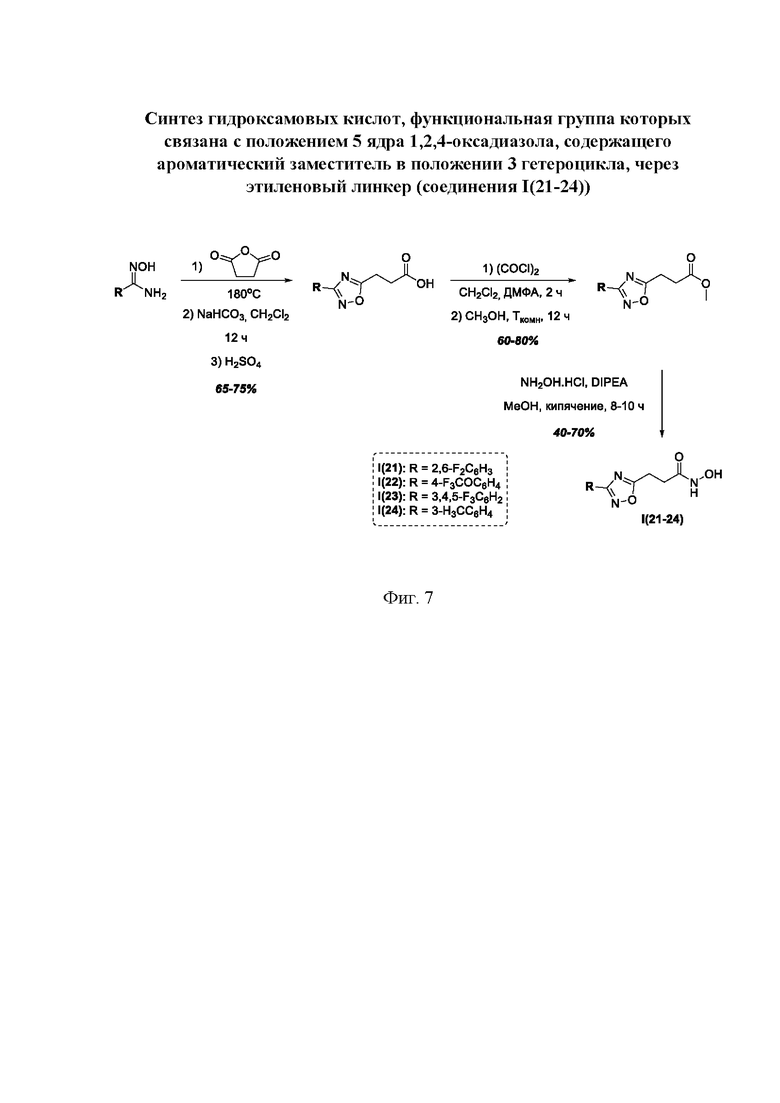
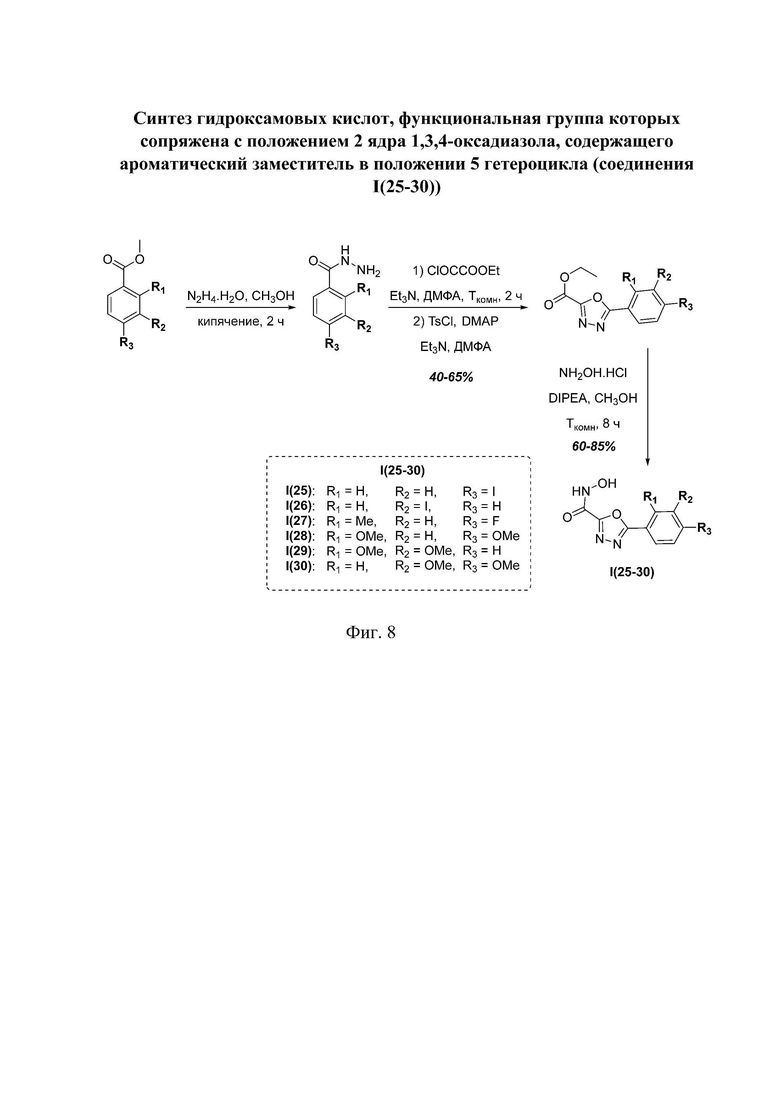
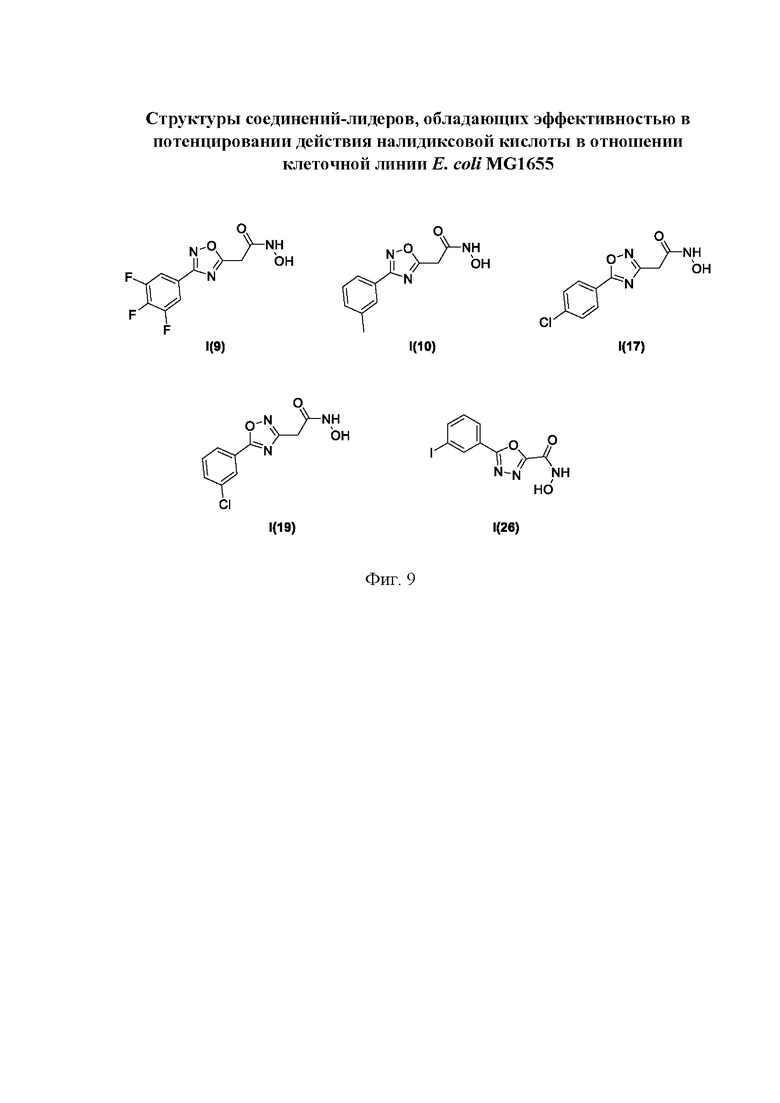
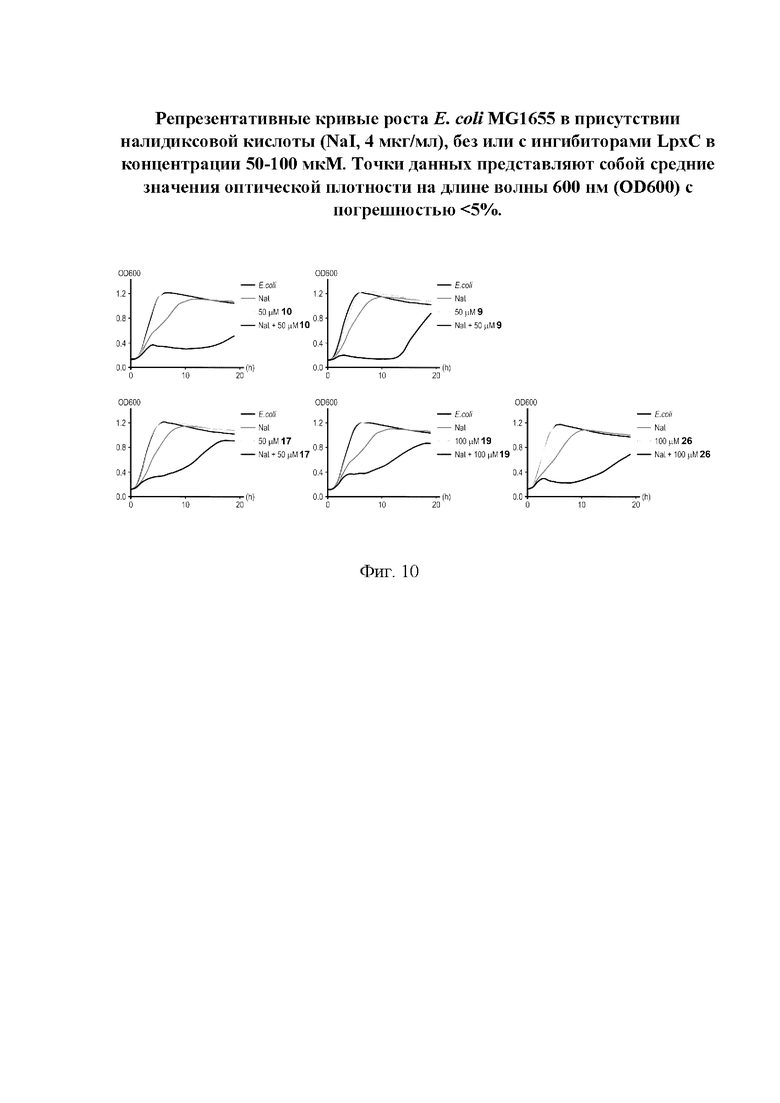
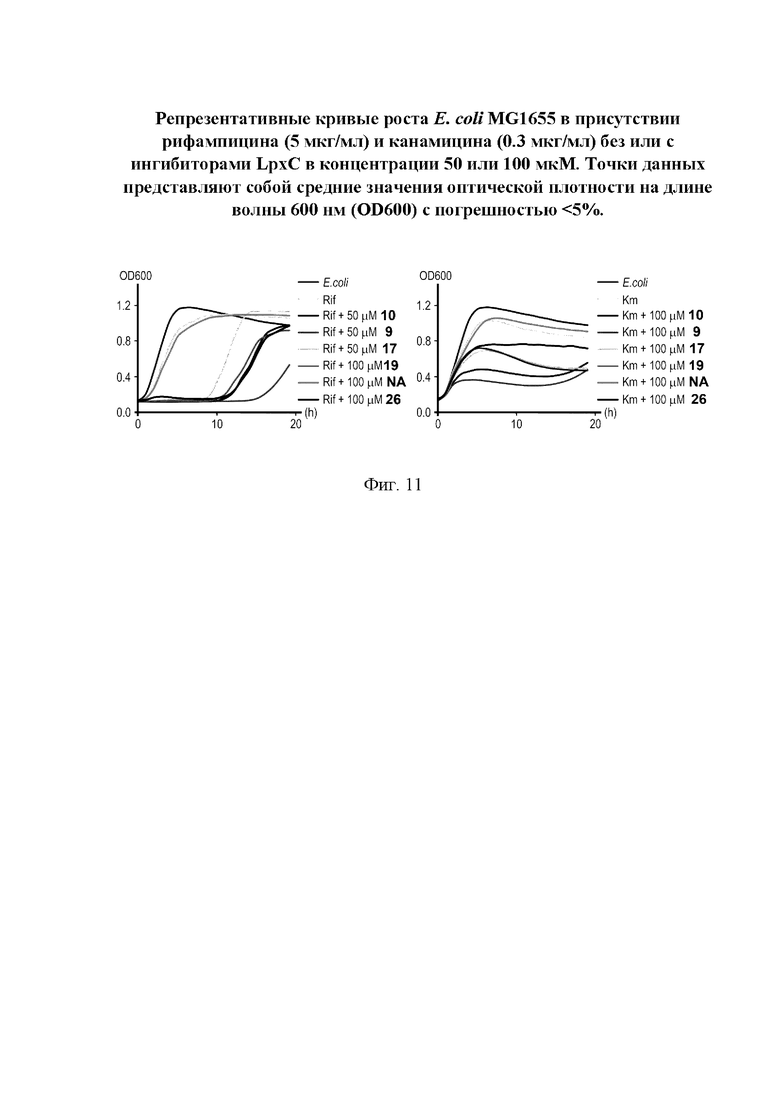
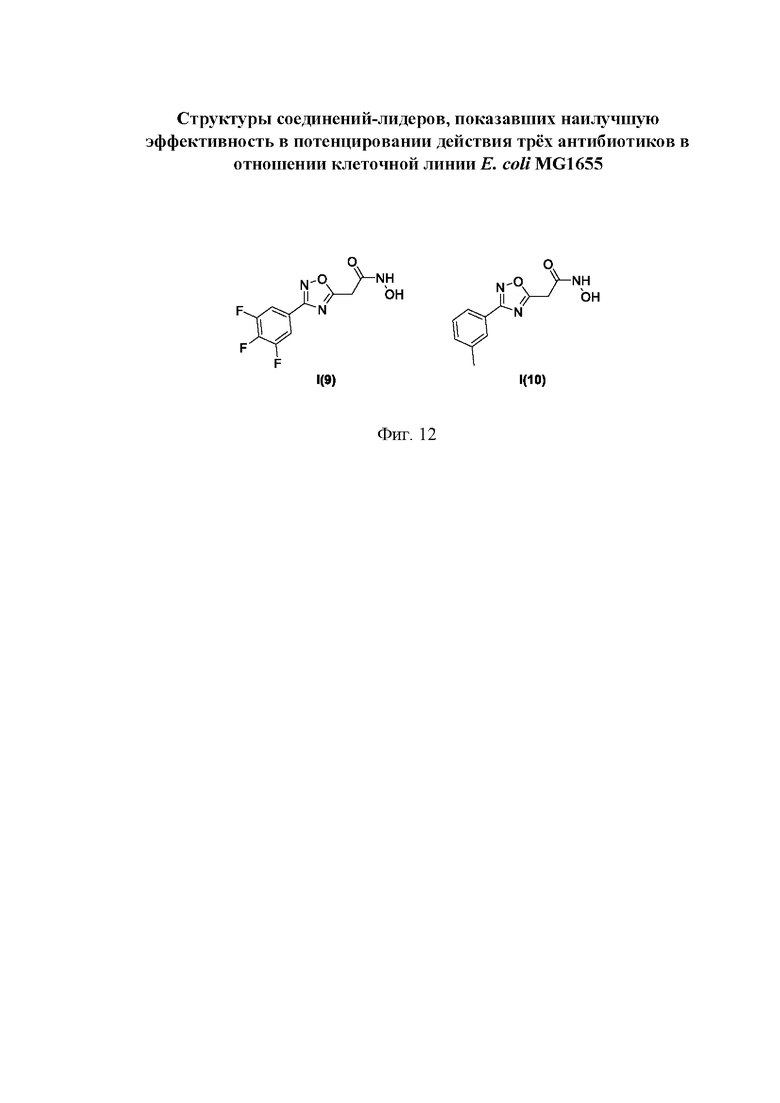
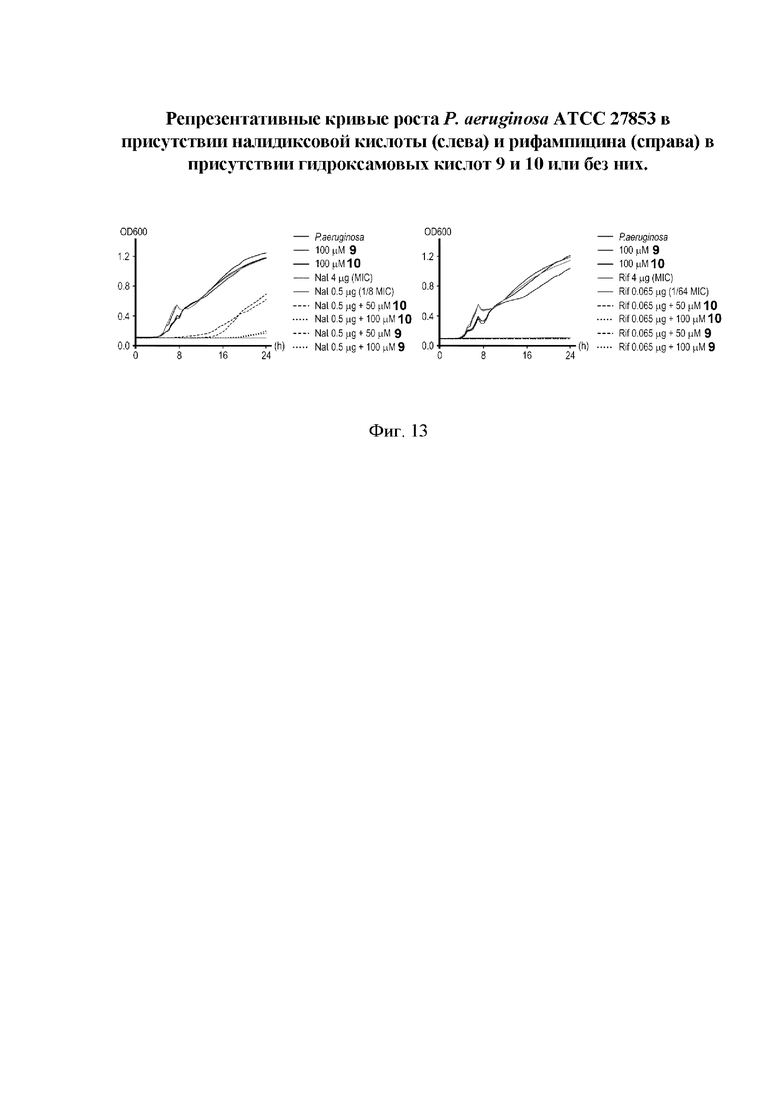
Изобретение относится к ингибиторам фермента уридиндифосфат-3-O-(гидроксимиристоил)-N-ацетилглюкозаминдеацетилазы (LpxC), имеющим формулу (I)
где А представляет собой 3,5-дизамещенное ядро 1,2,4-оксадиазола (в виде двух региоизомеров 1 и 2) или 2,5-дизамещенное ядро 1,3,4-оксадиазола 3:
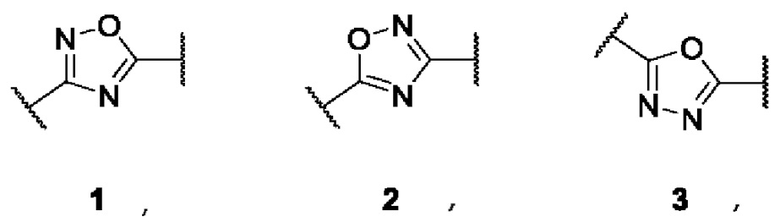
В представляет собой функциональную группу гидроксамовой кислоты (C(O)NHOH), свободную или сопряженную с алифатическим или ароматическим линкером:
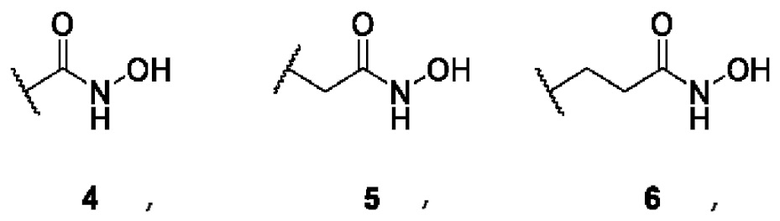
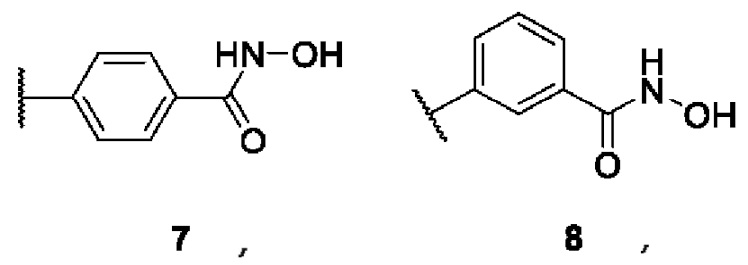
R1 представляет собой Н, F, Н3СО, Н3С; R2 представляет собой Н, F, C1, I, Н3СО, Н3С; R3 представляет собой Н, F, C1, I, Н3СО, F3CO; R4 представляет собой Н, F; R5 представляет собой Н, F. Технический результат: проведен синтез новых химических соединений, являющихся ингибиторами фермента LpxC, обладающих фармакологической активностью, показано уменьшение цитотоксичности синтезированных химических соединений, являющихся ингибиторами фермента LpxC, показано улучшение ADME-свойств синтезированных химических соединений, являющихся ингибиторами фермента LpxC. 34 з.п. ф-лы, 11 табл., 99 пр., 13 ил.
1. Соединение формулы (I)
где А представляет собой 3,5-дизамещенное ядро 1,2,4-оксадиазола (в виде двух региоизомеров 1 и 2) или 2,5-дизамещенное ядро 1,3,4-оксадиазола 3:
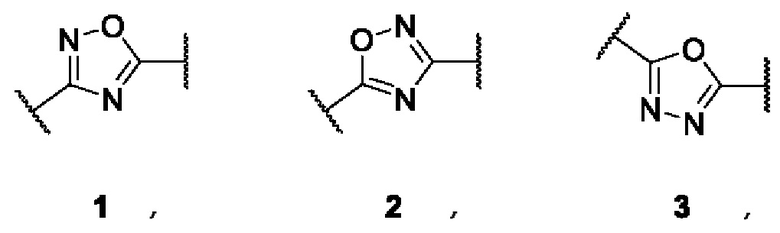
В представляет собой функциональную группу гидроксамовой кислоты (C(O)NHOH), свободную или сопряженную с алифатическим или ароматическим линкером:
R1 представляет собой Н, F, Н3СО, Н3С,
R2 представляет собой Н, F, C1, I, Н3СО, Н3С,
R3 представляет собой Н, F, C1, I, Н3СО, F3CO,
R4 представляет собой Н, F,
R5 представляет собой Н, F,
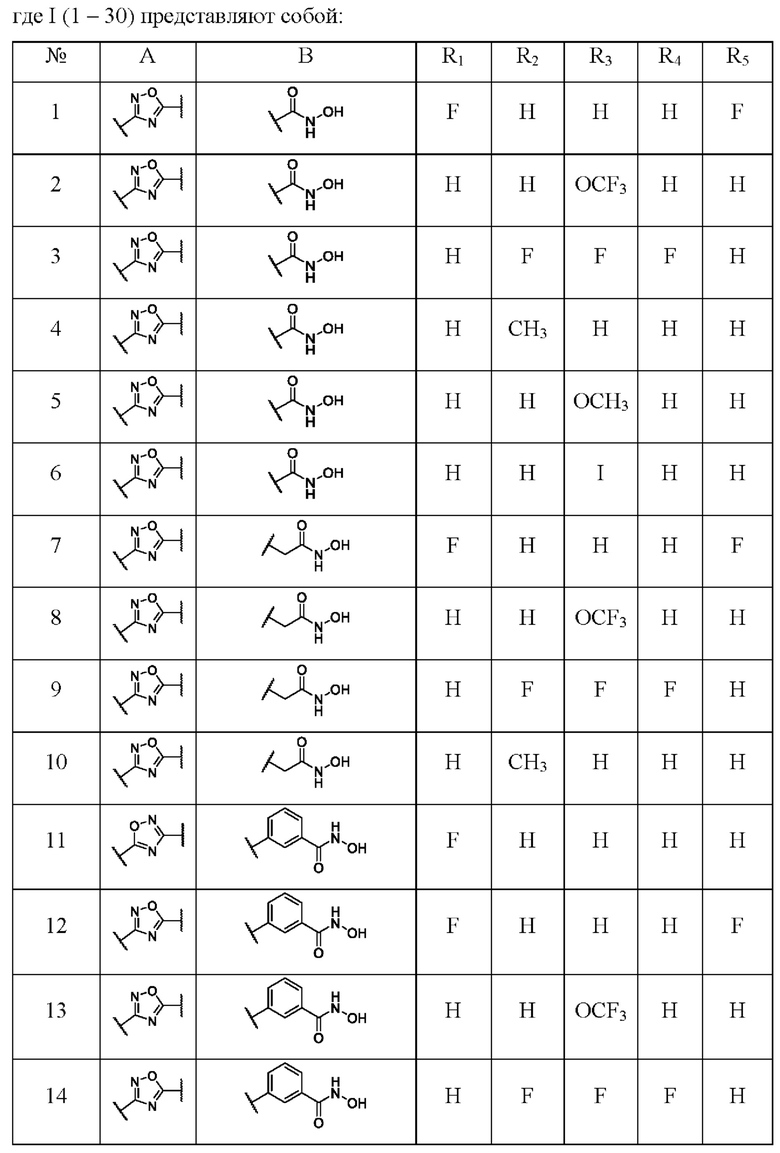
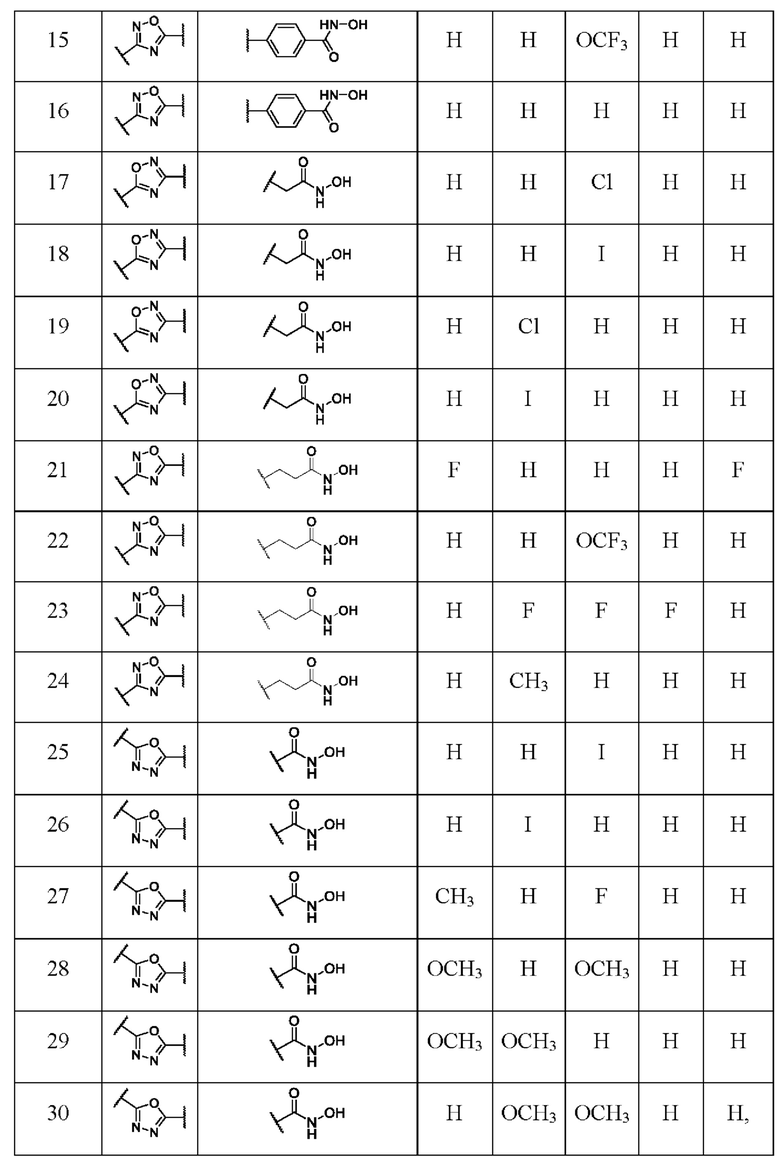
являющееся ингибитором фермента уридиндифосфат-3-O-(гидроксимиристоил)-N-ацетилглюкозаминдеацетилаза (LpxC) и обладающее эффективностью в потенцировании действия антибиотиков в отношении антибиотикорезистентных бактерий.
2. Соединение по п. 1, имеющее формулу
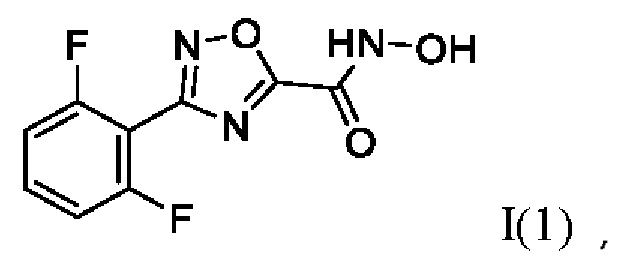
или его фармацевтически приемлемая соль.
3. Соединение по п. 1, имеющее формулу
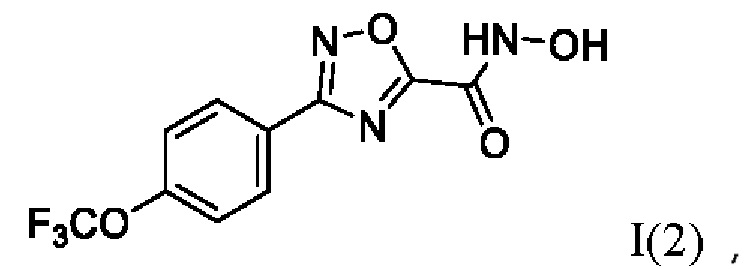
или его фармацевтически приемлемая соль.
4. Соединение по п. 1, имеющее формулу
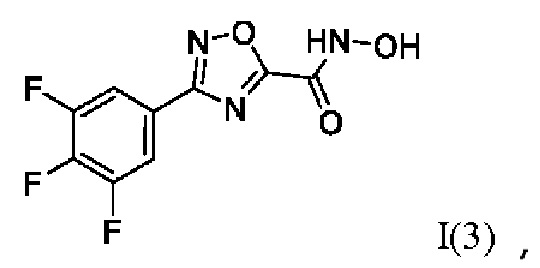
или его фармацевтически приемлемая соль.
5. Соединение по п. 1, имеющее формулу
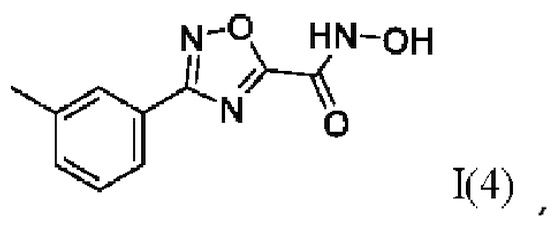
или его фармацевтически приемлемая соль.
6. Соединение по п. 1, имеющее формулу
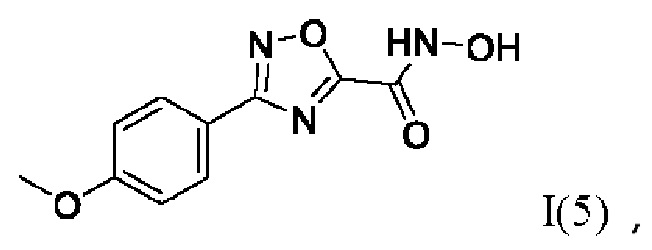
или его фармацевтически приемлемая соль.
7. Соединение по п. 1, имеющее формулу
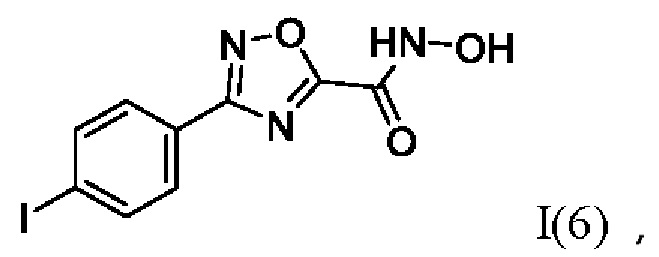
или его фармацевтически приемлемая соль.
8. Соединение по п. 1, имеющее формулу
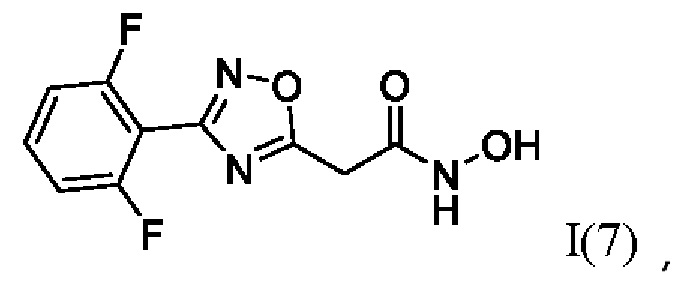
или его фармацевтически приемлемая соль.
9. Соединение по п. 1, имеющее формулу
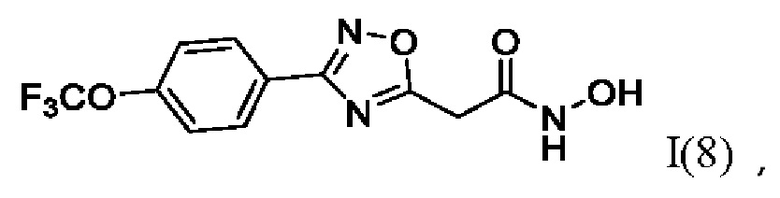
или его фармацевтически приемлемая соль.
10. Соединение по п. 1, имеющее формулу
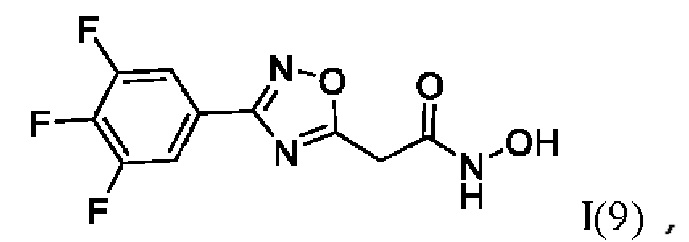
или его фармацевтически приемлемая соль.
11. Соединение по п. 1, имеющее формулу
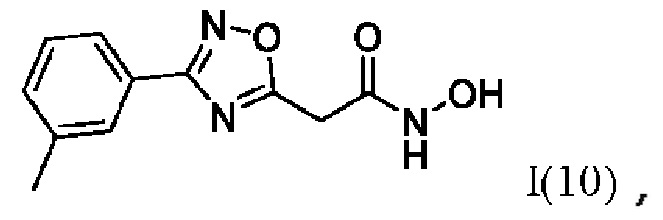
или его фармацевтически приемлемая соль.
12. Соединение по п. 1, имеющее формулу
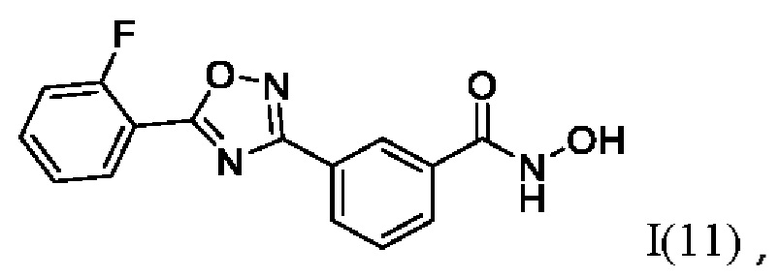
или его фармацевтически приемлемая соль.
13. Соединение по п. 1, имеющее формулу
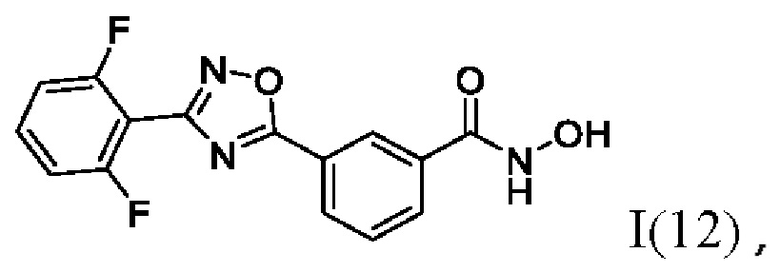
или его фармацевтически приемлемая соль.
14. Соединение по п. 1, имеющее формулу
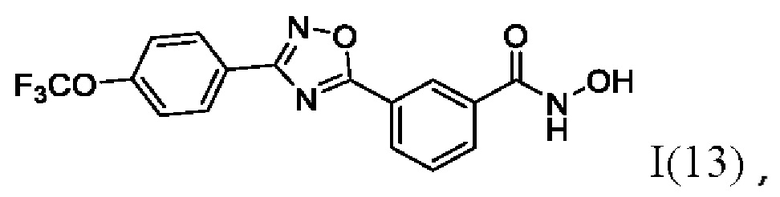
или его фармацевтически приемлемая соль.
15. Соединение по п. 1, имеющее формулу
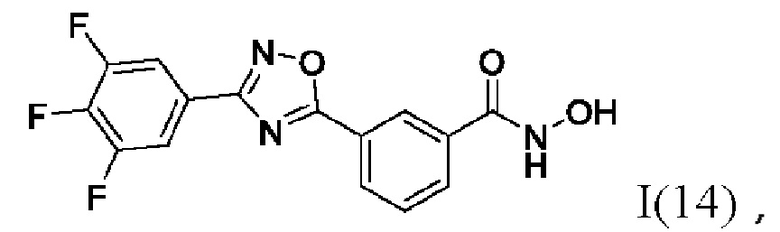
или его фармацевтически приемлемая соль.
16. Соединение по п. 1, имеющее формулу
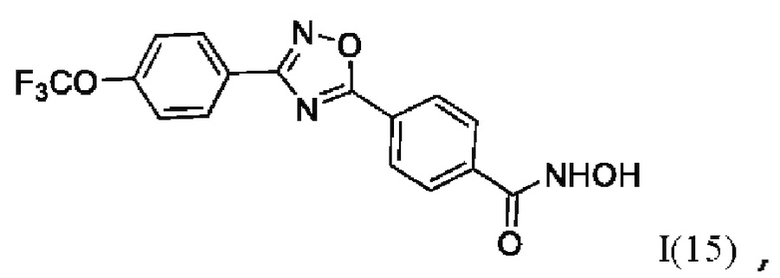
или его фармацевтически приемлемая соль.
17. Соединение по п. 1, имеющее формулу
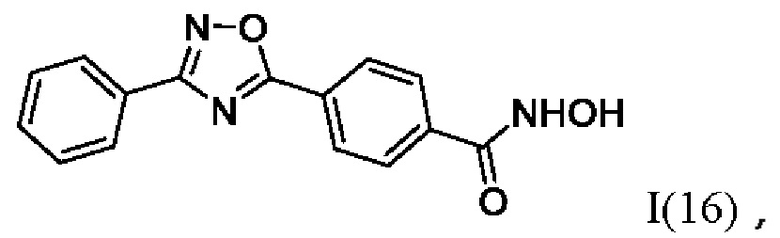
или его фармацевтически приемлемая соль.
18. Соединение по п. 1, имеющее формулу
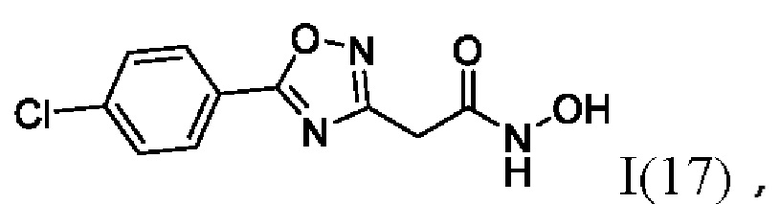
или его фармацевтически приемлемая соль.
19. Соединение по п. 1, имеющее формулу
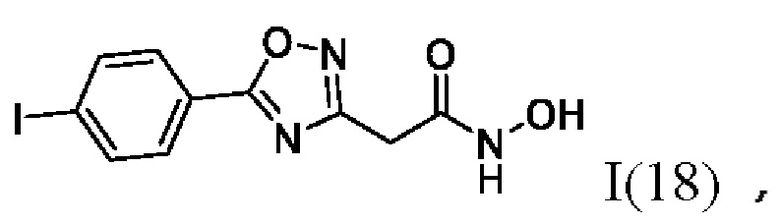
или его фармацевтически приемлемая соль.
20. Соединение по п. 1, имеющее формулу
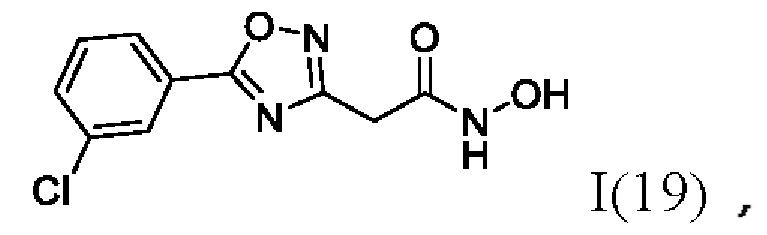
или его фармацевтически приемлемая соль.
21. Соединение по п. 1, имеющее формулу
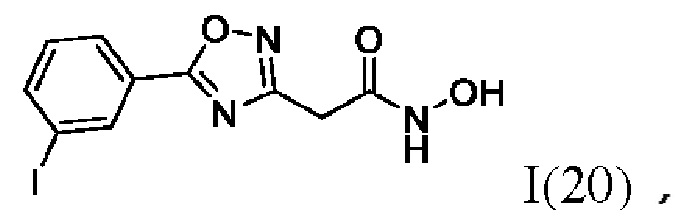
или его фармацевтически приемлемая соль.
22. Соединение по п. 1, имеющее формулу
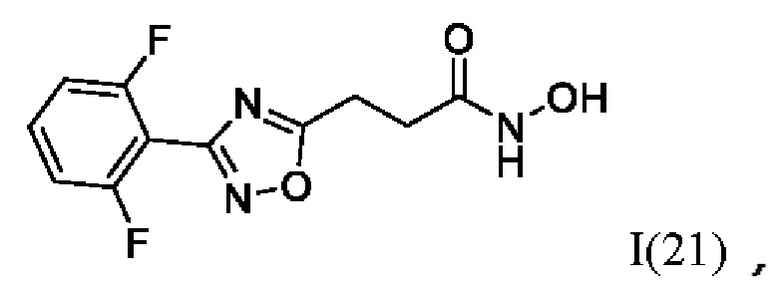
или его фармацевтически приемлемая соль.
23. Соединение по п. 1, имеющее формулу
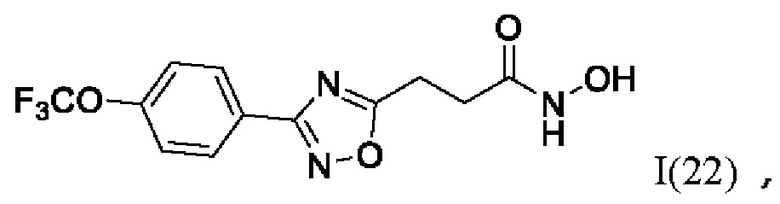
или его фармацевтически приемлемая соль.
24. Соединение по п. 1, имеющее формулу
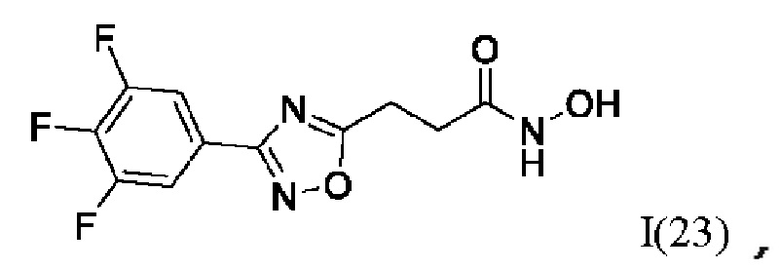
или его фармацевтически приемлемая соль.
25. Соединение по п. 1, имеющее формулу
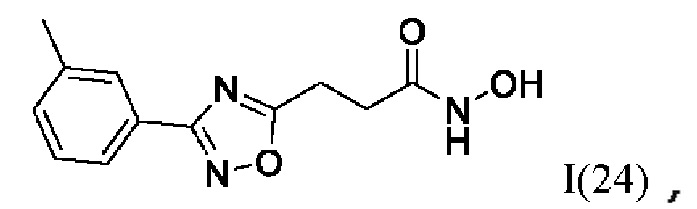
или его фармацевтически приемлемая соль.
26. Соединение по п. 1, имеющее формулу
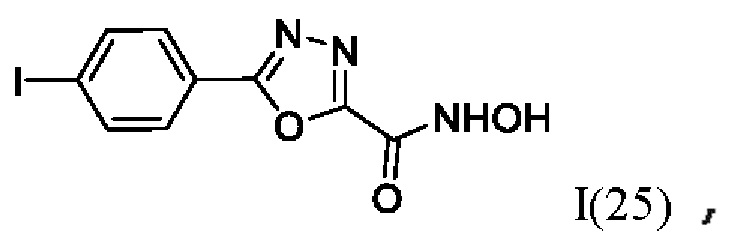
или его фармацевтически приемлемая соль.
27. Соединение по п. 1, имеющее формулу
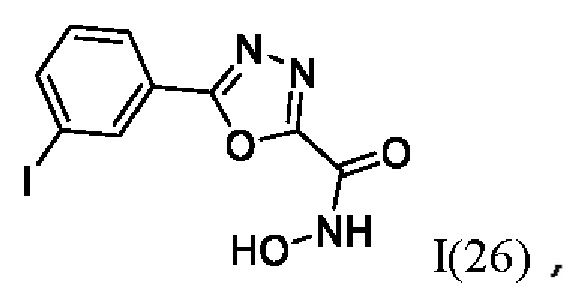
или его фармацевтически приемлемая соль.
28. Соединение по п. 1, имеющее формулу
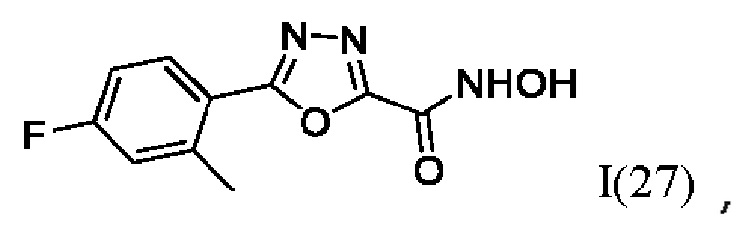
или его фармацевтически приемлемая соль.
29. Соединение по п. 1, имеющее формулу
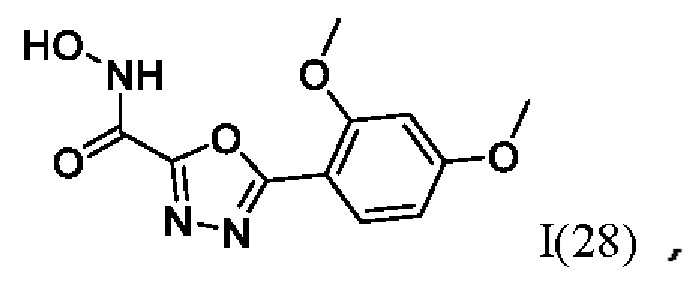
или его фармацевтически приемлемая соль.
30. Соединение по п. 1, имеющее формулу
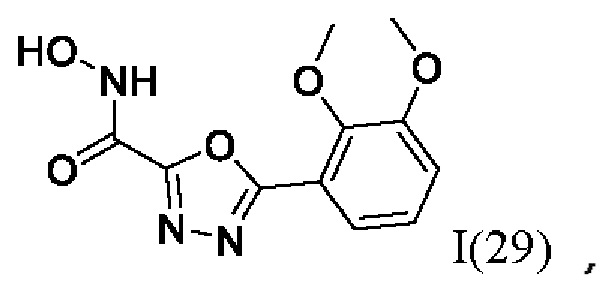
или его фармацевтически приемлемая соль.
31. Соединение по п. 1, имеющее формулу
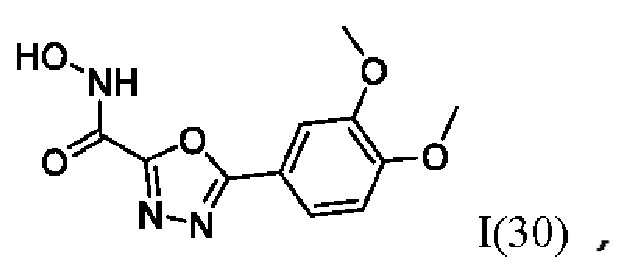
или его фармацевтически приемлемая соль.
32. Соединение по п. 1, представляющее собой функциональное производное гидроксамовой кислоты, содержащей ядро дизамещенного 1,2,4-оксадиазола или ядро дизамещенного 1,3,4-оксадиазола, являющееся ингибитором фермента уридиндифосфат-3-O-(гидроксимиристоил)-N-ацетилглюкозаминдеацетилаза (LpxC).
33. Соединение по п. 1, представляющее собой ингибитор биосинтеза структурного липида А, компонента липополисахарида, являющегося основным компонентом клеточной стенки грамотрицательных бактерий.
34. Соединение по п. 1, обладающее эффективностью в потенцировании действия в отношении грамотрицательных бактерий антибиотиков, выбираемых из группы: налидиксовая кислота, рифампицин, канамицин.
35. Соединение по п. 1, обладающее антибактериальной активностью и применяемое для лечения заболеваний, вызываемых антибиотикорезистентными грамотрицательными бактериями, выбираемыми из группы: Escherichia coli MG1655, Pseudomonas aeruginosa АТСС 27853.
| Yang K | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Приспособление для соединения пучка кисти с трубкою или втулкою, служащей для прикрепления ручки | 1915 |
|
SU66A1 |
| Ветряный двигатель с поворотными створчатыми рамами | 1920 |
|
SU2681A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Токарный резец | 1924 |
|
SU2016A1 |
| Пиридинон- и пиримидинон-фосфаты и боронаты, полезные в качестве антибактериальных агентов | 2019 |
|
RU2754177C1 |

