Перекрестная ссылка на родственную заявку
Настоящая заявка заявляет приоритет в соответствии с заявкой на патент Китая № CN 201611170508.1, поданной 16 декабря 2016 года, и с заявкой на патент Китая № CN 201710787583,0, поданной 4 сентября 2017 года, содержимое которых включено в настоящую заявку посредством ссылки в своей полноте.
Область изобретения
Настоящее изобретение относится к ряду соединений, действующих в качестве ингибиторов CDK4/6. Более конкретно, раскрыто соединение, представленной формулой (I), его фармацевтически приемлемая соль или его изомер, содержащая его фармацевтическая композиция, и его применение в изготовления лекарственного средства для лечения раковых заболеваний.
Предшествующий уровень техники
Клеточный цикл относится к непрерывному динамическому процессу, которому нормальные делящиеся клетки подвергаются с момента окончания предыдущего митоза до конца следующего митоза. Клеточный цикл млекопитающих состоит из четырех фаз: фаза G1 (фаза, предшествующая фазе синтеза ДНК), фаза S (фаза синтеза ДНК), фаза G2 (фаза, следующая за фазой синтеза ДНК) и фаза М (фаза митоза). Цитокинез начинается немедленно после фазы М, в результате чего образуются две дочерние клетки. Хотя рождающиеся клетки, произведенные циклом клеточного деления, повторно входят в клеточный цикл, в некоторый момент в поздней стадии G1 (называемый контрольной точкой или точкой R), регуляторный механизм клеточного цикла определяет конечную судьбу клеток: продолжать участвовать в клеточном цикле или отойти из активного пролиферативного состояния в статичное состояние (G0). На регуляцию клеточного цикла в основном влияет ряд серин/треонинкиназ. Ряд серин/треонинкиназ также называется циклин-зависимыми киназами (CDK), которые объединяются с соответствующими им регуляторными субъединицами циклинами для достижения цели регуляции клеточного цикла. На настоящий момент было обнаружено по меньшей мере 10 циклин-зависимых киназ (CDK) и 15 циклинов, которые могут образовывать парные комплексы следующим образом: CDK1 сопрягается с циклином А или В; CDK2 сопрягается с циклином А или Е; CDK3 сопрягается с неизвестным циклином; CDK4 сопрягается с циклином D (1-3); CDK5 сопрягается с циклином D или p35Nck5A; CDK6 сопрягается с циклином D; CDK7 сопрягается с циклином Н; CDK8 сопрягается с циклином С; CDK9 сопрягается с циклином Т.
Ненормальная пролиферация раковых клеток и дисрегуляция нормального клеточного цикла являются общими характеристиками всех типов рака. Таким образом, ингибиторы ключевых регуляторов клеточного цикла стали привлекательной новой противоопухолевой мишенью. В ранней фазе G1 клеточного цикла комплекс CDK4/6 и циклина D активируется внеклеточными факторами роста. Белок ретинобластомы (RB) фосфорилируется активированным комплексом, что приводит к высвобождению фактора трансприпции E2F, который тесно связан с комплексом в нефосфорилированном состоянии. E2F активирует дальнейшую транскрипцию и способствует прохождению клеточного цикла дальше точки R и продвижению из фазы G1 в фазу S. После прохождения точки R другие циклины последовательно активируются для регуляции всего клеточного цикла. Например, связывание CDK2 с циклином Е контролирует вхождение клетки в фазу S; связывание CDK2 с циклином А контролирует прохождение фазы S, и затем CDK1 связывает циклин А в фазе G2; наконец, связывание CDK1 с циклином В контролирует вхождение клеток в фазу митоза. Комплекс, образованный CDK4/6 и циклином D, представляет собой "главный выключатель" регуляции клеточного цикла, ингибируя CDK4/6 и предотвращая образование комплекса циклин D-CDK4/6, можно блокировать продвижение клеточного цикла из фазы G1 в фазу S для достижения цели, заключающейся в ингибировании пролиферации опухоли. Таким образом, CDK4/6 стал важной противораковой мишенью.
В последние годы несколько низкомолекулярных ингибиторов CDK4/6 вошли в фазу клинических исследований для лечения рака либо по отдельность, либо в комбинации. Основываясь на промежуточных данных Фазы II клинического исследования PALOMA-1, палбоциклиб был одобрен FDA (Управление по контролю за качеством продуктов и лекарств США) к выпуску в продажу в феврале 2015 и использованию в комбинации с летрозолом в качестве терапии первой линии для ER-положительного/HER2-отрицательного постклимактерического метастатического рака молочной железы. Кроме того, исследование палбоциклиба в лечении немелкоклеточного рака легкого также находится в Фазе III клинических исследований. Кроме того, основываясь на результатах фазы III клинического исследования MONALEESA-2, FDA предоставило статус терапии прорыва ингибитору CDK4/6 рибоциклибу (LEE-011) в августе 2016, который может быть комбинирован с летрозолом в качестве терапии первой линии лечения распространенного или метастатического положительного на рецепторы гормонов/HER2-отрицательного рака молочной железы. Ингибитор CDK4/6 абемациклиб (LY2835219) от компании Eli Lilly & Со. также находится в фазе III клинического испытания MONARCH 2, и получение конечных результатов клинического испытания MONARCH 2 ожидается в первой половине 2017 года. Кроме того, что эти низкомолекулрные гетероциклические соединения являются полезными в лечении рака молочной железы, они также являются клинически полезными в лечении различных видов других раковых заболеваний. Эти патенты включают WO 2014128588, WO 2012018540, WO 2012129344, WO 2011101409, WO 2011130232, WO 2010075074, WO 2009126584, WO 2008032157, WO 2005094830, WO 2005117980 и WO 2003062236.
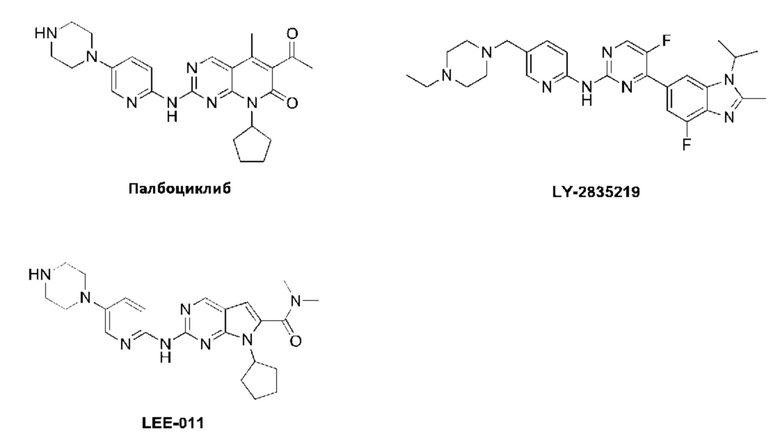
Несмотря на то, что были предприняты значительные усилия для разработки ингибиторов CDK4/6 для лечения рака и других заболеваний, только одно лекарственное средство (Палбоциклиб) для этой цели было выпущено на рынок до настоящего момента, и показанием для его применения является только ER-положительный/HER2-отрицательный постклимактерический метаститический рак молочной железы. Несмотря на то, что клинические исследования лечения рака легкого ингибиторами CDK4/6 продвинулись до фазы III клинических исследований, не существует лекарственных средств, выпущенных на рынок до настоящего времени. Таким образом, все еще существует срочная необходимость в разработке нового более безопасного и более эффективного ингибитора CDK4/6, который может лечить различные раковые заболевания, включая рак легкого. С другой стороны, несмотря на то, что палбоциклиб был одобрен для продажи, сообщалось, что его проходимость в головной мозг является низкой, что делает сложным проникновение через гемато-энцефалический барьер и приводит к неспособности лечить метастазы в головном мозге.
Содержание настоящего изобретения
В одном из аспектов в настоящем изобретении предложено соединение формулы (I), его фармацевтически приемлемая соль или его изомер,
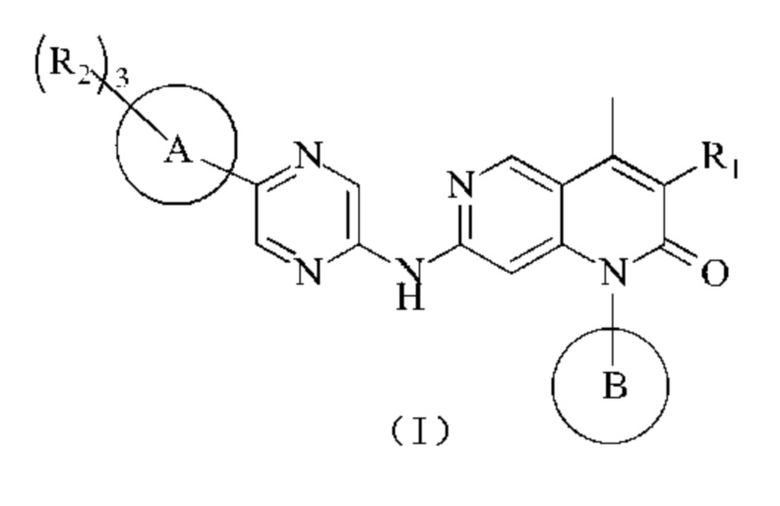
где
R1 представляет собой Н или выбран из группы, состоящей из С1-3алкила, С1-3гетероалкила,  , каждый из которых возможно замещен 1, 2 или 3 R;
, каждый из которых возможно замещен 1, 2 или 3 R;
каждый из R2 независимо представляет собой Н, ОН, CN, галоген или выбран из группы, состоящей из С1-5алкила, С1-5гетероалкила, С3-6циклоалкила и 3-6-членного гетероциклоалкила, каждый из которых возможно замещен 1, 2 или 3 R;
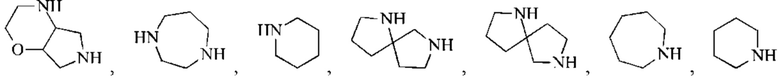
кольцо А представляет собой 4-11-членный гетероциклоалкил;
кольцо В выбрано из группы, состоящей из С3-6циклоалкила, 3-6-членного гетероциклоалкила, фенила и 5-6-членного гетероарила, каждый из которых возможно замещен 1, 2 или 3 R;
R представляет собой галоген, ОН, CN, NH2, NO2 или выбран из группы, состоящей из С1-3алкила и С1-3гетероалкила, каждый из которых возможно замещен 1, 2 или 3 R';
R' выбран из группы, состоящей из F, Cl, Br, I, ОН, CN и NH2;
каждый из "гетеро" в С1-3гетероалкиле, С1-5гетероалкиле, 3-6-членном гетероциклоалкиле, 4-11-членном гетероциклоалкиле и 5-6-членном гетероариле независимо выбран из группы, состоящей из N, -О-, -S-, -NH-, -(С=O)-, -(S=O)- и -(S=O)2-;
в любом из вышеуказанных случаев число гетероатомов или гетероатомных групп независимо представляет собой 1, 2 или 3.
В некоторых воплощениях настоящего изобретения вышеуказанный R выбран из F, Cl, Br, ОН, CN, NH2, СН3, СН3СН2, CH3O, CF3, CHF2, CH2F, и другие переменные являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанный R1 представляет собой Н или выбран из группы, состоящей из СН3, СН3СН2, СН3(С=O)-,  , каждый из которых возможно замещен 1, 2 или 3 R, и R и другие переменные являются такими, как определено в настоящем изобретении.
, каждый из которых возможно замещен 1, 2 или 3 R, и R и другие переменные являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанный R1 выбран из СН3, CHF2, СН3(С=O)-, 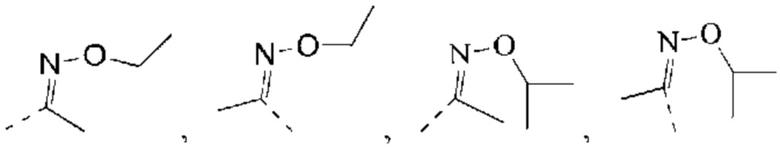 , и другие переменные являются такими, как определено в настоящем изобретении.
, и другие переменные являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанное кольцо В выбрано из группы, состоящей из циклобутила, циклопентила, циклогексила и фенила, каждый из которых возможно замещен 1, 2 или 3 R, и R и другие переменные являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанное кольцо В выбрано из циклопентила, циклогексила, фенила, и другие переменные являются такими, как определено в настоящем изобретении.
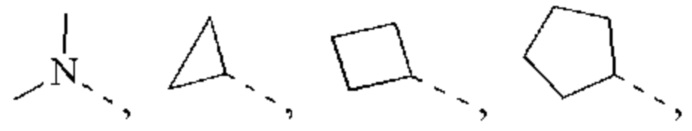
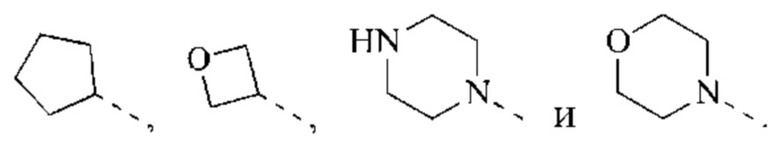
В некоторых воплощениях настоящего изобретения каждый из вышеуказанного R2 независимо выбран из Н, ОН, CN, F, Cl или выбран из группы, состоящей из СН3,  оксетанила, пиперазинила и морфолинила, каждый из которых возможно замещен 1, 2 или 3 R, и R и другие переменные являются такими, как определено в настоящем изобретении.
оксетанила, пиперазинила и морфолинила, каждый из которых возможно замещен 1, 2 или 3 R, и R и другие переменные являются такими, как определено в настоящем изобретении.
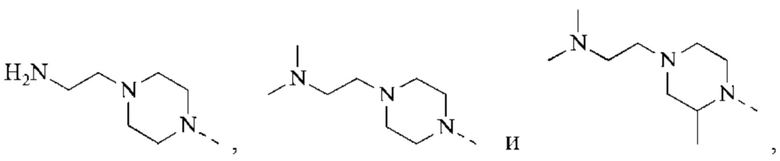
В некоторых воплощениях настоящего изобретения каждый из вышеуказанного R2 независимо представляет собой Н или выбран из группы, состоящей из СН3, 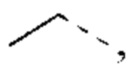
 и
и  каждый из которых возможно замещен 1, 2 или 3 R, и R и другие переменные являются такими, как определено в настоящем изобретении.
каждый из которых возможно замещен 1, 2 или 3 R, и R и другие переменные являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения каждый из вышеуказанного R2 независимо выбран из группы, состоящей из Н, СН3, 

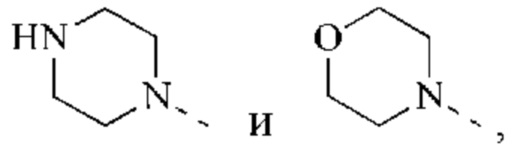 и другие переменные являются такими, как определено в настоящем изобретении.
и другие переменные являются такими, как определено в настоящем изобретении.
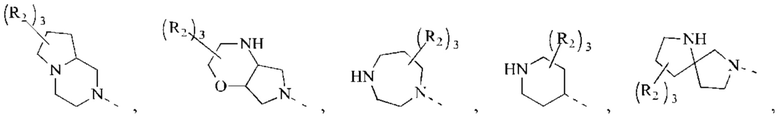
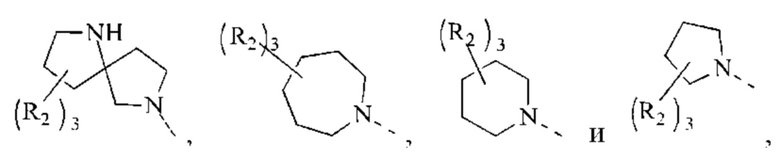
В некоторых воплощениях настоящего изобретения вышеуказанное кольцо А представляет собой 5-9-членный гетероциклоалкил, и другие переменные являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанная группировка 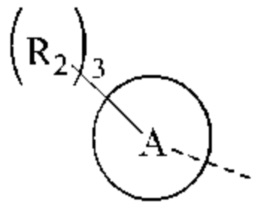 выбрана из группы, состоящей из
выбрана из группы, состоящей из 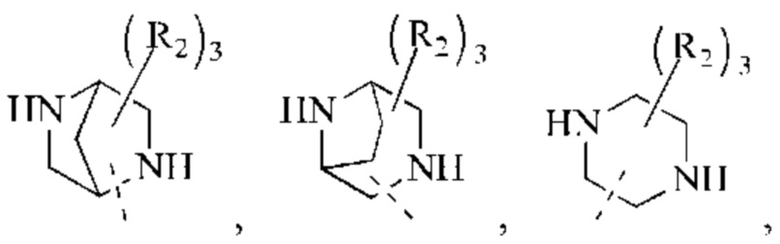
и R2 и другие переменные являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанная группировка 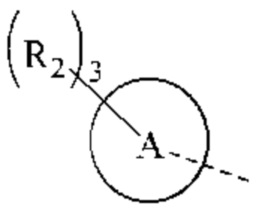 выбрана из группы, состоящей из
выбрана из группы, состоящей из 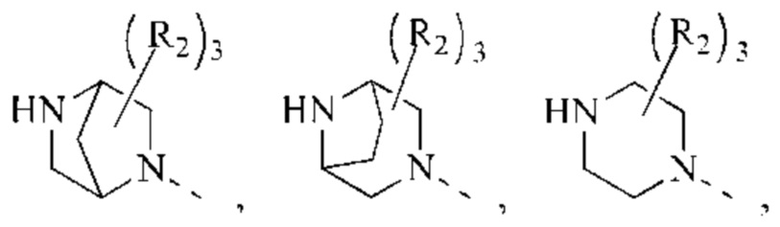
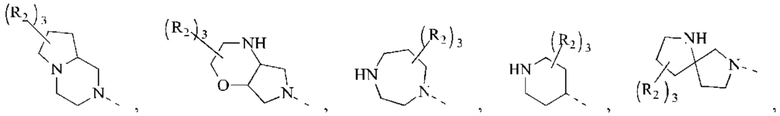
и R2 и другие переменные являются такими, как определено в настоящем изобретении.
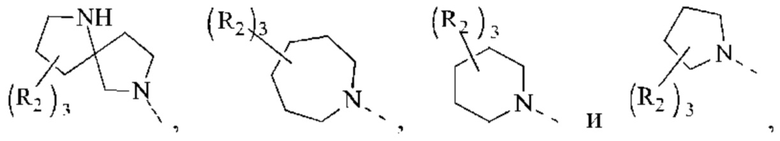
В некоторых воплощениях настоящего изобретения вышеуказанная группировка  выбрана из группы, состоящей из
выбрана из группы, состоящей из 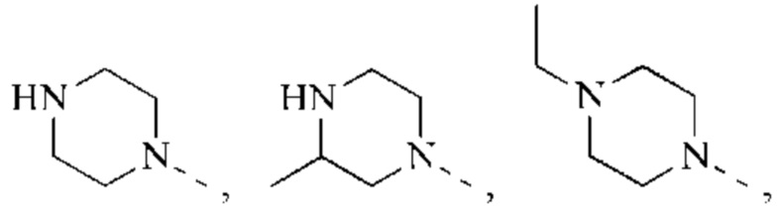
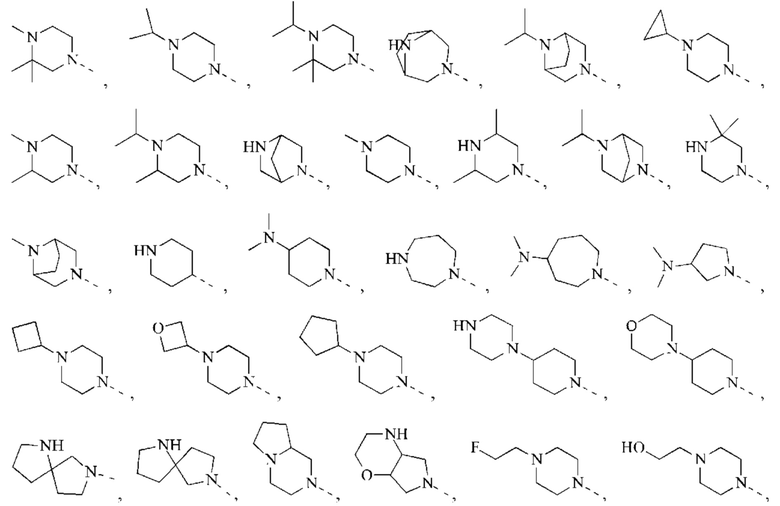
и другие переменные являются такими, как определено в настоящем изобретении.
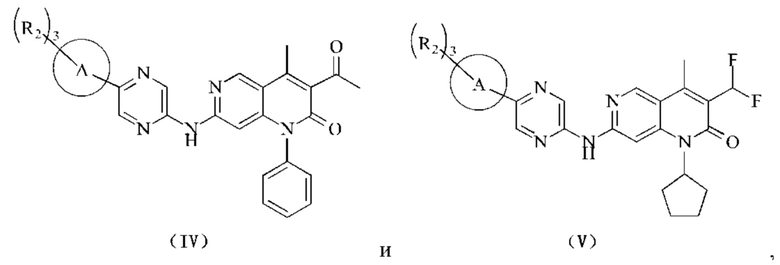
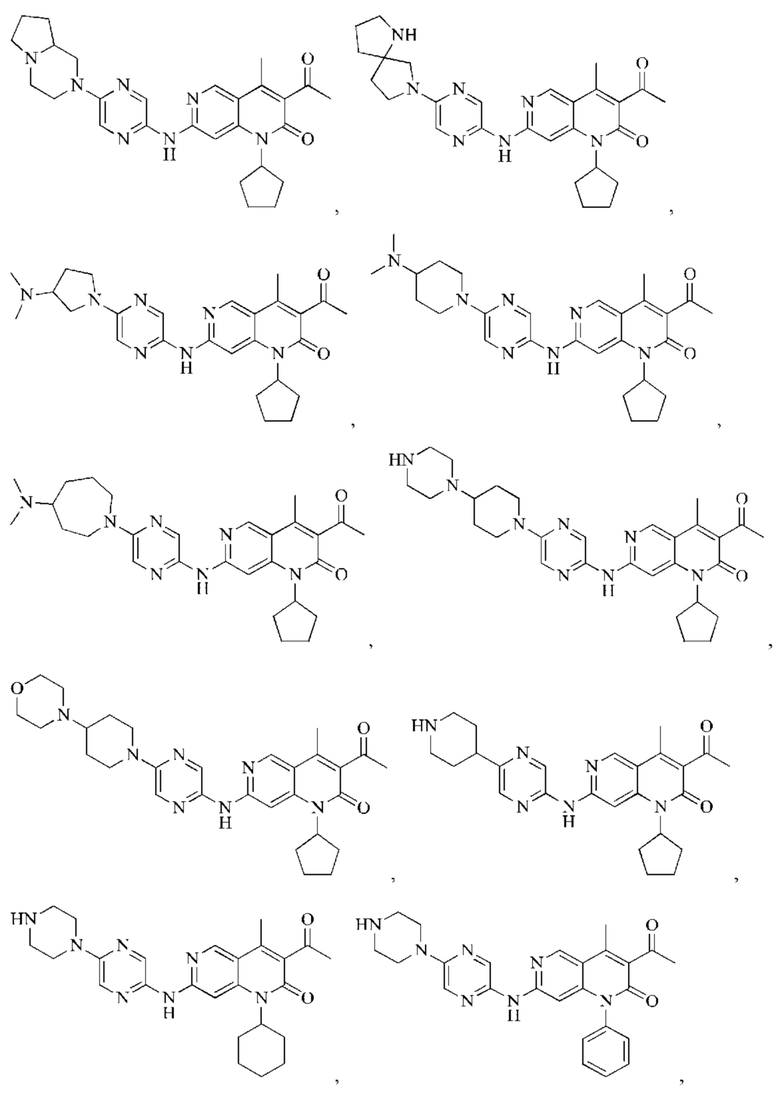
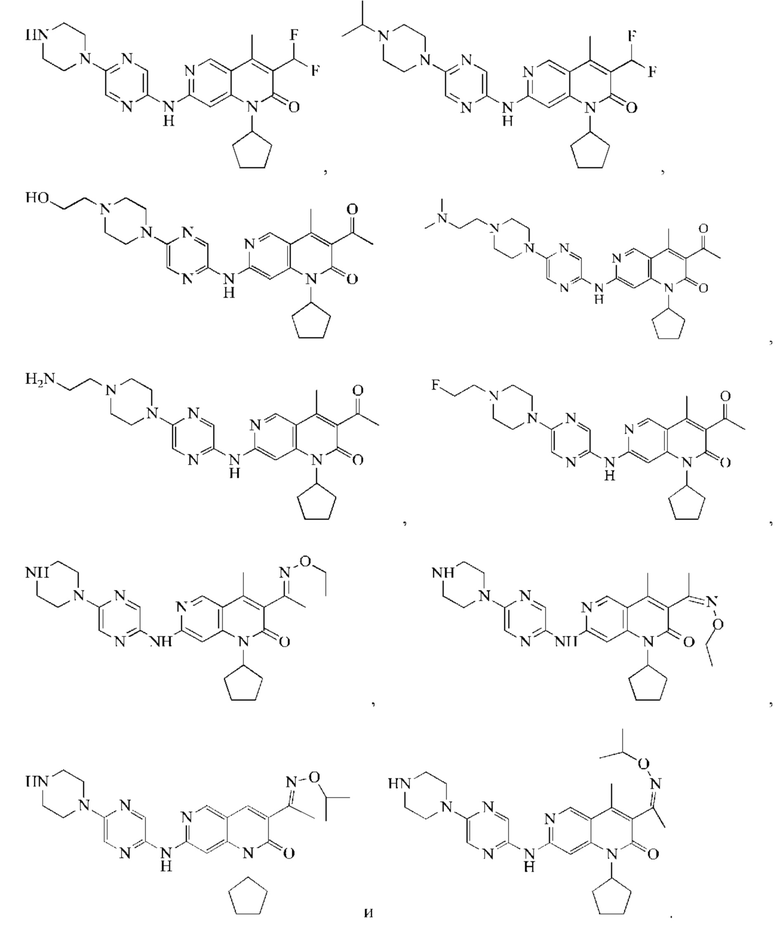
В некоторых воплощениях настоящего изобретения вышеуказанное соединение, его фармацевтически приемлемая соль или его изомер выбран из группы, состоящей из
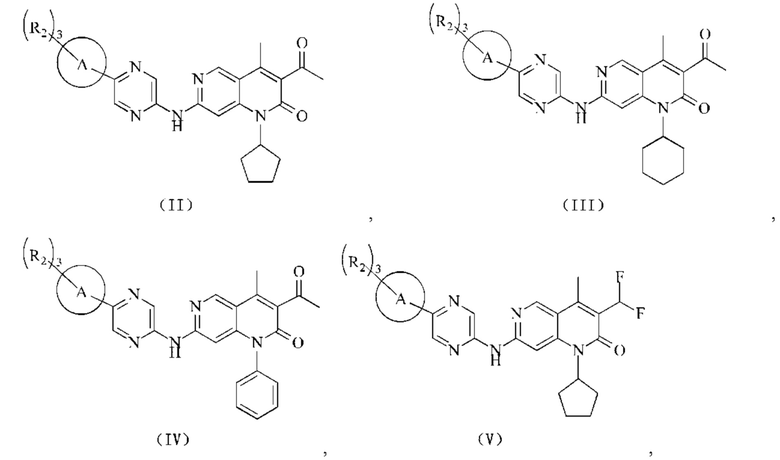
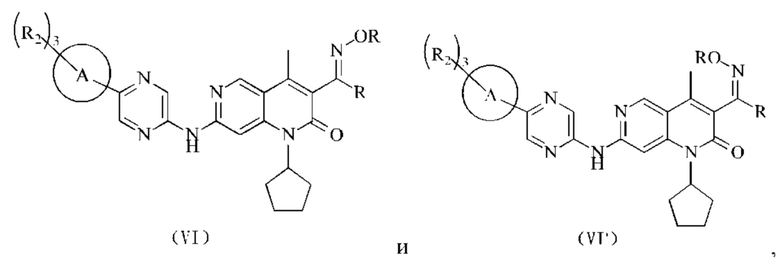 где R2, R и кольцо А являются такими, как определено в настоящем изобретении.
где R2, R и кольцо А являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанное соединение, его фармацевтически приемлемая соль или его изомер представляет собой
где R1 и R2 являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанное соединение, его фармацевтически приемлемая соль или его изомер представляет собой
где R2 является таким, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения R1 представляет собой Н или выбран из группы, состоящей из С1-3алкила и С1-3гетероалкила, каждый из которых возможно замещен 1, 2 или 3 R; каждый из R2 независимо выбран из Н, ОН, CN, галогена или выбран из группы, состоящей из С1-5алкила, С1-5гетероалкила, С3-6циклоалкила и 3-6-членного гетероциклоалкила, каждый из которых возможно замещен 1, 2 или 3 R;
кольцо А представляет собой 4-11-членный гетероциклогидрокарбил;
кольцо В выбрано из группы, состоящей из С3-6циклоалкила, 3-6-членного гетероциклоалкила, фенила и 5-6-членного гетероарила, каждый из которых возможно замещен 1, 2 или 3 R;
R выбран из галогена, ОН, CN, NH2 или выбран из группы, состоящей из С1-3алкила и С1-3гетероалкила, каждый из которых возможно замещен 1, 2 или 3 R';
R' выбран из группы, состоящей из F, Cl, Br, I, ОН, CN и NH2;
каждый из "гетеро" в С1-3гетероалкиле, С1-5гетероалкиле, 3-6-членном гетероциклоалкиле, 4-11-членном гетероциклогидрокарбиле и 5-6-членном гетероариле независимо выбран из группы, состоящей из N, -О-,=O, -S-, -NH-, -(С=O)-, -(S=O)- и -(S=O)2-;
в любом из вышеуказанных случаев число гетероатомом или гетероатомных групп независимо представляет собой 1, 2 или 3.
В некоторых воплощениях настоящего изобретения вышеуказанный R выбран из F, Cl, Br, ОН, CN, NH2, СН3, СН3СН2, CH3O, CF3, CHF2, CH2F, и другие переменные являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанный R1 представляет собой Н или выбран из группы, состоящей из СН3, СН3СН2 и СН3(С=O), каждый из которых возможно замещен 1, 2 или 3 R.
В некоторых воплощениях настоящего изобретения вышеуказанный R1 представляет собой СН3, CHF2 или СН3(С=O), и R и другие переменные являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанное кольцо В выбрано из группы, состоящей из циклобутила, циклопентила, циклогексила и фенила, каждый из которых возможно замещен 1, 2 или 3 R, и R и другие переменные являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанное кольцо В представляет собой циклопентил, циклогексил или фенил, и другие переменные являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанный R2 выбран из Н, ОН, CN, F, Cl или выбран из группы, состоящей из СН3, 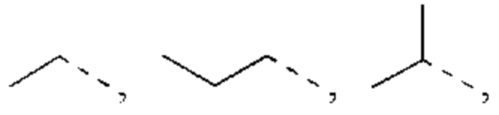
 оксетанила, пиперазинила и морфолинила, каждый из которых возможно замещен 1, 2 или 3 R, и R и другие переменные являются такими, как определено в настоящем изобретении.
оксетанила, пиперазинила и морфолинила, каждый из которых возможно замещен 1, 2 или 3 R, и R и другие переменные являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанный R2 представляет собой Н или выбран из группы, состоящей из СН3, 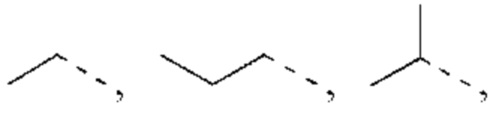
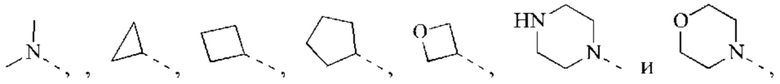 каждый из которых возможно замещен 1, 2 или 3 R, и R и другие переменные являются такими, как определено в настоящем изобретении.
каждый из которых возможно замещен 1, 2 или 3 R, и R и другие переменные являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанный R2 выбран из группы, состоящей из Н, СН3, 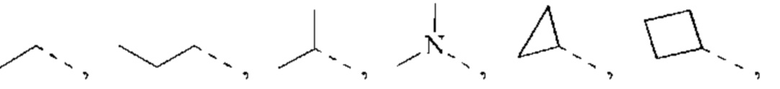
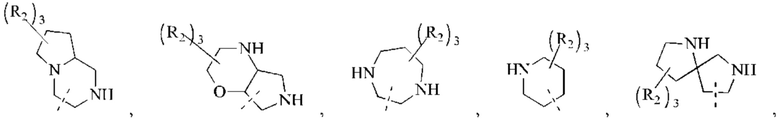
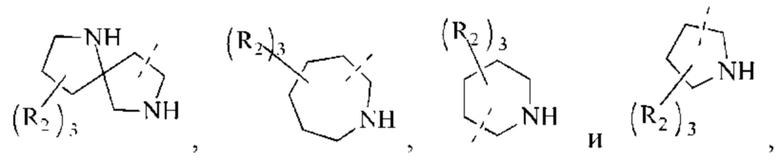
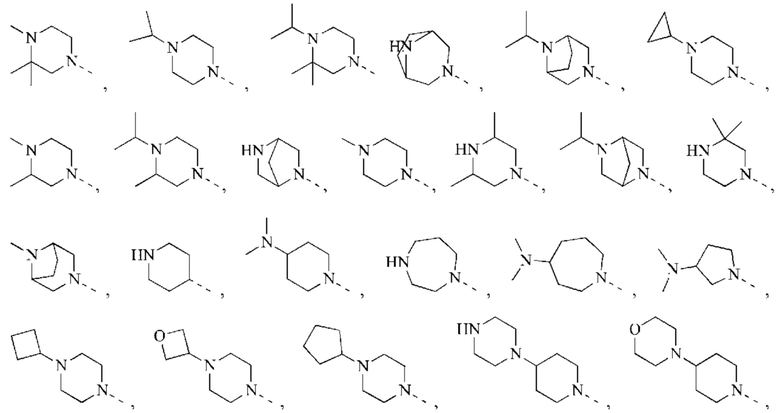
В некоторых воплощениях настоящего изобретения вышеуказанное кольцо А выбрано из группы, состоящей из 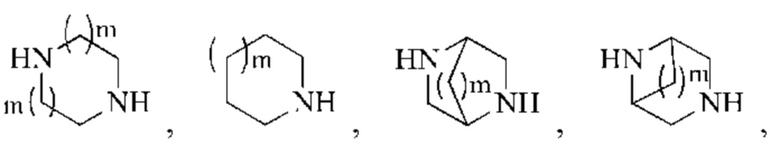
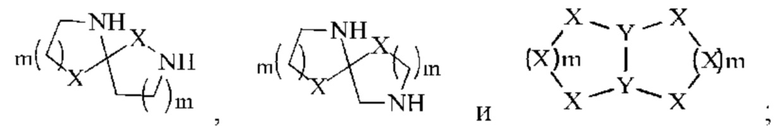 каждый из m независимо представляет собой 0, 1 или 2; каждый из X независимо представляет собой СН2, NH или О; каждый из Y независимо представляет собой СН или N, и другие переменные являются такими, как определено в настоящем изобретении.
каждый из m независимо представляет собой 0, 1 или 2; каждый из X независимо представляет собой СН2, NH или О; каждый из Y независимо представляет собой СН или N, и другие переменные являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанное кольцо А выбрано из группы, состоящей из 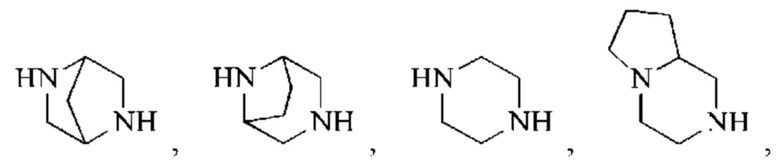
 и
и 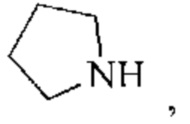 и другие переменные являются такими, как определено в настоящем изобретении.
и другие переменные являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанная группировка  выбрана из группы, состоящей из
выбрана из группы, состоящей из 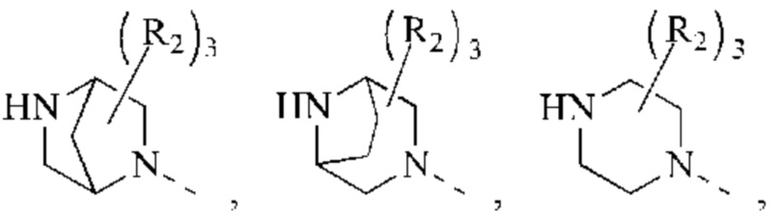
 и R2 и другие переменные являются такими, как определено в настоящем изобретении.
и R2 и другие переменные являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанная группировка 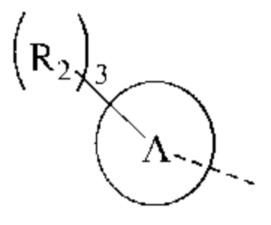 выбрана из группы, состоящей из
выбрана из группы, состоящей из 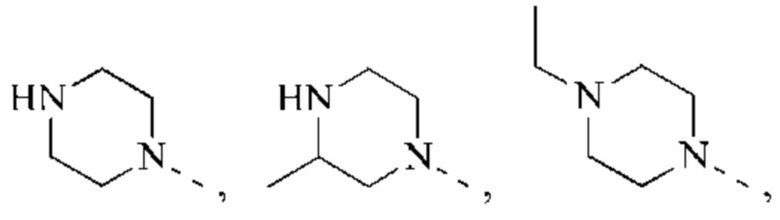
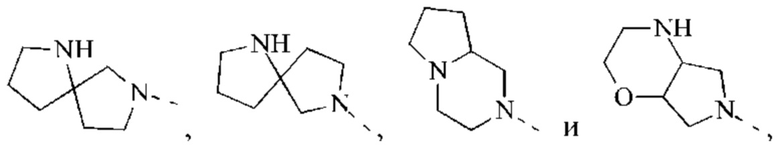 и другие переменные являются такими, как определено в настоящем изобретении.
и другие переменные являются такими, как определено в настоящем изобретении.
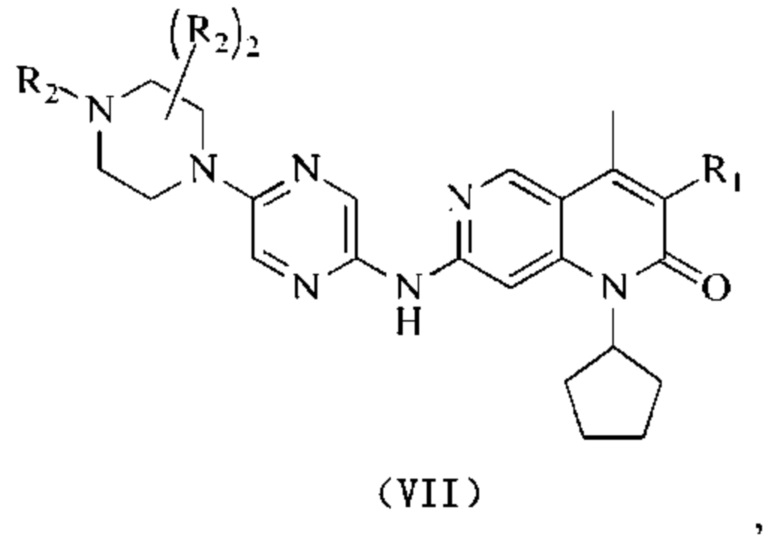
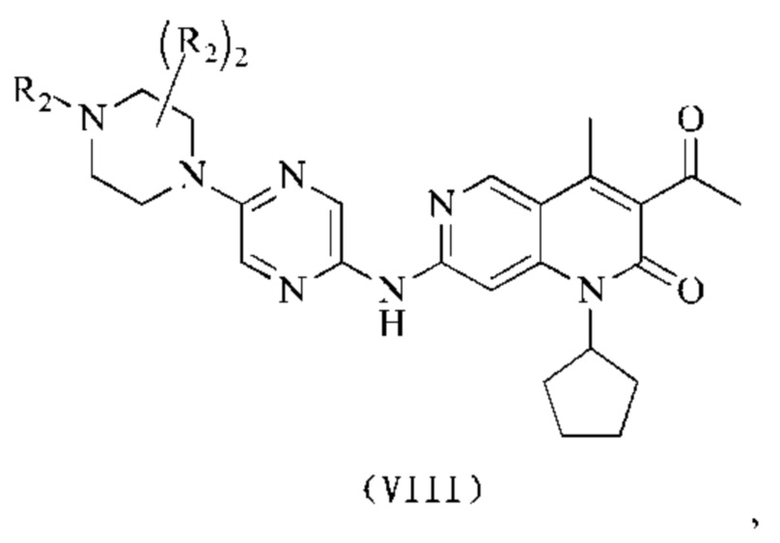
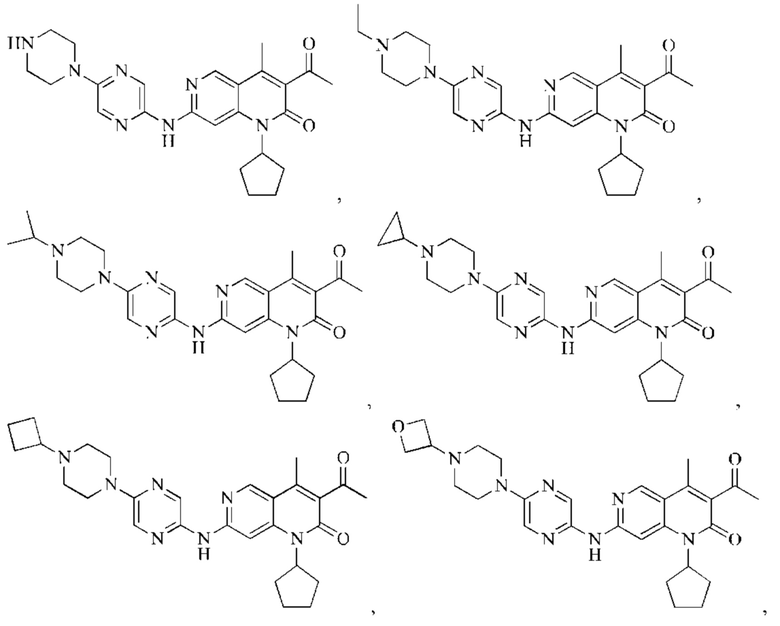
В некоторых воплощениях настоящего изобретения вышеуказанное соединение выбрано из группы, состоящей из
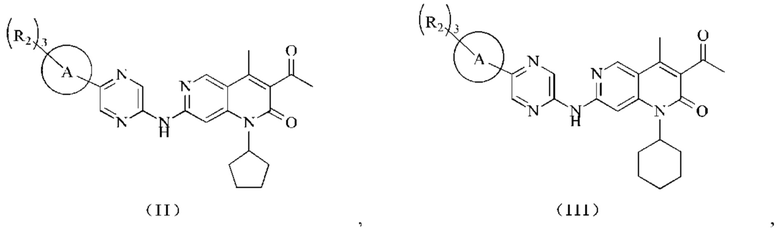
 где R2 и кольцо А являются такими, как определено в настоящем изобретении.
где R2 и кольцо А являются такими, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанное соединение выбрано из
где R2 является таким, как определено в настоящем изобретении.
В некоторых воплощениях настоящего изобретения вышеуказанное соединение, его фармацевтически приемлемая соль или его изомер выбран из группы, состоящей из
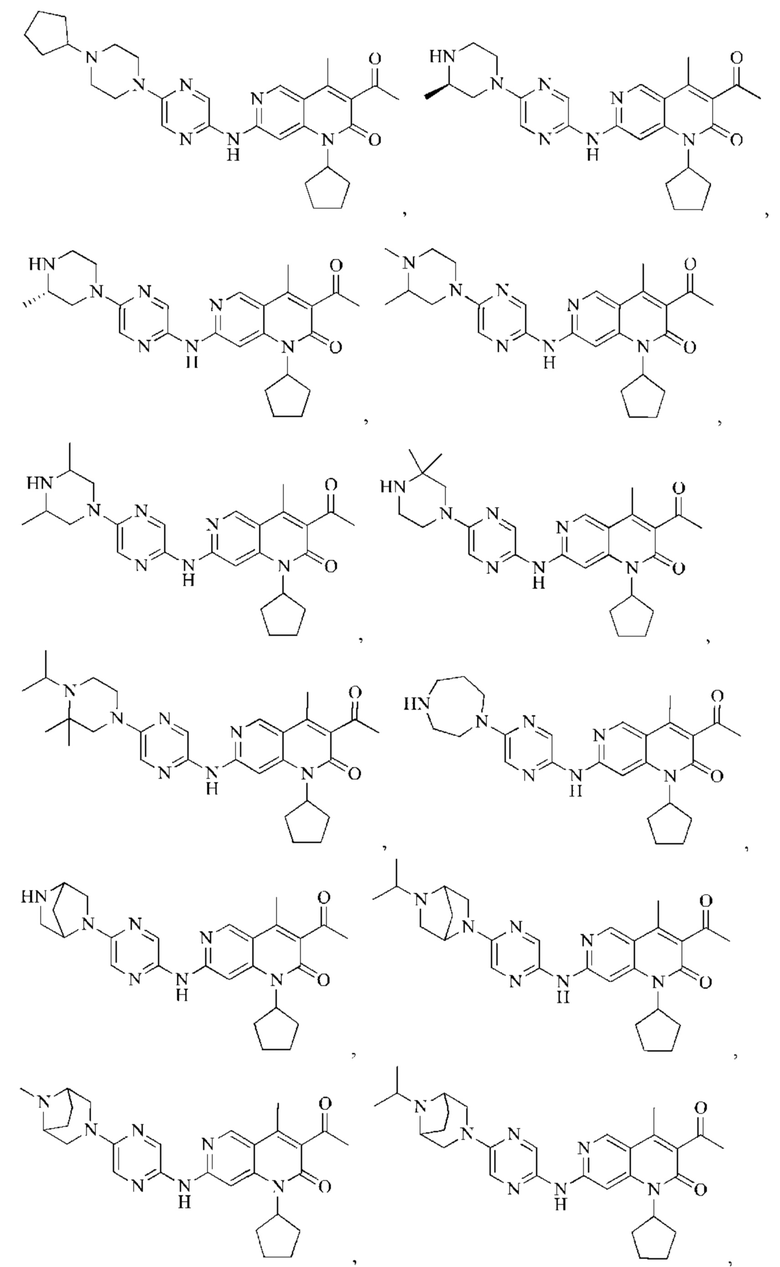
Другие воплощения настоящего изобретения могут быть получены путем произвольного комбинирования вышеуказанных переменных.
В настоящем изобретении также предложена фармацевтическая композиция, содержащая терапевтически эффективное количество вышеуказанного соединения, его фармацевтически приемлемой соли или его изомера и фармацевтически приемлемый носитель.
В настоящем изобретении также предложено применение вышеуказанного соединения, его фармацевтически приемлемой соли или его изомера в изготовлении лекарственного средства для лечения рака.
Определения и описание
Если не указано иное, следующие термины при их использовании в описаниях и формуле изобретения настоящего изобретения имеют следующие значения. Конкретный термин или фраза не должны считаться неопределенными или неясными в отсутствии конкретного определения, но должны пониматься в их обычном смысле. Когда в данной заявке упомянут товарный знак, он предназначен для обозначения соответствующему ему вещества или его активного игредиента. Термин "фармацевтически приемлемый" использован в данной заявке в отношении тех соединений, веществ, композиций и/или лекарственных форм, которые являются подходящими для применения в контакте с тканями животных и человека в пределах здравого медицинского суждения, без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соизмеримых с разумным соотношением выгода/риск.
Термин "фармацевтически приемлемая соль" относится к соли соединения по настоящему изобретению, которая получения путем приведения во взаимодействие соединения по настоящему изобретению, имеющего специфический заместитель, с относительно нетоксичной кислотой или основанием. Когда соединение по настоящему изобретению содержит относительно кислотную функциональную группу, соль добавления основания может быть получена путем приведения нейтральной формы соединения в контакт с достаточным количеством основания в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемая соль добавления основания включает соль натрия, калия, кальция, аммония, органического амина или магния или похожие соли. Когда соединение по настоящему изобретению содержит относительно основную функциональную группу, соль добавления кислоты может быть получена путем приведения нейтральной формы соединения в контакт с достаточным количеством кислоты в чистом растворе или в подходящем инертном растворителе. Примеры соли добавления фармацевтически приемлемой кислоты включают соль неорганической кислоты, где неорганическая кислота включает, например, соляную кислоту, бромистоводородную кислоту, азотную кислоту, угольную кислоту, бикарбонат, фосфорную кислоту, моногидрофосфат, дигидрофосфат, серную кислоту, гидросульфат, йодистоводородную кислоту, фосфористую кислоту и тому подообное; и соль органической кислоты, где органическая кислота включает, например, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфоновую кислоту, n-толуолсульфоновую кислоту, лимонную кислоту, винную кислоту и метансульфоновую кислоту и тому подобное; и соль аминокислоты (такой как аргинин и тому подобное), а также соль органической кислоты, такой как глюкуроновая кислота и тому подобное (см. Berge et al., "Pharmaceutical Salts", Journal of Pharmaceutical Science 66: 1-19 (1977)). Некоторые конкретные соединения по настоящему изобретению, которые содержат как основные, так и кислотные функциональные группы, могут быть превращены в любую соль добавления основания или кислоты.
Предпочтительно, в результате приведения соли в контакт с основанием или кислотой обычным способом с последующим отделением родительского соединения регенерируется нейтральная форма соединения. Разница между родительской формой соединения и его различными солевыми формами заключается в конкретных физических свойствах, таких как различная растворимость в полярном растворителе.
"Фармацевтически приемлемая соль", как использовано в настоящей заявке, относится к производному соединения по настоящему изобретению, где родительское соединение модифицируют путем образования соли с кислотой или основанием. Примеры фармацевтически приемлемой соли включают, но не ограничиваются этим, соль неорганической кислоты или органической кислоты с основной группировкой, такой как амин, соль щелочного металла, или органическую соль кислотной группировки, такой как карбоновая кислота, и тому подобное. Фармацевтически приемлемая соль включает обычную нетоксичную соль или четвертичную аммонийную соль родительского соединения, такую как соль, образованную нетоксичной неорганической кислотой или органической кислотой. Традиционная нетоксичная соль включает, но не ограничивается этим, соль, полученную из неорганической кислоты и органической кислоты, где неорганическая кислота или органическая кислоты выбрана из группы, состоящей из 2-ацетоксибензойной кислоты, 2-гидроксиэтансульфоновой кислоты, уксусной кислоты, аскорбиновой кислоты, бензолсульфоновой кислоты, бензойной кислоты, бикарбоната, угольной кислоты, лимонной кислоты, эдетовой кислоты, этандисульфоновой кислоты, этансульфоновой кислоты, фумаровой кислоты, глюкогептозы, глюконовой кислоты, глутаминовой кислоты, гликолевой кислоты, бромистоводородной кислоты, соляной кислоты, йодистоводородной кислоты, гидроксила, гидроксинафталина, изетионовой кислоты, молочной кислоты, лактозы, додецилсульфоновой кислоты, малеиновой кислоты, яблочной кислоты, миндальной кислоты, метансульфоновой кислоты, азотной кислоты, щавелевой кислоты, памовой кислоты, пантотеновой кислоты, фенилуксусной кислоты, фосфорной кислоты, полигалактаналевой кислоты, пропионовой кислоты, салициловой кислоты, стеариновой кислоты, субуксусной кислоты, янтарной кислоты, сульфаминовой кислоты, сульфаниловой кислоты, серной кислоты, таннина, винной кислоты и n-толуолсульфоновой кислоты.
Фармацевтически приемлемая соль по настоящему изобретению может быть получена из родительского соединения, которое содержит кислотную или основную группировку, традиционным химическим способом. Обычно такая соль может быть получена путем приведения во взаимодействие свободной кислотной или основной формы соединения со стехиометрическим количеством подходящего основания или кислоты в воде или в органическом растворителе или в их смеси. Обычно предпочтительными являются неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил.
Кроме солевой формы соединение, предложенное в настоящем изобретении, также существует в форме пролекарства. Пролекарство соединения, описанного в настоящей заявке, представляет собой соединение, которое без труда претерпевает химическое превращение в физиологических условиях с превращением в соединение по настоящему изобретению. Кроме того, пролекарство может быть превращено в соединение по настоящему изобретению химическим или биохимическим способом в среде in vivo.
Некоторые соединения по настоящему изобретению могут существовать в несольватированной или сольватированной форме, включая гидратированную форму. Обычно сольватированная форма является эквивалентом несольватированной формы, и обе они включены в объем настоящего изобретения.
Некоторые соединения по настоящему изобретению могут иметь асимметрический атом углерода (оптический центр) или двойную связь. Рацематы, диастереомеры, геометрические изомеры и индивидуальные изомеры включены в объем настоящего изобретения.
Если не указано иное, абсолютная конфигурация стереогенного центра представлена клиновидной сплошной связью  и клиновидной прерывистой связью
и клиновидной прерывистой связью  и относительная конфигурация стереогенного центра представлена прямой сплошной связью
и относительная конфигурация стереогенного центра представлена прямой сплошной связью  и прямой прерывистой связью
и прямой прерывистой связью  Волнистая линия
Волнистая линия 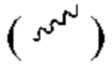 представляет собой клиновидную сплошную связь
представляет собой клиновидную сплошную связь  или клиновидную прерывистую связь
или клиновидную прерывистую связь 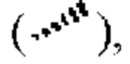 или представляет собой прямую сплошную связьили
или представляет собой прямую сплошную связьили  прямую прерывистую связь
прямую прерывистую связь 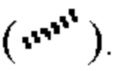
Когда соединение, описанное в данной заявке, содержит олефиновую двойную связь или другие геометрические асимметрические центры, Е- и Z-геометрические изомеры включены, если не указано иное. Аналогично, все таутомерные формы включены в объем настоящего изобретения.
Соединение по настоящему изобретению может иметь конкретную геометрическую или стереоизомерную форму. Настоящее изобретение охватывает все такие соединения, включая цис- и дарсшоизомер, (-)- и (+)-энантиомер, (R)- и (S)-энантиомер, диастереоизомер, (D)-изомер, (L)-изомер и рацемическую смесь и другие смеси, например, энантиомерно или диастереоизомерно обогащенную смесь, все из которых включены в объем настоящего изобретения. Заместители, такие как алкил, могут иметь дополнительный асимметрический атом углерода. Все эти изомеры и их смеси включены в объем настоящего изобретения.
Если не указано иное, термин "энантиомер" или "оптический изомер", относится к стереоизомерам, которые представляют собой зеркальные отображения друг друга.
Если не указано иное, термин "цис-транс-изомер" или "геометрический изомер" вызван неспособностью двойной связи или одинарной связи атомов углерода кольца свободно вращаться.
Если не указано иное, термин "диастереомер" относится к стереоизомерам, в которых молекулы имеют два или более хиральных центра и не представляют собой зеркальные отображения друг друга.
Если не указано иное, "(D)" или "(+)" обозначает правовращение, "(L)" или "(-)" обозначает левовращение, "(DL)" или "(±)" обозначает рацемизацию.
Соединения по изобретению могут быть представлены в конкретной форме. Если не указано иное, термины "таутомер" или "таутомерная форма" относятся к тому факту, что различные функциональные изомеры находятся в динамическом равновесии при комнатной температуре и могут быстро превращаться друг в друга. Если таутомеры являются возможными (например, в растворе), может быть достигнуто химическое равновесие таутомеров. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимные превращения в результате протонного переноса, такого как кето-енольная изомеризация и имин-енаминная изомеризация. Валентный таутомер включает взаимное превращение некоторых связывающих электронов. Конкретным примером кето-енольной таутомеризации является взаимное превращение между двумя таутомерами пентан-2,4-диона и 4-гидроксипент-3-ен-2-она.
Если не указано иное, термины "обогащенный одним изомером", "изомерно обогащенный", "обогащенный одним энантиомером" или "энантиомерно обогащенный" относятся тому, что содержание одного из изомеров или энантиомеров составляет менее 100%, и что содержание изомера или энантиомера составляет 60% или более, либо 70% или более, либо 80% или более, либо 90% или более, либо 95% или более, либо 96% или более, либо 97% или более, либо 98% или более, либо 99% или более, либо 99,5% или более, либо 99,6% или более, либо 99,7% или более, либо 99,8% или более, либо 99,9% или более.
Если не указано иное, термины "избыток изомера" или "избыток энантиомера" относятся к разнице между относительными процентными содержаниями двух изомеров или энантиомеров. Например, в случае, когда содержание одного из изомеров или энантиомеров составляет 90%, и содержание другого составляет 10%, тогда избыток изомера или энантиомера (значение ее (энантиомерного избытка)) составляет 80%.
Оптически активный (R)- и (S)-изомер, или D- и L-изомер может быть получен путем хирального синтеза или хиральных реагентов или других традиционных методик. Если требуется получить один тип энантиомера определенного соединения по настоящему изобретению, чистый целевой энантиомер может быть получен путем асимметрического синтеза или производного действия хиральной добавки с последующим разделением полученной диастереомерной смеси и отщеплением дополнительной группы. Альтернативно, когда молекула содержит основную функциональную группу (такую как амино) или кислотную функциональную группу (такую как карбоксил), соединение взаимодействует с подходящей оптически активной кислотой или основанием с образованием соли диастереомерного изомера, который затем подвергается диастереомерному разделению традиционным способом в данной области с получением чистого энантиомера. Кроме того, энантиомер и диастереоизомер обычно выделяют посредством хроматографии, в которой используют хиральную неподвижную фазу, и возможно комбинируют со способом химических производных (таким как карбамат, полученный из амина).
Соединение по настоящему изобретению может содержать неприродную пропорцию атомного изотопа в одном или более, чем одном атоме(ах), которые составляют соединение. Например, соединение может быть радиоактивно мечено радиоактивным изотопом, таким как тритий (3Н), йод-125 (125I) или С-14 (14С). В другом примере водород может быть заменен тяжелым водородом с образованием дейтерированного лекарственного средства, и связь, состоящая из бария и углерода, является более сильной, чем связь, состоящая из обычного водорода и углерода. По сравнению с недейтерированными лекарственными средствами, дейтерированные лекарственные средства обладают сниженными побочными эффектами и повышенной лекарственной стабильностью, усиленной эффективностью и длительный периодом биологического полувыведения лекарственного средства. Все изотопные вариации соединения по настоящему изобретению вне зависимости от того, является оно радиоактивным или нет, включены в объем настоящего изобретения.
Термин "фармацевтически приемлемый носитель" относится к любому агенту или среде носителя, которая способна доставлять эффективное количество активного вещества по настоящему изобретению, не вмешивается в биологическую активность активного вещества и не оказывает токсического побочного эффекта на хозяина или пациента. Репрезентативный носитель включает воду, масло, растительное и минеральное, кремовую основу, лосьонную основу, мазевую основу и тому подобное. Основание включает суспендирующий агент, загуститель, ускоритель проникновения и тому подобное. Эти препараты хорошо известны специалисту в косметической области или в области лекарственных средств для местного применения. Дополнительная информация о носителях может быть найдена в Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams & Wilkins (2005), содержание которой включено в данную заявку посредством ссылки.
Для лекарственного средства или фармакологически активного агента термин "эффективное количество" или "терапевтически эффективное количество" относится к нетоксичному, но достаточному количеству для достижения целевого эффекта лекарственного средства или агента. Для пероральной лекарственной формы по настоящему изобретению "эффективное количество" активного вещества в композиции относится к количеству, требуемому для достижения целевого эффекта при объединении с другим активным веществом в композиции. Эффективное количество варьируется от индивидуума к индивидууму и определяется в зависимости от возраста и общего состояния реципиента, а также конкретного активного вещества. Подходящее эффективное количество в индивидуальном случае может быть определено специалистом в данной области на основе рутинного эксперимента.
Термин "активный ингредиент", "терапевтический агент", "активное вещество" или "активный агент" относится к химическому веществу, которое может эффективно лечить целевое расстройство, заболевание или состояние.
"Возможный" или "возможно" означает, что последующее событие или условие может случаться, но не обязательно, так этот термин включает случай, при котором событие или условие случается, и случай, при котором событие или состояние не случается.
Термин "замещенный" означает, что один или более чем один атом водорода при конкретном атоме замещен заместителем, включая дейтерий и варианты водорода, до тех пор, пока валентность конкретного атома является нормальной и замещенное соединение является стабильным. Когда заместитель представляет собой кислород (т.е., =O), это означает, что замещены два атома водорода. Положения в ароматическом кольце не могут быть замещены кетоном. Термин "возможно замещенный" означает, что атом может быть или не быть замещен заместителем, если не указано иное, тип и количество заместителей может быть любым до тех пор, пока это является химически достижимым.
Когда любая переменная (такая как R) присутствует в составе или структуре соединения более чем один раз, определение производной при каждом появлении является независимым. Так, например, если группа замещена 0-2 R, эта группа возможно может быть замещена вплоть до двух R, где определение R при каждом появлении является независимым. Кроме того, комбинация заместителя и/или его варианта разрешена только тогда, когда результатом комбинации является стабильное соединение.
Когда число связывающих групп представляет собой 0, например, -(CRR)0-, это означает, что связывающая группа представляет собой одинарную связь.
Когда одна из переменных выбрана из одинарной связи, это означает, что две группы, связанные одинарной связью, соединены напрямую. Например, когда L в A-L-Z представляет собой одинарную связь, структура A-L-Z в действительности представляет собой A-Z.
Когда заместитель отсутствует, это означает, что заместителя не существует. Например, когда X отсутствует в А-Х, структура А-Х в действительности представляет собой А.
Когда связь заместителя может быть перекрестно связана с более, чем одним атомом в кольце, такой заместитель может быть связан с любым атомом кольца. Когда в перечисленном заместителе не указано, каким атомов он присоединен к соединению, включенное в общую химическую формулу, но не указанное конкретно, такой заместитель может быть связан любым из его атомов. Комбинация заместителей и/или их вариантов допустима только тогда, когда результатом такой комбинации является стабильное соединение. Например, структурная единица 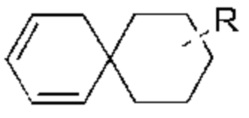 или
или 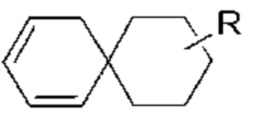 означает, что заместитель R может быть расположен в любом положении в циклогексиле или циклогексадиене.
означает, что заместитель R может быть расположен в любом положении в циклогексиле или циклогексадиене.
Когда перечисленная связывающая группа не указывает направление для связи, направление для связи является произвольным, например, связывающая группа L, содержащаяся в 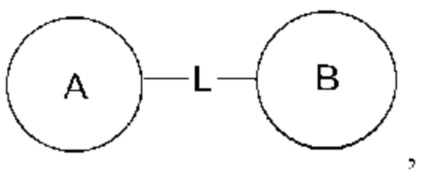 представляет собой -MW-, значит -MW- может связывать кольцо А и кольцо В с образованием
представляет собой -MW-, значит -MW- может связывать кольцо А и кольцо В с образованием 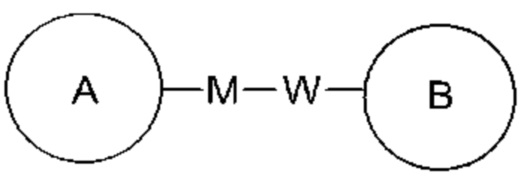 в направлении таком же, как направление чтения слева направо, и образовывать
в направлении таком же, как направление чтения слева направо, и образовывать 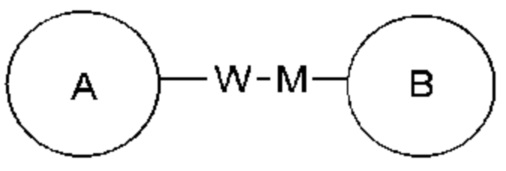 в направлении противоположном направлению чтения слева направо.
в направлении противоположном направлению чтения слева направо.
Если не указано иное, термин "гетеро" представляет собой гетероатом или гетероатомную группу (например, атомную группу, содержащую гетероатом), включая атом, отличны от углерода (С) и водорода (Н), и атомную группу, содержащую вышеуказанный гетероатом, например, включая кислород (О), азот (N), серу (S), кремний (Si), германий (Ge), алюминий (Al), бор (В), -O-, -S-, =O, =S, -С(=O)O-, -С(=O)-, -C(=S)-, -S(=O), -S(=O)2-, а также группу, состоящую из -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2N(H)- и -S(=O)N(H)-, каждая из которых является возможно замещенной.
Если не указано иное, термин "кольцо" относится к замещенному или незамещенному циклоалкилу, гетероциклоалкилу, циклоалкенилу, гетероциклоалкенилу, циклоалкинилу, гетероциклоалкинилу, арилу или гетероарилу. Так называемое кольцо включает отдельное кольцо, двойное кольцо, спиральное кольцо, конденсированное кольцо или мостиковое кольцо. Число атомов в кольце обычно определено как число членов в кольце, например, "5-7-членное кольцо" означает, что 5-7 атомов собраны в кольцо. Если не указано иное, кольцо возможно содержит 1-3 гетероатома. Таким образом, "5-7-членное кольцо" включает, например, фенил, пиридинил и пиперидинил; с другой стороны, термин "5-7-членное гетероциклоалкильное кольцо" включает пиридил и пиперидинил, но исключает фенил. Термин "кольцо" также включает кольцевую систему, содержащую по меньшей мере одно кольцо, где каждое кольцо независимо соответствует вышеуказанному определению.
Если не указано иное, термин "гетероцикл" или "гетероцикло" относится к стабильному моноциклическому, бициклическому или трициклическому кольцу, содержащему гетероатом или гетероатомную группу, которая может быть насыщенной, частично ненасыщенной или ненасыщенной (ароматической) и может содержать атомы углерода и 1, 2, 3 или 4 кольцевых гетероатомов, независимо выбранных из N, О и S, где любой из вышеуказанного гетероцикла может быть конденсирован с бензольным кольцом с образованием бициклического кольца. Атомы азота и серы возможно могут быть окислены (т.е., NO и S(O)p, p представляет собой 1 или 2). Атом азота может быть замещенным или незамещенным (т.е., N или NR, где R представляет собой Н или другой заместитель, уже определенный в данной заявке). Гетероцикл может быть присоединен к боковой группе любого гетероатома или атома углерода с образованием стабильной структуры. Если полученное соединение является стабильным, гетероцикл, описанный в данной заявке, может иметь заместитель при углероде или азоте. Атом азота в гетероцикле возможно может быть кватернизован. В предпочтительном воплощении, когда общее количество атомов S и О в гетероцикле составляет более чем 1, гетероатомы не являются смежными друг с другом. В другом предпочтительном воплощении общее число атомов S и О в гетероцикле составляет не более чем 1. Как использовано в данной заявке, термин "ароматическая гетероциклическая группа" или "гетероарил" относится к стабильному 5-, 6- или 7-членному моноциклическому или бициклическому или 7-, 8-, 9- или 10-членному бициклическому гетероциклическому ароматическому кольцу, которое содержит атомы углерода и 1, 2, 3 или 4 кольцевых гетероатома, независимо выбранных из N, О и S. Атом азота может быть замещенным или незамещенным (т.е., N или NR, где R представляет собой Н или другие заместители, уже определенные в данной заявке). Гетероатомы азота и серы возможно могут быть окислены (т.е., NO и S(O)p, p представляет собой 1 или 2). Следует отметить, что общее число атомов S и О в ароматическом гетероцикле составляет не более чем один. Мостиковое кольцо также включено в определение гетероцикла. Мостиковое кольцо образуется, когда один или более чем один атом (т.е., С, О, N или S) связывает два несмежных атома углерода или азота. Предпочтительное мостиковое кольцо включает, но не ограничивается этим, один атом углерода, два атома углерода, один атом азота, два атома азота и одну углерод-азотную группу. Следует отметить, что мостик всегда превращает моноциклическое кольцо в трициклическое кольцо. В мостиковом кольце заместитель на кольце также может присутствовать на мостике.
Примеры гетероциклического соединения включают, но не ограничиваются этим: акридинил, ацоцинил, бензимидазолил, бензофуранил, бензомеркаптофуранил, бензомеркаптофенил, бензоксазолил, бензоксазолинил, бензотиазолил, бензотриазолил, бензотетразолил, бензоизоксазолил, бензоизотиазолил, бензоимидазолинил, карбазолил, 4aH-карбазолил, карболинил, хроманил, хромен, циннолинил, декагидрохинолинил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2,3-b]тетрагидрофуранил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индазолил, индоленил, индолинил, индолизинил, индолил, 3H-индолил, изобензофуранил, изоиндолил, изоиндолинил, изохинолинил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидро-изохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, гидроксииндолил, пиримидинил, фенантридинил, фенантролинил, феназин, фенотиазин, бензоксантинил, фенолоксазинил, фталазинил, пиперазинил, пиперидинил, пиперидонил, 4-пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридо-оксазолил, пиридо-имидазолил, пиридо-тиазолил, пиридинил, пирролидинил, пирролинил, 2H-пирролил, пирролил, хиназолинил, хинолинил, 4H-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6H-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, изотиазолилтиенил, тиенооксазолил, тиенотиазолил, тиеноимидазолил, тиенил, триазинил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил и ксантенил. Также включены конденсированно-кольцевые соединения и спиро-соединения.
Если не указано иное, термин "гидрокарбил" или его гипонимы (например, алкил, алкенил, алкинил, и арил, и т.п.), отдельно или как часть другого заместителя, относится к линейному, разветвленному или циклическому углеводородному радикалу или любой их комбинации. Они могут быть полностью насыщенными (например, алкил), моно- или полиненасыщенными (например, алкенил, алкинил и арил), могут быть моно-, ди- или полизамещенными, могут быть одновалентными (например, метил), двухвалентными (например, метилен) или поливалентными (например, метенил), также могут включать двухвалентную или поливалентную группу, имеют конкретное число атомов углерода (например, С1-С12 обозначает от 1 до 12 атомов углерода, С1-12 выбран из C1, С2, С3, С4, С5, С6, С7, С8, С9, С10, С11 и С12; С3-12 выбран из С3, С4, С5, С6, С7, С8, С9, С10, С11 и С12). Термин "гидрокарбил" включает, но не ограничивается этим, алифатический гидрокарбил и ароматический гидрокарбил. Алифатический гидрокарбил включает линейный и циклический гидрокарбил, конкретно включает, но не ограничивается этим, алкил, алкенил и алкинил. Ароматический гидрокарбил включает, но не ограничивается этим, 6-12-членный ароматический гидрокарбил, такой как фенил, нафтил и тому подобное. В некоторых воплощениях термин "гидрокарбил" относится к линейной или разветвленной группе или их комбинации, которая может быть полностью насыщенной, моно- или полиненасыщенной и может включать двухвалентную или поливалентную группу. Примеры насыщенной гидрокарбильной группы включают, но не ограничиваются этим, метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, циклогексил, (циклогексил)метил, циклопропилметил и гомолог или изомер н-амила, н-гексила, н-гептила, н-октила и других атомных групп. Ненасыщенный гидрокарбил имеет одну или более чем одну двойную или тройную связь. Примеры ненасыщенного алкила включают, но не ограничиваются этим, винил, 2-пропенил, бутенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и высшие гомологи и изомеры.
Если не указано иное, термин "гетерогидрокарбил" или его гипонимы (такие как гетероалкил, гетероалкенил, гетероалкинил, и гетероарил, и т.п.), самостоятельно или как часть другого заместителя, относится к стабильной линейной, разветвленной или циклической углеводородной группе или к любым ее комбинациям, которая имеет определенное число атомов углерода и по меньшей мере один гетероатом. В некоторых воплощениях термин "гетероалкил" самостоятельно или в комбинации с другим термином относится к стабильному прямоцепочечному, разветвленному углеводородному радикалу или из комбинации, который имеет определенное число атомов углерода и по меньшей мере один гетероатом. В конкретном воплощении гетероатом выбран из В, О, N и S, где атомы азота и серы возможно окислены, и атом азота возможно кватернизован. Гетероатом или гетероатомная группа может быть расположена в любом внутреннем положении гетерогидрокарбила, включая положение, где гидрокарбил присоединяется к остальной части молекулы. Но термины "алкокси", "алкиламино" и "алкилтио" (или тиоалкил) использованы в традиционном значении и относятся к алкильной группе, присоединенной к остальной части молекулы атомом кислорода, амино или атомом серы, соответственно. Примеры включают, но не ограничиваются этим, -CH2-CH2-O-СН3, -CH2-CH2-NH-СН3, -CH2-CH2-N(СН3)-СН3, -CH2-S-CH2-СН3, -СН2-СН2, -S(O)-СН3, -CH2-CH2-S(O)2-СН3, -CH=CH-O-СН3, -CH2-CH=N-OCH3 и -CH=CH-N(СН3)-СН3. Может присутствовать вплоть до двух последовательных гетероатомов, например, -CH2-NH-OCH3.
Если не указано иное, термин "циклогидрокарбил", "гетероциклогидрокарбил" или его гипонимы (такие как арил, гетероарил, циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, циклоалкинил, гетероциклоалкинил и т.п.) самостоятельно или в комбинации с другим термином относятся к циклизованному "гидрокарбилу" или "гетерогидрокарбилу". Кроме того, для гетерогидрокарбила или гетероциклогидрокарбила (например, гетероалкила и гетероциклоалкила), один гетероатом может занимать положение, в котором гетероцикл присоединен к остальной части молекулы. Примеры циклоалкила включают, но не ограничиваются этим, циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и тому подобное. Неограничивающие примеры гетероциклоалкила включают 1-(1,2,5,6-тетрагидропиридил), 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, тетрагидротиофен-2-ил, тетрагидротиофен-3-ил, 1-пиперазинил и 2-пиперазинил.
Если не указано иное, термин "алкил" относится к линейной или разветвленной насыщенной углеводородной группе, которая может быть монозамещенной (например, -CH2F) или полизамещенной (например, -CF3), может быть одновалентной (например, метил), двухвалентной (например, метилен) или поливалентной (например, метенил). Примеры алкила включают метил (Me), этил (Et), пропил (такой как н-пропил и изопропил), бутил (такой как н-бутил, изобутил, втор-бутил, трет-бутил), пентил (такой как н-пентил, изопентил, неопентил) и тому подобное.
Если не указано иное, термин "алкенил" относится к алкильной группе, имеющей одну или более чем одну углерод-углеродную двойную связь в любом положении цепи, может быть монозамещенной или полизамещенной, и может быть одновалентной, двухвалентной или поливалентной. Примеры алкенила включают этенил, пропенил, бутенил, пентенил, гексенил, бутадиенил, пентадиенил, гексадиенил и тому подобное.
Если не указано иное, термин "алкинил" относится к алкильной группе, имеющей одну или более чем одну углерод-углеродную тройную связь в любом положении цепи, может быть монозамещенной или полизамещенной, и может быть одновалентной, двухвалентной или поливалентной. Примеры алкинила включают этинил, пропинил, бутинил, пентинил и тому подобное.
Если не указано иное, циклоалкил включает любой стабильный циклический или полициклический гидрокарбил, и любой атом углерода является насыщенным, может быть монозамещенным или полизамещенным, и может быть одновалентным, двухвалентным или поливалентным. Примеры циклоалкила включают, но не ограничиваются этим, циклопропил, норборнанил, [2.2.2]бициклооктан, [4,4,0]бициклодеканил и тому подобное.
Если не указано иное, циклоалкенил включает любой стабильный циклический или полициклический гидрокарбил, имеющий одну или более чем одну ненасыщенную углерод-углеродную одинарную связь в любом положении кольца, может быть монозамещенным или полизамещенным, и может быть одновалентным, двухвалентным или поливалентным. Примеры циклоалкенила включают, но не ограничиваются этим, циклопентенил, циклогексенил и тому подобное.
Если не указано иное, циклоалкинил включает любой стабильный циклический или полициклический гидрокарбил, имеющий одну или более углерод-углеродную тройную связь в любом положении кольца, может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или поливаленьным.
Если не указано иное, термин "галогено" или "галоген" самостоятельно или как часть другого заместителя относится к атому фтора, хлора, брома или йода. Кроме того, термин "галогеналкил" должен включать моногалогеналкил и полигалогеналкил. Например, термин "галоген(С1-С4)алкил" должен включать, но не ограничивается этим, трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и тому подобное. Примеры галогеналкила включают, но не ограничиваются этим, трифторметил, трихлорметил, пентафторэтил и пентахлорэтил.
Термин "алкокси" представляет собой любой алкил, определенный выше, имеющий конкретное число атомов углерода, присоединенных кислородным мостиком. Если не указано иное, С1-6алкокси включает C1, С2, С3, С4, С5 и С6 алкокси. Примеры алкокси включают, но не ограничиваются этим метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси и S-пентокси.
Если не указано иное, термин "арил" относится к полиненасыщенному ароматическому заместителю, может быть одно-, двух- или полизамещенным, может быть одновалентным, двухвалентным или поливалентным, может представлять собой отдельное кольцо или несколько колец (например, от одного до трех колец; где по меньшей мере одно кольцо является ароматическим), которые сконденсированы вместе или соединены ковалентно. Термин "гетероарил" относится к ар илу (или кольцу), содержащему от одного до четырех гетероатомов. В иллюстративном примере гетероатом выбран из В, О, N и S, где атомы азота и серы возможно окислены, и атом азота возможно кватернизован. Гетероарил может быть присоединен к остальной части молекулы через гетероатом. Неограничивающие примеры арила или гетероарила включают фенил, нафтил, бифенил, пирролил, пиразолил, имидазолил, пиразинил, оксазолил, фенил-оксазолил, изоксазолил, тиазолил, фуранил, тиенил, пиридил, пиримидинил, бензотиазолил, пуринил, бензимидазолил, индолинил, изохинолил, хиноксалинил, хинолил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фурил, 3-фурил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидил, 4-пиримидил, 5-бензотиазолил, пуринил, 2-бензимидазолил, 5-индолил, 1-изохинолил, 5-изохинолил, 2-хиноксалинил, 5-хиноксалинил, 3-хинолил и 6-хинолил. Заместитель любой из вышеуказанной арильной и гетероарильной кольцевой системы выбран из приемлемого заместителя, описанного ниже.
Если не указано иное, когда арил объединен с другими терминами (такими как арилокси, арилтио, арилалкил), арил включает арильное и гетероарильное кольцо, как определено выше. Таким образом, термин "аралкил" должен включать группу (например, бензил, фенэтил, пиридилметил и т.д.), в которой арил присоединен к алкилу, включая алкил, где атом углерода (например, метилен) был заменен таким атомом, как кислород, например, феноксиметил, 2-пиридилокси, 3-(1-нафтилокси)пропил и тому подобное.
Термин "уходящая группа" относится к функциональной группе или атому, который может быть заменен другой функциональной группой или атомом посредством реакции замещения (такой как аффинная реакция замещения). Например, репрезентативные уходящие группы включают трифлат; хлор, бром и йод; сульфонатную группу, такую как мезилат, тозилат, n-бромбензолсульфонат, n-толуолсульфонаты и тому подобное; ацилокси, такой как ацетокси, трифторацетокси и тому подобное.
Термин "защитная группа" включает, но не ограничивается этим, "защитную группу амино", "защитную группу гидрокси" или "защитную группу тио". Термин "защитная группа амино" относится к защитной группе, подходящей для блокирования побочной реакции азота в группе амино. Репрезентативные защитные группы амино включают, но не ограничиваются этим: формил; ацил, такой как алканоил (например, ацетил, трихлорацетил или трифторацетил); алкоксикарбонил, такой как дареда-бутоксикарбонил (Boc); арилметоксикарбонил, такой как бензилоксикарбонил (Cbz) и 9-флуоренилметоксикарбонил (Fmoc); арилметил, такой как бензил (Bn), тритил (Tr), 1,1-бис-(4'-метоксифенил)метил; силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS) и тому подобное. Термин "защитная группа гидрокси" относится к защитной группе, подходящей для блокирования побочной реакции гидрокси. Репрезентативные защитные группы гидрокси включают, но не ограничиваются этим: алкил, такой как метил, этил и трет-бутил; ацил, такой как алканоил (например, ацетил); арилметил, такой как бензил (Bn), n-метоксибензил (РМВ), 9-флуоренилметил (Fm), и дифенилметил (бензгидрил, DPM); силил, такой как триметилсилил (TMS) и трет-бутилдиметилсилил (TBS) и тому подобное.
Соединение по настоящему изобретению может быть получено различными синтетическими способами, хорошо известными специалисту в данной области, включая следование перечисленному воплощению, воплощению, полученному следующим перечисленным воплощением в комбинации с другими способами химического синтеза и эквивалентной заменой, хорошо известной специалисту в данной области. Предпочтительное воплощение включает, но не ограничивается этим, воплощение по настоящему изобретению.
Соединения называют вручную или с использованием программного обеспечения ChemDraw®, для доступных в продаже соединений использованы названия, данные их производителями.
Все растворители, использованные в настоящем изобретении, доступны в продаже. В настоящем изобретении приняты следующие сокращения: "MeCN" относится к ацетонитрилу; "DCM" относится к дихлорметану; "THF" относится к тетрагидрофурану; "АсОН" относится к уксусной кислоте; "TFA" относится к трифторуксусной кислоте; "DMF" относится к N,N-диметилформамиду; "Н2О" относится к воде; "Boc" относится к трет-бутоксикарбонилу, и "Bn" относится к бензилу, оба из которых представляют собой защитные группы амина; "DIPEA" относится к диизопропилэтиламину; "MnO2" относится к диоксиду марганца; "DIBAL-H" относится к диизобутилалюминия гидриду; "NaH" относится к гидриду натрия; "MeMgBr" относится к метилмагния бромиду; "LiHMDS" относится к лития гексаметилдисилазиду; "Pd2(dba)3" относится к трис(дибензилиденацетон)дипалладию; "Pd(dppf)Cl2" относится к [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладию; "Pd(OAc)2" относится к ацетату палладия; "Pd(PPh3)4" относится к тетракис(трифенилфосфин)палладию; "Pd(PPh3)2Cl2" представляет собой бис(трифенилфосфин)палладия дихлорид; "РО" относится к пероральному пути; "Xphos" относится к 2-дициклогексилфосфин-2',4',6'-триизопропилбифенилу; "BINAP" относится к (±)-2,2'-бис-(дифенилфосфино)-1,1'-бинафтилу; "Xphos-Pd-G1" относится к хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-аминоэтилфенил)]палладию(II); "Xphos-PD-G2" относится к хлор(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладию(II); "Xphos-Pd-G3" относится к метансульфоновой кислоте(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2'-амино-1,1'-бифенил)]палладию(II); "NIS" относится к N-йоддибутилимиду; "NBS" относится к N-бромсукцинимиду; "Br2" относится к жидкому брому; "NH2OH⋅HCl" относится к гидроксиламина гидрохлориду; "NaOAc" относится к ацетату натрия; "Cs2CO3" относится к карбонату цезия; "OsO4" относится к тетраоксиду осмия; "NaIO4" относится к перйодату натрия; "DAST" относится к диэтиламиносеры трифториду; "РО" относится к внутрижелудочному введению; "QD" относится к одному разу в сутки.
Технический эффект
Соединения по настоящему изобретению обладают значительной ингибирующей активностью в отношении киназ CDK4 и CDK6. В то же время, соединения по настоящему изобретению обладают значительной активностью ингибирования пролиферации в отношении клеток рака легкого Н358. Некоторые соединения по настоящему изобретению имеют большую ингибирующую активность в отношении пролиферации клеток NCI-Н358, чем референсное соединение палбоциклиб.
По сравнению с референсными соединениями палбоциклибом и LY2835219, соединения по настоящему изобретению обладают более высокой проникающей способностью, и абсорбция и транспорт in vivo с меньшей вероятностью подвержены влиянию эффлюксных транспортеров. Лучшая проникающая способность позволяет соединениям по настоящему изобретению более широко распространяться в in vivo тканях, таких как легкое, что приводит к лучшей противоопухолевой эффективности in vivo. Кроме того, лучшая проникающая способность делает возможным для соединений по настоящему изобретению проникать через гемато-энцефалический барьер и достигать цели лечения метастаз в головного мозге (в том числе при раке легкого).
Соединения по настоящему изобретению имеют более высокую кинетическую растворимость, чем палбоциклиб. Кинетическая растворимость может помочь лучше понимать данные биологических испытаний in vitro и in vivo. Кроме того, соединения по настоящему изобретению обладают улучшенной стабильностью в микросомах печени у человека, крыс и мышей, и скорость их клиренса является низкой. В модельном исследовании подкожно имплантированного колоректального рака НСТ-116, потеря массы у животных, которых лечили соединениями по настоящему изобретению, была меньшей, что указывало на то, что соединения по настоящему изобретению имеют лучшую безопасность.
Соединения по настоящему изобретению демонстрируют значительную противоопухолевую активность в отношении ксенографта опухолевой ткани (PDX), полученной от пациента с раком легкого LU-01-0393. Хотя некоторые соединения по настоящему изобретению имеют схожий эффект в ингибировании роста объема опухоли по сравнению с референсным соединением палбоциклибом, их дозировка составляет лишь 1/2 от дозировки референсного соединения. Это может указывать на то, что соединения по настоящему изобретению обладают более высокой противоопухолевой активностью при той же дозе. С точки зрения введения, возможно уменьшить дозировку лекарственного средства, используемую пациентом, и улучшить комплаентность. Кроме того, в модельном исследовании подкожно имплантированного немелкоклеточного рака легкого NCI-H358 масса животных, которых лечили соединением по настоящему изобретению, не только не уменьшалась значительно, но также постепенно увеличивалась при той же дозе, что указывало на то, что соединение по настоящему изобретению является более совершенным и обладает значительно большей безопасностью, чем соединения из предшествующего уровня техники. В заключение, соединения по настоящему изобретению обладают лучшими фармацевтическими перспективы, чем соединения согласно предшествующему уровню техники.
Подробное описание предпочтительного воплощения
Настоящее изобретение далее проиллюстрирвоано в следующих примерах, однако настоящее изобретение не ограничено ими. Настоящее изобретение было подробно описано в тексте настоящего описания, и также были раскрыты его конкретные воплощения, для специалиста в данной области очевидно то, как модифицировать или улучшать настоящее изобретение в пределелах объема и сущности настоящего изобретения.
Соединения по настоящему изобретению могут быть получены последовательностью синтетических процедур, где R1, R2, кольцо А и кольцо В являются такими, как определено выше.
Реакционная схема 1: Получение соединения формулы (I)
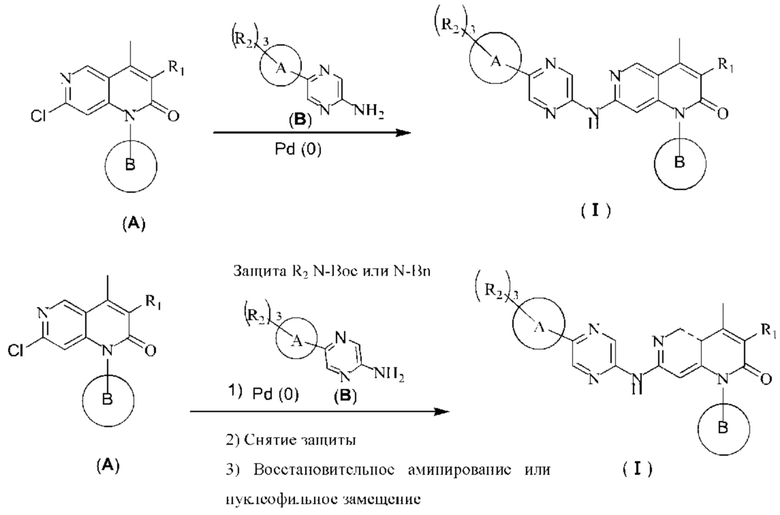
Когда в гетероциклическом ароматическом амине (В) не присутствует защитная группа N-Boc или N-Bn, соединение формулы (I) получают путем реакции 2-хлор-1,6-нафтиридин-2-она (А) и гетероциклического ароматического амина (В) согласно вышеуказанной реакции, показанной в Реакционной схеме 1. Эта реакция требует подходящего катализатора (такого как ацетат палладия), подходящего лиганда (такого как Xphos), подходящего основания (такого как карбонат цезия) и подходящего растворителя (такого как 1,4-диоксан). Согласно Реакционной схеме 1, реакцию осуществляют более подходящим образом при высокой температуре.
Когда защитная группа N-Boc или N-Bn защитная группа присутствует в гетероциклическом ароматическом амине (В), соединение формулы (I) по-прежнему может быть получено путем реакции 2-хлор-1,6-нафтиридин-2-она (А) и гетероциклического ароматического амина (В) согласно реакции ниже, показанной на Реакционной схеме 1. Группу Boc удаляют в условиях сильной кислоты (такой как трифторуксусная кислота), в то время как группу Bn удаляют в условиях восстановления (таких как палладий на углероде (смоченный водой)/формиате аммония). Конечное промежуточное соединение со снятой защитой подвергают восстановительному аминированию в условиях восстановления (таких как натрия цианоборгидрид) или реакции нуклеофильного замещения в основных условиях (таких как карбонат калия) с получением соединения формулы (I).
Реакционная схема 2: Получение 2-хлор-1,6-нафтиридин-2-она
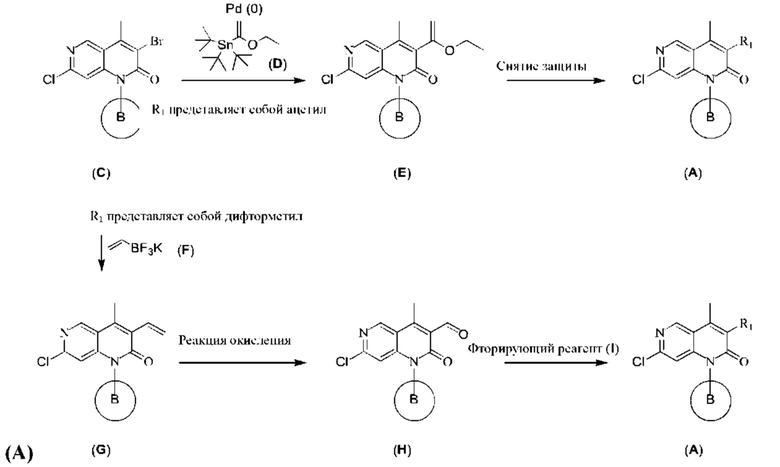
Когда R1 представляет собой ацетил, с точки зрения вышеуказанной реакции, показанной в Реакционной схеме 2, Соединение (Е) может быть получено путем реакции сочетания 2-хлор-3-бром-1,6-нафтиридин-2-она (С) и реагента олова (D). Эта реакция требует подходящего катализатора (такого как Pd(PPh3)4) и подходящего растворителя (такого как толуол). Согласно Реакционной схеме 2, реакцию более предпочтительно осуществляют при высокой температуре. После этого, с Соединения (Е) снимают защиту в условиях сильной кислоты (такой как трифторуксусная кислота) с получением 2-хлор-1,6-нафтиридин-2-она (А).
Когда R1 представляет собой дифторметил, с точки зрения нижеприведенной реакции, показанной на Реакционной схеме 2, Соединение (G) может быть получено путем реакции сочетания 2-хлор-3-бром-1,6-нафтиридин-2-она (С) и винилборного реагента (F). Реакция требует подходящего катализатора (такого как Pd(PPh3)2Cl2), подходящего основания (такого как карбонат цезия) и подходящего растворителя (такого как 1,4-диоксан/вода). Согласно Реакционной схеме 2, реакцию более предпочтительно осуществляют при высокой температуре. Соединение (Н) получают путем реакции окисления Соединения (G) в присутствии окислителя, и эта реакция требует подходящего окислителя (такого как перийодат натрия). После этого получают 2-хлор-1,6-нафтиридин-2-он (А) путем реакции Соединения (Н) со фторирующим реагентом (I), и эта реакция требует подходящего фторирующего реагента (такого как DAST).
Реакционная схема 3: Получение 2-хлор-3-бром-1,6-нафтиридин-2-она (С)
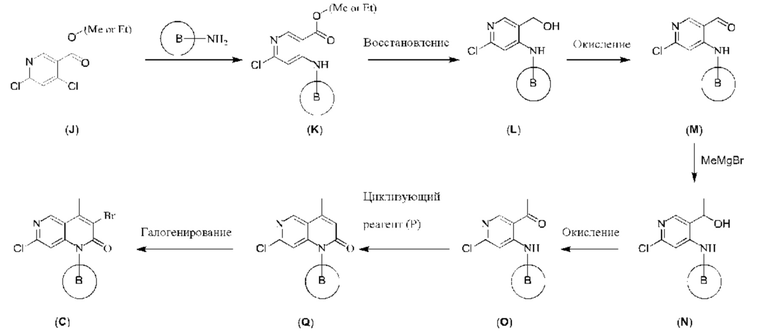
С точки зрения реакции, показанной в Реакционной схеме 3, Соединение (K) может быть получено путем реакции 4,6-дихлорникотината (J) с первичным амином, и эта реакция требует подходящего основания (такого как триэтиламин) и подходящего растворителя (такого как ацетонитрил). Соединение (K) подвергают реакции восстановления с получением Соединения (L). Эта реакция требует подходящего восстановителя (такого как DIBAL-H) и подходящего растворителя (такого как безводный тетрагидрофуран). Соединение (М) может быть получено путем реакции окисления Соединения (L), и реакция требует подходящего окислителя (такого как активный диоксид марганца). Соединение (М) и метилмагния бромид подвергают реакции нуклеофильного присоединения с получением Соединения (N), и реакция требует подходящего растворителя (такого как безводный тетрагидрофуран). Согласно Реакционной схеме 3, реакцию более предпочтительно осуществляют при низкой температуре. Соединение (N) подвергают реакции окисления с получением Соединения (О), и эта реакция требует подходящего окислителя (такого как активный диоксид марганца). Соединение (Q) может быть получено путем конденсации и реакции циклизации Соединения (О) с циклизующим реагентом (Р), и эта реакция требует подходящего циклизующего агента (такого как триэтилфосфорилацетат, этилацетат), подходящего основания (такого как гидрид натрия, LiHMDS) и подходящего растворителя (такого как тетрагидрофуран). Согласно Реакционной схеме 3, реакцию более предпочтительно осуществляют при высокой температуре. После этого Соединение (Q) подвергают реакции галогенирования с получением Соединения (С), и галогенирующий реагент может представлять собо Br2, NBS или NIS, и эта реакция требует подходящего растворителя (такого как N,N-диметилформамид, ацетонитрил).
Реакционная схема 4: Получение гетероциклического ароматического амина (В)
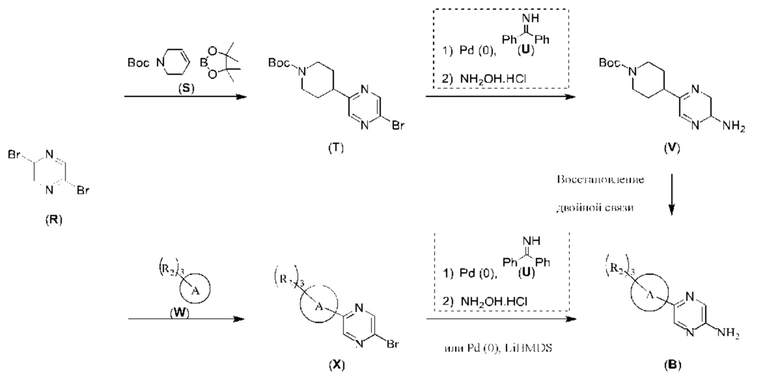
С точки зрения реакции, показанной в Реакционной схеме 4, гетероциклический ароматический амин (В) может быть получен двумя следующими способами: 1) атом брома в 2,5-дибромпиразине (R) и боратное соединение (S) подвергают палладий-катализированной реакции сочетания с получением Соединения (Т). Соединение (Т) подвергают взаимодействию с дифенилметилимином (U) в условиях катализа палладием, и затем подвергают взаимоддействию с гидроксиламина гидрохлоридом в щелочных условиях с получением Соединения (V). Наконец, Соединение (V) подвергают восстановлению двойной связи с получением гетероциклического ароматического амина (В); 2) атом брома в 2,5-дибромпиразине (R) замещают доступным в продаже или синтетическим амином (W) с получением Соединения (X). Гетероциклический ароматический амин (В) может быть получен следующими двумя способами через Соединение (X): 1) соединение (X) подвергают взаимодействию с дибензилимином (U) в условиях палладиевого катализа и затем подвергают взаимодействию с гидроксиламина гидрохлоридом в щелочных условиях с получением гетероциклического ароматического амина (В); 2) соединение (X) приводят во взаимодействие с LiHMDS в условиях палладиевого катализа с получением гетероциклического ароматического амина (В).
График А
Синтез Промежуточного соединения А и Промежуточного соединения В
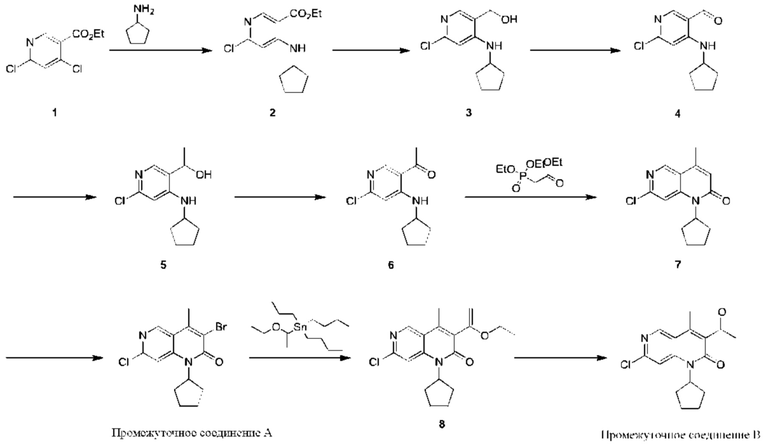
Стадия 1:
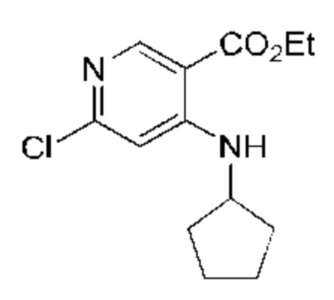
N,N-диизопропилэтиламин (17,62 г, 136,32 ммоль, 3,00 экв.) и циклопентиламин (3,87 г, 45,44 ммоль, 1,00 экв.) добавляли к раствору этил-4,6-дихлорникотината (Соединение 1) (10,00 г, 45,44 ммоль, 1,00 экв.) в ацетонитриле (100,00 мл). Реакционную смесь перемешивали при 25°С в течение 16 часов. TLC (тонкослойная хроматография) подтверждала наличие оставшегося исходного вещества, и затем реакционную смесь нагревали до 50°С и перемешивали в течение 8 часов. Завершение реакции подтверждали с помощью TLC (петролейный эфир : этилацетат = 10:1). Смесь концентрировали и полученный неочищенный продукт растворяли в этилацетате (100 мл), промывали насыщенным рассолом (50 мл × 2) и сушили над безводным сульфатом натрия, после чего фильтровали. Фильтрат концентрировали и полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 10:1) с получением указанного в заголовке соединения (Соединение 2) (9,50 г, 35,35 ммоль, выход: 77,80%). 1H-ЯМР (400 МГц, CDCl3) δ 8.67 (s, 1H), 8.20 (d, J=4,8 Гц, 1H), 6.58 (s, 1H), 4.35 (q, J=7,2 Гц, 2Н), 3.88-3.80 (m, 1H), 2.12-2.04 (m, 2Н), 1.82-1.75 (m, 2Н), 1.74-1.67 (m, 2Н), 1.63-1.57 (m, 2Н), 1,40 (t, J=7,2 Гц, 3Н); LCMS (ESI) m/z: 269,0 (М+1).
Стадия 2:
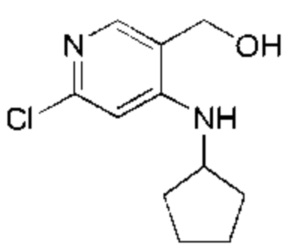
DIBAL-H (1М, 70,70 мл, 2,00 экв.) по каплям добавляли к раствору этил-6-хлор-4-(циклопентиламино)никотината (Соединение 2) (9,50 г, 35,35 ммоль, 1,00 экв.) в тетрагидрофуране (100,00 мл) при -30°С в атмосфере азота. После добавления по каплям реакционную смесь нагревали до 25°С и перемешивали в течение 16 часов. Завершение реакции подтверждали с помощью TLC (петролейный эфир : этилацетат = 5:1). Смесь охлаждали до 0°С, гасили насыщенным водным раствором сульфата натрия (50 мл) и экстрагировали этилацетатом (30 мл × 3). Объединенный органический слой промывали насыщенным рассолом (50 мл × 2) и сушили над безводным сульфатом натрия, после чего фильтровали. Фильтрат концентрировали с получением указанного в заголовке соединения (Соединение 3) (7,50 г, 33,08 ммоль, выход: 93,59%). 1Н-ЯМР (400 МГц, CDCl3) δ 7.71 (s, 1H), 6.51 (s, 1H), 5.57 (d, J=5,2 Гц, 1H), 4.60 (s, 2Н), 3.86-3.77 (m, 1H), 2.12-2.03 (m, 2Н), 1.82-1.62 (m, 4Н), 1.60-1.50 (m, 2Н).
Стадия 3:
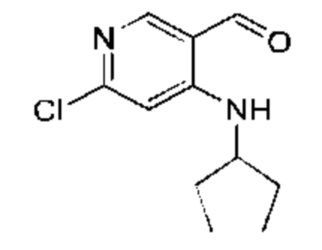
Активированный диоксид марганца (28,76 г, 330,80 ммоль, 10,00 экв.) добавляли к раствору (6-хлор-4-(циклопентиламино)-пиридин-3-ил)метанола (Соединение 3) (7,50 г, 33,08 ммоль, 1,00 экв.) в дихлорметане (80,00 мл). Реакционную смесь перемешивали при 25°С в течение 16 часов. Завершение реакции подтверждали с помощью TLC (петролейный эфир : этилацетат = 5:1). Реакционную смесь фильтровали и осадок на фильтре промывали дихлорметаном (50 мл). Фильтрат концентрировали с получением указанного в заголовке соединения (Соединение 4) (7,00 г, 31,15 ммоль, выход: 94,18%). 1Н-ЯМР (300 МГц, CDCl3) δ 9.75 (s, 1H), 8.57 (d, J=6,8 Гц, 1H), 8.20 (s, 1H), 6.53 (s, 1H), 3.85-3.73 (m, 1H), 2,05-1.94 (m, 2Н), 1.78-1.48 (m, 6Н).
Стадия 4:
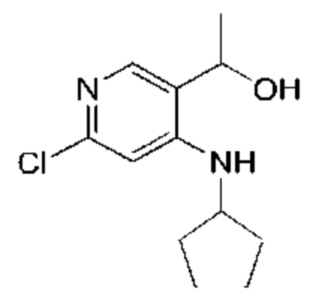
Метилмагния бромид (3М, 25,96 мл, 2,50 экв.) по каплям добавляли к раствору 6-хлор-4-(циклопентиламино)никотинальдегида (Соединение 4) (7,0 г, 31,15 ммоль, 1,00 экв.) в тетрагидрофуране (70,00 мл) при -10°С в атмосфере азота. После добавления по каплям смесь перемешивали при этой температуре в течение 1 часа. Завершение реакции подтверждали с помощью TLC (петролейный эфир : этилацетат = 5:1). Реакционную смесь гасили насыщенным водным раствором хлорида аммония (30 мл) и экстрагировали этилацетатом (50 мл × 3). Объединенный органический слой промывали насыщенным рассолом (80 мл × 2) и сушили над безводным сульфатом натрия, после чего фильтровали. Фильтрат концентрировали с получением указанного в заголовке соединения (Соединение 5) (6,70 г, 27,83 ммоль, выход: 89,35%). 1H-ЯМР (300 МГц, CDCl3) δ 7.50 (s, 1H), 6.37 (s, 1H), 6,01 (d, J=6,4 Гц, 1H), 4.76 (q, J=6,4 Гц, 1H), 3.75-3.64 (m, 1H), 1.97-1.90 (m, 2Н), 1.75-1.50 (m, 6Н), 1.46 (d, J=6,6 Гц, 3Н).
Стадия 5:
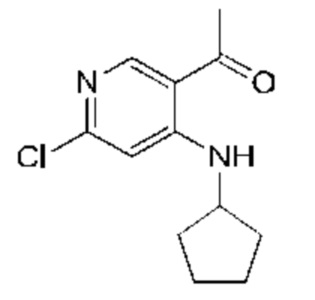
Активный диоксид марганца (24,20 г, 278,30 ммоль, 10,00 экв.) добавляли к раствору 1-(6-хлор-4-(циклопентиламино)пиридин-3-ил)этанола (Соединение 5) (6,70 г, 27,83 ммоль, 1,00 экв.) в дихлорметане (70,00 мл). Реакционную смесь перемешивали при 25°С в течение 16 часов. TLC подтверждала наличие оставшегося исходного вещества, и затем реакционную смесь нагревали до 50°С и перемешивали в течение 8 часов. Завершение реакции подтверждали с помощью TLC (петролейный эфир : этилацетат = 5:1). Реакционную смесь охлаждали до 20°С, после чего фильтровали. Осадок на фильтре промывали дихлорметаном (50 мл). Фильтрат концентрировали с получением указанного в заголовке соединения (Соединение 6) (6,00 г, 25,14 ммоль, выход: 90,32%). 1H-ЯМР (300 МГц, CDCl3) δ 9.22 (s, 1H), 8.59 (s, 1H), 6.60 (s, 1H), 3.90-3.79 (m, 1H), 2.58 (s, 3Н), 2.14-2.00 (m, 2Н), 1.87-1.67 (m, 4Н), 1.63-1.53 (m, 2Н).
Стадия 6:
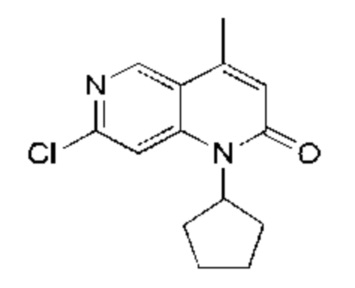
Гидрид натрия (2,61 г, 65,36 ммоль, 2,60 экв., 60%-ная чистота) порциями добавляли к раствору триэтилфосфорилацетата (14,65 г, 65,36 ммоль, 2,60 экв.) в тетрагидрофуране (60,00 мл) при 0°С в атмосфере азота. Реакционную смесь перемешивали при этой температуре в течение 20 минут и затем к реакционной смеси добавляли 1-(6-хлор-4-(циклопентиламино)пиридин-3-ил)этанон (Соединение 6) (6,00 г, 25,14 ммоль, 1,00 экв.). После добавления по каплям реакционную смесь нагревали до 70°С и перемешивали в течение 16 часов. Завершение реакции подтверждали с помощью TLC (петролейный эфир : этилацетат = 5:1). Смесь охлаждали до 25°С, гасили насыщенным водным раствором хлорида аммония (20 мл) и экстрагировали этилацетатом (30 мл × 3). Объединенный органический слой промывали насыщенным рассолом (50 мл × 2) и сушили над безводным сульфатом натрия, после чего фильтровали. Фильтрат концентрировали и полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = от 50:1 до 20:1) с получением указанного в заголовке соединения (Соединение 7) (5,00 г, 3,19 ммоль, выход: 75,70%). 1Н-ЯМР (400 МГц, CDCl3) δ 8.68 (s, 1H), 7.35 (s, 1H), 6.54 (s, 1H), 5,49 (q, J=9,2 Гц, 1H), 2.50 (s, 3Н), 2.24-2,00 (m, 6Н), 1.84-1.76 (m, 2Н); LCMS (ESI) m/z: 263.0 (M+1).
Стадия 7:
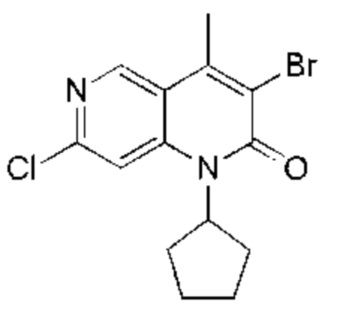
Промежуточное соединение А
Ацетат натрия (1,25 г, 15,22 ммоль, 4,00 экв.) и жидкий бром (1,22 г, 7,61 ммоль, 2,00 экв.) последовательно добавляли к раствору 7-хлор-1-циклопентил-4-метил-1,6-нафтиридин-2-она (Соединение 7) (1,00 г, 3,81 ммоль, 1,00 экв.) в уксусной кислоте (20,00 мл). Реакционную смесь нагревали до 70°С и перемешивали в течение 20 часов. Реакционную смесь концентрировали и полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 20:1) с получением указанного в заголовке соединения (Промежуточное соединение А) (1,10 г, 3,22 ммоль, выход: 84,51%). 1Н-ЯМР (400 МГц, CDCl3) δ 8.80 (s, 1H), 7,40 (s, 1H), 5,40-5.30 (m, 1H), 2.73 (s, 3Н), 2.29-2.12 (m, 4Н), 2.07-1.98 (m, 2Н), 1.81-1.75 (m, 2Н).
Стадия 8:
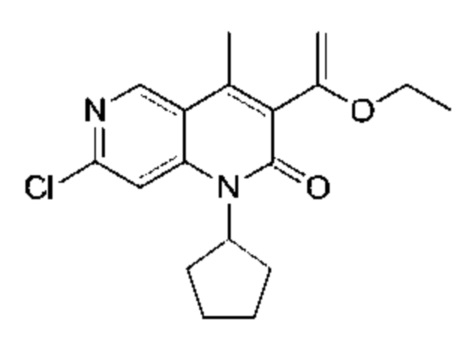
Трибутил(1-этоксивинил)олово (580,01 мг, 1,61 ммоль, 1,10 экв.) и Pd(PPh3)4 (168,71 мг, 146,00 мкмоль, 0,10 экв.) добавляли к раствору 3-бром-7-хлор-1-циклопентил-4-метил-1,6-нафтиридин-2-она (Соединение 8) (500,00 мг, 1,46 ммоль, 1,00 экв.) в толуоле (5,00 мл) в атмосфере азота. Реакционную смесь нагревали до 110°С и перемешивали в течение 16 часов. Завершение реакции подтверждали с помощью LCMS (жидкостная хроматография-масс-спектрометрия). Реакционный раствор концентрировали и полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 30:1) с получением указанного в заголовке соединения (Соединение 9) (400,00 мг, 1,20 ммоль, выход: 82,32%). 1H-ЯМР (300 МГц, CD3OD) δ 8.84 (s, 1H), 7.72 (s, 1H), 5,41-5.30 (m, 1H), 4.57 (d, J=2,4 Гц, 1H), 4.15 (d, J=2,4 Гц, 1H), 3.94 (q, J=7,2 Гц, 2Н), 2.56 (s, 3Н), 2.24-2,05 (m, 6Н), 1.84-1.78 (m, 2Н), 1.35 (t, J=6,8 Гц, 3Н); LCMS (ESI) m/z: 333.1 (M+1).
Стадия 9:
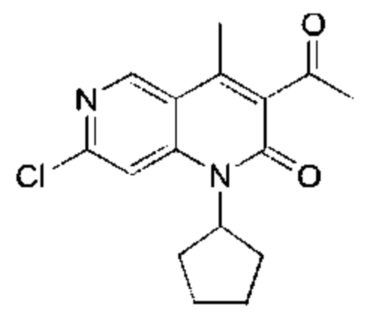
Промежуточное соединение В
Трифторуксусную кислоту (3,00 мл) добавляли к раствору 7-хлор-1-циклопентил-3-(1-этоксивинил)-4-метил-1,6-нафтиридин-2-она (Соединение 9) (400,00 мг, 1,20 ммоль, 1,00 экв.) в дихлорметане (5,00 мл). Реакционную смесь перемешивали при 25°С в течение 1 часа. Завершение реакции подтверждали с помощью TLC (петролейный эфир : этилацетат = 5:1) и LCMS. Реакционный раствор концентрировали, затем добавляли воду (5 мл) и экстрагировали этилацетатом (10 мл × 3). Объединенный органический слой промывали насыщенным рассолом (20 мл × 2) и сушили над безводным сульфатом натрия, после чего фильтровали. Фильтрат концентрировали и полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = от 20:1 до 10:1) с получением указанного в заголовке соединения (Промежуточное соединение В) (300 мг, 984,35 мкмоль, выход: 81,90%). LCMS (ESI) m/z: 305.2 (М+1).
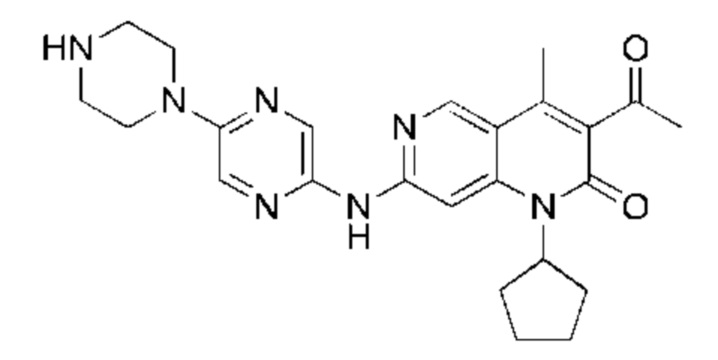
Пример 1
Стадия 1:
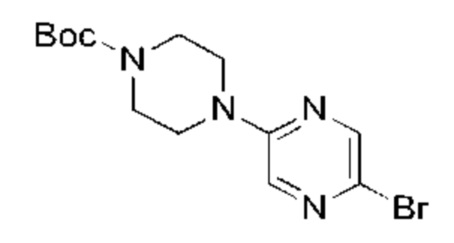
Трет-бутил пиперазин-1-карбоксилат (7,83 г, 42,04 ммоль, 1,00 экв.) и карбонат калия (8,72 г, 63,06 ммоль, 1,50 экв.) добавляли к раствору 2,5-дибромпиразина (10,00 г, 42,04 ммоль, 1,00 экв.) в 1-метилпирролидин-2-она (100,00 мл). Смесь нагревали до 100°С и перемешивали в течение 18 часов. Завершение реакции подтверждали с помощью TLC (петролейный эфир : этилацетат = 10:1). Реакционную смесь разбавляли водой (200 мл) и экстрагировали этилацетатом (200 мл × 2). Объединенный органический слой сушили над безводным сульфатом натрия, после чего фильтровали. Фильтрат концентрировали и полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = от 20:1 до 5:1) с получением указанного в заголовке соединения (11,00 г, 32,05 ммоль, выход: 76,24%). 1H-ЯМР (400 МГц, CDCl3) δ 8.15 (d, J=1,38 Гц, 1H), 7.87 (d, J=1,38 Гц, 1H), 3.56 (m, 8Н), 1.49 (s, 9Н).
Стадия 2:
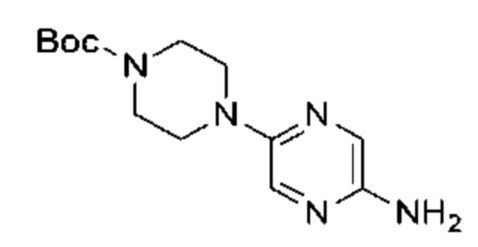
LiHMDS (1 M, 60,00 мл, 2,06 экв.) и Pd2(dba)3 (2,60 г, 2,84 ммоль, 0,10 экв.) добавляли к раствору трет-бутил-4-(5-бромпиразин-2-ил)пиперазин-1-карбоксилата (10,00 г, 29,14 ммоль, 1,00 экв.) и три-трет-бутилфосфония тетрафторбората (2,54 г, 8,74 ммоль, 0,30 экв.) в толуоле (100,00 мл) в атмосфере азота. Реакционную смесь нагревали до 65°С и перемешивали в течение 16 часов. Завершение реакции подтверждали с помощью LCMS. Реакционную смесь гасили водой (50 мл) и экстрагировали этилацетатом (100 мл × 3). Объединенный органический слой концентрировали и неочищенный продукт очищали посредством препаративной HPLC (высокоэффективная жидкостная хроматография) (щелочная) с получением указанного в заголовке соединения (5,00 г, 17,90 ммоль, выход: 61,43%). LCMS (ESI) m/z: 280.1 (М+1).
Стадия 3:
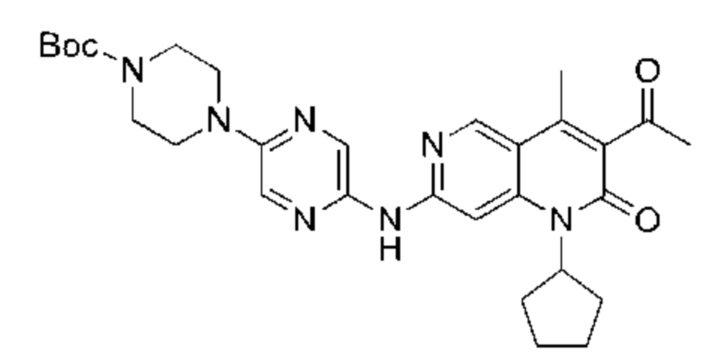
Xphos-Pd-G2 (25,82 мг, 32,81 мкмоль, 0,10 экв.) добавляли к раствору 3-ацетил-7-хлор-1-циклопентил-4-метил-1,6-нафтиридин-2-она (Промежуточное соединение В) (100,00 мг, 328,12 мкмоль, 1,00 экв.), 4-(5-аминопиразин-2-ил)пиперазин-1-карбоновой кислоты трет-бутилового сложного эфира (137,48 мг, 492,17 мкмоль, 1,50 экв.) и трет-бутоксида калия (110,45 мг, 984,35 мкмоль, 3,00 экв.) в тетрагидрофуране (2,00 мл). Смесь нагревали до 80°С и перемешивали в течение 16 часов. Полное превращение исходных веществ подтверждали с помощью TLC (петролейный эфир : этилацетат = 1:1). Реакционный раствор охлаждали до комнатной температуры и концентрировали и неочищенный продукт очищали посредством препаративной TLC (петролейный эфир : этилацетат = 1:1) с получением указанного в заголовке соединения (40,00 мг, 73,04 мкмоль, выход: 22,26%).
Стадия 4:
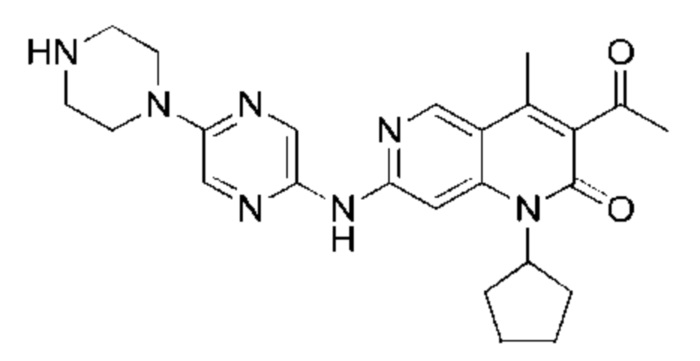
Трифторуксусную кислоту (0,5 мл) добавляли к раствору трет-бутил-4-(5-((3-ацетил-1-циклопентил-4-метил-2-оксо-1,2-дигидро-1,6-нафталин-7-ил)амино)пиразин-2-ил)пиперазин-1-карбоксилата (60,00 мг, 109,56 мкмоль, 1,00 экв.) в дихлорметане (1,00 мл) при 25°С и смесь перемешивали в течение 0,5 часа. Завершение реакции подтверждали с помощью LCMS. Реакционную смесь концентрировали и полученный неочищенный продукт очищали посредством препаративной HPLC (соляная кислота) с получением гидрохлоридной соли указанного в заголовке соединения (22,78 мг, 50,90 мкмоль, выход: 46,46%). 1Н-ЯМР (400 МГц, CD3OD) δ 8.78 (s, 1H), 8.27 (s, 1H), 8.19 (s, 1H), 7.30 (s, 1H), 5.44-5.32 (m, 1H), 3.93-3.88 (m, 4Н), 3.44-3.38 (m, 4Н), 2.51 (s, 3Н), 2,40 (s, 3Н), 2.31-2.16 (m, 4Н), 2.08 (d, J=8,0 Гц, 2Н), 1.82 (m, 2Н); LCMS (ESI) m/z: 448.1 (M+1).
Пример 2
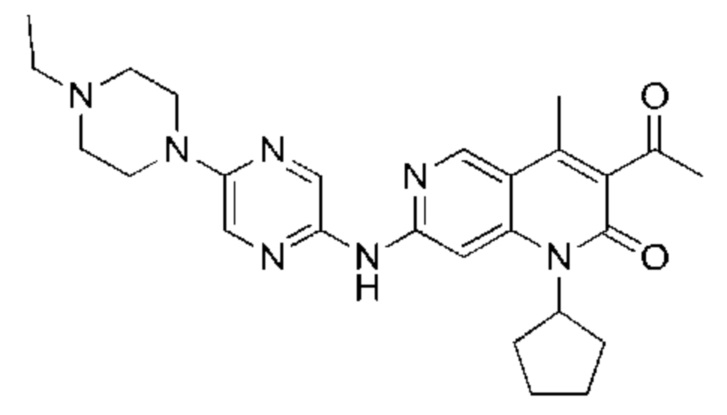
Раствор ацетальдегида (553,66 мг, 5,03 ммоль, 700,83 мкл, 15,00 экв.) и натрия триацетоксиборгидрида (213,11 мг, 1,01 ммоль, 3,00 экв.) добавляли к раствору 3-ацетил-1-циклопентил-4-метил-7-[(5-пиперазин-1-илпиразин-2-ил)амино]-1,6-нафтиридин-2-она (150,00 мг, 335,17 мкмоль, 1,00 экв.) в дихлорэтане (2,00 мл) при 25°С. Смесь перемешивали в течение 1 часа. Приблизительно 26% указанного в заголовке соединения детектировали с помощью LCMS. Смесь концентрировали и полученный неочищенный продукт очищали посредством препаративной HPLC (щелочная) с получением указанного в заголовке соединения (14,35 мг, 27,71 мкмоль, выход: 8,27%, чистота: 91,83%). 1H-ЯМР (400 МГц, CDCl3) δ 8.65 (s, 1H), 8.17 (d, J=1,3 Гц, 1H), 7.89-7.75 (m, 2Н), 7.65 (s, 1H), 5.73 (quin, J=9,3 Гц, 1H), 3.60-3.48 (m, 4Н), 2.65-2.58 (m, 4Н), 2.55 (s, 3Н), 2.49 (q, J=7,3 Гц, 2Н), 2.40 (s, 3Н), 2.29 (br dd, J=12,4, 7,3 Гц, 2H), 2.13 (br dd, J=7,9, 5,5 Гц, 2H), 2.02-1.95 (m, 2H), 1.82-1.73 (m, 2H), 1.15 (t, J=7,2 Гц, 3Н); LCMS (ESI) m/z: 492.3 (M+1).
Пример 3
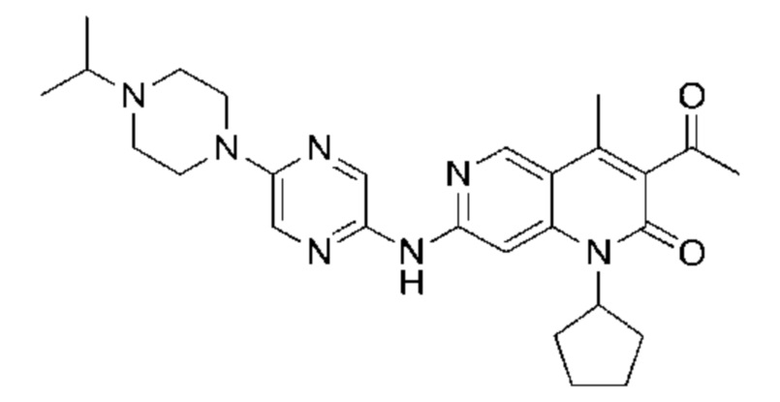
Синтез соединения по примеру 3 является таким же, как синтез соединения по примеру 2. 1H-ЯМР (400 МГц, CDCl3) δ 8.65 (s, 1H), 8.17 (d, J=1,3 Гц, 1H), 7.87-7.74 (m, 2Н), 7,46 (s, 1H), 5.74 (quin, J=9,3 Гц, 1H), 3.63-3.43 (m, 4Н), 2.77 (td, J=13,0, 6,5 Гц, 1H), 2.73-2.66 (m, 4Н), 2.56 (s, 3Н), 2.41 (s, 3Н), 2.35-2.24 (m, 2Н), 2.19-2,07 (m, 2Н), 2,03-1.95 (m, 2Н), 1.77 (br d, J=4,8 Гц, 2H), 1.11 (d, J=6,5 Гц, 6H); LCMS (ESI) m/z: 490.2 (M+1).
Пример 4
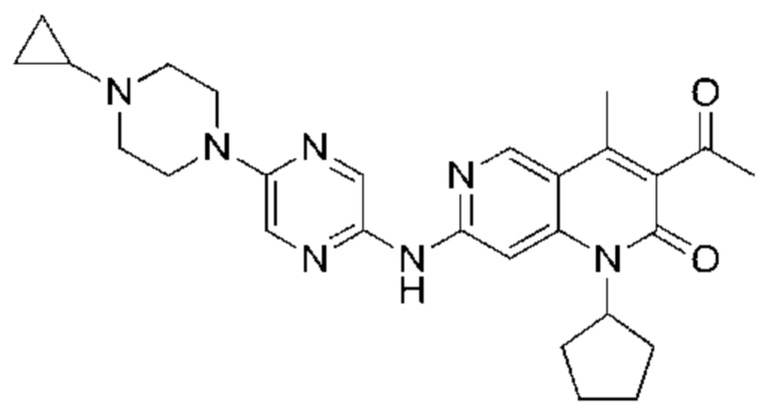
(1-этоксициклопропилокси)триметилсилан (292,12 мг, 1,68 ммоль, 335,77 мкл, 5,00 экв.) и натрия цианоборгидрид (63,19 мг, 1,01 ммоль, 3,00 экв.) добавляли к раствору 3-ацетил-1-циклопентил-4-метил-7-[(5-пиперазин-1-илпиразин-2-ил)амино]-1,6-нафтиридин-2-она (150,00 мг, 335,17 мкмоль, 1,00 экв.) в метаноле (2,00 мл) при 25°С. Смесь перемешивали при 25°С в течение 1 часа. Неполное превращение исходных веществ подтверждали с помощью LCMS. Реакционную смесь нагревали до 60°С и перемешивали в течение 18 часов. Указанное в заголовке соединение детектировали посредством LCMS. Реакционную смесь концентрировали и полученный неочищенный продукт очищали посредством препаративной HPLC (щелочной) с получением указанного в заголовке соединения (31,98 мг, 65,17 мкмоль, выход: 19,44%, чистота: 99,37%). 1Н-ЯМР (400 МГц, CDCl3) δ 8.66 (s, 1H), 8.17 (d, J=1,4 Гц, 1H), 7.84-7.77 (m, 2Н), 7,44 (s, 1H), 5.74 (quin, J=9,3 Гц, 1H), 3.54-3.44 (m, 4Н), 2.83-2.73 (m, 4Н), 2.56 (s, 3Н), 2.41 (s, 3Н), 2.35-2.24 (m, 2Н), 2.19-2,08 (m, 2Н), 2.04-1.93 (m, 2Н), 1.78 (br dd, J=10,3, 5,5 Гц, 2H), 0.56-0.45 (m, 4Н); LCMS (ESI) m/z: 488.3 (M+1).
Пример 5
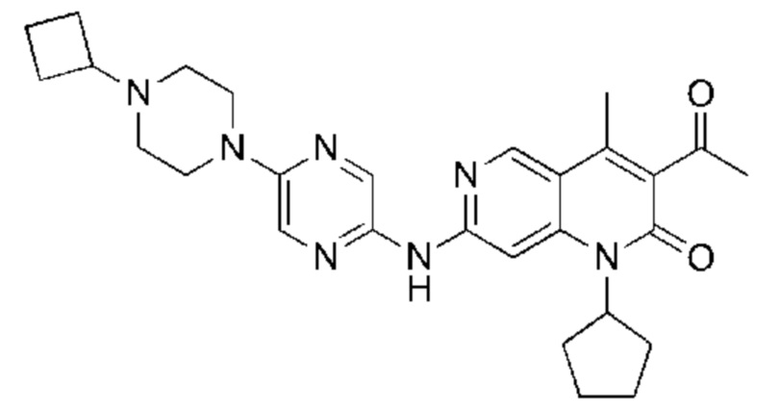
Синтез соединения по примеру 5 является таким же, как синтез соединения по примеру 2. 1Н-ЯМР (400 МГц, CDCl3) δ 8.65 (s, 1H), 8.17 (d, J=1,1 Гц, 1H), 7.88-7.73 (m, 2Н), 7.65 (s, 1H), 5.73 (quin, J=9,3 Гц, 1H), 3.57-3.48 (m, 4Н), 2.79 (quin, J=7,8 Гц, 1H), 2.55 (s, 3Н), 2.51-2.44 (m, 4Н), 2,40 (s, 3Н), 2.35-2.23 (m, 2Н), 2.18-2.04 (m, 4Н), 2,02-1.86 (m, 6Н), 1.83-1.70 (m, 2Н); LCMS (ESI) m/z: 502.3 (M+1).
Пример 6
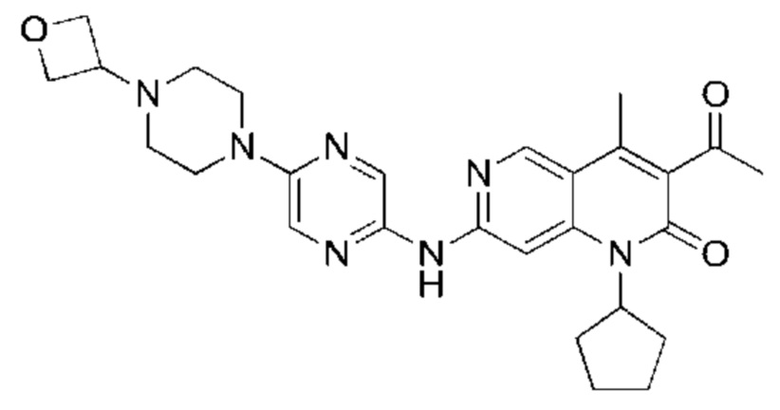
Синтез соединения по примеру 6 является таким же, как синтез соединения по примеру 2. 1Н-ЯМР (400 МГц, CDCl3) δ 8.66 (s, 1H), 8.19 (d, J=1,1 Гц, 1H), 7.82 (s, 2Н), 7.55 (s, 1H), 5.74 (quin, J=9,3 Гц, 1H), 4.70 (td, J=19,4, 6,4 Гц, 4H), 3.68-3.47 (m, 5H), 2.56 (s, 3H), 2.53-2.45 (m, 4H), 2,41 (s, 3H), 2.36-2.23 (m, 2H), 2.19-2.08 (m, 2H), 2.02-1.94 (m, 2H), 1.81-1.75 (m, 2H); LCMS (ESI) m/z: 504.2 (M+1)
Пример 7
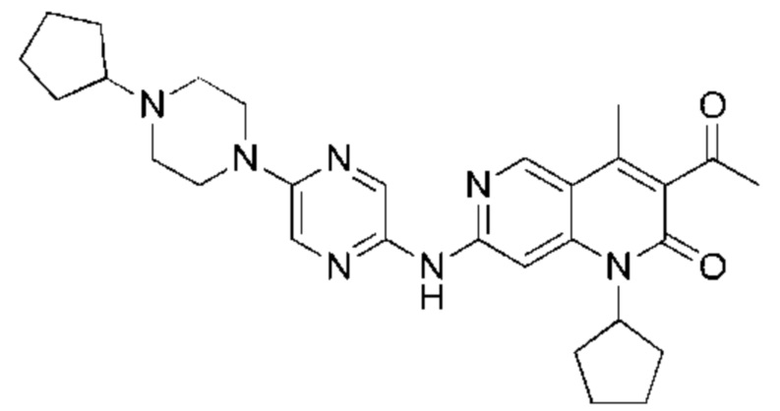
Синтез соединения по примеру 7 является таким же, как синтез соединения по примеру 2. 1H-ЯМР (400 МГц, CDCl3) δ 8.65 (s, 1H), 8.17 (d, J=1,4 Гц, 1H), 7.85-7.75 (m, 2Н), 7.70-7.58 (m, 1H), 5.73 (t, J=9,3 Гц, 1H), 3.61-3.45 (m, 4Н), 2.70-2.62 (m, 4Н), 2.61-2.48 (m, 4Н), 2.40 (s, 3Н), 2.34-2.22 (m, 2Н), 2.17-2.06 (m, 2Н), 1.98-1.87 (m, 4Н), 1.81-1.68 (m, 4Н), 1.64-1.53 (m, 2Н), 1.51-1.41 (m, 2Н); LCMS (ESI) m/z: 516.3 (M+1)
Пример 8
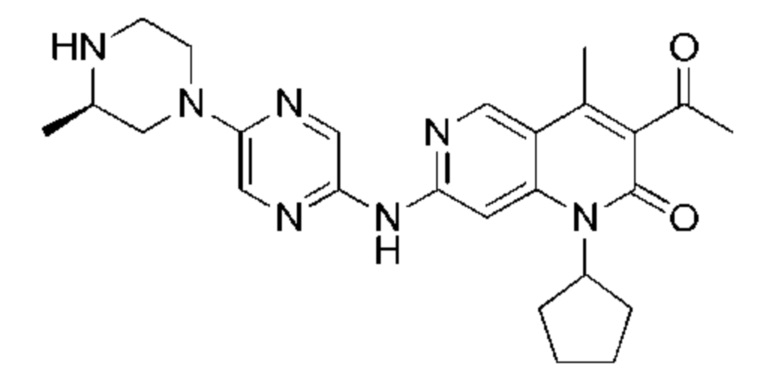
Стадия 1:
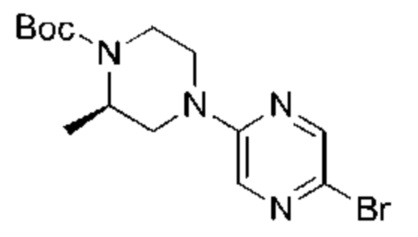
5-Дибромпиразин (1,00 г, 4,20 ммоль, 1,00 экв.) и карбонат калия (871,51 мг, 6,30 ммоль, 1,50 экв.) добавляли к раствору (2R)-трет-бутил-2-метилпиперазин-1-карбоксилата (841,94 мг, 4,20 ммоль, 1,00 экв.) в 1-метилпирролидин-2-оне (10,00 мл). Смесь нагревали до 100°С и перемешивали в течение 18 часов. Завершение реакции подтверждали с помощью LCMS. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой (100 мл) и экстрагировали этилацетатом (50 мл × 3). Объединенную органическую фазу промывали водой (50 мл × 3) и рассолом (50 мл) и концентрировали. Полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = 20:1) с получением указанного в заголовке соединения (900,00 мг, 2,52 ммоль, выход: 59,98%). 1Н-ЯМР (400 МГц, CDCl3) δ 8.12 (d, J=1,4 Гц, 1H), 7.84 (d, J=1,5 Гц, 1H), 4.35 (br s, 1H), 4.09-4.02 (m, 1H), 3.96 (td, J=13,2, 2,0 Гц, 2H), 3.32-3.21 (m, 2Н), 3.05 (dt, J=11,9, 3,8 Гц, 1H), 1.49 (s, 9Н), 1.19 (d, J=6,7 Гц, 3Н).
Стадия 2:
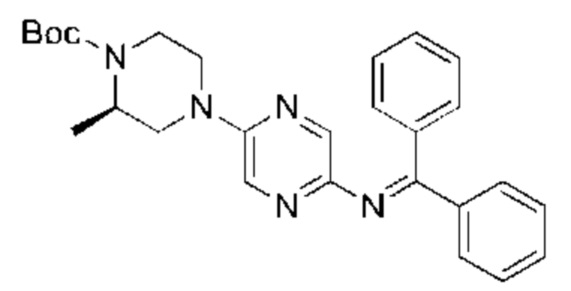
Дифенилметилимин (502,37 мг, 2,77 ммоль, 465,16 мкл, 1,00 экв.), карбонат цезия (1,64 г, 5,04 ммоль, 2,00 экв.), Pd(OAc)2 (56,56 мг, 252,00 мкмоль, 0,10 экв.) и BINAP (313,73 мг, 504,00 мкмоль, 0,20 экв.) добавляли к раствору (2R)-трет-бутил-4-(5-бромпиразин-2-ил)-2-метил-пиперазин-1-карбоксилата (900,00 мг, 2,52 ммоль, 1,00 экв.) в 1,4-диоксане (10,00 мл) в атмосфере азота. Смесь нагревали до 100°С и перемешивали в течение 18 часов. Завершение реакции подтверждали с помощью LCMS. Реакционную смесь охлаждали до комнатной температуры и затем разбавляли дихлорметаном (10 мл), после чего фильтровали. Фильтрат концентрировали и полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = от 40:1 до 10:1) с получением указанного в заголовке соединения (850,00 мг, 1,86 ммоль, выход: 73,72%). 1Н-ЯМР (400 МГц, CDCl3) δ 7.82-7.77 (m, 3Н), 7.55 (d, J=1,4 Гц, 1H), 7.51-7.47 (m, 1H), 7.44-7.38 (m, 2Н), 7.36-7.30 (m, 3Н), 7.21-7.15 (m, 2Н), 4.32 (br s, 1H), 4.01-3.83 (m, 3Н), 3.26-3.13 (m, 2Н), 2.93 (dt, J=12,0, 3,7 Гц, 1H), 1.49 (s, 9Н), 1.18 (d, J=6,8 Гц, 3Н).
Стадия 3:
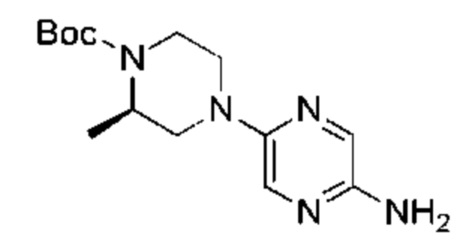
Ацетат натрия (183,09 мг, 2,23 ммоль) и гидроксиламина гидрохлорид (232,65 мг, 3,35 ммоль, 1,80 экв.) добавляли к раствору (2R)-трет-бутил-4-(5-(дифенилметиленамино)пиразин-2-ил)-2-метил-пиперазин-1-карбо ксилата (850,00 мг, 1,86 ммоль, 1,00 экв.) в метаноле (10,00 мл). Смесь перемешивали при 20°С в течение 30 минут. Завершение реакции подтверждали с помощью LCMS. Реакционную смесь концентрировали и полученное неочищенное вещество очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = от 10:1 до 3:1) с получением указанного в заголовке соединения (160,00 мг, 545,40 мкмоль, выход: 29,32%). 1Н-ЯМР (400 МГц, CDCl3) δ 7.69 (d, J=1,3 Гц, 1H), 7.64 (d, J=1,3 Гц, 1H), 4.36 (br s, 1H), 4.14-4.05 (m, 2Н), 3.96 (br d, J=13,4 Гц, 1H), 3.86 (br d, J=12,2 Гц, 1H), 3.73 (br d,J=12,3 Гц, 1H), 3.24 (dt, J=12.7, 3,5 Гц, 1H), 3.00 (dd, J=12,4, 4,0 Гц, 1H), 2.79 (dt, J=12,0, 3,6 Гц, 1H), 1.49 (s, 9H), 1.25 (d, J=7,0 Гц, 3Н).
Стадия 4:
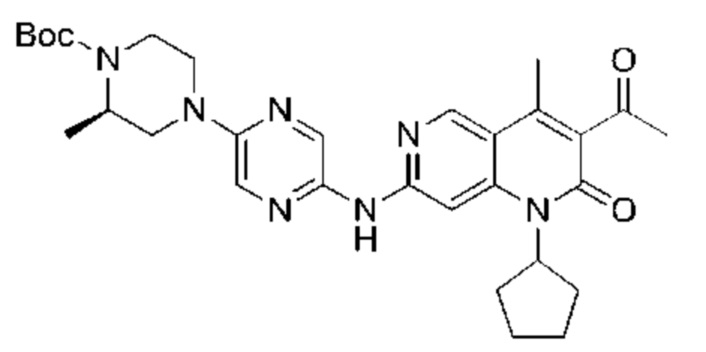
Xphos-Pd-G3 (46,17 мг, 54,54 мкмоль, 0,10 экв.) и трет-бутоксид калия (122,40 мг, 1,09 ммоль, 2,00 экв.) добавляли к раствору (2R)-трет-бутил-4-(5-аминопиразин-2-ил)-2-метил-пиперазин-1-карбоксилата (160,00 мг, 545,40 мкмоль, 1,00 экв.) и 3-ацетил-7-хлор-1-циклопентил-4-метил-1,6-нафтиридин-2-она (Промежуточное соединение В) (166,22 мг, 545,40 мкмоль, 1,00 экв.) в тетрагидрофуране (2,00 мл). Смесь нагревали до 70°С и перемешивали в течение 18 часов. Завершение реакции подтверждали с помощью LCMS. Реакционную смесь охлаждали до комнатной температуры и разбавляли дихлорметаном (5 мл), фильтровали и фильтрат концентрировали. Полученный неочищенный продукт очищали посредством препаративной TLC (петролейный эфир : этилацетат = 1:1) с получением указанного в заголовке соединения (200,00 мг, 313,35 мкмоль, выход: 57,45%, чистота: 88%). LCMS (ESI) m/z: 562.2 (M+1).
Стадия 5:
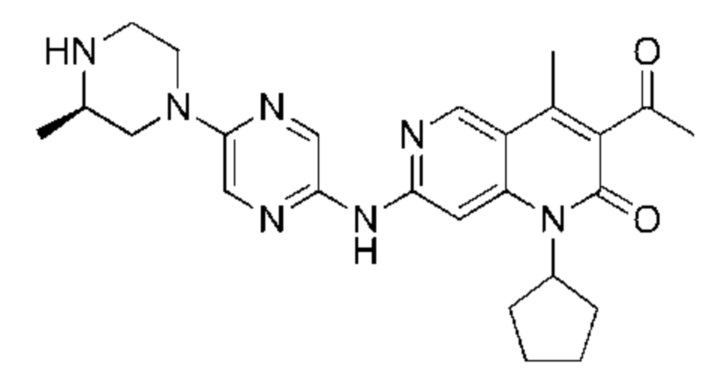
Трифторуксусную кислоту (1,00 мл) добавляли к раствору (2R)-трет-бутил-4-(5-((3-ацетил-1-циклопентил-4-метил-2-оксо-1,6-нафтиридин-7-ил)амино)пиразин-2-ил-2-метил-пиперазин-1-карбоксилата (200,00 мг, 313,35 мкмоль, 1,00 экв.) в дихлорметане (2,00 мл) при 20°С и перемешивали в течение 15 минут. Завершение реакции подтверждали с помощью LCMS. Реакционную смесь концентрировали досуха и полученный неочищенный продукт очищали посредством препаративной HPLC (соляная кислота) с получением гидрохлоридной соли указанного в заголовке соединения (66,91 мг, 123,94 мкмоль, выход: 39,55%, чистота: 99%). 1Н-ЯМР (400 МГц, CD3OD) δ 8.76 (s, 1H), 8.24 (d, J=1,1 Гц, 1H), 8.20 (s, 1H), 7.27 (s, 1H), 5.36 (quin, J=8,7 Гц, 1H), 4.50-4.39 (m, 2Н), 3.56-3.45 (m, 2Н), 3.31-3.21 (m, 2Н), 3.10 (dd, J=14,1, 10,8 Гц, 1H), 2.49 (s, 3Н), 2.38 (s, 3Н), 2.29-2.12 (m, 4Н), 2.10-1.99 (m, 2Н), 1.87-1.72 (m, 2Н), 1.45 (d, J=6,6 Гц, 3Н); LCMS (ESI) m/z: 462.1 (M+1); SFC (AD-3S_4_40_3ML колонка: Chiralpak AD-3 100 × 4,6 мм внутр. диам., 3 мкм; подвижная фаза А: 40% изопропанол (содержащий 0,05% диэтиламина); подвижная фаза В: CO2; скорость потока: 3 мл/мин; длина волны: 280 нм): RT=2,834 мин.
Пример 9
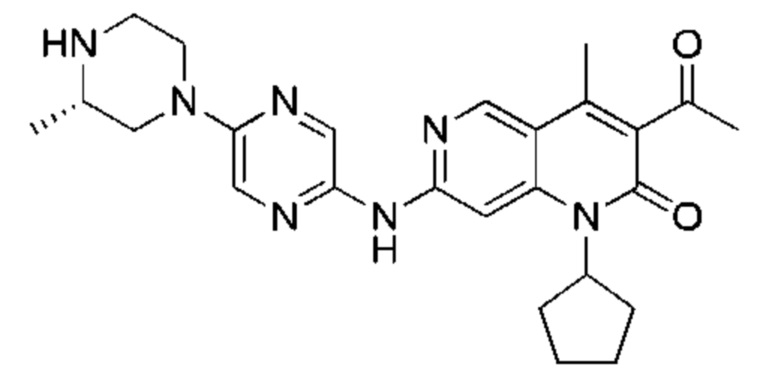
Исходное вещество примера 9 представляет собой (2)5)-трет-бутил-2-метилпиперазин-1-карбоксилат, способ синтеза которого является таким же, как синтез соединения по примеру 8. 1H-ЯМР (400 МГц, CD3OD) δ 8.76 (s, 1H), 8.24 (d, J=1,3 Гц, 1H), 8.20 (d, J=1,3 Гц, 1H), 7.26 (s, 1H), 5.37 (quin, J=8,7 Гц, 1H), 4.52-4.38 (m, 2Н), 3.58-3,44 (m, 2Н), 3.36-3.31 (m, 2Н), 3.09 (dd, J=14,1, 10,7 Гц, 1H), 2.49 (s, 3Н), 2.38 (s, 3Н), 2.30-2.12 (m, 4Н), 2.10-1.99 (m, 2Н), 1.86-1.74 (m, 2Н), 1,45 (d, J=6,5 Гц, 3Н); LCMS (ESI) m/z: 462.1 (M+1); SFC (AD-3S_4_40_3ML колонка: Chiralpak AD-3 100 × 4.6 мм внутр. диам., 3 мкм; подвижная фаза А: 40% изопропанол (содержащий 0,05% диэтиламина); подвижная фаза В: CO2; скорость потока: 3 мл/мин; длина волны: 280 нм): RT=3,284 мин.
Пример 10
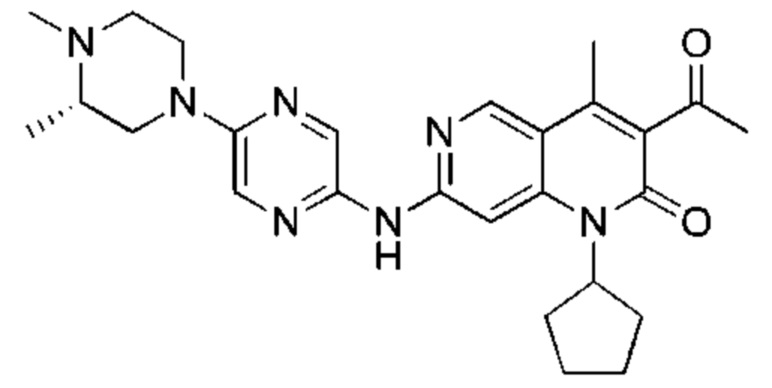
Раствор формальдегида (351,68 мг, 4,33 ммоль, 322,64 мкл, 10,00 экв.), уксусной кислоты (500,00 мкл) и палладия на углероде (смоченного водой) (100,00 мг) добавляли к раствору 3-ацетил-1-циклопентил-4-метил-7-[[5-[(3S)-3-метилпиперазин-1-ил]пиразин-2-ил]амино]-1,6-нафтиридин-2-она (200,00 мг, 433,31 мкмоль, 1,00 экв.) в метаноле (20,00 мл). Реакционную колбу трижды продували азотом и водородом последовательно. Давление водорода поддерживали при 50 фунтов на кв. дюйм (344,7 кПа) и реакционыый раствор нагревали до 50°С при перемешивании в течение 3 часов. Завершение реакции подтверждали с помощью LCMS. Реакционный раствор фильтровали и фильтрат концентрировали. Полученный неочищенный продукт очищали посредством препаративной HPLC (щелочной) с получением указанного в заголовке соединения (42,66 мг, 88,31 мкмоль, выход: 20,38%, чистота: 98,45%). 1Н-ЯМР (400 МГц, CDCl3) δ 8.65 (s, 1H), 8.16 (s, 1H), 7.81-7.79 (m,3H), 5.71 (quin, J=9,3 Гц, 1H), 3.98-3.93 (m, 2Н), 3.07 (dt, J=11,4, 3,0 Гц, 1H), 2.92 (td, J=11,4, 3,2 Гц, 1H), 2.70-2.6 (m, 1H), 2.56 (s, 3Н), 2.39 (s, 3Н), 2.38-2.37 (m, 1H), 2.35 (s, 3Н), 2.31-2.20 (m, 3Н), 2.17-2.04 (m, 3Н), 2,01-1.93 (m, 2H), 1.84-1.71 (m, 2H), 1.17 (d, J=5,6 Гц, 3Н); LCMS (ESI) m/z: 476.3 (M+1).
Пример 11
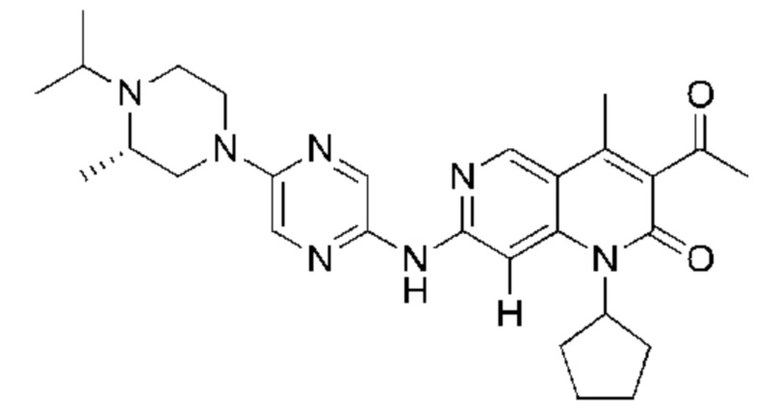
Карбонат калия (149,72 мг, 1,08 ммоль, 5,00 экв.) и 2-йодпропан (736,60 мг, 4,33 ммоль, 433,29 мкл, 20,00 экв.) добавляли к раствору 3-ацетил-1-циклопентил-4-метил-7-[[5-[(3S)-3-метилпиперазин-1-ил]пиразин-2-ил]амино]-1,6-нафтиридин-2-она (100,00 мг, 216,66 мкмоль, 1,00 экв.) в ацетонитриле (10,00 мл). Реакционную смесь нагревали до 80°С и перемешивали в течение 18 часов. Оставалось примерно 13,5% исходных веществ, и образовывалось примерно 75,5% указанного в заголовке соединения, как детектировали посредством LCMS. Реакционную смесь охлаждали до 20°С, после чего фильтровали. Фильтрат концентрировали и полученный неочищенный продукт очищали посредством препаративной HPLC (щелочной) с получением указанного в заголовке соединения (9,00 мг, 17,87 мкмоль, выход: 8,25%). 1Н-ЯМР (400 МГц, CDCl3) δ 8.65 (s, 1H), 8.16 (d, J=1,4 Гц, 1H), 7.81 (d, J=1,3 Гц, 1H), 7.77 (s, 1H), 7,40 (s, 1H), 5.74 (quin, J=9,3 Гц, 1H), 3.96-3.89 (m, 2Н), 3.33 (spt, J=6,5 Гц, 1H), 3,06 (dt, J=11,4, 3,0 Гц, 1H), 2.92 (td, J=11,4, 3,2 Гц, 1H), 2.85-2.70 (m, 2Н), 2.56 (s, 3Н), 2,49-2,47 (m, 1H), 2.41 (s, 3Н), 2.35-2.24 (m, 2Н), 2.19-2,07 (m, 2Н), 2,03-1.93 (m, 2Н), 1.84-1.75 (m, 2Н), 1.17 (dd, J=6,2, 4,2 Гц, 6Н), 0.93 (d, J=6,4 Гц, 3Н); LCMS (ESI) m/z: 504.3 (M+1).
Пример 12
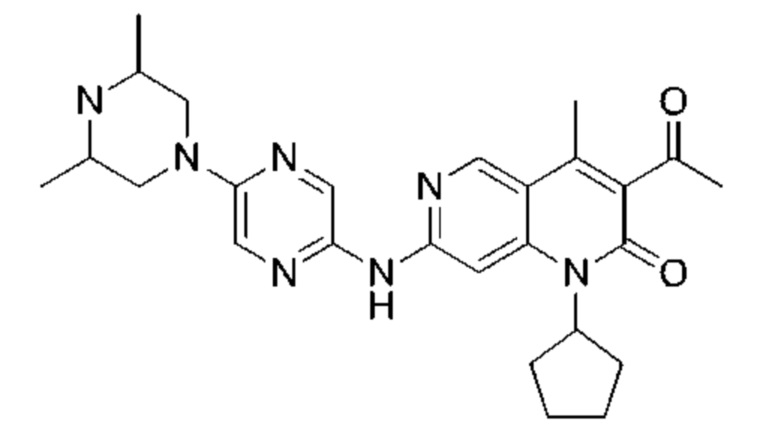
Синтез соединения по примеру 12 является таким же, как синтез соединения по примеру 1, с получением гидрохлоридной соли указанного в заголовке соединения. 1Н-ЯМР (400 МГц, CD3OD) δ 8.77 (s, 1H), 8.26 (d, J=1,2 Гц, 1H), 8.24 (d, J=1,2 Гц, 1H), 7.27 (s, 1H), 5,44-5.35 (m, 1H), 4.58-4.54 (m, 2Н), 3.55-5.45 (m, 2Н), 3,00-2.93 (m, 2Н), 2.51 (s, 3Н), 2,40 (s, 3Н), 2.30-2.15 (m, 4Н), 2.10-2,05 (m, 2Н), 1.84-1.78 (m, 2Н), 1.46 (d, J=7,2 Гц, 6Н); LCMS (ESI) m/z: 476.3 (M+1).
Пример 13
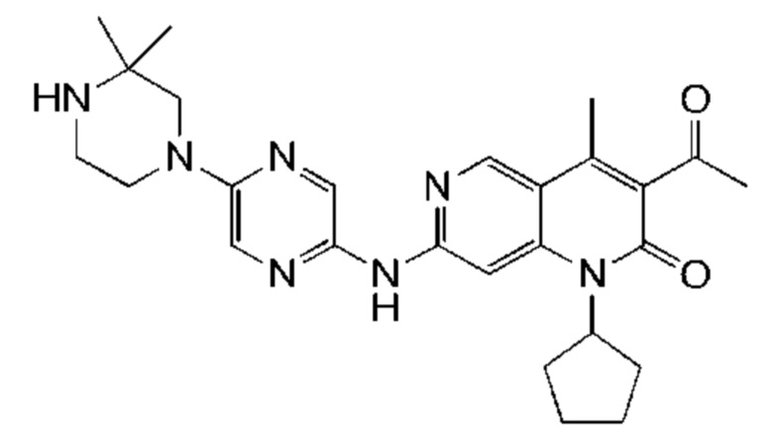
Синтез соединения по примеру 13 является таким же, как синтез соединения по примеру 1, с получением гидрохлоридной соли указанного в заголовке соединения. 1Н-ЯМР (400 МГц, CD3OD) δ 8.76 (s, 1H), 8.23 (d, J=8,9 Гц, 2Н), 7.26 (s, 1H), 5,43-5.29 (m, 1H), 3.99-3.83 (m, 2Н), 3.76 (s, 2Н), 3.50-3.39 (m, 2Н), 2.53-2.46 (m, 3Н), 2.39 (s, 3Н), 2.29-2.13 (m, 4Н), 2.11-2.01 (m, 2Н), 1.82 (br d, J=5,9 Гц, 2Н), 1.51 (s, 6Н); LCMS (ESI) m/z: 476.2 (M+1).
Пример 14
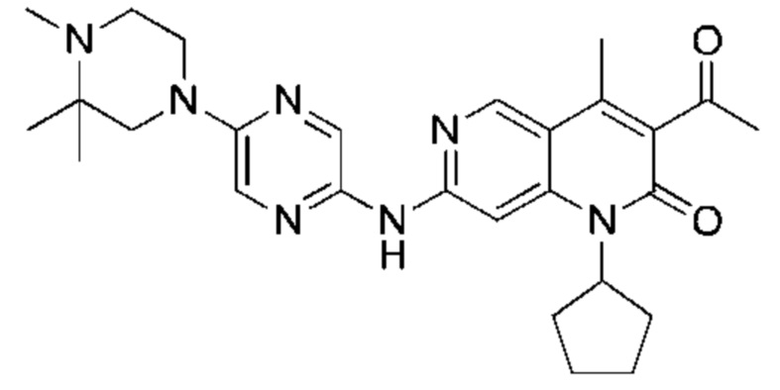
Триацетоксиборгидрид натрия (98,72 мг, 465,77 мкмоль, 2,50 экв.) добавляли к раствору 3-ацетил-1-циклопентил-7-[[5-(3,3-диметилпиперазин-1-ил)пиразин-2-ил]амино]-4-метил-1,6-нафтиридин-2-она (100,00 мг, 186,31 мкмоль, 1,00 экв.), раствору формальдегида (27,97 мг, 931,55 мкмоль, 25,66 мкл, 5,00 экв.) и уксусной кислоте (44,75 мг, 745,24 мкмоль, 42,62 мкл, 4,00 экв.) в дихлорэтане (1,00 мл). Реакционный раствор нагревали до 60°С и перемешивали в течение 16 часов. Завершение реакции подтверждали с помощью LCMS. Реакционный раствор концентрировали и насыщенный водный раствор бикарбоната натрия по каплям добавляли к полученному остатка для доведения рН до 9. Полученную водную фазу экстрагировали дихлорметаном (5 мл × 2). Объединенную органическую фазу сушили над безводным сульфатом натрия и фильтровали, фильтрат концентрировали и полученный неочищенный продукт очищали посредством препаративной HPLC (щелочной) с получением указанного в заголовке соединения (31,00 мг, 61,56 мкмоль, выход: 33,04%, чистота: 97,229%). 1Н-ЯМР (400 МГц, CDCl3) δ 8.64 (s, 1H), 8.14 (s, 1H), 7.80 (s, 1H), 7.73 (s, 1H), 7.53 (s, 1H), 5.84-5.59 (m, 1H), 3.62-3,44 (m, 2Н), 3.27 (s, 2Н), 2.68 (br t, J=5,0 Гц, 2Н), 2.55 (s, 3Н), 2.39 (s, 3Н), 2.29 (s, 3Н), 2.11 (br s, 2Н), 1.97 (br d, J=5,4 Гц, 2Н), 1.78 (br d, J=5,8 Гц, 4H), 1.10 (s, 6Н); LCMS (ESI) m/z: 490.2 (M+1).
Пример 15
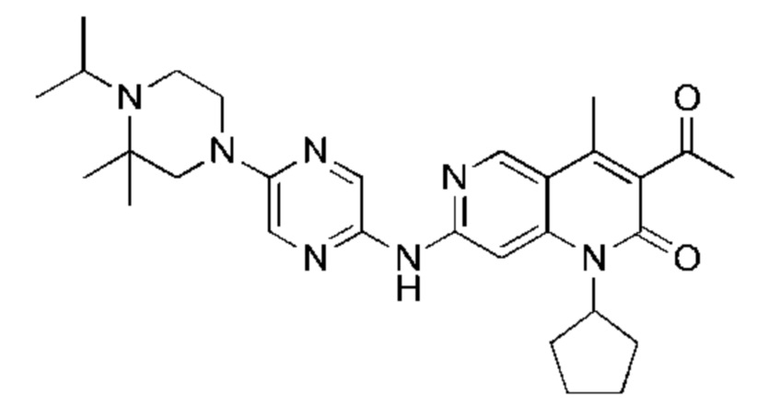
Синтез соединения по примеру 15 является таким же, как синтез соединения по примеру 11. 1Н-ЯМР (400 МГц, CDCl3) δ 8.64 (s, 1H), 8.12 (d, J=1,3 Гц, 1H), 7.79 (d, J=1,3 Гц, 1H), 7.70 (s, 1H), 7,44 (s, 1H), 5.86-5.63 (m, 1H), 3,49 (br s, 2Н), 3.39-3.28 (m, 1H), 3.23 (br s, 2Н), 2.82-2.69 (m, 2Н), 2.55 (s, 3Н), 2.39 (s, 3Н), 2.34-2.20 (m, 2Н), 2.17-2.05 (m, 2Н), 2.03-1.91 (m, 2Н), 1.76 (br dd, J=10,4, 5,6 Гц, 2H), 1.17 (s, 6H), 1.03 (br d, J=6,4 Гц, 6H); LCMS (ESI) m/z: 518.3 (M+1).
Пример 16
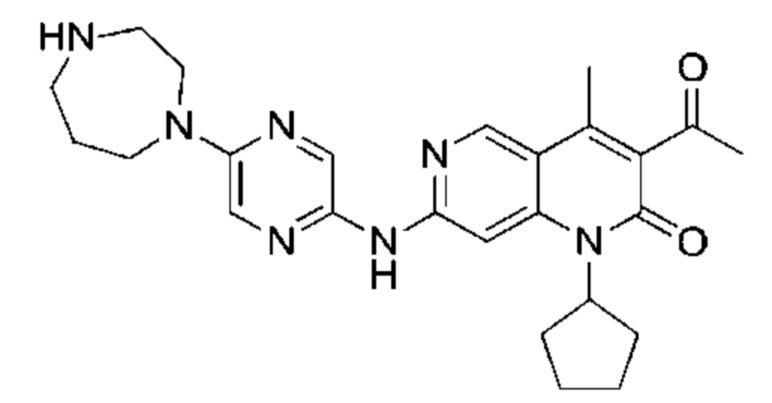
Синтез соединения по примеру 16 является таким же, как синтез соединения по примеру 1, с получением гидрохлоридной соли указанного в заголовке соединения. 1Н-ЯМР (400 МГц, CD3OD) δ 8.75 (s, 1H), 8.20 (s, 1H), 8,02 (s, 1H), 7.27 (s, 1H), 5.45-5.20 (m, 1H), 4.15-3.97 (m, 2Н), 3.81 (br t, J=5,8 Гц, 2Н), 3.44 (br d, J=4,4 Гц, 2H), 3.39-3.33 (m, 2Н), 2.48 (s, 3Н), 2.37 (s, 3Н), 2.31-2.13 (m, 6Н), 2.05 (br d, J=8,8 Гц, 2H), 1.79 (br s, 2H); LCMS (ESI) m/z: 462.2 (M+1).
Пример 17
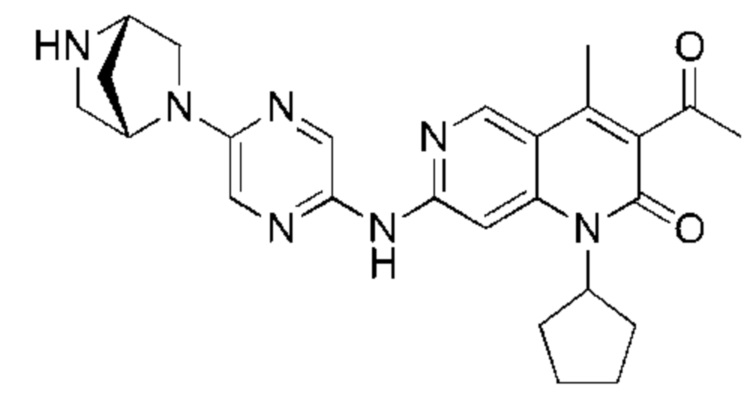
Синтез соединения по примеру 17 является таким же, как синтез соединения по примеру 1, с получением гидрохлоридной соли указанного в заголовке соединения. 1Н-ЯМР (400 МГц, CD3OD) δ 8.74 (s, 1H), 8.21 (s, 1H), 7.90 (s, 1H), 7.26 (s, 1H), 5.35 (t, J=8,3 Гц, 1H), 5.04 (br. s., 1H), 4.61 (s, 1H), 3.86-3.77 (m, 1H), 3.77-3.69 (m, 1H), 3.49-3.39 (m, 2Н), 2.49 (s, 3Н), 2.38 (s, 3Н), 2.32 (d, J=11,2 Гц, 2Н), 2.25-2.10 (m, 5Н), 2.05 (d, J=9,3 Гц, 2Н), 1.80 (br. s., 2Н); LCMS (ESI) m/z: 460.3 (M+1).
Пример 18
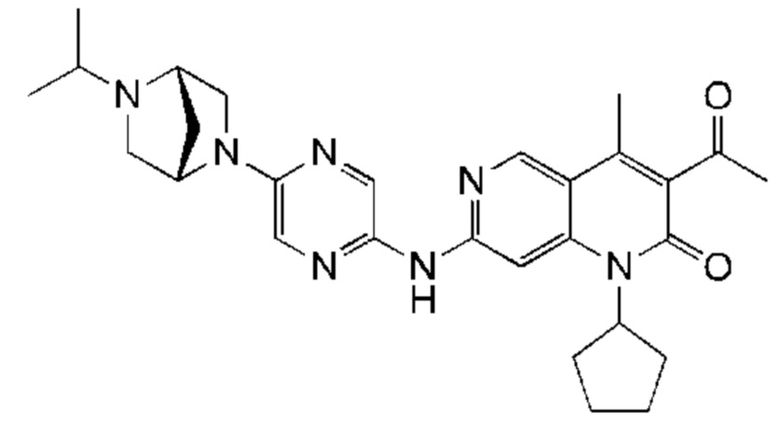
Синтез соединения по примеру 18 является таким же, как синтез соединения по примеру 11, с получением гидрохлоридной соли указанного в заголовке соединения. 1Н-ЯМР (400 МГц, CD3OD) δ 8.74 (d, J=7,9 Гц, 1H), 8.20 (d, J=4,1 Гц, 1H), 7.94-7.87 (m, 1H), 7.26 (d, J=4,6 Гц, 1H), 5.44-5.27 (m, 1H), 5.03 (br s, 1H), 4.82-4.72 (m, 1H), 4.00-3.88 (m, 1H), 3.85-3.67 (m, 2Н), 3.65-3.38 (m, 2Н), 2.49 (s, 3Н), 2.38 (s, 3Н), 2.36-2.25 (m, 2Н), 2.25-2.11 (m, 4Н), 2.05 (br d, J=8,9 Гц, 2H), 1.80 (br s, 2H), 1.51 (d, J=6,3 Гц, 2Н), 1.46 (br d, J=5,8 Гц, 1H), 1.43-1.35 (m, 1H), 1.39 (br d, J=6,3 Гц, 2H); LCMS (ESI) m/z: 502.2 (M+1).
Пример 19
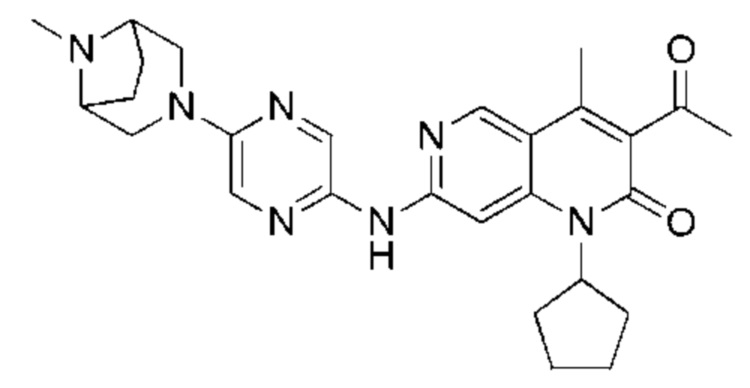
Синтез соединения по примеру 19 является таким же, как синтез соединения по примеру 1 и примера 2, с получением гидрохлоридной соли указанного в заголовке соединения. 1Н-ЯМР (400 МГц, CD3OD) δ 8.77 (s, 1H), 8.25 (s, 1H), 8.16-8.03 (m, 1H), 7.30 (s, 1H), 5.44-5.26 (m, 1H), 4.31 (br d, J=13,2 Гц, 2H), 4.21-4.06 (m, 2Н), 3.70-3.35 (m, 2Н), 2.93 (s, 3Н), 2.55-2.45 (m, 3Н), 2.38 (s, 3Н), 2.37-1.92 (m, 10Н), 1.80 (br s, 2H); LCMS (ESI) m/z: 488.1 (M+1).
Пример 20
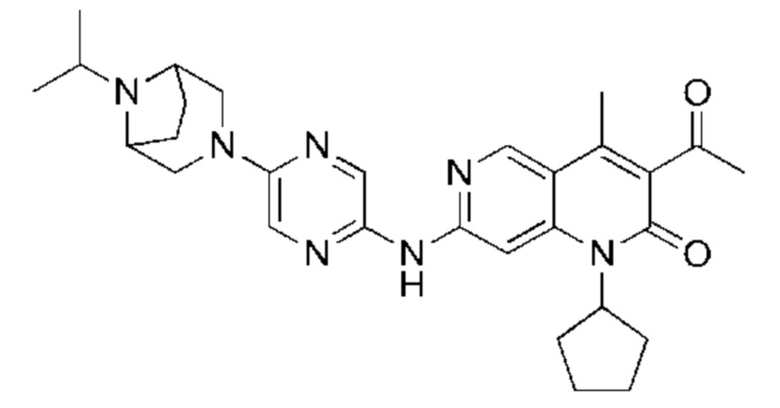
Синтез соединения по примеру 20 является таким же, как синтез соединения по примеру 11, с получением гидрохлоридной соли указанного в заголовке соединения. 1Н-ЯМР (400 МГц, CD3OD) δ 8.76 (s, 1H), 8.31-8.20 (m, 1H), 8.14-8.05 (m, 1H), 7.27 (s, 1H), 5.37 (br t, J=8,5 Гц, 1H), 4.54-4.35 (m, 2Н), 4.34-4.04 (m, 2Н), 3.67-3.45 (m, 2Н), 3.37 (td, J=12,6, 6,4 Гц, 1H), 2.54-2.45 (m, 3Н), 2.38 (s, 3Н), 2.34-2.26 (m, 2Н), 2.23 (br d, J=10,8 Гц, 2Н), 2.18 (br d, J=7,5 Гц, 2H), 2.14-2.08 (m, 2Н), 2.07-1.98 (m, 2Н), 1.86-1.73 (m, 1H), 1.80 (br s, 1H), 1.51 (d, J=6,4 Гц, 6Н); LCMS (ESI) m/z: 516.2 (M+1).
Пример 21
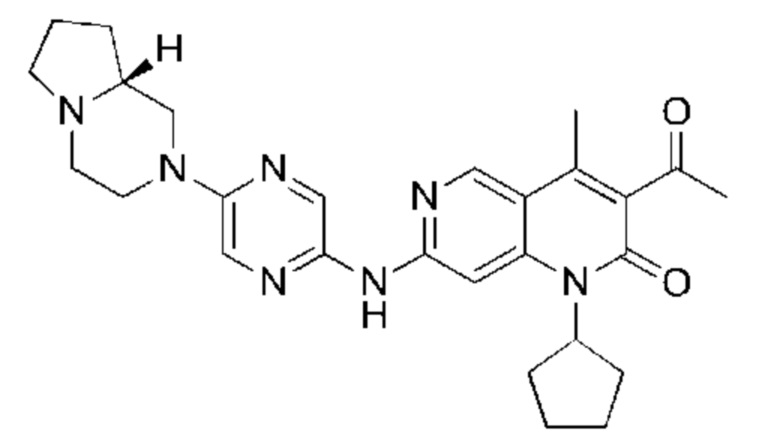
Синтез соединения по примеру 21 является таким же, как синтез соединения по примеру 1, с получением трифторацетата указанного в заголовке соединения. 1H-ЯМР (400 МГц, CD3OD) δ 8.71 (s, 1H), 8.24 (s, 1H), 8.07 (br s, 1H), 7.36 (s, 1H), 5.38 (quin, J=8,7 Гц, 1H), 4.92-4.47 (m, 2Н), 4.20-3.58 (m, 5Н), 3.31-3.01 (m, 2Н), 2.50 (s, 3Н), 2.37 (s, 3Н), 2.32-2.13 (m, 6Н), 2.09-2.01 (m, 2Н), 1.81 (br d, J=5,9 Гц, 2H); LCMS (ESI) m/z: 488.2 (M+1).
Пример 22
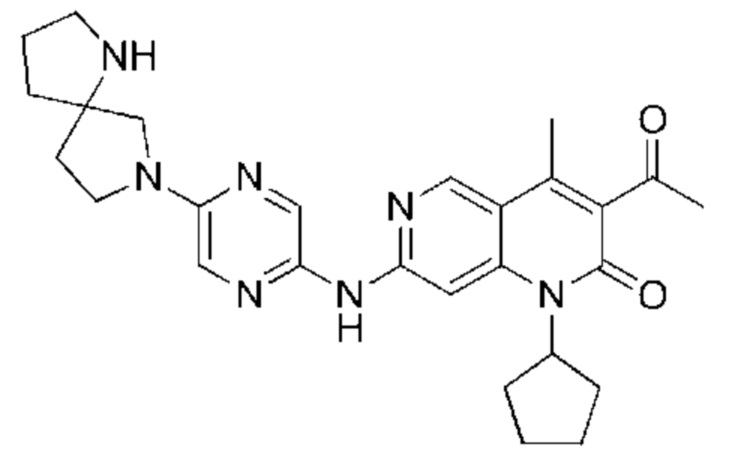
Стадия 1:
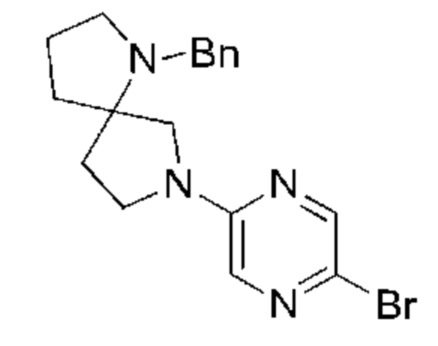
1-Бензил-1,7-диазаспиро[4,4]декан (908,55 мг, 4,20 ммоль, 1,00 экв.) и карбонат калия (870,72 мг, 6,30 ммоль, 1,50 экв.) добавляли к раствору 2,5-дибромпиразина (1,00 г, 4,20 ммоль, 1,00 экв.) в 1-метилпирролидоне (10,00 мл). Реакционную смесь нагревали до 100°С и перемешивали в течение 16 часов. Завершение реакции подтверждали с помощью LCMS. К реакционной смеси добавляли воду (15 мл), после чего экстрагировали этилацетатом (30 мл × 3). Объединенную органическую фазу промывали водой (20 мл × 4) и сушили над безводным сульфатом натрия, после чего фильтровали. Фильтрат концентрировали и полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = от 20:1 до 10:1) с получением указанного в заголовке соединения (952,00 мг, 2,37 ммоль, выход: 56,47%, чистота: 93%). 1Н-ЯМР (400 МГц, CDCl3) δ 8.04 (d, J=1,2 Гц, 1H), 7.63-7.51 (m, 1H), 7.29-7.20 (m, 4Н), 7.18-7.13 (m, 1H), 3.68-3.53 (m, 3Н), 3.52-3.32 (m, 2Н), 3.29-3.18 (m, 1H), 2.72-2.55 (m, 2Н), 2.28-2.14 (m, 1H), 1.94-1.68 (m, 5Н); LCMS (ESI) m/z: 375.0 (M+1).
Стадия 2:
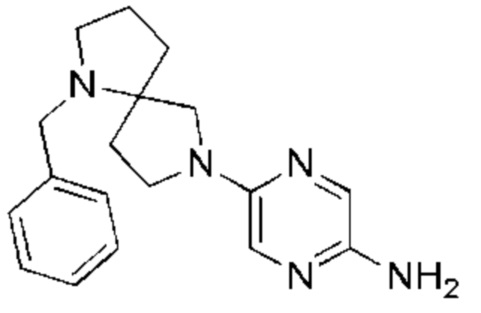
LiHMDS (1 M, 5,36 мл, 2,50 экв.), Pd2(dba)3 (196,25 мг, 214,31 мкмоль, 0,10 экв.) и три-трет-бутилфосфина тетрафторборат (186,53 мг, 642,93 мкмоль, 0,30 экв.) добавляли к раствору 1-бензил-7-(5-бромпиразин-2-ил)-диазаспиро[4,4]декана (800,00 мг, 2,14 ммоль, 1,00 экв.) в толуоле (10,00 мл) в атмосфере азота. Реакционную смесь нагревали до 65°С и перемешивали в течение 16 часов. Завершение реакции подтверждали с помощью TLC (петролейный эфир : этилацетат = 10:1). Реакционную смесь концентрировали и полученный остаток разбавляли этилацетатом (10 мл). Органическую фазу добавляли к насыщенному водному раствору фторида калия (10 мл) и полученную смесь перемешивали при 30°С в течение 16 часов. Вышеуказанную смесь экстрагировали этилацетатом (10 мл × 3). Органическую фазу концентрировали и полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = от 10:1 до дихлорметан : метанол = 20:1) с получением указанного в заголовке соединения (700,00 мг, неочищенный продукт). LCMS (ESI) m/z: 310.3 (M+1).
Стадия 3:
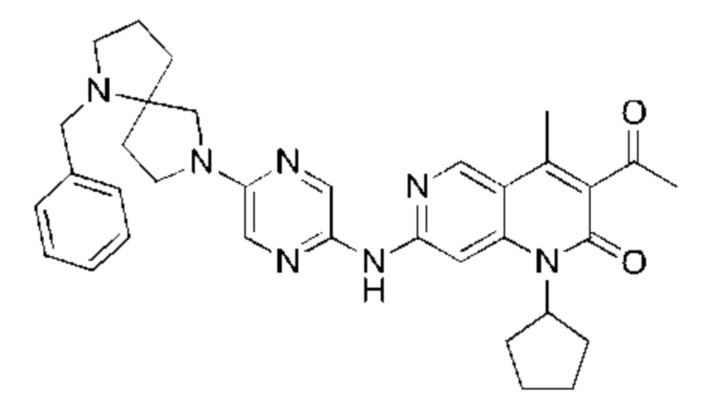
3-Ацетил-7-хлор-1-циклопентил-4-метил-1,6-нафтиридин-2-он (Промежуточное соединение В) (472,80 мг, 1,55 ммоль, 1,20 экв.), трет-бутоксид калия (435,19 мг, 3,88 ммоль, 3,00 экв.) и Xphos-Pd-G3 (191,01 мг, 258,56 мкмоль, 0,20 экв.) добавляли к раствору 5-(1-бензил-1,7-диазаспиро[4,4]декан-7-ил)пиразин-2-амина (400,00 мг, 1,29 ммоль, 1,00 экв.) в THF (5,00 мл). Реакционную смесь нагревали до 70°С и перемешивали в течение 16 часов. Завершение реакции подтверждали с помощью LCMS. Реакционную смесь концентрировали и полученный неочищенный продукт очищали посредством препаративной TLC (петролейный эфир : этилацетат = 3:1) с получением указанного в заголовке соединения (110,00 мг, неочищенный продукт). LCMS (ESI) m/z: 578.2 (M+1).
Стадия 4:
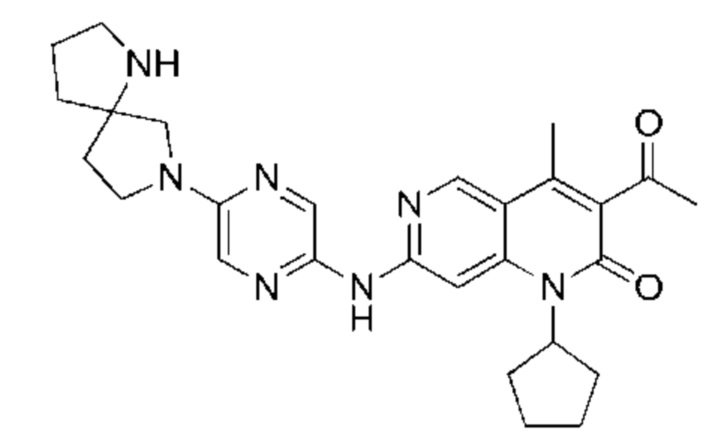
Палладий на углероде (5,00 мг) добавляли к смеси 3-ацетил-7-[[5-(1-бензил-1,7-диазаспиро[4,4]декан-7-ил)пиразин-2-ил]амино]-1-циклопентил-4-метил-1,6-нафтиридин-2-она (5,00 мг, 8,65 мкмоль, 1,00 экв.) в тетрагидрофуране (2,00 мл) и метаноле (2,00 мл). Давление водорода поддерживали при 15 фунтах на кв. дюйм (103,4 кПа) и реакционную смесь перемешивали при 30°С в течение 32 часов. Завершение реакции подтверждали с помощью LCMS. Реакционный раствор фильтровали и фильтрат концентрировали. Полученный неочищенный продукт очищали посредством препаративной HPLC (соляная кислота) с получением гидрохлоридной соли указанного в заголовке соединения (550,00 мкг, 1,05 мкмоль, выход: 12,13%, чистота: 93%). 1Н-ЯМР (400 МГц, CD3OD) δ 8.74 (s, 1H), 8.28-8.14 (m, 1H), 7.93-7.84 (m, 1H), 7.31-7.14 (m, 1H), 5.48-5.37 (m, 1H), 4.08-4.06 (m, 1H), 4.17-4.01 (m, 1H), 3.93-3.77 (m, 2Н), 3.73-3.63 (m, 1H), 3.55-3.50 (m, 1H), 2.96-2.88 (m, 1H), 2.51 (s, 5Н), 2.43-2.36 (m, 3Н), 2.31-2.13 (m, 8Н), 2.11-2.01 (m, 2Н), 1.89-1.78 (m, 2Н); LCMS (ESI) m/z: 488.2 (M+1).
Пример 23
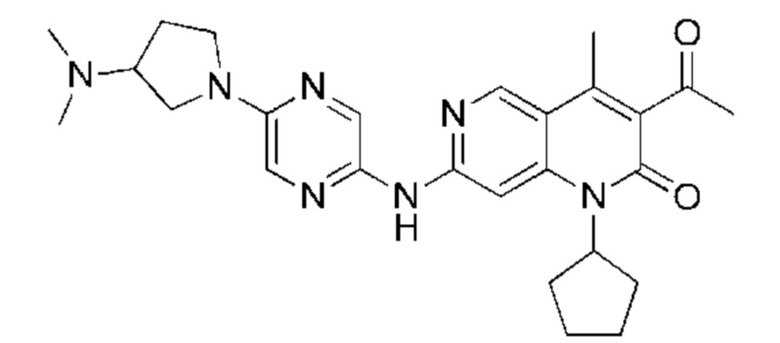
Синтез соединения по примеру 23 является таким же, как синтез соединения по примеру 1. 1Н-ЯМР (400 МГц, CDCl3) δ 8.63 (s, 1H), 8.14 (d, J=1,1 Гц, 1H), 7.63 (s, 1H), 7.58 (d, J=1,1 Гц, 1H), 7.46 (s, 1H), 5.84-5.59 (m, 1H), 3.77 (dd, J=9,8, 7,2 Гц, 1H), 3.72-3.63 (m, 1H), 3.46 (dt, J=9,9, 6,9 Гц, 1H), 3.37-3.25 (m, 1H), 2.92-2.79 (m, 1H), 2.59-2.51 (m, 3Н), 2.39 (s, 3Н), 2.34 (s, 6Н), 2.30-2.25 (m, 2Н), 2.08 (br d, J=4,9 Гц, 2H), 2.00-1.95 (m, 4Н), 1.77-1.74 (m, 2Н); LCMS (ESI) m/z: 476.2 (M+1).
Пример 24
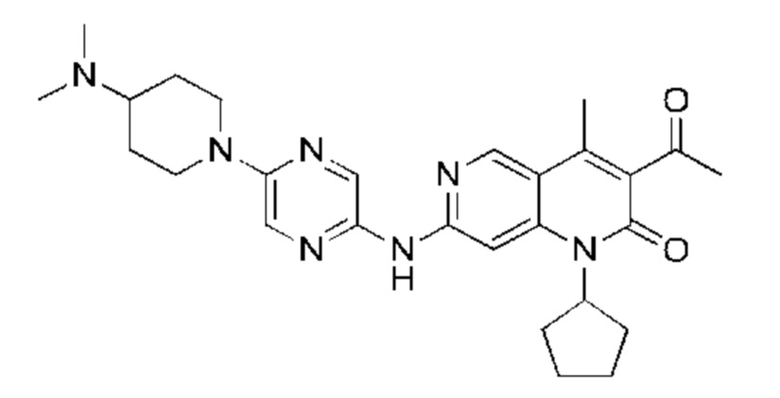
Синтез соединения по примеру 24 является таким же, как синтез соединения по примеру 8, с получением гидрохлоридной соли указанного в заголовке соединения. 1Н-ЯМР (400 МГц, CD3OD) δ 8.75 (s, 1H), 8.20 (s, 1H), 8.10 (br s, 1H), 7.27 (s, 1H), 5.40-5.25 (m, 1H), 4.52 (br d, J=12,3 Гц, 2H), 3.56 (br t, J=11,7 Гц, 1H), 3.01 (br t, J=12,5 Гц, 2H), 2.92 (s, 6H), 2.50 (s, 3H), 2.38 (s, 3H), 2.29-2.13 (m, 6H), 2.07 (br d, J=8,8 Гц, 2H), 1.80 (br d, J=7,8 Гц, 4H); LCMS (ESI) m/z: 490.2 (M+1).
Пример 25
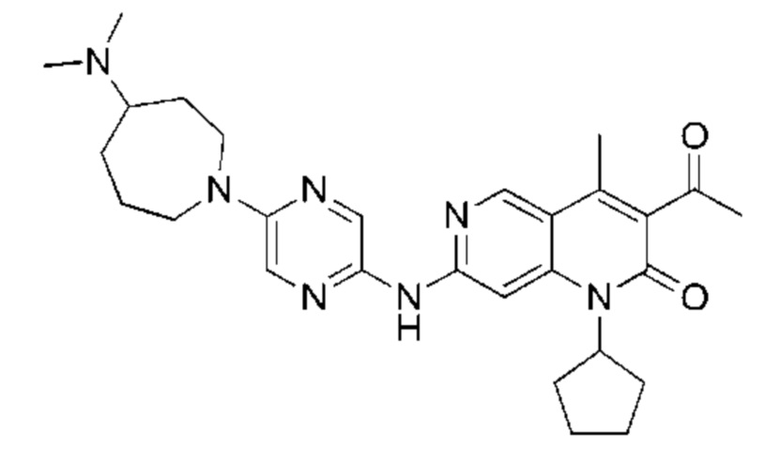
Синтез соединения по примеру 25 является таким же, как синтез соединения по примеру 1, с получением гидрохлоридной соли указанного в заголовке соединения. 1Н-ЯМР (400 МГц, CD3OD) δ 8.75 (s, 1H), 8.19 (d, J=1,0 Гц, 1H), 8.01 (s, 1H), 7.24 (s, 1H), 5.37 (quin, J=8,7 Гц, 1H), 4.06 (td, J=14,7, 4,8 Гц, 1H), 3.93-3.84 (m, 1H), 3.75-3.59 (m, 2Н), 3.51-3.40 (m, 1H), 2.85 (d, J=1,9 Гц, 6Н), 2.51 (s, 3Н), 2.39 (s, 4Н), 2.31-2.13 (m, 6Н), 2.12-1.97 (m, 3Н), 1.91-1.71 (m, 4Н); LCMS (ESI) m/z: 504.3 (M+1).
Пример 26
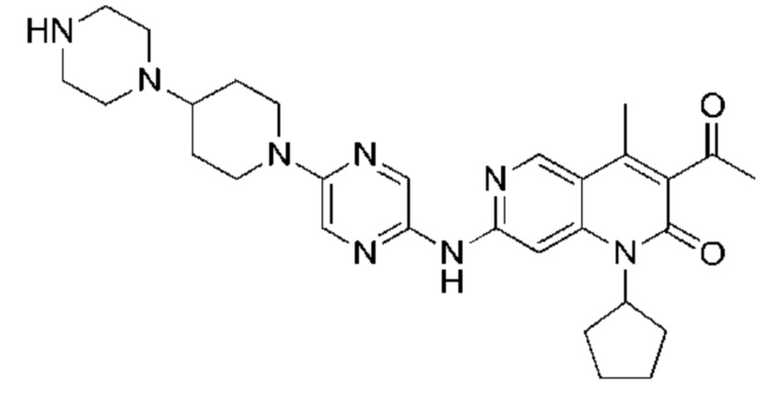
Стадия 1:
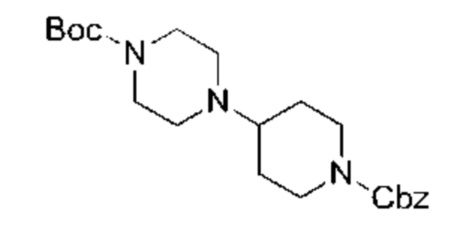
Триацетоксиборгидрид натрия (22,71 г, 107,18 ммоль, 2,50 экв.) добавляли к раствору бензил-4-пиперидон-1-карбоксилата (10,00 г, 42,87 ммоль, 8,55 мл, 1,00 экв.), трет-бутил-пиперазин-1-карбоксилата (9,58 г, 51,44 ммоль, 1.20 экв.) и уксусной кислоты (2,57 г, 42,87 ммоль, 2,45 мл, 1,00 экв.) в метаноле (100,00 мл) при 25°С. Реакционную смесь перемешивали в течение 20 часов. Образование указанного в заголовке соединения подтверждали с помощью LCMS. Реакционную смесь концентрировали и полученный остаток разбавляли этилацетатом. Полученную органическую фазу промывали водой (100 мл) и насыщенным рассолом (50 мл) последовательно и сушили над безводным сульфатом натрия, после чего фильтровали. Фильтрат концентрировали и полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = от 10:1 до 1:1) с получением указанного в заголовке соединения (14,00 г, 27,65 ммоль, выход: 64,50%, чистота: 79,7%). 1Н-ЯМР (400 МГц, CDCl3) δ 7.43-7.31 (m, 5Н), 5.16-5.12 (m, 2Н), 4.36-4.16 (m, 2Н), 4.02-3.81 (m, 1H), 3.49-3.40 (m, 4Н), 3.23-3.09 (m, 1H), 2.81 (br s, 2Н), 2.59-2.45 (m, 4Н), 1.81 (br d, J=10,5 Гц, 2H), 1.58-1.39 (m, 11Н).
Стадия 2:
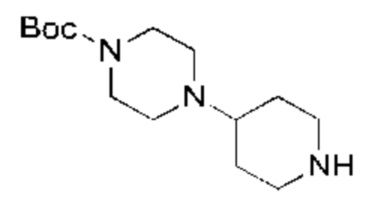
Палладий на углероде (1,00 г) добавляли к раствору трет-бутил-4-(1-бензилоксикарбонил-4-пиперидин)пиперазин-1-карбоксилата (9,00 г, 22,30 ммоль, 1,00 экв.) в тетрагидрофуране (90,00 мл) при 25°С. Суспензию дегазировали в вакууме и несколько раз продували водородом. Давление водорода поддерживали при 15 фунтах на кв. дюйм (103,4 кПа) и смесь перемешивали при 25°С в течение 16 часов. Полное превращение исходных веществ подтверждали с помощью LCMS (жидкостная хроматография-масс-спектрометрия) и регистрировали MS (масс-спектр) указанного в заголовке соединения. Реакционную смесь фильтровали и фильтрат концентрировали с получением указанного в заголовке соединения (5,25 г, 19,49 ммоль, выход: 87,40%). LCMS (ESI) m/z: 270.1 (M+1).
Стадия 3:
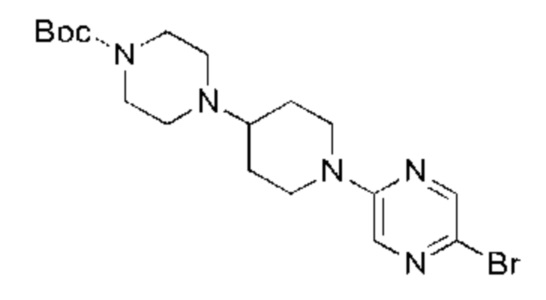
Карбонат калия (1,28 г, 9,25 ммоль, 1,10 экв.) добавляли к смеси трет-бутил-4-(4-пиперидинил)пиперазин-1-карбоксилата (2,27 г, 8,41 ммоль, 1,00 экв.) и 2,5-дибромпиразина (2,00 г, 8,41 ммоль, 1,00 экв.) в диметилсульфоксиде (30,00 мл) и воде (6,00 мл). Реакционную смесь нагревали до 90°С и перемешивали в течение 16 часов. Полное превращение исходных веществ подтверждали с помощью LCMS и регистрировали MS указанного в заголовке соединения. Реакционную смесь охлаждали до 25°С, затем добавляли воду (30 мл). Полученную смесь перемешивали в течение 30 минут, после чего фильтровали. Осадок на фильтре растворяли в дихлорметане (30 мл) и последовательно промывали водой (20 мл × 2) и насыщенным рассолом (20 мл × 2). Органическую фазу сушили над безводным сульфатом натрия, после чего фильтровали. Фильтрат концентрировали с получением указанного в заголовке соединения (1,92 г, 4,41 ммоль, выход: 52,41%, чистота: 97,88%). Продукт использовали на следующей стадии без очистки. 1Н-ЯМР (400 МГц, CDCl3) δ 8.10 (d, J=1,4 Гц, 1H), 7.87 (d, J=1,4 Гц, 1H), 4.28 (br d, J=13,3 Гц, 2H), 3.52-3.33 (m, 4Н), 2.98-2.80 (m, 2Н), 2.60-2.42 (m, 4Н), 1.92 (br d, J=12,3 Гц, 2H), 1.78 (br s, 1H), 1.59-1.50 (m, 2H), 1.46 (s, 9H); LCMS (ESI) m/z: 426,0 (M+1).
Стадия 4:
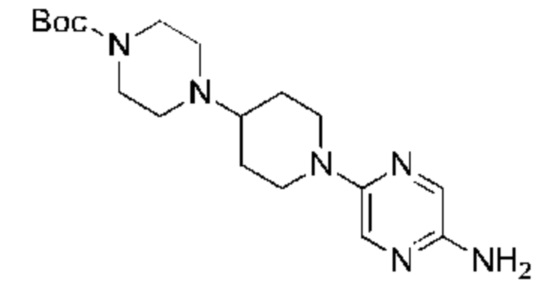
LiHMDS (1 M, 7,09 мл, 2,06 экв.) и Pd2(dba)3 (315,34 мг, 344,36 мкмоль, 0,10 экв.) добавляли к раствору трет-бутил-4-(1-(5-бромпиразин-2-ил)-4-пиперидинил]пиперазин-1-карбоксилата (1,50 г, 3,44 ммоль, 1,00 экв.) и три-трет-бутилфосфина тетрафторбората (299,73 мг, 1,03 ммоль, 0,30 экв.) в толуоле (15,00 мл) в атмосфере азота. Реакционную смесь нагревали до 65°С и перемешивали в течение 16 часов. Полное превращения большей части исходных веществ подтверждали с помощью LCMS и регистрировали MS целевого соединения. Реакционную смесь гасили насыщенным водным раствором хлорида аммония (10 мл) и экстрагировали этилацетатом (20 мл). Органическую фазу доводили до рН 2 с помощью 10%-ного водного раствора лимонной кислоты, после чего фазы разделяли. Водную фазу доводили до рН 9 с помощью 10%-ного раствора гидроксида натрия, после чего фильтровали. Полученный осадок на фильтре сушили в вакууме с получением указанного в заголовке соединения (672,00 мг, 1,85 ммоль, выход: 53,72%, чистота: 100%). Продукт использовали на следующей стадии без очистки. LCMS (ESI) m/z: 363.1 (M+1).
Стадия 5:
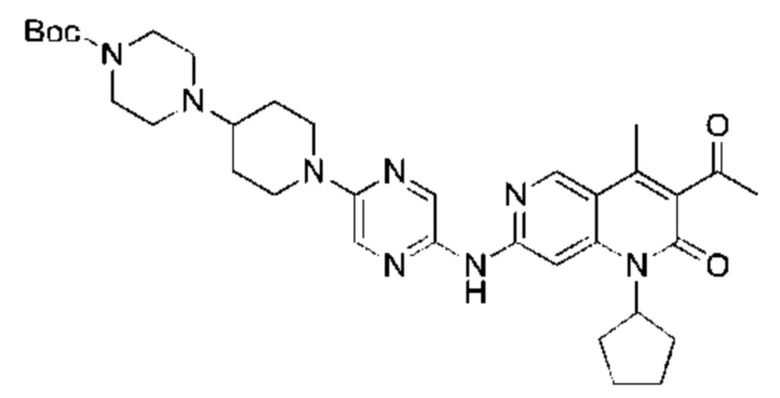
Xphos-Pd-G3 (35,03 мг, 41,38 мкмоль, 0,05 экв.) добавляли к раствору трет-бутил-4-(1-(5-аминопиразин-2-ил)-4-пиперидинил]пиперазин-1-карбоксилата (300,00 мг, 827,65 мкмоль, 1,00 экв.), 3-ацетил-7-хлор-1-циклопентил-4-метил-1,6-нафтиридин-2-она (Промежуточное соединение В) (252.24 мг, 827,65 мкмоль, 1,00 экв.) и калия трет-бутоксида (278,61 мг, 2,48 ммоль, 3,00 экв.) в тетрагидрофуране (20,00 мл) в атмосфере азота. Реакционную смесь нагревали до 80°С и перемешивали в течение 16 часов. Полное превращение большего количества исходных веществ подтверждали с помощью LCMS и регистрировали MS указанного в заголовке соединения. Реакционную смесь концентрировали. Полученный остаток растворяли в этилацетате (15 мл) и затем промывали водой (10 мл × 2). Органическую фазу сушили над безводным сульфатом натрия, после чего фильтровали. Неочищенный продукт очищали посредством препаративной TLC (дихлорметан : метанол = 10:1) с получением указанного в заголовке соединения (63,00 мг, 82,87 мкмоль, выход: 10,01%, чистота: 82,976%). LCMS (ESI) m/z: 631.3 (M+1).
Стадия 6:
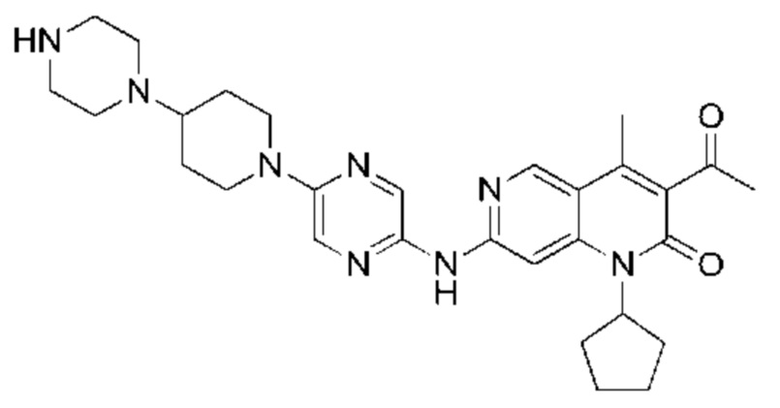
Трифторуксусную кислоту (1,54 г, 13,51 ммоль, 1,00 мл, 162,08 экв.) добавляли к раствору трет-бутил-4-[1-[5-[(3-ацетил-1-циклогексил-4-метил-2-оксо-1,6-нафтиридин-7-ил)амино]пиразин-2-ил]4-пиперидинил]пиперазин-1-карбоксилата (63,00 мг, 82,87 мкмоль, 1,00 экв.) в дихлорметане (3,00 мл) при 25°С. Реакционную смесь перемешивали в течение 30 минут. Полное превращение исходных веществ подтверждали с помощью LCMS и регистрировали MS указанного в заголовке соединения. Реакционную смесь концентрировали и полученный неочищенный продукт очищали посредством препаративной HPLC (соляная кислота) с получением гидрохлоридной соли указанного в заголовке соединения (13,00 мг, 19,34 мкмоль, выход: 23,34%, чистота: 95,231%). 1Н-ЯМР (400 МГц, CD3OD) δ 8.73 (s, 1H), 8.20 (s, 1H), 8.13 (s, 1H), 7.24 (s, 1H), 5.35 (quin, J=8,5 Гц, 1H), 4.56 (br d, J=13,1 Гц, 2Н), 4.05-3.43 (m, 9Н), 3.03 (br t, J=12.3 Гц, 2H), 2.49 (s, 3Н), 2.38 (s, 3Н), 2.34 (br s, 2H), 2.29-2.12 (m, 4H), 2.11-1.99 (m, 2H), 1.96-1.84 (m, 2H), 1.80 (br d, J=5,6 Гц, 2H); LCMS (ESI) m/z: 531.3 (M+1).
Пример 27
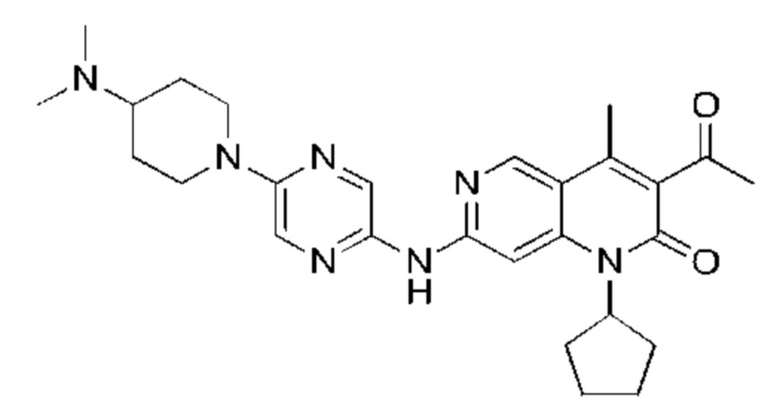
Синтез соединения по примеру 27 является таким же, как синтез соединения по примеру 26, с получением гидрохлоридной соли указанного в заголовке соединения. 1Н-ЯМР (400 МГц, CD3OD) δ 8.75 (s, 1H), 8.22 (s, 1H), 8.13 (s, 1H), 7.26 (s, 1H), 5.45-5.31 (m, 1H), 4.56 (br d, J=13,4 Гц, 2H), 4.12 (br d, J=10,1 Гц, 2H), 3.89 (br t, J=12,0 Гц, 2H), 3.58 (br d, J=12,2 Гц, 3Н), 3.30-3.20 (m, 2H), 3.02 (br t,J=12,6 Гц, 2H), 2.51 (s, 3H), 2.39 (s, 3H), 2.33 (brd, J=10,1 Гц, 2H), 2.15-2,03 (m, 3H), 2.26-2.00 (m, 3H), 1.90-1.74 (m, 4H); LCMS (ESI) m/z: 532.3 (M+1).
Пример 28
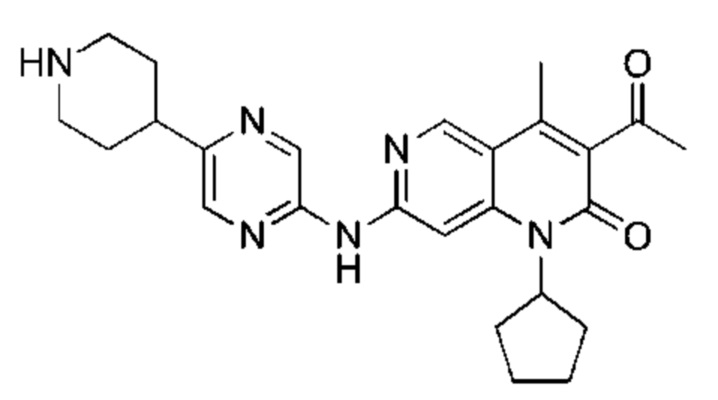
Стадия 1:
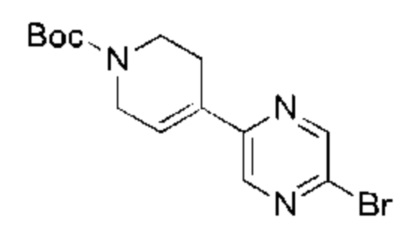
Трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксоЬогап-2-ил)-3,6-дигидро-2H-пиперидин-1-карбоксилат (2,60 г, 8,41 ммоль, 1,00 экв.), фосфат калия (3.57 г, 16,82 ммоль, 2,00 экв.) и Pd(dppf)Cl2 (307,60 мг, 420,50 мкмоль, 0,05 экв.) добавляли к смеси 2,5-дибромпиразина (2,00 г, 8,41 ммоль, 1,00 экв.) в 1,4-диоксане (20,00 мл) и воде (2,00 мл) в атмосфере азота. Реакционную смесь нагревали до 100°С и перемешивали в течение 16 часов. Завершение реакции подтверждали с помощью TLC (петролейный эфир : этилацетат = 20:1). Реакционный раствор концентрировали и полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир : этилацетат = от 20:1 до 10:1) с получением указанного в заголовке соединения (1,60 г, неочищенный продукт).
Стадия 2:
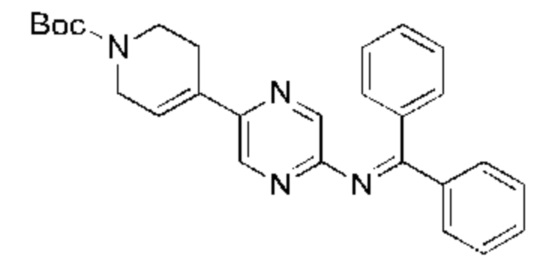
Дифенилметилимин (879,15 мг, 4,85 ммоль, 814,03 мкл, 1,10 экв.), карбонат цезия (2,87 г, 8,82 ммоль, 2,00 экв.), Pd(OAc)2 (99,01 мг, 441,00 мкмоль, 0,10 экв.) и BINAP (274,60 мг, 441,00 мкмоль, 0,10 экв.) добавляли к раствору трет-бутил-4-(5-бромпиразин-2-ил)-3,6-дигидро-2H-пиридин-1-карбоксилата (1,50 г, 4,41 ммоль, 1,00 экв.) в диоксане (20,00 мл) в атмосфере азота. Реакционный раствор нагревали до 100°С и перемешивали в течение 18 часов. Завершение реакции подтверждали с помощью LCMS. Реакционную смесь охлаждали до 20°С и разбавляли дихлорметаном (20 мл), после чего фильтровали. Фильтрат концентрировали и полученный неочищенный продукт очищали посредством препаративной HPLC (щелочной) с получением указанного в заголовке соединения (1,40 г, 3.14 ммоль, выход: 71,12%, чистота: 98,7%). 1Н-ЯМР (400 МГц, CDCl3) δ 8.36 (d, J=1,1 Гц, 1H), 7.88 (d, J=1,1 Гц, 1H), 7.82 (br d, J=6,7 Гц, 2H), 7.58-7.39 (m, 4Н), 7.30 (br d, J=7,1 Гц, 2H), 7.17 (br d,J=6,2 Гц, 2H), 6.55 (br s, 1H), 4.11 (br d, J=2,4 Гц, 2H), 3.62 (br t, J=5,5 Гц, 2H), 2.57 (br s, 2H), 1,48 (s, 9H).
Стадия 3:
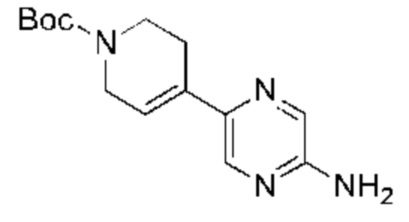
Ацетат натрия (110,27 мг, 1,34 ммоль, 1,20 экв.) и гидроксиламина гидрохлорид (140,12 мг, 2,02 ммоль, 1,80 экв.) добавляли к раствору трет-бутил-4-(5-(дифенилметиленамино)пиразин-2-ил]-3,6-дигидро-2H-пиридин-1-карбоксилата (500,00 мг, 1.12 ммоль, 1,00 экв.) в метаноле (10,00 мл) при 25°С и перемешивали в течение 30 минут. Завершение реакции подтверждали с помощью LCMS. Реакционный раствор концентрировали и полученный неочищенный продукт очищали посредством препаративной TLC (петролейный эфир : этилацетат = 1:1) с получением указанного в заголовке соединения (190,00 мг, 687,58 мкмоль, выход: 61,38%). 1Н-ЯМР (400 МГц, CDCl3) δ 8.10 (d, J=1,2 Гц, 1H), 7.95 (d, J=1,3 Гц, 1H), 6,42 (br s, 1H), 4.59 (br s, 2Н), 4.11 (br d, J=2,7 Гц, 2H), 3.64 (br t, J=5,6 Гц, 2H), 2.58 (br s, 2H), 1,49 (s, 9H).
Стадия 4:
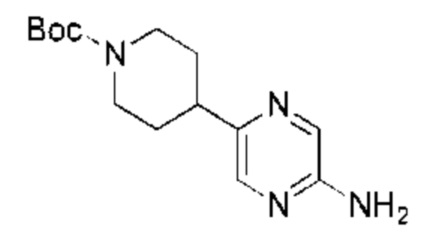
Палладий на углероде (100,00 мг, смоченный водой) добавляли к раствору трет-бутил-4-(5-аминопиразин-2-ил)-3,6-дигидро-2H-пиридин-1-карбоксилата (180,00 мг, 651,40 мкмоль, 1,00 экв.) в метаноле (3,00 мл). Давление водорода поддерживали при 15 фунтах на кв. дюйм (103,4 кПа) и реакционную смесь перемешивали при 25°С в течение 1 часа. Оставалось примерно 60% исходных веществ, что подтверждали с помощью LCMS, и реакционную смесь перемешивали в этих условиях в течение 18 часов. Завершение реакции подтверждали с помощью LCMS. Реакционный раствор фильтровали и фильтрат концентрировали с получением указанного в заголовке соединения (160,00 мг, 574,82 мкмоль, выход: 88,24%). 1Н-ЯМР (400 МГц, CDCl3) δ 7.94 (d, J=1,4 Гц, 1H), 7.87 (d, J=1,4 Гц, 1H), 4,46 (br s, 2Н), 4.25 (br s, 2H), 2.82 (br t, J=10,5 Гц, 2H), 2.78-2.69 (m, 1H), 1.85 (br d, J=13,2 Гц, 2H), 1.69 (dd, J=12,7, 4,1 Гц, 2H), 1,48 (s, 9H).
Стадия 5:
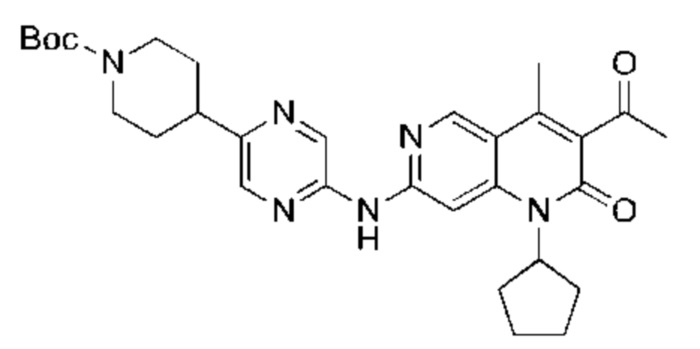
3-Ацетил-7-хлор-1-циклопентил-4-метил-1,6-нафтиридин-2-он (Промежуточное соединение В) (164,24 мг, 538,89 мкмоль, 1,00 экв.), трет-бутоксид калия (120,94 мг, 1,08 ммоль, 2,00 экв.) и Xphos-Pd-G3 (45,61 мг, 53,89 мкмоль, 0,10 экв.) добавляли к раствору трет-бутил-4-(5-аминопиразин-2-ил)пиперидин-1-карбоксилата (150,00 мг, 538,89 мкмоль, 1,00 экв.) в тетрагидрофуране (3,00 мл) в атмосфере азота. Реакционную смесь нагревали до 70°С и перемешивали в течение 18 часов. Завершение реакции подтверждали с помощью LCMS. Реакционную смесь охлаждали до 25°С и разбавляли дихлорметаном (10 мл), после чего фильтровали. Фильтрат концентрировали и полученный неочищенный продукт очищали посредством препаративной TLC (петролейный эфир : этилацетат = 1:2) с получением указанного в заголовке соединения (92,00 мг, 168,29 мкмоль, выход: 31,23%). 1Н-ЯМР (400 МГц, CDCl3) δ 8.70 (s, 1H), 8.40 (d, J=1,4 Гц, 1H), 8.27 (s, 1H), 8.29-8.23 (m, 1H), 8.09 (d, 7=1,3 Гц, 1H), 7.65 (s, 1H), 5.82 (quin, J=9,4 Гц, 1H), 4.29 (br s, 2Н), 2.87 (ddd, J=11,8, 8,4, 3,8 Гц, 3Н), 2.57 (s, 3Н), 2.43 (s, 3Н), 2.39-2.28 (m, 2Н), 2.28-2.16 (m, 2Н), 2.08-1.99 (m, 2Н), 1.95-1.88 (m, 2Н), 1.87-1.71 (m, 4Н), 1.49 (s, 8Н), 1.51-1.47 (m, 1Н).
Стадия 6:
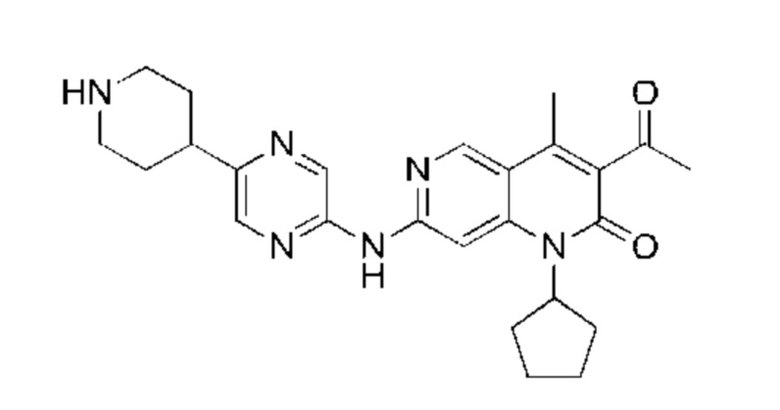
Трифторуксусную кислоту (500,00 мкл) добавляли к раствору трет-бутил-4-[5-[(3-ацетил-1-циклопентил-4-метил-2-оксо-1,6-нафтиридин-7-ил)амино]пиперазин-2-ил]пиперидин-1-карбоксилата (87,00 мг, 159,15 мкмоль, 1,00 экв.) в дихлорметане (1,00 мл). Реакционный раствор перемешивали при 30°С в течение 0,5 часа. Завершение реакции подтверждали с помощью LCMS. Реакционный раствор концентрировали и полученный неочищенный продукт очищали посредством препаративной HPLC (соляная кислота) с получением гидрохлоридной соли указанного в заголовке соединения (37,58 мг, 70,61 мкмоль, выход: 44,36%, чистота: 97,599%). 1Н-ЯМР (400 МГц, CD3OD) δ 8.86 (s, 1H), 8.62 (d, J = 0,8 Гц, 1H), 8.43 (s, 1H), 7,47 (s, 1H), 5.40 (quin, J = 8,6 Гц, 1H), 3.55 (br d, J = 12,8 Гц, 2Н), 3.29-3.17 (m, 3Н), 2.50 (s, 3Н), 2.41 (s, 3Н), 2.30-2.01 (m, 10Н), 1.86-1.74 (1H, 2Н); LCMS (ESI) m/z: 447.2 (М+1).
Пример 29
Синтез соединения по примеру 29 является таким же, как синтез соединения по примеру 1, с получением трифторацетата указанного в заголовке соединения. 1H-ЯМР (400 МГц, CD3OD) δ 8.69 (s, 1H), 8.32 (d, J = 1,1 Гц, 1H), 8.08 (d, J = 1,5 Гц, 1H), 7.70 (br s, 1H), 3.91-3.71 (m, 4Н), 4.88-4.87 (m, 1H), 3.42-3.35 (m, 4Н), 2.67-2.51 (m, 2Н), 2.48 (s, 3Н), 2.37 (s, 3Н), 2.04-1.93 (m, 2Н), 1.80 (br d, J = 11,8 Гц, 3Н), 1.54 (q, J = 13,0 Гц, 2Н), 1.0 (br t, J = 12,7 Гц, 1H); LCMS (ESI) m/z: 462.3 (M+1).
Пример 30
Синтез соединения по примеру 30 является таким же, как синтез соединения по примеру 1, с получением гидрохлоридной соли указанного в заголовке соединения. 1Н-ЯМР (400 МГц, CD3OD) δ 8.84 (s, 1H), 8.17 (d, J = 1,5 Гц, 1H), 8,05 (d, J = 1,5 Гц, 1H), 7.75-7.60 (m, 3Н), 7.47-7.36 (m, 2Н), 6.30 (s, 1H), 3.94-3.78 (m, 4Н), 3.41-3.34 (m, 4Н), 2.52 (s, 3Н), 2,49 (s, 3Н); LCMS (ESI) m/z: 456.1 (M+1).
График В
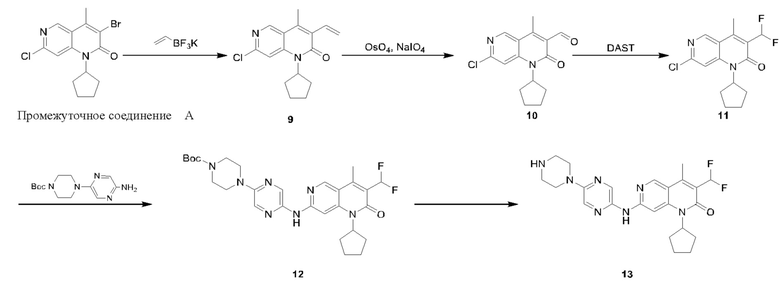
Пример 31
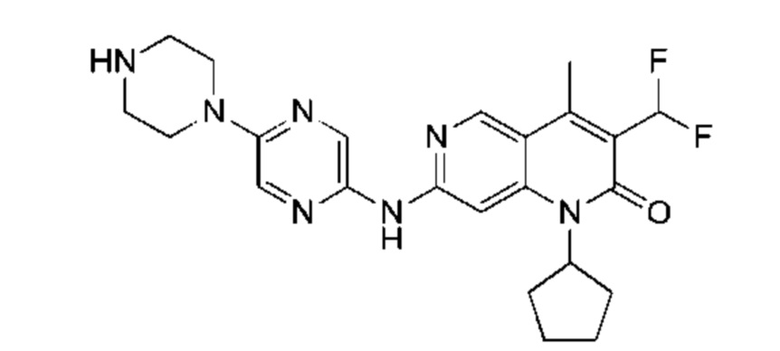
Стадия 1:
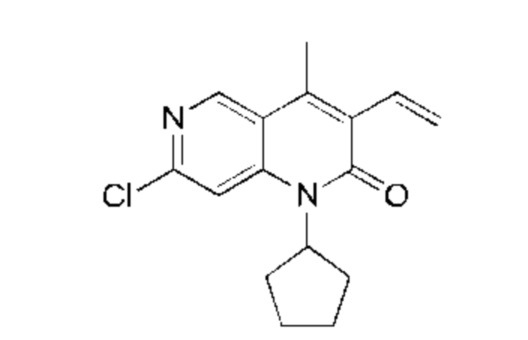
Pd(PPh3)2Cl2 (275,76 мг, 392,88 мкмоль, 0,05 экв.) добавляли к смеси 3-бром-7-хлор-1-циклопентил-4-метил-1,6-нафтиридин-2-она (Промежуточное соединение А) (3,00 г, 7,86 ммоль, 1,00 экв.), калия винилтрифторбората (1,26 г, 9,43 ммоль, 1,20 экв.) и карбоната цезия (5,12 г, 15,72 ммоль, 2,00 экв.) в диоксане (30,00 мл) и воде (6,00 мл) в атмосфере азота. Реакционную смесь нагревали до 110°С и перемешивали в течение 2 часов. Завершение реакции подтверждали с помощью LCMS. Реакционную смесь охлаждали до 25°С, затем добавляли воду (20 мл) и концентрировали для удаления диоксана. Водную фазу экстрагировали этилацетатом (30 мл × 3). Объединенную органическую фазу промывали насыщенным рассолом (30 мл × 2) и сушили над безводным сульфатом натрия, после чего фильтровали. Фильтрат концентрировали и полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир: этилацетат = 30:1) с получением указанного в заголовке соединения (Соединение 9) (1,00 г, 3,46 ммоль, выход: 44,03%). 1H-ЯМР (400 МГц, CDCl3) δ 8.77 (s, 1H), 7.34 (s, 1H), 6.82-6.72 (m, 1H), 5.75 (s, 1H), 5.71 (dd, J = 7,4, 1,9 Гц, 1H), 5.41 (quin, J = 8,9 Гц, 1H), 2.60 (s, 3Н), 2.28-2.18 (m, 2Н), 2.17-2.08 (m, 2Н), 2.05-1.97 (m, 2Н), 1.82-1.74 (m, 2Н); LCMS (ESI) m/z: 251.1 (M+1).
Стадия 2:
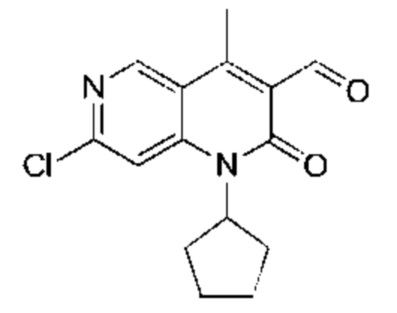
Перйодат натрия (1,56 г, 7,27 ммоль, 402,97 мкл, 3,00 экв.) и тетраоксид осмия (24,65 мг, 96,96 мкмоль, 5,03 мкл, 0,04 экв.) добавляли к смеси 7-хлор-1-циклопентил-4-метил-3-винил-1,6-нафтиридин-2-она (Соединение 9) (700,00 мг, 2,42 ммоль, 1,00 экв.) в диоксане (8,00 мл) и воде (2,00 мл). Смесь перемешивали при 30°С в течение 2 часов. Завершение реакции подтверждали с помощью TLC. Реакционную смесь разбавляли водой (10 мл), после чего фильтровали. Фильтрат экстрагировали этилацетатом (20 мл × 2). Объединенную органическую фазу промывали насыщенным рассолом (20 мл × 2) и сушили над безводным сульфатом натрия, после чего фильтровали. Фильтрат концентрировали и полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир: этилацетат = 20:1) с получением указанного в заголовке соединения (Соединение 10) (560,00 мг, 1,93 ммоль, выход: 79,62%). 1Н-ЯМР (400 МГц, CDCl3) δ 10,46 (s, 1H), 8.87 (s, 1H), 7.28 (s, 1H), 5.33 (quin, J = 8,9 Гц, 1H), 2.77 (s, 3Н), 2.15 (br dd, J = 12,1, 7,6 Гц, 2Н), 2.05 (br dd, J = 8,4, 5,4 Гц, 2H), 1.99-1.93 (m, 2Н), 1.76-1.69 (m, 2Н).
Стадия 3:
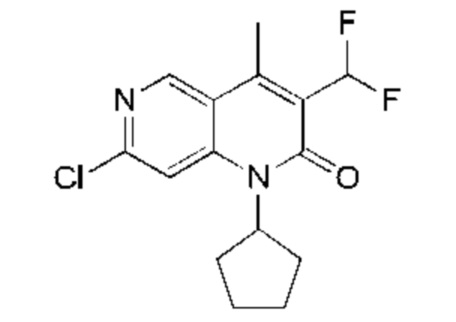
DAST (1,56 г, 9,65 ммоль, 1,27 мл, 5,00 экв.) добавляли к раствору дихлор-7-хлор-1-циклопентил-4-метил-2-оксо-1,6-нафтиридин-3-карбальдегида (Соединение 10) (560,00 мг, 1,93 ммоль, 1,00 экв.) в дихлорметане (6,00 мл) при 25°С. Смесь перемешивали в течение 16 часов. Завершение реакции подтверждали с помощью LCMS. Реакционный раствор концентрировали и полученный неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (петролейный эфир: этилацетат = 20:1) с получением указанного в заголовке соединения (Соединение 11) (500,00 мг, 1,60 ммоль, выход: 82,84%). 1Н-ЯМР (400 МГц, CDCl3) δ 10,46 (s, 1H), 8.87 (s, 1H), 7.28 (s, 1H), 5.33 (quin, J = 8,9 Гц, 1H), 2.77 (s, 3Н), 2.20-2.11 (m, 2Н), 2,08-2,01 (m, 2Н), 1.99-1.92 (m, 2Н), 1.78-1.70 (m, 2Н).
Стадия 4:
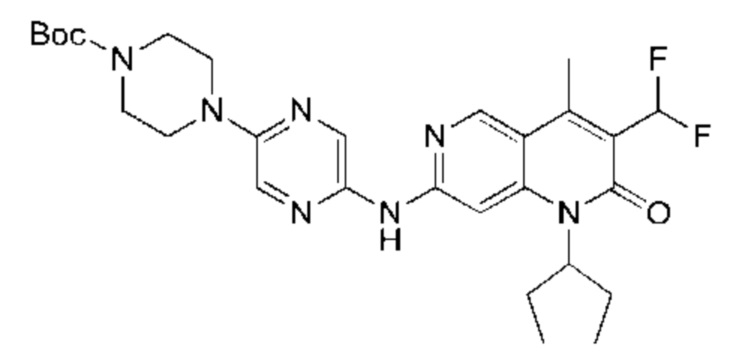
Карбонат цезия (208,36 мг, 639,51 мкмоль, 2,00 экв.), Xphos (15,24 мг, 31,98 мкмоль, 0,10 экв.) и Pd(OAc)2 (3,59 мг, 15,99 мкмоль, 0,05 экв.) добавляли к раствору 7-хлор-1-циклопентил-3-(дифторметил)-4-метил-1,6-нафтиридин-2-она (Соединение 11) (100,00 мг, 319,75 мкмоль, 1,00 экв.), трет-бутил-4-(5-аминопиразин-2-ил)пиперазин-1-карбоксилата (107,18 мг, 383,71 мкмоль, 1,20 экв.) в диоксане (2,00 мл) в атмосфере азота. Реакционную смесь нагревали до 100°С и перемешивали в течение 16 часов. Завершение реакции подтверждали с помощью LCMS. Реакционную смесь охлаждали до 30°С, после чего фильтровали. Фильтрат концентрировали и полученный неочищенный продукт очищали посредством препаративной TLC (петролейный эфир: этилацетат = 2:1) с получением указанного в заголовке соединения (Соединение 12) (16,70 мг, 30,06 мкмоль, выход: 9,40%). LCMS (ESI) m/z: 251.1 (М+1).
Стадия 5:
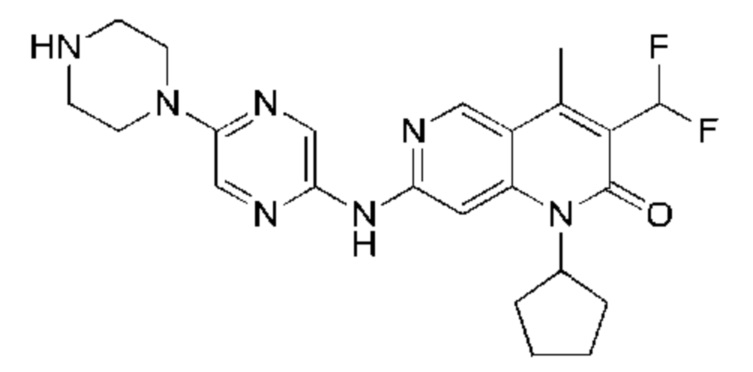
Трифторуксусную кислоту (1,00 мл) добавляли к раствору трет-бутил-4-[5-[[1-циклопентил-3-(дифторметил)-4-метил-2-оксо-1,6-нафтиридин-7-ил]амино]пиразин-2-пиперазин-1-карбоксилата (Соединение 12) (20,00 мг, 36,00 мкмоль, 1,00 экв.) в дихлорметане (2,00 мл) при 30°С. Реакционный раствор перемешивали в течение 0,5 часов. Завершение реакции подтверждали с помощью LCMS. Реакционный раствор концентрировали в пониженном давлении для удаления дихлорметане и трифторуксусной кислоты. Полученный неочищенный продукт очищали посредством препаративной HPLC (соляная кислота) с получением гидрохлоридной соли указанного в заголовке соединения (15,00 мг, 28,03 мкмоль, выход: 77,86%, чистота: 98,74%). 1H-ЯМР (400 МГц, CD3OD) δ 8.86 (s, 1H 1H), 8.28 (d, J = 0,9 Гц, 1H), 8.22 (s, 1H), 7.33-7.06 (m, 2Н), 5.45-5.34 (m, 1H), 3.96-3.88 (m, 4Н), 3.45-3.39 (m, 4Н), 2.70 (s, 3Н), 2.30-2.16 (m, 4Н), 2.12-2.02 (m, 2Н), 1.83 (br d, J=5,5 Гц, 2Н); LCMS (ESI) m/z: 251.1 (М+1).
Пример 32
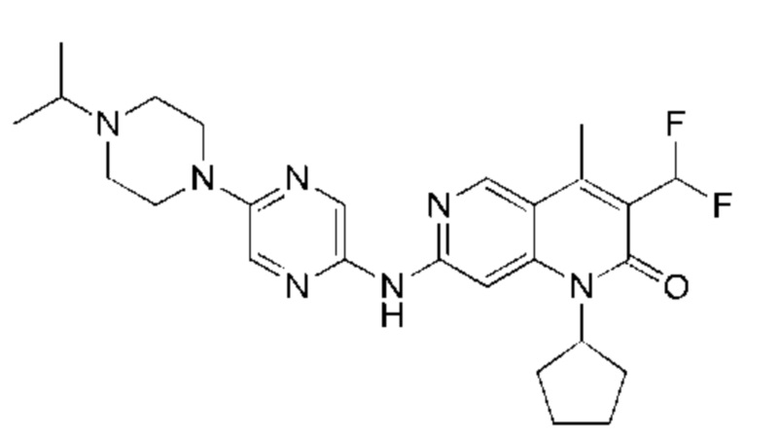
Ацетон (112,84 мг, 1,94 ммоль, 142,84 мкл, 5,00 экв.), триацетоксиборгидрид натрия (205,89 мг, 971,45 мкмоль, 2,50 экв.) и уксусную кислоту (46,67 мг, 777,16 мкмоль, 44,45 мкл, 2,00 экв.) добавляли к раствору 1-циклопентил-3-(дифторметил)-4-метил-7-[(5-пиперазин-1-илпиразин-2-ил)амино]-1,6-нафтиридине-2-она (177,00 мг, 388,58 мкмоль, 1,00 экв.) в дихлорэтане (5,00 мл). Реакционную смесь перемешивали при 30°С в течение 2 часов. Полное превращение исходных веществ подтверждали с помощью LCMS и регистрировали MS указанного в заголовке соединения. Реакционный раствор концентрировали в пониженном давлении и полученный неочищенный продукт очищали посредством препаративной HPLC (гидрохлорид) с получением гидрохлоридной соли указанного в заголовке соединения (49,67 мг, 85,49 мкмоль, выход: 22,00%, чистота: 98,19%). 1H-ЯМР (400 МГц, CD3OD) δ 8.86 (s, 1H), 8.27 (d, J = 1,1 Гц, 1H), 8.23 (d, J = 1,2 Гц, 1H), 7.34-7.07 (m, 1H), 7.24 (s, 1H), 5.45-5.33 (m, 1H), 4.58 (br d, J = 13,6 Гц, 2H), 3.68 (br d, J = 13,1 Гц, 2H), 3.72-3.66 (m, 1H), 3.43-3.35 (m, 2H), 3.32-3.22 (m, 2H), 2.70 (s, 3Н), 2.32-2.13 (m, 4H), 2.12-2.00 (m, 2H), 1.88-1.75 (m, 2H), 1.46 (d, J = 6,6 Гц, 6H); LCMS (ESI) m/z: 498.0 (M+1).
Пример 33
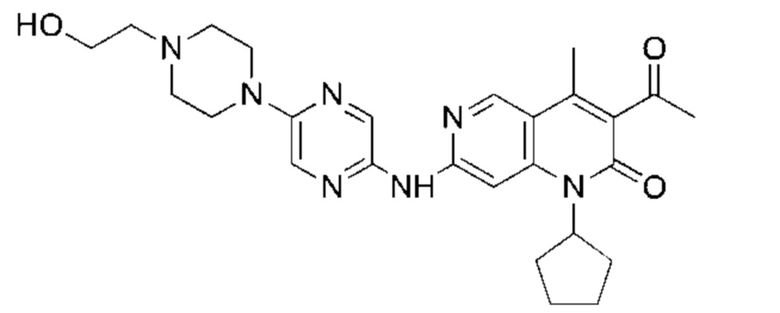
2-Бромэтанол (72,60 мг, 580,96 мкмоль, 41,25 мкл, 1,3 экв.) и карбонат натрия (142,10 мг, 1,34 ммоль, 3,0 экв.) добавляли к раствору 3 -ацетил-1-циклопентил-4-метил-7-[(5-пиперазин-1-илпиразин-2-ил)амино]-1,6-нафтиридин-2-она (0,2 г, 446,89 мкмоль, 1 экв.) в DMF (4 мл). Реакционную смесь нагревали до 80°С и перемешивали в течение 16 часов. Завершение реакции подтверждали с помощью LCMS. Реакционную смесь фильтровали и очищали с помощью препаративной HPLC (щелочной) с получением указанного в заголовке соединения. 1Н-ЯМР (400 МГц, DMSO-d6) δ 9.95 (br s, 1H), 8.75 (s, 1H), 8.54 (s, 1H), 7.97 (s, 1H), 7.63 (s, 1H), 5.57 (quin, J = 9,1 Гц, 1H), 3.55 (t, J = 6,2 Гц, 2H), 3.48-3.41 (m, 4Н), 2.57-2.53 (m, 4Н), 2.47-2.39 (m, 5Н), 2.34 (s, 3Н), 2.25-2.06 (m, 4Н), 1.94-1.82 (m, 2Н), 1.77-1.65 (m, 2Н); LCMS (ESI) m/z: 492.4 (M+1)
Пример 34
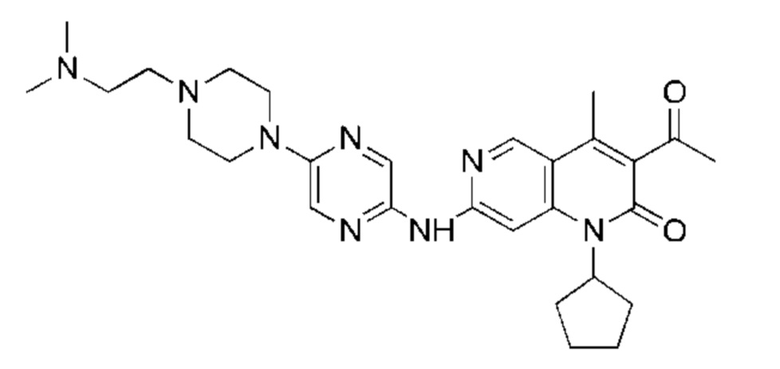
Йодид натрия (13,40 мг, 89,38 мкмоль, 0,2 экв.) добавляли к раствору 3-ацетил-1-циклопентил-4-метил-7-[(5-пиперазин-1-илпиразин-2-ил)амино]-1,6-нафтир идин-2-она (200 мг, 446,89 мкмоль, 1 экв.), 2-хлор-N,N-диметилэтиламина (64,37 мг, 446,89 мкмоль, 1 экв., гидрохлорид) и карбоната натрия (142,10 мг, 1,34 ммоль, 3 экв.) в DMF (5 мл). Реакционную смесь нагревали до 80°С и перемешивали в течение 16 часов. Завершение реакции подтверждали с помощью LCMS. Реакционную смесь фильтровали и фильтрат очищали посредством препаративной HPLC (соляная кислота) с получением гидрохлоридной соли указанного в заголовке соединения. 1H-ЯМР (400 МГц, CD3OD) δ 8.75 (s, 1H), 8.27 (d, J = 0,9 Гц, 1H), 8.22 (s, 1H), 7.25 (s, 1H), 5.41 (quin, J = 8,8 Гц, 1H), 3.82-3.70 (m, 4Н), 3.69-3.35 (m, 4Н), 3.33-3.31 (m, 4Н), 3.06 (s, 6Н), 2.51 (s, 3Н), 2.40 (s, 3Н), 2.32-2.16 (m, 4Н), 2.13-2.03 (m, 2Н), 1.83 (br d, J = 5,9 Гц, 2Н); LCMS (ESI) m/z: 519.5 (М+1).
Пример 35
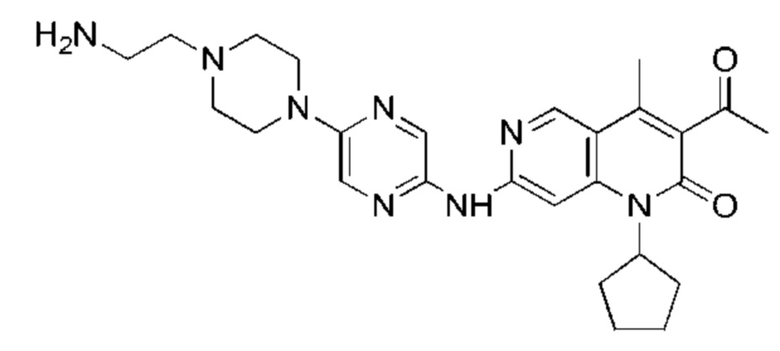
Стадия 1:
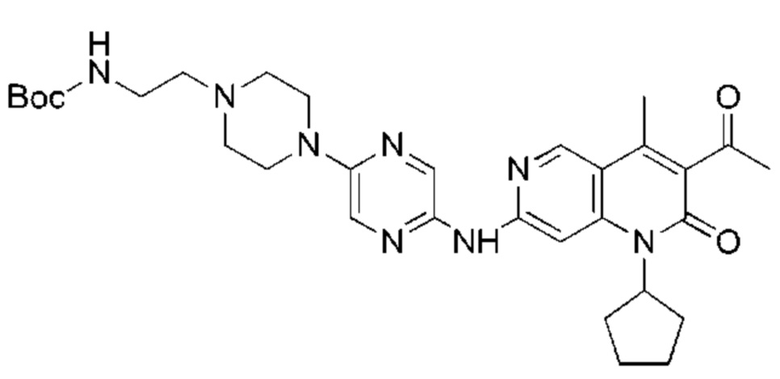
Трет-бутил-N-(2-бромэтил)карбамат (90,13 мг, 402,21 мкмоль, 1,2 экв.) и карбонат натрия (106,57 мг, 1,01 мкмоль, 3 экв.) добавляли к раствору 3-ацетил-1-циклопентил-4-метил-7-[(5-пиперазин-1-илпиразин-2-ил)амино]-1,6-нафтиридин-2-она (0,15 г, 335,17 мкмоль, 1 экв.) в DMF (3 мл). Реакционную смесь нагревали до 80°С и перемешивали в течение 16 часов. Полное превращение исходных веществ и образование продукта, указанного в заголовке, подтверждали с помощью LCMS. Реакционную смесь фильтровали с получением раствора указанного в заголовке соединения в DMF, который использовали непосредственно на следующей стадии без дальнейшей очистки. LCMS (ESI) m/z: 591.5 (M+1).
Стадия 2:
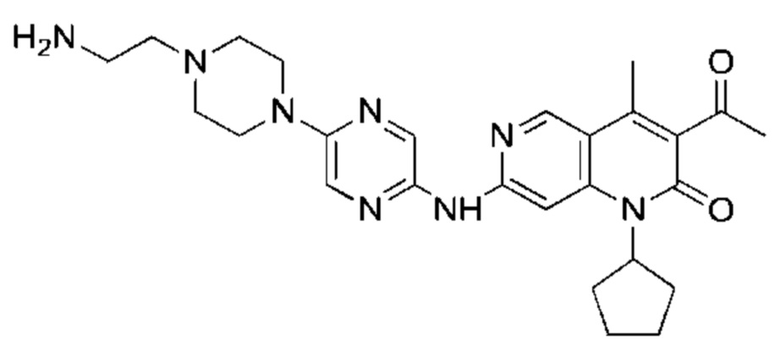
Трифторуксусную кислоту (1 мл) добавляли к раствору трет-бутил-N-[2-[4-[5-[(3-ацетил-1-циклопентил-4-метил-2-оксо-1,6-нафтиридин-7-ил)амино]пиразин-2-пиперазин-1-ил]этил]карбоксилата (197,99 мг, 335,17 ммоль, 1 экв.) в DMF (3 мл). Реакционную смесь перемешивали при 20°С в течение 15 часов. Неполное превращение исходного вещества подтверждали с помощью LCMS, но образовывалось большое количество продукта, указанного в заголовке. Реакционную смесь фильтровали и фильтрат очищали посредством препаративной HPLC (соляная кислота) с получением гидрохлоридной соли указанного в заголовке соединения. 1H-ЯМР (400 МГц, DMSO-d6) δ 11.52 (br s, 1H), 10.72 (br s, 1H), 8.78 (s, 1H), 8.57 (s, 1H), 8.46 (br s, 3H), 8.14 (d, J = 1,0 Гц, 1H), 7.67 (s, 1H), 5.53 (br t, J = 8,9 Гц, 1H), 4.34 (br s, 2H), 3.49-3.30 (m, 6H), 2.44 (s, 3H), 2.33 (s, 3H), 2.26-2.04 (m, 4H), 1.90 (br d, J = 8,6 Гц, 2H), 1.77-1.65 (m, 2H); LCMS (ESI) m/z: 491.4 (M+1).
Пример 36
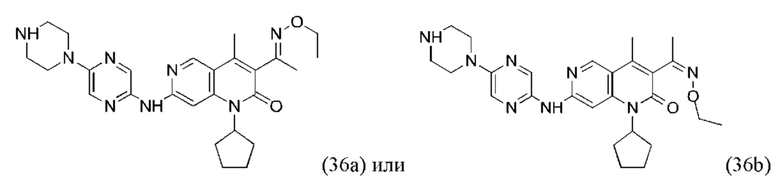
3-Ацетил-1-циклопентил-4-метил-7-[(5-пиперазин-1-илпиразин-2-ил)амино]-1,6-нафтиридин-2-он (0,1 г, 223,45 мкмоль, 1 экв.) добавляли к раствору О-этилгидроксиламина гидрохлорида (130,78 мг, 1,34 ммоль, 6 экв.) в пиридине (2 мл). Реакционную смесь нагревали до 70°С и перемешивали в течение 16 часов. Завершение реакции подтверждали с помощью LCMS. Реакционную смесь охлаждали до 20°С и концентрировали досуха. Полученный остаток очищали с помощью препаративной HPLC (соляная кислота) с получением гидрохлоридной соли указанного в заголовке соединения 36а или 36b. 1Н-ЯМР (400 МГц, CD3OD) δ 9,45 (br s, 2Н), 8.74 (s, 1H), 8.53 (s, 1H), 8.09 (d, J = 1,1 Гц, 1H), 7.64 (s, 1H), 5.57-5.46 (m, 1H), 4.11 (br d, J = 7,0 Гц, 4H), 3.76-3.71 (m, 4H), 3.21 (br s, 4H), 2.37 (s, 3H), 2.23-2.09 (m, 4H), 2.00 (s, 3H), 1.89 (br d, J = 9,0 Гц, 2H), 1.70 (br s, 2H), 1.24 (t, J = 7,0 Гц, 3H); LCMS (ESI) m/z: 491.3 (M+1); HPLC указанного в заголовке соединения: RT = 2,088 мин.
Фармакологический раздел
Соединения по настоящему изобретению представляют собой ингибиторы CDK4/6. Следующие экспериментальные результаты подтверждают, что соединения, перечисленные в настоящей заявке, действительно являются ингибиторами CDK4/6 и могут быть использованы в качестве потенциальных противораковых лекарственных препаратов. IC50, как использовано в данной заявке, относится к концентрации соответствующего реагента, которая требуется для вызывания 50% от максимального ингибирования.
Экспериментальный пример 1: Исследование ферментативной активности
Экспериментальные материалы:
CDK4/циклин D1, CDK6/циклин D1 (Life technology). ULight меченные полипептидные субстраты ULight-4E-BP1 и ULight-MBP (PerkinElmer). Меченное европием анти-миелоновое основное белковое антитело и меченное европием антитело, полученное от кролика (PerkinElmer). Envision Multi-label Analyzer (PerkinElmer) для детектирования сигнала.
Экспериментальные методы:
Тестируемые соединениея трехкратно разбавляли, включая 10 градиентов концентрации, и диапазон конечных концентраций составлял от 5 мкМ до 0,25 нМ.
Ферментативая реакционная система CDK4/циклина D1
Стандартную методику Lance Ultra осуществляли при помощи 10 мкл ферментативной реакционной системы, содержащей 0,3 нМ CDK4/белка циклина D1, 50 нМ полипептида ULight-4E-BP1 и 350 мкМ АТР. Их соответствующим образом растворяли в ферментативном буферном растворе, содержавшем: 50 мМ раствора гидроксиэтилпиперазинэтансульфоновой кислоты при рН 7,5, 1 мМ этилендиаминтетрауксусной кислоты, 10 мМ хлорида магния, 0,01% Brij-35, 2 мМ дитиотреитола. После начала реакции 384-луночный планшет OptiPlate герметизировали сверху термопленкой TopSeal-A и инкубировали при комнатной температуре в течение 180 минут.
Ферментативная реакционная система CDK6/циклин D1
Стандартную методику Lance Ultra осуществляли при помощи 10 мкл ферментативной реакционной системы, содержащей 0,8 нМ CDK6/белка циклина D1, 50 нМ полипептида ULight-4E-BP1 и 250 мкМ АТР. Их соответствующим образом растворяли в ферментативном буферном растворе, содержавшем: 50 мМ раствора гидроксиэтилпиперазинэтансульфоновой кислоты при рН 7,5, 1 мМ этилендиаминтетрауксусной кислоты, 10 мМ хлорида магния, 0,01% Brij-35, 2 мМ дитиотреитола. После начала реакции 384-луночный планшет OptiPlate герметизировали сверху термопленкой TopSeal-A и инкубировали при комнатной температуре в течение 180 минут.
Получали буферный раствор для завершения ферментативной реакции и EDTA растворяли в однократно разбавленном тестовом буферном растворе. Реакцию завершали и инкубировали при комнатной температуре в течение 5 минут. 5 мкл тестового смешанного раствора (полученного с меченного европием анти-миелинового белкового антитела и меченного европием антитела, полученного от кролика, соответственно) добавляли к реакционным смесям CDK4/циклина D1 и CDK6/циклина D1, соответственно, и инкубировали при комнатной температуре в течение 60 минут. Реакционный сигнал детектировали с помощью Envision в соответствии с теорией разрешенного во времени резонансного переноса энергии флуоресценции.
Анализ данных:
Необработанные данные конвертировали в степень ингибирования с помощью уравнения (Max-Ratio)/(Max-Min)*100%, и значение IC50 может быть получено путем аппроксимации кривой с четырьмя параметрами (Model 205 в XLFIT5, iDBS). В Таблице 1 представлена ингибирующая активность соединений по настоящему изобретению в отношении киназы CDK4/CDK6.
Результаты эксперимента: См. Таблицу 1.
Выводы эксперимента:
Соединения по настоящему изобретению обладают значительной ингибирующей активностью в отношении киназ CDK4 и CDK6.
Экспериментальный пример 2: Исследование жизнеспособности клеток
Экспериментальные материалы:
Среда RPMI 1640 (Invitrogen-22400089), фетальная бычья сыворотка (Gibco-10099141), антибиотик пенициллин/стрептомицин (Hyclone-SV30010), L-глутамин (Invitrogen-35050079). Клеточная линия NCI-H358 получена из банка клеток Департамента Биологии WuXi Apptec Co. Ltd,. Envision Multi-Label Analyzer (PerkinElmer).
Экспериментальные способы:
1) 100 мкл фосфатного буферного раствора добавляли в периферические лунки 384-луночного планшета и в другие лунки добавляли 40 мкл клеточной суспензии NCI-H358, содержавшей 250 клеток NCI-H358. Затем клеточный планшет помещали в инкубатор с диоксидом углерода и инкубировали в течение ночи.
2) К тестовым соединениям применяли трехкратное градиентное разбавление с использованием Echo. Каждое соединение разбавляли 10 градиентами концентрации (разбавляли от 25 мкМ до 1,27 нМ), 100 нл которого добавляли в соответствующие лунки клеточного планшета. Затем клеточный планшет помещали в инкубатор с диоксидом углерода и инкубировали в течение 7 суток.
3) 20 мкл реагента Promega CellTiter-Glo добавляли в каждую лунку клеточного планшета и встряхивали в течение 10 минут вдали от света при комнатной температуре для стабилизации сигнала люминесценции. Измерения осуществляли с использованием PerkinElmer Envision Multi-label Analyzer.
Анализ данных:
Исходные данные превращали в степень ингибирования с использованием уравнения (Max-Sample)/(Max-Min)*100%, и значение IC50 может быть получено путем аппроксимации кривой с четырьмя параметрами (рассчитанной по формуле log(ингибитор) относительно ответа - Variable slope в GraphPad Prism). В Таблице 1 представлена ингибирующая активность соединений по настоящему изобретению в отношении пролиферации клеток Н358.
Результаты эксперимента: См. Таблицу 1.
Выводы эксперимента:
Соединения по настоящему изобретению обладают лучшей ингибирующей активностью в отношении пролиферации клеток рака легкого NCI-H358 по сравнению с активностью референсного соединения палбоциклиба.
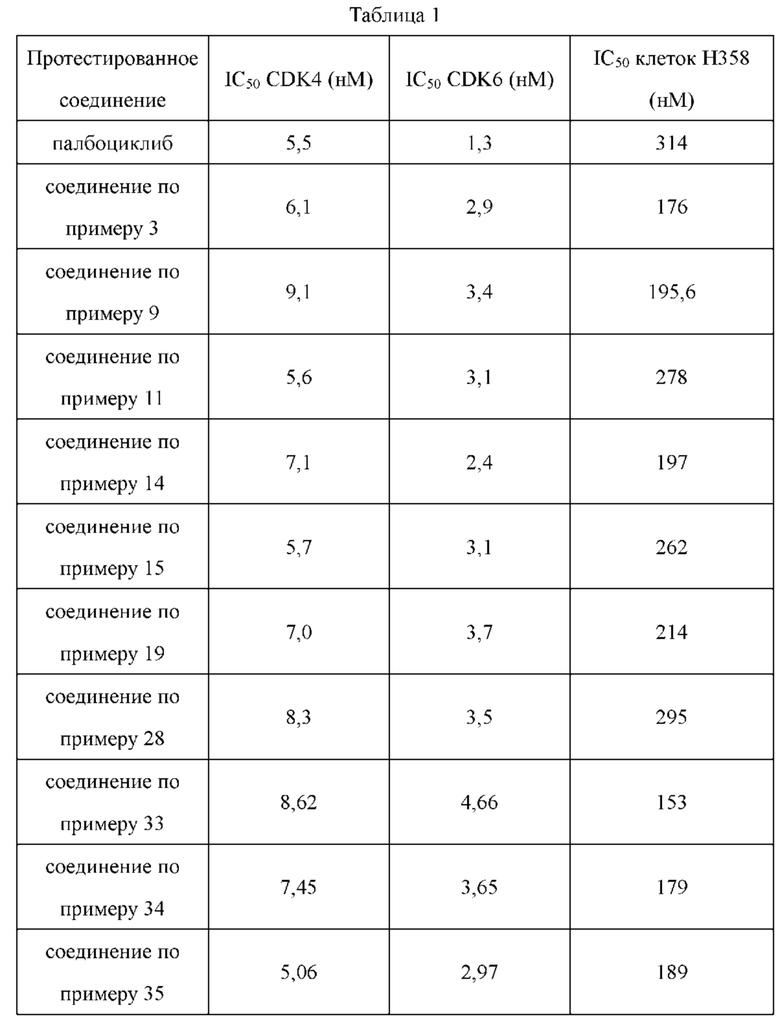
Экспериментальный пример 3: Исследование и оценка двусторонней проницаемости клеток Сасо-2
Цель эксперимента:
Клетки Сасо-2 представляют клетки человеческого рака толстой кишки, которые выступают в качестве модели in vitro, широко используемой в изучении кишечной абсорбции. Монослойная клеточная модель Сасо-2 широко используется в оценке процессов пассивного и активного транспорта в ходе кишечной абсорбции. Это исследование использовали для определения двухсторонней проницаемости посредством клеточной модели Сасо-2 соединений по настоящему изобретению и референсных соединений палбоциклиба и LY2835219.
Экспериментальные процедуры:
Стандартные условия эксперимента являются следующими:
- Тестовая концентрация: 2 мкм (DMSO ≤ 1%);
- Повторения: n = 2;
- Направление: двухсторонний транспорт, включая два направления: А→В (внутриклеточный→внеклеточный) и В→А (внеклеточный→внутриклеточный);
- Время инкубирования: одна временная точка, 2 часа;
- транспортный буферный раствор: HBSS, рН 7,4;
- условия инкубирования: 37°С, 5% CO2.
После инкубирования тестовые растворы в лунках дозирования и приемных лунках немедленно смешивали с холодным раствором ацетонитрила, содержавшим внутренний стандарт. Концентрацию тестированных соединений во всех образцах (включая исходный раствор для дозирования, надосадочную жидкость в лунках дозирования, и принимающий раствор) анализировали способом LC/MS/MS. Вычисляли коэффициент кажущейся проницаемости, скорость эффлюкса и другие параметры.
Результаты эксперимента:
См. Таблицу 2. Коэффициенты проницаемости через монослой клеток Сасо-2 соединений по настоящему изобретению и референсных соединений палбоциклиба и LY2835219 перечислены в Таблице 2.
Выводы эксперимента:
По сравнению с референсными соединениями палбоциклибом и LY2835219 соединения по настоящему изобретению обладают большей проницаемостью и с меньшей вероятностью затрагиваются эффлюксными переносчиками in vivo. Лучшая проницаемость позволяет соединениям по настоящему изобретению более широко распределяться в тканях in vivo, таких как легкое, что приводит к улучшенной противоопухолевой эффективности in vivo. Кроме того, лучшая проницаемость делает возможным для соединений по настоящему изобретению проникать через гемато-энцефалический барьер и достигать цели лечения метастаз в мозге при раке легкого.
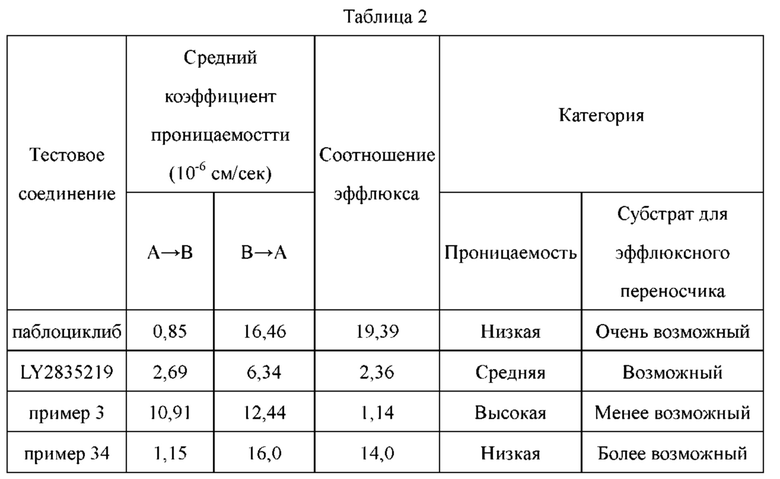
Экспериментальный пример 4: Тест на растворимость
(1) Исследование кинетической растворимости
Цель эксперимента:
Кинетичекую растворимость соединений определяли в аналитических условиях рутинного биологического скрининга.
Принцип тестирования:
Кинетическая растворимость зависит от рН, и рН тестового раствора обычно устанавливают на 7,4. Это исследование осуществляли методом встряхивания колбы и детектирования с помощью HPLC. Каждое соединение приготавливали в виде 10 мМ маточного раствора в DMSO (диметилсульфоксид) и разбавляли до теоретической концентрации 200 мкм (содержавшей 2% DMSO) в фосфатном буферном раствора. Смесь встряхивали при комнатной температуре в течение 24 часов, после чего фильтровали с водяным проточным насосом. Надосадочную жидкость собирали и анализировали с помощью HPLC-UV (высокоэффективная жидкостная хроматография с УФ детектором).
Экспериментальные процедуры:
Тестовый раствор кинетической растворимости
Буферный раствор (рН 7,4)
50 мМ фосфатный буферный раствор, рН 7,4.
Получение стандартных растворов:
50% ацетонитрильного раствора и 50% буферного раствора смешивали с получением разбавителя.
10 мМ (10 мкл/соединение) маточного раствора добавляли к 490 мкл разбавителя и смешивали в 200 мкм стандартного тестового раствора.
200 мкМ стандартного УФ тестового раствора разбавляли в 10 или 200 раз с получением 20 мкм или 1 мкм стандартного UV раствора.
Стандартные УФ растворы 1, 20 и 200 мкМ использовали в качестве стандартных растворов для исследования на кинетическую растворимость.
Способ:
Приготовление образца, встряхивание и фильтрация
Соединение растворяли в DMSO и изготавливали в виде 10 мМ маточного раствора. Количество маточного раствора представляет собой по меньшей мере 100 мкл. Амиодарона гидрохлорид, карбамазепин и хлорамфеникол использовали в качестве QC для исследования растворимости.
490 мкл среды растворения (буферный раствор) точно взвешивали в 2 мл 96-луночный планшет.
10 мкл тестового соединения и QC маточный раствор добавляли в среду для растворения (буферный раствор). Теоретическая максимальная концентрация тестового соединения, соответствующая кинетической растворимости в растворе при рН 7,4, составляла 200 мкМ при содержании 2% DMSO. Планшет закрывали крышкой. Теоретическая максимальная концентрация тестированного соединения составляет 200 мкМ. Если требуется  теоретическая максимальная концентрация, концентрация маточного раствора может быть увеличена.
теоретическая максимальная концентрация, концентрация маточного раствора может быть увеличена.
Планшет встряхивали на шейкере при 600 об./мин в течение 24 часов при комнатной температуре.
Образцы переносили в 96-луночный планшет для фильтрования, после чего фильтровали с помощью водяного вакуумного насоса.
Концентрацию фильтрата соединения определяли с помощью HPLC-UV.
QC образцы:
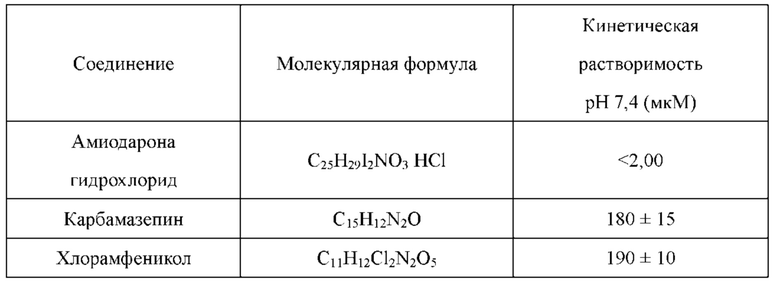
Анализ данных:
Три стандартных раствора для УФ-детектирования, имеющих концентрацию от низкой до высокой, инъецировали в HPLC, после чего инъецировали фильтрат тестируемого соединения в качестве тестового образца. Две иглы тестируемого образца вставляли параллельно. УФ-пики интегрировали. Получали стандартную кривую и вычисляли кинетическую растворимость образца.
Результаты эксперимента:
См. Таблицу 3-1. Данные кинетической растворимости соединений по настоящему изобретению и референсного соединения палбоциклиба приведены в Таблице 3-1.
Выводы эксперимента:
Соединения по настоящему изобретению обладают большей кинетической растворимостью, чем референсное соединение палбоциклиб.

(2) Исследование термодинамической растворимости
Цель эксперимента:
Термодинамическая растворимость соединения модет быть точно и достоверно определена методами фильтрации HPLC.
Принцип исследования:
Термодинамическую растворимость соединений определяли методом встряхивания колбы и HPLC. Растворимость соединений представляет собой важное свойство, которое влияет на лекарственный скрининг соединений и абсорбцию соединений у животных и людей. Сначала получали насыщенный раствор соединения и количественно анализировали с помощью HPLC с получением растворимости соединения.
Экспериментальные процедуры:
Раствор термодинамической растворимости
Буферный раствор (рН 7,4)
50 мМ фосфатный буферный раствор, рН 7,4.
Получение стандартных растворов:
50% раствора ацетонитрила и 50% буферного раствора смешивали с получением разбавителя.
10 мМ (10 мкл/соединение) маточного раствора добавляли к разбавителю (490 мкл/соединение) и смешивали в 200 мкМ стандартный УФ тестовый раствор.
200 мкм стандартного УФ тестового раствора разбавляли в 10 или 200 раз с получением 20 мкм или 2 мкм стандартного УФ раствора.
Стандартные УФ растворы 2, 20 и 200 мкм использовали в качестве стандартных растворов исследования кинетической растворимости.
Способ:
Приготовление образцов, встряхивание и фильтрация
Не менее чем 2 мг порошка образца взвешивали в виалу Whatman miniuniprep.Если требуется исследование термодинамической растворимости образца в нескольких буферных растворах, индивидуальная виала требуется для каждого исследования.
450 мкл буферного раствора (рН 7,4) добавляли в каждую виалу Whatman miniuniprep.
После добавления буферного раствора устанавливали крышку с фильтром для Whatman miniuniprep и помещали фильтр выше уровня жидкости так, чтобы фильтр мог контактировать с буферным раствором в ходе встряхивания.
Образец для исследования растворимости встряхивали на вихревой мешалке в течение 2 минут. Регистрировали визуальное состояние раствора.
Его встряхивали при 550 об./мин в течение 24 часов при комнатной температуре (примерно от 22 до 25°С).
Фильтр-крышку Whatman Miniunipreps опускали до дна с получением фильтрата раствора образца для исследования растворимости. Все виалы с образцами должны быть отфильтрованы до и после нерастворимых веществ и их утечки.
Буфер разбавляли в 50 раз с получением разбавителя образцов.
Три УФ стандарта, имеющих концентрацию от низкой до высокой, инъецировали в HPLC, после чего инъецировали разбавления и надосадочные жидкости тестируемых соединений. Тестовый образец инъецировали дважды.
УФ пики интегрировали. Получали стандартную кривую и вычисляли термодинамическую растворимость образца.
QC образцы:
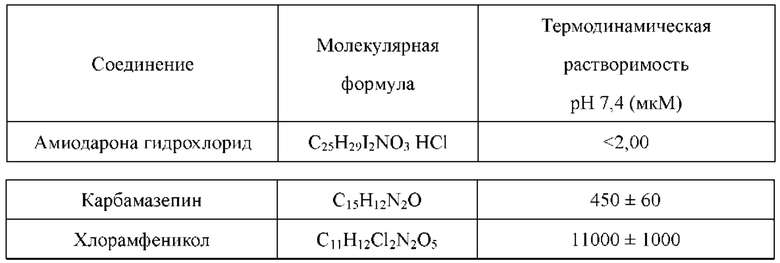
Результаты эксперимента:
См. Таблицу 3-2. Данные термодинамической растворимости соединения по настоящему изобретению и референсного соединения палбоциклиба приведены в Таблице 3-2.
Выводы эксперимента:
Соединения по настоящему изобретению обладают большей термодинамической растворимостью, чем референсное соединение палбоциклиб.

Экспериментальный пример 5: Исследование метаболической стабильности у крыс, мышей и в микросомах печени человека
Цель эксперимента:
Это исследование использовано для тестирования метаболической стабильности исследуемых соединений у крыс, мышей и в микросомах печени человека. Экспериментальные процедуры:
1) Тестируемое соединение с концентрацией 1 мкм совместно инкубировали с микросомами печени с концентрацией белка 0,5 мг/мл в регенерационной системе восстанавливительного коэнзима II на водной бане при 37°С.
2) Положительные контроли включают: тестостерон (субстрат 3А4), пропафенен (субстрат 2D6) и диклофенак (субстрат 2С9). Условия инкубирования положительных контролей соответствовали условиям инкубирования соединения.
3) Временные точки реакции составляли: 0, 5, 10, 20, 30 и 60 минут, и реакцию завершали в соответствующей временной точке с использованием раствора для завершения реакции, содержащего внутренний стандарт. Соединения также инкубировали с микросомами в течение 60 минут без регенерационной системы восстанавливительного коэнзима II, и это служило в качестве отрицательного контроля.
4) Каждый момент времени представлял собой одну точку (n = 1).
5) Образец определяли с помощью LC/MS/MS и концентрация соединения была показана как соотношение области пика соединения к области пика внутреннего стандарта(нестандарт).
6) В резюме проекта вычисляли время полувыведения и скорость клиренса.
7) Следующие формулы использовали для рассчета скорости клиренса:
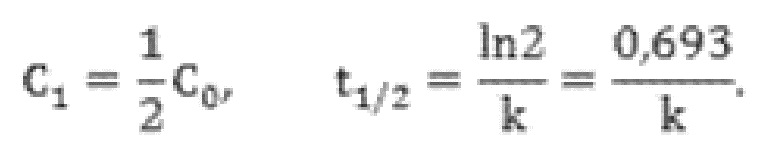

Примечание:
а) микросомный белок при инкубировании:
Массовое соотношение печени к телу: параметры крыс, мышей и человека составляли 40 г/кг, 88 г/кг и 20 г/кг, соответственно.
Скорость клиренса через печень вычисляли с помощью 
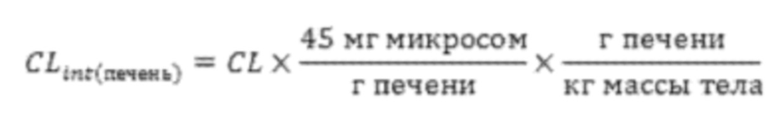
Примечание:
a) микросомы: микросомы;
b) печень: печень;
c) масса тела: масса тела;
Результаты эксперимента:
Результаты эксперимента показаны в Таблице 4.
Выводы эксперимента:
Соединения по настоящему изобретению обладают значительно улучшенной стабильностью в микросомах печени у людей, крыс и мышей по сравнению со стабильностью референсных соединений LY2835219 и палбоциклиба.
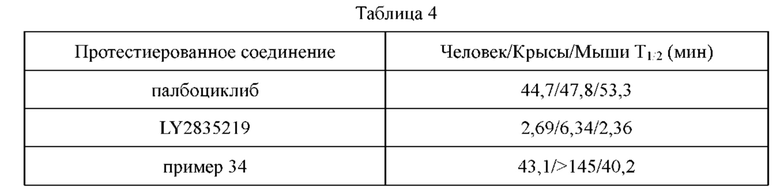
Экспериментальный пример 6: Фармакодинамическое исследование in vivo (1)
Фармакодинамические эксперименты in vivo проводили на «голых» мышах BALB/c, которым подкожно имплантировали ксенографт опухолевой ткани рака легкого LU-01-0393, полученной от пациента (PDX).
Экспериментальные процедуры:
«Голых» мышей BALB/c, самок, 6-8 недель, с массой приблизительно 17-21 г, помещали в одну вентилируемую клетку (5 мышей на клетку) в специальной среде, свободной от патогенов. Все клетки, подстилки и воду дезинфицировали перед использованием. Всем животным позволяли свободный доступ к стандартной сертифицированной лабораторной диете, доступной в продаже. Всего для исследования использовали 36 мышей, приобретенных в Vital River Laboratory Animal Co., LTD, Пекин. Каждой мыши подкожно имплантировали секцию FP4 опухоли LU-01-0393 (20-30 мм3) в правую часть спины для роста опухоли. Дозирование начинали тогда, когда средний объем опухоли достигал примерно 150-200 мм3. Тестируемое соединение перорально вводили ежедневно, и дозировка была такой, как показано в Таблице 2. Объем опухоли измеряли дважды в неделю с использованием штангенциркуля с двумя измерениями, и объем измеряли в мм3, вычисляя его согласно следующей формуле: V = 0.5 а × b2, где а и b представляли собой длинные и короткие диаметры опухоли, соответственно. Противоопухолевую эффективность определяли путем деления среднего увеличения объема опухоли животных, обрабатываемых соединением, на среднее увеличение объема опухоли у животных, которых не обрабатывали соединением.
Результаты эксперименты: См. Таблицу 5.
Выводы эксперимента:
Соединения по настоящему изобретению демонстрируют значительную противоопухолевую активность на модели ксенографта опухолевой ткани рака легкого LU-01-0393, полученной от пациента (PDX). Как показано в Таблице 5, по прошествии 20 суток с начала эксперимента объем опухоли группы животных, которых не обрабатывали, быстро увеличивался от исходных 144 мм3 до 437 мм3, в то время как объем опухоли группы животных, обрабатываемых соединением по примеру 1, медленно увеличивался от 144 мм3 до 212 мм3, и скорость увеличения была схожей со скоростью увеличения в случае референсного соединения палбоциклиба. Однако, дозировка соединения по примеру 1 (60 мг/кг) составляла лишь половину от дозировки референсного соединения палбоциклиба (120 мг/кг). Было показано, что противоопухолевая активность соединений по настоящему изобретению является большей, чем активность референсных соединений.
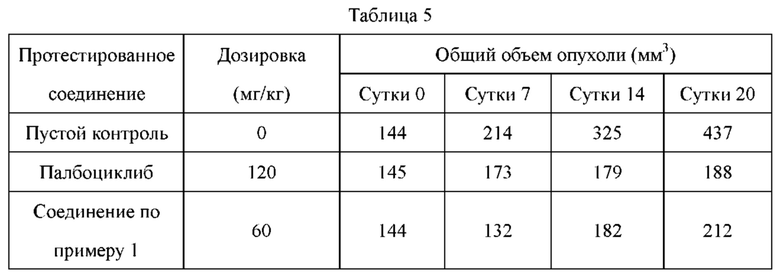
Экспериментальный пример 7: Фармакодинамические исследования in vivo (2)
Фармакодинамические эксперименты in vivo осуществляли на модели «голых» мышей BALB/c с подкожнно имплантированным немелкоклеточным раком легкого NCI-H358.
Экспериментальные процедуры:
Информация о животных в экспериментах с соединением по примеру 3 была следующей: «голых» мышей BALB/c, самок, 6-8 недель, с массой приблизительно 17-20 г, помещали в одну вентилируемую клетку (3-5 мыши на клетку) в специальной среде, свободной от патогенов. Все клетки, подстилки и воду дезинфицировали перед использованием. Всем животным позволяли свободный доступ к стандартной сертифицированной лабораторной диете, доступной в продаже. Всего для исследования использовали 86 мышей, приобретенных в Vital River Laboratory Animal Co., LTD, Пекин. Информация о животных в эксперименте с соединением по примеру 34 была следующей: голых мышей BALB/c, самок, 6-8 недель, массой приблизительно 16-18 г, помещали в одну вентилируемую клетку (4 мыши на клетку) в специальной среде, свободной от патогенов. Все клетки, подстилки и воду дезинфицировали перед использованием. Всем животным позволяли свободный доступ к стандартной сертифицированной лабораторной диете, доступной в продаже. Всего в исследовании использовали 56 мышей, приобретенных в Shanghai Lingchang Biotechnology Co., Ltd.
Каждой мыши подкожно имплантировали опухолевые клетки NCI-H358 в правую часть спины для роста опухоли. Дозирование начинали тогда, когда объем опухоли достигал примерно 100-200 мм3. Тестируемое соединение вводили перорально ежедневно и доизровки соединений по примеру 3 и по примеру 34 были такими, как показано в Таблице 6-1 и Таблице 6-2, соответственно. Объем опухоли измеряли дважды в неделю с использованием штангенциркуля с двумя измерениями, и объем измеряли в мм3 и вычисляли согласно следующей формуле: V = 0,5 а × b2, где а и b представляли собой длинный и короткий диаметры опухоли, соответственно. Противоопухолевую эффективность определяли путем деления среднего увеличения объема опухоли у животных, которых обрабатывали соединением, со средним увеличением объема опухоли животных, которых не обрабатывали, и безопасность соединения определяли как изменение массы тела животного, которого обрабатывали соединением.
Результаты эксперимента: См. Таблицу 6-1 и Таблицу 6-2.
Выводы эксперимента:
Соединения по настоящему изобретению демонстрируют значительную противоопухолевую активность в модели немелкоклеточного рака легкого NCI-H358 и лучшую стабильность. Кроме того, в этой модели противоопухолевый эффект соединений по изобретению имеет дозо-зависимую тенденцию.
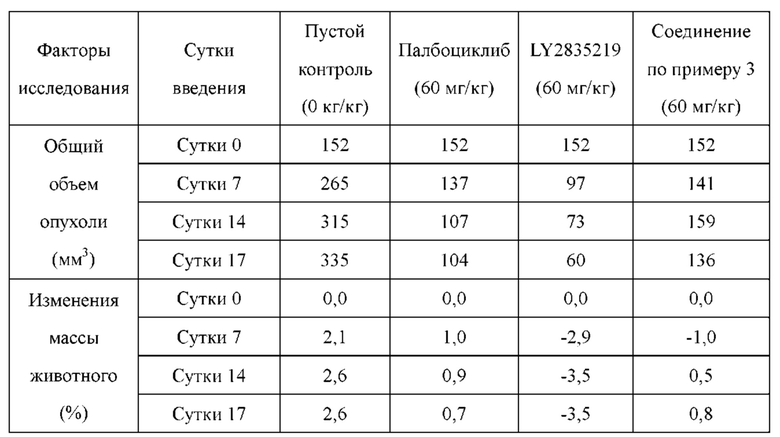
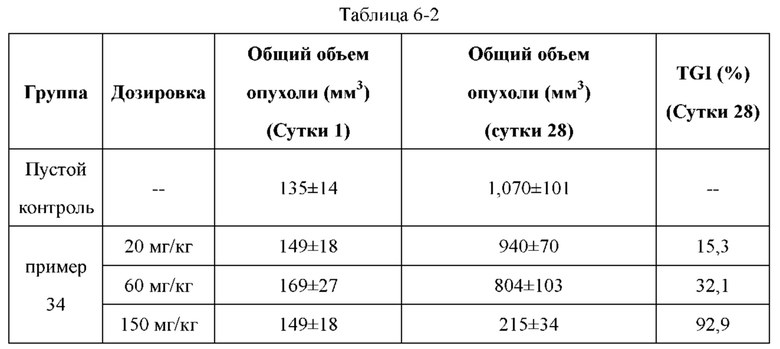
TGI: Ингибирование Роста Опухоли. TGI (%) = [(1-(средний объем опухоли в конце введения группе обработки - средний объем опухоли в начале введения группе обработки)) / (средний объем опухоли в конце обработки растворителем контрольной группы - средний объем опухоли в начале обработки растворителем контрольной группы)] × 100%
Экспериментальный пример 8: Фармакодинамическое исследование in vivo (3)
Фармакодинамические эксперименты in vivo осуществляли на модели «голых» мышей BALB/c, которым подкожно имплантировали колоректальный рак НСТ-116.
Экспериментальные процедуры:
«Голых» мышей BALB/c, самок, 6-8 недель, с массой приблизительно 18-22 г, помещали в одну вентилируемую клетку (3-5 мышей на клетку) в специальной среде, свободной от патогенов. Все клетки, подстилки и воду дезинфицировали перед использованием. Всем животным позволяли свободный доступ к стандартной сертифицированной лабораторной диете, доступной в продаже. Всего для исследования использовали 48 мышей, приобретенных в Shanghai Lingchang Biotechnology Co., Ltd. Каждой мыши подкожно имплантировали 0.2 мл 5 × 106 клеток НСТ-116 в правую часть спины для роста опухоли. Дозирование в группах начинали тогда, когда средний объем опухоли достигал примерно 132 мм3. Тестируемое соединение перорально вводили ежедневно, и дозировка была такой, как показано в Таблице 5. Объем опухоли измеряли дважды в неделю с использованием штангенциркуля с двумя измерениями, и объем измеряли в мм3, как было вычислено согласно следующей формуле: V = 0.5 а × b2, где а и b представляли собой длинный и короткий диаметры опухоли, соответственно. Противоопухолевую эффективность определяли путем деления среднего увеличения объема опухоли животных, которых обрабатывали соединением, на среднее увеличение объема опухоли животных, которых не обрабатывали соединением.
Результаты эксперимента: См. Таблицу 7.
Выводы эксперимента:
Соединения по настоящему изобретению демонстрируют лучшую противоопухолевую активность и  безопасность в модели колоректального рака НСТ-116.
безопасность в модели колоректального рака НСТ-116.
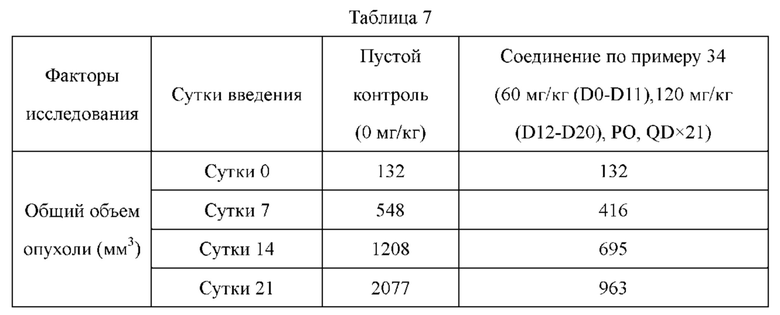
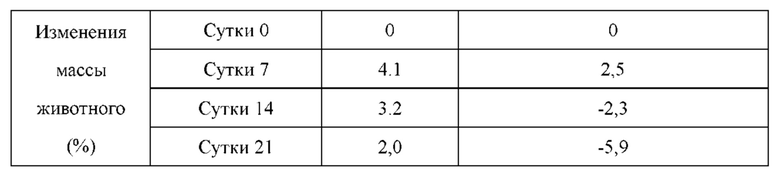
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ С КОНДЕНСИРОВАННЫМ КОЛЬЦОМ В КАЧЕСТВЕ ИНГИБИТОРА РЕЦЕПТОРА A | 2018 |
|
RU2748993C1 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
| СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ HDAC6, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2772274C2 |
| ТИОФЕНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2709473C1 |
| ЗАМЕЩЁННЫЕ ИНДОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ПОНИЖАЮЩИХ РЕГУЛЯТОРОВ РЕЦЕПТОРОВ ЭСТРОГЕНА | 2017 |
|
RU2722441C2 |
| ПРОИЗВОДНОЕ НА ОСНОВЕ ДИГИДРОПИРИМИДО-КОЛЬЦА В КАЧЕСТВЕ ИНГИБИТОРА HBV | 2015 |
|
RU2693897C2 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SMO | 2015 |
|
RU2695815C2 |
| ЦИКЛИЧЕСКИЕ ДИ-НУКЛЕОТИДНЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2790175C2 |
| ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2809631C2 |
Изобретение относится к соединениям формулы (I), их фармацевтически приемлемым солям или стереоизомерам, а также содержащим их фармацевтическим композициям, и их применению в изготовлении лекарственных средств для лечения раковых заболеваний. Технический результат: получены новые соединения, действующие в качестве ингибиторов CDK4/6 и пригодные для лечения опосредованных заболеваний. 4 н. и 9 з.п. ф-лы, 7 табл., 36 пр.
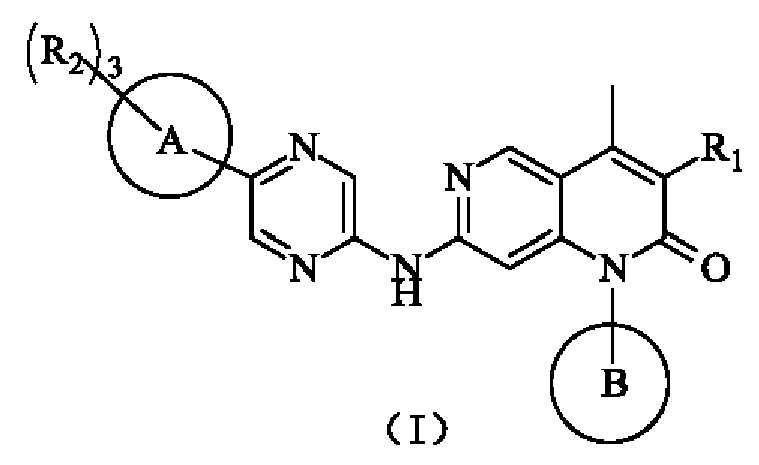
1. Соединение формулы (I), его фармацевтически приемлемая соль или его стереоизомер
где
R1 представляет собой CHF2, СН3(С=O)-, 
каждый из R2 независимо представляет собой Н, С1-3алкил, 
 С3-5циклоалкил или 4-6-членный гетероциклоалкил, содержащий 1-2 гетероатома, выбранных из N и О;
С3-5циклоалкил или 4-6-членный гетероциклоалкил, содержащий 1-2 гетероатома, выбранных из N и О;
кольцо А представляет собой 4-9-членный гетероциклоалкил, содержащий 1-2 гетероатома, выбранных из N и О;
кольцо В представляет собой С5-6циклоалкил или фенил.
2. Соединение формулы (I), его фармацевтически приемлемая соль или его стереоизомер по п. 1, где R2 независимо выбран из Н, СН3, 
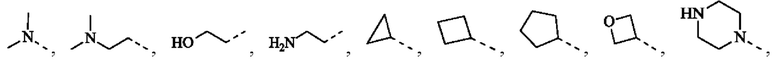
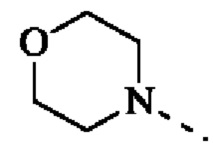
3. Соединение формулы (I), его фармацевтически приемлемая соль или его стереоизомер по п. 1, где кольцо А представляет собой 5-9-членный гетероциклоалкил, содержащий 1-2 гетероатома, выбранных из N и О.
4. Соединение формулы (I), его фармацевтически приемлемая соль или его стереоизомер по п. 3, где группировка  выбрана из группы, состоящей из
выбрана из группы, состоящей из 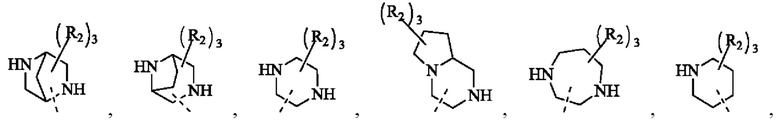
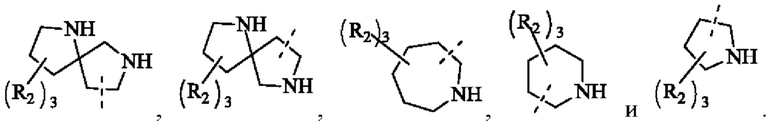
5. Соединение формулы (I), его фармацевтически приемлемая соль или его стереоизомер по п. 4, где группировка 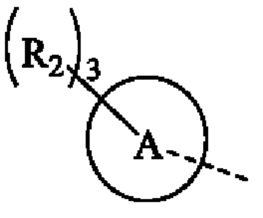 выбрана из группы, состоящей из
выбрана из группы, состоящей из 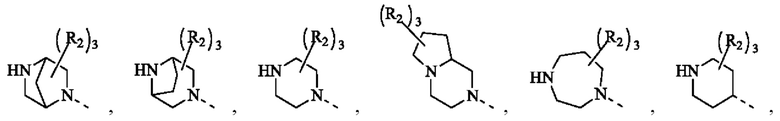
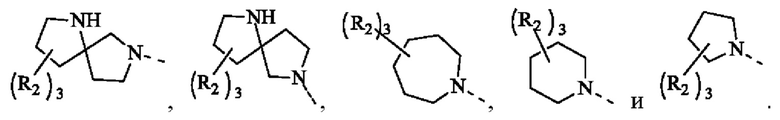
6. Соединение формулы (I), его фармацевтически приемлемая соль или его стереоизомер по п. 5, где группировка  выбрана из группы, состоящей из
выбрана из группы, состоящей из 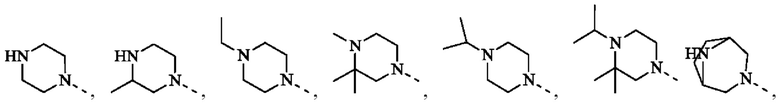
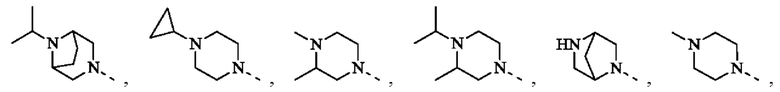
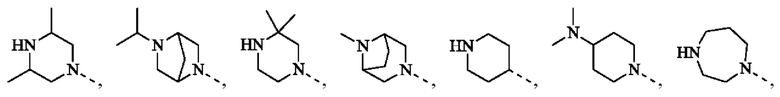
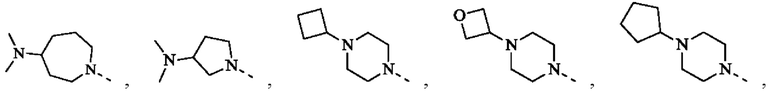
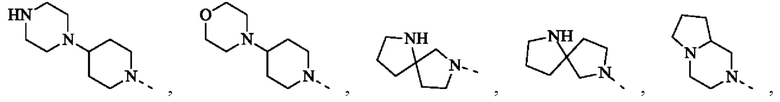
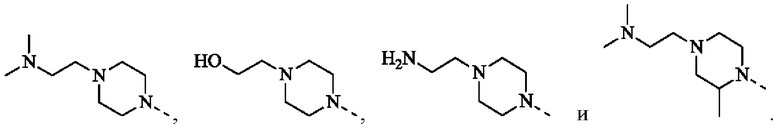
7. Соединение формулы (I), его фармацевтически приемлемая соль или его стереоизомер по п. 1, где соединение выбрано из группы, состоящей из
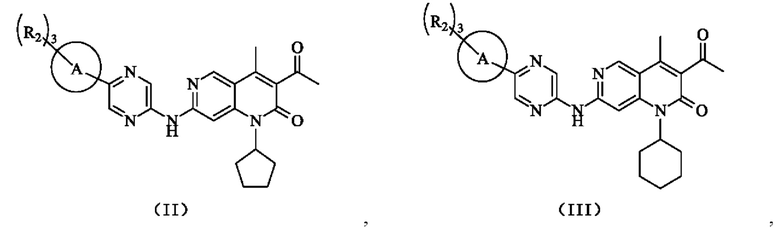
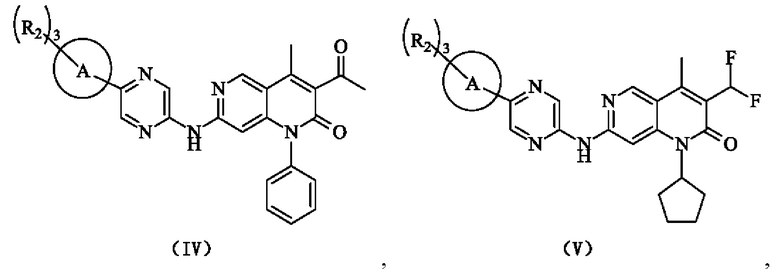
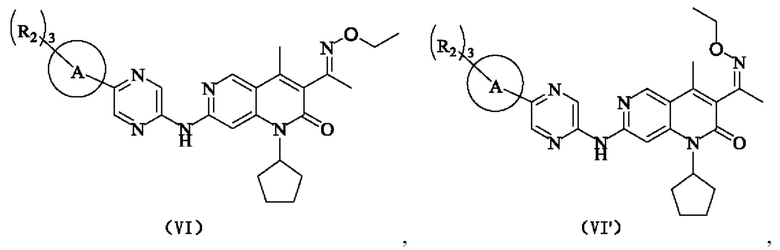
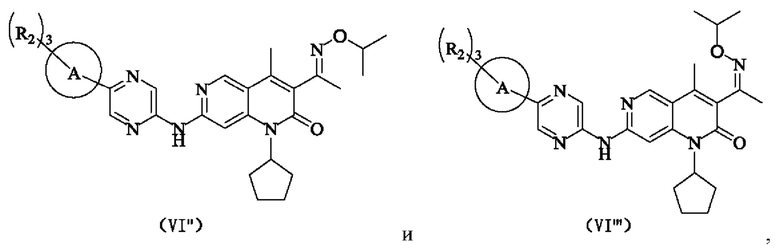
где R2 является таким, как определено в п. 1 или 2;
кольцо А является таким, как определено в пп. 1 или 3-6.
8. Соединение формулы (I), его фармацевтически приемлемая соль или его стереоизомер по п. 1, где соединение представляет собой
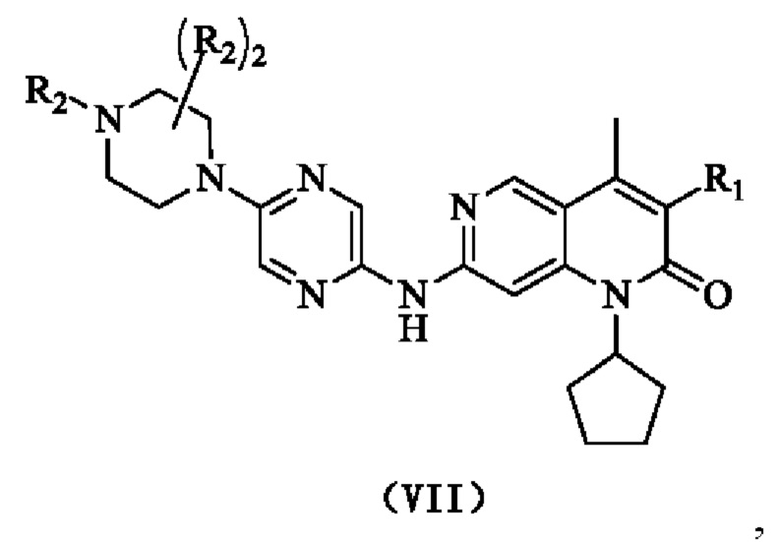
где R1 является таким, как определено в п. 1;
R2 является таким, как определено в п. 1 или 2.
9. Соединение формулы (I), его фармацевтически приемлемая соль или его стереоизомер по п. 1, где соединение представляет собой
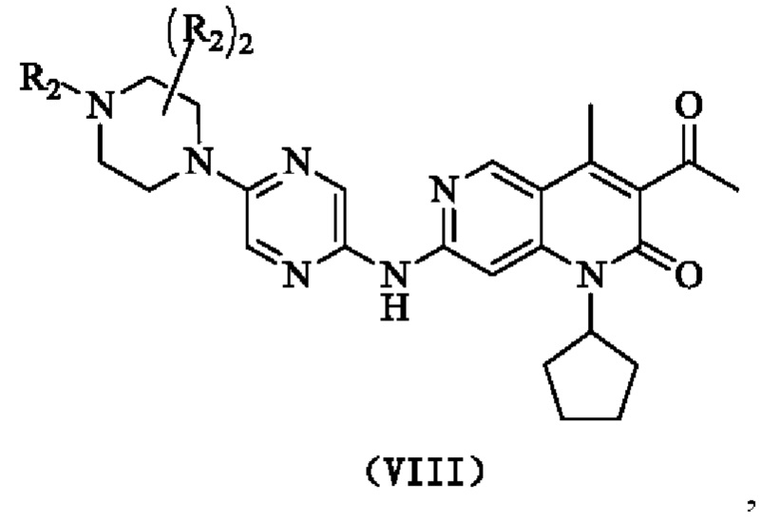
где R2 является таким, как определено в п. 1 или 2.
10. Соединение, его фармацевтически приемлемая соль или его стереоизомер, представленное следующими формулами:
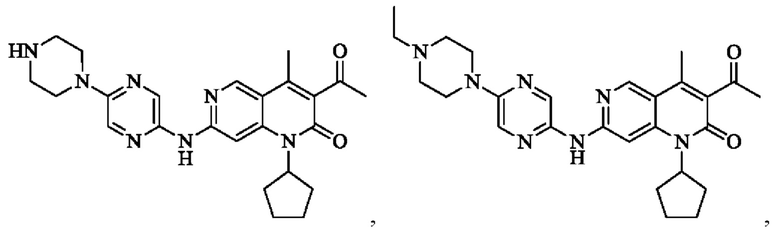
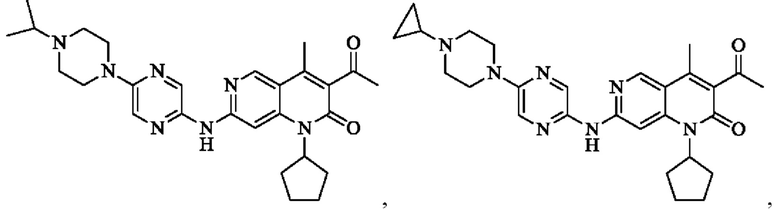
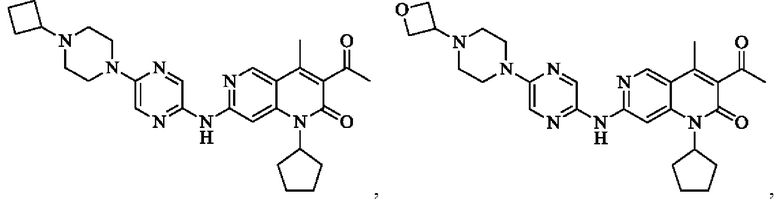
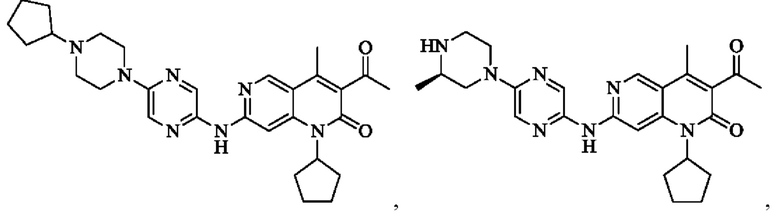
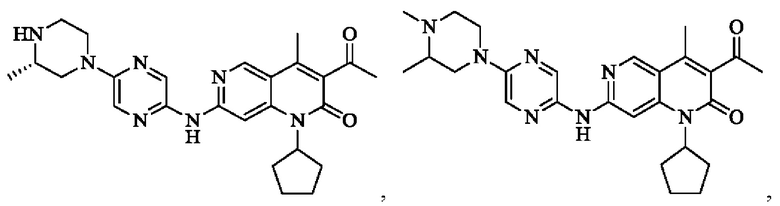
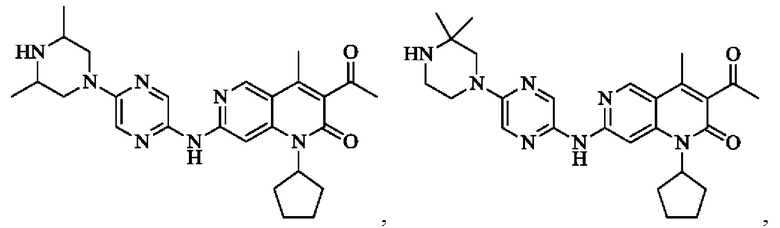
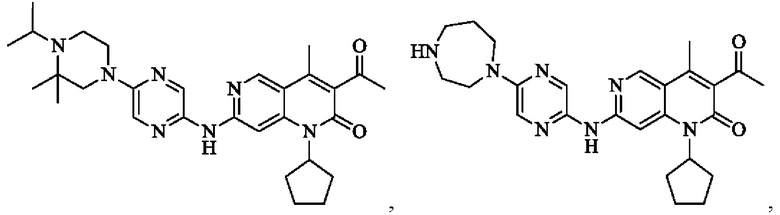
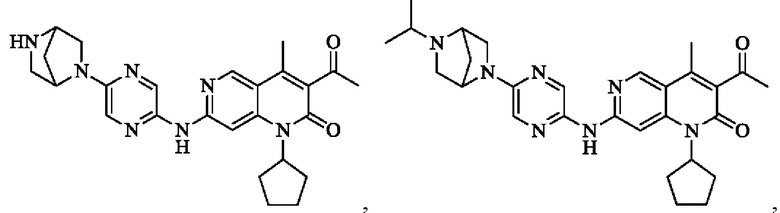
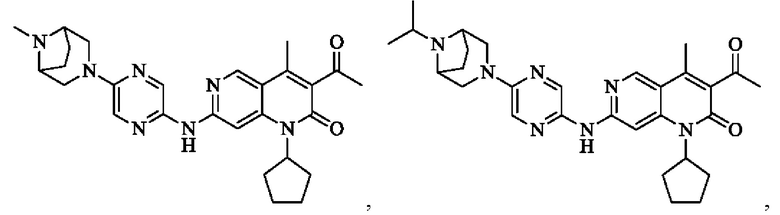
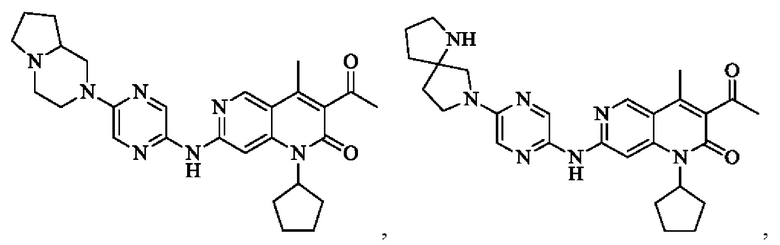
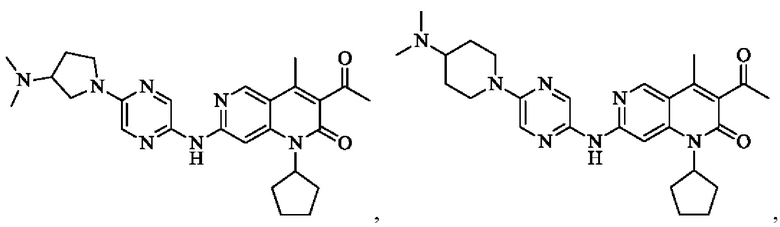
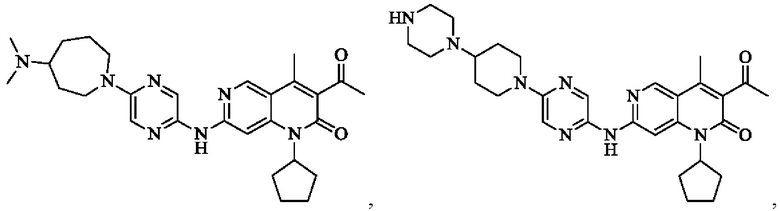
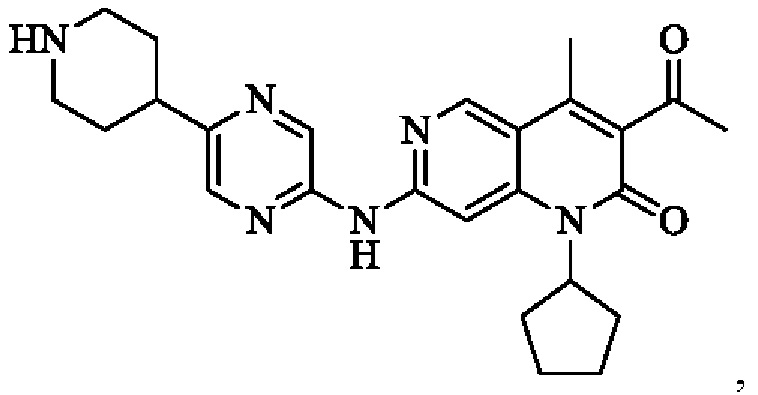
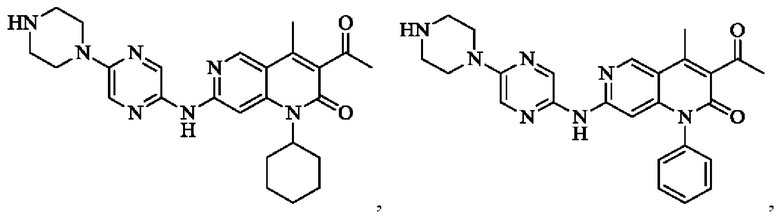
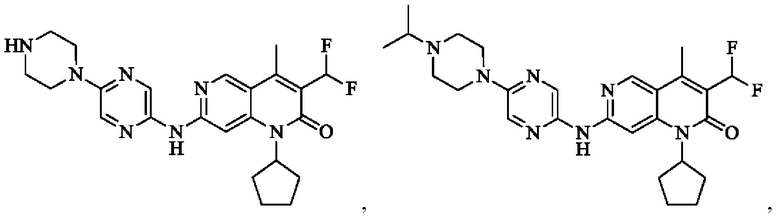
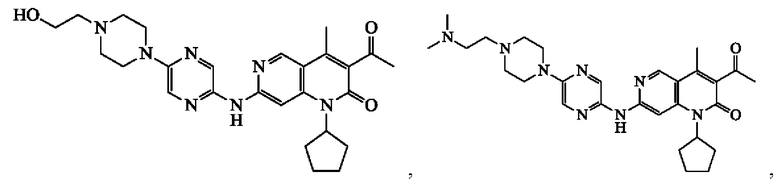
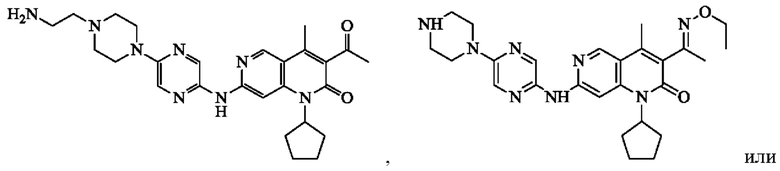
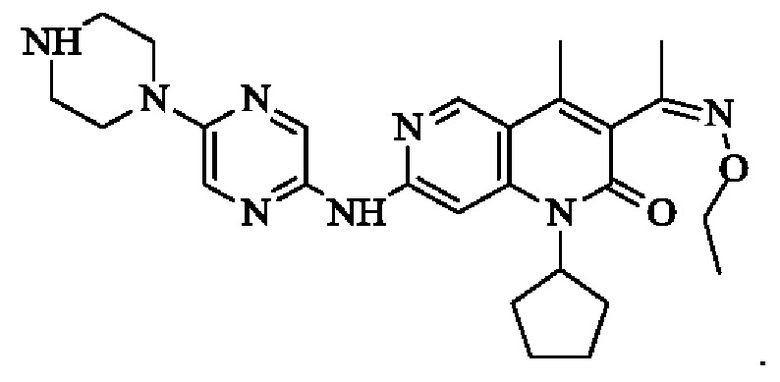
11. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении киназ CDK4/CDK6, содержащая терапевтически эффективное количество соединения, его фармацевтически приемлемой соли или его стереоизомера по п. 1 и фармацевтически приемлемый носитель.
12. Применение соединения, его фармацевтически приемлемой соли или его стереоизомера по п. 1 в изготовлении лекарственного средства для лечения раковых заболеваний, опосредованных ингибирующей активностью в отношении киназ CDK4 и CDK6.
13. Применение по п. 12, где раковое заболевание, опосредованное ингибирующей активностью в отношении киназ CDK4 и CDK6, представляет собой рак легкого или печени.
| CN 105130986 A, 09.12.2015 | |||
| WO 2014052365 A1, 20.11.2014 | |||
| WO 2014183520 A1, 20.11.2014 | |||
| 4-ОКСО-1-(3-ЗАМЕЩЕННЫЙ ФЕНИЛ)-1,4-ДИГИДРО-1,8-НАФТИРИДИН-3-КАРБОКСАМИДЫ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ И СПОСОБ УЛУЧШЕНИЯ ПОЗНАВАТЕЛЬНОЙ СПОСОБНОСТИ У ЗДОРОВОГО СУБЪЕКТА | 2003 |
|
RU2312865C2 |

