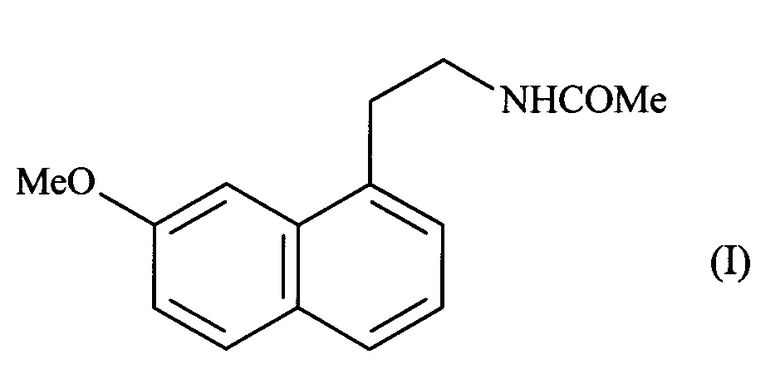
Настоящее изобретение относится к новому способу промышленного синтеза агомелатина или N-[2-(7-метокси-1-нафтил)этил]ацетамида формулы (I):
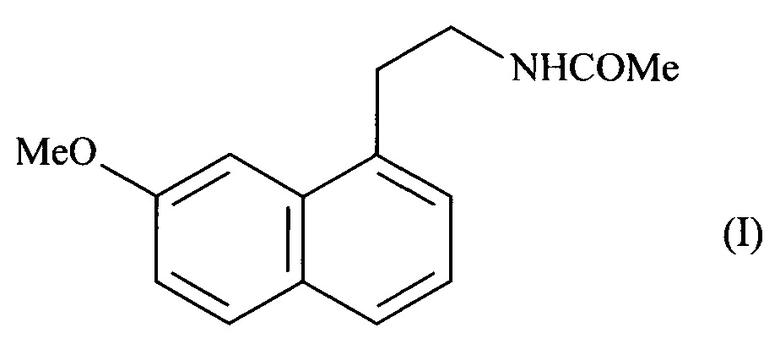
Агомелатин, или N-[2-(7-метокси-1-нафтил)этил]ацетамид, обладает ценными фармакологическими свойствами.
В действительности, он обладает двойственными свойствами, являясь, с одной стороны, агонистом рецепторов мелатонинэргической системы и, с другой стороны, антагонистом 5-НТ2С рецептора. Эти свойства придают ему активность в центральной нервной системе и, более конкретно в лечении глубокой депрессии, сезонных аффективных расстройств, нарушений сна, патологий сердечно-сосудистой системы, патологий пищеварительной системы, бессонницы и утомляемости вследствие нарушения биоритмов в связи с перелетом через несколько часовых поясов, нарушений аппетита и ожирения.
Агомелатин, его получение и терапевтическое применение описаны в Европейских патентах ЕР 0447285 и ЕР 1564202
Принимая во внимание фармацевтическую ценность данного соединения, важно, чтобы его получали при помощи высокопроизводительного способа промышленного синтеза, который легко переводится в промышленный масштаб и обеспечивает получение агомелатина с высоким выходом и с превосходной чистотой.
В описании патента ЕР 0447285 раскрыто получение агомелатина за восемь стадий, исходя из 7-метокси-1-тетралона. В описании патента ЕР 1564202, заявитель разработал намного более эффективный и применимый в промышленности способ синтеза всего лишь за четыре стадии, исходя из 7-метокси-1-тетралона, позволяя получить агомелатин чрезвычайно репродуктивным способом в хорошо определенной кристаллической форме. Тем не менее, все еще продолжаются поиски новых способов синтеза, в особенности исходя из исходных веществ менее дорогостоящих, нежели 7-метокси-1-тетралон.
Заявителем были продолжены его исследования, и был разработан новый способ синтеза агомелатина, исходя из 7-метокси-нафталин-2-ола: это новое исходное вещество обладает преимуществом в том, что его можно получать просто и без особого труда в больших количествах с меньшими затратами. Преимущество 7-метокси-нафталин-2-ола также заключается в том, что в своей структуре он имеет нафталиновую кольцевую систему, которая позволяет избежать в синтезе введения стадии ароматизации - стадии, которая всегда является проблематичной с промышленной точки зрения.
Кроме того, этот новый способ обеспечивает получение агомелатина воспроизводимым способом и без потребности в лабораторной очистке, с чистотой, которая сопоставима с его применением в качестве фармацевтического действующего вещества. http://www-iab.ujf-grenoble.fr/clientzone/plforme10/img/tomo_xfluo.jpg
Более конкретно, настоящее изобретение относится к способу промышленного синтеза соединения формулы (I):
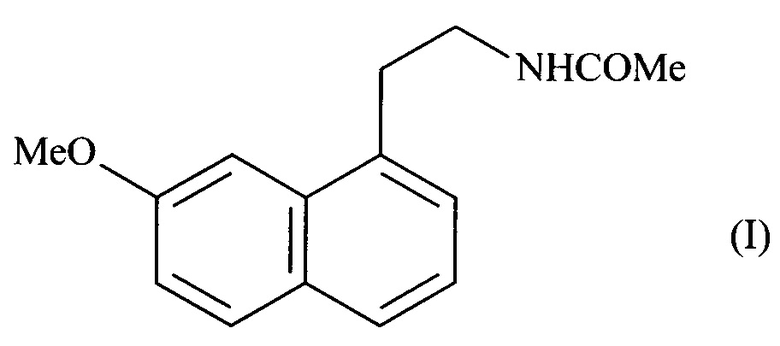
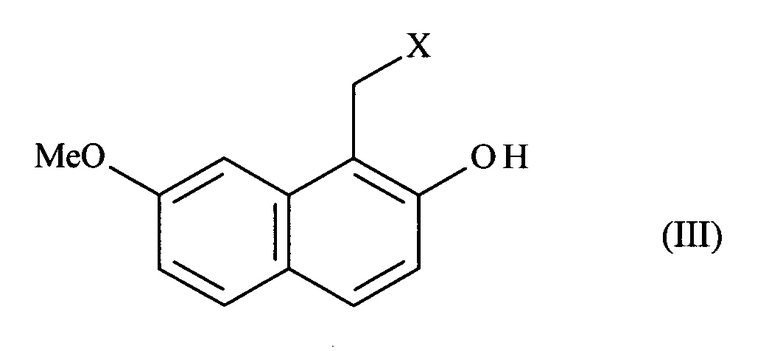
отличающемуся тем, что в реакцию вводят 7-метокси-нафталин-2-ол формулы (II):

в который в положении 1 соединения формулы (II) вводят группу -СН2-Х, в которой X представляет собой группу -N(CH3)2, -CO-N(CH2-Ph)2, -СН2-ОН, -СН=СН2 или -CO-NH2,
чтобы получить соединение формулы (III):

в которой X представляет собой -N(СН3)2, -CO-N(CH2-Ph)2, -СН2-ОН, -СН=СН2 или группу -CO-NH2;
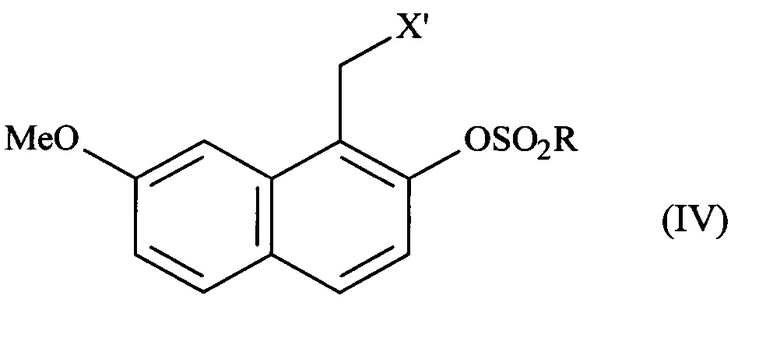
соединение формулы (III) подвергают реакции сульфонилирования по ароматическому спирту и заместитель X соединения формулы (III) является модифицированным, до или после стадии сульфонилирования ароматического спирта, при помощи обычных химических реакций с получением соединения формулы (IV):

в которой X' представляет собой -CN, -CO-NH2, -СН2-ОН, -CH2-N(CH2-Ph)2, -CH2-NH-CO-CH3, -СН(ОН)-СН2-ОН, -СНО или группу (2,5-диоксопирролидин-1-ил)метил и R представляет собой группу -СН3, -(СН2)2-СН3, -CF3 или толуил;
соединение формулы (IV) подвергают реакции дезоксигенирования в присутствии переходного металла и восстановителя с получением:
- или, если X' представляет собой группу -CH2-NH-CO-CH3, соединения формулы (I) непосредственно, которое выделяют в виде твердого вещества;
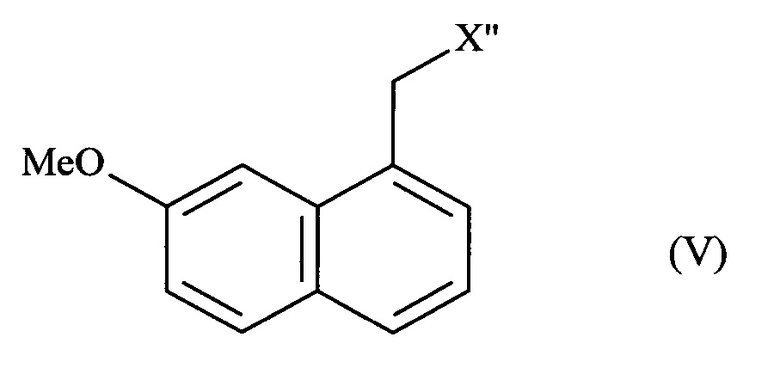
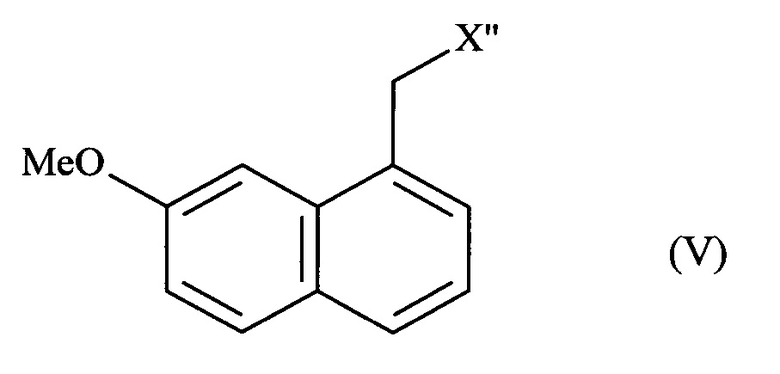
- или соединения формулы (V):
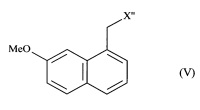
в которой X'' представляет собой группу -CN, -CH2-N(CH2-Ph)2, -СН2-ОН, -CO-NH2, -СН(ОН)-СН2-ОН или (2,5-диоксопирролидин-1-ил)метил;
затем соединение формулы (V) подвергают обычным химическим реакциям с получением соединения формулы (I), которое выделяют в виде твердого вещества.
Вариант способа промышленного синтеза состоит в том, что во время превращения соединения формулы (III) в соединение формулы (IV) группа X не модифицирована. Полученное соединение, сульфонилированное по его ароматическому спирту, затем подвергают реакции дезоксигенирования посредством действия переходного металла и восстановителя. Группу X впоследствии модифицируют при помощи обычных химических реакций с получением соединения формулы (I), которое выделяют в виде твердого вещества.
Соединение формулы (II) имеется в продаже или может быть легко получено специалистом в данной области техники, используя химические реакции, которые являются стандартными или описаны в литературных источниках.
В способе, предлагаемом в изобретении, превращение соединения формулы (II) в соединение формулы (III), в которой X представляет собой группу -N(CH3)2 осуществляют в соответствии с реакцией Манниха посредством действия формальдегида в присутствии диметиламина.
В способе, предлагаемом в изобретении, превращение соединения формулы (II) в соединение формулы (III), в которой X представляет собой группу -СН2-ОН, состоит в том, что соединение формулы (II) подвергают действию глиоксаля (или этан-1,2-диона), за которым следует действие восстановителя. Преимущественно, восстановитель представляет собой алюмогидрид лития, гидрид диизобутилалюминия, триэтилгидрид лития или диметилсульфид борана. Предпочтительно, восстановитель представляет собой алюмогидрид лития.
В способе, предлагаемом в изобретении, превращение соединения формулы (II) в соединение формулы (III), в которой X представляет собой группу -СО-NH2 или -CO-N(CH2-Ph)2 осуществляют посредством действия глиоксаля, за которым следует действие, в нагретой среде, соединения формулы NHR'R', в которой R' представляет собой Н или группу -СН2-Ph.
Указанную реакцию с глиоксалем, приводящую к образованию промежуточного лактона формулы (VI):

предпочтительно осуществляют в две стадии.
На первой стадии соединение формулы (II) растворяют в основной среде в присутствии глиоксаля. Предпочтительно основание представляет собой гидроксид натрия или гидроксид калия, и более конкретно гидроксид калия.
На второй стадии, промежуточный продукт, а именно 8-метокси-1,2-дигидронафто[2,1-б]фуран-1,2-диол, растворяют непосредственно в кислотной среде, предпочтительно хлористоводородной кислоте, с получением промежуточного лактона формулы (VI).
В способе, предлагаемом в изобретении, превращение соединения формулы (II) в соединение формулы (III), в которой X представляет собой группу -СН=СН2, осуществляют в соответствии с сигматропной перегруппировкой Клайзена посредством действия аллилбромида в основной среде, после чего следует термическая перегруппировка. Действие аллилбромида осуществляют в присутствии основания, такого как гидрид натрия, трет-бутилат калия, метилат натрия, гидроксид калия, гидроксид натрия, карбонат калия или карбонат натрия. Преимущественный вариант осуществления состоит в использовании карбоната калия в качестве основания в стадии, которая включает реакцию с аллилбромидом.
В способе, предлагаемом в изобретении, превращение соединения формулы (III) в соединение формулы (IV) состоит в том, что стадия сульфонилирования ароматического спирта сопровождается модификацией группы X при помощи обычных химических реакций, X определен как указано выше. В соответствии с другим преимущественным вариантом осуществления, превращение соединения формулы (III) в соединение формулы (IV) состоит в модификации группы X при помощи обычных химических реакций, после чего следует стадия сульфонилирования ароматического спирта, X определен как указано выше.
Указанную стадию сульфонилирования осуществляют посредством действия сульфонилхлорида, сульфонового ангидрида или сульфонимида. В соответствии с предпочтительным вариантом осуществления, стадию сульфонилирования осуществляют посредством действия тозилхлорида, н-пропилсульфонилхлорида, трифторметансульфонового ангидрида или фенилтрифлимида (или N,N-бис(трифторметилсульфонил)анилин).
В способе, предлагаемом в изобретении, превращение соединения формулы (IV) в соединение формулы (V) состоит из стадии дезоксигенирования в присутствии переходного металла и восстановителя.
Предпочтительно, переходный металл представляет собой никель, палладий или платину. Переходный металл может находиться или в виде соли, или в виде простого вещества. Предпочтительно, соль переходного металла представляет собой соль никеля или соль палладия, более предпочтительно соль никеля.
Преимущественно, восстановитель представляет собой или гидрид, такой как борогидрид натрия или алюмогидрид лития; или аминоборан, такой как диметиламин-боран; или алкоксисилан, такой как диметоксиметилсилан; или алкилсилан, такой как триэтилсилан; или щелочноземельный металл, такой как магний; или водород.
В соответствии с другим предпочтительным вариантом осуществления, превращение соединения формулы (IV) в соединение формулы (V) состоит из стадии дезоксигенирования в присутствии никеля, в частности соли никеля, и гидрида, предпочтительно борогидрида натрия.
В соответствии с другим предпочтительным вариантом осуществления, превращение соединения формулы (IV) в соединение формулы (V) состоит из стадии дезоксигенирования в присутствии палладия и водорода. Водород применяют непосредственно в его газообразном виде или его непрямо получают путем разложения формиата аммония.
В соответствии с другим предпочтительным вариантом осуществления, превращение соединения формулы (IV) в соединение формулы (V) состоит из стадии дезоксигенирования в присутствии палладия и щелочноземельного металла, предпочтительно магния.
В соответствии с другим предпочтительным вариантом осуществления, превращение соединения формулы (IV) в соединение формулы (V) состоит из стадии дезоксигенирования в присутствии переходного металла, восстановителя и лиганда. Лиганд предпочтительно представляет собой фосфиновый лиганд, и более конкретно трифенилфосфин.
В соответствии с конкретным вариантом осуществления, стадия дезоксигенирования соединения формулы (IV), в которой X' представляет собой группу -CH2-NH-CO-CH3, которую осуществляют:
- или в присутствии никеля, в частности соли никеля, и гидрида, предпочтительно борогидрида натрия;
- или в присутствии палладия и водорода;
- или в присутствии палладия и щелочноземельного металла; приводит непосредственно к образованию соединения формулы (I).
Этот способ является в особенности выгодным по нижеследующим причинам:
- обеспечивает возможность получения соединения формулы (I) в промышленных масштабах с хорошим выходом, исходя из простого и недорогого исходного вещества;
- дает возможность избежать реакции ароматизации - стадии, которая всегда является проблематичной с промышленной точки зрения - потому что в исходном субстрате присутствует нафталиновая кольцевая система;
- обеспечивает возможность получения агомелатина, исходя из 7-метокси-нафталин-2-ола с уменьшенным количеством стадий.
Соединения формул (III), (IV) и (VI), полученные в соответствии со способом, предлагаемым в изобретении, являются новыми и применимыми в качестве промежуточных соединений в синтезе агомелатина.
Соединения формулы (V), полученные в соответствии со способом, предлагаемым в изобретении, пригодны в качестве промежуточных соединений в синтезе агомелатина. Соединения формулы (V), полученные в соответствии со способом, предлагаемым в изобретении, являются новыми, за исключением (7-метоксинафталин-1-ил)ацетонитрила, N,N-дибензил-2-(7-метоксинафталин-1-ил)этанамида, 2-(7-метоксинафталин-1-ил)этанола и 2-(7-метоксинафталин-1-ил)ацетамида.
Предпочтительными соединениями формулы (III) являются следующие:
- 1-[(диметиламино)метил]-7-метоксинафталин-2-ол;
- N,N-дибензил-2-(2-гидрокси-7-метоксинафталин-1-ил)ацетамид;
- 1-(2-гидроксиэтил)-7-метоксинафталин-2-ол;
- 2-(2-гидрокси-7-метоксинафталин-1-ил)ацетамид;
- 7-метокси-1-(проп-2-ен-1-ил)нафталин-2-ол.
Предпочтительными соединениями формулы (IV) являются следующие:
- трифторметансульфонат 1-(цианометил)-7-метоксинафталин-2-ила;
- 4-метилбензолсульфонат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила;
- трифторметансульфонат 1-[2-(дибензиламино)этил]-7-метоксинафталин-2-ила;
- пропан-1-сульфонат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила;
- пропан-1-сульфонат 1-[2-(2,5-диоксопирролидин-1-ил)этил]-7-метоксинафталин-2-ила;
- 4-метилбензолсульфонат 1-(2-гидроксиэтил)-7-метоксинафталин-2-ила;
- 4-метилбензолсульфонат 1-(2-амино-2-оксоэтил)-7-метоксинафталин-2-ила;
- 4-метилбензолсульфонат 7-метокси-1-(2-оксоэтил)нафталин-2-ила;
- 4-метилбензолсульфонат 1-(2,3-дигидроксипропил)-7-метоксинафталин-2-ила.
(2-Гидрокси-7-метоксинафталин-1-ил)ацетонитрил, 4-метилбензолсульфонат 7-метокси-1-(2-{[(4-метилфенил)сульфонил]окси}этил)нафталин-2-ила, пропан-1-сульфонат 7-метокси-1-{2-[(пропилсульфонил)окси]этил}нафталин-2-ила и ацетат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила являются новыми и пригодны в качестве промежуточных соединений в синтезе агомелатина.
Приведенные ниже примеры демонстрируют изобретение, но никоим образом не ограничивают его.
Для того чтобы подтвердить правильность реакционных способов, промежуточные продукты синтеза были систематически выделены и охарактеризованы. Тем не менее, можно существенно оптимизировать процедуры путем ограничения количества выделенных промежуточных соединений.
Структуры описанных соединений были подтверждены при помощи стандартных спектроскопических методик: протонный ЯМР (s = синглет; bs = широкий синглет; d = дублет; t = триплет; dd = дублет дублетов; m = мультиплет); углеродный ЯМР (s = синглет; d = дублет; t = триплет; q = квадруплет); масс-спектрометрия с ионизацией электрораспылением (ESI).
Пример 1: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: 1-[(диметиламино)метил]-7-метоксинафталин-2-ол
К раствору 7-метокси-нафталин-2-ола (1.74 г; 10 ммоль) в этаноле (10 мл) при температуре окружающей среды добавляют диметиламин (40% в воде; 1.52 мл; 12 ммоль) и затем формальдегид (37% в воде; 0.78 мл; 10.5 ммоль). После перемешивания в течение одного часа, растворитель выпаривают. Полученный сырой продукт (количественный выход) применяют непосредственно в следующей стадии без дополнительной очистки.
1Н ЯМР спектроскопический анализ (ДМСО-d6, 300.13 МГц, δ в част. на млн.): 11.18 (bs, 1Н); 7.68 (d, J=8.8 Гц, 1Н); 7.6 (d, J=8.8 Гц, 1Н); 7.23 (d, J=2.3 Гц, 1Н); 6.93 (dd, J=8.8 и 2.3 Гц, 1H); 6.89 (d, J=8.8 Гц, 1Н); 3.96 (s, 2Н); 3.86 (s, 3Н); 2.3 (s, 6Н).
13С ЯМР спектроскопический анализ (ДМСО-d6, 75.5 МГц, δ в част. на млн.): 157.7 (s); 156.1 (s); 134.3 (s); 129.9 (d); 128.5 (d); 123.3 (s); 116.0 (d); 114.2 (d); 112.0 (s); 101.6 (d); 55.8 (t); 55.0 (q); 44.3 (2 x q).
Стадия В: (2-гидрокси-7-метоксинафталин-1-ил)ацетонитрил
Раствор продукта из стадии А выше (1.155 г; 5 ммоль) в диметилформамиде (5 мл) в присутствии цианида калия (390 мг; 6 ммоль) нагревают при 80°С в течение 30 часов. После разбавления с этилацетатом, добавляют 2М водный раствор HCl (5 мл). Смесь перемешивают и затем нейтрализуют посредством добавления раствора разбавленного NaHCO3. Две фазы разделяют и органическую фракцию промывают три раза рассолом, сушат над сульфатом натрия и фильтруют.После выпаривания растворителя получают сырой продукт, который затем очищают хроматографией на силикагелевой колонке (элюант : простой эфир/петролейный эфир 40/60) с получением целевого продукта.
1Н ЯМР спектроскопический анализ (ацетон-d6, 300.13 МГц, δ в част. на млн.): 9.25 (bs, 1Н, ОН); 7. 74 (d, J=9.1 Гц, 1Н); 7. 71 (d, J=8.9 Гц, 1Н); 7.31 (d, J=2.5 Гц, 1Н); 7.13 (d, J=8.9 Гц, 1Н); 7.03 (dd, J=9.1 и 2.5 Гц, 1Н); 4.21 (s, 2Н); 3.96 (s, 3Н).
13С ЯМР спектроскопический анализ (ацетон-d6, 75.5 МГц, δ в част. на млн.): 159.9 (s); 154.1 (s); 135.1 (s); 131.1 (d); 130.4 (d); 124.9 (s); 119.1 (s); 116.2 (d); 115. 7 (d); 108. 7 (s); 102.1 (d); 55.6 (q); 13.6 (t).
Стадия С: трифторметансульфонат 1-(цианометил)-7-метоксинафталин-2-ила
К раствору продукта из стадии В выше (120 мг; 0.52 ммоль) в дихлорметане (5 мл) добавляют N,N-бис(трифторметилсульфонил)анилин (204 мг; 0.571 ммоль) и триэтиламин (72 мкл; 0.52 ммоль). После перемешивания в течение 16 часов, растворители выпаривают, и остаток очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир, градиент от 10/90 до 20/80) с получением указанного в заголовке продукта (125 мг; 70%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.85-7.78 (m, 2Н); 7.3-7.22 (m, 3Н); 4.13 (s, 2Н); 3.98 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 159.9 (s); 145.6 (s); 133.2 (s); 131.2 (d); 130.8 (d); 128.0 (s); 125.0 (s); 120.3 (d); 118.6 (s, J=318 Гц); 116.9 (s); 116.8 (d); 116.0 (s); 102.3 (d); 55.6 (q); 15.2 (t). Масс-спектрометрия (ESI; m/z(%)): 345(45) [M]+•; 212(100); 184(15); 169(34); 140(18).
Стадия D: (7-метоксинафталин-1-ил)ацетонитрил
Раствор продукта из стадии С выше (73 мг; 0.212 ммоль) в абсолютном этаноле (4 мл) в присутствии палладия 10% на угле (4.5 мг; 0.004 ммоль) и триэтиламина (150 мкл) гидрируют (4 бар) при температуре окружающей среды в течение 20 часов. После фильтрации через целит, промывания этилацетатом и выпаривания растворителей, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 15/85) с получением указанного в заголовке продукта (28 мг; 67%).
Точка плавления: 86-87°С.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.78 (d, J=8.9 Гц, 1Н); 7.77 (d, J=7.8 Гц, 1Н); 7.52 (d, J=7.1 Гц, 1Н); 7.32 (dd, J=7.8 и 7.1 Гц, 1Н); 7.21 (dd, J=8.9 и 2.4 Гц, 1Н); 7.03 (d, J=2.4 Гц, 1Н); 4.0 (s, 2Н); 3.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.5 (s); 132.0 (s); 130.6 (d); 129.1 (s); 128.8 (d); 127.1 (d); 124.4 (s); 123.2 (d); 118.8 (d); 117.7 (s); 101.3 (d); 55.4 (q); 21.9 (t).
Стадия Е: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
136 г никеля Ренея, 2.06 л этанола и 0.23 л воды вводят в 8-литровый реактор. В ходе перемешивания при 70°С и под 30 бар водорода медленно добавляют соединение, полученное в стадии D выше (0.8 кг), растворенное в уксусном ангидриде (2.4 л). После завершения добавления реакционную смесь перемешивают в течение 1 часа под атмосферой водорода при 30 бар, и затем в реакторе сбрасывают давление, а жидкости фильтруют. После концентрирования смеси остаток кристаллизуют из смеси этанол/вода 35/65 с получением указанного в заголовке продукта с выходом в 89% и с химической чистотой более чем 99%.
Точка плавления: 108°С.
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1H); 7.74 (d, J=8.9 Гц, 1Н); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1H); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1Н); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 2: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: 4-метилбензолсульфонат 1-[(диметиламино)метил]-7-метоксинафталин-2-ила
К раствору продукта, полученного в стадии А примера 1 (2.042 г; 8.84 ммоль) в диметилформамиде (8 мл) добавляют трет-бутппат калия (1.091 г; 9.724 ммоль) и затем, через 5 минут, тозилхлорид (1.684 г; 8.84 ммоль). После перемешивания в течение 6 часов, раствор разбавляют с этилацетататом и два раза промывают водой, а затем рассолом. Органическую фазу высушивают над сульфатом натрия, и фильтруют, а растворитель выпаривают. Полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир, градиент от 30/70 до 70/30) с получением целевого продукта в виде твердого вещества (1.94 г; 57%).
Точка плавления: 115-118°С
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.78 (d, J=8.1 Гц, 2Н); 7.7 (d, J=9.0 Гц, 1Н); 7.62 (d, J=8.8 Гц, 1Н); 7.53 (d, J=2.3 Гц, 1Н); 7.34 (d, J=8.1 Гц, 2Н); 7.14 (dd, J=9.0 и 2.3 Гц, 1Н); 6.96 (d, J=8.8 Гц, 1Н); 3.93 (s, 3Н); 3.67 (s, 2Н); 2.47 (s, 3Н); 2.24 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.4 (s); 146.9 (s); 145.5 (s); 135.1 (s); 133.2 (s); 130.0 (2 x d); 129.9 (d); 129.1 (d); 128.6 (2 x d); 127.8 (s); 125.7 (s); 118.5 (d); 117.9 (d); 104.5 (d); 55.3 (q); 54.2 (t); 45.6 (2 x q); 21.9 (q).
Стадия В: 4-метилбензолсульфонат 1-(цианометил)-7-метоксинафталин-2-ила
Йодметан (267 мкл; 4.29 ммоль) добавляют к раствору продукта из стадии А выше (1.5 г; 9.9 ммоль) в диметилформамиде (8 мл). После перемешивания в течение 4 часов при температуре окружающей среды добавляют цианид калия (304 мг; 4.68 ммоль). Раствор перемешивают в течение еще 16 часов и затем разбавляют с этилацетататом, промывают водой и три раза рассолом, сушат над сульфатом натрия и фильтруют.Выпаривание растворителя обеспечивает сырой продукт, который затем очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 40/60) с получением целевого продукта (1.276 г; 89%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.77 (d, J=8.3 Гц, 2Н); 7.75 (d, J=8.9 Гц, 1Н); 7.69 (d, J=8.9 Гц, 1Н); 7.35 (d, J=8.3 Гц, 2Н); 7.22-7.15 (m, 2Н); 7.01 (d, J=8.9 Гц, 1Н); 3.98 (s, 2Н); 3.95 (s, 3Н); 2.45 (s, 3Н).
Стадия С: 4-метилбензолсульфонат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила
Продукт из стадии В выше (54 мг; 0.147 ммоль), хлорид никеля гексагидрат (35 мг; 0.147 ммоль), дихлорметан (1.5 мл) и метанол (1.5 мл) вводят в герметичную колбу. Затем аргон кипятят в растворе в течение 5 минут и после этого осторожно, маленькими порциями добавляют борогидрид натрия (100 мг; 2.94 ммоль). После перемешивания в течение 30 минут под аргоном и при температуре окружающей среды добавляют воду. После перемешивания в течение 15 минут, смесь фильтруют через целит, и затем промывают дихлорметаном. Органическую фракцию высушивают над сульфатом натрия и затем фильтруют. После выпаривания растворителей, полученный сырой продукт помещают в присутствии уксусного ангидрида (1 мл) и ацетата натрия (50 мг) при перемешивании при температуре окружающей среды в течение 30 минут. Смесь выливают в разбавленный раствор Na2CO3, и продукт экстрагируют три раза при помощи этилацетата. Органические фракции промывают рассолом, сушат над сульфатом натрия и фильтруют. Выпаривание растворителя обеспечивает сырой продукт, который затем очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением целевого продукта в виде твердого вещества (24 мг; 40%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.81 (d, J=8.2 Гц, 2Н); 7.68 (d, J=8.9 Гц, 1Н); 7.62 (d, J=2.4 Гц, 1Н); 7.55 (d, J=8.9 Гц, 1Н); 7.36 (d, J=8.2 Гц, 2Н); 7.14 (dd, J=8.9 и 2.4 Гц, 1Н); 6.89 (d, J=8.9 Гц, 1Н); 5.97 (m, 1H); 4.01 (s, 3Н); 3.53-3.46 (m, 2Н); 3.22-3.17 (m, 2Н); 2.46 (s, 3Н); 1.95 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 170.9 (s); 158.9 (s); 146.4 (s); 145.7 (s); 134.6 (s); 133.3 (s); 130.1 (2 x d); 130.0 (d); 128.5 (2 x d); 128.2 (d); 127. 7 (s); 126.2 (s); 119.2 (d); 117.8 (d); 103.3 (d); 55.8 (q); 39.4 (t); 26.4 (t); 23.4 (q); 21.9 (q).
Стадия D: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид Продукт из стадии С выше (83 мг; 0.2 ммоль), хлорид никеля (26 мг; 0.2 ммоль) и метанол (4 мл) вводят в герметичную колбу. Затем барботируют аргоном в растворе в течение 5 минут, и затем маленькими порциями осторожно добавляют натрия борогидрид (136 мг; 4 ммоль). После перемешивания в течение 30 минут под аргоном и при температуре окружающей среды, смесь фильтруют через целит, затем ее промывают при помощи этилацетата и растворители выпаривают. Полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением целевого продукта (39 мг; 80%).
1H ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1H); 7.74 (d, J=8.9 Гц, 1H); 7.65 (d, J=8.0 Гц, 1H); 7.52 (d, J=2.5 Гц, 1H); 7.31-7.2 (m, 2H); 7.11 (dd, J=8.9 и 2.5 Гц, 1H); 3.96 (s, 3H); 3.52-3.44 (m, 2H); 3.23-3.18 (m, 2H); 1.94 (s, 3H).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 3: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: 8-метоксинафто[2,1-b]фуран-2(1H)-он
Водный раствор 85% гидроксида калия (2.76 г; 40 ммоль) и 7-метокси-нафталин-2-ола (6.96 г; 40 ммоль) в воде (80 мл) добавляют по каплям при температуре окружающей среды к раствору глиоксаля (40% в воде; 28 мл; 240 ммоль). После перемешивания в течение 3 часов, осадок белого цвета собирают фильтрацией и промывают водой. Полученный твердый (8-метокси-1,2-дигидронафто[2,1-6]фуран-1,2-диол) растворяют в 1,2-дихлорэтан (160 мл) и затем добавляют водный раствор 3М HCl (300 мл). Гетерогенную смесь нагревают при 50°С с интенсивным перемешиванием. Через 1,5 часа все твердые вещества растворяют и две фазы разделяют. Органическую фазу собирают и растворители выпаривают. Сырой продукт сушат азеотропной перегонкой с толуолом с получением указанного в заголовке продукта (8.69 г), который будут использовать непосредственно в следующей стадии без дополнительной очистки.
1Н ЯМР спектроскопический анализ (ДМСО-d6, 300.13 МГц, δ в част. на млн.): 7.88 (d, J=8.8 Гц, 1Н); 7.85 (d, J=8.6 Гц, 1Н); 7.28 (d, J=8.8 Гц, 1Н); 7.12-7.06 (m, 2Н); 4.15 (s, 2Н); 3.89 (s, 3Н).
Стадия В: N,N-дибензил-2-(2-гидрокси-7-метоксинафталин-1-ил)ацетамид
Продукт из стадии А выше (1.976 г; 9.23 ммоль) и дибензиламин (4 мл; 20.3 ммоль) вводят в колбу и затем нагревают при 120°С в течение 2 часов. После охлаждения остаток разбавляют с этилацетататом (200 мл). Добавление водного раствора 2М HCl дает в осадке гидрохлоридную соль дибензиламина, которую затем фильтруют через целит. Органическую фазу промывают водным раствором 2М HCl, затем промывают рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителей, полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 30/70) с получением целевого продукта в виде твердого вещества (2.88 г; 76%).
Точка плавления: 155-157°С
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 9.85 (bs, 1Н); 7.66 (d, J=8.7 Гц, 1Н); 7.62 (d, J=8.7 Гц, 1Н); 7.4-7.24 (m, 6Н); 7.18-7.14 (m, 4Н); 7.08 (d, J=8.7 Гц, 1Н); 6.95-6.91 (m, 2Н); 4.72 (s, 2Н); 4.64 (s, 2Н); 4.21 (s, 2Н); 3.55 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 174.6 (s); 158.6 (s); 155.8 (s); 136.4 (s); 135.5 (s); 134.1 (s); 130.6 (d); 129.3 (d); 129.2 (2 x d); 128.9 (2 x d); 128.4 (2 x d); 128.1 (d); 127.8 (d); 126.4 (2 x d); 124.6 (s); 117.5 (d); 114.9 (d); 111.5 (s); 101.2 (d); 55.9 (q); 50.8 (t); 49.0 (t); 31.5 (t).
Стадия С: 1-[2-(дибензиламино)этил]-7-метоксинафталин-2-ол
1M раствор комплекса боран-тетрагидрофуран в тетрагидрофуране (1.7 мл; 1.7 ммоль) добавляют к раствору продукта из стадии В выше (230 мг; 0.56 ммоль) в тетрагидрофуране (10 мл). Смесь нагревают с обратным холодильником в течение 2 часов, и после чего добавляют водный раствор 2М HCl (10 мл). После перемешивания в течение ночи добавляют насыщенный раствор NaHCO3 для достижения нейтрального рН, и продукт экстрагируют три раза при помощи этилацетата. Органические фракции сушат над сульфатом натрия и фильтруют, а растворитель выпаривают. Полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 30/70) с получением целевого продукта (150 мг; 72%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 12.09 (bs, 1Н); 7.69 (d, J=8.9 Гц, 1Н); 7.61 (d, J=8.8 Гц, 1Н); 7.44-7.26 (m, 10Н); 7.14 (d, J=8.8 Гц, 1Н); 7.12 (d, J=2.4 Гц, 1Н); 7.0 (dd, J=8.8 и 2.4 Гц, 1Н); 3.93 (s, 3Н); 3.79 (s, 4Н); 3.21-3.18 (m, 2Н); 3.01-2.98 (m, 2Н).
Стадия D: трифторметансульфонат 1-[2-(дибензиламино)этил]-7-метоксинафталин-2-ила
Трифторметансульфоновый ангидрид (135 мкл; 0.801 ммоль) добавляют при 0°С к раствору продукта из стадии С выше (303 мг; 0.763 ммоль) в дихлорметане (10 мл). После перемешивания в течение 2 часов при этой температуре, растворитель выпаривают. Остаток ресуспендируют в смеси из диэтиловый простой эфир/водный полунасыщенный раствор NaHCO3. После разделения, органическую фазу высушивают над сульфатом натрия и фильтруют. После выпаривания растворителей, полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 10/90) с получением целевого продукта (306 мг; 76%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.73 (d, J=8.9 Гц, 1Н); 7.69 (d, J=8.9 Гц, 1Н); 7.44-7.4 (m, 4Н); 7.35-7.22 (m, 7Н); 7.15 (dd, J=8.9 и 2.3 Гц, 1Н); 6.99 (d, J=2.3 Гц, 1Н); 3.78 (s, 4Н); 3.6 (s, 3Н); 3.43-3.37 (m, 2Н); 2.91-2.86 (m, 2Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.9 (s); 146.1 (s); 139.8 (2 х s); 134.3 (s); 130.3 (d); 128.7 (4 x d); 128.6 (d); 128.3 (4 x d); 128.1 (s); 127.4 (s); 127.0 (2 x d); 119.5 (d); 118.7 (s, JC-F = 318 Гц); 116.8 (d); 102.8 (d); 58.3 (2 x t); 55.3 (q); 52.5 (t); 24.6 (t).
Стадия E: N,N-дибензил-2-(7-метоксинафталин-1-ил)этанамид
Продукт из стадии D выше (130 мг; 0.25 ммоль), ацетат палладия (5.6 мг; 0.025 ммоль), трифенилфосфин (20 мг; 0.075 ммоль), формиат аммония (142 мг; 2.25 ммоль) и диметилформамид (1 мл) вводят в колбу. После перемешивания в течение 16 часов при 60°С, раствор разбавляют с этилацетататом, промывают водой, два раза промывают рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителей, полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 10/90) с получением указанного в заголовке продукта в виде твердого вещества белого цвета (93 мг; 98%).
Точка плавления: 92-93°С.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.74 (d, J=8.9 Гц, 1Н); 7.65 (m, 1Н); 7.43-7.39 (m, 4Н); 7.35-7.22 (m, 8Н); 7.11 (dd, J=8.9 и 2.4 Гц, 1Н); 7.05 (d, J=2.4 Гц, 1H); 3. 76 (s, 4Н); 3.67 (s, 3Н); 3.29-3.23 (m, 2Н); 2.92-2.86 (m, 2Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 157.6 (s); 139.9 (2 х s); 135.3 (s); 133.2 (s); 130.3 (d); 129.3 (s); 128.7 (4 x d); 128.3 (4 x d); 127.3 (d); 127.0 (2 x d); 126.5 (d); 123.3(d); 118.2 (d); 102.2 (d); 58.6 (2 t); 55.2 (q); 54.2 (t); 31.5 (t).
Стадия F: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Продукт из стадии Е выше (66 мг; 0.173 ммоль), гидроксид палладия на угле (20% Pd, 60% влажность; 10 мг), этанол (2 мл) и этилацетат (2 мл) вводят в колбу, помещенную в автоклав. Автоклав заполняют водородом под давлением (5 бар), и смесь перемешивают в течение 30 часов. Раствор затем фильтруют через целит и промывают этанолом, а растворители выпаривают. К сырому реакционному продукту добавляют уксусный ангидрид (500 мкл) и ацетат натрия (100 мг). После перемешивания в течение 4 часов, смесь разбавляют с этилацетататом. Органическую фазу промывают два раза водным раствором 2М гидроксида натрия и рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителей, целевой продукт получают в чистом виде (41 мг; 98%).
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1Н); 7.74 (d, J=8.9 Гц, 1Н); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1Н); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1Н); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 4: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия A: 1-(2-гидроксиэтил)-7-метоксинафталин-2-ол
Продукт, полученный в стадии А примера 3 (8.96 г; 40 ммоль) растворяют в тетрагидрофуране (160 мл), и затем добавляют алюмогидрид лития (1.52; 40 ммоль) порциями при 0°С под потоком азота. Смесь перемешивают в течение 16 часов при температуре окружающей среды и затем реакцию останавливают при 0°С посредством добавления этилацетата, а потом и воды. Добавляют водный 1М раствор серной кислоты (80 мл). После перемешивания в течение одного часа, гетерогенную смесь фильтруют через целит, и промывают при помощи этилацетата. После сцеживания и разделения, органическую фазу промывают водой, а затем рассолом. Раствор снова фильтруют через тонкий слой кремнезема, и растворители выпаривают с получением указанного в заголовке продукта без дополнительной очистки (8.21 г; 94% исходя из 7-метокси-нафталин-2-ола).
1Н ЯМР спектроскопический анализ (ДМСО-d6, 300.13 МГц, δ в част. на млн.): 7.6 (d, J=8.8 Гц, 1Н); 7.49 (d, J=8.8 Гц, 1Н); 7.25 (d, J=2.4 Гц, 1Н); 6.94 (d, J=8.8 Гц, 1Н); 6.9 (dd, J=8.8 и 2.4 Гц, 1Н); 3.9 (s, 3Н); 3.79-3.73 (m, 2Н); 3.31-3.25 (m, 2Н).
Стадия В: 4-метилбензолсульфонат 7-метокси-1-(2-{[(4-метилфенил)сульфонил]окси}этил)нафталин-2-ила
Продукт из стадии А выше (436 мг; 2 ммоль), дихлорметан (10 мл), триэтиламин (670 мкл; 4.8 ммоль) и 4-диметиламинопиридин (12 мг; 0.1 ммоль) вводят в колбу. После охлаждения до 0°С, тозилхлорид (800 мг; 4 ммоль) добавляют за один раз, и раствор перемешивают в течение 2 часов при 0°С и затем в течение 14 часов при температуре окружающей среды. После выпаривание растворителя, остаток ресуспендируют в этилацетате и воде. Органическую фракцию промывают водой и рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир, градиент от 20/80 до 30/70) с получением указанного в заголовке продукта (728 мг; 69%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.78 (d, J=8.3 Гц, 2Н); 7.69 (d, J=9.1 Гц, 1Н); 7.66 (d, J=8.3 Гц, 2Н); 7.59 (d, J=8.9 Гц, 1Н); 7.35 (d, J=8.3 Гц, 2Н); 7.26 (d, J=2.4 Гц, 1Н); 7.23 (d, J=8.3 Гц, 2Н); 7.15 (dd, J=8.9 и 2.4 Гц, 1Н); 7.03 (d, J=9.1 Гц, 1Н); 4.2 (t, J=7.7 Гц, 2Н); 3.94 (s, 3Н); 3.34 (d, J=7.7 Гц, 2Н); 2.47 (s, 3Н); 2.39 (s, 3Н).
Стадия С: 4-метилбепзолсулъфонат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила
Продукт из стадии В выше (124 мг; 0.236 ммоль), ацетонитрил (1 мл) и 35% водный раствор аммиака (1 мл) вводят в герметичную колбу. Колбу помещают в баню, нагретую при 110°С, и раствор перемешивают в течение 3.5 часов. После охлаждения, раствор разбавляют с этилацетататом, промывают разбавленным раствором NaHCO3, промывают рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителей, сырой продукт растворяют в уксусном ангидриде (1 мл), и после чего добавляют ацетат натрия (300 мг). После перемешивания в течение 14 часов, смесь выливают в разбавленный раствор NaHCO3. После перемешивания в течение 30 минут, продукт экстрагируют три раза при помощи этилацетата. Органическую фазу промывают водой и рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителей, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением указанного в заголовке продукта (84 мг; 86%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.81 (d, J=8.2 Гц, 2Н); 7.68 (d, J=8.9 Гц, 1Н); 7.62 (d, J=2.4 Гц, 1Н); 7.55 (d, J=8.9 Гц, 1Н); 7.36 (d, J=8.2 Гц, 2Н); 7.14 (dd, J=8.9 и 2.4 Гц, 1Н); 6.89 (d, 8.9 Гц, 1H); 5.97 (m, 1Н); 4.01 (s, 3Н); 3.53-3.46 (m, 2Н); 3.22-3.17 (m, 2Н); 2.46 (s, 3Н); 1.95 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 170.9 (s); 158.9 (s); 146.4 (s); 145. 7 (s); 134.6 (s); 133.3 (s); 130.1 (2 x d); 130.0 (d); 128.5 (2 x d); 128.2 (d); 127.7 (s); 126.2 (s); 119.2 (d); 117.8 (d); 103.3 (d); 55.8 (q); 39.4 (t); 26.4 (t); 23.4 (q); 21.9 (q).
Стадия D: N-[2-(7-метоксинафталин-1-ил)этил)ацетамид
Указанный в заголовке продукт получают в соответствии со способом, описанным в стадии D примера 2, используя продукт из стадии С выше в качестве исходного реагента.
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1Н); 7.74 (d, J=8.9 Гц, 1H); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1H); 7.31-7.20 (m, 2H); 7.11 (dd, J=8.9 и 2.5 Гц, 1H); 3.96 (s, 3H); 3.52-3.44 (m, 2H); 3.23-3.18 (m, 2H); 1.94 (s, 3H).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 5: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: пропан-1-сульфонат 7-метокси-1-{2-[(пропилсулъфонил)окси]этил}нафталин-2-ила
Продукт, полученный в стадии А примера 4 (2.45 г; 11.238 ммоль), растворяют в дихлорметане (60 мл), и после чего добавляют триэтиламин (3.7 мл; 26.4 ммоль). После охлаждения до 0°С, по каплям добавляют n-пропилсульфонилхлорид (2.8 мл; 24.6 ммоль). После перемешивания в течение 2 часов при температуре окружающей среды, растворитель выпаривают. Полученный остаток ресуспендируют в простом диэтиловом эфире и воде. После разделения, органическую фазу промывают разбавленным водным раствором HCl, водой и рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 20/80) с получением указанного в заголовке продукта в виде твердого вещества (3.335 г; 66% в 3 стадии, исходя из 7-метокси-нафталин-2-ола).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.72 (m, 2Н); 7.37 (d, J=2.4 Гц, 1Н); 7.31 (d, J=9.0 Гц, 1Н); 7.17 (dd, J=9.0 и 2.4 Гц, 1Н); 4.48 (t, J=8.0 Гц, 2Н); 3.96 (s, 3Н); 3.6 (t, J=8.0 Гц, 2Н); 3.45-3.4 (m, 2Н); 3.04-2.98 (m, 2Н); 2.1 (m, 2Н); 1.8 (m, 2Н); 1.17 (t, J=7.3 Гц, 3Н); 0.99 (t, J=7.3 Гц, 3Н).
Стадия В: пропан-1-сульфонат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила
Продукт из стадии А выше (532 мг; 1.237 ммоль), ацетонитрил (18 мл) и водный 35% раствор аммиака (18 мл) вводят в герметичную колбу. Затем смесь помещают в баню, нагретую при 110°С в течение 3 часов. После этого растворители выпаривают под сниженным давлением, осуществляют азеотропную перегонку с этанолом. Сырой продукт растворяют в уксусном ангидриде (5 мл) и впоследствии добавляют ацетат натрия (500 мг). После перемешивания в течение одного часа, раствор разбавляют с этилацетататом и затем осторожно выливают в насыщенный водный раствор NaHCO3. После перемешивания в течение 15 минут две фазы разделяют и водную фазу экстрагируют два раза этилацетататом. Органические фракции объединяют, промывают водой и затем рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением указанного в заголовке продукта (289 мг; 64%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.73 (d, J=8.9 Гц, 1Н); 7.68 (d, J=8.9 Гц, 1Н); 7.62 (d, J=2.5 Гц, 1Н); 7.27 (d, J=8.9 Гц, 1Н); 7.16 (dd, J=8.9 и 2.5 Гц, 1Н); 5.95 (m, 1Н); 4.02 (s, 3Н); 3.62-3.54 (m, 2Н); 3.43-3.32 (m, 2Н); 2.09 (m, 2Н); 1.17 (t, J=7.4 Гц, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 170.9 (s); 159.0 (s); 145.6 (s); 134.7 (s); 130.2 (d); 128.6 (d); 127.7 (s); 126.0 (s); 119.2 (d); 118.1 (d); 103.3 (d); 55.9 (q); 53.6 (t); 39.4 (t); 26.5 (t); 23.4 (q); 17.6 (t); 13.1 (q).
Стадия С: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Продукт из стадии В выше (327 мг; 0.896 ммоль), ацетат аммония (1.19 г; 15.54 ммоль), палладий 10% на угле (95 мг; 0.09 ммоль), этанол (3 мл) и порошковый магний (83 мг; 3.584 ммоль) вводят в колбу. После перемешивания в течение 16 часов, гетерогенную смесь фильтруют через целит, и промывают этилацетататом. После выпаривания растворителей, полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением указанного в заголовке продукта (190 мг; 87%).
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1Н); 7.74 (d, J=8.9 Гц, 1H); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1Н); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1Н); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 6: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: пропан-1-сульфонат 1-[2-(2,5-диоксопирролидин-1-ил)этил]-7-метоксинафталин-2-ила
Продукт, полученный в стадии А примера 5 (240 мг; 0.558 ммоль), калия карбонат (231 мг; 1.674 ммоль), сукцинимид (66 мг; 0.67 ммоль) и диметилформамид (2 мл) вводят в колбу. После перемешивания в течение 16 часов при 100°С, раствор разбавляют с этилацетататом, промывают разбавленным водным раствором HCl, водой и рассолом и затем сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 70/30) с получением указанного в заголовке продукта (123 мг; 57%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.74 (d, J=9.0 Гц, 1Н); 7.7 (d, J=8.9 Гц, 1Н); 7.6 (d, J=2.4 Гц, 1Н); 7.38 (d, J=9.0 Гц, 1Н); 7.16 (dd, J=8.9 и 2.4 Гц, 1H); 4.05 (s, 3Н); 3.86-3.8 (m, 2Н); 3.58-3.52 (m, 2Н); 3.33-3.27 (m, 2Н); 2.72 (s, 4Н); 2.14 (m, 2Н); 1.2 (t, J=7.5 Гц, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 177.3 (2 х s); 159.1 (s); 145.3 (s); 134.4 (s); 130.4 (d); 128.8 (d); 127.7 (s); 124.2 (s); 119.2 (d); 118.4 (d); 102.4 (d); 55.8 (q); 53.5 (t); 37.7 (t); 28.4 (2 x t); 24. 7 (t); 17.5 (t); 13.1 (q).
Стадия В: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Продукт из стадии А выше (65 мг; 0.16 ммоль), ацетат аммония (240 мг; 3.12 ммоль), палладий 10% на угле (16.5 мг; 0.016 ммоль), метанол (0.7 мл) и порошковый магний (8 мг; 0.36 ммоль) одной порцией вводят в колбу. После перемешивания в течение 12 часов при 30°С, гетерогенную смесь фильтруют через целит, и затем промывают этилацетататом. После выпаривания растворителей, сырой продукт растворяют в этаноле (2 мл) в герметично закрытой пробирке, и затем добавляют водный раствор гидроксида натрия (180 мг; 4.5 ммоль в 1 мл воды). После этого смесь разбавляют с уксусным ангидрид (5 мл), и вводят ацетат натрия (500 мг). После перемешивания в течение одного часа, раствор разбавляют с этилацетататом и затем осторожно выливают в насыщенный водный раствор NaHCO3. После перемешивания в течение 15 минут, Две фазы разделяют и водную фазу экстрагируют два раза с этилацетататом. Органические фракции объединяют, промывают водой и рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением указанного в заголовке продукта (14.4 мг; 37%).
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1H); 7.74 (d, J=8.9 Гц, 1Н); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1Н); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1Н); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 7: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: 4-метилбензолсульфонат 1-(2-гидроксиэтил)-7-метоксинафталин-2-ила
К раствору продукта, полученного в стадии А примера 4 (109 мг; 0.5 ммоль) в тетрагидрофуране (2.5 мл) добавляют при 0°С трет-бутилат калия (56.4 мг; 0.5 ммоль) и затем, через 5 минут, тозилхлорид (95 мг; 0.5 ммоль). После перемешивания в течение 3 часов, раствору дают достичь температуры окружающей среды и перемешивают в течение еще 15 часов. Растворитель выпаривают, и полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 50/50) с получением смеси из двух диастереоизомеров (116 мг; 62%) в соотношении 88:12. Смесь применяют как таковую без дополнительного очищения.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.83 (d, J=8.2 Гц, 2Н); 7.71 (d, J=9.1 Гц, 1Н); 7.59 (d, J=8.9 Гц, 1Н); 7.35 (d, J=8.2 Гц, 2Н); 7.29 (d, J=2.4 Гц, 1Н); 7.15 (dd, J=8.9 и 2.4 Гц, 1H); 7.04 (d, J=9.1 Гц, 1H); 3.91 (s, 3Н); 3.87 (t, J=7.1 Гц, 2Н); 3.23 (t, J=7.1 Гц, 2Н); 2.46 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.6 (s); 146.7 (s); 145.7 (s); 134.5 (s); 133.3 (s); 130.3 (d); 130.1 (2 x d); 128.5 (2 x d); 128.2 (d); 127.8 (s); 125.7 (s); 118.5 (d); 118.3 (d); 103.4 (d); 62.1 (t); 55.5 (q); 29.7 (t); 21.9 (q).
Стадия В: 2-(7-метоксинафталин-1-ил)этанол
Смесь из диастереоизомеров, полученную в стадии А выше (80 мг; 0.176 ммоль соответствующего изомера), хлорид никеля гексагидрат (51 мг; 0.215 ммоль), дихлорметан (2 мл) и метанол (2 мл) вводят в герметичную колбу. Затем барботируют аргоном в растворе в течение 5 минут, и после этого маленькими порциями осторожно добавляют борогидрид натрия (146 мг; 4.3 ммоль). После перемешивания в течение одного часа при температуре окружающей среды под аргоном, добавляют разбавленный водный раствор HCl. После перемешивания в течение 4 часов, смесь фильтруют через целит и промывают этанолом. Растворитель выпаривают, и полученный сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 40/60) с получением указанного в заголовке продукта (22 мг; 62%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.72 (d, J=9.1 Гц, 1Н); 7.63 (d, J=8.1 Гц, 1Н); 7.3-7.21 (m, 3Н); 7.13 (dd, J=9.1 и 2.6 Гц, 1H); 3.93 (t, J=6.7 Гц, 2Н); 3.88 (s, 3Н); 3.24 (d, J=6.7 Гц, 2Н); 1.99 (bs, 1Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 157.8 (s); 133.2 (s); 133.0 (s); 130.4 (d); 129.4 (s); 127.7 (d); 127.1 (d); 123.3 (d); 118.1 (d); 102.5 (d); 62.7 (t); 55.4 (q); 36.4 (t).
Масс-спектрометрия (ESI; m/z(%)): 202(29) [M]+•; 171(100).
Стадия С: метансульфонат 2-(7-метоксинафталин-1-ил)этила
К раствору продукта из стадии В выше (275 мг; 1.361 ммоль) в дихлорметане (7 мл) добавляют при 0°С триэтиламин (227 мкл; 1.633 ммоль) и мезилсульфонилхлорид (116 мкл; 1.498 ммоль). После перемешивания в течение одного часа, растворители выпаривают и остаток ресуспендируют в простом диэтиловом эфире и воде. После разделения, органическую фазу промывают три раза водой и рассолом, сушат над сульфатом натрия и фильтруют. Выпаривание растворителя обеспечивает чистый целевой продукт без любой дополнительной очистки (356 мг; 93%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.72 (d, J=9.1 Гц, 1Н); 7.66 (d, J=8.9 Гц, 1Н); 7.31-7.21 (m, 3Н); 7.13 (dd, J=9.1 и 2.5 Гц, 1Н); 4.47 (t, J=7.4 Гц, 2Н); 3.92 (s, 3Н); 3.45 (d, J=7.4 Гц, 2Н); 2. 77 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.2 (s); 133.0 (s); 133.0 (s); 130.6 (s); 130.4 (d); 129.3 (s); 128.0 (d); 127.7 (d); 123.2 (d); 118.4 (d); 101.9 (d); 67.9 (t); 55.5 (q); 37.4 (q); 33.3 (t).
Стадия D: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Продукт из стадии С выше (356 мг; 1.271 ммоль), ацетонитрил (7 мл) и водный 35% раствор аммиака (5 мл) вводят в колбу. Колбу помещают в баню, нагретую до 110°С, и раствор перемешивают в течение 3 часов. Раствор разбавляют с этилацетататом и промывают водным 2М раствором гидроксида натрия, водой и рассолом, и затем сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, сырой продукт растворяют в уксусном ангидриде (2 мл) в присутствии ацетата натрия (500 мг). После перемешивания в течение одного часа добавляют воду и этилацетат и после перемешивания в течение 15 минут, органическую фазу промывают два раза водным 2М раствором гидроксида натрия, промывают водой и рассолом, сушат над сульфатом натрия и фильтруют. Растворитель выпаривают, и полученный сырой продукт затем очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением указанного в заголовке продукта (230 мг; 74%).
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1Н); 7.74 (d, J=8.9 Гц, 1Н); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1Н); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1Н); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 8: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: 2-(2-гидрокси-7-метоксинафталин-1-ил)ацетамид
7N метанольный раствор аммиака (10 мл; 70 ммоль) и продукт, полученный в стадии А примера 3 (1.5 г; 7 ммоль) вводят в герметичную колбу. После перемешивания в течение 18 часов при 100°С, растворитель выпаривают с получением сырого продукта, указанного в заголовке (1.58 г; 98%), который применяют непосредственно без всякой дополнительной очистки.
1Н ЯМР спектроскопический анализ (ДМСО-d6, 300.13 МГц, δ в част. на млн.): 9.87 (bs, 1H); 7.68 (d, J=9.0 Гц, 1Н); 7.59 (d, J=8.7 Гц, 1Н); 7.36 (bs, 1Н); 7.18 (d, J=2.4 Гц, 1Н); 7.01 (d, J=8.7 Гц, 1Н); 7.01 (bs, 1Н); 6.94 (dd, J=9.0 и 2.4 Гц, 1Н); 3.84 (s, 3Н); 3.79 (s, 2Н).
Стадия В: ацетат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила
Алюмогидрид лития (396 мг; 10.432 ммоль) добавляют к суспензии продукта из стадии А выше (1.205 г; 5.216 ммоль) в тетрагидрофуране (25 мл). После перемешивания при кипячении с обратным холодильником в течение δ часов, смесь охлаждают до 0°С и добавляют воду (40 мл), а после этого лимонную кислоту (10 г). После перемешивания в течение 14 часов, смесь нейтрализуют с использованием насыщенного водного раствора NaHCO3 и продукт экстрагируют три раза при помощи этилацетата. Органические фазы объединяют, промывают водой и рассолом, и затем сушат над сульфатом натрия и фильтруют. После выпаривания сырой продукт (802 мг) растворяют в уксусном ангидриде (3 мл) в присутствии ацетата натрия (500 мг). Смесь перемешивают в течение 16 часов и затем выливают в разбавленный водный раствор NaHCO3. Продукт экстрагируют три раза при помощи этилацетата и органические фазы объединяют, промывают водой и рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителей, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/метанол, градиент от 100/0 до 95/5) с получением указанного в заголовке продукта в виде твердого вещества белого цвета (337 мг; 21%). Точка плавления: 149-151°С
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.73 (d, J=8.9 Гц, 1Н); 7.68 (d, J=8.8 Гц, 1Н); 7.42 (d, J=2.2 Гц, 1Н); 7.14 (dd, J=8.8 и 2.2 Гц, 1Н); 7.01 (d, J=8.9 Гц, 1H); 5. 75 (bs, 1H); 3.97 (s, 3H); 3.52 (m, 2H); 3.19 (t, J=6.7 Гц, 2H); 2.39 (s, 3H); 1.9 (s, 3H).
l3C ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 170.7 (s); 170.6 (s); 158.6 (s); 147.5 (s); 134.3 (s); 130.3 (d); 128.2 (d); 127.5 (s); 124.1 (s); 119.0 (d); 118.2 (d); 102.9 (d); 55.6 (q); 39.1 (t); 25.9 (t); 23.4 (q); 21.1 (q).
Стадия С: 4-метилбензолсульфонат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила
Продукт из стадии В выше (230 мг; 0.764 ммоль) добавляют к раствору гидроксида натрия (61 мг; 1.528 ммоль) в абсолютном этаноле (10 мл). После перемешивания в течение одного часа, растворитель выпаривают. Остаток ресуспендируют в смеси из этилацетат/водный раствор разбавленной хлористоводородной кислоты. После разделения, органическую фазу промывают водой и рассолом, затем сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, сырой продукт растворяют в дихлорметане (4 мл) в присутствии триэтиламина (150 мкл; 2.47 ммоль) и тозилхлорид (174 мг; 0.917 ммоль). После перемешивания в течение 16 часов при температуре окружающей среды, растворитель выпаривают. Остаток ресуспендируют в этилацетате и воде. После разделения, органическую фазу промывают водой и рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением указанного в заголовке продукта (261 мг; 83%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.81 (d, J=8.2 Гц, 2Н); 7.68 (d, J=8.9 Гц, 1Н); 7.62 (d, J=2.4 Гц, 1Н); 7.55 (d, J=8.9 Гц, 1Н); 7.36 (d, J=8.2 Гц, 2Н); 7.14 (dd, J=8.9 и 2.4 Гц, 1Н); 6.89 (d, 8.9 Гц, 1Н); 5.97 (m, 1Н); 4.01 (s, 3Н); 3.53-3.46 (m, 2Н); 3.22-3.17 (m, 2Н); 2.46 (s, 3Н); 1.95 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 170.9 (s); 158.9 (s); 146.4 (s); 145.7 (s); 134.6 (s); 133.3 (s); 130.1 (2 x d); 130.0 (d); 128.5 (2xd); 128.2 (d); 127.7 (s); 126.2 (s); 119.2 (d); 117.8 (d); 103.3 (d); 55.8 (q); 39.4 (t); 26.4 (t); 23.4 (q); 21.9 (q).
Стадия D: N-[2-(7-метоксинафталии-1-ил)этил]ацетамид
Указанный в заголовке продукт получают в соответствии со способом, описанным в стадии D примера 2, используя продукт из стадии С выше в качестве исходного реагента.
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1Н); 7.74 (d, J=8.9 Гц, 1Н); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1Н); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1H); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 9: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: 4-метилбензолсульфонат 1-(2-амино-2-оксоэтил)-7-метоксинафталин-2-ила
Гидрид натрия (60% в минеральном масле; 78 мг; 1.953 ммоль) добавляют при 0°С к раствору продукта, полученного в стадии А примера δ (376 мг; 1.628 ммоль) в диметилформамиде (3 мл). После перемешивания в течение 30 минут, тозилхлорид (341 мг; 1.79 ммоль) вводят одной порцией. После перемешивания в течение 2 часов реакционную смесь разбавляют с этилацетататом, а после этого два раза промывают водой и два раза рассолом. Органическую фазу высушивают над сульфатом натрия и фильтруют. После выпаривания растворителей, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/дихлорметан 30/70) с получением указанного в заголовке продукта в виде твердого вещества белого цвета (260 мг; 40%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.89 (d, J=7.9 Гц, 2Н); 7.72 (d, J=9.0 Гц, 1Н); 7.66 (d, J=9.0 Гц, 1Н); 7.4 (d, J=7.9 Гц, 2Н); 7.37 (d, J=2.4 Гц, 1H); 7.17 (dd, J=9.0 и 2.4 Гц, 1Н); 7.01 (d, J=9.0 Гц, 1Н); 6.02 (bs, 1Н); 5.48 (bs, 1Н); 3.93 (s, 2Н); 3.91 (s, 3Н); 2.50 (s, 3Н).
Стадия В: 2-(7-метоксинафталин-1-ил)ацетамид
Продукт, полученный в стадии А выше (50 мг; 0.13 ммоль), хлорид никеля гексагидрат (31 мг; 0.13 ммоль), дихлорметан (1.3 мл) и метанол (1.3 мл) вводят в герметичную колбу. Затем барботируют аргоном в растворе в течение 5 минут и после чего маленькими порциями осторожно добавляют борогидрид натрия (88 мг; 2.6 ммоль). После перемешивания в течение 1 часа под аргоном при температуре окружающей среды, добавляют воду. После перемешивания в течение 15 минут, смесь фильтруют через целит, и затем промывают дихлорметаном и этилацетатом. Органическую фракцию высушивают над сульфатом натрия и затем фильтруют. После выпаривания растворителей, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением указанного в заголовке продукта (8 мг; 29%).
1Н ЯМР спектроскопический анализ (ДМСО-d6, 300.13 МГц, δ в част. на млн.): 7.84 (d, J=9.0 Гц, 1Н); 7.73 (d, J=8.0 Гц, 1Н); 7.62 (bs, 1Н); 7.41-7.36 (m, 2Н); 7.28 (dd, J=8.0 и 7.1 Гц, 1Н); 7.18 (dd, J=9.0 и 2.4 Гц, 1Н); 7.02 (bs, 1Н); 3.88 (s, 3Н); 3.82 (s, 2Н).
13С ЯМР спектроскопический анализ (ДМСО-d6, 75.5 МГц, δ в част. на млн.): 172.3 (s); 157.3 (s); 133.2 (s); 131.8 (s); 130.0 (d); 128.7 (s); 128.4 (d); 126.7 (d); 123.1 (d); 117.7 (d); 103.4 (d); 55.2 (q); 40.2 (t).
Стадия С: (7-метоксинафталин-1-ил)ацетонитрил
Указанный в заголовке продукт получают в соответствии с протоколом, описанным в стадии F получения 1 описания к патенту ЕР 0447285, используя продукт из стадии В выше в качестве исходного реагента.
Точка плавления: 86-87°С.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.78 (d, J=8.9 Гц, 1Н); 7.77 (d, J=7.8 Гц, 1Н); 7.52 (d, J=7.1 Гц, 1Н); 7.32 (dd, J=7.8 и 7.1 Гц, 1Н); 7.21 (dd, J=8.9 и 2.4 Гц, 1H); 7.03 (d, J=2.4 Гц, 1Н); 4.0 (s, 2Н); 3.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.5 (s); 132.0 (s); 130.6(d); 129.1 (s); 128.8 (d); 127.1 (d); 124.4 (s); 123.2 (d); 118.8 (d); 117.7 (s); 101,3 (d); 55.4 (q); 21.9 (t).
Стадия D: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Указанный в заголовке продукт получают в соответствии со способом, описанным в стадии Е примера 1, используя продукт из стадии С выше. Точка плавления: 108°С.
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1Н); 7.74 (d, J=8.9 Гц, 1Н); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1H); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1Н); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173,4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 10: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: 2-метокси-7-(проп-2-ен-1-илокси)нафталин
Аллилбромид (4.4 мл; 50.56 ммоль) добавляют к раствору 7-метокси-нафталин-2-ола (5.865 г; 33.71 ммоль) и карбоната калия (13.96 г; 101.12 ммоль) в ацетоне (33 мл). После перемешивания при 65°С в течение 16 часов, смесь охлаждают до температуры окружающей среды и добавляют воду (60 мл). После перемешивания в течение 3 часов, продукт экстрагируют три раза при помощи этилацетата. Органические фракции объединяют, два раза промывают водой и затем рассолом, сушат над сульфатом натрия и фильтруют. Выпаривание растворителей обеспечивает сырой продукт (7.963 г), который применяют непосредственно в следующей стадии без дополнительной очистки.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.68 (d, J=8.9 Гц, 1Н); 7.67 (d, J=8.9 Гц, 1Н); 7.08-6.99 (m, 3Н); 6.13 (m, 1Н); 5.48 (m, 1H); 5.34 (m, 1Н); 4.65 (m, 1Н); 3.92 (s, 3Н).
Стадия В: 7-метокси-1-(проп-2-ен-1-ил)нафталин-2-ол
Используя колбу, продукт из стадии А выше (7.963 г; 33.71 ммоль) помещают в баню, нагретую до 200°С в течение 2,5 часов. После охлаждения, сырой продукт применяют непосредственно в следующей стадии без дополнительной очистки.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.69 (d, J=8.9 Гц, 1Н); 7.6 (d, J=8.7 Гц, 1Н); 7.2 (d, J=2.4 Гц, 1Н); 7.03 (dd, J=8.9 и 2.4 Гц, 1H); 6.95 (d, J=8.7 Гц, 1Н); 6.08 (m, 1Н); 5.28 (s, 1Н); 5.14 (m, 1H); 5.1 (m, 1Н); 3.93 (s, 3Н); 3.8 (m, 1Н).
Стадия С: 4-метилбензолсульфонат 7-метокси-1-(проп-2-ен-1-ил)нафталин-2-ила
Триэтиламин (1.95 мл; 14.02 ммоль) и тозилхлорид (2.23 г; 11.68 ммоль) добавляют к раствору продукта из стадии В выше (2.5 г; 11.68 ммоль) в дихлорметане (22 мл). После перемешивания в течение 16 часов при температуре окружающей среды, растворитель выпаривают и остаток ресуспендируют в этилацетате и воде. После разделения, органическую фазу промывают водой и рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир, градиент от 10/90 до 20/80) с получением указанного в заголовке продукта (3.79 г; 88%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.8 (d, J=8.3 Гц, 2Н); 7.71 (d, J=8.9 Гц, 1Н); 7.6 (d, J=8.9 Гц, 1Н); 7.33 (d, J=8.3 Гц, 2Н); 7.24 (d, J=2.5 Гц, 1Н); 7.14 (dd, J=8.9 и 2.5 Гц); 7.1 (d, J=8.9 Гц, 1Н); 5. 79 (m, 1H); 5.02-4.95 (m, 2Н); 3.88 (s, 3Н); 3.68 (d, J=5.9 Гц, 2Н); 2.46 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.3 (s); 146.1 (s); 145.5 (s); 135.5 (d); 134.4 (s); 133.3 (s); 130.1 (d); 130.0 (2 x d); 128.5 (2 x d); 128.0 (d); 127.7 (s); 126.6 (s); 118.5 (d); 118.4 (d); 116.1 (t); 103.9 (d); 55.4 (q); 30.5 (t); 21.8 (q).
Стадия D: 4-метилбензолсульфонат 1-(2,3-дигидроксипропил)-7-метоксинафталин-2-ила
4-Метилморфолина N-оксид гидрат (1.666 г; 12.35 ммоль) и тетроксид осмия (4% в воде; 653 мкл; 0.01 ммоль) добавляют к раствору продукта из стадии С выше (3.786 г; 10.29 ммоль) в ацетоне (40 мл) и деионизированную воду (10 мл). После перемешивания при температуре окружающей среды в течение 20 часов, добавляют тиосульфат натрия пентагидрат (1 г). После еще дополнительного часа перемешивания растворители выпаривают. Остаток ресуспендируют в этилацетате и органическую фазу промывают рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат) с получением указанного в заголовке продукта в виде твердого вещества белого цвета (3.617 г; 87%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.81 (d, J=8.3 Гц, 2Н); 7.69 (d, J=9.0 Гц, 1Н); 7.58 (d, J=9.0 Гц, 1Н); 7.33 (d, J=8.3 Гц, 2Н); 7.13 (dd, J=9.0 и 2.4, 1Н); 7.02 (d, J=9.0 Гц, 1Н); 4.01 (m, 1Н); 3.88 (s, 3Н); 3.63-3.47 (m, 2Н); 3.11 (d, J=7.0 Гц, 2Н); 2.9 (bs, 1Н); 2.62 (bs, 1Н); 2.44 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.6 (s); 146.8 (s); 145.8 (s); 134.6 (s); 133.1 (s); 130.2 (d); 130.1 (2 x d); 128.4 (2 x d); 128.3 (d); 127.7 (s); 125.4 (s); 118.6 (d); 118.0 (d); 103.6 (d); 72.0 (d); 65.8 (t); 55.5 (q); 30.1 (t); 21.8 (q).
Стадия E: 4-метилбензолсульфонат 7-метокси-1-(2-оксоэтил)нафталин-2-ила
Периодат натрия (785 мг; 3.671 ммоль) и водный 2М раствор HCl (1.8 мл) добавляют к раствору продукта из стадии D выше (1.23 г; 3.06 ммоль) в тетрагидрофуране (12 мл) и воде (3 мл). Через 30 минут, смесь нейтрализуют разбавленным водным раствором NaHCO3, и продукт затем экстрагируют при помощи этилацетата. Органические фракции объединяют, сушат над сульфатом натрия и фильтруют. Выпаривание растворителей обеспечивает указанный в заголовке продукт в виде чистого твердого вещества белого цвета (1.13 г; 100%), которое применяют непосредственно в следующей стадии.
Точка плавления: 101-103°С.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 9.43 (t, J=2.6 Гц, 1Н); 7.78 (d, J=8.3 Гц, 2Н); 7. 74 (d, J=8.9 Гц, 1Н); 7.68 (d, J=8.9 Гц, 1Н); 7.35 (d, J=8.3 Гц, 2Н); 7.17 (dd, J=8.9 и 2.4 Гц, 1Н); 7.04 (d, J=8.9 Гц, 1Н); 6.97 (d, J=2.4 Гц, 1Н); 3.98 (d, J=2.6 Гц, 2Н); 3.88 (s, 3Н); 2.46 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 199.2 (d); 159.1 (s); 146.8 (s); 146.0 (s); 134.7 (s); 132.7 (s); 130.4 (d); 130.2 (2 x d); 129.4 (d); 128.6 (2 x d); 127.6 (s); 120.1 (s); 119.1 (d); 118.6 (d); 102.9 (d); 55.5 (q); 41.7 (t); 21.9 (q).
Стадия F: 2-(7-метоксинафталин-1-ил)этанол
К раствору продукта из стадии Е выше (950 мг; 2.567 ммоль) в метаноле (13 мл) добавляют под аргоном, маленькими порциями хлорид никеля (366 мг; 2.824 ммоль) и борогидрид натрия (1.3 г; 38.2 ммоль). После перемешивания в течение одного часа, смесь подвергают гидролизу водным раствором 2М HCl (80 мл) и затем перемешивают в течение 30 минут в присутствии этилацетата. Гетерогенный раствор фильтруют через целит, и промывают этилацетататом. Две фазы разделяют и органическую фракцию промывают рассолом, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 40/60) с получением указанного в заголовке продукта в виде твердого вещества белого цвета (442 мг; 85%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.72 (d, J=9.1 Гц, 1Н); 7.63 (d, J=8.1 Гц, 1Н); 7.3-7.21 (m, 3Н); 7.13 (dd, J=9.1 и 2.6 Гц, 1Н); 3.93 (t, J=6.7 Гц, 2Н); 3.88 (s, 3Н); 3.24 (d, J=6.7 Гц, 2Н); 1.99 (bs, 1H).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 157.8 (s); 133.2 (s); 133.0 (s); 130.4 (d); 129.4 (s); 127.7 (d); 127.1 (d); 123.3 (d); 118.1 (d); 102.5 (d); 62.7 (t); 55.4 (q); 36.4 (t).
Масс-спектрометрия (ESI; m/z(%)): 202(29) [M]+•; 171(100).
Стадия G: метансульфонат 2-(7-метоксинафталин-1-ил)этила
Указанный в заголовке продукт получают в соответствии со способом, описанным в стадии С примера 7, используя продукт из стадии F выше в качестве исходного реагента.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.72 (d, J=9.1 Гц, 1Н); 7.66 (d, J=8.9 Гц, 1Н); 7.31-7.21 (m, 3Н); 7.13 (dd, J=9.1 и 2.5 Гц, 1H); 4.47 (t, J=7.4 Гц, 2Н); 3.92 (s, 3Н); 3.45 (d, J=7.4 Гц, 2Н); 2.77 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.2 (s); 133.0 (s); 133.0 (s); 130.6 (s); 130.4 (d); 129.3 (s); 128.0 (d); 127.7 (d); 123.2 (d); 118.4 (d); 101.9 (d); 67.9 (t); 55.5 (q); 37.4 (q); 33.3 (t).
Стадия H: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Указанный в заголовке продукт получают в соответствии со способом, описанным в стадии D примера 7, используя продукт из стадии G выше в качестве исходного реагента.
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1Н); 7.74 (d, J=8.9 Гц, 1Н); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1Н); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1Н); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
ПРИМЕР 11: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Стадия А: 3-(7-метоксинафталин-1-ил)пропан-1,2-диол
Продукт, полученный в стадии D примера 10 (44 мг; 0.11 ммоль), хлорид никеля (16 мг; 0.12 ммоль) и метанол (1 мл) вводят, под аргоном, в герметичную колбу. После чего маленькими порциями осторожно добавляют борогидрид натрия (74 мг; 2.19 ммоль). После перемешивания в течение 2 часов, добавляют воду (1 мл) и затем пероксид водорода (35% в воде; 1 мл), и смесь перемешивают в течение 2 часов. После чего добавляют рассол, и водную фазу экстрагируют три раза при помощи этилацетата. Органические фазы объединяют, сушат над сульфатом натрия и фильтруют. Выпаривание растворителей обеспечивает чистый продукт (22 мг; 86%) без потребности в дополнительной очистке.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.75 (d, J=9.1 Гц, 1H); 7.68 (d, J=8.1 Гц, 1Н); 7.32-7.23 (m, 3Н); 7.16 (dd, J=9.1 и 2.4 Гц, 1H); 4.11 (m, 1H); 3.92 (s, 3Н); 3. 72-3.55 (m, 2Н); 3.25-3.1 (m, 2Н); 2.54 (bs, 2Н, ОН).
Стадия В: (7-метоксинафталин-1-ил)ацетальдегид
Указанный в заголовке продукт получают в соответствии со способом, описанным в стадии Е примера 10, используя продукт из стадии А выше вместо 4-метилбензолсульфоната 1-(2,3-дигидроксипропил)-7-метоксинафталин-2-ила.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 9.74 (t, J=2.6 Гц, 1Н); 7.74-7.84 (m, 2Н); 7.38 (d, J=6.0 Гц, 1Н); 7.32 (t, J=7.5 Гц, 1Н); 7.19 (dd, J=9.0 Гц и 2.4 Гц, 1Н); 7.1 (d, J=2.4 Гц, 1H); 4.05 (d, J=2.6 Гц, 2Н); 3.92 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 199.85; 158.29; 133.54; 130.44; 129.32; 129.07; 128.23; 126.83; 123.36; 118.63; 102.12; 55.36; 48.83.
Стадия С: 2-(7-метоксинафталин-1-ил)этанол
Борогидрид натрия (38 мг; 1 ммоль) добавляют к раствору продукта из стадии В выше (20 мг; 0.1 ммоль) в этаноле (1 мл). После перемешивания в течение 2 часов при температуре окружающей среды, смесь подвергают гидролизу посредством водного раствора 2М HCl (2 мл) и затем перемешивают в течение 30 минут в присутствии этилацетата (1 мл). Раствор экстрагируют 4 раза при помощи этилацетата. Органические фракции объединяют, сушат над сульфатом натрия и фильтруют. После выпаривания растворителя, сырой продукт очищают хроматографией на силикагелевой колонке (элюант : этилацетат/петролейный эфир 20/80) с получением указанного в заголовке продукта в виде твердого вещества белого цвета (19.8 мг; 98%).
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.72 (d, J=9.1 Гц, 1Н); 7.63 (d, J=8.1 Гц, 1H); 7.3-7.21 (m, 3Н); 7.13 (dd, J=9.1 и 2.6 Гц, 1Н); 3.93 (t, J=6.7 Гц, 2Н); 3.88 (s, 3Н); 3.24 (d, J=6.7 Гц, 2Н); 1.99 (bs, 1Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 157.8 (s); 133.2 (s); 133.0 (s); 130.4 (d); 129.4 (s); 127.7 (d); 127.1 (d); 123.3 (d); 118.1 (d); 102.5 (d); 62.7 (t); 55.4 (q); 36.4 (t).
Масс-спектрометрия (ESI; m/z(%)): 202(29) [M]+•; 171(100).
Стадия D: метансульфонат 2-(7-метоксинафталин-1-ил)этила
Указанный в заголовке продукт получают в соответствии со способом, описанным в стадии С примера 7, используя продукт из стадии С выше в качестве исходного реагента.
1Н ЯМР спектроскопический анализ (CDCl3, 300.13 МГц, δ в част. на млн.): 7.72 (d, J=9.1 Гц, 1H); 7.66 (d, J=8.9 Гц, 1Н); 7.31-7.21 (m, 3Н); 7.13 (dd, J=9.1 и 2.5 Гц, 1Н); 4.47 (t, J=7.4 Гц, 2Н); 3.92 (s, 3Н); 3.45 (d, J=7.4 Гц, 2Н); 2.77 (s, 3Н).
13С ЯМР спектроскопический анализ (CDCl3, 75.5 МГц, δ в част. на млн.): 158.2 (s); 133.0 (s); 133.0 (s); 130.6 (s); 130.4 (d); 129.3 (s); 128.0 (d); 127.7 (d); 123.2 (d); 118.4 (d); 101.9 (d); 67.9 (t); 55.5 (q); 37.4 (q); 33.3 (t).
Стадия E: N-[2-(7-метоксинафталин-1-ил)этил]ацетамид
Указанный в заголовке продукт получают в соответствии со способом, описанным в стадии D примера 7, используя продукт из стадии D выше в качестве исходного реагента.
1Н ЯМР спектроскопический анализ (CD3OD, 300.13 МГц, δ в част. на млн.): 8.21 (bs, 1Н); 7.74 (d, J=8.9 Гц, 1Н); 7.65 (d, J=8.0 Гц, 1Н); 7.52 (d, J=2.5 Гц, 1Н); 7.31-7.2 (m, 2Н); 7.11 (dd, J=8.9 и 2.5 Гц, 1Н); 3.96 (s, 3Н); 3.52-3.44 (m, 2Н); 3.23-3.18 (m, 2Н); 1.94 (s, 3Н).
13С ЯМР спектроскопический анализ (CD3OD, 75.5 МГц, δ в част. на млн.): 173.4 (s); 159.4 (s); 135.1 (s); 134.6 (s); 131.2 (d); 130.8 (s); 128.2 (d); 127.9 (d); 124.2 (d); 119.3 (d); 103.2 (d); 55.9 (q); 41.4 (t); 34.2 (t); 22.6 (q).
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ СПОСОБ СИНТЕЗА 7-МЕТОКСИ-НАФТАЛИН-1-КАРБАЛЬДЕГИДА И ЕГО ПРИМЕНЕНИЕ В СИНТЕЗЕ АГОМЕЛАТИНА | 2014 |
|
RU2680243C1 |
| Электрохимический способ получения производных тетрагидрохинолина, применение их в качестве фунгицидных средств и фунгицидные композиции на их основе | 2022 |
|
RU2784323C1 |
| ЛИГАНДЫ ДЛЯ АГРЕГИРОВАННЫХ МОЛЕКУЛ ТАУ-БЕЛКА | 2009 |
|
RU2518892C2 |
| НАФТИЛУКСУСНЫЕ КИСЛОТЫ | 2009 |
|
RU2539185C2 |
| АНТАГОНИСТЫ TLR7/8 И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2758686C2 |
| СОЕДИНЕНИЯ ГИДРОХИНОНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В ПРОТИВООПУХОЛЕВОЙ ИЛИ ИММУНОМОДУЛЯТОРНОЙ ТЕРАПИИ | 2017 |
|
RU2708464C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ, СОЕДИНЕНИЯ, КОМПЛЕКСНЫЙ МЕТАЛЛООРГАНИЧЕСКИЙ КАТАЛИЗАТОР | 2012 |
|
RU2652807C2 |
| ПРОИЗВОДНЫЕ 2-АМИНОПИРАЗИНА В КАЧЕСТВЕ ИНГИБИТОРОВ CSF-1R КИНАЗЫ | 2013 |
|
RU2642777C2 |
| Производное индола, используемое в качестве ингибитора CRTH2 | 2017 |
|
RU2756270C2 |
| СОЕДИНЕНИЕ НАФТИЛАМИДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2015 |
|
RU2655607C2 |
Изобретение относится к способу промышленного синтеза соединения формулы (I). Способ характеризуется тем, что в реакцию вводят 7-метоксинафталин-2-ол формулы (II), в который в положение 1 соединения формулы (II) вводят группу -СН2-Х, в которой X представляет собой -N(CH3)2, -CO-N(CH2-Ph)2, -СН2-ОН, -СН=СН2 или -CO-NH2, с получением соединения формулы (III), в которой X представляет собой группу -N(CH3)2, -CO-N(CH2-Ph)2, -СН2-ОН, -СН=СН2 или -CO-NH2. При этом соединение формулы (III) подвергают реакции сульфонилирования по ароматическому спирту и заместитель X соединения формулы (III) является модифицированным до или после стадии сульфонилирования ароматического спирта при помощи обычных химических реакций с получением соединения формулы (IV), в которой X' представляет собой группу -CN, -CO-NH2, -СН2-ОН, -СНО, -CH2-N(CH2-Ph)2, -CH2-NH-CO-CH3, -СН(ОН)-СН2-ОН или (2,5-диоксопирролидин-1-ил)метил и R представляет собой группу -СН3, -(СН2)2-СН3, -CF3 или толуил. Соединение формулы (IV) подвергают реакции дезоксигенирования в присутствии переходного металла и восстановителя с получением непосредственно соединения формулы (I), которое выделяют в виде твердого вещества, если X' представляет собой группу -CH2-NH-CO-CH3, или соединения формулы (V), в которой X'' представляет собой группу -CN, -CH2-N(CH2-Ph)2, -СН2-ОН, -СН(ОН)-СН2-ОН, -CO-NH2 или (2,5-диоксопирролидин-1-ил)метил. Соединение формулы (V) подвергают обычным химическим реакциям с получением соединения формулы (I), которое выделяют в виде твердого вещества. Предлагаемый способ позволяет получать агомелатин формулы (I) с высоким выходом и превосходной чистотой. Изобретение относится также к промежуточным соединениям и их применению в синтезе агомелатина формулы (I) указанным способом. 14 н. и 15 з.п. ф-лы, 11 пр.



1. Способ промышленного синтеза соединения формулы (I)
 ,
,
отличающийся тем, что в реакцию вводят 7-метоксинафталин-2-ол формулы (II)
 ,
,
в который в положение 1 соединения формулы (II) вводят группу -СН2-Х, в которой X представляет собой группу -N(CH3)2, -CO-N(CH2-Ph)2, -СН2-ОН, -СН=СН2 или -CO-NH2,
с получением соединения формулы (III)
 ,
,
в которой X представляет собой группу -N(CH3)2, -CO-N(CH2-Ph)2, -СН2-ОН, -СН=СН2 или -CO-NH2;
причем соединение формулы (III) подвергают реакции сульфонилирования по ароматическому спирту и заместитель X соединения формулы (III) является модифицированным до или после стадии сульфонилирования ароматического спирта при помощи обычных химических реакций с получением соединения формулы (IV)
 ,
,
в которой X' представляет собой группу -CN, -CO-NH2, -СН2-ОН, -СНО, -CH2-N(CH2-Ph)2, -CH2-NH-CO-CH3, -СН(ОН)-СН2-ОН или (2,5-диоксопирролидин-1-ил)метил и R представляет собой группу -СН3, -(СН2)2-СН3, -CF3 или толуил;
причем соединение формулы (IV) подвергают реакции дезоксигенирования в присутствии переходного металла и восстановителя с получением:
- или, если X' представляет собой группу -CH2-NH-CO-CH3, непосредственно соединения формулы (I), которое выделяют в виде твердого вещества;
- или соединения формулы (V)
 ,
,
в которой X'' представляет собой группу -CN, -CH2-N(CH2-Ph)2, -СН2-ОН, -СН(ОН)-СН2-ОН, -CO-NH2 или (2,5-диоксопирролидин-1-ил)метил;
причем соединение формулы (V) подвергают обычным химическим реакциям с получением соединения формулы (I), которое выделяют в виде твердого вещества.
2. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (II) в соединение формулы (III) осуществляют посредством действия формальдегида и диметиламина с получением соединения формулы (III), в которой X представляет собой -N(СН3)2.
3. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (II) в соединение формулы (III) осуществляют посредством действия глиоксаля с последующим действием восстановителя с получением соединения формулы (III), в которой X представляет собой -CH2-ОН.
4. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (II) в соединение формулы (III) осуществляют посредством действия глиоксаля с последующим действием соединения формулы NHR'R', в которой R' представляет собой Н или группу -CH2-Ph, с получением соединения формулы (III), в которой X представляет собой -CO-NH2 или -CO-N(CH2-Ph)2.
5. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (II) в соединение формулы (III) осуществляют посредством действия аллилбромида с последующей термической перегруппировкой с получением соединения формулы (III), в которой X представляет собой -СН=СН2.
6. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что при превращении соединения формулы (III) в соединение формулы (IV) стадию сульфонилирования осуществляют посредством действия сульфонилхлорида, сульфонового ангидрида или сульфонимида.
7. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (III) в соединение формулы (IV) состоит из стадии сульфонилирования ароматического спирта с последующей модификацией группы X при помощи обычных химических реакций, X является таким, как определен в формуле (III).
8. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (III) в соединение формулы (IV) состоит в модификации группы X при помощи обычных химических реакций, после чего следует стадия сульфонилирования ароматического спирта, X является таким, как определен в формуле (III).
9. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (IV) в соединение формулы (V) осуществляют в присутствии никеля и гидрида.
10. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (IV) в соединение формулы (V) осуществляют в присутствии палладия и водорода.
11. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (IV) в соединение формулы (V) осуществляют в присутствии палладия и щелочноземельного металла.
12. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (IV), в которой X' представляет собой группу -CH2-NH-CO-CH3, в соединение формулы (I) осуществляют в присутствии никеля и гидрида.
13. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (IV), в которой X' представляет собой группу -CH2-NH-CO-CH3, в соединение формулы (I) осуществляют в присутствии палладия и водорода.
14. Способ промышленного синтеза соединения формулы (I) по п.1, отличающийся тем, что превращение соединения формулы (IV), в которой X' представляет собой группу -CH2-NH-CO-CH3, в соединение формулы (I) осуществляют в присутствии палладия и щелочноземельного металла.
15. Соединение формулы (III)
 ,
,
в которой X представляет собой группу -N(СН3)2, -CO-N(CH2-Ph)2, -СН2-ОН, -СН=СН2 или -CO-NH2;
для применения в качестве промежуточного соединения в синтезе агомелатина формулы (I) по п.1.
16. Соединение формулы (III) по п.15, выбранное из следующих соединений:
- 1-[(диметиламино)метил]-7-метоксинафталин-2-ол;
- N,N-дибензил-2-(2-гидрокси-7-метоксинафталин-1-ил)ацетамид;
- 1-(2-гидроксиэтил)-7-метоксинафталин-2-ол;
- 2-(2-гидрокси-7-метоксинафталин-1-ил)ацетамид;
- 7-метокси-1-(проп-2-ен-1-ил)нафталин-2-ол.
17. Применение соединения формулы (III) по п.15 или 16 в синтезе агомелатина формулы (I) по п.1.
18. Соединение формулы (IV)
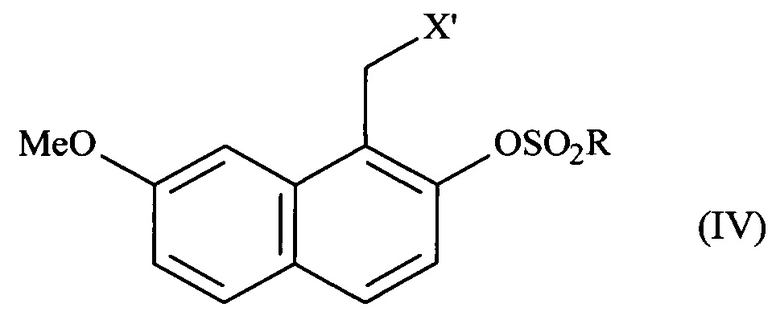 ,
,
в которой X' представляет собой группу -CN, -CO-NH2, -СН2-ОН, -СНО, -CH2-N(CH2-Ph)2, -CH2-NH-CO-CH3, -СН(ОН)-СН2-ОН или (2,5-диоксопирролидин-1-ил)метил и R представляет собой группу -СН3, -(СН2)2-СН3, -CF3 или толуил;
для применения в качестве промежуточного соединения в синтезе агомелатина формулы (I) по п.1.
19. Соединение формулы (IV) по п.18, выбранное из следующих соединений:
- трифторметансульфонат 1-(цианометил)-7-метоксинафталин-2-ила;
- 4-метилбензолсульфонат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила;
- трифторметансульфонат 1-[2-(дибензиламино)этил]-7-метоксинафталин-2-ила;
- пропан-1-сульфонат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила;
- пропан-1-сульфонат 1-[2-(2,5-диоксопирролидин-1-ил)этил]-7-метоксинафталин-2-ила;
- 4-метилбензолсульфонат 1-(2-гидроксиэтил)-7-метоксинафталин-2-ила;
- 4-метилбензолсульфонат 1-(2-амино-2-оксоэтил)-7-метоксинафталин-2-ила;
- 4-метилбензолсульфонат 7-метокси-1-(2-оксоэтил)нафталин-2-ила;
- 4-метилбензолсульфонат 1-(2,3-дигидроксипропил)-7-метоксинафталин-2-ила.
20. Применение соединения формулы (IV) по п.18 или 19 в синтезе агомелатина формулы (I) по п.1.
21. Соединение формулы (V)
 ,
,
в которой X'' представляет собой группу -СН(ОН)-СН2-ОН или (2,5-диоксопирролидин-1-ил)метил, выбранное из следующих соединений:
- 1-[2-(7-метоксинафталин-1-ил)этил]пирролидин-2,5-дион;
- 3-(7-метоксинафталин-1-ил)пропан-1,2-диол;
для применения в качестве промежуточных соединений в синтезе агомелатина формулы (I) по п.1.
22. Применение соединения формулы (V) по п.21 в синтезе агомелатина формулы (I) по п.1.
23. (2-Гидрокси-7-метоксинафталин-1-ил)ацетонитрил, 4-метилбензолсульфонат 7-метокси-1-(2-{[(4-метилфенил)сульфонил]окси}этил)нафталин-2-ила, пропан-1-сульфонат 7-метокси-1-{2-[(пропилсульфонил)окси]этил}нафталин-2-ила и ацетат 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила для применения в качестве промежуточных соединений в синтезе агомелатина формулы (I) по п.1.
24. Применение (2-гидрокси-7-метоксинафталин-1-ил)ацетонитрила, 4-метилбензолсульфоната 7-метокси-1-(2-{[(4-метилфенил)сульфонил]окси}этил)нафталин-2-ила, пропан-1-сульфоната 7-метокси-1-{2-[(пропилсульфонил)окси]этил}нафталин-2-ила и ацетата 1-[2-(ацетиламино)этил]-7-метоксинафталин-2-ила по п.23 в синтезе агомелатина формулы (I) по п.1.
25. Соединение формулы (VI)
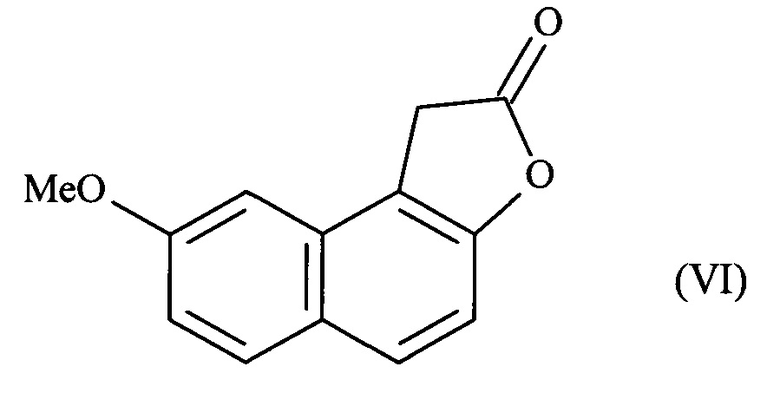
для применения в качестве промежуточного соединения в синтезе агомелатина формулы (I) по п.1.
26. Применение соединения формулы (VI) по п.25 в синтезе агомелатина формулы (I) по п.1.
27. Применение соединения формулы (II)

в синтезе агомелатина формулы (I) по п.1.
28. Способ синтеза агомелатина по п.1 исходя из соединения формулы (III), отличающийся тем, что соединение формулы (III) получают способом синтеза по любому из пп.2-5.
29. Способ синтеза агомелатина по п.1 исходя из соединения формулы (IV), отличающийся тем, что соединение формулы (IV) получают способом синтеза по любому из пп.2-8.
| Способ изготовления анода для электролиза воды | 1987 |
|
SU1564202A1 |
| СПОСОБ МОДЕЛИРОВАНИЯ ОСТРОГО ПЕРИТОНИТА | 1998 |
|
RU2151427C1 |
| СПОСОБ АНАЛИЗА НЕИЗВЕСТНЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ ОДНОЦЕПОЧЕЧНЫХ НУКЛЕИНОВЫХ КИСЛОТ | 2006 |
|
RU2322508C1 |
| Камнерезный станок | 1971 |
|
SU447285A1 |
| Приспособление для разбора наборных печатных форм | 1929 |
|
SU15545A1 |

