Изобретение относится к области аналитической химии, а именно к способам количественного определения примесных элементов в образцах вязких органических жидкостей спектральными методами, позволяющими проводить анализ сухого остатка жидкостей и растворов, и может быть использовано для контроля качества продукции, экологического мониторинга, токсикологии и других областей науки и техники.
Известны способы пробоподготовки [1, 2] для спектрального анализа вязких органических жидкостей путем экстракции элементов в виде комплексов в новую фазу с последующим определением их в получаемых экстрактах. Недостатками данных способов являются необходимость в большом количестве исходных образцов, использование токсичных растворителей, возможность потерь элементов вследствие адсорбции на стенках посуды, изменчивость коэффициентов экстракции элементов для различных по основному составу объектов, а также длительность и трудоемкость операции экстракции.
Известен способ [3] эмиссионного спектрального определения содержаний металлических примесей в органических образцах, заключающийся в предварительном разбавлении пробы органическим растворителем с целью снижения вязкости для последующего анализа разбавленных проб с построением градуировочных зависимостей по стандартным растворам на органической основе. Недостатками данного способа являются использование токсичных растворителей, необходимость приготовления стандартных растворов на органических растворителях и увеличение пределов обнаружения за счет разбавления пробы.
Известен способ количественного определения микроэлементов с помощью атомно-абсорбционной спектрометрии [4], наиболее близкий к заявленному изобретению и выбранный в качестве прототипа. Известный способ заключается в замораживании пробы после ее пробоотбора и последующем размораживании с одновременной гомогенизацией путем перемешивания. Далее отбирают 1-2 мл пробы в пробирку, куда помещают аналогичный объем концентрированной азотной кислоты, смесь выдерживают при комнатной температуре не менее 2,5 часов. Затем добавляют концентрированную перекись водорода в объеме, аналогичном объему пробы, нагревают полученную смесь 5-10 минут и далее охлаждают. Полученный минерализат доводят до объема 3 мл и анализируют методом атомно-абсорбционной спектрометрии, используя градуировочный график; для получения калибровочных растворов также используют смесь азотной кислоты и пероксида водорода в аналогичных количествах. Данный прототип позволяет полностью избавиться от матричных влияний, вызванных органической основой пробы, и проводить корректное количественное определение аналитов. Недостатками известного способа являются высокая трудоемкость и длительное время, затрачиваемое на пробоподготовку, а также высокие пределы обнаружения и, как следствие, низкая чувствительность, за счет разбавления пробы, кроме того, известный способ отличается относительно высокой стоимостью за счет использования относительно больших количеств анализируемой пробы и реагентов, а также возможными потерями элементов вследствие их адсорбции на стенках посуды.
Техническим результатом заявленного изобретения является сокращение времени и упрощение проводимых операций при пробоподготовке и анализе в целом, а также повышение чувствительности (снижение пределов обнаружения), снижение стоимости анализа, повышение достоверности получаемых результатов, отсутствие необходимости использования стандартных растворов на органических растворителях.
Техническая задача заявляемого изобретения состоит в снижении трудоемкости и сокращении времени проведения спектрального определения микросодержаний элементов в вязких органических жидкостях методами, позволяющими проводить анализ сухого остатка жидкостей и растворов, кроме того, следует значительно сократить перечень необходимых операций, а также необходимую навеску пробы и объем используемых реагентов, что предотвращает потери элементов и сохраняет низкие пределы обнаружения используемого метода спектрометрического анализа, а также делает предлагаемый способ экологичным, удовлетворяющим «зеленым технологиям».
Указанный технический результат достигается тем, что на этапе подготовки проб к анализу происходит нанесение небольшого объема (порядка 10 мкл) пробы с точно определяемой массой на торец (в форме лунки) стержневого электрода с диаметром от 3 до 6 мм (дальнейшее увеличение диаметра нежелательно, поскольку может привести к растеканию пробы по поверхности). Использование столь малых количеств пробы выгодно отличает предлагаемый способ в случае, когда образец имеется в ограниченном количестве или является высокотоксичным.
Затем производится небольшой нагрев пробы (либо путем помещения электрода на подставке на электроплитку, либо помещения электрода под излучение ИК-лампы), при этом происходит испарение легколетучей основы пробы и уменьшение капли в размерах. Далее на пробу наносится небольшой объем (5-20 мкл) концентрированной (65%) азотной кислоты ос. ч., после чего вновь производится ее умеренный нагрев (указанным выше способом). В результате под действием азотной кислоты происходит окисление органической составляющей пробы и самоудаление из нее газообразных продуктов реакции. При этом не происходит потерь элементов в результате адсорбции, поскольку минерализация осуществляется на электроде, с которого впоследствии будет осуществляться атомизация пробы. В результате указанной минерализации на торце электрода остается сухой остаток пробы в виде тонкой пленки. На этап разложения по указанной схеме затрачивается не более 10 минут. Кроме того, возможно осуществление пробоподготовки одновременно нескольких образцов на разных электродах. Уменьшение потребления реактивов выгодно отличает предлагаемый способ своей экологичностью, что особенно актуально с позиции «зеленой химии».
Если после реакции с азотной кислотой произошло неполное разложение органической матрицы, то операция нанесения азотной кислоты и последующего нагрева повторяется необходимое число раз (опытным путем установлено, что для эффективной минерализации подавляющего числа образцов достаточно не более пяти капель азотной кислоты по 10 мкл каждая). Вследствие того, что элементы пробы концентрируются на электроде в виде тонкого слоя сухого остатка, не происходит разбавления образца реагентами. В случае необходимости дополнительного снижения пределов обнаружения возможно последующее нанесение капель анализируемой пробы и пропорционального количества азотной кислоты.
Далее полученный сухой остаток пробы анализируется по известной методике эмиссионной спектрометрии [5-6] с сохранением всех ее достоинств (включая одновременное экспрессное определение малых концентраций большого набора элементов), либо прочими спектральными методами, позволяющими проводить анализ сухого остатка жидкостей и растворов [7-9]. Для построения градуировочных зависимостей используются стандартные водные растворы солей элементов, которые наносятся на электроды по аналогичной схеме для получения сухих остатков.
Заявленный способ был апробирован в лабораторных условиях СПбГУ, результаты апробации представлены в виде конкретных примеров реализаций.
Пример 1. Построение градуировочных зависимостей и определение метрологических характеристик метода.
В качестве головного раствора используется многоэлементный стандартный водный раствор солей элементов (или набор растворов), содержащий большое число элементов в высоких содержаниях. Допускается применение одноэлементных стандартных растворов, однако, в этом случае анализу следует подвергать каждый из них по очереди. Также, в случае отсутствия указанных растворов, допускается самостоятельное приготовление растворов из соединений элементов особой чистоты.
Из головного раствора методом последовательного разбавления с использованием деионизированной воды готовится серия рабочих растворов в диапазоне концентраций 10-7-1 г/л. Растворы с концентрацией менее 10-3 г/л готовятся в день анализа, растворы с большей концентрацией допускается хранить в течение 1 месяца.
Далее каждый калибровочный раствор наносится с помощью микрошприца (или иного дозирующего устройства) в виде капли объемом 10 мкл на подготовленный угольный электрод. Затем электрод с пробой устанавливается под включенной ИК-лампой и нагревается в течение 1 минуты. При этом происходит удаление водной основы. Далее на электрод с подсушенной пробой наносится 10 мкл концентрированной азотной кислоты ос. ч. Электрод вновь устанавливается под ИК-лампой и нагревается до получения тонкого слоя сухого остатка, на что затрачивается не более 1 минуты. Затем на полученный сухой остаток наносится 10 мкл водного раствора хлорида натрия ос. ч. (приготовленного с использованием деионизированной воды) с концентрацией 15 г/л, которые высушиваются по аналогичной схеме. Далее производится анализ полученного сухого остатка известным атомно-эмиссионным способом в дуге переменного тока [5-6]. Анализ производится по семи параллельным определениям. Дополнительно проводится анализ «холостой пробы» по описанной выше схеме за тем исключением, что вместо калибровочного раствора используется деионизированная вода.
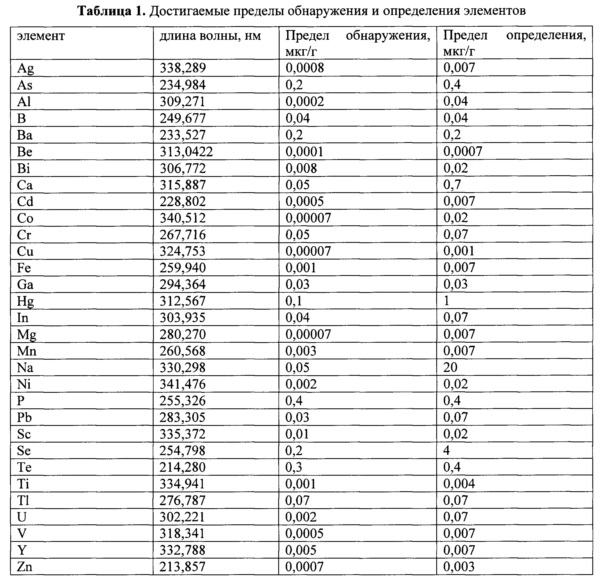
В зарегистрированных спектрах осуществляется поиск аналитических линий, в качестве которых в общем случае использовались наиболее чувствительные, по возможности свободные от перекрывания спектральные линии, градуировочные графики по которым обладают наибольшим диапазоном линейности, углом наклона, коэффициентом корреляции. При работе в спектральном диапазоне 193-344 нм перечень определяемых элементов и соответствующие им спектральные линии представлены в табл. 1. Затем для выбранных линий производится расчет их интенсивностей. В качестве аналитического сигнала использовался логарифм разности «чистых» (за вычетом минимального значения фонового излучения) интегральных (по всему контуру спектральной линии) суммарных (за все время экспозиции) интенсивностей спектральных линий из спектров стандартных растворов и «холостого опыта» [10-11]. Градуировочные графики строятся в координатах аналитический сигнал - логарифм массы элемента на торце электрода. На фиг. 1 в качестве примера представлена градуировочная зависимость для Cd по спектральной линии Cd 228,80 нм в координатах аналитический сигнал lgI - логарифм массы m элемента на электроде, иллюстрирующая возможности метода, из которого, в частности, видно, что диапазон линейности графика составляет 6 порядков величины содержания.
В табл. 1 также представлены пределы обнаружения метода из расчета нанесения 15 мг пробы (что примерно соответствует 10 мкл вязких органических жидкостей), рассчитанные по 3S-критерию (S - среднеквадратичное отклонение логарифма интенсивности «холостого опыта»), и пределы определения - минимальные концентрации, для которых относительное среднеквадратичное отклонение аналитического сигнала не превышает меньше 0,3. Достигаемые пределы достаточны для определения подавляющего числа элемента в реальных образцах. Воспроизводимость результатов находится на уровне 0,05-0,2, что является удовлетворительным при использовании дугового источника возбуждения спектра. Методика позволяет за один анализ определять весь перечень элементов, представленный в табл. 1.
Пример 2. Определение микроэлементов в реальных образцах медицинских клеев (используемых для ускорения заживления ран) и флокулянтов для очистки сточных вод (полимерных катионных веществ на основе полиакриламида и солей алюминия и железа).
Анализируемая проба (клея или флокулянта) наносится в виде капли объемом ≈10 мкл на подготовленный угольный электрод, находящийся на подставке (масса электрода с подставкой заранее известна), после чего взвешивается. По разнице значений определяется точная масса пробы. Затем электрод с пробой устанавливается под включенной ИК-лампой и нагревается в течение 1 минуты. При этом происходит удаление легколетучих компонентов пробы (водно-спиртовых в случае клея и водных в случае флокулянта). Далее на электрод с подсушенной пробой наносится 10 мкл концентрированной азотной кислоты ос. ч. Электрод вновь устанавливается под ИК-лампой и нагревается до получения тонкого слоя сухого остатка, на что затрачивается не более 5 минут. В случае анализа флокулянта добавляемого количества азотной кислоты достаточно для эффективного удаления органической матрицы. В случае медицинского клея требуется дополнительное последовательное нанесение еще четырех капель азотной кислоты аналогичного объема с их последующим высушиванием. Затем на полученный сухой остаток наносится 10 мкл водного раствора хлорида натрия ос. ч. (приготовленного с использованием деионизированной воды) с концентрацией 15 г/л, которые высушиваются по аналогичной схеме. Далее производится анализ полученного сухого остатка известным атомно-эмиссионным способом в дуге переменного тока [5-6]. Анализ производится по семи параллельным определениям. Количественная оценка содержаний элементов производится по предварительно построенным градуировочным графикам, полученным путем разбавления стандартного водного раствора солей элементов до концентраций, соответствующих анализируемым образцам. Со стандартными растворами проводятся все операции аналогично анализируемым пробам, как описано в Примере 1.
Проверка отсутствия матричных влияний проводилась посредством определения параметров плазмы по методу Орнштейна: температура определялась по спектральным линиям железа, концентрация электронов - по линиям магния. Как видно из табл. 2, параметры плазмы, определенные в случае возбуждения спектра сухого остатка стандартного раствора и анализируемых образцов, равны (в пределах погрешности измерения). Данный факт свидетельствует об отсутствии влияния макросостава проб на условия возбуждения их спектра.


Правильность результатов проводилась сравнением с результатами, полученными референтным методом, а именно: атомно-эмиссионной спектрометрией с индуктивно-связанной плазмой путем анализа минерализатов, полученных при микроволновом кислотном разложении образцов (Исследования проведены в Ресурсном образовательном центре по направлению Химия Научного парка СПбГУ). Полученные данные (на примере флокулянта) для некоторых определяемых элементов представлены в табл. 3, из которых видно, что в пределах погрешности оба метода дают сопоставимые результаты, что свидетельствует о правильности разработанного способа анализа вязких органических жидкостей.
Требуемый объем пробы составляет всего 10 мкл на одно параллельное определение, при этом на минерализацию затрачивается лишь 50 мкл азотной кислоты, что выгодно отличает метод с экономической точки зрения. Время, затрачиваемое на пробоподготовку пробы при 7 параллельных определениях, составляет всего 20 минут, при этом, возможно проводить минерализацию одновременно нескольких образцов. Общее время анализа каждого образца равно полчаса.
При необходимости достижения меньших пределов определения (менее указанных в табл. 1) возможно увеличение наносимого количества капель анализируемой пробы на торец электрода (с пропорциональным увеличением количества наносимой кислоты) с повторением всех необходимых процедур пробоподготовки. Экспериментальным путем удалось последовательно нанести на электрод не менее 5 капель пробы. При этом, поскольку использовалась азотная кислота особой чистоты, не происходило увеличения сигнала холостого опыта, вследствие чего пределы обнаружения и определения уменьшались пропорционально количеству наносимых капель.
Технико-экономическая эффективность заявленного способа состоит в существенном уменьшении времени, затрачиваемого на пробоподготовку, и упрощении процедуры минерализации образцов вязких органических жидкостей, а также в предотвращении потерь элементов (т.е. повышении достоверности результатов), уменьшении необходимой навески пробы и объемов используемых реагентов, что сохраняет низкие пределы обнаружения (на уровне сотых мкг/г) используемого метода спектрометрического анализа и снижает его стоимость. Заявленная методика пригодна для количественного определения микроэлементов в широком круге жидких образцов с органической (в т.ч. полимерной) основой, обладающих высокой вязкостью. За счет уменьшения потребления реактивов предлагаемый характеризуется экологичностью, что особенно актуально с позиции «зеленой химии».
Источники информации
1. Braun М., Menges М, Opoku F., Smith A.M. The relative contribution of calcium zinc and oxidation-based cross-links to the stiffness of Arion subfuscuc glue // The journal of experimental biology. 2013, v. 216, p. 1475-1483.
2. Nomngongo P.N., Ngila J.C. Functionalized nanometer-sized alumina supported micro-solid phase extraction coupled to inductively coupled plasma mass spectrometry for preconcentration and determination of trace metal ions in gasoline samples // RSC Advances. 2014, v. 4, is. 86, p. 46257-46264.
3. Кучумов В.А., Друженков В.В., Межов Э.А. Способ спектрально-эмиссионного определения содержаний металлических примесей в органических жидкостях. Патент на изобретение №2186368. Приоритет 31.07.2000. Опубликовано 27.07.2002.
4. Зайцева Н.В., Уланова Т.С., Леготкина Г.И., Стенно Е.В., Баканина М.А., Шардакова Ю.В. Способ количественного определения марганца, свинца и никеля в желчи методом атомно-абсорбционного анализа с атомизацией в пламени. Патент на изобретение №2410691. Приоритет 04.08.2009. Опубликовано 27.01.2011 бюл. №3 (прототип).
5. Дробышев А.И., Савинов С.С. Дуговой атомно-эмиссионный цифровой спектрографический анализ жидких биопроб с использованием МАЭС // Заводская лаборатория. Диагностика материалов. 2015, №1 (II), с. 142-145.
6. Савинов С.С., Дробышев А.И. Возможности атомно-эмиссионной цифровой спектрографии с дуговым возбуждением спектра в анализе жидких объектов // Вестник Санкт-Петербургского университета. Серия 4: физика, химия. 2013, №3, с. 98-102.
7. Зайдель А.Н. Основы спектрального анализа. М.: Наука, 1965 г., стр. 167.
8. Аполицкий В.Н. Способ анализа жидкости. Патент на изобретение №2280855. Приоритет 19.04.2004. Опубликовано 27.07.2006.
9. Танеев А.А., Потапов С.В., Усков К.Н., Крашенинников А.А. Масс-спектральное устройство для быстрого и прямого анализа проб. Патент на изобретение №2487434. Приоритет 26.01.2012. Опубликовано 10.07.2013.
10. Дробышев А.И., Савинов С.С. О некоторых особенностях регистрации спектра и фотометрирования спектральных линий с помощью цифрового спектрографа на базе МФС-МАЭС // Приборы и техника эксперимента. 2013, №6, с. 56-59.
11. Дробышев А.И., Савинов С.С. Экспериментальное исследование аппаратной функции и разрешающей способности оптического цифрового спектрографа на базе полихроматора МФС // Оптический журнал. 2014, т. 81, №1, с. 44-53.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ пробоподготовки растительных масел для определения их микроэлементного состава спектральными методами | 2018 |
|
RU2688840C1 |
| Способ спектрального определения микроэлементов в твердых восксодержащих пробах | 2022 |
|
RU2806045C1 |
| Способ измерений массовых концентраций мышьяка, кадмия, свинца, ртути в мясных и мясосодержащих продуктах методом масс-спектрометрии с индуктивно связанной плазмой | 2020 |
|
RU2738166C1 |
| Способ измерений массовых концентраций алюминия, мышьяка, стронция, кадмия, свинца, ртути в мукомольно-крупяных и хлебобулочных изделиях методом масс-спектрометрии с индуктивно связанной плазмой | 2021 |
|
RU2779425C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПРИМЕСЕЙ В НИКЕЛЕ, МЕДИ, КОБАЛЬТЕ И ИХ ПОРОШКАХ С ИСПОЛЬЗОВАНИЕМ СИНТЕТИЧЕСКИХ ОКСИДНЫХ СТАНДАРТНЫХ ОБРАЗЦОВ | 2000 |
|
RU2193183C2 |
| СПОСОБ ПРОБОПОДГОТОВКИ БИОЛОГИЧЕСКИХ ОБРАЗЦОВ ДЛЯ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЙОДА | 2023 |
|
RU2808066C1 |
| СПОСОБ ПРОБОПОДГОТОВКИ РАСТИТЕЛЬНОГО СЫРЬЯ С ВЫСОКИМ СОДЕРЖАНИЕМ МАСЛА ДЛЯ ЭЛЕКТРОТЕРМИЧЕСКОГО АТОМНО-АБСОРБЦИОННОГО АНАЛИЗА | 2016 |
|
RU2645995C2 |
| Способ определения суммарного содержания оловоорганических соединений в природных водах | 2022 |
|
RU2791423C1 |
| Способ атомно-эмиссионного определения олова в полимерах | 2020 |
|
RU2758435C1 |
| Способ подготовки проб для определения содержания свинца в пиролизной жидкости | 2018 |
|
RU2694355C1 |
Способ спектрального определения микроэлементного состава вязких органических жидкостей заключается в том, что анализу подвергается малый объем пробы, который предварительно минерализуется под действием малого объема концентрированной азотной кислоты при нагревании. Пробоподготовка производится на торце электрода с получением сухого остатка пробы, который анализируется спектральными методами. Количественная оценка производится по градуировочным графикам. Технический результат – сокращение времени и упрощение пробоподготовки, уменьшение массы анализируемой пробы и используемых реагентов, повышение чувствительности, повышение достоверности результатов. 1 з.п. ф-лы, 1 ил., 3 табл.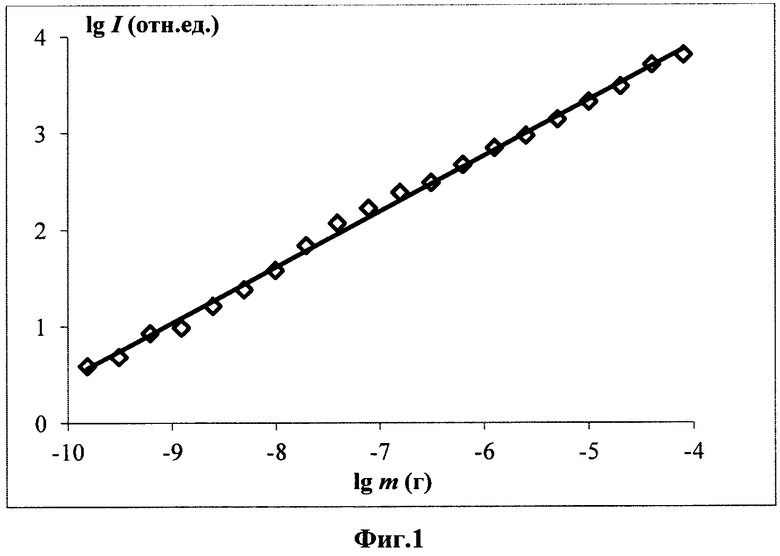
1. Способ спектрального определения микроэлементного состава вязких органических жидкостей, заключающийся в пробоподготовке при добавлении окислителей концентрированных азотной кислоты и пероксида водорода в химической посуде и последующем нагревании, разбавлении полученного минерализата и его последующем анализе, отличающийся тем, что пробоподготовку проводят на торце электрода, на который предварительно наносят анализируемую пробу в объеме 10 мкл, после чего ее высушивают, затем наносят концентрированную азотную кислоту в объеме 5-50 мкл и нагревают до получения сухого остатка, который анализируют на наличие в пробе микроэлементов, количественное определение элементов проводят на основе предварительно построенных градуировочных графиков с использованием калибровочных водных растворов солей элементов, которые проводят через все описанные этапы анализа.
2. Способ спектрального определения микроэлементного состава вязких органических жидкостей по п. 1, отличающийся тем, что сухой остаток анализируют известным спектральным методом, позволяющим проводить анализ сухого остатка жидкостей и растворов.
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ МАРГАНЦА, СВИНЦА И НИКЕЛЯ В ЖЕЛЧИ МЕТОДОМ АТОМНО-АБСОРБЦИОННОГО АНАЛИЗА С АТОМИЗАЦИЕЙ В ПЛАМЕНИ | 2009 |
|
RU2410691C1 |
| Дробышев А.И | |||
| и др | |||
| ОПТИМИЗАЦИЯ АНАЛИТИЧЕСКОЙ ЦИФРОВОЙ СПЕКТРОГРАФИИ ДЛЯ ОПРЕДЕЛЕНИЯ МИКРОСОДЕРЖАНИЙ ЭЛЕМЕНТОВ В ЖИДКИХ ОБРАЗЦАХ | |||
| Второй съезд аналитиков России | |||
| Тезисы докладов, 67, Москва, 27.09.2013 | |||
| Савинов С.С | |||
| Новые возможности дуговой атомно-эмиссионной спектрометрии для прямого анализа жидких биопроб | |||
| Диссертация на соискание ученой степени кандидата химических наук, Санкт-Петербург, 1-166, 2014 | |||
| И | |||
| Дробышев и др | |||
| О НЕКОТОРЫХ ОСОБЕННОСТЯХ РЕГИСТРАЦИИ СПЕКТРА И ФОТОМЕТРИРОВАНИЯ СПЕКТРАЛЬНЫХ ЛИНИЙ С ПОМОЩЬЮ ЦИФРОВОГО СПЕКТРОГРАФА НА БАЗЕ МФС-МАЭС | |||
| ПРИБОРЫ И ТЕХНИКА ЭКСПЕРИМЕНТА, N 6, с | |||
| Приспособление для разматывания лент с семенами при укладке их в почву | 1922 |
|
SU56A1 |
| Nomngongo P.N | |||
| et al | |||
| Functionalized nanometer-sized alumina supported micro-solid phase extraction coupled to inductively coupled plasma mass spectrometry for preconcentration and determination of trace metal ions in gasoline samples, RSC Advances | |||
| v | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Пюпитр для работы на пишущих машинах | 1922 |
|
SU86A1 |
| СПОСОБ ПЕРЕРАБОТКИ ХРОМАЛЯ ВО ФТОРИСТЫЕ СОЛИ АЛЮМИНИЯ И ХРОМА | 1935 |
|
SU46257A1 |

