Данное изобретение относится к применению замещенных нитрокатехолов формулы (I) в лечении расстройств центральной и периферической нервной системы в соответствии с определенным режимом введения (дозирования).
Основная причина применения ингибиторов СОМТ (катехол-О-метилтрансфераза) как вспомогательных средств к терапии леводопой/ингибитором декарбоксилазы ароматических L-аминокислот (AADCi) основана на их способности уменьшать метаболическое О-метилирование леводопы в 3-O-метил-леводопу (3-OMD). Продолжительность вызванного леводопой клинического улучшения является небольшой вследствие короткого периода полувыведения леводопы in vivo, который резко отличается от длительного периода полувыведения 3-OMD. Кроме того, 3-OMD конкурирует с леводопой за транспорт через гематоэнцефалический барьер (ВВВ), что означает, что только очень ограниченная часть перорально введенной дозы леводопы действительно достигает места действия, то есть мозга. Как правило, в течение всего лишь нескольких лет от начала терапии леводопой в обычном режиме введения, вызванное леводопой клиническое улучшение уменьшается к концу каждого цикла дозировки, приводя к так называемой картине «изнашивания» двигательных флуктуаций. Была описана тесная взаимосвязь между феноменом «изнашивания» и накоплением 3-OMD (Tohgi, Н., et al., Neurosci. Letters, 132:19-22, 1992). Было выдвинуто предположение, что это может происходить в результате нарушения проникновения леводопы в мозг вследствие конкуренции с 3-OMD за систему транспорта через ВВВ (Reches, А. et al., Neurology, 32:887-888, 1982) или, проще говоря, меньше леводопы доступно для того, чтобы достичь мозга (Nutt, J.G., Fellman, J.H., Clin. Neuropharmacol., 7:35-49, 1984). В действительности, ингибирование СОМТ защищает леводопу от метаболического разрушения O-метилированием на периферии и особенно в кишечнике, так что при повторных введениях доз леводопы средняя концентрация леводопы в плазме повышается. Дополнительно к уменьшению конкуренции за транспорт в мозг значительно более высокий процент от перорально введенной дозы леводопы получает возможность достичь места действия. Таким образом, ингибирование СОМТ служит для повышения биодоступности леводопы, и продолжительность антипаркинсонического действия увеличивается при однократных введениях леводопы (Nutt, J.G., Lancet, 351:1221-1222, 1998).
Наиболее мощными ингибиторами СОМТ, о которых сообщалось до сих пор, являются 3,4-дигидрокси-4'-метил-5-нитробензофенон (Толкапон, патент Австралии AU-B-69764/87) и (E)-2-циано-N,N-диэтил-3-(3,4-дигидрокси-5-нитрофенил)акриламид (Энтакапон, патент Германии DE 3740383 A1).
Несмотря на то что они имеют по существу одинаковый фармакофор, толкапон отличается от энтакапона тем, что он легко проникает в центральную нервную систему (CNS) и способен ингибировать как мозговую СОМТ, так и периферическую СОМТ. Вскоре после его выхода на рынок толкапон был изъят с рынка после того, как появились сообщения о нескольких случаях гепатотоксичности, включая три несчастных случая гибели от молниеносного гепатита с летальным исходом. Сегодня толкапон может применяться только у пациентов с болезнью Паркинсона, невосприимчивых к другим способам лечения и только при регулярном мониторинге функции печени, что дорого и неудобно для пациента. Хотя действительные механистические причины печеночной токсичности, связанной с толкапоном, неполностью понятны, исследования in vitro показали, что толкапон может метаболически восстанавливаться в активные промежуточные соединения, и было выдвинуто предположение, что они могут образовывать ковалентные аддукты с белками печени, приводя к повреждению клеток печени (Smith, K.S. et al, Chem. Res. Toxicol., 16:123-128, 2003).
Энтакапон, с другой стороны, хотя и имеет такой же нитрокатехоловый фармакофор, как и толкапон, не связан с печеночной токсичностью и обычно рассматривается как безопасное лекарственное средство. К сожалению, однако, энтакапон является значительно менее мощным ингибитором СОМТ, чем толкапон, и имеет гораздо более короткий период полувыведения in vivo. Это означает, что энтакапон оказывает очень ограниченный по продолжительности эффект и, как следствие, лекарственное средство должно быть введено в очень высоких дозах с каждой дозой леводопы, принимаемой пациентом. Как таковая, клиническая эффективность энтакапона была поставлена под вопрос, действительно, в недавнем исследовании (Parashos, S.A. et al., Clin. Neuropharmacol., 27(3): 119-123, 2004) было выявлено, что основная причина прекращения лечения энтакапоном пациентов с болезнью Паркинсона заключалась в ощутимом недостатке эффективности.
Более того, относительно короткий период полувыведения in vivo известных ингибиторов СОМТ требует длительных режимов лечения, обычно включающих введение нескольких доз в сутки, что многие пациенты сочтут обременительным. Например, толкапон должен быть введен три раза в сутки. Поэтому этот фактор может помешать соблюдению пациентом режима и схемы лечения и нанести ущерб качеству жизни пациента.
Соответственно, все еще существует потребность в ингибиторах СОМТ, проявляющих сбалансированные свойства биоактивности, биодоступности и безопасности. В частности, существует потребность в ингибиторах СОМТ, имеющих большую продолжительность периода полувыведения in vivo и, следовательно, продолжительное действие на СОМТ, позволяя меньшим количеством доз достичь желаемого терапевтического эффекта.
Заявитель ранее обнаружил соединения, которые, несмотря на то, что они имеют относительно короткий период полувыведения, являются очень мощными ингибиторами СОМТ, обладающими исключительно большой продолжительностью действия по сравнению с ингибиторами СОМТ из предшествующего уровня техники (см. WO 2007/013830).
Эти соединения, которые показаны ниже как соединения общей формулы (I), также заметно увеличивают биодоступность леводопы и увеличивают доставку леводопы к мозгу. Эти соединения значительно повышают уровни дофамина в мозге на продолжительный период времени.
Еще более удивительно то, что повышенные уровни леводопы сохраняются стабильными в течение продолжительных периодов времени. Эти устойчивые эффекты на активность СОМТ и на биодоступность леводопы после введения соединений общей формулы (I) заметно более сильны, чем эффекты, наблюдаемые для толкапона, единственного ингибитора СОМТ, для которого к настоящему времени известно, что он обладает относительно большой продолжительностью действия (толкапон имеет конечный период полувыведения примерно 2 ч и должен быть введен примерно 3 раза в сутки.) Более того, соединения общей формулы (I) дают стабильное увеличение доставки леводопы к мозгу на протяжении длительных периодов времени, что резко отличается от того, что наблюдается для толкапона, который склонен вызывать заметные колебания в доставке леводопы в мозг. Таким образом, более вероятно, что соединения общей формулы (I) будут обладать терапевтическими преимуществами вследствие устойчивого постоянного повышения уровней леводопы, в то время как вероятно, что применение толкапона вызывает нежелательные побочные эффекты, такие как дискинезия, вследствие резких повышений и понижений уровней леводопы.
Настоящее изобретение основано на режиме введения, обладающем неожиданными преимуществами для введения соединений формулы (I), который максимально увеличивает ингибиторный эффект данных соединений в отношении СОМТ.
Соответственно, в первом аспекте настоящее изобретение относится к соединению формулы (I)
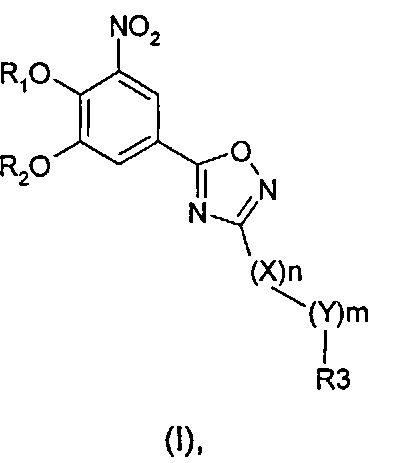
где R1 и R2 одинаковые или разные и обозначают атомы водорода, группы, гидролизуемые при физиологических условиях, или возможно замещенные алканоилы или ароилы; X обозначает метиленовую группу; Y представляет собой O, S или NH; n равно 0, 1, 2 или 3; m равно 0 или 1; R3 обозначает пиридиновую N-оксидную группу формулы A, B или C, которая присоединена, как обозначено "открытой" связью:
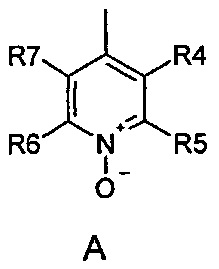

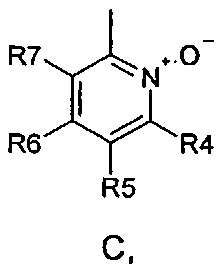
где R4, R5, R6 и R7 одинаковые или разные и обозначают водород, алкил, тиоалкил, алкокси, арилокси, тиоарил, алканоил, ароил, арил, амино, алкиламино, диалкиламино, циклоалкиламино, гетероциклоалкиламино, алкилсульфонил, арилсульфонил, галоген, галогеналкил, трифторметил, циано, нитро или гетероарил; или два или более чем два из R4, R5, R6 и R7 вместе взятые обозначают алифатические или гетероалифатические кольца или ароматические или гетероароматические кольца; термин "алкил", включая его вариант "алк-" в таких терминах, как "алкокси", "алканоил", обозначает углеродные остатки, прямые или разветвленные, содержащие от одного до шести атомов углерода; термин "арил" обозначает фенильную или нафтильную группу; термин "гетероциклоалкил" обозначает циклическое кольцо, от четырехчленного до восьмичленного, возможно включающее по меньшей мере один атом кислорода, серы или азота; термин "гетероарил" обозначает пятичленное или шестичленное кольцо, включающее по меньшей мере один атом серы, кислорода или азота; термин "галоген" обозначает фтор, хлор, бром или йод; и если R4, R5, R6 и R7 обозначают алкил или арил, они возможно замещены одной или более чем одной группой гидрокси, алкокси или галоген; или его фармацевтически приемлемой соли, сложному эфиру, карбамату или фосфату, для применения в профилактике или лечении расстройства центральной и периферической нервной системы, где соединение формулы (I) вводят до сна, до наступления времени сна или при наступлении времени сна.
Во втором аспекте настоящее изобретение относится к соединению формулы (I)
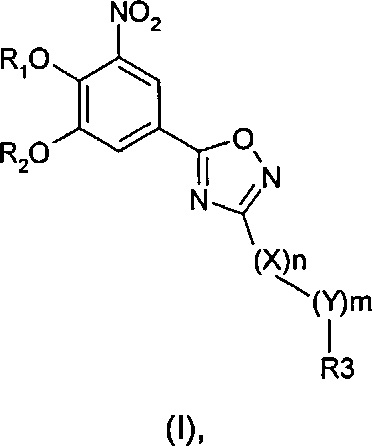
где R1 и R2 одинаковые или разные и обозначают атомы водорода, группы, гидролизуемые при физиологических условиях, или возможно замещенные алканоилы или ароилы; X обозначает метиленовую группу; Y представляет собой O, S или NH; n равно 0, 1, 2 или 3; m равно 0 или 1; R3 обозначает пиридиновую N-оксидную группу формулы A, B или C, которая присоединена, как обозначено, "открытой" связью:
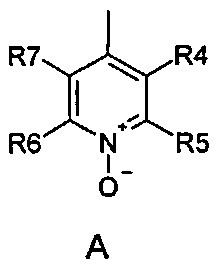
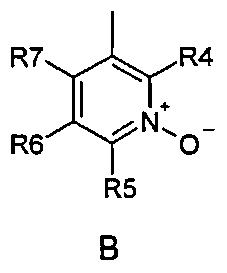
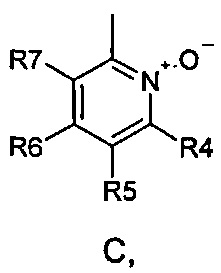
где R4, R5, R6 и R7 одинаковые или разные и обозначают водород, алкил, тиоалкил, алкокси, арилокси, тиоарил, алканоил, ароил, арил, амино, алкиламино, диалкиламино, циклоалкиламино, гетероциклоалкиламино, алкилсульфонил, арилсульфонил, галоген, галогеналкил, трифторметил, циано, нитро или гетероарил; или два или более чем два из R4, R5, R6 и R7 вместе взятые обозначают алифатические или гетероалифатические кольца или ароматические или гетероароматические кольца; термин "алкил", включая его вариант "алк-" в таких терминах, как "алкокси", "алканоил", обозначает углеродные остатки, прямые или разветвленные, содержащие от одного до шести атомов углерода; термин "арил" обозначает фенильную или нафтильную группу; термин "гетероциклоалкил" обозначает циклическое кольцо, от четырехчленного до восьмичленного, возможно включающее по меньшей мере один атом кислорода, серы или азота; термин "гетероарил" обозначает пятичленное или шестичленное кольцо, включающее по меньшей мере один атом серы, кислорода или азота; термин "галоген" обозначает фтор, хлор, бром или йод; и если R4, R5, R6 и R7 обозначают алкил или арил, они возможно замещены одной или более чем одной группой гидрокси, алкокси или галоген; или его фармацевтически приемлемой соли, сложному эфиру, карбамату или фосфату для применения в профилактике или лечении расстройства центральной и периферической нервной системы, где соединение формулы (I) вводят без пищи и/или между приемами пищи.
В третьем аспекте изобретение предусматривает применение соединения формулы (I) для изготовления лекарственного средства для применения в профилактике или лечении расстройства центральной и периферической нервной системы, где указанное соединение вводят без пищи, между приемами пищи, до сна, до наступления времени сна и/или при наступлении времени сна.
В четвертом аспекте изобретение предусматривает способ профилактики или лечения расстройства центральной и периферической нервной системы, включающий введение пациенту, страдающему от указанного заболевания, терапевтически эффективного количества соединения формулы (I) без пищи, между приемами пищи, до сна, до наступления времени сна и/или при наступлении времени сна.
Неожиданно заявитель обнаружил преимущество введения соединений формулы (I) пациенту, пищеварительная система которого не содержит пищи в той степени, насколько это возможно. Следовательно, соединения формулы (I) предпочтительно следует вводить пациенту без пищи и/или между приемами пищи, например между приемами пищи, перед сном, до наступления времени сна или при наступлении времени сна. Введение соединения таким образом приводит к улучшению активности соединений формулы (I), например более продолжительному и усиленному ингибированию СОМТ.
Далее, как упоминается, ингибиторы СОМТ формулы (I) применяются как вспомогательное средство в терапии катехоламинами с тем, чтобы снизить метаболизм катехоламинового лекарственного средства, вызванный СОМТ. Заявитель также неожиданно обнаружил, что, когда соединение формулы (I) вводят пациенту, уже принимающему катехоламин, такой как леводопа (L-DOPA), эффекты соединения формулы (I) улучшаются, если соединение формулы (I) вводят последовательно с катехоламином. В частности, неожиданно было обнаружено, что соединение формулы (I) неблагоприятно воздействует на биодоступность леводопы, и леводопа неблагоприятно воздействует на биодоступность соединения формулы (I). Следовательно, согласно изобретению соединение формулы (I) вводят перед сном, до наступления времени сна или при наступлении времени сна, до или после того как пациенту была дана последняя суточная доза леводопы и перед введением дозы леводопы следующих суток. Следовательно, соединение формулы (I) и катехоламиновое лекарственное средство не находятся одновременно в пищеварительной системе пациента и/или по существу не всасываются в одно и то же время.
Для целей данного изобретения, термины "последняя суточная доза", "последняя доза суток", "последнее суточное введение" и "последнее введение суток" имеют одинаковое значение и могут использоваться взаимозаменяемо.
Далее настоящее изобретение описано со ссылками на сопровождающие графические материалы.
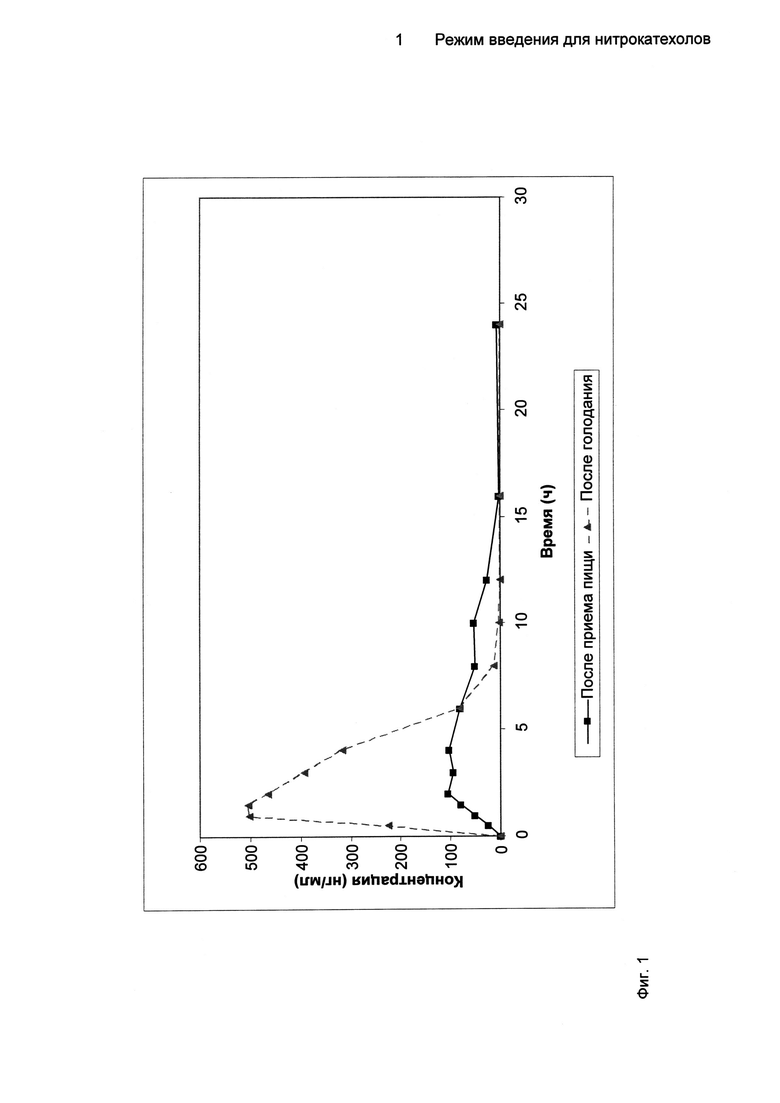
На Фиг.1 показан профиль зависимости средних концентраций 5-[3-(2,5-дихлор-4,6-диметил-1-оксипиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитробензол-1,2-диола (Соединение A) в плазме от времени в условиях голодания и приема пищи.
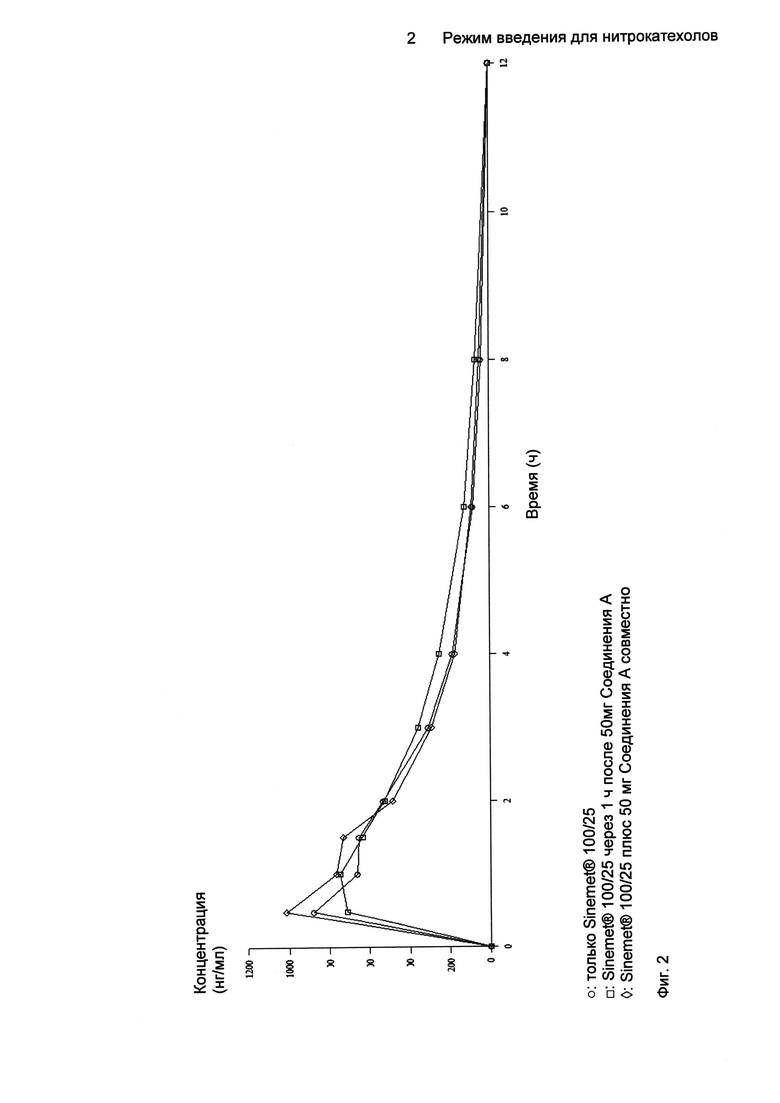
На Фиг.2 показаны профили зависимости средних концентраций леводопы в плазме от времени после однократного перорального введения 100/25 мг только Sinemet® (леводопа/карбидопа), 100/25 мг Sinemet®, введенного с 50 мг Соединения A с интервалом в 1 ч и введенного совместно с 50 мг Соединения A.
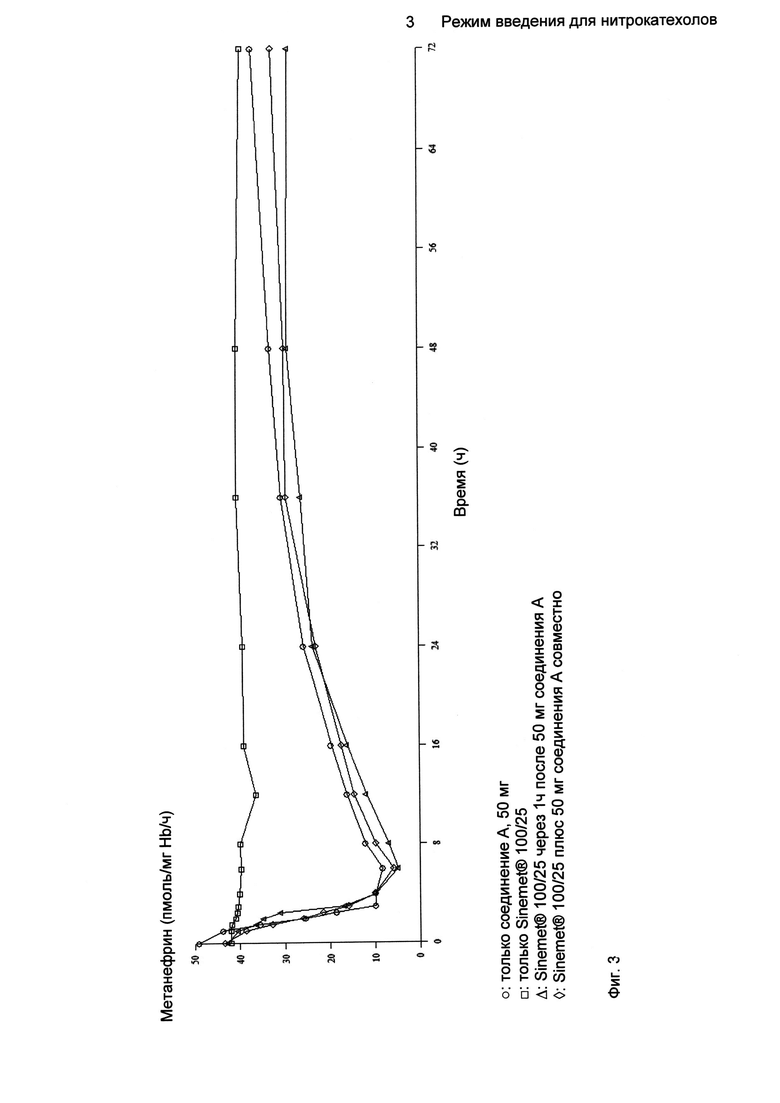
На Фиг.3 показаны профили средней активности S-COMT (образованный метанефрин, пмоль/мг белка/ч) от базового уровня (до введения дозы): после однократного перорального введения 100/25 мг только Sinemet®, 100/25 мг Sinemet®, введенного с 50 мг Соединения A с интервалом в 1 ч и введенного совместно с 50 мг Соединения A и после введения только Соединения A.
Как упоминалось, из-за того, что соединения формулы (I) являются очень мощными, они потенциально могут быть введены в виде однократной суточной дозы.
Соединения формулы (I) предпочтительно вводят перед сном, до наступления времени сна или при наступлении времени сна. Термин "перед сном" означает, что соединение формулы (I) вводят незадолго перед тем, как пациент ложится спать, например менее чем за 90 мин до сна, в частности менее чем за 1 ч до сна, менее чем за 30 мин до сна или непосредственно перед сном.
Термин "до наступления времени сна" (то есть перед тем, как лечь в постель) означает, в частности, менее чем за 90 мин до того, как лечь в постель, в частности менее чем за 60 мин до того, как лечь в постель, или менее чем за 30 мин до того, как лечь в постель. Термин "при наступлении времени сна" означает менее чем за 5 мин до наступления времени сна, например во время укладывания в постель.
Другими словами, пациент принимает соединение формулы (I) до того, как пациент ложится в постель (то есть до наступления времени сна или при наступлении времени сна), например менее чем за 90 мин до наступления времени сна, в частности менее чем за 60 мин до наступления времени сна, менее чем за 30 мин до наступления времени сна или менее чем за 5 мин до наступления времени сна.
Как будет ясно, в контексте данного изобретения, термин "перед сном" или "до наступления времени сна" не означает любое время дня перед сном или перед тем, как лечь в постель, и в частности не включает, например, 12 ч до сна или до того как лечь в постель. Скорее этот термин означает, что лекарственное средство принимают в период, близкий к тому, когда пациент ложится спать и, вероятно, как часть рутинной подготовки пациента ко сну.
В одном воплощении соединение формулы (I) вводят в форме комбинированной терапии с катехоламиновым лекарственным средством. Предпочтительно катехоламиновое лекарственное средство представляет собой леводопу.
Как таковые, режимы введения соединения формулы (I) и катехоламинового лекарственного средства могут различаться: каждый может быть введен в одно и то же время или в разное время. Поэтому будет ценным то, что соединения комбинации могут быть введены последовательно (например, до или после) или совместно - либо в одной и той же фармацевтической композиции (то есть вместе), либо в различных фармацевтических композициях (то есть раздельно). Одновременно в одной и той же композиции значит в единой композиции, в то время как одновременно в разных фармацевтических композициях значит не в единой. Режим введения каждого из двух или более чем двух соединений в комбинированной терапии также может отличаться в отношении пути введения.
Заявитель неожиданно обнаружил, что соединения формулы (I) и катехоламиновое лекарственное средство оказывают неблагоприятный эффект на биодоступность друг друга.
В частности, соединения формулы (I) вводят перед сном, до наступления времени сна или при наступлении времени сна, до или после последнего введения суток катехоламинового лекарственного средства и до первого введения катехоламинового лекарственного средства следующих суток. Следовательно, это позволяет избежать негативных последствий, которые каждое лекарственное средство оказывает на биодоступность другого. Предпочтительно, ингибиторная активность соединений формулы (I) в отношении СОМТ имеет место до введения катехоламинового лекарственного средства.
В одном воплощении соединение формулы (I) вводят за 30-150 мин до последнего суточного введения катехоламинового лекарственного средства или через 30-150 мин после последнего суточного введения катехоламинового лекарственного средства. Например, соединение формулы (I) вводят по меньшей мере за 30-50 мин, предпочтительно по меньшей мере за 1 ч, до последнего суточного введения катехоламинового лекарственного средства или через 30-50 мин, предпочтительно по меньшей мере через 1 ч после последнего суточного введения катехоламинового лекарственного средства.
В другом воплощении соединение формулы (I) вводят по меньшей мере за 1 ч до или через 1 ч после последнего суточного введения катехоламинового лекарственного средства, и предпочтительно соединение формулы (I) вводят один раз в сутки по меньшей мере за 1 ч до последнего суточного введения катехоламинового лекарственного средства или через 1 ч после последнего суточного введения катехоламинового лекарственного средства. В воплощениях изобретения рассматривают период по меньшей мере два, три, четыре, пять или шесть часов между введением катехоламина и соединений формулы (I).
Предпочтительно, при последовательном введении катехоламин вводят по меньшей мере через два, более предпочтительно через три и наиболее предпочтительно через по меньшей мере шесть часов после введения соединения формулы (I). Соответствующим образом, при последовательном введении катехоламин вводят через 12 ч или 23 ч после введения соединения формулы (I). Предпочтительно, последовательное введение катехоламинового лекарственного средства представляет собой первую суточную дозу катехоламинового лекарственного средства следующих суток.
Дополнительно, режим введения согласно изобретению включает введение соединения формулы (I), когда пищеварительная система пациента не содержит пищи. Заявитель обнаружил, что соединения формулы (I) имеют улучшенную биодоступность при введении пациенту, когда в пищеварительной системе пациента отсутствует пища. В частности, соединение формулы (I) следует вводить пациенту до сна, до наступления времени сна или при наступлении времени сна без пищи, после последнего на данные сутки приема пищи пациентом.
Термин "пищеварительная система не содержит пищи" означает, что часть пищеварительной системы, где происходит большая часть всасывания соединения формулы (I), например желудок, тонкая кишка (двенадцатиперстная кишка, тощая кишка, подвздошная кишка), не содержит пищи.
В одном воплощении изобретения соединение формулы (I) вводят по меньшей мере через 1 ч после последнего приема пищи и по меньшей мере за 1 ч до следующего приема пищи.
В одном воплощении изобретения соединение формулы (I) вводят через 0,25-12 ч, предпочтительно через 0,5-6 ч, более предпочтительно через 0,75-4 ч после приема пищи. В одном воплощении изобретения соединение формулы (I) вводят после 0,25-10 ч ночного голодания.
В одном воплощении изобретения соединение формулы (I) вводят за 0,25-2 ч до приема пищи, предпочтительно за 0,5-1,5 ч до приема пищи.
Предпочтительно соединение формулы (I) вводят до сна и более предпочтительно менее чем за 1 ч до сна.
Наиболее предпочтительно, с целью избежать взаимодействия между соединением формулы (I) и катехоламиновым лекарственным средством и также для введения соединения формулы (I) в условиях, когда пищеварительная система пациента не содержит пищи, соединение формулы (I) вводят один раз в сутки до сна, до наступления времени сна или при наступлении времени сна.
Как он используется здесь, термин «эффективная суточная доза» обозначает эффективное суточное количество введенного соединения, которое вводят в соответствии с периодичностью дозировки.
В настоящем изобретении эффективные суточные дозы соединений общей формулы (I) находятся в пределах от примерно 1 до примерно 1200 мг/сутки, предпочтительно от примерно 1 до примерно 900 мг/сутки, более предпочтительно от примерно 5 до примерно 400 мг/сутки, еще более предпочтительно от примерно 25 до примерно 300 мг/сутки, например конкретные суточные дозы в 1 мг, 3 мг, 5 мг, 10 мг, 15 мг, 20 мг, 25 мг, 30 мг, 50 мг, 100 мг, 200 мг, 400 мг, 800 мг или 1200 мг.
Как он используется здесь, термин «единица дозировки» относится к количеству соединения, введенного в каждом введении периодичности дозировки.
Предпочтительно, чтобы индивидуальные единицы дозировки соединений общей формулы (I) находились в пределах от примерно 1 до примерно 2400 мг, более предпочтительно от примерно 1 до примерно 1200 мг, еще более предпочтительно от примерно 1 до примерно 800 мг, например 1 мг, 3 мг, 5 мг, 10 мг, 15 мг, 20 мг, 25 мг, 30 мг, 50 мг, 100 мг, 200 мг, 400 мг, 800 мг или 1200 мг.
Как упоминалось выше, ингибиторы СОМТ часто применяются как вспомогательные средства к катехоламиновым соединениям, потому что они уменьшают их метаболическое О-метилирование. В частности, ингибиторы СОМТ часто применяются как вспомогательные средства к терапии леводопой/ингибитором декарбоксилазы ароматических L-аминокислот (AADCi), потому что они уменьшают метаболическое О-метилирование леводопы в 3-О-метил-леводопу (3-OMD).
Поэтому предпочтительно, чтобы патологические состояния, которые лечат этими соединениями, представляли собой расстройства, связанные с центральной и периферической нервной системой человека, на которые введение ингибитора СОМТ оказывает благоприятное воздействие.
Когда соединение формулы (I) вводят в комбинации с катехоламиновым лекарственным средством, возможно, что катехоламиновое лекарственное средство вводят последовательно или совместно с AADCi, в частности кардидопой или бенсеразидом.
Соединения общей формулы (I), катехоламиновое лекарственное средство и AADCi могут быть введены раздельно или в любой комбинации. Они могут быть введены совместно (например, одновременно) или последовательно и с одинаковой или разной периодичностью дозировки. Например, соединения общей формулы (I) можно вводить совместно или последовательно с катехоламиновым лекарственным средством.
Применение соединений согласно изобретению предназначено для профилактики или лечения расстройств центральной и периферической нервной системы. Расстройство центральной и периферической нервной системы представляет собой, например, аффективное расстройство, желудочно-кишечное расстройство, состояние формирования отека, гипертензию или двигательное расстройство. Предпочтительно, расстройства представляют собой двигательные расстройства, включая расстройства, включающие паркинсонизм, болезнь Паркинсона и синдром беспокойных ног. Наиболее предпочтительное расстройство центральной и периферической нервной системы представляет собой болезнь Паркинсона.
Как он используется здесь, термин "лечение" и такие его вариации, как "лечить" и "процесс лечения", относятся к любому режиму, который может быть благоприятным для человека или животного, не являющегося человеком. Дополнительно соединения формулы (I) можно применять для профилактики (профилактического лечения). Лечение может включать лечебное, облегчающее или ослабляющее симптомы действие, такие действия, относящиеся к одному или более чем одному симптому, связанному с расстройствами центральной и периферической нервной системы.
Одно конкретное воплощение изобретения, которое можно упомянуть, представляет собой соединение формулы (I), в частности 5-[3-(2,5-дихлор-4,6-диметил-1-оксипиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитробензол-1,2-диол и его фармакологически приемлемые соли, сложные эфиры, карбаматы и фосфаты, для применения в комбинации с катехоламиновым лекарственным средством, в частности леводопой, для профилактики или лечения расстройства центральной и периферической нервной системы, особенно двигательного расстройства, такого как болезнь Паркинсона, где соединение формулы (I) вводят перорально один раз в сутки по меньшей мере за 1 ч до приема последней суточной дозы катехоламинового лекарственного средства или через 1 ч после приема последней суточной дозы катехоламинового лекарственного средства и до сна, до наступления времени сна или при наступлении времени сна и/или без пищи, и/или между приемами пищи, и/или по меньшей мере через 1 ч после последнего приема пищи и по меньшей мере за 1 ч до следующего приема пищи.
Согласно другому аспекту данного изобретения предложен способ лечения по меньшей мере одного патологического состояния у пациента, нуждающегося в этом, включающий введение пациенту фармакологически эффективной дозы соединения общей формулы (I), как определено выше, без пищи, и/или между приемами пищи, и/или до сна, и/или до наступления времени сна, и/или при наступлении времени сна, и/или до или после введения катехоламинового лекарственного средства.
Согласно другому аспекту данного изобретения предложен способ ингибирования СОМТ у субъекта, включающий введение субъекту эффективной дозы соединения общей формулы (I), как определено выше, без пищи, и/или между приемами пищи, и/или до сна, и/или до наступления времени сна, и/или при наступлении времени сна, и/или до или после введения катехоламинового лекарственного средства.
Согласно другому аспекту данного изобретения предложен способ повышения уровней леводопы в мозге субъекта, которого лечат леводопой, включающий введение субъекту эффективной дозы соединения общей формулы (I), как определено выше, без пищи, и/или между приемами пищи, и/или до сна, и/или до наступления времени сна, и/или при наступлении времени сна, и/или до или после введения катехоламинового лекарственного средства.
Согласно другому аспекту данного изобретения предложен способ повышения уровней леводопы в плазме субъекта, которого лечат леводопой, включающий введение субъекту эффективной дозы соединения общей формулы (I), как определено выше, без пищи, и/или между приемами пищи, и/или до сна, и/или до наступления времени сна, и/или при наступлении времени сна, и/или до или после введения катехоламинового лекарственного средства.
Согласно другому аспекту данного изобретения предложен способ понижения уровней 3-О-метил-леводопы (3-OMD) в мозге субъекта, которого лечат леводопой, включающий введение субъекту эффективной дозы соединения общей формулы (I), как определено выше, без пищи, и/или между приемами пищи, и/или до сна, и/или до наступления времени сна, и/или при наступлении времени сна, и/или до или после введения катехоламинового лекарственного средства.
Согласно другому аспекту данного изобретения предложен способ понижения уровней 3-OMD в плазме субъекта, которого лечат леводопой, включающий введение субъекту эффективной дозы соединения общей формулы (I), как определено выше, без пищи и/или между приемами пищи и/или до сна, и/или до наступления времени сна, и/или при наступлении времени сна, и/или до или после введения катехоламинового лекарственного средства.
Согласно другому аспекту данного изобретения предложен способ увеличения биодоступности леводопы в мозге субъекта, которого лечат леводопой, включающий введение субъекту эффективной дозы соединения общей формулы (I), как определено выше, без пищи, и/или между приемами пищи, и/или до сна, и/или до наступления времени сна, и/или при наступлении времени сна, и/или до или после введения катехоламинового лекарственного средства.
Согласно другому аспекту данного изобретения предложен способ увеличения биодоступности леводопы в плазме субъекта, которого лечат леводопой, включающий введение субъекту эффективной дозы соединения общей формулы (I), как определено выше, без пищи, и/или между приемами пищи, и/или до сна, и/или до наступления времени сна, и/или при наступлении времени сна, и/или до или после введения катехоламинового лекарственного средства.
Настоящее изобретение также относится к упаковке, включающей фармацевтическую композицию соединения общей формулы (I) в комбинации с инструкциями по введению указанной композиции без пищи, и/или между приемами пищи, и/или до сна, и/или до наступления времени сна, и/или при наступлении времени сна, и/или до или после введения катехоламинового лекарственного средства.
Для соединений формулы (I) предпочтительны следующие определения. Группы, гидролизуемые при физиологических условиях, представляют собой группы, расщепляемые in vivo при физиологических значениях pH и температуры. На странице 1354 6-го издания "Foye's Principles of Medicinal Chemistry", 2006, изд. Wolter Kluwer, указаны величины pH для тканевых жидкостей. Примерами групп, гидролизуемых при физиологических условиях, до функциональной группы –OH, являются сложные эфиры, карбаматы и фосфаты. Другие примеры групп, гидролизуемых при физиологических условиях до функциональной группы -OH хорошо известны специалисту в данной области и могут быть найдены, например, на страницах 101-103 Korolkovas, "Essentials of Medicinal Chemistry", 2-е издание, 1988, изд. John Wiley & Sons и на странице 426 Krogsgaard-larsen и др., "Textbook of Drug Design and Discovery", 3-е издание, 2002, изд. Taylor & Francis.
Предпочтительно, R4, R5, R6 и R7 независимо друг от друга представляют собой водород, C1-С6алкил, C6-C12арил, C1-C6тиоалкил, C1-C6алкокси, C6-C10арилокси, C6-C10тиоарил, C1-C6алканоил, C7-C11ароил, амино, C1-C6алкиламино, ди-C1-C6алкиламино, C3-C12циклоалкиламино, C4-C8гетероциклоалкиламино, C1-C6алкилсульфонил, C6-C10арилсульфонил, галоген, C1-C6галогеналкил, трифторметил, циано, нитро или гетероарил.
Когда R4, R5, R6 и/или R7 представляют собой C1-C6алкильные остатки, предпочтительно R4, R5, R6 и/или R7 представляют собой метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, пентил или гексил.
Когда R4, R5, R6 и/или R7 представляют собой C6-C12арильные остатки, предпочтительно R4, R5, R6 и/или R7 представляют собой фенил или нафтил.
Когда R4, R5, R6 и/или R7 представляют собой C1-C6тиоалкильные остатки, предпочтительно R4, R5, R6 и/или R7 представляют собой тиометил, тиоэтил, тио-н-пропил, тио-изопропил, тио-н-бутил, тио-н-пентил или тио-н-гексил.
Когда R4, R5, R6 и/или R7 представляют собой C1-C6алкокси-остатки, предпочтительно R4, R5, R6 и/или R7 представляют собой метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси или трет-бутокси.
Когда R4, R5, R6 и/или R7 представляют собой C6-C10арилокси-остатки, предпочтительно R4, R5, R6 и/или R7 представляют собой фенокси или нафтокси.
Когда R4, R5, R6 и/или R7 представляют собой C6-C10тиоарильные остатки, предпочтительно R4, R5, R6 и/или R7 представляют собой тиофенил или тионафтил.
Когда R4, R5, R6 и/или R7 представляют собой C1-C6алканоильные остатки, предпочтительно R4, R5, R6 и/или R7 представляют собой метаноил, этаноил, пропаноил или бутаноил.
Когда R4, R5, R6 и/или R7 представляют собой C7-C11ароильные остатки, предпочтительно R4, R5, R6 и/или R7 представляют собой бензоил или нафтоил.
Когда R4, R5, R6 и/или R7 представляют собой C1-C6-алкиламино-остатки, предпочтительно R4, R5, R6 и/или R7 представляют собой метиламино, этиламино, н-пропиламино, изопропиламино или н-бутиламино.
Когда R4, R5, R6 и/или R7 представляют собой ди-C1-C6алкиламино-остатки, предпочтительно R4, R5, R6 и/или R7 представляют собой диметиламино, диэтиламино, ди-н-пропиламино, ди-н-бутиламино, диизопропиламино, метилэтиламино, метилпропиламино или этилпропиламино.
Когда R4, R5, R6 и/или R7 представляют собой C3-C12циклоалкиламино-остатки, предпочтительно R4, R5, R6 и/или R7 представляют собой пирролидино, пиперидино, циклогексиламино или дициклогексиламино.
Когда R4, R5, R6 и/или R7 представляют собой C4-C8гетероциклоалкиламино-остатки, предпочтительно R4, R5, R6 и/или R7 представляют собой морфолино, 2,6-диметилморфолино, 3,5-диметилморфолино, пиперазино, N-метилпиперазино или N-этилпиперазино.
Когда R4, R5, R6 и/или R7 представляют собой C1-C6алкилсульфонильные или C6-C10-арилсульфонильные остатки, предпочтительно R4, R5, R6 и/или R7 представляют собой метилсульфонил, этилсульфонил, фенилсульфонил или толилсульфонил.
Когда R4, R5, R6 и/или R7 представляют собой галогеновые остатки, предпочтительно R4, R5, R6 и/или R7 представляют собой хлор, бром, йод или фтор.
Когда R4, R5, R6 и/или R7 представляют собой C1-C6галогеналкильные остатки, предпочтительно R4, R5, R6 и/или R7 представляют собой хлорметил, фторметил, дихлорметил, дифторметил, трихлорметил или трифторметил.
Когда R4, R5, R6 и/или R7 представляют собой гетероарильные остатки, предпочтительно R4, R5, R6 и/или R7 представляют собой пиридил, пиримидил, изоксазолил, оксазолил, изоксадиазолил, оксадиазолил, триазолил или тетразолил.
Когда два или более чем два остатка R4, R5, R6 и R7 вместе взятые представляют собой алифатические или гетероалифатические кольца или ароматические или гетероароматические кольца, два или более чем два остатка предпочтительно представляют собой алифатические или гетероалифатические кольца или ароматические или гетероароматические кольца. Предпочтительные комбинированные остатки представляют собой индолизинил, изоиндолил, индолил, индазолил, пуринил, хинолизинил, нафтиридинил, изохинолил и хинолил.
Там, где они представляют собой арил или алкил, вышеупомянутые заместители R4, R5, R6 и R7 возможно могут быть замещены группами гидрокси, алкокси или галоген один или более чем один раз.
В настоящем описании медицинских показаний, лечений и режимов дозирования для фармацевтических композиций, содержащих соединения по изобретению, соответствующие общей формуле (I), наиболее предпочтительный пример соединения, соответствующего общей формуле (I), представляет собой 5-[3-(2,5-дихлор-4,6-диметил-1-оксипиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитробензол-1,2-диол, далее обозначенный как соединение A, и его фармакологически приемлемые соли, сложные эфиры, карбаматы и фосфаты. Период полувыведения соединения A относительно короткий, принимая во внимание большую продолжительность его действия.
Другие предпочтительные соединения приведенной выше общей формулы (I) для применения при последующих медицинских показаниях, лечениях и режимах дозирования включают 3-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-4-(трифторметил)пиридин-1-оксид, 2-хлор-3-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-4,6-диметилпиридин-1-оксид, 3-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-2-метил-6-(трифторметил)пиридин-1-оксид, 5-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-2-(трифторметил)пиридин-1-оксид, 5-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-2-метил-4-(трифторметил)пиридин-1-оксид, 3-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-2,6-диметил-4-(трифторметил)пиридин-1-оксид, 3,5-дихлор-4-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)пиридин-1-оксид, 3-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-6-метил-2-фенил-4-(трифторметил)пиридин-1-оксид, 2-бром-3-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-4,5,6-триметил пиридин-1-оксид, 2-хлор-3-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-4,5,6-триметилпиридин-1-оксид, 3-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-2-(трифторметил)пиридин-1-оксид, 2,5-дихлор-3-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-4,6-диметилпиридин-1-оксид, 3-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-5-(трифторметил)пиридин-1-оксид, 3-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-2-фторпиридин-1-оксид, 4-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-2-фторпиридин-1-оксид, 2-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-6-фторпиридин-1-оксид, 2-хлор-3-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-6-метилпиридин-1-оксид, 2-бром-3-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-6-метилпиридин-1-оксид и 2-бром-5-хлор-3-(3-(3,4-дигидрокси-5-нитрофенил)-1,2,4-оксадиазол-5-ил)-4,6-диметилпиридин-1-оксид и их фармакологически приемлемые соли, сложные эфиры, карбаматы или фосфаты.
Подробности получения соединений общей формулы (I) можно найти в WO 2007/013830 A1.
Соединения общей формулы (I) также могут быть представлены в форме их фармакологически приемлемых солей, сложных эфиров, карбаматов или фосфатов. Подходящие фармацевтически приемлемые противоионы известны из уровня техники.
Также возможно применять пролекарства соединений общей формулы (I) с целью изменить терапевтический профиль активного соединения.
Соединение общей формулы (I) вводят в виде фармацевтической композиции. Для изготовления фармацевтических композиций соединений общей формулы (I) инертные фармацевтически приемлемые носители смешивают с активными соединениями. Фармацевтически приемлемые носители могут быть твердыми или жидкими. Твердые лекарственные формы включают порошки, таблетки, диспергируемые гранулы и капсулы. Твердый носитель может представлять собой одно или более чем одно вещество, которое также может действовать как разбавитель, ароматический агент, растворитель, смазывающее вещество, суспендирующий агент, связующее вещество, скользящий агент или разрыхлитель; он также может быть инкапсулирующим материалом.
Предпочтительно, фармацевтическая композиция представлена в стандартной лекарственной форме, например в виде упакованной композиции, где упаковка содержит дискретные количества композиции, например упакованные таблетки, капсулы и порошки во флаконах или в ампулах.
Обычно соединение формулы (I) вводят перорально.
Соединение формулы (I) обычно вводят от одного раза в сутки до примерно одного раза в неделю.
Для того чтобы избежать сомнений, когда соединения формулы (I) вводят с периодичностью реже чем один раз в сутки (например, один раз в неделю), понятно, что их следует вводить до сна, до наступления времени сна или при наступлении времени сна, до или после приема последней суточной дозы леводопы того (тех) дня(ей) недели, когда следует вводить соединение (I) и не каждый день, как леводопу. Например, для режима введения один раз в неделю, если соединение (I) вводят в первый день недели, его будут вводить до сна, до наступления времени сна или при наступлении времени сна, до или после введения последней суточной дозы леводопы этого дня. Следующее введение будет происходить в первый день второй недели, до сна, до наступления времени сна или при наступлении времени сна, до или после введения последней суточной дозы леводопы этого дня и так далее. На протяжении этого периода леводопу вводят каждый день (в нескольких дозах).
Согласно способам, описанным выше, соединение формулы (I) обычно вводят от одного раза в сутки до примерно одного раза в неделю.
Другие аспекты изобретения являются такими, как определено в формуле изобретения.
Примеры
Пример 1: Получение соединения A
(5-[3-(2,5-дихлор-4,6-диметил-1-оксипиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитробензол-1,2-диол)
а) К перемешиваемому раствору 3,4-дибензилокси-5-нитробензойной кислоты (0,50 г, 1,319 ммоль) в диметилформамиде (5 мл) при комнатной температуре добавляли 1,1-карбонилдиимидазол (0,24 г, 1,45 ммоль) в виде одной порции. После перемешивания в течение девяноста минут добавляли 2,5-дихлор-N'-гидрокси-4,6-диметилникотинамид (0,40 г, 1,45 ммоль) в виде одной порции. Полученную смесь перемешивали при 135°C в течение пяти часов и затем при комнатной температуре в течение ночи. Реакционную смесь вливали в смесь лед - 2 н. HCl (100 мл) и полученный осадок отфильтровывали, промывали водой и высушивали на воздухе. Рекристаллизация из изопропанола давала бледно-желтое твердое вещество (0,55 г, 72%).
б) К перемешиваемому раствору твердого вещества, полученного выше (0,50 г, 0,866 ммоль), в дихлорметане (20 мл) добавляли комплекс присоединения мочевины и перекиси водорода (0,41 г, 4,33 ммоль) в виде одной порции. Смесь охлаждали на ледяной бане и по каплям добавляли трифторацетиловый ангидрид (0,73 г, 3,46 ммоль). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи, после чего нерастворимый материал отфильтровывали. Фильтрат промывали водой и соляным раствором, высушивали над безводным сульфатом магния, фильтровали и упаривали. Осадок кристаллизовали из изопропанола с получением бледно-желтого твердого вещества (0,35 г, 68%).
в) К перемешиваемому раствору твердого вещества, полученного выше (0,30 г, 0,5 ммоль), в дихлорметане (10 мл) при -78°C в атмосфере аргона по каплям добавляли трибромид бора (0,38 г, 1,5 ммоль). Полученную пурпурную суспензию оставляли перемешиваться при комнатной температуре в течение 1 ч, затем снова охлаждали до -78°C и осторожно гасили добавлением воды. После перемешивания при комнатной температуре в течение 1 ч осадок отфильтровывали, промывали водой и высушивали при 50°C в вакууме с получением желаемого соединения в виде желтых кристаллов (0,18 г, 87%) с т.пл. 237-240°C.
Пример 2: Фармацевтическая композиция
Фармацевтические композиции изготавливали следующим образом:
Капсула:
Капсула:
Таблетка:
Пример 3 - Режим дозирования
Пример 3a: Влияние пищи
Данное исследование представляло собой открытое, рандомизированное, перекрестное исследование с однократным введением, с двумя периодами, с двумя последовательностями, на 12 здоровых мужчинах с целью оценить влияние пищи на фармакокинетический (РК) профиль Соединения A и его метаболитов. Одиночную дозу Соединения A 50 мг (2 капсулы по 25 мг) вводили в условиях голодания и приема пищи.
Образцы крови брали до введения Соединения A и через 0,5; 1; 1,5; 2; 3; 4; 6; 8; 10; 12; 16 и 24 ч после введения Соединения A. Результаты фармакокинетических исследований:
После перорального введения одной 50 мг дозы (2 капсулы по 25 мг), биодоступность Соединения A не была эквивалентной в условиях голодания и приема пищи, что указывает на наличие влияния пищи на фармакокинетику Соединения A (Фиг.1).
Фармакокинетические параметры Соединения A в условиях голодания и приема пищи представлены в Таблице 1. Обобщение основных результатов РК исследований Соединения A в условиях голодания и приема пищи
Прием пищи уменьшал и задерживал всасывание Соединения A; введение Соединения A непосредственно после приема высококалорийной пищи с высоким содержанием жиров значительно уменьшало скорость и степень всасывания по сравнению с введением лекарственного средства в условиях голодания. Параметры Cmax и AUC были значительно ниже в состоянии после приема пищи по сравнению с этими параметрами в состоянии голодания (соотношение после приема пищи: при голодании 31,73% для Cmax, 47,11% для AUCT и 49,43% для AUC∞). Tmах значительно повышалось в присутствии пищи (4,00 ч в состоянии после приема пищи по сравнению с 1,50 ч в состоянии голодания).
Выводы
Результаты показывают, что введение Соединения A после приема пищи приводит к уменьшению его биодоступности. Присутствие пищи уменьшало скорость и степень всасывания Соединения A с задержанными пиками уровней в плазме по сравнению с этими показателями при введении лекарственного средства в условиях голодания.
Пример 3б: Введение леводопы и Соединения A совместно и с интервалом в 1 ч
Данное исследование представляло собой одноцентровое, открытое, рандомизированное, сбалансированное по полу, перекрестное исследование с четырьмя последовательными периодами лечения однократным введением для оценки взаимодействия PK-PD (фармакодинамика), когда 25/100 мг карбидопы/леводопы стандартного высвобождения вводят совместно с дозой Соединения A 50 мг или на 1 ч позже. Восемнадцать (18) субъектов осуществили 2 периода лечения, 17 субъектов осуществили 3 периода лечения и 16 субъектов осуществили все 4 периода лечения. Всего в этом исследовании было задействовано 18 мужчин [10 (55,6%)] и женщин [8 (44,4%)].
Лечение состояло из четырех периодов однократного введения. Однократное введение 50 мг Соединения A состояло из 2 капсул по 25 мг. Однократное введение 25 мг карбидопы/100 мг леводопы прямого/стандартного высвобождения состояло из 1 таблетки Sinemet® 100/25. В соответствии с последовательностью лечения, определенной путем рандомизации, субъектам вводили Соединение A и Sinemet® 100/25 совместно в одном периоде, Sinemet® 100/25 через 1 ч после введения Соединения A в другом периоде, только соединение A в еще одном периоде и только Sinemet® 100/25 в оставшемся периоде. Период отмывки между введениями составлял по меньшей мере 3 недели. В одном периоде лечения Соединение A и Sinemet® 100/25 следовало вводить совместно; в другом периоде лечения Sinemet® 100/25 следовало вводить через 1 ч после введения Соединения A; еще в одном периоде лечения следовало вводить только Соединение A; в оставшемся периоде лечения следовало вводить только Sinemet® 100/25.
Средних величин Cmax леводопы достигали через 0,5-1,0 ч после введения. Соответственно, концентрации леводопы в плазме уменьшались со средним периодом полувыведения (t1/2) в пределах от 1,94 ч (только Sinemet® 100/25) до 2,51 ч (Sinemet® 100/25 плюс 50 мг Соединения A совместно).
После введения Соединения A Cmax леводопы увеличивалась, и увеличение было больше, когда Соединение A вводили совместно, что позволяет предположить, что могло происходить взаимодействие в определенной степени между Соединением A и Sinemet® 100/25 во время фазы всасывания, приводящее к увеличению скорости потребления леводопы и менее плавному увеличению уровней леводопы.
Профили зависимости средней концентрации леводопы в плазме от времени после однократного перорального введения только Sinemet® 100/25, Sinemet® 100/25, введеного с 50 мг Соединения A с интервалом в 1 ч и введенного совместно с 50 мг Соединения A, проиллюстрированы на Фиг.2 (n равно 16 для совместного введения, n равно 17 для введения только Sinemet®, n равно 18 для введения с интервалом в 1 ч).
Точечные оценки (PE) и 90% доверительный интервал (CI) средних фармакокинетических параметров леводопы после совместного введения 50 мг Соединения A (Тест L1) и введения с интервалом в 1 ч с Sinemet® 100/25 (Тест L2) отражены в Таблице 2 (только Sinemet® 100/25 представлен как эталон):
Более значительное увеличение степени воздействия леводопы (как оценивали по AUC) имело место, когда Sinemet® 100/25 вводили через 1 ч после 50 мг Соединения A.
Увеличение AUC0-t леводопы находилось в пределах от 4,23%, когда Sinemet® 100/25 вводили совместно с 50 мг Соединения A (коэффициенты равны 104,23 [96,88; 112,14]; средние и 90% Cl) до 14,56%, когда Sinemet® 100/25 вводили через 1 ч после 50 мг Соединения A (коэффициенты равны 114,56 [106,65; 123,05]). Увеличение AUC0-∞ леводопы находилось в пределах от 3,13%, когда Sinemet® 100/25 вводили совместно с 50 мг Соединения A (коэффициенты равны 103,13 [94,02; 113,12]) до 9,85%, когда Sinemet® 100/25 вводили через 1 ч после 50 мг Соединения A (коэффициенты равны 109,85 [100,22; 120,41]).
Увеличение Cmax леводопы находилось в пределах от 2,96%, когда Sinemet® 100/25 вводили через 1 ч после 50 мг Соединения A (коэффициенты равны 102,96 [89,36; 118,62]) до 12,10%, когда Sinemet® 100/25 вводили совместно с 50 мг Соединения A (коэффициенты равны 112,10 [96,94; 129,64]), демонстрируя предпочтительно более устойчивое повышение уровней леводопы при введении Sinemet® 100/25 через 1 ч после Соединения A.
Cmax 3-OMD была ниже, когда Sinemet® 100/25 вводили через 1 ч после 50 мг Соединения A, чем когда Sinemet® 100/25 вводили совместно с 50 мг Соединения A. Существенное уменьшение как скорости (как оценивали по Cmax), так и степени (как оценивали по AUC) системной экспозиции 3-OMD имело место, когда Sinemet® 100/25 вводили совместно с 50 мг Соединения A и также через 1 ч после 50 мг Соединения A.
Cmax и AUC карбидопы были похожими при введении только Sinemet® 100/25 и при введении Sinemet® 100/25 и 50 мг Соединения A с интервалом в 1 ч. Увеличение Cmax карбидопы находилось в пределах от 5,33% при введении Sinemet® 100/25 через 1 ч после 50 мг Соединения A до 5,86% при введнии Sinemet® 100/25 совместно с 50 мг Соединения A. Увеличение AUC0-∞ карбидопы находилось в пределах от 5,42% при введении Sinemet® 100/25 через 1 ч после 50 мг Соединения A до 9,20% при введении Sinemet® 100/25 совместно с 50 мг Соединения A.
Средние величины Cmax Соединения A были достигнуты через 2,5-4,0 ч после введения. Соответственно, концентрации Соединения A в плазме уменьшались со средним периодом полувыведения (t1/2) в пределах от 1,14 ч (только Соединение A) до 1,28 ч (Соединение A с Sinemet® 100/25). Cmax Соединения A была ниже при введении Соединения A за 1 ч до Sinemet® 100/25, чем при введении совместно с Sinemet® 100/25, и даже ниже по сравнению с Cmax Соединения A при введении только соединения A. После введения с Sinemet® 100/25 Cmax Соединения A уменьшалась, а tmax увеличивалась, что позволяет предположить, что могло происходить взаимодействие в определенной степени между Соединением A и Sinemet® 100/25 во время фазы всасывания, приводящее к задержке в скорости усвоения Соединения A. Статистическое различие было обнаружено между tmax для 50 мг Соединения A при введении совместно с Sinemet® 100/25 и при введении только Соединения A (p равно 0,0020). Увеличение AUC0-∞ Соединения A было в пределах от 4,18% при введении Sinemet® 100/25 совместно с 50 мг Соединения A до 4,74% при введении Sinemet® 100/25 через 1 ч после 50 мг Соединения A. Наблюдали уменьшение Cmax Соединения A на 9,21% при введении Sinemet® 100/25 через 1 ч после 50 мг Соединения A.
Результаты фармакодинамических исследований
Профили зависимости средней активности S-COMT (образованный метанефрин, пмоль/мг белка/ч) относительно базового уровня (перед введением) после однократного перорального введения только Sinemet® 100/25, Sinemet® 100/25, введенного с 50 мг Соединения A с интервалом в 1 ч, введенного совместно с 50 мг Соединения A, и введения только Соединения A были следующими (Фиг.3) (n равно 16 для совместного введения, n равно 17 для введения только Sinemet®, n равно 18 для введения только Соединения A и для введения с интервалом в 1 ч).
Во всех случаях лечения Соединением A существенно уменьшались как максимальное значение, так и степень активности S-COMT по отношению к таковым при введении только Sinemet® 100/25. После введения Соединения A максимальное ингибирование S-COMT (Emax) имело место между 3,42 ч (только Соединение A) и 4,58 ч (при введении Sinemet® 100/25 через 1 ч после 50 мг Соединения A) после введения (tEmax) и находилось в пределах от 88,7% до 91,1% соответственно.
Выводы
Результаты множества выполненных исследований очень хорошо согласовались между собой. Во всех случаях лечения Соединением A уменьшались как максимальное значение, так и степень активности S-COMT по отношению к введению только Sinemet® 100/25. Совместное введение Соединения A с Sinemet® 100/25 показало влияние на всасывание как леводопы, так и Соединения A. В последнем случае наряду с небольшим уменьшением Cmax наблюдалось значительное увеличение tmax Соединения A. Что касается леводопы, при задержке введения Соединения A на 1 ч наблюдалось уменьшение Cmax (по сравнению с увеличением, наблюдаемым при совместном введении), что позволяет предположить, что возможное взаимодействие между Соединением A и Sinemet® 100/25 во время фазы всасывания уменьшалось при разделении обоих введений. Более того, при задержке на 1 ч введения леводопы по отношению к Соединению A наблюдалось увеличение системной экспозиции леводопы (как оценивали по AUC). Это можно было бы отнести на счет скорости всасывания Соединения A и, следовательно, ингибирования СОМТ. Действительно, и несмотря на значительное увеличение tEmax для обоих случаев лечения Соединением A и Sinemet® 100/25 относительно введения только соединения A, введение, разделенное интервалом в 1 ч, вызывало более устойчивую скорость всасывания Соединения A в отличие от прерывистого и замедленного всасывания, наблюдаемого при совместном введении. Это могло привести к раннему ингибированию СОМТ и последующему увеличению системной экспозиции к леводопе.
Пример 3в: Действие соединения A на степени воздействия леводопы на пациента после совместного введения L-DOPA и Соединения A, за которым следовало дополнительное введение L-DOPA (диоксифенилаланин) через 24 ч
Данное исследование представляло собой трехцентровое, двойное слепое, рандомизированное, с плацебо-контролем, перекрестное исследование, направленное на изучение устойчивости и действия однократного введения трех дозировок Соединения A (25, 50 и 100 мг) на фармакокинетику леводопы, двигательную реакцию и активность эритроцитарной растворимой катехол-О-метилтрансферазы у 10 пациентов с болезнью Паркинсона, которые получали лечение совместно леводопой/ингибитором декарбоксилазы DOPA.
Для исследования подходили субъекты, которые представили: диагноз PD (болезни Паркинсона) в соответствии с критериями диагностики UK PDS Brain Bank; предсказуемые признаки "истощения конца дозы", несмотря на "оптимальную" терапию леводопой/AADCi; прошли лечение в стабильном режиме 3-8 дозами леводопы/AADCi 100/25 мг стандарного высвобождения в сутки в течение по меньшей мере 1 недели перед рандомизацией; по модифицированной шкале Хен и Яра до 5 стадии в состоянии "выключения"; и/или средней продолжительностью стадии "выключения" большей или равной 1,5 ч на протяжении рабочего времени. Сопутствующее лечение антипаркинсоническими лекарственными средствами (кроме апоморфина, энтакапона или толкапона) допускалось в стабильных дозах в течение по меньшей мере 4 недель до рандомизации.
Регулирование дозы и частоты введения леводопы представляет собой обычный терапевтический подход при наступлении двигательных осложнений. Обычно это описывают как оптимизацию терапии леводопой. "Оптимальная" терапия леводопой/AADCi представляет собой режим дозирования и введения леводопы/AADCi, которые вызывают наилучший двигательный ответ у пациента, т.е. отсутствие или уменьшение до минимума "истощения конца дозы" (изнашивания) и/или двигательных осложнений.
Данное исследование состояло из четырех последовательных периодов лечения, соответствующих 4 различным вариантам лечения (Соединение A 25 мг, 50 мг, 100 мг или плацебо). В каждом из четырех периодов лечения субъекты должны были поступить на место исследования за 2 дня до получения введения Соединения A/Плацебо (Сутки 1) и оставаться госпитализированными ("стационарные больные") до истечения 48 ч после получения введения Соединения A/Плацебо. Период отмывки между введениями должен был составлять по меньшей мере 10 дней. Контрольное посещение должно было произойти приблизительно через 2 недели после последнего введения лечения или досрочного прекращения лечения. В течение каждого периода капсулы Соединения A/Плацебо следовало вводить совместно с утренней дозой леводопы/карбидопы 100/25 мг (1 таблетка Sinemet® 25/100) или леводопы/бенсеразида 100/25 мг (1 таблетка Madopar®/Restex® 125) в Сутки 3.
Всего в данном исследовании было задействовано 10 субъектов: 10 субъектов осуществили 3 периода лечения и 9 субъектов осуществили все 4 периода лечения. Средние (±SD) возраст, рост и вес составляли 58,40±10,24 (интервал: 42-70) лет, 1,69±0,14 (1,52-1,95) м, 71,5±15,06 (50-100) кг соответственно.
Результаты данного исследования можно видеть в Таблице 3 и в Таблице 4.
* значимо различны.
Пример 3г: Клинические исследования на пациентах с болезнью Паркинсона: дозировка до сна
В данном исследовании Соединение A тестируют как терапию исследования, и энтакапон и плацебо как "референсные терапии". Соединение A доступно в капсулах по 5 мг, 25 мг и 50 мг. Применяют таблетки энтакапона по 200 мг. Для обеспечения слепого метода в течение периода исследования двойным слепым методом (DB) капсулы Соединения A и таблетки энтакапона идентичным образом покрыты дополнительным покрытием. Капсулы плацебо приготавливают путем наполнения идентичных капсул наполнителем (также применяется как материал для заполнения). Все капсулы плацебо содержат в среднем 1 мг рибофлавина для маскировки изменения цвета мочи, наблюдаемого как безопасный побочный эффект энтакапона.
Схема дозировки
В течение DB периода субъекты принимают 1 капсулу "лечения" совместно с каждым дневным введением леводопы/AADCi (от 3 до 8 ежедневных введений). Дополнительное лечение (введение «до наступления времени сна»' или «до сна») вводят по меньшей мере через 1 ч после последнего введения суток леводопы/AADCi.
Для дневных введений (т.е. принимаемых совместно с каждым введением леводопы/AADCi) введение лечения представляет собой следующее:
- Группы Соединения A: плацебо.
- Группа энтакапона: 200 мг энтакапона.
- Группа плацебо: плацебо.
Для доз, принимаемых перед сном (по меньшей мере через 1 ч после последнего дневного введения леводопы/AADCi), введение лечения представляет собой следующее:
- Группы Соединения A: 5, 25 или 50 мг.
- Группа энтакапона: плацебо.
- Группа плацебо: плацебо.
Пример 3д: Клинические исследования на пациентах с болезнью Паркинсона: дозировка перед сном и через 1 час после приема пищи
В исследовании двойным слепым методом с плацебо-контролем пациентов с болезнью Паркинсона, поддерживаемых на леводопе/AADCi, лечат следующим образом. Пациенты принимают либо плацебо, либо соединение A (25 мг или 50 мг) вечером, по меньшей мере через 1 ч после введения последней дозы суток терапии леводопой/AADCi (доза (введение) перед сном).
Субъектам следует голодать в течение 1 ч до и по меньшей мере 1 ч после приема лечения.
Ожидается, что те пациенты, которые принимают Соединение A, продемонстрируют улучшенные эффекты по сравнению с пациентами, принимающими плацебо.
| название | год | авторы | номер документа |
|---|---|---|---|
| РЕЖИМ ВВЕДЕНИЯ НИТРОКАТЕХОЛОВ | 2009 |
|
RU2557532C2 |
| СПОСОБЫ ЛЕЧЕНИЯ БОЛЕЗНИ ПАРКИНСОНА | 2004 |
|
RU2342929C2 |
| РЕЖИМ ДОЗИРОВАНИЯ ИНГИБИТОРОВ КОМТ | 2007 |
|
RU2518483C2 |
| ПРОИЗВОДНЫЕ α-АМИНОАМИДА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ СИНДРОМА УСТАЛЫХ НОГ И ВЫЗЫВАЮЩИХ ПРИВЫКАНИЕ РАССТРОЙСТВ | 2005 |
|
RU2403030C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛИНДИОНА КАК ИНГИБИТОРЫ НАДФН-ОКСИДАЗЫ | 2010 |
|
RU2569855C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2777366C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2015 |
|
RU2728787C2 |
| ЗАМЕЩЕННЫЕ АРИЛСУЛЬФОНАМИДЫ КАК ПРОТИВОВИРУСНЫЕ СРЕДСТВА | 2007 |
|
RU2423352C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2015 |
|
RU2785871C2 |
| КОМПОЗИЦИИ ДЛЯ НЕПРЕРЫВНОГО ВВЕДЕНИЯ ИНГИБИТОРОВ ДОПА-ДЕКАРБОКСИЛАЗЫ | 2010 |
|
RU2678839C2 |
Изобретение относится к медицине, а именно к способу профилактики или лечения расстройства центральной и периферической нервной системы. Способ профилактики или лечения расстройства центральной и периферической нервной системы, включающий введение пациенту, страдающему от указанного расстройства, терапевтически эффективного количества соединения 5-[3-(2,5-дихлор-4,6-диметил-1-оксипиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитробензол-1,2-диол или его фармакологически приемлемых солей, сложных эфиров, карбаматов и фосфатов в комбинации с катехоламиновым лекарственным средством леводопой, где указанное соединение вводят в виде однократной суточной дозы по меньшей мере за час или через час после введения последней суточной дозы леводопы. Способ профилактики или лечения расстройства центральной и периферической нервной системы, включающий введение пациенту, страдающему от указанного расстройства, терапевтически эффективного количества соединения 5-[3-(2,5-дихлор-4,6-диметил-1-оксипиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитробензол-1,2-диол или его фармакологически приемлемых солей, сложных эфиров, карбаматов и фосфатов без пищи и/или между приемами пищи. Способ профилактики или лечения расстройства центральной и периферической нервной системы, включающий введение пациенту, страдающему от указанного расстройства, терапевтически эффективного количества соединения 5-[3-(2,5-дихлор-4,6-диметил-1-оксипиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитробензол-1,2-диол или его фармакологически приемлемых солей, сложных эфиров, карбаматов и фосфатов, в комбинации с катехоламиновым лекарственным средством леводопой, где указанное соединение вводят один раз в сутки по меньшей мере через 1 ч после введения последней суточной дозы леводопы и менее чем за 90 минут перед тем, как лечь в постель, или при наступлении времени сна. Способ профилактики или лечения болезни Паркинсона, включающий введение пациенту, страдающему от указанной болезни, терапевтически эффективного количества соединения 5-[3-(2,5-дихлор-4,6-диметил-1-оксипиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитробензол-1,2-диол или его фармакологически приемлемых солей, сложных эфиров, карбаматов и фосфатов в комбинации с катехоламиновым лекарственным средством леводопой, где указанное соединение вводят один раз в сутки по меньшей мере через 1 ч после введения последней суточной дозы катехоламинового лекарственного средства и менее чем за 90 минут перед тем, как лечь в постель, или при наступлении времени сна и по меньшей мере через 1 ч после последнего приема пищи и по меньшей мере за 1 ч до следующего приема пищи. Вышеописанные способы эффективны для профилактики или лечения расстройства центральной и периферической нервной системы. 4 н. и 15 з.п. ф-лы, 3 ил., 4 табл., 3 пр.
1. Способ профилактики или лечения расстройства центральной и периферической нервной системы, включающий введение пациенту, страдающему от указанного расстройства, терапевтически эффективного количества соединения 5-[3-(2,5-дихлор-4,6-диметил-1-оксипиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитробензол-1,2-диол или его фармакологически приемлемых солей, сложных эфиров, карбаматов и фосфатов в комбинации с катехоламиновым лекарственным средством леводопой, где указанное соединение вводят в виде однократной суточной дозы по меньшей мере за час или через час после введения последней суточной дозы леводопы.
2. Способ профилактики или лечения расстройства центральной и периферической нервной системы, включающий введение пациенту, страдающему от указанного расстройства, терапевтически эффективного количества соединения 5-[3-(2,5-дихлор-4,6-диметил-1-оксипиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитробензол-1,2-диол или его фармакологически приемлемых солей, сложных эфиров, карбаматов и фосфатов без пищи и/или между приемами пищи.
3. Способ по п. 2, где указанное соединение вводят в виде однократной суточной дозы.
4. Способ по п. 1 или 2, где указанное соединение вводят в эффективной суточной дозе от 25 до 300 мг/сутки.
5. Способ по п. 1 или 2, где указанное соединение вводят в конкретной суточной дозе 1 мг, 3 мг, 5 мг, 10 мг, 15 мг, 20 мг, 25 мг, 30 мг, 50 мг, 100 мг, 200 мг, 400 мг, 800 мг или 1200 мг.
6. Способ по п. 1 или 2, где указанное соединение вводят через 0,25-12 ч, предпочтительно через 0,5-6 ч, более предпочтительно через 0,75-4 ч после приема пищи.
7. Способ по п. 1 или 2, где указанное соединение вводят за 0,25-2 ч, предпочтительно за 0,5-1,5 ч до приема пищи.
8. Способ по п. 1 или 2, где указанное соединение вводят по меньшей мере через 1 ч после последнего приема пищи и по меньшей мере за 1 ч до следующего приема пищи.
9. Способ по п. 1 или 2, где указанное соединение вводят менее чем за 90 минут перед тем, как лечь в постель, или при наступлении времени сна.
10. Способ по п. 1 или 2, где указанное соединение вводят менее чем за 1 ч перед тем, как лечь в постель, или при наступлении времени сна.
11. Способ по п. 2, где указанное соединение вводят в комбинации с катехоламиновым лекарственным средством, таким как леводопа.
12. Способ по п. 11, где указанное соединение вводят последовательно с катехоламиновым лекарственным средством, например до или после катехоламинового лекарственного средства.
13. Способ по п. 1 или 11, где катехоламиновое лекарственное средство вводят последовательно или совместно с AADCi, таким как карбидопа или бенсеразид.
14. Способ по п. 1 или 2, где расстройство центральной и периферической нервной системы представляет собой аффективное расстройство, желудочно-кишечное расстройство, состояние формирования отека, гипертензию или двигательное расстройство, такое как болезнь Паркинсона.
15. Способ по п. 14, где двигательное расстройство представляет собой болезнь Паркинсона с двигательными флуктуациями.
16. Способ профилактики или лечения расстройства центральной и периферической нервной системы, включающий введение пациенту, страдающему от указанного расстройства, терапевтически эффективного количества соединения 5-[3-(2,5-дихлор-4,6-диметил-1-оксипиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитробензол-1,2-диол или его фармакологически приемлемых солей, сложных эфиров, карбаматов и фосфатов, в комбинации с катехоламиновым лекарственным средством леводопой, где указанное соединение вводят один раз в сутки по меньшей мере через 1 ч после введения последней суточной дозы леводопы и менее чем за 90 минут перед тем, как лечь в постель, или при наступлении времени сна.
17. Способ по п. 16, где расстройство центральной и периферической нервной системы представляет собой двигательное расстройство, такое как болезнь Паркинсона.
18. Способ по п. 16, где указанное соединение вводят без пищи и/или по меньшей мере через 1 ч после последнего приема пищи и по меньшей мере за 1 ч до следующего приема пищи пациентом.
19. Способ профилактики или лечения болезни Паркинсона, включающий введение пациенту, страдающему от указанной болезни, терапевтически эффективного количества соединения 5-[3-(2,5-дихлор-4,6-диметил-1-оксипиридин-3-ил)-[1,2,4]оксадиазол-5-ил]-3-нитробензол-1,2-диол или его фармакологически приемлемых солей, сложных эфиров, карбаматов и фосфатов в комбинации с катехоламиновым лекарственным средством леводопой, где указанное соединение вводят один раз в сутки, по меньшей мере через 1 ч после введения последней суточной дозы катехоламинового лекарственного средства и менее чем за 90 минут перед тем, как лечь в постель, или при наступлении времени сна и по меньшей мере через 1 ч после последнего приема пищи и по меньшей мере за 1 ч до следующего приема пищи.
| US 2010256194 A1, 07.10.2010 | |||
| US 20100256193 A1, 07.10.2010 | |||
| WO 2010014025 A1, 04.02.2010 | |||
| Регистр Лекарственных Средств России, Москва, 2002, часть 2, глава 2.9 | |||
| Лекарства и пища: когда до еды, когда после | |||
| WO 2009116882 A1, 24.09.2009 | |||
| US 20090054437 A1, 26.02.2009. |

