Настоящая международная заявка испрашивает приоритет предварительной заявки на патент США № 61/975610, поданной 4 апреля 2014 года, содержание которой включено в настоящую заявку посредством ссылки во всей полноте.
Область техники, к которой относится изобретение
[0001] Настоящее изобретение относится к определенным замещенным гетероцикл-конденсированным гамма-карболинам, в свободной форме, в форме фармацевтически приемлемой соли и/или в по существу чистой форме, как описано в настоящей заявке, их фармацевтическим композициям и способам применения для лечения заболеваний с вовлечением 5-HT2A рецептора, транспортера серотонина (SERT) и/или путей, включающих сигнальные системы D1/D2 дофаминовых рецепторов, например, заболеваний или расстройств, таких как тревожное расстройство, психоз, шизофрения, расстройства сна, сексуальные расстройства, мигрень, состояния, связанные с головной болью, социофобии, желудочно-кишечные расстройства, такие как дисфункция моторики желудочно-кишечного тракта и ожирение; депрессия и аффективные расстройства, связанные с психозом или болезнью Паркинсона; психоз, такой как шизофрения, связанный с депрессией; биполярное расстройство; и другие психиатрические и неврологические состояния, а также к комбинациям с другими средствами.
[0002] Психоз, в особенности шизофрения, поражает 1,1% населения во всем мире. Эта болезнь включает три фазы: продромальную фазу, активную фазу и остаточную фазу. Продромальная фаза представляет собой раннюю фазу, в которой наблюдают субклинические признаки и симптомы. Эти симптомы могут включать потерю интереса к обычным занятиям, попытки избегать общения с друзьями и членами семьи, спутанность сознания, проблемы с концентрацией, ощущение вялости и апатию. Активная фаза характеризуется обострениями позитивных симптомов, такими как бред, галлюцинации и мнительность. Остаточная фаза характеризуется негативными симптомами, такими как эмоциональное отчуждение, пассивная социальная самоизоляция и стереотипное мышление; и общими психопатологическими симптомами, включая активное социальное избегание, тревожное расстройство, напряженность и обеспокоенность соматическим здоровьем. Симптомы остаточной фазы также часто сопровождаются депрессией, когнитивной дисфункцией и бессонницей. В совокупности, эти симптомы остаточной фазы трудноизлечимы многими антипсихотическими препаратами, доступными в настоящее время на рынке, и поэтому их обычно наблюдают после того, как ослабевают симптомы активной фазы после лечения. Это фаза болезни, когда пациенты хотели бы вернуться к более продуктивной и полноценной жизни, но поскольку остаточные негативные симптомы и когнитивные нарушения не вылечены должным образом, то возвращение к такой функции становится невозможным. Сохраняется острая необходимость в антипсихотическом средстве, которое сможет вылечить не только симптомы активной или острой фазы, но также и симптомы остаточной фазы психоза, например, шизофрении. Кроме того, существует потребность в лекарственных препаратах для лечения этих симптомов, которые не имеют нежелательных побочных эффектов, вызываемых нецелевыми взаимодействиями с системами гистаминового H1 и ацетилхолинового мускаринового рецепторов.
Предпосылки создания изобретения
[0003] Известно, что замещенные гетероцикл-конденсированные гамма-карболины являются агонистами или антагонистами 5-HT2 рецепторов, в особенности 5-HT2A и 5-HT2C рецепторов, в лечении расстройств центральной нервной системы. Эти соединения раскрыты в патентах США № 6548493; 7238690; 6552017; 6713471; 7183282; U.S. RE39680 и U.S. RE39679 в качестве новых соединений, полезных для лечения расстройств, ассоциированных с модуляцией 5-HT2A рецепторов, таких как ожирение, тревожное расстройство, депрессия, психоз, шизофрения, расстройства сна, сексуальные расстройства, мигрень; состояния, связанные с головной болью; социофобии, желудочно-кишечные расстройства, такие как дисфункция моторики желудочно-кишечного тракта и ожирение.
[0004] PCT/US08/03340 (WO 2008/112280) и заявка на патент США серийный номер 10/786935 раскрывают способы получения замещенных гетероцикл-конденсированных гамма-карболинов и использование этих гамма-карболинов в качестве агонистов и антагонистов серотонина, полезных для контроля и профилактики расстройств центральной нервной системы, таких как аддиктивное поведение и расстройства сна.
[0005] WO/2009/145900 раскрывает использование определенных замещенных гетероцикл-конденсированных гамма-карболинов для лечения комбинации психоза и депрессивных расстройств, а также расстройств сна, депрессивных и/или аффективных расстройств у пациентов с психозом или болезнью Паркинсона. В дополнение к расстройствам, связанным с психозом и/или депрессией, данная заявка на патент раскрывает и заявляет применение этих соединений в низких дозах для селективного антагонистического действия на 5-HT2A рецепторы, не затрагивая или минимально затрагивая дофаминовые рецепторы D2, и поэтому полезных для лечения расстройств сна без побочных эффектов на дофаминовые D2 путей или побочных эффектов нп другие пути (например, ГАМКA (гамма-аминомасляная кислота) рецепторов), связанных с обычными седативно-снотворными средствами (например, бензодиазепинами), включая, но не ограничиваясь этим, развитие лекарственной зависимости, мышечную гипотонию, слабость, головную боль, затуманенное зрение, головокружение, тошноту, рвоту, эпигастральный дистресс, диарею, боли в суставах и боли в груди.
[0006] Кроме того, было обнаружено, что эти замещенные гетероцикл-конденсированные гамма-карболиновые соединения эффективны для лечения не только острых симптомов, но также и остаточных симптомов психоза. Поэтому были раскрыты способы применения этих замещенных гетероцикл-конденсированных гамма-карболиновых соединений, либо в отдельности, либо в качестве вспомогательной терапии, для лечения остаточных симптомов психоза, в особенности шизофрении. Смотри, например, заявку на патент PCT/US2014/68443.
[0007] WO 2009/114181 раскрывает способы получения кристаллов аддитивной соли толуолсульфоновой кислоты определенных замещенных гетероцикл-конденсированных гамма-карболинов, например, аддитивной соли толуолсульфоновой кислоты 4-((6bR,10aS)-3-метил-2,3,6b,9,10,10a-гексагидро-1H-пиридо[3',4':4,5]пирроло[1,2,3-de]хиноксалин-8(7H)-ил)-1-(4-фторфенил)-1-бутанона.
[0008] WO 2011/133224 раскрывает пролекарства/метаболиты замещенного гетероцикл-конденсированного гамма-карболина для усовершенствованной лекарственной формы, например, лекарственной формы длительного/контролируемого высвобождения. Эта заявка раскрывает, что гетероцикл-конденсированный гамма-карболин, N-замещенный 4-фторфенил(4-гидрокси)бутильной группой, как продемонстрировано, обладает высокой селективностью в отношении транспортера серотонина (SERT) по сравнению с гетероцикл-конденсированным гамма-карболином, содержащим 4-фторфенилбутанон. Гидрокси группа на этих соединениях, однако, взаимопреобразуется в кетон и из кетона в плазме и головном мозге, что позволяет им служить в качестве резервуара для 4-фторфенилбутанонового лекарственного средства. Несмотря на то, что замещенные гетероцикл-конденсированные гамма-карболины и их применение являются известными, авторы настоящего изобретения с удивлением обнаружили, что определенные замещенные гетероцикл-конденсированные гамма-карболины, несмотря на то, что являются менее активными в испытаниях in-vitro, являются взаимно преобразующимися между этими менее активными соединениями и высокоактивным кетоновым лекарственным средством в плазме и головном мозге. Авторы настоящего изобретения также обеспечили пролекарства специфических замещенных гетероцикл-конденсированных гамма-карболинов, которые имеют измененный фармакокинетический профиль, например, измененные механизмы и/или скорость абсорбции и дистрибуции, и поэтому могут быть полезны для усовершенствованной лекарственной формы и/или для контроля длительности эффекта лекарственного средства в организме (например, для длительного или контролируемого высвобождения).
[0009] WO 2013/155505 раскрывает соединения, которые блокируют взаимопреобразование в vivo между гидрокси и кетоном, путем введения алкильного заместителя на углеродсодержащую гидроксильную группу, приводя, таким образом, к получению соединений, которые антагонизируют 5-HT2A рецепторы и также ингибируют транспортер обратного захвата серотонина.
[00010] Основные пути метаболизма ранее раскрытых соединений, представляют собой N-деметилирование, катализируемое посредством CYP 3A4, и восстановление кетона, катализируемое кетон-редуктазой. Известно, что N-деалкилирование при помощи ферментов цитохромоксидазы происходит через первоначальное окисление одного или нескольких атомов углерода в альфа-положении по отношению к атому азота. Семейство ферментов, которые катализируют восстановление кетона, является большим и разнообразным, и механизм не был абсолютно выяснен. Интересно, что, механистически, восстановление кетона может происходить либо в форме енольного таутомера кетона, либо кето-таутомера.
Сущность изобретения
[00011] Не будучи связанным теорией, настоящее изобретение обеспечивает соединения, которые частично ограничивают метаболизм кетона и/или N-метильного заместителя, путем введения атомов дейтерия в различных положениях. Считается, что из-за очень схожих свойств атомов дейтерия (2Н) по сравнению с нормальными атомами водорода (1Н) считается, что лекарственные соединения, в которых водород замещен дейтерием, как правило, обладают такой же биологической активностью, как и недейтерированный аналог. Таким образом, настоящее изобретение обеспечивает соединения, содержащие тридейтерированный N-метил, моно- или ди-дейтерированный метилен, смежный с N-метилом, или моно- или ди-дейтерированный метилен, смежный с кетоном, или любую комбинацию этих дейтерирований. Эти новые соединения будут антагонизировать 5-HT2A рецепторы, ингибировать транспортер обратного захвата серотонина и модулировать дофаминэргическое фосфорилирование белка аналогичным образом, как их природные водородные аналоги, но будут обладать улучшенным метаболическим профилем. Авторы настоящего изобретения продемонстрировали, что дейтерирования некоторых метаболически лабильных положений улучшают стабильность в микросомах печени в vitro, тогда как дейтерирование 4-фторфенильного кольца (с получением соединения, содержащего 2,3,5,6-тетрадейтеро-4-фтор кольцо) не улучшает микросомальную стабильность.
[00012] В первом аспекте настоящее изобретение обеспечивает соединение формулы I:

где:
R1 представляет собой СH3 или CD3;
R2 и R3 каждый независимо представляют собой H или D;
R4 и R5 каждый независимо представляют собой H или D;
при условии, что R2, R3, R4 и R5 не все представляют собой H, когда R1 представляет собой СH3,
и где D представляет собой дейтерий;
в свободной форме или форме соли.
[0013] В следующем варианте осуществления первого аспекта, настоящее изобретение обеспечивает Соединение формулы I, как описано в следующих пунктах:
1.1 соединение формулы I, где R1 представляет собой СD3;
1.2 соединение формулы I, где R2 и R3 представляют собой D;
1.3 соединение формулы I, где R4 и R5 представляют собой D;
1.4 соединение формулы I, где R1 представляет собой СD3, и R2 и R3 оба представляют собой D;
1.5 соединение формулы I, где R1 представляет собой СD3, и R4 и R5 оба представляют собой D;
1.6 соединение формулы I, где R2 и R3 и R4 и R5 все представляют собой D;
1.7 соединение формулы I, где R1 представляет собой СD3, и R2 и R3 и R4 и R5 все представляют собой D;
1.8 соединение формулы I или в соответствии с любым из пунктов 1-1.7, где соединение представлено в по существу чистой диастереомерной форме (то есть, по существу свободно от других диастереоизомеров);
1.9 соединение формулы I или в соответствии с любым из пунктов 1-1.7, где соединение имеет диастереомерный избыток более чем 70%, предпочтительно более чем 80%, более предпочтительно более чем 90%, и наиболее предпочтительно более чем 95%;
в свободной форме или форме соли.
[0014] Во втором аспекте настоящее изобретение обеспечивает соединение формулы II:

где R1 - R5 такие, как определены выше в Формуле I, и где R6 - R9 каждый независимо выбран из H и D.
[0015] В следующем варианте осуществления первого аспекта, настоящее изобретение обеспечивает соединение формулы I в свободной форме или форме соли, как описано в следующих пунктах:
4.1 соединение формулы I или в соответствии с любым из пунктов 1-1.9, где соль выбрана из группы, состоящей из солей хлористоводородной, бромистоводородной, серной, сульфаминовой, фосфорной, азотной, уксусной, пропионовой, янтарной, гликолевой, стеариновой, молочной, яблочной, винной, лимонной, аскорбиновой, пальмоевой, малеиновой, гидроксималеиновой, фенилуксусной, глютаминовой, бензойной, салициловой, сульфаниловой, 2-ацетоксибензойной, фумаровой, толуолсульфоновой, метансульфоновой, этандисульфоновой, щавелевой, изетионовой и подобных кислот;
4.2 соединение формулы I или формулы 4.1, где соль представляет собой аддитивную соль фумаровой кислоты;
4.3 соединение формулы I или формулы 4.1, где соль представляет собой аддитивную соль фосфорной кислоты;
4.4 соединение формулы I или формулы 4.1, где соль представляет собой аддитивную соль толуолсульфоновой кислоты.
[0016] Во втором аспекте настоящее изобретение обеспечивает фармацевтическую композицию, включающую соединение формулы I или в соответствии с любым из пунктов 1-1.9 или 4.1-4.4 (соединения по настоящему изобретению), в свободной форме или форме фармацевтически приемлемой соли, в смеси с фармацевтически приемлемым растворителем или носителем, например, для обеспечения немедленного высвобождения или для обеспечения длительного или замедленного высвобождения.
[0017] В следующем варианте осуществления второго аспекта представлена фармацевтическая композиция по настоящему изобретению для длительного или замедленного высвобождения, например, лекарственная форма депо. В одном варианте осуществления лекарственная форма депо включает соединения по настоящему изобретению в полимерной матрице. В другом варианте осуществления соединения по настоящему изобретению диспергированы или растворены в полимерной матрице. В следующем варианте осуществления полимерная матрица включает стандартные полимеры, используемые в лекарственных формах депо, такие как полимеры, выбранные из полиэфира жирной гидрокси-кислоты и их производные, или полимер алкил альфа-цианоакрилата, полиалкиленоксалат, поли(орто-эфир), поликарбонат, полиорто-карбонат, поли(амино-кислота), сложный эфир гиалуроновой кислоты и их смеси. В следующем варианте осуществления полимер выбран из группы, состоящей из полилактида, поли d,l-лактида, полигликолида, PLGA 50:50, PLGA 75:25, PLGA 85:15 и PLGA 90:10 полимера. В другом варианте осуществления полимер выбран из поли(гликолевой кислоты), поли-D,L-молочной кислоты, поли-L-молочной кислоты, сополимеров вышеуказанных кислот, поли(алифатических карбоновых кислот), сополиоксалатов, поликапролактона, полидиоксонона, поли(орто-карбонатов), поли(ацеталей), поли(молочной кислоты-капролактона), полиортоэфиров, поли(гликолевой кислоты-капролактона), полиангидридов и природных полимеров, включая альбумин, казеин и воски, такие как, глицерин моно- и дистеарат, и подобных. В конкретном варианте осуществления полимерная матрица включает поли (d,l-лактид-co-гликолид). Любая из композиций, описанных выше в настоящей заявке, может представлять собой фармацевтическую композицию, где указанная композиция представлена в смеси с фармацевтически приемлемым растворителем или носителем.
[0018] (Фармацевтические) лекарственные формы депо, как описано выше в настоящей заявке, являются особенно полезными для длительного или замедленного высвобождения, где соединения по настоящему изобретению высвобождаются при распаде полимерной матрицы. Эти композиции можно формулировать для контролируемого и/или длительного высвобождения соединений по настоящему изобретению (например, в виде депо-композиции) в течение периода до 180 дней, например, от около 14 до около 30 до около 180 дней. Например, полимерная матрица может разрушаться и высвобождать соединения по настоящему изобретению в течение периода около 30, около 60 или около 90 дней. В другом примере полимерная матрица может разрушаться и высвобождать соединения по настоящему изобретению в течение периода около 120 или около 180 дней.
[0019] В еще одном дополнительном варианте осуществления фармацевтические композиции по настоящему изобретению, в частности, депо-композиции по настоящему изобретению, формулируют для введения путем инъекции.
[0020] В третьем аспекте настоящее изобретение обеспечивает соединения по настоящему изобретению, как описано выше в настоящей заявке, в лекарственной форме длительного или замедленного высвобождения для перорального применения. Например, настоящее изобретение обеспечивает систему пероральной доставки с осмотически контролируемым высвобождением (OROS) для доставки соединений по настоящему изобретению, например, аналогично системам, описанным в WO 2000/35419 и EP 1 539 115 (публикация США № 2009/0202631), содержание каждой из этих заявок включено во всей полноте посредством ссылки в настоящую заявку. Поэтому в одном варианте осуществления этого аспекта настоящее изобретение обеспечивает фармацевтическую композицию или устройство, включающее (a) желатиновую капсулу, содержащую Соединение по настоящему изобретению в свободной форме или форме фармацевтически приемлемой соли или Фармацевтическую композицию по настоящему изобретению, описанную выше в настоящей заявке; (b) многослойную оболочку, нанесенную на желатиновую капсулу, содержащую, в порядке от капсулы к наружной стороне: (i) барьерный слой, (ii) расширяемый слой и (iii) полупроницаемый слой; и (c) и отверстие, образованное или формируемое через оболочку. (Композиция P.1)
[0021] В другом варианте осуществления этого аспекта настоящее изобретение обеспечивает композицию, включающую желатиновую капсулу, содержащую жидкость, соединения по настоящему изобретению в свободной форме или форме фармацевтически приемлемой соли или фармацевтическую композицию по настоящему изобретению, описанную выше в настоящей заявке, при этом желатиновая капсула окружена композитной оболочкой, включающей барьерный слой, контактирующий с внешней поверхностью желатиновой капсулы, расширяемый слой, контактирующий с барьерным слоем, полупроницаемый слой, охватывающий расширяемый слой, и выходное отверстие, образованное или формируемое в оболочке. (Композиция P.2)
[0022] В еще одном варианте осуществления третьего аспекта настоящее изобретение обеспечивает композицию, включающую желатиновую капсулу, содержащую жидкость, соединение по настоящему изобретению в свободной форме или форме фармацевтически приемлемой соли или фармацевтическую композицию по настоящему изобретению, описанную выше в настоящей заявке, при этом желатиновая капсула окружена композитной оболочкой, включающей барьерный слой, контактирующий с внешней поверхностью желатиновой капсулы, расширяемый слой, контактирующий с барьерным слоем, полупроницаемый слой, охватывающий расширяемый слой, и выходное отверстие, образованное или формируемое в оболочке, где барьерный слой образует уплотнительную прокладку между расширяемым слоем и окружающей средой у выходного отверстия. (Композиция P.3)
[0023] В еще одном варианте осуществления третьего аспекта настоящее изобретение обеспечивает композицию, включающую желатиновую капсулу, содержащую жидкость, соединение по настоящему изобретению в свободной форме или форме фармацевтически приемлемой соли или фармацевтическую композицию по настоящему изобретению, описанную выше в настоящей заявке, при этом желатиновая капсула окружена барьерным слоем, контактирующим с внешней поверхностью желатиновой капсулы, расширяемым слоем, частично контактирующим с барьерным слоем, полупроницаемым слоем, охватывающим по меньшей мере расширяемый слой, с выходным отверстием, образованным или формируемым в лекарственной форме, проходящим от внешней поверхности желатиновой капсулы до окружающей среды. В которое ее используют. (Композиция P.4). Расширяемый слой может быть образован в одном или нескольких дискретных участках, таких как, например, два участка, расположенные на противоположных сторонах или концах желатиновой капсулы.
[0024] В конкретном варианте осуществления третьего аспекта, соединения по настоящему изобретению в системе пероральной доставки с осмотически контролируемым высвобождением (то есть, в Композиции P.1-P.4) представлены в жидкой лекарственной форме, при этом лекарственная форма может представлять собой чистое жидкое активное вещество, жидкое активное вещество в растворе, суспензии, эмульсии или самоэмульгирующуюся композицию или т.п.
[0025] Дополнительную информацию о системе пероральной доставки с осмотически контролируемым высвобождением, включая характеристики желатиновой капсулы, барьерного слоя, расширяемого слоя, полупроницаемого слоя и отверстия, можно найти в заявке WO 2000/35419, содержание которой включено в настоящую заявку посредством ссылки во всей полноте. Другую систему пероральной доставки с осмотически контролируемым высвобождением для соединения или фармацевтической композиции по настоящему изобретению можно найти в EP 1539115 (публикация США № 2009/0202631), содержание которой включено в настоящую заявку посредством ссылки во всей полноте.
[0026] Поэтому в другом варианте осуществления третьего аспекта, настоящее изобретение обеспечивает композицию или устройство, включающие (a) два или более слоев, где указанные два или более слоев включают первый слой и второй слой, при этом указанный первый слой включает соединение по настоящему изобретению в свободной форме или форме фармацевтически приемлемой соли или фармацевтическую композицию, описанную выше, указанный второй слой включает полимер; (b) внешнюю оболочку, окружающую указанные два или более слоев; и (c) отверстие в указанной внешней оболочке. (Композиция P.5)
[0027] В композиции P.5 предпочтительно используют полупроницаемую мембрану, окружающую трехслойное ядро: в этих вариантах осуществления первый слой называют первым лекарственным слоем, и он содержит низкое количество лекарственного средства (например, соединения по настоящему изобретению) и осмотический агент, такой как соль, средний слой, называемый вторым лекарственным слоем, содержит большее количество лекарственного средства, эксципиенты и не содержит соль; и третий слой, называемый слоем осмотического вещества, содержит осмотические агенты и не содержит лекарственное средство. По меньшей мере одно отверстие просверливается через мембрану на конце первого лекарственного слоя имеющей форму капсулы таблетки. (Композиция P.6)
[0028] Композиция P.5 или P.6 может включать мембрану, ограничивающую ячейку, при этом мембрана окружает внутренний защитный подслой, имеет по меньшей мере одно выходное отверстие, образованное или формируемое в ней, и по меньшей мере часть мембраны является полупроницаемой; расширяемый слой, расположенный в ячейке вдали от выходного отверстия и находящийся в жидкостной связи с полупроницаемой частью мембраны; первый лекарственный слой, расположенный рядом с выходным отверстием; и второй лекарственный слой, расположенный в ячейке между первым лекарственным слоем и расширяемым слоем, при этом лекарственные слои включают соединение по настоящему изобретению в свободной форме или форме фармацевтически приемлемой соли. В зависимости от относительной вязкости первого лекарственного слоя и второго лекарственного слоя получают различные профили высвобождения. Это необходимо для определения оптимальной вязкости для каждого слоя. В настоящем изобретении вязкость модулируют путем добавления соли, хлорида натрия. Профиль доставки из ядра зависит от массы, лекарственной формы и толщины каждого из лекарственных слоев. (Композиция P.7)
[0029] В конкретном варианте осуществления настоящее изобретение обеспечивает Композицию P.7, в которой первый лекарственный слой содержит соль, а второй лекарственный слой не содержит соль. Композиция P.5-P.7 необязательно может включать промотирующий текучесть слой между мембраной и лекарственными слоями. Композиции P.1-P.7 обычно называют композицией системы пероральной доставки с осмотически контролируемым высвобождением.
[0030] В четвертом аспекте настоящее изобретение обеспечивает способ (Способ I) для лечения или профилактики расстройства центральной нервной системы, включающий введение нуждающемуся в этом пациенту соединения формулы I или любой из формул 1-1.9, в свободной форме или форме фармацевтически приемлемой соли, описанного в любом из пунктов 4.1-4.4, или фармацевтической композиции, описанной выше.
[0032] В следующем варианте осуществления четвертого аспекта настоящее изобретение обеспечивает Способ I, который описан ниже в следующих формулах:
7.1 Способ I, где расстройство центральной нервной системы представляет собой одно или несколько расстройств, ассоциированных с деменцией, например, расстройства, ассоциированные с умеренными когнитивным нарушением и дементным состоянием, включая сенильную деменцию, болезнь Альцгеймера, болезнь Пика, фронто-темпоральную деменцию, супрануклеарный паралич, деменцию с тельцами Леви, сосудистую деменцию, болезнь Гентингтона, болезнь Паркинсона, рассеянный склероз, амиотрофический латеральный склероз, синдром Дауна, депрессию пожилых людей, синдром Вернике-Корсакова, кортико-базальные дегенерации и прионное заболевание, аутизм и синдром гиперактивности с дефицитом внимания, как раскрыто в WO 2013/155506, содержание которой включено в настоящую заявку посредством ссылки во всей полноте;
7.2 Способ I или 7.1, где расстройства, ассоциированные с деменцией, выбраны из группы, включающей (1) поведенческие или аффективные расстройства, такие как ажитацию/раздражение, агрессивное/буйное поведение, гнев, физические или эмоциональные всплески; (2) психоз; (3) депрессию; и (4) расстройства сна;
7.3 Способ I или 7.1, где расстройство центральной нервной системы представляет собой ажитацию/раздражение, агрессивное/буйное поведение, гнев, физические или эмоциональные всплески, как раскрыто в WO 2013/155504, содержание которой включено в настоящую заявку посредством ссылки во всей полноте;
7.4 Способ I, где расстройство центральной нервной системы представляет собой расстройство, выбранное из группы, включающей ожирение, тревожное расстройство, депрессию (например, рефрактерную депрессию и большое депрессивное расстройство (MDD)), психоз, шизофрению, расстройства сна (в частности, расстройства сна, связанные с шизофренией и другими психиатрическими и неврологическими заболеваниями), сексуальные расстройства, мигрень, состояния, связанные с головной болью, социофобии, ажитацию при деменции (например, ажитацию при болезни Альцгеймера), ажитацию при аутизме и родственных аутических расстройствах и желудочно-кишечные расстройства, такие как дисфункция моторики желудочно-кишечного тракта;
7.5 Способ I или любой из 7.1-7.4, где расстройство центральной нервной системы представляет собой расстройство с вовлечением серотонина 5-HT2A, системы дофаминовых D1/D2 рецепторов и/или путей транспортера обратного захвата серотонина (SERT), аналогично описанное в WO/2009/145900, содержание которого включено в настоящую заявку посредством ссылки во всей полноте;
7.6 Способ I или любой из 7.1-7.5, где расстройство центральной нервной системы представляет собой расстройство с вовлечением путей транспортера обратного захвата серотонина (SERT);
7.7 Способ I или любой из 7.1-7.6, где расстройство центральной нервной системы представляет собой расстройство, выбранное из следующих: (i) психоз, например, шизофрения, у пациента, страдающего депрессией; (2) депрессия у пациента, страдающего психозом, например, шизофрения; (3) аффективные расстройства, ассоциированные с психозом, например, шизофрения или болезнь Паркинсона; и (4) расстройства сна, связанные с психозом, например, шизофренией или болезнью Паркинсона; (5) депрессия; (6) тревожное расстройство; (7) посттравматическое стрессовое расстройство; или (8) расстройство импульсного контроля, например, интермиттирующее эксплозивное расстройство;
7.8 Способ I или любой из 7.1-7.7, где расстройство центральной нервной системы представляет собой психоз, например, шизофрению, и указанный пациент представляет собой пациента, страдающего депрессией;
7.9 Способ I или любой из 7.1-7.8, где указанный пациент неспособен переносить побочные эффекты обычных антипсихотических препаратов, например, хлорпромазина, галоперидола, дроперидола, флуфеназина, локсапина, мезоридазина, молиндона, перфеназина, пимозида, прохлорперазина, промазина, тиоридазина, тиотиксена, трифлуоперазина, клозапина, арипипразола, оланзапина, кветиапина, рисперидона и зипрасидона;
7.10 Способ I или любой из 7.1-7.9, где указанный пациент неспособен переносить побочные эффекты обычных антипсихотических препаратов, например, галоперидола, арипипразола, клозапина, оланзапина, кветиапина, рисперидона и зипрасидона;
7.11 Способ I или любой из 7.1-7.10, где указанное расстройство представляет собой депрессию, и указанный пациент представляет собой пациента, страдающего психозом, например, шизофренией, или болезнью Паркинсона;
7.12 Способ I или любой из 7.1-7.6, где указанное расстройство представляет собой расстройство сна, и указанный пациент страдает депрессией;
7.13 Способ I или любой из 7.1-7.6, где указанное одно или несколько расстройств представляет собой расстройство сна, и указанный пациент страдает психозом, например, шизофренией;
7.14 Способ I или любой из 7.1-7.6, где указанное одно или несколько расстройств представляет собой расстройство сна, и указанный пациент страдает болезнью Паркинсона;
7.15 Способ I или любой из 7.1-7.6, где указанное одно или несколько расстройств представляет собой расстройство сна, и указанный пациент страдает депрессией и психозом, например, шизофренией или болезнью Паркинсона;
7.16 Способ I или любой из 7.1-7.6, где расстройство центральной нервной системы представляет собой остаточные симптомы психоза, например, шизофрении (например, остаточный подтип), бредовое расстройство (например, соматический тип), глубокую депрессию с психозом, биполярное расстройство с психотическими симптомами, кратковременное психотическое расстройство, шизофреноформное расстройство, шизоаффективное расстройство или психоз, вызванный медицинским состоянием или употреблением психоактивных веществ. Предпочтительно, пациент страдает от остаточных симптомов шизофрении;
7.17 Способ I или любой из 7.1-7.6, где симптомы остаточной фазы включают: негативные симптомы, такие как притупленный аффект, эмоциональное отчуждение, малоконтактность, пассивное или апатическое социальное отчуждение, затруднение абстрактного мышления, отсутствие спонтанности и плавного течения беседы и стереотипное мышление; общие психопатологические симптомы, такие как обеспокоенность соматическим здоровьем, тревожное расстройство, чувство вины, напряженность, манерность и «застывание» в определенной позе, депрессия, двигательная заторможенность, неконтактность, необычное содержание мыслей, дезориентирование, слабое внимание, отсутствие здравого смысла и проницательности, нарушение силы воли, ослабление контроля импульсивности, озабоченность и активное социальное избегание; когнитивное нарушение и расстройства сна (например, бессонница);
7.18 Любой из вышеперечисленных способов, где эффективное количество составляет 1 мг-1000 мг, предпочтительно 2,5 мг-50 мг, еще предпочтительнее 1-40 мг, например, 1-10 мг, например, 10 мг, 20 мг, более 20 мг, например, 30 мг, 40 мг;
7.19 Любой из вышеперечисленных способов, где эффективное количество составляет 1 мг-100 мг в сутки, предпочтительно 2,5 мг-50 мг в сутки, еще предпочтительнее 1-40 мг/сутки, например, 1-10 мг/сутки, например, 10 мг/сутки, 20 мг/сутки, более 20 мг/сутки, например, 30 мг/сутки, 40 мг/сутки;
7.20 Любой из вышеперечисленных способов, где требующее лечения состояние представляет собой дискинезию, например, у пациента, получающего дофаминэргические лекарственные препараты, например, лекарственные препараты, выбранные из леводопы и вспомогательных средств для леводопы (карбидопа, ингибиторы COMT, ингибиторы MAO-B), дофаминовых агонистов и антихолинергических средств, например, леводопы;
7.21 Любой из вышеперечисленных способов, где пациент страдает болезнью Паркинсона;
7.22 Любой из вышеперечисленных способов, где пациент не отвечает на лечение селективным ингибитором обратного захвата серотонина, например, выбранным из одного или нескольких из следующих: циталопрам (Celexa, Cipramil, Cipram, Dalsan, Recital, Emocal, Sepram, Seropram, Citox, Cital); дапоксетин (Priligy); эсциталопрам (Lexapro, Cipralex, Seroplex, Esertia); флуоксетин (Depex, Prozac, Fontex, Seromex, Seronil, Sarafem, Ladose, Motivest, Flutop, Fluctin (EUR), Fluox (NZ), Depress (UZB), Lovan (AUS), Prodep (IND)); флувоксамин (Luvox, Fevarin, Faverin, Dumyrox, Favoxil, Movox); индалпин (Upstene); пароксетин (Paxil, Seroxat, Sereupin, Aropax, Deroxat, Divarius, Rexetin, Xetanor, Paroxat, Loxamine, Deparoc); сертралин (Zoloft, Lustral, Serlain, Asentra); вилазодон (Viibryd); или зимелидин (Zelmid, Normud);
7.23 Любой из вышеперечисленных способов, где пациент также принимает селективный ингибитор обратного захвата серотонина, например, выбранный из одного или нескольких из следующих: циталопрам (Celexa, Cipramil, Cipram, Dalsan, Recital, Emocal, Sepram, Seropram, Citox, Cital); дапоксетин (Priligy); эсциталопрам (Lexapro, Cipralex, Seroplex, Esertia); флуоксетин (Depex, Prozac, Fontex, Seromex, Seronil, Sarafem, Ladose, Motivest, Flutop, Fluctin (EUR), Fluox (NZ), Depress (UZB), Lovan (AUS), Prodep (IND)); флувоксамин (Luvox, Fevarin, Faverin, Dumyrox, Favoxil, Movox); индалпин (Upstene); пароксетин (Paxil, Seroxat, Sereupin, Aropax, Deroxat, Divarius, Rexetin, Xetanor, Paroxat, Loxamine, Deparoc); сертралин (Zoloft, Lustral, Serlain, Asentra); вилазодон (Viibryd); или зимелидин (Zelmid, Normud);
7.24 Любой из вышеперечисленных способов, где пациент страдает расстройством аутического спектра, например, аутизмом или синдромом Аспергера;
7.25 Любой из вышеперечисленных способов, где пациенты страдают деменцией, например, расстройствами, связанными с умеренными когнитивными нарушениями и дементным состоянием, включая сенильную деменцию, болезнь Альцгеймера, болезнь Пика, фронто-темпоральную деменцию, супрануклеарный паралич, деменцию с тельцами Леви, сосудистую деменцию, болезнь Гентингтона, болезнь Паркинсона, рассеянный склероз, амиотрофический латеральный склероз, синдром Дауна, депрессию пожилых людей, синдром Вернике-Корсакова, кортико-базальные дегенерации и прионное заболевание, аутизмом и синдромом гиперактивности с дефицитом внимания;
7.26 Любой из вышеперечисленных способов, где пациент также получает ингибитор холинэстеразы (например, ингибитор ацетилхолинэстеразы) или антагонист рецептора N-Метил D-Аспартата (NMDA), в свободной форме или форме фармацевтически приемлемой соли;
7.27 Способ 7.26, где ингибитор холинэстеразы (например, ингибитор ацетилхолинэстеразы) выбран из группы, включающей Такрин, ривастигмин (Exelon), донепезил (Aricept) и галантамин (Razadyne,, ранее называемый Reminyl)) в свободной форме или форме фармацевтически приемлемой соли;
7.28 Способ 7.26, где ингибитор холинэстеразы (например, ингибитор ацетилхолинэстеразы) представляет собой донепезил в свободной форме или форме фармацевтически приемлемой соли;
7.29 Способ 7.26, где антагонист NMDA-рецептора представляет собой мемантин в свободной форме или форме фармацевтически приемлемой соли;
7.30 Любой из вышеперечисленных способов, дополнительно включающий введение одного или нескольких других терапевтических средств, таких как дополнительные антипсихотические средства и/или антидепрессивные средства и/или снотворные средства;
7.31 Способ 7.30, где одно или несколько других терапевтических средств выбраны из антидепрессивных средств, таких как соединения, которые модулируют активность ГАМК (например, повышают активность и стимулируют трансмиссию ГАМК), агониста ГАМК-B, модулятора 5-HT (например, агониста 5-HT1A, антагониста 5-HT2A, обратного агониста 5-HT2A и т.д.), агониста мелатонина, модулятора ионных каналов (например, блокатора), антагониста и ингибитора обратного захвата серотонина-2 (SARIs), антагониста рецептора орексина, агониста H3, норадренергического антагониста, агониста галанина, антагониста CRH, гормона роста человека, агониста гормона роста, эстрогена, агониста эстрогена, нейрокинин-1 лекарственного средства; и антипсихотических средств, например, атипичных антипсихотических средств, в свободной форме или форме фармацевтически приемлемой соли;
7.32 Способ 7.30 или 7.31, где одно или несколько других терапевтических средств представляют собой антипсихотические средства, например, хлорпромазин, галоперидол, дроперидол, флуфеназин, локсапин, мезоридазин, молиндон, перфеназин, пимозид, прохлорперазин промазин, тиоридазин, тиотиксен, трифлуоперазин, клозапин, арипипразол, оланзапин, кветиапин, рисперидон, зипрасидон, палиперидон, азенапин, луразидон, илоперидон, карипразин, амисульприд, зотепин, сертиндол, где одно или несколько других терапевтических средств вводят в качестве вспомогательного средства к соединению формулы I, или соединение формулы I представляет собой вспомогательное средство к одному или нескольким другим терапевтическим средствам.
[0033] В конкретном варианте осуществления четвертого аспекта настоящее изобретение обеспечивает способ (Способ IP) для лечения или профилактики расстройства центральной нервной системы, описанного выше, включающий введение нуждающемуся в этом пациенту:
7.4P соединения формулы I или любой из формул 1-1.9, в свободной форме или в форме (фармацевтически приемлемой) соли, описанной в любом из пунктов 4.1-4.4;
7.8P фармацевтической или депо-композиции, описанной выше; или
7.11P композиции системы пероральной доставки с осмотически контролируемым высвобождением, описанной выше.
[0034] В следующем варианте осуществления четвертого аспекта настоящее изобретение обеспечивает Способ IP, который более подробно описан в любой из формул 7.1-7.32.
[0035] В конкретном варианте осуществления четвертого аспекта настоящее изобретение обеспечивает Способ I, IP или любой из 7.1-7.32, где расстройство представляет собой шизофрению или расстройство сна.
[0036] В конкретном варианте осуществления четвертого аспекта настоящее изобретение обеспечивает Способ I, IP или любой из 7.1-7.32, где расстройство представляет собой депрессию или тревожное расстройство.
[0037] В конкретном варианте осуществления четвертого аспекта настоящее изобретение обеспечивает Способ I, IP или любой из 7.1-7.32, где расстройство представляет собой посттравматическое стрессовое расстройство или расстройство импульсного контроля, например, интермиттирующее эксплозивное расстройство.
[0038] В конкретном варианте осуществления четвертого аспекта настоящее изобретение обеспечивает Способ I, IP или любой из 7.1-7.32, где расстройство представляет собой посттравматическое стрессовое расстройство или расстройство импульсного контроля, например, интермиттирующее эксплозивное расстройство у пациента, страдающего деменцией, например, сенильной деменцией, болезнью Альцгеймера, болезнью Пика, фронто-темпоральной деменцией, супрануклеарным параличом, деменцией с тельцами Леви, сосудистой деменцией, болезнью Гентингтона, болезнью Паркинсона, рассеянным склерозом, амиотрофическим латеральным склерозом, синдромом Дауна, депрессией пожилых людей, синдромом Вернике-Корсакова, кортико-базальными дегенерациями, прионным заболеванием, аутизмом и/или синдромом гиперактивности с дефицитом внимания.
[0039] В еще одном варианте осуществления четвертого аспекта настоящее изобретение обеспечивает Способ I, IP или любой из 7.1-7.32, где депо-композицию по настоящему изобретению вводят для контролируемого и/или длительного высвобождения соединений по настоящему изобретению в течение периода от около 14 дней, около 30 до около 180 дней, предпочтительно в течение периода около 30, около 60 или около 90 дней. Контролируемое и/или длительное высвобождение является особенно полезным, чтобы избежать преждевременного прекращения лечения, особенно для антипсихотической лекарственной терапии, где невыполнение или несоблюдение режима приема лекарств является обычным явлением.
[0040] В пятом аспекте настоящее изобретение обеспечивает способ (Способ II) для профилактики или лечения одного или нескольких расстройств сна, ажитации, агрессивного поведения, посттравматического стрессового расстройства и/или расстройства импульсного контроля, например, интермиттирующего эксплозивного расстройства, включающий введение нуждающемуся в этом пациенту соединения, описанного ниже:
8.1 соединения формулы I или в соответствии с любым из пунктов 1-1.9 в свободной форме или в форме (фармацевтически приемлемой) соли, описанной в любом из пунктов 4.1-4.4;
8.2 фармацевтической или депо-композиции, описанной выше;
8.3 композиции системы пероральной доставки с осмотически контролируемым высвобождением, описанной выше.
[0041] В одном варианте осуществления пятого аспекта настоящее изобретение обеспечивает Способ II или любой из 8.1-8.3, где расстройство представляет собой расстройства сна. В другом варианте осуществления пятого аспекта настоящее изобретение обеспечивает Способ II, где расстройство представляет собой ажитацию, агрессивное поведение, посттравматическое стрессовое расстройство и/или расстройство импульсного контроля, например, интермиттирующее эксплозивное расстройство.
[0042] В следующем варианте осуществления пятого аспекта настоящее изобретение обеспечивает Способ II, 8.1-8.3, где расстройство сна включает бессонницу поддержания сна, частые пробуждения и когда субъект просыпается неотдохнувшим;
8.11 Любой из вышеперечисленных способов, где расстройство сна представляет собой бессонницу поддержания сна;
8.12 Любой из вышеперечисленных способов, где эффективное количество составляет 1 мг-10 мг в сутки, например, 1-5 мг, предпочтительно 2,5-5 мг, в сутки, еще предпочтительнее 10 мг в сутки;
8.13 Любой из вышеперечисленных способов, где эффективное количество составляет 2,5 мг или 5 мг в сутки или 10 мг в сутки;
8.14 Любой из вышеперечисленных способов, где расстройство сна возникает у пациента, страдающего от или имеющего риск развития дискинезии, например, пациента, принимающего дофаминэргические лекарственные препараты, например, выбранные из леводопы и вспомогательных средств для леводопы (карбидопа, ингибиторы COMT, ингибиторы MAO-B), агонистов дофамина и антихолинергических средств, например, принимающего леводопу;
8.15 Любой из вышеперечисленных способов, где пациент страдает болезнью Паркинсона.
[0043] Соединения по настоящему изобретению обеспечивают эффективное лечение расстройств, связанных с 5-HT2A, SERT и/или D2 рецепторами без или с минимальными экстрапирамидальными побочными эффектами, как подробно раскрыто и заявлено в WO 2009/145900, содержание которой включено в настоящую заявку посредством ссылки во всей полноте. Поэтому соединения по настоящему изобретению, фармацевтические композиции по настоящему изобретению или депо-композиции по настоящему изобретению можно использовать в комбинации с вторым терапевтическим средством, в частности, при более низких дозах, чем те, когда отдельные средства используют в качестве монотерапии, для повышения терапевтических активностей комбинированных средств, не вызывая при этом нежелательных побочных эффектов, обычно возникающих при введении традиционной монотерапии. Поэтому соединения по настоящему изобретению можно вводить одновременно, последовательно или также продолжительно с другим антидепрессантом, антипсихотическим средством, другими снотворными средствами и/или средствами, используемыми для лечения болезни Паркинсона или аффективных расстройств или деменции. В другом примере, побочные эффекты можно уменьшить или минимизировать путем введения Соединения по настоящему изобретению в комбинации с одним или несколькими вторыми терапевтическими средствами в свободной форме или форме соли, где дозы (i) второго терапевтического средства(средств) или (ii) обоих Соединения по настоящему изобретению и второго терапевтического средства меньше, чем если бы такое средство/соединение вводили бы в качестве монотерапии. В конкретном варианте осуществления настоящего изобретения соединения по настоящему изобретению являются полезными для лечения дискинезии у пациента, получающего дофаминэргические лекарственные средства, например, выбранные из леводопа и вспомогательных средств для леводопы (карбидопа, ингибиторы COMT, ингибиторы MAO-B), дофаминовых агонистов и антихолинергических средств, например, таких, которые используют в лечении болезни Паркинсона.
[0044] Поэтому в шестом аспекте настоящее изобретение обеспечивает Способ I или IP или, например, в соответствии с любым из пунктов 7.1-7.32, или Способ II или в соответствии с любым из пунктов 8.1-8.15, дополнительно включающий одно или несколько терапевтических средств, выбранных из соединений, которые модулируют активность ГАМК (например, повышают активность и стимулируют трансмиссию ГАМК), агониста ГАМК-B, модулятора 5-HT (например, агониста 5-HT1A, антагониста 5-HT2A, обратного агониста 5-HT2A и т.д.), агониста мелатонина, модулятора ионных каналов (например, блокатора), антагониста/ингибитора обратного захвата серотонина-2 (SARIs), антагониста рецептора орексина, агониста или антагониста H3, норадренергического агониста или антагониста, агониста галанина, антагониста CRH, гормона роста человека, агониста гормона роста, эстрогена, агониста эстрогена, нейрокинин-1 лекарственного средства, антидепрессанта и антипсихотического средства, например, атипичного антипсихотического средство, в свободной форме или форме фармацевтически приемлемой соли (Способ I-A и II-A соответственно).
[0045] В другом варианте осуществления шестого аспекта Способ I-A и II-A, Способ I, Способ IP или, например, в соответствии с любым из пунктов 7.1-7.32, или Способ II или в соответствии с любым из пунктов 8.1-8.15, дополнительно включает одно или несколько терапевтических средств, выбранных из ингибитора холинэстеразы (например, ингибитора ацетилхолинэстеразы) или антагониста рецептора N-Метил D-Аспартата (NMDA), в свободной форме или форме фармацевтически приемлемой соли. В конкретном варианте осуществления ингибитор холинэстеразы (например, ингибитор ацетилхолинэстеразы) выбран из группы, включающей Такрин, ривастигмин (Exelon), донепезил (Aricept) и галантамин (Razadyn, ранее называемый Reminyl)) в свободной форме или форме фармацевтически приемлемой соли. В следующем варианте осуществления ингибитор холинэстеразы (например, ингибитор ацетилхолинэстеразы) представляет собой донепезил в свободной форме или форме фармацевтически приемлемой соли. В другом варианте осуществления антагонист NMDA-рецептора представляет собой мемантин в свободной форме или форме фармацевтически приемлемой соли.
[0046] В следующем варианте осуществления шестого аспекта настоящее изобретение обеспечивает Способ I-A или II-A, описанный ниже, дополнительно включающий одно или несколько терапевтических средств.
9.1 Способ I-A или II-A, где терапевтическое средство(средства) представляет собой соединения, которые модулируют активность ГАМК (например, повышают активность и стимулируют трансмиссию ГАМК);
9.2 Способ I-A или II-A или 9.1, где ГАМК соединение выбрано из группы, состоящей из одного или нескольких из доксепина, алпразолама, бромазепама, клобазама, клоназепама, клоразепата, диазепама, флунитразепама, фиуразепама, лоразепама, мидазолама, нитразепама, оксазепама, темазапама, триазолама, индиплона, зопиклона, эсзопиклона, залеплона, Золпидема, габаксадола, вигабатрина, тиагабина, EVT 201 (Evotec Pharmaceuticals) и эстазолама;
9.3 Способ I-A или II-A, где терапевтическое средство представляет собой дополнительный антагонист 5HT2A;
9.4 Способ I-A или II-A или 9.3, где указанный дополнительн антагонист 5HT2A выбран из одного или нескольких из кетансерина, рисперидона, эпливансерина, волинансерина (Sanofi-Aventis, France), прувансерина, MDL 100907 (Sanofi-Aventis, France), HY 10275 (Eli Lilly), APD 125 (Arena Pharmaceuticals, San Diego, CA) и AVE8488 (Sanofi-Aventis, France); Способ I-A или II-A, 9.3 или 9.4, дополнительно выбранный из пимавансерина (ACP-103) и пизотифена;
9.5 Способ I-A или II-A, где терапевтическое средство представляет собой агонист мелатонина;
9.6 Способ I-A или II-A или 9.5, где агонист мелатонина выбран из группы, состоящей из одного или нескольких из мелатонина, рамелтеона (ROZEREM®, Takeda Pharmaceuticals, Japan), VEC-162 (Vanda Pharmaceuticals, Rockville, MD), PD-6735 (Phase II Discovery) и агомелатина;
9.7 Способ I-A или II-A, где терапевтическое средство представляет собой блокатор ионных каналов;
9.8 Способ I-A или II-A или 9.7, где указанный блокатор ионных каналов представляет собой один или несколько из ламотригина, габапентина и прегабалина.
9.9 Способ I-A или II-A, где терапевтическое средство представляет собой антагонист рецептора орексина;
9.10 Способ I-A или II-A или 9.9, где антагонист рецептора орексина выбран из группы, состоящей из орексина, 1,3-биарилмочевины, SB-334867-a (GlaxoSmithKline, UK), GW649868 (GlaxoSmithKline) и бензамидного производного;
9.11 Способ I-A или II-A, где терапевтическое средство представляет собой антагонист/ингибитор обратного захвата серотонина-2 (SARI);
9.12 Способ I-A или II-A или 9.11, где антагонист/ ингибитор обратного захвата серотонина-2 (SARI) выбран из группы, состоящей из одного или нескольких из Org 50081 (Organon -Netherlands), ритансерина, нефазодона, серзона и тразодона;
9.13 Способ I-A или II-A, где терапевтическое средство является агонистом 5HT1a;
9.14 Способ I-A или II-A или 9.13, где агонист 5HT1a выбран из группы, состоящей из одного или нескольких из репинотана, саризотана, эптапирона, буспирона и MN-305 (MediciNova, San Diego, CA);
9.15 Способ I-A или II-A, где терапевтическое средство представляет собой нейрокинин-1 лекарственное средство;
9.16 Способ I-A или II-A или 9.15, где нейрокинин-1 лекарственное средство представляет собой Казопитант (GlaxoSmithKline);
9.17 Способ I-A или II-A, где терапевтическое средство представляет собой антипсихотическое средство;
9.18 Способ I-A или II-A или 9.17, где антипсихотическое средство выбрано из группы, состоящей из хлорпромазина, галоперидола, дроперидола, флуфеназина, локсапина, мезоридазина, молиндона, перфеназина, пимозида, прохлорперазина промазина, тиоридазина, тиотиксена, трифлуоперазина, клозапина, арипипразола, оланзапина, кветиапина, рисперидона, зипрасидона и палиперидона;
9.19 Способ I-A или II-A, где терапевтическое средство представляет собой антидепрессант;
9.20 Способ I-A или II-A или 9.19, где антидепрессант выбран из амитриптилина, амоксапина, бупропиона, циталопрама, кломипрамина, дезипрамина, доксепина, дулоксетина, эсциталопрама, флуоксетина, флувоксамина, имипрамина, изокарбоксазида, мапротилина, миртазапина, нефазодона, нортриптилина, пароксетина, фенелазин сульфата, протриптилина, сертралина, транилципромина, тразодона, тримипрамина и венлафаксина;
9.21 Способ I-A или II-A, 9.17 или 9.18, где антипсихотическое средство представляет собой атипичное антипсихотическое средство;
9.22 Способ I-A или II-A, или любой из 9.17-9.21, где атипичное антипсихотическое средство выбрано из группы, состоящей из клозапина, арипипразола, оланзапина, кветиапина, рисперидона, зипрасидона и палиперидона;
9.23 Способ I-A или II-A, где терапевтическое средство выбрано из любого из способов 9.1-9.22, например, выбрано из группы, состоящей из модафинила, армодафинила, доксепина, алпразолама, бромазепама, клобазама, клоназепама, клоразепата, диазепама, флунитразепама, флуразепама, лоразепама, мидазолама, нитразепама, оксазепама, темазапама, триазолама, индиплона, зопиклона, эсзопиклона, залеплона, Золпидема, габаксадола, вигабатрина, тиагабина, EVT 201 (Evotec Pharmaceuticals), эстазолама, кетансерина, рисперидона, эпливансерина, волинансерина (Sanofi-Aventis, France), прувансерина, MDL 100907 (Sanofi-Aventis, France), HY 10275 (Eli Lilly), APD 125 (Arena Pharmaceuticals, San Diego, CA), AVE8488 (Sanofi-Aventis, France), репинотана, саризотана, эптапирона, буспирона, MN-305 (MediciNova, San Diego, CA), мелатонина, рамелтеона (ROZEREM®, Takeda Pharmaceuticals, Japan), VEC-162 (Vanda Pharmaceuticals, Rockville, MD), PD-6735 (Phase II Discovery), агомелатина, ламотригина, габапентина, прегабалина, орексина, 1,3-биарилмочевины, SB-334867-a (GlaxoSmithKline, UK), GW649868 (GlaxoSmithKline), бензамидного производного, Org 50081 (Organon -Netherlands), ритансерина, нефазодона, серзона, тразодона, Казопитанта (GlaxoSmithKline), амитриптилина, амоксапина, бупропиона, циталопрама, кломипрамина, дезипрамина, доксепина, дулоксетина, эсциталопрама, флуоксетина, флувоксамина, имипрамина, изокарбоксазида, мапротилина, миртазапина, нефазодона, нортриптилина, пароксетина, фенелазина сульфата, протриптилина, сертралина, транилципромина, тразодона, тримипрамина, венлафаксина, хлорпромазина, галоперидола, дроперидола, флуфеназина, локсапина, мезоридазина молиндона, перфеназина, пимозида, прохлорперазина промазина, тиоридазина, тиотиксена, трифлуоперазина, клозапина, арипипразола, оланзапина, кветиапина, рисперидона, зипрасидона и палиперидона; В дополнение к терапевтическим средствам, перечисленным выше, Способ I-A или II-A, терапевтическое средство дополнительно выбрано из пимавансерина (ACP-103) и пизотифена;
9.24 Способ I-A или II-A где терапевтическое средство представляет собой агонист H3;
9.25 Способ I-A или II-A, где терапевтическое средство представляет собой антагонист H3;
9.26 Способ I-A или II-A, где терапевтическое средство представляет собой норадренергический агонист или антагонист;
9.27 Способ I-A или II-A, где терапевтическое средство представляет собой агонист галанина;
9.28 Способ I-A или II-A, где терапевтическое средство представляет собой антагонист CRH;
9.29 Способ I-A или II-A, где терапевтическое средство представляет собой гормон роста человека;
9.30 Способ I-A или II-A, где терапевтическое средство представляет собой агонист гормона роста;
9.31 Способ I-A или II-A, где терапевтическое средство представляет собой эстроген;
9.32 Способ I-A или II-A, где терапевтическое средство представляет собой агонист эстрогена;
9.33 Способ I-A или II-A, где терапевтическое средство представляет собой нейрокинин-1 лекарственное средство;
9.34 Способ I-A или II-A, где терапевтическое средство объединяют с соединениями формулы (I), и терапевтическое средство представляет собой средство для лечения болезни Паркинсона, такое как L-допа, ко-карелдопа, дуодопа, сталова, Симметрел, бензотропин, бипериден, бромокриптин, энтакапон, перголид, прамипексол, проциклидин, ропинирол, селегелин и толкапон;
9.35 Способ I-A или II-A, где соединения формулы (I) можно использовать для лечения расстройств сна, депрессии, психоза или любых их комбинаций у пациентов, страдающих перечисленными заболеваниями и/или болезнью Паркинсона;
9.36 Способ I-A или II-A, где расстройство выбрано из по меньшей мере одного или нескольких из психоза, например, шизофрении, депрессии, аффективных расстройств, расстройств сна (например, поддержания сна и/или наступления сна) или любой комбинации таких расстройств;
9.37 Любой из вышеперечисленных способов, где расстройство представляет собой расстройство сна;
9.38 Любой из вышеперечисленных способов, где расстройство представляет собой расстройство сна, связанное с психозом, например, шизофренией, или болезнью Паркинсона; в свободной форме или форме фармацевтически приемлемой соли.
[0047] В другом варианте осуществления шестого аспекта настоящее изобретение обеспечивает Способ IP или Способ II, описанный выше, дополнительно включающий одно или несколько терапевтических средств, выбранных из соединений, которые модулируют активность ГАМК (например, повышают активность и стимулируют трансмиссию ГАМК), агониста ГАМК-B, модулятора 5-HT (например, агониста 5-HT1A, антагониста 5-HT2A, обратного агониста 5-HT2A и т.д.), агониста мелатонина, модулятора ионных каналов (например, блокатора), антагониста/ингибитора обратного захвата серотонина-2 (SARIs), антагониста рецептора орексина, агониста или антагониста H3, норадренергического агониста или антагониста, агониста галанина, антагониста CRH, гормона роста человека, агониста гормона роста, эстрогена, агониста эстрогена, нейрокинин-1 лекарственного средства, антидепрессанта и антипсихотического средства, например, атипичного антипсихотического средства, в свободной форме или форме фармацевтически приемлемой соли (Способ IP-A и II-A соответственно). В следующем варианте осуществления этого аспекта настоящее изобретение обеспечивает Способ IP-A или II-A, аналогично описанный в любом из пунктов 9.1-9.38.
[0048] В еще одном варианте осуществления шестого аспекта Способ IP или Способ II, описанный выше, дополнительно включает одно или несколько терапевтических средств, выбранных из ингибитора холинэстеразы (например, ингибитора ацетилхолинэстеразы) или антагониста рецептора N-Метил D-Аспартата (NMDA), в свободной форме или форме фармацевтически приемлемой соли. В конкретном варианте осуществления, ингибитор холинэстеразы (например, ингибитор ацетилхолинэстеразы) выбран из группы, включающей Такрин, ривастигмин (Exelon), донепезил (Aricept) и галантамин (Razadyn, ранее называемый Reminyl)), в свободной форме или форме фармацевтически приемлемой соли. В следующем варианте осуществления, ингибитор холинэстеразы (например, ингибитор ацетилхолинэстеразы) представляет собой донепезил в свободной форме или форме фармацевтически приемлемой соли. В другом варианте осуществления антагонист NMDA-рецептора представляет собой мемантин в свободной форме или форме фармацевтически приемлемой соли.
[0049] В седьмом аспекте изобретения комбинацию Соединения по настоящему изобретению и одного или нескольких вторых терапевтических средств, описанных в Способах I-A, II-A или любом из 9.1-9.38, можно вводить в виде фармацевтической композиции или депо-композиции, описанных выше. Подобным образом, комбинацию Соединения по настоящему изобретению и одного или нескольких вторых терапевтических средств, описанных в Способах Ip-A, II-A или любом из 9.1-9.38, можно вводить в виде фармацевтической композиции или депо-композиции описанной выше. Комбинированные композиции могут включать смеси комбинированных лекарственных средств, а также две или более отдельные композиции лекарственных средств, при этом такие отдельные композиции можно, например, совместно вводить пациенту.
[0050] В конкретном варианте осуществления настоящего изобретения Способы I-A, II-A, Ip-A, II-A или любой из 9.1-9.38 включают введение пациенту, нуждающемуся в этом, Соединения по настоящему изобретению в комбинации с атипичным антипсихотическим средством, например, соединением, выбранным из клозапина, арипипразола, оланзапина, кветиапина, рисперидона, зипрасидона или палиперидона, в свободной форме или форме фармацевтически приемлемой соли, например, где дозу атипичного антипсихотического средства снижают и/или побочные эффекты уменьшаются.
[0051] В другом варианте осуществления Способы I-A, II-A, Способы Ip-A, II-A или любой из 9.1-9.38 включают введение пациенту, нуждающемуся в этом, Соединения по настоящему изобретению в комбинации с антидепрессантом, например, амитриптилином, амоксапином, бупропионом, циталопрамом, кломипрамином, дезипрамином, доксепином, дулоксетином, эсциталопрамом, флуоксетином, флувоксамином, имипрамином, изокарбоксазидом, мапротилином, миртазапином, нефазодоном, нортриптилином, пароксетином, фенелазин сульфатом, протриптилином, сертралином, транилципромином, тразодоном, тримипрамином или венлафаксином, в свободной форме или форме фармацевтически приемлемой соли. Альтернативно, антидепрессант можно использовать в качестве вспомогательного лекарственного средства в дополнение к соединению по настоящему изобретению.
[0052] В еще одном варианте осуществления, Способы I-A, II-A, Ip-A, II-A или любой из 9.1-9.38 включают введение пациенту, нуждающемуся в этом, Соединения по настоящему изобретению в комбинации с соединением, которое модулирует активность ГАМК, например, соединением, выбранным из доксепина, алпразолама, бромазепама, клобазама, клоназепама, клоразепата, диазепама, флунитразепама, флуразепама, лоразепама, мидазолама, нитразепама, оксазепама, темазапама, триазолама, индиплона, зопиклона, эсзопиклона, залеплона, Золпидема, габаксадола, вигабатрина, тиагабина, EVT 201 (Evotec Pharmaceuticals), эстазолама или любых их комбинаций, в свободной форме или форме фармацевтически приемлемой соли.
[0053] В другом конкретном варианте осуществления Способы I-A, II-A, Ip-A, II-A или любой из 9.1-9.38 включают введение пациенту, нуждающемуся в этом, Соединения по настоящему изобретению в комбинации с доксепином в свободной форме или форме фармацевтически приемлемой соли. Дозы доксепина могут варьировать в любых пределах, известных средним специалистам в данной области. В одном примере, 10 мг дозу доксепина можно использовать в комбинации с любой дозой соединения по настоящему изобретению.
[0054] В другом варианте осуществления, Способы I-A, II-A, Ip-A, II-A или любой из 9.1-9.38 включают введение пациенту, нуждающемуся в этом, Соединения по настоящему изобретению в комбинации (в том числе как часть ежедневного режима введения) с атипичным стимулирующим средством, например, модафинилом, адрафинилом или армодафинилом. Схема введения Соединения по настоящему изобретению с таким лекарственным средством способствует более регулярному сну и избегает возникновения побочных эффектов, таких как психоз или мания, связанных с более высокими уровнями таких лекарственных средств, например, в лечении биполярной депрессии, когнитивного расстройства, связанного с шизофренией, и чрезмерной сонливости и усталости при таких состояниях, как болезнь Паркинсона и рак.
[0055] В восьмом аспекте настоящее изобретение обеспечивает применение соединения, как описано в следующих пунктах:
11.1 Соединение формулы I или в соответствии с любым из пунктов 1-1.9 в свободной форме или форме фармацевтически приемлемой соли;
11.2 фармацевтическая композиция, описанная выше;
11.3 депо-композиция, описанная выше; или
11.4 композиции системы пероральной доставки с осмотически контролируемым высвобождением, описанная выше,
(для получения лекарственного средства) для лечения или профилактики одного или нескольких расстройств, раскрытых выше, например, в любом из Способа I, любом из 7.1-7.32, Способа II, любом из 8.1-8.15, Способов I-A, II-A, любом из 9.1-9.38, Способа IP, Способов IP-A, или в любых способах, описанных в шестом или седьмом аспекте настоящего изобретения.
[0056] В девятом аспекте настоящее изобретение обеспечивает фармацевтическую композицию, как описано, например, в следующих пунктах:
12.1 фармацевтическая композиция, описанная выше;
12.2 депо-композиция, описанная выше; или
12.3 композиции системы пероральной доставки с осмотически контролируемым высвобождением, описанная выше,
Для применения в лечении или профилактике одного или нескольких расстройств, раскрытых выше, например, в любом из Способа I, любом из 7.1-7.32, Способа II, любом из 8.1-8.15, Способов I-A, II-A, любом из 9.1-9.38, Способа IP, Способов IP-A, или в любых способах, описанных в шестом или седьмом аспекте настоящего изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0057] Если не указано или явно не следует из контекста иное, следующие термины, используемые в настоящей заявке, имеют следующие значения:
a. ʺОстаточные симптомыʺ, в контексте настоящего изобретения, включают негативные симптомы и основные психопатологические симптомы, описанные в шкале оценки позитивных и негативных симптомов (PANSS) для шизофрении, описанных в Kay et al., Schizophr. Bull. (1987) 13(2):261-276, содержание которого включено в настоящую заявку посредством ссылки во всей полноте. Негативные симптомы включают: притупленный аффект, эмоциональное отчуждение, малоконтактность, пассивное/апатическое социальное отчуждение, затруднение абстрактного мышления, отсутствие спонтанности и плавного течения беседы и стереотипное мышление. Общие психопатологические симптомы включают следующие: обеспокоенность соматическим здоровьем, тревожное расстройство, чувство вины, напряженность, манерность и «застывание» в определенной позе, депрессия, двигательная заторможенность, неконтактность, необычное содержание мысли, дезориентирование, слабое внимание, отсутствие здравого смысла и проницательности, нарушение силы воли, ослабление контроля импульсивности, озабоченность и активное социальное избегание. Остаточные симптомы также могут включать депрессию, когнитивное расстройство и расстройства сна (например, бессонницу). Из этих остаточных симптомов, соединения по настоящему изобретению являются особенно полезными для лечения пассивной социальной самоизоляции, стереотипного мышления, обеспокоенности соматическим здоровьем, тревожного расстройства, напряженности, активного социального избегания и депрессии. Поэтому соединения по настоящему изобретению являются особенно полезными для улучшения социальной интеграции и социальной функции у пациентов, страдающих шизофренией. Лечение этих остаточных симптомов также особенно эффективно у шизофренических пациентов, также страдающих депрессией.
[0058] Если не указано иное, соединения по настоящему изобретению, например, Соединения формулы I или любое из 1-1.9, или любое из 4.1-4.4 могут существовать в свободной форме или в форме соли, например, в форме кислотно-аддитивной соли. Кислотно-аддитивная соль соединения по настоящему изобретению, которое является достаточно щелочным, например, может представлять собой кислотно-аддитивную соль с, например, неорганической или органической кислотой, например, с хлористоводородной, бромистоводородной, серной, фосфорной, уксусной, трифторуксусной, лимонной, малеиновой, толуол сульфоновой, пропионовой, янтарной, гликолевой, стеариновой, молочной, яблочной, винной, лимонной, аскорбиновой, пальмовой, гидроксималеиновой, фенилуксусной, глютаминовой, бензойной, салициловой, сульфаниловой, 2-ацетоксибензойной, фумаровой, толуолсульфоновой, метансульфоновой, этандисульфоновой, щавелевой, изэтионовой кислотой и т.п. Кроме того, соль соединения по настоящему изобретению, которое является достаточно кислотным, представляет собой соль щелочного металла, например натриевую или калиевую соль, соль щелочноземельного металла, например кальциевую или магниевую соль, соль аммония или соль с органическим основанием, которое обеспечивает физиологически приемлемый катион, например, соль с метиламина, диметиламином, триметиламином, пиперидином, морфолином или трис-(2-гидроксиэтил)-амином. В конкретном варианте осуществления соль соединений по настоящему изобретению представляет собой аддитивную соль толуолсульфоновой кислоты. В другом конкретном варианте осуществления соль соединений по настоящему изобретению представляет собой аддитивную соль фумаровой кислоты. В конкретном варианте осуществления, соль соединений по настоящему изобретению представляет собой аддитивную соль фосфорной кислоты.
[0059] Соединения по настоящему изобретению предназначены для применения в качестве фармацевтических средств, поэтому фармацевтически приемлемые соли являются предпочтительными. Соли, которые на являются подходящими для фармацевтического применения, могут быть полезными, например, для выделения или очистки свободного Соединения по настоящему изобретению, и поэтому также включены.
[0060] Соединения по настоящему изобретению могут включать один или несколько хиральных атомов углерода. Соединения, таким образом, существуют в индивидуальной изомерной, например, энантиомерной или диастереомерной форме или в виде смесей индивидуальных форм, например, рацемических/диастереомерных смесей. Любой изомер может присутствовать, в котором асимметрический центр находится в (R)-, (S)- или (R,S)- конфигурации. Настоящее изобретение следует рассматривать как охватывающее как индивидуальные оптически активные изомеры, так и их смеси (например, рацемические/диастереомерные смеси). Соответственно, соединения по настоящему изобретению могут представлять собой рацемическую смесь, или могут быть преимущественно, например, в чистой или по существу чистой изомерной форме, например, энантиомерный/диастереомерный избыток (ʺэиʺ) больше чем 70%, предпочтительно больше чем 80% эи, более предпочтительно больше чем 90% эи, наиболее предпочтительно больше чем 95% эи. Очистку указанных изомеров и разделение указанных изомерных смесей можно осуществить стандартными способами, известными в данной области техники (например, колоночной хроматографией, препаративной ТСХ, препаративной ВЭЖХ, с использованием псевдодвижущего слоя и подобными).
[0061] Геометрические по природе изомеры заместителей вокруг двойной связи или кольца могут присутствовать в цис (Z) или транс (E) форме, и обе изомерные формы охватываются объемом настоящего изобретения.
[0062] Альтернативно и/или дополнительно, соединения по настоящему изобретению могут быть включены в виде депо композиции, например, путем диспергирования, растворения или инкапсулирования соединения по настоящему изобретению в полимерноей матрице, описанной во втором и третьем аспекте, так, чтобы соединение непрерывно высвобождалось по мере разложения полимера в течение продолжительного времени. Высвобождение соединений по настоящему изобретению из полимерной матрицы обеспечивает контролируемое- и/или замедленное- и/или длительное высвобождение соединений, например, из фармацевтической депо-композиции, в организм субъекта, например теплокровного животного, такого как человек, которому вводят фармацевтическую депо композицию. Таким образом, фармацевтический депо препарат доставляет соединения по настоящему изобретению субъекту при концентрациях, эффективных для лечения конкретного заболевания или медицинского состояния в течение длительного периода времени, например, 14-180 дней, предпочтительно около 30, около 60 или около 90 дней.
[0063] Полимеры, полезные для полимерной матрицы, в композиции по настоящему изобретению (например, депо-композиции по настоящему изобретению) могут включать полиэфир гидрокси-жирной кислоты и его производные или другие вещества, такие как полимолочная кислота, полигликолевая кислота, полилимонная кислота, полияблочная кислота, поли-бета.-гидроксияблочная кислота, полимер, полученный путем раскрытия эпсилон.-капро-лактонового кольца, сополимер молочной кислоты-гликолевой кислоты, сополимер 2-гидроксияблочной кислоты-гликолевой кислоты, сополимер полимолочной кислоты-полиэтиленгликоля или сополимер полигликолевой кислоты-полиэтиленгликоля), полимер алкилальфа-цианоакрилата (например поли(бутил 2-цианоакрилат)), полиалкиленоксалат (например политриметиленоксалат или политетраметиленоксалат), полиортоэфир, поликарбонат (например полиэтиленкарбонат или полиэтиленпропиленкарбонат), полиортокарбонат, полиаминокислота (например поли-гамма.-L-аланин, поли-.гамма.-бензил-L-глютаминовая кислота или поли-γ-метил-L-глютаминовая кислота), эфир гиалуроновой кислоты и подобные, и можно использовать один или несколько из этих полимеров.
[0064] Если полимеры представляют собой сополимеры, они могут представлять собой любой из статистических, блок- и/или графт-сополимеров. Когда указанные выше альфа-гидроксикарбоновые кислоты, гидроксидикарбоновые кислоты и гидрокситрикарбоновые кислоты обладают оптической активностью в их молекулах, можно использовать любой из D-изомеров, L-изомеров и/или DL-изомеров. Из них можно использовать полимер альфа-гидроксикарбоновой кислоты (предпочтительно полимер молочной кислоты-гликолевой кислоты), его сложный эфир, эфиры поли-альфа-цианоакриловой кислоты и т.д. и сополимер молочной кислоты-гликолевой кислоты (также называемый поли(лактид-альфа-гликолид) или поли(молочная-ко-гликолевая кислота) и далее указываемый как PLGA) являются предпочтительными. Таким образом, в одном аспекте полимер, полезный для полимерной матрицы, представляет собой PLGA. В контексте настоящего изобретения, термин PLGA включает полимеры молочной кислоты (также указываемые как полилактид, поли(молочная кислота) или PLA). Наиболее предпочтительно, полимер представляет собой биоразлагаемый полимер поли(d,l-лактид-ко-гликолид).
[0065] В предпочтительном варианте осуществления, полимерная матрица по настоящему изобретению представляет собой биосовместимое и биоразлагаемое полимерное вещество. Термин ʺбиосовместимыйʺ определяется как полимерное вещество, которое не является токсичным, не является карциногенным и не вызывает существенного воспаления в тканях организма. Вещество матрицы должно быть биоразлагаемым, при этом полимерное вещество должно разлагаться под воздействием процессов в организме на продукты, легко используемые организмом, и не должно накапливаться в организме. Продукты биоразложения также должны быть биосовместимыми с организмом в том смысле, что полимерная матрица является биосовместимой с организмом. Конкретные полезные примеры веществ полимерной матрицы включают поли(гликолевую кислоту), поли-D,L-молочную кислоту, поли-L-молочную кислоту, сополимеры вышеуказанных кислот, поли(алифатические карбоновые кислоты), сополиоксалаты, поликапролактон, полидиоксонон, поли(ортокарбонаты), поли(ацетали), поли(молочная кислота-капролактон), полиортоэфиры, поли(гликолевая кислота-капролактон), полиангидриды и природные полимеры, включая альбумин, казеин и воски, такие как, глицерин моно- и дистеарат и подобные. Предпочтительный полимер для использования при практическом осуществлении настоящего изобретения представляет собой dl-(полилактид-ко-гликолид). Предпочтительно, чтобы молярное отношение лактида к гликолиду в таком сополимере было в пределах от около 75:25 до 50:50.
[0066] Полезные PLGA полимеры могут иметь среднемассовую молекулярную массу от около 5000 до 500000 дальтон, предпочтительно около 150000 дальтон. В зависимости от скорости разложения, которая должна достигаться, можно использовать разную молекулярную массу полимеров. Для диффузионного механизма высвобождения лекарственного средства полимер должен оставаться в неизмененном виде до тех пор, пока все лекарственное средство не высвободится из полимерной матрицы, и затем разрушаться. Лекарственное средство также может высвобождаться из полимерной матрицы по мере биоразложения полимерного эксципиента.
[0067] PLGA можно получить любым традиционным способом, или может быть коммерчески доступным продуктом. Например, PLGA можно получить путем полимеризации с раскрытием кольца с использованием подходящего катализатора из циклического лактида, гликолида и т.д. (см. EP-0058481B2; Effects of polymerization variables on PLGA properties: molecular weight, composition and chain structure).
[0068] Считают, что PLGA является биоразлагаемым посредством разложения всей твердой полимерной композиции в результате разрыва гидролизуемых и ферментативно расщепляемых сложноэфирных связей в биологических условиях (например, в присутствии воды и биологических ферментов, присутствующих в тканях теплокровных животных, таких как человек) с образованием молочной кислоты и гликолевой кислоты. Как молочная кислота, так и гликолевая кислота являются водорастворимыми, нетоксичными продуктами нормального метаболизма, которые далее могут подвергаться биоразложению с образованием диоксид углерода и воды. Иными словами, считают, что PLGA разлагается посредством гидролиза сложноэфирных групп в присутствии воды, например в организме теплокровного животного, такого как человек, с образованием молочной кислоты и гликолевой кислоты и созданием кислотного микроклимата. Молочная и гликолевая кислота являются побочными продуктами различных метаболических путей в организме теплокровного животного, такого как человек, в нормальных физиологических условиях и поэтому поэтому являются хорошо переносимыми и приводят к минимальной системной токсичности.
[0069] В другом варианте осуществления полимерная матрица, полезная для настоящем изобретении, может включать звездчатый полимер, где структура полиэфира имеет звездчатую форму. Эти полиэфиры содержат один полиольный остаток в виде центральной группы, окруженной цепями кислотных остатков. Полиольная группа может представлять собой, например, глюкозу или, например, маннит. Эти эфиры известны и описаны в GB 2145422 и в Патент США № 5538739, содержание которых включено посредством ссылки.
[0070] Звездчатые полимеры можно получить с использованием полигидрокси соединений, например, полиола, например, глюкозы или маннита, в качестве инициатора. Полиол содержит по меньшей мере 3 гидрокси группы и имеет молекулярную массу до около 20000 Дальтон, при этом по меньшей мере 1, предпочтительно по меньшей мере 2, например, в среднем 3 из гидрокси групп полиола находятся в форме сложноэфирных групп, которые содержат полилактидные или ко-полилактидные цепи. Разветвленные полиэфиры, например, поли (d,l-лактид-ко-гликолид), содержат центральную глюкозную группу, имеющую лучи линейных полилактидных цепей.
[0071] Депо-композиция по настоящему изобретению, описанная выше, может включать полимер в форме микрочастиц или наночастиц, или в жидкой форме, где соединения по настоящему изобретению диспергированы или инкапсулированы в нем. ʺМикрочастицыʺ означают твердые частицы, которые содержат соединения по настоящему изобретению либо в растворе, либо в твердой форме, где такое соединение диспергировано или растворено в полимере, который служит в качестве матрицы частиц. Путем правильного выбора полимерных веществ можно получить композицию в форме микрочастиц, где полученные микрочастицы демонстрируют как свойства диффузионного высвобождения, так и высвобождения в результате биоразложения.
[0072] В конкретном варианте осуществления настоящего изобретения, соединение по настоящему изобретению формулируют в виде микрочастиц подходящего размера для обеспечения медленной кинетики высвобождения после внутримышечной инъекции.
[0073] Когда полимер имеет форму микрочастиц, такие микрочастицы можно получить с использованием любого подходящего способа, такого как способ выпаривания растворителя или способ экстракции растворителем. Например, в способе выпаривания растворителя соединения по настоящему изобретению и полимер могут быть растворены в летучем органическом растворителе (например, кетоне, таком как ацетон, галогенированном углеводороде, таком как хлороформ или метиленхлорид, галогенированном ароматическом углеводороде, циклическом эфире, таком как диоксан, сложном эфире, таком как этилацетат, нитриле, таком как ацетонитрил, или спирте, таком как этанол) и диспергированы в водной фазе, включающей подходящий стабилизатор эмульсии (например, поливиниловый спирт, PVA). Органический растворитель затем выпаривают с получением микрочастиц с соединением по настоящему изобретению, инкапсулированным в них. В способе экстракции растворителем соединения по настоящему изобретению и полимер можно растворить в полярном растворителе (таком как ацетонитрил, дихлорметан, метанол, этилацетат или метилформиат) и затем диспергировать в водной фазе (такой как раствор вода/PVA). Образуется эмульсия с получением микрочастиц с соединением по настоящему изобретению, инкапсулированным в них. Распылительная сушка представляет собой альтернативную технологию изготовления для получения микрочастиц.
[0074] Другой способ для получения микрочастиц по настоящему изобретению также описан в патенте США № 4389330 и патенте США № 4530840, содержание которых включено посредством ссылки.
[0075] Микрочастицы по настоящему изобретению можно получить любым способом, способным обеспечивать микрочастицы с размерами в пределах, приемлемых для использования в композициях для инъекций. Один предпочтительный способ получения описан в патенте США № 4389330. В этом способе активное вещество растворяют или диспергируют в подходящем растворителе. В среду, содержащую это вещество, добавляют полимерную матрицу в количестве по отношению к активному ингредиенту, которое обеспечивает продукт, имеющий желаемую нагрузку активного вещества. Необязательно, все ингредиенты продукта в форме микрочастиц могут быть смешаны вместе в растворяющей среде.
[0076] Растворители для соединений по настоящему изобретению и полимерной матрицы, которые можно использовать при практическом осуществлении настоящего изобретения, включают органические растворители, такие как ацетон; галогенированные углеводороды, такие как хлороформ, метиленхлорид и подобные; ароматические углеводородные соединения; галогенированные ароматические углеводородные соединения; циклические эфиры; спирты, такие как, бензиловый спирт; этилацетат; и подобные. В одном варианте осуществления растворитель для использования при практическом осуществлении настоящего изобретения может представлять собой смесь бензилового спирта и этилацетата. Дополнительную информацию, касающуюся получения микрочастиц, полезных для настоящего изобретения, можно найти в публикации патента США № 2008/0069885, содержание которой включен в настоящую заявку посредством ссылки во всей полноте.
[0077] Количество соединений по настоящему изобретению, включенных в микрочастицы, обычно находится в пределах от около 1% масс. до около 90% масс., предпочтительно от 30 до 50% масс., более предпочтительно от 35 до 40% масс. Под % масс. подразумеваются части соединений по настоящему изобретению в расчете на общую массу микрочастицы.
[0078] Фармацевтическое депо может включать фармацевтически приемлемый разбавитель или носитель, такой как смешиваемый с водой разбавитель или носитель.
[0079] Подробное описание композиции системы пероральной доставки с осмотически контролируемым высвобождением можно найти в EP 1 539 115 (Публикация США № 2009/0202631) и WO 2000/35419, содержание каждой из которых включено посредством ссылки во всей их полноте.
[0080] "Терапевтически эффективное количество" представляет собой любое количество соединений по настоящему изобретению (например, содержащееся в фармацевтическом депо), которое при введении субъекту, страдающему заболеванием или расстройством, является эффективным, чтобы вызвать ослабление, ремиссию или регрессию этого заболевания или расстройства за период времени, предусмотренный для лечения.
[0081] Дозы, используемые при практическом осуществлении настоящего изобретения, конечно будут варьироваться в зависимости, например, от конкретного заболевания или состояния, подлежащего лечению, конкретного используемого соединения по настоящему изобретению, способа введения и требуемой терапии.
[0082] Соединения по настоящему изобретению можно вводить любым подходящим путем, в том числе перорально, парентерально (внутривенно, внутримышечно или подкожно) или чрескожно, но предпочтительно их вводят перорально. В некоторых вариантах осуществления соединения по настоящему изобретению, например, в виде депо композиции, предпочтительно вводят парентерально, например, путем инъекции.
[0083] Как правило, удовлетворительные результаты для Способа I или любого из 7.1-7.32 или Способа IP или применения соединений по настоящему изобретению, описанного выше, например, для лечения комбинации заболеваний, такой как комбинация по меньшей мере депрессии, психоза, например, (1) психоза, например, шизофрении, у пациента, страдающего депрессией; (2) депрессии у пациента, страдающего психозом, например, шизофренией; (3) аффективных расстройств, ассоциированных с психозом, например, шизофренией, или болезнью Паркинсона; и (4) расстройств сна, связанных с психозом, например, шизофренией, или болезнью Паркинсона, как описано выше, как показано, достигаются при пероральном введении при дозах порядка от около l мг до 100 мг один раз в день, предпочтительно около 2,5 мг-50 мг, например, 2,5 мг, 5 мг, l0 мг, 20 мг, 30 мг, 40 мг или 50 мг, один раз в день, предпочтительно путем перорального введения.
[0084] Удовлетворительные результаты для Способа II или любого из 8.1-8.15, Способа II или применения соединений по настоящему изобретению, описанного выше, например, для лечения только расстройства сна или только ажитации, агрессивного поведения, посттравматического стрессового расстройства или расстройства импульсного контроля, например, только интермиттирующего эксплозивного расстройства, как показано, достигаются при пероральном введении при дозах порядка от около 1 мг до 10 мг один раз в день, например, около 2,5 мг-5 мг, например, 2,5 мг, 3 мг, 4 мг, 5 мг или 10 мг, соединения по настоящему изобретению, в свободной форме или форме фармацевтически приемлемой соли, один раз в день, предпочтительно путем перорального введения.
[0085] Удовлетворительные результаты для Способа I-A или любого из 9.1-9.38 или Способа IP-A, как показано, достигаются при дозе меньше чем 100мг, предпочтительно меньше чем 50 мг, например, меньше чем 40 мг, меньше чем 30 мг, меньше чем 20 мг, меньше чем 10 мг, меньше чем 5 мг, меньше чем 2,5 мг, один раз в день. Удовлетворительные результаты для Способа II-A или любого из 9.1-9.38, как показано, достигаются при дозе меньше чем 10 мг, например, меньше чем 5 мг, или предпочтительно меньше чем 2,5 мг.
[0086] Для лечения расстройств, раскрытых в настоящей заявке, где депо-композицию используют для достижения большей продолжительности действия, дозы должны быть выше по сравнению с композицией с более коротким периодом действия, например, выше чем 1-100 мг, например, 25 мг, 50 мг, 100 мг, 500 мг, 1000 мг, или выше чем 1000 мг. В конкретном варианте осуществления настоящего изобретения, схема введения для депо-композиции включает начальную пероральную дозу с мгновенным высвобождением вместе с депо высвобождением, чтобы обеспечить устойчивую концентрацию лекарственного средства в крови. Продолжительность действия соединений по настоящему изобретению можно контролировать, манипулируя с полимерной композицией, то есть, соотношением полимер:лекарственное средство и размером микрочастиц. Когда композиция по настоящему изобретению представляет собой депо-композицию, введение путем инъекции является предпочтительным.
[0087] Фармацевтически приемлемые соли соединений по настоящему изобретению можно синтезировать из исходного соединения, которое содержит щелочную или кислотную группу, традиционными химическими способами. Как правило, такие соли можно получить путем взаимодействия форм свободного основания этих соединений с стехиометрическим количеством подходящей кислоты в воде или в органическом растворителе, или в смеси таких двух растворителей; как правило, не-водные среды, такие как простой эфир, этилацетат, этанол, изопропанол или ацетонитрил, являются предпочтительными. Дополнительную информацию, касающуюся получения этих солей, например, соли толуолсульфоновой кислоты, в аморфной или кристаллической форме, можно найти в PCT/US08/03340 и/или Предварительной заявке США № 61/036069.
[0088] Фармацевтические композиции, включающие соединения по настоящему изобретению, можно получить с использованием обычных разбавителей или эксципиентов (например, включающих, но не ограничивающихся этим, кунжутное масло) и методов, известных в области получения галеновых препаратов. Таким образом, пероральные лекарственные формы могут включать таблетки, капсулы, растворы, суспензии и т.п.
[0089] Все имеющиеся в настоящей заявке ссылки на дозировку, размер доз или терапевтически эффективное количество соединения или композиции по настоящему изобретению указаны в расчете на свободное основание или соединение в форме фармацевтически приемлемой соли в дозе.
[0090] In-vitro метаболизм соединения формулы Q и его метаболиты исследуют с использованием субклеточных фракций и выделенных гепатоцитов. Результаты показывают, что соединение формулы Q подвергается N-деметилированию до соединения формулы R через изоформу 3A4 P450 цитохромоксидазы (CYP 3A4), и что оба соединение формулы Q и соединение формулы R подвергаются восстановлению кетона через фермент кетон-редуктаза с образованием соединения формулы S и T, соответственно. Эти два восстановления оба катализируются в обратном направлении (окисление) посредством CYP 3A4. Эти результаты обобщенно представлены на схеме ниже:

[0091] Кроме того, in-vivo метаболизм соединения формулы Q исследуют после перорального введения крысам, собакам и человеку. Уровни в плазме после введения соединений формулы Q - T определяют для всех трех видов. Результаты исследований показывают, что метаболизм соединения формулы Q быстрый, и что N-деметил соединения являются высокополярными и быстро выводятся из организма. Результаты исследований плазмы человека в день 8 после 7-дневного введения (120 мг, 4 дозы/день) соединения формулы Q показаны ниже:
Способы получения соединений по настоящему изобретению:
[0092] Промежуточные соединения соединений по настоящему изобретению можно получить, как описано в WO PCT/US08/03340 (WO 2008/112280); Заявке на патент США серийный номер 10/786935; патенте США № 6548493; 7238690; 6552017; 6713471; 7183282; U.S. RE39680 и U.S. RE39679, содержание которых включено в настоящую заявку посредством ссылки во всей полноте. Соли соединений по настоящему изобретению также можно получить аналогично тому, как описано в патенте США № 6548493; 7238690; 6552017; 6713471; 7183282; U.S. RE39680; U.S. RE39679; и WO 2009/114181, содержание каждого из которых включено в настоящую заявку посредством ссылки во всей полноте.
[0093] Выделение или очистку диастереомеров соединений по настоящему изобретению можно осуществить обычными способами, известными в данной области техники, например, колоночной очисткой, препаративной тонкослойной хроматографией, препаративной ВЭЖХ, кристаллизацией, растиранием в порошок, методами псевдоподвижных слоев и т.п.
[0094] Соединения формулы I можно получить стандартными способами, известными специалистам в данной области техники. Патент США № 8309722, который включен посредством ссылки во всей его полноте, раскрывает синтез соединения формулы Q и всех промежуточных соединений для его получения:
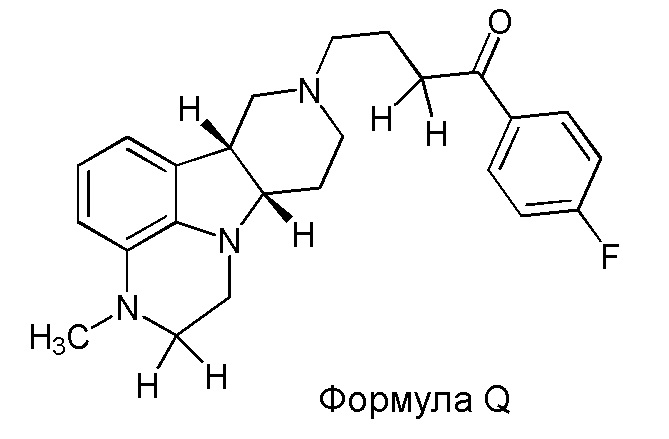
[0095] Соединения по настоящему изобретению синтезируют такими же способами, которые раскрыты для синтеза соединения формулы Q. Примеры этих способов синтеза представлены ниже.
[0096] Например, Соединение B можно получить из Соединения A (раскрыто в Патенте США № 8309722) путем взаимодействия с d3-иодметаном в присутствии основания, такого как карбонат калия, в подходящем растворителе, таком как ацетон. Следуя процедурам Патента США № 8309722, Соединение B затем можно преобразовать в Соединение формулы I, где R1 представляет собой СD3. Эту реакцию можно обобщенно представить на схеме реакций ниже:

[0097] Например, Соединение D можно получить из Соединения формулы C (раскрыто в Патенте США № 8309722 и в настоящей заявке) путем взаимодействия с комплексом d3-боран-THF в подходящем растворителе, таком как тетрагидрофуран. Следуя процедурам Патента США № 8309722, Соединение D затем можно преобразовать в Соединение формулы I, где R2 и R3 представляют собой D. Эту реакцию можно обобщенно представить на схеме реакций ниже:

[0098] Например, соединение формулы I, где R4 и R5 представляют собой D, можно получить из Соединения формулы F (раскрыто в Патенте США № 8309722 и в настоящей заявке) путем взаимодействия с Соединением H, определенным ниже, в присутствии иодида калия и основания, такого как карбонат калия и триэтиламин, в подходящем растворителе, таком как 3-пентанон. Следуя процедурам Патента США № 8309722, продукт может быть выделен и очищен. Эту реакцию можно обобщенно представить на схеме реакций ниже:

[0099] Соединение формулы H можно получить по существу в соответствии с процедурой, описанной J. R. Cabrero-Antonino (Chemistry: A European Journal, Vol 18, No. 35, p.11107-11114, 27 August 2012). Хлорид железа (III) и бисаминотрифлат серебра в диоксане перемешивают при комнатной температуре в течение 30 минут и затем добавляют 1-(4-хлорбут-1-ин-1-ил)-4-фторбензол и оксид дейтерия. Смесь нагревают при 80°C в течение 18 часов с получением Соединения H. Эту реакцию можно обобщенно представить на схеме реакций ниже:

[00100] Соединение формулы II, где R4 и R5 представляют собой H, и R6-R9 представляют собой D, можно получить из Соединения формулы F (раскрыто в Патенте США № 8309722 и в настоящей заявке) путем взаимодействия с Соединением J, определенным ниже, в присутствии иодида калия и основания, такого как карбонат калия и триэтиламин, в подходящем растворителе, таком как 3-пентанон. Следуя процедурам Патента США № 8309722, продукт может быть выделен и очищен. Эту реакцию можно обобщенно представить на схеме реакций ниже:

[00101] Соединение формулы J можно получить путем взаимодействия d5-фторбензола с 4-хлорбутаноилхлоридом в присутствии хлорида алюминия (III) в подходящем растворителе, таком как тетрахлорид углерода. Эту реакцию можно обобщенно представить на схеме реакций ниже:
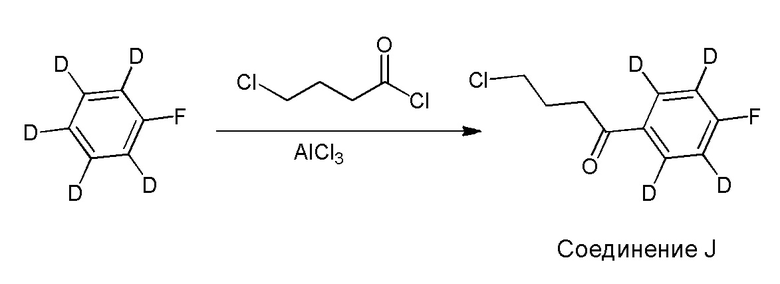
| название | год | авторы | номер документа |
|---|---|---|---|
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2015 |
|
RU2728787C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2017 |
|
RU2733975C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2017 |
|
RU2743513C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ РАССТРОЙСТВА ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | 2017 |
|
RU2776800C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2011 |
|
RU2591194C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2777366C2 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2780002C2 |
| НОВЫЕ СПОСОБЫ | 2014 |
|
RU2682658C1 |
| СЕЛЕКТИВНЫЕ ОБРАТНЫЕ АГОНИСТЫ СЕРОТОНИНОВЫХ РЕЦЕПТОРОВ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ПРИ ЗАБОЛЕВАНИИ | 2005 |
|
RU2442607C2 |
| ЛИГАНД С ШИРОКИМ СПЕКТРОМ ОДНОВРЕМЕННОЙ РЕЦЕПТОРНОЙ АКТИВНОСТИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2008 |
|
RU2374245C1 |
Изобретение относится к фармацевтической композиции для лечения заболевания с вовлечением одного из путей серотонина 5-HT2A, дофамина D2 и/или транспортера обратного захвата серотонина (SERT), включающей соединение формулы I:

где: R1 представляет собой СH3 или CD3; R2 и R3 каждый независимо представляют собой H или D; R4 и R5 каждый независимо представляют собой H или D; при условии, что R2, R3, R4 и R5 не все представляют собой H, когда R1 представляет собой СH3, и где D представляет собой дейтерий, в свободной форме или форме фармацевтически приемлемой соли, в комбинации или ассоциации с фармацевтически приемлемым растворителем или носителем, где композиция содержит терапевтически эффективное количество соединения формулы I от 1 до 100 мг, а также к способу лечения или профилактики расстройства центральной нервной системы с вовлечением 5-HT2A рецептора, транспортера серотонина (SERT) и/или путей, включающих сигнальные системы D1/D2 дофаминовых рецепторов, включающему введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I. Технический результат: изобретение обеспечивает соединения, которые частично ограничивают метаболизм кетона и/или N-метильного заместителя, путем введения атомов дейтерия в различных положениях. 2 н. и 7 з.п. ф-лы, 1 табл.
1. Фармацевтическая композиция для лечения заболевания с вовлечением одного из путей серотонина 5-HT2A, дофамина D2 и/или транспортера обратного захвата серотонина (SERT), включающая соединение формулы I:

где:
R1 представляет собой СH3 или CD3;
R2 и R3 каждый независимо представляют собой H или D;
R4 и R5 каждый независимо представляют собой H или D;
при условии, что R2, R3, R4 и R5 не все представляют собой H, когда R1 представляет собой СH3,
и где D представляет собой дейтерий,
в свободной форме или форме фармацевтически приемлемой соли, в комбинации или ассоциации с фармацевтически приемлемым растворителем или носителем, где композиция содержит терапевтически эффективное количество соединения формулы I от 1 до 100 мг.
2. Способ лечения или профилактики расстройства центральной нервной системы с вовлечением 5-HT2A рецептора, транспортера серотонина (SERT) и/или путей, включающих сигнальные системы D1/D2 дофаминовых рецепторов, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I:
где:
R1 представляет собой СH3 или CD3;
R2 и R3 каждый независимо представляют собой H или D;
R4 и R5 каждый независимо представляют собой H или D;
при условии, что R2, R3, R4 и R5 не все представляют собой H, когда R1 представляет собой СH3,
и где D представляет собой дейтерий,
в свободной форме или форме фармацевтически приемлемой соли, или фармацевтической композиции по п. 1;
где указанное расстройство выбрано из группы, состоящей из тревожного расстройства, депрессии, психоза, шизофрении, ажитации, ажитации при деменции, посттравматического стрессового расстройства, расстройств импульсного контроля, интермиттирующего эксплозивного расстройства, одного или нескольких расстройств, связанных с деменцией, и остаточных симптомов психоза.
3. Способ по п. 2, в котором указанное расстройство выбрано из группы, состоящей из тревожного расстройства, депрессии (например, рефрактерной депрессии и (MDD)), психоза, шизофрении, ажитации, ажитации при деменции (например, ажитации при болезни Альцгеймера), посттравматического стрессового расстройства, расстройств импульсного контроля, интермиттирующего эксплозивного расстройства.
4. Способ по п. 2, в котором указанное расстройство представляет собой одно или несколько расстройств, ассоциированных с деменцией, например, расстройства, ассоциированные с умеренными когнитивными нарушениями и дементным состоянием, включая сенильную деменцию, болезнь Альцгеймера, болезнь Пика, фронто-темпоральную деменцию, супрануклеарный паралич, деменцию с тельцами Леви, сосудистую деменцию, болезнь Гентингтона, болезнь Паркинсона, рассеянный склероз, амиотрофический латеральный склероз, синдром Дауна, депрессию пожилых людей, синдром Вернике-Корсакова, кортико-базальные дегенерации и прионное заболевание, аутизм и синдром гиперактивности с дефицитом внимания.
5. Способ по п. 2, в котором указанное расстройство представляет собой шизофрению.
6. Способ по п. 5, в котором лечат остаточные симптомы или негативные симптомы шизофрении.
7. Способ по п. 2, в котором указанное расстройство представляет собой депрессию.
8. Способ по п. 7, отличающийся тем, что депрессия представляет собой биполярную депрессию, рефрактерную депрессию или MDD (большое депрессивное расстройство).
9. Способ по п. 6, в котором указанные остаточные симптомы выбраны из негативных симптомов, таких как притупленный аффект, эмоциональное отчуждение, малоконтактность, пассивное или апатическое социальное отчуждение, затруднение абстрактного мышления, отсутствие спонтанности и плавного течения беседы и стереотипное мышление; общих психопатологических симптомов, таких как обеспокоенность соматическим здоровьем, тревожное расстройство, чувство вины, напряженность, манерность и «застывание» в определенной позе, депрессия, двигательная заторможенность, неконтактность, необычное содержание мысли, дезориентирование, слабое внимание, отсутствие здравого смысла и проницательности, нарушение силы воли, ослабление контроля импульсивности, озабоченность и активное социальное избегание; когнитивного нарушения и расстройств сна (например, бессонницы).
| WO 2013155504 A1, 17.10.2013 | |||
| WO 2009145900 A1, 03.12.2009 | |||
| Peng Li et al., Discovery of a Tetracyclic Quinoxaline Derivative as a Potent and Orally Active Multifunctional Drug Candidate for theTreatment of Neuropsychiatric and Neurological Disorders, J | |||
| Med.Chem., 57, 2670−2682, 2014 | |||
| L | |||
| E | |||
| Dyck, Effects of Deuterium Substitution on the |

