Область техники
Настоящее изобретение относится к новому трициклическому бензоксабороловому производному, способу его получения и применению антибиотиков, включающих его в качестве активного ингредиента, и предпочтительно к применению антибиотиков против грамотрицательных бактерий.
Предшествующий уровень техники
Терапевтические агенты против грамотрицательных бактерий активно разрабатывались в период с 1960-х по 1980-е годы. Однако, начиная с 1990-х грамотрицательные бактерии не изучались, так как возрос интерес к грамположительным бактериям из-за социальных последствий инфекции устойчивым к метициллину Staphylococcus aureus (MRSA). С конца 2000-х растет озабоченность в виду отсутствия терапевтических агентов против грамотрицательных бактерий с множественной лекарственной устойчивостью, и поэтому в последнее время им вновь уделяется огромное внимание.
В настоящее время имеется много лекарственных средств, используемых для лечения грамотрицательных бактериальных инфекций, однако они не эффективны против грамотрицательных бактерий с множественной лекарственной устойчивостью. Из-за растущей потребности в лекарственных средствах, которые эффективны против грамотрицательных бактерий, включая грамотрицательные бактерии с множественной лекарственной устойчивостью, многие фармацевтические кампании уделяют этому внимание, однако немного антибиотиков находится в разработке. Кроме того, число не поддающихся лечению штаммов, устойчивых к известным антибиотикам, увеличивается, приводя к серьезным социальным последствиям. Соответственно, существует необходимость в разработке новых антибиотиков широкого спектра действия.
При синтезе белка в бактериях аминокислота активируется посредством ATP (аденозинтрифосфата) с образованием аминоацил-АМР, который связывается с аминоацил-т-РНК-синтетазой, и эта аминокислота переносится на т-РНК. Таким образом, происходит загрузка т-РНК, и в этот момент фермент аминоацил-т-РНК-синтетаза может быть мишенью антибиотиков.
В 2010 году сообщили о новом механизме OBORT (оксаборол т-РНК захват) для лейцил-т-РНК-синтетазы. Это является новым механизмом, подразумевающим что оксабороловое соединение связывается с редактирующим доменом лейцил-т-РНК-синтетазы и связывается с А76 концом т-РНК через ковалентную связь для т-РНК захвата. Существует селективность вследствие структурного различия между эукариотическими и бактериальными редактирующими доменами. Таким образом, ингибитор лейцил-т-РНК-синтетазы может быть разработан в качестве лекарственного средства, эффективного против грамотрицательных бактерий.
Бензоксабороловое соединение не является продуктом ферментации, а представляет новый синтетический антибиотик, и его производные с различными структурами известны. Борсодержащие соединения, такие как оксаборол, известны в качестве полезных антибиотических веществ в US 2006/0234981 и US 2007/0155699. Кроме того, бензоксабороловые производные описаны в WO 2008/157726, WO 2009/140309, WO 2011/060196, WO 2012/033858 и WO 2013/093615.
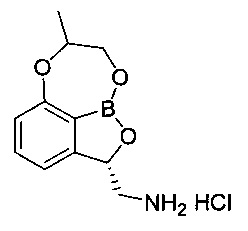
В WO 2008/157726 конкретно упоминается только производное (Соединение A), не имеющее заместителей по положениям 7,8 трициклического бензоксаборола, например, (7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамин. Это трициклическое бензоксабороловое соединение проявляет слабую антибактериальную активность против ряда грамотрицательных бактерий, а также слабую антибактериальную активность против Acinetobacter baumannii, в частности устойчивого к карбапенему Acinetobacter baumannii.
В WO 2013/093615 раскрыты только конкретные примеры синтеза соединения, имеющего заместитель по положению 8, и соединения, замещенного гидроксиметильной группой по положению 7 трициклического бензоксаборола.
Соответственно, существует острая необходимость в новых бензоксабороловых соединениях, которые селективно связываются с грамотрицательными бактериями с проявлением своей функциональной активности и минимизируют побочные эффекты, а также в терапевтическом агенте на их основе для лечения грамотрицательных бактериальных инфекций, в частности, против грамотрицательных бактерий с множественной лекарственной устойчивостью, которые вновь стали серьезной угрозой.
Подробное описание
Техническая задача
Задача настоящего изобретения заключается в том, чтобы предложить новое трициклическое бензоксабороловое соединение.
Еще одна задача настоящего изобретения заключается в том, чтобы предложить способ получения нового трициклического бензоксаборолового соединения.
Еще одна задача настоящего изобретения заключается в том, чтобы предложить антибиотик против грамотрицательных бактерий, в том числе грамотрицательных бактерий с множественной лекарственной устойчивостью, где указанный антибиотик включает новое трициклическое бензоксабороловое соединение в качестве активного ингредиента. Соединение по настоящему изобретению избирательно связывается с грамотрицательными бактериями с проявлением своей функциональной активности, тем самым минимизируя побочные эффекты. Дополнительная задача настоящего изобретения заключается в том, чтобы предложить антибактериальный, стерилизующий или бактерицидный способ в отношении грамотрицательных бактерий с использованием трициклического бензоксаборолового соединения по настоящему изобретению.
Еще одна задача настоящего изобретения заключается в том, чтобы предложить способ предупреждения или лечения инфекций, вызываемых грамотрицательными бактериями, включающий стадию введения субъекту терапевтически эффективного количества трициклического бензоксаборолового соединения по настоящему изобретению.
Еще одна задача настоящего изобретения заключается в том, чтобы предложить антибактериальное, стерилизующее или бактерицидное применение трициклического бензоксаборолового соединения по настоящему изобретению против грамотрицательных бактерий.
Еще одна задача настоящего изобретения заключается в том, чтобы предложить применение трициклического бензоксаборолового соединения по настоящему изобретению в предупреждении или лечении инфекций, вызываемых грамотрицательными бактериями.
Техническое решение
Согласно настоящему изобретению предложено трициклическое бензоксабороловое соединение, представленное химической формулой 1, его изомер или их фармацевтически приемлемая соль.
[Химическая формула 1]

В другом аспекте настоящего изобретения предложена антибиотическая фармацевтическая композиция против грамотрицательных бактерий, содержащая соединение химической формулы 1, его изомер или их фармацевтически приемлемую соль в качестве активного ингредиента. Предпочтительно, грамотрицательные бактерии могут представлять собой Acinetobacter baumannii, Citrobacter freundii, Escherichia coli, Enterobacter cloacae, Enterobacter aerogenes, Klebsiella pneumoniae, Klebsiella oxytoca, Morganella morganii, Pseudomonas aeruginosa, Proteus vulgaris, Proteus mirabilis, Neisseria gonorrhoeae или Serratia marcescens. Грамотрицательные бактерии могут представлять собой грамотрицательные бактерии с устойчивостью к карбапенему.
В настоящем изобретении авторы изучили бензоксабороловые соединения, проявляющие терапевтические эффекты в отношении бактериальных инфекций, и получили соединение, обладающее антибактериальным эффектом in vitro, эквивалентным или выше чем у известных веществ, и проявляющее превосходный антибактериальный эффект против грамотрицательных бактерий in vivo, а также обнаружили, что такое соединение может быть более эффективно использовано в качестве терапевтического агента против грамотрицательных бактериальных инфекций, тем самым создав настоящее изобретение.
Более конкретно, по сравнению с известным гидрохлоридом (8-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина, соединение по настоящему изобретению, как было обнаружено, проявляет сильную антибактериальную активность против множества грамотрицательных бактерий, в частности, устойчивого к карбапенему Acinetobacter baumannii, in vitro, и проявляет превосходные терапевтические эффекты в отношении бактериальных инфекций в модели тестирования эффективности in vivo. Кроме того, по сравнению с известным гидрохлоридом ((2S,8R)-2-(аминометил)-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-8-ил)метанола, соединение по настоящему изобретению, как обнаружено, проявляет сильную in vitro антибактериальную активность против большинства патогенных бактерий, включая Acinetobacter baumannii, и проявляет превосходные терапевтические эффекты в отношении бактериальных инфекций в модели тестирования эффективности in vivo.
Таким образом, согласно настоящему изобретению предложено новое бензоксабороловое соединение, которое селективно связывается с грамотрицательными бактериями с проявлением своей функциональной активности и минимизирует побочные эффекты, а также антибиотик и/или терапевтический агент на его основе для лечения инфекций, ассоциированных с грамотрицательными бактериями, включая Acinetobacter baumannii.
Используемые в данном описании термины кратко поясняются ниже.
Термин "фармацевтически приемлемая соль", как он использован здесь, относится к соли соединения, которая не вызывает значительного раздражения в организме, в который вводят соединение, и не нейтрализует биологическую активность и свойства этого соединения, и в настоящем изобретении он в собирательном значении относится к любой соли, которая сохраняет биологические эффективности и свойства соединения химической формулы 1 и является предпочтительной в контексте фармацевтических, биологических или других свойств. Фармацевтически приемлемая соль может включать соли присоединения кислот, образованные кислотами, способными образовывать нетоксичную соль присоединения кислоты, содержащую фармацевтически приемлемые анионы, например, неорганическими кислотами, такими как соляная кислота, серная кислота, азотная кислота, фосфорная кислота, бромоводородная кислота, йодоводородная кислота, и так далее; органическими карбоновыми кислотами, такими как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, миндальная кислота, фумаровая кислота, малеиновая кислота, салициловая кислота, и так далее; или сульфоновыми кислотами, такими как метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, пара-толуолсульфоновая кислота, и так далее. В качестве ее примера, соль присоединения кислоты соединения по одному воплощению может быть получена путем взаимодействия форм свободного основания этих соединений со стехиометрическим количеством подходящей кислоты.
В настоящее время это взаимодействие может быть осуществлено в воде, органическом растворителе или их смеси, и более конкретно, в неводной среде, такой как эфир, этилацетат, этанол, изопропанол, ацетонитрил, и так далее. Кроме того, в соответствии с формой фармацевтически приемлемой соли, каждая форма солей может быть получена типичной реакцией, которая очевидна специалистам в данной области техники. Кроме того, фармацевтически приемлемая соль может представлять собой соли щелочных металлов или соли щелочноземельных металлов, образуемых с литием, натрием, калием, кальцием, магнием, и так далее, соли с аминокислотами, такими как лизин, аргинин, гуанидин, и так далее, с органическими кислотами, такими как дициклогексиламин, N-метил-D-глюкамин, трис-(гидроксиметил)метиламин, диэтаноламин, холин, триэтиламин, и так далее.
Термин "изомер", как он использован здесь, означает соединение или его соль с такой же химической формулой или молекулярной формулой, но различающиеся оптически или геометрически. Изомер, его соль и их смесь (рацемическая смесь) также включены в объем настоящего изобретения.
Трициклическое бензоксабороловое соединение по настоящему изобретению может представлять собой рацемат соединения, его энантиомер, его диастереомер, смесь энантиомеров или смесь диастереомеров.
Более конкретно, соединение, представленное химической формулой 1, может иметь асимметрический углеродный центр. Когда соединение имеет асимметрический углеродный центр, оно может существовать в виде его энантиомера, его диастереомера или его рацемата. Все типы изомеров, включая их, могут быть также включены в объем соединения в соответствии с одним воплощением настоящего изобретения.
Соединение в соответствии с химической формулой 1 согласно настоящему изобретению или его фармацевтически приемлемая соль могут проявлять полиморфизм и существовать в форме сольвата (например гидрата, и так далее). Кроме того, индивидуальные соединения включают их стереоизомеры или их смеси.
Термин "фармацевтически эффективное количество", как он использован здесь, означает количество активного ингредиента, которое достаточно для получения желаемого фармацевтического эффекта, и в соответствии с обстоятельствами он означает концентрацию или дозу введения активного ингредиента в фармацевтической композиции, которая достаточна для желаемого фармацевтического эффекта.
Далее настоящее изобретение будет описано более подробно.
В одном воплощении настоящего изобретения предложено трициклическое бензоксабороловое соединение, представленное следующей химической формулой 1, его изомер или их фармацевтически приемлемая соль:
[Химическая формула 1]
Как используют здесь, изомер означает соединение или его соль, которые имеют одну и ту же химическую формулу или молекулярную формулу, но отличаются оптически или геометрически. Изомер, его соль и их смесь (рацемическая смесь) также включены в объем настоящего изобретения.
Более конкретно, изомер по настоящему изобретению или трициклическое бензоксабороловое соединение может представлять собой рацемат соединения, его энантиомер, его диастереомер, смесь энатиомеров или смесь диастереомеров.
В одном воплощении настоящего изобретения изомер может представлять собой энантиомер соединения химической формулы 1, стереоизомер или смесь изомеров (рацемическую смесь). В качестве энантиомера любой асимметрический атом углерода в соединении может существовать в любой форме (R)-, (S)- и (R,S)-конфигураций, и предпочтительно, в отдельной форме (R)- или (S)-конфигурации.
По меньшей мере один асимметрический атом углерода, выбранный из группы, состоящей из углеродов по положениям 2 и 7 трициклического бензоксаборолового кольца, может быть энантиомером, например (2S) изомером, (2R) изомером, (7S) изомером, (7R) изомером, (2S,7S) изомером, (2S,7R) изомером, (2R,7S) изомером или (2R,7R) изомером, но не ограничен ими.
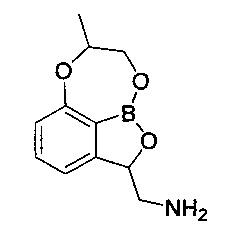
В одном конкретном воплощении настоящего изобретения изомер по настоящему изобретению может представлять собой (2S) изомер, представленный следующей химической формулой 2.
[Химическая формула 2]

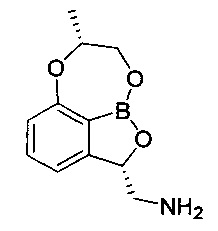
В другом конкретном воплощении настоящего изобретения изомер по настоящему изобретению может представлять собой (2S,7R) изомер, представленный следующей химической формулой 3.
[Химическая формула 3]
В качестве примера, согласно настоящему изобретению, соединение химической формулы 1 или его изомер могут быть выбраны из группы, состоящей из следующих соединений:
1) гидрохлорид (7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина;
2) гидрохлорид ((2S)-7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина;
3) гидрохлорид ((2S,7R)-7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина;
4) ((2S,7R)-7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамин; и
5) гидрохлорид ((2S,7S)-7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина.
В одном воплощении настоящего изобретения трициклическое бензоксабороловое соединение по настоящему изобретению, представленное химической формулой 1, может быть использовано в форме фармацевтически приемлемой соли. В качестве такой соли полезна соль присоединения кислоты, образуемая фармацевтически приемлемой кислотой. Свободная кислота может представлять собой неорганическую кислоту, такую как соляная кислота, серная кислота, азотная кислота, фосфорная кислота, бромоводородная кислота, йодоводородная кислота, и так далее; органическую карбоновую кислоту, такую как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, миндальная кислота, фумаровая кислота, малеиновая кислота, салициловая кислота, и так далее; или сульфоновую кислоту, такую как метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота и пара-толуолсульфоновая кислота, но не ограничена ими. Например, трициклическое бензоксабороловое соединение по настоящему изобретению может представлять собой соль присоединения фармацевтически приемлемой кислоты гидрохлорид.
Кроме того, трициклическое бензоксабороловое соединение по настоящему изобретению, представленное химической формулой 1, может включать все соли, гидраты и сольваты, которые могут быть получены традиционными способами, а также фармацевтически приемлемую соль.
Фармацевтически приемлемые соли согласно настоящему изобретению могут быть получены традиционным способом, например, путем растворения соединения химической формулы 1 в не смешивающемся с водой органическом растворителе, например ацетоне, метаноле, этаноле, ацетонитриле и других, добавления к нему избыточного количества органической кислоты или путем добавления к нему кислого водного раствора неорганической кислоты и затем осаждения или кристаллизации. Затем, получение может быть осуществлено путем выпаривания растворителя или избыточного количества кислоты из этой смеси и затем сушки с получением соли присоединения или вакуум-фильтрации осажденной соли.
Кроме того, в настоящем изобретении предложен способ получения трициклического бензоксаборолового соединения химической формулы 1. Трициклическое оксабороловое производное по настоящему изобретению может быть получено разными способами в соответствии с типом стереоизомера и способом, как проиллюстрировано ниже. Очевидно, что следующий способ получения приведен только для иллюстративных целей, и он может быть легко модифицирован специалистами в данной области техники в соответствии с целевым соединением. Таким образом, способ получения трициклического бензоксаборолового соединения по настоящему изобретению не предназначен ограничиваться следующим способом.
Способ получения соединения химической формулы 1 согласно настоящему изобретению может включать стадии:
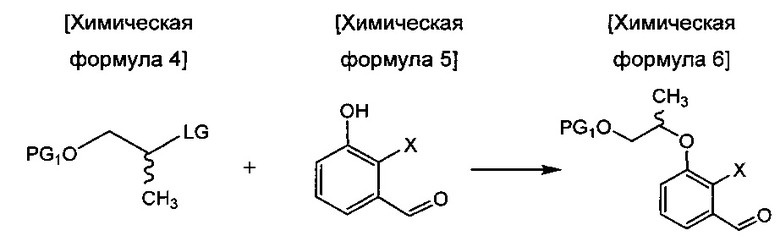
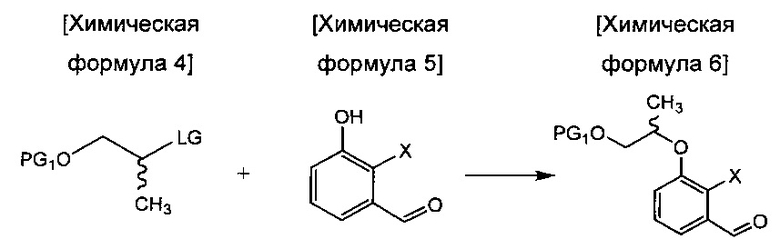
осуществления сочетания соединения химической формулы 4 и соединения химической формулы 5 с получением соединения химической формулы 6;
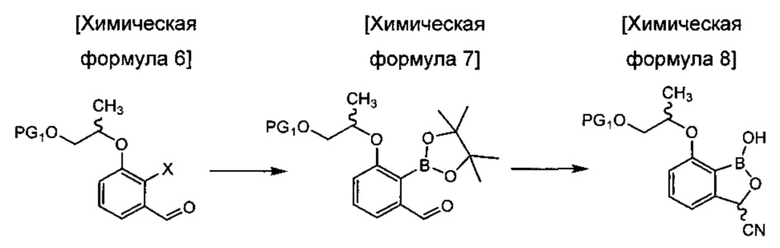
осуществления реакции борилирования соединения химической формулы 6 с получением соединения химической формулы 7 и затем осуществления цианирования с получением цианобензоксаборолового соединения химической формулы 8;
восстановления соединения химической формулы 8 с получением аминобензоксаборолового соединения химической формулы 9 путем замещения цианогруппы аминогруппой;

введения защитной группы (PG2) в аминогруппу соединения химической формулы 9 с получением соединения химической формулы 10, осуществления реакции конденсации соединения химической формулы 10 для удаления защитной группы (PG1) и затем осуществления реакции циклизации с получением трициклического бензоксаборолового соединения химической формулы 11; и
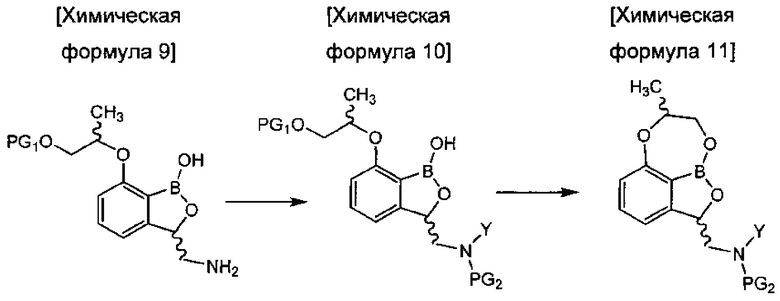
снятия защиты (PG2) с аминогруппы соединения химической формулы 11 с получением соединения химической формулы 1.
Кроме того, способ получения соединения химической формулы 1 согласно настоящему изобретению может включать стадии:
осуществления сочетания соединения химической формулы 4 и соединения химической формулы 5 с получением соединения химической формулы 6;
осуществления реакции нитрования соединения химической формулы 6 или использования хирального лиганда или хирального катализатора с получением соединения химической формулы 12 или его изомеров, восстановления соединения химической формулы 12 с получением соединения химической формулы 13 путем замещения нитрогруппы аминогруппой;
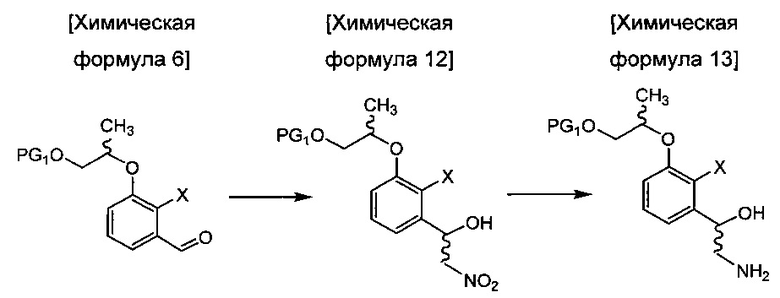
введения защитной группы (PG2) в аминогруппу соединения химической формулы 13 с получением соединения химической формулы 14 и осуществления реакции борилирования соединения химической формулы 14 с получением бензоксаборолового соединения химической формулы 10; и
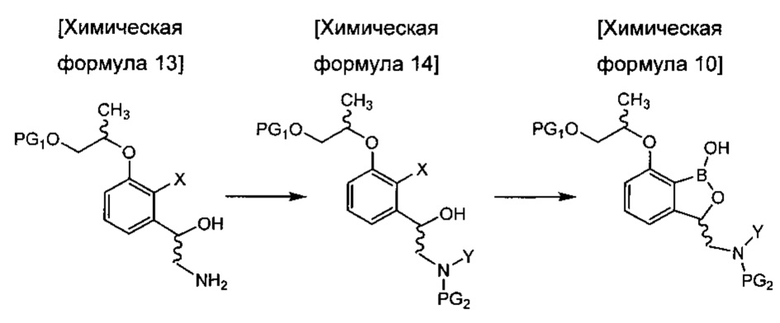
осуществления реакции внутренней циклизации соединения химической формулы 10 для удаления защитной группы (PG1) и затем осуществления реакции циклизации с получением трициклического бензоксаборолового соединения химической формулы 11; и
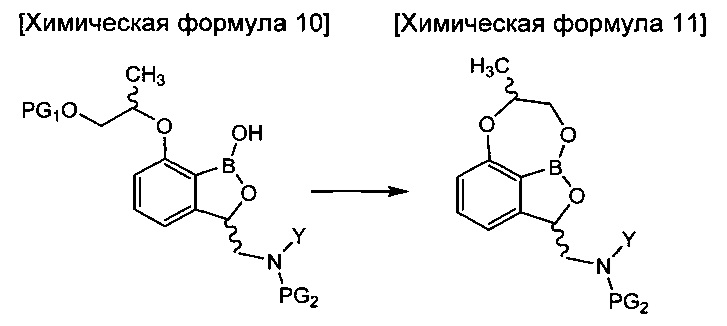
снятия защиты с аминогруппы соединения химической формулы 11 с получением соединения химической формулы 1.
в Химических формулах 4-14,
PG1 и PG2 представляют собой защитные группы для защиты активных групп и, каждая независимо, представляют собой бензил, трет-бутил, Boc (трет-бутилоксикарбонил), pmb (4-метоксибензил), Fmoc (флуоренилметилоксикарбонил), Ts (тозилат), MOM (метоксиметил), ТНР (тетрагидропиранил), TBDMS (трет-бутилдиметилсилил) или TBDPS (трет-бутилдиметилсилил),
LG представляет собой уходящую группу, которая удаляется во время реакции конденсации, и представляет собой галоген, пара-толуолсульфонильную группу или метансульфонильную группу,
X представляет собой водород, галоген или трифторметансульфонил, и
Y представляет собой водород или PG2.
В конкретном воплощении настоящего изобретения реакция борилирования может быть осуществлена с использованием бис(пинаколато)дибора или 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолана, но не ограничена ими.
В другом воплощении настоящего изобретения предложена фармацевтическая композиция, в частности фармацевтическая композиция антибактериального агента против грамотрицательных бактерий, включающая одно или более выбранных из группы, состоящей из соединения химической формулы 1, его изомеров и их фармацевтически приемлемых солей в качестве активного ингредиента.
В одном воплощении настоящего изобретения предложен способ предупреждения или лечения грамотрицательных бактериальных инфекций, включающий стадию введения терапевтически эффективного количества трициклического бензоксаборолового соединения по настоящему изобретению субъекту, нуждающемуся в предупреждении и/или лечении заболеваний, ассоциированных с грамотрицательной бактериальной инфекцией. Перед стадией введения способ может дополнительно включать стадию определения того, нуждается ли пациент в предупреждении и/или лечении заболеваний, ассоциированных с грамотрицательной бактериальной инфекцией.
В дополнительном воплощении настоящее изобретение относится к применению антибиотика против грамотрицательных бактерий, включающего одно или более выбранных из группы, состоящей из соединения химической формулы 1, его изомеров и их фармацевтически приемлемых солей в качестве активного ингредиента, или их применению для предупреждения и/или лечения грамотрицательных бактериальных инфекций.
Соединение химической формулы 1, его изомеры и их фармацевтически приемлемые соли, используемые в фармацевтической композиции, терапевтический способ и применение согласно настоящему изобретению являются такими, как описано выше.
Новое трициклическое бензоксабороловое соединение по настоящему изобретению представляет собой антибиотик широкого спектра действия против грамотрицательных бактерий, в частности грамотрицательных бактерий с множественной лекарственной устойчивостью, и например, оно проявляет превосходную антибактериальную активность против Acinetobacter baumannii, тем самым может эффективно использоваться в качестве нового антибиотического вещества. Грамотрицательные бактерии предпочтительны, более предпочтительны устойчивые к карбапенему грамотрицательные бактерии. Конкретные примеры грамотрицательных бактерий могут включать A. baumannii, C. freundii, E. coli, E. cloacae, E. aerogenes, К. pneumoniae, K. oxytoca, M. morganii, P. aeruginosa, P. vulgaris, P. mirabilis, N. gonorrhoeae или S. marcescens. Наиболее предпочтительно, настоящее изобретение относится к антибиотической фармацевтической композиции против устойчивого к карбапенему Acinetobacter baumannii (A.baumannii).
Фармацевтическая композиция, включающая соединение химической формулы 1, его изомер или их фармацевтически приемлемую соль в качестве активного ингредиента, может быть изготовлена в форме типичного лекарственного препарата. Например, лекарственный препарат может быть приготовлен в виде разнообразных препаратов для перорального или парентерального введения, и тип препарата может быть разным в зависимости от использования, способа введения, цели введения, и так далее.
При приготовлении в виде разнообразных препаратов для перорального или парентерального введения, он может быть изготовлен с использованием одного или более выбранных из группы, состоящей из разбавителей, эксципиентов и так далее, таких как типичный носитель, объемный наполнитель, связующий агент, увлажняющий агент, разрыхлитель или поверхностно-активное вещество.
Твердый препарат для перорального введения может включать таблетку, пилюлю, порошок, гранулу или капсулу, и эти твердые препараты получают путем смешивания активного ингредиента с по меньшей мере одним эксципиентом, например, выбранным из группы, состоящей из крахмала, карбоната кальция, сахарозы, лактозы, желатина, и так далее. В дополнение к простому эксципиенту, может быть использован смазывающий агент, такой как стеарат магния, тальк, и так далее. В качестве жидкого препарата для перорального введения могут быть использованы суспензия, жидкость для приема внутрь, эмульсия, сироп, и тому подобное. При приготовлении в виде жидкого препарата обычно используют простые разбавители, такие как вода и/или жидкий вазелин. Произвольным образом могут быть дополнительно использованы один или более выбранных из группы, состоящей из разнообразных других эксципиентов, например, увлажняющий агент, подсластитель, корригент, консервант, и так далее.
Парентеральное введение может быть осуществлено таким путем, как внутривенное, внутримышечное, подкожное, внутрибрюшное, интраназальное или чрескожное введение. Препарат для парентерального введения включает стерильные водные или неводные растворы, суспензии, эмульсии, лиофилизированные препараты, суппозитории, и так далее. В качестве неводного растворителя для получаемых неводных растворов или в качестве суспендирующего растворителя для получаемых суспензий могут быть использованы пропиленгликоль, полиэтиленгликоль, растительное масло, такое как оливковое масло, инъецируемый сложный эфир, такой как этиолат, и так далее. В качестве основы для суппозиториев могут быть использованы витепсол, макрогол, твин 61, масло какао, лауриновое масло, глицерожелатин, и так далее.
Содержание одного или более активных ингредиентов, выбранных из группы, состоящей из соединения химической формулы 1, его изомеров и их фармацевтически приемлемых солей, в фармацевтической композиции может составлять, например, от 0,001 до 99,9% по массе, от 0,01 до 90% по массе или от 0,1 до 50% по массе, но не ограничено ими. Оно может быть подходящим образом контролируемым, в зависимости от типа препарата, способа введения, цели введения и так далее.
Кроме того, фармацевтические эффективное количество фармацевтической композиции по настоящему изобретению, которая включает соединение химической формулы 1, его изомер и/или их фармацевтически приемлемую соль в качестве активного ингредиента, может находиться в диапазоне от приблизительно 0,1 до приблизительно 1000 мг/сутки. Фармацевтические эффективное количество может быть инъецировано или введено однократно или несколько раз в сутки, с учетом массы тела, возраста, пола, состояния здоровья и питания пациента, времени введения, способа введения, скорости выведения, тяжести заболевания и так далее, но не ограничено этим. Возможно введение композиции в различных дозах и способах введения.
Кроме того, если трициклическое бензоксабороловое производное по настоящему изобретению предназначено для перорального введения, соединение химической формулы 1 или его фармацевтические приемлемая соль могут быть включены в композицию в количестве от 1 до 95% по массе и предпочтительно от 1 до 70% по массе.
Пациенты могут представлять собой млекопитающих, например приматов, включая людей, грызунов, в том числе мышей, крыс и так далее, и в особенности людей. Например, пациент может представлять собой млекопитающее, например, людей, симптомы или заболевания у которых можно предотвратить, ослабить и/или лечить путем введения соединения по настоящему изобретению.
Область техники
Трициклическое бензоксабороловое соединение согласно настоящему изобретению имеет широкий спектр антибактериальной активности против устойчивых бактерий, низкую токсичность и превосходную антибактериальную активность против грамотрицательных бактерий, в частности, устойчивых к антибиотикам грамотрицательных бактерий, например Acinetobacter baumannii, и таким образом проявляет сильные антибактериальные эффекты в отношении широкого ряда патогенных бактерий у людей и животных. Таким образом, оно может быть эффективно использовано в качестве антибиотика против грамотрицательных бактерий или в предупреждении, ослаблении и/или лечении заболеваний, ассоциированных с инфекцией, вызываемой грамотрицательными бактериями.
Примеры
Далее для лучшего понимания предложены предпочтительные Примеры и Экспериментальные примеры. Однако, эти Примеры и Экспериментальные примеры приведены только для иллюстративных целей и не предназначены ограничивать данное изобретение.
Если не указано иное, все реагенты, используемые ниже, приобретали в Aldrich Korea, Acros, Lancaster, TCI, Alfa aesar, и других, и 1H ЯМР осуществляли на Varian 400 МГц, 600 МГц.
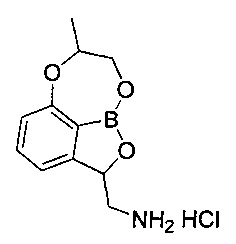
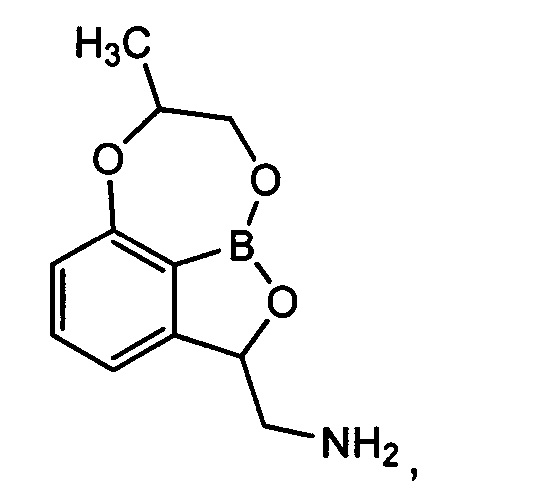
[ПРИМЕР 1] Получение гидрохлорида (7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина
[Стадия 1] Получение 1-(бензилокси)пропан-2-ола

1,2-Пропандиол (5 г, 65,7 ммоль) и NaH (3,29 г, 82,0 ммоль) растворяли в N,N-диметилформамиде (70 мл). Добавляли бензилбромид (7,82 мл, 65,7 ммоль) при 0°C с последующим перемешиванием в течение 2 часов. По окончании реакции проводили экстракцию водой и этилацетатом. Органический слой сушили над безводным сульфатом натрия и концентрировали. Остаток очищали посредством колоночной хроматографии. Получили указанное в заголовке соединение (4,45 г, 41%).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.15 (3H, d, J=6,4 Гц), 2.37 (1Н, brs), 3.28 (1Н, dd, J=9,4, 8,2 Гц), 3.47 (1Н, dd, J=9,4, 3,0 Гц), 3.98-4.02 (1Н, m), 4.56 (2Н, s), 7.25-7.38 (5Н, m).
[Стадия 2] Получение 1-(бензилокси)пропан-2-ил-метансульфоната
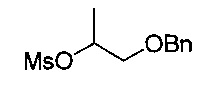
1-(Бензилокси)пропан-2-ол (4,5 г, 27,1 ммоль), полученный на стадии 1, растворяли в пиридине (50 мл). Добавляли метансульфонилхлорид (2,32 мл, 29,8 ммоль) при 0°C с последующим перемешиванием при комнатной температуре в течение 3 часов. По окончании реакции проводили экстракцию водой и этилацетатом. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии. Получили указанное в заголовке соединение (6,00 г, 91%).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.25 (3H, d, J=6,4 Гц), 3.01 (3H, s), 3.51-3.61 (2Н, m), 4.56 (2Н, d, J=2,0 Гц), 4.89-4.94 (1Н, m), 7.26-7.37 (5Н, m).
[Стадия 3] Получение 3-((1-(бензилокси)пропан-2-ил)окси)-2-бромбензальдегида
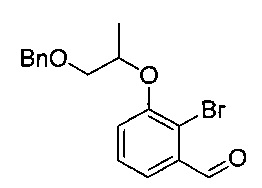
1-(Бензилокси)пропан-2-ил-метансульфонат (3,65 г, 14,9 ммоль), полученный на стадии 2, и 2-бром-3-гидроксибензальдегид (3,00 г, 14,9 ммоль) растворяли в N,N-диметилформамиде (50 мл). Добавляли K2CO3 (4,13 г, 29,8 ммоль) с последующим перемешиванием при кипячении с обратным холодильником при 100°C в течение 16 часов. После охлаждения температуры реакционной смеси до комнатной температуры проводили экстракцию водой и этилацетатом. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии. Получили указанное в заголовке соединение (4,06 г, 78%).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.40 (3H, d, J=6,4 Гц), 3.65 (1Н, dd, J=10,0, 4,0 Гц), 3.75 (1Н, dd, J=10,4, 6,4 Гц), 4.61-4.65 (3H, m), 7.23 (1Н, dd, J=8,2, 1,4 Гц), 7.28-7.35 (6Н, m), 7.52 (1Н, dd, J=8,2, 1,0 Гц), 10.43 (1Н, d, J=0,4 Гц).
[Стадия 4] Получение 3-((1-(бензилокси)пропан-2-ил)окси)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензальдегида
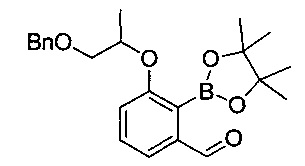
3-((1-(Бензилокси)пропан-2-ил)окси)-2-бромбензальдегид (3,00 г, 8,59 ммоль), полученный на стадии 3, растворяли в диоксане (60 мл). Добавляли 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий (314 мг, 0,43 ммоль), ацетат калия (1,68 г, 17,2 ммоль) и бис(пинаколато)дибор (4,36 г, 17,2 ммоль) с последующим перемешиванием при кипячении с обратным холодильником при 100°C в течение 1 часа. После охлаждения температуры реакционной смеси до комнатной температуры проводили экстракцию водой и этилацетатом. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии. Получили указанное в заголовке соединение (700 мг, 20%).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.35 (3H, d, J=6,0 Гц), 1.43 (12Н, s), 3.64 (1Н, dd, J=10,0, 5,2 Гц), 3.69 (1H, dd, J=10,0, 5,6 Гц), 4.55 (2H, d, J=3,2 Гц), 4.60 (1H, dd, J=12,0, 5,6 Гц), 7.18 (1H, d, J=8,0 Гц), 7.27-7.34 (5H, m), 7.38 (1Н, d, J=6,4 Гц), 7.43 (1H, t, J=7,8 Гц), 9.92 (1H, s).
[Стадия 5] Получение 7-((1-(бензилокси)пропан-2-ил)окси)-1-гидрокси-1,3-дигидробензо[с][1,2]оксаборол-3-карбонитрила
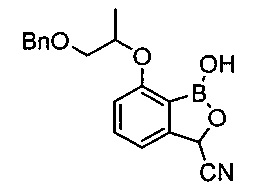
3-((1-(Бензилокси)пропан-2-ил)окси)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензальдегид (700 мг, 1,76 ммоль), полученный на стадии 4, растворяли в воде (1 мл) и тетрагидрофуране (1 мл). Добавляли цианид натрия (87 мг, 1,78 ммоль) при комнатной температуре. После перемешивания в течение 1 часа к реакционной смеси добавляли 2 н. соляную кислоту до достижения значения pH 1. По окончании реакции проводили экстракцию этилацетатом. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии. Получили указанное в заголовке соединение (500 мг, 88%).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.21-1.24 (3H, m), 3.59-3.70 (2Н, m), 4.30-4.42 (1Н, m), 4.43-4.52 (1Н, m), 4.66-4.77 (2Н, m), 5.79 (1Н, s), 5.82 (1Н, s), 7.01 (1Н, d, J=8,0 Гц), 7.25-7.39 (5Н, m), 7.53 (1Н, td, J=7,7, 1,9 Гц), 8.13 (1Н, s), 8.26 (1Н, s).
[Стадия 6] Получение трет-бутил-((7-((1-(бензилокси)пропан-2-ил)окси)-1-гидрокси-1,3-дигидробензо[с][1,2]оксаборол-3-ил)метил)-карбамата
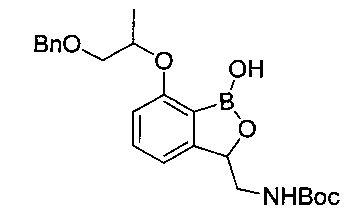
7-((1-(Бензилокси)пропан-2-ил)окси)-1-гидрокси-1,3-дигидробензо[с][1,2]оксаборол-3-карбонитрил (500 мг, 1,55 ммоль), полученный на стадии 5, растворяли в безводном тетрагидрофуране (10 мл). Медленно при комнатной температуре добавляли реагент, представляющий собой 1M боран-тетрагидрофурановый комплекс (3,1 мл). После этого реакционную смесь перемешивали при кипячении с обратным холодильником в течение 3 часов и затем температуру реакционной смеси понижали путем медленного охлаждения до комнатной температуры. Затем к реакционной смеси медленно добавляли метанол и осуществляли азеотропную перегонку с концентрированием при пониженном давлении. После трехкратного повторения вышеуказанного процесса с использованием метанола (10 мл) концентрированную реакционную смесь растворяли в тетрагидрофуране (10 мл). В реакционную смесь добавляли триэтиламин (0,42 мл) и ди-трет-бутил-дикарбонат (0,35 мл) по порядку и затем перемешивали при комнатной температуре в течение 4 часов. После добавления к реакционной смеси 2 н. соляной кислоты для подкисления проводили экстракцию этилацетатом дважды. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии. Получили указанное в заголовке соединение (340 мг, 51%).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.22 (3H, d, J=6,4 Гц), 1.42 (9Н, s), 2.98-3.08 (1Н, m), 3.58-3.69 (2Н, m), 3.82-3.95 (1Н, m), 4.36-4.43 (1Н, m), 4.64-4.76 (1Н, m), 4.96-5.08 (1Н, m), 5.19-5.24 (1Н, m), 6.89 (1Н, d, J=7,6 Гц), 7.09 (1Н, d, J=7,6 Гц), 7.31-7.46 (6Н, m), 7.53 (1Н, s), 7.60 (1Н, s).
[Стадия 7] Получение трет-бутил-((7-метил-7,8-дигидро-2Н-1,6.9-триокса-9а-борабензо[cd]азулен-2-ил)метил)карбамата
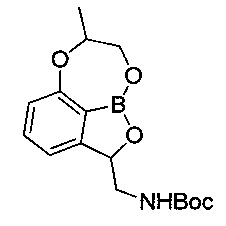
трет-Бутил-((7-((1-(бензилокси)пропан-2-ил)окси)-1-гидрокси-1,3-дигидробензо[с][1,2]оксаборол-3-ил)метил)карбамат (200 мг, 0,439 ммоль), полученный на стадии 6, и гидроксид палладия (12,3 мг, 0,088 ммоль) растворяли в метаноле (5 мл) с последующим перемешиванием с реакцией гидрирования в течение 1 часа. После фильтрования реакционной смеси через целит с использованием этилацетата остаточный раствор фильтровали при пониженном давлении. Получили указанное в заголовке соединение (140 мг, 89%).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.42 (9Н, s), 1.47 (3H, d, J=6,8 Гц), 2.98-3.08 (1Н, m), 3.80-3.93 (1Н, m), 4.22 (2Н, s), 4.18-4.42 (1Н, m), 4.90-5.18 (1Н, m), 5.30-5.37 (1Н, m), 6.85 (1Н, d, J=8,0 Гц), 6.98 (1Н, d, J=7,2 Гц), 7.42 (1Н, t, J=7,8 Гц).
[Стадия 8] Получение гидрохлорида (7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина
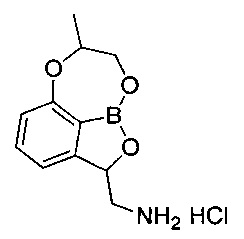
трет-Бутил-((7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метил)карбамат (140 мг, 0,439 ммоль), полученный на стадии 7, растворяли в диоксане (5 мл). Добавляли при 0°C раствор соляной кислоты (4 н. раствор диоксан, 3,29 мл, 13,1 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 16 часов, концентрировали при пониженном давлении для удаления растворителя и растворяли в этиловом эфире. Полученное твердое вещество отфильтровывали. Получили указанное в заголовке соединение (92,0 мг, 96%).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.43 (3H, s), 2.91-2.99 (1Н, m), 3.58 (1Н, td, J=13,0, 2,7 Гц), 4.15-4.42 (3H, m), 5.46 (1Н, t, J=10,4 Гц), 6.88 (1Н, d, J=8,0 Гц), 7.03 (1Н, d, J=7,2 Гц), 7.47 (1Н, t, J=7,8 Гц).
[ПРИМЕР 2] Получение гидрохлорида ((2S)-7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина
[Стадия 1] Получение 1-(бензилокси)пропан-2-ил-метансульфоната
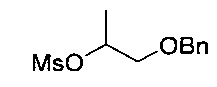
Исходное соединение 1-(бензилокси)пропан-2-ол (36 г, 217 ммоль) и диизопропилэтиламин (39,2 г, 303 ммоль) растворяли в толуоле (540 мл). Добавляли при 0°C метансульфонилхлорид (2,32 мл, 29,8 ммоль) с последующим перемешиванием в течение 2 часов и дополнительным перемешиванием при комнатной температуре в течение 1 часа. По окончании реакции проводили экстракцию водой (500 мл) и толуолом (200 мл, дважды). Органический слой экстрагировали насыщенным водным раствором хлорида аммония (200 мл) и насыщенным водным раствором хлорида натрия (200 мл) и промывали. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток сушили. Получили указанное в заголовке соединение (53 г).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.40 (3H, d, J=6,4 Гц), 3.01 (3H, s), 3.52-3.61 (2Н, m), 4.55 & 4.57 (2Н, ABq, JAB=11,8 Гц), 4.88-4.96 (1Н, m), 7.26-7.38 (5Н, m).
[Стадия 2] Получение 3-((1-(бензилокси)пропан-2-ил)окси)-бензальдегида
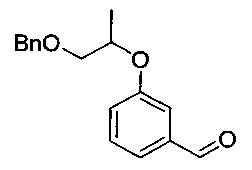
Исходное соединение 3-гидроксибензальдегид (26,5 г, 217 ммоль) и карбонат калия (36,0 г, 261 ммоль) растворяли в диметилформамиде (540 мл) с последующим перемешиванием при 0°C в течение 30 минут. Медленно добавляли 1-(бензилокси)пропан-2-ил-метансульфонат (53 г, 217 ммоль), полученный на стадии 1 ПРИМЕРА 1, с последующим перемешиванием при 100°C в течение 10 часов. Реакционную смесь охлаждали до комнатной температуры. Эту реакционную смесь экстрагировали ледяной водой (1 л) и гептаном (500 мл, дважды). Органический слой экстрагировали 0,02 н. водным раствором гидроксида натрия (200 мл, дважды), 0,01 н. водным раствором соляной кислоты (200 мл) и насыщенным водным раствором хлорида натрия (200 мл) и промывали. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток сушили. Получили указанное в заголовке соединение (46 г, 78%).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.35 (3H, d, J=6,0 Гц), 3.68 (1Н, dd,.7=10,4, 6,0 Гц), 3.59 (1Н, dd, J=10,0, 4,4 Гц), 4.59 (2Н, s), 4.63-4.71 (1Н, m), 7.18-7.21 (1Н, m), 7.28-7.36 (5Н, m), 7.40-7.46 (3H, m), 9.95 (1Н, s).
[Стадия 3] Получение (1S)-1-(3-((1-(бензилокси)пропан-2-ил)окси)фенил)-2-нитроэтан-1-ола
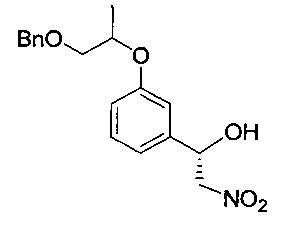
Моногидрат ацетата меди (0,739 г, 3,70 ммоль) и (1R)-1,7,7-триметил-N-(пиридин-2-илметил)бицикло[2.2.1]гептан-2-амин (0,994 г, 4,07 ммоль) растворяли в этаноле (110 мл) с последующим перемешиванием при комнатной температуре в течение 1 часа. После медленного добавления к реакционной смеси нитрометана (22,6 г, 370 ммоль) реакционную смесь перемешивали при -30°C в течение 30 минут. 3-((1-(Бензилокси)пропан-2-ил)окси)бензальдегид (20 г, 74 ммоль), полученный на стадии 1, разбавляли в этаноле (40 мл) и медленно в течение более 1 часа добавляли к реакционной смеси, поддерживая температуру -30°C. К перемешиваемой при -30°C реакционной смеси добавляли диизопропилэтиламин (1,29 мл, 7,40 ммоль), перемешивали при той же температуре в течение более 24 часов и медленно нагревали до комнатной температуры. Реакционную смесь экстрагировали 1 н. водным раствором соляной кислоты (300 мл) и дихлорметаном (300 мл) и водный слой экстрагировали дополнительным количеством дихлорметана (80 мл, дважды). Органический слой экстрагировали насыщенным водным раствором хлорида натрия (100 мл) и промывали. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток сушили. Получили указанное в заголовке соединение (25,3 г).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.33 (3H, m), 2.90 (1Н, brs), 3.54-3.59 (1Н, m), 3.61-3.69 (1Н, m), 4.45 (1Н, m), 4.50-4.62 (4Н, m), 5.38 (1Н, m), 6.86-6.99 (3H, m), 7.24-7.38 (6Н, m).
[Стадия 4] Получение (1S)-2-амино-1-(3-((1-(бензилокси)пропан-2-ил)окси)фенил)этан-1-ола
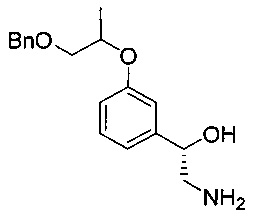
(1S)-1-(3-((1-(Бензилокси)пропан-ил)окси)фенил)-2-нитроэтан-1-ол (25,3 г, 76,0 ммоль), полученный на стадии 3, растворяли в этаноле (381 мл). Добавляли 5%-ный палладий на активированном углероде (4,06 г, 1,91 ммоль) и 5%-ную платину на активированном углероде (1,01 г, 0,259 ммоль) в качестве катализатора. После осуществления реакции гидрирования при комнатной температуре (приблизительно 25°C) при давлении примерно 50-60 фунт/кв. дюйм в течение более 9 часов проводили фильтрацию с использованием целита для удаления палладия и платины. Фильтрат сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток сушили. Получили указанное в заголовке соединение (22,2 г, 97%).
[Стадия 5] Получение гидрохлорида (1S)-1-(3-((1-(бензилокси)пропан-2-ил)окси)фенил)-2-(дибензиламино)этан-1-ола
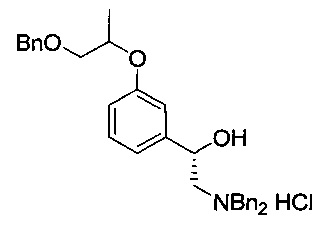
(1S)-2-Амино-1-(3-((1-(бензилокси)пропан-2-ил)окси)фенил)этан-1-ол (22,2 г, 73,6 ммоль), полученный на стадии 4, растворяли в этаноле (245 мл). Добавляли карбонат калия (22,4 г, 162 ммоль) с последующим перемешиванием при комнатной температуре в течение примерно 15 часов. К реакционной смеси добавляли метил-трет-бутиловый эфир (100 мл), охлажденный до 0°C, и перемешивали в течение 30 минут. После отфильтровывания полученного твердого осадка с использованием целита к фильтрату добавляли насыщенный раствор соляной кислоты (12,3 мл, 147 ммоль) и перемешивали в течение 30 минут. После удаления метил-трет-бутилового эфира и избытка соляной кислоты посредством концентрирования осуществляли азеотропную перегонку с использованием изопропанола (100 мл) для удаления оставшейся воды. После удаления воды путем повторной азеотропной перегонки два или три раза к остаточному твердому веществу добавляли изопропанол (45 мл) и перемешивали при 60°C в течение 2 часов до растворения этого твердого вещества. После повторного медленного охлаждения до комнатной температуры перемешивание осуществляли в течение 2 часов. Метил-трет-бутиловый эфир добавляли медленно при комнатной температуре в течение 1 часа, когда начинало образовываться твердое вещество, с последующим перемешиванием дополнительно в течение 2 часов. После отфильтровывания полученного белого твердого вещества, это отфильтрованное твердое вещество промывали метил-трет-бутиловым эфиром (50 мл). Это твердое вещество сушили при пониженном давлении. Указанное в заголовке соединение (20,4 г, 54%) получили в виде белой твердой фазы.
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.28 (3H, d, J=6,0 Гц), 2.98-3.03 (1Н, m), 3.13-3.22 (1Н, m), 3.53 (1Н, dd, J=10,2, 4,6 Гц), 3.59-3.64 (1Н, m), 4.15-4.18 (1Н, m), 4.34 (1Н, dd, J=13,2, 5,2 Гц), 4.46-4.55 (3H, m), 4.57 (2Н, s), 5.07 (1Н, d, J=9,6 Гц), 5.42 (1Н, s), 6.62 (1Н, d, J=6,6 Гц), 6.75 (1Н, brs), 6.80 (1Н, dd, J=8,0, 2,4 Гц), 7.15 (1Н, t, J=7,8 Гц), 7.26-7.35 (5Н, m), 7.44-7.51 (6Н, m), 7.61-7.63 (2Н, m), 7.67-7.72 (2Н, m), 12.06 (1Н, brs).
[Стадия 6] Получение (3S)-7-((1-(бензилокси)пропан-2-ил)окси)-3-((дибензиламино)метил)бензо[с][1,2]оксаборол-1(3H)-ола
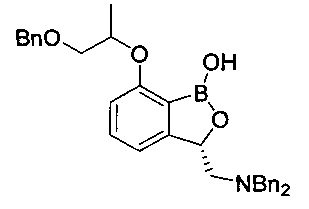
Гидрохлорид (1S)-1-(3-((1-(бензилокси)пропан-2-ил)окси)фенил)-2-(дибензиламино)этан-1-ола (10 г, 19,3 ммоль), полученный на стадии 4, растворяли в безводном толуоле (77 мл) в колбе (A), заполненной азотом. После того как реакционную смесь, которая не была полностью растворена, нагрели до 40-45°C, поддерживая заполненное азотом состояние, медленно в течение 1 часа добавляли н-бутиллитий (2,5М раствор в гексане, 8,49 мл, 21,2 ммоль). После перемешивания в течение 1 часа реакционную смесь охлаждали до -30°C, перемешивали и медленно в течение 1 часа добавляли н-бутиллитий (2,5М раствор в гексане, 37,82 мл, 94,3 ммоль), поддерживая заполненное азотом состояние. Температуре реакционной смеси не давали превышать -20°C. 2-Изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолан (31,2 мл, 154 ммоль) растворяли, используя тетрагидрофуран (9 мл) и безводный толуол (77 мл), в другой колбе (B), с последующим перемешиванием при -40°C. В реакционную колбу (B) медленно по каплям в течение 2 часов добавляли содержимое реакционной колбы (A). После перемешивания при той же температуре в течение еще одного часа реакционную смесь медленно в течение 1 часа нагревали до 10°C. К перемешиваемой реакционной смеси добавляли по каплям 5%-ный водный раствор бикарбоната натрия (150 мл). Реакционную суспензию фильтровали и фильтрат промывали этилацетатом (50 мл). После двукратной экстракции водного слоя из фильтрата этилацетатом (100 мл) органический слой промывали насыщенным водным раствором хлорида натрия (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии. Получили указанное в заголовке соединение (6,03 г, 62%).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.21 (3H, d, J=6,4 Гц), 2.67 (1Н, dd, J=14,0, 7,6 Гц), 2.99 (1Н, dd, J=14,4, 3,6 Гц), 3.56 (1Н, dd, J=10,0, 2,8 Гц), 3.63 (1Н, dd, J=10,0, 8,0 Гц), 3.74 (2Н, d, J=13,6 Гц) 3.90 (2Н, d, J=13,6 Гц), 4.36-4.42 (1Н, m), 4.65 & 4.72 (2Н, ABq, JAB=12,4 Гц), 5.35-5.38 (1Н, m), 6.76-6.83 (2Н, m), 7.19-7.38 (16Н, m).
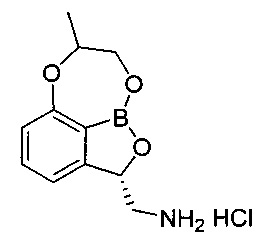
[Стадия 7] Получение гидрохлорида ((2S)-7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина
(3S)-7-((1-(Бензилокси)пропан-2-ил)окси)-3-((дибензиламино)метил)-бензо[с][1,2]оксаборол-1(3H)-ол (5,56 г, 11,0 ммоль) со Стадии 6 растворяли с использованием смешанного раствора 1 н. водного раствора соляной кислоты (13,2 мл, 13,2 ммоль) и метанола (110 мл) при комнатной температуре. К реакционной смеси добавляли катализатор 5%-ный палладий на активированном углероде. Реакционную смесь нагревали до 50°C и заполняли газообразным водородом под давлением примерно 50-60 фунт/кв. дюйм при перемешивании. После выдерживания реакционной смеси в вышеуказанном состоянии в течение более 10 часов ее фильтровали с использованием целита для удаления палладия. Отфильтрованный на целите слой промывали метанолом (10 мл) и затем фильтрат концентрировали. К фильтрату добавляли изопропанол (50 мл), осуществляли азеотропную перегонку для удаления воды. После удаления воды путем осуществления азеотропной перегонки два или три раза к полученному твердому веществу добавляли изопропанол (7 мл), перемешивали в течение более 5 часов и суспензию фильтровали. Полученное твердое вещество промывали изопропанолом (3 мл). Отфильтрованное твердое вещество, которое собрали, сушили при пониженном давлении. Получили указанное в заголовке соединение (2,4 г, 86%).
1H-ЯМР (DMSO-d6, Varian 400 МГц): δ 1.40 (3H, s), 2.82-2.93 (1Н, m), 3.45-3.57 (1Н, m), 4.14-4.30 (3H, m), 5.50-5.60 (1Н, m), 6.79-6.83 (1Н, m), 7.10-7.15 (1Н, m), 7.48 (1Н, t, J=7,8 Гц), 8.40 (3H, brs).
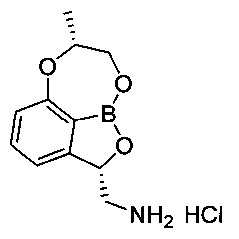
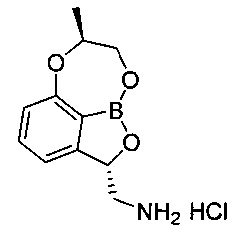
[ПРИМЕР 3] Получение гидрохлорида ((2S,7R)-7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина
[Стадия 1] Получение (S)-1-(бензилокси)пропан-2-ил-метансульфоната
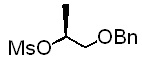
Исходное соединение (S)-1-(бензилокси)пропан-2-ол (36 г, 217 ммоль) и диизопропилэтиламин (39,2 г, 303 ммоль) растворяли в толуоле (540 мл). Добавляли при 0°C метансульфонилхлорид (2,32 мл, 29,8 ммоль) с последующим перемешиванием в течение 2 часов и перемешивали еще при комнатной температуре в течение 1 часа. По окончании реакции проводили экстракцию водой (500 мл) и толуолом (200 мл, дважды). Органический слой экстрагировали насыщенным водным раствором хлорида аммония (200 мл) и насыщенным водным раствором хлорида натрия (200 мл) и промывали. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток сушили. Получили указанное в заголовке соединение (53 г).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.40 (3H, d, J=6,4 Гц), 3.01 (3H, s), 3.52-3.61 (2Н, m), 4.55 & 4.57 (2Н, ABq, JAB=11,8 Гц), 4.88-4.96 (1Н, m), 7.26-7.38 (5Н, m).
[Стадия 2] Получение (R)-3-((1-(бензилокси)пропан-2-ил)окси)-бензальдегида
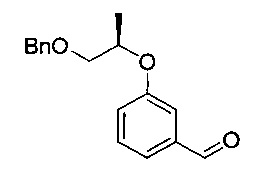
Исходное соединение 3-гидроксибензальдегид (26,5 г, 217 ммоль) и карбонат калия (36,0 г, 261 ммоль) растворяли в диметилформамиде (540 мл) с последующим перемешиванием при 0°C в течение 30 минут. Медленно добавляли (S)-1-(бензилокси)пропан-2-илметансульфонат (53 г, 217 ммоль), полученный на стадии 1, с последующим перемешиванием при 100°C в течение 10 часов. Реакционную смесь охлаждали до комнатной температуры. Реакционную смесь экстрагировали ледяной водой (1 л) и гептаном (500 мл, дважды). Органический слой экстрагировали 0,02 н. водным раствором гидроксида натрия (200 мл, дважды), 0,01 н. водным раствором соляной кислоты (200 мл) и насыщенным водным раствором хлорида натрия (200 мл) и промывали. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток сушили. Получили указанное в заголовке соединение (46 г, 78%).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.35 (3H, d, J=6,0 Гц), 3.68 (1Н, dd, J=10,4, 6,0 Гц), 3.59 (1Н, dd, J=10,0, 4,4 Гц), 4.59 (2Н, s), 4.63-4.71 (1Н, m), 7.18-7.21 (1Н, m), 7.28-7.36 (5Н, m), 7.40-7.46 (3H, m), 9.95 (1Н, s).
[Стадия 3] Получение (S)-1-(3-(((R)-1-(бензилокси)пропан-2-ил)окси)фенил)-2-нитроэтан-1-ола
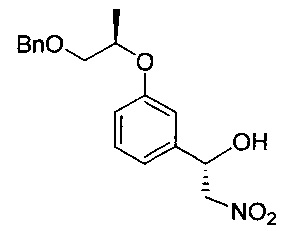
Моногидрат ацетата меди (0,739 г, 3,70 ммоль) и (1R)-1,7,7-триметил-N-(пиридин-2-илметил)бицикло[2.2.1]гептан-2-амин (0,994 г, 4,07 ммоль) растворяли в этаноле (110 мл) с последующим перемешиванием при комнатной температуре в течение 1 часа. После медленного добавления к реакционной смеси нитрометана (22,6 г, 370 ммоль) реакционную смесь перемешивали при -30°C в течение 30 минут. (R)-3-((1-(Бензилокси)пропан-2-ил)окси)бензальдегид (20 г, 74 ммоль), полученный на стадии 2, разбавляли в этаноле (40 мл) и медленно в течение более 1 часа добавляли к реакционной смеси, поддерживая температуру -30°C. К перемешиваемой при -30°C реакционной смеси добавляли диизопропилэтиламин (1,29 мл, 7,40 ммоль), перемешивали при той же температуре в течение более 24 часов и медленно нагревали до комнатной температуры. Реакционную смесь экстрагировали 1 н. водным раствором соляной кислоты (300 мл) и дихлорметаном (300 мл) и водный слой экстрагировали дополнительным количеством дихлорметана (80 мл, дважды). Органический слой экстрагировали насыщенным водным раствором хлорида натрия (100 мл) и промывали. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток сушили. Получили указанное в заголовке соединение (25,3 г).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.33 (3H, d, J=6,4 Гц), 2.82 (1Н, brs), 3.56 (1Н, dd, J=10,4, 4,4 Гц), 3.66 (1Н, dd, J=10,2, 6,2 Гц), 4.45 (1Н, dd, J=13,4, 3,0 Гц), 4.51-4.60 (2Н, m), 4.58 (2Н, s), 5.39 (1Н, dd, J=9,6, 2,8 Гц), 6.89-6.99 (3H, m), 7.26-7.37 (6Н, m).
[Стадия 4] Получение (S)-2-амино-1-(3-(((R)-1-(бензилокси)пропан-2-ил)окси)фенил)этан-1-ола
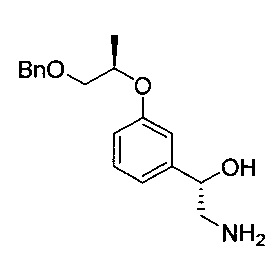
(S)-1-(3-(((R)-1-(бензилокси)пропан-2-ил)окси)фенил)-2-нитроэтан-1-ол (25,3 г, 76,0 ммоль), полученный на стадии 3, растворяли в этаноле (381 мл). Добавляли 5%-ный палладий на активированном углероде (4,06 г, 1,91 ммоль) и 5%-ную платину на активированном углероде (1,01 г, 0,259 ммоль) в качестве катализатора. После осуществления реакции гидрирования при комнатной температуре (приблизительно 25°C) при давлении примерно 50-60 фунт/кв. дюйм в течение более 9 часов проводили фильтрацию с использованием целита для удаления палладия и платины. Фильтрат сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток сушили. Получили указанное в заголовке соединение (22,2 г, 97%).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.32 (3H, d, J=6,0 Гц), 2.39 (3H, brs), 2.79 (1Н, dd, J=12,6, 7,8 Гц), 2.97 (1Н, dd, J=13,0, 3,8 Гц), 3.55 (1Н, dd, J=8,2, 3,0 Гц), 3.66 (1Н, dd, J=10,2, 5,8 Гц), 4.58 (2Н, s), 4.56-4.63 (2Н, m), 6.83 (1Н, dd, J=8,2, 1,8 Гц), 6.90 (1Н, d, J=7,6 Гц), 6.95-6.96 (1Н, m), 7.23 (1Н, t, J=7,8 Гц), 7.35-7.25 (5Н, m).
[Стадия 5] Получение гидрохлорида (S)-1-(3-(((R)-1-(бензилокси)-пропан-2-ил)окси)фенил)-2-(дибензиламино)этан-1-ола
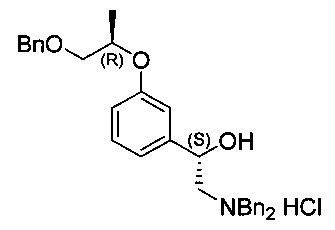
(S)-2-амино-1-(3-(((R)-1-(бензилокси)пропан-2-ил)окси)фенил)этан-1-ол (22,2 г, 73,6 ммоль), полученный на стадии 4, растворяли в этаноле (245 мл). Добавляли карбонат калия (22,4 г, 162 ммоль) с последующим перемешиванием при комнатной температуре в течение примерно 15 часов. К реакционной смеси добавляли метил-трет-бутиловый эфир (100 мл), охлажденный до 0°C, и перемешивали в течение 30 минут. После отфильтровывания полученного твердого осадка с использованием целита к фильтрату добавляли насыщенный раствор соляной кислоты (12,3 мл, 147 ммоль) и перемешивали в течение 30 минут. После удаления метил-трет-бутилового эфира и избытка соляной кислоты посредством концентрирования осуществляли азеотропную перегонку с использованием изопропанола (100 мл) для удаления оставшейся воды. После удаления воды путем повторной азеотропной перегонки два или три раза к остаточному твердому веществу добавляли изопропанол (45 мл) и перемешивали при 60°C в течение 2 часов до растворения этого твердого вещества. После повторного медленного охлаждения до комнатной температуры перемешивание осуществляли в течение 2 часов. Метил-трет-бутиловый эфир добавляли медленно при комнатной температуре в течение 1 часа, когда начинало образовываться твердое вещество, с последующим перемешиванием дополнительно в течение 2 часов. После отфильтровывания полученного белого твердого вещества, это отфильтрованное твердое вещество промывали метил-трет-бутиловым эфиром (50 мл). Это твердое вещество сушили при пониженном давлении. Указанное в заголовке соединение (20,4 г, 54%) получили в виде белой твердой фазы.
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.28 (3H, d, J=6,0 Гц), 2.98-3.02 (1Н, m), 3.15-3.21 (1Н, m), 3.53 (1Н, dd, J=10,2, 4,6 Гц), 3.62 (1Н, dd, J=10,4, 6,0 Гц), 4.17 (1Н, dd, J=13,4, 6,2 Гц), 4.33 (1Н, dd, J=13,2, 5,2 Гц), 4.47-4.55 (3H, m), 4.57 (2Н, s), 5.07 (1H,d, J=9,6 Гц), 5.42 (1Н, s), 6.62 (1Н, d, J=6,6 Гц), 6.75 (1Н, brs), 6.80 (1Н, dd, J=8,0, 2,4 Гц), 7.15 (1Н, t, J=7,8 Гц), 7.26-7.35 (5Н, m), 7.45-7.50 (6Н, m), 7.62-7.64 (2Н, m), 7.69-7.71 (2Н, m), 12.06 (1Н, brs).
[Стадия 6] Получение (S)-7-(((R)-(бензилокси)пропан-2-ил)окси)-3-((дибензиламино)метил)бензо[с][1,2]оксаборол-1(3H)-ола

Гидрохлорид (1S)-1-(3-((1-(бензилокси)пропан-2-ил)окси)фенил)-2-(дибензиламино)этан-1-ола (10 г, 19,3 ммоль), полученный на стадии 5, растворяли в безводном толуоле (77 мл) в колбе (A), заполненной азотом. После того как реакционную смесь, которая не была полностью растворена, нагрели до 40-45°C, поддерживая заполненное азотом состояние, медленно в течение 1 часа добавляли н-бутиллитий (2,5М раствор в гексане, 8,49 мл, 21,2 ммоль). После перемешивания в течение 1 часа реакционную смесь охлаждали до -30°C, перемешивали и медленно в течение 1 часа добавляли н-бутиллитий (2,5М раствор в гексане, 37,82 мл, 94,3 ммоль), поддерживая заполненное азотом состояние. Температуре реакционной смеси не давали превышать -20°C. 2-Изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолан (31,2 мл, 154 ммоль) растворяли, используя тетрагидрофуран (9 мл) и безводный толуол (77 мл), в другой колбе (В), с последующим перемешиванием при -40°C. В реакционную колбу (B) медленно по каплям в течение 2 часов добавляли содержимое реакционной колбы (А). После перемешивания при той же температуре в течение еще одного часа реакционную смесь медленно в течение 1 часа нагревали до 10°C. К перемешиваемой реакционной смеси добавляли по каплям 5%-ный водный раствор бикарбоната натрия (150 мл). Реакционную суспензию фильтровали и фильтрат промывали этилацетатом (50 мл). После двукратной экстракции водного слоя из фильтрата этилацетатом (100 мл) весь органический слой промывали насыщенным водным раствором хлорида натрия (100 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии. Получили указанное в заголовке соединение (6,03 г, 62%).
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.21 (3H, d, J=6,4 Гц), 2.67 (1Н, dd, J=14,0, 7,6 Гц), 2.99 (1Н, dd, J=14,4, 3,6 Гц), 3.56 (1Н, dd, J=10,0, 2,8 Гц), 3.63 (1Н, dd, J=10,0, 8,0 Гц), 3.74 (2Н, d, J=13,6 Гц) 3.89 (2Н, d, J=13,6 Гц), 4.36-4.41 (1Н, m), 4.65 & 4.72 (2Н, ABq, JAB=12,4 Гц), 5.35-5.38 (1Н, m), 6.78-6.83 (2Н, m), 7.20-7.39 (16Н, m).
[Стадия 7] Получение гидрохлорида ((2S,7R)-7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина
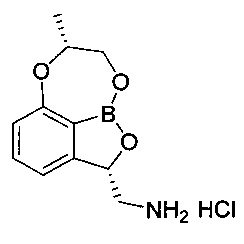
(3S)-7-((1-(Бензилокси)пропан-2-ил)окси)-3-((дибензиламино)метил)-бензо[с][1,2]оксаборол-1(3H)-ол (5,56 г, 11,0 ммоль) со Стадии 6 растворяли с использованием смешанного раствора 1 н. водного раствора соляной кислоты (13,2 мл, 13,2 ммоль) и метанола (110 мл) при комнатной температуре. К реакционной смеси добавляли катализатор 5%-ный палладий на активированном углероде. Реакционную смесь нагревали до 50°C и заполняли газообразным водородом под давлением примерно 50-60 фунт/кв. дюйм при перемешивании. После выдерживания реакционной смеси в вышеуказанном состоянии в течение более 10 часов ее фильтровали с использованием целита для удаления палладия. Отфильтрованный на целите слой промывали метанолом (10 мл) и затем фильтрат концентрировали. К фильтрату добавляли изопропанол (50 мл), осуществляли азеотропную перегонку для удаления воды. После удаления воды путем осуществления азеотропной перегонки два или три раза к полученному твердому веществу добавляли изопропанол (7 мл), перемешивали в течение более 5 часов и суспензию фильтровали. Полученное твердое вещество промывали изопропанолом (3 мл). Отфильтрованное твердое вещество, которое собрали, сушили при пониженном давлении. Получили указанное в заголовке соединение (2,4 г, 86%).
1H-ЯМР (DMSO-d6, Varian 400 МГц): δ 1.41 (3H, d, J=6,4 Гц), 2.87 (1Н, dd, J=13,2, 9,2 Гц), 3.52 (1Н, dd, J=13,2, 2,4 Гц), 4.15-4.26 (2Н, m), 4.41 (1Н, brs), 5.54-5.59 (1Н, m), 6.88 (1Н, d, J=8,4 Гц), 7.14 (1Н, d, J=7,2 Гц), 7.49 (1Н, t, J=7,8 Гц), 8.40 (3H, brs).
[ПРИМЕР 4] Получение ((2S,7R)-7-метил-7.8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина
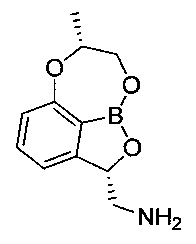
Гидрохлорид ((2S,7R)-7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина (1 г, 11,0 ммоль), полученный на стадии 7 ПРИМЕРА 3, растворяли в водном растворе бикарбоната натрия (80 мл) посредством нейтрализации. После удаления посредством однократной экстракции этилацетатом (100 мл) водный слой экстрагировали смешанным растворителем из метанола (20 мл) и дихлорметана (100 мл) с получением органического слоя. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток сушили в вакууме. Получили указанное в заголовке соединение (400 мг).
1H-ЯМР (CD3OD, Varian 400 МГц): δ 1.41 (3H, brs), 2.95-2.98 (1Н, m), 3.26-3.29 (1Н, m), 4.19 (2Н, brs), 4,42 (1Н, brs), 5.41-5.45 (1Н, m), 6.84-6.86 (1Н, m), 7.01 (1Н, brs), 7.44 (1Н, brs).
[ПРИМЕР 5] Получение гидрохлорида ((2S,7S)-7-метил-7.8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина
[Стадия 1] Получение (R)-1-(бензилокси)пропан-2-ил-метансульфоната
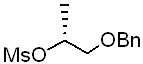
Указанное в заголовке соединение (27,96 г) получали по существу таким же способом, как описано для Стадии 1 Примера 2, за исключением того, что исходное соединение (R)-1-(бензилокси)пропан-2-ол (20 г, 120 ммоль) использовали вместо исходного соединения 1-(бензилокси)пропан-2-ола.
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.40 (3H, d, J=6,4 Гц), 3.01 (3H, s), 3.52-3.61 (2Н, m), 4.55 & 4.57 (2Н, ABq, JAB=11,8 Гц), 4.88-4.96 (1Н, m), 7.26-7.38 (5Н, m).
[Стадия 2] Получение (S)-3-((1-(бензилокси)пропан-2-ил)окси)-бензальдегида
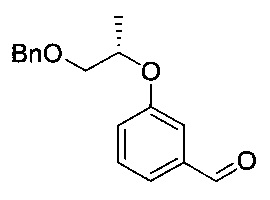
Указанное в заголовке соединение (19,14 г) получали по существу таким же способом, как описано для Стадии 2 Примера 2, за исключением того, что исходное соединение (R)-1-(бензилокси)пропан-2-ил-метансульфонат (27,96 г) использовали вместо исходного соединения 1-(бензилокси)пропан-2-ил-метансульфоната.
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.35 (3H, d, J=6,0 Гц), 3.68 (1Н, dd, J=10,4, 6,0 Гц), 3.59 (1Н, dd, J=10,0, 4,4 Гц), 4.59 (2Н, s), 4.63-4.71 (1Н, m), 7.18-7.21 (1Н, m), 7.28-7.36 (5Н, m), 7.40-7.46 (3H, m), 9.95 (1Н, s).
[Стадия 3] Получение (S)-1-(3-(((S)-(бензилокси)пропан-2-ил)окси)-фенил)-2-нитроэтан-1-ола
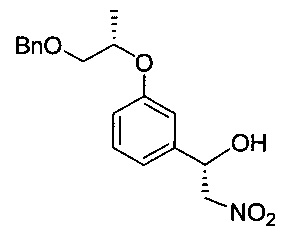
Указанное в заголовке соединение (20 г) получали по существу таким же способом, как описано для Стадии 3 Примера 2, за исключением того, что исходное соединение (S)-3-((1-(бензилокси)пропан-2-ил)окси)бензальдегид (19,14 г) использовали вместо исходного соединения 3-((1-(бензилокси)пропан-2-ил)окси)бензальдегида.
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.33 (3H, d, J=6,4 Гц), 2.90 (1Н, brs), 3.57 (1Н, dd, J=10,4, 4,4 Гц), 3.66 (1Н, dd, J=10,2, 6,2 Гц), 4.45 (1Н, dd, J=13,4, 3,0 Гц), 4.51-4.60 (2Н, m), 4.58 (2Н, s), 5.39 (1Н, dd, J=9,6, 2,8 Гц), 6.89-6.99 (3H, m), 7.24-7.38 (6Н, m).
[Стадия 4] Получение (S)-2-амино-1-(3-(((S)-1-(бензилокси)пропан-2-ил)окси)фенил)этан-1-ола
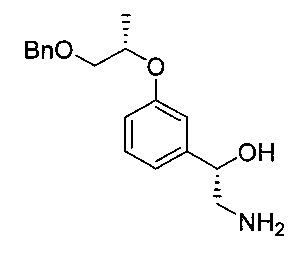
Указанное в заголовке соединение (13 г) получали по существу таким же способом, как описано для Стадии 4 Примера 2, за исключением того, что исходное соединение (S)-1-(3-(((S)-1-(бензилокси)пропан-2-ил)окси)фенил)-2-нитроэтан-1-ол (20 г, 60,4 ммоль) использовали вместо исходного соединения (1S)-1-(3-((1-(бензилокси)пропан-2-ил)окси)фенил)-2-нитроэтан-1-ола.
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.31 (3H, d, J=6,0 Гц), 1.88 (3H, brs), 2.77 (1Н, dd, J=12,6, 7,8 Гц), 2.96 (1Н, dd, J=13,0, 3,8 Гц), 3.54 (1Н, dd, J=8,2, 3,0 Гц), 3.65 (1Н, dd, J=10,2, 5,8 Гц), 4.57 (2Н, s), 4.56-4.62 (2Н, m), 6.82 (1Н, dd, J=8,2, 1,8 Гц), 6.90 (1Н, d, J=7,6 Гц), 6.95-6.96 (1Н, m), 7.22 (1Н, t, J=7,8 Гц), 7.35-7.25 (5Н, m).
[Стадия 5] Получение гидрохлорида (S)-1-(3-(((5)-1-(бензилокси)-пропан-2-ил)окси)фенил)-2-(дибензиламино)этан-1-ола
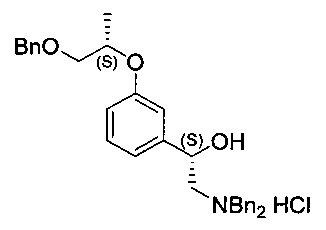
Указанное в заголовке соединение (10,4 г) получали по существу таким же способом, как описано для Стадии 5 Примера 2, за исключением того, что исходное соединение (S)-2-амино-1-(3-(((S)-1-(бензилокси)пропан-2-ил)окси)фенил)этан-1-ол (10,48 г, 34,8 ммоль) использовали вместо исходного соединения (1S)-2-амино-1-(3-((1-(бензилокси)-пропан-2-ил)окси)фенил)этан-1-ол.
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.27 (3H, d, J=6,0 Гц), 2.98-3.03 (1Н, m), 3.15-3.21 (1Н, m), 3.53 (1Н, dd, J=10,2, 4,6 Гц), 3.61 (1Н, dd, J=10,4, 6,0 Гц), 4.18 (1Н, dd, J=13,4, 6,2 Гц), 4.35 (1Н, dd, Л=13,2, 5,2 Гц), 4.48-4.55 (3H, m), 4.56 (2Н, s), 5.22 (1Н, d, J=9,6 Гц), 5.34 (1Н, brs), 6.64 (1Н, d, J=6,6 Гц), 6.72 (1Н, brs), 6.79 (1Н, dd, J=8,0, 2,4 Гц), 7.14 (1Н, t, J=7,8 Гц), 7.25-7.35 (5Н, m), 7.43-7.50 (6Н, m), 7.62-7.66 (2Н, m), 7.67-7.71 (2Н, m), 11.78 (1Н, brs).
[Стадия 6] Получение (S)-7-(((S)-1-(бензилокси)пропан-2-ил)окси)-3-((дибензиламино)метил)бензо[c][1,2]оксаборол-1(3H)-ола

Указанное в заголовке соединение (700 мг) получали по существу таким же способом, как описано для Стадии 6 Примера 2, за исключением того, что исходное соединение гидрохлорид (S)-1-(3-(((S)-1-(бензилокси)пропан-2-ил)окси)фенил)-2-(дибензиламино)этан-1-ол (7 г) использовали вместо исходного соединения гидрохлорида (1S)-1-(3-((1-(бензилокси)пропан-2-ил)окси)фенил)-2-(дибензиламино)этан-1-ола.
1H-ЯМР (CDCl3, Varian 400 МГц): δ 1.19 (3H, d, J=6,4 Гц), 1.60 (1Н, brs), 2.70 (1Н, dd, J=14,0, 7,6 Гц), 3.00 (1Н, dd, J=14,4, 3,6 Гц), 3.56 (1Н, dd, J=10,0, 2,8 Гц), 3.63 (1Н, dd, J=10,0, 8,0 Гц), 3.75 (2Н, d, J=13,6 Гц) 3.88 (2Н, d, J=13,6 Гц), 4.37- 4.41 (1Н, m), 4.66 & 4.72 (2Н, ABq, JAB=12,4 Гц), 5.34-5.38 (1Н, m), 6.70-6.84 (2Н, m), 7.20-7.39 (16Н, m).
[Стадия 7] Получение гидрохлорида ((2S,7S)-7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина

Указанное в заголовке соединение (250 мг) получали по существу таким же способом, как описано для Стадии 7 Примера 2, за исключением того, что исходное соединение (S)-7-(((S)-1-(бензил окси)пропан-2-ил)окси)-3-((дибензиламино)метил)бензо[с][1,2]оксаборол-1(3H)-ол (700 мг) использовали вместо исходного соединения (3S)-7-((1-(бензилокси)пропан-2-ил)окси)-3-((дибензиламино)метил)бензо[с][1,2]оксаборол-1(3H)-ола.
1H-ЯМР (DMSO-d6, Varian 400 МГц): δ 1.38 (3H, s), 2.82-2.91 (1Н, m), 3.44 (1Н, d, J=9,2 Гц), 4.14-4.24 (2Н, m), 4.42 (1Н, brs), 5.49-5.53 (1Н, m), 6.85 (1Н, d, J=5,2 Гц), 7.11 (1Н, d, J=4,8 Гц), 7.46 (1Н, t, J=5,2 Гц), 8.40 (3H, brs).
Хиральные свойства указанного в заголовке соединения анализировали в следующих условиях:

ЭКСПЕРИМЕНТАЛЬНЫЙ ПРИМЕР 1: антибактериальная активность in vitro
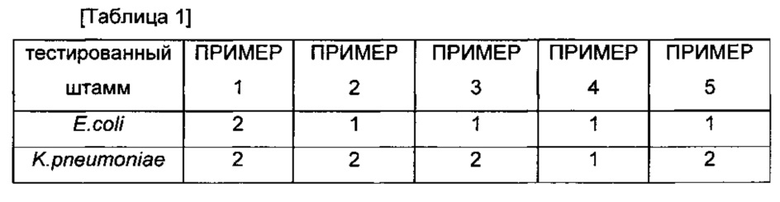
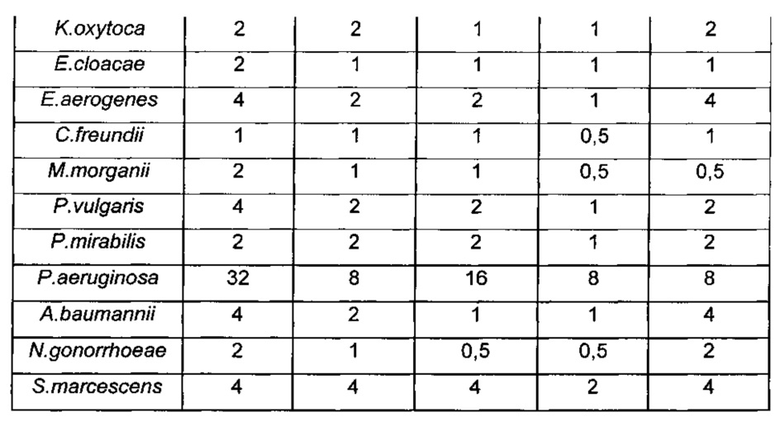
Антибактериальную активность новых производных, полученных в ПРИМЕРАХ 1-5, оценивали методом разведения на агаровых чашках in vitro с использованием агара Мюллера-Хинтона согласно NCCLS (Национальный Комитет по Клиническим Лабораторным Стандартам. 2000. Методы разведения в тестах на антимикробную чувствительность для бактерий, которые растут аэробно. Одобренный стандарт, NCCLS документ M7-A5, 5th ed, vol 20, no.2. National Committee for Clinical Laboratory Standards, Wayne, PA.). Тестированные штаммы представляли собой клинические изоляты от пациентов, находившихся в больнице общего профиля в Корее с 2010 года по 2013 год. Антибактериальную активность проверяли против грамотрицательных бактерий, включая устойчивый к карбапенему Acinetobacter baumannii (A. baumannii), устойчивый к карбапенему Pseudomonas aeruginosa (P. aeruginosa), Escherichia coli (E. coli) и Klebsiella pneumonia (K. pneumonia), и выражали в виде минимальных ингибирующих концентраций (MIC, мкг/мл) в Таблице 1.
Пример получения 1: Получение (7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина (соединение A)
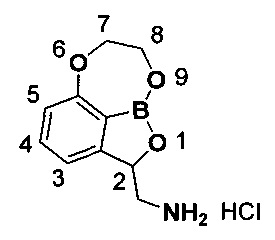
В качестве контрольного соединения, указанное в заголовке соединение A, 1,7 г, которое не имеет заместителя по положению 7 и 8, получили с использованием такого же способа, как описано в примере 24 в WO 2013/093615.
1H-ЯМР (DMSO-d6, Varian 400 МГц): δ 2.87-2.92 (1Н, m), 3.42-3.58 (1Н, m), 4.15-4.42 (3H, m), 4.62-4.76 (1Н, m), 5.45-5.87 (1Н, m), 6.92 (1Н, d, J=8,0 Гц), 7.15 (1Н, d, J=7,2 Гц), 7.50 (1Н, dd, J=8,0, 7,6 Гц), 8.26 (3H, brs).
Пример получения 2: Получение гидрохлорида (8-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]]азулен-2-ил)метанамина(соединение В)
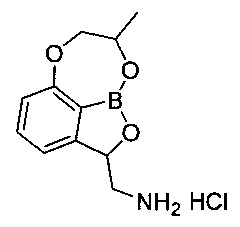
Указанное в заголовке соединение B, 45,0 мг, получили с использованием такого же способа, как описано в примере 9 в WO 2013/093615.
1H-ЯМР (CD3OD, Varian 400 МГц): δ 1.24-1.36 (3H, m), 2.88-3.00 (1Н, m), 3.55-3.64 (1Н, m), 4.14-4.23 (1Н, m), 4.46-4.57 (2Н, m), 5.42-5.48 (1Н, m), 6.86-6.96 (1Н, m), 7.06 (1Н, d, J=6,8 Гц), 7.48 (1Н, t, J=7,8 Гц).
Пример получения 3: Получение гидрохлорида ((2S,8R)-2-(аминометил)-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-8-ил)-метанола (соединение C)
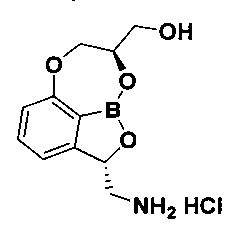
Указанное в заголовке соединение C, 50 мг, получили с использованием такого же способа, как описано в примере 3 в WO 2013/093615.
1H-ЯМР (DMSO-d6, Varian 400 МГц): δ 2.90-2.96 (1Н, m), 3.52-3.79 (3H, m), 4.03-4.36 (2Н, m), 4.72-4.75 (1Н, m), 5.01-5.18 (1Н, m), 5.49 (1Н, brs), 6.93 (1Н, d, J=7,6 Гц), 7.20 (1Н, d, J=7,2 Гц), 7.50 (1Н, t, J=7,8 Гц), 8.12 (3H, brs).
Пример получения 4: Получение гидрохлорида (2-(аминометил)-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-7-ил)-метанола (соединение D)
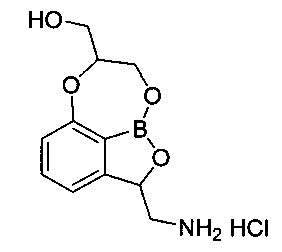
Указанное в заголовке соединение D, 12 мг, получили с использованием такого же способа, как описано в примере 11 в WO 2013/093615 предшествующего уровня техники.
1H-ЯМР (CD3OD, Varian 400 МГц): δ 2.97 (1Н, dd, J=12,8, 8,8 Гц), 3.52-3.62 (1Н, m), 3.72-3.92 (2Н, m), 4.18-4.28 (2Н, m), 4.42-4.52 (1Н, m), 5.46-5.52 (1Н, m), 6.95 (1Н, d, J=8,4 Гц), 7.05 (1Н, d, J=7,2 Гц), 7.49 (1Н, t, J=7,8 Гц).
ЭКСПЕРИМЕНТАЛЬНЫЙ ПРИМЕР 2: антибактериальная активность in vitro
Антибактериальную активность соединений (в Примерах получения 1-4) и меропенема (MEPM, антибиотик на основе карбапенема) проверяли в соответствии с такой же методикой, как показано в ЭКСПЕРИМЕНТАЛЬНОМ ПРИМЕРЕ 1. Антибактериальная активность (MIC, мкг/мл) в отношении грамотрицательных бактерий указана в Таблице 2.
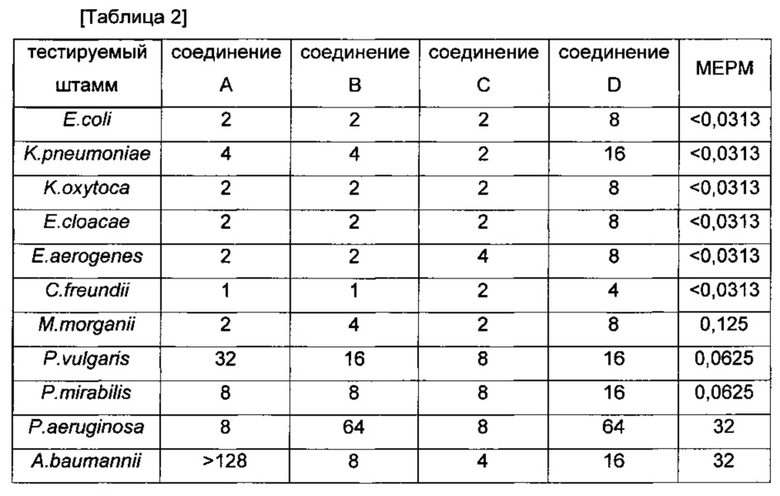
Как показано в Таблице 1 и Таблице 2, значения MIC трициклических бензоксабороловых соединений по настоящему изобретению были эквивалентны или ниже значений сравнительных соединений (соединение A, соединение B и соединение C). Трициклические бензоксабороловые соединения по настоящему изобретению продемонстрировали намного более превосходящие результаты MIC против многих грамотрицательных бактерий, по сравнению с соединением D, которое является сходным с соединением по настоящему изобретению, за исключением наличия гидроксиметила вместо метила по седьмому атому углерода.
Более того, в отношении Acinetobacter baumannii, который является типичным патогеном в больнице, соединения по настоящему изобретению продемонстрировали намного более превосходящие значения MIC по сравнению с соединением A, соединением B и соединением D, а также MEPM и показали результат MIC равный или выше чем для соединения С.Более конкретно, соединения ПРИМЕРОВ 2, 3 и 4 продемонстрировали более превосходящие значения MIC по сравнению с соединением С. Поскольку терапевтическая возможность ограничена в виду отсутствия эффективного антимикробного агента для инфекционного заболевания, вызываемого устойчивым к карбапенему Acinetobacter baumannii, трициклические бензоксабороловые соединения по настоящему изобретению могут представлять собой чрезвычайно эффективные терапевтические агенты против бактериального инфекционного заболевания.
ЭКСПЕРИМЕНТАЛЬНЫЙ ПРИМЕР 3: Антибактериальная активность в животной инфекционной модели
Антибактериальную активность соединений по настоящему изобретению исследовали in vivo в соответствии с методикой, описанной в S. Choi et al (Antimicrob. Agents Chemother 56 (9) 4713-4717. 2012).
Устойчивый к карбапенему Acinetobacter baumannii BAA-1605 использовали в качестве тестируемого штамма, и этот тестируемый штамм вводили мышам путем внутрибрюшинной инъекции для вызывания системной инъекции. Через 1 час инфицирования мышам вводили перорально соединения ПРИМЕРОВ 1, 2, 3, 5 и контрольные соединения. Затем наблюдали степени выживания на дозу в течение 7 суток и рассчитывали дозу, необходимую для степени выживания 50 процентов (ED50). Результат в отношении Acinetobacter baumannii BAA-1605 показан в Таблице 3.

Как показано в Таблице 3, пероральное введение соединений по настоящему изобретению продемонстрировало превосходный антибактериальный эффект в мышиной модели системной инфекции по сравнению с Соединением A, Соединением B и Соединением C. Значения ED50 соединений, полученных в ПРИМЕРАХ 1, 2 и 5, были в 5,18, 6,69 и 6,69 раз ниже, соответственно, чем значения ED50 для Соединения С. Значение ED50 для соединения ПРИМЕРА 3 было в 9,98 раз ниже значения ED50 для Соединения С.
На основании этих экспериментальных результатов, антибактериальный эффект соединений по настоящему изобретению против устойчивого к карбапенему Acinetobacter baumannii in vivo был намного более сильным, чем in vitro. Таким образом, соединения по настоящему изобретению могут быть чрезвычайно эффективными терапевтическими агентами против устойчивого к антибиотику бактериального инфекционного заболевания.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОНОЦИКЛИЧЕСКОЕ ПИРИДИНОВОЕ ПРОИЗВОДНОЕ | 2014 |
|
RU2645352C2 |
| Соединение, обладающее агонистической активностью в отношении GPR119, способ его получения и фармацевтическая композиция, содержащая его в качестве эффективного компонента | 2015 |
|
RU2670197C1 |
| Новые соединения пиридопиримидинона для модулирования каталитической активности гистонлизиндеметилаз (KDMS) | 2015 |
|
RU2684396C2 |
| БЕНЗОКСЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2557658C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ НЕЙРОДЕГЕНЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2008 |
|
RU2541430C2 |
| ПИПЕРИДИНОВЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЕЕ ПРИМЕНЕНИЕ | 2009 |
|
RU2514827C2 |
| ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ ХИНОЛИНА И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2012 |
|
RU2568258C2 |
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2021 |
|
RU2822180C1 |
| БИЦИКЛИЧЕСКИЕ ТЕТРАГИДРОТИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ НЕОПЛАСТИЧЕСКИХ И/ИЛИ ИНФЕКЦИОННЫХ ЗАБОЛЕВАНИЙ | 2016 |
|
RU2730524C2 |
| Производные изохинолинона, способ их получения и фармацевтическая композиция для профилактики или лечения заболеваний, связанных с поли(АДФ-рибоза)полимеразой-1, содержащая их в качестве активного ингредиента | 2020 |
|
RU2815480C1 |
Настоящее изобретение относится к новому трициклическому бензоксабороловому соединению, представленному химической формулой 1, его изомеру или их фармацевтически приемлемой соли. Химическая формула 1:

Также предложены способы получения указанного соединения и фармацевтическая композиция. Новое трициклическое бензоксабороловое соединение обладает антибиотической активностью в отношении грамотрицательных бактерий, в том числе в отношении грамотрицательных бактерий с множественной лекарственной устойчивостью. 4 н. и 10 з.п. ф-лы, 3 табл., 12 пр.
1. Трициклическое бензоксабороловое соединение, представленное химической формулой 1, его изомер или их фармацевтически приемлемая соль:
[Химическая формула 1]
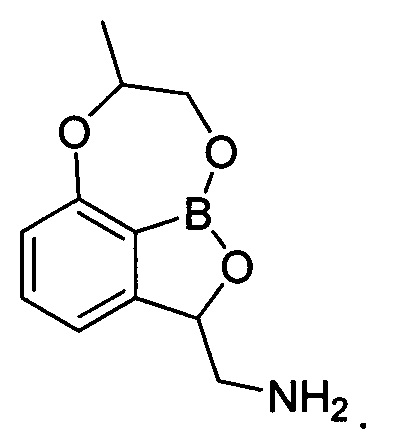
2. Трициклическое бензоксабороловое соединение, его изомер или их фармацевтически приемлемая соль по п. 1, где указанный изомер представлен химической формулой 2:
[Химическая формула 2]
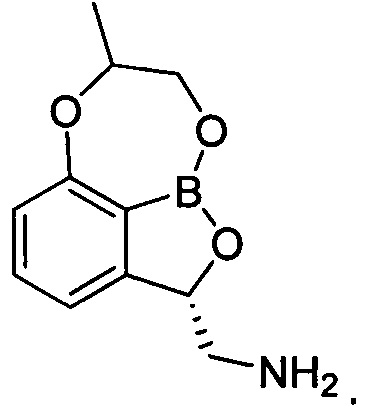
3. Трициклическое бензоксабороловое соединение или его фармацевтически приемлемая соль по п. 1, где указанный изомер представлен химической формулой 3:
[Химическая формула 3]
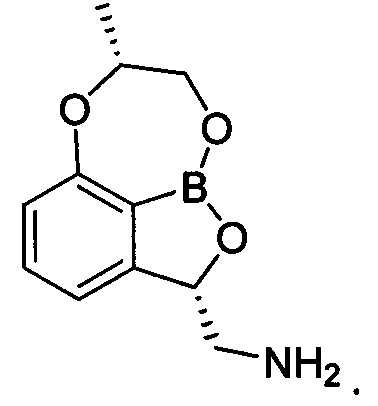
4. Трициклическое бензоксабороловое соединение, его изомер или их фармацевтически приемлемая соль по п. 1, где указанное соединение выбрано из группы, состоящей из:
1) гидрохлорида (7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина;
2) гидрохлорида ((2S)-7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина;
3) гидрохлорида ((2S,7R)-7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина;
4) ((2S,7R)-7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина; и
5) гидрохлорида ((2S,7S)-7-метил-7,8-дигидро-2Н-1,6,9-триокса-9а-борабензо[cd]азулен-2-ил)метанамина.
5. Трициклическое бензоксабороловое соединение, его изомер или их фармацевтически приемлемая соль по п. 1, где указанная фармацевтически приемлемая соль образована кислотой, выбранной из группы, состоящей из соляной кислоты, серной кислоты, азотной кислоты, фосфорной кислоты, бромоводородной кислоты, йодоводородной кислоты, винной кислоты, муравьиной кислоты, лимонной кислоты, уксусной кислоты, трихлоруксусной кислоты, трифторуксусной кислоты, глюконовой кислоты, бензойной кислоты, молочной кислоты, миндальной кислоты, фумаровой кислоты, малеиновой кислоты, салициловой кислоты, метансульфоновой кислоты, этансульфоновой кислоты, бензолсульфоновой кислоты и пара-толуолсульфоновой кислоты.
6. Способ получения трициклического бензоксаборолового соединения химической формулы 1 или его фармацевтически приемлемой соли, включающий стадии:
осуществления сочетания соединения химической формулы 4 и соединения химической формулы 5 с получением соединения химической формулы 6;
осуществления реакции борилирования соединения химической формулы 6 с получением соединения химической формулы 7 и затем осуществления цианирования с получением цианобензоксаборолового соединения химической формулы 8;
восстановления соединения химической формулы 8 до аминобензоксаборолового соединения с последующим введением защитной группы (PG2) в аминогруппу с получением соединения химической формулы 10, осуществления реакции конденсации соединения химической формулы 10 для удаления защитной группы (PG1) и затем осуществления реакции циклизации с последующим снятием защитной группы (PG2) с аминогруппы с получением соединения химической формулы 1,
[Химическая формула 4]
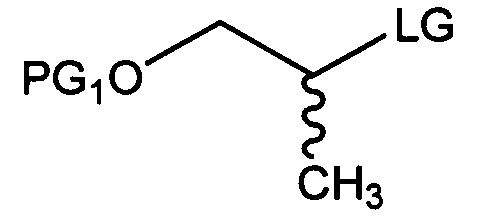
[Химическая формула 5]
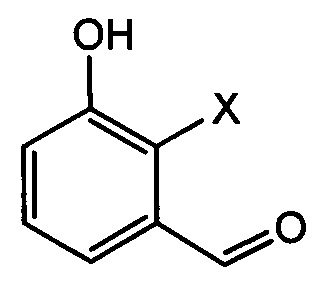
[Химическая формула 6]
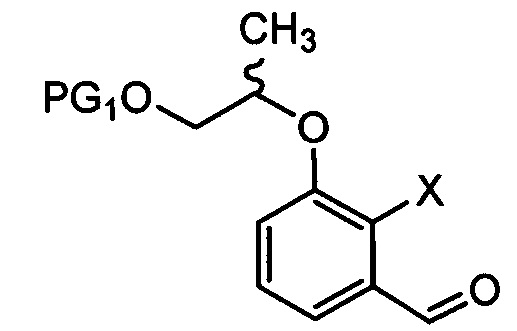
[Химическая формула 7]
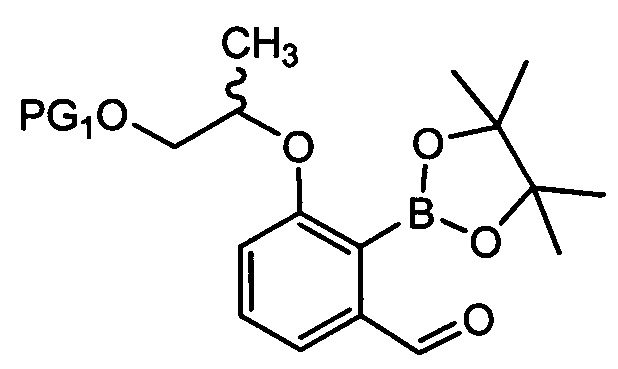
[Химическая формула 8]
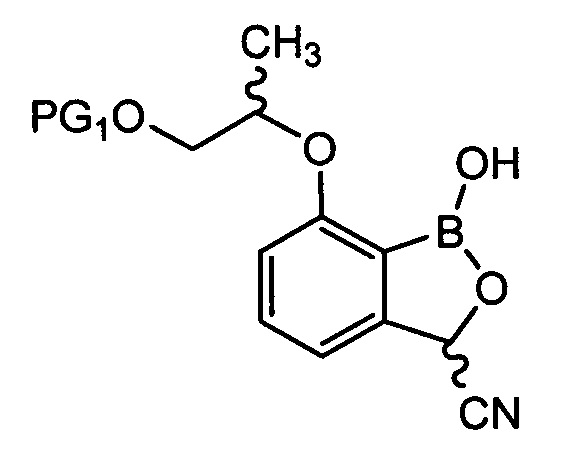
[Химическая формула 10]
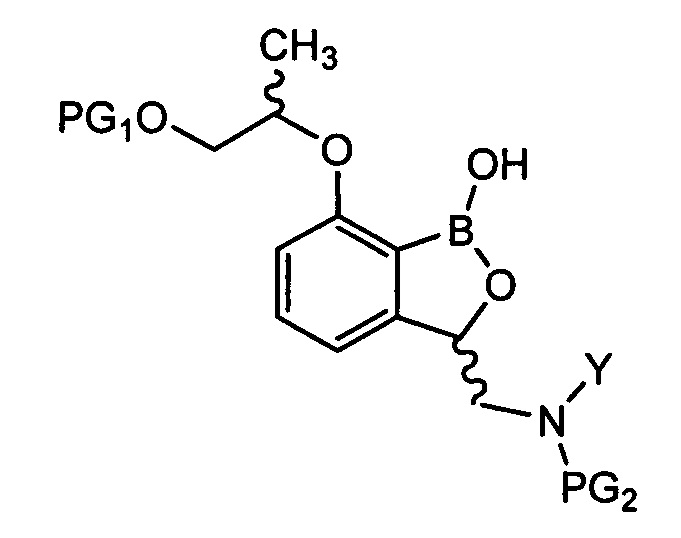
[Химическая формула 1]
где в химических формулах 4-8 и 10:
каждая из групп PG1 и PG2 независимо представляет собой бензил, трет-бутил, Boc (трет-бутилоксикарбонил), pmb (4-метоксибензил), Fmoc (флуоренилметилоксикарбонил), Ts (тозилат), MOM (метоксиметил), ТНР (тетрагидропиранил), TBDMS (трет-бутилдиметилсилил) или TBDPS (трет-бутилдиметилсилил),
LG представляет собой галоген, пара-толуолсульфонильную группу или метансульфонильную группу,
X представляет собой водород, галоген или трифторметансульфонил, и
Y представляет собой водород или PG2.
7. Способ получения трициклического бензоксаборолового соединения химической формулы 1, включающий стадии:
осуществления сочетания соединения химической формулы 4 и соединения химической формулы 5 с получением соединения химической формулы 6;
осуществления реакции нитрования соединения химической формулы 6 или осуществление реакции нитрования с использованием хирального лиганда или хирального катализатора с получением соединения химической формулы 12 или его изомеров, восстановления соединения химической формулы 12 с получением соединения химической формулы 13 путем замещения нитрогруппы аминогруппой;
введения защитной группы (PG2) в аминогруппу соединения химической формулы 13 с получением соединения химической формулы 14 и осуществления реакции борилирования соединения химической формулы 14 с получением бензоксаборолового соединения химической формулы 10;
осуществления реакции внутренней циклизации соединения химической формулы 10 путем удаления защитной группы (PG1) и затем осуществления реакции циклизации с последующим снятием защиты с аминогруппы с получением соединения химической формулы 1,
[Химическая формула 4]
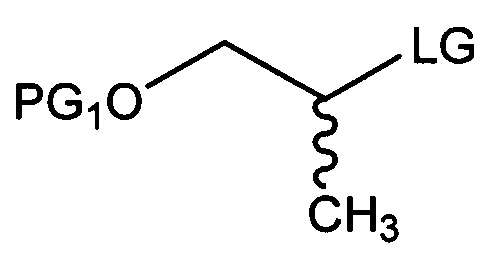
[Химическая формула 5]
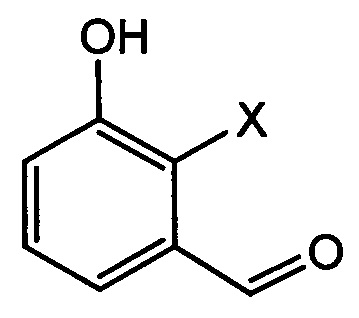
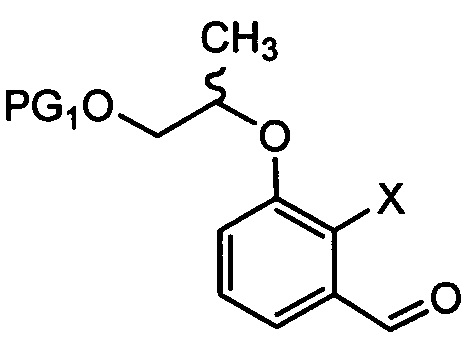
[Химическая формула 6]
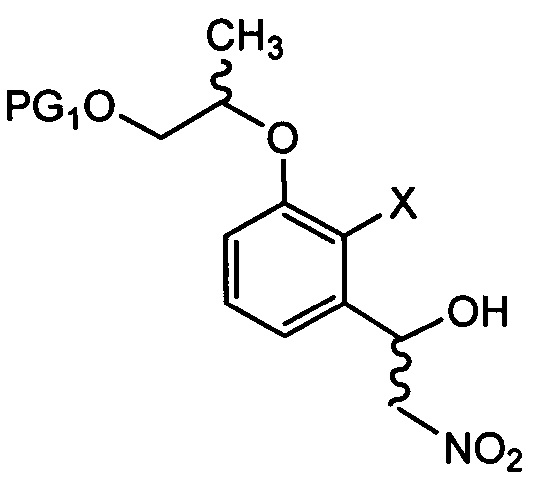
[Химическая формула 12]
[Химическая формула 13]
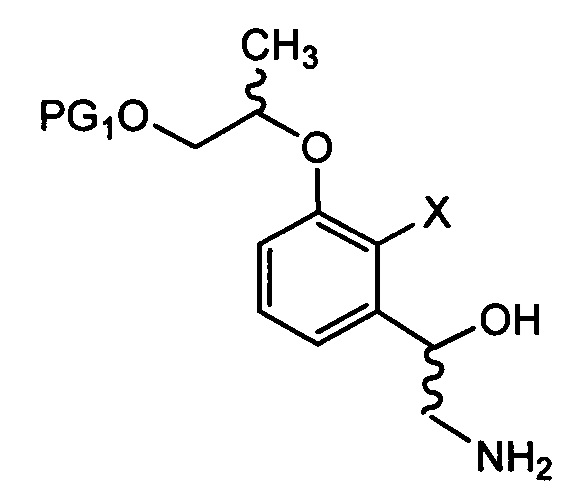
[Химическая формула 14]
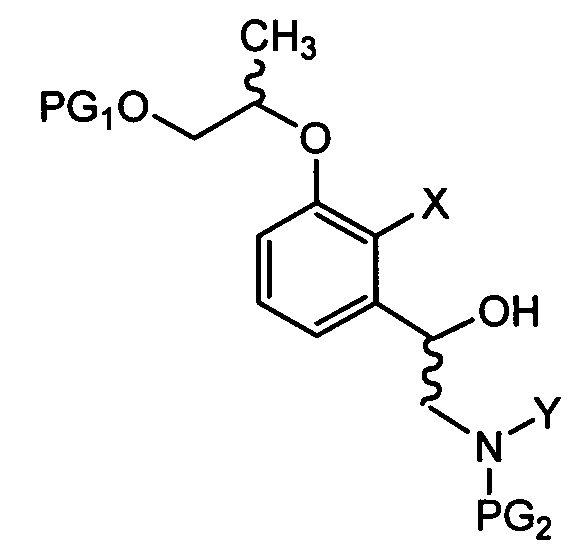
[Химическая формула 10]
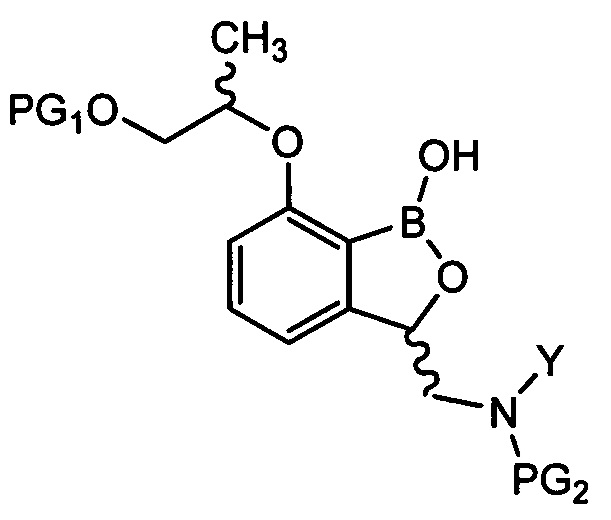
[Химическая формула 1]
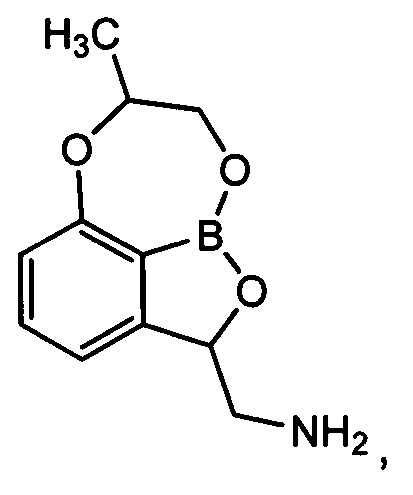
где в химических формулах 4-6 и 10 и 12-14:
каждая из групп PG1 и PG2 независимо представляет собой бензил, трет-бутил, Boc (трет-бутилоксикарбонил), pmb (4-метоксибензил), Fmoc (флуоренилметилоксикарбонил), Ts (тозилат), MOM (метоксиметил), ТНР (тетрагидропиранил), TBDMS (трет-бутилдиметилсилил) или TBDPS (трет-бутилдиметилсилил), LG представляет собой галоген, пара-толуолсульфонильную группу или метансульфонильную группу,
X представляет собой водород, галоген или трифторметансульфонил, и Y представляет собой водород или PG2.
8. Способ по п. 6 или 7, где реакцию борилирования осуществляют с использованием бис(пинаколато)дибора или 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолана.
9. Фармацевтическая композиция с антибиотической активностью в отношении грамотрицательных бактерий, содержащая соединение, его изомер или их фармацевтически приемлемую соль по любому из пп. 1-5 в качестве активного ингредиента.
10. Фармацевтическая композиция по п. 9, где грамотрицательная бактерия представляет собой грамотрицательную бактерию с множественной лекарственной устойчивостью.
11. Фармацевтическая композиция по п. 10, где грамотрицательная бактерия представляет собой грамотрицательную бактерию с устойчивостью к карбапенему.
12. Фармацевтическая композиция по п. 9, где грамотрицательная бактерия представляет A. baumannii, C. freundii, E. coli, E. cloacae, E. aerogenes, K. pneumoniae, K. oxytoca, M. morganii, P. aeruginosa, P. vulgaris, P. mirabilis, N. gonorrhoeae или S. marcescens.
13. Фармацевтическая композиция по п. 12, где грамотрицательная бактерия представляет собой грамотрицательную бактерию с множественной лекарственной устойчивостью.
14. Фармацевтическая композиция по п. 13, где грамотрицательная бактерия представляет собой грамотрицательную бактерию с устойчивостью к карбапенему.
| US 2013165411 A1, 27.06.2013 | |||
| WO 2008157726 A1, 24.12.2008 | |||
| WO 2007095638 A2, 23.08.2007 | |||
| АНТИБИОТИКИ, СОДЕРЖАЩИЕ КОМПЛЕКСЫ БОРИНОВОЙ КИСЛОТЫ, И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2397986C2 |

