Область техники
[0001] Настоящее изобретение относится к моноциклическому пиридиновому производному, обладающему ингибиторной активностью по отношению к FGFR, или его фармацевтически приемлемой соли, а также к их применению в медицине.
[0002] Все ссылки, цитированные в настоящем документе, тем самым включены посредством ссылки во всей их полноте.
Предшествующий уровень техники
[0003] FGF (фактор роста фибробластов) известен как фактор роста, контролирующий ряд физиологических функций, таких как рост клеток, миграция клеток, клеточная инфильтрация, выживаемость клеток, индукция дифференцировки, заживление ран и ангиогенез.
FGF контролирует различные физиологические функции посредством рецепторов FGF (FGFR: FGFR1, FGFR2, FGFR3 и FGFR4), то есть рецепторных тирозинкиназ. Каждый FGFR включает в себя три типа доменов - внеклеточный домен, трансмембранный домен и внутриклеточный тирозинкиназный домен. При связывании FGF с внеклеточным доменом из FGFR образуется димер рецептора. После этого активируется внутриклеточная тирозинкиназа, а затем внутриклеточный сигнал передается, главным образом, посредством пути MAPK (митоген-активируемой протеинкиназы)/ERK (регулируемой внеклеточным сигналом киназы) или пути PI3K (фосфатидилинозитол-3-киназы)/Akt.
[0004] При этом сообщалось, что различные виды рака, такие как рак молочной железы, рак мочевого пузыря, EMS (8р11 миелопролиферативный синдром), рак желудка, эндометриальный рак и рак предстательной железы, возникают в результате индукции патологии сигнала FGF/FGFR, сопровождаемой усилением продуцирования FGF, амплификацией гена FGFR, сверхэкспрессией FGFR, продуцированием слитого белка FGFR, мутацией FGFR и т.п. (Непатентная литература 1). Более того, следующие заболевания были зарегистрированы как виды рака, сопровождаемые патологией сигнала FGF/FGFR: немелкоклеточная карцинома легкого, мелкоклеточная карцинома легкого, рак яичника, саркома, рак толстой кишки, меланома, глиобластома, астроцитома и рак головы и шеи (непатентная литература 2 и 3), рак щитовидной железы (непатентная литература 4), рак поджелудочной железы (непатентная литература 5 и 6), рак печени (непатентная литература 7), рак кожи (непатентная литература 8), рак почки (непатентная литература 9) и плоскоклеточная карцинома легкого (непатентная литература 10, 11 и 12).
[0005] Кроме того, сигнал FGF/FGFR является одним из главных ангиогенных сигналов в эндотелиальных клетках вместе с сигналом VEGF (фактор роста эндотелия сосудов)/KDR (рецептор, имеющий в составе домен, содержащий киназу), и, как сообщалось, вовлекается во взаимодействие раковых стромальных клеток (фибробластов) и раковых клеток (непатентная литература 1).
Следовательно, предполагается, что ингибитор FGFR, нацеливающийся на сигнал FGF/FGFR, работает как противоопухолевое лекарственное средство против видов рака, сопровождаемых патологией сигнала FGF/FGFR, основанное на его ингибиторной активности против патологии сигнала и его ингибиторной активности против ангиогенного сигнала. Недавно сообщалось про селективный ингибитор FGFR, рассматриваемый как нечувствительный к поражению противостоящим эффектом другого сигнала, например, селективный ингибитор FGFR против FGFR1, FGFR2 или FGFR3, который очевидно отличается по структуре от соединения в соответствии с настоящим изобретением. При разработке в качестве противоопухолевого лекарственного средства для людей, однако, селективный ингибитор FGFR остается позади противоопухолевого лекарственного средства, одновременно нацеливающегося и на сигнал FGF/FGFR, и на сигнал VEGF/KDR, и еще не был представлен на рынке (непатентная литература 13 и 14; патентная литература 1 и 2). В патентной литературе 3 раскрываются пиримидиновые производные, но не раскрывается ингибиторная активность против сигнальной патологии сигнала FGF/FGFR. В патентной литературе 4 раскрываются пиридиновые производные или пиримидиновые производные, которые ингибируют ангиогенез, индуцируемый с помощью VEGF и FGF. Ни в одном из этих литературных источников, однако, не раскрываются соединения в соответствии с настоящим изобретением.
Перечень цитированных источников
Патентная литература
[0006]
[Патентная литература 1] Публикация международной заявки № WO 2008/075068.
[Патентная литература 2] Публикация международной заявки № WO 2006/000420.
[Патентная литература 3] Публикация международной заявки № WO 2002/032872.
[Патентная литература 4] Публикация международной заявки № WO 2004/020434.
Непатентная литература
[0007]
[Непатентная литература 1] Nicholas et al., "Fibroblast growth factor signalling: from development to cancer", Nature Reviews Cancer. 2010; 10: 116-129.
[Непатентная литература 2] Joergen WESCHE et al., Fibroblast growth factors and their receptors in cancer, Biochem J. 2011: 437; 199-213.
[Непатентная литература 3] Gennaro Daniele et al., FGF Receptor Inhibitors: Role in Cancer Therapy, Curr Oncol Rep. 2012; 14: 111-119.
[Непатентная литература 4] Rosanne St. Bernard et al., Fibroblast Growth Factor Receptors as Molecular Targets in Thyroid Carcinoma, Endocrinology. 2005; 146: 1145-1153.
[Непатентная литература 5] Toshiyuki Ishiwata et al., Enhanced Expression of Fibroblast Growth Factor Receptor 2 IIIс Promotes Human Pancreatic Cancer Cell Proliferation, Am J Pathol. 2012; 180: 1928-1941.
[Непатентная литература 6] G Chen et al., Inhibition of endogenous SPARC enhances pancreatic cancer cell growth: modulation by FGFR1-III isoform expression, Br J Cancer. 2010; 102: 188-195.
[Непатентная литература 7] Dorothy M. French et al., Targeting FGFR4 Inhibits Hepatocellular Carcinoma in Preclinical Mouse Models, PLoS One. 2012; 7: е36713.
[Непатентная литература 8] Armelle Logie et al., Activating mutations of the tyrosine kinase receptor FGFR3 are associated with benign skin tumors in mice and humans, Hum Mol Genet 2005; 14: 1153-1160.
[Непатентная литература 9] Tsimafeyeu I et al., Overexpression of fibroblast growth factor receptors FGFR1 and FGFR2 in renal cell carcinoma, Scand J Urol Nephrol 2011; 45: 190-195.
[Непатентная литература 10] Jonathan Weiss et al., Frequent and Focal FGFR1 Amplification Associates with Therapeutically Tractable FGFR1 Dependency in Squamous Cell Lung Cancer, Sci Transi Med. 2010; 2: issue 62 62-93.
[Непатентная литература 11] Hidefumi Sasaki et al., Increased FGFR1 copy number in lung squamous cell carcinomas, Mol Med Report. 2012; 5: 725-728.
[Непатентная литература 12] The Cancer Genome Atlas Research Network, Comprehensive genomic characterization of squamous cell lung cancers, Nature 2012; 489: 519-525.
[Непатентная литература 13] Paul R Gavine et al., AZD4547: An Orally Bioavailable, Potent, and Selective Inhibitor of the Fibroblast Growth Factor Receptor Tyrosine Kinase Family, Cancer Res. 2012; 72: 2045-2056.
[Непатентная литература 14] Vito Guagnano et al., Discovery of 3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyri midin-4-yl}-1-methyl-urea (NVP-BGJ398), A Potent and Selective Inhibitor of the Fibroblast Growth Factor Receptor Family of Receptor Tyrosine Kinase, J Med Chem. 2011; 54: 7066-7083.
Краткое описание изобретения
Техническая задача
[0008] При этих обстоятельствах целью настоящего изобретения является обеспечение нового соединения с ингибиторной активностью по отношению к FGFR или его фармацевтически приемлемой соли и фармацевтической композиции, содержащей его.
Решение задачи
[0009] Авторы настоящего изобретения провели серьезные исследования с учетом вышеупомянутых обстоятельств, и в результате удалось синтезировать новое моноциклическое пиридиновое производное, представленное следующей формулой (IA) (далее называемое соединением (IA) в соответствии с настоящим изобретением), и было обнаружено, что такое соединение обладает ингибиторной активностью по отношению к FGFR1 и ингибиторной активностью по отношению к FGFR2, и, таким образом, создали настоящее изобретение. Более того, авторы настоящего изобретения обнаружили, что соединение (IA) в соответствии с настоящим изобретением обладает действием селективного ингибитора сигнала FGF/FGFR по сравнению с сигналом VEGF/KDR, в частности, селективной ингибиторной активностью по отношению к FGFR1, FGFR2 или FGFR3.
[Химическая формула 1]

[0010] В частности, настоящее изобретение предусматривает следующее [1] - [26].
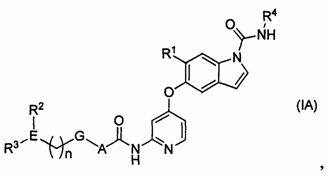
[1] Соединение, представленное следующей формулой (IA), или его фармацевтически приемлемая соль:
[Химическая формула 2]
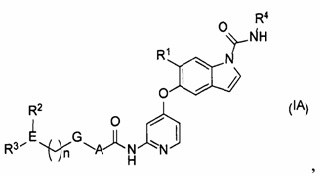
где
n равняется 0-2;
А представляет собой С6-10ариленовую группу или С3-5гетероариленовую группу;
G представляет собой одинарную связь, атом кислорода или -CH2-;
Е представляет собой содержащий азот неароматический С3-5гетероцикл;
R1 представляет собой цианогруппу, моно-C1-6алкиламиногруппу, ди-С1-6алкиламиногруппу, С2-6алкильную группу, необязательно замещенную 1-3 атомами галогена, C1-6алкоксигруппу, необязательно замещенную 1-3 атомами галогена, или одну гидроксильную группу, С1-6алкоксиС1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, или С1-6алкоксиС1-6алкоксигруппу, необязательно замещенную 1-3 атомами галогена;
R2 представляет собой атом водорода, атом галогена, гидроксильную группу, С2-6ацильную группу, необязательно замещенную одним заместителем, выбранным из группы S, описанной ниже, C1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, пздроксиС1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, или содержащую азот неароматическую С3-5 гетероциклическую группу;
R3 представляет собой атом водорода, оксогруппу, C1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, или C1-6алкоксигруппу, необязательно замещенную 1-3 атомами галогена;
R4 представляет собой C1-6алкильную группу при условии что, если Е представляет собой азетидиновое кольцо, а R2 или R3 находится на атоме азота в азетидиновом кольце, то R2 или R3 не является атомом водорода; и
группа S представляет собой группу, состоящую из гидроксильной группы, моно-С1-6алкиламиногруппы, ди-С1-6алкиламиногруппы, C1-6алкоксигруппы и содержащей азот неароматической С3-5 гетероциклической группы.
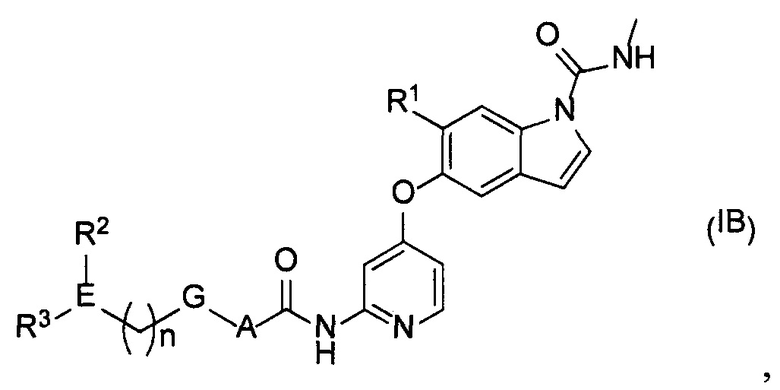
[2] Соединение или его фармацевтически приемлемая соль согласно [1], представленные следующей формулой (IB):
[Химическая формула 3]

где
n равняется 0-2;
А представляет собой С6-10ариленовую группу или С3-5гетероариленовую группу;
G представляет собой одинарную связь, атом кислорода или -СН2-;
Е представляет собой содержащий азот неароматический С3-5гетероцикл;
R1 представляет собой C1-6алкоксигруппу, необязательно замещенную 1-3 атомами галогена или одной гидроксильной группой, или С1-6алкоксиС1-6алкоксигруппу, необязательно замещенную 1-3 атомами галогена;
R2 представляет собой атом водорода, атом галогена, гидроксильную группу, C1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, гидрокси C1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, или содержащую азот неароматическую С3-5 гетероциклическую группу; и
R3 представляет собой атом водорода, оксогруппу, C1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, или C1-6алкоксигруппу, необязательно замещенную 1-3 атомами галогена при условии что, если Е представляет собой азетидиновое кольцо, а R2 или R3 находится на атоме азота в азетидиновом кольце, то R2 или R3 не является атомом водорода.
[3] Соединение или его фармацевтически приемлемая соль согласно [1], представленные следующей формулой (IB):
[Химическая формула 4]

где
n равняется 0-2;
А представляет собой С6-10ариленовую группу или С3-5гетероариленовую группу;
G представляет собой одинарную связь, атом кислорода или -СН2-;
Е представляет собой содержащий азот неароматический С3-5гетероцикл;
R1 представляет собой C1-6алкоксигруппу, необязательно замещенную 1-3 атомами галогена, или C1-6алкоксиC1-6алкоксигруппу, необязательно замещенную 1-3 атомами галогена;
R2 представляет собой атом водорода, атом галогена, гидроксильную группу, C1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, гидрокси C1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, или содержащую азот неароматическую С3-5 гетероциклическую группу; и
R3 представляет собой атом водорода, C1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, или C1-6алкоксигруппу, необязательно замещенную 1-3 атомами галогена при условии, что, если Е представляет собой азетидиновое кольцо, a R2 или R3 находится на атоме азота в азетидиновом кольце, то R2 или R3 не является атомом водорода.
[4] Соединение или его фармацевтически приемлемая соль согласно любой из [1]-[3], где А представляет собой С6-10ариленовую группу.
[5] Соединение или его фармацевтически приемлемая соль согласно любой из [1]-[4], где G представляет собой одинарную связь или атом кислорода.
[6] Соединение или его фармацевтически приемлемая соль согласно любой из [1]-[3], где
А представляет собой фениленовую группу, тиениленовую группу, пиразолиленовую группу или пиридиленовую группу; и
Е представляет собой азетидиновое кольцо, пирролидиновое кольцо, пиперидиновое кольцо или пиперазиновое кольцо.
[7] Соединение или его фармацевтически приемлемая соль согласно любой из [1]-[3], где
А представляет собой фениленовую группу; и
Е представляет собой азетидиновое кольцо или пиперидиновое кольцо.
[8] Соединение или его фармацевтически приемлемая соль согласно любой из [1]-[3], где
А представляет собой фениленовую группу; и
Е представляет собой пиперидиновое кольцо.
[9] Соединение или его фармацевтически приемлемая соль согласно любой из [6]-[8], где
n равняется 0; и
G представляет собой одинарную связь.
[10] Соединение или его фармацевтически приемлемая соль согласно любой из [1]-[9], где
R1 представляет собой C1-6алкоксигруппу или С1-6алкоксиС1-6алкоксигруппу;
R2 представляет собой атом водорода, гидроксильную группу, C1-6алкильную группу или гидроксиС1-6алкильную группу; и
R3 представляет собой атом водорода.
[11] Соединение или его фармацевтически приемлемая соль согласно любой из [1]-[10], где R1 представляет собой С1-6алкоксиС1-6алкоксигруппу.
[12] Соединение или его фармацевтически приемлемая соль согласно любой из [1]-[3], представленное следующей формулой (II):
[Химическая формула 5]

где
R1 представляет собой С1-6алкоксиС1-6алкоксигруппу; и
R2 представляет собой атом водорода, C1-6алкильную группу или гидроксиС2-6алкильную группу.
[13] Соединение или его фармацевтически приемлемая соль согласно любой из [1]-[3], представленное следующей формулой (III):
[Химическая формула 6]

где
R1 представляет собой С1-6алкоксиС1-6алкоксигруппу; и
R2 представляет собой C1-6алкильную группу или гидроксиС2-6алкильную группу.
[14]
5-((2-(4-(1-Этилпиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид, представленный следующей структурной формулой, или его фармацевтически приемлемая соль:
[Химическая формула 7]

[15] 6-(2-Метоксиэтокси)-N-метил-5-((2-(4-(1-метилпиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид, представленный следующей структурной формулой, или его фармацевтически приемлемая соль:
[Химическая формула 8]

[16]
5-((2-(4-(1-(2-Гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1Н-индол-1-карбоксамид, представленный следующей структурной формулой, или его фармацевтически приемлемая соль:
[Химическая формула 9]

[17] 6-(2-Этоксиэтокси)-5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-N-метил-1Н-индол-1-карбоксамид, представленный следующей структурной формулой, или его фармацевтически приемлемая соль:
[Химическая формула 10]

[18] 6-(2-Этоксиэтокси)-5-((2-(4-(1-этилазетидин-3-ил)бензамид)пиридин-4-ил)окси)-N-метил-1Н-индол-1-карбоксамид, представленный следующей структурной формулой, или его фармацевтически приемлемая соль:
[Химическая формула 11]
[19] Фармацевтическая композиция, содержащая соединение или его фармацевтически приемлемую соль согласно любой из [1]-[18].
[20] Терапевтическое средство против рака желудка, немелкоклеточной карциномы легкого, рака мочевого пузыря или рака эндометрия, содержащее соединение или его фармацевтически приемлемую соль согласно любой из [1]-[18] в качестве активного ингредиента.
[21] Способ лечения рака желудка, немелкоклеточной карциномы легкого, рака мочевого пузыря или рака эндометрия, включающий введение фармакологически эффективной дозы соединения или его фармацевтически приемлемой соли согласно любой из [1]-[18].
[22] Терапевтическое средство против немелкоклеточной карциномы легкого, содержащее соединение или его фармацевтически приемлемую соль согласно любой из [1]-[18] в качестве активного ингредиента.
[23] Терапевтическое средство против плоскоклеточной карциномы легкого, содержащее соединение или его фармацевтически приемлемую соль согласно любой из [1]-[18] в качестве активного ингредиента.
[24] Ингибитор FGFR для лечения немелкоклеточной карциномы легкого, содержащий соединение или его фармацевтически приемлемую соль согласно любой из [1]-[18] в качестве активного ингредиента.
[25] Соединение или его фармацевтически приемлемая соль согласно любой из [1]-[18] для применения в качестве терапевтического средства против рака желудка, немелкоклеточной карциномы легкого, рака мочевого пузыря или рака эндометрия.
[26] Применение соединения или его фармацевтически приемлемой соли согласно любой из [1]-[18] для изготовления терапевтического средства против рака желудка, немелкоклеточной карциномы легкого, рака мочевого пузыря или рака эндометрия.
Полезные эффекты настоящего изобретения
[0011] Соединение (IA) в соответствии с настоящим изобретением или его фармацевтически приемлемая соль обладает ингибиторной активностью по отношению к FGFR1, FGFR2 и FGFR3, как показано в данных активности, полученных в примерах фармакологических тестов, описанных ниже. Более того, соединение (IA) в соответствии с настоящим изобретением или его фармацевтически приемлемая соль обладает селективной ингибиторной активностью по отношению к FGFR1, FGFR2 или FGFR3, в отличие от ингибиторной активности по отношению к KDR или HUVEC. Следовательно, соединение (IA) в соответствии с настоящим изобретением или его фармацевтически приемлемая соль находит возможное применение для терапевтического средства против рака желудка, немелкоклеточной карциномы легкого, в том числе плоскоклеточной карциномы легкого, рака мочевого пузыря или рака эндометрия.
Описание вариантов осуществления
[0012] Далее настоящее изобретение описывается подробно путем определения символов и терминов, используемых в настоящем документе, и описания вариантов осуществления и т.п. настоящего изобретения.
[0013] В настоящем документе структурная формула соединения для удобства может представлять конкретный изомер, но соединение в соответствии с настоящим изобретением включает в себя изомеры, такие как все геометрические изомеры, структурно образованные из соединения, оптические изомеры на основе асимметричного углерода, стереоизомеры, ротамеры и таутомеры, а также смеси этих изомеров, и, следовательно, не ограничивается формулой, приведенной для удобства, но может быть любым из изомеров и смесей. Следовательно, соединение в соответствии с настоящим изобретением может иметь асимметричный(ые) атом(ы) углерода в молекуле и может быть оптически активным веществом и рацематом, и настоящее изобретение предусматривает все из упомянутого, но не ограничивается этим. Однако, следует понимать, что некоторые изомеры, рацематы и смеси изомеров могут демонстрировать более сильную активность, чем другие. Более того, может существовать кристаллический полиморфизм, который также не ограничивает настоящее изобретение, и соединение в соответствии с настоящим изобретением может иметь любую из форм монокристалла или смеси двух или более кристаллических форм, а также соединение в соответствии с настоящим изобретением включает в себя аморфную форму и охватывает ангидриды и сольваты, такие как гидрат.
[0014] Настоящее изобретение предусматривает меченное изотопом соединение (IА) в соответствии с настоящим изобретением и его фармацевтически приемлемую соль. Меченное изотопом соединение эквивалентно соединению, представленному формулой (IA) за исключением того, что один или несколько атомов замещены атомом(ами) с атомной массой или массовым числом, отличным от тех, которые обычно встречаются в природе. Примеры изотопа, который может быть встроен в соединение в соответствии с настоящим изобретением, включают в себя изотопы водорода, углерода, азота, кислорода, фосфора, фтора, йода, брома и хлора, такие как 2H, 3H, 11C, 13C, 14C, 18F, 35S, 123I и 125I.
[0015] Меченное изотопом соединение, такое как соединение, в которое встроен радиоактивный изотоп, например, 3H и/или 14С, применимо для анализа распределения в тканях, для медицинского препарата и/или матрицы. Изотопы 3H и 14С считаются применимыми, поскольку эти изотопы можно легко получать и выявлять. Изотопы 11С и 18F считаются применимыми для PET (позитрон-эмиссионной томографии); изотоп 125I считается применимым для SPECT (однофотонной эмиссионной компьютерной томографии); и могут быть применимы для картирования головного мозга. Замена на более тяжелый изотоп, такой как 2Н, обеспечивает за счет его более высокой метаболической стабильности некоторые преимущества в лечении, например, продление периода полувыведения in vivo или снижение необходимой дозы, и, поэтому, считается применимой при конкретных обстоятельствах. Меченное изотопом соединение можно получать аналогичным образом с использованием легкодоступного меченного изотопом реагента вместо немеченного изотопом реагента и путем осуществления способов, раскрытых в схемах и/или примерах, описанных ниже.
[0016] Соединение (IA) в соответствии с настоящим изобретением может быть использовано в качестве химического зонда для захвата целевого белка биологически активного низкомолекулярного соединения. В частности, соединение в соответствии с настоящим изобретением может быть превращено в зонд для аффинной хромагографии, фотоаффинный зонд и т.п. путем введения группы-метки, линкера или подобного в часть, отличную от структурной части, необходимой для проявления активности соединения, способом, описанным в J. Mass Spectrum. Soc. Jpn. Vol. 51, No. 5, 2003, p. 492-498, в WO 2007/139149 или подобном.
Примеры группы-метки, линкера или подобного, используемых в таком химическом зонде, включают в себя группы, принадлежащие следующим группам (1)-(5):
(1) группы-метки для белков, такие как фотоаффинные группы-метки (такие как бензоильная группа, бензофеноновая группа, азидогруппа, карбонилазидогруппа, диазиридиновая группа, еноновая группа, диазогруппа и нитрогруппа) и химические аффинные группы (такие как кетонная группа, в которой альфа-углеродный атом замещен атомом галогена, карбамоильная группа, сложноэфирная группа, алкилтиогруппа, рецептор Михаэля α,β-ненасыщенного кетона, сложного эфира и подобного, и оксирановая группа);
(2) отщепляемые линкеры, такие как -S-S-, -O-Si-O-, моносахарид (такой как глюкозная группа или галактозная группа) и дисахарид (такой как лактоза), а также олигопептидные линкеры, которые могут отщепляться ферментативной реакцией;
(3) группы с меткой для флуоресцентной in situ гибридизации, такие как биотин и 3-(4,4-дифтор-5,7-диметил-4Н-3а,4а-диаза-4-бора-s-индацен-3-ил)пропионильная группа;
(4) радиоактивные группы-метки, такие как 125I, 32Р, 3H и 14С; флуоресцентные группы-метки, такие как флуоресцеин, родамин, дансил, умбеллиферон, 7-нитрофуразанил и 3-(4,4-дифтор-5,7-диметил-4Н-3а,4а-дааза-4-бора-s-индецен-3-ил)пропионильная группа; хемилюминесцентные группы, такие как люциферин и люминол; а также выявляемые маркеры, такие как ионы тяжелых металлов, такие как ионы металлов-лантаноидов и ионы радия; и
(5) группы для связывания с твердофазным носителем, таким как стеклянные гранулы, стеклянная пластина, титрационный микропланшет, агарозные гранулы, агарозный слой, полистирольные гранулы, полистирольный слой, нейлоновые гранулы и нейлоновый слой.
[0018] Зонд, полученный при введении в соединение в соответствии с настоящим изобретением группы-метки и т.п., выбранной из описанных выше групп (1) - (5), способом, описанным в любом из вышеупомянутых литературных источников или подобном, может быть использован в качестве химического зонда для идентификации маркерного белка, применимого для исследования цели новых потенциальных лекарственных средств.
[0019] Используемый в настоящем документе "атом галогена" означает атом фтора, атом хлора, атом брома или атом йода.
[0020] Используемый в настоящем документе "гетероатом" означает атом азота, атом серы или атом кислорода.
[0021] Используемая в настоящем документе "C1-6алкильная группа" означает линейную или разветвленную алкильную группу с 1-6 атомами углерода, которая представляет собой одновалентную группу, получаемую удалением любого одного атома водорода из алифатического углеводорода с 1-6 атомами углерода. Примеры такой группы включают в себя метальную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, н-пентильную группу, изопентильную группу, втор-пентильную группу, неопентильную группу, 1-метилбутильную группу, 2-метилбутильную группу, 1,1-диметилпропильную группу, 1,2-диметилпропильную группу, н-гексильную группу, изогексильную группу, 1-метилпентильную группу, 2-метилпентильную группу, 3-метилпентильную группу, 1,1-диметилбутильную группу, 1,2-диметилбутильную группу, 2,2-диметилбутильную группу, 1,3-диметилбутильную группу, 2,3-диметилбутильную группу, 3,3-диметилбутильную группу, 1-этилбутильную группу, 2-этилбутильную группу, 1,1,2-триметилпропильную группу, 1,2,2-триметилпропильную группу, 1-этил-1-метилпропильную группу, 1-этил-2-метилпропильную группу и т.п. Более конкретно, она представляет собой метальную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу или подобное, и предпочтительно является метальной группой, этильной группой или изопропильной группой.
[0022] Используемая в настоящем документе "C1-6алкильная группа, необязательно замещенная 1-3 атомами галогена" означает вышеупомянутую C1-6алкильную группу, в которой любые 1-3 атома водорода могут быть замещены атомом галогена. Положение замещения атомом галогена специально не ограничивается, и конкретные примеры такой группы включают в себя монофторметильную группу, дифторметильную группу, трифторметильную группу, 2-фторэтильную группу, 2,2-дифторэтильную группу, 2,2,2-трифторэтильную группу, монохлорметильную группу, дихлорметильную группу, трихлорметильную группу, 2-хлорэтильную группу, 2,2-дихлорэтильную группу, 2,2,2-трихлорэтильную группу, 3-фторпропильную группу и т.п. В качестве атома галогена, используемого для замещения, предпочтительно используется, например, атом фтора, атом хлора или подобное, при этом атом фтора используется более предпочтительно.
[0023] Используемая в настоящем документе "С2-6алкильная группа" означает линейную или разветвленную алкильную группу с 2-6 атомами углерода, которая представляет собой одновалентную группу, получаемую удалением любого одного атома водорода из алифатического углеводорода с 2-6 атомами углерода. Примеры такой группы включают в себя этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, н-пентильную группу, изопентильную группу, втор-пентильную группу, неопентильную группу, 1-метилбутильную группу, 2-метилбутильную группу, 1,1-диметилпропильную группу, 1,2-диметилпропильную группу, н-гексильную группу, изогексильную группу, 1-метилпентильную группу, 2-метилпентильную группу, 3-метилпентильную группу, 1,1-диметилбутильную группу, 1,2-диметилбутильную группу, 2,2-диметилбутильную группу, 1,3-диметилбутильную группу, 2,3-диметилбутильную группу, 3,3-диметилбутильную группу, 1-этилбутильную группу, 2-этилбутильную группу, 1,1,2-триметилпропильную группу, 1,2,2-триметилпропильную группу, 1-этил-1-метилпропильную группу, 1-этил-2-метилпропильную группу. Более конкретно, она представляет собой этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, втор-бутильную группу или трет-бутильную группу, а предпочтительной является этильная группа.
[0024] Используемая в настоящем документе "С2-6алкильная группа, необязательно замещенная 1-3 атомами галогена" означает вышеупомянутую С2-6алкильную группу, в которой любые 1-3 атома водорода могут быть замещены атомом галогена. Положение замещения атомом галогена специально не ограничивается, и конкретные примеры такой группы включают в себя 2-фторэтильную группу, 2,2-дифторэтильную группу, 2,2,2-трифторэтильную группу, 2-хлорэтильную группу, 2,2-дихлорэтильную группу, 2,2,2-трихлорэтильную группу и 3-фторпропильную группу. В качестве атома галогена, используемого для замещения, предпочтительно используется, например, атом фтора, атом хлора или подобное, при этом атом фтора используется более предпочтительно.
[0025] Используемая в настоящем, документе "гидроксиC1-6алкильная группа" означает вышеупомянутую C1-6алкильную группу, в которой любой атом водорода замещен гидроксильной группой. Положение замещения гидроксильной группой специально не ограничивается, и конкретные примеры такой группы включают в себя гидроксиметильную группу, 2-гидроксиэтильную группу, 1-гидроксиэтильную группу, 3-гидроксипропильную группу, 2-гидроксипропильную группу, 1-гидроксипропильную группу, 2-гидрокси-2,2-диметилэтильную группу и т.п. Предпочтительной является 2-гидроксиэтильная группа, 2-гидроксипропильная группа или 2-гидрокси-2,2-диметилэтильная группа.
[0026] Используемая в настоящем документе "гидроксиС2-6алкильная группа" означает линейную или разветвленную алкильную группу с 2-6 атомами углерода, которая представляет собой одновалентную группу, получаемую удалением любого одного атома водорода из алифатического углеводорода с 2-6 атомами углерода, в котором любой атом водорода замещен гидроксильной группой. Положение замещения гидроксильной группой специально не ограничивается, и конкретные примеры такой группы включают в себя 2-гидроксиэтильную группу, 1-гидроксиэтильную группу, 3-гидроксипропильную группу, 2-гидроксипропильную группу, 1-гидроксипропильную группу, 2-гидрокси-2,2-диметилэтильную группу и т.п. Предпочтительной является 2-гидроксиэтильная группа, 2-гидроксипропильная группа или 2-гидрокси-2,2-диметилэтильная группа.
[0027] Используемая в настоящем документе "гидроксиC1-6алкильная группа, необязательно замещенная 1-3 атомами галогена" означает вышеупомянутую гидрокиС1-6алкильную группу, в которой любые 1-3 атома водорода могут быть замещены атомом галогена. Положение замещения атомом галогена специально не ограничивается. В качестве атома галогена, используемого для замещения, предпочтительно используется, например, атом фтора, атом хлора или подобное, при этом атом фтора используется более предпочтительно.
[0028] Используемая в настоящем документе "C1-6алкоксигруппа" означает вышеупомянутую "С1-6алкильную группу" с атомом кислорода, связанным с ее концом, и примеры такой группы включают в себя метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, н-бутоксигруппу, изобутоксигруппу, втор-бутоксигруппу, трет-бутоксигруппу, н-пентилоксигруппу, изопентилоксигруппу, втор-пентилоксигруппу, неопентилоксигруппу, 1-метилбутоксигруппу, 2-метилбутоксигруппу, 1,1-диметилпропоксшруппу, 1,2-диметилпропоксигруппу, н-гексилоксигруппу, изогексилоксигруппу, 1-метилпентилоксигруппу, 2-метилпентилоксигруппу, 3-метилпентилоксигруппу, 1,1-диметилбутоксигруппу, 1,2-диметилбутоксигруппу, 2,2-диметилбутоксигруппу, 1,3-диметилбутоксигруппу, 2,3-диметилбутоксигруппу, 3,3-диметилбутоксигруппу, 1-этилбутоксигруппу, 2-этилбутоксигруппу, 1,1,2-триметилпропоксигруппу, 1,2,2-триметилпропоксигруппу, 1-этил-1-метилпропоксигруппу, 1-этил-2-метилпропоксигруппу и т.п., и более конкретно представляет собой метоксигруппу этоксигруппу, н-пропоксигруппу, изопропоксигруппу, н-бутоксигруппу, изобутоксигруппу, втор-бутоксигруппу, н-пентилоксигруппу, изопентилоксигруппу, н-гексилоксигруппу, изогексилоксигруппу или подобное, и предпочтительной является метоксигруппа, этоксигруппа или изопропоксигруппа.
[0029] Используемая в настоящем документе "C1-6алкоксигруппа, необязательно замещенная 1-3 атомами галогена" означает вышеупомянутую С1-6алкоксигруппу, в которой любые 1-3 атома водорода могут быть замещены атомом галогена. Положение замещения атомом галогена специально не ограничивается, и конкретные примеры такой группы включают в себя монофторметоксигруппу, дифторметоксигруппу, трифторметоксигруппу, 2-фторэтоксигруппу, 2,2-дифторэтоксигруппу, 2,2,2-трифторэтоксигруппу, монохлорметоксигруппу, дихлорметоксигруппу, трихлорметоксигруппу, 2-хлорэтоксигруппу, 2,2-дихлорэтоксигруппу, 2,2,2-трихлорэтоксигруппу, 3-фторпропоксигруппу и т.п.
[0030] Используемая в настоящем документе "C1-6алкоксигруппа, необязательно замещенная одной гидроксильной группой" означает вышеупомянутую C1-6алкжсигруппу, в которой любой один атом водорода может быть замещен гидроксильной группой. Положение замещения гидроксильной группой специально не ограничивается, и конкретные примеры такой группы включают в себя 2-гидроксиэтоксигруппу, 2-гидроксипропоксигруппу и 3-гидроксипропоксигруппу. Предпочтительной является 2-гидроксиэтоксигруппа.
[0031] Используемая в настоящем документе "С1-6алкоксиС1-6алкоксигруппа" означает вышеупомянутую "C1-6алкоксигруппу", в которой любой один атом водорода замещен вышеупомянутой "C1-6алкоксигруппой", и конкретные примеры такой группы включают в себя метоксиметоксигруппу, этоксиметоксигруппу, н-пропоксиметоксигруппу, 2-метоксиэтоксигруппу, 2-этоксиэтоксигруппу, 3-метоксипропоксигруппу и т.п. Предпочтительной является 2-метоксиэтоксигруппа, 2-этоксиэтоксигруппа или 3-метоксипропоксигруппа.
[0032] Используемая в настоящем документе "С1-6алкоксиС1-6алкоксигруппа, необязательно замещенная 1-3 атомами галогена" означает вышеупомянутую "С1-6алкоксиС1-6алкоксигруппу", в которой любые 1-3 атома водорода могут быть замещены атомом галогена. Положение замещения атомом галогена специально не ограничивается, и конкретные примеры такой группы включают в себя монофторметоксиметоксигруппу, дифторметоксиметоксигруппу, монофторэтоксиметоксигруппу, дифторэтоксиметоксигруппу, монофторметоксиэтоксигруппу, трифторметоксиэтоксигруппу, дафторметоксиэтоксигруппу, монофторэтоксиэтоксигруппу, дифторэтоксиэтоксигруппу и т.п.
[0033] Используемая в настоящем документе "С1-6алкоксиС1-6алкильная группа" означает вышеупомянутую "С1-6алкильную группу", в которой любой один атом водорода замещен вышеупомянутой "С1-6алкоксигруппой", и конкретные примеры такой группы включают в себя метоксиметильную группу, этоксиметильную группу, н-пропоксиметильную группу, 2-метоксиэтильную группу, 2-этоксиэтильную группу и 3-метоксипропильную группу. Предпочтительной является метоксиметильная группа.
[0034] Используемая в настоящем документе "С1-6алкоксиС1-6алкильная группа, необязательно замещенная 1-3 атомами галогена" означает вышеупомянутую С1-6алкоксиС1-6алкильную группу, в которой любые 1-3 атома водорода могут быть замещены атомом галогена. Положение замещения атомом галогена специально не ограничивается, и конкретные примеры такой группы включают в себя монофторметоксиметильную группу, дифторметоксиметильную группу, монофторэтоксиметильную группу, дифторэтоксиметильную группу, монофторметоксиэтильную группу, трифторметоксиэтильную группу, дифторметоксиэтильную группу, монофторэтоксиэтильную группу и дифторэтоксиэтильную группу.
[0035] Используемая в настоящем документе "моно-C1-6алкиламиногруппа" означает аминогруппу, в которой один атом водорода замещен вышеупомянутой "С1-6алкильной группой", и конкретные примеры такой группы включают в себя метиламиногруппу, этиламиногруппу, н-пропиламиногруппу, изопропиламиногруппу, н-бутиламиногруппу, изобутиламиногруппу, втор-бутиламиногруппу, трет-бутиламиногруппу, н-пентиламиногруппу, изопентиламиногруппу, втор-пентиламиногруппу, неопентиламиногруппу, 1-метилбутиламиногруппу, 2-метилбутиламиногруппу, 1,1-диметилпропиламиногруппу, 1,2-диметилпропиламиногруппу, н-гексиламиногруппу, изогексиламиногруппу, 1-метилпентиламиногруппу, 2-метилпентиламиногруппу, 3-метилпентиламиногруппу, 1,1-диметилбутиламиногруппу, 1,2-диметилбутиламиногруппу, 2,2-диметилбутиламиногруппу, 1,3-диметилбутиламиногруппу, 2,3-диметилбутиламиногруппу, 3,3-диметилбутиламиногруппу, 1-этилбутиламиногруппу, 2-этилбутиламиногруппу, 1,1,2-триметилпропиламиногруппу, 1,2,2-триметилпропиламиногруппу, 1-этил-1-метилпропиламиногруппу и 1-этил-2-метилпропиламиногруппу, а предпочтительной является метиламиногруппа или подобное.
[0036] Используемая в настоящем документе "ди-С1-6алкиламиногруппа" означает аминогруппу, в которой каждый из двух атомов водорода замещен одинаковыми или отличными "С1-6алкильными группами", определенными выше, и конкретные примеры такой группы включают в себя N,N-диметиламиногруппу, N,N-диэтиламиногруппу, N,N-ди-н-пропиламиногруппу, N,N-ди-изопропиламиногруппу, N,N-ди-н-бутиламиногруппу, N,N-ди-изобутиламиногруппу, N,N-ди-втор-бутиламиногруппу, N,N-ди-трет-бутиламиногруппу, N-этил-N-метиламиногруппу, N-н-пропил-N-метиламиногруппу, N-изопропил-N-метиламиногруппу, N-н-бутил-N-метиламиногруппу, N-изобутил-N-метиламиногруппу, N-втор-бутил-N-метиламиногруппу и N-трет-бутил-N-метиламиногруппу, а предпочтительной является N,N-диметиламиногруппа или подобное.
[0037] Используемая в настоящем документе "С2-6ацильная группа" означает группу, содержащую группу атомов, получаемую путем исключения группы ОН из карбоксильной группы алифатической карбоновой кислоты с 2-6 атомами углерода, и конкретные примеры такой группы включают в себя ацетильную группу, пропионильную группу и бутилоильную группу.
[0038] Используемая в настоящем документе "оксогруппа" означает заместитель, в мотором атом кислорода связывается с атомом углерода или атомом азота, и конкретный пример структуры, в которой атом кислорода связывается с атомом углерода, включает в себя карбонильную группу, и конкретный пример группы, в которой атом кислорода связывается с атомом азота включает в себя N-оксид.
[0039] Используемая в настоящем документе "С3-5гетероариленовая группа" означает двухвалентную группу с 3-5 атомами углерода, образующими кольцо, которое получено путем удаления любых 2 атомов водорода из гетероароматического соединения с 1-2 гетероатомами в качестве атомов, образующих кольцо, и конкретные примеры такой группы включают в себя фуриленовую группу, тиениленовую группу, пирролиленовую группу, имидазолиленовую группу, тиазолиленовую группу, пиразолиленовую группу, оксазолиленовую группу, изооксазолиленовую группу, изотиазолиленовую группу, фуразаниленовую группу, пиридиленовую группу, пиразиниленовую группу, пиридазиниленовую группу, пиримидиниленовую группу и т.п., и предпочтительной является пиридиленовая группа, пиразолиленовая группа или тиениленовая группа.
[0040] Используемая в настоящем документе "Содержащая азот неароматическая С3-5 гетероциклическая группа" означает одновалентную неаромагическую циклическую группу с 3-5 атомами углерода, образующими кольцо, и с 1-2 атомами азота среди атомов, образующих кольцо, и конкретные примеры такой группы включают в себя азетидинильную группу, пирролидинильную группу, пиразолидинильную группу, имидазолидинильную группу, пиперидинильную группу, пиперазинильную группу, изооксазолидинильную группу, изотиазолидинильную группу, морфолинильную группу, тиоморфолинильную группу и т.п.
[0041] Используемый в настоящем документе "Содержащий азот неароматический С3-5 гетероцикл" означает неароматическое кольцо с 3-5 атомами углерода, образующими кольцо, и с 1-2 атомами азота среди атомов, образующих кольцо, и конкретные примеры такой группы включают в себя азетидиновое кольцо, пирролидиновое кольцо, пиразолидиновое кольцо, имидазолидиновое кольцо, пиперидиновое кольцо, пиперазиновое кольцо, изооксазолидиновое кольцо, изотиазолидиновое кольцо, морфолиновое кольцо, тиоморфолиновое кольцо и т.п.
[0042] Используемая в настоящем документе "С6-10ариленовая группа" означает двухвалентную группу, полученную путем удаления любых двух атомов водорода из ароматического углеводорода с 6-10 атомами углерода, и конкретные примеры такой группы включают в себя фениленовую группу, нафтиленовую группу, индениленовую группу, азупениленовую группу, гепталениленовую группу и т.п., и предпочтительной является фениленовая группа.
[0043] В используемой в настоящем документе формуле n равняется 0-2. Предпочтительно n равняется 0 или 1 и более предпочтительно n равняется 0.
[0044] В используемой в настоящем документе формуле А представляет собой С6-10ариленовую группу или С3-5гетероариленовую группу, предпочтительно фениленовую группу, тиениленовую группу, пиридиленовую группу или пиразолиленовую группу и более предпочтительно фениленовую группу.
[0045] В используемой в настоящем документе формуле G представляет собой одинарную связь, атом кислорода или -СН2-, предпочтительно одинарную связь или атом кислорода и более предпочтительно одинарную связь.
[0046] В используемой в настоящем документе формуле Е представляет собой содержащий азот неароматический С3-5 гетероцикл, описанный выше, и конкретно, например, азетидиновое кольцо, пирролидиновое кольцо, пиперидиновое кольцо или пиперазиновое кольцо, предпочтительно азетидиновое кольцо или пиперидиновое кольцо и более предпочтительно пиперидиновое кольцо.
[0047] В используемой в настоящем документе формуле R1 представляет собой цианогруппу, моно-C1-6 алкиламиногруппу, ди-С1-6алкиламиногруппу, С2-6алкильную группу, необязательно замещенную 1-3 атомами галогена, C1-6 алкоксигруппу, необязательно замещенную 1-3 атомами галогена или одной гидроксильной группой, С1-6алкоксиС1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, или С1-6алкоксиС1-6алкоксигруппу, необязательно замещенную 1-3 атомами галогена. Предпочтительной является С1-6алкоксигруппа или С1-6алкоксиС1-6алкоксигруппа. Конкретные примеры такой группы включают в себя цианогруппу, метиламиногруппу, N,N-диметиламиногруппу, этильную группу, н-пропильную группу, изопропильную группу, метоксигруппу, этоксигруппу, н-пропоксигруппу, изопропоксигруппу, н-бутоксигруппу, изобутоксигруппу, втор-бутоксигруппу, н-пентилоксигруппу, изопентилоксигруппу, н-гексилоксигруппу, изогексилоксигруппу, монофторметоксигруппу, дифторметоксигруппу, трифторметоксигруппу, 2-фторэтоксигруппу, 2,2-дифторэтоксигруппу, 3-фторпропоксигруппу, 2-гидроксиэтоксигруппу, метоксиметильную группу, этоксиметильную группу, н-пропоксиметильную группу, этоксиметоксигруппу, н-пропоксиметоксигруппу, 2-метоксиэтоксигруппу, 2-этоксиэтоксигруппу, 3-метоксипропоксигруппу, монофторметоксиметоксигруппу, дифторметоксиметоксигруппу, монофторэтоксиметоксигруппу, дафторэтоксиметоксигруппу, монофторметоксиэтоксигруппу, дифторметоксиэтоксигруппу, трифторметоксиэтоксигруппу, монофторэтоксиэтоксигруппу и дифторэтоксиэтоксигруппу. Предпочтительные примеры включают в себя цианогруппу, N,N-диметиламиногруппу, этильную группу, метоксигруппу, этоксигруппу, изопропоксигруппу, 3-фторпропоксигруппу, 2-гидроксиэтоксигруппу, метоксиметильную группу, 2-метоксиэтоксигруппу, 2-этоксиэтоксигруппу и 3-метоксипропоксигруппу, среди которых метоксигруппа, 2-метоксиэтоксигруппа, 2-этоксиэтоксигруппа и т.п. являются предпочтительными, а 2-метоксиэтоксигруппа и 2-этоксиэтоксигруппа являются более предпочтительными.
[0048] В используемой в настоящем документе формуле R2 представляет собой атом водорода, атом галогена, гидроксильную группу, С2-6ацильную группу, необязательно замещенную одним заместителем, выбранным из группы S, C1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, гидроксиС1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, или содержащую азот неароматическую С3-5 гетероциклическую группу. В настоящем документе группа S представляет собой группу, состоящую из гидроксильной группы, моно-C1-6алкиламиногруппы, ди-С1-6алкиламиногруппы, С1-6алкоксигруппы и содержащей азот неароматической С3-5 гетероциклической группы. Предпочтительными являются атом водорода, атом галогена, гидроксильная группа, C1-6алкильная группа или гидроксиС1-6алкильная группа, и более предпочтительными являются атом водорода, гидроксильная группа, C1-6алкильная группа или гидроксиС1-6алкильная группа. Конкретные примеры R2 включают в себя атом водорода, атом фтора, атом хлора, атом брома, атом йода, гидроксильную группу, метальную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутальную группу, изобутильную группу, втор-бутильную группу, трет-бутильную группу, монофторметильную группу, дифторметильную группу, трифторметильную группу, 2-фторэтильную группу, 2,2-дифторэтильную группу, 2,2,2-трифторэтильную группу, монохлорметильную группу, дихлорметильную группу, трихлорметильную группу, 2-хлорэтильную группу, 2,2-дихлорэтильную группу, 2,2,2-трихлорэтильную группу, 3-фторпропильную группу, гидроксиметильную группу, 2-гидроксиэтильную группу, 1-гидроксиэтильную группу, 3-гидроксипропильную группу, 2-гидроксипропильную группу, 1-гидроксипропильную группу, 2-гидрокси-2,2-диметилэтильную группу, азетидинильную группу, пирролидинильную группу, пиперидинильную группу и пиперазинильную группу. Предпочтительными являются атом водорода, атом фтора, гидроксильная группа, метальная группа, этильная группа, н-пропильная группа, изопропильная группа, гидроксиметильная группа, 2-гидроксиэтильная группа, 1-гидроксиэтильная группа, 3-гидроксршропильная группа, 2-гидроксипропильная группа, 1-гидроксипропильная группа, 2-гидрокси-2,2-диметилэтильная группа, азетидинильная группа, пирролидинильная группа или пиперидинильная группа, и более предпочтительной является метальная группа, этильная группа или 2-гидроксиэтильная группа.
[0049] В используемой в настоящем документе формуле R3 представляет собой атом водорода, оксогруппу, C1-6алкильную группу, необязательно замещенную 1-3 атомами галогена или C1-6алкоксигруппу, необязательно замещенную 1-3 атомами галогена. Предпочтительными являются атом водорода, C1-6лкильная группа или C1-6алкоксигруппа. В частности, предпочтительными являются атом водорода, метальная группа или этильная группа, и более предпочтительным является атом водорода.
[0050] В используемой в настоящем документе формуле R4 представляет собой С1-6алкильную группу. Предпочтительной является метальная группа.
[0051] Однако, если Е представляет собой азетадиновое кольцо, и R2 или R3 находится на атоме азота азетидинового кольца, этот R2 или R3 не является атомом водорода. Кроме того, каждый из R2 и R3 представляет собой атом или группу, замещенную в любом положении на Е, а именно в любом положении на содержащем азот неароматаческом С3-5 гетероцикле.
[0052] В следующей формуле (IB),
[Химическая формула 12]
частичная структура, представленная следующей формулой (IV), предпочтительно является частичной структурой, представленной следующей формулой (V) или (VI), и более предпочтительно частичной структурой, представленной формулой (V).
[Химическая формула 13]

[Химическая формула 14]
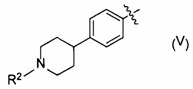
[Химическая формула 15]

[0053] В формуле (V) R2 представляет собой атом водорода, C1-6алкильную группу или гидроксиС2-6алкильную группу, и конкретные примеры включают в себя атом водорода, метальную группу, этильную группу, пропильную группу, изопропильную группу, 2-гидроксиэтильную группу, 3-гидроксипропильную группу, 2-гидроксипропильную группу, 2-гидрокси-2,2-диметилэтильную группу и т.п. Предпочтительной является метальная группа, этильная группа, 2-гидроксиэтильная группа, 2-гидроксипропильная группа или 2-гидрокси-2,2-диметилэтальная группа.
[0054] В формуле (VI) R2 представляет собой C1-6алкильную группу или гидроксиС2-6алкильную группу, и конкретные примеры включают в себя метальную группу, этильную группу, пропильную группу, изопропильную группу, 2-гидроксиэтильную группу, 3-гидроксипропильную группу, 2-гидроксипропильную группу, 2-гидрокси-2,2-диметилэтильную группу и т.п. Предпочтительной является метальная группа, этильная группа, 2-гидроксиэтильная группа, 2-гидроксипропильная группа или 2-гидрокси-2,2-диметилэтильная группа.
[0055] Соединением в соответствии с настоящим изобретением предпочтительно является любое из следующих соединений или подобное, или их фармацевтически приемлемые соли:
5-((2-(4-(1-этилпиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид;
6-(2-метоксиэтокси)-N-метил-5-((2-(4-(1-метилпиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид;
5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1Н-индол-1-карбоксамид;
6-(2-этоксиэтокси)-5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-N-метил-1Н-индол-1-карбоксамид;
6-(2-этоксиэтокси)-5-((2-(4-(1-этилазетидин-3-ил)бензамид)пиридин-4-ил)окси)-N-метил-1Н-индол-1-карбоксамид.
[0056] Примеры используемой в настоящем документе "соли" включают в себя соли неорганических кислот, соли органических кислот и соли кислых аминокислот, и особенно предпочтительными являются фармацевтически приемлемые соли. Кроме того, соль соединения в соответствии с настоящим изобретением охватывает ангидрид их фармацевтически приемлемой соли и сольват, такой как гидрат, его фармацевтически приемлемой соли.
[0057] Предпочтительные примеры соли неорганической кислоты включают в себя соли хлористоводородной кислоты, бромистоводородной кислоты, серной кислоты, азотной кислоты, фосфорной кислоты и т.п., и предпочтительные примеры соли органической кислоты включают в себя соли уксусной кислоты, янтарной кислоты, фумаровой кислоты, малеиновой кислоты, винной кислоты, лимонной кислоты, молочной кислоты, стеариновой кислоты, бензойной кислоты, метансульфоновой кислоты, этансульфоновой кислоты, п-толуолсульфоновой кислоты и т.п.
[0058] Предпочтительные примеры соли кислой аминокислоты включают соли аспарагиновой кислоты, глутаминовой кислоты и подобные.
[0059] Соединение (IА) в соответствии с настоящим изобретением или его фармацевтически приемлемая соль могут быть составлены общим способом, и готовой лекарственной формой может быть, например, пероральный состав (такой как таблетка, гранула, порошок, капсула, сироп или подобное), инъекционный состав (для внутривенного введения, внутримышечного введения, подкожного введения, внутрибрюшинного введения или т.п.) или наружный состав (такой как чрескожное абсорбируемое лекарственное средство (в том числе мазь, пластырь и т.п.), глазные капли, назальные капли, суппозиторий или подобное).
[0060] Для получения перорального твердого состава к соединению (IA) в соответствии с настоящим изобретением или его фармацевтически приемлемой соли при необходимости могут быть добавлены среда, связующее, разрыхлитель, скользящее вещество, краситель и т.п., и полученная в результате смесь может быть традиционным способом превращена в таблетки, гранулы, порошки или капсулы. Кроме того, таблетки, гранулы, порошки, капсулы или подобное при необходимости могут быть покрыты пленкой.
Примеры среды включают в себя лактозу, кукурузный крахмал, кристаллическую целлюлозу и т.п., примеры связующего включают в себя гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу и т.п., примеры разрыхлителя включают в себя кальция карбоксиметилцеллюлозу, натрия кроскармеллозу и т.п., примеры скользящего вещества включают в себя магния стеарат, кальция стеарат и т.п., пример красителя включает в себя титана оксид и т.п., и примеры пленкообразователя включают в себя гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, метилцеллюлозу и т.п., но эти компоненты не ограничиваются вышеупомянутыми примерами.
Твердый состав, такой как таблетка, капсула, гранула или порошок, может содержать соединение (IA) в соответствии с настоящим изобретением или его фармацевтически приемлемую соль, как правило, в количестве 0,001-99,5% по весу и, в частности, 0,001-90% по весу.
[0061] Для получения инъекционного состава (для внутривенного введения, внутримышечного введения, подкожного введения, внутрибрюшинного введения и т.п.) к соединению (IA) в соответствии с настоящим изобретением или его фармацевтически приемлемой соли при необходимости добавляют регулятор pH, буфер, суспендирующее средство, солюбилизирующее средство, антиоксидант, консервант (антисептическое средство), регулирующее тоничность средство и т.п., и из полученной в результате смеси традиционным способом может быть получен инъекционный состав. Кроме того, полученный в результате состав может быть высушен вымораживанием для использования в качестве лиофилизированного продукта, подлежащего растворению перед применением.
Примеры регулятора pH и буфера включают в себя органические кислоты или неорганические кислоты и/или их фармацевтически приемлемые соли, примеры суспендирующего средства включают в себя метилцеллюлозу, Polysorbate 80, натрия карбоксиметилцеллюлозу и т.п., примеры солюбилизирующего средства включают в себя Polysorbate 80, полиоксиэтиленсорбитанмонолаурат и т.п., пример антиоксиданта включает в себя α-токоферол и т.п., примеры консервантов включают в себя метилпараоксибензоат, этилпараоксибензоат и т.п., а примеры средства, регулирующего тоничность, включают в себя виноградный сахар, натрия хлорид, маннит и т.п., но эти компоненты не ограничиваются вышеупомянутыми примерами.
Такой инъекционный состав может содержать соединение (IА) в соответствии с настоящим изобретением или его фармацевтически приемлемую соль, как правило, в количестве 0,000001-99,5% по весу и, в частности, 0,00001-90% по весу
[0062] Для получения наружного состава к соединению (IA) в соответствии с настоящим изобретением или его фармацевтически приемлемой соли добавляют материал основы, а также туда при необходимости добавляют, например, консервант, стабилизатор, регулятор pH, антиоксидант, краситель и т.п., описанные выше, и из полученной в результате смеси традиционным способом получают, например, трансдермальное абсорбируемое лекарственное средство (такое как мазь или пластырь), глазные капли, назальные капли, суппозиторий или подобное.
В качестве используемого материала-основы могут применять различные материалы обычно используемые, например, для медицинских препаратов, лечебно-профилактической косметики и косметических средств. Конкретные примеры материала включают в себя масла животного и растительного происхождения, минеральные масла, сложноэфирные масла, воски, эмульгаторы, высшие спирты, жирные кислоты, силиконовые масла, поверхностно-активные вещества, фосфолипиды, спирты, многоатомные спирты, водорастворимые полимеры, глинистые минералы, очищенную воду и т.п.
Такой внешний состав может содержать соединение (IА) в соответствии с настоящим изобретением или его фармацевтически приемлемую соль, как правило, в количестве 0,000001-99,5% по весу и, в частности, 0,00001-90% по весу.
[0063] Доза соединения (IA) в соответствии с настоящим изобретением или его фармацевтически приемлемой соли зависит от степени тяжести симптома, возраста пациента, пола и веса, формы введения и типа соли, конкретного вида заболевания и т.п., и особенно не ограничивается, если не превышает максимальную дозу медицинского препарата, которая может быть принята без неприемлемой побочной реакции, и взрослому пациенту его вводят, за один раз или поделенным на несколько раз в день, при дозе для перорального введения, как правило, составляющей приблизительно 30 μг - 10 г, конкретнее 100 μг - 5 г и более конкретно 100 μг - 1 г, или при дозе для инъекционного введения, как правило, составляющей приблизительно 30 μг - 1 г, конкретнее 100 μг - 500 мг и более конкретно 100 μг - 300 мг.
[0064] [Общие способы синтеза]
Далее будет описан способ получения соединения (IA) в соответствии с настоящим изобретением. Соединение (IA) в соответствии с настоящим изобретением может быть синтезировано с использованием средств общего органического синтеза, и некоторые примеры соединения (IА) в соответствии с настоящим изобретением, например, соединений (1-1), (1-2), (1-3), (1-4), (1-5), (1-6), (1-7), (1-8), (1-9) и (1-10) могут быть синтезированы способами, описанными в [способе получения 1] или подобными описанными ниже. Если в способах получения, описанных в настоящем документе, используют защитную группу, то соответственно выбирают и вводят известные защитные группы, например, описанные в Green's PROTECTIVE GROUP IN ORGANIC CHEMISTRY, fourth edition, JOHN WILEY & SONS, INC., и снятие защиты, соответственно, может быть выполнено известным способом.
[0065] [Способ получения 1]
Типичный способ получения соединения (IА) в соответствии с настоящим изобретением
[Химическая формула 16]
где R1, R2, R3, A, E, G и n определяются выше.
[0066] [Способ получения 1-1]
Способ получения соединения (1-1), (1-2), (1-3), (1-6) или (1-7)
[Химическая формула 16]
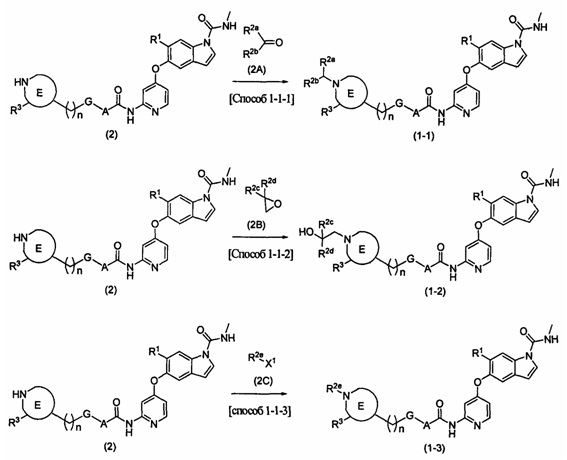
В вышеупомянутой формуле R1, R3 (оксогруппа исключается), R4, А, E, G и n определяются выше; каждый R2a и R2b представляет собой атом водорода, С1-5алкильную группу, необязательно замещенную 1-3 атомами галогена, или гидроксиС1-5алкильную группу, необязательно замещенную 1-3 атомами галогена;
каждый R2c и R2d представляет собой атом водорода или С1-4алкильную группу, необязательно замещенную 1-3 атомами галогена; R2e представляет собой С1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, или гидроксиС1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, если R2e содержит гидроксильную группу, то гидроксильная группа может быть защищена известной приемлемой защитной группой; X1 представляет собой атом галогена, такой как атом хлора, атом брома или атом йода, или уходящую группу сульфоната, такую как метансульфонат или п-толуолсульфонат; R2f представляет собой С1-5алкильную группу, необязательно замещенную C1-6алкоксигруппой; R2g представляет собой гидроксигруппу, моно-C1-6алкиламиногруппу или С1-5алкильную группу, необязательно замещенную ди-С1-6алкиламиногруппой, если R2g содержит гидроксильную группу или моно-C1-6алкиламиногруппу, то гидроксильная группа или моно-C1-6алкиламиногруппа может быть защищена известной приемлемой защитной группой.
[0067] Соединение (2) также может быть получено способом, описанным в примере получения какого-либо из примеров, описанных ниже, [способом получения 2] или подобным.
Соединения (2А), (2В), (2С), (2D) и (2Е) могут быть коммерчески доступными продуктами или могут быть получены из коммерчески доступных продуктов известными способами. В качестве альтернативы эти соединения могут быть получены способами, описанными в примерах получения какого-либо из примеров, описанных ниже, или подобными. Соединение (2А) может быть в любой форме от димерной до мультимерной.
[0068] [Способ 1-1-1]
В этом способе осуществляют реагирование соединения (2) и соединения (2А) друг с другом в присутствии восстанавливающего средства с получением соединения (1-1). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, спиртовой растворитель, такой как этанол, нитриловый растворитель, такой как ацетонитрил, растворитель на основе карбоновой кислоты, такой как уксусная кислота, растворитель на основе ароматического углеводорода, такой как бензол или толуол, амидный растворитель, такая как N,N-диметилформамид или N-метилпирролидинон, галогенированный углеводородный растворитель, такой как дихлорметан или хлороформ, вода или смешанный растворитель. В качестве восстанавливающего средства, используемого в этой реакции, может быть использовано металловодородное комплексное соединение, такое как натрия боргидрид, натрия цианоборгидрид или натрия триацетоксиборгидрид, или замещенный боран, такой как диборан, или пиридин-борановый комплекс. Кроме того, может быть использован катализатор каталитического восстановления, такой как палладий на угле, в атмосфере азота. Соединение (2А) может быть использовано в количестве 1-10 эквивалентов по отношению к соединению (2), а предпочтительно используется в количестве 1-2 эквивалентов. Восстанавливающее средство может быть использовано в количестве 1 эквивалента или более по отношению к соединению (2), а предпочтительно используется в количестве 1-5 эквивалентов. В этой реакции кислота, такая как уксусная кислота, может быть добавлена в количестве 0-10 эквивалентов. Температура реакции составляет от -20°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0069] [Способ 1-1-2]
В этом способе осуществляют реагирование соединения (2) и соединения (2В) друг с другом с получением соединения (1-2). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, спиртовой растворитель, такой как этанол, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, нитриловый растворитель, такой как ацетонитрил, растворитель на основе ароматического углеводорода, такой как толуол, галогенированный углеводородный растворитель, такой как дихлорметан или хлороформ, или смешанный растворитель. В этой реакции в качестве основания могут быть использованы алкиламин, такой как триэтиламин, ароматический амин, такой как пиридин, или неорганическое основание, такое как калия карбонат. Соединение (2В) может быть использовано в количестве 1 эквивалента или более по отношению к соединению (2), а предпочтительно используется в количестве 1-10 эквивалентов. Основание можно использовать в количестве от 0 до 10 эквивалентов по отношению к соединению (2). Температура реакции составляет от 0°С до 200°С, а время реакции составляет от 10 минут до 24 часов.
[0070] [Способ 1-1-3]
В этом способе осуществляют реагирование соединения (2) и соединения (2С) друг с другом с получением соединения (1-3). Выбор растворителя, используемого в реакции, специально не ограничивают; при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, нитриловый растворитель, такой как ацетонитрил, растворитель на основе ароматического углеводорода, такой как толуол, галогенированный углеводородный растворитель, такой как дихлорметан или хлороформ, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, диметилсульфоксид или смешанный растворитель. В этой реакции в качестве основания могут быть использованы алкиламин, такой как триэтиламин, ароматический амин, такой как пиридин, или неорганическое основание, такое как калия гидрокарбонат, калия карбонат или цезия карбонат. Соединение (2С) может быть использовано в количестве 1 эквивалента или более по отношению к соединению (2), а предпочтительно используется в количестве 1-3 эквивалентов. Основание можно использовать в количестве от 1 до 10 эквивалентов по отношению к соединению (2). Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. Гидроксильная группа R2e является защищенной, защита может быть снята известным способом.
[0071] [Способ 1-1-4]
В этом способе осуществляют реагирование соединения (2) и соединения (2D) друг с другом с получением соединения (1-6). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, сложноэфирный растворитель, такой как этилацетат, эфирный растворитель, такой как тетрагидрофуран, нитриловый растворитель, такой как ацетонитрил, растворитель на основе ароматического углеводорода, такой как толуол, галогенированный углеводородный растворитель, такой как дихлорметан или хлороформ, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, или смешанный растворитель. В этой реакции в качестве основания могут быть использованы алкиламин, такой как триэтиламин, ароматический амин, такой как пиридин, или неорганическое основание, такое как натрия гидрокарбонат, калия карбонат или цезия карбонат. Соединение (2D) может быть использовано в количестве 1 эквивалента или более по отношению к соединению (2), а предпочтительно используется в количестве 1-3 эквивалентов. Основание можно использовать в количестве от 1 до 10 эквивалентов по отношению к соединению (2). Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0072] [Способ 1-1-5]
В этом способе осуществляют реагирование соединения (2) и соединения (2Е) друг с другом с использованием средства конденсации с получением соединения (1-7). Выбор растворителя, используемого в реакции, специально не ограничивают; при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, сложноэфирный растворитель, такой как этилацетат, нитриловый растворитель, такой как ацетонитрил, растворитель на основе ароматического углеводорода, такой как толуол, галогенированный углеводородный растворитель, такой как дихлорметан или хлороформ, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, диметилсульфоксид или смешанный растворитель. В этой реакции может быть использовано средство конденсации, такое как O-(7-азабензсприазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат или 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид. Более того, в качестве добавки в реакции может быть использован 1-гидроксибензотриазол или подобное. В этой реакции в качестве основания могут быть использованы алкиламин, такой как триэтиламин или N,N-диизопропилэтиламин, ароматический амин, такой как 4-диметиламинопиридин, или неорганическое основание, такое как калия карбонат или цезия карбонат, или может быть использована комбинация таких оснований. Соединение (2Е) может быть использовано в количестве 1 эквивалента или более по отношению к соединению (2), а предпочтительно используется в количестве 1-3 эквивалентов, Средство конденсации может быть использовано в количестве стольких же эквивалентов, что и соединение (2Е), а основание может быть использовано в количестве 1-10 эквивалентов по отношению к соединению (2). Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. Если R2g защищена гидроксильной группой или моно-C1-6алкиламиногруппой, то защита может быть снята известным способом.
[0073] [Способ получения 1-2]
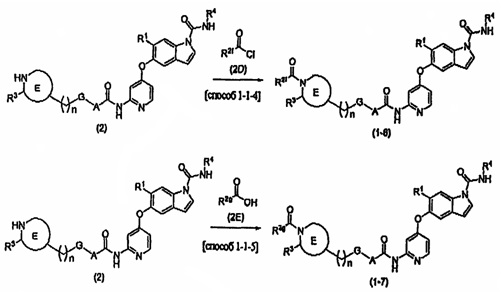
Способ получения соединения (1-4)
[Химическая формула 18]

В вышеупомянутой формуле R1, R2, R3, R4, А, Е, G и n определяются выше; и X2 представляет собой атом галогена, такой как атом хлора или атом брома, или уходящую группу сульфоната, такую как метансульфонат или п-толуолсульфонат.
Соединения (3) и (4) также могут быть получены способами, описанными в [способе получения 3], примерах получения какого-либо из примеров, описанных ниже, или подобном.
Соединения (3А) и (4-1) могут быть коммерчески доступными продуктами или могут быть получены из коммерчески доступных продуктов известными способами. В качестве альтернативы эти соединения могут быть получены способами, описанными в [способе получения 6], в примерах получения какого-либо из примеров, описанных ниже, или подобном.
[0074] [Способ 1-2-1]
В этом способе осуществляют реагирование соединения (4) и соединения (4-1) друг с другом с получением соединения (3). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, сложноэфирный растворитель, такой как этилацетат, нитриловый растворитель, такой как ацетонитрил, растворитель на основе ароматического углеводорода, такой как толуол, галогенированный углеводородный растворитель, такой как дихлорметан или хлороформ, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, или смешанный растворитель. В этой реакции в качестве основания могут быть использованы алкиламин, такой как триэтиламин или N,N-диизопропилэтиламин, ароматический амин, такой как 4-диметиламинопиридин, или неорганическое основание, такое как калия карбонат или цезия карбонат, или может быть использована комбинация таких оснований. Соединение (4-1) может быть использовано в количестве 1 эквивалента или более по отношению к соединению (4), а предпочтительно используется в количестве 2-3 эквивалентов. Основание можно использовать в количестве от 1 до 10 эквивалентов по отношению к соединению (4). Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. В этом способе может быть получена диацильная форма в результате реакции 2 эквивалентов соединения (4-1), и это может быть использовано непосредственно в следующей реакции.
[0075] [Способ 1-2-2]
В этом способе осуществляют реагирование соединения (3) и соединения (3А) друг с другом с получением соединения (1-4). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, растворитель на основе ароматического углеводорода, такой как толуол, галогенированный углеводородный растворитель, такой как дихлорметан или хлороформ, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, диметилсульфоксид или смешанный растворитель. В этой реакции в качестве основания могут быть использованы алкиламин, такой как триэтиламин или N,N-диизопропилэтиламин, или неорганическое основание, такое как калия карбонат или цезия карбонат. Соединение (3А) может быть использовано в количестве 1 эквивалента или более по отношению к соединению (3), а предпочтительно используется в количестве 1-10 эквивалентов. Основание можно использовать в количестве от 1 до 10 эквивалентов по отношению к соединению (3). Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0076] [Способ получения 1-3]
Способ получения соединения (1) или (1-5)
[Химическая формула 18]
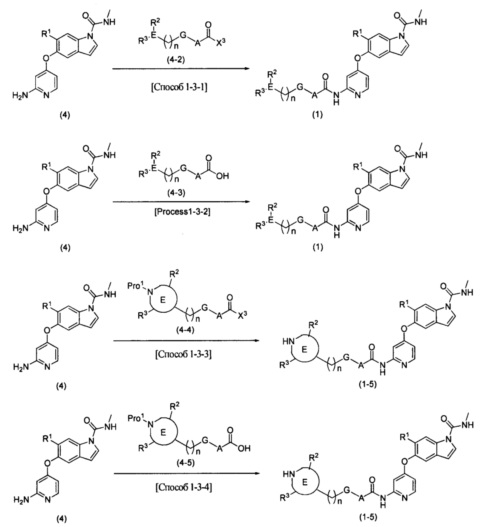
В вышеупомянутой формуле R1, R2, R3, R4, А, Е, G и n определяются выше; X3 представляет собой атом галогена, такой как атом хлора или атом брома; a Pro1 представляет собой известную защитную группу для атома азота, такую как трет-бутоксикарбонильная группа.
Соединение (4) может быть получено способом, описанным в примере получения какого-либо из примеров, описанных ниже, [способ получения 3] или подобным.
Соединения (4-2), (4-3), (4-4) и (4-5) могут быть коммерчески доступными продуктами или могут быть получены из коммерчески доступных продуктов известными способами. Эти соединения могут быть получены способами, описанными в [способе получения 5], в примерах получения какого-либо из примеров, описанных ниже, или подобным.
[0077] [Способ 1-3-1 или 1-3-3]
В этом способе осуществляют реагирование соединения (4) и соединения (4-2) или (4-4) друг с другом с получением соединения (1) или (1-5), соответственно. Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, сложноэфирный растворитель, такой как этилацетат, нитриловый растворитель, такой как ацетонитрил, растворитель на основе ароматического углеводорода, такой как толуол, галогенированный углеводородный растворитель, такой как дихлорметан или хлороформ, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, или смешанный растворитель. В этой реакции в качестве основания могут быть использованы алкиламин, такой как триэтиламин или N,N-диизопропилэтиламин, ароматический амин, такой как 4-диметиламинопиридин, или неорганическое основание, такое как калия карбонат или цезия карбонат, или может быть использована комбинация таких оснований. Соединение (4-2) или (4-4) может быть использовано в количестве 1 эквивалента или более по отношению к соединению (4), а предпочтительно используется в количестве 2-3 эквивалентов. Основание можно использовать в количестве от 1 до 10 эквивалентов по отношению к соединению (4). Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. В этом способе может быть получена диацильная форма в результате реакции 2 эквивалентов соединения (4-2) или (4-4). В этом случае диацильная форма может быть превращена в желаемую моноацильную форму путем обработки аммиаком, или первичным, или вторичным алкиламином, таким как метиламин или пиперидин.
В способе 1-3-3 защитная группа для атома азота может быть впоследствии удалена известным способом. Если Pro1 представляет собой, например, трет-бутоксикарбонильную группу, снятие защиты может быть выполнено с использованием растворителя, не подавляющего реакцию, например, галогенированного углеводородного растворителя, такого как дихлорметан, сложноэфирного растворителя, такого как этилацетат, спиртового растворителя, такого как метанол, или смешанный растворитель, полученный из вышеупомянутых, а также с использованием кислоты, такой как трифторуксусная кислота или хлористоводородная кислота.
[0078] [Способ 1-3-2 или 1-3-4]
В этом способе осуществляют реагирование соединения (4) и соединения (4-3) или (4-5) друг с другом с использованием средства конденсации с получением соединения (1) или (1-5). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, сложноэфирный растворитель, такой как этилацетат, нитриловый растворитель, такой как ацетонитрил, растворитель на основе ароматического углеводорода, такой как толуол, галогенированный углеводородный растворитель, такой как дихлорметан или хлороформ, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, диметилсульфоксид, или смешанный растворитель. В этой реакции может быть использовано средство конденсации, такое как O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат или 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид. Более того, в качестве добавки в реакции может быть использован 1-гидроксибензотриазол или подобное. В этой реакции в качестве основания могут быть использованы алкиламин, такой как триэтиламин или N,N-диизопропилэтиламин, ароматический амин, такой как 4-диметиламинопиридин, или неорганическое основание, такое как калия карбонат или цезия карбонат, или может быть использована комбинация таких оснований. Соединение (4-3) или (4-5) может быть использовано в количестве 1 эквивалента или более по отношению к соединению (4), а предпочтительно используется в количестве 2-3 эквивалентов. Средство конденсации может быть использовано в количестве стольких же эквивалентов, что и соединение (4-3) или (4-5), а основание может быть использовано в количестве 1-10 эквивалентов по отношению к соединению (4). Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. В этом способе может быть получена диацильная форма в результате реакции 2 эквивалентов соединения (4-3) или (4-5). В этом случае диацильная форма может быть превращена в желаемую моноацильную форму путем обработки аммиаком, или первичным, или вторичным алкиламином, таким как метиламин или пиперидин. Если R2 защищен известной защитной группой, то защитная группа удаляется известным способом.
В способе 1-3-4 защитная группа для атома азота может быть впоследствии удалена известным способом. Если Pro1 представляет собой, например, трет-бутоксикарбонильную группу, то снятие защиты может быть выполнено с использованием растворителя, не подавляющего реакцию, например, галогенированного углеводородного растворителя, такого как дихлорметан, сложноэфирного растворителя, такого как этилацетат, спиртового растворителя, такого как метанол, или смешанного растворителя, полученного из вышеупомянутых, а также с использованием кислоты, такой как трифторуксусная кислота или хлористоводородная кислота.
[0079] [Способ получения 1-4]
Способ получения соединения (1-8)
[Химическая формула 20]

В вышеупомянутой формуле R1, R2, R4, А, Е, G и n определяются выше.
Соединение (1-1-1) также может быть получено способом, описанным в примере получения какого-либо из примеров, описанных ниже, [способом получения 1-1] или подобным.
[0080] [Способ 1-4]
В этом способе осуществляют реагирование соединения (1-1-1) и окисляющего средства друг с другом с получением соединения (1-8). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, галогенированный углеводородный растворитель, такой как дихлорметан или хлороформ, спиртовой растворитель, такой как метанол или трет-бутанол, органическая кислота, такая как уксусная кислота, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции в качестве окисляющего средства могут быть использованы перекись водорода - вода, органический пероксид или органическая перкислота, такая как 3-хлорпероксибензойная кислота. Окисляющее средство может быть использовано в количестве 0,5-10 эквивалентов по отношению к соединению (1-1-1). Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0081] [Способ получения 1-5]
Способ получения соединения (1-10)
[Химическая формула 21]

В вышеупомянутой формуле R2, R4, А, Е, G и n определяются выше; и m представляет собой целое число от 2 до 6.
Соединение (1-9) также может быть получено способом, описанным в примере получения какого-либо из примеров, описанных ниже, [способом получения 1-1], [способом получения 1-2], [способом получения 1-3] или подобным.
[0082] [Способ 1-5]
В этом способе осуществляют реагирование соединения (1-9) и трехбромистого бора друг с другом с получением соединения (1-10). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, галогенированный углеводородный растворитель, такой как дихлорметан или хлороформ. Трехбромистый бор, используемый в этой реакции, может быть использован в количестве 1-10 эквивалентов по отношению к соединению (1-9). Температура реакции составляет от -20°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0083] [Способ получения 2]
Способ получения соединения (2)
[Химическая формула 22]
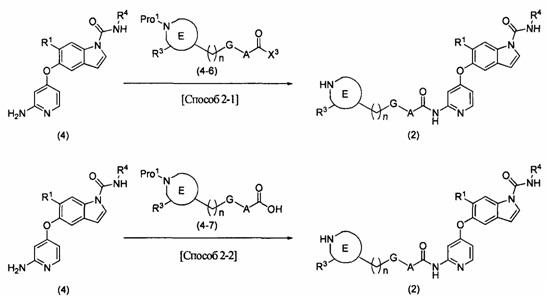
В вышеупомянутой формуле R1, R3, R4, А, Е, G и n определяются выше; X3 представляет собой атом галогена, такой как атом хлора или атом брома; a Pro1 представляет собой известную защитную группу для атома азота, такую как трет-бутоксикарбонильная группа.
Соединения (4-6) и (4-7) могут быть коммерчески доступными продуктами или могут быть получены из коммерчески доступных продуктов известными способами. Эти соединения могут быть получены способами, описанными в примерах получения какого-либо из примеров, описанных ниже, или подобными. Соединение (4-6) также может быть получено способами, описанными в [способе получения 5] или подобными.
[0084] [Способ 2-1]
В этом способе осуществляют реагирование соединения (4) и соединения (4-6) друг с другом с получением соединения (2). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, сложноэфирный растворитель, такой как этилацетат, нитриловый растворитель, такой как ацетонитрил, растворитель на основе ароматического углеводорода, такой как толуол, галогенированный углеводородный растворитель, такой как дихлорметан или хлороформ, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, или смешанный растворитель. В этой реакции в качестве основания могут быть использованы алкиламин, такой как триэтиламин или N,N-диизопропилэтиламин, ароматический амин, такой как 4-диметиламинопиридин, или неорганическое основание, такое как калия карбонат или цезия карбонат, или может быть использована комбинация таких оснований. Соединение (4-6) может быть использовано в количестве 1 эквивалента или более по отношению к соединению (4), а предпочтительно используется в количестве 2-3 эквивалентов. Основание можно использовать в количестве от 1 до 10 эквивалентов по отношению к соединению (4). Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. В этом способе может быть получена диацильная форма в результате реакции 2 эквивалентов соединения (4-6). В этом случае диацильная форма может быть превращена в желаемую моноацильную форму путем обработки аммиаком, или первичным или вторичным алкиламином, таким как метиламин или пиперидин. Защитная группа для атома азота может быть впоследствии удалена известным способом. Если Pro1 представляет собой, например, трет-бутоксикарбонильную группу, то снятие защиты может быть выполнено с использованием растворителя, не подавляющего реакцию, например, галогенированного углеводородного растворителя, такого как дихлорметан, сложноэфирного растворителя, такого как этилацетат, спиртового растворителя, такого как метанол, или смешанного растворителя, полученного из вышеупомянутых, а также с использованием кислоты, такой как трифторуксусная кислота или хлористоводородная кислота.
[0085] [Способ 2-2]
В этом способе осуществляют реагирование соединения (4) и соединения (4-7) друг с другом с получением соединения (2). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, сложноэфирный растворитель, такой как этилацетат, нитриловый растворитель, такой как ацетонитрил, растворитель на основе ароматического углеводорода, такой как толуол, галогенированный углеводородный растворитель, такой как дихлорметан или хлороформ, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, диметилсульфоксид, или смешанный растворитель. В этой реакции может быть использовано средство конденсации, такое как O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат или 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид. Более того, в качестве добавки в реакции может быть использован 1-гидроксибензотриазол или подобное. В этой реакции в качестве основания могут быть использованы алкиламин, такой как триэтиламин или N,N-диизопропилэтиламин, ароматический амин, такой как 4-диметиламинопиридин, или неорганическое основание, такое как калия карбонат или цезия карбонат, или может быть использована комбинация таких оснований. Соединение (4-7) может быть использовано в количестве 1 эквивалента или более по отношению к соединению (4), а предпочтительно используется в количестве 2-3 эквивалентов. Основание можно использовать в количестве от 1 до 10 эквивалентов по отношению к соединению (4). Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. В этом способе может быть получена диацильная форма в результате реакции 2 эквивалентов соединения (4-7). В этом случае диацильная форма может быть превращена в желаемую моноацильную форму путем обработки аммиаком, или первичным или вторичным алкиламином, таким как метиламин или пиперидин. Защитная группа для атома азота может быть впоследствии удалена известным способом. Если Pro1 представляет собой, например, трет-бутоксикарбонильную группу, то снятие защиты может быть выполнено с использованием растворителя, не подавляющего реакцию, например, галогенированного углеводородного растворителя, такого как дихлорметан, сложноэфирного растворителя, такого как этилацетат, спиртового растворителя, такого как метанол, или смешанного растворителя, полученного из вышеупомянутых, а также с использованием кислоты, такой как трифторуксусная кислота или хлористоводородная кислота.
[0086] [Способ получения 3]
Способ получения соединения (4)
[Химическая формула 23]

В вышеупомянутой формуле R1, R4 определены выше; Pro2 представляет собой защитную группу для атома азота, такую как ацетильная группа; Pro3 представляет собой защитная группу для фенольного атома кислорода, такую как бензильная группа; а X3 представляет собой атом галогена, такой как атом хлора или атом брома.
Соединения (7), (8), (9), (10) и (11) могут быть коммерчески доступными продуктами или могут быть получены из коммерчески доступных продуктов известными способами. Эти соединения могут быть получены способами, описанными в примерах получения какого-либо из примеров, описанных ниже. Соединения (5) и (6) также могут быть получены способами, описанными в примерах получения какого-либо из примеров, описанных ниже, или подобными.
Соединения (5А), (6А) и (11А) могут быть коммерчески доступными продуктами или могут быть получены из коммерчески доступных продуктов известными способами. В качестве альтернативы эти соединения могут быль получены способами, описанными в примерах получения какого-либо из примеров, описанных ниже, или подобными.
[0087] [Способ 3-1]
В этом способе осуществляют реагирование соединения (11) и соединения (11А) друг с другом с получением соединения (10). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, спиртовой растворитель, такой как этанол, нитриловый растворитель, такой как ацетонитрил, кетоновый растворитель, такой как ацетон, галогенированный углеводородный растворитель, такой как дихлорметан, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, диметилсульфоксид, или смешанный растворитель. В этой реакции соединение (11А) используется в количестве 1-5 эквивалентов по отношению к соединению (11) и может быть использовано 1-5 эквивалентов основания, такого как натрия гидрокарбонат, калия карбонат, натрия метоксид, натрия гидрид или диизопропилэтиламин. Кроме того, натрия йодид или калия йодид могут быть добавлены в качестве добавки. Температура реакции составляет от -20°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0088] [Способ 3-2]
В этом способе осуществляют реагирование соединения (10) и нитрометана друг с другом с получением соединения (9). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, спиртовой растворитель, такой как метанол, растворитель на основе органической кислоты, такой как уксусная кислота, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции нитрометан используется в количестве 1-10 эквивалентов, и в качестве добавки может быть использовано 0,1-10 эквивалентов аммония ацетата, этилендиамин-N,N'-диуксусной кислоты или подобное. Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 100 часов.
[0089] [Способ 3-3]
В этом способе соединение (9) нитрируют с получением соединения (8). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, галогенированный углеводородный растворитель, такой как дихлорметан, растворитель на основе органической кислоты, такой как уксусная кислота, серная кислота, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции используют дымящуюся азотную кислоту, концентрированную азотную кислоту или азотную кислоту, и в качестве добавки может быть добавлен уксусный ангидрид или подобное. Азотная кислота или подобное может использоваться в этой реакции в количестве 1-100 эквивалентов по отношению к соединению (9). Температура реакции составляет от -20°С до температуры флегмы, и время реакции составляет от 10 минут до 100 часов.
[0090] [Способ 3-4]
В этом способе соединение (8) циклизируют с получением соединения (7). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, спиртовой растворитель, такой как этанол, растворитель на основе ароматического углеводорода, такой как бензол или толуол, углеводородный растворитель, такой как циклогексан, вода, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции используется тяжелый металл, такой как железо или цинк, и в качестве добавки могут быть использованы кислота, такая как уксусная кислота, соль, такая как аммония хлорид, основание, такое как натрия гидроксид, неорганическое соединение, такое как силикагель, или смесь вышеупомянутых. Тяжелый металл, такой как железо, может использоваться в этой реакции в количестве 5-20 эквивалентов по отношению к соединению (8). Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0091] [Способ 3-5]
В этом способе Pro3 в соединении (7) снимают с получением соединения (6). Pro3 может быть снята известным способом, и если Pro3 является бензилом, то растворитель, используемый в этой реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, спиртовой растворитель, такой как этанол, растворитель на основе ароматического углеводорода, такой как бензол или толуол, уксусная кислота, вода или смешанный растворитель, полученный из вышеупомянутых. Эта реакция может быть выполнена в атмосфере азота с использованием в качестве катализатора катализатора каталитического восстановления, такого как палладий на угле. Давление водорода может составлять от нормального давления до 20 атм, а катализатор может использоваться в количестве 0,001-1 эквивалента по отношению к соединению (7). Кроме того, может быть добавлена кислота, такая как хлористоводородная кислота. Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0092] [Способ 3-6]
В этом способе соединение (8) циклизируют с получением соединения (6). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, спиртовой растворитель, такой как метанол, растворитель на основе ароматического углеводорода, такой как бензол или толуол, уксусная кислота, вода или смешанный растворитель. Эта реакция может быть выполнена в атмосфере азота с использованием в качестве катализатора катализатора каталитического восстановления, такого как палладий на угле. Давление водорода может составлять от нормального давления до 20 атм, а катализатор может использоваться в количестве 0,001-1 эквивалента по отношению к соединению (8). В качестве добавки может быть добавлена кислота, такая как хлористоводородная кислота. Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0093] [Способ 3-7]
В этом способе осуществляют реагирование соединения (6) и соединения (6А) друг с другом с получением соединения (5). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, диметилсульфоксид, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции в качестве основания может быть использовано основание, такое как калия карбонат, цезия карбонат или калия трет-бутоксид. Основание может быть использовано в количестве 1-10 эквивалентов по отношению к соединению (6), а предпочтительно используется в количестве 1-2 эквивалентов. Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. Pro2 может быть снята известным способом, и если Pro2 является ацетильной группой, то защита может быть снята с использованием растворителя, не подавляющего реакцию, такого как спиртовой растворитель, такой как метанол, и с использованием основания, такого как калия карбонат, натрия гидроксид или натрия метоксид, или с него может быть снята защита с использованием растворителя, не подавляющего реакцию, такого как эфирный растворитель, такой как 1,4-диоксан, и с использованием кислоты, такой как хлористоводородная кислота. В качестве альтернативы этот способ может быть выполнен с использованием соединения, не защищенного с помощью Pro2 такого, как соединение (6А).
[0094] [Способ 3-8]
В этом способе осуществляют реагирование соединения (5) и соединения (5А) друг с другом с получением соединения (4). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, диметилсульфоксид, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции в качестве основания может быть использовано основание, такое как натрия гидроксид или калия трет-бутоксид. Соединение (5А) может быть использовано в количестве 1 эквивалента или более по отношению к соединению (5), а предпочтительно используется в количестве 1-2 эквивалентов, Основание можно использовать в количестве от 1 до 2 эквивалентов по отношению к соединению (5). Температура реакции составляет от -20°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0095] [Способ получения 4]
Способ получения соединения (11-1)
[Химическая формула 24]

В вышеупомянутой формуле R1a представляет собой C1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, или C1-6алкоксиC1-6алкильную группу, необязательно замещенную 1-3 атомами галогена; а X3 представляет собой уходящую группу атома галогена, такого как атом брома или атом йода.
Соединение (12) может быть коммерчески доступным продуктом или может быть получено из коммерчески доступного продукта известным способом.
Соединение (12А) может быть коммерчески доступным продуктом или может быть получено из коммерчески доступного продукта известным способом.
[0096] [Способ 4-1]
В этом способе осуществляют реагирование соединения (12) и соединения (12А) друг с другом с получением соединения (11-1). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, спиртовой растворитель, такой как этанол, нитриловый растворитель, такой как ацетонитрил, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, диметилсульфоксид, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции соединение (12А) используют в количестве 0,5-1,5 эквивалента по отношению к соединению (12), и в качестве добавки может быть использовано 0,5-1,5 эквивалента основания, такого как натрия гидрокарбонат, калия карбонат, натрия метоксид или натрия гидрид. Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 100 часов.
[0097] [Способ получения 5]
Способ получения соединения (4-2), (4-4) или (4-6)
[Химическая формула 25]


В вышеупомянутой формуле R2, R3, А, Е, G и n определяются выше; Х3 представляет собой атом галогена, такой как атом хлора или атом брома; a Pro1 представляет собой известную защитную группу для атома азота, такую как трет-бутоксикарбонильная группа.
Соединения (4-3), (4-5) и (4-7) могут быть коммерчески доступными продуктами или могут быть получены из коммерчески доступных продуктов известными способами. В качестве альтернативы эти соединения также могут быть получены способами, описанными в примерах получения какого-либо из примеров, описанных ниже, или способом, описанном в [способе получения Т], [способе получения 9] или подобными.
[0098] [Способ 5-1,5-2 или 5-3]
В этом способе осуществляют реагирование соединения (4-3), (4-5) или (4-7) с получением соединения (4-2), (4-4) или (4-6), соответственно. Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, галогенированный углеводородный растворитель, такой как дихлорметан, эфирный растворитель, такой как тетрагидрофуран, нитриловый растворитель, такой как ацетонитрил, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции галоидангидрид, такой как оксалилхлорид, или неорганическое содержащее галоген соединение, такое как тионилхлорид, используется в количестве 1-10 эквивалентов по отношению к соединению (4-3), (4-5) или (4-7), и в качестве добавки может быть использовано 1-10 эквивалентов основания, такого как бензотриазол. Более того, может быть использовано каталитическое количество N,N-диметилформамида, N-метилпирролидинона или подобного. Температура реакции составляет от -20°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0099] [Способ получения 6]
Способ получения соединения (4-1)
[Химическая формула 26]
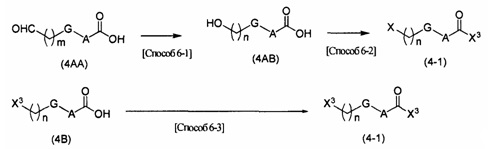
В вышеупомянутой формуле A, G и n определяются выше; m представляет собой 0 или 1; и X3 представляет собой атом галогена, такой как атом хлора или атом брома.
Соединения (4АА), (4АВ) и (4В) могут быть коммерчески доступными продуктами или могут быть получены из коммерчески доступных продуктов известными способами. В качестве альтернативы эти соединения могут быть получены способами, описанными в примерах получения какого-либо из примеров, описанных ниже, или подобными.
[0100] [Способ 6-1]
В этом способе осуществляют реагирование соединения (4АА) с получением соединения (4АВ). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, спиртовой растворитель, такой как этанол, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции может быть использован восстанавливающий реагент, такой как натрия боргидрид, в количестве 1-10 эквивалентов по отношению к соединению (4АА). Температура реакции составляет от -20°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0101] [Способ 6-2 или 6-3]
В этом способе осуществляют реагирование соединения (4АВ) или (4В) с получением соединения (4-1). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, галогенированный углеводородный растворитель, такой как дихлорметан, эфирный растворитель, такой как тетрагидрофуран, нитриловый растворитель, такой как ацетонитрил, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции галоидангидрид, такой как оксалилхлорид, или неорганическое содержащее галоген соединение, такое как тионилхлорид, может быть использовано в количестве 1-10 эквивалентов по отношению к соединению (4АВ) или (4В), и в качестве добавки может быть добавлено каталитическое количество N,N-диметилформамида, N-метилпирролидинона или подобного. Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0102] [Способ получения 7]
Способ получения соединения (4-3g)
[Химическая формула 27]
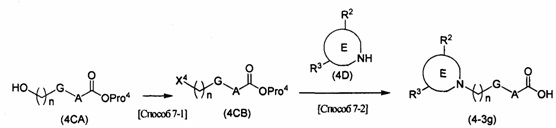
В вышеупомянутой формуле R2, R3, А, Е, G и n определяются выше; X4 представляет собой атом галогена, такой как атом хлора, атом брома или атом йода, или уходящую группу сульфоната, такую как метансульфонат, п-толуолсульфонат или трифторметансульфонат; a Pro4 представляет собой известную защитную группу для карбоновой кислоты, такую как этильная группа.
Соединение (4СВ) может быть коммерчески доступным продуктом или может быть получено из коммерчески доступного продукта известным способом. В качестве альтернативы эти соединения могут быть получены способом, описанным в примере получения какого-либо из примеров, описанных ниже, или подобным.
Соединение (4СА) или (4D) может быть коммерчески доступным продуктом или может быть получено из коммерчески доступного продукта известным способом.
[0103] [Способ 7-1]
В этом способе осуществляют реагирование соединения (4СА) с галогенирующим реагентом с получением соединения (4СВ) или реагирование с сульфонирующим реагентом с получением соединения (4СВ). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, нитриловый растворитель, такой как ацетонитрил, галогенированный углеводородный растворитель, такой как дихлорметан, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, вода или смешанный растворитель, полученный из вышеупомянутых. В реакции галогенирования может быть использовано избыточное количество галогенводородной кислоты, такой как хлористоводородная кислота или бромистоводородная кислота. В этом случае может быть использован неорганический хлорид, такой как цинка хлорид или лития бромид, катализатор фазового переноса или подобное. Кроме того, эта реакция может быть выполнена с использованием в качестве галогенирующего реагента соединения галогенированного фосфора, такого как фосфора трихлорид. В этом случае N,N-диметилформамид может быть использован в качестве иминиевой соли. В качестве альтернативы может быть использована комбинация органического фосфорсодержащего соединения, такого как трифенилфосфин и четырехгалоидный углерод в качестве галогенирующего реагента. В качестве альтернативы может быть использован галогенирующий реагент, такой как тионилхлорид. В этом случае в качестве добавки может быть использовано основание, такое как пиридин. Галогенирующий реагент может быть использован в количестве 1-10 эквивалентов. В реакции суньфонирования сульфонирующий реагент, такой как п-толуолсульфонилхлорид, может быть использован в количестве 1-10 эквивалентов, а основание, такое как триэтиламин или пиридин, может быть использовано в качестве добавки в количестве 1-10 эквивалентов. Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0104] [Способ 7-2]
В этом способе осуществляют реагирование соединения (4СВ) с соединением (4D) с получением соединения (4-3g). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, диметилсульфоксид, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции для конденсации может быть использовано органическое основание, такое как триэтиламин или неорганическое основание, такое как цезия карбонат. Соединение (4D) используется в количестве 1-10 эквивалентов по отношению к соединению (4-СВ), а основание может быть добавлено в количестве 0-10 эквивалентов. Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. Pro4 может быть снята известным способом снятия защиты для карбоновой кислоты.
[0105] [Способ получения 8]
Способ получения соединения (4СВ-1)
[Химическая формула 28]

В вышеупомянутой формуле А и n определяются выше; X3 представляет собой атом галогена, такой как атом хлора или атом брома; X5 представляет собой атом галогена, такой как атом брома или атом йода; и Pro4 представляет собой известную защитную группу для карбоновой кислоты, такую как этильная группа.
Соединения (4Е) и (4F) могут быть коммерчески доступными продуктами или могут быть получены из коммерчески доступных продуктов известными способами.
[0106] [Способ 8-1]
В этом способе осуществляют реагирование соединения (4Е) с соединением (4F) с получением соединения (4СВ-1). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, диметилсульфоксид, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции для конденсации может быть использовано органическое основание, такое как триэтиламин или неорганическое основание, такое как калия карбонат. Соединение (4F) используется в количестве 1-10 эквивалентов по отношению к соединению (4Е), а основание может быть добавлено в количестве 1-10 эквивалентов. Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. Pro4 также может быть снята известным способом снятия защиты для карбоновой кислоты.
[0107] [Способ получения 9]
Способ получения соединения (4-3b), (4-5a), (4-5b), (4-5c), (4-5d), (4-5e) или (4-5f)
[0108] [Способ получения 9-1]
Способ получения соединения (4-3b)
[Химическая формула 29]
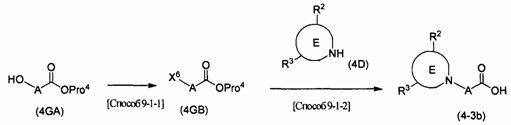
В вышеупомянутой формуле R2, R3, А и Е определяются выше; X6 представляет собой атом галогена, такой как атом хлора, атом брома или атом йода, или уходящую группу сульфоната, такую как трифторметансульфонат; и Pro4 представляет собой известную защитную группу для карбоновой кислоты, такую как этильная группа.
Соединения (4GA) и (4GB) могут быть коммерчески доступными продуктами или могут быть получены из коммерчески доступных продуктов известными способами. В качестве альтернативы соединение (4GB) может быть получено способом, описанным в примере получения какого-либо из примеров, описанных ниже, или подобном.
[0109] [Способ 9-1-1]
В этом способе осуществляют реагирование соединения (4GA) с галогенирующим реагентом с получением соединения (4GB) или реагирование с сульфонирующим реагентом с получением соединения (4GB). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, галогенированный углеводородный растворитель, такой как дихлорметан, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, нитриловый растворитель, такой как ацетонитрил, или смешанный растворитель, полученный из вышеупомянутых. В реакции галогенирования фосфора галид, такой как фосфора пентабромид, или галогенирующий реагент, такой как трифенилфосфиндибромид, могут быть использованы в количестве 1-5 эквивалентов. В реакции сульфонирования сульфонирующий реагент, такой как трифторметансульфоновый ангидрид, может быть использован в количестве 1-5 эквивалентов, а основание, такое как триэтиламин или пиридин, может быть использовано в качестве добавки в количестве 1-10 эквивалентов. Температура реакции составляет от -20°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0110] [Способ 9-1-2]
В этом способе осуществляют реагирование соединения (4GB) с соединением (4D) с получением соединения (4-3b). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, диметилсульфоксид, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции для конденсации может быть использовано органическое основание, такое как триэтиламин, или неорганическое основание, такое как цезия карбонат. Соединение (4D) может быть использовано в количестве 1-10 эквивалентов по отношению к соединению (4GB), а основание может быть добавлено в количестве 0-10 эквивалентов. Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. Более того, в этой реакции может быть добавлен тяжелый металл, такой как медь, а также может быть добавлен металлоорганический катализатор, содержащий палладий и фосфорорганический лиганд. Pro4 может быть снята известным способом снятия защиты для карбоновой кислоты.
[0111] [Способ получения 9-2]
Способ получения соединения (4-5а)
[Химическая формула 30]
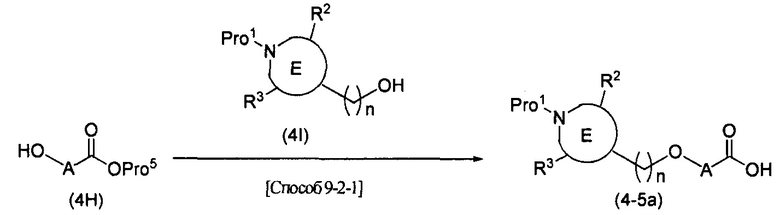
В вышеупомянутой формуле R2, R3, А, Е и n определяются выше; Pro1 представляет собой известную защитную группу для атома азота, такую как трет-бутоксикарбонильная группа; и Pro5 представляет собой известную защитную группу для карбоновой кислоты, такую как этильная группа или бензильная группа.
Соединения (4Н) и (4I) могут быть коммерчески доступными продуктами или могут быть получены из коммерчески доступных продуктов известными способами.
[0112] [Способ 9-2-1]
В этом способе осуществляют реагирование соединения (4Н) и соединения (4I) друг с другом с получением соединения (4-5а). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, растворитель на основе ароматического углеводорода, такой как толуол, галогенированный углеводородный растворитель, такой как дихлорметан, сложноэфирный растворитель, такой как этилацетат, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции можно использовать соединение (4Н) в количестве 0,5-2 эквивалентов по отношению к соединению (4Г). В этой реакции азодикарбоксилат, такой как диизопропилазодикарбоксилат, и трифенилфосфин могут быть использованы в количестве 1-5 эквивалентов, соответственно, по отношению к соединениям (4Н) и (4I). Температура реакции составляет от -20°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. Pro5 может быть снята известным способом снятия защиты для карбоновой кислоты.
[0113] [Способ получения 9-3]
Способ получения соединения (4-5b) или (4-5с)
[Химическая формула 31]
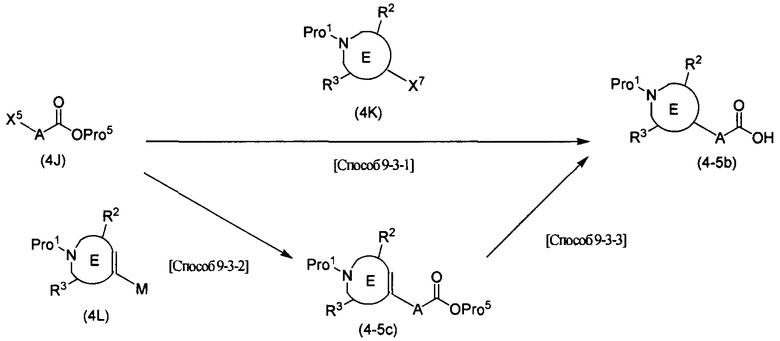
В вышеупомянутой формуле R2, R3, А и Е определяются выше; X5 представляет собой атом галогена, такой как атом брома или атом йода; X7 представляет собой атом галогена, такой как атом брома или атом йода, или уходящую группу сульфоната, такую как метансульфонат или трифторметансульфонат; N представляет собой уходящую группу сложного эфира борной кислоты или подобную; Pro1 представляет собой известную защитную группу для атома азота, такую как трет-бутоксикарбонильная группа; и Pro5 представляет собой известную защитную группу для карбоновой кислоты, такую как этильная группа или бензильная группа, и карбоновая кислота может быть защищенной или может не быть защищенной в этой реакции.
Соединения (4J), (4K) и (4L) могут быть коммерчески доступными продуктами или могут быть получены из коммерчески доступных продуктов известными способами.
[0114] [Способ 9-3-1]
В этом способе осуществляют реагирование соединения (4J) и соединения (4K) друг с другом с получением соединения (4-5b). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, растворитель на основе ароматического углеводорода, такой как толуол, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции можно использовать соединение (4К) в количестве 0,5-2 эквивалентов по отношению к соединению (4J). В этой реакции палладиевое комплексное соединение, содержащее органическое фосфорсодержащее соединение в качестве лиганда, может быть использовано в качестве катализатора. Катализатор может быть получен в реакционной системе. В этой реакции порошок цинка может быть использован в количестве 0,5-2 эквивалентов по отношению к соединению (4J). Кроме того, в качестве добавки могут быть добавлены меди галид, дигалогенированное алкильное соединение, галогенированное кремнийорганическое соединение или подобное. Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. Pro5 может быть снята известным способом снятия защиты для карбоновой кислоты.
[0115] [Способ 9-3-2]
В этом способе осуществляют реагирование соединения (4J) и соединения (4L) друг с другом с получением соединения (4-5с). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как 1,4-диоксан, растворитель на основе ароматического углеводорода, такой как толуол, спиртовой растворитель, такой как этанол, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции можно использовать соединение (4L) в количестве 0,5-2 эквивалентов по отношению к соединению (4J). В этой реакции палладиевое комплексное соединение, содержащее органическое фосфорсодержащее соединение в качестве лиганда, может быть использовано в качестве катализатора. Катализатор может быть получен в реакционной системе. В этой реакции основание, такое как натрия карбонат, может быть использовано в количестве 1-10 эквивалентов по отношению к соединению (4J). Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0116] [Способ 9-3-3]
В этом способе соединение (4-5с) восстанавливают и с него снимают защиту с получением соединения (4-5b). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, сложноэфирный растворитель, такой как этилацетат, спиртовой растворитель, такой как этанол, амидный растворитель, такой как N,N-димeтилфopмaмид или N-метилпирролидинон, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции катализатор каталитического восстановления, такой как палладий на угле, может быть использован в количестве 0,001-1 эквивалента по отношению к соединению (4-5с). Кроме того, реакция может быть выполнена в атмосфере водорода при давлении, варьирующемся от нормального давления до 20 атм. Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. Pro5 может быть снята известным способом снятия защиты для карбоновой кислоты.
[0117] [Способ получения 9-4]
Способ получения соединения (4-5d) или (4-5е)
[Химическая формула 32]

В вышеупомянутой формуле R3, А и Е определяются выше; Х8 представляет собой атом галогена, такой как атом фтора; Pro1 представляет собой известную защитную группу для атома азота, такую как трет-бутоксикарбонильная группа; a Pro4 представляет собой известную защитную группу для карбоновой кислоты, такую как этильная группа, и в этой реакции карбоновая кислота может быть защищена или может не быть защищена.
Соединение (4-5с) может быть коммерчески доступным продуктом или может быть получено из коммерчески доступного продукта известным способом. В качестве альтернативы эти соединения могут быть получены способом, описанным в примере получения какого-либо из примеров, описанных ниже, или подобным.
[0118] [Способ 9-4-1]
В этом способе осуществляют реагирование соединения (4-5с) с получением соединения (4-5d). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран. В этой реакции борановое комплексное соединение, такое как комплекс боран-метилсульфид, может быть использовано в количестве 1-2 эквивалентов по отношению к соединению (4-5с). Температура реакции составляет от -20°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. Затем с использованием водного раствора натрия гидроксида в количестве 1-10 эквивалентов и раствора перекиси водорода в количестве 1-10 эквивалентов может быть получена гидроксильная группа. Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
[0119] [Способ 9-4-2]
В этом способе гидроксильную группу соединения (4-5d) замещают галогеном с получением соединения (4-5е). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, галогенированный углеводородный растворитель, такой как дихлорметан, эфирный растворитель, такой как тетрагидрофуран, сложноэфирный растворитель, такой как этилацетат, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции галогенирующий реагент, такой как [бис-(2-метоксиэтил)-амино]серы трифторид, может быть использован в количестве 1-10 эквивалентов по отношению к соединению (4-5d). Температура реакции составляет от 0°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. Pro4 может быть снята известным способом снятия защиты для карбоновой кислоты.
[0120] [Способ получения 9-5]
Способ получения соединения (4-5f)
[Химическая формула 33]

В вышеупомянутой формуле R2, R3, А и Е определяются выше; Х7 представляет собой атом галогена, такой как атом брома или атом йода, или уходящую группу сульфоната, такую как метансульфонат или трифторметансульфонат; Pro1 представляет собой известную защитную группу для атома азота, такую как трет-бутоксикарбонильная группа; и Pro4 представляет собой известную защитную группу для карбоновой кислоты, такую как этильная группа, и в этой реакции карбоновая кислота может быть защищена или может не быть защищена.
Соединения (4-5b) и (4K) могут быть коммерчески доступными продуктами или могут быть получены из коммерчески доступных продуктов известными способами. В качестве альтернативы эти соединения могут быть получены способами, описанными в примерах получения какого-либо из примеров, описанных ниже, или подобными.
[0121] [Способ 9-5-1]
В этом способе осуществляют реагирование соединения (4-5b) и соединения (4K) друг с другом с получением соединения (4-5f). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, или смешанный растворитель, полученный из вышеупомянутых. В этой реакции можно использовать соединение (4К) в количестве 0,5-2 эквивалентов по отношению к соединению (4-5b). Кроме того, может быть использовано основание, такое как натрия гидрид или калия трет-бутоксид, в количестве 1-5 эквивалентов по отношению к соединению (4-5b). Температура реакции составляет от -20°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов. Pro4 может быть снята известным способом снятия защиты для карбоновой кислоты.
[0122] [Способ получения 10]
Способ получения соединения (4K)
[Химическая формула 34]

В вышеупомянутой формуле R2, R3 и Е определяются выше; Х7 представляет собой атом галогена, такой как атом брома или атом йода, или уходящую группу сульфоната, такую как метансульфонат или трифторметансульфонат; и Pro1 представляет собой известную защитную группу для атома азота, такую как трет-бутоксикарбонильная группа.
Соединение (5К) может быть коммерчески доступным продуктом или может быть получено из коммерчески доступного продукта известным способом.
[0123] [Способ 10]
В этом способе осуществляют реагирование соединения (5K) с галогенирующим реагентом с получением соединения (4K) или реагирование с сульфонирующим реагентом с получением соединения (4K). Выбор растворителя, используемого в реакции, специально не ограничивают, при условии, что он растворяет исходный материал до некоторой степени и не подавляет реакцию, и могут быть использованы, например, эфирный растворитель, такой как тетрагидрофуран, галогенированный углеводородный растворитель, такой как дихлорметан, амидный растворитель, такой как N,N-диметилформамид или N-метилпирролидинон, нитриловый растворитель, такой как ацетонитрил, или смешанный растворитель, полученный из вышеупомянутых. В реакции галогенирования фосфора галид, такой как фосфора пентабромид, или галогенирующий реагент, такой как трифенилфосфиндибромид, могут быть использованы в количестве 1-5 эквивалентов. В реакции сульфонирования сульфонирующий реагент, такой как метансульфонилхлорид или трифторметансульфоновый ангидрид, может быть использован в количестве 1-5 эквивалентов, а основание, такое как триэтиламин или пиридин, может быть использовано в качестве добавки в количестве 1-10 эквивалентов. Температура реакции составляет от -20°С до температуры флегмы, и время реакции составляет от 10 минут до 24 часов.
Пример
[0124] Соединения в соответствии с настоящим изобретением могут быть получены способами, описанными в примерах получения и примерах, описанных ниже, например. Однако, эти способы являются только лишь примерами, и поэтому соединения в соответствии с настоящим изобретением не ограничиваются теми, которые получены конкретными примерами, описанными ниже, в любых случаях.
[0125] В примерах получения и примерах Silica gel 60 (Kanto Chemicals) или Presep Silica Gel (WAKO) использовали в качестве силикагеля для очистки, используемого для колоночной хроматографии на силикагеле, если не указано иное. Кроме того, силикагель NH (Fuji Silysia Chemical LTD.) или Hi-Flash Column Amino (YAMAZENE CORPORATION) использовали в качестве силикагеля для очистки, используемого для колоночной хроматографии на силикагеле NH, a TLC Plates NH (20 см ×20 см, Fuji Silysia Chemical LTD.) использовали в качестве пластинки для тонкослойной хроматографии, используемой для TLC (тонкослойной хроматографии) на силикагеле NH.
[0126] Спектрометр Varian Mercury 400, Varian Mercury Plus 400, Varian INOVA 500 или Avance 600 MHz (Broker) использовали для измерения спектров протонного ядерного магнитного резонанса, и спектры протонного ядерного магнитного резонанса измеряли при 400 МГц, если не указано иное. Химические сдвиги спектров протонного ядерного магнитного резонанса регистрируют в единицах измерения δ (ppm) относительно тетраметилсилана, а константы взаимодействия регистрируют в единицах измерения герц (Гц). Сокращения паттернов расщепления являются следующими: s: синглет, d: дуплет, t: триплет, q: квартет, quin: квинтет, spt: септет, m: мультиплет и brs: широкий синглет.
[0127] Waters Micromass ZQ 2000, Waters SQ Detector 2 или Thermo Fisher Scientific LCQ использовали для измерения масс-спектров. В способе ионизации для измерения использовали метод ионизации электрораспылением (ESI).
[0128] В примерах получения и примерах соответственным образом использовали коммерчески доступные продукты в качестве коммерчески доступных соединений. В вышеприведенном описании термин ʺВОСʺ относится к трет-бутоксикарбонилу, а термин ʺТʺ относится к трет.
[0129] [Пример 1]
6-Метокси-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид
[Химическая формула 35]

Трет-бутил 4-(4-((4-((6-метокси-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)фенил)пиперидин-1-карбоксилат (3,42 г, 5,70 ммоля), описанный в примерах получения 1-8, растворяли в дихлорметане (45 мл) и добавляли трифторуксусную кислоту (15 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 60 минут, а затем концентрировали в вакууме, а затем остаток растворяли в дихлорметане-триэтиламине и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 97:3 - 4:1). Целевую фракцию концентрировали в вакууме, а затем осадок собирали фильтрацией и промывали диэтиловым эфиром и этилацетатом с получением названного соединения (2,61 г, 92%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,55-1,70 (2Н, m), 1,78-1,88 (2H, m), 2,62-2,80 (3H, m), 3,06 (3H, d, J=4,8 Гц), 3,15-3,24 (2H, m), 3,86 (3H, s), 5,52-5,61 (1H, m), 6,54 (1H, d, J=3,3 Гц), 6,60 (1H, dd, J=5,7, 2,4 Гц), 7,23 (1H, d, J=3,7 Гц), 7.28-7,35 (3H, m), 7,80 (2H, d, J=8,4 Гц), 7,91 (1H, d, J=2,2 Гц), 8,03 (1H, s), 8,10 (1H, d, J=5,9 Гц), 8,53 (1H, brs).
[0130] Исходный материал трет-бутил-4-(4-((4-((6-метокси-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)фенил)пиперидин-1-карбоксилат синтезировали следующим способом.
[0131] [Пример получения 1-1]
(Е)-2-(Бензилокси)-1-метокси-4-(2-нитровинил)бензол
[Химическая формула 36]

Коммерчески доступный 3-бензилокси-4-метоксибензальдегид (30 г, 124 ммоля) растворяли в уксусной кислоте (100 мл), затем добавляли аммония ацетат (10,8 г, 140 ммоль) и нитрометан (16,8 мл, 310 ммоля) в атмосфере азота при комнатной температуре и смесь нагревали и перемешивали при 130°С в течение 2 часов и 20 минут. Смесь охлаждали до комнатной температуры, затем осадок собирали фильтрацией и промывали этанолом с получением названного соединения (28,5 г, 81%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 3,95 (3H, s), 5,18 (2H, s), 6,93 (1H, d, J=8,1 Гц), 7,03 (1H, d, J=2,2 Гц), 7,17 (1H, dd, J=8,4, 2,2 Гц), 7,30-7,47 (6Н, m), 7,91 (1H, d, J=13,5 Гц).
[0132] [Пример получения 1-2]
(Е)-1-(Бензилокси)-2-метокси-4-нитро-5-(2-нитровинил)бензол
[Химическая формула 37]

69% азотную кислоту (10 мл, 155 ммоль) добавляли в смесь (Е)-2-(бензилокси)-1-метокси-4-(2-нитровинил)бензола (10 г, 35,1 ммоля), описанного в примере получения 1-1, и уксусной кислоты (70 мл) в атмосфере азота при 25°С и смесь перемешивали при 40°С в течение 3 часов. Реакционную смесь выливали на лед и осадок собирали фильтрацией, а затем промывали этанолом с получением названного соединения (10,5 г, 91%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 4,02 (3Н, s), 5,28 (2Н, s), 6,93 (1Н, s), 7,22 (1H, d, J=13,5 Гц), 7,35-7,48 (5H, m), 7,75 (1H, s), 8,58 (1H, d, J=13,2 Гц).
[0133] [Пример получения 1-3]
6-Метокси-1Н-индол-5-ол
[Химическая формула 38]

(Е)-1-(Бензилокси)-2-метокси-4-нитро-5-(2-нитровинил)бензол (9,4 г, 28,5 ммоля), описанный в примере получения 1-2, суспендировали в метаноле (300 мл), затем добавляли 10% палладий на угле (содержание воды 50%) (3,09 г) и смесь перемешивали в атмосфере водорода в течение 3 часов. Катализатор отфильтровывали через целит и промывали метанолом. Фильтрат концентрировали в вакууме и полученное очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 3:1 - 3:7). Целевую фракцию концентрировали в вакууме с получением названного соединения (2,12 г, 46%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 3,92 (3Н, s), 5,45 (1H, s), 6,39-6,44 (1H, m), 6,88 (1H, s), 7,08 (1H, t, J=2,9 Гц), 7,14 (1H, s), 7,95 (1H, brs).
[0134] [Пример получения 1-4]
4-((6-Метокси-1Н-индол-5-ил)окси)пиридин-2-амин
[Химическая формула 39]

6-Метокси-1Н-индол-5-ол (2,86 г, 17,6 ммоля), описанный в примере получения 1-3, N-(4-хлорпиридин-2-ил)ацетамид, описанный в примере получения 1-5 (8,98 г, 52,7 ммоля), и калия трет-бутоксид (3,94 г, 35,1 ммоля) растворяли в диметилсульфоксиде (45 мл) в атмосфере азота и смесь нагревали и перемешивали при 160°С в течение 12,5 часа. Реакционную жидкость охлаждали до комнатной температуры и добавляли воду и этилацетат для разделения. Водный слой дважды экстрагировали этилацетатом и объединенный органический слой дважды промывали водой, а затем насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния. Осушающее средство отфильтровывали, затем фильтрат концентрировали в вакууме и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 0:1 - этилацетат:метанол = 99:1 - 9:1). Смешанную фракцию концентрировали в вакууме, полученное очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 0:1 - этилацетат:метанол = 99:1 - 9:1), а затем целевую фракцию концентрировали в вакууме с получением неочищенного продукта (3,88 г).
Неочищенный продукт (3,88 г) растворяли в метаноле (75 мл), добавляли 28% натрия метоксид (14 мл, 68,6 ммоля) в атмосфере азота при комнатной температуре, а затем смесь нагревали и перемешивали при 70°С в течение 5,5 часа. Реакционную смесь охлаждали до комнатной температуры, а затем добавляли воду и этилацетат для разделения. Водный слой один раз экстрагировали этилацетатом и объединенный органический слой промывали насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния. Осушающее средство отфильтровывали, фильтрат концентрировали в вакууме и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:3 - 0:1 - этилацетат:метанол = 99:1 - 19:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (1,97 г, 44%).
1H-ЯМР спектр (CDCl3) δ (ppm): 3,82 (3H, s), 4,29 (2H, brs), 5,89-5,92 (1H, m), 6,30 (1H, dd, J=5,9, 2,2 Гц), 6,49 (1H, ddd, J=3,2, 2,2, 0,9 Гц), 7,01 (1H, s), 7,17 (1H, dd, J=3,1, 2,4 Гц), 7,33 (1H, s), 7,89 (1H, d, J=6,0 Гц), 8,19 (1H, brs).
[0135] [Пример получения 1-5]
N-(4-Хлорпиридин-2-ил)ацетамид
[Химическая формула 40]
Коммерчески доступный 2-амино-4-хлорпиридин (50 г, 389 ммоль) растворяли в уксусном ангидриде (500 мл), добавляли триэтиламин (271 мл, 1,94 моля) при 20°С и смесь перемешивали при 60°С в течение 12 часов. Смесь охлаждали до комнатной температуры, а затем растворитель выпаривали. Остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 4:1 - 1:1), а затем целевую фракцию концентрировали в вакууме с получением названного соединения (66 г, 99%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 2,21 (3H, s), 7.05 (1H, dd, J=5,4, 1,9 Гц), 8,15 (1H, d, J=5,4 Гц), 8,30 (2H, brs).
[0136] [Пример получения 1-6]
5-((2-Аминопиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 41]

4-((6-Метокси-1Н-индол-5-ил)окси)пиридин-2-амин (1,7 г, 6,66 ммоля), описанный в примере получения 1-4, растворяли в N,N-диметилформамиде (25 мл), добавляли 50-72% маслянистого натрия гидрида (424 мг) в атмосфере азота при 0°С, а затем смесь перемешивали при комнатной температуре в течение 45 минут. Смесь снова охлаждали до 0°С, добавляли фенилметилкарбамат (1,64 г, 10,8 ммоля), описанный в примере получения 1-7, и полученное вещество перемешивали при комнатной температуре в течение 3 часов. Насыщенный водный раствор аммония хлорида, воду и этилацетат добавляли в реакционную смесь для разделения. Водный слой один раз экстрагировали этилацетатом и объединенный органический слой промывали водой и насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния. Осушающее средство отфильтровывали, затем фильтрат концентрировали в вакууме и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:3 - 0:1 - этилацетат:метанол = 99:1 - 19:1). Целевую фракцию концентрировали в вакууме, а затем осадок собирали фильтрацией и промывали этилацетатом с получением названного соединения (1,37 г, 66%). Смешанную фракцию концентрировали в вакууме и очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 3:7 - 0:1 - этилацетат:метанол = 99:1 - 19:1). Целевую фракцию концентрировали в вакууме, а затем осадок собирали фильтрацией и дважды промывали этилацетатом с получением названного соединения (387 мг, 19%).
1H-ЯМР спектр (CDCl3) δ (ppm): 3,07 (3Н, d, J=4,6 Гц), 3,86 (3Н, s), 4,31 (2H, brs), 5,44-5,56 (1Н, m), 5,89 (1H, d, J=2,2 Гц), 6,27 (1H, dd, J=5,9, 2,2 Гц), 6,55 (1Н, d, J=3,7 Гц), 7,23 (1H, d, J=3,7 Гц), 7,25-7,28 (1H, m), 7,89 (1H, d, J=5,9 Гц), 8,01 (1H, brs).
[0137] Реагент фенилметилкарбамат синтезировали следующим способом.
[0138] [Пример получения 1-7]
Фенилметилкарбамат
[Химическая формула 42]
Смесь коммерчески доступного метиламина гидрохлорида (50 г, 0,74 моля), пиридина (124 мл, 1,53 моля) и N,N-диметилформамида (500 мл) перемешивали при 5°С и по каплям за 2 часа добавляли коммерчески доступный фенилхлоркарбонат (94 мл, 0,75 моля). После завершения добавления смесь перемешивали в атмосфере азота при комнатной температуре в течение 16 часов. Реакционную смесь добавляли в ледяную воду (2 л) и дважды экстрагировали этилацетатом (1,5 л). Органический слой промывали водой (1 л) и насыщенным солевым раствором (300 мл). Органический слой сушили над безводным сульфатом магния, а затем растворитель выпаривали. Добавляли н-гептан и этилацетат в концентрированный остаток, осадок собирали фильтрацией и промывали н-гептаном и трет-бутилметиловым эфиром с получением названного соединения (74,2 г, 66%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 2,90 (3H, d, J=4,9 Гц), 4,95 (1Н, brs), 7,08-7,16 (2H, m), 7,16-7,24 (1Н, m), 7,31-7,41 (2H, m)
[0139] [Пример получения 1-8]
Трет-бутил 4-(4-((4-((6-метокси-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)фенил)пиперидин-1-карбоксилат
[Химическая формула 43]
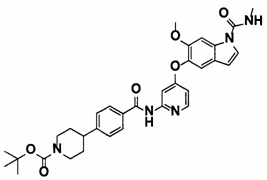
Бензотриазол (2,32 г, 19,5 ммоля) растворяли в дихлорметане (100 мл), добавляли тионилхлорид (1,4 мл, 19,2 ммоля) в атмосфере азота при комнатной температуре и смесь перемешивали в течение 5 минут. 4-(1-(трет-бутоксикарбонил)пиперидин-4-ил)бензойную кислоту (5,4 г, 17,7 ммоля), описанную в примере получения 1-12, добавляли в реакционную смесь при комнатной температуре, и смесь перемешивали в течение 25 минут. Реакционную смесь фильтровали через стеклянный фильтр, полностью покрытый безводным натрия сульфатом, а затем безводный натрия сульфат промывали дихлорметаном, затем фильтрат добавляли в смесь 5-((2-аминопиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамида (2,5 г, 8,01 ммоля), описанного в примере получения 1-6, триэтиламина (11 мл, 79,4 ммоля) и 4-диметиламинопиридина (101 мг, 0,827 ммоля) в тетрагидрофуране (80 мл) при 0°С. Полученное вещество перемешивали при комнатной температуре в течение 5 часов, а затем реакционную смесь концентрировали в вакууме. К остатку добавляли воду и этилацетат для разделения и органический слой промывали насыщенным солевым раствором, а затем сушили над безводным магния сульфатом и фильтровали. Фильтрат концентрировали в вакууме, остаток растворяли в тетрагидрофуране, добавляли избыточное количество 9,8 М раствора метиламинметанола при комнатной температуре и смесь перемешивали в течение 50 минут. Реакционную смесь концентрировали в вакууме, остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 1:3 - 0:1). Целевую фракцию концентрировали в вакууме, осадок собирали фильтрацией и промывали диэтиловым эфиром и этилацетатом с получением названного соединения (3,15 г, 66%). Фильтрат объединяли со смешанной фракцией, полученное вещество концентрировали в вакууме, растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 1:3 - 0:1). Целевую фракцию концентрировали в вакууме, осадок собирали фильтрацией и промывали диэтиловым эфиром и этилацетатом с получением названного соединения (264 мг, 5,5%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,48 (9H, s), 1,55-1,69 (2H, m), 1,77-1,87 (2H, m), 2,64-2,89 (3H, m), 3,02-3,07 (3H, m), 3,86 (3Н, s), 4,26 (2H, brs), 5,62 (1H, brs), 6,50-6,55 (1H, m), 6,61 (1H, dd, J=5,9, 2,2 Гц), 7,22 (1H, d, J=3,7 Гц), 7,27-7,33 (3Н, m), 7,80 (2H, d, J=8,4 Гц), 7,90 (1H, d, J=2,2 Гц), 8,04 (1H, s), 8,09 (1H, d, J=5,9 Гц), 8,54 (1H, brs).
[0140] 4-(1-(Трет-бутоксикарбонил)пиперидин-4-ил)бензойную кислоту синтезировали следующим способом.
[0141] [Пример получения 1-9]
1-(4-Фенилпиперидин-1-ил)этанон
[Химическая формула 44]
Смесь коммерчески доступного 4-фенилпиперидина (10 г, 62 ммоля), пиридина (5,7 мл, 70,5 ммоля) и тетрагидрофурана (80 мл) перемешивали при 0°С и затем за 10 минут по каплям добавляли смесь ацетилхлорида (5 мл, 70,3 ммоля) и тетрагидрофурана (20 мл). Смесь перемешивали в атмосфере азота при 25°С в течение 14 часов. Этилацетат (100 мл) и воду (100 мл) добавляли в реакционную жидкость для отделения. Водный слой экстрагировали этилацетатом (100 мл), затем органические слои объединяли и полученное вещество промывали насыщенным водным раствором натрия бикарбоната (100 мл), водой (100 мл), а затем насыщенным солевым раствором (50 мл). Органический слой сушили над безводным сульфатом магния, а затем растворитель выпаривали с получением названного соединения (12,3 г, 98%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,52-1,78 (2H, m), 1,81-1,99 (2H, m), 2,14 (3H, s), 2,63 (1H, td, J=12,9, 2,7 Гц), 2,74 (1Н, tt, J=12,1, 3,7 Гц), 3,17 (1Н, td, J=13,2, 2,6 Гц), 3,84-4,02 (1Н, m), 4,69-4,89 (1Н, m), 7,08-7,43 (5H, m).
[0142] [Пример получения 1-10]
4-(1-Ацетилпиперидин-4-ил)бензойная кислота
[Химическая формула 45]

Смесь алюминия хлорида (III) (26 г, 195 ммоль) и дихлорметана (200 мл) перемешивали при 0°С и за 10 минут по каплям добавляли оксалилхлорид (20 мл, 228 ммоль). Затем за 30 минут по каплям добавляли смесь 1-(4-фенилпиперидин-1-ил)этанона (12,3 г, 60,5 ммоля), описанного в примере получения 1-9, и дихлорметана (50 мл). Смесь перемешивали в атмосфере азота при 25°С в течение 14 часов. Реакционную жидкость выливали на лед и добавляли этилацетат (1 л) и воду (1 л) для отделения. Водный слой дважды экстрагировали этилацетатом (1 л), затем органический слой дважды промывали водой (1 л), а затем насыщенным солевым раствором (500 мл). Органический слой сушили над безводным сульфатом магния, а затем растворитель выпаривали. Этилацетат добавляли в концентрированный остаток, продукт собирали фильтрацией и промывали этилацетатом с получением названного соединения (9,09 г, 61%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,49-1,82 (2H, m), 1,92 (2H, t, J=13,2 Гц), 2,15 (3H, s), 2,65 (1Н, t, J=11,7 Гц), 2,75-2,94 (1Н, m), 3,08-3,30 (1Н, m), 3,97 (1Н, d, J=13,2 Гц), 4,82 (1Н, d, J=12,8 Гц), 7,30 (2H, d, J=8,4 Гц), 8,05 (2H, d, J=8,1 Гц).
[0143] [Пример получения 1-11]
4-(Пиперидин-4-ил)бензойной кислоты гидрохлорид
[Химическая формула 46]
Смесь 4-(1-ацетилпиперидин-4-ил)бензойной кислоты (4,50 г, 18,2 ммоля), описанной в примере получения 1-10, и 5 М хлористоводородной кислоты (50 мл, 250 ммоль) перемешивали в атмосфере азота при 140°С в течение 18 часов. Смесь охлаждали до комнатной температуры, а затем продукт собирали фильтрацией и промывали водой с получением названного соединения (3,77 г, 86%).
1Н-ЯМР спектр (DMSO-d6) δ (ppm): 1,60-2,15 (4H, m), 2,76-3,16 (3H, m), 3,27-3,45 (2H, m), 7,36 (2H, d, J=8,1 Гц), 7,92 (2H, d, J=8,1 Гц), 8,65-9,04 (2H, m), 12,89 (1H, brs).
[0144] [Пример получения 1-12]
4-(1-(Трет-бутоксикарбонил)пиперидин-4-ил)бензойная кислота
[Химическая формула 47]

Смесь 4-(пиперидин-4-ил)бензойной кислоты гидрохлорида (2,00 г, 8,27 ммоля), описанного в примере получения 1-11, 1 M раствора натрия гидроксида (25 мл, 25 ммоля) и ацетона (50 мл) перемешивали при 25°С и по каплям за 10 минут добавляли раствор ди-трет-бутилдикарбоната (1,9 г, 8,71 ммоля) в ацетоне (25 мл). Смесь перемешивали в атмосфере азота при 25°С в течение 18 часов. Добавляли 1 М хлористоводородную кислоту (17 мл) при охлаждении при 0°С. Смесь дважды экстрагировали этилацетатом (100 мл). Органический слой промывали насыщенным солевым раствором (50 мл). Органический слой сушили над безводным сульфатом магния, а затем концентрировали в вакууме. Добавляли н-гептан и трет-бутилметиловый эфир в концентрированный остаток, продукт собирали фильтрацией и промывали н-гептаном с получением названного соединения (2,30 г, 91%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,49 (9Н, s), 1,57-1,76 (2H, m), 1,84 (2H, d, J=13,5 Гц), 2,62-2,97 (3H, m), 4,27 (2H, brs), 7,28-7,36 (2H, m), 7,98-8,10 (2H, m).
[0145] [Пример 2]
5-((2-(4-(1-Этилпиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 48]
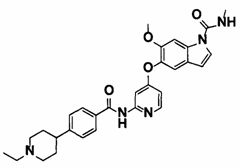
Ацетальдегид (0,677 мл, 12,1 ммоля) и натрия триацетоксиборгидрид (2,57 г, 12,1 ммоля) добавляли в смесь 6-метокси-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида (3,03 г, 6,06 ммоля), описанного в примере 1, и тетрагидрофурана (80 мл) и смесь перемешивали при комнатной температуре в течение 3 часов. Этилацетат, насыщенный водный раствор натрия бикарбоната и воду добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хромагографии на силикагеле NH (н-гептан:этилацетат = 1:4 - 0:1 - этилацетат:метанол = 99:1 - 9:1). Целевую фракцию концентрировали в вакууме, остаток собирали фильтрацией и промывали н-гептаном и диэтиловым эфиром с получением названного соединения (2,61 г, 82%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,12 (3H, t, J=7,1 Гц), 1,77-1,90 (4Н, m), 1,98-2,07 (2H, m), 2,45 (2H, q, J=7,1 Гц), 2,52-2,62 (1Н, m), 3,05-3,12 (5H, m), 3,83 (3H, s), 5,50-5,58 (1H, m), 6,55 (1Н, d, J=3,7 Гц), 6,60 (1Н, dd, J=5,9, 2,4 Гц), 7,23 (1Н, d, J=3,7 Гц), 7,30-7,35 (3H, m), 7,79 (2H, d, J=8,2 Гц), 7,91 (1Н, d, J=2,4 Гц), 8,03 (1Н, s), 8,10 (1Н, d, J=5,7 Гц), 8,50 (1H, brs).
[0146] [Пример 3]
5-((2-(4-(1-(2-Гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 49]

Коммерчески доступный 2-гидроксиацетальдегид (110 мг, 1,83 ммоля), натрия триацетоксиборгидрид (382 мг, 1,80 ммоля) и уксусную кислоту (103 μл, 1,80 ммоля) добавляли в смесь 6-метокси-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол -1-карбоксамида (300 мг, 0,601 ммоля), описанного в примере 1, и тетрагидрофурана (15 мл) в атмосфере азота, и смесь перемешивали при комнатой температуре в течение 15 часов. Насыщенный водный раствор натрия бикарбоната и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 99:1 - 4:1). Целевую фракцию и смешанную фракцию концентрировали в вакууме, затем остаток смешанной фракции очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 97:3 - 19:1 - 23:2). Объединенную целевую фракцию концентрировали в вакууме, осадок собирали фильтрацией и промывали диэтиловым эфиром и этилацетатом с получением названного соединения (209 мг, 64%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,70-1,90 (4H, m), 2,14-2,25 (2H, m), 2,53-2,65 (3H, m), 2,97-3,09 (2H, m), 3,06 (3H, d, J=4,4 Гц), 3,63 (2H, t, J=5,3 Гц), 3,86 (3H, s), 5,51-5,60 (1H, m), 6,54 (1H, d, J=3,7 Гц), 6,61 (1H, dd, J=5,7, 2,4 Гц), 7,23 (1Н, d, J=3,7 Гц), 7,29-7,35 (3H, m), 7,80 (2H, d, J=8,4 Гц), 7,91 (1H, d, J=2,6 Гц), 8,04 (1H, s), 8,10 (1H, d, J=5,9 Гц), 8,51 (1H, brs).
[0147] [Пример 4]
(S)-5-((2-(4-(1-(2-Гидроксипропил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 50]

Коммерчески доступный (S)-(-)-пропиленоксид (233 мг, 4,00 ммоля) добавляли в смесь 6-метокси-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида (400 мг, 0,801 ммоля), описанного в примере 1, и этанола (10 мл) и смесь нагревали и перемешивали в закупоренной пробирке при 60°С в течение 1 часа. Смесь охлаждали до комнатной температуры, а затем добавляли тетрагидрофуран (5,0 мл), смесь нагревали и перемешивали при 70°С в течение 2 часов. Реакционную смесь концентрировали в вакууме, полученный остаток растворяли в дихлорметане, и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:4-0:1 - этилацетат:метанол = 99:1 - 19:1). Целевую фракцию концентрировали в вакууме, осадок собирали фильтрацией и промывали диэтиловым эфиром и этилацетатом с получением названного соединения (347 мг, 78%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,14 (3H, d, J=6,2 Гц), 1,66-1,89 (4Н, m), 1,98-2,09 (1H, m), 2,21-2,45 (3H, m), 2,52-2,64 (1H, m), 2,88-2,96 (1H, m), 3,06 (3H, d, J=4,8 Гц), 3,10-3,17 (1H, m), 3,57 (1H, brs), 3,80-3,92 (1H, m), 3,86 (3H, s), 5,52-5,59 (1H, m), 6,54 (1H, d, J=3,7 Гц), 6,60 (1H, dd, J=5,9, 2,6 Гц), 7,23 (1H, d, J=3,7 Гц), 7,29-7,35 (3H, m), 7,78-7,83 (2H, m), 7,91 (1H, d, J=2,6 Гц), 8,03 (1H, s), 8,10 (1H, d, J=5,9 Гц), 8,53 (1H, brs).
[0148] [Пример 5]
5-((2-(4-(1-(3-Фторпропил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 51]
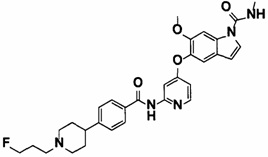
Триэтиламин (11,8 μл, 0,085 ммоля) и 3-фторпропил-4-метилбензолсульфонат (16,1 мг, 0,069 ммоля), описанный в примере получения 5-1, добавляли в смесь 6-метокси-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида (14,1 мг, 0,028 ммоля), описанного в примере 1, и N,N-диметилформамида (0,5 мл), смесь нагревали и перемешивали при 50°С в течение 1 часа, а затем перемешивали при комнатной температуре в течение 13 часов. Триэтиламин (11,8 μл, 0,085 ммоля) и 3-фторпропил-4-метилбензолсульфонат (16,1 мг, 0,069 ммоля) добавляли в реакционную смесь, смесь нагревали и перемешивали при 50°С в течение 3 часов. Полученное вещество охлаждали до комнатной температуры, а затем добавляли воду и этилацетат для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 2:3 - 0:1 этилацетат:метанол = 99:1 - 19:1 - 9:1). Целевую фракцию концентрировали в вакууме, осадок собирали фильтрацией и промывали диэтиловым эфиром с получением названного соединения (10,2 мг, 65%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,72-2,00 (6Н, m), 2,02-2,13 (2H, m), 2,47-2,62 (3H, m), 3,01-3,09 (2H, m), 3,06 (3H, d, J=4,8 Гц), 3,86 (3H, s), 4,47 (1H, t, J=6,0 Гц), 4,59 (1H, t, J=6,0 Гц), 5,48-5,58 (1H, m), 6,55 (1H, d, J=3,7 Гц), 6,60 (1H, dd, J=5,7, 2,4 Гц), 7,23 (1H, d, J=3,7 Гц), 7,30-7,34 (3H, m), 7,77-7,82 (2H, m), 7,91 (1H, d, J=2,6 Гц), 8,03 (1H, s), 8,10 (1H, d, J=6,2 Гц), 8,50 (1H, brs).
[0149] Реагент 3-фторпропил-4-метилбензолсульфонат синтезировали следующим способом.
[0150] [Пример получения 5-1]
3-Фторпропил-4-метилбензолсульфонат
[Химическая формула 52]
Триэтиламин (11 мл, 79,4 ммоля), 4-диметиламинопиридин (390 мг, 3,19 ммоля) и п-толуолсульфонил хлорид (13,4 г, 70,4 ммоля) добавляли в смесь коммерчески доступного 3-фторпропан-1-ола (5,0 г, 64 ммоля) и тетрагидрофурана (120 мл) в атмосфере азота при 0°С и смесь перемешивали при комнатной температуре в течение 90 часов. Воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным магния сульфатом и фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане и н-гептане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 10:1 - 2:1) с получением названного соединения (12,7 г, 85%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,97-2,15 (2Н, m), 2,46 (3H, s), 4,16 (2H, t, J=6,2 Гц), 4,49 (2H, dt, J=46,8, 5,6 Гц), 7,36 (2H, dd, J=8,4, 0,6 Гц), 7,75-7,85 (2H, m).
[0151] [Пример 6]
5-((2-(4-(3-Фторпиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 53]

Трет-бутил 3-фтор-4-(4-((4-((6-метокси-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)фенил)пиперидин-1-карбоксилат (29,1 мг, 0,047 ммоля), описанный в примере получения 6-5, растворяли в дихлорметане (4 мл), добавляли трифторуксусную кислоту (0,8 мл, 10,4 ммоля) при комнатной температуре, а затем смесь перемешивали в течение 30 минут. Растворитель выпаривали, полученный остаток растворяли в дихлорметане (4 мл) и добавляли триэтиламин для нейтрализации избыточной трифторуксусной кислоты. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:4 - 0:1 - этилацетат:метанол = 97:3 - 19:1) с получением названного соединения (16,8 мг, 69%).
1Н-ЯМР спектр (500 МГц, СDCl3) δ (ppm): 1,71-1,80 (2H, m), 1,89-1,97 (1H, m), 2,63-2,70 (1H, m), 2,70-2,76 (1H, m), 2,79-2,89 (1H, m), 3,05 (3H, d, J=4,9 Гц), 3,09 (1H, d, J=11,2 Гц), 3,48 (1H, dt, J=11,3, 4,1 Гц), 3,87 (3H, s), 4,58 (1H, dtd, J=50,0, 9,9, 4,6 Гц), 5,58 (1H, q, J=4,7 Гц), 6,54 (1H, d, J=3,4 Гц), 6,61 (1H, dd, J=5,6, 2,2 Гц), 7,22 (1H, d, J=3,9 Гц), 7,32 (1H, s), 7,38 (2H, d, J=8,3 Гц), 7,84 (2H, d, J=8,3 Гц), 7,91 (1H, d, J=2,4 Гц), 8,04 (1H, s), 8,10 (1H, d, J=5,9 Гц), 8,57 (1H, s).
[0152] Исходный материал трет-бутил 3-фтор-4-(4-((4-((6-метокси-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)фенил)пиперидин-1-карбоксилат синтезировали следующим способом.
[0153] [Пример получения 6-1]
Трет-бутил-4-(4-(метоксикарбонил)фенил)-5,6-дигидропиридин-1(2Н)-карбоксилат
[Химическая формула 54]

Толуол (20 мл), этанол (6 мл) и воду (2 мл) добавляли в коммерчески доступный 1-N-ВОС-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-3,6-дигидро-2Н-пиридин (1,00 г, 3,23 ммоля), метил-4-бромбензоат (695 мг, 3,23 ммоля), 2-дициклогексилфосфино-2',6'-диметоксибифенил (266 мг, 0,65 ммоля), палладия (II) ацетат (73 мг, 0,32 ммоля) и трикалия фосфат (2,06 г, 9,70 ммоля) в атмосфере азота и смесь перемешивали при 90°С в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры и насыщенный водный раствор натрия бикарбоната и этилацетат добавляли для разделения. Водный слой экстрагировали этилацетатом три раза, а затем органический слой сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 19:1 - 13:7) с получением названного соединения (0,99 г, 96%).
1Н-ЯМР спектр (500 МГц, CDCl3) δ (ppm): 1,49 (9H, s), 2,55 (2H, brs), 3,65 (2H, t, J=5,9 Гц), 3,92 (3Н, s), 4,10 (2H, brs), 6,16 (1H, brs), 7,43 (2H, d, J=8,3 Гц), 8,00 (2H, d, J=8,8 Гц).
[0154] [Пример получения 6-2]
Трет-бутил-3-гидрокси-4-(4-(метоксикарбонил)фенил)пиперидин-1-карбоксилат
[Химическая формула 55]

Трет-бутил-4-(4-(метоксикарбонил)фенил)-5,6-дигидропиридин-1(2Н)-карбоксилат (776 мг, 2,45 ммоля), описанный в примере получения 6-1, растворяли в тетрагидрофуране (10 мл), добавляли раствор комплекса боран-метилсульфид (0,269 мл, 2,69 ммоля) в тетрагидрофуране (10 мл) при комнатной температуре за 3 минуты и смесь перемешивали в течение 13 часов. Реакционную жидкость охлаждали до 0°С и добавляли 1 М раствор натрия гидроксида (6,12 мл, 6,12 ммоля) за 1 минуту. Добавляли 30% перекись водорода - воду (0,625 мл, 6,12 ммоля) и смесь перемешивали при комнатной температуре в течение 45 минут. Насыщенный водный раствор тиосульфата натрия (20 мл), воду и этилацетат последовательно добавляли в реакционную смесь. Водный слой экстрагировали этилацетатом три раза, а затем органические слои объединяли, сушили над безводным магния сульфатом и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 4:1 - 1:4) с получением названного соединения (518 мг, 63%).
1Н-ЯМР спектр (500 МГц, CDCl3) δ (ppm): 1,49 (9H, s), 1,59 (1H, d, J=2,9 Гц), 1,69-1,88 (2Н, m), 2,57-2,71 (2H, m), 2,77 (1H, brs), 3,67-3,82 (1H, m), 3,91 (3Н, s), 4,22 (1H, brs), 4,42 (1H, brs), 7,34 (2H, d, J=8,3 Гц), 8,02 (2H, d, J=8,3 Гц).
[0155] [Пример получения 6-3]
Трет-бутил 3-фтор-4-(4-(метоксикарбонил)фенил)пиперидин-1-карбоксилат
[Химическая формула 56]

3-гидрокси-4-(4-(метоксикарбонил)фенил)пиперидин-1-карбоксилат (518 мг, 1,54 ммоля), описанный в примере получения 6-2, растворяли в дихлорметане (15 мл), добавляли [бис-(2-метоксиэтил)амино]серы трифторид (0,313 мл, 1,70 ммоля) при -78°С за 3 минуты и смесь перемешивали в течение 1 часа при нагревании до комнатной температуры. Насыщенный водный раствор натрия бикарбоната и этилацетат последовательно добавляли в реакционную смесь. Водный слой экстрагировали этилацетатом три раза, а затем органические слои объединяли, сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 19:1 - 3:2) с получением названного соединения (402 мг, 77%).
1Н-ЯМР спектр (500 МГц, CDCl3) δ (ppm): 1,49 (9H, s), 1,64-1,81 (1H, m), 1,86-2,01 (1H, m), 2,72-2,94 (3H, m), 3,91 (3H, s), 4,19 (1H, brs), 4,44-4,66 (2H, m), 7,33 (2H, d, J=8,3 Гц), 8,02 (2Н, d, J=8,3 Гц).
[0156] [Пример получения 6-4]
4-(1-(Трет-бутоксикарбонил)-3-фторпиперидин-4-ил)бензойная кислота
[Химическая формула 57]
Трет-бутил-3-фтор-4-(4-(метоксикарбонил)фенил)пиперидин-1-карбоксилат (402 мг, 1,19 ммоля), описанный в примере получения 6-3, растворяли в тетрагидрофуране (18 мл) метаноле (6 мл) и воде (4 мл), затем добавляли 2 М водный раствор лития гидроксида (4,17 мл, 8,34 ммоля) при комнатной температуре, а затем смесь перемешивали в течение 3 часов. Тетрагидрофуран и метанол выпаривали в вакууме и добавляли 1 М хлористоводородную кислоту к остаточному раствору для нейтрализации. Водный слой экстрагировали этилацетатом четыре раза, органические слои объединяли, сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 4:1 - 1:4) с получением названного соединения (347 мг, 90%).
1Н-ЯМР спектр (500 МГц, CDCl3) δ (ppm): 1,50 (9H, s), 1,77 (1H, qd, J=13,0, 3,9 Гц), 1,92 (1H, d, J=14,1 Гц), 2,72-2,95 (3H, m), 4,21 (1H, brs), 4,46-4,67 (2H, m), 7,37 (2H, d, J=8,3 Гц), 8,09 (2Н, d, J=8,3 Гц).
[0157] [Пример получения 6-5]
Трет-бутил 3-фтор-4-(4-((4-((6-метокси-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)фенил)пиперидин-1-карбоксилат
[Химическая формула 58]
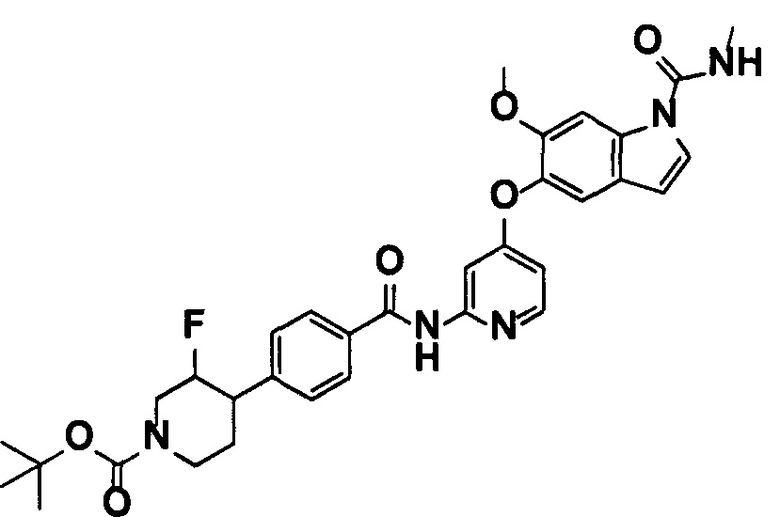
Тионилхлорид (30 μл, 0,319 ммоля) добавляли в смесь бензотриазола (49,3 мг, 0,414 ммоля) и дихлорметана (4 мл) в атмосфере азота, и смесь перемешивали при комнатной температуре в течение 5 минут. 4-(1-(Трет-бутоксикарбонил)-3-фторпиперидин-4-ил)бензойную кислоту (103 мг, 0,319 ммоля), описанную в примере получения 6-4, добавляли и смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную жидкость фильтровали через безводный натрия сульфат и безводный натрия сульфат промывали дихлорметаном (4 мл) с получением дихлорметанового раствора неочищенного продукта.
Дихлорметановый раствор вышеупомянутого неочищенного продукта (4 мл, 0,16 ммоля) добавляли в смесь 5-((2-аминопиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамида (25,0 мг, 0,080 ммоля), описанного в примере получения 1-6, триэтиламина (56 μл, 0,40 ммоля), 4-диметиламинопиридина (1,0 мг, 0,008 ммоля) и дихлорметана (1 мл) при 0°С, затем смесь перемешивали при 0°С в течение 10 минут, а затем при комнатной температуре в течение 3 часов. Добавляли 40% водный раствор метиламина (138 μл, 1,60 ммоля), и смесь далее перемешивали при комнатной температуре в течение 2 часов. Насыщенный водный раствор натрия бикарбоната и этилацетат добавляли в реакционную смесь для разделения. Водный слой экстрагировали этилацетатом три раза, а затем органические слои объединяли, сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 0:1) с получением названного соединения (29,1 мг, 59%).
1Н-ЯМР спектр (500 МГц, CDCl3) δ (ppm): 1,49 (9H, s), 1,65-1,82 (1H, m), 1,91 (1H, brs), 2,69-2,93 (3H, m), 3,04 (3H, d, J=4,9 Гц), 3,86 (3Н, s), 4,18 (1H, brs), 4,42-4,66 (2H, m), 5,65 (1H, q, J=4,4 Гц), 6,53 (1H, d, J=3,4 Гц), 6,62 (1H, dd, J=5,9, 2,4 Гц), 7,23 (1H, d, J=3,9 Гц), 7,31 (1Н, s), 7,36 (2H, d, J=8,3 Гц), 7,84 (2Н, d, J=8,3 Гц), 7,91 (1Н, d, J=2,0 Гц), 8,05 (1Н, s), 8,09 (1Н, d, J=5,9 Гц), 8,63 (1Н, s).
[0158] [Пример 7]
5-((2-(4-(4-Фторпиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 59]

Бензотриазол (33,2 мг, 0,278 ммоля) растворяли в дихлорметане (10 мл) в атмосфере азота, затем добавляли тионилхлорид (20 μл, 0,278 ммоля) и смесь перемешивали при 20°С в течение 5 минут. В реакционную смесь добавляли 4-(1-трет-бутоксикарбонил)-4-фторпиперидин-4-ил)бензойную кислоту (60 мг, 0,186 ммоля), описанную в примере получения 7-4, и смесь перемешивали при 20°С в течение 2 часов. Реакционную смесь фильтровали и дихлорметан выпаривали в вакууме, пока его количество не достигло приблизительно 5 мл. 5-((2-Аминопиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид (25 мг, 0,08 ммоля), описанный в примере получения 1-6, триэтиламин (55 μл, 0,40 ммоля) и 4-диметиламинопиридин (1,96 мг, 0,016 ммоля) последовательно добавляли в полученную реакционную смесь и смесь перемешивали при комнатной температуре в течение 2 часов. 2 М раствор метиламинтетрагидрофурана (120 μл, 0,24 ммоля) добавляли в реакционную смесь, затем смесь перемешивали при комнатной температуре в течение 1 часа, а затем этилацетат и насыщенный водный раствор натрия бикарбоната добавляли для разделения. Органический слой сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали в вакууме и полученный остаток растворяли в дихлорметане (5 мл), затем добавляли трифторуксусную кислоту (1 мл) при 0°С, а затем смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель выпаривали в вакууме, получали азеотропную смесь полученного остатка с толуолом, а затем смесь сушили в вакууме. Полученный остаток растворяли в смешанном растворителе из дихлорметана и триэтиламина, и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 1:0 - 17:3). Целевую фракцию концентрировали в вакууме с получением названного соединения (11,8 мг, 29%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,73-2,01 (4H, m), 2,91-3,23 (7H, m), 3,86 (3H, s), 5,76-5,91 (1H, m), 6,44-6,52 (1H, m), 6,64 (1H, dd, J=5,9, 2,2 Гц), 7,22 (1Н, d, J=3,7 Гц), 7,31 (1H, s), 7,48 (2H, d, J=8,4 Гц) 7,81-7,97 (3H, m), 8,02-8,14 (2H, m), 8,74 (1H, brs).
[0159] Исходный материал 4-(1-трет-бутоксикарбонил)-4-фторпиперидин-4-ил)бензойную кислоту синтезировали следующим способом.
[0160] [Пример получения 7-1]
1-Бензил-4-(4-(4,4-диметил-4,5-дигидрооксазол-2-ил)фенил)пиперидин-4-ол
[Химическая формула 60]
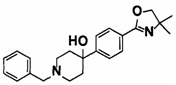
Магний (3,10 г, 127 ммоль), йод (135 мг, 0,531 ммоля) и 2-(4-бромфенил)-4,5-дигидро-4,4-диметилоксазол (27 г, 106 ммоль) добавляли в тетрагидрофуран (120 мл) и смесь нагревали с обратным холодильником в течение 1 часа. Эту реакционную смесь охлаждали до комнатной температуры, затем добавляли 1-бензил-4-пиперидон (21,7 мл, 117 ммоль) и смесь нагревали с обратным холодильником в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры, а затем насыщенный водный раствор аммония хлорида и этилацетат добавляли для разделения. Водный слой экстрагировали этилацетатом, а затем органические слои объединяли и последовательно промывали водой и насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния и фильтровали. Растворитель выпаривали в вакууме и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 3:1 - 1:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (13,4 г, 35%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,37 (6Н, s), 1,67-1,76 (2H, m), 2,16 (2H, td, J=13,1, 4,5 Гц), 2,47 (2Н, td, J=12,0, 2,5 Гц), 2,79 (2Н, d, J=11,4 Гц), 3,58 (2Н, s), 4,09 (2H, s), 7,23-7,40 (5H, m), 7,55 (2Н, d, J=8,8 Гц), 7,91 (2Н, d, J=8,6 Гц).
[0161] [Пример получения 7-2]
Этил-4-(1-бензил-4-гидроксипиперидин-4-ил)бензоат
[Химическая формула 61]

1-Бензил-4-(4-(4,4-диметил-4,5-дигидрооксазол-2-ил)фенил)пиперидин-4-ол (13,4 г, 36,8 ммоля), описанный в примере получения 7-1, растворяли в этаноле (300 мл), затем добавляли серную кислоту и смесь перемешивали при 90°С в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры, а затем растворитель выпаривали в вакууме. Полученный остаток растворяли в дихлорметане, полученное вещество промывали насыщенным водным раствором натрия бикарбоната, а затем органический слой сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали в вакууме и полученный остаток очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 4:1 - 1:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (7,23 г, 58%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,39 (3H, t, J=7,1 Гц), 1,59-1,85 (2Н, m), 2,17 (2Н, td, J=13,1, 4,5 Гц), 2,47 (2Н, td, J=12,0, 2,5 Гц), 2,72-2,93 (2Н, m), 3,59 (2Н, s), 4,37 (2Н, q, J=7,0 Гц), 7,14-7,44 (5H, m), 7,59 (2Н, d, J=8,4 Гц), 8,02 (2Н, d, J=8,4 Гц).
[0162] [Пример получения 7-3]
Трет-бутил 4-(4-(этоксикарбонил)фенил)-4-фторпиперидин-1-карбоксилат
[Химическая формула 62]
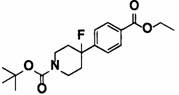
Этил-4-(1-бензил-4-гидроксипиперидин-4-ил)бензоат (6,23 г, 18,4 ммоля), описанный в примере получения 7-2, растворяли в дихлорметане (200 мл) в атмосфере азота, затем добавляли диэтиламиносеры трифторид (2,89 мл, 22,0 ммоля) при -78°С и смесь перемешивали при комнатной температуре в течение 3 часов. Дихлорметан добавляли в реакционную смесь и смесь промывали водой, а затем сушили над безводным магния сульфатом и фильтровали. Растворитель выпаривали в вакууме с получением неочищенного продукта А (6,13 г).
Неочищенный продукт А (6,13 г) растворяли в дихлорметане (100 мл) в атмосфере азота, затем добавляли 1-хлорэтилхлороформиат (2,13 мл, 19,8 ммоля) и смесь перемешивали при 20°С в течение 2 часов. Реакционную смесь концентрировали в вакууме, затем полученный остаток растворяли в метаноле (100 мл) и полученное вещество нагревали с обратным холодильником в течение 30 минут. Реакционную смесь концентрировали в вакууме, затем добавляли диэтиловый эфир в полученный остаток, осадок собирали фильтрацией, а затем полученное вещество промывали диэтиловым эфиром с получением неочищенного продукта В (4,72 г).
Неочищенный продукт В (4,72 г) растворяли в дихлорметане (50 мл), затем добавляли триэтиламин (5,24 мл, 37,6 ммоля) и ди-трет-бутилдикарбонат (4,92 г, 22,5 ммоля) и смесь перемешивали при комнатной температуре в течение 1 часа. Воду и этилацетат добавляли в реакционную смесь для разделения. Водный слой снова экстрагировали этилацетатом, а затем органические слои объединяли и промывали водой, а затем насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния и фильтровали. Растворитель выпаривали в вакууме и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 1:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (3,74 г, 58%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,24-1,34 (1H, m), 1,40 (3H, t, J=7,1 Гц), 1,50 (9Н, s), 1,86-2,14 (2H, m), 2,54 (1H, brs), 3,18 (1H, brs), 3,65 (1H, t, J=5,7 Гц), 4,00-4,26 (2Н, m), 4,38 (2H, qd, J=7,1, 3,1 Гц), 7,43 (2Н, dd, J=8,6, 1,3 Гц), 8,03 (2H, dd, J=18,1, 8,6 Гц).
[0163] [Пример получения 7-4]
4-(1-Трет-бутоксикарбонил)-4-фторпиперидин-4-ил)бензойная кислота
[Химическая формула 63]

Трет-бутил 4-(4-(этоксикарбонил)фенил)-4-фторпиперидин-1-карбоксилат (3,74 г, 10,6 ммоля), описанный в примере получения 7-3, растворяли в смешанном растворителе из тетрагидрофурана (10 мл) и метанола (5 мл), затем добавляли 1М раствор натрия гидроксида (42,6 мл) и смесь перемешивали при 50°С в течение 4 часов. 1 М хлористоводородную кислоту добавляли в реакционную смесь, осадок собирали фильтрацией и промывали водой, а затем полученное вещество сушили путем сушки со сквозным протоком с получением названного соединения (1,33 г, 39%).
1Н-ЯМР спектр (DMSO-d6) δ (ppm): 1,43 (9H, s), 1,83-2,15 (4H, m), 3,06 (2H, brs), 3,99 (2H, d, J=10,3 Гц), 7,50 (2H, d, J=8,4 Гц), 7,92 (2H, d, J=8,4 Гц).
[0164] [Пример 8]
6-Метокси-N-метил-5-((2-(4-(4-(пирролидин-1-ил)пиперидин-1-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид
[Химическая формула 64]

Оксалилхлорид (27 μл, 0,321 ммоля) и N,N-диметилформамид (1,24 μл, 0,016 ммоля) добавляли в раствор 4-(4-(пирролидин-1-ил)пиперидин-1-ил)бензойной кислоты (44 мг, 0,16 ммоля), описанной в примере получения 8-2, в дихлорметане (2 мл) при комнатной температуре. Реакционную жидкую смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную жидкую смесь концентрировали и последовательно добавляли триэтиламин (112 μл, 0,80 ммоля), 5-((2-аминопиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид (50 мг, 0,16 ммоля), описанный в примере получения 1-6, и 4-диметиламинопиридин (3,91 мг, 0,032 ммоля) в раствор остатка в дихлорметане (2 мл) при комнатной температуре. Реакционную жидкую смесь перемешивали при комнатной температуре. Насыщенный водный раствор натрия бикарбоната добавляли в реакционную жидкость, а затем для растворения добавляли этилацетат. Водный слой экстрагировали этилацетатом, затем объединенный органический слой сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 0:1) с получением названного соединения (3,6 мг, 4,0%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,18-1,38 (2H, m), 1,84 (4H, brs), 1,95-2,11 (2H, m), 2,24-2,38 (1H, m), 2,70 (4H, brs), 2,82-2,96 (2H, m), 3,06 (3H, d, J=4,8 Гц), 3,80-3,91 (5Н, m), 5,52-5,68 (1H, m), 6,54 (1H, d, J=3,3 Гц), 6,61 (1H, dd, J=5,9, 2,2 Гц), 6,89 (2H, d, J=8,8 Гц), 7,23 (1H, d, J=3,7 Гц), 7,32 (1H, s), 7,75 (2H, d, J=8,8 Гц), 7,90 (1H, d, J=2,2 Гц), 8,02 (1H, s), 8,09 (1H, d, J=5,9 Гц), 8,56 (1H, brs).
[0165] Исходный материал 4-(4-(пирролидин-1-ил)пиперидин-1-ил)бензойную кислоту синтезировали следующим способом.
[0166] [Пример получения 8-1]
Метил-4-(4-(пирролидин-1-ил)пиперидин-1-ил)бензоат
[Химическая формула 65]
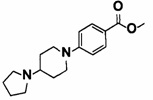
Коммерчески доступный метил-4-фторбензоат (1,5 г, 9,73 ммоля) добавляли в раствор коммерчески доступного 4-(1-пирролидинил)пиперидина (3,0 г, 19,5 ммоля) в диметилсульфоксиде (15 мл) при комнатной температуре, а затем смесь перемешивали в атмосфере азота и при микроволновом излучении при 150°С в течение 4 часов. Обеспечивали отстаивание реакционной жидкости с охлаждением до комнатой температуры, а затем разбавляли водой и этилацетатом. Органический слой промывали водой, а затем сушили традиционным способом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1) с получением названного соединения (2,06 г, 73%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,54-1,70 (2H, m), 1,74-1,88 (4H, m), 1,93-2,06 (2H, m), 2,12-2,26 (1H, m), 2,53-2,65 (4H, m), 2,90 (2H, td, J=12,4, 2,6 Гц), 3,74-3,97 (5Н, m), 6,86 (2H, d, J=9,2 Гц), 7,89 (2H, d, J=9,2 Гц).
[0167] [Пример получения 8-2]
4-(4-(Пирролидин-1-ил)пиперидин-1-ил)бензойная кислота
[Химическая формула 66]

5 М раствор натрия гидроксида (15 мл, 75,0 ммоля) добавляли в раствор метил-4-(4-(пирролидин-1-ил)пиперидин-1-ил)бензоата (2,06 г, 7,14 ммоля), описанного в примере получения 8-1, в тетрагидрофуране (10 мл) и метаноле (10 мл). Реакционную жидкость перемешивали при 50°С в течение 4 часов. Реакционную жидкость охлаждали до 0°С, а затем капали 5 М хлористоводородную кислоту до достижения рН 1. Жидкую смесь разбавляли этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме с получением названного соединения (1,61 г, 82%).
ESI-MS (масса/заряд): 297 [М+H]+.
[0168] [Пример 9]
5-((2-(4-((4-Гидроксипиперидин-1-ил)метил)бензамид)пиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 67]

Триэтиламин (1,6 мл, 11,5 ммоля) и коммерчески доступный 4-(хлорметил)бензоилхлорид (1,37 г, 7,25 ммоля) добавляли в смесь 5-((2-аминопиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамида (905 мг, 2,90 ммоля), описанного в примере получения 1-6, и тетрагидрофурана (28 мл) в атмосфере азота при 0°С. Смесь перемешивали при комнатной температуре в течение 100 минут, затем добавляли Триэтиламин (1,6 мл, 11,5 ммоля) и 4-(хлорметил)бензоилхлорид (0,90 г, 4,76 ммоля) в реакционную смесь при 0°С, а затем полученное вещество перемешивали при комнатной температуре в течение 1,5 часа. Воду, тетрагидрофуран и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным магния сульфатом. Полученное вещество фильтровали с помощью силикагеля NH (этилацетат), а затем концентрировали в вакууме с получением неочищенного продукта.
Неочищенный продукт растворяли в N,N-диметилформамиде (15 мл), коммерчески доступный 4-гидроксипиперидин (1,48 г, 14,6 ммоля) добавляли в атмосфере азота при комнатной температуре и смесь перемешивали в течение 16 часов. Добавляли воду и этилацетат в реакционную смесь для разделения и водный слой экстрагировали этилацетатом. Объединенный органический слой промывали водой и насыщенным солевым раствором, сушили над безводным магния сульфатом и фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 1:0 - 9:1 - 17:3 - 4:1). Целевой продукт собирали фильтрацией и промывали этилацетатом с получением названного соединения (1,30 г, 84%).
1Н-ЯМР спектр (DMSO-d6) δ (ppm): 1,32-1,44 (2H, m), 1,64-1,74 (2H, m), 1,97-2,09 (2H, m), 2,59-2,69 (2H, m), 2,86 (3H, d, J=4,2 Гц), 3,39-3,50 (3Н, m), 3,76 (3H, s), 4,54 (1H, d, J=4,2 Гц), 6,63 (1H, d, J=3,7 Гц), 6,65 (1H, dd, J=5,7, 2,2 Гц), 7,37 (2H, d, J=8,1 Гц), 7,44 (1H, s), 7,66 (1H, d, J=2,4 Гц), 7,78 (1H, d, J=3,7 Гц), 7,92 (2H, d, J=8,2 Гц), 8,10 (1H, s), 8,15-8,22 (2H, m), 10,68 (1H, s).
[0169] [Пример 10]
5-((2-(4-(((3S,4S)-3-Гидрокси-4-метоксипирролидин-1-ил)метил)бензамид)пиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 68]

Неочищенный продукт (20,2 мг, 0,033 ммоля), описанный в примере 9, растворяли в N,N-диметилформамиде (0,5 мл), затем добавляли (3S,4S)-4-метоксипирролидин-3-ол (55,9 мг, 0,477 ммоля) (описанный в ЕР 1375465) в атмосфере азота при комнатной температуре, а затем смесь перемешивали в течение 200 минут. Добавляли воду и этилацетат в реакционную смесь для разделения, органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отфильтровывали, растворитель выпаривали, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 1:0 - 19:1 - 9:1). Остаток промывали диэтиловым эфиром с получением названного соединения (13,8 мг, 77%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 2,32 (1H, dd, J=10,1, 4,2 Гц), 2,61-2,68 (1Н, m), 2,70-2,77 (1H, m), 3,03-3,16 (4H, m), 3,36 (3H, s), 3,62-3,76 (4H, m), 3,87 (3H, s), 4,12-4,19 (1H, m), 5,42-5,51 (1H, m), 6,56 (1H, d, J=3,7 Гц), 6,59-6,62 (1H, m), 7,23 (1H, d, J=3,7 Гц), 7,32 (1H, s), 7,42 (2H, d, J=8,4 Гц), 7,81 (2Н, d, J=8,1 Гц), 7,91 (1H, d, J=2,2 Гц), 8,03 (1H, s), 8,11 (1H, d, J=5,9 Гц), 8,48 (1H, brs).
[0170] [Пример 11]
5-((2-(4-(2-(4-Этилпиперазин-1-ил)этил)бензамид)пиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 69]

Тионилхлорид (0,116 мл, 1,60 ммоля) добавляли в смесь коммерчески доступной 4-(2-хлорэтил)бензойной кислоты (88 мг, 0,479 ммоля) и 1,2-дихлорэтана (1,0 мл) в атмосфере азота при комнатной температуре и смесь нагревали с обратным холодильником при 90°С в течение 1,5 часа. Смесь охлаждали до комнатной температуры, затем растворитель выпаривали, полученное вещество растворяли в тетрагидрофуране (1,0 мл) и таким образом получали раствор хлорангидрида. Триэтиламин (0,443 мл, 3,20 ммоля) добавляли в смесь 5-((2-аминопиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамида (49,9 мг, 0,160 ммоля), описанного в примере получения 1-6, и N,N-диметилформамида (0,5 мл) в атмосфере азота при комнатной температуре и смесь охлаждали до 0°С. Предварительно полученный тетрагидрофурановый раствор хлорангидрида добавляли при той же температуре и смесь перемешивали при комнатной температуре в течение 2,5 часа. Добавляли воду и этилацетат в реакционную смесь для разделения, затем органический слой промывали насыщенным солевым раствором и сушили над безводным натрия сульфатом. Растворитель выпаривали и полученный остаток растворяли в дихлорметане, полученное вещество очищали с помощью колоночной хромагографии на силикагеле NH (н-гептан:этилацетат = 3:1 - 0:1), а затем целевую фракцию концентрировали в вакууме с получением неочищенного продукта (48,9 мг).
Неочищенный продукт (48,9 мг) растворяли в N,N-диметилформамиде (0,25 мл), затем добавляли коммерчески доступный N-этилпиперазин (129 μл, 1,02 ммоля) и N,N-диизопропилэтиламин (26,6 μл, 0,153 ммоля) в атмосфере азота при комнатной температуре и смесь нагревали и перемешивали при 60°С в течение 16 часов. Смесь охлаждали до комнатной температуры, затем воду и этилацетат добавляли в реакционную смесь для разделения, а потом органический слой промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хромагографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 0:1 - этилацетат:метанол = 99:1 - 19:1). Смешанную фракцию концентрировали в вакууме и остаток очищали с помощью TLC на силикагеле NH (этилацетат) с получением названного соединения (7,03 мг, 16%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,10 (3Н, t, J=7,3 Гц), 2,44 (2Н, q, J=7,0 Гц), 2,35-2,70 (10Н, m), 2,81-2,90 (2Н, m), 3,07 (3Н, d, J=4,8 Гц), 3,86 (3Н, s), 5,43-5,51 (1H, m), 6,56 (1H, d, J=3,7 Гц), 6,60 (1H, dd, J=5,9, 2,6 Гц), 7,23 (1H, d, J=3,7 Гц), 7,28-7,33 (3Н, m), 7,78 (2Н, d, J=8,4 Гц), 7,91 (1H, d, J=2,2 Гц), 8,03 (1H, s), 8,10 (1H, d, J=5,9 Гц), 8,46 (1Н, brs).
[0171] [Пример 12]
5-((2-(4-(2-(4-Гидроксипиперидин-1-ил)этокси)бензамид)пиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 70]

Добавляли оксалилхлорид (44 μл, 0,513 ммоля) и каталитическое количество N,N-диметилформамида в смесь 4-(2-хлорэтокси)бензойной кислоты (51,2 мг, 0,255 ммоля), описанной в примере получения 12-1, и дихлорметана (2,0 мл) в атмосфере азота при комнатной температуре и смесь перемешивали в течение 45 минут. Добавляли оксалилхлорид (44 μл, 0,513 ммоля) в реакционную смесь при комнатой температуре и смесь перемешивали в течение 30 минут. Растворитель выпаривали и полученное вещество растворяли в тетрагидрофуране (0,5 мл) и таким образом получали раствор хлорангидрида. Триэтиламин (89 μл, 0,64 ммоля) добавляли в смесь 5-((2-аминопиридин-N-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамида (40 мг, 0,128 ммоля), описанного в примере получения 1-6, и N,N-диметилформамида (0,5 мл) в атмосфере азота при комнатной температуре. Предварительно полученный тетрагидрофурановый раствор хлорангидрида добавляли при той же температуре и смесь перемешивали при комнатной температуре в течение 80 минут. Насыщенный водный раствор натрия бикарбоната и этилацетат добавляли в реакционную смесь для разделения и органический слой сушили над безводным натрия сульфатом. Полученное вещество фильтровали с помощью силикагеля NH (этилацетат), а затем концентрировали в вакууме с получением неочищенного продукта (80,6 мг).
Неочищенный продукт (80,6 мг) растворяли в N,N-диметилформамиде (0,7 мл), добавляли коммерчески доступный 4-гидроксипиперидин (93 мг, 0,919 ммоля) в атмосфере азота при комнатной температуре и смесь нагревали и перемешивали при 80°С в течение 12 часов. Смесь охлаждали до комнатной температуры, воду и этилацетат добавляли в реакционную смесь для разделения и водный слой один раз экстрагировали этилацетатом. Объединенный органический слой промывали водой и насыщенным солевым раствором, сушили над безводным натрия сульфатом, а затем фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью TLC на силикагеле NH (этилацетат:метанол = 9:1). Полученный остаток очищали с помощью TLC на силикагеле NH (этилацетат) с получением названного соединения (10,8 мг, 30%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,55-1,67 (2Н, m), 1,86-1,97 (2H, m), 2,31 (2H, t, J=9,5 Гц), 2,78-2,92 (4Н, m), 3,06 (3H, d, J=4,4 Гц), 3,67-3,77 (1Н, m), 3,86 (3H, s), 4,14 (2H, t, J=5,7 Гц), 5,50-5,59 (1Н, m), 6,54 (1Н, d, J=3,3 Гц), 6,60 (1Н, dd, J=5,7, 2,4 Гц), 6,95 (2H, d, J=8,8 Гц), 7,22 (1Н, d, J=3,7 Гц), 7,32 (1Н, s), 7,81 (2H, d, J=8,8 Гц), 7,88 (1Н, d, J=1,8 Гц), 8,03 (1Н, s), 8,09 (1Н, d, J=5,9 Гц), 8,47 (1Н, brs).
[0172] Реагент 4-(2-хлорэтокси)бензойную кислоту синтезировали следующим способом.
[0173] [Пример получения 12-1] 4-(2-Хлорэтокси)бензойная кислота
[Химическая формула 71]

Калия карбонат (7,27 г, 52,6 ммоля) и коммерчески доступный 1-хлор-2-йодэтан (3,6 мл, 39,5 ммоля) добавляли в жидкую смесь коммерчески доступного метил-п-гидроксибензоата (2,0 г, 13,1 ммоля) и N,N-диметилформамида (50 мл) в атмосфере азота при комнатной температуре и смесь нагревали и перемешивали при 60°С в течение 12 часов. Смесь охлаждали до комнатной температуры, а затем насыщенный водный раствор аммония хлорида, воду и диэтиловый эфир добавляли в реакционную смесь для разделения. Органический слой промывали водой и насыщенным солевым раствором, затем сушили над безводным магния сульфатом, а затем фильтровали и концентрировали в вакууме. Тетрагидрофуран (25 мл), метанол (10 мл) и 2 М раствор натрия гидроксида (10 мл) добавляли к остатку при комнатной температуре и смесь нагревали и перемешивали при 80°С в течение 4 часов. Реакционную смесь охлаждали до 0°С, подкисляли 5 М хлористоводородной кислотой, а затем смесь разбавляли этилацетатом для разделения. Водный слой экстрагировали этилацетатом, объединенный органический слой промывали водой и насыщенным солевым раствором, затем сушили над безводным магния сульфатом и фильтровали. Растворитель выпаривали, осадок отделяли фильтрацией с жидкой смесью этилацетата и тетрагидрофурана, полученное вещество промывали и фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:эталацетат = 3:1 - 2:1 - 1:1) и целевую фракцию концентрировали в вакууме с получением названного соединения (278 мг, 11%).
1Н-ЯМР спектр (DMSO-d6) δ (ppm): 3,92-3,98 (2H, m), 4,26-4,35 (2H, m), 6,96-7,06 (2H, m), 7,82-7,91 (2H, m), 12,68 (1H, brs).
[0174] [Пример 13]
5-((2-(1-(1-Этилпиперидин-4-ил)-1Н-пиразол-4-карбоксамид)пиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 72]

Ацетальдегид (8,9 мг, 0,202 ммоля) и натрия триацетоксиборгидрид (43,9 мг, 0,207 ммоля) добавляли в смесь 6-метокси-N-метил-5-((2-(1-(пиперидин-4-ил)-1Н-пиразол-4-карбоксамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида (20,3 мг, 0,041 ммоля), описанного в примере получения 13-4, и тетрагидрофурана (1,0 мл) при комнатной температуре и смесь перемешивали в течение 2,5 часа. Насыщенный водный раствор натрия бикарбоната и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью TLC на силикагеле NH (этилацетат). Продукт промывали диэтиловым эфиром с получением названного соединения (9,3 мг, 43%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,11 (3H, t, J=7,1 Гц), 1,94-2,24 (6H, m), 2,46 (2H, q, J=7,4 Гц), 3,03-3,11 (5Н, m), 3,86 (3H, s), 4,09-4,19 (1H, m), 5,49-5,56 (1H, m), 6,54 (1H, d, J=2,9 Гц), 6,60 (1H, dd, J=5,9, 2,4 Гц), 7,22 (1H, d, J=3,7 Гц), 7,31 (1H, s), 7,81 (1H, d, J=2,0 Гц), 7,85 (1H, d, J=0,7 Гц), 7,96 (1H, s), 8,02 (1H, s), 8,08 (1H, d, J=5,9 Гц), 8,17 (1H, brs).
[0175] Исходный материал 6-метокси-N-метил-5-((2-(1-(пиперидин-4-ил)-1Н-пиразол-4-карбоксамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид синтезировали следующим способом.
[0176] [Пример получения 13-1]
Трет-бутил 4-((метилсульфонил)окси)пиперидин-1-карбоксилат
[Химическая формула 73]

Коммерчески доступный метансульфонилхлорид (6,35 мл, 82,0 ммоля) и триэтиламин (26,0 мл, 186 ммоля) добавляли в смесь коммерчески доступного трет-бутил 4-гидрокси-1-пиперидинкарбоксилата (15 г, 74,5 ммоля) и тетрагидрофурана (200 мл) при 0°С и смесь перемешивали в течение 30 минут. Воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой сушили и фильтровали традиционными способами. Растворитель выпаривали и остаток собирали фильтрацией и промывали н-гептаном с получением названного соединения (19,8 г, 95%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,46 (9H, s), 1,72-1,88 (2H, m), 1,90-2,04 (2H, m), 3,03 (3Н, s), 3,24-3,36 (2H, m), 3,64-3,77 (2H, m), 4,80-4,94 (1H, m).
[0177] [Пример получения 13-2]
Трет-бутил 4-(4-(этоксикарбонил)-1Н-пиразол-1-ил)пиперидин-1-карбоксилат
[Химическая формула 74]

Трет-бутил-4-((метилсульфонил)окси)пиперидин-1-карбоксилат (2,7 г, 9,67 ммоля), описанный в примере получения 13-1, и коммерчески доступный этил-4-пиразолкарбоксилат (1,49 г, 10,6 ммоля) растворяли в N,N-диметилформамиде (30 мл), добавляли 50-72% маслянистый натрия гидрид (570 мг) при 0°С и смесь нагревали и перемешивали при 60°С в течение 11 часов. Реакционную смесь охлаждали до комнатной температуры и добавляли воду и этилацетат для разделения. Органический слой дважды промывали водой, а затем сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане, полученное вещество очищали с помощью колоночной хромагографии на силикагеле (н-гептан:этилацетат = 4:1 - 1:1 - 1:3 - 0:1) и целевую фракцию концентрировали в вакууме с получением названного соединения (2,11 г, 68%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,34 (3Н, t, J=7,1 Гц), 1,47 (9Н, s), 1,82-1,96 (2H, m), 2,10-2,18 (2H, m), 2,82-2,96 (2H, m), 4,19-4,34 (3Н, m), 4,29 (2H, q, J=7,1 Гц), 7,91 (1Н, s), 7,92 (1H, s).
[0178] [Пример получения 13-3]
1-(1-(Трет-бутоксикарбонил)пиперидин-4-ил)-1Н-пиразол-4-карбоновая кислота
[Химическая формула 75]
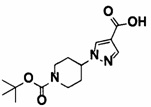
Трет-бутил-4-(4-(этоксикарбонил)-1Н-пиразол-1-ил)пиперидин-1-карбоксила т (2,11 г, 6,53 ммоля), описанный в примере получения 13-2, растворяли в метаноле (60 мл), затем добавляли калия гидроксид (1,46 г, 26,1 ммоля), растворенный в воде (16 мл), а затем смесь перемешивали в течение 27 часов. Реакционную смесь концентрировали в вакууме и диэтиловый эфир добавляли для разделения. Этилацетат добавляли в водный слой для разведения, затем 5% водный раствор калия гидросульфата добавляли для подкисления и смесь дважды экстрагировали этилацетатом. Объединенный органический слой сушили над безводным сульфатом магния и фильтровали. Растворитель выпаривали с получением названного соединения (1,58 г, 82%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,48 (9Н, s), 1,84-1,98 (2H, m), 2,11-2,20 (2H, m), 2,81-2,97 (2H, m), 4,18-4,36 (3Н, m), 7,97 (1Н, s), 7,98 (1Н, s).
[0179] [Пример получения 13-4]
6-Метокси-N-метил-5-((2-(1-(пиперидин-4-ил)-1Н-пиразол-4-карбоксамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид
[Химическая формула 76]
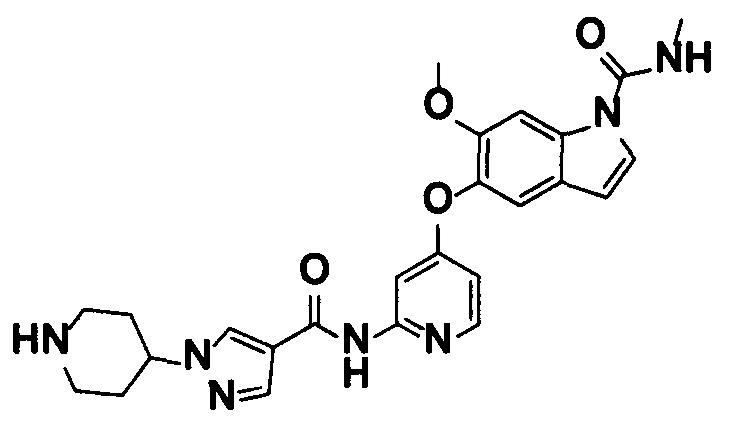
Бензотриазол (97 мг, 0,813 ммоля) растворяли в дихлорметане (4,0 мл), добавляли тионилхлорид (59 μл, 0,813 ммоля) при комнатной температуре и смесь перемешивали в течение 5 минут в атмосфере азота. Добавляли 1-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1Н-пиразол-4-карбоновую кислоту (200 мг, 0,677 ммоля), описанную в примере получения 13-3, в реакционную смесь при комнатной температуре и смесь перемешивали в течение 25 минут. Реакционную смесь фильтровали через стеклянный фильтр, полностью покрытый безводным натрия сульфатом, безводный натрия сульфат промывали дихлорметаном, затем фильтрат добавляли в смесь 5-((2-аминопиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамида (102 мг, 0,327 ммоля), описанного в примере получения 1-6, триэтиламина (0,453 мл, 3,27 ммоля) и 4-диметиламинопиридина (3,99 мг, 0,033 ммоля) в тетрагидрофуране (5,0 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 2 часов, затем воду и этилацетат добавляли в реакционную смесь для разделения, затем органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отфильтровывали и фильтрат концентрировали в вакууме. Остаток растворяли в тетрагидрофуране, добавляли избыточное количество 9,8 М раствора метиламинметанола при комнатной температуре и смесь перемешивали в течение 4 часов. Реакционную смесь концентрировали в вакууме, остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 1:9 - 0:1). Целевую фракцию концентрировали в вакууме с получением неочищенного продукта (170 мг).
Неочищенный продукт (170 мг) растворяли в дихлорметане (1,8 мл) и добавляли трифторуксусную кислоту (0,5 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 80 минут, затем концентрировали в вакууме и остаток растворяли в дихлорметан-триэтиламине, затем полученное вещество очищали с помощью колоночной хромагографии на силикагеле NH (этилацетат:метанол = 49:1 - 4:1), затем целевую фракцию концентрировали в вакууме с получением названного соединения (81,2 мг, 51%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,78-1,91 (2H, m), 2,11-2,19 (2H, m), 2,75 (2H, td, J=12,3, 2,3 Гц), 3,05 (3Н, d, J=4,6 Гц), 3,19-3,27 (2H, m), 3,85 (3Н, s), 4,16-4,26 (1H, m), 5,53-5,63 (1H, m), 6,53 (1H, d, J=3,5 Гц), 6,60 (1H, dd, J=5,8, 2,5 Гц), 7,22 (1H, d, J=3,8 Гц), 7,30 (1H, s), 7,81 (1H, d, J=2,4 Гц), 7,86 (1H, s), 7,96 (1H, s), 8,03 (1H, s), 8,08 (1H, d, J=5,9 Гц), 8,22 (1H, brs).
[0180] [Пример 14]
5-((2-(4-((1-(2-Гидроксиэтил)пиперидин-4-ил)окси)бензамид)пиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 77]

Бензотриазол (34,1 мг, 0,286 ммоля) растворяли в дихлорметане (1,5 мл), затем тионилхлорид (20 μл, 0,274 ммоля) добавляли в атмосфере азота при комнатной температуре, а затем смесь перемешивали в течение 5 минут. 4-((1-(Трет-бутоксикарбонил)пиперидин-4-ил)окси)бензойную кислоту (79,3 мг, 0,247 ммоля), описанную в примере получения 14-1, добавляли в реакционную смесь при комнатной температуре и смесь перемешивали в течение 25 минут. Реакционную смесь фильтровали через стеклянный фильтр, полностью покрытый безводным натрия сульфатом, а затем безводный натрия сульфат промывали дихлорметаном, затем фильтрат добавляли в смесь 5-((2-аминопиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамида, описываемого в примере получения 1-6, (35,6 мг, 0,114 ммоля), триэтиламина (0,158 мл, 1,14 ммоля) и 4-диметиламинопиридина (1,39 мг, 0,011 ммоля) в тетрагидрофуране (1,1 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часа, затем воду и этилацетат добавляли в реакционную смесь для разделения, затем органический слой промывали насыщенным солевым раствором и сушили над безводным натрия сульфатом. Осушающее средство отфильтровывали, фильтрат концентрировали в вакууме, остаток растворяли в тетрагидрофуране, добавляли избыточное количество 9,8 М раствора метиламинметанола при комнатной температуре и смесь перемешивали в течение 2 часов. Реакционную смесь концентрировали в вакууме, остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 1:3 - 0:1). Целевую фракцию концентрировали в вакууме с получением неочищенного продукта А (41,1 мг).
Неочищенный продукт А (41,1 мг) растворяли в дихлорметане (0,6 мл) и добавляли трифторуксусную кислоту (0,2 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 75 минут и полученное вещество концентрировали в вакууме, остаток растворяли в дихлорметан-триэтиламине, а затем полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 49:1 - 4:1), а затем целевую фракцию концентрировали в вакууме с получением неочищенного продукта В (34 мг).
Натрия триацетоксиборгидрид (17,5 мг, 0,082 ммоля) и коммерчески доступный 2-гидроксиацетальдегид (4,95 мг, 0,082 ммоля) добавляли в смесь неочищенного продукта В (8,5 мг, 0,016 ммоля) и тетрагидрофурана (0,5 мл) при комнатной температуре и смесь перемешивали в течение 2 часов. Насыщенный водный раствор натрия бикарбоната и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью TLC на силикагеле NH (этилацетат). Остаток промывали диэтиловым эфиром с получением названного соединения (6,4 мг, 40%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,78-1,90 (2H, m), 1,95-2,06 (2H, m), 2,35-2,47 (2H, m), 2,57 (2H, t, J=5,6 Гц), 2,72-2,83 (2H, m), 3,07 (3H, d, J=4,4 Гц), 3,62 (2H, t, J=5,3 Гц), 3,86 (3H, s), 4,38-4,48 (1H, m), 5,45-5,54 (1H, m), 6,55 (1H, d, J=3,7 Гц), 6,59 (1H, dd, J=5,7, 2,4 Гц), 6,94 (2H, d, J=8,8 Гц), 7,23 (1H, d, J=3,7 Гц), 7,32 (1H, s), 7,81 (2H, d, J=8,8 Гц), 7,89 (1H, d, J=2,6 Гц), 8,03 (1H, s), 8,09 (1H, d, J=5,5 Гц), 8,45 (1H, brs).
[0181] Исходный материал 4-((1-(трет-бутоксикарбонил)пиперидин-4-ил)окси)бензойную кислоту синтезировали следующим способом.
[0182] [Пример получения 14-1]
4-((1-(Трет-бутоксикарбонил)пиперидин-4-ил)окси)бензойная кислота
[Химическая формула 78]
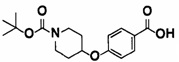
Диизопропилазодикарбоксилат (9,65 мл, 1,9 М, 18,3 ммоля) добавляли в смесь коммерчески доступного бензил-4-гидроксибензоата (3 г, 13,1 ммоля), коммерчески доступного трет-бутил-4-гидроксипиперидин-1-карбоксилата (2,77 г, 13,8 ммоля), трифенилфосфина (4,81 г, 18,3 ммоля) и тетрагидрофурана (150 мл) при 0°С и смесь перемешивали при комнатной температуре в течение ночи. Воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным водным раствором натрия бикарбоната, а затем насыщенным солевым раствором, затем сушили над безводным магния сульфатом и фильтровали. Растворитель выпаривали, полученный остаток очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1), затем целевую фракцию концентрировали в вакууме с получением неочищенного продукта (3,8 г).
Часть неочищенного продукта (500 мг, 1,22 ммоля) растворяли в этаноле (5 мл), добавляли 5 М раствор натрия гидроксида (0,729 мл, 3,65 ммоля) и смесь нагревали и перемешивали при 50°С в течение 7 часов. Реакционную смесь охлаждали до комнатной температуры, а затем 5 М хлористоводородную кислоту и этилацетат добавляли для разделения. Органический слой сушили и фильтровали традиционными способами. Растворитель выпаривали, а затем полученное вещество собирали фильтрацией и промывали н-гептаном с получением названного соединения (343 мг, 62%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,47 (9H, s), 1,65-1,84 (2H, m), 1,87-2,07 (2H, m), 3,30-3,51 (2H, m), 3,60-3,80 (2H, m), 4,50-4,66 (1H, m), 6,87-7,03 (2H, m), 7,98-8,14 (2H, m).
[0183] [Пример 15]
6-Этокси-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид
[Химическая формула 79]

4-(4-((4-((6-этокси-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)фенил)пиперидин-1-карбоксилат, описываемый в примере получения 15-8 (3,35 г, 5,46 ммоля), растворяли в дихлорметане (45 мл) и добавляли трифторуксусную кислоту (15 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 110 минут, затем концентрировали в вакууме и остаток растворяли в дихлорметан-триэтиламине, затем полученное очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 97:3 - 4:1) с получением названного соединения (2,41 г, 86%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,25 (3H, t, J=7,0 Гц), 1,50-1,70 (2Н, m), 1,79-1,88 (2H, m), 2,63-2,80 (3H, m), 3,06 (3H, d, J=4,8 Гц), 3,16-3,24 (2H, m), 4,11 (2H, q, J=7,0 Гц), 5,46-5,54 (1Н, m), 6,55 (1H, d, J=3,3 Гц), 6,60 (1H, dd, J=5,9, 2,6 Гц), 7,23 (1Н, d, J=3,7 Гц), 7,29-7,34 (3H, m), 7,78-7,83 (2H, m), 7,92 (1H, d, J=2,2 Гц), 8,00 (1H, s), 8,08-8,11 (1H, m), 8,50 (1H, brs).
[0184] Исходный материал трет-бутил 4-(4-((4-((6-этокси-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)фенил)пиперидин-1-карбоксилат синтезировали следующим способом.
[0185] [Пример получения 15-1]
4-Этокси-3-гидроксибензальдегид
[Химическая формула 80]

Коммерчески доступный 3,4-дигидроксибензальдегид (35,8 г, 259 ммоля) и калия карбонат (37,6 г, 272 ммоля) растворяли в N,N-диметилформамиде (150 мл), затем коммерчески доступный йодэтан (22 мл, 275 ммоля) добавляли в атмосфере азота при 0°С, а затем смесь перемешивали в течение 2 дней. Растворитель выпаривали в вакууме, полученное вещество охлаждали до 0°С, а затем 5 М хлористоводородную кислоту, этилацетат и воду добавляли для разделения. Водный слой экстрагировали этилацетатом, объединенный органический слой дважды промывали водой, а затем насыщенным солевым раствором и смесь сушили над безводным сульфатом магния, а затем фильтровали. Растворитель выпаривали, затем добавляли дихлорметан, а затем осадок собирали фильтрацией с получением названного соединения (25,8 г, 60%). Фильтрат очищали с помощью колоночной хромагографии на силикагеле (н-гептан:этилацетат = 9:1 - 4:1 - 1:1). Целевую фракцию концентрировали в вакууме, затем в остаток добавляли диэтиловый эфир и дихлорметан и осадок собирали фильтрацией с получением названного соединения (3,45 г, 8,0%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,50 (3H, t, J=7,1 Гц), 4,22 (2Н, q, J=7,0 Гц), 5,75 (1Н, s), 6,95 (1Н, d, J=8,1 Гц), 7,39-7,45 (2Н, m), 9,84 (1Н, s).
[0186] [Пример получения 15-2]
3-(Бензилокси)-4-этоксибензальдегид
[Химическая формула 81]
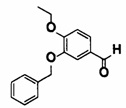
Калия карбонат (19,8 г, 143 ммоля) и бензилхлорид (16,5 мл, 143 ммоля) добавляли в суспензию 4-этокси-3-гидроксибензальдегида, описываемого в примере получения 15-1 (20 г, 120 ммоля), в этаноле (200 мл) в атмосфере азота при комнатной температуре и смесь нагревали и перемешивали при 90°С в течение 2,5 часа. Смесь охлаждали до 0°С и 2 М хлористоводородную кислоту, этилацетат, и воду добавляли для разделения. Органический слой промывали насыщенным солевым раствором, сушили над безводным магния сульфатом и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хромагографии на силикагеле (н-гептан:этилацетат = 9:1 - 4:1 - 3:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (28,5 г, 92%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,51 (3H, t, J=7,0 Гц), 4,20 (2Н, q, J=7,0 Гц), 5,19 (2Н, s), 6,98 (1Н, d, J=8,8 Гц), 7,29-7,34 (1Н, m), 7,35-7,41 (2Н, m), 7,43-7,49 (4H, m), 9,81 (1H, s).
[0187] [Пример получения 15-3]
(Е)-2-(Бензилокси)-1-этокси-4-(2-нитровинил)бензол
[Химическая формула 82]
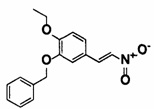
3-(Бензилокси)-4-этокси-бензальдегид, описываемый в примере получения 15-2 (14,5 г, 56,4 ммоля), растворяли в уксусной кислоте (45 мл), затем добавляли аммония ацетат (5,22 г, 67,7 ммоля) и нитрометан (7,5 мл, 138 ммоля) в атмосфере азота при комнатной температуре, смесь нагревали и перемешивали при 130°С в течение 2,5 часа. Смесь охламодали до комнатной температуры, осадок собирали фильтрацией и промывали этанолом с количественным получением названного соединения.
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,50 (3H, t, J=7,0 Гц), 4,17 (2Н, q, J=7,2 Гц), 5,17 (2Н, s), 6,92 (1Н, d, J=8,4 Гц), 7,04 (1Н, d, J=2,2 Гц), 7,16 (1Н, dd, J=8,6.2,0 Гц), 7,29-7,51 (6Н, m), 7,90 (1H, d, J=13,5 Гц).
[0188] [Пример получения 15-4]
6-Этокси-1Н-индол-5-ол
[Химическая формула 83]
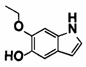
Дымящуюся азотную кислоту (13 мл, 289 ммоля) медленно добавляли в смесь (Е)-2-(бензилокси)-1-этокси-4-(2-нитровинил)бензола, описываемого в примере получения 15-3 (16,9 г, 56,6 ммоля), и уксусной кислоты (160 мл) при комнатной температуре и смесь перемешивали в течение 6 часов. Реакционную смесь выливали на лед, затем осадок собирали фильтрацией, а затем промывали жидкой смесью уксусной кислоты и этанола с получением неочищенного продукта (19,5 г).
Неочищенный продукт (19,5 г) суспендировали в метаноле (500 мл), затем добавляли 10% палладий на угле (содержание воды 50%) (6,85 г) и смесь перемешивали в атмосфере водорода в течение 17 часов. Катализатор отфильтровывали через целит и полученное вещество промывали метанолом. Фильтрат концентрировали в вакууме и полученное вещество растворяли в тетрагидрофуране и адсорбировали силикагелем. Адсорбированный силикагель концентрировали в вакууме и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 -13:7). Целевую фракцию концентрировали в вакууме, остаток собирали фильтрацией и промывали диэтиловым эфиром с получением названного соединения (3,78 г, 38%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,48 (3H, t, J=6,9 Гц), 4,13 (2Н, q, J=6,8 Гц), 5,50 (1Н, s), 6,39-6,43 (1Н, m), 6,87 (1Н, s), 7,05-7,09 (1Н, m), 7,13 (1Н, s), 7,91 (1Н, brs).
[0189] [Пример получения 15-5]
N-(4-((6-Этокси-1 Н-индол-5-ил)окси)пиридин-2-ил)ацетамид
[Химическая формула 84]

6-Этокси-1Н-индол-5-ол, описываемый в примере получения 15-4 (7,0 г, 39,5 ммоля), растворяли в диметилсульфоксиде (40 мл) в атмосфере азота, затем добавляли N-(4-хлорпиридин-2-ил)ацетамид, описываемый в примере получения 1-5 (8,09 г, 47,4 ммоля), и калия трет-бутоксид (4,88 г, 43,5 ммоля) при комнатной температуре и смесь нагревали и перемешивали при 160°С в течение 4 часов. Реакционную жидкость охлаждали до комнатной температуры и добавляли воду и этилацетат для разделения. Водный слой экстрагировали этилацетатом и объединенный органический слой дважды промывали водой, а затем насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния, затем фильтровали и фильтрат концентрировали в вакууме. Остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 3:1 - 2:1 - 3:2 - 1:3 - 1:4 - 0:1), а затем целевую фракцию концентрировали в вакууме с получением названного соединения (7,16 г, 58%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,23 (3Н, t, J=7,0 Гц), 2,14 (3Н, s), 4,02 (2H, q, J=7,0 Гц), 6,46-6,49 (1Н, m), 6,54 (1H, dd, J=5,9, 2,2 Гц), 7,01 (1Н. s), 7,13-7,16 (1H, m), 7,35 (1H, s), 7,74 (1H, brs), 7,87 (1H, brs), 8,02 (1H, d, J=5,9 Гц), 8,10 (1H, brs).
[0190] [Пример получения 15-6]
4-((6-Этокси-1Н-индол-5-ил)окси)пиридин-2-амин
[Химическая формула 85]

N-(4-((6-этокси-1Н-индол-5-ил)окси)пиридин-2-ил)ацетамид, описываемый в примере получения 15-5, (7,16 г, 23,0 ммоля) растворяли в метаноле (50 мл), добавляли 2 М раствор натрия гидроксида (50 мл) в атмосфере азота при комнатной температуре и смесь нагревали и перемешивали при 75°С в течение 2,5 часа. Реакционную смесь охлаждали до комнатной температуры, а затем добавляли воду и этилацетат для разделения. Водный слой экстрагировали этилацетатом и объединенный органический слой промывали насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния, затем полученное вещество фильтровали и концентрировали в вакууме. Жидкую смесь диэтилового эфира и этилацетата добавляли в остаток, смесь собирали фильтрацией и промывали с получением названного соединения (5,35 г, 86%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,28 (3Н, t, J=7,0 Гц), 4,02 (2H, q, J=7,0 Гц), 4,28 (2H, brs), 5,91 (1H, d, J=2,2 Гц), 6,29 (1H, dd, J=5,9, 2,2 Гц), 6,46-6,50 (1H, m), 7,00 (1H, s), 7,15-7,18 (1H, m), 7,33 (1H, s), 7,88 (1H, d, J=5,9 Гц), 8,13 (1H, brs).
[0191] [Пример получения 15-7]
5-((2-Аминопиридин-4-ил)окси)-6-этокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 86]
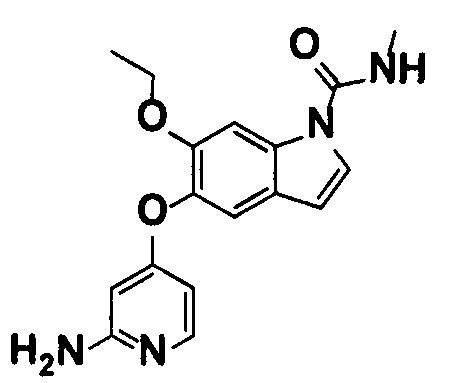
4-((6-Этокси-1Н-индол-5-ил)окси)пиридин-2-амин, описываемый в примере получения 15-6 (6,44 г, 23,9 ммоля), растворяли в N,N-диметилформамиде (80 мл), добавляли 50-72% маслянистый натрия гидрид (1,41 г) в атмосфере азота при 0°С и смесь перемешивали при комнатной температуре в течение 40 минут. Смесь снова охлаждали до 0°С, добавляли фенилметилкарбамат, описываемый в примере получения 1-7 (5,78 г, 38,3 ммоля), и смесь перемешивали при комнатной температуре в течение 3 часов. Насыщенный водный раствор аммония хлорида, воду и этилацетат добавляли в реакционную смесь для разделения. Водный слой экстрагировали этилацетатом и объединенный органический слой дважды промывали водой, а затем насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния, затем фильтровали, а потом фильтрат концентрировали в вакууме. Жидкую смесь диэтилового эфира и этилацетата добавляли в остаток, затем осадок собирали фильтрацией и полученное вещество снова промывали этилацетатом с получением названного соединения (4,24 г, 54%). Фильтрат очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 1:0-9:1), целевую фракцию концентрировали в вакууме, а затем остаток собирали фильтрацией и промывали жидкой смесью диэтилового эфира и этилацетата с получением названного соединения (1,58 г, 20%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,29 (3H, t, J=7,0 Гц), 3,07 (3Н, d, J=4,8 Гц), 4,09 (2Н, q, J=7,0 Гц), 4,29 (2Н, brs), 5,42-5,51 (1H, m), 5,90 (1H, d, J=2,2 Гц), 6,27 (1H, dd, J=6,2, 2,2 Гц), 6,55 (1H, dd, J=3,7, 0,7 Гц), 7,23 (1H, d, J=3,7 Гц), 7,27 (1H, s), 7,89 (1H, d, J=5,9 Гц), 7,98 (1Н, 8).
[0192] [Пример получения 15-8]
Трет-бутил
4-(4-((4-((6-этокси-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)фенил)пиперидин-1-карбоксилат
[Химическая формула 87]

Бензотриазол (1,92 г, 16,1 ммоля) растворяли в дихлорметане (80 мл), тионил хлорид (1,15 мл, 15,8 ммоля) добавляли в атмосфере азота при комнатной температуре и смесь перемешивали в течение 5 минут. 4-(1-(Трет-бутоксикарбонил)пиперидин-4-ил)бензойную кислоту (4,1 г, 13,4 ммоля), описанную в примере получения 1-12, добавляли в реакционную смесь при комнатной температуре, и смесь перемешивали в течение 25 минут. Реакционную смесь фильтровали через стеклянный фильтр, полностью покрытый безводным натрия сульфатом, безводный натрия сульфат промывали дихлорметаном, затем фильтрат добавляли в смесь 5-((2-аминопиридин-4-ил)окси)-6-этокси-N-метил-1Н-индол-1-карбоксамида (2 г, 6,13 ммоля), описанного в примере получения 15-7, триэтиламина (8,5 мл, 61,3 ммоля) и 4-диметиламинопиридина (75 мг, 0,613 ммоля) в тетрагидрофуране (40 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 14 часов, затем воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, затем полученное вещество сушили над безводным натрия сульфатом и фильтровали. Фильтрат концентрировали в вакууме, остаток растворяли в тетрагидрофуране, добавляли избыточное количество 9,8 М раствора метиламинметанола при комнатной температуре и смесь перемешивали в течение 5 часов. Реакционную смесь концентрировали в вакууме, дихлорметан добавляли к остатку, дополнительно добавляли диэтиловый эфир и этилацетат, а затем продукт собирали фильтрацией и промывали с получением названного соединения (3,08 г, 82%). Фильтрат концентрировали в вакууме, полученное вещество растворяли в дихлорметане и очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 1:9). Целевую фракцию концентрировали в вакууме с получением названного соединения (273 мг, 7,3%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,25 (3Н, t, J=7,0 Гц), 1,48 (9Н, s), 1,50-1,70 (2Н, m), 1,78-1,87 (2Н, m), 2,64-2,87 (3Н, m), 3,07 (3H, d, J=4,8 Гц), 4,11 (2Н, q, J=7,0 Гц), 4,16-4,33 (2Н, m), 5,45-5,52 (1H, m), 6,55 (1H, d, J=3,6 Гц), 6,60 (1H, dd, J=5,9, 2,3 Гц), 7,23 (1H, d, J=3,7 Гц), 7,28-7,33 (3H, m), 7,79-7,83 (2Н, m), 7,92 (1H, d, J=2,2 Гц), 8,00 (1H, s), 8,09 (1H, d, J=5,9 Гц), 8,51 (1H, brs).
[0193] [Пример 16]
6-этокси-5-((2-(4-(1-этилпиперидин-4-ил)бензамид)пиридин-4-ил)окси)-N-метил-1H-индол-1-карбоксамид
[Химическая формула 88]

Натрия триацетоксиборгидрид (1,36 г, 6,43 ммоля) добавляли в смесь 6-этокси-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида (2,2 г, 4,28 ммоля), описанного в примере 15, и тетрагидрофурана (33 мл) при комнатной температуре, затем добавляли тетрагидрофурановый раствор (11 мл) коммерчески доступного ацетальдегида (283 мг, 6,43 ммоля), а затем смесь перемешивали при комнатной температуре в течение 1,5 часа. Этилацетат и насыщенный водный раствор натрия бикарбоната добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали и таким образом полученный остаток объединяли с остатком, полученным из подобного исходного материала 6-этокси-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида (200 мг, 0,389 ммоля) подобным способом. Этилацетат добавляли к объединенному остатку и продукт собирали фильтрацией с получением названного соединения (2,20 г, 87%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,15 (3Н, t, J=7,2 Гц), 1,25 (3H, t, J=7,0 Гц), 1,82-1,92 (4Н, m), 2,01-2,13 (2Н, m), 2,43-2,62 (3H, m), 3,04 (3H, d, J=4,6 Гц), 3,08-3,17 (2Н, m), 4,10 (2Н, q, J=6,8 Гц), 5,55-5,62 (1Н, m), 6,53 (1H, d, J=3,7 Гц), 6,60 (1H, dd, J=5,7, 2,2 Гц), 7,23 (1H, d, J=3,7 Гц), 7,30-7,35 (3Н, m), 7,79 (2H, d, J=8,2 Гц), 7,91 (1H, d, J=2,2 Гц), 8,00 (1H, s), 8,09 (1H, d, J=5,7 Гц), 8,53 (1H, brs).
[0194] [Пример 17]
6-Изопропокси-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид
[Химическая формула 89]
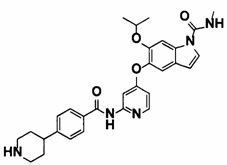
Бензотриазол (88,7 мг, 0,745 ммоля) растворяли в дихлорметане (4,0 мл), тионилхлорид (52 μл, 0,707 ммоля) добавляли в атмосфере азота при комнатной температуре и смесь перемешивали в течение 5 минут. 4-(1-(Трет-бутоксикарбонил)пиперидин-4-ил)бензойную кислоту (180 мг, 0,589 ммоля), описанную в примере получения 1-12, добавляли в реакционную смесь при комнатной температуре, и смесь перемешивали в течение 55 минут. Реакционную смесь фильтровали через стеклянный фильтр, полностью покрытый безводным натрия сульфатом, а затем безводный натрия сульфат промывали дихлорметаном, фильтрат добавляли в смесь 5-((2-аминопиридин-4-ил)окси)-6-изопропокси-N-метил-1Н-индол-1-карбоксамида (77 мг, 0,226 ммоля), описанного в примере получения 17-7, триэтиламина (0,314 мл, 2,26 ммоля) и 4-диметиламинопиридина (2,76 мг, 0,023 ммоля) в тетрагидрофуране (2,0 мл), дихлорметане (5,0 мл) и N,N-диметилформамиде (0,2 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 140 минут, а затем воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным натрия сульфатом и фильтровали. Фильтрат концентрировали в вакууме, остаток растворяли в тетрагидрофуране, добавляли избыточное количество тетрагидрофуранового раствора метиламина при комнатной температуре и смесь перемешивали в течение 30 минут. Реакционную смесь концентрировали в вакууме, остаток растворяли в дихлорметане, полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 1:3 - 0:1) и целевую фракцию концентрировали в вакууме с получением неочищенного продукта (111 мг).
Неочищенный продукт (111 мг) растворяли в дихлорметане (1,8 мл) и добавляли трифторуксусную кислоту (0,65 мл) при 0°С. Смесь перемешивали в течение 90 минут при комнатной температуре, а затем концентрировали в вакууме, остаток растворяли в дихлорметан-триэтиламине, а затем полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 1:0 - 17:3 - 4:1) с получением названного соединения (85,4 мг, 92%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,22 (6Н, d, J=6,0 Гц), 1,55-1,71 (2Н, m), 1,79-1,88 (2H, m), 2,62-2,80 (3H, m), 3,05 (3H, d, J=4,8 Гц), 3,15-3,25 (2H, m), 4,52-4,64 (1H, m), 5,49-5,57 (1H, m), 6,54 (1H, d, J=3,7 Гц), 6,58 (1H, dd, J=5,7, 2,4 Гц), 7,24 (1H, d, J=3,8 Гц), 7,29-7,35 (3H, m), 7,80 (2H, d, J=8,2 Гц), 7,93 (1H, d, J=2,4 Гц), 8,01 (1H, s), 8,08 (1H, d, J=5,9 Гц), 8,51 (1H, brs).
[0195] Исходный материал 5-((2-аминопиридин-4-ил)окси)-6-изопропокси-N-метил-1Н-индол-1-карбоксамид синтезировали следующим способом.
[0196] [Пример получения 17-1]
3-Гидрокси-4-изопропоксибензальдегид
[Химическая формула 90]
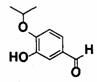
Коммерчески доступный 3,4-дигидроксибензальдегид (5 г, 36,2 ммоля) и калия карбонат (5,15 г, 37,3 ммоля) растворяли в N,N-диметилформамиде (20 мл), затем добавляли 2-бромпропан (3,5 мл, 37,3 ммоля) в атмосфере азота при комнатной температуре, смесь нагревали и перемешивали при 40°С в течение 2,5 часа. Реакционную смесь охлаждали до 0°С, а затем 2 М хлористоводородную кислоту, этилацетат и воду добавляли для разделения. Водный слой экстрагировали этилацетатом, объединенный органический слой промывали водой и насыщенным солевым раствором, и полученное вещество сушили над безводным сульфатом магния, а затем фильтровали. Растворитель выпаривали, дихлорметан добавляли к остатку, осадок отделяли фильтрацией и фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 19:1 - 13:7), а затем целевую фракцию концентрировали в вакууме с получением названного соединения (1,84 г, 28%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,42 (6Н, d, J=5,9 Гц), 4,73 (1Н, spt, J=6,1 Гц), 5,78 (1H, s), 6,95 (1Н, d, J=8,1 Гц), 7,41 (1Н, dd, J=8,2, 2,0 Гц), 7,44 (1Н, d, J=1,8 Гц), 9,83 (1H, s).
[0197] [Пример получения 17-2]
3-(Бензилокси)-4-изопропоксибензальдегид
[Химическая формула 91]
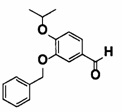
Калия карбонат (1,83 г, 13,2 ммоля) и бензилхлорид (1,55 мл, 13,5 ммоля) добавляли в суспензию 3-гидрокси-4-изопропоксибензальдегида (1,84 г, 10,2 ммоля), описанного в примере получения 17-1, в этаноле (20 мл) в атмосфере азота при комнатной температуре и смесь нагревали и перемешивали при 90°С в течение 2 часов. Смесь охлаждали до 0°С, а затем 2 М хлористоводородную кислоту, этилацетат и воду добавляли для разделения. Органический слой промывали насыщенным солевым раствором, полученное вещество сушили над безводным натрия сульфатом, а затем фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 19:1 - 3:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (2,59 г, 94%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,42 (6Н, d, J=6,0 Гц), 4,69 (1Н, spt, J=6,1 Гц), 5,18 (2Н, s), 7,00 (1Н, d, J=8,1 Гц), 7,29-7,41 (3Н, m), 7,43-7,48 (4H, m), 9,81 (1Н, s).
[0198] [Пример получения 17-3]
(Е)-2-(Бензилокси)-1-изопропокси-4-(2-нитровинил)бензол
[Химическая формула 92]

3-(Бензилокси)-4-изопропоксибензальдегид (2,59 г, 9,59 ммоля), описанный в примере получения 17-2, растворяли в уксусной кислоте (7,5 мл), затем добавляли аммония ацетат (887 мг, 11,5 ммоля) и нитрометан (1,25 мл, 23,1 ммоля) в атмосфере азота при комнатной температуре, смесь нагревали и перемешивали при 130°С в течение 2 часов. Смесь охлаждали до комнатной температуры, осадок собирали фильтрацией и промывали этанолом с получением названного соединения (2,20 г, 73%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,41 (6H, d, J=6,2 Гц), 4,55-4,71 (1Н, m), 5,15 (2H, s), 6,94 (1Н, d, J=8,4 Гц), 7,05 (1Н, d, J=1,8 Гц), 7,15 (1Н, dd, J=8,4, 1,8 Гц), 7,29-7,48 (6H, m), 7,90 (1Н, d, J=13,5 Гц).
[0199] [Пример получения 17-4]
6-Изопропокси-1Н-индол-5-ол
[Химическая формула 93]
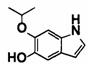
Дымящуюся азотную кислоту (1,5 мл, 33,3 ммоля) медленно добавляли в смесь (Е)-2-(бензилокси)-1-изопропокси-4-(2-нитровинил)бензола (2,20 г, 7,02 ммоля), описанного в примере получения 17-3, и уксусной кислоты (20 мл) при комнатной температуре и смесь перемешивали в течение 7,5 часа. Реакционную смесь выливали на лед, осадок собирали фильтрацией, а затем полученное вещество промывали жидкой смесью уксусной кислоты и этанола с получением неочищенного продукта (2,28 г).
Неочищенный продукт (2,28 г) суспендировали в метаноле (60 мл), добавляли 10% палладий на угле (содержание воды 50%) (677 мг) и смесь перемешивали в атмосфере водорода в течение 14,5 часа. Катализатор отфильтровывали через целит и полученное вещество промывали метанолом. Фильтрат концентрировали в вакууме и очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 3:2). Целевую фракцию концентрировали в вакууме с получением названного соединения (475 мг, 35%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,40 (6H, d, J=6,2 Гц), 4,57 (1Н, spt, J=6,1 Гц), 5,55 (1Н, s), 6,38-6,44 (1Н, m), 6,90 (1Н, s), 7,08 (1Н, t, J=2,7 Гц), 7,13 (1Н, s), 7,90 (1Н, brs).
[0200] [Пример получения 17-5]
N-(4-((6-изопропокси-1Н-индол-5-ил)окси)пиридин-2-ил)ацетамид
[Химическая формула 94]

6-Изопропокси-1Н-индол-5-ол (165 мг, 0,863 ммоля), описанный в примере получения 17-4, и N-(4-хлорпиридин-2-ил)ацетамид (442 мг, 2,59 ммоля), описанный в примере получения 1-5, растворяли в диметилсульфоксиде (2,0 мл) в атмосфере азота, затем калия трет-бутоксид (194 мг, 1,73 ммоля) добавляли при комнатной температуре, смесь нагревали и перемешивали при 160°С в течение 3 часов. Реакционную жидкость охлаждали до комнатной температуры и добавляли воду и этилацетат для разделения. Водный слой экстрагировали этилацетатом и объединенный органический слой промывали водой и насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния, затем фильтровали, а потом фильтрат концентрировали в вакууме. Остаток растворяли в дихлорметане, полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 3:2 - 1:3), а затем целевую фракцию концентрировали в вакууме с получением названного соединения (116 мг, 41%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,21 (6H, d, J=6,2 Гц), 2,14 (3Н, s), 4,34-4,48 (1Н, m), 6,45-6,53 (2H, m), 7,03 (1Н, s), 7,16 (1Н, t, J=2,7 Гц), 7,35 (1Н, s), 7,77 (1Н, brs), 7,85-8,05 (2H, m), 8,10 (1Н, brs).
[0201] [Пример получения 17-6]
4-((6-Изопропокси-1Н-индол-5-ил)окси)пиридин-2-амин
[Химическая формула 95]
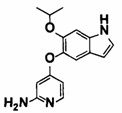
N-(4-((6-изопропокси-1Н-индол-5-ил)окси)пиридин-2-ил)ацетамид (116 мг, 0,357 ммоля), описанный в примере получения 17-5, растворяли в метаноле (2,5 мл), 28% натрия метоксид (0,728 мл) добавляли в атмосфере азота при комнатной температуре и смесь нагревали и перемешивали при 70°С в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры, а затем добавляли воду и этилацетат для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отфильтровывали и полученное вещество концентрировали в вакууме. Остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 3:7-0:1 - этилацетат:метанол = 99:1 - 9:1), затем целевую фракцию концентрировали в вакууме с получением названного соединения (66,3 мг, 66%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,22 (6H, d, J=5,9 Гц), 4,28 (2Н, brs), 4,3-4,47 (1H, m), 5,89 (1H, d, J=2,2 Гц), 6,29 (1H, dd, J=5,9, 2,2 Гц), 6,46-6,51 (1H, m), 7,04 (1H, s), 7,18 (1H, t, J=2,9 Гц), 7,34 (1H, s), 7,88 (1H, d, J=5,9 Гц), 8,12 (1H, brs).
[0202] [Пример получения 17-7]
5-((2-Аминопиридин-4-ил)окси)-6-изопропокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 96]
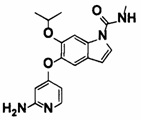
4-((6-Изопропокси-1Н-индол-5-ил)окси)пиридин-2-амин (65,6 мг, 0,232 ммоля), описанный в примере получения 17-6, растворяли в N,N-диметилформамиде (1,5 мл), добавляли 50-72% маслянистый натрия гидрид (16,7 мг) в атмосфере азота при 0°С и смесь перемешивали при комнатной температуре в течение 50 минут. Смесь снова охлаждали до 0°С, добавляли фенилметилкарбамат (69,2 мг, 0,458 ммоля), описанный в примере получения 1-7, и смесь перемешивали при комнатной температуре в течение 3 часов. Насыщенный водный раствор аммония хлорида, воду и этилацетат добавляли в реакционную смесь для разделения. Водный слой экстрагировали этилацетатом и объединенный органический слой промывали водой, а затем насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния, затем фильтровали, а потом фильтрат концентрировали в вакууме. Остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 3:7 - 0:1 - этилацетат:метанол = 99:1 - 19:1), затем целевую фракцию концентрировали в вакууме с получением названного соединения (77,0 мг, 98%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,24 (6Н, d, J=6,0 Гц), 3,06 (3Н, d, J=4,8 Гц), 4,32 (2Н, brs), 4,56 (1H, spt, J=6,1 Гц), 5,50-5,61 (1H, m), 5,89 (1H, d, J=2,2 Гц), 6,27 (1H, dd, J=5,9, 2,2 Гц), 6,55 (1H, dd, J=3,6, 0,6 Гц), 7,24-7,28 (2Н, m), 7,88 (1H, d, J=5,9 Гц), 8,00 (1H, s).
[0203] [Пример 18]
(R)-5-((2-(4-(1-(2-Гидроксипропил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-изопропокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 97]
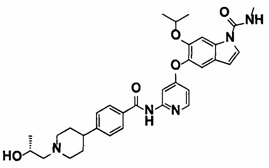
Коммерчески доступный (R)-(+)-пропиленоксид (20,5 мг, 0,353 ммоля) добавляли в смесь 6-изопропокси-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида (12,7 мг, 0,024 ммоля), описанного в примере 17, и этанола (0,5 мл), смесь нагревали и перемешивали в закупоренной пробирке при 80°С в течение 3 часов и 40 минут. Смесь охлаждали до комнатной температуры, затем реакционную смесь концентрировали в вакууме, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хромагографии на силикагеле NH (н-гептан:этилацетат = 1:4 - 0:1 этилацетат:метанол = 99:1 - 19:1). Целевую фракцию концентрировали в вакууме, осадок собирали фильтрацией и промывали диэтиловым эфиром с получением названного соединения (8,28 мг, 59%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,15 (3H, d, J=6,2 Гц), 1,22 (6Н, d, J=6,2 Гц), 1,70-1,90 (3H, m), 2,00-2,09 (1H, m), 2,21-2,46 (3H, m), 2,53-2,64 (1H, m), 2,92 (1H, d, J=11,0 Гц), 3,07 (3H, d, J=4,8 Гц), 3,14 (1H, d, J=11,3 Гц), 3,80-3,91 (1H, m), 4,53-4,64 (1H, m), 5,44-5,52 (1H, m), 6,55 (1H, d, J=3,3 Гц), 6,58 (1H, dd, J=5,7, 2,4 Гц), 7,25 (1H, d, J=3,7 Гц), 7,30-7,36 (3H, m), 7,81 (2H, d, J=8,4 Гц), 7,93 (1H, d, J=2,2 Гц), 8,01 (1H, s), 8,09 (1H, d, J=5,9 Гц), 8,49 (1H, s).
[0204] [Пример 19]
6-(Дифторметокси)-5-((2-(4-((4-гидроксипиперидин-1-ил)метил)бензамид)пиридин-4-ил)окси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 98]

Триэтиламин (17 μл, 0,123 ммоля) и коммерчески доступный 4-(хлорметил)бензоил хлорид (11,5 мг, 0,061 ммоля) добавляли в смесь 5-((2-аминопиридин-4-ил)окси)-6-(дифторметокси)-N-метил-1Н-индол-1-карбоксамида (4,3 мг, 0,012 ммоля), описанного в примере получения 19-7, и тетрагидрофурана (0,5 мл) в атмосфере азота при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 2,5 часов, воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом, полученное вещество фильтровали с помощью силикагеля NH (этилацетат) и концентрировали в вакууме с получением неочищенного продукта (6,18 мг).
Неочищенный продукт (6,18 мг) растворяли в N,N-диметилформамиде (0,5 мл), добавляли коммерчески доступный 4-гидроксипиперидин (17,7 мг, 0,175 ммоля) в атмосфере азота при комнатной температуре и смесь перемешивали в течение 14 часов. Воду и этилацетат добавляли в реакционную смесь для разделения и органический слой промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью TLC на силикагеле NH (этилацетат) с получением названного соединения (6,0 мг, 86%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,50-1,66 (2Н, m), 1,83-1,94 (2H, m), 2,16 (2H, t, J=9,7 Гц), 2,67-2,78 (2H, m), 3,08 (3H, d, J=4,4 Гц), 3,54 (2H, s), 3,62-3,77 (2H, m), 5,46-5,57 (1H, m), 6,53 (1H, t, J=73,9 Гц), 6,61 (1H, d, J=3,7 Гц), 6,64 (1H, dd, J=5,7, 2,4 Гц), 7,38-7,46 (4Н, m), 7,80 (2H, d, J=8,1 Гц), 7,91 (1H, d, J=2,2 Гц), 8,15 (1H, d, J=5,9 Гц), 8,24 (1H, s), 8,53 (1H, brs).
[0205] Исходный материал 5-((2-аминопиридин-4-ил)окси)-6-(дифторметокси)-N-метил-1Н-индол-1-карбоксамид синтезировали следующим способом.
[0206] [Пример получения 19-1]
4-(Дифторметокси)-3-гидроксибензальдегид
[Химическая формула 99]

Коммерчески доступный 3,4-дигидроксибензальдегид (5 г, 36,2 ммоля) и коммерчески доступный натрия хлордифторацетат (5,57 г, 36,5 ммоля) растворяли в N,N-диметилформамиде (45 мл) и воде (905 μл), а затем натрия гидроксид (1,48 г, 37,0 ммоля) добавляли при комнатной температуре, смесь нагревали и перемешивали при 120°С в течение 2 часов. Растворитель выпаривали в вакууме, остаток охлаждали до 0°С, 5 М хлористоводородную кислоту и диэтиловый эфир добавляли для разделения. Органический слой промывали водой и насыщенным солевым раствором, а затем растворитель выпаривали. Остаток растворяли в дихлорметане, полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 7:3), а затем целевую фракцию концентрировали в вакууме с получением названного соединения (2,66 г, 39%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 5,69-5,74 (1H, m), 6,65 (1H, t, J=72,5 Гц), 7,23-7,31 (1Н, m), 7,46 (1Н, dd, J=8,4,1,8 Гц), 7,54 (1Н, d, J=1,8 Гц), 9,92 (1Н, s).
[0207] [Пример получения 19-2]
3-(Бензилокси)-4-(дифторметокси)бензальдегид
[Химическая формула 100]
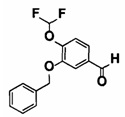
Калия карбонат (3,91 г, 28,3 ммоля) и бензилбромид (2,5 мл, 21,1 ммоля) добавляли в раствор 4-(дифторметокси)-3-гидроксибензальдегида (2,66 г, 14,2 ммоля), описанного в примере получения 19-1, в ацетонитриле (50 мл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь фильтровали, а затем растворитель выпаривали. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 3:2). Целевую фракцию концентрировали в вакууме, а затем количественно получали названное соединение.
1Н-ЯМР спектр (CDCl3) δ (ppm): 5,21 (2H, s), 6,68 (1Н, t, J=74,3 Гц), 7,31-7,51 (7Н, m), 7,57 (1Н, d, J=1,8 Гц), 9,92 (1Н, s).
[0208] [Пример получения 19-3]
(Е)-2-(Бензилокси)-1-(дифторметокси)-4-(2-нитровинил)бензол
[Химическая формула 101]

3-(Бензилокси)-4-(дифторметокси)бензальдегид (3,94 г, 14,2 ммоля), описанный в примере получения 19-2, растворяли в уксусной кислоте (11 мл), затем аммония ацетат (1,27 г, 16,4 ммоля) и нитрометан (1,9 мл, 35,1 ммоля) добавляли в атмосфере азота при комнатной температуре, смесь нагревали и перемешивали при 130°С в течение 2 часов. Смесь охлаждали до комнатной температуры, а затем нагревали и снова перемешивали при 130°С в течение 1 часа. Осадок собирали фильтрацией и промывали жидкой смесью уксусной кислоты и этанола с получением названного соединения (2,36 г, 52%). Фильтрат растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 19:1 - 4:1), а затем целевую фракцию концентрировали в вакууме. Осадок промывали этанолом с получением названного соединения (406 мг, 8,9%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 5,18 (2H, s), 6,65 (1H, t, J=74,3 Гц), 7,12-7,19 (2H, m), 7,21-7,29 (1H, m), 7,32-7,45 (5H, m), 7,48 (1H, d, J=13,9 Гц), 7,92 (1H, d, J=13,9 Гц).
[0209] [Пример получения 19-4]
5-(Бензилокси)-6-(дифторметокси)-1Н-индол
[Химическая формула 102]
Дымящуюся азотную кислоту (6,0 мл, 133 ммоля) медленно добавляли в смесь (Е)-2-(бензилокси)-1-(дифторметокси)-4-(2-нитровинил)бензола (2,77 г, 8,61 ммоля), описанного в примере получения 19-3, и уксусной кислоты (36 мл) в ледяной бане, и смесь перемешивали в течение 7,5 часа. Часть реакционной смеси нагревали и перемешивали при 50°С в течение 30 минут, затем при 70°С в течение 40 минут, а затем при 75°С в течение 165 минут. Часть реакционной смеси выливали на лед и полученное вещество разбавляли этилацетатом для разделения. Органический слой промывали водой и насыщенным солевым раствором и сушили над безводным магния сульфатом, а затем растворитель выпаривали. Остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 17:3 - 3:1), затем целевую фракцию концентрировали в вакууме, а затем остаток собирали фильтрацией и промывали диэтиловым эфиром с получением неочищенного продукта А (23,6 мг). Оставшуюся реакционную смесь нагревали и перемешивали при 65°С в течение 3 часов, затем полученное вещество выливали на лед, а затем осадок собирали фильтрацией и промывали жидкой смесью уксусной кислоты и этанола с получением неочищенного продукта В (603 мг). Этилацетат добавляли в фильтрат для разделения. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным магния сульфатом и фильтровали, а затем растворитель выпаривали. Остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 3:1), затем целевую фракцию концентрировали в вакууме, диэтиловый эфир добавляли к остатку и осадок собирали фильтрацией с получением неочищенного продукта С (73,3 мг).
Неочищенные продукты А, В и С (550 мг) суспендировали в этаноле (5,5 мл), уксусной кислоте (5,5 мл) и воде (676 μл), затем добавляли порошок железа (419 мг, 7,51 ммоля) в атмосфере азота, смесь нагревали и перемешивали при 70°С в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и добавляли этилацетат и водный раствор натрия дисульфита для разделения. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным магния сульфатом и фильтровали, а затем растворитель выпаривали. Остаток растворяли в дихлорметане, полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 9:1 - 3:2), а затем целевую фракцию концентрировали в вакууме с получением названного соединения (245 мг, 13%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 5,15 (2H, s), 6,45-6,49 (1H, m), 6,58 (1H, t, J=76,0 Гц), 7,19-7,22 (1H, m), 7,23 (1H, s), 7,24-7,27 (1H, m), 7,29-7,43 (3Н, m), 7,44-7,50 (2H, m), 8,12 (1H, brs).
[0210] [Пример получения 19-5]
6-(Дифторметокси)-1Н-индол-5-ол
[Химическая формула 103]
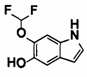
5-(Бензилокси)-6-(дифторметокси)-1Н-индол (245 мг, 0,847 ммоля), описанный в примере получения 19-4, растворяли в этаноле (8,0 мл), затем 10% палладий на угле (содержание воды, 50%) (90 мг) добавляли при комнатной температуре и смесь перемешивали в атмосфере водорода в течение 75 минут. Реакционную смесь разбавляли этилацетатом и катализатор отфильтровывали через целит. Фильтрат концентрировали в вакууме, а затем фильтровали с помощью силикагеля (этилацетат). Целевую фракцию концентрировали в вакууме с количественным получением названного соединения.
1Н-ЯМР спектр (CDCl3) δ (ppm): 5,15 (1H, s), 6,41-6,49 (1H, m), 6,53 (1H, t, J=74,1 Гц), 7,15-7,24 (3H, m), 8,06 (1H, brs).
[0211] [Пример получения 19-6]
4-((6-(Дифторметокси)-1Н-индол-5-ил)окси)пиридин-2-амин
[Химическая формула 104]
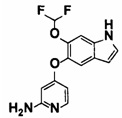
6-(Дифторметокси)-1Н-индол-5-ол (35,1 мг, 0,176 ммоля), описанный в примере получения 19-5, N-(4-хлорпиридин-2-ил)ацетамид (89,0 мг, 0,522 ммоля), описанный в примере получения 1-5, и калия трет-бутоксид (44,0 мг, 0,392 ммоля) растворяли в диметилсульфоксиде (500 μл) в атмосфере азота, и смесь нагревали и перемешивали при 160°С в течение 80 минут. Реакционную жидкость охлаждали до комнатной температуры и добавляли воду и этилацетат для разделения. Органический слой промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом, затем фильтровали, а потом фильтрат концентрировали в вакууме. Остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хромагографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 0:1 - этилацетат:метанол = 99:1 - 9:1), затем целевую фракцию концентрировали в вакууме с получением неочищенного продукта (12,5 мг).
Неочищенный продукт (12,5 мг) растворяли в метаноле (500 μл) добавляли 28% натрия метоксид (39 μл, 0,382 ммоля) в атмосфере азота при комнатной температуре, смесь нагревали и перемешивали при 65°С в течение 1 часа. Смесь охлаждали до комнатной температуры, добавляли 28% натрия метоксид (39 μл, 0,382 ммоля), смесь нагревали и перемешивали при 65°С в течение 2,5 часа. Воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отфильтровывали и полученное вещество концентрировали в вакууме. Остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 0:1 - этилацетат:метанол = 97:3 - 9:1), а затем целевую фракцию концентрировали в вакууме с получением названного соединения (4,4 мг, 8,6%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 4,48 (2H, brs), 5,91 (1H, d, J=1,8 Гц), 6,28 (1Н, dd, J=5,9, 1,8 Гц), 6,45 (1H, t, J=74,5 Гц), 6,52-6,58 (1H, m), 7,30 (1H, t, J=2,9 Гц), 7,36 (1H, s), 7,41 (1H, s), 7,90 (1H, d, J=5,9 Гц), 8,35 (1H, brs).
[0212] [Пример получения 19-7]
5-((2-Аминопиридин-4-ил)окси)-6-(дифторметокси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 105]
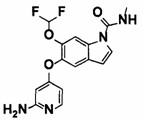
4-((6-(Дифторметокси)-1Н-индол-5-ил)окси)пиридин-2-амин (4,4 мг, 0,015 ммоля), описанный в примере получения 19-6, растворяли в N,N-диметилформамиде (500 μл), затем добавляли 50-72% маслянистый натрия гидрид (4,1 мг) в атмосфере азота при 0°С и смесь перемешивали при комнатной температуре в течение 30 минут. Смесь снова охлаждали до 0°С и добавляли фенилметилкарбамат (16,4 мг, 0,108 ммоля), описанный в примере получения 1-7, и смесь перемешивали при комнатной температуре в течение 50 минут. Воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, затем сушили над безводным натрия сульфатом, фильтровали, а потом фильтрат концентрировали в вакууме. Остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 0:1 - этилацетат:метанол = 99:1 - 9:1), а затем целевую фракцию концентрировали в вакууме с получением названного соединения (4,3 мг, 82%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 3,08 (3H, d, J=4,8 Гц), 4,44 (2H, brs), 5,49 (1H, brs), 5,91 (1H, d, J=1,8 Гц), 6,26 (1H, dd, J=6,2, 2,2 Гц), 6,50 (1H, t, J=74,0 Гц), 6,61 (1H, d, J=3,7 Гц), 7,35 (1H, s), 7,41 (1H, d, J=3,7 Гц), 7,92 (1H, d, J=5,9 Гц), 8,23 (1H, s).
[0213] [Пример 20]
6-(2-Метоксиэтокси)-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид
[Химическая формула 106]
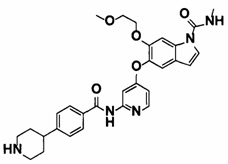
Бензотриазол (609 мг, 5,11 ммоля) растворяли в дихлорметане (25 мл), добавляли тионилхлорид (373 μл, 5,11 ммоля) в атмосфере азота при комнатной температуре и смесь перемешивали в течение 5 минут. 4-(1-(Трет-бутоксикарбонил)пиперидин-4-ил)бензойную кислоту (1,3 г, 4,26 ммоля), описанную в пример получения 1-12, добавляли в реакционную смесь при комнатной температуре, и смесь перемешивали в течение 30 минут. Реакционную смесь фильтровали через стеклянный фильтр, полностью покрытый безводным натрия сульфатом, и безводный натрия сульфат промывали дихлорметаном, фильтрат добавляли в смесь 5-((2-аминопиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1Н-индол-1-карбоксамида (0,95 г, 2,67 ммоля), описанного в примере получения 20-7, триэтиламина (1,86 мл, 13,3 ммоля) и 4-диметиламинопиридина (16 мг, 0,133 ммоля) в N,N-диметилформамиде (3 мл) и дихлорметане (20 мл) при 0°С за 5 минут и смесь промывали дихлорметаном (10 мл), а затем перемешивали при той же температуре в течение 5 минут. Смесь перемешивали при комнатной температуре в течение 2 часов, затем добавляли 40% водный раствор метиламина (2,3 мл, 26,7 ммоля), а затем смесь перемешивали при комнатной температуре в течение 1,5 часа. Насыщенный водный раствор натрия бикарбоната добавляли в реакционную смесь для разделения и водный слой три раза экстрагировали этилацетатом. Объединенный органический слой сушили над безводным натрия сульфатом. Осушающее средство отфильтровывали, затем фильтрат концентрировали в вакууме и полученное вещество очищали с помощью колоночной хромагографии на силикагеле (н-гептан:этилацетат = 1:1 - 0:1 - этилацетат:метанол = 49:1 - 23:2) с получением неочищенного продукта (1,11 г).
Неочищенный продукт (1,11 г) растворяли в дихлорметане (50 мл) и трифторуксусную кислоту (5,0 мл) добавляли при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 30 минут, затем полученное вещество концентрировали в вакууме, а затем остаток растворяли в дихлорметане и триэтиламине и полученное вещество концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 1:0 - 22:3) с получением названного соединения (829 мг, 57%).
1Н-ЯМР спектр (500 МГц, CDCl3) δ (ppm): 1,59-1,69 (2H, m), 1,83 (2H, d, J=14,1 Гц), 2,68 (1Н, tt, J=12,0, 3,6 Гц), 2,75 (2H, td, J=12,2, 2,4 Гц), 3,04 (3Н, d, J=4,9 Гц), 3,17-3,23 (2H, m), 3,26 (3Н, s), 3,55-3,61 (2H, m), 4,15-4,21 (2H, m), 5,57-5,65 (1Н, m), 6,53 (1Н, d, J=3,4 Гц), 6,62 (1Н, dd, J=5,8, 2,4 Гц), 7,25 (1Н, d, J=3,9 Гц), 7,30-7,34 (3Н, m), 7,77-7,82 (2H, m), 7,91 (1Н, d, J=2,4 Гц), 8,02 (1Н, s), 8,10 (1Н, d, J=5,9 Гц), 8,50 (1H, brs).
[0214] Исходный материал 5-((2-аминопиридин-4-ил)окси)-6-(2-метоксиэтокси)-Н-метил-1Н-индол-1-карбоксамид синтезировали следующим способом.
[0215] [Пример получения 20-1]
3-Гидрокси-4-(2-метоксиэтокси)бензальдегид
[Химическая формула 107]

Коммерчески доступный 3,4-дигидроксибензальдегид (39,3 г, 285 ммоля) и натрия карбонат (45,2 г, 427 ммоля) растворяли в N,N-диметилформамиде (400 мл), затем добавляли коммерчески доступный 2-бромэтилметиловый эфир (26,7 мл, 285 ммоля) в атмосфере азота при комнатной температуре и смесь перемешивали в течение 5 дней. Смесь охлаждали до 0°С, а затем 2 М хлористоводородную кислоту, этилацетат и воду добавляли для разделения. Водный слой экстрагировали этилацетатом, затем объединенный органический слой промывали насыщенным солевым раствором и сушили над безводным магния сульфатом, а затем фильтровали. Растворитель выпаривали, дихлорметан добавляли, осадок отделяли фильтрацией, а затем полученный фильтрат очищали с помощью колоночной хромагографии на силикагеле (н-гептан:этилацетат = 17:3-1:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (12,9 г, 23%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 3,47 (3H, s), 3,76-3,80 (2H, m), 4,25-4,29 (2H, m), 6,40 (1H, brs), 7,01 (1H, d, J=8,4 Гц), 7,41 (1H, dd, J=8,2, 2,0 Гц), 7,45 (1Н, d, J=1,8 Гц), 9,85 (1H, s).
[0216] [Пример получения 20-2]
3-(Бензилокси)-4-(2-метоксиэтокси)бензальдегид
[Химическая формула 108]
Калия карбонат (11,8 г, 85,7 ммоля) и бензилхлорид (10 мл, 86,9 ммоля) добавляли в жидкую смесь 3-гидрокси-4-(2-метоксиэтокси)бензальдегида (12,9 г, 65,9 ммоля), описанного в примере получения 20-1, в этаноле (130 мл) в атмосфере азота при комнатной температуре, смесь нагревали и перемешивали при 90°С в течение 2 часов. Смесь охлаждали до 0°С, а затем 2 М хлористоводородную кислоту, этилацетат и воду добавляли для разделения. Органический слой промывали насыщенным солевым раствором, сушили над безводным магния сульфатом, а затем фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1-1:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (17,6 г, 93%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 3,46 (3H, s), 3,79-3,85 (2H, m), 4,24-4,30 (2H, m), 5,18 (2H, s), 7,03 (1H, d, J=8,1 Гц), 7,29-7,35 (1H, m), 7,35-7,41 (2H, m), 7,43-7,50 (4H, m), 9,82 (1H, s).
[0217] [Пример получения 20-3]
(Е)-2-(Бензилокси)-1-(2-метоксиэтокси)-4-(2-нитровинил)бензол
[Химическая формула 109]
3-(Бензилокси)-4-(2-метоксиэтокси)бензальдегид (17,6 г, 61,5 ммоля), описанный в примере получения 20-2, растворяли в уксусной кислоте (49,3 мл), затем добавляли аммония ацетат (5,69 г, 73,8 ммоля) и нитрометан (8,32 мл, 154 ммоля) в атмосфере азота при комнатной температуре, смесь нагревали и перемешивали при 130°С в течение 2 часов. Смесь охлаждали до комнатной температуры, а затем осадок собирали фильтрацией и промывали этанолом с количественным получением названного соединения.
1Н-ЯМР спектр (CDCl3) δ (ppm): 3,46 (3H, s), 3,78-3,84 (2H, m), 4,21-4,27 (2H, m), 5,16 (2Н, s), 6,97 (1H, d, J=8,4 Гц), 7,06 (1Н, d, J=1,8 Гц), 7,16 (1Н, dd, J=8,4, 2,2 Гц), 7,30-7,48 (6Н, m), 7,91 (1Н, d, J=13,5 Гц).
[0218] [Пример получения 20-4]
6-(2-Метоксиэтокси)-1Н-индол-5-ол
[Химическая формула 110]

Добавляли 69% азотную кислоту (15 мл, 233 ммоля) в смесь (Е)-2-(бензилокси)-1-(2-метоксиэтокси)-4-(2-нитровинил)бензола (20,2 г, 61,5 ммоля), описанного в примере получения 20-3, и уксусной кислоты (120 мл) при 25°С и смесь перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь выливали на лед, осадок собирали фильтрацией, а затем промывали водой с получением неочищенного продукта (23,0 г).
Неочищенный продукт (23,0 г) суспендировали в метаноле (500 мл), затем добавляли 10% палладий на угле (содержание воды 50%) (8 г) при комнатной температуре и смесь перемешивали в атмосфере водорода в течение 6 часов. Катализатор отфильтровывали через целит, фильтрат концентрировали в вакууме и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 2:1 - 1:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (3,94 г, 31%).
1H-ЯМР спектр (CDCl3) δ (ppm): 3,48 (3H, s), 3,69-3,78 (2H, m), 4,16-4,23 (2H, m), 6,24 (1H, s), 6,41 (1H, ddd, J=3,1, 2,1, 0,8 Гц), 6,97 (1Н, s), 7,10 (1H, dd, J=3,2, 2,5 Гц), 7,15 (1H, s), 7,94 (1Н, brs).
[0219] [Пример получения 20-5]
N-(4-((6-(2-метоксиэтокси)-1Н-индол-5-ил)окси)пиридин-2-ил)ацетамид
[Химическая формула 111]

6-(2-Метоксиэтокси)-1Н-индол-5-ол (3,94 г, 19,0 ммоля), описанный в примере получения 20-4, и N-(4-хлорпиридин-2-ил)ацетамид (3,25 г, 19,0 ммоля), описанный в примере получения 1-5, растворяли в диметилсульфоксиде (25 мл), затем 97% калия трет-бутоксид (2,20 г, 19,0 ммоля) добавляли при комнатной температуре и смесь нагревали и перемешивали при 150°С в течение 13 часов. Воду и этилацетат добавляли в реакционную жидкость при комнатной температуре для разделения. Водный слой три раза экстрагировали этилацетатом и объединенный органический слой промывали водой. Органический слой сушили над безводным натрия сульфатом. Осушающее средство отфильтровывали, фильтрат концентрировали в вакууме и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 2:3 - 0:1 - этилацетат:метанол = 49:1 - 9:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (3,45 г, 53%).
1Н-ЯМР спектр (500 МГц, СDCl3) δ (ppm): 2,13 (3H, s), 3,27 (3H, s), 3,54-3,58 (2H, m), 4,07-4,11 (2H, m), 6,46-6,50 (1H, m), 6,54 (1H, dd, J=5,8, 1,9 Гц), 7,05 (1H, s), 7,14-7,17 (1H, m), 7,36 (1H, s), 7,75 (1H, brs), 8,02 (1H, d, J=5,8 Гц), 8,10 (1H, brs), 8,19 (1H, brs).
[0220] [Пример получения 20-6]
4-((6-(2-Метоксиэтокси)-1Н-индол-5-ил)окси)пиридин-2-амин
[Химическая формула 112]
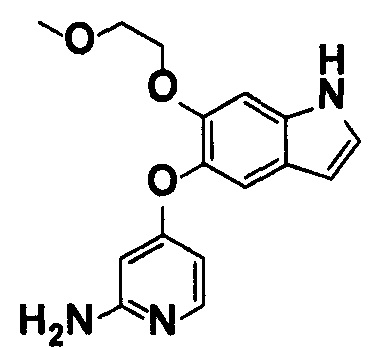
N-(4-((6-(2-метоксиэтокси)-1Н-индол-5-ил)окси)пиридин-2-ил)ацетамид (3,45 г, 10,1 ммоля), описанный в примере получения 20-5, растворяли в метаноле (50 мл), добавляли 2 М раствор натрия гидроксида (50 мл) при комнатной температуре, смесь нагревали и перемешивали при 70°С в течение 3 часов. Воду и этилацетат добавляли в реакционную смесь для разделения. Водный слой три раза экстрагировали этилацетатом и объединенный органический слой сушили над безводным натрия сульфатом. Осушающее средство отфильтровывали, фильтрат концентрировали в вакууме и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 3:7 - 0:1 - этилацетат:метанол = 49:1 - 24:1). Целевую фракцию и смешанную фракцию концентрировали в вакууме отдельно друг от друга, смешанную фракцию снова очищали с помощью колоночной хроматографии на силикагеле (этилацетат:метанол = 1:0 - 9:1), а затем полученное вещество объединяли с вышеупомянутой целевой фракцией с получением названного соединения (2,60 г, 86%).
1Н-ЯМР спектр (500 МГц, CDCl3) δ (ppm): 3,31 (3H, s), 3,58-3,63 (2H, m), 4,08-4,11 (2H, m), 4,28 (2H, brs), 5,90 (1H, d, J=2,4 Гц), 6,29 (1H, dd, J=6,1, 2,2 Гц), 6,44-6,52 (1Н, m), 7,06 (1H, s), 7,15-7,20 (1H, m), 7,34 (1H, s), 7,88 (1H, d, J=5,8 Гц), 8,22 (1H, brs).
[0221] [Пример получения 20-7]
5-((2-Аминопиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 113]

4-((6-(2-Метоксиэтокси)-1Н-индол-5-ил)окси)пиридин-2-амин (2,60 г, 8,67 ммоля), описанный в примере получения 20-6, растворяли в N,N-диметилформамиде (50 мл), затем добавляли 50-72% маслянистый натрия гидрид (499 мг) в атмосфере азота при комнатной температуре. Добавляли фенилметилкарбамат (1,97 г, 13,0 ммоля), описанный в примере получения 1-7, и смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь охлаждали до 0°С и добавляли этилацетат и воду для разделения. Водный слой дважды экстрагировали этилацетатом, добавляли натрия хлорид в водный слой и полученное вещество три раза экстрагировали этилацетатом. Объединенный органический слой сушили над безводным натрия сульфатом. Осушающее средство отфильтровывали, фильтрат концентрировали в вакууме и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:4 - 0:1 - этилацетат:метанол = 49:1 - 24:1). Целевую фракцию концентрировали в вакууме, добавляли этилацетат, осадок собирали фильтрацией и промывали с получением названного соединения (2,23 г, 72%).
1Н-ЯМР спектр (500 МГц, CDCl3) δ (ppm): 3,06 (3Н, d, J=4,9 Гц), 3,29 (3Н, s), 3,59-3,63 (2H, m), 4,14-4,17 (2H, m), 4,30 (2H, brs), 5,52-5,59 (1H, m), 5,89 (1H, d, J=2,4 Гц), 6,27 (1H, dd, J=5,8, 1,9 Гц), 6,55 (1H, d, J=3,9 Гц), 7,27-7,29 (2H, m), 7,89 (1H, d, J=5,9 Гц), 7,99 (1Н, s).
[0222] [Пример 21]
6-(2-Метоксиэтокси)-N-метил-5-((2-(4-(1-метилпиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид
[Химическая формула 114]
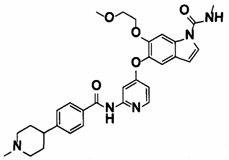
Добавляли 35% водный раствор формальдегида (186 μл, 2,36 ммоля) и натрия триацетоксиборгидрид (200 мг, 0,946 ммоля) в смесь 6-(2-метоксиэтокси)-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида (257 мг, 0,473 ммоля), описанного в примере 20, и тетрагидрофурана (30 мл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 3 минут. Добавляли уксусную кислоту (54 μл, 0,946 ммоля) в реакционную смесь и смесь перемешивали при комнатной температуре в течение 3 часов. Насыщенный водный раствор натрия бикарбоната и этилацетат добавляли в реакционную смесь для разделения. Водный слой три раза экстрагировали этилацетатом, объединенный органический слой сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 1:0 - 23:2). Целевую фракцию концентрировали в вакууме с количественным получением названного соединения.
1Н-ЯМР спектр (500 МГц, CDCl3) δ (ppm): 1,75-1,88 (4Н, m), 2,02-2,11 (2H, m), 2,33 (3H, s), 2,49-2,59 (1H, m), 2,99 (2H, d, J=11,2 Гц), 3,05 (3Н, d, J=4,9 Гц), 3,26 (3Н, s), 3,56-3,60 (2H, m), 4,15-4,21 (2H, m), 5,52-5,58 (1H, m), 6,54 (1H, d, J=3,9 Гц), 6,61 (1H, dd, J=5,8, 2,4 Гц), 7,25-7,27 (1H, m), 7,30-7,34 (3Н, m), 7,79 (2H, d, J=8,3 Гц), 7,91 (1H, d, J=2,4 Гц), 8,01 (1H, s), 8,09 (1H, d, J=5,8 Гц), 8,48 (1H, brs).
[0223] [Пример 22]
5-((2-(4-(1-(2-Гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 115]

Натрия триацетоксиборгидрид (286 мг, 1,35 ммоля) и коммерчески доступный 2-гидроксиацетальдегид (86,1 мг, 1,43 ммоля) добавляли в смесь 6-(2-метоксиэтокси)-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида (250 мг, 0,46 ммоля), описанного в примере 20, и тетрагидрофурана (10 мл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 3 часов и 45 минут. Насыщенный водный раствор натрия бикарбоната и этилацетат добавляли в реакционную смесь для разделения. Водный слой экстрагировали этилацетатом, объединенный органический слой промывали насыщенным солевым раствором, затем сушили над безводным натрия сульфатом, а потом фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 1:0 - 97:3 - 9:1). Целевую фракцию концентрировали в вакууме, затем осадок собирали фильтрацией и промывали жидкой смесью диэтилового эфира и н-гексана с получением названного соединения (236 мг, 87%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,70-1,92 (4Н, m), 2,14-2,25 (2H, m), 2,53-2,64 (3H, m), 3,01-3,08 (5H, m), 3,26 (3H, s), 3,56-3,60 (2H, m), 3,63 (2H, t, J=5,4 Гц), 4,15-4,20 (2H, m), 5,50-5,59 (1H, m), 6,54 (1H, d, J=3,7 Гц), 6,61 (1H, dd, J=5,7, 2,4 Гц), 7,24-7,28 (1Н, m), 7,30-7,35 (3H, m), 7,80 (2H, d, J=8,4 Гц), 7,91 (1H, d, J=2,2 Гц), 8,01 (1H, s), 8,09 (1H, d, J=5,9 Гц), 8,50 (1H, brs).
[0224] [Пример 23]
5-((2-(4-(1-Изопропилпиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 116]
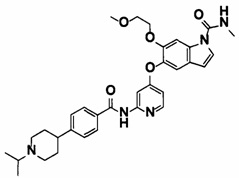
Ацетон (54 μл, 0,736 ммоля), натрия триацетоксиборгидрид (62,4 мг, 0,294 ммоля) и уксусную кислоту (17 μл, 0,294 ммоля) добавляли в раствор 6-(2-метоксиэтокси)-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида, описанного в примере 20 (20 мг, 0,037 ммоля), в тетрагидрофуране (3 мл) при комнатной температуре. Реакционную жидкость перемешивали при той же температуре в течение ночи. Насыщенный водный раствор натрия бикарбоната и этилацетат добавляли в реакционную жидкость при комнатной температуре для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем полученное вещество концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат) с получением названного соединения (6,6 мг, 31%).
1Н-ЯМР спектр (DMSO-d6) δ (ppm): 0,99 (6H, d, J=6,6 Гц), 1,54-1,68 (2Н, m), 1,70-1,81 (2H, m), 2,16-2,26 (2Н, m), 2,44-2,57 (1H, m), 2,66-2,76 (1H, m), 2,82-2,91 (5H, m), 3,12 (3H, s), 3,45-3,52 (2Н, m), 4,05-4,13 (2Н, m), 6,63 (1H, d, J=3,7 Гц), 6,67 (1H, dd, J=5,7, 2,4 Гц), 7,33 (2Н, d, J=8,4 Гц), 7,45 (1H, s), 7,69 (1H, d, J=2,6 Гц), 7,78 (1H, d, J=4,0 Гц), 7,89 (2Н, d, J=8,4 Гц), 8,08 (1H, s), 8,14-8,21 (2Н, m), 10,64 (1H, s).
[0225] [Пример 24]
6-(2-Этоксиэтокси)-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид
[Химическая формула 117]

Трифторуксусную кислоту (1,79 мл, 23,2 ммоля) добавляли в раствор трет-бутил 4-(4-((4-((6-(2-этоксиэтокси)-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)фенил)пиперидин-1-карбоксилата (382 мг, 0,581 ммоля), описанного в примере получения 24-9, в дихлорметане (10 мл) при комнатной температуре. Реакционную жидкость перемешивали при той же температуре в течение 1 часа. Реакционную жидкость концентрировали в вакууме и удаляли трифторуксусную кислоту. Остаток разбавляли дихлорметаном, а затем триэтиламин добавляли для нейтрализации трифторуксусной кислоты. Раствор концентрировали в вакууме, а затем остаток очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 49:1 - 17:3) с получением названного соединения (276 мг, 85%).
1Н-ЯМР спектр (DMSO-d6) δ (ppm): 0,94 (3H, t, J=7,0 Гц), 1,44-1,57 (2Н, m), 1,63-1,72 (2Н, m), 2,47-2,69 (3H, m), 2,85 (3H, d, J=4,4 Гц), 2,97-3,05 (2Н, m), 3,29 (2Н, q, J=7,0 Гц), 3,47-3,58 (2Н, m), 4,04-4,13 (2Н, m), 6,57-6,73 (2Н, m). 7,31 (2Н, d, J=8,1 Гц), 7,45 (1H, s), 7,70 (1H, d, J=2,2 Гц), 7,78 (1H, d, J=3,7 Гц), 7,90 (2Н, d, J=8,8 Гц), 8,08 (1H, s), 8,13-8,24 (2H, m), 10,64 (1H, s).
[0226] Исходный материал трет-бутил 4-(4-((4-((6-(2-этоксиэтокси)-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)фенил)пиперидин-1-карбоксилат синтезировали следующим способом.
[0227] [Пример получения 24-1]
4-(2-Этоксиэтокси)-3-гидроксибензальдегид
[Химическая формула 118]
Коммерчески доступный 2-бромэтилэтиловый эфир (33,8 г, 221 ммоля) добавляли в раствор коммерчески доступного 3,4-дигидроксибензальдегида (30,5 г, 221 ммоля) и натрия карбоната (35,1 г, 331 ммоля) в N,N-диметилформамиде (310 мл) в атмосфере азота при комнатной температуре. Жидкую смесь перемешивали при комнатной температуре в течение 5 дней. Реакционную жидкость охлаждали до 0°С и разбавляли 2 М хлористоводородной кислотой, этилацетатом и водой. Водный слой экстрагировали этилацетатом. Объединенный органический слой последовательно промывали водой и насыщенным солевым раствором, а затем сушили над безводным магния сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме. Нерастворимые вещества отделяли фильтрацией с помощью дихлорметана и неочищенный материал удаляли. Фильтрат очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 17:3 - 1:1) с получением названного соединения (13,2 г, 28%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,27 (3Н, t, J=7,0 Гц), 3,63 (2H, q, J=7,0 Гц), 3,80-3,85 (2H, m), 4,24-4,30 (2H, m), 6,65 (1H, s), 7,02 (1H, d, J=8,4 Гц), 7,38-7,42 (1H, m), 7,45 (1H, d, J=2,2 Гц), 9,85 (1H, s).
[0228] [Пример получения 24-2]
3-(Бензилокси)-4-(2-этоксиэтокси)бензальдегид
[Химическая формула 119]

Калия карбонат (11,3 г, 81,5 ммоля) и бензил хлорид (9,5 мл, 82,6 ммоля) добавляли в раствор 4-(2-этоксиэтокси)-3-гидроксибензальдегида (13,2 г, 62,7 ммоля), описанного в примере получения 24-1, в этаноле (130 мл) в атмосфере азота при комнатной температуре. Жидкую смесь перемешивали при температурном условии 90°С в течение 2 часов. Реакционную жидкость охлаждали до 0°С, а затем разбавляли 2 М хлористоводородной кислотой, этилацетатом и водой. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным магния сульфатом. Осушающее средство отделяли фильтрацией и фильтрат концентрировали в вакууме. Полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 1:1) с получением названного соединения (13,5 г, 72%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,22 (3Н, t, J=7,0 Гц), 3,63 (2Н, q, J=7,0 Гц), 3,82-3,90 (2Н, m), 4,23-4,30 (2Н, m), 5,18 (2Н, s), 7,03 (1H, d, J=7,7 Гц), 7,28-7,34 (1H, m), 7,35-7,42 (2Н, m), 7,43-7,50 (4H, m), 9,82 (1H, s).
[0229] [Пример получения 24-3]
(Е)-2-(Бензилокси)-1-(2-этоксиэтокси)-4-(2-нитровинил)бензол
[Химическая формула 120]

Аммония ацетат (4,16 г, 53,9 ммоля) и нитрометан (6,1 мл, 113 ммоля) добавляли в раствор 3-(бензилокси)-4-(2-этоксиэтокси)бензальдегида (13,5 г, 45,0 ммоля), описанного в примере получения 24-2, в уксусной кислоте (36 мл) в атмосфере азота при комнатной температуре. Жидкую смесь нагревали и перемешивали при нагревании с обратным холодильником при 130°С в течение 2 часов. Обеспечивали отстаивание реакционной жидкости для охлаждения до комнатной температуры. Осадок собирали фильтрацией и промывали этанолом с количественным получением названного соединения.
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,22 (3H, t, J=7,1 Гц), 3,62 (2Н, q, J=7,0 Гц), 3,82-3,88 (2Н, m), 4,21-4,26 (2Н, m), 5,16 (2Н, s), 6,97 (1H, d, J=8,3 Гц), 7,06 (1Н, d, J=2,2 Гц), 7,16 (1H, dd, J=8,3, 2,1 Гц), 7,29-7,50 (6Н, m), 7,91 (1H, d, J=13,6 Гц).
[0230] [Пример получения 24-4]
(Е)-1-(Бензилокси)-2-(2-этоксиэтокси)-4-нитро-5-(2-нитровинил)бензол
[Химическая формула 121]

Азотную кислоту (11 мл, 69%, 171 ммоль) добавляли в раствор (Е)-2-(бензилокси)-1-(2-этоксиэтокси)-4-(2-нитровинил)бензола (15,4 г, 44,9 ммоля), описанного в примере получения 24-3, и уксусной кислоты (100 мл) при 25°С и смесь перемешивали в течение 6 часов. Реакционную жидкость выливали на лед. Суспензию подвергали фильтрованию с отсасыванием и продукт промывали водой с количественным получением названного соединения.
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,23 (3H, t, J=7,0 Гц), 3,62 (2Н, q, J=6,9 Гц), 3,84-3,90 (2Н, m), 4,29-4,34 (2Н, m), 5,27 (2Н, s), 6,93 (1H, s), 7,23 (1H, d, J=13,6 Гц), 7,35-7,50 (5H, m), 7,84 (1H, s), 8,57 (1H, d, J=13,5 Гц).
[0231] [Пример получения 24-5]
6-(2-Этоксиэтокси)-1Н-индол-5-ол
[Химическая формула 122]
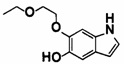
10% Палладий на угле (содержание воды 50%) (6 г) добавляли в раствор (Е)-1-(бензилокси)-2-(2-этоксиэтокси)-4-нитро-5-(2-нитровинил)бензола (17,5 г, 44,9 ммоля), описанного в примере получения 24-4, в метаноле (180 мл) при комнатной температуре. Реакционную жидкость перемешивали в атмосфере водорода при комнатной температуре. Через 6 часов катализатор отфильтровывали через целит. Фильтрат концентрировали в вакууме, а затем остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 2:1 - 1:1) с получением названного соединения (3,28 г, 33%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,29 (3H, t, J=7,0 Гц), 3,63 (2Н, q, J=7,0 Гц), 3,73-3,80 (2Н, m), 4,16-4,24 (2Н, m), 6,41 (1H, td, J=2,1, 1,1 Гц), 6,46 (1Н, s), 6,99 (1H, s), 7,10 (1H, dd, J=3,1, 2,4 Гц), 7,15 (1H, s), 7,93 (1H, brs).
[0232] [Пример получения 24-6]
N-(4-((6-(2-Этоксиэтокси)-1Н-индол-5-ил)окси)пиридин-2-ил)ацетамид
[Химическая формула 123]
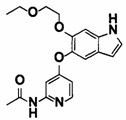
Диметилсульфоксид (20 мл) добавляли в смесь 6-(2-этоксиэтокси)-1Н-индол-5-ола (3,28 г, 14,8 ммоля), описанного в примере получения 24-5, N-(4-хлорпиридин-2-ил)ацетамида (2,78 г, 16,3 ммоля), описанного в примере получения 1-5, и калия трет-бутоксида (1,83 г, 16,3 ммоля) в атмосфере азота при комнатной температуре. Жидкую смесь перемешивали при 150°С в течение ночи. Обеспечивали отстаивание реакционной жидкости с охлаждением до комнатной температуры, а затем разбавляли водой и этилацетатом. Органический слой промывали насыщенным солевым раствором и сушили над безводным натрия сульфатом, а затем осушающее средство отделяли фильтрацией. Фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 1:1 - 0:1 - этилацетат:метанол = 99:1 - 19:1) с получением названного соединения (2,5 г, 48%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,09 (3H, t, J=7,0 Гц), 2,14 (3H, s), 3,41 (2Н, q, J=7,0 Гц), 3,56-3,66 (2Н, m), 4,05-4,13 (2Н, m), 6,44-6,50 (1H, m), 6,53 (1H, dd, J=5,9, 2,6 Гц), 7,05 (1H, d, J=0,7 Гц), 7,16 (1H, dd, J=3,3, 2,6 Гц), 7,36 (1H, s), 7,75 (1H, brs), 8,01 (1H, d, J=5,9 Гц), 8,11 (1H, brs), 8,19 (1H, brs).
[0233] [Пример получения 24-7]
4-((6-(2-Этоксиэтокси)-1Н-индол-5-ил)окси)пиридин-2-амин
[Химическая формула 124]

2 М раствор натрия гидроксида (20 мл) добавляли в раствор N-(4-((6-(2-этоксиэтокси)-1Н-индол-5-ил)окси)пиридин-2-ил)ацетамида (2,5 г, 7,04 ммоля), описанного в примере получения 24-6, в метаноле (20 мл) в атмосфере азота при комнатной температуре, смесь нагревали и перемешивали при нагревании с обратным холодильником при 75°С в течение 2 часов. Обеспечивали отстаивание реакционной жидкости с охлаждением до комнатной температуры, а затем разбавляли этилацетатом и водой. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 1:9 - 0:1 - этилацетат:метанол = 49:1 - 24:1) с получением названного соединения (1,92 г, 87%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,12 (3Н, t, J=7,0 Гц), 3,45 (2Н, q, J=7,2 Гц), 3,60-3,72 (2Н, m), 4,03-4,13 (2Н, m), 4,29 (2Н, s), 5,89 (1H, d, J=1,8 Гц), 6,29 (1Н, dd, J=6,2, 2,2 Гц), 6,44-6,54 (1H, m), 7,05 (1H, s), 7,18 (1H, dd, J=3,1, 2,4 Гц), 7,34 (1H, s), 7,88 (1H, d, J=5,9 Гц), 8,25 (1H, brs).
[0234] [Пример получения 24-8]
5-((2-Аминопиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 125]
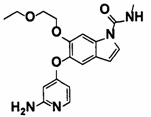
Добавляли 50-72% маслянистый натрия гидрид (265 мг) в раствор 4-((6-(2-этоксиэтокси)-1Н-индол-5-ил)окси)пиридин-2-амина (1,92 г, 6,13 ммоля), описанного в примере получения 24-7, в N,N-диметилформамиде (20 мл) в атмосфере азота при 0°С. Реакционную жидкость перемешивали при той же температуре в течение 10 минут. Фенилметилкарбамат (1,20 г, 7,97 ммоля), описанный в примере получения 1-7, добавляли в реакционную жидкость, а затем смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. Воду и этилацетат добавляли в реакционную жидкость. Органический слой последовательно промывали водой и насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле NH (раствор: н-гептан:этилацетат = 9:1 - 0:1) с получением названного соединения (1,97 г, 87%).
1Н-ЯМР спектр (DMSO-d6) δ (ppm): 1,01 (3Н, t, J=7,0 Гц), 2,84 (3Н, d, J=4,4 Гц), 3,37 (2Н, q, J=7,1 Гц), 3,54-3,59 (2H, m), 4,02-4,10 (2H, m), 5,69 (1H, d, J=2,2 Гц), 5,77 (2H, s), 6,09 (1H, dd, J=5,9, 2,2 Гц), 6,60 (1H, d, J=3,3 Гц), 7,35 (1H, s), 7,71-7,77 (2H, m), 8,04 (1H, s), 8,09-8,17 (1H, m).
[0235] [Пример получения 24-9]
Трет-бутил-4-(4-((4-((6-(2-этоксиэтокси)-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)фенил)пиперидин-1-карбоксилат
[Химическая формула 126]

Тионилхлорид (370 μл, 5,06 ммоля) добавляли в раствор бензотриазола (603 мг, 5,06 ммоля) в дихлорметане (20 мл) в атмосфере азота при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 5 минут, затем добавляли 4-(1-(трет-бутоксикарбонил)пиперидин-4-ил)бензойную кислоту (1,03 г, 3,38 ммоля), описанную в примере получения 1-12, и смесь перемешивали в течение 1 часа. Реакционную жидкость фильтровали через безводный натрия сульфат на стеклянном фильтре и безводный натрия сульфат промывали дихлорметаном. Триэтиламин (1,87 мл, 13,5 ммоля), 4-диметиламинопиридин (16,5 мг, 0,135 ммоля) и раствор 5-((2-аминопиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксамида (500 мг, 1,35 ммоля), описанного в примере получения 24-8, в тетрагидрофуране (5 мл) последовательно добавляли в полученный фильтрат в атмосфере азота при 0°С и смесь перемешивали при комнатной температуре в течение 4 часов. Избыточное количество метиламина добавляли в реакционную жидкость, а затем полученное вещество разбавляли этилацетатом и водой для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом и осушающее средство отделяли фильтрацией. Фильтрат концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 2:3 - 0:1) с получением названного соединения (383 мг, 43%).
1Н-ЯМР спектр (DMSO-d6) δ (ppm): 0,94 (3H, t, J=7,1 Гц), 1,42 (9Н, s), 1,43-1,60 (2H, m), 1,69-1,84 (2H, m), 2,75 (3H, brs), 2,85 (3H, d, J=4,4 Гц), 3,29 (2H, q, J=7,0 Гц), 3,47-3,58 (2H, m), 3,98-4,16 (4H, m), 6,56-6,71 (2H, m), 7,34 (2H, d, J=8,4 Гц), 7,45 (1H, s), 7,70 (1H, d, J=2,6 Гц), 7,78 (1H, d, J=3,7 Гц), 7,90 (2H, d, J=8,4 Гц), 8,08 (1H, s), 8,12-8,24 (2H, m), 10,66 (1H, s).
[0236] [Пример 25]
6-(2-Этоксиэтокси)-5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 127]
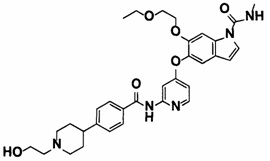
Коммерчески доступный 2-гидроксиацетальдегид (48,5 мг, 0,807 ммоля), натрия триацетоксиборгидрид (91 мг, 0,43 ммоля) и уксусную кислоту (25 μл, 0,43 ммоля) добавляли в суспензию 6-(2-этоксиэтокси)-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида (30 мг, 0,054 ммоля), описанного в примере 24 в тетрагидрофуране (3 мл) при комнатной температуре. Реакционную жидкость перемешивали при комнатной температуре в течение 2 часов. Насыщенный водный раствор натрия бикарбоната добавляли в реакционную жидкость при комнатной температуре и смесь разбавляли этилацетатом. Органический слой промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом и растворитель концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат) с получением названного соединения (20,1 мг, 62%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,07 (3Н, t, J=7,0 Гц), 1,67-1,99 (4Н, m), 2,20 (2Н, td, J=11,7, 2,6 Гц), 2,53-2,66 (3Н, m), 2,98-3,11 (5Н, m), 3,40 (2Н, q, J=7,0 Гц), 3,59-3,69 (4Н, m), 4,16-4,20 (2Н, m), 5,02 (1Н, s), 5,69-5,80 (1H, m), 6,51 (1Н, d, J=3,7 Гц), 6,62 (1Н, dd, J=5,7, 2,4 Гц), 7,25 (1Н, d, J=3,7 Гц), 7,28-7,36 (3Н, m), 7,76-7,83 (2Н, m), 7,91 (1Н, d, J=2,6 Гц), 8,02 (1Н, s), 8,09 (1Н, d, J=5,9 Гц), 8,62 (1Н, s).
[0237] [Пример 26]
6-(2-Этоксиэтокси)-5-((2-(4-(1-этилазетидин-3-ил)бензамид)пиридин-4-ил)окси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 128]

Натрия триацетоксиборгидрид (172 мг, 0,812 ммоля) и ацетальдегид (51,2 мг, 1,16 ммоля) добавляли в смесь 5-((2-(4-(азетидин-3-ил)бензамид)пиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксамида (215 мг, 0,406 ммоля), описанного в примере получения 26-6, и тетрагидрофурана (4,0 мл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 1,5 часа. Насыщенный водный раствор натрия бикарбоната и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, затем сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 2:3 - 0:1 - этилацетат:метанол = 99:1 - 19:1). Целевую фракцию концентрировали в вакууме, затем остаток собирали фильтрацией и промывали диэтиловым эфиром с получением названного соединения (180 мг, 79%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,00 (3H, t, J=7,1 Гц), 1,07 (3Н, t, J=7,0 Гц), 2,51 (2Н, q, J=7,1 Гц), 3,06 (3Н, d, J=4,8 Гц), 3,09-3,16 (2Н, m), 3,40 (2Н, q, J=7,1 Гц), 3,60-3,65 (2Н, m), 3,71-3,80 (3H, m), 4,15-4,20 (2Н, m), 5,47-5,57 (1H, m), 6,54 (1H, d, J=3,7 Гц), 6,61 (1H, dd, J=5,7, 2,4 Гц), 7,24-7,27 (1H, m), 7,34 (1H, s), 7,36-7,40 (2Н, m), 7,77-7,83 (2Н, m), 7,91 (1H, d, J=2,2 Гц), 8,01 (1H, s), 8,10 (1H, d, J=5,9 Гц), 8,47 (1H, brs).
[0238] Исходный материал 5-((2-(4-(азетидин-3-ил)бензамид)пиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксамид синтезировали следующим способом.
[0239] [Пример получения 26-1]
Трет-бутил
3-(4-((4-((6-(2-этоксиэтокси)-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)фенил)азетидин-1-карбоксилат
[Химическая формула 129]

Бензотриазол (335 мг, 2,81 ммоля) растворяли в дихлорметане (20 мл), добавляли тионилхлорид (200 μл, 2,74 ммоля) в атмосфере азота при комнатной температуре и смесь перемешивали в течение 5 минут. 4-(1-(Трет-бутоксикарбонил)азетидин-3-ил)бензойную кислоту (650 мг, 2,34 ммоля), описанную в примере получения 26-5, добавляли в реакционную смесь при комнатной температуре, и смесь перемешивали в течение 25 минут. Реакционную смесь фильтровали через стеклянный фильтр, полностью покрытый безводным натрия сульфатом, а затем безводный натрия сульфат промывали дихлорметаном, а затем фильтрат добавляли в смесь 5-((2-аминопиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксамид а (300 мг, 0,810 ммоля), описанного в примере получения 24-8, триэтиламина (1,3 мл, 9,38 ммоля) и 4-диметиламинопиридина (9,9 мг, 0,081 ммоля) в тетрагидрофуране (16 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 3 часов, затем воду и этилацетат добавляли в реакционную смесь для разделения, органический слой промывали насыщенным солевым раствором, а затем сушили над безводным магния сульфатом. Осушающее средство отфильтровывали, затем фильтрат концентрировали в вакууме, остаток растворяли в тетрагидрофуране, добавляли избыточное количество 9,8 М раствора метиламинметанола при комнатной температуре и смесь перемешивали в течение 75 минут. Реакционную смесь концентрировали в вакууме, остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 1:3 - 0:1 - этилацетат:метанол = 9:1). Фракцию смеси концентрировали в вакууме, остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 2:3 - 1:3 - 0:1 - этилацетат:метанол = 9:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (279 мг, 76%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,07 (3H, t, J=7,0 Гц), 1,47 (9Н, s), 2,81 (3H, d, J=4,8 Гц), 3,40 (2Н, q, J=6,8 Гц), 3,60-3,65 (2Н, m), 3,73-3,83 (1H, m), 3,93-4,01 (2H, m), 4,15-4,20 (2Н, m), 4,35 (2Н, t, J=8,6 Гц), 5,44-5,54 (1H, m), 6,56 (1H, d, J=3,7 Гц), 6,61 (1H, dd, J=5,9, 1,8 Гц), 7,23-7,29 (1H, m), 7,34 (1H, s), 7,42 (2Н, d, J=8,4 Гц), 7,85 (2Н, d, J=8,4 Гц), 7,91 (1H, d, J=2,2 Гц), 8,01 (1H, s), 8,09 (1H, d, J=5,9 Гц), 8,55 (1H, brs).
[0240] [Пример получения 26-2]
Трет-бутил 3-((метилсульфонил)окси)азетидин-1-карбоксилат
[Химическая формула 130]

Метансульфонилхлорид (2,57 мл, 33,3 ммоля) и триэтиламин (11,6 мл, 83,1 ммоля) добавляли в раствор коммерчески доступного N-BOC-3-гидроксиазетидина (4,8 г, 27,7 ммоля) в тетрагидрофуране (100 мл) в атмосфере азота при комнатной температуре. Реакционную жидкость перемешивали при комнатной температуре в течение 2 часов. Насыщенный водный раствор натрия бикарбоната добавляли в реакционную жидкость при комнатной температуре и смесь разбавляли этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 1:1) с количественным получением названного соединения.
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,45 (9H, s), 3,07 (3H, s), 4,03-4,18 (2H, m), 4,22-4,36 (2H, m), 5,12-5,27 (1H, m).
[0241] [Пример получения 26-3]
Трет-бутил 3-йодазетидин-1-карбоксилат
[Химическая формула 131]
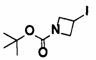
Калия йодид (51,0 г, 307 ммоль) добавляли в раствор трет-бутил-3-((метилсульфонил)окси)азетидин-1-карбоксилата (7,72 г, 30,7 ммоля), описанного в примере получения 26-2, в диметилсульфоксиде (80 мл) в атмосфере азота при комнатной температуре и смесь перемешивали при 140°С в течение 2 часов. Реакционную жидкость разбавляли диэтиловым эфиром и водой. Водный слой экстрагировали диэтиловым эфиром. Объединенный органический слой последовательно промывали водным раствором натрия пиросульфита и насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем полученное вещество концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 1:1) с получением названного соединения (5,91 г, 68%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,44 (9H, s), 4,25-4,33 (2H, m), 4,42-4,51 (1H, m), 4,61-4,69 (2H, m).
[0242] [Пример получения 26-4]
Трет-бутил-3-(4-(этоксикарбонил)фенил)азетидин-1-карбоксилат
[Химическая формула 132]
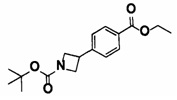
Добавляли 1,2-дибромэтан (0,286 мл, 3,32 ммоля) в суспензию порошка цинка (2,12 г, 32,4 ммоля) в тетрагидрофуране (10 мл) в атмосфере азота при комнатной температуре. Жидкую смесь перемешивали при 65°С в течение 10 минут. Обеспечивали отстаивание реакционной жидкости с охлаждением до комнатной температуры, затем добавляли хлортриметилсилан (0,400 мл, 3,13 ммоля) и смесь перемешивали при комнатной температуре в течение 30 минут. Раствор трет-бутил-3-йодазетидин-1-карбоксилата (5,91 г, 20,9 ммоля), описанного в примере получения 26-3, в тетрагидрофуране (10 мл) добавляли в реакционную жидкость за 5 минут и смесь перемешивали при комнатной температуре в течение 40 минут (раствор А). Раствор трис(дибензилиденацетон)дипалладия(0) (382 мг, 0,418 ммоля) и три-2-фурилфосфина (402 мг, 1,73 ммоля) в тетрагидрофуране (10 мл) перемешивали в атмосфере азота при комнатной температуре в течение 15 минут, а затем добавляли предварительно полученный раствор А при комнатной температуре. Затем раствор этил-4-йодбензоата (6,92 г, 25,1 ммоля) в тетрагидрофуране (18,5 мл) добавляли в атмосфере азота при комнатной температуре. Реакционную жидкость перемешивали при 65°С в течение ночи. Обеспечивали отстаивание реакционной жидкости с охлаждением до комнатной температуры, затем фильтровали через целит и полученное вещество промывали этилацетатом. Фильтрат последовательно промывали насыщенным водным раствором натрия бикарбоната и насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме. Остаток растворяли в дихлорметане, затем полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 4:1) с получением названного соединения (4,35 г, 68%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,40 (3Н, t, J=8,0 Гц), 1,47 (9Н, s), 3,71-3,84 (1H, m), 3,95-4,02 (2H, m), 4,32-4,44 (4H, m), 7,34-7,42 (2H, m), 7,98-8,07 (2H, m).
[0243] [Пример получения 26-5]
4-(1-(Трет-бутоксикарбонил)азетидин-3-ил)бензойная кислота
[Химическая формула 133]

2 М раствор натрия гидроксида (28,5 мл, 57,0 ммоля) добавляли в раствор трет-бутил-3-(4-(этоксикарбонил)фенил)азетидин-1-карбоксилата (4,35 г, 14,2 ммоля), описанного в примере получения 26-4, в тетрагидрофуране (32 мл) и метаноле (7 мл) при 25°С. Реакционную жидкость перемешивали при 60°С в течение 1 часа. 2 М хлористоводородную кислоту (28,5 мл) добавляли в реакционную жидкость и смесь разбавляли этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 1:1) с получением названного соединения (3,4 г, 86%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,48 (9Н, s), 3,71-3,88 (1H, m), 3,96-4,04 (2H, m), 4,37 (2H, t, J=8,6 Гц), 7,42 (2H, d, J=8,4 Гц), 8,09 (2H, d, J=8,3 Гц).
[0244] [Пример получения 26-6]
5-((2-(4-(Азетидин-3-ил)бензамид)пиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 134]

Трет-бутил-3-(4-((4-((6-(2-этоксиэтокси)-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)фенил)азетидин-1-карбоксилат (279 мг, 0,443 ммоля), описанный в примере получения 26-1, растворяли в дихлорметане (8,0 мл), и добавляли трифторуксусную кислоту (1,6 мл) при 0°С. Смесь перемешивали при комнатной температуре в течение 40 минут, а затем концентрировали в вакууме, остаток растворяли в дихлорметане и триэтиламине и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 97:3 - 4:1) с получением названного соединения (215 мг, 92%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,07 (3Н, t, J=7,0 Гц), 3,05 (3Н, d, J=4,8 Гц), 3,40 (2H, q, J=7,0 Гц), 3,61-3,65 (2H, m), 3,81 (2H, t, J=7,0 Гц), 3,93-4,09 (3Н, m), 4,15-4,20 (2H, m), 5,51-5,63 (1H, m), 6,53 (1H, d, J=3,7 Гц), 6,61 (1H, dd, J=5,9, 2,2 Гц), 7,24-7,28 (1H, m), 7,33 (1H, s), 7,40 (2H, d, J=8,1 Гц), 7,82 (2Н, d, J=8,1 Гц), 7,91 (1Н, d, J=2,2 Гц), 8,01 (1H, s), 8,09 (1H, d, J=5,5 Гц), 8,52 (1H, brs).
[0245] [Пример 27]
6-(2-Этоксиэтокси)-5-((2-(4-(1-(2-гидроксиэтил)азетидин-3-ил)бензамид)пиридин-4-ил)окси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 135]
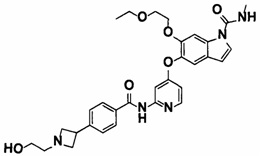
Коммерчески доступный 2-гидроксиацетальдегид (45,9 мг, 0,765 ммоля), натрия триацетоксиборгидрид (86 мг, 0,408 ммоля) и уксусную кислоту (23 μл, 0,408 ммоля) добавляли в раствор 5-((2-(4-(азетидин-3-ил)бензамид)пиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксамида (27 мг, 0,051 ммоля), описанного в примере получения 26-6, в тетрагидрофуране (2 мл) при комнатной температуре и смесь перемешивали при той же температуре в течение 2 часов. Насыщенный водный раствор натрия бикарбоната добавляли в реакционную жидкость при комнатной температуре и смесь разбавляли этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией и фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат) с получением названного соединения (20,0 мг, 68%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,08 (3H, t, J=7,0 Гц), 2,68-2,76 (2H, m), 3,06 (3H, d, J=4,8 Гц), 3,27-3,34 (2H, m), 3,40 (2H, q, J=7,0 Гц), 3,55-3,67 (4Н, m), 3,71-3,94 (3H, m), 4,15-4,22 (2H, m), 5,53-5,65 (1H, m), 6,55 (1H, d, J=3,7 Гц), 6,62 (1H, dd, J=5,9, 2,2 Гц), 7,23-7,29 (1H, m), 7,34 (1H, s), 7,38 (2H, d, J=8,4 Гц), 7,83 (2H, d, J=8,1 Гц), 7,91 (1H, d, J=2,6 Гц), 8,01 (1H, s), 8,09 (1H, d, J=5,9 Гц), 8,63 (1H, brs).
[0246] [Пример 28]
6-(2-Этоксиэтокси)-5-((2-(6-(1-этилпиперидин-4-ил)никотинамид)пиридин-4-ил)окси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 136]

Ацетальдегид (38 μл, 0,671 ммоля), уксусную кислоту (20 μл, 0,358 ммоля) и натрия триацетоксиборгидрид (76 мг, 0,358 ммоля) добавляли в раствор 6-(2-этоксиэтокси)-N-метил-5-((2-(6-(пиперидин-4-ил)никотинамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида (25 мг, 0,045 ммоля), описанного в примере получения 28-5, в тетрагидрофуране (3 мл) при комнатной температуре и смесь перемешивали в течение 1 часа. Насыщенный водный раствор натрия бикарбоната добавляли в реакционную жидкость при комнатной температуре и смесь разбавляли этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат) с получением названного соединения (13,6 мг, 52%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,07 (3Н, t, J=7,0 Гц), 1,13 (3Н, t, J=7,1 Гц), 1,78-1,93 (2Н, m), 1,94-2,15 (4H, m), 2,46 (2H, q, J=7,3 Гц), 2,74-2,86 (1Н, m), 3,00-3,17 (5H, m), 3,40 (2H, q, J=7,1 Гц), 3,63 (2H, t, J=4,8 Гц), 4,18 (2H, t, J=4,8 Гц), 5,45-5,57 (1Н, m), 6,56 (1Н, d, J=3,3 Гц), 6,61 (1Н, dd, J=5,9, 1,5 Гц), 7,20-7,39 (3Н, m), 7,84-7,92 (1Н, m), 8,02 (1Н, s), 8,05-8,15 (2H, m), 8,49 (1Н, s), 8,93-9,07 (1Н, m).
[0247] Исходный материал 6-(2-этоксиэтокси)-N-метил-5-((2-(6-(пиперидин-4-ил)никотинамид)пиридин-4-ил)окси)-1H-индол-1-карбоксамид синтезировали следующим способом.
[0248] [Пример получения 28-1]
1'-Трет-бутил 5-метил 5',6'-дигидро-[2,4'-бипиридин]-1',5(2'Н)-дикарбоксилат
[Химическая формула 137]
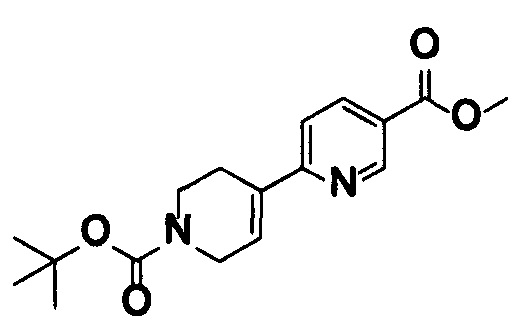
N,N-диметилформамид (100 мл) добавляли в коммерчески доступный 1-N-ВОС-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-3,6-дигидро-2Н-пиридин (4,68 г, 15,1 ммоля), коммерчески доступный метил 6-хлорникотинат (2,81 г, 16,4 ммоля), 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий (II) (1,17 г, 1,60 ммоля) и калия карбонат (7,02 г, 50,8 ммоля). Реакционную жидкость перемешивали в атмосфере азота при 100°С в течение 2 часов. Обеспечивали отстаивание реакционной жидкости с охлаждением до комнатной температуры, а затем разбавляли этилацетатом и водой. Водный слой экстрагировали этилацетатом. Объединенный органический слой последовательно промывали разбавленным водным раствором аммиака и насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 1:1) с получением названного соединения (1,07 г, 22%).
1Н-ЯМР спектр (СDl3) δ (ppm): 1,49 (9H, s), 2,59-2,73 (2H, m), 3,66 (2H, t, J=5,5 Гц), 3,95 (3Н, s), 4,17 (2H, d, J=2,9 Гц), 6,79 (1Н, dt, J=3,4, 1,8 Гц), 7,44 (1Н, d, J=8,4 Гц), 8,25 (1Н, dd, J=8,4, 2,2 Гц), 9,15 (1Н, dd, J=2,2, 0,7 Гц).
[0249] [Пример получения 28-2]
Метил-6-(1-(трет-бутоксикарбонил)пиперидин-4-ил)никотинат
[Химическая формула 138]
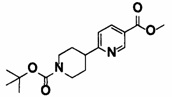
Добавляли 10% палладий на угле (содержание воды 50%) (213 мг) в раствор 1'-трет-бутил-5-метил 5',6'-дигидро-[2,4'-бипиридин]-1'5(2'H)-дикарбоксилата (1,06 г, 3,33 ммоля), описанного в примере получения 28-1, (1,06 г, 3,33 ммоля) в этаноле (71 мл) и тетрагидрофуране (12 мл) и смесь перемешивали в атмосфере водорода при комнатной температуре в течение 2 часов. Смесь фильтровали, затем фильтрат концентрировали в вакууме и остаток очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 9:1 - 1:1) с количественным получением названного соединения.
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,48 (9H, s), 1,73 (2H. qd, J=12,6, 4,4 Гц), 1,88-1,97 (2Н, m), 2,76-2,98 (3H, m), 3,94 (3H, s), 4,27 (2H, brs), 7,22-7,26 (1H, m), 8,23 (1H, dd, J=8,4, 2,2 Гц). 9,09-9,18 (1Н, m).
[0250] [Пример получения 28-3]
6-(1-(Трет-бутоксикарбонил)пиперидин-4-ил)никотиновая кислота
[Химическая формула 139]
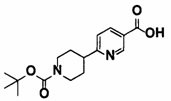
2 M раствор натрия гидроксида (20 мл) добавляли в раствор метил-6-(1-(трет-бутоксикарбонил)пиперидин-4-ил)никотината (1,07 г, 3,32 ммоля), описанного в примере получения 28-2, в этаноле (5 мл) и смесь перемешивали в течение 1 часа. 2 М хлористоводородную кислоту добавляли в реакционную жидкость при 0°С. Водный слой экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме с получением названного соединения (534 мг, 52%).
1Н-ЯМР спектр (DMSO-d6) δ (ppm): 1,42 (9H, s), 1,58 (2H, qd, J=12,6, 4,4 Гц), 1,77-1,89 (2H, m), 2,67-3,04 (3H, m), 3,95-4,17 (2H, m), 7,40 (1H, d, J=7,7 Гц), 8,16 (1H, dd, J=8,1, 2,2 Гц), 8,97 (1H, dd, J=2,2, 0,7 Гц).
[0251] [Пример получения 28-4]
Трет-бутил-4-(5-((4-((6-(2-этоксиэтокси)-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)пиридин-2-ил)пиперидин-1-карбоксилат
[Химическая формула 140]

Оксалилхлорид (74 μл, 0,864 ммоля) и одну каплю N,N-диметилформамида добавляли в раствор 6-(1-(трет-бутоксикарбонил)пиперидин-4-ил)никотиновой кислоты (80 мг, 0,216 ммоля), описанной в примере получения 28-3, в дихлорметане (2 мл) при 0°С и смесь перемешивали в течение 30 минут. Реакционную жидкость концентрировали в вакууме. Остаток растворяли в тетрагидрофуране (2 мл), затем добавляли триэтиламин (301 μл, 2,16 ммоля) и 5-((2-аминопиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксалата (80 мг, 0,216 ммоля), описанный в примере получения 24-8, и смесь перемешивали в течение 5 часов. Избыточное количество метиламина добавляли в реакционную жидкость, а затем смесь разбавляли водой и этилацетатом для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1) с получением названного соединения (80 мг, 56%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,04 (3H, t, J=7,0 Гц), 1,45 (9Н, s), 1,68 (2H, qd, J=12,6, 4,4 Гц), 1,83-1,93 (2H, m), 2,69-2,92 (3H, m), 2,96 (3H, d, J=4,8 Гц), 3,37 (2H, q, J=7,0 Гц), 3,56-3,63 (2H, m), 4,11-4,15 (2H, m), 4,21 (2H, brs), 6,10-6,20 (1H, m), 6,44 (1H, d, J=3,7 Гц), 6,58 (1H, dd, J=5,7, 2,4 Гц), 7,21 (1H, d, J=7,7 Гц), 7,24-7,31 (2H, m), 7,86 (1H, d, J=2,2 Гц), 7,95 (1H, d, J=5,9 Гц), 8,02 (1H, s), 8,07 (1H, dd, J=8,2, 2,4 Гц), 8,96-9,02 (1H, m), 9,20 (1H, brs).
[0252] [Пример получения 28-5]
6-(2-Этоксиэтокси)-N-метил-5-((2-(6-(пиперидин-4-ил)никотинамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид
[Химическая формула 141]
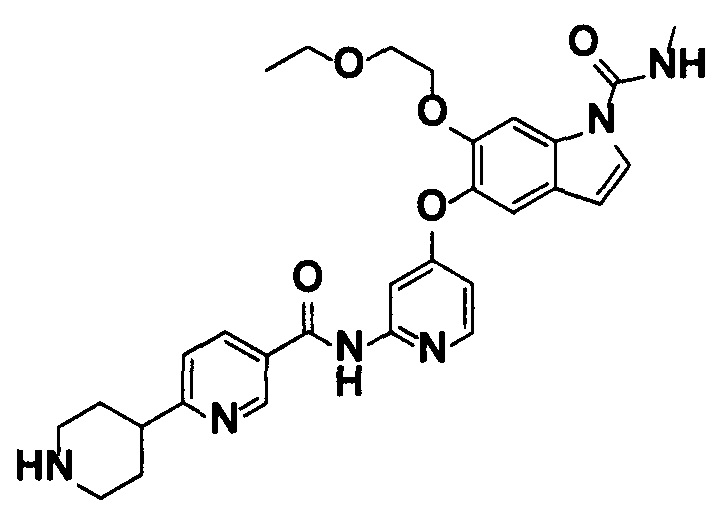
Трифторуксусную кислоту (374 μл, 4,86 ммоля) добавляли в раствор трет-бутил-4-(5-((4-((6-(2-этоксиэтокси)-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)пиридин-2-ил)пиперидин-1-карбоксилата (80 мг, 0,121 ммоля), описанного в примере получения 28-4, в дихлорметане (3 мл) при комнатной температуре и смесь перемешивали в течение 1,5 часа. Реакционную жидкость концентрировали в вакууме. Остаток растворяли в дихлорметане, а затем триэтиламин добавляли для нейтрализации трифторуксусной кислоты. Раствор концентрировали в вакууме, а затем остаток очищали с помощью колоночной хромагографии на силикагеле NH (этилацетат:метанол = 49:1 - 17:3) с получением названного соединения (51,3 мг, 76%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,07 (3H, t, J=7,1 Гц), 1,72 (2Н, qd, J=12,4, 4,0 Гц), 1,87-1,97 (2Н, m), 2,76 (2Н, td, J=12,3, 2,6 Гц), 2,84-2,95 (1Н, m), 2,99 (3H, d, J=4,4 Гц), 3,16-3,28 (2Н, m), 3,40 (2Н, q, J=7,1 Гц), 3,59-3,68 (2Н, m), 4,14-4,20 (2Н, m), 6,04-6,17 (1Н, m), 6,48 (1Н, d, J=3,7 Гц), 6,62 (1Н, dd, J=5,7, 2,4 Гц), 7,23-7,29 (2Н, m), 7,31 (1Н, s), 7,88 (1Н, d, J=2,2 Гц), 7,98-8,05 (2Н, m), 8,10 (1Н, dd, J=8,2, 2,4 Гц), 9,02 (1Н, dd, J=2,6, 0,7 Гц).
[0253] [Пример 29]
(S)-6-(2-Этоксиэтокси)-5-((2-(4-((2-гидроксиметил)пирролидин-1-ил)метил)бензамид)пиридин-4-ил)окси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 142]

Коммерчески доступный L-пролинол (31,3 мг, 0,309 ммоля) добавляли в смесь 5-((2-(4-(хлорметил)-N-(4-(хлорметил)бензоил)бензамид)пиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксамида (19,6 мг, 0,029 ммоля), описанного в примере получения 29-1, и N,N-диметилформамида (500 μл) при комнатной температуре и смесь перемешивали в атмосфере азота в течение 17,5 часа. Воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали, полученный остаток очищали с помощью TLC на силикагеле NH (этилацетат), продукт собирали фильтрацией и промывали диэтиловым эфиром с получением названного соединения (13,3 мг, 78%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,08 (3H, t, J=7,1 Гц), 1,65-1,99 (5Н, m), 2,23-2,32 (1H, m), 2,71-2,79 (1H, m), 2,92-2,99 (1H, m), 3,06 (3H, d, J=4,8 Гц), 3,37-3,48 (4H, m), 3,61-3,69 (3H, m), 4,03 (1H, d, J=13,5 Гц), 4,16-4,20 (2Н, m), 5,48-5,56 (1H, m), 6,55 (1H, d, J=3,7 Гц), 6,61 (1H, dd, J=5,9, 2,6 Гц), 7,24-7,28 (1H, m), 7,34 (1H, s), 7,41 (2Н, d, J=8,4 Гц), 7,79-7,84 (2Н, m), 7,91 (1H, d, J=2,2 Гц), 8,01 (1H, s), 8,10 (1H, d, J=5,5 Гц), 8,50 (1H, brs).
[0254] Исходный материал 5-((2-(4-(хлорметил)-N-(4-(хлорметил)бензоил)бензамид)пиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксамид синтезировали следующим способом.
[0255] [Пример получения 29-1]
5-((2-(4-(Хлорметил)-N-(4-(хлорметил)бензоил)бензамид)пиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 143]

Триэтиламин (300 μл, 2,16 ммоля) и коммерчески доступный 4-(хлорметил)бензоилхлорид (221 мг, 1,17 ммоля) добавляли в смесь 5-((2-аминопиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксамида (107 мг, 0,289 ммоля), описанного в примере получения 24-8, и тетрагидрофурана (8,0 мл) в атмосфере азота при 0°С. Смесь перемешивали при комнатной температуре в течение 1 часа, а затем воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, затем сушили над безводным натрия сульфатом, а потом фильтровали с помощью силикагеля NH. Фильтрат концентрировали в вакууме с количественным получением названного соединения.
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,11 (3H, t, J=7,0 Гц), 3,06 (3Н, d, J=4,8 Гц), 3,43 (2Н, q, J=7,0 Гц), 3,56-3,60 (2Н, m), 4,08-4,12 (2Н, m), 4,56 (4H, s), 5,41-5,49 (1H, m), 6,55 (1H, d, J=3,7 Гц), 6,67 (1H, d, J=2,2 Гц), 6,71 (1H, dd, J=5,9, 2,2 Гц), 7,24 (1H, s), 7,25-7,28 (1H, m), 7,34-7,39 (4H, m), 7,69-7,75 (4H, m), 7,98 (1H, s), 8,17 (1H, d, J=5,9 Гц).
[0256] [Пример 30]
5-((2-(4-(((3S,5R)-3,5-Диметилпиперазин-1-ил)метил)бензамид)пиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 144]

Коммерчески доступный цис-2,6-диметилпиперазин (32,5 мг, 0,285 ммоля) добавляли в смесь 5-((2-(4-(хлорметил)-N-(4-(хлорметил)бензоил)бензамид)пиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксамида (21,1 мг, 0,031 ммоля), описанного в примере получения 29-1, и N,N-диметилформамида (500 μл) при комнатной температуре и смесь перемешивали в атмосфере азота в течение 13 часов и 20 минут. Воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью TLC на силикагеле NH (этилацетат), затем продукт собирали фильтрацией и промывали диэтиловым эфиром с получением названного соединения (14,4 мг, 77%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,02 (6H, d, J=6,2 Гц), 1,07 (3Н, t, J=7,0 Гц), 1,63 (2Н, t, J=10,6 Гц), 2,69-2,76 (2Н, m), 2,89-2,99 (2Н, m), 3,07 (3Н, d, J=4,8 Гц), 3,40 (2Н, q, J=7,0 Гц), 3,52 (2Н, s), 3,61-3,65 (2Н, m), 4,16-4,20 (2Н, m), 5,45-5,52 (1H, m), 6,55 (1H, d, J=3,7 Гц), 6,60 (1H, dd, J=5,9,2, 2 Гц), 7,25-7,27 (1H, m), 7,34 (1H, s), 7,43 (2Н, d, J=8,1 Гц), 7,79-7,83 (2Н, m), 7,92 (1H, d, J=2,2 Гц), 8,01 (1H, s), 8,10 (1H, d, J=5,9 Гц), 8,47 (1H, brs).
[0257] [Пример 31]
(R)-6-(2-Этоксиэтокси)-5-((2-(5-((3-гидроксипирролидин-1-ил)метил)тиофен-2-карбоксамид)пиридин-4-ил)окси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 145]

Тионилхлорид (2,8 мл, 38,4 ммоля) и N,N-диметилформамид (5,87 μл, 0,076 ммоля) добавляли в 5-(гидроксиметил)тиофен-2-карбоновую кислоту (120 мг, 0,759 ммоля), описанную в примере получения 31-1, смесь нагревали и перемешивали при 90°С в течение 2 часов. Реакционную смесь выпаривали в вакууме с получением неочищенного продукта А (148 мг).
Триэтиламин (191 μл, 1,3 8 ммоля) и раствор тетрагидрофурана (1,0 мл) части неочищенного продукта А (74,0 мг, 0,379 ммоля) добавляли в смесь 5-((2-аминопиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксамида (51,1 мг, 0,138 ммоля), описанного в примере получения 24-8, и тетрагидрофурана (1,4 мл) в атмосфере азота при 0°С. Смесь перемешивали при комнатной температуре в течение 170 минут, а затем воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, затем сушили над безводным натрия сульфатом и фильтровали с помощью силикагеля NH, а затем полученное вещество концентрировали в вакууме с получением неочищенного продукта В (86,7 мг).
Часть неочищенного продукта В (17,3 мг) растворяли в N,N-диметилформамиде (1,0 мл), добавляли коммерчески доступный (R)-3-гидроксипирролидин (24,4 мг, 0,28 ммоля) в атмосфере азота при комнатной температуре и смесь перемешивали в течение 17 часов. Воду и этилацетат добавляли в реакционную смесь для разделения и органический слой промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью TLC на силикагеле NH (этилацетат), затем продукт собирали фильтрацией и промывали н-гексаном с получением названного соединения (9,0 мг, 56%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,07 (3H, t, J=6,8 Гц), 1,71-1,81 (1Н, m), 2,13-2,24 (1H, m), 2,34-2,43 (1H, m), 2,57-2,64 (1H, m), 2,69-2,76 (1H, m), 2,87-2,97 (1H, m), 3,06 (3H, d, J=4,8 Гц), 3,39 (2H, q, J=7,3 Гц), 3,59-3,64 (2Н, m), 3,84 (2H, s), 4,14-4,19 (2H, m), 4,31-4,37 (1H, m), 5,48-5,55 (1H, m), 6,54 (1H, d, J=3,7 Гц), 6,61 (1H, dd, J=5,9, 2,4 Гц), 6,91 (1H, d, J=3,5 Гц), 7,24-7,28 (1H, m), 7,32 (1H, s), 7,47 (1H, d, J=3,9 Гц), 7,78-7,82 (1H, m), 7,99 (1H, s), 8,08 (1H, d, J=5,7 Гц), 8,34 (1H, brs).
[0258] [Пример получения 31-1]
5-(Гидроксиметил)тиофен-2-карбоновая кислота
[Химическая формула 146]
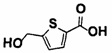
Натрия боргидрид (218 мг, 5,76 ммоля) добавляли в раствор коммерчески доступной 5-формил-2-тиофенкарбоновой кислоты (599 мг, 3,84 ммоля) в метаноле (19 мл) и смесь перемешивали в атмосфере азота при комнатной температуре в течение 4 часов и 30 минут. Ацетон добавляли в реакционную смесь и смесь концентрировали в вакууме. 2 М хлористоводородную кислоту и этилацетат добавляли к остатку для разделения и водный слой экстрагировали этилацетатом. Объединенный органический слой промывали водой, сушили над безводным натрия сульфатом и фильтровали. Фильтрат концентрировали в вакууме и осадок промывали диэтиловым эфиром и н-гексаном с получением названного соединения (529 мг, 87%).
1Н-ЯМР спектр (DMSO-d6) δ (ppm): 4,61-4,74 (2H, m), 5,65-72 (1H, m), 6,97-7,7,03 (1H, m), 7,55-7,62 (1H, m), 12,92 (1H, brs).
[0259] [Пример 32]
6-(3-Метоксипропокси)-N-метил-5-((2-(4-(1-метилазетидин-3-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид
[Химическая формула 147]

Формальдегид (12 μл, 0,425 ммоля), уксусную кислоту (13 μл, 0,227 ммоля) и натрия триацетоксиборгидрид (48,0 мг, 0,227 ммоля) добавляли в раствор 5-((2-(4-(азетидин-3-ил)бензамид)пиридин-N-ил)окси)-6-(3-метоксипропокси)-N-метил-1Н-индол-1-карбоксамида (15 мг, 0,028 ммоля), описанного в примере получения 32-10, в тетрагидрофуране (2 мл) при комнатной температуре. Реакционную жидкость перемешивали при комнатной температуре в течение 1 часа. Насыщенный водный раствор натрия бикарбоната и этилацетат добавляли в реакционную жидкость при комнатной температуре. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией и фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат) с получением названного соединения (8,8 мг, 57%).
1Н-ЯМР спектр (DMSO-d6) δ (ppm): 1,76 (2H, t, J=6,4 Гц), 2,25 (3Н, s), 2,85 (3H, d, J=4,4 Гц), 3,02-3,13 (7Н, m), 3,54-3,65 (3H, m), 3,99 (2H, t, J=6,2 Гц), 6,64 (1Н, d, J=3,3 Гц), 6,69 (1Н, dd, J=5,9, 2,6 Гц), 7,42 (2H, d, J=8,4 Гц), 7,46 (1Н, s), 7,69 (1Н, d, J=2,6 Гц), 7,77 (1Н, d, J=3,7 Гц), 7,92 (2H, d, J=8,4 Гц), 8,06 (1Н, s), 8,14-8,19 (1Н, m), 8,21 (1Н, d, J=5,9 Гц), 10,70 (1H, s).
[0260] Исходный материал 5-((2-(4-(азетидин-3-ил)бензамид)пиридин-4-ил)окси)-6-(3-метоксипропокси)-N-метил-1Н-индол-1-карбоксамид синтезировали следующим способом.
[0261] [Пример получения 32-1]
3-Гидрокси-4-(3-метоксипропокси)бензальдегид
[Химическая формула 148]
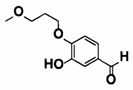
Коммерчески доступный 1-бром-3-метоксипропан (24,0 г, 157 ммоль) добавляли в раствор коммерчески доступного 3,4-дигидроксибензальдегида (21,7 г, 157 ммоль) и натрия карбоната (25,0 г, 236 ммоль) в N,N-диметилформамиде (50 мл) в атмосфере азота при комнатной температуре. Реакционную жидкость перемешивали при комнатной температуре в течение 3 дней и 4 часов. 2 М хлористоводородную кислоту, этилацетат и воду добавляли в реакционную жидкость при 0°С. Водный слой экстрагировали этилацетатом. Объединенный органический слой последовательно промывали водой и насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией и фильтрат концентрировали в вакууме. Нерастворимые вещества отделяли фильтрацией с дихлорметаном и фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 17:3 - 1:1) с получением названного соединения (17,2 г, 52%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 2,07-2,18 (2Н, m), 3,38 (3H, s), 3,59 (2H, t, J=5,9 Гц), 4,26 (2H, t, J=6,2 Гц), 6,21 (1Н, s), 7,00 (1H, d, J=8,1 Гц), 7,35-7,48 (2H, m), 9,85 (1H, s).
[0262] [Пример получения 32-2]
3-(Бензилокси)-4-(3-метоксипропокси)бензальдегид
[Химическая формула 149]

Калия карбонат (14,7 г, 106 ммоль) и бензилхлорид (12,2 мл, 106 ммоль) добавляли в раствор 3-гидрокси-4-(3-метоксипропокси)бензальдегида (17,2 г, 81,7 ммоля), описанного в примере получения 32-1, в этаноле (200 мл) в атмосфере азота при комнатной температуре и смесь перемешивали при температурном условии 90°С в течение 2 часов. Реакционную жидкость охлаждали до 0°С и смесь разбавляли 2 М хлористоводородной кислотой, этилацетатом и водой. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным магния сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 4:1 - 3:7) с получением названного соединения (19,8 г, 81%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 2,10-2,20 (2H, m), 3,35 (3H, d, J=0,7 Гц), 3,59 (2Н, t, J=6,0 Гц), 4,21 (2H, t, J=6,4 Гц), 5,18 (2H, s), 6,95-7,09 (1H, m), 7,28-7,50 (7H, m), 9,76-9,87 (1Н, m).
[0263] [Пример получения 32-3]
(Е)-2-(Бензилокси)-1-(3-метоксипропокси)-4-(2-нитровинил)бензол
[Химическая формула 150]
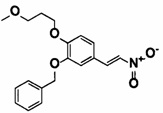
Аммония ацетат (6,10 г, 79,1 ммоля) и нитрометан (8,93 мл, 165 ммоль) добавляли в раствор 3-(бензилокси)-4-(3-метоксипропокси)бензальдегида (19,8 г, 65,9 ммоля), описанного в примере получения 32-2, в уксусной кислоте (52,8 мл) в атмосфере азота при комнатной температуре. Жидкую смесь перемешивали при температурном условии 130°С в течение 2 часов. Обеспечивали отстаивание реакционной жидкости с охлаждением до комнатной температуры, затем осадок собирали фильтрацией и промывали этанолом с количественным получением названного соединения.
1Н-ЯМР спектр (СDCl3) δ (ppm): 2,07-2,17 (2H, m), 3,35 (3H, s), 3,59 (2H, t, J=6,0 Гц), 4,19 (2H, t, J=6,4 Гц), 5,16 (2H, s), 6,96 (1H, d, J=8,4 Гц), 7,05 (1H, d, J=1,8 Гц), 7,16 (1H, dd, J=8,4, 1,8 Гц), 7,29-7,47 (6Н, m), 7,91 (1H, d, J=13,5 Гц).
[0264] [Пример получения 32-4]
(Е)-1-(Бензилокси)-2-(3-метоксипропокси)-4-нитро-5-(2-нитровинил)бензол
[Химическая формула 151]

69% азотную кислоту (16 мл, 249 ммоля) добавляли в жидкую смесь (Е)-2-(бензилокси)-1-(3-метоксипропокси)-4-(2-нитровинил)бензола (22,6 г, 65,9 ммоля), описанного в примере получения 32-3, в уксусной кислоте (150 мл) при комнатной температуре. Жидкую смесь перемешивали при комнатной температуре в течение 6 часов. Реакционную жидкость переносили в ледяную баню, затем осадок собирали фильтрацией и промывали водой с количественным получением названного соединения.
1Н-ЯМР спектр (CDCl3) δ (ppm): 2,10-2,13 (2H, m), 3,36 (3H, s), 3,59 (2H, t, J=5,9 Гц), 4,25 (2H, t, J=6,4 Гц), 5,27 (2H, s), 6,93 (1H, s), 7,24 (1H, d, J=13,6 Гц), 7,34-7,46 (5H, m), 7,78 (1H, s), 8,52-8,62 (1H, m).
[0265] [Пример получения 32-5]
6-(3-Метоксипропокси)-1Н-индол-5-ол
[Химическая формула 152]
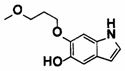
10% палладий на угле (содержание воды 50%) (8 г) добавляли в раствор (Е)-1-(бензилокси)-2-(3-метоксипропокси)-4-нитро-5-(2-нитровинил)бензола (19,6 г, 50,5 ммоля), описанного в примере получения 32-4, в метаноле (300 мл) при комнатной температуре. Жидкую смесь перемешивали в атмосфере водорода при комнатной температуре в течение 6 часов. Реакционную жидкость фильтровали через целит, а затем фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хромагографии на силикагеле (н-гептан:этилацетат = 2:1 - 1:1) с получением названного соединения (3,81 г, 34%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 2,05-2,13 (2H, m), 3,40 (3H, s), 3,61 (2H, t, J=6,0 Гц), 4,16 (2H, t, J=6,0 Гц), 5,97-6,05 (1H, m), 6,36-6,46 (1H, m), 6,92 (1H, s), 7,08 (1H, t, J=2,8 Гц), 7,14 (1H, s), 7,26 (1H, s), 7,82-8,06 (1H, m).
[0266] [Пример получения 32-6]
N-(4-((6-(3-метоксипропокси)-1Н-индол-5-ил)окси)пиридин-2-ил)ацетамид
[Химическая формула 153]
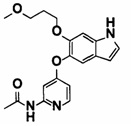
Диметилсульфоксид (25 мл) добавляли в смесь 6-(3-метоксипропокси)-1Н-ивдол-5-ола (3,81 г, 17,2 ммоля), описанного в примере получения 32-5, N-(4-хлорпиридин-2-ил)ацетамида (3,23 г, 18,9 ммоля), описанного в примере получения 1-5, и калия трет-бутоксида (2,12 г, 18,9 ммоля) в атмосфере азота при комнатной температуре. Жидкую смесь перемешивали при 150°С в течение 14 часов. Реакционную жидкость охлаждали до комнатной температуры, а затем добавляли этилацетат и воду. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 1:1 - 0:1 - этилацетат:метанол = 49:1) с получением названного соединения (2,83 г, 46%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,85-1,91 (2H, m), 2,13 (3H, s), 3,20 (3H, s), 3,24 (2H, t, J=6,2 Гц), 4,02 (2H, t, J=6,2 Гц), 6,44-6,49 (1H, m), 6,51-6,56 (1H, m), 6,95-7,02 (1H, m), 7,10-7,18 (1H, m), 7,31-7,38 (1H, m), 7,71-7,82 (1H, m), 7,97-8,06 (1H, m), 8,20-8,30 (1H, m), 8,36-8,52 (1H, m).
[0267] [Пример получения 32-7]
4-((6-(3-Метоксипропокси)-1Н-индол-5-ил)окси)пиридин-2-амин
[Химическая формула 154]
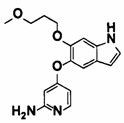
2 M раствор натрия гидроксида (30 мл) добавляли в раствор N-(4-((6-(3-метоксипропокси)-1Н-индол-5-ил)окси)пиридин-2-ил)ацетамида (2,8 г, 7,88 ммоля), описанного в примере получения 32-6, в метаноле (30 мл) в атмосфере азота при комнатной температуре. Жидкую смесь перемешивали при нагревании с обратным холодильником при температурном условии 75°С в течение 2 часов. Обеспечивали отстаивание реакционной жидкости с охлаждением до комнатной температуры, а затем добавляли этилацетат и воду. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 1:9 - 0:1 - этилацетат:метанол = 49:1) с получением названного соединения (2,24 г, 91%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,75-1,92 (2Н, m), 3,23 (3H, s), 3,28 (2H, t, J=6,0 Гц), 4,03 (2Н, t, J=6,0 Гц), 4,30 (2Н, s), 5,89 (1H, d, J=2,2 Гц), 6,29 (1Н, dd, J=5,9, 2,2 Гц), 6,46-6,53 (1H, m), 7,01 (1H, s), 7,16 (1H, dd, J=3,3, 2,6 Гц), 7,35 (1H, s), 7,88 (1H, d, J=5,9 Гц), 8,14-8,26 (1H, m).
[0268] [Пример получения 32-8]
5-((2-Аминопиридин-4-ил)окси)-6-(3-метоксипропокси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 155]

Добавляли 50-72% маслянистый натрия гидрид (308 мг) в раствор 4-((6-(3-метоксипропокси)-1Н-индол-5-ил)окси)пиридин-2-амина (2,23 г, 7,12 ммоля), описанного в примере получения 32-7, в N,N-диметилформамиде (30 мл) в атмосфере азота при 0°С. Реакционную жидкость перемешивали в течение 10 минут, затем добавляли фенилметилкарбамат (1,40 г, 9,25 ммоля), описанный в примере получения 1-7, и полученное вещество далее перемешивали при той же температуре в течение 30 минут. Воду и этилацетат добавляли в реакционную жидкость. Органический слой последовательно промывали водой и насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хромагографии на силикагеле NH (н-гептан:этилацетат = 9:1 - 0:1) с получением названного соединения (2,44 г, 93%).
1Н-ЯМР спектр (DMSO-d6) δ (ppm): 1,73-1,86 (2Н, m), 2,81-2,87 (3H, m), 3,12 (3H, d, J=1,8 Гц), 3,16-3,24 (2Н, m), 3,98 (2Н, t, J=5,7 Гц), 5,62-5,71 (1Н, m), 5,78 (2Н, s), 6,06-6,15 (1Н, m), 6,61 (1Н, dd, J=3,5, 1,7 Гц), 7,36 (1Н, d, J=1,8 Гц), 7,71-7,78 (2Н, m), 7,98-8,04 (1Н, m), 8,10-8,22 (1Н, m).
[0269] [Пример получения 32-9]
Трет-бутил 3-(4-((4-((6-(3-метоксипропокси)-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)фенил)азетидин-1 карбоксилат
[Химическая формула 156]

Тионилхлорид (118 μл, 1,62 ммоля) добавляли в раствор бензотриазола (193 мг, 1,62 ммоля) в дихлорметане (5 мл) в атмосфере азота при комнатной температуре и смесь перемешивали в течение 5 минут. Добавляли 4-(1-(трет-бутоксикарбонил)азетидин-3-ил)бензойную кислоту (300 мг, 1,08 ммоля), описанную в примере получения 26-5, в реакционную жидкость и смесь дополнительно перемешивали в течение 1 часа. Реакционную жидкость фильтровали через безводный натрия сульфат на стеклянном фильтре и безводный натрия сульфат промывали дихлорметаном. Раствор триэтиламина (748 μл, 5,40 ммоля), 4-диметиламинопиридина (6,60 мг, 0,054 ммоля), и 5-((2-аминопиридин-4-ил)окси)-6-(3-метоксипропокси)-N-метил-1Н-индол-1-карбоксамида (200 мг, 0,54 ммоля), описанного в примере получения 32-8, в тетрагидрофуране (10 мл) добавляли в полученный раствор дихлорметана в атмосфере азота при 0°С и смесь перемешивали в течение 4 часов и 20 минут. Реакционную жидкость разбавляли этилацетатом и водой. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отделяли фильтрацией, а затем фильтрат концентрировали в вакууме. Тетрагидрофуран и избыточное количество раствора метиламинтетрагидрофурана добавляли к остатку и смесь перемешивали при комнатной температуре. Жидкую смесь концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 3 - 0:1) с получением названного соединения (146 мг, 43%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,43-1,51 (9H, m), 1,80-1,95 (2H, m), 2,95 (3H, d, J=4,8 Гц), 3,17 (3H, s), 3,20-3,27 (2H, m), 3,69-3,82 (1H, m), 3,95 (2H, dd, J=8,6, 6,0 Гц), 4,05-4,17 (2H, m), 4,33 (2H, t, J=8,6 Гц), 6,13-6,25 (1H, m), 6,44 (1H, d, J=3,7 Гц), 6,63 (1H, dd, J=5,7, 2,4 Гц), 6,97 (1H, s), 7,24 (2H, d, J=3,7 Гц), 7,38 (2H, d, J=8,4 Гц), 7,83 (2H, d, J=8,4 Гц), 7,88 (1H, d, J=2,6 Гц), 8,01-8,09 (2H, m), 8,79-9,00 (1H, m).
[0270] [Пример получения 32-10]
5-((2-(4-(Азетидин-3-ил)бензамид)пиридин-4-ил)окси)-6-(3-метоксипропокси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 157]

Трифторуксусную кислоту (713 μл, 9,25 ммоля) добавляли в раствор трет-бутил-3-(4-((4-((6-(3-метоксипропокси)-1-(метилкарбамоил)-1Н-индол-5-ил)окси)пиридин-2-ил)карбамоил)фенил)азетидин-1-карбоксилата (146 мг, 0,231 ммоля), описанного в примере получения 32-9, в дихлорметане (5 мл) при комнатной температуре. Реакционную жидкость перемешивали в течение 1,5 часа, а затем полученное вещество концентрировали в вакууме. Остаток растворяли в дихлорметане и триэтиламине для нейтрализации трифторуксусной кислоты, а затем полученное вещество концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 49:1 - 17:3) с получением названного соединения (96,2 мг, 79%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,82-1,95 (2H, m), 3,06 (3H, d, J=4,8 Гц), 3,19 (3H, s), 3,24 (2H, t, J=6,4 Гц), 3,79-4,00 (5Н, m), 4,11 (2H, t, J=6,0 Гц), 5,56-5,64 (1H, m), 6,55 (1H, d, J=3,7 Гц), 6,61 (1H, dd, J=5,9, 2,2 Гц), 7,21-7,29 (1H, m), 7,34 (1H, s), 7,41 (2H, d, J=8,4 Гц), 7,84 (2Н, d, J=8,4 Гц), 7,92 (1Н, d, J=2,2 Гц), 8,00 (1Н, s), 8,10 (1H, d, J=5,9 Гц), 8,56-8,68 (1H, m).
[0271] [Пример 33]
5-((2-(4-(1-Этилпиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(3-фторпропокси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 158]

Бензотриазол (37,8 мг, 0,317 ммоля) растворяли в дихлорметане (2,0 мл), добавляли тионилхлорид (24 μл, 0,322 ммоля) в атмосфере азота при комнатной температуре и смесь перемешивали в течение 5 минут. Добавляли 4-(1-(трет-бутоксикарбонил)пиперидин-4-ил)бензойную кислоту (82 мг, 0,269 ммоля), описанную в примере получения 1-12, в реакционную смесь при комнатной температуре и смесь перемешивали в течение 20 минут. Реакционную смесь фильтровали через стеклянный фильтр, полностью покрытый безводным натрия сульфатом, а затем безводный натрия сульфат промывали дихлорметаном, фильтрат добавляли в смесь 5-((2-аминопиридин-4-ил)окси)-6-(3-фторпропокси)-N-метил-1Н-индол-1-карбоксамида (34,4 мг, 0,096 ммоля), описанного в примере получения 33-6, триэтиламина (133 μл, 0,960 ммоля) и 4-диметиламинопиридина (1,17 мг, 0,0096 ммоля) в тетрагидрофуране (1,5 мл) при 0°С и смесь перемешивали при комнатной температуре в течение 320 минут. Добавляли воду и этилацетат в реакционную смесь для разделения, затем органический слой промывали насыщенным солевым раствором и сушили над безводным натрия сульфатом. Полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH, затем фильтрат концентрировали в вакууме. Остаток растворяли в тетрагидрофуране, добавляли избыточное количество 9,8 М раствора метиламинметанола при комнатной температуре и смесь перемешивали в течение 50 минут. Реакционную смесь концентрировали в вакууме, остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 3:7 - 0:1). Целевую фракцию концентрировали в вакууме с получением неочищенного продукта А (46,3 мг).
Неочищенный продукт А (46,3 мг) растворяли в дихлорметане (1,25 мл) и добавляли трифторуксусную кислоту (250 μл) при 0°С. Смесь перемешивали при комнатной температуре в течение 20 минут, а затем концентрировали в вакууме, затем остаток растворяли в дихлорметан-триэтиламине и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 97:3 - 4:1). Целевую фракцию концентрировали в вакууме с получением неочищенного продукта В (35,8 мг).
Натрия триацетоксиборгидрид (10,4 мг, 0,049 ммоля) и ацетальдегид (2,17 мг, 0,049 ммоля) добавляли в смесь части неочищенного продукта В (9,0 мг, 0,016 ммоля) и тетрагидрофурана (500 μл) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 140 минут. Насыщенный водный раствор натрия бикарбоната и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью TLC на силикагеле NH (этилацетат). Полученное твердое вещество промывали диэтиловым эфиром с получением названного соединения (6,7 мг, 49%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,14 (3H, t, J=7,1 Гц), 1,76-1,91 (4Н, m), 1,93-2,12 (4H, m), 2,41-2,64 (3H, m), 3,03-3.17 (5H, m), 4,16 (2H, t, J=6,0 Гц), 4,24 (1Н, t, J=5,9 Гц), 4,36 (1Н, t, J=5,7 Гц), 5,46-5,57 (1Н, m), 6,55 (1Н, d, J=3,3 Гц), 6,58 (1Н, dd, J=5,9, 2,2 Гц), 7,24 (1Н, d, J=3,7 Гц), 7,31-7,36 (3H, m), 7,80 (2H, d, J=8,1 Гц), 7,90 (1Н, d, J=2,2 Гц), 8,02 (1Н, s), 8,10 (1Н, d, J=5,9 Гц), 8,49 (1Н, ors).
[0272] Исходный материал 5-((2-аминопиридин-4-ил)окси)-6-(3-фторпропокси)-N-метил-1Н-индол-1-карбоксамид синтезировали следующим способом.
[0273] [Пример получения 33-1]
4-(3-Фторпропокси)-3-гидроксибензальдегид
[Химическая формула 159]
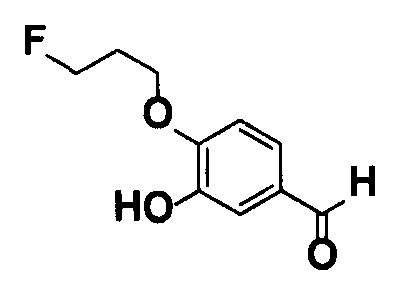
Коммерчески доступный 3,4-дигидроксибензальдегид (6,5 г, 47,1 ммоля) и калия карбонат (6,83 г, 49,4 ммоля) суспендировали в N,N-диметилформамиде (30 мл), затем 3-фторпропил-4-метилбензолсульфонат (11,3 г, 48,4 ммоля), описанный в примере получения 4-1, добавляли в атмосфере азота при комнатной температуре и смесь перемешивали в течение 37 часов. Смесь охлаждали до 0°С, а затем 2 М хлористоводородную кислоту, этилацетат и воду добавляли для разделения. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным магния сульфатом, а затем фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 4:1 - 2:1 - 1:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (4,62 г, 50%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 2,23 (1H, quin, J=5,9 Гц), 2,30 (1Н, quin, J=5,8 Гц), 4,31 (2Н, t, J=6,2 Гц), 4,60 (1H, t, J=5,6 Гц), 4,72 (1H, t, J=5,6 Гц), 5,70 (1H, s), 6,99 (1H, d, J=8,2 Гц), 7,41-7,47 (2Н, m), 9,85 (1H, s).
[0274] [Пример получения 33-2]
3-(Бензилокси)-4-(3-фторпропокси)бензальдегид
[Химическая формула 160]

Калия карбонат (3,87 г, 28,0 ммоля) и бензилхлорид (3,2 мл, 27,8 ммоля) добавляли в суспензию 4-(3-фторпропокси)-3-гидроксибензальдегида (4,62 г, 23,3 ммоля), описанного в примере получения 33-1, в этаноле (46 мл) в атмосфере азота при комнатной температуре и смесь перемешивали при 90°С в течение 1,5 часа. Реакционную смесь охлаждали до 0°С, затем 2 М хлористоводородной кислоты, этилацетат и воду добавляли для разделения. Органический слой промывали насыщенным солевым раствором, сушили над безводным магния сульфатом, а затем фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 3:2 - 1:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (6,14 г, 91%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 2,22 (1H, quin, J=5,9 Гц), 2,29 (1Н, quin, J=5,9 Гц), 4,25 (2Н, t, J=6,2 Гц), 4,62 (1H, t, J=5,7 Гц), 4,74 (1H, t, J=5,7 Гц), 5,18 (2Н, s), 7,02 (1H, d, J=8,1 Гц), 7,29-7,50 (7Н, m), 9,83 (1H, s).
[0275] [Пример получения 33-3]
(Е)-2-(Бензилокси)-1-(3-фторпропокси)-4-(2-нитровинил)бензол
[Химическая формула 161]
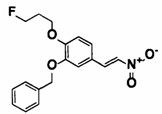
3-(Бензилокси)-4-(3-фторпропокси)бензальдегад (6,14 г, 21,3 ммоля), описанный в примере получения 33-2, растворяли в уксусной кислоте (17,0 мл), затем аммония ацетат (1,97 г, 25,6 ммоля) и нитрометан (2,8 мл, 51,7 ммоля) добавляли в атмосфере азота при комнатной температуре и смесь нагревали и перемешивали при 130°С в течение 2 часов. Смесь охлаждали до комнатной температуры, затем осадок собирали фильтрацией и промывали небольшим количеством этанола с количественным получением названного соединения.
1Н-ЯМР спектр (CDCl3) δ (ppm): 2,21 (1H, quin, J=6,0 Гц), 2,28 (1H, quin, J=5,9 Гц), 4,22 (2Н, t, J=6,0 Гц), 4,62 (1H, t, J=5,7 Гц), 4,73 (1H, t, J=5,7 Гц), 5,15 (2Н, s), 6,95 (1H, d, J=8,4 Гц), 7,06 (1H, d, J=1,8 Гц). 7,17 (1H, dd, J=8,2, 2,0 Гц), 7,29-7,49 (6Н, m), 7,91 (1H, d, J=13,5 Гц).
[0276] [Пример получения 33-4]
6-(3-Фторпропокси)-1Н-индол-5-ол
[Химическая формула 162]

Дымящуюся азотную кислоту (4,80 мл, 107 ммоля) добавляли в смесь (Е)-2-(бензилокси)-1-(3-фторпропокси)-4-(2-нитровинил)бензола (7,06 г, 21,3 ммоля), описанного в примере получения 33-3, и уксусной кислоты (61 мл) в ледяной бане и смесь перемешивали при комнатной температуре в течение 2,5 часа. Реакционную смесь выливали на лед, осадок собирали фильтрацией, а затем полученное вещество промывали жидкой смесью небольшого количества уксусной кислоты и этанола с получением неочищенного продукта (8,02 г).
Неочищенный продукт (8,02 г) суспендировали в метаноле (150 мл), затем добавляли 10% палладий на угле (содержание воды 50%) (2,27 г) и смесь перемешивали в атмосфере водорода в течение 6 часов. Внутреннюю среду реакционной системы замещали азотом, затем смесь разбавляли метанолом. Катализатор отфильтровывали через целит, затем фильтрат концентрировали в вакууме и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 1:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (1,47 г, 33%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 2,22 (1Н, quin, J=5,9 Гц), 2,29 (1H, quin, J=5,9 Гц), 4,23 (2H, t, J=6,0 Гц), 4,62 (1H, t, J=5,7 Гц), 4,74 (1H, t, J=5,7 Гц), 5,42 (1H, s), 6,42 (1H, ddd, J=3,1, 2,2, 0,9 Гц), 6,91 (1H, s), 7,09 (1H, dd, J=3,1, 2,4 Гц), 7,15 (1H, s), 7,94 (1Н, brs).
[0277] [Пример получения 33-5]
4-((6-(3-Фторпропокси)-1Н-индол-5-ил)окси)пиридин-2-амин
[Химическая 163]
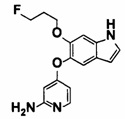
6-(3-Фторпропокси)-1Н-индол-5-ол (1,47 г, 7,04 ммоля), описанный в примере получения 33-4, М-(4-хлорпиридин-2-ил)ацетамид (1,32 г, 7,74 ммоля), описанный в примере получения 1-5, и калия трет-бутоксид (864 мг, 7,70 ммоля) растворяли в диметилсульфоксиде (7,0 мл), смесь нагревали и перемешивали в атмосфере азота при 160°С в течение 4,5 часа. Реакционную жидкость охлаждали до комнатной температуры, а затем добавляли воду и этилацетат для разделения. Водный слой снова экстрагировали этилацетатом, затем объединенный органический слой промывали водой и насыщенным солевым раствором, а затем сушили над безводным магния сульфатом. Осушающее средство отфильтровывали, фильтрат концентрировали в вакууме и полученное вещество очищали с помощью колоночной хромагографии на силикагеле NH (н-гептан:этилацетат = 4:1 - 1:1 - 0:1 - этилацетат:метанол = 99:1 - 9:1). Целевую фракцию концентрировали в вакууме с получением неочищенного продукта (177 мг).
Полученный неочищенный продукт (177 мг) растворяли в метаноле (2,5 мл), добавляли 2 М раствор натрия гидроксида (2,5 мл) в атмосфере азота при комнатной температуре и смесь нагревали и перемешивали при 70°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры, а затем добавляли воду и этилацетат для разделения. Органический слой промывали насыщенным солевым раствором и сушили над безводным натрия сульфатом. Осушающее средство отфильтровывали, затем фильтрат концентрировали в вакууме и полученное вещество очищали с помощью колоночной хромагографии на силикагеле (н-гептан:этилацетат = 2:3 - 1:4 - 0:1 - этилацетат:метанол = 19:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (49,4 мг, 2,3%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,95-2,11 (2H, m), 4,07 (2H, t, J=5,9 Гц), 4,28-4,47 (4Н, m), 5,88 (1H, d, J=2,2 Гц), 6,28 (1H, dd, J=5,9, 2,2 Гц), 6,48-6,52 (1H, m), 7,02 (1H, s), 7,16-7,20 (1H, m), 7,35 (1H, s), 7,88 (1H, d, J=5,9 Гц), 8,25 (1H, brs).
[0278] [Пример получения 33-6]
5-((2-Аминопиридин-4-ил)окси)-6-(3-фторпропокси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 164]

4-((6-(3-Фторпропокси)-1Н-индол-5-ил)окси)пиридин-2-амин (49,4 мг, 0,164 ммоля), описанный в примере получения 33-5, растворяли в N,N-диметилформамиде (1,6 мл), добавляли 50-72% маслянистый натрия гидрид (14,9 мг) в атмосфере азота при 0°С и смесь перемешивали при комнатной температуре в течение 15 минут. Смесь снова охлаждали до 0°С, добавляли фенилметилкарбамат (62,0 мг, 0,41 ммоля), описанный в примере получения 1-7, и смесь перемешивали при комнатной температуре в течение 3,5 часа. Насыщенный водный раствор аммония хлорида, воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом. Осушающее средство отфильтровывали, фильтрат концентрировали в вакууме и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:4 - 0:1 - этилацетат:метанол = 99:1 - 9:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (34,4 мг, 59%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,95-2,11 (2Н, m), 3,07 (3H, dd, J=4,7, 1,2), 4,12-4,17 (2H, m), 4,29 (1H, t, J=5,8 Гц), 4,38-4,52 (3H, m), 5,50 (1H, brs), 5,87 (1H, d, J=2,0 Гц), 6,26 (1H, dd, J=6,0, 2,1 Гц), 6,56 (1H, d, J=3,7 Гц), 7,23-7,27 (1H, m), 7,29 (1H, s), 7,87 (1H, d, J=6.0 Гц), 8,00 (1H, s).
[0279] Согласно примерам 1-33 иллюстративные соединения, представленные в таблицах 1 и 2, синтезировали из 5-((2-аминопиридин-4-ил)окси)-6-метокси-Н-метил-1Н-индол-1-карбоксамида, описанного в примере получения 1-6.

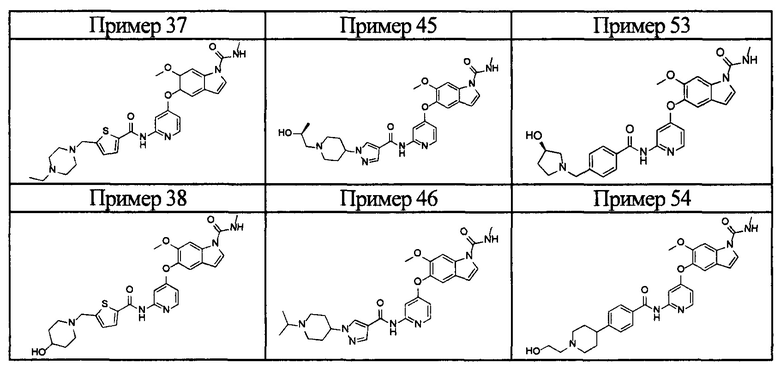
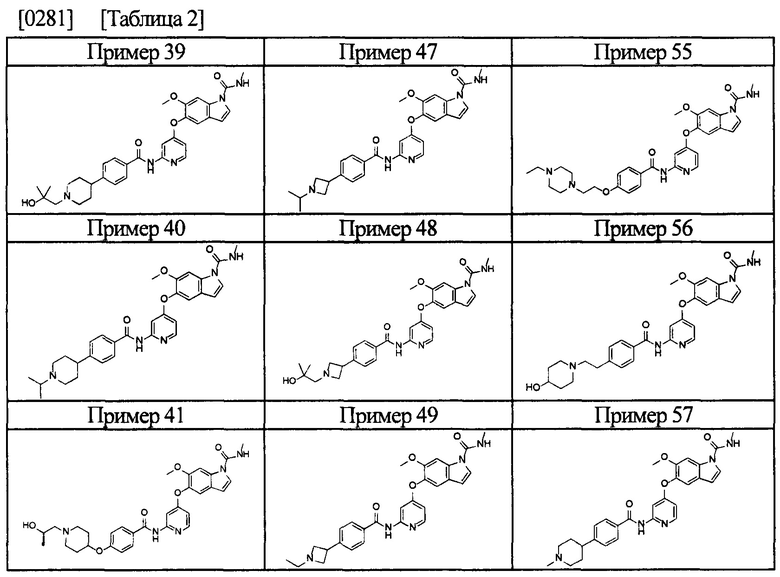
[0282] Согласно примерам 1-33 иллюстративные соединения, представленные в таблице 3, синтезировали из 5-((2-аминопиридин-4-ил)окси)-6-этокси-N-метил-1Н-индол-1-карбоксамида, описанного в примере получения 15-7.
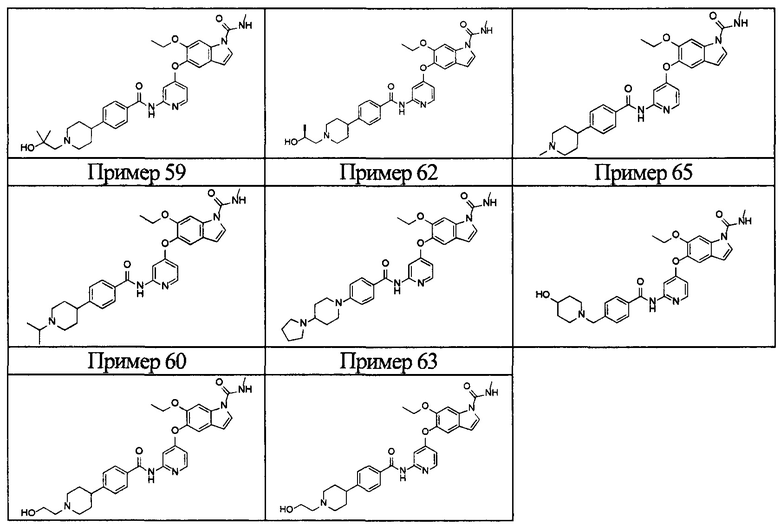
[0284] Согласно примерам 1-33 иллюстративные соединения, представленные в таблице 4, синтезировали из 5-((2-аминопиридин-4-ил)окси)-6-изопропокси-N-метил-1Н-индол-1-карбоксамида, описанного в примере получения 17-7.

[0286] Согласно примерам 1-33 иллюстративные соединения, представленные в таблице 5, синтезировали из 5-((2-аминопиридин-4-ил)окси)-6-(3-фторпропокси)-N-метил-1Н-индол-1-карбоксамида, описываемого в примере получения 33-6.


[0288] Согласно примерам 1-33 иллюстративные соединения, представленные в таблице 6, синтезировали из 5-((2-аминопиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1Н-индол-1-карбоксамида, описываемого в примере получения 20-7.
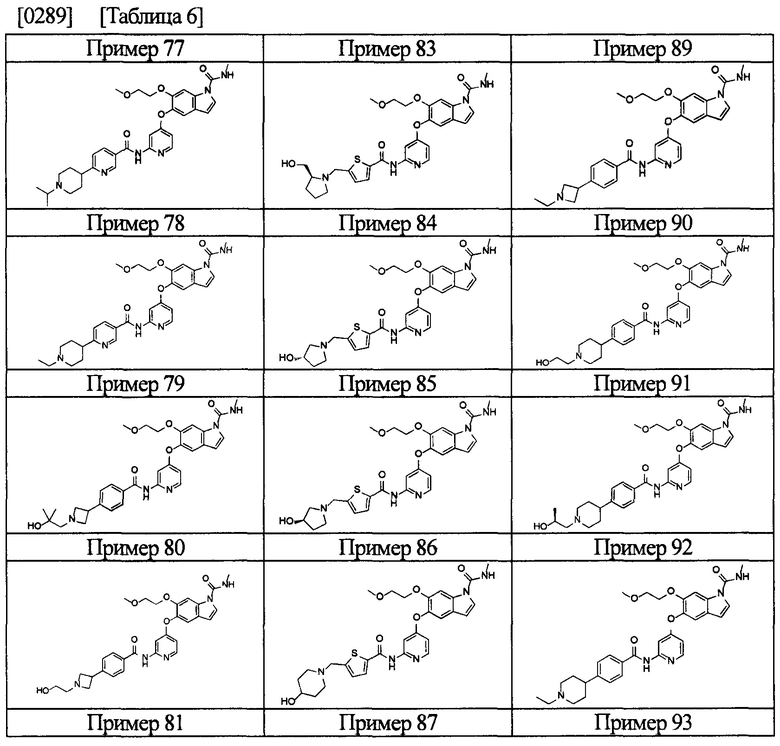
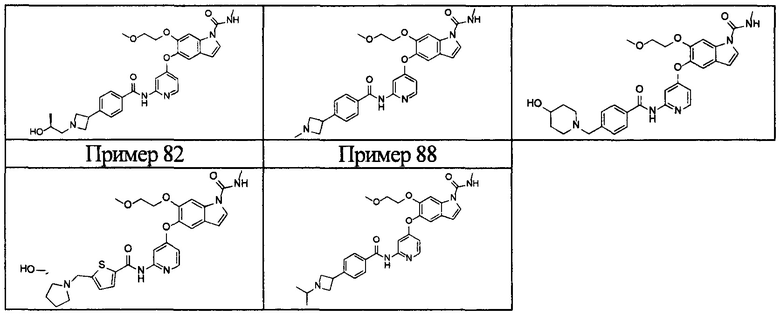
[0290] Согласно примерам 1-33 иллюстративные соединения, представленные в таблицах 7 и 8, синтезировали из 5-((2-аминопиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксамида, описываемого в примере получения 24-8.

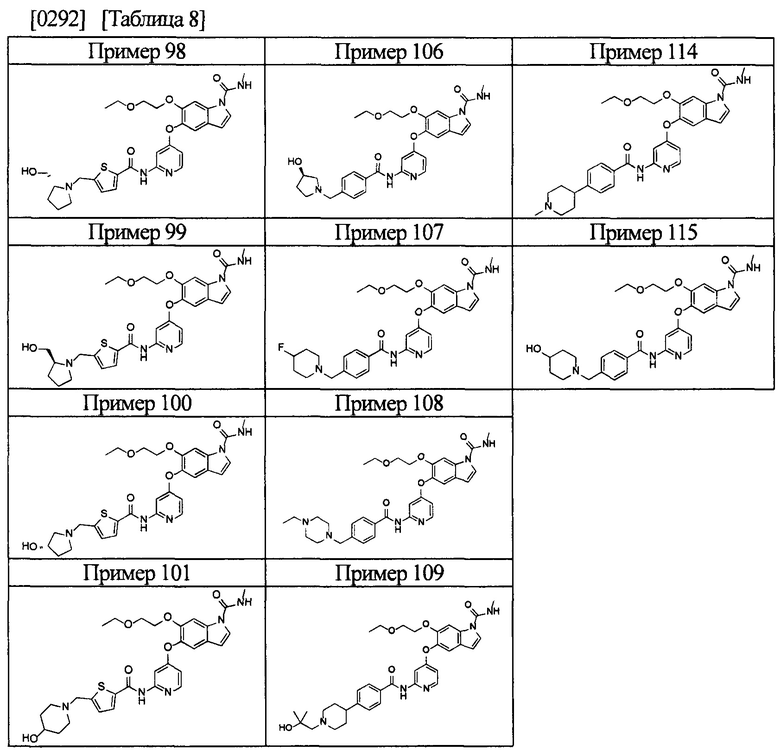
[0293] Согласно примерам 1-33 иллюстративные соединения, представленные в таблице 9, синтезировали из 5-((2-аминопиридин-4-ил)окси)-6-(3-метоксипропокси)-N-метил-1Н-индол-1-карбоксамида, описываемого в примере получения 32-8.


[0295] [Пример 131]
6-Метокси-5-((2-(4-(1-(2-метоксиацетил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 165]

Триэтиламин (33 μл, 0,237 ммоля) и метоксиацетил хлорид (12,5 мг, 0,115 ммоля) добавляли в смесь 6-метокси-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол -1-карбоксамида (13,4 мг, 0,027 ммоля), описываемого в примере 1, и тетрагидрофурана (1,0 мл) и смесь перемешивали при комнатной температуре в течение 3,5 часа. Воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 1:0-9:1). Целевую фракцию концентрировали в вакууме и твердое вещество промывали диэтиловым эфиром с получением названного соединения (10,2 мг, 67%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,50-1,73 (2Н, m), 1,87-1,98 (2H, m), 2,62-2,75 (1H, m), 2,76-2,89 (1H, m), 3,02-3,19 (4H, m), 3,45 (3H, s), 3,86 (3H, s), 3,97-4,03 (1H, m), 4,06-4,21 (2H, m), 4,71-4,81 (1H, m), 5,47-5,55 (1H, m), 6,55 (1H, d, J=3,7 Гц), 6,60 (1H, dd, J=5,8, 2,3 Гц), 7,23 (1H, d, J=3,8 Гц), 7,27-7,34 (3H, m), 7,81 (2H, d, J=8,4 Гц), 7,91 (1H, d, J=2,4 Гц), 8,03 (1H, s), 8,10 (1H, d, J=5,7 Гц), 8,49 (1H, brs).
[0296] Согласно примеру 131 иллюстративное соединение, представленное в таблице 10, синтезировали из 6-метокси-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида, описываемого в примере 1.
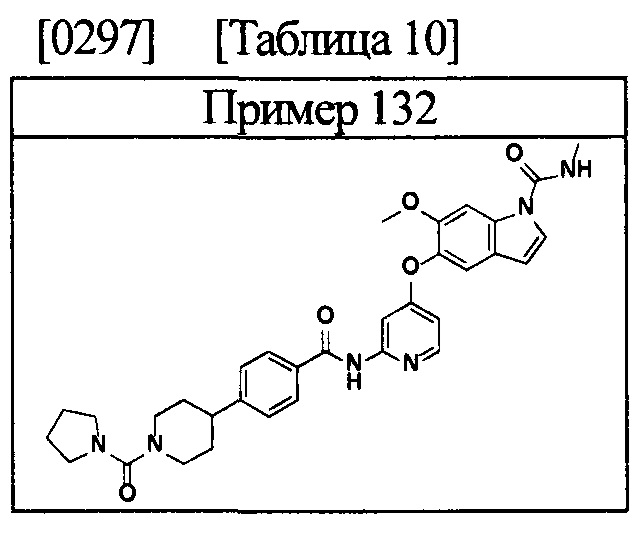
[0298] [Пример 133]
5-((2-(4-(1-(2-(Диметиламино)ацетил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-метокси-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 166]
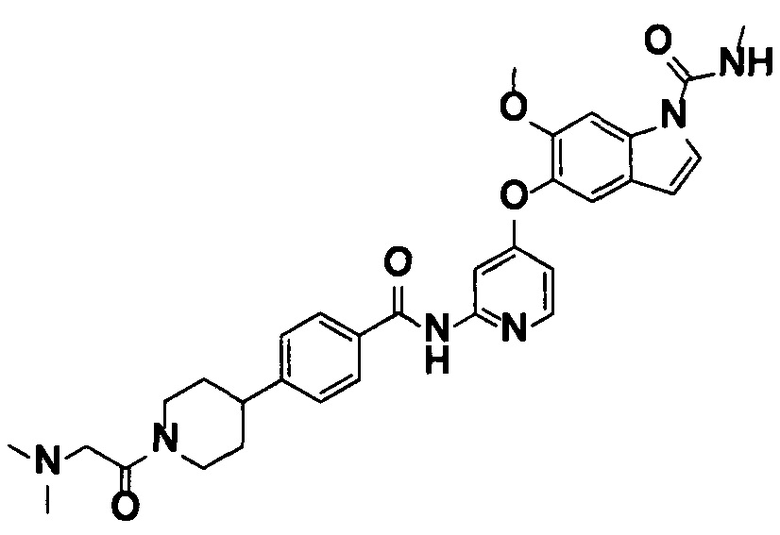
N,N-диметилформамид (0,5 мл) и N,N-диизопропилэтиламин (24 μл, 0,137 ммоля) добавляли в смесь 6-метокси-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида (13,7 мг, 0,027 ммоля), описываемого в примере 1, O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата (16,3 мг, 0,043 ммоля) и N,N-диметилглицина (5,5 мг, 0,053 ммоля) и смесь перемешивали при комнатной температуре в течение 2 часов. Воду и этидацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 1:0 - 9:1). Целевую фракцию концентрировали в вакууме и твердое вещество промывали диэтиловым эфиром с получением названного соединения (13,7 мг, 85%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,49-1,74 (2H, m), 1,85-1,97 (2H, m), 2,32 (6H, s), 2,60-2,71 (1H, m), 2,76-2,87 (1H, m), 3,04-3,26 (6H, m), 3,87 (3H, s), 4,20-4,32 (1H, m), 4,71-4,83 (1H, m), 5,45-5,54 (1H, m), 6,54-6,57 (1H, m), 6,60 (1H, dd, J=5,8, 2,3 Гц), 7,24 (1H, d, J=3,7 Гц), 7,28-7,34 (3H, m), 7,82 (2H, d, J=8,6 Гц), 7,92 (1H, d, J=2,4 Гц), 8,04 (1H, s), 8,10 (1H, d, J=5,7 Гц), 8,49 (1H, brs).
[0299] Согласно примеру 133 иллюстративное соединение, представленное в таблице 11, синтезировали из 6-метокси-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида, описываемого в примере 1.
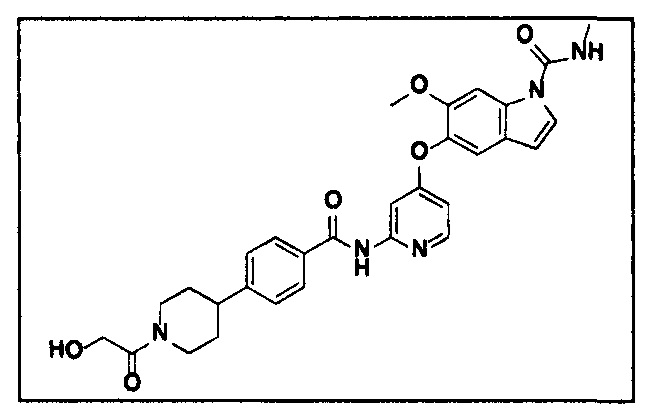
[0301] [Пример 135]
6-Этил-5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 167]
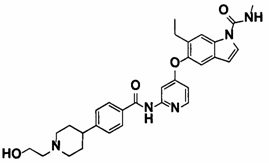
2-Гидроксиацетальдегид (9,41 мг, 0,157 ммоля), натрия триацетоксиборгидрид (33,2 мг, 0,157 ммоля) и уксусную кислоту (8,97 μл, 0,157 ммоля) добавляли в смесь 6-этил-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида (15,6 мг, 0,031 ммоля) и тетрагидрофурана (1,0 мл) и смесь перемешивали при комнатной температуре в течение 15 часов и 50 минут. Насыщенный водный раствор натрия бикарбоната и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 1:0 - 9:1 - 17:3). Целевую фракцию концентрировали в вакууме с получением названного соединения (13,2 мг, 78%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,24 (3Н, t, J=7,5 Гц), 1,71-1,92 (5Н, m), 2,16-2,26 (2H, m), 2,55-2,72 (5Н, m), 3,01-3,13 (5Н, m), 3,64 (2H, t, J=5,3 Гц), 5,46-5,53 (1Н, m), 6,52 (1H, dd, J-5,9, 2,2 Гц), 6,56 (1Н, d, J=3,7 Гц), 7,24-7,29 (1Н, m), 7,34 (2H, d, J=8,4 Гц), 7,37 (1Н, d, J=3,7 Гц), 7,82 (2H, d, J=8,1 Гц), 7,98 (1Н, d, J=2,2 Гц), 8,10 (1H, d, J=5,9 Гц), 8,12 (1H, s), 8,49 (1H, brs).
[0302] Исходный материал
6-этил-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид синтезировали следующим способом.
[0303] [Пример получения 135-1]
6-Этилиндолин
[Химическая формула 168]

Коммерчески доступный 3-этиланилин (10,1 г, 83,3 ммоля) растворяли в этаноле (40 мл), затем добавляли натрия гидрокарбонат (7,00 г, 83,3 ммоля) и бромацетальдегид диэтилацеталь (8,44 мл, 54,8 ммоля) и смесь нагревали и перемешивали при 80°С в течение 80 часов. Воду добавляли в реакционную смесь для разделения. Водный слой три раза экстрагировали этилацетатом, а затем объединенный органический слой промывали насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния, фильтровали, а затем концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 2:1). Целевую фракцию концентрировали в вакууме с получением неочищенного продукта А (11 г).
Часть полученного неочищенного продукта А (10 г) растворяли в трифторуксусной кислоте (45 мл), добавляли безводную трифторуксусную кислоту (45 мл) при 4°С и смесь перемешивали в течение 30 минут. Трифторуксусную кислоту (60 мл) добавляли в реакционную смесь и смесь перемешивали при 75°С в течение 2,5 часа. После концентрации в вакууме водный слой три раза экстрагировали этилацетатом, а затем объединенный органический слой промывали насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния, фильтровали, а затем концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 3:1). Целевую фракцию концентрировали в вакууме с получением неочищенного продукта В (3 г).
Часть полученного неочищенного продукта В (1,5 г) растворяли в уксусной кислоте (50 мл), добавляли натрия цианоборгидрид (1,30 г, 20,7 ммоля) при 0°С и смесь перемешивали при комнатной температуре в течение 1 часа. Насыщенный водный раствор натрия бикарбоната добавляли в реакционную смесь для разделения. Водный слой три раза экстрагировали этилацетатом, а затем объединенный органический слой промывали насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния, фильтровали, а затем концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хромагографии на силикагеле (н-гептан:этилацетат = 3:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (560 мг, 15%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,20 (3Н, t, J=7,6 Гц), 2,55 (2Н, q, J=7,7 Гц), 2,99 (2H, t, J=8,4 Гц), 3,54 (2H, t, J=8,3 Гц), 3,72 (1Н, brs), 6,43-6,61 (2H, m), 6,93-7,07 (1H, m).
[0304] [Пример получения 135-2]
6-Этил-1Н-индол-5-ол
[Химическая формула 169]
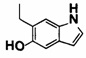
Калия нитрозодисульфонат (6,82 г, 25,4 ммоля) растворяли в 0,1 М калий-фосфатном буфере (450 мл), затем добавляли 6-этилиндолин (1,7 г, 11,5 ммоля), описываемый в примере получения 135-1, растворенный в ацетоне (150 мл) и смесь перемешивали при комнатной температуре в течение 12 часов. 2 М натрия гидроксид добавляли в реакционную смесь для разделения. Водный слой три раза экстрагировали этилацетатом, а затем объединенный органический слой промывали насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния, фильтровали, а затем концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хромагографии на силикагеле (н-гептан:этилацетат = 1:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (560 мг, 30%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,29 (3Н, t, J=7,5 Гц), 2,74 (2H, q, J=7,5 Гц), 4,50 (1H, s), 6,34-6,43 (1H, m), 6,99 (1H, s), 7,07-7,18 (2H, m), 7,95 (1H, brs).
[0305] Согласно примерам 1-33 иллюстративные соединения, представленные в таблице 12, включающей в себя исходный материал примера 135, синтезировали 6-этил-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид из 6-этил-1Н-индол-5-ола, описываемого в примере получения 135-2, и N-(4-хлорпиридин-2-ил)ацетамида, описываемого в примере получения 1-5.

[0307] [Пример 140]
5-((2-(4-((4-Гидроксипиперидин-1-ил)метил)бензамид)пиридин-4-ил)окси)-6-(метоксиметил)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 170]
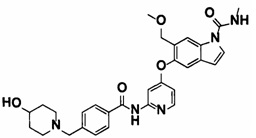
Триэтиламин (20 μл, 0,144 ммоля) и коммерчески доступный 4-(хлорметил)бензоил хлорид (16,6 мг, 0,088 ммоля) добавляли в смесь 5-((2-аминопиридин-4-ил)окси)-6-(метоксиметил)-N-метил-1Н-индол-1-карбоксамида (5,5 мг, 0,017 ммоля) и тетрагидрофурана (450 μл) при комнатной температуре в атмосфере азота и смесь перемешивали в течение 70 минут. Тетрагидрофуран, воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом, фильтровали с помощью силикагеля NH и полученное вещество концентрировали в вакууме с получением неочищенного продукта.
Полученный неочищенный продукт растворяли в N,N-диметилформамиде (500 μл), добавляли 4-гидроксипиперидин (16,5 мг, 0,163 ммоля) при комнатной температуре в атмосфере азота и смесь перемешивали в течение 12 часов и 50 минут. Этилацетат и воду добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом, фильтровали, а затем концентрировали в вакууме. Остаток растворяли в дихлорметане и полученное вещество очищали с помощью TLC на силикагеле NH (этилацетат) с получением названного соединения (7,6 мг, 83%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,38-1,67 (2H, m), 1,83-1,95 (2H, m), 2,10-2,25 (2H, m), 2,68-2,79 (2H, m), 3,10 (3H, d, J=4,8 Гц), 3,37 (3H, s), 3,56 (2H, brs), 3,62-3,77 (2H, m), 4,52 (2H, s), 5,60-5,69 (1H, m), 6,55 (1H, dd, J=5,9, 2,2 Гц), 6,58 (1H, dd, J=3,7, 0,7 Гц), 7,30 (1H, s), 7,44 (2H, d, J=8,1 Гц), 7,50 (1H, d, J=3,7 Гц), 7,82 (2H, d, J=8,1 Гц), 7,97 (1H, d, J=2,2 Гц), 8,09 (1H, d, J=5,5 Гц), 8,21 (1H, s), 8,60 (1H, brs).
[0308] Исходный материал 5-((2-аминопиридин-4-ил)окси)-6-(метоксиметил)-N-метил-1Н-индол-1-карбоксамид синтезировали следующим способом.
[0309] [Пример получения 140-1]
Бензил-5-амино-2-(бензилокси)-4-бромбензоат
[Химическая формула 171]
Бензил-5-амино-2-(бензилокси)бензоат (5 г, 15,0 ммоля), полученный путем бензилирования и восстановления коммерчески доступной 5-аминосалициловой кислоты традиционным способом растворяли в дихлорметане (100 мл) и метаноле (50 мл), затем тетра-н-бутиламмония трибромид (7,25 г, 15,0 ммоля) добавляли при комнатной температуре в атмосфере азота и смесь перемешивали в течение 4 часов. Водный раствор натрия гидросульфита и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, сушили над безводным магния сульфатом, а затем фильтровали. Фильтрат концентрировали в вакууме и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 4:1 - 3:2). Целевую фракцию концентрировали в вакууме с получением названного соединения (3,58 г, 58%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 3,88 (2H, s), 5,04 (2H, s), 5,31 (2H, s), 7,15 (1H, s), 7,27-7,43 (11H, m).
[0310] [Пример получения 140-2]
Бензил-5-амино-2-(бензилокси)-4-((триметилсилил)этинил)бензоат
[Химическая формула 172]
Бензил-5-амино-2-(бензилокси)-4-бромбензоат (3,58 г, 8,68 ммоля), описываемый в примере получения 140-1, растворяли в тетрагидрофуране (20 мл) и триэтиламине (40 мл), затем добавляли триметилсилилацетилен (2,5 мл, 17,7 ммоля), бис(трифенилфосфин)палладия (II) хлорид (304 мг, 0,434 ммоля) и меди йодид (I) (165 мг, 0,868 ммоля) при комнатной температуре в атмосфере азота, и смесь перемешивали при 70°С в течение 6 часов. Воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом, а затем фильтровали. Фильтрат концентрировали в вакууме, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хромагографии на силикагеле (н-гептан:этилацетат = 19:1 - 4:1 - 7:3). Целевую фракцию концентрировали в вакууме с получением названного соединения (2,75 г, 74%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 0,25-0,35 (9H, m), 4,03 (2H, s), 5,03 (2H, s), 5,31 (2H, s), 7,00 (1H, s), 7,20 (1H, s), 7,28-7,47 (10Н, m).
[0311] [Пример получения 140-3]
Бензил-5-(бензилокси)-1Н-индол-6-карбоксилат
[Химическая формула 173]

Бензил-5-амино-2-(бензилокси)-4-((триметилсилил)этинил)бензоат, описываемый в пример получения 140-2, (2,75 г, 6,40 ммоля) растворяли в N,N-диметилформамиде (30 мл), затем добавляли меди йодид (I) (610 мг, 3,20 ммоля) при комнатной температуре в атмосфере азота и смесь нагревали и перемешивали при 100°С в течение 3 часов. Этилацетат добавляли в реакционную смесь и полученную смесь фильтровали через целит. Фильтрат концентрировали в вакууме, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 17:3 - 3:2). Целевую фракцию концентрировали в вакууме с получением названного соединения (1,83 г, 80%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 5,18 (2H, s), 5,37 (2H, s), 6,44-6,50 (1H, m), 7,22 (1H, s), 7,27-7,38 (7H, m), 7,40-7,46 (2H, m), 7,47-7,53 (2H, m), 7,99 (1H, d, J=0,7 Гц), 8,25 (1H, brs).
[0312] [Пример получения 140-4]
(5-(Бензилокси)-1Н-индол-6-ил)метанол
[Химическая формула 174]
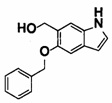
Лития алюминия гидрид (239 мг, 6,30 ммоля) суспендировали в тетрагидрофуране (22 мл), затем раствор бензил-5-(бензилокси)-1Н-индол-6-карбоксилата (1,5 г, 4,20 ммоля), описываемого в примере получения 140-3, растворенного в тетрагидрофуране (11 мл) добавляли при 0°С в атмосфере азота и смесь перемешивали при комнатной температуре в течение 3 часов. Воду (0,24 мл), 5 М водный раствор натрия гидроксида (0,24 мл) и воды (0,72 мл) добавляли в реакционную смесь и смесь перемешивали при комнатной температуре в течение 30 минут. Суспензию фильтровали через целит и промывали тетрагидрофураном. Фильтрат концентрировали в вакууме, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 4:1 - 1:1 - 1:3). Целевую фракцию концентрировали в вакууме с количественным получением названного соединения.
1Н-ЯМР спектр (CDCl3) δ (ppm): 2,95 (1H, t, J=6,4 Гц), 4,81 (2H, d, J=6,6 Гц), 5,17 (2H, s), 6,46-6,50 (1H, m), 7,16-7,21 (2H, m), 7,30-7,43 (4H, m), 7,44-7,50 (2H, m), 8,11 (1H, brs).
[0313] [Пример получения 140-5]
Трет-бутил 5-(бензилокси)-6-(гидроксиметил)-1Н-индол-1-карбоксилат
[Химическая формула 175]

Трет-бутилдиметилхлорсилан (280 мг, 1,86 ммоля) добавляли в раствор (5-(бензилокси)-1Н-индол-6-ил)метанола (392 мг, 1,55 ммоля), описываемого в примере получения 140-4, и имидазола (158 мг, 2,32 ммоля) в N,N-диметилформамиде (3,8 мл) при комнатной температуре в атмосфере азота и смесь перемешивали в течение 70 минут. Этилацетат и воду добавляли в реакционную смесь для разделения. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным натрия сульфатом, а затем фильтровали. Фильтрат концентрировали в вакууме с получением неочищенного продукта А.
Полученный неочищенный продукт А растворяли в дихлорметане (6,0 мл), добавляли ди-трет-бутилдикарбонат (538 мг, 2,47 ммоля) и 4-диметиламинопиридин (18,9 мг, 0,155 ммоля) при комнатной температуре в атмосфере азота и смесь перемешивали в течение 1,5 часа. Этилацетат и воду добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом, а затем фильтровали. Фильтрат концентрировали в вакууме с получением неочищенного продукта В.
Полученный неочищенный продукт В растворяли в тетрагидрофуране (6,0 мл), затем добавляли 1 М тетрабутиламмония фторид (3,0 мл, 3,00 ммоля) при комнатной температуре в атмосфере азота и смесь перемешивали в течение 6 часов. Этилацетат и воду добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом, а затем фильтровали. Фильтрат концентрировали в вакууме, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1-1:1). Целевую фракцию концентрировали в вакууме с количественным получением названного соединения.
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,66 (9Н, s), 2,42 (1H, t, J=6,6 Гц), 4,82 (2H, d, J=6,6 Гц), 5,17 (2H, s), 6,49 (1H, dd, J=3,7, 0,7 Гц), 7,09 (1H, s), 7,30-7,49 (5H, m), 7,56 (1H, d, J=3,7 Гц), 8,09 (1Н, brs).
[0314] [Пример получения 140-6]
Трет-бутил 5-(бензилокси)-6-(метоксиметил)-1Н-индол-1-карбоксилат
[Химическая формула 176]

Метилйодид (290 μл, 4,66 ммоля) и 50-72% маслянистый натрия гидрид (122 мг) добавляли в раствор трет-бутил-5-(бензилокси)-6-(гидроксиметил)-1Н-индол-1-карбоксилага (547 мг, 1,55 ммоля), описываемого в примере получения 140-5, в N,N-диметилформамиде (10 мл) при комнатной температуре в атмосфере азота и смесь перемешивали в течение 3 часов и 15 минут. Реакционную смесь гасили водой, а затем разбавленным этилацетатом для разделения. Органический слой дважды промывали водой, сушили над безводным натрия сульфатом, а затем фильтровали. Фильтрат концентрировали в вакууме, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хромагографии на силикагеле (н-гептан:этилацетат = 19:1 - 4:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (487 мг, 86%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,67 (9Н, s), 3,45 (3Н, s), 4,65 (2H, s), 5,14 (2H, s), 6,47 (1H, d, J=3,7 Гц), 7,06 (1H, s), 7,29-7,50 (5H, m), 7,55 (1H, d, J=3,7 Гц), 8,17 (1H, brs).
[0315] [Пример получения 140-7]
6-(Метоксиметил)-1Н-индол-5-ол
[Химическая формула 177]

28% натрия метоксид (4,0 мл, 19,6 ммоля) добавляли в раствор трет-бутил-5-(бензилокси)-6-(метоксиметил)-1Н-индол-1-карбоксилата (427 мг, 1,16 ммоля), описываемого в примере получения 140-6, в метаноле (4,0 мл) при комнатной температуре в атмосфере азота и смесь перемешивали в течение 2,5 часа. Воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом, а затем фильтровали. Фильтрат концентрировали в вакууме с получением неочищенного продукта (301 мг).
Полученный неочищенный продукт (301 мг) растворяли в метаноле (11 мл), добавляли 10% палладий на угле (содержание воды 50%) (12,0 мг) при комнатной температуре и смесь перемешивали в течение 3 часов в атмосфере водорода. Смесь разбавляли метанолом и катализатор отфильтровывали через целит. Фильтрат концентрировали в вакууме и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 4:1 - 2:3). Целевую фракцию концентрировали в вакууме с получением названного соединения (156 мг, 76%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 3,43 (3H, s), 4,73 (2H, s), 6,43-6,47 (1H, m), 6,91 (1H, s), 7,10 (1H, s), 7,13 (1H, s), 7,17 (1H, t, J=2,7 Гц), 8,00 (1H, brs).
[0316] Согласно примерам 1-33 иллюстративные соединения, представленные в таблице 13, включающей в себя исходный материал примера 140, 5-((2-аминопиридин-4-ил)окси)-6-(метоксиметил)-N-метил-1Н-индол-1-карбоксамид синтезировали из 6-(метоксиметил)-1Н-индол-5-ола, описываемого в примере получения 140-7, и N-(4-хлорпиридин-2-ил)ацетамида, описываемого в примере получения 1-5.

[0318] [Пример 143]
6-Циано-5-((2-(4-(1-этилпиперидин-4-ил)бензамид)пиридин-4-ил)окси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 178]
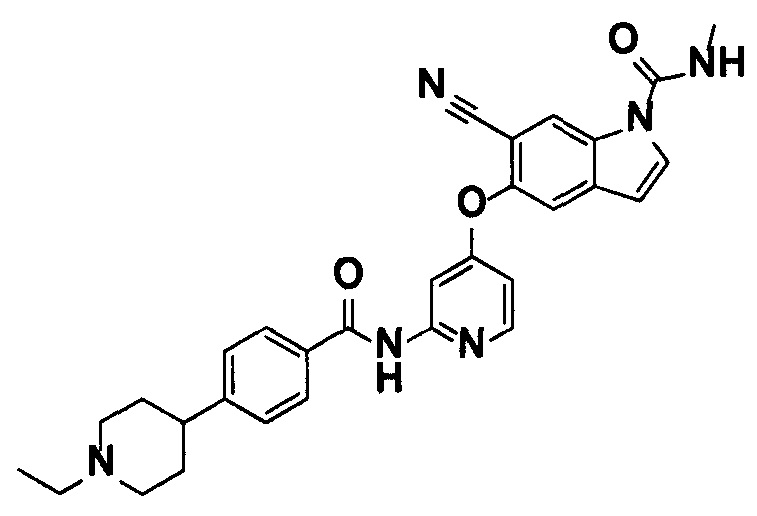
Натрия триацетоксиборгидрид (10,7 мг, 0,05 ммоля) и ацетальдегид (5,6 мг, 0,127 ммоля) добавляли в смесь 6-циано-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида (3,9 мг, 7,89 ммоля) и тетрагидрофурана (1,0 мл) при комнатной температуре в атмосфере азота и смесь перемешивали в течение 1 часа. Насыщенный водный раствор натрия бикарбоната и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью TLC на силикагеле NH (хлороформ:метанол = 49:1) с получением названного соединения (2,54 мг, 62%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,16 (3H, t, J=7,3 Гц), 1,81-1,96 (4H, m), 2,06-2,20 (2H, m), 2,47-2,65 (3H, m), 3,01-3,08 (3H, m), 3,16 (2H, d, J=11,7 Гц), 5,82 (1H, brs), 6,59-6,65 (1H, m), 6,76 (1H, dd, J=5,7, 2,4 Гц), 7,33 (2H, d, J=8,4 Гц), 7,41 (1H, s), 7,57 (1H, d, J=3,7 Гц), 7,78 (2H, d, J=8,1 Гц), 7,93 (1H, s), 8,20 (1H, d, J=5,5 Гц), 8,60-8,68 (2H, m).
[0319] Исходный материал 6-циано-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид синтезировали следующим способом.
[0320] [Пример получения 143-1]
1-(Бензилокси)-2-хлор-4-нитробензол
[Химическая формула 179]

Коммерчески доступный 2-хлор-4-нитрофенол (10 г, 57,6 ммоля) и бензил бромид (7,6 мл, 63,9 ммоля) растворяли в N,N-диметилформамиде (100 мл), добавляли калия карбонат (9,56 г, 69,1 ммоля) и смесь перемешивали при комнатной температуре в течение 3,5 часа. Добавляли воду в реакционную смесь и смесь перемешивали при комнатной температуре в течение 10 минут. Осажденное твердое вещество отфильтровывали, промывали трет-бутилметиловым эфиром и сушили путем сушки со сквозным протоком с количественным получением названного соединения.
1Н-ЯМР спектр (CDCl3) δ (ppm): 5,28 (2H, s), 7,03 (1H, d, J=9,2 Гц), 7,33-7,48 (5H, m), 8,12 (1H, dd, J=9,2, 2,9 Гц), 8,32 (1H, d, J=2,6 Гц).
[0321] [Пример получения 143-2]
2-(5-(Бензилокси)-4-хлор-2-нитрофенил)ацетонитрил
[Химическая формула 180]
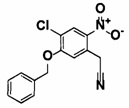
1-(Бензилокси)-2-хлор-4-нитробензол (5 г, 19,0 ммоля), описываемый в примере получения 143-1, и 4-хлорфеноксиацетонитрил (3,50 г, 20,9 ммоля) растворяли в N,N-диметилформамиде (35 мл), а затем добавляли калия трет-бутоксид (2 г, 17,8 ммоля) при от -30 до -40°С в атмосфере азота. Калия трет-бутоксид (2,32 г, 20,7 ммоля) добавляли при той же температуре. Смесь перемешивали при той же температуре в течение 45 минут, а затем добавляли 1 М хлористоводородную кислоту После осуществления экстракции этилацетатом органический слой дважды промывали водой и насыщенным солевым раствором, а затем сушили над безводным магния сульфатом. После осуществления фильтрации фильтрат концентрировали в вакууме, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 3:1 - 1:1). Смешанную фракцию концентрировали в вакууме, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 4:1 - 13:7). Полученное твердое вещество промывали диэтиловым эфиром с получением названного соединения (1,04 г, 18%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 4,24 (2H, s), 5,33 (2H, s), 7,29 (1H, s), 7,34-7,52 (5H, m), 8,34 (1H, s).
[0322] [Пример получения 143-3]
6-Хлор-1Н-индол-5-ол
[Химическая формула 181]
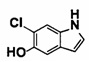
2-(5-(Бензилокси)-4-хлор-2-нитрофенил)ацетонитрил (1,04 г, 3,45 ммоля), описываемый в примере получения 143-2, суспендировали в этаноле (40 мл), добавляли 5% родий на угле (355 мг) при комнатной температуре и смесь перемешивали в течение 39 часов и 20 минут в атмосфере водорода. Метанол добавляли в реакционную смесь и полученную смесь фильтровали через целит. Фильтрат концентрировали в вакууме, полученное твердое вещество отфильтровывали и промывали этилацетатом. Фильтрат концентрировали, остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1 - 3:2) и целевую фракцию концентрировали в вакууме с получением неочищенного продукта.
Полученный неочищенный продукт растворяли в этаноле (20 мл), добавляли 10% палладий на угле (содержание воды 50%) (367 мг) при комнатной температуре и смесь перемешивали в течение 50 минут в атмосфере водорода. Метанол добавляли в реакционную смесь и полученную смесь фильтровали через целит. Фильтрат концентрировали в вакууме, полученное твердое вещество отфильтровывали и промывали дихлорметаном. Фильтрат концентрировали, остаток очищали с помощью колоночной хромагографии на силикагеле (н-гептан:этилацетат = 3:1 - 3:2) и целевую фракцию концентрировали в вакууме с получением названного соединения (193 мг, 33%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 5,25 (1Н, s), 6,41-6,47 (1H, m), 7,19 (1H, t, J=2,7 Гц), 7,24 (1H, s), 7,37 (1H, s), 8,02 (1H, brs).
[0323] [Пример получения 143-4]
5-((2-Аминопиридин-4-ил)окси)-1Н-индол-6-карбонитрил
[Химическая формула 182]

6-Хлор-1Н-индол-5-ол (137 мг, 0,817 ммоля), описываемый в примере получения 143-3, растворяли в диметилсульфоксиде (1,0 мл), N,N-(4-хлорпиридин-2-ил)ацетамид (181 мг, 1,06 ммоля), описываемый в примере получения 1-5, и калия трет-бутоксид (110 мг, 0,981 ммоля) добавляли при комнатой температуре в атмосфере азота и смесь нагревали и перемешивали при 150°С в течение 6,5 часа. Воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали водой и насыщенным солевым раствором, сушили над безводным натрия сульфатом, а затем фильтровали. Фильтрат концентрировали в вакууме, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 1:3 - 0:1). Целевую фракцию концентрировали в вакууме с получением неочищенного продукта А (96,6 мг).
Часть полученного неочищенного продукта А (57,4 мг) растворяли в метаноле (2 мл), добавляли 2 М раствор натрия гидроксида (2 мл) при комнатной температуре в атмосфере азота, смесь нагревали и перемешивали при 65°С в течение 2 часов. Воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом, а затем фильтровали. Фильтрат концентрировали в вакууме, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 1:1 - 0:1). Целевую фракцию концентрировали в вакууме с получением неочищенного продукта В (34,7 мг).
Часть полученного неочищенного продукта В (10,4 мг), бис(три-трет-бутилфосфин)палладий(0) (8,19 мг, 0,016 ммоля) и цинка цианид (9,4 мг, 0,08 ммоля) растворяли в N,N-диметилацетамиде (500 μл), смесь нагревали и перемешивали в атмосфере азота и при микроволновом излучении при 150°С в течение 1 часа. Этилацетат и воду добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, сушили над безводным натрия сульфатом, а затем фильтровали. Фильтрат концентрировали в вакууме, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью TLC на силикагеле (н-гептан:этилацетат = 1:3) с получением названного соединения (5,0 мг, 14%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 4,57 (2Н, brs), 5,97 (1H, d, J=2,2 Гц), 6,31 (1H, dd, J=6,2, 2,2 Гц), 6,56-6,63 (1H, m), 7,41 (1H, s), 7,48 (1H, t, J=3,0 Гц), 7,77 (1H, s), 7,92 (1H, d, J=5,9 Гц), 8,98 (1H, brs).
[0324] Согласно примерам 1-33 иллюстративное соединение, представленное в таблице 14, включающей в себя исходный материал примера 143, 6-циано-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид синтезировали из 5-((2-аминопиридин-4-ил)окси)-1Н-индол-6-карбонитрила, описываемого в примере получения 143-5.

[0326] [Пример 145]
6-(Диметиламино)-N-метил-5-((2-(4-(1-метилпиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид
[Химическая формула 183]

36,5% водный раствор формальдегида (7,95 μл, 0,105 ммоля), натрия триацетоксиборгидрид (14 мг, 0,066 ммоля) и уксусную кислоту (3,02 μл, 0,053 ммоля) добавляли в смесь 6-(диметиламино)-N-метил-5-((2-(4-(пиперидан-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамида (5,4 мг, 10,5 μмоля) и тетрагидрофурана (300 μл) при комнатной температуре и смесь перемешивали в течение 2 часов. Насыщенный водный раствор натрия бикарбоната и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, затем сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали, полученный остаток растворяли в хлороформе и полученное вещество очищали с помощью TLC на силикагеле NH (этилацетат). Полученное твердое вещество промывали диэтиловым эфиром с получением названного соединения (3,31 мг, 60%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 1,77-1,88 (4H, m), 2,01-2,11 (2H, m), 2,33 (3H, s), 2,48-2,60 (1H, m), 2,79 (6H, s), 2,95-3,03 (2H, m), 3,07 (3H, d, J=4,8 Гц), 5,48 (1H, brs), 6,47 (1H, dd, J-5,9, 2,6 Гц), 6,52 (1H, d, J=3,7 Гц), 7,22-7,25 (2H, m), 7,33 (2H, d, J=8,4 Гц), 7,78-7,84 (2H, m), 7,96 (1H, s), 7,99 (1H, d, J=2,2 Гц), 8,06 (1H, d, J=5,9 Гц), 8,49 (1H, brs).
[0327] Исходный материал 6-(диметиламино)-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид синтезировали следующим способом.
[0328] [Пример получения 145-1]
4-(Бензилокси)-3-нитроанилин
[Химическая формула 184]

Коммерчески доступный 4-амино-2-нитрофенол (5 г, 32,4 ммоля) и трифенилфосфин (10,2 г, 38,9 ммоля) растворяли в дихлорметане (200 мл), добавляли бензиновый спирт (4,0 мл, 38,7 ммоля) при 0°С, затем добавляли раствор диизопропилазодикарбоксилата (7,87 г, 38,9 ммоля) в дихлорметане (50 мл) и смесь перемешивали при комнатной температуре в течение 21,5 часа. Реакционную смесь концентрировали в вакууме, остаток растворяли в дихлорметане, полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 4:1 - 2:3) и целевую фракцию концентрировали в вакууме с получением названного соединения (7,26 г, 92%).
1Н-ЯМР спектр (DMSO-d6) δ (ppm): 5,09 (2H, s), 5,25 (2H, s), 6,81 (1H, dd, J=9,0, 2,7 Гц), 7,01 (1H, d, J=2,9 Гц), 7,13 (1H, d, J=9,2 Гц), 7,25-7,44 (5H, m).
[0329] [Пример получения 145-2]
4-(Бензилокси)-2-бром-5-нитроанилин
[Химическая формула 185]

4-(Бензилокси)-3-нитроанилин (2,9 г, 11,9 ммоля), описываемый в примере получения 145-1, растворяли в дихлорметане (80 мл) и метаноле (40 мл), затем добавляли тетра-N-бутиламмония трибромид (5,73 г, 11,9 ммоля) при комнатной температуре в атмосфере азота и смесь перемешивали в течение 50 минут. Реакционную смесь разбавляли водным раствором натрия гидросульфита и дихлорметаном. Органический слой промывали насыщенным солевым раствором и сушили над безводным магния сульфатом. После осуществления фильтрации фильтрат концентрировали в вакууме, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хромагографии на силикагеле (н-гептан:этилацетат = 9:1 - 3:2). Целевую фракцию концентрировали в вакууме с получением названного соединения (2,59 г, 67%) с чистотой 84%.
1Н-ЯМР спектр (CDCl3) δ (ppm): 4,06 (2H, brs), 5,12 (2H, s), 7,25 (1H, s), 7,31 (1H, s), 7,32-7,47 (5H, m).
[0330] [Пример получения 145-3]
4-(Бензилокси)-5-нитро-2-((триметилсилил)этинил)анилин
[Химическая формула 186]
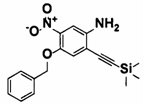
4-(Бензилокси)-2-бром-5-нитроанилин (2,59 г, 8,01 ммоля), описываемый в примере получения 145-2, растворяли в тетрагидрофуране (20 мл) и триэтиламине (40 мл), затем триметилсилилацетилен (2,26 мл, 16,0 ммоля), бис(трифенилфосфин)палладия (II) хлорид (291 мг, 0,415 ммоля) и меди йодид (I) (164 мг, 0,861 ммоля) добавляли при комнатной температуре, смесь нагревали и перемешивали при 60°С в атмосфере азота. Воду и этилацетат добавляли в реакционную смесь для разделения. Органический слой промывали насыщенным солевым раствором, а затем сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 9:1-3:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (2,19 г, 80%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 0,28 (9H, s), 4,19 (2H, brs), 5,11 (2H, s), 7,09 (1H, s), 7,21 (1H, s), 7,29-7,48 (5H, m).
[0331] [Пример получения 145-4]
5-(Бензилокси)-6-нитро-1Н-индол
[Химическая формула 187]

4-(Бензилокси)-5-нитро-2-(триметилсилил)этинил)анилин (2,19 г, 6,44 ммоля), описываемый в примере получения 145-3, растворяли в N,N-диметилформамиде (20 мл), добавляли меди йодид (I) (1,23 г, 6,44 ммоля) при комнатной температуре и смесь нагревали и перемешивали при 100°С. Воду и этилацетат добавляли в реакционную смесь для разделения. Водный слой экстрагировали этилацетатом, объединенный органический слой промывали водой и насыщенным солевым раствором, затем сушили над безводным магния сульфатом и фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 4:1 - 7:3 - 3:2). Целевую фракцию концентрировали в вакууме с получением названного соединения (1,23 г, 71%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 5,24 (2H, s), 6,49-6,56 (1H, m), 7,27-7,46 (5H, m), 7,49-7,57 (2H, m), 8,06 (1H, d, J=1,1 Гц), 8,39 (1H, brs).
[0332] [Пример получения 145-5]
6-Амино-1 Н-индол-5-ол
[Химическая формула 188]

5-(Бензилокси)-6-нитро-1Н-индол (400 мг, 1,49 ммоля), описываемый в примере получения 145-4, суспендировали в метаноле (10 мл) и тетрагидрофуране (5 мл), добавляли 10% палладий на угле (содержание воды 50%) (159 мг) при комнатной температуре и смесь перемешивали в течение 50 минут в атмосфере водорода. Метанол добавляли в реакционную смесь и полученную смесь фильтровали через целит. Фильтрат концентрировали в вакууме, а затем полученное твердое вещество промывали диэтиловым эфиром и этилацетатом с получением названного соединения (168 мг, 76%).
1Н-ЯМР спектр (DMSO-d6) δ (ppm): 4,31 (2H, brs), 6,05 (1H, t, J=2,0 Гц), 6,59 (1H, s), 6,74 (1H, s), 6,88 (1H, t, J=2,8 Гц), 8,48 (1H, brs), 10,25 (1H, bra).
[0333] [Пример получения 145-6]
6-(Диметиламино)-1Н-индол-5-ол
[Химическая формула 189]

6-Амино-1H-индол-5-ол (80 мг, 0,54 ммоля), описываемый в примере получения 145-5, растворяли в тетрагидрофуране (5,4 мл), добавляли 36,5% водный раствор формальдегида (122 μл, 1,62 ммоля), натрия триацетоксиборгидрид (343 мг, 1,62 ммоля) и уксусной кислоты (93 μл, 1,62 ммоля) при комнатной температуре и смесь перемешивали в течение 2 часов. Воду и этилацетат добавляли в реакционную смесь для разделения. Водный слой экстрагировали этилацетатом, объединенный органический слой промывали насыщенным солевым раствором, затем сушили над безводным магния сульфатом и фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане и полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 7:3 - 1:4). Целевую фракцию концентрировали в вакууме с получением названного соединения (62,0 мг, 65%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 2,70 (6Н, s), 6,42 (1H, ddd, J=3,0, 2,1, 1,1 Гц), 7,11-7,15 (2Н, m), 7,21 (1H, s), 7,94 (1H, brs).
[0334] Согласно примерам 1-33 иллюстративные соединения, представленные в таблице 15, включающей в себя исходный материал примера 145, синтезировали 6-(диметиламино)-N-метил-5-((2-(4-(пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-1Н-индол-1-карбоксамид из 6-(диметиламино)-1Н-индол-5-ола, описываемого в примере получения 145-6, и N-(4-хлорпиридин-2-ил)ацетамида, описываемого в примере получения 1-5.

[0336] [Пример 148]
5-((2-(4-(1-(2-Гидроксиэтил)-2-оксопиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 190]
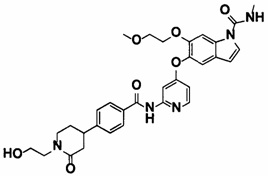
5-((2-Аминопиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1Н-индол-1-карбоксамид (460 мг, 1,29 ммоля), описываемый в примере получения 20-7, 4-(1-(2-((трет-бутилдиметилсилил)окси)этил)-2-оксопиперидин-4-ил)бензойную кислоту (585 мг, 1,55 ммоля), описываемую в примере получения 148-4, и 4-диметиламинопиридин (315 мг, 2,58 ммоля) растворяли в 1,2-диметоксиэтане (6 мл), добавляли триэтиламин (360 μл, 2,58 ммоля) и 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид (495 мг, 2,58 ммоля) и смесь перемешивали при комнатной температуре в течение 1 часа, а затем при 55°С в течение 2 часов. 2 М хлористоводородную кислоту (5 мл, 10,0 ммоля) добавляли в реакционную смесь и смесь перемешивали в течение 3 часов. Этилацетат добавляли в реакционную смесь, полученное вещество нейтрализовали 2 М раствором натрия гидроксида, а затем экстрагировали этилацетатом. Органический слой промывали насыщенным солевым раствором, а затем органический слой сушили над безводным сульфатом магния и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле NH (н-гептан:этилацетат = 1:1 - 0:1 - этилацетат:метанол = 24:1 - 9:1). Целевую фракцию концентрировали в вакууме, затем твердое вещество осаждали с использованием дихлорметана и трет-бутилметилового эфира и твердое вещество отфильтровывали с получением названного соединения (632 мг, 81%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,98-2,10 (1Н, m), 2,11-2,22 (1H, m), 2,54 (1H, dd, J=17,6, 11,0 Гц), 2,71-2,81 (1H, m), 3,06 (3H, d, J=4,4 Гц), 3,15-3,23 (2Н, m), 3,26 (3H, s), 3,39-3,48 (1H, m), 3,49-3,61 (4H, m), 3,63-3,71 (1H, m), 3,84 (2Н, d, J=4,8 Гц), 4,08-4,26 (2Н, m), 5,60 (1H, brs), 6,55 (1H, d, J=3,3 Гц), 6,62 (1H, dd, J=5,7, 2,4 Гц), 7,26-7,35 (4H, m), 7,85 (2Н, d, J=8,4 Гц), 7,91 (1H, d, J=2,2 Гц), 8,02 (1H, s), 8,09 (1H, d, J=5,9 Гц), 8,56 (1H, brs).
[0337] Исходный материал
4-(1-(2-((трет-бутилдиметилсилил)окси)этил)-2-оксопиперидин-4-ил)бензойную кислоту синтезировали следующим способом.
[0338] [Пример получения 148-1]
Трет-бутил 4-(4-(бензилокси)карбонил)фенил)пиперидин-1-карбоксилат
[Химическая формула 191]
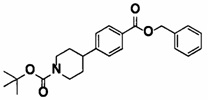
4-(1-(Трет-бутоксикарбонил)пиперидин-4-ил)бензойную кислоту (3,0 г, 9,82 ммоля), описываемую в примере получения 1-12, растворяли в N,N-диметилформамиде (15 мл), затем добавляли калия карбонат (1,63 г, 11,8 ммоля) и бензилбромид (1,29 мл, 10,8 ммоля) и смесь перемешивали при комнатной температуре в течение 24 часов. Воду и этилацетат добавляли в реакционную смесь для разделения. Водный слой экстрагировали этилацетатом и объединенный органический слой промывали насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 2:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (3,83 г, 99%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,48 (9H, s), 1,58-1,70 (2H, m), 1,82 (2H, d, J=12,4 Гц), 2,56-2,96 (3Н, m), 4,17-4,39 (2H, m), 5,36 (2H, s), 7,25-7,29 (3H, m), 7,31-7,50 (4H, m), 8,02 (2H, d, J=8,4 Гц).
[0339] [Пример получения 148-2]
Бензил-4-(2-оксопиперидин-4-ил)бензоат
[Химическая формула 192]
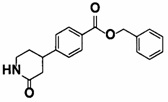
Трет-бутил-4-(4-((бензилокси)карбонил)фенил)пиперидин-1-карбоксилат (3,83 г, 9,68 ммоля), описываемый в примере получения 148-1, растворяли в этилацетате (90 мл), затем рутения оксида (IV) гидрат (44,0 мг, 0,29 ммоля) растворяли в воде (270 мл), добавляли натрия периодат (8,29 г, 38,7 ммоля) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 18 часов. Этилацетат добавляли в реакционную смесь для разделения, а затем органический слой промывали насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния и фильтровали. Растворитель выпаривали, полученный остаток растворяли в дихлорметане (30 мл), затем добавляли трифторуксусную кислоту (15 мл, 202 ммоля) и смесь перемешивали при комнатной температуре в течение 1 часа. Полученное вещество концентрировали в вакууме и насыщенный водный раствор натрия карбоната добавляли к остатку Этилацетат добавляли для разделения, а затем органический слой промывали насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния и фильтровали. Растворитель выпаривали и остаток промывали диэтиловым эфиром с получением названного соединения (2,03 г, 68%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 1,88-2,05 (1H, m), 2,06-2,17 (1H, m), 2,41-2,58 (1H, m), 2,64-2,78 (1H, m), 3,10-3,26 (1H, m), 3,33-3,52 (2H, m), 5,37 (2H, s), 5,71-5,83 (1H, m), 7,27-7,50 (7H, m), 8,01-8,10 (2H, m).
[0340] [Пример получения 148-3]
Бензил-4-(1-(2-((трет-бутилдиметилсилил)окси)этил)-2-оксопиперидин-4-ил)бензоат
[Химическая формула 193]
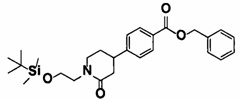
Бензил-4-(2-оксопиперидин-4-ил)бензоат (1,3 г, 4,20 ммоля), описываемый в примере получения 148-2, растворяли в диметилсульфоксиде (20 мл), затем добавляли 50-72% маслянистый натрия гидрид (218 мг) при комнатой температуре в атмосфере азота и смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавляли трет-бутил(2-йодэтокси)диметилсилан (1,92 г, 6,27 ммоля), растворенный в диметилсульфоксиде (10 мл) и смесь перемешивали при комнатной температуре в течение 20 часов. Насыщенный водный раствор аммония хлорида и этилацетат добавляли в реакционную смесь при 0°С для разделения. Водный слой экстрагировали этилацетатом и объединенный органический слой промывали насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 10:1 - 1:2). Целевую фракцию концентрировали в вакууме с получением названного соединения (920 мг, 47%).
1Н-ЯМР спектр (CDCl3) δ (ppm): 0,06 (6H, d, J=1,8 Гц), 0,89 (9H, s), 1,87-2,03 (1H, m), 2,07-2,18 (1H, m), 2,50 (1H, dd, J=17,4, 11,2 Гц), 2,63-2,81 (1H, m), 3,09-3,20 (1H, m), 3,46-3,59 (4H, m), 3,82 (2H, t, J=5,3 Гц), 5,36 (2Н, s), 7,27-7,50 (7H, m), 8,01-8,08 (2H, m).
[0341] [Пример получения 148-4]
4-(1-(2-((Трет-бутилдиметилсилил)окси)этил)-2-оксопиперидин-4-ил)бензойная кислота
[Химическая формула 194]

Бензил-4-(1-(2-((трет-бутилдиметилсилил)окси)этил)-2-оксопиперидин-4-ил) бензоат (920 мг, 1,97 ммоля), описываемый в примере получения 148-3, растворяли в метаноле (20 мл), затем добавляли 10% палладий на угле (содержание воды 50%) (209 мг) и смесь перемешивали в атмосфере водорода в течение 3 часов. Внутреннюю среду реакционной системы замещали азотом, затем катализатор отфильтровывали через целит и полученное вещество промывали метанолом. Фильтрат концентрировали в вакууме и полученное вещество очищали с помощью колоночной хроматографии на силикагеле (н-гептан:этилацетат = 2:1 - 0:1). Целевую фракцию концентрировали в вакууме с получением названного соединения (592 мг, 80%).
1Н-ЯМР спектр (СDCl3) δ (ppm): 0,07 (6Н, d, J=1,5 Гц), 0,90 (9Н, s), 1,96-2,02 (1H, m), 2,10-2,16 (1H, m), 2,52 (1H, dd, J=17,6, 11,0 Гц), 2,72-2,80 (1H, m), 3,10-3,24 (1H, m), 3,47-3,60 (4H, m), 3,83 (2H, t, J=5,5 Гц), 7,31 (2H, d, J=8,1 Гц), 8,06 (2H, d, J=8,4 Гц).
[0342] Согласно примерам 1-33 иллюстративное соединение, представленное в таблице 16, синтезировали из 5-((2-аминопиридин-4-ил)окси)-6-(2-этоксиэтокси)-N-метил-1Н-индол-1-карбоксамида, описываемого в примере получения 24-8.
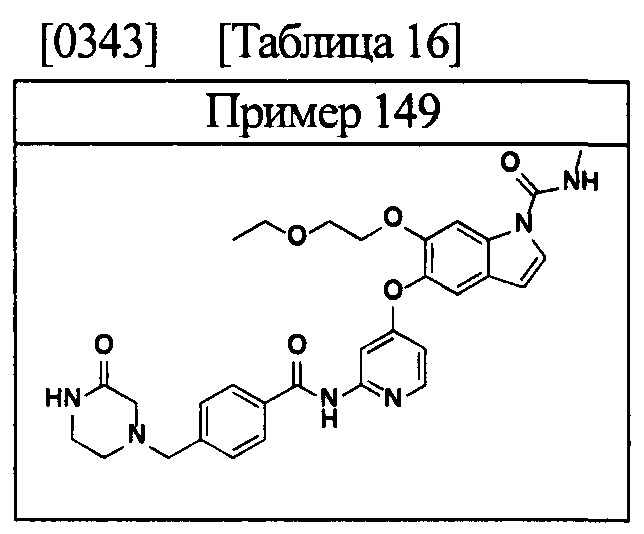
[0344] [Пример 150]
5-((2-(((4-(Цис-1-(2-гидроксиэтил)-1-оксидпиперидин-4-ил)фенил)карбонил)амино)пиридин-4-ил)окси)-6-(2-метоксизтокси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 195]

3-Хлорпероксибензойную кислоту (229 мг, 0,864 ммоля) (чистота: 65%) добавляли в смесь 5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1Н-индол-1-карбоксамида (508 мг, 0,864 ммоля), описываемого в примере 22, и дихлорметана (25 мл) в атмосфере азота и смесь перемешивали при комнатной температуре в течение 30 минут. Этилацетат и 10% водный раствор натрия сульфита добавляли в реакционную смесь для разделения. Водный слой насыщали натрия хлоридом и экстрагировали с использованием смешанного растворителя из этилацетата, тетрагидрофурана и метанола. Объединенный органический слой сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали и полученный остаток растворяли в метаноле и дихлорметане, а полученное вещество очищали с помощью колоночной хроматографии на силикагеле NH (хлороформ:метанол = 49:1 - 4:1). Целевую фракцию концентрировали в вакууме, а затем осажденное твердое вещество растворяли в этилацетате, тетрагидрофуране и метаноле. Осажденное твердое вещество отфильтровывали и промывали этилацетатом с получением фильтрата. Фильтрат концентрировали в вакууме (168 мг), суспендировали в тетрагидрофуране, этилацетате и изопропаноле и полученное вещество перемешивали при 60°С в течение 20 минут. Полученное вещество охлаждали до комнатной температуры, осажденное твердое вещество отфильтровывали и промывали этилацетатом с получением фильтрата. Фильтрат концентрировали в вакууме (62,4 мг) и часть фильтрата (10,4 мг) очищали ODS колоночной хроматографией (0,5% трифторуксусная кислота/вода:ацетонитрил = 37:13) для разделения ее на цис-форму (время удержания = 73-75 минут) и транс-форму (время удержания = 65-67 минут). Туда добавляли буферы, после экстракции с бутанолом, растворители концентрировали в вакууме, и полученное вещество очищали ODS колоночной хроматографией (метанол:вода = 1:9, ацетонитрил:вода = 1:9, последовательно элюировали ацетонитрилом и метанолом) с получением цис-формы названного соединения (1,1 мг) в виде высокополярного компонента и низкополярного компонента из примера 151.
цис-форма
1Н-ЯМР спектр (600 МГц, DMSO-d6) δ (ppm): 1,68 (2H, d, J=13,0 Гц), 2,32-2,63 (2H, m), 2,72-2,81 (1H, m), 2,85 (3H, d, J=4,2 Гц), 3,06-3,56 (11Н, m), 3,86-3,97 (2H, m), 4,03-4,14 (2H, m), 6,63 (1H, d, J=3,5 Гц), 6,67 (1H, dd, J=5,7, 2,4 Гц), 7,37 (2H, d, J=8,2 Гц), 7,45 (1H, s), 7,64-7,72 (1H, m), 7,75-7,82 (1H, m), 7,93 (2H, d, J=8,2 Гц), 8,08 (1H, s), 8,11-8,24 (2H, m), 10,65 (1H, s).
[0345] [Пример 151]
5-((2-(((4-(Транс-1-(2-гидроксиэтил)-1-оксидпиперидин-4-ил)фенил)карбонил)амино)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 196]

Трансформу (1,3 мг) получали в виде низкополярного компонента из примера 150.
трансформа
1Н-ЯМР спектр (600 МГц, DMSO-d6) δ (ppm): 1,85-2,03 (4H, m), 2,85 (3H, d, J=4,2 Гц), 2,90-3,02 (1H, m), 3,05-3,63 (11Н, m), 3,72-3,91 (2H, m), 3,99-4,18 (2H, m), 6,63 (1H, d, J=3,5 Гц), 6,68 (1H, dd, J=5,8, 2,1 Гц), 7,34-7,53 (3H, m), 7,68 (1H, d, J=2,1 Гц), 7,78 (1H, d, J=3,5 Гц), 7,92 (2H, d, J=8,2 Гц), 8,08 (1H, s), 8,12-8,25 (2H, m), 10,65 (1H, s).
[0346] [Пример 152]
6-(2-Гидроксиэтокси)-5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-N-метил-1Н-индол-1-карбоксамид
[Химическая формула 197]
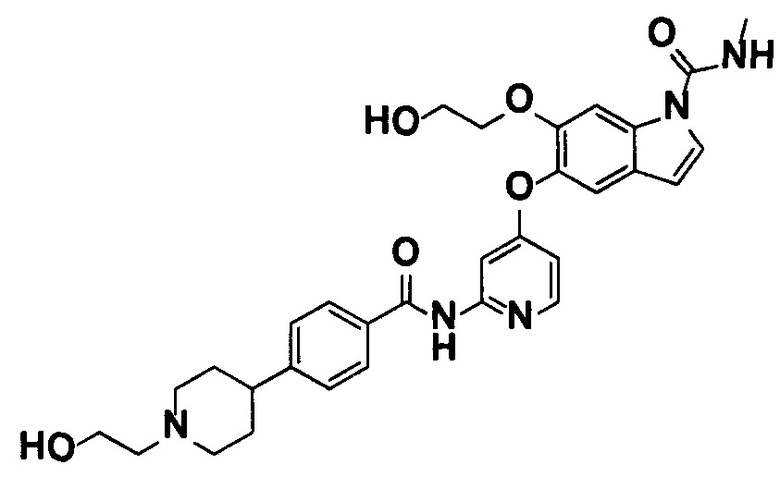
Трехбромистый бор (2,55 мл, 2,55 ммоля) добавляли в смесь 5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1Н-индол-1-карбоксамида (500 мг, 0,851 ммоля), описываемого в примере 22, и дихлорметана (5 мл) при 0°С в атмосфере азота и смесь перемешивали при комнатной температуре в течение 4 часов. Реакционную смесь охлаждали до 0°С, а затем метанол и насыщенный водный раствор натрия бикарбоната добавляли для разделения. Водный слой экстрагировали дихлорметаном, затем органические слои объединяли и полученное вещество промывали насыщенным солевым раствором. Органический слой сушили над безводным натрия сульфатом и фильтровали. Растворитель выпаривали и полученный остаток очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол = 1:0-5:1). Целевую фракцию концентрировали в вакууме и полученное твердое вещество промывали дихлорметаном с получением названного соединения (97 мг, 20%).
1Н-ЯМР спектр (DMSO-d6) δ (ppm): 1,58-1,79 (4Н, m), 2,00-2,11 (2H, m), 2,41 (2H, t, J=6,4 Гц), 2,52-2,59 (1Н, m), 2,85 (3H, d, J=4,4 Гц), 2,97 (2H, d, J=11,7 Гц), 3,46-3,58 (4Н, m), 3,96-4,01 (2H, m), 4,37 (1Н, t, J=5,3 Гц), 4,72 (1Н, t, J=5,5 Гц), 6,63 (1Н, d, J=3,5 Гц), 6,66 (1Н, m), 7,33 (2H, m), 7,43 (1Н, s), 7,70 (1Н, d, J=2,2 Гц), 7,78 (1Н, d, J=3,7 Гц), 7,85-7,96 (2H, m), 8,10 (1Н, s), 8,13-8,26 (2H, m), 10,65 (1Н, brs).
[0347] Масс-спектры (ESI-MS (масса/заряд)) соединений примеров 34-130, примера 132, примера 134, примеров 136-139, примера 141, примера 142, примера 144, примера 146, примера 147 и примера 149 проиллюстрированы в таблицах 17 и 18.


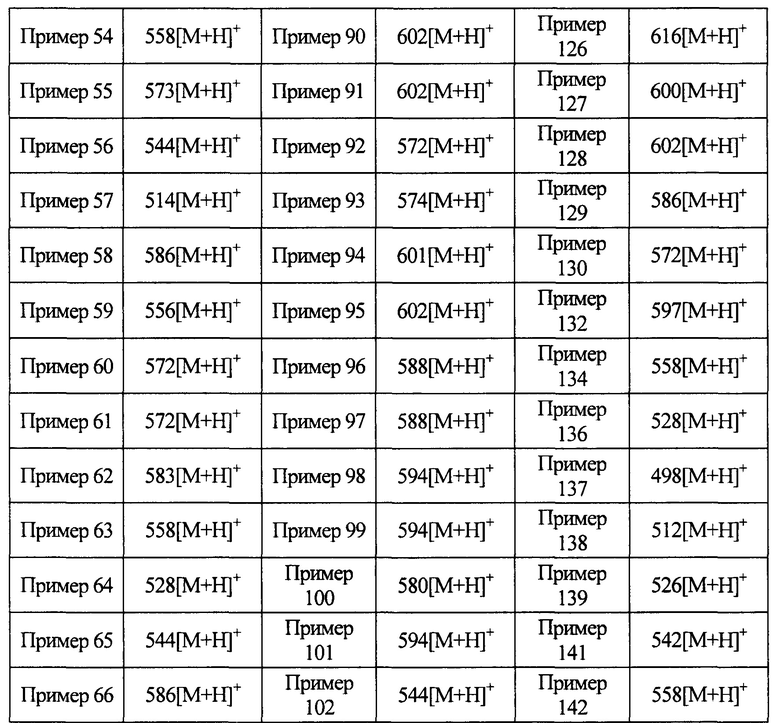

[0350] [Примеры фармакологических тестов]
1. Анализ киназной активности FGFR1
В данном анализе измеряли ингибиторную активность тестируемого вещества против тирозинкиназной активности белка FGFR1.
[0351] В каждую лунку белого 96-луночного планшета с плоским дном (Sumitomo Bakelite Co., Ltd., MS-8496W) добавляли 10 мкл раствора белка FGFR1 (Carna Biosciences, Inc., 08-133), разведенного до 1 мкг/мл с помощью буфера для анализа (20 мМ HEPES-NaOH, 0,01% Triton X-100, 2 мМ DTT и 5 мМ MgCl2), 10 мкл раствора буфера для анализа, содержащего субстрат CSK-tide (Ana Spec Inc., 63843) с конечной концентрацией 1000 нМ и АТР (Promega Corporation, V9102) с конечной концентрацией 58,3 мкМ, и 5 мкл тестируемого вещества, разведенного с помощью буфера для анализа, и реакцию осуществляли при комнатной температуре в течение 1 часа. Для измерения киназной активности использовали анализ киназной активности ADP-Glo (TM) (Promega Corporation, V9102). После окончания реакции в каждую лунку планшета добавляли 25 мкл реагента ADP-Glo, и реакцию осуществляли при комнатой температуре в течение 40 минут, киназную реакцию останавливали и оставшийся АТР удаляли. Далее добавляли реагент для обнаружения киназы и реакцию осуществляли при комнатной температуре в течение 40 минут, для того чтобы вызвать превращение ADP в АТР, сопряженную реакцию с люциферазой/люциферином и люминесцентную реакцию с помощью АТР. Количество люминесценции в каждой лунке измеряли с помощью Envision (TM) (PerkinElmer Co., Ltd.) для оценки активности фермента. Исходя из того, что количество люминесценции, наблюдаемое при добавлении белка-киназы без добавления тестируемого вещества, составляло 100%, и что количество люминесценции, наблюдаемое при отсутствии добавления как тестируемого вещества, так и белка-киназы, составляло 0%, получали показатель количества люминесценции, наблюдаемого в присутствии тестируемого вещества. На основании этого показателя количества люминесценции рассчитывали концентрацию тестируемого вещества, необходимую для ингибирования киназной активности на 50% (т.е. значение IC50), и рассчитанные таким образом значения IC50 для соответствующих тестируемых веществ показаны в таблицах 19, 20 и 21.
[0352] <Данные об ингибиторном действии на киназу FGFR1 вне клеток>

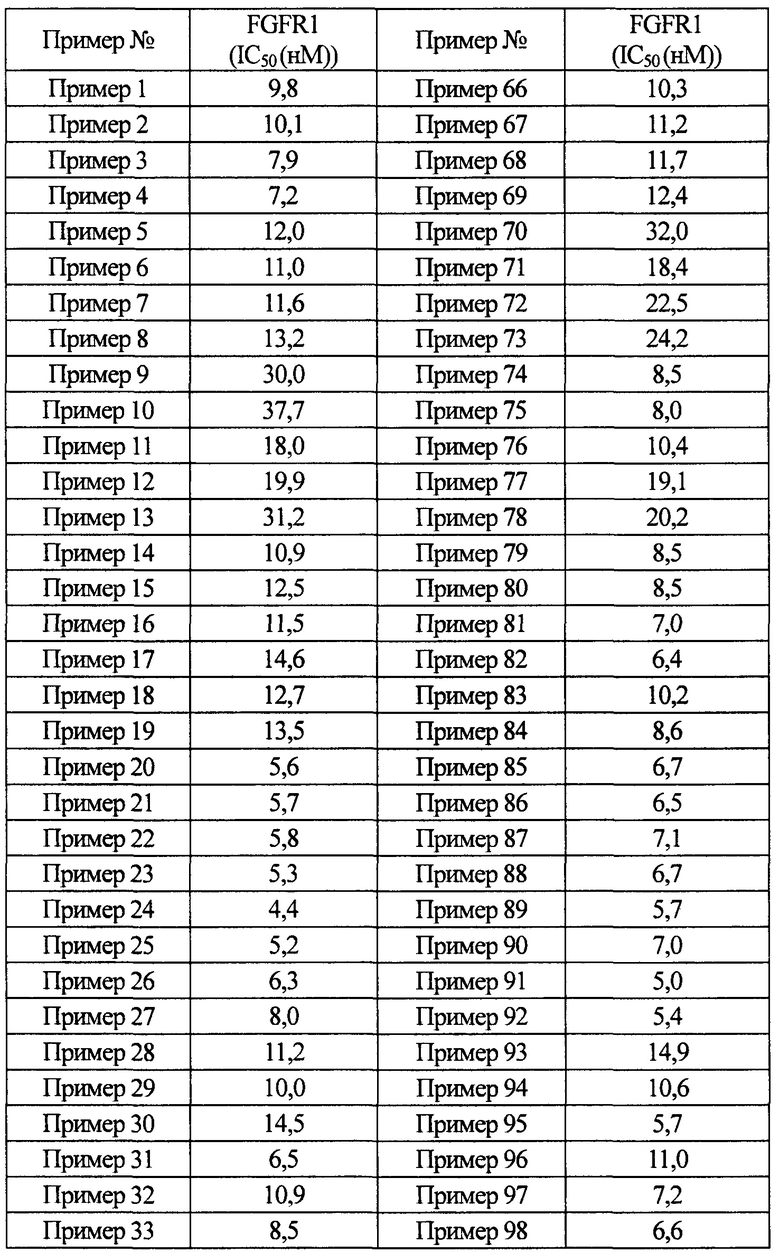



[0355] 2. Анализ киназной активности FGFR2
В данном анализе измеряли ингибиторную активность тестируемого вещества против тирозинкиназной активности белка FGFR2.
[0356] В каждую лунку белого 96-луночного планшета с плоским дном (Sumitomo Bakelite Co., Ltd., MS-8496W) добавляли 10 мкл раствора белка FGFR2 (Carna Biosciences, Inc., 08-134), разведенного до 1 мкг/мл с помощью буфера для анализа (20 мМ HEPES-NaOH, 0,01% Triton X-100, 2 мМ DTT и 5 мМ MgCl3), 10 мкл раствора буфера для анализа, содержащего субстрат CSK-tide (Ana Spec Inc., 63843) с конечной концентрацией 1000 нМ и АТР (Promega Corporation, V9102) с конечной концентрацией 35 мкМ, и 5 мкл тестируемого вещества, разведенного с помощью буфера для анализа, и реакцию осуществляли при комнатной температуре в течение 1 часа. Для измерения киназной активности использовали анализ киназной активности ADP-Glo (TM) (Promega Corporation, V9102). После окончания реакции в каждую лунку планшета добавляли 25 мкл реагента ADP-Glo, и реакцию осуществляли при комнатной температуре в течение 40 минут, киназную реакцию останавливали и оставшийся АТР удаляли. Далее добавляли реагент для обнаружения киназы и реакцию осуществляли при комнатной температуре в течение 40 минут, для того чтобы вызвать превращение ADP в АТР, сопряженную реакцию с люциферазой/люциферином и люминесцентную реакцию с помощью АТР. Количество люминесценции в каждой лунке измеряли с помощью Envision (TM) (PerkinElmer Co., Ltd.) для оценки активности фермента. Исходя из того, что количество люминесценции, наблюдаемое при добавлении белка-киназы без добавления тестируемого вещества, составляло 100%, и что количество люминесценции, наблюдаемое при отсутствии добавления как тестируемого вещества, так и белка-киназы, составляло 0%, получали показатель количества люминесценции, наблюдаемого в присутствии тестируемого вещества. На основании этого показателя количества люминесценции рассчитывали концентрацию тестируемого вещества, необходимую для ингибирования киназной активности на 50% (т.е. значение IC50), и рассчитанные таким образом значения IC50 для соответствующих тестируемых веществ показаны в таблице 22.
[0357] <Данные об ингибиторном действии на киназу FGFR2 вне клеток>
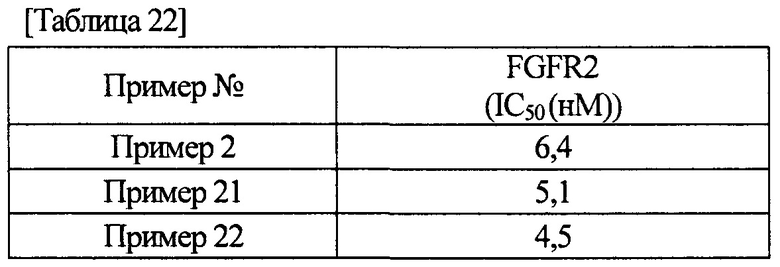
[0358] 3. Анализ киназной активности FGFR3
В данном анализе измеряли ингибиторную активность тестируемого вещества против тирозинкиназной активности белка FGFR3.
[0359] В каждую лунку белого 96-луночного планшета с плоским дном (Sumitomo Bakelite Co., Ltd., MS-8496W) добавляли 10 мкл раствора белка FGFR3 (Carna Biosciences, Inc., 08-135), разведенного до 1 мкг/мл с помощью буфера для анализа (20 мМ HEPES-NaOH, 0,01% Triton X-100, 2 мМ DTT и 5 мМ MgCl2), 10 мкл раствора буфера для анализа, содержащего субстрат CSK-tide (Ana Spec Inc., 63843) с конечной концентрацией 1000 нМ и ATP (Promega Corporation, V9102) с конечной концентрацией 16,7 мкМ, и 5 мкл тестируемого вещества, разведенного с помощью буфера для анализа, и реакцию осуществляли при комнатной температуре в течение 2 часа. Для измерения киназной активности использовали анализ киназной активности ADP-Glo (TM) (Promega Corporation, V9102). После окончания реакции в каждую лунку планшета добавляли 25 мкл реагента ADP-Glo, и реакцию осуществляли при комнатой температуре в течение 40 минут, киназную реакцию останавливали и оставшийся АТР удаляли. Далее добавляли реагент для обнаружения киназы и реакцию осуществляли при комнатной температуре в течение 40 минут, для того чтобы вызвать превращение ADP в АТР, сопряженную реакцию с люциферазой/люциферином и люминесцентную реакцию с помощью АТР. Количество люминесценции в каждой лунке измеряли с помощью Envision (ТМ) (PerkinElmer Co., Ltd.) для оценки активности фермента. Исходя из того, что количество люминесценции, наблюдаемое при добавлении белка-киназы без добавления тестируемого вещества, составляло 100%, и что количество люминесценции, наблюдаемое при отсутствии добавления как тестируемого вещества, так и белка-киназы, составляло 0%, получали показатель количества люминесценции, наблюдаемого в присутствии тестируемого вещества. На основании этого показателя количества люминесценции рассчитывали концентрацию тестируемого вещества, необходимую для ингибирования киназной активности на 50% (т.е. значение IC50), и рассчитанные таким образом значения IC50 для соответствующих тестируемых веществ показаны в таблице 23.
[0360] <Данные об ингибиторном действии на киназу FGFR3 вне клеток>

[0361] 4. Анализ киназной активности FGFR4
В данном анализе измеряли ингибиторную активность тестируемого вещества против тирозинкиназной активности белка FGFR4.
[0362] В каждую лунку белого 96-луночного планшета с плоским дном (Sumitomo Bakelite Co., Ltd., MS-8496W) добавляли 10 мкл раствора белка FGFR4 (Carna Biosciences, Inc., 08-136), разведенного до 1 мкг/мл с помощью буфера для анализа (20 мМ HEPES-NaOH, 0,01% Triton X-100,2 мМ DTT 5 мМ MgCl2 и 2 мМ MnCl2), 10 мкл раствора буфера для анализа, содержащего субстрат CSK-tide (Ana Spec Inc., 63843) с конечной концентрацией 1000 нМ и АТР (Promega Corporation, V9102) с конечной концентрацией 75 мкМ, и 5 мкл тестируемого вещества, разведенного с помощью буфера для анализа, и реакцию осуществляли при комнатной температуре в течение 2 часа. Для измерения киназной активности использовали анализ киназной активности ADP-Glo (ТМ) (Promega Corporation, V9102). После окончания реакции в каждую лунку планшета добавляли 25 мкл реагента ADP-Glo, и реакцию осуществляли при комнатной температуре в течение 40 минут, киназную реакцию останавливали и оставшийся АТР удаляли. Далее добавляли реагент для обнаружения киназы и реакцию осуществляли при комнатной температуре в течение 40 минут, для того чтобы вызвать превращение ADP в АТР, сопряженную реакцию с люциферазой/люциферином и люминесцентную реакцию с помощью АТР. Количество люминесценции в каждой лунке измеряли с помощью Envision (ТМ) (PerkinElmer Co., Ltd.) для оценки активности фермента. Исходя из того, что количество люминесценции, наблюдаемое при добавлении белка-киназы без добавления тестируемого вещества, составляло 100%, и что количество люминесценции, наблюдаемое при отсутствии добавления как тестируемого вещества, так и белка-киназы, составляло 0%, получали показатель количества люминесценции, наблюдаемого в присутствии тестируемого вещества. На основании этого показателя количества люминесценции рассчитывали концентрацию тестируемого вещества, необходимую для ингибирования киназной активности на 50% (т.е. значение IC50), и рассчитанные таким образом значения IC50 для соответствующих тестируемых веществ показаны в таблице 24.
[0363] <Данные об ингибиторном действии на киназу FGFR4 вне клеток>

[0364] 5. Анализ ингибирования роста SNU-16
В данном анализе измеряли активность ингибирования роста в клеточной линии рака желудка человека, характеризующейся амплификацией гена FGFR2, для тестируемого вещества.
[0365] Сообщалось, что клеточная линия рака желудка человека SNU-16 (номер в АТСС CRL-5974) характеризуется амплификацией гена FGFR2 (Cancer Res. 2008. 68: 2340-2348). Клетки SNU-16 культивировали для поддержания в инкубаторе с 5% СО2 (37°С) с использованием среды RPMI-1640 (Wako Pure Chemical Industries, Ltd., 187-02021), содержащей 10% FBS и пенициллин/стрептомицин (Wako Pure Chemical Industries, Ltd., 168-23191). В каждую лунку 96-луночного планшета (Becton, Dickinson and Company, 35-3075) добавляли 150 мкл суспензии клеток SNU-16, доведенной до концентрации 1×104 клеток/мл с использованием среды RPMI-1640, содержащей 10% FBS, и полученное культивировали в течение ночи в инкубаторе с 5% CO2 (37°С). На следующий день 50 мкл тестируемого вещества, разведенного с помощью среды RPMI-1640, содержащей 10% FBS, добавляли в каждую лунку и полученное культивировали в течение 3 дней в инкубаторе с 5% CO2 (37°С). В каждую лунку добавляли десять мкл Cell Counting Kit-8 (Dojindo Laboratories, CK04) и полученное культивировали в течение 1-2 часов в инкубаторе с 5% СO2 (37°С), чтобы вызвать цветную реакцию. ENVISION (TM) (PeridnElmer Co., Ltd.) использовали для измерения поглощения при 450 нм. Исходя из того, что поглощение, наблюдаемое без добавления тестируемого вещества, составляло 100%, и что поглощение, наблюдаемое в лунке, не содержащей клеток, составляло 0%, получали показатель поглощения, наблюдаемого в присутствии тестируемого вещества. Рассчитывали концентрацию тестируемого вещества, необходимую для ингибирования клеточного роста на 50% (т.е. значение IC50), и рассчитанные таким образом значения IC50 соответствующих тестируемых веществ показаны в таблицах 25, 26 и 27.
[0366] <Данные оценки ингибиторного действия на рост SNU-16>



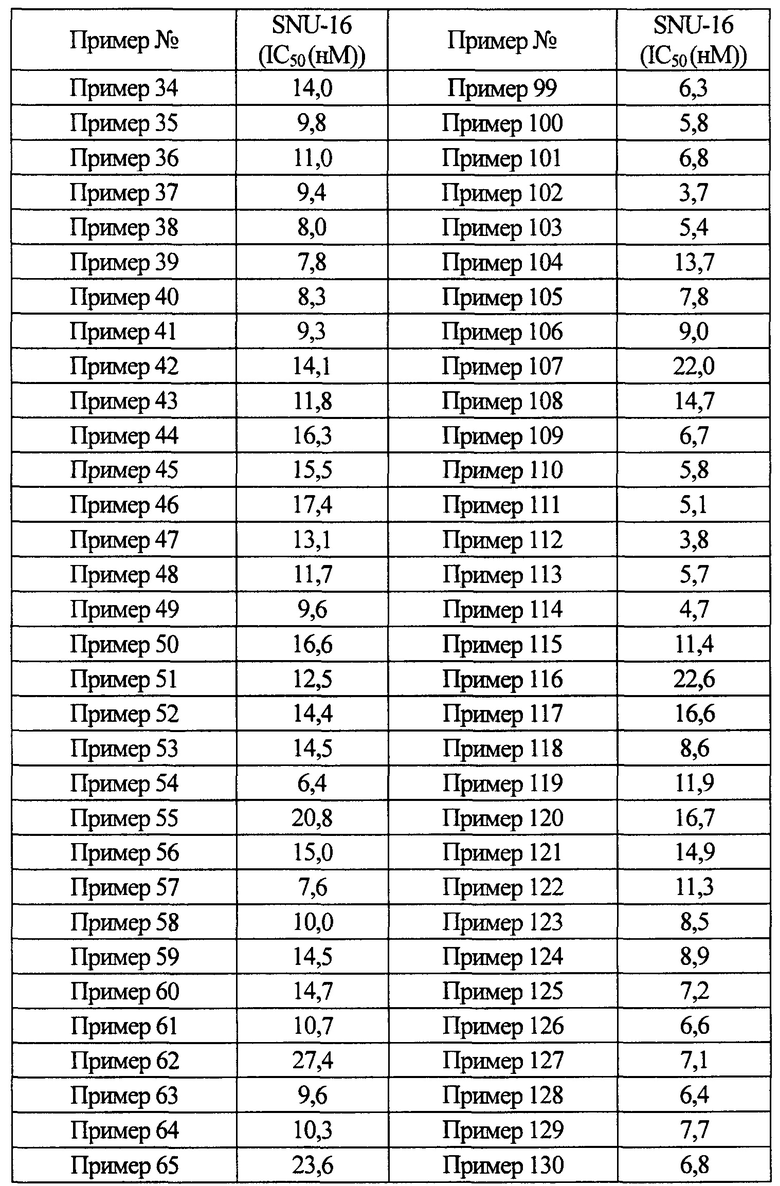


[0369] 6. Анализ ингибирования роста HUVEC
В данном анализе измеряли ингибиторную активность тестируемого вещества против роста эндотелиоцитов васкулярной стенки, индуцированного VEGF.
[0370] Нормальные эндотелиоциты пупочной вены человека (HUVEC) выделяли с помощью опубликованного способа (Shin Seikagaku Jikken Koza "Saibo Baiyo Gijutsu" (New Lectures on Biochemical Experiments "Cell Culture Techniques" (на японском), р. 197-202). Клетки культивировали до конфлюэнтности с использованием среды EGM-2 (LONZA Inc., CC-3162) в инкубаторе с 5% СО2 (37°С). В каждую лунку 96-луночного планшета (Becton, Dickinson and Company, 35-3075) добавляли 100 мкл суспензии клеток HUVEC, которая была доведена до концентрации 1,5×104 клеток/мл с использованием среды EGM-2, содержащей 2% фетальной бычьей сыворотки (FBS: Cell Culture Technologies, CC3008-504), и полученное was культивировали в течение ночи в инкубаторе с 5% CO2 (37°С). На следующий день тестируемое вещество, разведенное с помощью среды EGM-2, содержащей 2% FBS, и 50 мкл VEGF (R & D Systems, 293-VE-010), который был доведен до конечной концентрации 10 нг/мл с использованием среды EGM-2, содержащей 2% FBS, добавляли в каждую лунку и полученное культивировали в течение 3 дней в инкубаторе с 5% СO2 (37°С). В каждую лунку добавляли двадцать мкл Cell Counting Kit-8 (Dojindo Laboratories, CK04) и полученное культивировали в течение 3-4 часов в инкубаторе с 5% СО2 (37°С), чтобы вызвать цветную реакцию. ENVISION (ТМ) (PerkinElmer Co., Ltd.) использовали для измерения поглощения при 450 нм. Исходя из того, что поглощение, наблюдаемое без добавления тестируемого вещества, но с VEGF, составляло 100%, и что поглощение, наблюдаемое в лунке при отсутствии добавления тестируемого вещества и VEGF, составляло 0%, получали показатель поглощения, наблюдаемого в присутствии тестируемого вещества. На основании показателя поглощения рассчитывали концентрацию тестируемого вещества, необходимую для ингибирования роста HUVEC на 50% в присутствии VEGF (т.е. значение IC50), и рассчитанные таким образом значения IC50 values соответствующих тестируемых веществ показаны в таблицах 28, 29 и 30.
[0371] <Данные оценки ингибиторного действия на рост HUVEC>
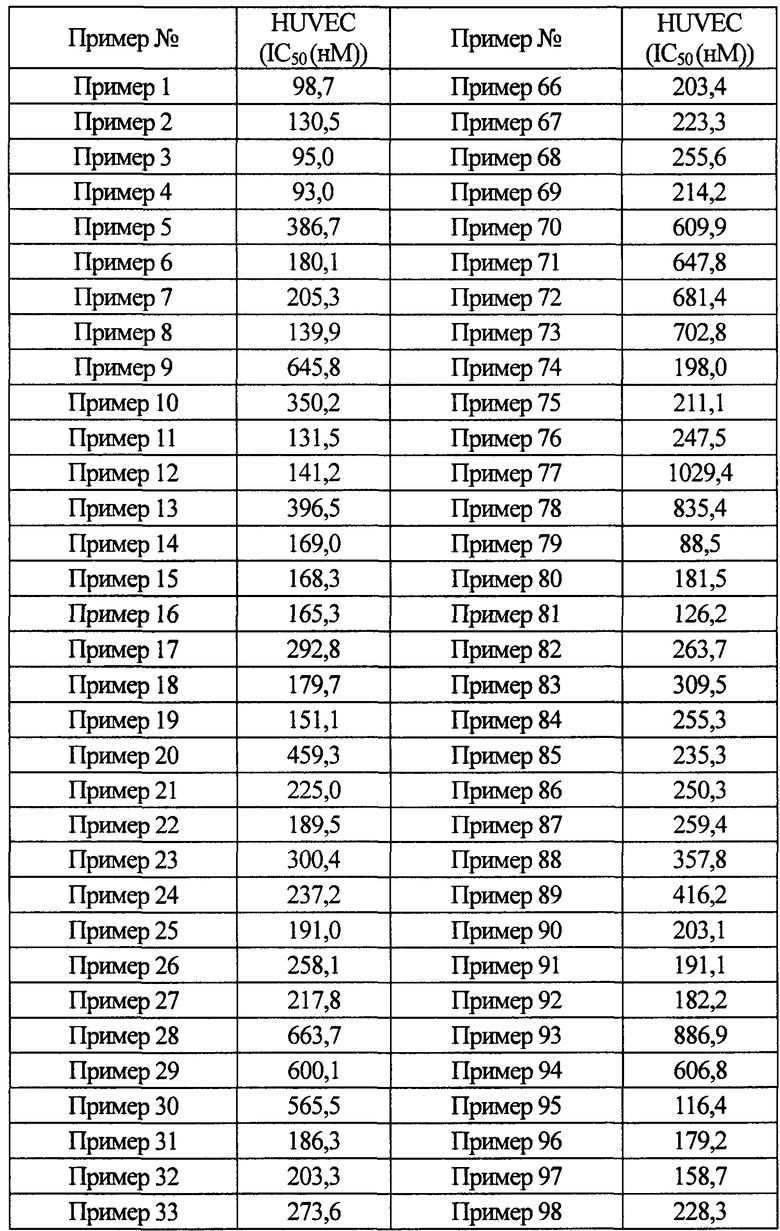

FP 12-0737-00
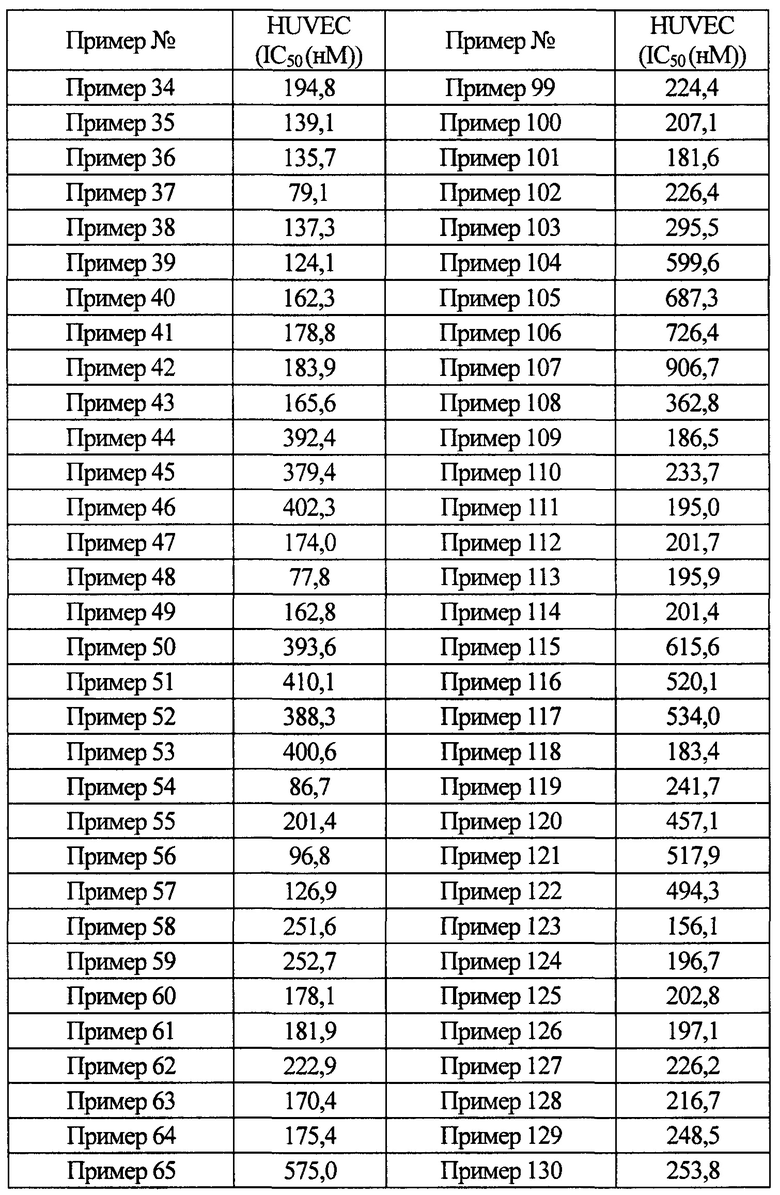

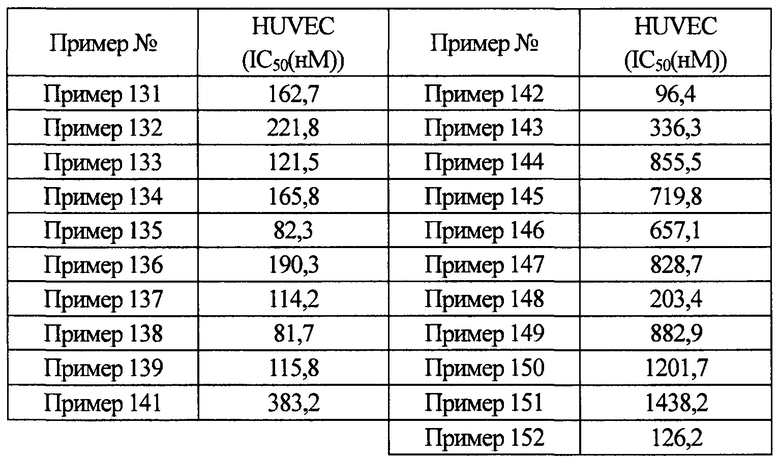
[0374] 7. Анализ ингибирования роста NCI-H1581
В данном анализе измеряли активность ингибирования роста в клеточной линии рака легкого человека с амплификацией гена FGFR1 для тестируемого вещества.
[0375] Сообщалось, что клеточная линия рака легкого человека NCI-H1581 (номер в АТСС CRL-5878) характеризуется амплификацией гена FGFR1 (PLoS One, 2011; 6: e20351, Sci Transl Med 2010; 2: 62ra93). Клетки NCI-H1581 культивировали для поддержания в инкубаторе с 5% CO2 (37°C) с использованием среды RPMI-1640, содержащей 10% FBS и пенициллин/стрептомицин. В каждую лунку 96-луночного планшета (Becton, Dickinson and Company, 35-3075) добавляли 150 мкл суспензии клеток NCI-H1581, доведенной до концентрации 1,3×104 клеток/мл с использованием среды RPMI-1640, содержащей 10% FBS, и полученное культивировали в течение ночи в инкубаторе с 5% СO2 (37°C). На следующий день 50 мкл тестируемого вещества, разведенного с помощью среды RPMI-1640, содержащей 10% FBS, добавляли в каждую лунку и полученное культивировали в течение 3 дней в инкубаторе с 5% CO2 (37°C). В каждую лунку добавляли десять мкл Cell Counting Kit-8 (Dojindo Laboratories, CK04) и полученное культивировали в течение 2-3 часов в инкубаторе с 5% СO2 (37°С), чтобы вызвать цветную реакцию. ENVISION (TM) (PerkinElmer Co., Ltd.) использовали для измерения поглощения при 450 нм. Исходя из того, что поглощение, наблюдаемое без добавления тестируемого вещества, составляло 100%, и что поглощение, наблюдаемое в лунке, не содержащей клеток, составляло 0%, получали показатель поглощения, наблюдаемого в присутствии тестируемого вещества. Рассчитывали концентрацию каждого тестируемого вещества, необходимую для ингибирования клеточного роста на 50% (т.е. значение IC50), и рассчитанные таким образом значения IC50 соответствующих тестируемых веществ показаны в таблице 31.
[0376] <Данные оценки ингибиторного действия на рост NCI-H1581>
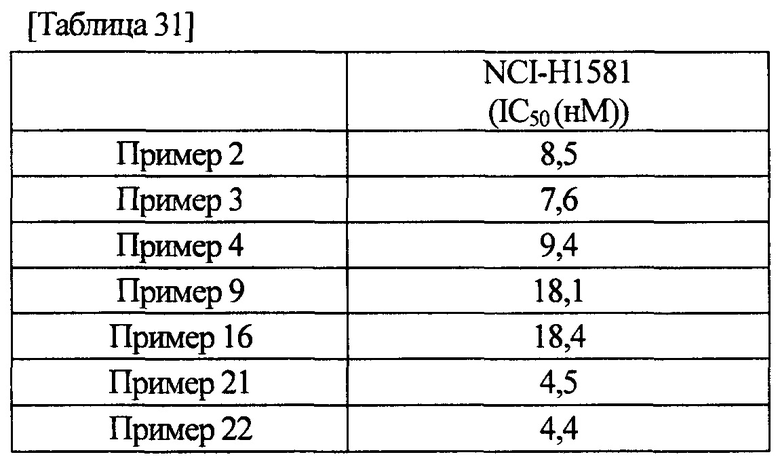
[0377] 8. Противоопухолевой эффект на мышиной модели с подкожно имплантированными SNU-16
Клетки клеточной линии рака желудка человека SNU-16, которые культивировали в среде RPMI-1640, содержащей 10% FBS и пенициллин/стрептомицин, получали в концентрации 1×108 клеток/мл с использованием сбалансированного солевого раствора Хенкса (GIBCO #24020) и полученное смешивали с MATRIGEL (BD Biosciences, № по кат. 354234) в соотношении 1:1 для получения суспензии клеток с концентрацией 5×107 клеток/мл. Суспензию имплантировали в объеме 100 мкл в подкожную часть правого бока каждой "голой" мыши (BALB/cAJcl-nu/nu, самка, Clea Japan Inc.) возрастом 6-7 недель. Через семь дней после имплантации у каждой мыши измеряли самый короткий диаметр и самый длинный диаметр вызванной таким образом опухоли использованием электронного цифрового штангенциркуля (штангенциркуль Digimatic ТМ, Mitutoyo Corporation), для того чтобы рассчитать объем опухоли согласно следующей формуле для расчета:
Объем опухоли (мм3) = самый длинный диаметр (мм)×самый короткий диаметр (мм)×самый короткий диаметр (мм)/2
На основании объемов опухолей, полученных в первый день введения, мышей распределяли по группам, так что средние значения объемов опухоли были практически равными у разных групп. Каждое тестируемое вещество растворяли в DMSO, к нему добавляли Tween 80 для получения раствора с 10-кратной концентрацией и приготовленный таким образом раствор замораживали для хранения. Непосредственно перед введением к нему добавляли 5% раствор глюкозы с получением конечного раствора для введения (в котором соотношение в % для DMSO, Tween 80 и 5% раствора глюкозы составляло 3,5:6,5:90). Каждый образец для оценки вводили перорально с дозой 20 мл/кг один раз в день на протяжении 11 дней, и у контрольной группы при тех же условиях перорально вводили растворитель для введения. В данном случае эксперимент проводили на группах, каждая из которых состояла из 5 мышей.
Для каждой из контрольной группы и групп введения тестируемого вещества рассчитывали соотношение веса, измеренного в последний день, к весу, измеренному в первый день (относительный вес тела: RBW). Если соотношение RBW группы введения тестируемого вещества/RBW контрольной группы составляет 0,9 или более, соответствующую группу введения тестируемого вещества определяли как группу с безопасным введением. У группы введения тестируемого вещества, таким образом определенной как группа с безопасным введением, рассчитывали соотношение (Т/С) (%) объема опухоли, наблюдаемого после введения тестируемого вещества, к объему опухоли у контрольной группы, полученному в последний день, и такие соотношения для соответствующих тестируемых веществ, рассчитанные таким образом, показаны в таблице 32.
[0378] <Данные оценки противоопухолевого эффекта на модели с подкожно имплантированными SNU-16>


[0379] 9. Противоопухолевой эффект на мышиной модели с подкожно имплантированными NCI-H1581
Клетки клеточной линии рака легкого человека NCI-H1581 (номер в АТСС CRL-5878), которые культивировали в среде RPMI-1640, содержащей 10% FBS, пенициллин и стрептомицин, получали в виде суспензии клеток с концентрацией 1×108 клеток/мл с использованием сбалансированного солевого раствора Хенкса (GIBCO #24020). Кроме того, полученную суспензию смешивали с MATRIGEL в соотношении 1:1 с получением суспензии клеток с концентрацией 5×107 клеток/мл. Суспензию клеток имплантировали в объеме 100 мкл в подкожную часть правого бока каждой "голой" мыши (BALB/cAJcl-nu/nu, самка, Clea Japan Inc.) возрастом 6-7 недель. Через десять-одиннадцать дней после имплантации у каждой мыши измеряли самый короткий диаметр и самый длинный диаметр вызванной таким образом опухоли использованием электронного цифрового штангенциркуля (штангенциркуль Digimatic ТМ, Mitutoyo Corporation), для того чтобы рассчитать объем опухоли согласно следующей формуле для расчета:
Объем опухоли (мм3) = самый длинный диаметр (мм)×самый короткий диаметр (мм)×самый короткий диаметр (мм)/2
На основании объемов опухолей, полученных в первый день введения, "голых" мышей распределяли по группам, так что средние значения объемов опухоли были практически равными у разных групп. Каждое тестируемое вещество растворяли в DMSO, к нему добавляли Tween 80 для получения раствора для хранения с 10-кратной концентрацией образца для оценки и приготовленный таким образом раствор замораживали для хранения до применения. Непосредственно перед введением раствор для хранения разбавляли с помощью 5% раствора глюкозы с получением образца для оценки (в котором соотношение в % для DMSO, Tween 80 и 5% раствора глюкозы составляло 3,5:6,5:90). В группе введения тестируемого вещества образец для оценки вводили перорально с дозой 0,4 мл на 20 г веса один раз в день на протяжении 11 дней, и у контрольной группы при тех же условиях перорально вводили растворитель для введения. В данном случае эксперимент проводили на группах, каждая из которых состояла из 5 мышей.
Для каждой из контрольной группы и групп введения тестируемого вещества рассчитывали соотношение веса, измеренного в последний день, к весу, измеренному в первый день (относительный вес тела: RBW). Если соотношение RBW группы введения тестируемого вещества/RBW контрольной группы составляет 0,9 или более, соответствующую группу введения тестируемого вещества определяли как группу с безопасным введением. У группы введения тестируемого вещества, таким образом определенной как группа с безопасным введением, рассчитывали соотношение (Т/С) (%) объема опухоли, наблюдаемого после введения тестируемого вещества, к объему опухоли у контрольной группы, полученному в последний день, и такие соотношения для соответствующих тестируемых веществ, рассчитанные таким образом, показаны в таблице 33.
[0380] <Данные оценки противоопухолевого эффекта на модели с подкожно имплантированными NCI-H1581>
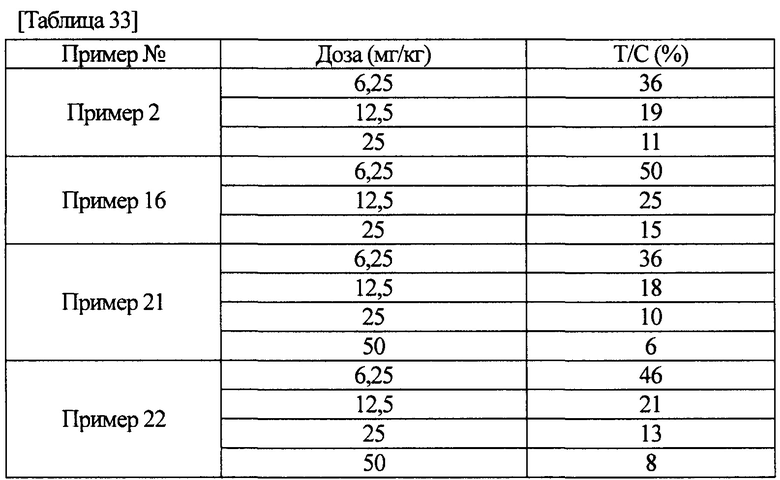
[0381] 10. Анализ ингибирования роста AN3CA
В данном анализе измеряли активность ингибирования роста в клеточной линии рака эндометрия человека, экспрессирующей N549K-вариант FGFR2, для тестируемого вещества.
[0382] Сообщалось, что клеточная линия рака эндометрия человека AN3CA (номер в АТСС НТВ-111) экспрессирует Н549K-вариант FGFR2 (Proc Natl Acad Sci USA, 2008, 105:8713-8717). Клетки AN3CA культивировали для поддержания в инкубаторе с 5% СО2 (37°С) с использованием среды RPMI-1640 (Wako Pure Chemical Industries, Ltd., 187-02021), содержащей 10% FBS, пенициллин и стрептомицин (Wako Pure Chemical Industries, Ltd., 168-23191). В каждую лунку 96-луночного планшета (Becton, Dickinson and Company, 35-3075) добавляли 150 мкл суспензии клеток AN3CA, доведенной до концентрации 1,3×104 клеток/мл с использованием среды RPMI-1640, содержащей 10% FBS, пенициллин и стрептомицин, и клетки культивировали в течение ночи в инкубаторе с 5% СО2 (37°С). На следующий день 50 мкл тестируемого вещества, разведенного с помощью среды RPMI-1640, содержащей 10% FBS, пенициллин и стрептомицин, добавляли в каждую лунку и полученное культивировали в течение 3 дней в инкубаторе с 5% CO2 (37°C). В каждую лунку добавляли десять мкл Cell Counting Kit-8 (Dojindo Laboratories, CK04) и полученное культивировали в течение 1-2 часов в инкубаторе с 5% СО2 (37°С), чтобы вызвать цветную реакцию. ENVISION (TM) (PerkinElmer Co., Ltd.) использовали для измерения поглощения при 450 нм. Исходя из того, что поглощение, наблюдаемое в лунке, содержащей клетки, но не содержащей тестируемое вещество, составляло 100%, и что поглощение, наблюдаемое в лунке, не содержащей клеток, составляло 0%, получали показатель поглощения, наблюдаемого в присутствии тестируемого вещества. Рассчитывали концентрацию тестируемого вещества, необходимую для ингибирования клеточного роста на 50% (т.е. значение IС50), и рассчитанные таким образом значения IC50 соответствующих тестируемых веществ показаны в таблице 34.
[0383] <Данные оценки ингибиторного действия на рост AN3CA>

[0384] 11. Анализ ингибирования роста MFE296
В данном анализе измеряли активность ингибирования роста в клеточной линии рака эндометрия человека, экспрессирующей N549K-вариант FGFR2, для тестируемого вещества.
[0385] Сообщалось, что клеточная линия рака эндометрия человека MFE296 (номер в DSMZ АСС-419) экспрессирует N549К-вариант FGFR2 (Proc Natl Acad Sci USA, 2008, 105:8713-8717). Клетки MFE296 культивировали для поддержания в инкубаторе с 5% СО2 (37°С) с использованием среды RPMI-1640 (Wako Pure Chemical Industries, Ltd., 187-02021), содержащей 10% FBS, пенициллин и стрептомицин (Wako Pure Chemical Industries, Ltd., 168-23191). В каждую лунку 96-луночного планшета (Becton, Dickinson and Company, 35-3075) добавляли 150 мкл суспензии клеток MFE296, доведенной до концентрации 1,3×104 клеток/мл с использованием среды RPMI-1640, содержащей 10% FBS, пенициллин и стрептомицин, и клетки культивировали в течение ночи в инкубаторе с 5% СО2 (37°С). На следующий день 50 мкл тестируемого вещества, разведенного с помощью среды RPMI-1640, содержащей 10% FBS, пенициллин и стрептомицин, добавляли в каждую лунку и полученное культивировали в течение 3 дней в инкубаторе с 5% СО2 (37°С). В каждую лунку добавляли десять мкл Cell Counting Kit-8 (Dojindo Laboratories, CK04) и полученное культивировали в течение 1-2 часов в инкубаторе с 5% СО2 (37°С), чтобы вызвать цветную реакцию. ENVISION (TM) (PerkinElmer Co., Ltd.) использовали для измерения поглощения при 450 нм. Исходя из того, что поглощение, наблюдаемое в лунке, содержащей клетки, но не содержащей тестируемое вещество, составляло 100%, и что поглощение, наблюдаемое в лунке, не содержащей клеток, составляло 0%, получали показатель поглощения, наблюдаемого в присутствии тестируемого вещества. Рассчитывали концентрацию тестируемого вещества, необходимую для ингибирования клеточного роста на 50% (т.е. значение IC50), и рассчитанные таким образом значения IC50 соответствующих тестируемых веществ показаны в таблице 35.
[0386] <Данные оценки ингибиторного действия на рост MFE296>

[0387] 12. Анализ ингибирования роста MFE280
В данном анализе измеряли активность ингибирования роста в клеточной линии рака эндометрия человека, экспрессирующей S252W-вариант FGFR2, для тестируемого вещества.
[0388] Сообщалось, что клеточная линия рака эндометрия человека MFE280 (номер в DMSZ АСС-410) экспрессирует S252W-вариант FGFR2 (Proc Natl Acad Sci USA, 2008, 105:8713-8717). Клетки MFE280 культивировали для поддержания в инкубаторе с 5% СO2 (37°С) с использованием среды RPMI-1640 (Wako Pure Chemical Industries, Ltd., 187-02021), содержащей 10% FBS, пенициллин и стрептомицин (Wako Pure Chemical Industries, Ltd., 168-23191). В каждую лунку 96-луночного планшета (Becton, Dickinson and Company, 35-3075) добавляли 150 мкл суспензии клеток MFE280, доведенной до концентрации 3,3×104 клеток/мл с использованием среды RPMI-1640, содержащей 10% FBS, пенициллин и стрептомицин, и клетки культивировали в течение ночи в инкубаторе с 5% СO2 (37°С). На следующий день 50 мкл тестируемого вещества, разведенного с помощью среды RPMI-1640, содержащей 10% FBS, пенициллин и стрептомицин, добавляли в каждую лунку и полученное культивировали в течение 3 дней в инкубаторе с 5% СO2 (37°С). В каждую лунку добавляли десять мкл Cell Counting Kit-8 (Dojindo Laboratories, CK04) и полученное культивировали в течение 1-2 часов в инкубаторе с 5% СO2 (37°С), чтобы вызвать цветную реакцию. ENVISION (TM) (PerkinElmer Co., Ltd.) использовали для измерения поглощения при 450 нм. Исходя из того, что поглощение, наблюдаемое в лунке, содержащей клетки, но не содержащей тестируемое вещество, составляло 100%, и что поглощение, наблюдаемое в лунке, не содержащей клеток, составляло 0%, получали показатель поглощения, наблюдаемого в присутствии тестируемого вещества. Рассчитывали концентрацию тестируемого вещества, необходимую для ингибирования клеточного роста на 50% (т.е. значение IC50), и рассчитанные таким образом значения IC50 соответствующих тестируемых веществ показаны в таблице 36.
[0389] <Данные оценки ингибиторного действия на рост MFE280>

[0390] 13. Противоопухолевой эффект на мышиной модели с подкожно имплантированными AN3CA
Клетки клеточной линии рака эндометрия человека AN3CA (номер в АТСС НТВ-111), которые культивировали в среде RPMI-1640, содержащей 10% FBS, пенициллин и стрептомицин, получали в виде суспензии клеток с концентрацией 1×108 клеток/мл с использованием сбалансированного солевого раствора Хенкса (GIBCO #24020). Полученную суспензию клеток смешивали с MATRIGEL (BD Biosciences, № по кат. 354234) в соотношении 1:1 с получением суспензии клеток с концентрацией 5×107 клеток/мл. Суспензию клеток имплантировали в объеме 100 мкл в подкожную часть правого бока каждой "голой" мыши (BALB/cAJcl-nu/nu, самка, Clea Japan Inc.) возрастом 7 недель. Через одиннадцать дней после имплантации у каждой мыши измеряли самый короткий диаметр и самый длинный диаметр вызванной таким образом опухоли использованием электронного цифрового штангенциркуля (штангенциркуль Digimatic TM, Mitutoyo Corporation), для того чтобы рассчитать объем опухоли согласно следующей формуле для расчета:
Объем опухоли (мм3) = самый длинный диаметр (мм)×самый короткий диаметр (мм)×самый короткий диаметр (мм)/2
На основании объемов опухолей, полученных в первый день введения, "голых" мышей распределяли по группам, так что средние значения объемов опухоли были практически равными у разных групп. Каждое тестируемое вещество растворяли в DMSO, к нему добавляли Tween 80 для получения раствора для хранения с 10-кратной концентрацией образца для оценки и приготовленный таким образом раствор замораживали для хранения до применения. Непосредственно перед введением раствор для хранения разбавляли с помощью 5% раствора глюкозы с получением образца для оценки (в котором соотношение в % для DMSO, Tween 80 и 5% раствора глюкозы составляло 3,5:6,5:90). В группе введения тестируемого вещества образец для оценки вводили перорально с дозой 0,4 мл на 20 г веса один раз в день на протяжении 14 дней, и у контрольной группы при тех же условиях перорально вводили растворитель для введения. В данном случае эксперимент проводили на группах, каждая из которых состояла из 5 мышей.
Для каждой из контрольной группы и групп введения тестируемого вещества рассчитывали соотношение веса, измеренного в последний день, к весу, измеренному в первый день (относительный вес тела: RBW). Если соотношение RBW группы введения тестируемого вещества/RBW контрольной группы составляет 0,9 или более, соответствующую группу введения тестируемого вещества определяли как группу с безопасным введением. У группы введения тестируемого вещества, таким образом определенной как группа с безопасным введением, рассчитывали соотношение (Т/С) (%) объема опухоли, наблюдаемого после введения тестируемого вещества, к объему опухоли у контрольной группы, полученному в последний день, и такие соотношения для соответствующих тестируемых веществ, рассчитанные таким образом, показаны в таблице 37.
[0391] <Данные оценки противоопухолевого эффекта на мышиной модели с подкожно имплантированными AN3CA>
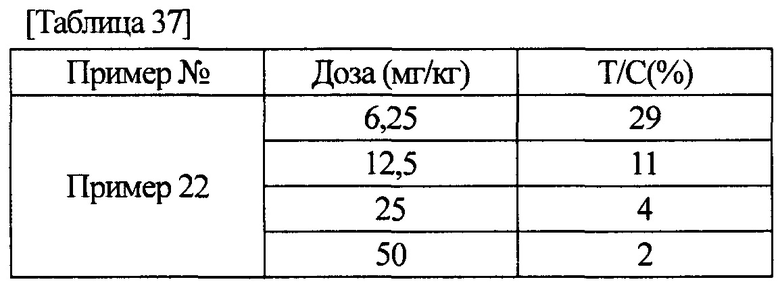
[0392] 14. Противоопухолевой эффект на мышиной модели с подкожно имплантированными MFE296
Клетки клеточной линии рака эндометрия человека MFE296 (номер в DSMZ АСС-419), которые культивировали в среде RPMI-1640, содержащей 10% FBS, пенициллин и стрептомицин, получали в виде суспензии клеток с концентрацией 1×108 клеток/мл с использованием сбалансированного солевого раствора Хенкса (GIBCO #24020). Полученную суспензию клеток смешивали с MATRIGEL (BD Biosciences, № по кат. 354234) в соотношении 1:1 с получением суспензии клеток с концентрацией 5×107 клеток/мл. Суспензию клеток имплантировали в объеме 100 мкл в подкожную часть правого бока каждой "голой" мыши (BALB/cAJcl-nu/nu, самка, Clea Japan Inc.) возрастом 7 недель. Через двенадцать дней после имплантации у каждой мыши измеряли самый короткий диаметр и самый длинный диаметр вызванной таким образом опухоли использованием электронного цифрового штангенциркуля (штангенциркуль Digimatic ТМ, Mitutoyo Corporation), для того чтобы рассчитать объем опухоли согласно следующей формуле для расчета:
Объем опухоли (мм3) = самый длинный диаметр (мм)×самый короткий диаметр (мм)×самый короткий диаметр (мм)/2
На основании объемов опухолей, полученных в первый день введения, "голых" мышей распределяли по группам, так что средние значения объемов опухоли были практически равными у разных групп. Каждое тестируемое вещество растворяли в DMSO, к нему добавляли Tween 80 для получения раствора для хранения с 10-кратной концентрацией образца для оценки и приготовленный таким образом раствор замораживали для хранения до применения. Непосредственно перед введением раствор для хранения разбавляли с помощью 5% раствора глюкозы с получением образца для оценки (в котором соотношение в % для DMSO, Tween 80 и 5% раствора глюкозы составляло 3,5:6,5:90). В группе введения тестируемого вещества образец для оценки вводили перорально с дозой 0,4 мл на 20 г веса один раз в день на протяжении 14 дней, и у контрольной группы при тех же условиях перорально вводили растворитель для введения. В данном случае эксперимент проводили на группах, каждая из которых состояла из 5 мышей.
Для каждой из контрольной группы и групп введения тестируемого вещества рассчитывали соотношение веса, измеренного в последний день, к весу, измеренному в первый день (относительный вес тела: RBW). Если соотношение RBW группы введения тестируемого вещества/RBW контрольной группы составляет 0,9 или более, соответствующую группу введения тестируемого вещества определяли как группу с безопасным введением. У группы введения тестируемого вещества, таким образом определенной как группа с безопасным введением, рассчитывали соотношение (Т/С) (%) объема опухоли, наблюдаемого после введения тестируемого вещества, к объему опухоли у контрольной группы, полученному в последний день, и такие соотношения для соответствующих тестируемых веществ, рассчитанные таким образом, показаны в таблице 38.
[0393] <Данные оценки противоопухолевого эффекта на мышиной модели с подкожно имплантированными MFE296>
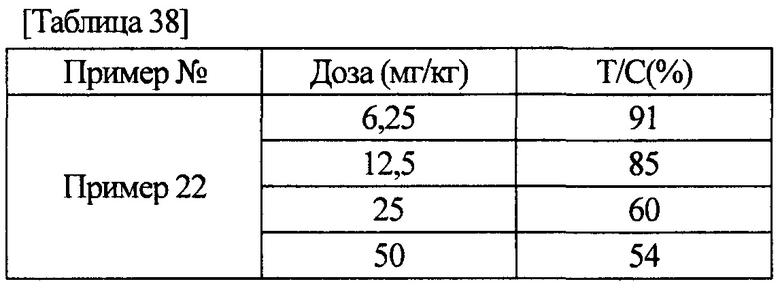
[0394] 15. Противоопухолевой эффект на мышиной модели с подкожно имплантированными MFE280
Клетки клеточной линии рака эндометрия человека MFE280 (номер в DMSZ АСС-410), которые культивировали в среде RPMI-1640, содержащей 10% FBS, пенициллин и стрептомицин, получали в виде суспензии клеток с концентрацией 4,7×107 клеток/мл с использованием сбалансированного солевого раствора Хенкса (GIBCO #24020). Полученную суспензию клеток смешивали с MATRIGEL (BD Biosciences, № по кат. 354234) в соотношении 1:1 с получением суспензии клеток с концентрацией 2,4×107 клеток/мл. Суспензию клеток имплантировали в объеме 100 мкл в подкожную часть правого бока каждой "голой" мыши (BALB/cAJcl-nu/nu, самка, Clea Japan Inc.) возрастом 7 недель. Через тридцать пять дней после имплантации у каждой мыши измеряли самый короткий диаметр и самый длинный диаметр вызванной таким образом опухоли использованием электронного цифрового штангенциркуля (штангенциркуль Digimatic TM, Mitutoyo Corporation), для того чтобы рассчитать объем опухоли согласно следующей формуле для расчета:
Объем опухоли (мм3) = самый длинный диаметр (мм)×самый короткий диаметр (мм)×самый короткий диаметр (мм)/2
На основании объемов опухолей, полученных в первый день введения, "голых" мышей распределяли по группам, так что средние значения объемов опухоли были практически равными у разных групп. Каждое тестируемое вещество растворяли в DMSO, к нему добавляли Tween 80 для получения раствора для хранения с 10-кратной концентрацией образца для оценки и приготовленный таким образом раствор замораживали для хранения до применения. Непосредственно перед введением раствор для хранения разбавляли с помощью 5% раствора глюкозы с получением образца для оценки (в котором соотношение в % для DMSO, Tween 80 и 5% раствора глюкозы составляло 3,5:6,5:90). В группе введения тестируемого вещества образец для оценки вводили перорально с дозой 0,4 мл на 20 г веса один раз в день на протяжении 14 дней, и у контрольной группы при тех же условиях перорально вводили растворитель для введения. В данном случае эксперимент проводили на группах, каждая из которых состояла из 5 мышей.
Для каждой из контрольной группы и групп введения тестируемого вещества рассчитывали соотношение веса, измеренного в последний день, к весу, измеренному в первый день (относительный вес тела: RBW). Если соотношение RBW группы введения тестируемого вещества/RBW контрольной группы составляет 0,9 или более, соответствующую группу введения тестируемого вещества определяли как группу с безопасным введением. У группы введения тестируемого вещества, таким образом определенной как группа с безопасным введением, рассчитывали соотношение (Т/С) (%) объема опухоли, наблюдаемого после введения тестируемого вещества, к объему опухоли у контрольной группы, полученному в последний день, и такие соотношения для соответствующих тестируемых веществ, рассчитанные таким образом, показаны в таблице 39.
[0395] <Данные оценки противоопухолевого эффекта на мышиной модели с подкожно имплантированными MFE280>

[0396] 16. Анализ ингибирования роста RT 112/84
В данном анализе измеряли активность ингибирования роста в клеточной линии рака мочевого пузыря, экспрессирующей слитый белок FGFR3-TACC3, для тестируемого вещества.
[0397] Клетки клеточной линии рака мочевого пузыря человека RT112/84 (номер в ECACC EC85061106-F0) культивировали для поддержания в инкубаторе с 5% СО2 (37°С) с использованием среды RPMI-1640 (Wako Pure Chemical Industries, Ltd., 187-02021), содержащей 10% FBS, пенициллин и стрептомицин (Wako Pure Chemical Industries, Ltd., 168-23191). В каждую лунку 96-луночного планшета (Becton, Dickinson and Company, 35-3075) добавляли 150 мкл суспензии клеток RT112/84, доведенной до концентрации 1,3×104 клеток/мл с использованием среды RPMI-1640, содержащей 10% FBS, пенициллин и стрептомицин, и клетки культивировали в течение ночи в инкубаторе с 5% СO2 (37°С). На следующий день 50 мкл раствора тестируемого вещества, разведенного с помощью среды RPMI-1640, содержащей 10% FBS, пенициллин и стрептомицин, добавляли в каждую лунку и полученное культивировали в течение 3 дней в инкубаторе с 5% СO2 (37°С). Затем в каждую лунку добавляли 10 мкл Cell Counting Kit-8 (Dojindo Laboratories, CK04) и полученное культивировали в течение 1-2 часов в инкубаторе с 5% СO2 (37°С), чтобы вызвать цветную реакцию. ENVISION (TM) (PerkinElmer Co., Ltd.) использовали для измерения поглощения при 450 нм. Исходя из того, что поглощение, наблюдаемое в лунке с суспензией клеток, содержащей клетки, но не содержащей тестируемое вещество, составляло 100%, и что поглощение, наблюдаемое в лунке, не содержащей клеток, составляло 0%, получали показатель поглощения, наблюдаемого в присутствии тестируемого вещества. Рассчитывали концентрацию тестируемого вещества, необходимую для ингибирования клеточного роста на 50% (т.е. значение IC50), и рассчитанные таким образом значения IC50 соответствующих тестируемых веществ показаны в таблице 40.
[0398] <Данные оценки ингибиторного действия на рост RT112/84>

[0399] 17. Анализ ингибирования роста SW780
В данном анализе измеряли активность ингибирования роста в клеточной линии рака мочевого пузыря, экспрессирующей слитый белок FGFR3-BAIAP2L1, для тестируемого вещества.
[0400] Сообщалось, что клеточная линия рака мочевого пузыря человека SW780 (номер в АТСС CRL-2169) экспрессирует слитый белок FGFR3-BAIAP2L1 (Hum Mol Genet. 2013, 22:795-803). Клетки SW780 культивировали для поддержания в инкубаторе с 5% СО2 (37°С) с использованием среды RPMI-1640 (Wako Pure Chemical Industries, Ltd., 187-02021), содержащей 10% FBS, пенициллин и стрептомицин (Wako Pure Chemical Industries, Ltd., 168-23191). В каждую лунку 96-луночного планшета (Becton, Dickinson and Company, 35-3075) добавляли 150 мкл суспензии клеток SW780, доведенной до концентрации 2,6×104 клеток/мл с использованием среды RPMI-1640, содержащей 1% FBS, пенициллин и стрептомицин, и клетки культивировали в течение ночи в инкубаторе с 5% СО2 (37°С). На следующий день 50 мкл тестируемого вещества, разведенного с помощью среды RPMI-1640, содержащей 1% FBS, пенициллин и стрептомицин, добавляли в каждую лунку и полученное культивировали в течение 3 дней в инкубаторе с 5% СО2 (37°С). В каждую лунку добавляли десять мкл Cell Counting Kit-8 (Dojindo Laboratories, CK04) и полученное культивировали в течение 1-2 часов в инкубаторе с 5% СО2 (37°С), чтобы вызвать цветную реакцию. ENVISION (TM) (PerkinElmer Co., Ltd.) использовали для измерения поглощения при 450 нм. Исходя из того, что поглощение, наблюдаемое в лунке, содержащей клетки, но не содержащей тестируемое вещество, составляло 100%, и что поглощение, наблюдаемое в лунке, не содержащей клеток, составляло 0%, получали показатель поглощения, наблюдаемого в присутствии тестируемого вещества. Рассчитывали концентрацию тестируемого вещества, необходимую для ингибирования клеточного роста на 50% (т.е. значение IC50), и рассчитанные таким образом значения IC50 соответствующих тестируемых веществ показаны в таблице 41.
[0401] <Данные оценки ингибиторного действия на рост SW780>

[0402] 18. Анализ ингибирования роста RT4
В данном анализе измеряли активность ингибирования роста в клеточной линии рака мочевого пузыря, экспрессирующей слитый белок FGFR3-TACC3, для тестируемого вещества.
[0403] Сообщалось, что клеточная линия рака мочевого пузыря человека RT4 (номер в АТСС НТВ-2) экспрессирует слитый белок FGFR3-TACC3 (Hum Mol Genet. 2013, 22:795-803). Клетки RT4 культивировали для поддержания в инкубаторе с 5% CO2 (37°С) с использованием среды RPMI-1640 (Wako Pure Chemical Industries, Ltd., 187-02021), содержащей 10% FBS, пенициллин и стрептомицин (Wako Pure Chemical Industries, Ltd., 168-23191). В каждую лунку 96-луночного планшета (Becton, Dickinson and Company, 35-3075) добавляли 150 мкл суспензии клеток RT4, доведенной до концентрации 2,6× 104 клеток/мл с использованием среды RPMI-1640, содержащей 10% FBS, пенициллин и стрептомицин, и клетки культивировали в течение ночи в инкубаторе с 5% СO2 (37°С). На следующий день 50 мкл тестируемого вещества, разведенного с помощью среды RPMI-1640, содержащей 10% FBS, пенициллин и стрептомицин, добавляли в каждую лунку и полученное культивировали в течение 3 дней в инкубаторе с 5% СO2 (37°С). В каждую лунку добавляли десять мкл Cell Counting Kit-8 (Dojindo Laboratories, CK04) и полученное культивировали в течение 1-2 часов в инкубаторе с 5% CO2 (37°С), чтобы вызвать цветную реакцию. ENVISION (TM) (PerkinElmer Co., Ltd.) использовали для измерения поглощения при 450 нм. Исходя из того, что поглощение, наблюдаемое в лунке, содержащей клетки, но не содержащей тестируемое вещество, составляло 100%, и что поглощение, наблюдаемое в лунке, не содержащей клеток, составляло 0%, получали показатель поглощения, наблюдаемого в присутствии тестируемого вещества. Рассчитывали концентрацию тестируемого вещества, необходимую для ингибирования клеточного роста на 50% (т.е. значение IC50), и рассчитанные таким образом значения IC50 соответствующих тестируемых веществ показаны в таблице 42.
[0404] <Данные оценки ингибиторного действия на рост RT4>

[0405] 19. Противоопухолевой эффект на мышиной модели с подкожно имплантированными RT112/84
Клетки клеточной линии рака человека RT112/84 (номер в ЕСАСС EC85061106-F0), которые культивировали в среде RPMI-1640, содержащей 10% FBS, пенициллин и стрептомицин, получали в виде суспензии клеток с концентрацией 1×108 клеток/мл с использованием сбалансированного солевого раствора Хенкса (GIBCO #24020). Полученную суспензию смешивали с MATRIGEL (BD Biosciences, № по кат. 354234) в соотношении 1:1 с получением суспензии клеток с концентрацией 5×107 клеток/мл. Суспензию клеток имплантировали в объеме 100 мкл в подкожную часть правого бока каждой "голой" мыши (BALB/cAJcl-nu/nu, самка, Clea Japan Inc.) возрастом 7 недель. Через десять дней после имплантации у каждой мыши измеряли самый короткий диаметр и самый длинный диаметр вызванной таким образом опухоли использованием электронного цифрового штангенциркуля (штангенциркуль Digimatic TM, Mitutoyo Corporation) для того, чтобы рассчитать объем опухоли согласно следующей формуле для расчета:
Объем опухоли (мм3) = самый длинный диаметр (мм)×самый короткий диаметр (мм)×самый короткий диаметр (мм)/2
На основании объемов опухолей, полученных в первый день введения, "голых" мышей распределяли по группам, так что средние значения объемов опухоли были практически равными у разных групп. Каждое тестируемое вещество растворяли в DMSO, к нему добавляли Tween 80 для получения раствора для хранения с 10-кратной концентрацией образца для оценки и приготовленный таким образом раствор замораживали для хранения до применения. Непосредственно перед введением раствор для хранения разбавляли с помощью 5% раствора глюкозы с получением образца для оценки (в котором соотношение в % для DMSO, Tween 80 и 5% раствора глюкозы составляло 3,5:6,5:90). В группе введения тестируемого вещества образец для оценки вводили перорально с дозой 0,4 мл на 20 г веса один раз в день на протяжении 14 дней, и у контрольной группы при тех же условиях перорально вводили растворитель для введения. В данном случае эксперимент проводили на группах, каждая из которых состояла из 5 мышей.
Для каждой из контрольной группы и групп введения тестируемого вещества рассчитывали соотношение веса, измеренного в последний день, к весу, измеренному в первый день (относительный вес тела: RBW). Если соотношение RBW группы введения тестируемого вещества/RBW контрольной группы составляет 0,9 или более, соответствующую группу введения тестируемого вещества определяли как группу с безопасным введением. У группы введения тестируемого вещества, таким образом определенной как группа с безопасным введением, рассчитывали соотношение (Т/С) (%) объема опухоли, наблюдаемого после введения тестируемого вещества, к объему опухоли у контрольной группы, полученному в последний день, и такие соотношения для соответствующих тестируемых веществ, рассчитанные таким образом, показаны в таблице 43.
[0406] <Данные оценки противоопухолевого эффекта на мышиной модели с подкожно имплантированными RT112/84>

| название | год | авторы | номер документа |
|---|---|---|---|
| СОЛЬ МОНОЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО ПИРИДИНА И ЕЕ КРИСТАЛЛ | 2015 |
|
RU2658821C1 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ОБЕСПЕЧИВАЮЩИЕ РАЗРУШЕНИЕ БЕЛКА IKAROS И БЕЛКА AIOLOS | 2020 |
|
RU2833608C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2667498C2 |
| ЗАМЕЩЕННЫЕ ЦИКЛОГЕКСИЛДИАМИНЫ | 2009 |
|
RU2526251C2 |
| (АЗА)ИНДОЛ-, БЕНЗОТИОФЕН- И БЕНЗОФУРАН-3-СУЛЬФОНАМИДЫ | 2017 |
|
RU2767904C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ ПИРИМИДИН-4(3H)-ОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2839367C1 |
| ПИРРОЛОПИРИМИДИНОВОЕ СОЕДИНЕНИЕ | 2015 |
|
RU2701206C2 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ ПО ОТНОШЕНИЮ К PDE9A, И ИХ ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2019 |
|
RU2788148C2 |
| ПИРИДОНОВЫЕ И АЗАПИРИДОНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2011 |
|
RU2617405C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРРОЛО[3,2-d]ПИРИМИДИН-4-ОНА И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2005 |
|
RU2577858C2 |
Изобретение относится к соединению, представленному следующей формулой (IA):
 ,
,
в которой n равняется 0-2; A представляет собой C6-10ариленовую группу или C3-5гетероариленовую группу, содержащую один или два атома азота или один атом серы; G представляет собой одинарную связь, атом кислорода или -CH2-; E представляет собой содержащий азот неароматический C3-5гетероцикл; R1 представляет собой цианогруппу, моно-C1-6алкиламиногруппу, ди-C1-6алкиламиногруппу, C2-6алкильную группу, C1-6алкоксигруппу, необязательно замещенную 1-3 атомами галогена или одной гидроксильной группой, C1-6алкоксиC1-6алкильную группу или C1-6алкоксиC1-6алкоксигруппу; R2 представляет собой атом водорода, атом галогена, гидроксильную группу, C2-6ацильную группу, необязательно замещенную одним заместителем, выбранным из группы S, описанной ниже, C1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, гидроксиC1-6алкильную группу или содержащую азот неароматическую C3-5 гетероциклическую группу; R3 представляет собой атом водорода, оксогруппу, C1-6алкильную группу или C1-6алкоксигруппу; R4 представляет собой C1-6алкильную группу; при условии, что, если E представляет собой азетидиновое кольцо, а R2 или R3 находится на атоме азота в азетидиновом кольце, то R2 или R3 не является атомом водорода; и группа S представляет собой группу, состоящую из гидроксильной группы, моно-C1-6алкиламиногруппы, ди-C1-6алкиламиногруппы, C1-6алкоксигруппы и содержащей азот неароматической C3-5 гетероциклической группы. Изобретение также относится к индивидуальным соединениям, фармацевтической композиции, терапевтическим средствам, способу лечения, ингибитору FGFR и к применению соединения для изготовления терапевтического средства. Технический результат: получены новые соединения формулы (IA), которые обладают ингибиторной активностью по отношению к FGFR и могут быть полезны при лечении рака желудка, карциномы легкого, рака мочевого пузыря или рака эндометрия. 13 н. и 12 з.п. ф-лы, 43 табл., 152 пр.
1. Соединение, представленное следующей формулой (IA), или его фармацевтически приемлемая соль:
 ,
,
где
n равняется 0-2;
A представляет собой C6-10ариленовую группу или C3-5гетероариленовую группу, содержащую один или два атома азота или один атом серы;
G представляет собой одинарную связь, атом кислорода или -CH2-;
E представляет собой содержащий азот неароматический C3-5гетероцикл;
R1 представляет собой цианогруппу, моно-C1-6алкиламиногруппу, ди-C1-6алкиламиногруппу, C2-6алкильную группу, C1-6алкоксигруппу, необязательно замещенную 1-3 атомами галогена или одной гидроксильной группой, C1-6алкоксиC1-6алкильную группу или C1-6алкоксиC1-6алкоксигруппу;
R2 представляет собой атом водорода, атом галогена, гидроксильную группу, C2-6ацильную группу, необязательно замещенную одним заместителем, выбранным из группы S, описанной ниже, C1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, гидроксиC1-6алкильную группу или содержащую азот неароматическую C3-5 гетероциклическую группу;
R3 представляет собой атом водорода, оксогруппу, C1-6алкильную группу или C1-6алкоксигруппу;
R4 представляет собой C1-6алкильную группу;
при условии, что, если E представляет собой азетидиновое кольцо, а R2 или R3 находится на атоме азота в азетидиновом кольце, то R2 или R3 не является атомом водорода; и
группа S представляет собой группу, состоящую из гидроксильной группы, моно-C1-6алкиламиногруппы, ди-C1-6алкиламиногруппы, C1-6алкоксигруппы и содержащей азот неароматической C3-5 гетероциклической группы.
2. Соединение или его фармацевтически приемлемая соль по п. 1, представленное следующей формулой (IB):
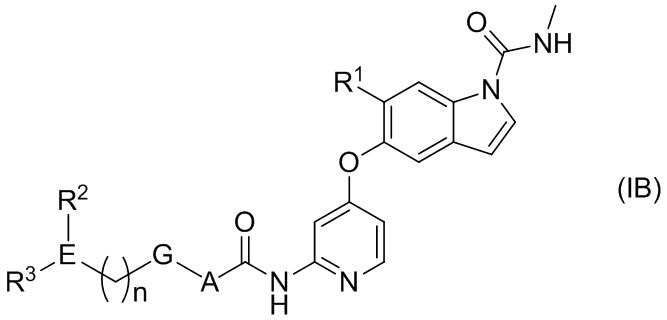 ,
,
где
n равняется 0-2;
A представляет собой C6-10ариленовую группу или C3-5гетероариленовую группу;
G представляет собой одинарную связь, атом кислорода или -CH2-;
E представляет собой содержащий азот неароматический C3-5гетероцикл;
R1 представляет собой C1-6алкоксигруппу, необязательно замещенную 1-3 атомами галогена или одной гидроксильной группой, или C1-6алкоксиC1-6алкоксигруппу;
R2 представляет собой атом водорода, атом галогена, гидроксильную группу, C1-6алкильную группу, необязательно замещенную 1-3 атомами галогена, гидрокси C1-6алкильную группу, или содержащую азот неароматическую C3-5 гетероциклическую группу; и
R3 представляет собой атом водорода, оксогруппу, C1-6алкильную группу или C1-6алкоксигруппу,
при условии, что, если E представляет собой азетидиновое кольцо, а R2 или R3 находится на атоме азота в азетидиновом кольце, то R2 или R3 не является атомом водорода.
3. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, где A представляет собой C6-10ариленовую группу.
4. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, где G представляет собой одинарную связь или атом кислорода.
5. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, где
A представляет собой фениленовую группу, тиениленовую группу, пиразолиленовую группу или пиридиленовую группу; и
E представляет собой азетидиновое кольцо, пирролидиновое кольцо, пиперидиновое кольцо или пиперазиновое кольцо.
6. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, где
A представляет собой фениленовую группу; и
E представляет собой азетидиновое кольцо или пиперидиновое кольцо.
7. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, где
A представляет собой фениленовую группу; и
E представляет собой пиперидиновое кольцо.
8. Соединение или его фармацевтически приемлемая соль по п. 5, где
n равняется 0; и
G представляет собой одинарную связь.
9. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, где
R1 представляет собой C1-6алкоксигруппу или C1-6алкоксиC1-6алкоксигруппу;
R2 представляет собой атом водорода, гидроксильную группу, C1-6алкильную группу или гидроксиC1-6алкильную группу; и
R3 представляет собой атом водорода.
10. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, где R1 представляет собой C1-6алкоксиC1-6алкоксигруппу.
11. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, представленное следующей формулой (II):
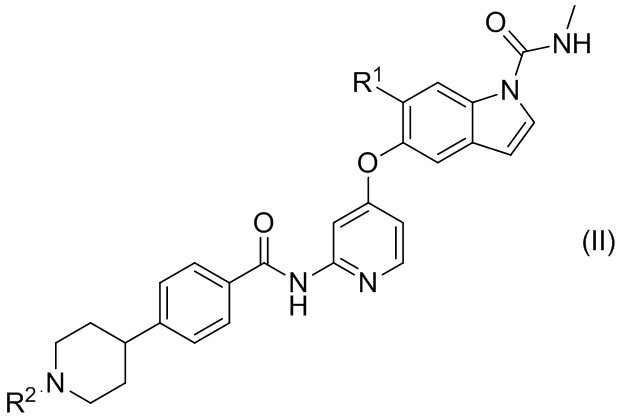 ,
,
где
R1 представляет собой C1-6алкоксиC1-6алкоксигруппу; и
R2 представляет собой атом водорода, C1-6алкильную группу или гидроксиC2-6алкильную группу.
12. Соединение или его фармацевтически приемлемая соль по п. 1 или 2, представленное следующей формулой (III):
 ,
,
где
R1 представляет собой C1-6алкоксиC1-6алкоксигруппу; и
R2 представляет собой C1-6алкильную группу или гидроксиC2-6алкильную группу.
13. 5-((2-(4-(1-Этилпиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-метокси-N-метил-1H-индол-1-карбоксамид, представленный следующей структурной формулой, или его фармацевтически приемлемая соль:
 .
.
14. 6-(2-Метоксиэтокси)-N-метил-5-((2-(4-(1-метилпиперидин-4-ил)бензамид) пиридин-4-ил)окси)-1H-индол-1-карбоксамид, представленный следующей структурной формулой, или его фармацевтически приемлемая соль:
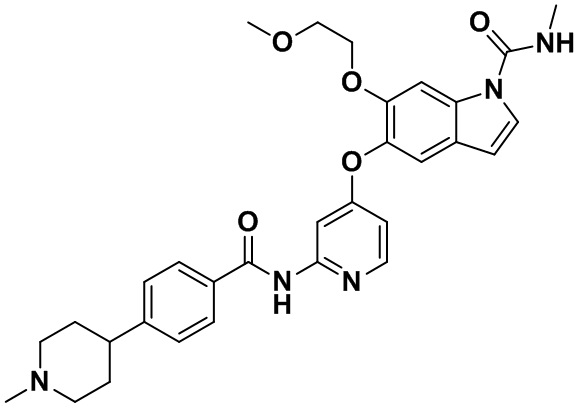 .
.
15. 5-((2-(4-(1-(2-Гидроксиэтил)пиперидин-4-ил)бензамид)пиридин-4-ил)окси)-6-(2-метоксиэтокси)-N-метил-1H-индол-1-карбоксамид, представленный следующей структурной формулой, или его фармацевтически приемлемая соль:
 .
.
16. 6-(2-Этоксиэтокси)-5-((2-(4-(1-(2-гидроксиэтил)пиперидин-4-ил)бензамид) пиридин-4-ил)окси)-N-метил-1H-индол-1-карбоксамид, представленный следующей структурной формулой, или его фармацевтически приемлемая соль:
 .
.
17. 6-(2-Этоксиэтокси)-5-((2-(4-(1-этилазетидин-3-ил)бензамид)пиридин-4-ил) окси)-N-метил-1H-индол-1-карбоксамид, представленный следующей структурной формулой, или его фармацевтически приемлемая соль:
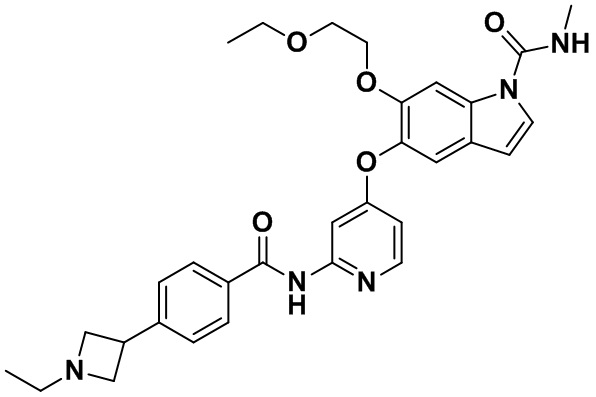 .
.
18. Фармацевтическая композиция для лечения рака желудка, немелкоклеточной карциномы легкого, рака мочевого пузыря или рака эндометрия, содержащая фармакологически эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп. 1-17.
19. Терапевтическое средство против рака желудка, немелкоклеточной карциномы легкого, рака мочевого пузыря или рака эндометрия, содержащее соединение или его фармацевтически приемлемую соль по любому из пп. 1-17 в качестве активного ингредиента.
20. Способ лечения рака желудка, немелкоклеточной карциномы легкого, рака мочевого пузыря или рака эндометрия, включающий введение фармакологически эффективной дозы соединения или его фармацевтически приемлемой соли по любому из пп. 1-17.
21. Терапевтическое средство против немелкоклеточной карциномы легкого, содержащее соединение или его фармацевтически приемлемую соль по любому из пп. 1-17 в качестве активного ингредиента.
22. Терапевтическое средство против плоскоклеточной карциномы легкого, содержащее соединение или его фармацевтически приемлемую соль по любому из пп. 1-17 в качестве активного ингредиента.
23. Ингибитор FGFR для лечения немелкоклеточной карциномы легкого, содержащий соединение или его фармацевтически приемлемую соль по любому из пп. 1-17 в качестве активного ингредиента.
24. Соединение или его фармацевтически приемлемая соль по любому из пп. 1, 2, 13-17 для применения в качестве терапевтического средства против рака желудка, немелкоклеточной карциномы легкого, рака мочевого пузыря или рака эндометрия.
25. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-17 для изготовления терапевтического средства против рака желудка, немелкоклеточной карциномы легкого, рака мочевого пузыря или рака эндометрия.
| АЗОТСОДЕРЖАЩИЕ АРОМАТИЧЕСКИЕ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ СОДЕРЖАЩАЯ, СПОСОБ ЛЕЧЕНИЯ И ПРИМЕНЕНИЕ | 2003 |
|
RU2310651C2 |
| ПРОИЗВОДНЫЕ ИНДОЛ-3-ИЛА | 2001 |
|
RU2257380C2 |
| WO 2009076602 A1, 18.06.2009 | |||
| WO 2013010380 A1, 24.01.2013. | |||

