Настоящее изобретение относится к конденсированным бициклическим производным азаиндазола и диазаиндазола, применяемым при лечении или предупреждении боли, в частности, боли, ассоциированной по меньшей мере с одним белком Trk.
Боль является термином, используемым для описания аномального и болезненного ощущения, испытываемого живым организмом и воспринимаемого головным мозгом. Это ощущение представляет собой неблагоприятный сенсорный и эмоциональный опыт, ассоциированный с повреждением ткани, или описываемый в данный момент или потенциально в отношении такого повреждения. Боль может быть острой или хронической, и обусловлена различными патологиями.
Описано несколько механизмов происхождения боли:
- Ноцицептивная боль: эта боль представляет собой сигнал тревоги в ответ на агрессию против организма, например, боль, вызванная ожогами, травмой, контузией, опоясывающим лишаем, хирургической операцией или удалением зуба. В головной мозг посылается сигнал, чтобы оповестить его об атаке.
- Воспалительная боль обусловлена острым или хроническим воспалением, поскольку она может обнаруживаться при инфекциях, артрите или болезни Крона.
- Невропатическая боль: представляет собой боль вследствие повреждения нервов. Это повреждение вызывает нарушение функции периферической или центральной нервной системы. Невропатическая боль может иметь центральное происхождение, например, вследствие повреждения или инфаркта головного мозга, или представлять собой периферическую невропатическую боль, такую как ишиалгия вследствие позвоночной грыжи.
- Идиопатическая боль: представляет собой болевой синдром, причины которого слабо объяснены. Тесты являются нормальными, но боль присутствует. Такая боль присутствует у пациентов, проявляющих, например, функциональные синдромы, такие как фибромиалгия или синдром раздраженной кишки.
- Психогенная боль: представляет собой боль психологического происхождения (симптомокомплекс, вызванный потерей близкого человека, депрессией, травмой и т.д.). В частности, она представляет собой хроническую боль, обусловленную депрессией.
Боль иногда имеет смешанное происхождение: ноцицептивное и воспалительное, как, например, при раках.
Анальгетики или болеутоляющие средства представляют собой лекарственные средства, применяемые для облегчения боли. Всемирной Организацией Здравоохранения (ВОЗ) они могут быть классифицированы на три уровня:
- Уровень 1 состоит из неопиоидных анальгетиков (парацетамола, противовоспалительных лекарственных средств). Их применяют для слабой или умеренной боли;
- Уровень 2 включает слабые опиоиды (например, кодеин). Их применяют при боли от умеренной до тяжелой степени или в тех случаях, когда болеутоляющие средства уровня 1 были неэффективны для купирования боли;
- Уровень 3 состоит из сильных опиоидов (например, морфина). Эти лекарственные средства применяют для острой боли или в тех случаях, когда анальгетики уровня 2 были неэффективны для купирования боли.
Для лечения определенной боли также применяют другие классы лекарственных средств, такие как нейролептики или антидепрессанты для невропатической боли или триптаны для головных болей по типу мигрени.
Необходимость в поиске новых терапевтических средств для лечения боли неизбежна с учетом ограниченной эффективности или побочных эффектов современных терапевтических средств, независимо от уровня анальгетика ВОЗ, типа или происхождения боли. Например, анальгетики уровня I согласно ВОЗ, включающие среди прочего парацетамол, аспирин, нестероидные противовоспалительные лекарственные препараты (НСПВЛ) и ингибиторы циклооксигеназы, как правило, неэффективны против интенсивной боли, но хорошо переносятся, хотя могут встречаться неблагоприятные желудочные эффекты. Тем не менее, селективные ингибиторы циклооксигеназы типа 2, такие как рофекоксиб (Vioxx®), показали серьезные сердечно-сосудистые риски. Опиаты, такие как морфин и его производные, эффективны в отношении определенных острых болей или тяжелой хронической боли, но могут вызывать сонливость, тошноту и рвоту в начале лечения, а затем констипацию или угнетение дыхания при передозировке. Повторное или хроническое применение опиатов может привести к толерантности к анальгезирующему эффекту, при которой требуются более высокие дозы, близкие к токсическим дозам, зависимости и синдрому отмены при перерыве в лечении. Опиаты и их производные не очень эффективны в отношении невропатической боли и хронической боли от низкой до умеренной интенсивности.
Протеинкиназы представляют собой ферменты, играющие ключевую роль в преобразовании клеточного сигнала. Они вовлечены, например, в такие физиологические процессы, как пролиферация, митоз, дифференцировка, клеточная инвазия, подвижность и апоптоз клеток.
Нарушение регуляции физиологических механизмов, контролируемых протеинкиназами, является центральным фактором для возникновения и развития многих патологий, в частности, включающих раки и боль.
Особый интерес в контексте изобретения представляют рецепторные тирозинкиназы из тропомиозин-родственных киназ (Trk; от англ. "tropomyosine-related kinases"), ассоциированные с острой или хронической болью. Trk представляют собой рецепторные тирозинкиназы, вовлеченные в развитие нервной системы. Семейство рецепторов Trk состоит из трех членов TrkA, TrkB и TrkC, активируемых специфичными лигандами, называемыми нейротрофинами. Описано, что белки Trk и ассоциированные с ними лиганды играют роль при развитии боли (Sah et al. 2003 Nat Rev Drug Discovery. 2: 460-472).
Например, мутации в гене рецептора TrkA описаны у пациентов с врожденной нечувствительностью к боли (Indo et al., 1996, Nat Genet. 13: 485-488; Indo et al., 2001, Hum Mutations, 18: 308-318). TrkA экспрессируется в ноцицептивных нейронах, то есть в нейронах, передающих болевые сигналы, и влияет на электрофизиологические свойства натриевых каналов, вовлеченных в передачу болевых сигналов (Fang et al., J. Neurosci. 25: 4868-4878). В нескольких обзорных статьях освещена роль TrkA и его лиганда, представляющего собой фактор роста нервов (NGF; от англ. "Nerve growth Factor"), при инициации боли на уровне ноцицептивных нейронов, в частности, при воспалительных болевых состояниях, например, при ревматических заболеваниях, таких как остеоартрит, боль в нижней части спины, люмбальная позвоночная грыжа и сдавление нервных корешков (Hefti et al., 2006; Trends Pharmacol Sci. 27: 85-91; Pezet and McMahon, 2006, Ann Rev Neurosci. 29: 507-538; Cheng and Ji, 2008, Neurochem Res. 33: 1970-1978; Seidel et al. 2010, Semin Arthritis Rheum. 40: 109-126).
TrkB или его лиганд, представляющий собой нейротрофический фактор головного мозга (BDNF; от англ. "Brain-Derived Neurotrophic factor"), также вовлечен в хроническую боль. BDNF синтезируется в первичных сенсорных нейронах и антероградно транспортируется в центральные окончания первичных афферентов в спинном мозге (Obata et al., 2006, Neurosci Res. 55: 1-10). BDNF, синтезируемый микроглией в спинном мозге, вызывает сдвиг в нейрональном анионном градиенте, приводящий к растормаживанию передачи боли после повреждения нервов (Coull et al., 2005, Nature, 438: 1017-1021). Этот результат указывает на то, что блокирование биохимического пути BDNF/TrkB полезно для лечения невропатической боли. Супраспинальный BDNF также играет роль при снятии боли (Guo et al., 2006, J Neurosci. 26: 126-137). Повышенная сывороточная концентрация BDNF также описана при фибромиалгии (Laske et al., 2006, J Psychiatric Res, 41: 600-605). Сделан обзор роли передачи сигнала BDNF/TrkB при модулировании боли (Merighi et al., 2008 85: 297-317).
Блокирование биохимического пути TrkA или TrkB может быть достигнуто за счет растворимых рецепторов Trk или нейтрализующих антител. Такие стратегии оценены для биохимического пути NGF/TrkA у животных, где они уменьшали боль, обусловленную раком кости (Sevcik et al., 2005, Pain 115: 128-141), или невоспалительную боль в суставах (McNamee et al., 2010 Pain 149: 386-392). Танезумаб, представляющий собой рекомбинантное гуманизированное моноклональное антитело против NGF, в настоящее время испытывают в качестве терапевтического средства у людей для лечения острой и хронической боли, обусловленной несколькими состояниями (Cattaneo, 2010, Curr Opin Mol Ther 12: 94-106). Тем не менее, такое терапевтическое средство необходимо вводить посредством повторных внутривенных инфузий. Весьма желательны другие формы лечения, такие как низкомолекулярные ингибиторные молекулы Trk с пероральной биодоступностью.
Число ингибиторов Trk, описанных в литературе, ограничено, и до сих пор их еще не применяют в качестве лекарственного средства против боли, хотя для ингибиторов Trk уже продемонстрирована анальгезирующая эффективность в экспериментальных доклинических моделях, в частности, в моделях боли при раке кости (Ghilardi et al. 2010, Mol Pain 6: 87) и хронической воспалительной боли (Winckler et al. 2009, 8th International Association for the Study of Pain Research Symposium, Poster # 348).
Таким образом, существует необходимость в соединениях, способных к ингибированию Trk.
Статья Wang et al. (Expert Opin. Ther. Patents 2009, 19(3), 305-319) представляет собой обзор заявок на патенты с 2002 года, относящихся к ингибиторам Trk и к их применению при лечении рака и боли. Ни одно из раскрытых соединений не соответствует соединению азаиндазола или диазаиндазола.
В WO 2008/112695 раскрыты соединения типа 5-азаиндазола или 5,7-диазаиндазола, замещенного в положении 6, в качестве ингибиторов протеинкиназ, таких как Trk. Тем не менее, в данной заявке отсутствует биологический результат, доказывающий ингибирование каких-либо протеинкиназ и, в частности, протеинкиназы Trk. Кроме того, в данной заявке нигде не указано, что эти соединения могут лечить или предупреждать боль.
В WO 2004/113303 раскрыты, в частности, соединения типа 5-азаиндазола, замещенного в положении 6, в качестве ингибиторов протеинкиназы JNK. Нигде не упомянуто, что такие соединения могли бы также ингибировать протеинкиназу Trk.
В WO 2007/023110 раскрыты, в частности, соединения типа азаиндазола или диазаиндазола в качестве ингибиторов протеинкиназы р38. Нигде не упомянуто, что такие соединения могли бы также ингибировать протеинкиназу Trk.
В WO 2008/089307 раскрыты соединения типа азаиндазола или диазаиндазола в качестве ингибиторов активности Δ5-десатуразы. Нигде не упомянуто, что такие соединения могли бы также ингибировать протеинкиназу Trk.
Соединения по настоящему изобретению обладают свойством ингибирования или модулирования ферментативной активности белков Trk. Следовательно, эти соединения можно применять в качестве лекарственных средств при лечении или предупреждении боли, в частности, боли, ассоциированной по меньшей мере с одним белком Trk.
Кроме того, соединения согласно изобретению представляют особый интерес, если авторы изобретения сумели показать, что они ингибируют или модулируют активность более чем одного белка Trk. Таким образом, они дают возможность лечить одновременно боли нескольких этиологий (например, сочетание воспалительных и ноцицептивных болей, что наблюдают в случае рака).
Таким образом, более конкретно объектом настоящего изобретения является соединение следующей общей формулы (I):

или его фармацевтически приемлемая соль или сольват, его таутомер, его стереоизомер или смесь стереоизомеров в любых соотношениях, такая как смесь энантиомеров, в частности, рацемическая смесь,
где:
- Y1 и Y4 каждый представляет собой, независимо друг от друга, группу СН или атом азота,
- Y2 представляет собой атом азота, либо группу СН или C-X-Ar,
- Y3 представляет собой атом азота, либо группу С-X-Ar или C-W,
при условии, что:
по меньшей мере одна и максимум две группы Y1, Y2, Y3 и Y4 представляют собой атом азота,
Y2 и Y4 не могут представлять собой атом азота одновременно,
когда Y2=C-X-Ar, Y3 представляет собой атом азота или группу C-W, и
когда Y2=CH или N, Y3 представляет собой группу C-X-Ar,
- Ar представляет собой арильную или гетероарильную группу, необязательно замещенную одной или более групп, выбранных из атома галогена, (C1-C6)алкила, (C1-C6)галогеналкила, (С1-С6)галогеналкокси, (C1-C6)галогентиоалкокси, CN, NO2, OR11, SR12, NR13R14, CO2R15, CONR16R17, SO2R18, SO2NR19R20, COR21, NR22COR23, NR24SO2R25 и R26NR27R28, и/или необязательно конденсированную с гетероциклом,
- X представляет собой двухвалентную группу, выбранную из O, S, S(O), S(O)2, NR4, S(NR4), S(O)(NR4), S(O)2(NR4), NR4S, NR4S(O), NR4S(O)2, CH2, CH2S, CH2S(O), CH2S(O)2, SCH2, S(O)CH2, S(O)2CH2, CH2CH2, CH=CH, C≡C, CH2O, OCH2, NR4CH2 и CH2NR4,
- W представляет собой группу R5, SR5, OR5 или NR5R6,
- U представляет собой группу CH2 или NH, один или более атомов водорода которой могут быть замещены (С1-С6)алкильной группой,
- V представляет собой C(O), C(S) или CH2,
- n равно 0 или 1,
- R1 представляет собой атом водорода, либо группу OR7 или NR7R8,
- R2 представляет собой атом водорода, необязательно замещенный гетероцикл, NO2, OR9 или NR9R10,
- R3, R4, R11-R25 и R27-R28 каждый представляет собой, независимо друг от друга, атом водорода или (С1-С6)алкильную группу,
- R5 и R6 каждый представляет собой, независимо друг от друга, атом водорода или (C1-C6)алкил, необязательно замещенную арильную или необязательно замещенную бензильную группу,
- R7, R8, R9 и R10 каждый представляет собой, независимо друг от друга, атом водорода или необязательно замещенную (С1-С6)алкильную или (C3-C12)циклоалкильную группу или необязательно замещенный гетероцикл, и
- R26 представляет собой (С1-С6)алкил,
применяемое при лечении или предупреждении боли, в частности, боли, ассоциированной по меньшей мере с одним белком Trk.
В приведенных выше определениях все комбинации заместителей или переменных возможны, если они приводят к стабильным соединениям.
Кроме того, следует отметить, что по меньшей мере один, но только один из Y2 и Y3 представляет собой группу C-X-Ar.
В предпочтительном воплощении изобретения объектом настоящего изобретения является соединение следующей общей формулы (I):

или его фармацевтически приемлемая соль или сольват, его таутомер, его стереоизомер или смесь стереоизомеров в любых соотношениях, такая как смесь энантиомеров, в частности, рацемическая смесь,
где:
- Y1 и Y4 каждый представляет собой, независимо друг от друга, группу CH или атом азота при условии, что по меньшей мере один из Y1 и Y4 представляет собой атом азота,
- Y2 представляет собой группу C-X-Ar,
- Y3 представляет собой группу C-W,
- Ar представляет собой арильную или гетероарильную группу, необязательно замещенную одной или более групп, выбранных из атома галогена, (С1-С6)алкила, (С1-С6)галогеналкила, (С1-С6)галогеналкокси, (C1-C6)галогентиоалкокси, CN, NO2, OR11, SR12, NR13R14, CO2R15, CONR16R17, SO2R18, SO2NR19R20, COR21, NR22COR23, NR24SO2R25 и R26NR27R28, и/или необязательно конденсированную с гетероциклом,
- X представляет собой двухвалентную группу, выбранную из O, S, S(O), S(O)2, NR4, S(NR4), S(O)(NR4), S(O)2(NR4), NR4S, NR4S(O), NR4S(O)2, CH2, CH2S, CH2S(O), CH2S(O)2, SCH2, S(O)CH2, S(O)2CH2, CH2CH2, CH=CH, C≡C, CH2O, OCH2, NR4CH2 и CH2NR4,
- W представляет собой группу R5, SR5, OR5 или NR5R6,
- U представляет собой группу CH2 или NH, один или более атомов водорода которой могут быть замещены (С1-С6)алкильной группой,
- V представляет собой C(О), C(S) или CH2,
- n равно 0 или 1,
- R1 представляет собой атом водорода, либо группу OR7 или NR7R8,
- R2 представляет собой атом водорода, необязательно замещенный гетероцикл, NO2, OR9 или NR9R10,
- R3, R4, R11-R25 и R27-R28 каждый представляет собой, независимо друг от друга, атом водорода или (С1-С6)алкильную группу,
- R5 и R6 каждый представляет собой, независимо друг от друга, атом водорода или (C1-C6)алкил, необязательно замещенную арильную или необязательно замещенную бензильную группу,
- R7, R8, R9 и R10 каждый представляет собой, независимо друг от друга, атом водорода или необязательно замещенную (С1-С6)алкильную или (C3-C12)циклоалкильную группу, либо необязательно замещенный гетероцикл, и
- R26 представляет собой (С1-С6)алкильную группу,
применяемое при лечении или предупреждении боли.
Термин "атом галогена" относится к атому фтора, хлора, брома или йода.
Термин "(C1-C6) алкил" относится к насыщенным нормальным или разветвленным углеводородным цепям, содержащим от 1 до 6 атомов углерода. Он может представлять собой метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, втор-бутильную, трет-бутильную, пентильную или гексильную группу.
Термин "(С1-С6)алкокси" относится к (С1-С6)алкильной цепи, связанной с остальной частью молекулы через атом кислорода. В качестве примера можно упомянуть метокси, этокси, пропокси, изопропокси, бутокси или трет-бутоксигруппы.
Термин "(C1-C6)тиоалкокси" относится к (С1-С6)алкильной цепи, связанной с остальной частью молекулы через атом серы. В качестве примера можно упомянуть тиометокси, тиоэтокси, тиопропокси, тиоизопропокси, тиобутокси или тио-трет-бутоксигруппы.
Термин "(C1-C6)галогеналкил" относится к (С1-С6)алкильной цепи, как определено выше, где один или более атомов водорода замещено атомом галогена, как определено выше. Он может представлять собой, в частности, тиофторметильную группу.
Термин "(С1-С6)галогеналкокси" относится к цепи (С1-С6)алкокси, как определено выше, где один или более атомов водорода замещено атомом галогена, как определено выше. Он может представлять собой, в частности, тиофторметоксигруппу.
Термин "(С1-С6)галогентиоалкокси" относится к цепи (С1-С6)тиоалкокси, как определено выше, где один или более атомов водорода замещено атомом галогена, как определено выше. Он может представлять собой, в частности, тиофтортиометокси группу.
Термин "(C3-C12)циклоалкил" относится к циклическим углеводородным системам, содержащим от 3 до 12 атомов углерода и содержащим одно или более колец, в частности, конденсированных колец. В качестве примера можно упомянуть адамантильную или циклогексильную группу.
Термин "арил" относится к ароматической углеводородной группе, предпочтительно содержащей от 6 до 14 атомов углерода и содержащей одно или более конденсированных колец, такую как, например, фенильная или нафтильная группа. Предпочтительно он представляет собой фенильную группу.
Термин "гетероарил" относится к циклической ароматической группе, содержащей от 5 до 7 атомов, включенных в кольцо, или к бициклической ароматической группе, содержащей от 8 до 11 атомов, включенных в кольца, где от 1 до 4 атомов, включенных в кольца, представляют собой гетероатом, независимо выбранный из атомов серы, азота и кислорода, и где другие атомы, включенные в кольца, представляют собой атомы углерода. Примеры гетероарильных групп включают фурильные, тиенильные, пиридинильные и бензотиенильные группы.
Термин "гетероцикл" относится либо к стабильному моноциклу, содержащему от 4 до 7 циклических атомов, либо к стабильному бициклу, содержащему от 8 до 11 циклических атомов, который может быть либо насыщенным, либо ненасыщенным, где от 1 до 4 циклических атомов представляют собой гетероатом, независимо выбранный из атомов серы, азота и кислорода, и где другие циклические атомы представляют собой атомы углерода. В качестве примера можно упомянуть фуран, пиррол, тиофен, тиазол, изотиазол, оксадиазол, имидазол, оксазол, изоксазол, пиридин, пиперидин, пиразин, пиперазин, тетрагидропиран, пиримидин, хиназолин, хинолин, хиноксалин, бензофуран, бензотиофен, индолин, индолизин, бензотиазол, бензотиенил, бензопиран, бензоксазол, бензо[1,3]диоксол, бензизоксазол, бензимидазол, хроман, хромен, дигидробензофуран, дигидробензотиенил, дигидроизоксазол, изохинолин, дигидробензо[1,4]диоксан, имидазо[1,2-a]пиридин, фуро[2,3-c]пиридин, 2.3-дигидро-1Н-инден, [1,3]диоксоло[4,5-с]пиридин, пирроло[1,2-c]пиримидин, пирроло[1,2-a]пиримидин, тетрагидронафталин, бензо[b][1,4]оксазин.
В контексте настоящего изобретения "необязательно замещенный" означает, что обсуждаемая группа необязательно замещена одним или более заместителей, которые могут быть выбраны, в частности, из атома галогена, (C1-C6)алкила, (C1-C6)галогеналкила, (С1-С6)галогеналкокси, (С1-С6)галогентиоалкокси, CN, NO2, OR11, SR12, NR13R14, CO2R15, CONR16R17, SO2R18, SO2NR19R20, COR21, NR22COR23, NR24SO2R25 и R26NR27R28, где R11-R28 являются такими, как определено выше.
В настоящем изобретении "фармацевтически приемлемый" относится к тому, что полезно при получении фармацевтической композиции, являющейся в целом безопасной, нетоксичной и ни биологически, ни иначе нежелательной, а также приемлемой для ветеринарного и медицинского фармацевтического применения.
"Фармацевтически приемлемая соль или сольват" соединения относится к солям и сольватам, являющимся фармацевтически приемлемыми, как определено в данном описании, и обладающим желаемой фармацевтической активностью исходного соединения.
Приемлемые соли для терапевтического применения соединений по настоящему изобретению включают традиционные нетоксичные соли соединений по изобретению, таких как соли, образованные из фармацевтически приемлемых органических или неорганических кислот или из фармацевтически приемлемых органических или неорганических оснований. В качестве примера можно упомянуть соли, образованные из неорганических кислот, таких как соляная кислота, бромисто-водородная кислота, фосфорная кислота и серная кислота, и соли, образованные из органических кислот, таких как уксусная кислота, трифторуксусная кислота, пропионовая кислота, янтарная кислота, фумаровая кислота, яблочная кислота, винная кислота, лимонная кислота, аскорбиновая кислота, малеиновая кислота, глутаминовая кислота, бензойная кислота, салициловая кислота, толуолсульфоновая кислота, метансульфоновая кислота, стеариновая кислота и молочная кислота. В качестве примера можно упомянуть соли, образованные из неорганических оснований, таких как сода, поташ или гидроксид кальция, и соли, образованные из органических оснований, таких как лизин или аргинин.
Эти соли можно синтезировать из соединений по изобретению, содержащих основную или кислотную часть, и соответствующих кислот или оснований в соответствии с традиционными химическими способами, хорошо известными специалистам в данной области техники.
Приемлемые сольваты для терапевтического применения соединений по настоящему изобретению включают традиционные сольваты, такие как сольваты, образованные на последней стадии получения соединений по изобретению за счет присутствия растворителей. В качестве примера можно упомянуть сольваты, образованные за счет присутствия воды или этанола.
В контексте настоящего изобретения "стереоизомер" относится к геометрическому изомеру или к оптическому изомеру.
Геометрические изомеры образуются в результате различного положения заместителей на двойной связи и могут, таким образом, иметь Z или Е конфигурацию.
Оптические изомеры образуются, в частности, в результате различного положения в пространстве заместителей на атоме углерода, содержащем четыре различных заместителя. Этот атом углерода составляет, таким образом, хиральный или асимметрический центр. Оптические изомеры включают диастереоизомеры и энантиомеры. Оптические изомеры, являющиеся зеркальными отображениями друг друга, но не налагаемыми друг на друга, представляют собой энантиомеры. Оптические изомеры, не являющиеся зеркальными отображениями друг друга, представляют собой диастереоизомеры.
В контексте настоящего изобретения "таутомер" относится к структурному изомеру соединения, полученному в результате прототропии, то есть в результате миграции атома водорода и изменения положения двойной связи. Различные таутомеры соединения, как правило, взаимно превращаемы и находятся в равновесии в растворе в соотношениях, которые могут варьировать в соответствии с используемым растворителем, температурой или pH.
Согласно первому воплощению изобретения Y4=N.
Предпочтительно Y2=C-X-Ar, и Y3 предпочтительно представляет собой группу C-W.
В частности:
- Y1=CH или N, и предпочтительно СН,
- Y2=C-X-Ar,
- Y3=C-W, и
- Y4=N.
Согласно второму воплощению изобретения Y1 и/или Y4 представляет собой атом азота.
В данном случае Y2 и Y3 предпочтительно представляет собой не атом азота.
В частности:
- Y1 и/или Y4=N,
- Y2=CH или С-X-Ar, и
- Y3=C-W или C-X-Ar.
В частности:
- Y1 представляет собой группу CH,
- Y4 представляет собой атом азота,
- Y2 представляет собой группу CH или C-X-Ar, и
- Y3 представляет собой группу C-X-Ar или C-W,
при условии, что:
когда Y2=C-X-Ar, Y3 представляет собой группу C-W, и
когда Y2=CH, Y2 представляет собой группу C-X-Ar.
Предпочтительно X представляет собой двухвалентную группу, выбранную из O, S, S(O), S(O)2, NR4, CH2, CH2S, CH2S(O), CH2S(O)2, NHS(O)2, SCH2, S(O)CH2, S(O)2CH2, S(O)2NH, CH2CH2, CH=CH, C≡C, CH2O, OCH2, NR4CH2 и CH2NR4.
В частности, X представляет собой двухвалентную группу, выбранную из S, S(O), S(O)2, NR4, CH2, CH2S, CH2S(O), CH2S(O)2, NHS(O)2, SCH2, S(O)CH2, S(O)2CH2, S(O)2NH, CH2CH2, C≡C, CH2O, OCH2, NR4CH2 и CH2NR4.
Более конкретно X может быть выбран из S, S(O), S(O)2, CH2, CH2S, CH2S(O), CH2S(O)2, NHS(O)2, SCH2, S(O)CH2, S(O)2CH2, S(O)2NH, CH2CH2, CH=CH и C≡C.
В частности, X может быть выбран из S, S(O)2, CH2, SCH2, S(O)2CH2, S(O)2NH, CH2S, CH2S(O)2, NHS(O)2, CH2CH2 и C≡C.
X может быть, в частности, выбран из S, S(O), S(O)2, NR4, CH2, SCH2, S(O)CH2, S(O)2CH2, S(O)2NH, CH2CH2, C≡C, OCH2 и NR4CH2; в частности, из S, S(O)2, CH2, SCH2, S(O)2CH2, S(O)2NH, CH2CH2 и C≡C, где первый атом этих групп связан с атомом C цепи C-X-Ar.
X может представлять собой, в частности, S, S(O)2, SCH2, S(O)2CH2, S(O)2NH, CH2S, CH2S(O)2 или NHS(O)2, и, в частности, S, S(O)2, SCH2, S(O)2CH2 или S(O)2NH, где первый атом этих групп связан с атомом C цепи C-X-Ar.
Предпочтительно Ar представляет собой гетероарильную группу, такую как пиридин, или арильную группу, такую как фенил, необязательно замещенную одной или более групп, выбранных из атома галогена, (С1-С6)алкила, (С1-С6)галогеналкила, (С1-С6)галогеналкокси, (С1-С6)галогентиоалкокси, CN, NO2, OR11, SR12, NR13R14, CO2R15, CONR16R17, SO2R18, SO2NR19R20, COR21, NR22COR23 и NR24SO2R25; и/или необязательно конденсированную с гетероциклом.
Более конкретно Ar может представлять собой арильную группу, такую как фенил, необязательно замещенную одной или более групп, выбранных из атома галогена, (С1-С6)алкила, (С1-С6)галогеналкила, (С1-С6)галогеналкокси, (C1-С6)галогентиоалкокси, CN, NO2, OR11, SR12, NR13R14, CO2R15, CONR16R17, SO2R18, SO2NR19R20, COR21, NR22COR23 и NR24SO2R25.
Ar может, в частности, представлять собой арильную группу, такую как фенил, необязательно замещенный одной или более групп, выбранных из атома галогена, (С1-С6)алкила, (С1-С6)галогеналкила и CONR16R17, и, в частности, из атома галогена, такого как атом фтора, (С1-С6)алкила, такого как метил, и CONR16R17, такого как CONH2.
Ar может также представлять собой пиридиновую группу.
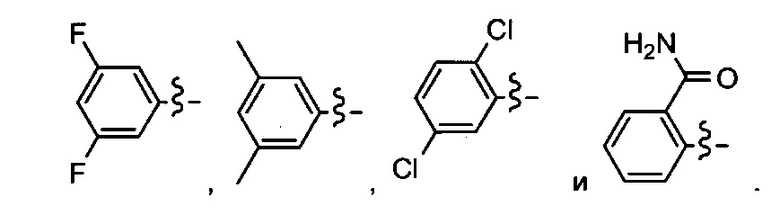
Ar может быть, в частности, выбран из следующих групп:

в частности, из следующих групп:
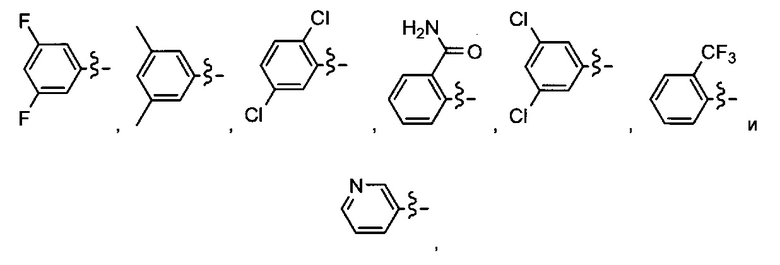
в частности, из следующих групп:
Ar может предпочтительно представлять собой группу:
 .
.
W может предпочтительно представлять собой группу R5, SR5, OR5 или NR5R6, и предпочтительно R5, OR5 или NR5R6, где R5 и R6 представляют собой, независимо друг от друга, атом водорода или (С1-С6)алкильную группу.
W может представлять собой, в частности, Н, OMe, Me, ОН или NH2, и, в частности, Н.
Предпочтительно R3 представляет собой атом водорода.
U может представлять собой более конкретно группу CH2 или NH.
Предпочтительно n может быть равно 0.
V может представлять собой более конкретно группу C(О) или C(S), и предпочтительно группу C(О).
Согласно конкретному воплощению изобретения:
- R3=H,
- U=CH2 или NH,
- V=C(O) или C(S), и, в частности, C(О), и
- n=0 или 1, и, в частности, 0.
Согласно другому конкретному воплощению изобретения:
- V=C(O) или C(S), и, в частности, C(О), и
- n=0.
Согласно другому конкретному воплощению изобретения:
- R3=H,
- V=C(O) или C(S), и, в частности, C(О), и
- n=0.
R1 может представлять собой более конкретно атом водорода или группу NR7R8, где R7, в частности, представляет собой атом водорода, а R8, в частности, представляет собой необязательно замещенную (C3-C12)циклоалкильную группу или необязательно замещенный гетероцикл.
(C3-C12)циклоалкильная группа может представлять собой, в частности, циклогексил. Она может быть замещена одним или более атомов галогена. Эта группа может, в частности, представлять собой группу:
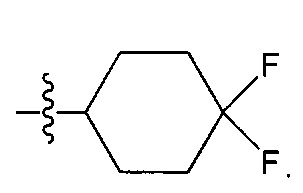
Гетероциклическая группа может представлять собой, в частности, тетрагидропиран, в частности, незамещенный. Таким образом, она может представлять собой следующую группу:
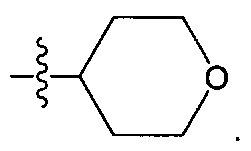
R1 может, таким образом, представлять собой более конкретно одну из следующих групп:
H,  и
и  ; и, в частности, Н и
; и, в частности, Н и  ; и предпочтительно
; и предпочтительно  .
.
R2 может представляет собой более конкретно необязательно замещенный гетероцикл (в частности, замещенный (С1-С6)алкилом или NH2), NO2 или NR9R10, где, в частности, R9=R10=H или иначе R9 и R10 каждый представляет собой H или необязательно замещенный (С1-С6)алкил.
R2 может представлять собой, в частности, необязательно замещенный гетероцикл, в частности, замещенный (С1-С6)алкилом или NH2. Гетероцикл может представлять собой, в частности, гетероцикл с 5 или 6 членами, включающими по меньшей мере один атом азота, и, в частности, один или два. Гетероцикл может быть, таким образом, выбран из пиперазина, пиперидина и пирролидина.
R2 может, в частности, представлять собой одну из следующих групп:
NH2, NH(CH2)3NMe2, NMe(CH2)3NMe2, NO2,  ,
,  и
и  ; и, в частности, NH2, NO2,
; и, в частности, NH2, NO2,  ,
,  и
и  ; и, в частности,
; и, в частности,  и
и  ; и более конкретно
; и более конкретно  .
.
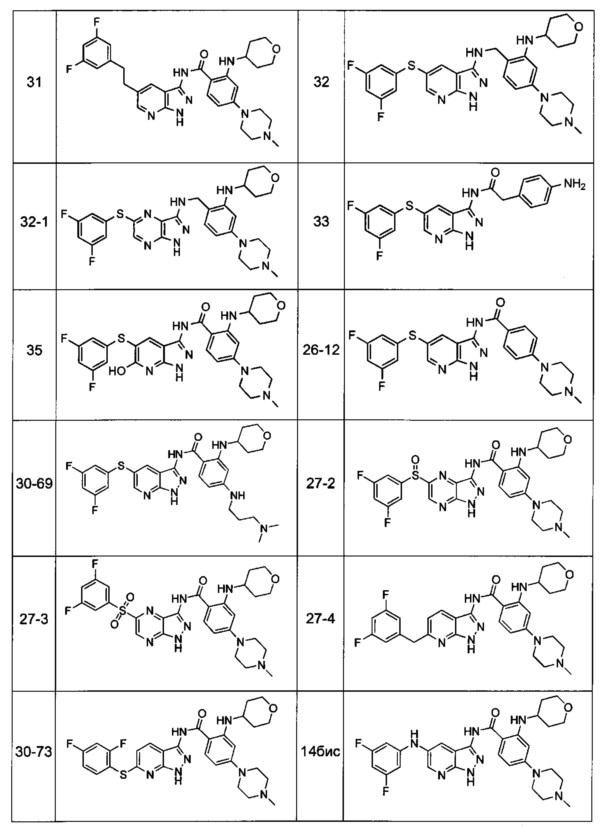
Соединения по настоящему изобретению могут быть выбраны из соединений, перечисленных в следующей таблице:


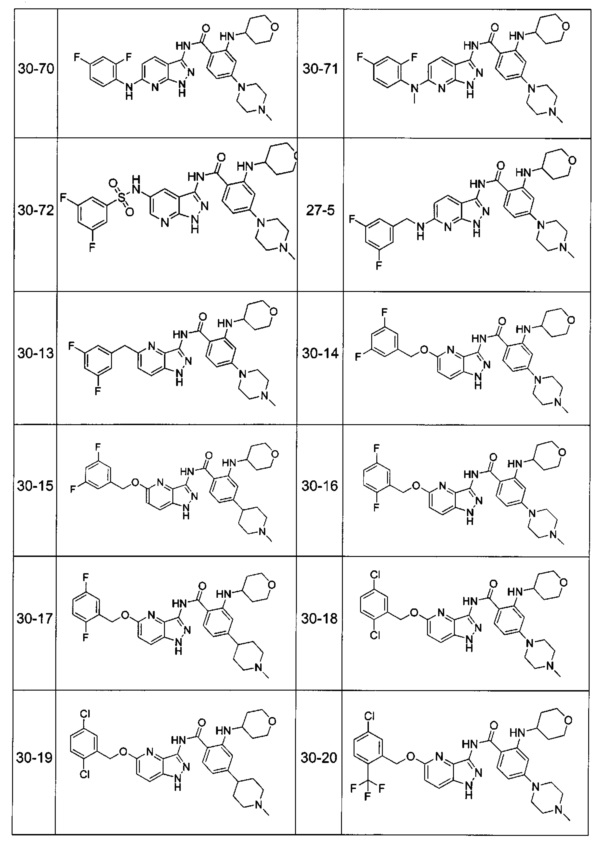

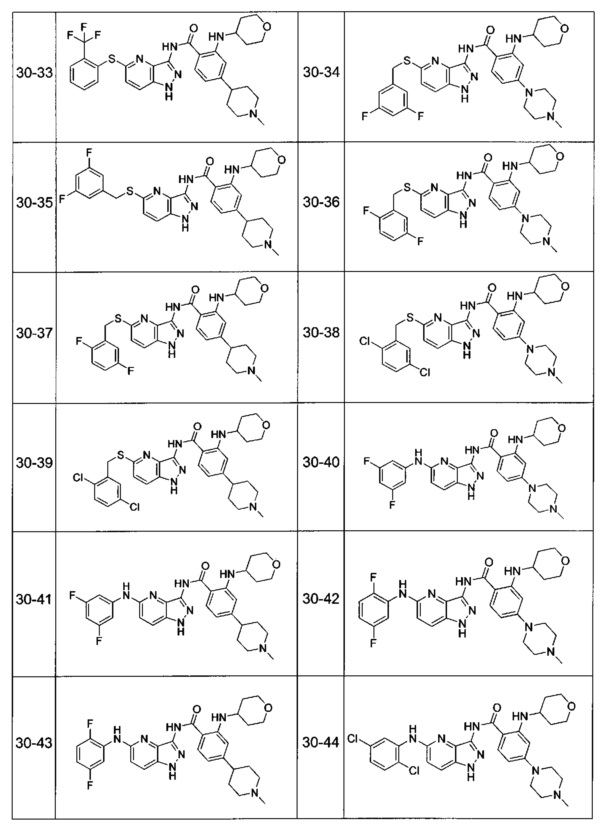



Настоящее изобретение также относится к применению соединения формулы (I), как определено выше, для получения лекарственного средства, в частности, предназначенного для лечения или предупреждения боли, в частности, боли, ассоциированной по меньшей мере с одним белком Trk.
Настоящее изобретение также относится к способу лечения или предупреждения боли, в частности, боли, ассоциированной по меньшей мере с одним белком Trk, включающему введение человеку, нуждающемуся в этом, эффективной дозы соединения формулы (I), как определено выше.
Настоящее изобретение также относится к фармацевтической композиции, содержащей по меньшей мере одно соединение формулы (I), как определено выше, и по меньшей мере один фармацевтически приемлемый эксципиент, применяемой при лечении или предупреждении боли, в частности, боли, ассоциированной по меньшей мере с одним белком Trk.
Фармацевтические композиции, применяемые при лечении или предупреждении боли, в частности, боли, ассоциированной по меньшей мере с одним белком Trk, согласно изобретению можно включать в препараты, в частности, для перорального введения или для инъекции, где данные композиции предназначены для млекопитающих, включая людей.
Активный ингредиент можно вводить в стандартных дозируемых формах введения, в смеси со стандартными фармацевтическими носителями, животным или людям. Соединения по изобретению в качестве активных ингредиентов можно применять в дозах, находящихся в диапазоне от 0,01 мг до 1000 мг в сутки, даваемых в однократной дозе один раз в сутки или вводимой в нескольких дозах на протяжении суток, например, дважды в сутки в равных дозах. Доза, вводимая в сутки, предпочтительно составляет от 5 мг до 500 мг, еще более предпочтительно от 10 мг до 200 мг. Может быть необходимым введение доз, находящихся вне этих диапазонов, как определено специалистами в данной области техники.
Фармацевтические композиции, применяемые при лечении или предупреждении боли, в частности, боли, ассоциированной по меньшей мере с одним белком Trk, согласно изобретению могут дополнительно содержать по меньшей мере один другой активный ингредиент, такой как, например, противораковое средство.
Объектом настоящего изобретения также является фармацевтическая композиция, применяемая при лечении или предупреждении боли, в частности, боли, ассоциированной по меньшей мере с одним белком Trk, содержащая:
a) по меньшей мере одно соединение формулы (I), как определено выше, и
b) по меньшей мере один другой активный ингредиент, такой как противораковое средство,
в виде комбинированного препарата для одновременного, отдельного или последовательного применения.
В контексте изобретения термин "лечение" означает устранение или облегчение боли.
Термин "боль" согласно изобретению означает любой вид боли, в частности, ноцицептивную боль, воспалительную боль, невропатическую боль, идиопатическую боль или психогенную боль, предпочтительно воспалительную или невропатическую боль. Боль согласно изобретению может представлять собой комбинацию двух или более из этих видов боли, например, комбинацию между воспалительной и ноцицептивной болью.
Боль в соответствии с изобретением может иметь любое происхождение. В воплощении изобретения боль в соответствии с изобретением является следствием рака, например, рака кости. В другом воплощении изобретения боль в соответствии с изобретением является следствием повреждения нервов, которое встречается, например, при невропатической боли. В другом воплощении изобретения боль в соответствии с изобретением является следствием воспалительного состояния, которое встречается, например, при ревматических заболеваниях, таких как остеоартрит, боль в нижней части спины, люмбальная позвоночная грыжа и сдавление нервных корешков. В другом воплощении изобретения боль в соответствии с изобретением обусловлена функциональными расстройствами, такими как, например, фибромиалгия.
Согласно изобретению "белок Trk" означает любой член семейства Trk, например, TrkA (в частности, описанный в GenBank под номером AB019488), TrkB (в частности, описанный в GenBank под номером ААВ33109.1) и TrkC (в частности, описанный в GenBank под номером САА12029.1), преимущественно TrkA.
Белок Trk в соответствии с изобретением может находиться в его нативной форме или в модифицированной форме. Под "модифицированной формой" подразумевают мутированную форму белка дикого типа. Мутация может представлять собой точечную мутацию, а также может представлять собой делецию или инсерцию одной или более аминокислот в последовательности белка Trk. Альтернативно модифицированный белок Trk в соответствии с изобретением может представлять собой слитый белок, например, полученный после хромосомной перестройки. Модифицированный белок Trk может быть также результатом альтернативного сплайсинга.
В соответствии с изобретением подразумевают, что выражение "ассоциированная по меньшей мере с одним белком Trk" означает, что боль, подлежащая лечению, ретранслируется биохимическими путями передачи сигнала, идущими через один или более белков Trk. В частности, боль считают ассоциированной с белком Trk, когда она представляет собой воспалительную или невропатическую боль. Биохимические пути передачи сигнала Trk хорошо известны специалистам в данной области техники.
Соединения согласно изобретению обладают свойством ингибирования или модулирования ферментативной активности одного или более белков Trk, предпочтительно более одного белка Trk.
Подразумевают, что ингибирование или модулирование активности одного или более белков Trk согласно изобретению означает, что соединение согласно изобретению способно модулировать активацию по меньшей мере одного белка Trk, что приводит в результате к снижению, возможно, к инактивации биохимического пути передачи сигнала Trk, которая, в свою очередь, приводит в результате к уменьшению ощущения боли. Соединения согласно изобретению дают возможность, например, снижения активности белка Trk приблизительно на 5% или более, в частности, приблизительно на 10% или более, в частности, примерно на 50% или более.
Соединения формулы (I) согласно настоящему изобретению могут быть получены различными способами, в частности, кратко изложенными на схемах 1а и 1b ниже.


Способ A:
Согласно способу A соединения формулы (I) получают путем предварительного синтеза соединений формулы (V), характеризующихся галогенированным гетеробициклическим кольцом, имеющим экзоциклический первичный амин. Эти соединения получают посредством синтеза промежуточных соединений общей формулы (II) или (III).
Способ A1:
Способ A1, представленный на схеме 2 (йодированные соединения) или 3 (бромированные соединения) ниже, описывает общий способ получения соединений общей формулы (V), где W определен, как в описании общей формулы (I), и, в частности, как Н, (C1-C6)алкил или арил, и Rj=H или N-защитная группа.
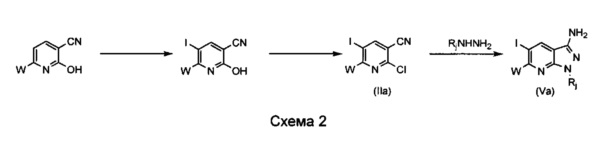
В контексте схемы 2 необязательно замещенный 2-хлор-5-йодникотинонитрил (IIa) получают из соответствующего гидроксиникотинонитрила путем последовательного использования агента йодирования, такого как N-йодсукцинимид (NIS), или молекулярного йода с неорганическим основанием, таким как, например, K2CO3 или Na2CO3, в частности, в полярном растворителе, таком как горячий N,N-диметилформамид (ДМФ), с последующей обработкой оксихлоридом фосфора, чистым или разведенным в неполярном растворителе с высокой температурой кипения, или любым другим эквивалентным агентом хлорирования, хорошо известным специалистам в данной области техники. Температуры реакции составляют от -20°C до 200°C. Затем полученное таким путем соединение (IIa) преобразуют в необязательно замещенный 5-йод-пиразоло[3,4-b]пиридин-3-амин (Va) путем его взаимодействия, предпочтительно при нагревании, в присутствии гидразина, необязательно несущего N-защитную группу, такую как тритил, трет-бутил или BOC.
Бромированные аналоги общей формулы (V), как описано на схеме 1а, могут быть получены путем использования способа, описанного в следующих ссылках: Witherington et al., Bioorg. Med. Chem. Lett., 2003, 13, 1577-1580 и Lijuan Chen et al., Bioorg. Med. Chem. Lett., 2010, 20, 4273-4278. Для удобства эти молекулы были получены путем использования последовательности реакций, представленных на следующей схеме 3.

Необязательно функционализированный 2-метокси-никотинонитрил получают, например, путем взаимодействия метанолята натрия в метаноле при температуре от -20°C до температуры кипения смеси. Альтернативно это соединение может быть получено путем метилирования 2-гидроксиникотинонитрила или другими способами, описанными выше. Бромирование 2-метокси-никотинонитрила в характерном случае проводят дибромом в уксусной кислоте при температуре, изменяющейся от 20°C до 110°C. Образование пиразола в характерном случае проводят путем взаимодействия избытка гидразина, несущего или не несущего функциональную группу, при температуре, изменяющейся от 20°C до 100°C, в присутствии полярного растворителя, такого как вода, этанол, тетрагидрофуран (ТГФ) или любой другой растворитель со сравнимыми свойствами. Альтернативно также возможно использование гидразина в физиологическом растворе или в гидратированной форме без растворителя.
Способ A2:
Способ A2 относится к синтезу функционализированных пиразолопиразинов, представленному на схеме 4 ниже, где Rj=H или N-защитная группа, Hal = атом галогена, и, в частности, W=H, (С1-С6)алкил или арил.

Необязательно функционализированные 3-амино-6-йодпиразин-2-карбоксамиды в характерном случае получают в две стадии из соответствующих метил-3-аминопиразин-2-карбоксилатов путем йодирования в присутствии N-йодсукцинимида или молекулярного йода, необязательно в присутствии кофактора, такого как KIO3, AgCO2CF3, Ag2SO4, AlCl3, CuCl2 или HgO, с последующей реакцией преобразования функциональной группы метилового эфира в карбоксамид, в частности, путем использования аммиака в полярном растворителе, таком как вода, метанол или ТГФ, при температурах, изменяющихся от 0°C до 100°C. Затем карбоксамидную функциональную группу необязательно функционализированного 3-амино-6-йодпиразин-2-карбоксамида преобразуют в нитрил путем использования агентов дегидратации, таких как, в частности, CCl4/PPh3, SOCl2, PhSO2Cl, P2O5, TsCl, COCl2, DCC/py (N,N'-дициклогексилкарбодиимид/пиридин) или (COCl)2, используемых в зависимости от обстоятельств, возможно, в присутствии органического основания, такого как пиридин. Предпочтительный способ включает использование оксихлорида фосфора в диметилформамиде (ДМФ). Удаление защиты функциональной группы диметилформимидамида проводят путем обработки кислотой, такой как водная соляная кислота, или другим реагентом с эквивалентными свойствами. Образование пиразольного кольца осуществляют путем реакции Зандмейера, хорошо известной специалистам в данной области техники, с последующим взаимодействием в присутствии гидразина, несущего или не несущего функциональную группу, в условиях, описанных в приведенных выше способах. Альтернативно диазониевую соль, являющуюся промежуточным соединением реакции Зандмейера, можно восстанавливать путем использования, например, хлорида олова в кислой среде или другого эквивалентного агента, с целью образования функциональной группы гидразина, которая может претерпевать внутримолекулярную циклизацию под действием нагревания.
Способ A3:
Способ A3 нацелен на получение производных общей формулы (V), отличающихся переменной функциональной группой в положении 6 пиразолопиридинового бицикла. Это подробно описано на схеме 5 ниже.

Взаимодействие цианотиоацетамида с этил-3-этоксиакрилатами, различно замещенными, согласно способам, описанным, в частности, Litrivnor et al. в Russ. Chem. Bull., 1999, 48(1), 195-196 и Tsann-Long Su et al. в J. Med. Chem., 1988, 31, 1209-1215 дает возможность получить в две стадии этил-5-циано-6-(метилтио)никотинаты, несущие переменную функциональную группу в положении 2. Эти синтезы в характерном случае проводят для первой стадии в безводном полярном растворителе, таком как, например, этанол, при температуре в диапазоне от 0°C до 70°C в присутствии органического основания, такого как метилморфолин, триэтиламин, DIPEA (N,N-диизопропилэтиламин) или DBU (1,8-диазабицикло[5,4,0]уедец-7-ен). Вторую стадию внутримолекулярной циклизации и алкилирования в характерном случае проводят путем нагревания до температуры в диапазоне от 20°C до 100°C раствора промежуточного тиоамидата в полярном растворителе, например, в этаноле, в присутствии подходящего алкилирующего агента, такого как алкилгалогенид или диалкилсульфат.
5-Циано-6-(метилтио)никотиновые кислоты, замещенные в положении 2, в характерном случае получают путем омыления соответствующих этиловых эфиров согласно способам, хорошо известным специалистам в данной области техники, в частности, путем использования горячего гидроксида лития. Декарбоксилирование этих соединений осуществляют путем обработки нагреванием в растворителе с высокой температурой кипения, таком как дифениловый эфир, при температуре в диапазоне от 150°C до 250°C.
Реакции галогенирования, в основном, нацелены на получение йодированных, бронированных или хлорированных производных, более конкретно йодированных производных. Последние в характерном случае получают путем обработки молекулярным йодом в присутствии соли серебра, такой как, например, Ag2SO4, в полярном растворителе, таком как этанол, при температуре в диапазоне от 0°C до 70°C. Рассмотрены также альтернативные способы, в частности, способы, основанные на других солях, таких как KIO3, AgCO2CF3, AlCl3, CuCl2 или HgO, или других йодирующих агентах, таких как N-йодсукцинимид. Возможные способы бромирования в характерном случае основаны на агентах, таких как N-бромсукцинимид или дибром, согласно способам, хорошо известным специалистам в данной области техники.
В случае, в котором W=OH (в характерном случае в результате использования диэтил-2-(этоксиметилен)малоната), соответствующие соединения защищают с помощью реакции алкилирования. Эту реакцию, в частности, проводят путем использования метилйодида или бромметана и карбоната серебра в диоксане, ТГФ, ацетонитриле или ацетоне, или любого эквивалентного агента, такого как диметилсульфат. Полученные 5-галоген-2-(метилтио)никотинонитрилы подвергают окислению их функциональной группы тиометокси, в характерном случае путем использования m-CPBA (m-хлорпербензойной кислоты), оксона или любого другого эквивалентного агента, что приводит к образованию соответствующего сульфоксида. Эти соединения, которые могут содержать различные количества соответствующего сульфона, вступают в реакцию в присутствии необязательно замещенного гидразина с образованием соответствующего 5-галоген-пиразоло[3,4-b]пиридин-3-амина, несущего переменную функциональную группу в положении 6.
Способ A4:
Способ A4 нацелен на получение производных общей формулы (V) из соединений общей формулы (III) посредством образования промежуточного соединения для соединения формулы (IV). Эти соединения в характерном случае получают путем, представленным на схеме 6. Используемый способ проиллюстрирован следующими ссылками: Gueiffier et al. Heterocycles, 1999, 51(7), 1661-1667; Gui-Dong Zhu et al. Bioorg. Med. Chem., 2007, 15, 2441-2452.

Соединения общей формулы (IIIa), предварительно ацетилированные одним или другим из способов, хорошо известных специалистам в данной области техники, подвергают действию изоамилнитрита, нитрита натрия или любого другого эквивалентного органического или неорганического нитрита в воде или в уксусной кислоте в течение периодов, в характерном случае варьирующих от 1 до 3 суток, при температурах, изменяющихся от 0°C до 40°C. Защиту полученных таким путем соединений общей формулы (IVa) удаляют в кислых условиях, например, путем использования соляной кислоты, после чего подвергают действию агентов нитрования, таких как концентрированная азотная кислота или нитрат калия, в серной кислоте при температурах, изменяющихся от 0°C до 25°C.
Следует отметить, что прямое преобразование соединений формулы (IIIa) в соединения (IVb) с удаленной защитой в целом возможно.
Полученные таким путем нитропиразолы в характерном случае восстанавливают до аминопиразолов общей формулы (Ve) путем использования SnCl2 в соляной кислоте. Альтернативные способы включают использование железа, цинка или олова в кислых условиях, и способы каталитической гидрогенизации в присутствии комплексов платины, никеля или Pd/C в атмосфере водорода или в присутствии эквивалентных агентов, таких как циклогексадиен, циклогексен, боргидрид натрия или гидразин.
Способ B:
Согласно способу B соединения формулы (I) получают путем предварительного синтеза соединений формулы (VI), характеризующихся функционализированным гетеробициклическим кольцом, несущим экзоциклический амин. Эти соединения получают посредством синтеза промежуточных соединений общей формулы (VI).
Способ B1:
Способ B1 представлен на схеме 7 ниже, где W, в частности, представляет собой Н, (С1-С6)алкил, арил или бензил.

Производные 3-нитро-6-тиоксо-1,6-дигидропиридин-2-карбонитрила и 3-нитро-6-тиоксо-1,6-дигидропиразин-2-карбонитрила, необязательно функционализированные в положении 5, в характерном случае получают из соответствующих 2,6-дихлор-3-нитропиридинов или 2,6-дихлор-3-нитропиразинов путем последовательных взаимодействий соли цианида, такой как цианид меди, в полярном растворителе с высокой температурой кипения, таком как N-метилпирролидон, при температурах в диапазоне от 100°C до 200°C; с последующим взаимодействием с водным гидросульфитом натрия в полярном растворителе. Затем эти соединения алкилируют, например, путем использования замещенного бензилбромида, в основной среде согласно способам, хорошо известным специалистам в данной области техники. Предпочтительный протокол включает использование апротонного и безводного полярного растворителя, такого как ацетон, при его температуре кипения и органического основания, такого как пиридин, триэтиламин или DIPEA, или неорганического основания, такого как карбонат натрия, калия или кальция. Реакции восстановления функциональной нитрогруппы в амине предпочтительно проводят путем использования SnCl2 в соляной кислоте. Альтернативные способы включают использование железа, цинка или олова в кислых условиях и способы каталитической гидрогенизации в присутствии комплексов палладия, никеля или Pd/C в атмосфере водорода или в присутствии эквивалентных агентов, таких как циклогексадиен, циклогексен, боргидрид натрия или гидразин.
В некоторых случаях продукт реакции восстановления в дополнение к первичной аминной имеет карбоксамидную функциональную группу, образующуюся в результате гидролиза нитрильной функциональной группы. В этом случае выделение соответствующих 3-аминопиколинонитрилов или 3-аминопиразин-2-карбонитрилов может быть выполнено путем дегидратации карбоксамида до нитрила путем использования оксихлорида фосфора в присутствии ДМФ или любого другого способа, хорошо известного специалистам в данной области техники. Наконец, образование аминопиразольного кольца проводят преимущественно путем образования диазония, полученного путем последовательного взаимодействия при низкой температуре изоамилнитрита, нитрита натрия или другого эквивалентного органического или неорганического нитрита в воде, соляной кислоте, уксусной кислоте или серной кислоте при температурах, изменяющихся от 0°C до 20°C, с его последующим восстановлением до гидразина и внутримолекулярной циклизацией, активируемой нагреванием реакционной смеси. Реакцию восстановления преимущественно проводят с хлоридом олова в кислых условиях, но его можно также проводить путем каталитической гидрогенизации или любым другим способом, хорошо известным специалистам в данной области техники. В качестве альтернативы этой последней стадии возможно, чтобы промежуточный диазоний претерпевал реакцию Зандмейера, в процессе которого эта функциональная группа замещается атомом галогена, таким как атом йода, путем взаимодействия адекватной соли, такой как NaI. Если данный вариант предпочтителен, образование аминопиразольного кольца проводят путем использования гидразина, несущего или не несущего функциональную группу, в полярном растворителе, таком как этанол, при температурах, изменяющихся от 25°C до 150°C.
Способ B2:
Альтернативно возможно использовать преимущество реакции ароматического нуклеофильного замещения для функционализации пиридинового или пиразинового кольца в положении 6. В этом случае используемыми нуклеофилами являются фенолы, тиофенолы, бензиловые спирты или тиобензиловые спирты, а также анилины или бензиламины, несущие или не несущие функциональную группу. Общая схема реакции 8а представлена ниже, в частности, где W=Н, (С1-С6)алкил, арил или бензил.
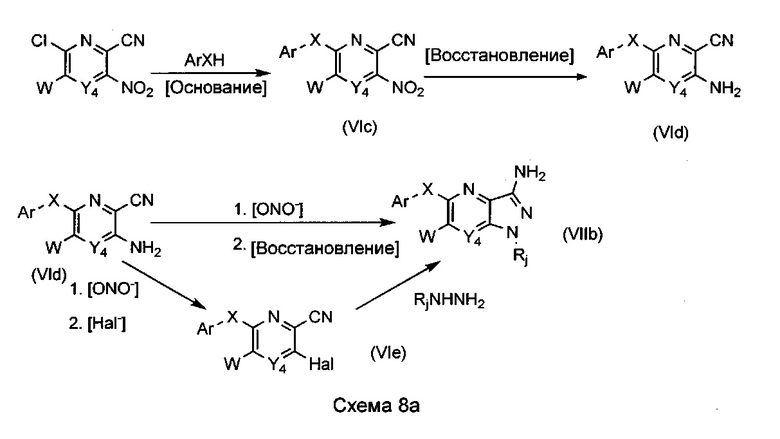
В случае, в котором X=O или S, 6-хлор-3-нитропиколинонитрилы и 6-хлор-3-нитропиразин-2-карбонитрилы, необязательно замещенные в положении 5, подвергают взаимодействию в присутствии подходящего нуклеофила, спирта или тиола, в полярном растворителе, таком как ацетонитрил, в присутствии неорганического основания, такого как карбонат калия или натрия. Могут быть также рассмотрены растворители, такие как ДМСО (диметилсульфоксид), ДМФ (диметилформамид), ацетон, ТГФ (тетрагидрофуран) или пиридин. При необходимости эти реакции можно катализировать под действием меди, а также можно проводить без растворителя. В характерном случае предпочтительный протокол включает температуры в диапазоне от 20°C до 150°C.
Альтернативно также возможно использование оснований, таких как пиридин, DIPEA, диизопропиламин, триэтиламин, DBU, трет-бутилат калия, NEt3 или NaH.
В случае, в котором X=N, толуол является предпочтительным растворителем, а основанием выбора является триэтиламин (NEt3).
Следующие стадии вплоть до соединений общей формулы (VIIb) идентичны стадиям, документированным в способе B1 выше.
Способ B3:
Способ B3, представленный на схеме 8b ниже, представляет собой вариант способа В2, отличающийся первой стадией, являющейся результатом реакции каталитического сочетания между бензилборонатом в кислотной или сложноэфирной форме и производного 6-хлор-3-нитропиколинонитрила или 6-хлор-3-нитропиразин-2-карбонитрила. Специалистам в данной области техники также хорошо известно, что возможны также реакции каталитического сочетания с использованием альтернативных катализаторов и бензильных производных. Среди прочего может быть рассмотрена реакция Стилла, основанная на комплексах олова, или реакции, основанные на цинкорганических соединениях.

Необязательно замещенный 2-бензил-4,4,5,5-тетраметил-1,3,2-диоксаборолан получают предварительно, например, из соответствующего бензилхлорида и октаметил-би-диоксаборолана в диоксане в присутствии ацетата калия и Pt(dppf)Cl2 (dppf=1,1'-бис(дифенилфосфино)ферроцен). Это соединение объединяют с 6-хлор-3-нитропиколинонитрилом, 6-хлор-3-нитропиразин-2-карбонитрилом, необязательно замещенным в положении 5 или 5-хлор-2-нитроникотинонитрилом, необязательно замещенным в положении 6, и палладиевым катализатором, таким как Pd(dppf)Cl2 или Pd(PPh3)4, с органическим основанием, таким как триэтиламин или алкоголят, или с неорганическим основанием, таким как карбонат натрия, калия или цезия, в растворителе, таком как толуол, бензол, ТГФ или диоксан. Предпочтительные температуры этой реакции составляют от 20°C до 100°C. Продукты этих реакций соответствуют замещенным производным 6-бензил-3-нитропиколинонитрила, 6-бензил-3-нитропиразин-2-карбонитрила или 5-бензил-2-нитроникотинонитрила, для которых следующие стадии преобразования воспроизводят из описанного выше способа B1.
Способ B4:
Способ B4, представленный на схеме 9 ниже, дает возможность получить пиразолопиридиновые и пиразолопиразиновые бициклы, характеризующиеся необязательно функционализированными арилсульфонамидными функциональными группами, где RI=(C1-C6)алкил и, в частности, W=Н, (C1-C6)алкил, арил или бензил.

Производные этил-2-хлор-5-(хлорсульфонил)никотината, необходимые для данной последовательности реакций, могут быть получены согласно способам, описанным Levett Р.С. et al., Org. Proc. Res. Dev., 2002, 6(6), 767-772; WO 01/98284 и WO 2008/010964.
Образование сульфонамидов в характерном случае проводят путем смешивания представляющего интерес 2-хлор-5-(хлорсульфонил)никотината с первичным или вторичным анилином, необязательно функционализированным, в апротонном растворителе, таком как дихлорметан, ТГФ, ацетон или ацетонитрил, в присутствии органического основания, такого как триэтиламин (NEt3), пиридин или DIPEA. Может быть также рассмотрено использование неорганического основания, такого как карбонат натрия или калия. Оптимальные температуры реакции составляют от 0°C до 70°C.
Реакция омыления полученного таким путем продукта, в частности, путем использования гидроксида лития в смеси ТГФ/вода, дает возможность получить 2-хлор-5-(N-фенилсульфамоил)никотиновые кислоты.
Соответствующие хлорангидриды получают путем обработки тионилхлоридом в толуоле с обратным холодильником или любым другим способом дегидрохлорирования, хорошо известным специалистам в данной области техники. Взаимодействие этих промежуточных соединений с водным аммиаком дает возможность образовать необязательно функционализированные 2-хлор-5-(N-фенилсульфамоил)никотинамиды, которые затем вступают в реакцию дегидратации, в частности, путем использования POCl3 при температуре в диапазоне от 75°C до 150°C. Может быть также рассмотрено альтернативное использование агентов, таких как P2O5 или трифторуксусный ангидрид и пиридин.
Наконец, эти производные общей формулы (VIh) подвергают взаимодействию в присутствии гидразина, несущего или не несущего функциональную группу, в полярном растворителе, таком как этанол, при температурах, изменяющихся от 25°C до 150°C, с образованием соответствующих производных общей формулы (VIId).
Способ В5:
Способ В5, представленный на схеме 10 ниже, дает возможность получить пиразолопиридиновые бициклы, характеризующиеся необязательно функционализированными бензилэфирными функциональными группами, в частности, где W=Н, (С1-С6)алкил, арил или бензил.
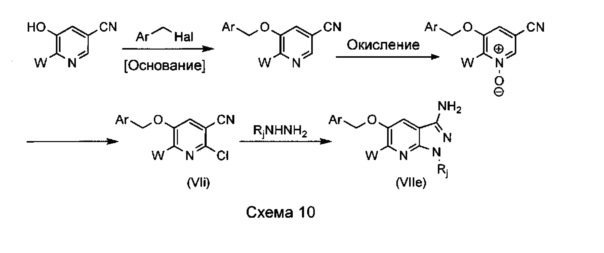
Способ, описанный ниже, основан на работе J. Baldwin et al., J. Heterocyclic. Chem., 1980, 17(3), 445-448. Производные 5-гидроксиникотинонитрила, необязательно функционализированные в положении 6, алкилируют, в характерном случае путем использования необязательно функционализированного бензилгалогенида в присутствии основания. Для предпочтительного способа требуется использование апротонного полярного растворителя, такого как ДМФ, и основания, такого как NaH. Оптимальные температуры реакции составляют от 20°C до 100°C. Альтернативно растворители, которые можно использовать, включают, например, ТГФ, ДМСО, диоксан, ацетонитрил, дихлорметан или ацетон, и основания, такие как tBuOK, DIPEA, пиридин, триэтиламин, DBU или карбонат натрия, калия или цезия.
Окисление пиридинового кольца до пиридин-N-оксида в характерном случае проводят путем использования m-CPBA в дихлорметане при комнатной температуре. Тем не менее, возможны многие альтернативные способы, в частности, основанные на использовании перкарбоната натрия в присутствии рениевого катализатора, пербората натрия в присутствии уксусной кислоты или комплекс мочевины с пероксидом водорода.
Обработка этих производных пиридин-N-оксида оксихлоридом фосфора приводит к образованию соответствующих 2-хлорникотинонитрилов (VI).
Взаимодействие этих соединений при нагревании с гидразином, несущим или не несущим функциональную группу, в полярном растворителе, таком как изопропанол или этанол, приводит к образованию искомых пиразолопиридиновых бициклов (VIIe).
Способ B6:
Способ B6, представленный на схеме 10а ниже, дает возможность получить необязательно функционализированные пиразолопиридиновые и пиразолопиразиновые бициклы, характеризующиеся обратимыми сульфонамидными функциональными группами, в частности, где W=Н, (C1-C6)алкил, арил или бензил.

Способ, описанный ниже, состоит в образовании сульфонамидной функциональной группы из ароматического амина и арилсульфонилгалогенида или любого другого эквивалентного реагента в присутствии основания, которое можно необязательно вводить в качестве растворителя или сорастворителя. Альтернативно арилсульфонилгалогенид или его эквивалент может быть образован in situ.
Эта реакция при нагревании с гидразином, несущим или не несущим функциональную группу, в полярном растворителе, таком как изопропанол или этанол, приводит к образованию желаемых пиразолопиридиновых и пиразолопиразиновых бициклов (VIIf).
Способ C:
Способ C нацелен на получение соединений формулы (XI), как описано на схеме 1.
Способ C1:
Способ C1, представленный на схеме 11 ниже, предназначен для получения пиразолопиридинов и пиразолопиразинов, функционализированных в положении 6, где Rn = атом галогена, мезилат, тозилат или трифлат, X=O, S, NH, N-(С1-C-)алкил и необязательно CH2 для (Xc) и (Xd), и Rj=Н или N-защитная группа.
Данный способ можно также использовать для выполнения синтеза молекул, содержащих двухатомную группу X, соответствующую, в частности, группе ArX, представляющую собой: -ArCH2NH-, -ArCH2N(R4)-, -ArCH2O-, -ArCH2S-, -ArCH2CH2-, -ArCHCH- или -ArCC-.

6-Гидрокси-2-(метилтио)никотинонитрилы или 5-гидрокси-3-(метилтио)пиразин-2-карбонитрилы подвергают реакции дегидрохлорирования, в характерном случае в присутствии оксихлорида фосфора, с растворителем или без растворителя, при температурах, изменяющихся от 70°C до 180°C. При использовании растворителя предпочтителен неполярный растворитель с высокой температурой кипения, такой как толуол или ксилол. Альтернативно возможно активировать 6-гидрокси-2-(метилтио)никотинонитрилы и 5-гидрокси-3-(метилтио)пиразин-2-карбонитрилы путем их преобразования в сульфоновые эфиры посредством образования соответствующих тозилатов, мезилатов или трифлатов. Если данный вариант предпочтителен, использование тозил-, мезил- или трифлилхлоридов в растворителе, таком как толуол, дихлорметан, ТГФ, ацетонитрил, ацетон или диоксан, в присутствии органического или неорганического основания с получением этих производных.
Затем соответственно полученные 6-хлор-2(метилтио)никотинонитрилы и 5-хлор-3-(метилтио)пиразин-2-карбонитрилы или их сульфоэфирные аналоги, если данный вариант предпочтителен, подвергают взаимодействию с нуклеофилом, таким как фенол, анилин или тиофенол, в контексте ароматического нуклеофильного замещения. В данном случае реакцию проводят в полярном растворителе, таком как ДМСО, ДМФ, ацетон, ТГФ или ацетонитрил, в присутствии основания, такого как трет-бутилат натрия или NaH. При необходимости эти реакции можно катализировать под действием меди, и можно также проводить без растворителя. В характерном случае предпочтительный протокол включает температуры в диапазоне от 20°C до 150°C.
Альтернативно также возможно использование органических оснований, таких как пиридин, диизопропиламин, триэтиламин или DBU, или неорганических оснований, таких как карбонат натрия или калия.
Альтернативно соединения формулы (IXb) могут вступать в каталитическую реакцию сочетания, такую как реакция Сузуки. В данном случае эти соединения объединяют с необязательно замещенным 2-бензил-4,4,5,5-тетраметил-1,3,2-диоксабороланом, уже описанным в предшествующем способе B3, палладиевым катализатором, таким как Pd(dppf)Cl2 или Pd(PPh3)4, органическим основанием, таким как триэтиламин или алкоголят, или неорганическим основанием, таким как карбонат натрия, калия или цезия, в растворителе, таком как толуол, бензол, ТГФ или диоксан. Предпочтительные температуры реакции составляют от 20°C до 100°C.
Затем производные, полученные одним или другим из этих способов, окисляют, в характерном случае путем использования m-СРВА или оксона с образованием соответствующих метилсульфоксидов или метилсульфонов. Эти соединения, иногда полученные в виде смесей, используют как таковые в реакции образования аминопиразольного кольца, путем использования необязательно замещенного гидразина в полярном растворителе, таком как этанол, при температурах, изменяющихся от 25°C до 150°C.
Альтернативно возможно модифицировать последовательность реакций, в частности, путем обратного порядка стадий синтеза.
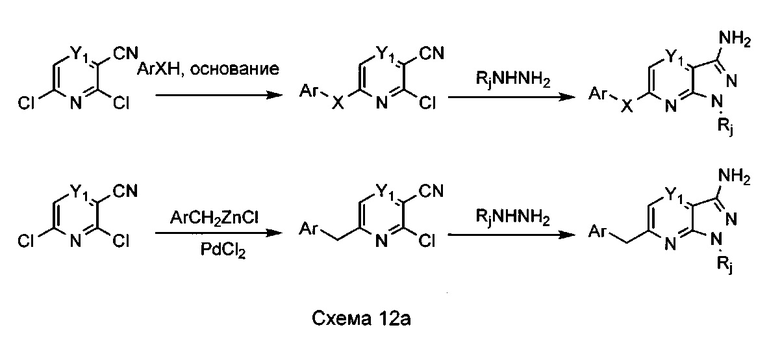
Способ C2:
Способ C2, представленный на схеме 12 ниже, предназначен для получения пиразолопиридинов и пиразолопиридазинов, функционализированных в положении 6, где X=О, S, NH, N-(C1-C-)алкил или CH2, и Rj=Н или N-защитная группа.

Производные 6-гидрокси-4-(метилтио)никотинонитрила или 6-гидрокси-4-(метилтио)пиридазин-3-карбонитрила окисляют, в характерном случае путем использования m-CPBA или оксона, с образованием соответствующих метилсульфоксидов или метилсульфонов. Эти соединения, иногда полученные в виде смесей, используют как таковые в реакции образования аминопиразольного кольца, путем использования необязательно замещенного гидразина в полярном растворителе, таком как этанол, при температурах, изменяющихся от 25°C до 150°C.
Полученные таким путем пиразолопиридины и пиразолопиридазины подвергают реакции дегидрохлорирования, в характерном случае в присутствии оксихлорида фосфора, с растворителем или без растворителя, при температурах, изменяющихся от 70°C до 180°C. При использовании растворителя предпочтителен неполярный растворитель с высокой температурой кипения, такой как толуол или ксилол. Затем соответственно полученный необязательно замещенный 6-хлор-пиразоло[4,3-c]пиридин-3-амин и 6-хлор-пиразоло[4,3-с]пиридазин-3-амин подвергают взаимодействию с нуклеофилом, таким как фенол, анилин или тиофенол, в контексте ароматического нуклеофильного замещения. В данном случае реакцию проводят в полярном растворителе, таком как ДМСО, ДМФ, ацетон, ТГФ или ацетонитрил, в присутствии основания, такого как трет-бутилат калия или NaH. При необходимости эти реакции можно катализировать под действием меди, и можно проводить без растворителя. В характерном случае предпочтительный протокол включает температуры в диапазоне от 20°C до 150°C.
Альтернативно возможно использование органических оснований, таких как пиридин, диизопропиламин, триэтиламин или DBU, или неорганических оснований, таких как карбонат натрия или калия.
Альтернативно соединения формулы (XIVa) могут вступать в каталитическую реакцию сочетания, такую как реакция Сузуки. В этом случае эти соединения объединяют с необязательно замещенным 2-бензил-4,4,5,5-тетраметил-1,3,2-диоксабороланом, описанным выше в предшествующем способе B3, палладиевым катализатором, таким как Pd(dppf)Cl2 или Pd(PPh3)4, органическим основанием, таким как триэтиламин или алкоголят, или неорганическим основанием, таким как карбонат натрия, калия или цезия, в растворителе, таком как толуол, бензол, ТГФ или диоксан. Предпочтительные температуры реакции составляют от 20°C до 100°C.
Способ C3:
Способ C3, представленный на схеме 12а ниже, представляет собой вариант способа C1, основанный на региоселективной функционалзации 2,6-дихлорникотинонитрила либо анионным нуклеофилом, таким как фенат или тиофенат, либо металлоорганическим соединением, таким как хлорид бензилцинка. В последнем случае реакцию катализируют, например, комплексом палладия(II). Преобразование полученного таким путем хлорникотинонитрила в соответствующий пиразолопиридин, в случае, где Y1=СН, выполняют, как описано выше в способе A1.
Способ D:
Объектом этих способов является синтез соединений формулы (I) или (VII) путем использования различных способов каталитического сочетания.
Способ D1:
В способе D1, представленном на схеме 13 ниже, используют каталитическую реакцию сочетания, как описано в J.A.C.S., 1984, 106, 158, между цинкорганическим соединением, полученным in situ, и арилбромидом, катализируемую палладиевыми комплексами.

Необязательно замещенные 3-амино-диазаиндазолы и 3-амино-азаиндазолы объединяют с необязательно замещенным хлоридом бензилцинка в апротонном полярном растворителе, таком как ТГФ или диоксан, в присутствии каталитического количества палладиевого комплекса, такого как (dppf)2PdCl2⋅CH2Cl2. Реакцию сочетания проводят при температурах в диапазоне от 25°C до 100°C.
Способ D2:
В способе D2, представленном на схеме 14 ниже, используют реакцию сочетания, как описано в Gueiffier A. et al., Tetrahedron, 2006, 62, 6042-6049, между тиолом, в частности, тиофенолом или бензилтиолом, и арилиодидом, катализируемую комплексами меди.

Эту реакцию в характерном случае проводят в полярном растворителе с высокой температурой кипения, таком как 2-пропанол, в присутствии каталитического количества полиэтиленгликоля, соли металла, такой как йодид меди (CuI), и избытка неорганического основания, такого как карбонат калия, карбонат кальция или карбонат натрия. Температуры реакции в характерном случае изменяются от 50°C до 100°C.
Способ D3:
В способе D3, представленном на схеме 15 ниже, используют реакцию сочетания, как описано в Sonogashira, K. et al. в Tetrahedron Lett., 1975, 16, 4467-4470, между производным ацетилена и арилгалогенидом, катализируемую комплексами меди и палладия.
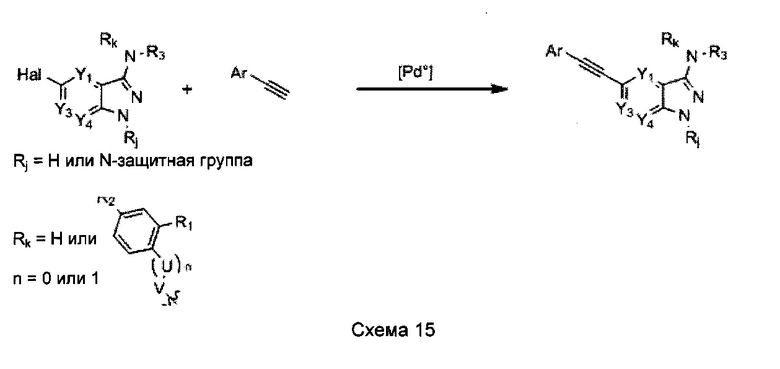
Такую реакцию в характерном случае проводят путем взаимодействия в инертной атмосфере между гетероарилгалогенидом и стехиометрическим количеством необязательно замещенного этинилбензола в присутствии каталитического количества палладиевого комплекса, например, PdCl2(PPh3)2 или Pd(PPh3)4, каталитического количества соли меди, например, CuI, и органического основания, такого как триэтиламин или DIPEA, или неорганического основания, такого как карбонат калия или цезия. Этот протокол обычно включает температуры реакции в диапазоне от 20°C до 45°C в растворителях, включающих ДМФ, ТГФ, диоксан или диэтиловый эфир.
Способ E:
Протоколы способа E нацелены на функционализацию экзоциклического амина аминопиразольных колец путем их взаимодействия с промежуточным соединением, характеризующимся электрофильной функциональной группой, необязательно образованной in situ, такой как хлорангидрид, изоцианат, изотиоцианат или альдегид.
Способ E1:
Способ E1, представленный на схеме 16 ниже, нацелен на преобразованием первичной экзоциклической аминной функциональной группы аминопиразольных соединений в амидную функциональную группу.

Эти соединения синтезируют посредством соответствующего 3-аминопиразола путем добавления адекватного хлорангидрида, полученного предварительно путем использования оксалилхлорида и каталитического количества ДМФ в растворителе, таком как тетрагидрофуран. Эти хлорангидриды могут быть получены путем использования альтернативных способов, таких как способы, основанные на использовании тионилхлорида или оксихлорида фосфора, хорошо известные специалистам в данной области техники. Конденсацию хлорангидридов на аминопиразолах в характерном случае проводят в апротонном растворителе, таком как тетрагидрофуран, толуол или дихлорметан, в присутствии основания, такого как DIPEA, пиридин или триэтиламин.
Альтернативно в качестве растворителя возможно использование основания, в частности, пиридина.
Альтернативно данный тип реакции можно проводить в двухфазной системе согласно хорошо известному способу Шоттена-Баумана.
Альтернативно образование амидной связи можно проводить из соответствующего 3-аминопиразола и представляющей интерес кислоты путем использования условий пептидного сочетания, используя реагенты, такие как HOBt (гидроксибензотриазол), TBTU (O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония тетрафторборат), HATU (2-(1Н-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат), EDCI (1-этил-3-(3-диметиламинопропил)карбодиимид) или карбонилдиимидазол, при температуре в диапазоне от -20°C до 100°C в апротонном растворителе, таком как тетрагидрофуран, диоксан, дихлорметан или любой растворитель с подобными свойствами.
Способ E2:
Производные, характеризующиеся присутствием вторичного амина в положении 3 аминопиразольного кольца, синтезируют путем реакции восстановительного аминирования согласно схеме 17 ниже.

Реакции восстановительного аминирования в характерном случае проводят путем смешивания адекватных стехиометрических количеств аминопиразола и альдегида в растворителе, таком как ДХЭ (дихлорэтан), ТГФ или ацетонитрил, необязательно в присутствии некоторого количества воды, ТФУ (трифторуксусной кислоты) или уксусной кислоты, путем добавления последовательных фракций восстанавливающего агента, такого как NaBH4, NaBH(OAc)3 или NaBH3CN. Эти реакции в характерном случае проводят при комнатной температуре.
Способ E3:
Производные, несущие функциональную группу 3-уреидо или 3-тиоуреидо, получают путем взаимодействия, представленного на схеме 18 ниже, аминопиразола с изоцианатом или изотиоцианатом, полученным согласно способам, хорошо известным специалистам в данной области техники.

При характерной реакции реакционную смесь получают в полярном или неполярном апротонном растворителе, таком как дихлорметан, ацетон, ДМФ, диметилацетамид (ДМА), ацетонитрил, ТГФ или диоксан, реакцию проводят при температурах, изменяющихся от 20°C до температуры кипения выбранного растворителя. При необходимости может быть необходим источник слабо нуклеофильного органического или неорганического основания. В данном случае возможным вариантом является гидрид натрия.
Способ F: постсинтетические удаления защиты и модификации
Способ F1: удаления защиты
Трифторацетатные защитные группы удаляют под действием органического основания, такого как триэтиламин или пиридин, в полярном растворителе, таком как метанол, этанол или ТГФ, при температурах образования флегмы используемых растворителей.
трет-Бутильные или тритильные защитные группы, которые несут пиразольные кольца, вытесняют под действием сильной кислоты, в характерном случае ТФУ, в неполярном растворителе, таком как дихлорметан или дихлорэтан (ДХЭ).
Способ F2: восстановления алкина

Реакции для восстановления диарилалкинов до диарилалканов в характерном случае проводят путем каталитической гидрогенизации под давлением водорода в присутствии катализаторов, таких как PtO2, Pt, Pd/C, Ni или Rh. Альтернативно возможно использование DIBAL-H (диизобутилалюмогидрида лития) в присутствии или в отсутствие катализатора, такого как Cp2TiCl2.
Способ F3: окисление сульфидов до сульфонов и сульфоксидов

Реакции окисления сульфидов до сульфоксидов в характерном случае проводят посредством использования оксона в смеси полярных растворителей, такой как ТГФ/MeOH или ДМФ/вода. Оптимальные температуры реакции в характерном случае составляют от 25°C до 50°C.
Доступны многие альтернативные способы, и некоторые дают возможность получения полуокисленных производных, в частности, сульфоксидов. Такие альтернативные способы включают использование m-CPBA, KMnO4/MnO2 в дихлорметане, H2O2 (30%) в двухфазной среде и присутствие катализатора фазового перехода или катализатора в форме мочевинного комплекса (UHP).
Комбинированное использование H2O2 и комплексов металлов, таких как Sc(OTf)3, способствует частично окисленным производным.
Другие известные способы включают, например, использование CAN/NaBrO3 (CAN = нитрат аммония-церия).
Следующие примеры и графические материалы иллюстрируют изобретение, никоим образом не ограничивая его объем.
Краткое описание графических материалов
Фиг. 1А представляет собой гистограмму эффекта ингибирования NGF-индуцированной гипералгезии (латентный период) Соединением 30, вводимым мыши в различных дозах интраперитонеальным путем.
Фиг.1B представляет собой представляет собой гистограмму эффекта ингибирования NGF-индуцированной гипералгезии (латентный период) Соединением 30, вводимым мыши в различных дозах пероральным путем.
Фиг. 2А представляет собой график, относящийся к числу спонтанных отдергиваний после введения крысе интраперитонеальным путем Соединения 30 в различных дозах.
Фиг. 2B представляет собой график, относящийся к мере механической аллодинии после перорального введения крысе Соединения 30 в различных дозах.
Фиг. 3A представляет собой гистограмму баллов использования конечности в зависимости от времени после введения физиологического раствора или Соединения 30 в дозе 2,5 мг/кг.
Фиг. 3B представляет собой гистограмму отдергиваний, спровоцированных легким касанием, в зависимости от времени после введения физиологического раствора или Соединения 30 в дозе 2,5 мг/кг.
Примеры
Использованы следующие сокращения:
I. Синтез соединений согласно изобретению
Примеры способа A1
Пример 1: 5-йод-1H-пиразоло[3,4-b]пиридин-3-амин

Пример 1а: 2-гидрокси-5-йодникотинонитрил
9 г (0,5 экв.) N-йодсукцинимида при комнатной температуре добавляли к раствору 10 г (83 ммоль) 2-гидроксиникотинонитрила в 150 мл безводного диметилформамида. Реакционную смесь перемешивали при 60°C. После 30 минут перемешивания добавляли 9 г (0,5 экв.) N-йодсукцинимида, а затем реакционную смесь перемешивали при 60°C в течение 5 часов. Растворитель выпаривали, и образовавшийся осадок фильтровали, промывали водой и диэтиловым эфиром, а затем высушивали в вакууме с получением 18,5 г (90%) 2-гидрокси-5-йодникотинонитрила в форме бежевого порошка.
ЖХМС (ЭР, m/z): (М+1) 246,93
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 12.79 (1Н, s, ОН), 8.36 (1Н, d, СНаром), 8.04 (1H, d, СНаром).
Пример 1b: 2-хлор-5-йодникотинонитрил
30,7 мл (329 ммоль) оксихлорида фосфора при 0°C и 6 капель серной кислоты добавляли к 9 г (6,6 ммоль) 2-гидрокси-5-йодникотинонитрила. Реакционную смесь нагревали при 110°C в течение 5 часов, а затем при комнатной температуре в течение ночи. Реакционную смесь наливали в химический стакан, содержащий лед и небольшое количество воды, и образовался осадок. Смеси давали постепенно вернуться к комнатной температуре, а затем фильтровали и промывали водой. Твердое вещество высушивали с получением 6,8 г (70%) 2-хлор-5-йодникотинонитрила.
ЖХМС (ЭР, m/z): (М+1) 265,45
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 9.61 (1Н, d, СНаром), 9.14 (1Н, d, СНаром).
Пример 1: 5-йод-1H-пиразоло[3,4-b]пиридин-3-амин
Гидразин (3,86 мл, 79 ммоль) добавляли при комнатной температуре к 7 г (26,5 ммоль) раствора 2-хлор-5-йодникотинонитрила в 25 мл пропан-2-ола. Реакционную смесь нагревали при 85°C в течение 7 часов, а затем при комнатной температуре в течение ночи. Суспендированное твердое вещество фильтровали, промывали изопропанолом, а затем эфиром и высушивали в печи при 50°C с получением 6 г (87%) 5-йод-1Н-пиразоло[3,4-b]пиридин-3-амина.
ЖХМС (ЭР, m/z): (М+1) 260,95
1H ЯМР: δH млн-1 (400 МГц, ДМСО): 12.12 (1Н, bs, NH), 8.51 (1Н, d, СНаром), 8.45 (1Н, d, СНаром), 5.64 (2Н, bs, NH2).
Следующие соединения были получены в соответствии с тем же способом.


Пример 2: 5-бром-1H-пиразоло[3,4-b]пиридин-3-амин

Пример 2а: 2-метокси-никотинонитрил
4,98 г (217 ммоль) натрия добавляли к 80 мл безводного метанола. Реакционную смесь перемешивали при комнатной температуре в течение 10 минут, а затем добавляли 10 г (72,2 ммоль) 2-хлорникотинонитрила при 0°C. Реакционную смесь перемешивали при 25°C в течение 16 часов. Реакционную смесь подвергали гидролизу путем медленного добавления воды при 0°C. После возврата к комнатной температуре полученный осадок фильтровали, промывали водой, а затем высушивали при 50°C с получением 7,85 г (81%) 2-метокси-никотинонитрила в форме желтого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 135,04
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.46-8.48 (1Н, dd, СНаром), 8.25-8.27 (1Н, dd, СНаром), 7.17-7.20 (1Н, dd, СНаром), 3.99 (3H, s, CH3).
Пример 2b: 5-бром-2-метокси-никотинонитрил
12,23 г (149 ммоль) ацетата натрия, а затем 7,66 мл (149 ммоль) брома при 0°C добавляли к 10 г (74,6 ммоль) раствора 2-метокси-никотинонитрила в 29 мл уксусной кислоты. Реакционную смесь нагревали при 70°C в течение ночи. После возврата к комнатной температуре реакционную смесь добавляли в ледяную баню, и полученный осадок фильтровали, промывали водой, а затем высушивали при 50°C с получением 11,6 г (73%) 5-бром-2-метокси-никотинонитрила в форме белого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 214,95
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.61 (1Н, d, СНаром), 8.60 (1Н, d, СНаром), 3.98 (3H, s, CH3)
Пример 2: 5-бром-1H-пиразоло[3,4-b]пиридин-3-амин
35 мл (23,47 ммоль) гидразина добавляли при комнатной температуре к 5 г (23,47 ммоль) 5-бром-2-метоксиникотинонитрила. Реакцию проводили при 100°C в течение 3 часов. После возврата к комнатной температуре полученный осадок фильтровали, промывали водой, а затем высушивали при 50°C с получением 3,6 г (72%) 5-бром-1Н-пиразоло[3,4-b]пиридин-3-амина в форме желтого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 214,05
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 12.18 (1Н, s, NH), 8.38 (1Н, d, СНаром), 8.37 (1Н, d, СНаром), 5.66 (2H,s, NH).
Примеры способа A2
Пример 3: 5-йод-1H-пиразоло[3,4-b]пиразин-3-амин
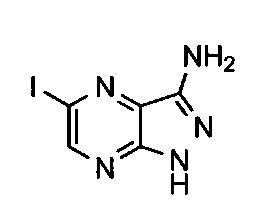
Пример 3a: метил-3-амино-6-йодпиразин-2-карбоксилат
1,5 эквивалента N-йодсукцинимида добавляли при комнатной температуре к раствору 5 г (32,7 ммоль) метил-3-аминопиразин-2-карбоксилата в 25 мл диметилформамида. Реакционную смесь нагревали при 65°C в течение 1 часа, объединяли с 0,5 эквивалентами N-йодсукцинимида и поддерживали при 65°C в течение 24 часов. После возврата к комнатной температуре растворитель выпаривали, а затем продукт несколько раз экстрагировали дихлорметаном. Органические фазы объединяли, промывали 10% раствором бисульфита натрия, высушивали на сульфате магния и концентрировали с получением 8 г (88%) метил-3-амино-6-йодпиразин-2-карбоксилата в форме желтого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 280
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.50 (1Н, s, СНаром), 7.50 (2Н, bs, NH2), 3.20 (3H, s, CH3).
Пример 3b: 3-амино-6-йодпиразин-2-карбоксамид
30 мл аммиака в воде добавляли при перемешивании с помощью магнитной мешалки 15 г (53,8 ммоль) раствора метил-3-амино-6-йодпиразин-2-карбоксилата в 150 мл метанола. Реакционную смесь перемешивали при 25°C в течение 48 часов. После выпаривания растворителей полученный осадок фильтровали, промывали водой, а затем высушивали при 50°C с получением 12,50 г 3-амино-6-йодпиразин-2-карбоксамида (88%) в форме бежевого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 265,02
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.35 (1Н, s, СНаром), 7.85 (1Н, bs, NH), 7.60 (3H, bs, NH), 3.25 (3H, s, CH3).
Пример 3c: N'-(3-циано-5-йодпиразин-2-ил)-N,N-диметилформимидамид
13,59 мл (146 ммоль) оксихлорида фосфора добавляли по каплям при 0°C к 11 г (41,7 ммоль) раствора 3-амино-6-йодпиразин-2-карбоксамида в 80 мл диметилформамида. Реакционную смесь перемешивали при комнатной температуре в течение ночи, а затем наливали в химический стакан, содержащий лед и небольшое количество воды. pH доводили до 8 1 н. раствором соды; образовался осадок. Смеси давали постепенно вернуться к комнатной температуре, а затем образовавшееся твердое вещество фильтровали, промывали водой и высушивали при 50°C с получением 10,50 г N'-(3-циано-5-йодпиразин-2-ил)-N,N-диметилформимидамида (84%) в форме бежевого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 302,07
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.69 (1Н, s, СНаром), 8.67 (1Н, s, СНэтил), 3.20 (3H, s, CH3), 3.11 (3H, s, CH3).
Пример 3d: 3-амино-6-йодпиразин-2-карбонитрил
77 мл (77 ммоль) 1 М раствора соляной кислоты добавляли к 7,7 г (25,6 ммоль) N'-(3-циано-5-йодпиразин-2-ил)-N,N-диметилформимидамида. Реакционную смесь нагревали при 50°C в течение 4 часов, а затем перемешивали при комнатной температуре в течение ночи. Образовавшийся осадок фильтровали, промывали водой и высушивали при 50°C с получением 6 г (95%) 3-амино-6-йодпиразин-2-карбонитрила в форме бежевого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 247,0
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.49 (1Н, s, СНаром), 7.53 (2Н, bs, NH2).
Пример 3e: 3-хлор-6-йодпиразин-2-карбонитрил
64,3 мл соляной кислоты добавляли при -5°C к 7,7 г (31,3 ммоль) 3-амино-6-йодпиразин-2-карбонитрила. При этой температуре к реакционной смеси добавляли раствор нитрита натрия (4,32 г, 62,6 ммоль), растворенного в 9 мл воды и перемешивали в течение 4 часов при -50°C, а затем при комнатной температуре в течение ночи. К реакционной смеси добавляли другой эквивалент нитрита натрия, и образовавшийся осадок фильтровали, промывали водой и высушивали при 50°C с получением 3,65 г (44%) 3-хлор-6-йодпиразин-2-карбонитрила в форме бежевого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 266,49
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 9.13 (1Н, s, СНаром)
Пример 3: 5-йод-1H-пиразоло[3,4-b]пиразин-3-амин
0,74 мл (9,8 ммоль) гидразина добавляли к 2,6 г (9,80 ммоль) раствора 3-хлор-6-йодпиразин-2-карбонитрила в 15 мл бутанола. Реакционную смесь нагревали при 110°C в течение 5 часов, а затем оставляли при комнатной температуре в течение ночи. Суспендированное твердое вещество фильтровали, промывали бутанолом, а затем высушивали в печи при 50°C с получением 2,2 г (86%) 5-йод-1Н-пиразоло[3,4-b]пиразин-3-амина в форме коричневого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 262,02
1H ЯМР: δH млн-1 (400 МГц, ДМСО): 12.59 (1Н, bs, NH), 8.60 (1Н, d, СНаром), 5.83 (2Н, bs, NH2).
Примеры способа A3
Пример 4: 5-йод-6-метокси-1H-пиразоло[3,4-b]пиридин-3-амин

Пример 4а: этил-5-циано-2-гидрокси-6-(метилтио)никотинат
Этил-5-циано-2-гидрокси-6-(метилтио)никотинат получают, следуя методу Ya. Yu. Yakunin et al., Russian Chemical Bulletin, 1999, 48(1), 195-6, при суммарном выходе 34%.
ЖХМС (ЭР, m/z): (M-1) 237,22
1H ЯМР: δH млн-1 (400 МГц, ДМСО): 12.72 (1Н, bs, ОН), 8.40 (1Н, s, СНаром), 4.29 (2Н, q, CH2), 2.64 (3H, s, CH3), 1.30 (3H, t, CH3).
Пример 4b: 5-циано-2-гидрокси-6-(метилтио)никотиновая кислота
4,16 г (2 экв.) моногидрата гидроксида лития добавляли при комнатной температуре к раствору 11,8 г (49,5 ммоль) этил-5-циано-2-гидрокси-6-(метилтио)никотината в 100 мл этанола и 100 мл воды. Реакционную смесь перемешивали при 60°C в течение 2 часов. Этанол выпаривали и добавляли 1 н. водный раствор соды. Водную фазу промывали этилацетатом, а затем снова подкисляли 1 н. водным раствором хлорида водорода (рН=1). Образовавшийся осадок фильтровали, промывали водой и диэтиловым эфиром, а затем высушивали в вакууме с получением 9,9 г (95%) 5-циано-2-гидрокси-6-(метилтио)никотиновой кислоты в форме коричневого порошка.
ЖХМС (ЭР, m/z): (M-1) 209,09
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.32 (1Н, s, СНаром), 2.61 (3H, s, CH3).
Пример 4c: 6-гидрокси-2-(метилтио)никотинонитрил
Раствор 6 г (28,5 ммоль) 5-циано-2-гидрокси-6-(метилтио)никотиновой кислоты в 35 мл дифенилового эфира перемешивали при 250°C в течение 4 часов. После возврата к комнатной температуре добавляли 100 мл циклогексана, и реакционную смесь растирали в течение 30 минут. Образовавшееся твердое вещество фильтровали, тщательно промывали циклогексаном, а затем высушивали в вакууме с получением 2,87 г (60%) 6-гидрокси-2-(метилтио)никотинонитрила в форме коричневого порошка.
ЖХМС (ЭР, m/z): (М+1) 167,12
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 12.16 (1Н, bs, ОН), 7.92 (1Н, d, СНаром), 6.46 (1Н, d, СНаром), 2.59 (3H, s, CH3).
Пример 4d: 6-гидрокси-5-йод-2-(метилтио)никотинонитрил
6 г (1,6 экв.) сульфата серебра и 4,58 г (1,5 экв.) йода последовательно добавляли к раствору 2 г (12 ммоль) 6-гидрокси-2-(метилтио)никотинонитрила в 200 мл этанола. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Твердое вещество фильтровали, и остаток тщательно промывали метанолом. Фильтрат выпаривали, а затем растворяли в этилацетате. Органическую фазу промывали водой три раза, высушивали на сульфате магния и выпаривали с получением 3,18 г (90%) 6-гидрокси-5-йод-2-(метилтио)никотинонитрила в форме желтого порошка.
ЖХМС (ЭР, m/z): (М+1) 292,93
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 12.96 (1Н, bs, ОН), 8.38 (1Н, s, СНаром), 2.62 (3H, s, CH3).
Пример 4е: 5-йод-6-метокси-2-(метилтио)никотинонитрил
905 мкл (2 экв.) метилиодида и 2,1 г (1,05 экв.) карбоната серебра последовательно добавляли к раствору 2,12 г (7,26 ммоль) 6-гидрокси-5-йод-2-(метилтио)никотинонитрила в 20 мл 1,4-диоксана. Реакционную смесь перемешивали при 60°C в течение 5 часов. Твердое вещество фильтровали, и остаток тщательно промывали метанолом. Фильтрат выпаривали, и остаток очищали колоночной хроматографией на силикагеле (4:6 дихлорметан/циклогексан в качестве элюента) с получением 1,52 г (68%) 5-йод-6-метокси-2-(метилтио)никотинонитрила в форме белого порошка.
ЖХМС (ЭР, m/z): (М+1) 306,95
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.50 (1Н, s, СНаром), 4.04 (3H, s, CH3), 2.63 (3H, s, CH3).
Пример 4f: 5-йод-6-метокси-2-(метилсульфинил)никотинонитрил
1,42 г (1,1 экв.) 70% 3-хлорпербензойной кислоты добавляли к раствору 1,6 г (5,23 ммоль) 5-йод-6-метокси-2-(метилтио)никотинонитрила в 20 мл дихлорметана. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли этилацетат, и органическую фазу промывали насыщенным раствором бикарбоната натрия, высушивали на сульфате магния, фильтровали и выпаривали с получением 1,63 г (97%) 5-йод-6-метокси-2-(метилсульфинил)никотинонитрила в форме белого порошка, который может также содержать 5-йод-6-метокси-2-(метилсульфонил)никотинонитрила в небольших долях (<20%). При необходимости смесь используют как таковую на следующих стадиях.
ЖХМС (ЭР, m/z): (М+1) 322,95
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.86 (1Н, s, СНаром), 4.05 (3H, s, CH3), 2.95 (3H, s, CH3).
Пример 4: 5-йод-6-метокси-1H-пиразоло[3,4-b]пиридин-3-амин
294 мкл (1,2 экв.) моногидрата гидразина добавляли к раствору 1,63 г (5,05 ммоль) 5-йод-6-метокси-2-(метилсульфинил)никотинонитрила в 30 мл 2-пропанола. Реакционную смесь перемешивали при 80°C в течение 9 часов. После возврата к комнатной температуре образовавшееся твердое вещество фильтровали и промывали 2-пропанолом с получением 1,14 г (78%) 5-йод-6-метокси-1Н-пиразоло[3,4-b]пиридин-3-амина в форме белого порошка.
ЖХМС (ЭР, m/z): (М+1) 291,00
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 11.87 (1Н, s, NH), 8.49 (1Н, s, СНаром), 5.49 (2Н, bs, NH2), 3.90 (3H, s, CH3).
Пример 5: 5-йод-1H-пиразоло[3,4-b]пиридин-3,6-диамин

Пример 5а: 4-метилморфолиния (2,4)-этил-5-амино-2,4-дициано-5-меркаптопента-2,4-диеноат
4-метилморфолиния (2,4)-этил-5-амино-2,4-дициано-5-меркаптопента-2,4-диеноат получают в соответствии с методом, описанным V.D. Dyachenko et al., Chemistry of Heterocyclic Compounds, 2005, 41 (4), 503-10 при выходе 50%.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 9.60 (1Н, bs, NH), 8.66 (1Н, s, СН), 8.33 (1Н, bs, NH), 7.43 (1Н, bs, NH), 4.08 (2Н, q, CH2), 3.82-4.02 (2Н, m, CH2), 3.55-3.78 (2Н, m, CH2), 3.24-3.42 (2Н, m, CH2), 3.98-3.17 (2Н, m, CH2), 2.81 (3H, s, CH3), 1.19 (3H, t, CH3).
Пример 5b: этил-2-амино-5-циано-6-(метилтио)никотинат
2,73 мл (1 экв.) метилиодида добавляли к раствору 14,2 г (43,8 ммоль) 4-метилморфолиния (2,4)-этил-5-амино-2,4-дициано-5-меркаптопента-2,4-диеноата в 78 мл N,N-диметилформамида. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, а затем при 75°C в течение 20 часов. После возврата к комнатной температуре добавляли воду, и образовавшееся твердое вещество фильтровали и высушивали в вакууме с получением 10,31 г (100%) этил-2-амино-5-циано-6-(метилтио)никотината в форме бежевого порошка.
ЖХМС (ЭР, m/z): (М+1) 238,20
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.25 (1Н, s, СНаром), 8.19 (1Н, bs, NH), 7.99 (1Н, bs, NH), 4.27 (2Н, q, CH2), 2.58 (3H, s, CH3), 1.31 (3H, t, CH3).
Пример 5c: 2-амино-5-циано-6-(метилтио)никотиновая кислота
3,08 г (2 экв.) моногидрата гидроксида лития добавляли при комнатной температуре к раствору 8,7 г (36,7 ммоль) этил-2-амино-5-циано-6-(метилтио)никотината в 87 мл этанола и 87 мл воды. Реакционную смесь перемешивали при 60°C в течение 2 часов. Этанол выпаривали и добавляли 1 н. водный раствор соды. Водную фазу промывали этилацетатом, а затем снова подкисляли добавлением 1 н. водного раствора хлорида водорода (pH=1). Образовавшийся осадок фильтровали, промывали водой и диэтиловым эфиром, а затем высушивали в вакууме с получением 7,67 г (количественно) 2-амино-5-циано-6-(метилтио)никотиновой кислоты в форме коричневого порошка.
ЖХМС (ЭР, m/z): (М+1) 210,16
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 13.28 (1Н, bs, CO2H), 8.21 (1Н, s, СНаром), 8.13 (2Н, bs, NH2), 2.57 (3H, s, CH3).
Пример 5d: 6-амино-2-(метилтио)никотинонитрил
Раствор 3 г (14,3 ммоль) 2-амино-5-циано-6-(метилтио)никотиновой кислоты в 30 мл дифенилового эфира перемешивали при 255°C в течение 60 часов. После возврата к комнатной температуре добавляли 60 мл циклогексана, и реакционную смесь растирали в течение 30 минут. Образовавшееся твердое вещество фильтровали, а затем тщательно промывали циклогексаном. Твердое вещество снова растворяли в этилацетате, а затем органическую фазу промывали водой, высушивали на сульфате магния, фильтровали, а затем выпаривали с получением 1,32 г (55%) 6-амино-2-(метилтио)никотинонитрила в форме коричневого порошка.
ЖХМС (ЭР, m/z): (М+1) 166,13
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 7.58 (1Н, d, СНаром), 7.12 (2Н, bs, NH2), 6.20 (1Н, d, СНаром), 2.51 (3H, s, CH3).
Пример 5е: 6-амино-5-йод-2-(метилтио)никотинонитрил
3,75 г (1,5 экв.) сульфата серебра и 2,85 г (1,4 экв.) йода последовательно добавляли к раствору 1,32 г (8,02 ммоль) 6-амино-2-(метилтио)никотинонитрила в 65 мл этанола. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Твердое вещество фильтровали, и остаток тщательно промывали метанолом. Фильтрат выпаривали и снова растворяли в этилацетате. Органическую фазу промывали водой три раза, высушивали на сульфате магния и выпаривали с получением 1,89 г (81%) 6-амино-5-йод-2-(метилтио)никотинонитрила в форме коричневого порошка.
ЖХМС (ЭР, m/z): (М+1) 291,99
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.13 (1Н, s, СНаром), 7.19 (1Н, широкий плоский синглет, NH2), 2.51 (3H, s, CH3).
Пример 5f: 6-амино-5-йод-2-(метилсульфинил)никотинонитрил
1,77 г (1,1 экв.) 70% 3-хлорпербензойной кислоты добавляли к раствору 1,89 г (6,51 ммоль) 6-амино-5-йод-2-(метилтио)никотинонитрила в 60 мл дихлорметана. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли этилацетат, и органическую фазу промывали насыщенным раствором бикарбоната натрия, высушивали на сульфате магния, фильтровали и выпаривали с получением 1,5 г (75%) 6-амино-5-йод-2-(метилсульфинил)никотинонитрила в форме белого порошка, который может также содержать 6-амино-5-йод-2-(метилсульфонил)никотинонитрил в небольших долях (<20%). При необходимости эту смесь используют как таковую на последующих стадиях.
ЖХМС (ЭР, m/z): (М+1) 307,98
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.45 (1Н, s, СНаром), 7.70 (2Н, широкий плоский синглет, NH2), 2.84 (3H, s, CH3).
Пример 5: 5-йод-1H-пиразоло[3,4-b]пиридин-3,6-диамин
275 мкл (2 экв.) моногидрата гидразина добавляли к раствору 872 мг (2,84 ммоль) 6-амино-5-йод-2-(метилсульфинил)никотинонитрила в 11 мл 2-пропанола. Реакционную смесь перемешивали при 80°C в течение 3 часов. Добавляли воду, и продукт экстрагировали этилацетатом. Органическую фазу высушивали на сульфате магния, фильтровали и выпаривали. Остаток растирали в минимальном количестве диизопропилового эфира. Твердое вещество фильтровали с получением 523 мг (67%) 5-йод-1Н-пиразоло[3,4-b]пиридин-3,6-диамина в форме коричневого порошка.
ЖХМС (ЭР, m/z): (М+1) 276,00
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 11.23 (1Н, s, NH), 8.26 (1Н, s, СНаром), 6.11 (2Н, bs, NH2), 5.25 (2Н, bs, NH2).
Примеры способа B1
Пример 6: 5-(3,5-дифторбензилтио)-1H-пиразоло[4,3-b]пиридин-3-амин
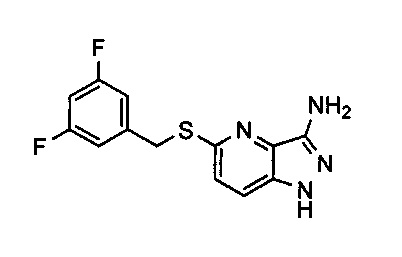
Пример 6а: 6-хлор-3-нитропиколинонитрил
2,6-Дихлор-3-нитропиридин (5,18 ммоль, 1 г) смешивали с 5 мл N-метил-2-пирролидинона в микроволновом реакторе. Реакционную смесь нагревали при 180°C в течение 15 минут (6 бар). Неочищенный продукт реакции растворяли в этилацетате, фильтровали и промывали несколько раз, используя водную фазу. Органическую фазу собирали, высушивали на сульфате магния и концентрировали до сухого состояния. Полученный таким путем неочищенный продукт очищали хроматографией на силикагеле (гептан/AcOEt) с получением после концентрирования 0,62 г (65%) коричневого масла.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.81 (1Н, d, СНаром), 8.18 (1Н, d, СНаром).
Пример 6b: 3-нитро-6-тиоксо-1,6-дигидропиридин-2-карбонитрил
Один эквивалент NaSH:H2O добавляли к раствору 6-хлор-3-нитропиколинонитрила (5,45 ммоль, 1 г) в 20 мл EtOH. Цвет становится оранжевым. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Затем неочищенный продукт реакции концентрировали, снова растворяли в этилацетате и экстрагировали несколько раз, используя кислую водную фазу (1 н. HCl), а затем нейтральную фазу. Органическую фазу концентрировали, и неочищенный продукт реакции перекристаллизовали в ацетоне с получением 0,64 г (79%) желтых кристаллов.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.71 (1Н, d, СНаром), 8.27 (1Н, d, СНаром).
Пример 6c: 6-(3,5-дифторбензилтио)-3-нитропиколинонитрил
Смесь 3-нитро-6-тиоксо-1,6-дигидропиридин-2-карбонитрила (4,42 ммоль, 1,34 г), 3,5-дифторбензилбензилбромида (8,83 ммоль, 1,828 г) и K2CO3 (11,04 ммоль, 1,525 г) в 5 мл ацетона нагревали при 70°C в течение 10 часов, а затем выпаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле (AcOEt/гептан) с получением 1,33 г (98%) ожидаемого продукта.
ЖХМС (ЭР-): m/z 306 (M-H+).
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.53 (1Н, d, СНаром), 7.91 (1Н, d, СНаром), 7.21 (2Н, m), 7.17 (1Н, m), 4.55 (2Н, CH2).
Пример 6d: 3-амино-6-(3,5-дифторбензилтио)пиколинамид
Смесь 6-(3,5-дифторбензилтио)-3-нитропиколинонитрила (0,05 г, 0,163 ммоль) и PtO2 (0,739 мг, 3,25 мкмоль) в 10 мл МеОН помещали при перемешивании при атмосферном давлении водорода в течение 2 часов. Катализатор фильтровали, раствор концентрировали, и полученный таким путем остаток очищали хроматографией на силикагеле (AcOEt/гептан) с получением после концентрирования 0,04 г (83%) белых кристаллов.
ЖХМС (ЭР+) m/z: 296 (МН+).
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 7.84 (1Н, широкий s, NH), 7.40 (1Н, широкий s, NH), 7.14 (1Н, d, СНаром), 7.08 (4Н, m, СНаром), 6.80 (2Н, широкий s, NH2), 4.43 (2Н, s, CH2).
Пример 6e: 3-амино-6-(3,5-дифторбензилтио)пиколинонитрил
Смесь 3-амино-6-(3,5-дифторбензилтио)пиколинамида (2,37 ммоль, 0,7 г) и P2Cl5 (9,48 ммоль, 1,346 г), 20 мл толуола и 1 мл ионного растворителя (1-бутил-3-метилимидазолия тетрафторборат) помещали в микроволновой реактор, а затем нагревали при 140°C в течение 30 минут. Затем неочищенный продукт реакции концентрировали при пониженном давлении, и полученные таким путем оранжевые кристаллы снова растворяли в этилацетате и промывали, используя насыщенный водный раствор NaHCO3. Органическую фазу высушивали на сульфате магния, а затем концентрировали с получением 0,7 г коричневого масла. Этот неочищенный продукт реакции очищали хроматографией на силикагеле (AcOEt/гептан + 0,1% NEt3) с получением после концентрирования 0,15 г (23%) оранжевых кристаллов.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 7.73 (1Н, d, СНаром), 7.25 (2Н, m, СНаром), 7.18 (1Н, m), 6.85 (1Н, d), 5.43 (2Н, CH2).
Пример 6: 5-(3,5-дифторбензилтио)-1H-пиразоло[4,3-b]пиридин-3-амин
Раствор NaNO2 в 3 мл воды, охлажденный до 0°C, добавляли по каплям при 0°C к 3-амино-6-(3,5-дифторбензилтио)пиколинонитрилу (1,587 ммоль, 0,44 г) в 15 мл 6 н. раствора HCl. Через 15 минут добавляли по каплям охлажденный до 0°C раствор SnCl2-2H20, разведенного в 4 мл 12 н. HCl. Затем реакционную смесь перемешивали при 25°C в течение 1 часа. Раствор экстрагировали этилацетатом, а затем промывали, используя насыщенный раствор NaHCO3, а затем насыщенный раствор NaCl. Органическую фазу собирали, высушивали на сульфате магния, а затем концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле (AcOEt/гептан) с получением после концентрирования органических фаз 0,07 г (15%) черных кристаллов.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 11.64 (1Н, s, NH), 7.63 (1Н, d, СНаром), 7.21 (2Н, m, СНаром), 7.13 (1Н, d, СНаром), 7.04 (1Н, m, СНаром), 5.38 (2Н, s, NH2), 4.51 (2Н, s, CH2).
Следующие соединения получены подобным способом:


Примеры способа B2
Пример 7: 5-(3,5-дихлорфенилтио)-1H-пиразоло[4,3-b]пиридин-3-амин

Пример 7а: 6-(3,5-дихлорфенилтио)-3-нитропиколинонитрил
Смесь 6-хлор-3-нитропиколинонитрила (3,70 г, 0,02 моль), 3,5-дихлорбензолтиола (3,60 г, 0,02 моль) и K2CO3 (5,6 г, 0,04 моль) в 100 мл ацетонитрила подвергают взаимодействию при 70°C в течение 16 часов. Неочищенный продукт реакции разбавляли в этилацетатной фракции и промывали, используя водную фазу. Органическую фазу высушивали сульфатом натрия, и остаток очищали хроматографией на силикагеле (AcOEt/петролейный эфир) с получением 5,4 г (80%) желтого твердого вещества.
Пример 7b: 3-амино-6-(3,5-дихлорфенилтио)пиколинонитрил
10 мл концентрированной HCl добавляли к раствору 6-(3,5-дихлорфенилтио)-3-нитропиколинонитрила (3,4 г, 0,01 моль) в 50 мл метанола при перемешивании. Реакционную смесь кипятили с обратным холодильником, добавляли 1,68 г (0,03 моль) железа и перемешивали в течение 10 минут. После возврата к комнатной температуре к реакционной смеси добавляли 100 мл этилацетата и 50 мл воды. Доводили pH до 10, используя 30% раствор соды, и органическую фазу экстрагировали, а затем высушивали на безводном сульфате натрия, а затем концентрировали. Остаток очищали хроматографией на силикагеле (этилацетат/петролейный эфир) с получением после концентрирования фракций 2,82 г (91%) желтого твердого вещества.
ЖХМС (m/e): 296 (М+Н+).
Пример 7: 5-(3,5-дихлорфенилтио)-1H-пиразоло[4,3-b]пиридин-3-амин
Раствор 350 мг NaNO2 (5,07 ммоль) в воде (2 мл) добавляли к перемешиваемому раствору 1,5 г 3-амино-6-(3,5-дихлорфенилтио)пиколинонитрила (5,07 ммоль) в 100 мл 50% серной кислоты при 0°C. Смесь перемешивали в течение 20 минут при 0-5°C. Затем добавляли раствор 2,9 г SnCl2⋅2H2O (12,7 ммоль, 2,5 экв.) в соляной кислоте (12 н. раствор, 10 мл), и этот раствор перемешивали в течение 1 часа при комнатной температуре. Образовавшееся твердое вещество фильтровали, а затем дважды промывали 20 мл воды. Твердое вещество суспендировали в 100 мл, и pH доводили до 10 добавлением 30% раствора соды. Органическую фазу отделяли, а затем высушивали на безводном сульфате натрия, а затем концентрировали в вакууме. Получили светло-желтое твердое вещество после перекристаллизации в этилацетате (470 мг, 34%).
ЖХМС m/z 311 (М+Н+).
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 11.91 (1Н, bs, NH), 7.79 (1Н, d, СНаром), 7.55 (1Н, s, СНаром), 7.36 (2Н, s, СНаром), 7.33 (1Н, m, СНаром), 5.42 (2Н, s, NH2).
Следующие соединения получены подобным способом:
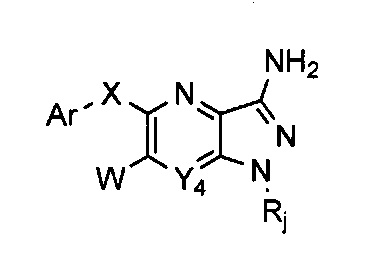



Пример 8: N5-(3,5-дифторфенил)-1H-пиразоло[4,3-b]пиридин-3,5-диамин

Пример 8а: 6-(3,5-дифторфениламино)-3-нитропиколинонитрил
Смесь 6,5 г 6-хлор-3-нитропиколинонитрила (0,065 моль) и 6,2 г 3,5-дифторанилина (0,048 моль) в 100 мл толуола нагревали при 70°C в течение 5 часов. Неочищенный продукт реакции разбавляли в этилацетатной фракции и промывали, используя насыщенный раствор NaCl. Органическую фазу высушивали сульфатом натрия, и остаток очищали хроматографией на силикагеле (AcOEt/петролейный эфир) с получением 3,9 г (33%) желтого твердого вещества.
Пример 8b: 3-амино-6-(3,5-дифторфениламино)пиколинонитрил
10 мл концентрированной HCl добавляли к раствору 6-(3,5-дихлорфенилтио)-3-нитропиколинонитрила (3,9 г, 0,0141 моль) в 150 мл этанола при перемешивании. Реакционную смесь кипятили с обратным холодильником, добавляли 2,4 г железа (0,0423 моль) и перемешивали при 80°C в течение 1 часа. После возврата к 0°C pH доводили до 8, используя 1 н. раствор соды, и реакционную смесь фильтровали на целлите. К реакционной смеси добавляли 100 мл этилацетата и 50 мл метанола. Органическую фазу экстрагировали, и водную фазу экстрагировали несколько раз этилацетатными фракциями. Органические фазы объединяли, а затем высушивали на безводном сульфате натрия, а затем концентрировали с получением после концентрирования 2,3 г (66%) коричневого твердого вещества.
Пример 8: 5-(3,5-дифторфениламино)-1H-пиразоло[4,3-b]пиридин-3-амин
Раствор 713 мг NaNO2 (10,3 ммоль) в воде (5 мл) добавляли по каплям к перемешиваемому раствору 2,3 г 3-амино-6-(3,5-дифторфениламино)пиколинонитрила (9,4 ммоль) в 100 мл 6 н. соляной кислоты при 0°C. Смесь перемешивали в течение 20 минут при 0-5°C. Затем добавляли по каплям раствор 5,3 г SnCl2⋅2H2O (23,5 ммоль, 2,5 экв.) в соляной кислоте (12 н. раствор, 30 мл), и раствор перемешивали в течение 1 часа при комнатной температуре. Затем реакционную смесь охлаждали при 0°C и подщелачивали до pH 8, используя 30% раствор соды. Смесь экстрагировали этилацетатом и промывали, используя насыщенный раствор NaCl, и органическую фазу высушивали на безводном сульфате натрия, после чего концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (AcOEt). Получили светло-желтое твердое вещество (530 мг, 22%).
ЖХМС: m/z 262 (М+Н+).
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 11.47 (s, 1Н), 9.45 (s, 1Н), 7.65 (m, 3H), 6.87 (d, 1Н, J=7.8 Гц), 6.60 (m, 1Н), 5.09 (s, 2Н).
Следующие соединения получили подобным способом:


Пример способа B3
Пример 9: 5-(3,5-дифторбензил)-1Н-пиразоло[4,3-b]пиридин-3-амин

Это соединение может быть получено из следующих промежуточных соединений согласно способу B3.
Пример 9а: 2-(3,5-дифторбензил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан
Пример 9b: 6-(3,5-дифторбензил)-3-нитропиколинонитрил
Пример 9c: 3-амино-6-(3,5-дифторбензил)пиколинонитрил
Пример способа B4
Пример 10: 3-амино-N-(3,5-дифторфенил)-1H-пиразоло[3,4-b]пиридин-5-сульфонамид

Пример 10а: 5-(N-(3,5-дифторфенил)сульфамоил)никотиновая кислота
2,74 г (9,64 ммоль) этил-2-хлор-5-(хлорсульфонил)никотината в растворе в 20 мл безводного дихлорметана добавляли по каплям при 0°C к смеси 623 мг (4,82 ммоль) 3,5-дифторанилина и 1,68 мл (12,05 ммоль) триэтиламина, разбавленного в смеси 10 мл безводного дихлорметана. Раствор перемешивали при комнатной температуре в течение 3 часов. Растворитель выпаривали с получением светло-коричневого твердого вещества. Твердое вещество растирали в 20 мл метанола, фильтровали, а затем промывали 3 мл метанола с получением 2,85 г белого твердого вещества.
Это твердое вещество снова растворяли в 25 мл тетрагидрофурана и добавляли раствор 0,421 г (10,04 ммоль) моногидрата гидроксида лития в 10 мл воды. Реакционную смесь оставляли при перемешивании на 3 часа при 35°C, а затем разбавляли в воде, подкисляли 1 н. соляной кислотой и экстрагировали этилацетатом. Органическую фазу собирали, высушивали на сульфате натрия, фильтровали и концентрировали с получением 1,12 г 5-(N-(3,5-дифторфенил)сульфамоил)никотиновой кислоты в форме оранжевого твердого вещества (выход=67%).
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.91 (1Н, s, СНаром), 8.51 (1Н, s, СНаром), 7.02 (1Н, dd, СНаром), 6.83 (2Н, d, СНаром).
Пример 10b: 2-хлор-5-(N-(3,5-дифторфенил)сульфамоил)никотинамид
0,288 мл (3,87 ммоль) тионилхлорида и каплю ДМФ последовательно добавляли к 0,450 г (1,29 ммоль) 2-хлор-5-(N-(3,5-
дифторфенил)сульфамоил)никотиновой кислоты в 5 мл безводного толуола. Смесь помещают при перемешивании в толуол, кипящий с обратным холодильником, на 2 часа. Затем хлорангидридную реакционную смесь добавляли по каплям к 4,5 мл 25% ледяного раствора гидроксида аммония. Наблюдали выделение газа. Реакционную смесь оставляли при перемешивании при комнатной температуре на 30 минут. Реакционную смесь экстрагировали несколько раз этилацетатом. Объединенные органические фазы высушивали на безводном сульфате натрия, а затем концентрировали. Получили 0,315 г 2-хлор-5-(N-(3,5-дифторфенил)сульфамоил)никотинамида в форме светло-коричневого твердого вещества (выход=72%).
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 11.18 (1Н, bs, NH), 8.86 (1Н, s, СНаром), 8.22 (1Н, s, СНаром), 8.21 (1Н, bs, NH), 7.98 (1Н, bs, NH), 6.96 (1Н, dd, СНаром), 6.79 (2Н, d, СНаром).
Пример 10c: 6-хлор-5-циано-N-(3,5-дифторфенил)пиридин-3-сульфонамид
3,4 мл (36,2 ммоль) фосфорилхлорида и каплю концентрированной серной кислоты добавляли к 0,315 г (0,906 ммоль) 2-хлор-5-(N-(3,5-дифторфенил)сульфамоил)никотинамида. Реакционную смесь перемешивали в течение 2 часов при 90°C, а затем добавляли по каплям в лед. Коричневое твердое вещество фильтровали, промывали водой, а затем высушивали в вакууме. Получили 0,217 г 6-хлор-5-циано-N-(3,5-дифторфенил)пиридин-3-сульфонамида в форме светло-коричневого твердого вещества (выход=72%).
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 11.34 (1Н, bs, NH), 9.04 (1Н, s, СНаром), 8.92 (1Н, s, СНаром), 7.03 (1Н, dd, СНаром), 6.85 (2Н, d, СНаром).
Пример 10: 3-амино-N-(3,5-дифторфенил)-1H-пиразоло[3,4-b]пиридин-5-сульфонамид
0,377 мл (2,63 ммоль) 35% гидразина добавляли к 0,217 г (0,658 ммоль) 6-хлор-5-циано-N-(3,5-дифторфенил)пиридин-3-сульфонамида, разбавленного в 6 мл изопропанола. Раствор нагревали при 75°C в течение 2 часов. Растворитель выпаривали с получением 0,214 г 3-амино-N-(3,5-дифторфенил)-1Н-пиразоло[3,4-b]пиридин-5-сульфонамида в форме желтого твердого вещества (выход=100%).
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.74 (1Н, d, СНаром), 8.68 (1Н, d, СНаром), 6.88 (1Н, dd, СНаром), 6.80 (2Н, d, СНаром), 6.04 (2Н, bs, NH).
Примеры способа В5
Пример 11: 5-(3,5-дифторбензилокси)-1H-пиразоло[3,4-b]пиридин-3-амин
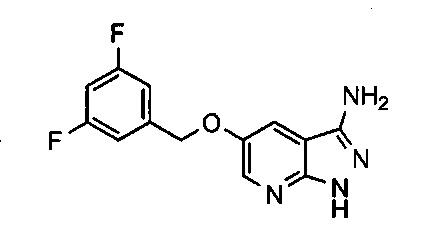
Это соединение может быть получено из следующих промежуточных соединений согласно способу В5.
Пример 11а: 5-гидроксиникотинонитрил
Смесь 1 г 5-метоксиникотинонитрила (7,46 ммоль) и 8,62 г пиридина гидрохлорида нагревали при 200°C в течение 2 часов. Неочищенный продукт реакции разбавляли в водной фракции несколько раз диэтиловым эфиром. Водную фазу подщелачивали добавлением бикарбоната натрия, а затем снова экстрагировали диэтиловым эфиром. Органическую фазу высушивали, а затем концентрировали с получением 850 мг 5-гидроксиникотинонитрила (95%) в форме бежевого твердого вещества.
ЖХМС: m/z 120,94 (М+Н+).
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 10.79 (s, 1Н), 8.46 (s, 1Н, СНаром), 8.42 (s, 1Н, СНаром), 7.60 (s, 1Н, СНаром).
Пример 11b: 5-(3,5-дифторбензилокси)никотинонитрил
876 мг (2 экв.) гидрида натрия постепенно добавляли при 0°C в атмосфере азота к раствору 865 мг 5-гидроксиникотинонитрила (7,2 ммоль) в 15 мл диметилацетамида. Смесь перемешивали 10 мин при 0°C, после чего добавляли 2,24 г (1,5 экв.) 3,5-дифторбензилбромида. Смесь помещали при перемешивании дополнительно на 2,5 часа, после чего разбавляли в этилацетатной фракции и промывали водными фракциями. Органические фазы выделяли, высушивали и концентрировали. Полученный твердый остаток перекристаллизовали в метаноле с получением 1,1 г (68% 5-(3,5-дифторбензилокси)никотинонитрила в форме бежевого порошка.
ЖХМС: m/z 247,11 (М+Н+).
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.69 (s, 1Н, СН), 8.65 (s, 1Н, СН), 8.08 (s, 5 1Н, СН), 7.26 (m, 3H, СН), 5.28 (d, 2Н, CH2).
Пример 11с: 3-циано-5-(3,5-дифторбензилокси)пиридин 1-оксид
224 мг m-CPBA добавляли при 0°C к раствору в ацетонитриле 250 мг 5-(3,5-дифторбензилокси)никотинонитрила. Реакционную смесь перемешивали в течение 20 часов, пока постепенно не образовался осадок. Затем это твердое вещество) фильтровали и промывали с получением 200 мг (75%) 3-циано-5-(3,5-дифторбензилокси)пиридин 1-оксида в форме белого порошка.
ЖХМС: m/z 263,06 (М+Н+).
Пример 11d: 2-хлор-5-(3,5-дифторбензилокси)никотинонитрил
К смеси 650 мг 3-циано-5-(3,5-дифторбензилокси)пиридина 1-оксида в 2,3 мл POCl3 добавляли несколько капель H2SO4 нагревали при 110°C в течение 1 ч 30 мин. Затем неочищенную реакционную смесь наливали в лед, и образовавшийся таким образом осадок выделяли фильтрованием и высушивали в вакууме с получением 600 мг бежевого твердого вещества в форме смеси региоизомеров, содержащей, в основном, желаемый 2-хлор-5-(3,5-дифторбензилокси)никотинонитрил, который использовали без дополнительной очистки.
ЖХМС: m/z 281,02 (M+H+).
Пример 11: 5-(3,5-дифторбензилокси)-1H-пиразоло[3,4-b]пиридин-3-амин
313 мг гидрата гидразина (5 экв.) добавляли к раствору 1,6 г 2-хлор-5-(3,5-дифторбензилокси)никотинонитрила (450 мкмоль) в 10 мл пропан-2-ола. Реакционную смесь нагревали при 100°C в течение 6 часов. После возврата к комнатной температуре, приводящего к осаждению, неочищенную реакционную смесь фильтровали, твердое вещество удаляли, и фильтрат выпаривали до сухого состояния. Затем его очищали хроматографией на колонке силикагеля, элюируя градиентом этилацетата и метанола, причем, фракцию большей полярности выделяли, концентрировали и снова суспендировали в небольшой фракции метанола при перемешивании. Полученное таким образом твердое вещество выделяли и высушивали с получением 221 мг 5-(3,5-дифторбензилокси)-1Н-пиразоло[3,4-b]пиридин-3-амина в форме бежевого твердого вещества, которое использовали без дополнительной очистки.
ЖХМС: m/z 277,07 (М+Н+).
Пример способа B6
Пример 11 бис: N-(3-амино-1Н-пиразоло[3,4-b]пиридин-5-ил)-3,5-дифторбензол-сульфонамид
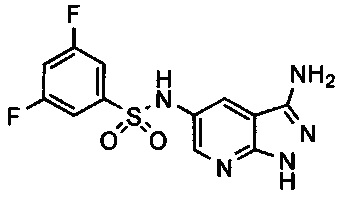
Пример 11бис-а: N-(6-хлор-5-цианопиридин-3-ил)-3,5-дифторбензол-сульфонамид
1,132 г (5,32 ммоль) 3,5-дифторбензол-1-сульфонилхлорида добавляли в атмосфере аргона к раствору 545 мг (3,55 ммоль) 5-амино-2-хлорникотинонитрила в 20 мл безводной смеси 1:1 ТГФ и пиридина. Реакционную смесь нагревали до 70°C в течение 3 часов и оставляли дополнительно на 12 часов при перемешивании при комнатной температуре. Растворитель выпаривали до сухого состояния, и неочищенный продукт реакции снова растворяли в этилацетате и промывали несколькими водными фракциями. Органическую фазу высушивали на сульфате магния, фильтровали, концентрировали, а затем очищали хроматографией на силикагеле с получением 784 мг (67%) N-(6-хлор-5-цианопиридин-3-ил)-3,5-дифторбензол-сульфонамида.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 11,39 (1Н, sl, NH), 8.34 (1Н, m, СНаром), 8.10 (1Н, m, СНаром), 7.67 (1Н, m, СНаром), 7.59 (2Н, m, СНаром).
Пример 11бис: N-(3-амино-1Н-пиразоло[3,4-b]пиридин-5-ил)-3,5-дифторбензол-сульфонамид
1,786 г (35,7 ммоль) гидрата гидразина добавляли в атмосфере аргона к раствору 784 мг (2,38 ммоль) N-(6-хлор-5-цианопиридин-3-ил)-3,5-дифторбензол-сульфонамида в 6 мл этанола. Раствор нагревали до 100°C в течение 20 часов, а затем охлаждали до комнатной температуры. Растворитель выпаривали с получением 810 мг N-(3-амино-1Н-пиразоло[3,4-b]пиридин-5-ил)-3,5-дифторбензол-сульфонамида (100%), который использовали без дополнительной очистки на следующей стадии.
ЖХМС: m/z 326,07 (М+Н+).
Пример способа C1
Пример 12: N6-(2,4-дифторфенил)-1H-пиразоло[3,4-b]пиридин-3,6-диамин
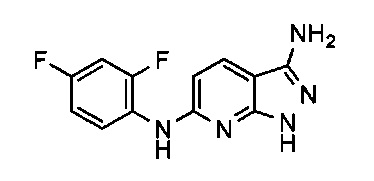
Это соединение может быть получено из следующих промежуточных соединений согласно способу C1.
Пример 12-а: 5-циано-6-(метилтио)пиридин-2-ила трифторметансульфонат
15,26 мл (1,2 экв.) 2-метилпропан-2-олята калия, а затем 9,03 г (1.2 экв.) 1,1,1-трифтор-N-фенил-N-(трифторметилсульфонил)метансульфонамида добавляли по каплям к раствору 3,5 г (21,06 ммоль) 6-гидрокси-2-(метилтио)никотинонитрила в 180 мл тетрагидрофурана в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч 45 мин. Добавляли воду, и продукт экстрагировали этилацетатом. Органическую фазу высушивали на безводном сульфате магния, фильтровали и выпаривали с получением оранжевого твердого вещества. Продукт очищали на колонке силикагеля (элюент: циклогексан/дихлорметан 5:5) с получением 5,31 г (85%) 5-циано-6-(метилтио)пиридин-2-ила трифторметансульфоната в форме желтого твердого вещества.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.57 (1Н, d, СН), 7.52 (1Н, d, СН), 2.59 (3H, s, CH3).
Пример 12-b: 6-(2,4-дифторфениламино)-2-(метилтио)никотинонитрил
0,81 мл (1,2 экв.) 2,4-дифторанилина и 1,53 г (1,4 экв.) карбоната цезия(I) добавляли в атмосфере азота к раствору 2 г (6,71 ммоль) 5-циано-6-(метилтио)пиридин-2-ила трифторметансульфоната в 30 мл 1,4-диоксана. Смесь дегазировали в течение 5 минут в атмосфере аргона, после чего добавляли 0,25 г (0,06 экв.) 2,2'-бис(дифенилфосфино)-1,1'-динафтила и 0,08 г (0,04 экв.) (1Е,4Е)-1,5-дифенилпента-1,4-диен-3-она, комплекса с палладием(II). Реакционную смесь перемешивали при 100°C в течение 2 часов. После возврата к комнатной температуре добавляли этилацетат и соляной раствор. Органическую фазу высушивали на безводном сульфате магния, фильтровали и выпаривали. Полученный остаток очищали хроматографией на силикагеле (элюент: циклогексан/этилацетат 8:2, затем 7:3) с получением 1,52 г (82%) 6-(2,4-дифторфениламино)-2-(метилтио)никотинонитрила в форме белого твердого вещества.
ЖХМС (ЭУ, m/z): (М+1) 278,06.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 9.57 (1Н, s, NH), 7.73-7.86 (2Н, m, СН), 7.28-7.44 (1Н, m, СН), 7.02-7.18 (1Н, m, СН), 6.60 (1Н, d, СН), 2.41 (3H, s, CH3).
Пример 12: N6-(2,4-дифторфенил)-1H-пиразоло[3,4-b]пиридин-3,6-диамин
769 мг (3,12 ммоль) мета-хлорпербензойной кислоты (mCPBA) добавляли в атмосфере аргона к перемешиваемому раствору 786 мг (2,83 ммоль) 6-(2,4-дифторфениламино)-2-(метилтио)никотинонитрила в 25 мл дихлорметана. Реакционную смесь перемешивали 1 час при комнатной температуре, после чего добавляли фракцию этилацетата и промывали эту органическую фазу насыщенным раствором NaHCO3. Объединенные органические фазы высушивали на сульфате магния и выпаривали до сухого состояния. Неочищенный продукт реакции снова растворяли в 10 мл пропанола и добавляли 2 эквивалента гидразина гидрохлорида в воде. Смесь нагревали при 90°C в течение 6 часов, после чего разбавляли в воде и экстрагировали этилацетатом. Органическую фазу высушивали на сульфате магния и выпаривали до сухого состояния, после чего очищали хроматографией на силикагеле с получением 495 мг N6-(2,4-дифторфенил)-1Н-пиразоло[3,4-b]пиридин-3,6-диамина в форме желто-оранжевого твердого вещества (67%).
ЖХМС (ЭУ, m/z): (М+1) 262,14.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 11.40 (1Н, s, NH), 8.76 (1Н, s, NH), 8.15 (1Н, m, СН), 7.81 (1Н, d, СН), 7.28 (1Н, m, СН), 7.06 (1Н, m, СН), 6.55 (1Н, d, СН), 5.24 (2Н, s, NH2).
Следующее соединение получили подобным способом:
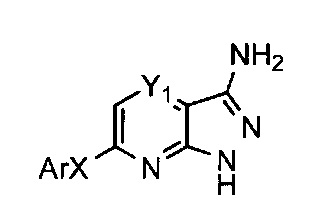

Пример 12бис: N6-(2,4-дифторфенил)N6-метил-1Н-пиразоло[3,4-b]пиридин-3,6-диамин

Пример 12бис-а: 6-((3,5-дифторфенил)(метил)амино)-2-(метилтио)никотинонитрил
3,05 мл (5,04 ммоль) 2-метилпропан-2-олята калия, а затем 286 мкл (1,8 экв.) йодметана добавляли по каплям в атмосфере азота к раствору 700 мг (2,52 ммоль) 6-(2,4-дифторфениламино)-2-(метилтио)никотинонитрила в 20 мл N,N-диметилформамида. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, а затем добавляли 126 мкл (0,8 экв., 2,02 ммоль) йодметана. Реакционную смесь перемешивали при комнатной температуре дополнительно в течение 2 часов. Добавляли воду, и продукт экстрагировали этилацетатом. Органическую фазу высушивали на безводном сульфате магния, фильтровали и выпаривали с получением 660 мг (90%) 6-((2,4-дифторфенил)(метил)амино)-2-(метилтио)никотинонитрила в форме коричневого твердого вещества.
ЖХМС (ЭУ, m/z): (М+1) 292,09.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 7.74-7.80 (1Н, m, СН), 7.55-7.63 (1Н, m, СН), 7.43-7.52 (1Н, m, СН), 7.18-7.27 (1Н, m, СН), 6.16-6.30 (1Н, m, СН), 3.43 (3H, s, CH3), 2.42 (3H, s, CH3).
Пример 12бис: N6-(2,4-дифторфенил)N6-метил-1Н-пиразоло[3,4-b]пиридин-3,6-диамин
452 мг (1,84 ммоль) mCPBA добавляли в атмосфере аргона к перемешиваемому раствору 486 мг (1,67 ммоль) 6-((2,4-дифторфенил)(метил)амино)-2-(метилтио)никотинонитрила в 15 мл дихлорметана. Реакционную смесь перемешивали 30 мин при комнатной температуре, после чего добавляли этилацетатную фракцию. Органическую фазу промывали насыщенным раствором NaHCO3, высушивали на сульфате магния и выпаривали до сухого состояния. Неочищенный продукт реакции снова растворяли в 6 мл пропанола и добавляли 164 мкл (3,38 ммоль) гидразина гидрохлорида в воде. Смесь нагревали при 90°C в течение 6 часов, после чего разбавляли в воде и экстрагировали этилацетатом. Органическую фазу высушивали на сульфате магния и выпаривали до сухого состояния, после чего очищали хроматографией на силикагеле с получением 328 мг N6-(2,4-дифторфенил)-N6-метил-1H-пиразоло[3,4-b]пиридин-3,6-диамина в форме желто-оранжевого твердого вещества (70%).
ЖХМС (ЭУ, m/z): (М+1) 276,15.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 11.41 (1Н, s, NH), 7.75 (1Н, d, СН), 7.51-7.55 (1Н, m, СН), 7.40-7.43 (1Н, m, СН), 7.17-7.22 (1Н, m, СН), 6.03 (1Н, d, СН), 5.23 (2Н, s, NH2), 3.28 (3H, s, CH3).
Пример способа C3
Пример 12-три: 6-(2,4-дифторфенилтио)-1H-пиразоло[3,4-b]пиридин-3-амин

Пример 12-три-а: 2-хлор-6-(2,4-дифторфенилтио)никотинонитрил
Раствор 362 мг (1,05 экв.) гидроксида калия в 10 мл этанола добавляли в атмосфере азота к раствору 698 мкл (6,16 ммоль) 2,4-дифторбензолтиола в 30 мл этанола. Реакционную смесь перемешивали при комнатной температуре в течение 15 минут, а затем охлаждали во льду, после чего добавляли раствор 1,015 г (0,95 экв.) 2,6-дихлорникотинонитрила в 30 мл этанола. Реакционную смесь перемешивали в течение 2 часов при 0-5°C. Для остановки реакции добавляли 63 мл 0,1 н. раствора HCl. Добавляли воду, и продукт экстрагировали этилацетатом. Органическую фазу высушивали на безводном сульфате магния, фильтровали и выпаривали. Остаток очищали хроматографией на силикагеле (элюент: циклогексан/этилацетат 94:6) с получением 1,09 г (66%) 2-хлор-6-(2,4-дифторфенилтио)-никотинонитрила в форме белого твердого вещества.
ЖХМС (ЭУ, m/z): (М+1) 282,98.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.24 (1Н, d, СН), 7.77-7.85 (1Н, m, СН), 7.52-7.63 (1Н, m, СН), 7.25-7.35 (2Н, m, СН), 2.41 (3H, s, CH3).
Пример 12-три: 6-(2,4-дифторфенилтио)-1H-пиразоло[3,4-b]пиридин-3-амин
0,561 мл (11,57 ммоль) моногидрата гидразина добавляли в атмосфере азота к перемешиваемому раствору 1,09 г (3,86 ммоль) 2-хлор-6-(2,4-дифторфенилтио)никотинонитрила в 15 мл пропанола. Реакционную смесь нагревали при 80°C в течение 4 часов. Осадок получали, когда реакционную смесь возвращали к комнатной температуре. Этот осадок фильтровали и промывали этанолом. Твердое вещество растворяли в этилацетатной фракции и промывали 1 н. раствором HCl. Органическую фазу высушивали на сульфате магния и выпаривали до сухого состояния с получением 420 мг (39%) 6-(2,4-дифторфенилтио)-1Н-пиразоло[3,4-b]пиридин-3-амина в форме желтого твердого вещества.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 12.10 (1Н, s, NH), 8.11 (1Н, d, СН), 7.82-7.89 (1H, m, СН), 7.58-7.63 (1Н, m, СН), 7.32-7.36 (1Н, m, СН), 6.86 (1Н, d, СН), 4.59 (2Н, s, NH2).
Следующее соединение получили подобным способом:
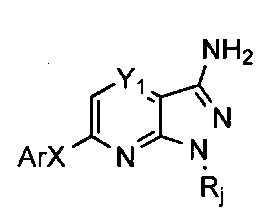
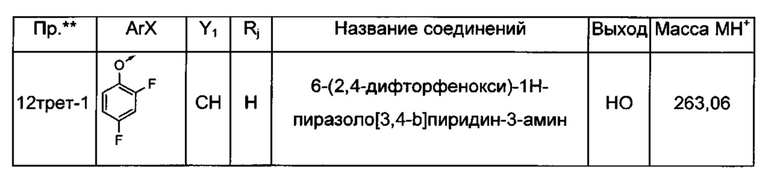
Пример 12-четыре: 6-(3,5-дифторбензил)-1H-пиразоло[3,4-b]пиридин-3-амин

17,35 мл 0,5 М раствора в ТГФ (3,5-дифторбензил)цинка хлорида (8,58 ммоль) добавляли в атмосфере аргона к раствору 416 мг хлорида палладия(II) (510 ммоль) и 883 мг 2,6-дихлорникотинонитрила (5,1 ммоль) в 2 мл безводного ТГФ. Реакционную смесь кипятили с обратным холодильником в течение 7 часов, затем охлаждали до комнатной температуры. Добавляли 1 н. водный раствор соды, и продукт экстрагировали несколькими последовательными этилацетатными фракциями. Органические фазы высушивали на сульфате магния и выпаривали до сухого состояния, после чего очищали хроматографией на силикагеле с получением 680 мг смеси 2-хлор-6-(3,5-дифторбензил)-никотинонитрила и побочных продуктов, которую использовали без дополнительной очистки на следующей стадии.
Предшествующую смесь растворяли в 10 мл изопропанола при перемешивании и добавляли 750 мкл 35% гидрата гидразина. Раствор нагревали при 80°C в течение 4 часов. Растворитель выпаривали до сухого состояния, и продукт очищали хроматографией на силикагеле (дихлорметан/метанол 9:1) с получением 290 мг 6-(3,5-дифторбензил)-1Н-пиразоло[3,4-b]пиридин-3-амина (64%).
ЖХМС (ЭУ, m/z): (М+1) 261,16.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 11.82 (1Н, s, NH), 8.01 (1Н, d, СН), 6.99-7.04 (3H, m, СН), 6.91 (1Н, d, СН), 5.49 (2Н, s, NH2), 4.12 (2Н, s, CH2).
Пример способа D1:
Пример 13: 5-(3,5-дифторбензил)-1H-пиразоло[3,4-b]пиридин-3-амин

0,575 г (0,704 ммоль) (dppf)2PdCl2⋅CH2Cl2 и 28 мл (14,08 ммоль) 3,5-дифторбензилцинка(II) хлорида добавляли к 1,5 г (7,04 ммоль) раствора 5-бром-1Н-пиразоло[3,4-b]пиридин-3-амина в 10 мл тетрагидрофурана. Реакционную смесь нагревали при 90°C в течение 18 часов. После возврата к комнатной температуре реакционную смесь подвергали гидролизу путем медленного добавления воды при 0°C. После фильтрования образовавшегося осадка твердое вещество промывали тетрагидрофураном, и водный фильтрат экстрагировали несколько раз этилацетатом. Органические фазы объединяли, высушивали на сульфате магния и концентрировали. Остаток очищали хроматографией на силикагеле (95:4,5:0,5, а затем 95:4:1 дихлорметан/метанол/аммиак в качестве элюента) с получением 1,7 г (93%) 5-(3,5-дифторбензил)-1Н-пиразоло[3,4-b]пиридин-3-амина в форме бежевого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 261,41.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 11.87 (1Н, s, NH), 8.31 (1Н, d, СНаром), 7.92 (1Н, d, СНаром), 6.98-7.08 (3H, m, СНаром), 5.47 (2Н, s, NH), 4.04 (2Н, s, CH2).
Следующие соединения получили подобным способом:

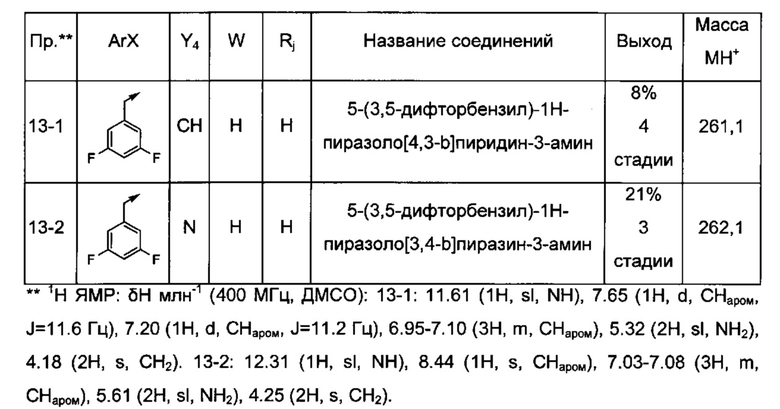
Примеры способа D2
Пример 14: 5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиразин-3-амин

0,7 г (2,68 ммоль) 5-йод-1H-пиразоло[3,4-b]пиридин-3-амина, 0,74 г (5,36 ммоль) безводного карбоната калия и 0,10 г йодида меди (0,536 ммоль) смешивали в круглодонной колбе на 50 мл. Затем добавляли 15 мл пропан-2-ола, 0,01 г (0,2 ммоль) полиэтиленгликоля и 0,43 г (2,95 ммоль) 3,5-дифтортиофенола. Реакционную смесь нагревали при 80°C в течение 2 часов. Растворитель выпаривали, и образовавшееся твердое вещество фильтровали, промывали водой, а затем пентаном и высушивали в печи при 50°C с получением 0,75 г (100%) 5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиразин-3-амина в форме коричневого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 280,03.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 12.65 (1Н, bs, NH), 8.52 (1Н, s, СНаром), 7.18 (1Н, t, СНаром), 7.05-7.18 (2Н, m, СНаром), 5.89 (2Н, s, NH).
Следующие производные получили в соответствии с тем же способом:
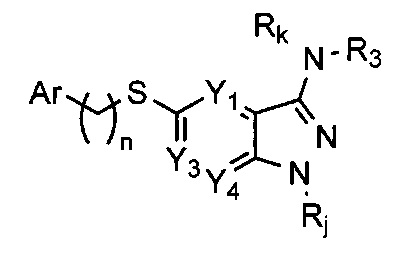

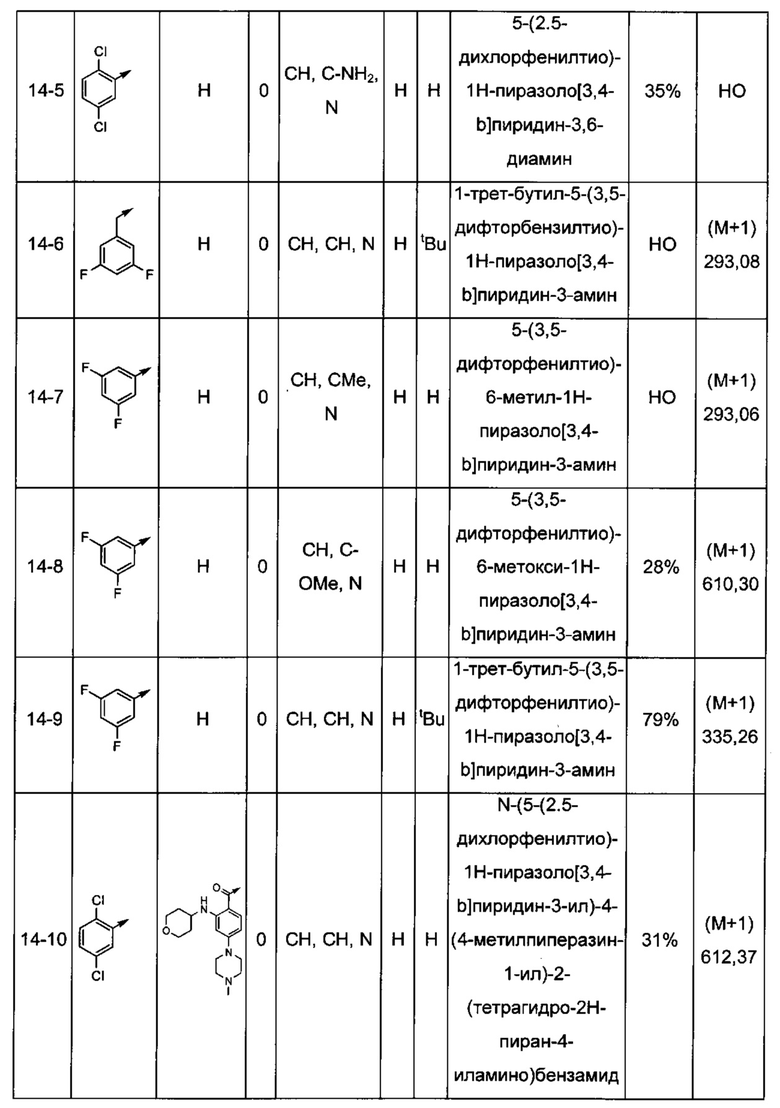

Пример 14бис: N-(5-(3,5-дифторфениламино)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамид

Раствор 225 мг N-(5-йод-1-тритил-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамида (0,25 ммоль), 36 мг дифторанилина (0,275 ммоль), 19 мг R-(+)-2,2'-бис(дифенилфосфино)-1,1'-динафтила (0,030 ммоль), 11 мг (0,013 ммоль) трис(дибензилиденацетон)дипалладия(0) и 75 мг (0,75 ммоль) трет-бутилата натрия в 10 мл ТГФ кипятили с обратным холодильником в атмосфере аргона в течение ночи. Неочищенную реакционную смесь охлаждали, экстрагировали этилацетатом и промывали водой. Органическую фазу высушивали на сульфате магния и очищали хроматографией на силикагеле с получением N-(5-(3,5-дифторфениламино)-1-тритил-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамида, который использовали в следующей стадии без дополнительной очистки.
Полученный таким образом продукт растворяли в 10 мл дихлорметана при 0°C и добавляли 56 мг (0,5 ммоль) ТФУ. Реакционную смесь перемешивали в течение 4 часов. Добавляли воду, и pH реакционной смеси доводили до 7 раствором NaHCO3. Водную фазу собирали, подщелачивали (pH 9-10) концентрированным раствором K2CO3 и экстрагировали дихлорметаном. Органическую фазу собирали, высушивали на сульфате магния и концентрировали до сухого состояния с получением 40 мг N-(5-(3,5-дифторфениламино)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамида.
ЖХМС (ЭУ, m/z): (М+1) 562,12.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 13.45 (1Н, sl, NH), 10.47 (1Н, sl, NH), 8.65 (1Н, s, СНаром), 8.55 (1Н, s, СНаром), 8.14 (1Н, d, NH), 7.77 (1Н, d, СНаром), 7.26 (2Н, m, СНаром), 7.05 (1Н, m, СНаром), 6.25 (1Н, d, СНаром), 6.14 (1Н, s, NH), 6.77 (1Н, s, NH), 3.82-3.84 (2Н, dt, СН), 3.72 (1Н, m, СН), 3.47-3.52 (2Н, m, СН), 3.28-3.34 (4Н, m, СН), 2.43 (4Н, m, СН), 2.23 (3H, s, CH3), 1.94-1.97 (2Н, m, СН), 1.37-1.39 (2Н, m, СН).
Примеры способа D3:
Пример 15: N-(5-((3,5-дифторфенил)ethynyl)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамид
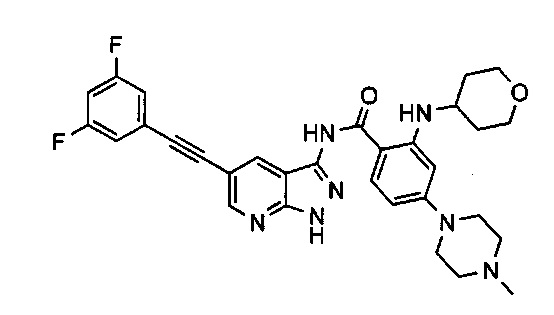
0,94 мг (0,926 ммоль) триэтиламина добавляли к 400 мг (0,712 ммоль) N-(5-йод-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамида, 67,8 мг (0,356 ммоль) CuI и 50 мг (0,071 ммоль) Pd(PPh3)2Cl2 в атмосфере аргона в 12 мл безводного диоксана при перемешивании. Реакционную смесь нагревали в течение 3,5 часов при 100°C. Реакционную смесь разбавляли 30 мл воды и экстрагировали этилацетатом. Органическую фазу высушивали на сульфате натрия, фильтровали и концентрировали. Полученный остаток очищали хроматографией на силикагеле (дихлорметан/метанол) с получением 152 мг N-(5-((3,5-дифторфенил)этинил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамида в форме желтого твердого вещества (выход=37%).
ЖХМС (ЭР, m/z): (М+1) 572,17.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 13.57 (1Н, bs, NH), 10.56 (1Н, bs, NH), 8.68 (1Н, s, СНаром), 8.43 (1Н, s, СНаром), 8.13 (1Н, d, NH), 7.80 (1Н, d, СНаром), 7.38 (2Н, m, СНаром), 6.27 (1Н, d, СНаром), 6.15 (1Н, d, СНаром), 3.84-3.82 (2Н, dt, СН), 3.70 (1Н, m, СН), 3.45-3.50 (2Н, m, СН), 3.21-3.33 (4Н, m, СН), 2.42-2.46 (4Н, m, СН), 2.28 (3H, s, CH3), 1.94-1.97 (2Н, m, СН), 1.37-1.39 (2Н, m, СН).
Следующее производное получили в соответствии с тем же способом:


Примеры способа E
Протоколы, включающие способ E, нацелены на функционализацию экзоциклического амина аминопиразольных колец путем их взаимодействия с промежуточными соединениями, характеризующимися электрофильной функциональной группой, необязательно образованными in situ, такими как хлорангидрид, изоцианат, изотиоцианат или альдегид.
Получение промежуточных соединений реакции
Пример 16: 2-(N-(4,4-дифторциклогексил)-2,2,2-трифторацетамидо)-4-(4-метилпиперазин-1-ил)бензойная кислота
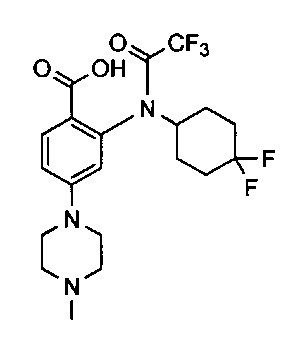
Пример 16а: трет-бутил-4-(4-метилпиперазин-1-ил)-2-нитробензоат
Данное соединение было описано ранее в WO 2008/74749. 5,28 мл (47,6 ммоль) 1-метилпиперазина добавляли к 4,1 г (17 ммоль) трет-бутил-4-фтор-2-нитробензоат. Реакционную смесь перемешивали без растворителя в течение 5 часов. В реакционную смесь добавляли 150 мл воды и перемешивали в течение 24 часов. Образовавшийся осадок фильтровали, промывали водой и высушивали в вакууме с получением 4,9 г (90%) трет-бутил-4-(4-метилпиперазин-1-ил)-2-нитробензоата в форме желтого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 322,37.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 7.69 (1Н, d, СНаром), 7.30 (1Н, d, СНаром), 7.20 (1Н, dd, СНаром), 3.38 (4Н, m, СН), 2.40 (4Н, m, СН), 2.22 (3H, s, CH3), 1.45 (9Н, s, CH3).
Пример 16b: трет-бутил-2-амино-4-(4-метилпиперазин-1-ил)бензоат
Данное соединение было описано ранее в WO 2008/74749.
0,160 г (1,500 ммоль) палладия на углероде (10%) и 15,19 мл (150 ммоль) циклогексена добавляли к раствору 4,82 г (15 ммоль) трет-бутил-4-(4-метилпиперазин-1-ил)-2-нитробензоата в 100 мл этанола. Реакционную смесь нагревали при температуре 80°C в течение 8 часов. Реакционную смесь фильтровали, а затем промывали этанолом с получением 4,2 г (выход=96%) трет-бутил-2-амино-4-(4-метилпиперазин-1-ил)бензоата в форме желтого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 292,39.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 7.44 (1Н, d, СНаром), 6.40 (2Н, bs, NH2), 6.19 (1Н, dd, СНаром), 6.12 (1Н, d, СНаром), 3.17 (4Н, m, СН), 2.40 (4Н, m, СН), 2.22 (3H, s, CH3), 1.49 (9Н, s, CH3).
Пример 16c: трет-бутил-2-(4,4-дифторциклогексиламино)-4-(4-метилпиперазин-1-ил)бензоат
1,045 мл (13,57 ммоль) трифторуксусной кислоты, 1 г (7,46 ммоль) 4,4-дифторциклогексанона и 2,158 г (8,20 ммоль) триацетоксиборгидрида тетраметиламмония добавляли к 1,521 г (5,22 ммоль) трет-бутил-2-амино-4-(4-метилпиперазин-1-ил)бензоата, растворенного в 60 мл дихлорметана. Реакционную смесь оставляли при перемешивании при комнатной температуре на 24 часа. Растворитель выпаривали, а затем неочищенный продукт реакции снова растворяли в 30 мл этилацетата. Раствор последовательно промывали 0,5 М раствором HCl, 0,5 М раствором соды и, наконец, насыщенным раствором NaHCO3. Органическую фазу высушивали на сульфате натрия, фильтровали и концентрировали с получением 2,2 г трет-бутил-2-(4,4-дифторциклогексиламино)-4-(4-метилпиперазин-1-ил)бензоата в форме светло-коричневой смолы (выход=72%).
ЖХМС (ЭР, m/z): (М+1) 410,3.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 7.73 (1Н, bs, NH), 7.58 (1Н, m, СНаром), 7.77 (1Н, m, СНаром), 6.09 (1Н, bs, СНаром), 3.37 (4Н, m, СН), 3.27 (4Н, m, СН), 2.47 (4Н, m, СН), 2.25 (3H, s, СН), 1.99 (4Н, s, СН), 1.40 (9Н, s, СН).
Пример 16d: трет-бутил-2-(N-(4,4-дифторциклогексил)-2,2,2-трифторацетамидо)-4-(4-метилпиперазин-1-ил)бензоат
0,99 мл (6,98 ммоль) трифторуксусного ангидрида и 1,12 мл (8,06 ммоль) триэтиламина добавляли к 2,2 г (5,3 ммоль) трет-бутил-2-(4,4-дифторциклогексиламино)-4-(4-метилпиперазин-1-ил)бензоата, растворенного в 40 мл дихлорметана. Реакцию оставляли при перемешивании при комнатной температуре в течение 3 часов. Растворитель выпаривали, а затем неочищенный продукт реакции растворяли в 30 мл этилацетата. Раствор промывали насыщенным раствором NaHCO3. Органическую фазу высушивали на сульфате натрия, фильтровали и концентрировали с получением 2,5 г трет-бутил-2-(N-(4,4-дифторциклогексил)-2,2,2-трифторацетамидо)-4-(4-метилпиперазин-1-ил)бензоата в форме светло-коричневой смолы (выход=92%).
ЖХМС (ЭР, m/z): (М+1) 506,26.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 7.84 (1Н, m, СНаром), 7.09 (1H, m, СНаром), 6.89 (1Н, bs, СНаром), 3.45-3.39 (8Н, m, СН), 2.83 (4Н, m, СН), 2.20 (4Н, m, СН), 2.05 (3H, s, СН), 1.46 (9Н, s, СН).
Пример 16: 2-(N-(4,4-дифторциклогексил)-2,2,2-трифторацетамидо)-4-(4-метилпиперазин-1-ил)бензойная кислота
7,62 мл (99 ммоль) трифторуксусной кислоты добавляли к 2,5 г (4,95 ммоль) трет-бутил-2-(N-(4,4-дифторциклогексил)-2,2,2-трифторацетамидо)-4-(4-метилпиперазин-1-ил)бензоата, растворенного в 30 мл дихлорметана. Реакционную смесь оставляли при перемешивании при комнатной температуре в течение ночи. Растворитель выпаривали, а затем неочищенный продукт реакции снова растворяли в 30 мл этилацетата. Растворители выпаривали, образовавшееся твердое вещество снова растворяли в этиловом эфире, и растворитель снова выпаривали. Эту процедуру повторяли три раза до получения светло-коричневого твердого вещества. Получили 2,2 г 2-(N-(4,4-дифторциклогексил)-2,2,2-трифторацетамидо)-4-(4-метилпиперазин-1-ил)бензойной кислоты в форме соли трифторуксусной кислоты (выход=79%).
ЖХМС (ЭР, m/z): (М+1) 450,1.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 10.01 (1Н, bs, ОН), 7.92 (1Н, m, СНаром), 7.13 (1Н, m, СНаром), 7.01 (1Н, bs, СНаром), 4.39 (1Н, m, СН), 3.12-3.52 (8Н, m, СН), 2.86 (3H, s, СН), 1.75-2.0 (8Н, m, СН).
Следующие соединения также получены этим способом:
4-(4-метилпиперазин-1-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил) ацетамидо)бензойная кислота.
Это соединение было описано ранее в WO 2008/74749, WO 2009/13126 и WO 2010/69966.
ЖХМС (ЭР, m/z): (М+1) 416,40.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 12.60 (1Н, bs, ОН), 10.08 (1Н, bs, ОН), 7.90 (1Н, d, СНаром), 7.13 (1Н, dd, СНаром), 6.90 (1Н, d, СНаром), 4.40 (1Н, m, СН), 4.10 (2Н, m, СН), 3.70-3.90 (2Н, m, СН), 3.59-3.62 (4Н, m, СН), 3.30-3.32 (4Н, m, СН), 2.87 (3H, s, CH3), 1.87-1.98 (1Н, m, СН), 1.59-1.60 (1Н, m, СН), 1.00-1.54 (2Н, m, СН).
4-((3-(диметиламино)пропил)(метил)амино)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензойная кислота.
Это соединение было описано ранее в WO 2009/13126 и WO 2008/74749.
Пример 17: (S)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)-4-(3-(2,2,2-трифторацетамидо)пирролидин-1-ил)бензойная кислота
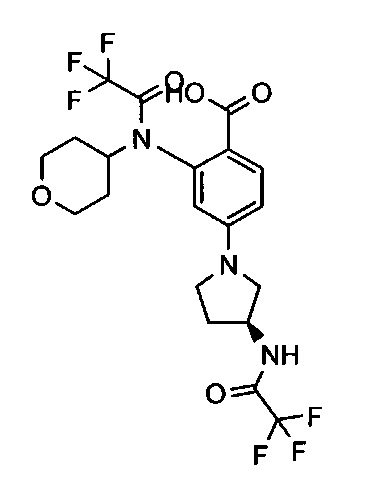
Пример 17а: трет-бутил-(S)-4-(3-(трет-бутоксикарбониламино)пирролидин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензоат
Это соединение было получено путем воспроизведения примера 16d, используя трет-бутил-(S)-пирролидин-3-илкарбамат.
Пример 17b: (S)-4-(3-аминопирролидин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензойная кислота
19,7 мл (25 экв.) трифторуксусной кислоты добавляли к раствору 4,72 г (10,23 ммоль) трет-бутил-(S)-4-(3-(трет-бутоксикарбониламино)пирролидин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензоата в 100 мл дихлорметана. Реакционную смесь перемешивали при комнатной температуре в течение 30 часов. Растворители выпаривали, и остаток снова растворяли в диэтиловом эфире и растирали до получения твердого вещества. Образовавшееся твердое вещество фильтровали и высушивали в вакууме с получением 4,3 г (100%) желтого порошка (S)-4-(3-аминопирролидин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензойной кислоты в форме соли трифторуксусной кислоты.
ЖХМС (ЭР, m/z): (М+1) 306,22.
Пример 17: (S)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)-4-(3-(2,2,2-трифторацетамидо)пирролидин-1-ил)бензойная кислота
1,74 мл (3,5 экв.) триэтиламина и 1,6 мл (2,1 экв.) трифторуксусного ангидрида добавляли к раствору 1,5 г (3,58 ммоль) (S)-4-(3-аминопирролидин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензойной кислоты в форме соли трифторуксусной кислоты в 40 мл дихлорметана при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Добавляли по каплям воду (10 мл), а затем органическую фазу промывали насыщенным раствором хлорида натрия, высушивали на сульфате магния, фильтровали и выпаривали. Остаток очищали хроматографией на силикагеле (96:4 дихлорметан/метанол в качестве элюента) с получением 250 мг (14%) (S)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)-4-(3-(2,2,2-трифторацетамидо)пирролидин-1-ил)бензойной кислоты в форме желтого порошка.
ЖХМС (ЭР, m/z): (М+1) 498,07.
Пример 18: 2-(2-фторэтокси)-4-(4-метилпиперазин-1-ил)бензойная кислота
Это соединение может быть получено из следующих промежуточных соединений.
Пример 18а: трет-бутил-4-фтор-2-(2-фторэтокси)бензоат
Пример 18b: трет-бутил-2-(2-фторэтокси)-4-(4-метилпиперазин-1-ил)бензоат
Следующее соединение было также получено этим способом: 2-(2-фторэтокси)-4-(4-(1-метилпиперидин-4-ил)пиперазин-1-ил)бензойная кислота.
Пример 19: 4-(4-метилпиперазин-1-ил)-2-(2,2,2-трифтор-N-(2-фторэтил)-ацетамидо)-бензойная кислота
Это соединение может быть получено из следующих промежуточных соединений.
Пример 19а: трет-бутил-4-фтор-2-(2-фторэтиламино)бензоат
Пример 19b: трет-бутил-4-фтор-2-(2,2,2-трифтор-N-(2-фторэтил)ацетамидо)бензоат
Пример 19c: трет-бутил-4-(4-метил пиперазин-1-ил)-2-(2,2,2-трифтор-N-(2-фторэтил)-ацетамидо)-бензоат
Следующее соединение было также получено этим способом:
4-((3-(диметиламино)пропил)(метил)амино)-2-(2,2,2-трифтор-N-(2-фторэтил)ацетамидо)бензойная кислота.
Пример 20: 4-(1-метилпиперидин-4-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензойной кислоты гидротрифторацетат

Это соединение может быть получено из следующих промежуточных соединений.
Пример 20а: трет-бутил-2-нитро-4-(пиридин-4-ил)бензоат
1,67 г бис(трифенилфосфин)палладия(II) хлорида (2,38 ммоль) и 15,8 г карбоната натрия (149 ммоль) добавляли к раствору 18 г трет-бутил-4-бром-2-нитробензоата (59,6 ммоль) и 10,98 г пиридин-4-илбороновой кислоты (89 ммоль) в смеси 200 мл диметоксиэтана и 100 мл воды. Реакционную смесь нагревали при 100°C в течение 24 часов, а затем концентрировали при пониженном давлении. Полученный остаток очищали флэш-хроматографией (CH2Cl2/AcOEt: 100:0-70:30, 30 мин). Продукт выделен в форме масла, которое кристаллизуется с получением 14,64 г (82%) кристаллов.
МС (m/z): (М+1) 301,0.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.73 (2Н, d, СНаром, J=6.0 Гц), 8.44 (1Н, s, СНаром), 8.24 (1Н, dd, СНаром, J=8.0 Гц), 7.97 (1Н, d, СНаром, J=8.0 Гц), 7.85 (2Н, dd, СНаром, J=4.4 Гц), 1.54 (9Н, s).
Пример 20b: 4-(4-(трет-бутоксикарбонил)-3-нитрофенил)-1-метилпиридиния йодид
7,55 мл йодметана (121 ммоль) добавляли к раствору 16,2 г трет-бутил-2-нитро-4-(пиридин-4-ил)бензоата (60,6 ммоль) в 20 мл ацетона. Реакционную смесь нагревали при 60°C в течение 4 часов, а затем при комнатной температуре в течение ночи. После концентрирования до сухого состояния выделили 27 г оранжевых кристаллов (100%).
МС (m/z): (М+1) 315,0.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 9.14 (2Н, d, СНаром, J=6.4 Гц), 8.71 (1Н, s, СНаром), 8.63 (2Н, d, СНаром, J=6.4 Гц), 8.47 (1Н, dd, СНаром, J=8.0 Гц), 8.08 (1Н, d, СНаром, J=8.0 Гц), 4.37 (3H, s, СН), 1.54 (9Н, s).
Пример 20c: трет-бутил-2-амино-4-(1-метилпиперидин-4-ил)бензоат
0,48 г оксида платины(IV) (2,12 ммоль) добавляли к раствору 26,8 г 4-(4-(трет-бутоксикарбонил)-3-нитрофенил)-1-метилпиридиния йодида (60,6 ммоль) в 200 мл метанола, помещенного в реактор, изготовленный из нержавеющей стали. Давление реакционной смеси доводили до 5 бар азота в течение 24 ч. Катализатор фильтровали, и фильтрат концентрировали при пониженном давлении с получением 24,8 г (98%) белых кристаллов.
МС (m/z): (М+1) 291,1.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 9.18 (1Н, s, HI), 7.60 (1Н, d, СНаром, J=8.4 Гц), 6.54-6.40 (3H, m, СНаром), 6.39 (1Н, d, СНаром, J=8.0 Гц), 3.48-3.53 (2Н, m, СН), 3.06 (2Н, t, СН), 2.81 (3H, s, СН), 2.60-2.70 (1Н, m, СН), 1.89-1.97 (2Н, m, СН), 1.70-1.80 (2Н, m, СН), 1.52 (9Н, s).
Пример 20d: трет-бутил-4-(1-метилпиперидин-4-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензоат
7,18 мл 2,2,2-трифторуксусной кислоты (93 ммоль), 4,11 мг дигидро-2Н-пиран-4(3H)-она (44,5 ммоль), а затем 14,5 г триацетоксиборгидрида тетраметиламмония (53,8 ммоль) последовательно добавляли к раствору 15 г трет-бутил-2-амино-4-(1-метилпиперидин-4-ил)бензоата в 200 мл дихлорметана при перемешивании. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, а затем растворяли в 1 н. растворе соды. Органическую фазу выделяли, высушивали на сульфате магния, а затем концентрировали до сухого состояния. Остаток всегда содержал HI. Поэтому его растворяли в дихлорметане и промывали 100 мл 1 н. раствора соды. Органическую фазу декантировали, высушивали на сульфате магния и концентрировали до сухого состояния с получением 14,6 г желтого твердого вещества (количественный выход).
МС (m/z): (М+1) 375,2.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 7.69 (1Н, d, СНаром, J=8.4 Гц), 7.63 (1Н, d, СНаром, J=7.6 Гц), 6.65 (1Н, s, СНаром), 6.44 (1Н, dd, СНаром, J=8.4 Гц), 3.74-3.86 (2Н, m, СН), 3.66-3.71 (1Н, m, СН), 3.51 (2Н, t, СН), 3.05-3.12 (2Н, m, СН), 2.6-2.5 (1Н, m, СН), 2.42 (3H, s, СН), 2.30-2.40 (2Н, m, СН), 1.89-1.97 (2Н, m, СН), 1.64-1.77 (4Н, m, СН), 1.52 (9Н, s), 1.33-1.45 (2Н, m, СН).
Пример 20e: трет-бутил-4-(1-метилпиперидин-4-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензоат
6,35 мл триэтиламина и 5,50 мл 2,2,2-трифторуксусного ангидрида (39,6 ммоль) добавляли при 0°C к раствору 11,4 г трет-бутил-4-(1-метилпиперидин-4-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензоата (30,4 ммоль) в 240 мл дихлорметана при перемешивании. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, а затем добавляли по каплям 100 мл воды. Органическую фазу декантировали, высушивали на сульфате магния и концентрировали до сухого состояния. Остаток растворяли в смеси этанол/диэтиловый эфир с получением твердого вещества, которое фильтровали на пористом диске, и выделили 12,06 г белых кристаллов. Фильтрат концентрировали (4,5 г), а затем очищали флэш-хроматографией на силикагеле (CH2Cl2/MeOH: 95:5 - 90:10, 20 мин). Полученный продукт перекристаллизовали в диэтиловом эфире с получением 1,04 г дополнительных белых кристаллов (общий выход=74%).
МС (m/z): (М+1) 471,1.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 9.45 (1Н, sl, NH+), 7.96 (1Н, d, СНаром, J=8 Гц), 7.51 (1Н, d, СНаром, J=8 Гц), 7.31 (1Н, s, СНаром), 4.6-4.5 (1Н, m, СН), 3.90-3.75 (2Н, m, СН), 3.5-3.35 (4Н, m, СН), 3.1-2.85 (3H, m, СН), 2.79 (3H, s, CH3), 2.1-1.95 (3H, 3, СН), 1.9-1.75 (2Н, m, СН), 1.55-1.40 (11Н, m), 1.0-0.85 (1Н, m, СН).
Пример 20: 4-(1-метилпиперидин-4-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензойной кислоты гидротрифторацетат
6,33 мл 2,2,2-трифторуксусной кислоты (82 ммоль) добавляли при перемешивании к раствору 3,2 г трет-бутил-4-(1-метилпиперидин-4-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензоата (5,47 ммоль) (в форме соли трифторуксусной кислоты) в 30 мл дихлорметана. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, а затем выпаривали при пониженном давлении. Остаток растворяли в этаноле, и образовавшееся белое твердое вещество фильтровали на пористом диске с получением 1,61 г (53%) белых кристаллов.
МС (m/z): (М+1) 415,1.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 13.39 (1Н, sl, СООН), 9.46 (1Н, sl, СООН ТФУ), 7.99 (1Н, d, СНаром, J=8.4 Гц), 7.49 (1Н, d, СНаром, J=8.4 Гц), 7.30 (1Н, s, СНаром), 4.53 (1Н, m, СН), 3.74-3.86 (2Н, m, СН), 3.35-3.45 (5Н, m, СН), 2.90-3.01 (3H, m, СН), 2.76 (3H, s, СН), 1.65-2.04 (5Н, m, СН), 1.44-1.54 (2Н, m, СН).
Пример 21: 1-(4-изотиоцианатофенил)-4-метилпиперазин
Это соединение было получено путем адаптации способа, описанного в EP 1215208.
Следующее соединение было также получено этим способом: трет-бутил-2-изотиоцианато-5-(4-метилпиперазин-1-ил)фенилкарбамат.
Пример 22: трет-бутил-2-изоцианато-5-(4-метилпиперазин-1-ил)фенилкарбамат
Это соединение может быть получено из следующих промежуточных соединений.
Пример 22а: трет-бутил-5-(4-метилпиперазин-1-ил)-2-нитрофенилкарбамат
Пример 22b: трет-бутил-2-амино-5-(4-метилпиперазин-1-ил)фенилкарбамат
Пример 22: трет-бутил-2-изоцианато-5-(4-метилпиперазин-1-ил)фенилкарбамат
Пример 23: 4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензальдегид

Пример 23а: (4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)фенил)метанол
500 мг (1,060 ммоль) 4-(4-метилпиперазин-1-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензойной кислоты, растворенной в 5 мл тетрагидрофурана, добавляли при 0°C к суспензии 201 мг (5,30 ммоль) LiAIH4 в 9 мл тетрагидрофурана. Реакционную смесь перемешивали при 0°C в течение 1 часа, а затем при комнатной температуре в течение 3 часов. Реакционную смесь охлаждали при 0°C, а затем добавляли по каплям 200 мкл воды, затем 200 мкл раствора соды (15 масс. %) и, наконец, 1 мл воды. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем фильтровали и промывали тетрагидрофураном. Фильтрат концентрировали с получением 250 мг (выход=77%) (4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)фенил)метанола в форме белого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 306,14.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 6.85 (1Н, d, СНаром), 6.20 (1Н, d, СНаром), 6.10 (1Н, d, СНаром), 4.95 (1Н, bs, ОН), 4.87 (1Н, d, NH), 4.37 (2Н, d, CH2), 3.83-3.86 (2Н, m, СН), 3.56 (1Н, m, СН), 3.46-3.56 (3H, m, СН), 3.45 (1Н, m, СН), 3.05-3.07 (4Н, m, СН), 2.41-2.44 (4Н, m, СН), 2.21 (3H, s, CH3), 1.89-1.92 (2Н, m, СН).
Пример 23: 4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензальдегид
85 мг (0,982 ммоль) диоксида марганца добавляли при комнатной температуре к раствору (4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)фенил)метанола (100 мг, 0,327 ммоль) в смеси этилацетата (10 мл) и дихлорметана (9 мл). Реакционную смесь помещали в ультразвуковую баню на 5 часов. Реакционную смесь фильтровали, растворители выпаривали, и неочищенный продукт очищали хроматографией с получением 50,0 мг (выход=50,3%) (4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензальдегида в форме белого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 304,19.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 9.43 (1Н, d, СН), 7.32 (1Н, d, СНаром), 6.36 (1Н, d, СНаром), 6.08 (1Н, d, СНаром), 3.94-3.99 (2Н, m, СН), 3.77 (1Н, m, СН), 3.61-3.63 (2Н, m, СН), 3.42-3.45 (4Н, m, СН), 2.57-2.60 (4Н, m, СН), 2.36 (3H, s, CH3), 2.04-2.08 (2Н, m, СН), 1.51-1.60 (2Н, m, СН).
Пример 24: 2-(4-(4-метилпиперазин-1-ил)фенил)уксусная кислота

Пример 24а: 2,2,2-трихлор-1-(4-(4-метилпиперазин-1-ил)фенил)этанол 1,0 мл (10,00 ммоль) трихлоруксусной кислоты и небольшими порциями 1,854 г (10 ммоль) 2,2,2-трихлорацетата натрия добавляли при комнатной температуре к раствору 1,362 г (6,67 ммоль) 4-(4-метилпиперазин-1-ил)бензальдегида в 13,5 мл диметилформамида. Реакционную смесь перемешивали в течение 3 часов при комнатной температуре. Растворитель концентрировали, и неочищенный продукт реакции экстрагировали этилацетатом. Органическую фазу промывали, используя насыщенный раствор бикарбоната натрия. Органические фазы объединяли, высушивали на сульфате магния, а затем концентрировали с получением 1,760 г (выход=82%) 2,2,2-трихлор-1-(4-(4-метилпиперазин-1-ил)фенил)этанола в форме белого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 324,04.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 7.41 (2Н, d, СНаром), 7.02 (1Н, bs, ОН), 6.90 (2Н, d, СНаром), 5.08 (1Н, bs, СН), 3.14-3.16 (4Н, m, СН), 2.42-2.47 (4Н, m, СН), 2.21 (3H, s, CH3).
Пример 24: 2-(4-(4-метилпиперазин-1-ил)фенил)уксусная кислота 0,559 г (14,77 ммоль) боргидрида натрия быстро добавляли к 2,294 г (7,35 ммоль) дибензилселенида в 28 мл этанола. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавляли 2,266 г (7 ммоль) 2,2,2-трихлор-1-(4-(4-метилпиперазин-1-ил)фенил)этанола и 1,680 г (42,0 ммоль) гидроксида натрия. Реакционную смесь перемешивали при 35°C в течение 24 часов. Растворитель концентрировали, и неочищенный продукт экстрагировали этилацетатом после доведения водной фазы до pH 5. Органические фазы объединяли, высушивали на сульфате магния, а затем концентрировали с получением 2-(4-(4-метилпиперазин-1-ил)фенил)уксусной кислоты, которую использовали без дополнительной очистки.
ЖХМС (ЭР, m/z): (М+1) 235,294.
Пример 25: 2-(4-(4-метилпиперазин-1-ил)-2-нитрофенил)уксусная кислота
Это соединение может быть получено из следующих промежуточных соединений.
Пример 25а: диэтил-2-(4-фтор-2-нитрофенил)малонат
Пример 25b: диэтил-2-(4-(4-метилпиперазин-1-ил)-2-нитрофенил)малонат
Пример способа E1:
Пример 26: N-(5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензамид

0,95 мл (11,21 ммоль) оксалилхлорида и 2 капли безводного диметилформамида добавляли к 2,97 г (5,61 ммоль) раствора 4-(4-метилпиперазин-1-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензойной кислоты в 95 мл дихлорметана. Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Растворители выпаривали, образовавшееся твердое вещество растворяли в толуоле, и растворитель выпаривали. Эту процедуру повторяли три раза до получения белого твердого вещества. Хлорангидрид растворяли в 35 мл безводного тетрагидрофурана при -20°C, а затем образовавшийся раствор добавляли к раствору, содержащему 1,56 г (5,61 ммоль) 5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиридин-3-амина и 3,71 мл (21,30 ммоль) N-этил-N-изопропилпропан-2-амина в 30 мл безводного тетрагидрофурана. Реакционную смесь перемешивали в течение 3 часов при -20°C, а затем в течение ночи при комнатной температуре. Полученный осадок фильтровали и промывали тетрагидрофураном и водой, а затем высушивали с получением 2 г (53%) N-(5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензамида.
ЖХМС (ЭР, m/z): (М+1) 676,20.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 13.66 (1Н, bs, NH), 11.08 (1Н, bs, NH), 8.61 (1Н, s, СНаром), 8.46 (1Н, s, СНаром), 7.83 (1Н, d, СНаром), 7.05-7.10 (2Н, m, СНаром), 6.83-6.89 (3H, m, СНаром), 4.39-4.44 (1Н, m, СН), 3.83-3.85 (1Н, m, СН), 3.69-3.72 (1Н, m, СН), 3.59-3.62 (1Н, m, СН), 3.30-3.32 (4Н, m, CH2), 2.30-2.44 (4Н, m, CH2), 2.27 (3H, s, CH3), 1.87-1.90 (1Н, m, СН), 1.59-1.60 (1Н, m, СН), 1.49-1.50 (1Н, m, СН), 1.20-1.40 (1Н, m, СН).
Следующие производные были получены в соответствии с тем же способом:


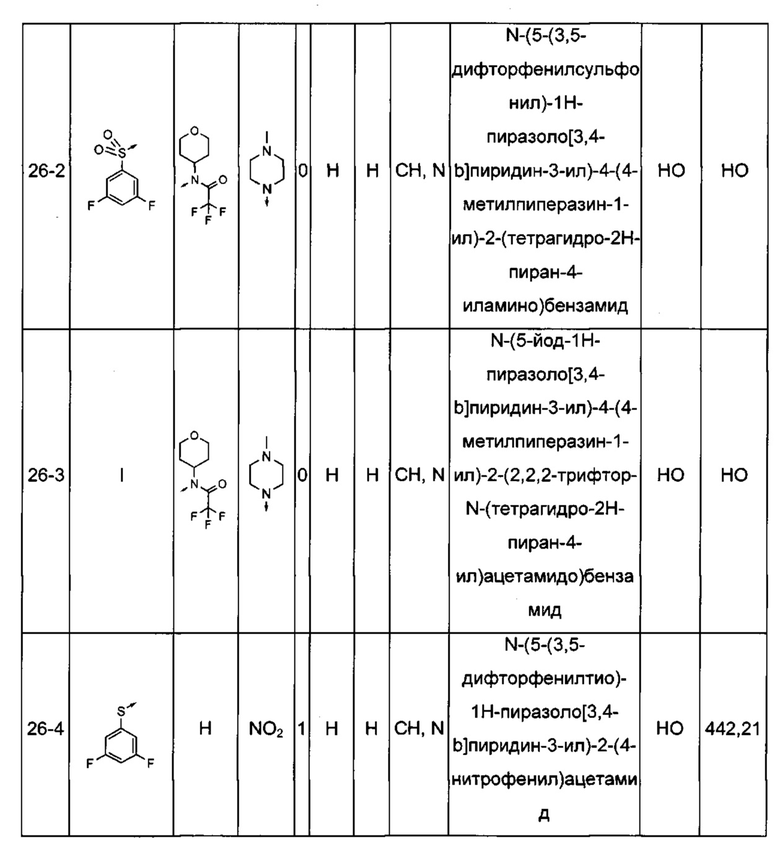
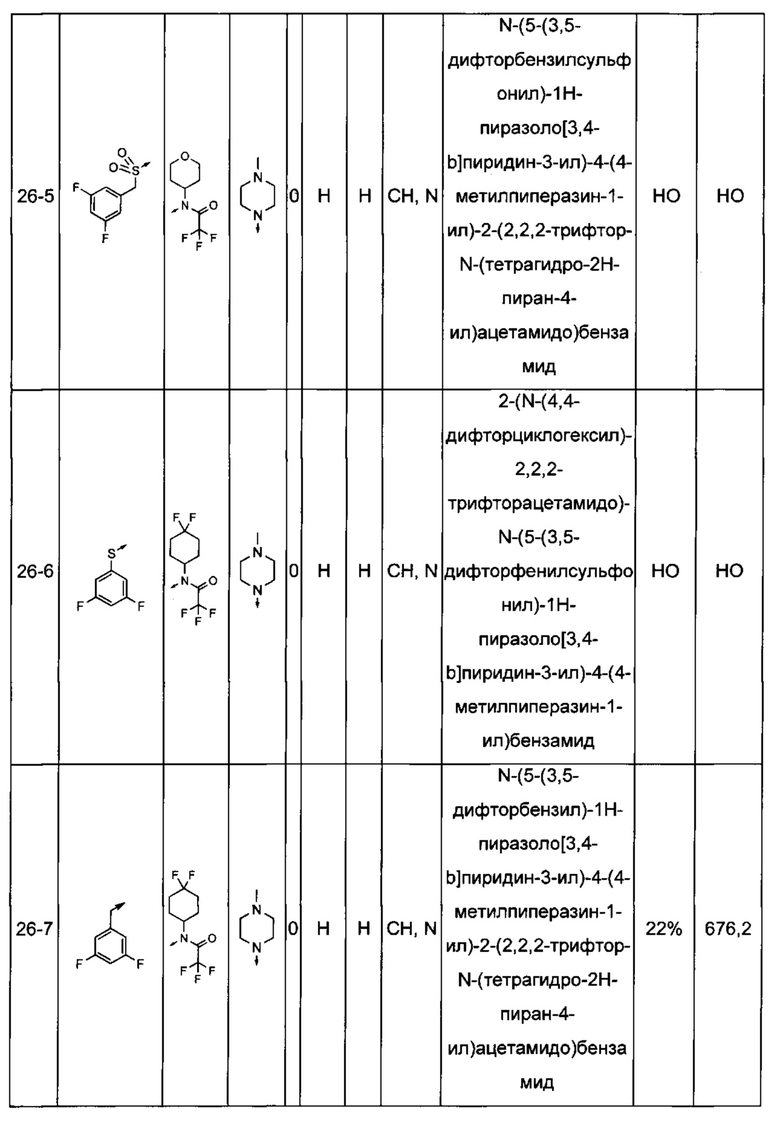

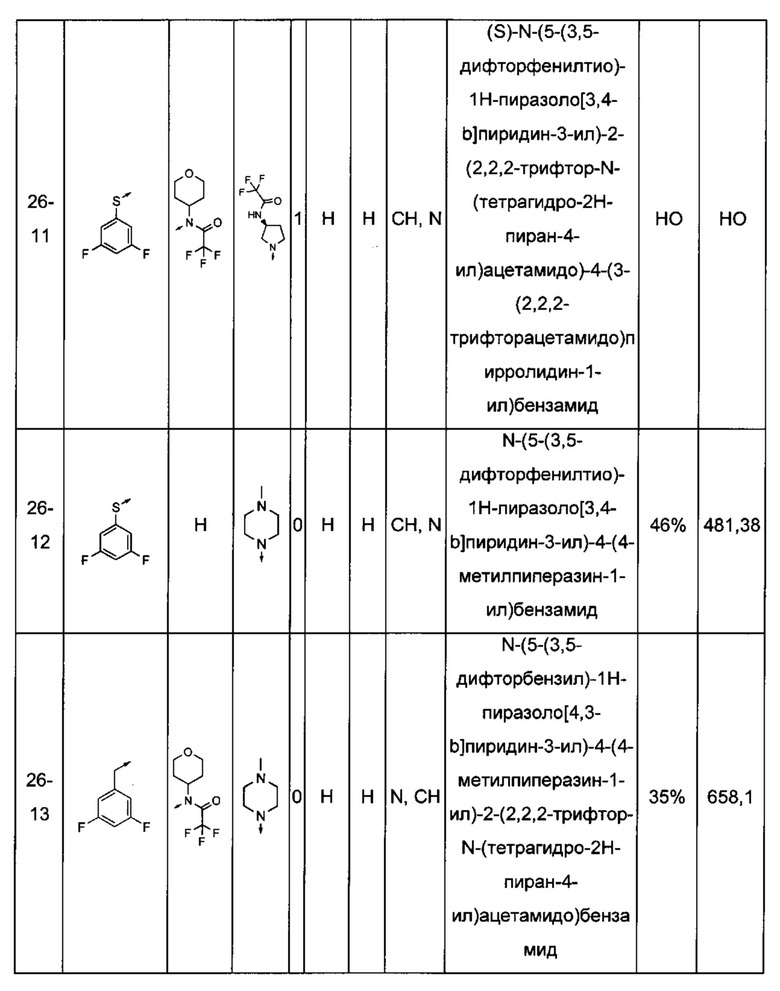


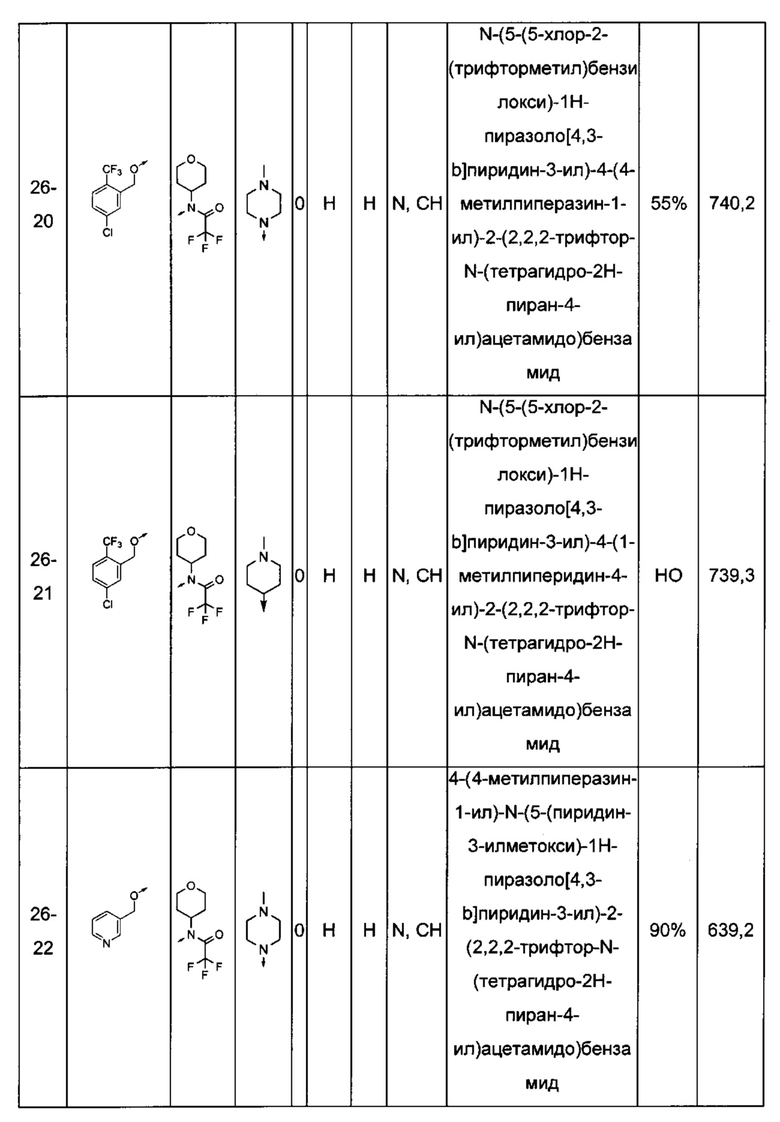
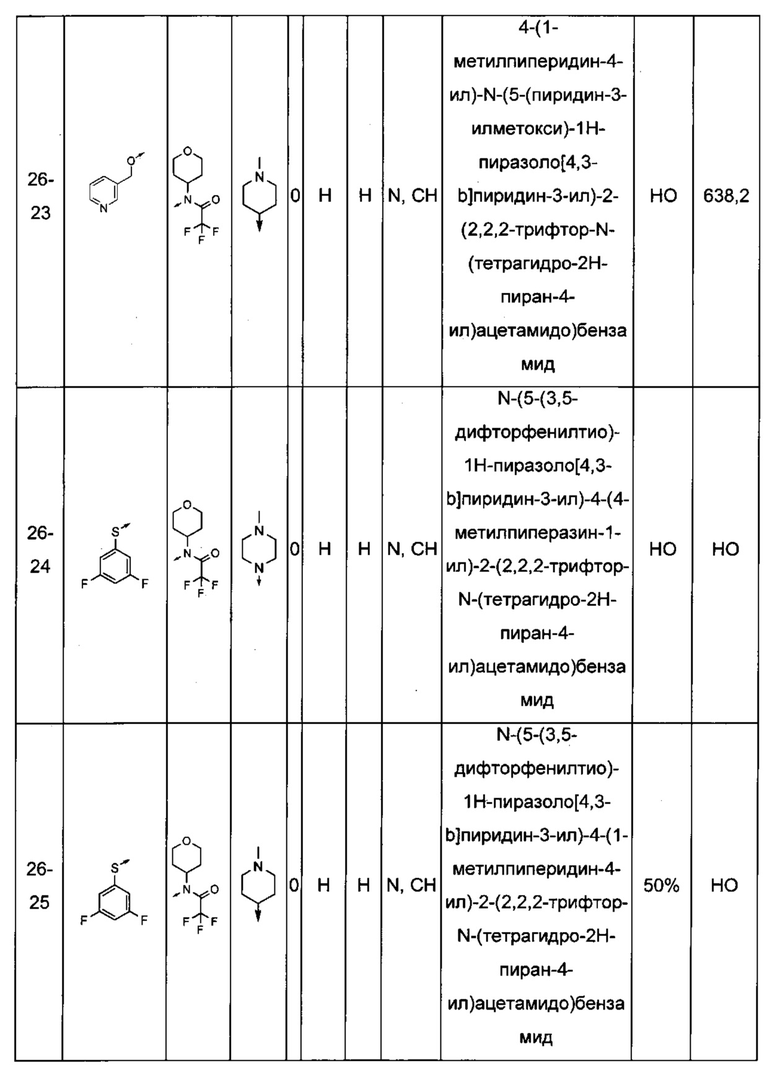



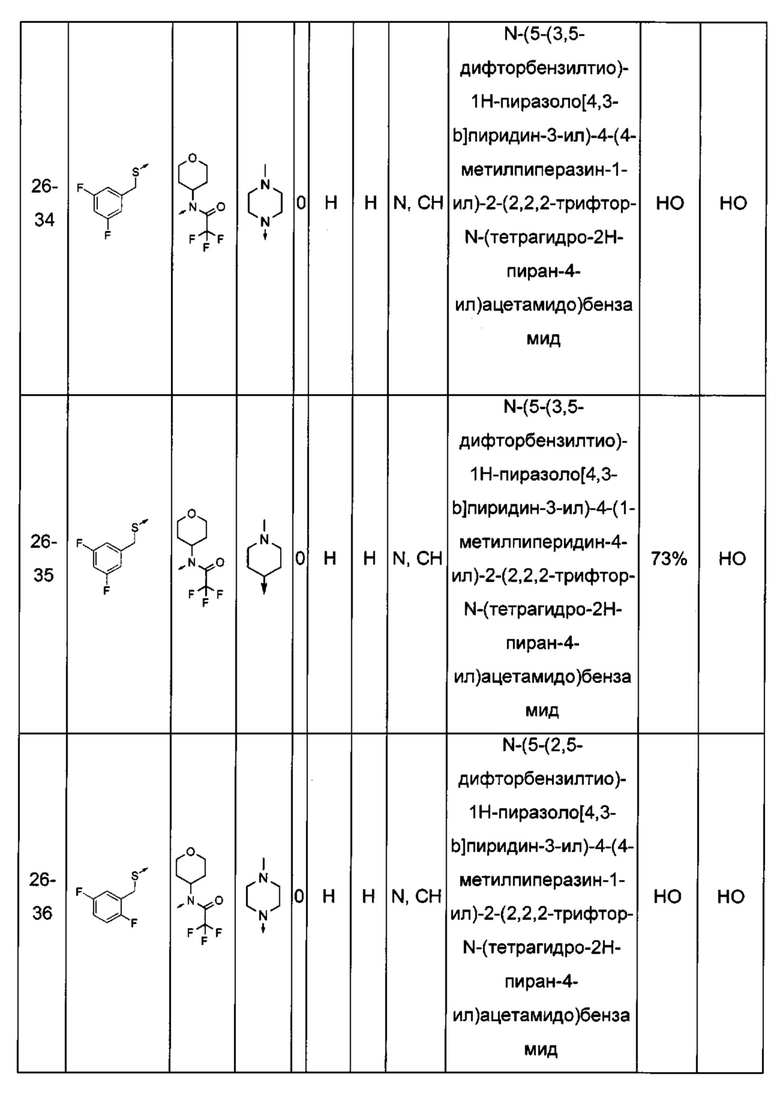



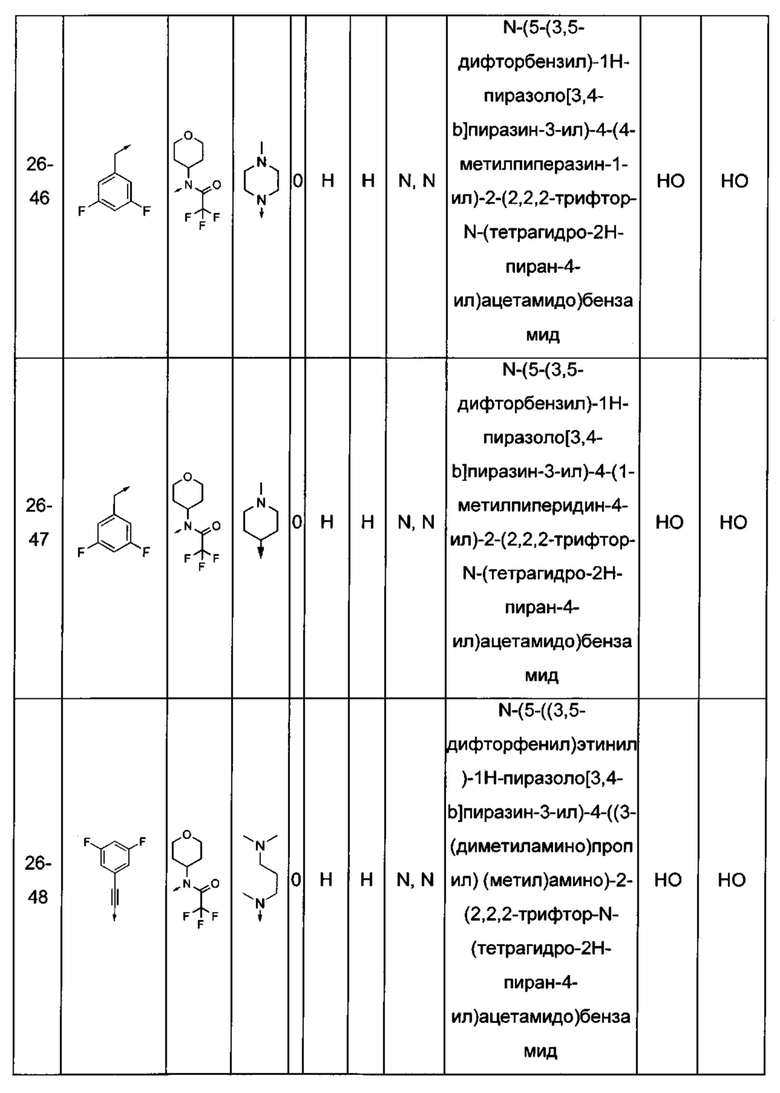
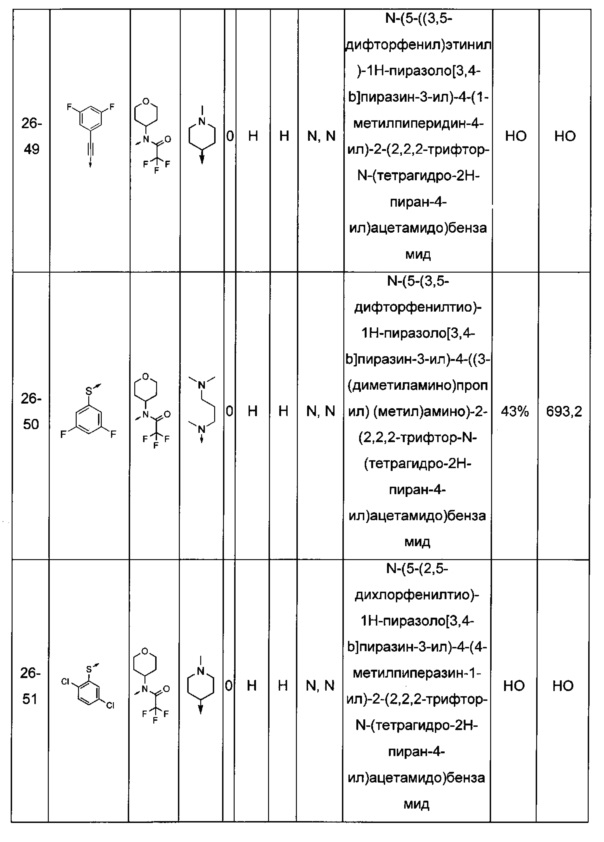


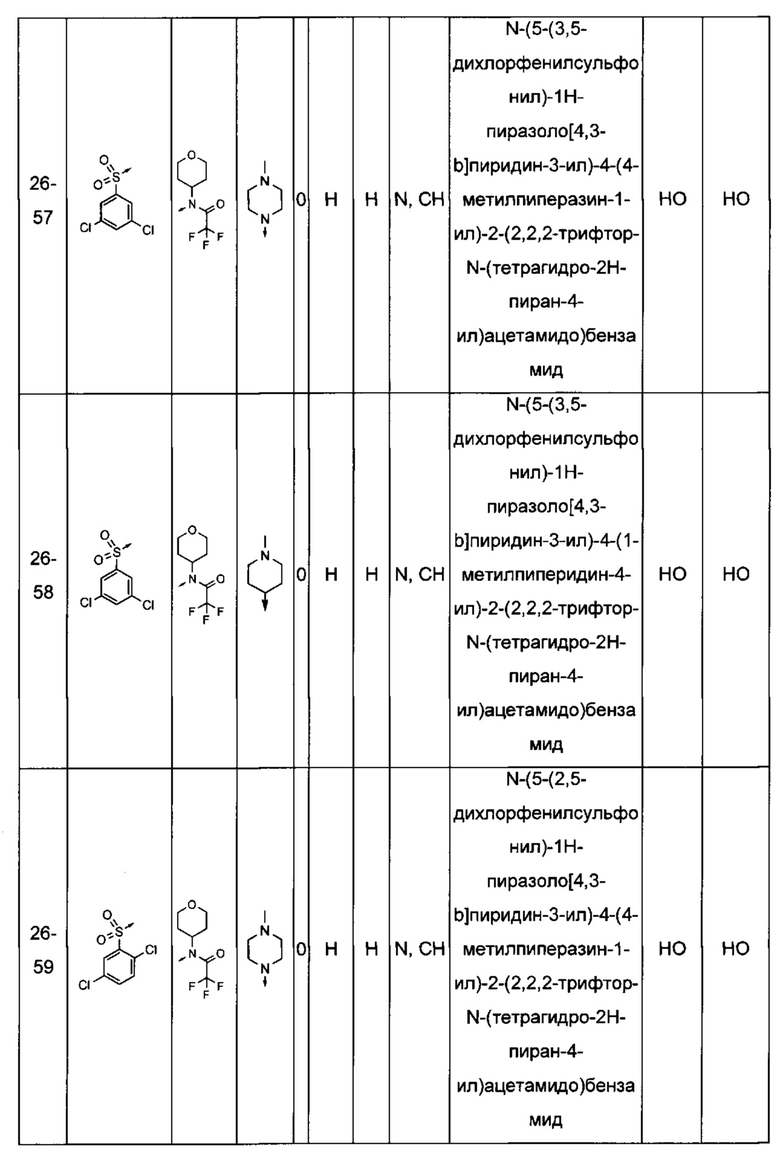


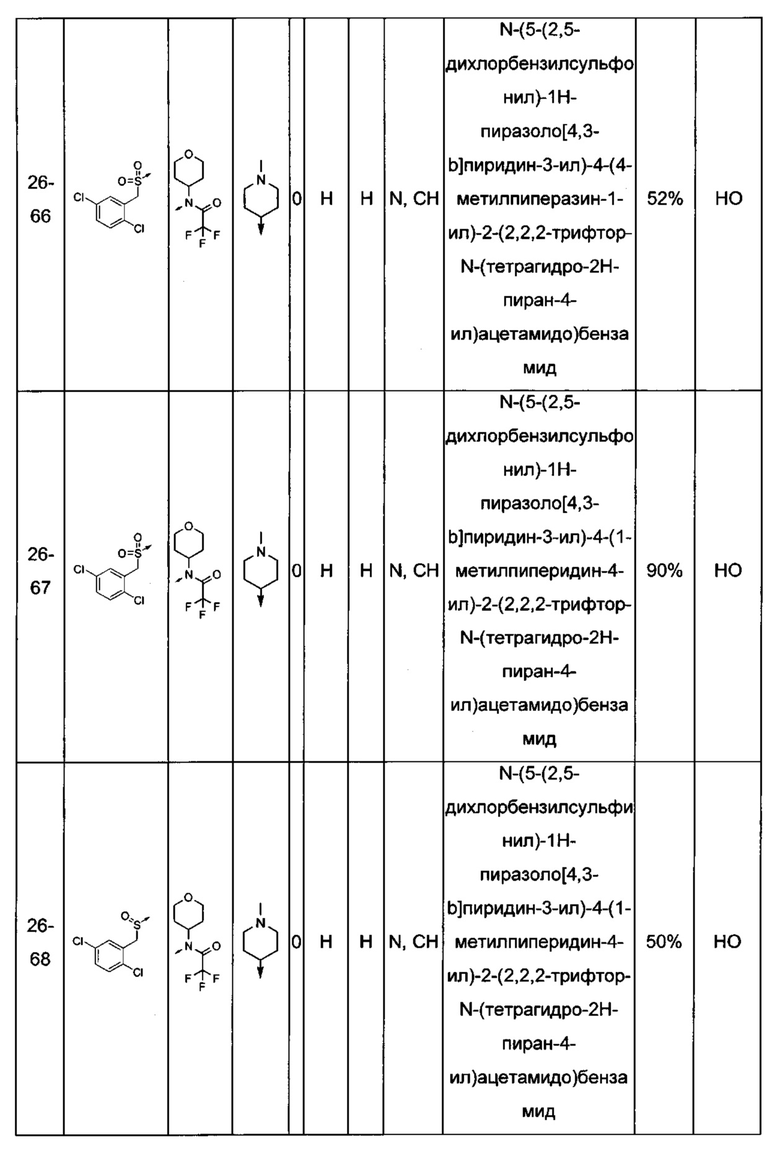
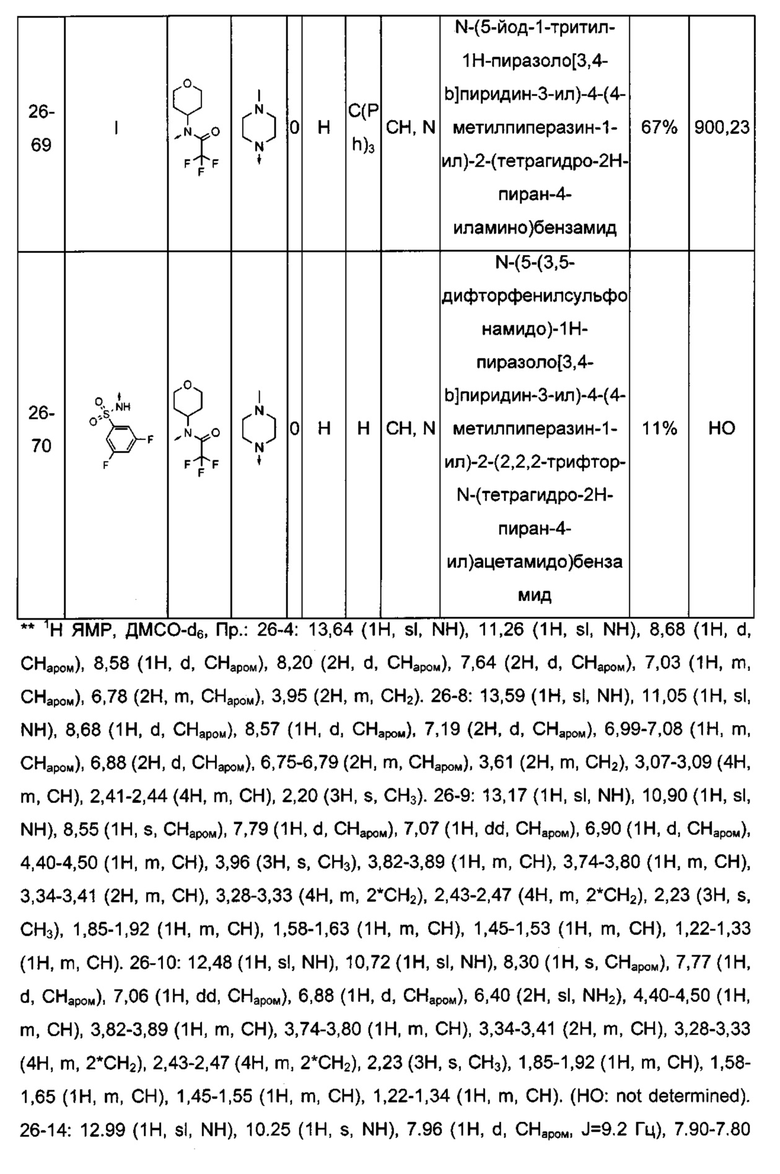

В некоторых случаях основной продукт этих реакций соответствует двузамещенному продукту, характеризующемуся дополнительной функционализацией пиразольного кольца. В этих случаях данный продукт выделяют и преобразуют в монозамещенный продукт путем обработки основанием, как описано ниже.
Пример 27: N-(5-(3,5-дифторфенилтио)-1-Н-пиразоло[3,4-b]пиразин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамид

Пример 27а: N-(5-(3,5-дифторфенилтио)-1-(4-(4-метилпиперазин-1-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензоил)-1Н-пиразоло[3,4-b]пиразин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензамид
1,51 мл (17,90 ммоль) оксалилхлорида и 2 капли безводного диметилформамида добавляли к 4,74 г (8,95 ммоль) раствора 4-(4-метилпиперазин-1-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензойной кислоты в 60 мл дихлорметана. Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Растворители выпаривали, образовавшееся твердое вещество растворяли в толуоле, и растворитель выпаривали; данную процедуру повторяли три раза до получения белого твердого вещества.
Хлорангидрид добавляли при 0°C небольшими фракциями к 1 г (3,58 ммоль) 5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиразин-3-амина, растворенного в 15 мл пиридина. Реакционную смесь перемешивали при 25°C в течение ночи при комнатной температуре. После выпаривания растворителя остаток очищали хроматографией на силикагеле (90:10 дихлорметан/метанол, а затем 90:9:1, затем 90:5:5 дихлорметан/метанол/аммиак в качестве элюента) с получением N-(5-(3,5-дифторфенилтио)-1-(4-(4-метилпиперазин-1-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензоил)-1Н-пиразоло[3,4-b]пиразин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензамида.
ЖХМС (ЭР, m/z): (М+1) 1074,64.
Пример 27: N-(5-(3, 5-дифторфенилтио)-1-Н-пиразоло[3,4-b]пиразин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамид
0,27 мл (1,95 ммоль) триэтиламина добавляли к 0,21 г (0,19 ммоль) раствора N-(5-(3,5-дифторфенилтио)-1-(4-(4-метилпиперазин-1-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензоил)-1Н-пиразоло[3,4-b]пиразин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензамида в 5 мл метанола. Реакционную смесь нагревали при 65°C в течение 4 часов, а затем в течение ночи при комнатной температуре. После выпаривания растворителя продукт экстрагировали несколько раз этилацетатом. Органические фазы объединяли, промывали насыщенным раствором бикарбоната натрия, высушивали на сульфате магния и концентрировали. Остаток очищали хроматографией на силикагеле (95:4:1 дихлорметан/метанол/аммиак в качестве элюента) с получением 0,065 г (57%) N-(5-(3,5-дифторфенилтио)-1-Н-пиразоло[3,4-b]пиразин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамида в форме желтого твердого вещества.
ЖХМС (ЭР, m/z): (M-1) 579,21.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 13.95 (1Н, bs, NH), 10.25 (1Н, bs, NH), 8.62 (1Н, s, СНаром), 8.27 (1Н, d, NH), 7.80 (1Н, d, СНаром), 7.17-7.27 (3H, m, СНаром), 6.27 (1Н, d, СНаром), 6.12 (1Н, d, СНаром), 3.79-3.82 (2Н, m, СН), 3.67 (1Н, m, СН), 3.45-3.50 (2Н, m, СН), 3.26-3.29 (4Н, m, СН), 2.42-2.44 (4Н, m, СН), 2.22 (3H, s, CH3), 1.90-1.93 (2Н, m, СН), 1.31-1.36 (2Н, m, СН).
Следующие соединения были получены тем же способом:



Реакции, проводимые в пиридине, часто дают возможность модифицировать региоизомерное распределение продуктов. Следующий пример является характеристическим для реакции этого типа.
Пример 27-бис: N-(5-(N-(3,5-дифторфенил)сульфамоил)-1H-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензамид
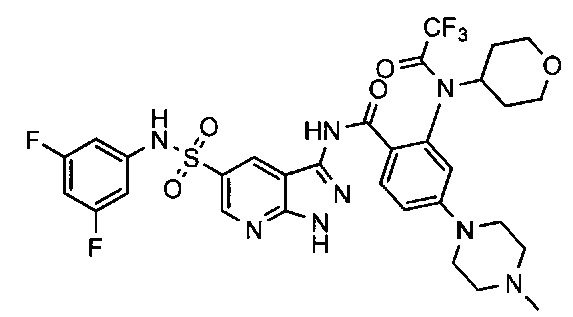
0,224 мл (2,63 ммоль) оксалилхлорида и 2 капли безводного диметилформамида добавляли к 0,697 г (1,316 ммоль) раствора 4-(4-метилпиперазин-1-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензойной кислоты в 20 мл дихлорметана. Реакционную смесь перемешивали в течение 2 часов при комнатной температуре. Растворители выпаривали, образовавшееся твердое вещество снова растворяли в толуоле, и растворитель выпаривали. Эту процедуру повторяли три раза до получения белого твердого вещества.
Хлорангидрид растворяли в 5 мл безводного пиридина, а затем образовавшийся раствор добавляли к раствору 0,214 г (0,658 ммоль) 3-амино-N-(3,5-дифторфенил)-1Н-пиразоло[3,4-b]пиридин-5-сульфонамида в 5 мл пиридина при 0°C. Реакционную смесь перемешивали в течение 3 часов при 0°C, а затем в течение ночи при комнатной температуре. Пиридин выпаривали, и неочищенный продукт реакции снова растворяли в толуоле, а затем концентрировали до сухого состояния. Реакционную смесь разбавляли насыщенным раствором NaHCO3 и экстрагировали этилацетатом. Органическую фазу высушивали на MgSO4, фильтровали и концентрировали, и неочищенный продукт использовали непосредственно в реакции удаления защиты без очистки или характеризации.
Следующие соединения были получены тем же способом:
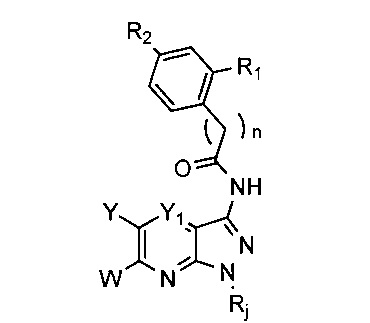

Пример способа E2:
Пример 28: 5-(3,5-дифторфенилтио)-N-(4-(4-метилпиперазин-1-ил)бензил)-1Н-пиразоло[3,4-b]пиридин-3-амин

41,5 мкл трифторуксусной кислоты (0,539 ммоль) и небольшими фракциями 129 мг (0,611 ммоль) триацетоксиборгидрида натрия добавляли к раствору 100 мг (0,35 ммоль) 5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиридин-3-амина и 81 мг (0,395 ммоль) 4-(4-метилпиперазин-1-ил)бензальдегида в 20 мл смеси 1:1 дихлорметана и тетрагидрофурана. Реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Добавляли дополнительную фракцию 125 мкл трифторуксусной кислоты и 388 мг триацетоксиборгидрида натрия, и реакционную смесь перемешивали дополнительно в течение 24 часов. Затем растворитель концентрировали, и реакционную смесь экстрагировали этилацетатом и промывали, используя насыщенный раствор бикарбоната натрия. Органические фазы объединяли, высушивали на сульфате магния, а затем концентрировали с получением желтого масла. В результате растирания этого масла в метаноле выделили 135 мг желтого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 467,57.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 12.43 (1Н, bs, NH), 8.49 (1Н, d, СНаром), 8.47 (1Н, d, СНаром), 7.25 (2Н, d, СНаром), 7.03-7.08 (1Н, m, СНаром), 6.89 (2Н, d, СНаром), 6.76-6.77 (3H, m, NH and СНаром), 4.34 (2Н, d, СН), 3.08 (4Н, m, СН), 2.44 (4Н, m, СН), 2.21 (3H, s, CH3).
Следующее производное было получено в соответствии с тем же способом:


Пример способа E3
Пример 29: 1-(5-(3,5-дифторфенилтио)-1H-пиразоло[3,4-b]пиридин-3-ил)-3-(4-(4-метилпиперазин-1-ил)фенил)тиомочевина
0,507 г (2,17 ммоль) 1-(4-изотиоцианатофенил)-4-метилпиперазина добавляли при 25°C к 0,540 г (2,17 ммоль) 3,5-дифторфенилтио-1Н-пиразоло[3,4-b]пиридин-3-амина, растворенного в 12 мл безводного диметилацетамида. Смесь оставляли при перемешивании в течение 15 часов при 85°C.Реакционную смесь обрабатывали добавлением 20 мл воды, а затем экстрагировали этилацетатом. Органическую фазу высушивали на сульфате натрия, фильтровали и концентрировали. Продукт очищали хроматографией на силикагеле (15:1 дихлорметан/метанол в качестве элюента) с получением 0,156 г (выход=15%) 1-(1-трет-бутил-5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиридйн-3-ил)-3-(4-(4-метилпиперазин-1-ил)фенил)тиомочевины в форме светло-коричневого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 512,08.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 13.69 (1Н, bs, NH), 11.50 (1Н, bs, NH), 11.19 (1Н, bs, NH), 8.96 (1H, d, СНаром), 8.66 (1H, d, СНаром), 7.41 (2Н, d, СНаром), 7.10 (1Н, ddd, СНаром), 6.95 (2Н, d, СНаром), 6.89 (2Н, bd, СНаром), 3.13-3.16 (4Н, m, СН), 2.45-2.47 (4Н, m, СН), 2.23 (3H, s, СН).
Пример 29-бис: 1-(5-(3,5-дифторфенилтио)-1H-пиразоло[3,4-b]пиридин-3-ил)-3-(4-(4-метилпиперазин-1-ил)фенил)мочевина

0,048 г (1,19 ммоль) гидрида натрия добавляли при 0°C к 0,200 г (0,598 ммоль) 1-трет-бутил-5-(3,5-дифторфенилтио)-1H-пиразоло[3,4-b]пиридин-3-амина, растворенного в 10 мл безводного диметилацетамида. Реакционную смесь оставляли при перемешивании на 10 минут. Затем добавляли 0,130 г (0,598 ммоль) 1-(4-изоцианатофенил)-4-метилпиперазина при 0°C. Смесь оставляли при перемешивании на 3 часа при комнатной температуре. Реакционную смесь обрабатывали добавлением по каплям 20 мл воды при 0°C, а затем экстрагировали этилацетатом. Органическую фазу высушивали на сульфате натрия, фильтровали и концентрировали. Продукт очищали хроматографией на силикагеле с получением 0,150 г (выход=45%) 1-(1-трет-бутил-5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиридин-3-ил)-3-(4-(4-метилпиперазин-1-ил)фенил)мочевины в форме светло-коричневого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 552,21.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 8.92 (1Н, bs, NH), 8.58 (1Н, bs, NH), 8.51 (1H, bs, СНаром), 8.30 (1H, bs, СНаром), 7.31 (2Н, d, СНаром), 7.05 (1Н, m, СНаром), 6.83-6.85 (2Н, m, СНаром), 3.03-3.08 (4Н, m, СН), 2.45-2.48 (4Н, m, СН), 2.21 (3H, s, СН), 1.76 (9Н, s, СН).
Раствор 0,150 г (0,272 ммоль) 1-(1-трет-бутил-5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиридин-3-ил)-3-(4-(4-метилпиперазин-1-ил)фенил)мочевины, растворенной в 20 мл ТФУ (трифторуксусной кислоты), кипятили с обратным холодильником в течение 3 часов. Растворитель выпаривали, и неочищенный продукт реакции разбавляли насыщенным раствором NaHCO3 и экстрагировали этилацетатом. Органическую фазу высушивали на MgSO4, фильтровали и концентрировали. Полученное твердое вещество растирали в метаноле, фильтровали и высушивали. Получили 110 мг (82%) 1-(5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиридин-3-ил)-3-(4-(4-метилпиперазин-1-ил)фенил)мочевины в
форме бежевого твердого вещества.
ЖХМС (ЭР, m/z): (М+1): 496,06.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 10.85 (1Н, bs, NH), 9.57 (1Н, bs, NH), 8.57 (1Н, bs, СНаром), 8.30 (1Н, bs, СНаром), 7.39 (2Н, d, СНаром), 6.99 (1Н, m, СНаром), 6.89 (2Н, d, СНаром), 6.70 (2Н, bd, СНаром), 3.03-3.08 (4Н, m, СН), 2.45-2.48 (4Н, m, СН), 2.21 (3H, s, СН).
Примеры способа F
Примеры способа F1: удаление защиты
Пример 30: N-(5-(3, 5-дифторфенилтио)-1-Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамид

9,08 мл (65,1 ммоль) триэтиламина добавляли к 2 г (2,96 ммоль) раствора N-(5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)бензамида в 65 мл метанола. Реакционную смесь нагревали при 65°C в течение 2 часов, а затем в течение ночи при комнатной температуре. Образовавшийся осадок фильтровали, промывали пентаном, водой, а затем диэтиловым эфиром, а затем высушивали в вакууме с получением 0,73 г (43%) (N-(5-(3,5-дифторфенилтио)-1-Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамида в форме белого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 580,23.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 13.59 (1Н, bs, NH), 10.56 (1Н, bs, NH), 8.61 (1Н, s, СНаром), 8.50 (1Н, s, СНаром), 8.17 (1Н, d, NH), 7.80 (1Н, d, СНаром), 7.07 (1Н, m, СНаром), 6.86 (2Н, m, СНаром), 6.23 (1Н, d, СНаром), 6.13 (1Н, d, СНаром), 3.79-3.82 (2Н, dt, СН), 3.60 (1Н, m, СН), 3.45-3.50 (2Н, m, СН), 3.21-3.33 (4Н, m, СН), 2.42-2.46 (4Н, m, СН), 2.22 (3H, s, CH3), 1.91-1.94 (2Н, m, СН), 1.35-1.38 (2Н, m, СН).
Следующие производные были получены в соответствии с тем же способом:
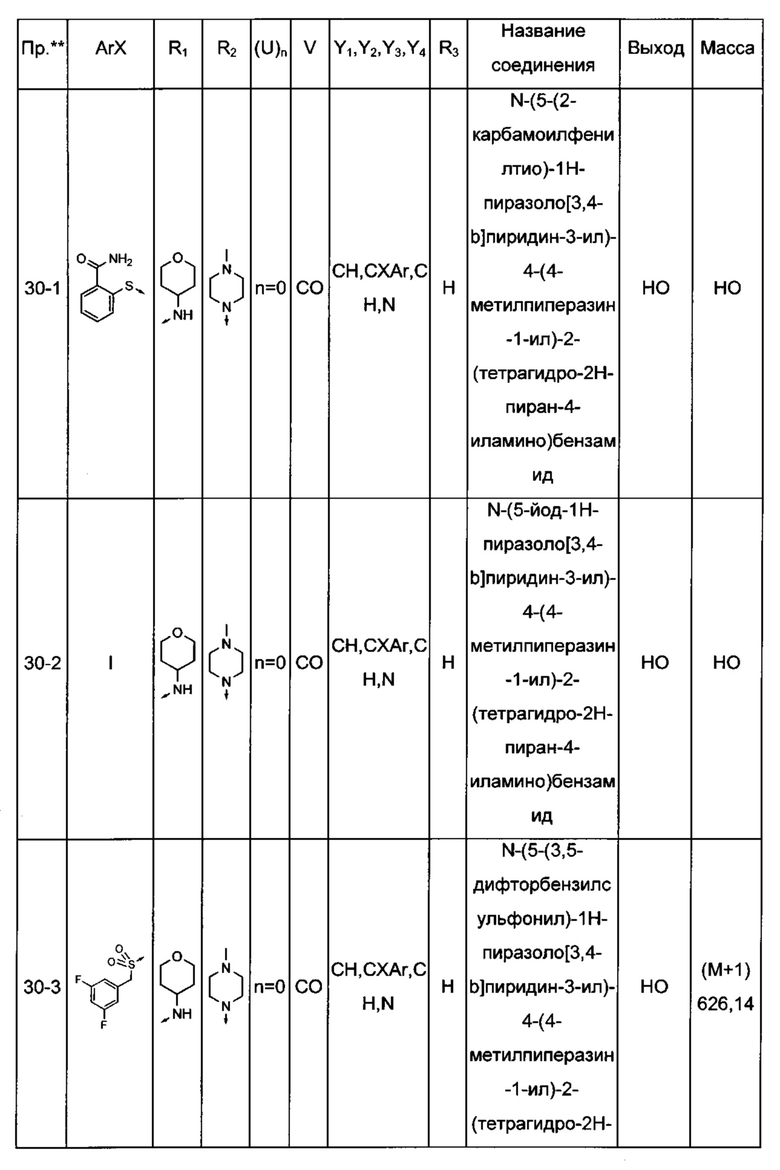

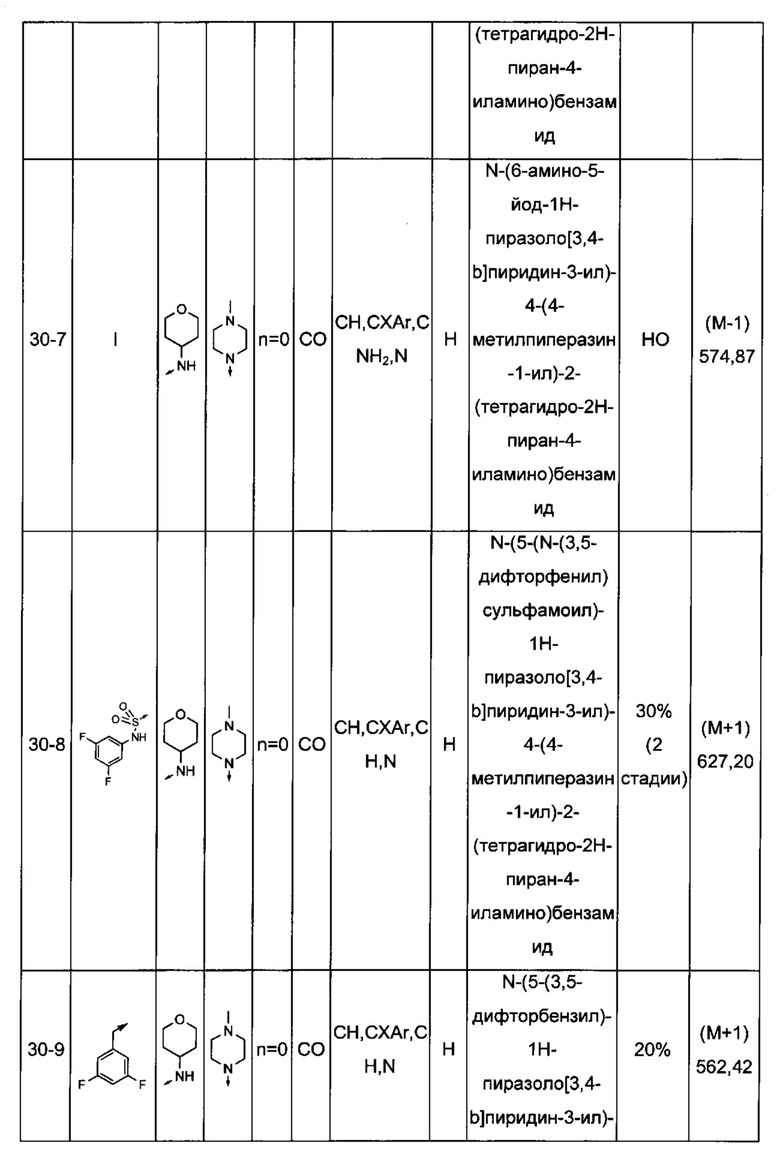

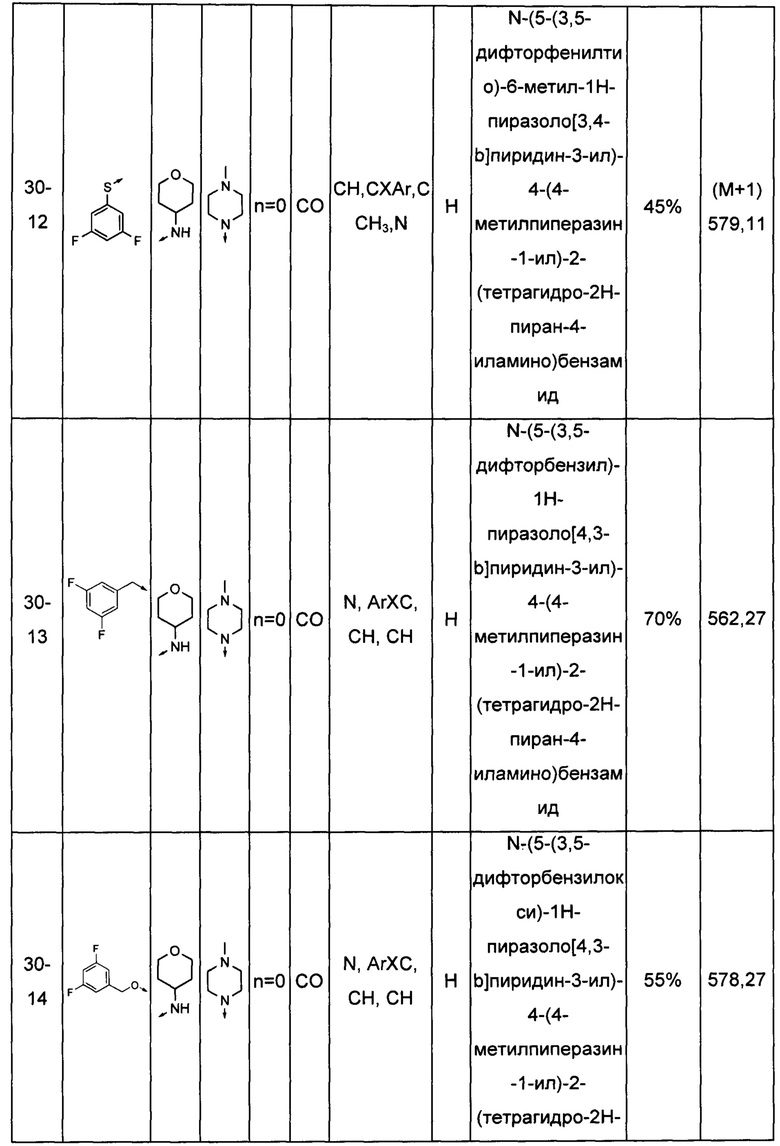
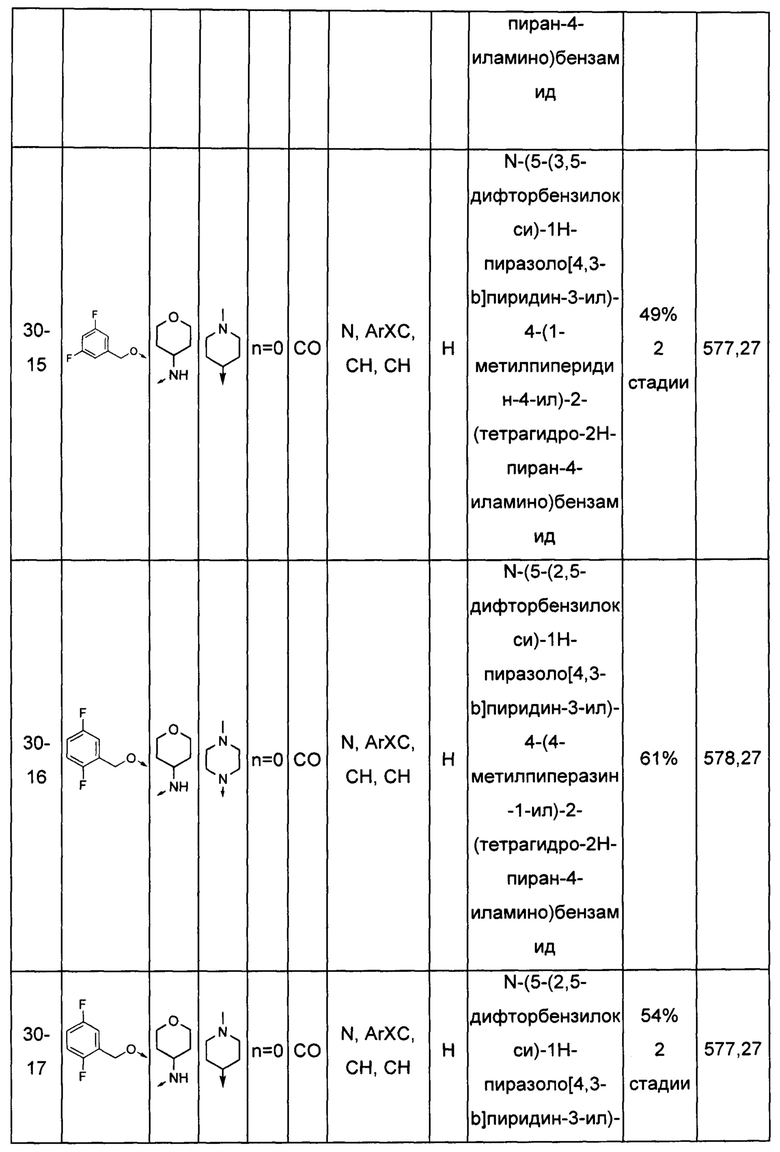
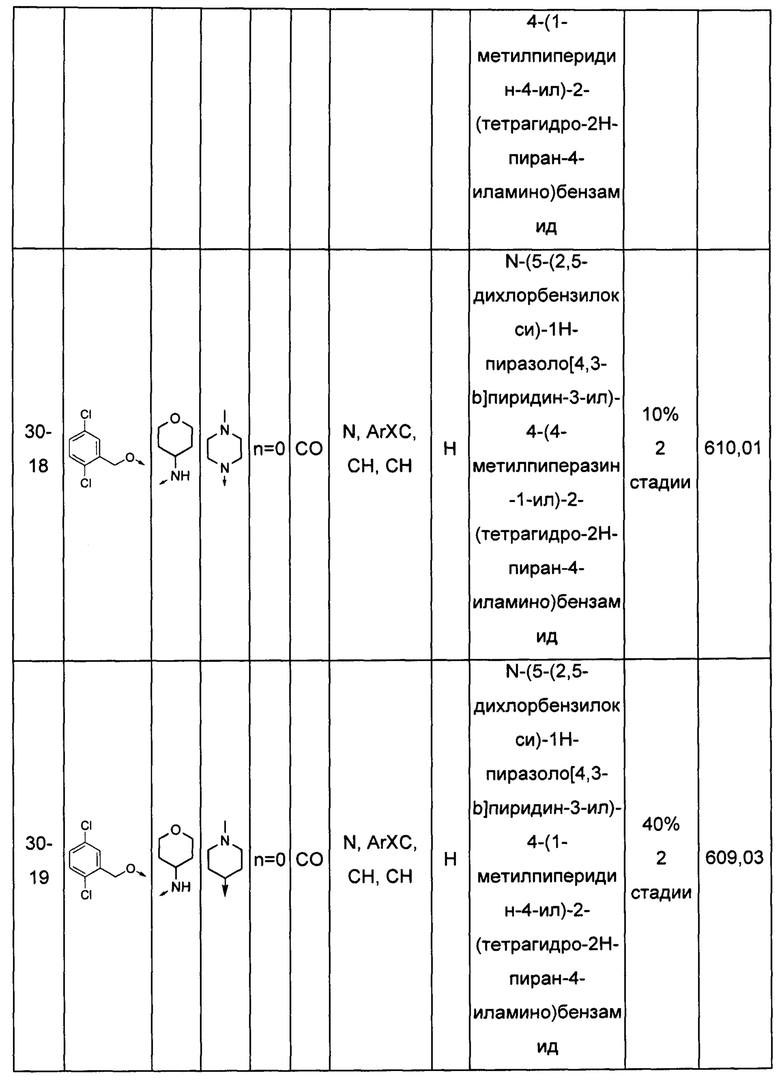


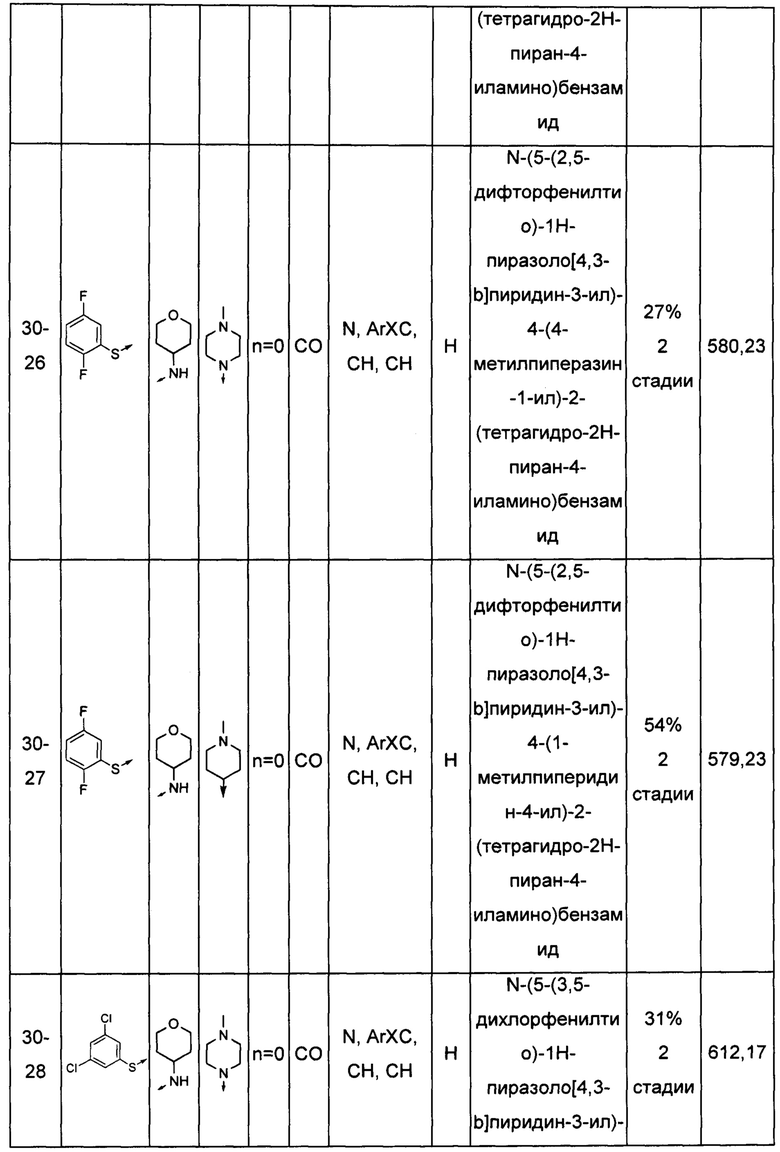

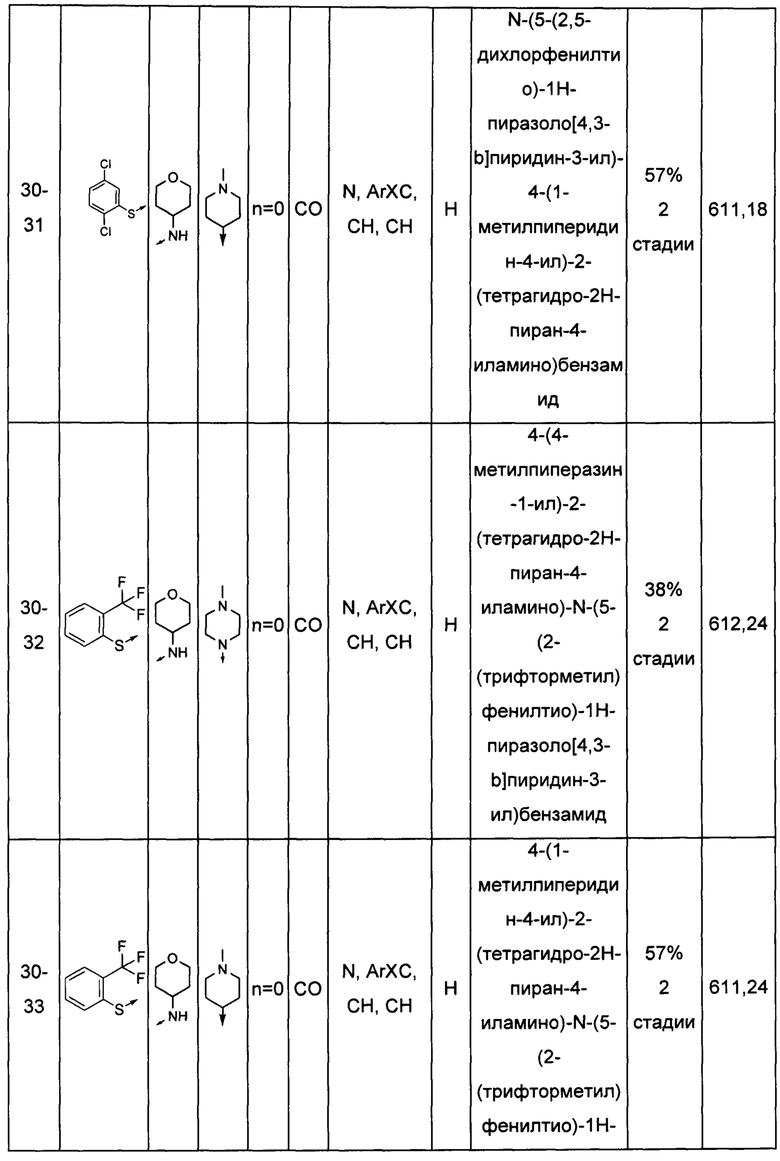
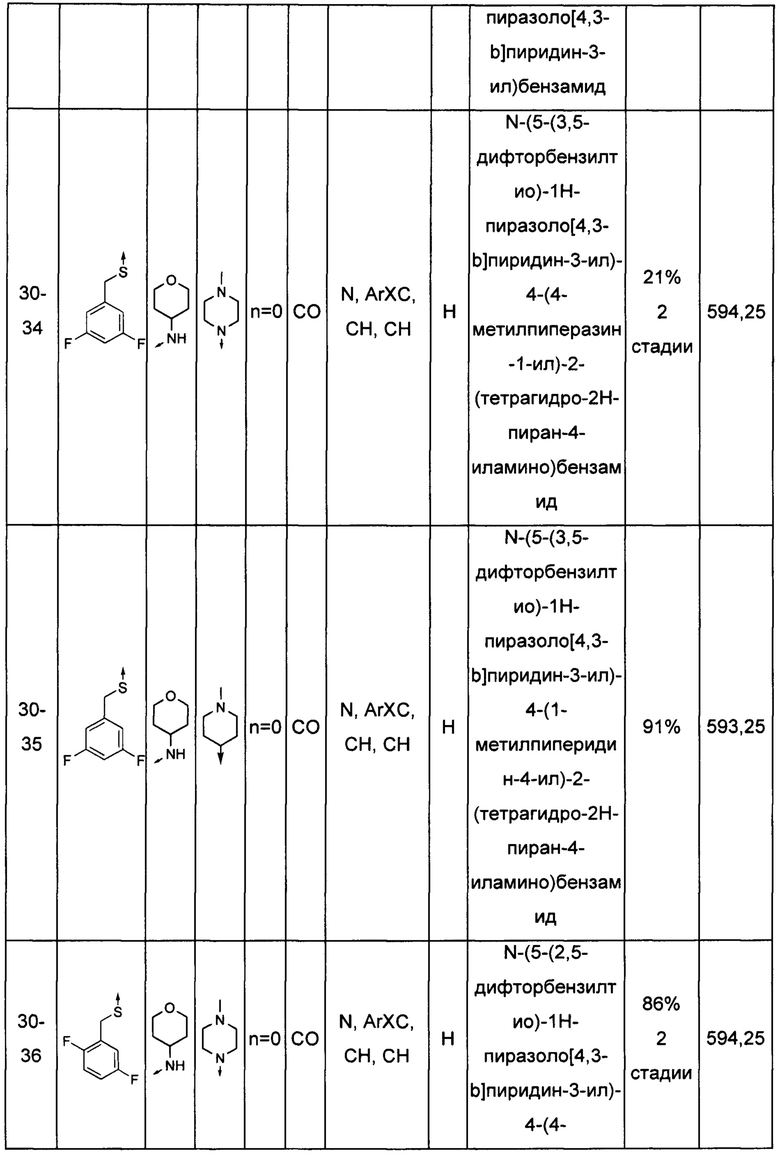



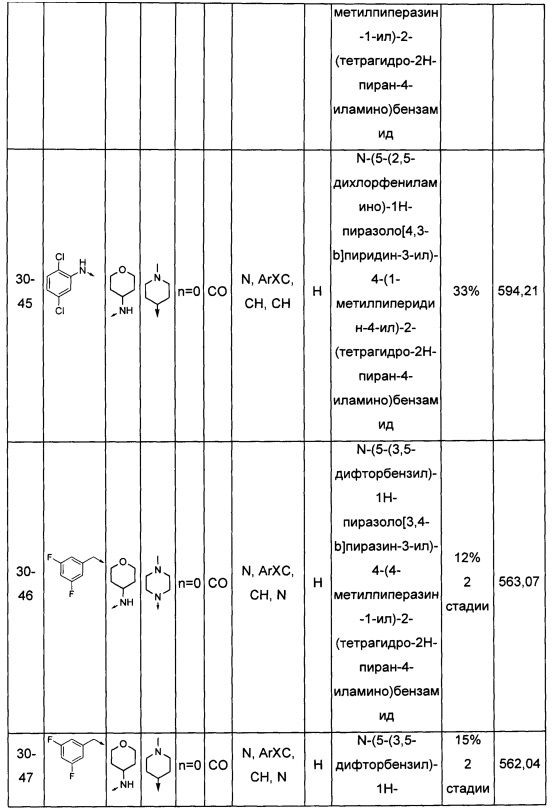


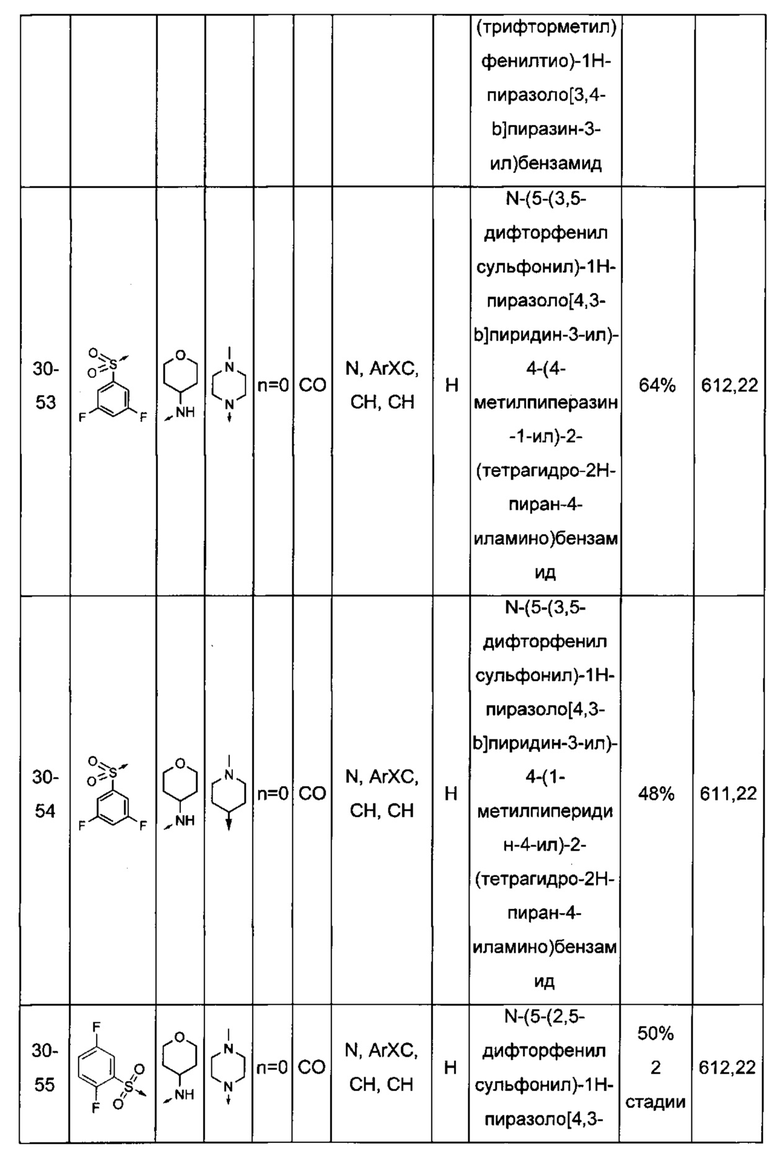

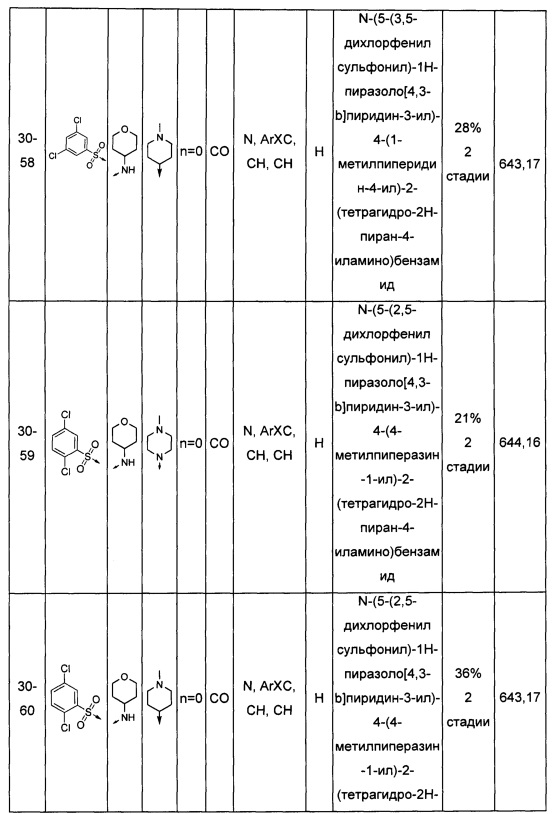
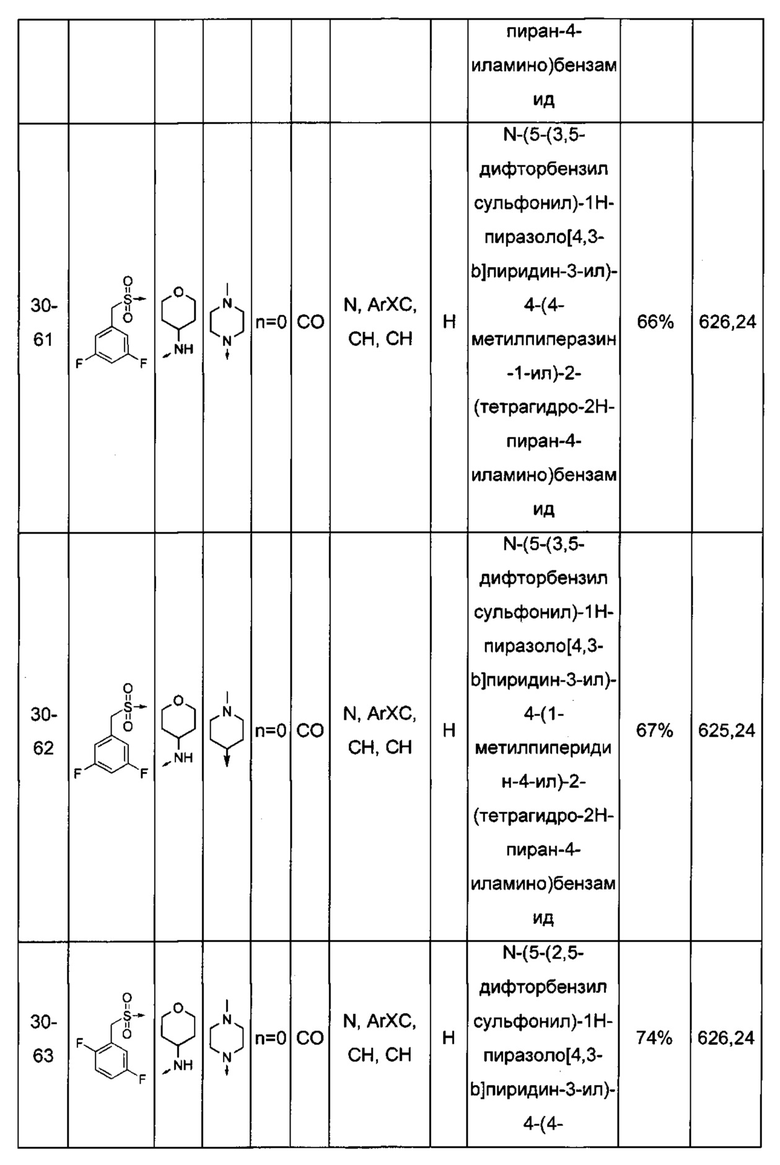


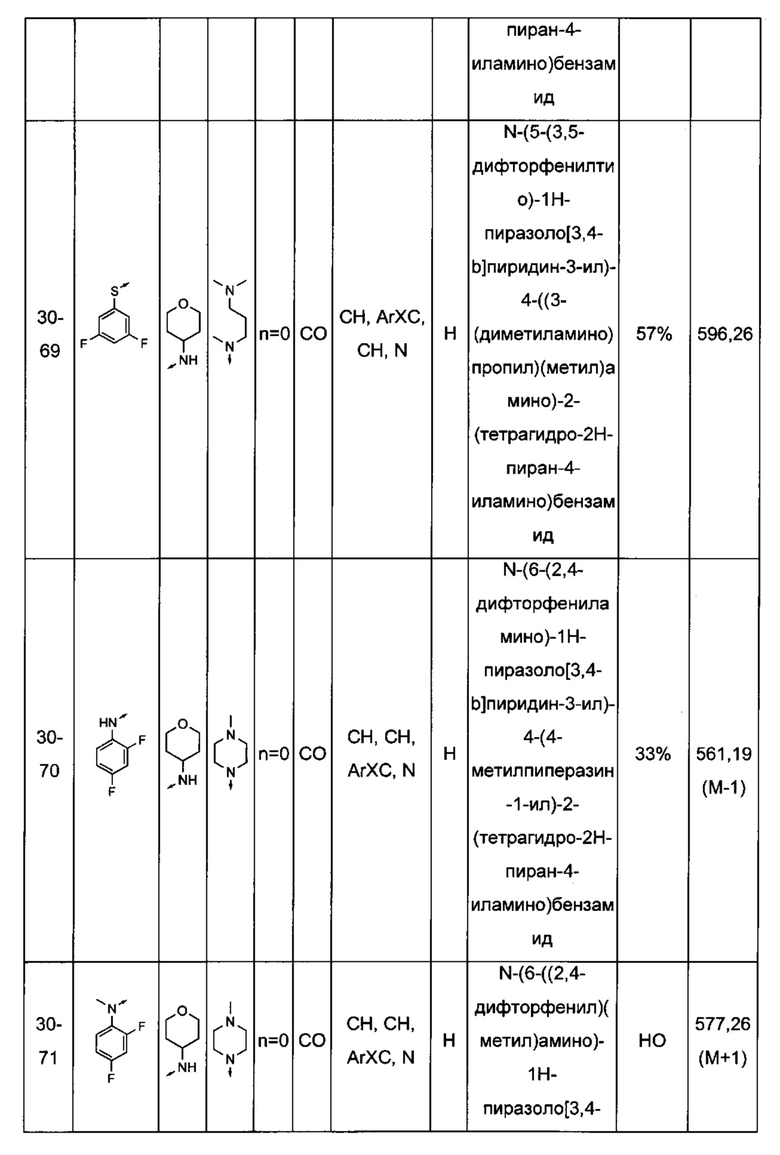










Пример 30-бис: (S)-4-(3-аминопирролидин-1-ил)-N-(5-(3,5-дифторфенилтио)-1H-пиразоло[3,4-b]пиридин-3-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамид

876 мкл (20 экв.) триэтиламина добавляли к раствору 238 мг (0,314 ммоль) (S)-N-(5-(3,5-дифторфенилтио)-1H-пиразоло[3,4-b]пиридин-3-ил)-2-(2,2,2-трифтор-N-(тетрагидро-2Н-пиран-4-ил)ацетамидо)-4-(3-(2,2,2-трифторацетамидо)пирролидин-1-ил)бензамида в 6 мл метанола. Реакционную смесь перемешивали при 65°C в течение 4 часов. После возврата к комнатной температуре добавляли 8 мл н-бутанола и 260 мг (6 экв.) карбоната калия. Реакционную смесь перемешивали при 80°C в течение 24 часов. После возврата к комнатной температуре растворители выпаривали, добавляли воду, и продукт экстрагировали дихлорметаном. Органическую фазу промывали насыщенным раствором хлорида натрия, высушивали на сульфате магния, фильтровали и выпаривали. Остаток очищали хроматографией на силикагеле (8:2 дихлорметан/метанол в качестве элюента) с получением 87 мг (выход=49%) (S)-4-(3-аминопирролидин-1-ил)-N-(5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиразин-3-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамида в форме коричневого порошка.
ЖХМС (ЭР, m/z): (М+1) 566,24.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 10.46 (1Н, bs, NH), 8.60 (1Н, s, СНаром), 8.50 (1Н, s, СНаром), 8.26 (1Н, d, NH), 7.78 (1Н, d, СНаром), 7.08 (1Н, t, СНаром), 6.86 (2Н, d, СНаром), 5.86 (1Н, dd, СНаром), 5.71 (1Н, d, СНаром), 3.80-3.88 (2Н, m, СН), 3.63-3.70 (2Н, m, СН), 3.40-3.55 (5Н, m, СН), 3.01-3.08 (1Н, m, CH), 2.08-2.13 (1Н, m, СН), 1.92-1.99 (2Н, m, CH3), 1.76-1.82 (1Н, m, СН), 1.30-1.41 (2Н, m, СНпиранон).
Примеры способа F2: восстановление
Пример 31: N-(5-(3,5-дифторфенетил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамид
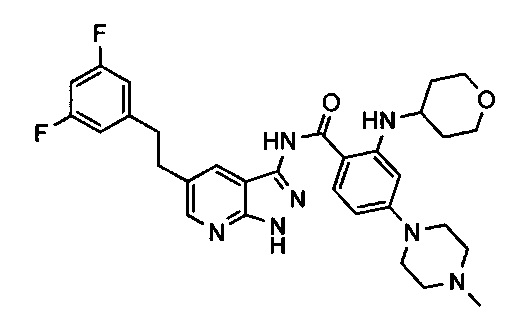
10 мг 10% Pd/C добавляли к 100 мг (0,175 ммоль) N-(5-((3,5-дифторфенил)этинил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамида в растворе в смеси 10 мл тетрагидрофурана и 5 мл метанола, после чего реакционную смесь помещали в атмосферу водорода. Реакционную смесь перемешивали в течение 12 часов при комнатной температуре, а затем фильтровали на целлите и концентрировали. Выделили 62 мг (выход=60%) N-(5-(3,5-дифторфенетил)-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамида в форме белого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 576,23.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 13.14 (1Н, bs, NH), 10.32 (1Н, bs, NH), 8.40 (1Н, d, СНаром), 8.22 (1Н, d, NH), 7.96 (1Н, d, СНаром), 7.80 (1Н, d, СНаром), 7.03-6.98 (3H, m, СНаром), 6.23 (1Н, d, СНаром), 6.16 (1Н, bs, СНаром), 3.84-3.81 (2Н, dt, СН), 3.70 (1Н, m, СН), 3.52-3.46 (2Н, m, СН), 3.04-2.93 (4Н, m, CH), 2.59-2.69 (4Н, m, СН), 2.42-2.46 (4Н, m, CH), 2.38 (3H, s, CH3), 1.96-1.93 (2Н, m, СН), 1.40-1.33 (2Н, m, CH).
Следующее производное получили в соответствии с тем же способом:


Пример 32: 5-(3,5-дифторфенилтио)-N-(4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензил)-1Н-пиразоло[3,4-b]пиридин-3-амин

100 мг (0,173 ммоль) N-(5-(3,5-дифторфенилтио)-1-Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамид добавляли небольшими фракциями к раствору 19,64 мг (0,518 ммоль) LiAIH4 в 3 мл безводного тетрагидрофурана в атмосфере аргона при 0°C. Реакционную смесь нагревали при 90°C в течение 15 часов. Затем добавляли дополнительную порцию 20 мг LiAIH4, и реакционную смесь перемешивали при 90°C в течение 5 часов. Затем к реакционной смеси добавляли 45 мкл воды при 0°C, а затем 45 мкл гидроксида натрия (15 масс. %) и, наконец, 120 мкл воды. Реакционную смесь перемешивали при 25°C в течение 1 часа, а затем фильтровали на дикалите. После выпаривания растворителей неочищенный продукт очищали хроматографией. Получили 16,80 мг (17%) 5-(3,5-дифторфенилтио)-N-(4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензил)-1Н-пиразоло[3,4-b]пиридин-3-амина в форме желтого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 566,68.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 12.57 (1Н, bs, NH), 8.45 (2Н, d, СНаром), 6.97-7.06 (2Н, m, СНаром), 6.73-6.75 (2Н, m, СНаром), 6.65 (1Н, t, NH), 6.13-6.19 (2Н, m, СНаром), 4.98 (1Н, d, NH), 4.30 (2Н, m, CH2), 3.73-3.77 (2Н, m, СН), 3.60 (1Н, m, СН), 3.45-3.50 (2Н, m, СН), 3.04 (4Н, m, СН), 2.42 (4Н, m, СН), 2.18 (3H, s, CH3), 1.80-1.83 (2Н, m, СН), 1.27-1.32 (2Н, m, СН).
Следующее производное получили в соответствии с тем же способом:

Пример 33: 2-(4-аминофенил)-N-(5-(3,5-дифторфенилтио)-1H-пиразоло[3,4-b]пиридин-3-ил)ацетамид
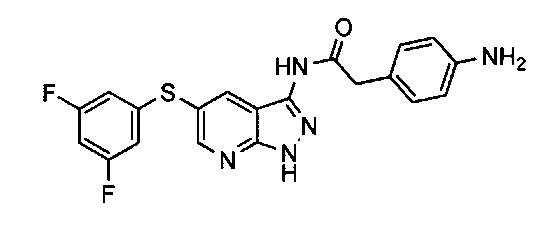
Раствор 152 мг (2,72 ммоль) железа и 70 мг (1,3 ммоль) хлорида аммония в 100 мкл воды добавляли к раствору 0,24 г (0,544 ммоль) N-(5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиридин-3-ил)-2-(4-нитрофенил)ацетамида в 10 мл смеси 2:1 этанол/вода. К этой смеси добавляли несколько капель уксусной кислоты и нагревали ее при 60°C в течение 4 часов. После охлаждения и концентрирования растворителей неочищенный продукт реакции экстрагировали этилацетатом и промывали насыщенным раствором бикарбоната натрия. Органические фазы объединяли, высушивали на сульфате магния, а затем концентрировали. Неочищенный продукт очищали хроматографией на силикагеле (ДХМ/МеОН) с получением 11 мг (4%) 2-(4-аминофенил)-N-(5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиридин-3-ил)ацетамида в форме коричневого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 412,09.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 13.60 (1Н, bs, NH), 10.96 (1Н, bs, NH), 8.68 (1Н, d, СНаром), 8.55 (1H, d, СНаром), 7.06 (1Н, m, СНаром), 6.98 (2Н, d, СНаром), 6.79 (2Н, m, СНаром), 6.50 (2Н, m, СНаром), 4.92 (2Н, s, NH), 3.51 (2Н, m, CH2).
Примеры способа F3: окисление сульфида
Пример 34: 5-(3,5-дифторфенилсульфонил)-1Н-пиразоло[3,4-b]пиридин-3-амин

Раствор 663 мг (1,078 ммоль) оксона в 1,1 мл воды добавляли к раствору 300 мг (1,078 ммоль) 5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиридин-3-амина в 10 мл смеси 1:1 тетрагидрофурана и метанола при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Затем добавляли дополнительную порцию 663 мг оксона при 0°C, и реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Растворители выпаривали, и реакционную смесь разбавляли раствором бикарбоната натрия, экстрагировали этилацетатом, высушивали на MgSO4, а затем концентрировали с получением 340 мг (81%) 5-(3,5-дифторфенилсульфонил)-1Н-пиразоло[3,4-b]пиридин-3-амина в форме желтого твердого вещества.
ЖХМС (ЭР, m/z): (М+1) 311,03.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 12.72 (1Н, bs, NH), 8.92 (1Н, d, СНаром), 8.84 (1Н, d, СНаром), 7.89-8.01 (1Н, d, СНаром), 7.62-7.80 (2Н, m, СНаром), 6.06 (2Н, bs, NH).
Следующие соединения были также получены этим способом:

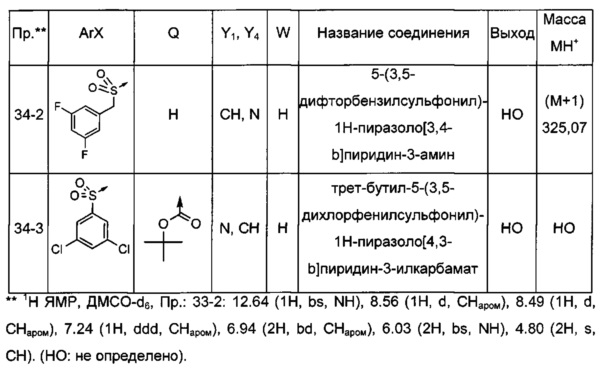
Альтернативно стадию защиты можно выполнять перед реакцией окисления с последующей стадией удаления защиты, которая может приводить к получению соответствующих сульфонов или сульфоксидов.
Пример 34-бис: 5-(3,5-дифторфенилсульфинил)-1Н-пиразоло[4,3-b]пиразин-3-амин

0,55 мл триэтиламина и 22 мг 4-диметиламинопиридина добавляли в атмосфере аргона к раствору 500 мг (1,790 ммоль) 5-(3,5-дифторфенилтио)-1Н-пиразоло[3,4-b]пиразин-3-амина в 10 мл тетрагидрофурана. Раствор перемешивали при 0°C и добавляли 0,915 мл ди-трет-бутилдикарбоната, и реакционную смесь перемешивали в течение ночи. К реакционной смеси добавляли водную фракцию, а затем экстрагировали ее этилацетатом. Органические фазы высушивали на MgSO4 и концентрировали в вакууме с получением неочищенного продукта, который использовали на стадии окисления без дополнительной очистки.
Полученный неочищенный продукт растворяли в 10 мл смеси 1:1 тетрагидрофурана и метанола при 0°C, а затем добавляли раствор 1,103 г (1,794 ммоль) оксона в 2 мл воды. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Затем добавляли дополнительную порцию 550 мг оксона, и реакционную смесь перемешивали при комнатной температуре в течение 5 часов. Растворители выпаривали, и реакционную смесь разбавляли раствором бикарбоната натрия, экстрагировали этилацетатом, высушивали на сульфате магния и концентрировали с получением смеси соответствующего сульфона и сульфоксида, который использовали на стадии окисления без дополнительной очистки.
0,373 мл ТФУ в 4 мл безводного ТГФ добавляли при 0°C к раствору 600 мг полученной ранее смеси в 6 мл дихлорметана. Смесь перемешивали в течение 1 часа при комнатной температуре и добавляли дополнительную порцию 4 эквивалента ТФУ в 4 мл ТГФ. После 1 часа перемешивания эту процедуру повторяли, и реакционную смесь перемешивали в течение суммарного времени 3 ч 45 мин. Растворители выпаривали, и реакционную смесь разбавляли раствором карбоната калия, экстрагировали этилацетатом, высушивали на сульфате магния и концентрировали с получением смеси 1:1 5-(3,5-дифторфенилсульфонил)-1Н-пиразоло[3,4-b]пиразин-3-амина и 5-(3,5-дифторфенил сульфинил)-1Н-пиразоло[3,4-b]пиразин-3-амина. Эту смесь использовали в следующей стадии без дополнительной очистки.
Следующие соединения были также получены этим способом:
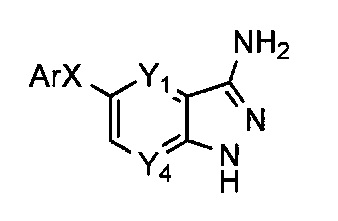
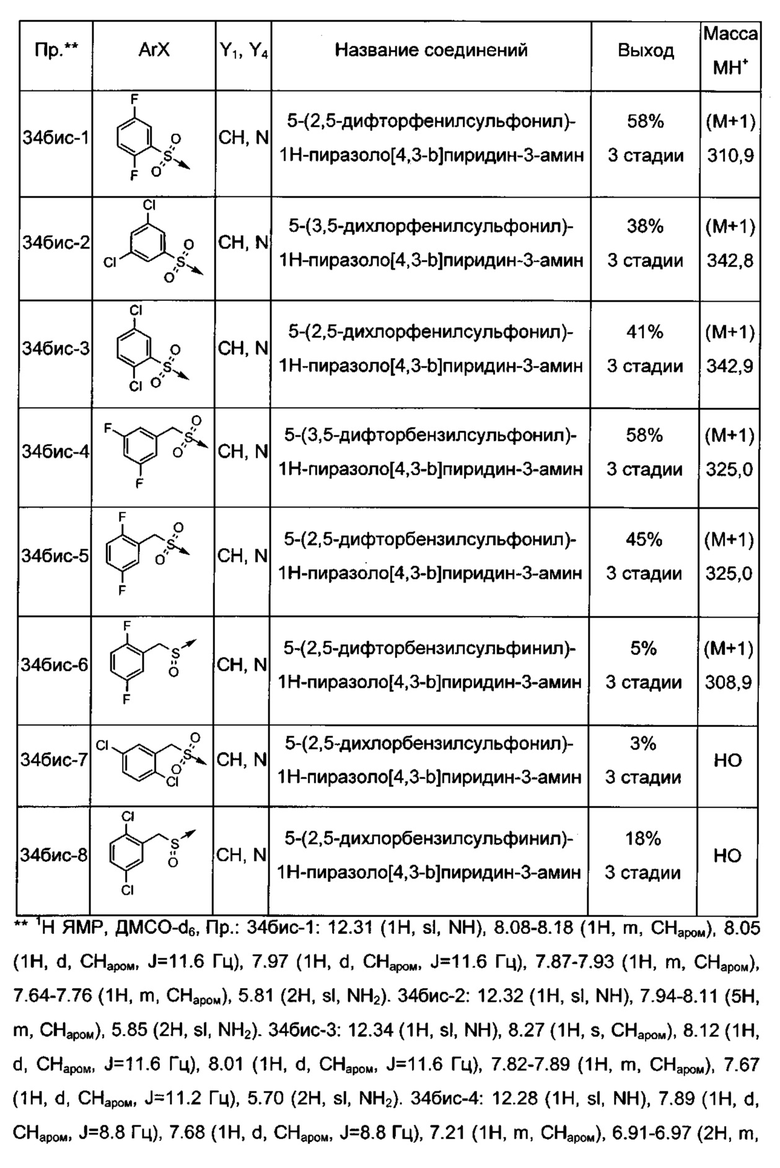

Пример способа F4: деметилирование
Пример 35: N-(5-(3,5-дифторфенилтио)-6-гидрокси-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамид

443 мкл (3 экв.) раствора 1 М трибромида бора в дихлорметане добавляли к раствору 90 мг (0,148 ммоль) N-(5-(3,5-дифторфенилтио)-6-метокси-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамида (пример 18) в 4 мл 1,2-дихлорэтана при 0°C. Реакционную смесь перемешивали при 60°C в течение 3 часов, а затем охлаждали в ледяной бане, после чего добавляли метанол. Растворители выпаривали, и остаток снова растворяли в смеси метанола и этилацетата. Образовавшееся твердое вещество фильтровали, снова растворяли в 3 мл тетрагидрофурана и добавляли к 1 н. раствору соды. Реакционную смесь перемешивали в течение 18 часов при комнатной температуре. pH раствора доводили до 8-9, и водную фазу экстрагировали этилацетатом. Органическую фазу высушивали на сульфате магния, и неочищенный продукт очищали на колонке силикагеля (дихлорметан/метанол в качестве элюента) с получением 21 мг (24%) N-(5-(3,5-дифторфенилтио)-6-гидрокси-1Н-пиразоло[3,4-b]пиридин-3-ил)-4-(4-метилпиперазин-1-ил)-2-(тетрагидро-2Н-пиран-4-иламино)бензамида в форме желтого порошка.
ЖХМС (ЭР, m/z): (М+1) 596,13.
1H ЯМР: δН млн-1 (400 МГц, ДМСО): 12.96 (1Н, широкий плоский синглет), 12.02 (1Н, широкий плоский синглет), 10.64 (1Н, bs, NH), 8.46 (1Н, bs), 8.09 (1Н, bs), 7.72 (1Н, d, СНаром), 6.97-7.10 (1Н, m, СНаром), 6.60-6.74 (2Н, m, СНаром), 6.28 (1Н, dd, СНаром), 6.13 (1Н, d, СНаром), 3.80-3.90 (2Н, m, СНпиранон), 3.65-3.77 (1Н, m, СНпиранон), 3.50 (2Н, t, СНпиранон), 3.25-3.32 (4Н, m, 2*CH2), 2.37-2.45 (4Н, m, 2*CH2), 2.22 (3H, s, CH3), 1.91-2.00 (2H, m, СНпиранон), 1.28-1.43 (2Н, m, СНпиранон).
II. Биологические тесты соединении согласно изобретению
Тест на измерение ингибирования киназы TrkA, TrkB или TrkC
Эти киназы изготавливаются фирмой Millipore или Kinome Scan (DiscoverX), и подвергают скринингу согласно протоколу изготовителя. Результаты представлены в таблице ниже:

Анальгезирующая активность in vivo против NGF-индуцированной гипералгезии Соединения общей формулы (I) и, в частности, соединение 30, протестированы in vivo у мыши, получающей внутриподошвенную инъекцию NGF. Данная обработка вызывает термическую гипералгезию (Schuligoi et al., 1998, Neuroscience Letters 252: 147-149). Кратко, самцы мышей получали подкожную инъекцию 10 мкл NGFβ (1 мкг; SIGMA) в поверхность подошвы правой задней лапы. После инъекции мышей возвращали в свои клетки. Через четыре часа после инъекции NGF мышей помещали в центр анальгезиметра с горячей пластинкой (BIO-HC2.00, Bioseb, Франция), и регистрировали латентный период до первого резкого поднятия или облизывания задней лапы или подпрыгивания. Для предотвращения повреждения ткани использовали максимальный порог отсечения времени, равный 60 сек. Результаты представлены в виде среднего ± стандартная погрешность средней величины (s.e.m.) латентного периода. Тестируемые соединения вводили за 30 мин до инъекции NGF.
На фиг. 1 показано, что соединение 30 дозозависимо ингибирует NGF-индуцированную гипералгезию, как при введении интраперитонеальным путем (фиг. 1А), так и пероральным путем (фиг. 1B). Полное или почти полное обратное развитие NGF-индуцированной гипералгезии происходило при дозах 10 мг/кг и ниже (*P менее 0,05 по сравнению с одним NGF на основании критерия ANOVA).
Анальгезирующая активность in vivo в формалиновом тесте у крыс
Способ представлял собой адаптированный способ из статей Wheeler-Aceto et al. (Pain, 1990, 40: 229-38) и Bardin et al. (2003, Pharmacology 67: 182-194). Bo второй фазе этого теста Соединение 30 ингибировало облизывание лапы на 44% по сравнению с животными, обработанными носителем (P менее 0,05 на основании критерия ANOVA).
Анальгезирующая активность in vivo против острой и хронической воспалительной боли, индуцированной полным адъювантом Фрейнда (ПАФ)
Использовали самцов мышей линии Спраг-Доули [Crl:OFA(SD) питомник Charles River, Лион, Франция] массой 160-180 г по прибытии. Сначала крысы привыкали в течение суток к содержанию и к аппарату для тестирования, используемому для моделей фон Фрея и горячей пластинки, за неделю до индукции воспаления. В следующий понедельник их подвергали ноцицептивным тестам для определения базальных баллов. Затем крысам вводят подкожную инъекцию 100 мкл ПАФ (1 мг/1 мл; Sigma) в поверхность подошвы правой задней лапы (сутки 0:D0). Физиологический раствор инъецировали тем же путем в группе плацебо.
Затем каждую крысу помещали на приподнятую пластмассовую сетку в прозрачной пластмассовой клетке и регистрировали число спонтанных отдергиваний (измерение спонтанной боли) в течение 10 мин. Механическую аллодинию оценивали, используя тест нитей фон Фрея. Каждую крысу помещали на приподнятую пластмассовую сетку в прозрачной пластмассовой клетке и давали возможность адаптироваться к окружающей среде тестирования в течение по меньшей мере 5 мин. Нити фон Фрея (монофиламенты Семмес-Вайнштейна, Stoelting IL, США; 0,6, 1,4, 2, 4, 6, 8, 10 и 15 г) прикладывали к поверхности подошвы инъецированной лапы снизу пола сетки на 1 с в возрастающем порядке. Порог определяли как самую низкую силу, вызывающую ответ избегания. Тест повторяли три раза с интервалом 5 мин между каждым из тестов. Балл отдергивания лапы в ответ на тактильный стимул определяли как среднее 3 измерений. Животные получали интраперитонеальные или пероральные инъекции тестируемых соединений.
Соединения общей формулы (I) проявляли анальгезирующую активность в данном тесте при дозах от 0,5 до 20 мг/кг, как против острой воспалительной боли (отдергивания), так и против механической аллодинии. Например, Соединение 30 дозозависимо ингибировало отдергивания после интраперитонеальной инъекции (фиг. 2А) и механическую аллодинию после перорального введения (фиг. 2B). Соединение вызывало полное или почти полное развитие спонтанной боли с 0,16 мг/кг и механической аллодинии с 2,5 мг/кг.
Анальгезирующая активность in vivo против хронической боли в модели боли при раке кости
Рак кости вызывает одно из наиболее болезненных состояний, поражающих людей и животных. Он также представляет собой наиболее распространенную боль у пациентов-людей с раком поздних стадий, и он остается трудным для лечения и вносит значительный вклад в повышенную заболеваемость и сниженное качество жизни. Разработано несколько моделей боли, вызванной остеосаркомой, у мышей (Pacharinsak and Beitz, Animal models of cancer pain. Comp Med. 2008 Jun; 58: 220-33).
Авторы изобретения индуцировали боль при раке кости путем имплантации 10 мкл фосфатно-солевого буфера (ФСБ), содержащего 2×105 клеток остеолитической фибросаркомы NCTC 2472 в бедро мышей C3H/HeNCrI 5-6 недельного возраста под анестезией изофлураном. Присутствие этих клеток индуцировало остеолитические повреждения в кости как вследствие увеличения числа остеокластов, так и вследствие их активации, что приводит к деструкции костной ткани и инвазии примыкающей мягкой ткани и к поведению, обусловленному болью. Поведение, обусловленное болью, оценивали как до инъекции, так и через 3, 7, 10, 14 и 17 суток после инъекции опухолевых клеток. Оценивали следующие виды поведения:
- Использование конечности во время спонтанного вставания по шкале 0-4, где 0 = нормальное использование и 4 = отсутствие использования инъецированной задней лапы;
- Боль, спровоцированная легким касанием. Отдергивание, вызванное легким касанием, измеряли после 1-мин периода неболевой стимуляции лапы, в которую инъецировали опухоль, маленькой кисточкой. Число отдергиваний регистрировали в течение 5 мин.
Для лекарственной терапии животных обрабатывали хронически тестируемым соединением или физиологическим раствором, вводимым за 30 минут до тестирования интраперитонеальным (i.p.) путем, начиная на сутки 7 после инъекции опухолевых клеток, когда поведение, обусловленное болью, явно присутствовало. В качестве примера, Соединение 30 в дозе 2,5 мг/кг значимо снижало балл использования конечности, начиная с суток 10 (фиг. 3A), и отдергивание, спровоцированное легким касанием, начиная с суток 8 (фиг. 3B).
Сравнительные эксперименты:
Анализ киназы in vitro на основе набора Kinase-glo plus (Promega) был разработан в формате 96-луночного планшета для оценки ингибирующих свойств соединений в отношении тропомиозин-родственных рецепторных киназ (Trk), используя растворимый внутриклеточный сегмент этих рецепторов, содержащий киназную активность (TrkA (или NTRK1; 400 нг/лунка; Millipore) и TrkB (или NTRK2; 200 нг/лунка; Invitrogen)). Этот анализ проводили в конечном объеме 50 мкл, содержащем 8 мМ 3-N-морфолин-пропансульфоновую кислоту (MOPS; от англ. "(3-(N-morpholino)propanesulfonic acid") (pH 7,0), 0,2 мМ этилендиаминтетрауксусную кислоту (ЭДТА), 40 мМ ацетат Mg, 25 мМ 2-глицерофосфат, 1 мМ дитиотрейтол (ДТП, 2 мМ ванадат Na, 0,01% Тритон Х-100, либо 1 (TrkA), либо 2,5 (TrkB) мг/мл поли EY (поли Glu-Tyr 4:1; субстрат) и 30 (Trk А) или 40 (Trk В) мкМ АТФ. Тестируемые соединения растворяли (10 мМ) и разводили в 100% ДМСО посредством серийного разведения, где тестируемые полулогарифмические концентрации, как правило, находились в диапазоне конечной концентрации от 1 до 30000 нМ (конечная концентрация 5% ДМСО), но эти концентрации могут быть адаптированы к эффективности соединения. Реакцию запускали добавлением АТФ, и планшеты инкубировали в течение 2 часов при 30°C. Планшеты извлекали из термостата, и в каждую лунку добавляли 50 мкл Kinase-glo plus с последующей 10-минутной инкубацией при комнатной температуре. Индуцированную люциферазой люминесценцию, пропорциональную АТФ, остающемуся в лунке, измеряли на люминометре. Контроли на каждом планшете включали лунки с одной киназой (максимальное потребление АТФ или 0% ингибирование), лунки без киназы (максимальный сигнал АТФ или 100% ингибирование), и лунки с 20 мкМ стауроспорином (положительный контроль ингибирования). Полную кривую ингибирования концентрация-ответ с K252a (полулогарифмические концентрации от 1 до 10000 нМ) также включали на каждом планшете. Значения pIC50 выводили на основании соответствия кривой потребления АТФ, используя диапазон люминесценции 0-100%, определенный контролями, с помощью сигмоидального уравнения доза-ответ: % ингибирования = 100-(100/(1+10(X+pIC50)*nH)), где X представляет собой логарифм концентраций тестируемых соединений. В этих условиях K252a имела pIC50 7,46±0,07 на TrkA (среднее ± SD; N=42; IC50=30 нМ), и 7,16±0,18 на TrkB (N=36; IC50=74 нМ), эти значения согласуются с литературными данными in vitro (Tapley et al (1992) Oncogene 7: 371-381, Tan et al (2007) Mol Pharmacol 72: 1440-1446).
Полученные результаты представлены ниже:
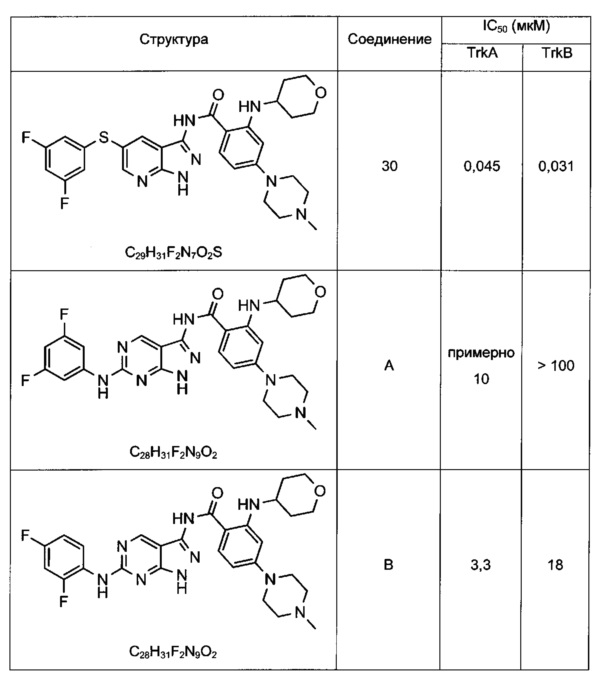

| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АЗАИНДАЗОЛА ИЛИ ДИАЗАИНДАЗОЛА В КАЧЕСТВЕ МЕДИКАМЕНТА | 2012 |
|
RU2600976C2 |
| СОДЕРЖАЩИЕ АЗОТ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ ИХ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2008 |
|
RU2465276C2 |
| Производные хромена в качестве ингибиторов фосфоинозитид-3-киназ | 2016 |
|
RU2722383C2 |
| НОВЫЕ ФОСФАТНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2617682C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2015 |
|
RU2733400C2 |
| НОВЫЕ ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2607788C2 |
| ПРОИЗВОДНЫЕ 2-(ИМИНОМЕТИЛ)АМИНОФЕНИЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ ВЕЩЕСТВА | 1998 |
|
RU2183211C2 |
| НОВЫЕ ИНДОЛИЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2693629C2 |
| ЗАМЕЩЕННЫЕ ФЕНИЛМОЧЕВИНЫ И ФЕНИЛАМИДЫ В КАЧЕСТВЕ ЛИГАНДОВ ВАНИЛЛОИДНЫХ РЕЦЕПТОРОВ | 2010 |
|
RU2553392C2 |
| НОВЫЕ МАКРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2018 |
|
RU2808689C2 |



Настоящее изобретение относится к применению соединения формулы (I) или к его фармацевтически приемлемых солей для лечения или предупреждения боли, ассоциированной по меньшей мере с одним белком Trk. Радикалы и символы в формуле (I) имеют определения, указанные в формуле изобретения. Изобретение также относится к фармацевтической композиции, содержащей в качестве активного вещества соединение формулы (I). 2 н. и 13 з.п. ф-лы, 6 ил., 1 табл., 35 пр.
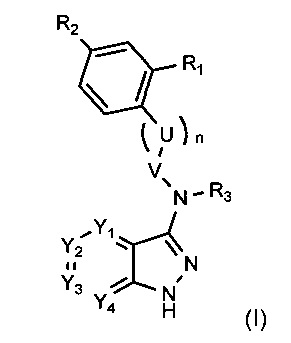
1. Применение соединения следующей общей формулы (I):

или его фармацевтически приемлемой соли,
где:
- Y1 и Y4 каждый представляет собой, независимо друг от друга, группу CH или атом азота при условии, что по меньшей мере один из Y1 и Y4 представляет собой атом азота,
- Y2 представляет собой группу C-X-Ar,
- Y3 представляет собой группу C-W,
- Ar представляет собой группу фенил или пиридинил, необязательно замещенную одной или более группами, выбранными из атома галогена, (C1-C6)алкила, (C1-C6)галогеналкила, CO2R15 и CONR16R17,
X представляет собой двухвалентную группу, выбранную из O, S, S(O), S(O)2, NR4, S(NR4), S(O)(NR4), S(O)2(NR4), NR4S, NR4S(O), NR4S(O)2, CH2, CH2S, CH2S(O), CH2S(O)2, SCH2, S(O)CH2, S(O)2CH2, CH2CH2, CH=CH, C≡C, CH2O, OCH2, NR4CH2 и CH2NR4,
- W представляет собой группу R5, SR5, OR5 или NR5R6,
- U представляет собой группу CH2 или NH,
- V представляет собой C(O), C(S) или CH2,
- n представляет собой 0 или 1,
- R1 представляет собой атом водорода, OR7 или NR7R8,
- R2 представляет собой 5- или 6-членный гетероцикл, замещенный (C1-C6)алкильной группой или NH2 и содержащий 1 или 2 гетероатома, представляющих собой N; NO2; OR9 или NR9R10,
- R3 и R4, каждый представляет собой, независимо друг от друга, атом водорода или (C1-C6)алкильную группу,
- R5 и R6 каждый представляет собой, независимо друг от друга, атом водорода или (C1-C6)алкил,
- R7 и R8 каждый представляет собой, независимо друг от друга, атом водорода или (C1-C6)алкильную или (С3-С12)циклоалкильную группу, замещенную атомами галогена, либо 6-членный гетероцикл, содержащий гетероатом, представляющий собой О,
- R9 и R10 каждый представляет собой, независимо друг от друга, атом водорода или (C1-C6)алкильную группу, необязательно замещенную группой NR13R14, и
- R13, R14, R16 и R17 каждый представляет собой, независимо друг от друга, атом водорода или (C1-C6)алкильную группу,
для лечения или предупреждения боли, ассоциированной по меньшей мере с одним белком Trk.
2. Применение по п. 1, отличающееся тем, что боль представляет собой ноцицептивную боль, воспалительную боль, невропатическую боль, идиопатическую боль или психогенную боль, предпочтительно воспалительную боль или невропатическую боль.
3. Применение по п. 1, где боль является следствием рака, повреждения нервов или ревматических заболеваний.
4. Применение по п. 1, отличающееся тем, что:
- Y1=СН или N, и
- Y4=N.
5. Применение по п. 1, отличающееся тем, что:
- Y1 представляет собой группу CH, и
- Y4 представляет собой атом азота.
6. Применение по п. 1, отличающееся тем, что X представляет собой двухвалентную группу, выбранную из S, S(O), S(O)2, NR4, СН2, CH2S, CH2S(O), CH2S(O)2, CH2O, CH2NR4, NHS(O)2, SCH2, S(O)CH2, S(O)2CH2, S(O)2NH, OCH2, NR4CH2, CH2CH2, CH=CH и C≡C; в частности, из S, S(O), S(O)2, NR4, CH2, SCH2, S(O)CH2, S(O)2CH2, S(O)2NH, CH2CH2, C≡C, OCH2 и NR4CH2; в частности, из S, S(O)2, CH2, SCH2, S(O)2CH2, S(O)2NH, CH2CH2 и C≡C, где первый атом этих групп связан с атомом С цепи C-X-Ar.
7. Применение по п. 1, отличающееся тем, что Ar представляет собой группу, выбранную из следующих групп:
 ,
,  ,
,  ,
,  ,
,  ,
,
 ,
, 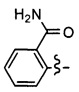 ,
, 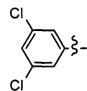 ,
, 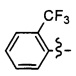 и
и 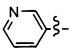 .
.
8. Применение по п. 1, отличающееся тем, что W представляет собой атом водорода.
9. Применение по п. 1, отличающееся тем, что:
- R3=H,
- U=CH2 или NH,
- V=C(O) или C(S), и в частности С(О), и
- n=0 или 1, и в частности 0.
10. Применение по п. 1, отличающееся тем, что R1 представляет собой атом водорода или группу NR7R8, где R7 представляет собой атом водорода и R8 представляет собой (С3-С12)циклоалкильную группу, замещенную атомами галогена, или 6-членный гетероцикл, содержащий гетероатом, представляющий собой O.
11. Применение по п. 10, отличающееся тем, что R1 представляет собой одну из следующих групп:
H,  и
и  .
.
12. Применение по п. 1, отличающееся тем, что R2 представляет собой NO2, NR9R10 или 5- или 6-гетероцикл, замещенный (C1-C6)алкилом или NH2 и содержащий 1 или 2 гетероатома, представляющих собой N.
13. Применение по п. 12, отличающееся тем, что R2 представляет собой одну из следующих групп:
NH2, NH(CH2)3NMe2, NMe(CH2)3NMe2, NO2,  ,
,  , и
, и 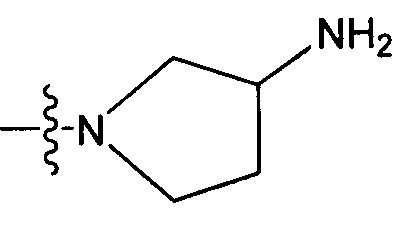 .
.
14. Применение по п. 1, отличающееся тем, что оно выбрано из следующих соединений:
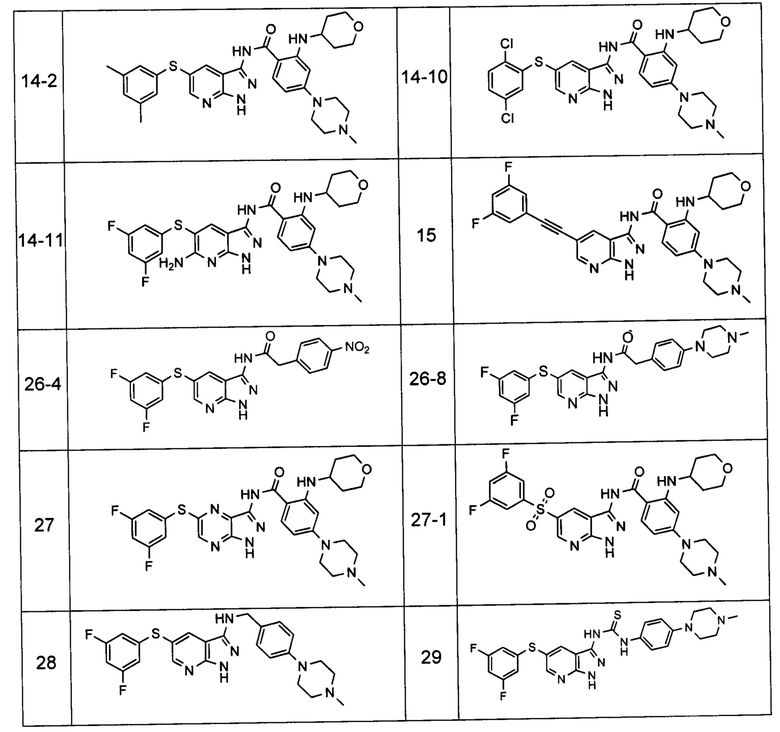

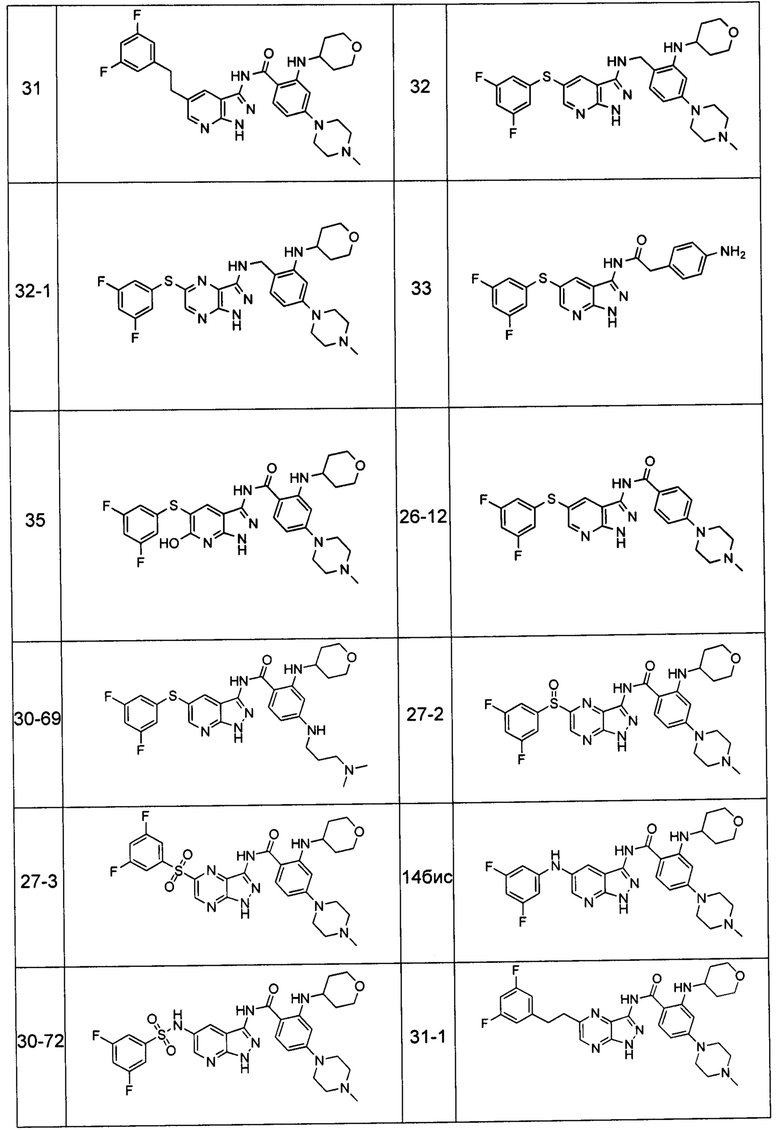


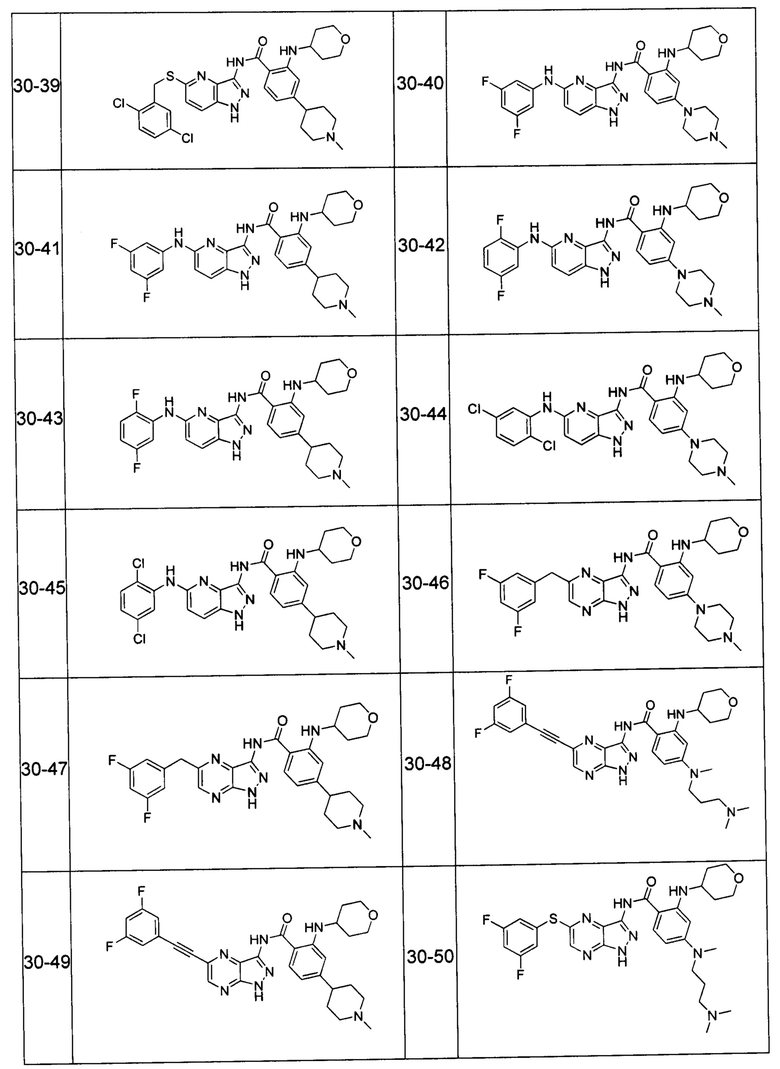
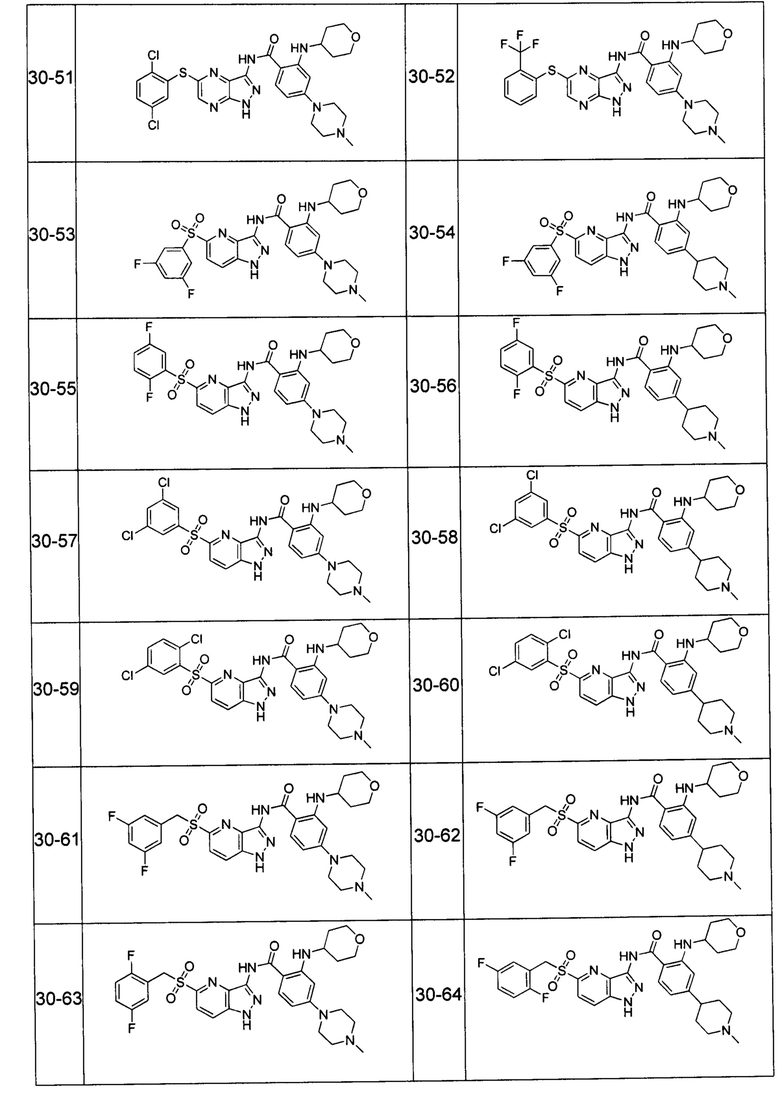

15. Фармацевтическая композиция, содержащая по меньшей мере один фармацевтически приемлемый эксципиент и одно соединение по любому из пп. 1-14 в качестве активного вещества, для применения при лечении или предупреждении боли, ассоциированной по меньшей мере с одним белком Trk.
| Крановое предохранительное приспособление для паровых котлов | 1925 |
|
SU2600A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| RU 2008110941 A, 27.09.2009 | |||
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| WANG T ET AL | |||
| Trk kinase inhibitors as new treatments for cancer and pain, EXPERT OPINION ON THERAPEUTIC PATENTS, 2009, vol | |||
| Способ изготовления электрических сопротивлений посредством осаждения слоя проводника на поверхности изолятора | 1921 |
|
SU19A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

