Настоящее изобретение относится к новым индолизиновым соединениям, к способу их получения и к фармацевтическим композициям, содержащим их.
Соединения настоящего изобретения являются новыми и обладают очень ценными фармакологическими характеристиками в области апоптоза и онкологии.
Апоптоз, или запрограммированная гибель клеток, является физиологическим процессом, который имеет решающее значение для эмбрионального развития и поддержания тканевого гомеостаза.
Гибель клеток по типу апоптоза вовлекает морфологические изменения, такие как конденсация ядра, фрагментация ДНК, а также биохимический феномен, такой как активация каспаз, что вызывает повреждение ключевых структурных компонентов клетки, таким образом вызывая ее разборку и смерть. Регуляция процесса апоптоза является комплексной и задействует активацию или репрессию нескольких внутриклеточных путей передачи сигналов (Cory S. и др., Nature Review Cancer, 2002, 2, 647-656).
Дерегулирование апоптоза вовлечено в определенные патологии. Повышенный апоптоз связан с нейродегенеративными заболеваниями, такими как болезнь Паркинсона, болезнь Альцгеймера и ишемия. Наоборот, недостаточности осуществления апоптоза играют важную роль в развитии злокачественных новообразований и их резистентности к химиотерапии, при аутоиммунных заболеваниях, воспалительных заболеваниях и вирусных инфекциях. Следовательно, отсутствие апоптоза является одним из характерных фенотипических признаков злокачественного новообразования (Hanahan D. и др., Cell 2000, 100, 57-70).
Антиапоптотические белки семейства Bcl-2 связаны с многочисленными патологиями. Задействование белков семейства Bcl-2 описано для многочисленных типов злокачественных новообразований, таких как колоректальный рак, рак молочной железы, мелкоклеточный рак легкого, немелкоклеточный рак легкого, рак мочевого пузыря, рак яичников, рак предстательной железы, хронический лимфоидный лейкоз, фолликулярная лимфома, миелома, рак предстательной железы и т.д. Сверхэкспрессия антиапоптотических белков семейства Bcl-2 связана с онкогенезом, с устойчивостью к химиотерапии и с клиническим прогнозом пациентов, страдающих злокачественным новообразованием. Таким образом, существует терапевтическая потребность в соединениях, которые ингибируют антиапоптотическую активность белков семейства Bcl-2.
Помимо того, что соединения настоящего изобретения являются новыми, они обладают проапоптотическими свойствами, что позволяет их применение при патологиях, в которые вовлечен дефект апоптоза, как, например, для лечения злокачественного новообразования, аутоиммунных заболеваний и заболеваний иммунной системы.
Более конкретно, настоящее изобретение относится к соединениям формулы (I):
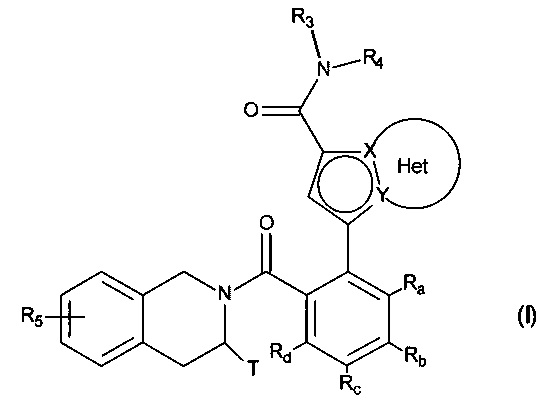
в которой:
♦ X и Y представляют собой атом углерода или атом азота, при этом предполагается, что они не могут одновременно представлять собой два атома углерода или два атома азота,
♦ Het фрагмент группы 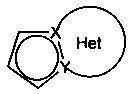 представляет собой необязательно замещенное, ароматическое или неароматическое кольцо, состоящее из 5, 6 или 7 кольцевых членов, которое может содержать, в дополнение к азоту, представленному посредством X или Y, от 1 до 3 гетероатомов, независимо выбранных из кислорода, серы и азота, при этом предполагается, что рассматриваемый азот может быть замещен группой, представляющей собой атом водорода, линейную или разветвленную (С1-С6)алкильную группу или группу -С(O)-O-Alk, где Alk означает линейную или разветвленную (C1-С6)алкильную группу,
представляет собой необязательно замещенное, ароматическое или неароматическое кольцо, состоящее из 5, 6 или 7 кольцевых членов, которое может содержать, в дополнение к азоту, представленному посредством X или Y, от 1 до 3 гетероатомов, независимо выбранных из кислорода, серы и азота, при этом предполагается, что рассматриваемый азот может быть замещен группой, представляющей собой атом водорода, линейную или разветвленную (С1-С6)алкильную группу или группу -С(O)-O-Alk, где Alk означает линейную или разветвленную (C1-С6)алкильную группу,
♦ Т представляет собой атом водорода, линейную или разветвленную (С1-С6)алкильную группу, необязательно замещенную 1-3 атомами галогена, группу (C2-C4)алкил-NR1R2, или группу (С1-С4)алкил-OR6,
♦ R1 и R2 независимо друг от друга представляют собой атом водорода или линейную или разветвленную (С1-С6)алкильную группу,
или R1 и R2 вместе с атомом азота, несущим их, образуют гетероциклоалкил,
♦ R3 представляет собой линейную или разветвленную (C1-С6)алкильную группу, линейную или разветвленную (С2-С6)алкенильную группу, линейную или разветвленную (С2-С6)алкинильную группу, циклоалкильную группу, (С3-С10)циклоалкил-(С1-С6)алкильную группу, в которой алкильный фрагмент является линейным или разветвленным, гетероциклоалкильную группу, арильную группу или гетероарильную группу, при этом предполагается, что один или несколько атомов углерода предшествующих групп или их возможных заместителей могут быть дейтерированы,
♦ R4 представляет собой арильную группу, гетероарильную группу, циклоалкильную группу или линейную или разветвленную (С1-С6)алкильную группу, при этом предполагается, что один или несколько атомов углерода предшествующих групп или их возможных заместителей могут быть дейтерированы,
♦ R5 представляет собой атом водорода или галогена, линейную или разветвленную (С1-С6)алкильную группу, или линейную или разветвленную (С1-С6)алкокси группу,
♦ R6 представляет собой атом водорода или линейную или разветвленную (С1-С6)алкильную группу,
♦ Ra, Rb, Rc и Rd, каждый независимо друг от друга, представляет собой R7, атом галогена, линейную или разветвленную (С1-С6)алкокси группу, гидрокси группу, линейную или разветвленную (С1-С6)полигалогеналкильную группу, трифторметокси группу, -NR7R7', нитро, R7-CO-(C0-C6)алкил-, R7-CO-NH-(С0-С6)алкил-, NR7R7'-CO-(C0-C6)алкил-, NR7R7'-CO-(C0-C6)алкил-O-, R7-SO2-NH-(С0-С6)алкил-, R7-NH-CO-NH-(C0-C6)алкил-, R7-O-CO-NH-(C0-C6)алкил-, гетероциклоалкильную группу, или заместители одной из пар (Ra,Rb), (Rb,Rc) или (Rc,Rd) вместе с атомами углерода, несущими их, образуют кольцо, состоящее из 5-7 кольцевых членов, которое может содержать от одного до двух гетероатомов, выбранных из кислорода и серы, при этом также предполагается, что один или несколько атомов углерода кольца, определенного выше, могут быть дейтерированы или замещены одной-тремя группами, выбранными из галогена и линейного или разветвленного (С1-С6)алкила,
♦ R7 и R7', каждый независимо друг от друга, представляет собой водород, линейную или разветвленную (С1-С6)алкильную, линейную или разветвленную (С2-С6)алкенильную, линейную или разветвленную (С2-С6)алкинильную, арильную или гетероарильную группу, или R7 и R7', вместе с атомом азота, несущим их, образуют гетероцикл, состоящий из 5-7 кольцевых членов,
при этом предполагается, что:
- "арил" означает фенильную, нафтильную, бифенильную или инденильную группу,
- "гетероарил" означает любую моно- или бициклическую группу, состоящую из 5-10 кольцевых членов, содержащую по меньшей мере один ароматический фрагмент и содержащую от 1 до 4 гетероатомов, выбранных из кислорода, серы и азота (включая четвертичный азот),
- "циклоалкил" означает любую моно- или бициклическую, неароматическую, карбоциклическую группу, содержащую от 3 до 10 кольцевых членов,
- "гетероциклоалкил" означает моно- или бициклическую, неароматическую, конденсированную или спиро-группу, содержащую от 3 до 10 кольцевых членов и содержащую от 1 до 3 гетероатомов, выбранных из кислорода, серы, SO, SO2 и азота,
причем арильные, гетероарильные, циклоалкильные и гетероциклоалкильные группы, определенные таким образом, и алкильные, алкенильные, алкинильные и алкокси группы могут быть замещены 1-3 группами, выбранными из необязательно замещенного, линейного или разветвленного (С1-С6)алкила, (С3-С6)спиро, необязательно замещенного, линейного или разветвленного (С1-С6)алкокси, (С1-С6)алкил-S-, гидрокси, оксо (или N-оксида, в соответствующих случаях), нитро, циано, -COOR', -OCOR', NR'R'', линейного или разветвленного (С1-С6)полигалогеналкила, трифторметокси, (С1-С6)алкилсульфонила, галогена, необязательно замещенного арила, гетероарила, арилокси, арилтио, циклоалкила, гетероциклоалкила, необязательно замещенного одним или несколькими атомами галогена или алкильными группами, при этом предполагается, что R' и R'', каждый независимо друг от друга, представляет собой атом водорода или необязательно замещенную, линейную или разветвленную (С1-С6)алкильную группу,
причем Het фрагмент группы 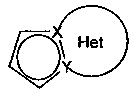 , определенный для формулы (I), может быть замещен 1-3 группами, выбранными из линейного или разветвленного (С1-С6)алкила, гидрокси, линейного или разветвленного (С1-С6)алкокси, NR1'R1'' и галогена, при этом предполагается, что R1' и R1'' являются такими, как определено для групп R' и R'', упомянутых выше,
, определенный для формулы (I), может быть замещен 1-3 группами, выбранными из линейного или разветвленного (С1-С6)алкила, гидрокси, линейного или разветвленного (С1-С6)алкокси, NR1'R1'' и галогена, при этом предполагается, что R1' и R1'' являются такими, как определено для групп R' и R'', упомянутых выше,
к их энантиомерам и диастереоизомерам, и к их солям присоединения с фармацевтически приемлемой кислотой или основанием.
Из числа фармацевтически приемлемых кислот могут быть упомянуты, без какого-либо ограничения, хлористоводородная кислота, бромистоводородная кислота, серная кислота, фосфоновая кислота, уксусная кислота, трифторуксусная кислота, молочная кислота, пировиноградная кислота, малоновая кислота, янтарная кислота, глутаровая кислота, фумаровая кислота, винная кислота, малеиновая кислота, лимонная кислота, аскорбиновая кислота, щавелевая кислота, метансульфоновая кислота, камфорная кислота и т.д.
Из числа фармацевтически приемлемых оснований могут быть упомянуты, без какого-либо ограничения, гидроксид натрия, гидроксид калия, триэтиламин, трет-бутиламин и т.д.
Предпочтительно, группа 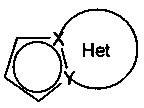 представляет собой одну из следующих групп: 5,6,7,8-тетрагидроиндолизин, необязательно замещенный аминогруппой; индолизин; 1,2,3,4-тетрагидропирроло[1,2-а]пиразин, необязательно замещенный метилом; пирроло[1,2-а]пиримидин. Группы 5,6,7,8-тетрагидроиндолизин и индолизин являются особенно предпочтительными.
представляет собой одну из следующих групп: 5,6,7,8-тетрагидроиндолизин, необязательно замещенный аминогруппой; индолизин; 1,2,3,4-тетрагидропирроло[1,2-а]пиразин, необязательно замещенный метилом; пирроло[1,2-а]пиримидин. Группы 5,6,7,8-тетрагидроиндолизин и индолизин являются особенно предпочтительными.
В предпочтительных соединениях изобретения Т представляет собой атом водорода, метильную группу (и более конкретно (R)-метил), группу 2-(морфолин-4-ил)этил, 3-(морфолин-4-ил)пропил, -СН2-ОН, 2-аминоэтил, 2-(3,3-дифторпиперидин-1-ил)этил, 2-[(2,2-дифторэтил)амино]этил или 2-(3-метоксиазетидин-1-ил)этил.
Предпочтительно Ra и Rd каждый представляет собой атом водорода и (Rb,Rc), вместе с атомами углерода, несущими их, образуют 1,3-диоксолановую группу или 1,4-диоксановую группу; или Ra, Rc и Rd каждый представляет собой атом водорода и Rb представляет собой водород, галоген, метил или метокси; или Ra, Rb и Rd каждый представляет собой атом водорода и Rc представляет собой гидрокси или метокси группу. Еще более предпочтительно Ra и Rd каждый представляет собой атом водорода и (Rb,Rc), вместе с атомами углерода, несущими их, образуют 1,3-диоксолановую группу; или Ra, Rc и Rd каждый представляет собой атом водорода и Rb представляет собой галоген.
Предпочтение отдают группе R4, представляющей собой 4-гидроксифенил.
В предпочтительных соединениях изобретения R3 представляет собой линейную (С1-С6)алкильную, арильную или гетероарильную группу, причем последние две группы могут быть замещены 1-3 группами, выбранными из галогена, линейного или разветвленного (С1-С6)алкила, линейного или разветвленного (С1-С6)алкокси, циано и гетероциклоалкил-(С1-С6)алкила, в котором алкильный фрагмент является линейным или разветвленным. Еще более предпочтительно R3 представляет собой гетероарильную группу, выбранную из следующей группы: 1H-индол, 2,3-дигидро-1H-индол, 1H-индазол, пиридин, 1Н-пирроло[2,3-b]пиридин, 1H-пиразол, имидазо[1,2-a]пиридин, пиразоло[1,5-а]пиримидин, [1,2,4]триазоло[1,5-а]пиримидин и 1H-пиразоло[3,4-b]пиридин, все из которых могут быть замещены линейной или разветвленной (C1-С6)алкильной группой.
Предпочтительные соединения согласно изобретению, включены в следующую группу:
- N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-{1-[2-(морфолин-4-ил)этил]-1Н-индол-5-ил}-5,6,7,8-тетрагидроиндолизин-1-карбоксамид,
- N-(4-гидроксифенил)-3-(6-{[(3S)-3-[2-(морфолин-4-ил)этил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамид,
- N-{3-фтор-4-[2-(морфолин-4-ил)этокси]фенил}-N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-карбоксамид,
- N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(пиридин-4-ил)индолизин-1-карбоксамид,
- N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(2-метилпиридин-4-ил)индолизин-1-карбоксамид,
- N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(1-метил-1H-пирроло[2,3-b]пиридин-5-ил)индолизин-1-карбоксамид,
- N-(4-гидроксифенил)-3-(6-{[(3R)-3-[3-(морфолин-4-ил)пропил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамид,
- N-(2,6-диметилпиридин-4-ил)-N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-карбоксамид,
- N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(пиридин-4-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид,
- 3-(5-хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-гидроксифенил)-N-(1-метил-1H-пирроло[2,3-b]пиридин-5-ил)индолизин-1-карбоксамид,
- N-(4-гидроксифенил)-N-(2-метоксипиридин-4-ил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-карбоксамид,
их энантиомеры и диастереоизомеры, и их соли присоединения с фармацевтически приемлемой кислотой или основанием.
Изобретение также относится к способу получения соединений формулы (I), который отличается тем, что в качестве исходного вещества применяют соединение формулы (II):
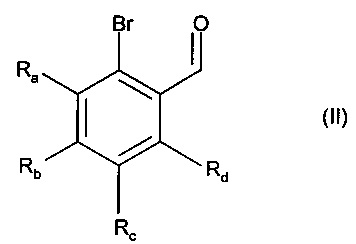
в которой Ra, Rb, Rc и Rd являются такими, как определено для формулы (I),
причем соединение формулы (II) подвергают реакции Хека, в водной или органической среде, в присутствии палладиевого катализатора, основания, фосфина и соединения формулы (III):
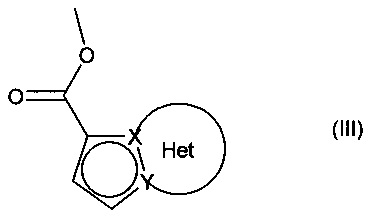
в которой группы X, Y и Het являются такими, как определено для формулы (I),
с получением соединения формулы (IV):
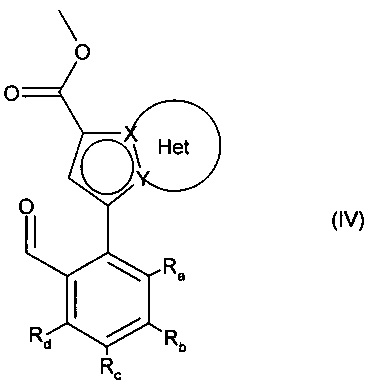
в которой Ra, Rb, Rc, Rd, X, Y и Het являются такими, как определено для формулы (I),
альдегидную функцию соединения формулы (IV) окисляют до карбоновой кислоты с образованием соединения формулы (V):
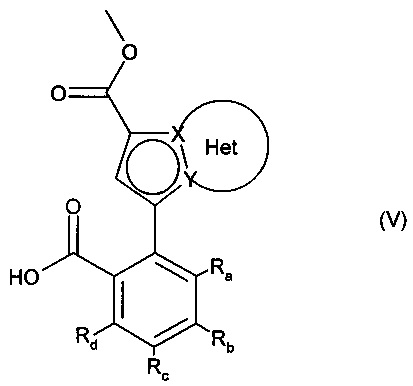
в которой Ra, Rb, Rc, Rd, X, Y и Het являются такими, как определено для формулы (I),
соединение формулы (V) затем подвергают пептидному сочетанию с соединением формулы (VI):

в которой Т и R5 являются такими, как определено для формулы (I),
с получением соединения формулы (VII):
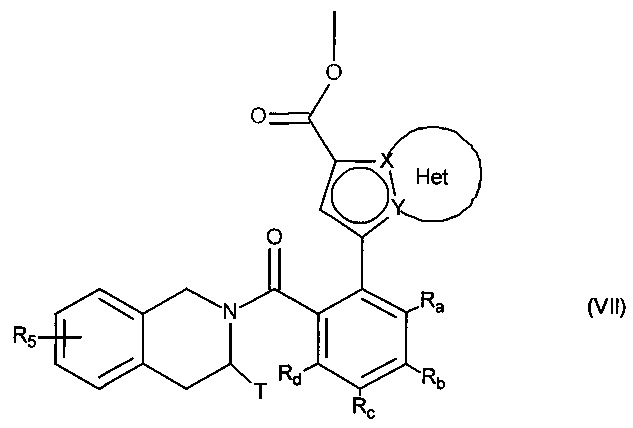
в которой Ra, Rb, Rc, Rd, T, R5, X, Y и Het являются такими, как определено для формулы (I),
сложноэфирную функцию соединения формулы (VII) гидролизуют с получением соответствующей карбоновой кислоты или карбоксилата, которую(-ый) можно превратить в производное кислоты, такое как соответствующий ацилхлорид или ангидрид, и затем осуществляют сочетание с амином NHR3R4, где R3 и R4 имеют те же значения, что и в случае формулы (I), с получением соединения формулы (I),
которое можно очистить в соответствии с обычными методиками разделения, которое превращают, при необходимости, в его соли присоединения с фармацевтически приемлемой кислотой или основанием и которое необязательно разделяют на его изомеры в соответствии с обычными методиками разделения,
при этом предполагается, что в любое время, признанное подходящим в ходе описанного выше способа, определенные группы (гидрокси, амино…) реагентов или промежуточных соединений синтеза можно защитить и затем снять с них защиту в соответствии с требованиями синтеза.
В частности, когда одна из групп R3 или R4 амина NHR3R4 замещена гидрокси функцией, последнюю можно подвергнуть реакции защиты заблаговременно перед каким-либо сочетанием с карбоновой кислотой, образованной из соединения формулы (VII), или с соответствующим производным таковой кислоты, где полученное в результате защищенное соединение формулы (I) затем подвергают реакции снятия защиты и затем необязательно превращают в одну из его солей присоединения с фармацевтически приемлемой кислотой или основанием.
Соединения формул (II), (III), (VI) и амин NHR3R4 либо доступны для приобретения, либо могут быть получены специалистом в данной области техники с использованием обычных химических реакций, описанных в литературе.
Фармакологическое исследование соединений изобретения показало, что они обладают проапоптотическими свойствами. Способность реактивировать апоптотический процесс в раковых клетках представляет большой терапевтический интерес для лечения злокачественных новообразований, аутоиммунных заболеваний и заболеваний иммунной системы.
Более конкретно, соединения в соответствии с изобретением будут полезны для лечения хемо- или радиорезистентных злокачественных новообразований, и в случае злокачественных заболеваний крови, и в случае мелкоклеточного рака легкого.
Из числа намеченных противоопухолевых терапий могут быть упомянуты, без какого-либо ограничения, терапия рака мочевого пузыря, головного мозга, молочной железы и матки, хронических лимфоидных лейкозов, колоректального рака, рака пищевода и печени, лимфобластных лейкозов, неходжкинских лимфом, меланом, злокачественных заболеваний крови, миелом, рака яичников, немелкоклеточного рака легкого, рака предстательной железы и мелкоклеточного рака легкого. Из числа неходжкинских лимфом, могут быть упомянуты более предпочтительно фолликулярные лимфомы, лимфомы из клеток мантийной зоны, диффузные В-крупноклеточные лимфомы, мелкоклеточные лимфоцитарные лимфомы и В-клеточные лимфомы из клеток маргинальной зоны.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим по меньшей мере одно соединение формулы (I) в комбинации с одним или несколькими фармацевтически приемлемыми наполнителями.
Из числа фармацевтических композиций в соответствии с изобретением могут быть упомянуты более конкретно те, которые подходят для перорального, парентерального, назального, чрес- или транскожного, ректального, перлингвального, офтальмологического или респираторного введения, в особенности таблетки или драже, сублингвальные таблетки, саше, пакеты, капсулы, глоссеты, пастилки, суппозитории, кремы, мази, гели для кожи и питьевые или инъекционные ампулы.
Дозировка варьируется в зависимости от пола, возраста и веса пациента, пути введения, природы терапевтического показания, или каких-либо сопутствующих лечений, и находится в диапазоне от 0.01 мг до 1 г в 24 часа за одно или несколько введений.
Кроме того, настоящее изобретение также относится к ассоциации соединения формулы (I) с противоопухолевым средством, выбранным из генотоксичных средств, митотических ядов, антиметаболитов, ингибиторов протеасом, ингибиторов киназ и антител, и также к фармацевтическим композициям, содержащим такой тип ассоциации, и их применению для изготовления лекарственного средства для применения для лечения злокачественного новообразования.
Соединения изобретения также можно применять в ассоциации с радиотерапией для лечения злокачественного новообразования.
В конечном счете, соединения изобретения могут быть связаны с моноклональными антителами или их фрагментами или связаны с каркасными белками, которые могут относиться или не относиться к моноклональным антителам.
Фрагменты антител следует понимать как фрагменты Fv, scFv, Fab, F(ab')2, F(ab'), scFv-Fc типа или диатела, которые обычно имеют такую же специфичность связывания, что и антитело, из которого они происходят. В соответствии с настоящим изобретением, фрагменты антител изобретения могут быть получены исходя из антител с помощью методов, таких как переваривание ферментами, такими как пепсин или папаин, и/или посредством расщепления дисульфидных мостиков с помощью химического восстановления. Другим путем, фрагменты антител, включенных в настоящее изобретение, могут быть получены с использованием методик генетической рекомбинации, также хорошо известных специалисту в данной области техники или даже посредством пептидного синтеза с помощью, например, автоматических пептидных синтезаторов, таких как те, которые поставляются компанией Applied Biosystems, и т.д.
Под каркасными белками, которые могут относиться или не относиться к моноклональным антителам, понимают белок, который содержит или не содержит укладку цепи иммуноглобулинов и который обеспечивает способность к связыванию, такую же, как и у моноклонального антитела. Специалисту в данной области техники известно, каким образом выбрать каркас белка. В частности, известно, что должен быть выбран такой каркас, который будет демонстрировать несколько следующих отличительных признаков (Skerra A., J. Mol. Recogn., 13, 2000, 167-187): хорошая филогенетическая консервативность, прочная архитектура с хорошо известной трехмерной молекулярной организацией (как, например, на основании кристаллографии или ЯМР), небольшой размер, отсутствие или лишь низкая степень посттрансляционных модификаций, простота получения, экспрессии и очистки. Таким каркасным белком может быть, без ограничения перечисленным, структура, выбранная из группы, состоящей из фибронектина и предпочтительно 10-го домена фибронектина типа III (FNfn10), липокалина, антикалина (Skerra A., J. Biotechnol., 2001, 74(4):257-75), белка Z, производного из домена В стафилококкового белка А, тиоредоксина А или любого белка с повторяющимся доменом, таким как "анкириновый повтор" (Kohl и др., PNAS, 2003, т. 100, №4, 1700-1705), "армадилло повтор", "богатый лейцином повтор" или "тетратрикопептидный повтор". Также можно упомянуть каркас, производный из токсинов (как, например, токсины скорпионов, насекомых, растений или моллюсков) или белковых ингибиторов нейрональной синтазы оксида азота (PIN).
Следующие Синтезы и Примеры иллюстрируют изобретение, но не ограничивают его каким-либо образом
Общие методики
Все реагенты и безводные растворители получали из коммерческих источников и использовали без дополнительной очистки или сушки. Флэш-хроматографию выполняли на приборе ISCO CombiFlash Rf 200i с предварительно набитыми силикагелевыми картриджами (SiliaSep™ F60 (40-63 мкм, 60 ). Тонкослойную хроматографию проводили на 5×10 см пластинах, покрытых силикагелем Merck Туре 60 F254. Нагрев микроволновым излучением выполняли в приборе СЕМ Discover® SP.
). Тонкослойную хроматографию проводили на 5×10 см пластинах, покрытых силикагелем Merck Туре 60 F254. Нагрев микроволновым излучением выполняли в приборе СЕМ Discover® SP.
Аналитическая ЖХ-МС
Соединения изобретения характеризовали с помощью жидкостной хроматографии высокого разрешения в сочетании с масс-спектроскопией (ЖХВР-МС), либо на приборе Agilent HP 1200 быстрого разрешения, соединенном с Mass detector 6140 с многомодовым источником (m/z диапазон 150-1000 атомных единиц массы или а.е.м.), либо на приборе Agilent HP1100, соединенном с Mass detector 1946D с электрораспылительным ионизационным источником (m/z диапазон 150-1000 а.е.м.). Условия и методы, перечисленные ниже, идентичны для обеих приборов.
Детектирование: УФ детектирование на длине волны 230, 254 и 270 нм.
Объем введенной пробы: 2 мкл
Подвижные фазы: А - Вода + 10 мМ / формиат аммония + 0.08% (об./об.) муравьиной кислоты при рН прибл. 3.5.
В - 95% Ацетонитрил + 5% А + 0.08% (об./об.) муравьиной кислоты
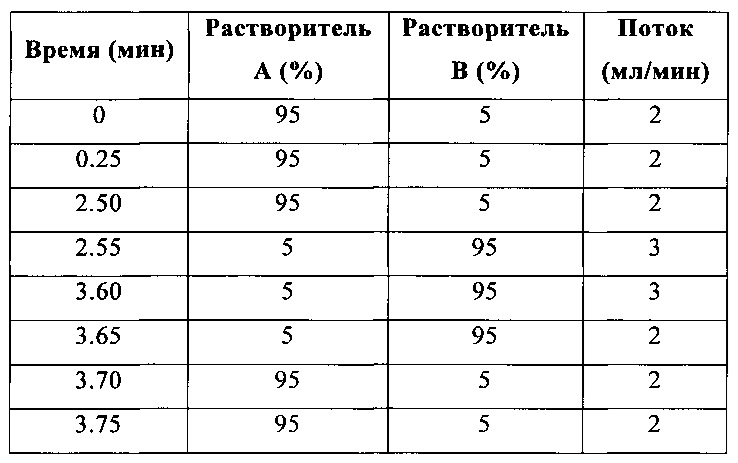
Метод А (3.75 мин; ионизация либо в режиме положительных ионов (положит.), либо в режиме положительных и отрицательных ионов (положит./отриц.))
Колонка: Gemini 5 мкм, С18, 30 мм × 4.6 мм (Phenomenex).
Температура: 35°С.
Градиент:
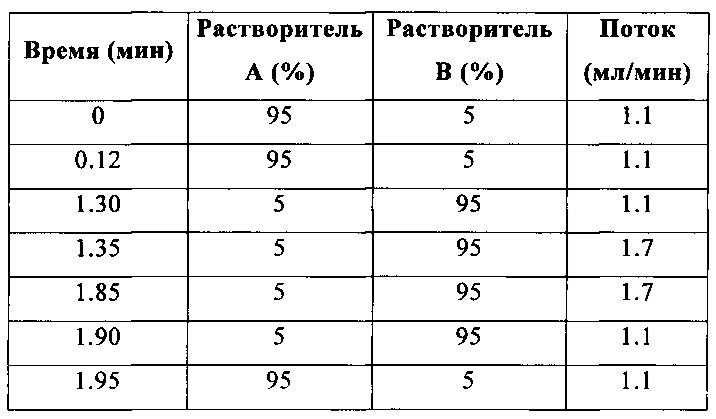
Метод В (1.9 мин; ионизация либо в режиме положительных ионов (положит.), либо в режиме положительных и отрицательных ионов (положит./отриц.))
Колонка: Gemini 5 мкм, С18, 30 мм × 4.6 мм (Phenomenex).
Температура: 35°С.
Градиент:
Синтез 1: 6-[1-(Метоксикарбонил)-5,6,7,8-тетрагидро-3-индолизинил]-1,3-бензодиоксол-5-карбоновая кислота
Стадия А: 1-Формил-2-пиперидинкарбоновая кислота
К раствору 40 г рацемической смеси 2-пиперидинкарбоновой кислоты (0.310 ммоль) в 300 мл муравьиной кислоты, охлажденному до 0°С, по каплям добавляют 200 мл (2.15 ммоль) ангидрида уксусной кислоты. Загрузку затем перемешивают при температуре окружающей среды в течение ночи. Затем реакционную смесь охлаждают до 0°С, гидролизуют путем добавления 250 мл воды, и перемешивают в течение получаса при 0°С и затем концентрируют досуха. Полученное таким образом масло вносят в 200 мл метанола и затем концентрируют досуха. Указанный в заголовке продукт получают в виде масла с выходом 98%. Его используют непосредственно, без очистки другим способом, на следующей стадии.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 13.0 (m, 1H ОН); 8.0-8.05 (2s, 1H альдегид); 4.9-4.5 (2d, 1Н α к N и СООН); 4.1-2.6 (m, 2Н α к N); 2.2-1.2 (m, 6Н пиперидин)
ИК: ν: -ОН: 2000-3000 см-1 кислота; ν: >С=O 1703 см-1 широкая полоса
Стадия В: Метил 5,6,7,8-тетрагидро-1-индолизинкарбоксилат
К раствору 10 г карбоновой кислоты, полученной на Стадии А, (63.6 ммоль) в 65 мл дихлорметана последовательно добавляют 13.4 г тозилхлорида (70.4 ммоль), 11.5 мл метил 2-хлоракрилата (113.5 ммоль) и затем, по каплям, 17.8 мл N,N,N-триэтиламина (127.2 ммоль). Реакционную смесь затем нагревают с обратным холодильником в течение 1 часа 30 минут. Смеси затем дают охладиться до температуры окружающей среды, и затем добавляют 5 мл метил 2-хлоракрилата (48.9 ммоль) и, по каплям, 9 мл N,N,N-триэтиламина (64 ммоль). Загрузку нагревают с обратным холодильником в течение ночи.
Затем реакционную смесь разбавляют метиленхлоридом, последовательно промывают 1 н. раствором HCl, насыщенным раствором NaHCO3 и затем насыщенным раствором NaCl до достижения нейтрального рН. Органическую фазу затем сушат над MgSO4, фильтруют, концентрируют досуха и очищают с помощью хроматографии на силикагеле (градиент гептан/AcOEt). Указанный в заголовке продукт получают в виде масла.
1Н ЯМР: δ (400 МГц; CDCl3; 300K): 6.55-6.40 (d, 2Н, тетрагидроиндолизин); 3.91 (t, 3Н сложный метиловый эфир); 3.78 (s, 3Н тетрагидроиндолизин); 3.08 (t, 2Н, тетрагидроиндолизин); 1.95-1.85 (m, 4Н, тетрагидроиндолизин)
ИК: ν: >C=O 1692 см-1 сложный эфир
Стадия С: Метил 3-(6-формил-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидро-1-индолизинкарбоксилат
К раствору 6.4 г сложного эфира, полученного на Стадии В, (35.7 ммоль) в 12 мл N,N-диметилацетамида последовательно добавляют 12.3 г 6-бром-1,3-бензодиоксол-5-карбальдегида (53.6 ммоль) и 7 г ацетата калия (71.4 ммоль), и затем загрузку перемешивают в атмосфере аргона в течение 20 минут. Затем добавляют 1.3 г палладиевого катализатора PdCl2(PPh3)2 (1.8 ммоль). Затем реакционную смесь нагревают при 130°С в течение одного часа и после этого добавляют 139 мкл Н2О. Нагревание при такой же температуре продолжают в течение ночи. Температуре смеси дают вернуться к температуре окружающей среды и затем смесь разбавляют AcOEt. Добавляют животный уголь (2 г на г продукта) и загрузку перемешивают при температуре окружающей среды в течение 1 часа и затем фильтруют. Органическую фазу затем промывают водой, сушат над сульфатом магния и концентрируют досуха. Полученный таким образом сырой продукт очищают на силикагеле (градиент гептан/ACOEt). Указанный в заголовке продукт получают в виде масла.
1Н ЯМР: δ: (400 МГц; ДМСО-d6; 353°K): 9.65 (s, 1Н, Н альдегид); 7.3-7.15 (2s, 2Н, ароматические Н); 6.45 (s, 1Н тетрагидроиндолизин); 6.20 (s, 2Н метилендиокси); 3.70 (s, 3Н сложный метиловый эфир); 3.5-4.0 (m, 2Н тетрагидроиндолизин); 3.05 (m, 2Н тетрагидроиндолизин); 1.85 (m, 4Н тетрагидроиндолизин)
ИК: ν: >С=O 1695 см-1 сложный эфир; ν: >С=O 1674 см-1
Стадия D: 6-[1-(Метоксикарбонил)-5,6,7,8-тетрагидро-3-индолизинил]-1,3-бензодиоксол-5-карбоновая кислота
Приготавливают раствор, содержащий 3.37 г соединения, полученного на Стадии С, (10.3 ммоль) в 9.3 мл ацетона и 8.8 мл (80.24 ммоль) 2-метил-2-бутена и охлаждают до 0°С. По каплям добавляют 9.3 мл водного раствора, содержащего смесь 3.3 г NaClO2 (36.05 ммоль) и 3.6 г Na2PO4 (25.75 ммоль). Загрузку затем перемешивают при температуре окружающей среды в течение 7 часов. Затем реакционную смесь концентрируют для удаления ацетона. Затем полученное твердое вещество отфильтровывают, промывают водой и затем сушат при 40°С в вакууме в течение ночи. Указанный в заголовке продукт получают в виде твердого вещества, которое затем используют без очистки каким-либо иным способом.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 12.10 (m, 1Н, Н карбоновая кислота); 7.40-6.88 (2s, 2Н, ароматические Н); 6.20 (s, 1Н, Н тетрагидроиндолизин); 6.18 (s, 2Н, Н метилендиокси); 3.70 (s, 3Н, сложный метиловый эфир); 3.55 (t, 2Н тетрагидроиндолизин); 3.00 (t, 2Н тетрагидроиндолизин); 1.80 (m, 4Н, Н тетрагидроиндолизин)
ИК: ν: -ОН: 3000-2000 см-1 кислота; ν: >С=O 1686-1676 см-1 сложный эфир + кислота; ν: >С=С< 1608 см-1
Синтез 2: 2-[2-(трет-Бутоксикарбонил)-8-(метоксикарбонил)-1,2,3,4-тетрагидропирроло[1,2-a]пиразин-6-ил]-4-хлорбензойная кислота
Стадия А: 1-трет-Бутил 3-метил 4-формил-1,3-пиперазиндикарбоксилат
К раствору пентафторфенола в 520 мл безводного эфира, охлажденного до 0°С, последовательно порциями добавляют 49 г 1-этил-3-(3'-диметиламинопропил)-карбодиимида (286 ммоль) и затем 12 мл муравьиной кислоты (312 ммоль). Загрузку перемешивают при температуре окружающей среды в течение 2 часов. Затем добавляют смесь 32 г 1-трет-бутил 3-метил 1,3-пиперазиндикарбоксилата (130 ммоль) и 18 мл триэтиламина (130 ммоль), растворенных в 520 мл CH2Cl2. Загрузку перемешивают в течение ночи при температуре окружающей среды. Реакционную смесь гидролизуют 1 н. водным раствором HCl и экстрагируют CH2Cl2. Органические фазы затем объединяют и промывают насыщенным водным раствором NaHCO3 и затем насыщенным водным раствором NaCl до нейтральной реакции среды. После сушки над MgSO4, фильтрования и концентрирования досуха, продукт выделяют с помощью хроматографии на силикагеле (градиент петролейный эфир/AcOEt: 0-30%). Указанный в заголовке продукт получают в виде масла.
ИК: ν: С=O: 1674-1745 см-1
m/z (C12H20N2O5): 272.1 (М+); 295.121 (M+Na)+; 567.253 (2M+Na)+
Стадия В: 4-(трет-Бутокеикарбонил)-1-формил-2-пиперазинкарбоксилат лития
К раствору 28 г соединения, полученного на Стадии А, (103 ммоль) в 515 мл диоксана добавляют 4.8 г LiOH (113 ммоль), растворенного в 100 мл Н2О. Загрузку перемешивают при температуре окружающей среды в течение 4 часов. Затем реакционную смесь концентрируют досуха и затем несколько раз упаривают совместно с этилацетатом. Указанный в заголовке продукт получают в виде твердого вещества и непосредственно используют на следующей стадии циклизации.
13С ЯМР: δ (500 МГц; ДМСО-d6; 300K): 46 (s, С пиперазин); 42-38 (m, С пиперазин); 58-53 (s, С пиперазин); 28.5 (s, С tBu)
ИК: ν: С=O: 1650 см-1; 2800 см-1
Стадия С: 2-трет-Бутил 8-метил 3,4-дигидропирроло[1,2-а]пиразин-2,8(1Н)-дикарбоксилат
К суспензии 29 г соединения, полученного на Стадии В, (103 ммоль) в 800 мл дихлорметана последовательно добавляют 24 г тозилхлорида (124 ммоль), 12.6 мл метил 2-хлоракрилата (124 ммоль) и затем 35 мл триэтиламина (247 ммоль). Загрузку перемешивают при нагревании с обратным холодильником в течение 2 часов. После охлаждения реакционную смесь разбавляют этилацетатом и органическую фазу промывают насыщенным раствором NaCl до нейтральной реакции среды. После сушки над MgSO4, фильтрования и концентрирования досуха, указанный в заголовке продукт выделяют с помощью хроматографии на силикагеле (градиент петролейный эфир/AcOEt: 0-20%) в виде твердого вещества.
1H ЯМР: δ (400 МГц; ДМСО-d6; 300K): 6.8-6.43 (m, 2Н, Н пиррол); 4.75-3.75 (m, 6Н, Н пиперазин); 3.73 (s, 3Н, Н СООСН3); 1.48 (s, 9Н, Н tBu)
ИК: ν: С=O (сопряженный сложный эфир): 1712 см-1; С=O (карбамат): 1677 см-1
Стадия D: 2-[2-(трет-Бутоксикарбонил)-8-(метоксикарбонил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-6-ил]-4-хлорбензойная кислота
Методика соответствует протоколу, описанному на Стадиях С и D Синтеза 1, с заменой 6-бром-1,3-бензодиоксол-5-карбальдегида, используемого на Стадии С, на 2-бром-4-хлорбензальдегид.
Синтез 3: 4-Хлор-2-[1-(метоксикарбонил)-5,6,7,8-тетрагидро-3-индолизинил]бензойная кислота
Методика соответствует способу Синтеза 1 с заменой 6-бром-1,3-бензодиоксол-5-карбальдегида, используемого на Стадии С, на 2-бром-4-хлорбензальдегид.
Синтез 4: 7-[2-(трет-Бутоксикарбонил)-8-(метоксикарбонил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-6-ил]-2,3-дигидро-1,4-бензодиоксин-6-карбоновая кислота
Стадия А: 2-трет-Бутил 8-метил 3,4-дигидропирроло[1,2-а]пиразин-2,8(1Н)-дикарбоксилат
Методика соответствует способу, описанному на Стадиях А-С Синтеза 2.
1H ЯМР: δ (400 МГц; ДМСО-d6; 300K): 6.8-6.43 (m, 2Н, Н пиррол); 4.75-3.75 (m, 6Н, Н пиперазин); 3.73 (s, 3Н, Н СООСН3); 1.48 (s, 9Н, Н tBu)
ИК: ν: С=O (сопряженный сложный эфир): 1712 см-1; С=O (карбамат): 1677 см-1
Стадия В: 7-[2-(трет-Бутоксикарбонил)-8-(метоксикарбонил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-6-ил]-2,3-дигидро-1,4-бензодиоксин-6-карбоновая кислота
Методика соответствует протоколу, описанному на Стадиях С и D Синтеза 1, с заменой 6-бром-1,3-бензодиоксол-5-карбальдегида, используемого на Стадии С, на 7-бром-2,3-дигидро-1,4-бензодиоксин-6-карбальдегид.
Синтез 5: 4-Хлор-2-[1-(метоксикарбонил)-3-индолизинил]бензойная кислота
Стадия А: Бромид 1-(карбоксиметил)-1,2-дигидропиридиния
К раствору 16.2 мл пиридина (200 ммоль) в 120 мл этилацетата порциями добавляют 27.8 г (200 ммоль) бромуксусной кислоты. Загрузку затем перемешивают при температуре окружающей среды в течение ночи. Полученный таким образом осадок отфильтровывают и затем промывают холодным этилацетатом. После сушки указанный в заголовке продукт получают в виде порошка, который используют непосредственно на следующей стадии.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 9.15 (d, 2Н, ароматические Н, пиридин); 8.7 (t, 1Н, ароматический Н); 8.25 (t, 2Н, ароматические Н); 5.65 (s, 2Н, Н СН2СООН)
ИК: ν: С=O: 1732 см-1; -ОН кислота: 2800 см-1
Стадия В: Метил 1-индолизинкарбоксилат
К суспензии 6.55 г соли пиридиния, полученной на Стадии А, (30 ммоль) в 240 мл толуола последовательно добавляют 16.7 мл метилакрилата (150 ммоль), 4.2 мл триэтиламина (30 ммоль) и затем, порциями, 20.9 г MnO2 (240 ммоль). Загрузку затем нагревают при 90°С в течение 3 часов. После охлаждения, реакционную смесь фильтруют через слой целита и концентрируют досуха. Указанный в заголовке продукт затем выделяют с помощью очистки на силикагеле (градиент гептан/AcOEt: 0-10%) в виде масла, которое кристаллизуется в холодном состоянии.
1Н ЯМР: δ (300 МГц; ДМСО-d6; 300K): 8.5 (d, 1Н, Н индолизин); 8.05 (d, 1Н, Н индолизин); 7.6 (s, 1Н, Н индолизин); 7.15 (m, 2Н, Н индолизин); 6.85 (m, 1Н, Н индолизин); 4.25 (q, 2Н, -С(O)СН2СН3); 1.35 (t, 3Н, -С(O)СН2СН3)
ИК: ν: С=O сложный эфир: 1675 см-1; ароматические С=С фрагменты: 1634 см-1
Стадия С: 4-Хлор-2-[1-(метоксикарбонил)-3-индолизинил]бензойная кислота
Методика соответствует протоколу, описанному на Стадиях С и D Синтеза 1, с заменой 6-бром-1,3-бензодиоксол-5-карбальдегида, используемого на Стадии С, на 2-бром-4-хлорбензальдегид.
Синтез 6: 2-[2-(трет-Бутоксикарбонил)-8-(метоксикарбонил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-6-ил]-4-фторбензойная кислота
Методика соответствует протоколу, описанному в Синтезе 2, с заменой 2-бром-4-хлорбензальдегида, используемого на Стадии D, на 2-бром-4-фторбензальдегид.
Синтез 7: 6-[2-(трет-Бутоксикарбонил)-8-(метоксикарбонил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-6-ил]-1,3-бензодиоксол-5-карбоновая кислота
Методика соответствует протоколу, описанному в Синтезе 2, с заменой 2-бром-4-хлорбензальдегида, используемого на Стадии D, на 6-бром-1,3-бензодиоксол-5-карбальдегид.
Синтез 8 6-[1'-(Метоксикарбонил)-5',6'-дигидро-8'H-спиро[1,3-диоксолан-2,7'-индолизин]-3'-ил]-1,3-бензодиоксол-5-карбоновая кислота
Стадия А: Метил 8-формил-1,4-диокса-8-азаспиро[4.5]декан-7-карбоксилат
24 г метил 1,4-диокса-8-азаспиро[4.5]декан-9-карбоксилата (111 ммоль) растворяют в 80 мл этилацетата и 80 мл дихлорметана. Добавляют 26 г (4-нитрофенил)формиата (155 ммоль) и загрузку перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь упаривают досуха и вносят в этилацетат. Органическую фазу затем последовательно промывают 1 н. раствором NaOH, водой и затем насыщенным раствором NH4Cl до достижения нейтрального рН. Смесь затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Полученное таким образом масло очищают с помощью флэш-хроматографии (градиент гептан/этилацетат). Указанный в заголовке продукт получают в виде масла.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 8.15 (s, 1H, СНО); 5.0-4.75 (m, 1H, третичный Н); 4.3-3.7 (m, 5Н, 4Н этилендиокси + 1Н алифатический, пиперидин); 3.70 (s, 3Н, Me); 3.4-2.9 (2m, 1Н, Н алифатический, пиперидин); 2.3-1.75 (m, 2Н, Н алифатический, пиперидин); 1.7-1.5 (m, 2Н, Н алифатический, пиперидин)
Стадия В: 8-Формил-1,4-диокса-8-азаспиро[4.5]декан-7-карбоновая кислота
15.25 г соединения, полученного на Стадии А, (62.7 ммоль) растворяют в 160 мл диоксана. По каплям добавляют 125 мл 1М раствора KOH и загрузку перемешивают при температуре окружающей среды в течение 1 часа. Затем добавляют 125 мл 1М HCl и соединение экстрагируют дихлорметаном. Органическую фазу сушат над MgSO4, фильтруют и концентрируют досуха. Указанный в заголовке продукт получают в виде порошка.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K) 13.5-12 (m, 1Н, ОН); 8.1+8.0 (2s, 1Н, СНО); 4.9+4.6 (2m, 1Н, третичный Н); 4.0-3.8 (m, 4Н, этилендиокси); 4.2+3.7 (2ms, 1Н, Н алифатический, пиперидин); 3.4+2.9 (2m, 1Н, Н алифатический, пиперидин); 2.4-1.5 (m, 4Н, Н алифатический, пиперидин)
ИК: ν: ОН: 3500-2000 см-1; -С=O (кислота + альдегид): 1731+1655 см-1
Стадия С: Метил 5',6'-дигидро-8'Н-спиро[1,3-диоксолан-2,7'-индолизин]-1'-карбоксилат
К раствору 13.5 г (62.7 ммоль) кислоты, полученной на Стадии В, в 380 мл дихлорметана последовательно добавляют 39.5 мл (238.4 ммоль) триэтиламина и затем, порциями, 12.5 г (65.6 ммоль) пара-толуолсульфонилхлорида и 23.7 мл (238.4 ммоль) метилхлоракрилата. Загрузку перемешивают при 80°С в течение 18 часов. Затем реакционную смесь фильтруют через целит. Фильтрат затем промывают насыщенным раствором NaHCO3 и затем насыщенным раствором NH4Cl. Органическую фазу сушат над MgSO4, фильтруют и концентрируют досуха. Полученное таким образом масло очищают с помощью флэш-хроматографии (градиент гептан/эти лацетат). Продукт получают в виде твердого вещества.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K) 6.70 (d, 1H, пиррол); 6.40 (d, 1H, пиррол); 4.05 (t, 2Н, Н алифатический, пиперидин); 4.00 (m, 4Н, этилендиокси); 3.70 (s, 3Н, метил); 3.15 (s, 2Н, Н алифатический, пиперидин); 2.05 (t, 2Н, Н алифатический, пиперидин)
ИК: ν: -C=O (сложный эфир): 1689 см-1
Стадия D: Метил 3'-(6-формил-1,3-бензодиоксол-5-ил)-5',6'-дигидро-8'Н-спиро[1,3-диоксолан-2,7'-индолизин]-1'-карбоксилат
Методика соответствует способу Стадии С Синтеза 1.
Стадия Е: 6-[1'-(Метоксикарбонил)-5',6'-дигидро-8'Н-спиро[1,3-диоксолан-2,7'-индолизин]-3'-ил]-1,3-бензодиоксол-5-карбоновая кислота
Методика соответствует способу Стадии D Синтеза 1.
Синтез 9: 6-[1-(Метоксикарбонил)-3-индолизинил]-1,3-бензодиоксол-5-карбоновая кислота
Методика соответствует протоколу, описанному в Синтезе 5, с заменой 2-бром-4-хлорбензальдегида, используемого на Стадии С, на 6-бром-1,3-бензодиоксол-5-карбальдегид.
Синтез 10: 4-Метил-2-[1-(метоксикарбонил)-5,6,7,8-тетрагидро-3-индолизинил]бензойная кислота
Методика соответствует протоколу, описанному в Синтезе 1, с заменой 6-бром-1,3-бензодиоксол-5-карбальдегида, используемого на Стадии С, на 2-бром-4-метилбензальдегид.
Синтез 11: 2-[1-(Метоксикарбонил)-5,6,7,8-тетрагидро-3-индолизинил]бензойная кислота
Методика соответствует протоколу, описанному в Синтезе 1, с заменой 6-бром-1,3-бензодиоксол-5-карбальдегида, используемого на Стадии С, на 2-бромбензальдегид.
Синтез 12: 6-[8-(Метоксикарбонил)пирроло[1,2-а]пиримидин-6-ил]-1,3-бензодиоксол-5-карбоновая кислота
Стадия А: Метил пирроло[1,2-а]пиримидин-8-карбоксилат
К раствору 6.2 г метил 2-пиримидин-2-илацетата (40.75 ммоль) в 250 мл ацетона последовательно добавляют 14.04 г (167 ммоль) NaHCO3 в виде порошка, 13.2 мл (203.75 ммоль) хлорацетальдегида и затем 3.54 г (40.75 ммоль) бромида лития. Загрузку нагревают при 60°С в течение 24 часов. Затем реакционную смесь концентрируют досуха, вносят в этилацетат, промывают водой, сушат над MgSO4, фильтруют и затем концентрируют досуха. Полученное таким образом твердое вещество затем очищают с помощью хроматографии на силикагеле (AcOEt). Ожидаемый продукт получают в виде масла.
Масс-спектр:
Эмпирическая формула: C8H8N2O2
ЖХ/МС: m/z = [М+Н]+ = 177
Стадия В: Метил 6-(6-формил-1,3-бензодиоксол-5-ил)пирроло[1,2-а]пиримидин-8-карбоксилат
К раствору 3.93 г соединения, полученного на Стадии А, (22.3 ммоль) в 80 мл безводного диметилацетамида добавляют 7.66 г (33.45 ммоль) 6-бром-1,3-бензодиоксол-5-карбальдегида и 4.4 г (44.6 ммоль) ацетата калия. Загрузку дегазируют в атмосфере азота в течение 15 минут. Затем добавляют 1.56 г (2.23 ммоль) катализатора PdCl2(PPh3)4. Реакционную смесь нагревают при 130°С в течение 16 часов в инертной атмосфере. После сушки остаток вносят в дихлорметан; загрузку фильтруют через слой целита и затем фильтрат промывают водой, сушат над MgSO4 и концентрируют досуха. Черное твердое вещество затем хроматографируют на силикагеле (CH2Cl2/МеОН 5%). Ожидаемый продукт получают в виде твердого вещества.
Масс-спектр:
Эмпирическая формула: C17H12N2O3
ЖХ/МС: m/z = [М+Н]+ = 325
Стадия С: 6-[8-(Метоксикарбонил)пирроло[1,2-а]пиримидин-6-ил]-1,3-бензодиоксол-5-карбоновая кислота
К раствору 2.91 г (8.97 ммоль) альдегида, полученного на Стадии В, в 140 мл ацетона, охлажденному до 0°С, добавляют 2-метилбутен и затем, по каплям, смесь 2.8 г (17.94 ммоль) NaH2PO4.2H2O и 2.84 г (31.4 ммоль) NaClO2, растворенных в 30 мл воды. Загрузку перемешивают при температуре окружающей среды в течение 4 часов. Затем реакционную смесь концентрируют в вакууме для удаления ацетона, охлаждают до 0°С и затем подкисляют до рН=2-3 путем добавления по каплям 5 н. раствора HCl. Наблюдают образование осадка, который отфильтровывают, промывают водой и затем диэтиловым эфиром, и сушат в вакууме.
1H ЯМР: δ (400 МГц; ДМСО-d6; 300K): 12.7 (m, 1Н, СООН); 8.45 (d, 1Н, ароматический Н, Н пирроло[1,2-а]пиримидин); 8.19 (d, 1Н, ароматический Н, Н пирроло[1,2-а]пиримидин); 6.9 (dd, 1Н, ароматический Н, Н пирроло[1,2-а]пиримидин); 7.51 (s, 1Н, ароматический Н); 7.21 (s, 1Н, ароматический Н); 7.07 (s, 1Н, ароматический Н); 6.2 (s, 2Н, алифатические Н, О-СН2-О); 3.8 (s, 3Н, алифатические Н, СООСН3)
ИК: ν -ОН-: 3300-1800 см-1; ν -СО-: 1705 см-1, ν >С=С<: 1616 см-1
Синтез 13: 4-Метокси-2-[1-(метоксикарбонил)-5,6,7,8-тетрагидро-3-индолизинил]бензойная кислота
Методика соответствует протоколу, описанному в Синтезе 1, с заменой 6-бром-1,3-бензодиоксол-5-карбальдегида, используемого на Стадии С, на 2-бром-4-метоксибензальдегид.
Синтез 14: 5-Метокси-2-[1-(метоксикарбонил)-5,6,7,8-тетрагидро-3-индолизинил]бензойная кислота
Методика соответствует протоколу, описанному в Синтезе 1, с заменой 6-бром-1,3-бензодиоксол-5-карбальдегида, используемого на Стадии С, на 2-бром-5-метоксибензальдегид.
Синтез 15: 7-[1-(Метоксикарбонил)-5,6,7,8-тетрагидро-3-индолизинил]-2,3-дигидро-1,4-бензодиоксин-6-карбоновая кислота
Методика соответствует способу Синтеза 1, с заменой 6-бром-1,3-бензодиоксол-5-карбальдегида, используемого на Стадии С, на 7-бром-2,3-дигидро-1,4-бензодиоксин-6-карбальдегид.
Синтез 16: 2-[1-(Метоксикарбонил)-3-индолизинил]бензойная кислота
Методика соответствует способу Синтеза 5 с заменой 2-бром-4-хлорбензальдегида, используемого на Стадии С, на 2-бромбензальдегид.
Синтез 17: 4-Фтор-2-[1-(метоксикарбонил)-3-индолизинил]бензойная кислота
Методика соответствует способу Синтеза 5 с заменой 2-бром-4-хлорбензальдегида, используемого на Стадии С, на 2-бром-4-фторбензальдегид.
Синтез 18: 4-Фтор-2-[1-(метоксикарбонил)-5,6,7,8-тетрагидро-3-индолизинил]бензойная кислота
Методика соответствует способу Синтеза 1 с заменой 6-бром-1,3-бензодиоксол-5-карбальдегида, используемого на Стадии С, на 2-бром-4-фторбензальдегид.
Синтез 1': Гидрохлорид (3R)-3-метил-1,2,3,4-тетрагидроизохинолина
Стадия А: {(3S)-2-[(4-Метилфенил)сульфонил]-1,2,3,4-тетрагидроизохинолин-3-ил}метил 4-метилбензолсульфонат
К раствору 30.2 г [(3S)-1,2,3,4-тетрагидроизохинолин-3-ил]метанола (185 ммоль) в 750 мл дихлорметана последовательно добавляют 91.71 г тозилхлорида (481 ммоль) и затем, по каплям, 122.3 мл N,N,N-триэтиламина (740 ммоль). Затем реакционную смесь перемешивают при температуре окружающей среды в течение 20 часов. Ее затем разбавляют дихлорметаном, последовательно промывают 1М раствором HCl, насыщенным раствором NaHCO3 и затем насыщенным раствором NaCl до нейтральной реакции среды. Органическую фазу затем сушат над MgSO4, фильтруют и концентрируют досуха. Полученное твердое вещество затем растворяют в минимальном объеме дихлорметана и затем до образования осадка добавляют циклогексан. Этот осадок затем отфильтровывают и промывают циклогексаном. После сушки указанный в заголовке продукт получают в виде кристаллов.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 7.75 (d, 2Н, ароматические Н, орто О-тозил); 7.6 (d, 2Н, ароматические Н, орто N-тозил); 7.5 (d, 2Н, ароматические Н, мета О-тозил); 7.3 (d, 2Н, ароматические Н, мета N-тозил); 7.15-6.9 (m, 4Н, ароматические Н, тетрагидроизохинолин); 4.4-4.15 (dd, 2Н, алифатические Н, тетрагидроизохинолин); 4.25 (m, 1Н, алифатический Н, тетрагидроизохинолин); 4.0-3.8 (2dd, 2Н, алифатические Н, СН2-O-тозил); 2.7 (2dd, 2Н, алифатические Н, тетрагидроизохинолин); 2.45 (s, 3Н, O-SO2-Ph- СН3); 2.35 (s, 3Н, N-SO2-Ph-СН3)
ИК: ν: -SO2: 1339-1165 см-1
Стадия В: (3R)-3-Метил-2-[(4-метилфенил)сульфонил]-1,2,3,4-тетрагидроизохинолин
К суспензии 8.15 г (214.8 ммоль) LiAlH4 в 800 мл метил трет-бутилового эфира (МТВЕ) добавляют 101.2 г дитозильного соединения, полученного на Стадии А, (214.8 ммоль), растворенного в 200 мл МТВЕ. Загрузку затем нагревают при 50°С в течение 2 часов. Смеси дают охладиться и затем при 0°С по каплям добавляют 12 мл 5 н. раствора NaOH. Загрузку перемешивают при температуре окружающей среды в течение 45 минут. Полученное таким образом твердое вещество затем отфильтровывают и промывают МТВЕ и затем дихлорметаном. Фильтрат затем концентрируют досуха. Указанный в заголовке продукт получают в виде твердого вещества.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 7.70 (d, 2Н, ароматические Н, орто N-тозил); 7.38 (d, 2Н, ароматические Н, мета N-тозил); 7.2-7.0 (m, 4Н, ароматические Н, тетрагидроизохинолин); 4.4 (m, 2Н, алифатические Н, тетрагидроизохинолин); 4.3 (m, 1Н, алифатический Н, тетрагидроизохинолин); 2.85-2.51 (2dd, 2Н, алифатические Н, тетрагидроизохинолин); 2.35 (s, 3Н, N-SO2-Ph-СН3); 0.90 (d, 3Н, тетрагидроизохинолин-СН3)
ИК: ν: -SO2: 1332-1154 см-1
Стадия С: (3R)-3-Метил-1,2,3,4-тетрагидроизохинолин
К раствору 31.15 г (103.15 ммоль) монотозильного соединения, полученного на Стадии В, в 500 мл безводного метанола порциями добавляют 3.92 г (161 ммоль) магниевых стружек. Загрузку перемешивают при обработке ультразвуком в течение 96 часов. Затем реакционную смесь фильтруют и твердое вещество промывают несколько раз метанолом. Фильтрат затем концентрируют досуха. После очистки с помощью колоночной хроматографии на силикагеле (дихлорметан/EtOH/NH4OH), указанный в заголовке продукт получают в виде масла.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 7.05 (m, 4Н, ароматические Н, тетрагидроизохинолин); 3.90 (m, 2Н, алифатические Н, тетрагидроизохинолин); 2.85 (m, 1Н, алифатический Н, тетрагидроизохинолин); 2.68-2.4 (2dd, 2Н, алифатические Н, тетрагидроизохинолин); 1.12 (d, 3Н, тетрагидроизохинолин-СН3); 2.9-2.3 (m, широкий, 1Н, HN (тетрагидроизохинолин))
ИК: ν: -NH: 3248 см-1
Стадия D: Гидрохлорид (3R)-3-метил-1,2,3,4-тетрагидроизохинолина
К раствору 14.3 г (97.20 ммоль) соединения, полученного на Стадии С, в 20 мл безводного этанола по каплям добавляют 100 мл 1М раствора HCl в эфире. Загрузку перемешивают при температуре окружающей среды в течение 1 часа и затем фильтруют. Полученные таким образом кристаллы промывают этиловым эфиром. После сушки указанный в заголовке продукт получают в виде кристаллов.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 9.57 (m, широкий, 2Н, NH2+ (тетрагидроизохинолин); 7.22 (m, 4Н, ароматические Н, тетрагидроизохинолин); 4.27 (s, 2Н, алифатические Н, тетрагидроизохинолин); 3.52 (m, 1Н, алифатический Н, тетрагидроизохинолин); 3.03-2.85 (2dd, 2Н, алифатические Н, тетрагидроизохинолин); 1.39 (d, 3Н, тетрагидроизохинолин-СН3)
ИК: ν: -NH2+: 3000-2300 см-1; ν: ароматический -СН: 766 см-1
Синтез 2': Гидрохлорид (3S)-3-[2-(морфолин-4-ил)этил]-1,2,3,4-тетрагидроизохинолина
Стадия А: трет-Бутил (3S)-3-(2-морфолино-2-оксоэтил)-3,4-дигидро-1Н-изохинолин-2-карбоксилат
К раствору 3 г (10.30 ммоль) [(3S)-2-(трет-бутоксикарбонил)-1,2,3,4-тетрагидроизохинолин-3-ил]уксусной кислоты в 100 мл дихлорметана по каплям добавляют 1.10 мл (11.32 ммоль) морфолина и, опять же по каплям, 4.3 мл (30.9 ммоль) триэтиламина, 2.20 г (12.40 ммоль) 1,2-дихлорметана и 1.70 г (1.68 ммоль) гидроксибензотриазола. Загрузку перемешивают при температуре окружающей среды в течение 15 часов. Затем реакционную смесь разбавляют дихлорметаном и последовательно промывают 1М раствором HCl, насыщенным раствором NaHCO3 и затем насыщенным раствором NaCl до нейтральной реакции среды. Органическую фазу затем сушат над MgSO4, фильтруют и концентрируют досуха. После очистки с помощью колоночной хроматографии на силикагеле (дихлорметан/МеОН), указанный в заголовке продукт получают в виде масла.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 7.20-7.10 (m, 4Н, ароматические Н, тетрагидроизохинолин); 4.70 (m, 1Н, алифатический Н, СН тетрагидроизохинолин); 4.75-4.20 (2m, 2Н, алифатические Н, СН2 альфа к N тетрагидроизохинолина); 3.60 (m, 8Н, алифатические Н, морфолин); 3.00 и 2.70 (2dd, 2Н, алифатические Н, тетрагидроизохинолин); 2.50-2.20 (2d, 2Н, алифатические Н, СН2СО); 1.40 (s, 9Н, tBu)
ИК: ν: С=O: 1687; 1625 см-1
Стадия В: Гидрохлорид 1-(морфолин-4-ил)-2-[(3S)-1,2,3,4-тетрагидроизохинолин-3-ил]этанона
К раствору 2.88 г (7.18 ммоль) соединения, полученного на Стадии А, в 16 мл дихлорметана по каплям добавляют 80 мл (80 ммоль) 1М раствора HCl в эфире. Загрузку перемешивают при температуре окружающей среды в течение 15 часов, и затем суспензию фильтруют и осадок промывают с эфиром. После сушки, указанный в заголовке продукт получают в виде твердого вещества.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 9.80-9.50 (m, 2Н, NH2+); 7.30-7.10 (m, 4Н, ароматические Н, тетрагидроизохинолин); 4.30 (m, 2Н, алифатические Н, СН2 альфа к N тетрагидроизохинолина); 3.80 (m, 1Н, алифатический Н, СН тетрагидроизохинолин); 3.70-3.40 (2m, 8Н, алифатические Н, морфолин); 3.15 и 2.8 (m, 4Н, алифатические Н, СН2 тетрагидроизохинолин и СН2СО)
ИК: ν: -NH2+: 2800-1900 см-1; ν: С=O: 1620 см-1
Стадия С: Гидрохлорид (3S)-3-[2-(морфолин-4-ил)этил]-1,2,3,4-тетрагидроизохинолина
Приготавливают раствор 2.2 г (7.44 ммоль) соединения, полученного на Стадии В, в 22 мл МТВЕ и 5 мл дихлорметана. После охлаждения на бане со льдом при 0°С к нему по каплям добавляют 15 мл (15 ммоль) 1М раствора LiAlH4 в тетрагидрофуране. Загрузку затем перемешивают при температуре окружающей среды в течение 6 часов. Смесь охлаждают до 0°С, и затем по каплям добавляют 1 мл 5 н. раствора NaOH. Загрузку перемешивают при температуре окружающей среды в течение 45 минут. Твердое вещество затем отфильтровывают и промывают МТВЕ и затем дихлорметаном, и фильтрат концентрируют досуха. Полученное таким образом масло разбавляют дихлорметаном и по каплям добавляют 6.3 мл 1М раствора HCl в эфире. Загрузку перемешивают при температуре окружающей среды в течение 1 часа и затем фильтруют. Полученные таким образом кристаллы промывают этиловым эфиром. После сушки указанный в заголовке продукт получают в виде твердого вещества.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 11.35+9.80 (2m, 2Н, NH2+); 10.00 (m, Н, NH+); 7.20 (m, 4Н, ароматические Н, тетрагидроизохинолин); 4.30 (s, 2Н, алифатические Н, СН2 альфа к N тетрагидроизохинолина); 4.00+3.85 (2m, 4Н, алифатические Н, СН2 альфа к N морфолина); 3.70 (m, 1Н, алифатический Н, СН тетрагидроизохинолин); 3.55-3.30 (m, 4Н, алифатические Н, СН альфа к О морфолина и СН2-морфолин); 3.15 (dd, 1Н, алифатический Н, СН2 тетрагидроизохинолин); 3.10 (m, 2Н, алифатические Н, СН альфа к О морфолина); 2.90 (dd, 1Н, алифатический H, СН2 тетрагидроизохинолин); 2.30+2.15 (2m, 2Н, алифатические Н, СН2-тетрагидроизохинолин)
ИК: ν: NH+ / -NH2+: между 3500 и 2250 см-1; ν: С=С: слабый 1593 см-1; ν: ароматическая С-Н: 765 см-1
Синтез 3': трет-Бутил {2-[(3S)-1,2,3,4-тетрагидроизохинолин-3-ил]этил}карбамат
Стадия А: Бензил (3S)-3-(2-гидроксиэтил)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают, исходя из (3S)-2-[(бензилокси)карбонил]-1,2,3,4-тетрагидроизохинолин-3-карбоновой кислоты на основании протокола из литературы (Jinlong Jiang и др. Bioorganic & Medicinal Chemistry Letters, 14, 1795, 2004).
Стадия В: Бензил (3S)-3-{2-[(метилсульфонил)окси]этил}-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору 10.6 г соединения Стадии А (35.6 ммоль) в 350 мл безводного CH2Cl2, охлажденному до 0°С, последовательно добавляют 10.1 мл триэтиламина (71.2 ммоль) и затем, по каплям, 3.1 мл метансульфонилхлорида (39 ммоль). Затем реакционную смесь перемешивают при температуре окружающей среды в течение 2 часов. Затем осуществляют гидролиз путем медленного добавления воды. Продукт экстрагируют несколько раз с помощью CH2Cl2. Органические фазы затем объединяют и последовательно промывают 1 н. раствором HCl, насыщенным раствором NaCl, насыщенным раствором NaHCO3 и насыщенным раствором NaCl до нейтральной реакции среды. Их затем сушат над MgSO4 и концентрируют досуха. После очистки с помощью хроматографии на силикагеле (градиент петролейный эфир/AcOEt), ожидаемый продукт получают в виде пены.
ЖХ/МС: m/z = (М+Н)+ = 375
Стадия С: Бензил (3S)-3-(цианометил)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору 15.4 г соединения, полученного на Стадии В, (41.02 ммоль) в 250 мл безводного ДМСО добавляют 22 г (449 ммоль) цианида натрия. Загрузку затем нагревают при 60°С в течение 12 часов. Реакционной смеси дают охладиться и затем разбавляют путем добавления этилацетата. Затем осуществляют гидролиз насыщенным раствором NaHCO3. После экстрагирования еще два раза этилацетатом, органические фазы объединяют, промывают Н2О, сушат над MgSO4 и концентрируют досуха. После очистки с помощью хроматографии на силикагеле (гексан/AcOEt 7/3), ожидаемый продукт получают в виде масла.
ЖХ/МС: m/z = [М+Н]+ = 307.1
Стадия D: Бензил (3S)-3-(2-аминоэтил)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору 15.4 г соединения, полученного на Стадии С, (50.3 ммоль) в 300 мл безводного ТГФ, охлажденному до 0°С, по каплям добавляют 1 н. раствор ВН3-ТГФ. Реакционной смеси понемногу дают вернуться до температуры окружающей среды, и затем загрузку перемешивают в течение 14 часов. Затем реакционную смесь гидролизуют путем медленного добавления насыщенного раствора NH4Cl. Затем дважды осуществляют экстрагирование этилацетатом, органические фазы объединяют и сушат над MgSO4. После концентрирования досуха ожидаемый продукт получают в виде пены, которую используют непосредственно без очистки на следующей стадии защиты.
Стадия Е: Бензил (3S)-3-{2-[(трет-бутоксикарбонил)амино]этил}-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору 15.6 г соединения, полученного на Стадии D, (50.3 ммоль) в 670 мл CH2Cl2 порциями последовательно добавляют 13.2 г (60.36 ммоль) Boc2O, 14 мл триэтиламина (100.6 ммоль) и DMAP в каталитическом количестве. Загрузку перемешивают при температуре окружающей среды в течение 5 часов. Затем реакционную смесь гидролизуют водой и два раза экстрагируют с помощью CH2Cl2. Органические фазы объединяют, промывают водой и сушат над MgSO4. После концентрирования досуха и очистки с помощью хроматографии на силикагеле (градиент гептан/AcOEt), ожидаемый продукт получают в виде масла.
ЖХ/МС: m/z = (М+Н)+ = 411
Стадия F: трет-Бутил {2-[(3S)-1,2,3,4-тетрагидроизохинолин-3-ил]этил}карбамат
К раствору 10.4 г соединения, полученного на Стадии Е, (25.5 ммоль) в 210 мл безводного МеОН добавляют 2.71 г (2.55 ммоль) Pd/C 10%. Загрузку дегазируют в течение 30 минут и затем перемешивают в атмосфере водорода в течение 16 часов. Затем реакционную смесь фильтруют и концентрируют досуха. Ожидаемый продукт получают в виде твердого вещества, которое вносят в смесь пентан/Et2O (90/10), растирают и отфильтровывают. После сушки, продукт получают в виде твердого вещества.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 7.1-6.98 (m, 4Н, ароматические Н, тетрагидроизохинолин); 6.83 (m, 1Н, CH2NHBoc); 3.85 (s, 2Н, алифатические Н, тетрагидроизохинолин); 3.09 (q, 2Н, CH2NHBoc); 2.73 (m, 1Н, алифатический Н, тетрагидроизохинолин); 2.70 и 2.39 (2m, 2Н, алифатические Н, тетрагидроизохинолин); 1.63 (m, 2Н, алифатические Н); 1.38 (s, 9Н, NHCOOtBu)
ИК: ν: >NH: 3378, -3201 см-1 (амин, амид); ν: >С=O: 1683 см-1 (амид); ν: >NH: 1524 см-1 (амид); ν: >С=O: 1168 см-1
ЖХ/МС: m/z = [М+Н]+ = 277
Синтез 4': (3R)-3-[3-(Морфолин-4-ил)пропил]-1,2,3,4-тетрагидроизохинолин
Стадия А: {(3S)-2-[(4-Метилфенил)сульфонил]-1,2,3,4-тетрагидроизохинолин-3-ил}метил 4-метилбензолсульфонат
Методика соответствует методике Стадии А Синтеза 1'.
Стадия В: трет-Бутил 2-({(3R)-2-[(4-метилфенил)сульфонил]-1,2,3,4-тетрагидроизохинолин-3-ил}метил)-3-(морфолин-4-ил)-3-оксопропаноат
К суспензии 1 г NaH (60%) (25.08 ммоль) в 30 мл МТВЕ по каплям добавляют раствор 5 г трет-бутил 3-морфолино-3-оксопропаноата (21.81 ммоль) в 20 мл безводного МТВЕ. Эту суспензию перемешивают при температуре окружающей среды в течение 1 часа и затем добавляют соединение, полученное на Стадии А, в виде порошка. Загрузку перемешивают при 60°С в течение 30 часов. Добавляют 100 мл насыщенного раствора хлорида аммония. Полученный в результате раствор экстрагируют дихлорметаном. Органическую фазу затем сушат над MgSO4, фильтруют и концентрируют досуха. После очистки с помощью колоночной хроматографии на силикагеле (дихлорметан/МеОН), ожидаемый продукт получают в виде масла.
1Н ЯМР (500 МГц; ДМСО-d6) δ м.д.: 7.63/7.59 (2d, 2Н), 7.3/7.26 (2d, 2Н); 7.13 (m, 2Н), 7.09/6.97 (2t, 2Н), 4.64/4.55/4.36/4.28 (2АВ, 2Н), 4.25/4.11 (2m, 1Н), 3.81 (m, 1Н), 3.73/3.48 (m, 4Н), 3.57-3.32 (m, 4Н), 2.51 (m, 2Н), 2.32/2.31 (2s, 3Н), 1.88/1.79 (2m, 2Н), 1.39/1.38 (2s, 9Н).
ИК (ATR) см-1: ν: >С=O: 1731 (сложный эфир); ν: >С=O: 1644 (амид); ν: -SO2: 1334-1156; ν: >С-O-С<: 1155; γ: >СН-Ar: 815-746-709
Стадия С: 2-({(3R)-2-[(4-Метилфенил)сульфонил]-1,2,3,4-тетрагидроизохинолин-3-ил}метил)-3-(морфолин-4-ил)-3-оксопропановая кислота
К раствору 9.5 г (17.97 ммоль) соединения, полученного на Стадии В, в 40 мл диоксана по каплям добавляют 20 мл 4М раствора HCl в диоксане. Загрузку перемешивают при температуре окружающей среды в течение 48 часов и затем раствор концентрируют досуха. После сушки, ожидаемый продукт получают в виде масла.
1Н ЯМР (400 МГц; ДМСО-d6) δ м.д.: 12.75 (m, 1Н), 7.6 (2*d, 2Н); 7.3 (2*d, 2Н), 7.1/6.95 (2*m, 4Н), 4.7/4.2 (d, 2Н), 4.25/4.12 (2*m, 1Н), 3.9-3.3 (m, 9Н), 2.55 (d, 2Н), 2.3 (2*s, 3Н), 1.8 (t, 2Н)
ИК (ATR) см-1: ν: -ОН: 3500-2000; ν: >С=O: 1727 (кислота); ν: >С=O: 1634 (амид); ν: -SO2: 1330-1155
Стадия D: 3-{(3R)-2-[(4-Метилфенил)сульфонил]-1,2,3,4-тетрагидроизохинолин-3-ил}-1-(морфолин-4-ил)пропан-1-он
К раствору 7.80 г (16.51 ммоль) соединения, полученного на Стадии С, в 100 мл ДМСО добавляют 1.16 г (19.83 ммоль) твердого хлорида натрия и затем, по каплям, 5 мл воды. Загрузку перемешивают при 130°С в течение 1 часа и затем раствор концентрируют до  . Затем реакционную смесь разбавляют дихлорметаном и последовательно промывают насыщенным раствором хлорида лития и затем насыщенным раствором NaCl. Органическую фазу затем сушат над MgSO4, фильтруют и концентрируют досуха. После очистки с помощью колоночной хроматографии на силикагеле (циклогексан/этилацетат), ожидаемый продукт получают в виде масла.
. Затем реакционную смесь разбавляют дихлорметаном и последовательно промывают насыщенным раствором хлорида лития и затем насыщенным раствором NaCl. Органическую фазу затем сушат над MgSO4, фильтруют и концентрируют досуха. После очистки с помощью колоночной хроматографии на силикагеле (циклогексан/этилацетат), ожидаемый продукт получают в виде масла.
1H ЯМР (400 МГц; ДМСО-d6) δ м.д.: 7.65 (d, 2Н), 7.3 (d, 2Н); 7.15/7 (2m, 4Н), 4.6 (d, 1Н), 4.25 (d, 1Н), 4.2 (m, 1Н), 3.5 (m, 4Н), 3.4 (2m, 4Н), 2.6 (2dd, 2Н), 2.35 (s, 3Н), 2.3 (m, 2Н), 1.5 (quad., 2Н)
ИК (ATR) см-1: ν: >С=O: 1639; ν: -SO2: 1331-1156; γ: >СН-Ar: 815-675
Стадия Е: (3R)-2-[(4-Метилфенил)сульфонил]-3-[3-(морфолин-4-ил)пропил]-1,2,3,4-тетрагидроизохинолин
К раствору 6.0 г (14.0 ммоль) соединения, полученного на Стадии D, в 60 мл МТВЕ и 14 мл дихлорметана порциями добавляют 1.06 г (28 ммоль) LAH в течение 5 минут. Загрузку перемешивают при температуре окружающей среды в течение 15 часов. По каплям добавляют 1.5 мл воды и перемешивание осуществляют в течение 15 минут. Затем по каплям добавляют 1.5 мл раствора 5М гидроксида натрия и перемешивание осуществляют в течение 15 минут. Затем реакционную смесь разбавляют МТВЕ и дихлорметаном. Суспензию затем фильтруют и осадок промывают МТВЕ и дихлорметаном. Органическую фазу затем сушат над MgSO4, фильтруют и концентрируют досуха. После очистки с помощью колоночной хроматографии на силикагеле (дихлорметан/EtOH/NH4OH), ожидаемый продукт получают в виде масла.
1Н ЯМР (400 МГц; ДМСО-d6) δ м.д.: 7.68 (d, 2Н), 7.32 (d, 2Н); 7.1 (неразрешенный пик, 4Н), 4.65/4.23 (АВ, 2Н), 4.2 (m, 1Н), 3.55 (t, 4Н), 2.7/2.6 (АВх, 2Н), 2.35 (s, 3Н), 2.25 (t, 4Н), 2.2 (t, 2Н), 1.4/1.3 (2m, 4Н)
ИК (ATR) см-1: ν: -SO2: 1333-1158
Стадия F: (3R)-3-[3-(Морфолин-4-ил)пропил]-1,2,3,4-тетрагидроизохинолин
К раствору 1.50 г (3.62 ммоль) соединения, полученного на Стадии Е, в 20 мл безводного метанола порциями добавляют 2.0 г (82.3 ммоль) магниевых стружек. Загрузку перемешивают при обработке ультразвуком в течение 96 часов. Затем реакционную смесь фильтруют, твердое вещество промывают несколько раз метанолом и фильтрат концентрируют досуха. После очистки с помощью колоночной хроматографии на силикагеле (дихлорметан/EtOH/NH4OH), ожидаемый продукт получают в виде масла.
1Н ЯМР (400 МГц; ДМСО-d6) δ м.д.: 7.3 (d, 2Н), 7.1 (t, 2Н); 7.1 (d + t, 3Н), 7 (d, 2Н), 3.9 (s, 2Н), 3.55 (t, 4Н), 2.75 (m, 1Н), 2.72/2.45 (dd, 2Н), 2.35 (t, 4Н), 2.25 (t, 2Н), 1.6 (m, 2Н), 1.45 (m, 2Н)
ИК (ATR) см-1: ν: >NH2+/NH+: 3500-2300; ν: >С-O-С<: 1115
Масс-спектроскопия высокого разрешения (ESI+-/FIA/BP):
Эмпирическая формула: C16H24N2O
[М+Н]+, рассчитано: 261.1961
[М+Н]+, найдено: 261.1959
Синтез 5': Гидрохлорид (3S)-3-[2-(3,3-дифторпиперидин-1-ил)этил]-1,2,3,4-тетрагидроизохинолина
Методика соответствует способу Синтеза 2' с заменой морфолина, используемого на Стадии А, на 3,3-дифтор-1-пиперидин.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 11.3 (m, 1Н, NH+); 10.2-9.8 (m, 2Н, NH2+); 7.25 (m, 4Н, ароматические Н, тетрагидроизохинолин); 4.3 (широкий s, 2Н, алифатические Н, СН тетрагидроизохинолин); 4.0-3.3 (m, 7Н, алифатические Н); 3.15-2.95 (dd, 2Н, алифатические Н, СН тетрагидроизохинолин); 2.4-1.9 (m, 6Н, алифатические Н, Н 3,3-дифтор-1-пиперидин)
ИК: ν: NH+/NH2+: между 300 и 2500 см-1; ν: C-F: 1204 см-1
Синтез 6': Гидрохлорид (3S)-3-[2-(3-метоксиазетидин-1-ил)этил]-1,2,3,4-тетрагидроизохинолина
Методика соответствует способу Синтеза 2' с заменой морфолина, используемого на Стадии А, на 3-метоксиазетидин.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 11.3 (m, 1Н, NH+); 10.00 (m, 2Н, NH2+); 7.20 (m, 4Н, ароматические Н, тетрагидроизохинолин); 4.4 (m, 1Н, алифатический Н, 3-метоксиазетидин); 4.30 (s, 2Н, алифатические Н, тетрагидроизохинолин); 4.2-3.45 (m, 4Н, 3-метоксиазетидин); 4.2-3.6 (m, 3Н, алифатические Н); 3.1 и 2.95 (dd, 2Н, алифатические Н); 3.25 (s, 3Н, ОСН3)
Синтез 7': Гидрохлорид (3S)-3-метил-1,2,3,4-тетрагидроизохинолина
Методика соответствует способу Синтеза 1' с заменой [(3S)-1,2,3,4-тетрагидроизохинолин-3-ил]метанола, используемого на Стадии А, на [(3R)-1,2,3,4-тетрагидроизохинолин-3-ил]метанол.
Синтез 1'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-1-метил-1H-пиразол-4-амин
Стадия А: 4-{[трет-Бутил(диметил)силил]окси}анилин
Указанное в заголовке соединение получают исходя из 4-аминофенола в ТГФ в присутствии имидазола и трет-бутил(диметил)силилхлорида в соответствии с протоколом, описанным в литературе (S. Knaggs и др., Organic & Biomolecular Chemistry, 3(21), 4002-4010; 2005).
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 6.45-6.55 (dd, 4Н, ароматические Н); 4.60 (m, 2Н, NH2-Ph); 0.90 (s, 9Н, Si (СН2)2СН(СН3)2); 0.10 (s, 6Н, Si (СН2)2СН(СН3)2)
ИК: ν: -NH2+: 3300-3400 см-1
Стадия В: N-[4-[трет-Бутил(диметил)силил]оксифенил]-1-метил-пиразол-4-амин
К раствору 30.8 г (0.137 моль) соединения Стадии А в 525 мл безводного толуола последовательно добавляют 29.8 г трет-бутилата натрия (0.310 моль), 4.55 г Pd2(dba)3 (также называемого трис(дибензилиденацетон)дипалладием(0)) (4.96 ммоль), 4.81 г 2-ди-трет-бутилфосфино-2',4',6'-триизопропил-1,1'-бифенила (9.91 ммоль) и 12.8 мл 4-бром-1-метил-1H-пиразола (0.124 моль). Загрузку дегазируют в атмосфере аргона в течение 30 минут и нагревают с обратным холодильником в течение 3 часов. Смеси дают охладиться. Реакционную смесь концентрируют досуха и затем вносят в дихлорметан, фильтруют через целит и затем снова концентрируют досуха. Остаток затем очищают с помощью хроматографии на силикагеле (градиент CH2Cl2/AcOEt) с обеспечением ожидаемого продукта в виде твердого вещества.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 7.55 (s, 1Н, пиразол); 7.23 (s, 1Н, пиразол); 7.18 (широкий s, 1Н, NH2-Ph); 6.64 (m, 4Н, ароматические Н); 3.77 (s, 3Н, СН3-пиразол); 0.90 (s, 9Н, Si (СН2)2СН(СН3)2); 0.12 (s, 6Н, Si (СН2)2СН(СН3)2)
ИК: ν -NH+: 3275 см-1; ν Ar и C=N: 1577 и 1502 см-1; ν -Si-C-: 1236 см-1;
ν -Si-O-: 898 см-1; ν -Si-C-: 828, 774 см-1
Синтез 2'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-1-метил-1H-индол-5-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 5-бром-1-метил-1H-индол.
Синтез 3'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-1-[2-(морфолин-4-ил)этил]-1H-индол-5-амин
Стадия А: 5-Бром-1-[2-(морфолин-4-ил)этил]-1Н-индол
К суспензии NaH (4.5 г; 112 ммоль) в безводном ТГФ (300 мл), охлажденной до 0°С, порциями добавляют 5-бром-1H-индол (10.4 г; 51 ммоль). После перемешивания в течение 20 минут при 0°С порциями добавляют гидрохлорид 4-(2-хлорэтил)морфолина (10.4 г; 56 ммоль) в течение 1 часа. После перемешивания в течение ночи при температуре окружающей среды реакционную смесь нагревают при 80°С в течение 5 часов. Ее затем выливают на смесь водного бикарбоната натрия и дихлорметана. Водную фазу экстрагируют дихлорметаном. Органическую фазу сушат над MgSO4 и концентрируют досуха, и остаток очищают с помощью хроматографии на силикагеле (градиент CH2Cl2/МеОН) с обеспечением ожидаемого продукта в виде масла.
1Н ЯМР: δ (400 МГц; CDCl3; 300K): 7.75 (d, 1Н); 7.30 (dd, 1H); 7.20 (d, 1Н); 7.15 (d, 1H); 6.40 (d, 1Н); 4.20 (t, 2Н); 3.70 (m, 4Н); 2.75 (t, 2Н); 2.45 (m, 4Н)
Стадия В: 5-Бром-1-[2-(морфолин-4-ил)этил]-1Н-индол
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на соединение, полученное на Стадии А.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 7.35 (d, 1Н); 7.15 (s, 1H); 6.85 (d, 3Н); 6.70 (d, 2Н); 7.30 (d, 1H); 6.25 (d, 1Н), 4.20 (t, 2Н); 3.55 (m, 4Н); 2.65 (t, 2Н); 2.45 (m, 4Н); 1.45 (s, 9Н), 0.15 (s, 6Н)
Синтез 4'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-1-[2-(морфолин-4-ил)этил]-2,3-дигидро-1H-индол-5-амин
Методика соответствует способу Синтеза 2'' с заменой 5-броминдола, используемого на Стадии А, на 5-бром-2,3-дигидро-1H-индол.
Синтез 5'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-4-фторанилин
Методика соответствует способу Синтеза 1' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 1-бром-4-фторбензол.
Синтез 6'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-3-фтор-4-метиланилин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 4-бром-2-фтор-1-метилбензол.
Синтез 7'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-1-метил-1H-индазол-5-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1H-пиразола, используемого на Стадии В, на 5-бром-1-метил-1H-индазол.
Синтез 8'': 4-{[трет-Бутил(диметил)силил]окси}-N-фениланилин
К раствору 12 г 4-анилинофенола (64.7 ммоль) в 200 мл ацетонитрила при температуре окружающей среды добавляют 6.7 г имидазола (97.05 ммоль) и 11.7 г трет-бутил(хлор)диметилсилана (77.64 ммоль). Загрузку перемешивают при 70°С в течение 4 часов. Затем реакционную смесь выливают в воду и экстрагируют эфиром. Органическую фазу затем сушат над сульфатом магния, затем фильтруют и упаривают досуха. Полученный таким образом сырой продукт затем очищают с помощью хроматографии на силикагеле (градиент петролейный эфир/дихлорметан). Указанный в заголовке продукт получают в виде порошка.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 7.84 (s, 1Н NH); 7.17 (t, 2Н анилин); 6.98 (d, 2Н фенокси); 6.94 (d, 2Н анилин); 6.76 (d, 2Н фенокси); 6.72 (t, 1Н анилин); 0.95 (s, 9Н трет-бутил); 0.15 (s, 6Н диметил)
ИК: ν: >NH: 3403 см-1; >Ar: 1597 см-1
Синтез 9'': 4-Бензилокси-N-фениланилин
К раствору 4-гидрокси-N-фениланилина (30 г; 162 ммоль) в ацетонитриле (400 мл) добавляют 58 г Cs2CO3 (178 ммоль) и перемешивание осуществляют в течение 15 минут при температуре окружающей среды. Затем по каплям добавляют бензилбромид (22.5 мл; 178 ммоль) и затем реакционную смесь нагревают с обратным холодильником в течение 4 часов. После фильтрования и промывки ацетонитрилом фильтрат концентрируют и хроматографируют на силикагеле (градиент петролейный эфир/AcOEt). Указанный в заголовке продукт получают в виде бесцветного твердого вещества.
Синтез 10'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-3-фтор-4-[2-(морфолин-4-ил)этокси]анилин
Методика соответствует способу Синтеза 3'' с заменой 5-бром-1H-индола, используемого на Стадии А, на 4-бром-2-фторфенол.
1Н ЯМР: 6 (400 МГц; ДМСО-d6; 300K): 7.75 (d, 1Н); 7 (dd, 1Н); 6.9 (d, 2Н); 6.75 (m, 3Н); 6.7 (ddd, 1Н); 4.05 (t, 2Н); 3.6 (t, 4Н); 2.65 (t, 2Н); 2.45 (t, 4Н); 0.95 (s, 9Н); 0.2 (s, 6Н)
Синтез 11'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)пиридин-4-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 4-бромпиридин.
ИК: ν -NH-: 3200 и 2500 см-1; ν -Si-O-: 902 см-1; ν -Si-C-: 820 см-1
Синтез 12'': 3-[(4-{[трет-Бутил(диметил)силил]окси}фенил)амино]бензонитрил
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 3-бромбензонитрил.
Синтез 13'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-3-фторанилин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 1-бром-3-фторбензол.
Синтез 14'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-3,4-дифторанилин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 4-бром-1,2-дифторбензол.
Синтез 15'': 4-{[трет-Бутил(диметил)силил]окси}-N-{4-[(3,3-дифторпиперидин-1-ил)метил]фенил}анилин
Стадия A: 1-(4-Бромбензил)-3,3-дифторпиперидин
К раствору 4-бромбензальдегида (500 мг; 2.7 ммоль) в 12 мл дихлорметана добавляют, в указанном порядке, гидрохлорид 3,3-дифторпиперидина (470 мг; 3 ммоль), триацетоксиборогидрид натрия (860 мг; 4 ммоль) и уксусную кислоту (0.17 мл; 3 ммоль). После перемешивания в течение 1 часа при температуре окружающей среды реакционную смесь выливают на смесь водного бикарбоната натрия и дихлорметана. Водную фазу экстрагируют дихлорметаном. Органическую фазу сушат над MgSO4, концентрируют досуха и остаток очищают с помощью хроматографии на силикагеле (градиент CH2Cl2/МеОН) с обеспечением ожидаемого продукта в виде масла.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K):7.55 (dd, 2Н); 7.25 (dd, 2Н); 3.55 (s, 2Н); 2.7 (t, 2Н); 2.35 (t, 2Н); 1.85 (m, 2Н); 1.65 (m, 2Н)
Стадия B: 4-{[трет-Бутил(диметил)силил]окси}-N-{4-[(3,3-дифторпиперидин-1-ил)метил]фенил}анилин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 1-[(4-бромфенил)метил]-3,3-дифторпиперидин.
Синтез 16'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)хинолин-6-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 6-бромхинолин.
ИК: ν -NH-: 3300 см-1
Синтез 17'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-2-метилпиридин-4-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 4-бром-2-метилпиридин.
ИК: ν -NH-: 3200 и 3100 см-1
Синтез 18'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-1-метил-1H-пирроло[2,3-b]пиридин-5-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 5-бром-1-метил-1H-пирроло[2,3-b]пиридин (полученный в соответствии с протоколом из литературы: Heterocycles, 60(4), 865, 2003).
ИК: ν: -NH-: 3278 см-1; ν: ароматические -С=С- фрагменты: 1605 см-1
Синтез 19'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)пиридин-3-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1H-пиразола, используемого на Стадии В, на 3-бромпиридин.
Синтез 20'': 4-{[трет-Бутил(диметил)силил]окси}-N-{4-[(3,3-дифторпиперидин-1-ил)этил]фенил}анилин
Стадия А: 2-(4-Бромфенил)-1-(3,3-дифторпиперидин-1-ил)этанон
К раствору 4-бромфенилуксусной кислоты (4 г; 18.6 ммоль) и гидрохлорида 3,3-дифторпиперидина (2.5 г; 20.4 ммоль) в дихлорметане (190 мл) добавляют EDC (3.8 г; 22.3 ммоль), HOBt (3 г; 22.3 ммоль) и триэтиламин (1.3 мл; 593 ммоль). Реакционную смесь перемешивают в течение 17 часов при температуре окружающей среды и затем выливают на смесь водного бикарбоната натрия и этилацетата. Водную фазу экстрагируют этилацетатом. Органическую фазу промывают 0.1 н. соляной кислотой, водой и солевым раствором и затем сушат над MgSO4 и концентрируют досуха. Остаток очищают с помощью хроматографии на силикагеле (градиент петролейный эфир/этилацетат) с обеспечением ожидаемого продукта в виде твердого вещества.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K):7.5 (d, 2Н); 7.2 (d, 2Н); 3.8 (t, 2Н); 3.7 (s, 3Н); 3.5 (t, 2Н); 2 (m, 2Н); 1.6 (m, 2Н)
Стадия В: 1-[2-(4-Бромфенил)этил]-3,3-дифторпиперидин
К раствору соединения Стадии А (4.6 г; 14.5 ммоль) в безводном ТГФ (145 мл) добавляют 1М раствор боран-диметилсульфида в ТГФ (14.5 мл; 14.5 ммоль). Реакционную смесь нагревают при 80°С в течение 3 часов и затем растворитель упаривают при пониженном давлении. Остаток обрабатывают метанолом (50 мл) и затем 5 н. HCl (5.8 мл). После перемешивания в течение ночи при температуре окружающей среды и нагревания с обратным холодильником в течение 3 часов, рН реакционной смеси доводят до 8 насыщенным раствором бикарбоната натрия; водную фазу затем экстрагируют дихлорметаном. Органическую фазу сушат над MgSO4 и концентрируют досуха, и остаток очищают с помощью хроматографии на силикагеле (градиент CH2Cl2/МеОН) с обеспечением ожидаемого продукта в виде масла.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K):7.45 (d, 2Н); 7.20 (d, 2Н); 2.71 (m, 2Н); 2.69 (t, 2Н); 2.58 (dd, 2Н); 2.45 (dd, 2Н); 1.86 (m, 2Н); 1.63 (m, 2Н)
Стадия C: 4-{[трет-Бутил(диметил)силил]окси}-N-{4-[(3,3-дифторпиперидин-1-ил)этил]фенил}анилин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на соединение Стадии В.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 7.7 (s, 1Н); 7.45 (d, 2Н); 7.39 (t, 2Н); 7.31 (t, 1Н); 7.0 (m, 4Н); 6.9 (d, 2Н); 6.81 (d, 2Н); 5.05 (s, 2Н); 2.7 (t, 2Н); 2.6 (t, 2Н); 2.5 (t, 2Н); 2.45 (t, 2Н); 1.89 (m, 2Н); 1.68 (m, 2Н)
Синтез 21'': 4-{[трет-Бутил(диметил)силил]окси}-N-{4-[2-(3,3-дифторпирролидин-1-ил)этил]фенил}анилин
Методика соответствует способу Синтеза 19'' с заменой гидрохлорида 3,3-дифторпиперидина на Стадии А на гидрохлорид 3,3-дифторпирролидина.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 7.7 (s, 1Н); 7.45 (d, 2Н); 7.35 (t, 2Н); 7.34 (t, 1Н); 7.05-6.85 (m, 8Н); 5.05 (s, 2Н); 2.9 (t, 2Н); 2.75-2.25 (m, 8Н)
Синтез 22'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-2,6-диметилпиридин-4-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 4-бром-2,6-диметилпиридин.
ИК: ν: -NH-: 3300 и 2700 см-1; ν: -Si-O-: 900 см-1; ν: -Si-C-: 823 см-1
Синтез 23'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-1-[2-(морфолин-4-ил)этил]-1H-пиразол-4-амин
Методика соответствует способу Синтеза 2'' с заменой 5-броминдола, используемого на Стадии А, на 4-бром-1H-пиразол.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 7.61 (s, 1Н); 7.25 (s, 1H); 7.18 (s, 1H); 6.65 (m, 4Н); 4.15 (t, 2Н); 3.55 (t, 4Н); 2.7 (t, 2Н); 2.4 (t, 4Н); 0.95 (s, 9Н); 0.15 (s, 6h)
Синтез 24'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-3-фторпиридин-4-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1H-пиразола, используемого на Стадии В, на 4-бром-3-фторпиридин.
ИК: ν -NH-: 3200 и 3000 см-1; ν -Si-O-: 900 см-1; ν -Si-C-: 820 см-1
Синтез 25'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)имидазо[1,2-а]пиридин-7-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1H-пиразола, используемого на Стадии В, на 7-бромимидазо[1,2-а]пиридин (полученный исходя из 4-бромпиридин-2-амина в соответствии с протоколом из литературы: WO 2008124323 А1).
ИК: ν -NH-: 3300-3000 см-1; ν -C=N-: 1652 см-1; ν -С=С-: 1610 см-1;
ν -Si-C-: 1236 см-1; ν -Si-O-: 898 см-1; ν -Si-C-: 828, 774 см-1
Синтез 26'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-2-метилимидазо[1,2-а]пиридин-7-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1H-пиразола, используемого на Стадии В, на 7-бром-2-метилимидазо[1,2-а]пиридин (полученный исходя из 4-бромпиридин-2-амина в соответствии с протоколом из литературы: A.J. Helliot и др. J. Heterocyclic Chemistry 19, 1437, 1982).
ИК: ν -NH-: 3300-3000 см-1; ν -C=N-: 1652 см-1; ν -С=С-: 1610 см-1;
ν -Si-C-: 1236 см-1; ν -Si-O-: 898 см-1; ν -Si-C-: 828, 774 см-1
Синтез 27'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-6-метилпиридин-3-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1H-пиразола, используемого на Стадии В, на 3-бром-6-метилпиридин.
ИК: ν -NH-: 3251 см-1; ν ароматические -С=С- фрагменты: 1605 см-1
Синтез 28'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-5-фторпиридин-3-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1H-пиразола, используемого на Стадии В, на 3-бром-5-фторпиридин.
ИК: ν -NH-: 3400-3000 см-1; ν -C-F-: 1245 см-1
Синтез 29'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-2-метоксипиридин-4-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 4-бром-2-метоксипиридин.
ИК: ν -NH-: 3200 и 3000 см-1; ν ароматические -С=С- фрагменты: 1618, 1601 см-1
Синтез 30'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-2-(пропан-2-ил)пиридин-4-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 4-бром-2-(пропан-2-ил)пиридин.
ИК: ν -NH-: 3300 и 3100 см-1
Синтез 31'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)пиразоло[1,5-а]пиримидин-6-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 6-бромпиразоло[1,5-а]пиримидин.
ИК: ν -NH-: 3272 см-1; ν -C=N-: 1634 см-1; ν -С=С-: 1616 см-1
Синтез 32'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-3,3а-дигидро[1,2,4]триазоло[1,5-а]пиримидин-6-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 6-бром-3,3а-дигидро[1,2,4]триазоло[1,5-а]пиримидин, полученный в соответствии с литературой (WO 2011015343) исходя из 4H-1,2,4-триазол-3-амина и 2-бромпропандиаля.
ИК: ν -NH-: 3244 см-1
Синтез 33'': N-(4-{[трет-Бутил(диметил)силил]окси}фенил)пиридин-4-амина 1-оксид
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1H-пиразола, используемого на Стадии В, на 4-бромпиридина 1-оксид, полученный в соответствии с литературой (WO 2009117269) исходя из 4-бромпиридина.
ИК: ν -NH-: 3246 см-1; ν ароматические -С=С- фрагменты: 1618 см-1
Масс-спектр:
Эмпирическая формула: C17H24N2O2Si
[М]+. найдено m/z: 316
[М-O]+. найдено m/z: 300
[М-С4Н9]+. найдено m/z: 259
Синтез 34'': Хлорид N-[4-[трет-бутил(диметил)силил]оксифенил]-1-метилпиридин-1-ия-4-амина
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на хлорид 4-бром-1-метилпиридин-1-ия, полученный в соответствии с литературой исходя из 4-бромпиридина.
Синтез 35'': N-(4-[трет-Бутил(диметил)силил]оксифенил]-1-метил-пиразоло[3,4-b]пиридин-5-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1H-пиразола, используемого на Стадии В, на 5-бром-1-метилпиразоло[3,4-b]пиридин, полученный в соответствии с литературой (WO 2006052568).
1H ЯМР (400 МГц, ДМСО-d6) δ м.д.: 8.33 (d, 1Н), 7.94 (bs, 1Н), 7.92 (s, 1Н), 7.71 (d, 1Н), 6.95 (d, 2Н), 6.76 (d, 2Н), 4.01 (s, 3Н), 0.95 (s, 9Н), 0.17 (s, 6Н)
ИК (ATR) см-1: 3290 ν >ОН; 1503 ν Ar; 1249 γ -Si-CH3
Синтез 36'': N-[4-[трет-Бутил(диметил)силил]оксифенил]-3-метил-пиразоло[1,5-а]пиримидин-6-амин
Методика соответствует способу Синтеза 1'' с заменой 4-бром-1-метил-1Н-пиразола, используемого на Стадии В, на 6-бром-3-метилпиразоло[1,5-а]пиримидин, полученный в соответствии с литературой (WO 2011015343 и WO 2011049917).
1Н ЯМР (400 МГц, ДМСО-d6) δ м.д.: 8.49 (d, 1Н), 8.4 (d, 1Н), 7.98 (m, 1Н), 7.87 (s, 1Н), 7 (d, 2Н), 6.81 (d, 2Н), 2.29 (s, 3Н), 0.98 (s, 9Н), 0.2 (s, 6Н)
ИК (ATR) см-1: 3257 ν >NH
Амины NHR3R4, где R3 и R4, каждый независимо друг от друга, представляет собой арильную или гетероарильную группу, получают в соответствии со способами, описанными в литературе (Surry D.S. и др., Chemical Science, 2011, 2, 27-50, Charles M.D. и др., Organic Letters, 2005, 7, 3965-3968). Реакцию защиты гидрокси функции 4-анилинофенола, описанную в Синтезе 8'', можно применять к различным вторичным аминам NHR3R4 (согласно определению выше), содержащим одну или несколько гидрокси функций, когда они доступны для приобретения. Альтернативно, вторичные амины, содержащие по меньшей мере один гидрокси заместитель, можно синтезировать непосредственно в защищенной форме, то есть исходя из реагентов, гидрокси функция которых была предварительно защищена. Среди защитных групп, трет-бутил(диметил)силилокси и бензилокси являются особенно предпочтительными.
Среди аминов NHR3R4, содержащих гидрокси заместитель, которые используются для синтеза соединений настоящего изобретения, могут быть упомянуты: 4-(4-толуидино)фенол, 4-(4-хлоранилино)фенол, 4-(3-фтор-4-метиланилино)фенол, 4-[4-(трифторметокси)анилино]фенол, 4-[4-гидроксианилино]фенол, {4-[(1-метил-1H-индол-6-ил)амино]фенил}метанол, 4-(2,3-дигидро-1H-индол-6-иламино)фенол, 4-[(1-метил-2,3-дигидро-1H-индол-6-ил)амино]фенол, 4-[(1-метил-1H-индол-6-ил)амино]фенол, 4-[(1-метил-1H-индол-6-ил)амино]циклогексанол, 4-[(1-метил-1,2,3,4-тетрагидро-6-хинолинил)амино]фенол, 4-[(4-метил-3,4-дигидро-2H-1,4-бензоксазин-7-ил)амино]фенол, 4-[4-(диэтиламино)анилино]фенол, 4-(2,3-дигидро-1H-инден-5-иламино)фенол, 4-[(1-метил-1H-индазол-5-ил)амино]фенол, 4-[(1'-метил-1',2'-дигидроспиро[циклопропан-1,3'-индол]-5'-ил)амино]фенол, 4-[(1,3,3-триметил-2,3-дигидро-1H-индол-5-ил)амино]фенол, 4-[4-метокси-3-(трифторметил)анилино]фенол, 4-[4-(метилсульфанил)-3-(трифторметил)анилино]фенол, 2-фтор-4-[(1-метил-1Н-индол-5-ил)амино]фенол, 4-[(1-этил-1H-индол-5-ил)амино]фенол, 4-[(1-этил-2,3-дигидро-1H-индол-5-ил)амино]фенол, 4-[(1-изопропил-2,3-дигидро-1H-индол-5-ил)амино]фенол, 4-(бутиламино)фенол, 3-[(1-метил-1H-индол-5-ил)амино]-1-пропанол, 4-[(1-метил-1H-индол-5-ил)амино]-1-бутанол, 4-[(3-фтор-4-метилфенил)амино]фенол, 4-[(3-хлор-4-метилфенил)амино]фенол, 4-[(4-фторфенил)амино]фенол, 4-[(1-метил-1H-пирроло[2,3-b]пиридин-5-ил)амино]фенол, 4-[(4-фторфенил)амино]фенол, 4-[(2-фторфенил)амино]фенол, 4-[(3-фторфенил)амино]фенол, 4-[(2,4-дифторфенил)амино]фенол, 4-[(3,4-дифторфенил)амино]фенол, 3-[(4-гидроксифенил)амино]бензонитрил, 4-[(3-метоксифенил)амино]фенол, 4-[(3,5-дифторфенил)амино]фенол, 4-[(3-метилфенил)амино]фенол, 4-[(4-гидроксифенил)амино]бензонитрил, 4-[(3-хлорфенил)амино]фенол, 4-(пиримидин-2-иламино)фенол, 4-[(циклобутилметил)амино]фенол, 2-[(4-гидроксифенил)амино]бензонитрил, 4-{[(1-метил-1H-пиразол-4-ил)метил]амино}фенол, 4-[(циклопропилметил)амино]фенол, 4-{[(1-метил-1H-пиразол-3-ил)метил]амино}фенол, 4-(бут-2-ин-1-иламино)фенол, 4-(пиразин-2-иламино)фенол, 4-(пиридин-2-иламино)фенол, 4-(пиридазин-3-иламино)фенол, 4-(пиримидин-5-иламино)фенол, 4-(пиридин-3-иламино)фенол, 4-[(3,5-дифтор-4-метоксифенил)амино]фенол, 4-(пиридин-4-иламино)фенол, 4-[(3-фтор-4-метоксифенил)амино]фенол, 2-(фениламино)пиримидин-5-ол, 5-[(4-гидроксифенил)амино]-2-метоксибензонитрил, 4-{[3-(трифторметил)фенил]амино}фенол, 4-(метиламино)фенол, 4-(этиламино)фенол и 4-(пропан-2-иламино)фенол.
Гидрокси функцию(-и) вторичных аминов, перечисленных выше, защищают заранее с помощью подходящей защитной группы перед каким-либо сочетанием с производным кислоты - соединения формулы (VII), как определено в предшествующем общем способе.
Пример 1. N-(4-Гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(1-метил-1H-индол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Стадия А: Метил 3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксилат
К раствору 2 г соединения Синтеза 1 в 20 мл дихлорметана при температуре окружающей среды добавляют 5.5 мл N,N,N-триэтиламина (6.96 ммоль), соединение Синтеза 1' (6.96 ммоль), и затем 0.94 г гидроксибензотриазола (НОВТ) и 1.34 г 1-этил-3-(3'-диметиламинопропил)-карбодиимида (EDC) (6.96 ммоль). Затем реакционную смесь перемешивают при температуре окружающей среды в течение ночи; ее затем вливают в раствор хлорида аммония и экстрагируют этилацетатом. Органическую фазу затем сушат над сульфатом магния, и затем фильтруют и упаривают досуха. Полученный таким образом сырой продукт затем очищают с помощью хроматографии на силикагеле (градиент гептан/AcOEt) с получением ожидаемого продукта.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 7.2-6.8 (m, 4Н, ароматические Н, Н тетрагидроизохинолин); 7.10 (s, 1Н, ароматический Н, бензодиоксол); 6.92 (s, 1Н, ароматический Н, бензодиоксол); 6.25 (m, 1Н, Н тетрагидроиндолизин); 6.10 (s, 2Н, алифатические Н, ОСН2О); 4.80 (m, 1Н, алифатический Н, Н тетрагидроизохинолин); 4.20 (m, 1Н, алифатический Н, Н тетрагидроизохинолин); 4.1-3.5 (m, 3Н); 3.60 (s, 3Н, СООСН3); 2.90 (m, 2Н, алифатические Н, Н тетрагидроиндолизин); 2.45 (m, 2Н, алифатические Н, Н тетрагидроизохинолин); 1.70 (m, 4Н, алифатические Н, Н тетрагидроиндолизин); 0.80 (m, 3Н, алифатические Н, CH3-THIQ).
ИК: ν: >С=O 1694 см-1 (сопряженный сложный эфир); ν: >С=O 1624 см-1 (амид); ν: >С-Ar 772-742 см-1
Стадия В: 3-(6-{[(3R)-3-Метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксилат лития
К раствору, содержащему 8.26 ммоль соединения Стадии А в 24 мл диоксана, добавляют раствор гидроксида лития (675 мг, 16.1 ммоль). Загрузку помещают в микроволновую печь при 140 Вт, 100°С на срок 2 часа 30 минут. Затем реакционную смесь фильтруют и упаривают. Полученное таким образом твердое вещество сушат при 40°С в печи в присутствии Р2О5.
Стадия С: N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(1-метил-1Н-индол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
К раствору, содержащему 4.73 ммоль соединения Стадии В в 47 мл дихлорметана, по каплям добавляют 1.2 мл оксалилхлорида при 0°С. Реакционную смесь перемешивают при температуре окружающей среды в течение 11 часов и затем упаривают несколько раз совместно с дихлорметаном. Полученный таким образом продукт суспендируют в 37 мл дихлорметана и затем добавляют к раствору, содержащему 7.1 ммоль соединения, полученного в Синтезе 2'', в 10 мл дихлорметана в присутствии 0.6 мл пиридина (7.1 ммоль). Загрузку перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь концентрируют, очищают с помощью хроматографии на силикагеле (градиент дихлорметан/метанол) с получением ожидаемого продукта.
Стадия D: N-(4-Гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(1-метил-1Н-индол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
К раствору, содержащему 2.3 ммоль соединения, полученного на Стадии С, в 4 мл метанола добавляют 0.646 г (11.5 ммоль) гидроксида калия, растворенного в 8 мл метанола. Загрузку перемешивают при температуре окружающей среды в течение 30 минут. Затем реакционную смесь разбавляют дихлорметаном и последовательно промывают 1 н. раствором HCl, насыщенным раствором NaHCO3 и затем насыщенным раствором NaCl до достижения нейтрального рН. Органическую фазу затем сушат над сульфатом магния, фильтруют и упаривают. Полученный таким образом сырой продукт очищают на силикагеле (градиент дихлорметан/метанол) и затем лиофилизуют с обеспечением ожидаемого продукта.
Масс-спектроскопия высокого разрешения (ESI+):
Эмпирическая формула: C42H38CN4O5
[М+Н]+, рассчитано: 679.2920
[М+Н]+, найдено: 679.2908
Пример 2. Гидрохлорид N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-{1-[2-(морфолин-4-ил)этил]-1H-индол-5-ил}-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Методика соответствует с способам, описанным на Стадиях A-D Примера 1, с использованием соответствующих реагентов. После стадии очистки на силикагеле (см. Стадия D), твердое вещество затем растворяют в дихлорметане и добавляют 2 мл 1 н. HCl в эфире. Всю загрузку перемешивают в течение 1 часа и затем упаривают досуха. Полученный таким образом гидрохлорид растворяют в смеси вода/ацетонитрил до полного растворения и затем лиофилизуют.
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=69.32:68.93; %Н=5.94:5.74; %N=8.6:8.51; %Cl-=4.35:4.6
Если не указано иное, соединения следующих Примеров синтезируют в соответствии со способом Примера 1, используя на Стадии A: (i) соответствующую кислоту, полученную в соответствии с одним из Синтезов 1-18, и (ii) соответствующее тетрагидроизохинолиновое соединение, полученное в соответствии с одним из Синтезом 1'-7', и на Стадии С: (iii) подходящий амин NHR3R4 (неполный перечень предложен в Синтезах 1''-36'').
Пример 3. Гидрохлорид 6-(5-хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-гидроксифенил)-N-(1-метил-1H-индол-5-ил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-8-карбоксамида
Методика соответствует способу Примера 1, с заменой, с одной стороны, соединения Синтеза 1, используемого на Стадии А, на соединение Синтеза 2 и, с другой стороны, соединения Синтеза 1'', используемого на Стадии С, на N-(4-{[трет-бутил(диметил)силил]окси}фенил)-1-метил-1H-индол-5-амин, при этом предполагается, что полученный таким образом продукт не подвергают стадии превращения в соль в присутствии HCl в эфире, как описано на Стадии D Примера 1. С полученного таким образом соединения защиту снимают в присутствии 10 эквивалентов трифторуксусной кислоты в дихлорметане (10 мл/ммоль) при температуре окружающей среды в течение ночи. Продукт затем выделяют путем концентрирования реакционной смеси досуха. В конечном счете, его подвергают стадии превращения в соль в присутствии HCl в эфире.
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=67.99:65.52; %Н=5.28:4.49; %N=9.91:9.24; %Cl=10.03:9.95; %Cl-=5.02:5.45
Масс-спектроскопия высокого разрешения (ESI+):
Эмпирическая формула: C40H36ClN5O3
[М+Н]+, рассчитано: 670.2585
[М+Н]+, найдено: 670.2587
Пример 4. 3-[5-Хлор-2-(3,4-дигидроизохинолин-2(1H)-илкарбонил)фенил]-N-(4-гидроксифенил)-N-{1-[2-(морфолин-4-ил)этил]-1H-индол-5-ил}-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Элементный микроанализ: % найдено (теоретический)
%С=70.85(71.65); %Н=5.39(5.88); %N=9.11(9.28); %Cl=4.48(4.7)
Пример 5. 3-[5-Хлор-2-(3,4-дигидроизохинолин-2(1H)-илкарбонил)фенил]-N-(4-гидроксифенил)-N-{1-[2-(морфолин-4-ил)этил]-2,3-дигидро-1H-индол-5-ил}-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Стадия А: N-(4-{[трет-Бутил(диметил)силил]окси)фенил)-3-[5-хлор-2-(3,4-дигидроизохинолин-2(1Н)-илкарбонил)фенил]-N-{1-[2-(морфолин-4-ил)этил]-2,3-дигидро-1Н-индол-5-ил}-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Методика соответствует протоколам, описанным на Стадиях А-С Примера 1, с использованием соединения Синтеза 3 и 1,2,3,4-тетрагидроизохинолина на Стадии А, и соединения Синтеза 4'' на Стадии С.
Стадия В: 3-[5-Хлор-2-(3,4-дигидроизохинолин-2(1Н)-илкарбонил)фенил]-N-(4-гидроксифенил)-N-{1-[2-(морфолин-4-ил)этил]-2,3-дигидро-1Н-индол-5-ил}-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
К раствору 1.3 г (1.45 ммоль) соединения Стадии А в 13 мл уксусной кислоты при температуре окружающей среды добавляют цианоборогидрид натрия (900 мг; 15 ммоль). После перемешивания в течение 2 часов, реакционную смесь концентрируют досуха, и затем разбавляют метанолом (8 мл) и обрабатывают 1М раствором гидроксида калия в метаноле (6.3 мл; 6.3 ммоль). После 1-часового выдерживания при температуре окружающей среды реакционную смесь концентрируют досуха, и затем хроматографируют на силикагеле (градиент дихлорметан/метанол) и затем лиофилизуют с обеспечением ожидаемого продукта в виде порошка.
Элементный микроанализ: % найдено (теоретический)
%С=70.74(71.46); %Н=5.74(6.13); %N=9(9.26); %Cl=4.46(4.69)
Пример 6. Гидрохлорид N-(4-гидроксифенил)-2-метил-6-(7-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-2,3-дигидро-1,4-бензодиоксин-6-ил)-N-(1-метил-1H-индол-5-ил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-8-карбоксамида
Методика аналогична методике, описанной на Стадии А Примера 7, с заменой соединения Синтеза 2 на соединение Синтеза 4.
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=69.39:69.13; %Н=5.69:4.98; %N=9.41:9.37; %Cl-=4.76:4.65
Пример 7. 6-(5-Хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-гидроксифенил)-2-метил-N-(1-метил-1H-индол-5-ил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-8-карбоксамид
Стадия А: трет-Бутил 8-[(4-{[трет-бутил(диметил)силил]окси}фенил)(1-метил-1Н-индол-5-ил)карбамоил]-6-(5-хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-3,4-дигидропирроло[1,2-а]пиразин-2(1Н)-карбоксилат
Методика соответствует протоколам, описанным на Стадиях А-С Примера 1, с использованием соединений Синтезов 2 и 1' на Стадии А, и соединения Синтеза 2'' на Стадии С.
Стадия В: трет-Бутил 6-(5-хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-8-[(4-гидроксифенил)(1-метил-1Н-индол-5-ил)карбамоил]-3,4-дигидропирроло[1,2-а]пиразин-2(1Н)-карбоксилат
К раствору соединения Стадии А (1.1 г; 1.25 ммоль) в метаноле (6 мл) добавляют 1М раствор гидроксида калия в метаноле (6.2 мл; 6.2 ммоль). После 2-часового выдерживания при температуре окружающей среды, метанол упаривают в вакууме и остаток вносят в смесь, состоящую из дихлорметана и насыщенного раствора бикарбоната натрия. Объединенные органические фазы сушат над MgSO4 и концентрируют досуха. Полученный остаток очищают с помощью хроматографии на силикагеле (градиент CH2Cl2/МеОН) с обеспечением ожидаемого продукта в виде твердого вещества.
ИК: ν: NH: 3450 см-1; ν: СО: 1745-1620 см-1
Стадия С: трет-Бутил 6-(5-хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-8-[{4-[(2,2-диметилпропаноил)окси]фенил}(1-метил-1Н-индол-5-ил)карбамоил]-3,4-дигидропирроло[1,2-а]пиразин-2(1Н)-карбоксилат
К раствору соединения Стадии В (0.7 г; 0.93 ммоль) в дихлорметане (7 мл) добавляют, при температуре окружающей среды, триэтиламин (0.2 мл; 1.39 ммоль) и затем пивалоилхлорид (0.11 мл; 0.93 ммоль). После перемешивания в течение 2 часов при температуре окружающей среды реакционную смесь промывают водой и солевым раствором, сушат над MgSO4 и концентрируют досуха. Полученный остаток используют в таком виде на следующей стадии без проведения его анализа.
Стадия D: 2,2-Диметил 4-[{[6-(5-хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-8-ил]карбонил}(1-метил-1Н-индол-5-ил)амино]фенил пропаноат
К раствору соединения предшествующей стадии (0.82 г; 0.93 ммоль) в дихлорметане (9 мл) при 0°С по каплям добавляют трифторуксусную кислоту (0.7 мл; 13.9 ммоль). После перемешивания в течение 15 часов при температуре окружающей среды к реакционной смеси медленно добавляют насыщенный раствор бикарбоната натрия и затем фазы разделяют. Водную фазу экстрагируют дихлорметаном. Объединенные органические фазы сушат над MgSO4 и концентрируют досуха. Полученный остаток очищают с помощью хроматографии на силикагеле (градиент CH2Cl2/МеОН) с обеспечением ожидаемого продукта в виде твердого вещества.
ЖХ/МС: m/z = [М+Н]+ = 754.30
Стадия Е: 2,2-Диметил 4-[{[6-(5-хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-2-метил-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-8-ил]карбонил}(1-метил-1Н-индол-5-ил)амино]фенил пропаноат
К раствору соединения предшествующей стадии (0.41 г; 0.54 ммоль) в дихлорметане (2 мл) добавляют, при температуре окружающей среды, формальдегид (48 мкл; 1.74 ммоль) и затем триацетоксиборогидрид натрия (161 мг; 0.76 ммоль). После перемешивания в течение 2 часов при температуре окружающей среды реакционную смесь разбавляют дихлорметаном и затем промывают насыщенным раствором бикарбоната натрия. Органическую фазу сушат над MgSO4 и концентрируют досуха. Полученный остаток очищают с помощью хроматографии на силикагеле (градиент CH2Cl2/МеОН). Ожидаемый продукт получают в виде твердого вещества.
ЖХ/МС: m/z = [М+Н]+ = 768.32
Стадия F: 6-(5-Хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-N-(4-гидроксифенил)-2-метил-N-(1-метил-1Н-индол-5-ил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-8-карбоксамид
К раствору соединения предшествующей стадии (0.25 г; 0.32 ммоль) в диоксане (1 мл) добавляют раствор гидроксида лития (27 мг; 0.65 ммоль) в воде (1 мл). После перемешивания в течение 5 часов при температуре окружающей среды реакционную смесь концентрируют и разбавляют насыщенным раствором бикарбоната натрия. Водную фазу экстрагируют CH2Cl2. Органическую фазу сушат над MgSO4 и концентрируют досуха. Полученный остаток очищают с помощью хроматографии на силикагеле (градиент CH2Cl2/МеОН). Ожидаемый продукт получают в виде твердого вещества.
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=71.97:71.51; %Н=5.6:5.25; %N=10.24:10.12
Пример 8. Гидрохлорид 3-(5-хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-гидроксифенил)-N-{1-[2-(морфолин-4-ил)этил]-1H-индол-5-ил}индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=69:69.16; %Н=5.41:4.82; %N=8.75:8.69; %Cl-=4.43:4.13
Пример 9. Гидрохлорид 6-(5-фтор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-фторфенил)-N-(4-гидроксифенил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-8-карбоксамида
Методика соответствует способу Примера 1, с заменой, с одной стороны, соединения Синтеза 1, используемого на Стадии А, на соединение Синтеза 6 и, с другой стороны, соединения Синтеза 1'', используемого на Стадии С, на соединение Синтеза 5'', при этом предполагается, что полученный таким образом продукт не подвергают стадии превращения в соль в присутствии HCl в эфире, как описано на Стадии D Примера 1. С полученного таким образом соединения защиту снимают в присутствии 10 эквивалентов трифторуксусной кислоты в дихлорметане (10 мл/ммоль) при температуре окружающей среды в течение ночи. Продукт затем выделяют путем концентрирования реакционной смеси досуха. В конечном счете, его подвергают стадии превращения в соль в присутствии HCl в эфире.
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=67.83:67.41; %Н=5.08:4.61; %N=8.55:8.39; %Cl-=5.41:5.28
Пример 10. Гидрохлорид 6-(5-фтор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(3-фтор-4-метилфенил)-N-(4-гидроксифенил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-8-карбоксамида
Методика соответствует способу Примера 1, с заменой, с одной стороны, соединения Синтеза 1, используемого на Стадии А, на соединение Синтеза 6 и, с другой стороны, соединения Синтеза 1'', используемого на Стадии С, на соединение Синтеза 6'', при этом предполагается, что полученный таким образом продукт не подвергают стадии превращения в соль в присутствии HCl в эфире, как описано на Стадии D Примера 1. С полученного таким образом соединения защиту снимают в присутствии 10 эквивалентов трифторуксусной кислоты в дихлорметане (10 мл/ммоль) при температуре окружающей среды в течение ночи. Продукт затем выделяют путем концентрирования реакционной смеси досуха. В конечном счете, его подвергают стадии превращения в соль в присутствии HCl в эфире.
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=68.21:68.29; %Н=5.27:4.91; %N=8.37:8.34; %Cl-=5.3:5.17
Пример 11. Гидрохлорид N-(4-гидроксифенил)-6-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(1-метил-1H-индазол-5-ил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-8-карбоксамида
Методика соответствует способу Примера 1, с заменой, с одной стороны, соединения Синтеза 1, используемого на Стадии А, на соединение Синтеза 7 и, с другой стороны, соединения Синтеза 1'', используемого на Стадии С, на N-(4-{[трет-бутил(диметил)силил]окси}фенил)-1-метил-1H-индазол-5-амин, при этом предполагается, что полученный таким образом продукт не подвергают стадии превращения в соль в присутствии HCl в эфире, как описано на Стадии D Примера 1. С полученного таким образом соединения защиту снимают в присутствии 10 эквивалентов трифторуксусной кислоты в дихлорметане (10 мл/ммоль) при температуре окружающей среды в течение ночи. Продукт затем выделяют путем концентрирования реакционной смеси досуха. В конечном счете, его подвергают стадии превращения в соль в присутствии HCl в эфире.
Элементный микроанализ: (%, теоретическое значение : найдено)
%Н=5.2:4.83; %N=11.72:11.64; %Cl-=4.94:5.34; %С=66.99:66.19
Пример 12. Гидрохлорид 6-(5-хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-гидроксифенил)-N-(1-метил-1H-индазол-5-ил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-8-карбоксамида
Методика соответствует способу Примера 1, с заменой, с одной стороны, соединения Синтеза 1, используемого на Стадии А, на соединение Синтеза 2 и, с другой стороны, соединения Синтеза 1'', используемого на Стадии С, на N-(4-{[трет-бутил(диметил)силил]окси}фенил)-1-метил-1H-индазол-5-амин, при этом предполагается, что полученный таким образом продукт не подвергают стадии превращения в соль в присутствии HCl в эфире, как описано на Стадии D Примера 1. С полученного таким образом соединения защиту снимают в присутствии 10 эквивалентов трифторуксусной кислоты в дихлорметане (10 мл/ммоль) при температуре окружающей среды в течение ночи. Продукт затем выделяют путем концентрирования реакционной смеси досуха. В конечном счете, его подвергают стадии превращения в соль в присутствии HCl в эфире.
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=66.19:65.83; %Н=5.13:4.99; %N=11.88:11.85; %Cl-=5.01:5.36
Пример 13. Гидрохлорид N-(4-гидроксифенил)-3-(6-{[(3S)-3-[2-(морфолин-4-ил)этил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=69.42:69.47; %Н=5.96:5.58; %N=7.36:7.36; %Cl-=4.66:4.42
Пример 14. Гидрохлорид N-(4-гидроксифенил)-N-(1-метил-1H-индазол-5-ил)-3-(6-{[(3S)-3-[2-(морфолин-4-ил)этил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=67.76:67.81; %Н=5.81:5.63; %N=10.31:10.13; %Cl-=4.35:4.22
Пример 15. Гидрохлорид 7-амино-N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Стадия A: Метил 3'-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5',6'-дигидро-8'Н-спиро[1,3-диоксолан-2,7'-индолизин]-1'-карбоксилат
Методика соответствует протоколу Стадии А Примера 1 с заменой соединения Синтеза 1 на соединение Синтеза 8.
Стадия В: Метил 3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-7-оксо-5,6,7,8-тетрагидроиндолизин-1-карбоксилат
4.47 ммоль соединения Стадии А, растворенного в 75 мл ТГФ, перемешивают в присутствии 37 мл 1М HCl при нагревании с обратным холодильником в течение 15 часов. К реакционной смеси добавляют 100 мл воды и 100 мл этилацетата. Затем добавляют 4 г NaHCO3 (4.7 ммоль) в виде порошка до основного значения рН. Соединение экстрагируют этилацетатом; органическую фазу сушат над MgSO4, фильтруют и концентрируют досуха.
Стадия С: Метил 7-гидрокси-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксилат
К раствору 4.47 ммоль соединения, полученного на Стадии В, в 30 мл метанола порциями добавляют 558 мг (14.75 ммоль) борогидрида натрия. Реакционную смесь перемешивают в течение 1 часа при температуре окружающей среды. Затем добавляют 50 мл 1М HCl и метанол упаривают. Водную фазу затем нейтрализуют, используя NaHCO3, и затем экстрагируют дихлорметаном. Органическую фазу последовательно промывают Н2О, сушат над MgSO4, фильтруют и концентрируют досуха. Полученное таким образом масло очищают с помощью флэш-хроматографии (градиент дихлорметан/этанол-аммиак) с получением ожидаемого продукта.
Стадия D: Метил 3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-7-(проп-2-ен-1-илокси)-5,6,7,8-тетрагидроиндолизин-1-карбоксилат
К суспензии 331 мг (8.26 ммоль) гидрида натрия в 15 мл безводного ТГФ, охлажденной до 0°С, добавляют 4.13 ммоль соединения, полученного на Стадии С. Полученную в результате суспензию перемешивают в течение 15 минут при 0°С и затем медленно добавляют (в течение 15 минут) раствор 790 мкл (9.1 ммоль) аллилбромида в 10 мл ТГФ. Реакционную смесь перемешивают в течение 1 часа при 0°С, и затем в течение 15 часов при температуре окружающей среды. Полученный в результате раствор гидролизуют насыщенным водным раствором NH4Cl. Соединение экстрагируют этилацетатом; органическую фазу сушат над MgSO4, фильтруют и концентрируют досуха. Полученное таким образом масло очищают с помощью флэш-хроматографии (градиент циклогексан/этилацетат) с получением ожидаемого продукта.
Стадия Е: N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-фенил-7-(проп-2-ен-1-илокси)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Методика соответствует способам, описанным на Стадиях В и С Примера 1, с использованием соответствующих реагентов.
Стадия F: N-(4-{[трет-Бутил(диметил)силил]окси}фенил)-7-гидрокси-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Затем осуществляют реакцию снятия защиты - аллильной группы в присутствии 1,3-диметилпиримидин-2,4,6(1H,3H,5H)-триона (также называемого диметилбарбитурат) и тетракис(трифенилфосфин)палладия в смеси метанола и дихлорметана.
Стадия G: 7-Азидо-N-(4-{[трет-бутил(диметил)силил]окси}фенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
К раствору соединения Стадии F (550 мг; 0.72 ммоль) в метиленхлориде (6 мл) добавляют, при температуре окружающей среды, триэтиламин (300 мкл; 1.8 ммоль) и мезилхлорид (0.14 мл; 1.8 ммоль). После перемешивания в течение 20 минут, реакционную смесь концентрируют досуха и затем разбавляют 10 мл ДМСО. К смеси добавляют 470 мг NaN3 в виде порошка (7.2 ммоль). Реакционную смесь выдерживают 20 часов при температуре окружающей среды и затем в течение 20 часов при 50°С. Ее затем вливают в смесь дихлорметана и воды. Органическую фазу промывают 3 раза водой и затем солевым раствором, сушат над MgSO4, и затем концентрируют досуха с получением ожидаемого продукта, который используют в таком виде на следующей стадии.
Стадия Н: Гидрохлорид 7-амино-N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
К раствору 550 мг соединения Стадии G (0.7 ммоль) в этаноле (10 мл) при температуре окружающей среды добавляют 20 мг Pd/C 10%. После перемешивания в течение 15 часов под давлением водорода 1 бар реакционную смесь пропускают через фильтр Ватмана и концентрируют досуха. После очистки с помощью колоночной хроматографии на силикагеле (градиент дихлорметан/метанол), твердое вещество затем растворяют в дихлорметане и добавляют 2 мл 1 н. HCl в эфире. Всю загрузку перемешивают в течение 1 часа и затем упаривают досуха. Полученный таким образом гидрохлорид растворяют в смеси вода/ацетонитрил до полного растворения и затем лиофилизуют с получением ожидаемого соединения в виде порошка.
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=69.17:68.68; %Н=5.51:5.09; %N=8.27:8.41; %Cl-=5.24:5.28
Пример 16. 3-(6-{[(3S)-3-(Гидроксиметил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(4-гидроксифенил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Стадия А: Метил 3-(6-{[(3S)-3-(гидроксиметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксилат
Методика соответствует способу Стадии А Примера 1 с использованием (3S)-1,2,3,4-тетрагидроизохинолин-3-илметанола.
Стадия В: Метил 3-(6-{[(3S)-3-[(проп-2-ен-1-илокси)метил]-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксилат
К суспензии NaH (703 мг; 17.6 ммоль) в ТГФ (20 мл) добавляют раствор 7.8 г соединения Стадии А (16 ммоль), растворенного в смеси ТГФ (50 мл) и ДМФА (30 мл). После перемешивания в течение 1 часа добавляют аллилбромид (1.7 мл; 19 ммоль). Реакционную смесь перемешивают в течение 48 часов при температуре окружающей среды и затем вливают в смесь этилацетата и воды. Органическую фазу промывают 3 раза водой и насыщенным раствором LiOH, сушат над MgSO4 и концентрируют досуха. После очистки с помощью хроматографии на силикагеле (градиент дихлорметан/метанол), ожидаемый продукт получают в виде твердого вещества.
1H ЯМР: δ: (500 МГц; ДМСО-d6; 300K): 7.2-6.9 (m, 4Н); 7.05 (m, 1Н); 6.9 (m, 1H); 6.45-6.1 (m, 1Н); 6.15 (m, 2Н); 5.9-5.65 (m, 1H); 5.2-5.0 (m, 2Н); 5.05-3.8 (m, 1Н); 4.85-4.25 (m, 2Н); 4.3-3.45 (m, 7Н); 3.4-2.4 (m, 6Н); 1.95-1.45 (m, 4Н)
Стадия С: N-[4-(Бензилокси)фенил]-N-фенил-3-(6-{[(3S)-3-[(проп-2-ен-1-илокси)метил]-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Методика соответствует способам Стадий В и С Примера 1 с использованием 4-(бензилокси)-N-фениланилина (см. Синтез 9'').
Стадия D: 3-(6-{[(3S)-3-(Гидроксиметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(4-гидроксифенил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
К суспензии 5.1 г (6.65 ммоль) соединения Стадии С в смеси дихлорметана (7 мл) и метанола (2 мл) добавляют диметилбарбитуровую кислоту (2.1 г; 13.3 ммоль) и тетракис(трифенилфосфин)палладий(0) (300 мг; 0.3 ммоль). После перемешивания в течение 15 часов при 45°С, реакционную смесь выливают на смесь этилацетата и воды. Органическую фазу дважды промывают водой, сушат над MgSO4, концентрируют досуха и разбавляют метанолом (5 мл). Загрузку затем перемешивают в течение 24 часов в атмосфере водорода в присутствии Pd/C (100 мг). Реакционную смесь затем пропускают через фильтр Ватмана, концентрируют досуха, затем хроматографируют на силикагеле (градиент дихлорметан/метанол) и в заключение лиофилизуют с получением ожидаемого продукта в виде порошка.
Элементный микроанализ: %, найдено (теоретический)
%С=72.38(73); %Н=5.22(5.5); %N=6.59(6.55)
Пример 17. Гидрохлорид N-{3-фтор-4-[2-(морфолин-4-ил)этокси]фенил}-N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=67.12:66.79; %Н=5.26:4.98; %N=6.96:7.17; %Cl-=4.4:4.77
Пример 18. Гидрохлорид 3-[6-(3,4-дигидроизохинолин-2(1H)-илкарбонил)-1,3-бензодиоксол-5-ил]-N-{3-фтор-4-[2-(морфолин-4-ил)этокси]фенил}-N-(4-гидроксифенил)индолизин-1-карбоксамида
Элементный микроанализ: %, найдено (теоретический)
%С=66.99(66.79); %Н=4.93(5.1); %N=7.11(7.08); %Сl-=4.46(4.48)
Пример 19. Гидрохлорид N-(4-гидроксифенил)-3-(5-метил-2-{[(3S)-3-[2-(морфолин-4-ил)этил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=72.26:72.51; %Н=6.48:6.13; %N=7.66:7.71; %Cl=4.85:4.95; %Cl-=4.85:4.64
Пример 20. Гидрохлорид N-(4-гидроксифенил)-3-(2-{[(3S)-3-[2-(морфолин-4-ил)этил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=72:71.11; %Н=6.32:5.94; %N=7.81:7.65; %Cl-=4.94:5.08
Пример 21. Гидрохлорид N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(пиридин-4-ил)индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=69.24:69.12; %Н=4.74:4.23; %N=8.5:8.45; %Cl-=5.38:5.2
Пример 22. Гидрохлорид N-(4-гидроксифенил)-6-(6-{[(3S)-3-[2-(морфолин-4-ил)этил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-фенилпирроло[1,2-а]пиримидин-8-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=68.11:66.66; %Н=5.32:4.93; %N=9.24:8.84; %Cl-=4.68:5.78
Масс-спектроскопия высокого разрешения (ESI+):
Эмпирическая формула: C43H39N5O6
[М+Н]+, рассчитано: 655.2915
[М+Н]+, найдено: 655.2915
Пример 23. Гидрохлорид N-(3-цианофенил)-N-(4-гидроксифенил)-3-(6{[(3S)-3-[2-(морфолин-4-ил)этил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=68.74:68.59; %Н=5.64:5.5; %N=8.91:8.98; %Cl-=4.51:4.48
Пример 24. Гидрохлорид N-(3-фторфенил)-N-(4-гидроксифенил)-3-(6-{[(3S)-3-[2-(морфолин-4-ил)этил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=67.81:67.45; %Н=5.69:5.61; %N=7.19:7.42; %Cl-=4.55:4.84
Пример 25. Гидрохлорид N-(3,4-дифторфенил)-N-(4-гидроксифенил)-3-(6-{[(3S)-3-[2-(морфолин-4-ил)этил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=66.28:66.56; %Н=5.44:5.25; %N=7.03:7.21; %Cl-=4.45:4.32
Пример 26. Гидрохлорид N-(3-фторфенил)-3-(6-{[(3S)-3-[2-(морфолин-4-ил)этил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=69.24:70.16; %Н=5.81:5.79; %N=7.34:7.47; %Cl-=4.64:4.58
Пример 27. Гидрохлорид 3-(5-хлор-2-{[(3S)-3-[2-(морфолин-4-ил)этил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(3-фторфенил)-N-(4-гидроксифенил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=67.1:67.68; %Н=5.63:5.4; %N=7.28:7.34; %Cl-=4.61:4.59
Пример 28. Гидрохлорид N-(4-гидроксифенил)-3-(5-метокси-2-{[(3S)-3-[2-(морфолин-4-ил)этил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=70.72:70.05; %Н=6.34:5.95; %N=7.5:7.33; %Cl-=4.74:4.74
Пример 29. Гидрохлорид N-(4-гидроксифенил)-3-(4-метокси-2-{[(3S)-3-[2-(морфолин-4-ил)этил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=70.72:68.96; %Н=6.34:5.78; %N=7.5:7.24; %Cl-=4.74:4.62
Масс-спектроскопия высокого разрешения (ESI+):
Эмпирическая формула: C44H46N4O5
[М+Н]+, рассчитано: 711.3546
[М+Н]+, найдено: 711.3540
Пример 30. Гидрохлорид N-{4-[(3,3-дифторпиперидин-1-ил)метил]фенил}-N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=68.31:69.12; %Н=5.22:4.93; %N=7.08:6.96; %Cl-=4.48:4.07
Пример 31. Гидрохлорид N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(хинолин-6-ил)индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=71.13:71.29; %Н=4.69:4.39; %N=7.9:8.14; %Cl-=5:4.5
Пример 32. Гидрохлорид N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(2-метилпиридин-4-ил)индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=69.59:69.81; %Н=4.94:4.53; %N=8.32:8.59; %Cl-=5.27:5.01
Пример 33. Гидрохлорид N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(1-метил-1H-пирроло[2,3-b]пиридин-5-ил)индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=69.14:70.09; %Н=4.81:4.55; %N=9.83:10.09; %Cl-=4.98:3.26
Пример 34. Гидрохлорид N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(пиридин-3-ил)индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=69.24:70.21; %Н=4.74:4.42; %N=8.5:8.51; %Cl-=5.38:3.33
Пример 35. Гидрохлорид N-{4-[2-(3,3-дифторпиперидин-1-ил)этил]фенил}-N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=68.61:67.96; %Н=5.38:5.14; %N=6.96:6.76; %Cl-=4.4:4.36
Пример 36. Гидрохлорид N-{4-[2-(3,3-дифторпирролидин-1-ил)этил]фенил}-N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=68.31:68.51; %Н=5.22:4.85; %N=7.08:6.83; %Cl-=4.48:4.48
Пример 37. Гидрохлорид 3-(6-{[(3S)-3-(2-аминоэтил)-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(4-гидроксифенил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Стадия А: Метил 3-(6-{[(3S)-3-{2-[(трет-бутоксикарбонил)амино]этил}-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксилат
К раствору 2 г соединения Синтеза 1 в 20 мл дихлорметана при температуре окружающей среды добавляют 5.5 мл N,N,N-триэтиламина (6.96 ммоль), соединение Синтеза 3' (6.96 ммоль), и затем 0.94 г гидроксибензотриазола (НОВТ) и 1.34 г 1-этил-3-(3'-диметиламинопропил)-карбодиимида (EDC) (6.96 ммоль). Затем реакционную смесь перемешивают при температуре окружающей среды в течение ночи, и затем вливают в раствор хлорида аммония и экстрагируют этилацетатом. Органическую фазу затем сушат над сульфатом магния, и затем фильтруют и упаривают досуха. Полученный таким образом сырой продукт затем очищают с помощью хроматографии на силикагеле (градиент гептан/AcOEt) с получением ожидаемого продукта.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 7.2-6.8 (m, 4Н, ароматические Н, Н тетрагидроизохинолин); 7.15-6.90 (m, 4Н, ароматические Н, тетрагидроизохинолин); 7.00-6.80 (m, 2Н, ароматические Н, бензодиоксол); 6.68+6.55+6.25 (m, 1Н, NH); 6.50-6.05 (m, 1Н, ароматический Н, тетрагидроиндолизин); 6.12 (m, 2Н, алифатические Н, OCH2O); 4.95+4.20+4.10 (m, 2Н, алифатические Н, CH2N тетрагидроизохинолин); 4.85+4.78+3.80 (m, 1Н, алифатический Н, СН тетрагидроизохинолин); 4.00-3.40 (m, 2Н, алифатические Н, CH2N тетрагидроиндолизин); 3.70-3.50 (m, 3Н, СООСН3); 2.95-2.45 (m, 2Н, алифатические Н, CH2NHBoc); 2.98-2.30 (m, 2Н, алифатические Н, СН2С тетрагидроиндолизин); 3.00+2.60+2.42 (m, 2Н, алифатические Н, СН2СН тетрагидроиндолизин); 1.95-1.40 (m, 4Н, алифатические Н, СН2СН2 тетрагидроиндолизин); 1.35-1.25 (m, 9Н, алифатические Н, tBu); 1.50-1.15 (m, 2Н, алифатические Н, CH2CH2NHBoc)
Стадия В: 3-(6-{[(3S)-3-{2-[(трет-Бутоксикарбонил)амино]этил}-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксилат лития
К раствору, содержащему 8.26 ммоль соединения Стадии А в 24 мл диоксана, добавляют раствор гидроксида лития (675 мг, 16.1 ммоль). Загрузку помещают в микроволновую печь при 140 Вт, 100°С на срок 2 часа 30 минут. Затем реакционную смесь фильтруют и упаривают. Полученное таким образом твердое вещество сушат при 40°С в печи в присутствии P2O5.
Стадия С: трет-Бутил (2-{(3S)-2-[(6-{1-[(4-{[трет-бутил(диметил)силил]окси}фенил)(фенил)карбамоил]-5,6,7,8-тетрагидроиндолизин-3-ил}-1,3-бензодиоксол-5-ил)карбонил]-1,2,3,4-тетрагидроизохинолин-3-ил}этил)карбамат
К раствору, содержащему 4.73 ммоль соединения Стадии В в 47 мл дихлорметана, по каплям добавляют 1.2 мл оксалилхлорида при 0°С. Реакционную смесь перемешивают при температуре окружающей среды в течение 11 часов и затем упаривают несколько раз совместно с дихлорметаном. Полученный таким образом продукт суспендируют в 37 мл дихлорметана и затем добавляют к раствору, содержащему 7.1 ммоль соединения, полученного в Синтезе 8'', в 10 мл дихлорметана в присутствии 0.6 мл пиридина (7.1 ммоль). Загрузку перемешивают при температуре окружающей среды в течение ночи.
Реакционную смесь концентрируют и очищают с помощью хроматографии на силикагеле (градиент дихлорметан/метанол) с получением ожидаемого продукта.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 7.0 (m, 11Н, ароматические Н, Ph + 4Н, тетрагидроизохинолин + 2Н, PhO); 6.80-6.65 (m, 2Н, ароматические Н, PhO); 6.95-6.85 (m, 2Н, ароматические Н, бензодиоксол); 6.70+6.40 (3tl, 1Н, NH); 6.10 (m, 2Н, алифатические Н, ОСН2О); 5.25-4.85 (m, 1Н, ароматический Н, тетрагидроиндолизин); 5.00+4.00 (m, 2Н, алифатические Н, CH2N тетрагидроизохинолин); 4.90-3.60 (m, 1Н, алифатический Н, СН тетрагидроизохинолин); 4.10-3.40 (m, 2Н, алифатические Н, CH2N тетрагидроиндолизин); 3.00-2.50 (m, 2Н, алифатические Н, СН2С тетрагидроиндолизин); 3.00+2.40 (m, 2Н, алифатические Н, СН2СН тетрагидроиндолизин); 3.00-2.50 (m, 2Н, алифатические Н, CH2NHBoc); 1.80-1.50 (m, 4Н, алифатические Н, СН2СН2 тетрагидроиндолизин); 1.50-1.30 (m, 2Н, алифатические Н, CH2CH2NHBoc); 1.35 (2s, 9Н, алифатические Н, tBu); 0.90 (s, 9Н, алифатические Н, tBu-Si); 0.10 (m, 6Н, алифатические Н, Me-Si)
Стадия D: Гидрохлорид 3-(6-{[(3S)-3-(2-аминоэтил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(4-гидроксифенил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
К раствору 800 мг (0.92 ммоль) соединения Стадии С в 10 мл метанола добавляют 258 мг (4.60 ммоль) KOH. После перемешивания в течение 3 часов при температуре окружающей среды реакционную смесь обрабатывают 4М раствором HCl в 6 мл диоксана. После перемешивания в течение 2 часов при температуре окружающей среды реакционную смесь концентрируют и обрабатывают насыщенным водным раствором NaHCO3 и экстрагируют метиленхлоридом. Органическую фазу затем сушат над сульфатом магния, и затем фильтруют и упаривают досуха. Полученный таким образом сырой продукт затем очищают с помощью хроматографии на силикагеле (градиент дихлорметан/метанол). Соединение затем растворяют в 5 мл дихлорметана, и добавляют 2.5 мл 1М HCl в эфире. Соединение отфильтровывают и сушат в вакууме. Ожидаемый продукт получают в виде пены.
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=69.51:69.53; %Н=5.69:5.27; %N=8.11:8.04; %Cl-=5.13:5.2
Масс-спектроскопия высокого разрешения (ESI+):
Эмпирическая формула: C40H38N4O5
[М+Н]+, рассчитано: 655.2915
[М+Н]+, найдено: 655.2915
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 9.55+9.45 (2s, 1Н, ОН); 7.80+7.75 (2s, 3Н, NH3+); 7.46-6.55 (m, 11Н, ароматические Н, Ph + 4Н, тетрагидроизохинолин + 2Н, PhO); 6.90-6.55 (m, 2Н, ароматические Н, PhO); 7.00-6.70 (несколько s, 2Н, ароматические Н, бензодиоксол); 5.35-5.00 (несколько s, 1Н, ароматический Н, тетрагидроиндолизин); 6.10 (несколько s, 2Н, алифатические Н, ОСН2О); 5.00-3.35 (несколько m, 4Н, алифатические Н, CH2N тетрагидроизохинолин + CH2N тетрагидроиндолизин); 4.85+4.75+3.60 (несколько m, 1Н, алифатический Н, СН тетрагидроизохинолин); 2.85-2.45 (несколько m, 2Н, алифатические Н, CH2NH2); 3.00-2.45 (несколько m, 2Н, алифатические Н, СН2С тетрагидроиндолизин); 3.05+2.30 (несколько m, 2Н, алифатические Н, СН2СН тетрагидроизохинолин); 1.85-1.40 (несколько m, 2Н, алифатические Н, СН2 тетрагидроизохинолин); 1.95-1.35 (несколько m, 2Н, алифатические Н, СН2 тетрагидроизохинолин); 1.75-1.40 (несколько m, 2Н, алифатические Н, CH2CH2NH2)
ИК: ν: -ОН: 3375 см-1 (фенол); ν: -NH3+: 3500-2300 см-1 (соль первичного амина), ν: >С=O 1612 см-1 + плечо (амид)
Пример 38. Гидрохлорид N-(4-гидроксифенил)-3-(6-{[(3R)-3-[3-(морфолин-4-ил)пропил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=69.71:69.62; %Н=6.11:5.67; %N=7.23:7.12; %Cl-=4.57:4.81
Пример 39. Гидрохлорид N-(2,6-диметилпиридин-4-ил)-N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=69.91:69.68; %Н=5.13:4.78; %N=8.15:8.03; %Cl-=5.16:5.16
Пример 40. N-(4-Гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=74.86:74.88; %Н=5.64:5.31; %N=6.72:6.78
Пример 41. Гидрохлорид 3-(6-{[(3S)-3-[2-(3,3-дифторпиперидин-1-ил)этил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(4-гидроксифенил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=67.96:68.34; %Н=5.7:5.4; %N=7.04:6.97; %Cl-=4.46:4.27
Пример 42. Гидрохлорид N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(пиридин-4-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=68.82:69.46; %Н=5.32:4.95; %N=8.45:8.48; %Cl-=5.35:4.6
Пример 43. 3-(6-{[(3S)-3-{2-[(2,2-Дифторэтил)амино]этил}-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(4-гидроксифенил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Стадия А: Этил 3-(6-{[(3S)-3-{2-[(трет-бутоксикарбонил)амино]этил}-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксилат
Способ аналогичен способу, описанному на Стадии А Примера 37.
Стадия В: Этил 3-(6-{[(3S)-3-{2-[(трет-бутоксикарбонил)(2,2-дифторэтил)амино]этил}-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксилат
К суспензии 337 мг NaH (60%) (8.41 ммоль) в 13 мл диметилформамида по каплям добавляют раствор 1.01 г (1.68 ммоль) соединения Стадии А в 13 мл диметилформамида. Полученную в результате суспензию перемешивают при температуре окружающей среды в течение 15 минут и затем добавляют 1.08 г (5.04 ммоль) 2,2-дифторэтилтрифторметансульфоната в 13 мл диметилформамида. Загрузку перемешивают при температуре окружающей среды в течение 2 часов. Добавляют 20 мл насыщенного раствора хлорида аммония. Раствор экстрагируют этилацетатом. Органическую фазу затем сушат над MgSO4, фильтруют и концентрируют досуха. После очистки с помощью колоночной хроматографии на силикагеле (циклогексан/этилацетат), ожидаемый продукт получают в виде масла.
Масс-спектроскопия высокого разрешения (ESI+):
Эмпирическая формула: C37H43CN3O7
[М+Н]+, рассчитано: 680.3142
[М+Н]+, найдено: 680.3145
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 7.25-6.90 (m, 4Н, ароматические Н, тетрагидроизохинолин); 7.10-6.75 (m, 2Н, ароматические Н, бензодиоксол);.40-6.05 (m, 1Н, ароматический Н, тетрагидроиндолизин); 6.10 (m, 2Н, алифатические Н, OCH2O); 6.25-5.90 (m, 1Н, алифатический Н, CHF2); 4.95-4.10 (m, 2Н, алифатические Н, CH2N тетрагидроизохинолин); 4.80+3.80 (2m, 1Н, алифатический Н, СН тетрагидроизохинолин); 4.10-4.00 (m, 2Н, СН2 Et); 4.05-3.40 (m, 2Н, алифатические Н, CH2N тетрагидроиндолизин); 3.60-2.60 (m, 4Н, алифатические Н, CH2CHF2 + CH2NBoc); 3.00-2.35 (m, 2Н, алифатические Н, СН2С тетрагидроиндолизин); 3.00+2.45 (m, 2Н, алифатические Н, СН2СН тетрагидроизохинолин); 1.95+1.40 (m, 4Н, алифатические Н, СН2СН2 тетрагидроиндолизин); 1.40 (m, 9Н, алифатические Н, tBu); 1.65-1.20 (m, 2Н, алифатические Н, CH2CH2NBoc); 1.18+1.10 (2t, 3Н, алифатические Н СН3Et)
Стадия С: трет-Бутил (2-{(3S)-2-[(6-{1-[(4-{[трет-бутил(диметил)силил]окси}фенил)(фенил)карбамоил]-5,6,7,8-тетрагидроиндолизин-3-ил}-1,3-бензодиоксол-5-ил)карбонил]-1,2,3,4-тетрагидроизохинолин-3-ил}этил)(2,2-дифторэтил)карбамат
Способ аналогичен способу, описанному на Стадиях В и С Примера 37.
1Н ЯМР: δ (400 МГц; ДМСО-d6; 300K): 7.30-6.60 (m, 9Н, ароматические Н, 4Н тетрагидроизохинолин + Ph); 6.90-6.70 (m, 2Н, ароматические Н, бензодиоксол); 6.80-6.60 (m, 4Н, PhO); 6.10 (m, 2Н, алифатические Н, OCH2O); 6.20-5.90 (m, 1Н, алифатический Н, CHF2); 5.50-4.80 (4s, 1Н, ароматический Н, тетрагидроиндолизин); 5.20-4.00 (m, 2Н, алифатические Н, CH2N тетрагидроизохинолин); 4.80+4.70+3.50 (3m, 1Н, алифатический Н, СН тетрагидроизохинолин); 4.20-3.40 (m, 2Н, алифатические Н, CH2N тетрагидроиндолизин); 3.60-3.10 (m, 4Н, алифатические Н, CH2CHF2 + CH2NBoc); 3.00+2.60 (m, 2Н, алифатические Н, СН2СН тетрагидроизохинолин); 3.00-2.50 (m, 2Н, алифатические Н, СН2С тетрагидроиндолизин); 1.80+1.50 (m, 4Н, алифатические Н, СН2СН2 тетрагидроиндолизин); 1.60-1.30 (m, 2Н, алифатические Н, CH2CH2NBoc); 1.40-1.30 (m, 9Н, алифатические Н, tBu); 0.90 (4s, 9Н, алифатические Н, tBu-Si); 0.10 (4s, 6Н, алифатические Н, Me-Si)
Стадия D: 3-(6-{[(3S)-3-{2-[(2,2-Дифторэтил)амино]этил}-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(4-гидроксифенил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
К раствору 933 мг (1.00 ммоль) соединения Стадии С в 10 мл метанола добавляют 280 мг (5.00 ммоль) KOH. После перемешивания в течение 3 часов при температуре окружающей среды реакционную смесь обрабатывают 4М раствором HCl в 6 мл диоксана. После перемешивания в течение 2 часов при температуре окружающей среды реакционную смесь концентрируют и обрабатывают водным насыщенным раствором NaHCO3 и затем экстрагируют метиленхлоридом. Органическую фазу затем сушат над сульфатом магния, и затем фильтруют и упаривают досуха. Полученный таким образом сырой продукт затем очищают с помощью хроматографии на силикагеле (градиент дихлорметан/метанол) с получением ожидаемого продукта в виде пены.
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=70.18:69.79; %Н=5.61:5.67; %N=7.79:7.7
Масс-спектроскопия высокого разрешения (ESI+):
Эмпирическая формула: C42H40F2N4O5
[М+Н]+, рассчитано: 655.2915
[М+Н]+, найдено: 655.2915
Пример 44. N-(4-Гидроксифенил)-3-(6-{[(3S)-3-[2-(3-метоксиазетидин-1-ил)этил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=72.91:72.73; %Н=6.12:5.67; %N=7.73:7.74
Пример 45. Гидрохлорид N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-{1-[2-(морфолин-4-ил)этил]-1H-пиразол-4-ил}индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=66.27:66.05; %Н=5.43:5.27; %N=11.04:11.07; %Cl-=4.66:4.61
Пример 46. Гидрохлорид N-(3-фторпиридин-4-ил)-N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-карбоксамида
Масс-спектроскопия высокого разрешения (ESI+):
Эмпирическая формула: C38H29FN4O5
[М+Н]+, рассчитано: 641.2195
[М+Н]+, найдено: 641.2195
Пример 47. 3-(5-Хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-гидроксифенил)-N-(1-метил-1H-пиразол-4-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=69.72:69.53; %Н=5.53:5.6; %N=11.29:10.85
Пример 48. N-(4-Гидроксифенил)-3-(7-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-2,3-дигидро-1,4-бензодиоксин-6-ил)-N-(1-метил-1H-пиразол-4-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=70.9:70.89; %Н=5.79:5.56; %N=10.88:10.8
Пример 49. Гидрохлорид 3-(5-хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-гидроксифенил)-N-(пиридин-4-ил)индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=68.42:68.17; %Н=4.65:4.48; %N=8.63:8.48; %Cl-=5.46:5.13
Пример 50. 3-(5-Хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-гидроксифенил)-N-(1-метил-1H-пирроло[2,3-b]пиридин-5-ил)индолизин-1-карбоксамид
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=72.12:71.58; %Н=4.84:4.84; %N=10.51:10.48
Пример 51. Гидрохлорид N-(4-гидроксифенил)-N-(имидазо[1,2-а]пиридин-7-ил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=68.81:68.28; %Н=4.62:4.59; %N=10.03:9.66; %Cl-=5.08:4.81
Пример 52. N-(4-Гидроксифенил)-3-(2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(1-метил-1H-пирроло[2,3-b]пиридин-5-ил)индолизин-1-карбоксамид
Элементный микроанализ: (%, теоретическое значение : найдено)
%C=76.05:75.88; %Н=5.26:5.24; %N=11.09:11.09
Пример 53. Гидрохлорид N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(2-метилимидазо[1,2-а]пиридин-7-ил)индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=69.14:69.65; %Н=4.81:4.75; %N=9.83:9.79; %Cl-=4.98:4.7
Пример 54. Гидрохлорид N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(6-метилпиридин-3-ил)индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%069.59:68.78; %Н=4.94:5; %N=8.32:8.33; %Cl-=5.27:5.18
Пример 55. N-(5-Фторпиридин-3-ил)-N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-карбоксамид
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=71.24:70.77; %Н=4.56:4.36; %N=8.75:8.82
Пример 56. N-(4-Гидроксифенил)-N-(2-метоксипиридин-4-ил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-карбоксамид
Масс-спектроскопия высокого разрешения (ESI+):
Эмпирическая формула: C39H32N4O6
[М+Н]+, рассчитано: 653.2395
[М+Н]+, найдено: 653.2385
Пример 57. 3-[6-(3,4-Дигидроизохинолин-2(1Н)-илкарбонил)-1,3-бензодиоксол-5-ил]-N-(4-гидроксифенил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Элементный микроанализ: %, найдено (теоретический)
%C=74.17(74.62); %Н=5.43(5.44); %N=6.87(6.87)
Пример 58. Гидрохлорид N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-[2-(пропан-2-ил)пиридин-4-ил]индолизин-1-карбоксамида
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=70.23:69.95; %Н=5.32:5.4; %N=7.99:7.99; %Cl-=5.06:4.92
Пример 59. N-(4-Гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(пиразоло[1,5-а]пиримидин-6-ил)индолизин-1-карбоксамид
Элементный микроанализ: (%, теоретическое значение : найдено)
%С=70.68:70.47; %Н=4.56:4.61; %N=12.68:12.45
Пример 60. 3-(5-Фтор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-гидроксифенил)-N-(1-метил-1H-пиразол-4-ил)индолизин-1-карбоксамид
Элементный микроанализ: %, найдено (теоретический)
%C=71.85(72.11); %Н=4.78(5.04); %N=10.79(11.68)
Пример 61. 3-(5-Фтор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-гидроксифенил)-N-(1-метил-1H-пиразол-4-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Элементный микроанализ: %, найдено (теоретический)
%С=72.31(71.62); %Н=5.6(5.68); %N=10.94(11.6)
Пример 62. 3-(5-Фтор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-гидроксифенил)-N-(пиридин-4-ил)индолизин-1-карбоксамид
Элементный микроанализ: %, найдено (теоретический)
%С=74.08(74.48); %Н=4.82(4.9); %N=8.59(9.39)
Пример 63. 3-(5-Фтор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-гидроксифенил)-N-метил-1H-пирроло[2,3-b]пиридин-5-ил)индолизин-1-карбоксамид
Элементный микроанализ: %, найдено (теоретический)
%С=73.14(73.95); %Н=4.83(4.96); %N=10.29(10.78)
Пример 64. 3-(5-Фтор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-гидроксифенил)-N-(пиридин-4-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Элементный микроанализ: %, найдено (теоретический)
%С=74.61(73.98); %Н=5.26(5.54); %N=8.94(9.33)
Пример 65. 3-(5-Фтор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-гидроксифенил)-N-(1-метил-1H-пирроло[2,3-b]пиридин-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
Элементный микроанализ: %, найдено (теоретический)
%C=73.59(73.49); %H=5.22(5.55); %N=9.93(10.71)
Пример 66. N-(4-Гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-([1,2,4]триазоло[1,5-а]пиримидин-6-ил)индолизин-1-карбоксамид
Элементный микроанализ: %, найдено (теоретический)
%С=68.57(68.77); %Н=3.92(4.4); %N=14.21(14.77)
Масс-спектроскопия высокого разрешения (ESI+):
Эмпирическая формула: C38H29N7O5
[М+Н]+, рассчитано: 664.2303
[М+Н]+, найдено: 664.2310
Пример 67. N-(4-Гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(1-оксидопиридин-4-ил)индолизин-1-карбоксамид
Элементный микроанализ: %, найдено (теоретический)
%С=69.7(71.46); %%Н=4.43(4.73); %N=8.54(8.77)
Масс-спектроскопия высокого разрешения (ESI+):
Эмпирическая формула: C38H30N4O6
[М+Н]+, рассчитано: 639.2238
[М+Н]+, найдено: 639.2234
Пример 68. Гидрохлорид N-(4-гидроксифенил)-3-(2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(пиридин-4-ил)индолизин-1-карбоксамида
Элементный микроанализ: %, найдено (теоретический)
%С=71.97(72.25); %Н=5.21(5.08); %N=8.99(9.11); %Cl-=5.32(5.76)
Масс-спектроскопия высокого разрешения (ESI+):
Эмпирическая формула: C37H30N4O3
[М+Н]+, рассчитано: 579.2391
[М+Н]+, найдено: 579.2403
Пример 69. Гидрохлорид N-(4-гидроксифенил)-N-(1-метил-1H-пирроло[2,3-b]пиридин-5-ил)-3-(6-{[(3R)-3-[3-(морфолин-4-ил)пропил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамида
Элементный микроанализ: %, найдено (теоретический)
%С=67.63(68.06); %Н=5.27(5.95); %N=10.08(10.13); %Cl-=4.53(4.27)
Масс-спектроскопия высокого разрешения (ESI+):
Эмпирическая формула: C47H48N6O6
[М+Н]+, рассчитано: 793.3708
[М+Н]+, найдено: 793.3704
Пример 70. N-(4-Гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(1-метил-1H-пиразоло[3,4-b]пиридин-5-ил)индолизин-1-карбоксамид
Стадия А: N-[4-[трет-Бутил(диметил)силил]оксифенил]-3-[6-[(3R)-3-метил-3,4-дигидро-1Н-изохинолин-2-карбонил]-1,3-бензодиоксол-5-ил]-N-(1-метил-1Н-пиразоло[3,4-b]пиридин-5-ил)индолизин-1-карбоксамид
Указанный в заголовке продукт получают в соответствии со способом Стадии А Примера 86 с заменой соединения Синтеза 36'' на соединение Синтеза 35''.
ЖХ-МС: [М+Н]+ = 791.4 в сравн. с рассчитанным значением 791.3
Стадия В: N-(4-Гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(1-метил-1Н-пиразоло[3,4-b]пиридин-5-ил)индолизин-1-карбоксамид
Методика соответствует протоколу, аналогичному описанному на Стадии D Примера 1. Полученный таким образом продукт подвергают стадии превращения в соль в присутствии HCl в эфире.
ИК (ATR) см-1: 2500-3000 ν -ОН, 1614 ν >С=O амиды, 1236 ν >С-O-С<,
740 γ >СН-Ar
Элементный микроанализ: %, найдено (теоретический)
%С=71.07(70.99); %Н=4.45(4.77); %N=12.37(12.42)
Масс-спектроскопия высокого разрешения (ESI+):
Эмпирическая формула: C40H32N6O5
[М+Н]+, рассчитано: 677.2507
[М+Н]+, найдено: 677.2510
Пример 71. Хлорид 4-[(4-гидроксифенил){[3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-ил]карбонил}амино]-1-метилпиридиния
Стадия А: Йодид 4-[(4-гидроксифенил){[3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-ил]карбонил}амино]-1-метилпиридиния
Соединение Примера 21 (311 мг, 0.5 ммоль) растворяют в дихлорметане и промывают насыщенным водным раствором гидрокарбоната натрия. После сушки органической фазы над сульфатом магния и упаривания досуха, остаток растворяют в этаноле (30 мл). Затем добавляют метилйодид (45 мкл, 0.7 ммоль) и реакционную смесь нагревают до 40°С. Таким образом полученный раствор упаривают досуха. Сырой реакционный продукт очищают на колонке с силикагелем, используя дихлорметан и метанол в качестве растворителей. Соединение получают в виде белого порошка, который используют непосредственно на следующей стадии.
1Н ЯМР (500 МГц, ДМСО-d6) δ м.д.: 9.95 (bs, 1Н), 8.6-8.45 (m, 2Н), 8.35-8.05 (несколько m, 1Н), 8.3-8 (несколько m, 1Н), 7.45-6.7 (несколько m, 8Н), 7.4-6.9 (несколько m, 4Н), 6.45-6.3 (несколько s, 1Н), 6.45-6.3 (m, 2Н), 6.15 (s, 2Н), 5.05-3.55 (несколько d, 2Н), 4.75/3.8 (m+m, 1Н), 4.15 (2*s, 3Н), 2.95-2.1 (несколько m, 2Н), 1-0.15 (несколько m, 3Н)
Стадия В: Хлорид 4-[(4-гидроксифенил){[3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-ил]карбонил}амино]-1-метилпиридиния
Соединение предшествующей стадии (320 мг, 0.42 ммоль) растворяют в метаноле (20 мл), и затем порциями добавляют карбонат серебра (173 мг, 0.628 ммоль) в течение 10 минут. Полученную в результате суспензию перемешивают в течение 1 часа при температуре окружающей среды; осадок отфильтровывают и промывают метанолом. Фильтрат концентрируют досуха, и затем обрабатывают 50 мл 2 н. раствора соляной кислоты, нагревают при 60°С в течение 30 минут и затем упаривают досуха. Конечный продукт получают после очистки на колонке с силикагелем С18, используя 0.1% раствор соляной кислоты и ацетонитрил в качестве растворителей. Указанное в заголовке соединение получают в виде белого порошка, который лиофилизуют из смеси вода/ацетонитрил.
ИК (ATR) см-1: 3388 ν -ОН фенол, 1650+1627 ν >С=O амиды
Масс-спектроскопия высокого разрешения (ESI+):
Эмпирическая формула: C39H33N4O5
[М]+, рассчитано = 637.2445.
[М]+, найдено = 637.2431
Соединения Примеров 72, 73, 77, 78-80, 84 и 85 синтезируют в соответствии со способом Примера 3, используя кислоту Синтеза 7, соответствующий 1,2,3,4-тетрагидроизохинолин или соответствующее соединение, полученное в соответствии с одним из Синтезов 1'-7', и подходящий амин NHR3R4.
Пример 72. N-(4-Гидроксифенил)-N-метил-6-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-8-карбоксамид
ЖХ/МС (C33H32N4O5) 565 [М+Н]+; RT 1.47 (Метод В), при этом предполагается, что RT означает время удержания
Пример 73. N-Этил-N-(4-гидроксифенил)-6-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-1,2,3,4-тетрагидропирроло[1,2-a]пиразин-8-карбоксамид
ЖХ/МС (C34H34N4O5) 579 [М+Н]+; RT 1.55 (Метод В)
Пример 74. 3-[6-(3,4-Дигидроизохинолин-2(1H)-илкарбонил)-1,3-бензодиоксол-5-ил]-N-(4-гидроксифенил)-N-метил-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
ЖХ/МС (C33H31N3O5) 550 [М+Н]+; RT 1.24 (Метод В)
Пример 75. 3-[6-(3,4-Дигидроизохинолин-2(1H)-илкарбонил)-1,3-бензодиоксол-5-ил]-N-этил-N-(4-гидроксифенил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
ЖХ/МС (C34H33N3O5) 564 [М+Н]+; RT 1.30 (Метод В)
Пример 76. N-Бутил-3-[6-(3,4-дигидроизохинолин-2(1H)-илкарбонил)-1,3-бензодиоксол-5-ил]-N-(4-гидроксифенил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
ЖХ/МС (C36H37N3O5) 592 [М+Н]+; RT 1.39 (Метод В)
Пример 77. N-Этил-N-(4-гидроксифенил)-6-(6-{[(3S)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-1,2,3,4-тетрагидропирроло[1,2-a]пиразин-8-карбоксамид
ЖХ/МС (C34H34N4O5) 579 [М+Н]+; RT 1.50 (Метод В)
Пример 78. N,N-Дибутил-6-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-8-карбоксамид
ЖХ/МС (C34H42N4O4) 571 [М+Н]+; RT 1.79 (Метод В)
Пример 79. N-Бутил-N-(4-гидроксифенил)-6-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-1,2,3,4-тетрагидропирроло[1,2-a]пиразин-8-карбоксамид
ЖХ/МС (C36H38N4O5) 607 [М+Н]+; RT 1.65 (Метод В)
Пример 80. N-(4-Гидроксифенил)-6-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(пропан-2-ил)-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-8-карбоксамид
ЖХ/МС (C35H36N4O5) 593 [М+Н]+; RT 1.58 (Метод В)
Пример 81. N-(4-Гидроксифенил)-N-метил-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
ЖХ/МС (C34H33N3O5) 564 [М+Н]+; RT 2.48 (Метод А)
Пример 82. N-(4-Гидроксифенил)-N-метил-3-(6-{[(3S)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид
ЖХ/МС (C34H33N3O5) 564 [М+Н]+; RT 2.55 (Метод А)
Пример 83. 3-[6-(3,4-Дигидроизохинолин-2(1H)-илкарбонил)-1,3-бензодиоксол-5-ил]-N-(4-гидроксифенил)-N-метилиндолизин-1-карбоксамид
ЖХ/МС (C33H27N3O5) 546 [М+Н]+; RT 2.40 (Метод А)
Пример 84. 6-[6-(3,4-Дигидроизохинолин-2(1H)-илкарбонил)-1,3-бензодиоксол-5-ил]-N-(4-гидроксифенил)-N-метил-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-8-карбоксамид
ЖХ/МС (C32H30N4O5) 551 [М+Н]+; RT 1.45 (Метод В)
Пример 85. 6-[6-(3,4-Дигидроизохинолин-2(1H)-илкарбонил)-1,3-бензодиоксол-5-ил]-N-этил-N-(4-гидроксифенил)-1,2,3,4-тетрагидропирроло[1,2-a]пиразин-8-карбоксамид
ЖХ/МС (C33H32N4O5) 565 [М+Н]+; RT 1.49 (Метод В)
Пример 86. Гидрохлорид N-(4-гидроксифенил)-3-[6-[(3R)-3-метил-3,4-дигидро-1H-изохинолин-2-карбонил]-1,3-бензодиоксол-5-ил]-N-(3-метилпиразоло[1,5-а]пиримидин-6-ил)индолизин-1-карбоксамида
Стадия A: N-[4-[трет-Бутил(диметил)силил]оксифенил]-3-[6-[(3R)-3-метил-3,4-дигидро-1Н-изохинолин-2-карбонил]-1,3-бензодиоксол-5-ил]-N-(3-метилпиразоло[1,5-а]пиримидин-6-ил)индолизин-1-карбоксамид
К раствору 0.6 г 3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-карбоновой кислоты (1.3 ммоль) в 6 мл дихлорэтана добавляют 0.18 мл 1-хлор-N,N,2-триметилпроп-1-ен-1-амина (2 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 2 часов и затем добавляют 0.8 г соединения Синтеза 36'' (2.2 ммоль). Загрузку нагревают с обратным холодильником в течение 20 часов и затем охлаждают и разбавляют смесью дихлорметана и насыщенного раствора NaHCO3. После разделения фаз, органическую фазу сушат над MgSO4 и концентрируют досуха. Полученный таким образом сырой продукт очищают с помощью хроматографии на силикагеле (градиент дихлорметан/метанол).
ЖХ/МС: [М+Н]+ = 791.4 в сравн. с рассчитанным значением 791.3
Стадия В: Гидрохлорид N-(4-гидроксифенил)-3-[6-[(3R)-3-метил-3,4-дигидро-1Н-изохинолин-2-карбонил]-1,3-бензодиоксол-5-ил]-N-(3-метилпиразоло[1,5-а]пиримидин-6-ил)индолизин-1-карбоксамида
Методика соответствует протоколу, аналогичному описанному на Стадии D Примера 1. Полученный таким образом продукт подвергают стадии превращения в соль в присутствии HCl в эфире.
ИК (ATR) см-1: 2500-3000 ν -ОН, 1614 ν >С=O амиды, 1236 ν >С-O-С<,
740 γ >СН-Ar
Масс-спектроскопия высокого разрешения (ESI+):
Эмпирическая формула: C40H32N6O5
[М+Н]+, рассчитано: 677.2507
[М+Н]+, найдено: 677.2506
ФАРМАКОЛОГИЧЕСКОЕ ИССЛЕДОВАНИЕ
ПРИМЕР А: Ингибирование Bcl-2 с использованием методики поляризации флуоресценции
Исследования с применением поляризации флуоресценции осуществляют на микропланшетах (384 луночных). Меченый белок Bcl-2 (histag-Bcl-2 такой, что Bcl-2 соответствует UniProtKB® первичному номеру доступа: Р10415), в конечной концентрации 2.50×10-8 М, смешивают с флуоресцентным пептидом (Fluorescein-REIGAQLRRMADDLNAQY), в конечной концентрации 1.00×10-8 М в буферном растворе (Hepes 10 мМ, NaCl 150 мМ, Tween20 0.05%, рН 7.4), в присутствии или отсутствии возрастающих концентраций исследуемых соединений. После инкубирования в течение 2 часов измеряют поляризацию флуоресценции.
Результаты выражены в IC50 (концентрация соединения, которая ингибирует поляризацию флуоресценции на 50%) и приведены в таблице 1 ниже.
Результаты показывают, что соединения изобретения ингибируют взаимодействие между белком Bcl-2 и флуоресцентным пептидом, описанным выше.
ПРИМЕР В: In vitro цитотоксичность
Исследования цитотоксичности осуществляют на линии клеток лейкозной опухоли RS4;11.
Клетки распределяют на микропланшетах и подвергают воздействию исследуемых соединений в течение 48 часов. Жизнеспособность клеток затем определяют количественно путем колориметрического анализа, анализа микрокультур с использованием тетразолия (Cancer Res., 1987, 47, 939-942).
Результаты выражены в IC50 (концентрация соединения, которая ингибирует жизнеспособность клеток на 50%) и приведены в таблице 1 ниже.
Результаты показывают, что соединения изобретения являются цитотоксичными.
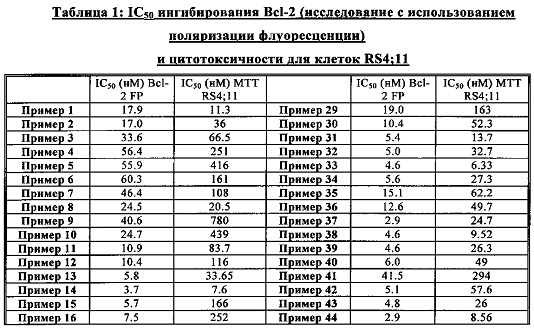
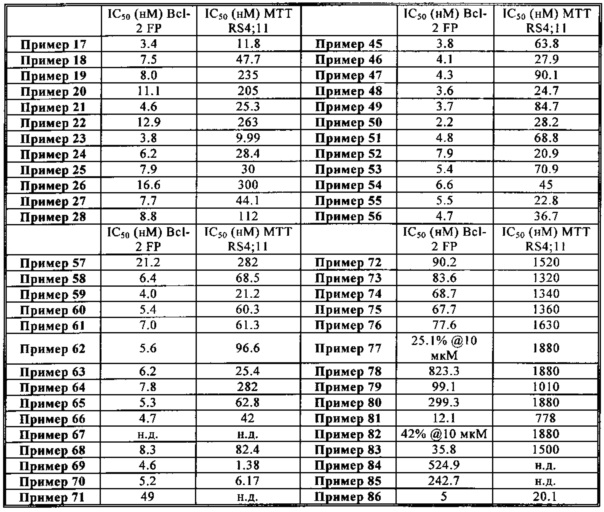
н.д.: не определено
Для частичных ингибиторов указано процентное значение ингибирования поляризации флуоресценции для приведенной концентрации исследуемого соединения. Соответственно, 25.1% @10 мкМ означает, что 25.1%-ное ингибирование поляризации флуоресценции наблюдается для концентрации исследуемого соединения, равной 10 мкМ.
ПРИМЕР С: Индукция активности каспазы in vivo.
Способность соединений изобретения активировать каспазу 3 оценивают на ксенотрансплантатной модели лейкозных клеток RS4;11.
1×107 Клеток RS4;11 трансплантируют под кожу иммуносупрессивным мышам (штамм SCID). Через 25-30 дней после трансплантации животные перорально получают лечение различными соединениями. Через шестнадцать часов после введения, опухолевые массы извлекают и лизируют, и в опухолевых лизатах измеряют активность каспазы 3.
Данное ферментативное измерение осуществляют путем анализа появления флуоригенного продукта расщепления (DEVD-азная активность, Promega). Оно выражается в виде фактора активации, который равняется соотношению между двумя активностями каспазы: активности у мышей, получающих лечение, деленной на активность у контрольных мышей.
Полученные результаты показывают, что соединения изобретения способны индуцировать апоптоз in vivo в опухолевых клетках RS4;11.
ПРИМЕР D: Количественное определение расщепленной формы каспазы 3 in vivo.
Способность соединений изобретения активировать каспазу 3 оценивают на ксенотрансплантатной модели лейкозных клеток RS4;11.
1×107 Клеток RS4; 11 трансплантируют под кожу иммуносупрессивным мышам (штамм SCID). Через 25-30 дней после трансплантации животные перорально получают лечение различными соединениями. После лечения, опухолевые массы извлекают (после периода времени Т) и лизируют, и количественно определяют расщепленную (активированную) форму каспазы 3 в опухолевых лизатах.
Количественное определение осуществляют с использованием исследования "Meso Scale Discovery (MSD) ELISA platform", которое предназначено для проведения специфического анализа на расщепленную форму каспазы 3. Оно выражается в виде фактора активации, который равняется соотношению между количеством расщепленной каспазы 3 у мышей, получающих лечение, и количеством расщепленной каспазы 3 у контрольных мышей.
Результаты показывают, что соединения изобретения способны индуцировать апоптоз in vivo в опухолевых клетках RS4;11.


ПРИМЕР Е: Антиопухолевая активность in vivo
Антиопухолевую активность соединений изобретения оценивают на ксенотрансплантатной модели лейкозных клеток RS4;11.
1×107 Клеток RS4;11 трансплантируют под кожу иммуносупрессивным мышам (штамм SCID). Через 25-30 дней после трансплантации, когда опухолевая масса достигнет приблизительно 150 мм3, мышей лечат пероральным путем различными соединениями в двух различных режимах (ежедневное введение в течение пяти дней в неделю в течение двух недель, или два введения в неделю в течение двух недель). Опухолевую массу измеряют дважды в неделю с начала лечения.
Полученные результаты, соответственно, показывают, что соединения изобретения способны индуцировать значительную регрессию опухолей в течение периода лечения.
ПРИМЕР Е: Фармацевтическая композиция: Таблетки
Соединение, выбранное из Примеров 1-86,
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ИНДОЛИЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2013 |
|
RU2646223C2 |
| НОВЫЕ ФОСФАТНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2617682C2 |
| НОВЫЕ ИЗОИНДОЛИНОВЫЕ ИЛИ ИЗОХИНОЛИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2689305C2 |
| НОВЫЕ ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2607788C2 |
| НОВЫЕ ИНДОЛЬНЫЕ И ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2693404C2 |
| НОВЫЕ МАКРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2018 |
|
RU2808689C2 |
| НОВЫЕ (ГЕТЕРО)АРИЛ-ЗАМЕЩЕННЫЕ ПИПЕРИДИНИЛЬНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2017 |
|
RU2742271C2 |
| ЗАМЕЩЕННЫЕ [1,2,4]ТРИАЗОЛО[1,5-a]ПИРИМИДИН-7-ИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE2 | 2015 |
|
RU2659070C9 |
| НОВЫЕ ПИПЕРИДИНИЛЬНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2017 |
|
RU2743163C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
Изобретение относится к области органической химии, а именно к гетероциклическому производному формулы (I) и к его энантиомеру и диастереоизомеру, к его соли присоединения с фармацевтически приемлемой кислотой, где X и Y представляют собой атом углерода или атом азота, при этом они не могут одновременно представлять собой два атома углерода или два атома азота; группа  представляет собой одну из следующих групп: 5,6,7,8-тетрагидроиндолизин, необязательно замещенный аминогруппой; индолизин; 1,2,3,4-тетрагидропирроло[1,2-а]пиразин, необязательно замещенный метилом; пирроло[1,2-а]пиримидин; Т представляет собой атом водорода, линейную или разветвленную (С1-С6)алкильную группу, группу (С2-С4)алкил-NR1R2, или группу (С1-С4)алкил-OR6; R1 и R2 независимо друг от друга представляют собой атом водорода или линейную или разветвленную (С1-С6)алкильную группу; или R1 и R2 вместе с атомом азота, несущим их, образуют гетероциклоалкил; R3 представляет собой линейную (С1-С6)алкильную, арильную или гетероарильную группу, причем последние две группы могут быть замещены 1-2 группами, выбранными из галогена, линейного или разветвленного (С1-С6)алкила, линейного или разветвленного (С1-С6)алкокси, циано и N-оксида; R4 представляет собой 4-гидроксифенильную группу; R5 представляет собой атом водорода; R6 представляет собой атом водорода; Ra и Rd каждый представляет собой атом водорода и (Rb,Rc), вместе с атомами углерода, несущими их, образуют 1,3-диоксолановую группу или 1,4-диоксановую группу; или Ra, Rc и Rd каждый представляет собой атом водорода и Rb представляет собой водород, галоген, метил или метокси; или Ra, Rb и Rd каждый представляет собой атом водорода и Rc представляет собой гидрокси или метокси группу; причем гетероциклоалкильные группы, определенные таким образом, и алкильные и алкокси группы могут быть замещены 1-2 группами, выбранными из линейного или разветвленного (С1-С6)алкила, линейного или разветвленного (С1-С6)алкокси, галогена, гетероциклоалкила, необязательно замещенного одним или несколькими атомами галогена, при этом "арил" означает фенил; "гетероарил" означает любую моно- или бициклическую группу, состоящую из 5-10 кольцевых членов, содержащую по меньшей мере один ароматический фрагмент и содержащую от 1 до 4 гетероатомов, выбранных из азота (включая четвертичный азот); "гетероциклоалкил" означает моноциклическую, неароматическую группу, содержащую от 4 до 6 кольцевых членов и содержащую от 1 до 2 гетероатомов, выбранных из кислорода и азота. Также изобретение относится к способу получения соединения формулы (I), фармацевтической композиции на основе соединения формулы (I) и его применению при лечении злокачественных новообразований, аутоиммунных заболеваний и заболеваний иммунной системы. Технический результат: получены новые соединения, обладающие ингибирующей активностью в отношении Bcl-2. 8 н. и 4 з.п. ф-лы, 2 табл., 92 пр.
представляет собой одну из следующих групп: 5,6,7,8-тетрагидроиндолизин, необязательно замещенный аминогруппой; индолизин; 1,2,3,4-тетрагидропирроло[1,2-а]пиразин, необязательно замещенный метилом; пирроло[1,2-а]пиримидин; Т представляет собой атом водорода, линейную или разветвленную (С1-С6)алкильную группу, группу (С2-С4)алкил-NR1R2, или группу (С1-С4)алкил-OR6; R1 и R2 независимо друг от друга представляют собой атом водорода или линейную или разветвленную (С1-С6)алкильную группу; или R1 и R2 вместе с атомом азота, несущим их, образуют гетероциклоалкил; R3 представляет собой линейную (С1-С6)алкильную, арильную или гетероарильную группу, причем последние две группы могут быть замещены 1-2 группами, выбранными из галогена, линейного или разветвленного (С1-С6)алкила, линейного или разветвленного (С1-С6)алкокси, циано и N-оксида; R4 представляет собой 4-гидроксифенильную группу; R5 представляет собой атом водорода; R6 представляет собой атом водорода; Ra и Rd каждый представляет собой атом водорода и (Rb,Rc), вместе с атомами углерода, несущими их, образуют 1,3-диоксолановую группу или 1,4-диоксановую группу; или Ra, Rc и Rd каждый представляет собой атом водорода и Rb представляет собой водород, галоген, метил или метокси; или Ra, Rb и Rd каждый представляет собой атом водорода и Rc представляет собой гидрокси или метокси группу; причем гетероциклоалкильные группы, определенные таким образом, и алкильные и алкокси группы могут быть замещены 1-2 группами, выбранными из линейного или разветвленного (С1-С6)алкила, линейного или разветвленного (С1-С6)алкокси, галогена, гетероциклоалкила, необязательно замещенного одним или несколькими атомами галогена, при этом "арил" означает фенил; "гетероарил" означает любую моно- или бициклическую группу, состоящую из 5-10 кольцевых членов, содержащую по меньшей мере один ароматический фрагмент и содержащую от 1 до 4 гетероатомов, выбранных из азота (включая четвертичный азот); "гетероциклоалкил" означает моноциклическую, неароматическую группу, содержащую от 4 до 6 кольцевых членов и содержащую от 1 до 2 гетероатомов, выбранных из кислорода и азота. Также изобретение относится к способу получения соединения формулы (I), фармацевтической композиции на основе соединения формулы (I) и его применению при лечении злокачественных новообразований, аутоиммунных заболеваний и заболеваний иммунной системы. Технический результат: получены новые соединения, обладающие ингибирующей активностью в отношении Bcl-2. 8 н. и 4 з.п. ф-лы, 2 табл., 92 пр.
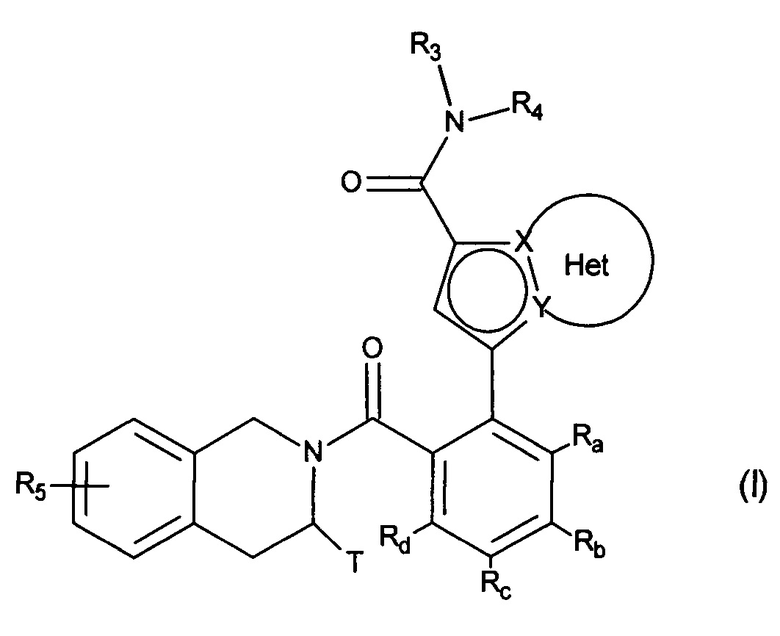
1. Соединение формулы (I):
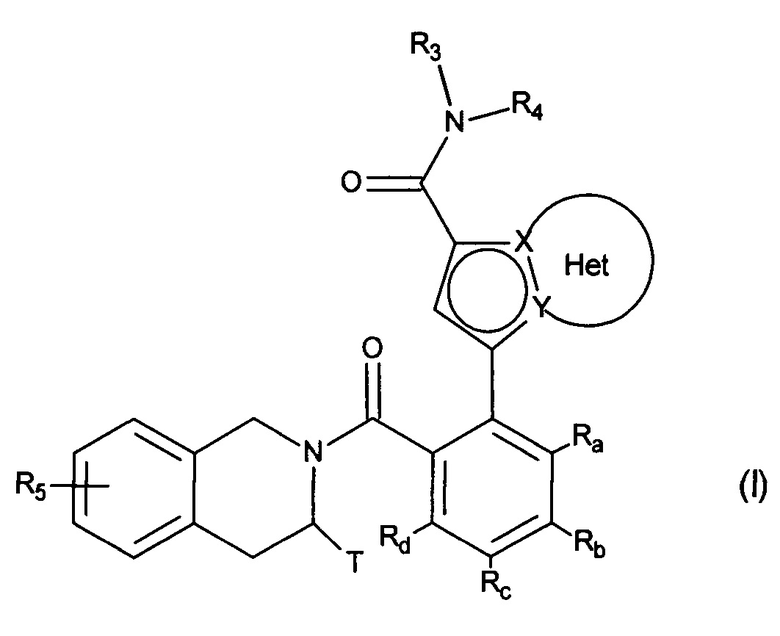 ,
,
в которой:
 X и Y представляют собой атом углерода или атом азота, при этом предполагается, что они не могут одновременно представлять собой два атома углерода или два атома азота,
X и Y представляют собой атом углерода или атом азота, при этом предполагается, что они не могут одновременно представлять собой два атома углерода или два атома азота,
группа 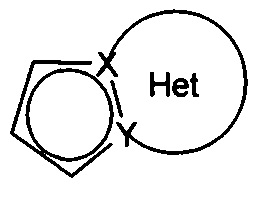 представляет собой одну из следующих групп: 5,6,7,8-тетрагидроиндолизин, необязательно замещенный аминогруппой; индолизин; 1,2,3,4-тетрагидропирроло[1,2-а]пиразин, необязательно замещенный метилом; пирроло[1,2-а]пиримидин,
представляет собой одну из следующих групп: 5,6,7,8-тетрагидроиндолизин, необязательно замещенный аминогруппой; индолизин; 1,2,3,4-тетрагидропирроло[1,2-а]пиразин, необязательно замещенный метилом; пирроло[1,2-а]пиримидин,
Т представляет собой атом водорода, линейную или разветвленную (С1-С6)алкильную группу, группу (С2-С4)алкил-NR1R2 или группу (С1-С4)алкил-OR6,
R1 и R2 независимо друг от друга представляют собой атом водорода или линейную или разветвленную (С1-С6)алкильную группу,
или R1 и R2 вместе с атомом азота, несущим их, образуют гетероциклоалкил,
R3 представляет собой линейную (С1-С6)алкильную, арильную или гетероарильную группу, причем последние две группы могут быть замещены 1-2 группами, выбранными из галогена, линейного или разветвленного (С1-С6)алкила, линейного или разветвленного (С1-С6)алкокси, циано и N-оксида,
R4 представляет собой 4-гидроксифенильную группу,
R5 представляет собой атом водорода,
R6 представляет собой атом водорода,
Ra и Rd каждый представляет собой атом водорода и (Rb,Rc), вместе с атомами углерода, несущими их, образуют 1,3-диоксолановую группу или 1,4-диоксановую группу; или Ra, Rc и Rd каждый представляет собой атом водорода и Rb представляет собой водород, галоген, метил или метокси; или Ra, Rb и Rd каждый представляет собой атом водорода и Rc представляет собой гидрокси или метокси группу,
причем гетероциклоалкильные группы, определенные таким образом, и алкильные и алкокси группы могут быть замещены 1-2 группами, выбранными из линейного или разветвленного (С1-С6)алкила, линейного или разветвленного (С1-С6)алкокси, галогена, гетероциклоалкила, необязательно замещенного одним или несколькими атомами галогена,
при этом предполагается, что:
- "арил" означает фенил,
- "гетероарил" означает любую моно- или бициклическую группу, состоящую из 5-10 кольцевых членов, содержащую по меньшей мере один ароматический фрагмент и содержащую от 1 до 4 гетероатомов, выбранных из азота (включая четвертичный азот),
- "гетероциклоалкил" означает моноциклическую, неароматическую группу, содержащую от 4 до 6 кольцевых членов и содержащую от 1 до 2 гетероатомов, выбранных из кислорода и азота,
их энантиомеры и диастереоизомеры, и их соли присоединения с фармацевтически приемлемой кислотой.
2. Соединение формулы (I) по п.1, в которой Т представляет собой атом водорода, метальную группу, группу 2-(морфолин-4-ил)этил, 3-(морфолин-4-ил)пропил, -СН2-ОН, 2-аминоэтил, 2-(3,3-дифторпиперидин-1-ил)этил, 2-[(2,2-дифторэтил)амино]этил или 2-(3-метоксиазетидин-1-ил)этил.
3. Соединение формулы (I) по п.1 или 2, в которой R3 представляет собой гетероарильную группу, выбранную из следующей группы: 1H-индол, 2,3-дигидро-1H-индол, 1H-индазол, пиридин, 1H-пирроло[2,3-b]пиридин, 1H-пиразол, имидазо[1,2-а]пиридин, пиразоло[1,5-а]пиримидин, [1,2,4]триазоло[1,5-а]пиримидин и 1H-пиразоло[3,4-b]пиридин, все из которых могут быть замещены линейной или разветвленной (С1-С6)алкильной группой.
4. Соединения формулы (I) по п.1, выбранные из следующей группы:
- N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-{1-[2-(морфолин-4-ил)этил]-1H-индол-5-ил}-5,6,7,8-тетрагидроиндолизин-1-карбоксамид,
- N-(4-гидроксифенил)-3-(6-{[(3S)-3-[2-(морфолин-4-ил)этил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамид,
- N-{3-фтор-4-[2-(морфолин-4-ил)этокси]фенил}-N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-карбоксамид,
- N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(пиридин-4-ил)индолизин-1-карбоксамид,
- N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(2-метилпиридин-4-ил)индолизин-1-карбоксамид,
- N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(1-метил-1H-пирроло[2,3-b]пиридин-5-ил)индолизин-1-карбоксамид,
- N-(4-гидроксифенил)-3-(6-{[(3R)-3-[3-(морфолин-4-ил)пропил]-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-фенил-5,6,7,8-тетрагидроиндолизин-1-карбоксамид,
- N-(2,6-диметилпиридин-4-ил)-N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-карбоксамид,
- N-(4-гидроксифенил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-1,3-бензодиоксол-5-ил)-N-(пиридин-4-ил)-5,6,7,8-тетрагидроиндолизин-1-карбоксамид,
- 3-(5-хлор-2-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}фенил)-N-(4-гидроксифенил)-N-(1-метил-1H-пирроло[2,3-b]пиридин-5-ил)индолизин-1-карбоксамид,
- N-(4-гидроксифенил)-N-(2-метоксипиридин-4-ил)-3-(6-{[(3R)-3-метил-3,4-дигидроизохинолин-2(1H)-ил]карбонил}-1,3-бензодиоксол-5-ил)индолизин-1-карбоксамид,
их энантиомеры и диастереоизомеры, и их соли присоединения с фармацевтически приемлемой кислотой.
5. Способ получения соединений формулы (I) по п.1, который отличается тем, что в качестве исходного вещества применяют соединение формулы (II):
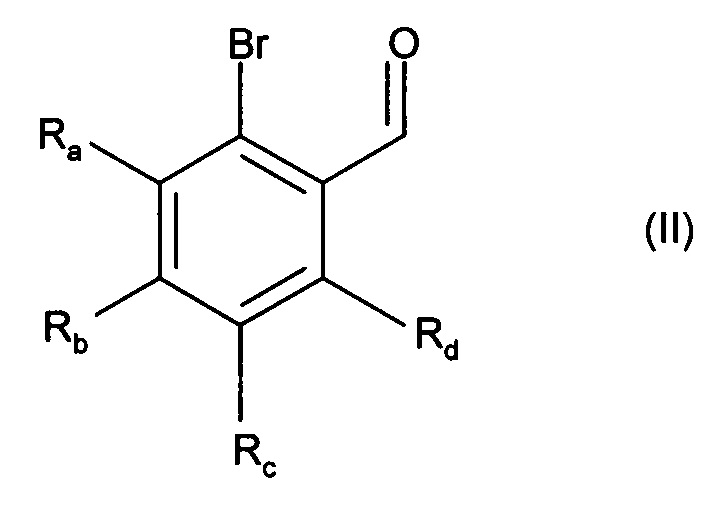 ,
,
в которой Ra, Rb, Rc и Rd являются такими, как определено для формулы (I),
причем соединение формулы (II) подвергают реакции Хека, в водной или органической среде, в присутствии палладиевого катализатора, основания, фосфина и соединения формулы (III):
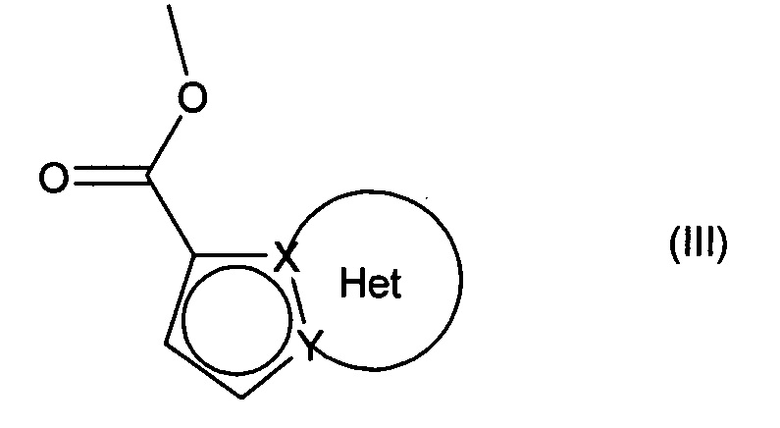 ,
,
в которой группы X, Y и Het являются такими, как определено для формулы (I),
с получением соединения формулы (IV):
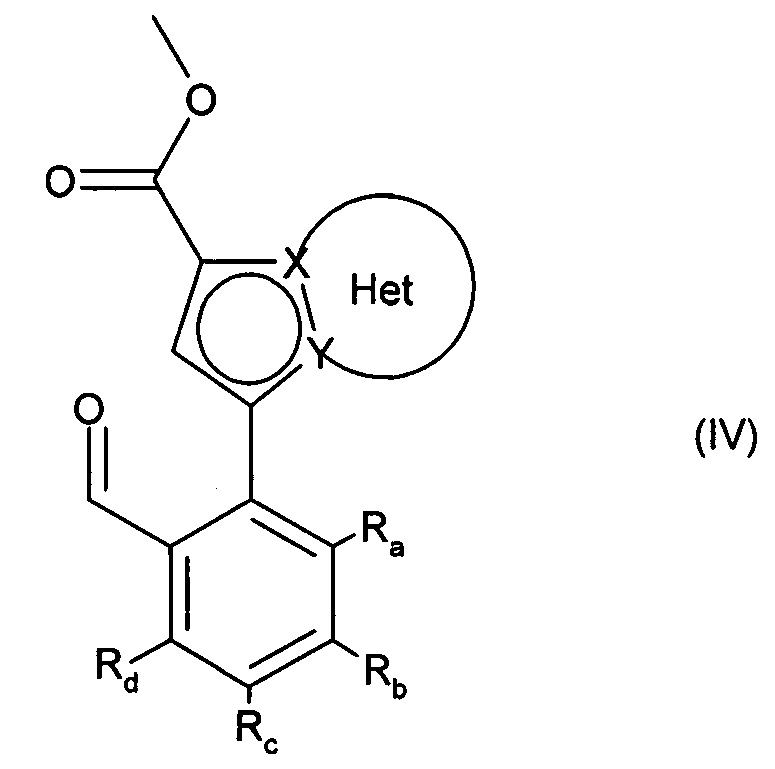 ,
,
в которой Ra, Rb, Rc, Rd, X, Y и Het являются такими, как определено для формулы (I),
альдегидную группу соединения формулы (IV) окисляют до карбоновой кислоты с образованием соединения формулы (V):
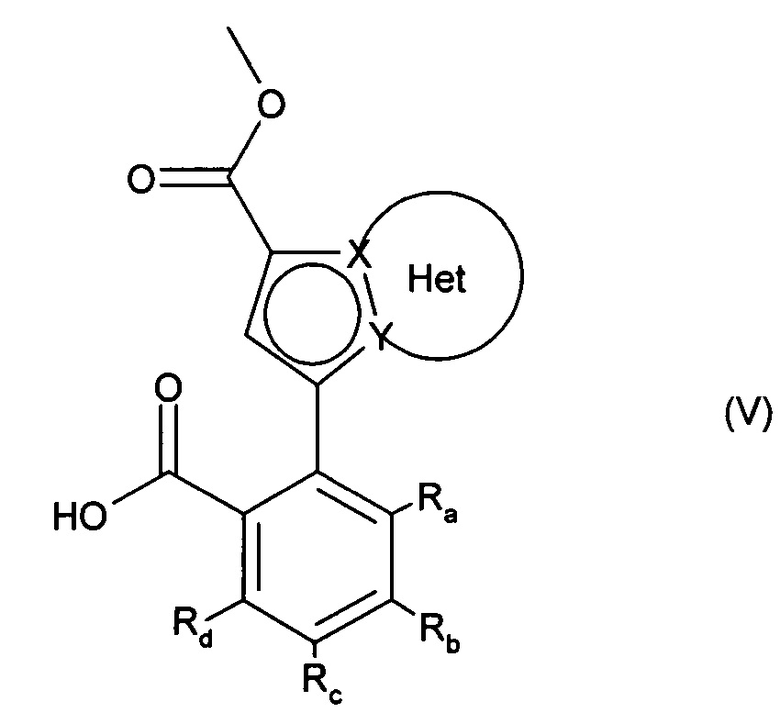 ,
,
в которой Ra, Rb, Rc, Rd, X, Y и Het являются такими, как определено для формулы (I),
соединение формулы (V) затем подвергают пептидному сочетанию с соединением формулы (VI):
 ,
,
в которой Т и R5 являются такими, как определено для формулы (I),
с получением соединения формулы (VII):
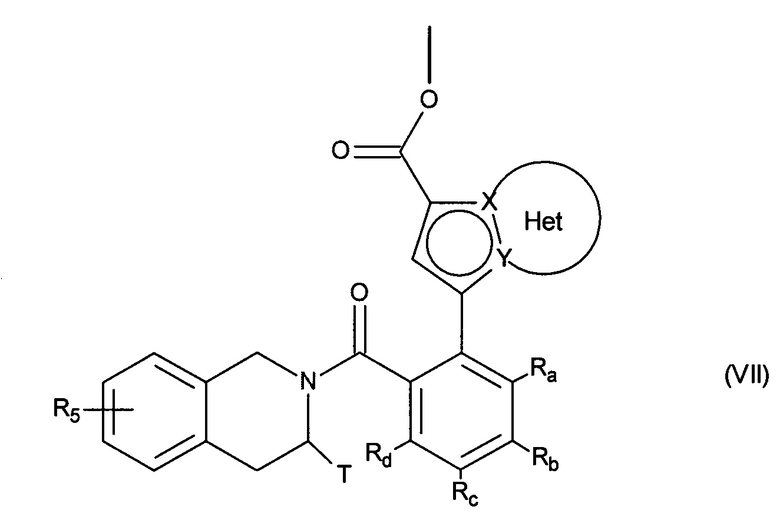 ,
,
в которой Ra, Rb, Rc, Rd, T, R5, X, Y и Het являются такими, как определено для формулы (I),
сложноэфирную группу соединения формулы (VII) гидролизуют с получением соответствующей карбоновой кислоты или карбоксилата, которую(-ый) можно превратить в производное кислоты, такое как соответствующий ацилхлорид или ангидрид, и затем осуществляют сочетание с амином NHR3R4, где R3 и R4 имеют те же значения, что и в случае формулы (I), с получением соединения формулы (I),
которое можно очистить в соответствии с обычными методиками разделения, которое превращают, при необходимости, в его соли присоединения с фармацевтически приемлемой кислотой и которое необязательно разделяют на его изомеры в соответствии с обычными методиками разделения.
6. Способ по п. 5 получения соединения формулы (I), в которой одна из групп R3 или R4 замещена гидрокси группой, отличающийся тем, что амин NHR3R4 подвергают реакции, защищающей гидрокси группу, заблаговременно перед каким-либо сочетанием с карбоновой кислотой, образованной из соединения формулы (VII), или с соответствующим производным таковой кислоты, где полученное в результате защищенное соединение формулы (I) затем подвергают реакции снятия защиты и затем необязательно превращают в одну из его солей присоединения с фармацевтически приемлемой кислотой.
7. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении Bcl-2, содержащая эффективное количество соединения формулы (I) по любому из пп.1-4 или его соль присоединения с фармацевтически приемлемой кислотой в комбинации с одним или несколькими фармацевтически приемлемыми наполнителями.
8. Применение фармацевтической композиции по п.7 для изготовления лекарственного средства для применения в качестве проапоптотического агента.
9. Применение фармацевтической композиции по п.7 для изготовления лекарственного средства, предназначенного для лечения злокачественных новообразований, аутоиммунных заболеваний и заболеваний иммунной системы.
10. Применение фармацевтической композиции по п.7 для изготовления лекарственного средства, предназначенного для лечения рака мочевого пузыря, головного мозга, молочной железы и матки, хронических лимфоидных лейкозов, колоректального рака, рака пищевода и печени, лимфобластных лейкозов, неходжкинских лимфом, меланом, злокачественных заболеваний крови, миелом, рака яичников, немелкоклеточного рака легкого, рака предстательной железы и мелкоклеточного рака легкого.
11. Применение соединения формулы (I) по любому из пп.1-4 или его соли присоединения с фармацевтически приемлемой кислотой для изготовления лекарственного средства, предназначенного для лечения рака мочевого пузыря, головного мозга, молочной железы и матки, хронических лимфоидных лейкозов, колоректального рака, рака пищевода и печени, лимфобластных лейкозов, неходжкинских лимфом, меланом, злокачественных заболеваний крови, миелом, рака яичников, немелкоклеточного рака легкого, рака предстательной железы и мелкоклеточного рака легкого.
12. Применение соединения формулы (I) по любому из пп.1-4 для изготовления лекарственного средства для применения в ассоциации с радиотерапией для лечения злокачественных новообразований.
| НОВЫЕ ИНДОЛИЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2013 |
|
RU2646223C2 |
| WO 2012162365 A1, 29.11.2012 | |||
| Приспособление к телеграфному аппарату Бодо для печатания строками | 1926 |
|
SU7983A1 |

