Настоящее изобретение относится к новым макроциклическим производным, к способу их получения и к содержащим их фармацевтическим композициям.
Предлагаемые в настоящем изобретении соединения являются новыми и обладают весьма перспективными фармакологическими характеристиками в области апоптоза и онкологии.
Апоптоз, или запрограммированная гибель клеток, является крайне важным физиологическим процессом для эмбрионального развития и поддержания гомеостаза в тканях.
Гибель клеток по типу апоптоза задействует морфологические изменения, такие как конденсация ядра, фрагментация ДНК, а также биохимический феномен, такой как активация каспаз, которые повреждают ключевые структурные компоненты клетки, чтобы вызвать ее разборку и смерть. Регуляция процесса апоптоза является сложной и вовлекает активацию или подавление нескольких внутриклеточных путей передачи сигналов (Cory S. et al., Nature Review Cancer, 2002, 2, 647-656).
Дерегулирование апоптоза вовлечено в некоторые патологии. Повышенный апоптоз связан с нейродегенеративными заболеваниями, такими как болезнь Паркинсона, болезнь Альцгеймера и ишемия. И наоборот, недостатки при осуществлении апоптоза играют важную роль в развитии злокачественных новообразований и их резистентности к химиотерапии, аутоиммунных заболеваний, воспалительных заболеваний и вирусных инфекций. Следовательно, отсутствие апоптоза является одним из характерных фенотипических признаков злокачественного новообразования (Hanahan D. et al., Cell 2000, 100, 57-70).
Антиапоптотические белки семейства Bcl-2 связаны с многочисленными патологиями. Причастность белков семейства Bcl-2 описана для многочисленных типов злокачественных новообразований, таких как рак прямой кишки, рак молочной железы, мелкоклеточный рак легкого, немелкоклеточный рак легкого, рак мочевого пузыря, рак яичников, рак предстательной железы, хронический, хронический лимфоцитарный лейкоз, фолликулярная лимфома, миелома… Сверхэкспрессия антиапоптотических белков семейства Bcl-2 связана с онкогенезом, с устойчивостью к химиотерапии и с клиническим прогнозом пациентов, заболевших раком. Таким образом, существует терапевтическая потребность в соединениях, которые ингибируют антиапоптотическую активность белков семейства Bcl-2.
Помимо их новизны, соединения в соответствии с настоящим изобретением обладают проапоптотическими свойствами, которые позволяют применять их при патологиях, связанных с дефектом апоптоза, например, в лечении рака, аутоиммунных заболеваний и иммунной системы.
В первом варианте осуществления (Е1) изобретение относится к соединениям формулы (I):

в которой:
 A1 и А2 независимо друг от друга представляют собой атом водорода или галогена, линейный или разветвленный (С1-С6)полигалогеналкил, линейную или разветвленную (С1-С6)алкильную группу или циклоалкильную группу,
A1 и А2 независимо друг от друга представляют собой атом водорода или галогена, линейный или разветвленный (С1-С6)полигалогеналкил, линейную или разветвленную (С1-С6)алкильную группу или циклоалкильную группу,
или A1 и А2 вместе с несущими их атомами образуют ароматический или неароматический гетероцикл Het, состоящий из 5, 6 или 7 кольцевых членов, и который, помимо азота, может содержать от одного до 3 гетероатомов, независимо выбранных из кислорода, серы и азота, при условии, что соответствующий азот может быть замещен группой, представляющей собой атом водорода, линейную или разветвленную (С1-С6)алкильную группу или группу С(O)-O-Alk, в которой Alk представляет собой линейную или разветвленную (С1-С6)алкильную группу,
G представляет собой группу -NR7-, группу 1,2,3,4-тетрагидроизохинолинлена, необязательно замещенного группой Т, группу 2,3-дигидро-1H-изоиндолилена, необязательно замещенного группой Т, или группу пиперидинилена,
Т представляет собой атом водорода, линейный или разветвленный (С1-С6)алкил, необязательно замещенный одним - тремя атомами галогена, группу (С1-С4)алкил-NR1NR2, или группу (С1-С4)алкил-OR6,
X представляет собой (С2-С8)алкиленовую группу, из которых от 1 до 3 кольцевых членов могут быть заменены гетероатомом, выбранным из кислорода, серы и N-R5, или группой арила или гетероарила,
Y представляет собой группу -СН2- или -СО-,
R1 и R2 независимо друг от друга представляют собой атом водорода или линейную или разветвленную (С1-С6)-алкильную группу,
или R1 и R2 с атомом азота, который несет их, образуют гетероциклоалкил,
R3 и R4 являются следующими:
- один из них представляет собой фенильную группу следующей формулы:

в которой W представляет собой гидроксигруппу или фосфатную группу, выбранную из -ОРО(ОМ)(ОМ'), -OPO(OM)(O-M1+), -OPO(O-M1+)(O-M2+), -ОРО(O-)(O-)М32+, -ОРО(ОМ)(O[CH2CH2O]nCH3), и -ОРО(O- М1+)(O[CH2CH2O]nCH3), в которых М и М' независимо друг от друга представляют собой атом водорода, линейную или разветвленную (C1-С6)алкильную группу, линейную или разветвленную (С2-С6)алкенильную группу, линейную или разветвленную (С2-С6)алкинильную группу, циклоалкил или гетероциклоалкил, оба состоят из 5-6 кольцевых членов, тогда как M1+ и М2+ независимо друг от друга представляют собой фармацевтически приемлемый одновалентный катион, М32+ представляет собой фармацевтически приемлемый двухвалентный катион и n означает целое число от 1 до 5,
- тогда как другая представляет собой арильную, гетероарильную, гетероциклоалкильную, циклоалкильную группу или линейную или разветвленную (С1-С6)алкильную группу, следует понимать, что один или несколько атомов углерода предшествующих групп или их возможных заместителей могут быть дейтерированными,
R5 представляет собой атом водорода или линейную или разветвленную (С1-С6)алкильную группу,
R6 и R7 независимо друг от друга представляют собой атом водорода или линейную или разветвленную (С1-С6)алкильную группу,
Ra, Rb, Rc и Rd независимо друг от друга представляют собой атом водорода, линейную или разветвленную (С1-С6)алкильную группу, атом галогена, линейную или разветвленную (С1-С6)алкоксигруппу, гидроксигруппу, линейную или разветвленную (С1-С6)полигалогеналкильную группу, трифторметокси-группу, или заменители одного из пары (Ra,Rb), (Rb,Rc) или (Rc,Rd) вместе с несущими их атомами углерода образуют кольцо, состоящее из 5-7 кольцевых членов, который может содержать от одного до 2 гетероатомов, выбранных из кислорода и серы, также при условии, что один или несколько атомов углерода определенного выше кольца могут быть дейтерированными или замещенными от 1 до 3 группами, выбранными из галогена или линейного или разветвленного (С1-С6)алкила,
при условии, что:
«арил» означает фенильную, нафтильную, бифенильную или инденильную группу,
- «гетероарил» означает любую моно- или бициклическую группу, состоящую из 5-10 кольцевых членов, содержащую по меньшей мере один ароматический фрагмент и содержащую от 1 до 4 гетероатомов, выбранных из кислорода, серы, азота и четвертичного азота,
- «циклоалкил» означает любую моно- или бициклическую неароматическую карбоциклическую группу, содержащую от 3 до 10 кольцевых членов,
- «гетероциклоалкил» означает любую моно- или бициклическую неароматическую, конденсированную или спиро-группу, содержащую от 3 до 10 кольцевых членов, и содержащую от 1 до 3 гетероатомов, выбранных из кислорода, серы, SO, SO2 или азота,
- арилен, гетероарилен, 1,2,3,4-тетрагидроизохинолинилен, 2,3-дигидро-1Н-изоиндолилен или пиперидинилен означают двухвалентную арильную, гетероарильную, 1,2,3,4-тетрагидроизохинолиновую или пиперидиновую группу,
определенные таким образом арильная, гетероарильная, циклоалкильная и гетероциклоалкильная группы и алкильная, алкенильная, алкинильная, алкоксигруппа, которые могут быть замещены 1-3 группами, выбраны из следующих: линейный или разветвленный (С1-С6)алкил, необязательно замещенный гидроксигруппой, морфолинил, 3-3-дифторпиперидинил или 3-3-дифторпирролидинил; (С3-С6)спиро; линейный или разветвленный (C1-С6)алкокси, необязательно замещенный группой морфолинила; (С1-С6)алкил-S-; гидрокси; оксо; N-оксид; нитро; циано; -COOR'; -OCOR'; NR'R''; линейный или разветвленный (С1-С6)полигалогеналкил; трифторметокси; (C1-С6)алкилсульфонил; галоген; арил, необязательно замещенный одним или несколькими атомами галогена; гетероарил; арилокси; арилтио; циклоалкил; гетероциклоалкил, необязательно замещенный одним или несколькими атомами галогена или линейными или разветвленными (С1-С6)алкильными группами; при условии, что R' и R'' независимо друг от друга представляют собой атом водорода или линейную или разветвленную (С1-С6)алкильную группу, необязательно замещенную метоксигруппой,
их энантиомеры, диастереоизомеры или их соли присоединения к кислоте или к фармацевтически приемлемому основанию.
В числе фармацевтически приемлемых кислот могут быть приведены, без какого-либо ограничения, хлористоводородная кислота, бромистоводородная кислота, серная кислота, фосфоновая кислота, уксусная кислота, трифторуксусная кислота, молочная кислота, пировиноградная кислота, малоновая кислота, янтарная кислота, глутаровая кислота, фумаровая кислота, винная кислота, малеиновая кислота, лимонная кислота, аскорбиновая кислота, щавелевая кислота, метансульфоновая кислота, камфорная кислота и т.д.
В числе фармацевтически приемлемых оснований могут быть приведены, без какого-либо ограничения, гидроксид натрия, гидроксид калия, триэтиламин, трет-бутиламин и т.д.
Различные конкретные варианты осуществления изобретения (Е) подробно описаны ниже. Следует отметить, что характеристики различных вариантов осуществления могут быть объединены друг с другом, чтобы создать новые варианты осуществления:
Е2. Соединение формулы (I) в соответствии с вариантом осуществления Е1, в котором A1 и А2 каждый представляют собой метальную группу, или одна из групп A1 или А2 представляет собой метил, тогда как другая представляет собой атом водорода.
Е3. Соединение формулы (I) в соответствии с вариантом осуществления Е1, в котором A1 и А2 вместе с несущими их атомами образуют гетероцикл, состоящий из 6 кольцевых членов.
Е4. Соединение формулы (I) в соответствии с одним из вариантов осуществления от Е1 до Е3, в котором Ra, Rc и Rd каждый представляет собой атом водорода и Rb представляет собой атом водорода или галогена.
Е5. Соединение формулы (I) в соответствии с одним из вариантов осуществления от Е1 до Е4, в котором Y представляет собой группу -СО-.
Е6. Соединение формулы (I) в соответствии с одним из вариантов осуществления от Е1 до Е5, в котором G представляет собой группу, выбранную из следующих групп:

где Т представляет собой метальную группу или (4-морфолинил)метильную группу.
Е7. Соединение формулы (I) в соответствии с одним из вариантов осуществления от Е1 до Е6, в котором X представляет собой группу, выбранную из следующих групп:

Е8. Соединение формулы (I) в соответствии с одним из вариантов осуществления от Е1 до Е7, в котором одна из групп R3 или R4 представляет собой 4-гидроксифенильную группу, тогда как другая представляет собой арильную, гетероарильную или линейную или разветвленную (С1-С6)алкильную группу.
Е9. Соединение формулы (I) в соответствии с вариантом осуществления Е8, в котором одна из групп R3 или R4 представляет собой 4-гидроксифенильную группу, тогда как другая представляет собой группу, выбранную из следующего списка:
- фенильная группа, необязательно замещенная цианогруппой,
- пиразолильна группа,
- 1-метил-1Н-пиразолильная группа,
- 1-(тетрагидрофуран-3-ил)-1H-пиразолильная группа,
- 5-метил-2-циано-1H-пирролильная группа,
- 1-метил-2-циано-1H-пирролильная группа,
- 1,2-диметил- 1H-пирролильная группа,
- 1,5-диметил-2-циано-1H-пирролильная группа,
- пиримидинильная группа,
- этильная группа,
- пиримидиниевая группа.
В некоторых вариантах осуществления изобретения R3 или R4 представляет собой 4-[(NaO)2OPO]фенильную группу.
Е10. Соединение формулы (I) в соответствии с вариантом осуществления Е9 в которой R4 представляет собой 4-гидроксифенильную группу.
Е11. Соединение формулы (I) в соответствии с вариантом осуществления Е9 в которой R3 представляет собой 4-гидроксифенильную группу.
Е12. Соединение формулы (I) в соответствии с вариантом осуществления Е11 в которой R3 представляет собой 4-гидроксифенильную группу и G представляет собой пиперидинильную группу.
Е13. Соединение формулы (I) в соответствии с вариантом осуществления Е1, выбранное из следующей группы:
- 6-хлор-14-(4-гидроксифенил)-10,11-диметил-2,13-диоксо-20,23-диокса-1,10,14-триазагексацикло[26.3.1.1~9,12~.1~15,19~.0~3,8~.0~24,29~]тетратриаконта-3,5,7,9(34), 11,15 (33), 16,18,24,26,28-ундекаен-17-карбонитрил,
- 11-хлор-4-(4-гидроксифенил)- 1,7,8-триметил-4,16,17,23,24,25-гексагидро-1Н,14Н-15,18-метано-6,9-(метено)дибензо[b,h]пиразоло[4,3-р][1,6,11,15]оксатриазациклоикозин-5,14(8Н)-дион,
- 6-хлор-14-(4-гидроксифенил)-10,11-диметил-2,13-диоксо-23-окса-1,10,14-триазагексацикло[26.3.1.1-9,12~.1-15,19~.0~3,8~.0~24,29~]тетратриаконта-3,5,7,9(34),11,15(33),16,18,24,26,28-ундекаен- 17-карбонитрил,
- 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-5,14-диоксо-4,5,8,16,17,23,24,25-октагидро-1Н,14Н-15,18-метано-6,9-(метено)дибензо[b,h]пирроло[3,2-р][1,6,11,15]оксатриазациклоикозин-2-карбонитрил,
- 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-5,14-диоксо-1,4,5,8,16,17,23,24-октагидро-14Н-15,18-метано-6,9-(метено)дибензо[l,r]пирроло[2,3-d][1,6,10,15]оксатриазациклононадецин-2-карбонитрил,
- 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-1,4,16,21,22,23-гексагидро-14Н-15,21-метано-6,9-(метено)дибензо[j,о]пиразоло[3,4-b][1,4,8,13]оксатриазациклононадецин-5,14(8Н)-дион,
(16S или R)-11-хлор-4-(4-гидроксифенил)-1,7,8,16-тетраметил-5,14-диоксо-4,5,8,16,17,23,24,25-октагидро-1Н,14Н-15,18-метано-6,9-(метено)дибензо[b,h]пирроло[3,2-р][1,6,11,15]оксатриазациклоикозин-2-карбонитрил,
- 4-[16-хлор-3-гидрокси-19,20-диметил-13,22-диоксо-6,7,8,9,10,11,19,22-октагидро-13Н,23Н-8,12-метано-21,18-(метено)дибензо[b,j][1,4,8,13]оксатриазациклононадецин-23-ил]-1,5-диметил-1Н-пиррол-2-карбонитрил,
- 10-фтор-2-(4-гидроксифенил)-5,6-диметил-24,27,30-триокса-2,6,15,32,35-пентаазагексацикло[29.2.2.1~4,7~.1~15,19~.0~8,13~.0~18,23~]гептатриаконта-1(33),4,7(37),8,10,12,18,20,22,31,34-ундекаен-3,14-дион,
- 10-хлор-2-(4-гидроксифенил)-5,6-диметил-24,30-диокса-2,6,15,32,35-пентаазагексацикло[29.2.2.1~4,7~.1-15,19~.0~8,13~.0~18,23~]гептатриаконта-1(33),4,7(37),8,10,12,18,20,22,31,34-ундекаен-3,14-дион,
- 10-хлор-2-(4-гидроксифенил)-5,6,27-триметил-24,30-диокса-2,6,15,27,32,35-гексаазагексацикло[29.2.2.1~4,7~.1-15,19~.0-8,13~.0-18,23~]гептатриаконта-1(33),4,7(37),8,10,12,18,20,22,31,34-ундекаен-3,14-дион,
- 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-1,4,16,17,23,24-гексагидро-14Н-15,18-метано-6,9-(метено)дибензо[l,r]пиразоло[3,4-d][1,6,10,15]оксатриазациклононадецин-5,14(8Н)-дион.
Е14. Способ получения соединения формулы (I) в соответствии с вариантом осуществления Е1, отличающийся тем, что в качестве исходного продукта используют соединение формулы (II):
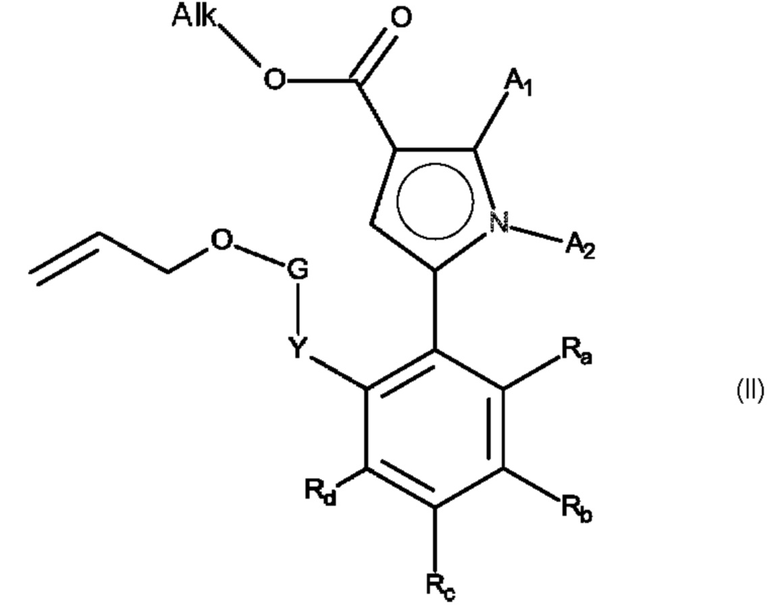
в котором A1, А2, Ra, Rb, Rc, Rd, Y и G имеют то же самое значение, как и в формуле (I), определенное в Е1, и Alk представляет собой линейную или разветвленную (С1-С6)алкильную группу,
соединение формулы (II), сложноэфирная функция -OAlk которого гидролизуется с образованием карбоновой кислоты или соответствующего карбоксилата, которые могут быть превращены в ацилхлорид или соответствующий ангидрид, перед сочетанием с амином NHR3AR4, где R4 имеет то же значение, что и в формуле (I), и R3A представляет собой:
- группу R3, как определено в формуле (I)
- или группу R3-O-Alk'-Z, R3-Alk'-Z или R3-Z, в которой Alk' представляет собой линейную или разветвленную (С1-С6)алкильную группу, и Z представляет собой атом галогена или группу -ОН,
чтобы образовать соединение формулы (III):

в которой A1, А2, Ra, Rb, Rc, Rd, R4, Y и G имеют то же самое значение, как и в формуле (I),
которое подвергают реакции снятия защиты со спиртовой функции с последующим или внутримолекулярным нуклеофильным замещением, или реакцией Мицунобу или ароматическим нуклеофильным замещением,
чтобы получить соединение формулы (I),
соединение формулы (I), которое может быть очищено в соответствии с обычной методикой разделения, которое может быть преобразовано в его соли присоединения к фармацевтически приемлемой кислоте или основанию, и которое необязательно разделяют на его изомеры в соответствии с обычной методикой разделения,
при условии, что в любое время, признанное подходящим в ходе описанного выше способа, гидрокси- или аминогруппы реагентов или промежуточные соединения синтеза могут быть защищены, а затем с них может быть снята защита для целей синтеза.
Е15. Способ получения соединения формулы (I) в соответствии с вариантом осуществления Е1, отличающийся тем, что в качестве исходного продукта применяют соединение формулы (IV):

в которой R3, R4 и X имеют то же самое значение, как и в формуле (I), и GA представляет собой группу, выбранную из следующего списка:

соединение формулы (IV), которое затем сочетают с соединением формулы (V):

в которой A1, А2, Ra, Rb, Rc, и Rd имеют то же самое значение, как и в формуле (I),
чтобы получить соединение формулы (VI):

в которой A1, А2, Ra, Rb, Rc, Rd, R3, R4 и X имеют то же самое значение, как и в формуле (I),
которое подвергают реакции снятия защиты с последующим внутримолекулярным сочетанием, чтобы получить соединение формулы (I),
соединение формулы (I) которое может быть очищено в соответствии с обычной методикой разделения, которое может быть преобразовано в его соли присоединения к фармацевтически приемлемой кислоте или основанию и которое необязательно разделяют на его изомеры в соответствии с обычной методикой разделения,
при условии, что в любое время, признанное подходящим в ходе описанного выше способа, гидрокси- или аминогруппы реагентов или промежуточные соединения синтеза могут быть защищены, а затем с них может быть снята защита для целей синтеза.
Е16. Фармацевтическая композиция, содержащая соединение формулы (I) в соответствии с одним из вариантов осуществления от Е1 до Е13, или одну из его солей присоединения к фармацевтически приемлемой кислоте или основанию, в сочетании с одним или несколькими фармацевтически приемлемыми наполнителями.
Е17. Фармацевтическая композиция в соответствии с вариантом осуществления Е16 для ее использования в качестве проапоптотического средства.
Е18. Фармацевтическая композиция в соответствии с вариантом осуществления Е16 для ее применения в лечении злокачественных новообразований, аутоиммунных заболеваний и иммунной системы.
Е19. Фармацевтическая композиция в соответствии с вариантом осуществления Е18, причем злокачественное новообразование выбирают из следующего списка: рак мочевого пузыря, головного мозга, молочной железы, матки, хронический лимфоцитарный лейкоз, рак прямой кишки, рак пищевода, печени, лимфобластный лейкоз, неходжкинская лимфома, меланома, злокачественная гемопатия, миелома, рак яичников, немелкоклеточный рак легкого, рак предстательной железы и мелкоклеточный рак легкого.
Е20. Применение соединения формулы (I) в соответствии с одним из вариантов осуществления от Е1 до Е13 для изготовления лекарственного средства, пригодного в качестве проапоптотического средства.
Е21. Применение соединения формулы (I) в соответствии с одним из вариантов осуществления от Е1 до Е13 для изготовления лекарственного средства, предназначенного для лечения злокачественных новообразований, иммунных и аутоиммунных заболеваний.
Е22. Применение соединения формулы (I) в соответствии с вариантом осуществления Е21, причем злокачественное новообразование выбирают из следующего списка: рак мочевого пузыря, головного мозга, молочной железы, матки, хронический лимфоцитарный лейкоз, рак прямой кишки, рак пищевода, печени, лимфобластный лейкоз, неходжкинская лимфома, меланома, злокачественная гемопатия, миелома, рак яичников, немелкоклеточный рак легкого, рак предстательной железы и мелкоклеточный рак легкого.
Е23. Комбинация соединения формулы (I) в соответствии с одним из вариантов осуществления от Е1 до Е13 с противораковым средством, выбранным из генотоксических средств, митотических ядов, антиметаболитов, ингибиторов протеасом, ингибиторов киназ или антител.
Е24. Фармацевтическая композиция, содержащая комбинацию в соответствии с вариантом осуществления Е23 в сочетании с одним или несколькими фармацевтически приемлемыми наполнителями.
Е25. Комбинация в соответствии с вариантом осуществления Е23 для ее применения в лечении злокачественных новообразований.
Е26. Применение комбинации в соответствии с вариантом осуществления Е23 для изготовления лекарственного средства, пригодного в лечении злокачественных новообразований.
Е27. Соединение формулы (I) в соответствии с одним из вариантов осуществления от Е1 до Е13 для его применения в комбинации с лучевой терапией в лечении злокачественных новообразований.
Из числа фармацевтических композиций в соответствии с изобретением могут быть приведены более конкретно те, которые пригодны для перорального, парентерального, назального, чрескожного или транскожного, ректального, перлингвального, офтальмологического или респираторного введения, а именно: обычные таблетки или драже, подъязычные таблетки, саше, пакетики, желатиновые капсулы, таблетки для рассасывания под языком, пастилки, суппозитории, кремы, мази, кожные гели и питьевые или инъекционные ампулы.
Дозировка варьируется в зависимости от пола, возраста и массы тела пациента, пути введения, характера терапевтического показания или, возможно, сопутствующего лечения и составляет от 0,01 мг до 1 г в сутки за один или несколько приемов.
В заключение, соединения в соответствии с изобретением могут быть связаны с моноклональными антителами или их фрагментами или связаны с каркасными белками, которые могут быть связаны и не связаны с моноклональными антителами.
Под фрагментами антител следует понимать фрагменты типа Fv, scFv, Fab, F(ab')2, F(ab'), scFv-Fc или диатела, которые обычно имеют такую же специфичность связывания, что и антитело, из которого они происходят. В соответствии с настоящим изобретением фрагменты антител в соответствии с изобретением могут быть получены исходя из антител с помощью методов, таких как переваривание ферментами, такими как пепсин или папаин и/или посредством расщепления дисульфидных мостиков с помощью химического восстановления. Иным способом, фрагменты антител, включенных в настоящее изобретение, могут быть получены с использованием методик генетической рекомбинации, также хорошо известных специалисту в данной области техники или посредством синтеза пептидов с использованием автоматических синтезаторов пептидов, например, таких, которые поставляет компания Applied Biosystems и т.д.
Под каркасными белками, которые могут быть связаны или не связаны с моноклональными антителами, понимают белок, который содержит или не содержит укладку цепи иммуноглобулинов и который обеспечивает способность к связыванию, сходную с таковой у моноклонального антитела. Специалисту в данной области техники известно, каким образом выбрать каркас белка. Более конкретно, известно, что должен быть выбран такой каркас, который будет проявлять несколько следующих отличительных признаков (Skerra A., J. Mol. Recogn. 13, 2000, 167-187): хорошая филогенетическая консервативность, прочное строение с хорошо известной трехмерной молекулярной организацией (такой как, например, кристаллография или ЯМР), маленький размер, отсутствие или незначительная посттрансляционная модификация(модификации), простота получения, экспрессии и очистки. Таким каркасным белком может быть, без ограничения перечисленным, структура, выбранная из группы, состоящей из фибронектина и предпочтительно десятого домена фибронектина типа III (FNfn10), липокалина, антикалина (Skerra A., J. Biotechnol., 2001, 74(4):257-75), белка Z, полученного из домена В белка А стафилококков, тиоредоксина А или любого белка с повторяющимся доменом, таким как «анкириновый повтор» (Kohl и al., PNAS, 2003, том 100, №4, 1700-1705), «повтор armadillo», «богатый лейцином повтор» или «тетратрикопептидный повтор». Также можно упомянуть каркас, полученный из токсинов (таких как, например, токсины скорпиона, насекомых, растений или моллюсков) или ингибирующие белки синтазы оксида азота (PIN).
Нижеследующие способы получения и примеры демонстрируют изобретение и никоим образом его не ограничивают.
Синтез 1а: 4-хлор-2-[4-(этоксикарбонил)-1,5-диметил-1Н-пиррол-2-ил]бензойная кислота
Стадия А: Этил 1,2-диметил-1Н-пиррол-3-карбоксилат
К раствору из этил 2-метил-1H-пиррол-3-карбоксилата (10 г; 65,3 ммоль) и йодистого метила (8,95 мл; 130,6 ммоль) в N,N-диметилформамиде (70 мл), помещенного при 0°С, добавляют, 3 порциями, 60% гидрид натрия (2,61 г; 65,3 ммоль). Затем все перемешивают при 0°С в течение 1 часа. Реакционную среду гидролизуют путем добавления ледяной воды (420 мл), затем разбавляют с этилацетатом. После декантации, органическую фазу последовательно промывают водным раствором 0,1 н. хлористоводородной кислоты, насыщенным водным раствором хлорида лития и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 6.65 (d, 1Н), 6.3 (1d, 1Н), 4.1 (1q, 2Н); 3.5 (s, 3Н), 2.4 (s, 3Н), 1.5 (1t, 3Н).
ИК: ν: >С=0: 1688 см-1; С-О-С: 1172 см-1.
Стадия В: этил 5-(5-хлор-2-формилфенил)-1,2-диметил-1Н-пиррол-3-карбоксилат
К раствору соединения, полученного в Стадии А (10 г; 62,8 ммоль) в N,N-диметилацетамиде (65 мл), добавляют последовательно 2-бром-4-хлорбензальдегид (15,2 г; 69 ммоль), ацетат калия (12,3 г; 125,6 ммоль), затем все перемешивают под аргоном в течение 20 минут. Потом добавляют дихлорбис(трифенилфосфин)палладий(II) (2,2 г; 3,14 ммоль). Реакционную среду после этого нагревают до 130°С в течение ночи. После достижения температуры окружающей среды, реакционную среду разбавляют в дихлорметане, затем туда добавляют костный уголь (2 г). Все перемешивают при температуре окружающей среды в течение 1 часа, затем фильтруют. Органическую фазу промывают затем водой, сушат над сульфатом магния и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и этанол в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 9.8 (s, 1H), 7.91-7.69-7,61 (d, 3H), 6.5 (s, 1H), 4.2 (q, 2Н,), 3.4 (s, 3H), 2.55 (s, 3H), 1.28 (t, 3H).
Стадия С: 4-хлор-2-[4-(этоксикарбонил)-1,5-диметил-1Н-пиррол-2-ил]бензойная кислота
Соединение, полученное в Стадии В (12,85 г; 42 ммоль) и 2-метил-2-бутен (35,7 мл; 336 ммоль) растворяют в смеси ацетона (20 мл) и тетрагидрофурана (20 мл). В нее добавляют по каплям 200 мл водного раствора, содержащего смесь хлорита натрия (13,3 г; 147 ммоль) и гидрофосфата натрия (14,5 г; 105 ммоль). Затем все перемешивают энергично при температуре окружающей среды в течение 7 часов. Реакционную среду концентрируют, чтобы удалить ацетон, затем разбавляют с этилацетатом. После декантации, органическую фазу промывают водой и концентрируют досуха. Затем остаток ресуспендируют в небольшом количестве этилового эфира. Затем полученное твердое вещество фильтруют, промывают этиловым эфиром, затем сушат в вакууме 40°С в течение одной ночи. Указанный в заголовке продукт используют позже без дальнейшей очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 13 (m, 1H), 7.85 (d, 1Н), 7.60 (dd, 1Н), 7.41 (d, 1H), 6.3 (s, 1Н), 4.15 (q, 2Н), 3.25 (s, 3H), 2.5 (s, 3H), 1.25 (t, 3H).
ИК: ν: -ОН: 3100-2500 см-1; >С=O: 1681 см-1.
Синтез 2а: 5-(5-хлор-2-формилфенил)-1,2-диметил-1Н-пиррол-3-карбоновая кислота
Стадия А: этил 1,2-диметил-1Н-пиррол-3-карбоксилат
К раствору этилацетатацетата (1000 г; 7,68 моль) в тетрагидрофуране (3 л) помещенного при -10°С, в течение 1 часа добавляют водный 50 мас. % раствор хлоруксусного альдегида (1206 г; 7,68 моль). После чего добавляют водный 40 мас. % раствор метиламина (1495 г; 19,2 моль) в течение 3 часов при -10°С. Затем реакционную смесь нагревают до 30°С в течение 1,5 часа и перемешивают при этой температуре в течение 16 часов. После достижения температуры окружающей среды, смесь разбавляют в этилацетате (3 л) и фазы разделяют. Основную водную фазу сохраняют для экстракции. Органическую фазу охлаждают до 10°С и добавляют водный раствор 1 н. соляной кислоты (2,5 л) в течение 15 минут. Фазы разделяют и органическую фазу снова промывают водным 1 н. раствором хлористоводородной кислоты (2,5 л), затем насыщенным водным раствором хлорида натрия (1 л). Водные основные и кислотные фазы объединяют и промывают с этилацетатом (1,5 л). Органические фазы объединяют и промывают насыщенным водным раствором хлорида натрия (1 л), сушат над сульфатом натрия, фильтруют и концентрируют досуха. Продукт очищают путем дистилляции в вакууме, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 6.50 (d, 1 Н), 6.45 (d, 1 Н), 4.25 (q, 2 Н), 3.51 (s, 3 Н), 2.49 (s, 3 Н), 1.33 (t, 3 Н).
Стадия В: 1,2-диметил-1Н-пиррол-3-карбоновая кислота
К раствору соединения, полученного в Стадии А (500 г; 2,99 моль) в воде (5 л) добавляют моногидрат гидроксида лития (251 г; 5,98 ммоль), и смесь нагревают до 100°С в течение 2 часов. После достижения температуры окружающей среды, реакционную смесь промывают толуолом (1 л) и метил-трет-бутиловый эфир (1 л). Водную фазу подкисляют водным раствором хлористоводородной кислоты (530 мл) до рН=1 при температуре между 10 и 15°С, перемешивают в течение 1 часа, затем фильтруют. Полученное твердое вещество промывают три раза водой и сушат в вакууме при температуре между 60 и 65°С в течение 36 часов.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 11.52 (s, 1 Н), 6.62 (d, 1 Н), 6.29 (d, 1 Н), 3.50 (s, 3 Н), 2.41 (s, 3 Н).
Стадия С: 5-(5-хлор-2-формилфенил)-1,2-диметил-1Н-пиррол-3-карбоновая кислота
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Синтеза 1а, используя 2-бром-4-хлорбензальдегидную кислоту и соединение, полученное в предыдущей Стадии.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 9.8 (s, 1 Н), 7.91 (d, 1 Н), 7.69 (dd, 1 Н), 7.61 (d, 1 Н), 6.5 (s, 1 Н), 4.2 (quad., 2 Н), 3.4 (s, 3 Н), 2.55 (s, 3 Н), 1.28 (t, 3 Н).
Синтез 1а': 5-(проп-2-ен-1-илокси)-1,2,3,4-тетрагидроизохинолин, гидрохлорид (1:1)
Стадия А: трет-бутил 5-гидрокси-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору из 5-гидроксиизохинолина (20 г; 137 ммоль) в уксусной кислоте (120 мл), добавляют диоксид платины (2 г; 8,8 ммоль). Все помещают под атмосферу водорода (2 бара) в течение 24 часов. Реакционную среду фильтруют, и катализатор промывают толуолом. Полученный таким образом фильтрат концентрируют досуха. Полученный остаток используют позже без дальнейшей очистки.
К раствору полученного остатка (1,95 г; 13 ммоль) в дихлорметане (110 мл), добавляют диизопропилэтиламин (9,7 мл; 57 ммоль) и ди-трет-бутилдикарбонат (3,69 г, 16,9 ммоль), затем все перемешивают в течение 2 часов при температуре окружающей среды. Реакционную среду разбавляют с водным насыщенным раствором хлорида аммония. После декантации, органическую фазу промывают водным насыщенным раствором гидрокарбоната натрия, затем насыщенным водным раствором хлорида натрия. После чего ее сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 9.41 (m, 1 Н), 6.97 (t, 1 Н), 6.64/6.54 (2d, 2 Н), 4.42 (m, 2 Н), 3.53 (t, 2 Н), 2.59 (t, 2 Н), 1.42 (s, 9 Н).
ИК: ν: -ОН: 3294 см-1; >С=O: 1652 см-1.
Стадия В: трет-бутил 5-(проп-2-ен-1-илокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Стадии А (10 г; 40,1 ммоль) в ацетонитриле (110 мл), добавляют аллилбромид (6 мл; 60,2 ммоль) и карбонат калия (14,6 г, 120,3 ммоль), затем все перемешивают в течение 20 часов при температуре окружающей среды. Реакционную среду разбавляют с этилацетатом. После декантации, органическую фазу промывают водой и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.8 (d, 1 Н), 7.12 (t, 1 Н), 6.75 (d, 1 Н), 6.05 (m, 1 Н), 5.41 (dquad, 1 Н), 5.28 (dquad, 1 Н), 4.58 (dt, 2 Н), 4.49 (s, 2 Н), 3.55 (t, 2 Н), 2.68 (t, 2 Н), 1.43 (s, 9 Н).
ИК: ν: >С=O: 1691 см-1;>С-O-С<: 1162 см-1.
Стадия С: 5-(проп-2-ен-1-илокси)-1,2,3,4-тетрагидроизохинолин, гидрохлорид (1:1)
К раствору соединения, полученного в Стадии В (9,7 г; 33,4 ммоль) в диоксане (30 мл), добавляют раствор 4 н. соляной кислоты в диоксане (33,4 мл; 133,5 ммоль), затем все перемешивают в течение 48 часов при температуре окружающей среды. Реакционную среду концентрируют досуха и полученный остаток ресуспендируют в этилацетате, затем фильтруют. Указанный в заголовке продукт получают в виде твердого вещества, которое используют без дальнейшей очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 9.4 (s, 2 Н), 7.21 (t, 1 Н), 6.92 (d, 1 Н), 6.8 (d, 1 Н), 6.08 (m, 1 Н), 5.41 (dt, 1 Н), 5.29 (dt, 1 Н), 4.62 (m, 2 Н), 4.25 (s, 2 Н), 3.35 (t, 2 Н), 2.86 (t, 2 Н).
ИК: ν:>NH2+: 3250-2250 см-1.
Синтез 2а': метил 1,2,3,4-тетрагидроизохинолин-4-илметанол
Стадия А: 1,2,3,4-тетрагидроизохинолин-4-карбоксилат
К раствору 1,2,3,4-тетрагидро-4-изохинолинкарбоновой кислоты (5 г; 28.2 ммоль) в метаноле (40 мл), добавляют триметилхлорсилан (5,4 мл; 42.3 ммоль), затем все перемешивают в течение 16 часов при температуре окружающей среды. После второго добавления триметилхлорсилана (4 мл; 31,35 ммоль), реакционную среду оставляют перемешиваться в течение 16 часов при температуре окружающей среды. Реакционную среду затем концентрируют досуха, и остаток ресуспендируют в метаноле, затем среду снова концентрируют. Это действие осуществляют два раза, чтобы обеспечить указанный в заголовке продукт, который используют позже без дальнейшей очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 9.75-9.6 (неразрешенный пик, 2 Н), 7.4-7.25 (неразрешенный пик, 4 Н), 4.3 (s, 2 Н), 4.29 (m, 1 Н), 3.72 (s, 3 Н), 3.65/3.55 (АВх, 2 Н).
ИК: ν: -NH2+: 3200-2150 см-1;>С=O: 1731 см-1.
Стадия В: 1,2,3,4-тетрагидроизохинолин-4-илметанол
К смеси алюмогидрида лития (1,24 г; 32,7 ммоль) в тетрагидрофуране (50 мл), помещенной при 0°С, добавляют по каплям при той же температуре соединение, полученное в Стадии А (3,47 г; 18,1 ммоль) в растворе в тетрагидрофуране (50 мл). Реакционную среду перемешивают 2 часа при 0°С, затем гидролизуют смесью воды (11 мл) и водного 1 н. раствора гидроксида натрия (15 мл). После добавления этилацетата, все оставляют перемешиваться в течение 16 часов при температуре окружающей среды. Нерастворимое вещество затем фильтруют, и фильтрат экстрагируют этилацетатом. Органические фазы объединяют и промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха, чтобы обеспечить указанный в заголовке продукт, который используют позже без дальнейшей очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.2-6.95 (m, 4 Н), 3.8 (s, 2 Н), 3.6 (d, 2 Н), 3.3 (m, 2 Н), 3.18/2.85 (2dd, 2 Н), 2.65 (m, 1 Н).
ИК: ν: -NH/-OH+: 3295 см-1;>С=С<: 1626 см-1.
Синтез 1а'': N-(4-{[трет-бутил(диметил)силил]окси}фенил)-3-(2-хлорэтокси) анилин
Стадия А: 1-бром-3-(2-хлорэтокси)бензол
К раствору из 3-бромфенола (5 г; 18,9 ммоль) и бромхлорэтан (3,7 мл; 43,3 ммоль) в ацетонитриле (80 мл), добавляют карбонат калия (12 г; 86,7 ммоль), затем все перемешивают при 80°С в течение 24 часов. Реакционную среду разбавляют со смесью из этилацетата и воды. После декантации, органическую фазу промывают водой, и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 7.15 (m, 2 Н), 7.09 (m, 1 Н), 6.85 (dt, 1 Н), 4.21 (t, 2 Н), 3.8 (t, 2 Н).
ИК: ν: Ar: 1588 см-1;>С-O-С<: 1227 см-1; γ:>СН-Ar: 764 и 678 см-1.
Стадия В: 4-{[трет-бутил(диметил)силил]окси}анилин
Указанное в заголовке соединение получают исходя из 4-аминофенола в тетрагидрофуране в присутствии имидазола и хлорида трет-бутил(диметил)силила в соответствии с протоколом, описанным в литературных источниках (S. Knaggs и al, Organic & Biomolecular Chemistry, 3(21), 4002-4010; 2005).
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 6.45-6.55 (dd, 4Н), 4.60 (m, 2Н), 0.90 (s, 9Н), 0.10 (s, 6Н). ИК: ν: -NH2+: 3300-3400 см-1
Стадия С: N-(4-{[трет-бутил(диметил)силил]окси}фенил)-3-(2-хлорэтокси) анилин
Раствор соединений, полученных в Стадии А (4,8 г; 20,8 ммоль) и в Стадии В (5,6 г; 24,9 ммоль) в толуоле (70 мл) дегазируют барботированием через аргон в течение 10 минут. В него добавляют трет-бутилат натрия (2,4 г; 24,9 ммоль) и хлор(2-ди-трет-бутилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2-аминоэтил)фенил]палладий(II) (0,7 г; 1 ммоль). Затем все перемешивают при 80°С в течение 1 часа. Реакционную среду фильтруют через Celite®. После промывания этилацетатом, фильтрат промывают водой и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1Н ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.88 (s, 1 Н), 7.08 (t, 1 Н), 7 (d, 2 Н), 6.79 (d, 2 Н), 6.54 (dd, 1 Н), 6.49 (t, 1 Н), 6.32 (dd, 1 Н), 4.19 (t, 2 Н), 3.91 (t, 2 Н), 0.95 (s, 9 Н), 0.2 (s, 6 Н)
ИК: ν:>NH: 3393 см-1; δ: Si-CH3: 1250 см-1.
Синтез 2а'': N-(4-{[трет-бутил(диметил)силил]окси}фенил)-3-(3-хлорпропокси)-анилин
Указанное в заголовке соединение получают в соответствии со способом Синтеза 1а'', заменяя бромхлорэтан на бромхлорпропан в Стадии А.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.85 (s, 1 Н), 7.05 (t, 1 Н), 6.99 (d, 2 Н), 6.77 (d, 2 Н), 6.52 (dd, 1 Н), 6.48 (t, 1 Н), 6.31 (dd, 1 Н), 4.02 (t, 2 Н), 3.8 (t, 2 Н), 2.13 (quint., 2 Н), 0.95 (s, 9 Н), 0.2 (s, 6 Н).
ИК: ν:>NH: 3398 см-1; δ: NH: 1504 см-1; δ: Si-CH3: 1250 см-1.
Синтез 3а'': N-(4-{[трет-бутил(диметил)силил]окси}фенил)-3-(4-хлорбутокси)анилин
Указанное в заголовке соединение получают в соответствии со способом Синтеза 1а'', заменяя бромхлорэтан на бромхлорбутан в Стадии А.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.81 (m, 1 Н), 7.05 (m, 1 Н), 7 (m, 2 Н), 6.77 (m, 2 Н), 6.5 (m, 1 Н), 6.47 (m, 1 Н), 6.3 (m, 1 Н), 3.92 (m, 2 Н), 3.7 (m, 2 Н), 1.8 (m, 4 Н), 0.97 (m, 9 Н), 0.2 (m, 6 Н).
ИК: ν:>NH: 3401 см-1.
Синтез 4а'': 3-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-5-(2-хлорэтокси)бензонитрил
Стадия А: (3-бром-5-метоксифенил)метанол
К раствору 3-бром-5-метоксибензойной кислоты (10 г; 43,3 ммоль) в тетрагидрофуране (280 мл), добавляют по каплям комплекс боран-диметилсульфид (32,5 мл; 64,9 ммоль), затем все перемешивают в течение 2 часов. Реакционную среду подкисляют по каплям водным 2 н. раствором соляной кислоты до рН=1. После экстракции простым эфиром, органическую фазу промывают водным 1 н. раствором гидроксида натрия и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Указанный в заголовке продукт получают в виде масла, которое используют в следующей стадии без очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.08 (m, 1 Н), 6.99 (m, 1 Н), 6.89 (m, 1 Н), 5.3 (br. s, 1 Н), 4.47 (s, 2 Н), 3.75 (s, 3 Н). "br." означает "широкий".
ИК: ν: -ОН: 1588 см-1;>С-O: 1268 и 1038 см-1; γ:>СН-Ar: 811 см-1.
Стадия В: 3-бром-5-метоксибензальдегид
К раствору соединения, полученного в Стадии А (8,6 г; 39,8 ммоль) в дихлорметане (400 мл), добавляют реагент Десса-Мартина (20,3 мл; 47,8 ммоль), затем все перемешивают в течение 2 часов. После добавления простого эфира, реакционную среду фильтруют через слой кремнезема. Фильтрат концентрируют, ресуспендируют в смеси гептана и этилацетата, затем фильтруют снова через слой кремнезема. После концентрации фильтрата, указанный в заголовке продукт получают в виде бледно-желтого твердого вещества, которое используют в следующей стадии без очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 9.5 (s, 1 Н), 7.69 (t, 1 Н), 7.5 (t, 1 Н), 7.42 (t, 1 Н), 3.85 (s, 3 Н).
ИК: ν:>С=0: 1691 см-1.
Стадия С: (Е)-1-(3-бром-5-метоксифенил)-N-гидроксиметанимин
К раствору соединения, полученного в Стадии В (7,8 г; 36,4 ммоль) в этаноле (10 мл), добавляют последовательно гидроксиламина гидрохлорид (12,6 г; 182 ммоль) и пиридин (6,27 мл; 87,4 ммоль), затем все перемешивают при 65°С в течение 1 часа. После достижения температуры окружающей среды, реакционную среду разбавляют со смесью из этилацетата и воды. После декантации, органическую фазу промывают водой и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Указанный в заголовке продукт получают в виде белого твердого вещества, которое используют в следующей стадии без очистки.
1Н ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 11.45 (s, 1 Н), 8.1 (s, 1 Н), 7.35 (t, 1 Н), 7.16 (d, 2 Н), 3.8 (s, 3 Н).
ИК: ν: -ОН: 3300-3000 см-1; Ar: 1600 и 1564 см-1;>С-O: 1220 и 1059 см-1; -N-O: 960 см-1; γ:>СН-Ar: 831 см-1.
Стадия D: 3-бром-5-метоксибензонитрил
К раствору соединения, полученного в Стадии С (8,1 г; 35,2 ммоль) в диоксане (70 мл), добавляют при 0°С пиридин (22 мл; 211 ммоль) и, по каплям, ангидрид трифторуксусной кислоты (1,4 мл; 70,4 ммоль), затем все перемешивают при температуре окружающей среды в течение 24 часов. Реакционную среду помещают при 0°С, затем добавляют по каплям вторую порцию трифторуксусного ангидрида (1,4 мл; 70,4 ммоль). Затем все перемешивают при температуре окружающей среды в течение 24 часов. Реакционную среду снова помещают при 0°С, затем добавляют по каплям третью порцию трифторуксусного ангидрида (1,4 мл; 70,4 ммоль). Все перемешивают при 60°С в течение 1 часа. После достижения температуры окружающей среды, реакционную среду разбавляют смесью дихлорметана и воды. После декантации, органическую фазу промывают водным 1 н. раствором хлористоводородной кислоты и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Указанный в заголовке продукт получают в виде бледно-желтого твердого вещества, которое используют в следующей стадии без очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.69 (t, 1 Н), 7.53 (dd, 1 Н), 7.5 (dd, 1 Н), 3.82 (s, 3 Н).
ИК: ν: -CN: 2232 см-1; Ar: 1597 и 1562 см-1;>С-O-С<: 1278 и 1051 см-1; γ:>СН-Ar: 848, 814 и 671 см-1.
Стадия Е: 3-бром-5-гидроксибензонитрил
К раствору соединения, полученного в Стадии D (5,9 г; 27,9 ммоль) в 2,4,6-коллидине (55 мл) добавляют йодид лития (11,2 г; 83,7 ммоль), затем все перемешивают при 150°С в течение 16 часов. После достижения температуры окружающей среды, реакционную среду выливают в ледяную воду. После экстракции дихлорметаном, органические фазы объединяют, промывают водой, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Указанный в заголовке продукт получают в виде оранжево-коричневого твердого вещества, которое используют в следующей стадии без очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 10.7 (br. s, 1 Н), 7.51 (t, 1 Н), 7.3 (t, 1 Н), 7.18 (dd, 1 Н).
ИК: ν: -ОН: 3283 см-1; -CN: 2245 см-1.
Стадия F: 3-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-5-(2-хлорэтокси)-бензонитрил
Указанное в заголовке соединение получают в соответствии со способами Стадий А и С Синтеза 1а'', используя соединение, полученное в предыдущей Стадии и бромхлорэтан в качестве исходных продуктов.
1Н ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 8.29 (s, 1 Н), 7.04 (d, 2 Н), 6.82 (d, 2 Н), 6.79/6.75/6.67 (3*m, 3 Н), 4.24 (dd, 2 Н), 3.91 (dd, 2 Н), 1.19 (s, 6 Н), 0.95 (s, 9 Н).
ИК: ν:>NH: 3332 см-1; -CN: 2232 см-1; Ar: 1595 и 1504 см-1;>С-O-С<: 1250 см-1; γ: -Si-C: 828 см-1.
Синтез 5а'': 3-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-5-(3-хлорпропокси)бензонитрил
Указанное в заголовке соединение получают в соответствии со способом Синтеза 4а'', заменяя бромхлорэтан на бромхлорпропан в Стадии F.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 8.3 (m, 1 Н), 7.05-6.85 (m, 4 Н), 6.8-6.6 (m, 3 Н), 4.1 (m, 2 Н), 3.8 (m, 2 Н), 2.05 (m, 2 Н), 0.95 (m, 9 Н), 0.2 (m, 6 Н).
ИК: ν:>NH: 3345 см-1; -CN: 2229 см-1.
Синтез 6а'': N-(4-{[трет-бутил(диметил)силил]окси}фенил)-5-(3-хлорпропил)-1-метил-1Н-пиразол-4-амин
Стадия А: 5-(3-{[трет-бутил(диметил)силил]окси}пропил)-1-метил-1Н-пиразол
К раствору N-метилпиразола (3,2 г; 39 ммоль) в тетрагидрофуране (65 мл), добавляют по каплям при -78°С раствор н-бутиллития в гексане (26,8 мл; 42,9 ммоль), затем все перемешивают в течение 1 часа до достижения температуры 0°С. Реакционную среду затем помещают при -78°С, и добавляют (3-бромпропокси)-трет-бутилдиметилсилан (10,6 мл; 46,8 ммоль). Все перемешивают при температуре окружающей среды в течение 16 часов и выливают в смесь ледяной воды и этилацетата. После экстрагирования этилацетатом, органическую фазу промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 7.35 (d, 1 Н), 7 (d, 1 Н), 3.8 (s, 3 Н), 3.65 (t, 2 Н), 2.7 (m, 2 Н), 1.85 (m, 2 Н), 0.9 (s, 9 Н), 0.5 (s, 6 Н).
ИК: γ: СН3: 1254 см-1; ν: -Si-O-: 1098 см-1; -Si-C-: 834 и 772 см-1.
Стадия В: 3-(4-бром-1-метил-1Н-пиразол-5-ил)пропан-1-ол
К раствору соединения, полученного в Стадии А (4,8 г; 18,9 ммоль) в метаноле (200 мл), при 0°С добавляют трибромид пиримидиния (6,6 г; 20,8 ммоль). Все перемешивают в течение 1 часа при 0°С, затем в течение 16 часов при температуре окружающей среды. После концентрации реакционной смеси, остаток ресуспендируют в смеси водного 10% раствора карбоната калия и дихлорметана. После экстракции дихлорметаном, органическую фазу промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и аммиак в метаноле в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.45 (s, 1 Н), 4.59 (t, 1 Н), 3.79 (s, 3 Н), 3.4 (quad, 2 Н), 2.7 (t, 2 Н), 1.65 (m, 2 Н).
ИК: ν: -ОН: 3348 см-1.
Стадия С: 4-бром-5-(3-хлорпропил)-1-метил-1Н-пиразол
К раствору соединения, полученного в Стадии В (3,9 г; 17,2 ммоль) в тетрагидрофуране (40 мл), добавляют по каплям при 0°С тионилхлорид (2,6 мл; 36,4 ммоль), затем все перемешивают в течение 1 часа при 50°С. После концентрации реакционной смеси, остаток ресуспендируют в смеси воды и этилацетата. После экстрагирования этилацетатом, органическую фазу промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и аммиак в метаноле в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.48 (s, 1 Н), 3.8 (s, 3 Н), 3.69 (t, 2 Н), 2.8 (t, 2 Н), 1.95 (m, 2 Н).
Стадия D: N-(4-{[трет-бутил(диметил)силил]окси}фенил)-5-(3-хлорпропил)-1-метил-1Н-пиразол-4-амин
Раствор соединений, полученных в Стадии С (3,6 г; 15,2 ммоль) и в Стадии 8 (3,4 г; 15,2 ммоль) Синтеза 1а'' в смеси толуола (25 мл) и тетрагидрофурана (25 мл) дегазируют барботированием через аргон в течение 10 минут. К нему добавляют трет-бутилат натрия (1,75 г; 18,2 ммоль) и хлор(2-ди-трет-бутилфосфино-2',4',6'-триизо пропил-1,1'-бифенил)[2-(2-аминоэтил)фенил]палладий(II) (0,5 г; 0,76 ммоль), затем все перемешивают при температуре окружающей среды в течение 2 часов. Реакционную среду фильтруют через Celite®, затем концентрируют после промывания тетрагидрофураном. Остаток ресуспендируют смесью воды и дихлорметана, затем экстрагируют дихлорметаном, сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают с помощью первой хроматографии на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, затем второй хроматографии, используя дихлорметан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.21 (s, 1 Н), 6.59 (d, 2 Н), 6.45 (d, 2 Н), 3.72 (s, 3 Н), 3.55 (t, 2 Н), 2.89 (quint, 2 Н), 2.65 (t, 2 Н), 0.91 (s, 9 Н), 0.1 (s, 6 Н).
Синтез 7а'': 4-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1-(4-хлорбутил)-5-метил-1Н-пиррол-2-карбонитрил
Стадия А: 5-метил-1Н-пиррол-2-карбонитрил
К раствору этилацетамидоцианоацетата (50,0 г; 0,29 моль) в этаноле (1,25 л), добавляют этаноат натрия (100 г; 1,47 моль), затем все перемешивают в течение 10 минут при 30°С, затем в течение 10 минут при 50°С. При этой температуре, добавляют по каплям в течение 2 часов раствор 1,4-дихлор-2-бутин (72,3 г; 0,587 моль) в этаноле (250 мл), затем все перемешивают при кипячении с обратным холодильником в течение 100 минут. После достижения температуры окружающей среды, добавляют водный раствор 2 н. соляной кислоты (588 мл), затем этанол концентрируют. После экстрагирования этилацетатом, органическую фазу промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Полученный остаток дистиллируют в вакууме, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 8.76 (br. s, 1 Н), 6.76 (t, 1 Н), 5.93-5.96 (m, 1 Н), 2.3 (s, 3 Н).
Стадия В: 4-бром-5-метил-1Н-пиррол-2-карбонитрил
К раствору соединения, полученного в Стадии А (40,7 г; 0,341 моль) в смеси уксусной кислоты (325 мл) и дихлорметана (122 мл), добавляют при 10°С в течение 75 минут раствор брома (59,9 г; 0,374 моль) в уксусной кислоте (163 мл). Затем все перемешивают в течение 30 минут при этой температуре, затем в течение 1 часа при температуре окружающей среды. Реакционную среду гидролизуют (200 мл) и дихлорметан концентрируют. После достижения температуры окружающей среды, добавляют воду (400 мл) и полученную суспензию перемешивают в течение 2 часов при 0°С.Осадок фильтруют и сушат в вакууме, чтобы получить указанный в заголовке продукт без очистки.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 9.37 (br. s, 1 Н), 6.78 (d, 1 Н), 2.27 (s, 3 Н).
Стадия С: 4-бром-1-(4-хлорбутил)-5-метил-1Н-пиррол-2-карбонитрил
К смеси 60% гидрида натрия в масле (0,95 г; 23,8 ммоль) в небольшом количестве N,N-диметилформамида, добавляют соединение, полученное в Стадии В (4 г; 21,6 ммоль) в N,N-диметилформамид (120 мл), затем все перемешивают в течение 15 минут при температуре окружающей среды перед тем, как добавить 1,4-дихлорбутан (4,7 мл; 43,2 моль). Реакционную среду оставляют перемешиваться при температуре окружающей среды в течение 3 дней, перед разбавлением в воде (1,5 л). Затем продукт экстрагируют этилацетатом. Органические фазы промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 6.76 (s, 1 Н), 4.05 (t, 2 Н), 3.56 (t, 2 Н), 2.27 (s, 3 Н), 1.87-1.95 (m, 2 Н).
Стадия D: 4-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1-(4-хлорбутил)-5-метил-1Н-пиррол-2-карбонитрил
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии С Синтеза 1а'', исходя из соединения, полученного в предыдущей стадии.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 6.64-6.69 (m, 3 Н), 6.46-6.51 (m, 2 Н), 4.67 (br. s, 1 Н), 4.04 (t, 2 Н), 3.57 (t, 2 Н), 2.15 (s, 3 Н), 1.89-1.99 (m, 2 Н), 1.79-1.88 (m, 2 Н), 0.96 (s, 9 Н), 0.15 (s, 6 Н).
Синтез 8а'': 4-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1-(3-хлорпропил)-5-метил-1Н-пиррол-2-карбонитрил
Указанное в заголовке соединение получают в соответствии со способом Синтеза 7а'', заменяя 1,4-дихлорбутан на бромхлорпропан в Стадии С.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 6.68 (s, 1 Н), 6.67 (d, 2 Н), 6.48 (d, 2 Н), 4.67 (br. s, 1 Н), 4.18 (t, 2 Н), 3.56 (t, 2 Н), 2.26 (quint., 2 Н), 2.18 (m, 3 Н), 0.96 (s, 9 Н), 0.15 (s, 6 Н).
Синтез 9а'': 3-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-5-(3-хлорпропил)бензонитрил
Стадия А: (3-бром-5-йодфенил)метанол
К раствору 3-бром-5-йодбензойной кислоты (10 г; 30,58 ммоль) в тетрагидрофуране (70 мл), добавляют по каплям при 0°С комплекс боран-тетрагидрофуран 1 M в тетрагидрофуране (61,1 мл; 61,1 ммоль), затем все перемешивают в течение 16 часов при температуре окружающей среды. Реакционную среду разбавляют в метаноле (10 мл) и гидролизуют водным 1 М раствором гидроксида натрия (100 мл). После экстракции дихлорметаном, органическую фазу промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Указанный в заголовке продукт получают в виде твердого вещества бледно-коричневого цвета, которое используют в следующей стадии без очистки.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 7.76-7.79 (m, 1 Н), 7.63-7.66 (m, 1 Н), 7.47-7.50 (m, 1 Н), 4.64 (s, 2 Н).
Стадия В: 3-бром-5-йодбензальдегид
К раствору соединения, полученного в Стадии А (7,89 г; 25,2 ммоль) в дихлорметане (80 мл), добавляют дихромат пиримидиния (12,3 г; 32,8 ммоль), затем все перемешивают в течение 16 часов при температуре окружающей среды. Реакционную среду фильтруют через силикагель, затем фильтрат концентрируют, чтобы обеспечить указанный в заголовке продукт без очистки.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 9.87 (s, 1 Н), 8.12 (t, 1 Н), 8.10 (t, 1 Н), 7.96 (t, 1 Н).
Стадия С: 3-бром-5-йодбензонитрил
К раствору соединения, полученного в Стадии В (6,95 г; 22,3 ммоль) в тетрагидрофуране (60 мл), добавляют водный раствор гидроксида аммония при 28% (30 мл) и йода (6,81 г; 26,8 ммоль), затем все перемешивают до исчезновения исходного соединения. Реакционную среду разбавляют водным раствором сульфита натрия до исчезновения оранжевого цвета. После экстрагирования этилацетатом, органическую фазу промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом натрия, фильтруют и концентрируют досуха с помощью кремнезема. Осажденный таким образом на силикагель продукт очищают с помощью хроматографии на силикагеле, используя дихлорметан и гептан в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 8.10 (s, 1 Н), 7.92 (s, 1 Н), 7.76 (s, 1 Н).
Стадия D: 3-бром-5-(3-оксопропил)бензонитрил
К раствору соединения, полученного в Стадии С (6,05 г; 19,6 ммоль) в N,N-диметилформамиде (85 мл), добавляют аллиловый спирт (2,78 мл; 39,3 ммоль), хлорид бензилтриэтиламмония (4,47 г; 19,6 ммоль) и бикарбонат натрия (3,30 г; 39,3 ммоль). После продувки азотом добавляют ацетат палладия(П) (0,13 г; 0,59 ммоль), затем все доводят до 40°С в течение 16 часов. Реакционную среду разбавляют в смеси воды (200 мл) и этилацетата (100 мл). После экстрагирования этилацетатом, органическую фазу промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом натрия, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя этилацетат и гептан в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 9.81 (t, 1 Н), 7.64 (t, 1 Н), 7.59-7.61 (m, 1 Н), 7.44 (t, 1 Н), 2.93-3.02 (m, 2 Н), 2.81-2.88 (m, 2 Н).
Стадия Е: 3-бром-5-(3-гидроксипропил)бензонитрил
К раствору соединения, полученного в Стадии D (2,98 г; 12,52 ммоль) в метаноле (30 мл), добавляют порциями боргидрид натрия (0,62 г; 16,27 ммоль). Все перемешивают при температуре окружающей среды в течение 30 минут. Реакционную среду разбавляют водным раствором 1 М гидроксида натрия (50 мл). После экстракции дихлорметаном, органическую фазу промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом натрия, фильтруют и концентрируют досуха, чтобы получить указанный в заголовке продукт без очистки.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 7.61-7.64 (m, 1 Н), 7.59-7.61 (m, 1 Н), 7.44 (t, 1 Н), 3.68 (t, 2 Н), 2.72-2.79 (m, 2 Н), 1.83-1.93 (m, 2 Н).
Стадия F: 3-бром-5-(3-хлорпропил)бензонитрил
К раствору соединения, полученного в Стадии Е (2,57 г; 10,7 ммоль), и триэтиламина (3,43 мл; 24,6 ммоль) в дихлорметане (30 мл) при 0°С, добавляют метансульфонилхлорид (1,65 мл; 21,4 ммоль). Все перемешивают при температуре окружающей среды в течение 2 часов, затем добавляют хлорид тетрабутиламмония (8,92 г; 32,1 ммоль). Реакционную среду перемешивают в течение 16 часов, затем разбавляют в смеси из воды и дихлорметана. После экстрагирования этилацетатом, органическую фазу промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом натрия, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя этилацетат и гептан в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1Н ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 7.65 (t, 1 Н), 7.60 (s, 1 Н), 7.44 (s, 1 Н), 3.53 (t, 2 Н), 2.78-2.86 (m, 2 Н), 2.04-2.14 (m, 2 Н).
Стадия G: 3-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-5-(3-хлорпропил)бензонитрил
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии С Синтеза 1а'', исходя из соединения, полученного в предыдущей стадии.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 6.78-7.07 (m, 7 Н), 5.60 (br. s, 1 Н), 3.52 (t, 2 Н), 2.71 (t, 2 Н), 2.00-2.10 (m, 2 Н), 0.99 (s, 9 Н), 0.27 (s, 6 Н).
Синтез 10а'': N-(4-{[трет-бутил(диметил)силил]окси}фенил)-1-метил-5-[2-(тетрагидро-2Н-пиран-2-илокси)этил]-1Н-пиразол-4-амин
Стадия А: 5-(2-{[трет-бутил(диметил)силил]окси}этил)-1-метил-1Н-пиразол
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии А Синтеза 6а'', заменяя (3-бромпропокси)-трет-бутилдиметилсилан на (2-бромэтокси)-трет-бутилдиметилсилан.
1H ЯМР (400/500 МГц, CDCl3, 300 K) δ част. на млн.: 7.35 (d, 1 Н), 7 (d, 1 Н), 3.8 (s, 3 Н), 3.65 (t, 2 Н), 2.7 (m, 2 Н), 1.85 (m, 2 Н), 0.9 (s, 9 Н), 0.5 (s, 6 Н)
ИК: ν: -Si-O-: 1098 см-1; -Si-C-: 834 и 772 см-1.
Стадия В: 2-(4-бром-1-метил-1Н-пиразол-5-ил)этанол
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Синтеза 6а''.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.45 (s, 1 Н), 4.59 (t, 1 Н), 3.79 (s, 3 Н), 3.4 (quad, 2 Н), 2.7 (t, 2 Н), 1.65 (m, 2 Н).
ИК: ν: -ОН: 3348 см-1.
Стадия С: 4-бром-1-метил-5-[2-(тетрагидро-2Н-пиран-2-илокси)этил]-1Н-пиразол
К раствору соединения, полученного в предыдущей Стадии (5,34 г; 2.4 ммоль) в дихлорметане (40 мл), добавляют 3,4-дигидро-2H-пиран (7 мл; 6 ммоль) и пара-толуолсульфоновую кислоту (4,6 г; 2,4 ммоль), затем все перемешивают в течение 16 часов. Реакционную среду разбавляют в водном насыщенном растворе гидрокарбоната натрия. После экстракции дихлорметаном, органическую фазу сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.42 (s, 1 Н), 4.55 (t, 1 Н), 3.8-3.3 (m, 4 Н), 3.8 (s, 3 Н), 2.71 (m, 2 Н), 1.78 (m, 2 Н), 1.7-1.4 (m, 6 Н).
Стадия D: N-(4-{[трет-бутил(диметил)силил]окси}фенил)-1-метил-5-[2-(тетрагидро-2Н-пиран-2-илокси)этил]-1Н-пиразол-4-амин
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии D Синтеза 6а'', исходя из бромированного соединения предыдущей стадии.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.2 (s, 1 Н), 6.6 (s, 1 Н), 6.55 (d, 2 Н), 6.45 (d, 2 Н), 4.4 (t, 1 Н), 3.7 (s, 3 Н), 3.65-3.2 (4 m, 4 Н), 2.58 (m, 2 Н), 1.68 (m, 2 Н), 1.6-1.3 (m, 6 Н), 0.92 (s, 9 Н), 0.1 (s, 6 Н).
ИК: ν:>NH: 3356 см-1; ->С-С-O-: 1240 см-1.
Синтез 11а'':
Стадия А: 4-(проп-2-ен-1-илокси)анилин
К раствору N-(аллилоксифенил)ацетамида (4 г; 20,9 ммоль) в этаноле (30 мл) добавляют концентрированный раствор гидроксида натрия (7 мл; 83,6 ммоль). Все перемешивают при 100°С в течение 24 часов. После достижения температуры окружающей среды, этанол реакционной смеси концентрируют, затем остаток ресуспендируют в воде (100 мл). После экстракции дихлорметаном, органические фазы сушат над сульфатом магния, фильтруют и концентрируют досуха, чтобы получить указанный в заголовке продукт без очистки.
1Н ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 6.66 (d, 2 Н), 6.49 (d, 2 Н), 6 (m, 1 Н), 5.34 (dq, 1 Н), 5.2 (dq, 1 Н), 4.59 (s, 2 Н), 4.4 (dt, 2 Н).
ИК: ν: -NH2: 3428, 3354 и 3220 см-1.
Стадия В: 4-фтор-3-{[4-(проп-2-ен-1-илокси)фенил]амино}бензонитрил
Указанное в заголовке соединение получают в соответствии со способом из Стадии С Синтеза 1а'', используя соединение, полученное в предыдущей Стадии и 3-бром-4-фторбензонитрил.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 8.08 (s, 1 Н), 7.36 (dd, 1 Н), 7.24 (dd, 1 Н), 7.19 (m, 1 Н), 7.12 (d, 2 Н), 6.96 (d, 2 Н), 6.05 (m, 1 Н), 5.4 (ddt, 1 Н), 5.26 (ddt, 1 Н), 4.56 (dt, 2 Н).
ИК: ν: -NH: 3327 см-1;>CN: 2235 см-1.
Синтез 1b: 5-[2-(трет-бутоксикарбонил)-5-хлофенил]-1,2-диметил-1Н-пиррол-3-карбоновая кислота
Стадия А: этил 1,2-диметил-1Н-пиррол-3-карбоксилат
Методика идентична описанной в Стадии А Синтеза 1а.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 6.50 (d, 1 Н), 6.45 (d, 1 Н), 4.25 (q, 2 Н), 3.51 (s, 3 Н), 2.49 (s, 3 Н), 1.33 (t, 3 Н).
Стадия В: кислота 1,2-диметил-1Н-пиррол-3-карбоновая
Методика идентична описанной в Стадии В Синтеза 1а.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 11.52 (s, 1 Н), 6.62 (d, 1 Н), 6.29 (d, 1 Н), 3.50 (s, 3 Н), 2.41 (s, 3 Н).
Стадия С: трет-бутил 2-бром-4-хлобензоат
К раствору из сульфата магния (1431 г; 11,9 моль) в дихлорметане (10,5 л), добавляют серную кислоту (287 г; 2,97 моль) в течение 30 минут, затем 2-бром-4-хлорбензойную кислоту (700 г; 2,97 моль) и 700 мл трет-бутанола. Реакционную смесь перемешивают в течение 4 дней при температуре окружающей среды, затем фильтруют. Фильтрат промывают водным раствором гидрокарбоната калия 5%, затем органическую фазу сушат над сульфатом натрия и концентрируют досуха, затем повторно концентрируют в гептане (1 л), чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 7.65 (d+d, 2 Н), 7.30 (dd, 1 Н), 1.65 (s, 9 Н).
Стадия D: 5-[2-(трет-бутоксикарбонил)-5-хлофенил]-1,2-диметил-1Н-пиррол-3-карбоновая кислота
К дегазированному раствору (барботированием азота в течение 15 минут) остатка, полученного в Стадии С (187,5 г; 0,643 моль) и соединения, полученного в Стадии В (89,6 г; 0,643 моль) в N,N-диметилформамиде (1,87 л), добавляют карбонат калия (178 г; 1,29 моль; предварительно растертый в порошок с помощью дробилки Ultra-Turrax®) в суспензию в этилацетате. Затем суспензию дегазируют в течение дополнительных 15 минут. Добавляют ацетат палладия(II) (7,2 г, 0,003 моль), затем суспензию нагревают до 100°С и перемешивают в течение 18 часов. После достижения температуры окружающей среды, реакционную смесь разбавляют водой (950 мл). Ту же самую процедуру осуществляют второй раз с таким же количеством соединения, полученного в Стадии С.
Два раствора объединяют и промывают метил-трет-бутиловым эфиром. Водные фазы с рН=10 подкисляют до рН=2 водным раствором 12 н. хлористоводородной кислоты при температуре между 10 и 20°С. Полученную суспензию охлаждают до 0°С, перемешивают в течение 1 часа затем фильтруют. Твердое вещество промывают водой (2 л), затем осушают в течение 1 часа. К раствору остатка, ресуспендированного в метаноле (12 л) добавляют уголь (375 г). Суспензию нагревают до 40°С и перемешивают в течение 2 часов. Смесь фильтруют через Celite® (375 г) и твердое вещество промывают метанолом. Фильтрат концентрируют досуха, и полученный остаток разбавляют в смеси этанола (1,3 л) и метанола (400 мл). Суспензию дистиллируют; 400 мл дистиллята собирают. Загружают этанол (1 л) для продолжения дистилляции до тех пор, пока не будет собран 1 л дистиллята. После достижения температуры окружающей среды, суспензию перемешивают в течение 16 часов, затем охлаждают до 0°С и снова перемешивают в течение 2 часов. Продукт фильтруют и промывают холодным этанолом, затем сушат в вакууме при 60°С в течение 16 часов. Указанный в заголовке продукт получают в виде твердого вещества белого цвета.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 10.87-12.20 (s, 1 Н), 7.77 (d, 1H), 7.57 (dd, 1 Н), 7.44 (d, 1 Н), 6.25 (s, 1 Н), 3.25 (s, 3 Н), 2.51 (s, 3 Н), 1.25 (t, 9 Н).
Синтез 2b: 5-[2-(трет-бутоксикарбонил)-5-фторфенил]-1,2-диметил-1Н-пиррол-3-карбоновая кислота
Указанное в заголовке соединение получают в соответствии со способом Синтеза 1b, используя 2-бром-4-фторбензойную кислоту в Стадии С.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 12.18 (s, 1 Н), 7.93 (dd, 1Н), 7.14 (td, 1 Н), 7.03 (dd, 1 Н), 6.52 (s, 1 Н), 3.27 (s, 3 Н), 2.60 (s, 3 Н), 1.33 (t, 9 Н).
Синтез 1b': трет-бутил 5-(3-гидроксипропил)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: метил (2Е)-3-(изохинолин-5-ил)проп-2-еноат
В запечатанной пробирке, содержащей раствор метилакрилата (5,4 мл; 60,2 ммоль), трифенилфосфина (0,63 г; 2,4 ммоль), триэтиламина (13,4 мл; 96,0 ммоль) и ацетата палладия(II) (0,27 г; 1,2 ммоль) в N,N-диметилформамиде (30 мл), добавляют 5-бромизохинолин (5 г; 24,1 ммоль). Азот барботируют в смеси в течение 10 минут, затем пробирку запечатывают и погружают в масляную баню при 120°С. Реакционную смесь перемешивают в течение 1,5 часов, затем гидролизуют после достижения температуры окружающей среды. Продукт экстрагируют этилацетатом, затем органические фазы промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 9.29 (s, 1 Н), 8.62 (d, 1H), 8.42 (d, 1 Н), 8.03 (d, 1 Н), 7.94-8.01 (m, 2 Н), 7.64 (t, 1 Н), 6.57 (d, 1 Н), 3.88 (s, 3 Н).
Стадия В: трет-бутил 5-[(1Е)-3-метокси-3-оксопроп-1-ен-1-ил]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Раствор соединения, полученного в Стадии А (7,24 г; 34 ммоль), и цианоборгидрида натрия (9,6 г; 152,8 ммоль) в метаноле (200 мл), помещают при 45°С. Диэтилэфират трифторида бора (18,9 мл; 152,8 ммоль) добавляют по каплям, затем реакционную смесь перемешивают в течение 20 минут при этой температуре, и добавляют ди-трет-бутилкарбонат (8,15 г; 37,4 ммоль) затем триэтиламин (14,2 мл; 101,9 ммоль). Реакционную среду перемешивают в течение 15 минут при 45°С. После достижения температуры окружающей среды, ее гидролизуют водой и водным раствором 1 н. гидроксида натрия. Продукт экстрагируют этилацетатом, затем органические фазы промывают водным раствором хлористоводородной кислоты и воды, сушат над сульфатом магния, фильтруют и концентрируют досуха, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 7.94 (d, 1 Н), 7.44 (d, 1 Н), 7,21 (t, 1 Н), 7.11-7.17 (m, 1 Н), 6.35 (d, 1 Н), 4.58 (s, 2 Н), 3.81 (s, 3 Н), 3.67 (t, 2 Н), 2.93 (t, 2 Н), 1.49 (s, 9 Н).
Стадия С: трет-бутил 5-[(1Е)-3-гидроксипроп-1-ен-1-ил]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К 25 мас. % раствору гидрида диизобутилалюминия в толуоле (50 мл; 74,7 ммоль), медленно при -78°С добавляют раствор соединения, полученного в Стадии В (10,78 г; 34 ммоль) в толуоле (200 мл). Реакционную смесь перемешивают в течение 20 минут при этой температуре, затем медленно добавляют метанол. После достижения температуры окружающей среды, реакционную среду гидролизуют водой и водный раствор 1 н. гидроксида натрия. Продукт экстрагируют этилацетатом, затем органические фазы промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют досуха, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 7.33 (d, 1 Н), 7.17 (t, 1 Н), 7,03 (d, 1 Н), 6.80 (d, 1 Н), 6.24 (dt, 1 Н), 4.54-4.60 (m, 2 Н), 4.35 (d, 2 Н), 3.65 (t, 2 Н), 2.84 (t, 2 Н), 1.49 (s, 9 Н).
Стадия D: трет-бутил 5-(3-гидроксипропил)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Стадии С (9,82 г; 34,0 ммоль) в метаноле (300 мл), добавляют палладий на угле (1 г; 10 мас. %). Реакционную смесь гидрируют в течение 16 часов затем фильтруют через Celite®. Фильтрат концентрируют досуха, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 7.32 (d, 1 Н), 7.16 (t, 1 Н), 7,02 (d, 1 Н), 6.79 (d, 1 Н), 6.24 (dt, 1 Н), 4.57 (s, 2 Н), 4.34 (d, 2 Н), 3.62-3.67 (m, 2 Н), 3.49 (s, 2 Н), 2.84 (t, 2 Н), 1.49 (s, 9 Н).
Синтез 2b': трет-бутил 5-(4-гидроксибутокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: трет-бутил 5-гидрокси-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору 5-гидрокси-изохинолина (20 г; 137 ммоль) в уксусной кислоте (120 мл) добавляют диоксид платины (2 г; 8,8 ммоль). Все помещают под атмосферу водорода (2 бара) в течение 24 часов. Реакционную среду фильтруют, и катализатор промывают толуолом. Полученный таким образом фильтрат концентрируют досуха. Указанный в заголовке продукт получают в виде масла, которое используют в дальнейшем без очистки.
К раствору полученного остатка (1,95 г; 13 ммоль) в дихлорметане (110 мл) добавляют диизопропилэтиламин (9,7 мл; 57 ммоль) и ди-трет-бутилдикарбонат (3,69 г, 16,9 ммоль), затем все перемешивают в течение 2 часов при температуре окружающей среды. Реакционную среду разбавляют с водным насыщенным раствором хлорида аммония. После декантации, органическую фазу промывают водным насыщенным раствором гидрокарбоната натрия, и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 9.41 (m, 1 Н), 6.97 (t, 1 Н), 6.64/6.54 (2d, 2 Н), 4.42 (m, 2 Н), 3.53 (t, 2 Н), 2.59 (t, 2 Н), 1.42 (s, 9 Н).
ИК: ν: -ОН: 3294 см-1;>С=O: 1652 см-1.
Стадия В: трет-бутил 5-[4-(тетрагидро-2Н-пиран-2-илокси)бутокси]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Стадии А (1 г; 4 ммоль) в ацетонитриле (15 мл), добавляют 2-(4-бромбутокси)-тетрагидропиран (0,77 мл; 4,2 ммоль) и карбонат цезия (1,4 г, 4,2 ммоль), затем все перемешивают в течение 18 часов при 70°С. Реакционную среду разбавляют с этилацетатом и водой. После декантации, органическую фазу промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.12 (dd, 1 Н), 6.79 (d, 1 Н), 6.72 (d, 1 Н), 4.55 (t, 1 Н), 4.45 (s, 2 Н), 3.98 (t, 2 Н), 3.74/3.42 (2*m, 2 Н), 3.68/3.41 (2*m, 2 Н), 3.54 (t, 2 Н), 2.63 (t, 2 Н), 1.79 (m, 2 Н), 1.74-1.4 (m, 6 Н), 1.68 (m, 2 Н), 1.42 (s, 9 Н).
ИК: ν:>С=0: 1693 см-1;>С-O-С<: 1033 см-1.
Стадия С: трет-бутил 5-(4-гидроксибутокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Стадии В (1,29 г; 3,18 ммоль) в метаноле (50 мл), добавляют пара-толуолсульфонат пиримидиния (0,16 г; 0,64 ммоль), затем все перемешивают в течение 8 часов при 60°С. Реакционную среду разбавляют дихлорметаном и водным насыщенным раствором хлорида аммония. После декантации, органическую фазу промывают водой, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.12 (dd, 1 Н), 6.78 (d, 1 Н), 6.72 (d, 1 Н), 4.45 (s, 2 Н), 4.42 (t, 1 Н), 3.96 (t, 2 Н), 3.54 (t, 2 Н), 3.45 (m, 2 Н), 2.62 (t, 2 Н), 1.75 (m, 2 Н), 1.58 (m, 2 Н), 1.42 (s, 9 Н).
ИК: ν: -ОН: 3600-3100 см-1;>С=0: 1693 см-1.
Синтез 3b': трет-бутил 5-гидрокси-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору 5-гидрокси-изохинолина (20 г; 137 ммоль) в уксусной кислоте (120 мл), добавляют диоксид платины (2 г; 8,8 ммоль). Все помещают под атмосферу водорода (2 бара) в течение 24 часов. Реакционную среду фильтруют, и катализатор промывают толуолом. Полученный таким образом фильтрат концентрируют досуха. Указанный в заголовке продукт получают в виде масла, которое используют в дальнейшем без очистки.
К раствору полученного остатка (1,95 г; 13 ммоль) в дихлорметане (110 мл), добавляют диизопропилэтиламин (9,7 мл; 57 ммоль) и ди-трет-бутилдикарбонат (3,69 г, 16,9 ммоль), затем все перемешивают в течение 2 часов при температуре окружающей среды. Реакционную среду разбавляют с водным насыщенным раствором хлорида аммония. После декантации, органическую фазу промывают водным насыщенным раствором гидрокарбоната натрия, и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 9.41 (m, 1 Н), 6.97 (t, 1 Н), 6.64/6.54 (2d, 2 Н), 4.42 (m, 2 Н), 3.53 (t, 2 Н), 2.59 (t, 2 Н), 1.42 (s, 9 Н).
ИК: ν: -ОН: 3294 см-1; >С=0: 1652 см-1.
Синтез 4b': трет-бутил 5-гидрокси-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: метил 3-метокси-2-метилбензоат
К раствору 2-метил-3-метоксибензойной кислоты (20 г; 0,12 моль) в метаноле (200 мл), при 0°С по каплям добавляют тионилхлорид (17,5 мл; 0,24 моль). Реакционную среду кипятят с обратным холодильником в течение 2 часов. После достижения температуры окружающей среды, реакционную среду концентрируют, затем разбавляют в смеси этилацетата и водного раствора 1 н. гидроксида натрия. После декантации, органическую фазу промывают водой и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Указанный в заголовке продукт получают в виде масла, которое применяют в следующей стадии без очистки.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 7.39 (dd, 1 Н), 7.19 (t, 1 Н), 6.98 (d, 1 Н), 3.89 (s, 3 Н), 3.84 (s, 3 Н), 2.42 (s, 3 Н).
ИК: ν:>С=0: 1719; >С-O-С 1254 и 1066 см-1.
Стадия В: метил 2-(бромметил)-3-метоксибензоат
К раствору соединения, полученного в Стадии А (19,8 г; 0,11 моль) в четыреххлористом углероде (100 мл), добавляют N-бромсукцинимид (19,56 г; 0,18 моль) и азоизобутиронитрил (2 г; 0,012 моль). Реакционную среду кипятят с обратным холодильником в течение 3 часов. После достижения температуры окружающей среды, реакционную среду разбавляют в смеси дихлорметана и воды. После декантации, органическую фазу промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом натрия, фильтруют и концентрируют досуха. Остаток ресуспендируют в дихлорметане, чтобы обеспечить указанный в заголовке продукт в виде твердого вещества белого цвета, которое используют в следующей стадии без очистки.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 7.51 (d, 1 Н), 7.31 (t, 1 Н), 7.08 (d, 1 Н), 5.05 (s, 2 Н), 3.91 (2s, 6 Н).
ИК: ν:>С=O: 1713 см-1.
Стадия С: диэтил (ацетиламино)[2-метокси-6-(метоксикарбонил)бензил]пропандиоат
К суспензии гидрида натрия (3,42 г; 85,6 ммоль) в N,N-диметилформамиде, при температуре ниже 30°С добавляют по каплям раствор диэтилацетамидомалоната (16,9 г; 77,8 ммоль) в N,N-диметилформамиде (100 мл). Реакционную среду перемешивают в течение 15 минут, затем добавляют по каплям при температуре окружающей среды раствор соединения, полученного в Стадии В (21,2 г; 81,67 ммоль). После контакта в течение 18 часов реакционную среду концентрируют, затем разбавляют в смеси этилацетата и водного насыщенного раствора гидрокарбоната натрия. После экстрагирования этилацетатом, органические фазы объединяют, промывают водой и насыщенным водным раствором хлорида лития, затем сушат над сульфатом натрия, фильтруют и концентрируют досуха. Остаток ресуспендируют в диизопропиловом эфире, чтобы обеспечить указанный в заголовке продукт в виде твердого вещества не совсем белого цвета, которое используют в следующей стадии без очистки.
1Н ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.68 (s, 1 Н), 7.3 (t, 1 Н), 7.1 (2d, 2 Н), 4.15/4.05 (2 m, 4 Н), 3.81 (s, 2 Н), 3.71/3.7 (2s, 6 Н), 1.79 (s, 3 Н), 1.15 (t, 6 Н).
ИК: ν: -NH: 3367;>С=O: 1755, 1732 и 1707;>С=O: 1668;>С=С<: 1600 см-1.
Стадия D: 5-метокси-1-оксо-1,2,3,4-тетрагидроизохинолин-3-карбоновая кислота
К раствору соединения, полученного в Стадии С (8,1 г; 20 ммоль) в водном растворе 5 н. соляной кислоты, добавляют уксусную кислоту (40 мл). Реакционную среду кипятят с обратным холодильником в течение 18 часов, затем ее фильтруют после достижения температуры окружающей среды. Осадок промывают водным раствором 5 н. хлористоводородной кислоты и толуолом. После сушки в вакууме, указанный в заголовке продукт получают в виде твердого вещества кремового цвета, которое используют в следующей стадии без очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 12.75 (s, 1 Н), 7.98 (d, 1 Н), 7.48 (d, 1 Н), 7.3 (t, 1 Н), 7.15 (d, 1 Н), 4.2 (m, 1 Н), 3.8 (s, 3 Н), 3.28/3.05 (m, 2 Н).
ИК: ν: -NH/OH: 3215 и 3000 до 2000;>С=0: 1715 и 1627 см-1.
Стадия Е: 5-метокси-3-(морфолин-4-илкарбонил)-3,4-дигидроизохинолин-1 (2Н)-он
К раствору соединения, полученного в Стадии D (11,1 г; 50 ммоль), в дихлорметане (150 мл), добавляют последовательно морфолин (4,4 мл; 50 ммоль), 1-гидроксибензотриазол (6,7 г; 50 ммоль), гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимид (9,6 г; 50 ммоль) и диизопропилэтиламин (20 мл; 115,2 ммоль). Затем все перемешивают в течение ночи при температуре окружающей среды. Реакционную среду разбавляют смесью дихлорметана и воды. После декантации, органическую фазу промывают с водным насыщенным раствором хлорида аммония и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток ресуспендируют в дихлорметане, фильтруют и промывают горячим изопропанолом, чтобы получить указанный в заголовке продукт, который используют в следующей стадии без очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.71 (d, 1 Н), 7.45 (d, 1 Н), 7.29 (t, 1 Н), 7.11 (d, 1 Н), 4.7 (m, 1 Н), 3.81 (s, 3 Н), 3.65-3.3 (неразрешенный пик, 8 Н), 3.05/2.95 (2*dd, 2 Н).
ИК: ν: -NH: 3284; >С=O: 1676; >С-O-С<: 1268 и 1248 см-1.
Стадия F: 5-метокси-3-(морфолин-4-илметил)-1,2,3,4-тетрагидроизохинолин
К раствору соединения, полученного в Стадии Е (5 г; 17,2 ммоль) в тетрагидрофуране (300 мл), добавляют по каплям 2 М раствор комплекса боран/диметилсульфид в тетрагидрофуране (43 мл; 86 ммоль). Затем все перемешивают при кипячении с обратным холодильником в течение 5 часов затем при температуре окружающей среды в течение ночи. Водный раствор 5 н. соляной кислоты добавляют по каплям и реакционную среду кипятят с обратным холодильником в течение 8 часов. В заключение, водный раствор гидроксида натрия добавляют при 0°С до достижения основного рН, и реакционную среду разбавляют дихлорметаном. После экстрагирования, органические фазы промывают водой, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и аммиак в этаноле в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.61 (d, 1 Н), 7.09 (t, 1 Н), 6.72 (d, 1 Н), 3.9 (s, 2 Н), 3.75 (s, 3 Н), 3.6 (t, 4 Н), 2.9 (m, 1 Н), 2.62/2.05 (2dd, 2 Н), 2.5-2.3 (m, 6 Н).
ИК: ν: -NH: 3203 см-1.
Стадия G: 3-(морфолин-4-илметил)-1,2,3,4-тетрагидроизохинолин-5-ол
К раствору соединения, полученного в Стадии F (5,6 г; 21 ммоль) в дихлорметане (60 мл), добавляют по каплям при температуре -10°С раствор 1 М трибромборана в дихлорметане (100 мл; 100 ммоль). Затем все перемешивают при этой температуре в течение 3 часов, затем подогревают до 10°С в течение 1 часа. Реакционную среду разбавляют при 0°С с дихлорметаном и водным насыщенным раствором гидрокарбоната натрия, затем фильтруют, чтобы получить твердое вещество белого цвета. После декантации, водную фазу экстрагируют этилацетатом, затем органические фазы концентрируют.Полученный остаток, а также белый осадок объединяют, чтобы получить указанный в заголовке продукт, который используют в следующей стадии без очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 9.2 (br. s, 1 Н), 6.89 (t, 1 Н), 6.59 (d, 1 Н), 6.48 (d, 1 Н), 3.81 (s, 2 Н), 3.6 (t, 4 Н), 2.89 (m, 1 Н), 2.6/2.02 (2dd, 2 Н), 2.5-2.25 (m, 6 Н).
ИК: ν: -NH/OH: 3412 и 3000 до 2500; >С=С<: 1615 см-1.
Стадия Н: трет-бутил 5-гидрокси-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Стадии G (4,1 г; 16,5 ммоль), в дихлорметане (100 мл), добавляют диизопропилэтиламин (7,4 мл; 72,6 ммоль) и ди-трет-бутилдикарбонат (7,9 г, 36,3 ммоль), затем все перемешивают в течение 18 часов при температуре окружающей среды. Реакционную среду разбавляют с водным насыщенным раствором хлорида аммония. После декантации, органическую фазу промывают водным насыщенным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток разбавляют в 1 М растворе поташа в метаноле. После контакта в течение 2 часов при температуре окружающей среды, реакционную среду разбавляют дихлорметаном и водным насыщенным раствором хлорида аммония. После экстракции дихлорметаном, органические фазы промывают водой, сушат над сульфатом магния, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, используя дихлорметан и метанол в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1Н ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 9.41 (br. s, 1 Н), 6.97 (t, 1 Н), 6.62 (d, 1 Н), 6.55 (d, 1 Н), 4.7-4.4 (d+m, 2 Н), 4.08 (m, 1 Н), 3.52 (m, 4 Н), 2.75-2.2 (m, 7 Н), 2.08 (dd, 1 Н), 1.42 (br. s, 9 Н).
ИК: ν: -ОН: 3295; >С=O: 1689 и 1656 см-1.
Синтез 5b': трет-бутил 4-(2-гидроксиэтил)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: 4-(проп-2-ен-1-ил)изохинолин
Раствор 4-бромизохинолина (25 г; 0,12 моль) и карбоната калия (50 г; 0,36 моль) в смеси воды (125 мл) и диметоксиэтана (375 мл) дегазируют с помощью струи азота. Затем добавляют тетракис(трифенилфосфин)палладий(0) (7 г; 0,006 моль) и пинаколат аллилбороновой кислоты (35 мл; 0,18 моль). Азот барботируют в смесь в течение 30 минут, затем ее кипятят с обратным холодильником и перемешивают в течение 18 часов. После достижения температуры окружающей среды, реакционную среду гидролизуют. Продукт экстрагируют этилацетатом, затем органические фазы промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 9.15 (s, 1 Н), 8.39 (s, 1 H), 7.98 (d, 2 Н), 7.71 (ddd, 1 Н), 7.60 (ddd, 1 Н), 6.08 (dddd, 1 Н), 5.05-5.15 (m, 2 Н), 3.78 (d, 2 Н).
Стадия В: 2-(изохинолин-4-ил)этанол
Соединение, полученное в Стадии А (14 г; 78 ммоль), растворяют в смеси дихлорметана (180 мл) и метанола (180 мл). Через полученный таким образом раствор барботируют озон с помощью газового диффузора при -78°С в течение 1,5 часа, затем воздух в течение 10 минут, и в заключение азот в течение того же времени.
Реакционную смесь поддерживают при 0°С, и порциями добавляют борогидрид натрия (8,83 г; 233 ммоль). После контакта в течение 18 часов при температуре окружающей среды, среду разбавляют в смеси воды и водного насыщенного раствора хлорида аммония. Продукт экстрагируют этилацетатом, затем органические фазы промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя этилацетат и метанол в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 9.00 (s, 1 Н), 8.37 (s, 1 H), 8.03 (d, 1 Н), 7.92 (d, 1 Н), 7.74 (ddd, 1 Н), 7.61 (ddd, 1 Н), 4.01 (t, 2 Н), 3.29 (t, 2 Н), 2.35 (s, 1 Н).
Стадия С: трет-бутил 4-(2-гидроксиэтил)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Синтеза 1b', используя соединение из предыдущей стадии в качестве исходного продукта.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.16-7.22 (т, 3 Н), 7.07-7.13 (m, 1Н), 4.87 (d, 1 Н), 4.32 (d, 1 Н), 4.19 (s, 1 Н), 3.73-3.87 (m, 2 Н), 3.19 (d, 1 Н), 2.97-3.04 (m, 1 Н), 1.81 (q, 2 Н), 1.65 (s, 1 Н), 1.50 (s, 9 Н).
Синтез 6b': трет-бутил 4-гидрокси-1,3-дигидро-2Н-изоиндол-2-карбоксилат
Стадия А: фуран-2-ил 2,2-диметилпропаноат
К раствору 2-(5H)-фуранона (22 г; 0,26 моль) и хлорида триметилацетила (38 г; 0,31 моль) в ацетонитриле (50 мл), добавляют раствор триэтиламина (43.5 мл; 0,31 моль) в ацетонитриле (11 мл). Реакционную смесь перемешивают в течение 3 дней при температуре окружающей среды, затем полученную суспензию фильтруют и промывают метил-трет-бутиловым эфиром. Органическую фазу промывают водным раствором насыщенного бикарбоната натрия и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Полученное масло дистиллируют в вакууме (15 торр, фракции собирают при 76-78°С), чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.05 (dd, 1 Н), 6.36 (dd, 1 Н), 5.86 (dd, 1 Н), 1.34 (s, 9 Н).
Стадия В: 1,3-диоксо-3,3а,7,7а-тетрагидро-4,7-эпокси-2-бензофуран-4(1Н)-ил 2,2-диметилпропаноат
К раствору свежемолотого в ступке малеинового ангидрида (24,9 г; 0,25 моль) в диэтиловом эфире (207 мл), добавляют соединение, полученное в Стадии А (38,76 г; 0,23 моль). Реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Полученную суспензию бежевого цвета фильтруют, и фильтрат концентрируют приблизительно до 50 мл, затем снова фильтруют. Полученные таким образом твердые вещества объединяют и сушат в вакууме, чтобы обеспечить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 6.77 (dd, 1 Н), 6.69 (d, 1 Н), 5.33 (d, 1 Н), 3.66 (s, 2 Н), 1.22 (s, 9 Н).
Стадия С: 4-гидрокси-2-бензофуран-1,3-дион
К раствору концентрированной серной кислоты (80 мл), охлажденной до -15°С, добавляют порциями соединение, полученное в Стадии В (36,5 г; 0,16 моль). Смесь перемешивают в течение 15 минут при -15°С, затем выливают в ледяную воду. Образованное твердое вещество затем фильтруют, промывают водой и сушат в вакууме, чтобы обеспечить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 11.72 (s, 1 Н), 7.77 (dd, 1 Н), 7.45 (d, 1 Н), 7.33 (d, 1 Н).
Стадия D: 4-гидрокси-2-(4-метоксибензил)-1Н-изоиндол-1,3(2Н)-дион
К раствору соединения, полученного в Стадии С (24,76 г; 0,15 моль) в уксусной кислоте (150 мл), добавляют 4-метоксибензиламин (21,7 мл; 0,17 моль), затем смесь кипятят с обратным холодильником в течение 5 часов. После достижения температуры окружающей среды, к смеси добавляют воду (200 мл), которую еще раз перемешивают в течение 1 часа. Суспензию фильтруют, а твердое вещество промывают водой. Сырой продукт растворяют в этилацетате, и органическую фазу промывают водным насыщенным раствором бикарбоната натрия, водным насыщенным раствором хлорида аммония и в заключение насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, чтобы обеспечить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.54 (t, 1 Н), 7.32-7.40 (m, 3 Н), 7.12 (d, 1 Н), 6.84 (d, 2 Н), 4.73 (s, 2 Н), 3.77 (s, 3 Н).
Стадия Е: 2-(4-метоксибензил)-2,3-дигидро-1Н-изоиндол-4-ол
К суспензии алюмогидрида лития (11,9 г; 0,31 моль) в тетрагидрофуране (150 мл) при 0°С, добавляют по каплям, при сохранении внутренней температуры смеси ниже 20°С, раствор соединения, полученного в Стадии D (35,5 г; 0,13 моль) в тетрагидрофуране (250 мл). После завершения добавления, смесь кипятят с обратным холодильником. Смесь перемешивают в течение 2 часов при кипячении с обратным холодильником, затем охлаждают до 0°С. Медленно добавляют этилацетат, при сохранении внутренней температуры смеси ниже 20°С. Когда больше не наблюдается экзотермический эффект при добавлении этилацетата, смесь разбавляют с этилацетатом и водным 1,5 н. раствором сегнетовой соли. Смесь интенсивно перемешивают в течение 2 часов при температуре окружающей среды. После декантации, водную фазу промывают с этилацетатом. Органические фазы объединяют и промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют в вакууме, чтобы обеспечить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.27-7.34 (m, 2 Н), 6.95 (t, 1 Н), 6.81-6.90 (m, 2 Н), 6.68 (d, 1 Н), 6.39 (d, 1 Н), 3.89 (d, 4 Н), 3.83 (s, 2 Н), 3.80 (s, 3 Н).
Стадия F: трет-бутил 4-гидрокси-1,3-дигидро-2Н-изоиндол-2-карбоксилат
К раствору соединения, полученного в Стадии Е (18,2 г; 71,3 ммоль) в метаноле (325 мл) и уксусной кислоте (8,2 мл), добавляют палладий на угле (10 мас. %). Реактор герметизируют и продувают азотом, а затем водородом.
Реакционную среду подвергают воздействию водорода 45 фунтов на квадратный дюйм и перемешивают в течение 4 часов при температуре окружающей среды. Реакционную смесь фильтруют и промывают метанолом, затем фильтрат концентрируют в вакууме. К раствору неочищенного продукта в метаноле (200 мл), добавляют триэтиламин (40 мл; 0,29 моль) и ди-трет-бутил дикарбонат (15,6 г; 71,3 ммоль). Среду перемешивают при температуре окружающей среды в течение 16 часов. Растворитель выпаривают в вакууме, и остаток разбавляют в этилацетате. Органическую фазу промывают водным раствором 2 н. соляной кислоты, водным насыщенным раствором бикарбоната натрия, и водным насыщенным раствором хлорида натрия. Затем, ее сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.11-7.17 (m, 1 Н), 7.02 (s, 0.5 Н), 6.68-6.84 (m, 2 Н), 5.98 (s, 0.5 Н), 4.63-4.85 (m, 4 Н), 1.51-1.55 (m, 9 Н).
Синтез 7b': трет-бутил 5-гидрокси-3-метил-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: 3-(бензилокси)-2-(проп-2-ен-1-ил)бензальдегид
К раствору 2-аллил-3-гидрокси-бензальдегида (20 г; 0,12 моль) в ацетонитриле (400 мл), добавляют бензилбромид (16 мл; 0,13 моль) и карбонат калия (18 г; 0,13 моль). Реакционную смесь перемешивают в течение 3 дней при температуре окружающей среды, затем выливают в смесь изо льда и водного насыщенного раствора гидрокарбоната натрия. После экстрагирования этилацетатом, органические фазы сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают хроматографией на силикагеле, используя дихлорметан в качестве растворителя для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 10.2 (s, 1 Н), 7.5-7.3 (m, 8 Н), 6 (ddt, 1 Н), 5.2 (s, 2 Н), 5-4.85 (m, 2 Н), 3.85 (dt, 2 Н).
ИК: ν: >С=O: 1681 см-1.
Стадия В: N-бензил-1-[3-(бензилокси)-2-(проп-2-ен-1-ил)фенил]метанамин
К раствору соединения, полученного в Стадии А (20 г; 0,078 моль) в дихлорметане (800 мл), добавляют бензиламин (10 мл; 0,078 моль) и порциями триацетоксиборгидрид натрия (25 г; 0,118 моль). Реакционную смесь перемешивают в течение 16 часов при температуре окружающей среды, затем добавляют водный раствор 1 н. гидроксида натрия и лед. После экстрагирования в дихлорметане, органические фазы промывают водным 1 н. раствором гидроксида натрия, затем насыщенным водным раствором хлорида натрия, и сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают хроматографией на силикагеле, используя дихлорметан и аммиак в этаноле в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.48-7.22 (m, 10 Н), 7.14 (t, 1 Н), 6.99 (d, 1 Н), 6.95 (d, 1 Н), 5.85 (m, 1 Н), 5.09 (s, 2 Н), 4.85 (m, 1 Н), 4.76 (m, 1 Н), 3.7 (s, 2 Н), 3.61 (s, 2 Н), 3.44 (d, 2 Н), 2.35 (br. s, 1 H).
Стадия С: 2-бензил-5-(бензилокси)-3-метил-1,2,3,4-тетрагидроизохинолин
К раствору соединения, полученного в Стадии В (18,9 г; 0,055 моль) в тетрагидрофуране (1 л), добавляют по каплям при 60°С раствор 1,5 н. н-бутиллития в гексане (40 мл; 0,06 моль). В конце добавления, реакционную смесь охлаждают до температуры окружающей среды, затем нейтрализуют водой. После экстракции простым эфиром, органические фазы промывают насыщенным водным раствором хлорида натрия и сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают хроматографией на силикагеле, используя гептан и аммиак в этаноле в качестве растворителя для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.5-7.2 (m, 5 Н), 7.5-7.2 (m, 5 Н), 7.03 (t, 1 Н), 6.83 (d, 1 Н), 6.58 (d, 1 Н), 5.1 (s, 2 Н), 3.76 (d, 1 Н), 3.53 (d, 1 Н), 3.58 (d, 1 Н), 3.49 (d, 1 Н), 3.05 (m, 1 Н), 2.8 (dd, 1 Н), 2.49 (dd, 1 Н), 1.09 (d, 3 Н).
Стадия D: 3-метил-1,2,3,4-тетрагидроизохинолин-5-ол
К раствору соединения, полученного в Стадии С (13,47 г; 39,22 ммоль) в метаноле (250 мл), добавляют водный раствор 1 н. соляной кислоты (58,8 мл; 58,8 ммоль), затем палладий на угле (10 мас. %). Колбу помещают под давление водорода, и реакционную среду перемешивают в течение 48 часов при температуре окружающей среды. Реакционную смесь фильтруют и промывают метанолом, затем фильтрат концентрируют в вакууме. Остаток ресуспендируют в этаноле затем фильтруют, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 9.75 (s, 1 Н), 9.3 (br. s, 1 Н), 7.05 (t, 1 Н), 6.75 (d, 1 Н), 6.65 (d, 1 Н), 4.21 (m, 2 Н), 3.48 (m, 1 Н), 2.95 (dd, 1 Н), 2.47 (dd, 1 Н), 1.4 (d, 3 Н).
ИК: ν: -ОН: 3226 см-1; -NH2+: 3300-3400 см-1.
Стадия Е: трет-бутил 5-гидрокси-3-метил-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Стадии D (5 г; 25,2 ммоль) в дихлорметане (250 мл), при 0°С добавляют триэтиламин (7,38 мл; 52,9 ммоль) и порциями ди-трет-бутил дикарбонат (5,5 г; 25,2 ммоль). Реакционную среду перемешивают при этой температуре в течение 3 часов, затем при температуре окружающей среды в течение 20 часов. Растворитель выпаривают в вакууме, и остаток разбавляют в этилацетате. Органическую фазу промывают с водным насыщенным раствором хлорида аммония, водой, насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток очищают хроматографией на силикагеле, используя циклогексан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 9.4 (br. s, 1 Н), 6.98 (t, 1 Н), 6.67 (d, 1 Н), 6.6 (d, 1 Н), 4.59 (d, 1 Н), 4.15 (d, 1 Н), 4.45 (m, 1 Н), 2.65 (d, 2 Н), 1.42 (s, 9 Н), 0.99 (d, 3 Н).
ИК: ν: -ОН: 3308 см-1; >С=O: 1655 см-1.
Синтез 8b': трет-бутил 5-(3-йодпропокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: трет-бутил 5-(3-хлорпропокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Синтезе 3b' (1 г; 4 ммоль) в ацетонитриле (20 мл), добавляют бром-хлорпропан (0,48 мл; 4,8 ммоль) и карбонат калия (1,1 г, 8 ммоль), затем все перемешивают в течение 18 часов при 70°С. Реакционную среду разбавляют с этилацетатом и водой. После декантации, органическую фазу промывают водой, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.14 (t, 1 Н), 6.82 (d, 1 Н), 6.75 (d, 1 Н), 4.46 (s, 2 Н), 4.08 (t, 2 Н), 3.82 (t, 2 Н), 3.55 (t, 2 Н), 2.64 (t, 2 Н), 2.18 (quint, 2 Н), 1.42 (s, 9 Н).
ИК: ν: >С=O: 1692 см-1; >С-O-С<: 1241/1164/1112 см-1.
Стадия В: трет-бутил 5-(3-йодпропокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Стадии А (1 г; 3,07 ммоль) в ацетоне (30 мл), добавляют йодид натрия (2,3 г; 15,3 ммоль), затем все перемешивают при кипячении с обратным холодильником в течение 24 часов. Реакционную среду концентрируют, затем разбавляют с этилацетатом и водой. После экстрагирования этилацетатом, органические фазы промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.12 (t, 1 Н), 6.8 (d, 1 Н), 6.72 (d, 1 Н), 4.45 (s, 2 Н), 4 (t, 2 Н), 3.55 (t, 2 Н), 3.41 (t, 2 Н), 2.63 (t, 2 Н), 2.2 (т, 2 Н), 1.41 (s, 9 Н).
ИК: ν: >С=O: 1692 см-1.
Синтез 9b': трет-бутил 5-[2-(2-гидроксиэтокси)этокси]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Синтезе 3b’ (5 г; 20 ммоль) в N,N-диметилформамиде (20 мл), добавляют 2-(2-хлорэтокси)этанол (6,25 мл; 60 ммоль) и карбонат калия (8,3 г, 60 ммоль), затем все перемешивают в течение 5 часов при 125°С. Реакционную среду разбавляют с этилацетатом и водой. После декантации, органическую фазу промывают насыщенным водным раствором хлорида лития, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.13 (t, 1 Н), 6.81 (d, 1 Н), 6.75 (d, 1 Н), 4.58 (m, 1 Н), 4.46 (s, 2 Н), 4.09 (m, 2 Н), 3.75 (m, 2 Н), 3.54 (t, 2 Н), 3.51 (неразрешенный пик, 4 Н), 2.63 (t, 2 Н), 1.42 (s, 9 Н).
ИК: ν: -ОН: 3450 см-1; >С=O: 1690 см-1.
Синтез 10b': трет-бутил 5-(бут-3-ин-1-ил)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: трет-бутил 5-(3-метокси-3-оксопропил)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Стадии В Синтеза 1b' (19,74 г; 62,3 ммоль) в метаноле (150 мл), добавляют палладий на угле (10 мас. %). Через суспензию барботируют водород в течение 10 минут, затем смесь перемешивают под атмосферой водорода (1 бар) в течение 18 часов. Реакционную смесь фильтруют через слой Celite® и фильтрат концентрируют досуха, чтобы обеспечить указанный в заголовке продукт, который используют в дальнейшем без очистки.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.10-7.16 (m, 1 Н), 7.04 (d, 1 Н), 6.98 (d, 1 Н), 4.56 (s, 2 Н), 3.68 (s, 3 Н), 3.66 (br. s, 2 Н), 2.90-2.96 (m, 2 Н), 2.80 (t, 2 Н), 2.55-2.61 (m, 2 Н), 1.48 (s, 9 Н).
Стадия В: трет-бутил 5-(бут-3-ин-1-ил)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору полученного в Стадии А продукта (10 г; 31,3 ммоль) в дихлорметане (100 мл) при -78°С, добавляют медленно раствор гидрида диизобутилалюминия при 25 мас. % в толуоле (25,2 мл, 37,6 ммоль) в течение 45 минут.Скорость добавления определяется таким образом, чтобы температура реакционной смеси была ниже -75°С. В конце добавления, смесь перемешивают при -78°С в течение 30 минут, затем к реакционной смеси медленно добавляют метанол (50 мл). После чего смесь постепенно нагревают до 0°С. Снова добавляют метанол (50 мл), а также карбонат калия (8,65 г; 62,6 ммоль), затем диметил-(1-диазо-2-оксопропил)фосфонат (7,22 г; 37,6 ммоль). Смесь перемешивают при температуре окружающей среды в течение 48 часов и разбавляют метил-трет-бутиловый эфир (400 мл). Затем добавляют водный раствор 1 н. тетрагидрат тартрата калия-натрия (250 мл). Фазы разделяют, и водную фазу промывают метил-трет-бутиловым эфиром. Органические фазы объединяют и промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и выпаривают в вакууме. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.12-7.18 (m, 1 Н), 7.09 (d, 1 Н), 7.00 (d, 1 Н), 4.57 (s, 2 Н), 3.61-3.73 (m, 2 Н), 2.78-2.89 (m, 4 Н), 2.44 (td, 2 Н), 1.98 (t, 1 Н), 1.49 (s, 9 Н).
Синтез 1b": трет-бутил 5-(3-{3-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-5-цианофенокси}пропил)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: трет-бутил 5-[3-(3-бром-5-цианофенокси)пропил]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Синтезе 1b' (9,89 г; 34,0 ммоль) и 3-бром-5-фторбензонитрила (27,2 г; 135,8 ммоль) в N,N-диметилформамиде (80 мл), добавляют порциями 60% гидрид натрия в масле (1,77 г; 44,1 ммоль). Затем все перемешивают в течение 45 минут при температуре окружающей среды затем гидролизуют медленно. После экстрагирования этилацетатом, органические фазы промывают водным насыщенным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.36 (t, 1 Н), 7.26-7.28 (m, 1 Н), 7.12-7.17 (m, 1 Н), 7,06-7.09 (m, 1 Н), 6.98-7.05 (m, 2 Н), 4.58 (s, 2 Н), 3.98 (t, 2 Н), 3.62-3.68 (m, 2 Н), 2.76-2.82 (m, 4 Н), 2.02-2.09 (m, 2 Н), 1.49 (s, 9 Н).
Стадия В: 4-{[трет-бутил(диметил)силил]окси}анилин
Указанное в заголовке соединение получают исходя из 4-аминофенола в тетрагидрофуране в присутствии имидазола и хлорида трет-бутил(диметил)силила в соответствии с протоколом, описанным в литературных источниках (S. Knaggs и соавт., Organic & Biomolecular Chemistry, 3(21), 4002-4010; 2005).
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 6.45-6.55 (dd, 4Н), 4.60 (m, 2Н), 0.90 (s, 9Н), 0.10 (s, 6Н). ИК: ν: -NH2+: 3300-3400 см-1
Стадия С: трет-бутил 5-(3-{3-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-5-цианофенокси}пропил)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Раствор соединений, полученных в Стадии А (8,11 г; 17,2 ммоль) и в Стадии В (4,23 г; 17,2 ммоль) в толуоле (110 мл) дегазируют аргоном в течение 10 минут. Добавляют mpem-бутилат натрия (1,82 г; 18,9 ммоль), 2-ди-трет-бутилфосфино-2',4',6'-триизопропил-1,1'-бифенил (0,73 г; 1,72 ммоль) и трис(дибензилиденацетон)дипалладий(0) (0,86 г; 0,86 ммоль), затем все перемешивают при 80°С в течение 30 минут.Реакционную среду фильтруют через Celite®. После промывания этилацетатом к фильтрату добавляют диоксид кремния, затем смесь концентрируют и очищают с помощью хроматографии на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.13 (t, 1 Н), 6.95-7.06 (m, 4 Н), 6.83 (d, 2 Н), 6.69 (br. s, 1 Н), 6.51-6.59 (m, 2 Н), 5.59 (br. s, 1 Н), 4.57 (s, 2 Н), 3.92 (t, 2 Н), 3.58-3.68 (m, 2 Н), 2.61-2.83 (m, 4 Н), 2.02 (quint., 2 Н), 1.49 (s, 9 Н), 1.00 (s, 9 Н), 0.21 (s, 6 Н).
Синтез 2b": трет-бутил 5-(4-{4-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1Н-пиразол-1-ил}бутокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: трет-бутил 5-[4-(4-бром-1Н-пиразол-1-ил)бутокси]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения Синтеза 2b' (0,786 г; 2,44 ммоль) и трифенилфосфина (0,768 мг; 2,93 ммоль) в тетрагидрофуране (5 мл), добавляют по каплям раствор 4-бромпиразола (0,359 мг; 2,44 ммоль) и азодикарбоксилат диизопропила (0,58 мл; 2,93 ммоль) в тетрагидрофуране (5 мл). Реакционную среду перемешивают при температуре окружающей среды в течение 2 часов, затем разбавляют в смеси этилацетата и воды. После экстрагирования водной фазы этилацетатом, органические фазы объединяют, промывают с водным насыщенным раствором хлорида аммония, насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают с помощью хроматографии на фазе Oasis®, используя ацетонитрил и воду в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 8.02 (s, 1 Н), 7.53 (s, 1 Н), 7.12 (dd, 1 Н), 6.81-6.68 (2*d, 2 Н), 4.46 (s, 2 Н), 4.16 (t, 2 Н), 3.95 (t, 2 Н), 3.54 (t, 2 Н), 2.61 (t, 2 Н), 1.93 (m, 2 Н), 1.65 (m, 2 Н), 1.41 (s, 9 Н).
ИК: ν: >С=O: 1688 см-1.
Стадия В: трет-бутил 5-(4-{4-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1Н-пиразол-1-ил}бутокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии С Синтеза 1b", используя соединение, полученное в предыдущей Стадии, соединение Стадии В Синтеза 1b" и хлор(2-ди-трет-бутилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2-аминоэтил)фенил]палладий(II) в качестве катализатора и лиганда.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.63 (d, 1 Н), 7.26 (d, 1 Н), 7.2 (s, 1 Н), 7.11 (t, 1 Н), 6.75 (dd, 2 Н), 6.64 (m, 4 Н), 4.45 (s, 2 Н), 4.1 (t, 2 Н), 3.96 (t, 2 Н), 3.53 (t, 2 Н), 2.62 (t, 2 Н), 1.93 (m, 2 Н), 1.68 (m, 2 Н), 1.41 (s, 9 Н), 0.93 (s, 9 Н), 0.12 (s, 6 Н).
ИК: ν: -NH: 3340 см-1; >С=O: 1690 см-1.
Синтез 3b": трет-бутил 5-(2-{4-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1-метил-1Н-пиразол-5-ил}этокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: 5-(2-{[трет-бутил(диметил)силил]окси}этил)-1-метил-1Н-пиразол
К раствору из N-метилпиразола (10,95 г; 133 ммоль) в тетрагидрофуране (200 мл), добавляют по каплям при -78°С раствор н-бутиллития в гексане (100 мл; 160 ммоль), затем температуру повышают до 0°С в течение 1 часа. Реакционную среду снова охлаждают до -78°С, и добавляют раствор (3-бромэтокси)-трет-бутилдиметилсилан (34,2 мл; 160 ммоль) в тетрагидрофуране (50 мл). Затем реакционную среду перемешивают при температуре окружающей среды в течение 18 часов и выливают в смесь из ледяной воды и этилацетата. После экстрагирования этилацетатом, органическую фазу промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.28 (d, 1 Н), 6.04 (d, 1 Н), 3.79 (t, 2 Н), 3.74 (s, 3 Н), 2.81 (t, 2 Н), 0.84 (s, 9 Н), 0.1 (s, 6 Н).
Стадия В: 2-(4-бром-1-метил-1Н-пиразол-5-ил)этанол
К раствору соединения, полученного в Стадии А (9,8 г; 41,1 ммоль) в метаноле (400 мл), добавляют при 0°С трибромид пиримидиния (14 г; 43 ммоль), затем все перемешивают в течение 1 часа при 0°С, затем в течение 2 часов при температуре окружающей среды. Затем реакционную среду концентрируют, и остаток ресуспендируют в смеси водного 10% раствора карбоната калия и дихлорметана. После экстракции дихлорметаном, органическую фазу промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и аммиак в метаноле в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.45 (s, 1 Н), 4.88 (t, 1 Н), 3.81 (s, 3 Н), 3.55 (q, 2 Н), 2.8 (t, 2 Н).
ИК: ν: -ОН: 3350 см-1; >С-С-O-: 1049 см-1.
Стадия С: трет-бутил 5-[2-(4-бром-1-метил-1Н-пиразол-5-ил)этокси]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии А Синтеза 2b", используя спирт, полученный в предыдущей Стадии и соединение из Синтеза 3b’.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.49 (s, 1 Н), 7.13 (t, 1 Н), 6.82 (d, 1 Н), 6.75 (d, 1 Н), 4.45 (s, 2 Н), 4.15 (t, 2 Н), 3.87 (s, 3 Н), 3.51 (t, 2 Н), 3.16 (t, 2 Н), 2.54 (t, 2 Н), 1.41 (s, 9 Н).
ИК: ν:>С=O: 1689 см-1.
Стадия D: трет-бутил 5-(2-{4-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1-метил-1Н-пиразол-5-ил}этокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии С Синтеза 1b", используя соединение, полученное в предыдущей Стадии, соединение Стадии В Синтеза 1b" и хлор(2-ди-трет-бутилфосфино-2',4',6'-триизо пропил-1,1'-бифенил)[2-(2-аминоэтил)фенил]палладий(II) в качестве катализатора и лиганда.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.28 (s, 1 Н), 7.06 (t, 1 Н), 6.71/6.67 (2*dd, 2 Н), 6.7 (s, 1 Н), 6.57 (d, 2 Н), 6.48 (d, 2 Н), 4.43 (s, 2 Н), 4.05 (t, 2 H), 3.82 (s, 3 H), 3.48 (t, 2 H), 3.03 (t, 2 H), 2.5 (t, 2 H), 1.4 (s, 9 H), 0.9 (s, 9 H), 0.09 (s, 6 H).
ИК: ν: >NH: 3321 см-1; >C=O: 1677 см-1.
Синтез 4b'': N-(4-{[трет-бутил(диметил)силил]окси}фенил)-1-метил-5-[3-(тетрагидро-2Н-пиран-2-илокси)пропил]-1Н-пиразол-4-амин
Стадия А: 5-(3-{[трет-бутил(диметил)силил]окси}пропил)-1-метил-1Н-пиразол
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии А Синтеза 3b", используя (3-бромпропокси)-трет-бутилдиметилсилан.
1H ЯМР (400/500 МГц, CDCl3, 300 K) δ част, на млн.: 7.35 (d, 1 Н), 7 (d, 1 Н), 3.8 (s, 3 Н), 3.65 (m, 2 Н), 2.7 (m, 2 Н), 1.85 (m, 2 Н), 0.9 (s, 9 Н), 0.5 (s, 6 Н).
ИК: ν: -Si-O-: 1098 см-1; -Si-C-: 834 и 772 см-1.
Стадия В: 3-(4-бром-1-метил-1Н-пиразол-5-ил)пропан-1-ол
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Синтеза 3b", используя соединение из предыдущей стадии.
1Н ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.45 (s, 1 Н), 4.59 (t, 1 Н), 3.79 (s, 3 Н), 3.4 (quad, 2 Н), 2.7 (m, 2 Н), 1.65 (m, 2 Н).
ИК: ν: -ОН: 3348 см-1.
Стадия С: 4-бром-1-метил-5-[3-(тетрагидро-2Н-пиран-2-илокси)пропил]-1Н-пиразол
К раствору соединения, полученного в Стадии В (5,34 г; 2.4 ммоль) в дихлорметане (40 мл), добавляют 3,4-дигидро-2H-пиран (7 мл; 6 ммоль) и пара-толуолсульфоновую кислоту (4,6 г; 2,4 ммоль), затем все перемешивают в течение 16 часов. Реакционную среду разбавляют в водном растворе насыщенного гидрокарбоната натрия. После экстракции дихлорметаном, органическую фазу сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1Н ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.42 (s, 1 Н), 4.55 (t, 1 Н), 3.8-3.3 (m, 4 Н), 3.8 (s, 3 Н), 2.71 (m, 2 Н), 1.78 (m, 2 Н), 1.7-1.4 (m, 6 Н).
Стадия D: N-(4-{[трет-бутил(диметил)силил]окси}фенил)-1-метил-5-[3-(тетрагидро-2Н-пиран-2-илокси)пропил]-1Н-пиразол-4-амин
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии D Синтеза 3b", используя соединение из предыдущей стадии.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.2 (s, 1 Н), 6.6 (s, 1 Н), 6.55 (d, 2 Н), 6.45 (d, 2 Н), 4.4 (t, 1 Н), 3.7 (s, 3 Н), 3.65-3.2 (4 m, 4 Н), 2.58 (m, 2 Н), 1.68 (m, 2 Н), 1.6-1.3 (m, 6 Н), 0.92 (s, 9 Н), 0.1 (s, 6 Н).
ИК: ν: >NH: 3356 см-1; ->С-С-O-: 1240 см-1.
Синтез 5b": трет-бутил 5-(3-{3-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-5-циано-1-метил-1Н-пиррол-2-ил}пропокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А:этил (2Е)-3-(1-метил-1Н-пиррол-2-ил)проп-2-еноат
К раствору из фосфоноацетата триэтила (14,2 мл; 71,5 ммоль) в тетрагидрофуране (300 мл), добавляют трет-бутоксид калия (9,25 г; 82,5 ммоль), затем все перемешивают при 0°С в течение 45 минут. Добавляют раствор N-метил-2-пирролкарбоксальдегида (6 г; 55,0 ммоль) в тетрагидрофуране (20 мл) и все перемешивают при температуре окружающей среды в течение 16 часов, затем растворитель концентрируют.Остаток разбавляют в воде и этилацетате. После экстрагирования этилацетатом, органическую фазу промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.59 (d, 1 Н), 6.73-6.76 (m, 1 Н), 6.65 (dd, 1 Н), 6.11-6.18 (m, 2 Н), 4.23 (q, 2 Н), 3.71 (s, 3 Н), 1.32 (t, 3 Н).
Стадия В: этил 3-(1-метил-1Н-пиррол-2-ил)пропаноат
К раствору соединения, полученного в Стадии А (8,1 г; 45,1 ммоль) в этаноле (70 мл), добавляют палладий на угле (10 мас. %), затем все гидрируют в течение 6 часов и 30 минут. Реакционную смесь фильтруют через Celite® и фильтрат концентрируют, чтобы получить указанный в заголовке продукт, который используют в дальнейшем без очистки.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 6.52-6.56 (m, 1 Н), 6.01-6.06 (m, 1 Н), 5.87 (ddt, 1 Н), 4.15 (q, 2 Н), 3.55 (s, 3 Н), 2.83-2.91 (m, 2 Н), 2.60-2.68 (m, 2 Н), 1.26 (t, 3 Н).
Стадия С: этил 3-(5-циано-1-метил-1Н-пиррол-2-ил)пропаноат
К раствору соединения, полученного в Стадии В (12 г; 66,2 ммоль) в ацетонитриле (300 мл) при -20°С, поддерживая эту температуру, добавляют по каплям изоцианат хлорсульфонила (6,92 мл; 79,5 ммоль), затем все перемешивают при -20°С в течение 30 минут. Добавляют N,N-диметилформамид (10,3 мл; 132,4 ммоль) затем триэтиламин (18,5 мл; 132,4 ммоль) при температуре, поддерживаемой на уровне -10°С, и все перемешивают до достижения температуры окружающей среды. Реакционную среду разбавляют в водном растворе 1 М соляной кислоты (500 мл). После экстрагирования этилацетатом, органическую фазу промывают водным раствором 1 М хлористоводородной кислоты, затем насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1Н ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 6.72 (d, 1 Н), 5.93 (d, 1 Н), 4.15 (q, 2 Н), 3.68 (s, 3 Н), 2.86-2.93 (m, 2 Н), 2.63-2.69 (m, 2 Н), 1.26 (t, 3 Н).
Стадия D: 4-бром-5-(3-гидроксипропил)-1-метил-1Н-пиррол-2-карбонитрил
К раствору соединения, полученного в Стадии С (9,74 г; 47,2 ммоль) в N,N-диметилформамиде (125 мл) при 0°С, добавляют порциями N-бромсукцинимид (8,82 г; 49,6 ммоль), затем все перемешивают при температуре окружающей среды в течение 30 минут. Реакционную среду разбавляют в воде и трет-бутилметиловом эфире. После экстрагирования трет-бутилметиловым эфиром, органическую фазу промывают водой и насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют досуха, чтобы получить продукт, который непосредственно используют в следующей стадии.
К раствору этого соединения (3,13 г; 10,97 ммоль) в тетрагидрофуране (30 мл) при 0°С, добавляют раствор 2 М боргидрида лития в тетрагидрофуране (11 мл; 21,95 ммоль), затем все перемешивают при температуре окружающей среды в течение 6 часов. Реакционную среду разбавляют медленно водным раствором гидроксида натрия 1 М (60 мл). После экстрагирования трет-бутилметиловым эфиром, органическую фазу промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 6.75 (s, 1 Н), 3.73 (s, 3 Н), 3.62-3.69 (m, 2 Н), 2.78 (t, 2 Н), 1.75-1.85 (m, 2 Н), 1.38-1.45 (m, 1 Н).
Стадия Е: трет-бутил 5-[3-(3-бром-5-циано-1-метил-1Н-пиррол-2-ил)пропокси]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии А Синтеза 2b", используя спирт, полученный в предыдущей Стадии, и соединение из Синтеза 3b’.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.11 (t, 1 Н), 7.06 (s, 1 Н), 6.75 (2d, 2 Н), 4.49 (s, 2 Н), 3.99 (t, 2 Н), 3.7 (s, 3 Н), 3.55 (t, 2 Н), 2.88 (t, 2 Н), 2.61 (t, 2 Н), 1.98 (m, 2 Н), 1.41 (s, 9 Н).
ИК: ν: >CN: 2218 см-1; ->С=O: 1681 см-1.
Стадия F: трет-бутил 5-(3-{3-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-5-циано-1-метил-1Н-пиррол-2-ил}пропокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии D Синтеза 3b", используя соединение из предыдущей стадии.
1Н ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.05 (t, 1 Н), 6.75 (s, 1 Н), 6.75 (s, 1 Н), 6.7/6.6 (2d, 2 Н), 6.52 (d, 2 Н), 6.45 (d, 2 Н), 4.42 (s, 2 Н), 3.88 (m, 2 Н), 3.68 (s, 3 Н), 3.5 (t, 2 Н), 2.75 (t, 2 Н), 2.51 (t, 2 Н), 1.89 (m, 2 Н), 1.41 (s, 9 Н), 0.9 (s, 9 Н), 0.1 (s, 6 Н).
ИК: ν: >NH: 3364 см-1; >CN: 2208 см-1; >С=O: 1690 см-1.
Синтез 6b": трет-бутил 5-(2-{3-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-5-циано-1-метил-1Н-пиррол-2-ил}этокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: (1-метил-1Н-пиррол-2-ил)(оксо)уксусная кислота
К раствору из оксалилхлорида (20,9 г; 0,25 моль) в дихлорметане при -10°С, добавляют N-метилпиррол (20 г; 0,25 моль), поддерживая более низкую температуру при 0°С, затем все перемешивают при 0°С в течение 1 часа. Реакционную среду разбавляют в водном 25% растворе гидроксида калия при 0°С. После декантации, водную фазу промывают дихлорметаном и подкисляют до рН=1 водным 20% раствором серной кислоты. Осадок фильтруют и сушат, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 8.01 (dd, 1 Н), 7.10 (t, 1 Н), 6.27 (dd, 1 Н), 3.99 (s, 3 Н).
Стадия В: (1-метил-1Н-пиррол-2-ил)уксусная кислота
К водному раствору моногидрата гидразина при 65% (15,5 мл; 0,21 моль), добавляют соединение, полученное в Стадии А (29 г; 0,19 моль), затем все перемешивают несколько минут. Медленно добавляют водный 20% раствор гидроксида натрия (326 мл) и все перемешивают при кипячении с обратным холодильником в течение 4 часов. После достижения температуры окружающей среды, добавляют водный 6 н. раствор соляной кислоты (25 мл). После экстракции дихлорметаном, органическую фазу промывают водой и насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют до объема в 100 мл. После медленного добавления гептана, осадок фильтруют, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 6.60-6.64 (m, 1 Н), 6.04-6.12 (т, 2 Н), 3.68 (s, 2 Н), 3.59 (s, 3 Н).
Стадия С: метил (1-метил-1Н-пиррол-2-ил)ацетат
К раствору соединения, полученного в Стадии В (10,2 г; 73.4 ммоль) и карбоната калия (15,2 г; 110,1 ммоль) в дихлорметане (100 мл), добавляют диметилсульфат (7 мл; 73,4 ммоль), затем все перемешивают интенсивно при 30°С в течение 4 часов. Добавляют водный 5% раствор гидроксида аммония (326 мл). После декантации, органическую фазу промывают водным 5% раствором гидроксида аммония и насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют досуха до объема в 100 мл. После медленного добавления гептана, осадок фильтруют, чтобы получить указанный в заголовке продукт. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1Н ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 6.58-6.62 (m, 1 Н), 6.06-6.09 (m, 1 Н), 6.03-6.06 (m, 1 Н), 3.71 (s, 3 Н), 3.64 (s, 2 Н), 3.58 (s, 3 Н).
Стадия D: метил (5-циано-1-метил-1Н-пиррол-2-ил)ацетат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии С Синтеза 5b", используя соединение из предыдущей стадии.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 6.74 (d, 1 Н), 6.08 (d, 1 Н), 3.73 (s, 3 Н), 3.69 (s, 3 Н), 3.66 (s, 2 Н).
Стадия Е: 5-(2-гидроксиэтил)-1-метил-1Н-пиррол-2-карбонитрил
К раствору соединения, полученного в Стадии D (11,4 г; 10,97 ммоль) в тетрагидрофуране (115 мл) при 0°С, добавляют раствор боргидрида лития 2M в тетрагидрофуране (47,7 мл; 95,4 ммоль) при температуре ниже 5°С, затем все перемешивают при температуре окружающей среды в течение 16 часов. Реакционную среду разбавляют медленно с водным насыщенным раствором хлорида аммония (150 мл). После экстрагирования mpem-бутилметиловым эфиром, органическую фазу промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 6.75 (d, 1 Н), 6.02 (d, 1 Н), 3.88 (t, 2 Н), 3.69 (s, 3 Н), 2.86 (t, 2 Н).
Стадия F: 4-бром-5-(2-гидроксиэтил)-1-метил-1Н-пиррол-2-карбонитрил
К раствору соединения, полученного в Стадии Е (4,25 г; 28,3 ммоль) в N,N-диметилформамиде (43 мл) при 0°С, добавляют порциями N-бромсукцинимид (5,04 г; 28,3 ммоль), затем все перемешивают при температуре окружающей среды в течение 3 часов. Реакционную среду разбавляют в воде и трет-бутилметиловым эфиром. После экстрагирования mpem-бутилметиловым эфиром, органическую фазу промывают водой и насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют досуха, чтобы получить указанный в заголовке продукт, который используют в дальнейшем без очистки.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 6.76 (d, 1 Н), 3.83 (t, 2 Н), 3.75 (s, 3 Н), 2.91 (t, 2 Н), 1.64 (s, 1 Н).
Стадия G: трет-бутил 5-[2-(3-бром-5-циано-1-метил-1Н-пиррол-2-ил)этокси]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии А Синтеза 2b", используя спирт, полученный в предыдущей Стадии и соединение из Синтеза 3b’.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.12 (t, 1 Н), 7.11 (s, 1 Н), 6.8 (d, 1 Н), 6.72 (d, 1 Н), 4.45 (s, 2 Н), 4.15 (t, 2 Н), 3.78 (s, 3 Н), 3.52 (t, 2 Н), 3.18 (t, 2 Н), 2.55 (t, 2 Н), 1.41 (s, 9 Н).
ИК: ν: >CN: 2215 см-1; ->С=O: 1686 см-1.
Стадия Н: трет-бутил 5-(2-{3-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-5-циано-1-метил-1Н-пиррол-2-ил}этокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии D Синтеза 3b", используя соединение из предыдущей стадии.
1Н ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.05 (t, 1 Н), 6.82 (s, 1 Н), 6.78 (s, 1 Н), 6.7 (2d, 2 Н), 6.55 (2d, 4 Н), 4.4 (s, 2 Н), 4.02 (t, 2 Н), 3.7 (s, 3 Н), 3.48 (t, 2 Н), 3.05 (t, 2 Н), 2.51 (t, 2 Н), 1.41 (s, 9 Н), 0.91 (s, 9 Н), 0.12 (s, 6 Н).
ИК: ν: >NH: 3315 см-1; >CN: 2212 см-1; >С=O: 1655 см-1.
Синтез 7b": трет-бутил 5-[2-({4-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1-метил-1Н-пиразол-5-ил}окси)этокси]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: трет-бутил 5-{2-[(4-бром-1-метил-1Н-пиразол-5-ил)окси]этокси}-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Синтезе 3b’ (4,06 г; 16,28 ммоль) в ацетонитриле (75 мл), добавляют бром-хлорэтан (2 мл; 24,43 ммоль) и карбонат калия (3,35 г; 24,43 ммоль), затем все перемешивают в течение 2 дней при 70°С. После концентрирования до 2/3 реакционной смеси, ее разбавляют с этилацетатом и водой. После экстрагирования этилацетатом, органические фазы промывают водным насыщенным раствором гидрокарбоната натрия, водой, затем насыщенным раствором хлорида натрия, и сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан в качестве растворителя для элюирования, чтобы получить бесцветное масло.
К раствору полученного таким образом остатка (4,4 г; 14,1 ммоль) в ацетоне (80 мл), добавляют йодид натрия (21,15 г; 141.1 ммоль), затем все перемешивают при кипячении с обратным холодильником в течение 5 дней. Реакционную среду концентрируют, затем разбавляют с этилацетатом и водой. После экстрагирования этилацетатом, органические фазы промывают раствором насыщенного хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха, чтобы получить масло коричневого цвета, которое применяют в следующей стадии без очистки.
К раствору полученного таким образом остатка (5,29 г; 13,13 ммоль) в ацетонитриле (100 мл), добавляют 5-гидрокси-N-метилпиразол (1,29 г; 13,13 ммоль), карбонат цезия (4,7 г, 24,43 ммоль) и йодид натрия (0,39 г; 2,6 ммоль), затем все перемешивают в течение 5 часов при 90°С. После концентрации реакционной смеси, остаток разбавляют с этилацетатом и водой. После экстрагирования этилацетатом, органические фазы промывают водным насыщенным раствором гидрокарбоната натрия, водой, насыщенным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить бесцветное масло.
К раствору полученного таким образом остатка (2,8 г; 7,5 ммоль) в метаноле (200 мл), добавляют порциями при 0°С трибромид пиримидиния (2,4 г; 7,5 ммоль). Все перемешивают в течение 1 часа при 0°С, затем в течение 2 часов при температуре окружающей среды. После концентрации реакционной смеси, остаток ресуспендируют в смеси воды и дихлорметана. После экстракции дихлорметаном, органические фазы промывают водным раствором 1 М хлористоводородной кислоты, водой, затем сушат над сульфатом натрия, фильтруют и концентрируют досуха. Остаток очищают с помощью хроматографии на фазе RP-18, используя ацетонитрил и воду в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.41 (s, 1 Н), 7.14 (m, 1 Н), 6.88 (d, 1 Н), 6.84 (d, 1 Н), 4.56/4.28 (2t, 4 Н), 4.45 (s, 2 Н), 3.62 (s, 3 Н), 3.52 (t, 2 Н), 2.55 (t, 2 Н), 1.42 (s, 9 Н).
ИК: ν: >С=O: 1689 см-1.
Стадия В: трет-бутил 5-[2-({4-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1-метил-1Н-пиразол-5-ил}окси)этокси]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Раствор соединения, полученного в Стадии А (2 г; 4,42 ммоль) и соединения, полученного в Стадии В Синтеза 1b" (1,48 г; 6,63 ммоль) в толуоле (15 мл) дегазируют аргоном в течение 10 минут. Добавляют трет-бутилат натрия (0,51 г; 5,3 ммоль), 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (0,187 г; 0,44 ммоль) и хлор(2-ди-трет-бутилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2-аминоэтил)фенил]палладий(II) (0,3 г; 0,44 ммоль), затем все перемешивают при 100°С в течение 2 часов в микроволновой печи (300 Вт). Реакционную среду фильтруют через Celite®. После промывания этилацетатом, фильтрат промывают водой и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, затем с помощью хроматографии на фазе RP-18, используя ацетонитрил и воду в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.18 (s, 1 Н), 7.1 (m, 1 Н), 6.75 (d, 2 Н), 6.7 (s, 1 Н), 6.6 (d, 2 Н), 6.48 (d, 2 Н), 4.45 (s, 2 Н), 4.42/4.15 (2t, 4 Н), 3.53 (s, 3 Н), 3.51 (t, 2 Н), 2.52 (t, 2 Н), 1.42 (s, 9 Н), 0.92 (s, 9 Н), 0.11 (s, 6 Н).
ИК: ν: >NH: 3485-3182; >С=O: 1689 см-1.
Синтез 8b": трет-бутил 4-[2-({4-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1-метил-1Н-пиразол-5-ил}окси)этил]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: 5-хлор-1-метил-4-нитро-1Н-пиразол
К раствору из 1-метил-4-нитро-1H-пиразола (5 г; 39,34 ммоль) в тетрагидрофуране (50 мл), добавляют по каплям при -78°С раствор 1,3 М бис(триметилсилил)амид лития в тетрагидрофуране (82 мл; 106,22 ммоль) и гексахлорэтан (14 г, 59,01 ммоль), затем все перемешивают в течение 1 часа при -78°С. Реакционную среду переносят в водный насыщенный раствор хлорида аммония и льда. Продукт экстрагируют дихлорметаном, затем органические фазы сушат над сульфатом натрия, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 8.41 (s, 1 Н), 3.90 (s, 3 Н).
ИК: ν: >СН: 3122; -NO2: 1521+1312 см-1.
Стадия В: трет-бутил 4-{2-[(1-метил-4-нитро-1Н-пиразол-5-ил)окси]этил}-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Синтезе 5b' (2,8 г; 10,09 ммоль) в тетрагидрофуране (50 мл), добавляют гидрид натрия при 60% (240 мг; 10,09 ммоль), затем все перемешивают в течение 1 часа при температуре окружающей среды. Добавляют раствор соединения, полученного в Стадии А (1,4 г; 8,66 ммоль) в тетрагидрофуране (25 мл), и реакционную среду перемешивают в течение 16 часов, затем переносят в водный насыщенный раствор хлорида аммония и льда. Продукт экстрагируют этилацетатом, затем органические фазы сушат над сульфатом натрия, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 8.15 (s, 1 Н), 7.20 (m, 4 Н), 4.80 (m, 1H), 4.51 (m, 2Н), 4.30 (d, 1H), 4.08 (m, 1H), 3.71 (s, 3Н), 3.25 (m, 1Н), 3.10 (m, 1Н), 2.03 (m, 1Н), 1.90 (m, 1H), 1.40 (s, 9 Н).
ИК: ν: >С=O: 1687; -NO2: 1567+1329 см-1.
Стадия С: трет-бутил 4-{2-[(4-амино-1-метил-1Н-пиразол-5-ил)окси]этил}-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Стадии В (2,4 г; 5,96 ммоль) в метаноле (75 мл), добавляют палладий на угле (15 мас. %), затем все гидрируют в течение 24 часов при температуре окружающей среды под давлением в 1 бар. Реакционную среду фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и аммиак в этаноле в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1Н ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.19 (гл, 4 Н), 6.88 (s, 1Н), 4.76 (m, 1Н), 4.39- 4.21 (m, 3Н), 4.01 (dd, 1Н), 3.52 (s, 3Н), 3.40 (s, 2Н), 3.24 (m, 1Н), 3.04 (m, 1Н), 1.91 (m, 1Н), 1.82 (m, 1Н), 1.42 (s, 9 Н).
ИК: ν: -NH2 и -NH: 3390, 3327 и 3240; >С=O: 1684 см-1.
Стадия D: трет-бутил(4-йодфенокси)диметилсилан
К раствору пара-йодфенола (20 г; 90 ммоль) в дихлорметане (50 мл), добавляют триэтиламин (15,2 мл; 109 ммоль) и хлорид трет-бутилдиметилхлорсилана (16,4 г; 109 ммоль), затем все перемешивают в течение 1 часа при температуре окружающей среды. После гидролиза, продукт экстрагируют этилацетатом, затем органические фазы промывают водой, насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.5 (d, 2Н), 6.67 (d, 2Н), 0.95 (s, 9Н), 0.20 (s, 6Н).
ИК: ν: >С-O-С<: 1252; -Si-O-C-: 905; -Si-C-: 822 см-1.
Стадия Е: трет-бутил 4-[2-({4-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1-метил-1Н-пиразол-5-ил}окси)этил]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Раствор соединения, полученного в Стадии С (1,05 г; 2,81 ммоль) и соединения, полученного в Стадии D (1,48 г; 3,14 ммоль) в толуоле (15 мл) дегазируют аргоном в течение 10 минут. Добавляют трет-бутилат натрия (300 мг; 3,1 ммоль) и хлор(2-ди-трет-бутилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2-аминоэтил)фенил]палладий(II) (387 мг; 0,56 ммоль), затем все перемешивают при 100°С в течение 2,5 часов в микроволновой печи (300 Вт). Реакционную среду фильтруют через Celite®. После промывки дихлорметаном, фильтрат концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1Н ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.2-7.20 (m, 5 Н), 6.60 (d, 2Н), 6.5 (d, 2Н), 6.4 (s, 1Н), 4.65 (d, 1Н), 4.25 (d, 1H), 4.20 (m, 2Н), 3.60 (s, 3Н), 3.30 (dd, 1H), 3.20 (dd, 1H), 2.88 (m, 1Н), 1.80 (m, 2Н), 1.40 (s, 9 Н), 0.9 (s, 9Н), 0.01 (s, 6Н).
ИК: ν: >NH: 3328; >С=O: 1693 см-1.
Синтез 9b": трет-бутил 5-(2-{4-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1-(тетрагидрофуран-3-ил)-1Н-пиразол-5-ил}этокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: тетрагидрофуран-3-ил метансульфонат
К раствору из 3-гидрокситетрагидрофурана (14,8 г; 0,25 моль) и триэтиламина (35 мл; 0,25 моль) в дихлорметане (200 мл), добавляют метансульфонилхлорид (14,6 мл; 0,18 моль), затем все перемешивают при температуре окружающей среды в течение 16 часов. Реакционную среду разбавляют в воде. После экстракции дихлорметаном, органические фазы промывают водой, затем сушат над сульфатом натрия, фильтруют и концентрируют досуха, чтобы получить указанный в заголовке продукт, который используют в следующей стадии без очистки.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 5.24-5.32 (m, 1 Н), 3.81-4.04 (m, 4 Н), 3.02 (s, 3 Н), 2.17-2.26 (m, 2 Н).
Стадия В: метил 1-(тетрагидрофуран-3-ил)-1Н-пиразол-5-карбоксилат
К раствору метил 1H-пиразол-5-карбоксилата (20 г; 0,16 моль) и соединения, полученного в Стадии А (28,9 г; 0,18 моль) в N,N-диметилформамиде (400 мл), добавляют карбонат калия (33 г; 0,24 моль), затем все перемешивают при 80°С в течение 48 часов. После достижения температуры окружающей среды, реакционную среду фильтруют и фильтрат концентрируют.Остаток ресуспендируют в смеси 15% этилацетата в гептане, затем фильтруют через силикагель. Фильтрат концентрируют и полученный остаток очищают с помощью хроматографии на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.50 (d, 1 Н), 6.83 (d, 1 Н), 5.85-5.94 (m, 1 Н), 4.08-4.20 (m, 2 Н), 4.01 (dd, 1 Н), 3.95 (td, 1 Н), 3.86 (s, 3 Н), 2.32-2.54 (m, 2 Н).
Стадия С: [1-(тетрагидрофуран-3-ил)-1Н-пиразол-5-ил]метанол
К суспензии алюмогидрида лития (3,38 г; 86,0 ммоль) в тетрагидрофуране (100 мл), охлажденной до 0°С, добавляют раствор соединения, полученного в Стадии В (8,43 г; 49,0 ммоль) в тетрагидрофуране (50 мл), затем все перемешивают при температуре окружающей среды в течение 16 часов. Реакционную среду охлаждают до 0°С и разбавляют медленно водой (3,4 мл), водным 15%раствором гидроксида натрия (6,8 мл) и в заключение водой (6,8 мл). К среде добавляют сульфат магния. После фильтрации и концентрирования фильтрата получают указанный в заголовке продукт, который используют в дальнейшем без очистки.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.43 (s, 1 Н), 6.17 (s, 1 Н), 5.05-5.18 (m, 1 Н), 4.69 (s, 2 Н), 4.19 (q, 1 Н), 4.06-4.13 (m, 1 Н), 3.91-4.02 (m, 2 Н), 2.32-2.50 (m, 2 Н), 2.10-2.32 (m, 1 Н).
Стадия D: [4-бром-1-(тетрагидрофуран-3-ил)-1Н-пиразол-5-ил]метанол
К раствору соединения, полученного в Стадии С (7,9 г; 47 ммоль) в дихлорметане (100 мл) при 0°С, добавляют порциями N-бромсукцинимид (8,78 г; 49,3 ммоль), затем все перемешивают при температуре окружающей среды в течение 1,25 часов. Реакционную среду разбавляют в водном растворе гидроксида натрия 1 М (100 мл). После экстракции дихлорметаном, органические фазы объединяют, промывают водой и сушат над сульфатом натрия, фильтруют и концентрируют досуха. Полученный остаток очищают с помощью хроматографии на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.44 (s, 1 Н), 5.08-5.19 (m, 1 Н), 4.69 (d, 2 Н), 4.19 (q, 1 Н), 4.09 (dd, 1 Н), 3.89-4.01 (m, 2 Н), 2.32-2.48 (m, 3 Н).
Стадия Е: 4-бром-5-(хлорметил)-1-(тетрагидрофуран-3-ил)-1Н-пиразол
К раствору соединения, полученного в Стадии D (8,93 г; 36,1 ммоль) и триэтиламин (7,5 мл; 54,2 ммоль) в дихлорметане (100 мл), добавляют метансульфонил хлорид (2,85 мл; 36,1 ммоль), затем все перемешивают при температуре окружающей среды в течение 16 часов. Реакционную среду разбавляют в воде. После экстракции дихлорметаном, органические фазы объединяют, промывают водой и сушат над сульфатом натрия, фильтруют и концентрируют досуха, чтобы получить указанный в заголовке продукт, который используют в дальнейшем без очистки.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.49 (s, 1 Н), 4.97-5.05 (m, 1 Н), 4.64 (s, 2 Н), 4.09-4.22 (m, 2 Н), 3.93-4.05 (m, 2 Н), 2.42 (q, 2 Н).
Стадия F: [4-бром-1-(тетрагидрофуран-3-ил)-1Н-пиразол-5-ил]ацетонитрил
К раствору соединения, полученного в Стадии Е (7,92 г; 29,8 ммоль) в смеси ацетонитрила (80 мл) и воды (80 мл), добавляют цианид калия (3,88 г; 59,7 ммоль), затем все перемешивают при 60°С в течение 16 часов. Ацетонитрил выпаривают, и водную фазу экстрагируют этилацетатом. Органические фазы объединяют, промывают водой, насыщенным водным раствором хлорида натрия, затем сушат над сульфатом натрия, фильтруют и концентрируют досуха. Полученный остаток очищают с помощью хроматографии на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.53 (s, 1 Н), 4.92-5.00 (m, 1 Н), 4.14-4.25 (m, 2 Н), 4.03-4.10 (m, 1 Н), 3.97 (td, 1 Н), 3.85 (d, 2 Н), 2.36-2.54 (m, 2 Н).
Стадия G: 2-[4-бром-1-(тетрагидрофуран-3-ил)-1Н-пиразол-5-ил]этанол
К раствору соединения, полученного в Стадии F (6,83 г; 26,7 ммоль) в дихлорметане (133 мл), при -78°С добавляют 1 М раствор гидрида диизобутилалюминия в дихлорметане (53,3 мл; 53,3 ммоль), затем все перемешивают при -78°С в течение 3 часов. После медленного добавления метанола (10 мл) и боргидрида натрия (3,04 г; 80,1 ммоль), реакционную среду перемешивают при температуре окружающей среды в течение 16 часов, затем разбавляют водным раствором хлористоводородной кислоты 1 N (50 мл). После контакта в течение 10 минут, растворители выпаривают, и продукт экстрагируют этилацетатом. Органические фазы объединяют, промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют досуха. Полученный остаток очищают с помощью хроматографии на силикагеле, используя дихлорметан и метанол в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.48 (s, 1 Н), 5.04 (tt, 1 Н), 4.13-4.21 (m, 1 Н), 4.06-4.12 (m, 1 Н), 3.91-4.00 (гл, 2 Н), 3.80-3.89 (m, 2 Н), 2.96 (t, 2 Н), 2.29-2.44 (m, 2 Н).
Стадия Н: трет-бутил 5-{2-[4-бром-1-(тетрагидрофуран-3-ил)-1Н-пиразол-5-ил]этокси}-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Готовят раствор трифенилфосфина (2,64 г; 10,1 ммоль) и диизопропила азодикарбоксилата (1,97 мл; 10,1 ммоль) в тетрагидрофуране при 0°С. После обесцвечивания и появления беловатого осадка (по истечении 5 минут), добавляют раствор соединения, полученного в Стадии G (2,02 г; 7,74 ммоль) в тетрагидрофуране (10 мл), затем все перемешивают при температуре окружающей среды в течение 1 часа. После добавления соединения Синтеза 3b’ (2,5 г; 10,1 ммоль) в растворе в тетрагидрофуране (10 мл), реакционную среду перемешивают при температуре окружающей среды в течение 4 часов, затем разбавляют диметилсульфоксидом (10 мл). После концентрации реакционной смеси, остаток очищают с помощью хроматографии на фазе RP-18, используя метанол и воду в качестве растворителей для элюирования, затем с помощью хроматографии на силикагеле, используя дихлорметан и метанол в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.49 (s, 1 Н), 7.12 (t, 1 Н), 6.74 (d, 1 Н), 6.66 (d, 1 Н), 4.99-5.09 (m, 1 Н), 4.54 (s, 2 Н), 4.12-4.23 (m, 3 Н), 4.09 (dd, 1 Н), 3.93-4.02 (m, 2 Н), 3.54-3.69 (m, 2 Н), 3.21 (td, 2 Н), 2.63-2.71 (m, 2 Н), 2.28-2.47 (m, 2 Н), 1.48 (s, 9 Н).
Стадия I: трет-бутил 5-(2-{4-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1-(тетрагидрофуран-3-ил)-1Н-пиразол-5-ил}этокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Синтеза 7b", используя соединение из предыдущей стадии.
1Н ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.48 (s, 1 Н), 7.08 (t, 1 Н), 6.73 (d, 1 Н), 6.62-6.67 (m, 2 Н), 6.56 (d, 1 Н),6.48-6.52 (m, 2 Н), 4.96-5.04 (m, 1 Н), 4.75 (s, 1 Н), 4.54 (s, 2 Н), 4.17-4.24 (m, 1 Н), 4.02-4.15 (m, 4 Н), 3.94-4.02 (m, 1 Н), 6.61 (br. s, 1 Н), 3.11 (t, 2 Н), 2.67 (br. s, 2 Н), 2.45-2.54 (m, 1 Н), 2.31-2.42 (m, 1 Н), 1.49 (s, 9 Н), 0.96 (s, 9 Н), 0.15 (s, 6 Н).
Синтез 10b": трет-бутил 4-(2-{4-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1-метил-1Н-пиразол-5-ил}этокси)-1,3-дигидро-2Н-изоиндол-2-карбоксилат
Стадия А: трет-бутил 4-[2-(4-бром-1-метил-1Н-пиразол-5-ил)этокси]-1,3-дигидро-2Н-изоиндол-2-карбоксилат
Раствор соединения, полученного в Стадии В Синтеза 3b" (1 г; 4,92 ммоль) и соединение Синтеза 6b' (1,54 г; 6,54 ммоль) в смеси тетрагидрофурана (10 мл) и толуола (10 мл) дегазируют аргоном в течение 10 минут.Добавляют цианометилен три-н-бутилфосфоран (2,58 мл; 9,84 ммоль), затем все герметизируют и перемешивают при 110°С в течение 48 часов. Реакционную среду концентрируют, затем разбавляют в смеси этилацетата и воды. После декантации, органическую фазу промывают водой и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток измельчают в циклогексане, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300K) δ част, на млн.: 7.49 (s, 1 Н), 7.24 (t, 1 Н), 6.89 (m, 2 Н), 4.55 (d, 2 Н), 4.41 (m, 2 Н), 4.19 (t, 2 Н), 3.87 (d, 3 Н), 3.16 (t, 2 Н), 1.45 (d, 9 Н).
ИК: ν:>С=С<: 1686 см-1.
Стадия В: трет-бутил 4-(2-{4-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1-метил-1Н-пиразол-5-ил}этокси)-1,3-дигидро-2Н-изоиндол-2-карбоксилат
Раствор соединения, полученного в Стадии А (0,95 г; 2,25 ммоль) и соединения, полученного в Стадии В Синтеза 1b" (0,74 г; 3,37 ммоль) в тетрагидрофуране (50 мл) дегазируют аргоном в течение 10 минут. Добавляют трет-бутилин натрия (281 мг; 2,9 ммоль) и хлор(2-ди-трет-бутилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2-аминоэтил)фенил]палладий(II) (154 мг; 0,22 ммоль), затем все перемешивают при 50°С в течение 3 часов. Реакционную среду фильтруют через Celite®. После промывания этилацетатом, продукт экстрагируют этилацетатом, затем органические фазы промывают водным насыщенным раствором хлорида аммония, водой и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.28 (s, 1 Н), 7.18 (t, 1 Н), 6.88 (2d, 1 Н), 6.75 (d, 1 Н), 6.69 (2s, 1 Н), 6.58 (d, 2 Н), 6.49 (d, 2 Н), 4.55 (d, 2 Н), 4.41 (s, 2 Н), 4.1 (t, 2 Н), 3.81 (s, 3 Н), 3.01 (t, 2 Н), 1.48 (s, 9 Н), 0.91 (s, 9 Н), 0.11 (s, 6 Н).
ИК: ν: >NH: 3308 см-1; >С=O: 1681 см-1; >С=С<: 1617 см-1.
Синтез 11b": трет-бутил (3R или 3S)-5-(3-{3-f(4-{fmpem-бутил(диметил)силил]окси}фенил)амино]-5-циано-1-метил-1Н-пиррол-2-ил}пропокси)-3-метил-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: трет-бутил 5-[3-(3-бром-5-циано-1-метил-1Н-пиррол-2-ил)пропокси]-3-метил-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Раствор соединения, полученного в Стадии D Синтеза 5b" (3,19 г; 13,12 ммоль) и соединения, полученного в Синтезе 7b' (3,14 г; 11,93 ммоль) в смеси тетрагидрофурана (30 мл) и толуола (30 мл) дегазируют аргоном. Добавляют цианометилен три-н-бутилфосфоран (6,26 мл; 23,86 ммоль). Колбу запечатывают и реакционную среду перемешивают при 110°С в течение 20 часов, затем концентрируют. Полученный таким образом остаток разбавляют в смеси этилацетата и воды. После экстрагирования водной фазы этилацетатом, органические фазы объединяют, промывают водой, насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.11 (t, 1 Н), 7.09 (s, 1 Н), 6.78 (t, 2 Н), 4.55 (d, 2 Н), 4.5 (m, 1 Н), 3.95 (т, 2 Н), 3.7 (т, 3 Н), 2.85 (t, 2 Н), 2.69 (d, 2 Н), 1.95 (t, 2 Н), 1.4 (s, 9 Н), 1 (d, 3 Н).
ИК: ν: -CN: 2215 см-1; >С=O: 1686 см-1.
Стадия В: трет-бутил 5-(3-{3-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-5-циано-1-метил-1Н-пиррол-2-ил}пропокси)-3-метил-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии D Синтеза 3b", используя соединение из предыдущей стадии.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.05 (t, 1 Н), 6.8 (m, 2 Н), 6.75 (d, 1 Н), 6.65 (d, 1 Н), 6.55 (d, 2 Н), 6.5 (d, 2 Н), 4.65 (d, 1 Н), 4.15 (d, 1 Н), 4.45 (m, 1 Н), 3.88 (t, 2 Н), 3.65 (s, 3 Н), 2.75 (t, 2 Н), 2.55 (d, 2 Н), 1.9 (m, 2 Н), 1.45 (s, 9 Н), 0.98 (d, 3 Н), 0.95 (s, 6 Н), 0.1 (s, 9 Н).
ИК: ν: >NH: 3400 см-1; -CN: 2208 см-1; >С=O: 1688 см-1.
Стадия С: трет-бутил (3R или 3S)-5-(3-{3-[(4-{[mpem-бутил(диметил)силил]окси}фенил)амино]-5-циано-1-метил-1Н-пиррол-2-ил}пропокси)-3-метил-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают с помощью хроматографии на хиральной фазе (S,S) Whelk-01, используя гептан и изопропанол в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.07 (t, 1 Н), 6.77 (s, 1 Н), 6.77 (m, 1 Н), 6.73 (d, 1 Н), 6.66 (d, 1 Н), 6.56 (d, 2 Н), 6.49 (d, 2 Н), 4.62 (d, 1 Н), 4.14 (d, 1 Н), 4.46 (m, 1 Н), 3.87 (t, 2 Н), 3.66 (s, 3 Н), 2.74 (t, 2 Н), 2.57 (m, 2 Н), 1.89 (m, 2 Н), 1.43 (s, 9 Н), 0.95 (d, 3 Н), 0.92 (s, 9 Н), 0.11 (s, 6 Н).
Синтез 12b": трет-бутил (3S или 3R)-5-(3-{3-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-5-циано-1-метил-1Н-пиррол-2-ил}пропокси)-3-метил-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом Синтеза 11b".
1Н ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.07 (t, 1 Н), 6.77 (s, 1 Н), 6.77 (m, 1 Н), 6.73 (d, 1 Н), 6.66 (d, 1 Н), 6.56 (d, 2 Н), 6.49 (d, 2 Н), 4.62 (d, 1 Н), 4.14 (d, 1 Н), 4.46 (m, 1 Н), 3.87 (t, 2 Н), 3.66 (s, 3 Н), 2.74 (t, 2 Н), 2.57 (m, 2 Н), 1.89 (m, 2 Н), 1.43 (s, 9 Н), 0.95 (d, 3 Н), 0.92 (s, 9 Н), 0.11 (s, 6 Н).
Синтез 13b": трет-бутил 4-(2-{5-(бензилокси)-2-[(5-циано-1,2-диметил-1Н-пиррол-3-ил)амино]фенокси}этил)пиперидин-1-карбоксилат
Стадия А: 4-(бензилокси)-2-фтор-1-нитробензол
К раствору из 3-фтор-4-нитрофенола (12,76 г; 81,2 ммоль) в ацетоне (165 мл), добавляют карбонат калия (13,47 г; 97,5 ммоль) и бензил бромид (9,75 мл; 82,0 ммоль). Реакционную среду перемешивают при кипячении с обратным холодильником в течение 14 часов, затем после достижения температуры окружающей среды, ее разбавляют в воде. После экстрагирования водной фазы этилацетатом, органические фазы объединяют, промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток ресуспендируют в небольшом количестве этилацетата и пентана, и полученный осадок фильтруют, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 8.15 (t, 1 Н), 7.25 (dd, 1 Н), 7.05 (dd, 1 Н), 5.25 (s, 2 Н), 7.3-7.5 (гл, 5 Н).
ИК: ν: -NO2: 1510 и 1500 см-1; -NO2: 1329 см-1.
Стадия В: трет-бутил 4-{2-[5-(бензилокси)-2-нитрофенокси]этил}пиперидин-1-карбоксилат
К смеси гидрида натрия (1,52 г; 38,1 ммоль) в тетрагидрофуране (30 мл) при 0°С, добавляют по каплям раствор трет-бутил 4-(2-гидроксиэтил)пиперидин-1-карбоксилата (8 г; 35 ммоль) в тетрагидрофуране (30 мл). Реакционную среду перемешивают в течение 15 минут при 0°С, затем в течение 30 минут при температуре окружающей среды. Реакционную среду снова охлаждают до 0°С, затем добавляют по каплям раствор соединения, полученного в Стадии А (7,86 г; 31,79 ммоль) в тетрагидрофуране (30 мл). Реакционную среду перемешивают при температуре окружающей среды в течение 17 часов, затем гидролизуют. После экстрагирования водной фазы этилацетатом, органические фазы объединяют, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.95 (d, 1 Н), 7.4 (m, 5 Н), 6.9 (d, 1 Н), 6.7 (dd, 1 Н), 5.25 (s, 2 Н), 4.2 (t, 2 Н), 3.9/2.7 (m, 4 Н), 1.7 (m, 3 Н), 1.7/1.05 (m, 4 Н), 1.4 (s, 9 Н).
ИК: ν:; >С=O: 1683 см-1; -NO2: 1513 и 1255 см-1.
Стадия С: трет-бутил 4-{2-[2-амино-5-(бензилокси)фенокси]этил}пиперидин-1-карбоксилат
К раствору соединения, полученного в Стадии В (9,4 г; 22 ммоль) в смеси тетрагидрофурана (70 мл) и ледяной уксусной кислоты (70 мл), добавляют железо (12,9 г; 23,1 ммоль), затем все нагревают до 65°С в течение 18 часов. Реакционную среду фильтруют через Celite®, затем после добавления водного раствора 5 н. гидроксида натрия для достижения значения рН=7, ее концентрируют наполовину. После экстрагирования водной фазы в дихлорметане, органические фазы объединяют, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.4-7.3 (m, 5 Н), 6.55 (m, 2 Н), 6.35 (dd, 1 Н), 4.95 (s, 2 Н), 4.22 (m, 2 Н), 3.95 (m, 4 Н), 2.7 (m, 2 Н), 1.65 (m, 3 Н), 1.65/1.05 (m, 4 Н), 1.4 (s, 9 Н).
ИК: ν:; -NH2: 3450-3365 см-1; >С=O: 1669 см-1.
Стадия D: 4-бром-1,5-диметил-1Н-пиррол-2-карбонитрил
К раствору 1,5-диметил-1H-пиррол-2-карбонитрила (15 г; 124,8 ммоль) в ледяной уксусной кислоте (300 мл), добавляют по каплям раствор брома (24 мл; 457,74 ммоль) в ледяной уксусной кислоте (60 мл). Реакционную среду перемешивают в течение 16 часов при температуре окружающей среды. После концентрации реакционной смеси наполовину, добавляют такое же количество воды (180 мл) и образованный осадок фильтруют, затем растворяют в дихлорметане. Органическую фазу промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.05 (s, 1 Н), 3.65 (s, 3 Н), 2.23 (s, 3 Н).
ИК: ν:; >СН: 3131 см-1; >CN: 2220 см-1.
Стадия Е: трет-бутил 4-(2-{5-(бензилокси)-2-[(5-циано-1,2-диметил-1Н-пиррол-3-ил)амино]фенокси}этил)пиперидин-1-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Синтеза 2b", используя соединение из предыдущей стадии.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.4 (d, 2 Н), 7.35 (t, 2 Н), 7.3 (f, 1 Н), 6.7 (s, 1 Н), 6.6 (d, 1 Н), 6.35 (dd, 1 Н), 6.3 (d, 1 Н), 5.85 (s, 1 Н), 4.95 (s, 2 Н), 4.05 (t, 2 Н), 3.9/2.7 (m+m, 2+2 Н), 3.6 (s, 3 Н), 2.05 (s, 3 Н), 1.7 (гл, 5 Н), 1.4 (s, 9 Н), 1.05 (m, 2 Н).
ИК: ν: >NH: 3404 см-1; >CN: 2207 см-1; >С=O: 1684 см-1.
Синтез 14b": трет-бутил 3-(2-{5-(бензилокси)-2-[(5-циано-1,2-диметил-1Н-пиррол-3-ил)амино]фенокси}этил)пиперидин-1-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом Синтеза 13b", используя трет-бутил 3-(2-гидроксиэтил)-пиперидин-1-карбоксилат в Стадии В.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.4 (d, 2 Н), 7.35 (t, 1 Н), 7.3 (t, 2 Н), 6.75 (s, 1 Н), 6.62 (d, 1 Н), 6.35 (dd, 1 Н), 6.25 (d, 1 Н), 5.85 (s, 1 Н), 4.95 (s, 2 Н), 4.05-2.5 (m, 4 Н), 4.05 (t, 2 Н), 3.63 (s, 3 Н), 2.1 (s, 3 Н), 1.8-1.3 (m, 7 Н), 1.35 (br. s, 9 Н).
ИК: ν:; >NH: 3400 см-1; >CN: 2207 см-1; >С=O: 1684 см-1.
Синтез 15b": йодид 1-(3-{[2-(трет-бутоксикарбонил)-1,2,3,4-тетрагидроизохинолин-5-ил]окси}пропил)-3-{[4-(проп-2-ен-1-илокси)фенил]амино}пиримидиния
Стадия А: 1-нитро-4-(проп-2-ен-1-илокси)бензол
К раствору 4-нитрофенола (20 г; 0,144 моль) в ацетонитриле (500 мл), добавляют аллилбромид (15 мл; 0,173 моль) и карбонат цезия (52 г, 0,158 моль), затем все перемешивают в течение 16 часов при температуре окружающей среды и в течение 2 часов при 70°С. После фильтрации нерастворимого вещества и концентрирования фильтрата, остаток разбавляют дихлорметаном и водой. После декантации, органическую фазу промывают водой, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя этилацетат и петролейный эфир в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 8.2 (d, 2 Н), 7 (d, 2 Н), 6 (m, 1 Н), 5.45 (tdd, 1 Н), 5.4 (tdd, 1 Н), 4.6 (m, 2 Н).
ИК: ν: >NO2: 1590 и 1331 см-1.
Стадия В: 4-(проп-2-ен-1-илокси)анилин
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии С Синтеза 13b", используя соединение из предыдущей стадии.
1Н ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 6.65 (d, 2 Н), 6.49 (d, 2 Н), 6 (m, 1 Н), 5.34/5.2 (dd, 2 Н), 4.59 (s, 2 Н), 4.4 (d, 2 Н).
ИК: ν: >NH2: 3429 и 3350 см-1.
Стадия С: N-[4-(проп-2-ен-1-илокси)фенил]пиридин-3-амин
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии D Синтеза 3b", используя 3-бромпиридин и соединение из предыдущей Стадии.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 8.22 (d, 1 Н), 8.05 (s, 1 Н), 7.92 (dd, 1 Н), 7.31-7.11 (2*dd, 2 Н), 7.05 (d, 2 Н), 6.91 (d, 2 Н), 6.04 (m, 1 Н), 5.46-5.19 (2*dd, 2 Н), 4.52 (d, 2 Н).
ИК: ν: >NH: 3250 и 3184 см-1.
Стадия D: йодид 1-(3-{[2-(трет-бутоксикарбонил)-1,2,3,4-тетрагидроизохинолин-5-ил]окси}пропил)-3-{[4-(проп-2-ен-1-илокси)фенил]амино}пиримидиния
К раствору соединения, полученного в Стадии С (740 мг; 3,27 ммоль), в диоксане (13 мл) добавляют соединение из Синтеза 8b' (1,05 г; 2,5 ммоль), затем все перемешивают в течение 18 часов при 70°С. После концентрирования остаток очищают хроматографией на силикагеле, используя дихлорметан и метанол в качестве растворителя для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 9.1 (s, 1 Н), 8.34 (т, 2 Н), 7.81 (m, 2 Н), 7.14 (t, 1 Н), 7.12 (d, 2 Н), 6.91 (d, 2 Н), 6.78 (t, 2 Н), 6.04 (m, 1 Н), 5.4/5.27 (2*dd, 2 Н), 4.67 (t, 2 Н), 4.54 (d, 2 Н), 4.46 (s, 2 Н), 4.04 (t, 2 Н), 3.5 (t, 2 Н), 2.5 (d, 2 Н), 2.37 (t, 2 Н), 1.42 (s, 9 Н).
ИК: ν: >NH: 3500-2700 см-1; >С=O: 1686 см-1.
Синтез 16b": 2-метил-N-[4-(проп-2-ен-1-илокси)фенил]-4-[2-(тетрагидро-2Н-пиран-2-илокси)этокси]пиримидин-5-амин
Стадия А: 4-хлор-2-метил-6-[2-(тетрагидро-2Н-пиран-2-илокси)этокси]пиримидин-5-амин
К смеси гидрида натрия (1,5 г; 36,4 ммоль) в тетрагидрофуране (10 мл) при 0°С, добавляют по каплям раствор 2-тетрагидропиран-2-илоксиэтанол (4,6 мл; 33,6 ммоль) в тетрагидрофуране (60 мл). Реакционную среду перемешивают 15 минут при 0°С, затем 30 минут при температуре окружающей среды. Реакционную среду снова охлаждают до 0°С, затем добавляют по каплям раствор 5-амино-4,6-дихлор-2-метилпиримидина (5 г; 28 ммоль) в тетрагидрофуране (70 мл). Реакционную среду перемешивают при температуре окружающей среды в течение 17 часов, затем гидролизуют. После экстрагирования водной фазы этилацетатом, органические фазы объединяют, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 5.01 (s, 2 Н), 4.66 (т, 1 Н), 4.49 (m, 2 Н), 3.94 (2*т, 2 Н), 3.77/3.44 (2*т, 2 Н), 2.34 (s, 3 Н), 1.76-1.35 (m, 4 Н), 1.61/1.45 (2*т, 2 Н).
ИК: ν: -NH2: 3464 и 3340 см-1.
Стадия В: 2-метил-4-[2-(тетрагидро-2Н-пиран-2-илокси)этокси]пиримидин-5-амин
К раствору соединения, полученного в Стадии А (6,9 г; 24,0 ммоль) в этаноле (120 мл), добавляют палладий на угле (10 мас. %), затем все гидрируют в течение 6 часов при температуре окружающей среды под 1 бар. Реакционную среду фильтруют и концентрируют досуха. Остаток разбавляют в смеси водного насыщенного раствора бикарбоната натрия и дихлорметана. После декантации и экстрагирования дихлорметаном, органические фазы объединяют, затем сушат над сульфатом магния, чтобы обеспечить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.76 (s, 1 Н), 4.76 (s, 2 Н), 4.65 (m, 1 Н), 4.46 (m, 2 Н), 3.93/3.75 (2 т, 2 Н), 3.75/3.44 (2 т, 2 Н), 2.34 (s, 3 Н), 1.75-1.35 (m, 6 Н).
ИК: ν: -NH2: 3600-3100 см-1.
Стадия С: 1-бром-4-(проп-2-ен-1-илокси)бензол
К раствору из 4-бромфенола (10 г; 57,8 ммоль) в ацетоне (290 мл), добавляют аллилбромид (5,5 мл; 63,6 ммоль) и карбонат калия (16 г, 116 ммоль), затем все перемешивают в течение 6 часов при 85°С, затем в течение 16 часов при температуре окружающей среды. После фильтрации нерастворимого вещества и концентрирования фильтрата, остаток разбавляют с этилацетатом и водой. После декантации, органическую фазу промывают водой и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя этилацетат и петролейный эфир в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част, на млн.: 7.36 (d, 2 Н), 6.79 (d, 2 Н), 6.01 (m, 1 Н), 5.4 (d, 1 Н), 5.3 (d, 1 Н), 4.5 (d, 2 Н).
ИК: ν: >СН-Аr: 821 см-1; >С=С<: 1590, 1578 и 1488 см-1.
Стадия D: 2-метил-М-[4-(проп-2-ен-1-илокси)фенил]-4-[2-(тетрагидро-2Н-пиран-2-илокси)этокси]пиримидин-5-амин
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Синтеза 2b", используя соединения, полученные в предшествующих Стадиях В и С.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 8.07 (s, 1 Н), 7.17 (s, 1 Н), 6.98 (d, 2 Н), 6.86 (d, 2 Н), 6.03 (m, 1 Н), 5.38/5.24 (2*dd, 2 Н), 4.62 (m, 1 Н), 4.52 (m, 2 Н), 4.5 (m, 2 Н), 3.94/3.76 (2*т, 2 Н), 3.74/3.4 (2*т, 2 Н), 2.43 (s, 3 Н), 1.61 (m, 2 Н), 1.42 (m, 4 Н).
ИК: ν:; >NH: 3415 см-1; >С=С<: 1649 см-1.
Синтез 17b": трет-бутил [2-(3-циано-5-{[4-(проп-2-ен-1-илокси)фенил]амино}фенокси)этил]метилкарбамат Стадия А: (3-бром-5-метоксифенил)метанол
К раствору из 3-бром-5-метоксибензойной кислоты (10 г; 43,3 ммоль) в тетрагидрофуране (280 мл), добавляют по каплям комплекс боран-диметилсульфид (32,5 мл; 64,9 ммоль), затем все перемешивают в течение 2 часов. Реакционную среду подкисляют по каплям водным раствором 2 н. соляной кислоты до рН=1. После экстракции простым эфиром, органическую фазу промывают водным 1 н. раствором гидроксида натрия и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Указанный в заголовке продукт получают в виде масла, которое применяют в следующей стадии без очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.08 (m, 1 Н), 6.99 (m, 1 Н), 6.89 (m, 1 Н), 5.3 (br. s, 1 Н), 4.47 (s, 2 Н), 3.75 (s, 3 Н).
ИК: ν: -ОН: 1588 см-1; >С-O: 1268 и 1038 см-1; γ: >СН-Ar: 811 см-1.
Стадия В: 3-бром-5-метоксибензальдегид
К раствору соединения, полученного в Стадии А (8,6 г; 39,8 ммоль) в дихлорметане (400 мл) добавляют реагент Десса-Мартина (20,3 мл; 47,8 ммоль), затем все перемешивают в течение 2 часов. После добавления простого эфира, реакционную среду фильтруют через слой кремнезема. Фильтрат концентрируют, ресуспендируют в смеси гептана и этилацетата, затем фильтруют снова через слой кремнезема. После концентрации фильтрата, указанный в заголовке продукт получают в виде твердого вещества бледно-желтого цвета, которое используют в следующей стадии без очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 9.5 (s, 1 Н), 7.69 (t, 1 Н), 7.5 (t, 1 Н), 7.42 (t, 1 Н), 3.85 (s, 3 Н).
ИК: ν: >С=O: 1691 см-1.
Стадия С: (Z)-1-(3-бром-5-метоксифенил)-N-гидроксиметанимин
К раствору соединения, полученного в Стадии В (7,8 г; 36,4 ммоль) в этаноле (10 мл), добавляют последовательно гидрохлорид гидроксиламина (12,6 г; 182 ммоль) и пиридин (6,27 мл; 87,4 ммоль), затем все перемешивают при 65°С в течение 1 часа. После достижения температуры окружающей среды, реакционную среду разбавляют со смесью из этилацетата и воды. После декантации, органическую фазу промывают водой и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Указанный в заголовке продукт получают в виде твердого вещества белого цвета, которое используют в следующей стадии без очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 11.45 (s, 1 Н), 8.1 (s, 1 Н), 7.35 (t, 1 Н), 7.16 (d, 2 Н), 3.8 (s, 3 Н).
ИК: ν: -ОН: 3300-3000 см-1; Ar: 1600 и 1564 см-1; >С-O: 1220 и 1059 см-1; -N-O: 960 см-1; γ: >СН-Ar: 831 см-1.
Стадия D: 3-бром-5-метоксибензонитрил
К раствору соединения, полученного в Стадии С (8,1 г; 35,2 ммоль), в диоксане (70 мл), при 0°С добавляют пиридин (22 мл; 211 ммоль) и по каплям трифторуксусный ангидрид (1,4 мл; 70,4 ммоль), затем все перемешивают при температуре окружающей среды в течение 24 часов. После возврата к 0°С, по каплям добавляют вторую порцию трифторуксусного ангидрида (1,4 мл; 70,4 ммоль), затем все перемешивают при температуре окружающей среды в течение 24 часов. После возврата к 0°С, по каплям добавляют третью порцию трифторуксусного ангидрида (1,4 мл; 70,4 ммоль), затем все перемешивают при 60°С в течение 1 часа. После достижения температуры окружающей среды, реакционную среду разбавляют смесью дихлорметана и воды. После декантации, органическую фазу промывают водным раствором 1 н. хлористоводородной кислоты и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Указанный в заголовке продукт получают в виде твердого вещества бледно-желтого цвета, которое используют в следующей стадии без очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.69 (t, 1 Н), 7.53 (dd, 1 Н), 7.5 (dd, 1 Н), 3.82 (s, 3 Н).
ИК: ν: -CN: 2232 см-1; Ar: 1597 и 1562 см-1; >С-O-С<: 1278 и 1051 см-1; γ: >СН-Ar: 848, 814 и 671 см-1.
Стадия Е: 3-бром-5-гидроксибензонитрил
К раствору соединения, полученного в Стадии D (5,9 г; 27,9 ммоль), в коллидине (55 мл), добавляют йодид лития (11,2 г; 83,7 ммоль), затем все перемешивают при 150°С в течение 16 часов. После достижения температуры окружающей среды, реакционную среду выливают в ледяную воду. После экстракции дихлорметаном, органические фазы объединяют, промывают водой, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Указанный в заголовке продукт получают в виде твердого вещества оранжево-коричневого цвета, которое используют в следующей стадии без очистки.
1Н ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 10.7 (br. s, 1 Н), 7.51 (t, 1 Н), 7.3 (t, 1 Н), 7.18 (dd, 1 Н).
ИК: ν: -ОН: 3283 см-1; -CN: 2245 см-1.
Стадия F: трет-бутил (2-гидроксиэтил)метилкарбамат
К раствору 2-(метиламино)этанола (30 г; 0,399 моль) в дихлорметане (800 мл), при температуре окружающей среды порциями добавляют ди-трет-бутил дикарбонат (87,3 г; 0,399 моль). Среду перемешивают при этой температуре в течение 16 часов. Растворитель выпаривают в вакууме и Остаток очищают хроматографией на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 3.75 (t, 2 Н), 3.39 (t, 2 Н), 2.91 (s, 3 Н), 2.85 (m, 1 Н), 1.45 (s, 9 Н).
ИК: v: -ОН: 3431 см-1; >C=O: 1692 и 1668 см-1.
Стадия G: трет-бутил [2-(3-бром-5-цианофенокси)этил]метилкарбамат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии Н Синтеза 9b'', используя соединения, полученные в предыдущих стадиях Е и F.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.7 (br. s, 1 Н), 7.52 (br. s, 1 Н), 7.5 (br. s, 1 Н), 4.2 (m, 2 Н), 3.5 (t, 2 Н), 2.88 (br. s, 3 Н), 1.35 (2br. s, 9 Н).
ИК: v: -CN: 2235 см-1; >C=O: 1687 см-1.
Стадия Н: трет-бутил [2-(3-циано-5-{[4-(проп-2-ен-1-илокси)фенил]амино}фенокси)этил]метилкарбамат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Синтеза 2b'', используя соединение из предыдущей стадии и соединение, полученное в Стадии В Синтеза 15b''.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.07 (d, 2 Н), 6.94 (d, 2 Н), 6.73 (dd, 2 Н), 6.63 (t, 1 Н), 6.05 (m, 1 Н), 5.4 (dd, 1 Н), 5.26 (dd, 1 Н), 4.54 (td, 2 Н), 4.06 (t, 2 Н), 3.5 (t, 2 Н), 2.83 (s, 3 Н), 1.34 (s, 9 Н).
ИК: v: -NH: 3344 см-1; -CN: 2231 см-1; >C=O: 1741 см-1; >C=O: 1673 см-1; >С=С<: 1591 см-1.
Синтез 18b'': трет-бутил [4-(3-циано-5-{[4-(проп-2-ен-1-илокси)фенил]амино}фенокси)бутил]метилкарбамат
Указанное в заголовке соединение получают в соответствии со способом Синтеза 17b'', используя 4-(метиламино)бутанол в Стадии F.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 8.23 (s, 1 Н), 7.07 (d, 2 Н), 6.94 (d, 2 Н), 6.73 (t, 1 Н), 6.68 (t, 1 Н), 6.63 (t, 1 Н), 6.05 (m, 1 Н), 5.39/5.26 (2dquad, 2 Н), 4.54 (dt, 2 Н), 3.96 (t, 2 Н), 3.19 (t, 2 Н), 2.76 (br. s, 3 Н), 1.59 (m, 4 Н), 1.37 (br. s, 9 Н).
ИК: v: -NH: 3340 см-1; -CN: 2229 см-1; >C=O: 1687 см-1; >C=O: 1670 см-1.
Синтез 19b'': трет-бутил 5-(2-{2-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]этокси}этокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: трет-бутил 5-[2-(2-оксоэтокси)этокси]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору оксалилхлорида (0,2 мл; 2,22 ммоль) в тетрагидрофуране (7 мл) при -78°C добавляют диметилсульфоксид (0,2 мл). После контакта в течение 30 минут при этой температуре, при -78°C по каплям добавляют раствор соединения, полученного в Синтезе 9b' (0,5 г; 1,48 ммоль) в тетрагидрофуране (8 мл) и, в тех же условиях триэтиламин (0,77 мл; 5,93 ммоль). Все перемешивают в течение 1 часа при -78°C, затем в течение 2 часов при 0°C. Реакционную среду разбавляют со смесью из этилацетата и воды. После декантации, органическую фазу промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Указанный в заголовке продукт получают в виде масла красновато-коричневого цвета, которое применяют в следующей стадии без очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 9.6 (s, 1 Н), 7.13 (t, 1 Н), 6.82 (d, 1 Н), 6.75 (d, 1 Н), 4.46 (br. s, 2 Н), 4.29 (s, 2 Н), 4.12 (m, 2 Н), 3.85 (m, 2 Н), 3.54 (m, 2 Н), 2.64 (t, 2 Н), 1.41 (s, 9 Н).
ИК: v: >C=O: 1689 см-1.
Стадия В: трет-бутил 5-(2-{2-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]этокси}этокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Стадии А (0,5 г; 1,49 ммоль) в дихлорметане (10 мл), добавляют соединение, полученное в Стадии В Синтеза 1b'' (0,4 г; 1,79 ммоль) и триацетокси-боргидрид натрия (0,63 г; 2,98 ммоль). Все перемешивают в течение 16 часов при температуре окружающей среды. Реакционную среду разбавляют в водный насыщенный раствор гидрокарбоната натрия. После экстракции дихлорметаном, органические фазы промывают насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя этилацетат и петролейный эфир в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.13 (t, 1 Н), 6.81 (d, 1 Н), 6.75 (d, 1 Н), 6.59 (d, 2 Н), 6.47 (d, 2 Н), 5.05 (t, 1 Н), 4.46 (s, 2 Н), 4.1 (t, 2 Н), 3.77 (t, 2 Н), 3.62 (t, 2 Н), 3.52 (t, 2 Н), 3.13 (quad, 2 Н), 2.63 (t, 2 Н), 1.42 (s, 9 Н), 0.92 (s, 9 Н), 0.11 (s, 6 Н).
ИК: v: >NH: 3387 см-1; >C=O: 1693 см-1.
Синтез 20b'': трет-бутил 5-[2-(1-{3-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-5-цианофенил}-1Н-1,2,3-triazol-4-ил)этил]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: 3-бром-5-нитробензонитрил
К раствору трет-бутилнитрита (8,19 мл; 62 ммоль) в N,N-диметилформамиде (35 мл) при 50°C, добавляют по каплям раствор 2-амино-3-бром-5-нитробензонитрила (10 г; 41,3 ммоль) в N,N-диметилформамиде (85 мл). Смесь перемешивают при 50°C, пока не прекратится выделение газа. Затем неочищенную смесь выливают в водный раствор 0,5 н. соляной кислоты (1 л), затем все экстрагируют метил-трет-бутиловым эфиром. Органические фазы объединяют, и промывают водным насыщенным раствором бикарбоната натрия, затем насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают хроматографией на силикагеле, используя этилацетат и гептан в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн. 8.61 (t, 1 Н), 8.47 (t, 1 Н), 8.12 (t, 1 Н).
Стадия В: 3-амино-5-бромбензонитрил
К раствору соединения, полученного в Стадии А (6,85 г; 30 ммоль) в этилацетате (20 мл) и этаноле (82 мл), порциями добавляют дигидрат хлорида олова (33,85 г; 150 ммоль). Реакционную смесь после этого нагревают до 70°C в течение 30 минут и концентрируют до объема приблизительно 40 мл, затем выливают на лед. Полученную таким образом смесь подщелачивают водным раствором 2 н. гидроксила натрия, и все перемешивают в течение 20 минут. Продукт экстрагируют этилацетатом. Органические фазы объединяют и промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают хроматографией на силикагеле, используя этилацетат и гептан в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 7.12 (t, 1 Н), 7.01 (t, 1 Н), 6.80-6.83 (m, 1 Н), 3.97 (br. s, 2 Н).
Стадия С: 3-азидо-5-бромбензонитрил
К раствору соединения, полученного в Стадии В (3,96 г; 20 ммоль) в этилацетате (60 мл) при 0°C, добавляют по каплям концентрированную хлористоводородную кислоту (12 мл) поддерживая температуру ниже 5°C, затем по каплям раствор нитрита натрия (1,66 г; 24 ммоль) в воде (25 мл), равным образом поддерживая температуру ниже 5°C. Через 1 час после перемешивания при 0°C, раствор азида натрия (1,56 г; 24 ммоль) в воде (25 мл) добавляют по каплям, поддерживая температуру ниже 5°C. Реакционную смесь перемешивают в течение 3 часов до достижения температуры окружающей среды, затем ее разбавляют водой. Продукт экстрагируют этилацетатом. Органические фазы объединяют и промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, чтобы обеспечить приведенное в заголовке соединение без очистки.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 7.55 (t, 1 Н), 7.41 (t, 1 Н), 7.20-7.24 (m, 1 Н).
Стадия D: трет-бутил 5-{2-[1-(3-бром-5-цианофенил)-1Н-1,2,3-triazol-4-ил]этил}-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Стадии С (1,56 г; 7 ммоль), и соединения, полученного в Синтезе 10b' (2 г; 7 ммоль) в тетрагидрофуране (30 мл), добавляют йодид меди (133 мг; 0,7 ммоль) и триэтиламин (1,16 мл; 8,4 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Затем реакционную смесь концентрируют до объема приблизительно 15 мл, и все разбавляют с этилацетатом. Органическую фазу промывают водным раствором 2 н. соляной кислоты, затем насыщенным водным раствором хлорида натрия. Органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают хроматографией на силикагеле, используя этилацетат и гептан в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 8.17 (s, 1 Н), 7.91-7.99 (m, 1 Н), 7.79-7.86 (m, 1 Н), 7.64 (br. s, 1 Н), 7.09-7.20 (т, 1 Н), 7.03 (dd, 2 Н), 4.58 (s, 2 Н), 3.65 (br. s, 2 Н), 3.06 (br. s, 4 Н), 2.82 (br. s, 2 H), 1.49 (s, 9 H).
Стадия Е: трет-бутил 5-[2-(1-{3-[(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-5-цианофенил}-1Н-1,2,3-triazol-4-ил)этил]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Синтеза 2b'', используя соединение из предыдущей стадии и соединение Стадии В Синтеза 1b''.
1H ЯМР (400 МГц, CDCl3, 300 K) δ част. на млн.: 7.40-7.76 (m, 2 Н), 7.19 (s, 1 Н), 7.05-7.16 (m, 4 Н), 6.95-7.05 (m, 2 Н), 6.82-6.90 (m, 2 Н), 6.67 (br. s, 1 Н), 4.57 (s, 2 Н), 3.58-3.67 (m, 2 Н), 2.96-3.07 (m, 4 Н), 2.70-2.81 (m, 2 Н), 1.47 (s, 9 Н), 1.00 (s, 9 Н), 0.22 (s, 6 Н).
Синтез 21b'': трет-бутил 4-(2-{2-[(5-{[4-(проп-2-ен-1-илокси)фенил]амино}пиримидин-2-ил)окси]этокси}этокси)-1,3-дигидро-2Н-изоиндол-2-карбоксилат
Стадия А: 5-бром-2-[2-(2-хлорэтокси)этокси]пиримидин
К раствору из 5-бром-2-хлорпиримидина (3,48 г; 18 ммоль) и 2-(2-хлорэтокси)-этанола (3,59 г; 28,8 ммоль) в ацетонитриле (50 мл), добавляют карбонат цезия (11,73 г, 36 ммоль), затем все перемешивают в течение 14 часов при 60°C. После охлаждения реакционную среду фильтруют, затем фильтрат концентрируют. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 8.75 (s, 2 Н), 4.41 (m, 2 Н), 3.8 (m, 2 Н), 3.72 (s, 4 Н).
Стадия В: трет-бутил 4-{2-[2-({5-[(4-гидроксифенил)амино]пиримидин-2-ил}окси)этокси]этокси}-1,3-дигидро-2Н-изоиндол-2-карбоксилат
К раствору соединения, полученного в Стадии А (2,81 г; 10 ммоль) и соединения Синтеза 6b' (2,5 г; 10 ммоль) в ацетонитриле (100 мл), добавляют карбонат цезия (3,9 г, 12 ммоль) и йодид калия (0,35 г; 2,1 ммоль), затем все перемешивают в течение 14 часов при 60°C. После достижения температуры окружающей среды, реакционную среду концентрируют. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования.
Раствор полученного таким образом остатка (3,3 г; 6,87 ммоль) и соединения Стадии В Синтеза 15b'' (1,44 г; 9,6 ммоль) в тетрагидрофуране (60 мл) дегазируют аргоном в течение 10 минут. Добавляют карбонат цезия (2,91 г; 8,9 ммоль) и хлор(2-ди-трет-бутилфосфино-2',4',6'-триизопропил-1,1'-бифенил)[2-(2-аминоэтил)фенил]палладий(II) (180 мг; 0,26 ммоль), затем все перемешивают при кипячении с обратным холодильником в течение 5 часов, затем при 50°C в течение 15 часов. После охлаждения, реакционную среду концентрируют. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 9.00 (1H, s), 8.22 (1Н, s), 8.21 (1Н, s), 7.65 (1H, s), 6.90 (1Н, d), 6.89 (1Н, d), 7.24 (1Н, t), 6.86 (2Н, m), 6.68 (2Н, m), 4.45-4.57 (4Н, m), 4.33 (2Н, m), 4.16 (2Н, m), 3.81 (4Н, m), 1.44 (9Н, s).
Стадия С: трет-бутил 4-(2-{2-[(5-{[4-(проп-2-ен-1-илокси)фенил]амино}пиримидин-2-ил)окси]этокси}этокси)-1,3-дигидро-2Н-изоиндол-2-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии С Синтеза 16b'', используя фенол из Стадии В.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 8.29/8.28 (s, 2 Н), 7.81 (brs, 1 Н), 7.24 (t, 1 Н), 6.93 (m, 2 Н), 6.9 (m, 1 Н), 6.88 (m, 1 Н), 6.86 (m, 2 Н), 6.03 (m, 1 Н), 5.37/5.24 (m+m, 2 Н), 4.57/4.55 (brs, 2 Н), 4.48/4.45 (brs, 2 Н), 4.38-3.78 (m, 8 Н), 1.44 (s, 9 Н).
Синтез 22b'': трет-бутил 5-(2-{2-[(5-{[4-(проп-2-ен-1-илокси)фенил]амино}пиримидин-2-ил)окси]этокси}этокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом Синтеза 21b'' заменяя в Стадии В соединение из Синтеза 6b' на соединение из Синтеза 3b'.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 8.29 (s, 2 Н), 7.82 (s, 1 Н), 7.12 (t, 1 Н), 6.94 (m, 2 Н), 6.86 (m, 2 Н), 6.8 (d, 1 Н), 6.73 (d, 1 Н), 6.02 (m, 1 Н), 5.37/5.23 (m+m, 2 Н), 4.49 (m, 2 Н), 4.45 (br., 2 Н), 4.36 (m, 2 Н), 4.1 (m, 2 Н), 3.81 (m, 2 Н), 3.81 (m, 2 Н), 3.51 (t, 2 Н), 2.61 (t, 2 Н), 1.4 (s, 9 Н).
Синтез 23b'': трет-бутил 5-({6-[(5-{[4-(проп-2-ен-1-илокси)фенил]амино}пиримидин-2-ил)окси]гексил}окси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом Синтеза 22b'' заменяя в Стадии А 2-(2-хлорэтокси)-этанол на 6-хлоргексанол.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C33H42N4O5
[М+Н]+ рассчитано 575.32,
[М+Н]+ измерено 575.4.
Синтез 24b'': трет-бутил 5-({5-[(5-{[4-(проп-2-ен-1-илокси)фенил]амино}пиримидин-2-ил)окси]пентил}окси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом Синтеза 22b'', заменяя в Стадии А 2-(2-хлорэтокси)-этанол на 6-хлорпентанол.
1H ЯМР (500 МГц, ДМСО-d6) δ част. на млн.: 8.29 (s, 2 Н), 7.79 (s, 1 Н), 7.12 (t, 1 Н), 6.93 (m, 2 Н), 6.86 (m, 2 Н), 6.79 (d, 1 Н), 6.72 (d, 1 Н), 6.03 (m, 1 Н), 5.38/5.24 (m, 2 Н), 4.49 (dt, 2 Н), 4.45 (br., 2 Н), 4.24 (t, 2 Н), 3.98 (t, 2 Н), 3.53 (t, 2 Н), 2.61 (t, 2 Н), 1.78 (m, 2 Н), 1.78 (m, 2 Н), 1.56 (m, 2 Н), 1.41 (s, 3 Н).
Синтез 25b'': трет-бутил 5-{4-[(5-{[4-(проп-2-ен-1-илокси)фенил]амино}пиримидин-2-ил)окси]бутокси}-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом Синтеза 22b'' заменяя в Стадии А 2-(2-хлорэтокси)-этанол на 6-хлорбутанол.
1H ЯМР (500 МГц, ДМСО-d6) δ част. на млн. 8.29 (s, 2 Н), 7.8 (s, 1 Н), 7.12 (t, 1 Н), 6.93 (d, 2 Н), 6.86 (d, 2 Н), 6.8 (d, 1 Н), 6.72 (d, 1 Н), 6.07-5.98 (m, 1 Н), 5.4-5.21 (m, 2 Н), 4.48 (dt, 2 Н), 4.45 (br, 2 Н), 4.28 (t, 2 Н), 4.02 (t, 2 Н), 3.53 (t, 2 Н), 2.62 (t, 2 Н), 1.87 (br, 4 Н), 1.4 (s, 9 Н).
Синтез 26b'': трет-бутил 3-[(2-{2-[(5-{[4-(проп-2-ен-1-илокси)фенил]амино}пиримидин-2-ил)окси]этокси}этокси)метил]пиперидин-1-карбоксилат
Стадия А: трет-бутил 3-({2-[2-(бензилокси)этокси]этокси}метил)пиперидин-1-карбоксилат
К раствору из трет-бутил 3-(гидроксиметил)пиперидин-1-карбоксилата (7,54 г; 35 ммоль) в N,N-диметилформамиду при 0°C, добавляют 60% гидрид натрия в масле (1,6 г; 40,2 ммоль). Затем все перемешивают в течение 60 минут. После достижения температуры окружающей среды добавляют раствор 2-(2-бензилоксиэтокси)этил-4-метилбензолсульфонат (12,26 г, 35 ммоль) в N,N-диметилформамиде (20 мл). Затем все перемешивают в течение 60 минут, затем гидролизуют медленно. После экстракции дихлорметаном, органические фазы выпаривают и Остаток очищают хроматографией на силикагеле, используя дихлорметан и метанол в качестве растворителя для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (500 МГц, ДМСО-d6) δ част. на млн. 7.36-7.29 (m, 4 Н), 7.27 (m, 1 Н), 4.48 (s, 2 Н), 4-3.77/2.7-2.36 (br, 2 Н), 3.75/2.74 (br+td, 2 Н), 3.59-3.44 (m, 8 Н), 3.27/3.21 (dd, 2 Н), 1.66/1.11 (br, 2 Н), 1.62 (br, 1 Н), 1.55/1.29 (m, 2 Н), 1.37 (s, 9 Н).
Стадия В: трет-бутил 3-{[2-(2-гидроксиэтокси)этокси]метил}пиперидин-1-карбоксилат
К раствору соединения, полученного в Стадии А (11,0 г; 27,95 ммоль) в этаноле (85 мл) добавляют палладий на угле (10 мас. %). Все гидрируют в течение 10 часов при температуре окружающей среды при 4 барах. Реакционную среду фильтруют, затем концентрируют досуха, чтобы получить указанный в заголовке продукт.
1H ЯМР (500 МГц, ДМСО-d6) δ част. на млн. 4.45 (t, 1 Н), 3.87/2.57 (brm+brs, 2 Н), 3.74/2.78 (m+m, 2 Н), 3.56-3.46 (m, 4 Н), 3.49 (m, 2 Н), 3.43 (t, 2 Н), 3.28/3.23 (dd+dd, 2 Н), 1.68/1.15 (m+m, 2 Н), 1.64 (m, 1 Н), 1.58/1.32 (m+m, 2 Н), 1.39 (s, 9 Н).
Стадия С: трет-бутил 3-[(2-{2-[(5-бромпиримидин-2-ил)окси]этокси}этокси)метил]пиперидин-1-карбоксилат
К раствору соединения, полученного в Стадии В (8,34 г, 27,5 ммоль), и 5-бром-2-хлорпиримидина (5,31 г; 27 ммоль) в ацетонитриле (160 мл), добавляют карбонат цезия (17,9 г, 55 ммоль), затем все перемешивают в течение 7 часов при 85°C. После охлаждения, реакционную среду фильтруют, затем фильтрат концентрируют. Остаток очищают хроматографией на силикагеле, используя гептан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (500 МГц, ДМСО-d6) δ част. на млн. 8.75 (s, 2 Н), 4.4 (m, 2 Н), 4.04-2.27 (brm, 4 Н), 3.75 (m, 2 Н), 3.57 (t, 2 Н), 3.49/3.47 (m+m, 2 Н), 3.25/3.2 (dd+dd, 2 Н), 1.64/1.1 (m+m, 2 Н), 1.6 (m, 1 Н), 1.55/1.29 (m+m, 2 Н), 1.37 (s, 9 Н).
Стадия D: трет-бутил 3-[(2-{2-[(5-{[4-(проп-2-ен-1-илокси)фенил]амино}пиримидин-2-ил)окси]этокси}этокси)метил]пиперидин-1-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Синтеза 2b'', используя соединение из предыдущей стадии и соединение, полученное в Стадии В Синтеза 15b''.
1H ЯМР (500 МГц, ДМСО-d6) δ част. на млн. 8.29 (s, 2 Н), 7.81 (s, 1 Н), 6.94 (m, 2 Н), 6.86 (m, 2 Н), 4.33 (m, 2 Н), 4-3.73/2.71-2.34 (br+br., 2 Н), 3.73/2.74 (br+m, 2 Н), 3.73 (m, 2 Н), 3.58 (t, 2 Н), 3.49 (m, 2 Н), 3.27/3.21 (dd+dd, 2 Н), 1.66/1.1 (brd+br., 2 Н), 1.62 (br., 1 Н), 1.55/1.29 (m+m, 2 Н), 1.37 (s, 9 Н).
Синтез 27b'': трет-бутил 4-[(2-{2-[(5-{[4-(проп-2-ен-1-илокси)фенил]амино}пиримидин-2-ил)окси]этокси}этокси)метил]пиперидин-1-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом Синтеза 26b'', заменяя в Стадии А трет-бутил 3-(гидроксиметил)пиперидин-1-карбоксилат на трет-бутил 4-(гидроксиметил)пиперидин-1-карбоксилат.
1H ЯМР (500 МГц, ДМСО-d6) δ част. на млн. 8.29 (s, 2 Н), 7.82 (s, 1 Н), 6.94 (m, 2 Н), 6.86 (m, 2 Н), 6.03 (m, 1 Н), 5.38/5.24 (m+m, 2 Н), 4.49 (m, 2 Н), 4.32 (m, 2 Н), 3.91/2.67 (br+br., 4 Н), 3.73 (m, 2 Н), 3.57 (m, 2 Н), 3.49 (m, 2 Н), 3.23 (d, 2 Н), 1.67 (m, 1 Н), 1.61/0.98 (brd+qd, 4 Н), 1.37 (s, 9 Н).
Синтез 28b'': трет-бутил 5-[2-(метил{2-[(5-{[4-(проп-2-ен-1-илокси)фенил]амино}пиримидин-2-ил)окси]этил}амино)этокси]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Стадия А: 2-{[2-(бензилокси)этил](метил)амино}этанол
К раствору 2-хлорэтоксиметилбензола (4,6 мл; 30 ммоль) и 2-(метиламино)этанола (3,1 мл; 39 ммоль) в N,N-диметилацетамиде (40 мл), добавляют йодид калия (1,0 г, 6 ммоль), затем добавляют триэтиламин (5,4 мл; 39 ммоль). Все перемешивают при 120°C в течение всей ночи. После фильтрации осадка, фильтрат концентрируют досуха, затем очищают с помощью хроматографии на силикагеле, используя дихлорметан и метанол в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (500 МГц, ДМСО-d6) δ част. на млн. 7.38-7.24 (m, 5 Н), 4.46 (s, 2 Н), 4.34 (brt, 1 Н), 3.51 (t, 2 Н), 3.45 (brt, 2 Н), 2.58 (t, 2 Н), 2.45 (t, 2 Н), 2.21 (s, 3 Н).
Стадия В: трет-бутил 5-(2-{[2-(бензилокси)этил](метил)амино}этокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Стадии А (6,28 г, 30 ммоль) и соединения, полученного в Синтезе 3b' (4,99 г, 20 ммоль) в тетрагидрофуране (150 мл), под атмосферой азота добавляют трифенилфосфин (7,87 г, 30 ммоль) при 0°C, затем по каплям 40% раствор диэтил азодикарбоксилата в толуоле (14 мл, 30 ммоль). После достижения температуры окружающей среды, смесь перемешивают в течение 1 часа. После выпаривания растворителей, остаток ресуспендируют 2 раза в диизопропиловом эфире, затем его очищают с помощью хроматографии на RP-18, используя ацетонитрил, воду и карбонат аммония в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (500 МГц, ДМСО-d6) δ част. на млн. 7.35-7.24 (m, 5 Н), 7.11 (t, 1 Н), 6.79 (dd, 1 Н), 6.72 (dd, 1 Н), 4.46 (s, 2 Н), 4.45 (s, 2 Н), 4.02 (t, 2 Н), 3.53 (t, 2 Н), 3.51 (t, 2 Н), 2.7 (t, 2 Н), 2.65 (t, 2 Н), 2.6 (t, 2 Н), 2.3 (s, 3 Н), 1.41 (s, 9 Н).
Стадия С: трет-бутил 5-{2-[(2-гидроксиэтил)(метил)амино]этокси}-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Стадии В (3,88 г; 8,8 ммоль) в тетрагидрофуране (50 мл) и этанол (80 мл), добавляют 4 М хлористоводородную кислоту в диоксане (2,4 мл), затем палладий на угле (570 мг). Смесь гидрируют при температуре окружающей среды при 4 барах в течение 15 часов. После фильтрации и выпаривания, остаток растворяют в воде, затем добавляют водный раствор 2 М гидроксида натрия до достижения рН=12. После экстрагирования этилацетатом и выпаривания растворителей получают указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6) δ част. на млн. 7.12 (t, 1 Н), 6.8 (dd, 1 Н), 6.73 (dd, 1 Н), 4.45 (s, 2 Н), 4.34 (t, 1 Н), 4.03 (t, 2 Н), 3.53 (t, 2 Н), 3.47 (t, 2 Н), 2.77 (t, 2 Н), 2.62 (t, 2 Н), 2.51 (t, 2 Н), 2.28 (s, 3 Н), 1.41 (s, 3 Н)
Стадия D: трет-бутил 5-{2-[{2-[(5-бромпиримидин-2-ил)окси]этил}(метил)амино]этокси}-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Синтеза 28b'', используя соединение из предыдущей Стадии и 5-бромпиримидин-2-ол.
1H ЯМР (500 МГц, ДМСО-d6) δ част. на млн. 8.72 (s, 1 Н), 7.11 (t, 1 Н), 6.78 (d, 1 Н), 6.72 (d, 1 Н), 4.45 (s, 2 Н), 4.38 (t, 2 Н), 4.03 (t, 2 Н), 3.49 (t, 2 Н), 2.84 (t, 2 Н), 2.83 (t, 2 Н), 2.57 (t, 2 Н), 2.35 (s, 3 Н), 1.41 (s, 9 Н)
Стадия Е: трет-бутил 5-[2-(метил{2-[(5-{[4-(проп-2-ен-1-илокси)фенил]амино}пиримидин-2-ил)окси]этил}амино)этокси]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии D Синтеза 26b'', используя соединение из предыдущей стадии.
1H ЯМР (500 МГц, ДМСО-d6) δ част. на млн. 8.28 (s, 2 Н), 7.8 (s, 1 Н), 7.11 (t, 1 Н), 6.93 (m, 2 Н), 6.85 (m, 2 Н), 6.8 (dd, 1 Н), 6.72 (dd, 1 Н), 6.03 (m, 1 Н), 5.38/5.24 (dq, 2 Н), 4.49 (dt, 2 Н), 4.44 (brs, 2 Н), 4.32 (t, 2 Н), 4.04 (t, 2 Н), 3.5 (t, 2 Н), 2.84 (t, 2 Н), 2.83 (t, 2 Н), 2.59 (t, 2 Н), 2.35 (s, 3 Н), 1.4 (s, 9 Н).
Пример 1: 6-хлор-14-(4-гидроксифенил)-10,11-диметил-20,23-диокса-1,10,14-триазагексацикло[26.3.1.1~9,12~.1~15,19~.0~3,8~.0~24,29~]тетратриаконта-3,5,7,9(34), 11,15(33), 16,18,24,26,28-ундекаен-2,13-дион
Стадия А: этил 5-(5-хлор-2-{[5-(проп-2-ен-1-илокси)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-1,2-диметил-1Н-пиррол-3-карбоксилат
К раствору соединения из Синтеза 1а (9,5 г; 29,5 ммоль) в дихлорметане (110 мл), последовательно добавляют соединение, полученное в Получении 1а' (7,32 г; 32,4 ммоль), 1-гидроксибензотриазол (4,78 г; 35,4 ммоль), гидрохлорид 1, (3-диметиламинопропил)-3-этилкарбодиимид (6,1 г; 35,4 ммоль) и триэтиламин (20,6 мл; 147,5 ммоль). Затем все перемешивают в течение ночи при температуре окружающей среды. Реакционную среду разбавляют смесью дихлорметана и воды. После декантации, органическую фазу промывают с водным насыщенным раствором хлорида аммония и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.54/7.45 (2m, 3 Н), 7.12/7 (2t, 1 Н), 6.75/6.45 (2d, 1 Н), 6.75/6.65 (2d, 1 Н), 6.35/6.25 (2s, 1 Н), 6.05 (m, 1 Н), 5.4 (m, 1 Н), 5.25 (m, 1 Н), 4.55-3.95/2.95 (m, 8 Н), 3.45/3.21 (2s, 3 Н), 2.6-2.2 (m, 2 Н), 2.49/2.05 (2s, 3 Н), 1.22 (t, 3 Н).
ИК: v: >C=O: 1695 см-1.
Стадия В: 5-(5-хлор-2-{[5-(проп-2-ен-1-илокси)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-1,2-диметил-1Н-пиррол-3-карбоновая кислота
К раствору соединения, полученного в Стадии А (10,8 г; 21,9 ммоль) в метаноле (100 мл), добавляют гидроксид лития (1,8 г; 43,8 ммоль) в растворе в 50 мл воды. Затем все перемешивают при 90°C в течение 48 часов. Реакционную среду концентрируют для удаления метанола, затем подкисляют до рН=4 путем добавления водного раствора соляной кислоты 1 н. и в заключение экстрагируют дихлорметаном. Органическую фазу сушат над сульфатом магния, фильтруют и концентрируют досуха. Указанный в заголовке продукт получают в виде порошка и используют в следующей Стадии без очистки.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 11.55 (br. s, 1 Н), 7.52/7.45 (m, 3 Н), 7.12/7 (2t, 1 Н), 6.78/6.7/6.48 (3d, 2 Н), 6.32/6.25 (2s, 1 Н), 6.05 (m, 1 Н), 5.4 (m, 1 Н), 5.25 (m, 1 Н), 4.8-3 (m, 6 Н), 3.45/3.2 (2s, 3 Н), 2.5 (m, 2 Н), 2.5/2.05 (2s, 3 Н).
Стадия С: N-(4-{[трет-бутил(диметил)силил]окси}фенил)-N-[3-(2-хлорэтокси)фенил]-5-(5-хлор-2-{[5-(проп-2-ен-1-илокси)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-1,2-диметил-1Н-пиррол-3-карбоксамид
К раствору соединения, полученного в Стадии В (1 г; 2,15 ммоль) в дихлорэтане (20 мл), добавляют 1-хлор-N,N,2-триметил-проп-1-ен-1-амин (0,31 мл; 2,4 ммоль). Реакционную среду перемешивают при температуре окружающей среды в течение 2 часов, затем добавляют соединение из Синтеза 1а'' (1,6 г; 4,3 ммоль). Все перемешивают при 80°C в течение 24 часов. Реакционную среду разбавляют в смеси дихлорметана и водного раствора насыщенного гидрокарбоната натрия. После экстрагирования водной фазы в дихлорметане органические фазы объединяют, сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и метанол в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.5-7.25 (m, 3 Н), 7.2 (m, 2 Н), 6.95/6.65 (m, 4 Н), 6.85-6.6 (m, 5 Н), 6.05 (m, 1 Н), 5.4/5.25 (m, 2 Н), 5.35-5.2 (m, 1 Н), 4.8-3.9 (m, 2 Н), 4.5 (m, 2 Н), 4.15 (m, 2 Н), 3.85 (m, 2 Н), 3.3-2.9 (m, 2 Н), 3.3-3.2 (m, 3 Н), 2.8-2.5 (m, 2 Н), 2.35-2.2 (т, 3 Н), 0.85 (m, 9 Н), 0.1 (m, 6 Н).
Стадия D: N-(4-{[трет-бутил(диметил)силил]окси}фенил)-N-[3-(2-хлорэтокси)фенил]-5-{5-хлор-2-[(5-гидрокси-3,4-дигидроизохинолин-2(1Н)-ил)карбонил]фенил}-1,2-диметил-1Н-пиррол-3-карбоксамид
К раствору соединения, полученного в Стадии С (1,3 г; 1,57 ммоль) в смеси дихлорметана (6 мл) и метанола (3 мл), добавляют 1,3-диметилбарбитуровую кислоту (0,49 г; 3,15 ммоль). Реакционную среду дегазируют путем барботирования через аргон в течение 10 минут и добавляют тетракис-(трифенилфосфин)палладий(0) (0,09 г; 0,07 ммоль). Все нагревают до 40°C в течение 19 часов. После концентрирования метанола, реакционную среду разбавляют в смеси этилацетата и воды. После декантации, органическую фазу промывают водным насыщенным раствором гидрокарбоната натрия и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и метанол в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 9.45 (m, 1 Н), 7.5-7.25 (m, 3 Н), 7.2 (m, 1 Н), 7-6.95 (m, 1 Н), 6.8-6.6 (m, 2 Н), 6.8-6.6 (m, 3 Н), 6.75-6.6 (m, 4 Н), 5.4-5.2 (m, 1 Н), 4.8-4.1 (m, 2 Н), 4.15 (m, 2 Н), 3.9 (m, 2 Н), 3.85 (m, 2 Н), 3.3-3.2 (m, 3 Н), 2.9-2.5 (m, 2 Н), 2.35-2.2 (m, 3 Н), 0.85 (m, 9 Н), 0.1 (m, 6 Н).
ИК: v: -ОН: 3260 см-1; >C=O: 1624 см-1.
Стадия Е: N-(4-{[трет-бутил(диметил)силил]окси}фенил)-5-{5-хлор-2-[(5-гидрокси-3,4-дигидроизохинолин-2(1Н)-ил)карбонил]фенил}-N-[3-(2-йодэтокси)фенил]-1,2-диметил-1Н-пиррол-3-карбоксамид
К раствору соединения, полученного в Стадии D (0,88 г; 1,17 ммоль) в ацетоне (15 мл), добавляют йодид натрия (0,35 г; 2,34 ммоль). Реакционную среду перемешивают при 80°C в течение 24 часов. После ее фильтрации фильтрат концентрируют. Полученный остаток ресуспендируют в смеси этилацетата и воды. После экстрагирования водной фазы этилацетатом, органические фазы объединяют, сушат над сульфатом магния, фильтруют и концентрируют досуха. Полученный остаток непосредственно используют в следующей стадии без очистки.
Стадия F: 14-(4-{[трет-бутил(диметил)силил]окси}фенил)-6-хлор-10,11-диметил-20,23-диокса-1,10,14-триазагексацикло[26.3.1.1~9,12~.1~15,19~.0~3,8~.0~24,29~]тетратриаконта-3,5,7,9(34), 11,15(33), 16,18,24,26,28-ундекаен-2,13-дион
К раствору соединения, полученного в Стадии Е (0,98 г; 1,12 ммоль) в ацетонитриле (112 мл), тремя порциями добавляют с интервалом в один час и при температуре окружающей среды карбонат цезия (0,36 г; 1,12 ммоль). Реакционную среду после этого нагревают до 50°C в течение 4 часов, затем концентрируют. Полученный остаток ресуспендируют в смеси дихлорметана и воды. После декантации, органическую фазу промывают водой и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют. Полученный остаток используют непосредственно в следующей стадии без очистки.
Стадия G: 6-хлор-14-(4-гидроксифенил)-10,11-диметил-20,23-диокса-1,10,14-триазагексацикло[26.3.1.1-9,12~.1~15,19~.0~3,8~.0~24,29~]тетратриаконта-3,5,7,9(34),11,15(33),16,18,24,26,28-ундекаен-2,13-дион
К раствору соединения, полученного в Стадии F (0,5 г; 0,67 ммоль) в тетрагидрофуране (10 мл), добавляют раствор 1 н. фторида тетрабутиламмония в тетрагидрофуране (1 мл; 1,01 ммоль). Все перемешивают при температуре окружающей среды в течение 1 часа. Реакционную среду затем разбавляют в смеси дихлорметана и воды. После декантации, органическую фазу промывают водой, и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют. Полученный таким образом остаток очищают с помощью хроматографии на силикагеле, используя дихлорметан и аммиак в метаноле в качестве растворителей для элюирования, с последующей хиральной колоночной хроматографией IC, используя ацетонитрил, изопропанол и диэтиламин. Полученное таким образом твердое вещество растворяют в смеси вода / ацетонитрил, фильтруют, затем лиофилизируют.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C37H32CIN3O5
[М+Н]+ рассчитано 634,2103,
[М+Н]+ измерено 634,2102.
Пример 2: 6-хлор-14-(4-гидроксифенил)-10,11-диметил-20,24-диокса-1,10,14-триазагексацикло[27.3.1.1~9,12~.1~15,19~.0~3,8~.0~25,30~]пентатриаконта-3,5,7,9(35),11,15(34),16,18,25,27,29-ундекаен-2,13-дион

Указанное в заголовке соединение получают в соответствии со способом из Примера 1, используя кислоту из Синтеза 1а и соединение из Синтеза 1а' в Стадии А, также как и соединение из Синтеза 2а'' в Стадии С.
Элементарный микроанализ: (теоретический %: измеренный)
%С=70.42: 69.92; %Н=5.29: 5.00; %N=6.48: 6.48.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C38H34ClN3O5
[М+Н]+ рассчитано 648,2259,
[М+Н]+ измерено 648,2260.
Пример 3: 6-хлор-14-(4-гидроксифенил)-10,11-диметил-20,25-диокса-1,10,14-триазагексацикло[28.3.1.1~9,12~.1~15,19~.0~3,8~.0~26,31~]гексатриаконта-3,5,7,9(36),11,15(35),16,18,26,28,30-ундекаен-2,13-дион
Указанное в заголовке соединение получают в соответствии со способом из Примера 1, используя кислоту из Синтеза 1а и соединение из Синтеза 1а' в Стадии А, также как и соединение из Синтеза 3а'' в Стадии С.
Элементарный микроанализ: (теоретический %: измеренный)
%С=70.74: 70.22; %Н=5.48: 5.37; %N=6.35: 6.27.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C39H36ClN3O5
[М+Н]+ рассчитано 662,2416,
[М+Н]+ измерено 662,2422.
Пример 4: 6-хлор-14-(4-гидроксифенил)-10,11-диметил-2,13-диоксо-20,23-диокса-1,10,14-триазагексацикло[26.3.1.1~9,12~.1~15,19~.0~3,8~.0~24,29~]тетратриаконта-3,5,7,9(34),11,15(33),16,18,24,26,28-ундекаен-17-карбонитрил

Указанное в заголовке соединение получают в соответствии со способом из Примера 1, используя кислоту из Синтеза 1а и соединение из Синтеза 1а' в Стадии А, также как и соединение из Синтеза 4а'' в Стадии С.
Элементарный микроанализ: (теоретический %: измеренный) %С=69.24: 68.29; %Н=4.74: 4.68; %N=8.50: 8.70; %Cl=5.38: 5.33.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C38H31ClN4O5
[М+Н]+ рассчитано 659,2057,
[М+Н]+ измерено 659,2056.
Пример 5: 6-хлор-14-(4-гидроксифенил)-10,11-диметил-2,13-диоксо-20,24-диокса-1,10,14-триазагексацикло[27.3.1.1~9,12~.1~15,19~.0~3,8~.0~25,30~]пентатриаконта-3,5,7,9(35),11,15(34),16,18,25,27,29-ундекаен-17-карбонитрил

Указанное в заголовке соединение получают в соответствии со способом из Примера 1, используя кислоту из Синтеза 1а и соединение из Синтеза 1а' в Стадии А, также как и соединение из Синтеза 5а'' в Стадии С.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C39H33ClN4O5
[М+Н]+ рассчитано 673,2212,
[М+Н]+ измерено 673,2211.
Пример 6: 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-4,16,17,23,24,25-гексагидро-1Н,14Н-15,18-метано-6,9-(метено)дибензо[b,h]пиразоло[4,3-р][1,6,11,15]оксатриазациклоикозин-5,14(8Н)-дион

Стадия А: N-(4-{[трет-бутил(диметил)силил]окси}фенил)-5-(5-хлор-2-{[5-(проп-2-ен-1-илокси)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-N-[5-(3-хлорпропил)-1-метил-1Н-пиразол-4-ил]-1,2-диметил-1Н-пиррол-3-карбоксамид
К раствору соединения, полученного в Стадии В Примера 1 (0,75 г; 1,6 ммоль) в дихлорэтане (40 мл), добавляют оксалилхлорид (0,28 мл; 3.2 ммоль). Реакционную среду перемешивают при температуре окружающей среды в течение 1 часа, затем концентрируют. Остаток ресуспендируют в дихлорэтане затем концентрируют, эту процедуру осуществляют дважды. Конечный остаток ресуспендируют в дихлорэтане (20 мл), затем добавляют к раствору соединение из Синтеза 6а'' (0,61 г; 1,6 ммоль) и пиридин (0,4 мл; 4,8 ммоль) в дихлорэтане (10 мл). Все перемешивают при 110°C в течение 16 часов. Реакционную среду концентрируют, затем остаток ресуспендируют в смеси дихлорметана и водного раствора насыщенного гидрокарбоната натрия. После экстрагирования водной фазы дихлорметаном, органические фазы объединяют, сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.5-7.25 (m, 3 Н), 7.2-7.1 (m, 1 Н), 7.15 (m, 1 Н), 7.15-6.93 (m, 2 Н), 6.82-6.7 (m, 2 Н), 6.8 (m, 1 Н), 6.62-6.5 (m, 1 Н), 6.08 (m, 1 Н), 5.5-5.2 (m, 2 Н), 5.38-5.2 (m, 1 Н), 4.75-2.9 (m, 2 Н), 4.55 (m, 2 Н), 4.25-3.9 (m, 2 Н), 3.73 (m, 3 Н), 3.55 (m, 2 Н), 3.3-3.15 (m, 3 Н), 2.7-2.3 (m, 2 Н), 2.6 (m, 2 Н), 2.3-2.15 (m, 3 Н), 1.6 (m, 2 Н), 0.8 (m, 9 Н), 0.1 (m, 6 Н).
Стадия В: 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-4,16,17,23,24,25-гексагидро-1Н,14Н-15,18-метано-6,9-(метено)дибензо[b,h]пиразоло[4,3-р][1,6,11,15]оксатриазациклоикозин-5,14(8Н)-дион
Указанное в заголовке соединение получают в соответствии со способами, описанными в Стадиях D, Е, F и G Примера 1,, используя соединение из предыдущей стадии в качестве исходного продукта.
Элементарный микроанализ: (теоретический %: измеренный)
%С=67.97: 68.05; %Н=5.39: 5.25; %N=11.01: 10.77.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C36H34ClN5O4
[М+Н]+ рассчитано 636,2372,
[М+Н]+ измерено 636,2369.
Пример 7: 6-хлор-14-(4-гидроксифенил)-10,11,33-триметил-2,13-диоксо-23-окса-1,10,14,18-тетраазагексацикло[26.3.1.1~9,12~.1~15,18~.0~3,8~.0~24,29~]тетратриаконта-3,5,7,9(34),11,15(33),16,24,26,28-декаен-17-карбонитрил

Указанное в заголовке соединение получают в соответствии со способом из Примера 6, используя кислоту, полученную в Стадии В Примера 1 и амин из Синтеза 7а''.
Элементарный микроанализ: (теоретический %: измеренный)
%С=69.48: 69.01; %Н=5.38: 5.34; %N=10.39: 10.19.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C39H36ClN5O4
[М+Н]+ рассчитано 674,2529,
[М+Н]+ измерено 674,2515.
Пример 8: 6-хлор-14-(4-гидроксифенил)-10,11,32-триметил-2,13-диоксо-22-окса-1,10,14,18-тетраазагексацикло[25.3.1.1~9,12~.1~15,18~.0~3,8~.0~23,28~]тритриаконта-3,5,7,9(33),11,15(32),16,23,25,27-декаен-17-карбонитрил

Указанное в заголовке соединение получают в соответствии со способом из Примера 6, используя кислоту, полученную в Стадии В Примера 1 и амин из Синтеза 8а''.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C38H34ClN5O4
[М+Н]+ рассчитано 660.2372,
[М+Н]+ измерено 660.2377.
Пример 9: 6-хлор-14-(4-гидроксифенил)-10,11-диметил-2,13-диоксо-23-окса-1,10,14-триазагексацикло[26.3.1.1~9,12~1.~15,19~.0~3,8~.0~24,29~]тетратриаконта-3,5,7,9(34),11,15(33),16,18,24,26,28-ундекаен-17-карбонитрил

Указанное в заголовке соединение получают в соответствии со способом из Примера 6, используя кислоту, полученную в Стадии В Примера 1 и амин из Синтеза 9а''.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C39H33ClN4O4
[М+Н]+ рассчитано 657.2263,
[М+Н]+ измерено 657.2267.
Пример 10: Гидрохлорид 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-1,4,16,17,23,24-гексагидро-14Н-15,18-метано-6,9-(метено)дибензо[1,r]пиразоло[3,4-d][1,6,10,15]оксатриазациклононадецин-5(8Н)-она

Стадия А: этил 5-(5-хлор-2-{[5-(проп-2-ен-1-илокси)-3,4-дигидроизохинолин-2(1Н)-ил]метил}фенил)-1,2-диметил-1Н-пиррол-3-карбоксилат
К раствору соединения из Стадии В Синтеза 1а (2 г; 6,54 ммоль) в дихлорметане (25 мл), добавляют последовательно соединение, полученное в Получении 1а' (1,5 г; 6,54 ммоль) и триацетоксиборгидрид натрия (2,2 г; 10 ммоль). Затем все перемешивают в течение ночи при температуре окружающей среды. Реакционную среду выливают в смесь из воды и хлорида аммония. После декантации, органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя гептан и аммиак в этаноле в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.6 (d, 1 Н), 7.47 (dd, 1 Н), 7.3 (d, 1 Н), 7.03 (t, 1 Н), 6.74 (d, 1 Н), 6.57 (d, 1 Н), 6.43 (s, 1 Н), 6.03 (m, 1 Н), 5.38 (m, 1 Н), 5.23 (m, 1 Н), 4.54 (m, 2 Н), 4.16 (quad, 2 Н), 3.42 (s, 2 Н), 3.4 (s, 2 Н), 3.22 (s, 3 Н), 2.64 (m, 2 Н), 2.56 (m, 2 Н), 2.49 (s, 3 Н), 1.25 (t, 3 Н).
ИК: v:>C=O: 1692 см-1; >С-O-С<: 1063 см-1
Стадия В: 5-(5-хлор-2-{[5-(проп-2-ен-1-илокси)-3,4-дигидроизохинолин-2(1Н)-ил]метил}фенил)-1,2-диметил-1Н-пиррол-3-карбоновая кислота
К раствору соединения, полученного в Стадии А (2,1 г; 4,4 ммоль) в диоксане (12 мл), добавляют водный раствор 1 н. гидроксида лития (13 мл; 13,1 ммоль). Все перемешивают в микроволновой печи (300 Вт) при 120°C в течение 4,5 часов. Реакционную среду выливают в смесь из водного раствора 1 н. соляной кислоты и льда. Продукт экстрагируют дихлорметаном. Органические фазы объединяют, сушат над сульфатом натрия, фильтруют и концентрируют, чтобы без дальнейшей очистки обеспечить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.61 (d, 1 Н), 7.45 (dd, 1 Н), 7.27 (br. s, 1 Н), 7.03 (t, 1 Н), 6.73 (d, 1 Н), 6.57 (d, 1 Н), 6.33 (s, 1 Н), 6.04 (m, 1 Н), 5.39 (d, 1 Н), 5.23 (d, 1 Н), 4.53 (m, 2 Н), 3.45 (s, 2 Н), 3.39 (s, 2 Н), 3.2 (s, 3 Н), 2.63 (m, 2 Н), 2.55 (m, 2 Н), 2.5 (s, 3 Н).
ИК: v: -ОН: 3300-2200 см-1; >C=O: 1661 см-1; >С-O-С<: 1258 см-1.
Стадия С: N-(4-{[трет-бутил(диметил)силил]окси}фенил)-5-(5-хлор-2-{[5-(проп-2-ен-1-илокси)-3,4-дигидроизохинолин-2(1Н)-ил]метил}фенил)-1,2-диметил-N-{1-метил-5-[2-(тетрагидро-2Н-пиран-2-илокси)этил]-1Н-пиразол-4-ил}-1Н-пиррол-3-карбоксамид
К раствору соединения, полученного в Стадии В (2 г; 4,43 ммоль) в дихлорэтане (50 мл), добавляют 1-хлор-N,N,2-триметил-проп-1-ен-1-амин (0,59 мл; 4,87 ммоль). Реакционную среду перемешивают при температуре окружающей среды в течение 2 часов, затем добавляют соединение из Синтеза 10а'' (3,2 г; 7,4 ммоль) и пиридин (1,8 мл; 22,1 ммоль). Все перемешивают при 80°C в течение 24 часов. Реакционную среду выливают в смесь водного раствора насыщенного гидрокарбоната натрия и льда. После экстрагирования водной фазы дихлорметаном, органические фазы объединяют, сушат над сульфатом натрия, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и изопропанол в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.45 (d, 1 Н), 7.3 (dd, 1 Н), 7.15 (s, 1 Н), 7.05 (d, 1 Н), 7.02 (d, 1 Н), 6.97 (t, 1 Н), 6.9/6.6 (ab, 4 Н), 6.5 (d, 1 Н), 5.95 (m, 1 Н), 5.3 (s, 1 Н), 5.3/5.15 (m, 2 Н), 4.48 (m, 2 Н), 4.3 (m, 1 Н), 3.65 (s, 3 Н), 3.5-3.25 (m, 6 Н), 3.2-3.1 (m, 4 Н), 3.08 (s, 3 Н), 2.8-2.5 (m, 2 Н), 2.5 (m, 2 Н), 2.3 (s, 3 Н), 1.6-1.2 (m, 6 Н), 0.87-0.8 (s, 9 Н), 0.1-0 (s, 6 Н).
Стадия D: N-(4-{[трет-бутил(диметил)силил]окси}фенил)-5-{5-хлор-2-[(5-гидрокси-3,4-дигидроизохинолин-2(1Н)-ил)метил]фенил}-N-[5-(2-гидроксиэтил)-1-метил-1Н-пиразол-4-ил]-1,2-диметил-1Н-пиррол-3-карбоксамид
Указанное в заголовке соединение получают в два этапа. Соединение Стадии С сначала подвергают реакции снятия защиты в соответствии со способом, описанным в Стадии D Примера 1, и используют в следующей стадии.
К раствору полученного таким образом продукта (430 мг; 0,52 ммоль) в метаноле (15 мл), добавляют пара-толуолсульфонат пиридина (140 мг; 0,52 ммоль). Реакционную среду перемешивают при 80°C в течение ночи, затем концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и метанол в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 9.2 (s, 1 Н), 4.62 (s, 1 Н), 7.44 (d, 1 Н), 7.3 (d, 1 Н), 7.15 (s, 1 Н), 7.02 (s, 1 Н), 6.94-6.6 (m, 4 Н), 6.8 (t, 1 Н), 6.5 (d, 1 Н), 6.35 (d, 1 Н), 5.7-5.3 (s, 1 Н), 3.65 (s, 3 Н), 3.3 (m, 2 Н), 3.2 (m, 2 Н), 3.1 (m, 2 Н), 3.05 (s, 3 Н), 2.6 (m, 2 Н), 2.5 (m, 2 Н), 2.48 (m, 2 Н), 2.3 (s, 3 Н), 0.8 (s, 9 Н), 0.15 (s, 6 Н).
Стадия Е: Гидрохлорид 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-1,4,16,17,23,24-гексагидро-14Н-15,18-метано-6,9-(метено)дибензо[1,r]пиразоло[3,4-d][1,6,10,15]оксатриазациклононадецин-5(8Н)-она
Раствор соединения, полученного в Стадии D (250 мг; 0,34 ммоль) в смеси тетрагидрофурана (80 мл) и толуола (80 мл) дегазируют аргоном в течение 10 минут. К нему добавляют цианометилен три-н-бутилфосфоран (0,18 мл; 0,75 ммоль), затем все герметизируют и перемешивают при 110°C в течение 48 часов. Реакционную среду концентрируют, затем разбавляют в смеси этилацетата и воды. После декантации, органическую фазу промывают водой и водный раствор насыщенного гидрокарбоната натрия, сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя аммиак в дихлорметане в качестве растворителя для элюирования, затем используют в следующей стадии.
К раствору полученного продукта в метаноле (5 мл), добавляют раствор поташа в метаноле 1 М (0,34 мл; 0,34 ммоль). Реакционную среду перемешивают при температуре окружающей среды в течение 2 часов, затем выливают в смесь из водного раствора насыщенного гидрокарбоната натрия и льда. После экстрагирования водной фазы дихлорметаном, органические фазы объединяют, сушат над сульфатом натрия, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и аммиак в этаноле в качестве растворителей для элюирования. Полученное таким образом твердое вещество растворяют в смеси водного раствора 1 н. соляной кислоты и ацетонитрила, фильтруют, затем лиофилизируют, чтобы получить указанный в заголовке продукт.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C35H34ClN5O3. HCl
[М+Н]+ рассчитано 608.2423,
[М+Н]+ измерено 608.2425.
Пример 11: 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-4,16,17,23,24,25-гексагидро-1Н,14Н-15,18-метано-6,9-(метено)дибензо[b,h]пиразоло[4,3-р][1,6,11,15]оксатриазациклоикозин-5(8Н)-он

Стадия А: N-(4-{[трет-бутил(диметил)силил]окси}фенил)-5-(5-хлор-2-{[5-(проп-2-ен-1-илокси)-3,4-дигидроизохинолин-2(1Н)-ил]метил}фенил)-N-[5-(3-хлорпропил)-1-метил-1Н-пиразол-4-ил]-1,2-диметил-1Н-пиррол-3-карбоксамид
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии А Примера 6, используя кислоту из Стадии В Примера 10 и амин из Синтеза 6а''.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.53 (d, 1 Н), 7.41 (dd, 1 Н), 7.28 (s, 1 Н), 7.1 (d, 1 Н), 7.05 (t, 1 Н), 6.99 (d, 2 Н), 6.75 (d, 1 Н), 6.69 (d, 2 Н), 6.59 (d, 1 Н), 6.04 (m, 1 Н), 5.4/5.24 (m+m, 1+1 Н), 5.4 (s, 1 Н), 4.55 (d, 2 Н), 3.7 (s, 3 Н), 3.59 (t, 2 Н), 3.34 (s, 2 Н), 3.22 (br. s, 2 Н), 3.15 (s, 3 Н), 2.63 (m, 2 Н), 2.62 (m, 2 Н), 2.5 (m, 2 Н), 2.38 (s, 3 Н), 1.73 (m, 2 Н), 0.87 (s, 9 Н), 0.08 (s, 6 Н).
Стадия В: N-(4-{[трет-бутил(диметил)силил]окси}фенил)-5-{5-хлор-2-[(5-гидрокси-3,4-дигидроизохинолин-2(1Н)-ил)метил]фенил}-N-[5-(3-хлорпропил)-1-метил-1Н-пиразол-4-ил]-1,2-диметил-1Н-пиррол-3-карбоксамид
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии D Примера 1, исходя из соединения, полученного в предыдущей стадии.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн. 7.55 (d, 1 Н), 7.4 (dd, 1 Н), 7.3 (s, 1 Н), 7.1 (d, 1 Н), 7 (d, 2 Н), 6.9 (t, 1 Н), 6.7 (d, 2 Н), 6.6 (d, 1 Н), 6.4 (d, 1 Н), 5.4 (s, 1 Н), 3.7 (s, 3 Н), 3.6 (t, 2 Н), 3.3 (s, 2 Н), 3.2 (br. s, 2 Н), 3.15 (s, 3 Н), 2.6 (m, 2 Н), 2.55 (m, 2 Н), 2.5 (m, 2 Н), 2.35 (s, 3 Н), 1.7 (quint, 2 Н), 0.85 (s, 9 Н), 0.1 (s, 6 Н).
ИК: v: -ОН: 3500-2500 см-1; >C=O: 1635 и 1622 см-1.
Стадия С: N-(4-{[трет-бутил(диметил)силил]окси}фенил)-5-{5-хлор-2-[(5-гидрокси-3,4-дигидроизохинолин-2(1Н)-ил)метил]фенил}-N-[5-(3-йодпропил)-1-метил-1Н-пиразол-4-ил]-1,2-диметил-1Н-пиррол-3-карбоксамид
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии Е Примера 1, исходя из соединения, полученного в предыдущей стадии. Полученный остаток используют непосредственно в следующей стадии без очистки.
Стадия D: 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-4,16,17,23,24,25-гексагидро-1Н,14Н-15,18-метано-6,9-(метено)дибензо[b,h]пиразоло[4,3-р][1,6,11,15]оксатриазациклоикозин-5(8Н)-он
Указанное в заголовке соединение получают в соответствии со способами, описанными в Стадиях F и G Примера 1, исходя из соединения, полученного в предыдущей стадии.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C36H36ClN5O3
[М+Н]+ рассчитано 622.2579,
[М+Н]+ измерено 622.2573.
Пример 12: Динатрийфосфат 4-[6-хлор-17-циано-10,11-диметил-2,13-диоксо-20,23-диокса-1,10,14-триазагексацикло[26.3.1.1~9,12~.1~15,19~.0~3,8~.0~24,29~]тетратриаконта-3,5,7,9(34),11,15(33),16,18,24,26,28-ундекаен-14-ил]фенила

К раствору соединения Примера 4 (75 мг; 0,113 ммоль) в дихлорметане (1,5 мл) добавляют 1,8-диазабицикло[5.4.0]ундец-7-ен (34 мкл; 0,227 ммоль), затем хлорид бис(диметиламино)фосфорил (16,5 мкл; 0,113 ммоль). Реакционную среду перемешивают при температуре окружающей среды в течение 16 часов, затем разбавляют в смеси этилацетата и водного раствора насыщенного гидрокарбоната натрия. После экстракции дихлорметаном, органические фазы объединяют и сушат над сульфатом магния, фильтруют и концентрируют досуха.
К раствору полученного продукта в ацетонитриле (2 мл), добавляют раствор трифторуксусной кислоты (1 мл) в воде (0,5 мл). Реакционную среду перемешивают при температуре окружающей среды в течение 24 часов затем концентрируют досуха. Полученный остаток ресуспендируют в ацетонитриле, затем добавляют по каплям водный насыщенный раствор бикарбоната натрия до рН=7. Среду концентрируют досуха, затем ресуспендируют в этаноле и фильтруют. Фильтрат концентрируют, затем полученный остаток очищают с помощью хроматографии на фазе Oasis®, используя ацетонитрил и воду в качестве растворителей для элюирования перед лиофилизацией, чтобы получить указанный в заголовке продукт.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C38H30ClN4O8P. 2 Na
[М+Н]+ рассчитано 783.1358,
[М+Н]+ измерено 783.1361.
Пример 13: 12-хлор-5-(4-гидроксифенил)-8,9-диметил-6,15-диоксо-6,9,22,23-тетрагидро-5Н,15Н,17Н-16,22-метано-7,10-(метено)трибензо[b,j,о][1,4,8,13]оксатриазациклооктадецин-3-карбонитрил

Стадия А: этил 5-(2-{[4-({[трет-бутил(диметил)силил]окси}метил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-5-хлофенил)-1,2-диметил-1Н-пиррол-3-карбоксилат
К раствору из Синтеза 2а' (2,95 г; 18,1 ммоль) в дихлорметане (30 мл), добавляют имидазол (3,08 г; 45,25 ммоль), 4-(диметил)-амино-пиридин (0,11 г; 0,9 ммоль) и хлор-трет-бутил-диметилсилан (3,28 г; 21,72 ммоль). Все перемешивают при температуре окружающей среды в течение 2 часов. После гидролиза органическую фазу промывают водой и сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и метанол в качестве растворителей для элюирования, затем используют в следующей стадии без очистки.
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии А Пример 1, используя вышеуказанный промежуточный остаток и кислоту из Синтеза 1а.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.6-7.35 (m, 3 Н), 7.2-6.85 (m, 4 Н), 6.45-5.9 (4s, 1 Н), 5.05-2.7 (m, 4 Н), 4.1/3.95 (2m, 2 Н), 3.85-3.2 (m, 2 Н), 3.45/3.3 (2s, 3 Н), 2.9/2.6 (2m, 1 Н), 2.45/2.35/2.2 (3s, 3 Н), 1.2/1.05 (2m, 3 Н), 0.85/0.7 (2br. s, 9 Н), 0.05-0 (3br.s, 6 Н)
ИК: v: >C=O: 1697 и 1634 см-1.
Стадия В: 5-(5-хлор-2-{[4-(гидроксиметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-1,2-диметил-1Н-пиррол-3-карбоновая кислота
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Примера 1, используя сложный эфир в предшествующей Стадии.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.6-7.4 (m, 3 Н), 7.2-7 (m, 4 Н), 6.5-6.2 (4br. s, 1 Н), 5.05/4.3-3.95 (5m, 2 Н), 4.3-2.8 (6m, 2 Н), 3.75-2.8 (m, 2 Н), 3.47/3.3 (2s, 3 Н), 2.85/2.6 (3m, 1 Н), 2.5/2.35/2.25 (3s, 3 Н).
ИК: v: >C=O: 1662 и 1613 см-1.
Стадия С: 5-(2-{[4-({[трет-бутил(диметил)силил]окси}метил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-5-хлофенил)-1,2-диметил-1Н-пиррол-3-карбоновая кислота
К раствору соединения, полученного в Стадии В (3,65 г; 8,32 ммоль) в ацетонитриле (10 мл) добавляют хлор-трет-бутил-диметилсилан (3 г; 20 ммоль) и 4-(диметил)-амино-пиридин (0,11 г; 0,9 ммоль), затем все перемешивают при температуре окружающей среды в течение 15 минут. Затем 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-а]азепин (1,65 мл; 10,8 ммоль) добавляют по каплям при 0°C. Реакционную среду перемешивают при 70°C в течение 24 часов. После добавления водного раствора 0,1 н. соляной кислоты (10 мл), реакционную среду оставляют перемешиваться в течение 3 часов. Реакционную среду разбавляют в смеси из воды и этилацетата. После экстрагирования этилацетатом, органические фазы объединяют, сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан, метанол и уксусную кислоту в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.5-7.1 (m, 7 Н), 6.3 (m, 1 Н), 3.6-3.4 (m, 2 Н), 3.5 (m, 4 Н), 3.4 (m, 3 Н), 3 (s, 1 Н), 2.5-2.3 (m, 3 Н), 0.9 (s, 9 Н), 0.1 (s, 6 Н).
Стадия D: 5-(2-{[4-({[трет-бутил(диметил)силил]окси}метил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}-5-хлофенил)-N-(5-циано-2-фторфенил)-1,2-диметил-N-[4-(проп-2-ен-1-илокси)фенил]-1Н-пиррол-3-карбоксамид
Соединение получают в соответствии со способом, описанным в Стадии А Примера 6, используя полученную в предшествующей Стадии кислоту и амин из Синтеза 11а''. Полученный остаток используют непосредственно в следующей стадии без очистки.
Стадия Е: 5-(5-хлор-2-{[4-(гидроксиметил)-3,4-дигидроизохинолин-2(1Н)-ил]карбонил}фенил)-N-(5-циано-2-фторфенил)-1,2-диметил-N-[4-(проп-2-ен-1-илокси)фенил]-1Н-пиррол-3-карбоксамид
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии G Примера 1, используя соединение, полученное предыдущей стадии. Полученный остаток используют непосредственно в следующей стадии без очистки.
Стадия F: 12-хлор-5-(4-гидроксифенил)-8,9-диметил-6,15-диоксо-6,9,22,23-тетрагидро-5Н,15Н,17Н-16,22-метано-7,10-(метено)трибензо[b,j,о][1,4,8,13]оксатриазациклооктадецин-3-карбонитрил
К раствору соединения, полученного в Стадии Е (40 мг; 0,058 ммоль) в тетрагидрофуране (20 мл), добавляют при 0°C гидрид натрия (4,64 мг; 0,012 ммоль), затем смесь перемешивают при 40°C в течение 16 часов. Одинаковое количество гидрида натрия добавляют еще два раза, затем реакционную среду перемешивают при 40°C в течение 16 часов перед разбавлением в водном насыщенном растворе хлорида аммония. После экстрагирования этилацетатом, органические фазы объединяют, промывают насыщенным водным раствором хлорида натрия, сушат над сульфатом магния, фильтруют и концентрируют досуха. Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии D Примера 1, используя указанный выше остаток.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C37H29ClN4O4
[М+Н]+ рассчитано 629.195,
[М+Н]+ измерено 629.194.
Пример 14: 6-хлор-14-(4-гидроксифенил)-10,11-диметил-2,13-диоксо-20-окса-1,10,14-триазагексацикло[26.3.1.1~9,12~.1~15,19~.0~3,8~.0~24,29~]тетратриаконта-3,5,7,9(34),11,15(33),16,18,24,26,28-ундекаен-17-карбонитрил
Стадия А: трет-бутил 5-[3-(3-циано-5-{[4-(проп-2-ен-1-илокси)фенил]амино}фенокси)пропил]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения Синтеза 1b'' (1 г; 1,63 ммоль) в тетрагидрофуране (10 мл) при 0°C, добавляют раствор фторида тетрабутиламмония в тетрагидрофуране 1 н. (2,45 мл; 2,45 ммоль). Все перемешивают при температуре окружающей среды в течение 30 минут. Реакционную среду концентрируют, затем разбавляют в смеси этилацетата и воды. После декантации, органическую фазу промывают водой и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют.
К раствору полученного остатка в ацетонитриле, добавляют аллилбромид (142 мкл; 1,63 ммоль) затем карбонат цезия (0,53 г: 1,63 ммоль). Все перемешивают при температуре окружающей среды в течение 16 часов, затем в течение 2 часов при 60°C после добавления 0,2 эквивалента бромида аллила. Реакционную среду концентрируют, затем разбавляют в смеси этилацетата и воды. После декантации, органическую фазу промывают водой, и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют. Полученный таким образом остаток очищают с помощью хроматографии на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 8.24 (s, 1 Н), 7.1 (dd, 1 Н), 7.06 (dd, 2 Н), 7 (d, 2 Н), 6.94 (d, 2 Н), 6.77-6.6 (3dd, 3 Н), 6.04 (m, 1 Н), 5.4/5.26 (2*d, 2 Н), 4.54 (d, 2 Н), 4.47 (s, 2 Н), 3.96 (t, 2 Н), 3.52 (t, 2 Н), 2.76-2.64 (m, 4 Н), 1.92 (m, 2 Н), 1.42 (s, 9 Н).
ИК: v: >NH: 3340 см-1; -CN: 2225 см-1; >C=O: 1624 см-1.
Стадия В: трет-бутил 5-[3-(3-{({5-[2-(трет-бутоксикарбонил)-5-хлофенил]-1,2-диметил-1Н-пиррол-3-ил}карбонил)[4-(проп-2-ен-1-илокси)фенил]амино}-5-цианофенокси)пропил]-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Синтезе 1b (0,7 г; 2 ммоль) в дихлорметане (20 мл) и N,N-диметилформамиде (5 капель), добавляют 1-хлор-N,N-2-триметил-проп-1-ен-1-амин (0,53 мл; 4 ммоль). Реакционную среду перемешивают при температуре окружающей среды в течение 1 часа, затем концентрируют. Остаток ресуспендируют в дихлорэтане затем концентрируют, эту процедуру осуществляют два раза. Конечный остаток ресуспендируют в дихлорэтане (10 мл), затем добавляют к раствору соединения, полученного в Стадии А (1,08 г; 2 ммоль) и пиридина (0,48 мл; 6 ммоль) в дихлорэтане (10 мл). Все нагревают до 80°C в течение 16 часов. Реакционную среду разбавляют в смеси дихлорметана и насыщенного водного раствора хлорида натрия. После экстрагирования водной фазы дихлорметаном, органические фазы объединяют, сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя петролейный эфир и этилацетат в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1Н ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.8 (d, 1 Н), 7.55 (d, 1 Н), 7.25 (s, 1 Н), 7.15 (d, 2 Н), 7.15/7.05/7 (3s, 3 Н), 7.1-6.95 (m, 3 Н), 6.9 (d, 2 Н), 6 (m, 1 Н), 5.35/5.2 (d+d, 1+1 Н), 5.25 (s, 1 Н), 4.55 (d, 2 Н), 4.45 (br. s, 2 Н), 4 (t, 2 Н), 3.5 (t, 2 Н), 3.15 (s, 3 Н), 2.7 (m, 4 Н), 2.45 (s, 3 Н), 1.9 (quint, 2 Н), 1.4 (s, 9 Н), 1.3 (s, 9 Н).
ИК: v: -CN: 2230 см-1; >C=O: 1695 см-1; >C=O: 1645 см-1.
Стадия С: 6-хлор-14-(4-гидроксифенил)-10,11-диметил-2,13-диоксо-20-окса-1,10,14-триазагексацикло[26.3.1.1~9,12~.1~15,19~.0~3,8~.0~24,29~]тетратриаконта-3,5,7,9(34),11,15(33),16,18,24,26,28-ундекаен-17-карбонитрил
К раствору соединения, полученного в Стадии В (0,58 г; 0,67 ммоль) в диоксане (15 мл), добавляют триэтиламин (1,38 мл; 10 ммоль) и трифторметансульфонат триметилсилила (1,8 мл; 10 ммоль). Реакционную среду перемешивают при кипячении с обратным холодильником в течение 2 часов, затем выливают в ледяную воду. После экстрагирования этилацетатом, органические фазы объединяют, сушат над сульфатом магния, фильтруют и концентрируют.
К раствору полученного таким образом остатка в дихлорметане (200 мл), добавляют последовательно 1-гидроксибензотриазол (0,108 г; 0,67 ммоль), гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (0,153 г; 0,8 ммоль) и диизопропилэтиламин (0,57 мл; 3,33 ммоль). Затем все перемешивают в течение 24 часов при температуре окружающей среды. Реакционную среду разбавляют смесью дихлорметана и воды. После декантации, органическую фазу сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и этилацетат в качестве растворителей для элюирования.
К раствору полученного таким образом остатка в смеси дихлорметана (10 мл) и метанола (5 мл), добавляют 1,3-диметилбарбитуровую кислоту (0,036 г; 0,23 ммоль). Реакционную среду дегазируют аргоном в течение 10 минут и добавляют тетракис-(трифенилфосфин)палладий(0) (0,013 г; 0,01 ммоль). Реакционную среду нагревают до 40°C в течение 45 минут. После концентрирования метанола, реакционную среду разбавляют в смеси этилацетата и воды. После декантации, органическую фазу сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают с помощью хроматографии на фазе Oasis®, используя ацетонитрил и воду в качестве растворителей для элюирования. Полученное таким твердое вещество растворяют в смеси воды и ацетонитрила, фильтруют затем лиофилизируют, чтобы получить указанный в заголовке продукт.
Элементарный микроанализ: (теоретический %: измеренный)
%С=71.28: 70.81; %Н=5.06: 4.94; %N=8.53: 8.51.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C39H33ClN4O5
[М+Н]+ рассчитано 673,2212,
[М+Н]+ измерено 673,2211.
Пример 15: 6-хлор-14-(4-гидроксифенил)-10,11-диметил-23-окса-1,10,14,17,18-пентаазагексацикло[26.3.1.1~9,12~.1~15,18~.0~3,8~.0~24,29~]тетратриаконта-3,5,7,9(34),11,15(33),16,24,26,28-декаен-2,13-дион

Стадия А: трет-бутил 5-(4-{4-[({5-[2-(трет-бутоксикарбонил)-5-хлофенил]-1,2-диметил-1Н-пиррол-3-ил}карбонил)(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1Н-пиразол-1-ил}бутокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Примера 14,, используя соединение из Синтеза 1b (кислота) и соединение из Синтеза 2b'' (амин).
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.83 (s, 1 Н), 7.73 (d, 1 Н), 7.51 (dd, 1 Н), 7.15 (s, 1 Н), 7.14-7.06 (m, 3 Н), 7.1 (s, 1 Н), 6.82 (d, 2 Н), 6.73 (dd, 2 Н), 4.97 (s, 1 Н), 4.46 (s, 2 Н), 4.1 (t, 2 Н), 3.91 (t, 2 Н), 3.53 (t, 2 Н), 3.15 (s, 3 Н), 2.59 (t, 2 Н), 2.47 (s, 3 Н), 1.9 (m, 2 Н), 1.64 (m, 2 Н), 1.41 (s, 9 Н), 1.27 (s, 9 Н), 0.86 (s, 9 Н), 0.07 (s, 6 Н).
ИК: v: >C=O: 1695 см-1; >C=O: 1633 см-1.
Стадия В: 6-хлор-14-(4-гидроксифенил)-10,11-диметил-23-окса-1,10,14,17,18-пентаазагексацикло[26.3.1.1~9,12~.1~15,18~.0~3,8~.0~24,29~]тетратриаконта-3,5,7,9(34),11,15(33),16,24,26,28-декаен-2,13-дион
К раствору соединения, полученного в Стадии А (0,13 г; 0,14 ммоль) в диоксане (5 мл), добавляют триэтиламин (0,1 мл; 0,71 ммоль) и трифторметансульфонат триметилсилила (0,13 мл; 0,71 ммоль). Реакционную среду перемешивают при кипячении с обратным холодильником в течение 1 часа затем выливают в ледяную воду. После экстрагирования этилацетатом, органические фазы объединяют, сушат над сульфатом магния, фильтруют и концентрируют.
К раствору полученного остатка в дихлорметане (85 мл), добавляют последовательно 1-гидроксибензотриазол (0,046 г; 0,34 ммоль), гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (0,065 г; 0,34 ммоль) и диизопропилэтиламин (0,243 мл; 1,42 ммоль). Затем все перемешивают в течение 20 часов при температуре окружающей среды. Реакционную среду разбавляют смесью дихлорметана и воды. После декантации, органическую фазу сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и метанол в качестве растворителей для элюирования.
К раствору полученного остатка в метаноле (3 мл), добавляют раствор 1 н. поташа в метаноле (0,11 мл; 0,11 ммоль). Затем все перемешивают в течение 18 часов при температуре окружающей среды. После концентрирования метанола, реакционную среду разбавляют в смеси дихлорметана и воды. После декантации, органическую фазу сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают с помощью хроматографии на фазе Oasis®, используя ацетонитрил и воду в качестве растворителей для элюирования. Полученное таким образом твердое вещество растворяют в смеси воды и ацетонитрила, затем лиофилизируют, чтобы получить указанный в заголовке продукт.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C36H34ClN5O4
[М+Н]+ рассчитано 636,2372,
[М+Н]+ измерено 636,2375.
Пример 16: 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-1,4,16,17,23,24-гексагидро-14Н-15,18-метано-6,9-(метено)дибензо[1,r]пиразоло[3,4-d][1,6,10,15]оксатриазациклононадецин-5,14(8Н)-дион

Указанное в заголовке соединение получают в соответствии со способом из Примера 15, используя кислоту из Синтеза 1b и амин из Синтеза 3b''.
Элементарный микроанализ: (теоретический %: измеренный)
%С=67.57: 67.70; %Н=5.18: 4.89; %N=11.26: 11.16.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C35H32ClN5O4
[М+Н]+ рассчитано 622,2209,
[М+Н]+ измерено 622,2216.
Пример 17: 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-16-(морфолин-4-илметил)-4,16,17,23,24,25-гексагидро-1Н,14Н-15,18-метано-6,9-(метено)дибензо[b,h]пиразоло[4,3-р][1,6,11,15]оксатриазациклоикозин-5,14(8Н)-дион
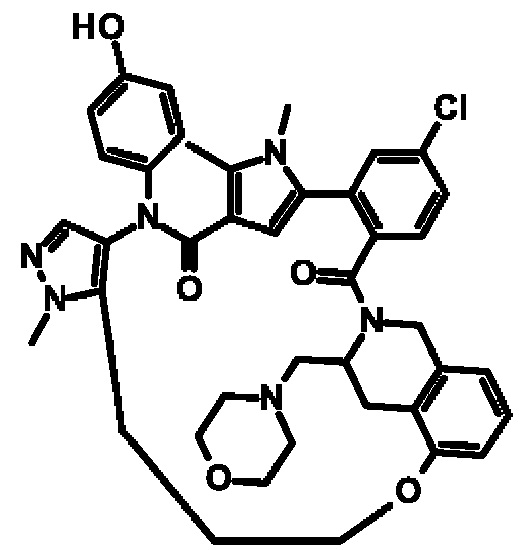
Стадия А: трет-бутил 2-{4-[(4-{[трет-бутил(диметил)силил]окси}фенил){1-метил-5-[3-(тетрагидро-2Н-пиран-2-илокси)пропил]-1Н-пиразол-4-ил}карбамоил]-1,5-диметил-1Н-пиррол-2-ил}-4-хлобензоат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Примера 14, используя кислоту из Синтеза 1b и амин из Синтеза 4b''.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.75 (d, 1 Н), 7.52 (dd, 1 Н), 7.21 (s, 1 Н), 7.1 (d, 1 Н), 7.02 (d, 2 Н), 6.71 (d, 2 Н), 6.19 (s, 1 Н), 4.5 (t, 1 Н), 3.71 (s, 3 Н), 3.7-3.2 (4m, 4 Н), 3.15 (s, 3 Н), 2.55 (m, 2 Н), 2.41 (s, 3 Н), 1.7-1.2 (m, 8 Н), 1.3 (s, 9 Н), 0.85 (s, 9 Н), 0.1 (s, 6 Н).
ИК: v: >C=O: 1706 см-1; >C=O: 1640 см-1.
Стадия В: трет-бутил 2-(4-{(4-{[трет-бутил(диметил)силил]окси}фенил)[5-(3-гидроксипропил)-1-метил-1Н-пиразол-4-ил]карбамоил}-1,5-диметил-1Н-пиррол-2-ил)-4-хлобензоат
К раствору соединения, полученного в Стадии А (3,28 г; 4,21 ммоль) в метаноле (30 мл), добавляют пара-толуолсульфонат пиримидиния (1,5 г; 0,42 ммоль), затем все перемешивают в течение 4 часов при 80°C. После достижения температуры окружающей среды, реакционную среду разбавляют дихлорметаном и водным раствором насыщенного хлорида аммония. После декантации, органическую фазу промывают водой, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и метанол в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1Н ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.75 (d, 1 Н), 7.59 (dd, 1 Н), 7.22 (s, 1 Н), 7.12 (d, 1 Н), 7.05 (d, 2 Н), 6.75 (d, 2 Н), 5.15 (s, 1 Н), 4.5 (m, 1 Н), 3.71 (s, 3 Н), 3.3 (t, 2 Н), 3.2 (s, 3 Н), 2.5 (t, 2 Н), 2.45 (s, 3 Н), 1.5 (m, 2 Н), 1.31 (s, 9 Н), 0.9 (s, 9 Н), 0.1 (s, 6 Н).
ИК: v: -ОН: 3421 см-1; >C=O: 1708 см-1; >C=O: 1634 см-1.
Стадия С: трет-бутил 5-(3-{4-[({5-[2-(трет-бутоксикарбонил)-5-хлофенил]-1,2-диметил-1Н-пиррол-3-ил}карбонил)(4-{[трет-бутил(диметил)силил]окси}фенил)амино]-1-метил-1Н-пиразол-5-ил}пропокси)-3-(морфолин-4-илметил)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Стадии В (1,67 г; 2,9 ммоль), трифенилфосфина (1,26 г; 3,5 ммоль) и соединения Синтеза 4' (0,84 г; 2,9 ммоль) в тетрагидрофуране (20 мл), добавляют по каплям раствор азодикарбоксилат диизопропила (1 мл; 3,5 ммоль) в тетрагидрофуране (20 мл). Реакционную среду перемешивают при температуре окружающей среды в течение 2 часов, затем разбавляют в смеси этилацетата и воды. После экстрагирования водной фазы этилацетатом, органические фазы объединяют, промывают с водным насыщенным раствором хлорида аммония, насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают с помощью хроматографии на фазе RP-18, используя ацетонитрил и воду в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.74 (d, 1 Н), 7.54 (d, 1 Н), 7.26 (s, 1 Н), 7.26 (s, 1 Н), 7.1 (t, 1 Н), 7.03 (d, 2 Н), 6.74 (d, 1 Н), 6.73 (d, 1 Н), 6.69 (d, 2 Н), 5.15 (s, 1 Н), 4.64/4.08 (d+m, 1+1 Н), 4.59/4.46 (m+m, 1 Н), 3.9 (m, 2 Н), 3.74 (s, 3 Н), 3.49 (m, 4 Н), 3.16 (s, 3 Н), 2.73/2.58 (m+m, 1+1 Н), 2.69 (m, 2 Н), 2.5-2.2 (m, 4 H), 2.42 (s, 3 H), 2.24/1.98 (m+m, 1+1 H), 1.79 (m, 2 H), 1.42 (s, 9 H), 1.25 (s, 9 H), 0.85 (s, 9 H), 0.07 (s, 6 H).
ИК: v: >C=O: 1693 см-1; >C=O: 1641 см-1.
Стадия D: 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-16-(морфолин-4-илметил)-4,16,17,23,24,25-гексагидро-1Н,14Н-15,18-метано-6,9-(метено)дибензо[b,h]пиразоло[4,3-р][1,6,11,15]оксатриазациклоикозин-5,14(8Н)-дион
К раствору соединения, полученного в Стадии С (0,315 г; 0,31 ммоль) в дихлорэтане (12,5 мл), добавляют триэтиламин (0,13 мл; 0,92 ммоль) и бромид цинка (0,34 г; 1,53 ммоль). Реакционную среду перемешивают при 120°C в течение 2 часов в микроволновой печи (150 Вт) три раза, затем ее выливают в смесь дихлорметана и ледяной воды. После экстракции дихлорметаном, органические фазы объединяют, сушат над сульфатом магния, фильтруют и концентрируют.
К раствору полученного таким образом остатка в дихлорметане (15 мл), добавляют последовательно 1-гидроксибензотриазол (0,052 г; 0,23 ммоль), гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (0,072 г; 0,23 ммоль) и диизопропилэтиламин (0,2 мл; 0,6 ммоль). Затем все перемешивают в течение 16 часов при температуре окружающей среды. Реакционную среду разбавляют смесью дихлорметана и воды. После декантации, органическую фазу сушат над сульфатом магния, фильтруют и концентрируют досуха.
К раствору полученного таким образом остатка в тетрагидрофуране (14 мл), добавляют раствор 1 М фторида тетрабутиламмония в тетрагидрофуране (0,5 мл; 0,5 ммоль). Затем все перемешивают в течение 30 минут при температуре окружающей среды. После концентрирования, остаток ресуспендируют в смеси этилацетата и насыщенного водного раствора хлорида натрия. После декантации, органическую фазу сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и аммиак в метаноле в качестве растворителей для элюирования. Полученное таким образом твердое вещество растворяют в смеси воды и ацетонитрила, затем лиофилизируют, чтобы получить указанный в заголовке продукт.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C41H43ClN6O5
[М+Н]+ рассчитано 735,3056,
[М+Н]+ измерено 735,3061.
Пример 18: 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-5,14-диоксо-4,5,8,16,17,23,24,25-октагидро-1Н,14Н-15,18-метано-6,9-(метено)дибензо[b,h]пирроло[3,2-р][1,6,11,15]оксатриазациклоикозин-2-карбонитрил
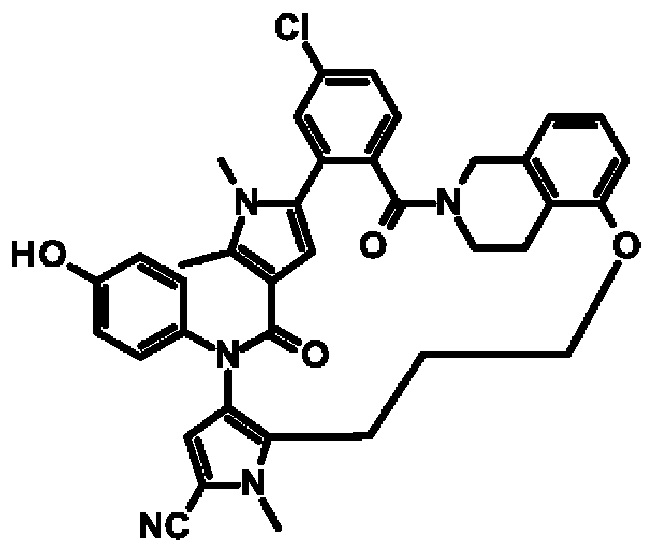
Указанное в заголовке соединение получают в соответствии со способом из Примера 15, используя кислоту из Синтеза 1b и амин из Синтеза 5b''.
Элементарный микроанализ: (теоретический %: измеренный)
%С=69.14: 68.70; %Н=5.19: 5.16; %N=10.61: 9.97.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C38H34ClN5O4
[М+Н]+ рассчитано 660,2372,
[М+Н]+ измерено 660,2374.
Пример 19: 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-5,14-диоксо-1,4,5,8,16,17,23,24-октагидро-14Н-15,18-метано-6,9-(метено)дибензо[1,r]пирроло[2,3-d][1,6,10,15]оксатриазациклононадецин-2-карбонитрил
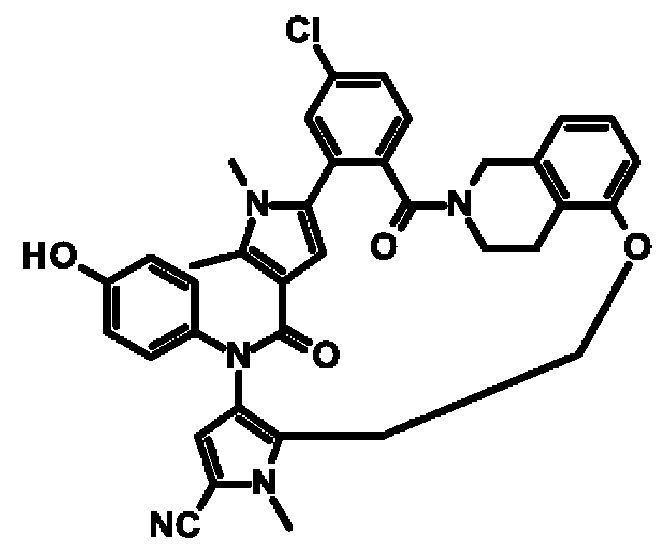
Указанное в заголовке соединение получают в соответствии со способом из Примера 15, используя кислоту из Синтеза 1b и амин из Синтеза 6b''.
Элементарный микроанализ: (теоретический %: измеренный)
%С=68.78: 68.64; %Н=4.99: 5.01; %N=10.84: 10.66.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C37H32ClN5O4
[М+Н]+ рассчитано 646,2217,
[М+Н]+ измерено 646,2216.
Пример 20: 11-фтор-4-(4-гидроксифенил)-1,7,8-триметил-1,4,16,17,23,24-гексагидро-14Н-15,18-метано-6,9-(метено)дибензо[l,r]пиразоло[3,4-d][1,6,10,15]оксатриазациклононадецин-5,14(8Н)-дион

Указанное в заголовке соединение получают в соответствии со способом из Примера 15, используя кислоту из Синтеза 2b и амин из Синтеза 3b''.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C35H33FN5O4
[М+Н]+ рассчитано 606.2511,
[М+Н]+ измерено 606.2517.
Пример 21: 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-1,4,16,17,23,24-гексагидро-14Н-15,18-метано-6,9-(метено)дибензо[m,s]пиразоло[3,4-е][1,4,7,11,16]диоксатриазациклоикозин-5,14(8Н)-дион
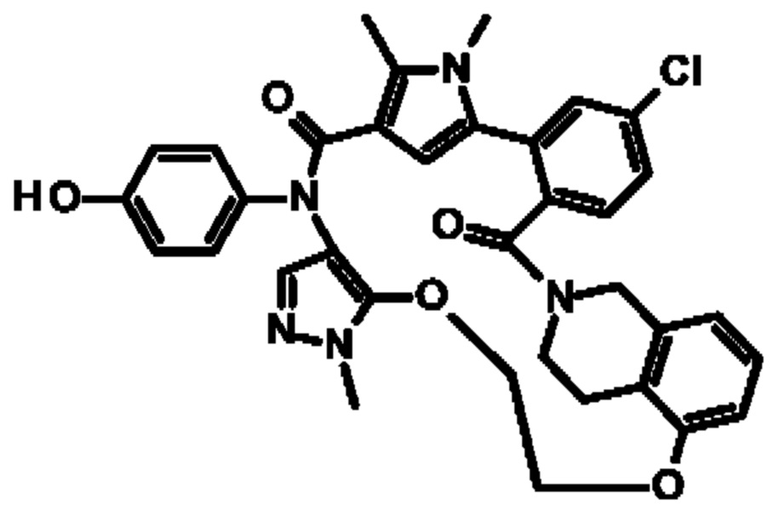
Указанное в заголовке соединение получают в соответствии со способом из Примера 15, используя кислоту из Синтеза 1b и амин из Синтеза 7b''.
Элементарный микроанализ: (теоретический %: измеренный)
%С=65.88: 65.61; %Н=5.05: 4.98; %N=10.60: 10.94.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C35H32ClN5O5
[М+Н]+ рассчитано 638,2165,
[М+Н]+ измерено 638,2166.
Пример 22: 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-1,4,16,21,22,23-гексагидро-14Н-15,21-метано-6,9-(метено)дибензо[j,о]пиразоло[3,4-b][1,4,8,13]оксатриазациклононадецин-5,14(8Н)-дион

Указанное в заголовке соединение получают в соответствии со способом из Примера 15, используя кислоту из Синтеза 1b и амин из Синтеза 8b''.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C35H32ClN5O4
[М+Н]+ рассчитано 622.2216,
[М+Н]+ измерено 622.2208.
Пример 23: 11-хлор-4-(4-гидроксифенил)-7,8-диметил-1-(тетрагидрофуран-3-ил)-1,4,16,17,23,24-гексагидро-14Н-15,18-метано-6,9-(метено)дибензо[l,r]пиразоло[3,4-d][1,6,10,15]оксатриазациклононадецин-5,14(8Н)-дион

Указанное в заголовке соединение получают в соответствии со способом из Примера 15, используя кислоту из Синтеза 1b и амин из Синтеза 9b''.
Элементарный микроанализ: (теоретический %: измеренный)
%С=67.30: 67.12; %Н=5.35: 5.08; %N=10.33: 10.31.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C38H36ClN5O5
[М+Н]+ рассчитано 678,2478,
[М+Н]+ измерено 678,2481.
Пример 24: 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-1,4,22,23-тетрагидро-14Н,16Н-15,17-метано-6,9-(метено)дибензо[b,г]пиразоло[4,3-о][1,5,10,14]оксатриазациклооктадецин-5,14(8Н)-дион

Указанное в заголовке соединение получают в соответствии со способом из Примера 15, используя кислоту из Синтеза 1b и амин из Синтеза 10b''.
Элементарный микроанализ: (теоретический %: измеренный)
%С=67.16: 67.31; %Н=4.97: 4.91; %N=11.52: 11.30.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C34H30ClN5O4
[М+Н]+ рассчитано 608,2059,
[М+Н]+ измерено 608,2074.
Пример 25: (16R или S)-11-хлор-4-(4-гидроксифенил)-1,7,8,16-тетраметил-5,14-диоксо-4,5,8,16,17,23,24,25-октагидро-1Н,14Н-15,18-метано-6,9-(метено)дибензо[b,h]пирроло[3,2-р][1,6,11,15]оксатриазациклоикозин-2-карбонитрил

Указанное в заголовке соединение получают в соответствии со способом из Примера 15, используя кислоту из Синтеза 1b и амин из Синтеза 11b''.
Элементарный микроанализ: (теоретический %: измеренный)
%С=69.48: 69.13; %Н=5.38: 5.37; %N=10.39: 10.05.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C39H36ClN5O4
[М+Н]+ рассчитано 674,2529,
[М+Н]+ измерено 674,253.
Пример 26: (16S или R)-11-хлор-4-(4-гидроксифенил)-1,7,8,16-тетраметил-5,14-диоксо-4,5,8,16,17,23,24,25-октагидро-1Н,14Н-15,18-метано-6,9-(метено)дибензо[b,h]пирроло[3,2-р][1,6,11,15]оксатриазациклоикозин-2-карбонитрил
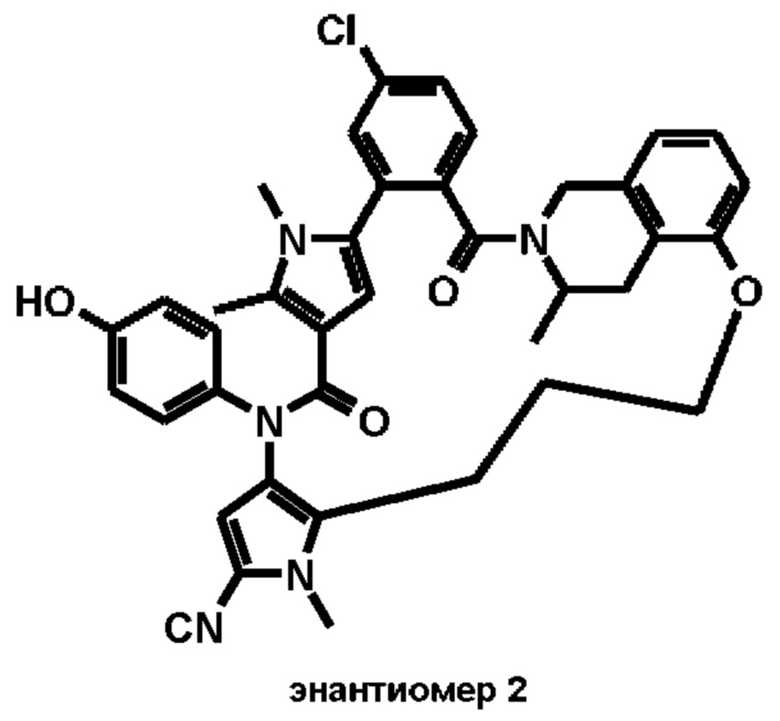
Указанное в заголовке соединение получают в соответствии со способом из Примера 15, используя кислоту из Синтеза 1b и амин из Синтеза 12b''.
Элементарный микроанализ: (теоретический %: измеренный)
%С=69.48: 68.73; %Н=5.38: 5.47; %N=10.39: 10.13.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C39H36ClN5O4
[М+Н]+ рассчитано 674,2529,
[М+Н]+ измерено 674,2532.
Пример 27: 4-[15-хлор-3-гидрокси-18,19-диметил-12,21-диоксо-7,8,9,10,18,21-гексагидро-6Н,12Н,22Н-8,11-этано-20,17-(метено)дибензо[b,j}[1,4,8,13]оксатриазациклооктадецин-22-ил]-1,5-диметил-1Н-пиррол-2-карбонитрил

Стадия А: трет-бутил 4-(2-{5-(бензилокси)-2-[({5-[2-(трет-бутоксикарбонил)-5-хлофенил]-1,2-диметил-1Н-пиррол-3-ил}карбонил)(5-циано-1,2-диметил-1Н-пиррол-3-ил)амино]фенокси}этил)пиперидин-1-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Примера 14, используя кислоту из Синтеза 1b и амин из Синтеза 13b''.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част, на млн.: 7.8 (d, 1 Н), 7.55 (dd, 1 Н), 7.45-7.25 (т, 5 Н), 7.05 (d, 1 Н), 7.05 (s, 1 Н), 6.65 (br. s, 1 Н), 6.65 (s, 1 Н), 6.5 (d, 1 Н), 5.35 (br. s, 1 Н), 5.05 (s, 2 Н), 3.95 (br. s, 2 Н), 3.85/2.55 (2br. s, 4Н), 3.55 (s, 3 Н), 3.15 (s, 3 Н), 2.35 (s, 3 Н), 2.1 (br. s, 3 H), 1.6/0.95 (m+m, 2+2 H), 1.55 (m, 1 H), 1.55 (m, 2 H), 1.35 (s, 9 H), 1.25 (s, 9 H).
ИК: ν: -CN: 2209 см-1; >C=0: 1715, 1688 и 1640 см-1.
Стадия В: 4-[15-хлор-3-гидрокси-18,19-диметил-12,21-диоксо-7,8,9,10,18,21-гексагидро-6Н,12Н,22Н-8,11-этано-20,17-(метено)дибензо[b,j][1,4,8,13]оксатриазациклооктадецин-22-ил]-1,5-диметил-1Н-пиррол-2-карбонитрил
К раствору соединения, полученного в Стадии А (1,3 г; 1,29 ммоль) в дихлорметане (12 мл) добавляют бромид цинка (1,45 г; 6,44 ммоль). Реакционную среду перемешивают при кипячении с обратным холодильником в течение 17 часов, затем выливают в воде. Реакционную смесь затем перемешивают в течение 2 часов и экстрагируют дихлорметаном. Органические фазы объединяют, сушат над сульфатом магния, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, используя дихлорметан и метанол в качестве растворителей для элюирования.
К раствору полученного остатка в дихлорметане (470 мл), добавляют последовательно 1-гидроксибензотриазол (0,252 г; 1,86 ммоль), гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (0,320 г; 1,86 ммоль) и диизопропилэтиламин (1,28 мл; 7,77 ммоль). Затем все перемешивают в течение 20 часов при температуре окружающей среды. Реакционную среду разбавляют смесью дихлорметана и воды. После декантации, органическую фазу сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и метанол в качестве растворителей для элюирования.
К раствору полученного таким образом остатка в смеси метанола (25 мл) и этилацетата (25 мл), добавляют палладий на угле (15 мас. %), затем все гидрируют в течение 4 часов при температуре окружающей среды под 0,6 бар. Реакционную среду фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и метанол в качестве растворителей для элюирования, затем с помощью хроматографии на сверхкритической фазе RP-18, используя этанол с 0,1% диэтиламином в качестве растворителя для элюирования. Полученное таким образом твердое вещество растворяют в смеси воды и ацетонитрила, затем лиофилизируют, чтобы получить указанный в заголовке продукт.
Элементарный микроанализ: (теоретический %: измеренный)
%С=66.71: 66.25; %Н=5.60: 5.52; %N=11.44: 11.37.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C34H34ClN5O4
[М+Н]+рассчитано 612,2367,
[М+Н]+измерено 612,2372.
Пример 28: 4-[3-гидрокси-18,19-диметил-12,21-диоксо-7,8,9,10,18,21-гексагидро-6Н,12Н,22Н-8,11-этано-20,17-(метено)дибензо[b,j][1,4,8,13]оксатриазациклооктадецин-22-ил]-1,5-диметил-1Н-пиррол-2-карбонитрил
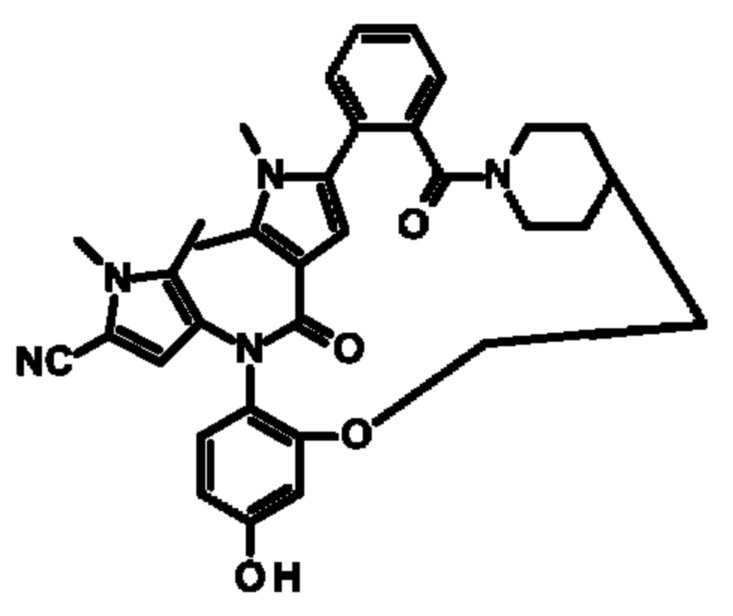
Указанное в заголовке соединение получают в качестве вторичного продукта в Стадии В Примера 27.
Элементарный микроанализ: (теоретический %: измеренный)
%С=70.69: 70.15; %Н=6.11: 6.13; %N=12.12: 12.86.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C34H35N5O4
[М+Н]+ рассчитано 578,2773,
[М+Н]+ измерено 578,2762.
Пример 29: 4-[16-хлор-3-гидрокси-19,20-диметил-13,22-диоксо-6,7,8,9,10,11,19,22-октагидро-13Н,23Н-8,12-метано-21,18-(метено)дибензо[b,j][1,4,8,13]оксатриазациклононадецин-23-ил]-1,5-диметил-1Н-пиррол-2-карбонитрил

Указанное в заголовке соединение получают в соответствии со способом из Примера 27, используя кислоту из Синтеза 1b и амин из Синтеза 14b''.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C34H34ClN5O4
[М+Н]+ рассчитано 612.2372,
[М+Н]+ измерено 612.2367.
Пример 30: 6-хлор-14-(4-гидроксифенил)-10,11-диметил-2,13-диоксо-23-окса-1,10,14-триаза-19-азониагексацикло [26.3.1.1~9,12~.1~15,19~.0~3,8~.0~24,29~]тетратриаконта-3,5,7,9(34), 11,15(33),16,18,24,26,28-ундекаен трифторметилсульфонат

Стадия А: 3-{({5-[2-(трет-бутоксикарбонил)-5-хлофенил]-1,2-диметил-1Н-пиррол-3-ил}карбонил)[4-(проп-2-ен-1-илокси)фенил]амино}-1-(3-{[2-(трет-бутоксикарбонил)-1,2,3,4-тетрагидроизохинолин-5-ил]окси}пропил)пиримидиния
К раствору соединения, полученного в Синтезе 1b (0,47 г; 1,36 ммоль) в дихлорметане (34 мл), добавляют 1-хлор-N,N-2-триметил-проп-1-ен-1-амин (0,183 мл; 1,49 ммоль). Реакционную среду перемешивают при температуре окружающей среды в течение 1 часа, затем концентрируют. Остаток ресуспендируют в тетрагидрофуране (14 мл).
Параллельно, готовят раствор гидрида натрия (65 мг; 1,63 ммоль) в тетрагидрофуране (5 мл) и соединения из Синтеза 15b'' (960 мг; 1,49 ммоль) в тетрагидрофуране (15 мл), который перемешивают при температуре окружающей среды в течение 30 минут. К нему добавляют полученный ранее остаток, и все перемешивают в течение 3 часов. Реакционную среду разбавляют в смеси дихлорметана и воды. После экстрагирования водной фазы дихлорметаном, органические фазы объединяют, сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и метанол в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част, на млн.: 9.14 (br. s, 1 Н), 8.87 (d, 1 Н), 8.18 (d, 1 Н), 8.06 (dd, 1 Н), 7.81 (d, 1 Н), 7.58 (d, 1 Н), 7.17 (d, 2 Н), 7.13 (t, 1 Н), 7.01 (s, 1 Н), 6.95 (d, 2 Н), 6.77 (d, 1 Н), 6.77 (d, 1 Н), 6.01 (m, 1 Н), 5.34 (d, 1 Н), 5.21 (d, 1 Н), 5.2 (s, 1 Н), 4.8 (t, 2 Н), 4.57 (d, 2 Н), 4.45 (br. s, 2 Н), 4.05 (t, 2 Н), 3.48 (т, 2 Н), 3.16 (s, 3 Н), 2.46 (т, 2 Н), 2.46 (s, 3 Н), 2.4 (т, 2 Н), 1.41/1.3 (2s, 18 Н).
Стадия В: 6-хлор-14-(4-гидроксифенил)-10,11-диметил-2,13-диоксо-23-окса-1,10,14-триаза-19-азониагексацикло[26.3.1.1~9,12~.1~15,19~.0~3,8~.0~24,29~]тетратриаконта-3,5,7,9(34),11,15(33),16,18,24,26,28-ундекаен трифторметилсульфонат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии С Примера 14, используя соединение, полученное в предыдущей Стадии.
Элементарный микроанализ: (теоретический %: измеренный)
%С=58.27: 58.74; %Н=4.38: 4.35; %N=7.15: 7.71.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C38H34ClF3N4O7S
[M-CF3SO3]+ рассчитано 633,2249,
[M-CF3SO3]+ измерено 633,2263.
Пример 31: 6-хлор-14-(4-гидроксифенил)-10,11,18-триметил-22-окса-1,10,14,17,19-пентаазагексацикло[25.3.1.1~9,12~.1~15,19~.0~3,8~.0~23,28~]тритриаконта-3,5,7,9(33),11,15,17,23,25,27-декаен-2,13,32-трион

Стадия А: трет-бутил 4-хлор-2-[1,5-диметил-4-({2-метил-4-[2-(тетрагидро-2Н-пиран-2-илокси)этокси]пиримидин-5-ил}[4-(проп-2-ен-1-илокси)фенил]карбамоил)-1Н-пиррол-2-ил]бензоат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии А Примера 30, используя соединение из Синтеза 1b и соединение из Синтеза 16b''.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 8.36 (s, 1 Н), 7.79 (d, 1 Н), 7.56 (dd, 1 Н), 7.21 (d, 2 Н), 7.05 (d, 1 Н), 6.87 (d, 2 Н), 5.99 (m, 1 Н), 5.33 (d, 1 Н), 5.29 (s, 1 Н), 5.2 (d, 1 Н), 4.56 (m, 1 Н), 4.53 (d, 2 Н), 4.45 (m, 2 Н), 3.83/3.61 (2m, 2 Н), 3.69/3.35 (2m, 2 Н), 3.15 (s, 3 Н), 2.5 (s, 3 Н), 2.39 (s, 3 Н), 1.63/1.37 (2m, 2 Н), 1.51/1.38 (2m, 2 Н), 1.42/1.32 (2m, 2 Н), 1.29 (s, 9 Н).
ИК: ν: >С=O: 1703 см-1; >С=O: 1644 см-1.
Стадия В: трет-бутил 5-{2-[5-{({5-[2-(трет-бутоксикарбонил)-5-хлофенил]-1,2-диметил-1Н-пиррол-3-ил}карбонил)[4-(проп-2-ен-1-илокси)фенил]амино}-2-метил-6-оксопиримидин-1(6Н)-ил]этокси}-3,4-дигидроизохинолин-2(1Н)-карбоксилат
К раствору соединения, полученного в Стадии А (0,97 г; 1,35 ммоль), в метаноле (7 мл), добавляют пара-толуолсульфонат пиримидиния (0,34 г; 1,35 ммоль), затем все перемешивают в течение 4 часов при 80°С. После достижения температуры окружающей среды, реакционную среду концентрируют, затем разбавляют с этилацетатом и водой. После декантации, органическую фазу промывают водой и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха.
К раствору полученного остатка (0,579 г; 0,915 ммоль) в дихлорметане (5 мл), добавляют тозилхлорид (0,349 г; 1,83 ммоль) и триэтиламин (514 мкл; 3,66 ммоль), затем все перемешивают в течение 16 часов при температуре окружающей среды. Реакционную среду разбавляют дихлорметаном и водой. После декантации, органическую фазу промывают водой и насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют досуха.
К раствору полученного остатка (0,7 г; 0,752 ммоль) в N,N-диметилформамиде (5 мл), добавляют соединение из Синтеза 3b' (0,225 г; 0,903 ммоль) и карбонат калия (0,313 г; 2,26 ммоль), затем все перемешивают в течение 2 часов при 125°С. Реакционную среду разбавляют с этилацетатом и водой. После декантации, органическую фазу промывают водой и насыщенным водным раствором хлорида лития, затем сушат над сульфатом магния, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, используя дихлорметан и метанол в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.88 (s, 1 Н), 7.72 (d, 1 Н), 7.5 (dd, 1 Н), 7.15 (d, 2 Н), 7.08 (t, 1 Н), 7.07 (d, 1 Н), 6.84 (d, 2 Н), 6.74 (d, 1 Н), 6.74 (d, 1 Н), 5.99 (m, 1 Н), 5.36 (s, 1 Н), 5.33 (d, 1 Н), 5.2 (d, 1 Н), 4.52 (d, 2 Н), 4.43 (br. s, 2 Н), 4.39 (t, 2 Н), 4.2 (t, 2 Н), 3.48 (t, 2 Н), 3.11 (s, 3 Н), 2.62 (s, 3 Н), 2.47 (t, 2 Н), 2.36 (s, 3 Н), 1.41/1.28 (2s, 18 Н).
ИК: ν:>С=O: 1682 см-1; >С=O: 1639 см-1.
Стадия С: 6-хлор-14-(4-гидроксифенил)-10,11,18-триметил-22-окса-1,10,14,17,19-пентаазагексацикло[25.3.1.1~9,12~.1~15,19~.0~3,8~.0~23,28~]тритриаконта-3,5,7,9(33),11,15,17,23,25,27-декаен-2,13,32-трион
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии С Примера 14, используя соединение из предыдущей стадии.
Элементарный микроанализ: (теоретический %: измеренный)
%С=66.51: 66.29; %Н=4.96: 4.95; %N=10.77: 10.38.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C36H32ClN5O5
[М+Н]+ рассчитано 650,2169,
[М+Н]+ измерено 650,2165.
Пример 32: 18-хлор-11-(4-гидроксифенил)-2,14,15-триметил-1,12-диоксо-1,2,3,4,12,15-гексагидро-11Н-6,10:13,16-ди(метено)-5,2,11,15-бензоксатриазациклооктадецин-8-карбонитрил

Стадия А: трет-бутил 2-(4-{(3-{2-[(трет-бутоксикарбонил)(метил)амино]этокси}-5-цианофенил)[4-(проп-2-ен-1-илокси)фенил]карбамоил}-1,5-диметил-1Н-пиррол-2-ил)-4-хлобензоат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Примера 14, используя соединение из Синтеза 1b и соединение из Синтеза 17b''.
1H ЯМР (400 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.78 (d, 1 Н), 7.56 (dd, 1 Н), 7.28 (br. s, 1 Н), 7.19 (br. s, 1 Н), 7.14 (d, 2 Н), 7.07 (d, 1 Н), 6.99 (неразрешенный пик, 1 Н), 6.92 (d, 2 Н), 6 (m, 1 Н), 5.34/5.21 (2dquad, 2 Н), 5.26 (s, 1 Н), 4.55 (m, 2 Н), 4.11 (m, 2 Н), 3.48 (t, 2 Н), 3.16 (s, 3 Н), 2.8 (s, 3 Н), 2.43 (s, 3 Н), 1.3 (m, 18 Н).
ИК: ν: >CN: 2230 см-1; >С=O: 1697 и 1645 см-1.
Стадия В: 18-хлор-11-(4-гидроксифенил)-2,14,15-триметил-1,12-диоксо-1,2,3,4,12,15-гексагидро-11Н-6,10:13,16-ди(метено)-5,2,11,15-бензоксатриазациклооктадецин-8-карбонитрил
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии С Примера 14, используя соединение из предыдущей стадии.
Элементарный микроанализ: (теоретический %: измеренный)
%С=66.60: 65.92; %Н=4.66: 4.62; %N=10.36: 10.08.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C30H25ClN4O4
[М+Н]+ рассчитано 541,1642,
[М+Н]+ измерено 541,1637.
Пример 33: 21-хлор-6-(4-гидроксифенил)-2,3,17-триметил-5,18-диоксо-5,6,13,14,15,16,17,18-октагидро-2Н-4,1:11,7-ди(метено)-12,2,6,17-бензоксатриазациклоикозин-9-карбонитрил

Указанное в заголовке соединение получают в соответствии со способом из Примера 32, используя кислоту из Синтеза 1b и амин из Синтеза 18b''.
Элементарный микроанализ: (теоретический %: измеренный)
%С=67.54: 67.16; %Н=5.14: 5.05; %N=9.85: 9.27.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C32H29ClN4O4
[М+Н]+ рассчитано 569,1955,
[М+Н]+ измерено 569,1950.
Пример 34: 18-хлор-11-(4-гидроксифенил)-14,15-диметил-6,7,10,11,23,24-гексагидро-9Н, 21Н-1,22-метано-13,16-(метено)дибензо[m,s][1,4,7,11,16]диоксатриазациклоикозин-12,21(15Н)-дион

Стадия А: трет-бутил 5-(2-{2-[({5-[2-(трет-бутоксикарбонил)-5-хлофенил]-1,2-диметил-1Н-пиррол-3-ил}карбонил)(4-{[трет-бутил(диметил)силил]окси}фенил)амино]этокси}этокси)-3,4-дигидроизохинолин-2(1Н)-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Примера 14, используя кислоту из Синтеза 1b и амин из Синтеза 19b''.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 7.7 (d, 1 Н), 7.5 (dd, 1 Н), 7.11 (d, 1 Н), 7.05 (m, 2 Н), 6.8 (t, 1 Н), 6.7 (d, 2 Н), 6.65 (m, 2 Н), 4.85 (s, 1 Н), 4.42 (s, 2 Н), 4.05 (m, 2 Н), 3.87 (m, 2 Н), 3.7 (m, 2 Н), 3.65 (m, 2 Н), 3.45 (m, 2 Н), 3.15 (s, 3 Н), 2.6 (m, 2 Н), 2.4 (s, 3 Н), 1.4-0.8 (s, 27 Н), 0.1 (s, 6 Н).
Стадия В: 18-хлор-11-(4-гидроксифенил)-14,15-диметил-6,7,10,11,23,24-гексагидро-9Н, 21Н-1,22-метано-13,16-(метено)дибензо[m,s][1,4,7,11,16]диоксатриазациклоикозин-12,21(15Н)-дион
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии D Примера 17, используя соединение из предыдущей стадии.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C33H32ClN3O5
[М+Н]+ рассчитано 586.2103,
[М+Н]+ измерено 586.2124.
Пример 35: 21-хлор-29-(4-гидроксифенил)-25,26-диметил-17,28-диоксо-2,3,4,16,25,29-гексаазагептацикло[28.3.1.1~2,5~.1-12,16~.1-24,27~.0-8,13~.0-18,23~]гептатриаконта-1(34), 3,5(37),8,10,12,18,20,22,24(35),26,30,32-тридекаен-32-карбонитрил

Указанное в заголовке соединение получают в соответствии со способом из Примера 34, используя кислоту из Синтеза 1b и амин из Синтеза 20b''.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C40H32ClN7O3
[М+Н]+ рассчитано 694.2328,
[М+Н]+ измерено 694.233.
Пример 36: 10-хлор-29-(4-гидроксифенил)-5,6-диметил-23,26,29-триоксо-2,6,15,31,34-пентаазагексацикло[28.3.1.1~4,7~.1~15,18~.0-8,13~.0-8,13~.0~17,22~]гексатриаконта-1(32),4,7(36),8,10,12,17,19,21,30,33-ундекаен-3,14-дион

Стадия А: трет-бутил 4-(2-{2-[(5-{({5-[2-(трет-бутоксикарбонил)-5-хлофенил]-1,2-диметил-1Н-пиррол-3-ил}карбонил)[4-(проп-2-ен-1-илокси)фенил]амино}пиримидин-2-ил)окси]этокси}этокси)-1,3-дигидро-2Н-изоиндол-2-карбоксилат
Указанное в заголовке соединение получают в соответствии со способом, описанным в Стадии В Примера 14, используя кислоту из Синтеза 1b и амин из Синтеза 21b''.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част. на млн.: 8.46/8.45 (s, 2 Н), 7.77 (d, 1 Н), 7.54 (dd, 1 Н), 7.26-6.83 (m, 3 Н), 7.21 (m, 2 Н), 7.08 (d, 1 Н), 6.92 (m, 2 Н), 5.99 (m, 1 Н), 5.33/5.2 (m+m, 2 Н), 5.23/5.22 (s, 1 Н), 4.6-4.41 (m, 4 Н), 4.54 (m, 2 Н), 4.45-3.75 (m, 8 Н), 3.15 (s, 3 Н), 2.43 (s, 3 Н), 1.46-1.25 (s, 18 Н).
Стадия В: 4-хлор-2-(4-{(2-{2-[2-(2,3-дигидро-1Н-изоиндол-4-илокси)этокси]этокси}пиримидин-5-ил)[4-(проп-2-ен-1-илокси)фенил]карбамоил}-1,5-диметил-1Н-пиррол-2-ил)бензойная кислота
К раствору соединения, полученного в Стадии А (1,2 г; 1,36 ммоль) в диоксане (18 мл), добавляют триэтиламин (1,9 мл; 13,6 ммоль) и трифторметансульфонат триметилсилила (2,46 мл; 13,6 ммоль). Реакционную среду перемешивают при температуре окружающей среды в течение 20 минут, затем выливают в ледяную воду. Полученный осадок фильтруют, затем очищают с помощью хроматографии на фазе RP-18, используя ацетонитрил, воду и ацетат аммония в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
1H ЯМР (500 МГц, ДМСО-d6, 300 K) δ част, на млн.: 10.98 (brs, 2 Н), 8.36 (s, 2 Н), 7.58 (d, 1 Н), 7.3 (dd, 1 Н), 7.28 (t, 1 Н), 7.16 (m, 2 Н), 6.97 (d, 1 Н), 6.95 (m, 2 Н), 6.95 (d, 1 Н), 6.92 (d, 1 Н), 6.03 (m, 1 Н), 5.41 (s, 1 Н), 5.38/5.25 (m+m, 2 Н), 4.57 (m, 2 Н), 4.41 (brs, 2 Н), 4.4-3.72 (m, 8 Н), 4.33 (brs, 2 Н), 3.15 (s, 3 Н), 2.31 (s, 3 Н).
Стадия С: 10-хлор-2-(4-гидроксифенил)-5,6-диметил-23,26,29-триокса-2,6,15,31,34-пентаазагексацикло[28.2.2.1~4,7~.1~15,18~.0~8,13~.0~17,22~]гексатриаконта-1(32),4,7(36),8,10,12,17,19,21,30,33-ундекаен-3,14-дион
К раствору соединения, полученного в Стадии В (370 мг; 0,61 ммоль) в дихлорметане (50 мл), добавляют тетпафторборат 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метил-морфолин-4-ия (237 мг; 0,7 ммоль). Реакционную среду перемешивают при температуре окружающей среды в течение 1 часа. После промывания водой органическую фазу сушат над сульфатом магния, фильтруют и концентрируют.
К раствору полученного таким образом остатка в смеси дихлорметана (10 мл) и метанола (5 мл), добавляют 1,3-диметилбарбитуровую кислоту (0,159 г; 1 ммоль). Реакционную среду дегазируют аргоном в течение 10 минут, и добавляют тетракис-(трифенилфосфин)палладий(0) (0,030 г; 0,02 ммоль). Смесь затем перемешивают в течение 16 часов при температуре окружающей среды. После концентрирования растворителей, остаток очищают с помощью хроматографии на фазе RP-18, используя ацетонитрил, воду и ацетат аммония в качестве растворителей для элюирования, чтобы получить указанный в заголовке продукт.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C36H32ClN5O6
[М+Н]+ рассчитано 666.2198,
[М+Н]+ измерено 666.2108.
Пример 37: 10-фтор-2-(4-гидроксифенил)-5,6-диметил-24,27,30-триокса-2,6,15,32,35-пентаазагексацикло[29.2.2.1~4,7~.1~15,19~.0~8,13~.0~18,23~]гептатриаконта-1(33),4,7(37),8,10,12,18,20,22,31,34-ундекаен-3,14-дион

Указанное в заголовке соединение получают в соответствии со способом из Примера 36, используя кислоту из Синтеза 2b и амин из Синтеза 22b''.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C37H34FN5O6
[М+Н]+ рассчитано 664.2559,
[М+Н]+ измерено 664.2571.
Пример 38: 10-хлор-2-(4-гидроксифенил)-5,6-диметил-24,27,30-триокса-2,6,15,32,35-пентаазагексацикло[29.2.2.1~4,7~.1~15,19~.0~8,13~.0~18,23~]гептатриаконта-1(33),4,7(37),8,10,12,18,20,22,31,34-ундекаен-3,14-дион

Указанное в заголовке соединение получают в соответствии со способом из Примера 36, используя кислоту из Синтеза 1b и амин из Синтеза 22b''.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C37H34ClN5O6
[М+Н]+ рассчитано 680.2276,
[М+Н]+ измерено 680.2280.
Пример 39: 10-хлор-2-(4-гидроксифенил)-5,6-диметил-24,31-диокса-2,6,15,33,36-пентаазагексацикло[29.2.2.1~4,7~.1~15,19~.0~8,13~.0~18,23~]октатриаконта-1(34),4,7(38),8,10,12,18,20,22,32,35-ундекаен-3,14-дион
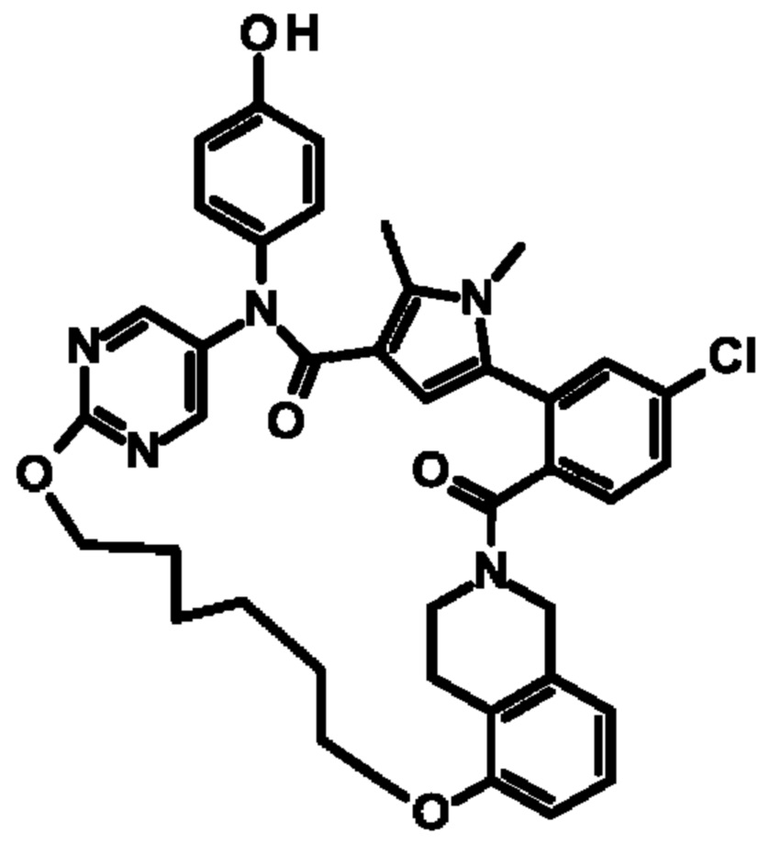
Указанное в заголовке соединение получают в соответствии со способом из Примера 38, используя амин из Синтеза 23b''.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C39H38ClN5O5
[М+Н]+ рассчитано 692.2640,
[М+Н]+ измерено 692.2646.
Пример 40: 10-хлор-2-(4-гидроксифенил)-5,6-диметил-24,30-диокса-2,6,15,32,35-пентаазагексацикло[29.2.2.1~4,7~.1~15,19~.0~8,13~.0~18,23~]гептатриаконта-1(33),4,7(37),8,10,12,18,20,22,31,34-ундекаен-3,14-дион

Указанное в заголовке соединение получают в соответствии со способом из Примера 38, используя амин из Синтеза 24b''.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C38H36ClN5O5
[М+Н]+ рассчитано 678.2484,
[М+Н]+ измерено: 678.2488.
Пример 41: 10-хлор-2-(4-гидроксифенил)-5,6-диметил-24,29-диокса-2,6,15,31,34-пентаазагексацикло[29.2.2.1~4,7~.1~15,19~.0~8,13~.0~18,23~]гептатриаконта-1(32),4,7(36),8,10,12,18,20,22,30,33-ундекаен-3,14-дион

Указанное в заголовке соединение получают в соответствии со способом из Примера 38, используя амин из Синтеза 25b''.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C37H34ClN5O5
[М+Н]+ рассчитано: 664.2328
[М+Н]+ измерено: 664.2331
Пример 42: 10-хлор-2-(4-гидроксифенил)-5,6-диметил-21,24,27-триокса-2,6,15,29,32-пентаазапентацикло[26.2.2.1~4,7~.1~15,19~.0~8,13~]тетратриаконта-1(30),4,7(34),8,10,12,28,31-октаен-3,14-дион

Указанное в заголовке соединение получают в соответствии со способом из Примера 38, используя амин из Синтеза 26b''.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C34H36ClN5O6
[М+Н]+ рассчитано: 646.2433
[М+Н]+ измерено: 646.2427.
Пример 43: 6-хлор-14-(4-гидроксифенил)-10,11-диметил-19,22,25-триокса-1,10,14,17,32-пентаазапентацикло[25.2.2.2~15,18~.1~9,12~.0~3,8~]тетратриаконта-3,5,7,9(34),11,15,17,32-октаен-2,13-дион
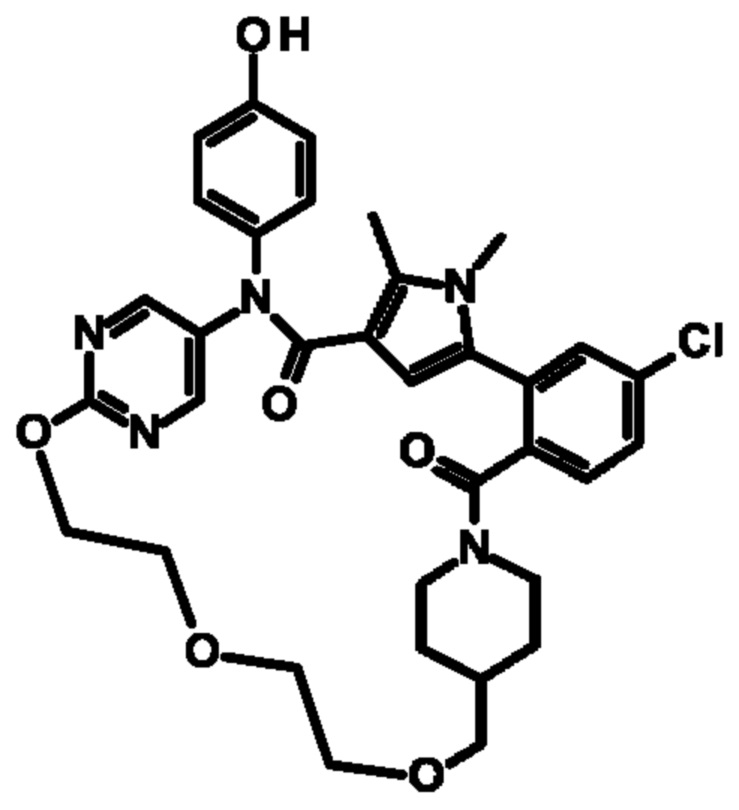
Указанное в заголовке соединение получают в соответствии со способом из Примера 38, используя амин из Синтеза 27b''.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C34H36ClN5O6
[М+Н]+ рассчитано: 646.2433
[М+Н]+ измерено: 646.2426.
Пример 44: 10-хлор-2-(4-гидроксифенил)-5,6,27-триметил-24,30-диокса-2,6,15,27,32,35-гексаазагексацикло[29.2.2.1~4,7~.1~15,19~.0~8,13~.0~18,23~]гептатриаконта-1(33),4,7(37),8,10,12,18,20,22,31,34-ундекаен-3,14-дион

Указанное в заголовке соединение получают в соответствии со способом из Примера 38, используя амин из Синтеза 28b''.
Масс-спектрометрия высокого разрешения (ЭРИ+):
Эмпирическая формула: C38H37ClN6O5
[М+Н]+ рассчитано: 693.2593
[М+Н]+ измерено: 693.2583.
Фармакологическое исследование
ПРИМЕР А: Ингибирование Bcl-2 методом флуоресцентной поляризации
Флуоресцентные поляризационные тесты проводили на микропланшетах (384 лунки). Меченный белок Bcl-2 (histag-Bcl-2, такой, что Bcl-2 соответствует первичному порядковому номеру UniProtKB® Р10415), в конечной концентрации 5×10-9 М, смешивают с флуоресцентным пептидом (флуоресцеин-REIGAQLRRMADDLNAQY), в конечной концентрации 1,00×10-9 М в буферном растворе (Hepes 10 мМ, NaCl 150 мМ, Tween20 0,05%, рН 7,4), в присутствии или в отсутствие возрастающих концентраций исследуемых соединений. После инкубации в течение 2 часов измеряют поляризацию флуоресценции.
Результаты выражены в IC50 (концентрация соединения, которое на 50% ингибирует поляризацию флуоресценции) и представлены в таблице 1 ниже.
Результаты показывают, что соединения в соответствии с изобретением ингибируют взаимодействие между белком Bcl-2 и описанным выше флуоресцентным пептидом.
ПРИМЕР В: Цитотоксичность in vitro.
Исследования цитотоксичности проводили на линии опухолей лейкемии RS4; 11.
Клетки распределяют по микропланшетам и подвергают воздействию исследуемых соединений в течение 48 часов. Затем жизнеспособность клеток определяют количественно колориметрическим анализом, анализом на микрокультуру тетразолия (Cancer Res., 1987, 47, 939-942).
Результаты выражены в IC50 (концентрация соединения, которое ингибирует жизнеспособность клеток на 50%) и представлены в таблице 1 ниже.
Результаты показывают, что соединения по изобретению являются цитотоксичными.


ПРИМЕР С: Количественная оценка расщепленной формы каспазы 3 in vivo.
Способность соединений в соответствии с изобретением активировать каспазу 3 оценивают на модели ксенотрансплантата клеток лейкемии RS4; 11.
1.107 клеток RS4; 11 подкожно прививают мышам с иммуносупрессией (штамм SCID). Через 25-30 дней после подсадки животных перорально обрабатывают различными соединениями. Через 2 часа обработки опухолевые массы извлекают и лизируют, и расщепленную форму (активированную) каспазы 3 количественно определяют в лизатах опухоли.
Это количественное определение проводят с использованием анализа «Meso Scale Discovery (MSD) ELISA platform», в котором конкретно анализируется расщепленная форма каспазы 3. Она выражается в форме фактора активации, соответствующего соотношению между количеством расщепленной каспазы 3 у обработанных мышей, разделенным на количество расщепленной каспазы 3 у контрольных мышей.
Результаты показывают, что соединения в соответствии с изобретением способны индуцировать апоптоз в опухолевых клетках RS4; 11 in vivo.

ПРИМЕР D: Фармацевтическая композиция: таблетки
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ИНДОЛИЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2693629C2 |
| НОВЫЕ ИНДОЛЬНЫЕ И ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2693404C2 |
| НОВЫЕ ФОСФАТНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2617682C2 |
| НОВЫЕ ПИРРОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2607788C2 |
| НОВЫЕ ИНДОЛИЗИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2013 |
|
RU2646223C2 |
| ФЕНИКОЛОВЫЕ ПРОТИВОБАКТЕРИАЛЬНЫЕ СРЕДСТВА | 2013 |
|
RU2593204C2 |
| Композиции для лечения легочного фиброза | 2017 |
|
RU2747801C2 |
| НОВЫЙ СПОСОБ СИНТЕЗА АГОМЕЛАТИНА | 2014 |
|
RU2683279C1 |
| Соединения формул (I) и (A), фармацевтическая композиция, лекарственное средство, применение и способ получения соединения формулы (I) | 2018 |
|
RU2822758C2 |
| НОВЫЕ ИЗОИНДОЛИНОВЫЕ ИЛИ ИЗОХИНОЛИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2689305C2 |

Изобретение относится к соединению формулы (I), фармацевтической композиции, а также применению соединения формулы (I) для изготовления лекарственного средства для лечения злокачественного новообразования, опосредованного Bcl-2. Технический результат: получены соединения, которые обладают ингибирующей активностью в отношении белка Bcl-2 и могут быть применимы для лечения злокачественного новообразования, опосредованного Bcl-2. 5 н. и 10 з.п. ф-лы, 2 табл., 48 пр., 1 ил.

1. Соединение формулы (I):

в которой:
♦ A1 и A2 каждый представляют собой метальную группу,
♦ G представляет собой группу -NR7-, группу 1,2,3,4-тетрагидроизохинолинилена, необязательно замещенного группой Т, группу 2,3-дигидро-1H-изоиндолилена, необязательно замещенного группой Т, или группу пиперидинилена,
♦ Т представляет собой атом водорода, линейный (С1-С6)алкил или группу (C1-C4)алкил-NR1R2,
♦ X представляет собой группу (С2-С8)алкилен, в которой от 1 до 3 атомов углерода могут быть заменены гетероатомом, выбранным из кислорода, серы и N-R5, или группой 5-6-членного гетероарилена, содержащего от 1 до 3 атомов азота,
♦ Y представляет собой группу СН2- или СО-,
♦ R1 и R2 с атомом азота, который несет их, образуют 6-членный гетероциклоалкил, содержащий 2 гетероатома, выбранных из N и О,
♦ R3 и R4 являются следующими:
- в Варианте 1: R4 представляет собой фенильную группу следующей формулы:
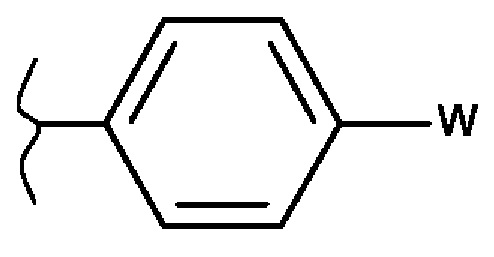
в которой W представляет собой гидроксигруппу или фосфатную группу, которая представляет собой -OPO(О-M1+)(О-M2+), где M1+ и М2+ независимо друг от друга представляют собой фармацевтически приемлемый одновалентный катион,
и R3 представляет собой фенилен, необязательно замещенный циано, 5-6-членный гетероарилен, содержащий от 1 до 2 атомов азота, необязательно замещенный 1-2 заместителями, выбранными из (С1-С6)алкильной, циано, тетрагидрофуранильной группы и оксогруппы; или линейную или разветвленную (С1-С6)-алкиленовую группу,
- в Варианте 2: R3 представляет собой фениленовую группу следующей формулы:
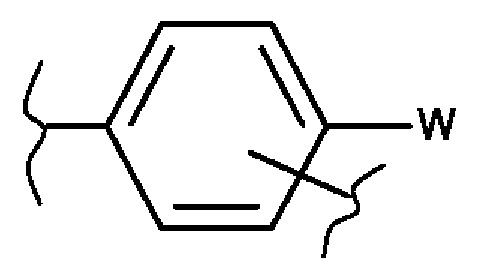
в которой W представляет собой гидроксигруппу или фосфатную группу, которая представляет собой -OPO(О-M1+)(О-M2+), где M1+ и М2+ независимо друг от друга представляют собой фармацевтически приемлемый одновалентный катион,
и R4 представляет собой 5-членный гетероарил, содержащий 1 атом азота, необязательно замещенный 1-3 заместителями, выбранными из (C1-С6)алкила и циано,
♦ R5 представляет собой атом водорода или линейную (С1-С6)алкильную группу,
♦ R6 и R7 независимо друг от друга представляют собой атом водорода или линейную (С1-С6)алкильную группу,
♦ Ra, Rc и Rd каждый представляют собой атом водорода, и Rb представляет собой атом водорода или атом галогена,
его энантиомеры, или его соли присоединения к фармацевтически приемлемой кислоте или основанию.
2. Соединение формулы (I) по п. 1, в котором Y представляет собой группу -СО-.
3. Соединение формулы (I) по п. 1 или 2, в котором G представляет собой группу, выбранную из следующих групп:


где Т представляет собой группу метила или группу (4-морфолинил)метила.
4. Соединение формулы (I) по одному из пп. 1-3, в котором X представляет собой группу, выбранную из следующих групп:


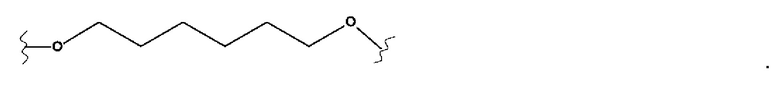
5. Соединение формулы (I) по одному из пп. 1-4, в котором:
в Варианте 1: R4 представляет собой группу 4-гидроксифенила, тогда как R3 представляет собой группу, выбранную из нижеследующего списка:
- фениленовая группа, необязательно замещенная цианогруппой,
- пиразолиленовая группа,
- 1-метил-1Н-пиразолиленовая группа,
- 1-(тетрагидрофуран-3-ил)-1Н-пиразолиленовая группа,
- 5-метил-2-циано-1H-пирролиленовая группа,
- 1-метил-2-циано-1H-пирролиленовая группа,
- 1,2-диметил-1H-пирролиленовая группа,
- 1,5-диметил-2-циано-1H-пирролиленовая группа,
- пиримидиниленовая группа,
- этиленовая группа,
- пиримидиниевая группа;
в Варианте 2: R4 представляет собой 1,5-диметил-2-циано-1Н-пирролиленовую группу, a R3 представляет собой 4-гидроксифениленовую группу.
6. Соединение формулы (I) по п. 5, в котором R4 представляет собой 4-гидроксифенильную группу.
7. Соединение формулы (I) по п. 5, в котором R3 представляет собой 4-гидроксифениленовую группу.
8. Соединение формулы (I) по п. 7, в котором R3 представляет собой 4-гидроксифениленовую группу и G представляет собой пиперидиниленовую группу.
9. Соединение формулы (I) по п. 1, выбранное из следующей группы:
- 6-хлор-14-(4-гидроксифенил)-10,11-диметил-2,13-диоксо-20,23-диокса-1,10,14-триазагексацикло[26.3.1.1~9,12~.1~15,19~.0~3,8~.0~24,~]тетратриаконта-3,5,7,9(34),11,15(33),16,18,24,26,28-ундекаен-17-карбонитрил,
- 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-4,16,17,23,24,25-гексагидро-1Н,14Н-15,18-метано-6,9-(метено)дибензо[b,h]пиразоло[4,3-р][1,6,11,15]оксатриазациклоикозин-5,14(8Н)-дион,
- 6-хлор-14-(4-гидроксифенил)-10,11-диметил-2,13-диоксо-23-окса-1,10,14-триазагексацикло[26.3.1.1-9,12-1-15,19-0-3,8-0-24,29-]тетратриаконта-3,5,7,9(34),11,15(33),16,18,24,26,28-ундекаен-17-карбонитрил,
- 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-5,14-диоксо-4,5,8,16,17,23,24,25-октагидро-1Н,14Н-15,18-метано-6,9-(метено)дибензо[b,h]пирроло[3,2-р][1,6,11,15]оксатриазациклоикозин-2-карбонитрил,
- 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-5,14-диоксо-1,4,5,8,16,17,23,24-октагидро-14Н-15,18-метано-6,9-(метено)дибензо[l,r]пирроло[2,3-(1][1,6,10,15]оксатриазациклононадецин-2-карбонитрил,
- 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-1,4,16,21,22,23-гексагидро-14Н-15,21-метано-6,9-(метено)дибензо[j,о]пиразоло[3,4-b][1,4,8,13]оксатриазациклононадецин-5,14(8Н)-дион,
- (16S или R)-11-хлор-4-(4-гидроксифенил)-1,7,8,16-тетраметил-5,14-диоксо-4,5,8,16,17,23,24,25-октагидро-1Н,14Н-15,18-метано-6,9-(метено)дибензо[b,h]пирроло[3,2-р][1,6,11,15]оксатриазациклоикозин-2-карбонитрил,
- 4-[16-хлор-3-гидрокси-19,20-диметил-13,22-диоксо-6,7,8,9,10,11,19,22-октагидро-13Н,23Н-8,12-метано-21,18-(метено)дибензо[b,j][1,4,8,13]оксатриазациклононадецин-23-ил]-1,5-диметил-1Н-пиррол-2-карбонитрил,
- 10-фтор-2-(4-гидроксифенил)-5,6-диметил-24,27,30-триокса-2,6,15,32,35-пентаазагексацикло[29.2.2.1~4,7~.1~15,19~.0~8,13~.0~18,23~]гептатриаконта-1(33),4,7(37),8,10,12,18,20,22,31,34-ундекаен-3,14-дион,
- 10-хлор-2-(4-гидроксифенил)-5,6-диметил-24,30-диокса-2,6,15,32,35-пентаазагексацикло[29.2.2.1~4,7~.1-15,19~.0-8,13-.0-18,23-]гептатриаконта-1(33),4,7(37),8,10,12,18,20,22,31,34-ундекаен-3,14-дион,
- 10-хлор-2-(4-гидроксифенил)-5,6,27-триметил-24,30-диокса-2,6,15,27,32,35-гексаазагексацикло[29.2.2.1~4,7~.1-15,19-.0-8,13~.0~18,23~]гептатриаконта-1(33),4,7(37),8,10,12,18,20,22,31,34-ундекаен-3,14-дион,
- 11-хлор-4-(4-гидроксифенил)-1,7,8-триметил-1,4,16,17,23,24-гексагидро-14Н-15,18-метано-6,9-(метено)дибензо[l,r]пиразоло[3,4-d][1,6,10,15]оксатриазациклононадецин-5,14(8Н)-дион.
10. Способ получения соединения формулы (I) по п. 1, отличающийся тем, что в качестве исходного продукта используют соединение формулы (II):

в котором A1, А2, Ra, Rb, Rc, Rd, Y и G имеют то же самое значение, как и в формуле (I), определенное в п. 1, и Alk представляет собой линейную или разветвленную (С1-С6)алкильную группу,
сложноэфирная функция -OAlk соединения формулы (II) гидролизуется с образованием соответствующей карбоновой кислоты или карбоксилата, которые могут быть превращены в соответствующий ацилхлорид или ангидрид, перед сочетанием с амином NHR3AR4, где R4 имеет то же значение, что и в формуле (I), a R3A представляет собой:
- группу R3, как определено в формуле (I)
- или группу R3-O-Alk'-Z, R3-Alk'-Z или R3-Z, где Alk' представляет собой линейную или разветвленную (С1-С6)алкильную группу, и Z представляет собой атом галогена или группу -ОН,
чтобы образовать соединение формулы (III):

в котором A1, А2, Ra, Rb, Rc, Rd, R4, Y и G имеют то же самое значение, как и в формуле (I),
которое подвергают реакции снятия защиты со спиртовой функции с последующим внутримолекулярным нуклеофильным замещением или реакцией Мицунобу или ароматическим нуклеофильным замещением,
чтобы получить соединение формулы (I),
где соединение формулы (I) может быть очищено в соответствии с обычной методикой разделения, которое может быть преобразовано в его соли присоединения к фармацевтически приемлемой кислоте или основанию и которое необязательно разделяют на его изомеры в соответствии с обычной методикой разделения,
при условии, что в любое время, признанное подходящим в ходе описанного выше способа, гидрокси- и аминогруппы реагентов или промежуточные соединения синтеза могут быть защищены, а затем с них может быть снята защита для целей синтеза.
11. Способ получения соединения формулы (I) по п. 1, отличающийся тем, что в качестве исходного продукта используют соединение формулы (IV):
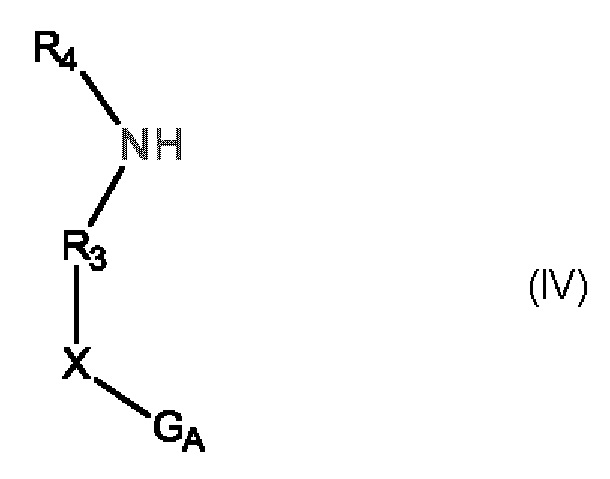
в которой R3, R4 и X имеют то же самое значение, как и в формуле (I), и GA представляет собой группу, выбранную из следующего списка:


где соединение формулы (IV) затем сочетают с соединением формулы (V):

в которой A1, А2, Ra, Rb, Rc, и Rd имеют то же самое значение, как и в формуле (I),
чтобы получить соединение формулы (VI):

в котором A1, А2, Ra, Rb, Rc, Rd, R3, R4 и X имеют то же самое значение, как и в формуле (I),
которое подвергают реакции снятия защиты с последующим внутримолекулярным сочетанием, чтобы получить соединение формулы (I),
где соединение формулы (I) может быть очищено в соответствии с обычной методикой разделения, которое может быть преобразовано в его соли присоединения к фармацевтически приемлемой кислоте или основанию и которое необязательно разделяют на его изомеры в соответствии с обычной методикой разделения,
при условии, что в любое время, признанное подходящим в ходе описанного выше способа, гидрокси- и аминогруппы реагентов или промежуточные соединения синтеза могут быть защищены, а затем с них может быть снята защита для целей синтеза.
12. Фармацевтическая композиция, ингибирующая
антиапоптотическую активность белка Bcl-2 и содержащая соединение формулы (I) по любому из пп. 1-9, или его соль присоединения к фармацевтически приемлемой кислоте или основанию, в сочетании с одним или несколькими фармацевтически приемлемыми наполнителями.
13. Фармацевтическая композиция по п. 12 для применения в лечении злокачественного новообразования, опосредованного Bcl-2.
14. Фармацевтическая композиция по п. 13, причем злокачественное новообразование, опосредованное Bcl-2, выбирают из следующего списка: рак мочевого пузыря, головного мозга, молочной железы, матки, хронический лимфоцитарный лейкоз, рак прямой кишки, рак пищевода, печени, лимфобластный лейкоз, неходжкинская лимфома, меланома, злокачественная гемопатия, миелома, рак яичников, немелкоклеточный рак легкого, рак предстательной железы и мелкоклеточный рак легкого.
15. Применение соединения формулы (I) по любому из пп. 1-9, для изготовления лекарственного средства для лечения злокачественного новообразования, опосредованного Bcl-2, выбранного из следующего списка: рак мочевого пузыря, головного мозга, молочной железы, матки, хронический лимфоцитарный лейкоз, рак прямой кишки, рак пищевода, печени, лимфобластный лейкоз, неходжкинская лимфома, меланома, злокачественная гемопатия, миелома, рак яичников, немелкоклеточный рак легкого, рак предстательной железы и мелкоклеточный рак легкого.
| WO2014022752A1, 06.02.2014 | |||
| RU2016105999A, 28.08.2017 | |||
| МАКРОЦИКЛИЧЕСКИЕ АМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕАЗЫ | 2013 |
|
RU2625796C2 |
