В общем, настоящее изобретение относится к применению соединений для получения композиций, применяемых для лечения рака.
В большей части видов рака смертность обусловлена не первичной опухолью, а скорее вторичными метастазами. Данное злокачественное развитие, которое приводит к инвазии опухоли и клинически определяется появлением метастазов, представляет собой окончательный результат первичной потери клеточной адгезии и повышения клеточной подвижности, которые вместе позволяют инвазивной клетке покидать исходное место опухоли и колонизировать разные ткани-мишени.
Метастазы рассматривают, как признак повторно возникающего неконтролируемого злокачественного развития рака. Во время данного процесса опухолевые клетки завершают свою злокачественную трансформацию путем повышения своей способности к миграции. Раковые клетки могут затем распространяться и образовывать опухолевые очаги в отдаленных местах. Распространение раковых клеток в организме представляет собой результат ряда событий, называемого "метастатическим каскадом": инвазия тканей вокруг опухоли, венозная или лимфатическая интравазация, миграция и установление в удаленном месте новой колонии, которая устойчива ко всем защитным механизмам организма.
Метастатическая инвазия, против которой не существует эффективного возможного способа лечения, доступного в настоящее время, представляет собой бесспорно главную причину смерти. В связи с частотой видов рака, диагностируемых на метастатической стадии, и безвыходным положением с терапевтической точки зрения, которое они представляют, разработка молекул, которые специфично нацелены на метастатическую инвазию, таким образом, представляет собой крайне важное требование к качественному изменению в способах лечения рака.
В документе WO 2009/087238 описаны соединения, которые могут быть применимы для лечения рака. Как видно ниже в данном документе, в примере 17 приведены сравнительные данные, где близкое соединение, как описано в указанном документе, неожиданно является менее активным в тесте инвазии, чем заявленное соединение.
В настоящее время обнаружили, что производные формулы (I), как определено ниже в данном документе, способны предотвращать, как проиллюстрировано в экспериментальных данных ниже в данном документе, инвазивное развитие метастатических видов рака, и на основе такой активности соединения применимы для лечении рака.
Вследствие этого, настоящее изобретение относится к соединениям формулы (I) и их фармацевтически приемлемым солям как таковым, как определено ниже.
Кроме того, настоящее изобретение относится к соединениям формулы (I), как определено ниже, для применения в качестве лекарственных средств и более конкретно для применения для предупреждения и/или ингибирования, и/или лечения рака.
Кроме того, настоящее изобретение относится к способу предупреждения, ингибирования или лечения рака, который включает по меньшей мере одну стадию, заключающуюся во введении пациенту, нуждающемуся в этом, эффективного количества соединения, как определено ниже в формуле (I), или одной из его фармацевтически приемлемых солей.
Настоящее изобретение дополнительно относится к способу получения указанных соединений формулы (I).
Согласно настоящему изобретению также предложены фармацевтические композиции, содержащие по меньшей мере одно из указанных соединений формулы (I).
Согласно одному аспекту предмет настоящего изобретения относится к соединению формулы (I):
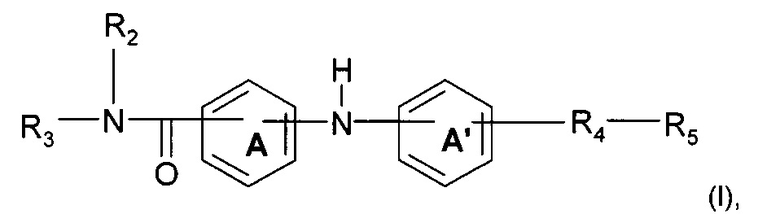
где А и А' независимо представляют собой фениленовую группу или пиридиленовую группу;
R2 представляет собой атом водорода или алкильную группу (C1-C4);
R3 представляет собой 2-пиридильную группу, 3-пиридильную группу, 4-пиридильную группу, 2-пиримидинильную группу, 4-пиримидинильную группу или 5-пиримидинильную группу;
R4 представляет собой карбонильную группу или сульфонильную группу; и
R5 представляет собой группу NH-(CH2)a-NR6R7 или 4-метилпиперазинильную группу, причем а представляет собой целое число от 1 до 4, R6 и R7 независимо представляют собой алкильную группу (С2-С4) или R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероциклическую группу, которую выбирают из 4-метилпиперазинильной группы, морфолиногруппы, пирролидинильной группы и пиперидиногруппы;
или любой из его фармацевтически приемлемых солей.
Согласно предпочтительному воплощению настоящее изобретение относится к соединению формулы (I), где группа -NH- между А и A' и группа -R4-R5 находятся в мета-положении друг от друга относительно A'.
Согласно предпочтительному воплощению настоящее изобретение относится к соединению формулы (А1):
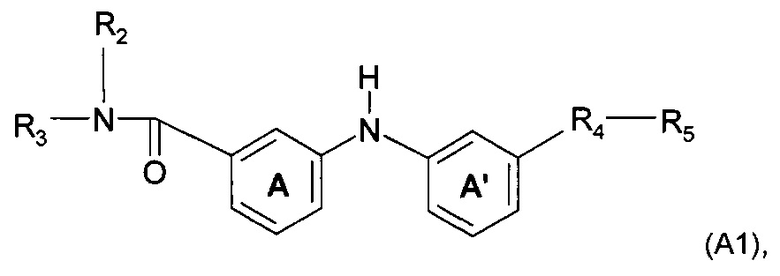
где A, A', R2, R3, R4 и R5 являются такими, как определено выше.
Настоящее изобретение охватывает воплощения, которые описаны ниже в данном документе, где положения замещающих групп в положениях А и A' соответствуют структуре формулы (А1), как описано выше, то есть мета-положению в отношении А и мета-положению в отношении A'.
Согласно другому предпочтительному аспекту настоящее изобретение относится к соединению формулы (I), как определено выше, где
А и A' независимо представляют собой фениленовую группу или пиридиленовую группу;
R2 представляет собой атом водорода или метильную группу;
R3 представляет собой 2-пиридильную группу, 4-пиридильную группу или 4-пиримидинильную группу;
R4 представляет собой карбонильную группу или сульфонильную группу; и
R5 представляет собой группу -NH-(CH2)a-NR6R7 или 4-метилпиперазинильную группу, причем а представляет собой целое число от 2 до 3, R6 и R7 представляют собой этильную группу или R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероциклическую группу, которую выбирают из 4-метилпиперазильной группы, морфолиногруппы, пирролидинильной группы и пиперидиногруппы;
или любой из его фармацевтически приемлемых солей.
Согласно более предпочтительному аспекту настоящее изобретение относится к соединению формулы (I'):
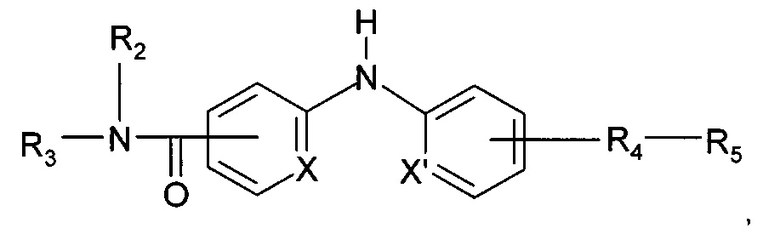
где X и X' независимо представляют собой СН или N;
R2 представляет собой атом водорода или метильную группу;
R3 представляет собой 2-пиридильную группу, 4-пиридильную группу или 4-пиримидинильную группу;
R4 представляет собой карбонильную группу или сульфонильную группу; и
R5 представляет собой группу -NH-(CH2)a-NR6R7 или 4-метилпиперазинильную группу, причем а представляет собой целое число от 2 до 3, R6 и R7 представляют собой этильную группу или R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероциклическую группу, которую выбирают из 4-метилпиперазинильной группы, морфолиногруппы, пирролидинильной группы и пиперидиногруппы;
или любой из его фармацевтически приемлемых солей.
Согласно конкретному воплощению дополнительный предмет настоящего изобретения представляет собой соединение формулы (Iа):
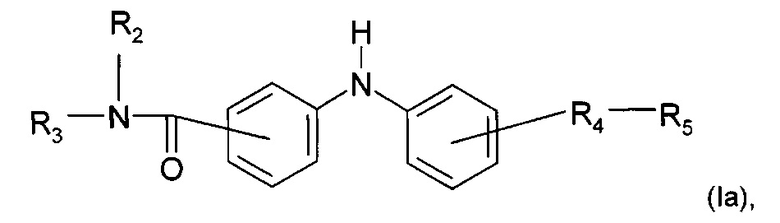
где R2, R3, R4 и R5 являются такими, как определено выше;
или любую из его фармацевтически приемлемых солей.
Согласно предпочтительному воплощению настоящее изобретение относится к соединению формулы (Iа), где группа -R4-R5 находится в мета-положении относительно группы -NH- между двумя фенильными группами.
Согласно более предпочтительному воплощению настоящее изобретение относится к соединению формулы (Iа), как описано выше, где R4 представляет собой карбонильную группу, и R2, R3 и R5 являются такими, как определено выше,
или любой из его фармацевтически приемлемых солей.
Также описано соединение формулы (Ib):
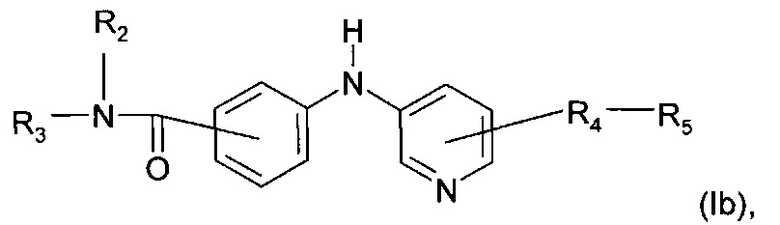
где R2, R3, R4 и R5 являются такими, как определено выше;
или любая из его фармацевтически приемлемых солей.
Согласно предпочтительному воплощению настоящее изобретение относится к соединению формулы (Ib), где группа -R4-R5 находится в мета-положение относительно группы -NH- между фенильной группой и пиридиновой группой.
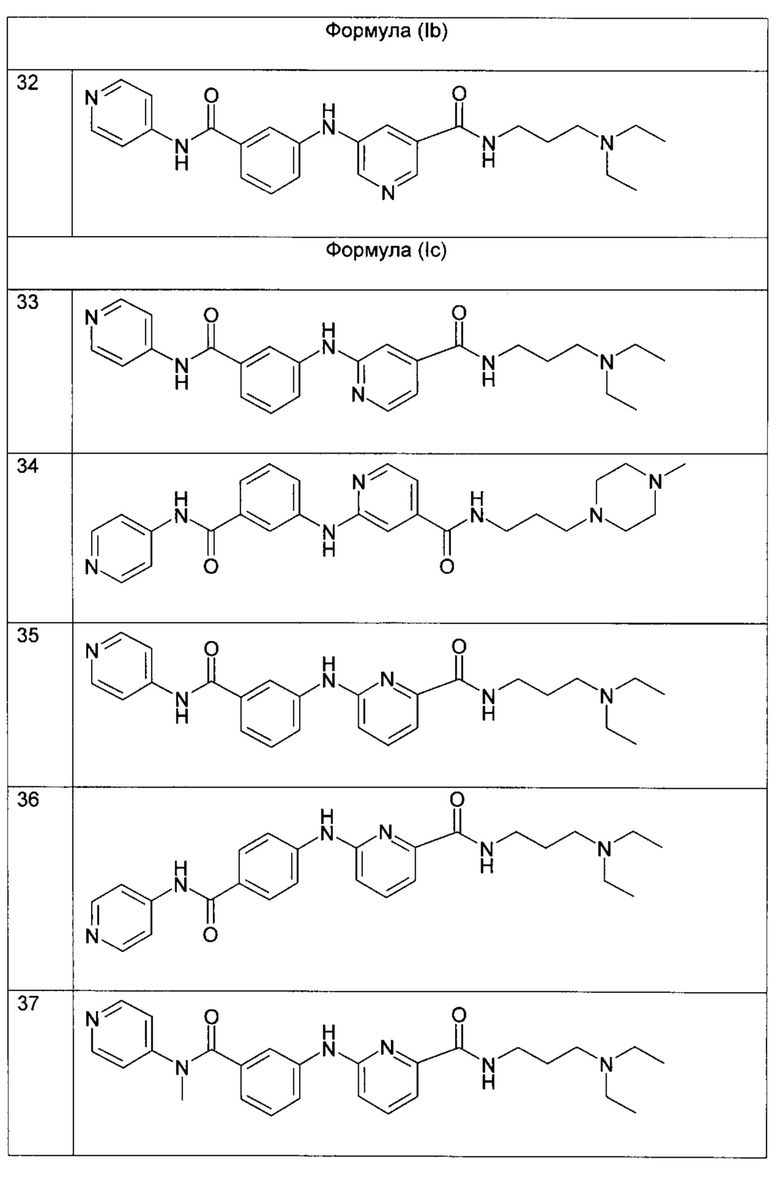
Более предпочтительно, в формуле (Ib), R2 представляет собой атом водорода; R3 представляет собой 4-пиридильную группу; R4 представляет собой карбонильную группу; и R5 представляет собой группу NH-(CH2)a-NR6R7, причем а представляет собой целое число 3, R6 и R7 представляют собой этильную группу; или любой из его фармацевтически приемлемых солей.
Согласно другому конкретному воплощению дополнительно настоящее изобретение относитися к соединению формулы (Iс):
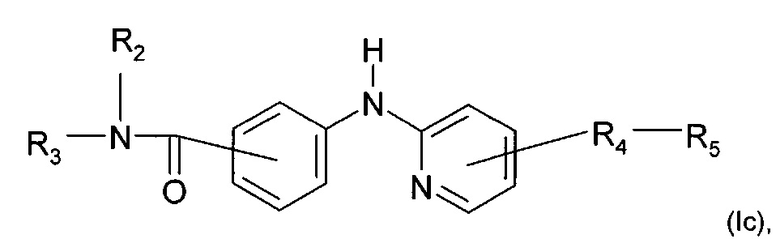
где R2, R3, R4 и R5 являются такими, как определено выше;
или любой из его фармацевтически приемлемых солей.
Согласно предпочтительному воплощению настоящее изобретение относится к соединению формулы (Iс), где группа -R4-R5 находится в мета-положении относительно группы -NH- между фенильной группой и пиридиновой группой.
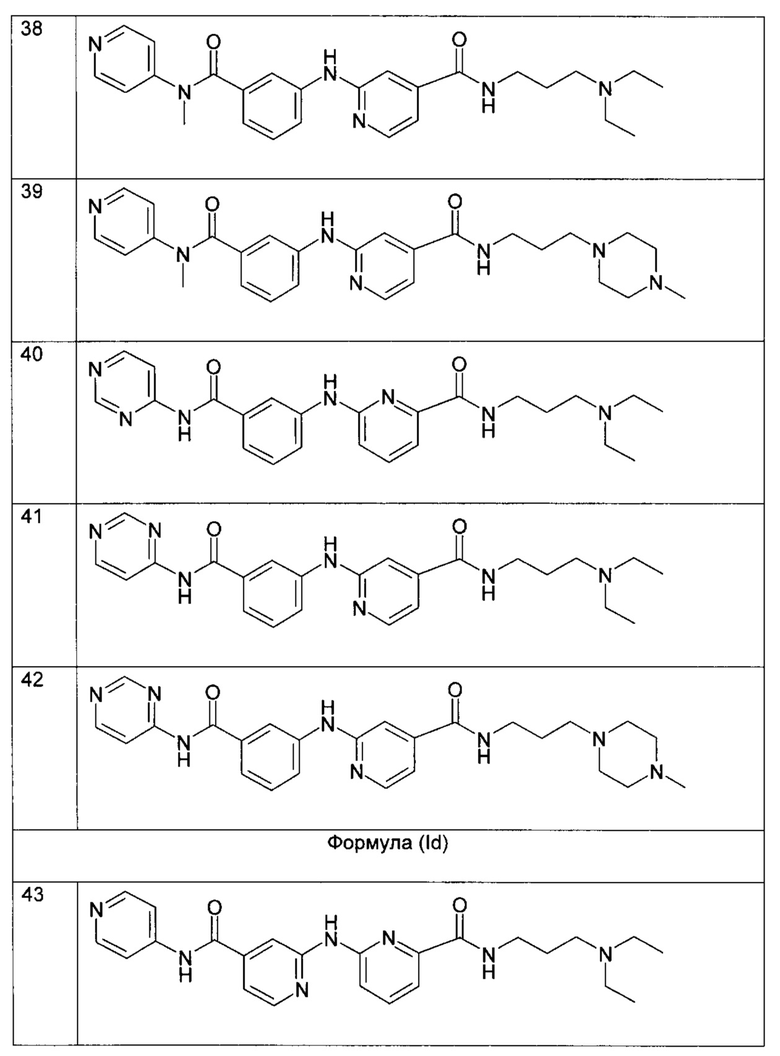
Согласно более предпочтительному воплощению настоящее изобретение относится к соединению формулы (Iс), как описано выше, где R2 представляет собой атом водорода или метильную группу; R3 представляет собой 4-пиридильную группу или 4-пиримидинильную группу; R4 представляет собой карбонильную группу; и R5 представляет собой группу NH-(CH2)a-NR6R7, причем а представляет собой целое число 3, R6 и R7 представляют собой этильную группу; или R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероциклическую группу, которая представляет собой 4-метилпиперазинильную группу; или любой из его фармацевтически приемлемых солей.
Согласно другому конкретному воплощению дополнительным объектом настоящего изобретения является соединение формулы (Id):
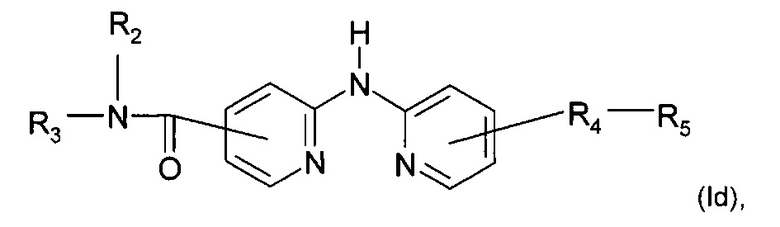
где R2, R3, R4 и R5 являются такими, как определено выше;
или любая из его фармацевтически приемлемых солей.
Согласно предпочтительному воплощению настоящее изобретение относится к соединению формулы (Id), где группа -R4-R5 находится в мета-положении относительно группы -NH- между двумя пиридиновыми группами.
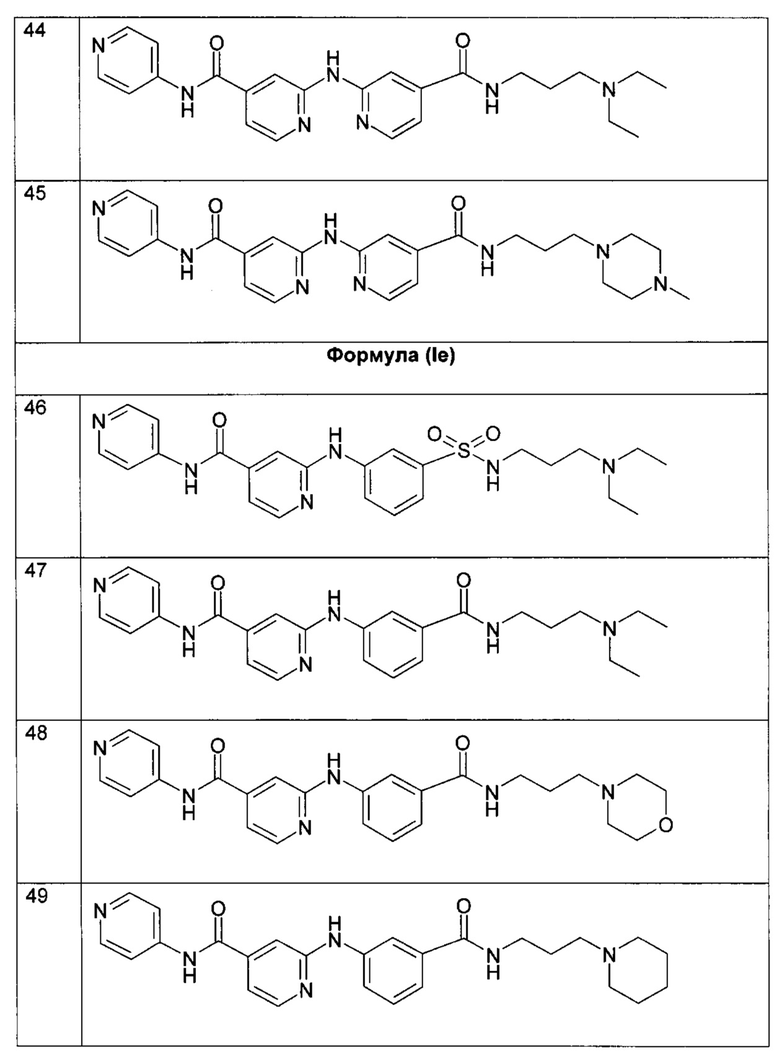
Согласно более предпочтительному воплощению настоящее изобретение относится к соединению формулы (Id), как определено выше, где R2 представляет собой атом водорода; R3 представляет собой 4-пиридильную группу; R4 представляет собой карбонильную группу; и R5 представляет собой группу NH-(CH2)a-NR6R7, причем а представляет собой целое число 3, R6 и R7 представляют собой этильную группу или R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероциклическую группу, которая представляет собой 4-метилпиперазинильную группу; или любой из его фармацевтически приемлемых солей.
Согласно другому конкретному воплощению дополнительным объектом настоящего изобретения является соединение формулы (Iе):
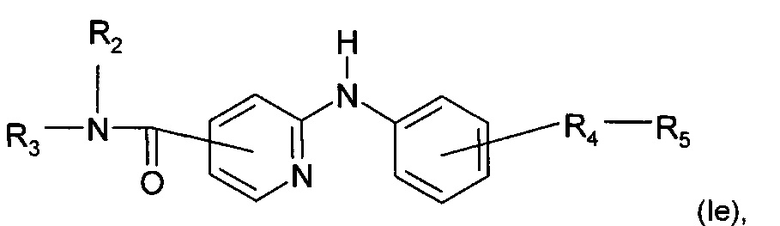
где R2, R3, R4 и R5 являются такими, как определено выше;
или любая из его фармацевтически приемлемых солей.
Согласно предпочтительному воплощению настоящее изобретение относится к соединению формулы (Ib), где группа -R4-R5 находится в мета-положении относительно группы -NH- между фенильной группой и пиридиновой группой.
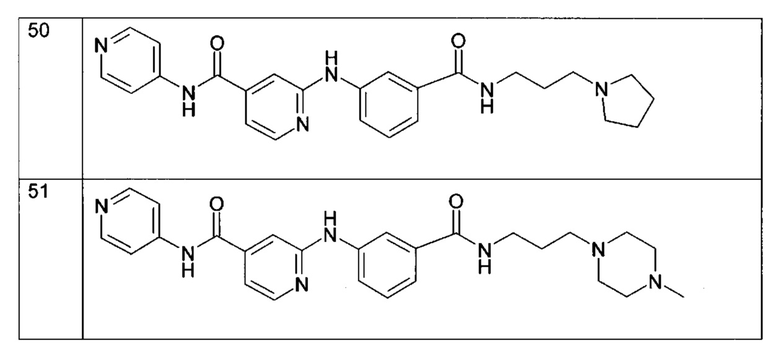
Согласно более предпочтительному воплощению настоящее изобретение относится к соединению формулы (Iе), как определено выше, где R2 представляет собой атом водорода; R3 представляет собой 4-пиридильную группу; R4 представляет собой карбонильную группу или сульфонильную группу; и R5 представляет собой группу -NH-(CH2)a-NR6R7, причем а представляет собой целое число 3, R6 и R7 представляют собой этильную группу или R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероциклическую группу, которую выбирают из 4-метилпиперазинильной группы; морфолиногруппы, пирролидинильной группы и пиперидиногруппы; или любой из его фармацевтически приемлемых солей.
Согласно предпочтительному воплощению настоящего изобретения соединение формулы (I) выбрано из следующих соединений:
(1) N-(3-(диэтиламино)пропил)-3-((3-(пиридин-4-илкарбамоил)фенил)амино)бензамид
(2) 3-((4-((3-(диэтиламино)пропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)бензамид
(3) N-(3-морфолинопропил)-3-((3-(пиридин-4-илкарбамоил)фенил)амино)бензамид
(4) N-(пиридин-4-ил)-3-((3-((3-(пирролидин-1-ил)пропил)карбамоил) фенил)амино)бензамид
(5) 3-((3-(N-(3-(диэтиламино)пропил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)бензамид
(6) N-(3-(4-метилпиперазин-1-ил)пропил)-3-((3-(пиридин-4-илкарбамоил) фенил)амино)бензамид
(7) N-(3-(пиперидин-1-ил)пропил)-3-((3-(пиридин-4-илкарбамоил)фенил)амино)бензамид
(8) 3-((3-(4-метилпиперазин-1-карбонил)фенил)амино)-N-(пиридин-4-ил)бензамид
(9) 3-((3-(N-(3-(пиперидин-1-ил)пропил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)бензамид
(10) 3-((3-(N-(2-(пиперидин-1-ил)этил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)бензамид
(11) N-(3-(диэтиламино)пропил)-3-((3-(пиридин-2-илкарбамоил)фенил)амино)бензамид
(12) 3-((3-(N-(3-морфолинопропил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)бензамид
(13) N-(3-(диэтиламино)пропил)-3-((4-(пиридин-4-илкарбамоил)фенил)амино)бензамид
(14) N-(3-морфолинопропил)-3-((4-(пиридин-4-илкарбамоил)фенил)амино)бензамид
(15) 4-((3-(N-(3-морфолинопропил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)бензамид
(16) N-(пиридин-4-ил)-4-((3-(N-(2-(пирролидин-1-ил)этил)сульфамоил)фенил)амино)бензамид
(17) 3-((3-((3-(диэтиламино)пропил)карбамоил)фенил)амино)-N-метил-N-(пиридин-4-ил)бензамид
(18) N-метил-N-(пиридин-4-ил)-3-((3-((3-(пирролидин-1-ил)пропил)карбамоил)фенил)амино)бензамид
(19) 3-((3-(N-(3-(диэтиламино)пропил)сульфамоил)фенил)амино)-N-метил-N-(пиридин-4-ил)бензамид
(20) N-метил-3-((3-((3-(4-метилпиперазин-1-ил)пропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)бензамид
(21) N-метил-3-((3-((3-(пиперидин-1-ил)пропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)бензамид
(22) N-метил-3-((3-((3-морфолинопропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)бензамид
(23) N-метил-3-((3-(N-(3-морфолинопропил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)бензамид
(24) N-метил-3-((3-(N-(3-(пиперидин-1-ил)пропил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)бензамид
(25) N-(3-(диэтиламино)пропил)-3-((3-(пиримидин-4-илкарбамоил)фенил) амино)бензамид
(26) 3-((3-(N-(3-(диэтиламино)пропил)сульфамоил)фенил)амино)-N-(пиримидин-4-ил)бензамид
(27) 3-((3-(N-(3-(пиперидин-1-ил)пропил)сульфамоил)фенил)амино)-N-(пиримидин-4ил)бензамид
(28) N-(пиримидин-4-ил)-3-((3-((3-(пирролидин-1-ил)пропил)карбамоил)фенил)амино)бензамид
(29) N-(3-(пиперидин-1-ил)пропил)-3-((3-(пиримидин-4-илкарбамоил) фенил)амино)бензамид
(30) N-(3-морфолинопропил)-3-((3-(пиримидин-4-илкарбамоил)фенил)амино)бензамид
(31) N-(3-(4-метилпиперазин-1-ил)пропил)-3-((3-(пиримидин-4-илкарбамоил)фенил)амино)бензамид
(32) N-(3-(диэтиламино)пропил)-5-((3-(пиридин-4-илкарбамоил)фенил)амино)никотинамид
(33) N-(3-(диэтиламино)пропил)-2-((3-(пиридин-4-илкарбамоил)фенил)амино)изоникотинамид
(34) N-(3-(4-метилпиперазин-1-ил)пропил)-2-((3-(пиридин-4-ил)карбамоил)-фенил)амино)изоникотинамид
(35) N-(3-(диэтиламино)пропил)-6-((3-(пиридин-4-илкарбамоил)фенил)амино)пиколинамид
(36) N-(3-(диэтиламино)пропил)-6-((4-(пиридин-4-илкарбамоил)фенил)амино)пиколинамид
(37) N-(3-(диэтиламино)пропил)-6-((3-(метил(пиридин-4-ил)карбамоил)фенил)амино)пиколинамид
(38) N-(3-(диэтиламино)пропил)-2-((3-(метил(пиридин-4-ил)карбамоил)фенил)амино)изоникотинамид
(39) 2-((3-(метил(пиридин-4-ил)карбамоил)фенил)амино)-N-(3-(4-метилпиперазин-1-ил)пропил)изоникотинамид
(40) N-(3-(диэтиламино)пропил)-6-((3-(пиримидин-4-илкарбамоил)фенил)амино)пиколинамид
(41) N-(3-(диэтиламино)пропил)-2-((3-(пиримидин-4-илкарбамоил)фенил)амино)изоникотинамид
(42) N-(3-(4-метилпиперазин-1-ил)пропил)-2-((3-(пиримидин-4-илкарбамоил)фенил)амино)изоникотинамид
(43) N-(3-(диэтиламино)пропил)-6-((4-(пиридин-4-илкарбамоил)пиридин-2-ил)амино)пиколинамид
(44) N-(3-(диэтиламино)пропил)-2-((4-(пиридин-4-илкарбамоил)пиридин-2-ил)амино)изоникотинамид
(45) N-(3-(4-метилпиперазин-1-ил)пропил)-2-((4-(пиридин-4-илкарбамоил)пиридин-2-ил)амино)изоникотинамид
(46) 2-((3-(N-(3-(диэтиламино)пропил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)изоникотинамид
(47) 2-((3-((3-(диэтиламино)пропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)изоникотинамид
(48) 2-((3-((3-морфолинопропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)изоникотинамид
(49) N-(3-(пиперидин-1-ил)пропил)-3-((3-(пиридин-4-илкарбамоил)фенил)амино)бензамид
(50) N-(пиридин-4-ил)-2-((3-((3-(пирролидин-1-ил)пропил)карбамоил)фенил)амино)изоникотинамид
(51) 2-((3-((3-(4-метилпиперазин-1-ил)пропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)изоникотинамид
Соединения по изобретению могут существовать в форме свободных оснований или солей присоединения с фармацевтически приемлемыми кислотами.
Подходящие физиологически приемлемые соли присоединения кислоты соединений формулы (I) включают гидробромид, тартрат, цитрат, трифторацетат, аскорбат, гидрохлорид, тартрат, трифлат, малеат, мезилат, формиат, ацетат и фумарат.
Соединения формулы (I), (I'), (Ia), (Ib), (с), (Id) и (Ie) или их соли могут образовывать сольваты (например, гидраты), и изобретение включает все такие сольваты.
Вследствие этого, настоящее изобретение распространяется на соединения от (1) до (51), их фармацевтически приемлемые соли, их сольваты и их гидраты, как таковые.
В контексте настоящего изобретения термин:
- "алкил (C1-C4)", в том виде, как он используется в данном документе, соответственно относится к нормальному, вторичному или третичному насыщенному углеводороду C1-C4. Примерами являются метил, этил, 1-пропил, 2-пропил, бутил, изобутил, трет-бутил, но они не ограничиваются ими, и
- термин "пациент" может распространяться на людей или млекопитающих, таких как кошки или собаки.
Соединение формул (I), (I'), (Ia), (Ib), (Ic), (Id) и (Ie) может содержать один или более асимметрических атомов углерода. Они могут таким образом существовать в форме энантиомеров или диастереоизомеров. Данные энантиомеры, диастереоизомеры и их смеси, включая рацемические смеси, включены в объем настоящего изобретения.
Согласно другому аспекту настоящее изобретение относится к соединению формул (I), (I'), (Ia), (Ib), (Ic), (Id) и (Iе) для применения в качестве лекарственного средства.
Согласно другому аспекту настоящее изобретение относится к соединению формул (I), (I'), (Ia), (Ib), (Ic), (Id) и (Iе) для применения для предупреждения и/или ингибирования, и/или лечения рака.
Согласно настоящему изобретению термин "предотвращение" или "предупреждение" означает снижение риска начала или замедление возникновения данного явления, а именно рака.
Соединения по настоящему изобретению могут быть получены традиционными способами органического синтеза, применяемыми на практике специалистами в данной области техники. Общие последовательности реакций, изложенные ниже, представляют общий способ, применимый для получения соединений по настоящему изобретению, и не являются ограничивающими в объеме или применимости.
Соединения общей формулы (I) могут быть получены согласно нижеприведенной схеме 1.
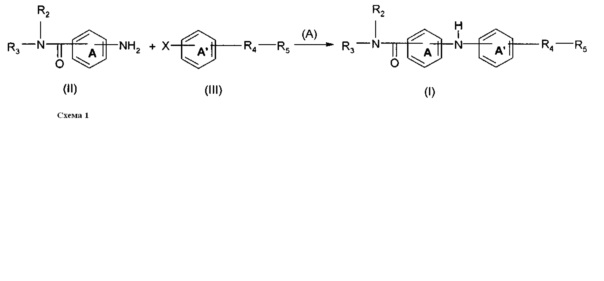
Синтез основан на реакции сочетания, начинающейся с галогенароматического соединения формулы (III), где R4 и R5 являются такими, как определено выше, и X представляет собой атом хлора, атом йода или атом брома.
В соответствии с путем (А) соединение формулы (III) помещают в протонный растворитель, такой как трет-бутанол. Затем добавляют соединение формулы (II), в котором R2, R3 и А являются такими, как определено выше, в молярном соотношении, находящемся в интервале от 1 до 1,5, относительно соединения формулы (III) в присутствии неорганического основания, такого как Cs2CO3 или K2CO3, в молярном соотношении, находящемся в интервале от 1 до 2, в присутствии дифосфина, такого как ксантфос (Xantphos) (4,5-бис(дифенилфосфино)-9,9-диметилксантен) или X-Phos (2-Дициклогексилфосфино-2',4',6'-триизопропилбифенил), в количестве, находящемся в интервале от 2 мол. % до 10 мол. %, относительно общего количества соединения формулы (III), и в присутствии органометаллического катализатора, такого как Pd(OAc)2 или Pd2dba3, в количестве от 2 мол. % до 10 мол. %, относительно общего количества соединения формулы (III). Реакционную смесь затем можно нагревать при температуре, находящейся в интервале от 80 до 120°C, например, при 90°C, и перемешивать в течение времени, находящегося в интервале от 15 до 25 часов, например, в течение 20 часов, в атмосфере инертного газа и, например, аргона. Реакционную смесь можно концентрировать под сниженным давлением, и остаток можно разбавлять органическим растворителем, таким как этилацетат. Органическую фазу можно промывать водой, декантировать и высушивать над сульфатом магния. Наконец, твердое вещество можно высушивать под вакуумом в течение ночи с получением продукта (I).
Исходные соединения формулы (II) и (III) доступны или могут быть получены согласно способам, известным специалисту в данной области техники.
Более конкретно, соединения формулы (II) (т.е. соответственно (IIа) и (IIс)), при использовании для получения соединений формул (Ia) и (Ic), могут быть получены согласно приведенной ниже схеме 2.
Получение промежуточных соединений формулы (II) для соединений формул (Ia) и (Ic), причем один из X1 или Х2 представляет собой N, и оставшийся из и Х2 представляет собой СН (R3 представляет собой пиридильную группу).
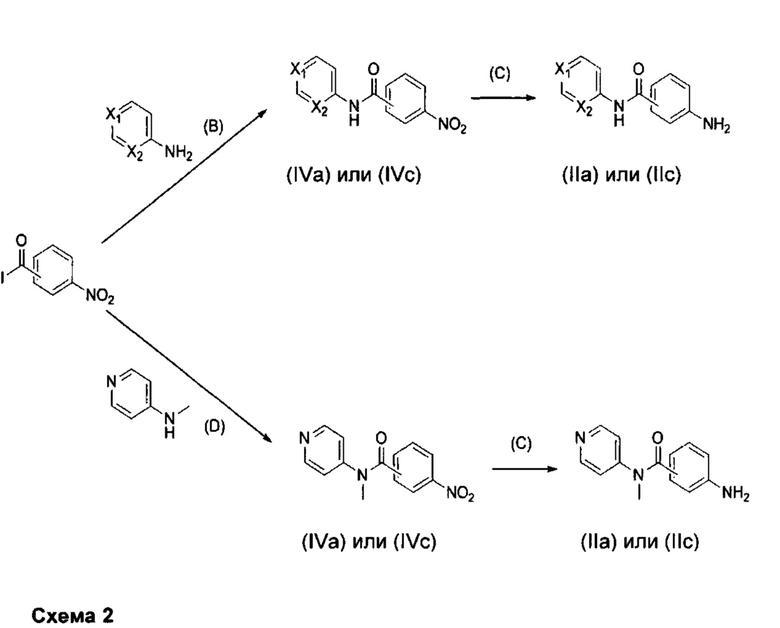
Как показано на схеме 2, промежуточные соединения формул (IIа) и (IVa) применимы для получения соединений формулы (Iа) согласно изобретению, и промежуточные соединения формул (IIс) и (IVc) применимы для получения соединений формулы (Iв) согласно изобретению.
В соответствии с путем (В) аминопиридин, добавляемый в молярном соотношении, находящемся в интервале от 1 до 1,5, относительно нитробензоилхлорида, помещают в водный раствор неорганического основания, такого как гидроксид натрия, в молярной концентрации, находящейся в интервале от 2 М до 5 М. К раствору добавляют полярный апротонный растворитель, такой как дихлорметан, реакционную смесь можно охлаждать до 0°C с помощью ледяной бани, и раствор нитробензоилхлорида в полярном апротонном растворителе, таком как дихлорметан, можно добавлять по каплям. Затем реакционную смесь можно перемешивать при комнатной температуре в течение времени, находящегося в интервале от 15 до 24 часов, например, 18 часов, в атмосфере инертного газа, например, аргона. Полученный осадок можно фильтровать, промывать водой и дихлорметаном и высушивать под вакуумом в течение ночи с получением продукта (IVa) или (IVc).
В соответствии с путем (С) соединение формулы (IVa) или (IVc) и 10% Pd/C в соотношении, находящемся в интервале от 2% до 10% относительно количества бензамида, помещают в протонный растворитель, такой как этанол. Затем реакционную смесь можно перемешивать при комнатной температуре в течение времени, находящегося в интервале от 5 до 20 часов, например, 16 часов, в атмосфере H2. Затем реакционную смесь можно фильтровать, и фильтрат можно концентрировать при пониженном давлении с получением продукта (IIa) или (IIс).
В соответствии с путем (D) 4-(метиламино)пиридин помещают в полярный апротонный растворитель, такой как дихлорметан. Нитробензоилхлорид затем добавляют в молярном соотношении, находящемся в интервале от 1 до 1,5, относительно 4-(метиламино)пиридина, в присутствии органического основания, такого как N,N-диизопропилэтиламин или триэтиламин, в молярном соотношении, находящемся в интервале от 1 до 2, в присутствии нуклеофильного катализатора, такого как диметиламинопиридин, в молярном соотношении, находящемся в интервале от 0,1 до 1. Реакционную смесь можно затем перемешивать при комнатной температуре в течение времени, находящегося в интервале от 5 до 20 часов, например, 18 часов, в атмосфере инертного газа, например, аргона. Органическую фазу можно промывать водой, декантировать и высушивать над сульфатом магния. Наконец, твердое вещество можно высушивать под вакуумом, в течение ночи с получением продукта (IVa) или (IVc).
Более конкретно, соединения формулы (II), при применении для получения соединений формулы (Ia) и (Ic) в одном случае или (Id) и (Ie) в другом случае, можно получать согласно приведенной ниже схеме 3.
Получение промежуточных соединений формулы (II) для соединений формул (Ia) и (Ic), причем X: представляет собой СН и X2 представляет собой N (R2 представляет собой пиримидинильную группу), и для соединений формул (Id) и (Ie), причем X1 представляет собой N и Х2 представляет собой СН (R3 представляет собой пиридильную группу).
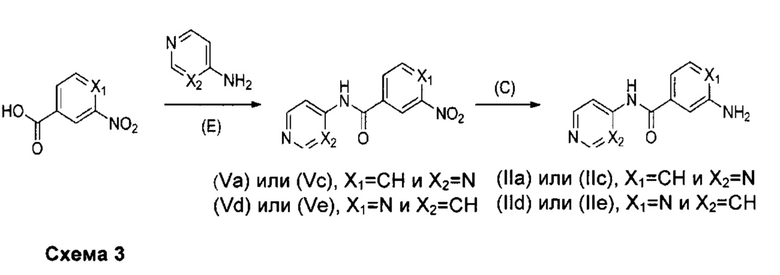
Как показано на схеме 3, промежуточные соединения формул (IIа) и (Va) применимы для получения соединений формулы (Iа) согласно изобретению, промежуточные соединения формул (IIс) и (Vc) применимы для получения соединений формулы (Iс) согласно изобретению, промежуточные соединения формул (IId) и (Vd) применимы для получения соединений формулы (Id) согласно изобретению и промежуточные соединения формул (IIе) и (Ve) применимы для получения соединений формулы (Iе) согласно изобретению.
В соответствии с путем (Е) производное карбоксильной кислоты помещают в полярный апротонный растворитель, такой как дихлорметан. Аминопроизводное затем добавляют в молярном соотношении от 1 до 1,5, относительно группировки карбоновой кислоты, в присутствии агента реакции сочетания, такого как EDCI.HCl (1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид), в молярном соотношении, находящемся в интервале от 1 до 3, в присутствии органического основания, такого как N,N-диизопропилэтиламин или триэтиламин, в молярном соотношении, находящемся в интервале от 1 до 3, и в присутствии нуклеофильного катализатора, такого как диметиламинопиридин в молярном отношении, находящемся от 0,1 до 1. Затем реакционную смесь можно перемешивать при комнатной температуре в течение времени, находящегося в интервале от 5 до 20 часов, например, 18 часов, в атмосфере инертного газа, например, аргона. Полученный осадок можно фильтровать и промывать водой и дихлорметаном. Органический фильтрат можно промывать водой, декантировать и высушивать над сульфатом магния. Наконец, твердые остатки можно собирать и высушивать под вакуумом в течение ночи с получением продукта (Va), (Vc), (Vd) или (Ve).
Аналогично, для получения соединений формулы (Ib) можно использовать или схему 2, или схему 3.
Химические структуры и спектроскопические данные для некоторых соединений формулы (I) изобретения проиллюстрированы соответственно в следующей таблице I и таблице II.
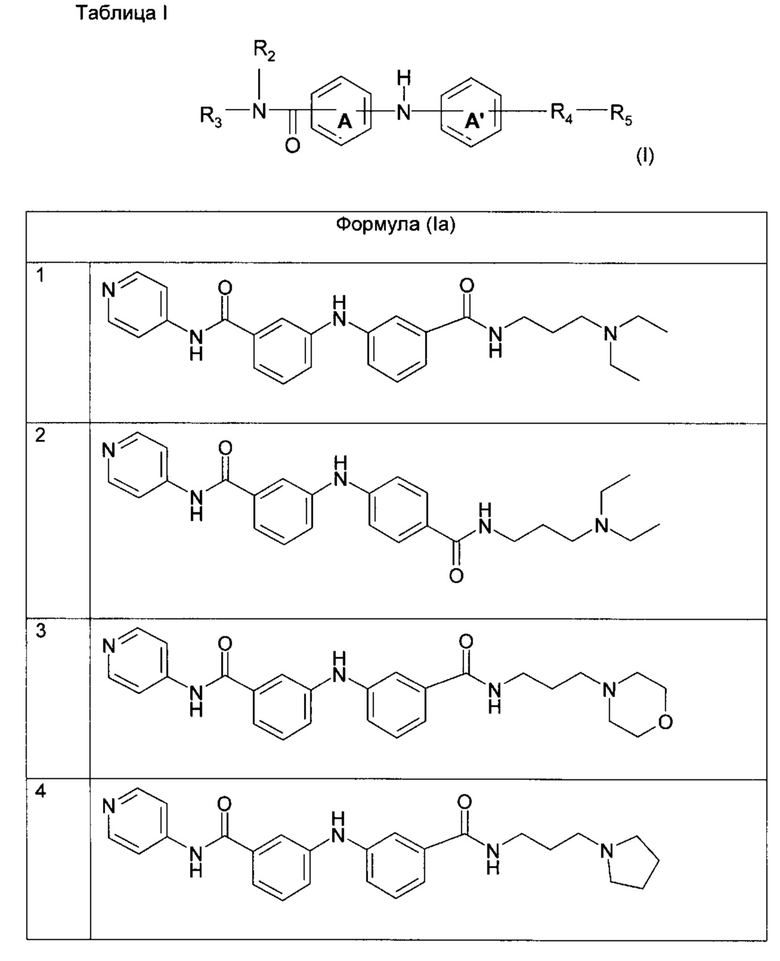
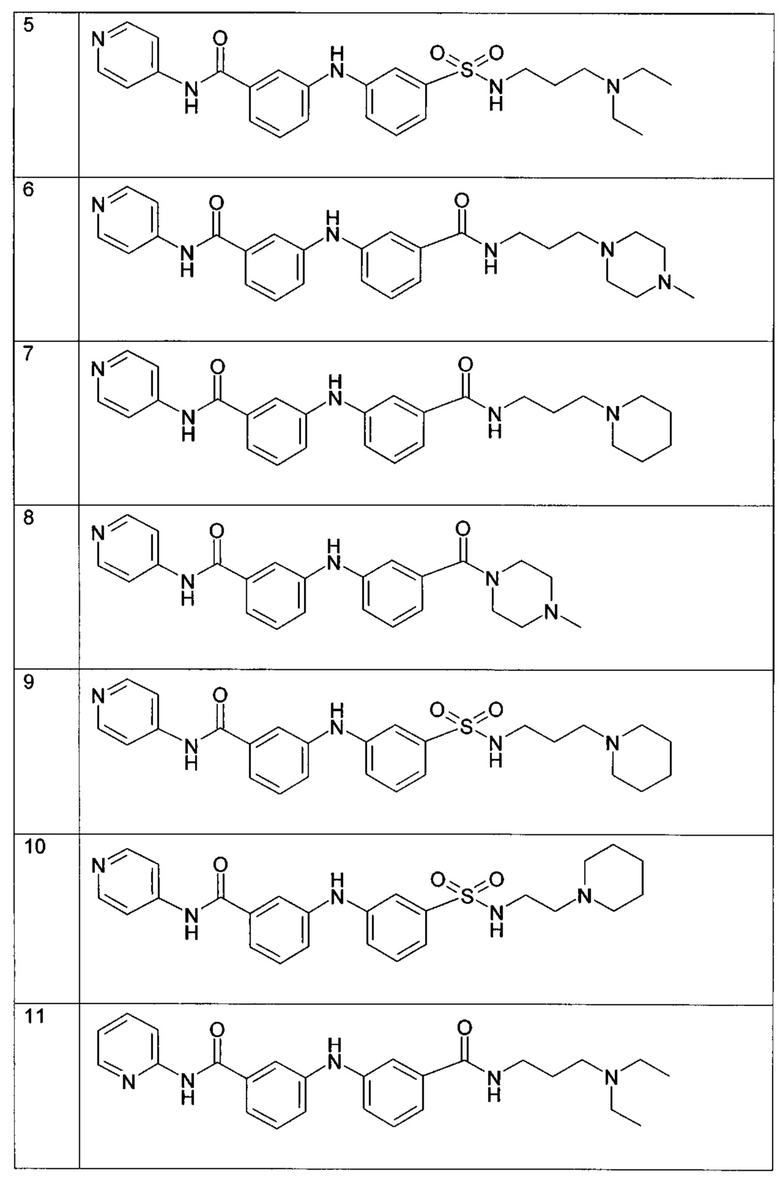
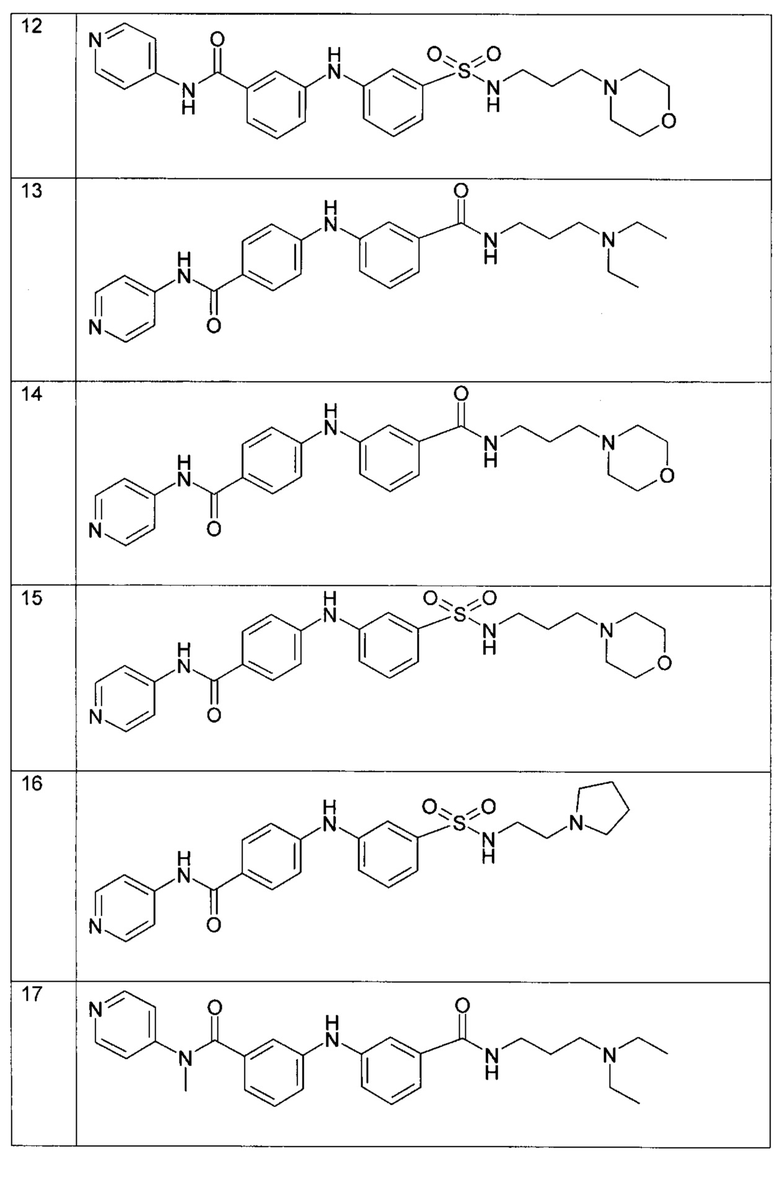
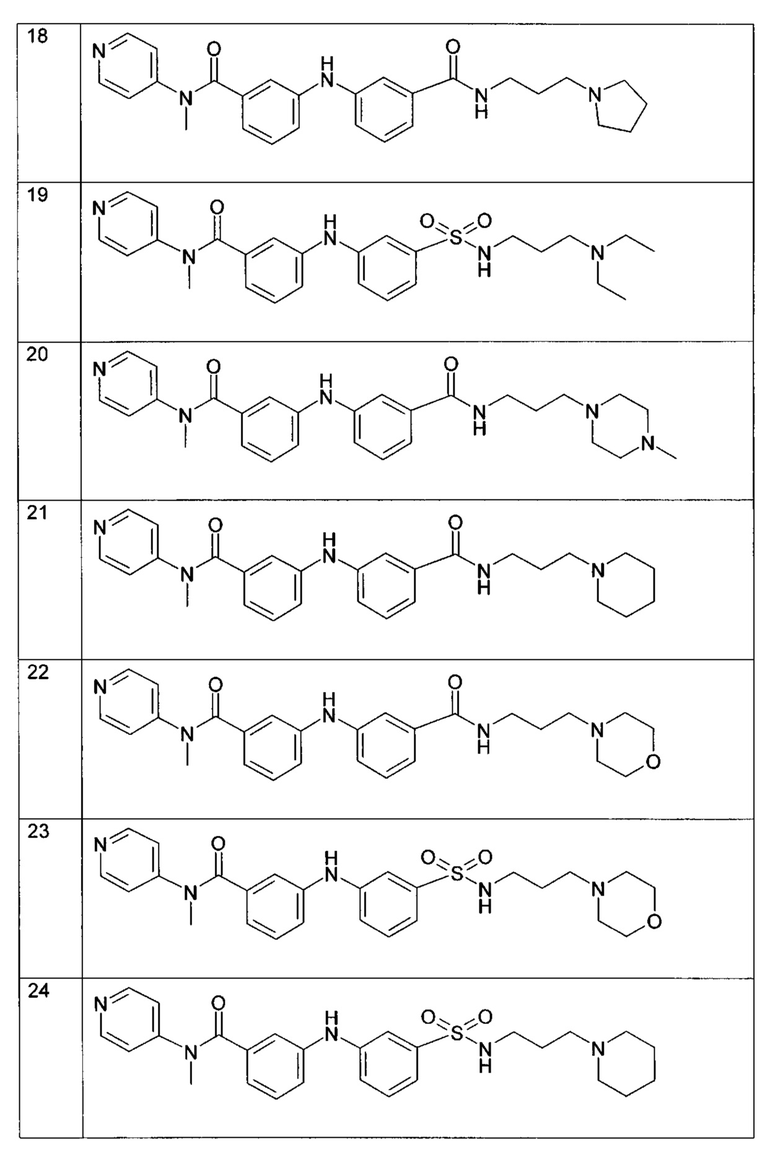
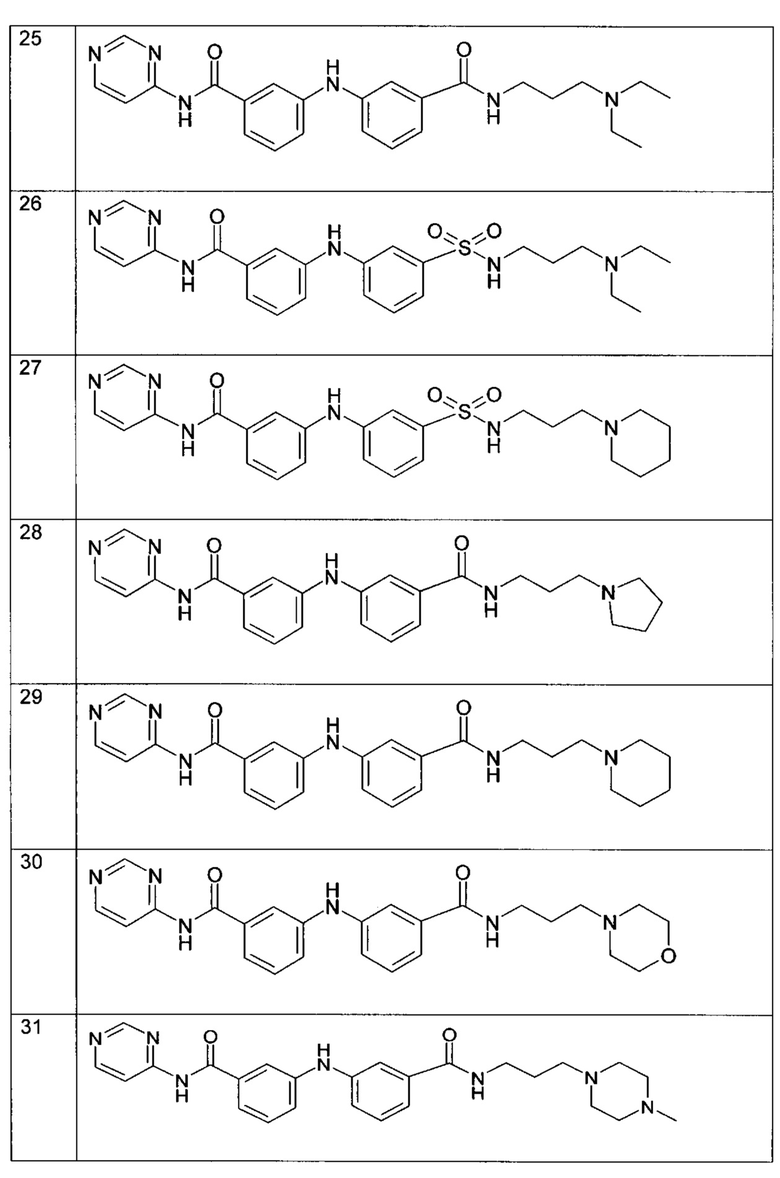








Среди указанных соединений формулы (I) соединения (1), (3), (4), (5), (6), (7), (9), (10), (12), (20), (25), (28), (29), (30), (31), (34), (35), (40), (41), (42), (44), (45), (48), (49) и (51) или одна из их фармацевтически приемлемых солей представляют особый интерес.
Следующие примеры подробно иллюстрируют получение соединений (1), (4), (5), (6) (7), (20), (25), (31), (35), (39), (41), (44), (47) и (51) по изобретению. Структуры полученных продуктов подтверждали по меньшей мере посредством спектров ЯМР.
Пример 1: соединение (1) в таблице I
В соответствии с путем (В) 4-аминопиридин (4,2 г, 44 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (56 мл), и к раствору добавляли дихлорметан (24 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор 3-нитробензоилхлорида (7,4 г, 40 ммоль, 1 эквивалент (экв.)) в дихлорметане (40 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. Полученный осадок фильтровали и промывали водой и дихлорметаном с получением 3-нитро-N-(пиридин-4-ил)бензамида (2,5 г, 26%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,91 (s, 1Н), 8,80 (s, 1Н), 8,52 (d, J=5,5 Гц, 2Н), 8,47 (d, J=7,9 Гц, 1Н), 8,41 (d, J=7,9 Гц, 1Н), 7,86 (t, J=7,9 Гц, 1Н), 7,79 (d, J=5,3 Гц, 2Н).
В соответствии с путем (С) 3-нитро-N-(пиридин-4-ил)бензамид (1,5 г, 6,2 ммоль, 1 экв.) и 10% Pd/C (250 мг) помещали в EtOH (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере H2. Реакционную смесь затем фильтровали на целите, промывали EtOH, и фильтрат концентрировали при пониженном давлении с получением 3-амино-N-(пиридин-4-ил)бензамида (1,24 г, 94%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,44 (s, 1Н), 8,44 (d, J=6,3 Гц, 2Н), 7,77 (d, J=6,3 Гц, 2Н), 7,18 (t, J=7,9 Гц, 1Н), 7,12-7,03 (m, 2Н), 6,78 (d, J=7,9 Гц, 1Н), 5,38 (s, 2Н).
N,N-диэтилпропилендиамин (8,7 мл, 55 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (71 мл), и к раствору добавляли дихлорметан (30 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор 3-бромбензоилхлорида (6,6 мл, 50 ммоль, 1 экв.) в дихлорметане (50 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. При декантации органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 3-бром-N-(3-диэтиламино-пропил)бензамида (15,6 г, 100%).
1Н ЯМР (300 МГц, CDCl3) δ 9,15 (br s, 1Н), 7,91 (s, 1Н), 7,75 (d, J=7,9 Гц, 1Н), 7,58 (d, J=7,9 Гц, 1Н), 7,29 (t, J=7,9 Гц, 1Н), 3,56 (dd, J=10,3, 5,8 Гц, 2Н), 2,67-2,53 (m, 6Н), 1,74 (квинт., J=5,7 Гц, 2Н), 1,04 (t, J=7,1 Гц, 6Н).
В соответствии с путем (А) реакционную смесь 3-бром-N-(3-диэтиламино-пропил)бензамида (291 мг, 0,9 ммоль, 1 экв.), 3-амино-N-(пиридин-4-ил)бензамида (300 мг, 1,4 ммоль, 1,5 экв.), Pd2(dba)3 (42 мг, 0,046 ммоль, 5 мол. %), XPhos (44 мг, 0,09 ммоль, 10 мол. %) и K2CO3 (514 мг, 3,72 ммоль, 4 экв.) в трет-BuOH (4 мл) нагревали при 90°C и перемешивали в течение 20 часов в инертной атмосфере аргона. Реакционную смесь затем концентрировали при пониженном давлении, и полученный остаток разводили этилацетатом. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный осадок очищали посредством колоночной хроматографии на силикагеле с получением N-(3-(диэтиламино)пропил)-3-((3-(пиридин-4-илкарбамоил)фенил)амино)бензамида (1) (230 мг, 57%).
Пример 2: соединение (4) в таблице I
В соответствии с путем (В) 4-аминопиридин (4,2 г, 44 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (56 мл), и к раствору добавляли дихлорметан (24 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор 3-нитробензоилхлорида (7,4 г, 40 ммоль, 1 экв.) в дихлорметане (40 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. Полученный осадок фильтровали и промывали водой и дихлорметаном с получением 3-нитро-N-(пиридин-4-ил)бензамида (2,5 г, 26%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,91 (s, 1Н), 8,80 (s, 1Н), 8,52 (d, J=5,5 Гц, 2Н), 8,47 (d, J=7,9 Гц, 1Н), 8,41 (d, J=7,9 Гц, 1Н), 7,86 (t, J=7,9 Гц, 1Н), 7,79 (d, J=5,3 Гц, 2Н).
В соответствии с путем (С) 3-нитро-N-(пиридин-4-ил)бензамид (994 мг, 4,1 ммоль, 1 экв.) и 10% Pd/C (218 мг) помещали в EtOH (20,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере H2. Реакционную смесь затем фильтровали на целите, промывали EtOH, и фильтрат концентрировали при пониженном давлении с получением 3-амино-N-(пиридин-4-ил)бензамида (900 мг, 100%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,42 (s, 1Н), 8,44 (d, J=6,3 Гц, 2Н), 7,75 (d, J=6,3 Гц, 2Н), 7,16 (t, J=7,9 Гц, 1Н), 7,10-7,01 (m, 2Н), 6,76 (d, J=7,9 Гц, 1Н), 5,36 (s, 2Н).
3-(Пирролидин-1-ил)пропиламин (1,4 мл, 11 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (14 мл), и к раствору добавляли дихлорметан (6 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор 3-бромбензоилхлорида (1,3 мл, 10 ммоль, 1 экв.) в дихлорметане (10 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. При декантации органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 3-бром-N-(3-пирролидин-1-ил-пропил)бензамида (2,9 г, 94%).
1Н ЯМР (300 МГц, CDCl3) δ 9,19 (s, 1Н), 7,82 (s, 1Н), 7,77 (d, J=7,9 Hz, 1Н), 7,58 (d, J=7,9 Гц, 1H), 7,29 (t, J=7,9 Гц, 1H), 3,57 (dd, J=9,4, 4,8 Гц, 2H), 2,72 (t, J=4,8 Гц, 2H), 2,58 (s, 4Н), 1,86 (s, 4Н), 1,78 (t, J=4,8 Гц, 2H).
В соответствии с путем (А) реакционную смесь 3-бром-N-(3-диэтиламино-пропил)бензамида (611 мг, 1,97 ммоль, 1 экв.), 3-амино-N-(пиридин-4-ил)бензамида (630 мг, 2,96 ммоль, 1,5 экв.), Pd2(dba)3 (90 мг, 0,095 ммоль, 5 мол. %), XPhos (94 мг, 0,19 ммоль, 10 мол. %) и K2CO3 (1,1 мг, 7,88 ммоль, 4 экв.) в трет-BuOH (8 мл) нагревали при 90°C и перемешивали в течение 20 часов в инертной атмосфере аргона. Реакционную смесь затем концентрировали при пониженном давлении, и полученный остаток разводили этилацетатом. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением N-(пиридин-4-ил)-3-((3-((3-(пирролидин-1-ил(пропил)карбамоил)фенил)амино)бензамида (4) (427 мг, 49%).
Пример 3: соединение (5) в таблице I
В соответствии с путем (В) 4-аминопиридин (4,2 г, 44 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (56 мл), и к раствору добавляли дихлорметан (24 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор 3-нитробензоилхлорида (7,4 г, 40 ммоль, 1 экв.) в дихлорметане (40 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. Полученный осадок фильтровали и промывали водой и дихлорметаном с получением 3-нитро-N-(пиридин-4-ил)бензамида (994 мг, 20%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,91 (s, 1Н), 8,80 (s, 1Н), 8,52 (d, J=5,5 Гц, 2Н), 8,47 (d, J=7,9 Гц, 1Н), 8,41 (d, J=7,9 Гц, 1H), 7,86 (t, J=7,9 Гц, 1H), 7,79 (d, J=5,3 Гц, 2H).
В соответствии с путем (С) 3-нитро-N-(пиридин-4-ил)бензамид (994 мг, 4,1 ммоль, 1 экв.) и 10% Pd/C (218 мг) помещали в EtOH (20,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере H2. Реакционную смесь затем фильтровали на целите, промывали EtOH, и фильтрат концентрировали при пониженном давлении с получением 3-амино-N-(пиридин-4-ил)бензамида (900 мг, 100%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,42 (s, 1Н), 8,44 (d, J=6,3 Гц, 2Н), 7,75 (d, J=6,3 Гц, 2Н), 7,16 (t, J=7,9 Гц, 1Н), 7,10-7,01 (m, 2Н), 6,76 (d, J=7,9 Гц, 1Н), 5,36 (s, 2Н).
3-Бромбензолсульфонилхлорид (0,56 мл, 3,9 ммоль, 1 экв.) и N,N-диизопропилэтиламин (1,02 мл, 5,9 ммоль, 1,5 экв.) помещали в безводный дихлорметан (20 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли N,N-диэтилпропилендиамин (1,23 мл, 7,8 ммоль, 2 экв.). Реакционную смесь затем перемешивали при 0°C в течение 2 часов в инертной атмосфере аргона. Смесь промывали насыщенными водными растворами NH4Cl и затем NaCl. Водные фазы экстрагировали дихлорметаном. Органические фазы собирали, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 3-бром-N-(3-диэтиламинопропил)бензолсульфоамида (524 мг, 38%).
1Н ЯМР (300 МГц, CDCl) δ 7,98 (s, 1Н), 7,78 (d, J=7,9 Гц, 1Н), 7,66 (d, J=8,0 Гц, 1Н), 7,37 (t, J=7,9 Гц, 1Н), 3,05 (t, J=5,4 Гц, 2Н), 2,63-2,47 (m, 6Н), 1,68 (t, J=5,4 Гц, 2Н), 1,06 (t, J=7,1 Гц, 6Н).
В соответствии с путем (А) реакционную смесь 3-бром-N-(3-диэтиламино-пропил)бензолсульфонамида (153 мг, 0,44 ммоль, 1 экв.), 3-амино-N-(пиридин-4-ил)бензамида (103 мг, 0,48 ммоль, 1,1 экв.), Pd2(dba)3 (20 мг, 0,022 ммоль, 5 мол. %), XPhos (21 мг, 0,044 ммоль, 10 мол. %) и K2CO3 (243 мг, 1,76 ммоль, 4 экв.) в трет-BuOH (2 мл) нагревали при 90°C и перемешивали в течение 20 часов в инертной атмосфере аргона. Реакционную смесь затем концентрировали при пониженном давлении, и полученный остаток разводили этилацетатом. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением 3-((3-(N-(3-(диэтиламино)пропил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)бензамида (5) (97 мг, 46%).
Пример 4: соединение (6) в таблице I
В соответствии с путем (В) 4-аминопиридин (2,1 г, 22 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (28 мл), и к раствору добавляли дихлорметан (12 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор 3-нитробензоилхлорида (3,7 г, 20 ммоль, 1 экв.) в дихлорметане (20 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. Полученный осадок фильтровали и промывали водой и дихлорметаном с получением 3-нитро-N-(пиридин-4-ил)бензамида (2,4 г, 50%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,98 (s, 1Н), 8,80 (s, 1Н), 8,51 (d, J=6,2 Гц, 2Н), 8,47 (d, J=7,9 Гц, 1Н), 8,42 (d, J=7,9 Гц, 1Н), 7,86 (t, J=7,9 Гц, 1Н), 7,80 (d, J=6,2 Гц, 2Н).
В соответствии с путем (С) 3-нитро-N-(пиридин-4-ил)бензамид (1 г, 4,1 ммоль, 1 экв.) и 10% Pd/C (150 мг) помещали в EtOH (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере H2. Реакционную смесь затем фильтровали на целите, промывали EtOH, и фильтрат концентрировали при пониженном давлении с получением 3-амино-N-(пиридин-4-ил)бензамида (660 мг, 75%).
1Н ЯМР (300 МГц, DMSO) δ 10,46 (s, 1Н), 8,45 (dd, J=5,0, 1,3 Гц, 2Н), 7,77 (dd, J=5,0, 1,3 Гц, 2Н), 7,17 (t, J=7,9 Гц, 1Н), 7,12-7,03 (m, 2Н), 6,77 (dd, J=7,9, 1,2 Гц, 1Н), 5,38 (s, 2Н).
3-(4-метилпиперазин-1-ил)пропиламин (1,9 мл, 11 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (14 мл), и к раствору добавляли дихлорметан (6 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор 3-бромбензоилхлорида (1,3 мл, 10 ммоль, 1 экв.) в дихлорметане (10 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. При декантации органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 3-бром-N-(3-метилпиперазин-1-ил-пропил)бензамида (2,7 г, 80%).
1Н ЯМР (300 МГц, CDCl3) δ 8,61 (br s, 1Н), 7,92 (s, 1Н), 7,82 (d, J=7,9 Гц, 1Н), 7,62 (d, J=7,9 Гц, 1Н), 7,32 (t, J=7,9 Гц, 1Н), 3,57 (q, J=5,2 Гц, 2Н), 2,79-2,35 (m, 10Н), 2,33 (s, 3Н), 1,78 (квинт., J=5,2 Гц, 2Н).
В соответствии с путем (А) реакционную смесь 3-бром-N-(4-метилпиперазин-1-ил-пропил)бензамида (170 мг, 0,5 ммоль, 1 экв.), 3-амино-N-(пиридин-4-ил)бензамида (117 мг, 0,55 ммоль, 1,1 экв.), Pd2(dba)3 (23 мг, 0,025 ммоль, 5 мол. %), XPhos (24 мг, 0,05 ммоль, 10 мол. %) и К2CO3 (276 мг, 2 ммоль, 4 экв.) в трет-BuOH (2 мл) нагревали при 90°C и перемешивали в течение 20 часов в инертной атмосфере аргона. Реакционную смесь затем концентрировали при пониженном давлении, и полученный остаток разводили этилацетатом. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением N-(3-(4-метилпиперазин-1-ил)пропил)-3-((3-(пиридин-4-илкарбамоил)фенил)амино)бензамида (6) (52 мг, 22%).
Пример 5: соединение (7) в таблице I
В соответствии с путем (В) 4-аминопиридин (4,2 г, 44 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (56 мл), и к раствору добавляли дихлорметан (24 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор 3-нитробензоилхлорида (7,4 г, 40 ммоль, 1 экв.) в дихлорметане (40 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. Полученный осадок фильтровали и промывали водой и дихлорметаном с получением 3-нитро-N-(пиридин-4-ил)бензамида (2,5 г, 26%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,91 (s, 1Н), 8,80 (s, 1Н), 8,52 (d, J=5,5 Гц, 2Н), 8,47 (d, J=7,9 Гц, 1Н), 8,41 (d, J=7,9 Гц, 1Н), 7,86 (t, J=7,9 Гц, 1Н), 7,79 (d, J=5,3 Гц, 2Н).
В соответствии с путем (С) 3-нитро-N-(пиридин-4-ил)бензамид (1,5 г, 6,2 ммоль, 1 экв.) и 10% Pd/C (250 мг) помещали в EtOH (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере H2. Реакционную смесь затем фильтровали на целите, промывали EtOH, и фильтрат концентрировали при пониженном давлении с получением 3-амино-N-(пиридин-4-ил)бензамида (1,24 мг, 94%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,44 (s, 1Н), 8,44 (d, J=6,3 Гц, 2Н), 7,77 (d, J=6,3 Гц, 2Н), 7,18 (t, J=7,9 Гц, 1Н), 7,12-7,03 (m, 2Н), 6,78 (d, J=7,9 Гц, 1Н), 5,38 (s, 2Н).
3-(Пиперидин-1-ил)пропиламин (1,7 мл, 11 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (14 мл), и к раствору добавляли дихлорметан (6 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор 3-бромбензоилхлорида (1,3 мл, 10 ммоль, 1 экв.) в дихлорметане (10 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. При декантации органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 3-бром-N-(пиперидин-1-ил-пропил)бензамида (3,24 г, 100%).
1Н ЯМР (300 МГц, CDCl3) δ 9,02 (s, 1Н), 7,97 (s, 1Н), 7,83 (d, J=7,9 Гц, 1Н), 7,60 (d, J=7,9 Гц, 1Н), 7,31 (t, J=7,9 Гц, 1Н), 3,56 (dd, J=9,8, 4,8 Гц, 2Н), 2,53 (t, J=4,8 Гц, 2Н), 2,44 (s, 4Н), 1,76 (t, J=4,8 Гц, 2Н), 1,62 (t, J=4,8 Гц, 4Н), 1,50 (s, 2Н).
В соответствии с путем (А) реакционную смесь 3-бром-N-(пиперидин-1-ил-пропил)бензамида (162 мг, 0,5 ммоль, 1 экв.), 3-амино-N-(пиридин-4-ил)бензамида (117 мг, 0,55 ммоль, 1,1 экв.), Pd2(dba)3 (23 мг, 0,025 ммоль, 5 мол. %), XPhos (24 мг, 0,05 ммоль, 10 мол. %) и K2CO3 (276 мг, 2 ммоль, 4 экв.) в трет-BuOH (2 мл) нагревали при 90°C и перемешивали в течение 20 часов в инертной атмосфере аргона. Реакционную смесь затем концентрировали при пониженном давлении, и полученный остаток разводили этилацетатом. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением N-(3-(пиперидин-1-ил)пропил)-3-((3-(пиридин-4-илкарбамоил)фенил)амино)бензамида (7) (115 мг, 50%).
Пример 6: соединение (20) в таблице I
В соответствии с путем (D) реакционную смесь 4-(метиламино)пиридина (1,25 г, 11,6 ммоль, 1,0 экв.), 3-нитробензоилхлорида (2,57 г, 13,9 ммоль, 1,2 экв.), N,N-диизопропилэтиламина (3,02 мл, 17,3 ммоль, 1,5 экв.) и диметиламинопиридина (103 мг, 1,41 ммоль, 1 экв.) в дихлорметане (25 мл) перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением N-метил-3-нитро-N-(пиридин-4-ил)бензамида (2,96 г, 100%).
1Н ЯМР (300 МГц, CDCl3) δ 8,50 (dd, J=4,6, 1,6 Гц, 2Н), 8,25 (s, 1Н), 8,21 (d, J=7,9 Гц, 1Н), 7,62 (d, J=7,9 Гц, 1Н), 7,45 (t, J=7,9 Гц, 1Н), 6,98 (dd, J=4,6, 1,6 Гц, 2Н), 3,56 (s, 3Н).
В соответствии с путем (С) N-метил-3-нитро-N-(пиридин-4-ил)бензамид (2,96 г, 11,5 ммоль, 1 экв.) и 10% Pd/C (450 мг) помещали в EtOH (100 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере H2. Реакционную смесь затем фильтровали на целите, промывали EtOH, и фильтрат концентрировали при пониженном давлении с получением 3-амино-N-метил-N-(пиридин-4-ил)бензамида (2,5 г, 96%).
1Н ЯМР (300 МГц, d6-DMSO) δ 8,40 (dd, J=4,6, 1,6 Гц, 2Н), 7,14 (dd, J=4,6, 1,6 Гц, 2Н), 6,89 (t, J=7,9 Гц, 1Н), 6,59 (s, 1Н), 6,53 (d, J=7,9 Гц, 1Н), 6,34 (d, J=7,9 Гц, 1Н), 5,22 (s, 2Н), 3,37 (s, 3Н).
3-(4-Метилпиперазин-1-ил)пропиламин (1,9 мл, 11 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (14 мл), и к раствору добавляли дихлорметан (6 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор 3-бромбензоилхлорида (1,3 мл, 10 ммоль, 1 экв.) в дихлорметане (10 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. При декантации органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 3-бром-N-(4-метилпиперазин-1-ил-пропил)бензамида (2,7 г, 80%).
1Н ЯМР (300 МГц, CDCl3) δ 8,61 (br s, 1Н), 7,92 (s, 1Н), 7,82 (d, J=7,9 Гц, 1H), 7,62 (d, J=7,9 Гц, 1H), 7,32 (t, J=7,9 Гц, 1H), 3,57 (q, J=5,2 Гц, 2H), 2,79-2,35 (m, 10Н), 2,33 (s, 3Н), 1,78 (квинт., J=5,2 Гц, 2Н).
В соответствии с путем (А) реакционную смесь 3-бром-N-(4-метилпиперазин-1-ил-пропил)бензамида (170 мг, 0,5 ммоль, 1 экв.), 3-амино-N-метил-N-(пиридин-4-ил)бензамида (125 мг, 0,55 ммоль, 1,1 экв.), Pd2(dba)3 (23 мг, 0,025 ммоль, 5 мол. %), XPhos (24 мг, 0,05 ммоль, 10 мол. %) и K2CO3 (276 мг, 2 ммоль, 4 экв.) в трет-BuOH (2 мл) нагревали при 90°C и перемешивали в течение 20 часов в инертной атмосфере аргона. Реакционную смесь затем концентрировали при пониженном давлении, и полученный остаток разводили этилацетатом. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением N-метил-3-((3-((3-(4-метилпиперазин-1-ил)пропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)бензамида (20) (34 мг, 14%).
Пример 7: соединение (25) в таблице I
В соответствии с путем (Е) реакционную смесь 4-аминопиримидина (885 мг, 9,3 ммоль, 1,1 экв.), 3-нитробензойной кислоты (1,4 г, 8,4 ммоль, 1 экв.), EDCI.HCl (2,4 г, 12,6 ммоль, 1,5 экв.), триэтиламина (1,3 мл, 9,3 ммоль, 1,1 экв.) и диметиламинопиридина (103 мг, 0,8 ммоль, 0,1 экв.) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. Полученный осадок фильтровали и промывали водой и дихлорметаном. Органический фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле. Первый осадок и очищенное соединение собирали с получением 3-нитро-N-(пиримидин-4-ил)бензамида (1,35 г, 66%).
1Н ЯМР (300 МГц, d6-DMSO) δ 11,69 (s, 1Н), 8,99 (s, 1Н), 8,83 (s, 1Н), 8,76 (d, J=5,6 Гц, 1Н), 8,50-8,40 (m, 2Н), 8,22 (dt, J=5,6, 1,2 Гц, 1Н), 7,83 (t, J=7,9 Гц, 1Н).
В соответствии с путем (С) 3-нитро-N-(пиримидин-4-ил)бензамид (1,35 г, 5,5 ммоль, 1 экв.) и 10% Pd/C (303 мг) помещали в EtOH (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере H2. Реакционную смесь затем фильтровали на целите, промывали EtOH, и фильтрат концентрировали при пониженном давлении с получением 3-амино-N-(пиримидин-4-ил)бензамида (1,2 г, 100%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,93 (br s, 1Н), 8,92 (dd, J=1,3, 0,5 Гц, 1Н), 8,69 (dd, J=5,8, 0,5 Гц, 1Н), 8,18 (dd, J=5,8, 1,3 Гц, 1Н), 7,20-7,12 (m, 3Н), 6,78 (dt, J=4,1, 2,3 Гц, 1Н), 5,35 (s, 2Н).
N,N-диэтилпропилендиамин (8,7 мл, 55 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (71 мл), и к раствору добавляли дихлорметан (30 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор 3-бромбензоилхлорида (6,6 мл, 50 ммоль, 1 экв.) в дихлорметане (50 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. При декантации органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 3-бром-N-(3-диэтиламино-пропил)бензамида (15,6 г, 100%).
1Н ЯМР (300 МГц, CDCl3) δ 9,15 (br s, 1Н), 7,91 (s, 1Н), 7,75 (d, J=7,9 Гц, 1H), 7,58 (d, J=7,9 Гц, 1H), 7,29 (t, J=7,9 Гц, 1H), 3,56 (dd, J=10,3, 5,8 Гц, 2H), 2,67-2,53 (m, 6Н), 1,74 (квинт,, J=5,7 Гц, 2Н), 1,04 (t, J=7,1 Гц, 6Н).
В соответствии с путем (А) реакционную смесь 3-бром-N-(4-диэтиламино-пропил)бензамида (156 мг, 0,5 ммоль, 1 экв.), 3-амино-N-(пиримидин-4-ил)бензамида (118 мг, 0,55 ммоль, 1,1 экв.), Pd2(dba)3 (23 мг, 0,025 ммоль, 5 мол. %), XPhos (24 мг, 0,05 ммоль, 10 мол. %) и K2CO3 (276 мг, 2 ммоль, 4 экв.) в трет-BuOH (2 мл) нагревали при 90°C и перемешивали в течение 20 часов в инертной атмосфере аргона. Реакционную смесь затем концентрировали при пониженном давлении, и полученный остаток разводили этилацетатом. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением N-метил-3-((3-((3-(4-метилпиперазин-1-ил)пропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)бензамида (25) (16 мг, 7%).
Пример 8: соединение (31) в таблице I
В соответствии с путем (Е) реакционную смесь 4-аминопиримидина (1,0 г, 10,5 ммоль, 1 экв.), 3-нитробензойной кислоты (1,76 г, 10,5 ммоль, 1 экв.), EDCI.HCl (3,0 г, 15,8 ммоль, 1,5 экв.), триэтиламина (1,6 мл, 11,6 ммоль, 1,1 экв.) и диметиламинопиридина (129 мг, 1,05 ммоль, 0,1 экв.) в дихлорметане (12 мл) перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. Полученный осадок фильтровали и промывали водой и дихлорметаном. Органический фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле. Первый осадок и очищенное соединение собирали с получением 3-нитро-N-(пиримидин-4-ил)бензамида (2,5 г, 97%).
1Н ЯМР (300 МГц, d6-DMSO) δ 11,69 (s, 1Н), 9,00 (s, 1Н), 8,83 (t, J=2,0 Гц, 1Н), 8,77 (d, J=5,7 Гц, 1Н), 8,51-8,41 (m, 2Н), 8,22 (dd, J=5,7, 1,1 Гц, 1Н), 7,84 (t, J=8,0 Гц, 1Н).
В соответствии с путем (С) 3-нитро-N-(пиримидин-4-ил)бензамид (3,3 г, 13,5 ммоль, 1 экв.) и 10% Pd/C (719 мг) помещали в EtOH (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере H2. Реакционную смесь затем фильтровали на целите, промывали EtOH, и фильтрат концентрировали при пониженном давлении с получением 3-амино-N-(пиримидин-4-ил)бензамида (1,6 г, 55%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,94 (s, 1Н), 8,92 (dd, J=1,3, 0,5 Гц, 1Н), 8,69 (dd, J=5,8, 0,5 Гц, 1Н), 8,18 (dd, J=5,8, 1,3 Гц, 1Н), 7,20-7,10 (m, 3Н), 6,78 (dt, J=4,1, 2,3 Гц, 1Н), 5,35 (s, 2Н).
3-(4-Метилпиперазин-1-ил)пропиламин (1,9 мл, 11 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (14 мл), и к раствору добавляли дихлорметан (6 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор 3-бромбензоилхлорида (1,3 мл, 10 ммоль, 1 экв.) в дихлорметане (10 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. При декантации органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 3-бром-N-(4-метилпиперазин-1-ил-пропил)бензамида (2,7 г, 80%).
1Н ЯМР (300 МГц, CDCl3) δ 8,61 (br s, 1Н), 7,92 (s, 1Н), 7,82 (d, J=7,9 Гц, 1Н), 7,62 (d, J=7,9 Гц, 1Н), 7,32 (t, J=7,9 Гц, 1Н), 3,57 (q, J=5,2 Гц, 2Н), 2,79-2,35 (m, 10Н), 2,33 (s, 3Н), 1,78 (квинт., J=5,2 Гц, 2Н).
В соответствии с путем (А) реакционную смесь 3-бром-N-(4-метилпиперазин-1-ил-пропил)бензамида (576 мг, 1,7 ммоль, 1 экв.), 3-амино-N-(пиримидин-4-ил)бензамида (400 мг, 1,87 ммоль, 1,1 экв.), Pd2(dba)3 (78 мг, 0,085 ммоль, 5 мол. %), XPhos (81 мг, 0,17 ммоль, 10 мол. %) и K2CO3 (940 мг, 6,8 ммоль, 4 экв.) в трет-BuOH (7 мл) нагревали при 90°C и перемешивали в течение 20 часов в инертной атмосфере аргона. Реакционную смесь затем концентрировали при пониженном давлении, и полученный остаток разводили этилацетатом. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный осадок очищали посредством колоночной хроматографии на силикагеле с получением N-(3-(4-метилпиперазин-1-ил)пропил)-3-((3-(пиримидин-4-илкарбамоил)фенил)амино)бензамида (31) (54 мг, 7%).
Пример 9: соединение (35) в таблице I
Согласно способу (В) 4-аминопиридин (2,1 г, 22 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (28 мл), и к раствору добавляли дихлорметан (12 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор 3-нитробензоилхлорида (3,7 г, 20 ммоль, 1 экв.) в дихлорметане (20 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. Полученный осадок фильтровали и промывали водой и дихлорметаном с получением 3-нитро-N-(пиридин-4-ил)бензамида (2,4 г, 50%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,98 (s, 1Н), 8,80 (s, 1Н), 8,51 (d, J=6,2 Гц, 2Н), 8,47 (d, J=7,9 Гц, 1Н), 8,42 (d, J=7,9 Гц, 1Н), 7,86 (t, J=7,9 Гц, 1Н), 7,80 (d, J=6,2 Гц, 2Н).
В соответствии с путем (С) 3-нитро-N-(пиридин-4-ил)бензамид (1 г, 4,1 ммоль, 1 экв.) и 10% Pd/C (150 мг) помещали в EtOH (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере H2. Реакционную смесь затем фильтровали на целите, промывали EtOH, и фильтрат концентрировали при пониженном давлении с получением 3-амино-N-(пиридин-4-ил)бензамида (660 мг, 75%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,46 (s, 1Н), 8,45 (dd, J=5,0, 1,3 Гц, 2Н), 7,77 (dd, J=5,0, 1,3 Гц, 2Н), 7,17 (t, J=7,9 Гц, 1Н), 7,12-7,03 (m, 2Н), 6,77 (dd, J=7,9, 1,2 Гц, 1Н), 5,38 (s, 2Н).
6-Хлор-пиридин-2-карбоновую кислоту (4,4 г, 27,9 ммоль, 1,1 экв.) помещали в инертную атмосферу аргона. Медленно добавляли тионилхлорид (8,1 мл, 111,6 ммоль, 4 экв.). Реакционную смесь нагревали с обратным холодильником и перемешивали в течение 48 часов. При охлаждении до комнатной температуры реакционную смесь концентрировали при пониженном давлении. N,N-диэтилпропилендиамин (2,5 мл, 15,7 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (20 мл), и к раствору добавляли дихлорметан (10 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор остатка 6-хлорпиридин-2-карбонилхлорида (2,5 г, 14,3 ммоль, 1 экв.) в дихлорметане (13 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. При декантации органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением (3-диэтиламино-пропил)амида 6-хлор-пиридин-2-карбоновой кислоты (2,7 г, 70%).
1Н ЯМР (300 МГц, CDCl3) δ 9,22 (s, 1Н), 8,10 (d, J=7,9 Гц, 1Н), 7,77 (t, J=7,9 Гц, 1Н), 7,40 (d, J=7,9 Гц, 1Н), 3,53 (dd, J=12,1, 5,8 Гц, 2Н), 2,65-2,49 (m, 6Н), 1,82-1,68 (m, 2Н), 1,06 (t, J=7,1 Гц, 6Н).
В соответствии с путем (А) реакционную смесь (3-диэтиламино-пропил)амида 6-хлор-пиридин-2-карбоновой кислоты (135 мг, 0,5 ммоль, 1 экв.), 3-амино-N-(пиридин-4-ил)бензамида (117 мг, 0,55 ммоль, 1,1 экв.), Pd2(dba)3 (23 мг, 0,025 ммоль, 5 мол. %), XPhos (24 мг, 0,05 ммоль, 10 мол. %) и K2CO3 (276 мг, 2 ммоль, 4 экв.) в трет-BuOH (2 мл) нагревали при 90°C и перемешивали в течение 20 часов в инертной атмосфере аргона. Реакционную смесь затем концентрировали при пониженном давлении, и полученный остаток разводили этилацетатом. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением N-(3-(диэтиламино)пропил)-6-((3-(пиридин-4-илкарбамоил)фенил)амино)пиколинамида (35) (79 мг, 35%).
Пример 10: соединение (39) в таблице I
В соответствии с путем (D) реакционную смесь 4-(метиламино)пиридина (1,25 г, 11,6 ммоль, 1,0 экв.), 3-нитробензоилхлорида (2,57 г, 13,9 ммоль, 1,2 экв.), N,N-диизопропилэтиламина (3,02 мл, 17,3 ммоль, 1,5 экв.) и диметиламинопиридина (103 мг, 1,41 ммоль, 1 экв.) в дихлорметане (25 мл) перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением N-метил-3-нитро-N-(пиридин-4-ил)бензамида (2,96 г, 100%).
1Н ЯМР (300 МГц, CDCl3) δ 8,50 (dd, J=4,6, 1,6 Гц, 2Н), 8,25 (s, 1Н), 8,21 (d, J=7,9 Гц, 1Н), 7,62 (d, J=7,9 Гц, 1Н), 7,45 (t, J=7,9 Гц, 1Н), 6,98 (dd, J=4,6, 1,6 Гц, 2Н), 3,56 (s, 3Н).
В соответствии с путем (С) N-метил-3-нитро-N-(пиридин-4-ил)бензамид (2,96 г, 11,5 ммоль, 1 экв.) и 10% Pd/C (450 мг) помещали в EtOH (100 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере H2. Реакционную смесь затем фильтровали на целите, промывали EtOH, и фильтрат концентрировали при пониженном давлении с получением 3-амино-N-метил-N-(пиридин-4-ил)бензамида (2,5 г, 96%).
1Н ЯМР (300 МГц, d6-DMSO) δ 8,40 (dd, J=4,6, 1,6 Гц, 2Н), 7,14 (dd, J=4,6, 1,6 Гц, 2Н), 6,89 (t, J=7,9 Гц, 1Н), 6,59 (s, 1Н), 6,53 (d, J=7,9 Гц, 1Н), 6,34 (d, J=7,9 Гц, 1Н), 5,22 (s, 2Н), 3,37 (s, 3Н).
2-Хлор-изоникотиновую кислоту (2,0 г, 12,7 ммоль, 1 экв.) помещали в ацетонитрил (25,4 мл) в инертной атмосфере аргона. Медленно добавляли тионилхлорид (1,2 мл, 16,5 ммоль, 1,3 экв.) и DMF (диметилфумарат) (100 мкл, 1,27 ммоль, 0,1 экв.). Реакционную смесь нагревали с обратным холодильником и перемешивали в течение 1 часа. При охлаждении до комнатной температуры реакционную смесь концентрировали при пониженном давлении. 3-(4-Метилпиперазин-1-ил)пропиламин (2,7 мл, 15,7 ммоль,1,2 экв.) помещали в 3 н. водный раствор NaOH (20 мл), и к раствору добавляли дихлорметан (10 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор остатка 2-хлор-изоникотиноилхлорида (12,7 ммоль, 1 экв.) в дихлорметане (13 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. При декантации органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 2-хлор-N-[(3-(4-метил-пиперазин-1-ил)-пропил]-изоникотинамида (1,8 г, 43%).
1Н ЯМР (300 МГц, CDCl3) δ 9,03 (s, 1Н), 8,51 (d, J=4,9 Гц, 1Н), 7,71-7,64 (m, 2Н), 3,58 (dd, J=10,8, 5,0 Гц, 2Н), 2,66-2,40 (m, 10Н), 2,32 (s, 3Н), 1,84-1,73 (m, 2Н).
В соответствии с путем (А) реакционную смесь 2-хлор-N-[3-(4-метилпиперазин-1-ил)-пропил]-изоникотинамида (148 мг, 0,5 ммоль, 1 экв.), 3-амино-N-метил-N-(пиридин-4-ил)бензамида (125 мг, 0,55 ммоль, 1,1 экв.), Pd2(dba)3 (23 мг, 0,025 ммоль, 5 мол. %), XPhos (24 мг, 0,05 ммоль, 10 мол. %) и К2CO3 (276 мг, 2 ммоль, 4 экв.) в трет-BuOH (2 мл) нагревали при 90°C и перемешивали в течение 20 часов в инертной атмосфере аргона. Реакционную смесь затем концентрировали при пониженном давлении, и полученный остаток разводили этилацетатом. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением 2-((3-(метил(пиридин-4-ил)карбамоил)фенил)амино)-N-(3-(4-метилпиперазин-1-ил)пропил)изоникотинамида (39) (43 мг, 18%).
Пример 11: соединение (41) в таблице I
В соответствии с путем (Е) реакционную смесь 4-аминопиримидина (885 мг, 9,3 ммоль, 1,1 экв.), 3-нитробензойной кислоты (1,4 г, 8,4 ммоль, 1 экв.), EDCI.HCl (2,4 г, 12,6 ммоль, 1,5 экв.), триэтиламина (1,3 мл, 9,3 ммоль, 1,1 экв.) и диметиламинопиридина (103 мг, 0,8 ммоль, 0,1 экв.) в дихлорметане (10 мл) перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. Полученный осадок фильтровали и промывали водой и дихлорметаном. Органический фильтрат концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле. Первый осадок и очищенное соединение собирали с получением 3-нитро-N-(пиримидин-4-ил)бензамида (1,35 г, 66%).
1Н ЯМР (300 МГц, d6-DMSO) δ 11,69 (s, 1Н), 8,99 (s, 1Н), 8,83 (s, 1Н), 8,76 (d, J=5,6 Гц, 1Н), 8,50-8,40 (m, 2Н), 8,22 (dt, J=5,6, 1,2 Гц, 1Н), 7,83 (t, J=7,9 Гц, 1Н).
В соответствии с путем (С) 3-нитро-N-(пиримидин-4-ил)бензамид (1,35 г, 5,5 ммоль, 1 экв.) и 10% Pd/C (303 мг) помещали в EtOH (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере H2. Реакционную смесь затем фильтровали на целите, промывали EtOH, и фильтрат концентрировали при пониженном давлении с получением 3-амино-N-(пиримидин-4-ил)бензамида (1,2 г, 100%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,93 (br s, 1Н), 8,92 (dd, J=1,3, 0,5 Гц, 1H), 8,69 (dd, J=5,8, 0,5 Гц, 1H), 8,18 (dd, J=5,8, 1,3 Гц, 1H), 7,20-7,12 (m, 3Н), 6,78 (dt, J=4,1, 2,3 Гц, 1H), 5,35 (s, 2H).
2-Хлор-изоникотиновую кислоту (2,0 г, 12,7 ммоль, 1,1 экв.) помещали в ацетонитрил (25,4 мл) в инертной атмосфере аргона. Медленно добавляли тионилхлорид (1,2 мл, 16,5 ммоль, 1,3 экв.) и DMF (100 мкл, 1,27 ммоль, 0,1 экв.). Реакционную смесь нагревали с обратным холодильником и перемешивали в течение 1 часа. При охлаждении до комнатной температуры реакционную смесь концентрировали при пониженном давлении. N,N-диэтилпропилендиамин (2,5 мл, 15,7 ммоль, 1,2 экв.) помещали в 3 н. водный раствор NaOH (20 мл), и к раствору добавляли дихлорметан (10 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор остатка 2-хлор-изоникотиноилхлорида (12,7 ммоль, 1 экв.) в дихлорметане (13 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. При декантации органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 2-хлор-N-(3-диэтиламино-пропил)изоникотинамида (1,8 г, 47%).
1Н ЯМР (300 МГц, CDCl3) δ 9,62 (s, 1Н), 8,49 (d, J=5,0 Гц, 1Н), 7,66 (s, 1Н), 7,58 (d, J=5,1 Гц, 1Н), 3,59 (dd, J=10,4, 5,0 Гц, 2Н), 2,70-2,55 (m, 6Н), 1,81-1,72 (m, 2Н), 1,06 (t, J=7,1 Гц, 6Н).
Согласно способу (А) реакционную смесь 2-хлор-N-(3-диэтиламино-пропил)изоникотинамида (135 мг, 0,5 ммоль, 1 экв.), 3-амино-N-(пиримидин-4-ил)бензамида (118 мг, 0,55 ммоль, 1,1 экв.), Pd2(dba)3 (23 мг, 0,025 ммоль, 5 мол. %), XPhos (24 мг, 0,05 ммоль, 10 мол. %) и K2CO3 (276 мг, 2 ммоль, 4 экв.) в трет-BuOH (2 мл) нагревали при 90°C и перемешивали в течение 20 часов в инертной атмосфере аргона. Реакционную смесь затем концентрировали при пониженном давлении, и полученный остаток разводили этилацетатом. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением N-(3-(диэтиламино)пропил)-2-((3-(пиримидин-4-илкарбамоил)фенил)амино)изоникотинамида (41) (79 мг, 35%).
Пример 12: соединение (44) в таблице I
В соответствии с путем (Е) реакционную смесь 4-аминопиридина (837 мг, 8,9 ммоль, 1,3 экв.), 2-нитро-изоникотиновой кислоты (1,15 г, 6,8 ммоль, 1 экв.), EDCI.HCl (1,7 г, 8,9 ммоль, 1,3 экв.), N,N-диизопропилэтиламина (3,0 мл, 17,1 ммоль, 2,5 экв.) и диметиламинопиридина (272 мг, 2,2 ммоль, 0,25 экв.) в дихлорметане (7 мл) перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный осадок очищали посредством колоночной хроматографии на силикагеле с получением 2-нитро-N-пиридин-4-ил-изоникотинамида (835 мг, 50%).
1Н ЯМР (300 МГц, MeOD) δ 8,86-8,80 (m, 2Н), 8,50 (dd, J=5,0, 1,6 Гц, 2Н), 8,31 (dd, J=4,8, 1,5 Гц, 1Н), 7,88 (dd, J=5,0, 1,6 Гц, 2Н).
В соответствии с путем (С) 2-нитро-N-пиридин-4-ил-бизоникотинамид (835 мг, 3,4 ммоль, 1 экв.) и 10% Pd/C (150 мг) помещали в EtOH (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере H2. Реакционную смесь затем фильтровали на целите, промывали EtOH, и фильтрат концентрировали при пониженном давлении с получением 2-амино-N-пиридин-4-ил-изоникотинамида (727 мг, 99%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,61 (s, 1Н), 8,48 (dd, J=4,8, 1,5 Гц, 2Н), 8,07 (d, J=5,3 Гц, 1Н), 7,75 (dd, J=4,8, 1,5 Гц, 2Н), 6,91 (d, J=5,3 Гц, 1Н), 6,86 (s, 1Н), 6,28 (s, 2Н).
2-Хлор-изоникотиновую кислоту (2,0 г, 12,7 ммоль, 1,1 экв.) помещали в ацетонитрил (25,4 мл) в инертной атмосфере аргона. Медленно добавляли тионилхлорид (1,2 мл, 16,5 ммоль, 1,3 экв.) и DMF (100 мкл, 1,27 ммоль, 0,1 экв.). Реакционную смесь нагревали с обратным холодильником и перемешивали в течение 1 часа. При охлаждении до комнатной температуры реакционную смесь концентрировали при пониженном давлении. N,N-диэтилпропилендиамин (2,5 мл, 15,7 ммоль, 1,2 экв.) помещали в 3 н. водный раствор NaOH (20 мл), и к раствору добавляли дихлорметан (10 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор остатка 2-хлор-изоникотиноилхлорида (12,7 ммоль, 1 экв.) в дихлорметане (13 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. При декантации органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 2-хлор-N-(3-диэтиламино-пропил)изоникотинамида (1,8 г, 47%).
1Н ЯМР (300 МГц, CDCl3) δ 9,62 (s, 1Н), 8,49 (d, J=5,0 Гц, 1Н), 7,66 (s, 1Н), 7,58 (d, J=5,1 Гц, 1Н), 3,59 (dd, J=10,4, 5,0 Гц, 2Н), 2,70-2,55 (m, 6Н), 1,81-1,72 (m, 2Н), 1,06 (t, J=7,1 Гц, 6Н).
В соответствии с путем (А) реакционную смесь 2-хлор-N-(3-диэтиламино-пропил)изоникотинамида (135 мг, 0,5 ммоль, 1 экв.), 2-амино-N-пиридин-4-ил-изоникотинамида (118 мг, 0,55 ммоль, 1,1 экв.), Pd2(dba)3 (23 мг, 0,025 ммоль, 5 мол. %), XPhos (24 мг, 0,05 ммоль, 10 мол. %) и K2CO3 (276 мг, 2 ммоль, 4 экв.) в трет-BuOH (2 мл) нагревали при 90°C и перемешивали в течение 20 часов в инертной атмосфере аргона. Реакционную смесь затем концентрировали при пониженном давлении, и полученный остаток разводили этилацетатом. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением N-(3-(диэтиламино)пропил)-2-((4-(пиридин-4-илкарбамоил)пиридин-2-ил)амино)изоникотинамида (44) (70 мг, 31%).
Пример 13: соединение (47) в таблице I
В соответствии с путем (Е) реакционную смесь 4-аминопиридина (837 мг, 8,9 ммоль, 1,3 экв.), 2-нитро-изоникотиновой кислоты (1,15 г, 6,8 ммоль, 1 экв.), EDCI.HCl (1,7 г, 8,9 ммоль, 1,3 экв.), N,N-диизопропилэтиламина (3,0 мл, 17,1 ммоль, 2,5 экв.) и диметиламинопиридина (272 мг, 2,2 ммоль, 0,25 экв.) в дихлорметане (7 мл) перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением 2-нитро-N-пиридин-4-ил-изоникотинамида (835 мг, 50%).
1Н ЯМР (300 МГц, MeOD) δ 8,86-8,80 (m, 2Н), 8,50 (dd, J=5,0, 1,6 Гц, 2Н), 8,31 (dd, J=4,8, 1,5 Гц, 1Н), 7,88 (dd, J=5,0, 1,6 Гц, 2Н).
В соответствии с путем (С) 2-нитро-N-пиридин-4-ил-изоникотинамид (835 мг, 3,4 ммоль, 1 экв.) и 10% Pd/C (150 мг) помещали в EtOH (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере H2. Реакционную смесь затем фильтровали на целите, промывали EtOH, и фильтрат концентрировали при пониженном давлении с получением 2-амино-N-пиридин-4-ил-изоникотинамида (727 мг, 99%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,61 (s, 1Н), 8,48 (dd, J=4,8, 1,5 Гц, 2Н), 8,07 (d, J=5,3 Гц, 1Н), 7,75 (dd, J=4,8, 1,5 Гц, 2Н), 6,91 (d, J=5,3 Гц, 1Н), 6,86 (s, 1Н), 6,28 (s, 2Н).
N,N-диэтилпропилендиамин (8,7 мл, 55 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (71 мл), и к раствору добавляли дихлорметан (30 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор 3-бромбензоилхлорида (6,6 мл, 50 ммоль, 1 экв.) в дихлорметане (50 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. При декантации органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 2-бром-N-(3-диэтиламино-пропил)бензамида (14,6 г, 94%).
1Н ЯМР (300 МГц, CDCl3) δ 9,16 (br s, 1Н), 7,91 (t, J=1,8 Гц, 1Н), 7,75 (d, J=7,9 Гц, 1Н), 7,58 (d, J=7,9 Гц, 1Н), 7,29 (t, J=7,9 Гц, 1Н), 3,56 (dd, J=10,1, 5,7 Гц, 2Н), 2,72-2,50 (m, 6Н), 1,75 (квинт., J=5,7 Гц, 2Н), 1,05 (t, J=7,1 Гц, 6Н)
В соответствии с путем (А) реакционную смесь 3-бром-N-(3-диэтиламино-пропил)бензамида (156 мг, 0,5 ммоль, 1 экв.), 2-амино-N-пиридин-4-ил-изоникотинамида (118 мг, 0,55 ммоль, 1,1 экв.), Pd2(dba)3 (23 мг, 0,025 ммоль, 5 мол. %), XPhos (24 мг, 0,05 ммоль, 10 мол. %) и K2CO3 (276 мг, 2 ммоль, 4 экв.) в трет-BuOH (2 мл) нагревали при 90°C и перемешивали в течение 20 часов в инертной атмосфере аргона. Реакционную смесь затем концентрировали при пониженном давлении, и полученный остаток разводили этилацетатом. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный осадок очищали посредством колоночной хроматографии на силикагеле с получением 2-((3-((3-(диэтиламино)пропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)изоникотинамида (47) (52 мг, 23%).
Пример 14: соединение (51) в таблице I
В соответствии с путем (Е) реакционную смесь 4-аминопиридина (1,57 г, 16,7 ммоль, 1,3 экв.), 2-нитро-изоникотиновой кислоты (2,16 г, 12,9 ммоль, 1 экв.), EDCI.HCl (3,69 г, 19,3 ммоль, 1,5 экв.), N,N-диизопропилэтиламина (5,3 мл, 32,1 ммоль, 2,5 экв.) и диметиламинопиридина (392 мг, 3,2 ммоль, 0,25 экв.) в дихлорметане (15 мл) перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением 2-нитро-N-пиридин-4-ил-изоникотинамида (1,68 г, 54%).
1Н ЯМР (300 МГц, MeOD) δ 8,86-8,80 (m, 2Н), 8,50 (dd, J=5,0, 1,6 Гц, 2Н), 8,31 (dd, J=4,8, 1,5 Гц, 1Н), 7,88 (dd, J=5,0, 1,6 Гц, 2Н).
В соответствии с путем (С) 2-нитро-N-пиридин-4-ил-изоникотинамид (1,1 г, 4,5 ммоль, 1 экв.) и 10% Pd/C (240 мг) помещали в EtOH (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов в атмосфере H2. Реакционную смесь затем фильтровали на целите, промывали EtOH, и фильтрат концентрировали при пониженном давлении с получением 2-амино-N-пиридин-4-ил-изоникотинамида (898 мг, 93%).
1Н ЯМР (300 МГц, d6-DMSO) δ 10,68 (s, 1Н), 8,48 (d, J=5,9 Гц, 2Н), 8,07 (d, J=5,2 Гц, 1Н), 7,77 (d, J=5,9 Гц, 2Н), 6,93 (d, J=5,2 Гц, 1Н), 6,87 (s, 1Н), 6,27 (s, 2Н).
3-(4-Метилпиперазин-1-ил)пропиламин (1,9 мл, 11 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (14 мл), и к раствору добавляли дихлорметан (6 мл). Реакционную смесь охлаждали до 0°C с помощью ледяной бани, и по каплям добавляли раствор 3-бромбензоилхлорида (1,3 мл, 10 ммоль, 1 экв.) в дихлорметане (10 мл). Реакционную смесь затем перемешивали при комнатной температуре в течение 18 часов в инертной атмосфере аргона. При декантации органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 3-бром-N-(4-метилпиперазин-1-ил-пропил)бензамида (2,7 г, 80%).
1Н ЯМР (300 МГц, CDCl3) δ 8,61 (br s, 1Н), 7,92 (s, 1Н), 7,82 (d, J=7,9 Гц, 1H), 7,62 (d, J=7,9 Гц, 1H), 7,32 (t, J=7,9 Гц, 1H), 3,57 (q, J=5,2 Гц, 2H), 2,79-2,35 (m, 10Н), 2,33 (s, 3Н), 1,78 (квинт., J=5,2 Гц, 2Н).
В соответствии с путем (А) реакционную смесь 3-бром-N-(4-метилпиперазин-1-ил-пропил)бензамида (123 мг, 0,36 ммоль, 1 экв.), 2-амино-N-(пиридин-4-ил-изоникотинамида (96 мг, 0,45 ммоль, 1,1 экв.), Pd2(dba)3 (17 мг, 0,018 ммоль, 5 мол. %), XPhos (17 мг, 0,036 ммоль, 10 мол. %) и K2CO3 (200 мг, 1,44 ммоль, 4 экв.) в трет-BuOH (1,4 мл) нагревали при 90°C и перемешивали в течение 48 часов в инертной атмосфере аргона. Реакционную смесь затем концентрировали при пониженном давлении, и полученный остаток разводили этилацетатом. Органическую фазу промывали водой, высушивали над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением 2-((3-((3-(4-метилпиперазин-1-ил)пропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)изоникотинамида (51) (34 мг, 20%).
Пример 15: Фармакологические данные
Стандартная рабочая процедура:
Эффект лекарственных соединений на инвазию клеток MDA-MB231-D3H2LN в коллаген
Предварительная информация:
Ключевой стадией в образовании опухолевого метастаза является инвазия опухолевых клеток во внеклеточный матрикс, главным компонентом которого является коллаген. Вследствие этого, инвазия опухолевых клеток в коллаген in vitro может являться показателем образования метастаза in vivo. Например, клетки рака молочной железы мыши MDA-MB231-luc-D3H2LN демонстрируют в действительности как более высокую инвазию в коллаген in vitro, так и более высокий метастатический потенциал in vivo, по сравнению с клетками MDA-MB231 (из которых они были получены). Используя данные клетки MDA-MB231-luc-D3H2LN в качестве модели, целью эксперимента, описанного в данном документе, является идентификация лекарственных соединений, которые ингибируют инвазию опухолевых клеток в коллаген in vitro, вследствие этого потенциально также ингибируя образование опухолевого метастаза in vivo.
Принцип анализа:
Стадия 1: Получение клеток на дне коллагенового геля: клетки суспендируют в жидком коллагеновом растворе (4°C), распределяют в лунки, покрытые BSA (бычий сывороточный альбумин), и затем собирают на дне лунок посредством центрифугирования. Затем коллаген затвердевает в результате инкубации при 37°C. BSA-покрытие улучшает адгезию коллагенового геля.
Стадия 2: Предварительная обработка соединениями, подлежащими тестированию: концентрированные растворы лекарственных средств затем добавляют поверх коллагена, и клетки предварительно инкубируют в течение 48 ч с лекарственными средствами в условиях низкой концентрации сыворотки (0,025% FBS (фетальная телячья сыворотка)).
Стадия 3: Стимуляция инвазии: затем среду с 5% FBS добавляют для стимуляции инвазии клеток в коллагеновый гель.
Стадия 4: Анализ жизнеспособности, фиксация и окрашивание: после еще 24 ч инкубации непосредственно на клетках в коллагене проводят анализ MTS. Затем фиксируют клетки и окрашивают ядра.
Стадия 5: Анализ: Наконец, планшеты анализируют с использованием автоматизированного микроскопа. Флуоресцентные гранулы, которые были включены в BSA-покрытие, служат для детекции дна лунок. Фотографии окрашенных ядер делают на том же уровне (0 мкм), а также выше 25 мкм и 50 мкм.
Внимание:
Для детекции возможных токсичных эффектов все соединения параллельно тестируют в анализе жизнеспособности. Анализ жизнеспособности проводят параллельно на бессывороточных клетках (как в анализе инвазии) по сравнению с клетками, находящимися в нормальных условиях культивирования (10% FBS).
Материалы:
Оборудование общего назначения: Морозильная камера (-20°C), холодильник (4°C), ледогенератор, водяная баня (37°C), инкубатор (37°C/5% CO2), бокс для клеточных культур, вортекс, вакуумный насос, микроскоп, пипеточный дозатор, микропипетки, (для отмеривания пипеткой 1-1000 мкл), мультиканальные пипетки (для отмеривания пипеткой 20-200 мкл), стандартная центрифуга для клеточных культур, центрифуга с охлаждением для 96-луночных планшетов.
Расходные материалы общего назначения: Стерильные пробирки (1,5/15/50 мл), стерильные пипетки (5/10/25 мл), стерильные наконечники для микропипеток (для отмеривания пипеткой 1-1000 мкл), стерильные пипетки Пастера, стерильные резервуары для реагентов.
Продукты общего назначения: Стерильный PBS (фосфатно-солевой буферный раствор), стерильная вода Milli-Q, DMSO, FBS без дополнений (замороженные аликвоты), 0,1 н. NaOH, 1 М Hepes (N-2-гидроксиэтилпиперазин-N'-2-этансульфоновая кислота), MEM без сыворотки (не старее, чем 1 месяц), 2,5×MEM без сыворотки (не старее, чем 1 месяц), MEM с 10% FBS (не старее, чем один месяц), раствор 0,25% трипсин/1 мМ EDTA (этилендиаминтетрауксусная кислота), 37% раствор формальдегида.
Специальное оборудование:
планшет-ридер: Tecan Infinite F200
автоматизированный микроскоп: Cellomics ArrayScan VTI HCS Reader
Специальные расходные материалы:
стерильные черные 96-луночные планшеты (для анализа инвазии): Perkin Elmer ViewPlate-96 F ТС, кат. №. 6005225
Специальные продукты:
коллаген крысиного хвоста, тип 1: BD Biosciences, кат. №354236 (внимание: каждая новая партия должна быть валидирована).
красные флуоресцентные гранулы (диаметр 1 мкм): Invitrogen, кат. №F13083
Y-27632 (5 мМ водный раствор): Calbiochem, кат. №688001 (в растворе) или 688000 (сухой порошок)
BSA без жирных кислот (стерильный-фильтрованный 4% водный раствор): Sigma, кат. №А8806 (сухой порошок)
ядерный краситель Hoechst 33342 (10 мг/мл): Invitrogen, кат. №Н3570
реагент MTS: Promega CellTiter CellTiter 96® AQueous One Solution Reagent, кат. №G3581
Лекарственные соединения, подлежащие тестированию: обычно 50 мМ в 100% DMSO (аликвоты, хранящиеся при -20°C, затем при 4°C в течение макс. 3 месяцев)
Клетки MDA-MB231-luc-D3H2LN:
Ограничения для клеточных культур, которые следует использовать в анализах:
общее количество пассажей: макс. 30
последний пассаж: от 2 до 4 суток, от 1:3 до 1:20
плотность клеток: от 50 до 90% (оптимально 70%) (от 1 до 2×106 клеток на 100 мм чашку)
Порядок проведения экспериментов:
Общие соображения: Контроли и схемы планшетов:
Отрицательный контроль: без лекарственного средства (только DMSO в эквивалентной концентрации). Положительный контроль: 10 мМ Y-27632. Для того, чтобы избежать краевых эффектов, клетки добавляют только к 60 центральным лункам B2-G11; горизонтальные ряды A и Н получают только коллаген без клеток (холостая проба для анализа MTS), вертикальные ряды 1 и 12 остаются свободными. Каждое лекарственное средство тестируют по меньшей мере в трехкратной повторности. Положительный и отрицательный контроли следует тестировать в нескольких трехкратных повторностях при разных положениях на каждом планшете. Типичная схема планшета (- = отрицательный контроль, + = положительный контроль, 1-12=12 разные условия тестирования, то есть разные лекарственные соединения или концентрации):
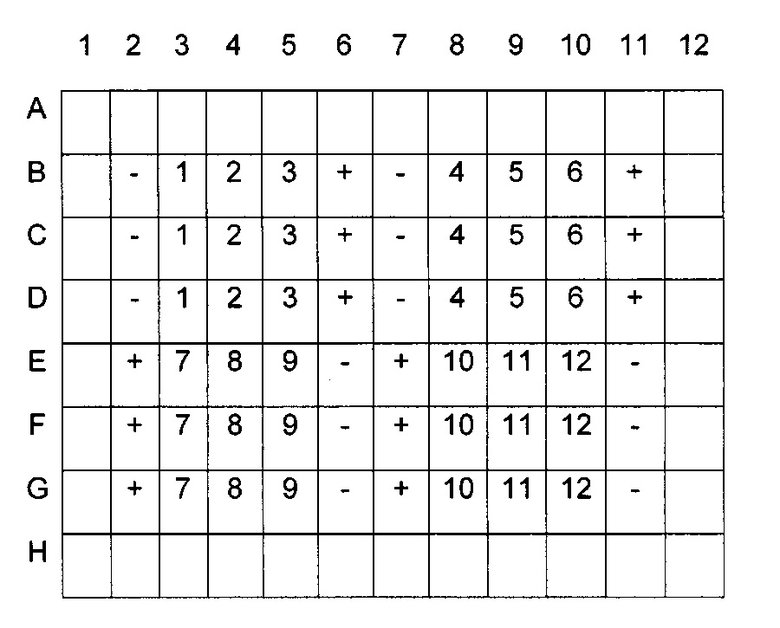
Объемы или другие количественные показатели, указанные ниже, требуются для четырех 96-луночных планшетов согласно указанной выше схеме планшета. В соответствии с количеством тестируемых соединений следует адаптировать объемы и другие количественные показатели.
Сутки 1. Получение и обработка клеток (все стадии проводят в боксе для клеточных культур):
Получение 4 × концентрированных растворов лекарственных средств в MEM + 0,1% FBS + 2% Lutrol Е-400 + 0,8% DMSO: смешать по 620 мкл MEM + 0,1% FBS + 2% Lutrol Е-400 с DMSO по 4 мкл + 50 мМ стоковыми растворами соединения по 1 мкл (с получением концентрации соединения 20 мкМ и 0,8% DMSO). Если требуемая конечная концентрация соединения составляет менее 5 мкМ, тогда дополнительно разбавить в MEM + 0,1% FBS + 0,8% DMSO. Отрицательный контроль: MEM + 0,1% FBS + 2% Lutrol E-400 + 0,8% DMSO без какого-либо лекарственного средства. Получение положительного контроля: Смешать 4,5 мл MEM + 0,1% FBS + 2% Lutrol E-400 + 0,8% FBS с 36 мкл 5 мМ Y-27632 (только что размороженная аликвота) (с получением 40 мкМ).
Покрытие планшетов для анализа инвазии:
Смешать 38 мл MEM без сыворотки + 2 мл 4% BSA без жирных кислот + 4 мкл перемешенных на вортексе флуоресцентных гранул (то есть разбавление 1:10000), перемешать на вортексе, распределить 100 мкл/лунку в планшет для анализа инвазии.
Центрифугировать 30 мин при 1800×g при 4°C (например, 3000 об/мин на центрифуге Jouan GR412).
Удалить супернатанты посредством аспирации.
Получение 10×106 клеток/мл клеточной суспензии (во время центрифугирования планшетов):
Удалить среду, промыть клетки приблизительно 10 мл/чашку PBS, добавить 1 мл/чашку 0,25% трипсина/1 мМ EDTA
Инкубировать 30-60 с при 37°C.
Добавить 5-10 мл/чашку предварительно нагретой MEM + 10% FBS. Гомогенизировать путем пипетирования с использованием 10 мл пипетки, все объединить.
Сосчитать клетки.
Центрифугировать 3×106 (или более) клеток в течение 5 мин при 150×g при КТ (комнатная температура) (850 об/мин. на стандартной центрифуге для клеточных культур).
Удалить супернатант, ресуспендировать массу клеток в 0,3 мл (или более, соответственно) MEM без сыворотки, с получением 10×106 клеток/мл.
Подготовка анализа инвазии (на льду: начать во время центрифугирования клеток):
Смешать на льду в заранее охлажденной пробирке: пример для 4,01 мг/мл стокового раствора коллагена; объемы коллагена и воды, подлежащие адаптации, в соответствии со стоковой концентрацией каждой партии коллагена:
16 мл 2,5 × MEM
5,452 мл воды
0,8 мл 1 М Hepes
0,39 мл 1 н. NaOH
16,958 мл 4,01 мг/мл стокового раствора коллагена.
Гомогенизировать путем осторожного пипетирования туда и обратно (держать на льду).
К 29,7 мл данной смеси добавить 300 мкл 10×106 клеток/мл клеточной суспензии, гомогенизировать посредством осторожного пипетирования туда и обратно (приводит к получению 0,1×106 клеток/мл в 1,7 мг/мл коллагена в 30 мл 1 × MEM + 20 мкМ Hepes) (держать на льду). К оставшимся 9,9 мл добавить 100 мкл MEM без сыворотки, гомогенизировать посредством осторожного пипетирования (приводит к получению 1,7 мг/мл коллагена в 10 мл 1 × MEM + 20 мкМ Hepes без клеток) (держать на льду).
Распределить 100 мкл/лунку в покрытые лунки (все на льду) согласно приведенной выше схеме планшета (горизонтальные ряды A и Н: коллаген без клеток, горизонтальные ряды В-G: коллаген с клетками: 10000 клеток/лунка).
Центрифугировать 5 мин при 200×g при 4°C (например, 1000 об/мин на центрифуге Jouan GR412).
Добавить 200 мкл/лунку PBS ко всем свободным лункам (вертикальные ряды 1 и 12).
Инкубировать 30 мин при 37°C/5% CO2 (затвердевание коллагена).
Обработка лекарственными средствами:
Добавить по 33 мкл/лунку 4 × концентрированных растворов лекарственных средств в MEM + 0,1% FBS + 2% Lutrol Е-400 + 0,8% DMSO к соответствующим лункам (приводит к получению в итоге 1 × концентрированных лекарственных средств в MEM + 0,025% FBS + 0,5% Lutrol Е-400 + 0,2% DMSO).
Инкубировать 48 ч при 37°C/5% CO2.
Сутки 3: Добавление FBS для стимуляции инвазии:
Получить MEM + 5% FBS: 19 мл MEM без сыворотки + 1 мл FBS (только что размороженная аликвота).
Добавить 33 мкл/лунку ко всем лункам.
Инкубировать 24 ч при 37°C/5% CO2.
Сутки 4: Анализ жизнеспособности, фиксация и окрашивание:
Анализ жизнеспособности: анализ MTS:
Добавить по 33 мкл/лунку реагента MTS, инкубировать 3-4 ч при 37°C/5% CO2.
Снять данные по поглощению при 490 нм (пропорционально жизнеспособности).
Рассчитать сигналы с поправкой на фон путем вычитания средних значений фоновых сигналов в отсутствии клеток из соответствующих сигналов в присутствии клеток.
Нормировать сигналы с поправкой на фон относительно среднего сигнала отрицательных контролей (без лекарственного средства) (жизнеспособность, таким образом, выражается в "% от контроля").
Фиксация и окрашивание (с формальдегидом операции нужно проводить в вытяжном шкафу):
Получить свежеприготовленный 1 мкг/мл Hoechst 33342 в 18,5% формальдегиде: 10 мл PBS (не обязательно стерильный) + 10 мл 37% формальдегида + 2 мкл 10 мг/мл Hoechst 33342.
Добавить 50 мкл/лунку ко всем лункам с клетками (приводит к получению в итоге 3,7% формальдегида).
Плотно закрыть черной пленкой (предоставлена с планшетами).
Инкубировать по меньшей мере 7 ч при КТ.
Сутки 5 (обычно): (минимум 7 ч/максимум 2 недели после Фиксации и окрашивания): Анализ тестирования инвазии:
Рекомендация по использованию ридера Cellomics ArrayScan VTI HCS:
Биоприложение: SpotDetector.V3
Тип планшета: 96-луночный Perkin Elmer
Параметры протокола анализа:
объектив: 10 × (NA.45)
апотом: есть (получающийся в результате оптический срез: 11,7 мкм)
областей на лунку: 6-8
автофокусировка в каждой области
канал автофокусировки: 1
канал 1 (автофокусировка включена, и фотография флуоресцентных гранул на дне лунок): фильтр: XF93 - TRITC; время экспозиции: обычно от 0,002 до 0,01 с;
канал 2 (фотография окрашенных клеток на том же уровне, как в случае флуоресцентных гранул): фильтр: XF93 - Hoechst; время экспозиции: обычно от 0,02 до 0,1 с; смещение по оси z: 0 мкм;
канал 3 (фотография окрашенных клеток 25 мкм выше флуоресцентных гранул): фильтр: XF93 - Hoechst; время экспозиции: обычно от 0,02 до 0,1 с; смещение по оси z: - 25 мкм;
канал 4 (фотография флуоресцентных клеток 50 мкм выше флуоресцентных гранул): фильтр: XF93 - Hoechst; время экспозиции: обычно от 0,02 до 0,1 с; смещение по оси z: - 50 мкм;

Анализ результатов сканирования с использованием vHCS Viewer:
экспортировать результаты: для каждой лунки:
число валидных областей
число объектов в каждой валидной области в каждом из каналов 2, 3 и 4 ("подробное описание")
среднее число объектов на валидную область для каждой лунки, в каждом из каналов 2, 3 и 4
исключить из дальнейшего анализа лунки с менее чем 6 валидными областями на лунку
визуально проверить все фотографии на предмет каких-либо очевидных проблем, таких как плохая фокусировка или очевидно негомогенная коллагеновая структура («пузырьки», …), …; в случае очевидных проблем: задокументировать, затем исключить соответствующие лунки из дальнейшего анализа
Дальнейший анализ результатов анализа инвазии (с использованием, например. Excel):
Для каждой лунки рассчитать среднее расстояние инвазии подсчитанных клеток: (25 мкм × число клеток на уровне 25 мкм + 50 мкм × число клеток на уровне 50 мкм)/сумма клеток на уровне 0, 25 и 50 мкм.
Для всех четырех параметров (число клеток на уровне 0 мкм, число клеток на уровне 25 мкм, число клеток на уровне 50 мкм, среднее расстояние инвазии подсчитанных клеток), рассчитать средние значения, SD (стандартное отклонение) и CV (коэффициент вариации) повторностей.
Не считать действительной любую повторность с CV≥50% (соединение, подлежащее повторному тестированию, или анализ, подлежащий повторному проведению, если CV≥50% в случае необработанного отрицательного контроля или положительного контроля, обработанного соединением Y-27632). Y27632 представляет собой селективный ингибитор Rho-ассоциированной протеинкиназы p160ROCK следующей формулы:
Признать анализ действительным, только если среднее расстояние инвазии клеток, обработанных 10 мкМ Y-27632 (положительный контроль), снижено на ≥40%, по сравнению с необработанным отрицательным контролем.
Итоговый анализ: определить концентрацию, при которой данное соединение обладает 50% антиинвазивного эффекта положительного контроля (10 мкМ Y-27632). Определить токсичность (равно потери жизнеспособности) соединения в данных условиях.
Анализы токсичности (в нормальных условиях культивирования клеток):
Соединения получали, как в случае анализа инвазии, и затем добавляли либо к клеткам MDA-MB231-luc-D3H2LN (2000/лунка) в нормальных условиях культивирования (MEM + 10% FBS), либо РВМС человека (75000/лунка, в RPMI + 10% FBS + IL-2 (интерлейкин-2)), в стандартных 96-луночных планшетах для тканевой культуры. После 72 ч инкубации проводили стандартный анализ MTS согласно инструкциям изготовителя (Promega, кат. №G3581). Соединения тестировали при разных концентрациях (кривые концентрация-ответ) для определения концентраций, при которых получают 50% токсичности.
Ингибирование канала hERG:
Выполнено Porsolt и Partners (Z.A. de Glatigné, 53940 Le Genest-Saint-Isle, Франция). Кратко, по 3 hERG-трансфицированные клетки НЕК293 обрызгивали соединениями в концентрации 1 и 10 мкМ, и ток канала hERG измеряли посредством электрофизиологии, как описано Crumb et al., J. Pharmacol. Exp. Ther. 2000.
Результаты
В приведенной ниже таблице показана токсичность в отношении MDA-МВ231, в отношении РВМС, антиинвазивный эффект и ингибирование канала hERG.
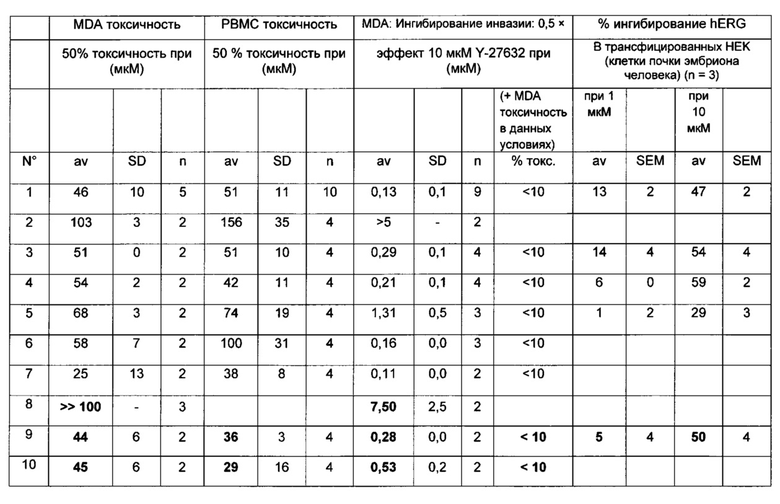
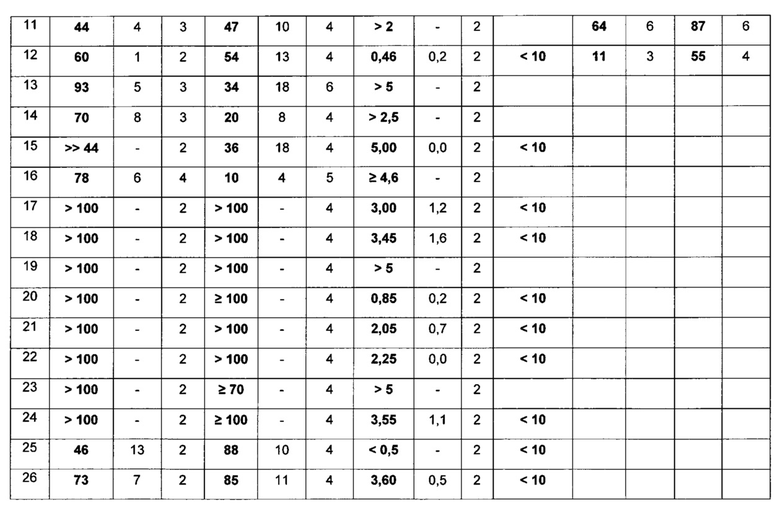
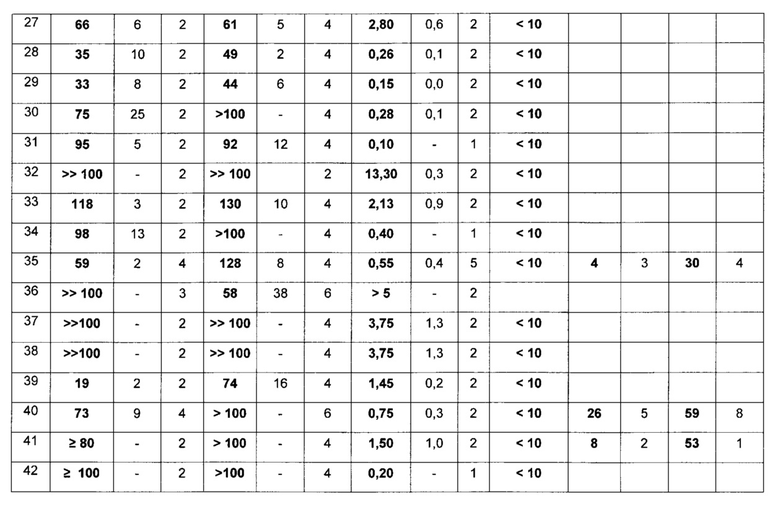
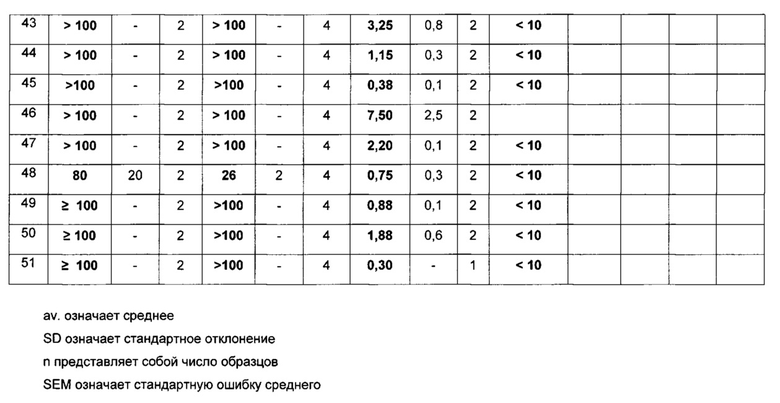
Пример 16: Сравнительные данные
Три соединения по настоящему изобретению (соединения 1, 3 и 4) соответственно сравнивают (токсичность MDA, токсичность РВМС, ингибирование инвазивности и ингибирование hERG) с тремя соединениями, которые уже конкретно или в общем описаны в WO 2009087238.
Более конкретно:
- соединение 1 по изобретению сравнивают с соединением под номером С88 в WO 2009087238 (на странице 66):
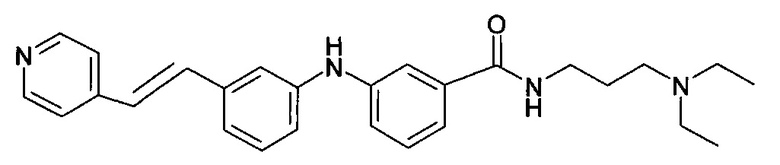
(соединение С88 в WO 2009087238, ниже в данном документе под номером 112)
- соединение 3 по изобретению сравнивают с соединением под номером 583 ниже в данном документе, которое соответствует следующей формуле:
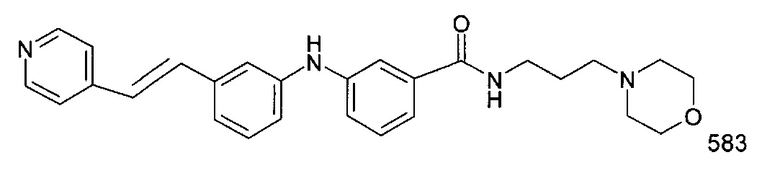
- соединение 4 по изобретению сравнивают с соединением под номером 585 ниже в данном документе, которое соответствует следующей формуле:
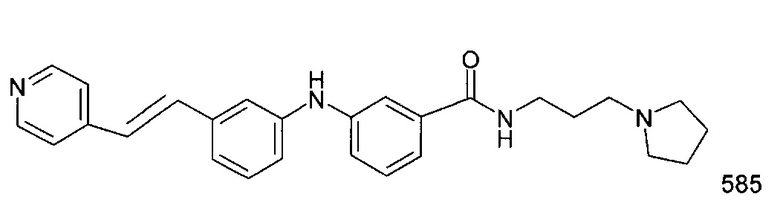
В приведенной ниже таблице показана токсичность в отношении MDA-МВ231, в отношении РВМС, антиинвазивный эффект и ингибирование канала hERG.
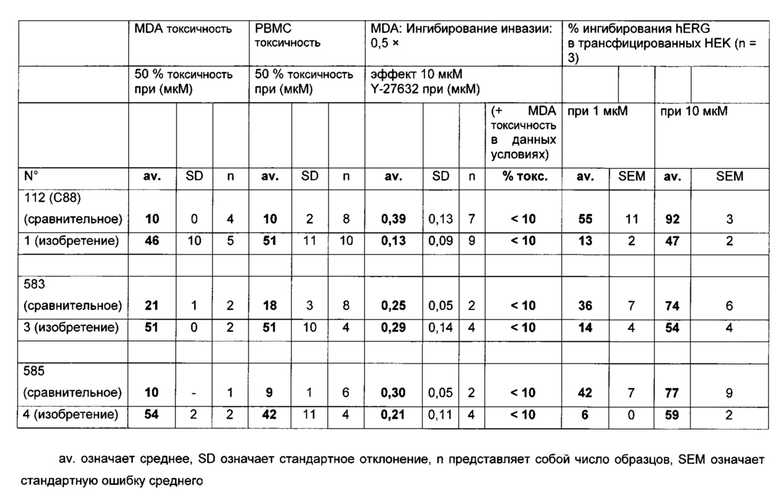
В данной таблице показано, что заявленные соединения (I) обладают улучшенными свойствами, по сравнению с ранее известными соединениями.
Более конкретно, соединение 1 по настоящему изобретению является менее токсичным в отношении MDA-MB231 и в отношении РВМС, более сильным в ингибировании инвазивности и демонстрирует более низкий уровень ингибирования hERG, чем соединение С88 (112).
Более конкретно, соединение 3 по настоящему изобретению является менее токсичным в отношении MDA-MB231 и в отношении РВМС и демонстрирует более низкий уровень ингибирования hERG, чем соединение 583.
Более конкретно, соединение 4 по настоящему изобретению является менее токсичным в отношении MDA-MB231 и в отношении РВМС, более сильным в ингибировании инвазивности и демонстрирует более низкий уровень ингибирования hERG, чем соединение 585.
Пример 17: сравнительные данные
Проводили 2 серии экспериментов на одном сравнительном соединении, как конкретно описано в WO 2009/087238, и одном заявленном соединении с использованием эталонного соединения "С88", как описано в WO 2009/087238 и как упомянуто в примере 16.
Указанное соединение С88 использовали в качестве эталона для сравнения эффективности ингибирования инвазивности между двумя сериями соединений.
Сначала соединение С88 сравнивали с FMMB46.1, как описано в WO 2009/087238 (на странице 95), формулы:
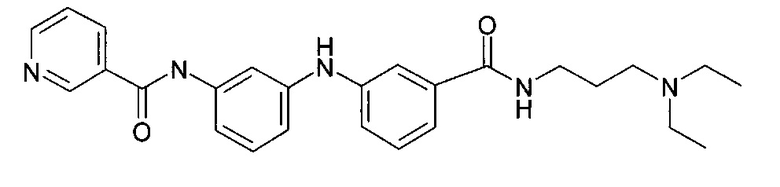
с использованием процедуры 1. Считается, что соединение С88 обладает антиинвазивными свойствами.
Затем соединение С88 сравнивали с соединением 1 по настоящему изобретению с использованием процедуры 2.
Следует отметить, что обе процедуры 1 и 2 представляют собой анализы инвазии для тестирования эффекта лекарственных соединений на инвазию. Они обе абсолютно идентичны, за исключением времени инкубации с лекарственным средством.
В первой серии экспериментов антиинвазивную активность детектировали, при обработке клеток соединением С88, а эффекта с FMMB46.1 не наблюдали (Таблица 1).
Во второй серии экспериментов антиинвазивную активность детектируют, при обработке клеток соединениями С88 и соединением 1.
Первая серия экспериментов - Результаты:
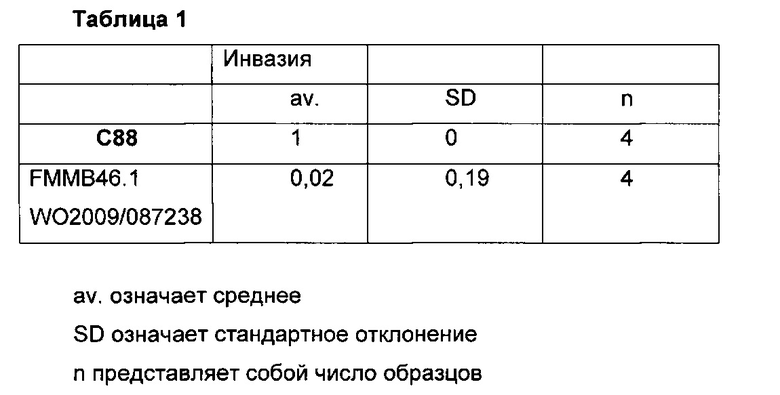
Здесь инвазия (в коллаген) основана на числе клеток на валидную область на уровне 50 мкм и выражена как кратность ингибирующего эффекта, по сравнению с 10 мкМ Y-27632.
Стандартная рабочая процедура 1:
Указанная процедура представляет собой такую, как описано в примере 15, за исключением того, что касается (i) последней стадии во время суток 1 "подготовка анализа инвазии" и "обработка лекарственными средствами" и суток 2 и 3, которые конкретизированы ниже в данном документе, и (ii) факта того, что в конце не определяют ни концентрации, ни токсичности.
Разные стадии выглядят следующим образом:
Подготовка анализа инвазии (на льду; начать во время центрифугирования клеток):
Смешать на льду в заранее охлажденной пробирке: пример для 3,4 мг/мл стокового раствора коллагена; объемы коллагена и воды, подлежащие адаптации в соответствии со стоковой концентрацией каждой партии коллагена:
2,8 мл 2,5 × MEM
441 мкл воды
140 мкл 1 М Hepes
49 мкл 1 н. NaOH
3,5 мл 3,4 мг/мл стокового раствора коллагена (с получением 1,7 мг/мл коллагена в 7 мл).
Гомогенизировать путем осторожного пипетирования (держать на льду).
Добавить 70 мкл 10×106 клеток/мл клеточной суспензии, гомогенизировать путем осторожного пипетирования (с получением 0,1×106 клеток/мл в 1,7 мг/мл коллагена в 7 мл 1 × MEM + 20 мкМ Hepes) (держать на льду).
Распределить 100 мкл/лунку (то есть 10000 клеток/лунку) в покрытые лунки планшета для анализа инвазии (все на льду).
Центрифугировать 5 мин при 200×g при 4°C (например, 1000 об/мин на центрифуге Jouan GR412).
Добавить 200 мкл/лунку PBS ко всем свободным лункам.
Инкубировать 30 мин при 37°C/5% СO2 (затвердевание коллагена).
Обработка лекарственными средствами:
Добавить по 33 мкл/лунку 4 × концентрированных растворов лекарственных средств в MEM + 0,1% FBS к соответствующим лункам во всех трех планшетах согласно приведенным выше схемам планшетов.
Инкубировать 24 ч при 37°C/5% СO2.
Сутки 2: Добавление FBS для стимуляции инвазии:
Наблюдение под микроскопом после 24 ч обработки:
исследовать клетки анализов жизнеспособности
Добавление FBS (в боксе для культивирования клеток):
получить MEM + 5% FBS: 7,2 мл MEM без сыворотки + 0,8 мл FBS (только что размороженная аликвота или оставшаяся часть аликвоты, размороженной за сутки до этого, при хранении при 4°C)
добавить 33 мкл/лунку во все лунки анализов инвазии и жизнеспособности
инкубировать 24 ч при 37°C/5% CO2
Сутки 3: Завершение:
Наблюдение под микроскопом после 48 ч обработки:
исследовать клетки анализов жизнеспособности
Анализы инвазии: фиксация и окрашивание (операции с формальдегидом нужно проводить в вытяжном шкафу):
Получить свежий 1 мкг/мл Hoechst 33342 в 18,5% формальдегиде: 5 мл PBS (не обязательно стерильный) + 5 мл 37% формальдегида + 1 мкл 10 мг/мл Hoechst 33342 (внимание: для одного планшета было бы достаточно меньшего объема, но минимальный отбираемый пипеткой объем не должен быть меньше 1 мкл).
Добавить 50 мкл/лунку ко всем лункам анализа инвазии (с получением в итоге 4,3% формальдегида).
Плотно закрыть черной пленкой (предоставлена с планшетами). Инкубировать по меньшей мере 7 ч при КТ.
Анализ, такой, как проведен в сутки 5 в примере 15, проводят точно в таких же условиях в сутки 3 в рамках настоящей процедуры 1.
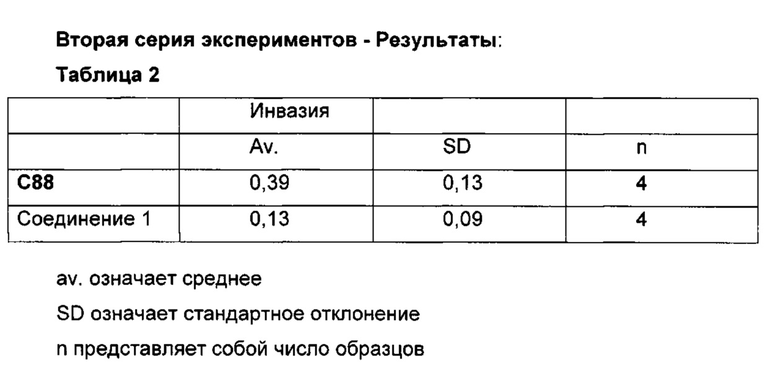
Здесь, инвазия (в коллаген) представляет собой концентрацию соединения, необходимую для достижения 50% ингибирующего эффекта, по сравнению с 10 мкМ Y-27632.
Стандартная рабочая процедура 2 является точно такой же, как процедура, описанная в примере 15.
Заключение: поскольку соединение 1 демонстрирует более хорошие результаты по инвазии, чем соединение С88, которое само по себе демонстрирует более хорошие результаты по инвазии, чем соединение FMMB46.1, без какого-либо руководства идеями WO 2009/087238 можно сделать вывод, что авторами изобретения открыты неожиданные свойства.
Соединения по настоящему изобретению демонстрируют антиинвазивный эффект, предсказывающий их противораковую активность.
Вследствие этого, результаты тестов, проведенных на соединениях, описанных в настоящем изобретении, показывают, что указанные соединения могут быть применимы для ингибирования и/или предупреждения и/или лечения рака. Более конкретно, посредством соединений по настоящему изобретению можно лечить следующий тип рака: колоректальный рак, рак поджелудочной железы, рак легкого, включая немелкоклеточный рак легкого, рак молочной железы, рак мочевого пузыря, рак желчного пузыря, рак щитовидной железы, меланома, рак печени, рак матки/шейки матки, рак пищевода, рак почки, рак яичника, рак предстательной железы, рак головы и шеи, и рак желудка и т.д.
Для данной цели эффективное количество указанного соединения можно вводить пациенту, страдающему от рака.
Настоящее изобретение также относится к применению по меньшей мере соединения, выбранного из любого соединения формулы (I), (I'), (Ia), (Ib), (Ic), (Id) и (Ie), как определено выше, и соединений (1)-(51), как определено выше, или одной из его фармацевтически приемлемых солей по настоящему изобретению для получения фармацевтической композиции, предназначенной для лечения рака.
Настоящее изобретение также охватывает фармацевтические композиции, содержащие по меньшей мере соединение, выбранное из соединений формул (I), (I'), (Ia), (Ib), (Ic), (Id) и (Iе), как определено выше, и соединений (1)-(51), как определено выше, или его любую фармацевтически приемлемую соль.
Таким образом, данные фармацевтические композиции содержат эффективное количество указанного соединения и один или более фармацевтических эксципиентов.
Упомянутые выше эксципиенты выбраны согласно лекарственной форме и желательному способу введения.
В данном контексте они могут быть представлены в любой фармацевтической форме, которая подходит для энтерального и парентерального введения, совместно с соответствующими эксципиентами, например, в форме плоских или покрытых оболочкой таблеток, твердого желатина, капсул в мягкой оболочке или других капсул, суппозиториев или напитка, такого как суспензии, сиропы, или вводимых путем инъекции растворов или суспензий, в дозах, которые делают возможным введение от 0,1 до 1000 мг активного вещества в сутки.
Настоящее изобретение также относится к применению соединения любой из формул (I), (I'), (Ia), (Ib), (Ic), (Id) и (Ie), как определено выше, и соединений (1)-(51), как определено выше, или одной из его фармацевтически приемлемых солей по настоящему изобретению для получения фармацевтической композиции, предназначенной для ингибирования и/или предупреждения и/или лечения рака.
Настоящее изобретение дополнительно относится к способу лечения пациентов, страдающих от рака, который включает по меньшей мере стадию введения пациенту, страдающему от него, эффективного количества соединения любой из формул (I), (I'), (Ia), (Ib), (Ic), (Id) и (Ie), как определено выше, и (1)-(51) или одной из его фармацевтически приемлемых солей.
| название | год | авторы | номер документа |
|---|---|---|---|
| АРИЛ-N-АРИЛЬНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РНК-ВИРУСНОЙ ИНФЕКЦИИ | 2020 |
|
RU2821414C2 |
| АРИЛ-N-АРИЛЬНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РНК-ВИРУСНОЙ ИНФЕКЦИИ | 2019 |
|
RU2815493C2 |
| МОДУЛЯТОРЫ АКТИВНОСТИ НЕС1 И СПОСОБЫ ДЛЯ НИХ | 2011 |
|
RU2576036C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРА FLT3 | 2015 |
|
RU2710928C2 |
| ПРОИЗВОДНЫЕ АДАМАНТИЛА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ JNK-ОПОСРЕДОВАННОГО РАССТРОЙСТВА | 2012 |
|
RU2626890C2 |
| ЗАМЕЩЕННЫЕ ДИАЛКИЛ (ОКСИДО)-Λ-СУЛЬФАНИЛИДЕН НИКОТИНАМИД ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2014 |
|
RU2711749C2 |
| Соединения, активные по отношению к бромодоменам | 2015 |
|
RU2743074C2 |
| СУЛЬФОНИЛМОЧЕВИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2739356C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ МОДУЛЯЦИИ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ, СПОСОБ ПОЛУЧЕНИЯ ЭТИХ СОЕДИНЕНИЙ | 2000 |
|
RU2255937C2 |
| СУЛЬФОНИЛМОЧЕВИНЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2795512C2 |
Изобретение относится к соединениям формулы (I) или их фармацевтически приемлемым солям, которые могут найти применение для предупреждения, и/или ингибирования, и/или лечения рака. В формуле (I) А и А' независимо представляют собой фениленовую группу или пиридиленовую группу; R2 представляет собой атом водорода или алкильную группу (С1-С4); R3 представляет собой 2-пиридильную группу, 3-пиридильную группу, 4-пиридильную группу, 2-пиримидинильную группу, 4-пиримидинильную группу или 5-пиримидинильную группу; R4 представляет собой карбонильную группу или сульфонильную группу; R5 представляет собой группу -NH-(CH2)a-NR6R7 или 4-метилпиперазинильную группу, причем а представляет собой целое число от 1 до 4, R6 и R7 независимо представляют собой алкильную группу (С1-С4) или R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероциклическую группу, которую выбирают из 4-метилпиперазинильной группы, морфолиногруппы, пирролидинильной группы и пиперидиногруппы. Изобретение относится также к способу получения соединений формулы (I) и содержащей их фармацевтической композиции. 3 н. и 13 з.п. ф-лы, 6 табл., 17 пр.
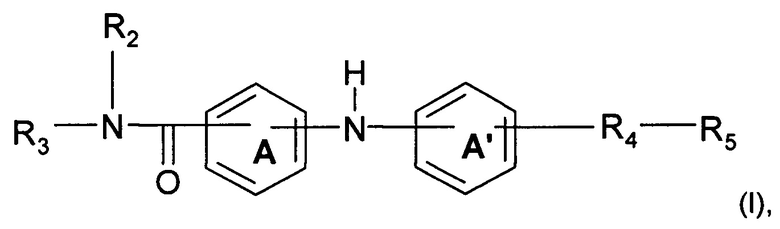
1. Соединение формулы (I)
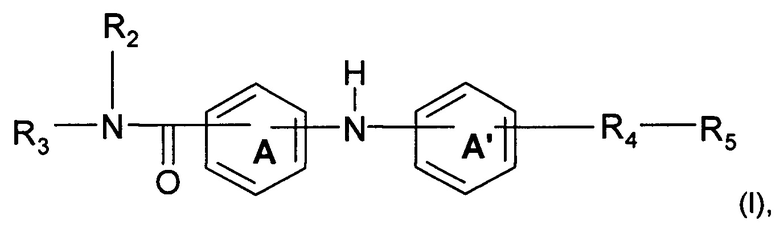
где А и А' независимо представляют собой фениленовую группу или пиридиленовую группу;
R2 представляет собой атом водорода или алкильную группу (С1-С4);
R3 представляет собой 2-пиридильную группу, 3-пиридильную группу, 4-пиридильную группу, 2-пиримидинильную группу, 4-пиримидинильную группу или 5-пиримидинильную группу;
R4 представляет собой карбонильную группу или сульфонильную группу; и
R5 представляет собой группу -NH-(CH2)a-NR6R7 или 4-метилпиперазинильную группу, причем а представляет собой целое число от 1 до 4, R6 и R7 независимо представляют собой алкильную группу (С1-С4) или R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероциклическую группу, которую выбирают из 4-метилпиперазинильной группы, морфолиногруппы, пирролидинильной группы и пиперидиногруппы;
или любая из его фармацевтически приемлемых солей.
2. Соединение формулы (I) по п.1,
где А и А' независимо представляют собой фениленовую группу или пиридиленовую группу;
R2 представляет собой атом водорода или метильную группу;
R3 представляет собой 2-пиридильную группу, 4-пиридильную группу или 4-пиримидинильную группу;
R4 представляет собой карбонильную группу или сульфонильную группу; и
R5 представляет собой группу -NH-(CH2)a-NR6R7 или 4-метилпиперазинильную группу, причем а представляет собой целое число от 2 до 3, R6 и R7 представляют собой этильную группу или R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероциклическую группу, которую выбирают из 4-метилпиперазинильной группы, морфолиногруппы, пирролидинильной группы и пиперидиногруппы;
или любая из его фармацевтически приемлемых солей.
3. Соединение формулы (I) по п.1 или 2, где группа -NH- между А и А' и группа -R4-R5 находятся в мета-положении друг от друга относительно А'.
4. Соединение формулы (I') по п.1
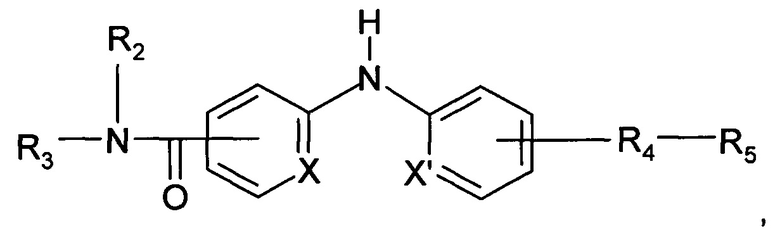
где X и X' независимо представляют собой СН или N;
R2 представляет собой атом водорода или метильную группу;
R3 представляет собой 2-пиридильную группу, 4-пиридильную группу или 4-пиримидинильную группу;
R4 представляет собой карбонильную группу или сульфонильную группу; и
R5 представляет собой группу -NH-(CH2)a-NR6R7 или 4-метилпиперазинильную группу, причем а представляет собой целое число от 2 до 3, R6 и R7 представляют собой этильную группу или R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероциклическую группу, которую выбирают из 4-метилпиперазинильной группы, морфолиногруппы, пирролидинильной группы и пиперидиногруппы;
или любая из его фармацевтически приемлемых солей.
5. Соединение формулы (Ia) по п.1
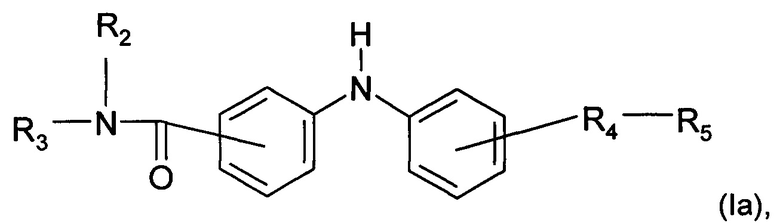
где R2, R3, R4 и R5 являются такими, как определено в п.1;
или любая из его фармацевтически приемлемых солей.
6. Соединение формулы (Ia) по п.5, где R4 представляет собой карбонильную группу и R2, R3 и R5 являются такими, как определено в п.1;
или любая из его фармацевтически приемлемых солей.
7. Соединение формулы (Ic) по п.1
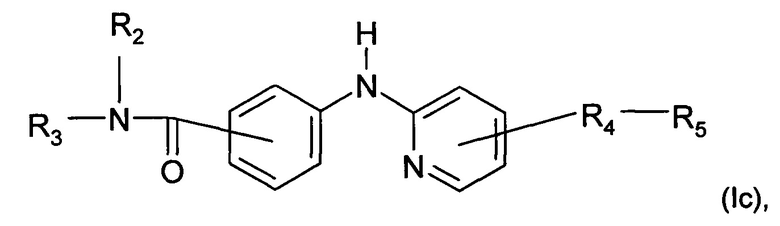
где R2, R3, R4 и R5 являются такими, как определено в п.1;
или любая из его фармацевтически приемлемых солей.
8. Соединение формулы (Ic) по п.7, где R2 представляет собой атом водорода или метильную группу; R3 представляет собой 4-пиридильную группу или 4-пиримидинильную группу; R4 представляет собой карбонильную группу; и R5 представляет собой группу -NH-(CH2)a-NR6R7, причем а представляет собой целое число 3, R6 и R7 представляют собой этильную группу или R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероциклическую группу, которая представляет собой 4-метилпиперазинильную группу;
или любая из его фармацевтически приемлемых солей.
9. Соединение формулы (Id) по п.1
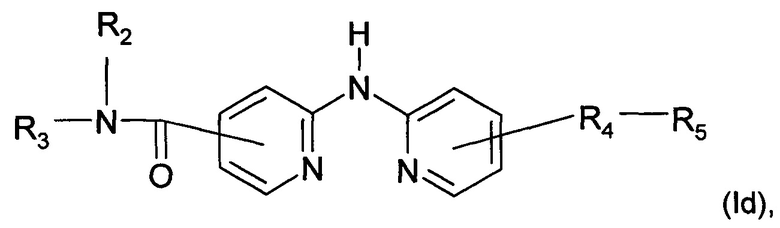
где R2, R3, R4 и R5 являются такими, как определено в п.1;
или любая из его фармацевтически приемлемых солей.
10. Соединение формулы (Id) по п.9, где R2 представляет собой атом водорода; R3 представляет собой 4-пиридильную группу; R4 представляет собой карбонильную группу; и R5 представляет собой группу -NH-(CH2)a-NR6R7, причем а представляет собой целое число 3, R6 и R7 представляют собой этильную группу или R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероциклическую группу, которая представляет собой 4-метилпиперазинильную группу;
или любая из его фармацевтически приемлемых солей.
11. Соединение формулы (Ie) по п.1
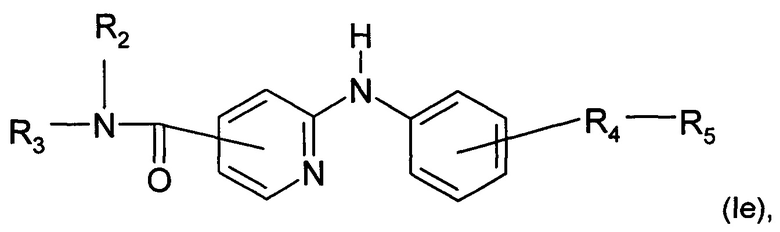
где R2, R3, R4 и R5 являются такими, как определено в п.1;
или любая из его фармацевтически приемлемых солей.
12. Соединение формулы (Ie) по п.11, где R2 представляет собой атом водорода; R3 представляет собой 4-пиридильную группу; R4 представляет собой карбонильную группу или сульфонильную группу; и R5 представляет собой группу -NH-(CH2)a-NR6R7, причем а представляет собой целое число 3, R6 и R7 представляют собой этильную группу или R6 и R7 вместе с атомом азота, с которым они связаны, образуют гетероциклическую группу, которую выбирают из 4-метилпиперазинильной группы, морфолиногруппы, пирролидинильной группы и пиперидиногруппы;
или любая из его фармацевтически приемлемых солей.
13. Соединение формулы (I) по п.1, выбранное из следующих соединений:
(1) N-(3-(диэтиламино)пропил)-3-((3-(пиридин-4-илкарбамоил)фенил)амино)бензамид,
(2) 3-((4-((3-(диэтиламино)пропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)бензамид,
(3) N-(3-морфолинопропил)-3-((3-(пиридин-4-илкарбамоил)фенил)амино)бензамид,
(4) N-(пиридин-4-ил)-3-((3-((3-(пирролидин-1-ил)пропил)карбамоил)фенил)амино)бензамид,
(5) 3-((3-(N-(3-(диэтиламино)пропил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)бензамид,
(6) N-(3-(4-метилпиперазин-1-ил)пропил)-3-((3-(пиридин-4-илкарбамоил)фенил)амино)бензамид,
(7) N-(3-(пиперидин-1-ил)пропил)-3-((3-(пиридин-4-илкарбамоил)фенил)амино)бензамид,
(8) 3-((3-(4-метилпиперазин-1-карбонил)фенил)амино)-N-(пиридин-4-ил)бензамид,
(9) 3-((3-(N-(3-(пиперидин-1-ил)пропил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)бензамид,
(10) 3-((3-(N-(2-(пиперидин-1-ил)этил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)бензамид,
(11) N-(3-(диэтиламино)пропил)-3-((3-(пиридин-2-илкарбамоил)фенил)амино)бензамид,
(12) 3-((3-(N-(3-морфолинопропил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)бензамид,
(13) N-(3-(диэтиламино)пропил)-3-((4-(пиридин-4-илкарбамоил)фенил)амино)бензамид,
(14) N-(3-морфолинопропил)-3-((4-(пиридин-4-илкарбамоил)фенил)амино)бензамид,
(15) 4-((3-(N-(3-морфолинопропил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)бензамид,
(16) N-(пиридин-4-ил)-4-((3-(N-(2-(пирролидин-1-ил)этил)сульфамоил)фенил)амино)бензамид,
(17) 3-((3-((3-(диэтиламино)пропил)карбамоил)фенил)амино)-N-метил-N-(пиридин-4-ил)бензамид,
(18) N-метил-N-(пиридин-4-ил)-3-((3-((3-(пирролидин-1-ил)пропил)карбамоил)фенил)амино)бензамид,
(19) 3-((3-(N-(3-(диэтиламино)пропил)сульфамоил)фенил)амино)-N-метил-N-(пиридин-4-ил)бензамид,
(20) N-метил-3-((3-((3-(4-метилпиперазин-1-ил)пропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)бензамид,
(21) N-метил-3-((3-((3-(пиперидин-1-ил)пропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)бензамид,
(22) N-метил-3-((3-((3-морфолинопропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)бензамид,
(23) N-метил-3-((3-(N-(3-морфолинопропил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)бензамид,
(24) N-метил-3-((3-(N-(3-(пиперидин-1-ил)пропил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)бензамид,
(25) N-(3-(диэтиламино)пропил)-3-((3-(пиримидин-4-илкарбамоил)фенил)амино)бензамид,
(26) 3-((3-(N-(3-(диэтиламино)пропил)сульфамоил)фенил)амино)-N-(пиримидин-4-ил)бензамид,
(27) 3-((3-(N-(3-(пиперидин-1-ил)пропил)сульфамоил)фенил)амино)-N-(пиримидин-4-ил)бензамид,
(28) N-(пиримидин-4-ил)-3-((3-((3-(пирролидин-1-ил)пропил)карбамоил)фенил)амино)бензамид,
(29) N-(3-(пиперидин-1-ил)пропил)-3-((3-(пиримидин-4-илкарбамоил)фенил)амино)бензамид,
(30) N-(3-морфолинопропил)-3-((3-(пиримидин-4-илкарбамоил)фенил)амино)бензамид,
(31) N-(3-(4-метилпиперазин-1-ил)пропил)-3-((3-(пиримидин-4-илкарбамоил)фенил)амино)бензамид,
(32) N-(3-(диэтиламино)пропил)-5-((3-(пиридин-4-илкарбамоил)фенил)амино)никотинамид,
(33) N-(3-(диэтиламино)пропил)-2-((3-(пиридин-4-илкарбамоил)фенил)амино)изоникотинамид,
(34) N-(3-(4-метилпиперазин-1-ил)пропил)-2-((3-(пиридин-4-илкарбамоил)фенил)амино)изоникотинамид,
(35) N-(3-(диэтиламино)пропил)-6-((3-(пиридин-4-илкарбамоил)фенил)амино)пиколинамид,
(36) N-(3-(диэтиламино)пропил)-6-((4-(пиридин-4-илкарбамоил)фенил)амино)пиколинамид,
(37) N-(3-(диэтиламино)пропил)-6-((3-(метил(пиридин-4-ил)карбамоил)фенил)амино)пиколинамид,
(38) N-(3-(диэтиламино)пропил)-2-((3-(метил(пиридин-4-ил)карбамоил)фенил)амино)изоникотинамид,
(39) 2-((3-(метил(пиридин-4-ил)карбамоил)фенил)амино)-N-(3-(4-метилпиперазин-1-ил)пропил)изоникотинамид,
(40) N-(3-(диэтиламино)пропил)-6-((3-(пиримидин-4-илкарбамоил)фенил)амино)пиколинамид,
(41) N-(3-(диэтиламино)пропил)-2-((3-(пиримидин-4-илкарбамоил)фенил)амино)изоникотинамид,
(42) N-(3-(4-метилпиперазин-1-ил)пропил)-2-((3-(пиримидин-4-илкарбамоил)фенил)амино)изоникотинамид,
(43) N-(3-(диэтиламино)пропил)-6-((4-(пиридин-4-илкарбамоил)пиридин-2-ил)амино)пиколинамид,
(44) N-(3-(диэтиламино)пропил)-2-((4-(пиридин-4-илкарбамоил)пиридин-2-ил)амино)изоникотинамид,
(45) N-(3-(4-метилпиперазин-1-ил)пропил)-2-((4-(пиридин-4-илкарбамоил)пиридин-2-ил)амино)изоникотинамид,
(46) 2-((3-(N-(3-(диэтиламино)пропил)сульфамоил)фенил)амино)-N-(пиридин-4-ил)изоникотинамид,
(47) 2-((3-((3-(диэтиламино)пропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)изоникотинамид,
(48) 2-((3-((3-морфолинопропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)изоникотинамид,
(49) N-(3-(пиперидин-1-ил)пропил)-3-((3-(пиридин-4-илкарбамоил)фенил)амино)бензамид,
(50) N-(пиридин-4-ил)-2-((3-((3-(пирролидин-1-ил)пропил)карбамоил)фенил)амино)изоникотинамид,
(51) 2-((3-((3-(4-метилпиперазин-1-ил)пропил)карбамоил)фенил)амино)-N-(пиридин-4-ил)изоникотинамид,
и их фармацевтически приемлемые соли.
14. Способ получения соединений формулы (I) по п.1, включающий взаимодействие соединения формулы (II)
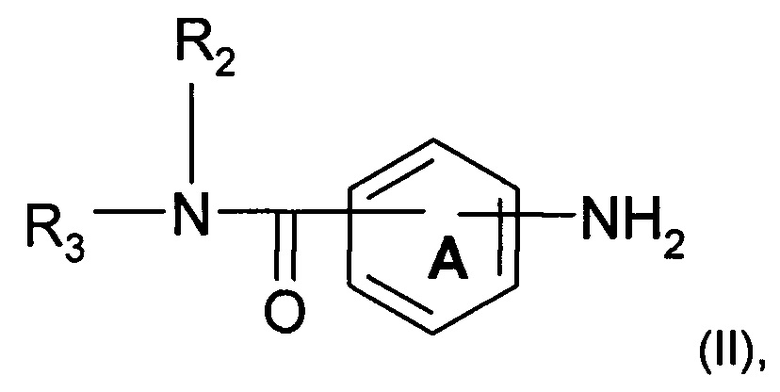
где R2, R3 и А являются такими, как определено в п.1, с соединением формулы (III)
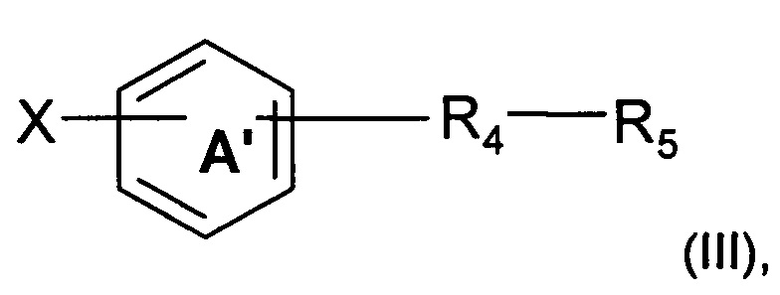
где X представляет собой атом хлора, атом йода или атом брома и R4, R5 и А' являются такими, как определено в п.1.
15. Соединение формулы (I) по п.1 для применения для предупреждения, и/или ингибирования, и/или лечения рака.
16. Фармацевтическая композиция для применения для предупреждения, и/или ингибирования, и/или лечения рака, содержащая в эффективном количестве по меньшей мере одно соединение формулы (I) по любому из пп.1-13 и фармацевтически приемлемый эксципиент.
| Колосоуборка | 1923 |
|
SU2009A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| НОВЫЕ АНТРАНИЛАМИДОПИРИДИНМОЧЕВИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ РЕЦЕПТОРА VEGF | 2005 |
|
RU2415850C2 |

