Настоящее изобретение относится к соединениям, применимым для предупреждения и/или лечения РНК-вирусной инфекции, и наиболее предпочтительно РНК-вирусной инфекции, вызванной РНК-вирусами, принадлежащими к группе IV или V классификации по Балтимору.
Настоящее изобретение также относится к некоторым новым соединениям, в частности, применимым для предупреждения и/или лечения РНК-вирусной инфекции, и наиболее предпочтительно РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору.
Оно также относится к фармацевтическим композициям, содержащим указанные новые соединения, и к способам химического синтеза для их получения.
Уровень техники
Вирусы являются одной из основных причин заболеваний во всем мире. Вирусы в общем случае определены как маленькие, неживые возбудители инфекции, которые реплицируются только в живых клетках, поскольку они не обладают механизмом полностью автономной репликации. Несмотря на разнообразие форм и размеров, они обычно состоят из вирусной частицы (известной как «вирион»), состоящей из белковой оболочки, которая содержит по меньшей мере одну молекулу нуклеиновой кислоты и, необязательно, в зависимости от типа вируса, один или более белков или нуклеопротеинов.
Поскольку вирусы не обладают механизмом полностью автономной репликации, они неизбежно должны полагаться на аппарат и метаболизм инфицированной клетки или хозяина для репликации и продуцирования множества их копий.
Даже несмотря на то, что их цикл репликации существенно варьирует среди разных видов, общепризнано, что жизненный цикл вирусов включает шесть основных стадий: прикрепление, проникновение, сбрасывание оболочки, репликация, сборка и высвобождение.
В зависимости от природы вирусов-мишеней были сконструированы терапевтические молекулы, которые могут вмешиваться в один или более из данных механизмов.
Среди них стадия репликации включает не только размножение вирусного генома, но также синтез вирусной матричной РНК, белка вируса и модуляцию механизма транскрипции или трансляции хозяина. Однако, также понятно, что тип генома (одноцепочечный, двухцепочечный, РНК, ДНК…) в значительной мере характеризует данную стадию репликации. Например, большинство ДНК-вирусов собираются в ядре, в то время как большинство РНК-вирусов развиваются исключительно в цитоплазме. Кроме того, появляется все больше данных о том, что вирусы с одноцепочечной РНК, такие как вирус гриппа, используют механизм сплайсинга и созревания РНК хозяина.
Соответственно, и принимая во внимание значение данного типа генома в стадии репликации, была разработана классификация вирусов по Балтимору. Данная классификация объединяет вирусы в семейства (или «группы»), в зависимости от их типа генома. Текущая классификация вирусов на 2018 год включает семь разных групп:
- Группа I: вирусы с двухцепочечной ДНК (дцДНК);
- Группа II: вирусы с одноцепочечной ДНК (оцДНК);
- Группа III: вирусы с двухцепочечной РНК (дцРНК);
- Группа IV: вирусы с положительно-полярной или смысловой цепью РНК ((+)оцРНК);
- Группа V: вирусы с отрицательно-полярной или антисмысловой цепью РНК ((-)оцРНК);
- Группа VI: одноцепочечные РНК-вирусы, реплицирующиеся через стадию ДНК (оцРНК-ОТ);
- Группа VII: двухцепочечные ДНК-вирусы, реплицирующиеся через стадию РНК (дцДНК-ОТ).
В соответствии с данной классификацией, вирусы, принадлежащие к группе VI, не являются, в строгом смысле, РНК-вирусами. По тем же причинам вирусы, принадлежащие к группе VII, не являются, в строгом смысле, ДНК-вирусами. Одним хорошо изученным примером семейства вирусов, принадлежащих к группе VI, является семейство Retroviridae (ретровирусы), которое включает ВИЧ (вирус иммунодефицита человека). Одним хорошо изученных примером семейства вирусов, принадлежащих к группе VII, является семейство Hepadnaviridae, которое включает вирус гепатита B (HBV).
В качестве представителя вирусов, принадлежащих к группе IV, можно привести пикорнавирусы (которое является семейством вирусов, включающим хорошо известные вирусы, такие как вирус гепатита A, энтеровирусы, риновирусы, полиовирусы и вирус ящура), вирус SARS, вирус гепатита C, вирус желтой лихорадки и вирус краснухи. Семейство Togaviridae также принадлежит к группе IV, и их известный род представляет собой альфавирус, включающий вирус Чикунгунья. Flaviridae также является семейством, принадлежащим к группе IV, охватывающим известный вирус, передаваемый комарами, а именно, вирус Денге.
В качестве представителя вирусов, принадлежащих к группе V, можно привести семейство вирусов Filoviridae, охватывающее вирус Эбола, семейство Paramyxoviridae, охватывающее респираторно-синцитиальный вирус (RSV), семейство Rhabdoviridae, семейство Orthomyxoviridae, охватывающее вирус гриппа A, вирус гриппа B и вирус гриппа C.
Группы в семействах вирусов, которым уделено особое внимание в рамках настоящего изобретения, представляют собой группы, охватывающие РНК-вирусы, особенно одноцепочечные РНК-вирусы и более конкретно РНК-вирусы, принадлежащие к группе IV и группе V классификации по Балтимору.
Существует мало способов лечения заболеваний, вызываемых РНК-вирусными инфекциями, в частности, вирусами с одноцепочечной РНК, и более конкретно РНК-вирусными инфекциями, вызываемыми вирусами, принадлежащими к группе IV и V классификации по Балтимору. Лечение направлено на облегчение симптомов. Таким образом, остается необходимость в идентификации новых противовирусных лекарственных средств для лечения РНК-вирусных инфекций, таких как инфекция РНК-вирусом группы IV и V, в частности, малых химических молекул.
Определения
В том виде, в котором он используется в настоящем документе, термин «пациент» относится или к животному, такому как животное, ценное с точки зрения разведения, содержания в качестве компаньона или сохранения вида, или предпочтительно взрослому человеку или ребенку, который поражен или может быть пораженным одним или более заболеваниями и состояниями, описанными в настоящем документе.
В частности, термин «пациент», в том виде, в котором он используется в настоящей заявке, относится к млекопитающему, такому как грызун, кошка, собака, примат или человек, предпочтительно указанный субъект представляет собой человека и также распространяется на птиц.
Идентификация данных пациентов, которые нуждаются в лечении описанных в настоящем документе заболеваний и состояний, находится в пределах компетентности и знаний специалиста в данной области. Ветеринар или лечащий врач, являющийся специалистом в данной области, может легко идентифицировать, посредством применения клинических исследований, медицинского осмотра, истории болезни/семейного анамнеза или биологических и диагностических тестов, тех пациентов, которые нуждаются в таком лечении.
В контексте изобретения термин «осуществление лечения» или «лечение», в том виде, в котором он используется в настоящем документе, означает обращение вспять, облегчение, подавление прогрессирования или предупреждение заболевания, являющегося результатом РНК-вирусной инфекции, и более конкретно, инфекции, вызванной РНК-вирусом из группы IV или V, или одного или более симптомов такого заболевания.
В том виде, в котором он используется в настоящем документе, термин «эффективное количество» относится к количеству соединения согласно настоящему изобретению, которое является эффективным в предупреждении, уменьшении, устранении, лечении или осуществлении контроля симптомов описанных в настоящем документе заболеваний и состояний, а именно, РНК-вирусной инфекции, и более конкретно инфекции, вызванной РНК-вирусом из группы IV или V. Подразумевается, что термин «осуществление контроля» относится ко всем процессам, где может иметь место замедление, прерывание, купирование или прекращение прогрессирования заболеваний и состояний, описанных в настоящем документе, но не обязательно указывает на полную ликвидацию всех симптомов заболевания и состояния, и подразумевает в том числе профилактическое лечение.
Термин «эффективное количество» включает «профилактически эффективное количество», а также «терапевтически эффективное количество».
Термин «предупреждение», в том виде, в котором он используется в настоящем документе, означает снижение риска начала или замедление возникновения данного явления, а именно, в настоящем изобретении, заболевания, возникающего в результате РНК-вирусной инфекции, и более конкретно инфекции, вызванной РНК-вирусом из группы IV или V.
В том виде, в котором он используется в настоящем документе, термин «предупреждение» также охватывает «уменьшение вероятности возникновения» или «уменьшение вероятности повторного возникновения».
Термин «профилактически эффективное количество» относится к концентрации соединения согласно настоящему изобретению, которая является эффективной в ингибировании, предупреждении, уменьшении вероятности заболевания, вызванного РНК-вирусами, и более конкретно РНК-вирусом из группы IV или V классификации по Балтимору, или предупреждении РНК-вирусной инфекции, и более конкретно инфекции, вызванной РНК-вирусом из группы IV или V, или предупреждении отсроченного начала заболевания, вызванного РНК-вирусом и более конкретно РНК-вирусом из группы IV или V, при введении до инфицирования, т. е. до, во время и/или немного позже периода воздействия РНК-вируса и, в частности, РНК-вируса из группы IV или V.
Аналогично, термин «терапевтически эффективное количество» относится к концентрации соединения, которая является эффективной в лечении РНК-вирусной инфекции, например, приводит к уменьшению РНК-вирусной инфекции, после исследования при введении после возникновения инфекции.
В том виде, в котором он используется в настоящем документе, термин «фармацевтически приемлемый» относится к таким соединениям, веществам, эксципиентам, композициям или лекарственным формам, которые, в пределах объема тщательной медицинской оценки, подходят для контакта с тканями человека и животного без излишней токсичности, раздражения, аллергической реакции или других проблематичных осложнений, в соответствии с разумным отношением «польза/риск».
В том виде, в котором он используется в настоящем документе, термин «вирусная инфекция или связанное с ней состояние» относится к инфекции или состоянию, связанным с вирусом, более конкретно, где указанный вирус имеет РНК-геном, и особенно с РНК-вирусом, принадлежим к группе IV или V в соответствии с классификацией по Балтимору. Вирусы могут быть дополнительно подразделены на различные семейства, порядки и роды.
Для справки, содержание «Классификации вирусов по Балтимору», о которой сообщается в настоящем документе, содержит дополнительные ссылки на таксономию вирусов, как изложено в базе данных Международного комитета по таксономии вирусов (ICTV) 2017 года, опубликованной в электронном виде 12 марта 2018 года по адресу http://ictvonline.org/virusTaxonomy.asp. Данная таксономия полностью включена в настоящий документ.
В изобретении могут, в частности, рассматриваться альфавирусы, они относятся к РНК-вирусам группы IV и семейству Togaviridae, которые могут быть определены как вирусы с одноцепочечной смысловой РНК или (+)оцРНК-вирусы. Их порядок представляет собой «не назначенный» в соответствии с таксономией вирусов 2017 года. Семейство Togaviridae включает род Alphavirus и Rubivirus.
Примеры альфавирусов, которые рассматриваются в изобретении, включают: вирус леса Барма, вирус Чикунгунья, вирус Майяро, вирус О'Ньонг-Ньонг, вирус Росс-Ривер, вирус леса Семлики, вирус Уна, вирус восточного энцефалита лошадей, вирус Тонате, вирус венесуэльского энцефалита лошадей и вирус западного энцефалита лошадей.
Наиболее предпочтительно альфавирусная инфекция или состояние, связанное с альфавирусом, согласно изобретению представляет собой вирусную инфекцию Чикунгунья или состояние, связанное с вирусом Чикунгунья.
Более конкретно, вирус Чикунгунья (CHIKV) представляет собой РНК-вирус, который принадлежит к роду альфавирусов, который, в свою очередь, принадлежит к семейству Togaviridae, т. е. группе IV классификации по Балтимору. Чикунгунья представляет собой вирусное заболевание, передаваемое комарами, впервые описанное во время вспышки на юге Танзании в 1952 году. CHIKV представляет собой оболочечный вирус с одноцепочечной смысловой РНК, длиной приблизительно 12 т.п.н. (тысяч пар нуклеотидов). Геном CHIKV организован следующим образом: 5'-cap-nsPl-nsP2-nsP3-nsP4-(область соединения)-C-E3-E2-6k-El-poly(A)-3'c, где первые четыре белка (nsPl-4) представляют собой неструктурные белки, и структурными белками являются белки капсида (C) и оболочки (E). Отсутствует четко выраженное различие серотипов среди CHIKV, выделенных из Африки, Азии и островов Индийского океана. На основании филогенетических анализов, основанных на последовательностях гена El, можно подразделить CHIKV на три генотипа (линии): азиатский, восточно-/центрально-/южно-африканский (ECSA) и западноафриканский. Азиатский генотип отличался от ECSA и западноафриканского генотипов уровнями нуклеотидов на -5% и -15%, соответственно. Африканские генотипы (ECSA в сравнении с западноафриканским) расходились на -15%. Идентичности по аминокислотному составу у данных трех генотипов варьировали от 95,2 до 99,8%.
Вирус Чикунгунья может вызывать вспышки, связанные с тяжелым течением заболевания.
Чикунгунья представляет собой вирусное заболевание, передающееся человеку от инфицированных комаров. К крупным вспышкам Чикунгуньи имели отношение как Ae. aegypti, так и Ae. albopictus. В то время как среда обитания Ae. aegypti ограничивается тропиками и субтропиками, Ae. albopictus также встречается в областях с умеренным и даже умеренно-холодным климатом. За последние несколько десятилетий Ae. albopictus распространился за пределы Азии и обосновался в регионах Африки, Европы и стран Южной и Северной Америки.
После инфицирования вирусом Чикунгунья имеет место инкубационный период, длящийся в среднем 2-4 суток с последующим появлением симптомов заболевания. Среди таки симптомов можно привести лихорадку и сильную боль в суставах. Другие симптомы включают боль в мышцах, головную боль, тошноту боль в спине, слабость, мышечную боль и сыпь. Могут встречаться тяжелые клинические проявления инфекции Чикунгунья, например, геморрагическая лихорадка, конъюнктивиты, светобоязнь, гепатит, стоматит. Также сообщается о неврологических проявлениях, таких как энцефалит, фебрильные судороги, менингеальный синдром и острая энцефалопатия.
Боль в суставах часто является изнурительной и может различаться по продолжительности.
Близость мест выплода комаров к месту проживания человека является значительным фактором риска для Чикунгуньи.
Вирус Чикунгунья в основном распространен в Африке, Индии и Юго-Восточной Азии. За последние десятилетия комары-переносчики Чикунгуньи распространились в Европу и страны Южной и Северной Америки. В 2007 году впервые была зарегистрирована передача заболевания в виде локальной вспышки в северо-восточной Италии. С тех пор вспышки были зарегистрированы во Франции и Хорватии.
В изобретении также могут рассматриваться вирусы Денге, которые демонстрируют различные серотипы и относятся к РНК-вирусам группы IV и семейству Flaviviridae, которые можно определить как вирусы с одноцепочечной смысловой РНК или (+)оцРНК-вирусы. Более конкретно, вирус Денге представляет собой (+)оц-РНК-вирус, принадлежащий к группе IV классификации по Балтимору. Он является частью рода Flavivirus, который принадлежит к семейству Flaviviridae. Другие вирусы, принадлежащие к семейству Flaviviridae, представляют собой вирус гепатита C и вирус желтой лихорадки.
В изобретении также, в частности, рассматриваются вирусы порядка Mononegavirales. Порядок Mononegavirales включает вирусы, принадлежащие к группе V классификации по Балтимору. По состоянию на 2018 год данный порядок включает главным образом следующие семейства вирусов: Bornaviridae, Mymonaviridae, Filoviridae, Nyamiviridae, Paramyxoviridae, Pneumoviridae, Rhabdoviridae и Sunviridae.
Респираторно-синцитиальный вирус человека (HRSV) представляет собой синцитиальный вирус, который вызывает инфекции дыхательных путей. Он является основной причиной инфекций нижних дыхательных путей и посещений больниц в период младенчества и детства. В изобретении может, в частности, рассматриваться вирус HRSV, он относится к группе V РНК-вирусов. Более конкретно, вирус RSV представляет собой (-)оцРНК-вирус, принадлежащий к группе V классификации по Балтимору. Он является пневмовирусом, который является частью семейства Paramyxoviridae, которое принадлежит к порядку Mononegavirales. К другим вирусам порядка Mononegavirales, которые, в частности, рассматриваются в изобретении, относятся: вирус кори, вирус эпидемического паротита, вирус Нипах, вирус краснухи и вирус парагриппа человека (который включает HPIV-1, HPIV-2, HPIV-3 и HPIV-4). Следует отметить, подсемейство Paramyxovirinae было условно объединено с семейством Paramyxoviridae, исходя из таксономии порядка Mononegavirales, обновленной в 2016 году.
Роды вирусов в пределах семейства Paramyxoviridae, которые, в частности, рассматриваются в изобретении, включают: род Aquaparamyxovirus, Avulavirus, Ferlavirus, Henipavirus, Morbillivirus, Respirovirus и Rubulavirus.
В изобретении также, в частности, рассматриваются вирусы семейства Orthomyxoviridae. Согласно Таксономии вирусов 2017 г. семейство Orthomyxoviridae относится к «не назначенному» порядку. Роды вирусов в пределах семейства Orthomyxoviridae, которые, в частности, рассматриваются, включают: Alphainfluenzavirus, Betainfluenzavirus, Deltainfluenzavirus, Gammainfluenzavirus, Isavirus, Quaranjavirus и Thogotovirus.
В изобретении могут, в частности, рассматриваться вирус гриппа A, вирус гриппа B, вирус гриппа C, они относятся к РНК-вирусам группы V и семейству Orthomyxoviridae, которые могут быть определены как вирусы с одноцепочечной антисмысловой РНК или (-)оцРНК-вирусы. Изавирус и тоготовирус также принадлежат к порядку Orthomyxoviridae.
Подробное описание изобретения
Авторы изобретения неожиданно обнаружили, что арил-N-арильные соединения обладают активностью широкого спектра против РНК-вирусов и, в частности, одноцепочечных РНК-вирусов, принадлежащих к группе IV или V классификации по Балтимору. Группы IV и V включают соответственно (+)оцРНК-вирусы и (-)оцРНК-вирусы; которые также относятся к вирусам с одноцепочечной смысловой РНК и вирусам с одноцепочечной антисмысловой РНК.
Для справки, содержание «классификации по Балтимору» рассматривается в свете Классификации и номенклатуры вирусов, изложенной в 10-м отчете по таксономии вирусов от 2017 г.
В настоящем документе раскрыто соединение формулы (I)
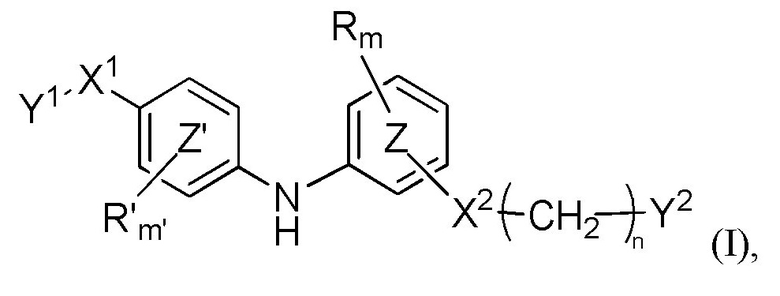
где:
кольцо  и кольцо
и кольцо  независимо означают фениленовую или пиридиленовую группу,
независимо означают фениленовую или пиридиленовую группу,
где группа  находится в мета- или пара-положении на кольце
находится в мета- или пара-положении на кольце  , в частности, в мета-положении, относительно группы -NH-,
, в частности, в мета-положении, относительно группы -NH-,
X1 представляет собой алкениленовую группу, в частности, этениленовую группу, группу -NH-CO-, группу -CO-NH-, группу -CRaRbO-,
Y1 представляет собой арильную группу, выбранную из 2-пиридильной группы или пиримидинильной группы, причем один из атомов азота пиримидинильной группы находится в орто-положении относительно X1,
или, в качестве альтернативы, X1-Y1 представляет собой группу (A) формулы

X2 представляет собой группу -CO-NH-, группу -NH-CO-NH-, группу -OCH2-, группу -NH-CO- или группу -SO2-NH-,
n равен 0, 1, 2 или 3,
m и m'c независимо равны 0, 1 или 2,
Y2 представляет собой атом водорода, гидроксильную группу или группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода, атом фтора или (C1-C4)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C8)циклоалкильную группу, причем указанная (C3-C8)циклоалкильная группа необязательно замещена одной или двумя (C1-C4)алкильными группами, атомами галогена или (C1-C4)алкоксигруппами, и указанная (C3-C8)циклоалкильная группа необязательно прервана на указанном R1 и/или R2 атомом кислорода,
R и R'c независимо представляют собой атом галогена, (C1-C4)алкильную группу, (C3-C6)циклоалкильную группу, (C1-C5)алкоксигруппу, группу -SO2-NRaRb, группу -SO3H, группу -OH, группу -O-SO2-ORc или группу -O-P(=O)-(ORc)(ORd),
Ra, Rb, Rc и Rd независимо представляют собой атом водорода или (C1-C4)алкильную группу,
при условии, что когда X1 представляет собой группу -CRaRbO-, Y1 может дополнительно представлять собой 3-пиридильную, 4-пиридильную или фенильную группу, необязательно замещенную одним или двумя заместителями, выбранными из атома галогена, (C1-C4)алкильной группы, цианогруппы, (C1-C5)алкоксигруппы, трифторметильной группы, трифторметоксигруппы, группы -SO2-NRaRb, группы -SO3H, группы -OH, группы -O-SO2-ORc или группы -O-P(=O)-(ORc)(ORd),
или любая из его фармацевтически приемлемых солей,
для применения для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, и, в частности, вирусной инфекции Чикунгунья, вирусной инфекции Денге, вирусной инфекции гриппа или вирусной инфекции RSV, или связанного с вирусом состояния.
Согласно первому аспекту настоящее изобретение относится к соединению формулы (I)

где
кольцо  и кольцо
и кольцо 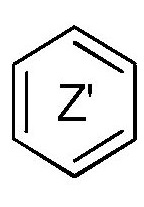 независимо означают фениленовую или пиридиленовую группу,
независимо означают фениленовую или пиридиленовую группу,
X1 представляет собой алкениленовую группу, группу -NH-CO-, группу -CO-NH-,
Y1 представляет собой арильную группу, выбранную из пиридильной группы, пиразинильной группы или пиримидинильной группы,
X2 представляет собой
группу -O-,
группу -CO-NH-,
группу -NH-CO-NH-,
группу -OCH2-,
группу -NH-CO-,
двухвалентное 5-членное гетероароматическое кольцо, содержащее 1, 2, 3 или 4 гетероатома, такое как триазол или оксадиазол,
или
группу -SO2-NH-,
n равен 0, 1, 2 или 3,
m и m'c независимо равен 0, 1 или 2,
Y2 представляет собой
атом водорода,
гидроксильную группу,
морфолинильную группу,
пиперидинильную группу, необязательно замещенную (C1-C4)алкильной группой,
пиперазинильную группу, необязательно замещенную (C1-C4)алкильной группой,
или
группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода, атом фтора или (C1-C4)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C8)циклоалкильную группу, причем указанная (C3-C8)циклоалкильная группа необязательно замещена одной или двумя (C1-C4)алкильными группами, атомами галогена или (C1-C4)алкоксигруппами, и указанная (C3-C8)циклоалкильная группа необязательно прервана на указанном R1 и/или R2 атомом кислорода,
или, в качестве альтернативы, X2-Y2 представляет собой группу -C(=O)-NRcRd, где Rc и Rd образуют вместе с атомом азота насыщенное гетероциклическое кольцо, необязательно замещенное одной или двумя (C1-C4)алкильными группами, циклопентильной группой, образуя, таким образом, спироциклопентильное производное, или трифторметильной группой,
R и R'c независимо представляют собой
(C1-C4)алкильную группу,
(C3-C6)циклоалкильную группу,
атом галогена,
(C1-C5)алкоксигруппу,
группу -SO2-NRaRb,
группу -SO3H,
группу -OH или
группу -O-SO2-ORc,
при условии, что когда X1 представляет собой группу -NH-CO-, Y1 может дополнительно представлять собой фенильную группу, необязательно замещенную одним или двумя заместителями, выбранными из атома галогена, (C1-C4)алкильной группы, цианогруппы, (C1-C5)алкоксигруппы, трифторметильной группы, трифторметоксигруппы, группы -SO2-NRaRb, группы -SO3H, группы -OH, группы -O-SO2-ORc или группы -O-P(=O)-(ORc)(ORd),
или любой из его фармацевтически приемлемых солей,
для применения для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору.
Согласно второму аспекту настоящее изобретение относится к соединениям формулы (I), как определено выше, для применения для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, в частности, вирусной инфекции Чикунгунья, вирусной инфекции Денге, вирусной инфекции гриппа или вирусной инфекции RSV, или связанного с вирусом состояния, и даже более конкретно вирусной инфекции RSV, вирусной инфекции Чикунгунья и вирусной инфекции Денге, или связанного с вирусом состояния.
Согласно третьему аспекту настоящее изобретение относится к соединению формулы (Ia),

где
Y1, R, R'c, m, m'c, кольцо  , X2, n и Y2 представляют собой такие, как определено выше,
, X2, n и Y2 представляют собой такие, как определено выше,
или любой из его фармацевтически приемлемых солей,
для применения для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, и в частности, вирусной инфекции Чикунгунья, вирусной инфекции Денге, вирусной инфекции гриппа или вирусной инфекции RSV, или связанного с вирусом состояния.
Кроме того, согласно данному третьему аспекту настоящее изобретение относится к соединению формулы (Ia) для применения, как определено выше,
где
кольцо  представляет собой фениленовую группу или пиридиленовую группу,
представляет собой фениленовую группу или пиридиленовую группу,
Y1 представляет собой 2-пиридильную группу, 3-пиридильную группу или пиразинильную группу,
n равен 1, 2 или 3, m равен 0,
R'c представляет собой атом галогена, (C1-C2)алкоксигруппу или (C1-C2)алкильную группу,
X2 представляет собой группу -CO-NH-, группу -SO2NH- или двухвалентную триазольную группу,
Y2 представляет собой
морфолинильную группу, пиперидинильную группу или пиперазинильную группу, необязательно замещенную (C1-C4)алкильной группой,
группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода или (C1-C2)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу,
или, в качестве альтернативы, X2-Y2 представляет собой группу -C(=O)-NRcRd, где Rc и Rd образуют вместе с атомом азота насыщенное гетероциклическое кольцо, необязательно замещенное одной или двумя (C1-C4)алкильными группами, циклопентильной группой, образуя, таким образом, спироциклопентильное производное, или трифторметильной группой,
или любой из его фармацевтически приемлемых солей.
Кроме того, согласно указанному третьему аспекту настоящее изобретение дополнительно относится к соединениям формулы (Ia), как определено выше, где
кольцо представляет собой фениленовую группу или пиридиленовую группу,
Y1 представляет собой 2-пиридильную группу,
n равен 2, m равен 0, R'c представляет собой атом галогена, (C1-C2)алкоксигруппу или (C1-C2)алкильную группу,
Y2 представляет собой группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода или (C1-C2)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу,
или любой из их фармацевтически приемлемых солей,
для применения для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, и, в частности, вирусной инфекции Чикунгунья, вирусной инфекции Денге, вирусной инфекции гриппа или вирусной инфекции RSV, или связанного с вирусом состояния.
Согласно четвертому аспекту настоящее изобретение относится к соединению формулы (Ib)

где
Y1, R, R'c, m, m'c, кольцо, X2, n и Y2 представляют собой такие, как определено выше,
или любой из его фармацевтически приемлемых солей,
для применения для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, и, в частности, вирусной инфекции Чикунгунья, вирусной инфекции Денге, вирусной инфекции гриппа или вирусной инфекции RSV, или связанного с вирусом состояния.
Кроме того, согласно данному четвертому аспекту настоящее изобретение относится к соединению формулы (Ib) для применения, как определено выше, где:
кольцо представляет собой фениленовую группу,
Y1 представляет собой фенильную группу, 2-пиридильную группу или пиримидинильную группу, причем один из атомов азота пиримидинильной группы находится в орто-положении относительно группы -NH-CO-,
n равен 1, 2 или 3, m равен 0, m'c равен 0 или 1,
R'c представляет собой (C3-C6)циклоалкильную группу,
X2 представляет собой группу -CO-NH-, группу -O- или двухвалентный триазол,
Y2 представляет собой
гидроксильную группу,
морфолинильную группу, пиперидинильную группу или пиперазинильную группу, необязательно замещенную (C1-C4)алкильной группой,
группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода или (C1-C2)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу, причем указанная (C3-C6)циклоалкильная группа необязательно прервана на указанном R1 и/или R2 атомом кислорода,
или любой из его фармацевтически приемлемых солей.
Кроме того, согласно указанному четвертому аспекту настоящее изобретение дополнительно относится к соединениям формулы (Ib), как определено выше,
где кольцо  представляет собой фениленовую группу,
представляет собой фениленовую группу,
Y1 представляет собой 2-пиридильную группу или пиримидинильную группу, причем один из атомов азота пиримидинильной группы находится в орто-положении относительно группы -NH-CO-,
n равен 1 или 2, m и m'c равны 0,
X2 представляет собой группу -CO-NH-,
Y2 представляет собой группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода или (C1-C2)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу, причем указанная (C3-C6)циклоалкильная группа необязательно прервана на указанном R1 и/или R2 атомом кислорода,
или любой из их фармацевтически приемлемых солей,
для применения для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, и, в частности, вирусной инфекции Чикунгунья, вирусной инфекции Денге, вирусной инфекции гриппа или вирусной инфекции RSV, или связанного с вирусом состояния.
Согласно пятому аспекту настоящее изобретение относится к соединению формулы (Id)

где
Y1, R, R'c, m, m'c, кольцо 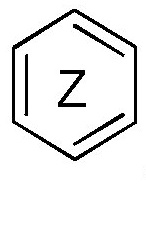 , X2, n и Y2 представляют собой такие, как определено выше,
, X2, n и Y2 представляют собой такие, как определено выше,
или любой из его фармацевтически приемлемых солей,
для применения для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, и, в частности, вирусной инфекции Чикунгунья, вирусной инфекции Денге, вирусной инфекции гриппа или вирусной инфекции RSV, или связанного с вирусом состояния.
Кроме того, согласно данному пятому аспекту настоящее изобретение относится к соединению формулы (Id) для применения, как определено выше, где
кольцо  представляет собой фениленовую группу,
представляет собой фениленовую группу,
Y1 представляет собой 2-пиридильную группу или 3-пиридильную группу,
X2 представляет собой группу -CO-NH-, группу -SO2-NH- или двухвалентный триазол,
m'c и m равны 0, n равен 1, 2 или 3,
Y2 представляет собой гидроксил или группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода или (C1-C2)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу,
или любой из его фармацевтических приемлемых солей.
Кроме того, согласно указанному пятому аспекту настоящее изобретение дополнительно относится к соединениям формулы (Id), где кольцо 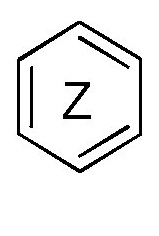 представляет собой фениленовую группу,
представляет собой фениленовую группу,
группа  находится в мета-положении на кольце
находится в мета-положении на кольце  относительно группы -NH-,
относительно группы -NH-,
Y1 представляет собой 2-пиридильную группу,
m'c и m равны 0, n равен 2,
Y2 представляет собой группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода или (C1-C2)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу,
или любой из их фармацевтических приемлемых солей
для применения для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, и, в частности, вирусной инфекции Чикунгунья, вирусной инфекции Денге, вирусной инфекции гриппа или вирусной инфекции RSV, или связанного с вирусом состояния.
Упомянутые выше соединения (I), (Ia), (Ib) и (Id) являются особенно подходящими для лечения или предупреждения вирусной инфекции или связанного с ней состояния, в частности, РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, или связанного состояния, и наиболее предпочтительно вирусной инфекции Чикунгунья, вирусной инфекции Денге, вирусной инфекции гриппа или вирусной инфекции RSV, или связанного с вирусом состояния.
Упомянутые выше соединения даже более предпочтительно подходят для лечения или предупреждения вирусной инфекции Чикунгунья, вирусной инфекции Денге или вирусной инфекции RSV, или связанного с вирусом состояния, наиболее предпочтительно вирусной инфекции RSV.
Дополнительные аспекты настоящего изобретения будут описаны далее в настоящем документе, как например, применение новых соединений формулы (I), (Ia), (Ib) и (Id) в качестве лекарственного средства, фармацевтическая композиция и способ синтеза.
Согласно частному варианту осуществления в настоящем документе описано соединение формулы (I), как определено выше, где алкениленовая группа представляет собой (E)-алкениленовую группу,
m и m'c независимо равны 0 или 1,
Y2 представляет собой группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода, атом фтора или (C1-C2)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу, причем указанная (C3-C6)циклоалкильная группа необязательно замещена одним или двумя атомами галогена, и указанная (C3-C6)циклоалкильная группа необязательно прервана на указанном R1 и/или R2 атомом кислорода,
R и R'c независимо представляют собой атом галогена, (C1-C2)алкильную группу, (C3-C6)циклоалкильную группу или (C1-C2)алкоксигруппу,
или любая из его фармацевтически приемлемых солей,
для применения для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, и, в частности, вирусной инфекции Чикунгунья, вирусной инфекции Денге, вирусной инфекции гриппа или вирусной инфекции RSV, или связанного с вирусом состояния.
Согласно еще одному варианту осуществления в настоящем документе описано соединение формулы (I):

где:
кольцо  и кольцо независимо означают фениленовую и пиридиленовую группу,
и кольцо независимо означают фениленовую и пиридиленовую группу,
где группа  находится в мета- или пара-положении на кольце относительно группы -NH-,
находится в мета- или пара-положении на кольце относительно группы -NH-,
X1 представляет собой алкениленовую группу, группу -NH-CO-, группу -CO-NH-, группу -CRaRbO-,
Y1 представляет собой арильную группу, выбранную из 2-пиридильной группы или пиримидинильной группы, причем один из атомов азота пиримидинильной группы находится в орто-положении относительно X1,
или, в качестве альтернативы, X1-Y1 представляет собой группу (A) формулы:
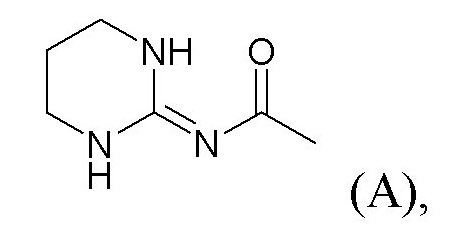
X2 представляет собой группу -CO-NH-, группу -NH-CO-NH-, группу -OCH2-, группу -NH-CO- или группу -SO2-NH-,
n равен 0, 1, 2 или 3,
m и m'c независимо равны 0, 1 или 2,
Y2 представляет собой атом водорода, гидроксильную группу или группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода, атом фтора или (C1-C4)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C8)циклоалкильную группу, причем указанная (C3-C8)циклоалкильная группа необязательно замещена одной или двумя (C1-C4)алкильными группами, атомами галогена или (C1-C4)алкоксигруппами, и указанная (C3-C8)циклоалкильная группа необязательно прервана на указанном R1 и/или R2 атомом кислорода,
R и R'c независимо представляют собой атом галогена, (C1-C4)алкильную группу, (C3-C6)циклоалкильную группу, (C1-C5)алкоксигруппу, группу -SO2-NRaRb, группу -SO3H, группу -OH, группу -O-SO2-ORc или группу -O-P(=O)-(ORc)(ORd),
Ra, Rb, Rc и Rd независимо представляют собой атом водорода или (C1-C4)алкильную группу,
при условии, что когда X1 представляет собой группу -CRaRbO-, Y1 может дополнительно представлять собой 3-пиридильную, 4-пиридильную или фенильную группу, необязательно замещенную одним или двумя заместителями, выбранными из атома галогена, (C1-C4)алкильной группы, цианогруппы, (C1-C5)алкоксигруппы, трифторметильной группы, трифторметоксигруппы, группы -SO2-NRaRb, группы -SO3H, группы -OH, группы -O-SO2-ORc или группы -O-P(=O)-(ORc)(ORd),
и при условии, что, когда Y1-X1 представляет собой 2-пиридилэтениленовую группу, X2 представляет собой группу -CO-NH- и Y2 представляет собой группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода или (C1-C4)алкильную группу, и m'c отличается от 0,
или любая из его фармацевтически приемлемых солей,
для применения для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору.
Кроме того, согласно частному варианту осуществления настоящее изобретение относится к новым соединениям, охватываемым формулой (I).
Указанные новые соединения, которые могут также находиться в форме приемлемых солей, выбраны из следующих соединений:
(1) соединение формулы (Ia), как определено выше для формулы (I),
где
Y1 представляет собой 2-пиридильную группу, 3-пиридильную или пиразинильную группу,
группа  находится в мета-положении на кольце
находится в мета-положении на кольце  относительно группы -NH-,
относительно группы -NH-,
X2, Y2, n, R, R'c, m и m'c представляют собой такие, как определено выше,
кольцо представляет собой фениленовую группу, и
при условии, что соединения 1 и 2, как определено ниже в настоящем документе, исключены,
(2) соединение формулы (Ib), как определено выше для формулы (I),
где
Y1 представляет собой фенильную группу, 2-пиридильную группу или пиримидинильную группу, причем один из атомов азота пиримидинильной группы находится в орто-положении относительно X1,
X2, Y2, n, R, R'c, кольцо , m и m'c представляют собой такие, как определено выше, и
(4) соединение формулы (Id), как определено выше для формулы (I),
где
Y1 представляет собой 2-пиридильную группу или 3-пиридильную группу, и
R, R'c, m, m'c, кольцо , X2, n и Y2 представляют собой такие, как определено выше.
Согласно еще одному аспекту настоящее изобретение относится к новому соединению формулы (Ia), (Ib) и (Id), как определено непосредственно выше, или любой из его фармацевтически приемлемых солей, и любому из соединений (3) - (18), (32) - (35), (91) - (122), как определено ниже в настоящем документе, или любой из их фармацевтически приемлемых солей, для применения в качестве лекарственного средства.
Любая комбинация определенных выше вариантов осуществления для R, R'c, m, m'c, кольца , кольца  , X1, X2, n, Y1, Y2 друг с другом является частью настоящего изобретения.
, X1, X2, n, Y1, Y2 друг с другом является частью настоящего изобретения.
Согласно частному варианту осуществления настоящее изобретение относится к соединению формулы (I) для применения, как определено выше, где
кольцо и кольцо оба представляют собой фениленовую группу.
Согласно частному варианту осуществления в настоящем документе описано соединение формулы (Ia), как определено выше,
где
алкениленовая группа представляет собой (E)-алкениленовую группу,
m и m'c независимо равны 0 или 1,
Y2 представляет собой группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода, атом фтора или (C1-C2)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу, причем указанная (C3-C6)циклоалкильная группа необязательно замещена одним или двумя атомами галогена, и указанная (C3-C6)циклоалкильная группа необязательно прервана на указанном R1 и/или R2 атомом кислорода,
R и R'c независимо представляют собой атом галогена, (C1-C2)алкильную группу, (C3-C6)циклоалкильную группу или (C1-C2)алкоксигруппу,
или любая из его фармацевтически приемлемых солей,
для применения для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, и, в частности, вирусной инфекции Чикунгунья, вирусной инфекции Денге, вирусной инфекции гриппа или вирусной инфекции RSV, или связанного с вирусом состояния.
Согласно частному варианту осуществления в настоящем документе описано соединение формулы (Ib), как определено выше,
где
m и m'c независимо равны 0 или 1,
Y2 представляет собой группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода, атом фтора или (C1-C2)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу, причем указанная (C3-C6)циклоалкильная группа необязательно замещена одним или двумя атомами галогена, и указанная (C3-C6)циклоалкильная группа необязательно прервана на указанном R1 и/или R2 атомом кислорода,
R и R'c независимо представляют собой атом галогена, (C1-C2)алкильную группу, (C3-C6)циклоалкильную группу или (C1-C2)алкоксигруппу,
или любая из его фармацевтически приемлемых солей,
для применения для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, и, в частности, вирусной инфекции Чикунгунья, вирусной инфекции Денге, вирусной инфекции гриппа или вирусной инфекции RSV, или связанного с вирусом состояния.
Согласно частному варианту осуществления в настоящем документе описано соединение формулы (Id), как определено выше,
где
m и m'c независимо равны 0 или 1,
Y2 представляет собой группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода, атом фтора или (C1-C2)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу, причем указанная (C3-C6)циклоалкильная группа необязательно замещена одним или двумя атомами галогена, и указанная (C3-C6)циклоалкильная группа необязательно прервана на указанном R1 и/или R2 атомом кислорода,
R и R'c независимо представляют собой атом галогена, (C1-C2)алкильную группу, (C3-C6)циклоалкильную группу или (C1-C2)алкоксигруппу,
или любая из его фармацевтически приемлемых солей,
для применения для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, и, в частности, вирусной инфекции Чикунгунья, вирусной инфекции Денге, вирусной инфекции гриппа или вирусной инфекции RSV, или связанного с вирусом состояния.
В еще одном варианте осуществления настоящее изобретение относится к соединениям формулы (I) для применения, как определено выше,
где
Y1 представляет собой
2-пиридинильную группу или 3-пиридинильную группу,
пиримидинильную группу или пиразинильную группу, где один из атомов азота находится в орто-положении относительно X1,
при условии, что когда X1 представляет собой группу -NH-CO-, Y1 может дополнительно представлять собой фенильную группу,
или любой из их фармацевтически приемлемых солей.
В еще одном варианте осуществления предложены соединения формулы (I) для применения, как определено выше, где
X2 представляет собой
группу -O-,
группу -CO-NH-,
двухвалентный триазол
или
группу -SO2-NH-,
или любая из их фармацевтически приемлемых солей.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы (I) для применения, как определено выше, где
Y2 представляет собой
гидроксильную группу,
морфолинильную группу,
пиперидинильную группу, необязательно замещенную (C1-C4)алкильной группой,
пиперазинильную группу, необязательно замещенную (C1-C4)алкильной группой,
или
группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода, атом фтора или (C1-C4)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C8)циклоалкильную группу,
или любой из его фармацевтически приемлемых солей.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы (I) для применения, как определено выше, где
R и R'c независимо представляют собой
(C1-C4)алкильную группу,
(C3-C6)циклоалкильную группу,
атом галогена или
(C1-C5)алкоксигруппу,
или любой из его фармацевтически приемлемых солей.
Любая комбинация определенных выше вариантов осуществления для R, R'c, m, m'c, кольца  , кольца
, кольца  , X1, X2, n, Y1, Y2 друг с другом является частью настоящего изобретения.
, X1, X2, n, Y1, Y2 друг с другом является частью настоящего изобретения.
В еще одном варианте осуществления настоящее изобретение относится к соединению формулы (I) для применения, как определено выше, где
кольцо  и кольцо
и кольцо 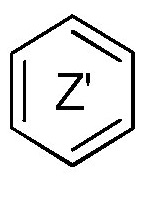 оба представляют фениленовую группу,
оба представляют фениленовую группу,
R” представляет собой атом водорода,
Y1 представляет собой 2-пиридинильную группу, при условии, что когда X1 представляет собой группу -NH-CO-, Y1 может дополнительно представлять собой фенильную группу,
X2 представляет собой группу -O-, группу -CO-NH-,
Y2 представляет собой группу -CR1R2R3, где R1 и R2 образуют вместе с несущим их атомом углерода (C3-C8)циклоалкильную группу, и R3 представляет собой атом водорода или (C1-C4)алкильную группу, и
R и R'c представляет собой атом водорода или (C3-C6)циклоалкильную группу, такую как циклопропильная группа,
или любой из его фармацевтически приемлемых солей.
Согласно предпочтительному варианту осуществления настоящего изобретения соединение формулы (I) выбрано из:
- (1) (E)-N-изопентил-3-((4-(2-(пиридин-2-ил)винил)фенил)амино)бензамида
- (2) (E)-N-изопентил-4-((4-(2-(пиридин-2-ил)винил)фенил)амино)бензамида
- (3) (E)-N-изопентил-3-((3-метокси-4-(2-(пиридин-2-ил)винил)фенил)амино)бензамида
- (4) (E)-N-(2-циклогексилэтил)-3-((4-(2-(пиридин-2-ил)винил)фенил)амино)бензамида
- (5) (E)-N-неопентил-3-((4-(2-(пиридин-2-ил)винил)фенил)амино)бензамида
- (6) (E)-N-(2-циклопентилэтил)-3-((4-(2-(пиридин-2-ил)винил)фенил)амино)бензамида
- (7) (E)-N-изопентил-3-((4-(2-(пиридин-2-ил)винил)фенил)амино)бензолсульфонамида
- (8) (E)-N-(2-циклопропилэтил)-3-((4-(2-(пиридин-2-ил)винил)фенил)амино)бензамида
- (9) (E)-N-(2-циклобутилэтил)-3-((4-(2-(пиридин-2-ил)винил)фенил)амино)бензамида
- (10) (E)-N-(2-циклопентилэтил)-6-((4-(2-(пиридин-2-ил)винил)фенил)амино)пиколинамида
- (11) (E)-N-(2-циклогексилэтил)-3-((3-фтор-4-(2-(пиридин-2-ил)винил)фенил)амино)бензамида
- (12) (E)-N-(2-циклопентилэтил)-3-((2-метил-4-(2-(пиридин-2-ил)винил)фенил)амино)бензамида
- (13) (E)-N-изопентил-3-((2-метил-4-(2-(пиридин-2-ил)винил)фенил)амино)бензамида
- (14) N-изопентил-3-((4-(пиридин-2-илкарбамоил)фенил)амино)бензамида
- (15) N-(2-циклопентилэтил)-3-((4-(пиридин-2-илкарбамоил)фенил)амино)бензамида
- (16) N-(2-циклогексилэтил)-3-((4-(пиридин-2-илкарбамоил)фенил)амино)бензамида
- (17) N-(2-циклопентилэтил)-3-((4-(пиримидин-4-илкарбамоил)фенил)амино)бензамида
- (18) N-изопентил-3-((4-(пиримидин-2-илкарбамоил)фенил)амино)бензамида
- (32) N-(4-((3-(изопентилкарбамоил)фенил)амино)фенил)пиколинамида
- (33) N-(4-((3-(неопентилкарбамоил)фенил)амино)фенил)пиколинамида
- (34) N-(4-((3-(N-изопентилсульфамоил)фенил)амино)фенил)пиколинамида
- (35) N-(4-((3-((2-циклопентилэтил)карбамоил)фенил)амино)фенил)пиколинамида
- (91) 3-({3-метокси-4-[(E)-2-(пиридин-3-ил)этенил]фенил}амино)-N-[3-(морфолин-4-ил)пропил]бензамида
- (92) 3-({2-метил-4-[(E)-2-(пиридин-3-ил)этенил]фенил}амино)-N-[3-(морфолин-4-ил)пропил]бензамида
- (93) N-(2-циклопентилэтил)-3-({4-[(E)-2-(пиридин-3-ил)этенил]фенил}амино)бензол-1-сульфонамида
- (94) 4-[(E)-2-(пиридин-2-ил)этенил]-N-{3-[4-(трифторметил)пиперидин-1-карбонил]фенил}анилина
- (95) N-(3-метилбутил)-3-({4-[(E)-2-(пиразин-2-ил)этенил]фенил}амино)бензамида
- (96) N-(2-циклопентилэтил)-3-({4-[(E)-2-(пиразин-2-ил)этенил]фенил}амино)бензамида
- (97) N-[3-(4,4-диметилпиперидин-1-карбонил)фенил]-4-[(E)-2-(пиридин-2-ил)этенил]анилина
- (98) N-(3-{8-азаспиро[4.5]декан-8-карбонил}фенил)-4-[(E)-2-(пиридин-2-ил)этенил]анилина
- (99) 3-[4-(2-метилпропил)-1H-1,2,3-триазол-1-ил]-N-{4-[(E)-2-(пиридин-2-ил)этенил]фенил}анилина
- (100) 3-[4-(2-метилпропил)-1H-1,2,3-триазол-1-ил]-N-{4-[(E)-2-(пиридин-3-ил)этенил]фенил}анилина
- (101) N-(3-метилбутил)-3-({4-[(E)-2-(пиридин-3-ил)этенил]фенил}амино)бензамида
- (102) N-(3-метилбутил)-3-({4-[(E)-2-(пиридин-3-ил)этенил]фенил}амино)бензол-1-сульфонамида
- (103) N-(2-циклопентилэтил)-3-({4-[(E)-2-(пиридин-3-ил)этенил]фенил}амино)бензамида
- (104) N-(2,2-диметилпропил)-3-({4-[(E)-2-(пиридин-3-ил)этенил]фенил}амино)бензамида
- (105) N-(2-циклогексилэтил)-3-({4-[(E)-2-(пиридин-3-ил)этенил]фенил}амино)бензамида
- (106) N-[3-(морфолин-4-ил)пропил]-3-({4-[(E)-2-(пиридин-2-ил)этенил]фенил}амино)бензол-1-сульфонамида
- (107) N-[3-(морфолин-4-ил)пропил]-2-({4-[(E)-2-(пиридин-2-ил)этенил]фенил}амино)пиридин-4-карбоксамида
- (108) N-[3-(морфолин-4-ил)пропил]-6-({4-[(E)-2-(пиридин-2-ил)этенил]фенил}амино)пиридин-2-карбоксамида
- (109) N-[3-(4-метилпиперазин-1-ил)пропил]-2-({4-[(E)-2-(пиридин-2-ил)этенил]фенил}амино)пиридин-4-карбоксамида
- (110) N-[3-(4-метилпиперазин-1-ил)пропил]-6-({4-[(E)-2-(пиридин-2-ил)этенил]фенил}амино)пиридин-2-карбоксамида
- (111) N-[3-(морфолин-4-ил)пропил]-3-({4-[(E)-2-(пиридин-2-ил)этенил]фенил}амино)бензамида
- (112) N-[3-(пиперидин-1-ил)пропил]-3-({4-[(E)-2-(пиридин-2-ил)этенил]фенил}амино)бензамида
- (113) N-[3-(4-метилпиперазин-1-ил)пропил]-3-({4-[(E)-2-(пиридин-2-ил)этенил]фенил}амино)бензамида
- (114) 3-{4-[(диэтиламино)метил]-1H-1,2,3-триазол-1-ил}-N-{4-[(E)-2-(пиридин-2-ил)этенил]фенил}анилина
- (115) 4-{[3-(3-циклогексилпропокси)фенил]амино}-3-циклопропил-N-фенилбензамида
- (116) 3-циклопропил-4-({2-[(3-метилбутил)карбамоил]фенил}амино)-N-(пиримидин-2-ил)бензамида
- (117) 3-циклопропил-N-(пиримидин-2-ил)-4-({2-[(3,3,3-трифторпропил)карбамоил]фенил}амино)бензамида
- (118) 4-[(3-{[3-(морфолин-4-ил)пропил]карбамоил}фенил)амино]-N-(пиридин-2-ил)бензамида
- (119) 4-[(3-{[3-(пиперидин-1-ил)пропил]карбамоил}фенил)амино]-N-(пиридин-2-ил)бензамида
- (120) 4-[(3-{[3-(4-метилпиперазин-1-ил)пропил]карбамоил}фенил)амино]-N-(пиридин-2-ил)бензамида
- (121) 4-({3-[4-(гидроксиметил)-1H-1,2,3-триазол-1-ил]фенил}амино)-N-(пиридин-2-ил)бензамида
- (122) N-[4-({3-[4-(2-метилпропил)-1H-1,2,3-триазол-1-ил]фенил}амино)фенил]пиридин-2-карбоксамида
и их фармацевтически приемлемых солей.
Настоящее изобретение распространяется на соединения (3) - (18), (32) - (35), (91) - (122) и их фармацевтически приемлемые соли, такие как гидробромид, тартрат, цитрат, трифторацетат, аскорбат, гидрохлорид, тозилат, трифлат, малеат, мезилат, формиат, ацетат и фумарат.
Согласно еще одному аспекту предмет настоящего изобретения относится к соединению формулы (I), (Ia), (Ib) и (Id), как определено выше, или любой из его фармацевтически приемлемых солей, и любому из соединений (1) - (18), (32) - (35) и (91) - (122) или любой из его фармацевтически приемлемых солей, для применения в качестве средства для предупреждения, ингибирования или лечения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору.
Соединения формулы (I), как определено выше, для которых
кольцо  и кольцо оба представляют собой фениленовую группу,
и кольцо оба представляют собой фениленовую группу,
Y1 представляет собой 2-пиридинильную группу, при условии, что когда X1 представляет собой группу -NH-CO-, Y1 может дополнительно представлять собой фенильную группу,
X2 представляет собой группу -O-, группу -CO-NH-,
Y2 представляет собой
группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода или (C1-C4)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C8)циклоалкильную группу, и R3 представляет собой атом водорода или (C1-C4)алкильную группу, или
морфолинильную группу, и
R и R'c независимо представляют собой атом водорода, (C1-C4)алкильную группу, такую как метильная группа, или (C3-C6)циклоалкильную группу, такую как циклопропильная группа,
или любая из их фармацевтически приемлемых солей, могут особенно подходить для применения в качестве средства для предупреждения, ингибирования или лечения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежим к группе IV или V классификации по Балтимору.
Соединения (12), (13), (15) и (35) или любая из их фармацевтически приемлемых солей могут быть особенно подходящими для предупреждения, ингибирования или лечения инфекции Денге.
Соединения (12), (13), (16) и (115) или любая из их фармацевтически приемлемых солей могут быть особенно подходящими для предупреждения, ингибирования или лечения инфекции RSV.
Соединения (1), (2), (4), (6), (9), (12), (13), (14), (15), (16), (32), (35) или любая из их фармацевтически приемлемых солей могут быть особенно подходящими для предупреждения, ингибирования или лечения инфекции Чикунгунья.
Соединения согласно изобретению могут существовать в форме свободных оснований или солей присоединения фармацевтически приемлемых кислот.
Выражение «его фармацевтически приемлемая соль» относится к солям, которые образованы из солей присоединения кислоты, образованных с неорганическими кислотами (например, соляной кислотой, бромистоводородной кислотой, серной кислотой, ортофосфорной кислотой, азотной кислотой и т. п.), а также солей, образованных с органическими кислотами, такими как уксусная кислота, щавелевая кислота, винная кислота, янтарная кислота, яблочная кислота, фумаровая кислота, малеиновая кислота, аскорбиновая кислота, бензойная кислота, дубильная кислота, пальмитиновая кислота, альгиновая кислота, полиглутаминовая кислота, нафталинсульфоновая кислота, нафталиндисульфоновая кислота и полигалактуроновая кислота.
Подходящие физиологически приемлемые соли присоединения кислоты соединений формулы (I) включают гидробромид, тартрат, цитрат, трифторацетат, аскорбат, гидрохлорид, тозилат, трифлат, малеат, формиат, ацетат и фумарат.
Соединения формулы (I), (Ia), (Ib) и (Id) и любые из соединений (1) - (18), (32) - (35) и (91) - (122) или любая из их фармацевтически приемлемых солей могут образовывать сольваты или гидраты, и изобретение включает все такие сольваты и гидраты.
Термины «гидраты» и «сольваты» просто означают, что соединения (I), (Ia), (Ib) и (Id) согласно изобретению могут находиться в форме гидрата или сольвата, т. е., быть объединены или ассоциированы с одной или более молекулами воды или растворителя. Это лишь химическая характеристика таких соединений, которую можно применять в отношении всех органических соединений данного типа.
В контексте настоящего изобретения термин:
- «галоген» подразумевает хлор, фтор, бром или иод и, в частности, обозначает хлор, фтор или бром,
- «(C1-Cx)алкил», в том виде, в котором он используется в настоящем документе, соответственно относится к C1-Cx нормальному, вторичному или третичному насыщенному углеводороду, например, (C1-C6)алкилу. Примеры представляют собой метил, этил, 1-пропил, 2-пропил, бутил, пентил, но не ограничиваются ими,
- «алкенилен» означает двухвалентную (C1-Cx)алкильную группу, содержащую двойную связь, и более конкретно этениленовую группу, также известную как винилен или 1,2-этендиил,
- «(C3-C6)циклоалкил», в том виде, в котором он используется в настоящем документе, относится к циклическому насыщенному углеводороду. Примеры представляют собой циклопропил, циклобутил, циклопентил, циклогексил, но не ограничиваются ими,
- «(C3-C6)циклоалкенил», в том виде, в котором он используется в настоящем документе, относится к циклическому неароматическому углеводороду, содержащему по меньшей мере одну ненасыщенную связь. Примеры представляют собой циклопентенил и циклогексенил, но не ограничиваются ими,
- «(C1-Cx)алкокси», в том виде, в котором он используется в настоящем документе, относится к O-(C1-Cx)алкильному фрагменту, где алкил представляет собой такой, как определено выше, например, (C1-C6)алкокси. Примеры представляют собой метокси, этокси, 1-пропокси, 2-пропокси, бутокси, пентокси, но не ограничиваются ими.
- «арил», в том виде, в котором он используется в настоящем документе, относится к моноциклической ароматической группе, содержащей 6 атомов углерода и содержащей от 0 до 2 гетероатомов, таких как азот, кислород или сера и, в частности, азот. В качестве примеров арильных групп можно упомянуть следующие: фенил, пиридин, пиримидин, пиридазин, пиразин и т. п., не ограничиваясь перечисленным. В рамках настоящего изобретения арил предпочтительно представляет собой фенил, пиридазин, пиразин, пиридин, такой как 2-пиридин или 3-пиридин, и пиримидин. Арил еще более предпочтительно представляет собой фенил и пиридин, такой как 2-пиридин или 3-пиридин.
- выражение «двухвалентное 5-членное гетероароматическое кольцо, содержащее 1, 2, 3 или 4 гетероатома», в том виде, в котором оно используется в настоящем документе, означает двухвалентное кольцо, состоящее из ароматического кольца, содержащего 5 цепей и 1, 2, 3 или 4 гетероатома, выбранных из атомов азота и кислорода. В одном варианте осуществления оно содержит по меньшей мере 1 гетероатом и предпочтительно по меньшей мере один атом азота. В еще одном варианте осуществления оно содержит по меньшей мере 2 гетероатома, например, по меньшей мере с одним атомом азота. Согласно еще одному варианту осуществления оно содержит 2, 3 или 4 атома азота, предпочтительно 3 атома азота. Согласно еще одному варианту осуществления оно содержит один атом азота и один атом кислорода или два атома азота и один атом кислорода. Примеры представляют собой двухвалентный триазол, такой как 1,2,3- или 1,2,4-триазолы, оксадиазолы, такие как 1,2,4-оксадиазол или 1,2,3-оксадиазол, и двухвалентные диазолы, такие как двухвалентный диазол и двухвалентный имидазол, но не ограничиваются ими. Согласно предпочтительному варианту осуществления такое двухвалентное 5-членное гетероароматическое кольцо, содержащее 2 или 3 гетероатома, представляет собой двухвалентный триазол.
Соединения формулы (I), (Ia), (Ib) и (Id) могут содержать один или более ассиметрических атомов углерода. Они могут, таким образом, существовать в форме энантиомеров или диастереоизомеров. Данные энантиомеры, диастереоизомеры и их смеси, включая рацемические смеси, входят в объем настоящего изобретения.
Соединения согласно настоящему изобретению могут быть получены традиционными способами органического синтеза, применяемыми на практике специалистами в данной области. Общие последовательности реакций, изложенные ниже, представляют собой общий способ, применимый для получения соединений согласно настоящему изобретению, и не предназначены для ограничения объема или области применения.
Соединения общей формулы (I) могут быть получены в соответствии с нижеследующей схемой 1.

Схема 1
Синтез основан на реакции сочетания, начинающейся с галогенароматического соединения формулы (III), где R, R'c, m, m'c, кольцо , кольцо , X1, X2, n, Y1, Y2 представляют собой такие, как определено выше, и X представляет собой атом хлора, атом иода или атом брома.
Согласно одному варианту осуществления предпочтительно может быть использован способ (A1), когда группа  находится в мета- или пара-положении на кольце относительно группы -NH-.
находится в мета- или пара-положении на кольце относительно группы -NH-.
Согласно способу (A1) соединение формулы (III) может быть помещено в протонный растворитель, такой как трет-бутанол. Затем может быть добавлено соединение формулы (II), например, в молярном соотношении, находящемся в интервале от 1 до 1,5 относительно соединения формулы (III), в присутствии неорганического основания, такого как Cs2CO3 или K2CO3, например, в молярном соотношении, находящемся в интервале от 1 до 5 опять же относительно соединения формулы (III), в присутствии дифосфина, такого как Xantphos (4,5-бис(дифенилфосфино)-9,9-диметилксантен) или X-Phos (2-дициклогексилфосфино-2'c,4'c,6'c-триизопропилбифенил), в частности, в количестве, находящемся в интервале от 2 мол.% до 15 мол.% относительно общего количества соединения формулы (III), и в присутствии металлорганического катализатора, такого как Pd(OAc)2 или Pd2dba3, в количестве, находящемся в интервале от 2 мол.% до 25 мол.% относительно общего количества соединения формулы (III). Затем реакционная смесь может быть нагрета при температуре, находящейся в интервале от 80 до 130°C, например, при 90°C, и перемешиваться в течение от 15 до 25 часов, например, в течение 20 часов, в атмосфере инертного газа и, например, аргона. Реакционная смесь может быть концентрирована при пониженном давлении, и осадок может быть разведен органическим растворителем, таким как этилацетат. Органическая фаза может быть промыта водой, подвергнута декантации, высушена над сульфатом магния, отфильтрована и затем концентрирована при пониженном давлении с получением соединения формулы (I).
Согласно одному варианту осуществления предпочтительно может быть использован способ (A2), когда группа  находится в орто-положении на кольце
находится в орто-положении на кольце  относительно группы -NH-.
относительно группы -NH-.
Согласно способу (A2) соединение формулы (II) может быть помещено в полярный апротонный растворитель, такой как диметилсульфоксид. Затем может быть добавлено соединение формулы (III), например, в молярном соотношении, находящемся в интервале от 1 до 1,5 относительно соединения формулы (II) в присутствии неорганического основания, такого как Cs2CO3 или K2CO3, например, в молярном соотношении, находящемся в интервале от 1 до 5 опять же относительно соединения формулы (II), в присутствии лиганда, такого как L-пролин, в частности, в количестве, находящемся в интервале от 2 мол.% до 25 мол.% относительно общего количества соединения формулы (II), и в присутствии металлорганического катализатора, такого как CuI, в количестве, находящемся в интервале от 2 мол.% до 25 мол.% относительно общего количества соединения формулы (II). Затем реакционная смесь может быть нагрета при температуре, находящейся в интервале от 80 до 130°C, например, при 90°C, и перемешиваться в течение от 15 до 25 часов, например, в течение 20 часов, в атмосфере инертного газа и, например, аргона. Реакционная смесь может быть разведена органическим растворителем, таким как этилацетат. Органическая фаза может быть промыта водой, подвергнута декантации, высушена над сульфатом магния, отфильтрована и затем концентрирована при пониженном давлении с получением соединения формулы (I).
Исходные соединения формулы (II), (III) доступны или могут быть получены в соответствии со способами, известными специалисту в данной области.
Соответственно, настоящее изобретение дополнительно относится к способу синтеза для производства новых соединений формулы (I), (I), (Ia), (Ib) и (Id), как определено выше, более конкретно новых соединений формулы (Ia), (Ib) или (Id), включающему по меньшей мере стадию сочетания соединения формулы (II)

где X1, Y1, R, R'c, m, m'c, кольцо  , кольцо
, кольцо  , X2, Y2 представляют собой такие, как определено выше, и X представляет собой атом хлора, атом иода или атом брома, в присутствии неорганического основания и лиганда и в присутствии металлорганического катализатора, с получением новых соединений формулы (I), (I), (Ia), (Ib) или (Id), более конкретно новых соединений формулы (Ia), (Ib) или (Id).
, X2, Y2 представляют собой такие, как определено выше, и X представляет собой атом хлора, атом иода или атом брома, в присутствии неорганического основания и лиганда и в присутствии металлорганического катализатора, с получением новых соединений формулы (I), (I), (Ia), (Ib) или (Id), более конкретно новых соединений формулы (Ia), (Ib) или (Id).
Более конкретно, соединения формулы (IIa), при использовании для получения соединений формулы (Ia), могут быть получены в соответствии с нижеследующей схемой 2.

Промежуточные соединения формул (IIa) и (IVa) применимы для получения соединений формулы (Ia) согласно изобретению.
Согласно способу (B) производное бромнитробензола может быть помещено в полярный растворитель, такой как N,N-диметилформамид. Затем может быть добавлен виниларил, причем арил имеет такое же значение, как определено выше, например, в молярном соотношении, находящемся в интервале от 1 до 1,5 относительно производного бромнитробензола, в присутствии неорганического основания, такого как ацетат натрия или ацетат калия, в частности, в молярном соотношении, находящемся в интервале от 1 до 3 опять же относительно соединения формулы (III), в присутствии фосфина, такого как трифенилфисфин, например, в количестве, находящемся в интервале от 5 мол.% до 15 мол.% относительно количества производного бромнитробензола, и в присутствии металлорганического катализатора, такого как Pd(OAc)2 или Pd2dba3, в количестве, находящемся в интервале от 2 мол.% до 10 мол.% относительно количества производного бромнитробензола. Затем реакционная смесь может быть нагрета при температуре, находящейся в интервале от 80 до 140°C, например, при 135°C, и перемешиваться в течение от 15 до 30 часов, например, 24 часов, в атмосфере инертного газа, например, аргона. Реакционная смесь может быть концентрирована при пониженном давлении, и осадок может быть разведен органическим растворителем, таким как этилацетат. Органическая фаза может быть промыта водой, подвергнута декантации, высушена над сульфатом магния, отфильтрована и затем концентрирована при пониженном давлении с получением соединения формулы (IVa).
Согласно способу (C) соединение формулы (IVa) и дигидрат хлорида олова (II) в соотношении, находящемся в интервале от 3 до 8 эквивалентов, могут быть помещены в протонный растворитель, такой как этанол. Затем реакционная смесь может быть нагрета при температуре, находящейся в интервале от 40 до 80°C, например, при 60°C, и перемешиваться в течение от 15 до 25 часов, например, в течение 20 часов. Смесь может быть вылита в 1 н. раствор водный NaOH и экстрагирована органическим растворителем, таким как этилацетат. Органическая фаза затем может быть промыта водой и насыщенным водным солевым раствором, высушена над сульфатом магния, отфильтрована и затем концентрирована при пониженном давлении с получением соединения формулы (IIa).
Более конкретно, промежуточные соединения формулы (IIb) (в которых по меньшей мере один из W1 или W2 представляет собой CH), при использовании для получения соединений формулы (Ib), могут быть получены в соответствии с нижеследующей схемой 3.
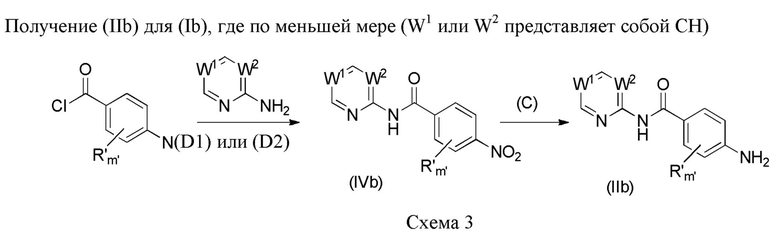
Промежуточные соединения формул (IIb) и (IVb) применимы для получения соединений формулы (Ib) согласно изобретению.
Согласно способу (D1) аминопиридин (W1 и W2 представляют собой CH), добавляемый, например, в молярном соотношении, находящемся в интервале от 1 до 1,5 относительно производного нитробензоилхлорида, может быть помещен в водный раствор неорганического основания, такого как гидроксид натрия, например, в молярной концентрации, находящейся в интервале от 2 M до 5 M. К раствору может быть добавлен полярный апротонный растворитель, такой как дихлорметан, реакционная смесь может быть охлаждена до 0°C посредством ледяной бани, и может быть по каплям добавлен раствор производного нитробензоилхлорида в полярном апротонном растворителе, таком как дихлорметан. Затем реакционная смесь может перемешиваться при комнатной температуре в течение от 15 до 24 часов, например, 18 часов, в атмосфере инертного газа, например, аргона. Полученный осадок может быть отфильтрован, промыт водой и дихлорметаном и высушен под вакуумом в течение ночи с получением соединения формулы (IVb).
Согласно способу (D2) аминопиримидин (W1 или W2 представляет собой N, а другой W1 или W2 представляет собой CH) может быть помещен в полярный апротонный растворитель, такой как дихлорметан. Затем может быть добавлено производное нитробензоилхлорида, например, в молярном соотношении, находящемся в интервале от 1 до 1,5 относительно аминопиримидина в присутствии органического основания, такого как N,N-диизопропилэтиламин или триэтиламин, например, в молярном соотношении, находящемся в интервале от 1 до 2 опять же относительно аминопиримидина, в присутствии нуклеофильного катализатора, такого как диметиламинопиридин, например, в молярном соотношении, находящемся в интервале от 0,1 до 1 опять же относительно аминопиримидина. Реакционная смесь затем может перемешиваться при комнатной температуре в течение от 5 до 20 часов, например, 18 часов, в атмосфере инертного газа и, например, аргона. Органическая фаза может быть промыта водой, и полученный осадок может быть отфильтрован, промыт водой и дихлорметаном и высушен под вакуумом в течение ночи с получением соединения формулы (IVb).
Согласно способу (C) соединение формулы (IVb) и дигидрат хлорида олова (II) в соотношении, находящемся в интервале от 3 до 8 эквивалентов, могут быть помещены в протонный растворитель, такой как этанол. Затем реакционная смесь может быть нагрета при температуре, находящейся в интервале от 40 до 80°C, например, при 60°C, и перемешиваться в течение от 15 до 25 часов, например, в течение 20 часов. Смесь может быть вылита в 1 н. водный раствор NaOH и экстрагирована органическим растворителем, таким как этилацетат. Органическая фаза затем может быть промыта водой и насыщенным водным солевым раствором, высушена над сульфатом магния, отфильтрована и концентрирована при пониженном давлении с получением соединения формулы (IIb).
В случае, когда X1 представляет собой группу -NH-CO-, можно следовать другому пути для получения соединений формулы (Ib), и он изображен ниже на схеме X.
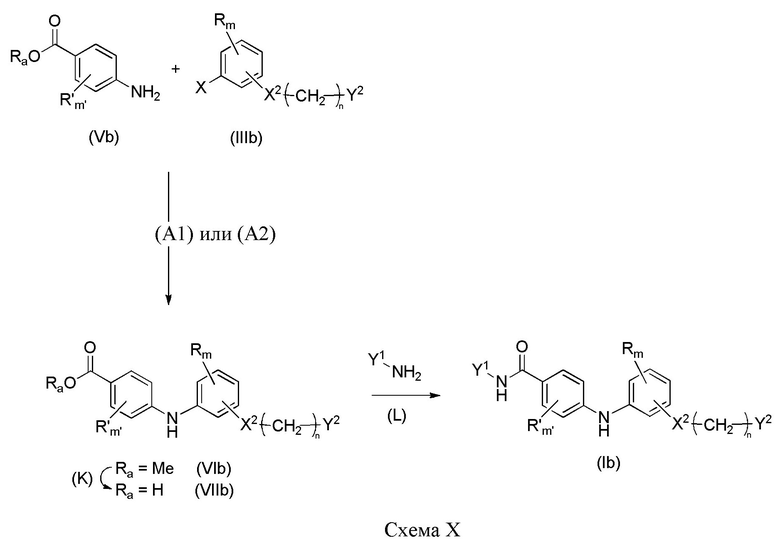
Синтез начинается с реакции сочетания галогенароматического соединения формулы (IIIb) с производным анилина (Vb), где R, R'c, m, m'c, X1, X2, n, Y1, Y2 представляют собой такие, как определено выше, и X представляет собой атом хлора, атом иода или атом брома, в соответствии со способом (A1) или (A2).
Согласно способу (K) соединение формулы (VIb) может быть помещено в протонный растворитель, такой как метанол, и может быть добавлен водный раствор 2 M NaOH в соотношении, находящемся в интервале от 3 до 10 эквивалентов. Реакционная смесь затем может быть нагрета при температуре, находящейся в интервале от 50 до 90°C, например, при 80°C, и перемешиваться в течение от 1 до 24 часов, например, в течение 3 часов. Смесь может быть концентрирована при пониженном давлении и, после добавления водного раствора 2 M HCl, экстрагирована органическим растворителем, таким как дихлорметан. Объединенные органические фазы затем могут быть высушены над сульфатом магния, отфильтрованы и концентрированы при пониженном давлении с получением соединения формулы (VIIb).
Согласно способу (L) соединение формулы (VIIb) и производное амина Y1-NH2, в соотношении, находящемся в интервале от 1,0 до 3 эквивалентов, например, 1,2 эквивалента, могут быть помещены в безводный полярный растворитель, такой как N,N-диметилформамид. Затем может быть добавлен агент реакции сочетания, такой как HATU, например, в молярном соотношении, находящемся в интервале от 1 до 2 относительно соединения (VIIb), в присутствии органического основания, такого как триэтиламин или N,N-диизопропилэтиламин, например, в молярном соотношении, находящемся в интервале от 2 до 5 опять же относительно соединения (VIIb). Реакционную смесь затем можно перемешивать при комнатной температуре в течение от 5 до 20 часов, например, 16 часов. Реакция может быть остановлена при добавлении водного раствора 1 M соляной кислоты, и смесь может быть экстрагирована органическим растворителем, таким как этилацетат. Объединенные органические фазы затем могут быть высушены над сульфатом магния, отфильтрованы и концентрированы при пониженном давлении с получением соединения формулы (Ib).
Более конкретно, соединения формулы (IId), при использовании для получения соединений формулы (Id), могут быть получены в соответствии с нижеследующей схемой 5.

Промежуточные соединения формул (IId), (IVd) и (Vd) применимы для получения соединений формулы (Id) согласно изобретению.
Согласно способу (F) производное 1,4-бензолдиамина может быть помещено в смесь полярных растворителей, таких как N,N-диметилформамид и тетрагидрофуран, например, в соотношении, находящемся в интервале от 1/4 до 1/2. Затем по каплям может быть добавлен Boc2O (ди-трет-бутилдикарбонат), например, в молярном соотношении, находящемся в интервале от 0,25 до 0,75 относительно производного 1,4-бензолдиамина, в присутствии неорганического основания, такого как Cs2CO3 или K2CO3, водного раствора с концентрацией, находящейся в интервале от 2 M до 3 M, и, например, в молярном соотношении, находящемся в интервале от 0,5 до 1 опять же относительно 1,4-бензолдиамина. Затем реакционную смесь можно перемешивать при комнатной температуре в течение от 10 до 70 часов, например, в течение 64 часов, в атмосфере инертного газа и, например, аргона. Реакционная смесь может быть концентрирована при пониженном давлении, и остаток может быть разведен органическим растворителем, таким как этилацетат. Органическая фаза может быть промыта водой, подвергнута декантации, высушена над сульфатом магния, отфильтрована и концентрирована при пониженном давлении с получением соединения формулы (IVd).
Согласно способу (G) аминопроизводное (IVd) может быть помещено в полярный апротонный растворитель, такой как дихлорметан. Затем может быть добавлено производное карбоновой кислоты, например, в молярном соотношении, находящемся в интервале от 1 до 1,5 относительно аминопроизводного (IVd), в присутствии агента реакции сочетания, такого как EDCI.HCl (1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид), например, в молярном соотношении, находящемся в интервале от 1 до 3, в присутствии органического основания, такого как N,N-диизопропилэтиламин или триэтиламин, например, в молярном соотношении, находящемся в интервале от 1 до 5 опять же относительно аминопроизводного (IVd), и в присутствии HOBt (1-гидроксибензотриазолгидрат), например, в молярном соотношении, находящемся в интервале от 1 до 3 опять же относительно аминопроизводного (IVd). Затем реакционную смесь можно перемешивать при комнатной температуре в течение от 5 до 30 часов, например, 24 часов, в атмосфере инертного газа и, например, аргона. Органическая фаза может быть промыта водой, подвергнута декантации, высушена над сульфатом магния, отфильтрована и концентрирована при пониженном давлении с получением соединения формулы (Vd).
Согласно способу (H) соединение формулы (Vd) может быть помещено в полярный апротонный растворитель, такой как дихлорметан. Затем может быть добавлена трифторуксусная кислота, например, в молярном соотношении, находящемся в интервале от 10 до 30 относительно аминопроизводного (IVd). Затем реакционная смесь может перемешиваться при комнатной температуре в течение от 1 до 7 часов, и затем может быть охлаждена до 0°C посредством ледяной бани. Могут быть добавлены вода и неорганическое основание, такое как карбонат натрия или карбонат калия, до достижения pH выше 7. Водная фаза может быть экстрагирована органическим растворителем, таким как дихлорметан. Органические фазы могут быть собраны, высушены над сульфатом магния, отфильтрованы и концентрированы при пониженном давлении с получением соединения формулы (IId).
Химические структуры и спектроскопические данные некоторых соединений формулы (I) согласно изобретению проиллюстрированы соответственно в следующей таблице I и таблице II.
Таблица I
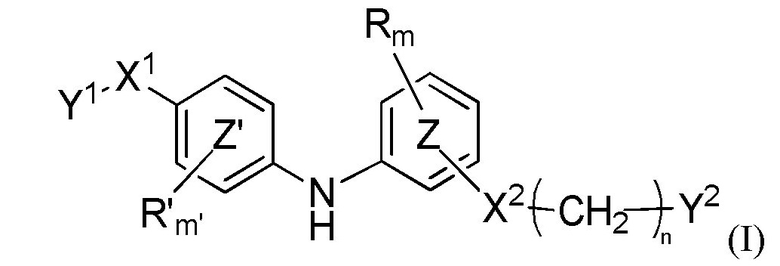





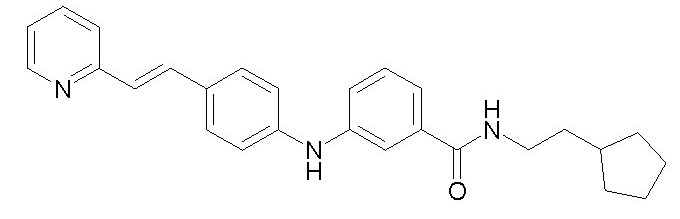

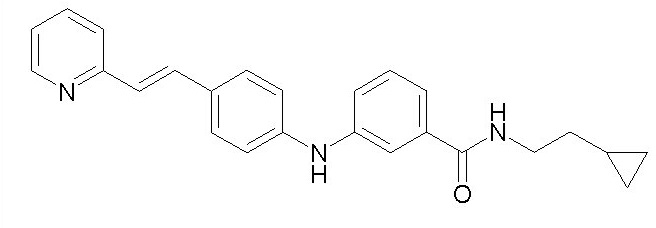



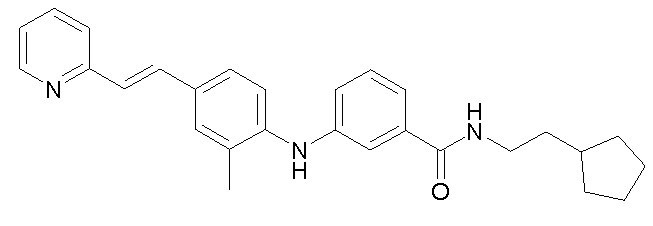
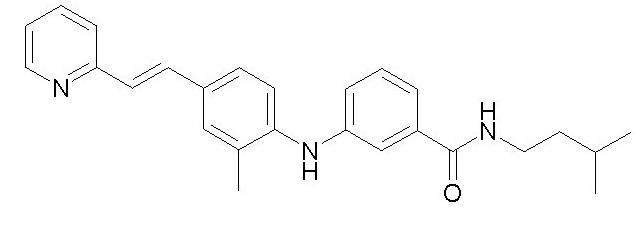
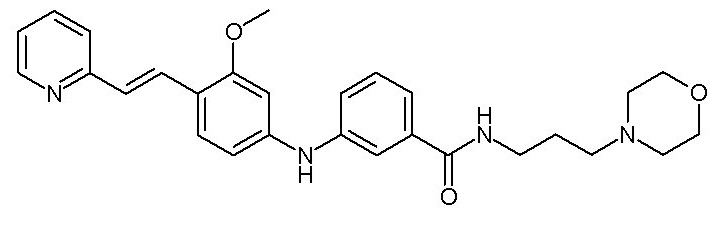

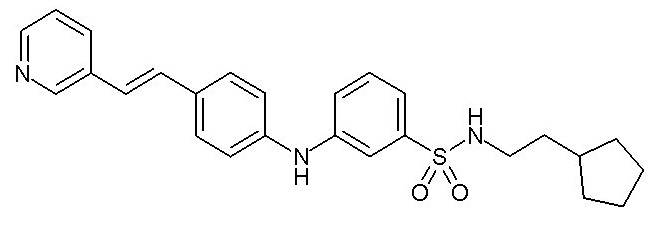









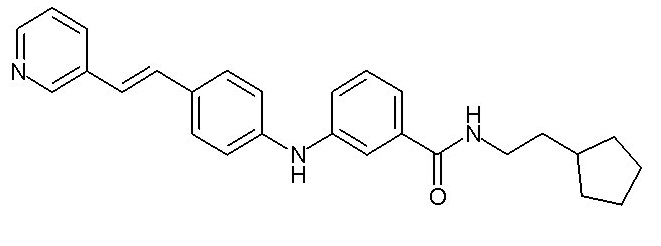

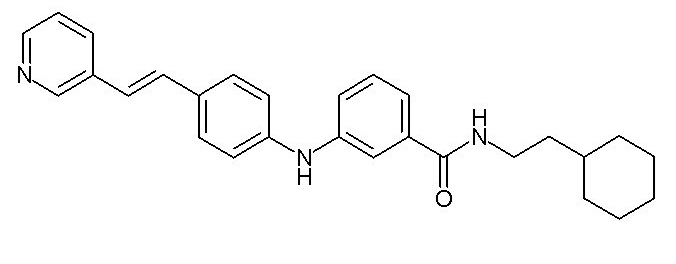



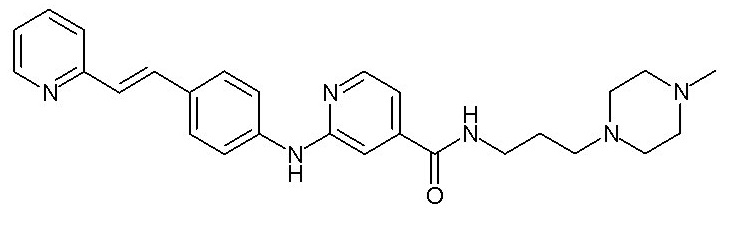
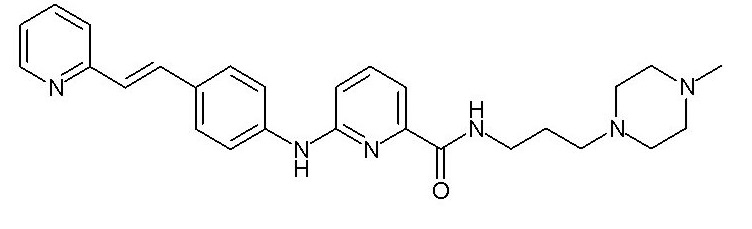
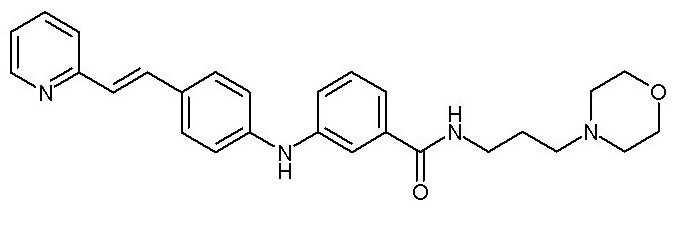



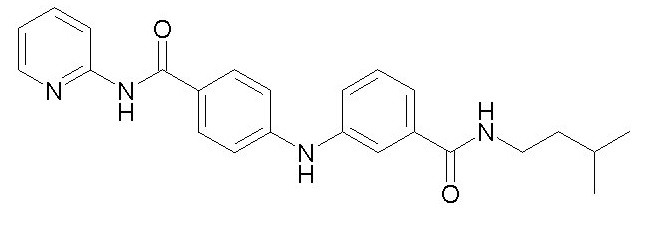


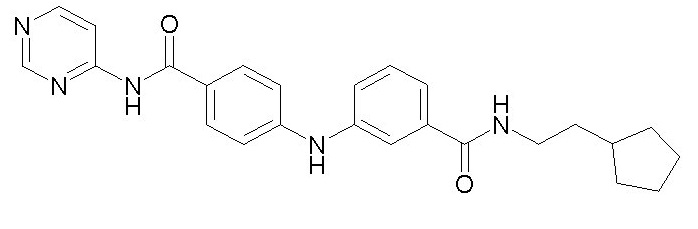










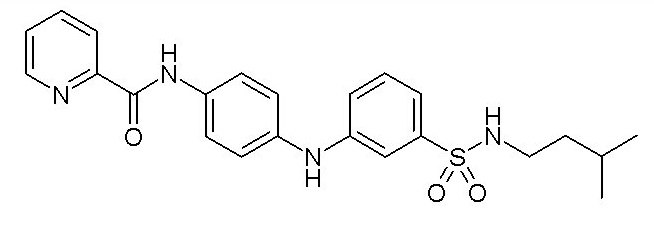

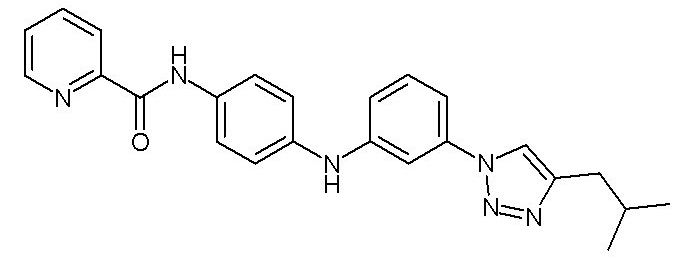
Таблица II
13C ЯМР (75 МГц, d6-ДМСО) δ 164,2, 153,6, 147,5, 141,6, 140,9, 134,8, 134,1, 130,1, 127,2, 126,4, 126,0, 122,8, 119,9, 119,8, 117,8, 116,8, 114,5, 114,2, 77,9, 35,5, 33,6, 30,3, 22,8
[M+H]+ = 412,3
[M+H]+ = 398,0
[M+H]+ = 426,4
[M+H]+ = 400,3
[M+H]+ = 473,4
[M+H]+ = 448,3
[M+H]+ = 387,0
[M+H]+ = 403,1
13C ЯМР (75 МГц, d6-ДМСО) 164,2, 163,5, 150,8, 146,1, 145,3, 140,1, 136,2, 134,3, 128,1, 127,5, 122,3, 119,3, 118,1, 117,6, 117,5, 115,7, 112,8, 112,7, 35,7, 33,7, 30,5, 23,0
[M+H]+ = 429,1
[M+H]+ = 430,0
[M+H]+ = 404,0
13C ЯМР (151 МГц, d6-ДМСО) δ 165,6, 160,0, 146,2, 144,9, 139,9, 131,8, 130,2, 128,9, 126,8, 126,7, 126,4, 123,7, 120,9, 116,2, 111,4, 107,5, 105,3, 68,0, 37,2, 33,7, 33,3, 26,6, 26,6, 26,3, 11,6, 7,8
[M+H]+ = 469,2
[M+H]+ = 444,4
[M+H]+ = 470,3
[M+H]+ = 470,3
[M+H]+ = 403,1
[M+H]+ = 403,1
[M+H]+ = 439,1
13C ЯМР (75 МГц, d6-ДМСО) δ 164,9, 160,5, 148,7, 146,9, 142,6, 137,6, 136,6, 134,5, 130,0, 127,6, 125,3, 120,8, 120,0, 116,7, 116,4, 116,1, 113,1, 35,9, 34,0, 30,7, 23,2
[M+H]+ = 429,1
Следующие примеры предложены в качестве иллюстраций и никаким образом не ограничивают объем данного изобретения.
Следующие примеры подробно иллюстрируют получение некоторых соединений согласно изобретению. Структуры полученных продуктов были подтверждены посредством анализов ЯМР (ядерный магнитный резонанс).
Примеры
Пример 1: соединение (6) в таблице I
Согласно способу (B) 2-винилпиридин (2,32 мл, 22 ммоль, 1,1 экв.) помещали в N,N-диметилформамид (20 мл) с 1-бром-4-нитробензолом (4 г, 20 ммоль, 1 экв.), NaOAc (3,3 г, 40 ммоль, 2 экв.), Pd(OAc)2 (450 мг, 2 ммоль, 10 мол.%), PPh3 (525 мг, 2 ммоль, 10 мол.%). Реакционную смесь нагревали при 135°C и перемешивали в течение 16 часов в атмосфере инертного газа аргона. При охлаждении до комнатной температуры реакционную смесь концентрировали при пониженном давлении, и полученный осадок делили между этилацетатом и водой. После декантации водную фазу дополнительно экстрагировали дихлорметаном. Органические фазы дополнительно промывали водой, сушили над MgSO4, фильтровали, собирали и концентрировали при пониженном давлении с получением (E)-2-[2-(4-нитрофенил)этенил]пиридина (1,9 г, 42%).
1H ЯМР (300 МГц, CDCl3) δ 8,65 (d, J = 4,1 Гц, 1H), 8,24 (d, J = 9,0 Гц, 2H), 7,75 - 7,69 (m, 4H), 7,42 (d, J = 7,8 Гц, 1H), 7,30 (d, J = 16,2 Гц, 1H), 7,27 - 7,20 (m, 1H).
Согласно способу (C) (E)-2-[2-(4-нитрофенил)этенил]пиридин (1,9 г, 8,4 ммоль, 1 экв.) и дигидрат хлорида олова (II) (9,5 г, 42 ммоль, 5 экв.) помещали в EtOH (84 мл). Реакционную смесь нагревали при 60°C и перемешивали в течение 88 часов в атмосфере инертного газа аргона. Затем реакционную смесь концентрировали при пониженном давлении, и полученный остаток разводили этилацетатом. Органическую фазу промывали 1 н. водным раствором NaOH, затем насыщенным водным солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением (E)-4-[2-(пиридин-2-ил)этенил]анилина (1,59 г, 96%).
1H ЯМР (300 МГц, CDCl3) δ 8,56 (d, J = 4,5 Гц, 1H), 7,62 (td, J = 7,9, 1,8 Гц, 1H), 7,53 (d, J = 16,1 Гц, 1H), 7,40 (d, J = 8,5 Гц, 2H), 7,33 (d, J = 7,9 Гц, 1H), 7,09 (dd, J = 7,0, 4,5 Гц, 1H), 6,98 (d, J = 16,1 Гц, 1H), 6,68 (d, J = 8,5 Гц, 2H), 3,80 (s, 2H).
2-Циклопентилэтан-1-амина гидрохлорид (3,0 г, 19,1 ммоль, 1,1 экв.) помещади в 3 н. водный раствор NaOH (13 мл), и к раствору добавляли дихлорметан (3,2 мл). Реакционную смесь охлаждали до 0°C посредством ледяной бани и по каплям добавляли раствор 3-бромбензоилхлорида (2,3 мл, 17,4 ммоль, 1 экв.) в дихлорметане (5,5 мл). Затем реакционную смесь перемешивали при комнатной температуре в течение 18 часов в атмосфере инертного газа аргона. После декантации органическую фазу промывали насыщенным водным солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 3-бром-N-(2-циклопентилэтил)бензамида (5,1 г, 99%).
1H ЯМР (300 МГц, CDCl3) δ 7,89 (t, J = 1,7 Гц, 1H), 7,67 (d, J = 8,0 Гц, 1H), 7,62 (d, J = 8,0 Гц, 1H), 7,30 (t, J = 8,0 Гц, 1H), 6,07 (s, 1H), 3,46 (dt, J = 7,4, 5,9 Гц, 2H), 1,93 - 1,77 (m, 3H), 1,67 - 1,52 (m, 6H), 1,25 - 1,06 (m, 2H).
Согласно способу (A) реакционную смесь 3-бром-N-(2-циклопентенилэтил)бензамида (755 мг, 2,55 ммоль, 1 экв.), (E)-4-[2-(пиридин-2-ил)этенил]анилина (500 мг, 2,55 ммоль, 1 экв.), Pd2(dba)3 (233 мг, 255 мкмоль, 10 мол.%), XPhos (243 мг, 510 мкмоль, 20 мол.%) и K2CO3 (1,41 г, 10,2 ммоль, 4 экв.) в t-BuOH (10,2 мл) нагревали при 90°C и перемешивали в течение 20 часов в атмосфере инертного газа аргона. Затем реакционную смесь концентрировали при пониженном давлении, и полученный остаток разводили дихлорметаном. Органическую фазу промывали насыщенным водным солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением фракции, которая, после измельчения в порошок в простом диэтиловом эфире, дала (E)-N-(2-циклопентилэтил)-3-((4-(2-(пиридин-2-ил)винил)фенил)амино)бензамид (6) (613 мг, 58%).
1H ЯМР (300 МГц, d6-ДМСО) δ 8,59 (s, 1H), 8,54 (d, J = 3,8 Гц, 1H), 8,40 (t, J = 5,5 Гц, 1H), 7,75 (td, J = 7,7, 1,6 Гц, 1H), 7,60 (dd, J = 8,5, 7,7 Гц, 2H), 7,56 (d, J = 8,5 Гц, 2H), 7,48 (d, J = 7,9 Гц, 1H), 7,36 - 7,30 (m, 2H), 7,21 (ddd, J = 12,2, 5,5, 1,6 Гц, 2H), 7,14 (s, 1H), 7,11 (d, J = 8,5 Гц, 2H), 3,26 (dd, J = 13,8, 6,2 Гц, 2H), 1,85 - 1,72 (m, 3H), 1,66 - 1,42 (m, 6H), 1,18 - 0,99 (m, 2H).
13C ЯМР (75 МГц, d6-ДМСО) δ 164,2, 153,6, 147,5, 141,6, 140,9, 134,8, 134,1, 130,1, 127,2, 126,4, 126,0, 122,8, 119,9, 119,8, 117,8, 116,8, 114,5, 114,2, 77,9, 35,5, 33,6, 30,3, 22,8
[M+H]+ = 412,3
Пример 2: соединение (15) в таблице I
Согласно способу (D1) 2-пиридинамин (4,7 г, 50 ммоль, 1,1 экв.) помещали в водный раствор 3 н. NaOH (56 мл), и к раствору добавляли дихлорметан (24 мл). Реакционную смесь охлаждали до 0°C посредством ледяной бани, и по каплям добавляли раствор 4-нитробензоилхлорида (8,4 г, 45 ммоль, 1 экв.) в дихлорметане (40 мл). Затем реакционную смесь перемешивали при комнатной температуре в течение 18 часов в атмосфере инертного газа аргона. Полученный осадок фильтровали и промывали водой и дихлорметаном с получением 4-нитро-N-(пиридин-2-ил)бензамида (6,7 г, 61%).
1H ЯМР (300 МГц, d6-ДМСО) δ 10,91 (s, 1H), 8,80 (s, 1H), 8,52 (d, J = 5,5 Гц, 2H), 8,47 (d, J = 7,9 Гц, 1H), 8,41 (d, J = 7,9 Гц, 1H), 7,86 (t, J = 7,9 Гц, 1H), 7,79 (d, J = 5,5 Гц, 2H).
Согласно способу (C) 4-нитро-N-(пиридин-2-ил)бензамид (1,5 г, 6,2 ммоль, 1 экв.) и дигидрат хлорида олова (II) (7,0 г, 30,8 ммоль, 5 экв.) помещали в EtOH (62 мл). Реакционную смесь нагревали при 60°C и перемешивали в течение 16 часов в атмосфере инертного газа аргона. Затем реакционную смесь концентрировали при пониженном давлении, и полученный остаток разводили этилацетатом. Органическую фазу промывали 1 н. водным раствором NaOH, затем водой, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 4-амино-N-(пиридин-2-ил)бензамида (273 мг, 21%).
1H ЯМР (300 МГц, d6-ДМСО) δ 10,44 (s, 1H), 8,44 (d, J = 6,3 Гц, 2H), 7,77 (d, J = 6,3 Гц, 2H), 7,18 (t, J = 7,9 Гц, 1H), 7,12 - 7,03 (m, 2H), 6,78 (d, J = 7,9 Гц, 1H), 5,38 (s, 2H).
2-Циклопентилэтан-1-амина гидрохлорид (1,0 г, 7 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (4,6 мл), и к раствору добавляли дихлорметан (1,1 мл). Реакционную смесь охлаждали до 0°C посредством ледяной бани, и по каплям добавляли раствор 3-бромбензоилхлорида (0,8 мл, 6 ммоль, 1 экв.) в дихлорметане (1,9 мл). Затем реакционную смесь перемешивали при комнатной температуре в течение 18 часов в атмосфере инертного газа аргона. После декантации органическую фазу промывали насыщенным водным солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 3-бром-N-(2-циклопентилэтил)бензамида (1,77 г, 98%).
1H ЯМР (300 МГц, CDCl3) δ 7,89 (t, J = 1,7 Гц, 1H), 7,67 (d, J = 7,9 Гц, 1H), 7,62 (d, J = 7,9 Гц, 1H), 7,30 (t, J = 7,9 Гц, 1H), 6,07 (s, 1H), 3,46 (dt, J = 7,4, 5,9 Гц, 2H), 1,93 - 1,77 (m, 3H), 1,67 - 1,52 (m, 6H), 1,25 - 1,06 (m, 2H).
Согласно способу (A) реакционную смесь 3-бром-N-(2-циклопентилэтил)бензамида (296 мг, 1,0 ммоль, 1 экв.), 4-амино-N-(пиридин-2-ил)бензамида (213 мг, 1,0 ммоль, 1 экв.), Pd2(dba)3 (92 мг, 0,1 ммоль, 10 мол.%), XPhos (95 мг, 0,2 ммоль, 20 мол.%) и K2CO3 (553 мг, 4,0 ммоль, 4 экв.) в t-BuOH (4 мл) нагревали в микроволновом реакторе при 120°C в течение 60 минут. Затем реакционную смесь концентрировали при пониженном давлении, и полученный осадок разводили этилацетатом. Органическую фазу промывали водой, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный осадок очищали посредством колоночной хроматографии на силикагеле с получением N-(2-циклопентилэтил)-3-((4-(пиридин-2-илкарбамоил)фенил)амино)бензамида (15) (30 мг, 7%).
1H ЯМР (300 МГц, d6-ДМСО) δ 10,46 (s, 1H), 8,83 (s, 1H), 8,44 (t, J = 5,5 Гц, 1H), 8,37 (d, J = 3,8 Гц, 1H), 8,19 (d, J = 8,4 Гц, 1H), 7,98 (d, J = 8,7 Гц, 2H), 7,87 - 7,77 (m, 1H), 7,65 (s, 1H), 7,45 - 7,36 (m, 2H), 7,30 (dt, J = 6,5, 2,2 Гц, 1H), 7,15 (d, J = 5,5 Гц, 1H), 7,11 (d, J = 8,7 Гц, 2H), 3,26 (dd, J = 13,7, 6,4 Гц, 2H), 1,87 - 1,72 (m, 3H), 1,67 - 1,40 (m, 6H), 1,07 (m, 2H).
13C ЯМР (75 МГц, d6-ДМСО) δ 164,2, 163,5, 150,8, 146,1, 145,3, 140,1, 136,2, 134,3, 128,1, 127,5, 122,3, 119,3, 118,1, 117,6, 117,5, 115,7, 112,8, 112,7, 35,7, 33,7, 30,5, 23,0
[M+H]+ = 429,1
Пример 3: соединение (16) в таблице I
Согласно способу (D1) 2-пиридинамин (5,0 г, 53,1 ммоль, 1 экв.) помещали в 3 н. водный раствор NaOH (65,5 мл), и к раствору добавляли дихлорметан (5 мл). Реакционную смесь охлаждали до 0°C посредством ледяной бани, и по каплям добавляли раствор 4-нитробензоилхлорида (9,8 г, 53,1 ммоль, 1 экв.) в дихлорметане (70 мл). Затем реакционную смесь перемешивали при комнатной температуре в течение 18 часов в атмосфере инертного газа аргона. Полученный осадок фильтровали и промывали водой и дихлорметаном с получением 4-нитро-N-(пиридин-2-ил)бензамида (5,8 г, 45%).
1H ЯМР (300 МГц, d6-ДМСО) δ 11,16 (s, 1H), 8,40 (dd, J = 4,8, 1,6 Гц, 1H), 8,32 (d, J = 8,9 Гц, 2H), 8,21 (d, J = 9,0 Гц, 2H), 8,17 (d, J = 8,4 Гц, 1H), 7,90 - 7,82 (m, 1H), 7,23 - 7,15 (m, 1H).
Согласно способу (C) 4-нитро-N-(пиридин-2-ил)бензамид (5,8 г, 23,8 ммоль, 1 экв.) и дигидрат хлорида олова (II) (27 г, 119 ммоль, 5 экв.) помещали в EtOH (240 мл). Реакционную смесь нагревали при 60°C и перемешивали в течение 16 часов в атмосфере инертного газа аргона. Затем реакционную смесь концентрировали при пониженном давлении и полученный осадок разводили дихлорметаном. Органическую фазу промывали 1 н. водным раствором NaOH, затем насыщенным водным солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 4-амино-N-(пиридин-2-ил)бензамида (955 мг, 19%).
1H ЯМР (300 МГц, d6-ДМСО) δ 10,17 (s, 1H), 8,34 (d, J = 3,8 Гц, 1H), 8,15 (d, J = 8,4 Гц, 1H), 7,80 - 7,75 (m, 3H), 7,09 (dd, J = 6,4, 5,0 Гц, 1H), 6,57 (d, J = 8,6 Гц, 2H), 5,82 (br s, 2H).
2-Циклогексилэтан-1-амин (1,1 мл, 7,9 ммоль, 1,1 экв.) помещали в 3 н. водный раствор NaOH (5,3 мл), и к раствору добавляли дихлорметан (1 мл). Реакционную смесь охлаждали до 0°C посредством водяной бани, и по каплям добавляли раствор 3-бромбензоилхлорида (945 мкл, 7,1 ммоль, 1 экв.) в дихлорметане (2,5 мл). Затем реакционную смесь перемешивали при комнатной температуре в течение 18 часов в атмосфере инертного газа аргона. После декантации органическую фазу промывали насыщенным водным солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 3-бром-N-(2-циклогексилэтил)бензамида (2,0 г, 90%).
1H ЯМР (300 МГц, CDCl3) δ 7,89 (t, J = 1,7 Гц, 1H), 7,67 (d, J = 7,8 Гц, 1H), 7,62 (d, J = 7,8 Гц, 1H), 7,31 (t, J = 7,9 Гц, 1H), 6,00 (bs, 1H), 3,47 (dt, J = 7,5, 5,8 Гц, 2H), 1,81 - 1,62 (m, 4H), 1,51 (dd, J = 14,6, 7,1 Гц, 2H), 1,31 - 1,12 (m, 5H), 1,04 - 0,85 (m, 2H).
Согласно способу (A) реакционную смесь 3-бром-N-(2-циклогексилэтил)бензамида (310 мг, 1,0 ммоль, 1 экв.), 4-амино-N-(пиридин-2-ил)бензамида (213 мг, 1,0 ммоль, 1 экв.), Pd2(dba)3 (92 мг, 0,1 ммоль, 10 мол.%), XPhos (95 мг, 0,2 ммоль, 20 мол.%) и K2CO3 (553 мг, 4,0 ммоль, 4 экв.) в t-BuOH (4 мл) нагревали в микроволновом реакторе при 120°C в течение 60 минут. Затем реакционную смесь концентрировали при пониженном давлении, и полученный осадок разводили этилацетатом. Органическую фазу промывали насыщенным водным солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный осадок очищали посредством колоночной хроматографии на силикагеле и затем перетирали с простым диэтиловым эфиром с получением N-(2-циклогексилэтил)-3-((4-(пиридин-2-илкарбомоил)фенил)амино)бензамида (16) (78 мг, 18%).
1H ЯМР (300 МГц, d6-ДМСО) δ 10,46 (s, 1H), 8,83 (s, 1H), 8,42-8,37 (m, 2H), 8,19 (d, J = 8,4 Гц, 1H), 7,98 (d, J = 8,7 Гц, 2H), 7,82 (t, J = 8,8 Гц, 1H), 7,64 (s, 1H), 7,42 - 7,35 (m, 2H), 7,32-7..29 (m 1H), 7,17 - 7,13 (m, 1H), 7,10 (d, J = 8,6 Гц, 2H), 3,28 (dd, J = 13,3, 6,8 Гц, 2H), 1,78 - 1,57 (m, 5H), 1,43 (dd, J = 14,2, 6,9 Гц, 2H), 1,34 - 1,12 (m, 4H), 0,99 - 0,82 (m, 2H).
13C ЯМР (75 МГц, d6-ДМСО) δ 166,4, 165,7, 152,9, 148,3, 147,5, 142,3, 138,4, 136,5, 130,2, 129,7, 124,5, 121,5, 120,3, 119,8, 117,9, 115,0, 114,9, 37,5, 37,1, 35,3, 33,2, 26,5, 26,2
[M+H]+ = 429,1
Пример 4: соединение (115) в таблице I
Согласно способу (J) раствор метил-4-амино-3-бромбензоата (3,00 г, 12,8 ммоль, 1 экв.) и циклопропилтрифторбората калия (2,84 г, 19,2 ммоль, 1,5 экв.) в толуоле (52,5 мл) и воде (13,5 мл) дегазировали посредством аргона в течение 5 минут, затем добавляли фосфат трикалия (6,88 г, 31,9 ммоль, 2,5 экв.), RuPhos (239 мг, 511 мкмоль, 0,04 экв.) и ацетат палладия (II) (57,9 мг, 256 мкмоль, 0,02 экв.). Реакционную смесь нагревали при 110°C и перемешивали в течение 2 ч 30 мин в атмосфере инертного газа. При охлаждении до комнатной температуры ее фильтровали через слой целита и промывали этот слой EtOAc. Затем к фильтрату добавляли насыщенный водный солевой раствор, и смесь экстрагировали EtOAc. Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный осадок очищали посредством колоночной хроматографии на силикагеле с получением метил-4-амино-3-циклопропилбензоата (2,02 г, 81%).
1H ЯМР (400 МГц, d6-ДМСО) δ 7,53 (dd, J = 8,4, 2,0 Гц, 1H), 7,42 (d, J = 1,9 Гц, 1H), 6,63 (d, J = 8,4 Гц, 1H), 5,87 (s, 2H), 3,73 (s, 3H), 1,65 (tt, J = 8,3, 5,4 Гц, 1H), 0,95 - 0,82 (m, 2H), 0,54 - 0,40 (m, 2H).
3-Бромфенол (701 мг, 3,97 ммоль, 1,2 экв.) помещали в N,N-диметилформамид (4 мл) с Cs2CO3 (1,30 г, 3,97 ммоль, 1,2 экв.). После добавления (3-бромпропил)циклогексана (715 мг, 3,31 ммоль, 1,0 экв.) реакционную смесь перемешивали в течение 16 часов в атмосфере инертного газа аргона. Реакционную смесь разделяли между этилацетатом и насыщенным водным раствором NaHCO3. После экстракции этилацетатом объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением 1-бром-3-(3-циклогексилпропокси)бензола (882 мг, 90%).
1H ЯМР (500 МГц, d6-ДМСО) δ 7,22 (t, J = 8,1 Гц, 1H), 7,14 - 7,08 (m, 2H), 6,93 (dd, J = 8,3, 2,3 Гц, 1H), 3,95 (t, J = 6,5 Гц, 2H), 1,68 (tt, J = 15,1, 9,2 Гц, 7H), 1,32 - 1,06 (m, 6H), 0,92 - 0,82 (m, 2H).
Согласно способу (A) реакционную смесь 1-бром-3-(3-циклогексилпропокси)бензола (547 мг, 1,84 ммоль, 1,1 экв.), метил-4-амино-3-циклопропил-бензоата (320 мг, 1,67 ммоль, 1 экв.), BrettPhos Pd G3 (31,9 мг, 33,5 мкмоль, 0,02 экв.) и Cs2CO3 (818 мг, 2,51 ммоль, 1,5 экв.) в безводном ДМФА (диметилформамид) (8 мл) дегазировали посредством аргона и нагревали при 80°C в течение 75 минут в атмосфере инертного газа. Затем реакционную смесь охлаждали до комнатной температуры, фильтровали через слой целита и промывали этот слой EtOAc. Затем к фильтрату добавляли солевой раствор, и смесь экстрагировали EtOAc. Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением метил-4-{[3-(3-циклогексилпропокси)фенил]амино}-3-циклопропилбензоата (1,35 г, 80%).
1H ЯМР (400 МГц, d6-ДМСО) δ 7,82 (s, 1H), 7,66 (dd, J = 8,5, 2,0 Гц, 1H), 7,54 (d, J = 2,0 Гц, 1H), 7,24 - 7,14 (m, 2H), 6,76 (d, J = 7,9 Гц, 1H), 6,73 (t, J = 2,1 Гц, 1H), 6,56 (dd, J = 8,1, 2,2 Гц, 1H), 3,92 (t, J = 6,5 Гц, 2H), 3,78 (s, 3H), 1,94 (ddd, J = 13,8, 8,3, 5,4 Гц, 1H), 1,75 - 1,58 (m, 7H), 1,35 - 1,08 (m, 6H), 1,04 - 0,94 (m, 2H), 0,88 (q, J = 10,0, 9,3 Гц, 2H), 0,65 - 0,56 (m, 2H).
Согласно способу (E) метил-4-{[3-(3-циклогексилпропокси)фенил]амино}-3-циклопропилбензоат (575 мг, 1,34 ммоль, 1 экв.) помещали в метанол (10 мл) и добавляли 2 M водный раствор NaOH (4,7 мл, 9,4 ммоль, 7 экв.). Реакционную смесь нагревали при 80°C и перемешивали в течение 3 часов. Затем ее концентрировали при пониженном давлении, и после добавления 2 M водного раствора HCl (7,0 мл, 14 ммоль, 10,5 экв.) экстрагировали дихлорметаном. Объединенные органические фазы сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением 4-{[3-(3-циклогексилпропокси)фенил]амино}-3-циклопропилбензойной кислоты (540 мг, 97%).
1H ЯМР (400 МГц, d6-ДМСО) δ 12,37 (s, 1H), 7,76 (s, 1H), 7,64 (dd, J = 8,5, 2,0 Гц, 1H), 7,52 (d, J = 1,9 Гц, 1H), 7,18 (t, J = 8,6 Гц, 2H), 6,74 (d, J = 7,9 Гц, 1H), 6,71 (d, J = 2,1 Гц, 1H), 6,53 (dd, J = 8,1, 2,1 Гц, 1H), 3,91 (t, J = 6,5 Гц, 2H), 1,94 (ddd, J = 13,6, 8,4, 5,4 Гц, 1H), 1,75 - 1,58 (m, 7H), 1,35 - 1,09 (m, 6H), 0,98 (dd, J = 4,0, 2,0 Гц, 2H), 0,88 (q, J = 10,1, 9,3 Гц, 2H), 0,65 - 0,56 (m, 2H).
Согласно способу (F) 4-{[3-(3-циклогексилпропокси)фенил]амино}-3-циклопропилбензойную кислоту (50,0 мг, 127 мкмоль, 1 экв.) и анилин (12,2 мкл, 133 мкмоль, 1,05 экв.) помещали в безводный N,N-диметилформамид (650 мкл). Добавляли HATU (48,3 мг, 127 мкмоль, 1 экв.) и DIPEA (44,4 мкл, 254 мкмоль, 2 экв.) и перемешивали полученную реакционную смесь при комнатной температуре в течение 16 часов. Реакцию останавливали 1 M водным раствором соляной кислоты и экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением 4-{[3-(3-циклогексилпропокси)фенил]амино}-3-циклопропил-N-фенилбензамида (115) (42,0 мг, 67%).
1H ЯМР (400 МГц, d6-ДМСО) δ 9,96 (s, 1H), 7,78 - 7,67 (m, 4H), 7,57 (d, J = 2,0 Гц, 1H), 7,33 (t, J = 7,9 Гц, 2H), 7,23 (d, J = 8,5 Гц, 1H), 7,16 (t, J = 8,1 Гц, 1H), 7,07 (t, J = 7,4 Гц, 1H), 6,74 - 6,68 (m, 1H), 6,67 (t, J = 2,1 Гц, 1H), 6,48 (dd, J = 8,1, 2,1 Гц, 1H), 3,91 (t, J = 6,5 Гц, 2H), 2,04 - 1,92 (m, 1H), 1,75 - 1,57 (m, 7H), 1,35 - 1,09 (m, 6H), 1,04 - 0,95 (m, 2H), 0,88 (q, J = 10,1, 9,5 Гц, 2H), 0,76 - 0,67 (m, 2H).
13C ЯМР (151 МГц, d6-ДМСО) δ 165,6, 160,0, 146,2, 144,9, 139,9, 131,8, 130,2, 128,9, 126,8, 126,7, 126,4, 123,7, 120,9, 116,2, 111,4, 107,5, 105,3, 68,0, 37,2, 33,7, 33,3, 26,6, 26,6, 26,3, 11,6, 7,8
[M+H]+ = 469,2
Пример 5: соединение (35) в таблице I
Согласно способу (F) бензол-1,4-диамин (4,0 г, 37 ммоль, 3,0 экв.) помещали в тетрагидрофуран (37 мл) и N,N-диметилформамид (12 мл). К раствору добавляли 2,2 M водный раствор K2CO3 (6,8 мл, 13,6 ммоль, 1,1 экв.), и затем по каплям добавляли Boc2O (2,6 мл, 12,3 ммоль, 1,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 64 часов. Затем реакционную смесь концентрировали при пониженном давлении, и полученный осадок разводили этилацетатом. Органическую фазу промывали водой, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный осадок очищали посредством колоночной хроматографии на силикагеле с получением трет-бутил-(4-аминофенил)карбамата (1,6 г, 62%).
1H ЯМР (300 МГц, CDCl3) δ 7,13 (d, J = 8,5 Гц, 2H), 6,63 (d, J = 8,5 Гц, 2H), 6,32 (s, 1H), 3,54 (s, 2H), 1,50 (s, 9H).
Согласно способу (G) реакционную смесь трет-бутил-(4-аминофенил)карбамата (1,6 г, 7,7 ммоль, 1,0 экв.), 2-пиридинкарбоновой кислоты (1,0 г, 8,5 ммоль, 1,1 экв.), EDCI.HCl (1,6 г, 8,5 ммоль, 1,1 экв.), N,N-диизопропилэтиламина (3,8 мл, 23 ммоль, 3,0 экв.) и HOBt (1,1 г, 8,5 ммоль, 1,1 экв.) в безводном дихлорметане (15 мл) перемешивали при комнатной температуре в течение 24 часов в атмосфере инертного газа аргона. Органическую фазу промывали водой, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали посредством колоночной хроматографии на силикагеле с получением трет-бутил-N-[4-(пиридин-2-амидо)фенил]карбамата (983 мг, 41%).
1H ЯМР (300 МГц, CDCl3) δ 9,98 (s, 1H), 8,61 (d, J = 4,5 Гц, 1H), 8,29 (d, J = 7,7 Гц, 1H), 7,91 (td, J = 7,7, 1,5 Гц, 1H), 7,72 (d, J = 8,8 Гц, 2H), 7,48 (ddd, J = 7,7, 4,5, 1,5 Гц, 1H), 7,40 (d, J = 8,8 Гц, 2H), 6,61 (s, 1H), 1,52 (s, 9H).
Согласно способу (H) трет-бутил-N-[4-(пиридин-2-амидо)фенил]карбамат (983 мг, 3,1 ммоль, 1 экв.) помещали в дихлорметан (6,3 мл), и к раствору добавляли трифторуксусную кислоту (5,1 мл, 68,2 ммоль, 22 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и затем охлаждали до 0°C посредством ледяной бани. Добавляли воду и затем K2CO3 до достижения pH выше 7. Водный раствор экстрагировали дихлорметаном. Органические фазы собирали, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением N-(4-аминофенил)пиридин-2-карбоксамида (659 мг, 99%).
1H ЯМР (300 МГц, CDCl3) δ 9,85 (s, 1H), 8,60 (d, J = 4,5 Гц, 1H), 8,29 (d, J = 7,7 Гц, 1H), 7,89 (td, J = 7,7, 1,5 Гц, 1H), 7,57 (d, J = 8,7 Гц, 2H), 7,46 (ddd, J = 7,7, 4,5, 1,5 Гц, 1H), 6,72 (d, J = 8,7 Гц, 2H), 3,63 (s, 2H).
2-Циклопентилэтан-1-амина гидрохлорид (1,0 г, 7 ммоль, 1,1 экв.) помещали в водный 3 н. раствор NaOH (4,6 мл), и к раствору добавляли дихлорметан (1,1 мл). Реакционную смесь охлаждали до 0°C посредством ледяной бани, и по каплям добавляли раствор 3-бромбензоилхлорида (0,8 мл, 6 ммоль, 1 экв.) в дихлорметане (1,9 мл). Затем реакционную смесь перемешивали при комнатной температуре на протяжении 18 часов в атмосфере инертного газа аргона. После декантации органическую фазу промывали насыщенным водным солевым раствором, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением 3-бром-N-(2-циклопентилэтил)бензамида (1,77 г, 98%).
1H ЯМР (300 МГц, CDCl3) δ 7,89 (t, J = 1,7 Гц, 1H), 7,67 (d, J = 7,9 Гц, 1H), 7,62 (d, J = 7,9 Гц, 1H), 7,30 (t, J = 7,9 Гц, 1H), 6,07 (s, 1H), 3,46 (dt, J = 7,4, 5,9 Гц, 2H), 1,93 - 1,77 (m, 3H), 1,67 - 1,52 (m, 6H), 1,25 - 1,06 (m, 2H).
Согласно способу (A) реакционную смесь 3-бром-N-(2-циклопентилэтил)бензамида (296 мг, 1,0 ммоль, 1 экв.), N-(4-аминофенил)пиридин-2-карбоксамида (213 мг, 1,0 ммоль, 1 экв.), Pd2(dba)3 (92 мг, 0,1 ммоль, 10 мол.%), XPhos (95 мг, 0,2 ммоль, 20 мол.%) и K2CO3 (553 мг, 4,0 ммоль, 4 экв.) в t-BuOH (4 мл) нагревали в микроволновом реакторе при 120°C в течение 60 минут. Затем реакционную смесь концентрировали при пониженном давлении, и полученный осадок разводили этилацетатом. Органическую фазу промывали водой, сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Полученный осадок очищали посредством колоночной хроматографии на силикагеле с получением, после перекристаллизации, N-(4-((3-((2-циклопентилэтил)карбамоил)фенил)амино)фенил)пиколинамида (35) (89 мг, 21%).
1H ЯМР (300 МГц, d6-ДМСО) δ 10,54 (s, 1H), 8,74 (d, J = 4,4 Гц, 1H), 8,36 (t, J = 5,4 Гц, 1H), 8,29 (s, 1H), 8,16 (d, J = 7,7 Гц, 1H), 8,07 (td, J = 7,7, 1,5 Гц, 1H), 7,82 (d, J = 8,8 Гц, 2H), 7,71 - 7,62 (m, 1H), 7,51 (s, 1H), 7,34 - 7,20 (m, 2H), 7,16 - 7,10 (m, 3H), 3,25 (dd, J = 13,4, 6,5 Гц, 2H), 1,82 - 1,72 (m, 3H), 1,68 - 1,45 (m, 6H), 1,12 - 1,08 (m, 2H).
13C ЯМР (75 MГц, d6-ДМСО) δ 164,9, 160,5, 148,7, 146,9, 142,6, 137,6, 136,6, 134,5, 130,0, 127,6, 125,3, 120,8, 120,0, 116,7, 116,4, 116,1, 113,1, 35,9, 34,0, 30,7, 23,2
[M+H]+ = 429,1
Фармакологические данные
Пример 6: вирус Чикунгунья
Соединения согласно изобретению были предметом фармакологических тестов, которые продемонстрировали их применимость в качестве активных веществ в терапии и, в частности, для предупреждения, ингибирования или лечения инфекции вирусом Чикунгунья.
Материалы и методы
Ингибирование продукции вируса Чикунгунья (CHIKV) в инфицированной клеточной линии HEK293T.
Способность соединений ингибировать репликацию вируса оценивали посредством эксперимента, в котором инфицированные клетки обрабатывали соединениями формулы (I) в концентрации 1 мкМ. В качестве положительного контроля для ингибирования Чикунгуньи использовали рибавирин. Параллельно оценивали токсичность соединений.
Амплификация клеток
Клетки эмбриональной почки человека 293T (HEK293T, CRL-11268) поддерживали в среде Игла, модифицированной по Дульбекко (DMEM, 31966-021, Thermo Fisher Scientific) с добавлением 10% фетальной телячьей сыворотки (FBS), пенициллина и стрептомицина. После удаления среды клетки промывали солевым раствором, не содержащим Ca2+ и Mg2+, для удаления всех следов сыворотки. После удаления отсасыванием промывочного раствора клетки диссоциировали 0,25% раствором трипсина-ЭДТА и инкубировали в течение по меньшей мере 30 с в инкубаторе при 37°C. Концентрацию клеточной суспензии определяли посредством автоматического счетчика клеток (EVE, NanoEntek) и, при необходимости, доводили до 0,33 × 106 клеток/мл средой DMEM с добавлением 10% FBS.
Подготовка соединений
100 мкл клеточной суспензии помещали в ViewPlate-96 Black (6005182, PerkinElmer) и в прозрачный 96-луночный планшет для клеточной культуры (655180, Greiner bio-one). После инкубации в течение 24 ч при 37°C при 5% CO2 добавляли соединения в нужной концентрации.
Скрининг при 1 мкМ
Из исходного раствора готовили промежуточное с ДМСО (D8418, Sigma) в концентрации 2 мМ в 96-луночном микропланшете с V-образным дном:
Смесь 1 мкл 50 мМ исходной библиотеки в 25 мкл ДМСО.
Смесь 2 мкл 25 мМ исходной библиотеки в 25 мкл ДМСО.
Определение значений IC50
Из исходного раствора готовили промежуточное разведение с ДМСО (D8418, Sigma) в концентрации 25 мМ в 96-луночном микропланшете с V-образным дном:
Смесь 2 мкл 50 мМ исходной библиотеки в 2 мкл ДМСО.
Выполняли последовательное разведение в 2 мкл ДМСО 13 раз до достижения 0,0015 мМ. Продолжали, как указано в таблице III:

Как для скрининга, так и для определения IC50 (концентрация полумаксимального ингибирования), 1 мкл каждого раствора добавляли в лунки плашета Masterblock, 96-луночный с объемом лунки 1 мл (Greiner bio-one, 780261), содержащие 1 мл среды DMEM. В качестве положительного контроля 5 мкл 80 мМ раствора рибавирина (R9644, Sigma) добавляли к 1 мл DMEM. С другой стороны, в качестве отрицательного контроля использовали ДМСО.
Инфицирование
Клетки инфицировали 30 мкл штамма CHIKV вспышки на острове Реюньон (LR2006-OPY1) с GFP-модификацией на 5'c (CHIK 5'cLR) (Tsetsarkin K, Higgs S, McGee CE, De Lamballerie X, Charrel RN, Vanlandingham DL. Infectious Clones of Chikungunya Virus (La Réunion Isolate - Ref-SKU: 001N-EVA249 (PMID: 17187566), доступна по адресу: https://www.european-virus-archive.com/nucleic-acid/chikv-lr-5gfp-infectious-clone) for Vector Competence Studies. Vector Borne Zoonotic Dis. 2006; 6(4)). Данный модифицированный вирус использовали для инфицирования клеток при MOI (множественность инфекции) 0,1. Штамм LR2006-OPY1 CHIKV (CHIKV-LR) был получен от Международного справочного центра по арбовирусам при отделении медицины Техасского университета, Галвестон, Техас. Данный штамм исходно был выделен из сыворотки французского пациента с лихорадкой, вернувшегося с острова Реюньон.
Лизис клеток
Среду удаляли спустя 22 ч при 37°C при 5% CO2, и клетки промывали, как описано выше. 60 мкл буфера RIPA (50 мМ Tris-HCl pH 8, 100 мМ NaCl, 1 мМ MgCl2, 1% Triton X-100) добавляли к клеткам и инкубировали в течение по меньшей мере 20 мин перед считыванием сигнала флуоресценции. Для нормирования сигнала флуоресценции по количеству белка использовали реагент для анализа белков при 660 нм Pierce 660nm Protein Assay Reagent (22660, Thermo scientific).
Набор CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) (G3581, Promega) использовали для проверки токсичности данных соединений. Авторы изобретения добавляли 20 мкл раствора MTS и считывали значение поглощения при 492 нм, спустя один час.
Результаты
- Был проведен первый цикл экспериментов, где результаты выражены в виде процента ингибирования, который рассчитывали следующим образом, посредством следующих стадий:
1. Интенсивность флуоресценции (FI) / Поглощение при 660 нм (A660) = A
Данное отношение позволяет учитывать инфицирование (вирус с GFP) относительно количества белка.
2. A'c = A - фоновый шум неинфицированного планшета,
3. B = Интенсивность флуоресценции (FI) / Поглощение при 660 нм (A660) инфицированных, но не обработанных планшетов,
4. C = A'c / B, которое затем переводят в процент инфицирования после обработки, по сравнению с необработанным образцом, и впоследствии в процент инфицирования. Например, величина 100 в приведенной ниже таблице IV означает, что после обработки сигнал, связанный с флуоресценцией GFP, устраняется, что коррелирует с отсутствием инфекции.
5. C'c = 100 - C
Данное значение соответствует проценту ингибирования.
Следующая таблица IV содержит указанное значение C'c для некоторых соединений, рассчитанное, как указано выше, со средним значением для 2 экспериментов и соответствующим среднеквадратическим отклонением.
Некоторые значения исходно были выше 100. В данных случаях значение было снижено до 100. Это означает, что некоторые молекулы также влияют на жизнеспособность клеток. Иными словами, значение A может быть ниже, чем фоновый шум.
Кроме того, для каждого измерения проводили тест с рибавирином в качестве контроля. Проверяли значение процента ингибирования при 100%.
Таблица IV
- Проводили второй цикл экспериментов, результаты были представлены в виде значений IC50.
Значения IC50 находятся в интервале от 0,1 нМ до 1 мкМ, в частности, от 0,5 до 500 нМ, и даже более конкретно от 1 до 400 нМ, например, от 1 до 200 нМ, и в частности для соединений 6, 14, 15, 16, 32 и 35 значения IC50 находятся в интервале от 200 до 500 нМ. Например, соединения 12 и 13 имеют значение IC50, находящееся в интервале от 1 до 200 нМ.
Вывод
На основе полученных выше результатов можно сделать вывод о том, что соединения формулы (I) представляют собой подходящие химические соединения для лечения и/или предупреждения РНК-вирусных инфекций, вызванных РНК-вирусами группы IV, в частности, альфавирусных инфекций и, в частности, вирусных инфекций Чикунгунья.
Пример 7: вирус RSV
Соединения согласно изобретению были предметом фармакологических тестов, которые продемонстрировали их применимость в качестве активных веществ в терапии и, в частности, для предупреждения, ингибирования или лечения вирусной инфекции RSV.
Материалы и методы
Протокол скрининга противовирусных соединений в отношении ингибирования RSV и цитотоксичности с использованием анализа Viral ToxGlo
Клетки HEp-2 поддерживали в минимальной необходимой среде Игла (EMEM) с BSS (сбалансированный солевой раствор) Эрла, модифицированной таким образом, чтобы она содержала 2 мМ L-глутамина, 10% фетальной телячьей сыворотки, 100 ЕД/мл пенициллина и 100 мкг/мл стрептомицина. В целях скринингового анализа их выращивали до 90% конфлюэнтности, трипсинизировали и выделяли. Трипсин нейтрализовали средой для культивирования клеток и центрифугировали клетки при 150 x g на протяжении 5 минут перед отбрасыванием супернатанта и ресуспендированием клеточного осадка в среде для анализа (EMEM с BSS Эрла, модифицированная таким образом, чтобы она содержала 2 мМ L-глутамина, 2% фетальной телячьей сыворотки и 100 ЕД/мл пенициллина и 100 мкг/мл стрептомицина). Клетки высевали в белые планшеты для клеточной культуры с прозрачным дном при плотности 1,5x104 клеток/лунка в 50 мкл и 4x103 клеток/лунка в 25 мкл в случае 96-луночных планшетов и 384-луночных планшетов, соответственно. В случае колоночного анализа среды/фонового контроля добавляли только среду для анализа. Планшеты для клеток помещали во влажную камеру и инкубировали в течение ночи при 37°C/5% CO2. После инкубации в течение ночи клетки проверяли в отношении конфлюэнтности и нормальности на вид.
Тестируемые соединения были приготовлены при 10-кратной тестируемой концентрации в максимальной концентрации ДМСО 10% (конечная максимальная концентрация для анализа ДМСО 1%) и добавляли в планшеты для клеток в объемах 10 мкл в случае 96-луночных планшетов и 5 мкл - в случае 384-луночных планшетов. В случае лунок с клеточным контролем и вирусным контролем добавляли только растворитель тестируемого соединения. Вирус или среду для анализа в случае лунок для анализа на цитотоксичность и контрольных лунок со средой/клетками добавляли сразу после тестируемых соединений при MOI 0,5, 40 или 20 мкл в случае 96- и 384-луночных планшетов, соответственно. Вирусную суспензию получали посредством размораживания замороженных исходных материалов RSV A2 и разведения до требуемой концентрации бляшкообразующих единиц в среде анализа на льду.
Планшеты для клеток дополнительно инкубировали во влажной камере в течение 72 ч после введения при 37°C/5%CO2. После периода инкубирования клетки наблюдали под микроскопом для проверки на наличие характерного цитопатического эффекта в лунках с вирусным контролем и нормальных клеток в лунках с клеточным контролем. После доведения планшетов до комнатной температуры в каждую лунку 384/96-луночных планшетов для клеток добавляли 20/40 мкл Viral ToxGlo (Promega). Планшеты инкубировали при комнатной температуре, защищая от света, на качалке для планшетов в течение 20 минут перед измерением люминесценции на спектрофотометре (Biotek Synergy HTX).
Ингибирование RSV рассчитывали в виде процента ингибирования цитопатического эффекта относительно вирусного контроля, а цитотоксичность - в виде процента выживаемости клеток относительно лунок с клеточным контролем. Это позволило рассчитать значения EC50 (полумаксимальная эффективная концентрация) для каждого тестируемого соединения, где идентифицировали ингибирование вируса или цитотоксический дозозависимый эффект. Были обнаружены значения EC50, находящиеся в интервале от 0,001 мкМ до 2,5 мкМ, и, в частности, для соединения 12, 13, 16 и 115.
Таблица V
Вывод
На основе полученных выше результатов можно сделать вывод о том, что соединения формулы (I) представляют собой подходящие химические соединения для лечения и/или предупреждения РНК-вирусных инфекций, вызванных РНК-вирусами группы V, в частности, пневмовирусных инфекций и, в частности, вирусных инфекций RSV.
Пример 8: вирус Денге 2
Соединения согласно изобретению были предметом фармакологических тестов, которые продемонстрировали их применимость в качестве активных веществ в терапии и, в частности, для предупреждения, ингибирования или лечения вирусной инфекции Денге 2.
Материалы и методы
Протокол скрининга противовирусных соединений в отношении ингибирования DENV-2 и цитотоксичности с использованием анализа Viral ToxGlo
Клетки A549 поддерживали в среде Игла, модифицированной по Дульбекко (DMEM), с добавлением 10% фетальной телячьей сыворотки, 100 ЕД/мл пенициллина и 100 мкг/мл стрептомицина. В целях скринингового анализа их выращивали до 90% конфлюэнтности, трипсинизировали и выделяли. Трипсин нейтрализовали средой для культивирования клеток и центрифугировали клетки при 150 x g на протяжении 5 минут перед отбрасыванием супернатанта и ресуспендированием клеточного осадка в среде для анализа (DMEM с добавлением 2% фетальной телячьей сыворотки и 100 ЕД/мл пенициллина и 100 мкг/мл стрептомицина). Клетки высевали в белые 96-луночные планшеты для клеточной культуры с прозрачным дном при плотности 1,0x104 клеток/лунка в 50 мкл. В случае колоночного анализа среды/фонового контроля добавляли только среду для анализа. Планшеты для клеток помещали во влажную камеру и инкубировали в течение ночи при 37°C/5% CO2. После инкубации в течение ночи клетки проверяли в отношении конфлюэнтности и нормальности на вид.
Тестируемые соединения получали в конечной концентрации 10 мкМ при максимальной концентрации ДМСО 1% (конечная максимальная концентрация анализа ДМСО 0,1%) и добавляли в планшеты для клеток в объемах 10 мкл. В случае лунок с клеточным контролем и вирусным контролем добавляли только растворитель тестируемого соединения. В качестве положительного контроля ингибирования в 3 лунки добавляли 7-деаза-2'c-C-метиладенозин в концентрации 100 мкМ. Вирус (DENV-2, штамм 16681) или среду для анализа в случае лунок для анализа на цитотоксичность и контрольных лунок со средой/клетками добавляли сразу после тестируемых соединений при MOI 0,5, 40 в случае 96-луночных планшетов, соответственно. Вирусную суспензию получали посредством размораживания замороженных исходных материалов DENV-2 и разведения до требуемой концентрации бляшкообразующих единиц в среде для анализа.
Планшет для клеток дополнительно инкубировали во влажной камере в течение 5 суток после введения при 37°C/5%CO2. После периода инкубирования клетки наблюдали под микроскопом для проверки на наличие характерного цитопатического эффекта в лунках с вирусным контролем и нормальных клеток в лунках с клеточным контролем. После доведения планшетов до комнатной температуры в каждую лунку 96-луночных планшетов для клеток добавляли 20 мкл Viral ToxGlo (Promega). Планшеты инкубировали при комнатной температуре в течение 5 минут перед измерением люминесценции на спектрофотометре (Envision, PerkinElmer).
Ингибирование DENV-2 рассчитывали в виде процента ингибирования цитопатического эффекта относительно вирусного контроля, а цитотоксичность - в виде процента выживаемости клеток относительно лунок с клеточным контролем.
Таблица VI
среднее (n = 3)
Вывод
На основе полученных выше результатов можно сделать вывод о том, что соединения формулы (I) представляют собой подходящие химические соединения для лечения и/или предупреждения РНК-вирусных инфекций, вызванных РНК-вирусами группы IV, в частности, флавивирусных инфекций и, в частности, вирусных инфекций Денге 2.
Кроме того, настоящее изобретение относится к фармацевтической композиции, содержащей по меньшей мере одно новое соединение, как определено выше, или любую из его фармацевтически приемлемых солей, или по меньшей мере любое из соединенией (3) - (18), (32) - (35), (91) - (125), как определено выше, или любую из их фармацевтически приемлемых солей, и также по меньшей мере один фармацевтически приемлемый эксципиент.
Фармацевтические композиции согласно изобретению могут содержать одно или более соединений согласно изобретению в любой форме, описанной в настоящем документе.
Еще одним объектом настоящего изобретения является применение по меньшей мере одного соединения формулы (I), как определено выше, и соединений (1) - (18) и (32) - (35) и (91) - (122), как определено выше, или одной из их фармацевтически приемлемых солей согласно настоящему изобретению для получения лекарственного средства для предупреждения или лечения у субъекта РНК-вирусной инфекции, вызванной РНК-вирусом группы IV или группы V в соответствии с классификацией по Балтимору, и, например, инфекции Чикунгунья, инфекции Денге, инфекции гриппа или инфекции RSV.
Таким образом, настоящее изобретение относится к одному соединению формулы (I), как определено выше, и соединениям (1) - (18) и (32) - (35) и (91) - (122) или одной из их приемлемых солей в качестве средства для ингибирования, предупреждения или лечения РНК-вирусной инфекции, и наиболее предпочтительно инфекции РНК-вирусом из группы IV или V, и, например, инфекции Чикунгунья, инфекции Денге, инфекции гриппа или инфекции RSV.
Согласно конкретному варианту осуществления лечение является непрерывным или проводится с перерывами.
«Непрерывное лечение» означает длительное лечение, которое может осуществляться с разной частотой введения, такой как один раз в сутки, один раз в трое суток, один раз в неделю или один раз в две недели, или один раз в месяц.
Согласно одному варианту осуществления соединение формулы (I) или любую из его фармацевтически приемлемых солей вводят в дозе, варьирующей от 0,1 до 1000 мг, в частности, варьирующей от 0,1 до 10 мг, или, например, варьирующей от 10 до 200 мг, или, например, варьирующей от 200 до 1000 мг.
Еще один объект изобретения относится к терапевтическому способу лечения и/или предупреждения у субъекта РНК-вирусной инфекции и наиболее предпочтительно РНК-вирусной инфекции, вызванной вирусом, принадлежащим к группе IV или V классификации по Балтимору, включающему введение терапевтически эффективного количества соединения формулы (I), соединений (1) - (18) и (32) - (35) и (91) - (122), как определено выше, или одной из их приемлемых солей.
В частном варианте осуществления изобретение относится к применению соединения формулы (I) согласно изобретению или его фармацевтически приемлемой соли или его фармацевтически активного производного, или способа согласно изобретению, где соединение формулы (I) подлежит введению в комбинации с дополнительным средством, применимым для лечения указанной РНК-вирусной инфекции, и наиболее предпочтительно указанной инфекции РНК-вирусом из группы IV или V, и, например, инфекции Чикунгунья, инфекции Денге, инфекции гриппа или инфекции RSV.
Соединения могут быть введены любым способом введения, таким как, например, внутримышечный, внутривенный, интраназальный или пероральный способ и т. д.
Соединения согласно настоящему изобретению могут в соответствующих случаях быть введены в виде пролекарств, таких как сложные эфиры, соединений, к которым относится данное изобретение. «Пролекарство» означает соединение, которое может быть превращено in vivo посредством метаболических процессов (например, посредством гидролиза, восстановления или окисления) в соединение по настоящему изобретению. Например, сложноэфирное пролекарство соединения согласно настоящему изобретению может превращаться посредством гидролиза in vivo в исходную молекулу. Подходящие сложные эфиры соединений согласно настоящему изобретению представляют собой, например, ацетаты, цитраты, лактаты, тартраты, малонаты, оксалаты, салицилаты, пропионаты, сукцинаты, фумараты, малеаты, метилен-бис-β-гидроксинафтоаты, гентизаты, изетионаты, ди-пара-толуоилтартраты, метансульфонаты, этансульфонаты, бензолсульфонаты, пара-толуолсульфонаты, циклогексилсульфаматы и хинаты. Примеры сложноэфирных пролекарств представляют собой такие, как описано в F. J. Leinweber, Drug Metab. Res., 1987, 18, 379. В том виде, в котором они используются в данном документе, ссылки на соединения согласно настоящему изобретению также включают любые формы пролекарств или метаболитов.
Композиция согласно изобретению может дополнительно включать одну или более добавок, таких как разбавители, эксципиенты, стабилизаторы и консерванты. Такие добавки хорошо известны специалистам в данной области и описаны, в частности, в «Ullmann'cs Encyclopedia of Industrial Chemistry, 6th Ed.» (разные ред., 1989-1998, Marcel Dekker) и в «Pharmaceutical Dosage Forms and Drug Delivery Systems» (ANSEL et al., 1994, WILLIAMS & WILKINS).
Упомянутые выше эксципиенты выбирают в соответствии с лекарственной формой и желаемым способом введения.
Согласно еще одному варианту осуществления фармацевтически приемлемые композиции согласно данному изобретению могут быть введены человеку и другим животным перорально, ректально, парентерально, интрацистернально, внутривагинально, внутрибрюшинно, местно (в виде порошков, мазей или капель), буккально, в виде перорального или назального спрея, или тому подобное, в зависимости от тяжести инфекции, подлежащей лечению.
Композиции согласно настоящему изобретению могут быть введены перорально, парентерально, посредством спрея для ингаляций, местно, ректально, назально, буккально, вагинально или посредством вживляемого резервуара. Термин «парентеральный», в том виде, в котором он используется в настоящем документе, включает подкожные, внутривенные, внутримышечные, внутрисуставные, интрасиновиальные, внутригрудинные, интратекальные, внутрипеченочные, внутриочаговые и внутричерепные методы инъекции или инфузии. Предпочтительно композиции вводят перорально, внутрибрюшинно или внутривенно. Стерильные инъецируемые формы композиций согласно данному изобретению могут представлять собой водную или масляную суспензию. Данные суспензии могут быть приготовлены в соответствии с методиками, известными в данной области, с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильный инъецируемый препарат может также представлять собой стерильный инъецируемый раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. К приемлемым носителям и растворителям, которые могут быть использованы, относятся вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные нелетучие масла.
Композиции согласно данному изобретению могут быть введены любым способом, включая пероральное, парентеральное, подъязычное, трансдермальное, вагинальное, ректальное, трансмукозальное, местное, интраназальное посредством ингаляции, буккальное или интраназальное введение или их комбинации, но не ограничиваясь ими. Парентеральное введение включает внутривенное, внутриартериальное, внутрибрюшинное, подкожное, внутримышечное, интратекальное и внутрисуставное введение, но не ограничивается ими. Композиции согласно данному изобретению могут также быть введены в форме имплантата, который делает возможным медленное высвобождение композиций, а также путем медленной контролируемой в/в (внутривенной) инфузии.
Например, соединение формулы (I) может находиться в любой фармацевтической форме, которая подходит для энтерального или парентерального введения, совместно с подходящими эксципиентами, например, в форме простых таблеток или таблеток с оболочкой, твердых желатиновых капсул, капсул в мягкой желатиновой оболочке и других капсул, суппозиториев или питьевых препаратов, таких как суспензии, сиропы или инъецируемые растворы или суспензии, в дозах, которые обеспечивают ежесуточное введение от 0,1 до 1000 мг активного вещества.
В частном варианте осуществления соединение формулы (I) согласно данному изобретению вводят перорально.
Пероральный путь введения является особенно предпочтительным в аспекте данного изобретения, относящемся к профилактике или лечению.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФЕНИЛ/ПИРИДИЛ-N-ФЕНИЛ/ПИРИДИЛЬНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РНК-ВИРУСНОЙ ИНФЕКЦИИ | 2019 |
|
RU2790838C2 |
| ФЕНИЛ/ПИРИДИЛ-N-ФЕНИЛ/ПИРИДИЛЬНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РНК-ВИРУСНОЙ ИНФЕКЦИИ | 2019 |
|
RU2803216C2 |
| ПРОИЗВОДНЫЕ ФЕНИЛ-N-ХИНОЛИНА ДЛЯ ЛЕЧЕНИЯ РНК-ВИРУСНОЙ ИНФЕКЦИИ | 2019 |
|
RU2805064C2 |
| АРИЛ-N-АРИЛЬНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РНК-ВИРУСНОЙ ИНФЕКЦИИ | 2020 |
|
RU2821414C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА ДЛЯ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ | 2015 |
|
RU2760686C2 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ СПИДА | 2010 |
|
RU2575845C2 |
| НОВЫЕ АНТИИНВАЗИВНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2641650C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИН-2-АМИНА, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ СПИДа | 2011 |
|
RU2598845C2 |
| Соединения, активные по отношению к бромодоменам | 2015 |
|
RU2743074C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ РЕСПИРАТОРНО-СИНЦИТИАЛЬНОЙ ВИРУСНОЙ ИНФЕКЦИИ | 2012 |
|
RU2612530C2 |
Изобретение относится к соединению формулы (I) для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, выбранной из вирусной инфекции Чикунгунья, RSV (респираторно-синцитиальный вирус), Денге и гриппа, где кольцо  и кольцо
и кольцо  независимо означают фениленовую или пиридиленовую группу, X1 представляет собой этениленовую группу, группу -NH-CO-, группу -CO-NH-, Y1 представляет собой арильную группу, выбранную из пиридильной группы, пиразинильной группы или пиримидинильной группы, X2 представляет собой группу -O-, -CO-NH-, триазол или -SO2-NH-, n равен 0, 1, 2 или 3, m и m’независимо равны 0 или 1, Y2 представляет собой гидроксильную группу, морфолинильную группу, пиперидинильную группу, пиперазинильную группу, необязательно замещенную (C1-C4)алкильной группой, или группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода, атом фтора или (C1-C4)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу, или, в качестве альтернативы, X2-Y2 представляет собой группу -C(=O)-NRcRd, где Rc и Rd образуют вместе с атомом азота насыщенное гетероциклическое кольцо, необязательно замещенное одной или двумя (C1-C4)алкильными группами, циклопентильной группой, образуя, таким образом, спироциклопентильное производное, или трифторметильной группой, R и R’ независимо представляют собой (C1-C4)алкильную группу, (C3-C6)циклоалкильную группу, атом галогена, (C1-C5)алкоксигруппу, при условии, что когда X1 представляет собой группу -NH-CO-, Y1 может дополнительно представлять собой фенильную группу. Изобретение также относится к способу получения указанного соединения и фармацевтической композиции на его основе. Технический результат - получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине для лечения и/или предупреждения РНК-вирусной инфекции, вызванной вирусами Чикунгунья, RSV, Денге или гриппа. 8 н. и 9 з.п. ф-лы, 6 табл., 8 пр.
независимо означают фениленовую или пиридиленовую группу, X1 представляет собой этениленовую группу, группу -NH-CO-, группу -CO-NH-, Y1 представляет собой арильную группу, выбранную из пиридильной группы, пиразинильной группы или пиримидинильной группы, X2 представляет собой группу -O-, -CO-NH-, триазол или -SO2-NH-, n равен 0, 1, 2 или 3, m и m’независимо равны 0 или 1, Y2 представляет собой гидроксильную группу, морфолинильную группу, пиперидинильную группу, пиперазинильную группу, необязательно замещенную (C1-C4)алкильной группой, или группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода, атом фтора или (C1-C4)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу, или, в качестве альтернативы, X2-Y2 представляет собой группу -C(=O)-NRcRd, где Rc и Rd образуют вместе с атомом азота насыщенное гетероциклическое кольцо, необязательно замещенное одной или двумя (C1-C4)алкильными группами, циклопентильной группой, образуя, таким образом, спироциклопентильное производное, или трифторметильной группой, R и R’ независимо представляют собой (C1-C4)алкильную группу, (C3-C6)циклоалкильную группу, атом галогена, (C1-C5)алкоксигруппу, при условии, что когда X1 представляет собой группу -NH-CO-, Y1 может дополнительно представлять собой фенильную группу. Изобретение также относится к способу получения указанного соединения и фармацевтической композиции на его основе. Технический результат - получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине для лечения и/или предупреждения РНК-вирусной инфекции, вызванной вирусами Чикунгунья, RSV, Денге или гриппа. 8 н. и 9 з.п. ф-лы, 6 табл., 8 пр.  (I)
(I)
1. Применение соединения формулы (I) для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, выбранной из вирусной инфекции Чикунгунья, RSV (респираторно-синцитиальный вирус), Денге и гриппа

где:
кольцо  и кольцо
и кольцо  независимо означают фениленовую или пиридиленовую группу,
независимо означают фениленовую или пиридиленовую группу,
X1 представляет собой этениленовую группу, группу -NH-CO-, группу -CO-NH-,
Y1 представляет собой арильную группу, выбранную из пиридильной группы, пиразинильной группы или пиримидинильной группы,
X2 представляет собой
группу -O-,
группу -CO-NH-,
триазол,
или
группу -SO2-NH-,
n равен 0, 1, 2 или 3,
m и m’независимо равны 0 или 1,
Y2 представляет собой
гидроксильную группу,
морфолинильную группу,
пиперидинильную группу,
пиперазинильную группу, необязательно замещенную (C1-C4)алкильной группой,
или
группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода, атом фтора или (C1-C4)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу,
или, в качестве альтернативы, X2-Y2 представляет собой группу -C(=O)-NRcRd, где Rc и Rd образуют вместе с атомом азота насыщенное гетероциклическое кольцо, необязательно замещенное одной или двумя (C1-C4)алкильными группами, циклопентильной группой, образуя, таким образом, спироциклопентильное производное, или трифторметильной группой,
R и R’ независимо представляют собой
(C1-C4)алкильную группу,
(C3-C6)циклоалкильную группу,
атом галогена,
(C1-C5)алкоксигруппу,
при условии, что когда X1 представляет собой группу -NH-CO-, Y1 может дополнительно представлять собой фенильную группу.
2. Применение соединения формулы (I) по п. 1, где:
кольцо 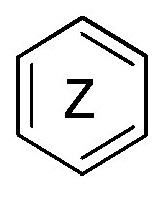 и кольцо
и кольцо  оба представляют собой фениленовую группу, или кольцо
оба представляют собой фениленовую группу, или кольцо  представляет собой пиридиленовую группу, и кольцо представляет собой фениленовую группу.
представляет собой пиридиленовую группу, и кольцо представляет собой фениленовую группу.
3. Применение соединения формулы (I) по п. 1 или 2, где
Y1 представляет собой
2-пиридинильную группу или 3-пиридинильную группу,
пиримидинильную группу или пиразинильную группу, где один из атомов азота находится в орто-положении относительно X1,
при условии, что когда X1 представляет собой группу -NH-CO-, Y1 может дополнительно представлять собой фенильную группу.
4. Применение соединения формулы (I) по любому из пп. 1-3, где
кольцо  и кольцо
и кольцо 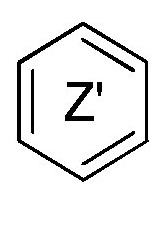 оба представляют собой фениленовую группу,
оба представляют собой фениленовую группу,
Y1 представляет собой 2-пиридинильную группу, при условии, что когда X1 представляет собой группу -NH-CO-, Y1 может дополнительно представлять собой фенильную группу,
X2 представляет собой группу -O-, группу -CO-NH-,
Y2 представляет собой
группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода или (C1-C4)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу, и R3 представляет собой атом водорода или (C1-C4)алкильную группу, или
морфолинильную группу, и
R и R’ независимо представляют собой атом водорода, (C1-C4)алкильную группу, такую как метильная группа, или (C3-C6)циклоалкильную группу, такую как циклопропильная группа.
5. Применение соединения формулы (Ia) для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, выбранной из вирусной инфекции Чикунгунья, RSV, Денге и гриппа

где
Y1, R, R’, m, m’, кольцо 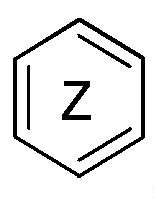 , X2, n и Y2 представляют собой такие, как определено в любом из пп. 1-4.
, X2, n и Y2 представляют собой такие, как определено в любом из пп. 1-4.
6. Применение соединения формулы (Ia) по п. 5, где
кольцо представляет собой фениленовую группу или пиридиленовую группу,
Y1 представляет собой 2-пиридильную группу, 3-пиридильную группу или пиразинильную группу,
n равен 1, 2 или 3, m равен 0,
R’ представляет собой атом галогена, (C1-C2)алкоксигруппу или (C1-C2)алкильную группу,
X2 представляет собой группу -CO-NH-, группу -SO2NH- или двухвалентный триазол,
Y2 представляет собой
морфолинильную группу, пиперидинильную группу или пиперазинильную группу, необязательно замещенную (C1-C4)алкильной группой,
группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода или (C1-C2)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу,
или, в качестве альтернативы, X2-Y2 представляет собой группу -C(=O)-NRcRd, где Rc и Rd образуют вместе с атомом азота насыщенное гетероциклическое кольцо, необязательно замещенное одной или двумя (C1-C4)алкильными группами, циклопентильной группой, образуя, таким образом, спироциклопентильное производное, или трифторметильной группой.
7. Применение соединения формулы (Ib) для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, выбранной из вирусной инфекции Чикунгунья, RSV, Денге и гриппа
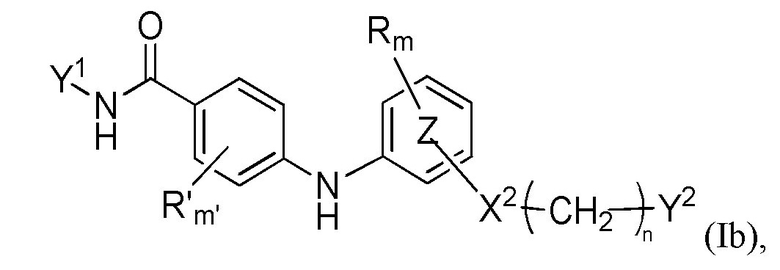
где
Y1, R, R’, m, m’, кольцо  , X2, n и Y2 представляют собой такие, как определено в любом из пп. 1-4.
, X2, n и Y2 представляют собой такие, как определено в любом из пп. 1-4.
8. Применение соединения формулы (Ib) по п. 7, где
кольцо представляет собой фениленовую группу,
Y1 представляет собой фенильную группу, 2-пиридильную группу или пиримидинильную группу, причем один из атомов азота пиримидинильной группы находится в орто-положении относительно группы -NH-CO-,
n равен 1, 2 или 3, m равен 0, m’ равен 0 или 1,
R’ представляет собой (C3-C6)циклоалкильную группу,
X2 представляет собой группу -CO-NH-, группу -O- или двухвалентный триазол,
Y2 представляет собой
гидроксильную группу,
морфолинильную группу, пиперидинильную группу или пиперазинильную группу, необязательно замещенную (C1-C4)алкильной группой,
группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода или (C1-C2)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу.
9. Применение соединения формулы (Id) для лечения и/или предупреждения РНК-вирусной инфекции, вызванной РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, выбранной из вирусной инфекции Чикунгунья, RSV, Денге и гриппа:

где
Y1, R, R’, m, m’, кольцо  , X2, n и Y2 представляют собой такие, как определено в п. 1 или 2.
, X2, n и Y2 представляют собой такие, как определено в п. 1 или 2.
10. Применение соединения формулы (Id) по п. 9,
где кольцо  представляет собой фениленовую группу,
представляет собой фениленовую группу,
Y1 представляет собой 2-пиридильную группу или 3-пиридильную группу,
X2 представляет собой группу -CO-NH-, группу -SO2-NH- или двухвалентный триазол,
m’ и m равны 0, n равен 1, 2 или 3,
Y2 представляет собой гидроксил или группу -CR1R2R3, где R1, R2 и R3 независимо представляют собой атом водорода или (C1-C2)алкильную группу, при этом подразумевается, что не больше чем один из R1, R2 и R3 представляет собой атом водорода, или R1 и R2 образуют вместе с несущим их атомом углерода (C3-C6)циклоалкильную группу.
11. Применение соединения формулы (I) по п. 1, где соединение формулы (I) выбрано из



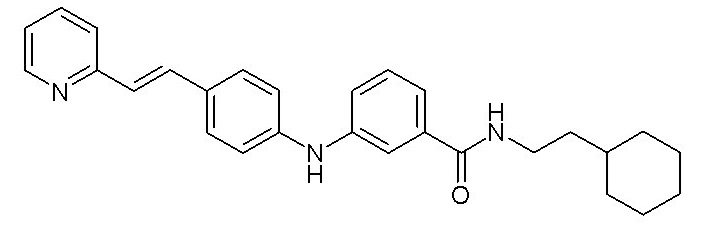

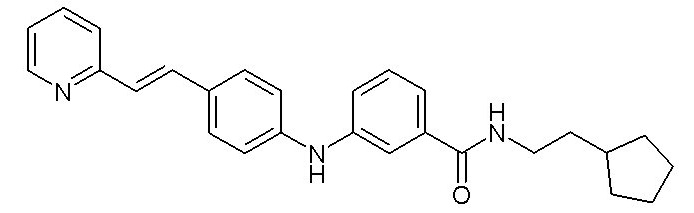


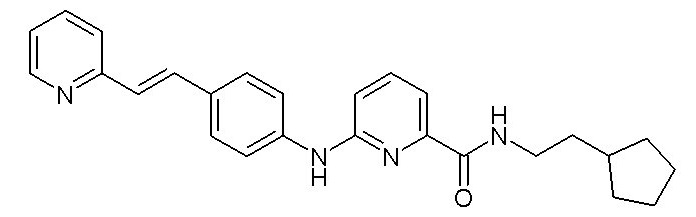
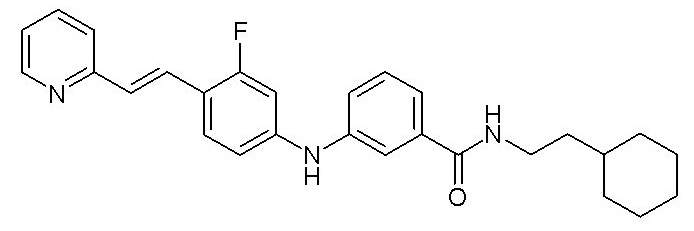



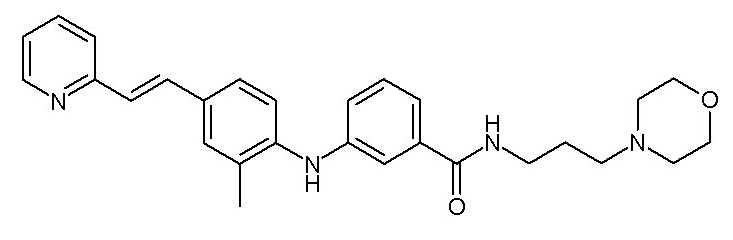



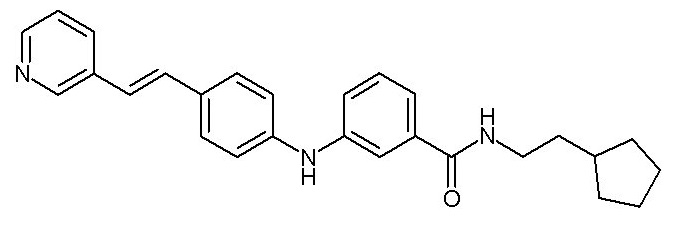



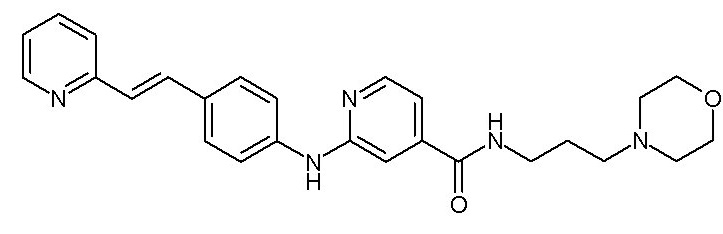
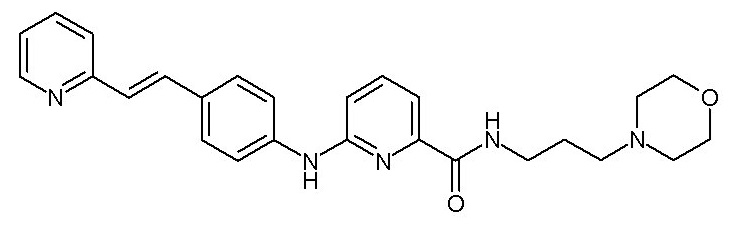





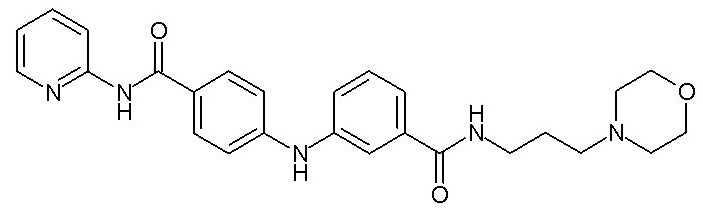
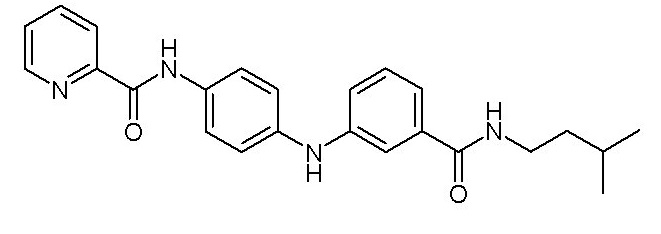



12. Применение соединения формулы (I) по любому из пп. 1-4, где РНК-вирусная инфекция, вызванная РНК-вирусом, принадлежащим к группе IV или V классификации по Балтимору, выбрана из вирусной инфекции RSV, вирусной инфекции Чикунгунья и вирусной инфекции Денге.
13. Соединение формулы (I), как определено в любом из пп. 1-4, выбранное из любого из следующих соединений:
(1) соединение формулы (Iа), как определено в п. 5 или 6,

где
Y1 представляет собой 2-пиридильную группу, 3-пиридильную группу или пиразинильную группу,
группа 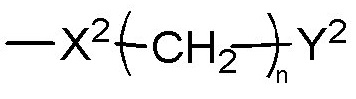 находится в мета-положении на кольце
находится в мета-положении на кольце  относительно группы -NH-,
относительно группы -NH-,
кольцо  представляет собой фениленовую группу, и
представляет собой фениленовую группу, и
при условии, что соединения 1 и 2, как определено в п. 11, исключены,
(2) соединение формулы (Ib), как определено в п. 7 или 8,

где
Y1 представляет собой фенильную группу, 2-пиридильную группу или пиримидинильную группу, причем один из атомов азота пиримидинильной группы находится в орто-положении относительно группы -NH-CO-,
(4) соединение формулы (Id), как определено в п. 9 или 10,

где
Y1 представляет собой 2-пиридильную группу.
14. Применение соединения формулы (I) по п. 1, где соединение формулы (I) выбрано из соединения формулы (Ia), (Ib) и (Id), как определено в п. 13, и любого из соединений (3)-(6), (8)-(16), (32), (33), (35), (91), (92), (95), (96), (101), (103)-(108), (111), (115), (118), (122), как определено в п. 11:

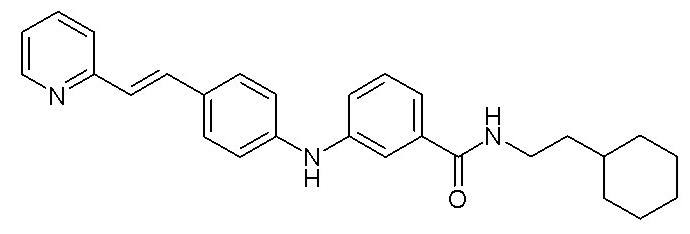


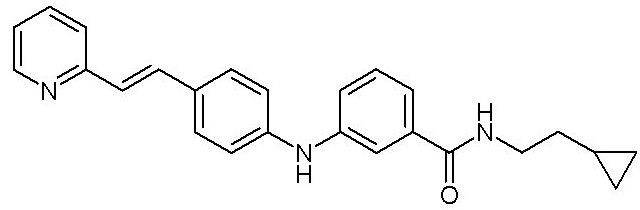
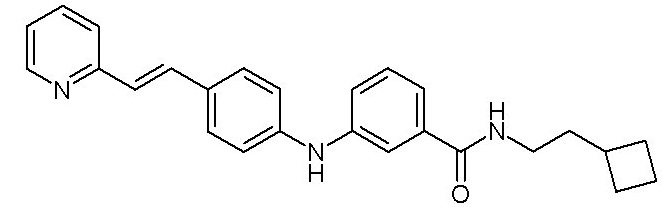





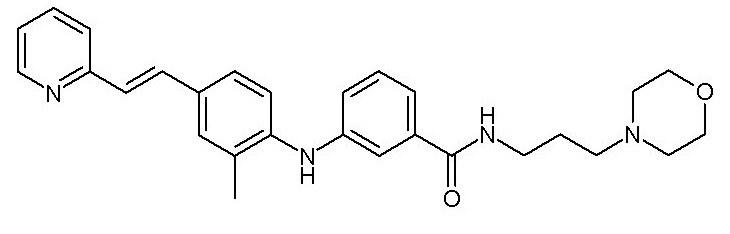
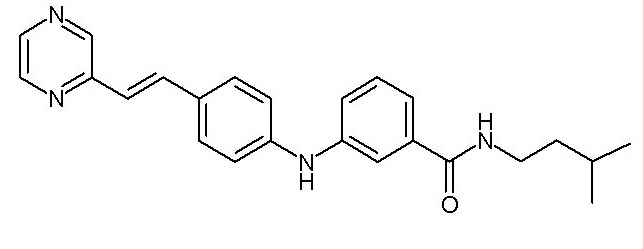
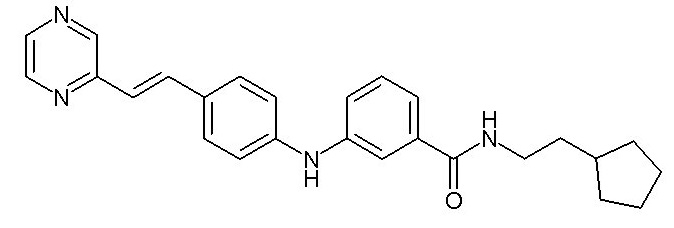
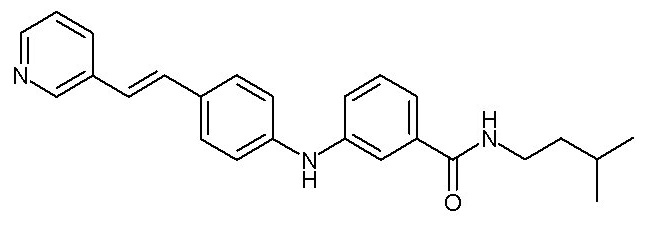
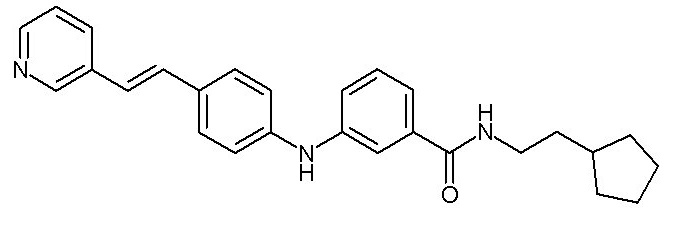

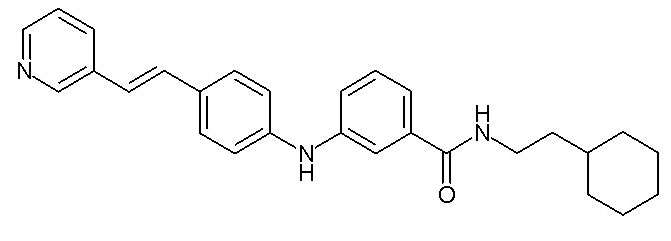
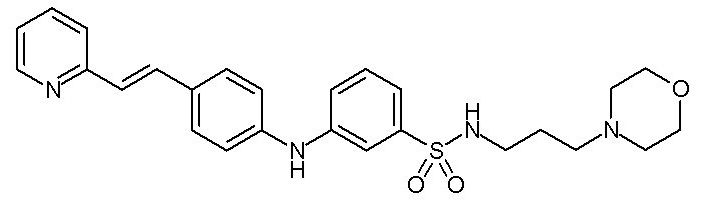
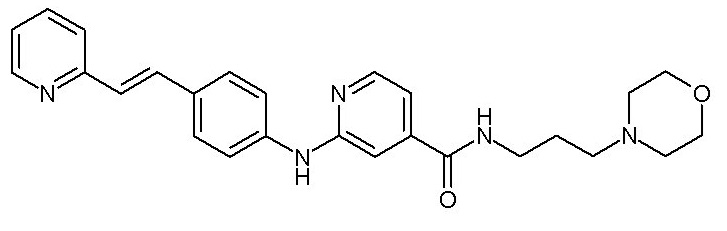
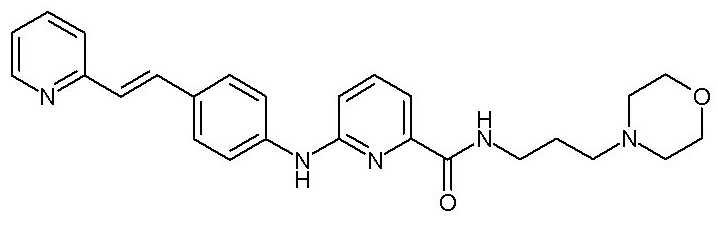

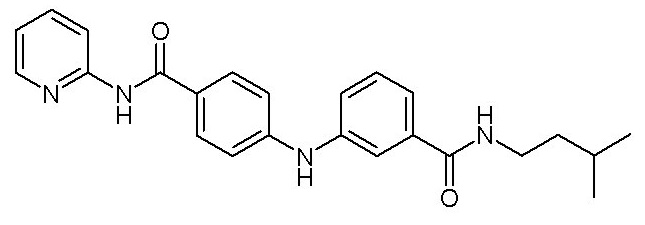


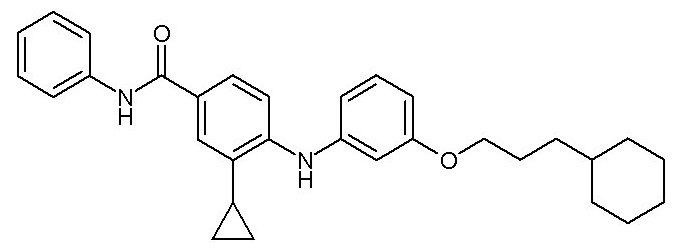
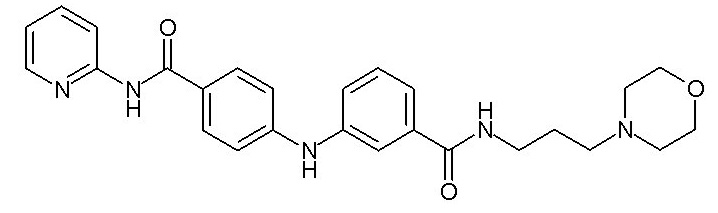



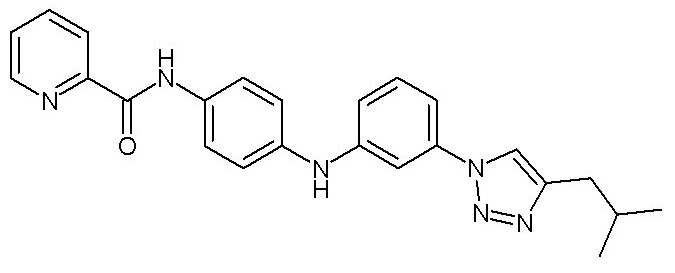
15. Соединение, как определено в п. 11, за исключением соединений № 1 и 2:

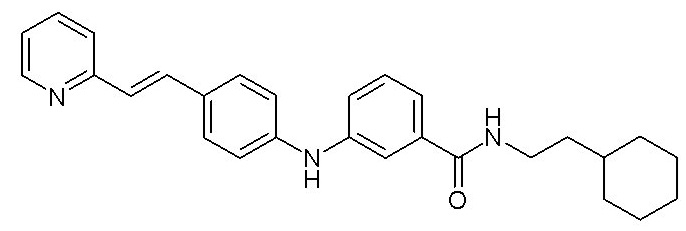
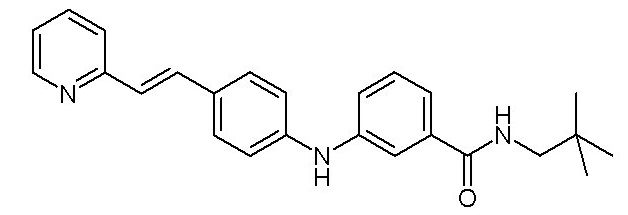

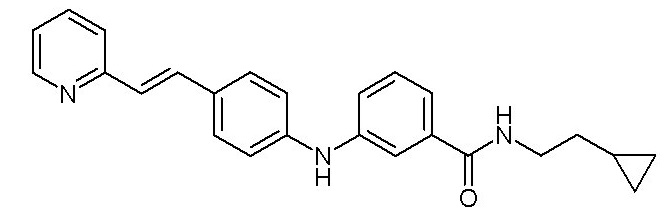
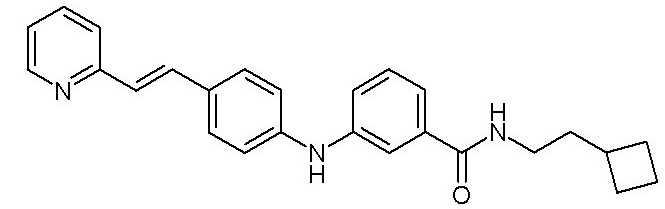



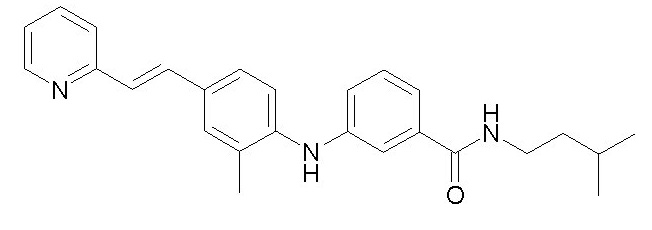
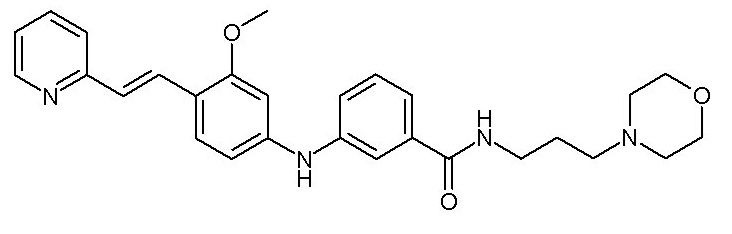

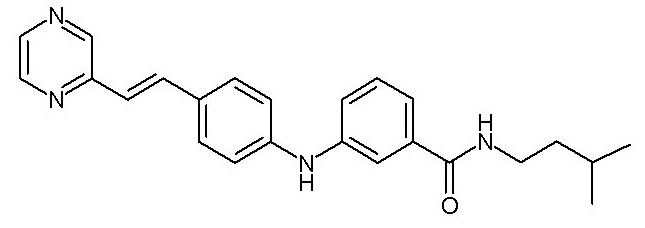



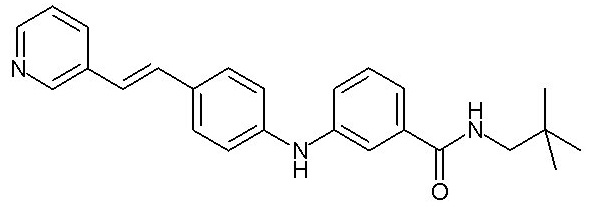
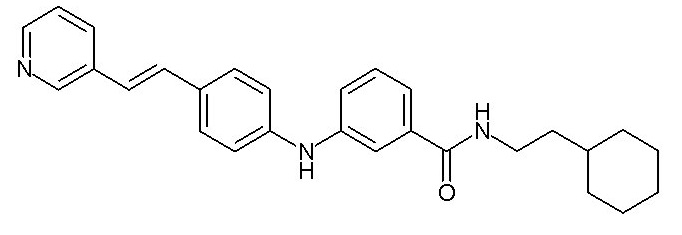







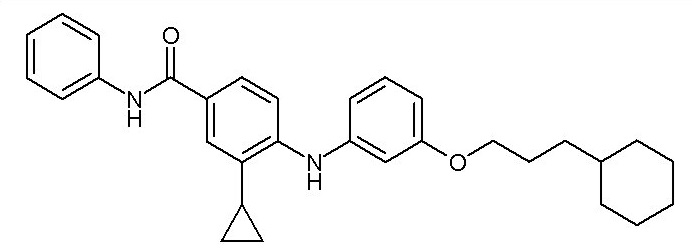
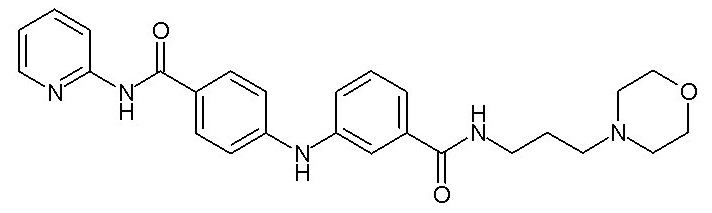




16. Фармацевтическая композиция для лечения и/или предотвращения инфекции, вызванной вирусом Чикугунья, вирусом RSV, вирусом Денге или вирусом гриппа, содержащая в эффективном количестве по меньшей мере одно соединение, как определено в п. 13, или по меньшей мере любое из соединений (3)-(6), (8)-(16), (32), (33), (35), (91), (92), (95), (96), (101), (103)-(108), (111), (115), (118), (122), как определено в п. 15, и также по меньшей мере один фармацевтически приемлемый эксципиент:





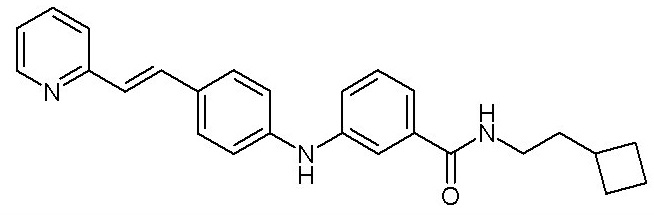

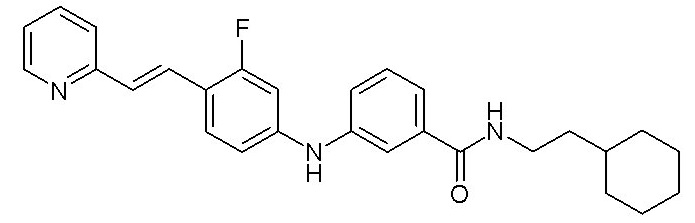

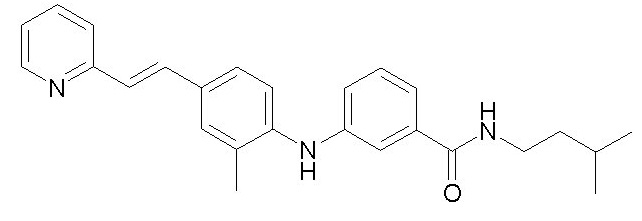
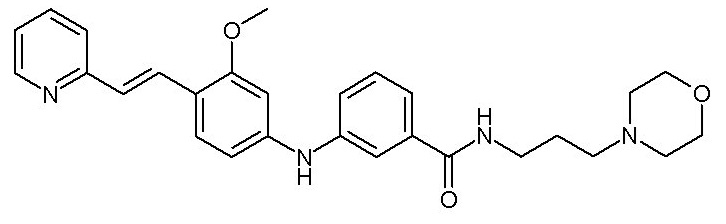


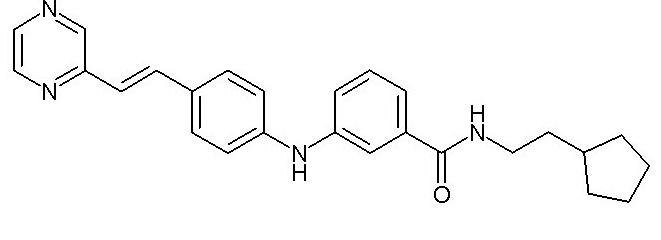
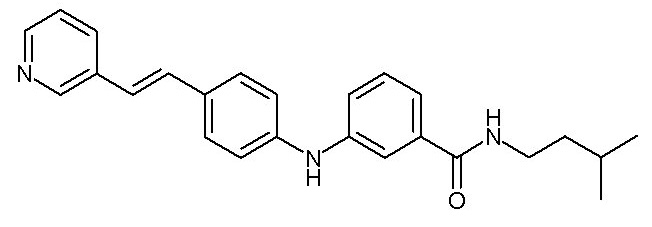





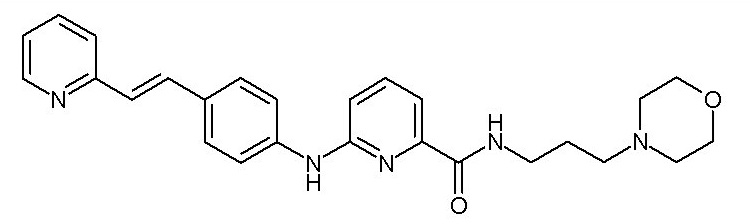
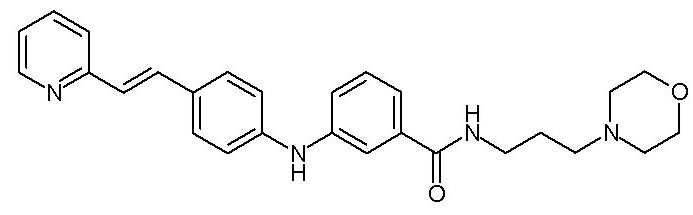
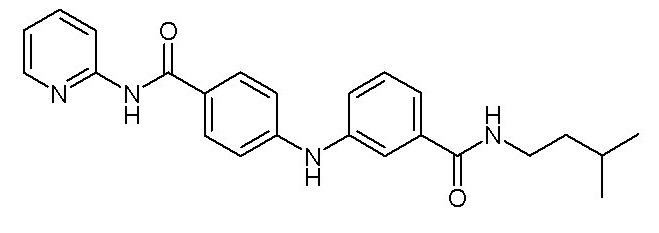



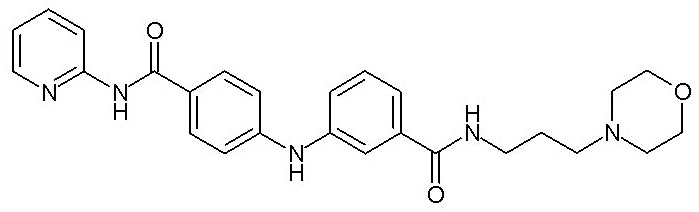



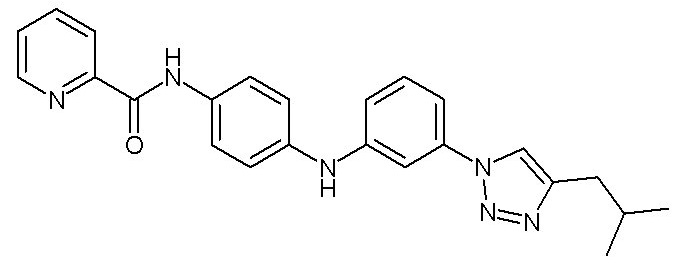
17. Способ синтеза для производства соединений формулы (Ia), (Ib) и (Id), как определено в п. 13, включающий по меньшей мере стадию сочетания соединения формулы (II)

где X1, Y1, R, R’, m, m’, n, кольцо  , кольцо
, кольцо  , X2, Y2 представляют собой такие, как определено в п. 13, и X представляет собой атом хлора, атом иода или атом брома, в присутствии неорганического основания и дифосфина и в присутствии металлорганического катализатора, с получением соединения формулы (Ia), (Ib) или (Id), как определено в п. 13.
, X2, Y2 представляют собой такие, как определено в п. 13, и X представляет собой атом хлора, атом иода или атом брома, в присутствии неорганического основания и дифосфина и в присутствии металлорганического катализатора, с получением соединения формулы (Ia), (Ib) или (Id), как определено в п. 13.
| WO 2009087238 A9, 16.07.2009 | |||
| WO 2012131656 A2, 04.10.2012 | |||
| WO 2010143168 A2, 16.12.2010 | |||
| RU 2016103217 А, 10.08.2017. |
