Изобретение относится к фармации, а именно к фармацевтической химии.
Микофенолат натрия - это иммуносупрессивное средство, подавляющее рост и деление лимфоцитов. Согласно международным и отечественным рекомендациям данный препарат используется при пересадке почек и легких для предупреждения отторжения трансплантата в комбинации с глюкокортикостероидами, циклоспорином и другими препаратами. Для индивидуального подбора дозировки и проведения исследований сравнительной фармакокинетики необходимо измерение концентрации микофеноловой кислоты в плазме крови. Для лаборатории, которая проводит данные исследования также важна высокая производительность процесса подготовки проб и аналитических процедур, а также высокая точность измерений. Конъюгат микофеноловой кислоты с глюкуроновой кислотой обладает способностью к обратной конверсии после отбора крови, что может привести к завышенным результатам количественного определения. Требования к точности биоаналитических методик для оценки биоэквивалентности оригинального и воспроизведенного препарата и терапевтического лекарственного мониторинга строго регламентированы: она должна находиться в диапазоне 85-115% от истинного значения. В связи с этим большое значение имеет разработка новых точных и экспрессных способов определения концентрации микофеноловой кислоты в плазме крови.
Известен способ количественного определения микофеноловой кислоты в плазме крови человека, заключавшийся в том, что плазму крови, полученную от пациента, подвергают депротеинизации раствором внутреннего стандарта дейтерированной микофеноловой кислоты в метаноле, затем после центрифугирования в надосадочной жидкости проводят измерение концентрации микофеноловой кислоты в диапазоне от 0,015 до 15 мкг/мл методом высокоэффективной жидкостной хроматографии с тандемным масс-спектрометрическим детектированием (Upadhyay V., Trivedi V., Shah G. et. al., J. Pharm. Anal., V. 4, №3, pp. 205-216, 2014). Недостатком данного способа является низкая точность.
Наиболее близким аналогом (прототипом) предлагаемого решения является способ, заключавшийся в том, что плазму крови, полученную от пациента, подвергают депротеинизации смесью раствора внутреннего стандарта дейтерированной микофеноловой кислоты в метаноле с водным раствором цинка сульфата, затем после центрифугирования в надосадочной жидкости проводят измерение концентрации микофеноловой кислоты в диапазоне от 1 до 15 мкг/мл методом высокоэффективной жидкостной хроматографии с тандемным масс-спектрометрическим детектированием при следующих условиях хроматографического разделения (Decavele А.С., Favoreel N., Heyden V.F. et. al., Clin. Chem. Lab. Med., V. 49, №7, pp. 1159-1165, 2011):
Колонка: Zorbax RP-C18, (2,1*30 мм, 2,6 мкм);
Скорость потока: 0,5 мл/мин;
Условия градиентного элюирования:
сначала анализа и до 1 мин анализа содержание раствора формиата аммония 2 ммоль/л в воде в подвижной фазе составляет 90%, содержание содержание раствора формиата аммония 2 ммоль/л в метаноле - 10%;
с 1,5 мин до 9,5 мин анализа содержание раствора формиата аммония 2 ммоль/л в воде линейно понижается до 5%, содержание раствора формиата аммония 2 ммоль/л в метаноле линейно повышается до 95%;
с 9,5 мин до 12 мин анализа содержание раствора формиата аммония 2 ммоль/л в воде линейно повышается до 90%, содержание раствора формиата аммония 2 ммоль/л в метаноле линейно понижается до 10%.
Недостатком данного способа является низкая точность.
Цель предлагаемого способа - повышение точности количественного определения микофеноловой кислоты в плазме крови.
Поставленная цель достигается тем, что хроматографическое разделение компонентов матрицы проводят с использованием хроматографической колонки Phenomenex Kinetex C18 (30×4,6 мм, 2,6 мкм) при скорости потока 0,4 мл/мин и следующих условиях градиентного элюирования:
сначала анализа и до 1 мин анализа содержание ацетонитрила в подвижной фазе составляет 40%, содержание воды - 60%;
с 1 мин до 1,5 мин анализа содержание ацетонитрила линейно повышается до 65%, содержание воды линейно понижается до 35%;
с 1,5 мин до 2,0 мин анализа содержание ацетонитрила линейно повышается до 90%, содержание воды линейно понижается до 10%.
с 2,0 мин до 2,5 мин анализа содержание ацетонитрила составляет 90%, содержание воды - 10%;
с 2,5 мин до 3,0 мин анализа содержание ацетонитрила линейно понижается до 65%, содержание воды линейно повышается до 35%.
с 3,0 мин до 3,5 мин анализа содержание ацетонитрила линейно понижается до 40%, содержание воды линейно повышается до 60%.
с 3,5 мин до конца анализа содержание ацетонитрила составляет 40%, содержание воды - 60%.
Технические решения, имеющие признаки, совпадающие с отличительными признаками предлагаемого нами способа, не выявлены, что позволяет сделать вывод о соответствии предлагаемого способа критерию «изобретательский уровень».
Предлагаемый способ осуществляется следующим образом: из плазмы крови отбирают аликвоту объемом 50 мкл, переносят ее в пробирку, добавляют 450 мкл раствора внутреннего стандарта дейтерированной микофеноловой в концентрации 1,5 мкг/мл, затем пробирку закрывают, полученную смесь перемешивают на вортексе в течение 30 сек при частоте около 1800 мин-1, подвергают центрифугированию в течение 10 мин при 2500 об/мин. Затем проводят хромато-масс-спектрометрическое определение при следующих условиях:
Колонка: Phenomenex Kinetex C18 (30×4,6 мм, 2,6 мкм)
Скорость потока: 0,5 мл/мин;
Температура термостата: комнатная;
Объем вводимой пробы: 5 мкл
Способ ионизации: электрораспылительная ионизация с термофокусировкой, полярность отрицательная;
Условия градиентного элюирования:
сначала анализа и до 1 мин анализа содержание ацетонитрила в подвижной фазе составляет 40%, содержание воды - 60%;
с 1 мин до 1,5 мин анализа содержание ацетонитрила линейно повышается до 65%, содержание воды линейно понижается до 35%;
с 1,5 мин до 2,0 мин анализа содержание ацетонитрила линейно повышается до 90%, содержание воды линейно понижается до 10%,
с 2,0 мин до 2,5 мин анализа содержание ацетонитрила составляет 90%, содержание воды - 10%;
с 2,5 мин до 3,0 мин анализа содержание ацетонитрила линейно понижается до 65%, содержание воды линейно повышается до 35%.
с 3,0 мин до 3,5 мин анализа содержание ацетонитрила линейно понижается до 40%, содержание воды линейно повышается до 60%.
с 3,5 мин до конца анализа содержание ацетонитрила составляет 40%, содержание воды - 60%.
Для детекции аналита и внутреннего стандарта были подобраны следующие параметры масс-спектрометрического детектора:
Поток распыляющего газа: 30 условных единиц;
Поток фокусирующего газа: 20 условных единиц;
Поток вихревого газа: 2 условных единицы;
Напряжение электроспрея: 3250 Вт;
Температура капилляра: 222°C;
Температура испарения: 324°C
Давление газа в ячейке соударений: 1,5 мТорр;
Режим детектирования микофеноловой кислоты: MRM m/z -319,10→191,05+205,10;
Режим детектирования внутреннего стандарта: MRM - m/z - 322,10→191,05+205,10.
Данные параметры индивидуальны для каждой модели масс-спектрометрического детектора.
Расчет концентрации микофеноловой кислоты проводят путем сравнения соотношения площадей пиков микофеноловой кислоты и дейтерированного внутреннего стандарта микофеноловой кислоты со значениями данных соотношений в калибровочной зависимости, которая построена предварительно перед проведением анализа на модельных смесях в диапазоне от 0,5 до 30 мкг/мл, и если полученное соотношение площадей пиков укладывается в диапазон значений соотношений калибровочной зависимости, то концентрация микофеноловой кислоты в плазме рассчитывается по линейному уравнению данной зависимости:
x=(y-b)/а, где
x - измеренная концентрация метилдопы в плазме;
y - соотношение площадей пиков «аналит-внутренний стандарт»;
a - наклон калибровочной кривой;
b - свободный член.
В процессе разработки способа была проведена его валидация на модельных смесях по показателям селективность, нижний предел количественного определения, линейность калибровочных кривых, внутрисерийная и межсерийная прецизионность и точность, эффект переноса предыдущей пробы, степень извлечения, тест на разведение образца, оценка эффекта матрицы, стабильность согласно руководству ЕМЕА (Guideline on bioanalytical method validation, ЕМЕА, 192217, 2009) и руководству по экспертизе лекарственных средств (А.Н. Миронов (ред.), Руководство по экспертизе лекарственных средств, т. 1, Москва, 2013). Данный способ соответствует всем установленным требованиям.
В рамках валидации проводилась оценка показателей «калибровочная кривая» и «нижний предел количественного определения» путем анализа модельных смесей плазмы, содержащей микофеноловую кислоту, на 8 уровнях концентраций в диапазоне от 0,5 до 30 мкг/мл. Для приготовления данных модельных смесей к 950 мкл плазмы добавляли 50 мкл стандартного раствора микофеноловой кислоты в ацетонитриле. Концентрации стандартных растворов представлены в таблице 1.
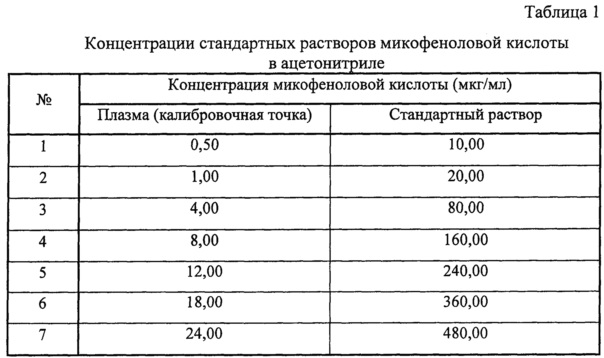

Каждую концентрацию анализировали 6 раз: измеряли соотношение площадей пиков «аналит / внутренний стандарт». По средним значениям данных соотношений был построен калибровочный график, уравнение которого: x=(y+0,00125)/0,05351, где y - соотношение площадей пиков «аналит-внутренний стандарт», x - измеренная концентрация микофеноловой кислоты в плазме. По данному уравнению рассчитывались концентрации данных градуировочных образцов: значения не менее 75% концентраций должны быть в пределах ±15% от номинальных значений, (для нижнего предела количественного определения в пределах ±20%), коэффициент вариации (CV) не должен превышать 15% (для нижнего предела количественного определения - 20%). Полученные результаты представлены в таблице 2.

Из данных, приведенных в таблицы 2, видно, что способ соответствует критериям приемлемости по показателям «калибровочная кривая» и «нижний предел количественного определения».
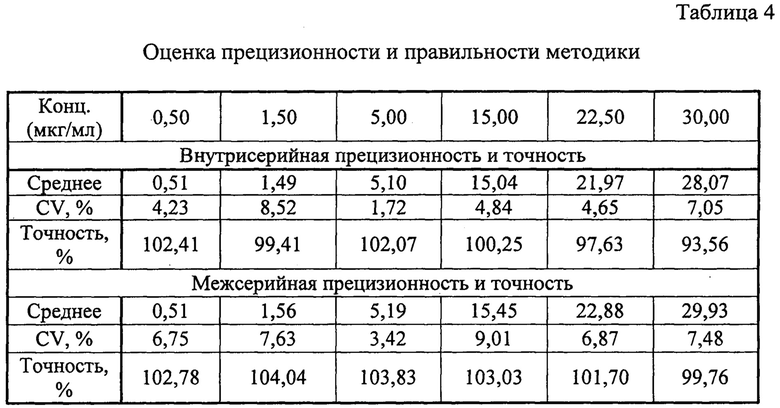
Оценка внутрисерийной и межсерийной точности и прецизионности способа оценивалась на 6 уровнях концентрациях: 0,5; 1,5; 5,0; 15,0; 22,5 и 30,0 мкг/мл. Для приготовления данных модельных смесей к 950 мкл плазмы добавляли 50 мкл стандартного раствора метилдопы в ацетонитриле. Концентрации стандартных растворов представлены в таблице 3.

При выполнении оценки межсерийной прецизионности и точности результаты тестов, проведенных в разные дни разными лаборантами, сравнивались между собой. Прецизионность метода выражалась коэффициентами вариации. Точность оценивалась путем соотношения значения полученного по результатам измерения и номинального значения. Расчет концентраций выполнялся по калибровочному графику, уравнение которого: x=(y+0,00125)/0,05351, где y - соотношение площадей пиков «аналит-внутренний стандарт», x - измеренная концентрация микофеноловой кислоты в плазме. Значения измеренных концентраций должны быть в пределах ±15% от номинальных значений, (для нижнего предела количественного определения в пределах ±20%), коэффициент вариации (CV) не должен превышать 15% (для нижнего предела количественного определения - 20%). Полученные результаты представлены в таблице 4.
Из данных, приведенных в таблице 4, видно, что способ соответствует критериям приемлемости по показателям внутрисерийная и межсерийная прецизионность и точность.
Для оценки влияния обратной конверсии основного метаболита на точность количественного определения бланковая плазма с добавлением фенольного глюкуронида микофеноловой кислоты в концентрации 100 мкг/мл исследована в исходный момент времени и после 24 часов хранения при лабораторной температуре. Для приготовления данных образцов к 950 мкл плазмы добавляли 50 мкл стандартного раствора микофеноловой кислоты в ацетонитриле в концентрации 20000 мкг/мл. Выполнялось сравнение откликов микофеноловой кислоты в свежеприготовленных образцах, содержащих ее глюкуронид, и аналогичных образцах после 24 часов хранения. Полученные результаты представлены в таблице 5.

Из данных, приведенных в таблице 5, видно, что значимой обратной конверсии метаболита в исходное соединение через 24 часа не наблюдалось: полученные значения концентрации микофеноловой кислоты составили 2,58% от нижнего предела количественного определения. Таким образом, данное явление не может повлиять на точность количественного определения.
Предлагаемый способ иллюстрируется следующими примерами:
Пример 1. Испытуемый А., 28 лет принял 1 таблетку «Майфортик», содержащую микофенолат натрия в дозировке 360 мг. Затем у него проводился забор крови из вены предплечья через установленный катетер через 15 мин, 30 мин, 45 мин, 1 ч, 1 ч 30 мин, 2 ч, 3 ч, 4 ч, 6 ч, 8 ч, 12 ч, 18 ч, 24 ч, 48 ч после приема лекарственного препарата. После отбора крови пробирки направляли на центрифугирование для отделения плазмы. Затем из данных образцах отбирали аликвоту объемом 50 мкл, переносили ее в пробирку, добавляли 450 мкл раствора внутреннего стандарта дейтерированной микофеноловой кислоты в концентрации 1,5 мкг/мл, затем пробирку закрывали, полученную смесь перемешивали на вортексе в течение 30 сек при частоте около 1800 мин-1, подвергали центрифугированию в течение 10 мин при 2500 об/мин. Хромато-масс-спектрометрическое определение концентрации микофеноловой кислоты в надосадочной жидкости проводили с использованием хроматографической колонки Phenomenex Kinetex C18 (30×4,6 мм, 2,6 мкм) при скорости потока 0,4 мл/мин и градиентном режиме элюирования. Для расчета концентрации микофеноловой кислоты использовался калибровочный график, уравнение которого: x=(y+0,00125)/0,05351, где y - соотношение площадей пиков «аналит-внутренний стандарт», x - измеренная концентрация микофеноловой кислоты в плазме. Полученные значения концентраций представлены в таблице 6.

Часть результатов определения концентраций микофеноловой кислоты в плазме была подтверждена с помощью способа-прототипа (Decavele А.С., Favoreel N., Heyden V. F. et. al., Clin. Chem. Lab. Med., V. 49, №7, pp. 1159-1165, 2011). Так, концентрация аналита через 1 час после отбора составила 5,61 мкг/мл - расхождение составило 1,4%, через 8 часов - 5,40 мкг/мл - расхождение составило 1,3%, что укладывается в допустимый диапазон точности: ±15% от измеренной концентрации.
Предлагаемый способ определения концентрации микофеноловой кислоты в плазме крови применен в лаборатории ООО «Квинта-Аналитика Ярославль».
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИИ МЕТИЛДОПЫ В ПЛАЗМЕ КРОВИ ЧЕЛОВЕКА | 2016 |
|
RU2642593C1 |
| Способ определения амиодарона и его основного метаболита дезэтиламиодарона в сыворотке крови человека | 2020 |
|
RU2749566C1 |
| Способ количественного определения леводопы в плазме крови | 2017 |
|
RU2665164C1 |
| Способ определения топирамата в плазме крови | 2016 |
|
RU2631613C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КАРНОЗИНА В БИОЛОГИЧЕСКИХ МАТЕРИАЛАХ | 2015 |
|
RU2585115C1 |
| Способ количественного определения окадаиковой кислоты в морепродуктах | 2017 |
|
RU2666247C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОКСИМА ПИНОСТРОБИНА В ПЛАЗМЕ КРОВИ | 2015 |
|
RU2568876C1 |
| Способ количественного определения амантадина в плазме крови | 2017 |
|
RU2650968C1 |
| Способ контроля содержания противотуберкулёзных препаратов основного ряда и их токсичных метаболитов в плазме крови | 2018 |
|
RU2702998C1 |
| Способ количественного определения салицилатов в плазме крови | 2016 |
|
RU2622996C1 |
Изобретение относится к фармации, а именно к фармацевтической химии.
Способ определения концентрации микофеноловой кислоты в плазме крови человека отличается тем, что хроматографическое разделение компонентов матрицы проводят с использованием хроматографической колонки Phenomenex Kinetex C18 (30×4,6 мм, 2,6 мкм) при скорости потока 0,4 мл/мин и следующих условиях градиентного элюирования: сначала анализа и до 1 мин анализа содержание ацетонитрила в подвижной фазе составляет 40%, содержание воды - 60%; с 1 мин до 1,5 мин анализа содержание ацетонитрила линейно повышается до 65%, содержание воды линейно понижается до 35%; с 1,5 мин до 2,0 мин анализа содержание ацетонитрила линейно повышается до 90%, содержание воды линейно понижается до 10%; с 2,0 мин до 2,5 мин анализа содержание ацетонитрила составляет 90%, содержание воды - 10%; с 2,5 мин до 3,0 мин анализа содержание ацетонитрила линейно понижается до 65%, содержание воды линейно повышается до 35%; с 3,0 мин до 3,5 мин анализа содержание ацетонитрила линейно понижается до 40%, содержание воды линейно повышается до 60%; с 3,5 мин до конца анализа содержание ацетонитрила составляет 40%, содержание воды - 60%. 6 табл., 1 пр.
Способ определения концентрации микофеноловой кислоты в плазме крови человека, заключающийся в том, что из плазмы отбирают аликвоту, подвергают ее депротеинизации раствором внутреннего стандарта дейтерированной микофеноловой кислоты в ацетонитриле, полученную смесь центрифугируют, в надосадочной жидкости измеряют концентрацию микофеноловой кислоты методом высокоэффективной жидкостной хроматографии с тандемным масс-спектрометрическим детектированием в диапазоне от 0,5 до 30 мкг/мл по калибровочному графику, отличающийся тем, что хроматографическое разделение компонентов матрицы проводят с использованием хроматографической колонки Phenomenex Kinetex C18 (30×4,6 мм, 2,6 мкм) при скорости потока 0,4 мл/мин и следующих условиях градиентного элюирования:
сначала анализа и до 1 мин анализа содержание ацетонитрила в подвижной фазе составляет 40%, содержание воды - 60%;
с 1 мин до 1,5 мин анализа содержание ацетонитрила линейно повышается до 65%, содержание воды линейно понижается до 35%;
с 1,5 мин до 2,0 мин анализа содержание ацетонитрила линейно повышается до 90%, содержание воды линейно понижается до 10%;
с 2,0 мин до 2,5 мин анализа содержание ацетонитрила составляет 90%, содержание воды - 10%;
с 2,5 мин до 3,0 мин анализа содержание ацетонитрила линейно понижается до 65%, содержание воды линейно повышается до 35%;
с 3,0 мин до 3,5 мин анализа содержание ацетонитрила линейно понижается до 40%, содержание воды линейно повышается до 60%;
с 3,5 мин до конца анализа содержание ацетонитрила составляет 40%, содержание воды - 60%.
| A.C | |||
| Decavele et al | |||
| Способ фотографической записи звуковых колебаний | 1922 |
|
SU400A1 |
| V.Upadhyay et al | |||
| Determinationofmycophenolicacidinhuman plasma byultraperformanceliquid chromatographytandemmassspectrometry /Journal ofPharmaceuticalAnalysis, 2014; 4(3), pages 205-216 | |||
| P.Džodić et al | |||
| VALIDATION OF HPLC METHOD FOR THE DETERMINATION OF MYCOPHENOLIC ACID IN HUMAN PLASMA OBTAINED FROM RENAL TRANSPLANT RECIPIENTS / Acta Medica Medianae, 2016, vol.55(4), pages 28-36 | |||
| И.Г.Зенкевич и др | |||
| ОСОБЕННОСТИ ВЭЖХ-МС ОПРЕДЕЛЕНИЯ МОНОЭТАНОЛАМИНА В ВОДНЫХ РАСТВОРАХ МЕТОДОМ СТАНДАРТНОЙ ДОБАВКИ / Аналитика и контроль, 2012, т | |||
| Устройство для электрической сигнализации | 1918 |
|
SU16A1 |
| Водяные лыжи | 1919 |
|
SU181A1 |

