Настоящее изобретение относится к фармации, фармакологии и клинической фармакологии, исследованию биологических материалов, в частности к способам определения амиодарона, его основного метаболита дезэтиламиодарона в сыворотке крови человека методом высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием (ВЭЖХ-МС/МС) при терапевтическом лекарственном мониторинге и исследованиях фармакокинетики.
Уровень техники
Амиодарон является самым эффективным из существующих антиаритмических препаратов. В клинической практике замена оригинального препарата амиодарона на воспроизведенные часто приводит к изменению концентрации действующего вещества и/или его активного метаболита в крови и серьезным клиническим последствиям, в виде аритмогенных проявлений. Определение концентрации амиодарона и его активного метаболита в крови важно для обеспечения эффективности и безопасности применения воспроизведенных препаратов амиодарона. При этом отсутствует простой эффективный, чувствительный, селективный, точный и воспроизводимый способ пробоподготовки для определения амиодарона и его активного метаболита дезэтиламиодарона в сыворотке крови человека.
Из уровня техники известен способ определения амиодарона и дезэтиламиодарона в сыворотке крови человека методом высокоэффективной жидкостной хроматографии с хемилюминесцентным детектированием [1]. В данной работе к образцам сыворотки крови (1000 мкл) добавляют ацетонитрил в качестве осадителя белков (2000 мкл), затем встряхивают и центрифугируют, после чего надосадочную жидкость отбирают и высушивают в токе азота, сухой остаток растворяют в 250 мкл метанола. Хроматографическое разделение проводят с использованием колонки Mediterranea С18, 4,6 мм × 150 мм, диаметр частиц сорбента 5 мкм (Teknokroma), элюирование осуществляют в изократическом режиме подвижной фазой состоящей из смеси метанол: 0,017 моль/л раствор аммония сульфата рН 6,8 (92:8, v/v). Для детектирования используется оригинальная установка, позволяющая проводить постколоночную дериватизацию образца и измерять хемилюминесценцию при участии комплексных соединений рутения.
Недостатками данного способа являются:
- использование специфического оборудования для хемилюминесцентного детектирования;
- используется редкий и дорогостоящий реагент для постколоночной дериватизации (комплексное соединение рутения);
- использование изократического режима элюирования удлиняет время анализа проб до 12 минут;
- недостаточный нижний предел количественного определения (НПКО) дезэтиламиодарона (0,5 мкг/мл).
В другом известном способе определение амиодарона в плазме крови человека проводят методом высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием (ВЭЖХ МС/МС) [3]. Пробоподготовку осуществляют методом жидкость-жидкостной экстракции (ЖЖЭ) диэтиловым эфиром с последующим упариванием отобранного органического слоя и перерастворением сухого остатка в метаноле. Хроматографическое разделение проводится в режиме изокритического элюирования на колонке Hypersil GOLD, 4,5 мм × 50 мм, диаметр частиц сорбента 1,9 мкм с масс-спектрометрическим детектором. Подвижная фаза состояла из двух растворов: 10 мМ ацетат аммония (раствор А) и ацетонитрил - 10 мМ ацетат аммония (90:10) (раствор Б), взятых в соотношении 10%:90% соответственно.
К недостаткам этого метода следует отнести:
- невозможность определения дезэтиламиодарона совместно с амиодароном;
- использование диэтилового эфира в пробоподготовке;
- изократическое элюирование, увеличивающее время анализа до 8 минут;
- необходимость оборудования, обеспечивающего работу при сверхвысоком давлении, из-за использования сорбента с диаметром частиц 1,8 мкм.
В качестве прототипа выбран известный способ определения амиодарона и его метаболита в плазме и сыворотке крови человека [2] предложена схема пробоподготовки методом твердофазной экстракции (ТФЭ). Хроматографическое разделение проводится в режиме градиентного элюирования с подвижной фазой А (раствор 0,1% муравьиной кислоты и 2 ммоль/л аммония ацетата в воде)/ В (раствор 0,1% муравьиной кислоты и 2 ммоль/л аммония ацетата в метаноле) на колонке Phenomenex Hydro-RP 2×20 мм с диаметром частиц сорбента 2,5 мкм. Амиодарон и его метаболит определяли с помощью масс-спектрометрического детектора в режиме мониторинга множественных реакций (multiple reaction monitoring - MRM).
К недостаткам прототипа следует отнести:
- использование дорогостоящего и времязатратного метода пробоподготовки -твердофазной экстракции;
- избыточное количество калибровочных образцов и необоснованно широкий аналитический диапазон до 40 мкг/мл, тогда как в плазме крови обычно определяются концентрации амиодарона и дезэтиламиодарона на порядок ниже;
- использование дорогостоящих дейтерированных аналогов амиодарона и дезэтиламиодарона в роли внутренних стандартов;
- недостаточное хроматографическое разделение аналитов.
Таким образом, существует потребность в разработке простого, эффективного, чувствительного, селективного, точного и воспроизводимого способа для определения амиодарона и его активного метаболита дезэтиламиодарона в сыворотке крови методом высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием.
Сущность изобретения
Технической задачей заявленного изобретения является разработка простого способа пробоподготовки с отказом от применения ЖЖЭ, ТФЭ и специфического оборудования, для совместного определения амиодарона и дезэтиламиодарона в диапазоне, соответствующем средним терапевтическим концентрациям, достижения высокой эффективности, точности и воспроизводимости при определении амиодарона, его активного метаболита дезэтиламиодарона, применение единого внутреннего стандарта, а также расширение спектра аналитических методик, пригодных для количественного определения изучаемых веществ.
Техническим решением предлагаемого изобретения является то, что заявленный способ характеризуется эффективностью, селективностью, чувствительностью, точностью, с широким линейным диапазоном, позволяющим определять минимальные количества анализируемых веществ при совместном присутствии в сыворотке крови человека.
Кроме того, настоящее техническое решение - является эффективным для применения в области клинической фармакологии для проведения терапевтического лекарственного мониторинга амиодарона и дезэтиламиодарона путем исследования сыворотки крови пациента.
Технический результат предложенного решения достигается благодаря тому, что:
- используют унифицированную схему пробоподготовки образцов для всех анализируемых веществ с использованием осадителя белков, представляющий собой неразбавленный ацетонитрил;
- применяют цинакальцет, в качестве единого внутреннего стандарта для всех анализируемых веществ при определении их в сыворотке крови, имеющего сходные с анализируемыми веществами физико-химические свойства и, как следствие, удерживание на сорбенте;
применяют градиентное элюирование адаптировано для определения высоколипофильньгх соединений за короткое время анализа без ущерба для эффективности хроматографического разделения;
- проводят хроматографическое разделение на сорбенте с диаметром частиц 5 мкм с целью снижения рабочего давления, что позволяет расширить спектр аналитических методик, пригодных для количественного определения амиодарона и дезэтиламиодарона в сыворотке крови;
- при проведении пробоподготовки центрифугируют пробу один раз;
- детектируют при положительной ионизации анализируемых компонентов.
Описание изобретения.
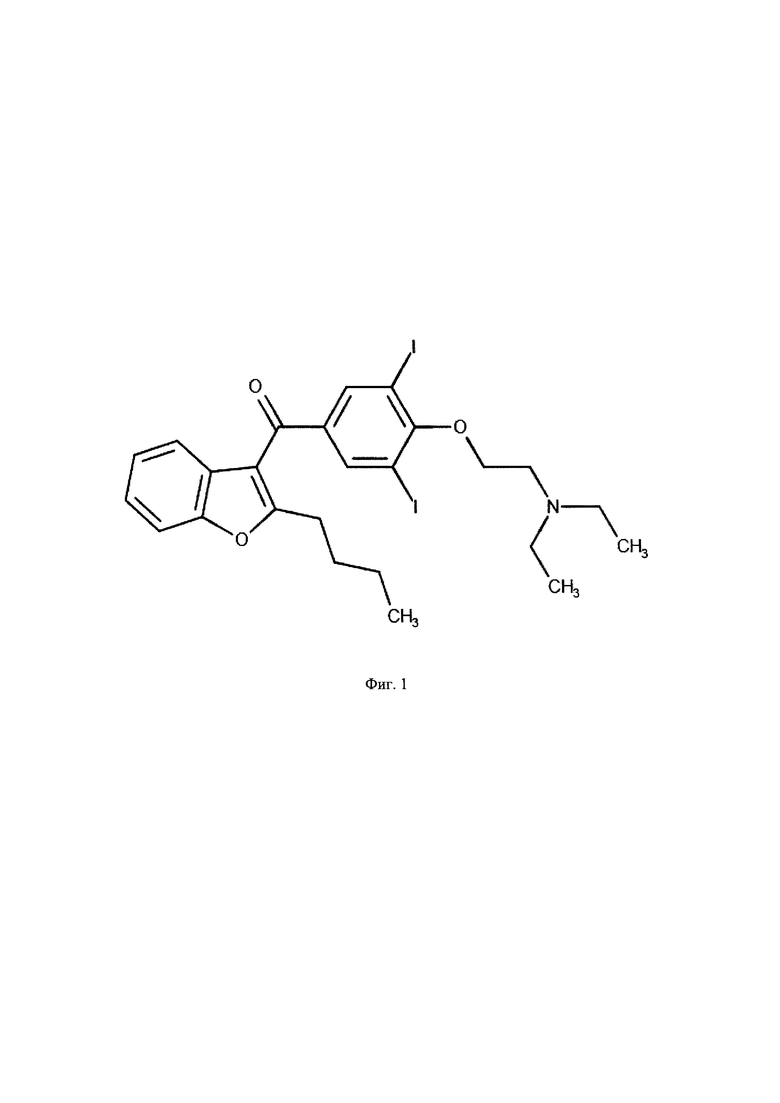
В заявленном способе для определения амиодарона и дезэтиламиодарона (Фиг. 1-2) при их совместном присутствии в сыворотке крови человека осуществляется методом высокоэффективной жидкостной хроматографии (далее - ВЭЖХ МС/МС). Для определения концентраций требуется приготовление набора стандартных растворов определяемых веществ, то есть, растворов с точно известной концентрацией, для дальнейшего приготовления из них калибровочных растворов, которые необходимы для построения калибровочной кривой, по которой определяются концентрации аналитов исследуемых образцов.
Калибровочные образцы готовят путем прибавления к 195 мкл интактной сыворотки крови 5 мкл стандартных растворов амиодарона и дезэтиламиодарона до получения концентраций в диапазоне от 30 нг/мл до 3000 нг/мл.
Пробоподготовку калибровочных и анализируемых образцов сыворотки крови человека проводят методом осаждения белков следующим образом: к 200 мл биологического образца, помещенным в центрифужные микропробирки типа «эппендорф» вместимостью 2 мл, прибавляют 10 мкл рабочего раствора внутреннего стандарта цинакальцета с концентрацией 500,00 нг/мл, затем прибавляют 1000 мкл неразбавленного ацетонитрила от 99,9% мас. до 100% мас.; перемешивают в смесителе типа «вортекс» в течение не менее 15 с до однородного состояния (до полного смешивания), далее центрифугируют до разделения содержимого на твердую и жидкую фазы, с центробежным ускорением 15294 g в течение от 10 минут до 20 минут, предпочтительнее в течение 15 минут. Затем надосадочную жидкость (супернатант), в объеме (количестве) 500 мкл, переносят в хроматографические виалы, далее помещают в автоматический пробоотборник хроматографа.
Неожиданно было обнаружено, что для определения амиодарона и его метаболита дезэтиламиодарона в сыворотке крови, целесообразно использовать неразбавленный ацетонитрил в качестве осадителя белков, который позволяет эффективно извлекать наибольшее количество аналитов анализируемых веществ из сыворотки крови человека.
Использование неразбавленного ацетонитрила, в качестве осадителя белков для определения анализируемой группы аналитов амиодарона и его метаболита дезэтиламиодарона, приводит к более эффективному, точному и воспроизводимому анализу, характеризующемуся способностью, наиболее эффективно дифференцировать анализируемую группу аналитов в сыворотке крови человека.
Также, неожиданно было обнаружено, что применение неразбавленного ацетонитрила, в качестве осадителя белка, при совместно присутствующих в сыворотке крови человека амиодарона и его метаболита дезэтиламиодарона и раствора цинакальцета, в качестве внутреннего стандарта, приводят к уменьшению времени пробоподготовки, так как позволяют использовать единую пробоподготку для сыворотки крови человека, и таким образом позволяют экономить время при проведении анализа.
Кроме того, неожиданно было обнаружено, что настоящее техническое решение с заявленным техническим результатом - является эффективным для применения в области клинической фармакологии при проведении терапевтического лекарственного мониторинга путем исследования сыворотки крови пациента.
Условия хроматографического разделения и детектирования Количественное определение амиодарона и дезэтиламиодарона проводят на высокоэффективном жидкостном хроматографе, оснащенном градиентным насосом, термостатом колонок, дегазатором, термостатируемым автоматическим пробоотборником и тандемным масс-спектрометрическим детектором (тройным квадруполем). Обработку первичных данных проводят при помощи соответствующего программного обеспечения.
Колонка (неподвижная фаза): для проведения ВЭЖХ МС/МС выбрана колонка размером 150 × 2,1 мм с привитой фазой С18 из группы Zorbax и сорбентом с диаметром частиц 5 мкм, с универсальной предколонкой С 18,4 × 3,0 мм, что обеспечивает рабочее давление в системе не более 17 МПа.
Температура термостата колонок: 40°С.
Подвижная фаза:
- Элюент А: 0,1% (об.) раствор муравьиная кислота/деионизированная вода
- Элюент В: 0,1% (об.) раствор муравьиная кислота/ ацетонитрил
Скорость потока подвижной фазы: 0,75 мл/мин.
Градиентный режим элюирования по составу подвижной фазы и представлен в таблице 1.

Объем вводимой пробы: 1 мкл.
Время регистрации хроматограммы по масс-спектрометрическому детектору: 0-3,5 мин.
Время удерживания цинакальцета: 0,79±0,02 мин
Время удерживания амиодарона: 1,83±0,02 мин
Время удерживания дезэтиламиодарона: 1,38±0,02 мин
Параметры источника ионизации: распыляющий газ 3 л/мин, осушающий газ 20 л/мин, блок нагрева 400°С, линия десольвации 250°С, напряжение на капилляре +5 кВ.
Условия детектирования сигнала для амиодарона, его основного метаболита дезэтиламиодарона и внутреннего стандарта, в виде раствора цинакальцета, в режиме мониторинга множественных реакций представлены в таблице 2.


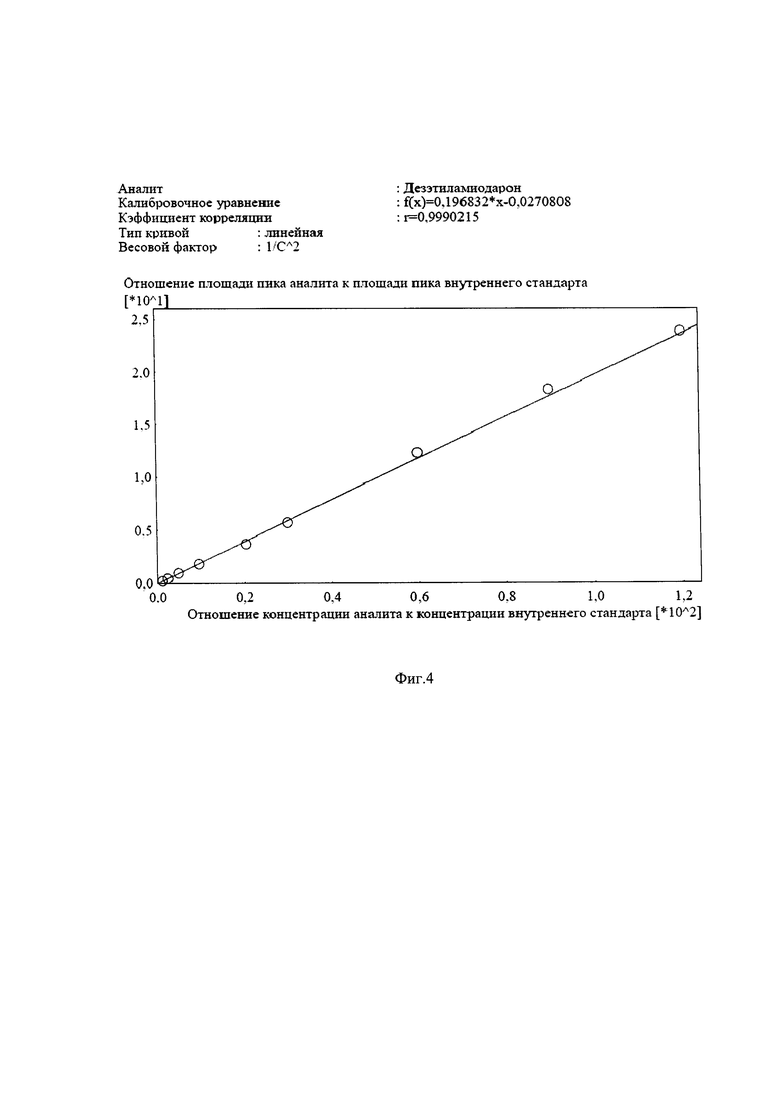
Полученные калибровочные кривые имеют линейный характер в диапазоне концентраций, для каждого анализируемого активного вещества от 30 нг/мл до 3000 нг/мл в сыворотке крови человека, с коэффициентами корреляции 0,997 и 0,999 по амиодарону и дезэтиламиодарону (Фиг. 3-4), соответственно. Точность результатов, под которой следует понимать правильность и прецизионность, рассчитываются в соответствии с требованиями Руководства по экспертизе лекарственных средств [4]. Правильность измерений (Е,%), не превышает 3,92% по амиодарону и не превышает 4,15% по дезэтиламиодарону. Относительное стандартное отклонение, позволяющее оценить прецизионность (RSD %) результатов, имеет значение не более 5,47% по амиодарону и не более 5,71% по дезэтиламиодарону.
Предложенный способ позволяет:
1. Отказаться от использования ЖЖЭ и ТФЭ при проведении пробоподготовки и использовать метод осаждения белков ацетонитрилом.
2. Получить аналитические диапазоны, для каждого анализируемого вещества от 30 нг/мл до 3000 нг/мл за счет унификации пробоподготовки.
3. Повысить эффективность чувствительность, селективность, точность и воспроизводимость предлагаемого способа для определения амиодарона и дезэтиламиодарона в сыворотке крови человека.
4. Использовать цинакальцет в качестве единого и доступного внутреннего стандарта.
5. Расширить спектр аналитических методик для определения амиодарона и дезэтиламиодарона в сыворотке крови человека за счет проведения хроматографического разделения на сорбенте с размером частиц 5 мкм, что обеспечивает рабочее давление в системе не более 17МПа при сохранении скорости анализа.
Краткое описание чертежей и иных материалов (Приложения 1-6).
Фиг. 1. Структурная формула амиодарона.
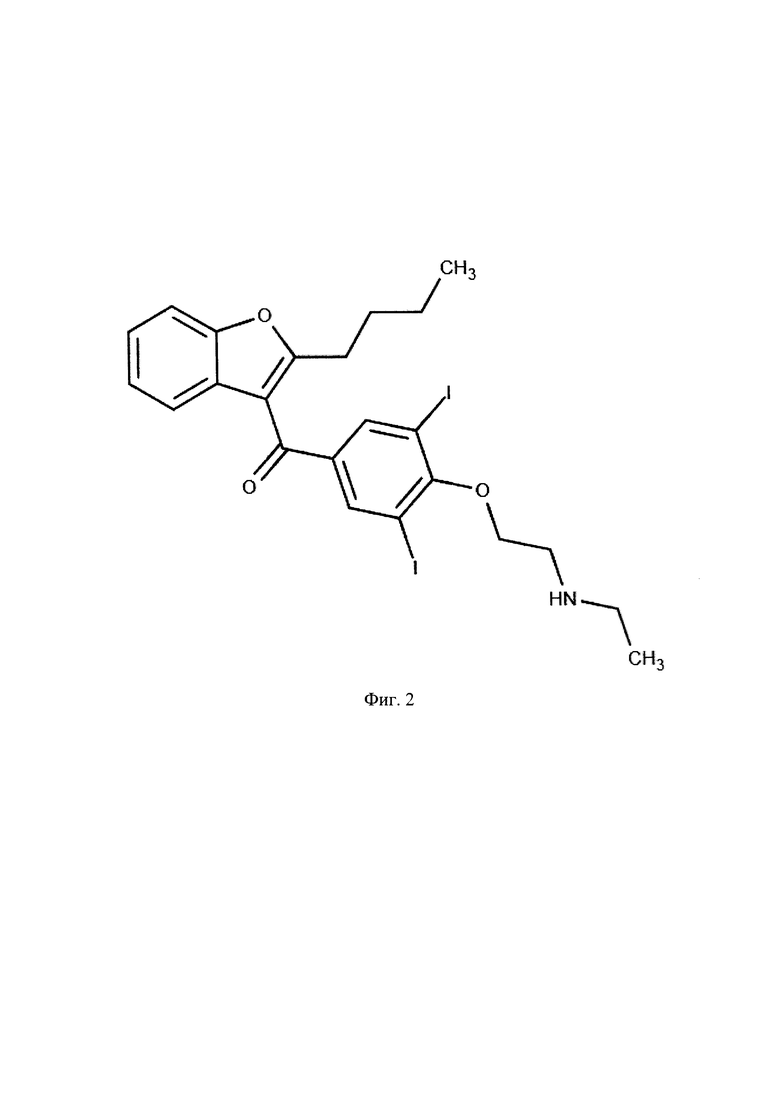
Фиг. 2. Структурная формула активного метаболита амиодарона дезэтиламиодарона.
Фиг. 3. Калибровочная зависимость отношения площади пика амиодарона к площади пика цинакальцета от отношения концентрации амиодарона к концентрации цинакальцета в сыворотке крови.
Фиг. 4. Калибровочная зависимость отношения площади пика дезэтиламиодарона к площади пика цинакальцета от отношения концентрации дезэтиламиодарона к концентрации цинакальцета в сыворотке крови.
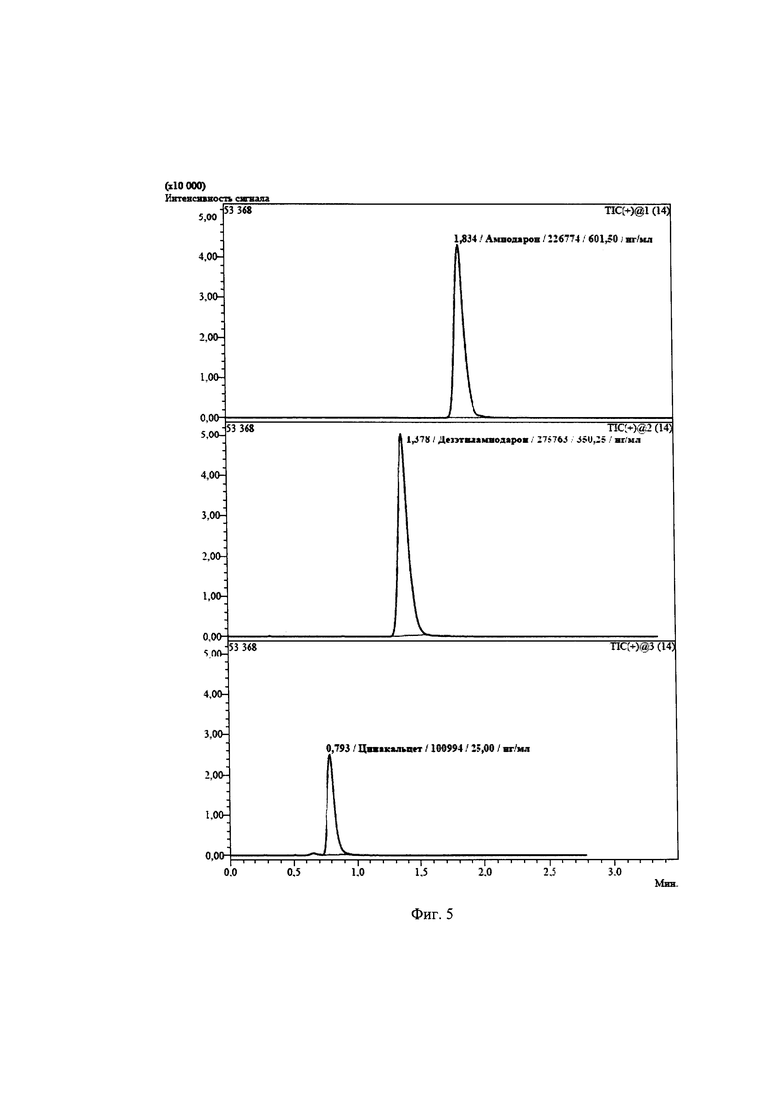
Фиг. 5. Хроматограмма образца сыворотки крови пациента с содержанием амиодарона 601,50 нг/мл, активного метаболита дезэтиламиодарона 350,25 нг/мл, и рабочего стандартного раствора цинакальцета в концентрации 25,0 нг/мл.
Фиг. 6. Хроматограмма образца интактной сыворотки крови человека не содержащего амиодарон, дезэтиламиодарон и цинакальцет.
Возможность осуществления заявляемого изобретения раскрыта в следующих примерах.
Пример 1. Определение содержания амиодарона и дезэтиламиодарона в сыворотке крови человека.
Пациент, включенный в исследование, находился на постоянной терапии амиодароном с терапевтической эффективной дозировкой 600 мг в сутки. Отбор крови производили непосредственно перед приемом амиодарона для определения значения минимальной стационарной концентрации амиодарона и дезэтиламиодарона в сыворотке крови человека.
Проведение анализа.
1. Получение сыворотки крови человека. Забор крови производят в вакуумные пробирки. Кровь, взятую у пациента, выдерживают при комнатной температуре в течение 30 мин. По истечении 30 минут пробирки центрифугируют не менее 15 мин при 3000 об/мин. Затем аликвоту полученной сыворотки крови переносят в пробирки типа Eppendorf Save Lock вместимостью 2 мл для дальнейшей пробоподготовки.
2. Калибровочные образцы готовят путем прибавления к интактной сыворотке крови 5 мкл стандартных растворов амиодарона и дезэтиламиодарона до получения концентраций в анализируемом диапазоне от 30 нг/мл до 3000 нг/мл.
3. Пробоподготовку калибровочных и анализируемых образцов сыворотки крови проводят методом осаждения белков следующим образом: к 200 мкл биологического образца, помещенным в центрифужные микропробирки типа «эппендорф» вместимостью 2 мл, прибавляют 10 мкл рабочего раствора внутреннего стандарта цинакальцета с концентрацией 500 нг/мл, затем прибавляют 1000 мкл неразбавленного ацетонитрила, перемешивают в смесителе типа «вортекс» до однородного состояния (полное смешивание) не менее 15 с, затем центрифугируют с центробежным ускорением 15294 g в течение от 10 минут до 20 минут, наиболее предпочтительно в течение 15 минут, до разделения содержимого на твердую и жидкую фазы. Затем 500 мкл переносят надосадочную жидкость в хроматографические виалы и помещают в автоматический пробоотборник хроматографа.
4. Количественное определение амиодарона и дезэтиламиодарона проводят на высокоэффективном жидкостном хроматографе, оснащенном градиентным насосом, термостатом колонок, дегазатором, термостатируемым автоматическим пробоотборником и тандемным масс-спектрометрическим детектором (тройным квадруполем). Обработку первичных данных проводят при помощи соответствующего программного обеспечения.
Колонка (неподвижная фаза): для проведения ВЭЖХ МС/МС выбрана колонка размером 150 × 2,1 мм с привитой фазой С18 из группы Zorbax и сорбентом с диаметром частиц 5 мкм, с универсальной предколонкой С18, 4 × 3,0 мм, что обеспечивает рабочее давление в системе, не превышающем 17 МПа.
Скорость потока подвижной фазы: 0,75 мл/мин.
Объем вводимой пробы: 1 мкл.
Время регистрации хроматограммы по масс-спектрометрическому детектору: 0-3,5 мин.
Время удерживания цинакальцета: 0,79±0,02 мин
Время удерживания амиодарона: 1,83±0,02 мин
Время удерживания дезэтиламиодарона: 1,38±0,02 мин
Параметры источника ионизации: распыляющий газ 3 л/мин, осушающий газ 20 л/мин, блок нагрева 400°С, линия десольвации 250°С, напряжение на капилляре +5 кВ.
Условия детектирования амиодарона, его основного метаболита дезэтиламиодарона и цинакальцета, взятого в виде внутреннего стандарта, в режиме мониторинга множественных реакций представлены в Таблице 3.
Обнаружено амиодарона 601,50 нг/мл и дезэтиламиодарона 350,25 нг/мл (Фиг. 5). От момента забора крови до предоставления результатов затрачивается не более 1 ч.
Данный пример демонстрирует возможность определения амиодарона и его активного метаболита дезэтиламиодарона, при совместном присутствии в сыворотке крови человека заявленным способом.
Пример 2. Определение содержания амиодарона и дезэтиламиодарона в интактной сыворотке крови.
Определение содержания амиодарона и дезэтиламиодарона в интактной сыворотке крови человека, достоверно не содержащей эти препараты, проводят способом, описанным в Примере 1.
На хроматограммах полученных образцов отсутствуют пики (Фиг. 6), соответствующие по времени удерживания амиодарону и дезэтиламиодарону.
Таким образом, данный пример демонстрирует эффективность, селективность заявленного способа и отсутствие риска предоставления ложноположительных результатов количественного определения амиодарона и дезэтиламиодарона в сыворотке крови.
Пример 3. Определение точности (правильности и прецизионности) результатов определения амиодарона и его метаболита дезэтиламиодарона в сыворотке крови человека. Оценку точности проводили в диапазоне от 30 нг/мг до 3000 нг/мл, путем анализа образцов контроля качества, содержащих амиодарон и дезэтиламиодарон на уровнях 30 нг/мл, 90 нг/мл, 900 нг/мл, 2100 нг/мл, 3000 нг/мл. Анализ проводили в рамках 3 циклов, включавших по 5 образцов для каждого уровня концентраций. Правильность и прецизионность определяли внутри аналитического цикла и между циклами (таблицы 3-5). Правильность измерений (Е,%), и относительное стандартное отклонение (RSD,%), позволяющее оценить прецизионность результатов, соответствовали нормам.
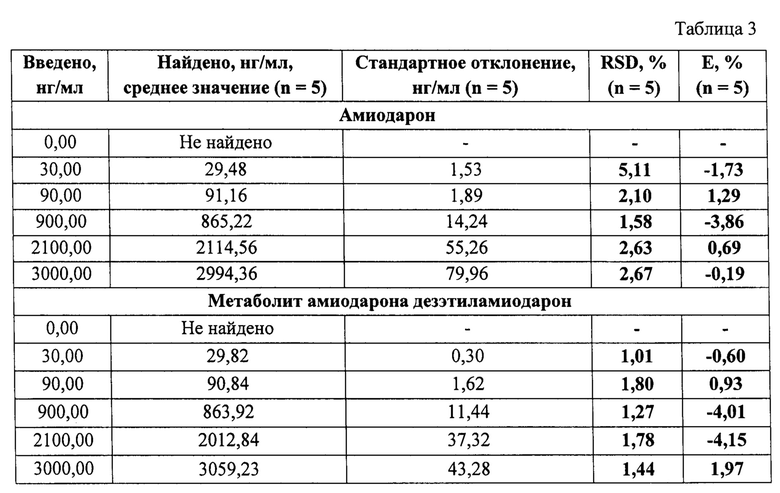

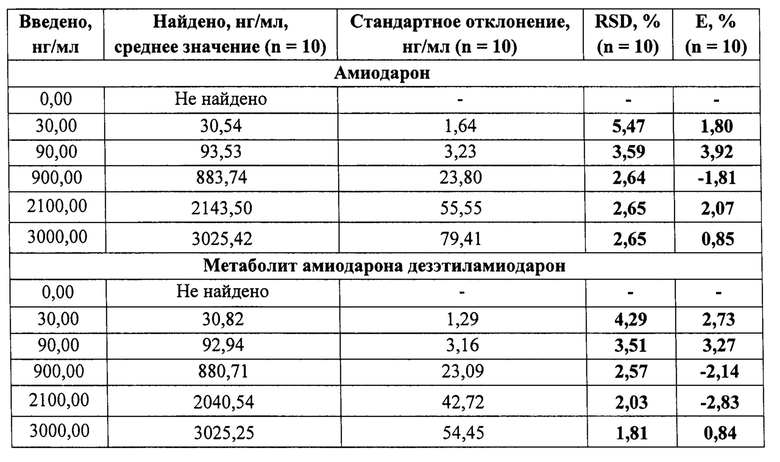
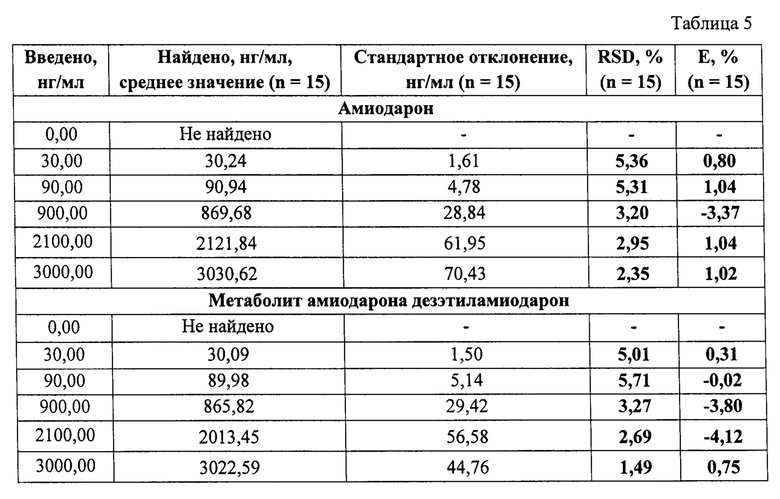
Данный пример раскрывает возможность определения амиодарона и дезэтиламиодарона в сыворотке крови человека с приемлемой точностью (правильностью и прецизионностью) в концентрациях от 30 нг/мл до 3000 нг/мл, что отвечает заявленным линейным аналитическим диапазонам.
Представленные примеры не ограничивают объем притязаний настоящего изобретения и служат только для цели иллюстрации. Промышленная применимость
Все приведенные примеры, подтверждают эффективность, чувствительность, селективность, точность и воспроизводимость заявленного способа.
Таким образом, поставленная техническая задача, а именно, разработка эффективного, чувствительного, селективного, точного и воспроизводимого заявленного способа для определения амиодарона и его метаболита в сыворотке крови, достигнута, что подтверждается приведенными примерами.
Применение заявленного способа в клинической фармакологии для проведения терапевтического лекарственного мониторинга путем исследования сыворотки крови пациента, является эффективным.
Список литературы
1. Perez-Ruiz Т., Martinez-Lozano С, Garcia-Martmez М. D. Simultaneous determination of amiodarone and its metabolite desethylamiodarone by high-performance liquid chromatography with chemiluminescent detection //Analytica chimica acta. - 2008. - T. 623. -№. l. -C. 89-95.
2. Абаимов Д. А. и др. Открытое, рандомизированное, перекрестное исследование сравнительной фармакокинетики и биоэквивалентности препаратов Сантодарон и Кордарон //Фармакокинетика и фармакодинамика. - 2012. - №. 2.
3. Kuhn J., Gotting С, Kleesiek К. Simultaneous measurement of amiodarone and desethylamiodarone in human plasma and serum by stable isotope dilution liquid chromatography-tandem mass spectrometry assay //Journal of pharmaceutical and biomedical analysis.-2010.-T. 51.-№. 1. - C. 210-216.
4. Руководство по экспертизе лекарственных средств. Под ред. проф. А.Н. Миронова. Том I. М.: Гриф и К, 2013. С. 201-207.
5. Руководство по валидации аналитических методик проведения испытаний лекарственных средств. Решение Коллегии Евразийского экономической комиссии от 17 июля 2018 №113.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ определения лозартана, его основного метаболита лозартан карбоновой кислоты и глибенкламида в сыворотке крови и моче человека | 2020 |
|
RU2749567C1 |
| Способ определения дабигатрана в сыворотке крови человека | 2018 |
|
RU2683032C1 |
| Способ количественного определения амиодарона в плазме крови человека | 2024 |
|
RU2835337C1 |
| Определение полисорбата 80 в биологических лекарственных препаратах | 2023 |
|
RU2812788C1 |
| СПОСОБ ИЗМЕРЕНИЯ КОНЦЕНТРАЦИИ ПОЛИГЛУТАМАТОВ МЕТОТРЕКСАТА МЕТОДОМ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ С МАСС-СПЕКТРОМЕТРИЧЕСКИМ ДЕТЕКТИРОВАНИЕМ (ВЭЖХ-МС-МС) | 2020 |
|
RU2752805C1 |
| Определение стабилизаторов углеводной природы в биологически активных препаратах | 2023 |
|
RU2816030C1 |
| Способ определения летучих компонентов в лекарственных препаратах | 2022 |
|
RU2790000C1 |
| Способ количественного определения антигипертензивных лекарственных веществ в плазме крови | 2022 |
|
RU2803887C1 |
| Способ количественного определения амантадина в плазме крови | 2017 |
|
RU2650968C1 |
| Способ контроля содержания противотуберкулёзных препаратов основного ряда и их токсичных метаболитов в плазме крови | 2018 |
|
RU2702998C1 |


Изобретение относится к медицине, а именно к фармакологии, и может быть использовано для пробоподготовки при определении амиодарона и его метаболита дезэтиламиодарона высокоэффективной жидкостной хроматографией с масс-спектрометрическим детектированием (ВЭЖХ-МС/МС) в сыворотке крови человека. В предварительно приготовленные калибровочные и анализируемые образцы, представляющие собой сыворотку крови человека, добавляют эффективное количество внутреннего стандарта, в виде раствора цинакальцета в концентрации 500 нг/мл. Далее добавляют эффективное количество осадителя белка, представляющего собой неразбавленный ацетонитрил. Затем перемешивают до однородного состояния, с последующим центрифугированием при центробежном ускорении 15294 g до разделения на твердую и жидкую фазы, с дальнейшим отбором эффективного количества супернатанта. Проводят хроматографическое разделение компонентов пробы в режиме градиентного элюирования с последующим детектированием сигнала при положительной ионизации для амиодарона, его метаболита дезэтиламиодарона и внутреннего стандарта цинакальцета. Данный способ применяют для определения амиодарона и его метаболита дезэтиламиодарона в сыворотке крови человека. Способ обеспечивает возможность осуществления пробоподготовки для определения амиодарона и дезэтиламиодарона при совместном присутствии в сыворотке крови человека за счет использования унифицированной схемы пробоподготовки образцов для всех анализируемых веществ с применением неразбавленного ацетонитрила, центрифугирования пробы один раз, применения цинакальцета, в качестве единого внутреннего стандарта для всех анализируемых веществ, применения градиентного элюирования. 2 н. и 7 з.п. ф-лы, 5 табл., 3 пр., 6 ил.
1. Способ пробоподготовки для определения амиодарона и его метаболита дезэтиламиодарона высокоэффективной жидкостной хроматографией с масс-спектрометрическим детектированием (ВЭЖХ-МС/МС) в сыворотке крови человека, характеризующийся тем, что в предварительно приготовленные калибровочные и анализируемые образцы, представляющие собой сыворотку крови человека, добавляют эффективное количество внутреннего стандарта, в виде раствора цинакальцета в концентрации 500 нг/мл; далее добавляют эффективное количество осадителя белка, представляющего собой неразбавленный ацетонитрил, затем перемешивают до однородного состояния, с последующим центрифугированием при центробежном ускорении 15294 g до разделения на твердую и жидкую фазы, с дальнейшим отбором эффективного количества супернатанта; с последующим хроматографическим разделением компонентов пробы в режиме градиентного элюирования, далее с последующим детектированием сигнала при положительной ионизации для амиодарона, его метаболита дезэтиламиодарона и внутреннего стандарта цинакальцета.
2. Способ по п. 1, дополнительно характеризующийся тем, что аналитический диапазон для амиодарона и его метаболита дезэтиламиодарона составляет от 30 нг/мл до 3000 нг/мл.
3. Способ по п. 1, дополнительно характеризующийся тем, что осаждают белки неразбавленным ацетонитрилом в соотношении к сыворотке крови 5:1.
4. Способ по п. 1, дополнительно характеризующийся тем, что предварительно подготовленная сыворотка крови содержит эффективное количество амиодарона и его метаболита дезэтиламиодарона.
5. Способ по п. 1, дополнительно характеризующийся тем, что используют сорбент из группы Zorbax с привитой фазой С 18 с размером частиц 5 мкм.
6. Способ по п. 1, дополнительно характеризующийся тем, что хроматографируют пробу при рабочем давлении не более 17 МПа при сохранении скорости анализа.
7. Способ по п. 1, дополнительно характеризующийся тем, что центрифугируют в течение 15 минут.
8. Способ по п. 1, дополнительно характеризующийся тем, что детектированный сигнал получают с помощью масс-спектрометрического детектора в режиме мониторинга множественных реакций.
9. Применение способа по п. 1 для определения амиодарона и его метаболита дезэтиламиодарона в сыворотке крови человека.
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ АМИОДАРОНА | 2009 |
|
RU2413937C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ АМИОДАРОНА (КОРДАРОНА) МЕТОДОМ ИНВЕРСИОННОЙ ВОЛЬТАМПЕРОМЕТРИИ | 2003 |
|
RU2246722C1 |
| АБАИМОВ Д.А | |||
| и др | |||
| Открытое, рандомизированное, перекрёстное исследование сравнительной фармакокинетики и биоэквивалентности препаратов Сантодарон и Кордарон | |||
| Фармакокинетика и фармакодинамика | |||
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| BUNSE C | |||
| et al | |||
| Fully Automated online sample preparation and quanti¬ cation | |||

