Область техники
Настоящее изобретение относится к способу получения бромида умеклидиния и к способам получения промежуточных соединений, используемых при получении бромида умеклидиния.
Уровень техники
В международной патентной публикации WO 2005/104745 (Glaxo Group Limited), поданной 27 апреля 2005, описаны антагонисты мускариновых ацетилхолиновых рецепторов. В частности, в WO 2005/104745 описан 4-[гидрокси(дифенил)метил]-1-{2-[(фенил метил)окси]этил}-1-азонийбицикло[2.2.2]октан бромид формулы (I) и способ получения этого соединения (пример 84):
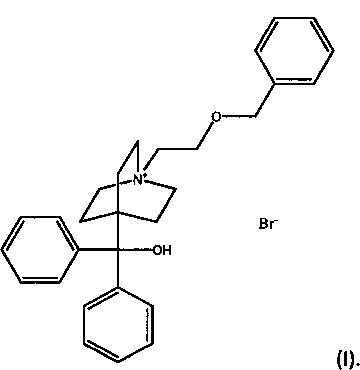
4-[гидрокси(дифенил)метил]-1-{2-[(фенилметил)окси]этил}-1-азонийбицикло[2.2.2]октан бромид также может упоминаться как бромид умеклидиния.
В международной патентной публикации WO 2011/029896 (Glaxo Group Limited), поданной 10 сентября 2010, описано альтернативное получение начального промежуточного соединения - этил-1-азабицикло[2.2.2]октан-4-карбоксилата - для многостадийного синтеза бромида умеклидиния.
Существует потребность в альтернативном способе получения бромида умеклидиния. В частности, необходим способ, который дает преимущества по сравнению с ранее описанными в WO 2005/104745 и WO 2011/029896 способами. Преимущества могут включать в себя, но не быть ограниченными следующими преимуществами: улучшение безопасности, контроля (т.е. конечная форма продукта и физические характеристики), выхода, эксплуатационных качеств, манипулирования, масштабируемости и эффективности.
Сущность изобретения
Настоящее изобретение относится, согласно первому аспекту, к способу получения бромида умеклидиния, включающему:
a) взаимодействие ((2-бромэтокси)метил)бензола формулы (II)
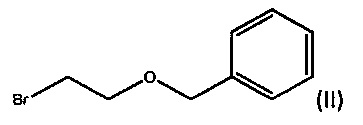
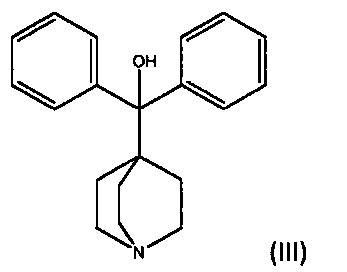
с дифенил(хинуклидин-4-ил)метанолом формулы III
в биполярном апротонном растворителе с температурой кипения более чем примерно 90°C или спирте с температурой кипения выше чем примерно 80°C; и возможно
b) перекристаллизацию продукта, полученного на стадии (а).
Настоящее изобретение дополнительно относится к промежуточным соединениям, используемым при получении соединения формулы (III), и, следовательно, бромида умеклидиния. Описанный способ обеспечивает ряд преимуществ по сравнению с известными способами, описанными в WO 2005/104745 и WO 2011/029896.
Краткое описание чертежей
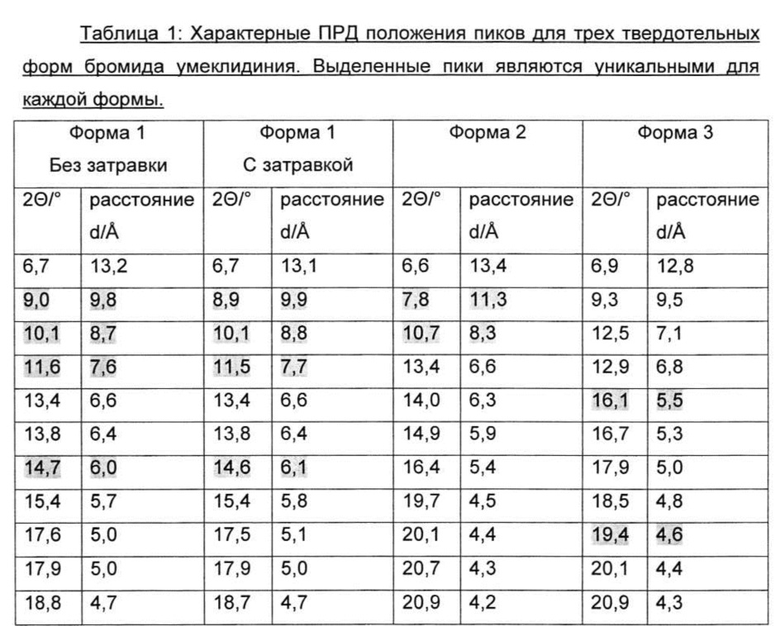
Фиг. 1: данные порошковой рентгеновской дифракции (ПРД) кристаллической формы 1 бромида умеклидиния, полученного в примере 8.
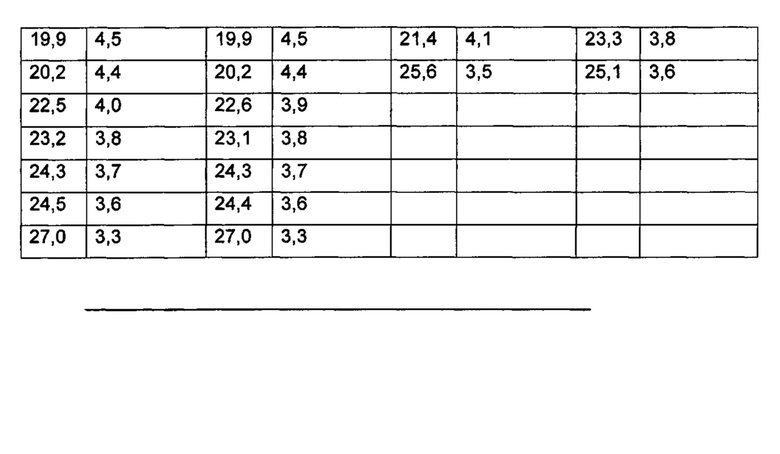
Фиг. 2: данные ПРД кристаллической формы 1 бромида умеклидиния, полученного в примере 7.
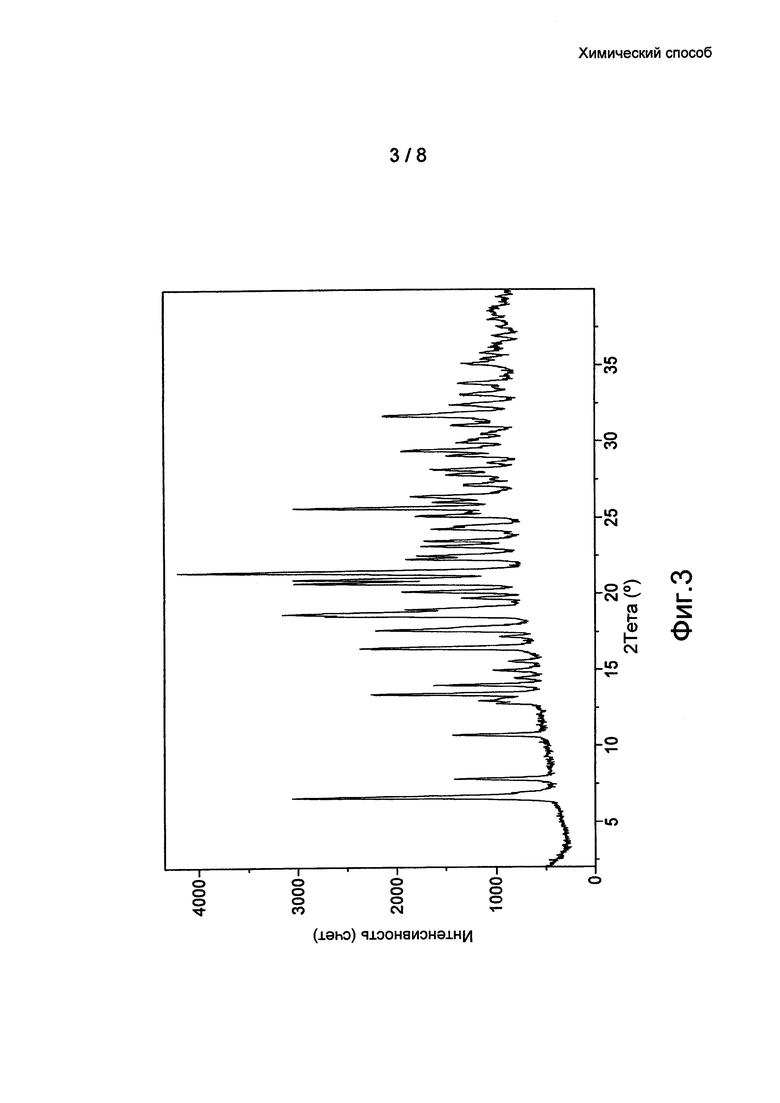
Фиг. 3: данные ПРД кристаллической формы 2 бромида умеклидиния, полученного в примере 9.
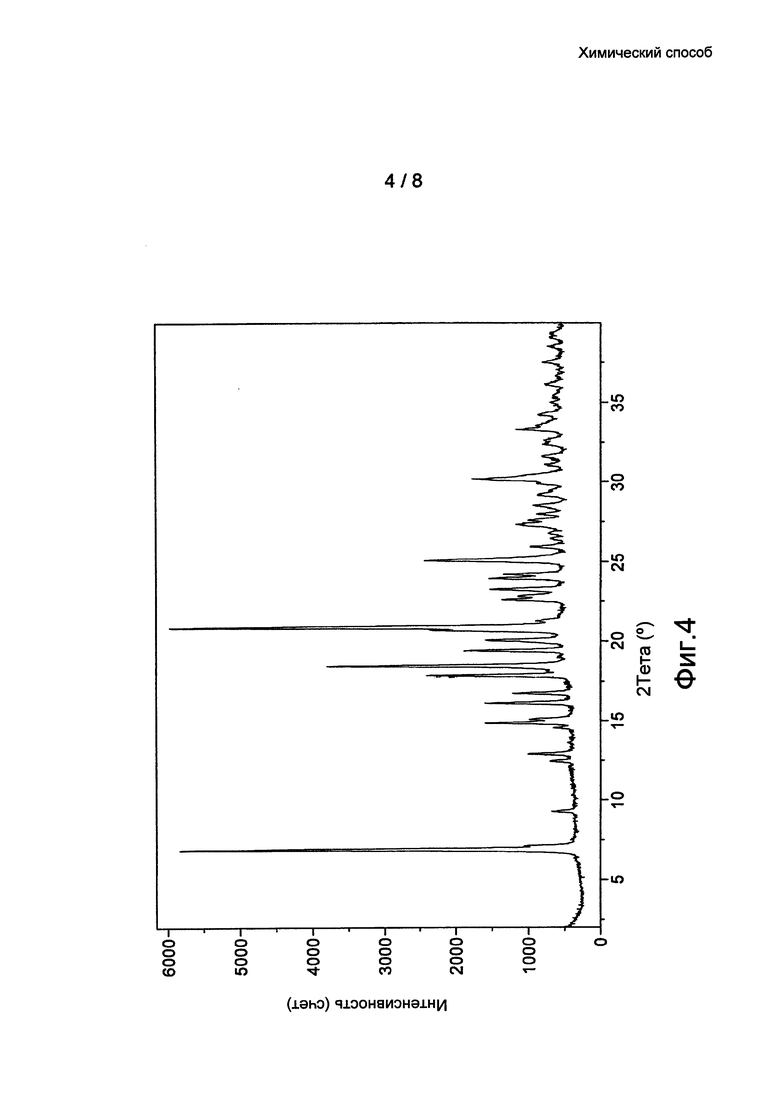
Фиг. 4: данные ПРД кристаллической формы 3 бромида умеклидиния, полученного в примере 10.
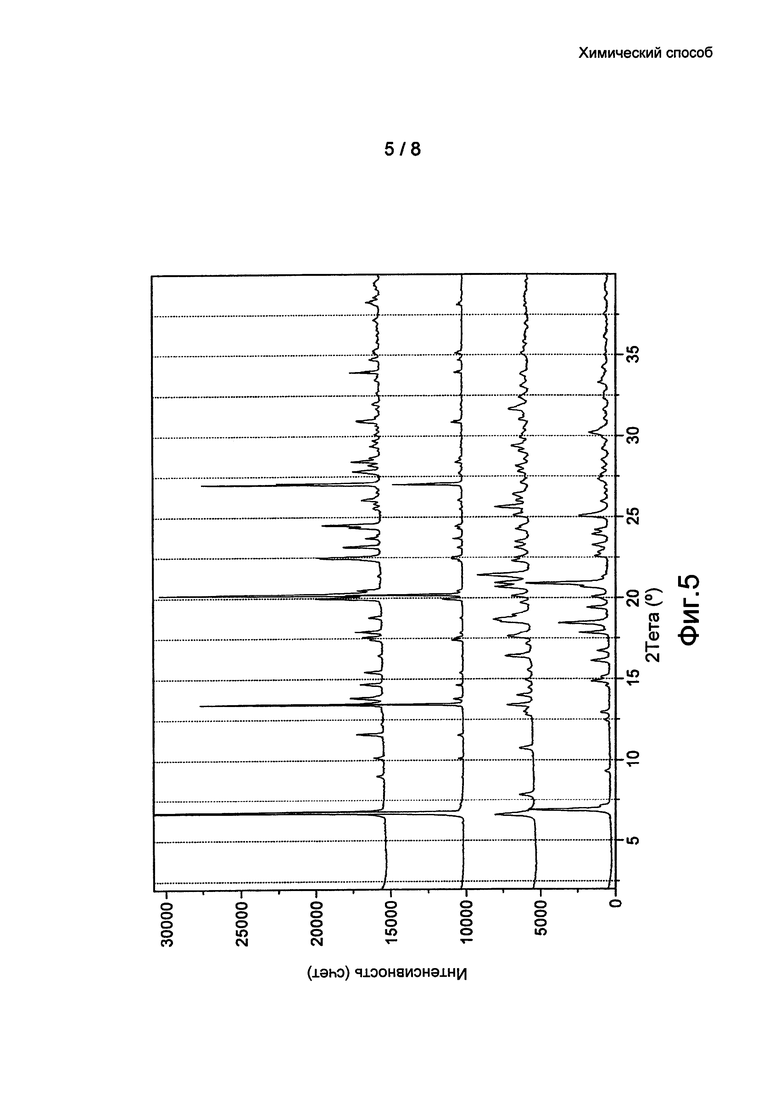
Фиг. 5: Наложение данных ПРД кристаллических форм 1 (с посевом и без) 2 и 3 бромида умеклидиния, полученного в примерах 7-10.
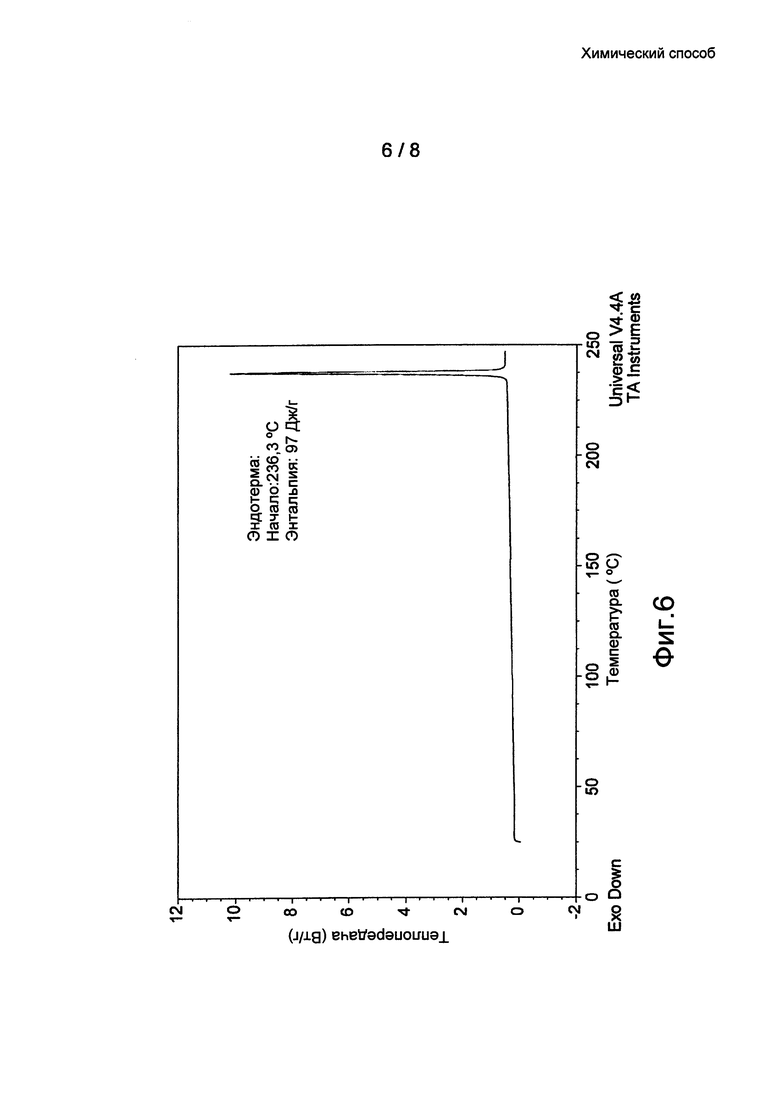
Фиг. 6: термограмма дифференциальной сканирующей калориметрии (ДСК) кристаллической формы 1 бромида умеклидиния, полученного в примере 8.
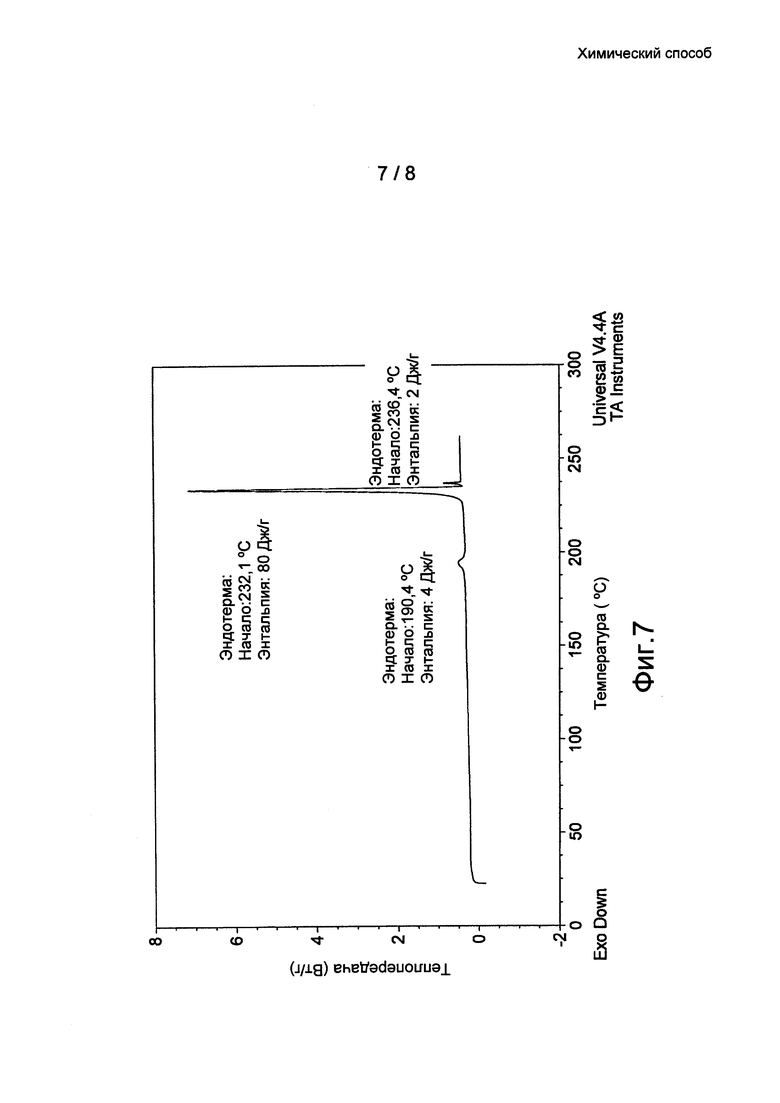
Фиг. 7: термограмма ДСК кристаллической формы 2 бромида умеклидиния, полученного в примере 9.
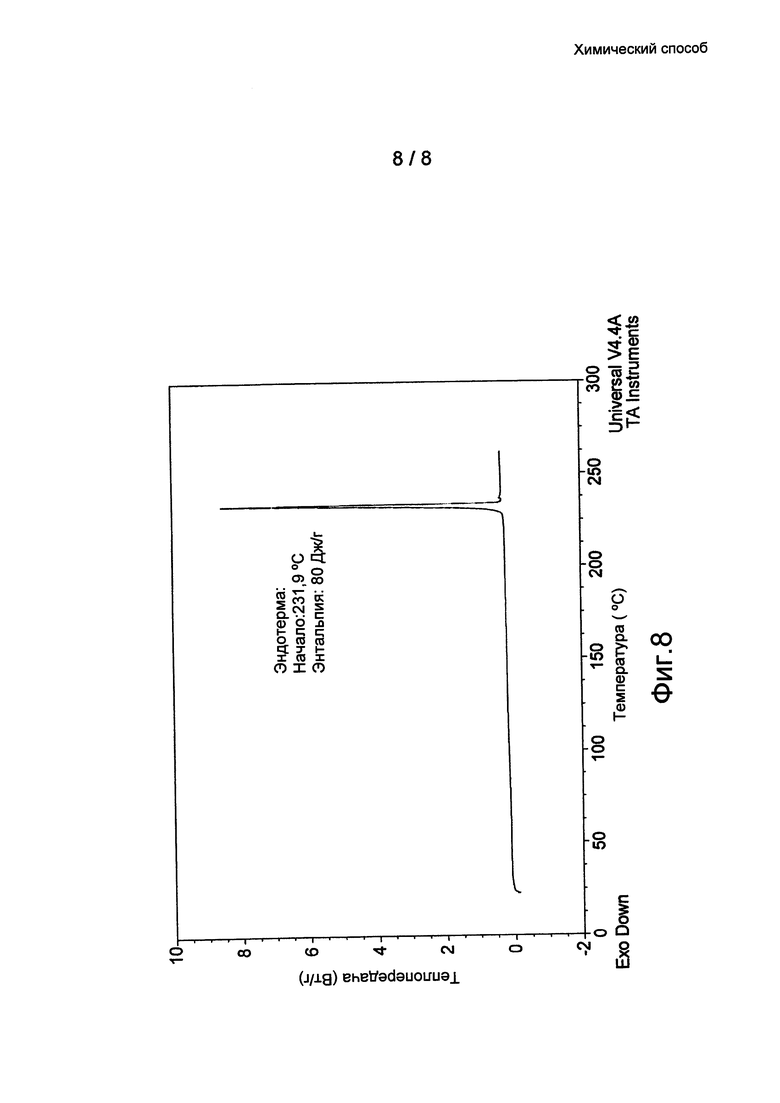
Фиг. 8: термограмма ДСК кристаллической формы 3 бромида умеклидиния, полученного в примере 10.
Подробное описание изобретения
Настоящее изобретение относится, согласно первому аспекту, к способу получения бромида умеклидиния, включающему:
с) взаимодействие ((2-бромэтокси)метил)бензола формулы (II)
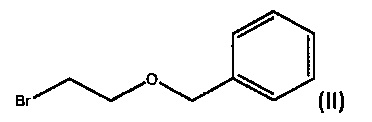
с дифенил(хинуклидин-4-ил)метанолом формулы III
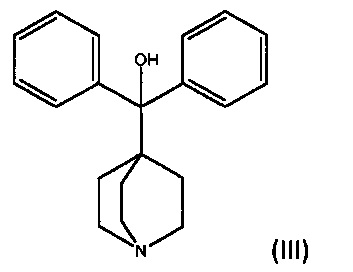
в биполярном апротонном растворителе с температурой кипения более чем примерно 90°C или спирте с температурой кипения выше чем примерно 80°C; и возможно
d) перекристаллизацию продукта, полученного на стадии (а).
Согласно одному аспекту, спирт, используемый при получении бромида умеклидиния, может быть, например, любым изомером пропанола или бутанола, например, н-пропанолом. Альтернативно, реакцию можно проводить в присутствии биполярного апротонного растворителя, имеющего температуру кипения выше чем 90°C, включая ДМФА, ДМА, ДМСО или НМП, но не ограничиваясь ими.
Диапазон температур, при которых осуществляют стадию (а), может быть определен на основе растворимости соединения формулы (III) в выбранном растворителе и температурой кипения указанного растворителя. Например, реакцию в н-пропаноле можно осуществлять между приблизительно 60°C и 97°C.
Бромид умеклидиния получают в виде несольватированной формы для лечения респираторных заболеваний, таких как астма и ХОБЛ. Следовательно, необходим эффективный, коммерчески выгодный способ получения этой формы, которая оказалась сложной в получении из-за способности соединения образовывать сольваты. На сегодняшний день было идентифицировано множество сольватов бромида умеклидиния, включая сольваты с метанолом, этанолом, изо-пропанолом, изо-бутанолом, хлорбензолом и п-ксилолом. Неожиданно было обнаружено, что при взаимодействии соединений формул (II) и (III) в н-пропаноле сводится к минимуму риск образования сольвата и, таким образом, отпадает необходимость в повторном суспендировании в этилацетате, метаноле и воде, которое ранее было необходимо (Пример 84, способ В, WO 2005/104745).
Проведение реакции в н-пропаноле также приводит к значительно более высокой скорости превращения по сравнению со способом предшествующего уровня техники WO 2005/104745 (43,3% против ~90% в примере 5), и является более безопасным благодаря отсутствию хлороформа и ацетонитрила (пример 84, WO 2005/104745). Кроме того, время реакции также было значительно снижено (с 16 часов до 3 часов в н-пропаноле).
Продукт, полученный на стадии (а), может быть перекристаллизован (стадия (b)) с использованием стандартных методик, известных в данной области техники, например, кристаллизация при охлаждении или кристаллизация при добавлении антирастворителя. При кристаллизации при охлаждении реакционную смесь, содержащую раствор продукта, полученного на стадии (а), медленно охлаждают и возможно с затравкой, что приводит к образованию кристаллов бромида умеклидиния, которые будут отделяться от раствора.
Неожиданно оказалось, что использование растворяющей смеси н-пропанола с водой для перекристаллизации (при кристаллизации при охлаждении) позволяет надежно контролировать конечную несольватированную форму и физические свойства бромида умеклидиния. По другому аспекту настоящего изобретения перекристаллизацию проводят в водном растворе н-пропанола, в котором отношение воды к н-пропанолу составляет 2:1.
После кристаллизации кристаллы могут быть выделены путем фильтрации, промывки с использованием подходящего растворителя, такого как охлажденный н-пропанол, и высушены в вакууме.
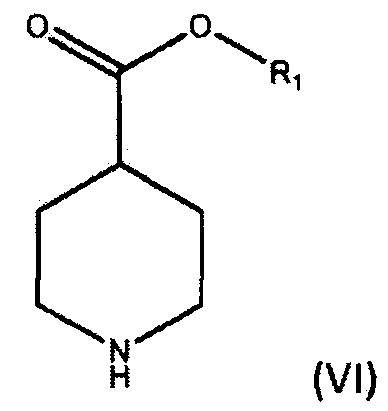
По дополнительному аспекту настоящее изобретение относится к способу получения соединения формулы (III), который включает взаимодействие фениллития в дибутиловом эфире с соединением формулы (IV).
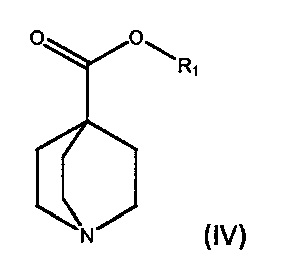
где R1 является С1-6алкилом, арилом или бензилом, в подходящем растворителе.
По дополнительному аспекту настоящее изобретение относится к способу получения бромида умеклидиния, который включает получение соединения формулы (III), включая стадию взаимодействия фениллития в дибутиловом эфире с соединением формулы (IV),
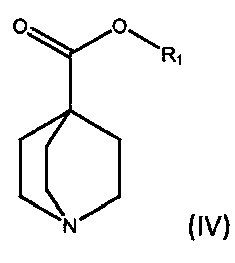
где R1 является C1-6алкилом, арилом или бензилом, в подходящем растворителе, а затем превращение соединения формулы (III) в бромид умеклидиния.
По одному аспекту R1 является этилом.
В данном описании термин «алкил» относится к насыщенной углеводородной цепи, имеющей указанное число атомов. Например, C1-6алкил относится к алкильной группе, имеющей от 1 до 6 атомов. Алкильные группы могут быть линейными или разветвленными. Типичные разветвленные алкильные группы имеют одну, две или три ветви. Алкил включает метил, этил, пропил (н-пропил и изопропил), бутил (н-бутил, изобутил и трет-бутил), пентил (н-пентил, изопентил и неопентил) и гексил.
В данном описании термин «арил» относится к фенилу или нафтилу. Арильные группы могут быть возможно замещенными одним или более чем одним заместителем, таким как галоген, C1-6алкил и C1-6алкоксил.
Реакцию между фениллитием и соединением формулы (IV) осуществляют в подходящем растворителе, например, апротонном растворителе, таком как толуол, ТГФ, МеТГФ, МТБЭ, или алкане, таком как гексан и циклогексан. По другому аспекту настоящего изобретения реакцию проводят в толуоле в качестве растворителя и/или при температуре 0°C.
Синтез соединения формулы (III), описанный в WO 2005/104745, необходимо проводить при криогенных температурах (например, -30°C) из-за конкретного реагента и используемой системы растворителей. Следовательно, способ, описанный здесь, преимущественно обеспечивает улучшенную производительность по сравнению с известным способом в данной области техники и, таким образом, лучше подходит для больших масштабов и коммерческого производства. Кроме того, время реакции было значительно сокращено (например, с 16 часов до 1 часа в толуоле при 0°C).
Побочные продукты реакции, образующиеся в ходе получения соединения формулы (III), могут быть удалены путем обработки водой. Например, гидроксид лития может быть удален путем добавления воды и полярного с высокой температурой кипения несмешивающегося с водой растворителя, такого как изомеры бутанола и пентанола, к реакционной смеси, нагревания до нужной температуры и затем выполнения жидкость-жидкостной экстракции. По одному аспекту растворителем является н-бутанол, а температура может быть от 79 до 85°C. Реализация, при которой неорганические побочные продукты могут быть удалены с использованием полярного с высокой температурой кипения несмешивающегося с водой растворителя, такого как н-бутанол, дает дополнительное преимущество на следующей стадии способа, когда неожиданно было обнаружено, что присутствие этих примесей замедляет реакцию. Таким образом, надлежащее удаление неорганических побочных продуктов повышает эффективность процесса.
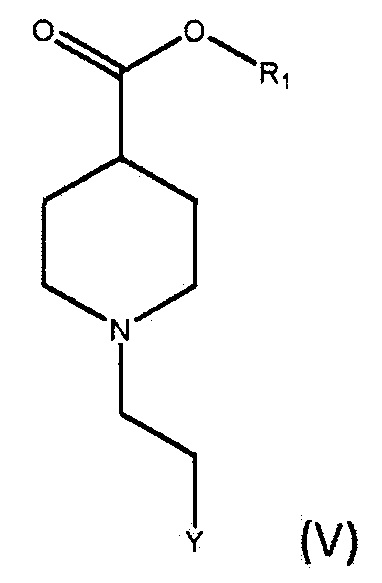
По дополнительному аспекту настоящее изобретение относится к способу получения соединения формулы (IV), включающему взаимодействие соединения формулы (V),
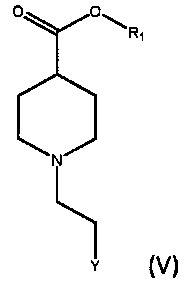
где R1 является C1-6алкилом, арилом или бензилом, и Y представляет собой уходящую группу, с подходящим основанием, выбранным из КГМДС, LiГМДС и NaГМДС, в подходящем растворителе.
По дополнительному аспекту настоящее изобретение относится к способу получения бромида умеклидиния, включающему получение соединения формулы (IV), которое включает стадию взаимодействия соединения формулы (V)
где R1 является C1-6 алкилом, арилом или бензилом, и Y представляет собой уходящую группу, с подходящим основанием, выбранным из КГМДС, LiГМДС и NaГМДС, в подходящем растворителе, а затем превращение соединения формулы (IV) в бромид умеклидиния.
По одному аспекту R1 является этилом.
По одному аспекту основанием является КГМДС.
Пример для уходящей группы Y включает, но не ограничивается ими, -OTs, -OMs, -OTf, Cl или Br. По одному аспекту Y является Cl.
Подходящие растворители для получения соединения формулы (IV) включают в себя, но не ограничиваются ими, апротонные растворители, такие как толуол, ТГФ, МеТГФ, МТБЭ, или алканы, такие как гексан и циклогексан. По одному аспекту растворитель представляет собой толуол.
Получение соединения формулы (IV), описанное в WO 2005/104745, включает реакцию соединения формулы (V) с ЛДА (основание) в ТГФ, и этот способ снова требует использования криогенных температур (-50°C). Неожиданно обнаружено, что условия способа, описанные здесь, позволяют осуществлять реакцию примерно при 50°C. Замена ЛДА, например, КГМДС и замена тетрагидрофурана, например, толуолом также благоприятно приводит к сокращению времени реакции (с 16 часов до примерно 1 часа в условиях, непосредственно изложенных выше).
По дополнительному аспекту настоящее изобретение относится к способу получения соединения формулы (V), включающему:
а) взаимодействие соединения формулы (VI),
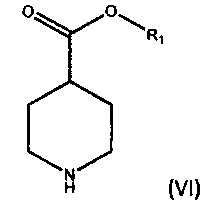
где R1 является C1-6 алкилом, арилом или бензилом, с соединением формулы (VII),
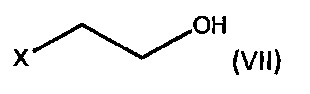
где X является уходящей группой, в присутствии подходящего основания; и b) превращение продукта, полученного на стадии (а), в соединение формулы (V) с соответствующим реагентом.
По дополнительному аспекту настоящее изобретение относится к способу получения бромида умеклидиния, включающему получение соединения формулы (V), которое включает:
а) взаимодействие соединения формулы (VI),
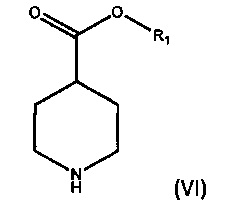
где R1 является С1-6 алкилом, арилом или бензилом, с соединением формулы (VII),
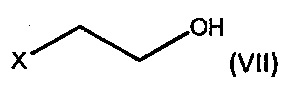
где X является уходящей группой, в присутствии подходящего основания, и b) превращение продукта, полученного на стадии (а), в соединение формулы (V) с соответствующим реагентом; а затем превращение соединения формулы (V) в бромид умеклидиния.
По одному аспекту X является Cl или Br, и/или R1 является этилом.
Стадию (а) реакции проводят в присутствии подходящего основания, которое включает, но не ограничивается ими, карбонат калия и ДБУ. По дополнительному аспекту настоящего изобретения обеспечивается сочетание хлорэтанола (X является Cl) в присутствии ДБУ, что обеспечивает приемлемую скорость реакции и превосходный выход.
Превращение на стадии (b) проводят с подходящим реагентом, например, выбранным из группы, состоящей из тионилхлорида, сульфонилгалогенидов, таких как сульфонилхлориды, сульфонильные ангидриды и галогениды фосфора.
По одному аспекту реагент на стадии (b) выбирают из группы, состоящей из тионилхлорида, п-толуолсульфонилхлорида, метансульфонилхлорида, метансульфонового ангидрида, п-толуолсульфонового ангидрида, трифторметансульфонового ангидрида, фосфорилхлорида, трибромида фосфора и пентахлорида фосфора.
По еще одному дополнительному аспекту реагент является тионилхлоридом.
Этот двухстадийный способ предпочтительно может обеспечить соединение формулы (V), по существу, с более высоким выходом, чем в способе, описанном в WO 2005/104745. Например, способ, раскрытый в настоящем описании, с использованием бромэтанола и карбоната калия в качестве реагентов обеспечивает приблизительно 80% конверсию (смотри Пример 1). Напротив, способ, описанный в WO 2005/104745, обеспечивает выход, равный 38,6% (Пример 1: Этил 1-(2-хлорэтил)-4-пиперидин карбоксилат).
Приведенный низкий выход в WO 2005/104745 в значительной степени является результатом образования димерной примеси - диэтил 1,1'-(этан-1,2-диил)бис(пиперидин-4-карбоксилата), которая должна быть впоследствии отделена от интересующего соединения с помощью хроматографии, при этом увеличивается присутствие высокотоксичного соединения формулы (V). Способ по настоящему изобретению исключает образование этой димерной основной примеси, устраняя необходимость в стадии разделения с высокой экспозицией, и, таким образом, обеспечивает гораздо более безопасную альтернативу.
В WO 2011/029896 описан альтернативный способ получения этил-хинуклидин-4-карбоксилата (соединение формулы (IV)) через этил-4-(2-хлорэтил)-пиперидин-4-карбоксилат (сравнительный пример 9), который исключает образование высокотоксичного промежуточного соединения (соединение формулы (V)). Тем не менее, этот альтернативный способ получения также приводит к очень низким выходам (т.е. 1,71-45,56% для предшественника этил-4-(2-хлорэтил)пиперидин-4-карбоксилата, сравнительный пример 1-7 в WO 2011/029896).
Обе стадии в получении соединения формулы (V) удобно проводить в присутствии подходящего растворителя, такого как толуол. Различные растворители или смеси растворителей могут быть использованы для каждой стадии реакции.
Получение соединения формулы (IV), исходя из соединения формулы (VI), может быть выполнено в виде серий отдельных реакций, при этом каждое промежуточное соединение изолируют, или может быть выполнено в виде телескопического синтеза (telescopic synthesis).
Бромид умеклидиния существует в нескольких кристаллических формах, которые могут быть охарактеризованы и дифференцированы с использованием ряда обычных аналитических методов, в том числе, но не ограничиваясь ими, рентгеновской порошковой дифракции (ПРД), инфракрасной спектроскопии (ИК), спектроскопии комбинационного рассеяния, дифференциальной сканирующей калориметрии (ДСК), термогравиметрического анализа (ТГА) и твердотельного ядерного магнитного резонанса (ттЯМР).
По одному аспекту настоящее изобретение обеспечивает бромид умеклидиния, находящийся в форме кристаллического твердого тела.
По дополнительному аспекту предложена кристаллическая твердотельная форма бромида умеклидиния, характеризующаяся рентгеновской порошковой дифрактограммой (ПРД), по существу, как показано на фиг. 1, 2, 3 или 4, и/или имеющей значительные дифракционные пики при значениях 20, приведенных в таблице 1.
По дополнительному аспекту настоящее изобретение относится к кристаллической твердотельной форме бромида умеклидиния, характеризующейся рентгеновской порошковой дифрактограммой, имеющей дифракционные пики при значениях 2Θ, экспериментальная ошибка ±0,10° 2Θ: 6,7, 8,9, 10,1, 11,5, 13,4, 13,8, 14,6, 15,4, 17,5, 17,9, 18,7, 19,9, 20,2, 22,6, 23,1, 24,3, 24,4 и/или 27,0 (Форма 1).
По дополнительному аспекту настоящее изобретение относится к кристаллической твердотельной форме бромида умеклидиния, характеризующейся рентгеновской порошковой дифрактограммой, имеющей дифракционные пики при значениях 2Θ, экспериментальная ошибка ±0,10° 2Θ: 6,6, 7,8, 10,7, 13,4, 14,0, 14,9, 16,4, 19,7, 20,1, 20,7, 20,9, 21,4 и/или 25,6 (Форма 2).
По дополнительному аспекту настоящее изобретение относится к кристаллической твердотельной форме бромида умеклидиния, характеризующейся рентгеновской порошковой дифрактограммой, имеющей дифракционные пики при значениях 2Θ, экспериментальная ошибка ±0,10° 2Θ: 6,9, 9,3, 12,5, 12,9, 16,1, 16,7, 17,9, 18,5, 19,4, 20,1, 20,9, 23,3 и/или 25,1 (Форма 3).
Кристаллические формы (Формы 1, 2 и 3) бромида умеклидиния дополнительно характеризуются кривыми дифференциальной сканирующей калориметрии (ДСК). По дополнительному аспекту настоящее изобретение относится к кристаллической твердотельной форме бромида умеклидиния, характеризующейся кривой ДСК с начальной температурой приблизительно 236°C (Форма 1, без затравки), 232°C (Форма 2) и 232°C (Форма 3).
Экспериментальная часть
Сокращения
Изобретение проиллюстрировано следующими примерами.
Пример 1: Получение этил 1-(2-хлорэтил)-4-пиперидинкарбоксилата
Этилизонипекотат (400,1 г), порошок карбоната калия (448,7 г) и 2-бромэтанол (256 мл) в толуоле (4000 мл) нагревали с обратным холодильником (около 110°C) и перемешивали в течение 160 минут. Реакционную смесь охладили до 60°C и добавили воды (1200 мл), после чего охладили до 20°C. После перемешивания водный слой отделили и экстрагировали толуолом (2000 мл). Объединенные органические слои сконцентрировали до 4200 мл путем вакуумной перегонки. Часть этой реакционной смеси (4050 мл) нагрели до 50°C и добавили тионилхлорид (193 мл). После перемешивания в течение 1 часа добавили этилацетат (4600 мл) при 40°C, а затем засеяли этил 1-(2-хлорэтил)-4-пиперидинкарбоксилатом (0,4 г). Суспензию выдерживали в течение 35 мин, затем охладили до 20°C и выдерживали в течение 40 мин. Продукт отфильтровали, промыли этилацетатом (1500 мл) и сушили в вакууме при 45°C, получая белое твердое вещество (502,7 г, 80%). EI-MS (масс-спектрометрия с ионизацией электронным ударом) m/z 220 (М+Н+) Rt (2,1 мин).
1Н ЯМР (400 МГц; ДМСО-d6): 4,14-4,01 (4Н, м), 3,57-3,42 (4Н, м), 3,01 (2Н, м), 2,59 (1Н, м), 2,05 (2Н, м), 1,88 (2Н, м), 1,19 (3Н, т).
Пример 2: Получение 1-азабицикло[2.2.2]окт-4-ил(дифенил)метанола
Этил-1-(2-хлорэтил)-4-пиперидинкарбоксилат (199 г, например, полученный, как в примере 1) в воде (800 мл) добавили к толуолу (2000 мл), затем раствор карбоната калия (118 г) в воде (800 мл) и сполоснули водой (400 мл). Смесь перемешивали до тех пор, пока не получили двухфазный раствор. Водный слой отделили и экстрагировали толуолом (3000 мл). Объединенные органические слои сконцентрировали до 4000 мл путем вакуумной перегонки. Этот раствор был добавлен к 0,5 М гексаметилдисилазиду калия в толуоле (1700 мл) и толуолу (2000 мл) при 40°C. Затем добавили уксусную кислоту (178 мл), смесь сконцентрировали до 4000 мл с помощью вакуумной перегонки и добавили к 18% масс, водному раствору карбоната калия (2432 г). Слои разделили, органический слой сконцентрировали до 3000 мл путем вакуумной перегонки и добавили толуол (1000 мл). Раствор охладили до -15°C и добавили 2 М раствор фениллития в дибутиловом эфире (800 мл). Затем добавили воду (2000 мл) и н-бутанол (700 мл) и смесь нагрели до 75°C. Водный слой удалили, а органический слой промыли водой (1000 мл). Затем добавили толуол (1000 мл) и смесь перегоняли, пока 3000 мл растворителя не были удалены, а затем охладили до 20°C и перемешивали в течение ночи. Продукт отфильтровали, промыли толуолом (2×200 мл) и сушили в вакууме при 40°C, получая белое твердое вещество (131 г, 57%). EI-MS m/z 294 (М+Н+) Rt (3,6 мин).
1Н-ЯМР (400 МГц; MeOD): 7,55 (4Н, м), 7,27 (4Н, м), 7,18 (2Н, м), 2,84 (6Н, м), 1,83 (6Н, м).
Пример 3: Получение 1-азабицикло[2.2.2]окт-4-ил(дифенил)метанола
Этилизонипекотат (300 г), порошок карбоната калия (330 г) и 2-бромэтанол (150 мл) в толуоле (2700 мл) нагревали с обратным холодильником в условиях Дина-Старка в течение 4 ч. Реакционную смесь охладили до 20°C и добавили воду (900 мл). После перемешивания водный слой отделили и экстрагировали толуолом (1500 мл). Объединенные органические слои сконцентрировали до 2700 мл путем вакуумной перегонки. Реакционную смесь нагрели до 60°C и добавили тионилхлорид (150 мл). После перемешивания в течение 90 мин смесь охладили до 20°C, перемешивали в течение 30 мин и добавили толуол (1800 мл). Затем добавили воду (900 мл) и 26% масс. водный раствор карбоната калия (2028 г). Слои разделили, а водный слой экстрагировали толуолом (7500 мл). Объединенные органические слои промыли водой (300 мл) и сушили с добавлением толуола (3000 мл) и концентрирования до 4800 мл путем вакуумной перегонки. Добавили 0,5 М гексаметилдисилазид калия в толуоле (4200 мл) при 40°C и смесь перемешивали в течение 1 часа. Затем добавили этанол (192 мл) и уксусную кислоту (426 мл) и смесь перемешивали при 40°C в течение 2 часов. Добавили 26% масс. водный раствор карбоната калия (4038 г) и слои разделили. Водный слой экстрагировали толуолом (2500 мл). Объединенные органические слои промыли водой (300 мл) и сконцентрировали до 4500 мл путем вакуумной перегонки. Раствор охладили до 0-5°C и добавили 2 М раствор фениллития в дибутиловом эфире (1920 мл). Через один час добавили воду (1500 мл) и н-бутанол (1680 мл) и смесь нагрели до 78°C. Водный слой удалили, а органический слой промыли водой (1500 мл). Органическую фазу сконцентрировали до 6000 мл с помощью перегонки с добавлением толуола, затем охладили до 20°C и перемешивали в течение ночи. Продукт отфильтровали, промыли толуолом (2×600 мл) и сушили в вакууме, получая белое твердое вещество (300 г, 53%). EI-MS m/z 294 (М+Н+) Rt (3,7 мин).
1Н-ЯМР (400 МГц; MeOD): 7,54 (4Н, м), 7,26 (4Н, м), 7,17 (2Н, м), 2,83 (6Н, м), 1,82 (6Н, м).
Пример 4: Получение 1-азабицикло[2.2.2]окт-4-ил(дифенил)метанола
Этилизонипекотат (600 г) и ДБУ (600 мл) растворили в толуоле (3000 мл) и нагрели до 100°C. Добавляли 2-хлороэтанол (330 мл) в течение 2 часов и смесь перемешивали при 109°C в течение еще 4,5 часов. Температуру довели до 55-65°C и добавили тионилхлорид (378 мл). После перемешивания в течение 1 часа смесь охладили до 30°C, добавили воду (1800 мл) и 40% масс. водный раствор карбоната калия (3350 г). Слои разделили, а водный слой экстрагировали толуолом (3000 мл). Объединенные органические слои промыли водой (600 мл) и сушили с добавлением толуола (2000 мл) и концентрирования до 6000 мл путем вакуумной перегонки. Анализ раствора показал 94%-ную конверсию по этилизонипекотату. Добавили 0,5 М гексаметилдисилазид калия в толуоле (8400 мл) при 45-50°C и смесь перемешивали в течение 2 часов. Добавили еще 0,5 М гексаметилдисилазида калия в толуоле (1260 мл) и смесь перемешивали в течение 30 минут. Затем добавили этанол (390 мл) и смесь сконцентрировали до 8400 мл путем вакуумной перегонки. Добавили уксусную кислоту (850 мл) и смесь перемешивали при 45°C в течение 15 часов. Затем добавили 26% масс, водный раствор карбоната калия (8110 г) и слои разделили. Водный слой экстрагировали толуолом (4800 мл). Объединенные органические слои разделили на две половины и профильтровали через фильтр (38 г Celite или Harborlite), снова объединили и сконцентрировали до 6000 мл путем вакуумной перегонки. Раствор охладили до 0-5°C и добавили 2 М раствор фениллития в дибутиловом эфире (3840 мл). Через один час добавили воду (3000 мл) и н-бутанол (3960 мл) и смесь нагрели до 83°C. Водный слой удалили, а органический слой промыли водой (3000 мл). Органическую фазу сконцентрировали до 12000 мл путем перегонки с добавлением толуола, затем охладили до 20°C и перемешивали в течение ночи. Продукт отфильтровали, промыли толуолом (2×1200 мл) и сушили в вакууме при 70°C, получая белое твердое вещество (561 г, 50%). EI-MS m/z 294 (М+Н+) Rt (3,7 мин).
1Н-ЯМР (400 МГц; ДМСО-d6): 7,51 (4Н, м), 7,25 (4Н, м), 7,15 (2Н, м), 2,65 (6Н, м), 1,60 (6Н, м).
Пример 5: Получение 4-[гидрокси(дифенил)метил]-1-{2-[(фенилметил)окси]этил}-1-азонийбицикло[2.2.2]октанбромида (средний класс (Grade) - без стадии засевания)
Раствор 1-азабицикло[2.2.2]окт-4-ил(дифенил)метанола (31,7 кг) и бензил 2-бромэтилового эфира (25,7 кг) в н-пропаноле (257,5 кг) нагревали с обратным холодильником в течение 13 часов. Раствор охлаждали до 50-55°C в течение не менее 1 часа и перемешивали в течение 40 минут, чтобы вызвать кристаллизацию. Суспензию охлаждали до 17-23°C в течение не менее 1 часа и перемешивали в течение 60 минут. Затем суспензию охлаждали до 0-5°C в течение не менее 1 часа и выдерживали в течение 2 часов. Продукт отфильтровали и дважды промыли н-пропанолом (34,8 кг и 33,8 кг). После сушки в вакууме при 50°C получили белое вещество (47,95 кг, 87%).
Пример 6 Получение 4-[гидрокси(дифенил)метил]-1-{2-[(фенилметил)окси]этил}-1-азонийбицикло[2.2.2]октан бромида (средний класс)
Раствор 1-азабицикло[2.2.2]окт-4-ил(дифенил)метанола (445,6 г, например, полученный, как в примере 4) и бензил 2-бромэтилового эфира (360,9 г) в н-пропаноле (4456 мл) нагревали с обратным холодильником в течение 3 часов. Раствор охладили до 87°C, внесли затравку бромида умеклидиния (Форма 1) (0,44 г), охладили далее до 82°C и выдерживали в течение 1 часа. Суспензию охлаждали до 0-5°C в течение 2,5 часов и выдерживали в течение 1 часа. Продукт отфильтровали и промыли н-пропанолом (2×900 мл). После сушки в вакууме при 50°C получили белое твердое вещество (690 г, 89%). EI-MS m/z 428 (М+) Rt (4,7 мин).
1Н ЯМР (400 МГц; ДMCO-d6): 7,57 (4Н, д), 7,40-7,30 (9Н, м), 7,26 (2Н, т), 5,94 (1Н, с), 4,52 (2Н, с), 3,84 (2Н, м), 3,49 (6Н, т), 3,38 (2Н, м), 2,02 (6Н, т).
Пример 7: Получение кристаллической Формы 1 4-[гидрокси(дифенил)метил]-{2-(фенилметил)окси]этил}-1-азонийбииикло[2.2.2]октан бромида
4-[гидрокси(дифенил)метил]-1-{2-[(фенилметил)окси]этил}-1-азонийбицикло[2.2.2]октан бромид (165 г) растворили в смеси н-пропанола (495 мл) и воды (990 мл) при 80°C. Полученный раствор охладили до 50°C, добавили затравку Формы 1 (0,825 г) в н-пропаноле (2,8 мл) и сполоснули дополнительно н-пропанолом (5,5 мл). После выдержки в течение одного часа при 50°C суспензию охлаждали до 40°C в течение 80 мин, затем охлаждали далее до 0-5°C в течение 105 мин. Образец суспензии выдерживали в течение 3 ч при 0-5°C, отфильтровали и продукт промыли н-пропанолом (2×330 мл). После вакуумной сушки при 60°C получили белое твердое вещество (146 г, 88%). Оно охарактеризовано ПРД (см. Фиг. 2 и таблицу 1).
Пример 8: Получение кристаллической Формы 1 4-[гидрокси(дифенил)метил]-1-{2-[(фенилметил)окси]этил}-1-азонийбицикло[2.2.2]октан бромида (альтернатива примеру 6, без стадии засевания)
4-[гидрокси(дифенил)метил]-1-{2-[(фенилметил)окси]этил}-1-азонийбицикло[2.2.2]октан бромид (20 г) растворили в смеси н-пропанола (60 мл) и воды (120 мл) при 80°C, при этом смесь стала прозрачной. Полученный раствор охладили до 45°C и выдерживали в течение 2 часов. Полученную густую суспензию охлаждали до 0-5°C в течение 3 часов. Образец суспензии выдерживали в течение 1 ч при 0-5°C, отфильтровали и продукт промыли н-пропанолом (2×40 мл). После вакуумной сушки при 50°C получили белое твердое вещество (16 г, 80%). Оно охарактеризовано ПРД (см. Фиг. 1 и таблицу 1) и ДСК (см. Фиг. 6).
Пример 9: Получение кристаллической Формы 2 4-[гидрокси(дифенил)метил]-1-{2-[(фенилметил)окси]этил}-1-азонийбицикло[2.2.2]октан бромида
Нитрометан (105 мл) и н-пропанол (45 мл) добавили к 4-[гидрокси(дифенил)метил]-1-{2-[(фенилметил)окси]этил}-1-азонийбицикло[2.2.2]октан бромиду (6 г) при 21°C. Полученную смесь перемешивали при комнатной температуре (примерно 21°C) в течение 30,5 часов. Мутную смесь отфильтровали под действием силы тяжести с помощью стеклянной воронки и фильтровальной бумаги. Прозрачный раствор выдерживали в вакууме в роторном испарителе (0,6-0,7 кПа (6-7 мбар)) в течение 15 минут и полученное твердое вещество сушили в вакууме при 50°C. Выход: 5,8 г (97%). Оно охарактеризовано ПРД (см. Фиг. 3 и таблицу 1) и ДСК (см. Фиг. 7).
Пример 10: Получение кристаллической формы 3 4-[гидрокси(дифенил)метил]-1-{2-[(фенилметил)окси]этил}-1-азонийбицикло[2.2.2]октан бромида
Дихлорметан (105 мл) и 1-пентанол (45 мл) добавили к 4-[гидрокси(дифенил)метил]-1-{2-[(фенилметил)окси]этил}-1-азонийбицикло[2.2.2]октан бромиду (6 г) при 21°C. Полученную смесь перемешивали при комнатной температуре (примерно 21°C) в течение 30 часов. Мутную смесь отфильтровали под действием силы тяжести с помощью стеклянной воронки и фильтровальной бумаги. Прозрачный раствор выдерживали в вакууме в роторном испарителе (0,6-0,7 кПа (6-7 мбар)) в течение 20 минут, пока не получили суспензию. Белое твердое вещество отфильтровали под вакуумом и высушили при 50°C. Выход: 5,1 г (85%). Оно охарактеризовано ПРД (см. Фиг. 4 и таблицу 1) и ДСК (см. Фиг. 8).
Инструментальные параметры
Экспериментальные условия LC-MS (жидкостная хроматомасс-спектрометрия)
Колонка: 5 см × 2,1 мм, 3 мкм, Luna (С18)
Подвижная фаза: вода/ацетонитрил + 0,05% об/об ТФУК
Изменение концентрации ацетонитрила от 0% до 95% в течение 8 минут.
Общее время работы: 10 минут
Скорость потока: 1,0 мл/мин
Температура колонки: 40°C
Диапазон масс: от 100 до 1000 Дальтон
Диапазон длин волн: от 205 до 400 нм
Н1 ЯМР
ЯМР-спектры регистрировали на спектрометре Bruker DPX400, 400 МГц, в MeOD или ДМСО-d6.
Рентгеновская порошковая дифракция (ПРД)
Данные ПРД получали на порошковом дифрактометре PANalytical X'Pert Pro, оснащенном детектором X'Celerator. Условия измерения составляли: излучение: Cu Kα, напряжение генератора: 40 кВ, ток генератора: 45 мА, начальный угол: 2,0° 2Θ, конечный угол: 40,0° 2Θ, размер шага: 0,0167° 2Θ. Время шага составляло 31,750 сек. Образец получали путем помещения нескольких миллиграмм образца на кремниевую пластину с нулевым фоном, в результате чего получался тонкий слой порошка. Характерные положения пиков и соответствующие расстояния d приведены в таблице 1. Они были рассчитаны из необработанных данных с использованием программного обеспечения PANalytical HighScore. Экспериментальная ошибка в положениях пиков составляет примерно ±0,10° 2Θ. Относительные интенсивности пиков могут варьироваться в зависимости от преимущественной ориентации и, следовательно, не учитываются.
Дифференциальная сканирующая калориметрия (ДСК)
ДСК-термограмма была получена с использованием калориметра ТА Instruments Q2000. Образец взвешивали в алюминиевой кювете; крышку кюветы поместили сверху и слегка обжали без герметизации кюветы. Эксперимент проводили, используя скорость нагрева 10°C мин-1.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЛЬ ОМЕКАМТИВА МЕКАРБИЛА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2014 |
|
RU2663663C2 |
| ГИДРОХЛОРИД (1S,2S,4R)-4-{ 4-[(1S)-2,3-ДИГИДРО-1Н-ИНДЕН-1-ИЛАМИНО]-7Н-ПИРРОЛО [2,3-d]ПИРИМИДИН-7-ИЛ} -2-ГИДРОКСИЦИКЛОПЕНТИЛ)МЕТИЛ СУЛЬФАМАТА | 2010 |
|
RU2562245C2 |
| АЛКАЛОИДНЫЕ ПРОИЗВОДНЫЕ НА ОСНОВЕ СЛОЖНЫХ АМИНОЭФИРОВ И КОМПОЗИЦИИ ЛЕКАРСТВЕННЫХ СРЕДСТВ, СОДЕРЖАЩИЕ ИХ | 2011 |
|
RU2580835C2 |
| ПРОИЗВОДНЫЕ ХИНУКЛИДИНАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, КОМБИНИРОВАННЫЙ ПРОДУКТ И СПОСОБ ИНГИБИРОВАНИЯ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2003 |
|
RU2314306C2 |
| ПРОИЗВОДНЫЕ ГЛИЦИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2011 |
|
RU2585767C2 |
| ЭФФЕКТИВНЫЙ ПРОЦЕСС ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 6-КАРБОКСИБЕНЗОКСАЗОЛА | 2020 |
|
RU2810493C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИЛИРОВАННЫХ ЦИКЛОПЕНТАПИРИМИДИНОВЫХ СОЕДИНЕНИЙ И ИХ СОЛЕЙ | 2013 |
|
RU2642311C2 |
| АНСА-ЦИРКОНОЦЕНЫ, ФУНКЦИОНАЛИЗИРОВАННЫЕ ПО ЦИКЛОСИЛАНОВОМУ МОСТИКУ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1999 |
|
RU2160277C1 |
| Агрегированные частицы | 2013 |
|
RU2666963C2 |
| ПРОИЗВОДНЫЕ СЛОЖНОГО АМИНОЭФИРА АЛКАЛОИДА И ИХ ЛЕКАРСТВЕННЫЕ КОМПОЗИЦИИ | 2011 |
|
RU2567548C2 |
Изобретение относится к способу получения соединения формулы (V),
где R1 является C1-6 алкилом, арилом или бензилом, и Y является уходящей группой, который включает:
a) взаимодействие соединения формулы (VI),
с 2-хлорэтанолом в присутствии подходящего основания;
и
b) превращение продукта, полученного на стадии (а), в соединение формулы (V) с использованием реагента, выбранного из группы, состоящей из тионилхлорида, сульфонилгалогенидов, таких как сульфонилхлориды, сульфонильные ангидриды и галогениды фосфора. 6 з.п. ф-лы., 8 ил.,1 табл., 10 пр.
1. Способ получения соединения формулы (V),
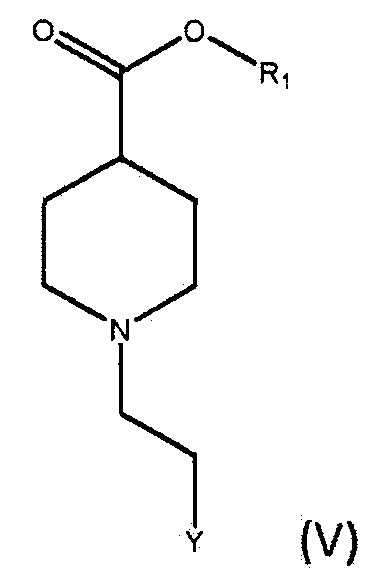
где R1 является C1-6 алкилом, арилом или бензилом, и Y является уходящей группой, который включает:
a) взаимодействие соединения формулы (VI),
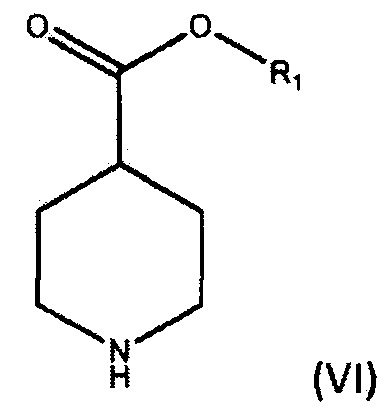
с 2-хлорэтанолом в присутствии подходящего основания;
и
b) превращение продукта, полученного на стадии (а), в соединение формулы (V) с использованием реагента, выбранного из группы, состоящей из тионилхлорида, сульфонилгалогенидов, таких как сульфонилхлориды, сульфонильные ангидриды и галогениды фосфора.
2. Способ по п. 1, где R1 представляет собой этил.
3. Способ по п. 1, где реагент на стадии (b) выбран из группы, состоящей из тионилхлорида, п-толуолсульфонилхлорида, метансульфонилхлорида, метансульфонового ангидрида, п-толуолсульфонового ангидрида, трифторметансульфонового ангидрида, фосфорилхлорида, трибромида фосфора и пентахлорида фосфора.
4. Способ по п. 3, где реагент представляет собой тионилхлорид.
5. Способ по п. 1, где стадии (а) и (b) осуществляют в подходящем растворителе.
6. Способ по п. 5, где растворитель представляет собой толуол.
7. Способ по любому из пп. 1-6, где основание представляет собой 1,8-диазабицикло(5.4.0)ундец-7-ен (ДБУ).
| WO2011029896 A1 17.03.2011 | |||
| EA200601991 A1 2007-02-27 | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |

