Перекрестная ссылка на родственные заявки
[0001] В настоящей заявке испрашивается приоритет предварительной заявки на патент США № 61/785763, поданной 14 марта 2014 года, содержание которой полностью включено в настоящую заявку посредством ссылки.
Область техники
[0002] Предложены полиморфные формы дигидрохлорида омекамтива мекарбила, способы получения омекамтива мекарбила, включая полиморфные формы дигидрохлорида омекамтива мекарбила, композиции, содержащие полиморфные формы дигидрохлорида омекамтива мекарбила, и способы применения полиморфных форм дигидрохлоридной соли омекамтива мекарбила.
Уровень техники
[0003] Саркомер сердечной мышцы представляет собой основную функциональную единицу сокращения мышцы сердца. Саркомер сердечной мышцы представляет собой высокоупорядоченную цитоскелетную структуру, состоящую из миозина, актина и группы регуляторных белков сердечной мышцы. Открытие и разработка низкомолекулярных активаторов миозина сердечной мышцы обеспечит перспективные средства лечения острой и хронической сердечной недостаточности. Миозин сердечной мышцы представляет собой цитоскелетный движущий белок в клетке сердечной мышцы. Он непосредственно отвечает за превращение химической энергии в механическую силу, что приводит к сокращению сердечной мышцы.
[0004] Существующие положительные инотропные агенты, такие как агонисты бета-адренергического рецептора или ингибиторы активности фосфодиэстеразы, повышают концентрацию внутриклеточного кальция, увеличивая, таким образом, сократимость саркомера сердечной мышцы. Однако увеличение уровней кальция повышает скорость сокращения сердечной мышцы и сокращает систолическое время изгнания, что связано с потенциально опасными для жизни побочными эффектами. Напротив, активаторы миозина сердечной мышцы действуют по механизму, который напрямую стимулирует активность движущего белка миозина сердечной мышцы, не увеличивая внутриклеточную концентрацию кальция. Они ускоряют лимитирующую стадию ферментативного цикла миозина и сдвигают ее в сторону состояния выработки силы. Вместо увеличения скорости сердечного сокращения, этот механизм скорее увеличивает время систолического изгнания, что приводит к повышению сократимости сердечной мышцы и сердечному выбросу с потенциально более эффективной утилизацией кислорода.
[0005] В патенте США № 7507735, включенном в настоящий документ посредством ссылки, описан класс соединений, включающий омекамтива мекарбил (AMG 423, CK-1827452), имеющий структуру:
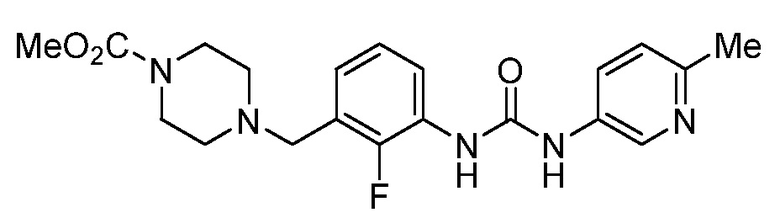
[0006] Омекамтива мекарбил является первым в своей группе прямым активатором сердечного миозина, движущего белка, который вызывает сердечное сокращение. В настоящее время его исследуют в качестве потенциального средства для лечения сердечной недостаточности во внутривенных и пероральных препаратах для создания нового спектра средств лечения пациентов как в стационарных, так и в амбулаторных условиях.
[0007] Поскольку постоянно необходимы лекарственные соединения, обладающие, например, улучшенной стабильностью, растворимостью, сроком годности и фармакологией in vivo, существует непрерывная потребность в новых или чистых солях, гидратах, сольватах и полиморфных кристаллических формах молекул существующих лекарств. Кристаллические формы омекамтива мекарбила, описанные в настоящем документе, способствуют удовлетворению этой и других потребностей.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0008] Предложена дигидрохлоридная форма омекамтива мекарбила.
[0009] Предложен также гидрат дигидрохлорида омекамтива мекарбила.
[0010] Предложена также кристаллическая форма дигидрохлоридной формы омекамтива мекарбила.
[0011] Предложена также Форма A гидрата дигидрохлорида омекамтива мекарбила.
[0012] Предложен также безводный дигидрохлорид омекамтива мекарбила.
[0013] Предложена также Форма B безводного дигидрохлорида омекамтива мекарбила.
[0014] Предложена также Форма C безводного дигидрохлорида омекамтива мекарбила.
[0015] Предложены также композиции и фармацевтические композиции, содержащие дигидрохлоридную форму омекамтива мекарбила.
[0016] Предложен также способ получения омекамтива мекарбила, включающий:
смешивание метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилата и фенил-(6-метилпиридин-3-ил)карбамата в присутствии триалкиламинного основания с получением омекамтива мекарбила.
[0017] Предложен также способ получения гидрата дигидрохлорида омекамтива мекарбила, включающий:
(a) гидрирование метил-4-(2-фтор-3-нитробензил)пиперазин-1-карбоксилата в присутствии катализатора гидрирования с получением метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилата;
(b) смешивание метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилата и фенил-(6-метилпиридин-3-ил)карбамата в присутствии триалкиламинного основания с получением омекамтива мекарбила в виде свободного основания; и
(c) кристаллизацию свободного основания омекамтива мекарбила в присутствии водного раствора хлористоводородной кислоты и спиртового растворителя с получением гидрата дигидрохлоридной соли омекамтива мекарбила.
ОПИСАНИЕ ЧЕРТЕЖЕЙ
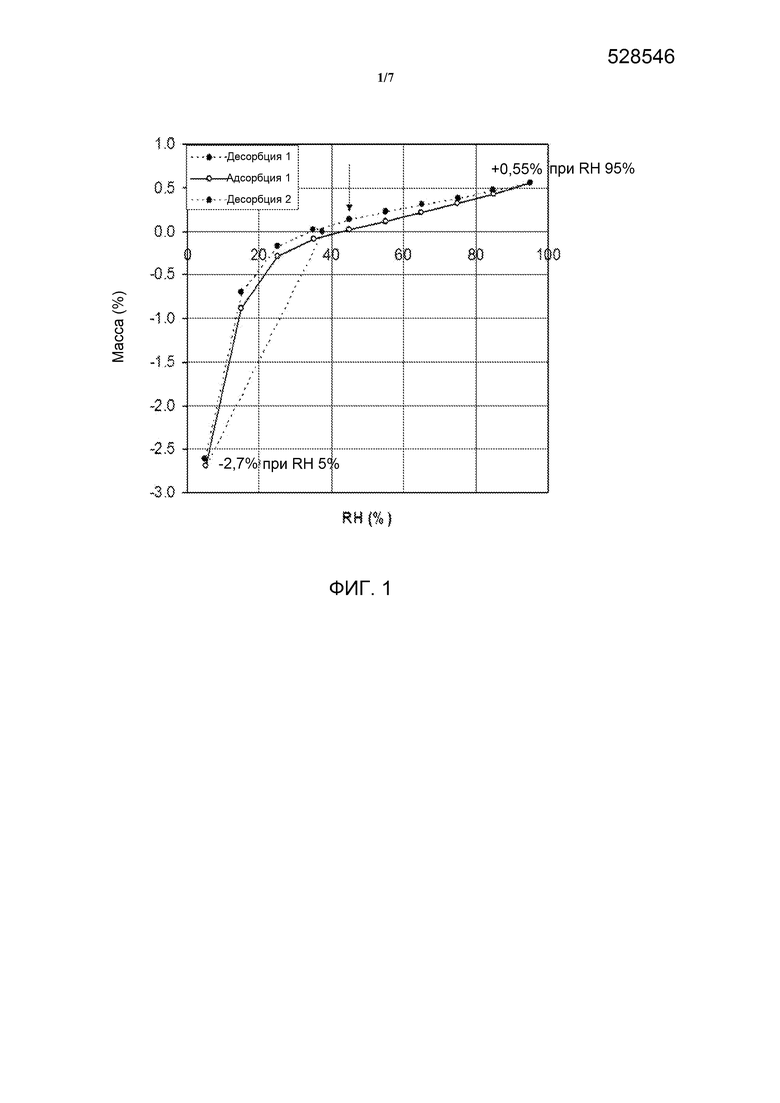
[0018] На фиг. 1 представлена динамическая сорбция паров Формы А гидратной формы дигидрохлорида омекамтива мекарбила.
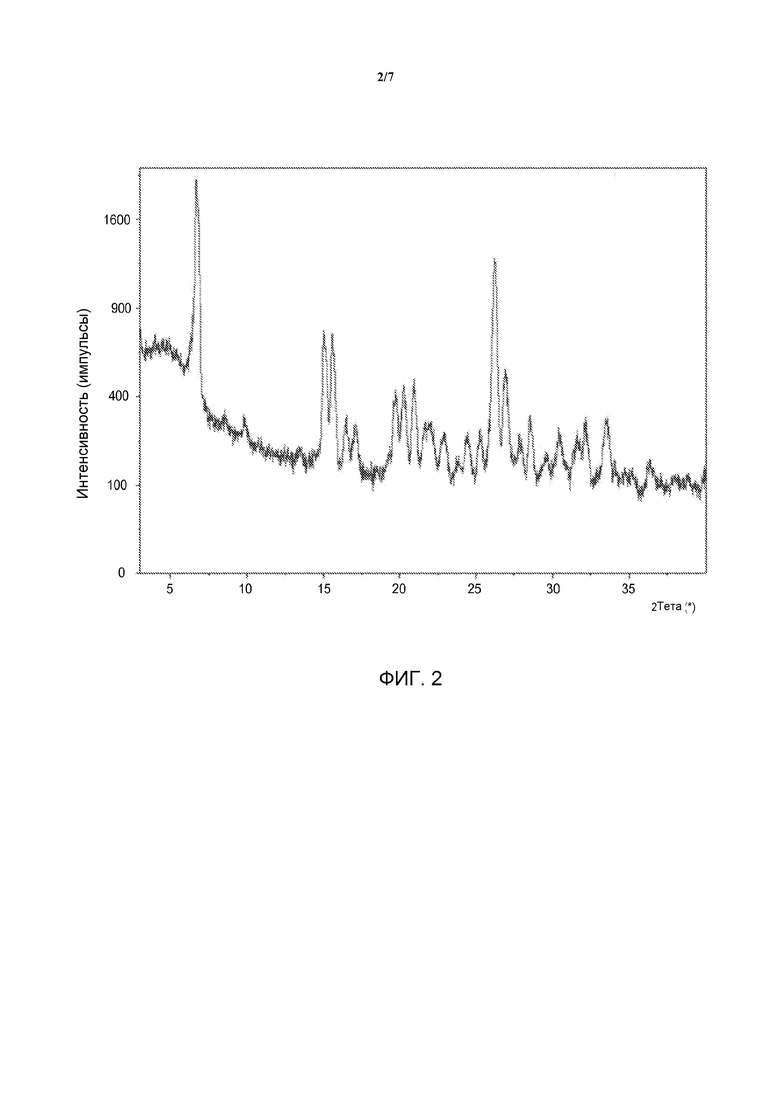
[0019] На фиг. 2 представлена диаграмма порошковой рентгеновской дифракции (ПРД) Формы А.
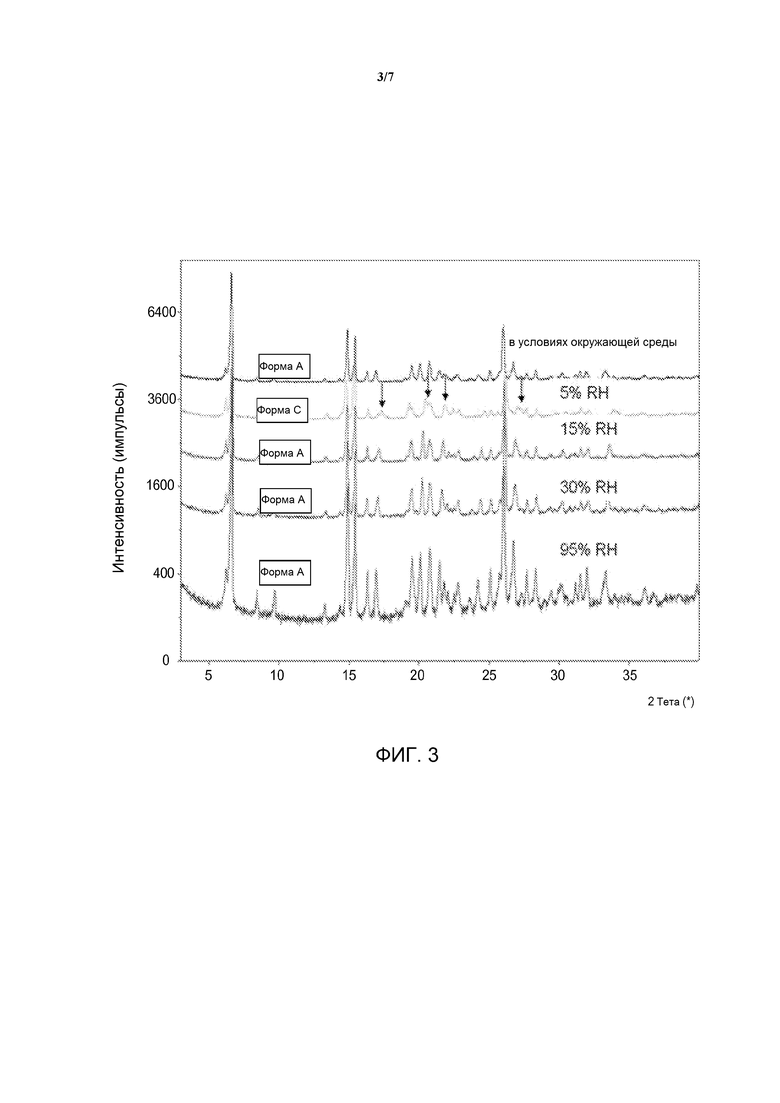
[0020] На фиг. 3 представлена ПРД гидратной формы дигидрохлоридной соли омекамтива мекарбила в условиях различной относительной влажности.
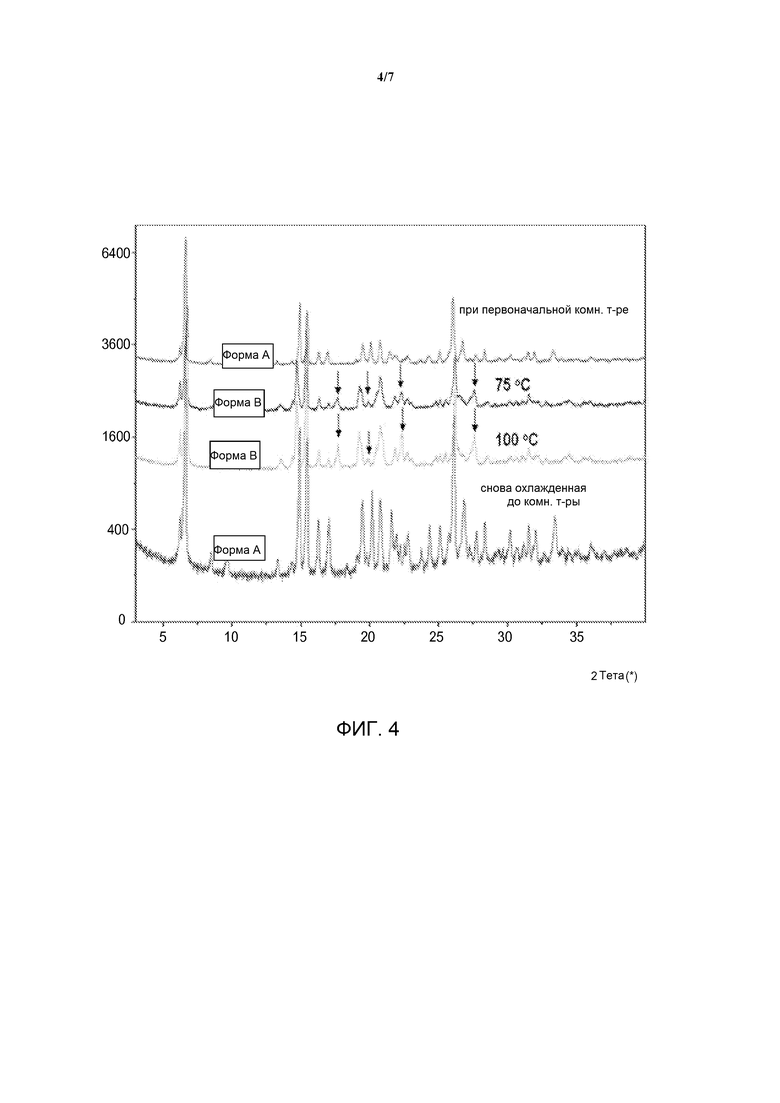
[0021] На фиг. 4 представлена ПРД гидратной формы дигидрохлоридной соли омекамтива мекарбила при различных температурах.
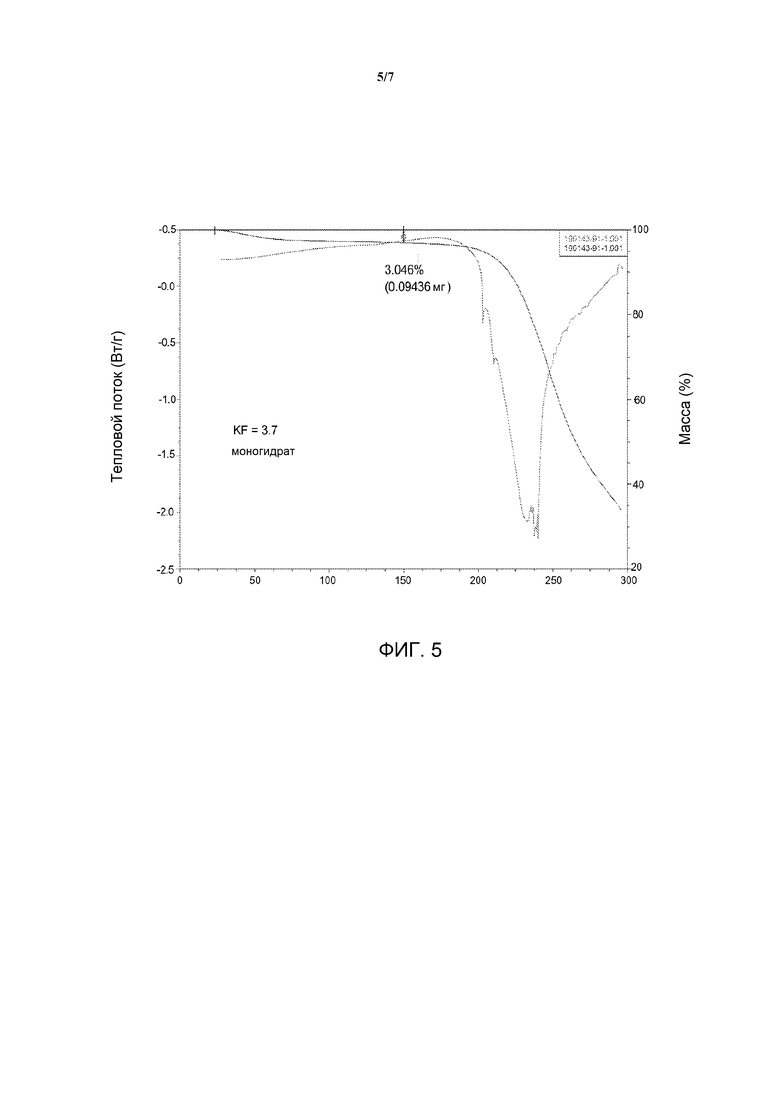
[0022] На фиг. 5 представлена термограмма дифференциальной сканирующей калориметрии и термогравиметрический анализ Формы А.
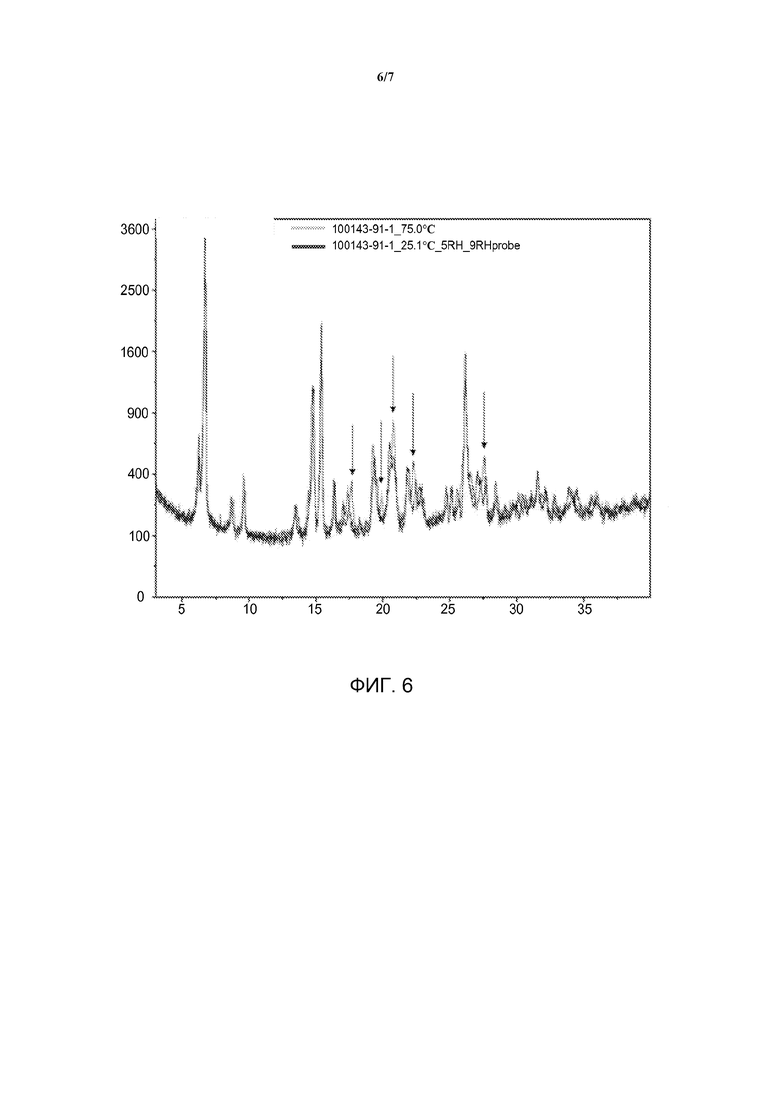
[0023] На фиг. 6 представлены наложенные друг на друга диаграммы ПРД Форм А, В и С дигидрохлоридной соли омекамтива мекарбила.
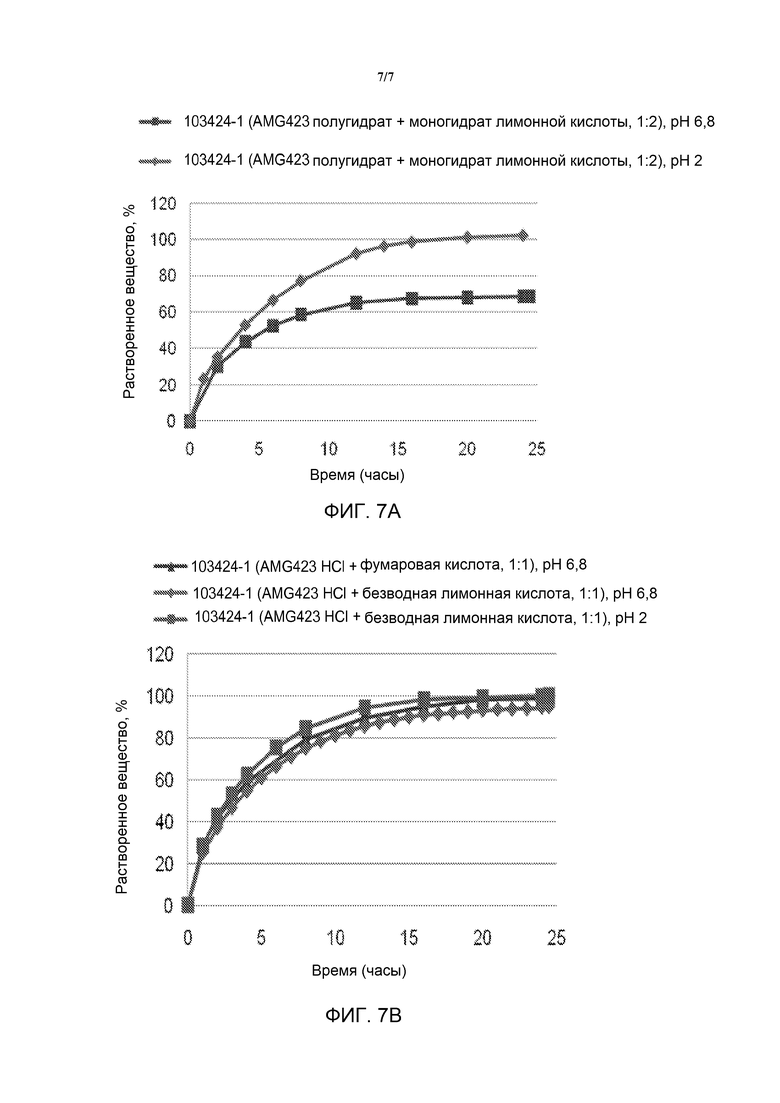
[0024] На фиг. 7 представлено высвобождение лекарства при двух различных рН (2 и 6,8) из препарата свободного основания омекамтива мекарбила (сверху) и Формы А гидратной формы дигидрохлоридной соли омекамтива мекарбила (снизу).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0025] Если не указано иное, то к терминам, содержащимся в описании и формуле изобретения, применяют следующие определения:
[0026] «Лечение» или «лечить» означает любое лечение болезни у пациента, включая: a) предупреждение заболевания, то есть предотвращение развития клинических симптомов заболевания; b) подавление заболевания; c) замедление или остановку развития клинических симптомов; и/или d) облегчение заболевания, то есть инициацию регрессии клинических симптомов. В настоящем документе предполагается, что лечение заболеваний и расстройств включает также профилактическое введение фармацевтического препарата, описанного в настоящем документе, субъекту (т.е. животному, предпочтительно млекопитающему, наиболее предпочтительно человеку), который, предположительно, нуждается в превентивном лечении, как, например, в случае хронической сердечной недостаточности.
[0027] Термин «терапевтически эффективное количество» означает количество, эффективное при введении человеку или пациенту, не являющемуся человеком, для лечения заболевания, например, терапевтически эффективное количество может быть количеством, достаточным для лечения заболевания или расстройства, восприимчивого к активации миозина. Терапевтически эффективное количество может быть определено экспериментально, например, анализом концентрации химического вещества в крови, или теоретически, т.е. путем расчета биодоступности.
[0028] «Фармацевтически приемлемые соли» включают, но не ограничиваются ими, соли с неорганическими кислотами, такие как гидрохлоратные (т.е. гидрохлоридные), фосфатные, дифосфатные, гидроброматные, сульфатные, сульфинатные, нитратные и подобные соли; а также соли с органическими кислотами, такие как малатные, малеатные, фумаратные, тартратные, сукцинатные, цитратные, ацетатные, лактатные, метансульфонатные, п-толуолсульфонатные, 2-гидроксиэтилсульфонатные, бензоатные, салицилатные, стеаратные и алканоатные, такие как ацетатные, HOOC--(CH2)n--COOH, где n равен 0-4, и подобные соли. Аналогично фармацевтически приемлемые катионы включают, но не ограничиваются ими, натрий, калий, кальций, алюминий, литий и аммоний. Специалистам в данной области техники известны различные методики синтеза, которые могут быть использованы для получения нетоксичных фармацевтически приемлемых солей присоединения.
[0029] При использовании в настоящем документе термин «полиморфы» или «полиморфные формы» относится к кристаллическим формам одной и той же молекулы. Различные полиморфные формы молекулы имеют разные физические свойства, как результат расположения или конформации молекул в кристаллической решетке. Некоторые из указанных различных физических свойств включают температуру плавления, теплоту плавления, растворимость, скорость растворения и/или колебательные спектры. Физическая форма конкретного соединения особенно важна при использовании соединения в фармацевтическом препарате, поскольку разные твердые формы соединения обуславливают различные свойства лекарственного препарата.
[0030] Полиморфы молекулы могут быть получены многочисленными способами, известными в данной области техники, такими как, например, перекристаллизация из расплава, охлаждение расплава, перекристаллизация из растворителя, десольватация, быстрое испарение, быстрое охлаждение, медленное охлаждение, диффузия из паровой фазы и сублимация. Методы, характеризующие полиморф, включают порошковую рентгеновскую дифракцию (ПРД), рентгеновскую дифракцию монокристалла (РД), дифференциальную сканирующую калориметрию (ДСК), колебательную спектроскопию (например, ИК-спектроскопию и Раман-спектроскопию), твердотельный ядерный магнитный резонанс (ттЯМР), оптическую микроскопию в горячем состоянии, сканирующую электронную микроскопию (СЭМ), электронную кристаллографию и количественный анализ, анализ размера частиц (АРЧ), анализ площади поверхности, исследования растворимости и исследования растворения.
[0031] Термин «гидрат» относится к химической структуре, образованной в результате взаимодействия воды и соединения.
[0032] При использовании в настоящем документе термин «моногидрат» относится к гидрату, который содержит одну молекулу воды на одну молекулу субстрата.
[0033] При использовании в настоящем документе термин «кристаллический» относится к твердому веществу, в котором составные атомы, молекулы или ионы расположены симметрично упорядоченным, повторяющимся трехмерным образом.
[0034] Настоящее описание и формула изобретения содержат перечень элементов с применением выражения «выбран из … и …» и «представляет собой … или …» (иногда упомянуты как группы Маркуша). При использовании такого выражения в настоящей заявке, если не указано иное, оно включает эту группу в целом или любой из ее отдельных членов, или любую ее подгруппу. Данное выражение используют лишь для краткости, и оно никоим образом не означает ограничение удаления отдельных элементов или подгрупп при необходимости.
[0035] Предложена гидратная форма дигидрохлорида омекамтива мекарбила. В различных вариантах воплощения указанного аспекта гидратная форма дигидрохлорида омекамтива мекарбила является кристаллической (Форма А). Варианты воплощения гидратной формы дигидрохлорида омекамтива мекарбила могут быть описаны при помощи одного или более параметров, более подробно описанных ниже.
[0036] Гидратная форма дигидрохлорида омекамтива мекарбила имеет растворимость в воде более 40 мг/мл при рН в диапазоне приблизительно 3,5. Кроме того, Форма А является негигроскопичной. Например, при испытании динамической сорбции паров Форма А демонстрировала суммарное увеличение массы, равное приблизительно 0,55 масс.% при относительной влажности (RH) от приблизительно 40% до приблизительно 95%, и потерю массы приблизительно 2,7 масс.% при RH от приблизительно 30% до приблизительно 5%. В некоторых вариантах воплощения изобретения гидратная форма дигидрохлорида омекамтива мекарбила имеет, по существу, такой профиль динамической сорбции паров, как показан на фиг. 1, где под термином «по существу» подразумевается, что указанные характеристики ДСП могут варьироваться в пределах приблизительно ±5% RH.
[0037] Динамическая сорбция паров указывает, что данная соль теряет воду при высушивании до относительной влажности 5%, но практически полностью регидратируется при относительной влажности 15%. При относительной влажности более 15% образец является негигроскопичным, демонстрируя изменение массы лишь приблизительно 1,0% по достижении относительной влажности 95%. После испытания сорбции паров, по данным ПРД, не было обнаружено фазового перехода.
[0038] Определили, что растворимость Формы A в воде превышает 40 мг/мл (рН=3,5) при отсутствии фазового перехода в течение 24-часового испытания суспензии, по результатам ПРД. Кроме того, Форма A устойчива в условиях ускоренного испытания стабильности. Например, Форма A остается, по существу, в одной и той же физической форме в течение 6 месяцев при 40°С и 75% RH.
[0039] В различных вариантах воплощения изобретения Форма A может быть описана диаграммой порошковой рентгеновской дифракции, полученной так, как описано в Примерах, имеющей пики при приблизительно 6,6, 14,9, 20,1, 21,4 и 26,8±0,2° 2θ с применением излучения Cu Kα. Форма A, необязательно, может быть дополнительно описана диаграммой порошковой рентгеновской дифракции, имеющей дополнительные пики при приблизительно 8,4, 24,2, 26,0, 33,3±0,2° 2θ с применением излучения Cu Kα. Форма A, необязательно, может быть дополнительно описана диаграммой порошковой рентгеновской дифракции, имеющей дополнительные пики при приблизительно 6,2, 9,7, 13,2, 14,3, 15,4, 16,3, 16,9, 18,9, 19,5, 20,7, 21,8, 22,8, 23,6, 25,1, 27,3, 27,7, 28,4, 29,4, 30,2, 31,2, 31,5, 31,9, 33,9, 34,5, 34,9, 36,1, 36,8, 37,7, 38,5 и 39,7±0,2° 2θ с применением излучения Cu Kα. В разных случаях Форма A может быть описана диаграммой ПРД, имеющей пики при приблизительно 6,2, 6,6, 8,4, 9,7, 13,2, 14,3, 14,9, 15,4, 16,3, 16,9, 18,9, 19,5, 20,1, 20,7, 21,4, 21,8, 22,8, 23,6, 24,3, 25,1, 26,0, 26,8, 27,3, 27,7, 28,4, 29,4, 30,2, 31,2, 31,5, 31,9, 33,3, 33,9, 34,5, 34,9, 36,1, 36,8, 37,7, 38,5 и 39,7±0,2° 2θ с применением излучения Cu Kα. В некоторых вариантах воплощения изобретения Форма A имеет, по существу, такую диаграмму порошковой рентгеновской дифракции, как показана на фиг. 2, где под термином «по существу» подразумевается, что указанные пики могут варьироваться в пределах приблизительно ±0,2°. В области ПРД хорошо известно, что, хотя относительные интенсивности пиков в спектрах зависят от ряда факторов, таких как получение образца и геометрия прибора, положения пиков относительно нечувствительны к особенностям эксперимента.
[0040] Полиморфы омекамтива мекарбила Формы B и Формы C представляют собой метастабильные безводные дигидрохлоридные формы и они могут быть получены в различных условиях гидратации, указанных на фиг. 3, 4 и 6. Характеристические значения 2-тета Формы B включают 6,8, 8,8, 14,7, 17,7 и 22,3±0,2° 2θ с применением излучения Cu Kα и могут дополнительно включать пики при 9,6, 13,5, 19,2, 26,2±0,2° 2θ с применением излучения Cu Kα. Форма B может быть описана пиками диаграммы ПРД при 6,2, 6,8, 8,8, 9,6, 13,5, 14,4, 14,7, 15,4, 16,3, 17,0, 17,7, 18,3, 19,2, 19,9, 20,5, 20,8, 21,8, 22,3, 22,7, 23,0, 24,8, 25,1, 25,5, 26,2, 26,4, 26,8, 27,5, 28,5, 30,2, 30,6, 31,1, 31,5, 32,1, 32,7, 34,1, 34,4, 35,5, 35,9, 38,1, 38,9±0,2° 2θ с применением излучения Cu Kα. Характеристические значения 2-тета Формы C включают 6,7, 14,8, 17,4, 20,6 и 26,2±0,2° 2θ с применением излучения Cu Kα и могут дополнительно включать пики при 8,7, 22,0, 27,1 и 27,7±0,2° 2θ с применением излучения Cu Kα. Форма C может быть описана пиками диаграммы ПРД при 6,2, 6,7, 8,7, 9,6, 13,5, 14,5, 14,8, 15,4, 16,4, 17,1, 17,4, 18,4, 19,3, 19,5, 19,9, 20,6, 20,8, 21,8, 22,0, 22,5, 22,8, 24,3, 24,7, 25,1, 25,6, 26,2, 26,5, 27,1, 27,3, 27,7, 28,5, 30,0, 30,5, 31,0, 31,5, 32,2, 32,8, 34,1, 35,2, 36,0, 36,9 и 38,8±0,2° 2θ с применением излучения Cu Kα. В некоторых вариантах воплощения изобретения Формы B и C имеют, по существу, такую диаграмму порошковой рентгеновской дифракции, как показана на фиг. 6, где под термином «по существу» подразумевается, что указанные пики могут варьироваться в пределах приблизительно ±0,2°.
[0041] В различных вариантах воплощения изобретения Форма A может быть описана диаграммой рентгеновской дифракции монокристалла (РД), полученной так, как описано в разделе «Примеры», где Форма A имеет триклинную пространственную группу P-1 и параметры элементарной ячейки приблизительно a = 5,9979(4) Ǻ, b = 13,4375(9) Ǻ, c = 14,4250(9) Ǻ, α = 97,617(4)°, β = 93,285(4)° и γ = 94,585(5)°. Форма A, необязательно, может быть дополнительно описана параметрами РД, представленными ниже в таблице.
α = 97,617(4)°
b = 13,4375(9) Å
β = 93,285(4)°
c = 14,4250(9) Å
γ = 94,585(5)°
[0042] Для Формы A получили термограммы ДСК. Кривая ДСК показывает эндотермический переход, который, по-видимому, обусловлен плавлением/разложением при температуре приблизительно 235°С. Таким образом, в некоторых вариантах воплощения изобретения Форма A может быть описана термограммой ДСК, имеющей эндотерму разложения с началом в диапазоне от приблизительно 230°С до приблизительно 240°С при нагревании Формы A в открытом алюминиевом тигле. Например, в тех вариантах воплощения, в которых Форму A нагревают от приблизительно 25°С со скоростью приблизительно 10°С/мин, Форма A может быть описана термограммой ДСК, имеющей эндотерму разложения с началом при приблизительно 235°С, как показано на фиг. 5.
[0043] Форма A также может быть описана при помощи термогравиметрического анализа (ТГА). Так, Форма А может быть описана по потере массы в диапазоне от приблизительно 2% до приблизительно 5% с температурой начала в диапазоне от приблизительно 100°С до приблизительно 150°С. Например, Форма А может быть описана по потере приблизительно 3% массы до 150°С. В некоторых вариантах воплощения Форма А имеет, по существу, такие результаты термогравиметрического анализа, как показаны на фиг. 5, где под термином «по существу» подразумевается, что указанные характеристики ТГА могут варьироваться в пределах приблизительно ±5°С. При помощи анализа Карла Фишера (KF) определили, что указанная потеря массы относится к воде. Анализ KF показал, что содержание воды в Форме А может составлять приблизительно 3,7, что соответствует моногидрату.
[0044] Форма А может быть описана при помощи РПД при различных температурах и РПД при различной относительной влажности. Данные РПД при различных температурах представлены на фиг. 4. Указанные данные демонстрируют, что при нагревании гидрата Формы А до температуры выше точки десольватации, показанной на кривой ТГА (приблизительно 75°С), материал превращается в новую дегидратированную фазу, Форму B. При последующем охлаждении материала до условий окружающей среды Форма В повторно сорбирует воду из атмосферы и превращается обратно в гидрат Формы А. Данные РПД при различной относительной влажности представлены на фиг. 3. Указанные данные демонстрируют, что при воздействии на гидрат Формы А относительной влажности 5% материал превращается в новую дегидратированную фазу, Форму С. При воздействии на материал относительной влажности 15% и более Форма С повторно сорбирует воду из окружающей среды и превращается обратно в гидрат Формы А. Полученные данные согласуются с экспериментом сорбции паров. Наложенные друг на друга данные для Формы В и Формы С представлены на фиг. 6. Стрелками отмечены существенные отраженные сигналы двух порошковых диаграмм, указывающие на индивидуальность двух фаз.
[0045] Предложены также композиции, содержащие гидратную форму дигидрохлорида омекамтива мекарбила. В некоторых вариантах воплощения изобретения композиции содержат по меньшей мере приблизительно 50, приблизительно 60, приблизительно 70, приблизительно 80, приблизительно 90, приблизительно 95, приблизительно 96, приблизительно 97, приблизительно 98 или приблизительно 99% по массе гидратной формы дигидрохлорида омекамтива мекарбила. В некоторых вариантах воплощения изобретения композиции содержат по меньшей мере приблизительно 50, приблизительно 60, приблизительно 70, приблизительно 80, приблизительно 90, приблизительно 95, приблизительно 96, приблизительно 97, приблизительно 98 или приблизительно 99% по массе Формы A гидратной формы дигидрохлорида омекамтива мекарбила. В некоторых вариантах воплощения изобретения композиции содержат смесь двух или более Форм A, B и C.
[0046] Предложены также фармацевтические препараты, содержащие гидратную форму дигидрохлорида омекамтива мекарбила и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество. В некоторых вариантах воплощения изобретения препараты содержат по меньшей мере приблизительно 50, приблизительно 60, приблизительно 70, приблизительно 80, приблизительно 90, приблизительно 95, приблизительно 96, приблизительно 97, приблизительно 98 или приблизительно 99% по массе гидратной формы дигидрохлорида омекамтива мекарбила. В некоторых вариантах воплощения изобретения препараты содержат по меньшей мере приблизительно 50, приблизительно 60, приблизительно 70, приблизительно 80, приблизительно 90, приблизительно 95, приблизительно 96, приблизительно 97, приблизительно 98 или приблизительно 99% по массе Формы A гидратной формы дигидрохлорида омекамтива мекарбила. В некоторых вариантах воплощения изобретения препараты содержат смесь двух или более Форм A, B и C.
[0047] Предложен также способ применения указанных фармацевтических препаратов для лечения сердечной недостаточности, включая, но не ограничиваясь ими: острую (или декомпенсированную) застойную сердечную недостаточность и хроническую застойную сердечную недостаточность; в частности, заболевания, связанные с систолической сердечной дисфункцией.
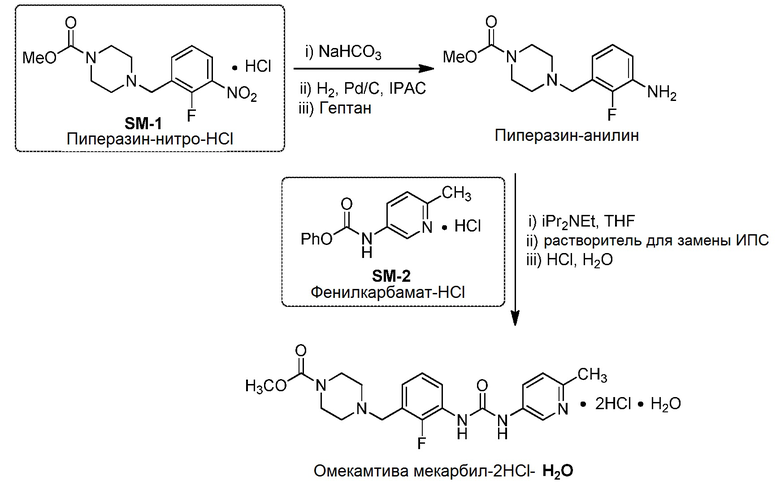
[0048] Предложен также синтез омекамтива мекарбила, включающий: смешивание метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилата и фенил-(6-метилпиридин-3-ил)карбамата в присутствии триалкиламинного основания с получением омекамтива мекарбила.
[0049] В некоторых вариантах воплощения изобретения массовое отношение гидрохлорида фенил-(6-метилпиридин-3-ил)карбамата (т.е. исходного вещества SM-2 или фенилкарбамата) к метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилату (т.е. исходному веществу SM-1 или пиперазину-нитро) составляет от приблизительно 1,1 до 1,5. В некоторых вариантах воплощения массовое отношение гидрохлорида фенил-(6-метилпиридин-3-ил)карбамата к метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилату составляет приблизительно 1,2.
[0050] В некоторых вариантах воплощения изобретения смешивание выполняют в присутствии апротонного растворителя. В некоторых вариантах воплощения растворитель представляет собой ТГФ.
[0051] В некоторых вариантах воплощения изобретения триалкиламинное основание представляет собой триэтиламин, диизопропилэтиламин или их комбинацию. В некоторых вариантах воплощения изобретения триалкиламинное основание содержит диизопропилэтиламин.
[0052] В некоторых вариантах воплощения изобретения используют избыток триалкиламинного основания. В некоторых вариантах воплощения используют от приблизительно 1,1 до 1,5 эквивалента триалкиламинного основания. В некоторых вариантах воплощения изобретения используют приблизительно 1,3 эквивалента триалкиламинного основания.
[0053] В некоторых вариантах воплощения изобретения смешивание выполняют при 65°С.
[0054] В некоторых вариантах воплощения изобретения указанный способ дополнительно включает кристаллизацию омекамтива мекарбила в присутствии водного раствора хлористоводородной кислоты и спиртового растворителя с получением гидрата дигидрохлорида омекамтива мекарбила.
[0055] В некоторых вариантах воплощения изобретения спиртовой растворитель содержит изопропиловый спирт.
[0056] В некоторых вариантах воплощения изобретения водный раствор хлористоводородной кислоты содержит 6 н. HCl.
[0057] В некоторых вариантах воплощения изобретения указанный способ дополнительно включает смешивание гидрата дигидрохлорида омекамтива мекарбила с по меньшей мере фармацевтически приемлемым вспомогательным веществом с получением фармацевтического препарата.
[0058] В некоторых вариантах воплощения изобретения фармацевтический препарат содержит гидрат дигидрохлорида омекамтива мекарбила; слой набухающего вещества; и покрытие из полупроницаемой мембраны, имеющее по меньшей мере одно впускное отверстие. Общие свойства лекарственного слоя и слоя набухающего вещества представлены в публикации патента США 2011/0182947, включенной в настоящий документ посредством ссылки.
[0059] В некоторых вариантах воплощения изобретения фармацевтический препарат представляет собой таблетку из матрицы с модифицированным высвобождением, содержащую гидрат дигидрохлорида омекамтива мекарбила; агент для регулирования высвобождения; агент для изменения рН; наполнитель и смазывающее вещество.
[0060] В некоторых вариантах воплощения изобретения метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилат получают способом, включающим: гидрирование метил-4-(2-фтор-3-нитробензил)пиперазин-1-карбоксилата в присутствии катализатора гидрирования с получением метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилата.
[0061] В некоторых вариантах воплощения изобретения катализатор гидрирования содержит палладий. В некоторых вариантах воплощения изобретения катализатор гидрирования представляет собой палладий на углероде.
[0062] Предложен также способ получения гидрата дигидрохлорида омекамтива мекарбила, включающий кристаллизацию омекамтива мекарбила в присутствии водного раствора хлористоводородной кислоты и спиртового растворителя с получением гидрата дигидрохлорида омекамтива мекарбила.
[0063] В некоторых вариантах воплощения изобретения спиртовой растворитель содержит изопропиловый спирт.
[0064] Предложен также способ получения гидрата дигидрохлорида омекамтива мекарбила, включающий:
(a) гидрирование метил-4-(2-фтор-3-нитробензил)пиперазин-1-карбоксилата в присутствии катализатора гидрирования с получением метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилата;
(б) смешивание метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилата и фенил-(6-метилпиридин-3-ил)карбамата в присутствии триалкиламинного основания с получением омекамтива мекарбила в виде свободного основания; и
(в) кристаллизацию свободного основания омекамтива мекарбила в присутствии водного раствора хлористоводородной кислоты и спиртового растворителя с получением гидрата дигидрохлоридной соли омекамтива мекарбила.
[0065] Указанный синтез обеспечивает высокий общий выход (более 70%). Кроме того, дигидрохлоридная соль, которая образуется в результате указанных стадий, при кристаллизации может быть получена в виде длинных стержней, имеющих улучшенные объемные свойства, время фильтрации, составляющее несколько минут (в отличие от нескольких дней для формы свободного основания), и высокую растворимость (более 40 мг/мл при рН 3,8). В разных случаях полученная соль представляет собой гидрат дигидрохлорида Формы A.
ПРИМЕРЫ
Общие способы
[0066] Реагенты и растворители использовали в таком виде, в котором они были приобретены из коммерческих источников. Спектры 1H ЯМР записывали на 400 МГц спектрометре. Химические сдвиги записаны в м.д. от тетраметилсилана с резонансом растворителя в качестве внутреннего стандарта (CDCl3, ДМСО-d6). Данные записаны следующим образом: химический сдвиг, мультиплетность (с = синглет, д = дублет, т = триплет, к = квартет, ш = широкий, м = мультиплет), константы связывания (Гц) и интеграция. Спектры 13C ЯМР записывали на 100 МГц спектрометре с полным отщеплением протонов. Химические сдвиги записаны в м.д. от тетраметилсилана с растворителем в качестве внутреннего стандарта (CDCl3, ДМСО-d6). Все количества растворителей записаны относительно исходного 2-фтор-3-нитротолуола.
[0067] Данные порошковой рентгеновской дифракции получили при помощи автоматического порошкового рентгеновского дифрактометра Phillips (X’Pert), оснащенного щелью фиксированного размера. Использовали излучение Cu Kα (1,541837 Å), а напряжение и сила тока составили 45 кВ и 40 мА, соответственно. Данные записывали при комнатной температуре от 3,000 до 40,009 градусов 2-тета; величина шага составила 0,008 градусов; время счета составило 15,240 секунд. Образцы массой 5-40 мг помещали на держатель образца и вращали предметный столик со временем одного оборота 2,000 секунды.
[0068] Термические свойства бис-HCl соли омекамтива мекарбила исследовали при помощи дифференциального сканирующего калориметра модели DSC Q 1000 или DSC Q 100, TA Instruments , и термогравиметрического анализатора Q 500, TA Instruments. Анализ данных выполняли при помощи программного обеспечения Universal Analysis 2000, TA Instruments. Для дифференциальной сканирующей калориметрии и термогравиметрического анализа во всех диапазонах температур использовали скорость нагревания 10°С/мин. Для анализа ДСК образцы массой в диапазоне <1-5 мг помещали в опрессованные, герметичные или открытые алюминиевые тигли.
[0069] Данные водного баланса получали при помощи симметричного паросорбционного анализатора VTI SGA 100. В цикле адсорбции относительную влажность изменяли с приращениями, составляющими 5%, в диапазоне от относительной влажности 5% до 95%, а в цикле десорбции в диапазоне от относительной влажности 95% до 5%. Критерий равновесия установили при изменении массы 0,01% в течение 1 минуты с максимальным временем уравновешивания 180 минут. Использовали приблизительно 1-15 мг образца.
[0070] Для рентгеновского кристаллографического анализа использовали бесцветную пластинку из C20H28Cl2FN5O4 с приблизительными размерами 0,03 мм × 0,12 мм × 0,50 мм. Данные об интенсивности рентгеновского излучения измеряли при 100(2) K на дифрактометре Bruker Kappa APEX II, оснащенном графитовым монохроматором и остро сфокусированной запаянной трубкой, излучающей CuKα (λ=1,54178Å), работающем при мощности 1,2 кВт (40 кВ, 30 мА). Детектор установили на расстоянии 5,0 см от кристалла.
[0071] Получили в целом 7824 кадра при ширине развертки 0,5° в ω и ϕ и времени воздействия 90 с/кадр. Общее время получения данных составило 260 часов. Кадры интегрировали при помощи пакета программного обеспечения Bruker SAINT, используя узкокадровый алгоритм интегрирования. В результате интегрирования данных с применением триклинной ячейки получили в целом 12349 отражений до максимального угла θ 69,57° (разрешение 0,83 Å), из которых 4046 были независимыми (избыточность 3,06, полнота = 93,6%, Rint = 5,13%, Rsig = 5,18%) и 3351 (82,8%) были больше >2сигма(I) σ (F2). Окончательные постоянные параметры ячейки, a=5,9979(4) Å, b=13,4375(9) Å, c=14,4250(9) Å, α=97,617(4)°, β=93,285(4)°, γ=94,585(5)°, объем=1145,95(13) Å3, основаны на улучшении XYZ-центроидов 4790 отражений свыше 20 σ(I) с 6,196°<2θ<138,239°. Анализ данных показал незначительное разложение в ходе сбора данных. Данные скорректировали по абсорбционному эффекту, используя метод многократного сканирования (SADABS). Отношение минимального к максимальному кажущемуся пропусканию составило 0,350. Рассчитанные коэффициенты минимального и максимального пропускания (на основании размера кристалла) составили 0,3206 и 0,9168.
[0072] Структуру определили и уточнили при помощи пакета программного обеспечения Bruker SHELXTL (версии 6.1), используя для формульной единицы C20H28Cl2FN5O4 пространственную группу P-1 с Z=2. Окончательное уточнение по методу наименьших квадратов с анизотропной полной матрицей на F2 с 320 переменными сошлось при R1=6,43% для наблюдаемых данных и wR2=19,18% для всех данных. Критерий согласия составил 1,067. Наибольший пик на окончательной карте электронной плотности составил 1,084 e-/Å3, а наибольшая дырка составила -0,527 e-/Å3 со среднеквадратическим отклонением 0,101 e-/Å3. На основании окончательной модели рассчитанная плотность составила 1,427 г/см3 и F(000) 516 e-.
[0073] В данной структуре были найдены и уточнены два положения, которые могут быть частично заняты водой. Занятость водой независимо уточнили до 53% и 41% для общего содержания воды 0,94 эквивалента воды на молекулу омекамтива мекарбила. Это согласуется с другими измерениями содержания воды в данной форме указанного соединения. Атомы водорода одной из сольватирующих молекул воды, имеющей занятость 41%, были обнаружены на карте разности электронной плотности и были уточнены как имеющие длину связи 1,01 Å. Были обнаружены и оставлены для изотропного уточнения атомы водорода у N3, C4 и N4. Все остальные атомы водорода поместили в идеализированные положения и уточнили в режиме Riding mode.
[0074] Данные порошковой рентгеновской дифракции (ПРД) получили на дифрактометре PANalytical X’Pert PRO (PANalytical, Алмело, Нидерланды), оснащенном многополосным детектором, работающим в реальном времени (RTMS). Использовали излучение CuKα (1,54 Å), а напряжение и силу тока установили на 45 кВ и 40 мА соответственно. Данные получали при комнатной температуре от 5 до 45 градусов 2-тета с величиной каждого шага 0,0334 градуса. Образцы поместили в держатель образца с низким фоном и установили на предметный столик, который вращали со временем одного оборота 2 секунды.
[0075] Альтернативно данные ПРД получили на дифрактометре PANalytical X’Pert PRO (PANalytical, Алмело, Нидерланды), оснащенном многополосным детектором RTMS. Использовали излучение CuKα(1,54 Å), а напряжение и силу тока установили на 45 кВ и 40 мА соответственно. Данные получали при комнатной температуре от 5 до 40 градусов 2-тета с величиной каждого шага 0,0334 градуса. Образцы поместили в держатель образца с низким фоном и установили на предметный столик, который вращали со временем одного оборота 2 секунды.
[0076] Альтернативно данные ПРД получили на дифрактометре PANalytical X’Pert PRO (PANalytical, Алмело, Нидерланды), оснащенном многополосным детектором RTMS. Использовали излучение CuKα(1,54 Å), а напряжение и силу тока установили на 45 кВ и 40 мА соответственно. Данные получали при комнатной температуре от 5 до 40 градусов 2-тета с величиной каждого шага 0,0167 градуса. Образцы поместили в держатель образца с низким фоном и установили на предметный столик, который вращали со временем одного оборота 2 секунды.
[0077] Альтернативно данные ПРД получили на дифрактометре PANalytical X’Pert Pro (PANalytical, Алмело, Нидерланды), оснащенном многополосным детектором RTMS. Использовали излучение CuKα(1,54 Å), а напряжение и силу тока установили на 45 кВ и 40 мА соответственно. Данные получали при комнатной температуре от 3 до 40 градусов 2-тета с величиной шага 0,008 градуса. Образцы поместили в держатель образца с низким фоном и установили на предметный столик со временем одного оборота 2 секунды.
[0078] Альтернативно данные ПРД получили на рентгенодифракционной системе Bruker D8 Discover (Bruker, Биллерика, штат Массачусетс), оснащенной xyz предметным столиком с электроприводом и детектором площади GADDS. Использовали излучение CuKα(1,54 Å), а напряжение и силу тока установили на 45 кВ и 40 мА соответственно. Твердые образцы картировали на плоской стеклянной пластинке и для каждого образца сканировали площадь 1 мм2 в режиме генерации 3 минуты от 5 до 48 градусов 2-тета.
[0079] Данные дифференциальной сканирующей калориметрии (ДСК) получили в стандартном режиме ДСК (DSC Q200, TA Instruments, Нью-Касл, штат Делавэр). Использовали скорость нагревания 10°С/мин в температурном диапазоне от 40°С до 300°С. Анализ выполняли под азотом, а образцы помещали в стандартные, герметично закрытые алюминиевые тигли. В качестве калибровочного стандарта использовали индий.
[0080] Альтернативно данные ДСК получили в режиме ДСК с модуляцией по температуре (DSC Q200, TA Instruments, Нью-Касл, штат Делавэр). После уравновешивания образца при 20°С в течение пяти минут использовали скорость нагревания 3°С/мин с модуляцией +/- 0,75°С/мин в температурном диапазоне от 20°С до 200°С. Анализ выполняли под азотом, а образцы помещали в стандартные, неопрессованные алюминиевые тигли. В качестве калибровочного стандарта использовали индий.
Получение гидрата дигидрохлорида омекамтива мекарбила
Синтез омекамтива мекарбила
Синтез исходного материала АФИ пиперазин-нитро-HCl
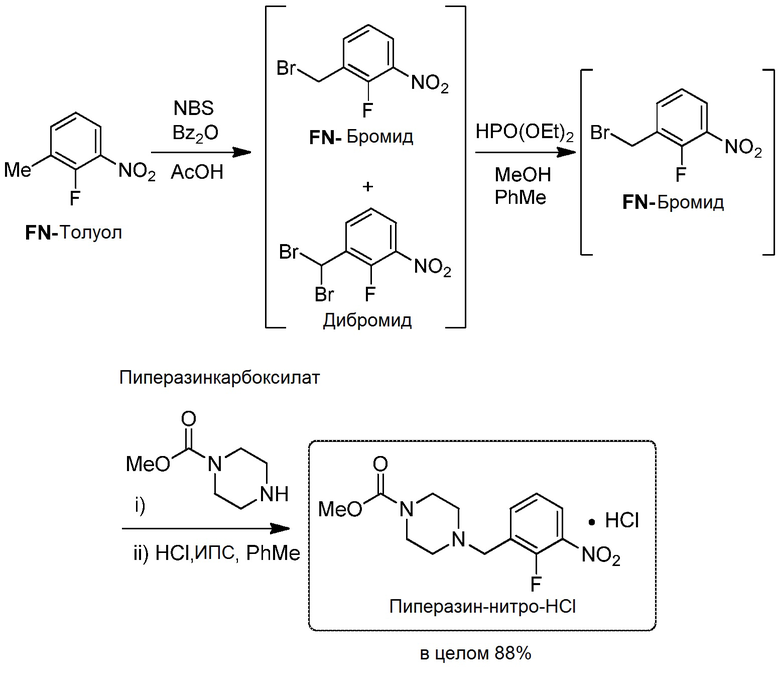
[0081] В реактор объемом 60 л (не содержащий открытой нержавеющей стали, Hastelloy® или других металлических деталей), оснащенный обратным конденсатором/обратным холодильником и скруббером, загрузили 5 н. раствор NaOH, механически перемешанную смесь FN-толуола (2,0 кг, 12,89 моль, 1,0 экв.), N-бромсукцинимида (3,9 кг, 21,92 моль, 1,70 экв.), перекиси бензоила (125,0 г, 0,03 экв., 0,39 моль, содержащей 25 масс.% воды) и уксусной кислоты (7,0 л, 3,5 объема) и нагревали до 85°С в атмосфере азота в течение 7 часов. Добавили приготовленный в отдельной емкости раствор H3PO3 (106,0 г, 1,29 моль, 0,1 экв.) и уксусной кислоты (200 мл, 0,1 объема). Реакционную смесь перемешивали в течение 0,5 часа и выполнили анализ аликвоты, который подтвердил завершение разложения перекиси бензоила (не обнаруживалась, ВЭЖХ254 нм). Реакционную смесь охладили до 22°С. Загрузили деионизированную воду (8,0 л, 4 объема) и толуол (16,0 л, 8 объемов); двухфазную смесь перемешивали (20 минут) и разделили слои. К органическому слою добавляли 1,6 н. водный раствор NaOH (14,0 л, 7,0 объемов) со скоростью, обеспечивающей возможность сохранения температуры смеси ниже 25°С, и измерили рН полученной водной фазы (≥11). Двухфазную смесь отфильтровали через 5 мкм слой картриджа Teflon® и разделили слои. Фильтровальный слой дополнительно промыли 2 л толуола.
[0082] Результаты анализа показали наличие 2,5% FN-толуола, 62,3% FN-бромида и 30,0% дибромида. Толуольный раствор не содержал перекиси бензоила, сукцинимида или α-бромуксусной кислоты, а содержание воды по KF титрованию составило 1030 м.д. (данный раствор можно хранить под азотом при комнатной температуре в течение >12 часов без изменения результатов анализа).
[0083] К полученному раствору при комнатной температуре добавили диизопропилэтиламин (880,0 г, 6,63 моль, 0,53 экв.), затем метанол (460 мл, 11,28 моль, 0,88 экв.) и нагрели до 40°С. Приготовили раствор диэтилфосфита (820,0 г, 5,63 моль, 0,46 экв.) в метаноле (460 мл, 11,28 моль, 0,88 экв.) и добавляли к реакционной смеси при 40°С через капельную воронку в течение 1 часа с такой скоростью, чтобы температура смеси составляла 40±5°С. Содержимое перемешивали при 40°С в течение 3 часов от начала добавления и охладили до комнатной температуры, и выдерживали в атмосфере азота в течение 12 часов. Результаты анализа показали, что реакционная смесь содержит 2,5% FN-толуола, 92,0% FN-бромида и 0,2% дибромида. Полученный раствор использовали в таком виде на стадии алкилирования.
[0084] Характеристики компонентов конечной смеси продуктов (полученные для чистых соединений).
[0085] 2-Фтор-3-нитротолуол (FN-толуол): 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ м.д. 2,37 (с, 1H), 7,13-7,20 (м, 1H), 7,45-7,51 (м, 1H), 7,79-7,85 (м, 1H). 13C ЯМР (100 МГц, хлороформ-d) δ м.д. 14,3 (д, J=5 Гц), 123,3 (д, J=3 Гц), 123,6 (д, J=5 Гц), 128,2 (д, J=16 Гц), 136,7 (д, J=5 Гц), 137,5 (широкий), 153,7 (д, J=261 Гц); 1-(бромметил)-2-фтор-3-нитробензол (FN-бромид): 1H ЯМР (400 МГц, хлороформ-d) δ м.д. 4,56 (с, 1H), 7,28-7,34 (м, 1H), 7,69-7,76 (м, 1H), 7,98-8,05 (м, 1H). 13C ЯМР (100 МГц, хлороформ-d) δ м.д. 23,6 (д, J=5 Гц), 124,5 (д, J=5 Гц), 126,1 (д, J=3 Гц), 128,5 (д, J=14 Гц), 136,5 (д, J=4 Гц), 137,7 (широкий), 153,3 (д, J=265 Гц). ДСК: однократное плавление при 53,59°С. Точная масса [C7H5BrFNO2 + H]+: расч. = 233,9566, измеренная = 233,9561; 1-(дибромметил)-2-фтор-3-нитробензол (дибромид): 1H ЯМР (400 МГц, хлороформ-d) δ м.д. 6,97 (с, 1H), 7,39-7,45 (м, 1H), 8,03-8,10 (м, 1H), 8,16-8,21 (м, 1H). 13C ЯМР (100 МГц, хлороформ-d) δ м.д. 29,2 (д, J=7 Гц), 124,9 (д, J=5 Гц), 127,1 (д, J=2 Гц), 132,1 (д, J=11 Гц), 135,7 (д, J=2 Гц), 137,2 (широкий), 149,8 (д, J=266 Гц). ДСК: однократное плавление при 49,03°С. Точная масса [C7H4Br2FNO2 + H]+: расч. = 311,8671, измеренная = 311,8666.
Пиперазин-нитро-HCl:
[0086] К механически перемешанному толуольному раствору (9 объемов) FN-бромида (полученного на предыдущей стадии) в реакторе объемом 60 л при 22°С в атмосфере азота добавили диизопропилэтиламин (1,90 кг, 14,69 моль, 1,14 экв.). К полученной смеси добавили раствор метилового эфира пиперазинкарбоксилата (пиперазинкарбоксилат) (2,03 кг, 14,05 моль, 1,09 экв.) в толуоле (1,0 л, 0,5 объема) со скоростью, обеспечивающей возможность сохранения температуры смеси ниже 30,0°С (экзотермический эффект). Во время добавления температуру рубашки довели до 5°С для поддержания температуры смеси ниже 30°С. Смесь перемешивали при 22°С в течение 3 часов и выполнили анализ аликвоты, который подтвердил завершение реакции алкилирования (процент площади ЖХ (LCAP) FN-бромида <1,0, ВЭЖХ254 нм). Реакционную смесь обработали водным раствором NH4Cl (20 масс.%, 10,0 л, 5 объемов; полученным из 2,0 кг NH4Cl и 10,0 л деионизированной воды), двухфазную смесь перемешивали (30 минут) и разделили слои. Затем органический слой промыли водным раствором NaHCO3 (9 масс.%, 10,0 л, 5 объемов; полученным из 0,90 кг NaHCO3 и 10,0 л деионизированной воды). Органический слой отфильтровали через 5 мкм слой картриджа Teflon® и перенесли в барабан, дополнительно промыли слой фильтра 1,0 л толуола, взвесили объединенный толуольный раствор (10,0 объемов) и анализировали (ВЭЖХ) для количественного определения свободного основания пиперазин-нитро. Анализ показал содержание свободного основания пиперазин-нитро 89,0%, FN-толуола 2,5% и FN-бромида 0,2% при отсутствии обнаруживаемого FN-бромида. Общие потери продукта из-за промывания водой составили <1,0%. Полученный раствор устойчив в атмосфере азота в течение более 12 часов.
[0087] К механически перемешанному толуольному раствору свободного основания пиперазин-нитро, полученному так, как описано выше, при 22°С в реакторе объемом 60 л в атмосфере азота добавили ИПС (19,4 л, 9,7 объемов) и деионизированную воду (1,0 л, 0,5 объема). Смесь нагрели до 55°С и добавили 20% 1,4 экв. концентрированной HCl (перед использованием оттировали и определили общее количество по значению титра; 276,0 мл, 3,21 моль). Содержимое перемешивали в течение 15 минут и добавили затравочные кристаллы пиперазин-нитро-HCl (130,0 г, 0,39 моль, 0,03 экв.) в виде суспензии в ИПС (400 мл, 0,2 объема). Смесь перемешивали в течение 30 минут и за 4 часа добавили остальное количество концентрированной HCl (80% от общего количества, 1,10 л, 12,82 моль). Смесь перемешивали при 55°С в течение 1 часа, линейно охладили до 20°С за 1,5 часа и перемешивали при этой температуре в течение 12 часов. Измерили концентрацию надосадочного раствора пиперазин-нитро-HCl (2,8 мг/г). Смесь отфильтровали через фильтр Aurora с 5 мкм тканью Teflon®. Маточный раствор перенесли в чистый барабан и выполнили анализ. Осадок на фильтре дважды промыли ИПС (11,2 л, 5,6 объемов) и высушили до постоянной массы (определенной как потеря массы ≤1,0% в ходе 2 последовательных измерений ТГА в течение 2 часов) на фильтре под вакуумом и продувая азотом (14 часов). Суммарные потери пиперазин-нитро-HCl в маточных растворах и промывочных растворах составили 2,5%. Масса выделенного пиперазин-нитро-HCl составила 3,59 кг с выходом 87,6%, поправленным на >99,5 масс.%, и чистотой по LCAP 99,0%.
[0088] Метил-4-(2-фтор-3-нитробензил)пиперазин-1-карбоксилата гидрохлорид (пиперазин-нитро-HCl): 1H ЯМР (300 МГц, ДМСО-d) δ м.д. 3,25 (ш с, 3H), 3,52-3,66 (м, 8H), 4,47 (с, 2H), 7,44-7,63 (т, 1H, J=8 Гц), 7,98-8,15 (м, 1H), 8,17-8,34 (м, 1H). 13C ЯМР (75 МГц, ДМСО-d) δ м.д. 50,3, 51,4, 52,8, 119,6 (д, J=14 Гц), 125,1 (д, J=5 Гц), 127,9, 137,4 (д, J=8 Гц), 139,8 (д, J=3 Гц), 152,2, 154,7, 155,7. ДСК: начало плавления при 248,4°С. Точная масса [C13H16FN3O4 + H]+: рассчитанная = 298,1203, измеренная = 298,1198.
Альтернативный способ синтеза пиперазин-нитро:
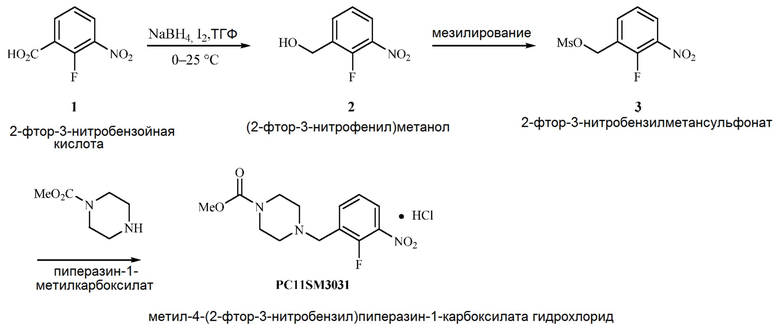
[0089] Смесь NaBH4 (1,7 г, 44 ммоль) в ТГФ (68 мл) обработали 2-фтор-3-нитробензойной кислотой (3,4 г, 18,4 ммоль) и охладили до 0–5°C. Затем по каплям добавляли раствор йода (4,7 г, 18,4 ммоль) в ТГФ (12 мл) с такой скоростью, чтобы регулировать выделение газа. Ход реакции анализировали по ВЭЖХ. Через 2 часа анализ ВЭЖХ показал наличие остаточной 2-фтор-3-нитробензойной кислоты с AUC 4%. Смесь погасили, вылив в 1 M раствор HCl (30 мл) и экстрагировали МТБЭ (5 мл). Затем органический слой промыли 20% водным раствором KOH и 10% раствором тиосульфата натрия. Органический слой высушили при помощи Na2SO4, отфильтровали через целит и концентрировали с получением (2-фтор-3-нитрофенил)метанола (2,8 г, 88%, AUC 89% по ВЭЖХ).
[0090] Раствор (2-фтор-3-нитрофенил)метанола (2,8 г, 16 ммоль) в 2-MeТГФ (26 мл) обработали триэтиламином (4,5 мл, 32 ммоль) и охладили до 0–5°C. Затем раствор обработали метансульфонилхлоридом (1,6 мл, 21 ммоль). Ход реакции анализировали по ВЭЖХ. Через 30 минут при 0–5°C реакцию считали завершенной. Смесь погасили водой (14 мл) и разделили фазы. Органическую фазу промыли насыщенным солевым раствором, высушили при помощи Na2SO4, отфильтровали через целит и концентрировали с получением 2-фтор-3-нитробензилметансульфоната (3,3 г, 83,1%, AUC 81% по ВЭЖХ) в виде желтого маслянистого вещества.
[0091] Раствор 2-фтор-3-нитробензилметансульфоната (3,3 г, 13 ммоль, AMRI, партия № 46DAT067B) в толуоле (33 мл) обработали одной порцией диизопропилэтиламина (2,7 мл, 15 ммоль). Через шприц медленно добавили раствор метилпиперазин-1-карбоксилата (2,1 г, 15 ммоль) в толуоле (1,1 мл), чтобы поддерживать температуру в диапазоне 23–29°C. После добавления реакционную смесь перемешивали в течение 16 часов. Через это время анализ ВЭЖХ показал завершение реакции. Добавили 20% водный раствор NH4Cl (11 мл) при 20–25°C. Двухфазную смесь перемешивали в течение 15 минут и разделили фазы. Этот процесс повторили с использованием 9% водного раствора бикарбоната натрия (11 мл). Затем толуольный слой отфильтровали через целит при 20–25°C. К толуольному раствору добавили 2-пропанол (50 мл) и воду (1,1 мл) и нагрели смесь до 55–60°C. Затем смесь обрабатывали 37 масс.% раствором HCl (1,6 мл, 18,7 ммоль) в течение 20 минут. После добавления заметили осадок. После завершения добавления смесь оставили постепенно остывать до 20–25°С и перемешивали несколько часов, а затем отфильтровали и промыли ИПС (2 объема слоя).
[0092] Затем осадок на фильтре высушили под вакуумом с получением 4-(2-фтор-3-нитробензил)пиперазин-1-карбоксилата гидрохлорида (2,41 г, 54%, AUC 90% по ВЭЖХ, 88 масс.% по ВЭЖХ).
Свободное основание пиперазин-нитро:
[0093] В реакторе объемом 60 л, оснащенном обратным конденсатором/обратным холодильником, механически перемешивали смесь пиперазин-нитро-HCl (2,0 кг, 5,99 моль, 1,0 экв.) и изопропилацетата (6,0 л, 3,0 объема) при комнатной температуре в атмосфере азота. Добавили раствор бикарбоната натрия (629 г, 7,49 моль, 1,25 экв.) в воде (7,5 л, 3,75 объема), полученный в отдельной емкости. Двухфазную смесь перемешивали (15 минут) и разделили слои. Верхний органический слой (содержащий продукт) перенесли в делительную воронку, а реактор промыли водой и изопропанолом. Затем органический слой пропустили через подключенный 5 мкм картридж Teflon® и вернули в чистый реактор объемом 60 л. Фильтровальный слой промыли 4,0 л (2,0 объема) изопропанола в указанный реактор объемом 60 л. В реактор объемом 60 л дополнительно добавили 12,0 л (6,0 объемов) изопропанола и нагрели до 40°С. Под пониженным давлением (50 Торр) смесь концентрировали приблизительно до 6 л (3,0 объема). Раствор линейно охладили с 27°С до 20°С за 10 минут. В течение 30 минут добавляли воду (4,0 л, 2,0 объема) при 20°С, а затем добавляли затравочные кристаллы свободного основания пиперазин-нитро (18 г, 0,06 моль, 0,01 экв.). Смесь выстаивали в течение 5 минут и за 90 минут добавили остальное количество воды (24,0 л, 12,0 объемов). После выдерживания в течение ночи при 20°С измерили концентрацию свободного основания пиперазин-нитро в надосадочном растворе (<10 мг/мл). Смесь отфильтровали через фильтр Aurora, оснащенный 12 мкм вкладышем Teflon®. Осадок на фильтре промыли смесью воды (3,3 л, 1,65 объема) и изопропанола (700 мл, 0,35 объема) и высушили до постоянной массы (определенной как потеря массы ≤1,0% в ходе 2 последовательных измерений ТГА в течение 2 часов) на фильтре под вакуумом и продувая азотом (48 часов). Суммарные потери свободного основания пиперазин-нитро в маточном растворе и промывочных растворах составили приблизительно 7,5%. Масса выделенного свободного основания пиперазин-нитро составила 1,67 кг с выходом 92,5%, поправленным на 100,0 масс.%, и чистотой по LCAP 99,4%.
Синтез исходного материала АФИ фенилкарбамат-HCl
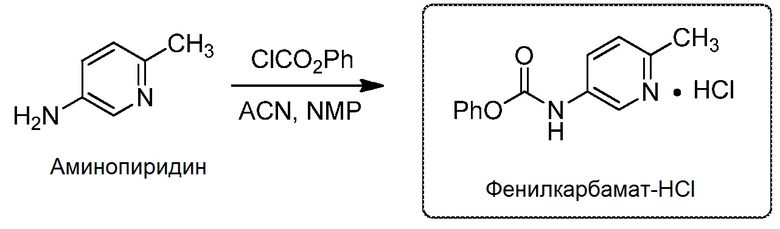
[0094] В эмалированный реактор объемом 60 л, оснащенный рубашкой, установленной на 20°С, в атмосфере азота с продуванием через скруббер (содержащий 5 н. раствор NaOH) загрузили 2,5 кг аминопиридина (1,0 экв., 23,1 моль), затем 25 л (19,6 кг, 10 объемов) ацетонитрила. После начала перемешивания и (эндотермического) растворения аминопиридина в реактор загрузили 12,5 л N-метил-2-пирролидинона (12,8 кг, 5 объемов). В капельную воронку поместили 1,8 л (0,6 экв., 13,9 моль) фенилхлорформиата, который затем добавляли в течение 68 минут к раствору аминопиридина, поддерживая внутреннюю температуру ≤30°C. Реакционную смесь перемешивали в течение >30 минут при внутренней температуре 20±5°C. Затем в реактор загрузили 61±1 г затравочных кристаллов в виде суспензии в 200 мл ацетонитрила и выстаивали в течение ≥30 минут. В капельную воронку поместили 1,25 л (0,45 экв., 9,7 моль) фенилхлорформиата, который затем добавляли в течение 53 минут к реакционной суспензии, снова поддерживая температуру ≤30°C. Содержимое реактора выстаивали в течение ≥30 часов при 20±5°C. После анализа надосадочного раствора (≤15 мг/г продукта и исходного материала) твердые вещества отфильтровали через фильтр Aurora, оснащенный 12 мкм вкладышем Teflon. Маточный раствор слили во второй эмалированный реактор объемом 60 л, оснащенный рубашкой. Реактор и осадок на фильтре промыли 1×10 л смеси 5:10 NMP/ACN и 1×10 л ACN. Промывочные растворы также слили во второй реактор. Осадок на фильтре высушили под вакуумом, продувая азотом в течение ≥24 часов, с получением 5,65 кг (выход 90,2%) продукта фенилкарбамат-HCl в виде грязновато-белого твердого вещества с 98,8 масс.%, с чистотой по LCAP 99,2%.
[0095] Фенил-(6-метилпиридин-3-ил)карбамата гидрохлорид (фенилкарбамат-HCl) 1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 11,24 (с, 1H), 8,81 (с, 1H), 8,41 (д, 1H, J=8,8 Гц), 7,85 (д, 1H, J=8,8 Гц), 7,48-7,44 (м, 2H), 7,32-7,26 (м, 3H), 2.69 (с, 3H); 13C ЯМР (100 МГц, ДМСО-d6) δ м.д. 151,66, 150,01, 147,51, 136,14, 133,79, 129,99, 129,49, 127,75, 125,87, 121,70, 18,55: МС-ВР: рассчитано для C13H12N2O2: 228,0899, M + H+ = 229,0972; наблюдаемая масса: 229,0961
Альтернативный синтез фенилкарбамат-HCl
[0096] 5-Амино-2-метилпиридин (53,2 кг, 1,0 экв.) и ацетонитрил (334 кг, 8,0 мл/г) загрузили в продутый азотом эмалированный реактор. Содержимое реактора перемешивали при нагревании до 25–30°С. Затем смесь рециркулировали через фильтр, заполненный активированным углем (11 кг, 20 масс.%) с интервалами в 3 часа, поддерживая температуру 25–30°С. После каждого 3-часового интервала анализировали цвет образца смеси, сравнивая его с цветовым стандартом и УФ-поглощением при 440 нм. По достижении удовлетворительного результата содержимое фильтра выдули в реактор и промыли фильтр ацетонитрилом (85 кг, 2,0 мл/г). Промывочный ацетонитрил перенесли в реакционную смесь. В реакционную смесь в эмалированном реакторе добавили 1-метил-2-пирролидинон (274 кг, 5,0 мл/г). К смеси медленно добавляли фенилхлорформиат (46,6 кг, 0,6 экв.), поддерживая температуру 15–30°C (обычно в течение 60–70 мин). Реакционную смесь перемешивали приблизительно 60 минут, поддерживая температуру 20–25°C. В перемешиваемую смесь добавили затравочные кристаллы фенил-(6-метилпиридин-3-ил)карбамата гидрохлорида (0,58 кг, 0,010 экв.). Затем суспензию перемешивали приблизительно 4 часа при 20±5°C. К суспензии медленно добавили фенилхлорформиат (33,4 кг, 0,45 экв.), поддерживая температуру 15–30°С. Затем смесь оставили созревать, перемешивая в течение 8±1 час, после чего сразу проверили концентрацию 5-амино-2-метилпиридина (требуемое значение ≤15 мг/мл) и фенил-(6-метилпиридин-3-ил)карбамата гидрохлорида (требуемое значение ≤15 мг/мл) по ВЭЖХ. Затем смесь отфильтровали под вакуумом и промыли смесью ацетонитрила (112 кг, 2,68 мл/г) и 1-метил-2-пирролидинона (72 кг, 1,32 мл/г), затем трижды промыли ацетонитрилом (167 кг, 4,0 мл/г). Твердые вещества высушили, затем перенесли в полочную сушилку, поддерживаемую при температуре в диапазоне 20–40°C и абсолютном давлении 8,96-4,48 кПа (1,3–0,65 фунт/квадратный дюйм), до достижения потерь при сушке <1 масс.%, после чего сразу убрали фенил-(6-метилпиридин-3-ил)карбамата гидрохлорид, 106,3 кг (выход 81,6%) из сушилки.
Метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилат (пиперазин-анилин)
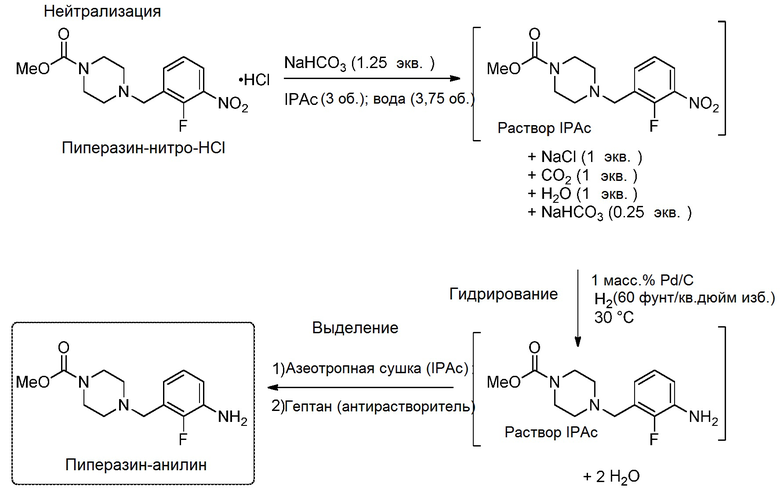
[0097] В эмалированный реактор объемом 100 л, оснащенный рубашкой, добавили метил-4-(2-фтор-3-нитробензил)пиперазин-1-карбоксилата гидрохлорид (2,00 кг, 1,00 экв.) и изопропилацетат (6,00 л, 3,00 объема относительно исходного материала). Полученную суспензию перемешивали, продувая азотом. За 45±30 минут к смеси по каплям добавили: 7,7% масса/массу водного раствора бикарбоната натрия (629 г, 1,25 экв. бикарбоната натрия, растворенного в 7,50 л воды), поддерживая внутреннюю температуру 20±5°C, регулируя рубашку (примечание: добавление является эндотермическим и может приводить к выделению до 1 экв. газообразного диоксида углерода). Смесь перемешивали в течение ≥15 минут с получением прозрачной двухфазной смеси. Перемешивание прекратили и оставили слои отстаиваться.
[0098] Нижний (водный) слой слили и анализировали при помощи рН-индикаторной бумаги, чтобы убедиться, что рН слоя >6. Количественный ВЭЖХ анализ верхнего (органического) слоя показал аналитический выход 97-100% свободного основания метил-4-(2-фтор-3-нитробензил)пиперазин-1-карбоксилата (1,73-1,78 кг). Верхний (органический) слой перенесли через подключенный фильтр в 20 л реактор для гидрирования Hastelloy® и промыли 100 л реактор и фильтровальные слои дополнительной аликвотой изопропилацетата (2,00 л, 1,00 объем). Реактор для гидрирования продули азотом и сбросили давление до атмосферного. К реакционной смеси добавили суспензию 5,0 масс.% палладия на углероде (20,0 г, Strem/BASF Escat™ 1421, приблизительно 50% воды) в изопропилацетате (400 мл), затем еще 400 мл для промывания. Полученную реакционную смесь разбавили дополнительной аликвотой изопропилацетата (1,2 л; общее количество изопропилацетата 10,0 л, 5,00 объемов). Реактор для гидрирования три раза продули азотом [сжатым до 414±69 кПа (60±10 фунт/кв. дюйм) изб., затем сбросили давление до атмосферного], затем закачали водород до давления 414±35 кПа (60±5 фунт/кв. дюйм) изб. Реакционную смесь перемешивали при <100 оборотов/мин при 30±5°C, поддерживая давление водорода 414±35 кПа (60±5 фунт/кв. дюйм) изб., в течение >2 часов до момента, когда реакцию считали завершенной. Указанная температура и давление соответствуют измеренному значению kLa, равному приблизительно 0,40 в реакторе для гидрирования объемом 20 л. Окончание реакции установили по резкому снижению расхода водорода, сопровождающемуся снижением выделения теплоты реакции. Для устранения возможных димерных примесей реакцию продолжали в течение по меньшей мере 30 минут после указанного изменения профиля реакции и выполнили анализ ВЭЖХ для подтверждения достижения превращения гидроксиламина в анилин >99,5%.
[0099] По окончании реакции реактор для гидрирования дважды продули азотом [сжатым до 414±69 кПа (60±10 фунт/кв. дюйм) изб., затем сбросили давление до атмосферного]. Неочищенную реакционную смесь отфильтровали через 5 мкм фильтр, затем последовательно через 0,45 мкм фильтр, в эмалированный реактор объемом 40 л. Реактор для гидрирования и фильтровальные вкладыши промыли дополнительной аликвотой изопропилацетата (2,00 л). Количественный ВЭЖХ анализ неочищенной реакционной смеси показал аналитический выход 95-100% (1,52-1,60 кг анилинового продукта). Реакционную смесь перегоняли под пониженным давлением (обычно 250–300 мбар) при температуре смеси 50±5°C до достижения общего реакционного объема приблизительно 8,00 л (4,00 объема). Смесь перегоняли при постоянном объеме при 50±5°C, 250–300 мбар, добавляя гептан для регулирования общего объема смеси. Затем добавили приблизительно 8,00 л (4,00 объема) гептана, анализ ГХ показал, что растворитель содержит приблизительно 50% изопропилацетата, 50% гептана. Вакуум сняли, а внутреннюю температуру смеси поддерживали при 50±5°C. К реакционной смеси добавили суспензию затравочных кристаллов (20,0 грамм продукта метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилата в смеси растворителей из 80 мл гептана и 20 мл изопропилацетата). Полученную суспензию оставили перемешиваться при 50±5°C в течение 2±1 час, затем охладили до 20±5°С за 2,5±1,0 часа. В течение 2 часов по каплям добавляли дополнительное количество гептана (24,0 л, 12,0 объемов) и оставили смесь перемешиваться при 20±5°C в течение ≥1 часа (обычно в течение ночи). Количественный ВЭЖХ анализ отфильтрованного надосадочного раствора показал содержание продукта в растворе <5 мг/мл, а кристаллы продукта представляли собой двулучепреломляющие стержни размером 50–400 мкм. Реакционную суспензию отфильтровали при 20°С через фильтрующую ткань и выполнили промывку осадка на фильтре вытеснением гептаном (6,00 л, 2,00 объемов). Осадок высушивали на фильтре под потоком азота при комнатной температуре в течение >4 часов до подтверждения сухости образца в анализе потерь при высушивании (потери составили <1,0 масс.%). Продукт метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилат (1,56 кг) выделили в виде бледно-желтого порошка с выходом 86% при 99,8 масс.% по ВЭЖХ, со значением LCAP210 100,0. [Анализ объединенных фильтратов и промывочных растворов показал, что в потери продукта в маточных растворах составили 108 грамм (7,0%). Остальная часть массового баланса состоит из массы продукта в реакторе (налет)]. 1H ЯМР (ДМСО-d6, 400 МГц) δ: 6,81 (дд, J=7,53, 7,82 Гц, 1H), 6,67 (м, 1H), 6,49 (м, 1H), 5,04 (с, 2H), 3,58 (с, 3H), 3,45 (м, 2H), 3,34 (м, 4H), 2,33 (м, 4H). 19F ЯМР (d6-ДМСО, 376 МГц) δ: -140,2. 13C ЯМР (d6-ДМСО, 125 МГц) δ: 155,0, 150,5, 148,2, 136,2 (м), 123,7 (м), 117,6, 115,1, 73,7, 54,9 (м), 52,1 (м), 43,4. Тпл. = 89,2°C.
Альтернативный способ получения пиперазин-анилина
[0100] В эмалированный реактор, оснащенный рубашкой, добавили метил-4-(2-фтор-3-нитробензил)пиперазин-1-карбоксилата гидрохлорид (46,00 кг, 1,00 экв.) и изопропилацетат (200 кг, 5,0 мл/г). Полученную суспензию перемешивали, продувая азотом. К смеси добавили 7,4% масса/массу водный раствор бикарбоната натрия (1,25 экв.), поддерживая внутреннюю температуру 25±5°C. Смесь перемешивали в течение ≥30 минут с получением прозрачной двухфазной смеси. Перемешивание прекратили и слили нижний (водный) слой. Анализ водного слоя показал pH ≥6. К органическому слою добавили воду (92 кг, 2,0 мл/г) и перемешивали в течение ≥15 минут. Затем перемешивание прекратили и слили нижний (водный промывочный) слой. К органическому слою добавили воду (92 кг, 2,0 мл/г) и перемешивали в течение ≥15 минут. Затем перемешивание прекратили и слили нижний (водный промывочный) слой. Смесь перегоняли под пониженным давлением, поддерживая температуру смеси в диапазоне 40–50°С. В ходе перегонки поддерживали постоянный объем смеси путем непрерывного добавления изопропилацетата. Когда содержание воды в смеси составило <1500 м.д., раствор пропустили через встроенный фильтр в реактор из сплава Хастеллой, содержащий 5,0 масс.% палладия на углероде (BASF Escat 1421, 0,69 кг, 1,5 масс.%). Эмалированный реактор, оснащенный рубашкой, промыли изопропилацетатом (100 кг, 2,5 мл/г) и добавили промывочный раствор в реактор из сплава Хастеллой через встроенный фильтр.
[0101] Смесь довели до температуры приблизительно 25–35°С (предпочтительно 30°С) и добавили газообразный водород, поддерживая приблизительно 4 бар изб. при энергичном перемешивании. После прекращения расхода водорода гидрирование продолжали в течение 1 часа с достижением превращения ≥99,0% по ВЭЖХ. Катализатор палладий на углероде собрали фильтрацией, а надосадочный раствор собрали в реактор. В реактор из сплава Хастеллой загрузили изопропилацетат (40 мг, 1,0 мл/г) и перенесли через фильтр в эмалированный реактор с рубашкой.
[0102] Смесь концентрировали под пониженным давлением, поддерживая температуру смеси в диапазоне 35–55°С до конечного объема приблизительно 4,0 мл/г. В эмалированный реактор с рубашкой добавили гептан (219 кг, 7,0 мл/г), поддерживая температуру смеси в диапазоне 50–60°С до достижения концентрации 20–25% изопропилацетата в гептане, измеренной при помощи ГХ. Раствор охладили до 40–50°С и внесли затравочные кристаллы метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилата (0,46 кг, 1,0 масс.%) в виде суспензии в гептане (6,4 кг, 0,20 мл/г). Суспензию выдерживали приблизительно 2 часа и сразу после этого перегоняли смесь под пониженным давлением, поддерживая температуру смеси в диапазоне 35–45°С. Во время перегонки поддерживали постоянный объем смеси путем непрерывного добавления гептана (219 кг, 7,0 мл/г). Затем смесь охлаждали до 15–25°С в течение приблизительно 3 часов. При помощи ВЭЖХ измерили концентрацию метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилата в надосадочном растворе, которая составила ≤5 мг/мл.
[0103] Смесь отфильтровали, а полученные твердые вещества последовательно промыли гептаном (63 кг, 2,0 мл/г), затем снова гептаном (94 кг, 3,0 мл/г). Твердые вещества высушили на фильтре потоком сухого азота с вакуумом до достижения потерь при высушивании ≤1 масс.%, после чего из осушителя фильтра получили 33,88 кг (выход 90,7%) вещества.
Способ получения гидрата дигидрохлорида омекамтива мекарбила
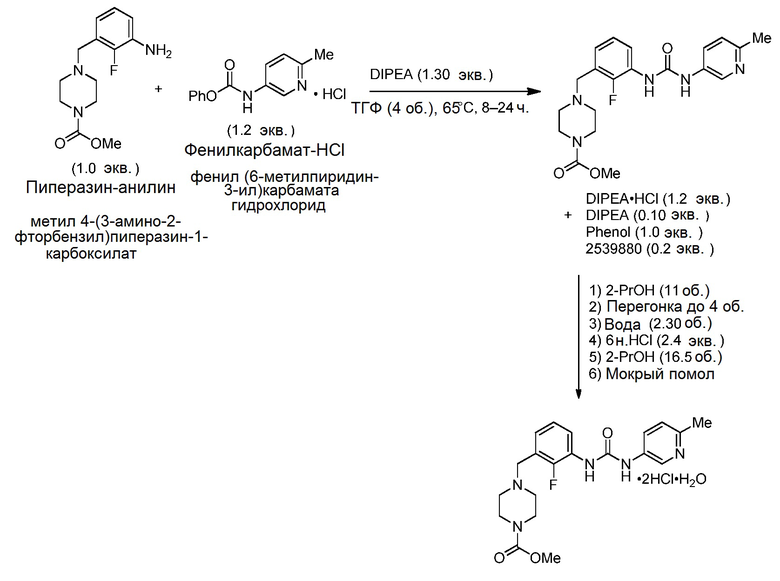
[0104] В эмалированный реактор объемом 15 л загрузили метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилат (1202 г, 4,50 моль), фенил-(6-метилпиридин-3-ил)карбамата гидрохлорид (1444 г, 5,40 моль) и тетрагидрофуран (4,81 л). Полученную суспензию перемешивали под потоком азота, а затем к суспензии добавили N,N-диизопропилэтиламин (1019 л, 5,85 моль) с получением коричневого раствора. Температуру раствора повысили до 65°С и перемешивали в течение 22 часов до AUC остаточного пиперазин-анилина <1% по анализу ВЭЖХ.
[0100] Смесь охладили до 50°С и перегнали под пониженным давлением, поддерживая внутреннюю температуру реактора ниже 50°С, регулируя давление вакуума. С остаточным вакуумом добавили 2-пропанол с такой скоростью, чтобы поддерживать постоянный объем в реакторе объемом 15 л. В целом потребовалось 10,5 кг 2-пропанола для достижения <5% ТГФ по ГХ. Затем в реактор загрузили воду (2,77 кг), после чего добавили 6 н. раствор HCl (1,98 кг) с такой скоростью, чтобы поддерживать внутреннюю температуру ниже 60°С. Давление в реакторе сбросили до атмосферного, продувая азотом. Затем раствор нагрели до 60°С и перенесли в эмалированный реактор объемом 60 л через встроенный фильтр. Затем промыли 15 л реактор смесью 1:1 воды/2-пропанола (1,2 л), которую направили через встроенный фильтр в реактор объемом 60 л.
[0101] Температуру в реакторе объемом 60 л довели до 45°С и добавили в реактор суспензию затравочных кристаллов (114 г, 0,23 моль) в 2-пропаноле (0,35 л) с получением суспензии. Смесь выстаивали при 45°С в течение 1 часа, затем в течение 2 часов добавляли 2-пропанол (3,97 кг) через встроенный фильтр. Смесь нагревали до 55°С в течение 1 часа и выдерживали в течение 0,25 часа, затем охладили до 45°С за 1 час и выдерживали в течение ночи при 45°С. Затем в смесь через встроенный фильтр добавляли 2-пропанол (11,71 кг) в течение 3 часов. Смесь выстаивали в течение 1 часа, а затем охлаждали до 20°С в течение 2 часов и выдерживали при 20°С в течение 0,5 часа. Затем смесь рециркулировали через мельницу влажного помола, оснащенную 1 ротором-статором для среднего помола и 2 роторами-статорами для тонкого помола, эксплуатируемую при 56 Гц в течение 2,15 часа, до момента, когда под микроскопом перестали наблюдать уменьшение размера частиц.
[0102] Затем смесь отфильтровали через 20” фильтр Hastelloy®, оснащенный 12 мкм фильтровальной тканью, под вакуумом 500 Торр. Через встроенный фильтр в реактор объемом 60 л пропустили промывочный раствор 95:5 2-пропанола:воды (1,82 л), затем на фильтр. Через встроенный фильтр в реактор объемом 60 л пропустили второй промывочный 2-пропанол (2,85 л), затем на фильтр. Затем смесь высушили под давлением влажного азота 35 кПа (5 фунтов/кв. дюйм) до остаточного содержания 2-пропанола <5000 м.д. и воды 2,5–5%. Полученное твердое вещество сняли с фильтра с получением 2,09 кг метил-4-(2-фтор-3-(3-(6-метилпиридин-3-ил)уреидо)бензил)пиперазин-1-карбоксилата в виде грязновато-белого кристаллического твердого вещества с выходом 89% при 99,88 масс.% по ВЭЖХ, AUC 100,0%. Общие потери в растворах составили 0,10 кг (4,7%).
[0103] ДСК: Tначала = 61,7°С, Tmax = 95,0°С; ТГА = 2,2%, начало разложения = 222°С; 1H ЯМР (D2O, 500 МГц) δ 8,87 (с, 1H), 8,18 (д, J=8,9 Гц, 1H), 7,83 (т, J=7,5 Гц, 1H), 7,71 (д, J=8,8 Гц, 1H), 7,35–7,29 (м, 2H), 4,48 (с, 2H), 4,24 (ш с, 2H), 3,73 (с, 3H), 3,31 (ш с, 6H), 2,68 (с, 3H); 13C ЯМР (D2O, 150 МГц) δ 156,8, 154,2, 153,9 (J=249 Гц), 147,8, 136,3, 136,1, 130,1, 129,4, 128,0, 127,2, 125,5 (J=11,8 Гц), 125,1 (J=4,2 Гц), 116,1 (J=13,5 Гц), 53,54, 53,52, 53,49, 50,9, 40,5, 18,2.
Альтернативный способ сочетания (анилинфенилкарбамат)
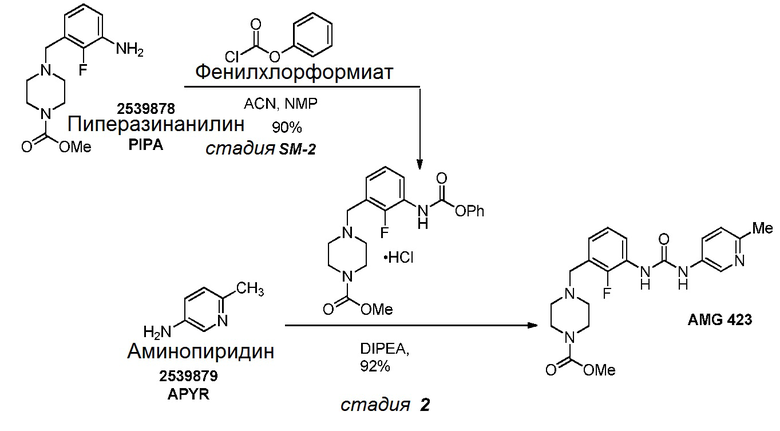
[0104] В реакционный сосуд загрузили метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилат (2,5 г, 1,0 экв.), ацетонитрил (25,0 мл, 10,0 мл/г) и 1-метил-2-пирролидинон (12,5 мл, 5,0 мл/г). Смесь охладили до 0°С, после чего добавляли фенилхлорформиат (1,20 мл, 1,02 экв.) в течение приблизительно 5 минут. Через 45 минут полученную суспензию оставили нагреваться до 20°С. Твердые вещества собрали фильтрацией и дважды промыли ацетонитрилом (10,0 мл, 4,0 мл/г). Твердые вещества высушили под потоком сухого азота с получением метил-4-(2-фтор-3-((феноксикарбонил)амино)бензил)пиперазин-1-карбоксилата гидрохлорида, 2,8 г (выход 71%), в виде белого твердого вещества.
[0105] 4-(2-фтор-3-((феноксикарбонил)амино)бензил)пиперазин-1-карбоксилата гидрохлорид: 1H ЯМР (400 МГц, ДМСО-d6) δ м.д. 3,08 (ш с, 2H), 3,24-3,52 (м, 4H), 3,62 (с, 3H), 4,03 (д, J=11,25 Гц, 2H), 4,38 (ш с, 2H), 7,11-7,35 (м, 4H), 7,35-7,49 (м, 2H), 7,49-7,66 (м, 1H), 7,80 (с, 1H), 10,12 (ш с, 1H), 11,79 (ш с, 1H); МСВР = 388,1676 найденная, 388,1667 рассчитанная.
[0106] В реакционный сосуд загрузили метил-4-(2-фтор-3-((феноксикарбонил)амино)бензил)пиперазин-1-карбоксилата гидрохлорид (0,50 г, 1,0 экв.), 6-метилпиридин-3-амин (0,15 г, 1,2 экв.), тетрагидрофуран (2,0 мл, 4,0 мл/г) и N,N-диизопропилэтиламин (0,23 мл, 1,1 экв.). Смесь нагревали до 65°С в течение 22 часов, после чего анализ количественной ВЭЖХ показал 0,438 г (аналитический выход 92%) омекамтива мекарбила.
Альтернативный способ получения гидрата дигидрохлорида омекамтива мекарбила
[0107] Омекамтива мекарбил, свободное основание (3,0 кг, 1,0 экв.), загрузили в продутый азотом реактор, оснащенный рубашкой, затем добавили воду (4,6 л, 1,5 мл/г) и 2-пропанол (6,1 л, 2,60 мл/г). Суспензию перемешивали и нагревали приблизительно до 40°С, после чего в суспензию добавили 6 н. раствор HCl (2,6 л, 2,10 экв.) с получением бесцветного однородного раствора. Раствор нагревали до 60–65°С и перенесли через встроенный фильтр в реактор объемом 60 л, предварительно нагретый до 60°С. Смесь охладили до 45°С, после чего в реактор добавили гидрат дигидрохлорида омекамтива мекарбила (150 г, 5,0 масс.%) в виде суспензии в 95:5 (объем/объем) смеси 2-пропанола/воды (600 мл, 0,20 мл/г). Полученную суспензию поддерживали при 45°С в течение 0,5 часа, затем охладили приблизительно до 20°С, затем выдерживали в течение 3–16 часов. В течение ≥2 часов добавляли 2-пропанол (33,0 л, 11,0 мл/г), затем выдерживали при постоянной температуре в течение ≥1 часа приблизительно при 20°С (рН надосадочного раствора ≤7).
[0108] Смесь 5–10 раз рециркулировали через мельницу влажного помола до достижения достаточного снижения размера частиц, по сравнению с автономно калиброванным эталоном визуальной микроскопии. Суспензию отфильтровали под вакуумом, а полученные твердые вещества промыли два раза смесью 95:5 (объем/объем) 2-пропанола/воды (3,0 л, 1,0 мл/г) с окончательным промыванием осадка 2-пропанолом (6,0 л, 2,0 мл/г). Осадок высушили на фильтре, продувая влажный азот через осадок до достижения остаточного содержания 2-пропанола ≤5000 м.д. и воды 2,5–5%, измеренных в анализе ГХ и KF соответственно. Гидрат дигидрохлорида омекамтива мекарбила выделили в виде бесцветного кристаллического твердого вещества (3,40 кг, выход 93%).
рН-зависимые профили высвобождения
[0109] Препарат полугидрата омекамтива мекарбила (свободного основания) и гидрата дигидрохлорида (Формы A) получили из следующих компонентов, содержание которых указано в масс./масс.%:
Активная грануляция свободного основания (матричная таблетка 75 мг): 15,37% свободного основания; 30% гипромеллозы, ГПМЦ K100 MPrem CR; 10% моногидрата лимонной кислоты; 11,88% микрокристаллической целлюлозы, Avicel PH 101; 6,75% моногидрата лактозы, FastFlo 316; 12,5% очищенной воды; и грануляция лимонной кислоты: 20% моногидрата лимонной кислоты; 5% микрокристаллической целлюлозы, Avicel PH 101; и 1% стеарата магния, не из крупного рогатого скота. Интрагрануляция Формы A (матричная таблетка 75 мг): 18,37% Формы A; 30% гипромеллозы, ГПМЦ K100 MPrem CR; 0,50% стеарата магния; и экстрагрануляция: 16,88% микрокристаллической целлюлозы, Avicel PH 101; 18,37% безводной лимонной кислоты; и 0,5% стеарата магния, не из крупного рогатого скота.
[0110] Препараты испытывали при рН 2 и рН 6,8 и измеряли количество высвобожденного с течением времени лекарства. Результаты профилей высвобождения лекарства представлены на фиг. 6.
[0111] Изложенное выше описание является лишь иллюстрацией настоящего изобретения и не подразумевает ограничения настоящего изобретения до описанных солей или полиморфов. Варианты и изменения, которые очевидны для специалистов в данной области техники, подразумеваются входящими в границы объема и сущность настоящего изобретения, которые определены в прилагаемой формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| 2-АМИНОБЕНЗОКСАЗОЛКАРБОКСАМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ 5-НТ3 | 2007 |
|
RU2448105C2 |
| Твердые формы селективного ингибитора CDK4/6 | 2014 |
|
RU2619944C2 |
| КРИСТАЛЛИЧЕСКИЙ ИНГИБИТОР FGFR4 И ЕГО ПРИМЕНЕНИЕ | 2016 |
|
RU2763328C2 |
| ГИДРОХЛОРИД (1S,2S,4R)-4-{ 4-[(1S)-2,3-ДИГИДРО-1Н-ИНДЕН-1-ИЛАМИНО]-7Н-ПИРРОЛО [2,3-d]ПИРИМИДИН-7-ИЛ} -2-ГИДРОКСИЦИКЛОПЕНТИЛ)МЕТИЛ СУЛЬФАМАТА | 2010 |
|
RU2562245C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИЛИРОВАННЫХ ЦИКЛОПЕНТАПИРИМИДИНОВЫХ СОЕДИНЕНИЙ И ИХ СОЛЕЙ | 2013 |
|
RU2642311C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2005 |
|
RU2409582C2 |
| ИНГИБИТОРЫ ПРОЛИЛГИДРОКСИЛАЗЫ | 2010 |
|
RU2518071C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ КОЛИТА | 2010 |
|
RU2518416C2 |
| ПРОИЗВОДНЫЕ ПИРИДОНА | 2013 |
|
RU2632885C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2014 |
|
RU2696310C1 |
Изобретение относится к моногидрату дигидрохлоридной соли омекамтива мекарбила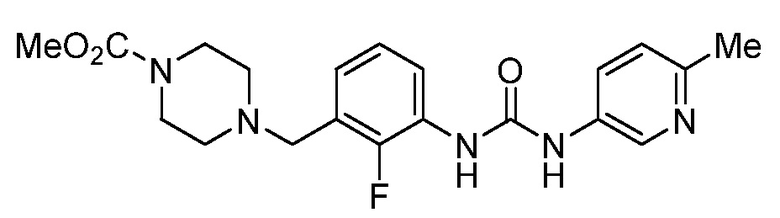 , а также к способу его получения. Технический результат: получен моногидрат дигидрохлоридной соли омекамтива мекарбила, который имеет более желательный профиль высвобождения лекарственного средства, а также обладает улучшенной стабильностью. 3 н. и 22 з.п. ф-лы, 7 ил., 1 табл.
, а также к способу его получения. Технический результат: получен моногидрат дигидрохлоридной соли омекамтива мекарбила, который имеет более желательный профиль высвобождения лекарственного средства, а также обладает улучшенной стабильностью. 3 н. и 22 з.п. ф-лы, 7 ил., 1 табл.
1. Моногидрат дигидрохлоридной соли омекамтива мекарбила.
2. Соль по п. 1, отличающаяся тем, что указанная соль является кристаллической.
3. Соль по п. 1, отличающаяся тем, что указанная соль характеризуется диаграммой порошковой рентгеновской дифракции, содержащей пики при приблизительно 6,6, 14,9, 20,1, 21,4 и 26,8±0,2° 2θ с применением излучения Cu Kα.
4. Соль по п. 3, отличающаяся тем, что диаграмма порошковой рентгеновской дифракции дополнительно содержит пики при приблизительно 8,4, 24,2, 26,0 и 33,3±0,2° 2θ с применением излучения Cu Kα.
5. Соль по п. 3, отличающаяся тем, что диаграмма порошковой рентгеновской дифракции дополнительно содержит пики при приблизительно 6,2, 9,7, 13,2, 14,3, 15,4, 16,3, 16,9, 18,9, 19,5, 20,7, 21,8, 22,8, 23,6, 25,1, 27,3, 27,7, 28,4, 29,4, 30,2, 31,2, 31,5, 31.9, 33,9, 34,5, 34,9, 36,1, 36,8, 37,7, 38,5 и 39,7±0,2° 2θ с применением излучения Cu Kα.
6. Соль по п. 1, отличающаяся тем, что имеет, по существу, такую диаграмму порошковой рентгеновской дифракции, как показана на фиг. 2.
7. Соль по п. 1, отличающаяся тем, что имеет эндотермический переход при температуре от приблизительно 230°С до приблизительно 240°С, измеренный при помощи дифференциальной сканирующей калориметрии.
8. Соль по п. 7, отличающаяся тем, что переход происходит при приблизительно 235°С.
9. Способ получения моногидрата дигидрохлоридной соли омекамтива мекарбила, включающий:
(a) гидрирование метил-4-(2-фтор-3-нитробензил)пиперазин-1-карбоксилата в присутствии катализатора гидрирования с получением метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилата;
(б) смешивание метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилата и фенил-(6-метилпиридин-3-ил)карбамата в присутствии триалкиламинного основания с получением омекамтива мекарбила в виде свободного основания; и
(в) кристаллизацию свободного основания омекамтива мекарбила в присутствии водного раствора хлористоводородной кислоты и спиртового растворителя с получением моногидрата дигидрохлоридной соли омекамтива мекарбила.
10. Способ по п. 9, отличающийся тем, что дополнительно включает получение препарата моногидрата дигидрохлоридной соли омекамтива мекарбила.
11. Способ по п. 9, отличающийся тем, что катализатор гидрирования содержит палладий.
12. Способ по п. 11, отличающийся тем, что катализатор гидрирования представляет собой палладий на углероде.
13. Способ по любому из пп. 9-12, отличающийся тем, что триалкиламинное основание представляет собой триэтиламин, диизопропилэтиламин или их комбинацию.
14. Способ по любому из пп. 9-12, отличающийся тем, что триалкиламинное основание содержит диизопропилэтиламин.
15. Способ по любому из пп. 9-12, отличающийся тем, что спиртовой растворитель содержит изопропиловый спирт.
16. Способ по любому из пп. 9-12, отличающийся тем, что моногидрат дигидрохлоридной соли омекамтива мекарбила имеет диаграмму порошковой рентгеновской дифракции (ПРД), содержащую пики при приблизительно 6,6, 14,9, 20,1, 21,4 и 26,8±0,2° 2θ с применением излучения Cu Kα.
17. Способ по п. 16, отличающийся тем, что диаграмма ПРД дополнительно содержит пики при приблизительно 8,4, 24,2, 26,0 и 33,3±0,2° 2θ с применением излучения Cu Kα.
18. Способ по п. 16, отличающийся тем, что диаграмма ПРД дополнительно содержит пики при приблизительно 6,2, 9,7, 13,2, 14,3, 15,4, 16,3, 16,9, 18,9, 19,5, 20,7, 21,8, 22,8, 23,6, 25,1, 27,3, 27,7, 28,4, 29,4, 30,2, 31,2, 31,5, 31,9, 33,9, 34,5, 34,9, 36,1, 36,8, 37,7, 38,5 и 39,7±0,2° 2θ с применением излучения Cu Kα.
19. Способ получения омекамтива мекарбила, включающий:
смешивание метил-4-(3-амино-2-фторбензил)пиперазин-1-карбоксилата и фенил-(6-метилпиридин-3-ил)карбамата в присутствии триалкиламинного основания с получением омекамтива мекарбила.
20. Способ по п. 19, отличающийся тем, что триалкиламинное основание содержит диизопропилэтиламин.
21. Способ по п. 19 или 20, отличающийся тем, что дополнительно включает кристаллизацию омекамтива мекарбила в присутствии водного раствора хлористоводородной кислоты и спиртового растворителя с получением моногидрата дигидрохлоридной соли омекамтива мекарбила.
22. Способ по п. 21, отличающийся тем, что спиртовой растворитель содержит изопропиловый спирт.
23. Способ по п. 21, отличающийся тем, что моногидрат дигидрохлоридной соли омекамтива мекарбила имеет диаграмму порошковой рентгеновской дифракции (ПРД), содержащую пики при приблизительно 6,6, 14,9, 20,1, 21,4 и 26,8±0,2° 2θ с применением излучения Cu Kα.
24. Способ по п. 23, отличающийся тем, что диаграмма ПРД дополнительно содержит пики при приблизительно 8,4, 24,2, 26,0 и 33,3±0,2° 2θ с применением излучения Cu Kα.
25. Способ по п. 23, отличающийся тем, что диаграмма ПРД дополнительно содержит пики при приблизительно 6,2, 9,7, 13,2, 14,3, 15,4, 16,3, 16,9, 18,9, 19,5, 20,7, 21,8, 22,8, 23,6, 25,1, 27,3, 27,7, 28,4, 29,4, 30,2, 31,2, 31,5, 31,9, 33,9, 34,5, 34,9, 36,1, 36,8, 37,7, 38,5 и 39,7±0,2° 2θ с применением излучения Cu Kα.
| RU 2007101619 A, 10.08.2008 | |||
| WO 2009138438 A1, 19.11.2009 | |||
| WO 2007070683 A2, 21.06.2007. |

