Область техники
Настоящее изобретение относится к способу получения стабилизатора транстиретина на основе производного бензоксазола или его фармацевтически приемлемой соли. В частности, настоящее изобретение относится к способу получения производного 2-(3,5-дихлорфенил)-1,3-бензоксазол-6-карбоновой кислоты или его фармацевтически приемлемой соли путем взаимодействия 4-амино-3-гидроксибензойной кислоты или ее карбоксил-защищенного производного с подходящим соединением 3,5-дихлорфенилортоэфира. Способы по изобретению особенно полезны для получения 2-(3,5-дихлорфенил)-1,3-бензоксазол-6-карбоновой кислоты или ее фармацевтически приемлемой соли, которые полезны для стабилизации транстиретина, ингибирования неправильного сворачивания транстиретина, протеолиза и лечения амилоидных заболеваний, связанных с ним.
Уровень техники
Транстиретин (TTR) представляет собой 55 кДа гомотетрамерный белок, присутствующий в сыворотке и спинномозговой жидкости и функционирующий как транспортер L-тироксина (Т4) и голоретинолсвязывающего белка (RBP). Было обнаружено, что TTR представляет собой амилоидогенный белок, который при определенных условиях может трансформироваться в фибриллы и другие агрегаты, которые могут приводить к болезненной патологии, такой как полиневропатия или кардиомиопатия у людей.
Патенты США №№ 7,214,695; 7,214,696; 7,560,488; 8,168,683; и 8,653,119, каждый из которых включен в настоящий документ в качестве ссылки, описывают производные бензоксазола, которые действуют как стабилизаторы транстиретина и имеют формулу
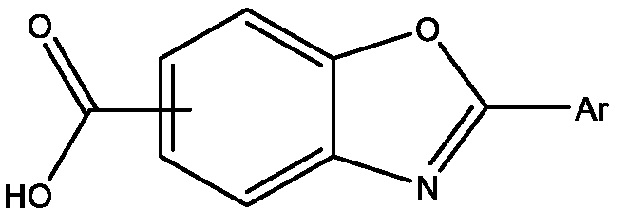
или их фармацевтически приемлемую соль; где Ar представляет собой 3,5-дифторфенил, 2,6-дифторфенил, 3,5-дихлорфенил, 2,6-дихлорфенил, 2-(трифторметил)фенил или 3-(трифторметил)фенил, и способы получения этих соединений. В частности, там описаны 2-(3,5-дихлорфенил)-1,3-бензоксазол-6-карбоновая кислота (тафамидис) формулы

и способы ее получения. Тафамидис представляет собой перорально активный стабилизатор транстиретина, который ингибирует диссоциацию тетрамера и протеолиз, который был одобрен в некоторых юрисдикциях для лечения транстиретиновой полинейропатии (TTR-PN) и для лечения транстиретиновой кардиомиопатии (TTR-CM). Патент США № 9,249,112 и публикация заявки на патент США № US 2019/0119226, также включенные в настоящий документ посредством ссылки, описывают полиморфные формы меглуминовой соли 2-(3,5-дихлорфенил)-1,3-бензоксазол-6-карбоновой кислоты (меглумин тафамидис). Патент США № 9,770,441 описывает полиморфные формы свободной кислоты 2-(3,5-дихлорфенил)-1,3-бензоксазол-6-карбоновой кислоты (тафамидиса), который также включен в настоящий документ посредством ссылки. Существует постоянная потребность в разработке эффективных способов синтеза производных 2-(3,5-дихлорфенил)-1,3-бензоксазол-6-карбоновой кислоты, их фармацевтически приемлемых солей и полиморфных форм производных 2-(3,5-дихлорфенил)-1,3-бензоксазол-6-карбоновой кислоты и их фармацевтически приемлемых солей.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
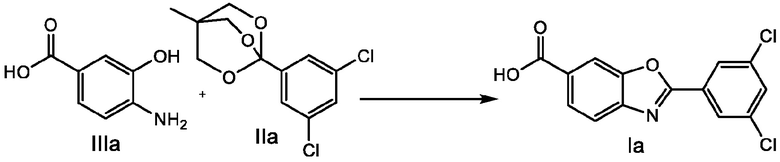
В настоящем изобретении предложен способ получения соединения 6-карбокси-2-(3,5-дихлорфенил)бензоксазола формулы I.

где способ включает стадию взаимодействия соединения 4-амино-3-гидроксибензойной кислоты формулы III с соединением 3,5-дихлорфенилортоэфира формулы II с получением соединения формулы I
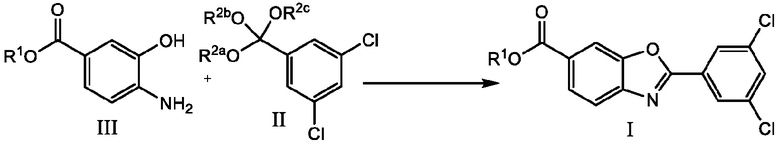
где R1 представляет собой водород или карбоксильную защитную группу; и R2a, R2b и R2c каждый независимо представляет собой C1-C6алкил или любые два из R2a, R2b и R2c, взятые вместе представляют собой C1-C6алкандиил, или R2a, R2b и R2c, взятые вместе представляют собой C3-C10алкантриил. Способ получения соединений формулы I по изобретению можно проводить в подходящем растворителе, или в некоторых случаях, когда соединение формулы II представляет собой масло, может не потребоваться дополнительный растворитель. Способ по изобретению можно проводить в присутствии кислотного катализатора, основного катализатора или без катализатора, и процесс можно проводить при температуре от 0°C до температуры кипения используемого растворителя, при этом процесс проводят в течение периода от 15 минут до нескольких дней. Способы по изобретению также включают дополнительные стадии, такие как выделение соединений формулы I или Ia и получение полиморфных форм соединений формулы I или Ia.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фигура 1: Спектр ПРД кристаллического 1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октана.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Первый вариант осуществления по настоящему изобретению, обозначенный E1, представляет собой способ получения соединения 6-карбокси-2-(3,5-дихлорфенил)бензоксазола формулы I
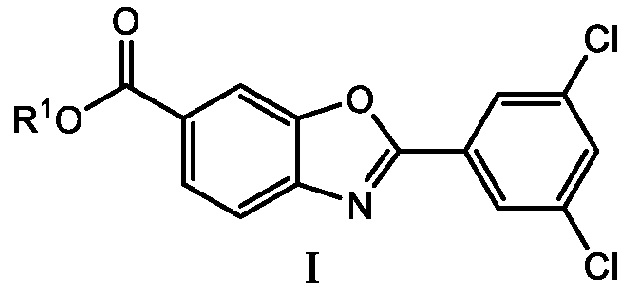
способ, включающий взаимодействие соединения 4-амино-3-гидроксибензойной кислоты формулы III с соединением ортоэфира 3,5-дихлорфенила формулы II с получением соединения формулы I

где R1 представляет собой водород или карбоксильную защитную группу; и R2a, R2b и R2c каждый независимо представляет собой C1-C6алкил или любые два из R2a, R2b и R2c, взятые вместе представляют собой C1-C6алкандиил, или R2a, R2b и R2c, взятые вместе представляют собой C3-C10алкантриил.
Дополнительные варианты осуществления настоящего изобретения описаны ниже как варианты осуществления 2-46, которые обозначены как E2-E46, соответственно.
E2 представляет собой процесс по E1, в котором реакцию соединения формулы III с соединением формулы II с получением соединения формулы I проводят в растворителе.
E3 представляет собой процесс по E2, в котором растворитель выбран из группы, состоящей из метанола, этанола, изопропанола, н-бутанола, ацетона, метилэтилкетона, тетрагидрофурана, 1,4-диоксана, трет-бутилметилового эфира, анизола, этилацетата, хлороформа, хлорбензола, гептана, циклогексана, толуола, ацетонитрила и 1,2-диметоксиэтана.
E4 представляет собой процесс по E3, в котором растворитель выбран из группы, состоящей из метанола, изопропанола, ацетонитрила, этилацетата, 1,2-диметоксиэтана, тетрагидрофурана, трет-бутилметилового эфира и 1,4-диоксана.
E5 представляет собой процесс по любому из E1-E4, где реакцию соединения формулы III с соединением формулы II с получением соединения формулы I проводят в присутствии кислотного катализатора.
E6 представляет собой процесс по E5, в котором кислотный катализатор выбран из группы, состоящей из трифторуксусной кислоты, уксусной кислоты, хлористоводородной кислоты и метансульфоновой кислоты.
E7 представляет собой процесс по любому из E1-E4, в котором реакцию соединения формулы III с соединением формулы II с получением соединения формулы I проводят в присутствии основного катализатора.
E8 представляет собой процесс по E7, в котором основным катализатором является триэтиламин.
E9 представляет собой процесс по любому из E1-E8, где реакцию соединения формулы III с соединением формулы II с получением соединения формулы I проводят при температуре от примерно комнатной температуры до примерно 100°C.
E10 представляет собой процесс по E9, где температура составляет от примерно комнатной температуры до примерно 65°C.
E11 представляет собой способ по любому из E1-E10, где реакцию соединения формулы III с соединением формулы II с получением соединения формулы I проводят в течение периода от примерно 0,25 часа до примерно 40 часов.
E12 представляет собой процесс по любому из E1-E11, где R1 представляет собой водород.
E13 представляет собой процесс по любому из E1-E12, где каждый из R2a, R2b и R2c независимо представляет собой C1-C6алкил.
E14 представляет собой процесс по E13, где каждый из R2a, R2b и R2c представляет собой метил.
E15 представляет собой процесс по любому из E1-E12, где R2a, R2b и R2c вместе представляют собой C3-C10алкантриил.
E16 представляет собой процесс по E15, где соединение формулы II представляет собой
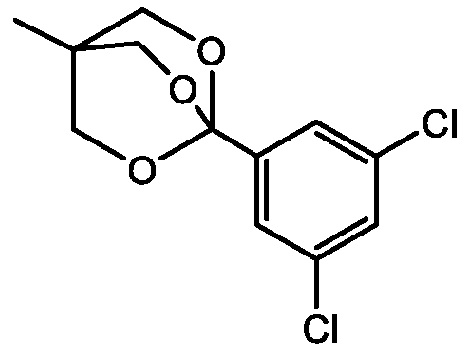 .
.
E17 представляет собой процесс по любому из E1-E16, дополнительно включающий стадию выделения соединения формулы I.
E18 представляет собой процесс по E17, где соединение формулы I выделяют фильтрованием.
E19 представляет собой способ получения 6-карбокси-2-(3,5-дихлорфенил)бензоксазола формулы Ia

где способ включает взаимодействие 4-амино-3-гидроксибензойной кислоты формулы IIIa с соединением ортоэфира 3,5-дихлорфенила формулы II с получением 6-карбокси-2-(3,5-дихлорфенил)бензоксазола формулы Ia

где R2a, R2b и R2c каждый независимо представляет собой C1-C6 алкил, или любые два из R2a, R2b и R2c, взятые вместе, представляют собой C1-C6алкандиил, или R2a, R2b и R2c, взятые вместе представляют собой C3-C10алкантриил.
E20 представляет собой процесс по E19, где реакцию соединения формулы IIIa с соединением формулы II с получением соединения формулы Ia проводят в растворителе.
E21 представляет собой процесс по E20, где растворитель выбирают из группы, состоящей из метанола, этанола, изопропанола, н-бутанола, ацетона, метилэтилкетона, тетрагидрофурана, 1,4-диоксана, трет-бутилметилового эфира, анизола, этилацетата, хлороформа, хлорбензола, гептана, циклогексана, толуола, ацетонитрила и 1,2-диметоксиэтана.
E22 представляет собой процесс по E21, где растворитель выбран из группы, состоящей из метанола, изопропанола, ацетонитрила, этилацетата, 1,2-диметоксиэтана, тетрагидрофурана, трет-бутилметилового эфира и 1,4-диоксана.
E23 представляет собой процесс по любому из E19-E22, где реакцию соединения формулы IIIa с соединением формулы II с получением соединения формулы Ia проводят в присутствии кислотного катализатора.
E24 представляет собой процесс по E23, где кислотный катализатор выбран из группы, состоящей из трифторуксусной кислоты, уксусной кислоты, хлористоводородной кислоты и метансульфоновой кислоты.
E25 представляет собой процесс по E24, где кислотным катализатором является трифторуксусная кислота.
E26 представляет собой процесс по любому из E19-E22, где реакцию соединения формулы IIIa с соединением формулы II с получением соединения формулы Ia проводят в присутствии основного катализатора.
E27 представляет собой процесс по E26, где основным катализатором является триэтиламин.
E28 представляет собой процесс по любому из E19-E27, где реакцию соединения формулы III с соединением формулы II с получением соединения формулы I проводят при температуре от примерно комнатной температуры до примерно 100°C.
E29 представляет собой процесс по E28, где температура составляет от примерно комнатной до примерно 65°C.
E30 представляет собой процесс по любому из E19-E29, где реакцию соединения формулы III с соединением формулы II с получением соединения формулы I проводят в течение периода от примерно 0,25 часа до примерно 40 часов.
E31 представляет собой процесс по любому из E19-E30, где каждый из R2a, R2b и R2c независимо представляет собой C1-C6алкил.
E32 представляет собой процесс по E31, где каждый из R2a, R2b и R2c представляет собой метил.
E33 представляет собой процесс по любому из E19-E30, где R2a, R2b и R2c вместе представляют собой C3-C10алкантриил.
E34 представляет собой процесс по E33, где соединение формулы II представляет собой
 .
.
E35 представляет собой процесс по любому из E19-E34, дополнительно включающий стадию выделения соединения формулы Ia.
E36 представляет собой процесс по E35, где соединение формулы Ia выделяют фильтрованием.
E37 представляет собой процесс по любому из E19-E36, дополнительно включающий стадию взаимодействия 6-карбокси-2-(3,5-дихлорфенил)бензоксазола формулы Ia с фармацевтически приемлемым основанием с получением фармацевтически приемлемой соли 6-карбокси -2-(3,5-дихлорфенил)бензоксазола.
E38 представляет собой процесс по E37, где 6-карбокси-2-(3,5-дихлорфенил)бензоксазол взаимодействует с меглумином в подходящем растворителе с получением соли меглумина 6-карбокси-2-(3,5-дихлорфенил)бензоксазола.
E39 представляет собой процесс по E38, где 6-карбокси-2-(3,5-дихлорфенил)бензоксазол подвергают взаимодействию при комнатной температуре с меглумином в растворителе, выбранном из метилизобутилкетона, МТБЭ и EtOAc, и полученное твердое вещество выделяют и сушат, чтобы получить полиморфную форму E соли меглумина 6-карбокси-2-(3,5-дихлорфенил)бензоксазола.
E40 представляет собой процесс по E38, где 6-карбокси-2-(3,5-дихлорфенил)бензоксазол взаимодействует с меглумином в смеси изопропилового спирта и воды, и полученное твердое вещество выделяют и сушат с получением полиморфной формы M соли меглумина 6-карбокси-2-(3,5-дихлорфенил)бензоксазола.
E41 представляет собой процесс по E35, дополнительно включающий стадию перемешивания соединения формулы Ia в смеси воды и ИПС с последующим выделением и сушкой полученного твердого вещества с получением полиморфной формы 1 6-карбокси-2-(3,5-дихлорфенил)бензоксазола.
E42 представляет собой способ получения 6-карбокси-2-(3,5-дихлорфенил)бензоксазола формулы Ia

где способ включает взаимодействие примерно одного молярного эквивалента 4-амино-3-гидроксибензойной кислоты формулы IIIa с примерно одним молярным эквивалентом сложного ортоэфира 3,5-дихлорфенила формулы IIa в подходящем растворителе с получением 6-карбокси-2- (3,5-дихлорфенил)бензоксазола формулы Ia
 .
.
E43 представляет собой процесс по E42, где растворитель выбран из группы, состоящей из метанола, этанола, изопропанола, н-бутанола, ацетона, метилэтилкетона, тетрагидрофурана, 1,4-диоксана, трет-бутилметилового эфира, анизола, этилацетата, хлороформа, хлорбензола, гептана, циклогексана, толуола, ацетонитрила и 1,2-диметоксиэтана.
E44 представляет собой процесс по E43, где растворитель выбран из группы, состоящей из метанола, изопропанола, ацетонитрила, этилацетата, 1,2-диметоксиэтана, тетрагидрофурана, трет-бутилметилового эфира и 1,4-диоксана.
E45 представляет собой процесс по E44, где процесс проводят с использованием кислотного катализатора, выбранного из группы, состоящей из трифторуксусной кислоты, уксусной кислоты, хлористоводородной кислоты и метансульфоновой кислоты.
E46 представляет собой процесс по E44, где кислотным катализатором является трифторуксусная кислота, и растворителем является изопропанол.
E47 представляет собой способ получения соединения 6-карбокси-2-(3,5-дихлорфенил)бензоксазола формулы I

где способ включает взаимодействие соединения 4-амино-3-гидроксибензойной кислоты формулы III с соединением ортоэфира 3,5-дихлорфенила формулы II с получением соединения формулы I

где R1 представляет собой водород или карбокси защитную группу; и R2a, R2b и R2c каждый независимо представляет собой C1-C6алкил или любые два из R2a, R2b и R2c, взятые вместе, представляют собой C1-C8алкандиил или R2a, R2b и R2c, взятые вместе представляют собой C3-C12алкантриил, где C1-C8алкандиил и C3-C12алкантриил каждый необязательно замещен фенилом, который необязательно замещен одной-двумя группами, независимо выбранными из галогена, C1-С3алкил и С1-С3алкокси.
E48 представляет собой процесс по E47, где соединение формулы III представляет собой 4-амино-3-гидроксибензойную кислоту, и соединение формулы II выбрано из группы, состоящей из 1,3-дихлор-5-(триметоксиметил)бензола; 1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октана; 1-(3,5-дихлорфенил)-4-этил-2,6,7-триоксабицикло[2.2.2]октана; 1-(3,5-Дихлорфенил)-4-фенил-2,6,7-триоксабицикло[2.2.2]октана; 2-(3,5-дихлорфенил)-2-метокси-1,3-диоксолана; 1-(3,5-дихлорфенил)-2,7,8-триоксабицикло[3.2.1]октана; 3-(3,5-дихлорфенил)-2,4,10-триоксаадамантана; 1-(3,5-дихлорфенил)-4-изопропил-2,6,7-триоксабицикло[2.2.2]октана; и 4-(трет-бутил)-1-(3,5-дихлорфенил)-2,6,7-триоксабицикло[2.2.2]октана.
E49 представляет собой процесс по E48, где реакцию соединения формулы II с формулой III проводят в ИПС в качестве растворителя в присутствии МСК в качестве кислотного катализатора.
Е50 представляет собой соединение, выбранное из группы, состоящей из 1,3-дихлор-5-(триметоксиметил)бензола; 1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октана; 1-(3,5-дихлорфенил)-4-этил-2,6,7-триоксабицикло[2.2.2]октана; 1-(3,5-дихлорфенил)-4-фенил-2,6,7-триоксабицикло[2.2.2]октана; 2-(3,5-дихлорфенил)-2-метокси-1,3-диоксолана; 1-(3,5-дихлорфенил)-2,7,8-триоксабицикло[3.2.1]октана; 3-(3,5-дихлорфенил)-2,4,10-триоксаадамантана; 1-(3,5-дихлорфенил)-4-изопропил-2,6,7-триоксабицикло[2.2.2]октана; и 4-(трет-бутил)-1-(3,5-дихлорфенил)-2,6,7-триоксабицикло[2.2.2]октана.
E51 представляет собой соединение по E50, которое представляет собой 1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октан. E52 представляет собой соединение по E51, которое представляет собой кристаллическую форму 1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октана. E53 представляет собой соединение по E52, которое характеризуется пиками ПРД при 21,2 и 19,7 2-тета, каждый ±0,2 2-тета. E54 представляет собой соединение по E53, которое характеризуется пиками ПРД при 21,2, 19,7 и 14,4 2-тета, каждый ±0,2 2-тета. E55 представляет собой соединение по E54, которое характеризуется пиками ПРД при 21,2, 19,7, 14,4 и 23,7 2-тета, каждый ±0,2 2-тета. E56 представляет собой соединение по E55, которое характеризуется пиками ПРД при 21,2, 19,7, 14,4, 23,7 и 24,2 2-тета, каждый ±0,2 2-тета. E57 представляет собой соединение по E56, которое характеризуется пиками ПРД при 21,2, 19,7, 14,4, 23,7, 24,2 и 30,6 2-тета, каждый ±0,2 2-тета.
E58 представляет собой процесс получения соединения формулы IIa-2

где R3 выбран из группы, состоящей из изопропила, трет-бутила и неопентила, включающий взаимодействие соединения формулы IV с 3,5-дихлорбензойной кислотой

в соответствующем растворителе в присутствии соответствующего кислотного катализатора.
E59 представляет собой процесс по E58, где R3 представляет собой изопропил или трет-бутил. E60 представляет собой процесс по E59, где растворителем является толуол, и кислотным катализатором является метансульфоновая кислота.
В описанных выше процессах по настоящему изобретению, соединение формулы III представляет собой либо 4-амино-3-гидроксибензойную кислоту (когда R1 представляет собой Н), либо соединение, в котором карбоксильная группа защищена (когда R1 представляет собой карбоксильную защитную группу). В данной области техники известны многочисленные карбоксильные защитные группы, и их можно использовать для соединений формулы III, где R1 представляет собой карбоксильную защитную группу. Глава 5 Protective Groups in Organic Synthesis, Third Edition. Theodora W. Greene, Peter G.M. Wuts, Copyright 1999, John Wiley & Sons, Inc. описывает различные классы карбоксильных защитных групп. Tan J, Akakura M, Yamamoto H. “The supersilyl group as a carboxylic acid protecting group: application to highly stereoselective aldol and Mannich reactions” Angew Chem Int Ed Engl. 2013;52(28):7198-7202. doi:10.1002/anie.201300102 описывает использование суперсилила (EtSi)3Si- в качестве универсальной защитной группы карбоксила. Обычно используемые карбоксильные защитные группы, которые можно использовать для соединения формулы III, включают, но не ограничены ими, сложные эфирные защитные группы, такие как метил, этил, трет-бутил, 2-цианоэтил, 2,2,2-трихлорэтил, аллил, (2,2-диметил)аллил, фенил, бензил, пара-метоксибензил и триметилсилил в дополнение к суперсилилу. Также могут быть использованы другие эквивалентные карбоксильные защитные группы, такие как использование сложного тиоэфира (т.е. группа C(O)OR1 вместо этого может представлять собой C(O)S(C1-C6алкил) или где вся группа C(O)OR1 вместо этого представляет собой оксазолиновую группу. Карбоксильные защитные группы могут быть удалены способами, известными в данной области техники, такими как обработка кислотой, основанием или гидрирование, в зависимости от конкретной защитной группы карбоксила, используемой для получения соединений, в которых R1 представляет собой H.
Ортоэфиры и циклические ортоэфиры, такие как соединения формулы II, используемые в настоящих способах, могут быть получены в соответствии со способами, аналогичными тем, которые описаны в E. J. Corey and N. Raju, Tetrahedron Letters, 1983, 24(50), 5571-5574; P. Wipf et. al. Pure Appl. Chem. 1999, 71(3), 415-421; S. Tange et. al. Synthesis, 2008, 3219-3222; M. Noe et. al. Green Chem. 2013, 15, 2252; европейской патентной заявке № 0279698 и японском патенте 5419545, выданном на основе японской патентной заявки № 2010-270091.
Предпочтительными соединениями, полученными способом по изобретению, являются соединения формулы I и, более конкретно, соединения формулы Ia или их фармацевтически приемлемые соли. Соединение формулы Ia, 6-карбокси-1-(3,5-дихлорфенил)бензоксазол или 2-(3,5-дихлорфенил)-1,3-бензоксазол-6-карбоновая кислота, также известное под наименованием USAN тафамидис, несет группу карбоксильной кислоты в положении 6 своего бензоксазольного кольца. Этот фрагмент карбоновой кислоты может легко образовывать соли с подходящими основаниями, такими как меглумин, с получением фармацевтически приемлемых солей соединений формулы Ia.
Способы по настоящему изобретению включают получение соединений формулы I или Ia в форме их соответствующих солей, полученных из неорганических или органических оснований. Конкретная соль соединения формулы I или Ia может быть предпочтительной благодаря одному или нескольким физическим свойствам соли, таким как повышенная фармацевтическая стабильность при различных температурах и влажности, или желаемая растворимость в воде или масле. В некоторых случаях, соль соединения также может быть использована в качестве вспомогательного средства при выделении, очистке и/или разделении соединения в способах по настоящему изобретению.
Если соль предназначена для введения пациенту (в отличие, например, от использования в контексте in vitro), соль предпочтительно является фармацевтически приемлемой. Термин «фармацевтически приемлемая соль» относится к соли, полученной объединением соединения Формулы I или Ia с основанием, катион которого обычно считается подходящим для потребления человеком. Фармацевтически приемлемые соли особенно полезны в качестве продуктов процессов по настоящему изобретению из-за их большей растворимости в воде по сравнению с исходным соединением. Для использования в медицине, соли соединений, полученных способами по настоящему изобретению, представляют собой нетоксичные «фармацевтически приемлемые соли». Соли, охватываемые термином «фармацевтически приемлемые соли», относятся к нетоксичным солям соединений по настоящему изобретению, которые обычно получают реакцией свободной кислоты соединения формулы I или Ia с подходящим органическим или неорганическим основанием.
Поскольку соединения, полученные способами по изобретению, могут нести кислотную группу (т.е. формула I, где R1 представляет собой Н), их подходящие фармацевтически приемлемые соли могут включать более легкие соли щелочных металлов, т.е. соли натрия или калия; соли щелочноземельных металлов, например соли кальция или магния; и соли, образованные с подходящими органическими лигандами, например соли четвертичного аммония. В другом варианте осуществления, основные соли образуются из оснований, которые образуют нетоксичные соли, включая соли алюминия, аргинина, бензатина, холина, диэтиламина, диоламина, глицина, лизина, меглумина, оламина, трометамина и цинка.
Органические соли могут быть получены из вторичных, третичных или четвертичных аминов, таких как трометамин, диэтиламин, N,N'-дибензилэтилендиамин, хлорпрокаин, холин, диэтаноламин, этилендиамин, меглумин (N-метилглюкамин) и прокаин. Основные азотсодержащие группы могут быть кватернизованы такими агентами, как галогениды низших алкилов (C1-C6) (например, хлориды, бромиды и йодиды метила, этила, пропила и бутила), диалкилсульфаты (например, сульфаты диметила, диэтила, дибутила и диамила), галогениды с длинной цепью (например, хлориды, бромиды и йодиды децила, лаурила, миристила и стеарила), арилалкилгалогениды (например, бромиды бензила и фенетила) и другие. Предпочтительной солью, полученной способами по настоящему изобретению, является меглуминовая соль тафамидиса.
Гемисоли кислот (т.е. соединения формулы I или Ia) также могут быть получены способами по настоящему изобретению, например, гемисульфаты и гемикальциевые соли тафамидиса.
Специалисту в данной области техники будет понятно, что вышеупомянутые соли включают соли, в которых противоион является оптически активным, например хиральные аминовые основания, такие как меглумин, который также известен как (2R,3R,4R,5S)-6-(метиламино)гексан-1, 2,3,4,5-пентол или N-метил-D-глюкамин.
Обзор подходящих солей см. в «Handbook of Pharmaceutical Salts: Properties, Selection, and Use» by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
Фармацевтически приемлемые соли соединений формул I и Ia могут быть получены одним или несколькими из трех способов:
(i) взаимодействием соединения формулы I или Ia с желаемым основанием;
(ii) удалением основно-лабильной защитной группы из подходящего предшественника соединения формулы I или Ia с использованием желаемого основания; или
(iii) превращением одной соли соединения формулы I или Ia в другую путем реакции с соответствующим основанием или с помощью подходящей ионообменной колонки.
Все три реакции обычно проводят в растворе. Полученная соль может выпадать в осадок и собираться фильтрованием или может быть выделена выпариванием растворителя. Степень ионизации в полученной соли может варьироваться от полностью ионизированной до почти неионизированной. Предпочтительной солью тафамидиса, которую можно получить, является меглумин тафамидиса.
Соединения формулы I или их фармацевтически приемлемые соли, полученные способами по настоящему изобретению, могут существовать как в не сольватированной, так и в сольватированной формах. Термин «сольват» используется здесь для описания молекулярного комплекса, включающего соединение формулы I или его фармацевтически приемлемую соль и одну или несколько молекул фармацевтически приемлемого растворителя, например, этанола. Термин «гидрат» используется, когда указанный растворитель представляет собой воду. Фармацевтически приемлемые сольваты по изобретению включают сольваты, в которых растворитель кристаллизации может быть изотопно замещен, например D2O, d6-ацетон и d6-ДМСО.
В настоящее время принятой системой классификации органических гидратов является та, которая определяет гидраты с выделенным сайтом, каналом или координированными ионами металла - см. Polymorphism in Pharmaceutical Solids by K. R. Morris (Ed. H. G. Brittain, Marcel Dekker, 1995), включенную в настоящее описание посредством ссылки. Гидраты изолированных сайтов представляют собой такие, в которых молекулы воды изолированы от прямого контакта друг с другом за счет промежуточных органических молекул. В канальных гидратах молекулы воды лежат в каналах решетки, где они находятся рядом с другими молекулами воды. В гидратах, координированных с ионами металлов, молекулы воды связаны с ионами металлов.
Когда растворитель или вода прочно связаны, комплекс будет иметь четко определенную стехиометрию, не зависящую от влажности. Когда, однако, растворитель или вода слабо связаны, как в канальных сольватах и гигроскопичных соединениях, содержание воды/растворителя будет зависеть от влажности и условий сушки. В таких случаях, не стехиометрия будет нормой.
В объем изобретения также входят способы получения многокомпонентных комплексов (отличных от солей и сольватов) соединений формулы I или их фармацевтически приемлемых солей, в которых лекарственное средство (т.е. соединение формулы I или Ia (тафамидис)) и, по меньшей мере, один другой компонент присутствует в стехиометрических или не стехиометрическмх количествах. Комплексы этого типа включают клатраты (комплексы включения лекарственное средство-хозяин) и сокристаллы. Последние обычно определяются как кристаллические комплексы нейтральных молекулярных составляющих, которые связаны друг с другом посредством нековалентных взаимодействий, но также могут представлять собой комплекс нейтральной молекулы с солью. Сокристаллы, содержащие соединение формулы I, могут быть получены кристаллизацией из расплава, перекристаллизацией из растворителей или физическим измельчением компонентов вместе - см. Chem Commun, 17, 1889-1896, by O. Almarsson and M. J. Zaworotko (2004), включенной в настоящий документ посредством ссылки. Общий обзор многокомпонентных комплексов см. в J Pharm Sci, 64 (8), 1269-1288, by Haleblian (August 1975), включенной в настоящий документ посредством ссылки.
Соединения, полученные способами по изобретению, могут существовать в континууме твердых состояний в диапазоне от полностью аморфного до полностью кристаллического. Термин «аморфный» относится к состоянию, в котором в материале отсутствует дальний порядок на молекулярном уровне и, в зависимости от температуры, он может проявлять физические свойства твердого тела или жидкости. Обычно такие материалы не дают отчетливых дифрактограмм и, хотя и проявляют свойства твердого тела, более формально описываются как жидкости. При нагревании происходит переход от твердых к жидким свойствам, который характеризуется изменением состояния, как правило, второго порядка («стеклование»). Термин «кристаллический» относится к твердой фазе, в которой материал имеет правильную упорядоченную внутреннюю структуру на молекулярном уровне и дает отчетливую дифрактограмму с определенными пиками. Такие материалы при достаточном нагревании также будут проявлять свойства жидкости, но переход от твердого состояния к жидкому характеризуется фазовым переходом, обычно первого порядка («точка плавления»). Предпочтительные формы соединения формулы Ia, тафамидиса (свободной кислоты), полученные способами по настоящему изобретению, включают полиморфные формы, как описано в патенте США № 9,770,441, и, в частности, полиморф формы 1 свободной кислоты тафамидиса, как описано в настоящем документе. Предпочтительные формы соединения формулы Ia, меглумина тафамидиас (меглуминовой соли тафамидиса), полученные способами по настоящему изобретению, включают полиморфные формы меглумина тафамидиса, как описано в патенте США № 9,249,112 и публикации заявки на патент США № US 2019/0119226. Особенно предпочтительной формой меглумина тафамидиса, полученной способами по настоящему изобретению, является полиморфная форма М, описанная в патенте США № 9,249,112. Другой формой меглумина тафамидиса, которую можно получить с использованием способов по настоящему изобретению, является полиморфная форма E меглумина тафамидиса, как описано в публикации заявки на патент США № US 2019/0119226.
Соединения формулы I или Ia, полученные способом по изобретению, могут также существовать в мезоморфном состоянии (мезофазе или жидком кристалле) при воздействии на них подходящих условий. Мезоморфное состояние является промежуточным между истинным кристаллическим состоянием и истинным жидким состоянием (расплавом или раствором). Мезоморфизм, возникающий в результате изменения температуры, описывается как «термотропный», и возникающий в результате добавления второго компонента, такого как вода или другой растворитель, описывается как «лиотропный». Соединения, способные образовывать лиотропные мезофазы, описываются как «амфифильные» и состоят из молекул, обладающих ионными (такими как COO-Na+, -COO-K+ или -COO-меглумин+) полярными головными группами, как в случае с тафамидисом. Для получения дополнительной информации см. Crystals and the Polarizing Microscope by N. H. Hartshorne and A. Stuart, 4th Edition (Edward Arnold, 1970), включенную в настоящий документ посредством ссылки.
СОКРАЩЕНИЯ
Используют следующие сокращения:
AcOH=уксусная кислота
AHBA=4-амино-3-гидроксибензойная кислота
BF3⋅Et2O=эфират трехфтористого бора
13C=углерод 13
°C=градусы Цельсия
CDCl3=дейтеро хлороформ
см-1=обратный сантиметр
д=дублет
дд=дублет дублетов
ДХМ=дихлорметан
1,2-ДМЭ=1,2-диметоксиэтан
ДМСО-d6=дейтеро диметилсульфоксид
EtOAc=этилацетат
EtOH=этанол
экв.=эквиваленты
г=грамм
ч=час
H=атом водорода
HCl=хлористоводородная кислота
ВЭЖХ=жидкостная хроматография высокого давления
Гц=герц
ИПС=изопропиловый спирт
iPrOAC=изопропилацетат
J=константа сочетания
КОН=гидроксид калия
л=литр
м=мультиплет
мм=миллиметр
М=моль
мбар=миллибар
МЭК=метилэтилкетон
МеОН=метанол
мг=миллиграмм
МГц=мегагерц
мкл=микролитр
мл=миллилитр
ммоль=миллимоль
моль=моль
МСК или MSOH=метансульфоновая кислота
МТБЭ=метил-трет-бутиловый эфир
NaOMe=метоксид натрия
н-BuOH=н-бутанол
ЯМР=ядерный магнитный резонанс
PTSA=пара-толуолсульфоновая кислота
КТ=комнатная температура
с=синглет
т=триплет
ТЭА=триэтиламин
ТГФ=тетрагидрофуран
ТФК=трифторуксусная кислота
ПОЛУЧЕНИЕ
Получение 1: 1,3-дихлор-5-(триметоксиметил)бензол
1,3-Дихлор-5-(триметоксиметил)бензол получают в соответствии со способом, изображенным на схеме реакции Р1 и как описано ниже.
Схема реакции P1
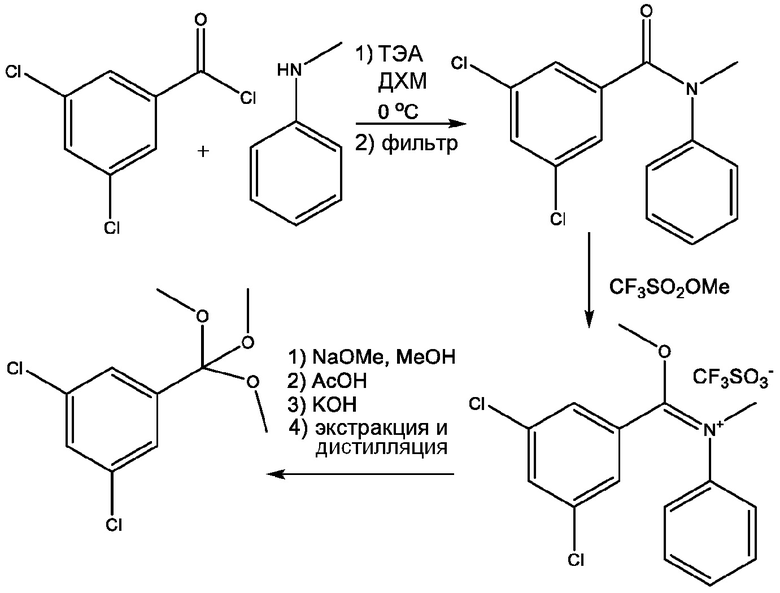
Стадия 1: Получение (Е)-[(3,5-дихлорфенил)метоксиметилен]метилфениламмония; трифторметансульфоната
К перемешиваемому раствору N-метиланилина (150 г, 1399,9 ммоль) в ДХМ (1200 мл, 1590 г) добавляют ТЭА (141,7 г, 1400 ммоль), и полученный раствор охлаждают до 0°С в течение 30 минут. К этому раствору добавляют раствор 3,5-дихлорбензоилхлорида (308 г, 1470,4 ммоль, 1,05 экв.) в ДХМ (300 мл, 398 г) в течение 60 минут, поддерживая температуру <5°C. Реакционную смесь перемешивают в течение 30 минут при 0°С, затем фильтруют для удаления гидрохлорида триэтиламина. Полученный фильтрат охлаждают до 0°С и к нему добавляют метилтрифторметансульфонат (288,3 г, 1687 ммоль, 1,205 экв.) в течение 60 минут. Реакционную смесь нагревают до 52°С и перемешивают в течение 12 ч, затем концентрируют дистилляцией до объема 900 мл. Смесь охлаждают до 20°С в течение 20 минут и к ней добавляют МТБЭ (1500 мл, 1113 г) в течение 60 минут. Смесь охлаждают до 0°С в течение 30 минут и перемешивают при 0°С в течение 30 минут. Реакционную смесь фильтруют, и осадок на фильтре промывают МТБЭ (300 мл, 223 г). Полученное твердое вещество сушат в течение ночи в вакууме, с получением 450 г (Е)-[(3,5-дихлорфенил)метоксиметилен]метилфениламмония; трифторметансульфоната (выход 72%).
Стадия 2: Получение 1,3-дихлор-5-(триметоксиметил)бензола
К раствору метоксида натрия в метаноле (25% масс. в метаноле, 364,9 г, 386,1 мл, 1520 ммоль, 1,5 экв.) при 0°C добавляют раствор (Е)-[(3,5-дихлорфенил)-метоксиметилен]метилфениламмония; трифторметансульфоната (450 г, 1013 ммоль) в MeOH (3000 мл) в течение 60 минут при поддержании температуры <5°C. Реакционную смесь перемешивают в течение 30 минут, затем к ней добавляют AcOH (206,4 мл, 216,3 г, 3,2 экв.) в течение 30 минут, поддерживая температуру <5°C. Затем эту реакционную смесь добавляют в течение 60 минут в реактор, содержащий 4М водный раствор гидроксида калия (1317 г, 1126 мл, 4 экв.), поддерживая температуру на уровне 20°C. Затем реакционную смесь нагревают и концентрируют дистилляцией до объема 1500 мл. Реакционную смесь доводят до 20°С, и к ней добавляют воду (1250 мл) и ДХМ (1250 мл). Перемешивание смеси прекращают и дают слоям разделиться, и затем собирают нижний органический слой. К оставшемуся водному слою добавляют ДХМ (1250 мл), и смесь перемешивают в течение 10 минут, перемешивание прекращают, дают слоям разделиться и собирают нижний органический слой. Затем объединенные органические слои дистиллируют, сначала при атмосферном давлении, отбрасывая фракцию, кипящую до 100°С, и затем дистиллируют в вакууме при 5-10 мбар, повышая температуру реактора с 95°С до 160°С в течение 2 ч. Желаемый ортоэфир, 1,3-дихлор-5-(триметоксиметил)бензол, собирают в виде бледно-желтого масла (201,9 г, 803,4 ммоль, выход 79,3%). 1H ЯМР (500 МГц, CDCl3) δ 7,48 (д, J=1,9 Гц, 2H), 7,36 (т, J=1,9 Гц, 1H), 3,15 (с, 9H). 13С ЯМР (126 МГц, CDCl3) δ 140,4, 134,9, 129,0, 126,2, 113,7, 49,9. FTIR (чистый): 1568,2, 1418,8, 1256,3, 1094,5, 987,2, 862,7, 796,0, 656,6, 517,3 см-1.
Получение 2: 1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октан
1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октан получают в соответствии со способом, изображенным на схеме реакции Р2, и как описано ниже.
Схема реакции P2

Стадия 1: Синтез (3-метилоксетан-3-ил)метил 3,5-дихлорбензоата
К раствору 3-метил-3-оксетанметанола (54,3 г, 0,53 моль) в ДХМ (400 мл) при температуре ~0°С добавляют раствор 3,5-дихлорбензоилхлорида (111,4 г, 0,53 моль) в ДХМ (100 мл) в течение 45 минут. Смесь перемешивают при ~0°С в течение 1,5 ч, затем промывают водой (3х200 мл). Затем органический слой концентрируют для удаления ДХМ. В результате получают 143,7 г (выход неочищенного продукта 98,6%) бесцветного вязкого масла, которое кристаллизуется при выстаивании. Половину этого материала переносят на следующую стадию, и другую половину этого материала перекристаллизовывают из изопропилового спирта (250 мл) с получением 55,0 г (3-метилоксетан-3-ил)метил-3,5-дихлорбензоата в виде бесцветного кристаллического твердого вещества. 1H ЯМР (44 МГц, CDCl3) δ 7,85 (д, 2H), 7,55 (т, 1H), 4,75-4,28 (м, 6H), 1,45 (с, 3H).
Стадия 2: Синтез 1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октана
Раствор (3-метилоксетан-3-ил)метил-3,5-дихлорбензоата (71,85 г, 0,261 моль) в ДХМ (435 мл) охлаждают до -5°С на бане ацетон/лед. К этому раствору добавляют эфират трехфтористого бора (BF3•Et2O, 9,4 г, 66,25 ммоль) и раствор перемешивают в течение ночи при нагревании до КТ с получением желтого раствора. К этому раствору добавляют ТЭА (25,0 г, 0,247 моль) и смесь перемешивают в течение 1 часа при КТ, затем к ней добавляют МТБЭ (520 мл). Поскольку осадок не образуется, смесь концентрируют для удаления растворителя, что дает оранжево-желтое твердое вещество (сырая масса ~85 г), которое перекристаллизовывают в ИПС (250 мл) с получением 41,6 г (выход 57%) желаемого продукта. Еще две порции получают концентрированием маточного раствора и затравкой материалом из 1 порции с получением дополнительных 5,0 г и 3,46 г, соответственно, желаемого продукта 1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октана. Общий полученный выход составляет 50,06 г (68%).
Время удерживания при ВЭЖХ 5,062 минуты; 1H ЯМР (44 МГц, CDCl3) δ 7,83-7,13 (м, 3H), 4,15 (с, 6H), 0,97 (с, 3H).
Кристаллический 1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октан оценивают с помощью ПРД, и собранный спектр пика представлен на фиг. 1. Значения 2-тета представлены в ПРД таблице 1 ниже, и составляют ±0,2 2-тета. Кристаллическая форма 1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октана характеризуется пиками ПРД при 21,2 и 19,7 2-тета, каждый ±0,2 2-тета; 21,2, 19,7 и 14,4 2-тета, каждый ±0,2 2-тета; 21,2, 19,7, 14,4 и 23,7 2-тета, каждый ±0,2 2-тета; 21,2, 19,7, 14,4, 23,7 и 24,2 2-тета, каждый ±0,2 2-тета; 21,2, 19,7, 14,4, 23,7, 24,2 и 30,6 2-тета, каждый ±0,2 2-тета.
ПРД таблица 1
Получение 3: 1-(3,5-Дихлорфенил)-4-этил-2,6,7-триоксабицикло[2.2.2]октан

К раствору 1,3-дихлор-5-(триметоксиметил)бензола (10,04 г, 40,0 ммоль) в безводном ДХМ (40 мл) добавляют 2-этил-2-(гидроксиметил)-1,3-пропандиол (5,375 г, 40,0 ммоль), затем ТФК (0,3 мл, 3,9 ммоль), и смесь перемешивают при комнатной температуре в течение 65 часов. Добавляют безводный K2CO3 (2,2 г), смесь перемешивают в течение 3 ч, затем фильтруют. Фильтрат концентрируют на роторном испарителе, и остаток растворяют в горячем толуоле (70 мл), охлаждают до комнатной температуры и фильтруют через ПТФЭ мембрану, затем концентрируют в вакууме с получением остатка (12,40 г), который затвердевает. Остаток кристаллизуют из гептана: добавляют гептан (50 мл), твердое вещество растворяют с обратным холодильником и затем раствор выдерживают при -5°С в течение 3 часов. Твердый осадок отфильтровывают, промывают охлажденными гептанами и сушат на воздухе; это дает первую порцию сырого продукта (6,12 г). Объединенный фильтрат выпаривают до половины исходного объема, затем оставляют на ночь при -5°С; вторую порцию (1,45 г) вещества собирают фильтрованием. Обе части содержат желаемый 2,6,7-триоксабицикло[2.2.2]октан, смешанный с побочным продуктом, 2,2-бис(гидроксиметил)бутил 3,5-дихлорбензоатом. Первая порция имеет мольное соотношение желаемый продукт:побочный продукт 1:0,25, и вторая порция имеет соотношение 1:0,37.
Обе части полученного неочищенного материала объединяют с другой партией, полученной аналогичным образом (7,58 г, соотношение желаемого продукта:побочного продукта 1:0,18).
Объединенный неочищенный продукт (15,15 г) перекристаллизовывают из i-PrOAc (35 мл). Твердое вещество растворяют при нагревании ниже температуры кипения с обратным холодильником, и затем раствор оставляют при комнатной температуре на 2 часа. Осажденное твердое вещество отфильтровывают, промывают холодным i-PrOAc и сушат на воздухе; таким образом, получают продукт с соотношением продукт/побочный продукт 1:5,2 (0,77 г).
Фильтрат оставляют при -5°C на ночь, выпавшее в осадок твердое вещество отделяют, и фильтрат ступенчато выпаривают/охлаждают. Эти манипуляции дают четыре партии продукта недостаточной чистоты. Их объединяют в одну партию (11,02 г, соотношение продукт/побочный продукт 1:0,11) и повторно кристаллизуют: партию растворяют в i-PrOAc (25 мл), выдерживают при комнатной температуре в течение 1 ч, затем охлаждают до -5°С и подвергнут кратковременной сонификации. Это вызывает спонтанную кристаллизацию. Немедленная фильтрация дает кристаллический продукт в количестве 2,46 г после вакуумной сушки, партия 1, анализ 97,4% (кЯМР).
Фильтрат дает много кристаллов недостаточного качества; затем кристаллические культуры повторно объединяют.
Собранное сырье (8,50 г) перекристаллизовывают из изопропанола: образец растворяют в 45 мл i-PrOH при нагревании, добавляют дополнительно 45 мл i-PrOH, раствор охлаждают до комнатной температуры и выдерживают при этой температуре в течение 1 ч, затем ставят в холодильник при -5°С на 10 мин. В это время появляются первые кристаллы. Далее раствор выдерживают при 0°С в течение 1 ч, после чего кристаллическое вещество собирают фильтрованием, промывают охлажденным i-PrOH и сушат под вакуумом. Указанное в заголовке соединение получают в количестве 6,67 г, партия 2, анализ 95,4% (кЯМР).
Фильтрат выпаривают до половины исходного объема и оставляют при 0°С в течение ночи. Следующую порцию кристаллов собирают фильтрованием, промывают охлажденным i-PrOH и сушат в вакууме. Целевое вещество получают в количестве 1,81 г, партия 3, анализ 97,2% (кЯМР).
Для партий 1-3: каждая партия имеет т.пл. 76°С; каждая партия имеет следующие 1H ЯМР и 13C ЯМР:
1H ЯМР (400 МГц, CDCl3), δ, ч./млн.: 7,52 (д, J=2,0 Гц, 2H), 7,33 (т, J=2,0 Гц, 1H), 4,09 (с, 6H), 1,32 (кв, J=7,7 Гц, 2H), 0,88 (т, J=7,7 Гц).
13C ЯМР (101 МГц, CDCl3) δ, ч./млн.: 140,7, 134,8, 129,3, 124,9, 106,7, 72,0, 33,7, 22,5, 7,7.
HRMS: ЭР+ m/z, (%): рассчитано для C13H15Cl2O3 [M+1]+ 289,0398; партия 1, найдено 289,0410; партия 2, найдено 289,0407; и партия 3, найдено 289,0404.
Получение 4: 1-(3,5-Дихлорфенил)-4-фенил-2,6,7-триоксабицикло[2.2.2]октан

Стадия 1: Получение 1,1,1-трис(гидроксиметил)фенилметана
К суспензии параформальдегида (12,8 г, 0,43 моль) в безводном ТГФ (160 мл) добавляют фенилацетальдегид (12,4 мл, 0,11 моль) и Ca(OH)2 (31,5 г, 0,43 моль). Реакционную смесь перемешивают при 60-65°С (температура бани) в течение 4 дней.
После охлаждения до комнатной температуры, реакционную смесь фильтруют через целит, и фильтровальную лепешку промывают ДХМ. Объединенные фильтраты концентрируют на роторном испарителе. Остаточное масло обрабатывают этилацетатом, затравливают (твердым веществом из предыдущих экспериментов, выделенным хроматографией) и оставляют в холодильнике в течение ночи.
Образовавшееся кристаллическое твердое вещество собирают фильтрованием, промывают охлажденным EtOAc и сушат с получением целевого соединения триола (9,8 г, выход 49%).
Стадия 2: Получение 1-(3,5-дихлорфенил)-4-фенил-2,6,7-триоксабицикло[2.2.2]октана
К 1,3-дихлор-5-(триметоксиметил)бензолу (6,3 г, 0,025 моль) и 1,1,1-трис(гидроксиметил)фенилметану (4,6 г, 0,025 моль), суспендированным в безводном ДХМ (40 мл), добавляют 0,19 мл (0,0025 моль) ТФК. Суспензия становится прозрачным раствором, и ее оставляют при комнатной температуре на 48 часов, затем добавляют K2CO3 и смесь перемешивают в течение 3 часов. Смесь фильтруют, и растворитель выпаривают. Попытка растворить остаток (~9 г) в кипящем толуоле (25 мл) приводит к неполному растворению. После охлаждения до температуры окружающей среды, раствор декантируют и охлаждают. Образовавшееся твердое вещество собирают фильтрованием и кристаллизуют из н-гептана с получением белого мягкого вещества (5,2 г).
Другой эксперимент, проведенный в том же масштабе, дает 5,0 г продукта.
Объединенные партии кристаллизуют из изопропилацетата с получением 5,78 г 2,6,7-триоксабицикло[2.2.2]октана; партия 1, анализ 99,6% (кЯМР);
Фильтрат из партии 1 охлаждают и хранят при 0°С в течение ночи. Затем фильтрованием собирают дополнительные 1,85 г 2,6,7-триоксабицикло[2.2.2]октана. Партия 2, анализ 98,0% (кЯМР). Общий выход перекристаллизованного продукта составляет 45%.
Партия 1 имеет т. пл. 139°С; партия 2 имеет т. пл. 138°С.
Каждая партия 1 и партия 2 имеет
1H ЯМР (400 МГц, CDCl3), δ, ч./млн.: 7,59 (д, J=2,0 Гц, 2H), 7,39-7,45 (м, 2H), 7,33, 7,39 (м, 2H), 7,17-7,23 (м, 2H), 4,15 (с, 6H).
Спектр 13С ЯМР (101 МГц, CDCl3) δ, ч./млн.: 140,4, 135,6, 134,9, 129,5, 129,4, 128,4, 125,4, 125,0, 107,2, 72,4, 37,1.
HRMS ЭР+ m/z, (%): рассчитано для C17H15Cl2O3 [M+1]+ 337,0398; найдено 337,0406.
Получение 5: 2-(3,5-дихлорфенил)-2-метокси-1,3-диоксолан

К раствору 1,3-дихлор-5-(триметоксиметил)бензола (10,04 г, 40,0 ммоль) в безводном ДХМ (40 мл) добавляют этиленгликоль (2,7 мл, 48,3 ммоль, 1,2 экв.), затем ТФК (0,3 мл, 3,9 ммоль) и смесь перемешивают при комнатной температуре в течение 72 часов. Добавляют безводный K2CO3 (1,5 г), смесь перемешивают в течение 2 ч, затем фильтруют. Фильтрат выпаривают досуха и получают желтое масло (10,76 г). Полученное масло подвергают вакуумной дистилляции, собирая фракцию, кипящую при 160-185°С/16 мбар. Собранную фракцию (мутное зеленоватое масло) дополнительно очищают повторной дистилляцией, собирая дистиллят при 180-183°С/16 мбар. Перегонка дает желаемый продукт (5,63 г) в виде желтого масла, анализ 96,2% (кЯМР).
Спектр 1H ЯМР (400 МГц, CDCl3), δ, ч./млн.: 7,47 (д, J=1,9 Гц, 2H), 7,34 (т, J=1,9 Гц, 1H), 4,19-4,30 (м, 2H), 4,03-4,13 (м, 2Н), 3,28 (с, 3Н).
Спектр 13C ЯМР (101 МГц, CDCl3), ч./млн.: δ 141,4, 135,0, 129,1, 125,0, 120,1, 65,6, 50,4.
ГХМС m/z, (%): 217 (100) [M-OMe]+, 173 (71) [C7H3Cl2O]+, 145 (30) [C6H3Cl2]+
Получение 6: 1-(3,5-Дихлорфенил)-2,7,8-триоксабицикло[3.2.1]октан
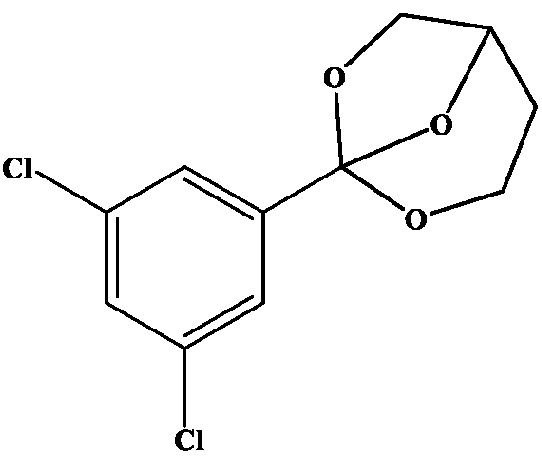
К раствору 1,3-дихлор-5-(триметоксиметил)бензола (10,04 г, 40,0 ммоль) в безводном ДХМ (50 мл) добавляют 1,2,4-бутантриол (3,57 мл, 40,0 ммоль) с последующим добавлением ТФК (0,3 мл, 3,9 ммоль), и смесь перемешивают при комнатной температуре в течение 18 часов. Добавляют безводный K2CO3 (1,5 г), смесь перемешивают в течение 2 ч, затем фильтруют. Фильтрат выпаривают досуха, давая желтое масло (10,6 г), которое медленно затвердевает.
Неочищенный продукт растворяют в толуоле (30 мл), фильтруют через мембрану из ПТФЭ, затем выпаривают при пониженном давлении. К остатку добавляют гептан (80 мл), кипятят с обратным холодильником, затем охлаждают и декантируют верхний слой (гексановый раствор). Нижний слой (масло) неоднократно обрабатывают горячим гептаном. Растворы в гептане объединяют, уменьшают объем до ~25 мл и выдерживают при 4°С в течение 3 ч. Белый осадок отфильтровывают, промывают холодным гептаном и оставляют сушиться на воздухе в течение ночи.
Выделенный 2,7,8-триоксабицикло[3.2.1]октан (4,27 г) содержит примерно 11% моль побочных продуктов с открытой цепью (дигидроксибутил-3,5-дихлорбензоаты). Этот продукт объединяют с другим образцом, полученным аналогичным образом (4,03 г, загрязнение бензоатами с открытой цепью примерно 7% моль), для дальнейшей очистки.
Объединенную партию (всего 8,30 г) растворяют в гептане (30 мл) при нагревании, раствор фильтруют горячим и выдерживают при комнатной температуре в течение 5 часов. Осажденное твердое вещество собирают фильтрованием и сушат на воздухе. Это дает 6,78 г кристаллического продукта. Фильтрат частично выпаривают (примерно до половины исходного объема) и хранят при -5°С в течение ночи. Это дает дополнительное количество (вторую порцию) твердого вещества в количестве 1,03 г и качество, близкое к качеству основной порции. Вторая порция содержит примерно 6% моль дигидроксибутилбензоатов. Оба образца объединяют и перекристаллизовывают из i-PrOAc следующим образом. Объединенный образец (7,84 г) растворяют в горячем изопропилацетате (10 мл), раствор охлаждают, затем оставляют при -5°С в течение ночи. Образовавшееся белое кристаллическое вещество собирают фильтрованием, промывают охлажденным i-PrOAc и сушат в вакууме при комнатной температуре, чтобы получить указанное в заголовке соединение (5,30 г). Анализ 96,2% (кЯМР), т. пл. 81-84°C.
Спектр 1H ЯМР (400 МГц, CDCl3), δ, ч./млн.: 7,53 (д, J=2,0 Гц, 2H), 7,36 (т, J=2,0 Гц, 1H), 4,83 (тд, J=3,4, 1,6 Гц, 1H), 4,27 (дт, J=11,7, 4,2 Гц, 1H), 4,24 (д, J=7,2 Гц, 1H), 4,12 (ддд, J=7,3, 4,8, 1,6 Гц, 1H), 4,03 (ддд, J=11,7, 6,7, 1,0 Гц, 1H), 2,41 (дддд, J=13,8, 12,2, 6,7, 3,4, 1,8 Гц, 1H), 1,51 (дддд, J=13,8, 4,2, 2,0, 0,5 Гц, 1H).
Спектр 13C ЯМР (101 МГц, CDCl3), δ, ч./млн.: δ 139,7, 134,9, 129,6, 125,0, 117,2, 73,9, 69,6, 59,5, 28,2.
HRMS ЭР+ m/z, (%): рассчитано для C11H11Cl2O3 [M+1]+ 261,0085; найдено 261,0094.
Получение 7: 3-(3,5-Дихлорфенил)-2,4,10-триоксаадамантан

К раствору 1,3-дихлор-5-(триметоксиметил)бензола (6,42 г, 25,57 ммоль) в безводном ДХМ (50 мл) добавляют цис-флороглюцит (2,92 г, 22,16 ммоль) с последующим добавлением по каплям BF3•OEt2 (0,28 мл, 2,21 ммоль, ~0,1 экв.) в суспензию при перемешивании. Смесь перемешивают при комнатной температуре в течение 34 ч, затем отфильтровывают остаточный флороглюцит (0,14 г) и раствор выпаривают при пониженном давлении. К остатку добавляют ТБМЭ (40 мл), и смесь кратковременно кипятят с обратным холодильником, полного растворения не достигают. Смесь охлаждают, выдерживают при 0°С 1 ч, затем фильтруют, собирая белый осадок (5,03 г).
Выпаривание фитрата до четверти первоначального объема дает дополнительный выход твердого вещества (0,50 г).
Аналогичный эксперимент начинают с 1,61 г (6,41 ммоль) 1,3-дихлор-5-(триметоксиметил)бензола и 0,85 г (6,44 ммоль) цис-флороглюцита, что дает 1,27 г неочищенного твердого вещества.
Все твердые вещества объединяют и растворяют в МЭК (30 мл) при кипячении с обратным холодильником. Раствор фильтруют горячим и выдерживают в течение ночи при комнатной температуре, затем кристаллическое твердое вещество отфильтровывают, промывают небольшим объемом холодного МЭК и сушат в вакууме при комнатной температуре; это дает указанное в заголовке соединение в количестве 5,586 г (выход 61%*), анализ 99,8% (кЯМР), т. пл. 188°C.
Фильтрат выпаривают до  объема и хранят при -5°С в течение 4 ч с получением дополнительной порции кристаллов того же качества, 0,79 г (выход 9%*), анализ 99,8% (кЯМР), т. пл. 188°C.
объема и хранят при -5°С в течение 4 ч с получением дополнительной порции кристаллов того же качества, 0,79 г (выход 9%*), анализ 99,8% (кЯМР), т. пл. 188°C.
* выход рассчитан по общему количеству исходного ортоэфира в обоих опытах.
1H ЯМР (400 МГц, CDCl3), δ, ч./млн.: 7,57 (д, J=1,9 Гц, 2H), 7,33 (т, J=1,9 Гц, 1H), 4,56 (м, 3H), 3,11-2,49 (м, 3H), 1,81 (ддт, J=12,9, 2,1, 1,2 Гц, 2H)
13C ЯМР (101 МГц, CDCl3) δ, ч./млн.: 142,5, 134,8, 129,3, 124,4, 108,1, 69,3, 33,0.
HRMS ЭР+ m/z, (%): рассчитано для C13H13Cl2O3 [M+1]+ 287,0242; найдено 287,0253.
Получение 8: 1-(3,5-дихлорфенил)-4-изопропил-2,6,7-триоксабицикло[2.2.2]октан

Стадия 1: Синтез 2-изопропил-2-гидроксиметил-1,3-пропандиола
Изовалеральдегид (83 г, 1,0 моль) добавляют в течение 0,5 ч при комнатной температуре к раствору гидроксида натрия (60 г, 1,5 моль) в воде (1200 мл), содержащему 37% раствор формальдегида (324 г, 4,0 моль). Затем раствор перемешивают при 50-55°C в течение 3 ч и оставляют в течение ночи при комнатной температуре. Его отфильтровывают от небольшого количества (2,7 г) кристаллического твердого вещества, и раствор взвешивают (1725 г). Из них берут 15% (259 г) и экстрагируют 4 раза ДХМ (200 мл); экстракты концентрируют с получением 7,0 г масла, которое кристаллизуется. Полученное твердое вещество, как показано 1H ЯМР, представляет собой димер простого эфира, а именно (2,2'-(оксибис(метилен))бис(2-изопропилпропан-1,3-диол)).
Водный раствор, оставшийся после предыдущей экстракции ДХМ, обрабатывают твердым хлоридом натрия (40 г) и экстрагируют изопропилацетатом (4х200 мл). Объединенные экстракты изопропилацетата концентрируют в вакууме с получением 14,5 г (97,8 ммоль) желаемого 2-изопропил-2-гидроксиметил-1,3-пропандиола (чистого, но содержащего небольшое количество изопропилацетата по 1H ЯМР).
Стадия 2: Синтез 1-(3,5-дихлорфенил)-4-изопропил-2,6,7-триоксабицикло[2.2.2]октана
Загружают толуол (150 мл), затем 3,5-дихлорбензойную кислоту (DCBA, 17,0 г, 89 ммоль) и метансульфоновую кислоту (МСА, 0,8 г). Смесь кипятят с обратным холодильником с ловушкой Дина-Старка в течение 6 ч; собирают 1,9 г воды (теоретическое количество 3,2 г) и выстаивают в течение ночи. Смесь экстрагируют 10% раствором КОН (100 мл и 20 мл) при 60-70°С с последующей промывкой водой при той же температуре. Подкисление водного экстракта с последующим фильтрованием и сушкой полученного белого твердого вещества дает 8,8 г (53% от исходного) DCBA.
Раствор в толуоле концентрируют с получением 14,5 г масла, которое, по данным ЯМР, содержит примерно 30% желаемого ортоэфира. При растворении в метаноле (100 мл) и обработке твердым КОН (5,0 г, 89 ммоль) получают раствор, из которого при выстаивании осаждаются кристаллы. Фильтрация и промывка (метанол) дают чистый 1-(3,5-дихлорфенил)-4-изопропил-2,6,7-триоксабицикло[2.2.2]октан (1,6 г, 13% от прореагировавшей DCBA); 1H ЯМР: δ 7,4 м (2H); 4,0 (с, 6Н); 1,4 м (1Н) и 0,8 д (6Н).
Альтернативно, при выполнении стадии 2 с использованием пара-толуолсульфоновой кислоты (PTSA) в качестве катализатора вместо МСК, наблюдают очень плохое превращение в желаемый орто-эфир (90% восстановление DCBA и образуется 30% теоретического количества воды).
Дополнительную партию желаемого орто-эфира получают следующим образом. Берут 388 г 1725 г раствора (со стадии 1 выше) (22,5% от общего количества) и обрабатывают таким же образом, как описано выше: 4 экстракции с ДХМ (250 мл) для удаления димера тетраола, затем добавление NaCl (60 г) и 4 экстракции изопропилацетатом (200 мл) с получением 17,5 г (118,2 ммоль) чистого (2-(гидроксиметил)-2-изопропилпропан-1,3-диола) в виде полутвердого вещества. К этому полутвердому веществу загружают 3,5-дихлорбензойную кислоту (16,5 г, 86,39 ммоль), затем толуол (200 мл) и МСК (1,2 г, 12,5 ммоль). Раствор кипятят с обратным холодильником при ловушке Дина-Старка в течение 6 ч, охлаждают до температуры окружающей среды. Ловушка содержит 2,3 г воды. Добавляют дополнительное количество МСК (1,2 г) и кипятят с обратным холодильником еще в течение 6 часов, в результате чего получают дополнительные 0,5 мл воды. Партии дают остыть до температуры окружающей среды, обрабатывают 100 мл 6% раствора КОН при 40-50°С с последующей промывкой 100 мл воды при той же температуре. Толуол удаляют, оставляя 28 г масла. Это масло растворяют в метаноле (150 мл) и воде (10 мл) и обрабатывают гидроксидом калия (6 г, 107 ммоль) при температуре окружающей среды (при этом нормальный эфир превращается в растворимый триол и 3,5-дихлорбензоат калия). Затем ортоэфир начинает быстро кристаллизоваться; суспензию охлаждают до 0-10°C в течение 0,5 ч, фильтруют, промывают метанолом и сушат с получением 1-(3,5-дихлорфенил)-4-изопропил-2,6,7-триоксабицикло[2.2.2]октана (5,6 г, 21,5%) в виде белого кристаллического порошка.
Получение 9: 4-(трет-бутил)-1-(3,5-дихлорфенил)-2,6,7-триоксабицикло[2.2.2]октан
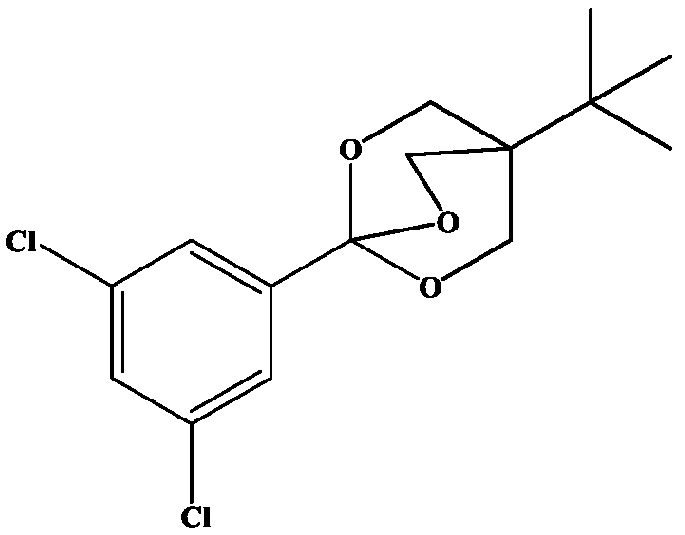
4-(трет-бутил)-1-(3,5-дихлорфенил)-2,6,7-триоксабицикло[2.2.2]октан получают аналогично соединению из Препаративного примера 8, начиная с 3,3-диметилбутанальдегида (1,0 моль), который добавляют в течение 0,5 ч при КТ к раствору гидроксида натрия (60 г, 1,5 моль) в воде (1200 мл), содержащему 37% раствор формальдегида (324 г, 4,0 моль). Затем осуществляют последовательность реакций способом, аналогичным получению 8, с получением желаемого 4-(трет-бутил)-1-(3,5-дихлорфенил)-2,6,7-триоксабицикло[2.2.2]октана. На стадии 2 синтеза, в дополнение к использованию метансульфоновой кислоты можно использовать эфират трифторида бора аналогичным образом в качестве альтернативного кислотного катализатора.
ПРИМЕРЫ
Примеры 1-47 были осуществляют для изучения влияния различных параметров, как описано в настоящем документе ниже, на реакцию 4-амино-3-гидроксибензойной кислоты с 1,3-дихлор- 5-(триметоксиметил)бензолом с получением 6-карбокси-1-(3,5-дихлорфенил)бензоксазола (тафамидиса), изображенного ниже.

ПРИМЕР 1
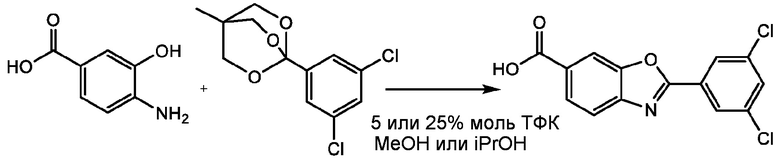
4-Амино-3-гидроксибензойную кислоту (0,500 г, 3,26 ммоль, 1,0 экв.) растворяют в 8,00 мл МеОН и туда добавляют 1,3-дихлор-5-(триметоксиметил)бензол (0,86 г, 3,4 ммоль, 1,05 экв.) в 2,00 мл МеОН. Реакционную смесь перемешивают при КТ в течение 6 ч, затем нагревают до 60°C и перемешивают в течение ночи. Реакционная смесь становится густой суспензией. Через 23 часа ВЭЖХ-анализ реакционной смеси показывает 77,29% площади желаемого продукта. Реакционной смеси дают остыть и фильтруют. Полученные твердые вещества промывают МеОН (10 мл) и сушат в вакууме (65°С, ~50 мбар) с получением 0,531 г (выход при выделении 52,8%) желаемого продукта тафамидиса в виде желто-розового твердого вещества.
ПРИМЕР 2
4-Амино-3-гидроксибензойную кислоту (0,500 г, 3,26 ммоль, 1,0 экв.) растворяют в 8,00 мл МеОН и к ней добавляют ТФК (25,0 мкл, 0,33 ммоль, 0,1 экв.), и затем 1,3-дихлор-5-(триметоксиметил)бензол (0,86 г, 3,4 ммоль, 1,05 экв.) в 2,00 мл MeOH. Реакционную смесь перемешивают при комнатной температуре в течение 6 ч, затем нагревают до 60°С и перемешивают в течение ночи. Реакционная смесь превращается в густую суспензию. Через 23 ч ВЭЖХ анализ реакционной смеси показала 92,19% площади желаемого продукта. Реакционную смесь охлаждают и фильтруют. Полученные твердые вещества промывают МеОН (10 мл) и сушат в вакууме (65°С, ~50 мбар) с получением 0,834 г (выход при выделении 82,9%) желаемого продукта тафамидиса в виде розового твердого вещества.
ПРИМЕР 3
4-Амино-3-гидроксибензойную кислоту (0,500 г, 3,26 ммоль, 1,0 экв.) растворяют в 8,00 мл МеОН и к ней добавляют ТФК (63,0 мкл, 0,82 ммоль, 0,25 экв.), и затем 1,3-дихлор-5-(триметоксиметил)бензол (0,86 г, 3,4 ммоль, 1,05 экв.) в 2,00 мл MeOH. Реакционную смесь перемешивают при комнатной температуре в течение 6 ч, затем нагревают до 60°С и перемешивают в течение ночи. Реакционная смесь превращается в густую суспензию. Через 23 ч ВЭЖХ анализ реакционной смеси показывает 91,29% площади желаемого продукта. Реакционную смесь охлаждают и фильтруют. Полученные твердые вещества промывают МеОН (10 мл) и сушат в вакууме (65°С, ~50 мбар) с получением 0,818 г (выход при выделении 81,3%) желаемого продукта тафамидиса в виде розового твердого вещества.
ПРИМЕР 4
4-Амино-3-гидроксибензойную кислоту (0,500 г, 3,26 ммоль, 1,0 экв.) растворяют в 8,00 мл МеОН и к ней добавляют ТФК (63,0 мкл, 0,82 ммоль, 0,25 экв.), и затем 1,3-дихлор-5-(триметоксиметил)бензол (0,86 г, 3,4 ммоль, 1,05 экв.) в 2,00 мл MeOH. В реакционную смесь вносят затравку путем добавления тафамидиса (50 мг, 0,16 ммоль, 0,05 экв.). Реакционную смесь перемешивают при комнатной температуре в течение 6 ч, затем нагревают до 60°С и перемешивают в течение ночи. Реакционная смесь превращается в суспензию, которую легче перемешивать, чем суспензию, полученную в примерах 1-3. Через 23 часа ВЭЖХ анализ реакционной смеси показывает 92,36% площади желаемого продукта. Реакционную смесь охлаждают и фильтруют. Полученные твердые вещества промывают МеОН (10 мл) и сушат в вакууме (65°С, ~50 мбар) с получением 0,831 г (выход при выделении 82,7%) желаемого продукта в виде твердого вещества розового цвета.
Реакции для примеров 1-3 контролируют через 1 час, 2 часа, 4 часа, 7 часов и 23 часа с использованием высокоэффективной жидкостной хроматографии (ВЭЖХ) с УФ-обнаружением. Берут образец 25 мл реакционной смеси и разводят 975 мл ДМСО.
Условия хроматографии ВЭЖХ (способ А), использованные для контроля реакции, следующие:
Колонка: Agilent Zorbax SB-C18, 1,8 мм, 3,0×50 мм; Температура колонки: 50°С; Подвижная фаза А (МРА): 0,05% ТФК в воде; подвижная фаза B (MPB): ацетонитрил;
Градиент [Время (мин), (%MPA/%MPB)]: 0 (95,0/5,0); 1 (95,0/5,0); 9 (0/100); 11,5 (0/100); 11,6 (95,0/5,0); 12,0 (95,0/5,0);
УФ обнаружение; Объем впрыска: 1 мл; Время сбора данных: 12 мин с 2 мин после сбора данных.
Результаты хроматографии показывают, что реакции, катализируемые ТФК, протекают намного быстрее, чем не катализируемая реакция, и по существу завершаются через 4 ч, в то время как не катализируемая реакция не завершается через 23 часа (см. таблицу 1 ниже). Затравка катализируемой реакции тафамидисом дает суспензию реакционной смеси, которую легче перемешивать, чем реакционные смеси без затравки.
Продукт, полученный в результате процесса, анализируют с помощью 1H, 13C ЯМР и с помощью ЖХМС. Полученные результаты указывают на то, что желаемый продукт тафамидис получен.
1H ЯМР (500 МГц, ДМСО-d6) δ 1H ЯМР (500 МГц, ДМСО-d6) δ 13,20 (с, 1H), 8,18 (дд, J=1,5, 0,6 Гц, 1H), 8,02 (д, J=1,9 Гц, 2H), 7,97 (дд, J=8,4, 1,5 Гц, 1Н), 7,86-7,81 (м, 2Н).
13С ЯМР (126 МГц, ДМСО-d6) δ 167,07, 162,35, 150,42, 145,05, 135,58, 132,06, 129,47, 129,16, 126,89, 126,23, 120,36, 112,59.
Анализ SQ-ЖХМС продукта, полученного в настоящем способе, сравнивают со стандартным образцом свободной кислоты тафамидиса, и оценивают время удержания, наблюдаемый молекулярный ион (m/z 308,10 (M+H)) и фрагментацию продукта этого процесса, которые соответствуют свободной кислоте тафамидиса.
ПРИМЕРЫ 5-23
Реакцию 4-амино-3-гидроксибензойной кислоты (AHBA) с 1,3-дихлор-5-(триметоксиметил)бензолом (ортоэфир=«OE») в присутствии 0,25 экв. ТФК в качестве катализатора оценивают в различных растворителях согласно следующей общей процедуре.
AHBA (0,100 г, 0,653 ммоль, 1,0 экв.) смешивают в 1,50 мл растворителя в течение приблизительно 1 ч, к нему добавляют ТФК (12,6 мкл, 0,163 ммоль, 0,25 экв.) и эту смесь перемешивают в течение 10 минут. Затем к смеси AHBA добавляют OE (0,18 г, 0,718 ммоль, 1,1 экв.) в 0,50 мл растворителя. Реакционную смесь нагревают при 60°С. Образцы реакционной смеси отбирают через 1 час, 5 часов и 21,5 часа и анализируют с помощью ВЭЖХ с использованием ранее описанного способа А ВЭЖХ, и определяют % площади желаемого продукта тафамидиса (см. таблицу 2 ниже).
Обнаружено, что реакция быстро протекает в нескольких растворителях. В MeOH, EtOH, ИПС, n-BuOH, ацетонитриле, ацетоне, МЭК, EtOAc и 1,2-ДМЭ реакция, по-видимому, завершалась за один час или меньше. Однако в реакциях, проведенных в EtOH, ИПС, n-BuOH, ацетоне и МЭК, наблюдают примеси. Примесями в реакциях, где растворителями являются EtOH, ИПС или н-BuOH, могут быть соответствующие этиловый, изопропиловый или н-бутиловый эфиры 3,5-дихлорбензойной кислоты, соответственно. Обнаружено, что реакция протекает медленнее в эфирных растворителях ТГФ, 1,4-диоксане или МТБЭ, но значительных уровней примесей не наблюдается. В менее полярных растворителях анизоле, хлороформе, хлорбензоле, гептане, циклогексане и толуоле, реакция протекает значительно медленнее, и в каждой из этих реакций наблюдается значительное количество примеси со временем удерживания 6,1 мин. Вода не является идеальным растворителем, поскольку 1,3-дихлор-5-(триметоксиметил)бензол нерастворим в воде и легко гидролизуется.
ПРИМЕРЫ 24-28
В примерах 24-28 оценивают влияние различных количеств ортоэфира, 1,3-дихлор-5-(триметоксиметил)бензола (1,0, 1,5, 2,0, 5,0 или 10,0 эквивалентов) на реакцию образования тафамидиса.
Общая методика
К раствору ТФК (0,0050 мл, 0,065 ммоль) в MeOH (4 мл) добавляют 4-амино-3-гидроксибензойную кислоту, AHBA (0,200 г, 1,31 ммоль) с последующим добавлением 1,3-дихлор-5-( триметоксиметил)-бензола (ортоэфира). Реакционную смесь перемешивают и нагревают при 60°С. Образцы реакционной смеси отбирают через 1 час, 2 часа, 5 часов и 22 часа 40 минут и анализируют с помощью ВЭЖХ с использованием ранее описанного способа ВЭЖХ А, и определяют % площади желаемого продукта тафамидиса. Все реакции протекают быстро, осадок образовывается в течение 15 минут. За развитием реакции следят по исчезновению пика AHBA. Чем больше ортоэфира использовано, тем быстрее завершается реакция. Пример 24, где используют 1,0 экв. ортоэфира, не был полностью завершен, вероятно, в результате гидролиза небольшого количества ортоэфира до метилового эфира 3,5-дихлорбензойной кислоты во время реакции. Реакционные смеси из примеров 27 и 28, в которых используют 5,0 и 10,0 экв. ортоэфира, становятся фиолетовым. Желаемый продукт, тафамидис, выделяют фильтрованием, и осадок на фильтре промывают метанолом. Выделенный продукт, полученный из примеров 27 и 28, является слегка розовым.
Пример 24: Использованное количество 1,3-дихлор-5-(триметоксиметил)бензола составляет 0,260 мл, 1,31 ммоль, 1,0 экв.
Пример 25: Использованное количество 1,3-дихлор-5-(триметоксиметил)бензола составляет 0,390 мл, 1,96 ммоль, 1,5 экв.
Пример 26: Использованное количество 1,3-дихлор-5-(триметоксиметил)бензола составляет 0,521 мл, 2,61 ммоль, 2,0 экв.
Пример 27: Использованное количество 1,3-дихлор-5-(триметоксиметил)бензола составляет 1,30 мл, 6,52 ммоль, 5,0 экв.
Пример 28: Использованное количество 1,3-дихлор-5-(триметоксиметил)бензола составляет 2,60 мл, 13,1 ммоль, 10,0 экв.
Результаты для примеров 24-28 представлены в таблице 3 ниже.
ПРИМЕРЫ 29-33
Примеры 29-33 проводят для определения влияния различных количеств (5, 10, 25, 50 и 100% моль по отношению к АНВА) трифторуксусной кислоты (ТФК) на реакцию АНВА с 1,3-дихлор-5-(триметоксиметил)бензолом.
Общая методика: готовят маточный раствор ТФК (1,263 мл) в МеОН (50 мл). MeOH добавляют к 0,200 г AHBA (1,31 ммоль) с последующим добавлением исходного раствора ТФК в MeOH, чтобы получить общий объем 4,0 мл. К этому раствору добавляют 1,3-дихлор-5-(триметоксиметил)бензол (0,286 мл, 1,44 ммоль, 1,10 экв.). Реакционную смесь нагревают до 60°С и выдерживают при этой температуре в течение ночи. Образцы реакционной смеси отбирают через 0,5 ч, 1,0 ч, 2,0 ч, 5,0 ч и 21,25 ч и анализируют с помощью способа А ВЭЖХ, и определяют % площади желаемого продукта тафамидиса (см. Таблицу 4 ниже).
(5% моль)
(10% моль)
(25% моль)
(50% моль)
(100% моль)
Реакции из примеров 29-33 протекают быстро с образованием осадка при нагревании реакционных смесей, что в конечном итоге приводит к получению реакционных смесей, которые становятся неперемешиваемыми суспензиями. Реакции из примеров 29 и 30 первоначально протекают с меньшей скоростью, чем реакции из примеров 31-33, но по существу доходят до конца, тогда как реакции из примеров 31-33 первоначально имеют более высокую скорость реакции, чем реакции из примеров 29-30, но все реакции из примеров 31-33 содержат непрореагировавшую AHBA, присутствующую в 21,25 ч.
ПРИМЕРЫ 34-46
Примеры 34-46 проводят для определения влияния отсутствия кислоты, 10% моль или 25% моль различных кислот (по отношению к АНВА) или 0,75, 1,00 или 1,25 эквивалента триэтиламина на реакцию АНВА с 1,3-дихлор-5-(триметоксиметил)бензолом.
Общая методика. К 150 мг AHBA (0,979 ммоль, 1,0 экв.) добавляют 2,50 мл MeOH. Туда либо не добавляют кислоту, либо добавляют 10% моль кислоты, 25% моль кислоты, либо 0,75, 1,0 или 1,25 эквивалента ТЭА (примеры см. ниже). Туда добавляют 1,3-дихлор-5-триметоксиметилбензол (0,246 г, 0,980 ммоль, 1,00 эквивалента) в 0,50 мл МеОН. Реакции из примеров 34-43 перемешивают при КТ, и реакции из примеров 44-47 перемешивают при КТ в течение 6 часов, затем нагревают при 60°С в течение ночи. Образцы реакционной смеси отбирают через 1 ч, 6 ч и как указано в таблице 5, и анализируют с помощью способа А ВЭЖХ, и определяют % площади желаемого продукта тафамидиса (см. таблицу 5 ниже). Реакционные смеси из примеров 34-39 и 42-43 фильтруют через 24,25 ч, твердые вещества промывают МеОН (3 мл) и сушат в вакууме (65°С, ~50 мбар) с получением тафамидиса в виде твердого вещества розового цвета (выход представлен в таблице 5).
Пример 34: Кислота: ТФК, 0,0075 мл, 0,098 ммоль, 10% моль.
Пример 35: Кислота: ТФК, 0,0188 мл, 0,246 ммоль, 25% моль.
Пример 36: Кислота: HCl в воде (12,2 М) 0,0080 мл, 0,098 ммоль, 10% моль.
Пример 37: Кислота: HCl в воде (12,2 М) 0,0201 мл, 0,245 ммоль, 25% моль.
Пример 38: Кислота: HCl в MeOH (1,0 М) 0,098 мл, 0,098 ммоль, 10% моль.
Пример 39: Кислота: HCl в MeOH (1,0 М) 0,24 мл, 0,24 ммоль, 25% моль.
Пример 40: Кислота: АсОН, 0,0056 мл, 0,098 ммоль, 10% моль.
Пример 41: Кислота: АсОН, 0,0140 мл, 0,244 ммоль, 25% моль.
Пример 42: Кислота: MsOH, 0,0064 мл, 0,098 ммоль, 10% моль.
Пример 43: Кислота: MsOH, 0,0161 мл, 0,246 ммоль, 25% моль.
Пример 44: Основание: ТЭА, 0,102 мл, 0,732 ммоль, 75% моль.
Пример 45: Основание: ТЭА, 0,137 мл, 0,983 ммоль, 100% моль.
Пример 46: Основание: ТЭА, 0,171 мл, 1,23 ммоль, 125% моль.
Пример 47: без добавления кислоты или основания.
5,7 через 30 ч
8,8 через 47 ч
7,2 через 30 ч
10,9 через 47 ч
10,9 через 23 ч
17,8 через 40 ч
6,6 через 23 ч
10,8 через 40 ч
4,4 через 23 ч
7,1 через 40 ч
5,0 через 30 ч
7,7 через 47 ч
ПРИМЕРЫ 48-51
Методика: готовят растворы ТФК (316,25 мкл) в МеОН (50 мл) и ТФК (316,25 мкл) в ИПС (50 мл). В примере 48, 3,20 мл MeOH добавляют к AHBA (0,200 г, 1,31 ммоль, 1,0 экв.) с последующим добавлением 0,80 мл ТФК в растворе MeOH (0,065 ммоль, 5% моль), приготовленном выше. В примере 49, 4,0 мл раствора ТФК в MeOH (0,327 ммоль, 25% моль) добавляют к AHBA (0,200 г, 1,31 ммоль, 1,0 экв.). В примере 50, 3,20 мл ИПС добавляют к AHBA (0,200 г, 1,31 ммоль, 1,0 экв.) с последующим добавлением 0,80 мл ТФК в растворе ИПС (0,065 ммоль, 5% моль), приготовленном выше. В примере 51, 4,0 мл раствора ТФК в ИПС (0,327 ммоль, 25% моль) добавляют к AHBA (0,200 г, 1,31 ммоль, 1,0 экв.). К каждой из смесей в примерах 48-51 добавляют циклический ортоэфир, 1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октан (0,395 г, 1,44 ммоль, 1,10 экв.). Реакционные смеси нагревают до 60°С и отбирают образцы через 1, 2, 4 и 19,75 ч, и % площади тафамидиса определяют с помощью ВЭЖХ (см. таблицу 6 ниже). В примере 51, реакционную смесь фильтруют, твердые вещества промывают изопропиловым спиртом (4 мл) и сушат в вакуумной печи (65°С, ~50 мбар) в течение ночи с получением желаемого продукта тафамидиса (0,298 г, выход 74,1%) в виде слегка розового твердого вещества. 1H ЯМР и ВЭЖХ твердого вещества, полученного в результате реакции, согласуются с тафамидисом.
Реакции в ИПС протекают быстрее, чем реакции в МеОН, при этом реакции в ИПС близки к завершению через 19,75 ч.
ПРИМЕР 52
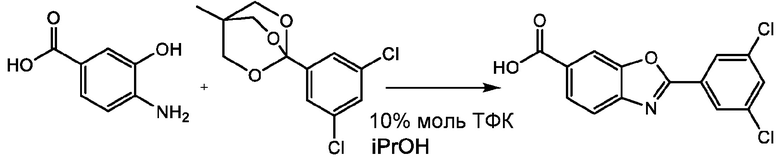
К 8 мл ИПС при 20°С при перемешивании добавляют 4-амино-3-гидроксибензойную кислоту (401 мг, 2,62 ммоль) и 1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октан (795 мг, 2,89 ммоль), затем ТФК (30 мкл, 0,388 ммоль). Реакционную смесь нагревают до 85°С и перемешивают при этой температуре в течение 12 часов. Затем смесь охлаждают до 20°С и периодически гранулируют при 20°С в течение 2 часов. Полученное твердое вещество собирают фильтрованием и промывают ИПС, которым промывают реакционный сосуд (2х4 мл). Полученное твердое вещество розового цвета сушат в вакууме (50 мбар) при 60°С в течение 12 ч с получением тафамидиса (637 мг, 2,068 ммоль, выход 79%). Получают данные 1H ЯМР, ЖХ-МС и ЖХ-МС/МС при сравнении со стандартом, которые соответствуют требуемому продукту тафамидиса. ЖХ-МС, использованная для контроля завершения реакции, показывает 76,7% площади пика тафамидиса до выделения.
ПРИМЕР 53

К 8 мл ИПС при 20°С при перемешивании добавляют 4-амино-3-гидроксибензойную кислоту (402 мг, 2,62 ммоль) и 1-(3,5-дихлорфенил)-4-этил-2,6,7-триоксабицикло[2.2.2]октан (832 мг, 2,88 ммоль), затем ТФК (30 мкл, 0,388 ммоль). Реакционную смесь нагревают до 85°С и перемешивают при этой температуре в течение 12 часов. Затем смесь охлаждают до 20°С и периодически гранулируют при 20°С в течение 2 часов. Полученное твердое вещество собирают фильтрованием и промывают ИПС, которым промывают реакционный сосуд (2х4 мл). Полученное твердое вещество розового цвета сушат в вакууме (50 мбар) при 60°С в течение 12 ч с получением тафамидиса (564 мг, 1,83 ммоль, выход 69,9%). 1H ЯМР, ЖХ-МС и ЖХ-МС/МС при сравнении со стандартом соответствуют желаемому продукту тафамидиса. ЖХ-МС, использованная для контроля завершения реакции, показывает 65,4% площади пика тафамидиса до выделения.
ПРИМЕР 54

К 8 мл ИПС при 20°С при перемешивании добавляют 4-амино-3-гидроксибензойную кислоту (405 мг, 2,65 ммоль) и 1-(3,5-дихлорфенил)-4-фенил-2,6,7-триоксабицикло[2.2.2]октан (974 мг, 2,89 ммоль), затем ТФК (30 мкл, 0,388 ммоль). Реакционную смесь нагревают до 85°С и перемешивают при этой температуре в течение 12 часов. Затем смесь охлаждают до 20°С и периодически гранулируют при 20°С в течение 2 часов. Полученное твердое вещество собирают фильтрованием и промывают ИПС, которым промывают реакционный сосуд (2х4 мл). Полученное твердое вещество розового цвета сушат в вакууме (50 мбар) при 60°С в течение 12 ч с получением тафамидиса (591 мг, 1,92 ммоль, выход 72,5%). 1H ЯМР, ЖХ-МС и ЖХ-МС/МС при сравнении со стандартом соответствуют желаемому продукту тафамидиса. ЖХ-МС, использованная для контроля завершения реакции, показывает 65,5% площади пика тафамидиса до выделения.
ПРИМЕР 55

К 8 мл ИПС при 20°С при перемешивании добавляют 4-амино-3-гидроксибензойную кислоту (402 мг, 2,62 ммоль) и 2-(3,5-дихлорфенил)-2-метокси-1,3-диоксолан (735 мг, 2,95 ммоль), затем ТФК (30 мкл, 0,388 ммоль). Реакционную смесь нагревают до 85°С и перемешивают при этой температуре в течение 12 часов. Затем смесь охлаждают до 20°С и периодически гранулируют при 20°С в течение 2 часов. Полученное твердое вещество собирают фильтрованием и промывают ИПС, которым промывают реакционный сосуд (2х4 мл). Полученное твердое вещество розового цвета сушат в вакууме (50 мбар) при 60°С в течение 12 ч с получением тафамидиса (701 мг, 2,28 ммоль, выход 86,7%). 1H ЯМР, ЖХ-МС и ЖХ-МС/МС при сравнении со стандартом соответствуют желаемому продукту тафамидиса. ЖХ-МС, использованная для контроля завершения реакции, показывает 88,9% площади пика тафамидиса до выделения.
ПРИМЕР 56

К 8 мл ИПС при 20°С при перемешивании добавляют 4-амино-3-гидроксибензойную кислоту (402 мг, 2,65 ммоль) и 1-(3,5-дихлорфенил)-2,7,8-триоксабицикло[3.2.1]октан (761 мг, 2,91 ммоль), затем ТФК (30 мкл, 0,388 ммоль). Реакционную смесь нагревают до 85°С и перемешивают при этой температуре в течение 12 часов. Затем смесь охлаждают до 20°С и периодически гранулируют при 20°С в течение 2 часов. Полученное твердое вещество собирают фильтрованием и промывают ИПС, которым промывают реакционный сосуд (2х4 мл). Полученное твердое вещество розового цвета сушат в вакууме (50 мбар) при 60°С в течение 12 ч с получением тафамидиса (679 мг, 2,20 ммоль, выход 84,0%). 1H ЯМР, ЖХ-МС и ЖХ-МС/МС при сравнении со стандартом соответствуют желаемому продукту тафамидиса. ЖХ-МС, использованная для контроля завершения реакции, показывает 78,5% площади пика тафамидиса до выделения.
ПРИМЕР 57
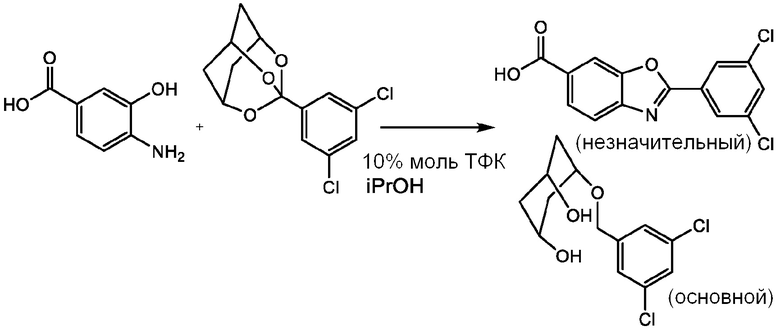
К 8 мл ИПС при 20°С при перемешивании добавляют 4-амино-3-гидроксибензойную кислоту (400 мг, 2,61 ммоль) и 3-(3,5-дихлорфенил)-2,4,10-триоксаадамантан (842 мг, 2,93 ммоль), затем ТФК (30 мкл, 0,388 ммоль). Реакционную смесь нагревают до 85°С и перемешивают при этой температуре в течение 12 часов. Затем смесь охлаждают до 20°С и периодически гранулируют при 20°С в течение 2 часов. Полученное твердое вещество собирают фильтрованием и промывают ИПС, которым промывают реакционный сосуд (2х4 мл). Полученное твердое вещество сушат в вакууме (50 мбар) при 60°С в течение 12 часов с получением твердого вещества (692 мг). Анализ этого твердого вещества с помощью 1H ЯМР и ЖХ-МС показывает, что в основном оно является 3-(3,5-дихлорфенил)-2,4,10-триоксаадамантаном с раскрытым кольцом, указанным выше в качестве основного компонента, и анализ ЖХ-МС/МС показывает, что желаемый продукт тафамидиса присутствует в качестве незначительного компонента. Предположительно, напряжение кольца исходного 3-(3,5-дихлорфенил)-2,4,10-триоксаадамантана приводит к гидролизу этого материала, благоприятному для реакции, приводящей к образованию тафамидиса.
ПРИМЕР 58

К 8 мл ИПС при 20°С при перемешивании добавляют 4-амино-3-гидроксибензойную кислоту (402 мг, 2,62 ммоль) и 1-(3,5-дихлорфенил)-4-изопропил-2,6,7-триоксабицикло[2.2.2]октан (2,88 ммоль), затем ТФК (30 мкл, 0,388 ммоль). Реакционную смесь нагревают до 85°С и перемешивают при этой температуре в течение 12 часов. Смесь охлаждают до 20°С и периодически гранулируют при 20°С в течение 2 часов. Полученное твердое вещество собирают фильтрованием и промывают ИПС, которым промывают реакционный сосуд (2х4 мл). Полученное твердое вещество сушат в вакууме (50 мбар) при 60°С в течение 12 часов с получением тафамидиса. 1H ЯМР, ЖХ-МС и ЖХ-МС/МС сравнивают со стандартным образцом тафамидиса.
ПРИМЕР 59

К 8 мл ИПС при 20°С при перемешивании добавляют 4-амино-3-гидроксибензойную кислоту (402 мг, 2,62 ммоль) и 4-(трет-бутил)-1-(3,5-дихлорфенил)-2,6,7-триоксабицикло[2.2.2]октан (2,88 ммоль), затем ТФК (30 мкл, 0,388 ммоль). Реакционную смесь нагревают до 85°С и перемешивают при этой температуре в течение 12 часов. Смесь охлаждают до 20°С и периодически гранулируют при 20°С в течение 2 часов. Полученное твердое вещество собирают фильтрованием и промывают ИПС, которым промывают реакционный сосуд (2х4 мл). Полученное твердое вещество сушат в вакууме (50 мбар) при 60°С в течение 12 часов с получением тафамидиса. 1H ЯМР, ЖХ-МС и ЖХ-МС/МС сравнивают со стандартным образцом тафамидиса.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1,4-БИС-ЗАМЕЩЕННЫХ 2,6,7-ТРИОКСАБИЦИКЛО [2.2.2] ОКТАНОВ | 1992 |
|
RU2051152C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОБИЦИКЛОАЛКАНОВ | 1991 |
|
RU2034847C1 |
| АЗОЛИНОВЫЕ СОЕДИНЕНИЯ, ЗАМЕЩЕННЫЕ КОНДЕНСИРОВАННОЙ КОЛЬЦЕВОЙ СИСТЕМОЙ | 2015 |
|
RU2742767C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ИНГИБИРОВАНИЯ ПУТИ JAK | 2011 |
|
RU2672100C2 |
| ПИРИМИДОПИРИДАЗИНОВЫЕ ПРОИЗВОДНЫЕ, ПРИГОДНЫЕ В КАЧЕСТВЕ Р38 МАРК | 2010 |
|
RU2516466C2 |
| Способ получения 1,4-бис-замещенных 2,6,7-триоксабицикло (2,2,2)октанов | 1985 |
|
SU1792417A3 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРИРОВАННОГО СОЕДИНЕНИЯ | 2017 |
|
RU2743037C2 |
| Азолины | 2015 |
|
RU2727307C2 |
| ЦИКЛОПРОПИЛПИПЕРИДИНОВЫЕ ИНГИБИТОРЫ ТРАНСПОРТЕРА ГЛИЦИНА | 2005 |
|
RU2387644C2 |
| СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ TRPM8 | 2012 |
|
RU2563030C2 |
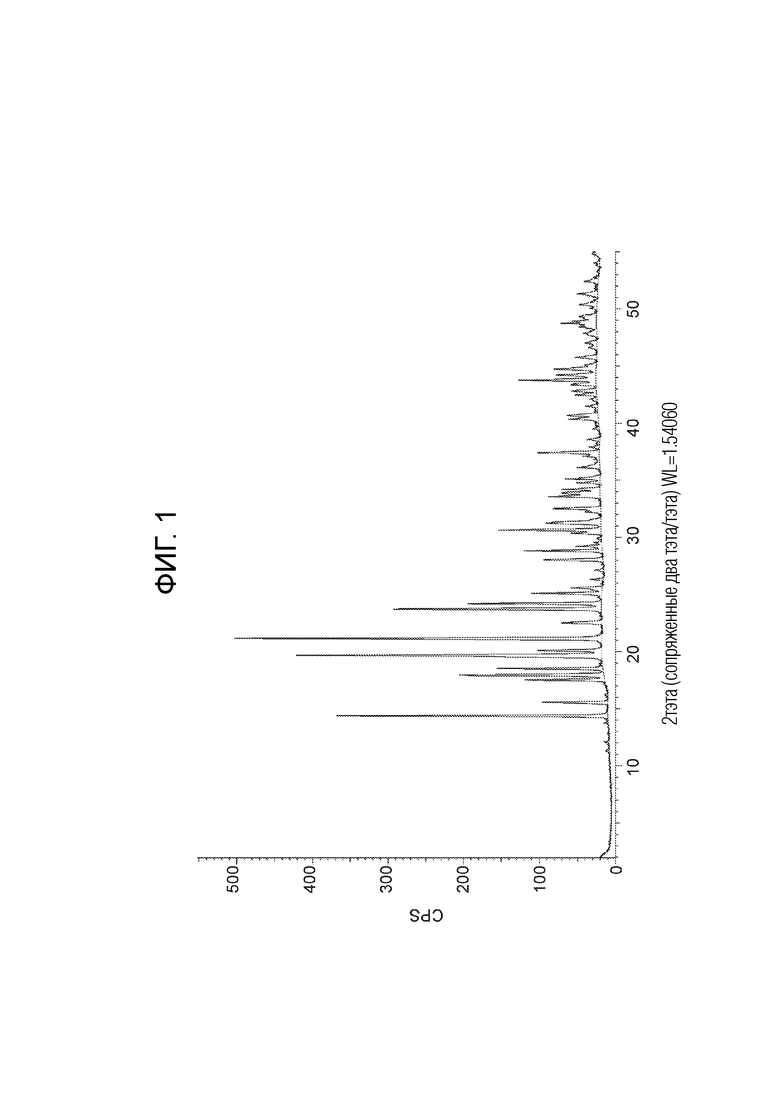
Изобретение относится к способу получения соединения 6-карбокси-2-(3,5-дихлорфенил)бензоксазола формулы I, причем способ включает взаимодействие соединения 4-амино-3-гидроксибензойной кислоты формулы III с соединением ортоэфира 3,5-дихлорфенила формулы II в присутствии кислотного или основного катализатора или в отсутствие катализатора с получением соединения формулы I, где R1 представляет собой водород или карбоксильную защитную группу; и R2a, R2b и R2c каждый независимо представляет собой C1-C6алкил или любые два из R2a, R2b и R2c, взятые вместе, представляют собой C1-C6алкандиил, или R2a, R2b и R2c, взятые вместе, представляют собой C3-C10алкантриил. Изобретение также относится к промежуточным соединениям формулы II и способу их получения. Технический результат – разработан новый эффективный способ получения соединения формулы I, которое находит свое применение в медицине для лечения амилоидных заболеваний. 6 н. и 47 з.п. ф-лы, 1 ил., 6 табл., 59 пр.

1. Способ получения соединения 6-карбокси-2-(3,5-дихлорфенил)бензоксазола формулы I

где способ включает взаимодействие соединения 4-амино-3-гидроксибензойной кислоты формулы III с соединением ортоэфира 3,5-дихлорфенила формулы II в присутствии кислотного или основного катализатора или в отсутствие катализатора с получением соединения формулы I

где R1 представляет собой водород или карбоксильную защитную группу; и R2a, R2b и R2c каждый независимо представляет собой C1-C6алкил или любые два из R2a, R2b и R2c, взятые вместе, представляют собой C1-C6алкандиил, или R2a, R2b и R2c, взятые вместе, представляют собой C3-C10алкантриил.
2. Способ по п.1, отличающийся тем, что реакцию соединения формулы III с соединением формулы II с получением соединения формулы I проводят в растворителе.
3. Способ по п.2, отличающийся тем, что растворитель выбран из группы, состоящей из метанола, этанола, изопропанола, н-бутанола, ацетона, метилэтилкетона, тетрагидрофурана, 1,4-диоксана, трет-бутилметилового эфира, анизола, этилацетата, хлороформа, хлорбензола, гептана, циклогексана, толуола, ацетонитрила и 1,2-диметоксиэтана.
4. Способ по п.3, отличающийся тем, что растворитель выбран из группы, состоящей из метанола, изопропанола, ацетонитрила, этилацетата, 1,2-диметоксиэтана, тетрагидрофурана, трет-бутилметилового эфира и 1,4-диоксана.
5. Способ по п.1, отличающийся тем, что кислотный катализатор выбран из группы, состоящей из трифторуксусной кислоты, уксусной кислоты, хлористоводородной кислоты и метансульфоновой кислоты.
6. Способ по п.1, отличающийся тем, что основным катализатором является триэтиламин.
7. Способ по любому из пп.1-4, отличающийся тем, что реакцию соединения формулы III с соединением формулы II с получением соединения формулы I проводят при температуре от комнатной температуры до 100°C.
8. Способ по п.7, отличающийся тем, что температура составляет от комнатной температуры до 65°C.
9. Способ по любому из пп.1-4, отличающийся тем, что реакцию соединения формулы III с соединением формулы II с получением соединения формулы I проводят в течение периода от 0,25 часа до 40 часов.
10. Способ по любому из пп.1-4, отличающийся тем, что R1 представляет собой водород.
11. Способ по любому из пп.1-4, отличающийся тем, что каждый из R2a, R2b и R2c независимо представляет собой C1-C6алкил.
12. Способ по п.11, отличающийся тем, что каждый из R2a, R2b и R2c представляет собой метил.
13. Способ по любому из пп.1-4, отличающийся тем, что R2a, R2b и R2c вместе представляют собой C3-C10алкантриил.
14. Способ по п.13, отличающийся тем, что соединение формулы II представляет собой
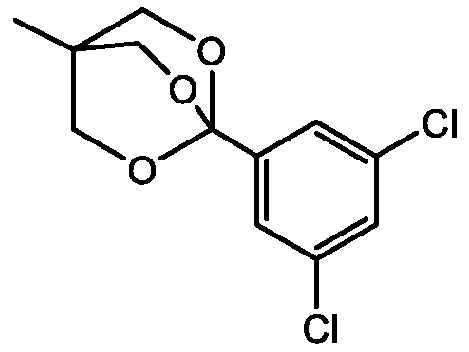 .
.
15. Способ по любому из пп.1-4, дополнительно включающий стадию выделения соединения формулы I.
16. Способ по п.15, отличающийся тем, что соединение формулы I выделяют фильтрованием.
17. Способ получения 6-карбокси-2-(3,5-дихлорфенил)бензоксазола формулы Ia

где способ включает взаимодействие 4-амино-3-гидроксибензойной кислоты формулы IIIa с соединением ортоэфира 3,5-дихлорфенила формулы II в присутствии кислотного или основного катализатора с получением 6-карбокси-2-(3,5-дихлорфенил)бензоксазола формулы Ia

отличающийся тем, что R2a, R2b и R2c каждый независимо представляет собой C1-C6 алкил, или любые два из R2a, R2b и R2c, взятые вместе, представляют собой C1-C6алкандиил, или R2a, R2b и R2c, взятые вместе, представляют собой C3-C10алкантриил.
18. Способ по п.17, отличающийся тем, что реакцию соединения формулы IIIa с соединением формулы II с получением соединения формулы Ia проводят в растворителе.
19. Способ по п.18, отличающийся тем, что растворитель выбирают из группы, состоящей из метанола, этанола, изопропанола, н-бутанола, ацетона, метилэтилкетона, тетрагидрофурана, 1,4-диоксана, трет-бутилметилового эфира, анизола, этилацетата, хлороформа, хлорбензола, гептана, циклогексана, толуола, ацетонитрила и 1,2-диметоксиэтана.
20. Способ по п.19, отличающийся тем, что растворитель выбран из группы, состоящей из метанола, изопропанола, ацетонитрила, этилацетата, 1,2-диметоксиэтана, тетрагидрофурана, трет-бутилметилового эфира и 1,4-диоксана.
21. Способ по п.17, отличающийся тем, что кислотный катализатор выбран из группы, состоящей из трифторуксусной кислоты, уксусной кислоты, хлористоводородной кислоты и метансульфоновой кислоты.
22. Способ по п.21, отличающийся тем, что кислотным катализатором является трифторуксусная кислота.
23. Способ по п.17, отличающийся тем, что основным катализатором является триэтиламин.
24. Способ по любому из пп.17-23, отличающийся тем, что реакцию соединения формулы III с соединением формулы II с получением соединения формулы I проводят при температуре от комнатной температуры до 100°C.
25. Способ по п.24, отличающийся тем, что температура составляет от комнатной до 65°C.
26. Способ по любому из пп.17-20, отличающийся тем, что реакцию соединения формулы III с соединением формулы II с получением соединения формулы I проводят в течение периода от 0,25 часа до 40 часов.
27. Способ по любому из пп.17-20, отличающийся тем, что каждый из R2a, R2b и R2c независимо представляет собой C1-C6алкил.
28. Способ по п.27, отличающийся тем, что каждый из R2a, R2b и R2c представляет собой метил.
29. Способ по любому из пп.17-20, отличающийся тем, что R2a, R2b и R2c вместе представляют собой C3-C10алкантриил.
30. Способ по п.29, отличающийся тем, что соединение формулы II представляет собой
 .
.
31. Способ по любому из пп.17-20, дополнительно включающий стадию выделения соединения формулы Ia.
32. Способ по п.31, отличающийся тем, что соединение формулы Ia выделяют фильтрованием.
33. Способ по любому из пп.17-20, дополнительно включающий стадию взаимодействия 6-карбокси-2-(3,5-дихлорфенил)бензоксазола формулы Ia с фармацевтически приемлемым основанием с получением фармацевтически приемлемой соли 6-карбокси-2-(3,5-дихлорфенил)бензоксазола.
34. Способ по п.33, отличающийся тем, что 6-карбокси-2-(3,5-дихлорфенил)бензоксазол взаимодействует с меглумином в подходящем растворителе с получением соли меглумина 6-карбокси-2-(3,5-дихлорфенил)бензоксазола.
35. Способ по п.34, отличающийся тем, что 6-карбокси-2-(3,5-дихлорфенил)бензоксазол подвергают взаимодействию при комнатной температуре с меглумином в растворителе, выбранном из метилизобутилкетона, метил-трет-бутилового эфира (МТБЭ) и этилацетата (EtOAc), и полученное твердое вещество выделяют и сушат, чтобы получить полиморфную форму E соли меглумина 6-карбокси-2-(3,5-дихлорфенил)бензоксазола.
36. Способ по п.34, отличающийся тем, что 6-карбокси-2-(3,5-дихлорфенил)бензоксазол взаимодействует с меглумином в смеси изопропилового спирта (ИПС) и воды, и полученное твердое вещество выделяют и сушат с получением полиморфной формы M соли меглумина 6-карбокси-2-(3,5-дихлорфенил)бензоксазола.
37. Способ по п.31, дополнительно включающий стадию перемешивания соединения формулы Ia в смеси воды и изопропилового спирта (ИПС) с последующим выделением и сушкой полученного твердого вещества с получением полиморфной формы 1 6-карбокси-2-(3,5-дихлорфенил)бензоксазола.
38. Способ получения 6-карбокси-2-(3,5-дихлорфенил)бензоксазола формулы Ia
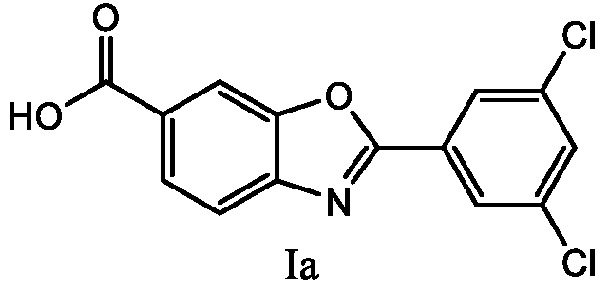
где способ включает взаимодействие одного молярного эквивалента 4-амино-3-гидроксибензойной кислоты формулы IIIa с одним молярным эквивалентом сложного ортоэфира 3,5-дихлорфенила формулы IIa в растворителе с получением 6-карбокси-2-(3,5-дихлорфенил)бензоксазола формулы Ia
 .
.
39. Способ по п.38, отличающийся тем, что растворитель выбран из группы, состоящей из метанола, этанола, изопропанола, н-бутанола, ацетона, метилэтилкетона, тетрагидрофурана, 1,4-диоксана, трет-бутилметилового эфира, анизола, этилацетата, хлороформа, хлорбензола, гептана, циклогексана, толуола, ацетонитрила и 1,2-диметоксиэтана.
40. Способ по п.39, отличающийся тем, что растворитель выбран из группы, состоящей из метанола, изопропанола, ацетонитрила, этилацетата, 1,2-диметоксиэтана, тетрагидрофурана, трет-бутилметилового эфира и 1,4-диоксана.
41. Способ по п.40, отличающийся тем, что способ проводят с использованием кислотного катализатора, выбранного из группы, состоящей из трифторуксусной кислоты, уксусной кислоты, хлористоводородной кислоты и метансульфоновой кислоты.
42. Способ по п.40, отличающийся тем, что кислотным катализатором является трифторуксусная кислота, и растворителем является изопропанол.
43. Способ получения соединения 6-карбокси-2-(3,5-дихлорфенил)бензоксазола формулы I
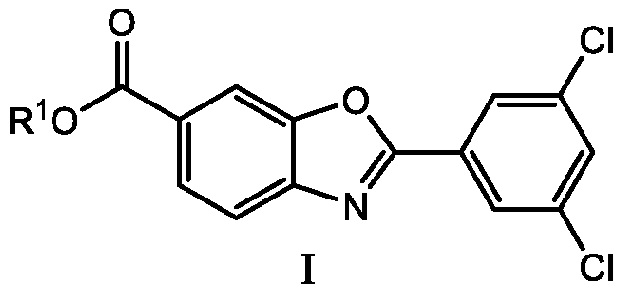
где способ включает взаимодействие соединения 4-амино-3-гидроксибензойной кислоты формулы III с соединением ортоэфира 3,5-дихлорфенила формулы II в ИПС (изопропиловый спирт) в качестве растворителя в присутствии метансульфоновой кислоты (МСК) в качестве кислотного катализатора с получением соединения формулы I

где R1 представляет собой водород или карбокси защитную группу; и R2a, R2b и R2c каждый независимо представляет собой C1-C6алкил или любые два из R2a, R2b and R2c, взятые вместе, представляют собой C1-C8алкандиил, или R2a, R2b и R2c, взятые вместе, представляют собой C3-C12алкантриил, отличающийся тем, что C1-C8алкандиил и C3-C12алкантриил каждый необязательно замещен фенилом, который необязательно замещен одной-двумя группами, независимо выбранными из галогена, C1-С3алкил и С1-С3алкокси.
44. Способ по п.43, отличающийся тем, что соединение формулы III представляет собой 4-амино-3-гидроксибензойную кислоту, и соединение формулы II выбрано из группы, состоящей из
1,3-дихлор-5-(триметоксиметил)бензола;
1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октана;
1-(3,5-дихлорфенил)-4-этил-2,6,7-триоксабицикло[2.2.2]октана;
1-(3,5-дихлорфенил)-4-фенил-2,6,7-триоксабицикло[2.2.2]октана;
2-(3,5-дихлорфенил)-2-метокси-1,3-диоксолана;
1-(3,5-дихлорфенил)-2,7,8-триоксабицикло[3.2.1]октана;
3-(3,5-дихлорфенил)-2,4,10-триоксаадамантана;
1-(3,5-дихлорфенил)-4-изопропил-2,6,7-триоксабицикло[2.2.2]октана; и
4-(трет-бутил)-1-(3,5-дихлорфенил)-2,6,7-триоксабицикло[2.2.2]октана.
45. Соединение, выбранное из группы, состоящей из
1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октана;
1-(3,5-дихлорфенил)-4-этил-2,6,7-триоксабицикло[2.2.2]октана;
1-(3,5-дихлорфенил)-4-фенил-2,6,7-триоксабицикло[2.2.2]октана;
2-(3,5-дихлорфенил)-2-метокси-1,3-диоксолана;
1-(3,5-дихлорфенил)-2,7,8-триоксабицикло[3.2.1]октана;
3-(3,5-дихлорфенил)-2,4,10-триоксаадамантана;
1-(3,5-дихлорфенил)-4-изопропил-2,6,7-триоксабицикло[2.2.2]октана; и
4-(трет-бутил)-1-(3,5-дихлорфенил)-2,6,7-триоксабицикло[2.2.2]октана.
46. Соединение по п.45, которое представляет собой 1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октан.
47. Соединение по п.46, которое представляет собой кристаллическую форму 1-(3,5-дихлорфенил)-4-метил-2,6,7-триоксабицикло[2.2.2]октана, которая характеризуется пиками порошковой рентгеновской дифрактограммы (ПРД) при 21,2, 19,7 и 14,4 градусов 2-тета, каждый ±0,2 градуса 2-тета.
48. Соединение по п.47, которое характеризуется пиками ПРД при 21,2, 19,7, 14,4 и 23,7 градусов 2-тета, каждый ±0,2 градуса 2-тета.
49. Соединение по п.48, которое характеризуется пиками ПРД при 21,2, 19,7, 14,4, 23,7 и 24,2 градусов 2-тета, каждый ±0,2 градуса 2-тета.
50. Соединение по п.49, которое характеризуется пиками ПРД при 21,2, 19,7, 14,4, 23,7, 24,2 и 30,6 градусов 2-тета, каждый ±0,2 градуса 2-тета.
51. Способ получения соединения формулы IIa-2

отличающийся тем, что R3 выбран из группы, состоящей из изопропила, трет-бутила и неопентила, включающий взаимодействие соединения формулы IV с 3,5-дихлорбензойной кислотой

в растворителе в присутствии кислотного катализатора.
52. Способ по п.51, отличающийся тем, что R3 представляет собой изопропил или трет-бутил.
53. Способ по п.52, отличающийся тем, что растворителем является толуол, и кислотным катализатором является метансульфоновая кислота.
| WO 2004056315 A2, 08.07.2004 | |||
| WO 2013168014 A1, 14.11.2013 | |||
| SHI YAJIE ET AL, Tetrahedron Letters, vol.60, no | |||
| Приспособление с иглой для прочистки кухонь типа "Примус" | 1923 |
|
SU40A1 |
| YONG ZHANG ET AL, European Journal of Organic Chemistry, vol | |||
| Станок для придания концам круглых радиаторных трубок шестигранного сечения | 1924 |
|
SU2019A1 |
| Железобетонный фасонный камень для кладки стен | 1920 |
|
SU45A1 |
| ПОДДЕРЖКА ДЛЯ ГРУЗОВОГО И ВЕДУЩЕГО ТЕЛЕЖКУ КАНАТОВ В КАБЕЛЬНЫХ ПОДЪЕМНЫХ КРАНАХ | 1926 |
|
SU7506A1 |
| TIM H | |||
| M | |||
| JONCKERS ET AL, Bioorganic & Medicinal Chemistry Letters, vol | |||
| Машина для добывания торфа и т.п. | 1922 |
|
SU22A1 |
| Прибор для нагревания перетягиваемых бандажей подвижного состава | 1917 |
|
SU15A1 |
| КЛОЗЕТНЫЙ БАК | 1926 |
|
SU4998A1 |
| GULLUZAR | |||
