Описание изобретения
Способ окисления аммиака и система, подходящая для его осуществления
Настоящее изобретение относится к улучшенному способу окисления аммиака до оксидов азота, который используется, в частности, в производстве азотной кислоты и капролактама. Настоящее изобретение также относится к улучшенной системе для получения продуктов окисления аммиака.
Уровень техники и постановка задачи
Каталитическое окисление аммиака (NH3) до оксидов азота NOx часто оказывается основной реакцией в крупнотоннажном производстве азотсодержащих основных материалов для химической промышленности. Здесь следует упомянуть производство азотной кислоты (HNO3) в качестве исходного материала, например, для производства азотсодержащих удобрений или производства гидроксиламина или гидроксиламмониевых солей для получения капролактама и, таким образом, полиамидов.
Следующие утверждения в отношении известного уровня техники, в качестве примера, относятся к производству HNO3 посредством каталитического окисления NH3.
Производство азотной кислоты является одним из самых хорошо разработанных способов химической технологии, который был доведен до промышленной пригодности после применения В. Оствальдом способа Габера-Боша для синтеза аммиака с применением платиновых катализаторов и того технического решения, которое все еще лежит в основе современного производства HNO3 даже в наши дни.
Первая промышленная система с применением платинового катализатора (гофрированных лент платиновой фольги) для производства 1500 тонн в год нитрата аммония была построена еще в 1906 г. в Герте под Бохумом. Вскоре (в 1909 г.) были выданы первые патенты на применение в качестве катализаторов тканых сеток на основе платины. Чуть позже для таковых стали применять сплавы с родием. И хотя применение этого катализатора сопряжено с большими инвестиционными расходами и безвозвратным расходом катализатора в способе окисления NH3 (платина выгорает), эти каталитические системы используют до настоящего времени, а в модифицированном виде (заказные тканые материалы) они все еще составляют уровень техники (смотри, Winnacker  , Chemische Technik - Prozesse und Produkte, 5th Edition, Volume 3, Chapter 3, p. 248-275, Wiley-VCH Verlag GmbH & Co. KGaA).
, Chemische Technik - Prozesse und Produkte, 5th Edition, Volume 3, Chapter 3, p. 248-275, Wiley-VCH Verlag GmbH & Co. KGaA).
В последнее время увеличилось применение сеток на основе металла платиновой группы с высоким содержанием Pd, поскольку они обеспечивают не только определенное снижение стоимости, но и уменьшают содержание веселящего газа (N2O), образование которого является нежелательным при окислении NH3, и который вызывает парниковый эффект.
Обычные размеры диаметра сетки на основе металла платиновой группы, которые натянуты внутри реактора окисления аммиака над широкой областью, часто называемой "горелкой", находятся в диапазоне от 0,5 до 5 м. Толщина сетчатой насадки обычно составляет от нескольких миллиметров до примерно двух сантиметров в зависимости от числа применяемых сеток.
Газовая смесь, обычно содержащая от примерно 9 до 12% по объему NH3 и воздух, проходит сквозь сетки, на которых устанавливается температура от примерно 800 до 950°С вследствие экзотермической природы реакции окисления. Посредством этого NH3 окисляется очень селективно до монооксида азота (NO) (см. ниже схема реакции 1), который затем окисляется в ходе протекания дальнейшего процесса до диоксида азота (NO2) (схема реакции 2) и, наконец, превращается в HNO3 при взаимодействии с водой в абсорбционной колонне (схема реакции 3).
Первичное окисление NH3 - целевая реакция:
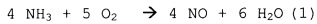 .
.
Окисление NO:
 .
.
Образование HNO3:
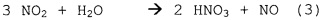 .
.
В результате брутто-реакция имеет вид:
 .
.
Несмотря на то, что содержание O2 в воздухе для горения составляет 21% по объему и формально его вполне хватает, чтобы обеспечить полное превращение 10% по объему NH3 в HNO3, при промышленном производстве HNO3 после каталитического окисления NH3, но при этом перед подачей в абсорбционную колонну, в технологический газ вводят дополнительное количество атмосферного кислорода (вторичный воздух), чтобы увеличить скорость окисления NO и, следовательно, скорость образования HNO3 в абсорбционной колонне. Остаточное содержание кислорода в отходящем газе на выходе из абсорбционной колонны обычно составляет от примерно 1 до 5% по объему.
Согласно современным представлениям о реакции первичного окисления (см. Handbook of Heterogeneous Catalysis, 2nd Edition, Volume 5, 2008, Chapter 12.2.7.1, p. 2582, WILEY-VCH Verlag GmbH & Co. KGaA, 2008) при горении NH3 на поверхности катализатора необходимо высокое парциальное давление кислорода, чтобы подавить образование азота и веселящего газа, не представляющих ценности вторичных продуктов. Данное наблюдение согласуется со стехиометрическими соотношениями образования N2 и N2O (см. схемы реакции 5 и 6 ниже), для которых необходимо меньше кислорода по сравнению с образованием NO (схема реакции 1).
Первичное окисление NH3 - вторичные реакции:
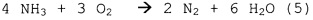 ;
;
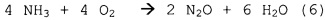 .
.
Образование NO2, для которого согласно схеме реакции (7) потребовалось бы повышенное количество кислорода, не происходит на катализаторе на основе металла платиновой группы.
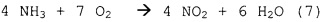
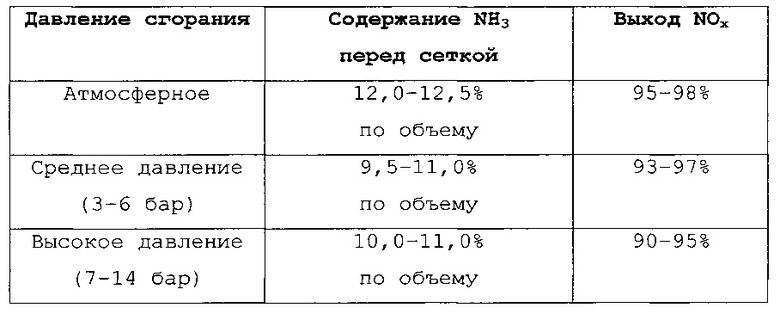
Селективность образования вторичных продуктов или NOx зависит также от общего рабочего давления при окислении NH3. Чем выше давление, тем ниже выход NOx. Значения выхода NOx, которые удается достичь в уровне техники с применением различных вариантов способа (при различных значениях давления сгорания), приведены в следующей таблице (взятой из  , Chemische Technik - Prozesse und Produkte, 5th Edition, Volume 3, Chapter 3, p. 248-275, Wiley-VCH Verlag GmbH & Co. KGaA).
, Chemische Technik - Prozesse und Produkte, 5th Edition, Volume 3, Chapter 3, p. 248-275, Wiley-VCH Verlag GmbH & Co. KGaA).
Однако единственным недостатком катализаторов на основе металла платиновой группы является их низкая стойкость при высокой рабочей температуре примерно 900°С. При широко применяемом давлении сгорания вследствие выгорания благородного металла расход катализатора составляет от примерно 0,04 до 0,4 г Pt/т HNO3, так что катализатор приходится периодически обновлять через определенные промежутки времени каждые от примерно 3 до 15 месяцев в зависимости от давления сгорания. Это приводит к довольно значительным затратам, даже если часть выгоревшей платины извлекают с применением различных систем улавливания (например, сеток на основе Pd).
Чтобы устранить эти недостатки, неоднократно пытались разработать альтернативные каталитические материалы на основе оксидов металлов, в частности, с целью экономии платины. Обзор множества разных попыток применять оксидные катализаторы приведен в Sadykov et al., Appl. Catal. General A: 204 (2000), p. 59-87. Так, в Восточной Европе применяли каталитические системы на основе легированных оксидов железа, нередко в комбинации с сетками на основе металла платиновой группы, при этом в Западном полушарии предпочтительно применяли системы на основе оксида кобальта.
Однако ни одна из этих попыток создать не содержащие металлической платины катализаторы окисления NH3 так до настоящего времени и не получила признания в промышленности, поскольку подобные катализаторы обладают низкой селективностью в отношении образования NO по сравнению с высоко селективными катализаторами на основе металла платиновой группы, а в современных системах производства HNO3 стоимость продукта более чем на 70% складывается из стоимости NH3.
Во многих случаях потенциально активные не содержащие благородный металл катализаторы на основе оксидов переходных металлов также с течением времени подвергаются значительной деактивации в условиях их применения, которая обусловлена не только эффектами спекания под действием высоких термическим напряжений, но нередко и (частичным) восстановлением оксидов посредством NH3 до соответствующих оксидов с более низкой валентностью, которые обычно обладают низкой активностью и селективностью в отношении образования NO. Можно упомянуть, например, восстановление MnO2 и Mn2O3 до Mn3O4, восстановление CuO2 до CuO, восстановление α-Fe2O3 до Fe3O4 и FeO или хорошо известное восстановление высокоактивного Co3O4 до менее активного СоО.
Чтобы предотвратить подобную деактивацию в случае промышленного применения катализаторов на основе Co3O4 для окисления NH3 в ректоре с неподвижным слоем катализатора от Incitec Ltd., Австралия, слой катализатора периодически перекладывали, чтобы остаточным кислородом из задней части слоя катализатора провести обратное окисление катализатора, подвергшегося восстановлению под действием высокой концентрации NH3 в передней части слоя катализатора. Та же самая идея также рассматривается в соответствующих работах авторов Schmidt-Szalowski et al. (см. статью: Appl. Catal. А: General 177 (1998), p. 147-157), которые сообщали об окислении NH3 на катализаторах на основе Co3O4 в псевдоожиженном слое. Они считали, что при вихревом перемешивании частиц катализатора в нижней части псевдоожиженного слоя под действием кислорода постоянно происходит обратное окисление образующегося СоО.
Неоднократно подвергалась исследованию еще одна возможность подавить деактивирующее восстановление оксидов посредством их легирования, иначе говоря, посредством стабилизации вышеупомянутых бинарных оксидов иными оксидами металлов, которые с трудом подаются восстановлению, однако это легирование часто сопровождается понижением удельной активности, как описано Sadykov et al. в статье: Appl. Catal. General A: 204 (2000) p. 59-87. Можно упомянуть в качестве примера легирование α-Fe2O3 с применением Al2O3, которое послужило основой для создания двухступенчатых каталитических систем, разработанных в 1970-ых годах в СССР для окисления NH3 в комбинации с пониженным количеством обычных сетчатых катализаторов на основе Pt/Rh. Оксиды переходных металлов можно также превратить посредством легирования иными оксидами металлов в тройные смешанные оксиды, имеющие разную кристаллическую структуру, в которой высшие степени окисления переходных металлов обладают, в принципе, низкой способностью к восстановлению. Можно упомянуть, в частности, перовскитовые структуры, которые отличаются высокой активностью в отношении образования NO и высокой химической стойкостью.
Например, в US 4812300 А заявляют катализаторы на основе смешанного оксида для окисления аммиака перовскитового типа, имеющие общую формулу АВО3±δ, где А представляет собой щелочные металлы, щелочноземельные металлы, лантаноиды или актиноиды, В представляет собой один или несколько элементов из IB, IVB-VIIB и VIII групп. Считают, что для этих катализаторов характерны равновесные парциальные давления кислорода более 10-15 бар при 1000°С, так что возможен хороший перенос решеточного кислорода к молекуле NH3 без нарушения структурной целостности перовскита. Испытание катализаторов было проведено в данном документе в аппарате или в условиях температурно-программируемого восстановления (TPR) при атмосферном давлении, а также при концентрации NH3 3,3% по объему и содержании кислорода 6,7% по объему в гелии. Особенно предпочтительные перовскитовые катализаторы содержат лантан и/или стронций в качестве элемента положения А и кобальт, никель и/или марганец в качестве элемента положения В.
В WO-99/25650 А1 описано устройство для окисления NH3, в котором предпочтительно используют катализаторы на основе смешанного оксида, образованные из редкоземельных металлов и кобальта. В качестве примера описано окисление 10% NH3 по объему в воздухе при атмосферном давлении с применением смешанного оксида лантана/церия/кобальта (атомное отношение La:Се:Со=8:2:10).
В US 3888792 А для окисления NH3 описано применение оксида Co3O4, легированного редкоземельными металлами, который, считается, обладает более высокой селективностью и более высокой стойкостью по сравнению с чистым Co3O4. Испытание выбранных образцов проводили при объемном отношении NH3/воздух, равном 1/10, и атмосферном давлении. При длительном испытании в течение свыше 900 часов с применением легированного Се Co3O4, при котором промежуточное давление также повышали до 7 бар, при этом выход NOx всегда составлял более 90%.
В WO 2009/028949 А1 описаны катализаторы на основе смешанного оксида для получения NO посредством проведения реакции в газовой смеси, состоящей из NH3 и O2, причем эти катализаторы имеют общую формулу А3-хВх09-y. А и В выбирают из группы, в состав которой входят металлы Mn, Cr, Со, Fe и Al. Эти катализаторы подвергали испытанию при атмосферном давлении с применением газовой смеси с составом, включающим 10% NH3 в воздухе по объему или 10% NH3 по объему, 18% O2 по объему и 72% аргона по объему. Максимальное значение селективности в отношении образования NOx, составляющее 96%, достигали при применении смешанного оксида, имеющего состав Mn1,5Co1,5О4.
В качестве дополнительного примера можно привести US 3962138 А. В нем заявляются катализаторы для окисления NH3, которые состоят из 60-95% Со3O4, 5-15% Al2O3 и 0-25% оксида тория, церия, цинка или кадмия. Формованные катализаторы подвергали испытанию в реакторе с диаметром 10 см под давлением 4-5 бар с применением газовой смеси, содержащей 10% NH3 в воздухе по объему. При применении самых лучших катализаторов, каждый из которых содержал 10% ThO2, после эксплуатации в течение 400 часов был достигнут выход NOx, равный примерно 93-95%. Добавление Al2O3 и ThO2 привело к значительному увеличению выхода NOx и срока службы катализатора.
В DE 102012000419 А1 раскрыто низкотемпературное окисление аммиака при производстве азотной кислоты посредством пропускания содержащего аммиак и кислород потока газа над нагретым до температуры менее 500°С неподвижным слоем частиц катализатора на основе оксида LaSrCo и последующего охлаждения газового потока, содержащего оксид азота. Эта реакция описана на примере реакции, протекающей в потоке газа, который содержит 5% по объему диоксида углерода, 5% по объему воды, 10% по объему кислорода, 200 частей на миллион аммиака и азот в качестве остальной части.
В заявке WO 2006/010904 А1 приведено описание способов окисления, которые проводят на селективных перовскитных катализаторах. Эти катализаторы содержат висмут и/или лантаноиды, за исключением самого лантана. В качестве модельной реакции описано окисление аммиака в воздухе.
В DE 19903616 А1 описан способ получения оксидов азота, характеризующихся низкой степенью окисления, посредством каталитического окисления аммиака в смеси с воздухом и паром на катализаторе окисления. Упоминаются катализаторы, содержащие благородные металлы, или катализаторы, содержащие оксиды металлов.
В заявке WO 01/49603 А1 описан катализатор, содержащий оксид церия и оксид марганца, а также оксид магния, алюминия, цинка или кальция и активатор, для селективного окисления аммиака кислородом до динитрооксида N2O. Реакция протекает при относительно низкой температуре, не превышающей 250°С.
В DE 2148707 А описан катализатор окисления аммиака до оксидов азота. Этот катализатор состоит, главным образом, из оксида кобальта и характеризуется удельной площадью поверхности в диапазоне от 0,1 до 7 м2/г и пористостью при отношении объем/вес в диапазоне от 1 до 15%.
В US-A-5849257 описан способ получения оксидов азота, при котором осуществляют реакцию аммиака с кислородом в присутствии пара на катализаторе на основе оксида меди/марганца. Этот катализатор характеризуется определенным рентгеновским спектром.
В ЕР 0384563 В1 описан способ окисления аммиака в присутствии катализатора на основе оксида кобальта, легированного литием.
В US 2013/0039828 A1 описана структура катализатора, который подходит для окисления аммиака и отличается гибким размещением катализаторных блоков. Катализаторы могут содержать металлы платиновой группы или другие металлы.
В научной публикации [J. Catal. 276 (2010) 306-313] авторы Biausque и Schuurmann описывают механизм высокотемпературного окисления NH3 до NO на катализаторе на основе LaCoO3. Для его изучения были проведены различные исследования, в том числе с изменением содержания O2 и NH3, причем в одной из серий испытаний, начиная с концентрации NH3 3% по объему, меняли содержание кислорода в диапазоне от 10% до 40% по объему, а в другой серии испытаний, начиная с концентрации кислорода 20% по объему, меняли содержание NH3 в диапазоне от 1% до 5% по объему. Что касается достигаемого выхода NOx, то было обнаружено отрицательное влияние на него парциального давления O2 и положительное влияние парциального давления NH3. Иначе говоря, по мере увеличения парциального давления O2 и уменьшения парциального давления NH3 наблюдалось увеличение образования N2 и N2O, что противоречит характеру поведения платиновых катализаторов для окисления NH3.
В статье Catal. Lett. (2011) 141: 1215-8 авторы Tianfeng Hou et al. описывают каталитическое окисление аммиака до монооксида азота в присутствии перовскитовых катализаторов типа на основе LaMnO3 и LaVO4.
Во многих случаях из приведенного выше описания уровня техники исследуют окисление NH3 в воздухе, как в традиционном способе Оствальда, или в примерах практического осуществления устанавливают соответствующее объемное отношение O2/NH3 величиной по меньшей мере 1,9. Кроме того, почти во всех случаях проведенные исследования или опубликованные данные ограничиваются атмосферными условиями, при которых обеспечивают гораздо большую селективность в отношении образования NO, которую следовало бы ожидать при повышенных давлениях.
Тем не менее, высокие значения выхода NOx, ожидавшиеся от применения Pt/Rh сетчатых катализаторов, не достигнуты. Так же обстоит дело, в частности, и с высокими значениями пропускной способности по NH3, иначе говоря, при высокой начальной концентрации величиной 10% по объему и повышенном рабочем давлении, которые выгодны и привычны для промышленного производства благодаря уменьшению размеров аппарата и оптимальной адаптации к последующей абсорбции NO/NO2. Таким образом, выход NOx обычно уменьшается при повышении концентрации или при повышении (парциального) давления аммиака. В частности, так обстоит дело и для известных катализаторов на основе оксида, такого как, например, Co3O4 (смотри, например, Andrew, S.P.S.; Chinchen, G.C., "The loss in selectivity of a cobalt oxide ammonia oxidation catalyst" in "Studies in surface science and catalysis"; 6 (1980), p. 141-148, (Catalyst deactivation : proceedings of an international symposium, Antwerp, October 13-15, 1980)), который по сравнению с катализаторами на основе металла платиновой группы проявляет значительно меньшую активность. Высокое парциальное давление аммиака способствует протеканию нежелательных вторичных и последующих реакций, которые приводят к образованию N2 или N2O.
Несмотря на все усилия, катализаторы на основе оксидов переходных металлов для окисления NH3 не нашли применения в крупнотоннажном производстве, за исключением упомянутой удачной комбинации катализаторов на основе оксидов железа с сетками на основе благородного металла.
Сетчатые катализаторы на основе Pt/Rh все еще используют, за редким исключением. Как было упомянуто выше, различные варианты осуществления способа или системы могут отличаться друг от друга по рабочему давлению при горении NH3 (атмосферное давление/среднее давление/высокое давление) и по превалирующему уровню давления при абсорбции NOx в абсорбционной колонне (см. также , Chemische Technik - Prozesse und Produkte, 5th Edition, Volume 3, Chapter 3, p. 248-275, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005; Thiemann, M., Scheibler, E., Wiegand, K. W. Nitric Acid, Nitrous Acid, and Nitrogen Oxides, Wiley-VCH Verlag GmbH & Co. KGaA, 2000).
В настоящее время особое значение приобрели так называемый способ единого давления, при котором среднее давление или высокое давление используют как при горении NH3, так и при абсорбции NOx, и так называемый способ двойного давления, при котором горение NH3 осуществляют при среднем давлении, а абсорбцию NOx осуществляют при высоком давлении. Ранее применяемые традиционные системы, в которых горение проводили при атмосферном давлении, а абсорбцию при среднем давлении, в настоящее время почти полностью вытеснены способами с единым давлением и двойным давлением, которые более экономичны в случае большой мощности производства.
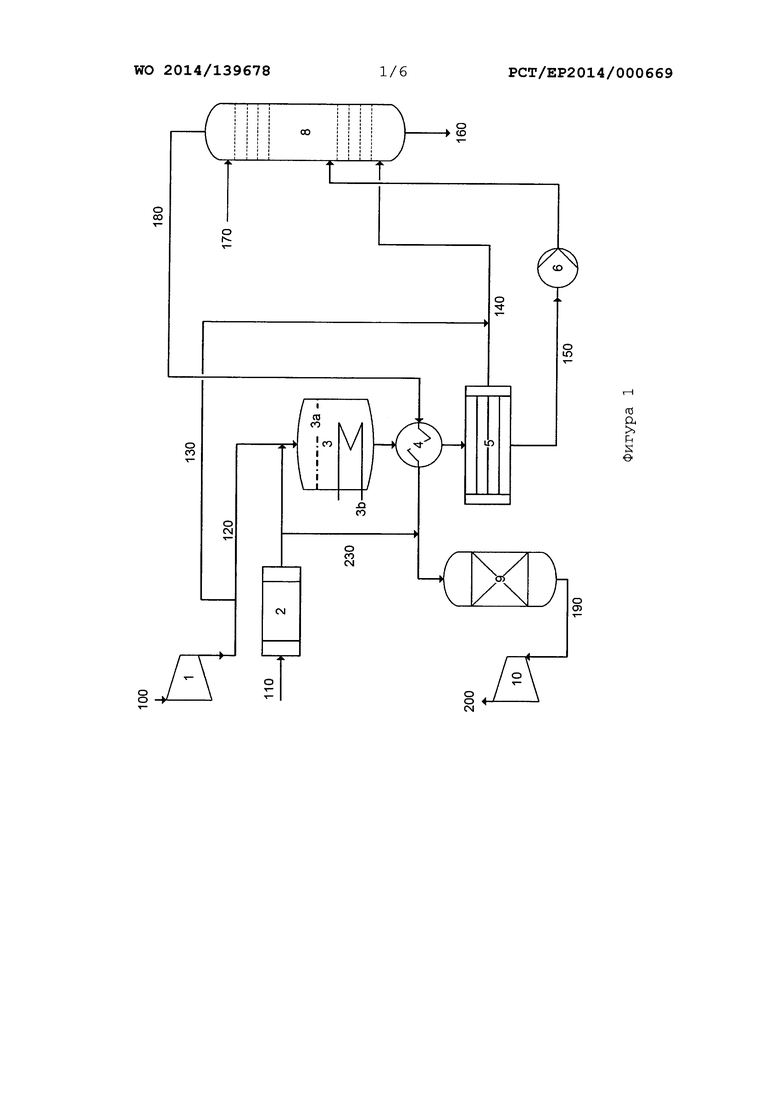
На фиг. 1 показана упрощенная технологическая схема типичной системы единого среднего давления.
Системы для производства HNO3 обычно содержат испаритель NH3, предназначенный для получения газообразного NH3, воздушный компрессор, предназначенный для получения сжатого воздуха, реактор окисления NH3, предназначенный для установки сетчатых катализаторов на основе Pt, со встроенным охладителем технологического газа, различные теплообменники или охладители и конденсаторы, предназначенные для дальнейшего охлаждения технологического газа или для нагревания остаточного газа, выходящего из абсорбционной колонны, абсорбционную колонну, предназначенную для абсорбции NOx и образования HNO3, реактор, предназначенный для (каталитического) удаления остаточного NOx, и необязательно N2O из остаточного газа, и турбину остаточного газа, предназначенную для регенерации энергии из остаточного газа при его расширении и выбросе в атмосферу. В системах двойного давления между реактором окисления NH3 и абсорбционной колонной находится дополнительная ступень сжатия, предназначенная для сжатия технологического газа до давления, при котором производят абсорбцию.
Цель изобретения
Целью настоящего изобретения является обеспечение улучшенного способа окисления NH3 с применением катализаторов на основе оксидов переходных металлов и системы, подходящей для его осуществления, которые отличаются более высокими выходами NOx по сравнению с выходами, обеспечиваемыми на подобных катализаторах до настоящего времени. Кроме того, указанный способ отличается более продолжительным сроком службы катализаторов и более низкой стоимостью катализаторов.
Описание изобретения
Указанная цель достигается посредством обеспечения способа окисления аммиака кислородом, при котором соотношение O2 к NH3 в реакционной газовой смеси, подаваемой на катализатор, регулируют до значения, значительно меньше традиционного отношения 1,9 моль/моль, чтобы обеспечить высокий выход NOx, и в котором используют катализаторы, отличающиеся от коммерчески доступных катализаторов на основе металла платиновой группы, применяемых до настоящего времени.
Неожиданно было обнаружено, что при применении катализаторов на основе металлов, не относящихся к платиновой группе, иначе говоря, катализаторов на основе выбранных оксидов переходных металлов, например, таких, как LaCoO3 или LaMnO3, выход полезного продукта NOx может значительно возрасти, если содержание кислорода или отношение O2/NH3 в реакционной газовой смеси отрегулировать так, чтобы почти весь кислород вступал в реакцию с аммиаком согласно реакциям первичного окисления (схемы реакции 1, 5, 6 и 7) и чтобы в полученной газовой смеси оставалось лишь небольшое количество остаточного кислорода или не оставалось его совсем. Большой избыток кислорода, обычно создававшийся до настоящего времени, оказывает в данном случае отрицательное воздействие.
Было обнаружено, что в результате уменьшения содержания кислорода или молярного отношения кислорода к аммиаку перед подачей реакционной газовой смеси в слой катализатора можно увеличить выход NOx до значений, которые удавалось обеспечить только при применении катализаторов на основе металла платиновой группы (сеток на основе Pt/Rh), даже при повышенном парциальном давлении NH3, то есть, при повышенном полном давлении, или при повышенной концентрации NH3.
Соответственно, настоящее изобретение относится к способу окисления аммиака кислородом в присутствии катализаторов, содержащих по меньшей мере один оксид переходного металла, который не является оксидом металла платиновой группы, при этом отношение молярных количеств кислорода к аммиаку на входе для реакционной газовой смеси в слой катализатора регулируют до значений, меньше или равных 1,75 моль О2/моль NH3.
Обычно молярное отношение кислорода к аммиаку, которое подлежит регуляции согласно настоящему изобретению, находится в диапазоне от 1,25 до 1,75 моль О2/моль NH3. В частности, молярное отношение составляет в диапазоне от 1,30 до 1,75 моль О2/моль NH3, особенно предпочтительно отношение регулируют в диапазоне от 1,35 до 1,60 моль О2/моль NH3, наиболее предпочтительно отношение регулируют в диапазоне от 1,35 до 1,50 моль О2/моль NH3.
При реализации предпочтительного варианта осуществления способа согласно настоящему изобретению отношение молярных количеств кислорода к аммиаку в реакционной газовой смеси на входе в слой катализатора выбирают так, чтобы оно превышало в диапазоне от 0,1 моль O2/моль NH3 до 0,4 моль О2/моль NH3 оптимальное молярное отношение, причем оптимальное молярное отношение представляет собой отношение молярных количеств кислорода к аммиаку реакционной газовой смеси на входе в слой катализатора, при котором обеспечивается максимальный выход NOx.
Особенно предпочтительно отношение молярных количеств кислорода к аммиаку на входе в слой катализатора превышает на от 0,05 моль O2/моль NH3 до 0,3 моль О2/моль NH3 оптимальное отношение, особенно предпочтительно превышает на от 0,025 моль O2/моль NH3 до 0,25 моль O2/моль NH3 оптимальное отношение.
Оптимальное молярное отношение O2/NH3 или оптимальное содержание кислорода можно определить посредством проведения серии соответствующих испытаний в конкретно заданных условиях способа, то есть с применением выбранного катализатора в определенной системе при определенной объемной скорости и скорости потока, при определенной температуре на входе и на выходе, при определенном давлении и с применением определенной реакционной среды, содержащей кислород, и определенное количество аммиака. При этом для постоянной концентрации NH3 концентрацию кислорода на входе в слой катализатора выбирают так, чтобы соответствующее молярное отношение O2/NH3 находилось в диапазоне между минимальным значением отношения O2/NH3, например, 1,25 моль/моль, и максимальным значением отношения O2/NH3, например, 1,75 моль/моль, это отношение предпочтительно меняют с выбранным шагом, например, с шагом, меньше или равным 0,1 моль O2/моль NH3, и в каждом случае определяют достигаемый выход NOx. Молярное отношение кислорода к аммиаку, при котором обеспечивается максимальный выход NOx при иных постоянных условиях проведения реакции, представляет собой оптимальное отношение кислорода к аммиаку.
В другом предпочтительном варианте осуществления способа согласно настоящему изобретению отношение молярных количеств кислорода к аммиаку реакционной газовой смеси на входе в слой катализатора регулируют до значений, меньших или равных 1,75 моль O2/моль NH3, предпочтительно до значений, меньших или равных 1,60 моль O2/моль NH3 и особенно предпочтительно до значений, меньших или равных 1,50 моль O2/моль NH3, причем содержание кислорода в полученном газе на выходе из слоя катализатора составляет по меньшей мере 0,3% по объему, предпочтительно по меньшей мере 0,4% по объему и особенно предпочтительно по меньшей мере 0,5% по объему.
В предпочтительном варианте осуществления этого предпочтительного варианта способа согласно настоящему изобретению молярное отношение O2/NH3, которое пригодно согласно настоящему изобретению, меньше или равно 1,75 моль O2/моль NH3 или содержание кислорода в реакционной газовой смеси на входе в слой катализатора, которое пригодно согласно настоящему изобретению, обусловлено содержанием кислорода в полученном газе на выходе из слоя катализатора в том смысле, что молярное отношение O2/NH3, которое пригодно согласно настоящему изобретению, или - при заданной концентрации NH3 на входе - содержание кислорода на входе в слой катализатора, которое пригодно согласно настоящему изобретению, выбирают так, чтобы содержание кислорода в полученной газовой смеси на выходе из слоя катализатора находилось в диапазоне от 0,3% по объему до 10,0% по объему, предпочтительно в диапазоне от 0,4% по объему до 6,0% по объему и особенно предпочтительно в диапазоне от 0,5 по объему до 4,0% по объему, наиболее предпочтительно в диапазоне от 0,3% по объему до 2,0% по объему, в частности, от 0,4% по объему до 2,0% по объему, наиболее предпочтительно в диапазоне от 0,5% по объему до 1,5% по объему.
Отношение O2/NH3, которое пригодно согласно настоящему изобретению, можно практически отрегулировать для конкретно заданных условий способа, то есть при применении выбранного катализатора в определенной системе при определенной объемной скорости и скорости потока, при определенной температуре на входе и на выходе, при определенном давлении и с применением определенной реакционной среды, содержащей кислород и аммиак, таким образом, например, что, начиная от заранее установленного отношения O2/NH3, например, начиная от ранее принятого отношения O2/NH3 1,9 моль/моль или, в частности, начиная с отношения O2/NH3 1,75 моль/моль, при фиксированном количестве аммиака, уменьшают содержание кислорода на входе в слой катализатора до тех пор, пока в полученном газе на выходе из слоя катализатора не будут присутствовать или не будут обнаружены упомянутые выше низкие значения содержания кислорода.
Содержание NOx и O2 на выходе из слоя катализатора можно определять способами, известными специалистам в данной области. Например, содержание NOx можно определять с помощью ИК/УФ-анализаторов с применением измерительных ячеек для нагретого газа. Подходящими анализаторами являются, например, многокомпонентные FT-IR анализаторы или однокомпонентные системы со множеством ИК- или УФ-каналов. В качестве альтернативы, можно измерять содержание NOx с помощью хемилюминесцентного анализатора, оснащенного на входе конвертором для восстановления NO2 до NO. Содержание кислорода предпочтительно в том числе можно измерять нагретым анализатором для измерения парамагнетизма или с помощью циркониевого датчика.
Содержание кислорода в полученном газе на выходе из слоя катализатора можно также определить арифметически как разность между содержанием кислорода в реакционной газовой смеси на входе в слой катализатора и расходом кислорода в слое катализатора. Расход кислорода в слое катализатора находят исходя из концентраций компонентов N2, NO, NO2 и N2O на выходе или по значениям селективностям образования этих продуктов из аммиака и концентрации NH3 на входе с применением соответствующих стехиометрических отношений O2/NH3 согласно уравнениям (1), (5), (6) и (7).
Способ согласно настоящему изобретению предпочтительно осуществляют при значениях абсолютного давления от 1 до 10 бар, особенно предпочтительно при значениях абсолютного давления от 1,5 до 6 бар, наиболее предпочтительно при значениях абсолютного давления от 2 до 5 бар.
Концентрация NH3 на входе в реактор окисления по способу согласно настоящему изобретению предпочтительно составляет от 1 до 17% по объему, особенно предпочтительно от 4 до 15% по объему, в частности, составляет от 7 до 14% по объему. Верхний предел содержания NH3 предпочтительно определяется нижним пределом детонации смесей NH3/кислород, который зависит также от других возможных компонентов газа, таких, например, как пар.
При химическом превращении на катализаторах каждого объемного процента аммиака в газовой смеси по объему высвобождается определенное количество тепла, которое в случае реакционной смеси NH3 с воздухом в адиабатических условиях соответствует повышению температуры между реакционной смесью и получаемой смесью примерно на 68 K. В том случае, когда температура подаваемой на вход смеси, содержащей аммиак и кислород, задается системой, следовательно, температура в потоке технологического газа на выходе из катализатора окисления зависит от концентрации аммиака в газовой смеси на входе в катализатор окисления.
Катализатор, применяемый согласно настоящему изобретению, проявляет свою наивысшую способность предпочтительно в более низком температурном диапазоне, чем катализаторы на основе металла платиновой группы. Температура на выходе из слоя катализатора, например, из упаковки формованных изделий катализатора, подлежит регуляции согласно настоящему изобретению предпочтительно от 700°С до 950°С, особенно предпочтительно от 750°С до 850°С (ее измеряют на выходе газовой смеси из слоя катализатора, в случае нескольких слоев катализатора на выходе из последнего слоя катализатора). Это может достигаться посредством регулирования температуры реакционной газовой смеси на входе и/или концентрации аммиака в реакционной газовой смеси.
Более свободное пространственное расположение в упаковке формованных изделий катализатора, применяемой согласно настоящему изобретению по сравнению с каталитическими сетками на основе металла платиновой группы обеспечивает политропный режим работы в результате пространственного рассеяния или отвода теплоты реакции. Это может достигаться, например, посредством охлаждения стенок реактора или посредством размещения интегрированных охлаждающих устройств в упаковке катализатора.
Как уже упоминалось, высокая активность формованных изделий катализатора позволяет понизить температуру реакции или температуру «проскока» и тем самым понизить температуру реакционной газовой смеси, содержащей NH3 и кислород, на входе в (первый) слой катализатора, например, в упаковку формованных изделий катализатора. Данная температура на входе может составлять от 20°С до 300°С, предпочтительно от 50°С до 200°С, особенно предпочтительно от 50°С до 150°С.
Катализаторы
По способу согласно настоящему изобретению в принципе можно применять любые катализаторы, которые содержат в качестве активного компонента по меньшей мере один оксид переходного металла, который не является оксидом металла платиновой группы.
В контексте данного описания металл платиновой группы представляет собой элемент из 8-10 групп 5-го и 6-го периодов Периодической системы, то есть элементы из группы Ru, Rh, Pd, Os, Ir и Pt.
В тех рабочих условиях, в которых осуществляют способ согласно настоящему изобретению, то есть при отношении NH3/O2, отрегулированном согласно настоящему изобретению при значении, которое превышает на от 0,1 моль O2/моль NH3 до 0,4 моль O2/моль NH3 оптимальное молярное отношение, катализаторы, применяемые согласно настоящему изобретению, неожиданно обеспечивают более высокий выход NOx по сравнению с выходом NOx, достигаемым при применении традиционных способов окисления аммиака, при которых отношение молярных количеств кислорода и аммиака реакционной газовой смеси на входе в катализатор составляет по меньшей мере 1,9 моль O2/моль NH3.
Соответственно, в тех рабочих условиях, в которых осуществляют способ согласно настоящему изобретению, катализаторы, применяемые согласно настоящему изобретению, обеспечивают выход NOx, который сопоставим или даже превышает выход NOx для способа Оствальда при применении коммерчески доступных катализаторов на основе металла платиновой группы.
Особенно пригодными являются катализаторы, которые содержат оксиды переходных металлов, которые не являются оксидами металлов платиновой группы и не подвергаются необратимому восстановлению до менее активных низковалентных оксидов при указанных выше условиях.
В частности, можно применять катализаторы, которые содержат стабилизированные, то есть легированные, оксиды переходных металлов, которые не являются оксидами металлов платиновой группы, или которые содержат смешанные оксиды таких переходных металлов. Примерами легированных оксидов переходных металлов являются оксиды железа, легированные, например, оксидом висмута, оксидом хрома или оксидом марганца.
Применяемые смешанные оксиды предпочтительно имеют структуру шпинели, делафоссита или особенно предпочтительно перовскита или браунмиллерита.
Перовскиты, применяемые согласно настоящему изобретению, предпочтительно имеют структуру АВО3-δ, где А представляет собой одновалентные, двухвалентные или трехвалентные катионы, В представляет собой трехвалентные, четырехвалентные или пятивалентные катионы, ионный радиус катионов А больше ионного радиуса катионов В, а δ представляет собой число от 0,001 до 1,5, предпочтительно от 0,01 до 0,09 и особенно предпочтительно от 0,01 до 0,5. В перовскитах, применяемых согласно настоящему изобретению, могут также присутствовать смеси разных катионов А и/или катионов В.
Браунмиллериты, применяемые согласно настоящему изобретению, обычно имеют структуру А2В2O5-δ, где А, В и δ имеют такие же значения, как было указано выше. В браунмиллеритах, применяемых согласно настоящему изобретению, могут также присутствовать смеси разных катионов А и/или катионов В.
Катионы В могут присутствовать в соединении предпочтительно в нескольких степенях окисления. Некоторые или все катионы типа В могут представлять собой также трехвалентные катионы или катионы с более высокой валентностью с постоянной степенью окисления.
Особенно предпочтительным является применение перовскитов, имеющих общую эмпирическую формулу АВО3±δ, и/или браунмиллеритов, имеющих общую эмпирическую формулу A2B2O5±δ, в которых положение А занято одним или несколькими элементами, выбранными из группы редкоземельных металлов и щелочноземельных металлов, более чем на 50%, предпочтительно более чем на 80%, особенно предпочтительно более чем на 95%, и в которых положение В занято одним или несколькими элементами, выбранными из группы Cr, Mn, Fe, Со, Ni, более чем на 50%, предпочтительно более чем на 80%, особенно предпочтительно более чем на 95%. Из этих элементов особенно предпочтительным является Со. Особенно пригодным соединением на основе перовскита является LaCoO3±δ, где δ составляет от 0,01 до 0,5.
Дополнительными подходящими легирующими средствами предпочтительно являются переходные металлы, оксиды которых предпочтительно находятся в четырехвалентном состоянии, такие, например, как Се или Mn.
Конечно, к катализаторам, применяемым согласно настоящему изобретению, можно добавлять металлы платиновой группы или оксиды металлов платиновой группы в небольших количествах, например, до 10% по весу, предпочтительно до 5% по весу, исходя из активного компонента(компонентов) катализатора. Могут присутствовать и другие металлы (оксиды), которые обычно используют в качестве легирующих средств. Примерами таких легирующих средств являются щелочные и/или щелочноземельные металлы. Эти легирующие средства, если присутствуют, также присутствуют в небольших количествах, например, в количестве до 10% по весу, в частности, в количестве до 5% по весу, исходя из активного компонента(компонентов) катализатора.
Получение каталитически активных компонентов или оксидов переходных металлов, применяемых по способу согласно настоящему изобретению, и их формование будут обсуждаться только в качестве примера, поскольку специалисту в данной области известно множество способов получения из практики. Ниже в данном документе будут рассмотрены технологии, соответствующие известному уровню техники, которые можно применять для получения каталитически активных компонентов.
Каталитически активные компоненты, применяемые по способу согласно указанному способу, можно получать посредством твердофазной реакции. Для этой цели в качестве исходных материалов обычно используют смеси оксидов, двойных оксидов или предшественников оксидов, например, карбонатов. Исходные материалы тщательно перемешивают в композицию, приспособленную для целевой фазы, а затем прокаливают. При прокаливании образуются каталитически активные фазы в виде кристаллитов.
Чтобы повысить однородность исходной смеси, часто проводят интенсивный помол исходных материалов, необязательно с добавлением вспомогательных веществ, таких, например, как вода. Чтобы увеличить степень превращения в требуемую кристаллическую фазу, можно провести несколько стадий помола и прокаливания. Когда будет определена достаточная степень кристалличности целевой фазы, обеспечивают размер частиц, пригодный для стадий дополнительной обработки формованием для получения формованных изделий катализатора, таких как экструзия или прессование, например, посредством сухого помола.
Дополнительным подходящим способом получения является техника осаждения, в которой исходные материалы получают из растворов посредством осаждения. В качестве предшественников для оксидов металлов можно применять, например, гидроксиды металлов или комплексные катионы металлов, например, в форме цитратов или оксалатов, которые можно осаждать из растворов солей металлов, содержащих лишь один первичный компонент или первичный и вторичный компоненты, с помощью осаждающих реагентов. Подходящими осаждающими реагентами являются, например, щелочи, такие как аммиак, или карбонат аммония.
Для получения предшественников оксидов металлов можно также применять гидролиз алкоксидов. Особенной техникой, которая может применяться, является так называемый золь-гель синтез, когда вместо растворов в качестве исходного материала используют стабильную коллоидную систему. В случае алкоксидов можно применять в качестве гидролизирующих средств, например, воду или различные спирты. Стехиометрию получаемых металлооксидных фаз задают посредством подходящего выбора относительных соотношений исходных соединений, например, солей металлов или алкоксидов.
Полученные подобным образом предшественники оксидов металлов обрабатывают посредством фильтрации, промывки и высушивания. На последующей стадии прокаливания образуются металлооксидные фазы, которые можно дополнительно гомогенизировать посредством чередования стадий размола и прокаливания. Затем проводят дополнительную обработку порошков, в частности, размолом и фракционированием, чтобы получить в результате порошки, пригодные для формования, например, посредством экструзии или прессования.
Каталитически активные компоненты можно также получать посредством реакций пиролиза. Для этой цели металлсодержащие исходные материалы, например, соли металлов, металлорганические соединения или продукты, полученные посредством осаждения, подвергают высоко экзотермической реакции при высокой температуре, например, до 1000°С. При получении по данному типу к исходным материалам можно добавлять окислители, например, нитрат аммония, и горючие органические вещества, такие как мочевина, лимонная кислота или глицин. Реакцию пиролиза можно инициировать исходя из растворов, суспензий или твердых веществ. В данном способе стехиометрическое соотношение целевой фазы можно регулировать посредством обеспечения соответствующих исходных материалов. Полученные порошки характеризуются высокой фазовой чистотой и большой или даже очень высокой удельной поверхностью.
Для формования полученных порошков оксида переходного металла эти и другие активные компоненты или сопутствующие компоненты можно внедрять или заключать в какую-либо подходящую, предпочтительно керамическую, матрицу или наносить на какую-либо подходящую, предпочтительно керамическую, подложку.
Предпочтение отдают керамическим материалам на основе оксидов, карбидов или нитридов элементов, выбранных из группы Si, Al, Mg, Zr и В, особое предпочтение отдают таким керамическим материалам, как кордиерит, муллит, оксид магния, или наиболее предпочтительно карбиду кремния, который отличается высокой химической стойкостью, механической прочностью и превосходной теплопроводностью.
Применение твердых катализаторов, то есть формованных изделий из катализаторов, которые в основном состоят из каталитически активных материалов, также является особенно подходящим и предпочтительным в рамках настоящего изобретения. Формованные изделия из катализаторов, таким образом, состоят из более 70%, предпочтительно более 80%, особенно предпочтительно более 85% каталитически активного материала, исходя из общего веса формованного изделия.
Формованные изделия из катализаторов могут иметь любую геометрическую форму и размер, предпочтительно геометрические формы, которые характеризуются большим значением отношения площади поверхности к объему и в которых возникает минимальная потеря давления при протекании через них газовой среды. Предпочтение отдают формованным изделиям, в которых отношение площади поверхности к объему имеет значение от 0,5 до 10 мм-1, в частности, от 1 до 5 мм-1. Они могут иметь обычную геометрическую форму, применяемую в катализе, например, форму цилиндров, полых цилиндров, цилиндров с несколькими отверстиями, колец, дробленых гранул, структуру трилистника или сот. Особое предпочтение отдают монолитам в форме сот или так называемым минилитам, то есть очень мелким изделиям в форме сот, которые обычно используют в качестве сыпучего материала. Формованные изделия можно изготовлять способами формования, применяемыми для керамики, такими, например, как сухое прессование, гранулирование или экструзия.
Упаковку формованных изделий катализатора можно производить, например, посредством засыпки или упорядоченной укладки.
Реактор
Реактор окисления аммиака, применяемый согласно настоящему изобретению, может быть выполнен по типу традиционного реактора окисления аммиака или «горелки». Это особенно выгодно в случае переоснащения существующих систем, поскольку почти не приходится вносить изменения в аппаратуру. Сетки на основе Pt/Rh часто расположены на рыхлой засыпке из керамических колец. В способе согласно настоящему изобретению формованное изделие катализатора затем можно поместить в реактор вместо Pt/Rh сеток и керамических колец, как уже упоминалось выше, в виде засыпки или упорядоченной упаковки, например, из формованных изделий в форме сот. Особые меры предосторожности следует принимать на кромке реактора, чтобы предотвратить проскок содержащей аммиак/кислород реакционной газовой смеси. Такие меры предосторожности могут представлять собой, например, укладку газонепроницаемых, стойких к высокой температуре полос, соединенных со стенками реактора, на которые частично опирается каталитическая засыпка или наружные элементы упорядоченной каталитической насадки.
В случае новых систем можно отдать предпочтение альтернативным конструкциям традиционных реакторов окисления аммиака, в которых каталитическая насадка обладает большим диаметром и очень маленькой высотой в направлении течения. Посредством уменьшения поперечного сечения потока можно избежать возможных затруднений, связанных с равномерностью распределения поступающей газовой смеси. Особенно предпочтительным является быстрое протекание через слой катализатора с коротким временем пребывания в нем, поскольку посредством этого удается подавить нежелательные вторичные реакции, такие как каталитическое разложение образовавшегося NO, и, кроме того, можно создать обеспечивающую экономию пространства компактную конструкцию реактора окисления аммиака. Дополнительные возможные конфигурации реактора окисления аммиака, содержащего катализатор согласно настоящему изобретению, раскрыты в WO 2008/148487 А1.
Способ согласно настоящему изобретению предпочтительно осуществляют при объемной скорости в диапазоне от 50000 ч-1 до 500000 ч-1, особенно предпочтительно в диапазоне от 100000 ч-1 до 300000 ч-1. В контексте данного описания выражение «объемная скорость» обозначает частное от деления значений объемной доли газовой смеси (измеренной при 273,15 K и давлении 1,01325 бар) в час на объемную долю катализатора, то есть на объем засыпки или насадки. Соответственно, объемную скорость можно регулировать посредством регуляции объемного расхода газа и/или объема катализатора или его количества.
Независимо от того, какую конструкцию предпочтительно выбрать для конкретного применения, реактор окисления аммиака по способу согласно настоящему изобретению предпочтительно оснащают устройством для запуска реакции на катализаторе. Например, для этой цели можно применять водородное пламя с подвижного копья, выдвигаемого в сторону поступления газа в формованное изделие катализатора.
Процедура
Молярное отношение O2/NH3 согласно настоящему изобретению в газовом потоке на входе в катализатор окисления можно технически обеспечивать различными путями.
Проще всего вводить газообразный NH3 в поток воздуха в таком количестве, чтобы получить требуемое молярное отношение O2/NH3. В этом случае содержание NH3 от 14,4% по объему до 10,7% по объему соответствует отношению от 1,25 до 1,75 моль O2/моль NH3, содержание NH3 от 13,9% по объему до 10,7% по объему соответствует отношению от 1,3 до 1,75 моль O2/моль NH3, и содержание NH3 от 13,5% по объему до 11,6% по объему соответствует отношению от 1,35 до 1,6 моль O2/моль NH3.
Другая возможная стадия регулирования молярного отношения O2/NH3 согласно настоящему изобретению заключается, например, в подаче на сжигание NH3 вместе с воздухом или вместо воздуха газового потока, при этом газовый поток содержит менее 20% по объему, предпочтительно менее 10% по объему, особенно предпочтительно менее 5% по объему кислорода.
Когда способ окисления NH3 согласно настоящему изобретению интегрирован в способ производства азотной кислоты или капролактама, на сжигание NH3 вместе с воздухом или вместо воздуха предпочтительно можно подавать некоторую долю остаточного газа с низким содержанием кислорода, например, удаленного после реактора для очистки остаточного газа посредством восстановления N2O и NOx. Это показано в качестве примера на фиг. 2 для системы двойного давления, предназначенной для производства HNO3. Здесь поток очищенного остаточного газа (210) подвергают расширению до уровня давления, соответствующего горению NH3, посредством турбины (11) перед подачей на сжигание NH3.
Подвергаемый рециркуляции очищенный остаточный газ должен иметь содержание кислорода <5% по объему, в частности, <3% по объему, наиболее предпочтительно <2% по объему. Остаточное содержание NOx должно составлять <20 частей на миллион по объему, предпочтительно <10 частей на миллион по объему, особенно предпочтительно <5 частей на миллион по объему.
Можно также разделить поток воздуха, подаваемый на сжигание NH3, на обедненный O2 частичный поток и на обогащенный O2 частичный поток, например, посредством проведения адсорбции с перемежающимся давлением, криогенного разложения или с помощью мембран, например, с помощью керамических мембран, пропускающих анионы кислорода. Подобная конфигурация показана в качестве примера на фиг. 3. Затем подлежащий сжиганию NH3 с концентрацией, например, 10% по объему, вводят в обедненный O2 частичный поток с содержанием O2, например, 13% по объему O2, при этом обогащенный O2 частичный поток вводят в технологический газ после первичного окисления NH3.
Отношение O2/NH3 согласно настоящему изобретению дополнительно можно регулировать перед подачей NH3 в катализатор окисления NH3 и перед обеспечением контакта с ним посредством разбавления содержащего кислород газового потока паром. Затем пар можно снова конденсировать после сжигания NH3 посредством охлаждения потока технологического газа перед его поступлением в абсорбционную колонну с образованием разбавленной кислоты.
Можно также вводить и другие инертные газовые компоненты для разбавления содержащего кислород газового потока.
Упомянутые выше возможности для регуляции молярного отношения O2/NH3 согласно настоящему изобретению не ограничиваются окончательным перечнем, и они могут дополнительно применяться в любых необходимых комбинациях.
Системы
Настоящее изобретение относится также к системе для окисления аммиака, содержащей:
A) реактор (3) для окисления аммиака, оснащенный по меньшей мере одной линией подачи реакционной газовой смеси и по меньшей мере одной линией выпуска для технологического газа,
B) катализатор (3а) внутри реактора (3), содержащий по меньшей мере один оксид переходного металла, который не является металлом платиновой группы, и
C) устройство для регуляции молярного отношения кислорода к аммиаку в реакционной газовой смеси, меньшего или равного 1,75 моль/моль, посредством смешивания содержащего кислород газового потока с содержанием O2<20% по объему с выбранным количеством аммиака, причем содержащий кислород газовый поток образуется:
c1) с помощью устройства для разбавления воздушного потока газовым потоком, который содержит менее 20% по объему, предпочтительно менее 10% по объему, особенно предпочтительно менее 5% по объему кислорода, или
с2) с помощью устройства для уменьшения содержания кислорода в содержащей кислород газовой смеси, предпочтительно в воздухе, или
с3) с помощью комбинации средств c1 и с2.
Система согласно настоящему изобретению может работать при повышенном давлении. В этом варианте осуществления система содержит по меньшей мере один компрессор (1) посредством которого содержащий кислород газовый поток, например, воздух, подвергается сжатию и подается в реактор (3) для окисления аммиака. Аммиак можно подавать в реактор (3) посредством введения аммиака в поток сжатого содержащего кислород газа.
Расширение технологических газов из реактора (3) или из частей системы после реактора (3) до атмосферного давления производят после их выхода из реактора (3) или частей системы после реактора (3) с применением подходящих средств, известных специалистам в данной области. Если систему согласно настоящему изобретению используют, например, для производства азотной кислоты, то содержащий кислород вторичный газ, например, вторичный воздух, добавляют к оксиду азота, полученному в реакторе (3), окисляют его до NO2 и подают в абсорбционную колонну (8), где происходит превращение NO2 в азотную кислоту при взаимодействии с водой. Содержащий оксид азота остаточный газ из абсорбционной колонны (8) подают на очистку (9) остаточного газа, а затем очищенный остаточный газ подают в турбину (10) остаточного газа, где он расширяется с обеспечением регенерации энергии и выбрасывается в атмосферу.
Система согласно настоящему изобретению предпочтительно содержит по меньшей мере еще один компрессор, предназначенный для сжатия потока содержащего кислород вторичного газа перед вводом в абсорбционную колонну (8), в которой полученный оксид азота взаимодействует с водой.
В одном конкретном варианте осуществления разбавление воздушного потока согласно c1) проводят с применением пара и/или азота с содержанием O2<5% по объему.
В дополнительном конкретном варианте осуществления уменьшение содержания кислорода согласно с2) в содержащей кислород газовой смеси, предпочтительно в воздухе, проводят посредством адсорбции с перемежающимся давлением, криогенного разложения или с помощью мембран.
Настоящее изобретение относится также к системе для окисления аммиака и последующей абсорбции NOx, содержащей:
A) реактор (3) для окисления аммиака, оснащенный по меньшей мере одной линией подачи реакционной газовой смеси и по меньшей мере одной линией выпуска технологического газа,
B) катализатор (3а) внутри реактора (3), содержащий по меньшей мере один оксид переходного металла, который не является оксидом металла платиновой группы,
C) устройство для регуляции молярного отношения кислорода к аммиаку в реакционной газовой смеси, меньше или равного 1,75 моль/моль, посредством смешивания содержащего кислород газового потока с содержанием O2<20% по объему с выбранным количеством аммиака, где содержащий кислород газовый поток образуется
c1) с помощью устройства для разбавления воздушного потока газовым потоком, который содержит менее 20% по объему, предпочтительно менее 10% по объему, особенно предпочтительно менее 5% по объему кислорода, или
с2) с помощью устройства для уменьшения содержания кислорода в содержащей кислород газовой смеси, предпочтительно в воздухе, или
с3) с помощью комбинации средств c1 и с2,
D) абсорбционную колонну (8) для абсорбции NOx и образования HNO3, HNO2 или растворов нитратов или нитритов и
E) устройство, расположенное между реактором (3) для окисления аммиака и абсорбционной колонной (8), для объединения содержащего NOx потока технологического газа с содержащим кислород газовым потоком, который содержит более 25%, предпочтительно более 30%, особенно предпочтительно более 40% кислорода.
В одном конкретном варианте осуществления разбавление воздушного потока согласно c1) проводят паром или потоком азота, содержащим менее 20% по объему, предпочтительно менее 10% по объему, особенно предпочтительно менее 5% по объему кислорода. Особенно предпочтительно поток азота с содержанием O2<5% по объему удаляют из линии остаточного газа после абсорбционной колонны.
В дополнительном конкретном варианте осуществления уменьшение содержания кислорода согласно с2) в содержащей кислород газовой смеси, предпочтительно в воздухе, проводят посредством адсорбции с перемежающимся давлением, криогенного разложения или с помощью мембран.
Получение содержащего кислород газового потока, который объединяют согласно Е) с потоком технологического газа, содержащего NOx, предпочтительно проводят посредством обогащения кислородом воздуха с применением адсорбции с перемежающимся давлением, криогенного разложения или с помощью мембран.
Кроме того, предпочтительно проводят добавление потока, содержащего пероксид, в верхней части абсорбционной колонны (8). Этот поток может представлять собой поток жидкости, содержащей растворенный пероксид. Примерами могут служить растворы, содержащие неорганические перекисные соединения, такие как пероксид или перборат водорода, или растворы, содержащие органическое перекисное соединение, такое как органический пероксид, органический гидропероксид или органическая перкарбоновая кислота или ее сложные эфиры.
Упомянутые выше системы для окисления аммиака предпочтительно интегрируют в систему для производства азотной кислоты или капролактама.
На фиг. 1-3, с одной стороны, показана известная система (фиг. 1), а с другой стороны, проиллюстрированы, в качестве примера, различные варианты осуществления системы согласно настоящему изобретению (фиг. 2 и 3) с применением примера системы для производства азотной кислоты. При этом на фигурах представлено следующее.
На фиг. 1 приведено упрощенное схематическое изображение традиционной системы единого давления для производства азотной кислоты.
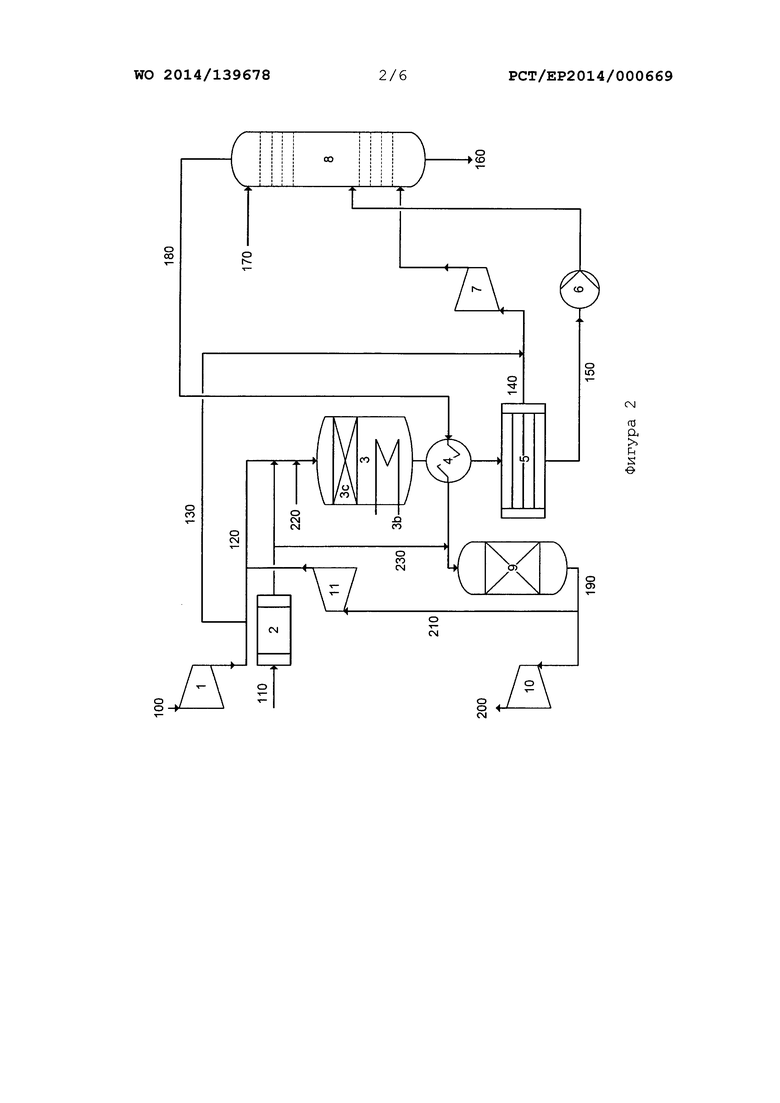
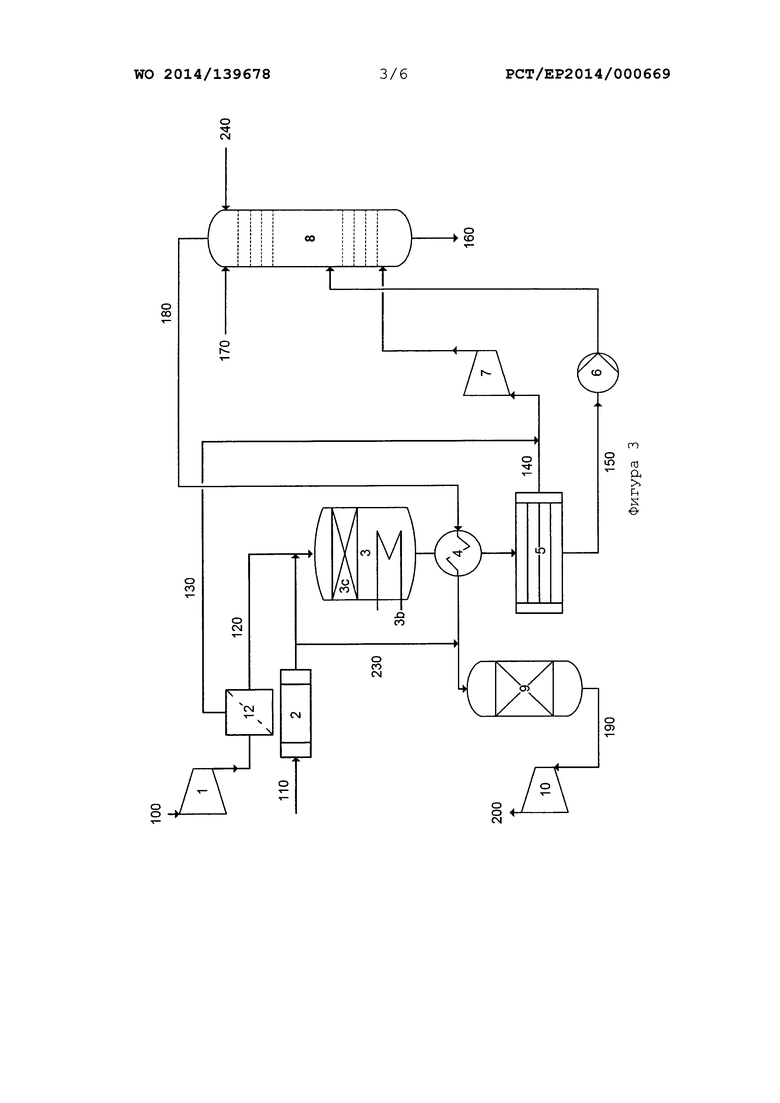
На фиг. 2 и 3 приведено схематическое изображение вариантов осуществления способа/системы согласно настоящему изобретению, интегрированной в систему двойного давления для производства азотной кислоты.
На фиг. 4, 5 и 6 показана зависимость выхода NOx от содержания кислорода в реакционной газовой смеси или от молярного отношения кислорода к аммиаку в реакционной газовой смеси для трех вариантов осуществления способа согласно настоящему изобретению.
На фиг. 1 приведена упрощенная технологическая схема традиционной системы единого давления. Поток воздуха, поступающий по линии 100, подвергают сжатию в воздушном компрессоре (1) и подают по линии 120 в реактор (3) для окисления аммиака. Прежде чем сжатый воздух поступит в реактор (3), в него подают по линии 120 газообразный аммиак, который предварительно подавали в жидком виде по линии 110 в испаритель (2) аммиака. Кроме того, часть сжатого воздуха отводят из линии 120 и подают по линии 130 в качестве так называемого вторичного воздуха в технологический газ перед тем, как подавать его в абсорбционную колонну (8). В реакторе (3), в котором над широкой областью натянуты сетки на основе металла платиновой группы в качестве катализатора (3а), происходит окисление аммиака, большая часть которого окисляется до NO и Н2О. Полученная газовая смесь отдает первую часть теплоты реакции в теплообменник (3b) в теплоутилизационной части реактора (3), выходит из реактора (3), и при этом в ней продолжает происходить окисление образовавшегося NO под действием остатков атмосферного кислорода или атмосферного кислорода, подаваемого по линии 130, и проходит через последующие теплообменники (4) для дальнейшего охлаждения технологического газа перед тем, как подавать его в абсорбционную колонну (8). По меньшей мере один теплообменник представляет собой конденсатор (5), в котором часть полученных NOx и H2O отделяется в виде конденсата кислоты, который в свою очередь подают по линии 150 в абсорбционную колонну (8) насосом (6). Остальную газовую смесь, которая все еще содержит большую часть NOx, смешивают со вторичным воздухом из линии 130, а затем подают по линии 140 в абсорбционную колонну (8). Дополнительное количество воздуха, введенное в технологический газ, используется для дополнительного окисления NO, содержащегося в технологическом газе, до NO2. В абсорбционной колонне (8) протекает реакция NOx с водой, а образовавшаяся азотная кислота выходит из абсорбционной колонны (8) по линии 160. Требуемое количество воды подают в абсорбционную колонну (8) по линии 170. Остаточный газ, содержащий оксид азота, выходит из абсорбционной колонны (8) по линии 180, проходит через теплообменник (4), где подвергается нагреванию, и поступает на очистку (9) остаточного газа. В современных системах производят каталитическое разложение N2O, содержащегося в остаточном газе, и оксидов азота NOx до азота и кислорода или до азота и воды при добавлении газообразного аммиака (по линии 230). После очистки (9) остаточный газ, который содержит, главным образом, азот и небольшое количество воды и кислорода, необязательно следы остаточных оксидов азота, подают по линии 190 в турбину остаточного газа (10), где он расширяется, обеспечивая регенерацию энергии, выходит по линии 200 и выбрасывается в атмосферу.
Технологическая схема типичной системы двойного давления для производства HNO3 отличается от системы единого давления, показанной на фиг. 1, наличием дополнительной ступени сжатия, которая расположена на линии 140 между вводом потока вторичного воздуха 130 и подачей в абсорбционную колонну (8).
На фиг. 1 не показан также функциональный блок для обесцвечивания полученной кислоты потоком вторичного воздуха. Он может быть встроен в нижнюю часть абсорбционной колонны или может представлять собой отдельную колонну, которая в системе двойного давления расположена между вводом потока 130 вторичного воздуха в линию 140 потока технологического газа и упомянутый выше ступенью сжатия технологического газа.
На фиг. 2 приведена в качестве примера технологическая схема одного или нескольких вариантов осуществления способа согласно настоящему изобретению и одного или нескольких вариантов осуществления системы согласно настоящему изобретению с применением системы двойного давления для производства азотной кислоты.
Воздушный компрессор (1), испаритель аммиака (2), реактор (3), теплообменники (3b, 4), конденсатор (5), насос (6), абсорбционная колонна (8), очистка (9) остаточного газа и турбина (10) остаточного газа, а также линии 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200 и 230 соответствуют элементам, показанным на фиг. 1, с точки зрения функционального назначения и схемы соединения друг с другом. Поскольку речь идет о системе двойного давления, в отличие от фиг. 1 дополнительно показан компрессор (7), в котором технологический газ из линии 140 подвергается сжатию перед тем, как поступать в абсорбционную колонну (8).
Кроме того, в отличие от фиг. 1, катализатор (3с) представляет собой не сетку на основе металла платиновой группы, а упаковку или засыпку формованных изделий катализатора, который содержит оксид переходного металла, который не является оксидом металла платиновой группы.
В системе на фиг. 2 дополнительно имеется линия 220, по которой в реакционную газовую смесь можно вводить газообразную разбавляющую среду, содержащую кислород в количестве <20% по объему, например, обедненный кислородом воздушный поток или пар, перед тем, как подавать ее в реактор (3). Кроме того, можно подавать часть очищенного остаточного газа (содержащего главным образом, азот), поступающего с очистки (9) остаточного газа, в расширитель (11) перед тем, как подавать его в турбину (10) остаточного газа, и расширять его в достаточной степени, чтобы можно было подавать его в сжатый воздух в линии 120.
Благодаря этим действиям (подаче разбавляющей среды, содержащей кислород в количестве <20% по объему, по линии 220 или частичной рециркуляции очищенного остаточного газа по линии 210), принимаемым по отдельности или совместно, можно проводить регулирование отношения кислорода к аммиаку в реакционной газовой смеси в определенном диапазоне согласно настоящему изобретению. С помощью системы, показанной на фиг. 2, содержащей линии 210 и 220 подачи или не содержащей их, можно также регулировать отношение кислорода к аммиаку в реакционной газовой смеси в определенном диапазоне согласно настоящему изобретению, уменьшая поток первичного воздуха 120 и увеличивая поток вторичного воздуха 130 по сравнению с обычными способами производства HNO3.
На фиг. 3 показаны дополнительные варианты осуществления способа согласно настоящему изобретению и системы согласно настоящему изобретению на примере системы двойного давления для производства азотной кислоты. Воздушный компрессор (1), испаритель аммиака (2), реактор (3), слой катализатора (3с), теплообменники (3b, 4), конденсатор (5), насос (6), компрессор технологического газа (7), абсорбционная колонна (8), очистка (9) остаточного газа и турбина остаточного газа (10), а также линии 100, 110, 140, 150, 160, 170, 180, 190, 200 и 230 соответствуют элементам, показанным на фиг. 2, с точки зрения функционального назначения и схемы соединения друг с другом. В системе на фиг. 3 дополнительно имеется устройство (12), предназначенное для разделения воздуха, в которое подают сжатый воздух из воздушного компрессора (1). В устройстве (12) проводится (частичное) разделение воздуха на часть, имеющую пониженное содержание кислорода, и часть, имеющую повышенное содержание кислорода. Газовую смесь, имеющую повышенное содержание азота, загружают в линию 120 и подают в реактор (3). Газовую смесь, имеющую повышенное содержание кислорода, загружают в линию 130 и подают через линии 140 в газовую смесь, поступающую из конденсатора (5) и содержащую главным образом NOx. Благодаря этим действиям также можно регулировать отношение кислорода к аммиаку в реакционной газовой смеси в определенном диапазоне.
Кроме того, на фиг. 3 показана линия 240, по которой можно подавать в абсорбционную колонну (8) поток жидкости, содержащий пероксид. Подача этого потока обеспечивает еще одну возможность для введения кислорода, необходимого для окисления NO в абсорбционной колонне. Эту меру можно принимать вместо или вместе с упомянутой выше мерой Е, то есть, с устройством, расположенным между реактором (3) для окисления аммиака и абсорбционной колонной (8), которое предназначено для смешивания потока технологического газа, содержащего NOx, с газовым потоком, содержащего кислород.
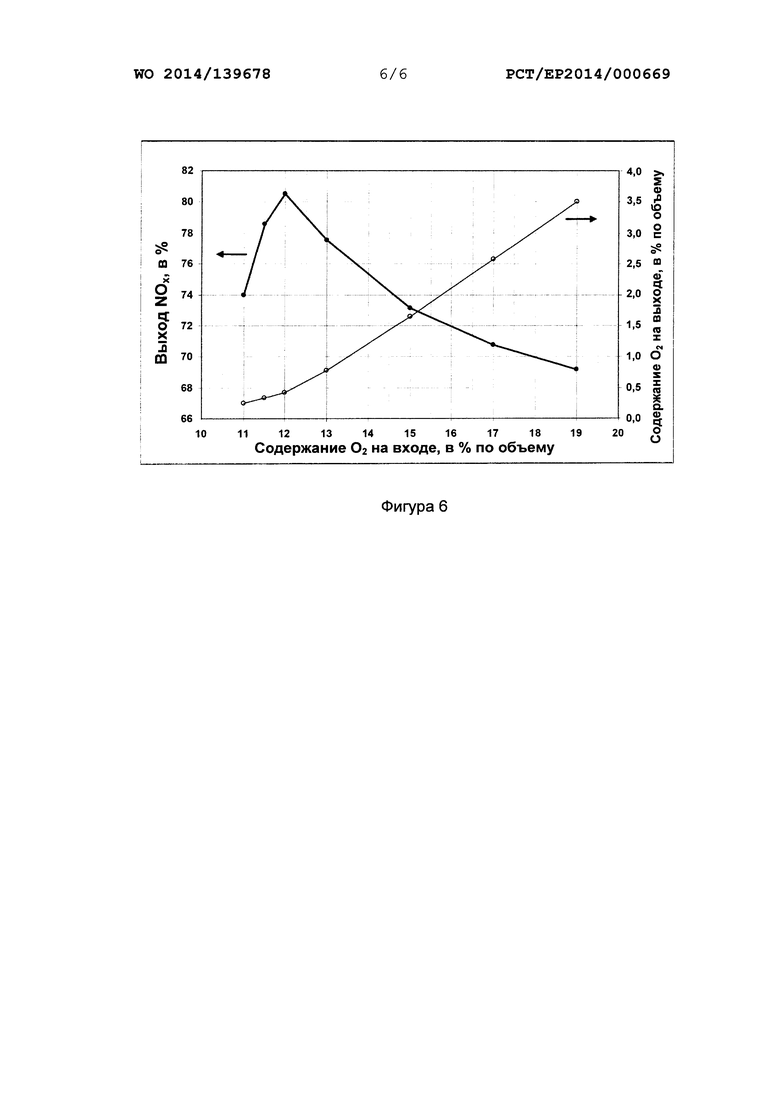
На фиг. 4-6 для выбранных в качестве примера катализаторов на основе оксида переходного металла (на фиг. 4 и 5 для активного компонента LaCoO3, а на фиг. 6 для активного компонента LaMnO3) показано, как при окислении NH3 в лабораторных условиях можно увеличить выход NOx с применением различных вариантов осуществления способа согласно настоящему изобретению, понижая содержание кислорода в газовой смеси, содержащей аммиак и кислород, то есть регулируя согласно настоящему изобретению отношение молярных количеств кислорода и аммиака на входе реакционной газовой смеси в слой катализатора.
С этой целью катализаторы в форме сот, имеющие длину 1 см, диаметр примерно 18 мм и плотность упаковки 200 ячеек на кв. дюйм или 400 ячеек на кв. дюйм, вставляли в трубчатый реактор из кварцевого стекла, имеющий внутренний диаметр 20 мм, и пропускали через него синтетическую газовую смесь аммиака, кислорода и азота. Анализ газовых потоков проводили с применением термических массовых расходомеров, причем при заданной концентрации аммиака 5% по объему (фиг. 4) и 10% по объему (фиг. 5 и 6) меняли концентрацию кислорода в диапазоне от 20 до 6% по объему (фиг. 4) и в диапазоне от 19 до 12% по объему (фиг. 5) или в диапазоне от 19 до 11% по объему (фиг. 6). Объемная скорость во всех случаях составляла 100000 ч-1. Регулируя давление на выходе из реактора, создавали рабочее давление 4,5 бар (фиг. 4 и 6) или 2,0 бар (фиг. 5). Отдельные параметры примеров осуществления, показанных на фиг. 4-6, перечислены в следующей таблице.
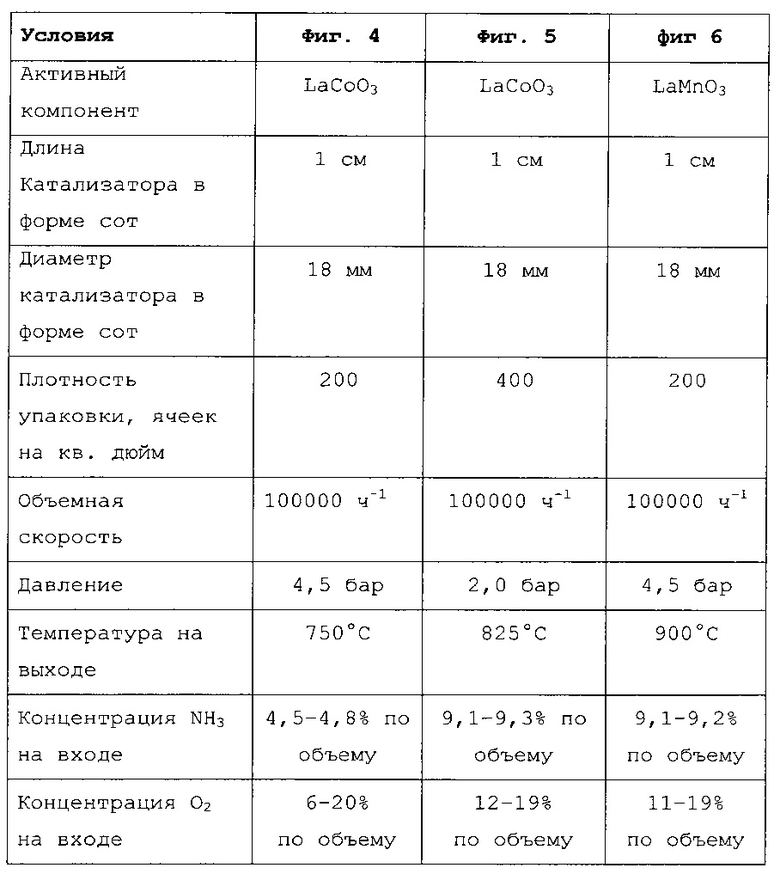
Чтобы компенсировать потери тепла, реакционную трубу окружили металлическим кожухом и поместили в две трубчатые печи, расположенные друг над другом. Достигаемую в результате температуру реакции измеряли термоэлементом примерно на 1 мм ниже катализатора в форме сот (Твых.). Эта температура на выходе составляла примерно 750°С в случае концентрации аммиака на входе 5% по объему (фиг. 4) и 825°С (фиг. 5) или около 900°С (фиг. 6) в случае концентрации аммиака на входе 10% по объему. Анализ рабочего газа проводили, попеременно подавая в анализатор пробы со входа и выхода реактора. Анализатор, использовавшийся для определения объемной концентрации аммиака, монооксида азота (NO), диоксида азота (NO2) и воды, представлял собой FT-IR спектрометр (модели "6700 Advanced Gold" от Thermo-Nicolet), оснащенный нагреваемой газовой кюветой длиной 15 см. Концентрацию O2 определяли газоанализатором OXYMAT 6, в версии с нагревом, от Siemens.
Указанные объемные концентрации соответствуют в первом приближении молярным концентрациям отдельных компонентов. При каждой регулировке новой концентрации кислорода на входе ожидали достижение стационарного режима работы на основании результатов анализа газа на выходе из реактора. На фиг. 4-6 приведены графики зависимости выхода суммарного параметра NOx (= NO + NO2), исходя из концентрации аммиака на входе, в зависимости от концентрации кислорода на входе, при этом для расчета выхода NOx по молярным концентрациям учитывались изменения объема, связанные с отдельными первичными реакциями (схемы 1, 5, 6 и 7).
В каждом случае можно видеть четко выраженный максимум выхода NOx, соответствующий плотности точек измерения, который на фиг. 4 приходится на содержание O2 величиной 7% по объему, что соответствует отношению молярных концентраций O2 и NH3 на входе величиной 1,4 моль/моль, на фиг. 5 приходится на содержание O2 величиной 13% по объему, что соответствует отношению молярных концентраций O2 и NH3 на входе величиной 1,3 моль/моль, а на фиг. 6 приходится на содержание O2 величиной 12% по объему, что соответствует отношению молярных концентраций O2 и NH3 на входе величиной 1,2 моль/моль.
Можно также четко видеть, что при отношении молярных концентраций О2 и NH3 на входе величиной 1,9 моль/моль, которое не соответствует настоящему изобретению и используется, как правило, в обычных промышленных способах окисления аммиака (10% аммиака в воздухе по объему), обеспечивается значительно более низкий выход NOx, чей в диапазоне, выбранном согласно настоящему изобретению вблизи оптимального молярного отношения кислорода к аммиаку.
Если это отношение не достигает упомянутого выше диапазона, выход NOx значительно снижается, как можно видеть на фиг. 4, при содержании O2 величиной 6% по объему, соответствующему отношению молярных концентраций O2 и NH3 величиной 1,2 моль/моль, на фиг. 5 при содержании O2 величиной 12% по объему, соответствующему отношению молярных концентраций О2 и NH3 величиной 1,2 моль/моль, а на фиг. 6 при содержании О2 величиной 11% по объему, соответствующему отношению молярных концентраций О2 и NH3 величиной 1,1 моль/моль.
Изделия в форме сот были изготовлены экструзией из обработанных соответствующим образом порошков LaMnO3 или LaCoO3 с добавлением соответствующих связующих и пластификаторов и последующими сушкой и прокаливанием. Исходные порошки для подвергаемых испытанию катализаторов получали посредством осаждения щелочью из стехиометрически полученных растворов соответствующих солей металлов, фильтрации, промывки и прокаливания полученных осадков. Образование фаз и чистоту соответствующих перовскитовых фаз контролировали с применением XRD (рентгеновской дифрактометрии).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ УДАЛЕНИЯ NO И NO В ПРОЦЕССЕ ПОЛУЧЕНИЯ АЗОТНОЙ КИСЛОТЫ И УСТРОЙСТВО ДЛЯ ОСУЩЕСТВЛЕНИЯ ЭТОГО СПОСОБА | 2012 |
|
RU2600753C2 |
| НИЗКОФОСФОРИСТЫЕ ХАБАЗИТЫ | 2012 |
|
RU2634702C2 |
| СПОСОБ ПРОИЗВОДСТВА СЕРНОЙ КИСЛОТЫ | 2007 |
|
RU2458857C9 |
| КАТАЛИЗАТОРЫ ДЛЯ УМЕНЬШЕНИЯ ВЫБРОСОВ АММИАКА С ВЫХЛОПНЫМИ ГАЗАМИ ОТ СЖИГАНИЯ БОГАТОЙ ТОПЛИВНОЙ СМЕСИ | 2012 |
|
RU2593293C2 |
| КАТАЛИЗАТОР ДЛЯ ОБРАБОТКИ ВЫХЛОПНЫХ ГАЗОВ | 2014 |
|
RU2675821C2 |
| СПОСОБ УДАЛЕНИЯ ОКСИДОВ АЗОТА | 2003 |
|
RU2320400C2 |
| СПОСОБЫ СЕЛЕКТИВНОГО КАТАЛИТИЧЕСКОГО ВОССТАНОВЛЕНИЯ С ИСПОЛЬЗОВАНИЕМ ЛЕГИРОВАННЫХ ОКСИДОВ ЦЕРИЯ(IV) | 2014 |
|
RU2664905C2 |
| КАТАЛИЗАТОР ДЛЯ РАЗЛОЖЕНИЯ NO ЕГО ПРИМЕНЕНИЕ, А ТАКЖЕ СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2001 |
|
RU2258030C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЗОТНОЙ КИСЛОТЫ | 1998 |
|
RU2151736C1 |
| СПОСОБ ПРОИЗВОДСТВА НЕКОНЦЕНТРИРОВАННОЙ АЗОТНОЙ КИСЛОТЫ | 2005 |
|
RU2296706C1 |


Группа изобретений относится к неорганической химии и может быть использовано в химической промышленности для получения продуктов окисления аммиака. Система для окисления аммиака содержит реактор (3), оснащенный линией (120) подачи газовоздушной смеси и линией выпуска технологического газа. Внутри реактора (3) расположен катализатор (3с), содержащий по меньшей мере один оксид переходного металла, который не является оксидом платиновой группы. Система также содержит устройство для регуляции молярного отношения кислорода к аммиаку в реакционной газовой смеси до значений от 1,25 до 1,75 моль/моль посредством смешивания газового потока с содержанием О2<20% по объему с выбранным количеством аммиака. Содержащий кислород газовый поток может быть образован 1) с помощью устройства для разбавления воздушного потока газовым потоком (линия 220); 2) с помощью устройства для уменьшения содержания кислорода в содержащей кислород газовой смеси, предпочтительно в воздухе (линия 210); 3) при их комбинации. Температура полученного газа на выходе из слоя катализатора составляет от 700°С до 950°С. 2 н. и 28 з.п. ф-лы, 6 ил., 2 табл.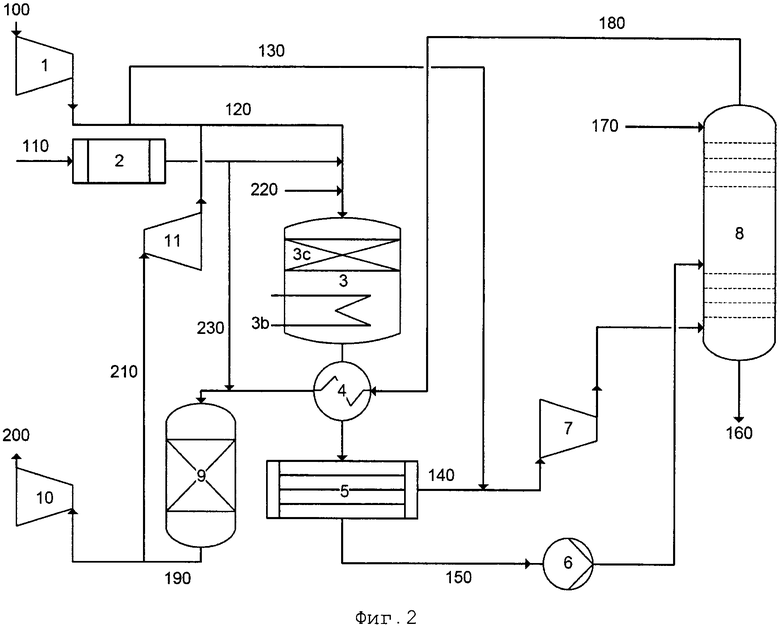
1. Способ окисления аммиака кислородом в присутствии катализаторов, содержащих по меньшей мере один оксид переходного металла, который не является оксидом металла платиновой группы, где отношение молярных количеств кислорода к аммиаку для реакционной газовой смеси на входе в слой катализатора регулируют до значений от 1,25 до 1,75 моль О2/моль NH3, и температура полученного газа на выходе из слоя катализатора составляет от 700°С до 950°С.
2. Способ по п. 1, отличающийся тем, что отношение молярных количеств О2 к NH3 в реакционной газовой смеси на входе в слой катализатора составляет от 1,30 до 1,75 моль О2/моль NH3, особенно преимущественно от 1,35 до 1,60 моль О2/моль NH3 и наиболее преимущественно от 1,35 до 1,50 моль О2/моль NH3.
3. Способ по п. 1, отличающийся тем, что отношение молярных количеств О2 к NH3 в реакционной газовой смеси на входе в слой катализатора выбирают так, чтобы оно находилось в диапазоне от 0,1 моль О2/моль NH3 ниже до 0,4 моль O2/моль NH3 выше оптимального молярного отношения, где оптимальное молярное отношение представляет собой отношение кислорода к аммиаку реакционной газовой смеси на входе в слой катализатора, при котором достигается максимальный выход NOx.
4. Способ по меньшей мере по п. 1, отличающийся тем, что отношение молярных количеств кислорода к аммиаку в реакционной газовой смеси на входе в слой катализатора представляет собой от 0,05 моль О2/моль NH3 ниже до 0,3 моль О2/моль NH3 выше оптимального молярного отношения, особенно преимущественно от 0,025 моль О2/моль NH3 ниже до 0,25 моль O2/моль NH3 выше оптимального молярного отношения.
5. Способ по меньшей мере по п. 1, отличающийся тем, что оптимальное молярное отношение кислорода к аммиаку определяют посредством проведения серии испытаний при заданных условиях способа с применением выбранного катализатора и с применением реакционной среды, содержащей кислород и аммиак, где концентрацию кислорода на входе в слой катализатора выбирают так, чтобы соответствующее молярное отношение O2/NH3 изменялось в диапазоне между минимальным молярным отношением О2/NH3, предпочтительно минимальным молярным отношением O2/NH3, составляющим 1,25 моль/моль, и максимальным молярным отношением O2/NH3, предпочтительно максимальным молярным отношением О2/NH3, составляющим 1,75 моль/моль, посредством определения выхода NOx, который достигается в каждом случае, а затем посредством определения молярного отношения кислорода к аммиаку, которое обеспечивает максимальный выход NOx при постоянных прочих условиях реакции.
6. Способ по п. 1, отличающийся тем, что отношение молярных количеств кислорода к аммиаку реакционной газовой смеси на входе в слой катализатора регулируют до значений, меньше или равных 1,75 моль О2/моль NH3, предпочтительно до значений, меньше или равных 1,60 моль O2/моль NH3, особенно предпочтительно до значений, меньше или равных 1,50 моль О2/моль NH3, и где содержание кислорода в полученной газовой смеси на выходе из слоя катализатора составляет по меньшей мере 0,3% по объему, предпочтительно по меньшей мере 0,4% по объему и особенно предпочтительно по меньшей мере 0,5% по объему.
7. Способ по п. 6, отличающийся тем, что отношение молярных количеств кислорода к аммиаку реакционной газовой смеси на входе в слой катализатора выбирают так, чтобы полученное в результате содержание кислорода в полученной газовой смеси на выходе из слоя катализатора составляло от 0,3% по объему до 10,0% по объему, предпочтительно от 0,4% по объему до 6,0% по объему, особенно предпочтительно от 0,5 по объему до 4,0% по объему.
8. Способ по п. 7, отличающийся тем, что отношение молярных количеств кислорода к аммиаку реакционной газовой смеси на входе в слой катализатора выбирают так, чтобы полученное в результате содержание кислорода в полученном газе на выходе из слоя катализатора составляло от 0,3% по объему до 2,0% по объему, предпочтительно от 0,4% по объему до 2,0% по объему, особенно предпочтительно от 0,5% по объему до 1,5% по объему.
9. Способ по п. 1, отличающийся тем, что концентрация NH3 на входе в реактор окисления составляет от 1 до 17% по объему, особенно предпочтительно от 4 до 15% по объему, в частности от 7 до 14% по объему.
10. Способ по п. 1, отличающийся тем, что температура полученного газа на выходе из слоя катализатора составляет от 750°С до 850°С.
11. Способ по п. 1, отличающийся тем, что часть теплоты реакции рассеивают, в частности, посредством охлаждения стенок реактора и/или посредством размещения интегрированных охлаждающих устройств в упаковке катализатора.
12. Способ по п. 1, отличающийся тем, что температура реакционной газовой смеси, содержащей NH3 и кислород, на входе в слой катализатора составляет от 20°С до 300°С, предпочтительно от 50°С до 200°С и особенно предпочтительно от 50°С до 150°С.
13. Способ по п. 1, отличающийся тем, что объем потока реакционной газовой смеси и/или объем катализатора регулируют так, чтобы полученная в результате объемная скорость составляла от 50000 ч-1 до 500000 ч-1, предпочтительно от 100000 ч-1 до 300000 ч-1.
14. Способ по п. 1, отличающийся тем, что катализатор содержит легированные оксиды переходных металлов, которые не являются оксидами металлов платиновой группы, и/или смешанные оксиды таких оксидов переходных металлов.
15. Способ по п. 14, отличающийся тем, что смешанные оксиды имеют структуру шпинели, делафоссита, перовскита или браунмиллерита.
16. Способ по п. 15, отличающийся тем, что смешанный оксид, имеющий структуру перовскита, характеризуется общей эмпирической формулой ABO3±δ, и/или что смешанный оксид, имеющий структуру браунмиллерита, характеризуется общей эмпирической формулой А2B2O5±δ, где δ принимает значение от 0,01 до 0,5, А представляет собой одно-, двух- или трехвалентные катионы, В представляет собой трех-, четырех- или пятивалентные катионы, и ионные радиусы А больше ионных радиусов В.
17. Способ по п. 16, отличающийся тем, что положение А перовскитов и/или браунмиллеритов занято на более чем 50%, предпочтительно на более чем 80%, особенно предпочтительно на более чем 95% одним или несколькими элементами, выбранными из группы редкоземельных металлов и щелочноземельных металлов, особенно предпочтительно La, а положение В перовскитов и/или браунмиллеритов занято на более чем 50%, предпочтительно на более чем 80%, особенно предпочтительно на более чем 95% одним или несколькими элементами, выбранными из группы Cr, Mn, Fe, Со, Ni, особенно предпочтительно Со и Mn, наиболее предпочтительно Со.
18. Способ по п. 17, отличающийся тем, что перовскит имеет состав LaCoO3±δ.
19. Способ по п. 1, отличающийся тем, что катализатор содержит оксиды переходных металлов, которые не являются оксидами металлов платиновой группы, и необязательно другие активные компоненты и/или сопутствующие компоненты, внедренные или заключенные в матрицу, предпочтительно в керамическую матрицу, или содержит оксиды переходных металлов, которые не являются оксидами металлов платиновой группы, и необязательно другие активные компоненты и/или сопутствующие компоненты, нанесенные на подложку.
20. Способ по п. 1, отличающийся тем, что катализатор представляет собой твердый катализатор, то есть формованное изделие, которое главным образом состоит из каталитически активного материала.
21. Способ по п. 1, отличающийся тем, что молярное отношение O2/NH3 в реакционной газовой смеси на входе в слой катализатора регулируют посредством добавления в воздушный поток газообразного аммиака.
22. Способ по п. 1, отличающийся тем, что молярное отношение O2/NH3 в реакционной газовой смеси на входе в слой катализатора регулируют посредством добавления газообразного аммиака и посредством добавления газа с содержанием кислорода менее 20%, предпочтительно менее 10%, особенно предпочтительно <5%, в воздушный поток.
23. Система для окисления аммиака, содержащая:
А) реактор (3) для окисления аммиака, оснащенный по меньшей мере одной линией подачи реакционной газовой смеси и по меньшей мере одной линией выпуска технологического газа,
B) катализатор (3 с) внутри реактора (3), содержащий по меньшей мере один оксид переходного металла, который не является оксидом металла платиновой группы,
C) устройство для регуляции молярного отношения кислорода к аммиаку в реакционной газовой смеси до значений от 1,25 до 1,75 моль/моль посредством смешивания содержащего кислород газового потока с содержанием O2 <20% по объему с выбранным количеством аммиака, где содержащий кислород газовый поток образуется
c1) с помощью устройства для разбавления воздушного потока газовым потоком, который содержит менее 20% по объему, предпочтительно менее 10% по объему, особенно предпочтительно менее 5% по объему кислорода, или
с2) с помощью устройства для уменьшения содержания кислорода в содержащей кислород газовой смеси, предпочтительно в воздухе, или
с3) с помощью комбинации средств c1 и с2.
24. Система для окисления аммиака по п. 23 с последующей абсорбцией NOx, содержащая элементы А), В) и С) по п. 23, а также
D) абсорбционную колонну (8) для абсорбции NOx и образования HNO3, HNO2 или растворов нитратов или нитритов и
E) устройство, расположенное между реактором (3) для окисления аммиака и абсорбционной колонной (8), для объединения содержащего NOx потока технологического газа с газовым потоком, который содержит более 25%, предпочтительно более 30%, особенно предпочтительно более 40% кислорода, и/или линию (240), сообщающуюся с абсорбционной колонной (8), для введения содержащего пероксид потока.
25. Система для окисления аммиака по п. 23, отличающаяся тем, что она интегрирована в систему для производства азотной кислоты или капролактама.
26. Система по п. 23, отличающаяся тем, что воздушный поток согласно c1) разбавляется паром или потоком азота, который содержит менее 20% по объему, предпочтительно менее 10% по объему, особенно предпочтительно менее 5% по объему кислорода.
27. Система по п. 23, отличающаяся тем, что воздушный поток разбавляется согласно c1) газовым потоком, удаленным из остаточного газа после абсорбционной колонны (8).
28. Система по п. 23, отличающаяся тем, что уменьшение содержания кислорода в содержащей кислород газовой смеси, предпочтительно в воздухе, согласно с2) происходит посредством адсорбции с перемежающимся давлением, криогенного разложения или с помощью мембран.
29. Система по п. 24, отличающаяся тем, что обогащение кислородом содержащего кислород газового потока согласно Е) происходит посредством обогащения кислородом воздуха путем адсорбции с перемежающимся давлением, криогенного разложения или с помощью мембран.
30. Система по п. 23, отличающаяся тем, что она содержит по меньшей мере один компрессор (1), посредством которого содержащий кислород газовый поток подвергается сжатию и подается в реактор (3) для окисления аммиака.
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| СПОСОБ ПРОИЗВОДСТВА НЕКОНЦЕНТРИРОВАННОЙ АЗОТНОЙ КИСЛОТЫ | 2005 |
|
RU2296706C1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| УСТРОЙСТВО И СПОСОБ ДЛЯ КАТАЛИТИЧЕСКИХ ГАЗОФАЗНЫХ РЕАКЦИЙ, А ТАКЖЕ ИХ ПРИМЕНЕНИЕ | 2008 |
|
RU2474469C2 |
| Способ регенерации угольного адсорбентаи уСТРОйСТВО для ЕгО ОСущЕСТВлЕНия | 1979 |
|
SU799792A1 |
| МИЩЕНКО К.П., РАВДЕЛЬ А.А., Краткий справочник физико-химических величин, Ленинград, Химия, 1972, с.с | |||
| Подъемник для выгрузки и нагрузки барж сплавными бревнами, дровами и т.п. | 1919 |
|
SU149A1 |
| Парный автоматический сцепной прибор для железнодорожных вагонов | 0 |
|
SU78A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

