ОБЛАСТЬ ПРИМЕНЕНИЯ ИЗОБРЕТЕНИЯ
Область техники, к которой относится изобретение
[0002] Настоящее описание относится к кристаллической форме (R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он диводород фосфата и способам применения кристаллической формы. Кристаллическая форма может быть использована в качестве фармацевтически активного соединения в композициях, которые являются полезными для ингибирования роста бактерий или для лечения пациентов, страдающих от бактериальных инфекций.
Описание уровня техники
[0003] Патентная публикация US 20070155798, которая приводится здесь для ссылки во всей ее целостности, раскрывает ряд сильнодействующих антибактериальных оксазолидинонов, включающий
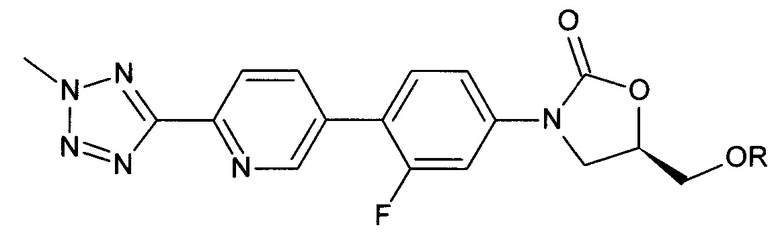
где R=Н, РО(ОН)2 и PO(ONa)2.
[0004] Хотя эта патентная заявка раскрывает способы получения соединений, таких как свободная кислота (где R=РО(ОН)2) и динатриевая соль (где R=PO(ONa)2), в ней отсутствует информация о том, что любое из этих соединений было стабильно в кристаллической или очищенной форме. Дополнительно, эти способы включают использование реагентов, которые являются химически агрессивными, например, трифторуксусная кислота или взрывоопасными, например, этиловый эфир, и таким образом не подходят для коммерческого использования. Как обсуждается в деталях ниже, попытки кристаллизовать динатриевую соль авторами настоящего изобретения привели к получению высоко гигроскопичной, нестабильной формы соли, которая стала аморфной при высушивании.
[0005] В уровне техники существует необходимость в стабильной, не гигроскопичной кристаллической форме свободной кислоты (где R=РО(ОН)2) или ее соли, которая может быть аккуратно перенесена и взвешена для применения в фармацевтических средствах. Также предпочтительно, чтобы кристаллическая форма не образовывала большое число полиморфных модификаций, поскольку некоторые из полиморфов препятствуют способности воспроизвести идентичные полиморфы в процессе получения. Получение конкретной кристаллической формы, обладающей этими свойствами, это эмпирический процесс, и специалист в данной области не сможет предсказать, какая из форм свободной кислоты фармацевтического соединения или одной из соответствующих солей будет кристаллизоваться, если будет, в указанных условиях кристаллизации. Дополнительно специалист в данной области не может предсказать, какая из кристаллических форм будет обладать лучшими свойствами стабильности, способностью к переносу, отсутствию гигроскопичности и воспроизводимости.
[0006] Дополнительно улучшенные способы получения свободной кислоты раскрыты в патентной заявке US 12/577,089, переданной Траюс Терапьютикс, Инк., которая приводится здесь для ссылки во всей ее целостности. Существуют сложности в фильтрации кристаллического материала и переработке кристаллического материала в дозированные формы, такие как таблетки, потому что свободная кислота образует мелкие частицы, которые удлиняют время процесса. Таким образом, в уровне техники существует необходимость в кристаллической форме соединения и соответствующих способах, которые преодолевают эти сложности.
[0007] Дополнительным преимуществом было бы иметь очищенное соединение, которое подходило для применения в фармацевтических композициях.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
[0008] Неожиданно (R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он диводород фосфат 1 (R=РО(ОН)2), оказался более стабильным и не гигроскопичным, чем уже проверенные солевые формы. Дополнительно в отличии от обычной кристаллизации, где условия кристаллизации, например, растворитель и температура, определяют конкретную кристаллическую форму, одна и та же кристаллическая форма 1 (R=РО(ОН)2), была получена, используя различные растворители и условия кристаллизации. Таким образом, эта кристаллическая форма была очень стабильной, воспроизводимой и идеальной для коммерческого производства, поскольку она уменьшила количество шансов, что другие полиморфы образуют загрязняющие примеси во время производства. Однако, во всех предварительных испытаниях свободная кислота кристаллизуется в виде мелких частиц, затрудняя фильтрование и переработку.
[0009] Для преодоления трудностей с фильтрованием и переработкой кристаллического (R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он диводород фосфата 1 (R=РО(ОН)2), процессы, описанные здесь, приводят к значительному снижению времени фильтрования, помогают избежать использования токсичных растворителей, и значительно увеличивают легкость получения дозированных форм, таких как таблетки. Было обнаружено, что осуществление различных процессов можно контролировать с помощью распределения по размерам частиц полученного материала, что является полезным для получения кристаллической формы, и для коммерческого и фармацевтического использования. Неожиданно было установлено, что процесс увеличения размера частиц уменьшает количество примеси димера в сравнении с процессом получения свободной кислоты, описанной в патентной заявке США 12/577,089. Таким образом, различные способы получения и использования кристаллической формы также представлены.
[0010] Дополнительно, используя способы получения свободной кислоты, описанные в патентной заявке США 12/577,089, права на которую переданы тому же патентообладателю, что и в настоящей заявке, и применяя способы кристаллизации, описанные здесь, кристаллическая свободная кислота, имеющая, по меньшей мере, 96% чистоты по весу может быть получена в форме, которая содержит соединение, имеющее следующую формулу:
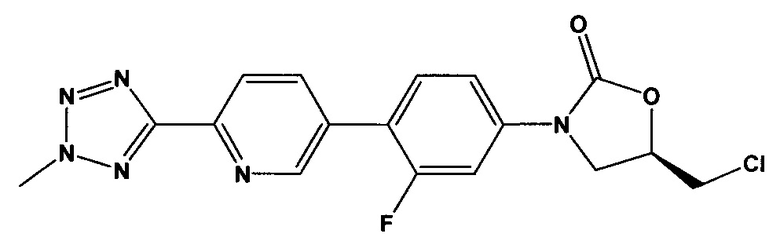
(здесь и далее «хлор содержащие примеси», то есть (R)-5-(хлорметил)-3-(3-фтор-4-(6-(2-метил-2Н-тетразол-5-ил)пиридин-3-ил)фенил)оксазолидин-2-он в количестве менее 1%.
[0011] Подобным образом, используя способы получения свободной кислоты, раскрытой в патентной заявке США 12/577,089, права на которую переданы тому же правообладателю, что и настоящая заявка, и используя способы кристаллизации, описанные здесь, кристаллическая свободная кислота, имеющая, по меньшей мере, 96% чистоту по весу может быть получена в форме, которая содержит соединение, имеющую следующую формулу:
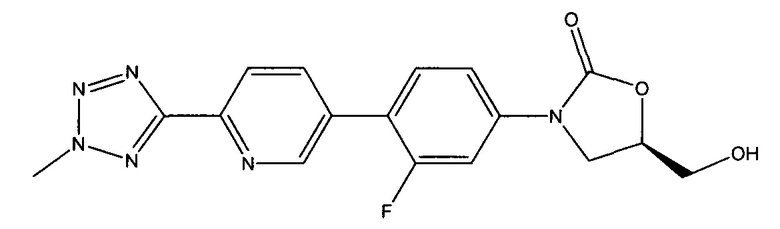
(здесь и далее "TR-700"), то есть, 5R)-3-{3-фтор-4-[6-(2-метил-2Н-1,2,3,4-тетразол-5-ил)-пиридин-3-ил]-фенил}-5-гидроксиметил-1,3-оксазолидин-2-он, в количестве, меньше 1%.
[0012] Кристаллическая свободная кислота может иметь один или более атрибутов, описанных здесь.
[0013] В некоторых аспектах, очищенный кристаллический (R)-3-(4-(2-(2-метил-теразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он диводород фосфат, то есть свободная кислота имеет чистоту, по меньшей мере, около 96% по весу. В некоторых вариантах осуществления изобретения, кристаллическая свободная кислота имеет средний диаметр, по меньшей мере, около 1.0 мкм.
[0014] В некоторых вариантах осуществления изобретения, фармацевтическая композиция содержит свободную кислоту или соль и, по меньшей мере, один фармацевтически приемлемый носитель, экципиент или дилюент.
[0015] В некоторых вариантах осуществления изобретения способ лечения бактериальной инфекции включает введение эффективного количества кристаллической свободной кислоты или ее соли субъекту, которому это необходимо. Способы могут также включать лечение бактериальной инфекции, включающей введение свободной кислоты, фармацевтической композиции или соли субъекту, которому это необходимо.
[0016] В некоторых аспектах способы получения свободной кислоты включают высушивание кристаллического (R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он диводород фосфата или фармацевтической композиции, содержащей его соль.
[0017] Эти и другие варианты осуществления изобретения описаны в деталях ниже.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
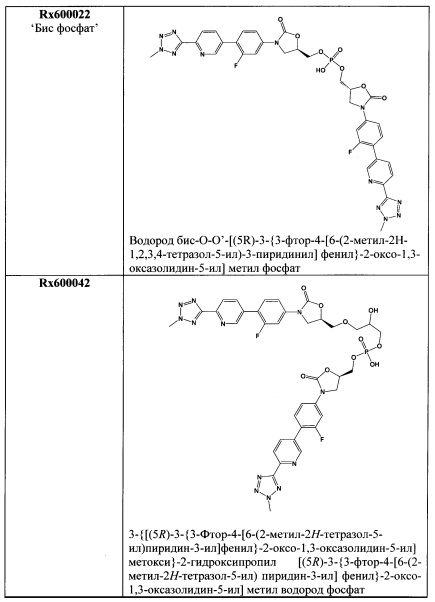
[0018] Фигура 1 представляет собой Фурье-Раман спектр кристаллического соединения 1 (R=РО(ОН)2).
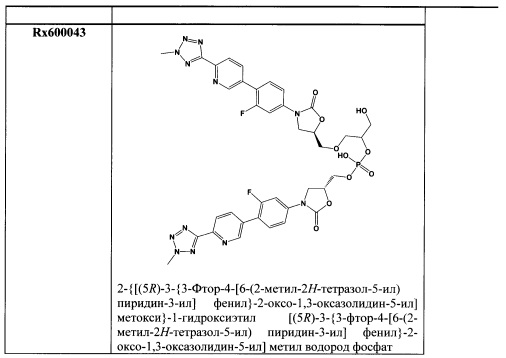
[0019] Фигура 2 представляет собой рентгенограмму кристаллического соединения 1 (R=РО(ОН)2).
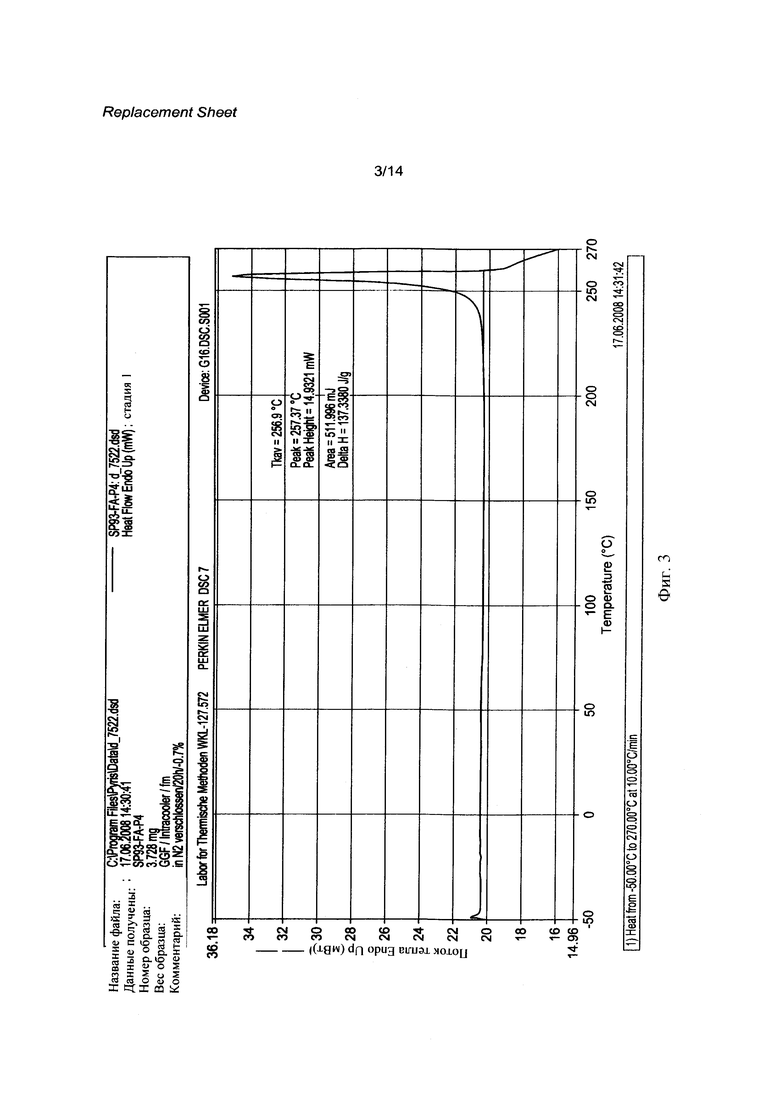
[0020] Фигура 3 представляет собой термограмму дифференциальной сканирующей калориметрии (DSC) кристаллического соединения 1 (R=РО(ОН)2).
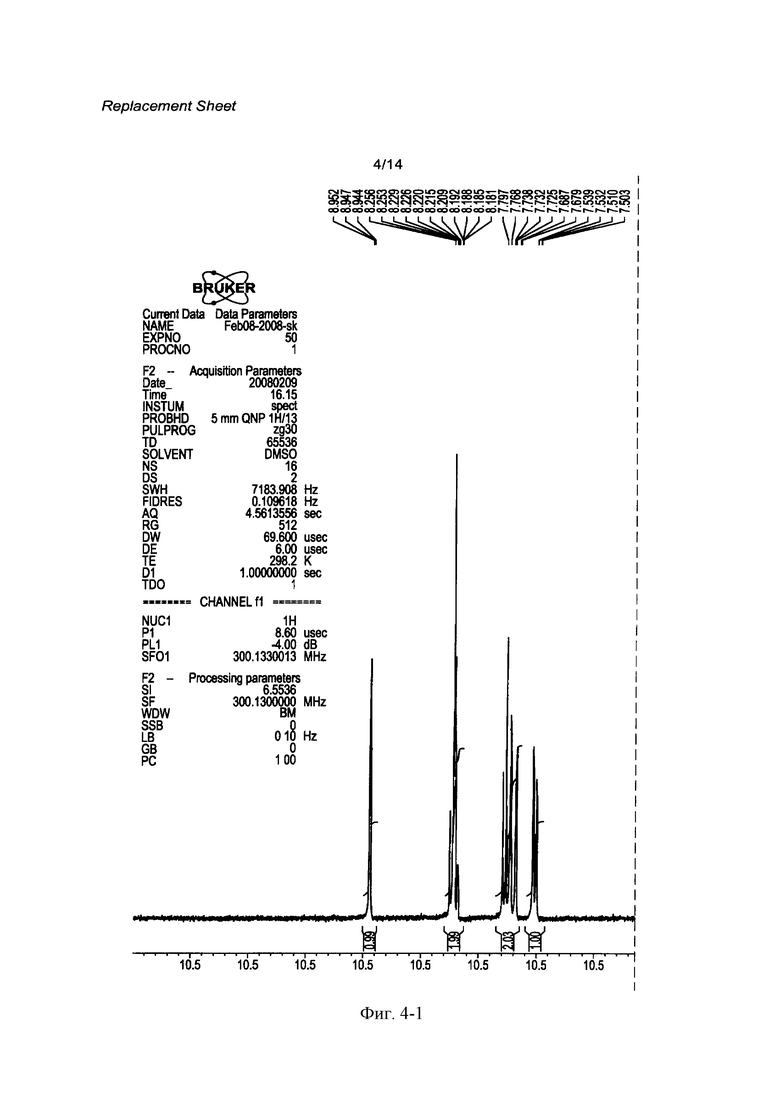
[0021] Фигура 4 демонстрирует 1Н ЯМР спектр соединения 1 (R=РО(ОН)2).
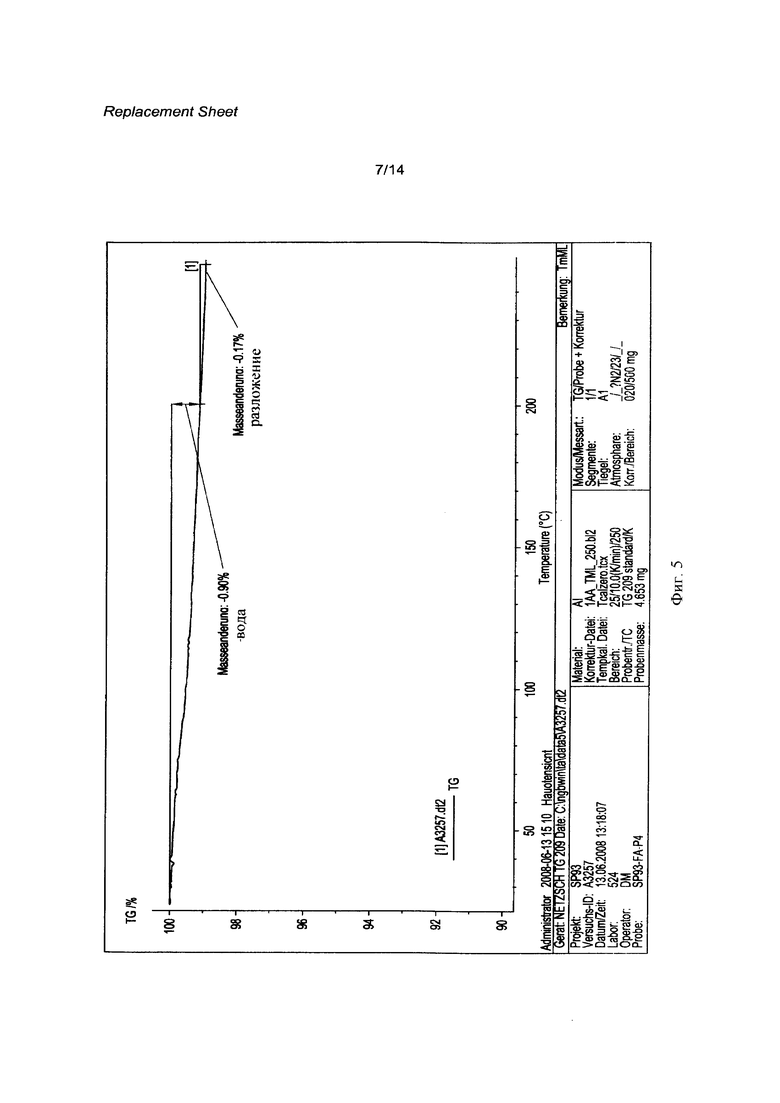
[0022] На Фигуре 5 изображена диаграмма кристаллического соединения 1 (R=РО(ОН)2) методами термогравиметрии и инфракрасной спектроскопии.
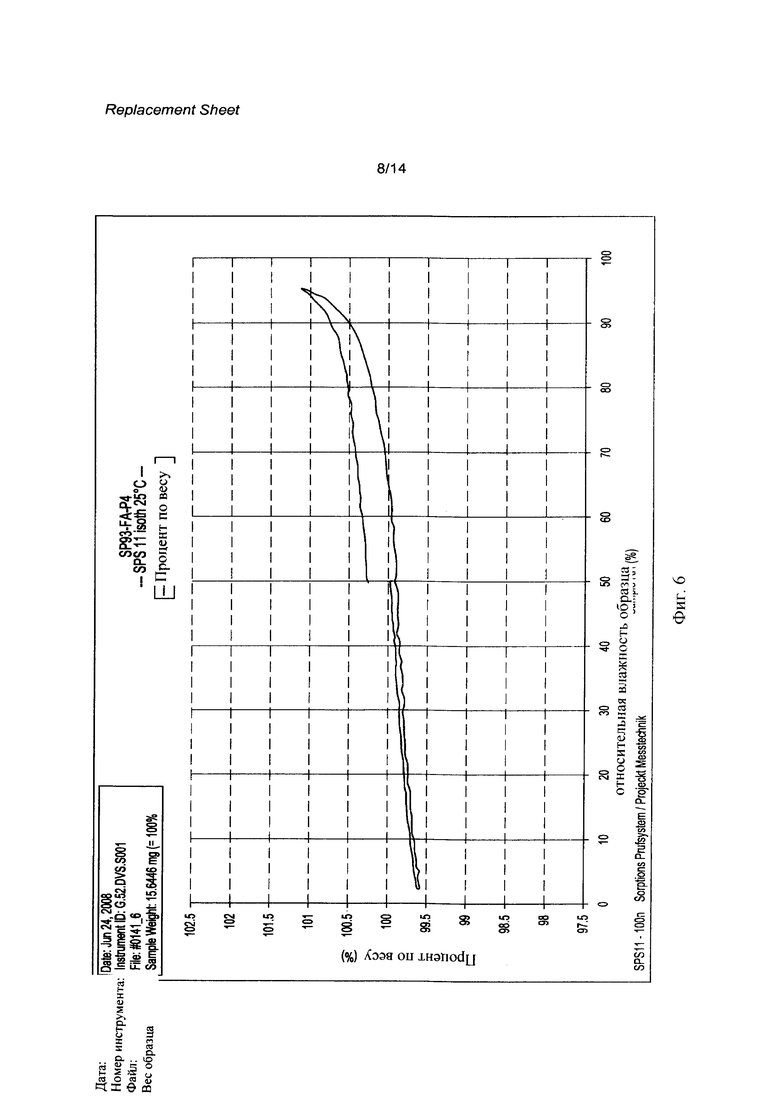
[0023] Фигура 6 представляет собой диаграмму, демонстрирующую свойства динамической сорбции паров (DVS) кристаллического соединения 1 (R=РО(ОН)2).
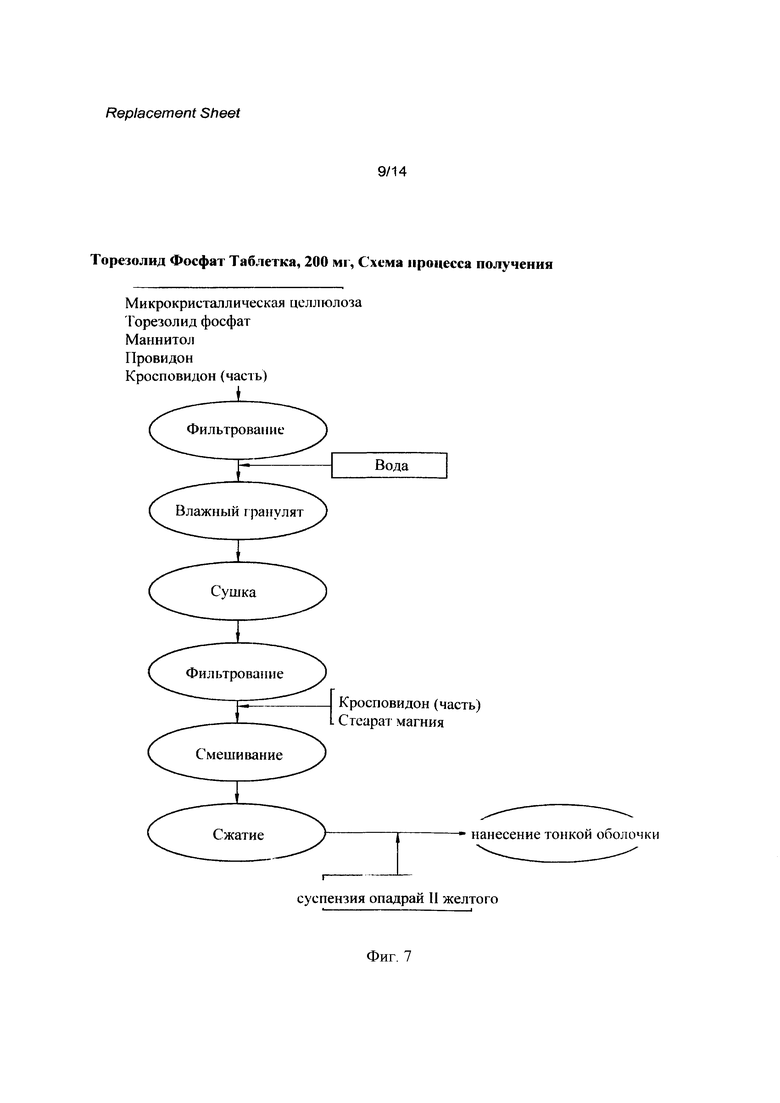
[0024] Фигура 7 представляет собой схематический процесс получения соединения 1 (R=РО(ОН)2) (TR-701 FA) в форме таблеток.
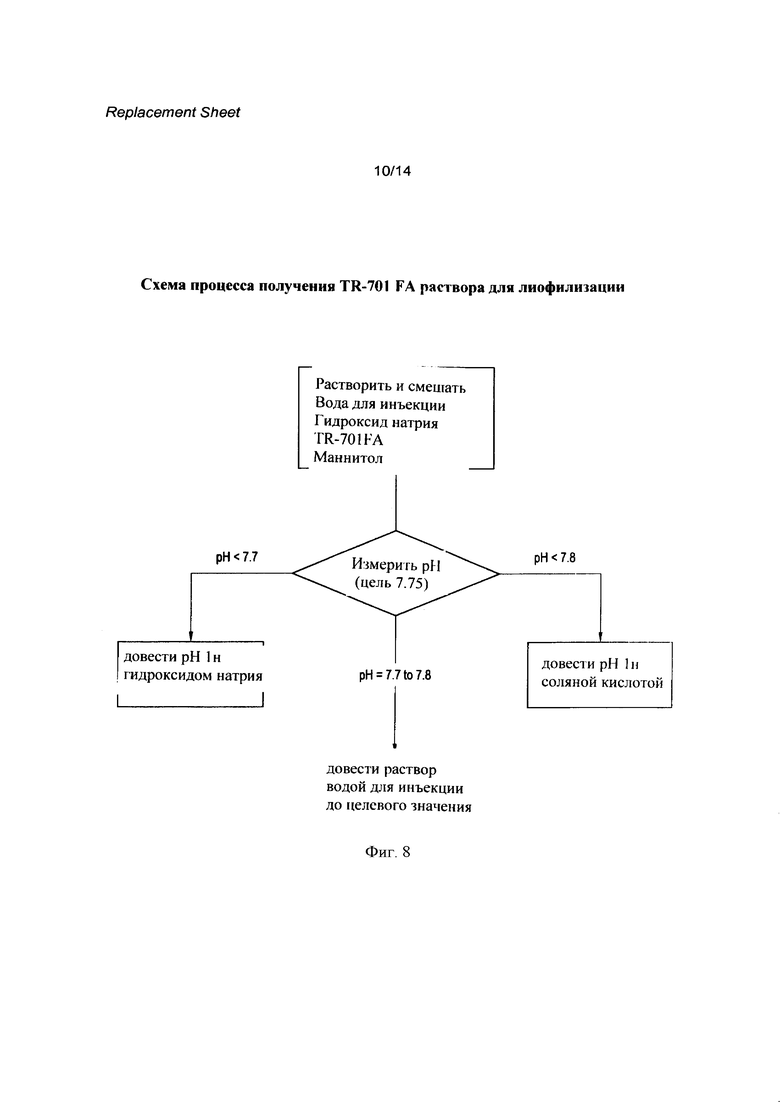
[0025] Фигура 8 представляет собой схематический процесс получения соединения 1 (R=РО(ОН)2) (TR-701 FA) в виде раствора для лиофилизации.
[0026] Фигура 9 представляет собой схематический процесс получения соединения 1 (R=РО(ОН)2) (TR-701 FA) для инъекции, 200 мг/ампула: стерильная фильтрация, наполнение и лиофилизация.
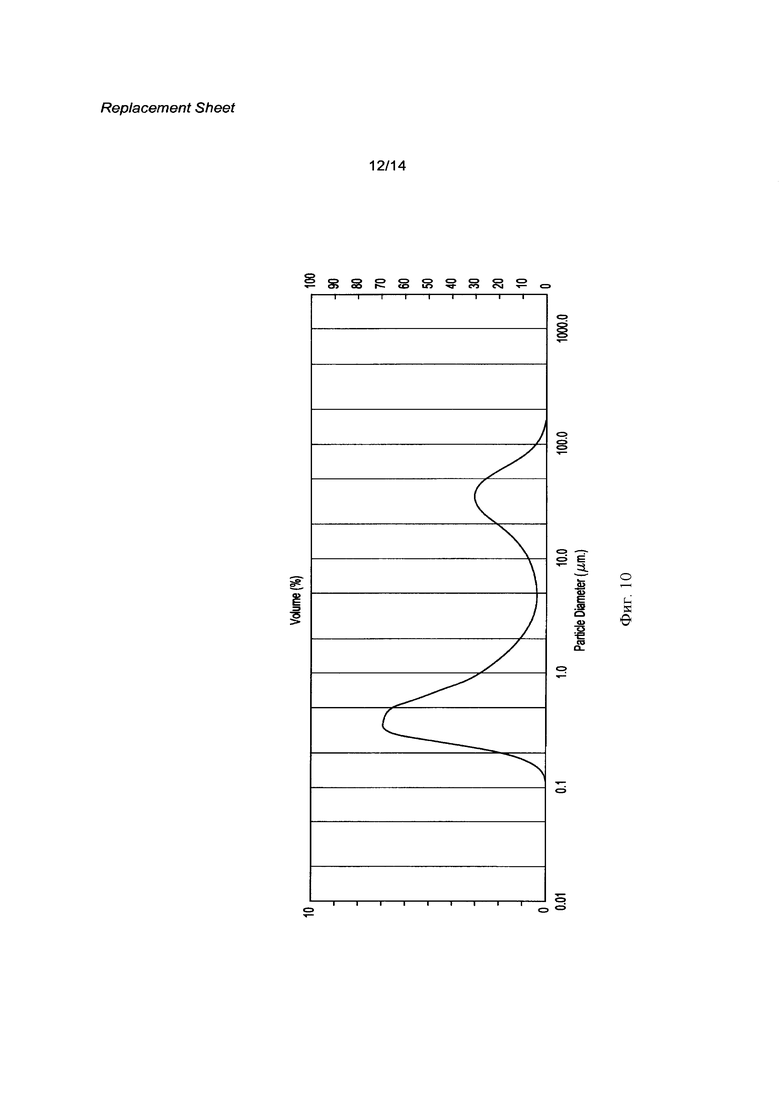
[0027] Фигура 10 представляет собой распределение частиц по размерам кристаллической свободной кислоты без контролирования размера частиц как описано здесь.
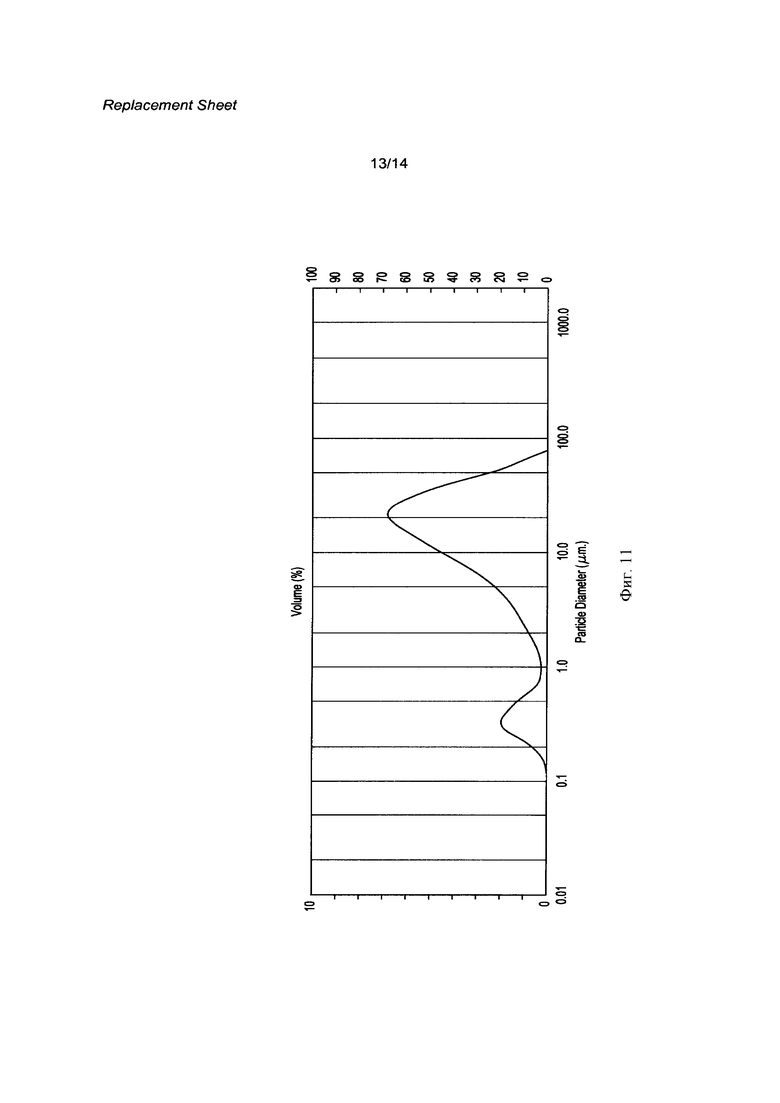
[0028] Фигура 11 представляет собой распределение частиц по размерам кристаллической свободной кислоты, выполненное используя лабораторные процессы для контроля размера частиц, представленное здесь.
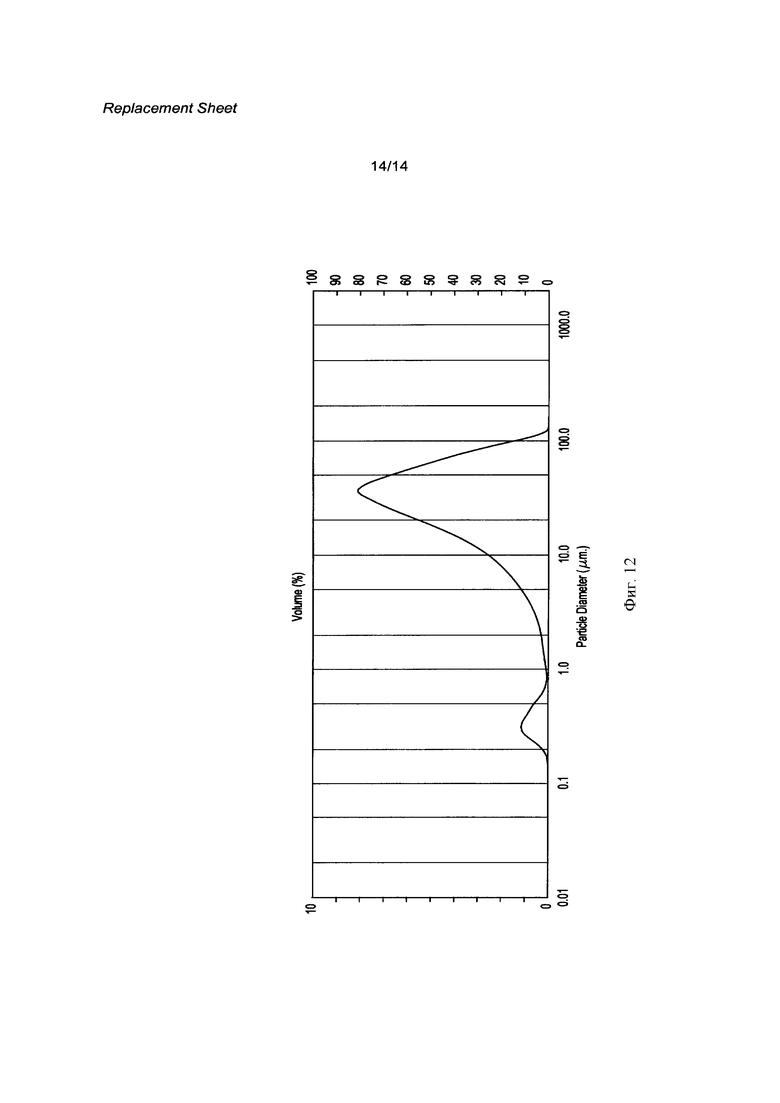
[0029] Фигура 12 распределение частиц по размерам кристаллической свободной кислоты, используя увеличенный в масштабе производственный процесс для контроля размера частиц, описанный здесь.
ДЕТАЛЬНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ.
[0030] R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он диводород фосфат 1 (R=РО(ОН)2), который иногда именуется как «свободная кислота» или "TR-701 FA" и некоторые его соли были получены в различных условиях кристаллизации для определения какой из материалов образует самое стабильное и наименее гигроскопичное кристаллическое соединение. Эмпирический процесс получения кристаллических форм свободной кислоты и ее солей привел к отбору кристаллической свободной кислоты, которая дополнительно к лучшей стабильности и отсутствию гигроскопичности, могла быть воспроизведена в различных условиях кристаллизации, затем эта форма была очищена и высушена.
[0031] Большинство из солей, которые были проверены, было трудно получить в кристаллической форме или они были нестабильны в очищенной или высушенной форме. Например, в отношении моно-натриевой соли, получение стабильного гидрата не было обнаружено. Также материал содержал свыше 10% по весу воды и таким образом материал был очень гигроскопичным, и при этом не стабильным для желаемого использования.
[0032] Кристаллический гидрат динатриевой соли был получен, но он оказался нестабильным и содержал 19.6% воды по весу. Однако динатриевая соль оказалась очень хорошо растворимой. В результате высушивания гидрата аморфные образцы были получены. Содержание воды в аморфном образце составило 6.2% по весу.
[0033] Кристаллический твердый материал не был выделен для дикалиевой соли.
[0034] Полу-кальциевая соль была получена в виде кристалла, однако, она была слишком гигроскопичной.
[0035] Кристаллический материал полу-магниевой соли был получен, но как оказалось, он содержал различные гидраты соли, а присутствие различных полиморфов является нежелательным для применения в лекарственных формах. В одном эксперименте, магниевая соль имела температуру плавления 152.8°С, что указывает на меньшую стабильность этого материала по сравнению со свободной кислотой.
[0036] Полученные кристаллы свободной кислоты, которые не были гигроскопичными при фильтровании и высушивании, продемонстрировали растворимость в 0.1 мг/мл (рН=3.2 насыщенного раствора). Температура плавления кристаллического материала составила 255-258°С, и таким образом материал был очень стабилен при относительно высокой температуре.
[0037] В целом условия кристаллизации являются обычно критическими для формирования конкретного полиморфа; однако, неожиданно оказалось, что один и тот полиморф свободной кислоты был получен в различных условиях, в которых кристаллическая свободная кислота образовывалась.
[0038] В некоторых вариантах осуществления изобретения, кристаллический материал является не гигроскопичным, поэтому он не поглощает и удерживает воду из атмосферного воздуха. В некоторых вариантах осуществления изобретения, «не гигроскопичный» материал имеет содержание воды менее приблизительно 5%. 4%, 3%, 2%, 1%, 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2% или 0.1% воды по весу.
[0039] Преимущественно, свободная кислота может быть использована как для получения твердой лекарственной формы, так и формы для внутривенного (IV) введения. При анализе было обнаружено, что динатриевая соль, хотя и является нестабильной для твердых форм, таких как таблетки, была очень хорошо растворимой и подходящей для форм IV введения. При этом в других вариантах осуществления изобретения, стерильный лиофилизированный порошок для инъекций был получен из динатриевой соли in situ с гидроксидом натрия и последующей лиофилизацией полученного раствора. Динатриевая соль является хорошо растворимой, и поэтому лучше восстанавливать ее стерильной водой с получением раствора. В некоторых вариантах осуществления изобретения полученный раствор может быть добавлен в емкость для внутривенного введения. Емкость может содержать изотонический раствор, такой как 0.9% хлорид натрия или 5% декстрозу.
[0040] В некоторых вариантах осуществления изобретения, солевой раствор, такой как динатриевая или мононатриевая соль, может быть лиофилизован путем замораживания раствора в лиофилизаторе до приблизительно -50 -30°С со скоростью около 0.1-1 градуса/минуту и выдерживания в течение 200-700 минут, в этой точке в камере лиофилизатора создается давление около 200-250 миллиторр и температура увеличивается до -30 -10°С со скоростью 0.5-3 градуса/минуту. Продукт выдерживается при -30 -10°С в течение приблизительно 1000-2500 минут и затем температуру увеличивают до приблизительно 21-35°С со скоростью 0.1-1 градус/минуту и выдерживают в течение 1000-2500 минут с получением желаемого продукта.
[0041] В вариантах некоторых способов получения, кристаллическая свободная кислота (R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он диводород фосфат 1 (R=РО(ОН)2) может быть получена подкислением водного раствора соответствующей соли, такой как динатриевая соль 1 (R=PO(ONa)2).
[0042] Любая соль свободной кислоты 1 (R=РО(ОН)2) может быть использована для восстановления свободной кислоты с помощью подкисления. В некоторых вариантах осуществления, соль представляет собой соль щелочного или щелочноземельного металла. В других вариантах осуществления изобретения, соль представляет собой соль щелочного металла, такая как динатриевая соль 1 (R=РО(ОН)2).
[0043] Было обнаружено, что выбор кислоты не является критическим. Любая кислота является кислотообразующей в достаточной степени для двойного протонирования динатриевого фосфата 1 (R=PO(ONa)2), или другой соли с получением свободной кислоты 1 (R=РО(ОН)2). В некоторых вариантах осуществления изобретения кислота представляет собой HCl, HBr или H2SO4.
[0044] После растворения соли (R)-3-(4-(2-(2-метил-тетразол-5-ил)-пиридин-5-ил)-3-фтор-фенил)-5-гидрокси-метил оксазолидин-2-он диводород фосфата, и затем, подкисления солевого раствора с образованием кристаллов, кристаллы могут быть отфильтрованы из супернатанта. В некоторых вариантах осуществления изобретения, влажные кристаллы могут быть высушены, например, используя вакуум или лиофилизацию кристаллов.
[0045] В некоторых вариантах осуществления изобретения термин кристаллический относится к однородному кристаллическому материалу кристаллического (R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он диводород фосфата, таким как по существу чистые кристаллы.
[0046] Термины "приблизительно, "около" и "по существу" при их использовании здесь для указания количества, близкого к заявляемому, при котором все еще выполняется желаемая функция или достигается желаемый результат. Например, термины "приблизительно, "около" и "по существу" могут относиться к количеству, которое отличается от заявляемого значения в пределах менее 10%, менее 5%, менее 1%, менее 0.1% и менее 0.01%.
[0047] Например, в фармацевтической промышленности, стандартной практикой является обеспечение по существу чистого материала при изготовлении фармацевтических композиций. Таким образом, в некоторых вариантах осуществления изобретения «по существу чистый» относится к степени чистоты, необходимой для получения лекарственных средств, которые могут включать, например, небольшое количество аморфного или другого материала, при этом материал все еще обладает достаточной способности к переносу, не является гигроскопичным и имеет чистоту, подходящую для фармацевтического применения. В некоторых вариантах осуществления изобретения кристаллическая свободная кислота, которая является по существу чистой, содержит, по меньшей мере, около 96% кристаллической свободной кислоты по весу, например, по меньшей мере, приблизительно 96.1%, 96.2%, 96.3%, 96.4%, 96.5%, 96.6%, 96.7%, 96.8%, 96.9%, 97%, 97.1%, 97.2%, 97.3%, 97.4%, 97.5%, 97.6%, 97.7%, 97.8%. 97.9%, 98%, 98.1%, 98.2%, 98.3%, 98.4%, 98.5%, 98.6%, 98.7%, 98.8%, 98.9%, 99%, 99.1%, 99.2%, 99.3%, 99.4%, 99.5%, 99.6%, 99.7%, 99.8%, 99.9% или 100% кристаллической свободной кислоты по весу. В некоторых вариантах осуществления изобретения ди- или мононатриевая соль в фармацевтических составах, описанных здесь, содержит, по меньшей мере, 96%, 96.1%, 96.2%, 96.3%, 96.4%, 96.5%, 96.6%, 96.7%, 96.8%, 96.9%. 97%, 97.1%, 97.2%, 97.3%, 97.4%, 97.5%, 97.6%, 97.7%, 97.8%, 97.9%, 98%, 98.1%, 98.2%, 98.3%, 98.4%, 98.5%, 98.6%, 98.7%, 98.8%, 98.9%, 99%, 99.1%, 99.2%, 99.3%, 99.4%, 99.5%, 99.6%, 99.7%, 99.8%, 99.9% или 100% кристаллической соли по весу. В полученных лекарственных средствах является полезным обеспечить не вязкий твердый кристаллический (R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он диводород фосфат, который может быть перенесен и аккуратно взвешен для применения, например, в таблетках и капсулах. Таким образом, в некоторых вариантах осуществления изобретения, кристаллический материал находится в переносимой форме так, что частицы не сильно прилипают одна к другой или к сосуду, в котором они находятся так, что возможно однородно перенести материал из сосуда.
[0048] Получение свободной кислоты (R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он диводород фосфата 1 (R=РО(ОН)2) и его динатриевой соли 1 (R=РО(ONa)2) описано в патентной публикации No. 2007/0155798 и патентной заявке США No. 12/577,089, права на последнюю из указанных заявок переданы тому же правообладателю, что и у настоящей заявки.
[0049] В вариантах осуществления изобретения в некоторых способах получения, кристаллическая свободная кислота (R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он диводород фосфат 1 (R=РО(ОН)2) может быть получена подкислением водного раствора соответствующей соли, например, динатриевой соли 1 (R=PO(ONa)2).
[0050] Любая соль свободной кислоты 1 (R=РО(ОН)2) может быть использована для восстановления свободной кислоты подкислением. В некоторых вариантах осуществления изобретения, соль представляет собой соль щелочного или щелочноземельного металла. В других вариантах осуществления изобретения, соль представляет собой соль щелочного металла, такого как динатриевая соль 1 (R=РО(ОН)2).
[0051] В дополнительных вариантах осуществления изобретения сама по себе свободная кислота может быть использована для получения кристаллической формы например, растворением в диполярном апротонном растворителе: диметил сульфоксиде (ДМСО) или 1-метил-2-пирролидоне (NMP); а затем следует добавление растворителя, вызывающего кристаллизацию, такого как этанол, ацетон, ацетонитрил, диоксан, гептан, изопропиловый спирт, метанол, тетрагдрофуран, толуол, вода, дихлорметан, метил изобутил кетон и этилацетат. В некоторых вариантах осуществления изобретения растворители, применяемые для растворения или для кристаллизации, могут представлять собой чистый растворитель или смесь чистых растворителей или могут быть в форме жидкости, пара или второго слоя. В некоторых вариантах осуществления изобретения из последних двух случаев, растворитель, вызывающий кристаллизацию может быть использован согласно способу выращивания кристаллов диффузией из паровой фазы, или способом расслоения растворителей, оба из которых хорошо известны в уровне техники.
[0052] В дополнительных способах получения, свободная кислота может быть растворена, по меньшей мере, в одном диполярном апротонном растворителе, таком как ДМСО или NMP при повышенной температуре, и кристаллическая свободная кислота 1 (R=PO(OH)2) получается охлаждением раствора согласно хорошо известным способам из уровня техники. Растворитель может быть чистым сам по себе или представлять собой смесь чистых растворителей.
[0053] При получении лекарственных средств полезно обеспечить твердое кристаллическое соединение, которое может быть легко переведено в дозированные формы, например, таблетки. Также желательно сократить промежуток времени, необходимый для получения соединения. В некоторых вариантах осуществления изобретения для того, чтобы удовлетворить эти потребности, описан способ получения кристаллического соединения 1 (R=РО(ОН)2), который приводит к получению большого размера частиц, что значительно сокращает время фильтрования, поскольку, как известно, мелкие частицы замедляют процесс фильтрования. В других вариантах осуществления изобретения кристаллическое вещество 1 (R=РО(ОН)2) имеет определенное распределение частиц по размерам, что напрямую следует из способа получения вне зависимости от фильтрования материала для получения такого распределения.
[0054] В некоторых вариантах осуществления изобретения больший размер частиц кристаллов соединения 1 (R=РО(ОН)2) может быть получен, используя процедуру осаждения при высокой температуре. Дополнительно, в вариантах, где кислота используется для получения свободной кислоты из соли, было обнаружено, что увеличение скорости, при которой реакционную смесь добавляют к кислоте, влияет на размер частиц и делает частицы больше. При этом реакционная смесь может взаимодействовать с раствором кислоты так быстро, как это возможно так, что по существу происходит незамедлительный контакт с раствором кислоты. В обычных способах реакционная смесь взаимодействует с раствором кислоты более медленно, потому что раствор кислоты добавляют к реакционной смеси и таким образом реакционная смесь не может взаимодействовать с раствором кислоты до некоторого времени после добавления раствора кислоты, что приводит к гораздо более маленькому размеру частиц. Было обнаружено, что действие обратное этой стадии, то есть добавление реакционной смеси к раствору кислоты позволяет реакционной смеси практически незамедлительно взаимодействовать за время введения реакционной смеси в раствор кислоты, что приводит к получению материала с большим размером частиц. В некоторых вариантах осуществления изобретения незамедлительный контакт осуществляют добавлением реакционной смеси к раствору кислоты. Реакционная смесь может быть прокачана через раствор кислоты за промежуток времени, например, за несколько часов, например 1-4 часа.
[0055] В некоторых вариантах осуществления изобретения, водный этанол- или ТГФ-содержащий раствор TR-701FA может быть получен добавлением раствора бикарбоната натрия, например, 2-10% раствора по весу, такого как 5% раствор. В некоторых вариантах осуществления изобретения, раствор может быть добавлен к водному раствору кислоты и этанолу или ТГФ с образованием свободной кислоты. В некоторых вариантах осуществления изобретения приблизительно от 0.5-10, приблизительно 1.5-3.0 или приблизительно 2.2 эквивалента 1 М HCl может быть использовано. Дополнительно в некоторых вариантах осуществления изобретения, приблизительно 1-10 объемов, 2-6 объемов или 4 объема этанола могут быть использованы. ТГФ также может быть использован. В некоторых вариантах осуществления изобретения раствор, содержащий соляную кислоту и этанол, может поддерживаться при температуре около 40-100°С, около 60-70°С или около 65-70°С. Содержание кислоты и спирта могут быть приведены в соответствие. TR-701FA закристаллизовался во время этого добавления с получением меньшего количества небольших кристаллов в продукте по сравнению с ранее раскрытыми способами.
[0056] В некоторых вариантах осуществления изобретения этанол или ТГФ препятствуют гелеобразованию свободной кислоты во время процесса.
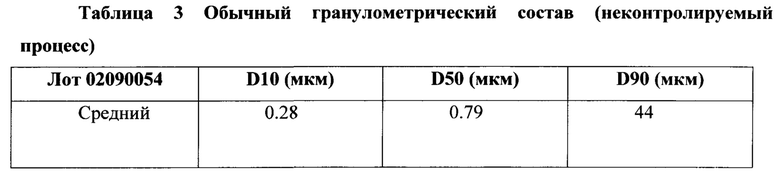
[0057] Обычно распределение частиц по размерам измеряется, используя анализатор лазерной дифракции размера частиц, а именно Malvern Mastersizer. D10 (мкм) представляет собой диаметр, ниже которого лежит 10% общего объема частиц. D50 (мкм) представляет собой медианный диаметр. D90 (мкм) представляет собой диаметр, ниже которого лежит 90% общего объема частиц.
[0058] В некоторых вариантах осуществления изобретения, когда размер частиц не контролируется, 10% общего объема частиц может иметь диаметр меньше приблизительно 0.28 мкм, медианный диаметр может быть около 0.79 мкм, и 90% общего объема частиц может иметь диаметр меньше приблизительно 0.44 мкм. Контроль (за увеличением) размера частиц, используя способы, описанные здесь, приводит к тому, что частицы становятся значительно больше по размеру в целом.
[0059] В некоторых вариантах осуществления изобретения, когда размер частиц контролируется, используя способы, описанные здесь, для увеличения размера частиц, 10% общего объема частиц может иметь средний диаметр, по меньшей мере, около 0.5 мкм, и/или медианный диаметр может быть, по меньшей мере, около 1.0 мкм, и/или 90% общего объема частиц могут иметь средний диаметр, по меньшей мере, около 45 мкм. В некоторых вариантах осуществления изобретения, когда размер частиц контролируется (для увеличения размера частиц), 10% общего объема может иметь средний диаметр около 0.5-10 мкм, например, около 1-5 мкм. Например, когда размер частиц контролируется (для увеличения размера частиц), 10% общего объема может иметь средний диаметр около 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2.0, 2.1, 2.2, 2.3, 2.4, 2.5, 2.6, 2.7, 2.8, 2.9, 3.0, 3.1, 3.2, 3.3, 3.4, 3.5, 3.6, 3.7, 3.8, 3.9, 4.0, 4.1, 4.2, 4.3, 4.4, 4.5, 4.6, 4.7, 4.8, 4.9, 5.0, 5.1, 5.2, 5.3, 5.4, 5.5, 5.6, 5.7, 5.8, 5.9, 6.0, 6.1, 6.2, 6.3, 6.4, 6.5, 6.6, 6.7, 6.8, 6.9, 7.0, 7.1, 7.2, 7.3, 7.4, 7.5, 7.6, 7.7, 7.8, 7.9, 8.0, 8.1, 8.2, 8.3, 8.4, 8.5, 8.6, 8.7, 8.8, 8.9, 9.0, 9.1, 9.2, 9.3, 9.4, 9.5, 9.6, 9.7, 9.8, 9.9 или 10.0 мкм.
[0060] В некоторых вариантах осуществления изобретения, когда размер частиц контролируется (для увеличения размера частиц), медианный диаметр может быть больше, чем приблизительно 1.0 мкм и иметь средний медианный диаметр приблизительно 1-44 мкм, приблизительно 1-40 мкм, приблизительно 10-35 мкм, приблизительно 20-30 мкм или около 25-29, например, около 27 мкм. В некоторых вариантах осуществления изобретения, когда размер частиц контролируется для увеличения размера частиц, средний медианный диаметр может быть приблизительно равен 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 38, 39, 40, 41, 42, 43 или 44 мкм. Например, средний медианный диаметр может быть равен приблизительно 25, 25.1, 25.2, 25.3, 25.4, 25.5, 25.6, 25.7, 25.8, 25.9, 26, 26.1, 26.2, 26.3, 26.4, 26.5, 26.6, 26.7, 26.8, 26.9, 27, 27.1, 27.2, 27.3, 27.4, 27.5, 27.6, 27.7, 27.8, 27.9, 28, 28.1, 28.2, 28.3, 28.4, 28.5, 28.6, 28.7, 28.8, 28.9 или 29 мкм.
[0061] В некоторых вариантах осуществления изобретения, когда размер частиц контролируется, 90% общего объема частиц может иметь средний диаметр, по меньшей мере, около 45 мкм, такой как приблизительно 45-100, приблизительно 45-80, приблизительно 55-75 или приблизительно 64-68, например, приблизительно 66. В некоторых вариантах осуществления изобретения, когда размер частиц контролируется, 90% общего объема частиц может иметь средний диаметр около 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99 или 100 мкм. Например, 90% 90% общего объема частиц может иметь средний диаметр приблизительно 64, 64.1, 64.2, 64.3, 64.4, 64.5, 64.6, 64.7, 64.8, 64.9, 65, 65.1, 65.2, 65.3, 65.4, 65.5, 65.6, 65.7, 65.8, 65.9, 66, 66.1, 66.2, 66.3, 66.4, 66.5, 66.6, 66.7, 66.8, 66.9, 67, 67.1, 67.2, 67.3, 67.4, 67.5, 67.6, 67.7, 67.8, 67.9 или 68 мкм.
[0062] Кристаллическая свободная кислота 1 (R=РО(ОН)2) может быть охарактеризована Фурье-Раман спектром, как показано на Фигуре 1 и рентгенограммой, как показано на Фигуре 2, с соответствующими данными, представленными в Таблице 1 и 2 соответственно. На Фигуре 3, Фигуре 4, Фигуре 5 и Фигуре 6 показаны термограмма дифференциальной сканирующей калориметрии ⋅ (DSC), 1Н ЯМР спектр, диаграмма, полученная методами термогравиметрии и инфракрасной спектроскопии, и диаграмма, демонстрирующая свойства динамической сорбции паров (DVS) соответственно.
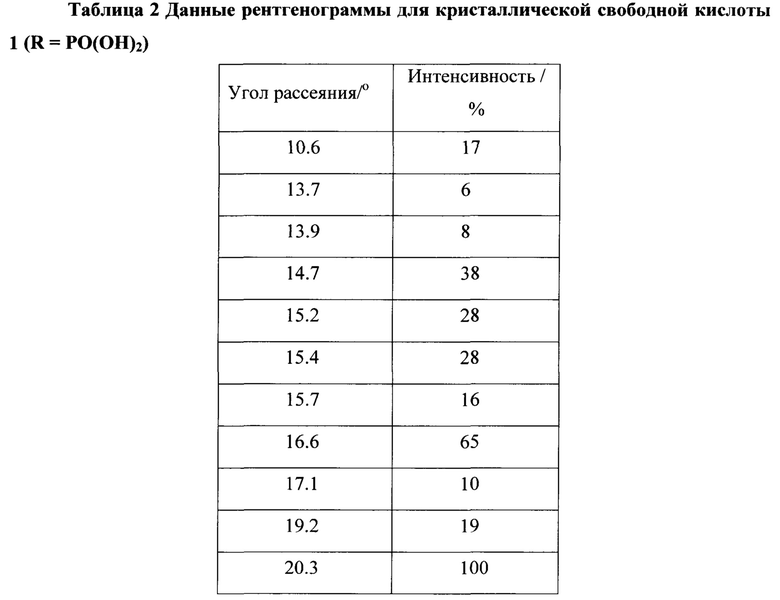
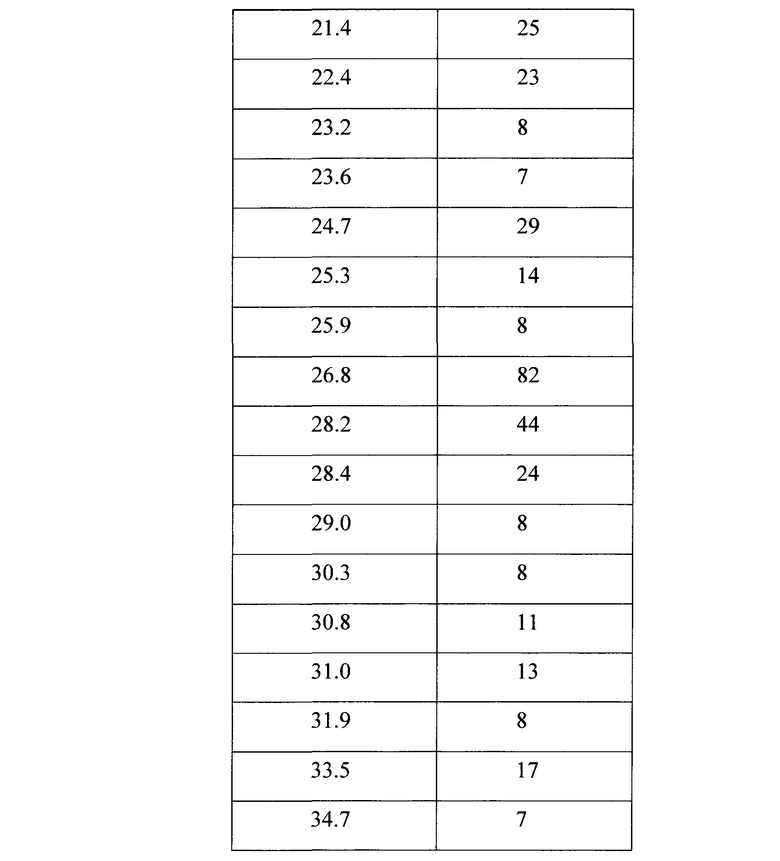
[0063] В некоторых вариантах осуществления изобретения, отличительные пики для кристаллической свободной кислоты включают следующие пики: 14.7°, 15.2°, 16.6°, 20.3°, 26.8° и 28.2°.
[0064] В других вариантах осуществления изобретения, отличительные пики для кристаллической свободной кислоты включают следующие пики: 10.6°, 13.9°, 14.7°, 15.2°, 16.6°, 20.3°, 26.8° и 28.2°.
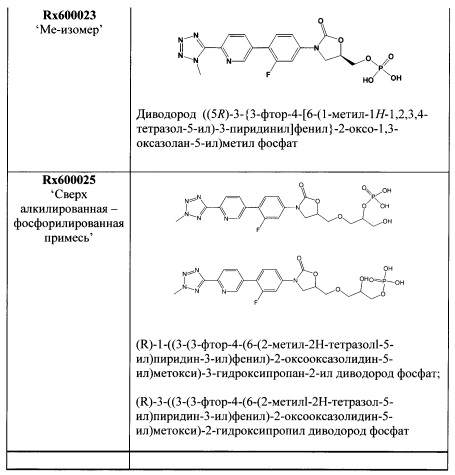
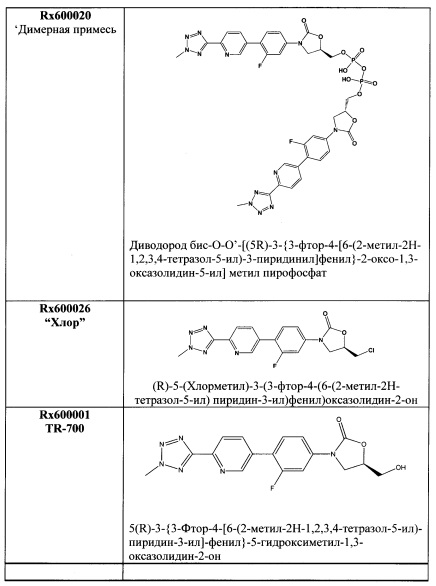
[0065] В некоторых вариантах осуществления изобретения кристаллическая свободная кислота содержит примеси, которые присутствуют в количестве меньше 1% очищенной кристаллической свободной кислоты. Эти примеси включают
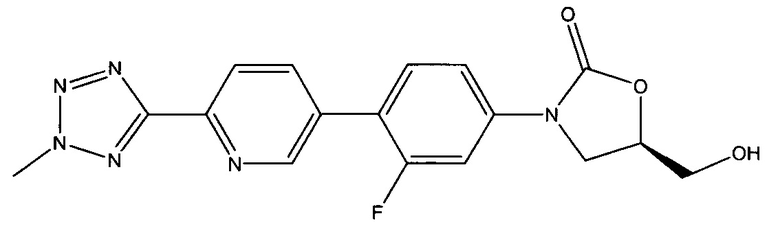
то есть 5(R)-3-{3-фтор-4-[6-(2-метил-2Н-1,2,3,4-тетразол-5-ил)-пиридин-3-ил]-фенил}-5-гидроксиметил-1,3-оксазолидин-2-он ("TR-700") и/или
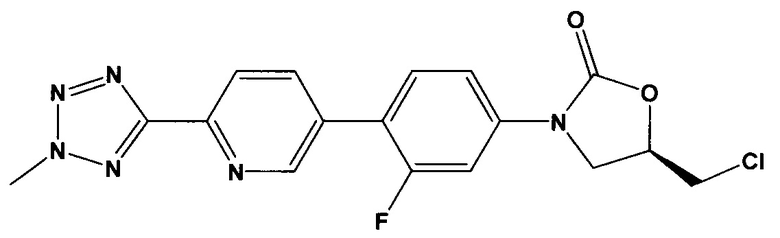
то есть (R)-5-(хлорметил)-3-(3-фтор-4-(6-(2-метил-2Н-тетразол-5-ил)пиридин-3-ил)фенил)оксазолидин-2-он ("хлор содержащая примесь").
[0066] Из обычно получаемого материала, имеющего примеси, которые были идентифицированы, используя ВЭЖХ в Примере 15, по меньшей мере, 2% по весу хлор содержащей примеси присутствует. В очищенной кристаллической свободной кислоте, полученной, используя способ получения свободной кислоты, описанной в патентной заявке США 12/577,089, права на которую были переданы тому же правообладателю, что и по настоящей заявке, и способы кристаллизации, описанные здесь, хлор содержащая примесь присутствует в количестве приблизительно меньше 1% по весу, например, меньше приблизительно 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2% или 0.1% по весу кристаллической свободной кислоты. В некоторых вариантах осуществления изобретения количество хлор содержащей примеси может быть снижено до 0.1% по весу, например, менее чем приблизительно 0.09%, 0.08%, 0.07%, 0.06%, 0.05%, 0.04%, 0.03%, 0.02% или 0.01% по весу кристаллической свободной кислоты. В некоторых вариантах осуществления изобретения очищенная кристаллическая свободная кислота вообще не содержит этой примеси.
[0067] Из обычно получаемого материала, имеющего примеси, которые были идентифицированы, используя ВЭЖХ в Примере 15, по меньшей мере, 1% по весу TR-700 примеси присутствует. В очищенной кристаллической свободной кислоте, полученной, используя способ получения свободной кислоты, описанный в патентной заявке США 12/577,089, права на которую были переданы тому же правообладателю, что и по настоящей заявке, и способы кристаллизации, описанные здесь, TR-700 примесь присутствует в количестве приблизительно меньше 1% по весу. В некоторых вариантах осуществления изобретения кристаллическая свободная кислота содержит менее 0.9%, 0.8%, 0.7%, 0.6%, 0.5%, 0.4%, 0.3%, 0.2% или 0.1% по весу TR-700 примеси. В некоторых вариантах осуществления изобретения очищенная кристаллическая свободная кислота вообще не содержит TR-700 примеси.
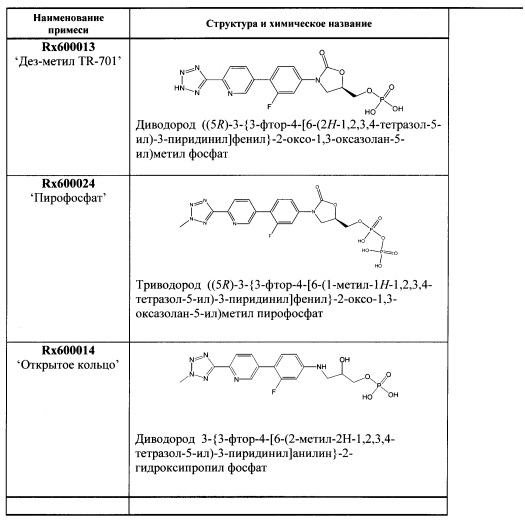
[0068] Дополнительно, очищенная кристаллическая свободная кислота, полученная, используя способ получения свободной кислоты, описанный в патентной заявке США 12/577,089, права на которую были переданы тому же правообладателю, что и по настоящей заявке, и способы кристаллизации, описанные здесь, может также отличаться от кристаллической свободной кислоты, полученной обычным способом присутствием следующих соединений. Например, следующие примеси не были обнаружены в образце кристаллической свободной кислоты, полученной обычным способом, как показано в Примере 15:
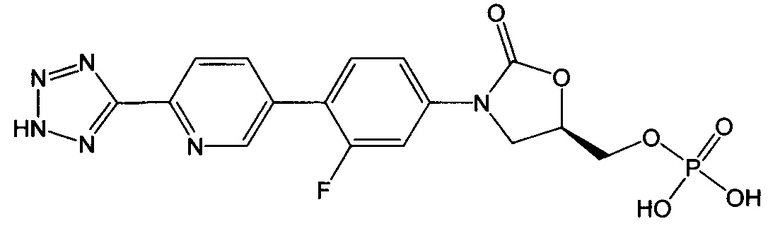
(здесь и далее "дез-метил TR-701"), то есть диводород ((5R)-3-{3-фтор-4-[6-(2H-1,2,3,4-тетразол-5-ил)-3-пиридинил]фенил}-2-оксо-1,3-оксазолан-5-ил)метил фосфат;
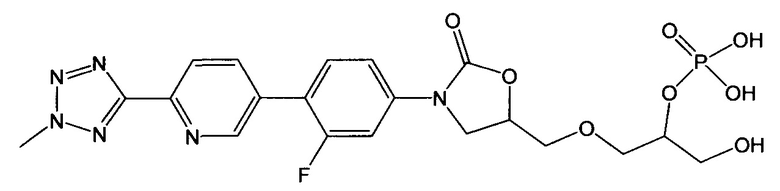
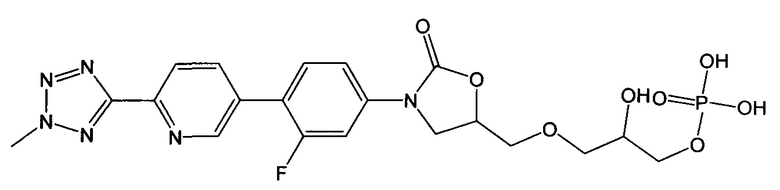
(здесь и далее "пералкилированная-фосфорилированная примесь"), то есть, 5-((3-(3-фтор-4-(6-(2-метил-2Н-тетразол-5-ил)пиридин-3-ил)фенил)-2-оксаоксазолидин-5-ил)метокси)-3-гидроксипропан-2-ил диводород фосфат и, 3-((3-(3-фтор-4-(6-(2-метил-2Н-тетразол-5-ил)пиридин-3-ил)фенил)-2-оксаоксазолидин-5-ил)метокси)-2-гидроксипропил диводород фосфат;
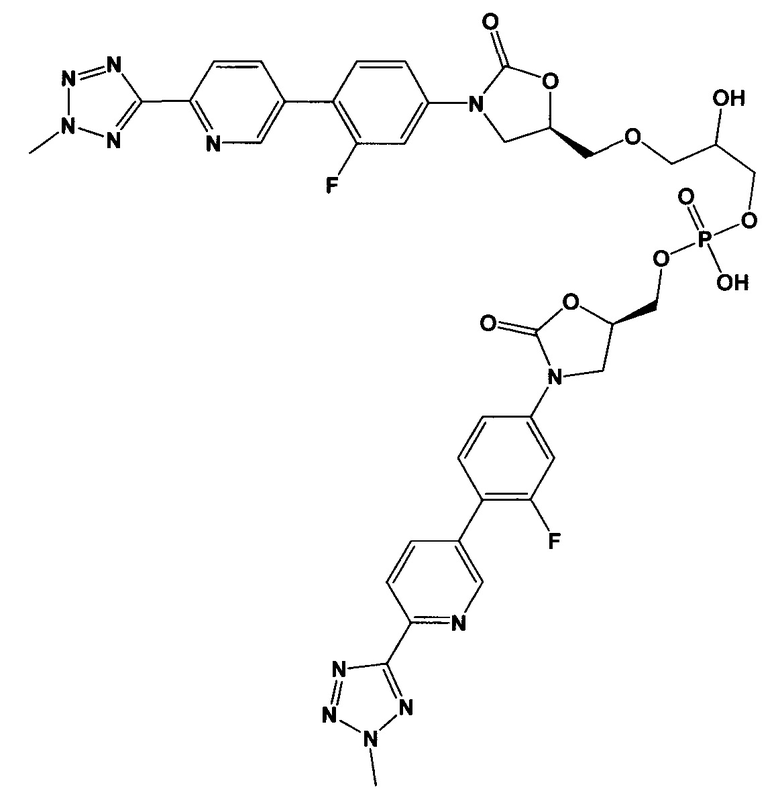
(здесь и далее "один из ОА-700 смешанных диэфиров"), то есть, 3-{[(5R)-3-{3-фтор-4-[6-(2-метил-2Н-тетразол-5-ил)пиридин-3-ил]фенил}-2-оксо-1,3-оксазолидин-5-ил]метокси}-2-гидроксипропил [(5R)-3-{3-фтор-4-[6-(2-метил-2Н-тетразол-5-ил)пиридин-3-ил]фенил}-2-оксо-1,3-оксазолидин-5-ил]метил водород фосфат; и/или
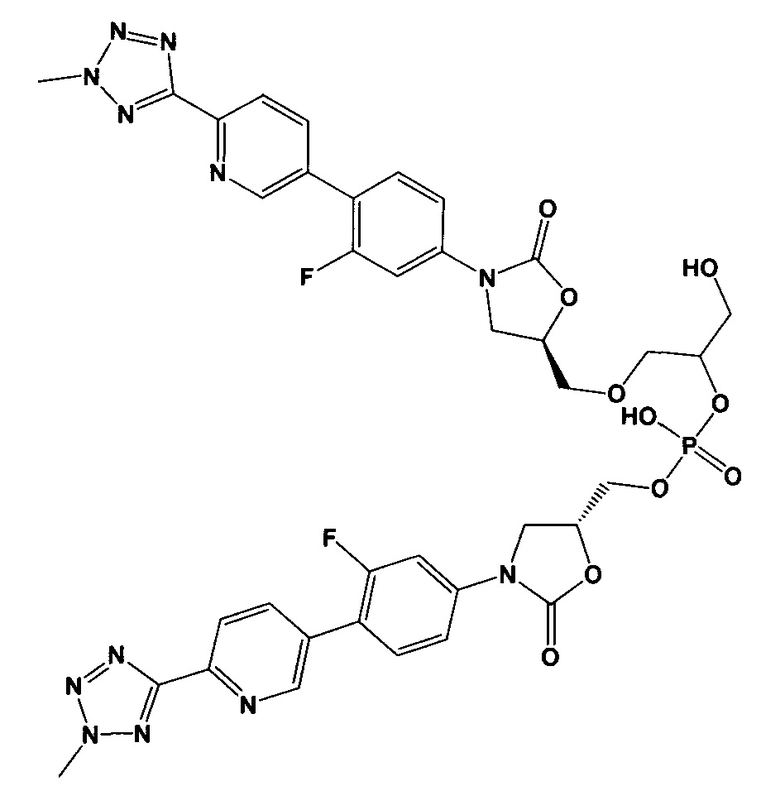
(здесь и далее "другой из ОА-700 смешанных диэфиров"), то есть 2-{[(5R)-3-{3-фтор-4-[6-(2-метил-2Н-тетразол-5-ил)пиридин-3-ил]фенил}-2-оксо-1,3-оксазолидин-5-ил]метокси}-1-гидроксиэтил [(5R)-3-{3-фтор-4-[6-(2-метил-2Н-тетразол-5-ил)пиридин-3-ил]фенил}-2-оксо-1,3-оксазолидин-5-ил]метил водород фосфат.
[0069] Специалисты в данной области определят, что различные замещенные изотопами варианты (например, через замещение водорода дейтерием, углерода - 13С, азота - 15N, или фосфора - 32Р) также могут быть легко получены. Все такие варианты включены в объем притязаний по изобретению.
[0070] В различных вариантах осуществления изобретения очищенная кристаллическая свободная кислота, описанная здесь, может быть использована отдельно, в комбинации с другими соединениями, описанными здесь, или в комбинации с одним или более другими агентами, активными в терапевтической области, описанной здесь.
[0071] В другом аспекте настоящее описание относится к фармацевтической композиции, содержащей один или несколько физиологически приемлемых поверхностно-активных агентов, дополнительных носителей, дилюентов, эксципиентов, разглаживающих компонентов, суспензионных агентов, агентов, образующих пленку и вспомогательных веществ, для образования оболочки или их комбинации и композиции, описанной здесь. Дополнительные приемлемые носители или дилюенты для терапевтического использования хорошо известны в фармацевтической области, и описаны, например, в Remington's Pharmaceutical Sciences, 18th Ed., Mack Publishing Co., Easton, PA (1990), который приводится здесь для ссылки во всей его целостности. Консерванты, стабилизаторы, красители, подсластители, отдушки, ароматизаторы и тому подобное могут быть представлены в фармацевтической композиции. Например, бензоат натрия, аскорбиновая кислота и эфиры п-гидроксибензойной кислоты могут быть добавлены в качестве консервантов. Дополнительно, антиоксиданты и суспендирующие агенты могут быть использованы. В различных вариантах осуществления изобретения спирты, эфиры, сульфурированные алифатические спирты и тому подобное могут быть использованы в качестве поверхностно-активных агентов; сахароза, глюкоза, лактоза, крахмал, микрокристаллическая целлюлоза, кристаллическая целлюлоза, маннитол, светлый безводный силикат, алюминат магния, метасиликат алюминат магния, синтетический силикат алюминия, карбонат кальция, гидрокарбнат натрия, гидрофосфат кальция, кальций карбоксиметил целлюлоза, и тому подобное могут быть использованы в качестве экципиентов; стеарат магния, тальк, закаленное масло и тому подобное могут быть использованы в качестве разглаживающих агентов; кокосовое масло, оливковое масло, кунжутное масло, арахисовое масло, соевое масло могут быть использованы в качестве суспензионных агентов или лубрикантов; фталат ацетата целлюлозы в виде производного углевода, такого как целлюлоза или сахароза или метилацетат-метакрилат сополимер в виде производного поливинила может быть использован в качестве суспензионного агента; и пластификаторы, такие как эфир фталаты и тому подобное могут быть в качестве суспензионных агентов.
[0072] Термин "фармацевтическая композиция" относится к смеси соединения, описанного здесь с другими химическими компонентами, такими как дилюенты или дополнительные носители. Фармацевтическая композиция облегчает введение соединения в организм. Многочисленные техники введения фармацевтической композиции существуют в области техники, включая, без ограничения, пероральный, инъекционный, аэрозольный, парентеральный и локальный. Фармацевтические композиции могут также быть получены взаимодействием свободной кислоты с неорганическим или органическим основанием, таким как гидроксид натрия или гидроксид магния. В некоторых вариантах осуществления изобретения фармацевтически приемлемые соли соединений, описанных здесь (например, полученных in situ во время изготовления внутривенной фармацевтической формы) представлены. В некоторых вариантах осуществления изобретения гидроксид натрия используется для получения лиофилизированного порошка, который содержит соль свободной кислоты, получаемой in situ.
[0073] Термин «носитель» относится к химическому соединению, которое облегчает введение соединения в клетки или ткани.
[0074] Термин "дилюент" относится к химическим соединениям, разбавленным водой, которые растворяют целевую композицию, а также стабилизируют биологически активную форму соединения. Соли, растворенные в буферных растворах, используются в качестве дилюентов в уровне техники. Одним из широко используемых буферных растворов является солевой раствор фосфатного буфера, потому что он имитирует состав солей в человеческой крови. Поскольку буферные соли могут контролировать рН раствора при низкой концентрации, буферный дилюент редко изменяет биологическую активность соединения. Как используется здесь, термин «экципиент» относится к инертному веществу, которое придает композиции, без ограничения, объем, устойчивость, стабильность, связывающую способность, увлажнение, способность к распаду. "Дилюент" представляет собой вид экципиента.
[0075] Термин "физиологически приемлемый" относится к носителю или дилюенту, который не изменяет биологическую активность или свойства соединения.
[0076] Фармацевтические соединения, описанные здесь, могут быть введены человеку (пациенту) самостоятельно или в составе фармацевтической композиции, где они смешиваются с другими активными ингредиентами, в качестве комбинированной терапии или подходящими носителями или экципиентом (экципиентами). Техники изготовления и введения соединений по настоящей заявке могут быть обнаружены в "Remington's Pharmaceutical Sciences," Mack Publishing Co., Easton, PA, 18th edition, 1990.
[0077] Подходящие способы введения могут включать, например, пероральный, ректальный, введение через слизистую оболочку, локальный или интестинальный способ; парентеральную доставку, включая внутримышечную, внутривенную, интрамедуллярную инъекцию, также интратекальную, прямую интравентрикулярную, интраперитонеальную, интраназальную или внутриглазную инъекцию. Соединение может быть введено в виде дозированной формы с продолжительным или контролируемым высвобождением, включая, инъекцию веществ замедленного всасывания, осмотические насосы, таблетки, трансдермальные (включая электротранспорт) участки и тому подобное, для пролонгированного и/или временного, импульсного введения с заранее определенной скоростью.
[0078] Фармацевтические композиции по настоящему изобретению могут быть изготовлены известным способом, например с помощью обычного перемешивания, растворения, гранулирования, изготовления драже, растирания в порошок, эмульсификации, капсулирования или таблетирования.
[0079] Фармацевтические композиции могут быть получены обычным способом, используя один или несколько физиологически приемлемых носителей, включающие экципиенты и вспомогательные вещества, которые облегчают подготовку активных соединений для включения в форму, которая может быть использована фармацевтически. Состав зависит от выбранного способа введения. Любая из хорошо известных техник, дилюентов, носителей и эксципиентов могут быть использованы в качестве подходящих и понятных, например, указанные в Remington's Pharmaceutical Sciences, смотреть выше.
[0080] Инъецируемые вещества могут быть получены в обычных формах, или в виде жидких растворов или суспензий, твердых формах, подходящих для растворения или суспендирования в жидкости до инъекции или в виде эмульсий. Подходящие эксципиенты представляют собой, например, воду, солевой раствор, декстрозу, маннитол, лактозу, лецитин, альбумин, глютамат натрия, гидрохлорид цистеина и тому подобное. Дополнительно, если желательно, инъецируемые фармацевтические композиции могут содержать небольшие количества нетоксичных вспомогательных веществ, таких как увлажняющие агенты, рН буферные агенты и тому подобное. Физиологически совместимые буферы включают, без ограничения, раствор Хэнкса (Hanks), раствор Ринджера (Ringer) или физиологического солевого раствора. Если желательно, абсорбция, улучшающая изготовление может быть использована.
[0081] Для введение через слизистую оболочку, проникающие агенты, необходимые для проникновения через барьер, могут быть использованы в формах.
[0082] Фармацевтические формы для парентерального введения, например, с помощью болюсной инъекции или непрерывным вливанием, включает водные растворы активных соединений в водорастворимой форме. Дополнительно, суспензии активных соединений могут быть получены в виде подходящих масляных инъекционных суспензий. Водные инъекционные суспензии могут содержать вещества, которые увеличивают вязкость суспензии, такие как натрий карбоксиметилцеллюлоза, сорбитол или декстран. Необязательно суспензия может также содержать подходящие стабилизаторы или агенты, которые увеличивают растворимость соединений, позволяющих получить высоко концентрированные растворы. Составы для инъекций могут быть представлены в единичной дозированной форме, например, в ампулах или контейнерах, содержащих несколько доз, с добавлением консерванта. Композиции могут содержать такие формы в виде суспензий, растворов или эмульсий в масляном или водном наполнителе, и могут содержать агенты для получения состава, например, суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно, перед применением активный ингредиент может быть в виде порошка для получения лекарственной формы с подходящим наполнителем, например, стерильной апирогенной водой.
[0083] Для перорального введения композиция может быть составлена комбинированием представляющей интерес композиции и фармацевтически приемлемых носителей, хорошо известных в уровне техники. Такие носители могут быть использованы дополнительно к катионному полимерному носителю, что дает возможность приготовить композиции по изобретению в виде таблеток, пилюль, драже, жидкостей, гелей, сиропов, суспензий и тому подобное для перорального приема пациентов, нуждающимся в лечении. Фармацевтические формы для перорального использования могут быть получены комбинированием активных соединений с твердым экципиентом, необязательным измельчением полученной смеси, и обработкой смеси гранул после добавления подходящих вспомогательных веществ, если желательно, получение таблеток или ядрышек драже. Подходящие эксципиенты представляют собой, в частности, наполнители, такие как сахара, включая лактозу, сахарозу, маннитол или сорбитол; целлюлозные формы, такие как кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовая камедь, метилцеллюлоза, гидроксипропилметил-целлюлоза, натрий карбоксиметилцеллюлоза и/или поливинилпирролидон (PVP), например, Povidone. Если желательно, дезинтегрирующие агенты могут быть добавлены, такие как кросс-сшитый поливинилпирролидон (например, Crospovidone), агар или альгиновая кислота или ее соль, такая как альгинат натрия. Ядрышки драже покрываются подходящей оболочкой. Для этой цели концентрированные растворы сахаров могут быть использованы, которые необязательно содержат гуммиарабик, тальк, поливинилпирролидон, карбополь гель, полиэтиленгликоль, и/или диоксид титана, лаковые растворы и подходящие органические растворители или смеси растворителей. Красители или пигменты могут быть добавлены к оболочке таблеток или драже для идентификации или для характеристики различных комбинаций доз активных соединений. Для этой цели концентрированные растворы сахаров могут быть использованы, которые могут необязательно содержать, гуммиарабик, тальк, поливинилпирролидон, карбополь гель, полиэтиленгликоль, и/или диоксид титана, лаковые растворы и подходящие органические растворители или смеси растворителей. Красители или пигменты могут быть добавлены к оболочке таблеток или драже для идентификации или для характеристики различных комбинаций доз активных соединений.
[0084] Фармацевтические формы, которые могут быть использованы перорально, включают push-fit капсулы (твердые капсулы из двух частей), выполненные из желатина, а также мягкие, герметичные капсулы из желатина и пластификатора, такого как глицерол или сорбитол. Твердые капсулы могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связующим веществом, таким как крахмалы и/или лубриканты, такие как тальк или стеарат магния и необязательно стабилизаторы. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, жидкие парафины или жидкие полиэтиленгликоли. Дополнительно стабилизаторы могут быть добавлены. Все составы для перорального введения должны быть в дозировке, подходящей для такого введения.
[0085] Для буккального введения, композиции могут быть выполнены в форме таблеток или пастилок обычным способом.
[0086] Для введения ингаляцией, композиция может быть обычным образом доставлена в форме аэрозоля из баллончика под давлением или распылителя, с использованием подходящего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, углекислого газа или другого подходящего газа. В случае аэрозоля под давлением дозированная единица может быть определена присутствием клапана для доставки фиксированного количества. Капсулы и картриджи, например, из желатина для использования в ингаляторе или инсуффляторе могут содержать порошковую смесь соединения и подходящей основы, например, лактоза или крахмал.
[0087] Также описываемые здесь различные фармацевтические композиции включают хорошо известные в фармацевтической области интраокулярную, интраназальную и интрааурикулярную доставку. Подходящие проникающие агенты для этого применения хорошо известны в уровне техники. Такие подходящие фармацевтические формы чаще всего и предпочтительнее составляют, чтобы они были стерильные, изотонические и буферные для стабильности и комфорта. Фармацевтические композиции для интраназальной доставки могут также включать капли и спреи, часто подготовленные для имитации назальной секреции для того, чтобы гарантировать поддержание нормального цилиарного действия. Как описано в Remington's Pharmaceutical Sciences, 18th Ed., Mack Publishing Co., Easton, PA (1990), который приводится здесь для ссылки во всей его целостности и хорошо известен специалистам в данной области, подходящие фармацевтические формы являются чаще всего и предпочтительнее всего изотоническими, буферными для сохранения рН 5.5-6.5, и наиболее часто и предпочтительно содержат антимикробные консерванты и соответствующие стабилизаторы лекарственных средств. Фармацевтические формы для интрааурикулярной доставки включают суспензии и мази для местного применения в ухе. Наиболее распространенными растворителями для таких ушных форм являются глицерин и вода.
[0088] Композиции могут также представлять собой ректальные композиции, такие как суппозитории или удерживающая клизма, например, содержащая обычную основу для суппозитория, такую как масло какао или другие глицериды.
[0089] Дополнительно к формам, описанным предварительно, композиции могут быть также составлены в виде форм с веществами замедленного всасывания. Такие долго действующие формы могут быть введены имплантацией (например, подкожно или внутримышечно) или внутримышечной инъекцией. При этом, например, соединения могут быть смешаны с подходящими полимерными или гидрофобными материалами (например, в виде эмульсии в приемлемом масле) или ион обменными смолами или трудно растворимыми производными, например, в виде трудно растворимой соли.
[0090] Для гидрофобных соединений, подходящий фармацевтический носитель может представлять собой систему сорастворителей, содержащую бензиловый спирт, неполярное поверхностно-активное вещество (ПАВ), водорастворимый органический полимер и водную фазу. Распространенная система представляет собой систему сорастворителей VPD, которая содержит раствор 3% вес/объем бензилового спирта, 8% вес/объем неполярного ПАВ POLYSORBATE 80™ и 65% вес/объем полиэтиленгликоля 300, доведенных до объема абсолютным этанолом. Естественно пропорции системы сорастворителей могут значительно изменяться без нарушения характеристик их растворимости и токсичности. Более того, идентичность компонентов со-растворителей может изменяться: например, другие низко токсичные ПАВ могут использоваться вместо POLYSORBATE 80™; количество полиэтиленгликоля может изменяться; другие биосовместимые полимеры могут заменить полиэтиленгликоль, например, поливинилпирролидон и другие сахара или полисахариды могут заменить декстрозу.
[0091] Способы лечения бактериальных инфекций могут включать введение терапевтически эффективного количества соединения, описанного здесь. Лечение бактериальной инфекции может также включать профилактическое введение терапевтических соединений для предотвращения инфекции или распространения инфекции у пациента при неминуемом риске, например, у пациента, только что перенесшего хирургическое вмешательство или у пациента с ослабленным иммунитетом или пациента в зоне риска, если соединение не было введено. Соединения демонстрируют ингибиторную активность против широкого спектра бактерий, против метициллин резистентной Staphylococcus aureus (MRSA) и ванкомицин резистентной Enterococci (VRE) и имеют относительную антибиотическую активность с относительно низкой концентрацией или in vivo. Также соединения по настоящему изобретению могут проявлять потенциально антибактериальную активность против различных человеческих и животных патогенов, включая Грам-положительные бактерии, такие как Staphylococi, Enterococci и Streptococi, анаэробные микроорганизмы, такие как бактериоды и Clostridia, и кислотостойкие микроорганизмы, такие как Mycobacterium tuberculosis и Mycobacterium avium. В одном варианте осуществления изобретения, бактериальная инфекция, которая может быть подвергнута лечению или состояние пациента с этой инфекцией может быть улучшено, представляет собой MRSA.
[0092] Композиции или фармацевтические композиции, описанные здесь, могут быть введены пациенту без каких-либо подходящих средств. Примеры, не ограничивающие объем притязаний, включают среди прочего (а) введение через пероральные способы, когда вводится капсула, таблетка, гранула, спрей, сироп или другие такие формы; (b) введение не пероральными способами, такие как ректальный, вагинальный, интрауретральный, интраокулярный, интраназальный или интрааурикулярный, где вводится водная суспензия или масляная форма или тому подобное или капли, спрей, суппозиторий, бальзам, мазь или тому подобное; (с) введение через инъекцию, подкожно, интраперитонеально (внутрибрюшинно), внутривенно, внутримышечно, внутрикожно, интраорбитально, внутрисуставно, интраспинально, интрастернально или тому подобное, включая доставку инфузионным насосом; также (d) локальное введение; как понятно специалисту в данной области для приведения активного соединения в контакт с живой тканью.
[0093] Фармацевтические композиции, подходящие для введения, включают композиции, где активные ингредиенты присутствуют в количестве эффективном для достижения заявленного назначения. Терапевтически эффективное количество соединений, описанное здесь и необходимое в качестве дозы, будет зависеть от способа введения, вида животного, включая человека, которое необходимо подвергнуть лечению и физических характеристик конкретного рассматриваемого животного. Дозировка может быть подобрана для достижения желаемого эффекта, но она также будет зависеть от таких факторов, как вес, диета, параллельный медицинский курс и другие факторы, которые специалист в данной области легко выявит. В частности, терапевтически эффективное количество означает количество соединения, эффективное для предотвращения или облегчения симптомов заболевания или улучшения состояния или продления жизни пациента. Определение терапевтически эффективного количества находится в рамках возможностей специалиста в данной области, в частности в свете детального описания, представленного здесь.
[0094] Как очевидно специалисту в данной области, полезная in vivo доза для введения и конкретный режим введения будут изменяться в зависимости от возраста, веса и вида млекопитающих, конкретных используемых соединений и конкретного назначения, для которого эти соединения используются. Определение уровня эффективной дозы, необходимой для достижения желаемого результата, может быть осуществлено специалистом в данной области, используя обычные фармакологические способы. Обычно клинические испытания на человеке начинают с низким уровнем доз, с постепенным увеличением до достижения желаемого эффекта. Альтернативно, приемлемые исследования in vitro могут быть использованы для установления полезных доз и способов введения композиций, идентифицируемых настоящими методами, используя установленные фармакологические методы.
[0095] У животных (не человека), испытания потенциальных продуктов осуществляют с дозами более высокого уровня, с постепенным снижением доз до уровня, до тех пор пока желаемый эффект больше не будет достигаться, а противоположный эффект не исчезнет. Дозировка может изменяться достаточно широко, в зависимости от желаемых эффектов и терапевтического назначения. Обычно дозировки могут быть равны приблизительно 10 микрограмм/кг до приблизительно 100 мг/кг веса тела, предпочтительно 100 микрограмм/кг до приблизительно 10 мг/кг веса тела. Дозировки могут быть основаны и рассчитаны исходя из площади поверхности пациента, что понятно специалисту в данной области.
[0096] Точная форма, способ введения и дозировка фармацевтической композиции по настоящему изобретению может быть выбрана индивидуальным медицинским специалистом с точки зрения состояния пациента. (Смотреть, например, Fingl и другие, 1975, в "The Pharmacological Basis of Therapeutics", которая приводится здесь для ссылки во всей ее целостности, с конкретной отсылкой на Главу 1, страницу 1). Обычно диапазон доз композиции, вводимой пациенту, может составлять приблизительно от 0.5 до 1000 мг/кг веса тела пациента. Дозировка может быть единичной или представлять собой серию из двух или более приемов в течение одного или более дней, как необходимо пациенту. В случае, когда дозировка для человека была установлена, по меньшей мере, для некоторого состояния, настоящее изобретение будет использовать те же самые дозы или дозы, которые составляют от 0.1% до приблизительно 500%, более предпочтительно от 25% до 250% установленной человеческой дозы. Там, где доза для человека не установлена, поскольку бывает случаи новых фармацевтических композиций, подходящая человеческая дозировка может быть подобрана от ED50 или ID50 значений или других подходящих значений, выведенных из in vitro или in vivo исследований, как определено исследованиями токсичности и эффективности у животных.
[0097] Необходимо отметить, что практикующий врач знает, как и когда прекратить, прервать или изменить лечение из-за токсичности или дисфункции органов. Наоборот практикующий врач также будет знать, как изменить лечение до более высокого уровня доз, если клинический ответ не является ожидаемым (исключая токсичность). Величина вводимой дозы будет изменяться в зависимости от серьезности состояния больного и способа введения. Серьезность состояния может, например, быть оценена частично, стандартными прогностическими методами оценки. Доза и возможно частота доз будут также изменяться согласно возрасту, весу тела и отклику индивидуального пациента. Программа, сравнимая с обсуждаемой выше, может быть использована в ветеринарии.
[0098] Хотя точная дозировка будет определена только в зависимости от конкретного лекарственного средства, в большинстве случаев некоторые общие правила, касающиеся дозировки могут быть сформулированы. Режим ежедневного приема для взрослого пациента может представлять, например, пероральную дозу приблизительно от 0.1 мг - 2000 мг каждого активного ингредиента, предпочтительно от 1 мг до 500 мг, например, от 5 до 200 мг. В других вариантах осуществления изобретения, внутривенная, подкожная или внутримышечная доза каждого активного ингредиента составляет приблизительно от 0.01 мг - 100 мг, предпочтительно от 0.1 мг до 60 мг, например, от 1 до 40 мг. В случаях введения фармацевтически приемлемой соли, дозировки могут быть рассчитаны для свободного основания. В некоторых вариантах осуществления изобретения композиция вводится от 1 до 4 раз в день. Альтернативно композиции по изобретению могут быть введены непрерывным внутривенным вливанием, предпочтительно с дозой каждого активного ингредиента до 1000 мг в день. Как понятно специалисту в данной области, в некоторых ситуациях необходимо ввести соединения, описанные здесь, в количествах, которые превышают или даже значительно превышают, указанный выше, предпочтительный диапазон доз для того, чтобы эффективно и интенсивно вылечить конкретное заболевание или инфекцию. В некоторых вариантах осуществления изобретения соединения будут вводиться в течение периода непрерывной терапии, например, в течение недели или более или месяцев или лет.
[0099] Количество и интервал могут быть подобраны индивидуально для обеспечения уровней плазмы активного фрагмента, которые являются достаточными для поддержания модулирующих эффектов или минимальной эффективной концентрации (МЕС). МЕС будет изменяться для каждого соединения, но может быть оценена на основании данных in vitro. Дозы, необходимые для достижения МЕС будут зависеть от индивидуальных характеристик и способа введения. Однако ВЭЖХ исследования или биоисследования могут быть использованы для определения концентраций в плазме.
[0100] Диапазон доз может быть также определен, используя значение МЕС. Композиции должны быть введены, используя режим, который поддерживает уровни в плазме выше МЕС в течение 10-90% времени, предпочтительно между 30-90% и наиболее предпочтительно 50-90%.
[0101] В случаях местного введения или селективного потребления, эффективная локальная концентрация лекарственного средства может не относиться к концентрации в плазме.
[0102] Количество введенной композиции может зависеть от пациента, от веса пациента, серьезности инфекции, способа введения и назначения предписанного врачом.
[0103] Композиции, описанные здесь, могут быть оценены на эффективность и токсичность, используя известные способы. Например, токсикология соединения может быть установлена определением in vitro токсичности по отношению к клеточной линии, например, млекопитающего или предпочтительно человека. Результаты таких исследований часто прогнозируют токсичность у животных, таких как млекопитающие или в частности у человека. Альтернативно, токсичность конкретных соединений на животной модели, такой как мыши, крысы, кролики или обезьяны может быть определена, используя известные способы. Эффективность конкретного соединения может быть установлена, используя различные признанные способы, такие как in vitro способы, животные модели или клинические исследования на человеке. Признанные in vitro модели существуют почти для каждого класса состояний. Подобным образом, приемлемые животные модели могут быть использованы для установления эффективности химических соединений для лечения таких состояний. При выборе модели для определения эффективности, специалист сможет руководствоваться состоянием уровня техники для выбора соответствующей модели, дозы и способа введения, режима. Конечно, клинические испытания на человеке могут быть использованы для определения эффективности соединения на человеке.
[0104] Композиции, если желательно, могут быть представлены в упаковке или дозаторе, который может содержать одну или более дозированную форму с активным ингредиентом. Упаковка может содержать, например, металлическую или пластиковую фольгу, например, блистерная упаковка. Упаковка или дозатор может содержать инструкцию по введению. Упаковка или дозатор может также содержать уведомление, связанное с контейнером в форме, предписанной правительственным агентством, регулирующим производство, использование или продажу лекарственного средства, это уведомление отражает одобрение формы лекарственного средства для введения человеку или его применение в ветеринарии. Такое уведомление, например, может быть маркировкой, одобренной Администрацией США по надзору в сфере лекарств и пищевых продуктов или одобренным продуктом вкладышем. Композиции, содержащие соединение по изобретению, с совместимым фармацевтическим носителем могут быть также получены, помещены в соответствующий контейнер и маркировка для лечения указанного состояния должна быть нанесена.
А. Примеры
1. Использование инструментов, приборов
[0105] Микроскопия Рамана была осуществлена на Renishaw System 1000, со стабилизированным диодным лазером 385 нм возбуждением и ближней ИК-областью с охлаждением с помощью эффекта Пелтье и камерой на приборе с зарядовой связью в качестве детектора. Измерения были осуществлены с 50х или более рабочем расстоянии на 20х объективе с диапазоном частоты 2000-100 см-1.
[0106] Фурье-Раман спектр был получен на Bruker RFS100 спектрометре с Nd:YAG 1064 нм возбуждением, 100 мВ лазерной мощностью и Ge детектором. 64 сканограммы было зарегистрировано в диапазоне 25-3500 см-1, при разрешении 2 см-1.
[0107] Bruker D8; Bragg-Brentano, геометрия отражения; Copper K(alpha) излучение, 40 кВ/ 40 мА; переменная дивергенционная щель; LynxEye детектор с 3° окошком; шаг, 0.02-°2; время, 37 с. Образцы вращались (0.5 оборота в секунду) во время измерения.
[0108] Подготовка образца: Образцы были подготовлены без специальной обработки, кроме применения слабого давления для получения гладкой поверхности. Виды образцов держателей из силиконового монокристалла: а) стандартный держатель для проверки полиморфов, 0.1 мм глубины, необходим образец менее 20 мг; b) 0.5 мм глубина, 12 мм диаметр полости, необходим образец 40 мг; с) 1.0 мм глубины, 12 мм диаметр полости, необходим образец 80 мг. Обычно образцы измерялись непокрытыми. Наличие колпачков из каптоновой пленки или РММА "купола" всегда указывалось на диффрактограмме с идентификацией образца.
1. Получение кристаллической свободной кислоты 1 (R=РО(ОН)2)
Пример 1
[0109] Раствор 1 (R=PO(ONa)2) в H2O был подготовлен, и 1 М HCl была добавлена для получения тонкой суспензии, которая после добавления тетрагидрофурана (ТГФ) была перемешана и отфильтрована. Полученный кристаллический твердый осадок 1 (R=РО(ОН)2) был высушен в вакууме и охарактеризован Фурье-Раман спектром (FTR) (Фигура 1), рентгенограммой (XRPD, Malvern Mastersizer) (Фигура 2), диаграммой, полученной методами термогравиметрии и инфракрасной спектроскопии (TG-FTIR) и термограммой дифференциальной сканирующей калориметрии (DSC). Измерение DSC показало температуру плавления в 256.9°С, затем последовало разложение образца (Фигура 3).
Пример 2
[0110] К 1 (R=PO(ONa)2) (2 г), растворенному в 10 мл H2O, была медленно добавлена HCI (6 мл; 1 М) с получением тонкой суспензии светло-желтого твердого вещества. После добавления дополнительного количества 5 мл H2O и 20 мл ТГФ, суспензия была отфильтрована и высушена под вакуумом.
Пример 3
[0111] К 1 (R=PO(ONa)2) (2 г), растворенному в 10 мл H2O была медленно добавлена HCl (8 мл; 1 М) с получением тонкой суспензии светло-желтого твердого вещества, к которому еще 25 мл H2O было добавлено. Твердое вещество было отфильтровано, промыто 10 мл 0.1 М HCl и 100 мл воды и высушено под вакуумом.
Пример 4
[0112] К 1 (R=PO(ONa)2) (5 г), растворенному в 30 мл воды, была добавлена 15 мл HCI (1 М) и 30 мл ТГФ для получения светло-желтой суспензии, которая была перемешана 30 мин при комнатной температуре и отфильтрована. Полученный твердый осадок был суспендирован в 150 мл воды и перемешан в течение 60 минут при комнатной температуре. Затем 50 мл ТГФ было добавлено, и суспензия была перемешана 18 часов. Суспензия была отфильтрована, и твердый осадок был промыт 10 мл HCl (0.1 М) и 100 мл воды и высушен под вакуумом (15 часов).
Пример 5
[0113] К 1 (R=PO(ONa)2) (2 г), растворенному в 15 мл воды, была медленно добавлена HCl (6 мл; 1 М) с получением светло-желтой суспензии. После добавления 20 мл ТГФ и 60 мл воды, суспензия была перемешана 18 часов, отфильтрована, и твердое вещество было перемешано вновь в 6 мл HCI (1М) в течение 15 минут. Затем суспензия была отфильтрована, и твердое вещество было высушено под вакуумом.
Пример 6
[0114] К 1 (R=РО(ONa)2) (3 г), растворенному в 35 мл воды была добавлена HCI (9 мл; 1М) с получением светло-желтой суспензии. После добавления 20 мл ТГФ суспензия была перемешана 30 минут при комнатной температуре и затем отфильтрована. Полученное твердое вещество было промыто 20 мл HCl (0.1М) и водой, и высушено под вакуумом.
Пример 7
[0115] Твердый диводород фосфат был добавлен к объему ДМСО или N-метилпирролидона при 50°С до тех пор, пока соль не перестала растворяться. Раствор, содержащий суспендированную соль, был нагрет, до тех пока остатки твердого вещества не растворились, раствор был отфильтрован в горячем состоянии и оставлен охлаждаться, наблюдалось появление кристаллов диводород фосфата.
Пример 8
[0116] Раствор диводород фосфата был подготовлен в ДМСО или N-метилпирролидоне и отфильтрован. К отфильтрованному раствору был добавлен этанол с перемешиванием, до тех пор раствор не стал мутным. Перемешивание было остановлено, и слой этанола осторожно помещен на поверхность мутного раствора, который был оставлен, наблюдалось появление кристаллов диводород фосфата.
Пример 9
[0117] Раствор диводород фосфата был подготовлен в ДМСО или N-метилпирролидоне и отфильтрован. Отфильтрованный раствор был подвергнут воздействию паров этанола, например, помещением открытого контейнера раствора и открытого контейнера с этанолом вместе в герметичном сосуде так, чтобы оба контейнера разделяли общее пространство внутри сосуда. Спустя какое-то время в контейнере с раствором появляются кристаллы диводород фосфата.
Пример 10
[0118] Раствор соли диводород фосфата, такой как моно- или динатрий фосфат, был подготовлен. Такой раствор может быть подготовлен такими методами, как простое растворение образца твердого фосфата динатрия в воде или добавлением диводород фосфата к водному раствору основания, достаточно сильного депротонирования диводород фосфата. Идентификация соответствующего основания это обычное дело для практикующего химика. Обычно полученный раствор соли диводород фосфата отфильтровывается, к фильтрату добавляется кислота для репротонирования соли и для начала кристаллизации диводород фосфата. В обычном примере, диводород фосфат добавляют к водному раствору, содержащему NaOH или Na2CO3 с получением раствора динатрий фосфата, к которому после фильтрации добавляют водный или газообразный HCl для восстановления диводород фосфата, который выпадает в виде кристаллов.
[0119] Для фармацевтических целей преимущественно использовать фармацевтически приемлемые кислоты и основания в этом процессе, например, те, которые указаны в Handbook of Pharmaceutical Salts: Properties, Selection and Use. (P. Heinrich Stahl and Camille G. Wermuth, eds.) International Union of Pure and Applied Chemistry, Wiley-VCH 2002 and L.D. Bighley, S.M. Berge, D.C. Monkhouse, в "Encyclopedia of Pharmaceutical Technology'. Eds. J. Swarbrick and J.C. Boylan, Vol. 13, Marcel Dekker, Inc., New York, Basel, Hong Kong 1995, стр. 453-499, такие соли обсуждаются детально.
[0120] Как понятно специалисту в данной области, элементы этих способов могут быть комбинированы. Например, раствор диводород фосфата в ДМСО или N-метилпирролидоне может быть приготовлен при одной температуре, второй растворитель, такой как, этанол может быть добавлен, и полученный раствор оставлен охлаждаться. Подобным образом смеси растворителей могут быть использованы вместо чистых растворителей, что также хорошо известно специалистам по кристаллизации соединений. Более того, другие растворители и смеси могут также быть использованы.
[0121] Элементный анализ для C17H16FN6O6P (измерено/рассчитано) С 43.9 (44.8); Н 3.6 (3.7); N 18.1 (18.4); О 21.2 (22.1); F 4.2 (4.2); Р 6.7 (6.8).
Пример 11
[0122] Размер частиц был измерен, используя Malvern Mastersizer. Инструкции по подготовке образцов, которые совпадали с инструкциями производителя прибора, были выполнены. Образец был подготовлен суспендированием в 1-2 мл деионизированной воды и обработкой ультразвуком в течение 3 минут.
[0123] Примерное распределение частиц по размерам кристаллического материала, такого как описан в Примерах 1-10 выше, показан на Фигуре 10 и в Таблице 3 ниже:
Пример 12 Эксперимент по Регулированию Размера Частиц
[0124] В 22-л реактор была загружена 1 М HCl (1.95 л, 2.2 эквивалента) и этанол (1.6 л, 4 объема), и раствор был нагрет до 70°С. В отдельный 12-л реактор, снабженный барботером для отслеживания выделения газа, был загружен TR-701FA [0.4 кг, AMRI лот # DUG-AH-166(2)], вода (2.8 л, 7 объемов) и этанол (0.4 л, 1 объем). Суспензия была перемешана при температуре окружающей среды, и 5% по весу водный раствор NaHCO3 был добавлен через перистальтический насос за 30 минут. Пенообразование не наблюдалось, однако выделение газа было очень интенсивным через барботер. По завершении добавления, рН чистого желтого раствора составило 6.6. Водный раствор TR-701 был добавлен через перистальтический насос к этанол/HCl раствору за 90 минут. По завершении добавления рН реакционной смеси была 1.9, смесь была охлаждена до 30°С. Образец суспензии был отобран для анализа оптической микроскопией. Суспензия была отфильтрована через полипропиленовую фильтрующую ткань, реактор и осадок на фильтре были промыты водой (5 объемов) и ацетоном (5 объемов). Общее время фильтрации, включая промывание, составило 12 минут. Твердый осадок был высушен под глубоким вакуумом при 50°C с получением 391.7 г повторно осажденного TR-701FA (98% выход). Анализ 1Н ЯМР соответствовал указанной структуре. ВЭЖХ анализ (Способ А): 98.8% (AUC) tR=5.2 мин. Уровень остаточного этанола 1Н ЯМР анализом составил 0.03%, содержание воды было 0.15% по титрованию Карла Фишера, и содержание натрия было 5 мд.
[0125] Размер частиц был измерен, используя Malvern Mastersizer лазерную рассеивающую микроскопию. Инструкции по подготовке образцов, соответствующие инструкциям производителя инструмента, были четко соблюдены. Образец был получен суспендированием в 1-2 мл деионизированной воды и обработкой ультразвуком в течение 3 минут. Данные по лазерной дифракции представлены на Фигуре 11 и в Таблице 4 ниже.

[0126] В другом эксперименте распределение по размерам частиц, используя способ с контролем, такой как описан в этом примере, представлен на Фигуре 12 и в Таблице 5 ниже:

[0127] Лекарственная форма с быстрым высвобождением и форма для внутривенной инъекции, описанные в Примерах 13-14 ниже, были подготовлены, используя кристаллическую свободную кислоту, где размер частиц контролировался.
Пример 13 Форма с быстрым высвобождением
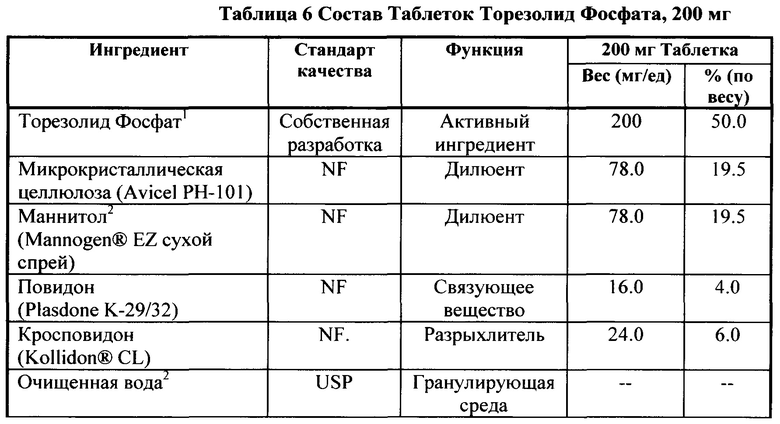
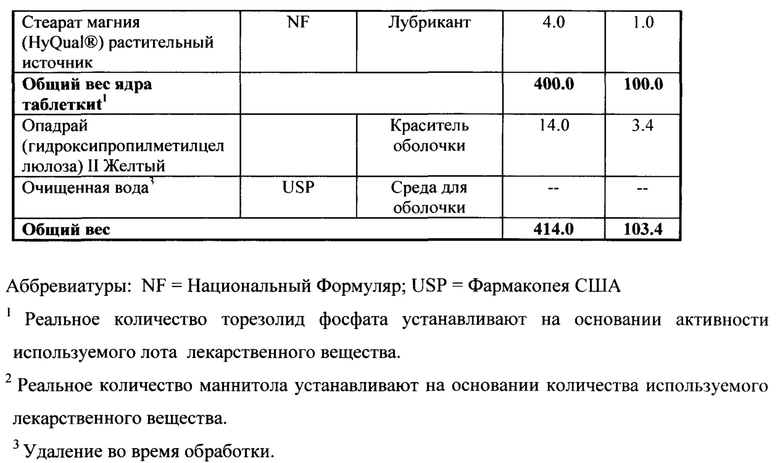
[0128] Качественный и количественный состав таблеток с быстрым высвобождением (R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он диводород фосфата 1 (R=РО(ОН)2) ("Таблетки Торезолид Фосфата"), 200 мг, представлен в Таблице 6. Все компоненты, используемые в подготовке, перечислены с указанием стандарта качества, их функции, процента по весу для каждого индивидуального компонента. Список включает все материалы, используемые при получении лекарственного средства, вне зависимости от их присутствия в конечном продукте.
Пример 14 Порошок и форма для инъекции
[0129] (R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он диводород фосфат 1 (R=РО(ОН)2) ("Торезолид Фосфат для инъекции" или "TR-701 FA для инъекции"), 200 мг/ампула, был получен в виде стерильного лиофилизированного порошка для инъекции. TR-701 FA для инъекции подготавливают in situ в виде динатриевой соли, используя гидроксид натрия из-за его более высокой растворимости в воде (>130 мг/мл).
[0130] TR-701 FA для инъекции, 200 мг/ампула, восстанавливают 4 мл стерильной воды для инъекции (WFI), USP с получением 50 мг/мл раствора. Соответствующий объем клинической дозы отбирают из ампулы и добавляют в нон-ди(2-этилгексил)фталатный (DEHP) контейнер для внутривенного (IV) введения содержащий или 0.9% инъекцию хлорида натрия, USP (солевой раствор) или 5% инъекцию декстрозы, USP (декстроза). Полученный IV раствор вводят, используя комплект нон-DEHP раствора с 0.22 мкм встроенным фильтром.
[0131] Состав TR-701 FA раствора для лиофилизации представлен в Таблице 7 и состав TR-701 FA для инъекции, 200 мг/ампула представлен в Таблице 8.
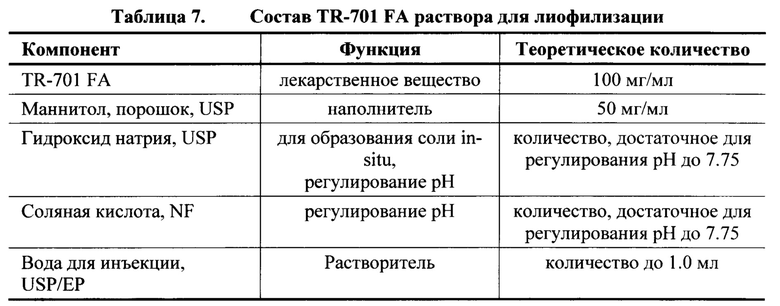
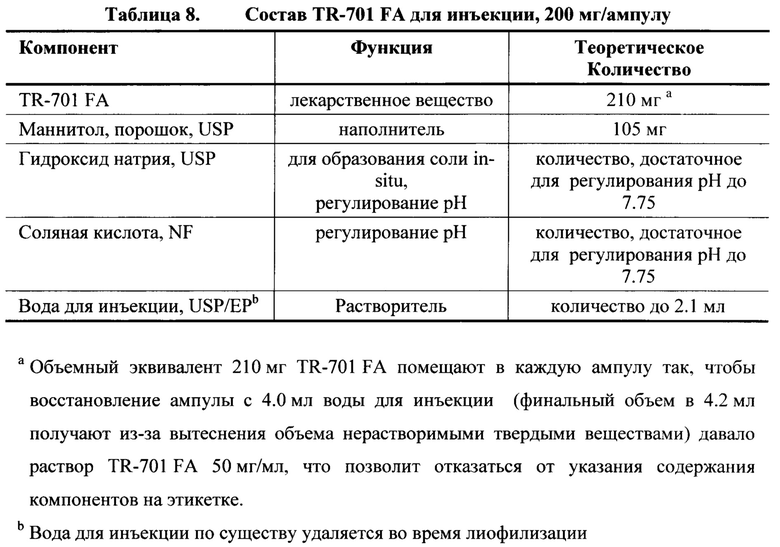
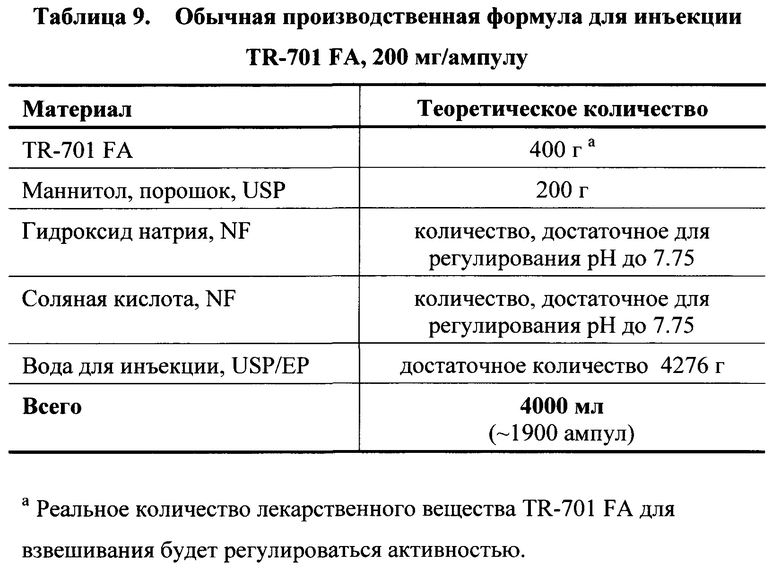
[0132] Обычная производственная формула для инъекции TR-701 FA, 200 мг/ампула представлена в Таблице 9.
[0133] Краткое описание производственного процесса представлено ниже, схематическое изображение производственного процесса для получения раствора, стерильной фильтрации, наполнения и лиофилизации представлено на Фигурах 8 и 9.
Получение раствора
[0134] Раствор получают в соответствии со следующей схемой:
[0135] Добавить приблизительно 50% общего количества воды для инъекции в сосуд с целевым соединением.
[0136] Добавить TR-701 FA и медленно нейтрализовать раствором гидроксида натрия при перемешивании.
[0137] Добавить и растворить маннитол при перемешивании.
[0138] Измерить рН полученного раствора. Если раствор находится за пределами целевого диапазона рН 7.70-7.80, довести рН, используя или 1н гидроксид натрия или 1н соляную кислоту.
[0139] Добавить воду для инъекции до финального объема и перемешать.
Стерильное Фильтрование/Наполнение/Лиофилизация
[0140] Отфильтровать полученный раствор через 2 проверенных на отсутствие повреждений 0.22 мкм фильтра последовательно в стерильный сосуд.
[0141] Наполнить целевым раствором 20 мл ампулы в асептических условиях.
[0142] Частично ввести лиофилизационные пробки в ампулы.
[0143] Провести лиофилизацию ампул в соответствии с определенным циклом.
[0144] В конце цикла лиофилизации, заполнить камеру азотом и закрыть ампулы под частичным вакуумом.
[0145] Герметично закрыть ампулы колпачками флип-офф (обжимной колпачок с резиновой пробкой, которая укрыта съемной пластиковой крышкой для обеспечения места прокола).
Пример 15
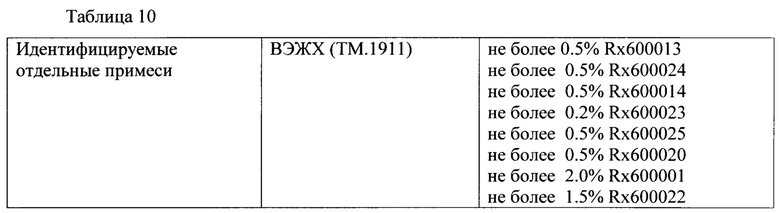
[0146] Образец кристаллической свободной кислоты, который был получен в соответствии со способом получения свободной кислоты, описанным в заявке на патент США №12/577,089, права по которой переданы тому же правообладателю, что и по настоящей заявке, был кристаллизован в соответствии со способами, описанными в настоящей заявке, и был охарактеризован, используя ВЭЖХ, образец содержит различные количества примесей, такие как описаны в таблице 10 ниже:
[0147] Дополнительно, по существу чистый образец кристаллической свободной кислоты, который был получен в соответствии с процессами, не раскрытыми в опубликованной заявке на патент США №20070155798 и был кристаллизован в соответствии со способами, описанными здесь (здесь и далее именуемый как «наши») был сравнен с образцом материала, полученным Dong-A Pharm. Со. (здесь и далее именуемый как "Dong-A материал"), который был предоставлен компании Траюс Терапьютикс Инк. приблизительно в 2007. Содержание активного вещества в Dong-A материале составило приблизительно 84% по весу образца по сравнению с чистым образцом, однако, чистота кристаллической свободной кислоты была 94.1% по весу материала, идентифицированного ВЭЖХ, как указано ниже. Таким образом, приблизительно 10% примесей в Dong-A материале не было идентифицировано ВЭЖХ. Сравнение характеристик чистоты представлено в Таблице 11 ниже:
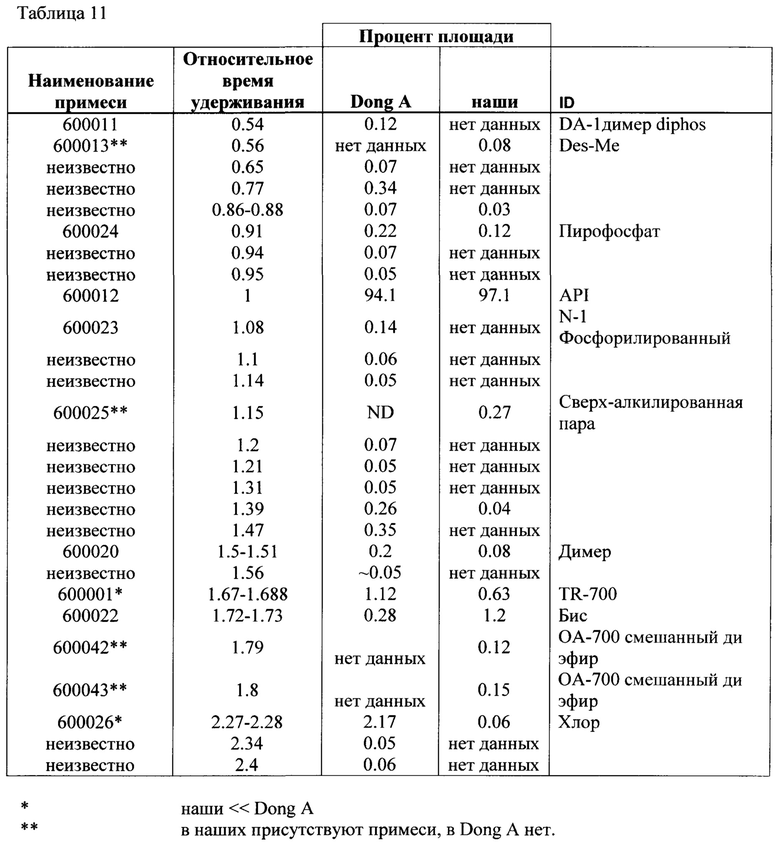
Органические примеси в TR-701 FA лекарственном веществе
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОЛУЧЕНИЯ ОКСАЗОЛИДИНОНОВ И СОДЕРЖАЩИХ ИХ КОМПОЗИЦИЙ | 2009 |
|
RU2556234C2 |
| ОКСАЗОЛИДИНОНСОДЕРЖАЩИЕ ДИМЕРНЫЕ СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИСПОЛЬЗОВАНИЯ | 2010 |
|
RU2557910C2 |
| ОКСАЗОЛИДИНОНЫ И СПОСОБ ИХ ОЧИСТКИ | 2009 |
|
RU2659792C1 |
| КОМБИНИРОВАННЫЕ ЛЕКАРСТВЕННЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2008 |
|
RU2488394C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ОКСАЗОЛИДИНОНА | 2004 |
|
RU2414469C2 |
| БИАРИЛЬНЫЕ МОНОБАКТАМНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2016 |
|
RU2746129C2 |
| ПРОИЗВОДНЫЕ МАННОЗЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2014 |
|
RU2678327C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ОКСАЗОЛИДИНОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2004 |
|
RU2559620C2 |
| НАЦЕЛЕННЫЕ КОНЪЮГАТЫ И ЧАСТИЦЫ И ИХ СОСТАВЫ | 2015 |
|
RU2695220C2 |
| ПРОИЗВОДНЫЕ 1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-7-ОНА И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2012 |
|
RU2570423C2 |


Предложена кристаллические частицы, содержащие (R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он диводород фосфат, имеющие рентгенограмму, характеризующуюся пиками при 14.7°, 15.2°, 16.6°, 20.3°, 26.8° и 28.2°, а также составы на их основе, пригодные для использования в фармацевтической промышленности. Предложены новые кристаллические частицы, характеризующиеся средним диаметром от 1,0 мкм до 44,0 мкм, содержанием помимо основного вещества воды и как минимум одного соединения, выбранного из:
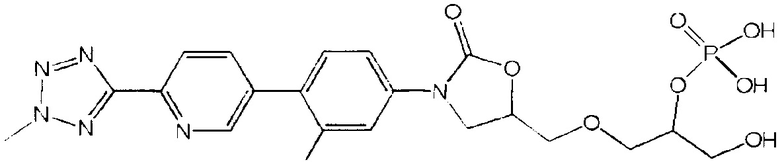
и
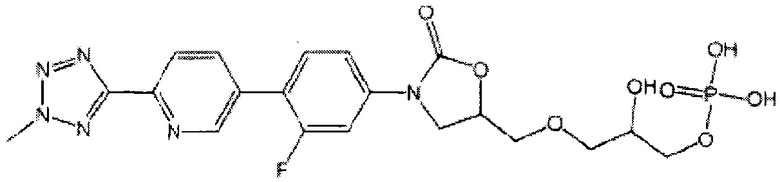 ,
,
которые обладают повышенной стабильностью при изготовлении твердых лекарственных форм для лечения бактериальных инфекций. 3 н. и 8 з.п. ф-лы, 12 ил., 12 табл., 15 пр.
1. Кристаллические частицы для приготовления твердых лекарственных форм для лечения бактериальных инфекций, характеризующиеся средним диаметром от 1,0 мкм до 44,0 мкм, содержанием воды от 0,1% до 4% по весу, имеющие рентгенограмму, содержащую пики при 14.7°, 15.2°, 16.6°, 20.3°, 26.8° и 28.2°, и дополнительно характеризующиеся тем, что они содержат
(R)-3-(4-(2-(2-метилтетразол-5-ил)пиридин-5-ил)-3-фторфенил)-5-гидроксиметил оксазолидин-2-он диводород фосфат в количестве не менее 96% по весу,
как минимум одно соединение, которое выбирают из группы, состоящей из:
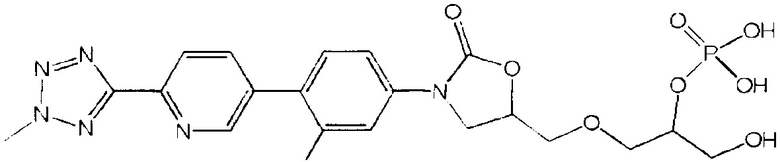
и
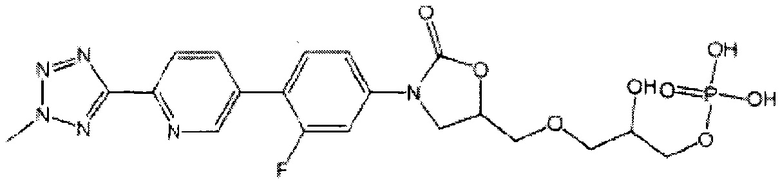
и необязательные примеси.
2. Кристаллические частицы по п. 1, характеризующиеся тем, что указанные частицы дополнительно содержат как минимум одно соединение, которое выбирают из группы, состоящей из:
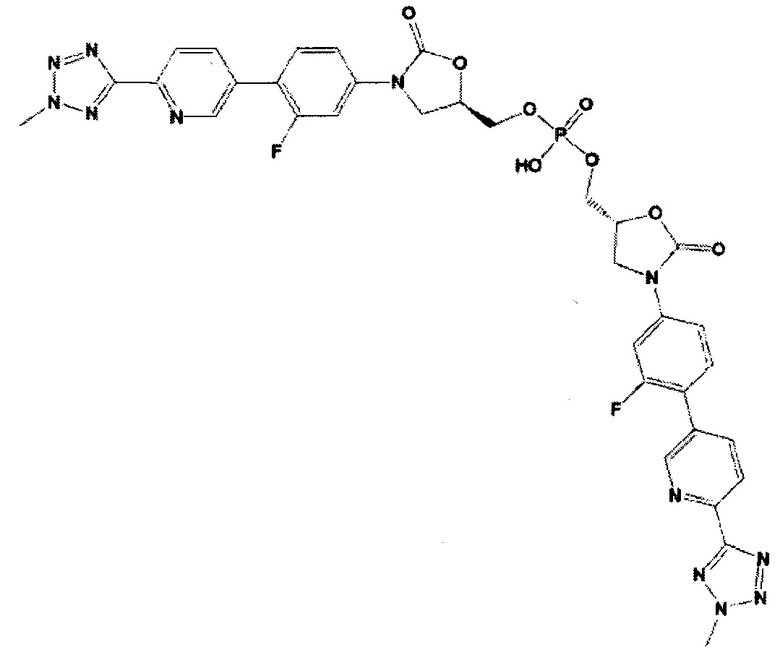 и
и
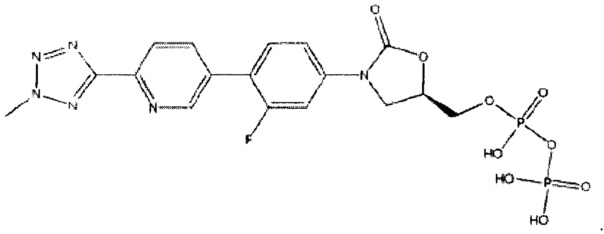
3. Кристаллические частицы по п. 2, характеризующиеся тем, что указанные частицы дополнительно содержат как минимум одно соединение, представляющее собой:
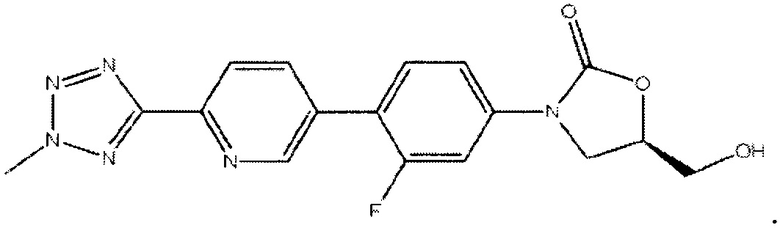
4. Кристаллические частицы по п. 3, характеризующиеся тем, что указанные частицы дополнительно содержат как минимум одно соединение, которое выбирают из группы, состоящей из:
 и
и
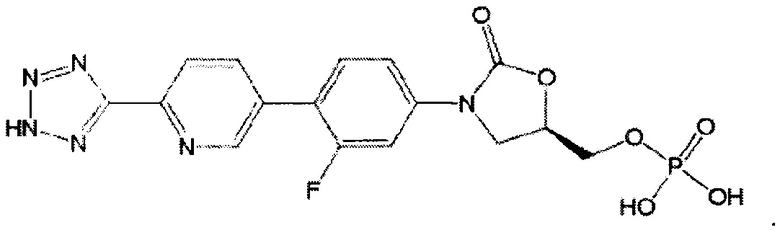
5. Кристаллические частицы по п. 4, характеризующиеся тем, что указанные частицы дополнительно содержат как минимум одно соединение, представляющее собой:
6. Кристаллические частицы по п. 1, характеризующиеся содержанием воды от 0,1% до 3% по весу.
7. Кристаллические частицы по п. 1, характеризующиеся средним диаметром от 10,0 мкм до 35,0 мкм.
8. Кристаллические частицы по п. 1, характеризующиеся средним диаметром от 1,0 мкм до 40.0 мкм.
9. Реакционная смесь для приготовления твердых лекарственных форм для лечения бактериальных инфекций, содержащая кристаллические частицы по любому из пп. 1-8 и гидроксид натрия.
10. Фармацевтическая композиция для лечения бактериальных инфекций, содержащая кристаллические частицы по п. 1 и как минимум один фармацевтически приемлемый носитель или наполнитель.
11. Фармацевтическая композиция по п. 10, характеризующаяся тем, что указанный фармацевтически приемлемый носитель или наполнитель представляет собой как минимум одно вещество, которое выбирают из группы, состоящей из маннитола, поливинипирролидона и стеарата магния.
| WO2005058886 A1, 30.06.2005 | |||
| LUCIO VERA-CABRERA et al., Antimicrobial agents and chemotherapy, 2006, vol | |||
| Устройство для выпрямления многофазного тока | 1923 |
|
SU50A1 |
| ПОРШЕНЬ ДЛЯ БЫСТРОХОДНЫХ ДВИГАТЕЛЕЙ ВНУТРЕННЕГО ГОРЕНИЯ | 1926 |
|
SU7490A1 |

