Область техники
Заявляемое изобретение относится к фармацевтике, медицине и ветеринарии, в частности, к разработке синтетического анальгетика пептидной природы и способу купирования боли с его помощью.
Заявляемое изобретение в части, касающейся состава анальгетического средства, может быть использовано для производства анальгетиков пептидной природы. Заявляемое изобретение может быть применено в медицине и в ветеринарии для терапии болевого синдрома различной этиологии.
Уровень техники
Анальгетическими средствами, или анальгетиками (от греч. algos - боль и an - без), называют лекарственные средства, обладающие специфической способностью ослаблять или устранять чувство боли [Машковский М.Д. Лекарственные средства. 14 издание. М.: Новая волна, 2003, т. 1, с. 145].
Анальгетическое (болеутоляющее) действие могут оказывать не только собственно анальгетики, но и другие вещества, относящиеся к разным фармакологическим группам. Так, анальгетическим эффектом могут обладать препараты, применяемые для наркоза (общего обезболивания), и некоторые из них в соответствующих концентрациях и дозах (например, трихлорэтилен, закись азота) используются специально для анальгезии.
Местные анестетики также являются анальгетическими средствами, особенно при периферических болях. При резорбтивном действии местных анестетиков они могут влиять на ЦНС: это проявляется в беспокойстве, треморе, судорогах, а в более высоких дозах они негативно влияют на дыхательный и сосудодвигательный центры. Местные анестетики, оказывая блокирующее влияние на потенциалочувствительные натриевые каналы, угнетают сократимость миокарда, расширяют кровеносные сосуды, угнетающе влияют на симпатическую иннервацию, снижают артериальное давление. Наиболее важным свойством местных анестетиков является их способность блокировать ноцицепторы и чувствительные нервные волокна других модальностей. В связи с этим их используют для местного обезболивания (местной анестезии), в частности, при хирургических операциях.
Под анальгетическими в собственном смысле слова подразумеваются средства, доминирующим эффектом которых является анальгезия, не сопровождающаяся в терапевтических дозах выключением сознания и выраженным нарушением двигательных функций.
Усиливать обезболивающий эффект могут дополнительные препараты (адъюванты), которые, оказывая косвенное влияние на ноцицептивную систему мозга, ослабляют негативное влияние патологического состояния клеток тканей или органов, вызывающих боль. Болеутоляющее действие, например, могут оказать спазмолитические и холинолитические средства.
Всемирная организация здравоохранения (ВОЗ) утвердила трехступенчатый подход к лечению онкологической боли, который широко применяется в настоящее время. Он подразумевает использование анальгетиков перорально по часам, в зависимости от длительности действия препарата. На первом этапе используются неопиоидные анальгетики, такие как парацетамол (ацетаминофен), или нестероидные противовоспалительные препараты (НПВП). При недостаточном эффекте добавляются слабые опиоиды, например, кодеин, декстропропоксифен или трамадол, (вторая ступень). Если и это не позволяет купировать боль, то на последнем этапе слабый опиоид меняется на сильный, подобный морфину (например, морфин, гидроморфон, оксикодон или фентанил).
Эта «лестница лечения боли ВОЗ» применяется с 1986 года. Данный подход затем был экстраполирован на неонкологическую боль, в том числе для лечения хронической боли различной этиологии и локализации. Дополнительные препараты используются при этом и для снижения тревожности и других негативных побочных эффектов.
По химической природе, характеру и механизмам фармакологической активности современные анальгетики делятся на две группы.
Основными представителями анальгетиков первой группы являются производные салициловой кислоты (натрия салицилат, ацетилсалициловая кислота, салициламид и др.), производные пиразолона - антипирин, амидопиприн, анальгин, производные пара-аминофенола (или анилина) - фенацетин, парацетамол [Машковский М.Д. Лекарственные средства. 14 издание. М.: Новая волна, 2003, т. 1, с. 146]. Указанные анальгетики являются синтетическими лекарственными средствами.
Анальгезирующая активность лекарственных средств, относящихся к первой группе, проявляется при определенных видах болевых ощущений, главным образом при невралгических, мышечных, суставных болях, при головной и зубной боли. При сильной боли, связанной с травмами, полостными оперативными вмешательствами и т.п., они практически неэффективны.
Указанные средства проявляют жаропонижающее действие при лихорадочных состояниях, и противовоспалительное действие, выраженное в разной степени у разных анальгетиков этой группы.
При применении этих анальгетиков отсутствуют угнетающее влияние на дыхательный и кашлевой центры, а также явлений эйфории, психической и физической зависимости.
В механизме действия анальгетиков первой группы определенную роль играет влияние на таламические центры, которое приводит к торможению проведения болевых импульсов к коре головного мозга. В механизме действия салицилатов важную роль играют ингибирование биосинтеза простагландинов, а также стимулирующее влияние на «ось» гипофиз - надпочечники, способствующее высвобождению кортикостероидов. Существенное значение в действии анальгетиков первой группы имеет их влияние на кининовую систему (антагонизм с альгезирующим действием брадикинина и др.).
Анальгетики второй группы включают природные соединения - морфин и близкие к нему алкалоиды (опиаты), и синтетические соединения, обладающие опиатоподобными свойствами (опиоиды). Для них характерна сильная анальгезирующая активность, обеспечивающая возможность их использования при травмах (операционные вмешательства, ранения и др.) и при заболеваниях, сопровождающихся выраженным болевым синдромом (злокачественные новообразования, инфаркт миокарда, наркотическая «ломка» и др.). Поэтому их также называют большими анальгетиками.
По источникам получения и химическому строению к анальгетикам второй группы относятся природные алкалоиды - морфин и кодеин, содержащиеся в снотворном маке; полусинтетические соединения, полученные путем химической модификации молекулы морфина (этилморфин); синтетические соединения (промедол, фентанил, пентазоцин, налбуфин, буторфанол, трамадол и др.). Большинство синтетических препаратов получено по принципу воспроизведения упрощенной структуры морфина. Путем химической модификации молекулы морфина получены также соединения, являющиеся его фармакологическими антагонистами (налоксон, налтрексон). Для уменьшения побочных явлений часто назначают вместе с морфином атропин, метацин или другие холинолитики [Машковский М.Д. Лекарственные средства. 14 издание. М.: Новая волна, 2003, т. 1, с. 145].
Указанные средства обладают особым влиянием на центральную нервную систему человека, выражающимся в развитии эйфории и появлении при повторном применении синдромов психической и физической зависимости (лекарственной зависимости), что ограничивает возможность длительного применения этих препаратов. У пациентов с развившимся синдромом физической зависимости при лишении их анальгетического препарата может развиться болезненное состояние - абстинентный синдром.
Анальгетики второй группы вызывают острые токсические явления, такие как угнетение дыхания, нарушение сердечной деятельности. Для их снятия применяются специальные лекарственные средства -антагонисты, например, налоксон или налтрексон.
При повторном применении больших анальгетиков обычно развивается толерантность, то есть ослабление действия назначенной текущей дозы препарата, когда для получения анальгезирующего эффекта требуются все более высокие дозы препарата.
Действие указанных анальгетиков не ограничивается болеутоляющим эффектом. В той или иной степени, они оказывают снотворное действие, угнетают дыхание и кашлевой рефлекс, повышают тонус кишечника и мочевого пузыря.
В то же время они могут вызвать тошноту, понос и другие побочные явления.
В связи с выраженным наркогенным потенциалом анальгетики второй группы подлежат хранению, назначению и отпуску из аптек согласно особым правилам. В последние годы ряд применявшихся больших анальгетиков, обладающих выраженным наркогенным потенциалом (текодин, гидрокодона фосфат, фенадон, некоторые готовые лекарственные формы, содержащие опий и кодеин), исключен из номенклатуры лекарственных средств в Российской Федерации [Машковский М.Д. Лекарственные средства. 14 издание. М.: Новая волна, 2003, т. 1, с. 145].
Нейрофизиологические исследования свидетельствуют об угнетении указанными анальгетиками таламических центров болевой чувствительности и блокировании передачи болевых импульсов к коре головного мозга. Однако по своему действию эти анальгетики отличаются от представителей первой группы, так как влияют на способность центральной нервной системы к суммации подпороговых импульсов.
В последнее время получены данные о наличии в мозге и других органах специфических «опиатных» рецепторов [Машковский М.Д. Лекарственные средства. 14 издание. М.: Новая волна, 2003, т. 1, с. 146]. Эндогенными лигандами, то есть связывающимися с этими рецепторами специфическими, образующимися в организме физиологически активными соединениями, являются нейропептиды - прежде всего, энкефалины и эндорфины и недавно открытые в США проф. Д. Задиной эндоморфины [Zadina J.E., Hackler L., Ge L., Kastin A. A potent and selective endogenous agonist for the μ-opiate receptor // Nature 1997, V. 386. Р. 499-502]. Эндогенные лиганды оказывают анальгетическое действие и их эффект блокируется специфическими антагонистами опиатов. Связывание морфина с этими рецепторами обеспечивается тем, что определенная часть его молекулы имеет структурное, в том числе и конформационное, сходство с тирозиновым остатком энкефалинов и эндорфинов. Таким образом, экзогенный анальгетик морфин (так же как и другие близкие к нему по структуре опиаты и опиоиды) при введении в организм взаимодействует с теми же рецепторами, которые предназначены для связывания эндогенных лигандов. Кроме того, не исключено, что действие экзогенных анальгетиков связано также со стабилизацией эндогенных лигандов путем инактивации разрушающих энкефалины ферментов - энкефалиназ.
Показано, что опиатные рецепторы существуют в виде разных субпопуляций (подгрупп): μ (мю) (или ОР3 по системе IUPHAR), 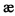 (каппа) (ОР2), σ (сигма), δ (дельта) (OP1), имеющих различную функциональную значимость. Полагают, что мю-рецепторы опосредуют супраспинальную анальгезию, эйфорию, угнетение дыхания и физическую зависимость, каппа-рецепторы опосредуют спинальную анальгезию, миоз, седативный эффект. Разные эндогенные лиганды и большие анальгетики могут связываться преимущественно с той или иной подгруппой рецепторов, что может определять особенности их фармакологического действия.
(каппа) (ОР2), σ (сигма), δ (дельта) (OP1), имеющих различную функциональную значимость. Полагают, что мю-рецепторы опосредуют супраспинальную анальгезию, эйфорию, угнетение дыхания и физическую зависимость, каппа-рецепторы опосредуют спинальную анальгезию, миоз, седативный эффект. Разные эндогенные лиганды и большие анальгетики могут связываться преимущественно с той или иной подгруппой рецепторов, что может определять особенности их фармакологического действия.
Разнообразные большие анальгетики различаются также по характеру связывания с опиатными рецепторами. Одни из них (морфин, промедол, фентанил и др.) являются «чистыми» полными агонистами; связываясь с рецепторами, они оказывают характерное для эндогенных лигандов физиологическое (фармакологическое) действие. Другие являются «чистыми» антагонистами (налоксон, налтрексон). Связываясь с рецепторами, они блокируют действие эндогенных лигандов и экзогенных опиатов. В третью группу входят препараты смешанного типа действия (агонисты-антагонисты), по-разному связывающиеся с разными подгруппами опиатных рецепторов и оказывающие в связи с этим по одним видам действия агонистический эффект, по другим - антагонистический (трамадол, налорфин, пентазоцин, нальбуфин и др.).
Действие больших анальгетиков на периферические органы (кишечник и др.) также связано с взаимодействием с локализующимися в них опиатными рецепторами.
Выраженного местноанестезирующего действия у большинства опиатов не отмечается. Вместе с тем в последние годы обнаружено, что они оказывают сильное обезболивающее действие при эпидуральном и субарахноидальном введении. Этот эффект связан с непосредственным воздействием на нейрональные системы спинного мозга, участвующие в формировании болевого потока импульсов. Этот способ введения опиатов находит применение для купирования тяжелых острых и хронических болей.
Из сказанного выше следует, что, с точки зрения современных воззрений на механизм действия больших анальгетиков, наиболее эффективными из них являются представители агонистов. Однако из-за вызываемых ими побочных эффектов, прежде всего сильной лекарственной (по сути наркотической) зависимости и интоксикации организма, их применение строго ограничено в зарубежных странах и запрещено в Российской Федерации. В настоящее время не известны синтетические аналоги агонистов, прежде всего, морфина, которые не вызывали бы указанных побочных эффектов.
Синтетический анальгетик последнего поколения, относящийся к группе агонистов-антагонистов опиатных рецепторов, - трамадол (tramadol), представляет собой ±-транс-2-[(диметиламино)метил]-1-(м-метоксифенил)циклогексанола гидрохлорид [Машковский М.Д. Лекарственные средства. 14 издание. М.: Новая волна, 2003, т. 1, с. 157]. Препарат обладает высокой анальгетической активностью, дает быстрый и длительный эффект. Трамадол уступает по активности морфину, поэтому применяется соответственно в больших дозах. Трамадол относительно хорошо переносится, не вызывает в обычных дозах выраженного угнетения дыхания и существенно не влияет на кровообращение и желудочно-кишечный тракт. Трамадол, однако, может вызывать тошноту, рвоту, головокружение, потливость; повышенное артериальное давление может несколько снижаться.
Недостатком известного средства является то, что трамадол не является «чистым» агонистом, поэтому по эффективности объективно уступает морфину. Так же, как и морфин, при повторных приемах он вызывает привыкание и интоксикацию организма у большинства пациентов.
Из уровня техники известно применение соединений пептидной природы в качестве анальгетических средств.
Так в патенте RU 2286169 (приоритет от 18.04.2005, дата публикации 27.10.2006) раскрыто получение и применение пептидов формулы (I), обладающих анальгетической активностью, для создания новых лекарственных препаратов. Пептиды обладают повышенной активностью по сравнению с пентальгином, анальгином, морфином, не токсичны.
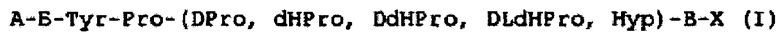
где А - О, - Ala, -Asp, -Glu, -Phe, -Gly, -His, -He, -Lys, -Leu, -Met, -Pro, -Arg, -Ser, -Thr, -Val, -Trp, -Tyr; Б - О, - Ala, -Asp, -Glu, -Phe, -Gly, -His, -He, -Lys, -Leu, -Met, -Pro, -Arg, -Ser, -Thr, -Val, -Trp, -Tyr; В - О, - Ala, -Asp, -Glu, -Phe, -Gly, -His, -He, -Lys, -Leu, -Met, -Pro, -Arg, -Ser, -Thr, -Val, -Trp, -Tyr; X - OH, -OCH3, -NH2
Однако в материалах данного патента не представлен молекулярный механизм связывания указанных субстанций со специфической мишенью - мембранным рецептором. Скорее всего анальгетический эффект указанных агентов пептидной природы обусловлен их воздействием на один из классов опиоидных рецепторов. В таком случае их действие не может быть абсолютно безопасным, поскольку все агонисты опиоидных рецепторов, как было отмечено выше, вызывают большое количество негативных побочных эффектов в организме человека. Следует отметить, что в описании изобретения данные, указывающие на безопасность применения указанных агентов, отсутствуют. В первую очередь это относится к таким негативным побочным эффектам, как привыкание и токсичность.
Наиболее близким к заявляемому решению является трипептид Ac-RER-NH2, [Молекулярный механизм модуляции трипептидом возбудимости мембраны ноцицептивного нейрона, авт. Шелых Т.Н., Рогачевский И.В., Ноздрачев А.Д., Веселкина О.С., Подзорова С.А., Крылов Б.В., Плахова В.Б., доклады академии наук, Том 466, №6, 2016, с. 734]. В данной работе указано, что механизм действия данного трипептида обусловлен модуляцией каналов NaV1.8. Кроме того, описана способность аргининсодержащих пептидов Ac-RER-NH2, Ac-RR-NH2 и свободной молекулы Arg модулировать возбудимость мембраны ноцицепторов. Внеклеточный Ac-RER-NH2 при взаимодействии с наружной мембраной ноцицептивного нейрона вызывает незначительное снижение величины Zeff активационной воротной системы каналов NaV1.8, что позволило в данной публикации предложить использование трипептида Ас-RER-NH2 в качестве анальгетического средства.
Однако, в настоящее время существуют указания на то, что трипептиды могут взаимодействовать непосредственно с ДНК живой клетки благодаря своим малым размерам и способности проникать через цитоплазматическую и ядерную мембраны [Khavinson V, Linkova N, Kukanova E, et al. Neuroprotective Effect of EDR Peptide in Mouse Model of Huntington Disease. J Neurol, and Neurosci. 2016, 8: 1-10]. Следствием этого могут стать негативные нарушения функционирования генетического аппарата, что, в свою очередь, может проявляться в отложенных во времени негативных действиях испытываемого препарата.
Актуальной является задача синтеза новых пептидов, обладающих анальгетическими свойствами для разработки на их основе эффективных и безопасных лекарственных препаратов, лишенных недостатков, характеризующих перечисленных выше аналоги.
Раскрытие изобретения
Задачей настоящей группы изобретений является создание нового высокоэффективного синтетического пептида, обладающего анальгетическим свойством, не вызывающего негативных побочных клинических эффектов, прежде всего интоксикации и привыкания, присущих известным аналогам, новой фармацевтической композиции, включающей данный пептид, и способа купирования хронической и острой боли.
Поставленная задача решается созданием пептидного соединения, обладающего анальгетическим свойством, общей формулы:
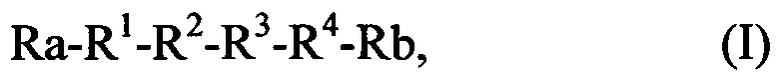
где
- Ra представляет собой N-концевую первичную аминную группу аминокислоты R1, либо свободную, либо замещенную аминозащитной группой,
- Rb представляет собой гидроксильную группу С-концевой карбоксильной группы аминокислоты R4, либо свободную, либо замещенную гидроксизащитной группой, и
- последовательность R1-R2-R3-R4 представляет собой аминокислотную последовательность, выбранную из группы: RERR, RERK, REKR, REKK, KERR, KEKR, KERK, KEKK, RDRR, RDRK, RDKR, RDKK, KDRR, KDRK, KDKR, KDKK, RRER, KRER, RKER, KKER, RREK, RKEK, KREK, KKEK, RRDR, KRDR, RKDR, KKDR, RRDK, KRDK, RKDK, KKDK.
Аминокислоты, входящие в состав последовательности R1-R2-R3-R4, являются левовращающими или правовращающими. При этом пептидное соединение может представлять собой водный раствор, или лиофилизат.
Поставленная задача решается также созданием фармацевтической композиции, обладающей анальгетическим действием, включающей активный компонент - пептидное соединение указанной формулы в эффективном количестве и фармацевтически приемлемый носитель.
Активный компонент и носитель могут быть взяты в следующем соотношении компонентов, мас. %: активный компонент - 1-95, носитель - остальное. Содержание активного компонента в однократной дозе может составлять составляет 10 - 400 мг.
Композиция может быть выполнена в форме для внутривенного, внутримышечного, спинального, эпидурального, мукозального, чрескожного, парентерального, перорального, энтерального, интраназального или ректального введения, например, в форме порошка, пластыря, свечи, раствора, суспензии, крема, мази, таблетированной или капсулированной форме.
Носитель может быть выбран из группы: вода, этанол, глицерол, физиологический раствор. Носитель дополнительно может содержать эмульгирующий агент и/или буферный агент. Носитель может быть выбран из группы: полиэтиленгликоль, лактоза, магнезиум карбонат, тальк.
Поставленная задача решается также созданием способа купирования болевого синдрома, включающего введение фармацевтической композиции в терапевтически приемлемом количестве. Композиция может быть использована в разовой дозировке с содержанием активного компонента не более 400 мг, при этом суточная дозировка активного компонента составляет не более 600 мг для пациентов с массой тела 50-100 кг.
Совокупность существенных признаков заявляемого синтетического пептида и анальгетического средства на его основе позволяет достичь следующего технического результата. Создано высокоэффективное синтетическое анальгетическое средство типа большого анальгетика, способного к применению в замещающей терапии, при этом не вызывающее вредных побочных фармакологических эффектов, прежде всего, интоксикации и привыкания. Поскольку нет привыкания и побочных вредных действий, в отличие, например, от трамадола, а также отсутствует вероятность возникновения отложенных негативных эффектов из-за воздействия на геном, как это могло бы иметь место при действии трипептидов (RER), то заявляемое средство, можно начинать принимать в максимальных для данного средства однократных дозах (от 1 до 60 мг в сутки) и практически сразу снимать острую боль (1 фаза), а затем и тоническую (2 фаза). Указанный эффект достигается в результате тонкого механизма специфического воздействия указанной субстанции пептидной природы на ее молекулярную мишень - медленные натриевые каналы (NaV1.8), ответственные за импульсное кодирование болевых сигналов и неожиданного проявления коротким пептидом уникальной способности обеспечить купирование хронической боли в течение длительного периода времени (не менее четырех-шести часов при однократном воздействии), сравнимого по своей длительности с эффектом больших анальгетиков.
Удлинение аминокислотной последовательности до уровня тетрапептидов позволяет обойти проблему потери специфичности действия разрабатываемого анальгетика. Ее решение было осуществлено следующим образом. В процессе исследования методом локальной фиксации потенциала (patch-clamp method) в конфигурации целой клетки (whole cell) была изучена возможность проявления положительно заряженными аргининсодержащими тетрапептидами способности модулировать функционирование медленных натриевых каналов NaV1.8, ответственных за кодирование и передачу в центральную нервную систему ноцицептивной информации. Из литературных данных известно [Молекулярный механизм модуляции трипептидом возбудимости мембраны ноцицептивного нейрона, авт. Шелых Т.Н., Рогачевский И.В., Ноздрачев А.Д., Веселкина О.С., Подзорова С.А., Крылов Б.В., Плахова В.Б., доклады академии наук, Том 466, №6, 2016, с. 734], что свободная молекула аргинина (R) и молекула дипептида RR не оказали влияния на электрофизиологические характеристики каналов NaV1.8, в то время как молекула трипептида RER, приложенная в форме Ac-RER-NH2 в концентрации 1 мкмоль/л, вызывала достоверное снижение величины эффективного заряда их активационного воротного устройства от Zeff=6.6±0.3 (n=24) до Zeff=4.6±0.4 (n=14). По-видимому, данный пептид связывается непосредственно с аминокислотной последовательностью этого канала.
Однако, как было отмечено выше, действие этого трипептида может вызвать негативные побочные эффекты из-за прямого (неспецифического) воздействия на геном. Для устранения этого недостатка были проведены соответствующие исследования.
Указанный трипептид был заменен на тетрапептид путем добавления к аминокислотной последовательности еще одного аргинильного остатка. В результате было обнаружено, что тетрапептид Ac-RERR-NH2, характеризующийся увеличенной длиной аминокислотной последовательности атакующего пептида до четырех аминокислот, вызывает достоверное снижение величины Zeff до 4.6±0.3 (n=23) в концентрации 100 нмоль/л, что на порядок эффективнее, чем действие Ac-RER-NH2. Это свидетельствует о более высокой специфичности действия заявляемого тетрапептида при связывании со своей молекулярной мишенью (каналами NaV1.8) по сравнению с молекулой Ac-RER-NH2. Очевидно, что чем ниже действующая концентрация атакующей молекулы, тем менее вероятно ее взаимодействие с другими белками, активация которых может стать причиной негативных побочных эффектов.
Таким образом, возможно использование тетрапептида в более низких концентрациях при сохранении терапевтического эффекта, что в свою очередь снижает вероятность его взаимодействия с геном, поскольку он уже задействован в связывании со своей мишенью - ионным каналом.
Помимо этого, удлинение аминокислотной последовательности делает невозможным прямое действие на геном указанной молекулы в примененной концентрации. В отличие от эффектов трипептидов, в мировой литературе отсутствуют доказательства изменения экспрессии генов или фенотипа клетки, вызванных механизмами, обусловленными действием тетрапептидов на последовательности ДНК.
Аргинильные остатки в составе молекулы Ac-RERR-NH2 непосредственно ответственны за высокоспецифическое связывание атакующей молекулы с ее молекулярной мишенью (каналом NaV1.8) за счет ион-ионных взаимодействий. Остаток глутаминовой кислоты отвечает за поддержание продуктивной конформации тетрапептида. Это означает, что замена любого из аргининов (R) на лизин (K), несущий положительный заряд в боковой цепи на сравнимом расстоянии от пептидного скелета, равно как и замена глутаминовой кислоты (Е) на также отрицательно заряженную аспарагиновую кислоту (D) приводит к получению тетрапептида, проявляющего эффект, аналогичный наблюдаемому для Ac-RERR-NH2. Инверсия аминокислотной последовательности тетрапептида с одновременной заменой составляющих его аминокислот на их правовращающие стереоизомеры также сохраняет проявляемый эффект. К тому же, природа защитных групп на N- и С-концах короткого пептида не влияет на его способность модулировать функционирование каналов NaV1.8, если эти группы не обладают большим объемом и, тем самым, не препятствуют образованию лиганд-рецепторного комплекса. Основной вклад в энергетику образования лиганд-рецепторного комплекса вносят высокоэнергетические ион-ионные связи между положительно заряженными гуанидиновыми группами боковых цепей двух аргинильных остатков молекулы Ac-RERR-NH2 и отрицательно заряженными нуклеофильными группами в составе молекулярной мишени - указанного ионного канала. Поскольку свободная молекула аргинина и дипептид Ac-RR-NH2 не проявили эффекта при исследовании методом локальной фиксации потенциала, можно сделать вывод, что для связывания с каналом NaV1.8 необходимо наличие как минимум двух положительно заряженных групп в составе атакующей молекулы, причем данные группы должны быть соответствующим образом ориентированы и располагаться на определенном расстоянии друг от друга для обеспечения возможности наиболее энергетически выгодного связывания тетрапептида по условной модели «вилка-розетка». Трипептид Ac-RER-NH2, удовлетворяет этому требованию, но не самым оптимальным образом, что проявляется в необходимости применения указанного агента в относительно высокой концентрации для достижения анальгетического эффекта. Эта высокая концентрация является причиной того, что трипептид Ac-RER-NH2 может преодолеть все мембранные барьеры и осуществить взаимодействие с молекулой ДНК, вызывая негативные побочные эффекты. Тетрапептид Ac-RERR-NH2 лишен этого недостатка благодаря своей молекулярной структуре.
Анализ структуры молекулы Ac-RERR-NH2 позволяет предположить, что указанное расстояние, за которое было принято расстояние между центральными атомами углерода гуанидиновых групп, находится в интервале 14±6.  . Такая модель «вилка-розетка» с указанным интервалом расстояний между центральными атомами углерода гуанидиновых групп является неочевидной, не следующей из уровня техники.
. Такая модель «вилка-розетка» с указанным интервалом расстояний между центральными атомами углерода гуанидиновых групп является неочевидной, не следующей из уровня техники.
Обобщая вышеприведенные данные, можно заключить, что анальгетическими свойствами обладают следующие тетрапептиды, характеризующиеся набором защитных групп на N- и С-концах, а также незащищенных концевых групп:
1. RERR, RERK, REKR, REKK, KERR, KEKR, KERK, KEKK, RDRR, RDRK, RDKR, RDKK, KDRR, KDRK, KDKR, KDKK.
2. также инвертированные цепочки, состоящие из D-аминокислот: RRER, KRER, RKER, KKER, RREK, RKEK, KREK, KKEK, RRDR, KRDR, RKDR, KKDR, RRDK, KRDK, RKDK, KKDK.
Заявляемая композиция (средство) с использованием тетрапептида может быть выполнена в любой форме для систематического приема - как для наружного, так и внутреннего применения в виде раствора, порошка, крема, геля, капсулы, пластыря, свечей. Средство может быть использовано для парентерального, орального, инвазивного (в том числе пролонгированного) приема, ингаляций, распыления на кожу и т.д. Заявляемое средство способно воздействовать однократно (одномоментно) и может быть приготовлено в форме для пролонгированного действия (пластырь, свеча, имплантат).
Заявляемое средство нетоксично, так как нетоксично его активное начало, имеющее эндогенную пептидную природу. Активное начало для заявляемого средства технологически доступно. Его синтез из аминокислот является рутинной процедурой. Кроме того, возможен более дешевый микробиологический способ получения тетрапептида.
Заявляемое средство отличается от известных аналогов, прежде всего морфина и трамадола, существенными признаками, связанными с эндогенной природой активного начала (короткий пептид). Способность короткого пептида к анальгетической активности была открыта автором изобретения случайно. Уникальный механизм его действия, который по сути, отрицает привыкание к препарату, раскрыт так же автором изобретения. Следует подчеркнуть, что исследователям до сих пор не удавалось разработать большие анальгетики, которые сами по себе не вызывают привыкания.
Заявляемый способ купирования боли позволяет достичь абсолютно безопасного эффекта долговременного облегчения хронической и острой боли, длящегося в течении 4-6 часов после однократного введения действующей субстанции, что сравнимо с известными эффектами действия опиатов. Заявляемый способ купирования боли является щадящим, так как используемое в нем средство безопасно, а механизм его воздействия сугубо избирателен. Использование средства не вызывает привыкания и, следовательно, необходимости постоянного увеличения дозы лекарства. Заявляемый способ отличается от известного рядом существенных признаков: использованием нового анальгетического средства в определенных дозах, универсальными возможностями применения лекарства - при самых разных видах приема: наружно, в том числе в виде крема (геля), имплантатов (подшитых капсул), пластырей, и в виде приема внутрь (порошки, таблетки, уколы, ингаляции). Из уровня техники не известно способов лечения хронической боли, при реализации которых происходит подобное тонкое избирательное воздействие на соответствующие каналы и активация сигнального пути передачи ноцицептивной информации, приводящая к купированию хронической боли без интоксикации, привыкания.
Краткое описание чертежей
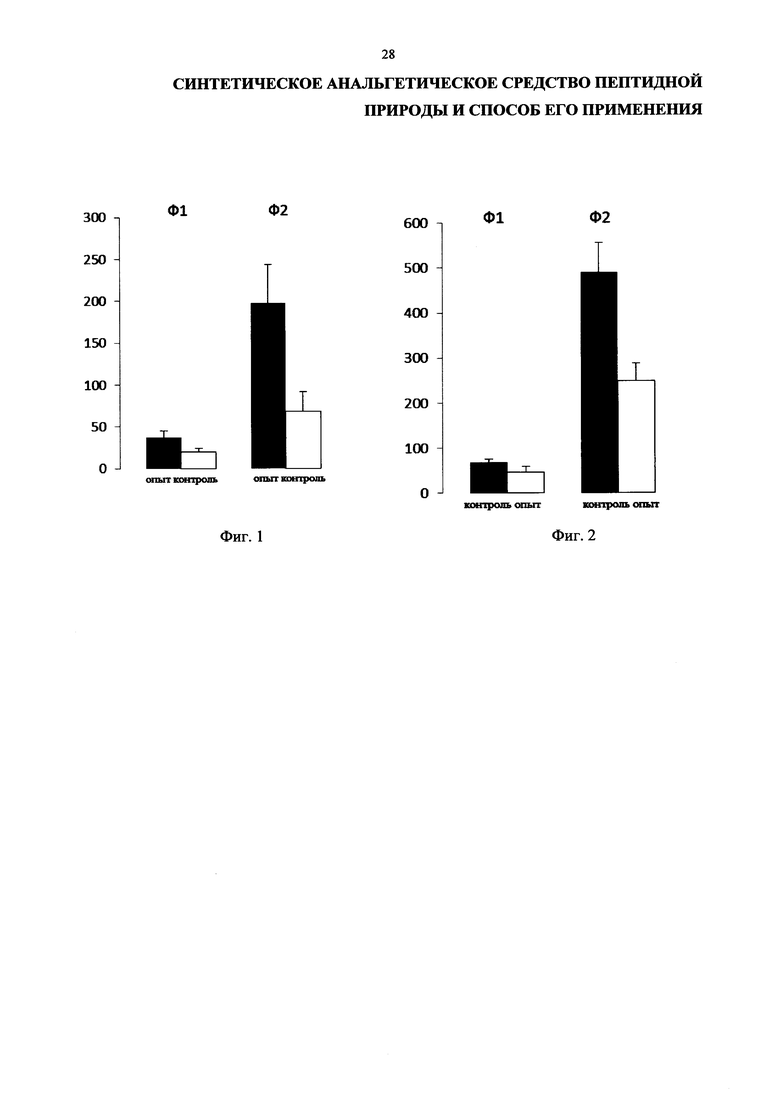
На фиг. 1 представлены результаты формалинового теста, а именно интенсивность паттернов вылизывания, подтверждающие достижение анальгетического эффекта.
На фиг. 2 представлены результаты формалинового теста, а именно паттернов сгибаний + встряхиваний, подтверждающие достижение анальгетического эффекта.
Осуществление изобретения
Синтез заявляемого синтетического тетрапептида из аминокислот является рутинной процедурой. Также возможен более дешевый микробиологический способ получения тетрапептидов.
Для приготовления фармацевтической композиции (средства), пептид формулы (I) смешивают, по крайней мере, с одним фармацевтически пригодным наполнителем (носителем). Подходящие наполнители и носители хорошо известны в данной области техники (см., например, ''Handbook of Pharmaceutical Excipients''' 5th Edition by Raymond C. Rowe, Paul J. Sheskey, Sian C. Owen (2005), APhA Publications).
Фармацевтическая композиция в соответствии с настоящим изобретением может быть представлена в твердой или жидкой форме, например, в виде таблеток, капсул, жидкостей, инфузий и суппозиториев, изготовленных по известной технологии (см., например, "Pharmaceutical Formulation Development of Compounds" by Sven Frokjaer (1999), CRC; "Handbook of Pharmaceutical Manufacturing Formulations" by Sarfaraz K. Niazi (2004), CRC.
Для приготовления твердых форм композиций активное начало соединяют с, по меньшей мере, одним наполнителем, выбранным из группы: лактоза, магнезиум карбонат, целлюлоза и ее производные (например, микрокристаллическая целлюлоза); полиолы (лактоза и ее производные, сахароза, мальтоза, фруктоза); сорбит; маннит; поливинилпирролидон и его сополимеры; соли неорганических соединений (например, кальция карбонат); кальция фосфат, кальция дигидрофосфат, кальция гидрофосфат дигидрат, кальция сульфат дигидрат) и т.д. При приготовлении твердых форм могут быть использованы вспомогательные вещества с сорбционной способностью: коллоидный кремний (аэросил, Syloid® (Grace, США)), алюмометасиликат магния (Neusilin®, Fuji, Япония).
В качестве связывающего вещества может быть использовано, по меньшей мере, одно, выбранное из группы: крахмальный клейстер, растворы: карбоксиметилцеллюлозы, оксиэтилцеллюлозы, оксипропилметилцеллюлозы; поливинилового спирта, поливинилпирролидона, натрия альгината, метилцеллюлозы, гидроксипропилметилцеллюлозы. В качестве растворителя для приготовления раствора связывающего вещества может быть использовано, по меньшей мере, одно, выбранное из группы: вода очищенная, спирт этиловый, хлороформ.
В случае необходимости добавляют разрыхляющие вещества. В качестве разрыхляющего вещества может быть использовано, по меньшей мере, одно, выбранное из группы: крахмал (пшеничный, картофельный, кукурузный); пектин, натрий карбоксиметилцеллюлоза; поливинилпирролидон, кроскармеллоза натрия, крахмал прежелатизированный, карбоксиметилированный).
В качестве поверхностно-активного вещества (ПАВ) может быть использовано, по меньшей мере, одно, выбранное из группы: олеат натрия, лаурилсульфат натрия.
В качестве вспомогательного вещества, предотвращающего налипание и обеспечивающего выталкивание из матрицы (антифрикционное вещество), может быть использовано, по меньшей мере, одно, выбранное из группы: стеариновая кислота и/или ее соли, коллоидный кремний (аэросил).
Фармацевтическая композиция в ТЛФ в виде таблеток или капсул, получают методом прямого прессования, влажного и сухого гранулирования, а также пеллетизацией.
Жидкую композицию приготавливают растворением или суспендированием активного начала в воде, физиологическом растворе, глицероле, этаноле и т.д. с добавлением при необходимости эмульгирующих агентов, рН буферных агентов. Как правило, используют 1-95%-ный водный раствор активного начала.
В соответствии с предпочтительным воплощением настоящего изобретения композиция может дополнительно включать, по крайней мере, один дополнительный фармацевтически активный компонент. Например, антиоксиданты, такие как витамины, могут рассматриваться как дальнейшие активные компоненты, поскольку антиоксиданты ингибируют окисление или подавляют реакции, стимулируемые кислородом, свободными радикалами кислорода, реакционноспособными формами кислорода, включая пероксиды. Антиоксиданты, пригодные в настоящем изобретении, предпочтительно являются антиоксидантными витаминами, которые могут быть выбраны из группы, состоящей из всех форм витамина А, включая ретиналь и 3,4-дидегидроретиналь, все формы каротина, такие как α-каротин, β-каротин, γ-каротин, δ-каротин, все формы витамина С (D-аскорбиновую кислоту, L-аскорбиновую кислоту), все формы токоферола, такие как витамин Е (α-токоферол, 3,4-дигидро-2,5,7,8-тетраметил-2-(4,8,12-триметилтридецил)-2Н-1-бензопиран-6-ол), β-токоферол, γ-токоферол, δ-токоферол, токохинон, токотриенол и эфиры витамина Е, которые легко подвергаются гидролизу до витамина Е, такие как витамин Е ацетат и витамин Е сукцинат, и фармацевтически пригодные соли витамина Е, такие как витамин Е фосфат, пролекарства витамина А, каротина, витамина С и витамина Е, фармацевтически приемлемые соли витамина А, каротина, витамина С и витамина Е и т.п. и их смесей.
Для подтверждения анальгетического действия заявляемого синтетического пептида и отсутствия побочных фармакологических эффектов, прежде всего интоксикации и привыкания были проведены эксперименты, где в качестве тестируемого тетрапептида использовалось соединение формулы Ac-RERR-NH2.
Для подтверждения соответствия заявляемого решения требованию «промышленная применимость», а также для лучшего понимания сущности изобретения ниже представлены примеры его конкретной реализации, которые не ограничивают заявляемое изобретение.
Пример 1. Способ получения тетрапептида формулы Ac-RERR-NH2.
Пептид заявляемой формулы был получен методом классического пептидного синтеза (Х.-Д. Якубке, X. Ешкайт. Аминокислоты. Пептиды. Белки. Москва, Мир, 1985. 339 с.) с применением реактивов и производных аминокислот фирм "Sigma Chemical Co." (США), "Iris Biotech GmbH" (Германия), растворителей производства компаний "Экос-1" и "Криохром" (Россия). Конечный продукт характеризовали с помощью аналитической ВЭЖХ (чистота >95%) и масс-спектрометрии.
Пример 2. Способ получения фармацевтической композиций, содержащей в качестве активного компонента тетрапептид формулы Ac-RERR-NH2, представленной в форме крема, и подтверждение его анальгетического действия.
Для чрезкожной доставки действующей пептидной субстанций был подобран оптимальный состав кремовой основы с учетом совместимости ее компонентов. Пептиды способны проникнуть через эпидермис, если помещены в амфифильную базу. Стандартная эмульсия является амфифильной базой и работает как система доставки пептида к глубоким слоям рогового слоя эпидермиса. Использование осмолитиков, таких как увлажнители (карбопол, гиалуроновая кислота), может обеспечить проникновение пептида до зернистого слоя эпидермиса. Здесь использован компонент Карбопол, который является проводником активных компонентов через эпидермис и не оказывает влияния на свойства активного компонента (тетрапептида), в сочетании с Триэтаноламином. Сначала была замешана кремовая основа, проверена ее стабильность в течение 1 (Одного) месяца (отсутствие изменения показателя рН более чем на 0,3 единицы, проверка коллоидной и термической стабильности и микробиологической чистоты). Затем в кремовую основу был введен тетрапептид (в количестве 1.0%). Никаких изменений внешнего вида рецептуры не произошло. Кремовая композиция, содержащая тетрапептид, была использована для получения клинических впечатлений после получения разрешительных документов (исходя из требований Технического Регламента Таможенного Союза 009/2011) на использование кремовой композиции в качестве косметического средства. Разработанная рецептура представлена в таблице 1.
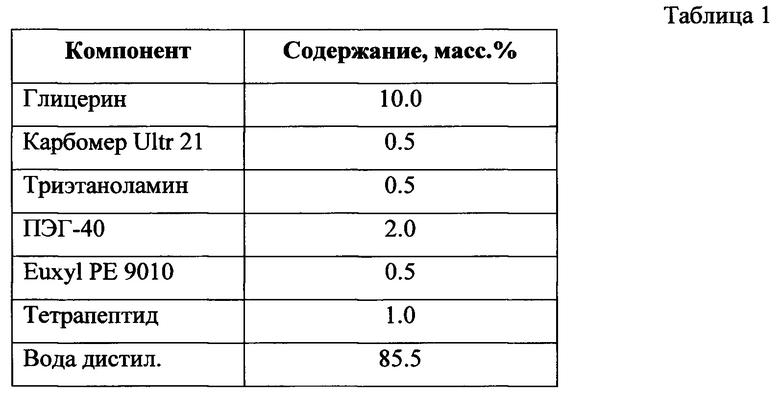
Физико-химические параметры рецептуры крема-геля с пептидной субстанцией приведены в таблице 2.
После проверки стабильности и значения водородного показателя рецептуры в кремовую основу был введен тетрапептид (в количестве 1.0%). При введении тетрапептида не произошло изменения внешнего вида и значения водородного показателя.
В клинических подразделениях было проведено исследования анальгетического эффекта тетрапептида Ac-RERR-NH2. Тетрапептид поступил для клинической апробации в виде стерильного 1% геля. Изучение его анальгетических свойств было выполнено на модели боли, связанной с травмой кожных покровов, а именно разреза кожи (послеоперационная боль). После обработки раны 70% раствором этилового спирта гель объемом около 1 мл втирали в рану семи пациентам после получения их информированного согласия. Все семь испытуемых отметили положительный эффект разной степени выраженности, т.е. интенсивность боли в ответ на давление на края раны становилась ниже после применения тетрапептида, чем до втирания его в рану, на 1-2 единицы (5-балльная вербальная шкала оценки боли (Frank A. J. М., Moll J. М. Н., Hort J.F., A comparison of three ways of measuring pain. Rheumatology and Rehabilitation 21, 211-217, 1982). В контрольной группе, состоящей из такого же числа испытуемых, интенсивность боли снизилась на единицу лишь у двух пациентов в ответ на воздействие стерильного геля, не содержащего активный компонент (тетрапептид).
Таким образом, полученный результат говорит о явном анальгетическом эффекте тетрапептида Ac-RERR-NH2.
Пример 3. Фармацевтические композиции, содержащие в качестве активного компонента тетрапептиды формул, представленные в различных фармацевтических формах.
1. Порошок, включающий активное начало - короткий пептид - в количестве от 10 мг (1 мас. %) и наполнитель - лактозу.
2. Порошок, включающий активное начало - короткий пептид - в количестве 100 мг (95 мас. %) и наполнитель - лактозу.
3. Порошок, включающий активное начало - короткий пептид - в количестве 50 мг (10 мас. %) и наполнитель - лактозу.
4. Порошок, включающий активное начало - короткий пептид - в количестве 10 мг (1 мас.%) и наполнитель - полиэтиленгликоль.
5. Порошок, включающий активное начало - короткий пептид - в количестве от 100 мг (90 мас. %) и наполнитель - магнезиум карбонат.
6. Таблетированная форма, включающая активное начало - короткий пептид - в количестве от 100 мг (80 мас. %) и наполнитель - магнезиум карбонат.
7. Пластырь, с нанесенным на него заявляемым средством, включающим активное начало - короткий пептид - в количестве 400 мг (70 мас. %).
8. Капсула, включающая активное начало - короткий пептид - в количестве от 400 мг (70 мас. %).
9. 0,5%-ный водный раствор для инъекций, включающий активное начало - короткий пептид - в количестве от 100 мг.
10. 0,5%-ный раствор в физиологическом растворе для ингаляций, включающий активное начало - короткий пептид - в количестве 100 мг.
11. 0,5%-ную суспензию в глицероле для опрыскивания, включающую активное начало - короткий пептид - в количестве 100 мг.
Указанные составы испытаны на животных - крысах в однократных дозах и суточных дозах, определенных в приведенных выше экспериментах. Аллергических реакций не наблюдалось.
Эффект снятия боли после каждого применения длился не менее 4-6 часов, до 24 часов.
Показано, что устойчивый эффект купирования боли наблюдался после применения заявляемого средства в течение месяца - 50 дней и даже более - без возникновения лекарственной зависимости.
При лечении заявляемыми средствами у животных не наблюдалась присущая аналогам интоксикация организма, они не испытывают рвоты.
Эффективная суточная доза заявляемого средства была пересчитана по итогам опытов на крысах в соответствии с методикой, описанной в [Руководство по экспериментальному (доклиническому) изучению новых фармакологических веществ. Министерство здравоохранения РФ. Фармкомитет РФ. М., 2000]. Так, суточную дозу, использованную для крысы массой 250 г, для перевода в дозу для человека массой 70 кг следует разделить на коэффициент, равный 7. Как указывалось выше, в эксперименте с крысами была использована оптимальная суточная_доза от 34 до 40 мг короткого пептида/кг массы крысы. Пересчет показывает верхний предел суточной дозы (позволяет ввести начальную безопасную по опыту работы с другими анальгетиками точку отсчета для человека для проведения дальнейших доклинических испытаний по действию и концентрациям в ту или другую сторону) для человека - до 6 мг короткого пептида/кг. Этот подход коррелирует с данными по анальгетикам, полученными в работе [Leeling J.L., Evans J.V., Helms R.J., Ryerson B.A. Disposition and metabolism of codorphone in the rat, dog, and man // Drug Metab Dispos. 1982. Nov-Dec. V. 10(6) P. 649-653], где показаны дозы для человека в 10 раз меньшие, чем у животных, из-за более быстрого метаболизма животных.
Таким образом, эффективная суточная доза для человека составляет до 6 мг активного начала на 1 кг живой массы, что обеспечивает терапевтический эффективный ее уровень в крови. Предпочтительная доза составляет от 4,8 до 5,7 мг на 1 кг живой массы в один или несколько дневных приемов. Определяемая врачом доза может быть получена пациентом за один прием или за несколько, а также в виде продолжительного применения.
При использовании для лечения людей с массой 50-100 кг дневной прием с учетом массы тела может составлять до 300-600 мг заявляемого средства, что решается сугубо индивидуально в зависимости от состояния пациента.
Оптимальная суточная доза для постоянного применения в виде уколов каждодневно может составлять до 4-6 мг на 1 кг массы.
Прием заявляемого средства может быть прописан пациенту в любом виде и в любой форме: орально, ректально и т.д.
Возможно лечение при имплантации лекарственной субстанции внутрь организма или через кожные покровы.
Возможен прием лекарства в виде порошка - назально, или в виде ингаляций аэрозолей.
Следует подчеркнуть, что дозы заявляемого средства не превосходят дозы известного препарата - трамадола. Суточные дозы в рамках заявленных могут назначаться в широких пределах в соответствии с индивидуальными особенностями и рекомендациями врачей.
Возможность реализации изобретения не ограничивается приведенными выше примерами.
Согласно результатам испытаний, получено высокоэффективное синтетическое анальгетическое средство, не вызывающее побочных фармакологических эффектов, прежде всего интоксикации и привыкания, присущих известным аналогам (морфину, трамадолу), и метод лечения хронической боли, а также метод реабилитации при наркотической и лекарственной зависимости с помощью этого средства, протекающий быстрее, чем у аналогов, и приводящий к долговременному купированию хронической боли, а также к снятию абстинентного синдрома и к значительному уменьшению рецидивов и летальных исходов.
Многократное повторение экспериментов показало хорошую воспроизводимость результатов.
Нижние рамки интервальных параметров допустимых концентраций короткого пептида распространяются и на гомеопатические дозы; выход за верхние рамки доз возможен, но не целесообразен с точки зрения передозировки.
Пример 4. Подтверждение анальгетического эффекта заявляемого пептида.
4.1 Измерение эффективного заряда активационной воротной системы каналов NaV1.8 методом локальной фиксации потенциала.
Эксперименты выполнены на культивируемых изолированных нейронах спинальных ганглиев новорожденных крысят линии Wistar. Для выделения нейронов был применен модифицированный метод краткосрочного культивирования (Elliott A.A., Elliott J.R. Characterization of TTX-sensitive and TTX-resistant sodium currents in small cells from adult rat dorsal root ganglia // J. Physiol. (Lond.) 1993. V. 463. P. 39-56.), обеспечивающий высокий выход жизнеспособных клеток. В работе использовались следующие стандартные растворы (концентрации представлены в ммоль/л). Внеклеточный раствор: NaCl - 65, CaCl2 - 2, MgCl2 - 2, Choline Cl - 70, HEPES-Na - 10, TTX (тетродотоксин) -0.0001, рН=7.4. Внутриклеточный раствор: CsF - 100, NaCl - 10, CsCl - 40, MgCl2 - 2, HEPES-Na - 10, pH=7.2. Исключение из вне- и внутриклеточного растворов ионов калия позволило избавиться от всех компонентов калиевого тока, а ионы фтора во внутриклеточном растворе обеспечивали блокирование кальциевых токов (Kostyuk P.G., Krishtal О.A., Pidoplichko V.I. Effect of internal fluoride and phosphate on membrane currents during intracellular dialysis of nerve cells // Nature. 1975. V. 257. N 5528. P. 691-693). Наличие во внеклеточном растворе ионов тетродотоксина в низкой концентрации обеспечивало блокирование быстрых тетродотоксинчувствительных каналов, что делало возможным регистрировать характеристики только одной популяции натриевых каналов - медленных (тетродотоксиннечувствительных) каналов NaV1.8. В работе использованы реактивы фирмы "Sigma". Все опыты проведены при комнатной температуре 22-24°С.
Трансмембранные ионные токи регистрировали с помощью метода фиксации потенциала на целой клетке (Hamill О.P., Marty A., Neher Е., Sakmann В., and Sigworth F. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches // Pfliigers Arch. 1981. V. 391. N 1. P. 85-100) в условиях плотного контакта (метод patch-clamp в конфигурации «whole-cell»). Эксперименты проводили с использованием аппаратно-программного комплекса на основе усилителя «L/М ЕРС-7», ПЭВМ и системы автоматизации. Система автоматизации предназначена для выполнения следующих функций. Она включает в себя формирование программы эксперимента, генерацию ступенек напряжения, подаваемых на мембрану в соответствии с указанной программой, регистрацию ионных токов, визуальный контроль ионных токов, их экспресс-обработку, уточнение программы в ритме эксперимента в зависимости от характеристик исследуемого нейрона. Регистрация семейств натриевых токов и их дальнейшая обработка проводилась общепринятым методом (Hodgkin A. L., Huxley A. F. Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo // J. Physiol. 1952. V. 116. N 4. P. 449-472).
С помощью метода локальной фиксации потенциала в конфигурации регистрации активности целой клетки была исследована способность молекулы Ac-RERR-NH2 модулировать возбудимость мембраны ноцицепторов благодаря снижению потенциалочувствительности ответственных за кодирование болевых сигналов медленных натриевых каналов NaV1.8.
Наиболее важным стационарным параметром, определяющим процесс первичного кодирования ноцицептивной информации, служит эффективный заряд Zeff активационной воротной системы каналов NaV1.8. Было обнаружено снижение величины Zeff было при воздействии тетрапептида Ac-RERR-NH2 в концентрации 100 нмоль/л. Этот параметр снижался с контрольного значения Zeff=6.41±0.31 (n=17) до нмоль/л и до 4.63±0.32 (n=23) при приложении тетрапептида в концентрации 100 нмоль/л.
4.2 Формалиновый тест проведен по известной методике (Dubuisson D., Dennis S.G. The formalin test: a quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats // Pain. 1977. V. 4. P. 161-174; Tjolsen A., Berge O.-G., Hunskaar S., Rosland J.H., Hole K. The formalin test: an evaluation of the method // Pain. 1992. V. 51. P. 5-17, а также Henry J.L., Yashpal K., Pitcher G.M., Coderre T.J. Physiological evidence that the "interphase" in the formalin test is due to active inhibition // Pain. 1999. V. 89. P. 57-63; Dallel R., Raboisson P., Clavelou P. Evidence for a peripheral origin of the tonic nociceptive response to subcutaneous formalin // Pain. 1995. V. 61. P. 11-16; Abbott F.V., Melzack R, Samuel C. Morphine analgesia in the tail-test and formalin pain tests is mediated by different neural systems // Exp. Neurol. 1982. V. 75. P. 644-651; Abbott F.V., Ocvirk R., Najafee R. et al. Improving the efficiency of the formalin test // Pain. 1999. V. 83. P. 561-569). Опыты проведены на 7 опытных и 7 контрольных взрослых самцах крыс линии Вистар (средний вес животных 210 г). Введение тетрапептида опытным крысам в дозе 1,4 мг/кг (в растворе Хенкса) и физиологического раствора контрольным крысам в объеме 1 мл производили внутрибрюшинно за 5 мин до инъекции раствора формалина (2.5%, 50 мкл, подкожно) в подошву задней конечности.
В стандартных условиях эксперимента в контрольной и экспериментальной группах наблюдают два вида боли: 1) острая боль - первая фаза, обычно длящаяся менее 5 минут, 2) тоническая боль - вторая фаза, наблюдающаяся в течение 90 минут. Между первой и второй фазами существует интерфаза, продолжающаяся ~ 10 минут, во время которой ответов животного не наблюдается.
В протоколе отражали количество и продолжительность «вылизываний», «сгибаний», «встряхиваний» левой больной лапой. Оценивали межфазные интервалы и поведенческие реакции: исследовательское поведение, груминг, реакцию избегания, реакцию привыкания, реакцию интоксикации. В контрольных экспериментах вся последовательность событий соблюдалась: после введения формалина животное в течение 5 либо 7 минут находилось в темноте под колпаком. Интенсивность паттернов сгибаний + встряхиваний определяется их числом - это спинальный уровень ответа на формалин. Интенсивность паттернов вылизывания, определяемая их продолжительностью в секундах, отражает супраспинальный уровень ответа. Продолжительность первой и второй фазы, а также межфазного интервала отражали в минутах.
Регистрацию специфических паттернов ответа на инъекцию формалина - сгибание + встряхивание (спинальный уровень) и вылизывание (супраспинальный уровень) - проводили в течение часа после инъекции ноцицептивного химического раздражителя.
Данные формалинового теста. Фаза I (острая фаза).
Полученные данные показали в паттернах вылизывания тенденцию к снижению длительности вылизывания у всех экспериментальных животных (19,4±4,3) по сравнению с контрольными (37,3±7,4) (фиг. 1).

t=2,0715 р=0,05946 (тенденция),
при значении р=0.5 значения не различаются статистически достоверно.
В паттернах сгибание + встряхивание обнаружена значительная тенденция к снижению длительности острой фазы ответов, однако статистически достоверных различий не выявлено: длительность ответов опытных животных сократилась (46,6±14,0) по сравнению с длительностью ответов контрольных животных (68,7±7,2) (фиг. 2).
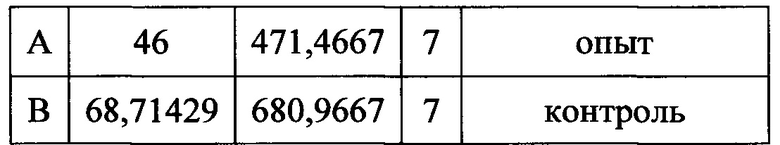
t=2,60961 р=0,17527
при значении р=0.5 значения не различаются статистически достоверно
Фаза II (тоническая)
Полученные данные показали достоверные различия в паттернах вылизывания у всех экспериментальных животных (68,5±23,9) по сравнению с контрольными (197,3±45,6).
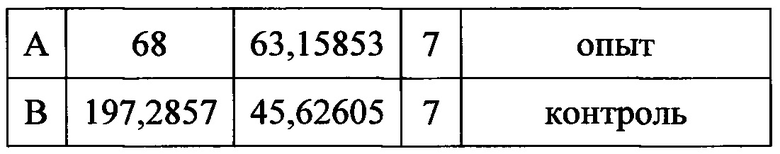
t=2,61084 р=0,02737
при значении р=0.5 значения различаются статистически достоверно
В паттернах сгибание + встряхивание также выявлены достоверные различия: у опытных животных (248,3±40,4), у контрольных (490,3±66,2).
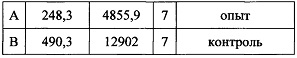
t=3,19432 р=0,00886
при значении р=0.5 значения различаются статистически достоверно
Проведенные поведенческие эксперименты позволяют сделать следующие выводы:
1. Инъекция тетрапептида крысам в дозе 1,4 мг/кг вызывает статистически достоверный анальгетический эффект на супраспинальном уровне. На спинальном уровне наблюдается тенденция к снижению болевого эффекта.
2. Наблюдения за экспериментальными животными показали, что ни в одном случае не наблюдалась реакция привыкания к действию исследуемой субстанции.
Пример 5. Подтверждение безопасности применения заявляемого пептида. Метод органотипической культуры ткани.
С помощью метода органотипической культуры ткани исследовали влияние тетрапептида Ac-RERR-NH2 на рост нейритов сенсорных ганглиев 10-12-дневных куриных эмбрионов. Культивирование эксплантатов осуществляли в соответствии со стандартной методикой (Чалисова Н.И., Мелькишев В.Ф., Акоев Г.Н., Людыно М.И., Куренкова Т.К. Стимулирующее влияние пролактина на рост нейритов чувствительных нейронов в органотипической культуре. // Цитология. - 1991. - Т. 33, №2. - С. 29-31).
Эксперименты были выполнены на культивируемых изолированных нейронах спинальных ганглиев крыс линии Wistar. Дорзальные ганглии выделяли из областей L5 - S1 спинного мозга и помещали в раствор Хенкса. Ферментативную обработку [10] проводили в течение 5-12 мин. (в зависимости от возраста крыс) при 37°С в растворе следующего состава: 1 мл раствора Хенкса, 1 мл среды Игла, 2 мг/мл коллагеназы (тип 1А), 1 мг/мл проназы-Е. В качестве буфера использовали 10 ммоль/л Hepes Na (рН 7.4). После ферментативной обработки ганглии тщательно отмывали путем центрифугирования (1 мин., 900 об/мин.) и последующей сменой надосадочной жидкости. Для отмывки и культивирования использовалась среда на основе среды Игла (ЕМЕМ) с добавлением глютамина (2 ммоль/л), эмбриональной сыворотки коровы (10%), глюкозы (0.6%), гентамицина 40 ед./мл. Механическую диссоциацию производили путем пипетирования. К полученной клеточной суспензии добавляли культуральную среду до достижения соответствующей плотности клеток в объеме чашки Петри. Культуру клеток, состоящую преимущественно из нейронов спинальных ганглиев, получали с помощью предварительного осаждения нейрональных клеток (шванновских клеток и фибробластов) на пластиковой поверхности 90-мм чашки Петри в течение 25 мин. при 37°С. Далее клетки культивировали на покрытой коллагеном поверхности 40-мм чашек Петри. Коллаген получали из хвостов крыс. Регистрацию электрической активности нейронов производили спустя 1-2 часа после завершения культивирования. Клетки использовали в опытах в течение одних суток.
Тетрапептид был применен в концентрациях 0,1 пкМ, 10 пкМ, 0,1 нМ, 1 нМ, 10 нМ и 100 нМ. В концентрациях 0,1 пкМ и 10 пкМ исследуемый агент практически не влиял на рост нервной ткани. Индекс площади (ИП) эксплантатов спинальных ганглиев не отличался от своего контрольного значения. В концентрации 0,1 нМ исследуемый тетрапептид проявлял нейрит-стимулирующее действие. Его введение в питательную среду в этой концентрации вызывало достоверную стимуляцию роста нейритов в среднем на 34% по отношению к контрольным значениям. При добавлении в культуральную среду тетрапептида в концентрации 1 нМ наблюдали недостоверную стимуляцию роста нейритов. ИП экспериментальных эксплантатов был выше на 20% по сравнению с контролем. В концентрациях 10 нМ и 100 нМ он практически не влиял на рост нейритов спинальных ганглиев. ИП экспериментальных эксплантатов не отличался от контроля. Таким образом, полученные данные свидетельствуют, что тетрапептид Ac-RERR-NH2 обладает выраженными нейрит-стимулирующими свойствами в очень низкой концентрации (0,1 нМ).
Отсутствие эффекта ингибирования роста нейритов позволяет сделать важнейший вывод о том, что тетрапептид Ac-RERR-NH2 не имеет нейротоксических свойств и является безопасным агентом, воздействующим на нервные клетки живого организма.
По аналогии были проведены исследования и с другими тетрапептидами, выбранными из перечисленной выше группы с получением сопоставимых результатов.
| название | год | авторы | номер документа |
|---|---|---|---|
| Анальгетическая субстанция эндогенной природы, фармацевтическая композиция на ее основе и способы их применения | 2023 |
|
RU2823100C2 |
| СРЕДСТВО ДЛЯ ЭФФЕКТИВНОГО КУПИРОВАНИЯ ОСТРОГО И/ИЛИ ХРОНИЧЕСКОГО БОЛЕВОГО СИНДРОМА И СПОСОБ ЕГО ПРИМЕНЕНИЯ | 2016 |
|
RU2622980C1 |
| СЕМЕЙСТВО ПЕПТИДОВ, ОБЛАДАЮЩЕЕ АНАЛЬГЕТИЧЕСКОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2286169C1 |
| СОСТАВ И СПОСОБ ПОЛУЧЕНИЯ АНАЛЬГЕТИЧЕСКОГО СРЕДСТВА ПЕПТИДНОЙ СТРУКТУРЫ | 2017 |
|
RU2669386C1 |
| АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО ПЕПТИДНОЙ СТРУКТУРЫ НА ОСНОВЕ ТРИДЕКАПЕПТИДА, СОДЕРЖАЩЕГО D-ОКТААРГИНИНОВЫЙ ВЕКТОР | 2013 |
|
RU2538727C1 |
| СИНТЕТИЧЕСКОЕ АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО И СПОСОБ ЛЕЧЕНИЯ НА ОСНОВЕ ЭТОГО СРЕДСТВА | 2006 |
|
RU2322977C1 |
| АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО И СПОСОБ ЛЕЧЕНИЯ БОЛЕВОГО СИНДРОМА РАЗЛИЧНОЙ ЭТИОЛОГИИ С ПОМОЩЬЮ ЭТОГО СРЕДСТВА | 2007 |
|
RU2367432C2 |
| СРЕДСТВО ПЕПТИДНОЙ СТРУКТУРЫ, ОБЛАДАЮЩЕЕ АНАЛЬГЕТИЧЕСКИМ, ПРОТИВОВОСПАЛИТЕЛЬНЫМ ДЕЙСТВИЕМ, И ЛЕКАРСТВЕННЫЕ ФОРМЫ НА ЕГО ОСНОВЕ | 2012 |
|
RU2528094C2 |
| АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО ПЕПТИДНОЙ СТРУКТУРЫ НА ОСНОВЕ УНДЕКАПЕПТИДА, СОДЕРЖАЩЕГО D-ОКТААРГИНИНОВЫЙ ВЕКТОР | 2013 |
|
RU2541127C1 |
| Суппозитории нефопама для лечения острого и хронического болевого синдрома на липофильной основе и способ их получения | 2017 |
|
RU2661618C1 |
Изобретение относится к фармацевтике, медицине и ветеринарии, в частности к разработке синтетического анальгетика пептидной природы и способу купирования боли с его помощью. Предложено пептидное соединение, представляющее собой Ac-RERR-NH2, обладающее анальгетическим действием, фармацевтическая композиция, содержащая указанное пептидное соединение, и способ купирования болевого синдрома. 3 н. и 9 з.п. ф-лы, 2 ил., 5 табл., 4 пр.
1. Пептидное соединение, представляющее собой Ac-RERR-NH2.
2. Фармацевтическая композиция, обладающая анальгетическим действием, включающая активный компонент - пептидное соединение по п. 1 в эффективном количестве и фармацевтически приемлемый носитель.
3. Композиция по п. 3, характеризующаяся тем, что активный компонент и носитель взяты в следующем соотношении компонентов, маc. %:
активный компонент - 1-95,
носитель – остальное.
4. Композиция по п. 3, характеризующаяся тем, что содержание активного компонента в однократной дозе составляет 10-400 мг.
5. Композиция по п. 3, характеризующаяся тем, что она выполнена в форме для внутривенного, внутримышечного, спинального, эпидурального, мукозального, чрескожного, парентерального, перорального, энтерального, интраназального или ректального введения.
6. Композиция по п. 3, характеризующаяся тем, что она выполнена в форме порошка, пластыря, свечи, раствора, суспензии, крема, мази, таблетированной капсулированной форме.
7. Композиция по п. 3, характеризующаяся тем, что носитель выбран из группы: вода, этанол, глицерол, физиологический раствор.
8. Композиция по п. 3, характеризующаяся тем, что носитель дополнительно содержит эмульгирующий агент и/или буферный агент.
9. Композиция по п. 3, характеризующаяся тем, что носитель выбран из группы: полиэтиленгликоль, лактоза, магнезиум карбонат, тальк.
10. Способ купирования болевого синдрома, включающий введение фармацевтической композиции по п. 3 в терапевтически приемлемом количестве.
11. Способ по п. 10, характеризующийся тем, что композицию используют в форме для внутривенного, внутримышечного, спинального, эпидурального, мукозального, чрескожного, парентерального, перорального, энтерального, интраназального или ректального введения.
12. Способ по п. 10, характеризующийся тем, что композицию вводят в разовой дозировке с содержанием активного компонента не более 400 мг, при этом суточная дозировка активного компонента составляет не более 600 мг для пациентов с массой тела 50-100 кг.
| US 2006172933 A1, 03.08.2006 | |||
| FR 2858772 A1, 18.02.2005 | |||
| WO 2002047719 A2, 20.06.2002 | |||
| 0 |
|
SU269389A1 | |
| WO 2010018136 A1, 18.02.2010 | |||
| US 20100322862 A1, 23.12.2010 | |||
| WO 03097089 A2, 27.11.2003 | |||
| WO 2005100557 A1, 27.10.2005 | |||
| WO 9638164 A1, 05.12.1996 | |||
| Mohri, Hiroshi et al "Synthetic peptides inhibit the interaction of von Willebrand factor-platelet membrane glycoproteins." Peptides, 1993, 14(2), 125-129 | |||
| Mazoyer, Elisabeth et al "KRDS, a new peptide derived from human lactotransferrin, inhibits platelet aggregation and release reaction." European Journal of Biochemistry, 1990, 194(1), 43-49 | |||
| Demina, O | |||
| V | |||
| et al "Modelling of prebiotic synthesis and selection of peptides under isothermal conditions and thermal cycling mode." Russian Chemical Bulletin, 2012, 61(2), 422-441 | |||
| СЕМЕЙСТВО ПЕПТИДОВ, ОБЛАДАЮЩЕЕ АНАЛЬГЕТИЧЕСКОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2286169C1 |

