Изобретение относится к медицине и ветеринарии, в частности к лекарственным препаратам, и может быть использовано для лечения острых и хронических болевых синдромов.
Боль - одна из самых распространенных жалоб, заставляющая больного обращаться к врачу и почти всегда свидетельствующая о наличии патологического процесса. Болевой синдром приводит людей к нетрудоспособности, снижает их активность, вызывает психоэмоциональные расстройства, регионарные и системные нарушения микроциркуляции, является причиной вторичных иммунных депрессий и нарушений деятельности висцеральных систем.
В настоящее время болевой синдром - серьезная клиническая проблема, требующая значительных усилий со стороны врачей по улучшению качества и эффективности ее терапии. Согласно статистическим данным служб здравоохранения, хроническим болевым синдромом, являющимся чаще следствием некупированного острого болевого синдрома, в США страдают около 20 млн человек. Неудивительно, что анальгетики представляют собой одну из наиболее востребованных категорий лекарств.
По данным Всемирной Организации Здравоохранения (ВОЗ), ежегодно в мире умирают свыше 4 млн онкологических больных, не получивших адекватного обезболивания в последние недели жизни [Ruiz-Lopez, 1993]. Столь неудовлетворительные результаты объясняют недостаточным использованием обезболивающих средств и преувеличенным страхом развития наркомании у смертельно больных людей. В терминальной стадии заболевания от боли страдают от 55 до 95% онкологических больных [Grond, 1991]. При этом боль средней интенсивности испытывают 40-50%, очень сильную и невыносимую - 25-30% пациентов [Осипова, 1998].
В то же время проведенное в Европе крупномасштабное исследование по изучению хронической боли доказало и высокую распространенность выраженной хронической неонкологической боли (умеренной и сильной, более 5 баллов по 10-балльной шкале) в популяции. Наиболее частые источники боли - артриты и остеоартроз (35%), и боли в спине (24%) [Breivik et al., 2006]. Лечение таких хронических болевых синдромов остается одной из самых актуальных и сложных задач современной фармакологии, так как длительное болевое воздействие запускает патологические реакции в периферической и центральной нервной системе, вследствие чего развивается резистентность к медикаментозным воздействиям.
В настоящее время опиатные анальгетики стоят в первом ряду средств борьбы с сильной болью, так как они обладают самой высокой анальгетической активностью. Основная область применения опиатов - соматогенные болевые синдромы, возникающие вследствие активации ноцицепторов при травме, воспалении, ишемии, отеке тканей и других повреждениях. На системном уровне анальгетическое действие опиатов происходит за счет глубокого угнетения процессов поверхностной и временной суммации подпороговых болевых раздражений на всех уровнях проведения болевого импульса, что собственно и составляет физиологическую основу опиоидной аналгезии. Также наличие противотревожного и эйфоризирующего действия, которое подавляет ожидание боли, сглаживает восприятие и оценку болевых ощущений.
Очевидно, что основное показание к применению опиатов является острый и хронический болевой синдром. Вместе с тем опиатным анальгетикам присущ ряд дополнительных фармакологических эффектов, включающих сонливость, угнетение дыхания, перемену настроения, развитие толерантности, затемнение сознания без конечной потери сознания, физическую зависимость. Наличие указанных побочных эффектов накладывает некоторые ограничения на широкое использование опиатов в клинической практике.
Кроме опиатов, существует класс соединений, активирующих опиоидные рецепторы, - эндогенные опиоидноподобные пептиды, которые присутствуют в областях центральной нервной системы, связанных с ощущением боли, с движением, настроением и поведением, а также с регуляцией нейроэндокринных функций [Михайлович, Игнатов, 1990]. Классифицируются эндогенные пептиды по агонистической активности к определенным типам рецепторов. В настоящее время известны следующие группы эндогенных пептидов: МОР (энкефалин, β-эндорфин, эндоморфин-1, эндоморфин-2; DOP (энкефалин, β-эндорфин); KОР (динорфин А, В, α-неоэндорфин) и NOP (ноцицептин, орфанин FQ). Образование эндогенных эндорфинов начинается в передней доле гипофиза, где молекула предшественника проопиомеланокортин (ПОМК) расщепляется на АКТГ и β-липотропин, которые секретируются в плазму. Небольшая часть (около 15%) β-липотропина расщепляется с образованием β-эндорфина. В других областях головного мозга, а у животных и в промежуточной доле гипофиза, в β-эндорфин превращается основная часть ПОМК. Эндогенный опиоид динорфин продуцируется нейронами тех же крупноклеточных ядер гипоталамуса, которые синтезируют вазопрессин, и накапливается вместе с вазопрессином в задней доле гипофиза.
Кроме эндогенных опиоидов, существуют пептидные соединения, полученные из других источников. Наиболее изученным из них является дерморфин (Tyr-D-Ala-Phe-Gly-Tyr-Pro-Ser-NH2), выделенный из кожи жабы Phyllomedusa sauvagei, обитающей в Южной Америке. Дерморфин является одним из самых мощных опиоидных пептидов с высокой селективностью к µ-опиоидным рецепторам [Montecucchi et al., 1981]. С другой стороны, полученный из того же источника делторфин (Tyr-D-Met-Phe-His-Leu-Met-Asp-NH2), обладает высоким сродством к δ-опиоидным рецепторам. В отличие от опиоидных пептидов млекопитающих, дерморфин и делторфин содержат D-аминокислоту во втором положении, что делает эти пептиды относительно устойчивыми к действию ферментов.
В середине 80-х годов прошлого века были синтезированы различные пептиды, содержащие аминокислотные последовательности, характерные для дерморфина. Четыре N-концевые аминокислоты дерморфина - минимальная последовательность, необходимая для проявления опиоидергической активности. Было показано, что аналоги энкефалинов, имеющие в своем составе последовательность Tyr-D-Arg2, обладают мощным антиноцицептивным действием [Sasaki et al., 1985], и что структурные аналоги этих соединений - тетрапептиды - обладают анальгетической активностью, в несколько раз превышающей таковую у морфина. Так, в 1988 году экспериментально было продемонстрировано, что антиноцицептивный эффект синтетического тетрапептида Tyr-D-Arg-Phe-Gly-NH2 в 6 раз превышает активность морфина [Chaki et al., 1988a]. Однако выраженное обезболивающее действие тетрапептидов было сопряжено с развитием толерантности и физической зависимости, что, по-видимому, ограничило их дальнейшее продвижение в качестве лекарственных средств [Chaki et al., 1988b].
В последующие годы велись работы по модификации структуры пептидов, близких к дерморфину. Так, например, в 1997 получен патент на способы изменения физиологической активности дерморфина [Емельянова и др., 1999] путем химической модификации входящего в состав аминокислотного остатка Pro. В конкретном случае химическую модификацию осуществляли путем замены аминокислотного остатка L-Pro на D-Pro, или Dh-Pro, или Dh-D-Pro, где Dh-Pro-3,4-дегидропролин. Но существенных успехов достигнуто не было: снижение побочных эффектов, как правило, вело и к снижению анальгетической активности.
Одним из направлений конструирования пептидов с анальгетической активностью является создание соединений, устойчивых к действию пептидаз. Это достигается рядом модификаций молекулы энкефалинов: метилированием N-терминали, замещением Gly во втором положении на D-Ala, блокадой С-терминали правовращающей аминокислотой (D-Met, D-Leu) и др. В большинстве случаев с помощью модификаций энкефалинов успешно решается проблема их стабилизации, но не улучшается их проникновение в головной мозг. Это ограничивает применение опиоидных пептидов рамками лабораторных исследований, но делает их важным инструментом анализа опиоидергических механизмов на рецепторном уровне. Наибольшим аффинитетом к µ-опиатным рецепторам обладают стабильные аналоги энкефалина ДАГО (О-Ala-2-N-Met-Plie4-Gly-ol5-энкефалин), FK 33-824, PLO-17, TPIMU-4. Синтезирован ряд правовращающих аналогов энкефалина, рассматриваемых в качестве экзогенных лигандов δ-опиатных рецепторов (DADL, DPLP, DSLFT, DTTL, DTLKT). Наиболее избирательными агонистами κ-рецепторов считаются короткие фрагменты пептида динорфина и такие соединения, как тифлуадом, MR 2034 и, особенно, U 50.488 Н [Михайлович, Игнатов, 1990].
В настоящее время, по-прежнему, весьма активно ведется создание синтетических аналогов эндогенных опиоидных пептидов, обладающих заданным спектром рецепторного действия и лишенных недостатков, присущих энкефалинам и эндорфину: плохой проницаемостью через гематоэнцефалический барьер и высокой скоростью разрушения. Так, в Институте молекулярной генетики РАН совместно с Институтом фармакологии имени В.В. Закусова РАМН с целью устранения отмеченных недостатков предложена новая группа пептидных последовательностей общей формулы: A-B-Tyr-Pro(DPro, dHPro, DdHPro, DLdHPro, Hyp)-B-X [Алфеева и др., 2006]. Синтезированы дипептиды формулы Tyr-Pro, Tyr-Pro-ОСН3, Tyr-Pro-NH2, трипептиды Tyr-Pro-Ser-ОСН3, Tyr-Pro-Ser-NH2, Tyr-Hyp-Ser-ОСН3, Tyr-Hyp-Ser-NH2, Tyr-DPro-Ser-ОСН3, Tyr-DPro-Ser-NH2, Tyr-DLdHPro-Ser-ОСН3, Tyr-dHPro-Ser-NH2, Tyr-Pro-Lys-NH2, Tyr-Pro-Ala-NH2, тетрапептиды: Tyr-Pro-Gly-Pro-NH2, Tyr-Tyr-Pro-Ser-NH2, Ala-Tyr-DPro-Ser-NH2, DAla-Tyr-DPro-Ser-NH2, Glu-Tyr-DPro-Ser-NH2, Met-Tyr-DPro-Ser-NH2, отвечающие вышеприведенной общей формуле и обладающие анальгетической активностью. Эти аминокислотные последовательности с антиноцицептивным действием обладают общей закономерностью, а именно наличием в структуре пептидной последовательности Tyr-Pro (DPro, dHPro, DdHPro, DLdHPro, Hyp) в 1,2 или 2,3-положениях.
Таким образом, довольно активно ведется поиск новых пептидных соединений, обладающих высокой избирательностью действия и лишенных недостатков, присущих уже известным пептидам.
Вместе с тем изыскание опиоидных пептидов в качестве анальгетических лекарственных средств лимитировано фармакокинетическими характеристиками, включая низкую стабильность и плохую проницаемость через гематоэнцефалический барьер (ГЭБ), а в некоторых случаях развитием побочных эффектов, присущих опиатам [Mizoguchi et al., 2011]. Однако проведенные исследования показали, что катионизация эндоморфина-2 добавлением гуанидина увеличивает метаболическую стабильность и проницаемость через ГЭБ при сохранении анальгетической активности [Hau et al., 2002]. Также было продемонстрированно, что добавление олигоаргининового вектора может увеличивать доставку молекул внутрь клетки [Furuhata et al., 2006; Futaki et al., 2006; Yang et al., 2008] и через ГЭБ [Pham et al., 2005]. В других исследованиях добавление D-формы аргинина к известному тетрапептиду дерморфину в N-концевом положении кардинально увеличивало анальгетическую активность синтезированного пептида в 1100 раз и 9,1 раз по сравнению с морфином при интратекальном и подкожном введении соответственно [Sakurada et al., 2000; Sato et al., 1999]. Но при этом возрастало количество побочных эффектов, таких как дыхательная депрессия, каталепсия, толерантность и физическая зависимость [Mizoguchi et al., 2010].
Полиаргинин известен как сильнодействующий ингибитор фурина, протеолитического фермента, относящегося к сериновым протеазам клеток животных, расположенных в аппарате Гольджи. Как и L-форма аргинина, показывающая низкую ингибиторную константу Ki около 114 нМ, D-форма полиаргинина обладает схожей величиной константы ингибирования [Cameronet al., 2000]. Более того, D-форма полиаргинина показала ингибиторную активность в отношении экзотоксина A Pseudomonas aeruginosa [Sarac et al., 2002] и при токсемии сибирской язвы [Sarac et al., 2004]. Kacprzak с коллегами на основании предыдущих исследований и собственных данных сделали вывод о том, что D-форма полиаргинина является высокостабильной, низкотоксичной, имеющий низкий молекулярный вес и низкую ингибиторную константу Ki в отношении протеолитических ферментов (например, фурина) [Kacprzak et al., 2004].
Октааргининовые пептиды известны своей способностью проникать внутрь клетки через мембрану при помощи различных молекулярных механизмов [Miklán et al., 2011]. По всей видимости, один из механизмов, при помощи которого полиаргинин проникает в клеточное пространство и увеличивает проницаемость ГЭБ, заключается в том, что клеточная поверхность содержит гепаран-сульфат протеогликан, включающий в себя синдеканы, которые в свою очередь являются основным компонентом экстрацеллюлярной матрицы, и создают некий якорь для различных внешних молекул и патогенов на поверхности клетки-мишени [Esclatine et al., 2001; Yoneda et al., 2003]. После первичного электростатического взаимодействия лиганда с экстрацеллюлярной матрицей наступает дальнейшая кластеризация этих ″якорей″, что вызывает ремоделирование цитоскелета при помощи активации протеинкиназы С (ПКС) и Rho/Rac-ГТФазы, которые контролируют динамику микродомена липидного бислоя клеточной мембраны (липидного ″рафта″), обогащенного холестерином, сфинголипидами и насыщенными фосфолипидами. Образно говоря, это участок плотно-упакованного липида, ″плавающего″ на поверхности ″жидкого″ фосфолипида. Дальнейшее встраивание специфических мембранных белков в липидный рафт приводит к его стабилизации, а последующее связывание лигандов с рецепторами или гликосфинголипидами, локализующимся в таких рафтах, приводит к их слиянию и запускает передачу внутриклеточного сигнала [Dehio et al., 1998; Couchman. 2003; Tkachenko et al., 2004]. При помощи данного механизма и происходит связывание лигандов и клеточный эндоцитоз. Но некоторые исследователи считают [Richard, 2003], что есть несколько путей проникновения полиаргинина в клетку в зависимости от свойств носителя пептидов, его концентрации и клеток мишеней. Так Nakase и его коллеги считают, что интернализация полиаргинина осуществляется через механизмы макропиноцитоза [Nakase et al., 2004]. Данная группа ученых в режиме реального времени при использовании ядерно-магнитного резонанса выяснила, что меченный октааргинин показал самую высокую динамику в отличие от других полиаргининов (тетрааргинин и гескадекааргинин). Затем при ингибировании процесса макропитоцитоза этил-лизопропиламилоридом (EIPA) и циточалазином Д, который является ингибитором Ф-актин полимеризации, было выявлено достоверное угнетение поглощения октааргинина HeLa клетками [Nakase et al., 2004].
Таким образом, выбранная D-форма октааргинина представляется крайне перспективным вектором доставки пептидов внутрь клетки, увеличением их стабильности, путем блокады действия протеаз и облегчения проницаемости ГЭБ, что ведет к увеличению устойчивости молекулы к ферментативному метаболизму и, тем самым, увеличивает стабильность и фармако кинетические свойства пептида при периферическом введении.
Задачей настоящего изобретения является создание анальгетического средства удобного и эффективного в применении с целью купирования болевых синдромов различной интенсивности.
Предметом заявки является применение ундекапептида H-Tyr-Pro-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-Gly-OH·9HCl (далее по тексту H-Tyr-Pro-(D-Arg)8-Gly-OH-9HCl) в качестве анальгетического средства. Кроме того, для достижения поставленного результата предлагается лекарственная форма, содержащая в качестве действующего вещества ундекапептид H-Tyr-Pro-(D-Arg)8-Gly-OH·9HCl для лечения острых и хронических болевых синдромов; предпочтительные, но не обязательные варианты такой формы предполагают ее выполнение в виде одной из следующих лекарственных форм: раствор для инъекций, назальный спрей, назальные капли, сублингвальные таблетки, суббукальные пластыри, ректальные суппозитории, кишечнорастворимые таблетки и капсулы, трансдермальные системы для резорбтивного применения.
В качестве иллюстративных материалов к описанию приложены результаты масс спектр анализа, а также рис.1 (влияние дипептида при разных путях введения на ноцицептивный ответ типа ″корчи″) и рис.2 (влияние ундекапептида при разных путях введения на ноцицептивный ответ типа ″корчи″).
Фармакологические свойства и способ получения заявляемого в настоящем изобретении ундекапептида H-Tyr-Pro-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-Gly-OH·9HCl в литературе не описаны.
Синтез ундекапептида H-Tyr-Pro-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-Gly-OH·9HCl осуществляли по нижеследующей методике. Список использованных сокращений представлен в таблице 1.
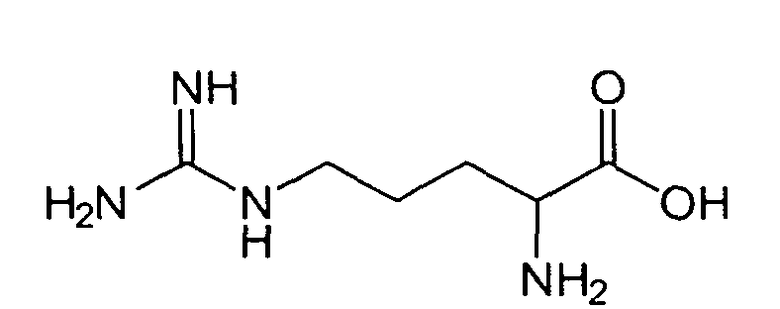
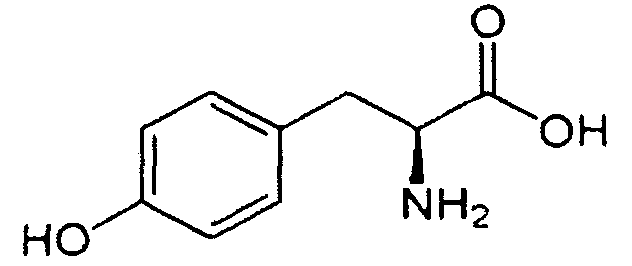
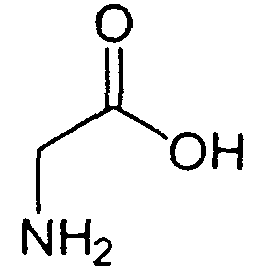
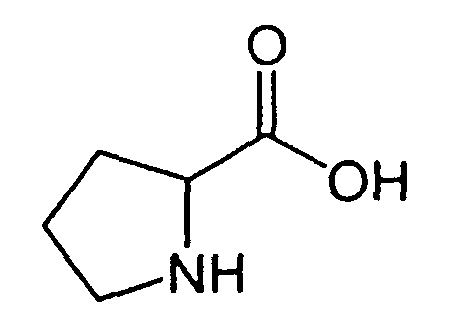
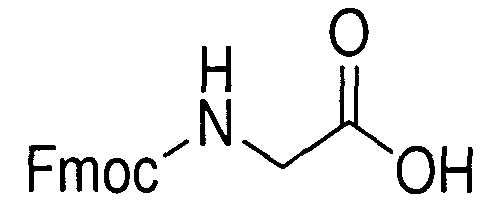
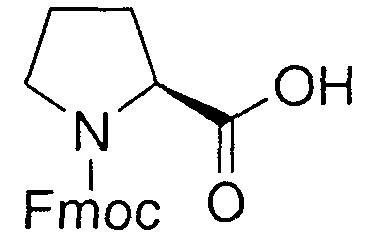
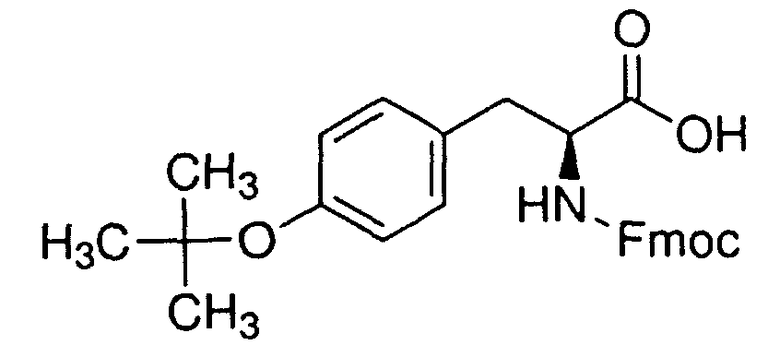

В реакторе для твердофазного синтеза суспендировали 2.0 г 2-хлортритильной смолы (емкость 1.5 ммоль/г) в 15 мл DCM, выдерживали в течение 5 мин, смолу отфильтровывали, промывали 2×10 мл DCM. К смоле добавляли раствор 0.98 г (3.3 ммоль) Fmoc-Gly-OH и 2 мл (12 ммоль) DIPEA в 10 мл DCM, перемешивали в течение 60 мин при комнатной температуре. Смолу отфильтровывали, промывали 2×10 мл DCM, обрабатывали 2×10 мл смеси DCM/метанол/DIPEA (17:2:1) в течение 10 мин, промывали 2×10 мл DCM и 3×10 мл DMF. В реактор загружали 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°C) раствор 3.89 г (6.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 0.98 г (7.2 ммоль) HOBt и 1.12 мл (7.2 ммоль) DIC в 10 мл DMF, перемешивали в течение 2 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°C) раствор 3.89 г (6.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 0.98 г (7.2 ммоль) HOBt и 1.12 мл (7.2 ммоль) DIC в 10 мл DMF, перемешивали в течение 4 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°C) раствор 5.19 г (8.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 1.35 г (10.0 ммоль) HOBt и 1.56 мл (10.0 ммоль) DIC в 10 мл DMF, перемешивали в течение 3 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°C) раствор 5.19 г (8.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 1.35 г (10.0 ммоль) HOBt и 1.56 мл (10.0 ммоль) DIC в 10 мл DMF, перемешивали в течение 4 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°C) раствор 5.19 г (8.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 1.35 г (10.0 ммоль) HOBt и 1.56 мл (10.0 ммоль) DIC в 10 мл DMF, перемешивали в течение 10 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°C) раствор 6.48 г (10.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 1.62 г (12.0 ммоль) HOBt и 1.88 мл (12.0 ммоль) DIC в 10 мл DMF, перемешивали в течение 12 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°C) раствор 6.48 г (10.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 1.62 г (12.0 ммоль) HOBt и 1.88 мл (12.0 ммоль) DIC в 10 мл DMF, перемешивали в течение 12 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°C) раствор 6.48 г (10.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 1.62 г (12.0 ммоль) HOBt и 1.88 мл (12.0 ммоль) DIC в 10 мл DMF, перемешивали в течение 16 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°C) раствор 3.38 г (10.0 ммоль) Fmoc-Pro-OH, 1.62 г (12.0 ммоль) HOBt и 1.88 мл (12.0 ммоль) DIC в 10 мл DMF, перемешивали в течение 16 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°C) раствор 4.59 г (10.0 ммоль) Fmoc-Tyr(tBu)-ОН, 1.62 г (12.0 ммоль) HOBt и 1.88 мл (12.0 ммоль) DIC в 10 мл DMF, перемешивали в течение 12 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
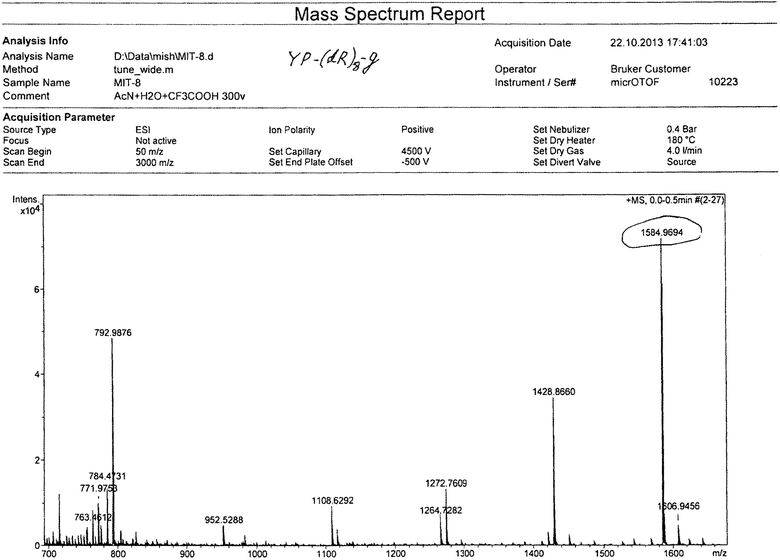
Смолу промывали 4×10 мл DCM, сушили, добавляли 15 мл смеси TFA/TIS/EDT/H2O (37:1:1:1), выдерживали в течение 4 часов при комнатной температуре, отфильтровывали, промывали 3×10 мл трифторуксусной кислотой, объединенные фильтраты упаривали до объема ~10 мл, к остатку добавляли 40 мл сухого эфира. Выпавший осадок отфильтровывали, промывали на фильтре эфиром, сушили. Полученный продукт растворяли в 50 мл воды, раствор замораживали и лиофилизовали. Лиофилизат растворяли в 30 мл воды и наносили на колонку с ионообменной смолой Amberlite IRA-400 (Cl- форма). Колонку промывали водой, фракции, содержащие продукт, объединяли, упаривали до объема ~40 мл и наносили на обращеннофазную колонку Waters X-Bridge C18, 10 мкм, 127 Å, 50×250 мм. Элюирование проводили при скорости потока элюента 50 мл/мин. Фаза А: 0,1% HCl/H2O, В:/ацетонитрил. Градиент: 0%(В)-70%(В) за 70 минут. Фракции, содержащие основной продукт, объединяли, упаривали до объема ~50 мл, замораживали и лиофилизовали. Получено 1.1 г (19,2%) продукта с чистотой (ВЭЖХ) 99.0%. Масс спектр: вычислено M+ 1584.88, получено M+ 1584.97. (см. приложение 1). Аминокислотный анализ: аргинин 8.0 (8), тирозин 1,10 (1), пролин 1.00 (1), глицин 0.95 (1).
Нижеследующие примеры фармакологических исследований подтверждают возможность применения заявленного ундекапептида Н-Tyr-Pro-(D-Arg)8-Gly-OH·9HCl в качестве средства с анальгетической активностью.
Пример 1. Изучение анальгетической активности дипептида Н-Tyr-Pro-ОН и ундекапептида H-Tyr-Pro-(D-Arg)g-Gly-OH·9HCl в тесте отдергивания хвоста (tail flick).
Болевая чувствительность оценивалась на белых беспородных мышах методом термического раздражения корня хвоста (″tail-flick″) [D′Amour, Smith, 1941]. Для проведения теста подвижность животного ограничивается, после чего на хвост животного (1 см от основания) направляется сфокусированное световое излучение (мощность 300 Вт). С помощью фоточувствительного датчика фиксировалось время от включения источника излучения до отдергивания хвоста (латентный период). Для предотвращения повреждения тканей предельная продолжительность воздействия ограничивалась интервалом в 15 сек.
Исходная болевая чувствительность определялась как среднее по двум измерениям. Мыши с исходным значением латентного периода более 8 с были исключены из эксперимента. Мышей тестировали перед введением препаратов (исходная болевая чувствительность) и через 5, 15, 30, 60 и 120 минут после введения. При сохранении анальгетического эффекта через 120 мин после введения болевая чувствительность измерялась и через 180 минут после введения.
При обработке полученных результатов были определены средние значения латентных периодов реакции в каждый момент времени и степень анальгезии в процентах, которая рассчитывается как процент от максимально возможного эффекта по формуле:
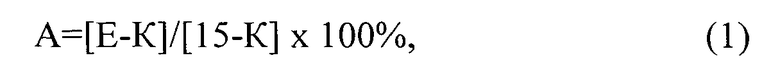
где: A - степень аналгезии, E - латентный период реакции в секундах в эксперименте (после введения препаратов), K - исходный латентный период данного животного (до введения препарата), 15 - максимальное время термического воздействия, в секундах.
Результаты исследования анальгетической активности дипептида Н-Tyr-Pro-ОН и ундекапептида H-Tyr-Pro-(D-Arg)8-Gly-OH·9HCl в тесте отдергивания хвоста (tail flick) представлены в таблицах 2 и 3.
Исходные (до введения пептидов) латентные периоды отдергивания хвоста не отличались между группами (p=0,85).
Далее анализировалось влияние пептидов на ЛП в сравнении с группой, получавшей физиологический раствор. Двухфакторный дисперсионный анализ с повторными измерениями показал значимое влияние дипептида (F1,14=12,16; p<0,01) и ундекапептида (F1,15=5,78; p<0,05) на ЛП отдергивания хвоста.
Однофакторный дисперсионный анализ с повторными измерениями показал значимое влияние дипептида (F1,14=16,2; p<0,001) и ундекапептида (F1,15=10,35; p<0,01) на степень аналгезии.
Таким образом, дипептид и ундекапептид обладали антиноцицептивным действием в тесте термического раздражения корня хвоста. При этом обезболивающее действие ундекапептида было умеренным, однако максимально продолжительным. Анальгетический эффект ундекапептида продолжался вплоть до конца наблюдения (180 минут), тогда как дипептид сохранял свои антиноцицептивные свойства в течение 60 минут эксперимента.
Пример 2. Изучение анальгетической активности дипептида Н-Tyr-Pro-ОН и ундекапептида H-Tyr-Pro-(D-Arg)8-Gly-OH·9HCl в тесте химического раздражения брюшины (тест ″корчи″ - ″Writhing test″).
Внутрибрюшинное введение химических раздражающих агентов позволяет моделировать висцеральную боль [Le Bars et al., 2001]. Метод химического раздражения брюшины (тест ″корчей″) заключается во внутрибрюшинном введении (в левую половину живота) 1% раствора уксусной кислоты в объеме 0.1 мл на животное. После инъекции мыши помещались в специальные индивидуальные боксы по 4 животных в одном блоке, где в течение 10 минут осуществляли визуальное наблюдение за животными. Регистрировали количество специфических ноцицептивных ответов типа ″корчи″, возникающих в ответ на введение химического раздражающего агента и проявляющихся характерными потягиваниями. Препараты вводились однократно за 20 мин до инъекции уксусной кислоты. Анальгетическое действие оценивается по уменьшению количества ″корчей″ за период регистрации данной поведенческой реакции.
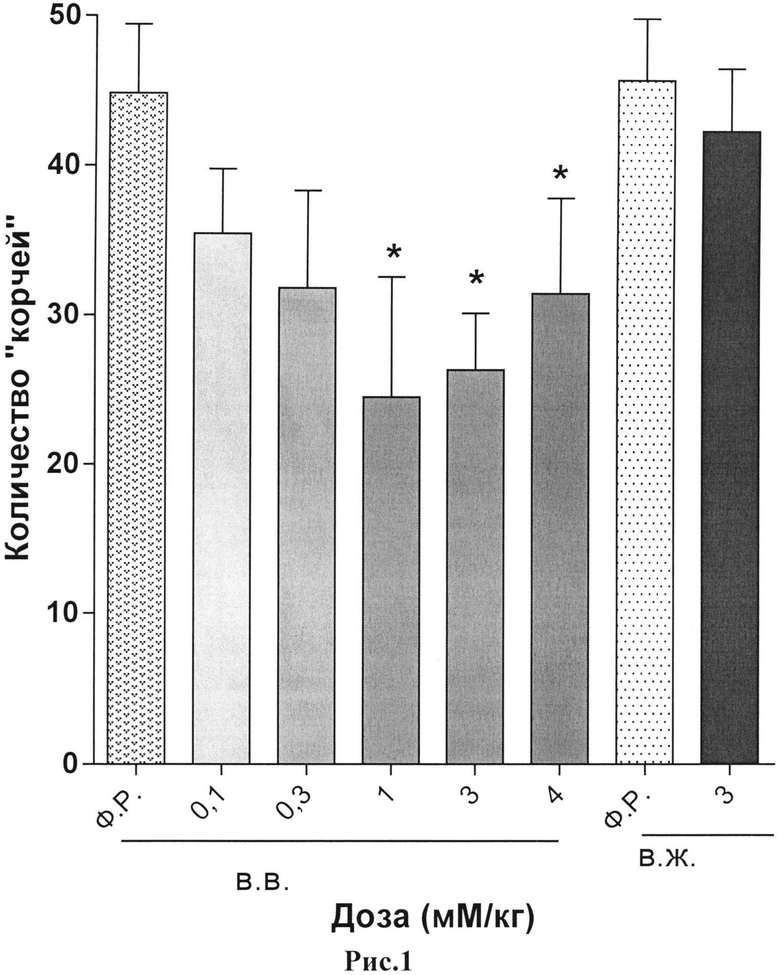
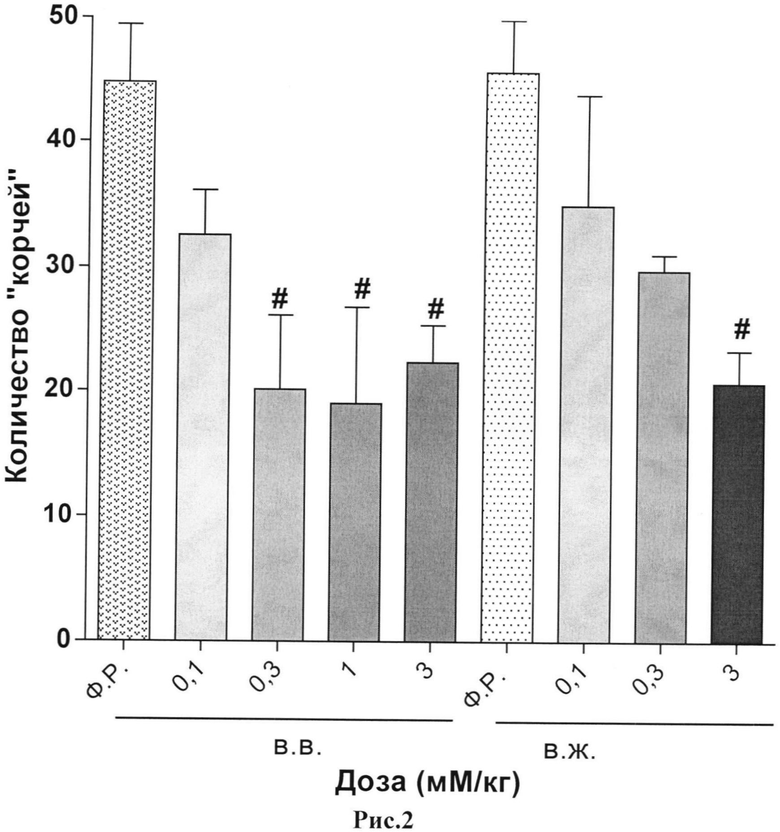
Результаты исследования анальгетической активности дипептида H-Tyr-Pro-ОН и ундекапептида H-Tyr-Pro-(D-Arg)8-Gly-OH·9HCl в тесте химического раздражения брюшины представлены в таблицах 4 и 5 и на рисунках 1 и 2.
Пояснения к рис.1. Данные представлены как среднее количество ″корчей″ (M±m) за 10-минутный период наблюдения.
Примечание:
* - статистически значимое отличие от группы, получавшей физиологический раствор, (попарное сравнение с контролем, критерий Манна-Уитни), p<0,05.
Ф.Р. - физиологический раствор;
в.в. - внутривенно;
в.ж. - внутрижелудочно.
Пояснения к рис.2. Данные представлены как среднее количество ″корчей″ (M±m) за 10-минутный период наблюдения.
Примечание:
#- - статистически значимое отличие от группы, получавшей физиологический раствор, (критерий Крускала-Уолисса, непараметрическая ANOVA, тест Данна), p<0,05.
Ф.Р. - физиологический раствор;
в.в. - внутривенно;
в.ж. - внутрижелудочно.
Проведенные экспериментальные исследования анальгетической активности нового пептидного соединения показали, что исследуемое соединение обладает антиноцицептивным эффектом.
В тесте термического прижигания корня хвоста наличие антиноцицептивной активности было зафиксировано для дипептида и его аналога с D-формой октааргинина - ундекапептида. Действие дипептида было умеренным и непродолжительным, тогда как умеренное анальгетическое действие ундекапептида продолжалась в течение всего периода наблюдения (до 180 минут).
В тесте химического раздражения брюшины был исследован дипептид и его аналог с добавлением D-формы октааргинина - ундекапептид в широком диапазоне доз при различных путях введения.
При внутривенном пути введения ундекапептида была отмечена умеренная антиноцицептивная активность на фоне низкой токсичности. Картина обезболивающего действия была схожа с внутривенным введением дипептида.
При внутрибрюшинном пути введения было подтверждено действие исследуемого пептида на ноцицептивный ответ. Картина анальгетического действия была схожа с внутривенным путем введения.
При внутрижелудочном применении исследуемых пептидов дипептид утратил свою анальгетическую активность. Так введение дипептида, обладающего умеренной степенью обезболивания, не имело достоверного антиноцицептивного действия в максимальной эффективной дозе, как при внутривенном введение. Напротив, применение пептида с D-формой октааргинина - ундекапептида - показало дозозависимое сохранение анальгезии схожей с внутривенным и внутрибрюшинным путями введения. Данное наблюдение позволяет сделать вывод о том, что включение октааргинина в молекулу пептида позволяет повысить устойчивость пептида к действию протеаз при внутрижелудочном пути введение, тем самым сохраняя антиноцицептивную активность молекулы. В данном случае аналог дипептида с добавлением D-формы октааргинина ундекапептид показал устойчивый и продолжительный анальгетический эффект как при парентеральном, так и при пероральном путях введения.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО ПЕПТИДНОЙ СТРУКТУРЫ НА ОСНОВЕ ТРИДЕКАПЕПТИДА, СОДЕРЖАЩЕГО D-ОКТААРГИНИНОВЫЙ ВЕКТОР | 2013 |
|
RU2538727C1 |
| СЕМЕЙСТВО ПЕПТИДОВ, ОБЛАДАЮЩЕЕ АНАЛЬГЕТИЧЕСКОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2286169C1 |
| ПЕПТИД ДЛЯ ЛЕЧЕНИЯ САХАРНОГО ДИАБЕТА 2-ГО ТИПА И ЕГО ОСЛОЖНЕНИЙ | 2014 |
|
RU2573933C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДА ЭКСЕНАТИДА | 2011 |
|
RU2458066C1 |
| СРЕДСТВО ПЕПТИДНОЙ СТРУКТУРЫ, ИНГИБИРУЮЩЕЕ ДИПЕПТИДИЛПЕПТИДАЗУ-4, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2015 |
|
RU2589258C1 |
| ГИПОГЛИКЕМИЧЕСКОЕ СРЕДСТВО ПЕПТИДНОЙ СТРУКТУРЫ, ИНГИБИРУЮЩЕЕ ДИПЕПТИДИЛПЕПТИДАЗУ-4 | 2015 |
|
RU2600810C1 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2087480C1 |
| Аналог альфа-конотоксина RgIA для лечения боли | 2016 |
|
RU2731217C2 |
| ИНГИБИТОРЫ ФАКТОРА ХА | 1995 |
|
RU2152954C1 |
| АГОНИСТЫ ОКСИТОЦИНОВЫХ РЕЦЕПТОРОВ | 2010 |
|
RU2539692C2 |
Группа изобретений относится к медицине и касается применения ундекапептида - H-Tyr-Pro-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-Gly-OH·9HCl в качестве анальгетического средства. Группа изобретений также касается лекарственной формы, содержащей в качестве действующего вещества указанный пептид для лечения острых и хронических болевых синдромов. Изобретение обеспечивает устойчивый и продолжительный анальгетический эффект как при парентеральном, так и при пероральном путях введения. 2 н. и 1 з.п. ф-лы, 2 пр., 2 ил., 5 табл.
1. Применение ундекапептида - H-Tyr-Pro-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-Gly-OH 9НС1 в качестве анальгетического средства.
2. Лекарственная форма, содержащая в качестве действующего вещества - Н-Tyr-Pro-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-Gly-OH·9НСl для лечения острых и хронических болевых синдромов.
3. Форма по п.2, выполненная в виде одной из следующих лекарственных форм, пригодных для парентерального или перорального введения: раствор для инъекций, назальный спрей, назальные капли, сублингвальные таблетки, суббукальный пластырь, ректальные суппозитории, кишечнорастворимые таблетки и капсулы, трансдермальные системы для резорбтивного применения.
| MIZOGUCHI H., et al., Dermorphin tetrapeptide analogs as potent and long-lasting analgesics with pharmacological profiles distinct from morphine | |||
| Peptides | |||
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| WOLFE D., et al., Engineering an endomorphin-2 gene for use in neuropathic pain therapy | |||
| Pain | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |

