Заявляемое решение относится к фармацевтике, космецевтике, медицине и ветеринарии и может быть использовано для производства анальгетика для терапии болевого синдрома различной этиологии, что способствует излечению и от хронической боли.
Анальгетическими средствами, или анальгетиками (от греч. algos - боль и an - без), называют лекарственные средства, обладающие специфической способностью ослаблять или устранять чувство боли [Машковский М.Д. Лекарственные средства. 14 издание. М: Новая волна, 2003. Т.1. С.145].
Анальгетическое (болеутоляющее) действие могут оказывать не только собственно анальгетики, но и другие вещества, относящиеся к разным фармакологическим группам. Так, анальгетическим эффектом могут обладать препараты, применяемые для наркоза (общего обезболивания), и некоторые из них в соответствующих концентрациях и дозах (например, трихлорэтилен, закись азота) используются специально для анальгезии.
Местные анестетики также являются анальгетическими средствами, особенно при периферических болях. При резорбтивном действии местных анестетиков они могут влиять на ЦНС: это проявляется в беспокойстве, треморе, судорогах; а в более высоких дозах они негативно влияют на дыхательный и сосудодвигательный центры. Местные анестетики, оказывая блокирующее влияние на потенциалочувствительные натриевые каналы, угнетают сократимость миокарда, расширяют кровеносные сосуды, угнетающе влияют на симпатическую иннервацию, снижают артериальное давление. Наиболее важным свойством местных анестетиков является их способность блокировать ноцицепторы и чувствительные нервные волокна других модальностей. В связи с этим их используют для местного обезболивания (местной анестезии), в частности, при хирургических операциях.
Под анальгетическими в собственном смысле слова подразумеваются средства, доминирующим эффектом которых является анальгезия, не сопровождающаяся в терапевтических дозах выключением сознания и выраженным нарушением двигательных и сенсорных функций. Влиять на обезболивающий эффект средства, а также стабилизировать его лекарственную форму, могут вспомогательные вещества, например, гидрокарбонат натрия.
Всемирная организация здравоохранения (ВОЗ) утвердила трехступенчатый подход к лечению онкологической боли, который широко применяется в настоящее время. Он подразумевает использование анальгетиков перорально по часам, в зависимости от длительности действия препарата. На первом этапе используются неопиоидные анальгетики, такие как парацетамол (ацетаминофен) или нестероидные противовоспалительные препараты (НПВП). При недостаточном эффекте добавляются слабые опиоиды, например, кодеин, декстропропоксифен или трамадол (вторая ступень). Если и это не позволяет купировать боль, то на последнем, третьем этапе слабый опиоид меняется на сильный, подобный морфину (например, морфин, гидроморфон, оксикодон или фентанил).
Эта «лестница лечения боли ВОЗ» применяется с 1986 года. Данный подход затем был экстраполирован на неонкологическую боль, в том числе, для лечения хронической боли различной этиологии и локализации. Дополнительные препараты используются при этом для снижения тревожности и других негативных побочных эффектов.
По химической природе, характеру и механизмам фармакологической активности современные анальгетики делятся на две группы.
Основными представителями анальгетиков первой группы являются производные салициловой кислоты (натрия салицилат, ацетилсалициловая кислота, салициламид и др.), производные пиразолона - антипирин, амидопирин, анальгин, производные пара-аминофенола (или анилина) - фенацетин, парацетамол [там же. С.146]. Указанные анальгетики являются синтетическими лекарственными средствами. Анальгезирующая активность лекарственных средств, относящихся к первой группе, проявляется при определенных видах болевых ощущений, главным образом, при невралгических, мышечных, суставных болях, при головной и зубной боли. При сильной боли, связанной с травмами, полостными оперативными вмешательствами и т.п., они практически неэффективны. Указанные средства проявляют жаропонижающее действие при лихорадочных состояниях и противовоспалительное действие, выраженное в разной степени у разных анальгетиков данной группы. При применении этих анальгетиков отсутствуют угнетающее влияние на дыхательный и кашлевой центры, а также явления эйфории, психической и физической зависимости. В механизме действия анальгетиков первой группы определенную роль играет влияние на таламические центры, которое приводит к торможению проведения болевых импульсов к коре головного мозга. В механизме действия салицилатов важную роль играют ингибирование биосинтеза простагландинов, а также стимулирующее влияние на «ось» гипофиз - надпочечники, способствующее высвобождению кортикостероидов. Существенное значение в действии анальгетиков первой группы имеет их влияние на кининовую систему. Однако длительное применение анальгетиков данной группы приводит к сильным негативным побочным эффектам и продолжению болевых ощущений, что требует применения анальгетиков второй группы.
Анальгетики второй группы (также называемые большими анальгетиками) включают природные соединения - морфин и близкие к нему алкалоиды (опиаты), и синтетические соединения, обладающие опиатоподобными свойствами (опиоиды). Для них характерна сильная анальгезирующая активность, обеспечивающая возможность их использования при травмах (операционные вмешательства, ранения и др.) и при заболеваниях, сопровождающихся выраженным болевым синдромом (злокачественные новообразования, инфаркт миокарда, наркотическая «ломка» и др.). По источникам получения и химическому строению к анальгетикам второй группы относятся природные алкалоиды - морфин и кодеин, содержащиеся в снотворном маке; полусинтетические соединения, полученные путем химической модификации молекулы морфина (этилморфин); синтетические соединения (промедол, фентанил, пентазоцин, налбуфин, буторфанол, трамадол и др.). Большинство синтетических препаратов получено по принципу воспроизведения упрощенной структуры морфина. Для уменьшения побочных явлений вместе с морфином часто назначают атропин, метацин или другие холинолитики [там же. С.145]. Указанные средства обладают особым влиянием на центральную нервную систему человека, выражающемся в развитии эйфории и появлении при повторном применении синдромов психической и физической зависимости (лекарственной зависимости), что ограничивает возможность длительного применения этих препаратов. У пациентов с развившимся синдромом физической зависимости при лишении их анальгетического препарата может развиться болезненное состояние -абстинентный синдром. Анальгетики второй группы вызывают острые токсические явления, такие как угнетение дыхания, нарушение сердечной деятельности. Для их снятия применяются специальные лекарственные средства - антагонисты, например, налоксон или налтрексон, полученные путем химической модификации молекулы морфина. При повторном применении больших анальгетиков обычно развивается толерантность, то есть ослабление действия назначенной текущей дозы препарата, когда для получения анальгезирующего эффекта требуются все более высокие дозы препарата. Действие указанных анальгетиков не ограничивается болеутоляющим эффектом. В той или иной степени, они оказывают снотворное действие, угнетают дыхание и кашлевой рефлекс, повышают тонус кишечника и мочевого пузыря, могут вызвать тошноту, понос и другие побочные явления. В связи с выраженным наркогенным потенциалом анальгетики второй группы подлежат хранению, назначению и отпуску из аптек согласно особым правилам. В последние годы ряд применявшихся больших анальгетиков, обладающих выраженным наркогенным потенциалом (текодин, гидрокодона фосфат, фенадон, некоторые готовые лекарственные формы, содержащие опий и кодеин), исключен из номенклатуры лекарственных средств в Российской Федерации [там же. С.145]. Нейрофизиологические исследования свидетельствуют об угнетении указанными анальгетиками таламических центров болевой чувствительности и блокировании передачи болевых импульсов к коре головного мозга. Однако по своему действию эти анальгетики отличаются от представителей первой группы, так как влияют на способность центральной нервной системы к суммации подпороговых импульсов.
Известно, что в мозге и других органах присутствуют специфические опиатные рецепторы [там же. С.146]. Их эндогенными лигандами, то есть образующимися в организме физиологически активными соединениями, специфически связывающимися с этими рецепторами, являются нейропептиды - прежде всего, энкефалины, эндорфины и открытые в США проф. Д. Задиной эндоморфины [Zadina J.E., Hackler L., Ge L., Kastin A. A potent and selective endogenous agonist for the u-opiate receptor // Nature 1997, V.386. P.499-502]. Эндогенные лиганды оказывают анальгетическое действие и их эффект блокируется специфическими антагонистами опиатов. Экзогенный анальгетик морфин (равно как и другие близкие ему по структуре опиаты и опиоиды) при введении в организм взаимодействует с теми же рецепторами, которые предназначены для связывания эндогенных лигандов. Кроме того, не исключено, что действие экзогенных анальгетиков связано также со стабилизацией эндогенных лигандов путем инактивации разрушающих энкефалины ферментов - энкефалиназ.
Показано, что опиатные рецепторы существуют в виде разных субпопуляций (подгрупп): μ (мю) (или ОР3 по системе IUPHAR), κ (каппа) (ОР2), σ (сигма), δ (дельта) (OP1), имеющих различную функциональную значимость. Полагают, что мю-рецепторы опосредуют супраспинальную анальгезию, эйфорию, угнетение дыхания и физическую зависимость, каппа-рецепторы опосредуют спинальную анальгезию, миоз, седативный эффект. Разные эндогенные лиганды и большие анальгетики могут связываться преимущественно с той или иной подгруппой рецепторов, что может определять особенности их фармакологического действия.
Разнообразные большие анальгетики различаются также по характеру связывания с опиатными рецепторами. Одни из них (морфин, промедол, фентанил и др.) являются «чистыми» полными агонистами; связываясь с рецепторами, они оказывают характерное для эндогенных лигандов физиологическое (фармакологическое) действие. Другие являются «чистыми» антагонистами (налоксон, налтрексон). Связываясь с рецепторами, они блокируют действие как эндогенных лигандов, так и экзогенных опиатов. И, наконец, препараты смешанного типа действия (агонисты-антагонисты), по-разному связывающиеся с разными подгруппами опиатных рецепторов и оказывающие в связи с этим по одним видам действия агонистический эффект, по другим - антагонистический (трамадол, налорфин, пентазоцин, нальбуфин и др.). Действие больших анальгетиков на периферические органы (кишечник и др.) также связано с взаимодействием с локализующимися в них опиатными рецепторами.
Выраженного местноанестезирующего действия у большинства опиатов не отмечается. Вместе с тем в последние годы обнаружено, что они оказывают сильное обезболивающее действие при эпидуральном и субарахноидальном введении. Этот эффект связан с непосредственным воздействием на нейрональные системы спинного мозга, участвующие в формировании потока ноцицептивных импульсов. Этот способ введения опиатов находит применение для купирования тяжелых острых и хронических болей.
Из сказанного выше следует, что, с точки зрения современных воззрений на механизм действия больших анальгетиков, наиболее эффективными из них являются представители агонистов. Однако из-за вызываемых ими побочных эффектов, прежде всего сильной лекарственной (по сути, наркотической) зависимости и интоксикации организма, их применение строго ограничено.
Синтетический анальгетик последнего поколения, относящийся к группе агонистов-антагонистов опиатных рецепторов, - трамадол (tramadol), представляет собой±-отранс-2-[(диметиламино)метил]-1-(м-метоксифенил) циклогексанола гидрохлорид [там же. С.157]. Препарат обладает высокой анальгетической активностью, дает быстрый и длительный эффект. Трамадол относительно хорошо переносится, не вызывает в обычных дозах выраженного угнетения дыхания и существенно не влияет на кровообращение и желудочно-кишечный тракт. Трамадол, однако, может вызывать тошноту, рвоту, головокружение, потливость; повышенное артериальное давление может несколько снижаться.
Недостатком указанного средства является то, что трамадол не является «чистым» агонистом, трамадол уступает по активности морфину и применяется соответственно в больших дозах. Как и морфин, при повторных приемах он вызывает привыкание и интоксикацию организма у большинства пациентов.
При разработке принципиально новых безопасных анальгетиков, не обладающих указанными выше многочисленными негативными побочными эффектами и способных заменить опиаты, следует обратить внимание на субстанции эндогенной природы, способных создать принципиально новую по своей химической природе, характеру и механизмам фармакологической активности, третью группу анальгетических средств. Как правило, это относится к применению соединений пептидной природы.
Из уровня техники известны соединения пептидной (эндогенной) природы, потенциально применимые в качестве анальгетических средств. Патент RU 2286169 (приоритет от 18.04.2005, дата публикации 27.10.2006) «Семейство пептидов, обладающее анальгетической активностью» относится к химико-фармацевтической промышленности и описывает получение пептидов следующей формулы для создания новых лекарственных препаратов:
А-Б-Tyr-Pro- (DPro, dHPro, DdPro, DLdHPro,Hyp,) - B-X,
где А - О, -Ala, -Asp, -Glu, -Phe, -Gly, -His, -Ile, -Lys, -Leu, -Met, -Pro, -Arg, -Ser, -Thr, -Val, -Trp, -Tyr; Б - О, -Ala, -Asp, -Glu, -Phe, -Gly, -His, -Ile, -Lys, -Leu, -Met, -Pro, -Arg, -Ser, -Thr, -Val, -Trp, -Tyr; В -О, -Ala, -Asp, -Glu, -Phe, -Gly, -His, -Ile, -Lys, -Leu, -Met, -Pro, -Arg, -Ser, -Thr, -Val, -Trp, -Tyr; X - OH, -OCH3, -NH2.
По мнению авторов данного патента, все многочисленные описанные в нем пептиды обладают повышенной активностью по сравнению с пенталгином, анальгином, морфином.
Однако в материалах данного патента, в отличие от нашего подхода, не представлен молекулярный механизм связывания указанных субстанций со специфической мишенью - мембранным рецептором. Скорее всего, именно по нижеследующей причине: поскольку препаратом сравнения был морфин, анальгетический эффект указанных агентов пептидной природы обусловлен их воздействием на один из классов опиоидных рецепторов, которые эндогенно активируются еще и другими эндогенными пептидными субстанциями - например, энкефалинами и эндорфинами. Экзогенное применение последних с целью заменить опиаты в клинической практике остается до настоящего времени невозможным, поскольку их применение не может быть абсолютно безопасным: все агонисты опиоидных рецепторов, как было отмечено выше, вызывают большое количество негативных побочных эффектов в организме человека.
Следует отметить, что в описании изобретения, защищенного патентом RU 2286169, отсутствуют данные, свидетельствующие о безопасности длительного применения рассматриваемых агентов пептидной природы для лечения хронической боли. Также полностью отсутствуют данные об их действии на организм человека. Не приведены доказательства отсутствия у человека вследствие применения вышеуказанных пептидных субстанций таких негативных побочных эффектов, как привыкание и токсичность. С момента публикации патента RU 2286169 в мировой литературе не появилось данных, свидетельствующих о том, что на основе хотя бы одного из сотен упомянутых в этом патенте коротких пептидов было разработано принципиально новое безопасное и эффективное анальгетическое средство, способное заменить опиаты и опиоиды. По нашему мнению, этому есть ряд объяснений. Полученные на животных данные, особенно при использовании пептидных субстанций, могут сильно отличаться от результатов клинических исследований ввиду того, что эффективность реализации механизмов деструктивного действия пептидаз на экзогенно приложенные пептидные молекулы заметно различается у грызунов и у человека. В последнем случае, видимо, не осуществляется эффективная доставка действующей пептидной субстанции до своей молекулярной мишени в мембране ноцицептивного нейрона нервной системы человека.
Мы считаем, что описанные в патенте RU 2286169 субстанции могут быть приняты в качестве прототипа нашего изобретения благодаря своей эндогенной (пептидной) природе и попытке авторов цитируемого патента использовать их по назначению в качестве анальгетиков.
Хроническая боль заставляет страдать более 20% людей в мире, и это количество постоянно увеличивается. В этой связи, главная проблема, которая требует решения, заключается в получении контроля над механизмами трансформации «боли как симптома», выполняющей защитную функцию, в «боль как болезнь». Около 3,5 млн. онкологических больных ежедневно страдают от болей разной интенсивности и около 40% больных с промежуточными стадиями онкологического процесса и 60-90% с генерализацией заболевания испытывают боли в диапазоне от умеренных до сильных. Применение наркотических лекарственных препаратов в этих случаях сопряжено с серьезными опасностями, обусловленными многими побочными эффектами опиатов, включая угрожающую жизни пациента депрессию ЦНС с угнетением и даже остановкой дыхания. В первую очередь, значимость настоящего изобретения может проявиться при создании новых подходов к лечению хронической боли, требующих длительного применения анальгетических лекарственных средств. Хроническая боль определяется как «боль, которая продолжается сверх нормального периода заживления», причем срок ее существования составляет не менее 3 мес. К сожалению, в арсенале практической медицины до настоящего времени отсутствуют безопасные и эффективные анальгетики, способные заменить опиаты и опиоиды на второй и третьей ступенях анальгетической лестницы ВОЗ, т.е. в тех случаях, когда для купирования хронической боли необходимо применять опиаты. Именно поэтому становится остро необходимым создание принципиально новых анальгетиков, которые благодаря своей эндогенной природе и очень низким действующим концентрациям, были бы способны заменить опиаты и опиоиды при длительном системном клиническом применении для лечения хронической и острой боли, не вызывая при этом негативных побочных эффектов.
Подойти к решению этой технической проблемы станет возможным только в том случае, если удастся обнаружить новые молекулярные механизмы переработки сигналов в ноцицептивной системе (причем эти механизмы по своей физиологической эффективности должны быть сравнимы с эффективностью опиоидергической системы мозга), а также разработать лекарственную субстанцию эндогенной природы, которая позволила бы получить фармакологический контроль над подобными физиологическими механизмами без возникновения негативных побочных эффектов.
Молекулярные механизмы сигнализации, вызываемые активацией метаботропных рецепторов, в частности, опиоидных, как правило, включают G-белки в качестве трансдукторов сигнала. В случае экзогенной активации опиоидергической системы это имеет своим неизбежным следствием привыкание к опиатам и опиоидам, модулирующим указанные механизмы, и проявление иных негативных побочных эффектов. Как стало относительно недавно известно, альтернативой G-белкам может являться молекула Na,K-АТФазы, которая способна выполнять не только свою насосную функцию, но и выступать в качестве трансдуктора сигнала. Также известно, что модуляция трансдукторной функции Na,K-АТФазы осуществляется рядом соединений непептидной природы, в частности, эндогенного происхождения, относящихся к классу кардиотонических стероидов. Поэтому нами выдвинута совершенно неочевидная гипотеза, что эта функция может быть вовлечена и в регуляцию ответов ноцицептивной системы мозга человека. Заявляемые результаты получены посредством комплексного изучения различных представителей этого класса.
Наиболее близким по составу к заявляемой лекарственной субстанции является хорошо известный препарат «Строфантин Г», включающий в свой состав уабаин -кардиотонический стероид, эндогенно синтезируемый в организме человека. Препарат «Строфантин Г» не обладает анальгетическим эффектом и используется по иному медицинскому назначению. «Строфантин Г» - это инъекционная лекарственная форма (раствор для внутривенного введения, 0,25 мг/мл [VIDAL, Справочник лекарственных средств (https://www.vidal.ru/drugs/strophanthin-g)]). Вспомогательные вещества: лимонной кислоты моногидрат, натрия гидроксид, вода для инъекций. Доза подбирается индивидуально в зависимости от нозологии и реакции больного на терапию. При среднем темпе дигитализации в период насыщения вводят по 1 мл (0,25 мг) 2 раза в сутки (с интервалом 12 ч). Длительность периода насыщения составляет в среднем 2 дня. При необходимости можно ввести дополнительную дозу - 0,1-0,15 мг с интервалом от 0,5 до 2 ч. Суточная доза не должна превышать 1 мг, что соответствует 4 мл раствора для инъекций. Поддерживающая доза, как правило, не превышает 0,25 мг/сут. Связь с белками плазмы - 40%, не подвержен метаболизму, выводится почками в неизмененном виде. Кардиотоническое, антиаритмическое средство, блокирует насосную функцию Na,K-АТФазы клеточной мембраны кардиомиоцитов. Повышает силу и скорость сердечных сокращений (положительный ионотропный эффект), снижает атриовентрикулярную (AV) проводимость (отрицательный дромотропный эффект), стимулирует (в субтоксических и токсических дозах) образование гетеротопных импульсов вследствие снижения порога возбудимости и снижает частоту сердечных сокращений (ЧСС) - отрицательный хронотропный эффект. Негативные побочные эффекты препарата «Строфантин Г» проявляются как нарушения сердечного ритма, тошнота, рвота, диарея, мезентериальный инфаркт (редко), головная боль, повышенная утомляемость, бессонница, депрессия, галлюцинации, психоз, нарушения зрения, редко - гинекомастия. Побочные эффекты данного препарата в основном обусловлены его применением в относительно высоких концентрациях, превышающих эндогенные, или повышенной чувствительностью к этому препарату. Слишком быстрое введение может привести к развитию брадиаритмии, желудочковой тахикардии, AV-блокады, фибрилляции желудочков, остановке сердца. Еще раз отметим, что в мировой литературе отсутствуют результаты исследований, указывающих на потенциальную способность препарата «Строфантин Г» или других лекарственных средств на основе кардиотонических стероидов проявлять анальгетический эффект.
Сущность настоящего изобретения заключается в выяснении физиологической роли хелатных комплексов кардиотонических стероидов (CTS) с двухзарядными неорганическими катионами (М2+), поскольку, согласно нашей гипотезе, эндогенные кардиотонические стероиды, обнаруженные в тканях головного мозга и в кровотоке человека в чрезвычайно низких (наномолярных) концентрациях, в физиологически адекватных условиях находятся в молекулярной форме хелатного комплекса общей формулы CTS-M2+. Частным случаем подобного комплекса является рассматриваемый ниже кальциевый хелатный комплекс молекулы уабаина (ЭУ).
Нами обнаружены принципиальные различия между эффектами применения ЭУ и препарата «Строфантин Г» (пример 3). Последний содержит, фактически, свободную молекулу уабаина в относительно высокой концентрации, что приводит к частичному ингибированию насосной функции Na,K-АТФазы, не приводящему к проявлению анальгетического эффекта. ЭУ (кальциевый хелатный комплекс уабаина) в кратно более низкой концентрации активирует трансдукторную функцию Na,K-АТФазы, результатом чего является проявление у данной субстанции анальгетического эффекта. Перспективность и принципиальное отличие разрабатываемого нами подхода обусловлены тем фактом, что предлагаемые нами эндогенные субстанции непептидной природы активируют не опиоидные рецепторы, а воздействуют на совершенно другую молекулярную мембранную мишень - молекулу Na,K-АТФазы, выполняющую в мембране ноцицептивного нейрона не насосную, а трансдукторную функцию. Активация трансдукторной (ненасосной) функции Na,K-АТФазы приводит к модуляции медленных натриевых каналов Navl.8, кодирующих ноцицептивную (болевую) информацию. Достижение фармакологического контроля над этим механизмом легло в основу создания заявляемой анальгетической субстанции, фармакологической композиции на ее основе и способов их применения, лишенного недостатков, характеризующих прототип. Разработка лекарственной субстанции на основе ЭУ позволит найти ему применение в качестве эффективного и безопасного анальгетика, способного заменить опиаты в клинической практике благодаря активации нового молекулярного механизма мембранной сигнализации в ноцицептивном нейроне.
Нами разработана анальгетическая лекарственная субстанция на основе ЭУ, примененного в таком диапазоне концентраций, который обеспечивает доставку субстанции к своей мишени, к молекуле Na,K-АТФазы мембраны ноцицептивного нейрона, именно в эндогенных, т.е. наномолярных концентрациях, но не вызывает частичного ингибирования насосной функции Na,K-АТФазы, что достижимо при кратном снижении концентрации активного компонента по сравнению с препаратом «Строфантин Г». В этом случае, согласно нашим данным (примеры 3, 4), ЭУ обладает сильными анальгетическими свойствами, а также является безопасным для клинического применения благодаря своей эндогенной природе и низким действующим концентрациям. Подчеркнем, что наше решение основано на том, что инкубация уабаина в водном растворе, содержащем двухзарядные катионы кальция, приводит к образованию устойчивого комплексного соединения (ЭУ), которое, по сути, и является действующей лекарственной субстанцией. Молекула уабаина сначала координационно связывает (хелатирует) свободный катион кальция, тем самым образуя лекарственную субстанцию, а затем взаимодействие этой экзогенно приложенной субстанции с молекулой Na,K-АТФазы приводит к активации трансдукторной функции последней, что вызывает снижение функциональной активности медленных натриевых каналов Navl.8. В результате происходит купирование боли на спинальном и супраспинальном уровнях (пример 3), что подтверждается результатами клинических исследований (пример 4).
Настоящим изобретением описывается создание новой высокоэффективной анальгетической лекарственной субстанции на основе кардиотонических стероидов (CTS), в частности, ЭУ, не вызывающей негативных побочных клинических эффектов, новой фармацевтической композиции на основе указанной субстанции, и способов их применения для купирования хронической и острой боли.
Заявляемая анальгетическая субстанция является хелатным комплексом общей формулы CTS-M2+ (в частности, ЭУ, представляющий собой хелатный комплекс молекулы уабаина с катионом Са2+),
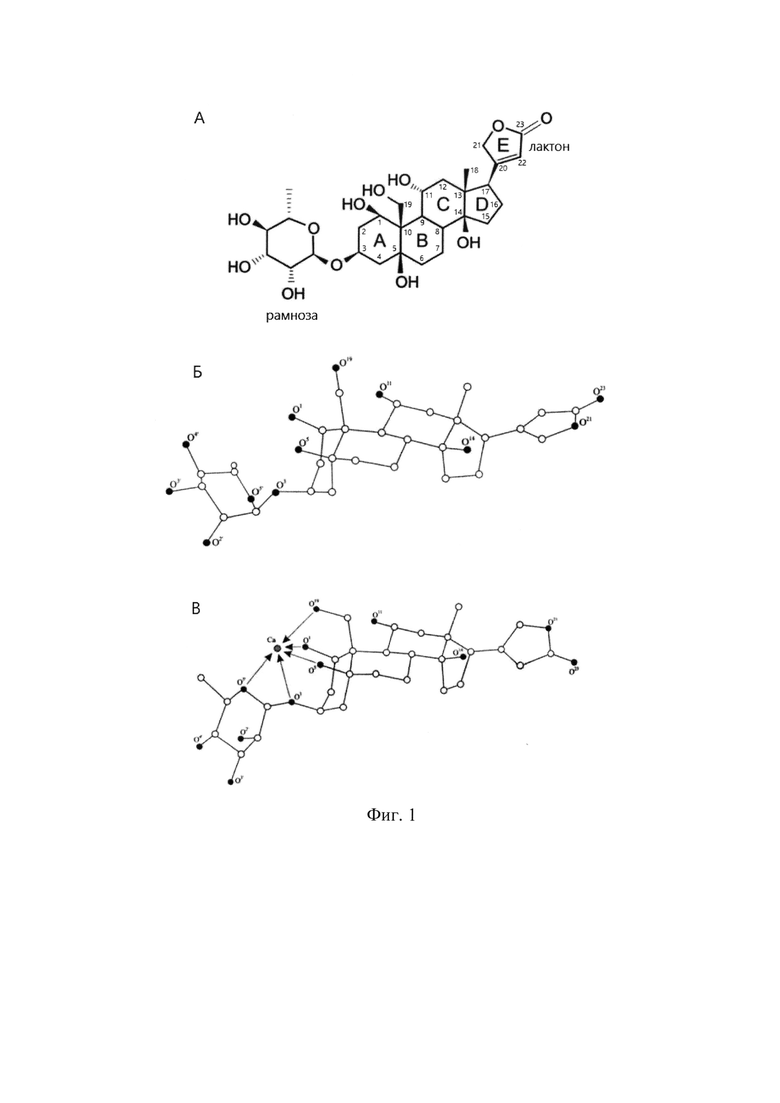
где CTS (в частности, молекула уабаина) представляет собой молекулу кардиотонического стероида, обладающего нижеуказанными структурными свойствами (фиг.1):
1) стероидный скелет молекулы представляет собой циклопентанпергидрофенантреновую структуру, содержащую 17 атомов углерода, дополненную метальными группами в положениях 10 и 13, в которой сочленение колец АВ и CD осуществляется в цис-положении, а колец ВС - в транс-положении;
2) в положении 17 стероидного скелета находится пяти- или шестичленное лактоновое кольцо Е (карденолиды и буфадиенолиды, соответственно);
3) в положении 3 стероидного скелета может присутствовать моносахаридный остаток или олигосахаридная цепь;
4) на атомах углерода колец А и В стероидного скелета (атомы С1-С10 согласно общепринятой номенклатуре кардиотонических стероидов), включая атом углерода С19, присутствуют не менее двух гидроксильных заместителей, участвующих в хелатировании катиона М2+, в дополнение к гидроксильному заместителю, моносахаридному остатку или олигосахаридной цепи в положении 3; М2+- любой двухзарядный неорганический катион (в частности, Са2+).
Заявляемая фармацевтическая композиция обладает анальгетическим действием и включает активный компонент - хелатный комплекс общей формулы CTS-M2+, а также фармацевтически приемлемый носитель (носитель, необходимый для доставки анальгетической субстанции до мембранной молекулы-мишени, но не влияющий на механизм хелатирования катиона М2+). Носитель может быть выбран из группы: вода для инъекций, полиэтиленгликоль, глицерол, глицерин. Композиция может быть выполнена в любой форме для систематического приема - как для наружного, так и внутреннего применения в форме пластыря, свечи, раствора, суспензии, крема, мази, геля, в ампулированной форме; она способна воздействовать однократно (одномоментно) и может быть приготовлена в форме для пролонгированного действия. Использование композиции не вызывает привыкания и, следовательно, отсутствует необходимость постоянного увеличения дозы лекарственной субстанции. Компоненты для создания фармацевтической композиции технологически доступны.
Заявляемые способы применения вышеуказанной фармацевтической композиции включают ее введение в терапевтически приемлемом количестве для купирования болевого синдрома. Однократная доза активного компонента не должна превышать 0,1 мг, при этом суточная дозировка активного компонента составляет не более 0,4 мг для пациентов с массой тела 50 - 100 кг. Композиция может быть введена внутривенным, эпидуральным, мукозальным, накожным, парентеральным, пероральным, энтеральным, интраназальным или ректальным способами. Заявляемые способы позволяют достичь абсолютно безопасного эффекта долговременного облегчения хронической и острой боли, длящегося в течении 4-6 ч после однократного введения фармакологической композиции, что сравнимо по продолжительности действия с известными эффектами действия опиатов и опиоидов. Заявляемые способы являются щадящими, так как все компоненты используемой в них фармацевтической композиции обладают эндогенной природой, а механизм ее воздействия сугубо избирателен. Заявляемые способы отличаются от известного рядом существенных признаков: использованием новой анальгетической субстанции эндогенной природы в определенной ограниченной терапевтической дозе, универсальными возможностями его применения при самых разных видах приема: внутривенном, эпидуральном, мукозальном, накожном, парентеральном, пероральном, энтеральном, интраназальном или ректальном. Из уровня техники не известно способов лечения хронической и острой боли, при реализации которых происходит подобное тонкое избирательное воздействие на соответствующие сигнальные пути передачи ноцицептивной информации, приводящее к купированию хронической и острой боли без интоксикации и привыкания.
Совокупность существенных признаков заявляемой группы изобретений позволяет достичь следующего технического результата. Создано высокоэффективное анальгетическое средство типа большого анальгетика общей формулы CTS-M2+(в частности, ЭУ), способное заменить опиаты в клинической практике и при этом не вызывающее негативных побочных фармакологических эффектов, таких как интоксикация и привыкание. По этой причине заявляемую лекарственную субстанцию можно начинать применять в максимальных терапевтических суточных дозах (до 0,4 мг в сутки) и практически сразу добиваться эффекта купирования острой боли (фаза 1), а затем и тонической боли (фаза 2) (фиг.2). Указанный эффект не затрагивает опиоидергическую систему молекулярной сигнализации и достигается в результате активации тонкого механизма специфического воздействия ЭУ на свою молекулярную мишень - молекулу Na,K-АТФазы, выполняющую функцию трансдуктора (усилителя и передатчика) сигнала. Этот сигнал снижает функциональную активность медленных натриевых каналов Nay 1.8, ответственных за импульсное кодирование болевых стимулов. В результате неожиданно проявляется уникальная способность ЭУ купировать хроническую боль в течение длительного периода времени (не менее четырех-шести часов при однократном воздействии), сравнимого по своей длительности с эффектом больших анальгетиков.
На фиг.1 приведены структурные данные молекул уабаина и ЭУ. (А) Структурная формула молекулы уабаина с обозначением колец стероидного скелета и нумерацией атомов углерода. (В) Пространственное строение наиболее низкоэнергетической конформации молекулы уабаина по результатам полного неэмпирического конформационного анализа методом RHF/6-31G* и нумерация атомов кислорода. Атомы углерода представлены в виде белых шаров, атомы кислорода - в виде черных шаров. Атомы водорода не приведены. (С) Пространственное строение наиболее низкоэнергетической конформации молекулы хелатного комплекса уабаина с катионом кальция (ЭУ) по результатам полного неэмпирического конформационного анализа методом RHF/6-31G* и нумерация атомов кислорода. Атомы углерода представлены в виде белых шаров, атомы кислорода - в виде черных шаров, атом кальция - в виде серого шара. Координационные связи Са-O показаны стрелками. Атомы водорода не приведены.
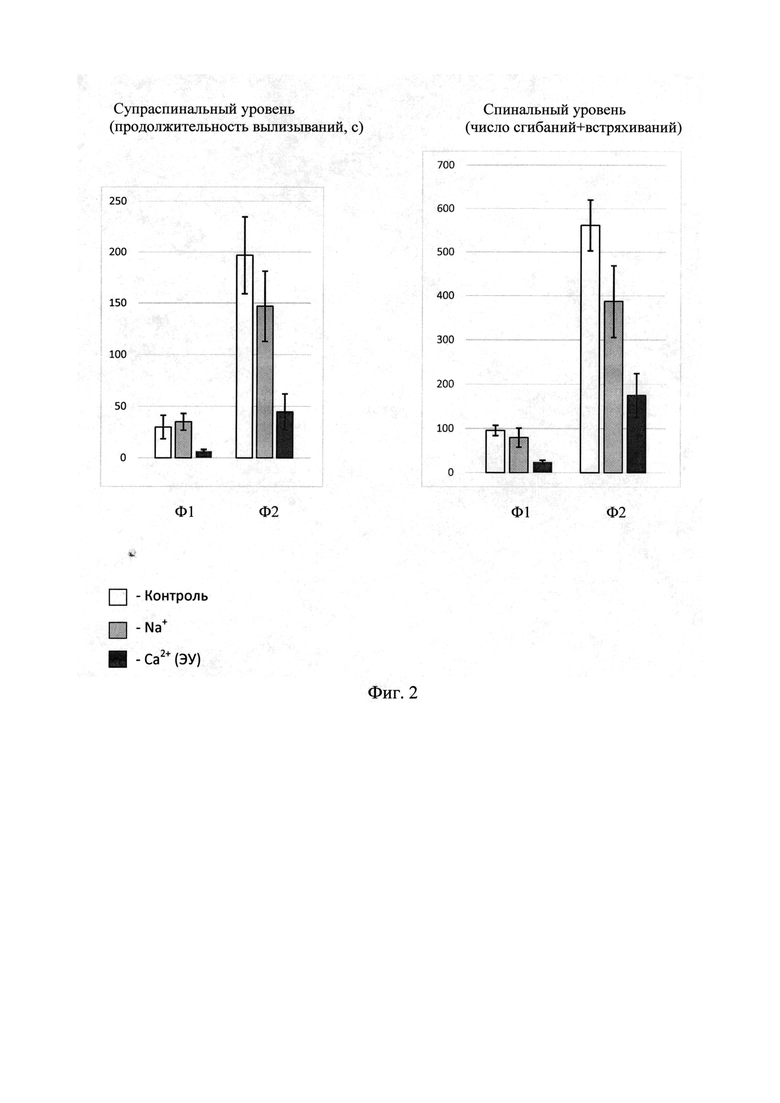
Фиг. 2 - анальгетическое действие кальциевого хелатного комплекса уабаина (ЭУ) на супраспинальном и спинальном уровнях по результатам формалинового теста.
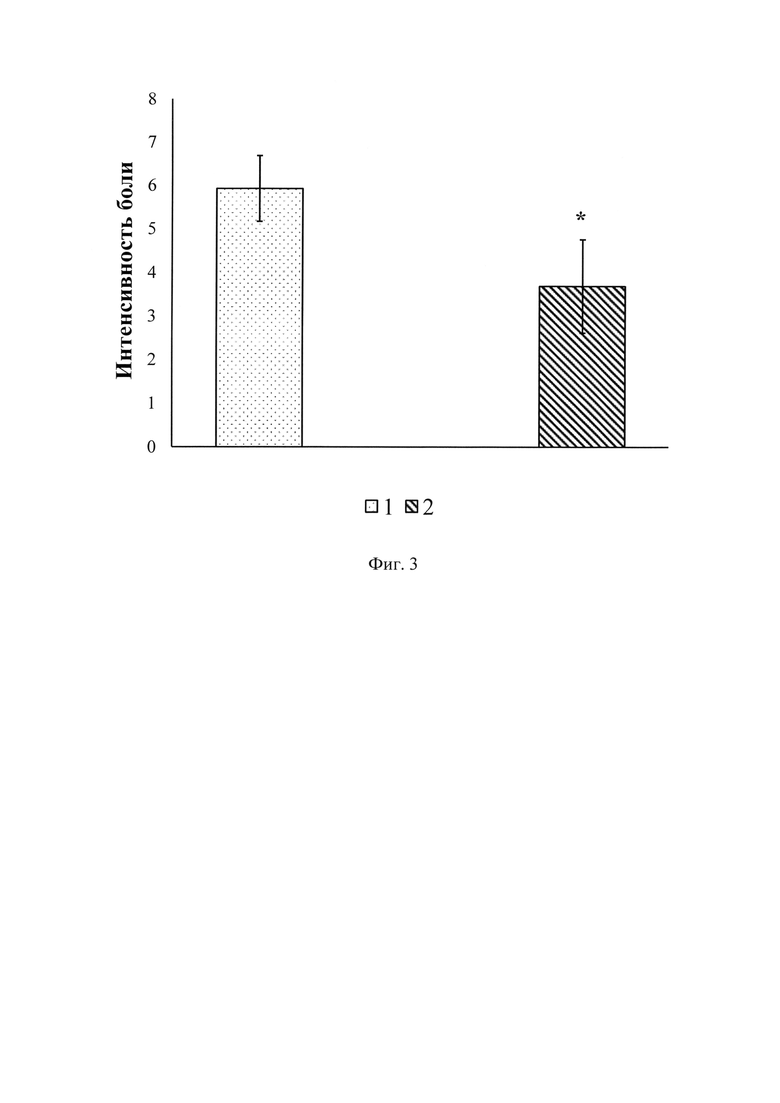
Фиг. 3 - анальгетическое действие кальциевого хелатного комплекса уабаина (ЭУ), входящего в состав геля при накожном применении.
1 - Интенсивность боли до нанесения геля на кожу;
2 - Интенсивность боли после нанесения геля на кожу.
* - р<0.05 (t-критерий Стьюдента).
Для подтверждения соответствия заявляемого решения требованию «промышленная применимость», а также для лучшего понимания сущности изобретения ниже представлены примеры его конкретной реализации, которые не ограничивают заявляемое изобретение.
Пример 1. Инъекционная форма. Способ получения фармацевтической композиции, содержащей в качестве активного компонента ЭУ, в форме раствора
Анальгетическая субстанция (ЭУ) представляет собой двухкомпонентный водный раствор уабаина и гидрокарбоната кальция, причем гидрокарбонат кальция взят в избытке по сравнению с уабаином. Молекула кальциевого хелатного комплекса уабаина образуется непосредственно в водном растворе, что является основой инъекционной формы.
Описание: Прозрачная жидкость (3 мг/мл) без цвета и запаха
Состав:
Активное вещество: 3 мг/мл.
Кальциевый хелатный комплекс 3 г уабаина (ЭУ).
Вспомогательные вещества:
Кальция гидрокарбонат 5,5 г.
Вода для инъекций до 1 л.
Форма выпуска
Ампулы по 1, 2 или 5 мл, содержащие 3 мг/мл препарата.
По 5 или 10 ампул в блистере, упакованных в картонную коробку.
Пример 2. Способ получения фармацевтической композиции в форме геля, содержащей в качестве активного компонента ЭУ
Для трансдермальной доставки ЭУ был подобран оптимальный состав гелевой основы с учетом совместимости ее компонентов. Субстанции эндогенной природы способны проникнуть через эпидермис, если помещены в амфифильную базу. Стандартная эмульсия является амфифильной базой и работает как система доставки ЭУ к глубоким слоям рогового слоя эпидермиса. Сначала была замешана гелевая основа, проверена ее стабильность в течение одного месяца (отсутствие изменения показателя рН более чем на 0,3 единицы, проверка коллоидной и термической стабильности и микробиологической чистоты). Затем в гелевую основу был введен ЭУ (в количестве 0,3%). Никаких изменений внешнего вида рецептуры не произошло. Гелевая композиция, содержащая ЭУ, была использована для получения клинических впечатлений после получения разрешительных документов (исходя из требований Технического Регламента Таможенного союза 009/2011) на использование гелевой композиции в качестве косметического средства.
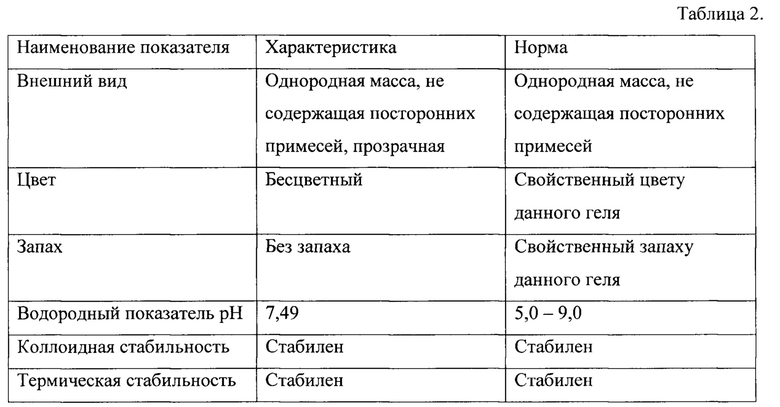
Разработанная рецептура представлена в таблице 1.

Физико-химические параметры рецептуры геля с ЭУ приведены в таблице 2.
Пример 3. Формалиновый тест
Формалиновый тест широко применяется во многих исследованиях при изучении эффектов экзогенных и эндогенных веществ на болевую чувствительность, а также при оценке анальгетической мощности препаратов разной природы. Формалиновый тест проведен по известной методике (Dubuisson D., Dennis S.G. The formalin test: a quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats // Pain. 1977. V. 4. P. 161-174; Tjolsen A., Berge O.-G., Hunskaar S., Rosland J.H., Hole K. The formalin test: an evaluation of the method // Pain. 1992. V.51. P. 5-17; Henry J.L., Yashpal K., Pitcher G.M., Coderre T.J. Physiological evidence that the "interphase" in the formalin test is due to active inhibition // Pain. 1999. V.89. P. 57-63; Dallel R., Raboisson P., Clavelou P. Evidence for a peripheral origin of the tonic nociceptive response to subcutaneous formalin // Pain. 1995. V.61. P. 11-16; Abbott F.V., Melzack R., Samuel C. Morphine analgesia in the tail-test and formalin pain tests is mediated by different neural systems // Exp. Neurol. 1982. V.75. P. 644-651; Abbott F.V., Ocvirk R., Najafee R. et al. Improving the efficiency of the formalin test // Pain. 1999. V.83. P. 561-569).
Исследования выполнены на 14 опытных самцах крыс линии Вистар (7 - кальциевый хелатный комплекс уабаина (ЭУ) и 7 - препарат «Строфантин Г», содержащий в качестве вспомогательного катиона не Са2+, a Na+. Контрольная группа состояла из семи взрослых самцов крыс. Средний вес животных составил 360 г. Введение препаратов опытным крысам в дозе 0,3 мг/кг и физиологического раствора контрольным крысам в объеме 1 мл производили внутрибрюшинно за 10 мин до инъекции раствора формалина (2,5%, 50 мкл, подкожно в подошву левой задней конечности). Регистрацию специфических реакций крысы в ответ на инъекцию формалина - сгибание + встряхивание (спинальный уровень) и вылизывание (супраспинальный уровень) - проводили в течение часа после инъекции ноцицептивного химического раздражителя.
В стандартных условиях эксперимента в контрольной и экспериментальной группах наблюдают два вида боли: 1) острая боль - первая короткая (менее 5 мин) фаза, вызванная непосредственным влиянием формалина на периферические ноцицептивные окончания, 2) тоническая боль - вторая продолжительная фаза, которую определяют процессы сенситизации в спинном мозгу, вызванные первой фазой, и афферентная сигнализация из очага воспаления; вторая фаза продолжается визуально 60 мин. Между первой и второй фазами существует интерфаза, не превышающая 10 мин, во время которой реакции на воспалительный агент у животного не наблюдается.
В протоколе отражали число «сгибаний», «встряхиваний» и продолжительность «вылизываний» левой инъецированной конечности. Оценивали межфазные интервалы и поведенческие реакции: исследовательское поведение, груминг, реакцию избегания, реакцию привыкания. Для контрольных крыс соблюдалась аналогичная для экспериментальных животных вся последовательность событий: внутрибрюшинная инъекция физиологического раствора, через 10 мин - инъекция формалина в подошву левой задней конечности, помещение крысы в камеру для наблюдения поведенческих реакций, регистрация реакций сгибаний + встряхиваний и продолжительности реакций вылизывания (с) в течение часа.
Результаты формалинового теста
Фаза 1 (острая фаза)
Полученные данные показали значимые различия в паттернах вылизывания у опытных животных, которые были подвергнуты воздействию кальциевого хелатного комплекса уабаина (ЭУ): среднее значение составляло 6,6±1,6 с. Среднее значение ответов контрольной группы животных было значимо выше: 30,0±11,4 с. Это свидетельствует о сильном анальгетическом эффекте заявляемой субстанции на супраспинальном уровне. Важно подчеркнуть, что при воздействии препарата «Строфантин Г» ответы экспериментальных животных (уабаин с Na+ был введен в той же концентрации 0,3 мг/кг) и животных контрольной группы значимо не отличались: 35,1±8,1 vs 30,0±11,4 с.
В паттернах сгибание+встряхивание также выявлены значимые различия между контрольными животными (95,7±21,5) и опытными животными после воздействия кальциевого хелатного комплекса уабаина (ЭУ) (24,3±3,1). Это свидетельствует о его сильном анальгетическом эффекте на спинальном уровне. При воздействии препарата «Строфантин Г» ответы экспериментальных животных и животных контрольной группы значимо не различались.
Итак, изучение острой боли (фаза 1) показало, что кальциевый хелатный комплекс уабаина (ЭУ) проявляет свойства сильного анальгетика как на спинальном, так и супраспинальном уровнях.
Фаза 2 (тоническая фаза)
Полученные данные показали значимые различия в паттернах вылизывания между контрольными животными (196,8±37,8) и опытными животными после воздействия кальциевого хелатного комплекса уабаина (ЭУ) (44,8±17,3). При воздействии препарата «Строфантин Г» ответы экспериментальных животных и животных контрольной группы значимо не различались. Итак, на супраспинальном уровне заявляемая субстанция обладает сильным анальгетическим эффектом.
В паттернах сгибание + встряхивание также выявлены значимые различия: между контрольными животными (561,5±58,4) и опытными животными после воздействия кальциевого хелатного комплекса уабаина (ЭУ) (175,0±49,3), что свидетельствует о его сильном анальгетическом эффекте на спинальном уровне. При воздействии препарата «Строфантин Г» наблюдался слабый анальгетический эффект при сравнении с ответами экспериментальных животных. Этот эффект был в два раза слабее по сравнению с сильным анальгетическим действием кальциевого хелатного комплекса уабаина (ЭУ).
Изучение тонической боли (фаза 2) показало, что кальциевый хелатный комплекс уабаина (ЭУ) проявляет сильные анальгезирующие свойства как на спинальном, так и на супраспинальном уровне.
Проведенные поведенческие эксперименты позволяют сделать следующие выводы:
1. Инъекция кальциевого хелатного комплекса уабаина (ЭУ) крысам в дозе 0,3 мг/кг, в отличие от инъекции препарата «Строфантин Г», вызывает сильный анальгетический эффект как на спинальном, так и на супраспинальном уровнях.
2. Сильный анальгетический эффект ЭУ наблюдается при возникновении как острой, так и тонической боли.
3. Наблюдения за экспериментальными животными показали, что ни в одном случае не проявлялась реакция привыкания к действию исследуемой субстанции.
Пример 4. Инициативное клиническое исследование анальгетического эффекта кальциевого хелатного комплекса уабаина (ЭУ)
Для решения поставленных задач была сформирована группа пациентов в количестве 30 человек, страдающих хроническим вариантом неспецифической боли в спине или болью в конечностях без уточнения нозологической формы и в том, и в другом случае. Все пациенты жаловались на болевые ощущения средней или тяжелой степени интенсивности, оцененные при помощи вербально-цифровой шкалы. Исследование одобрено локальным этическим комитетом.
После включения пациента в группу исследования опрашивали пациента с целью охарактеризовать болевые ощущения и оценки их интенсивности. Затем 1-2 г разрешенного к применению геля, содержащего ЭУ, наносили на кожные покровы и немного втирали. Нанесение осуществляли в проекции боли, где определялась максимальная ее интенсивность. Через 60 и 120 мин пациент повторно прицельно опрашивался с целью выявления изменений в характеристиках болевых ощущений. Также оценивали изменение интенсивности боли с помощью вербально-цифровой шкалы. Кроме того, производили осмотр места нанесения геля для выявления местных изменений на коже и выявляли чувствительность кожи в месте нанесения геля на прикосновение (неноцицептивное тактильное ощущение). Повторное нанесение геля производили через 48 ч; на этом этапе оценивали местные изменения на кожных покровах в месте первичного проведения процедуры. Следующее нанесение геля осуществляли еще через 48-72 ч. Заключительную оценку интенсивности боли с помощью вербально-цифровой шкалы выполняли через 48 ч после третьего нанесения геля.
Осмотр пациентов перед накожным нанесением геля, содержащего ЭУ, позволил выяснить, что при хронической неспецифической боли в спине и боли в конечностях преобладает ноющий вариант боли. В двух случаях была обнаружена жгучая боль, и в обоих этих случаях боль локализовалась в спине. После нанесения геля интенсивность боли в подавляющем большинстве случаев снижалась. При сравнении значений интенсивности боли до нанесения геля и через 60 мин после была выявлена статистически достоверная разница (t=8,1; р<0,05 - t - критерий Стьюдента для связанных выборок). Тот же результат был обнаружен и через 120 мин после нанесения геля на кожу (t=8,1; р<0,05 - t - критерий Стьюдента для связанных выборок). Прикосновение к участку кожи, на который был нанесен гель, вызывало ослабленное неноцицептивное тактильное ощущение в сравнении с другими областями кожных покровов, т.е. наблюдалась гипестезия. При осмотре кожных покровов в месте нанесения геля через 60 мин у двух пациентов наблюдали слабо выраженную гиперемию (переполнение кровью сосудов кровеносной системы области кожи), которая не определялась через 48 ч и не сказалась на дальнейшем плане проведения этого исследования. Других изменений (зуда, шелушения и пр.) также не было выявлено. Жалоб на какие-либо кожные изменения у остальных пациентов не было. Анализ данных, полученных после курса воздействия на протяжении недели, состоящего из трехкратного нанесения на кожу геля, содержащего ЭУ, позволил выявить статистически достоверную разницу величин интенсивности болевых ощущений между исходными значениями и данными, полученными после завершения курса (фиг.3).
В результате выполненного исследования стало очевидно, что гель, содержащий ЭУ, при нанесении на кожные покровы способен снизить интенсивность хронической боли в спине и конечностях.
Результаты, полученные с помощью вербально-цифровой шкалы после недельного курса воздействия, состоящего из трехкратного нанесения на кожу геля, содержащего ЭУ, доказывают статистически достоверную разницу величин интенсивности болевых ощущений между исходными значениями и значениями после завершения курса.
Полученные данные указывают на перспективность замены анальгетических препаратов, обладающих негативными побочными эффектами при системном применении на высокоэффективный кальциевый хелатный комплекс уабаина (ЭУ), низкие (эндогенные) концентрации которого обеспечивают безопасность его применения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИНТЕТИЧЕСКОЕ АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО И СПОСОБ ЛЕЧЕНИЯ НА ОСНОВЕ ЭТОГО СРЕДСТВА | 2006 |
|
RU2322977C1 |
| СИНТЕТИЧЕСКОЕ АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО ПЕПТИДНОЙ ПРИРОДЫ И СПОСОБ ЕГО ПРИМЕНЕНИЯ | 2017 |
|
RU2656188C1 |
| АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО И СПОСОБ ЛЕЧЕНИЯ БОЛЕВОГО СИНДРОМА РАЗЛИЧНОЙ ЭТИОЛОГИИ С ПОМОЩЬЮ ЭТОГО СРЕДСТВА | 2007 |
|
RU2367432C2 |
| 6-(3,3- диметил-3,4-дигидроизохинолин-1-ил) аминогексановая кислота и фармацевтическая композиция на ёе основе, обладающие анальгетической активностью | 2016 |
|
RU2648445C1 |
| Средство для купирования опиоидного абстинентного синдрома | 2017 |
|
RU2643588C1 |
| Суппозитории нефопама для лечения острого и хронического болевого синдрома на липофильной основе и способ их получения | 2017 |
|
RU2661618C1 |
| СПОСОБ ЛАЗЕРНОЙ КОРРЕКЦИИ БОЛЕВОГО ОЩУЩЕНИЯ | 2005 |
|
RU2296596C2 |
| АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО ПЕПТИДНОЙ СТРУКТУРЫ НА ОСНОВЕ УНДЕКАПЕПТИДА, СОДЕРЖАЩЕГО D-ОКТААРГИНИНОВЫЙ ВЕКТОР | 2013 |
|
RU2541127C1 |
| СРЕДСТВО ДЛЯ ЭФФЕКТИВНОГО КУПИРОВАНИЯ ОСТРОГО И/ИЛИ ХРОНИЧЕСКОГО БОЛЕВОГО СИНДРОМА И СПОСОБ ЕГО ПРИМЕНЕНИЯ | 2016 |
|
RU2622980C1 |
| АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО ПЕПТИДНОЙ СТРУКТУРЫ НА ОСНОВЕ ТРИДЕКАПЕПТИДА, СОДЕРЖАЩЕГО D-ОКТААРГИНИНОВЫЙ ВЕКТОР | 2013 |
|
RU2538727C1 |
Группа изобретений относится к области фармацевтики и медицины. 1 объект представляет собой фармацевтическую композицию, обладающую анальгетическим действием, включающую активный компонент – анальгетическую субстанцию, являющуюся хелатным комплексом уабаин-Са2+, и фармацевтически приемлемый носитель. 2 и 3 объекты – способы купирования болевого синдрома, заключающиеся в активации трансдукторной ненасосной функции Na,K-АТФазы, включающие введение фармацевтической композиции. Технический результат заключается в эффективности анальгетического средства, являющегося хелатным комплексом уабаин-Са2+, способного заменить опиаты в клинической практике и при этом не вызывающего негативных побочных фармакологических эффектов, таких как интоксикация и привыкание. 3 н. и 5 з.п. ф-лы, 3 ил., 2 табл., 4 пр.
1. Фармацевтическая композиция, обладающая анальгетическим действием, включающая активный компонент - анальгетическую субстанцию, являющуюся хелатным комплексом уабаин-Са2+, где уабаин представляет собой молекулу кардиотонического стероида с нижеуказанными структурными свойствами:
1) стероидный скелет молекулы представляет собой циклопентанпергидрофенантреновую структуру, содержащую 17 атомов углерода, дополненную метильными группами в положениях 10 и 13, в которой сочленение колец АВ и CD осуществляется в цис-положении, а колец ВС - в транс-положении;
2) в положении 17 стероидного скелета находится пятичленное лактоновое кольцо Е;
3) в положении 3 стероидного скелета присутствует моносахаридный остаток;
4) на атомах углерода 1-10 колец А и В стероидного скелета согласно номенклатуре стероидов и атоме углерода С19 присутствуют не менее двух гидроксильных заместителей, участвующих в хелатировании катиона Са2+, в дополнение к моносахаридному остатку в положении 3;
и фармацевтически приемлемый носитель.
2. Композиция по п. 1, характеризующаяся тем, что она выполнена в форме для инъекций или трансдермального введения.
3. Композиция по п. 1, характеризующаяся тем, что она выполнена в форме раствора или геля.
4. Композиция по п. 1, характеризующаяся тем, что носитель выбран из группы: полиэтиленгликоль, вода для инъекций, глицерин.
5. Способ купирования болевого синдрома, заключающийся в активации трансдукторной ненасосной функции Na,K-АТФазы, причем способ включает введение фармацевтической композиции по п. 1, обладающей анальгетическим действием, в форме для инъекций или трансдермального введения.
6. Способ по п. 5, характеризующийся тем, что композиция выполнена в форме раствора или геля.
7. Способ купирования болевого синдрома, заключающийся в активации трансдукторной ненасосной функции Na,K-АТФазы, причем способ включает введение фармацевтической композиции по п. 1, обладающей анальгетическим действием, характеризующийся тем, что композицию вводят в разовой дозировке с содержанием активного компонента не более 0,1 мг, при этом суточная дозировка активного компонента составляет не более 0,4 мг для пациентов с массой тела 50-100 кг.
8. Способ по п. 7, характеризующийся тем, что композиция выполнена в форме раствора или геля.
| PLAKHOVA V.B | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| [электронный ресурс] | |||
| Усилитель двойного действия с одновременным усилением высокой и низкой частоты | 1923 |
|
SU1807A1 |
| JP 2007186477 A, 26.07.2007 | |||
| ПЕННИЯЙНЕН В.А | |||
| и др | |||
| Возможные | |||

