Изобретение относится к медицине и ветеринарии, в частности к лекарственным препаратам, и может быть использовано для лечения острого и хронического болевого синдрома.
Боль является одной из наиболее распространенных причин обращения пациентов за врачебной помощью. Болевые ощущения приводят к значительному снижению качества жизни населения и к прямым экономическим потерям за счет снижения трудоспособности людей. В настоящее время болевой синдром представляет собой серьезную клиническую проблему, требующую значительных усилий со стороны врачей по улучшению качества и эффективности ее терапии.
Онкологические заболевания - одна из четырех главных медико-социальных проблем современного общества, ведущих к заболеваемости, смертности и инвалидизации как в России, так и во всем мире. По данным Всемирной Организации Здравоохранения (ВОЗ), ежегодно в мире умирают свыше 4 млн онкологических больных. В России ежегодно регистрируется более 500000 новых случаев злокачественных новообразований, умирают ежегодно около 300 тыс человек, не получающих адекватного обезболивания в последние недели жизни.
В терминальной стадии онкологического заболевания от боли страдают от 55 до 95% больных. При этом боль средней интенсивности испытывают 40-50%, очень сильную и невыносимую - 25-30% пациентов. В то же время проведенное в Европе крупномасштабное исследование по изучению хронической боли доказало и высокую распространенность выраженной хронической неонкологической боли (умеренной и сильной, более 5 баллов по 10-балльной шкале) в популяции. Лечение хронических болевых синдромов остается одной из самых актуальных и сложных задач современной фармакологии, так как длительное болевое воздействие запускает патологические реакции в периферической и центральной нервной системе, вследствие чего развивается резистентность к медикаментозным воздействиям.
В настоящее время опиатные анальгетики стоят в первом ряду средств борьбы с сильной болью. Для этой группы препаратов характерна высокая анальгезирующая активность, обеспечивающая возможность их применения в качестве высокоэффективных болеутоляющих средств в разных областях медицины, особенно при травмах, оперативных вмешательствах, ранениях и т.д. и при заболеваниях, сопровождающихся выраженным болевым синдромом (злокачественные новообразования, инфаркт миокарда и др.). Оказывая специфическое влияние на центральную нервную систему (ЦНС), опиаты вызывают эйфорию, изменение эмоциональной окраски боли и реакции на нее. Наиболее существенным их недостатком является опасность развития психической и физической зависимости, а также угнетение дыхания.
К группе опиатных анальгетиков относят природные алкалоиды (морфин, кодеин) и синтетические соединения (тримеперидин, фентанил, трамадол, налбуфин и др.). Большинство синтетических препаратов получено по принципу модификации молекулы морфина с сохранением элементов его структуры или ее упрощением. Путем химической модификации молекулы морфина получены также вещества, являющиеся его антагонистами (налоксон, налтрексон). По выраженности анальгетического действия и побочным эффектам препараты различаются между собой, что обусловлено их химической структурой и физико-химическими свойствами и, соответственно, взаимодействием с рецепторами, вовлеченными в осуществление их фармакологических эффектов.
В понимании нейрохимических механизмов действия опиатов большую роль сыграло открытие в 70-х годах специфических опиатных рецепторов и их эндогенных пептидных лигандов - энкефалинов и эндорфинов. Опиатные рецепторы сконцентрированы в основном в ЦНС, но содержатся также в периферических органах и тканях. В мозге опиатные рецепторы находятся в основном в структурах, имеющих непосредственное отношение к передаче и кодированию болевых сигналов. В зависимости от чувствительности к разным лигандам среди опиатных рецепторов выделяют субпопуляции: 1 - (мю), 2 - (каппа), 3 - (дельта), 4 - (сигма), 5 - (эпсилон), имеющие различную функциональную значимость.
Прогресс в области синтеза биологически активных пептидов создал основу для получения химическим путем не только самих эндогенных пептидов, но и их многочисленных аналогов. Последние нередко обладают более избирательной биологической активностью, большей стабильностью и продолжительностью действия.
Одним из таких аналогов лейцин-энкефалина явился гексапептид, получивший название даларгин. Он был синтезирован в лаборатории синтеза пептидов ВКНЦ АМН СССР под руководством профессора М.И. Титова. В даларгине в отличие от эндогенного лейцин-энкефалина глицин заменен на D-аланин, что ведет к замедлению расщепления пептида энкефалиназами, а к С-терминальной части молекулы энкефалина был присоединен аргинин (положительно заряженный аргинин вводился для устранения проникновения даларгина в высшие отделы центральной нервной системы - ЦНС): Tyr-Gly-Gly-Phe-Leu - лейцин-энкефалин, Tyr-D-Ala-Gly-Phe-Leu-Arg - даларгин.
Предварительные проведенные исследования показали, что даларгин относится к опиоидным пептидам периферического действия со своеобразным спектром влияния на опиатные рецепторы висцеральных органов. Он преимущественно связывается с δ-рецепторами и в меньшей степени с µ-рецепторами. Даларгин практически не проникает через гематоэнцефалический барьер, не вызывает привыкания, физической зависимости и толерантности. Этими качествами он выгодно отличается от многих опиоидных соединений [Булгаков С.А., 2011].
В середине 80-х годов прошлого века было синтезировано много различных пептидов, содержащих аминокислотные последовательности, характерные для опиоидных пептидов. В ранних работах было установлено, что С-концевые аминокислоты дерморфина (Tyr-D-Ala-Phe-Gly) - минимальная последовательность, необходимая для проявления опиоидергической активности [Montecucchi et al., 1981].
Кроме того, было показано, что аналоги энкефалинов, имеющие в своем составе последовательность Tyr-D-Arg2, обладают мощным антиноцицептивным действием [Sasaki et al., 1985], и что структурные аналоги этих соединений - тетрапептиды - обладают анальгетической активностью, в несколько раз превышающей таковую у морфина.
Так, в 1988 году экспериментально было продемонстрировано, что антиноцицептивный эффект синтетического тетрапептида Tyr-D-Arg-Phe-Gly-NH2 в 6 раз превышает активность морфина [Chaki et al., 1988a]. Однако выраженное обезболивающее действие тетрапептида было сопряжено с развитием толерантности и физической зависимости, что ограничило его дальнейшее продвижение в качестве лекарственного вещества [Chaki et al., 1988b]. Системное введение тетрапептида (Tyr-D-Arg-Phe-Gly-NH2) в течение 5 дней мышам вело к развитию толерантности к анальгетическому эффекту пептида [Chaki et al., 1990]. При хроническом подкожном введении тетрапептида крысам развивалась физическая зависимость. Однако признаки развития зависимости были несколько менее выражены, чем при развитии физической зависимости к морфину [Chaki et al., 1988].
В настоящее время весьма активно ведется создание синтетических аналогов эндогенных опиоидных пептидов, обладающих заданным спектром рецепторного действия и лишенных недостатков, присущих энкефалинам и эндорфинам: плохой проницаемостью через гематоэнцефалический барьер и высокой скоростью разрушения. В 2004 году Kacprzak и его коллеги на основании предыдущих исследований и собственных данных сделали вывод, что D-формы полиаргинина, будучи высокостабильными, низкотоксичными молекулами, являются ингибиторами фурина, протеолитического фермента, относящегося к сериновым протеазам клеток млекопитающих, расположенных в аппарате Гольджи [Kacprzak et al., 2004]. Соответственно, включение полиаргининового вектора в структуру пептидной молекулы могло бы препятствовать действию эндопротеаз и способствовать увеличению продолжительности действия пептида.
Кроме того, для октааргининовых пептидов установлена способность проникать внутрь клетки через мембрану при помощи различных молекулярных механизмов [Miklan et al., 2011]. Так Nakase и его коллеги выяснили, что интернализация полиаргинина осуществляется через механизмы макропиноцитоза [Nakase et al., 2004]. Другие исследования показали, что октааргинин проникает в клеточное пространство и увеличивает проницаемость ГЭБ при помощи связи с синдеканами клеточной матрицы [Esclatine et al., 2001; Yoneda et al., 2003], которые создают некий якорь для различных внешних молекул и патогенов на поверхности клетки-мишени и входят в состав механизма липидного «рафта» [Dehio et al., 1998; Couchman, 2003; Tkachenko et al., 2004].
Таким образом, было высказано предположение о том, что включение D-формы полиаргинина в структуру пептидной молекулы, обладающей анальгетическим спектром действия, может в значительной степени увеличить биодоступность соединения без существенной потери фармакологической эффективности.
Задачей настоящего изобретения является создание высокоактивного болеутоляющего средства для перорального и парентерального введения, предназначенного для купирования острого болевого синдрома в экстремальной медицине и терапии хронического болевого синдрома, в том числе и на поздних стадиях онкологических заболеваний.
Предметом заявки является применение тридекапептида со следующей структурой - H-Tyr-D-Arg-Phe-Gly-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-Gly-OH·10HCl (далее по тексту H-Tyr-D-Arg-Phe-Gly-(D-Arg)8-Gly-OH 10HCl) в качестве анальгетического средства.
Кроме того, для достижения поставленного результата предлагается лекарственная форма, содержащая в качестве действующего вещества тридекапептид H-Tyr-D-Arg-Phe-Gly-(D-Arg)8-Gly-OH 10HCl. Форма для приема препарата может быть выполнена в виде одной из следующих лекарственных форм: раствор для инъекций, назальный спрей, назальные капли, сублингвальные таблетки, суббукальные пластыри, ректальные суппозитории, кишечнорастворимые таблетки и капсулы, трансдермальные терапевтические системы для резорбтивного применения.
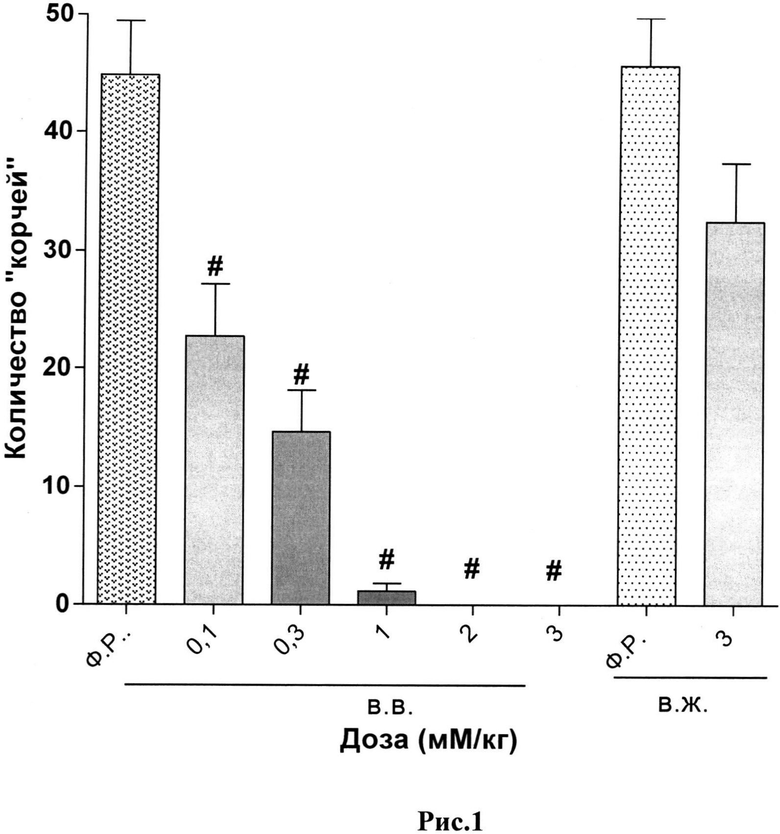
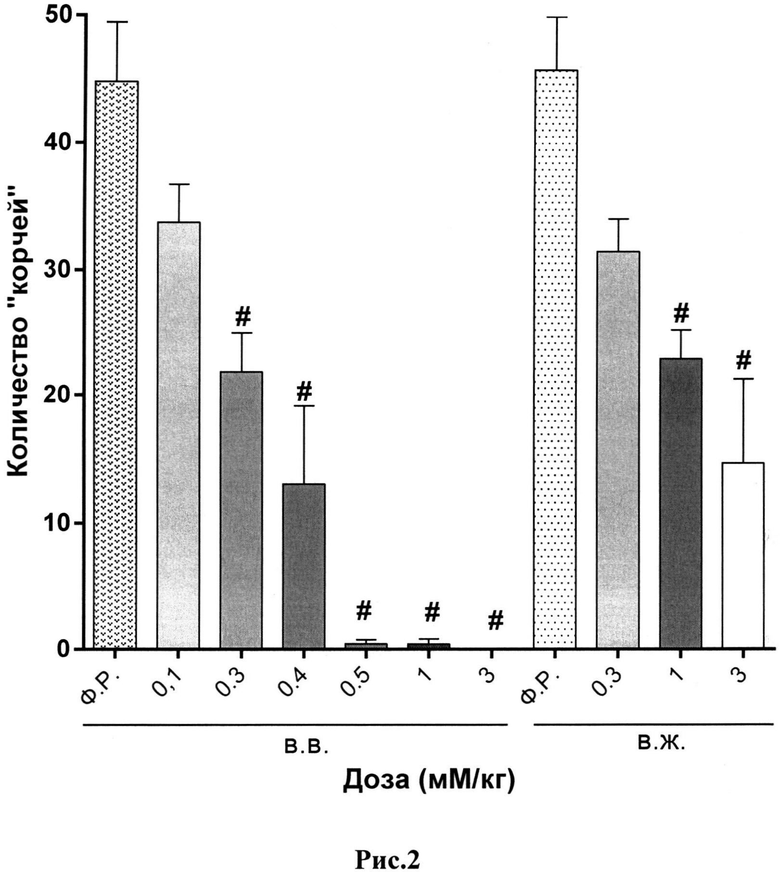
В качестве иллюстративных материалов к описанию приложены результаты масс-спектр анализа, а также рис.1 (влияние тетрапептида при разных путях введения на ноцицептивный ответ типа «корчи») и рис.2 (влияние тридекапептида при разных путях введения на ноцицептивный ответ типа «корчи»).
Способ получения заявляемого в настоящем изобретении пептида Н-Tyr-D-Arg-Phe-Gly-(D-Arg)8-Gly-OH 10HCl в литературе не описан.
Синтез пептида H-Tyr-D-Arg-Phe-Gly-(D-Arg)8-Gly-OH 10HCl осуществляли по изложенной ниже методике. Список сокращений представлен в таблице 1.
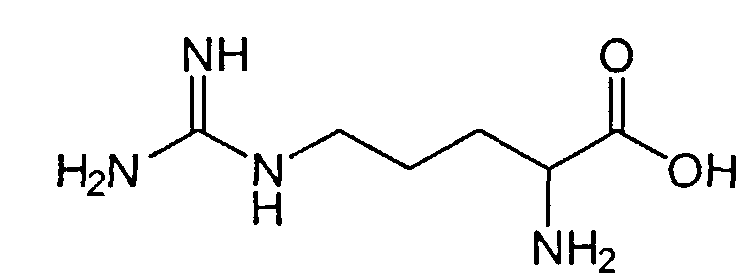
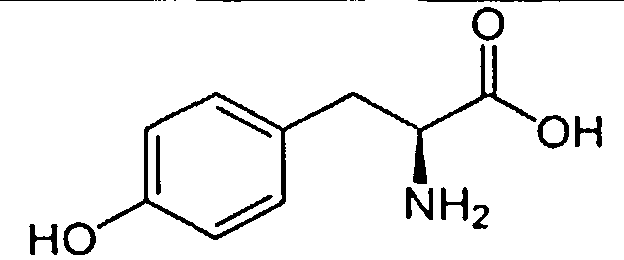
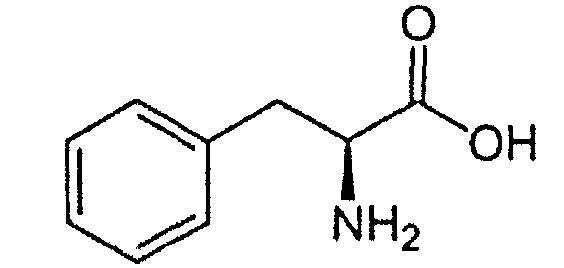
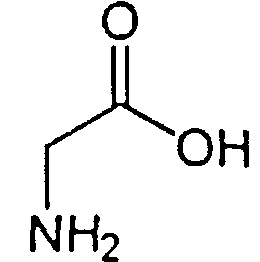
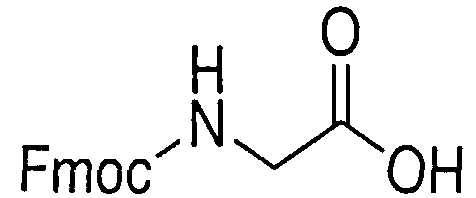
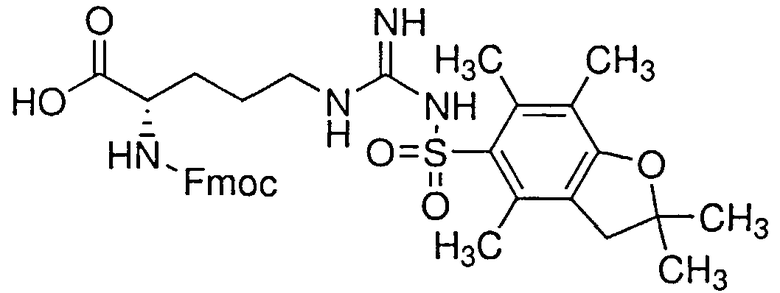
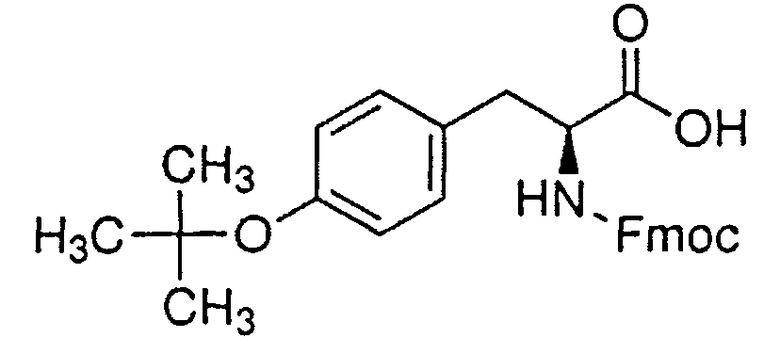
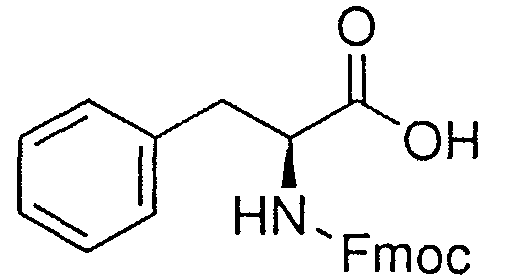
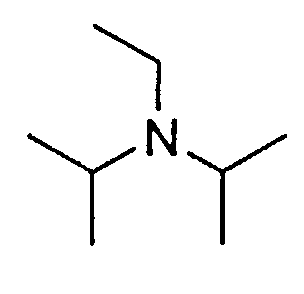
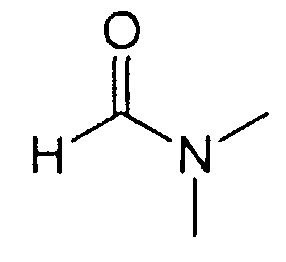
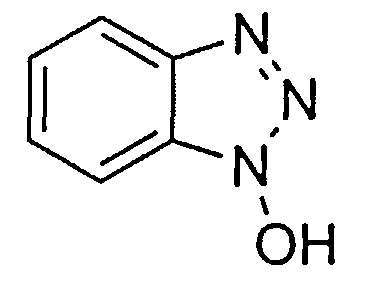
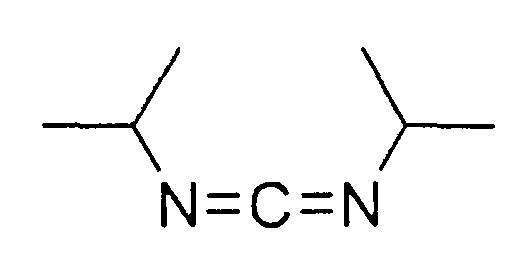
В реакторе для твердофазного синтеза суспендировали 6.0 г 2-хлортритильной смолы (емкость 1.5 ммоль/г) в 45 мл DCM, выдерживали в течение 5 мин, смолу отфильтровывали, промывали 2×30 мл DCM. К смоле добавляли раствор 2/95 г (9.9 ммоль) Fmoc-Gly-OH и 6 мл (36 ммоль) DIPEA в 30 мл DCM, перемешивали в течение 60 мин при комнатной температуре. Смолу отфильтровывали, промывали 2×30 мл DCM, обрабатывали 2×30 мл смеси DCM/метанол/DIPEA (17:2:1) в течение 10 мин, промывали 2×30 мл DCM и 3×30 мл DMF. В реактор загружали 30 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×30 мл DMF, добавляли 30 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×30 мл DMF.
В реактор загружали охлажденный (4°С) раствор 7.75 г (20.0 ммоль) Fmoc-Phe-OH, 2.98 г (22.0 ммоль) HOBt и 3.42 мл (22.0 ммоль) DIC в 30 мл DMF, перемешивали в течение 2 час при комнатной температуре. Смолу отфильтровывали, промывали 6×30 мл DMF, добавляли 30 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×30 мл DMF, добавляли 30 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×30 мл DMF.
В реактор загружали охлажденный (4°С) раствор 12.98 г (20.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 2.98 г (22.0 ммоль) HOBt и 3.42 мл (22.0 ммоль) DIC в 30 мл DMF, перемешивали в течение 2 час при комнатной температуре. Смолу отфильтровывали, промывали 6×30 мл DMF, добавляли 30 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×30 мл DMF, добавляли 30 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×30 мл DMF.
В реактор загружали охлажденный (4°С) раствор 9.19 г (20.0 ммоль) Fmoc-Tyr(tBu)-OH, 2.98 г (22.0 ммоль) HOBt и 3.42 мл (22.0 ммоль) DIC в 30 мл DMF, перемешивали в течение 2 час при комнатной температуре. Смолу отфильтровывали, промывали 6×30 мл DMF, 3×30 мл DCM. Затем смолу обрабатывали 10×30 мл 1% раствором трифторуксусной кислоты в DCM, получаемые растворы объединяли в колбе, содержащей 30 мл 10% раствора пиридина в метаноле. Смесь упаривали до объема ~50 мл, к остатку добавляли 200 мл воды. Выпавший осадок отфильтровывали, промывали водой, сушили. Получено 9.1 г продукта с чистотой по ВЭЖХ 98%.
В реакторе для твердофазного синтеза суспендировали 2.0 г 2-хлортритильной смолы (емкость 1.5 ммоль/г) в 15 мл DCM, выдерживали в течение 5 мин, смолу отфильтровывали, промывали 2×10 мл DCM. К смоле добавляли раствор 0.98 г (3.3 ммоль) Fmoc-Gly-OH и 2 мл (12 ммоль) DIPEA в 10 мл DCM, перемешивали в течение 60 мин при комнатной температуре. Смолу отфильтровывали, промывали 2×10 мл DCM, обрабатывали 2×10 мл смеси DCM/метанол/DIPEA (17:2:1) в течение 10 мин, промывали 2×10 мл DCM и 3×10 мл DMF. В реактор загружали 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°С) раствор 3.89 г (6.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 0.98 г (7.2 ммоль) HOBt и 1.12 мл (7.2 ммоль) DIC в 10 мл DMF, перемешивали в течение 2 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°С) раствор 3.89 г (6.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 0.98 г (7.2 ммоль) HOBt и 1.12 мл (7.2 ммоль) DIC в 10 мл DMF, перемешивали в течение 4 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°С) раствор 5.19 г (8.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 1.35 г (10.0 ммоль) HOBt и 1.56 мл (10.0 ммоль) DIC в 10 мл DMF, перемешивали в течение 3 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°С) раствор 5.19 г (8.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 1.35 г (10.0 ммоль) HOBt и 1.56 мл (10.0 ммоль) DIC в 10 мл DMF, перемешивали в течение 4 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°С) раствор 5.19 г (8.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 1.35 г (10.0 ммоль) HOBt и 1.56 мл (10.0 ммоль) DIC в 10 мл DMF, перемешивали в течение 10 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°С) раствор 6.48 г (10.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 1.62 г (12.0 ммоль) HOBt и 1.88 мл (12.0 ммоль) DIC в 10 мл DMF, перемешивали в течение 12 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°С) раствор 6.48 г (10.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 1.62 г (12.0 ммоль) HOBt и 1.88 мл (12.0 ммоль) DIC в 10 мл DMF, перемешивали в течение 12 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
В реактор загружали охлажденный (4°С) раствор 6.48 г (10.0 ммоль) Fmoc-D-Arg(Pbf)-OH, 1.62 г (12.0 ммоль) HOBt и 1.88 мл (12.0 ммоль) DIC в 10 мл DMF, перемешивали в течение 16 час при комнатной температуре. Смолу отфильтровывали, промывали 6×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×10 мл DMF, добавляли 10 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×10 мл DMF.
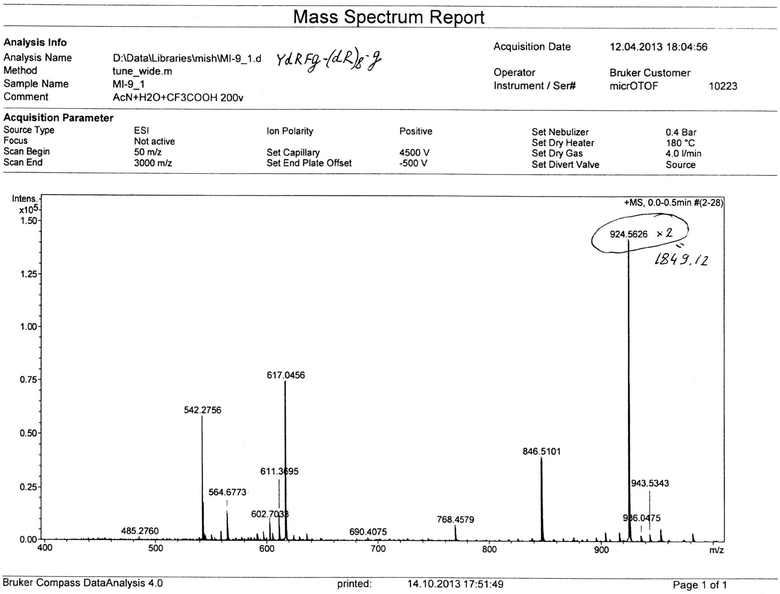
В реактор загружали охлажденный (4°С) раствор 8.50 г (8.0 ммоль) Fmoc-Tyr(tBu)-D-Arg(Pbf)-Phe-Gly-OH, 1.62 г (12.0 ммоль) HOBt и 1.88 мл (12.0 ммоль) DIC в 30 мл DMF, перемешивали в течение 12 час при комнатной температуре. Смолу отфильтровывали, промывали 6×20 мл DMF, добавляли 20 мл 20% раствора диэтиламина в DMF, перемешивали в течение 5 мин, отфильтровывали, промывали 3×20 мл DMF, добавляли 20 мл 20% раствора диэтиламина в DMF, выдерживали в течение 20 мин, отфильтровывали, промывали 5×20 мл DMF. Смолу промывали 4×20 мл DCM, сушили, добавляли 30 мл смеси TFA/TIS/EDT/H2O (37:1:1:1), выдерживали в течение 4 час при комнатной температуре, отфильтровывали, промывали 3×20 мл трифторуксусной кислотой, объединенные фильтраты упаривали до объема ~20 мл, к остатку добавляли 60 мл сухого эфира. Выпавший осадок отфильтровывали, промывали на фильтре эфиром, сушили. Полученный продукт растворяли в 50 мл воды, раствор замораживали и лиофилизовали. Лиофилизат растворяли в 30 мл воды и наносили на колонку с ионообменной смолой Amberlite IRA-400 (Cl- форма). Колонку промывали водой, фракции, содержащие продукт, объединяли, упаривали до объема ~40 мл и наносили на обращеннофазную колонку Waters X-Bridge C18, 10 мкм, 127Å, 50×250 мм. Элюирование проводили при скорости потока элюента 50 мл/мин. Фаза А: 0,1% HCl/Н2О, В: /ацетонитрил. Градиент: 0%(В)-70%(В) за 70 минут. Фракции, содержащие основной продукт, объединяли, упаривали до объема ~50 мл, замораживали и лиофилизовали. Получено 1.2 г (17%) продукта с чистотой (ВЭЖХ) 97.5%. Масс-спектр: вычислено M+ 1848.18, получено M+ 1849.12 (приложение 1). Аминокислотный анализ: аргинин 9.0 (9), тирозин 0.95 (1), фенилаланин 1.05 (1), глицин 1.05 (1).
Нижеследующие примеры фармакологических исследований подтверждают возможность применения заявленного пептида H-Tyr-D-Arg-Phe-Gly-(D-Arg)8-Gly-OH 10HCl в качестве средства с анальгетической активностью, эффективного при парентеральном и пероральном введении.
Пример 1. Изучение анальгетической активности тетрапептида Tyr-D-Arg-Phe-Gly-NH2 и тридекапептида H-Tyr-D-Arg-Phe-Gly-(D-Arg)8-Gly-OH 10HCl в тесте отдергивания хвоста (tail flick).
Болевая чувствительность оценивалась на белых беспородных мышах методом термического раздражения корня хвоста («tail-flick») [D′Amour, Smith, 1941]. Для проведения теста подвижность животного ограничивается, после чего на хвост животного (1 см от основания) направляется сфокусированное световое излучение (мощность 300 Вт). С помощью фоточувствительного датчика фиксировалось время от включения источника излучения до отдергивания хвоста (латентный период). Для предотвращения повреждения тканей предельная продолжительность воздействия ограничивалась интервалом в 15 сек.
Исходная болевая чувствительность определялась как среднее по двум измерениям. Мыши с исходным значением латентного периода более 8 с были исключены из эксперимента. Мышей тестировали перед введением препаратов (исходная болевая чувствительность) и через 5, 15, 30, 60 и 120 минут после введения. При сохранении анальгетического эффекта через 120 мин после введения болевая чувствительность измерялась и через 180 минут после введения.
При обработке полученных результатов были определены средние значения латентных периодов реакции в каждый момент времени и степень анальгезии в процентах, которая рассчитывается как процент от максимально возможного эффекта по формуле:
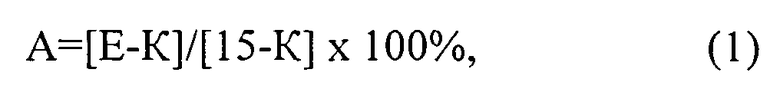
где: А - степень аналгезии, Е - латентный период реакции в секундах в эксперименте (после введения препаратов), К - исходный латентный период данного животного (до введения препарата), 15 - максимальное время термического воздействия, в секундах.
Результаты исследования анальгетической активности тетрапептида Tyr-D-Arg-Phe-Gly-NH2 и тридекапептида H-Tyr-D-Arg-Phe-Gly-(D-Arg)8-Gly-OH 10HCl в тесте отдергивания хвоста представлены в таблицах 2 и 3.
Влияние исследуемых пептидов (тетрапептида и тридекапептида в дозе 2 мМ/кг и 0,5 мМ/кг, соответственно, внутривенно) на латентный период (ЛП, в секундах) отдергивания хвоста (М±m)
Исходные (до введения пептидов) латентные периоды отдергивания хвоста не отличались между группами (р=0,85).
Далее анализировалось влияние тетрапептида на ЛП в сравнении с группой, получавшей физиологический раствор. Двухфакторный дисперсионный анализ с повторными измерениями показал значимое влияние тетрапептида (F1,15=118,0; р<0,0001) и тридекапептида (F1.15=4,67; p<0,05) на ЛП отдергивания хвоста.
*
1*
*
6*
2*
3*
4*
7*
0*
* - р<0,05, статистически значимое отличие от группы, получавшей физиологический раствор в соответствующий временной интервал (тест Бонферрони).
н.д. - нет данных.
Двухфакторный дисперсионный анализ с повторными измерениями показал значимое влияние тетрапептида (F1,15=115,49; р<0,0001) и тридекапептида (F1,15=11,91; р=0,004) на степень аналгезии.
Пример 2. Изучение анальгетической активности тетрапептида Tyr-D-Arg-Phe-Gly-NH2 и тридекапептида H-Tyr-D-Arg-Phe-Gly-(D-Arg)8-Gly-OH 10HCl в тесте химического раздражения брюшины (тест «корчи» - «Writhing test»).
Внутрибрюшинное введение химических раздражающих агентов позволяет моделировать висцеральную боль [Le Bars et al., 2001]. Метод химического раздражения брюшины (тест "корчей") заключается во внутрибрюшинном введении (в левую половину живота) 1% раствора уксусной кислоты в объеме 0.1 мл на животное. После инъекции мыши помещались в специальные индивидуальные боксы по 4 животных в одном блоке, где в течение 10 минут осуществляли визуальное наблюдение за животными. Регистрировали количество специфических ноцицептивных ответов типа "корчи", возникающих в ответ на введение химического раздражающего агента и проявляющихся характерными потягиваниями. Препараты вводились однократно за 20 мин до инъекции уксусной кислоты. Анальгетическое действие оценивается по уменьшению количества "корчей" за период регистрации данной поведенческой реакции.
Результаты исследования анальгетической активности тетрапептида Tyr-D-Arg-Phe-Gly-NH2 и тридекапептида H-Tyr-D-Arg-Phe-Gly-(D-Arg)8-Gly-OH 10HCl представлены в таблицах 4 и 5 и на рисунках 1 и 2.
Пояснения к рис.1. Данные представлены как среднее количество «корчей» (M±m) за 10-минутный период наблюдения.
Примечание:
#- - статистически значимое отличие от группы, получавшей физиологический раствор, (критерий Крускала-Уолисса, непараметрическая ANOVA, тест Данна), р<0,05.
Ф.Р. - физиологический раствор;
в.в. - внутривенно;
в.ж. - внутрижелудочно.
Пояснения к рис.2. Данные представлены как среднее количество «корчей» (M±m) за 10-минутный период наблюдения.
Примечание:
#- - статистически значимое отличие от группы, получавшей физиологический раствор, (критерий Крускала-Уолисса, непараметрическая ANOVA, тест Данна), р<0,05.
Ф.Р. - физиологический раствор;
в.в. - внутривенно;
в.ж. - внутрижелудочно.
Проведенные экспериментальные исследования анальгетической активности нового тридекапептида H-Tyr-D-Arg-Phe-Gly-(D-Arg)8-Gly-OH 10HCl показали, что исследуемое соединение обладает антиноцицептивным эффектом.
В тесте термического прижигания корня хвоста наличие антиноцицептивной активности было зафиксировано как у тетрапептида, так и у его аналога с D-октааргинином (тридекапептидом). Действие тридекапептида было умеренным и продолжалось в течение 30 минут.
В тесте химического раздражения брюшины тетрапептид и тридекапептид были исследованы в широком диапазоне доз при различных путях введения.
При внутривенном пути введения наибольшую анальгетическую активность, как и ожидалось, показал тетрапептид. Однако при введении тетрапептида в максимальных дозах была отмечена 100% смертность экспериментальных животных, которой предшествовала дыхательная депрессия и каталепсия.
При внутрибрюшинном пути введения было подтверждено действие исследуемых пептида на ноцицептивный ответ. Картина анальгетического действия была схожа с внутривенным путем введения.
При внутрижелудочном введении тетрапептид (пептид, не содержащий D-формы октааргинина) утратил свою анальгетическую активность. Так, введение тетрапептида, обладающего самой высокой степенью обезболивания, не имело достоверного антиноцицептивного действия в максимальной эффективной дозе, как при внутривенном введение. Напротив, применение тридекапептида (пептид, содержащий в составе молекулы D-форму октааргинина) показало дозозависимое сохранение анальгезии, схожей с внутривенным и внутрибрюшинным путями введения. Данное наблюдение позволяет сделать вывод о том, что введение октааргинина в состав пептидной молекулы оказывает защитное действие в отношении протеолитических ферментов при внутрижелудочном пути введения, тем самым, сохраняя антиноцицептивную активность молекулы. В данном случае аналог тетрапептида с добавлением D формы октааргинина-тридекапептид проявил выраженный анальгетический эффект при пероральном пути введения.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНАЛЬГЕТИЧЕСКОЕ СРЕДСТВО ПЕПТИДНОЙ СТРУКТУРЫ НА ОСНОВЕ УНДЕКАПЕПТИДА, СОДЕРЖАЩЕГО D-ОКТААРГИНИНОВЫЙ ВЕКТОР | 2013 |
|
RU2541127C1 |
| ПЕПТИД ДЛЯ ЛЕЧЕНИЯ САХАРНОГО ДИАБЕТА 2-ГО ТИПА И ЕГО ОСЛОЖНЕНИЙ | 2014 |
|
RU2573933C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕПТИДА ЭКСЕНАТИДА | 2011 |
|
RU2458066C1 |
| СПОСОБ ИММУНОФЕРМЕНТНОГО АНАЛИЗА ДЛЯ ОПРЕДЕЛЕНИЯ АУТОАНТИТЕЛ К β-АДРЕНОРЕЦЕПТОРУ В ПЛАЗМЕ И СЫВОРОТКЕ КРОВИ ЧЕЛОВЕКА | 2011 |
|
RU2452964C1 |
| СИНТЕТИЧЕСКИЙ АНТИГЕН, ОБЛАДАЮЩИЙ СПОСОБНОСТЬЮ СВЯЗЫВАТЬ АУТОАНТИТЕЛА К β-АДРЕНОРЕЦЕПТОРУ | 2007 |
|
RU2356576C1 |
| СОСТАВ И СПОСОБ ПОЛУЧЕНИЯ АНАЛЬГЕТИЧЕСКОГО СРЕДСТВА ПЕПТИДНОЙ СТРУКТУРЫ | 2017 |
|
RU2669386C1 |
| ГИПОГЛИКЕМИЧЕСКОЕ СРЕДСТВО ПЕПТИДНОЙ СТРУКТУРЫ, ИНГИБИРУЮЩЕЕ ДИПЕПТИДИЛПЕПТИДАЗУ-4 | 2015 |
|
RU2600810C1 |
| СЕМЕЙСТВО ПЕПТИДОВ, ОБЛАДАЮЩЕЕ АНАЛЬГЕТИЧЕСКОЙ АКТИВНОСТЬЮ | 2005 |
|
RU2286169C1 |
| СРЕДСТВО ПЕПТИДНОЙ СТРУКТУРЫ, ИНГИБИРУЮЩЕЕ ДИПЕПТИДИЛПЕПТИДАЗУ-4, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2015 |
|
RU2589258C1 |
| СОЕДИНЕНИЯ, ИНГИБИРУЮЩИЕ MASP, И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2840441C2 |
Группа изобретений относится к применению тридекапептида H-Tyr-D-Arg-Phe-Gly-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-Gly-OH·10HCl в качестве анальгетического средства, а также к лекарственной форме, содержащей тридекапептид H-Tyr-D-Arg-Phe-Gly-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-Gly-OH·10HCl. Заявленная группа изобретений является эффективной в купировании острого болевого синдрома, в экстремальной медицине и терапии хронического болевого синдрома, в частности на поздних стадиях онкологических заболеваний. 2 н. и 1 з.п. ф-лы, 3 ил., 5 табл.
1. Применение тридекапептида - H-Tyr-D-Arg-Phe-Gly-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-Gly-OH·10HCl в качестве анальгетического средства.
2. Лекарственная форма, содержащая в качестве действующего вещества - Н-Tyr-D-Arg-Phe-Gly-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-D-Arg-Gly-ОН·10HCl для лечения острых и хронических болевых синдромов.
3. Форма по п.2, выполненная в виде одной из следующих лекарственных форм, пригодных для парентерального и/или перорального введения: раствор для инъекций, назальный спрей, назальные капли, сублингвальные таблетки, суббукальный пластырь, ректальные суппозитории, кишечнорастворимые таблетки и капсулы, трансдермальные системы для резорбтивного применения.
| US2010317564 A1, 16.12.2010 | |||
| US2003087810 A1, 08.05.2003 | |||
| ПЕПТИД, ОБЛАДАЮЩИЙ СТРЕССПРОТЕКТОРНЫМ ДЕЙСТВИЕМ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ И СПОСОБ ЕЕ ПРИМЕНЕНИЯ | 2006 |
|
RU2304444C1 |
| ЗАМЕЩЕННЫЕ АМИНОСОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1998 |
|
RU2197474C2 |

