Настоящее изобретение относится к способу получения биофармацевтического лекарственного продукта, содержащего интересующую биомолекулу, причем способ включает: (а) первую фазу приготовления лекарственной субстанции из представляющей интерес биомолекулы, причем указанная первая фаза включает по меньшей мере один этап обработки, выбранный из (а1) сбора, (а2) очистки, (а3) ребуферизации и (а4) обогащения, где указанный по меньшей мере один этап обработки в этой первой фазе проводят в присутствии композиции, содержащей по меньшей мере три аминокислоты, где комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию; и (b) вторую фазу дальнейшей обработки лекарственной субстанции, полученной на стадии (а), для получения биофармацевтического лекарственного продукта, причем указанная вторая фаза включает по меньшей мере один этап обработки, выбранный из (b1) ребуферизации, (b2) замораживания, (b3) размораживания, и (b4) фасовки; где указанный по меньшей мере один этап обработки на этой второй фазе проводят в присутствии композиции, содержащей (i) по меньшей мере три аминокислоты, при этом комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу при по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию; и (ii) один или несколько сахаров; при отношении аминокислота: сахар от 10:1 до 1:100 (по массе). Настоящее изобретение также относится к биофармацевтическому лекарственному продукту, полученному или получаемому способом по изобретению.
В этом описании цитируется ряд документов, в том числе патентные заявки и руководства производителя. Раскрытие этих документов, хотя и не считается значимым для патентоспособности данного изобретения, полностью включено в настоящее описание посредством ссылки. Более конкретно, все ссылочные документы включены посредством ссылки в той же степени, как если бы каждый отдельный документ был конкретно и индивидуально указан для включения посредством ссылки.
Область биофармацевтических продуктов, а также область способов их производства стремительно расширяется. Как правило, после крупномасштабного производства интересующей биомолекулы, биофармацевтический лекарственный продукт получают двумя основными фазами последующей обработки. На ранней первой стадии готовят так называемую лекарственную субстанцию (также называемую здесь как нерасфасованная субстанция или нерасфасованная лекарственная субстанция). Следовательно, термин «лекарственная субстанция» в данном описании представляет собой не только активный фармацевтический ингредиент, но и переработанную массу, также содержащую, например, буферы, соли и стабилизаторы. На второй поздней фазе указанную лекарственную субстанцию обрабатывают с получением биофармацевтического лекарственного продукта.
Одним из важнейших аспектов при производстве биофармацевтических лекарственных продуктов является стабильность используемых биомолекул. Известно, что фармацевтические белки и пептиды, а также более сложные биомолекулярные частицы, содержащие различные виды молекул, такие как нуклеиновые кислоты, полипептиды, белки, полисахариды, а также фосфолипиды в случае вирусов с оболочкой или вирусоподобных частиц, подвергаются физическому и химическому стрессу во время каждого этапа обработки. Эти стрессы могут привести к нежелательным молекулярным изменениям, которые, в свою очередь, часто приводят к потере функции, а в некоторых случаях даже к серьезным проблемам безопасности. Стабильность готовых биофармацевтических лекарственных продуктов дополнительно подвергается воздействию процессов старения в зависимости от соответствующих условий хранения.
Молекулярные изменения, которые происходят в таких биофармацевтических лекарственных продуктах в течение всего процесса производства, являются кумулятивными. Другими словами, объединенные изменения в ходе отдельных этапов производства для получения исходной лекарственной субстанции, ее хранения и отгрузки, последующих этапов разработки от лекарственной субстанции до лекарственного продукта, включая процедуры фасовки и упаковки, последующую отгрузку и хранение лекарственного продукта, а также этапов, необходимых для его окончательной подготовки к применению - все они вносят вклад в ряд нежелательных молекулярных изменений в лекарственном продукте. Таким образом, предотвращение таких молекулярных изменений является важной целью не только на стадии изготовления готового лекарственного продукта, но также и на более ранней стадии производства лекарственной субстанции.
Процесс производства лекарственной субстанции обычно начинается с сбора интересующей биомолекулы в качестве первого этапа, например, либо из продуцирующей клеточной линии, либо, в случае секретированных биомолекул, из культуральной среды. Биомолекулу, собранную из клеточной культуры или среды, содержащей нерасфасованную лекарственную субстанцию из неочищенной биомолекулы, затем дополнительно очищают и оценивают.Как правило, раствор, содержащий лекарственную субстанцию, проходит этапы обработки, такие как ультрацентрифугирование или несколько этапов хроматографии в стандартных буферах. Эти буферы обычно оптимизированы для обеспечения удовлетворительной очистки с градиентом плотности или хроматографической очистки, но они обычно не содержат каких-либо стабилизирующих вспомогательных веществ, которые специально выбраны для отдельной биомолекулы. Из-за этих процедур уже на этой ранней стадии производства биофармацевтических препаратов биомолекула или лекарственная субстанция обычно подвергаются физическому и химическому стрессу. В частности, этапы очистки обычно связаны с огромными физическими и химическими стрессами. Соответственно, в целом существует необходимость в обеспечении максимальной стабилизации как можно раньше, предпочтительно во время сбора и/или очистки.
Следующий этап оценки собранной биомолекулы может быть выполнен посредством одной (или нескольких) из некоторых возможных аналитических методик. Для работоспособности этих аналитических методик важно, чтобы буфер, в котором присутствует собранная биомолекула или лекарственная субстанция, не содержал компонентов, которые могли бы помешать аналитическим процедурам и могли бы привести к неправильной интерпретации молекулярной целостности и чистоты биомолекулы или лекарственной субстанции. Таким образом, важно избегать всех вспомогательных веществ, которые не требуются для последующих этапов процесса изготовления лекарственного продукта, в то же время чтобы максимальная стабильность лекарственной субстанции достигалась как можно раньше во время или после сбора, очистки и/или оценки.
После подтверждения того, что лекарственная субстанция соответствует требованиям молекулярной целостности и чистоты, очищенную и охарактеризованную лекарственную субстанцию затем распределяют во вспомогательных веществах, которые предназначены для поддержания качества и целостности продукта на последующих этапах обработки лекарственной субстанции, таких как фильтрация, фасовка, лиофилизация, упаковка, хранение и транспортировка.
Как правило, лекарственную субстанцию хранят в виде замороженного материала. Недостатком замораживания является то, что холодовая денатурация (Privalov PL. Crit. Rev. Biochem. Mol. Biol., 1990) и эффекты развертывания белка могут происходить во время этих процедур замораживания-размораживания. Несмотря на то, что замораживание-размораживание нерасфасованной субстанции дает многочисленные преимущества в отношении качества работы и качества продукции, оно также может оказаться вредным для стабильности лекарственной субстанции из-за механизмов криоконцентрирования. Такие механизмы включают изменения pH (Pikal-Cleland KA et al., J. Pharm. Sci., 2002) и неконтролируемое постепенное обогащение вспомогательными веществами и биомолекулами, которые могут приводить к модификациям структуры биомолекул (Rathore N и Rajan RS. Biotechnol. Prog., 2008: Webb SD et al., BioPharm, 2002; Lashmar UT et al. BioProcess Int, 2007; Glaser V. Gen. Eng. Biotechnol. News, 2005). Кроме того, замороженные нерасфасованные субстанции необходимо обязательно размораживать перед дальнейшей обработкой. Размораживание может вызвать дополнительный стресс и повреждение лекарственной субстанции, например, на границах раздела между льдом и жидкостью и во время перекристаллизации. Во многих случаях дополнительные процессы смешивания включены во время процедуры размораживания. В этих случаях параметры смешивания должны быть тщательно отрегулированы, чтобы избежать дальнейшего повреждения биомолекулы из-за напряжения сдвига, пенообразования и/или образования пузырьков воздуха, приводящих к повреждению лекарственной субстанции на границе раздела жидкость-воздух и т.Д. (Rathore N and Rajan RS. Biotechnol. Prog., 2008).
Вышеописанные эффекты замораживания-размораживания нерасфасованной субстанции на любом конкретном продукте являются специфическими для соответствующей используемой биомолекулы, и таким образом, могут влиять на качество продукта для некоторых препаратов лекарственной субстанции, но не для других. Поэтому перед крупномасштабным производственным процессом целесообразно оценить влияние нескольких циклов замораживания-размораживания на качество продукта для каждого отдельного интересующего продукта. Обычно это проводят в экспериментах с уменьшением масштаба со стандартизованными параметрами, чтобы имитировать крупномасштабные процессы, и часто это включает дальнейшие систематические этапы отбора для определения наиболее подходящих стабилизирующих вспомогательных веществ.
Как только лекарственная субстанция получена, ее далее обрабатывают с получением лекарственного продукта. Эти последующие этапы обработки, также часто называемые «этапами приготовления», включают, например, изменение концентрации выбранных вспомогательных веществ, регуляцию рН, а также регуляцию электропроводности и концентрации биомолекулы (т.е. обогащение биофармацевтического продукта), как необходимо (Scott C., BioProcess Int., 2006). На этой стадии разработки дальнейшие процессы могут также включать такие этапы, как этапы разбавления или замены буфера (ребуферизацию). Для замены буфера обычно проводят операции ультрафильтрации или диафильтрации, которые выбирают с целью ограничения взаимодействия биомолекул и растворенных веществ, которые, как известно, приводят к событиям адсорбции на поверхностях и последующей потере молекулярной целостности лекарственной субстанции (Stoner MR et al., J. Pharm. Sci, 2004). Таким образом, еще одной целью при замене буфера или ребуферизации, например, с использованием диализа, должно быть уменьшение или предотвращение известной потери молекулярной целостности и адсорбции лекарственной субстанции при взаимодействии жидкости с мембраной.
В случае белков дополнительные этапы обработки, такие как стерилизующая фильтрация и фасовка лекарственного продукта, часто подвергают биомолекулы лекарственной субстанции воздействию высокого напряжения сдвига и адсорбции на поверхностях, которые могут вызвать развертывание белка (Maa Y и Hsu CC. Biotechnol. Bioeng., 1997). Для терапевтических антител сообщалось о значительных уровнях агрегации и осаждения белка вследствие сдвига в присутствии границ раздела твердое вещество-жидкость (Biddlecombe et al., Biotechnol. Prog., 2007). Кроме того, когда процессы фасовки не проводятся в атмосфере азота, они могут быть связаны с окислением и дезамидированием биомолекулы (Sharma B., Biotechnol. Adv., 2007).
Для оптимизации хранения и достижения приемлемого срока годности биофармацевтические лекарственные продукты часто лиофилизируют.Лиофилизация включает три основных этапа, а именно замораживание, первичную сушку и вторичную сушку: каждый из этих этапов может привести к нестабильности, которая может вызвать необратимое изменение структуры и/или более высокие уровни агрегации биомолекул биофармацевтического лекарственного продукта. Например, удаление основного объема воды из окружения биомолекулы может уменьшить величину гидрофобного эффекта, который обычно сохраняет структуры биомолекул в их должным образом сложенной форме. Кроме того, адсорбция биомолекулы в биофармацевтической лекарственной субстанции или лекарственном продукте на поверхности раздела лед/вода может привести к денатурации (Strambini GB и Gabellieri E., Biophys. J., 1996). Поэтому во избежание значительного повреждения биомолекулы в биофармацевтической лекарственной субстанции или лекарственном продукте важно выбрать подходящие фармацевтические вспомогательные вещества и подходящие параметры цикла лиофилизации. Помимо проблем со стабильностью, формирование и восстановление лиофилизата являются важными параметрами для успешного производства лекарственных препаратов (Rathore N and Rajan RS. Biotechnol. Prog., 2008).
Физические стрессы также могут возникать во время последующей обработки, такой как фасовка, упаковка и маркировка, в частности, когда маркировку выполняют без надлежащего контроля температуры, или если образец подвергается механическому воздействию во время маркировки, хранения, транспортировки и доставки/введения индивидууму. В частности, напряжение сдвига, тепловой стресс и ограниченная фотостабильность во время хранения и транспортировки являются серьезной логистической и экономической проблемой, особенно для мест доставки с проблемами холодовой цепи. Таким образом, высокие температуры могут подвергать биомолекулы термическому стрессу, который приводит к тепловому развертыванию и агрегации биомолекул на основе белка. Кроме того, присутствие света в сочетании с растворенным кислородом может привести к образованию пероксидных радикалов, что может привести к фотодеградации пептидного каркаса (Davies MJ and Dean RT., Oxford University Press, 1997). Физические стрессы, которые могут возникнуть во время этих этапов обработки, должны рассматриваться в зависимости от того, являются ли составы жидкими или сухими составами: обычно сухие составы более стабильны, чем жидкие составы.
Наконец, для применения биофармацевтических лекарственных продуктов в медицине, включая здоровье человека или животных, предполагаемый путь введения должен быть принят во внимание при выборе состава готового биофармацевтического лекарственного продукта, включая выбор подходящих вспомогательных веществ. Например, внутривенное, трансдермальное, внутрикожное, подкожное или внутримышечное введение высококонцентрированных терапевтических антител требует соответствующих условий, таких как достаточная возможность введения через шприц, пригодность для инъекции, адекватная осмоляльность и низкая вязкость, чтобы обеспечить легкое и безболезненное введение. С другой стороны, при рассмотрении перорального, легочного или интраназального введения применяются другие требования. Например, пероральное введение требует композиции, которая позволяет лекарственному продукту проходить через желудочно-кишечный тракт без потери активности при расщеплении молекул, тогда как легочное введение требует сухой композиции, которая стабильна при прохождении через верхние и нижние дыхательные пути, а при растворении обеспечивает прохождение через соответствующую слизистую оболочку.
Международная заявка WO 2005/007185 направлена на стабилизацию белковых фармацевтических препаратов без добавления часто используемого стабилизатора человеческого сывороточного альбумина (ЧСА). Вместо этого стабилизирующий раствор содержит (i) поверхностно-активное вещество, которое предпочтительно представляет собой неионный детергент, т.е. сурфактант; и (ii) смесь по меньшей мере двух аминокислот, где по меньшей мере две аминокислоты представляют собой глутамат и глутамин или аспартат и аспарагин. Целью данной заявки является, прежде всего, стабилизация низкоконцентрированного фармацевтического соединения при хранении, в частности, при длительном хранении в течение более шести месяцев при повышенных температурах. Однако конкретная стабилизация при обработке и изготовлении не описана.
В международной заявке WO 2008/000780 порошок, высушенный распылением, содержащий белок, стабилизируется и описывается как обладающий предпочтительным аэродинамическим поведением, когда включено по меньшей мере 30% или по меньшей мере 40% фенилаланина. Благодаря добавлению фенилаланина в порошок, когезивнные и адгезионные свойства порошка изменяются, чтобы уменьшить взаимодействие между частицами. За счет того, что поверхность частиц порошка становится более гидрофобной, аэродинамические свойства порошка улучшаются, что делает его более подходящим для легочного применения. Соответственно, целью WO 2008/000780 является, прежде всего, корректировка конечного продукта, готового к применению, в то время как общая стабилизация во время приготовления и улучшенные процедуры производства не обсуждаются.
В заявке на европейский патент EP 1789019 описаны высушенные распылением порошки белковых лекарств для легочного применения, которые стабилизируют путем добавления новых смесей олигосахаридов. Явные защитные эффекты комбинаций аминокислот во время обработки не рассматриваются. Вместо этого, эта заявка направлена на стабилизацию или оптимизацию указанных порошков, высушенных распылением, для того, чтобы сделать их пригодными для легочного введения.
Международная заявка WO 2010/151703 раскрывает фармацевтическую композицию для повышения стабильности, уменьшения агрегации или снижения иммуногенности пептида или полипептида, включающую по меньшей мере один алкилгликозид. Композиции, содержащие аминокислоты или конкретные комбинации аминокислот для применения при обработке фармацевтических композиций, не описаны.
Заявка на патент США US 2014/0127227 описывает белковые составы, содержащие по меньшей мере одну аминокислоту, для обеспечения стабильности и вязкости даже для высококонцентрированных составов. Заявка фокусируется на стабильности составов коммерческих биофармацевтических продуктов, в то время как воздействие вспомогательных веществ на биофармацевтические препараты на ранних стадиях разработки и во время приготовления лекарственных субстанций не рассматривается.
Международная заявка WO 2013/001044 описывает преимущество композиций на основе аминокислот для предотвращения развертывания и обеспечения эффективного повторного свертывания даже сложных биомолекул, таких как антитела IgM, во время сушки и восстановления. Международная заявка WO 2010/115835 описывает преимущество содержащих аминокислоты композиций для защиты биомолекул, иммобилизованных на поверхностях материала, даже во время облучения и терминальной стерилизации. Также в WO 2010/112576 раскрыто преимущество композиций, содержащих аминокислоты, для защиты биомолекул во время облучения и терминальной стерилизации, для биомолекул в закрытом контейнере. В международной заявке WO 2013/001034 описано преимущество содержащих аминокислоты композиций для защиты живых вирусов при хранении и транспортировке. Наконец, в WO 2015/059284 описано преимущество содержащих аминокислоты композиций для защиты коммерческих вакцин, например, против гриппа, во время термического стресса, распылительной сушки и терминальной стерилизации. Однако стабилизация во время различных этапов обработки для производства антитела, включая ранние этапы обработки, и во время окончательного формирования лекарственного продукта не рассматривается ни в одной из этих заявок.
Таким образом, большинство доступных на данный момент подходов к стабилизации фокусируется на одном конкретном этапе производственного процесса, на улучшенных условиях хранения или на оптимизированных составах для предполагаемого пути введения. До сих пор не было уделено внимания предотвращению ранних молекулярных изменений при производстве биофармацевтических лекарственных субстанций и лекарственных продуктов и предотвращению потенциального умножения этих ранних нестабильностей во время дальнейшей обработки. Более того, ни один из этих подходов не был разработан с учетом общего процесса приготовления, то есть с целью обеспечения улучшенной защиты на протяжении большей части или даже на всех этапах производства, последующей обработки и приготовления, при одновременной минимизации количества различных стабилизирующих композиций, необходимых для этих этапов. Соответственно, все еще существует потребность в улучшении существующей конструкции состава для усовершенствования производства биофармацевтической лекарственной субстанции и лекарственного продукта.
Эта задача решается путем обеспечения вариантов осуществления, охарактеризованных в формуле изобретения.
Соответственно, настоящее изобретение относится к способу получения биофармацевтического лекарственного продукта, содержащего интересующую биомолекулу, причем способ включает: (а) первую фазу приготовления лекарственной субстанции из интересующей биомолекулы, причем указанная первая фаза включает по меньшей мере один этап обработки, выбранный из (а1) сбора, (а2) очистки, (а3) ребуферизации и (а4) обогащения, где указанный по меньшей мере один этап обработки на этой первой фазе проводят в присутствии композиции, содержащей по меньшей мере три аминокислоты, где комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию; и (b) вторую фазу дальнейшей обработки лекарственной субстанции, приготовленной на стадиии (а), для получения биофармацевтического лекарственного продукта, причем указанная вторая фаза включает по меньшей мере один этап обработки, выбранный из (b1) ребуферизации, (b2) замораживания, (b3 размораживания и (b4) фасовки; где указанный по меньшей мере один этап обработки на этой второй фазе проводят в присутствии композиции, содержащей (i) по меньшей мере три аминокислоты, при этом комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию; и (ii) один или несколько сахаров; при отношении аминокислота: сахар от 10:1 до 1:100 (по массе).
Используемый в настоящей заявке термин «биофармацевтический лекарственный продукт» хорошо известен и относится к фармацевтическому лекарственному продукту, где указанный лекарственный продукт основан на одной или нескольких биомолекулах (также называемый здесь фармацевтическим продуктом на основе биомолекул). Указанный термин охватывает любой фармацевтический продукт на основе биомолекул, изготовленный, экстрагированный или полусинтетический, полученный из биологических источников или синтезированный, например, химически синтезированный или с помощью систем in vitro, таких как, например, транслированные in vitro белки и т.д. Термин «биофармацевтические продукты» также используется взаимозаменяемо с терминами «биофармацевтические препараты», «лекарственный продукт», «биологические медицинские продукты», «биологические средства» или «биологические препараты».
Биофармацевтический лекарственный продукт содержит интересующую биомолекулу. Используемый в настоящей заявке термин «биомолекула» относится к любой молекуле, которая обычно присутствует в живых организмах. Предпочтительными биомолекулами являются крупные макромолекулы, такие как белки, углеводы, липиды и нуклеиновые кислоты, а также небольшие молекулы, такие как первичные метаболиты, вторичные метаболиты и натуральные продукты. Понятно, что термин «биомолекула» не ограничивается одним типом биомолекулы, но может также охватывать более одной биомолекулы, то есть он также относится к «одной или нескольким биомолекулам».
Способ по настоящему изобретению относится к производству такого биофармацевтического лекарственного продукта, где производство включает две фазы. На первой фазе готовят лекарственную субстанцию из интересующей биомолекулы. На второй фазе указанную лекарственную субстанцию дополнительно обрабатывают для получения биофармацевтического лекарственного продукта.
Термин «лекарственная субстанция» используется в настоящей заявке взаимозаменяемо с терминами «нерасфасованная субстанция» или «нерасфасованная лекарственная субстанция». Эти термины хорошо известны в данной области техники и относятся к любому веществу, которое представляется для использования в лекарственном средстве и которое при использовании при производстве, обработке или упаковке лекарственного средства становится активным ингредиентом или готовой лекарственной формой. Согласно определению Управления по контролю продуктов питания и лекарственных средств США, этот термин не включает промежуточные продукты, используемые в синтезе таких субстанций.
Указанная первая фаза способа получения по настоящему изобретению включает по меньшей мере один этап обработки, выбранный из (а1) сбора, (а2) очистки, (а3) ребуферизации и (а4) обогащения.
Используемый в настоящей заявке термин «включающий» означает, что дополнительные этапы и/или компоненты могут быть включены в дополнение к перечисленным этапам и/или компонентам. Например, в предпочтительном варианте осуществления этот термин охватывает то, что сахар присутствует в композиции, такой как композиция этапа (а). Однако этот термин также охватывает то, что заявленный предмет обсуждения состоит именно из перечисленных этапов и/или компонентов.
Термин «по меньшей мере», используемый в настоящем документе, относится к конкретно указанному количеству или числу, но также к более чем конкретно указанному количеству или числу. Например, термин «по меньшей мере один» охватывает также по меньшей мере 2, по меньшей мере 3, по меньшей мере 4, по меньшей мере 5, по меньшей мере 6, по меньшей мере 7, по меньшей мере 8, по меньшей мере 9, по меньшей мере 10, такой как по меньшей мере 20, по меньшей мере 30, по меньшей мере 40, по меньшей мере 50 и т.д. Кроме того, этот термин также включает в себя ровно 1, ровно 2, ровно 3, ровно 4, ровно 5, ровно 6, ровно 7, ровно 8, ровно 9, ровно 10, ровно 20, ровно 30, ровно 40, ровно 50 и т.д. В контексте перечисленных этапов обработки термин «по меньшей мере один этап обработки, выбранный из», охватывает то, что один, два, три, четыре или пять из указанных этапов обработки выполняются, но также и то, что выполняются все шесть этапов обработки. Понятно, что порядок перечисления этих этапов обработки не является особенно ограничивающим, хотя предпочтительно, чтобы в тех случаях, когда выполняется более одного этапа, указанные этапы выполнялись в указанном порядке. Кроме того, следует понимать, что определенные этапы обработки, такие как, например, замена буфера, могут проводиться более одного раза в процессе приготовления лекарственной субстанции в указанной первой фазе способа по изобретению.
Используемый в настоящей заявке термин «сбор» относится к этапу процесса получения интересующей биомолекулы из источника, который ее производит.Большинство коммерческих терапевтических белков, таких как рекомбинантный человеческий инсулин, человеческий гормон роста, эритропоэтин (ЭПО), факторы свертывания крови, моноклональные антитела и интерфероны, например, получают путем крупномасштабной ферментации с использованием любых микроорганизмов, таких как Bacillus subtilis и Escherichia coli, дрожжи, и другие грибы или клетки млекопитающих в качестве источников. Яркими примерами клеточных культур млекопитающих в качестве важных источников терапевтических белков являются клетки яичника китайского хомячка (СНО) и клетки почки детеныша хомяка (ВНК). Указанные источники могут либо секретировать биомолекулу в культуральную среду, либо могут экспрессировать биомолекулу внутриклеточно. В последнем случае процедура сбора, как правило, является более сложной, поскольку существует дополнительное требование разрушения клеток для сбора белка. Кроме того, биомолекулы также можно собирать из таких источников, как ткани животных, биологические жидкости и растения.
Типичные способы, используемые в процессе сбора, включают центрифугирование, фильтрацию и микрофильтрацию, а также хроматографию. Эти способы хорошо известны в данной области техники.
Используемый в настоящей заявке термин «очистка» относится к способам, используемым для выделения интересующей биомолекулы. Очистку обычно проводят на ранней стадии производства биофармацевтических препаратов, чтобы извлечь высокоочищенную лекарственную субстанцию для дальнейшей обработки, то есть продукт, лишенный или по существу лишенный каких-либо других веществ, кроме представляющей интерес биомолекулы. Способы и этапы, обычно выполняемые для очистки биомолекулы, могут включать, например, концентрирование биомолекулы и/или осветление для удаления чужеродных белков (из клеток-хозяев), например, с помощью центрифугирования, осаждения, фильтрации/ультрацентрифугирования или хроматографических способов, таких как ионообменная хроматография, аффинная хроматография, хроматография гидрофобного взаимодействия и эксклюзионная хроматография; а также дальнейшие этапы дополнительной очистки, например, для удаления продуктов разложения, производных продукта, таких как окисленные, дезамидированные или деградированные формы продукта, и загрязняющих веществ, таких как пирогенные вещества, например, с помощью эксклюзионной хроматографии.
Используемый в настоящей заявке термин «ребуферизация» относится к способам модификации существующего препарата для получения адаптированной или оптимизированной среды для лекарственной субстанции или готового лекарственного продукта. Одним из возможных способов ребуферизации является разбавление существующего состава путем добавления, например, воды или буферов. Альтернативно, существующий состав может быть модифицирован путем добавления определенных вспомогательных веществ, таких как, например, вспомогательные вещества, описанные здесь ниже. Особенно предпочтительным способом проведения ребуферизации является диализ. Диализ является хорошо известным в данной области техники способом, в котором для обеспечения диффузии растворов низкомолекулярных веществ через мембрану используются полупроницаемые диализные мембраны, в результате чего компоненты жидкостей обмениваются, а биомолекулы остаются в диализной кассете в зависимости от молекулярной массы и предела отсечения по молекулярной массе мембраны.
Используемый в настоящей заявке термин «обогащение» относится к увеличению концентрации (концентраций) соответствующей молекулы (молекул) (например, биомолекулы, лекарственной субстанции или биофармацевтического лекарственного вещества или лекарственного продукта, в зависимости от стадии производства). Предпочтительно, концентрацию увеличивают до уровней, которые соответствуют конечной концентрации и дозировке, при которой должен использоваться соответствующий обогащенный продукт.
Эту первую фазу способа получения по настоящему изобретению проводят в присутствии специальной композиции, а именно композиции, содержащей по меньшей мере три аминокислоты, где комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию. Эта композиция также упоминается здесь как «композиция первой фазы» или «композиция ранней фазы».
Указанная композиция характеризуется наличием по меньшей мере трех аминокислот.Эти три аминокислоты выбраны так, что они обеспечивают указанные четыре функциональные группы. Понятно, что термин «по меньшей мере три аминокислоты» относится к трем различным аминокислотам.
Используемый в настоящей заявке термин «аминокислота» хорошо известен в данной области техники. Аминокислоты являются основными строительными блоками белков. В соответствии с настоящим изобретением термин «аминокислота» относится к свободным аминокислотам, которые не связаны друг с другом с образованием олиго- или полимеров, таких как дипептиды, трипептиды, олигопептиды или белки (также называемые здесь полипептидами). Их можно классифицировать на характерные группы вспомогательных веществ с неполярными, алифатическими; полярными, незаряженными; положительно и/или отрицательно заряженными и/или ароматическими R группами (Nelson D.L. & Cox M.M., «Lehninger Biochemie» (2005), pp.122-127).
Аминокислоты в соответствии с настоящим изобретением могут быть выбраны из встречающихся в природе аминокислот, а также из искусственных аминокислот или производных этих встречающихся в природе или искусственных аминокислот.Встречающиеся в природе аминокислоты представляют собой, например, 20 протеиногенных аминокислот: глицин, пролин, аргинин, аланин, аспарагин, аспарагиновую кислоту, глутаминовую кислоту, глутамин, цистеин, фенилаланин, лизин, лейцин, изолейцин, гистидин, метионин, серин, валин, тирозин, треонин и триптофан. Другими природными аминокислотами являются, например, карнитин, креатин, креатинин, гуанидиноуксусная кислота, орнитин, гидроксипролин, гомоцистеин, цитруллин, гидроксилизин или бета-аланин. Искусственные аминокислоты представляют собой аминокислоты, которые имеют другую длину боковой цепи и/или структуру боковой цепи и/или имеют аминогруппу в участке, отличном от альфа-С-атома. Производные аминокислот включают н-ацетил-триптофан, фосфоносерин, фосфонотреонин, фосфонотирозин, меланин, аргинино-янтарную кислоту и ее соли и ДОФА, но не ограничиваются ими. В контексте настоящего изобретения все термины также включают соли соответствующих аминокислот.
Аминокислоты, которые обеспечивают положительно заряженную функциональную группу, то есть через их соответствующую боковую цепь, хорошо известны в данной области техники и включают, например, лизин, аргинин, гистидин и не-протеиногенные аминокислоты, такие как, например, орнитин.
Используемый в настоящей заявке термин «аминокислоты, которые обеспечивают осмолитическую функцию» относится к аминокислотам, которые обеспечивают осмолитическое свойство. Такие аминокислоты также хорошо известны в данной области техники и включают, например, глицин, аланин и глутаминовую кислоту, а также производные протеиногенных и не-протеиногенных аминокислот, соответственно, такие как, например, бетаин, карнитин, креатин, креатинин и бета-аланин.
Используемый в настоящей заявке термин «аминокислоты, которые обеспечивают антиоксидантную функциональную группу», относится к аминокислотам, которые обеспечивают антиоксидантное свойство через (одну из) их боковую цепь (цепи). Такие аминокислоты также хорошо известны в данной области техники и включают, например, метионин, цистеин, гистидин, триптофан, фенилаланин и тирозин, а также производные протеиногенных и не-протеиногенных аминокислот, такие как, например, N-ацетил- триптофан, N-ацетил-гистидин или карнозин.
Термин «аминокислоты, которые обеспечивают буферную функцию» относится к аминокислотам, которые обеспечивают буферную способность через одну или несколько их функциональных групп.Такие аминокислоты также хорошо известны в данной области и включают, например, глицин, аргинин и гистидин.
Понятно, что одна аминокислота может также объединять несколько из указанных функциональных групп и/или функций, таких как, например, две, три или даже все четыре функциональные группы и функции, соответственно. Здесь также предусмотрено, что аминокислоты могут перекрываться при обеспечении таких функциональных групп и/или функций, то есть аминокислота, обеспечивающая антиоксидантную функциональную группу, может также обеспечивать буферную функцию, например, гистидин.
В определенных вариантах осуществления, т.е. когда композиция состоит ровно из трех аминокислот, требуется, чтобы все четыре функциональные группы и функции, соответственно, обеспечивались указанными тремя аминокислотами. Другими словами, по меньшей мере одна из аминокислот обеспечивает две (или более) функциональных группы и функции, соответственно. Например, глицин обеспечивает осмолитическую функцию, а также буферную функцию, в то время как гистидин обеспечивает антиоксидантную функциональную группу, а также буферную функцию.
В предпочтительном варианте осуществления эта композиция первой фазы состоит только из аминокислот, то есть она не содержит никаких других вспомогательных веществ, таких как, например, сахара (включая сахароспирты), хелатирующие агенты и антиокислительные агенты, кроме аминокислот, сурфактанты, стабилизирующие белки или пептиды. Еще более предпочтительно, композиция первой фазы состоит из ровно трех аминокислот, обеспечивающих четыре перечисленные функциональные группы и функции, соответственно.
В альтернативном предпочтительном варианте осуществления эта композиция первой фазы содержит по меньшей мере один сахар. В более предпочтительном варианте осуществления композиция первой фазы состоит из аминокислот, как указано выше, и по меньшей мере одного сахара.
Предпочтительные количества по меньшей мере трех аминокислот, которые должны содержаться в композиции первой фазы в соответствии с изобретением, составляют от 5 до 100 мг/мл, более предпочтительно от 10 мг/мл до 75 мг/мл, еще более предпочтительно от 15 мг/мл и 50 мг/мл, и наиболее предпочтительно количество составляет около 20 мг/мл. Понятно, что эти предпочтительные количества относятся к сумме всех аминокислот, присутствующих в растворе.
Используемый в настоящей заявке термин «примерно» охватывает явно указанные значения, а также небольшие отклонения от них. Другими словами, количество аминокислот «около 20 мг/мл» включает, но не обязательно должно быть точно указанным количеством 20 мг/мл, но может отличаться на несколько мг/мл, включая, например, 21 мг/мл или 19 мг/мл.
Специалисту в данной области техники известно, что такие значения являются относительными значениями, которые не требуют полной точности, если значения приблизительно соответствуют приведенным значениям. Соответственно, отклонение от приведенного значения, например, 15%, более предпочтительно 10%, и наиболее предпочтительно 5%, охватывается термином «примерно». Эти отклонения 15%, более предпочтительно 10% и наиболее предпочтительно 5% сохраняются для всех вариантов осуществления, относящихся к данному изобретению, где используется термин «примерно».
Способ по настоящему изобретению требует, чтобы указанный этап (этапы) обработки в первой фазе выполнялся/ выполнялись «в присутствии» этого состава первой фазы. Другими словами, нерасфасованную лекарственную субстанцию приводят в контакт с композицией первой фазы. Это может быть достигнуто, например, если интересующую биомолекулу собирают непосредственно в композицию первой фазы; путем замены существующего растворителя композицией первой фазы; или путем добавления по меньшей мере трех аминокислот к существующему растворителю, например, во время первой очистки на колонке в случае антител, или во время ультрацентрифугирования в случае вирусных векторов.
Лекарственную субстанцию, полученную на этой первой фазе, затем дополнительно подвергают второй фазе дальнейших этапов обработки для превращения лекарственной субстанции в биофармацевтический лекарственный продукт.На этой второй фазе выполняют по меньшей мере один этап обработки, выбранный из (b1) ребуферизации, (b2) замораживания, (b3) размораживания и (b4) фасовки.
Определения и предпочтительные варианты осуществления, приведенные выше в отношении первой фазы, применяются с учетом необходимых изменений, если не указано иное. Например, термины «включающий», «по меньшей мере», «ребуферизация», «диализ», «аминокислоты», «в присутствии» и т.д. являются такими, как определено выше.
Используемый в настоящей заявке термин «замораживание» относится к процессу перевода образца в твердое замороженное состояние. Замораживание обычно используется для подготовки образцов к хранению, так как риск, например, контаминации уменьшается в этом состоянии.
Термин «размораживание», используемый в настоящей заявке, относится к процессу перевода образца из твердого замороженного состояния в незамороженное состояние. В большинстве случаев размороженный образец будет присутствовать в жидкой фазе, но в тех случаях, когда сухой продукт был заморожен, размороженный продукт будет возвращен в сухое незамороженное состояние, которое впоследствии может быть восстановлено для дальнейшей обработки. После размораживания продукт становится доступным для дальнейших процессов разработки или производства, таких как, например, фасовка.
Используемый в настоящей заявке термин «фасовка» относится к процессу переноса жидких или высушенных продуктов в (а) специальный контейнер (контейнеры) для дальнейшей обработки или - в качестве готового продукта - для транспортировки, хранения и/или применения.
Эту вторую фазу способа получения по настоящему изобретению вновь проводят в присутствии конкретной композиции, в этом случае композиции, содержащей по меньшей мере три аминокислоты и один или несколько сахаров. Эта композиция также упоминается здесь как «композиция второй фазы».
Указанная композиция характеризуется присутствием по меньшей мере трех аминокислот, причем функциональные группы и функции, которые обязательно присутствуют, являются такими, как определено для композиции первой фазы. Однако фактический выбор аминокислот не ограничен теми же аминокислотами, что и в композиции первой фазы; вместо этого некоторые или все аминокислоты могут отличаться от аминокислот композиции первой фазы. Здесь также охватывается то, что по меньшей мере три аминокислоты композиции второй фазы идентичны по меньшей мере трем аминокислотам композиции первой фазы.
Кроме того, один или несколько сахаров присутствуют в композиции второй фазы.
Используемый в настоящей заявке термин «сахар» относится к любым типам сахаров, то есть к моносахаридным, дисахаридным или олигосахаридным формам углеводов, а также к сахароспиртам или производным сахаров, таким как аминосахара, например, глюкозамин или н-ацетилглюкозамин. Примеры подходящих сахаров включают трегалозу, сахарозу, маннитол и сорбитол, но не ограничиваются ими.
Предпочтительные количества сахаров, которые должны содержаться в растворе по изобретению, составляют от 5 до 200 мг/мл, более предпочтительно, от 10 до 100 мг/мл, еще более предпочтительно, от 15 до 80 мг/мл, и наиболее предпочтительно, количество составляет около 30 мг/мл. Когда используют смесь различных типов сахаров, эти предпочтительные количества относятся к сумме всех сахаров в растворе.
В соответствии с настоящим изобретением отношение между указанными аминокислотами и сахаром (сахарами), присутствующими в композиции второй фазы, составляет от 10:1 до 1:100. Это отношение относится к концентрации аминокислот и сахара (сахаров), которая обычно представлена в мг/мл. Предпочтительно отношение составляет от 5:1 до 1:50, более предпочтительно от 2,5:1 до 1:25, и наиболее предпочтительно от 1:1 до 1:2.
В соответствии с настоящим изобретением был разработан усовершенствованный способ производства биофармацевтических лекарственных препаратов. Этот способ был разработан с акцентом на обеспечение простой, но эффективной защиты на протяжении всего производственного процесса в сочетании с уменьшенной потребностью в повторяющихся, зависящих от этапа изменениях в поддерживающей композиции. С этой целью на ранней стадии способа используют простую аминокислотную композицию. Удивительно, что эта аминокислотная композиция оказалась достаточной для стабилизации лекарственной субстанции сразу после сбора и на начальных этапах очистки. Более того, было обнаружено, что стабилизация лекарственной субстанции уже на этих ранних стадиях приводит к улучшению качества и стабильности продукта на протяжении всего процесса производства, хранения и применения.
Способ обеспечивает дополнительное преимущество, заключающееся в том, что стабилизирующая композиция на ранней фазе способа во время производства лекарственной субстанции требует наличия только небольшого количества аминокислот, то есть трех аминокислот (или, при необходимости, большего количества), которые не вызывают нарушений во время обычно необходимых аналитических процедур при разработке лекарственной субстанции.
Эти результаты особенно удивительны, так как предыдущие работы, такие как, например, WO 2013/001044, WO 2010/115835, WO 2010/112576, WO 2013/001034 или WO 2015/059284 показали, что различные комбинации аминокислот - с дополнительными стабилизирующими вспомогательными веществами или без них - обеспечивают защитные эффекты в отношении трехмерной структуры определенных биомолекул в разных стрессовых условиях. Этими стрессовыми условиями были, например, сушка и/или восстановление биомолекул, хранение при высоких температурах, а также стерилизация биомолекул. Однако, хотя эти композиции хорошо работают в этих стрессовых условиях, здесь было неожиданно обнаружено, что не все эти композиции обеспечивают такие же превосходные эффекты, как и «композиция ранней фазы» согласно изобретению, когда применяется уже на ранней фазе приготовления лекарственной субстанции, например, непосредственно после сбора из систем культивирования клеток и, например, после первого этапа ультрацентрифугирования.
Как показано, например, в Примере 2, приведенном ниже, раннее добавление стабилизирующей композиции «ранней фазы» может оказывать сильное влияние на стабильность биофармацевтического лекарственного продукта в течение всей процедуры его приготовления. Таким образом, было обнаружено, что раннее применение стабилизирующей композиции согласно изобретению оказывает выраженный стабилизирующий эффект на конкретную биомолекулу в течение всего процесса производства.
Эти выводы дополнительно подтверждаются, например, в примерах 3-5 и 7, которые показывают стабилизирующую эффективность нескольких композиций согласно изобретению в отношении воздействия стрессов, обычно возникающих на этапах обработки в процессе производства, таких как, например, замена буфера с использованием диализа или концентрирование итогового терапевтического состава антител. Было обнаружено, что как терапевтические составы антител в низкой концентрации (примеры 3 и 4), так и высококонцентрированные составы терапевтических антител (пример 5) демонстрируют пониженное образование агрегатов при получении в различных композициях согласно изобретению и по сравнению с аналогичным этапом приготовления в оригинальном составе поставщика.
Кроме того, как показано в примере 7, увеличение концентрации коммерческой жидкой композиции трастузумаба до концентрации 200 мг/мл для приготовления высококонцентрированных жидких терапевтических композиций антител приводило к нежелательному увеличению образования агрегатов. Напротив, концентрация этого терапевтического антитела после дополнительного этапа ребуферизации в композициях согласно настоящему изобретению была достаточной, чтобы избежать такого увеличения образования агрегатов. В примерах 8, 9 и 10 приведены дополнительные данные, подтверждающие стабилизирующую эффективность композиций по данному изобретению на этапах производства и обработки биофармацевтических препаратов. Примеры показывают, что композиции по изобретению превосходили оригинальный жидкий состав поставщика, когда моноклональное антитело трастузумаб подвергали стрессу во время замены ребуферизацииа и последующих этапов концентрирования и перемешивания в качестве модели механического стресса.
Упомянутые выше примеры в качестве моделей для ребуферизации, диализа и концентрирования во время различных этапов получения лекарственной субстанции показывают, что стабилизирующая эффективность композиций по изобретению на этой ранней фазе не особенно зависит от соотношений концентраций аминокислот и сахаров. Удивительно, что на более поздней последующей технологической фазе соотношения концентраций аминокислот и сахара (или смеси сахаров) и/или соотношения концентраций аминокислот и лекарственной субстанции обеспечивают сильную эффективность в отношении стабилизации лекарственного продукта, например, во время хранения жидкости, хранения жидкости при повышенных температурах (см., например, примеры 3-7) и в отношении вязкости, особенно в случае высококонцентрированных составов антител (см., например, примеры 5 и 7).
Простота стабилизирующей аминокислотной композиции на ранней фазе способа имеет дополнительное преимущество, заключающееся в том, что ее можно легко отрегулировать в соответствии с потребностями лекарственной субстанции во время дальнейших этапов обработки для получения соответствующего лекарственного продукта. Модификации исходной простой стабилизирующей композиции, например, с помощью добавления других вспомогательных веществ, таких как сахара и/или смеси сахаров, позволяет легко регулировать итоговый состав, избегая или ограничивая дополнительные этапы ребуферизации. Таким образом, требуется меньше этапов обработки и меньше стрессовых технологических этапов на протяжении всего процесса производства и приготовления; тем самым уменьшается прилагаемый стресс и повышается стабильность биофармацевтического лекарственного продукта, но также уменьшается работа и стоимость, связанная с производством биофармацевтической лекарственной субстанции и лекарственного продукта.
Кроме того, использование композиций, описанных в настоящем документе, во время заявленного процесса (процессов) производства приводит к осмоляльности готового биофармацевтического лекарственного продукта ниже 450 мОсмоль/кг.Несколько исследователей сообщили, что высокая осмоляльность связана с болью и побочными эффектами в месте инъекции. Таким образом, общепринято, что осмоляльность парентерально применяемого раствора должна быть ниже 450 мОсмоль/кг, предпочтительно близко к физиологическому диапазону от 275 до 320 мОсмоль/кг.Лекарственный продукт, полученный способом по настоящему изобретению, удовлетворяет этому требованию в отношении фармацевтических лекарственных продуктов для введения людям и животным.
Таким образом, авторы настоящего изобретения неожиданно обнаружили, что применение композиции ранней фазы в соответствии с настоящим изобретением, которая основана на очень простой аминокислотной композиции, обеспечивает идеальную отправную точку для последующих этапов обработки, требуемых при приготовлении биофармацевтической лекарственной субстанции и лекарственного продукта. При использовании этой композиции ранней фазы в качестве основы возможен модульный подход и специфичный для фазы разработки подход к составу, который требует только минимальных корректировок композиций, в то же время достигая хорошо сбалансированных стабилизирующих эффектов. Таким образом, сбалансированные составы могут быть специально адаптированы в соответствии с конкретными требованиями последующих этапов, такими как различные цели хранения и/или введения для конкретного биофармацевтического лекарственного продукта.
В соответствии со всеми вариантами осуществления способа по изобретению, предоставленного в настоящем документе, предпочтительные композиции, которые следует использовать на ранней стадии получения лекарственной субстанции в соответствии со способом по настоящему изобретению, содержат либо три аминокислоты, выбранные из аминокислот аргинина, глицина, триптофана, и гистидина, или комбинацию указанных четырех аминокислот.Кроме того, в соответствии со всеми вариантами осуществления способа по настоящему изобретению, предложенного в настоящем документе, предпочтительные композиции поздней фазы содержат либо три аминокислоты, выбранные из аргинина, глицина, триптофана и гистидина, либо комбинацию указанных четырех аминокислот, в то время как аминокислоты находятся в комбинации с сахаром или смесью сахаров, таких как трегалоза и сахароза. При необходимости могут быть добавлены хелатирующие агенты и/или антиоксиданты. Более предпочтительные композиции для поздней фазы производства дополнительно включают хелатирующий агент ЭДТА и/или антиоксидант аскорбиновую кислоту.
В предпочтительном варианте осуществления способа по изобретению биофармацевтический лекарственный продукт, полученный на стадии (b), дополнительно обрабатывают для хранения и/или введения в виде жидкой композиции.
«Хранение» в соответствии с настоящим изобретением означает, что лекарственную субстанцию или лекарственный продукт, который не используют немедленно для последующих стадий обработки или введения субъекту, хранят в определенных условиях. Соответственно, термин «хранение», как используется в данном документе, конкретно не ограничен и охватывает, например, хранение лекарственной субстанции или лекарственного продукта на производственном участке, в исследовательской лаборатории, в медицинском институте или на практике перед использованием, при транспортировке/перевозке лекарственной субстанции или лекарственного продукта, а также на подготовительных этапах, такие как, например, аликвотирование биофармацевтического продукта.
Условия хранения зависят от типа лекарственной субстанции или лекарственного продукта, а также от предполагаемого пути введения, если продукт предназначен для введения. Например, стерильность и стабильность лекарственного продукта должны учитываться и контролироваться. Многие лекарственные субстанции должны храниться в холодном и/или темном месте, чтобы предотвратить процессы деградации, связанные с температурой или УФ-излучением, соответственно. Кроме того, лекарственные продукты, которые должны вводиться в виде жидких композиций, предпочтительно хранят в виде жидкости до использования, чтобы избежать необходимости выполнять дополнительный этап восстановления перед применением.
Биофармацевтический лекарственный продукт может быть обработан для хранения и/или применения с помощью любого подходящего этапа обработки. Неограничивающие примеры таких этапов обработки включают, например, асептическую фасовку, то есть фасовку, при которой состав переносят в предварительно приготовленные стерильные контейнеры, так что биофармацевтический лекарственный продукт подходит для последующих процедур применения.
В случае жидкой композиции концентрацию биофармацевтического лекарственного продукта, например, при фасовке, предпочтительно выбирают таким образом, чтобы она соответствовала конечной концентрации, необходимой для введения. Более того, особенно предпочтительно выбирать композицию таким образом, чтобы она стабилизировала продукт и, таким образом, позволяла избежать или свести к минимуму потерю молекулярной целостности и функции во время фасовки, хранения и применения.
В случае сухой композиции обработка для хранения и/или введения включает то, что концентрация лекарственного продукта должна быть отрегулирована таким образом, чтобы при последующем растворении с небольшими объемами растворов (например, воды для инъекций; ВДИ) была достигнута конечная дозировка посредством простой процедуры, предпочтительно без замены буфера и/или регулирования концентрации с помощью любых дополнительных этапов обработки.
В случае, если биофармацевтический лекарственный продукт присутствует в виде замороженного продукта, указанный замороженный продукт необходимо разморозить перед фасовкой. Полученную жидкость необходимо разделить на порции в конечной дозировке. На этой стадии необходимо учитывать, что замораживание и размораживание могут привести к потере молекулярной целостности и функциональности, поэтому, возможно, придется корректировать конечную дозировку соответствующим образом.
В соответствии с этим предпочтительным вариантом осуществления способа по изобретению биофармацевтический лекарственный продукт перерабатывают в жидкую композицию. Указанную жидкую композицию можно хранить и/или предоставлять для введения в любом подходящем флаконе, контейнере или носителе, например, таких как экспериментальные пробирки или пробирки для замораживания, шприцы, микроиглы, дозаторы, трансдермальные пластыри и т.д.
Как правило, хранение биофармацевтических лекарственных препаратов осуществляют в определенных условиях. Такие определенные условия включают, например, конкретные температурные профили, влажность и другие условия хранения, как, например, предписано инструкциями по «надлежащей практике хранения» Управления по контролю за продуктами питания и лекарственными средствами США (FDA) и указаниями Международного совета по гармонизации технических требований к лекарственным препаратам для человека (ICH). Однако могут возникнуть проблемы при транспортировке/перевозке биофармацевтического лекарственного продукта, а также во время введения, когда не всегда возможно соблюсти такие строгие условия. Например, холодовая цепь может прерываться при транспортировке, в частности в страны третьего мира, образцы могут подвергаться воздействию света или механическим воздействиям из-за перемешивания. Это имеет особое значение в отношении жидких композиций, которые более подвержены повреждению вследствие таких неблагоприятных условий, чем высушенные композиции.
Стабильность биофармацевтических лекарственных продуктов во время хранения частично зависит от соблюдения вышеописанных условий хранения, но также зависит от наличия соответствующих стабилизирующих вспомогательных веществ, а также от природы и концентрации самого биофармацевтического лекарственного продукта. Таким образом, обеспечение биофармацевтического лекарственного продукта в соответствующей жидкой композиции может защитить биофармацевтический лекарственный продукт от неблагоприятных условий, тем самым повышая его стабильность.
Соответственно, в предпочтительном варианте осуществления способа по изобретению жидкая композиция предназначена для хранения и/или применения биофармацевтического лекарственного продукта в концентрации в диапазоне от 0,001 до<100 мг/мл, и композиция характеризуется тем, что включает (i) по меньшей мере три аминокислоты, где комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию; и (ii) один или несколько сахаров, где отношение между аминокислотами и сахаром установлено в диапазоне от 4:1 до 1 2 (по массе).
Этот предпочтительный вариант осуществления относится к хранению биофармацевтического лекарственного продукта в жидкой форме в низкой концентрации. Как определено здесь выше, такими низкими концентрациями являются концентрации биофармацевтического лекарственного продукта, которые ниже 100 мг/мл.
Чтобы обеспечить улучшенную стабильность таких жидких композиций с низкой концентрацией, композицию корректируют таким образом, чтобы она включала по меньшей мере три аминокислоты и один или несколько сахаров. Определения и предпочтительные варианты осуществления для «по меньшей мере трех аминокислот» и «сахара (сахаров)» являются такими, как приведенные здесь выше в отношении способа по изобретению. Однако фактический выбор аминокислот не ограничен теми же аминокислотами, что и в композиции первой и/или второй фазы, определенной в настоящем документе выше; вместо этого некоторые или все аминокислоты могут отличаться от аминокислот композиции первой и/или второй фазы. Здесь также охватывается то, что по меньшей мере три аминокислоты этого предпочтительного варианта осуществления идентичны по меньшей мере трем аминокислотам композиции первой и/или второй фазы. То же самое относится и к сахару (сахарам).
Важно отметить, что отношение между аминокислотами и сахаром должно быть доведено до 4:1 - 1:2 (по массе). В данной области техники хорошо известно, как может быть выполнена такая корректировка. Предпочтительно, корректировку осуществляют путем регуляции массовых отношений между аминокислотами и сахаром. Основываясь на знании количества вспомогательных веществ, уже присутствующих в растворе, и их известной молекулярной массы, можно рассчитать, сколько дополнительных вспомогательных веществ необходимо добавить для получения указанного соотношения, используемого в разведениях.
Как показано в примерах 3 и 4, соответствующих нижеприведенным композициям терапевтических антител с низкой концентрацией, неожиданно было обнаружено, что сочетание лекарственной субстанции, полученной способом по изобретению, с указанными по меньшей мере тремя аминокислотами и сахаром при отношении аминокислот к сахару от 4:1 до 1:2 (по массе) обеспечивает превосходные результаты как на ранних этапах производства, так и при последующем хранении жидкости при повышенной температуре в отношении агрегации и фрагментации. Эта стабилизирующая композиция также показана в примере 2 для предотвращения потери функции и молекулярной целостности на ранних этапах процесса производства, а также после нескольких циклов замораживания и размораживания, имитирующих события замораживания и размораживания на различных стадиях процесса производства. Благодаря тому, что на этих стадиях обработки избегают потери продукта с помощью составов на основе аминокислот в сочетании с сахаром, можно избежать дополнительных этапов ребуферизации и диализа.
В альтернативном предпочтительном варианте осуществления способа по изобретению жидкая композиция предназначена для хранения и/или введения биофармацевтического лекарственного продукта в высокой концентрации в диапазоне от 100 до 500 мг/мл, и где композиция характеризуется тем, что она содержит следующие вспомогательные вещества: (i) по меньшей мере три аминокислоты, где комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию; и (ii) один или несколько сахаров; и где отношение между аминокислотами и сахаром регулируют от 4:1 до 1:1 (по массе).
Этот альтернативный предпочтительный вариант осуществления относится к хранению биофармацевтического лекарственного продукта в жидкой форме в высокой концентрации. Как определено здесь выше, такими высокими концентрациями являются концентрации биофармацевтического лекарственного продукта, которые находятся в диапазоне от 100 до 500 мг/мл.
Чтобы обеспечить улучшенную стабильность таких высококонцентрированных жидких составов, состав подбирают таким образом, чтобы он содержал по меньшей мере три аминокислоты и один или несколько сахаров. Определения и предпочтительные варианты осуществления для «по меньшей мере трех аминокислот» и «сахара (сахаров)» являются такими, как приведенные здесь выше в отношении способа по изобретению.
Опять же, фактический выбор аминокислот не ограничен теми же аминокислотами, что и в композициях, определенных здесь выше; вместо этого некоторые или все аминокислоты могут отличаться от аминокислот из указанных выше композиций. Здесь также охватывается то, что по меньшей мере три аминокислоты этого предпочтительного варианта осуществления идентичны по меньшей мере трем аминокислотам одной из указанных выше композиций. То же самое относится и к сахару (сахарам).
Важно отметить, что отношение между аминокислотой и сахаром должно быть отрегулировано в пределах от 4:1 до 1:1 (по массе), включая, например, 3:1 и 2:1 (по массе). Наиболее предпочтительно, отношение составляет 1:1 (по массе). Способы корректировки отношения известны в данной области техники, как обсуждалось выше. Предпочтительно, корректировку осуществляют путем регуляции массовых отношений аминокислот и сахара. Основываясь на знании количества вспомогательных веществ, уже присутствующих в растворе, и их известной молекулярной массы, можно рассчитать, сколько дополнительного вспомогательного вещества необходимо добавить для получения указанного отношения, используемого в разведениях.
В соответствии с этим предпочтительным вариантом осуществления способа по изобретению также предусматривается, что в жидкую композицию могут входить дополнительные вспомогательные вещества. Такие дополнительные вспомогательные вещества предпочтительно выбирают из хелатирующих агентов, дополнительных антиокислительных агентов и сурфактантов.
Используемый в настоящей заявке термин «хелатирующие агенты» относится к вспомогательным веществам, которые улавливают ионы металлов в составах, чтобы избежать, например, катализируемых ионами металлов окислительных реакций в составе. Неограничивающие примеры хелатирующих агентов включают десферал, диэтилтриаминпентаактиновую кислоту (DTPA), этилендиаминтетрауксусную кислоту (ЭДТА) или дефероксамин (DFO). Такие хелатирующие агенты обычно используются в хелатной терапии для детоксикации ядовитых металлических агентов, таких как ртуть [Hg], мышьяк [As] и свинец [Pb], превращая их в химически инертную форму, которая может выводиться из организма без дальнейшего взаимодействия с организмом. Понятно, что хелатирующие агенты используются в соответствии с настоящим изобретением в низких концентрациях, например, от 0,3 до 0,5 мг/мл, что не вызывает терапевтического эффекта, а скорее стабилизирует биофармацевтические продукты, например, при хранении.
Термин «дополнительные антиоксидантные агенты», как он используется в настоящем документе, относится к метионину, цистеину, глутатиону, триптофану, гистидину, аскорбиновой кислоте и любым производным перечисленных здесь агентов, не ограничиваясь ими.
Термин «сурфактанты», используемый в настоящей заявке, относится к поверхностно-активным агентам. Этот термин также включает смачивающие агенты, эмульгирующие агенты и суспендирующие агенты, в зависимости от их свойств и использования. Поверхностно-активные агенты представляют собой вещества, которые при низких концентрациях адсорбируются на поверхностях или границах раздела системы и изменяют поверхностную или межфазную свободную энергию и поверхностное или межфазное натяжение. Поскольку они растворимы как в органических растворителях, так и в воде, их называют «амфифильными». Предпочтительные сурфактанты в соответствии с настоящим изобретением включают полисорбат 20 (Твин 20) и полисорбат 80 (Твин 80), но не ограничиваются ими.
Как показано в примерах 5-7 ниже, неожиданно было обнаружено, что объединение биофармацевтического лекарственного продукта, например высококонцентрированного терапевтического состава антител, полученного способом по изобретению, с указанными по меньшей мере тремя аминокислотами и сахаром при отношении аминокислот к сахару от 4:1 до 1:1 (по массе) приводит к меньшей агрегации во время обработки терапевтических антител, особенно во время ребуферизации посредством диализа и/или концентрирования, по сравнению с оригинальным составом поставщика. Кроме того, этот состав уменьшал агрегацию, а также фрагментацию при последующем хранении жидкости при повышенной температуре. Примеры 8 и 9 дополнительно подтверждают этот вывод. Например, по сравнению с предпочтительным препаратом, описанным в патенте США US 9,364,542 B2, мы подчеркнули превосходную эффективность стабилизации наших составов в соответствии с настоящим изобретением, а также по сравнению с исходным поставщиком жидкости.
Кроме того, количественный статистический анализ кинетики времени хранения жидкости во время хранения показал, что стабилизирующие эффекты, обусловленные композициями по настоящему изобретению, были статистически значимыми.
Наиболее важно, что такие жидкие составы высококонцентрированного биофармацевтического продукта неожиданно были обнаружены в примерах 5 и 7 с особенно низкой вязкостью, что является важным фактором, например, для возможности введения готового продукта с помощью шприца. В частности, Пример 5 показал высококонцентрированную терапевтическую композицию антител, соответствующую настоящему изобретению и содержащую соответствующее антитело в концентрациях 120 мг/мл с вязкостью, значительно меньшей, чем 4 мПа*с, по сравнению с измеренной вязкостью в оригинальном составе поставщика (приблизительно 5 мПа*с). Кроме того, в Примере 7 высококонцентрированные составы терапевтических антител, соответствующие настоящему изобретению и содержащие антитело в концентрациях 200 и 220 мг/мл, показали вязкость, значительно меньшую, чем 20 мПа*с.В целом во всех вариантах было обнаружено, что вязкость ниже, чем в оригинальных составах поставщика. Таким образом, эти значения ниже, чем в соответствующем составе предшествующего уровня техники.
Соответственно, в особенно предпочтительных вариантах осуществления этого варианта хранения биофармацевтического лекарственного продукта в жидкой форме в высокой концентрации вязкости высококонцентрированных биофармацевтических лекарственных продуктов составляют менее 20 мПа*с; при концентрации высококонцентрированного биофармацевтического лекарственного продукта от 100 до 120 мг/мл вязкость составляет<4; при концентрации высококонцентрированного биофармацевтического лекарственного продукта от 120 до 150 мг/мл вязкость составляет<8 мПа*с; и при концентрации высококонцентрированного биофармацевтического лекарственного продукта от 150 до 220 мг/мл вязкость составляет<20 мПа*с.
В еще более предпочтительном варианте осуществления этого способа по изобретению жидкую композицию дополнительно корректируют таким образом, чтобы отношение между представляющей интерес биомолекулой и по меньшей мере тремя аминокислотами (i) составляло от 3,5:1 до 1:2 (по массе). Регулирование отношения предпочтительно выполняют по массовому отношению. Подробности, приведенные выше относительно регулирования отношения между интересующей биомолекулой и сахаром (сахарами), применяются с учетом необходимых изменений к этому варианту осуществления в отношении дополнительной корректировки отношения между представляющей интерес биомолекулой и по меньшей мере тремя аминокислотами.
В предпочтительном варианте осуществления этого способа изобретения жидкая композиция для высококонцентрированных лекарственных субстанций не содержит пролина. Пример 8 подтвердил, что стабилизирующая эффективность по изобретению была выше, чем у составов, содержащих пролин, в соответствии с патентом США US 9,364,542 B2. Это было подтверждено ограниченной химической деградацией при анализе с помощью катионообменной ВЭЖХ. В предпочтительном варианте осуществления отношение между аминокислотами и сахаром составляет от 10:1 до 1:100. Более того, предпочтительное отношение между биомолекулами и вспомогательными веществами составляет от 1:1 до 1:500. Эти отношения также являются предпочтительными в других вариантах осуществления.
В еще одном предпочтительном варианте осуществления способа по изобретению способ дополнительно включает (с) третий этап сушки биофармацевтической лекарственной субстанции, полученной на стадии (b), для получения высушенного биофармацевтического лекарственного продукта, где указанный этап сушки на этой третьей фазе проводят в присутствии композиции, содержащей (i) по меньшей мере три аминокислоты, где комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию; и (ii) один или несколько сахаров; и где отношение между интересующей биомолекулой и суммой вспомогательных веществ устанавливают в пределах от 1:1 до 1:10 (по массе).
В соответствии с этим предпочтительным вариантом осуществления биофармацевтический лекарственный продукт, полученный способом по настоящему изобретению, дополнительно подвергают дополнительному этапу сушки, чтобы получить высушенный биофармацевтический лекарственный продукт.Биофармацевтический лекарственный продукт считается сухим, если содержание жидкости было удалено или уменьшено до менее чем 20% объема, например, менее 10%, например, менее 5%, более предпочтительно менее 3% объема, например, менее 2% или менее 1%. Наиболее предпочтительно, жидкость уменьшают до 0,5% или менее. Подходящие способы сушки включают лиофилизацию (сублимационную сушку), распылительную сушку, распылительную сублимационную сушку, конвекционную сушку, кондуктивную сушку, сушку газовым потоком, сушку в барабане, вакуумную сушку, диэлектрическую сушку (например, радиочастотными волнами или микроволнами), сушку поверхности, воздушную сушку или пенную сушку, но не ограничиваются ими.
Указанный этап сушки в соответствии с этим вариантом осуществления способа по настоящему изобретению проводят в присутствии композиции, содержащей (i) по меньшей мере три аминокислоты и (ii) один или несколько сахаров. Опять же, определения и предпочтительные варианты осуществления для «по меньшей мере трех аминокислот» и «сахара (сахаров)» являются такими, как приведенные здесь выше в отношении способа по изобретению. Также, опять же, фактический выбор аминокислот не ограничен теми же аминокислотами, что и в композициях, определенных здесь выше; вместо этого некоторые или все аминокислоты могут отличаться от аминокислот из указанных выше композиций. Здесь также охватывается то, что по меньшей мере три аминокислоты этого предпочтительного варианта осуществления идентичны по меньшей мере трем аминокислотам из одной из указанных выше композиций. То же самое относится и к сахару (сахарам).
Важно отметить, что отношение между интересующей биомолекулой и суммой вспомогательных веществ должно быть от 1:1 до 1:10 (по массе), включая, например, также отношения между 1:2, 1:5, 1:8 (по массе) и т.д. Наиболее предпочтительно, отношение составляет 1:2 (по массе). Способы регулирования отношения известны в данной области техники, как обсуждалось выше. Также, что касается этого предпочтительного варианта осуществления, предпочтительно осуществлять регуляцию путем корректировки массового отношения представляющей интерес биомолекулы и суммы вспомогательных веществ. Основываясь на знании количества вспомогательных веществ, уже присутствующих в растворе, и их известной молекулярной массы, можно рассчитать, сколько дополнительного вспомогательного вещества необходимо добавить для получения указанного отношения, используемого в разведениях.
В соответствии с этим предпочтительным вариантом осуществления способа по изобретению также предусматривается, что в композицию, используемую для этапа сушки, могут быть включены дополнительные вспомогательные вещества. Такие дополнительные вспомогательные вещества предпочтительно выбирают из хелатирующих агентов, дополнительных антиоксидантных агентов и сурфактантов. Определения и предпочтительные варианты осуществления, приведенные выше для указанных дополнительных вспомогательных веществ, применяются с учетом необходимых изменений.
Как показано в примере 1 ниже, было неожиданно обнаружено, что объединение биофармацевтической лекарственной субстанции, полученной способом по изобретению, с перечисленными по меньшей мере тремя аминокислотами и сахаром при отношении интересующей биомолекулы и суммы вспомогательных веществ между 1:1 и 1:10 (по массе) обеспечивает превосходную стабильность для интересующей высушенной биомолекулы. В примере 1 комбинация препарата аденовирусного вектора с композициями согласно настоящему изобретению уже на ранних стадиях последующих этапов производства и последующей лиофильной сушки приводила к полному сохранению титра инфекции и гидродинамических радиусов вирусных частиц. Напротив, лиофилизация соответствующих препаратов аденовирусного вектора в оригинальном составе поставщика привела к значительной потере инфекционности и увеличению размера частиц. Подобная процедура в сочетании с обычным фосфатно-солевым буферным раствором (PBS) привела к полной потере инфекционности и значительному увеличению размера частиц уже после сублимационной сушки. Наиболее важно, что такие высушенные составы биофармацевтических лекарственных субстанций или лекарственных продуктов подходят для различных дополнительных этапов обработки, таких как аликвотирование, распределение, отгрузка, хранение и т.д.
В еще более предпочтительном варианте осуществления способа по изобретению сушку биофармацевтического лекарственного продукта, полученного на стадии (b), осуществляют сублимационной сушкой, распылительной сушкой или распылительной сублимационной сушкой.
Сублимационная сушка, также называемая лиофилизацией, хорошо известна в данной области техники и включает этапы замораживания образца и последующего снижения окружающего давления при добавлении достаточного количества тепла, чтобы позволить замерзшей воде в материале испаряться непосредственно из твердой фазы до паровой фазы с последующей вторичной фазой сушки. Предпочтительно лиофилизированный препарат затем герметизируют, чтобы предотвратить повторное поглощение влаги.
Распылительная сушка также хорошо известна в данной области техники и представляет собой способ превращения раствора, суспензии или эмульсии в твердый порошок за один этап процесса. Обычно концентрат жидкого продукта подают насосом в распылительное устройство, где он разбивается на маленькие капли. Эти капли подвергаются воздействию потока горячего воздуха и очень быстро теряют влагу, оставаясь при этом взвешенными в сушильном воздухе. Сухой порошок отделяют от влажного воздуха в циклонах центробежным действием, то есть частицы плотного порошка направляются к стенкам циклона, в то время как более легкий влажный воздух направляется через выводящие трубы.
Распылительная сушка часто является предпочтительным способом, так как она позволяет избежать этапа замораживания и требует меньших энергетических затрат по сравнению с лиофилизацией. Было также показано, что распылительная сушка является особенно выгодной процедурой сушки, которая подходит для биомолекул, из-за короткого времени контакта с высокой температурой и специального контроля процесса. Таким образом, поскольку распылительная сушка приводит к диспергируемому сухому порошку всего за один этап, она часто предпочтительнее лиофилизации, когда речь идет о методиках сушки для биомолекул.
Распылительная сублимационная сушка также хорошо известна в данной области техники и представляет собой способ, который объединяет этапы обработки, общие для лиофилизации и распылительной сушки. Предоставленный образец распыляется в криогенную среду (такую как, например, жидкий азот), которая генерирует дисперсию капель, подвергнутых шоковой заморозке. Эту дисперсию затем сушат в лиофильной сушилке.
В следующем предпочтительном варианте осуществления способа по изобретению высушенный биофармацевтический лекарственный продукт, полученный на этапе (с), стерилизуют, предпочтительно стерилизуют на конечной стадии.
Термин «стерилизованный на конечной стадии», используемый в данном документе, относится к процессу стерилизации полученного продукта на этапе (с), в котором указанный процесс стерилизации является последним (т.е. конечным) процессом при обработке этого образца перед его подготовкой к назначенному применению, такому как восстановление для обеспечения, например, введения субъекту, как обсуждается здесь ниже.
Средства и способы стерилизации биофармацевтического продукта хорошо известны в данной области техники. Например, высушенный образец может присутствовать или может быть введен в контейнер или флакон, затем контейнер или флакон укупоривают и подвергают условиям стерилизации в течение периода времени, достаточного для существенной инактивации патогенов, особенно бактерий и вирусов. Неограничивающие примеры условий стерилизации включают облучение, такое как бета-, рентгеновское или гамма-облучение, обработку этиленоксидом, тепловую инактивацию, автоклавирование и плазменную стерилизацию.
Предпочтительно стерилизацию проводят облучением или обработкой этиленоксидом.
В другом предпочтительном варианте осуществления способа по изобретению способ дополнительно включает этап восстановления высушенного биофармацевтического лекарственного продукта, полученного на этапе (с), для получения жидкой композиции, отличающийся тем, что высушенный биофармацевтический лекарственный продукт восстанавливают для получения жидкого биофармацевтического лекарственного продукта в композиции, содержащей (i) по меньшей мере три аминокислоты, где комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию, и по меньшей мере одну буферную функцию; и (ii) один или несколько сахаров; при отношении между аминокислотами и сахаром от 4:1 до 1:1 (по массе).
Соответственно, высушенный биофармацевтический лекарственный продукт, полученный в соответствии с предпочтительным вариантом осуществления способа по настоящему изобретению, впоследствии восстанавливают, чтобы получить жидкую композицию. Восстановление осуществляют в растворе, который приводит к композиции, содержащей по меньшей мере три аминокислоты, один или несколько сахаров. Опять же, определения и предпочтительные варианты для степени вязкости, «по меньшей мере трех аминокислот» и «сахара (сахаров)» являются такими, как приведено здесь выше в отношении способа по изобретению, но фактический выбор аминокислот не ограничивается теми же аминокислотами, что и в композициях, определенных в настоящем документе выше; вместо этого некоторые или все аминокислоты могут отличаться от аминокислот из указанных выше композиций. Здесь также охватывается то, что по меньшей мере три аминокислоты этого предпочтительного варианта осуществления идентичны по меньшей мере трем аминокислотам одной из указанных выше композиций. То же самое относится и к сахару (сахарам).
Важно отметить, что отношение между аминокислотой и сахаром составляет от 4:1 до 1:1 (по массе), включая, например, также отношения между 3:1 или 2:1 (по массе). Наиболее предпочтительно, отношение составляет 1:1 (по массе). Способы корректировки отношения известны в данной области техники, как обсуждалось выше. Также, что касается этого предпочтительного варианта осуществления, предпочтительно, чтобы корректировка осуществлялась путем регуляции массового отношения аминокислоты и сахара. Основываясь на знании количества вспомогательных веществ, уже присутствующих в растворе, и их известной молекулярной массы, можно рассчитать, сколько дополнительного вспомогательного вещества необходимо добавить для получения указанного соотношения, используемого в разведениях.
В соответствии с этим предпочтительным вариантом осуществления способа по изобретению также предусматривается, что в композицию могут быть включены дополнительные вспомогательные вещества, используемые на этапе сушки,. Такие дополнительные вспомогательные вещества предпочтительно выбирают из хелатирующих агентов, дополнительных антиоксидантных агентов и сурфактантов. Определения и предпочтительные варианты осуществления, приведенные выше для указанных дополнительных вспомогательных веществ, применяются с учетом необходимых изменений. Такие дополнительные вспомогательные вещества могут быть выбраны так, чтобы они были идентичны дополнительным вспомогательным веществам (если таковые имеются), используемым на одном из предыдущих этапов, но также могут быть выбраны независимо от них, так что они могут отличаться.
В предпочтительном варианте осуществления способа по настоящему изобретению композиция на этапе (а) содержит от 0,5 мг/мл до 10 мг/мл триптофана и от 0,5 мг/мл до 30 мг/мл гистидина.
Как показано в примерах 2-7, растворы в сбалансированном массовом отношении триптофана и гистидина приводили к увеличению стабильности лекарственной субстанции, и в сочетании с сахаром увеличивали стабильность лекарственного продукта.
В другом предпочтительном варианте осуществления способа по настоящему изобретению представляющую интерес биомолекулу выбирают из группы, состоящей из белков и пептидов, а также их смесей.
Термин «пептид», как используется в настоящей заявке, описывает группу молекул, состоящую из до 30 аминокислот, тогда как «белки» состоят из более чем 30 аминокислот. Пептиды и белки могут дополнительно образовывать димеры, тримеры и высшие олигомеры, то есть состоять из более чем одной молекулы, которая может быть одинаковой или неидентичной. Соответствующие структуры более высокого порядка, следовательно, называются гомо- или гетеродимерами, гомо- или гетеротримерами и т.д. Термины «пептид» и «белок» (где «белок» взаимозаменяемо используется с «полипептидом») также относятся к естественно модифицированным пептидам/белкам, в которых осуществляется модификация, например, гликозилированием, ацетилированием, фосфорилированием и тому подобным. Такие модификации хорошо известны в данной области техники. Кроме того, пептидомиметики из таких пептидов и белков, в которых аминокислота (аминокислоты) и/или пептидная связь (связи) заменены функциональными аналогами, также включены в данный документ. Такие функциональные аналоги включают все известные аминокислоты, кроме 20 кодируемых генами аминокислот, такие как селеноцистеин. Конкретные предпочтительные примеры подходящих белков или пептидов подробно описаны ниже.
Предпочтительными примерами белков и пептидов являются антитела и гормоны.
Антитело в соответствии с настоящим изобретением может быть, например, поликлональным или моноклональным антителом. Используемый в настоящей заявке термин «антитело» также включает варианты осуществления, такие как химерные (человеческий константный домен, вариабельный домен, не являющийся человеческим), одноцепочечные и гуманизированные (человеческое антитело, за исключением CDR, не являющегося человеческим) антитела, а также фрагменты антител, такие как, среди прочего, Fab, Fab', Fd, F(ab')2, Fv или scFv фрагменты или нанотела, то есть одиночные мономерные вариабельные домены антител; см., например, Harlow and Lane «Antibodies, A Laboratory Manual», Cold Spring Harbor Laboratory Press, 1988; и Harlow and Lane «Using Antibodies: A Laboratory Manual» Cold Spring Harbor Laboratory Press, 1999.
Способы получения антител хорошо известны в данной области техники, и описаны, например, в Harlow and Lane (1988) и (1999), процитированных выше.
Предпочтительно антитело представляет собой антитело, способное вызывать терапевтические эффекты. Неограничивающие примеры предпочтительных антител включают Инфликсимаб, Бевацизумаб, Ранибизумаб, Цетуксимаб, Ранибизумаб, Паливизумаб, Абаговомаб, Абциксимаб, Актоксумаб, Адалимумаб, Афелимомаб, Афутузумаб, Алацизумаб, Алацизумаб пегол, ALD518, Алемтузумаб, Алирокумаб, Алемтузумаб, Алтумомаб, Аматуксимаб, Анатумомаб мафенатокс, Анрукинзумаб, Аполизумаб, Арцитумомаб, Аселизумаб, Алтинумаб, Атлизумаб, Аторолимумаб, Тоцилизумаб, Бапинейзумаб, Базиликсимаб, Бавитуксимаб, Бектумомаб, Белимумаб, Бенрализумаб, Бертилимумаб, Бесилесомаб, Бевацизумаб, Безлотоксумаб, Бициромаб, Биватузумаб, Биватузумаб мертансин, Блинатумомаб, Блосозумаб, Брентуксимаб ведотин, Бриакинумаб, Бродалумаб, Канакинумаб, Кантузумаб мертансин, Каплацизумаб, Капромаб пендетид, Карлумаб, Катумаксомаб, CC49, Цеделизумаб, Цертолизумаб пегол, Цетуксимаб, Цитатузумаб богатокс, Циксутумумаб, Клазакизумаб, Кленоликсимаб, Кливатузумб тераксетан, Конатумумаб, Кренезумаб, CR6261, Дацетузумаб, Даклизумаб, Далотузумаб, Даратумумаб, Демцизумаб, Деносумаб, Детумомаб, Дорлимомаб аритокс, Дрозитумаб, Дулиготумаб, Дупилумаб, Экромексимаб, Экулизумаб, Эдобакомаб, Эдреколомаб, Эфализумаб, Эфунгумаб, Элотузумаб, Элсилимомаб, Энаватузумаб, Энлимомаб пегол, Энокизумаб, Энотикумаб, Энситуксимаб, Эпитумомаб цитуксетан, Эпратузумаб, Эрлизумаб, Эртумаксомаб, Этарацизумаб, Этролизумаб, Эксбивирумаб, Фанолесомаб, Фаралимомаб, Фарлетузумаб, Фасинумаб, FBTA05, Фелвизумаб, Фезакинумаб, Фиклатузумаб, Фигитумумаб, Фланвотумаб, Фонтолизумаб, Форалумаб, Форавирумаб, Фресолимумаб, Фулранумаб, Футуксимаб, Галиксимаб, Ганитумаб, Гантенерумаб, Гавилимомаб, Гемтузумаб озогамицин, Гевокизумаб, Гирентуксимаб, Глембатумумаб ведотин, Голимумаб, Гомиликсимаб, GS6624, Ибализумаб, Ибритумомаб тиуксетан, Икрукумаб, Иговомаб, Имциромаб, Имгатузумаб, Инклакумаб, Индатуксимаб равтансин, Инфликсимаб, Интетумумаб, Инолимомаб, Инотузумаб озогамицин, Ипилимумаб, Иратумумаб, Итолизумаб, Иксекизумаб, Келиксимаб, Лабетузумаб, Лебрикизумаб, Лемалесомаб, Лерделимумаб, Лексатумумаб, Либивирумаб, Лигелизумаб, Линтузумаб, Лирилумаб, Лорвотузумаб мертансин, Лукатумумаб, Люмиликсимаб, Мапатумумаб, Маслимомаб, Маврилимумаб, Матузумаб, Меполизумаб, Метелимумаб, Милатузумаб, Минретумомаб, Митумомаб, Могамулизумаб, Моролимумаб, Мотавизумаб, Моксетумомаб пасудотокс, Муромонаб-CD3, Наколомаб тафенатокс, Намилумаб, Наптумомаб эстафенатокс, Нарнатумаб, Натализумаб, Небакумаб, Нецитумумаб, Нерелимомаб, Несвакумаб, Нимотузумаб, Ниволумаб, Нофетумомаб мерпентан, Окаратузумаб, Окрелизумаб, Одулимомаб, Офатумумаб, Оларатумаб, Олокизумаб, Омализумаб, Онартузумаб, Опортузумаб монатокс, Ореговомаб, Ортикумаб, Отелизизумаб, Окселумаб, Озанезумаб, Озорализумаб, Пагибаксимаб, Паливизумаб, Панитумумаб, Панобакумаб, Парсатузумаб, Пасколизумаб, Пателизумаб, Патритумаб, Пемтумомаб, Перакизумаб, Пертузумаб, Пекселизумаб, Пидилизумаб, Пинтумомаб, Плакулумаб, Понезумаб, Приликсимаб, Притумумаб, PRO140, Квилизумаб, Ракотумомаб, Радретумаб, Рафивирумаб, Рамуцирумаб, Ранибизумаб, Раксибакумаб, Регавирумаб, Реслизумаб, Рилотумумаб, Ритуксимаб, Робатумумаб, Роледумаб, Ромосозумаб, Ронтализумаб, Ровелизумаб, Руплизумаб, Самализумаб, Сарилумаб, Сатумомаб пендетид, Секукинумаб, Севирумаб, Сибротузумаб, Сифалимумаб, Силтуксимаб, Симтузумаб, Сиплизумаб, Сирукумаб, Соланезумаб, Солитомаб, Сонепцизумаб, Сонтузумаб, Стамулумаб, Сулесомаб, Сувизумаб, Табалумаб, Такатузумаб тетраксетан, Тадоцизумаб, Тализумаб, Танезумаб, Таплитумомаб паптокс, Тефибазумаб, Телимомаб аритокс, Тенатумомаб, Тефибазумаб, Телимомаб аритокс, Тенатумомаб, Тенеликсимаб, Теплизумаб, Тепротумумаб, TGN1412, Тремелимумаб, Тицилимумаб, Тилдракизумаб, Тигатузумаб, TNX-650, Тоцилизумаб, Торализумаб, Тозитумомаб, Тралокинумаб, Трастузумаб, TRBS07, Трегализумаб, Тремелимумаб, Тукотузумаб целмолейкин, Тувирумаб, Ублитуксимаб, Урелумаб, Уртоксазумаб, Устекинумаб, Вапаликсимаб, Вателизумаб, Ведолизумаб, Велтузумаб, Вепалимомаб, Весенкумаб, Висилизумаб, Волоциксимаб, Ворсетузумаб мафодотин, Вотумумаб, Залутумумаб, Занолимумаб, Затуксимаб, Зиралимумаб и Золимомаб аритокс.
Используемый в настоящей заявке термин «гормоны» хорошо известен в данной области техники и относится к группе терапевтических биомолекул, используемых для лечения нарушений обмена веществ. Неограничивающие примеры включают терипаратид или эстроген. Терипаратид является рекомбинантной формой паратиреоидного гормона, который обычно используется для лечения нарушения метаболизма костей, такого как остеопороз. Эстроген обычно используется для лечения менопаузальных расстройств и вводится в сочетании с прогестероном для снижения риска развития рака матки.
В более предпочтительном варианте осуществления способа по настоящему изобретению представляющая интерес биомолекула представляет собой антиген, такой как, например, антиген для использования в качестве вакцин.
Используемый в настоящей заявке термин «антигены» относится к молекулам, способным индуцировать иммунный ответ в организме хозяина. Как правило, антигенами являются белки и полисахариды. Однако в сочетании с белками и полисахаридами также липиды или нуклеиновые кислоты могут быть антигенными. Антигены часто получают из частей бактерий, вирусов и других микроорганизмов, таких как, например, их оболочки, капсулы, клеточные стенки, жгутики, фимбрии или токсины. Антигены также могут быть немикробными, такими как, например, аутоантигены или экзогенные антигены (не аутоантигены), такие как пыльца, яичный белок или белки из трансплантированных тканей/органов или с поверхности перелитых клеток крови. В соответствии с настоящим изобретением термин «антигены» включает, без ограничения, (i) антигены, представленные одним конкретным молекулярным типом антигена, таким как, например, один конкретный белок; (ii) смеси антигенов различных молекулярных типов антигена, таких как, например, смесь разных белков или смесь белков с полисахаридами; а также (iii) антигенные препараты, содержащие дополнительные компоненты, такие как, например, антигены расщепленного вируса, которые представляют собой препараты, в которых вирус был разрушен, например, с помощью детергента, или другим способом, без дальнейшего удаления других вирусных компонентов.
Предпочтительно, антигены предназначены для использования в качестве вакцин. Подходящие антигены для приготовления вакцин хорошо известны в данной области техники, и соображения по выбору антигена для производства вакцин, обычно применяемые в данной области техники, применяются с учетом необходимых изменений в отношении выбора подходящего антигена для применения в качестве вакцины в соответствии с настоящим изобретением. Соответственно, могут использоваться антигены, уже имеющиеся в данной области техники, а также новые антигены.
Особо предпочтительными примерами антигенов являются субъединичные антигены или вирусные векторы, включая, например, вирусоподобные частицы и живые вирусы.
«Вирусные векторы» представляют собой сложные супрамолекулярные ансамбли макромолекул, которые подвержены различным химическим и физическим путям деградации при производстве, хранении и распространении. Термин «вирусный вектор» в соответствии с настоящим изобретением относится к носителю, то есть «вектору», который происходит от вируса. «Вирусные векторы» в соответствии с настоящим изобретением включают векторы, полученные из встречающихся в природе или модифицированных вирусов, а также вирусоподобных частиц (VLP). Когда вирусы являются исходным материалом для разработки вектора, необходимо выполнить определенные требования, такие как безопасность и специфичность, чтобы обеспечить их пригодность для клинического использования у животных или у пациентов. Одним из важных аспектов является предотвращение неконтролируемой репликации вирусного вектора. Обычно это достигается удалением части вирусного генома, критического для репликации вируса. Такой вирус может инфицировать клетки-мишени без последующей продукции новых вирионов. Более того, вирусный вектор не должен оказывать никакого влияния или только минимальное влияние на физиологию клетки-мишени, и перестройка генома вирусного вектора не должна происходить. Такие вирусные векторы, полученные из встречающихся в природе или модифицированных вирусов, хорошо известны в данной области техники, и неограничивающие примеры обычно используемых вирусных векторов включают, например, например, Модифицированный вирус осповакцины Анкара (MVA) или аденовирус.
Также векторы, полученные из вирусоподобных частиц, хорошо известны в данной области техники и описаны, например, в Tegerstedt et al. (Tegerstedt et al. (2005), «Murine polyomavirus virus-like particles (VLPs) as vectors for gene and immune therapy and vaccines against viral infections and cancer». Anticancer Res. 25(4):2601-8). Одним из основных преимуществ VLP является то, что они не связаны с каким-либо риском повторной сборки, который возможен, когда живые аттенуированные вирусы используются в качестве вирусных векторов и, как таковые, они представляют «вирусные векторы с дефицитом репликации» в соответствии с настоящим изобретением. Производство VLP имеет дополнительное преимущество, заключающееся в том, что оно может быть начато раньше, чем производство традиционных вакцин, как только станет доступна генетическая последовательность конкретного интересующего штамма вируса.
VLP содержат повторяющиеся дисплеи высокой плотности вирусных поверхностных белков, которые представляют собой конформационные вирусные эпитопы, которые могут вызывать сильные иммунные ответы Т-клеток и В-клеток. VLP уже использовались для разработки одобренных FDA вакцин против гепатита В и вируса папилломы человека, и, кроме того, VLP использовались для разработки доклинической вакцины против вируса чикунгунья. Данные также свидетельствуют о том, что VLP вакцины против вируса гриппа могут превосходить по защите от вирусов гриппа другие вакцины. В ранних клинических испытаниях VLP вакцины против гриппа, по-видимому, обеспечивали полную защиту как от вируса гриппа A подтипа H5N1, так и от гриппа 1918 года.
В другом предпочтительном варианте осуществления способа по изобретению готовую биофармацевтическую композицию дополнительно регулируют для внутримышечного, подкожного, внутрикожного, трансдермального, орального, перорального, назального и/или ингаляционного применения. При рассмотрении перорального, легочного или интраназального введения применяются разные требования. Например, пероральное введение требует композиции, которая позволяет лекарственному продукту проходить через желудочно-кишечный тракт без потери активности при расщеплении молекул, тогда как легочное введение требует сухой композиции, которая стабильна при прохождении через верхние и нижние дыхательные пути и при растворении проходит через соответствующую слизистую оболочку. Для других способов введения, таких как подкожная или внутримышечная инъекция, необходимо учитывать осмоляльность, вязкость, пригодность для инъекции и введения шприцем. Например, низкое количество вспомогательных веществ является предпочтительным для ограничения осмоляльности и вязкости, например, для уменьшения боли и побочных эффектов в месте инъекции.
Как обсуждалось здесь выше, использование композиций, описанных в настоящей заявке, во время заявленного способа(ов) производства приводит к осмоляльности конечного биофармацевтического лекарственного продукта ниже 450 мОсмоль/кг.Несколько исследователей сообщили, что высокая осмоляльность связана с болью и побочными эффектами в месте инъекции. Таким образом, общепринято, что осмоляльность парентерально вводимого раствора должна быть ниже 450 мОсмоль/кг, предпочтительно близко к физиологическому диапазону от 275 до 320 мОсмоль/кг.Лекарственный продукт, полученный способом по настоящему изобретению, удовлетворяет этому требованию в отношении фармацевтических лекарственных продуктов для введения людям и животным.
Настоящее изобретение также относится к биофармацевтическому лекарственному продукту, полученному или получаемому способом по изобретению.
В предпочтительном варианте осуществления биофармацевтического лекарственного продукта по изобретению указанный продукт предназначен для внутримышечного, подкожного, внутрикожного, трансдермального, орального, перорального, назального и/или ингаляционного применения. В другом предпочтительном варианте осуществления биофармацевтического лекарственного продукта по изобретению указанный продукт предназначен для исследовательских, терапевтических и/или профилактических целей.
В соответствии с предпочтительным вариантом осуществления изобретения лекарственный продукт предназначен для использования при вакцинации. Понятно, что лекарственные препараты для использования при вакцинации могут использоваться как таковые, т.е. сами по себе или в комбинации с адъювантом, который может вводиться одновременно с лекарственным препаратом или отдельно, т.е. до или после введения лекарственного продукта. Используемый в настоящей заявке термин «адъювант» относится к одному или нескольким соединениям, которые усиливают иммунный ответ реципиента на вакцину. Адъюванты часто добавляют для стимуляции более раннего, более сильного ответа и/или более стойкого иммунного ответа на вакцину, что часто позволяет снизить дозу вакцины. Неограничивающие примеры адъювантов включают, например, гидроксид алюминия и фосфат алюминия, органическое соединение сквален, а также такие соединения, как, например, лиганды толл-подобных рецепторов, QS21, производные гидроксида алюминия, масляные иммерсии, липид A и его производные (например, монофосфорил-липид A (MPL), мотивы CpG, поли I:C дцРНК, мурамилдипептид (MDP), полный адъювант Фрейнда (FCA) (только для использования у животных), неполный адъювант Фрейнда (FIA, только для использования у животных) или MF59C. Такие адъюванты хорошо известны в данной области техники.
Лекарственные препараты-вакцины, изготовленные в соответствии с данным изобретением, обладают превосходной термостабильностью, и следовательно, могут подвергаться длительному хранению и транспортировке даже в ситуациях, когда холодовая цепь не гарантируется. Кроме того, более высокая стабильность лекарственного препарата-вакцины позволяет уменьшить количество необходимых адъювантов или может даже сделать ненужными адъюванты. Это обеспечивает дополнительное преимущество, так как адъюванты обычно рассматриваются в данной области техники как существенные для достаточных эффектов вакцинации, но которые также часто вызывают серьезные побочные эффекты.
Настоящее изобретение относится в альтернативе к способу получения биофармацевтической лекарственной субстанции, содержащей интересующую биомолекулу, причем указанный способ включает по меньшей мере один этап обработки, выбранный из (а1) сбора, (а2) очистки, (а3) ребуферизации и (а4) обогащения, при котором указанный по меньшей мере один этап обработки проводят в присутствии композиции, содержащей по меньшей мере три аминокислоты, при этом комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию.
Все определения и предпочтительные варианты осуществления, представленные здесь выше в отношении способа по изобретению, применяются с учетом необходимых изменений к этому альтернативному способу по изобретению. Например, вышеупомянутые предпочтительные варианты осуществления для выбора и/или количества аминокислот, присутствующих в композиции, предпочтительного выбора биомолекулы и т.д., в равной степени применимы к этому альтернативному способу.
Этот изобретательский этап представляет особый интерес, поскольку стабилизация биомолекулы во время последующей обработки обычно рассматривается на этапе фасовки и окончательной обработки, но не на ранних этапах после сбора.
В соответствии с этим альтернативным способом изобретения способ включает в предпочтительном варианте осуществления второй этап дополнительной обработки лекарственной субстанции для получения биофармацевтического лекарственного продукта, причем указанный второй этап включает по меньшей мере один этап обработки, выбранный из (b1) замены буфера, (b2) замораживания, (b3) размораживания и (b4) фасовки; где указанный по меньшей мере один этап обработки проводят в присутствии композиции, содержащей (i) по меньшей мере три аминокислоты, при этом комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию; и (ii) один или несколько сахаров; при отношении аминокислота: сахар от 10:1 до 1:100 (по массе).
Все определения и предпочтительные варианты осуществления, представленные здесь выше в отношении способа по изобретению, применяются с учетом необходимых изменений к этому альтернативному способу по изобретению. Например, вышеупомянутые предпочтительные варианты осуществления для выбора и/или количества аминокислот, присутствующих в композиции, предпочтительного выбора биомолекулы, предпочтительных соотношений и т.д., в равной степени применимы к этому альтернативному способу.
Если не указано иное, все технические и научные термины, используемые в данной заявке, имеют то же значение, которое обычно принято для специалистов в области техники, к которой относится данное изобретение. В случае конфликта, описание патента, включая определения, будет иметь преимущественную силу.
Что касается вариантов осуществления, охарактеризованных в этом описании, в частности в формуле изобретения, предполагается, что каждый вариант, упомянутый в зависимом пункте формулы изобретения, объединяется с каждым вариантом осуществления каждого пункта формулы (независимого или зависимого), от которого зависит указанный зависимый пункт формулы. Например, в случае независимого пункта формулы 1, указывающего 3 альтернативы A, B и C, зависимого пункта формулы 2, указывающего 3 альтернативы D, E и F, и пункта формулы 3, зависимого от пунктов 1 и 2, и указывающего 3 альтернативы G, H и I, следует понимать, что описание однозначно раскрывает варианты осуществления, соответствующие комбинациям A, D, G; A, D, H; A, D, I; A, E, G; A, E, H; A, E, I; A, F, G; A, F, H; A, F, I; B, D, G; B, D, H; B, D, I; B, E, G; B, E, H; B, E, I; B, F, G; B, F, H; B, F, I; C, D, G; C, D, H; C, D, I; C, E, G; C, E, H; C, E, I; C, F, G; C, F, H; C, F, I, если специально не указано иное.
Аналогичным образом, а также в тех случаях, когда независимые и/или зависимые пункты формулы изобретения не содержат альтернатив, подразумевается, что если зависимые пункты формулы изобретения ссылаются на множество предыдущих пунктов формулы изобретения, любая комбинация предмета, охватываемого ими, считается явно раскрытой. Например, в случае независимого пункта формулы 1, зависимого пункта формулы 2, ссылающейся на пункт 1, и зависимого пункта формулы 3, относящегося к обоим пунктам 2 и 1, следует, что комбинация предмета пунктов 3 и 1 формулы изобретения ясно и недвусмысленно раскрыта, как и комбинация предмета пунктов формулы изобретения 3, 2 и 1. В случае наличия дополнительного зависимого пункта формулы изобретения 4, который относится к любому одному из пунктов формулы изобретения 1-3, следует, что комбинация предмета обсуждения пунктов 4 и 1, пунктов 4, 2 и 1, пунктов 4, 3 и 1, а также пунктов 4, 3, 2 и 1 четко и недвусмысленно раскрыта.
Приведенные выше соображения применяются с учетом необходимых изменений ко всем прилагаемым пунктам формулы изобретения. Чтобы привести неограничивающий пример, комбинация пунктов формулы изобретения 10, 6 и 1 четко и однозначно предусмотрена с точки зрения структуры формулы изобретения. То же самое относится, например, к комбинации пунктов 10, 2 и 1 и т.д.
Описание фигур
Фигура 1: Определение посредством динамического рассеяния света (DLS) гидродинамических радиусов аденовирусных векторных композиций перед лиофилизацией в качестве модели оценки стабильности лекарственной субстанции. (A) Оценка корреляционной функции, записанной в экспериментах DLS, с использованием подбора регуляризации с помощью программного обеспечения DynaPro DLS исходного аденовирусного раствора в качестве контроля; (B) оценка корреляционной функции препарата аденовирусного вектора непосредственно после смешивания с композицией 1 путем разбавления; и (C) оценка корреляционной функции препарата аденовирусного вектора непосредственно после смешивания с композицией 2 путем разбавления. Рассчитанные гидродинамические радиусы препаратов аденовирусного вектора в композициях 1 и 2 соответствуют измеренным радиусам аденовирусных частиц в необработанном исходном растворе и значениям, известным из литературы.
Фигура 2: Определение посредством динамического рассеяния света (DLS) гидродинамических радиусов композиций аденовирусного вектора перед лиофилизацией в качестве модели оценки стабильности лекарственной субстанции. (A) Оценка корреляционной функции, записанной в экспериментах DLS, с использованием подбора регуляризации с помощью программного обеспечения DynaPro DLS препарата аденовирусного вектора непосредственно после смешивания с оригинальным составом поставщика; и (B) оценка корреляционной функции препарата аденовирусного вектора непосредственно после смешивания с PBS. В отличие от предыдущей фигуры, гидродинамические радиусы аденовирусных частиц после смешивания с оригинальным составом поставщика и с PBS увеличиваются по сравнению с необработанным исходным раствором.
Фигура 3: Инфекционность in vitro аденовирусных векторов после лиофилизации в различных композициях в качестве модели оценки стабильности лекарственной субстанции. Препараты аденовирусного вектора готовили путем разведения и затем лиофилизировали в композициях 1 и 2. После восстановления лиофилизированных векторов проводили анализ инфекционности in vitro в клетках НЕК 293 с использованием колориметрической детекции аденовирусного гексонового белка на основе антител, указывающей на успешную амплификацию аденовируса в инфицированных клетках. Наблюдалось полное сохранение инфекционных титров препаратов аденовирусного вектора, полученных в композициях 1 и 2 (количество инфекционных единиц на мл по сравнению с положительным контролем; изображено пунктирной линией). Напротив, лиофилизация аденовирусных векторов, разведенных в оригинальном составе поставщика, привела к значительной потере инфекционных титров, а лиофилизация аденовирусных векторов, разведенных в PBS, привела к полной потере соответствующих инфекционных титров.
Фигура 4: Определение посредством динамического рассеяния света (DLS) гидродинамических радиусов аденовирусных частиц в соответствующих аденовирусных векторных препаратах после лиофилизации в качестве модели оценки стабильности лекарственной субстанции. (A) Оценка корреляционной функции, записанной в эксперименте DLS, с использованием подбора регуляризации с помощью программного обеспечения DynaPro DLS препарата аденовирусного вектора после лиофилизации в композиции 1; (B) оценка препарата аденовирусного вектора после лиофилизации в композиции 2. Рассчитанные гидродинамические радиусы препаратов аденовирусного вектора в композициях 1 и 2 соответствуют измеренным радиусам аденовирусных частиц в необработанном исходном растворе (фиг.1А) и значениям, известным из литературы.
Фигура 5: Определение посредством динамического рассеяния света (DLS) гидродинамических радиусов аденовирусных частиц в соответствующих аденовирусных векторных препаратах после лиофилизации с помощью оценочной модели стабильности лекарственной субстанции. (A) Оценка корреляционной функции, записанной в эксперименте DLS, с использованием подбора регуляризации с помощью программного обеспечения DynaPro DLS препарата аденовирусного вектора непосредственно после лиофилизации в оригинальном составе поставщика; и (B) оценка корреляционной функции препарата аденовирусного вектора непосредственно после лиофилизации в PBS. В отличие от предыдущей фигуры, гидродинамические радиусы аденовирусных частиц после лиофилизации в оригинальном составе поставщика и в PBS увеличиваются, что связано с образованием агрегатов более высокого порядка по сравнению с необработанным исходным раствором (фиг.1А).
Фигура 6: Инфекционность in vitro аденовирусных векторов после лиофильной сушки в различных составах и последующего хранения высушенных составов при повышенных температурах в качестве модели оценки стабильности лекарственной субстанции. t=0 день (черные столбцы слева) показывает инфекционность in vitro непосредственно после лиофилизации и восстановления перед хранением. Пунктирная линия показывает соответствующий инфекционный титр необработанного положительного контроля. (A) Инфекционность in vitro композиций аденовирусного вектора после замены буфера путем разбавления в композициях 1 и 2 и последующего хранения лиофилизированных составов в течение 21 дня (набор столбцов в середине) и 42 дней (набор столбцов справа) при 25°С и остаточной влажности 60% по сравнению с оригинальным буфером поставщика и PBS. (B) Инфекционность in vitro композиций аденовирусного вектора после замены буфера путем разбавления в композициях 1 и 2 и последующего хранения лиофилизированных составов в течение 7 дней (набор столбцов посередине) и 28 дней (набор столбцов справа) при 40°C и остаточной влажности 75% по сравнению с оригинальным буфером поставщика и PBS. Полное сохранение аденовирусной инфекционности наблюдалось в образцах, приготовленных в композициях 1 и 2, тогда как хранение в оригинальном буфере поставщика или в PBS приводило к полной потере аденовирусной инфекционности.
Фигура 7: Определение посредством динамического рассеяния света (DLS) гидродинамических радиусов аденовирусных частиц в соответствующих препаратах аденовирусного вектора после лиофилизации и последующего хранения в течение 14 дней при 40°C в качестве модели оценки стабильности лекарственной субстанции. (A) Оценка корреляционной функции, записанной в эксперименте DLS, с использованием подбора регуляризации с помощью программного обеспечения DynaPro DLS препарата аденовирусного вектора после хранения высушенных составов в течение 14 дней при 40°C в композиции 1; и (B) оценка препарата аденовирусного вектора после хранения высушенных составов в течение 14 дней при 40°С в композиции 2. Рассчитанные гидродинамические радиусы препаратов аденовирусного вектора в композициях 1 и 2 соответствуют измеренным радиусам частиц аденовируса в необработанном исходном растворе (Фигура 1 А) и значениям, известным из литературы.
Фигура 8: Определение посредством динамического рассеяния света (DLS) гидродинамических радиусов аденовирусных частиц в соответствующих препаратах аденовирусного вектора после лиофилизации и последующего хранения в течение 14 дней при 40°C в качестве модели оценки стабильности лекарственной субстанции. (A) Оценка корреляционной функции, записанной в эксперименте DLS, с использованием подбора регуляризации с помощью программного обеспечения DynaPro DLS для приготовления аденовирусного вектора после лиофилизации и последующего хранения в течение 14 дней при 40°C в оригинальном составе поставщика; и (B) оценка корреляционной функции препарата аденовирусного вектора после лиофилизации и последующего хранения в течение 14 дней при 40°C в PBS. В отличие от предыдущей фигуры, гидродинамические радиусы аденовирусных частиц после лиофилизации и последующего хранения при повышенной температуре в оригинальном составе поставщика и в PBS увеличены по сравнению с необработанным исходным раствором, что связано с образованием агрегатов более высокого порядка.
Фигура 9: Инфекционность in vitro препаратов аденовирусного вектора после получения в композициях 1 и 2, стабилизирующих лекарственную субстанцию, полученных на этапе 1 процесса или этапе 2 процесса, в качестве модели оценки стабильности лекарственной субстанции. В аденовирусных препаратах проводили замену буфера путем диализа в композициях 1 и 2, соответственно, либо непосредственно после этапа очистки ультрацентрифугированием в градиенте плотности CsCl (этап процесса 1), либо позже в процессе приготовления (этап процесса 2). На этапе 1 процесса наблюдалось полное сохранение инфекционного титра после диализа в обеих композициях по сравнению с положительным контролем (изображено пунктирной линией). Напротив, диализ во время этапа 2 процесса приводил к заметной потере инфекционных титров почти на два log-уровня при проведении в композиции 2, тогда как диализ в композиции 1 приводил к полному сохранению инфекционного титра, аналогично результатам, полученным для этапа 1 процесса.
Фигура 10: DLS определение гидродинамических радиусов аденовирусных частиц в соответствующих аденовирусных векторных препаратах лекарственной субстанции в стабилизирующих композициях 1 и 2 во время либо этапа 1 процесса, либо этапа 2 процесса в качестве модели оценки стабильности лекарственной субстанции. Ребуферизация препаратов частиц аденовирусного вектора в композиции 1 с использованием диализа на этапе 1 или 2 процесса привела к сохранению гидродинамических радиусов частиц (А) и (С). Ребуферизация аденовирусных частиц на композицию 2 во время приготовления на этапе процесса 1 привела к полному сохранению гидродинамического радиуса аденовирусного вектора (B). Напротив, ребуферизация аденовирусных частиц на композицию 2 во время приготовления на этапе 2 процесса привела к увеличению гидродинамического радиуса частиц и к связанному с ним образованию крупных агрегатов (D).
Фигура 11: Инфекционность in vitro препаратов аденовирусного вектора после многократного применения циклов замораживания и размораживания в качестве модели оценки стабильности лекарственной субстанции. (A) Ребуферизация препаратов аденовирусного вектора путем диализа во время приготовления на этапе 1 процесса. (B) Ребуферизация препаратов аденовирусного вектора путем диализа во время приготовления на этапе 2 процесса. В обеих процедурах получения (этап 1 и 2 процесса) ребуферизация композиции 1 привела к полному сохранению инфекционности непосредственно после диализа (начальный титр) и после применения 5 и 10 циклов замораживания и размораживания (А) и (В) по сравнению с положительным контролем, изображенным в виде пунктирной линии. Ребуферизация композиции 2 во время более раннего этапа процесса приготовления (этап 1 процесса) также приводила к полному сохранению инфекционности непосредственно после диализа (начальный титр; A, левый набор столбцов) и незначительной потере инфекционного титра после применения повторных циклов замораживания и размораживания (A). Напротив, ребуферизация композиции 2 во время приготовления на этапе процесса 2 привела к значительному снижению инфекционного титра уже непосредственно после диализа (B; левый набор столбцов). Дальнейшее применение повторяющихся циклов замораживания и замораживания привело к дальнейшему значительному снижению инфекционного титра (B; средний и правый набор столбцов).
Фигура 12: Динамическое рассеяние света (DLS). Определение гидродинамических радиусов аденовирусных частиц в стабилизирующих композициях 1 после применения пяти или десяти циклов замораживания и размораживания в качестве модели оценки стабильности лекарственной субстанции. Ребуферизация препаратов аденовирусных векторных частиц в композиции 1 с использованием диализа на этапе 2 процесса привела к сохранению гидродинамических радиусов частиц (A) после применения пяти циклов замораживания и размораживания; и (B) после применения десяти циклов замораживания и размораживания.
Фигура 13: Динамическое рассеяние света (DLS). Определение гидродинамических радиусов аденовирусных частиц в стабилизирующих композициях 1 и 2 на этапе 1 или этапе 2 процесса после применения пяти циклов замораживания и размораживания в качестве модели оценки стабильности лекарственной субстанции. Ребуферизация препаратов частиц аденовирусного вектора в композиции 1 с использованием диализа на этапе 1 или 2 процесса привела к сохранению гидродинамических радиусов частиц после применения пяти циклов замораживания и размораживания (A) и (B). Напротив, ребуферизация аденовирусных частиц в композиции 2 во время приготовления на этапе 1 или 2 процесса привела к увеличению гидродинамических радиусов частиц и соответствующему образованию агрегатов более высокого порядка после применения пяти циклов замораживания и размораживания (С) и (D).
Фигура 14: Полученные посредством эксклюзионной ВЭЖХ хроматограммы низкоконцентрированных композиций трастузумаба после ребуферизации в качестве модели процесса производства лекарственного продукта из лекарственной субстанции. Лиофилизированные препараты трастузумаба (Герцептин®) восстанавливали и затем подвергали ребуферизации путем диализа либо в композиции оригинального состава поставщика лиофилизированного продукта, либо в композициях по изобретению Her_1 или Her_9 (A) и Her_1 или Her_2 (В), соответственно (трастузумаб - 25 мг/мл). Основной пик при времени элюции приблизительно 16 мин соответствует структурно интактным молекулам мономеров антитела, небольшой пик, элюирующийся раньше при времени элюирования 14 мин, соответствует агрегатам, в частности димерам, а более ранние пики в оригинальном составе поставщика (черная линия) на 11 мин представлены агрегатами более высокого порядка. Ребуферизация на оригинальный состав поставщика привела к увеличению образования агрегатов в форме димеров и даже агрегатов более высокого порядка. Напротив, ребуферизации на композиции по изобретению приводила к сопоставимому количеству агрегатов по сравнению с трастузумабом в качестве стандарта, и к четкому разделению на базовой линии между пиком димера и пиком мономеров антитела.
Фигура 15: Полученные посредством эксклюзионной ВЭЖХ хроматограммы низкоконцентрированных композиций трастузумаба после ребуферизации в качестве модели для процесса производства лекарственного продукта из лекарственной субстанции. Лиофилизированные препараты трастузумаба (Герцептин®) восстанавливали и затем подвергали ребуферизации путем диализа в композиции жидкого оригинального состава поставщика. Аликвоты восстановленного лиофилизированного трастузумаба (Герцептин®), хранящегося при -80°С, подвергали ребуферизации путем диализа в композициях по изобретению 11_1 и 11_1+трегалоза, соответственно (трастузумаб - 20 мг/мл). Основной пик при времени элюции приблизительно 17 мин соответствует структурно интактным молекулам мономеров антитела, небольшой пик, элюирующийся раньше при времени элюирования 14,5 мин, соответствует агрегатам, в частности димерам. Применение жидкого оригинального состава обычно уменьшало склонность к агрегации антитела во время процедуры приготовления с использованием диализа, по сравнению с оригинальным составом поставщика лиофилизированного продукта (Пример 3; Фигура 14). Но замена буфера антитела на композиции 11_1 и 11_1+трегалоза приводила к дальнейшему небольшому снижению образования агрегатов.
Фигура 16: Профили, полученные посредством эксклюзионной ВЭЖХ высококонцентрированных композиций трастузумаба после ребуферизации в качестве модели для процесса производства лекарственного продукта из лекарственной субстанции. Профили, полученные путем эксклюзионной ВЭЖХ необработанных образцов жидких препаратов трастузумаба (Герцептин®; трастузумаб - 120 мг/мл) непосредственно из исходного контейнера, анализировали по сравнению с образцами после ребуферизации жидкой оригинальной композиции трастузумаба на композиции 3 и 4, и были получены сопоставимые профили пиков. Основной пик при времени элюции около 16,5 мин соответствует структурно интактным молекулам мономеров антитела, небольшой пик, элюирующийся раньше при времени элюирования 14 мин, соответствует агрегатам, в частности димерам. Следы фрагментов элюируются при времени элюции 20 мин. Полученные профили эксклюзионной ВЭЖХ для антитела в композициях 3 и 4 полностью сопоставимы с соответствующими хроматограммами антитела в необработанном оригинальном препарате поставщика.
Фигура 17: Профили эксклюзионной ВЭЖХ для высококонцентрированных композиций трастузумаба после ребуферизации и/или концентрирования в качестве модели для процесса производства лекарственного продукта из лекарственной субстанции. Концентрирование жидкой коммерческой композиции трастузумаба (Герцептин®; трастузумаб - 120 мг/мл) до концентрации 200 мг/мл приводило к значительному увеличению образования агрегатов по сравнению с необработанным исходным материалом, как видно из выраженного плеча элюции при времени элюции 15 мин перед элюированием основного пика при 16,5 мин. Напротив, ребуферизация жидкой композиции трастузумаба на любую из трех композиций по изобретению (композиция 4_3, 4_4 или 4_5) и последующее концентрирование до 200 мг/мл привели к полному сохранению хроматографического профиля необработанного исходного жидкого состава трастузумаба (фиг.8) и четкому исходному разделению между пиком через 13 мин, соответствующим димерам антитела и пиком мономеров при времени элюирования 16 мин. Следы фрагментов элюируются при времени элюции 20 мин.
Фигура 18: Профили, полученные путем эксклюзионной ВЭЖХ низкоконцентрированных композиций трастузумаба в момент времени t=0 непосредственно после диализа и после последующего хранения в течение 21 и 28 дней при 45°C в качестве модели оценки стабильности лекарственного продукта. Хранение низкоконцентрированной, жидкой терапевтической композиции антител (трастузумаб - 25 мг/мл) после ребуферизации с использованием диализа на (A) оригинальный состав поставщика, (B) композицию Her_1, (C) композицию Her_2 и (D) композицию Her_9. Хранение в оригинальном составе поставщика приводило к повышенному образованию агрегатов (элюирование через 13 минут) и фрагментов (элюирование через>20 минут), по сравнению с хранением антитела в композициях согласно изобретению.
Фигура 19: Профили эксклюзионной ВЭЖХ высококонцентрированных композиций трастузумаба в момент времени t=0 и после хранения жидкости при повышенных температурах в указанные аналитические моменты времени в качестве модели оценки стабильности лекарственного продукта. Хранение в жидкости высококонцентрированного трастузумаба - 120 мг/мл в течение 1,5 дней при 40°C (A), в течение 12 дней при 40°C (B) или в течение 21 дня при 30°C (C) после ребуферизации с использованием диализа на стабилизирующие композиции 3 и 4, по сравнению с параллельным хранением жидкости необработанного оригинального состава поставщика (Герцептин®; трастузумаб - 120 мг/мл). Хранение в композициях согласно изобретению снижало склонность антитела к агрегации (элюирование агрегатов через 14 минут), и в случае композиции 4, дополнительно наблюдалось незначительное уменьшение фрагментации (элюирование фрагментов через 21 минуту).
Фигура 20: Динамическая вязкость высококонцентрированных композиций трастузумаба после ребуферизации в композициях 3 и 4 с использованием диализа по сравнению с необработанной жидким оригинальным составом трастутумаба (Герцептин®; 120 мг/мл) в качестве модели оценки вязкости лекарственного продукта. Ребуферизация жидкой оригинальной композиции трастузумаба (Герцептин®; трастузумаб - 120 мг/мл) в композициях 3 и 4 (трастузумаб - 120 мг/мл) приводила к заметному снижению вязкости по сравнению с измеренной вязкостью высококонцентрированного трастузумаба в необработанном оригинальном жидком составе поставщика, особенно в композиции 4, также в композиции 3, но в незначительной степени.
Фигура 21: Профили эксклюзионной ВЭЖХ высококонцентрированных композиций трастузумаба после хранения жидкости в качестве модели оценки стабильности лекарственного продукта. Лиофилизированные препараты трастузумаба (Герцептин®) восстанавливали подвергали ребуферизации посредством диализа в композиции исходного жидкого состава поставщика и в композициях 3, 4_1 и 4_2, соответственно, и впоследствии концентрировали до трастузумаба - 135 мг/мл в исходных жидких составах поставщика, до трастузумаба - 145 мг/мл в композиции 3, до трастузумаба - 150 мг/мл в композиции 4_1 и до трастузумаба - 151 мг/мл в композиции 4_2. (A) Профили эксклюзионной ВЭЖХ составов трастузумаба после хранения жидкости в течение 8 дней при 40°C и (B) после хранения жидкости в течение одного месяца при 30°C. Хранение трастузумаба в жидкости в течение указанных периодов времени при 30 и 40°С в оригинальном жидком составе поставщика приводило к увеличению образования агрегатов, элюирующихся с временем 14 минут, с образованием плеча между пиком агрегата и пиком мономеров при 16 мин и образованием фрагментов с плечом между пиком мономеров и слегка увеличенным пиком фрагмента, элюирующимся с временем 20 мин. Напротив, соответствующее хранение антитела в течение 8 дней при 40°C и одного месяца при 30°C в композициях 3, 4_1 и 4_2 приводило к снижению агрегации с четким исходным разделением между пиком агрегата и пиком мономеров и уменьшением фрагментации.
Фигура 22: Профили эксклюзионной ВЭЖХ высококонцентрированных композиций трастузумаба после хранения жидкости в качестве модели оценки стабильности лекарственного продукта. Лиофилизированные препараты трастузумаба (Герцептин®) восстанавливали, подвергали ребуферизации посредством диализа вкомпозиции оригинального жидкого состава поставщика и композициях 3, 4_1 и 4_2, соответственно, и впоследствии концентрировали до трастузумаба - 135 мг/мл в оригинальном жидком составе поставщика, до трастузумаба - 145 мг/мл в композиции 3, до трастузумаба - 150 мг/мл в композиции 4_1 и до трастузумаба - 151 мг/мл в композиции 4_2. (A) Профили эксклюзионной ВЭЖХ составов трастузумаба после хранения в жидкости в течение шести месяцев при 25°C и (B) после хранения жидкости в течение шести месяцев при 2-8°C. Хранение антител в жидкости в оригинальном жидком составе поставщика в течение указанных периодов времени при 2-8°С и 25°С привело к повышенному образованию агрегатов, элюирующихся с временем 14 мин в виде димеров и в виде плеча между пиком димера и пиком мономеров на 16 мин, и к усиленному образованию фрагментов, элюирующих в виде плеча между пиком мономеров на 17 мин и 20 мин, особенно в случае 25°С, по сравнению с хранением антитела в жидкости в композициях 3, 4_1 и 4_2. Профили эксклюзионной ВЭЖХ антитела, изготовленного в композициях 3, 4_1 и 4_2, показали четкое разделение на базовой линии между пиками агрегатов и пиками мономеров. В случае хранения жидкости в течение шести месяцев при 2-8°C фрагментация была незначительной во всех составах.
Фигура 23: Профили эксклюзионной ВЭЖХ высококонцентрированных композиций трастузумаба после хранения в жидкости при 40°C в качестве модели для оценки стабильности лекарственного продукта. Концентрирование жидкости, коммерческой жидкой композиции трастузумаба (Герцептин®; трастузумаб - 120 мг/мл) до концентрации 200 мг/мл и ребуферизация жидкой композиции трастузумаба (Герцептин®; трастузумаб - 120 мг/мл) на любую из трех композиций по изобретению (композиция 4_3, 4_4 или 4_5) и последующее концентрирование до 200 мг/мл. (A) Профили эксклюзионной ВЭЖХ после хранения жидкости в течение 3 дней при 40°C; и (B) после хранения жидкости в течение 14 дней при 40°C. Фрагментация была незначительной при хранении антитела в таких высоких концентрациях. В композициях согласно изобретению склонность к агрегации была сильно снижена, по сравнению с оригинальным составом поставщика, и дополнительно наблюдалось четкое разделение на базовой линии между пиком агрегата с временем элюции 14 минут, и пиком мономеров с временем элюции 16 минут.
Фигура 24: Профили эксклюзионной ВЭЖХ высококонцентрированных композиций трастузумаба после хранения жидкости при повышенных температурах в качестве модели оценки стабильности лекарственного продукта. Концентрирование жидкости, коммерческой жидкой композиции трастузумаба (Герцептин®; трастузумаб - 120 мг/мл) до концентрации 200 мг/мл и ребуферизация жидкой композиции трастузумаба (Герцептин®; трастузумаб - 120 мг/мл) на любую из трех композиций по изобретению (композиция 4_3, 4_4 или 4_5) и последующее концентрирование до 200 мг/мл. (A) Профили эксклюзионной ВЭЖХ после хранения жидкости в течение 42 дней при 30°C; и (B) после хранения жидкости в течение 3 месяцев при 25°C. Фрагментация была незначительной при хранении антитела в таких высоких концентрациях. В композициях согласно изобретению склонность к агрегации была сильно снижена по сравнению с оригинальным составом поставщика, а также наблюдалось четкое разделение на базовой линии между пиком агрегата с временем элюции 14 минут, и пиком мономеров с временем элюции 16 минут.
Фигура 25: Динамическая вязкость высококонцентрированных композиций трастузумаба в качестве модели оценки вязкости лекарственного средства. Концентрирование жидкого оригинального состава поставщика трастузумаба (Герцептин®; трастузумаб - 120 мг/мл) до 220 мг/мл, ребуферизация жидкого оригинального состава поставщика трастузумаба (Герцептин®; трастузумаб - 120 мг/мл) посредством диализа, и последующее концентрирование до 220 и 200 мг/мл в композициях 4_3, 4_4 и 4_5. Динамические вязкости высококонцентрированных составов трастузумаба измеряли в двух разных концентрациях: 200 мг/мл (белые столбцы) и 220 мг/мл (серые столбцы); для оригинального состава поставщика измеряли только вязкость препарата антител с концентрацией 220 мг/мл. Динамические вязкости составов антител, соответствующих композициям 4_3 и 4_4 при концентрации антител 220 мг/мл, были заметно снижены по сравнению с динамическими вязкостями оригинального состава поставщика при той же концентрации. В композиции 4_4 измеренная динамическая вязкость немного увеличивалась. Аналогичная тенденция была показана в оцененных динамических вязкостях образцов с концентрацией антител 200 мг/мл.
Фигура 26. Анализ посредством эксклюзионной ВЭЖХ высококонцентрированного трастузумаба во время хранения жидкости. Показаны относительные AUC пиков агрегатов (вверху), пиков мономеров (в центре) и пиков фрагментов (внизу), полученных с помощью эксклюзионной ВЭЖХ. (A) Относительные AUC эксклюзионной ВЭЖХ пиков 120 мг/мл трастузумаба при хранении жидкости в течение 3 месяцев при 30°C в F2-1 и F2-2 по сравнению с оригинальным составом. (B) Относительные AUC эксклюзионной ВЭЖХ пиков 150 мг/л трастузумаба при хранении жидкости в течение 6 месяцев при 25°C в F2-3 и F2-4 по сравнению с оригинальным составом. (C) Относительные AUC эксклюзионной ВЭЖХ пиков 200 мг/мл трастузумаба при хранении жидкости в течение 3 месяцев при 25°C в F2-5, F2-6 и F2-7 по сравнению с оригинальным жидким составом. Оригинал: (120 мг/мл) состав F0 (F0).
Фигура 27. Анализ посредством катионообменной хроматографии (КО-ВЭЖХ) трастузумаба при хранении жидкости. (A) Относительные AUC пиков КО-ВЭЖХ 120 мг/мл трастузумаба при хранении жидкости в течение 3 месяцев при 30°C в F2-1 и F2-2 по сравнению с оригинальным составом. (B) Относительные AUC пиков КО-ВЭЖХ 150 мг/мл трастузумаба при хранении жидкости в течение 6 месяцев при 25°C в F2-3 и F2-4 по сравнению с оригинальным составом. (C) Относительные AUC пиков КО-ВЭЖХ 200 мг/мл трастузумаба при хранении жидкости в течение 3 месяцев при 25°C в F2-5, F2-6 и F2-7, по сравнению с оригинальным жидким составом. Оригинал: (120 мг/мл) состав F0 (F0). (A-C) кислые виды (вверху), виды главных пиков (середина) и основные виды (внизу).
Фигура 28: Профили эксклюзионной ВЭЖХ высококонцентрированных композиций трастузумаба после ребуферизации и последующего концентрирования до концентрации антител 200 мг/мл в качестве модели для процесса производства лекарственного продукта из лекарственной субстанции. Лиофилизированные препараты трастузумаба (Герцептин®) восстанавливали, проводили ребуферизацию посредством диализа в композиции оригинального жидкого состава поставщика и композициях 4_1, 4_3 и 4_5, соответственно, а затем концентрировали до концентраций трастузумаба 200 мг/мл. Концентрирование антитела в исходном жидком составе поставщика приводило к повышенному образованию агрегатов, элюирующихся с временем удерживания 14 минут, по сравнению с концентрацией антитела, приготовленного в композициях 4_1, 4_3 и 4_5.
Фигура 29: Профили эксклюзионной ВЭЖХ высококонцентрированных композиций трастузумаба после хранения жидкости в качестве модели оценки стабильности лекарственного продукта. Лиофилизированные препараты трастузумаба (Герцептин®) восстанавливали, проводили ребуферизацию посредством диализа в композиции оригинального жидкого состава поставщика, и в оригинальном составе поставщика лиофилизированного продукта, где этот состав дополнительно содержал аминокислоты глицин и пролин в концентрациях, описанных в патенте США US 9,364,542 B2 (Пример 16). Для сравнения в восстановленном трастузумабе (Герцептин®) проводили ребуферизацию путем диализа в композициях 4_1, 4_3 и 4_5, соответственно. Все композиции впоследствии концентрировали до концентраций трастузумаба 200 мг/мл. (A) и (B) Профили эксклюзионной ВЭЖХ трастузумаба после хранения жидкости в течение 24 часов при 55°C, при получении в композициях 4_1, 4_3 и 4_5, по сравнению с оригинальным жидким составом поставщика (А), и по сравнению с оригинальными составами поставщика с глицином и пролином (В), соответственно. (C) и (D) Профили эксклюзионной ВЭЖХ трастузумаба после хранения жидкости в течение 14 дней при 40°C, при изготовлении в композициях 4_1, 4_3 и 4_5, по сравнению с оригинальным жидким составом поставщика (C) и по сравнению с оригинальными составами поставщика с глицином и пролином (D), соответственно. (E) и (F) Профили эксклюзионной ВЭЖХ трастузумаба после хранения жидкости в течение 28 дней при 40°C, при изготовлении в композициях 4_1, 4_3 и 4_5, по сравнению с оригинальным жидким составом поставщика (E) и по сравнению с оригинальными составами поставщика с глицином и пролином (F), соответственно. Изображенные хроматограммы эксклюзионной ВЭЖХ после хранения жидкости в течение различных периодов времени при повышенных температурах показали, что композиции 4_1, 4_3 и 4_4 эффективно предотвращали образование агрегатов по сравнению, в частности, с оригинальными составами лиофилизированного продукта с добавками глицина и пролина в соответствии с указанным патентом США.
Фигура 30. Анализ посредством КО-ВЭЖХ высококонцентрированных композиций трастузумаба после получения жидкости в качестве модели оценки стабильности лекарственного продукта. Лиофилизированные препараты трастузумаба (Герцептин®) восстанавливали, проводили ребуферизацию посредством диализа в композиции оригинального жидкого состава поставщика, и в оригинальном составе поставщика лиофилизированного продукта, где этот состав дополнительно содержал аминокислоты глицин и пролин в концентрациях, описанных в патенте США US 9,364,542 B2 (Пример 16). Для сравнения в восстановленном трастузумабе (Герцептин®) проводили ребуферизацию путем диализа в композициях 4_1, 4_3 и 4_5, соответственно. Все составы были впоследствии сконцентрированы до концентрации трастузумаба 200 мг/мл. Курс увеличения относительного процента площадей пиков, соответствующих образованию вариантов с кислым зарядом, показан (A) в указанные моменты времени анализа при хранении жидкости при 55°C, (B) в указанные моменты времени анализа при хранении жидкости при 40°C и (С) в указанные моменты времени анализа при хранении жидкости при 25°С.Изображенный курс образования вариантов кислого заряда молекулы антитела дополнительно подчеркивает стабилизирующую эффективность композиций 4_1, 4_3 и 4_5 против химических изменений в молекуле антитела. В частности, стабилизация против образования вариантов с кислым зарядом предполагает более высокую стабилизирующую эффективность композиций 4_1, 4_3 и 4_5 против дезамидирования, которое, как известно, является основным химическим изменением в молекуле трастузумаба, и вносит основной вклад в величину образования вариантов с кислым зарядом при хранении жидкости при повышенных температурах. Напротив, оригинальный состав поставщика лиофилизированного продукта с аминокислотами глицином и пролином в качестве добавок в соответствии с примером патента США показал значительно более высокую тенденцию к образованию вариантов с кислым зарядом в процессе хранения жидкости при повышенных температурах при высоких концентрациях антител.
Примеры иллюстрируют изобретение.
Пример 1. Исследование функциональной и структурной целостности in vitro лиофилизированных и впоследствии хранившихся аденовирусных векторов показало, что композиции на основе аминокислот и сахара стабилизируют аденовирусные векторы на модели оценки стабильности лекарственной субстанции на ранней стадии производственного процесса.
1.1 Материалы и методы
Композиции 1 и 2 содержали 7 аминокислот аланин, аргинин, глицин, глутаминовую кислоту, лизин, гистидин и триптофан в концентрации, соответствующей сумме аминокислот 40 г/л. Но в композиции 1 5-кратное увеличение концентрации триптофана и 1,67-кратное увеличение концентрации гистидина и глутаминовой кислоты при снижении концентраций других аминокислот аргинина, глицина, лизина и сохранении концентрации аланина по сравнению с композицией 2 приводит к той же концентрации в соответствии с суммой аминокислот 40 г/л. Кроме того, дополнительный сурфактант полисорбат 80 в концентрации 0,05 г/л был добавлен к композиции 1 в отличие от композиции 2. Обе композиции содержали трегалозу в качестве соответствующего сахара при отношении аминокислоты к трегалозе 1:2 (по массе). Значение рН доводили во всех композициях до 7.
Использовали исходный аденовирусный раствор, хранящийся при -80°C с концентрацией 7,5*1010 IFU/мл в оригинальном составе поставщика (Firma Sirion; Мартинсрид/Мюнхен, Германия).
1.1.1 Приготовление образцов и лиофилизация
В исходном растворе аденовирусного вектора проводили ребуферизацию путем разбавления исходного раствора до концентрации 1*108 IFU/мл либо композицией 1, либо композицией 2. Для сравнения исходный раствор разбавляли либо оригинальным составом поставщика, либо PBS до одинаковых концентраций.
Для приготовления образцов для лиофилизации, различные аденовирусные составы аликвотировали в объемах 500 мкл в 2R флаконах для лиофилизации (Schott AG; Майнц; Германия), а затем лиофилизировали с использованием следующих параметров сушки:
После лиофилизации образцы визуально оценивали, и одну часть образцов хранили в течение короткого времени при 2-8°C до анализа исходного инфекционного титра в момент времени t=0. Другую часть образцов хранили в соответствии с рекомендациями Международного совета по гармонизации (ICH) в течение 21 или 42 дней при 25°C в условиях окружающей среды с остаточной влажностью 60% или в течение 7 или 28 дней при 40°C при условиях окружающей среды с остаточной влажностью 75%.
1.1.2 Определение инфекционных титров для аденовирусных векторов в клеточной культуре
Чтобы проанализировать инфекционный титр составов аденовирусного вектора, применяли эксперимент по титрованию вируса на основе антител в культуре клеток HEK 293 с использованием детекции аденовирусного гексонового белка после успешной амплификации аденовируса в инфицированных клетках. Клетки в количестве 2,5*105 НЕК 293 (CCS) (Firma Sirion; Мартинсрид/Мюнхен; Германия) высевали на лунку 24-луночного планшета для микротитрования в объеме 500 мкл. Композиции аденовирусного вектора восстанавливали либо непосредственно после лиофилизации, либо в указанные моменты времени при хранении при 25°С и при 40°С. В качестве положительного контроля использовали аликвоту исходного аденовирусного раствора, хранящегося при температуре -80°C с концентрацией 7,5*1010 IFU/мл в оригинальном составе поставщика (Firma Sirion; Мартинсрид/Мюнхен; Германия). Затем готовили серийные разведения образцов аденовируса, и 50 мкл полученных разведений на лунку использовали для инфицирования клеток. Планшеты инкубировали в течение 42 часов при 37°С.После инфицирования клетки фиксировали метанолом, инкубировали с первичным антителом против гексонового белка (Santa Cruz Biotechnology, Inc.; Даллас; Техас, США), затем инкубировали с вторичным антителом к иммуноглобулину мыши, конъюгированным с пероксидазой хрена (HRP) (Cell Signaling Technology; Дэнвер; Массучусетс, США), специфичным для первичного антитела, и проводили ферментативную реакцию HRP с диаминобензидином (Carl Roth GmbH and Co.KG; Графрат; Германия), где коричневая окраска указывала на инфицированные клетки. Количество инфицированных клеток определяли количественно путем подсчета клеток коричневого цвета под микроскопом, где каждая инфицированная клетка считалась одной инфекционной вирусной частицей.
1.1.3 Измерение динамического рассеяния света (DLS)
DLS проводили на образцах, отобранных до лиофилизации непосредственно после замены буфера, по сравнению с необработанным положительным контролем, соответствующим аликвоте исходного аденовирусного раствора, хранящегося при -80°C, а также на образцах после восстановления составов аденовирусного вектора. В последнем случае DLS проводили либо сразу после лиофилизации (t=0), либо в соответствующие моменты времени при хранении при 25°C (21 день, 42 дня) и при 40°C (7 дней, 28 дней).
Для этого 5 мкл образцов отбирали пипеткой в специальную кювету DLS и анализировали в приборе DynaPro Nanostar DLS (Wyatt Technology Europe GmbH; Дернбах; Германия). Для каждой экспериментальной композиции измерение холостой пробы проводили в тех же самых условиях. Измерения DLS выполняли со временем сбора данных от 20 до 40 секунд в 10 или 20 циклах. Полученные кривые корреляции анализировали с использованием программного обеспечения DynaPro DLS.
1.2 Результаты
Интересно, что оценка корреляционных функций, записанных в экспериментах DLS непосредственно после смешивания препаратов аденовирусного вектора с растворами согласно изобретению (композицией 1 или композицией 2), позволила сделать предположение о полном сохранении гидродинамических радиусов аденовирусных векторов (фиг.1 B и C) по сравнению с необработанными аденовирусными частицами в оригинальном исходном растворе (фиг.1А). Подобное смешивание исходного аденовирусного раствора путем разбавления с оригинальным составом поставщика или с PBS во время процесса приготовления образцов перед лиофилизацией уже привело к значительному увеличению измеренных гидродинамических радиусов аденовирусных векторов (фиг.2A и B) по сравнению с необработанным аденовирусным вектором (фиг.1 А).
Анализ инфекционности in vitro после лиофилизации показал, что состав препаратов аденовирусного вектора в стабилизирующих композициях 1 и 2 уже на ранней фазе уменьшения масштаба и последующей лиофилизации приводил к инфекционным титрам, которые соответствуют титрам положительного контроля, обозначенным пунктирной линией на фиг.1. Таким образом, полное сохранение инфекционных титров наблюдалось после лиофилизации. Напротив, когда аденовирусные векторы со сменой буфера на оригинальный состав поставщика лиофилизировали, наблюдалась заметная потеря инфекционных титров, и лиофилизация в PBS даже приводила к полной потере соответствующих инфекционных титров (фиг.3).
Результаты исследования инфекционности in vitro аденовирусных препаратов после восстановления высушенных продуктов хорошо согласуются с результатами параллельного определения гидродинамических радиусов в экспериментах по динамическому рассеянию света. Комбинация препарата аденовирусного вектора с композицией 1 и 2 согласно настоящему изобретению уже на ранней фазе этапов уменьшения масштаба и последующей лиофилизации привела к полному сохранению гидродинамических радиусов вирусных частиц (пример 1; фиг.4 A и B). Напротив, лиофилизация соответствующих препаратов аденовирусного вектора в оригинальном составе поставщика приводила к увеличению размеров частиц (пример 1; фиг.5А). Подобная процедура приготовления образца в сочетании с обычным фосфатно-солевым буферным раствором (PBS) привела к значительному увеличению размера частиц (пример 1, фиг.5B) и образованию значительных количеств агрегатов более высокого порядка уже после лиофилизации.
Эти различия были еще более значительными после хранения лиофилизированных препаратов. Наблюдалась полная потеря функции вирусных векторов, лиофилизированных в оригинальном составе поставщика (фиг.6), аналогично результатам, полученным в PBS. Напротив, даже после хранения при 25°С или даже при 40°С лиофилизированные аденовирусные векторные композиции, которые были приготовлены в стабилизирующих композициях 1 и 2 на ранних стадиях процесса получения, сохраняли почти ту же вирусную активность, что и положительный контроль, т.е. аденовирусный вектор перед лиофилизацией (изображен в виде пунктирной линии на диаграмме на фиг.6.
Эти результаты экспериментов по инфекционности in vitro хорошо соответствуют экспериментам DLS, проводимым параллельно. В качестве примеров, оценка записанной корреляционной функции DLS после хранения высушенных аденовирусных векторных композиций либо в композиции 1 и 2 согласно изобретению, либо в оригинальном составе поставщика и PBS, соответственно, после хранения в течение 14 дней при 40°C, изображена на фиг.7 и 8. Хранение высушенных препаратов аденовирусных векторов в стабилизирующей композиции 1 и 2 привело к сохранению определенных гидродинамических радиусов аденовирусных частиц (пример 1; фиг.7А и В), в отличие от аденовирусных частиц, хранившихся в оригинальном составе поставщика и в PBS (пример 1; рис.8 A и 8 B).
Пример 2. Исследование in vitro функциональной и структурной целостности различных препаратов аденовирусных векторов после замораживания и размораживания показало, что конкретные аминокислотные композиции демонстрируют превосходную стабилизирующую эффективность, по сравнению с другими, на модели для оценки стабильности лекарственной субстанции на ранней фазе производственного процесса.
2.1 Материалы и методы
2.1.1 Приготовление образцов и дальнейшая обработка
Высокие титры исходных препаратов аденовирусных векторов типа 5, содержащих кодирующую ДНК для белка eGFP 5*108 клеток НЕК293, трансдуцировали аденовирусными частицами. Через 48 часа после трансдукции клетки собирали, и высвобождение вирусных частиц осуществляли путем обработки Na-дезоксихолатом и ДНК-азой I. Вирусные частицы очищали ультрацентрифугированием в градиенте CsCl, обычно с последующим заменой буфера на оригинальный состав поставщика на колонках PD10 и последующим определением инфекционного титра. Полученные запасы аденовируса с высоким титром затем разделяли на аликвоты и хранили при -80°С.
Приготовление образца - этап процесса 1. Композиции аденовирусного вектора готовили путем ребуферизации препаратов аденовирусного вектора сразу после ультрацентрифугирования в градиенте CsCl. Полученную аденовирусную векторную полосу собирали и диализовали при 2-8°С в композиции 1 или 2 (как описано в 1.1). Полученные составы разделяли на аликвоты и хранили при -80°С.
Подготовка образца - этап 2 процесса: замороженные (-80°C) исходные аденовирусные растворы (7,5*1010 IFU/мл; Sirion, Мартинсрид/Мюнхен, Германия) размораживали (комнатная температура; RT) в оригинальном буфере поставщика и затем диализовали при 2-8°С в композициях 1 и 2.
2.1.2. Повторные циклы замораживания и размораживания с образцами аденовирусов, полученных на этапе 1 и этапе 2.
Чтобы проанализировать стабильность препаратов аденовирусного вектора во время последующих стрессовых условий, 50 мкл аденовирусных векторов, приготовленных в композиции 1 или 2, подвергали повторным циклам замораживания (-80°C) и размораживания (RT). Инфекционность in vitro (описанную в 1.1.2) определяли в начальный момент времени t=0 и после 5 и 10 циклов замораживания-размораживания путем титрования вируса в клеточных культурах НЕК 293 (описано в 1.1.2). Параллельно гидродинамические радиусы аденовирусных частиц измеряли с помощью DLS (описано в 1.1.3).
2.2 Результаты
Анализ инфекционности in vitro показал, что композиция 1 полностью сохранила инфекционные титры обоих препаратов аденовирусного вектора с этапа 1 и этапа 2 процесса (фиг.9) по сравнению с положительным контролем (пунктирная линия на фиг.9). Ребуферизация препаратов аденовирусного вектора сразу после этапа ультрацентрифугирования (этап процесса 1) в композиции 2 также полностью сохраняла инфекционность препарата аденовирусного вектора. Интересно, что композиция 2, использованная после этапа 2 процесса, привела к потере приблизительно двух логарифмических уровней исходного титра (Фигура 9).
После дополнительных циклов замораживания и размораживания (пяти и десяти) композиция 1 сохраняла полный инфекционный титр, независимо от этапа производственного процесса и момента ребуферизации (Фиг.11 A и B). Напротив, композиция 2 приводила к удивительно различным эффектам при приготовлении на двух разных этапах процесса 1 и 2. Инфекционные титры образцов в композиции 2, полученных согласно этапу процесса 2, значительно снижались после пяти и даже сильнее после десяти циклов замораживания и размораживания (Фиг.11 B). Когда аденовирусные векторы были приготовлены на более раннем этапе процесса 1 в композиции 2, только небольшая потеря титра наблюдалась после пяти циклов замораживания и размораживания. Десять циклов замораживания и размораживания привели к более сильному уменьшению, но в незначительной степени по сравнению с приготовлением на этапе 2 процесса (фиг.11А).
Параллельно с определением инфекционных титров до и после повторных циклов замораживания и размораживания анализировали гидродинамические радиусы соответствующих аденовирусных частиц с использованием динамического рассеяния света (DLS) (фиг.10, 12 и 13). Ребуферизация препарата аденовирусного вектора непосредственно после этапа очистки с использованием ультрацентрифугирования (этап приготовления 1) привела к полному сохранению гидродинамических радиусов вирусных частиц в обеих композициях (фиг.10 A и B), подтверждая полное сохранение соответствующей инфекционности in vitro (Фиг.9). В случае композиции 1 после замены буфера препарата аденовирусного вектора в соответствии с этапом 2 процесса наблюдалось небольшое увеличение радиусов гидродинамических частиц (фиг.10 C), что соответствует результатам инфекционности, показанным на фиг.9. Напротив, замена буфера препарата аденовирусного вектора на композицию 2, соответствующая этапу обработки 2, привела к значительному увеличению гидродинамического радиуса частиц аденовируса (фиг.10 D), сопровождающемуся образованием агрегатов более высокого порядка, которые могут объяснить потерю функции в тестах на инфекционность in vitro (Фиг.9).
После пяти и десяти повторяющихся циклов замораживания и размораживания изменения гидродинамических радиусов вирусных частиц, особенно в композиции 2, измеряли с помощью DLS. Никакого заметного увеличения не наблюдалось в композиции 1, при приготовлении на этапах 1 и 2 процесса. В качестве примера результаты DLS для размера аденовирусных частиц в композиции 1 после применения пяти и десяти циклов замораживания и размораживания изображены на фиг.12. Когда композицию 2 использовали во время этапа 1 процесса, гидродинамические радиусы уже после пяти циклов замораживания и размораживания были заметно увеличены в связи с образованием агрегатов более высокого порядка, и не могли быть измерены после десяти циклов замораживания и размораживания и при использовании на этапе 2 процесса из-за дальнейшего увеличения радиусов и агрегатов более высокого порядка, которые были вне предела измерения DSL (фиг.13 C и D). Поведение размера частиц аденовируса в композициях 1 и 2, полученных на этапе 1 или 2 процесса после применения пяти циклов замораживания и размораживания, изображено на фиг.13A-D).
Подводя итог, можно сказать, что композиция 1, как правило, демонстрирует превосходную стабилизирующую эффективность для частиц аденовирусного вектора во время обоих примененных ранних этапов получения. Напротив, хотя композиция 2 показала стабилизирующую эффективность при использовании непосредственно после ультрацентрифугирования, сниженная стабилизирующая эффективность наблюдалась при использовании позже в процессе производства по сравнению с композицией 1.
Данные DLS коррелируют с данными по инфекционности in vitro, показанными в примере 1. Это приводит к заключению, что использование специально адаптированных стабилизирующих композиций на основе аминокислот на ранних этапах процесса получения композиций вирусных векторов важно для стабильности во время дальнейших этапов обработки в биофармацевтическом производстве. Кроме того, стабилизация композиций на основе вирусного вектора с точки зрения уменьшения полидисперсности раствора приводит к растворам с высокой инфекционностью in vitro.
Пример 3. Анализ молекулярной целостности низкоконцентрированных терапевтических составов антител во время обработки и хранения жидкости показал, что специфические аминокислотные и сахарные композиции снижали склонность к агрегации, и в частности, фрагментацию антител на модели процесса производства лекарственного продукта из лекарственной субстанции.
3.1 Материалы и методы
Композиции Her_1 и Her_2 содержали 7 аминокислот аланин, аргинин, глицин, глутаминовую кислоту, лизин, гистидин и триптофан в концентрациях согласно сумме аминокислот 30,6 г/л в сочетании с 9,4 г/л трегалозы в случае композиции Her_1 и 9,4 г/л метионина в случае композиции Her_2, в результате чего общая концентрация вспомогательных веществ составляла 40 г/л. Отношение суммы аминокислот к трегалозе составило 3,25:1. Композиция Her_9 содержала только 4 основные аминокислоты: аргинин, глицин, триптофан в более высокой концентрации и гистидин в буферной концентрации в концентрации согласно сумме аминокислот 20 г/л в сочетании с 10 г/л трегалозы и 10 г/л метионина. Соответствующее отношение суммы 4 основных аминокислот к трегалозе составляло 2:1. Отношение IgG к наполнителю (по массе) во всех примерах составляло 1:1,6. Отношение IgG к наполнителю в случае оригинального состава поставщика составляло 1:1. Значение рН доводили во всех композициях до 6.
В качестве модельного белка использовали коммерческий лиофилизированный Герцептин® (Roche; Базель; Швейцария), терапевтическое гуманизированное моноклональное антитело IgG1 (трастузумаб). Восстановление лиофилизированного лекарственного средства в необходимом объеме воды приводило к концентрации антител 25 мг/мл IgG1 в качестве оригинального состава поставщика (24 г/л трегалозы; 0,9 мг/мл гистидинового буфера, приблизительно 5 мМ; 0,1 г/л полисорбата 20; рН 6).
3.1.1 Приготовление образца
После диализа полученного состава при 2-8°С в композиции из оригинального состава поставщика, а также в различных композициях на основе аминокислот Her_1, Her_2 и Her_9 все составы были подвергнуты стерилизующей фильтрации и аликвотированы в стерильные флаконы для хранения жидкости. Образцы хранили при 37°C и 45°C, а агрегацию и фрагментацию белка анализировали перед хранением, непосредственно после приготовления образца и в указанные моменты времени, используя эксклюзионную хроматографию (SEC).
3.1.2 Эксклюзионная хроматография (SEC)
Агрегацию и фрагментацию белка количественно определяли посредством SEC. Анализ был выполнен на системе УВЭЖХ ULTIMate3000 (Thermo Scientific; Дармштадт; Германия), оборудованной детектором УФ-280 нм и колонкой TSK-gel G3000SWXL 7,8 x 300 мм (Tosoh Bioscience, Токио, Япония) при 30°C и скорости потока 0,5 мл/мин. Перед анализом SEC образцы, содержащие антитело 25 мг/мл или более высокие концентрации в соответствии с другими примерами, разбавляли до достижения концентрации 2,5 мг/мл IgG с использованием PBS буфера для разделения, и делили на аликвоты в специальных пробирках для ВЭЖХ. Объем введения составлял 25 мкл. Рабочим буфером для SEC был PBS Дульбекко, рН 7,1 (PAA Laboratories, Пашинг, Австрия). В каждой последовательности использовали стандарты молекулярной массы (BSA, Thermo Scientific; Уоэлтем, Массачусетс, США) и буфер-плацебо. Количественную оценку агрегации и фрагментации в % определяли путем сравнения площади под кривыми пиков мономеров, суммы высокомолекулярных частиц и суммы низкомолекулярных частиц с использованием программного обеспечения Chromeleon 7 Chromatography Data (Thermo Scientific, Германия).
3.2 Результаты
Приготовление образцов
Структура пиков в анализе эксклюзионной хроматографии (SEC) непосредственно после диализа показала увеличение образования агрегатов в исходной композиции до процента площади агрегатного пика около 1,55%, и даже образование агрегатов с более высокой молекулярной массой. Обычное содержание агрегатов в соответствующем стандарте трастузумаба составляло около 0,4%. Процент площади пика мономеров, соответствующего молекулам интактного антитела, одновременно уменьшился до примерно 98,44% в оригинальном составе.
Напротив, замена буфера исходного состава IgG1 с использованием диализа в различных композициях на основе аминокислот Her_1, Her_2 и Her_9, соответственно, приводила к сохранению количества агрегатов примерно 0,4% без образования агрегатов с более высокой молекулярной массой, соответствующих необработанному стандартному составу трастузумаба. Процент площади соответствующего пика мономеров был соответственно сохранен до примерно 99,6% (Фигура 14).
Хранение жидкости при 45°C
Через 21 день хранения антитела в жидкой форме в оригинальном составе поставщика было обнаружено увеличение относительной площади пика агрегата до 1,76%, а через 28 дней образование агрегатов было дополнительно увеличено примерно до 2,53% (для сравнения, в необработанном стандарте трастузумаба из того же исходного раствора содержалось 0,4% агрегатов). Более того, фрагментация увеличилась через 21 день до примерно 1,92%, а через 28 дней примерно до 2,78%. Кинетика агрегации и фрагментации была сходной в течение срока хранения при 45°С. Все композиции на основе аминокислот Her_1, Her_2 и Her_9 снижали степень агрегации и фрагментации антитела во время хранения жидкости при 45°C, однако в разной степени. Композиции, содержащие 7 аминокислот в комбинации с трегалозой и/или метионином (композиции Her_1 и Her_2), заметно снижали агрегацию антитела во время хранения жидкости при 45°C. Например, композиция Her_2, состоящая из 7 аминокислот в сочетании с метионином, снижала образование агрегатов во время хранения в течение 21 дня примерно до 0,56% (по сравнению с 1,76% в исходном составе). Фрагментация с 1,32% была несколько более выраженной после хранения в течение 21 дня при 45°C, но значительно снижена по сравнению с хранением в оригинальном составе (1,92%). После дальнейшего хранения в течение 28 дней агрегация не изменилась (0,52%; по сравнению с 2,53% в оригинальном составе), и фрагменты слегка повышались до 1,49% по сравнению с 2,78% в оригинальном составе. Снижение содержания аминокислот до 4 основных аминокислот аргинина, глицина, триптофана и гистидина в буферной концентрации в сочетании с трегалозой (отношение аминокислот к трегалозе составляло 2:1) и метионином приводило к сопоставимому количеству агрегатов после 21 дня хранения при 45°С, но с пониженной склонностью антитела к образованию фрагментов (1,02%). После хранения в течение 28 дней при 45°C содержание агрегатов было дополнительно снижено до примерно 0,39%, но фрагментация увеличилась до примерно 1,56% (Фигура 18).
Таким образом, уменьшение количества аминокислот с 7 аминокислот до 4 аминокислот, корректировка отношения аминокислот к трегалозе с 3,25:1 (7 аминокислот и трегалоза) до 2:1 (4 аминокислоты и трегалоза) и добавление метионина, действующего в качестве антиоксиданта, уменьшало склонность к агрегации, и в частности, к фрагментации антитела во время хранения при повышенной температуре.
Пример 4. Анализ молекулярной целостности низкоконцентрированных терапевтических составов антител во время обработки и хранения жидкости показал, что специально подобранные аминокислотные и сахарные композиции снижали склонность к агрегации, и в частности, к фрагментации антител на процесса производства лекарственного продукта из лекарственной субстанции.
4.1. Материалы и методы
Композиция 11_1 содержала 6 аминокислот аргинин, глицин, глутаминовую кислоту, триптофан, метионин и бета-аланин в концентрации, соответствующей сумме аминокислот 48 г/л, а в случае композиции 11_1+трегалоза в сочетании с 96 г/л трегалозы. Соотношение аминокислоты к трегалозе в этой композиции составляло 1:2. Значение рН доводили до 6. Отношение антитела к вспомогательным веществам составило 1:2,4 (по массе) без добавления трегалозы и 1: 7,2 после добавления трегалозы. Отношение антитела к вспомогательным веществам составило 1:3,4 в случае оригинального жидкого состава поставщика.
В качестве модельного белка использовали коммерческий лиофилизированный Герцептин® (Roche; Базель; Швейцария), терапевтическое гуманизированное моноклональное антитело IgG1 (трастузумаб). Восстановление лиофилизированного лекарственного средства в необходимом объеме воды приводило к концентрации антител 50 мг/мл IgG1 в оригинальном составе поставщика (48 г/л трегалозы; 1,8 мг/мл гистидинового буфера, приблизительно 10 мМ; 0,2 г/л полисорбата 20; рН 6).
4.1.1 Приготовление образца
Ребуферизация полученного состава с использованием диализа при 2-8°С в композиции оригинального жидкого состава поставщика (79,45 г/л трегалозы; 3,13 г/л гистидинового буфера 20 мМ; 1,49 г/л метионина; 0,4 г/л полисорбата 20; рН 6) и частичное последующее разбавление привели к получению составов в оригинальном жидком составе поставщика с конечными концентрациями антител 25 г/л и 50 г/л.
Параллельно проводили ребуферизацию с использованием диализа в композициях на основе аминокислот 11_1 и 11_1+трегалоза после размораживания аликвот восстановленного лиофилизированного коммерческого Герцептина® при концентрации антител 20 мг/мл, аликвотировали и хранили при -80°C.
Агрегацию и фрагментацию белка анализировали непосредственно после приготовления образца с использованием эксклюзионной хроматографии (SEC).
4.1.2. Эксклюзионная хроматография (SEC)
SEC выполняли в соответствии с пунктом 3.1.2.
4.2 Результаты
Приготовление образца
После диализа в оригинальном жидком составе поставщика образование агрегатов было уменьшено по сравнению с первым примером (0,4% агрегатов). Но после диализа в других композициях на основе аминокислот 11_1 без добавления и с добавлением трегалозы при отношении аминокислоты к трегалозе 1:2 это привело к дальнейшему снижению агрегации примерно до 0,3% (фигура 15).
Хранение жидкости
В течение всего примера добавление трегалозы к составу на основе аминокислот (композиция 11_1) при отношении аминокислот к трегалозе 1: 2 привело к значительному уменьшению относительной площади фрагментов при хранении при 37°С по сравнению с соответствующей композицией без трегалозы, и к слегка сниженной склонности к агрегации в присутствии трегалозы (данные не показаны).
Таким образом, последние два примера уже показали, что комбинация аминокислот с трегалозой, очевидно, является предпосылкой для снижения образования агрегатов, и в частности, снижения фрагментации антитела при хранении жидкости при повышенных температурах.
Воздействие отношения антитело: вспомогательное вещество на агрегацию и фрагментацию антитела уже наблюдалось в ходе этого эксперимента по хранению с низкоконцентрированными жидкими композициями трастузумаба. При концентрации IgG 50 мг/мл и составе в композиции на основе аминокислот, содержащей 4 основных аминокислоты в сочетании с трегалозой и антиоксидантом в отношениях, сопоставимых с композицией Her_9 в предыдущем эксперименте, и в концентрациях 25 г/л, 50 г/л и 75 г/л было продемонстрировано уменьшение образования агрегатов с увеличением концентрации вспомогательного вещества, и увеличение фрагментации с увеличением концентрации вспомогательного вещества после хранения в течение 42 дней при 37°С.Наивысшее отношение антитела к вспомогательному веществу 2:1 приводило к наименьшему образованию фрагментов и наивысшему образованию агрегатов (данные не показаны).
Вместе эти результаты примеров 3 и 4 показывают, что уменьшение числа аминокислот с 7 аминокислот до 4 основных аминокислот в сочетании со сбалансированным отношением к трегалозе, добавлением антиоксиданта, например, метионина и сбалансированным отношение антител к вспомогательным веществам оказывает влияние как на агрегацию, так и на фрагментацию антитела в ходе хранения жидкости при повышенной температуре.
Пример 5: Анализ молекулярной целостности и вязкости высококонцентрированных терапевтических составов антител во время производства и хранения жидкости показал, что специфические аминокислотные и сахарные композиции снижали склонность к агрегации, и в частности, к фрагментации антител на модели процесса производства лекарственного продукта из лекарственной субстанции.
5.1 Материалы и методы
Композиции 3 и 4 содержали 4 основные аминокислоты аргинин, глицин, триптофан и гистидин в концентрации, соответствующей сумме аминокислот до 50 г/л. В случае композиции 3 аминокислотная композиция была в комбинации с 80 г/л трегалозы, а в случае композиции 4 в комбинации с 32,2 г/л трегалозы. В качестве дополнительных соединений композиции 3 и 4 содержали 1,5 г/л метионина и 0,4 г/л полисорбата 20. Композиция 4 содержала два дополнительных соединения, хелатирующий агент ЭДТА и антиоксидант аскорбиновую кислоту. Полученная сумма вспомогательных веществ составила 131,5 г/л в случае композиции 3 и 85 г/л в случае композиции 4. Отношение суммы основных аминокислот к трегалозе в композиции 3 составило 1:1,55 (по массе) с избытком трегалозы, тогда как в композиции 4 отношение основных аминокислот к трегалозе составляло 1,6:1 (по массе) с избытком аминокислот.Отношение антитела к сумме вспомогательных веществ составило 1:0,9 (по массе) в композиции 3 и 1:1,4 (по массе) в композиции 4. Значение рН доводили до 5,5.
В качестве модельного белка использовали коммерчески доступное жидкое терапевтическое высококонцентрированное антитело Герцептин® (Roche; Базель; Швейцария), содержащее трастузумаб в концентрации 120 мг/мл в оригинальном составе поставщика (79,45 г/л трегалозы; 3,13 г/л гистидинового буфера, 20 мМ; 1,49 г/л метионина; 0,4 г/л полисорбата 20; 0,024 г/л rHuPh20 (рекомбинантная человеческая гиалуронидаза), рН 5,5).
5.1.1 Приготовление образца
Образцы необработанного препарата антител в исходном оригинальном жидком составе поставщика были непосредственно разделены на аликвоты в стерильные флаконы для ВЭЖХ из исходного контейнера для хранения при 25°С, 30°С и 40°С. В другой части оригинального жидкого состава поставщика трастузумаба с концентрацией антител 120 мг/мл проводили ребуферизацию с использованием диализа при 2-8°C на композиции согласно изобретению. Полученные составы подвергали стерилизующей фильтрации, разделяли на аликвоты в стерильных флаконах для ВЭЖХ и хранили при 25°С, 30°С и 40°С.Агрегацию и фрагментацию перед хранением, непосредственно после подготовки образца и в указанные моменты времени при хранении анализировали с использованием SEC.
5.1.2. Эксклюзионная хроматография (SEC)
SEC выполняли в соответствии с пунктом 3.1.2.
5.1.3 Вискозиметрия
После приготовления образца в соответствии с пунктом 5.1.1 вязкости высококонцентрированных составов антител на основе аминокислотных композиций 3 и 4 по сравнению с вязкостью необработанного жидкой оригинального состава поставщика определяли с использованием вискозиметра с падающим шариком (Anton Paar GmbH; Ostfildern-Schamhausen; Германия). После определения плотности высококонцентрированного образца белка (120 мг/мл) и калибровки капилляра водой при 20°C с использованием угла падения 70° шарик вводили в капилляр, и приблизительно 500 мкл составов антител осторожно вводили в капилляр. Заполненный капилляр вставляли в капиллярный блок прибора, и образцы измеряли в виде десяти отдельных анализов при 20°C и угле падения 70°.
5.2 Результаты
Приготовление образцов
Профиль SEC необработанного жидкого состава трастузумаба из исходного контейнера показал только небольшой пик агрегатов 0,19%, пик мономеров 99,77% и пик фрагментов 0,04%. После ребуферизации этого состава в композиции на основе аминокислот 3 и 4 согласно пункту 5.1 были выявлены сопоставимые профили SEC, что свидетельствует о стабилизирующем воздействии композиций на основе аминокислот на антитело во время процесса ребуферизации (фиг.16), которое было более выражено во время последующего хранения при повышенных температурах (фиг.19 и следующий параграф).
Хранение жидкости
Уже после 1,5 дней хранения при 40°C в исходном необработанном жидком составе поставщика было определено увеличение образования агрегатов (0,22%) и было обнаружено небольшое увеличение фрагментации (0,09%). Пик мономеров слегка уменьшился до 99,69%. Напротив, хранение в течение 1,5 дней при 40°C антитела в двух разных композициях на основе аминокислот 3 и 4 в соответствии с пунктом 5.1 показало сниженную склонность антитела к агрегации и в незначительной степени к фрагментации (фиг.19A). В композиции 3 избыток трегалозы по сравнению с аминокислотами с отношением аминокислоты к трегалозе 1:1,55 (по массе) приводил к содержанию агрегатов после хранения в течение 1,5 дней при 40°С с фрагментами примерно 0,16% и 0,06%, Хранение в течение 1,5 дней при 40°С в композиции 4, содержащей отношение аминокислот к трегалозе 1,6:1 с избытком аминокислот, привело к образованию 0,16% агрегатов и 0,05% фрагментов. После хранения в течение 12 дней при 40°С в оригинальном жидком составе поставщика агрегаты и фрагменты еще больше увеличились (0,26% агрегатов и 0,27% фрагментов). В композиции 3 агрегаты увеличились после хранения в течение 12 дней при 40°С только до 0,20%, а фрагменты - до 0,29%. В композиции 4 содержание агрегатов составляло только 0,18%, а содержание фрагментов увеличивалось только до 0,20% (фиг.19 B). После хранения в течение 21 дня при 30°C содержание агрегатов в оригинальном составе поставщика также увеличилось до 0,26%, а фрагментация составила 0,13%. Хранение в течение 21 дня при 30°C в композиции 3, а также в композиции 4 показало сниженную склонность к агрегации антитела (0,18% в композиции 3 и 0,16% в композиции 4). Увеличение фрагментации антитела в композиции 3 (0,15%) было сопоставимо с исходной композицией, а в композиции 4 содержание фрагментов уменьшилось до 0,09% (фиг.19 C). Данные после более длительных периодов хранения при 40°С подтвердили эти наблюдения. После хранения в течение 42 дней при 40°С в оригинальном составе агрегация увеличилась до 0,53%, а фрагментация до 0,8%. Напротив, в композиции 3 и композиции 4 содержание агрегатов после этого периода хранения составляло 0,37% и 0,36%. Далее, фрагментация составила всего 0,72% и 0,66%, соответственно.
Эти результаты позволяют предположить, что составы на основе аминокислот имеют более сильную стабилизирующую эффективность, чем оригинальный состав, при хранении при 40°С и 30°С как против образования агрегатов, так и, в частности, в композиции 4 против образования фрагментов. Результаты также показывают, что, в частности, композиция 4 с избытком аминокислот по сравнению с трегалозой показала лучшую стабилизацию против агрегации и фрагментации по сравнению с композицией 3. Это наблюдение было подтверждено после хранения в течение 3 месяцев при 30°C. В оригинальном составе содержание агрегатов составляло 0,35%, тогда как в композиции 3 содержание агрегатов составило 0,26%, а в композиции 4 содержание агрегатов составило 0,20%. Фрагментация в оригинальном составе составила 0,40%, в композиции 3 - 0,46%, а в композиции 4 - только 0,33%. Количественный статистический анализ курса хранения жидкости высококонцентрированных составов антител (120 мг/мл) в условиях ускоренного старения в течение 3 месяцев при 30°C дополнительно подтвердил вышеуказанные выводы. Ускоренное старение (фиг.26) в оригинальном жидкой составе продемонстрировало значительно (p<0,01) более высокую степень агрегации по сравнению с композицией 3 и композицией 4 (фиг.26А), что соответствует вязкости композиции (см. ниже и фиг.26). Напротив, образование фрагментов во время хранения было сходным между оригинальным составом и композицией 3, но заметно уменьшилось (р<0,05) в композиции 4 (фиг.26А). Связанное с этим уменьшение пика мономеров было ограничено в композиции 3 (р<0,01), и было еще меньше (р<0,01) в композиции 4 (фиг.26А).
Стабилизирующая эффективность композиции 4 с отношением аминокислот к трегалозе 1,6:1 (по массе) была подтверждена следующими примерами.
Была проанализирована стабилизирующая эффективность дополнительной композиции 1, полученной из композиции 11_1 согласно п.4.1 примера 4, содержащей только аминокислоты аргинин, глицин, глутаминовую кислоту, триптофан, метионин и бета-аланин в сочетании с дипептидом без сахара. В каждый момент времени анализа, как упомянуто выше, агрегация антитела также заметно снижалась, но антитело демонстрировало более сильную склонность к фрагментации по сравнению с композицией 3 и особенно с композицией 4 в соответствии с параграфом 5.1, подтверждая наблюдение, что сбалансированное отношение между аминокислотами и трегалозой приводит к снижению агрегации и фрагментации (данные не показаны).
Кроме того, анализ характера химической деградации при хранении жидкости высококонцентрированных составов антител (120 мг/мл) в условиях ускоренного старения в течение 3 месяцев при 30°C в композициях 3 и 4 по сравнению с исходной композицией с использованием катионообменной ВЭЖХ дополнительно подчеркнул полезный эффект корректировки основных аминокислотных композиций, например, путем добавления сахара при соответствующем отношении, и одного или нескольких антиоксидантов. Как и в случае высококонцентрированного (120 мг/мл) трастузумаба, композиция 3 привела к снижению основных видов (фиг.27; р<0,05). Таким образом, балансировка отношения аминокислота: сахар позволила ограничить основные продукты химической деградации при хранении жидкости высококонцентрированного трастузумаба.
Эти данные дополнительно подтверждают заявленное изобретение в том, что корректировка применяемой композиции основных аминокислот, включающей по меньшей мере три аминокислоты аргинин, глицин, гистидин и/или триптофан, используемых на ранних этапах обработки лекарственной субстанции, в соответствии с требованиями конкретной биомолекулы (например, конечной концентрацией и вязкостью лекарственного продукта) стабилизирует биомолекулу во время дальнейшей обработки, такой как фасовка, лиофилизация, хранение высушенного или жидкого продукта.
Измерение вязкости
Было обнаружено, что измеренные динамические вязкости в высококонцентрированных составах антител на основе аминокислот значительно снижались, по сравнению с соответствующей вязкостью необработанного жидкого оригинального состава поставщика. Динамическая вязкость в композиции 3 составила 4 мПа*с, а в композиции 4 составила 3,5 мПа*с.Напротив, динамическая вязкость в высококонцентрированном жидком оригинальном составе поставщика составила 4,8 мПа*с (фиг.20).
Композиция 4 и оригинальный состав поставщика содержали антитело в приблизительно сопоставимом отношении антитела к вспомогательному веществу 1,4:1 (по массе). Однако в композиции 4 корректировка отношения аминокислоты к трегалозе и сопутствующего отношения соответствующего антитела к вспомогательному веществу привела к влиянию как на стабилизирующую эффективность при хранении жидкости при повышенных температурах, так и на заметное уменьшение вязкости состава по сравнению с оригинальным составом. Уже корректировка отношения аминокислоты к трегалозе в композиции 3 и полученного отношения антитела к вспомогательному веществу привела к повышению эффективности стабилизации и снижению вязкости состава по сравнению с оригинальным составом, но в незначительной степени по сравнению с дальнейшими корректировками, приводящими к эффектам композиции 4.
Таким образом, эти данные дополнительно подтверждают вывод о том, что комбинация аминокислот с трегалозой в сбалансированном отношении и одновременная корректировка отношения антитела к сумме вспомогательных веществ оказывает сильное влияние на стабилизирующую эффективность композиции в отношении агрегации, и в частности, фрагментации антитела при хранении жидкости при повышенных температурах. Кроме того, помимо вышеупомянутой стабилизирующей эффективности корректировка композиций таким способом приводит к значительному снижению вязкости высококонцентрированных композиций терапевтических антител.
Пример 6. Анализ молекулярной целостности высококонцентрированных терапевтических составов антител во время обработки и хранения жидкости показал, что специфические композиции аминокислот и сахаров, а также отношения аминокислот к сахару (по массе) снижают склонность к агрегации, и в частности, к фрагментации антитела на модели стабильности лекарственного продукта.
6.1 Материалы и методы
Композиции 3 и 4_1 являются аналогичными композициями, применяемыми в Примере 5 в соответствии с пунктом 5.1. Но в случае композиции 4_1 доведение рН до 5,5 проводили с использованием HCl вместо лимонной кислоты. Обе композиции содержали сходные отношения суммы аминокислот к трегалозе в соответствии с пунктом 5.1 в Примере 5. Композиция 4_2 была также вариантом композиции 4 в соответствии с пунктом 5.1 Примера 5. Композиция 4_2 содержала 4 основные аминокислоты аргинин, глицин, триптофан и гистидин при добавлении дополнительной аминокислоты аланина. В композиции 4_2 сахарная фракция представляла собой смесь трегалозы и сахарозы в отношении 3:1 (по массе). Количество метионина было слегка увеличено до 3,5 г/л, а также были добавлены дополнительные вспомогательные вещества, например, хелатирующий агент ЭДТА и аскорбиновая кислота.
Отношение аминокислот к сахару было несколько снижено в композиции от 4_2 до 1:1 (по массе). В исходной композиции отношение антитела к вспомогательному веществу составило 1,6:1 (по массе), в композиции 3 - 1:1,1 (по массе); в композиции 4_1 - 1,76:1 (по массе), а в композиции 4_2 - 1,12:1 (по массе). Значение рН доводили до 5,5.
В качестве модельного белка использовали коммерческий лиофилизированный Герцептин® (Roche; Базель; Швейцария) в соответствии с пунктами 3.1 и 4.1. Подготовку образцов проводили путем разведения коммерческого лиофилизированного Герцептина® в необходимом объеме воды.
6.1.1 Приготовление образца
Полученную композицию подвергали диализу при 2-8°C против композиции из оригинального жидкого состава (79,45 г/л трегалозы; 3,13 г/л гистидинового буфера (20 мМ); 1,49 г/л метионина; 0,4 г/л полисорбата 20; рН 5,5) и против композиций на основе аминокислот согласно пункту 6.1. Последующее концентрирование полученных составов IgG проводили с получением 135 мг/мл антитела в исходной композиции, 145 мг/мл антитела в композиции 3, 150 мг/мл антитела в композиции 4_1 и 151 мг/мл антитела в композиции 4_2. Затем составы подвергали стерилизующей фильтрации, разделяли на аликвоты в стерильных флаконах для ВЭЖХ и хранили при 5°С, 25°С, 30°С и 40°С.Агрегацию и фрагментацию перед хранением, непосредственно после подготовки образца и в указанные моменты времени хранения анализировали с использованием SEC.
6.1.2. Эксклюзионная хроматография
SEC выполняли в соответствии с пунктом 3.1.2.
6.2 Результаты
Хранение жидкости
Уже после первоначального времени хранения, равного 8 дням, при 40°С в оригинальном составе образование агрегатов и фрагментов было заметно увеличено (агрегаты 0,77% и фрагменты 0,75% против агрегатов 0,35% и отсутствия фрагментов до хранения жидкости, соответственно). Напротив, при хранении в течение 8 дней при 40°C во всех испытанных составах на основе аминокислот четко ограничивалась агрегация и фрагментация. В композиции 3 агрегация составила 0,39%, а фрагментация - 0,51%. В композиции 4_1 агрегация составила 0,38%, и что интересно, фрагментация была дополнительно снижена до 0,31%. В композиции 4_2 было обнаружено небольшое дальнейшее снижение агрегации и фрагментации (0,36% агрегатов и 0,28% фрагментов), как показано на фиг.21 А.
Эти данные подтвердили результаты предыдущего эксперимента, касающиеся эффективности композиции 4 (композиция 4_1 в этом примере 6) для дальнейшего снижения, в частности, образования фрагментов во время хранения жидкости.
Сопоставимые результаты были получены после хранения в течение 1 месяца при 30°С.Хранение в исходной рецептуре дало 0,53% агрегатов и 0,50% фрагментов. Соответствующий анализ SEC для композиции 3 выявил 0,4% агрегатов и 0,51% фрагментов. Хранение жидкости в течение 1 месяца при 30°C в композиции 4_1 привело к заметному снижению образования агрегатов (0,35%) и образования фрагментов (0,30%). Сопоставимые результаты были показаны в композиции 4_2 с образованием агрегатов 0,32% и образованием фрагментов 0,31% (фиг.21 В).
После хранения жидкости в течение 6 месяцев при 25°C агрегация антитела в исходной композиции увеличивалась до 0,98%, а фрагментация достигала 1,00%. В композиции 3 было показано меньшее увеличение агрегации до 0,71% и фрагментации до 0,9%. В обеих композициях 4_1 и 4_2 была обнаружена только почти половина агрегации и фрагментации по сравнению с исходной композицией; композиция 4_1: 0,58% агрегатов и 0,53% фрагментов, и композиция 4_2: 0,58% агрегатов и 0,56% фрагментов (фиг.22А).
Сопоставимые результаты были получены после хранения жидкости в течение 6 месяцев при 2-8°C с меньшими изменениями в агрегации и фрагментации по сравнению с хранением при 25°C (фиг.22 B).
Количественный статистический анализ всего курса хранения высококонцентрированных составов антител (150 мг/мл) в течение 6 месяцев при 25°С в композициях 4_1 и 4_2 по сравнению с исходным составом выявил значительно уменьшенные агрегаты и фрагменты (р<0,01) и сохранение стабильного пика мономеров во время хранения в течение 6 месяцев при 25°C (фиг.26B).
Дополнительный анализ профиля химической деградации высококонцентрированных составов антител во время хранения в течение шести месяцев при 25°С в композициях 4_1 и 4_2 по сравнению с исходными составами с использованием катионообменной ВЭЖХ подчеркнул предыдущие результаты SEC и результаты предыдущих примеров. После шести месяцев хранения 150 мг/мл трастузумаба при 25°C для композиций 4_1 и 4_2 наблюдались меньшие количества основных видов (р<0,05, р<0,01) по сравнению с оригинальным составом (фиг.27 В). Интересно, что в сочетании с ограниченным увеличением основных видов, те же составы, композиции 4_1 и 4_2 также ограничивали увеличение кислых видов по сравнению с композициями 3 и 4, как показано в примере 5. Как следствие, относительное значение AUC основного пика было стабилизировано при хранении высококонцентрированного трастузумаба, особенно композицией 4_2.
Таким образом, эти результаты подтверждают, что сбалансированная смесь аминокислот и сахара необходима для предотвращения агрегации и фрагментации антител во время хранения жидкости, и что регулирование отношения аминокислот к сахару, по меньшей мере с избытком аминокислот, и более предпочтительно с отношением аминокислоты и сахара примерно 1: 1 (по массе) приводило к дальнейшему повышению эффективности стабилизации.
Пример 7. Анализ молекулярной целостности и вязкости высококонцентрированных терапевтических составов антител во время обработки и хранения в жидком состоянии показал, что конкретные композиции аминокислот и сахаров, а также отношения аминокислот к сахару (по массе), а также корректировка концентраций и отношений триптофана и гистидина (по массе) снижали склонность к агрегации, и в частности, к фрагментации антител на модели оценки стабильности лекарственного продукта.
7.1 Материалы и методы
Композиции 4_3, 4_4 и 4_5 были получены путем концентрирования композиции 4_2 из предыдущего примера. Композиция 4_3 аналогична композиции 4_2 в соответствии с пунктом 6.1 в Примере 6, но сумма вспомогательных веществ была уменьшена с 135 г/л до 90 г/л в композиции 4_3. Отношение смеси аминокислоты и сахара сохранялось до 1:1 (по массе), а отношение антитела к вспомогательному веществу составило 2,22:1 (по массе). В композиции 4_4 использовали аналогичную смесь аминокислот и смеси сахара по сравнению с композицией 4_2 в пункте 6.1 в примере 6 и композицией 4_3 в этом примере 7, но отношение аминокислот и сахара было увеличено до 3,4:1 (по массе), а отношение антитела к вспомогательному веществу было увеличено до 3,33:1 (по массе). В композиции 4_5 также использовали ту же смесь вспомогательных веществ, но концентрация аминокислоты гистидина была увеличена, а концентрация триптофана была снижена. Отношение трегалозы к сахарозе было снижено до 2:1 (по массе) по сравнению с 3:1 (по массе) в предыдущих экспериментах, а отношение аминокислоты к сахару было уменьшено до 1,5:1 (по массе). Отношение антитело/ вспомогательное вещество было сопоставимо с композицией 4_3, доведенной до 2,22:1 (по массе). Отношение антитела к вспомогательному веществу составило 2,4:1 (по массе) в случае оригинального жидкого состава поставщика. Значение рН доводили до 5,5.
В качестве модельного белка использовали коммерческое жидкое терапевтическое высококонцентрированное антитело Герцептин® (Roche; Базель; Швейцария) согласно пункту 5.1 в Примере 5.
7.1.1 Приготовление образца
Чтобы получить более концентрированные препараты трастузумаба по сравнению с примерами 5 и 6, антитело в оригинальном жидком составе поставщика концентрировали до получения 200 мг/мл, а антитело в композициях 4_3, 4_4 и 4_5 концентрировали после дополнительного этапа диализа при 2-8°С. Высококонцентрированные составы готовили для последующего эксперимента по хранению путем стерилизующей фильтрации и последующего разделения на аликвоты в стерильных флаконах для ВЭЖХ. Высококонцентрированные образцы хранили при 5°С, 25°С, 30°С и 40°С.Агрегацию и фрагментацию анализировали перед хранением, непосредственно после подготовки образца и в указанные моменты времени хранения с использованием SEC.
7.1.2. Эксклюзионная хроматография
SEC выполняли в соответствии с пунктом 3.1.2.
7.1.3 Измерение вязкости высококонцентрированных составов антител.
Вязкости высококонцентрированных составов антител в соответствии с этим примером измеряли с использованием вискозиметра с падающим шариком в соответствии с пунктом 5.1.3 в примере 5.
7.2 Результаты
Приготовление образца
Профиль SEC необработанного жидкого состава трастузумаба из исходного контейнера показал только небольшой пик агрегатов 0,19%, пик мономеров 99,77% и пик фрагментов 0,04% (пример 5; фиг.16). Напротив, соответствующий профиль SEC после концентрирования этого коммерческого жидкого терапевтического высококонцентрированного Герцептина® до концентрации антител около 200 мг/мл (0,04 мг/мл rHuPH20) показал заметно другой профиль SEC концентрированного образца. Образование агрегатов было увеличено до относительной площади около 1,9% для пиков, соответствующих агрегатам. Относительная площадь соответствующего пика мономеров была соответственно уменьшена до 98,07%. Фрагментация не изменилась. Сходная концентрация антитела, полученного в стабилизирующих композициях на основе аминокислот после дополнительной стадии замены буфера с использованием диализа, привела к профилям SEC, сравнимым с необработанным оригинальным жидким препаратом трастузумаба с агрегатами от 0,16 до 0,19% и без изменений в фрагментации. Относительная площадь пика мономеров составила около 99,8%. Более того, соответствующие хроматограммы SEC показали четкое разделение на базовой линии между пиком с временем элюирования 14 мин, соответствующим агрегатам (димерам) антитела, и основным пиком с временем элюирования около 16,5 мин, соответствующим мономерам антител с интактной структурой (фиг.17).
Хранение жидкости
Интересно, что в течение всего периода хранения жидкости этих конкретных высококонцентрированных составов при различных температурах фрагментация антитела была лишь незначительной в оригинальном составе, а также в композициях на основе аминокислот согласно изобретению. Этот эффект может быть результатом повышенного отношения антител к вспомогательному веществу, и уже наблюдался в предыдущем эксперименте с низкоконцентрированными композициями трастузумаба (пример 4; данные не показаны).
Хранение жидкости в течение 3 дней при 40°C привело к увеличению агрегации в исходной композиции до 2,11% агрегатов и 0,1% фрагментов. В композиции 4_3 этот период хранения приводил к агрегации около 0,22% и фрагментации около 0,07%. Хранение в течение 3 дней при 40°С в композиции 4_4 приводило к образованию агрегатов примерно до 0,26% и образованию фрагментов примерно 0,06%. В композиции 4_5 содержание агрегатов составляло всего 0,18%, а фрагментация составила примерно 0,08% (фиг.23А).
Дальнейшее хранение жидкости в течение 14 дней при 40°C привело к содержанию агрегатов около 2,28% в оригинальном составе и к фрагментации 0,33%. В композиции 4_3 агрегация составила всего 0,43%, а фрагментация была аналогичной 0,33%. Сопоставимые результаты были получены в композиции 4_4 с агрегацией около 0,40% и фрагментацией около 0,33%. Также в композиции 4_5 сопоставимые результаты были обнаружены для образования агрегатов (0,27%) и образования фрагментов на 0,34% (фиг.23 В).
После длительного хранения жидкости в течение 1,5 месяцев при 30°С в исходной композиции образование агрегатов составило 1,84%, а образование фрагментов 0,25%. В композиции 4_3 агрегация составила всего 0,28%, а фрагментация 0,23%, что сопоставимо с исходной композицией. В композиции 4_4 образование агрегатов слегка увеличилось до 0,38%, а образование фрагментов составило около 0,22%. В композиции 4_5 агрегация после хранения в течение 1,5 месяцев при 30°C составила только 0,22%, а фрагментация достигала 0,25% (фиг.24 A).
Хранение жидкости в режиме реального времени в течение 3 месяцев при 25°C привело к образованию агрегатов около 1,89% в оригинальном составе и к фрагментации 0,25%. В композиции 4_3 при длительном хранении при 25°С формировалось 0,35% агрегатов и 0,22% фрагментов. Агрегация была слегка увеличена в композиции 4_4 после 3 месяцев хранения при 25°С до 0,40%, а образование фрагментов сохранялось равным 0,22%. В композиции 4_5 была обнаружена самая низкая агрегация 0,27%, и была достигнута сопоставимая фрагментация с 0,25% (фиг.24 B).
Во все моменты времени анализа при хранении жидкости антитела при различных температурах композиция 4_5 показала лучшую стабилизирующую эффективность в отношении агрегации. В композиции 4_3 и композиции 4_4 склонность к агрегации антитела во время хранения жидкости при различных температурах была более или менее сопоставимой, тогда как в композиции 4_5 антитело показало самое низкое образование агрегатов в указанные моменты времени анализа. Между композицией 4_3 и композицией 4_4 первая показала немного лучшую стабилизирующую эффективность.
Оценка склонности антитела к агрегации и фрагментации и к связанной с ними потере пика мономеров при хранении жидкости высококонцентрированных составов антител (200 мг/мл) в композициях 4_3, 4_4 и 4_5 по сравнению с оригинальным составом в течение 3 месяцев при 25°С также подтвердила приведенные выше подробные результаты.
Пики агрегатов (время элюирования ≈ 14 минут), соответствующие димерам антител, были значительно снижены в композиции 4_5 (р<0,01; р<0,0001) и в незначительной степени в композиции 4_3 и композиции 4_4 (р<0,01, р<0,001), по сравнению с оригинальным составом (фиг.26C). В композиции 4_4 (самое высокое отношение антитело: вспомогательное вещество 3,33:1) была обнаружена слегка повышенная агрегация, связанная с увеличением вязкости состава, по сравнению с композицией 4_3 и особенно с композицией 4_5 (фиг.26C; фиг.4). Не наблюдалось соответствующей фрагментации с 200 мг/мл (фиг.26C), что может быть связано с повышенным отношением антител к вспомогательному веществу, как это уже наблюдалось для низкоконцентрированных составов (см. ниже и фиг.25). Пик мономеров в составах на основе аминокислот практически полностью сохранялся при хранении жидкости при 25°С.Композиции 4_3 и 4_4 продемонстрировали самое низкое увеличение фрагментации по сравнению с оригинальным составом и композицией 4_5 (p>0,05; Фиг.26C).
Эти результаты позволяют предположить, что как изменения в составе, так и изменения концентрации гистидина и триптофана, а также изменение отношения трегалозы к сахарозе оказали положительное влияние на стабилизирующую эффективность, особенно на агрегацию. Фрагментация была несколько более снижена в композициях на основе аминокислот, содержащих более высокие концентрации триптофана и гистидина в концентрации буфера (см. предыдущие примеры и композиции 4_3 и 4_4 по сравнению с композицией 4_5). Отношение смеси аминокислот: трегалозы/ сахарозы было немного увеличено в композиции 4_4 (1,5: 1 (по массе) по сравнению с 1:1 (по массе) в композиции 4_3, полученной из композиции 4_2 из предыдущего эксперимента.
Таким образом, корректировка концентраций триптофана и гистидина вместе с корректировкой отношения аминокислоты к сахару важна для предотвращения агрегации и фрагментации совместно при хранении антитела в жидкости. Кроме того, сильное увеличение отношения антитела к вспомогательному веществу до 3,3:1 (по массе) в композиции 4_4 привело к незначительному увеличению склонности антитела к агрегации во время хранения жидкости. Было показано, что параллельное регулирование отношения антитела к вспомогательному веществу оказывает влияние на стабилизирующую эффективность композиций на основе аминокислот согласно изобретению.
Аналогичные наблюдения были сделаны при анализе химической деградации антитела во время хранения жидкости при повышенной температуре. Для этого образцы хранили в течение 3 месяцев при 25°C и анализировали с использованием CEX. Композиции 4_4 и 4_5 привели к незначительному увеличению образования вариантов кислотного заряда (самая низкая степень в композиции 4_5), но к уменьшенному образованию основных вариантов заряда, особенно в случае композиций 4_3 и 4_4 (р<0,05) по сравнению с исходной композицией. Потеря площади основного пика была частично предотвращена композицией 4_3 и 4_4 (р>0,05) и была сопоставима с оригинальным составом в композиции 4_5 (фигура 27C). Массовое отношение между двумя выбранными аминокислотами гистидином и триптофаном несколько раз изменяли, что приводило к измененному образованию вариантов кислого и основного заряда (фиг.27C).
Измерение вязкости
Для анализа динамической вязкости высококонцентрированных составов антител по сравнению с примером 5 концентрации составов были скорректированы до 200 мг/мл и 220 мг/мл в соответствии со способом подготовки образца в пункте 7.1.1 этого примера (фиг.25). Динамическая вязкость высококонцентрированной композиции антител в исходной жидкой композиции поставщика (220 мг/мл) составила до 20,53 мПа*с. В композициях согласно изобретению 4_3 и 4_5, содержащих концентрацию антител 220 мг/мл, измеренные динамические вязкости были заметно снижены до 15,2 мПа*с в композиции 4_3 и 17,6 мПа*с в композиции 4_5. Небольшое увеличение вязкости было оценено в случае композиции 4_4 с 22,4 мПа*с. Это может быть результатом высокого отношения антитела к наполнителю 3,7:1 в этой композиции по сравнению с композициями 4_3 и 4_5. Измерения вязкости соответствующих составов с концентрациями антител 200 мг/мл также показали явное снижение вязкости в композиции 4_3, 11,2 мПа*с, в композиции 4_4 15,4 мПа*с и в композиции 4_5 11,6 мПа*с. В случае концентрации антител 200 мг/мл композиция 4_4 также показала слегка повышенную вязкость по сравнению с другими препаратами, что указывает на такую же тенденцию, оцененную в композиции 4_4 с концентрацией антитела 220 мг/мл. Вместе со слегка сниженной стабилизирующей эффективностью композиции 4_4 во время хранения в жидкой форме высококонцентрированного антитела (200 мг/мл) эти данные дополнительно подтверждают вывод о том, что помимо регуляции отношения аминокислоты и сахара, также регуляция отношения антитела к вспомогательным веществам оказывает существенное влияние как на стабилизирующую эффективность, так и на вязкость композиции.
Пример 8. Анализ как молекулярной целостности, так и химической стабильности во время обработки и во время последующего хранения в жидкой форме высококонцентрированных составов антител показал, что конкретные аминокислотные и сахарные композиции, не содержащие пролина, были способны снижать склонность к агрегации во время обработки, а также при последующем хранении жидкости. В частности, химические изменения были значительно снижены по сравнению с оригинальным составом лиофилизированного продукта трастузумаба, содержащего глицин и пролин.
8.1 Материалы и методы
Композиция 4_1 соответствует составу, применяемому в Примере 6, а композиции 4_3 и 4_5 соответствуют составам, применяемым в Примере 7. Стабилизирующий эффект этих составов сравнивали с оригинальным жидким составом поставщика (79,45 г/л трегалозы; 3,13 г/л гистидинового буфера 20 мМ; 1,49 г/л метионина; 0,4 г/л полисорбата 20; рН 5,5). В этих составах рН доводили до 5,5. Кроме того, стабилизирующий эффект композиций по изобретению сравнивали с оригинальным составом поставщика лиофилизированного продукта (20 г/л трегалозы; 0,9 мг/мл гистидинового буфера, приблизительно 5 мМ; 0,1 г/л полисорбата 20; рН 6) с добавлением аминокислот глицина и пролина в соответствии с учением патента США US 9,364,542 B2 (пример 16; фиг.29 и 30).
В качестве модельного лекарственного средства использовали коммерческий лиофилизированный Герцептин® (Roche; Базель; Швейцария), терапевтическое гуманизированное моноклональное антитело IgG1 (трастузумаб). Восстановление лиофилизированного лекарственного средства в необходимом объеме воды привело к концентрации антител 21 мг/мл в оригинальном составе поставщика (20 г/л трегалозы; 0,9 мг/мл гистидинового буфера, приблизительно 5 мМ; 0,1 г/л полисорбата 20; рН 6).
8.1.1 Приготовление образца
Полученную композицию подвергали диализу при 2-8°C против композиции из оригинального жидкого состава (79,45 г/л трегалозы; 3,13 г/л гистидинового буфера, приблизительно 20 мМ; 1,49 г/л метионина; 0,4 г/л полисорбата 20; pH 5,5), и оригинального состава поставщика лиофилизированного продукта (20 г/л трегалозы; 0,9 мг/мл гистидинового буфера, приблизительно 5 мМ; 0,1 г/л полисорбата 20; pH 6), где этот состав дополнительно содержал аминокислоты глицин и пролин в концентрациях, описанных в патенте США US 9,364,542 B2 (пример 16).
Параллельно проводили диализ против композиций на основе аминокислот по настоящему изобретению, как подробно описано в пункте 8.1. Последующее концентрирование полученных составов IgG проводили для получения 200 мг/мл антитела. Затем составы подвергали стерилизующей фильтрации, разделяли на аликвоты в стерильных флаконах для ВЭЖХ и хранили либо при 25°С, либо при 40°С, либо для кратковременного хранения при 55°С, как описано в патенте США US 9,364,542 В2 (пример 16; фиг.29 и 30). Агрегацию и фрагментацию перед хранением, непосредственно после приготовления образца и в указанные моменты времени хранения анализировали с использованием эксклюзионной ВЭЖХ. Химические изменения в белковых молекулах при хранении жидкости анализировали с использованием катионообменной ВЭЖХ.
8.1.2. Эксклюзионная хроматография
SEC выполняли, как описано в разделе 3.1.2 выше.
8.1.3 Катионообменная хроматография
Катионообменную ВЭЖХ (детектор УФ-280 нм; UHPLC UltiMate3000 Thermo Scientific, Германия) и катионообменную колонку TSK-гель CM-STAT 4,5 x 100 нм (Tosoh Bioscience, Токио, Япония) использовали при 45°C и со скоростью потока 0,8 мл/мин (объем введения 25 мкл). Перед анализом катионообменной ВЭЖХ образцы разбавляли до 2,5 мг/мл IgG в разделительном буфере A (10 мМ натрий-фосфатный буфер pH 7,5). Иммобилизованные молекулы трастузумаба элюировали в градиенте хлорида натрия, используя от 0% до 30% буфера В (10 мМ натрий-фосфатный буфер рН 7,5; 100 мМ хлорида натрия). Относительные площади под кривыми (% AUC) определяли с помощью программного обеспечения Chromeleon 7 Chromatography Data (Thermo Scientific).
8.2 Результаты
Приготовление образца
В ходе приготовления образца с использованием диализа и последующего концентрирования, чтобы получить концентрацию антител 200 мг/мл, ранее полученные результаты примеров 3, 5 и 7 были подтверждены. В частности, когда процесс получения проводили в одной из композиций по настоящему изобретению (композиция_4_1, композиция_4_3 или композиция_4_5), было получено сохранение количества агрегатов и мономеров, которое сопоставимо со стандартом трастузумаба (0,65-0,70% агрегатов; 99,30-99,35% мономеров). Эти результаты позволяют предположить, что сохранение структурной целостности антитела, полученного в композициях по изобретению, сопоставимо с уровнями структурно интактных антител перед приготовлением (фиг.28).
С другой стороны, процесс получения высококонцентрированного антитела в оригинальном жидком составе поставщика приводил к увеличению относительной площади пика, соответствующего димерам антитела (т.е. при времени удерживания 14 минут), до примерно 0,84%, и соответствующему снижению пика мономеров до примерно 99,16%. Кроме того, соответствующий процесс приготовления высококонцентрированного трастузумаба в исходной рецептуре поставщика лиофилизированного продукта в сочетании с дополнительными аминокислотами глицином и пролином также привел к сильному увеличению образования агрегатов примерно до 0,79% и, следовательно, к снижению пика мономеров до 99,21%.
Хранение жидкости - эксклюзионная ВЭЖХ
Кратковременное хранение антитела в жидком состоянии в экстремальных (то есть физиологически и фармацевтически неприемлемо высоких) температурных условиях (24 часа при 55°С), как было проведено в патенте США 9364542 В2, выявило эффективное стабилизирующее действие композиций по изобретению против агрегации и фрагментации по сравнению с оригинальным жидким составом поставщика, а также оригинальным составом поставщика лиофилизированного продукта с глицином и пролином в качестве добавок. Относительные площади пиков, соответствующие образованию агрегатов, были почти полностью сохранены в композициях по изобретению, особенно в композиции 4_3 и композиции 4_5, по сравнению со стандартом трастузумаба, хранящимся при -80°С (0,65-0,70% агрегатов; 99,30-99,35% мономеров).
Напротив, кратковременное хранение в течение 24 часов при 55°C в оригинальном составе жидком составе поставщика привело к заметному увеличению агрегации с более выраженным снижением относительной площади пика мономеров (фиг.29; таблица 1; 2; t=0). В результате кратковременного хранения в течение 24 ч при 55°C фрагментация в оригинальном жидком составе поставщика, а также в оригинальном составе поставщика лиофилизированного продукта с глицином и пролином в качестве добавок также была немного увеличена по сравнению с антителом, полученным в композициях по изобретению (таблица 1).
Хранение жидкости в течение 7 дней, 14 дней, 28 и 42 дней, соответственно, при 40°С/ относительной влажности 75% выявило повышенную склонность антитела как к агрегации, так и к фрагментации во всех композициях, но в разной степени. Наиболее поразительно, что получение антитела в композициях по изобретению (композиция_4_1, композиция_4_3 и композиция_4_5) приводило к заметному снижению агрегации и фрагментации по сравнению с оригинальным жидким составом поставщика, а также с оригинальным составом поставщика в сочетании с аминокислотами глицином и пролином (фигуры 29C и D; таблицы 1 и 2).
Кроме того, хранение жидкости в течение 14 дней и 28 дней при 25°C дополнительно подтверждает наблюдение, что растворы по настоящему изобретению способны предотвращать агрегацию и фрагментацию при хранении при температуре окружающей среды и повышенных температурах и даже при экстремальных температурных условиях, таких как 55°C (Таблица 3, 4).
Хранение жидкости - Катионообменная ВЭЖХ
В патенте США US 9,364,542 B2 проводили только анализ образования макроскопических нерастворимых агрегатов антитела с использованием измерений мутности, и в некоторых примерах, анализ образования растворимых агрегатов с помощью эксклюзионной ВЭЖХ при кратковременном хранении в физиологически и фармацевтически неприемлемых высокотемпературных условиях, таких как 55°C. Здесь дополнительно анализировали степень химических изменений в молекуле антитела при кратковременном хранении при 55°С, а также при длительном хранении при 40°С и 25°С. Уже краткосрочное хранение трастузумаба в течение 24 ч при 55°С вызвало химические изменения во всех составах в разной степени.
В оригинальном составе поставщика лиофилизированного продукта с добавлением аминокислот глицина и пролина (Оригинал+Г/П) значительно увеличился процент вариантов кислого заряда антитела (>30%) при кратковременном хранении при 55°С.Это наблюдение было связано, как следствие, с заметным уменьшением относительной площади основного пика (53,23%), как показано в таблице 5.
Напротив, кратковременное хранение антитела в композициях согласно изобретению приводило к заметному уменьшению образования вариантов кислого заряда (например,<23% в композиции 4-5). Увеличение вариантов кислого заряда свидетельствует о дезамидировании или гликировании белка и является важным критерием для отрицательного выбора тестируемых составов в промышленных производственных стандартах. Таким образом, различные композиции в соответствии с изобретением были испытаны по сравнению с Оригиналом+Г/П для изучения модификаций вариантов кислого заряда при трехдневном хранении при 55°C, до 21 дня при 40°C и до двух месяцев при 25°C (фиг.30). Все тестируемые композиции в соответствии с настоящим изобретением обычно приводили к значительно более низким вариантам кислого заряда, чем при применении Оригинала+Г/П во все моменты времени анализа и при всех температурах хранения. Как следствие, основные пики были соответственно выше во всех составах согласно настоящему изобретению во все моменты времени анализа и температуры (таблица 5).
Пример 9. Анализ молекулярной целостности после диализа, концентрирования и последующей эксклюзионной ВЭЖХ показывает значимость по меньшей мере трех комбинаций аминокислот по отдельности или в сочетании с трегалозой для ограничения увеличения агрегатов на важных для производства этапах обработки и последующего хранения.
9.1 Материалы и методы
Композиция 5 содержала две основные аминокислоты гистидин и метионин, и была аналогична композиции 8, которая соответствует оригинальному составу поставщика (гистидин, метионин, трегалоза), за исключением того, что она не содержит сахара. Композиция 6 содержала три аминокислоты гистидин, метионин и глицин без сахара, тогда как композиция 9 была аналогичной, но содержала трегалозу. Композиция 7 содержала пять аминокислот: гистидин, метионин, аланин, аргинин и триптофан, тогда как композиция 10 была аналогичной, но содержала трегалозу.
В качестве модельного белка использовали коммерческое жидкое терапевтическое высококонцентрированное антитело Герцептин® (Roche; Базель; Швейцария), содержащее трастузумаб в концентрации 120 мг/мл в оригинальном составе поставщика (79,45 г/л трегалозы; 3,13 г/л гистидинового буфера, 20 мМ; 1,49 г/л метионина; 0,4 г/л полисорбата 20; 0,024 г/л rHuPh20 (рекомбинантная человеческая гиалуронидаза), рН 5,5).
9.1.1 Приготовление образца
Жидкое терапевтическое высококонцентрированное антитело Герцептин® подвергали диализу при 2-8°С против 5 мМ гистидинового буфера, рН 5,5. После определения концентрации белка и последующей корректировки концентрации белка до 100 мг/мл антитело вводили в состав композиций в соответствии с пунктом 9.1, а также в оригинальный состав поставщика путем разведения 1 на 5 диализированного высококонцентрированного антитела с использованием 1,25-кратно концентрированных составов до концентрации антител 20 мг/мл. Для экспериментов с высококонцентрированными составами антител избранные композиции и антитело, приготовленное в оригинальном жидком составе поставщика, концентрировали до 200 мг/мл. Затем составы подвергали стерилизующей фильтрации и делили на аликвоты в стерильных пробирках для ВЭЖХ и хранили при 45°С до 14 дней. Агрегацию и фрагментацию перед хранением, непосредственно после диализа, непосредственно после этапа концентрирования и после хранения в течение 7 дней и 14 дней при хранении жидкости анализировали с использованием эксклюзионной ВЭЖХ.
9.1.2. Эксклюзионная хроматография
SEC проводили в соответствии с пунктом 3.1.2.
9.2 Результаты
Хранение жидкости - эксклюзионная ВЭЖХ
Как показано в таблице 6, повышенная агрегация наблюдалась после семи дней хранения при 45°C после предыдущих стадий диализа и концентрации. Неожиданно было обнаружено, что составы с двумя аминокислотами и без сахара в целом демонстрировали самые высокие значения (например, 0,65% через семь дней; 1,29% через 14 дней) по сравнению с композициями, содержащими три аминокислоты (0,53% после семи дней; 1,11% после 14 дней) по сравнению с композициями, содержащими пять аминокислот (0,4% через семь дней; 0,88% через 14 дней). Основные пики были соответственно стабилизированы (таблица 6). Это наблюдение было подтверждено, когда те же аминокислотные комбинации были дополнены трегалозой. В частности, составы с двумя аминокислотами с трегалозой (соответствующие оригинальному составу) в целом демонстрировали самые высокие значения (например, 0,57% через семь дней; 1,07% через 14 дней) по сравнению с составами, содержащими три аминокислоты (0,43% через семь дней; 0,93 через 14 дней) и по сравнению с композициями, содержащими пять аминокислот (0,34% через семь дней; 0,8% через 14 дней). Основные пики были соответственно стабилизированы (таблица 6).
Пример 10
Анализ молекулярной целостности трастузумаба (20 мг/мл) после механического стресса на модели с постоянным перемешиванием в течение 14 дней с помощью эксклюзионной ВЭЖХ показывает эффективность по меньшей мере трех специально подобранных комбинаций аминокислот для ограничения повышения агрегации на соответствующих производственных этапах обработки, и их преимущество по сравнению с оригинальным составом поставщика.
10.1 Материалы и методы
Оригинальный состав поставщика содержал гистидин, метионин и трегалозу. Композиция 11 содержала две основные аминокислоты гистидин и метионин без трегалозы. Композиция 12 содержала три аминокислоты: гистидин, метионин и аланин. Композиция 13 содержала пять аминокислот: гистидин, метионин, аланин, аргинин и триптофан.
Составы антител подвергали механическому воздействию в определенной модели перемешивания. В этой модели образцы переносили в стерильные пробирки для ПЦР и перемешивали со скоростью 750 оборотов в минуту до 14 дней в темноте.
В качестве модельного белка использовали коммерческое жидкое терапевтическое высококонцентрированное антитело Герцептин® (Roche; Базель; Швейцария), содержащее трастузумаб в концентрации 120 мг/мл в оригинальном составе поставщика (79,45 г/л трегалозы; 3,13 г/л гистидинового буфера, 20 мМ; 1,49 г/л метионина; 0,4 г/л полисорбата 20; 0,024 г/л rHuPh20 (рекомбинантная человеческая гиалуронидаза), рН 5,5).
10.1.1 Приготовление образцов
Жидкое терапевтическое высококонцентрированное антитело Герцептин® подвергали диализу при 2-8°С против композиции из оригинального жидкого состава (79,45 г/л трегалозы; 3,13 г/л гистидинового буфера (20 мМ); 1,49 г/л метионина; 0,4 г/л полисорбата 20; рН 5,5) и против 5 мМ гистидинового буфера, рН 5,5. После определения концентрации белка и последующей корректировки концентрации белка до 20 мг/мл антитело вводили в композиции в соответствии с пунктом 10.1. Затем составы переносили во флаконы для ПЦР и перемешивали в соответствии со способом, описанным в 10.1. Значения агрегации и фрагментации перед перемешиванием и после перемешивания в течение 14 дней, полученные с помощью эксклюзионной ВЭЖХ, показаны в таблице 7.
10.1.2. Эксклюзионная хроматография
Эксклюзионную хроматографию проводили в соответствии с пунктом 3.1.2.
10.2 Результаты
Хранение жидкости - эксклюзионная ВЭЖХ
Как показано в таблице 7, повышенная агрегация наблюдалась после 14 дней перемешивания. Неожиданно было обнаружено, что составы с одной или двумя аминокислотами и без сахара в целом демонстрируют самые высокие значения (например, 0,32% в оригинальном составе, который содержит гистидин, метионин и трегалозу; 0,36% в композиции Х, включающей гистидин и метионин, без трегалозы).
Композиции, содержащие три аминокислоты, демонстрируют более низкие значения агрегации по сравнению с композициями с двумя аминокислотами. Как репрезентативно показано в таблице 7, композиция №Х с гистидином, метионином и аланином привела к значениям агрегации 0,62%. Композиции, содержащие пять аминокислот, приводили к значениям агрегации 0,24%. Основные пики были соответственно стабилизированы (таблица 7).
Таблицы
Таблица 1. Анализ посредством эксклюзионной ВЭЖХ высококонцентрированного трастузумаба непосредственно после приготовления образца (t=0) и в указанные моменты времени при хранении жидкости при 55°С и 40°С при приготовлении в оригинальном жидком составе поставщика и в оригинальном составе поставщика лиофилизированного продукта с добавлением аминокислот глицина и пролина - количественное определение агрегатов, мономеров и фрагментов, выраженных в относительной площади соответствующих пиков на хроматограммах эксклюзионной ВЭЖХ
40°C
Таблица 2. Анализ посредством эксклюзионной ВЭЖХ высококонцентрированного трастузумаба после приготовления образца (t=0) и в указанные моменты времени при хранении жидкости при 55°С и 40°С при получении композиций по изобретению - количественное определение агрегатов, мономеров и фрагментов, выраженных в относительной площади соответствующих пиков на хроматограммах эксклюзионной ВЭЖХ
Таблица 3. Анализ посредством эксклюзионной ВЭЖХ высококонцентрированного трастузумаба после приготовления образца (t=0) и в указанные моменты времени при хранении жидкости при 25°С при получении в оригинальном жидком составе поставщика и в оригинальном составе поставщика лиофилизированного продукта с добавлением аминокислот глицина и пролина - количественное определение агрегатов, мономеров и фрагментов, выраженных в относительной площади соответствующих пиков на хроматограммах эксклюзионной ВЭЖХ
Таблица 4. Анализ посредством эксклюзионной ВЭЖХ высококонцентрированного трастузумаба непосредственно после приготовления образца (t=0) и в указанные моменты времени при хранении жидкости при 25°С при получении в композициях в соответствии с настоящим изобретением - количественное определение агрегатов, мономеров и фрагментов, выраженных в относительной площади соответствующих пиков на хроматограммах эксклюзионной ВЭЖХ
Таблица 5. Главные пики на хроматограммах катионообменной ВЭЖХ высококонцентрированного трастузумаба после приготовления образца (t=0) и в указанные моменты при хранении жидкости при 55°С, 40°С и 25°С при приготовлении в оригинальном составе поставщика лиофилизированного продукта с добавлением аминокислот глицина и пролина (оригинал+Г/П) и в композициях 4_1, 4_3 и 4_5 в соответствии с настоящим изобретением. Главные пики выражены в виде относительной площади пиков соответствующих хроматограмм
Таблица 6. Анализ посредством эксклюзионной ВЭЖХ высококонцентрированного трастузумаба непосредственно после приготовления образца (t=0) и в указанные моменты времени при хранении жидкости при 45°С при приготовлении в композициях в соответствии с изобретением - количественное определение агрегатов, мономеров и фрагментов, выраженных в относительной площади соответствующих пиков на хроматограммах эксклюзионной ВЭЖХ
(после диализа)
Таблица 7. Анализ посредством эксклюзионной ВЭЖХ трастузумаба (20 мг/мл) непосредственно после приготовления образца (t=0) и спустя 14 дней перемешивания при приготовлении в оригинальном составе поставщика или в композициях в соответствии с изобретением - количественное определение агрегатов, мономеров и фрагментов, выраженных в относительной площади соответствующих пиков на хроматограммах эксклюзионной ВЭЖХ.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЖИДКИЙ СОСТАВ ПОЛИПЕПТИДОВ, СОДЕРЖАЩИХ FC-ДОМЕН ИММУНОГЛОБУЛИНА | 2011 |
|
RU2600847C2 |
| ПОРОШКОВЫЕ БЕЛКОВЫЕ КОМПОЗИЦИИ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2009 |
|
RU2537139C2 |
| СТАБИЛЬНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2017 |
|
RU2736830C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ БИОФАРМАЦЕВТИЧЕСКОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2011 |
|
RU2587056C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АДАЛИМУМАБ | 2014 |
|
RU2664736C2 |
| ВЫСОКОКОНЦЕНТРИРОВАННЫЕ СОСТАВЫ МОНОКЛОНАЛЬНЫХ АНТИТЕЛ | 2013 |
|
RU2711089C2 |
| СТАБИЛИЗИРОВАННЫЕ СОДЕРЖАЩИЕ АНТИТЕЛА ЖИДКИЕ КОМПОЗИЦИИ | 2011 |
|
RU2653447C2 |
| ЖИДКАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2015 |
|
RU2719431C2 |
| КОМПОЗИЦИЯ АНТИТЕЛ ПРОТИВ CTLA-4 | 2006 |
|
RU2356579C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КОНЪЮГАТА АНТИТЕЛА К HER2-ЛЕКАРСТВЕННОГО СРЕДСТВА | 2020 |
|
RU2795196C2 |
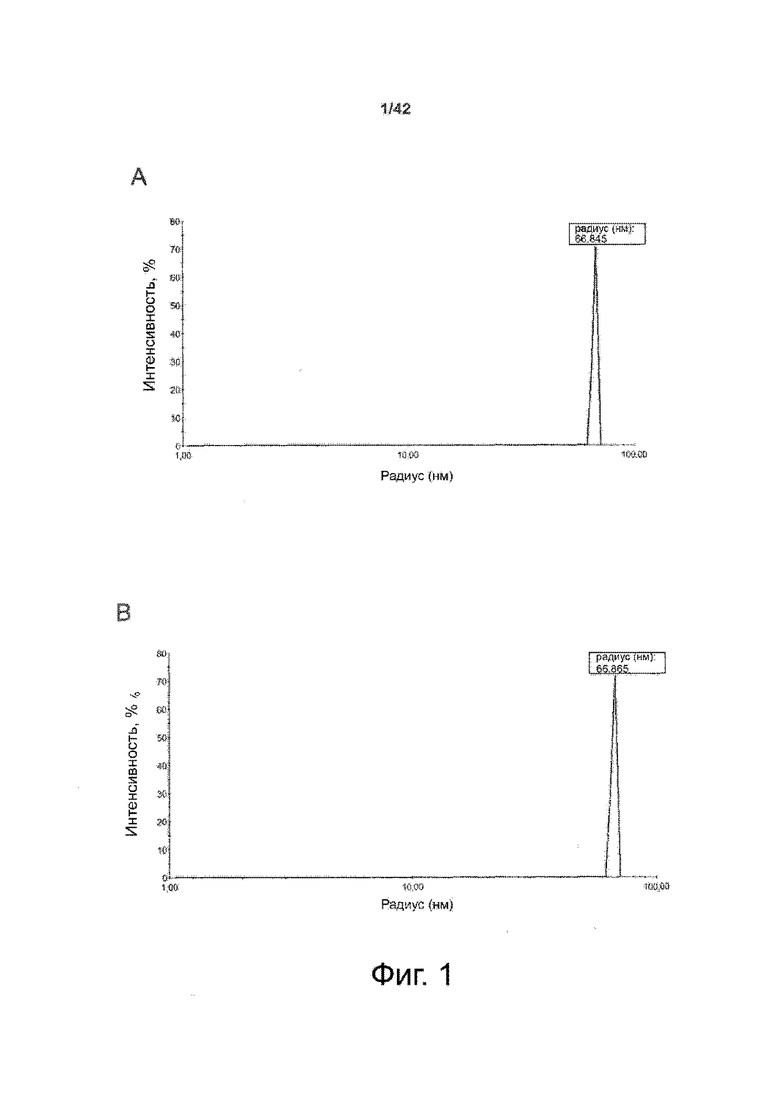

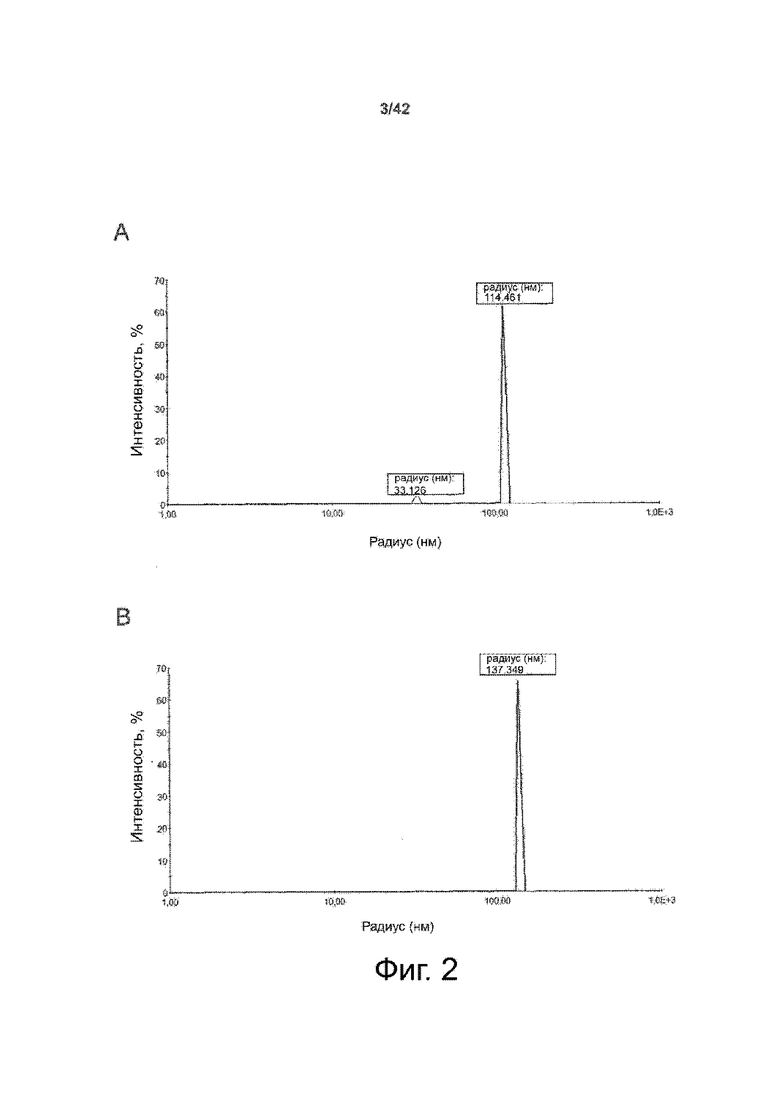
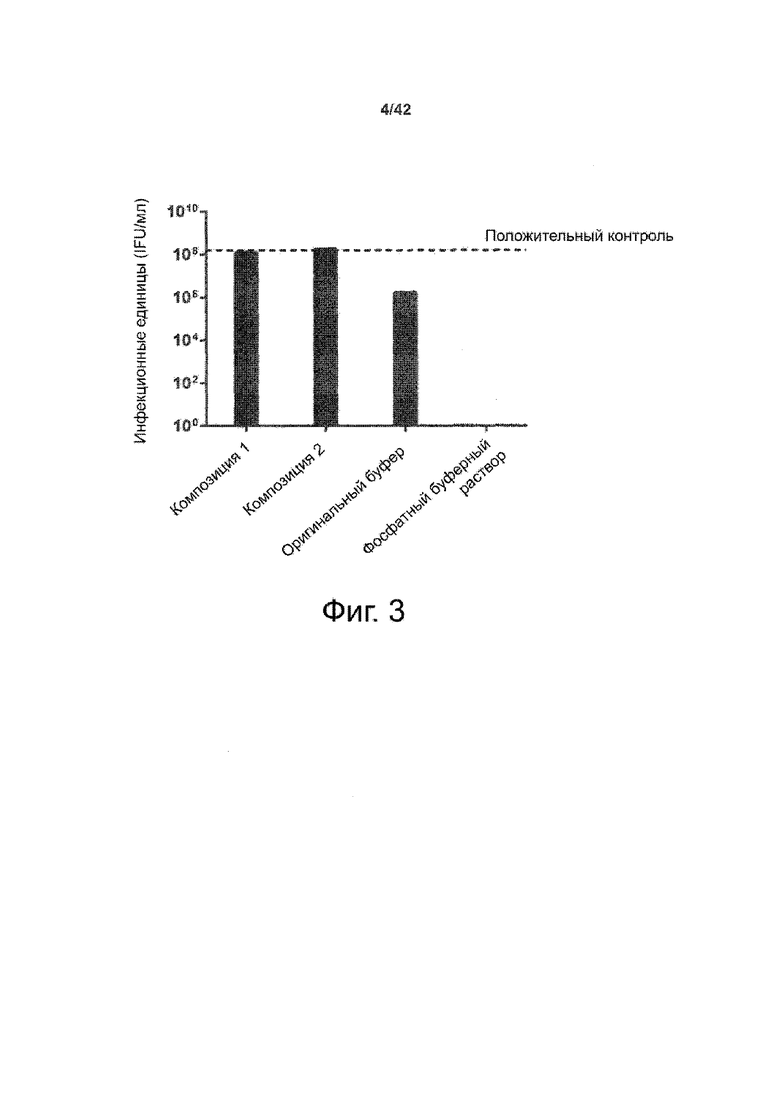
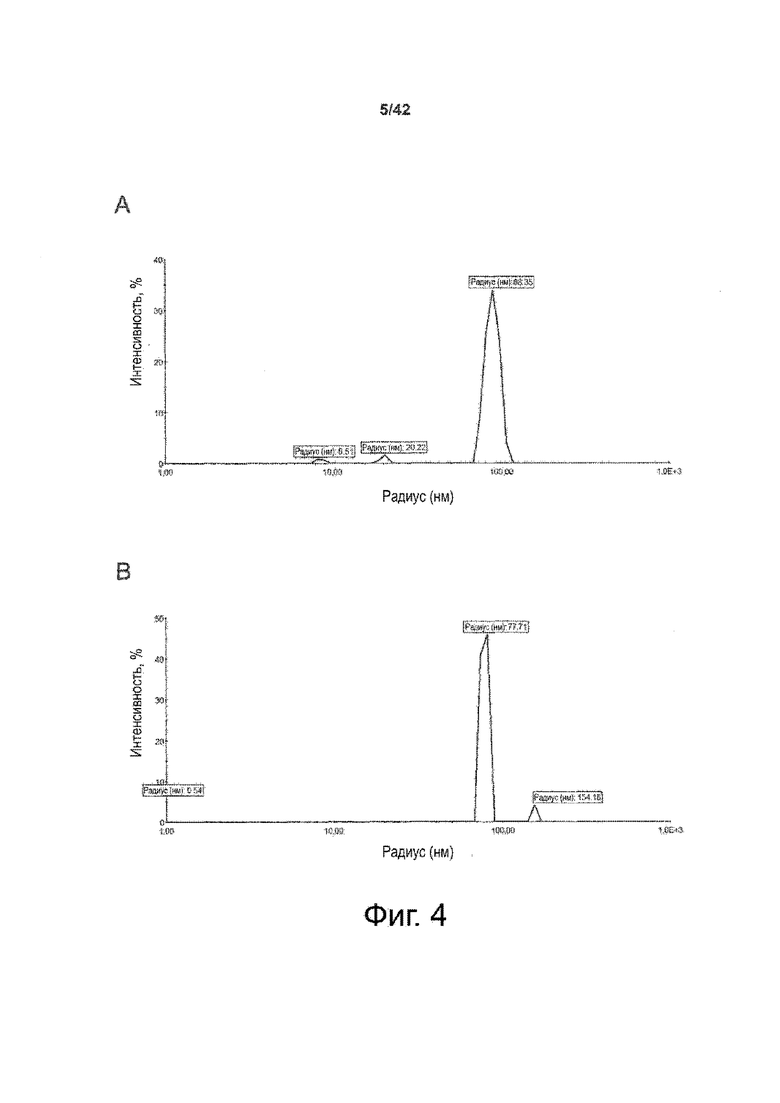
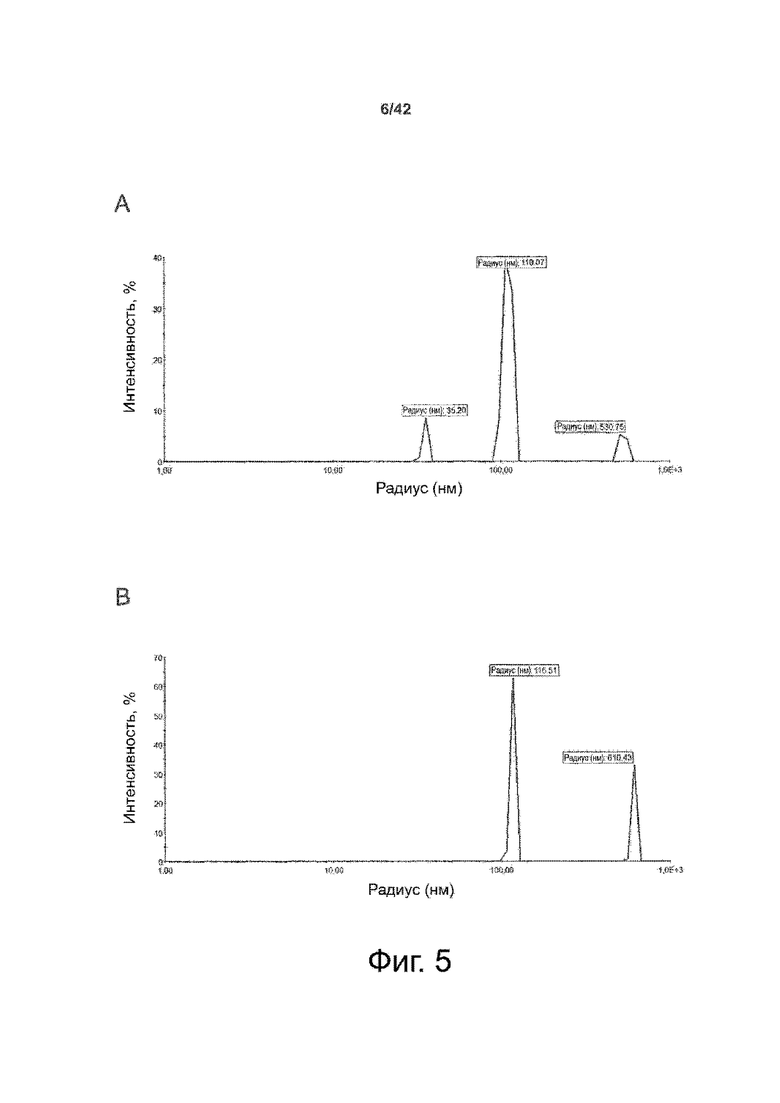
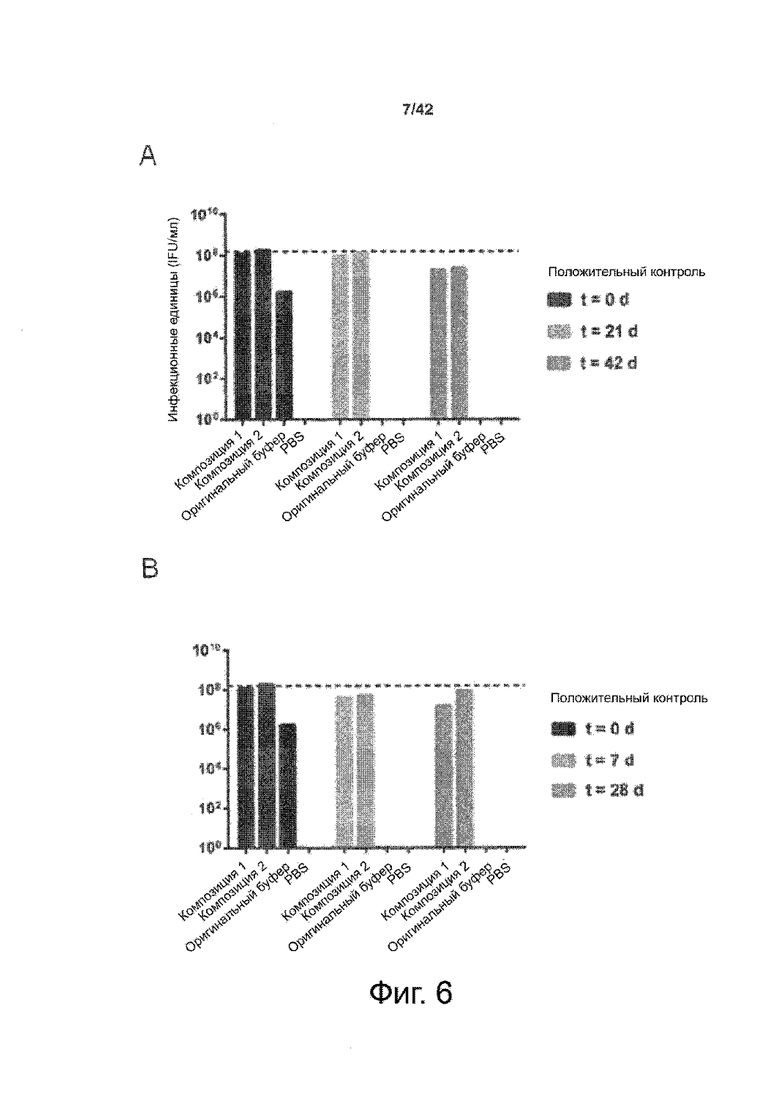
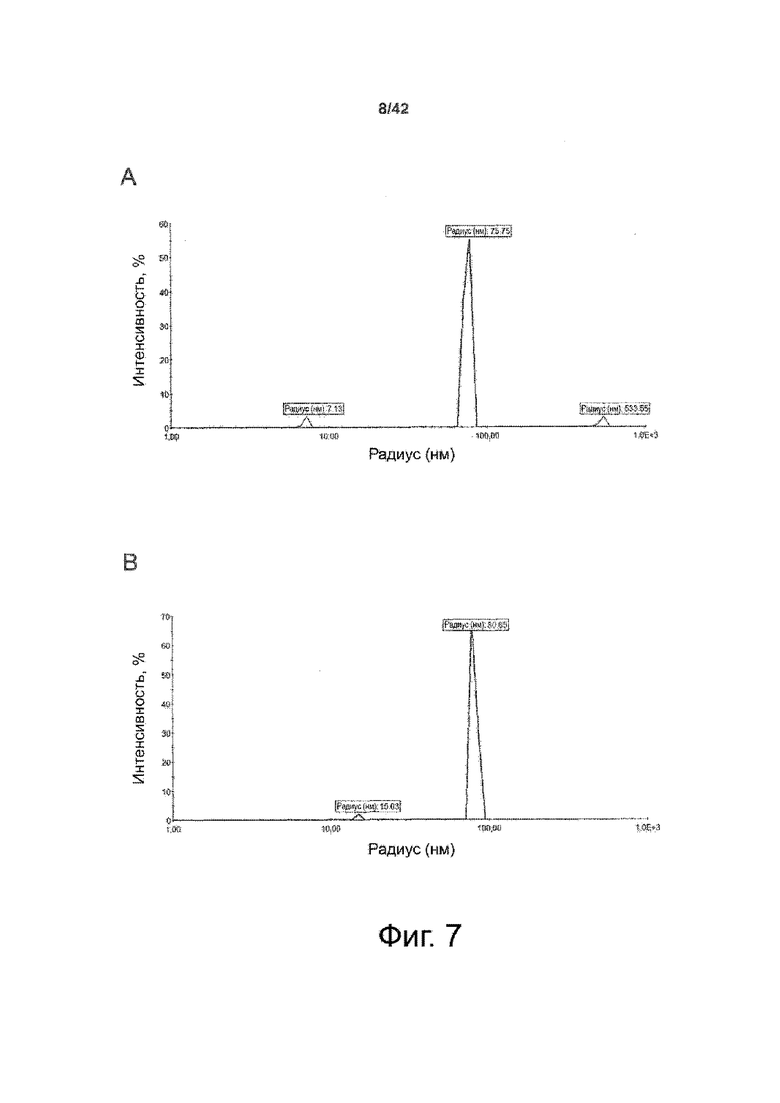
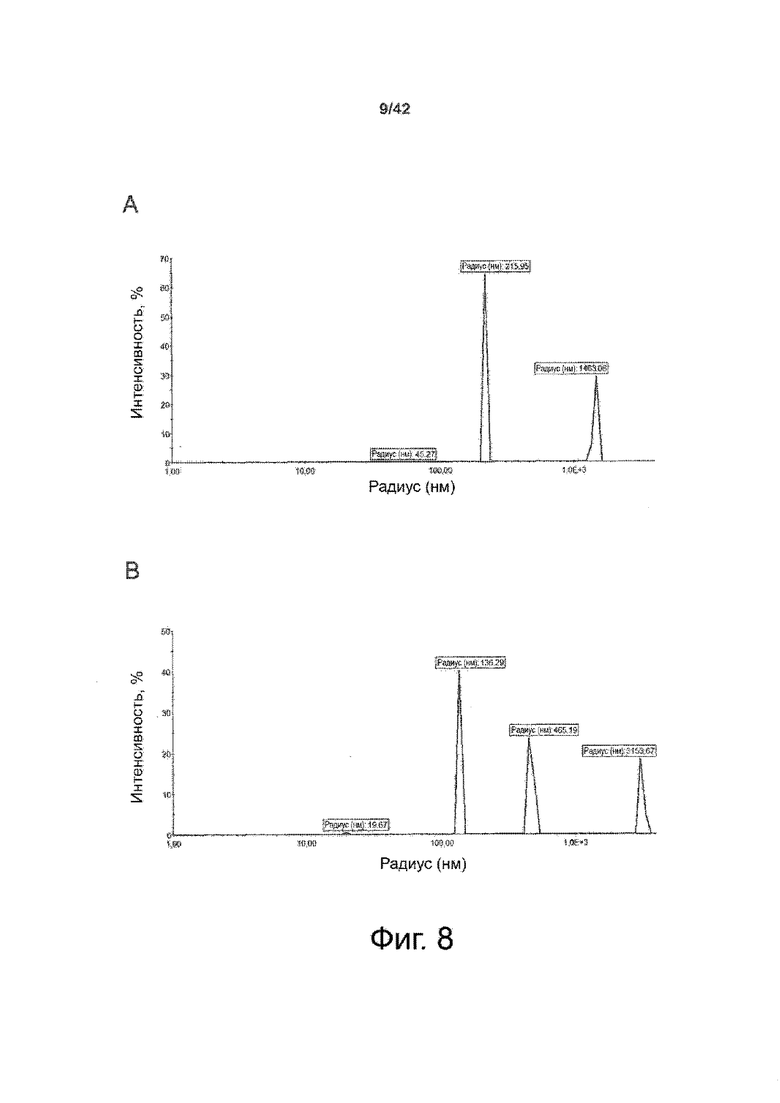
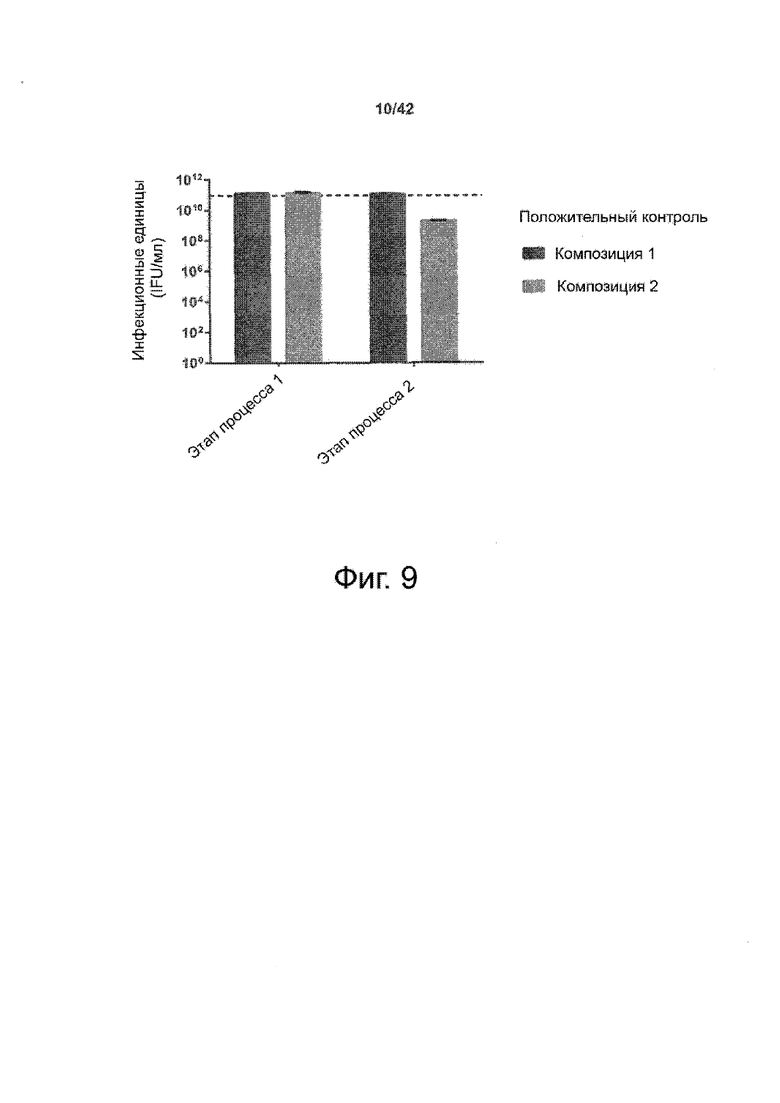
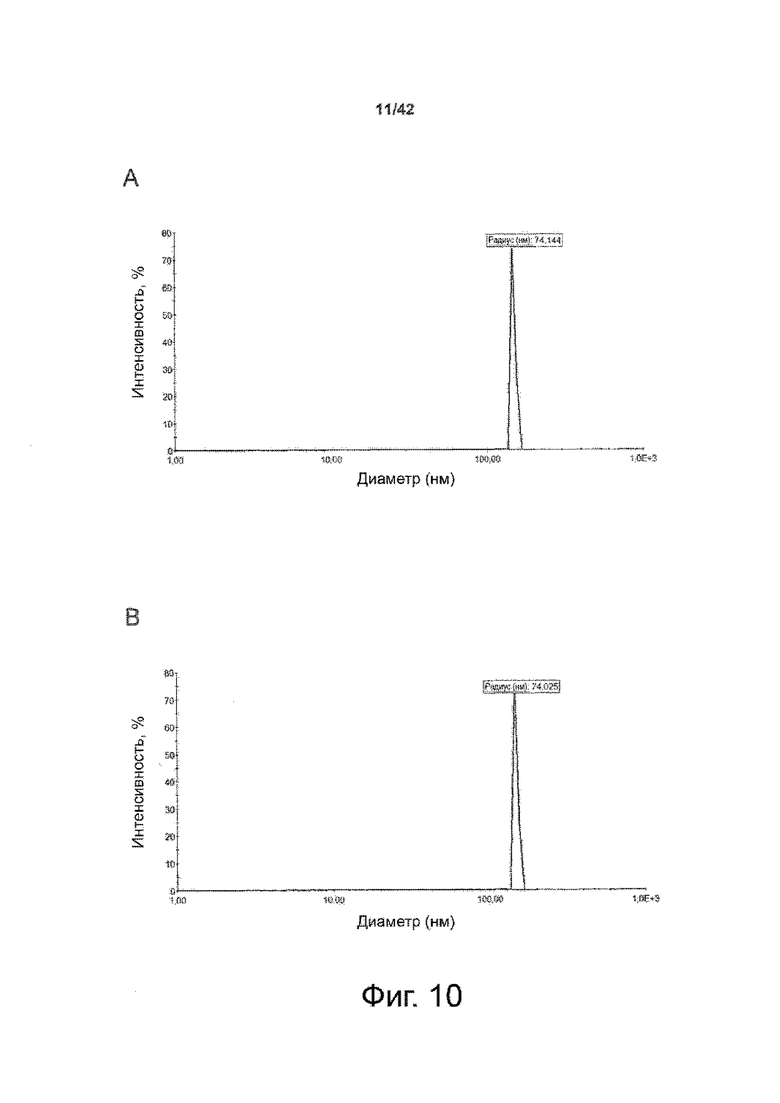
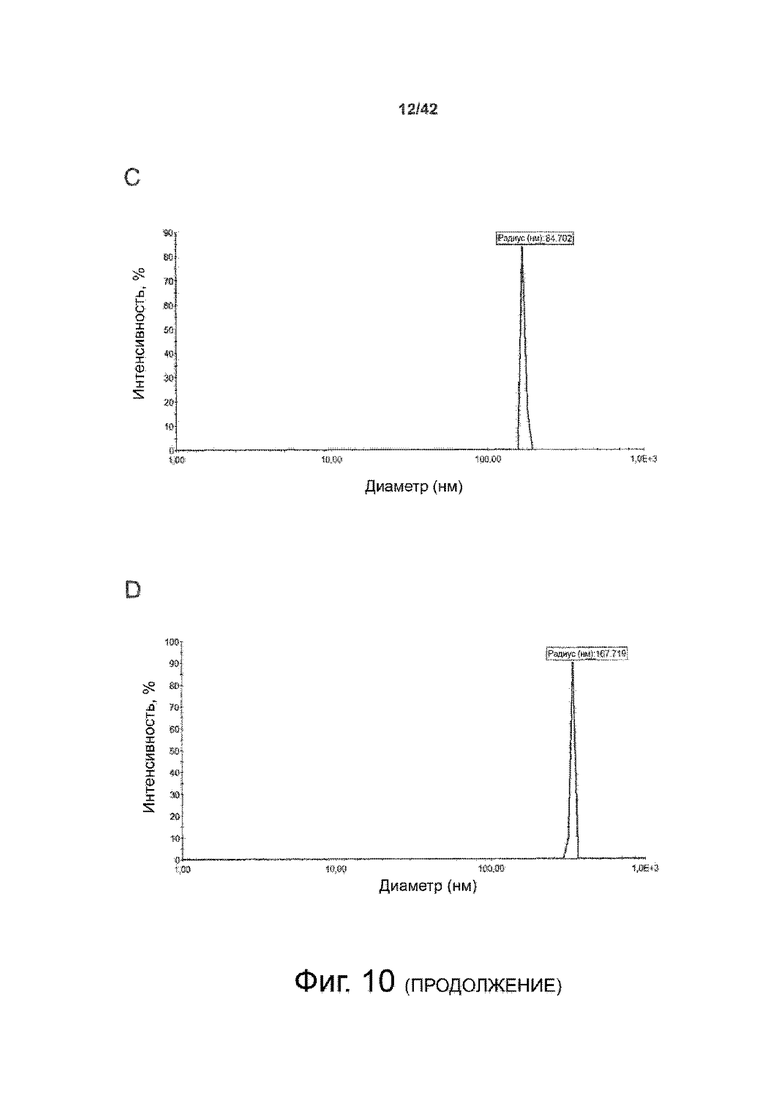
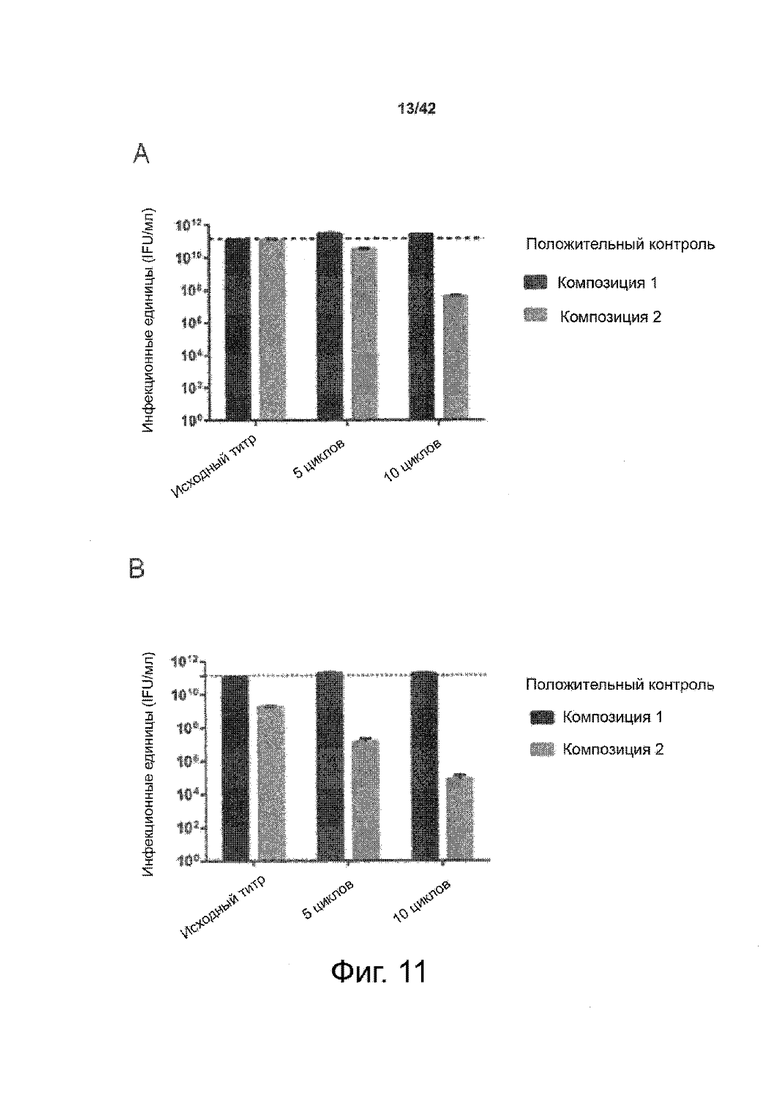
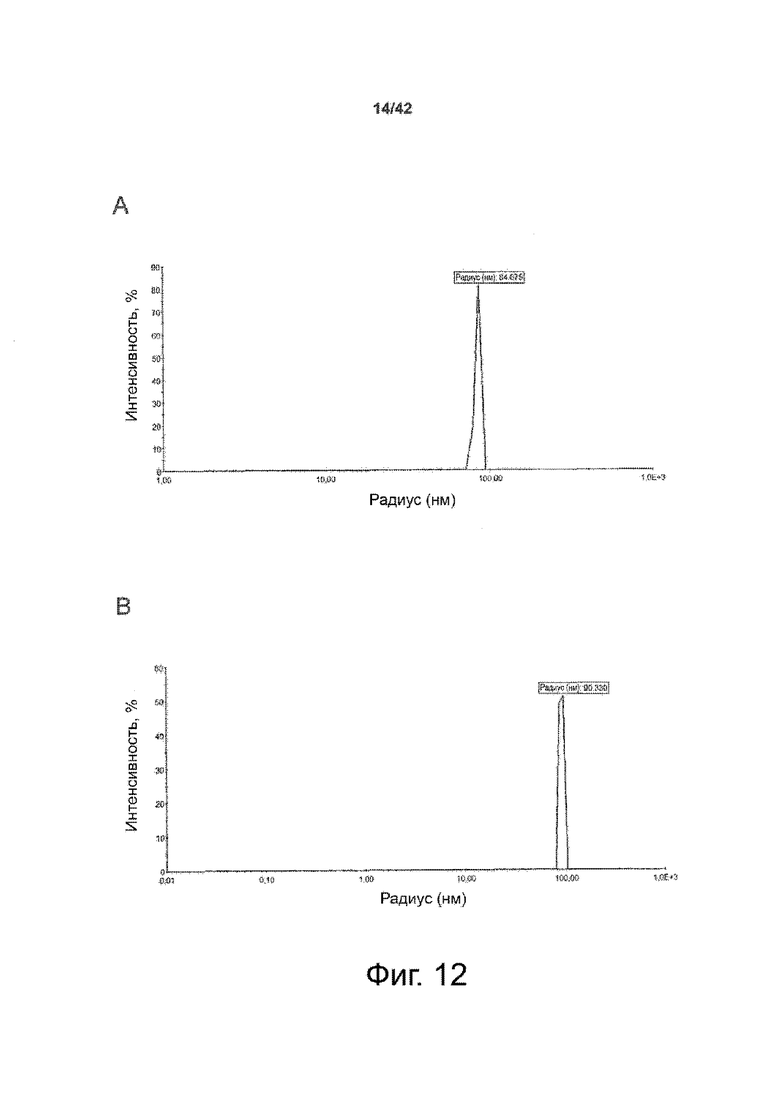

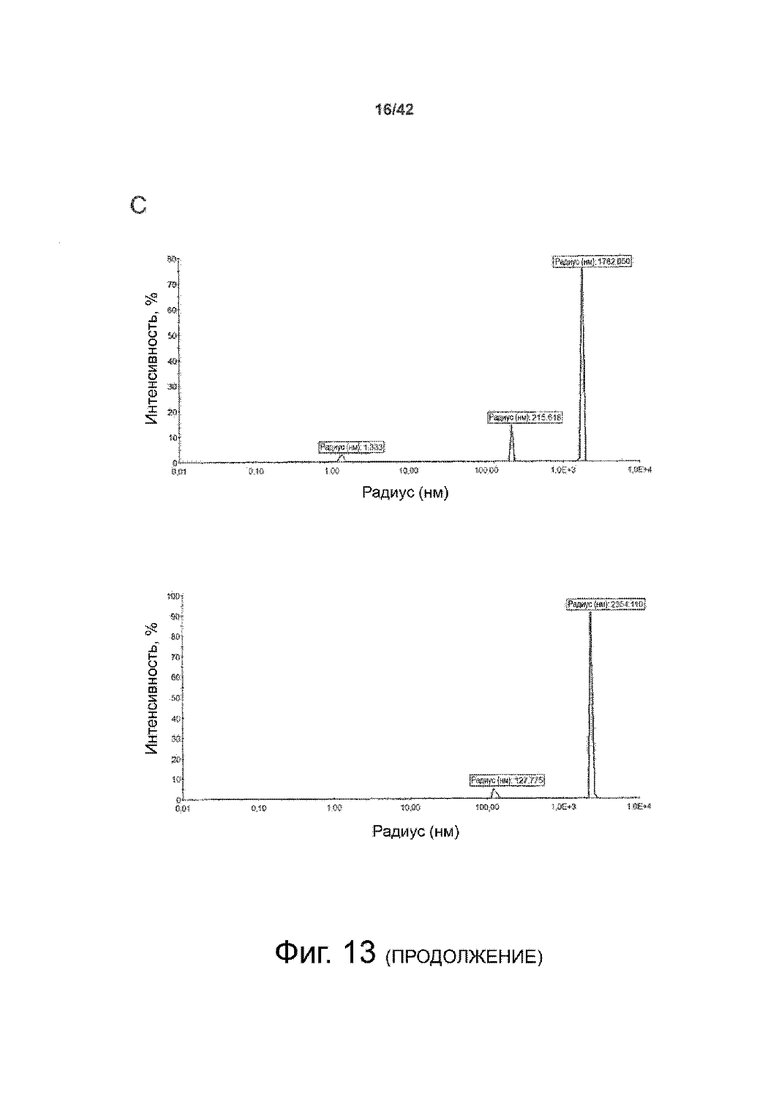
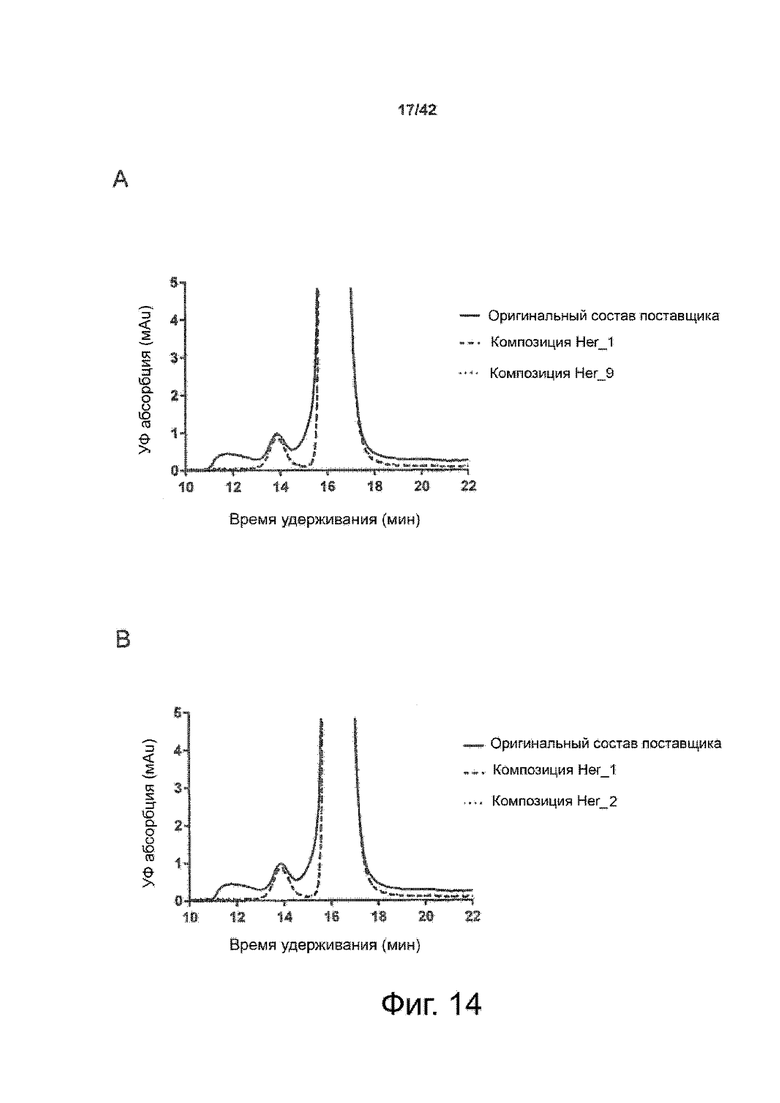
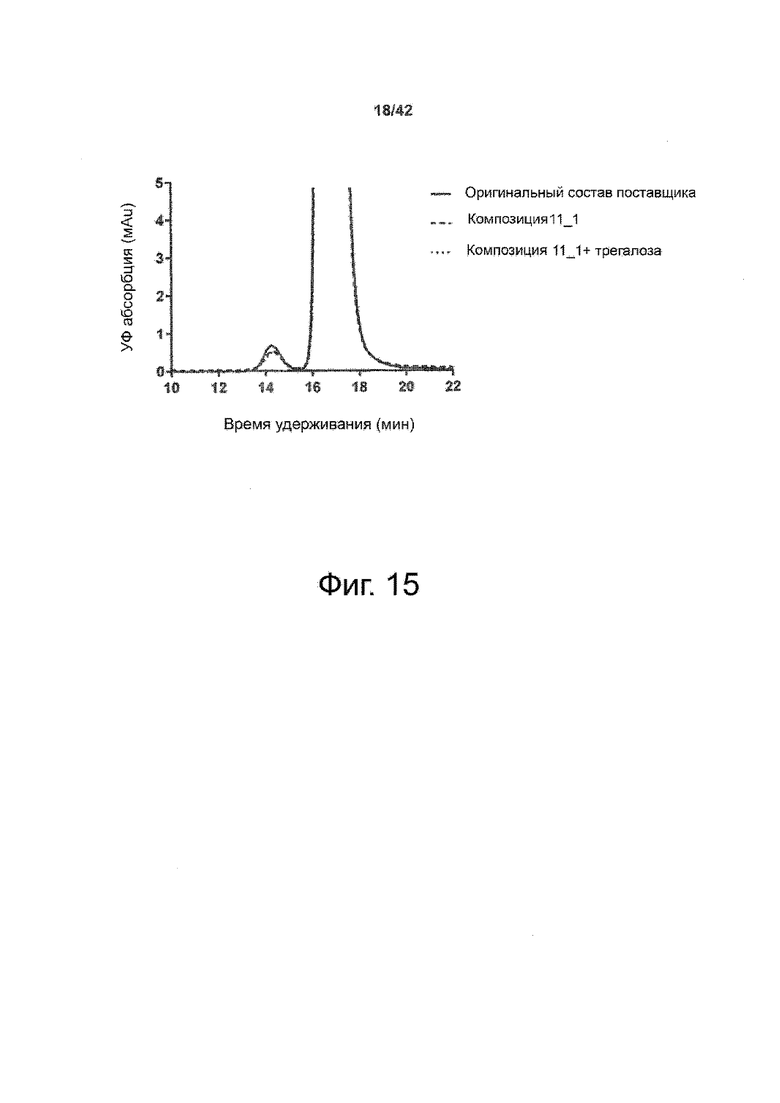
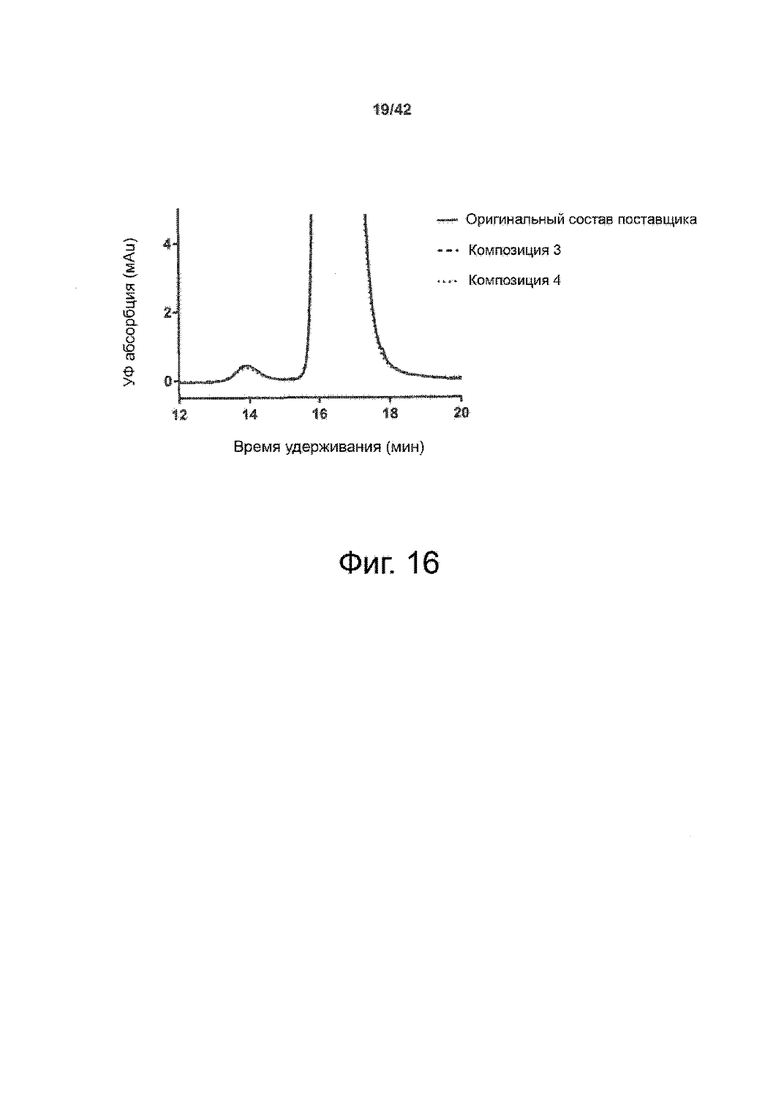
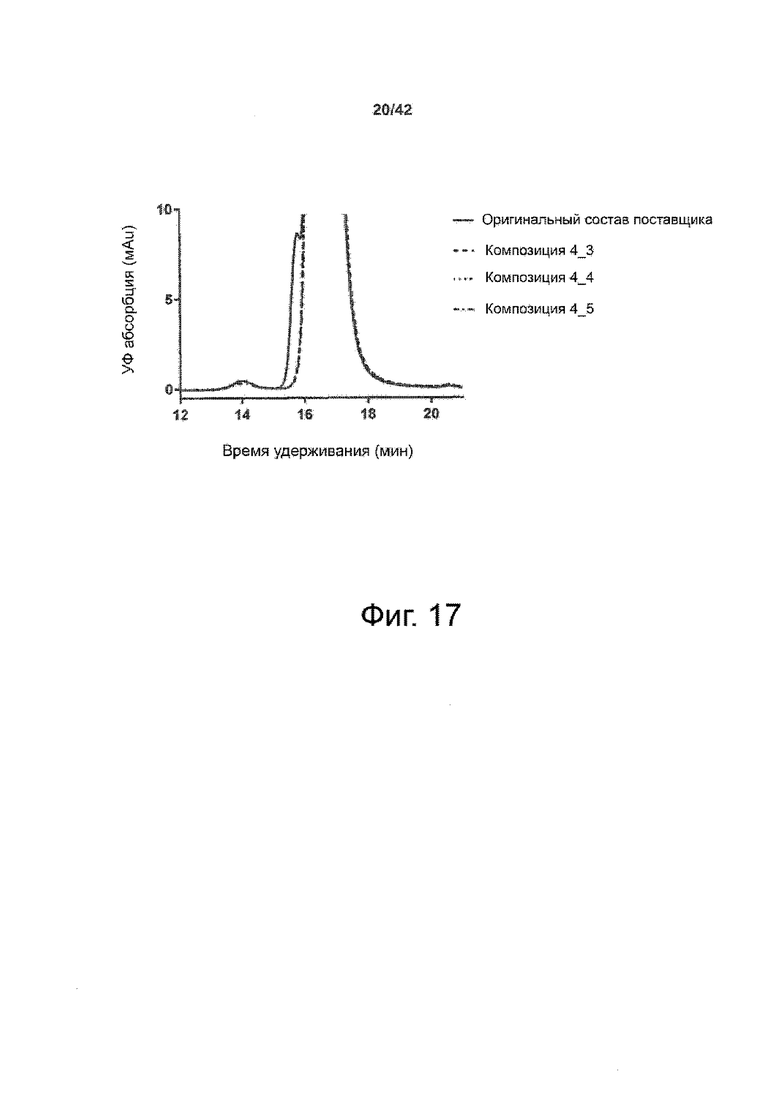
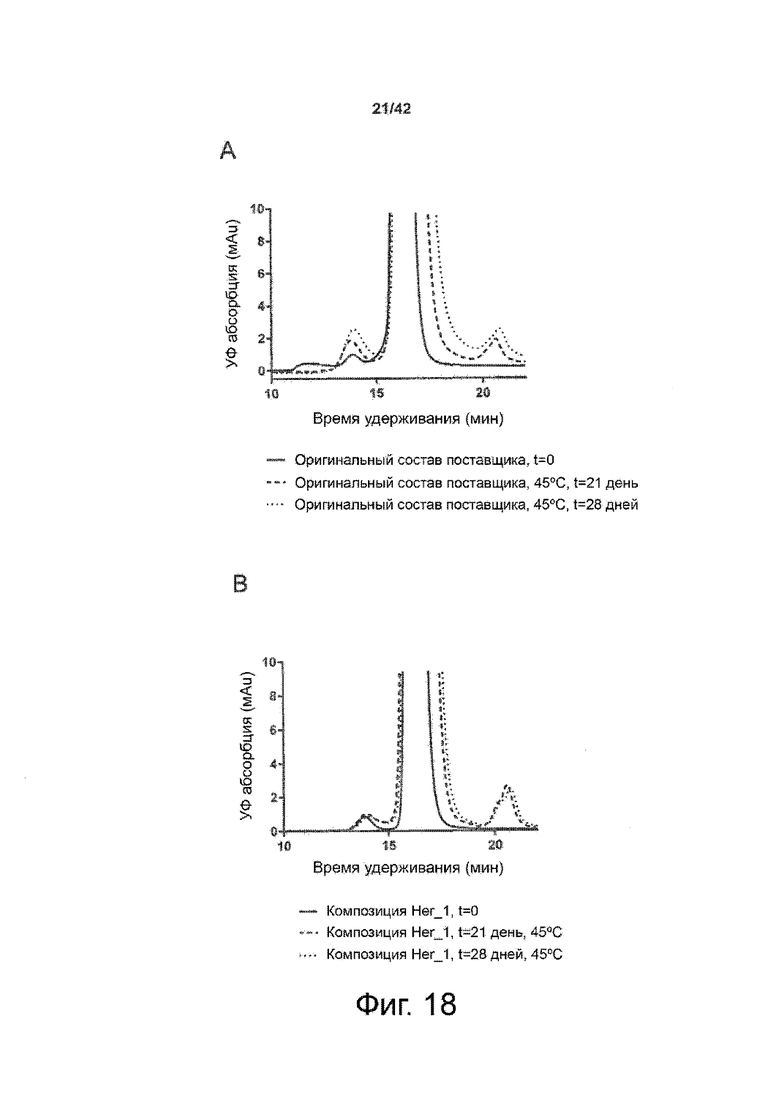
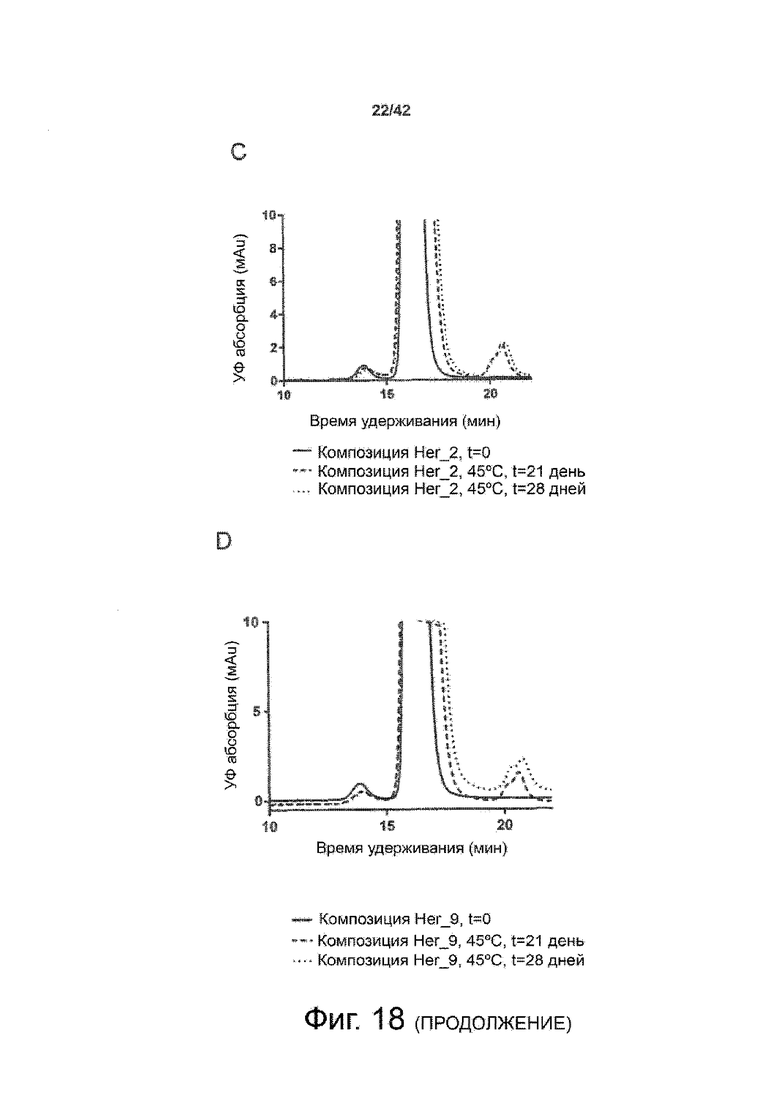
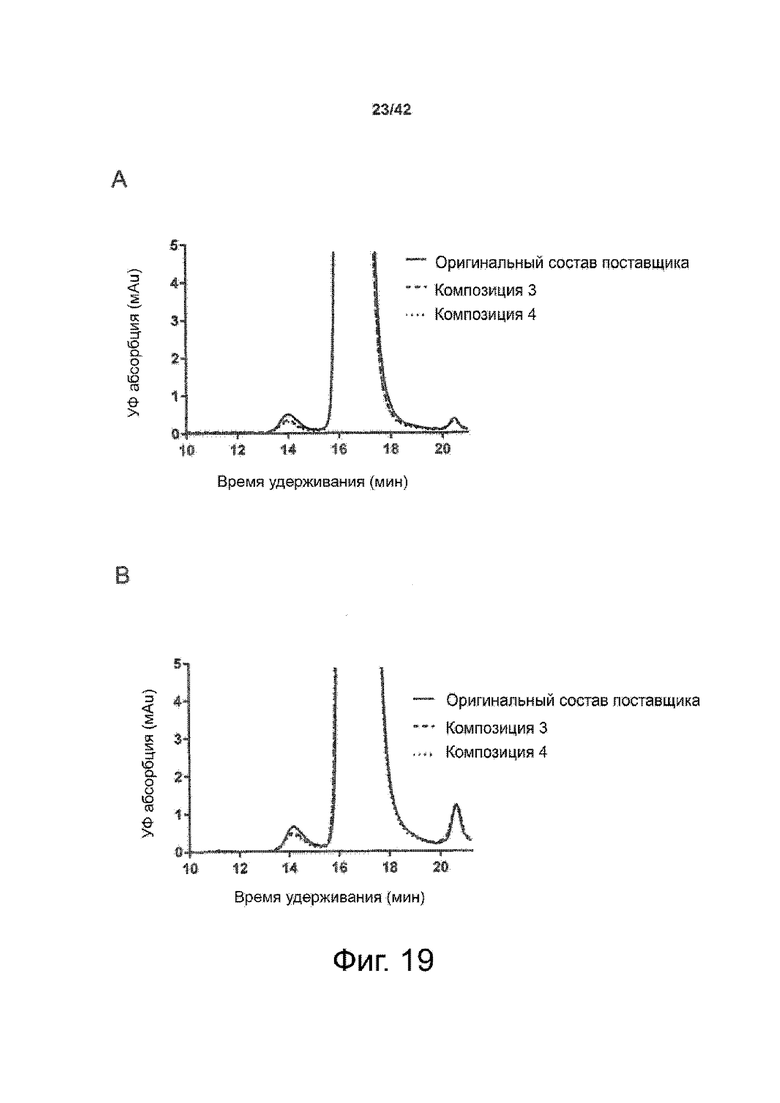
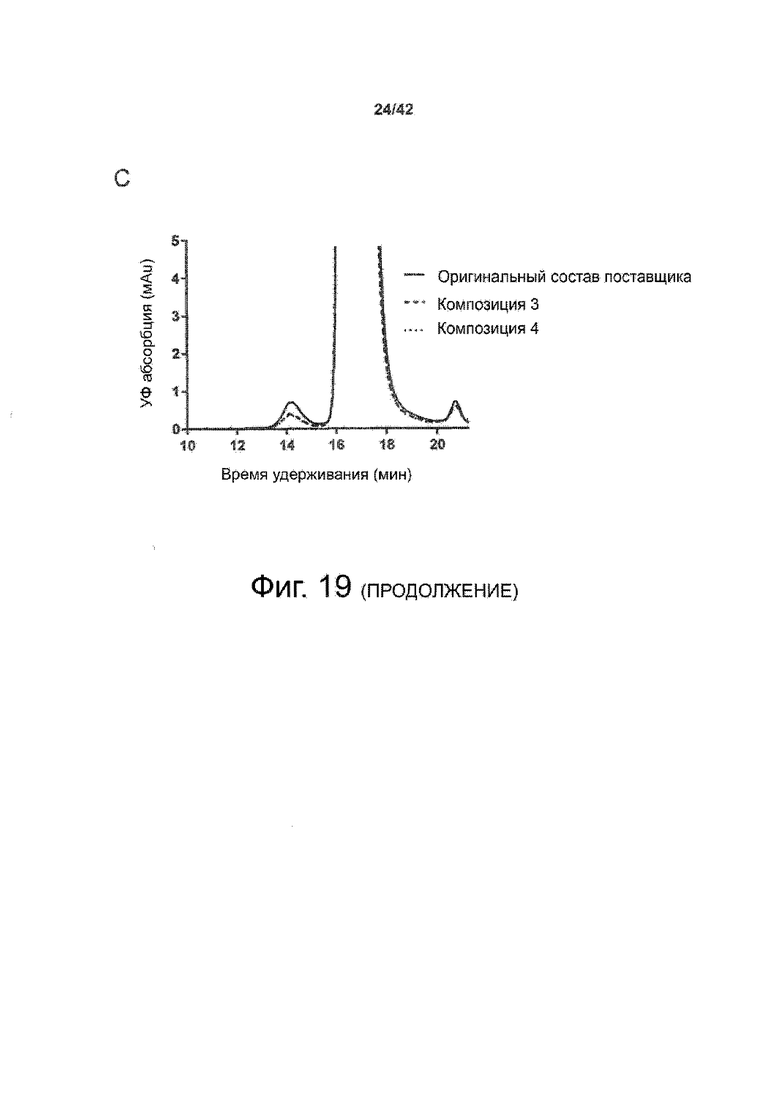
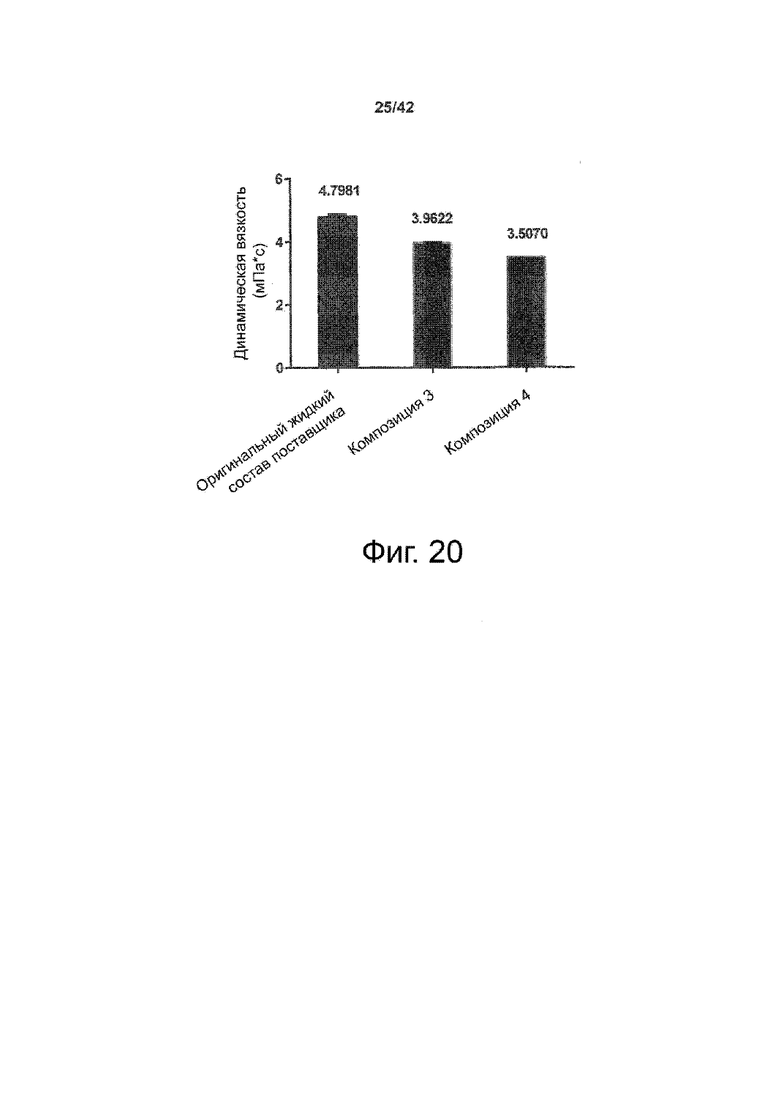
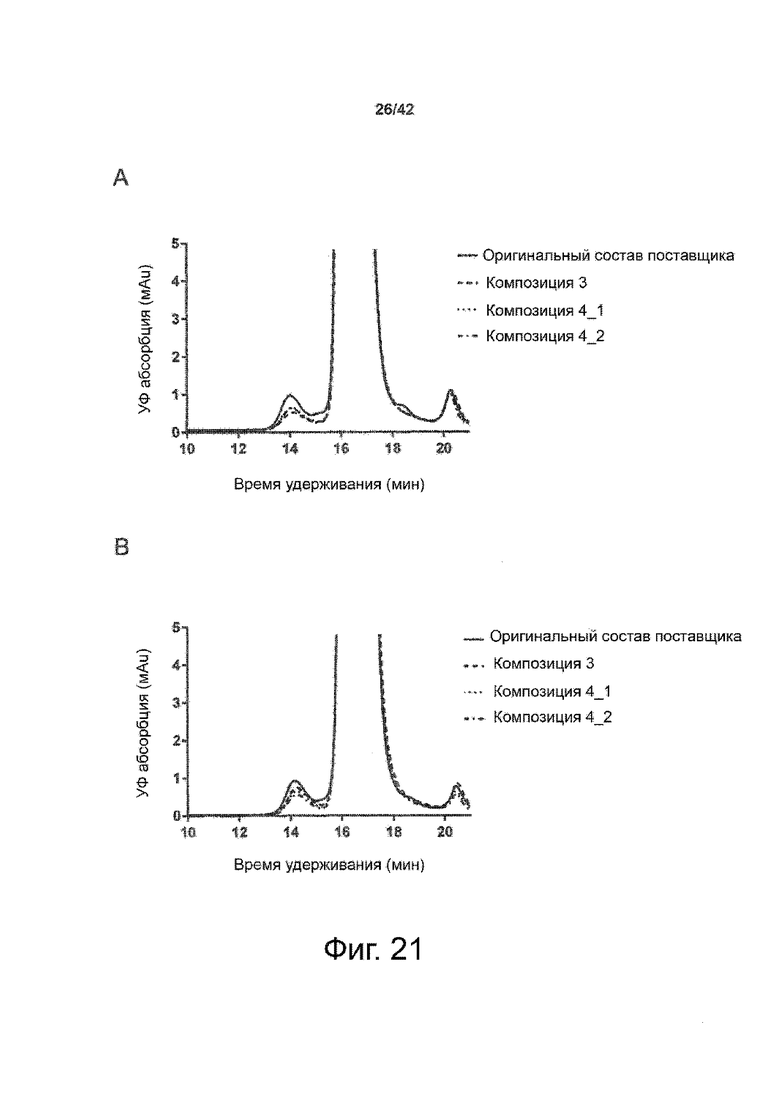
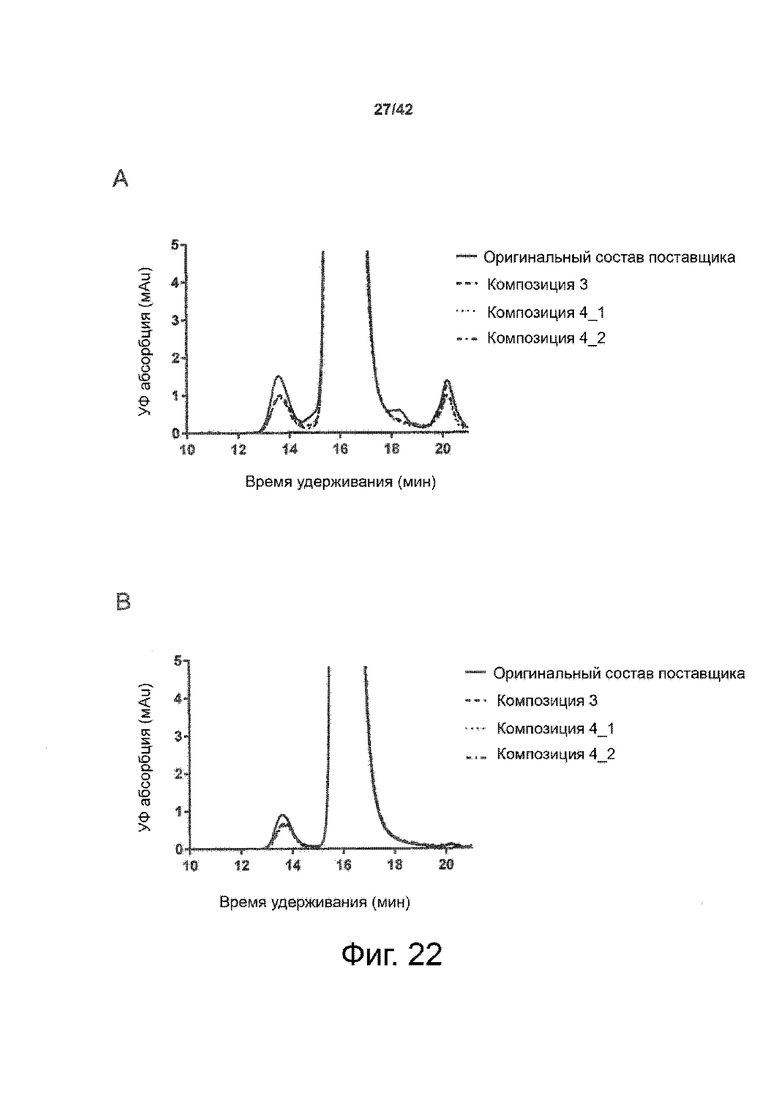
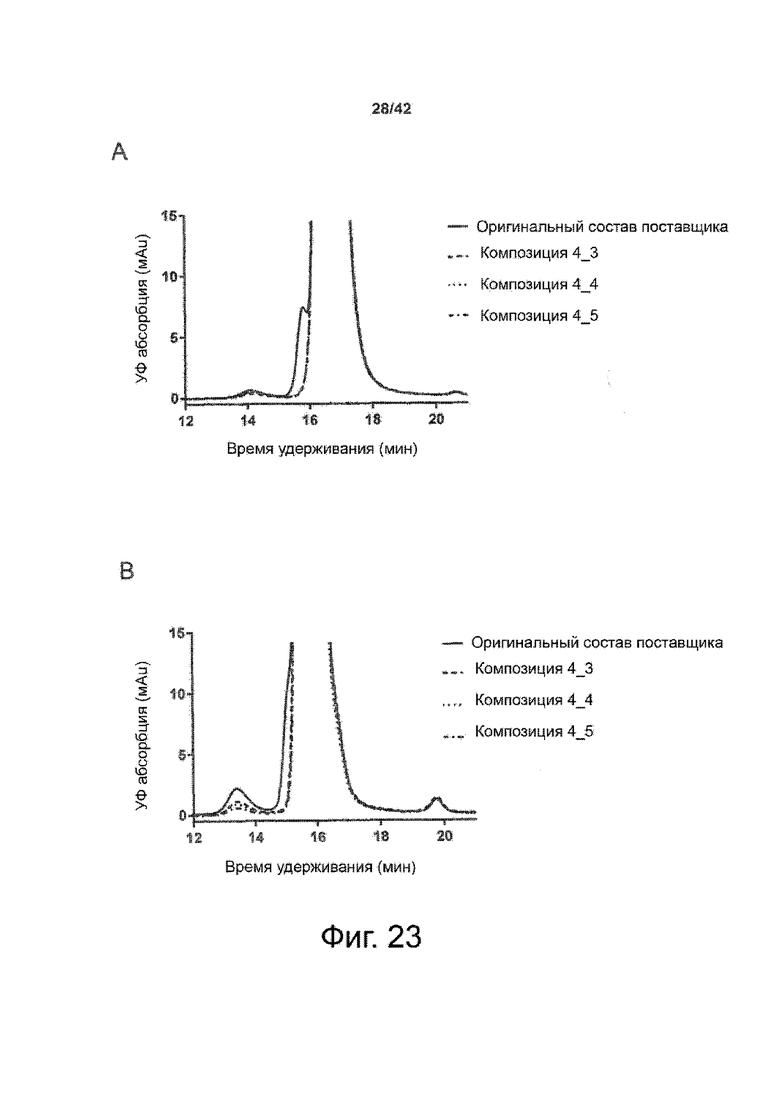
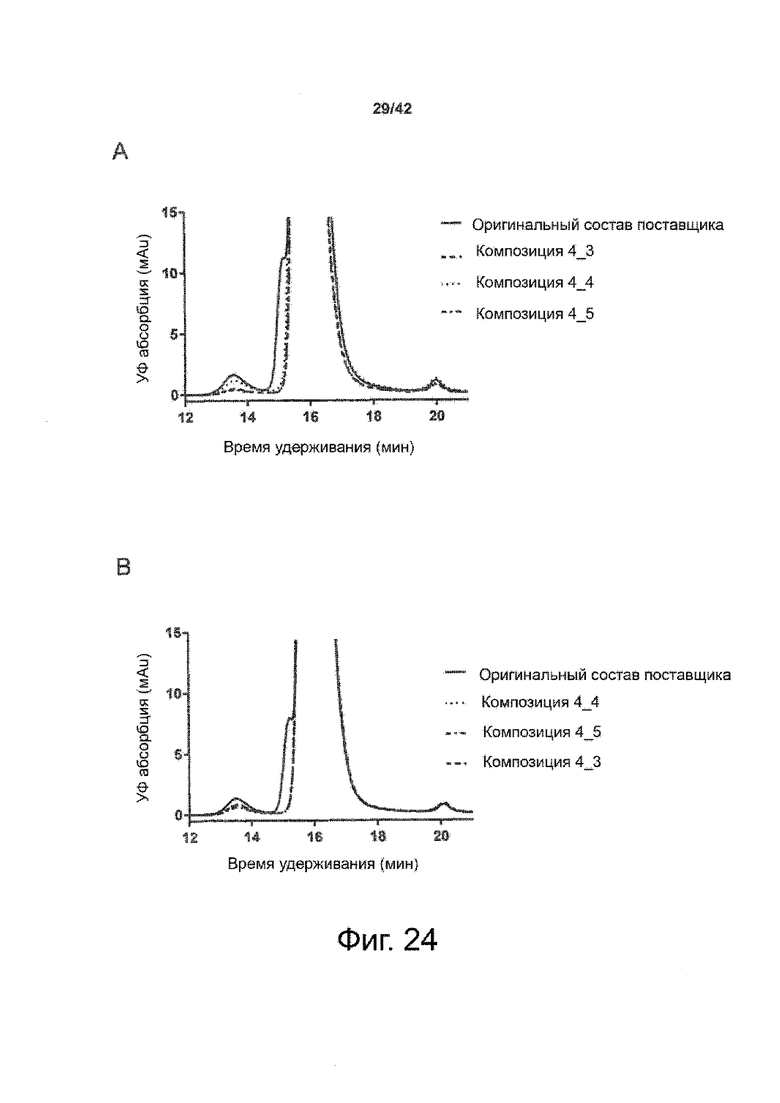
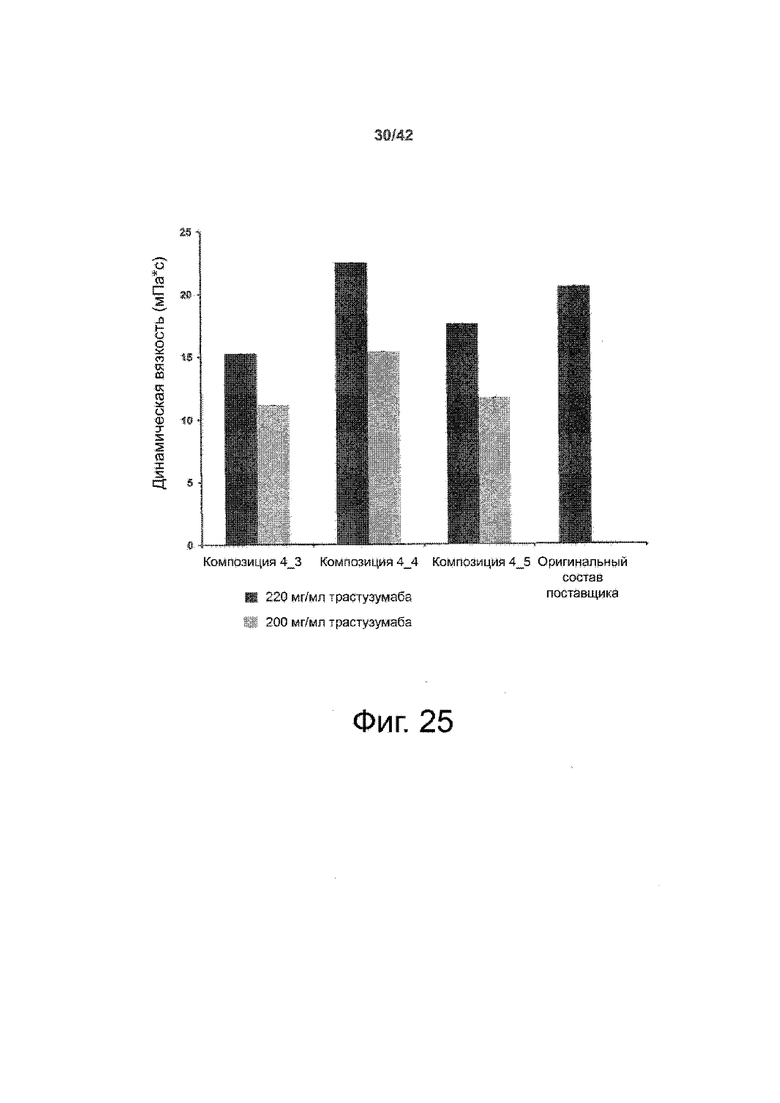
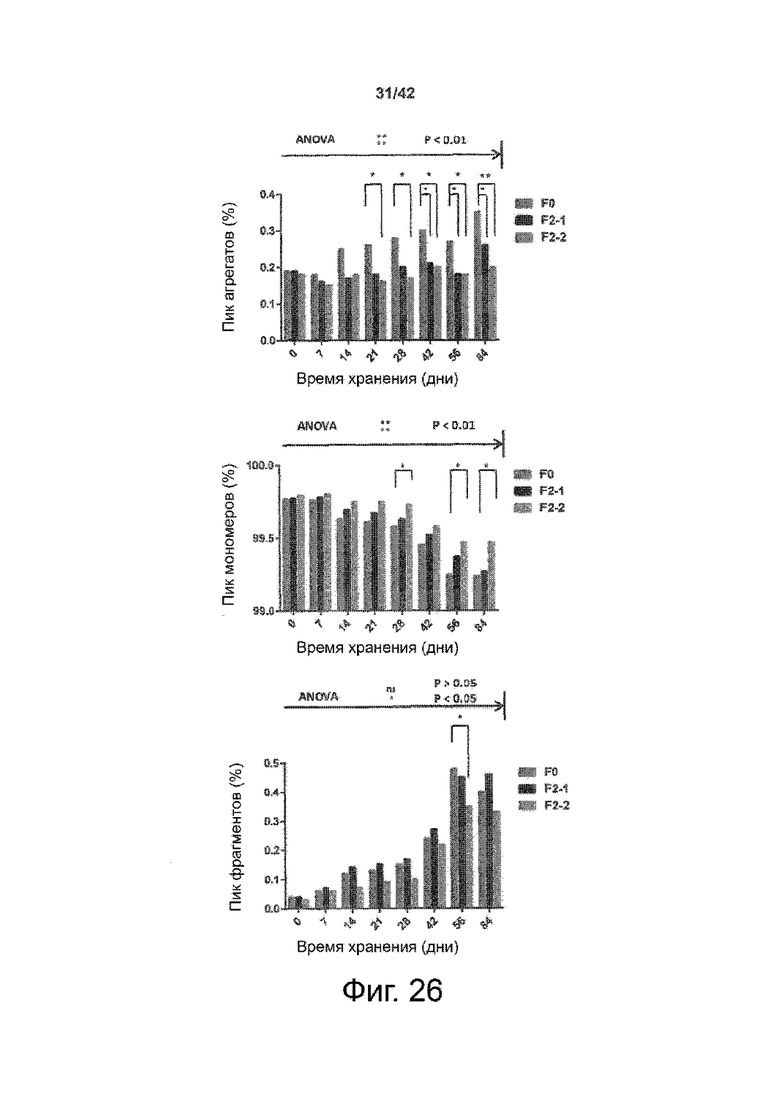
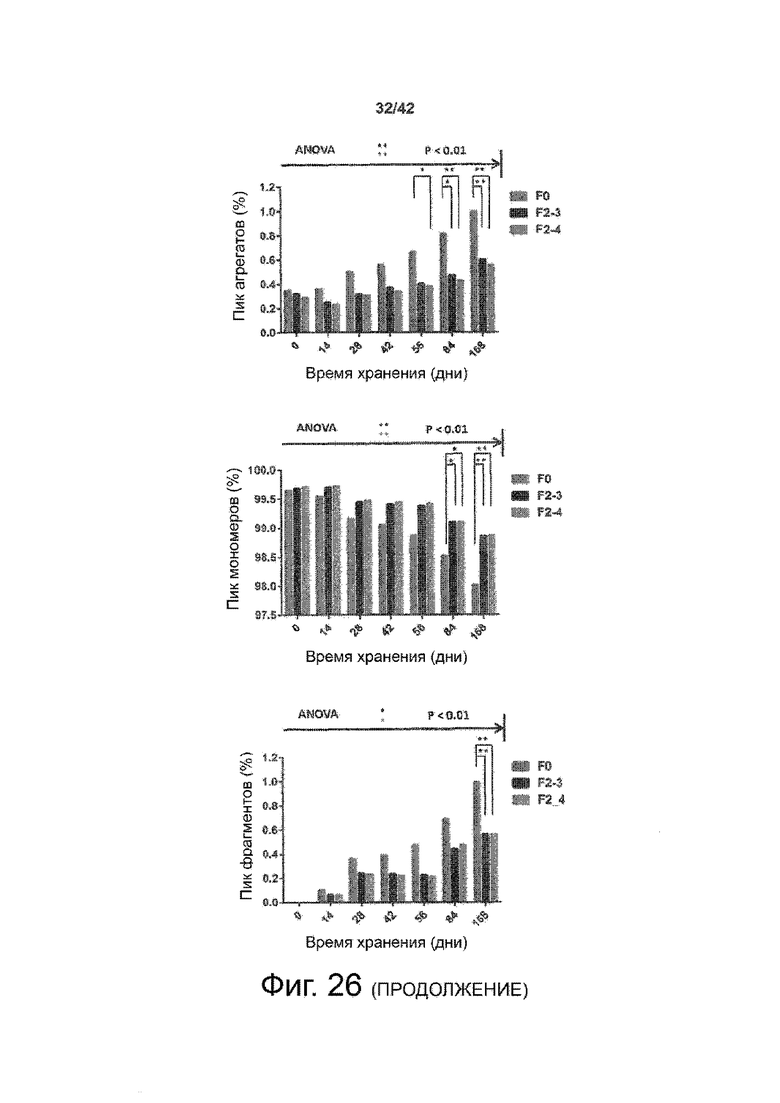
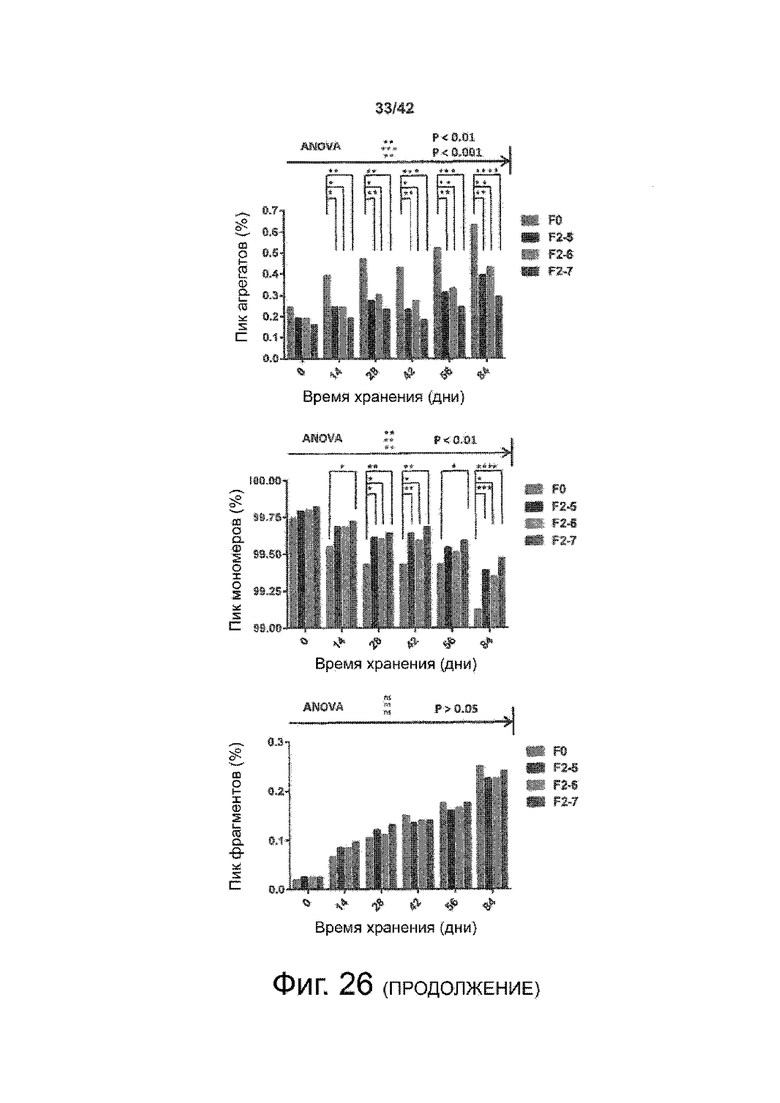
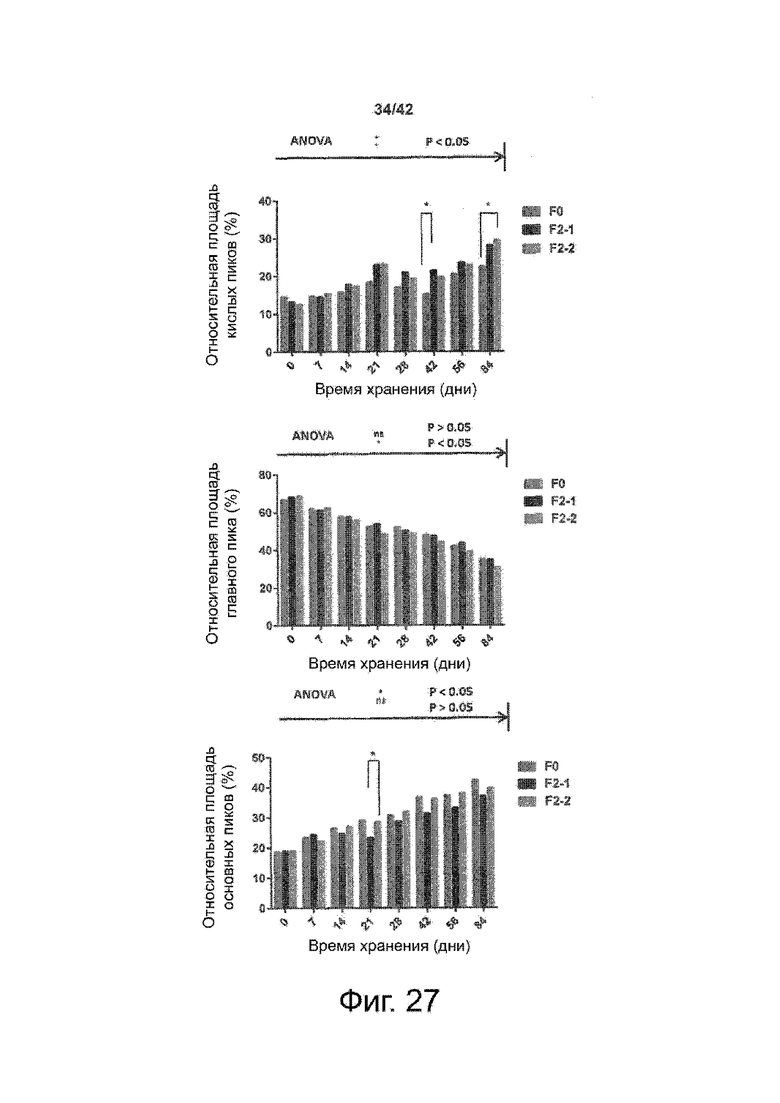
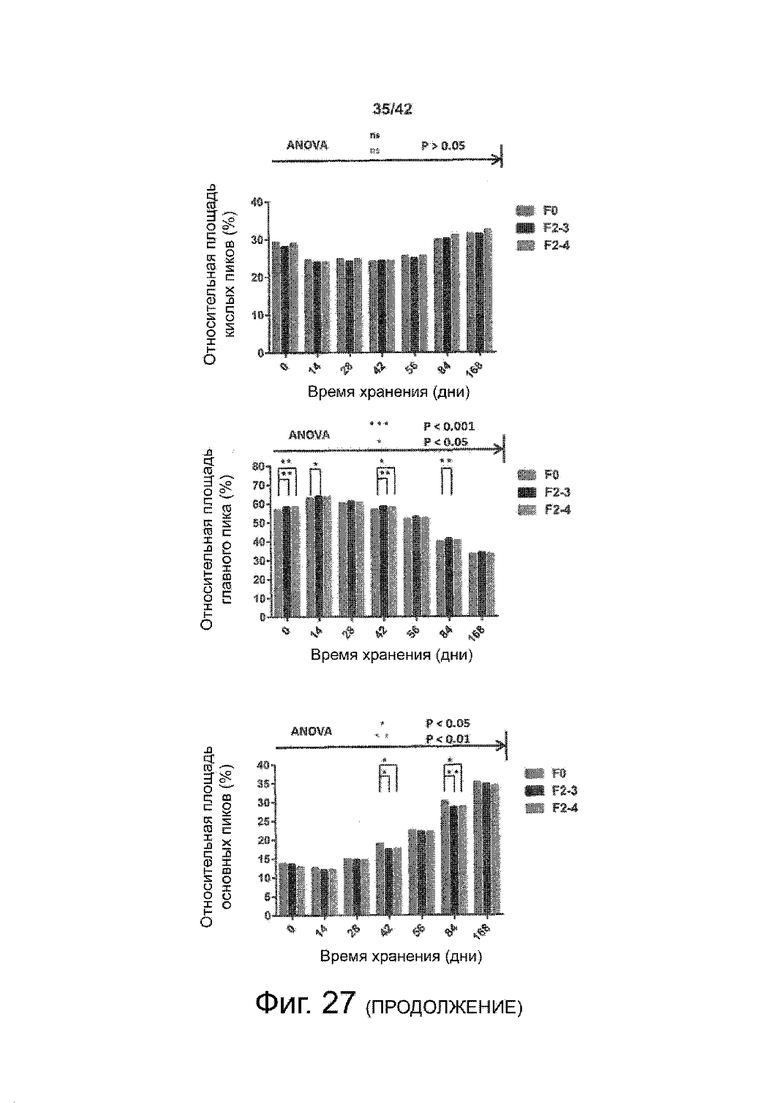
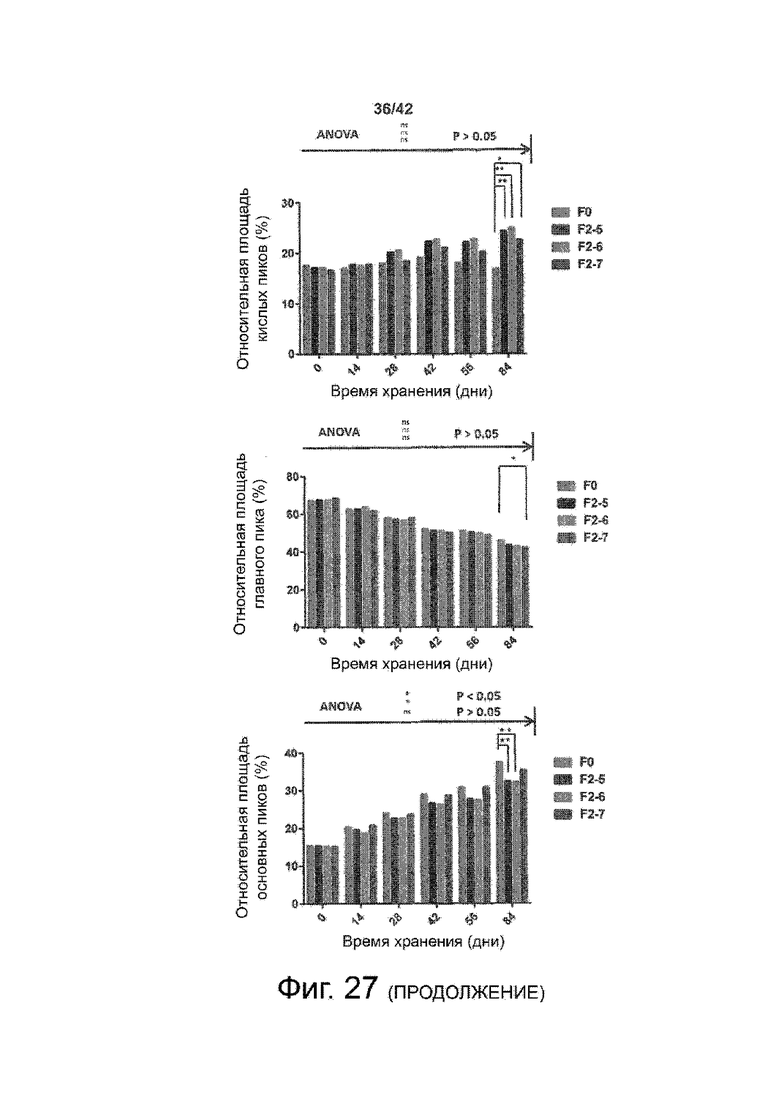
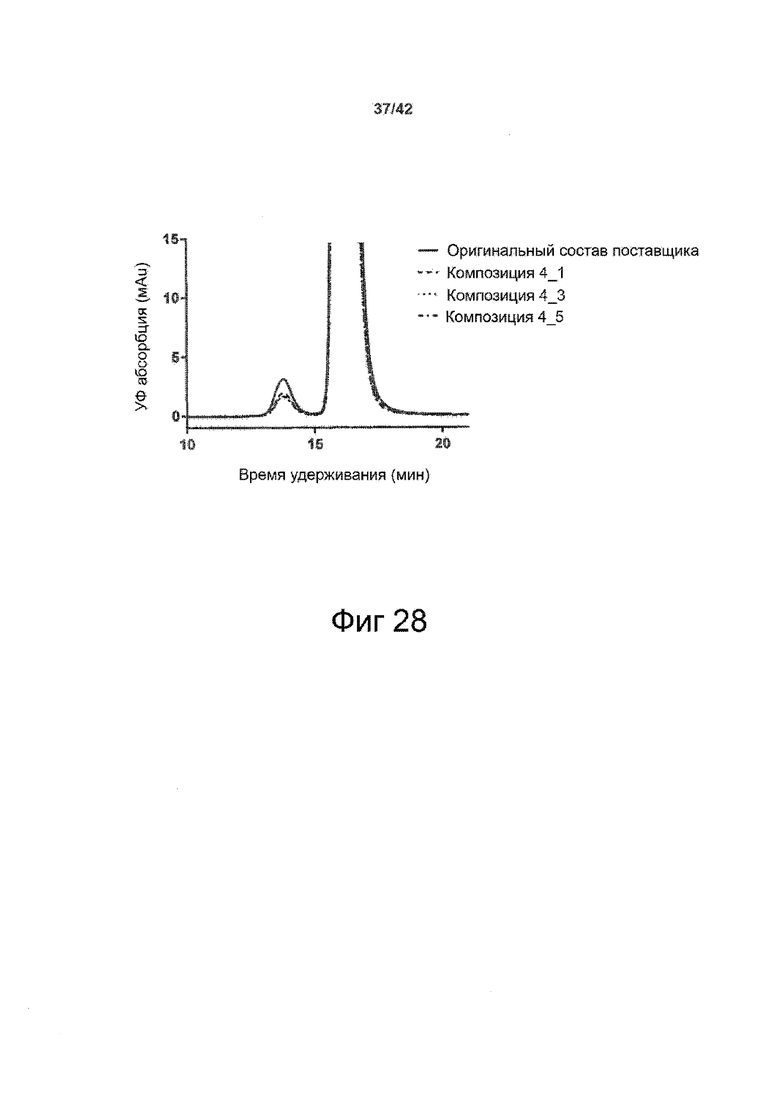
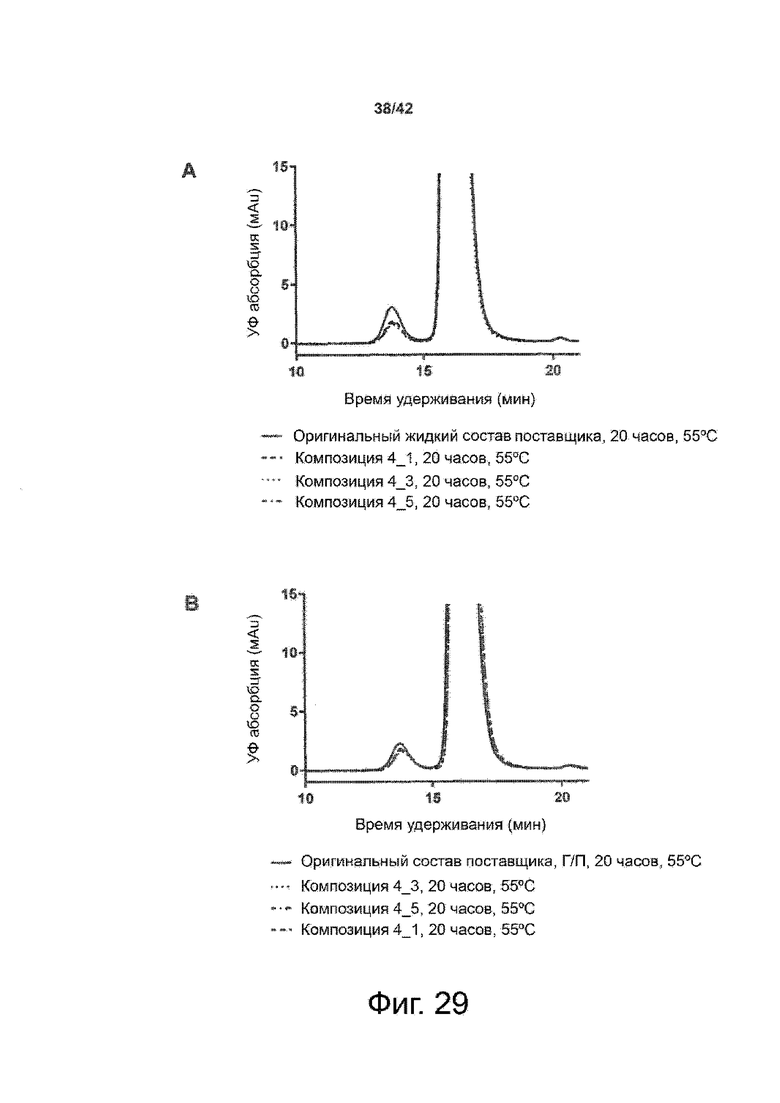
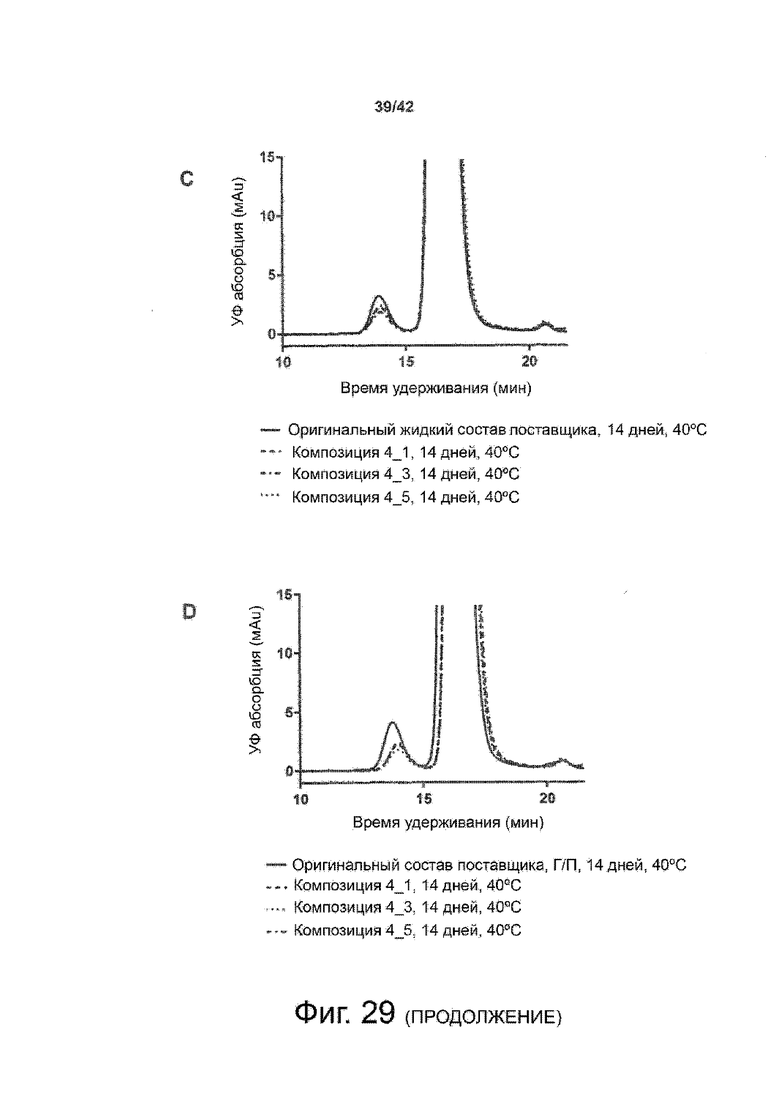
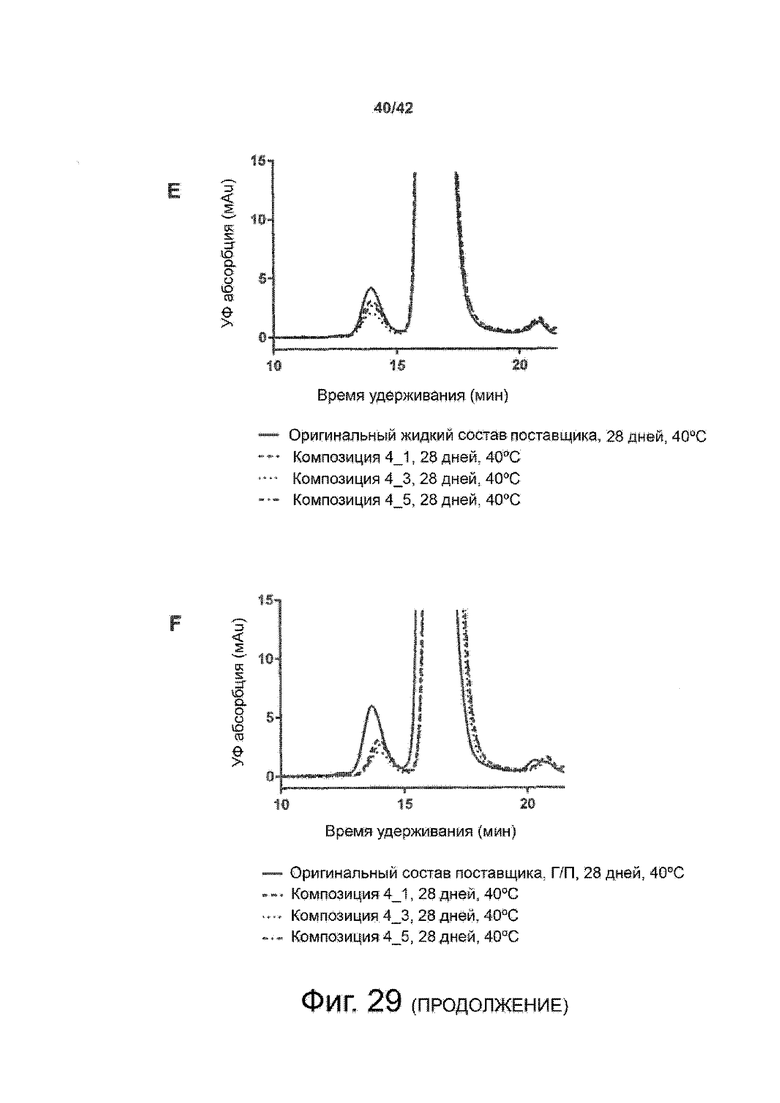
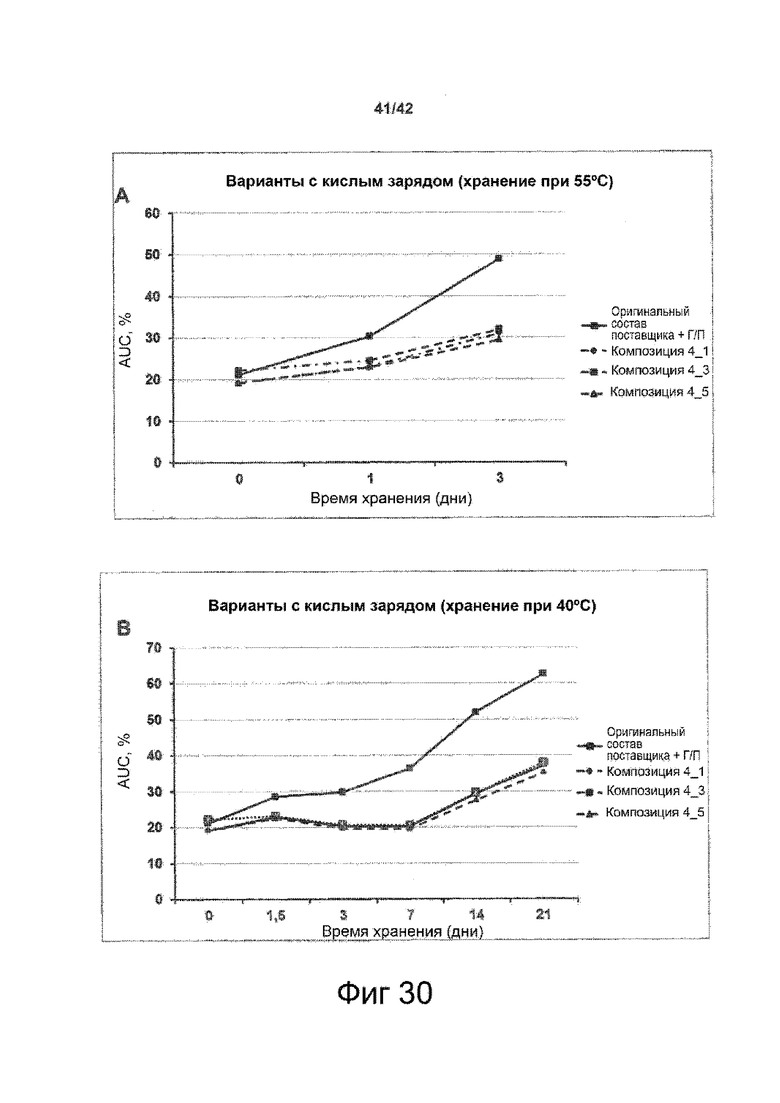
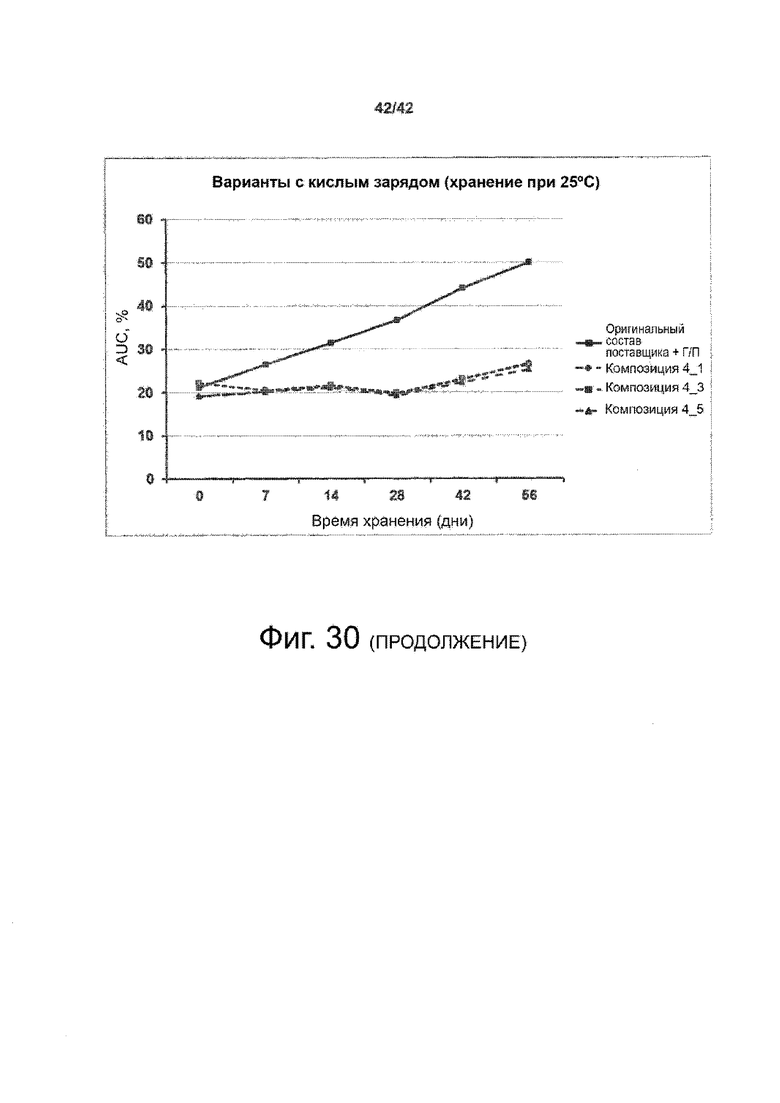
Настоящее изобретение относится к способу получения биофармацевтического лекарственного продукта, содержащего интересующую биомолекулу, причем способ включает: (а) первую фазу приготовления лекарственной субстанции из представляющей интерес биомолекулы, причем указанная первая фаза включает по меньшей мере один этап обработки, выбранный из сбора, обогащения, где указанный по меньшей мере один этап обработки в этой первой фазе проводят в присутствии композиции, содержащей по меньшей мере три аминокислоты, где комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию; и (b) вторую фазу дальнейшей обработки лекарственной субстанции, полученной в (а), для получения биофармацевтического лекарственного продукта, причем указанная вторая фаза включает по меньшей мере один этап обработки, выбранный из (b1) замены буфера, (b2) замораживания, (b3) размораживания и (b4) фасовки; где указанный по меньшей мере один этап обработки на этой второй фазе проводят в присутствии композиции, содержащей (i) по меньшей мере три аминокислоты, при этом комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию; и (ii) один или несколько сахаров; при отношении аминокислота: сахар от 10:1 до 1:100 (по массе). 13 з.п. ф-лы, 7 табл., 30 ил.
1. Способ получения биофармацевтического лекарственного продукта, включающего представляющую интерес биомолекулу, причем способ включает:
(а) первую фазу приготовления лекарственной субстанции из представляющей интерес биомолекулы, причем указанная первая фаза включает этап:
(а4) обогащения,
где указанный по меньшей мере один этап обработки в этой первой фазе проводят в присутствии композиции, содержащей по меньшей мере три аминокислоты, при этом комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию; а также
(b) вторую фазу дальнейшей обработки лекарственной субстанции, полученной на фазе (а), для получения биофармацевтического лекарственного продукта, причем указанная вторая фаза включает по меньшей мере один этап обработки, выбранный из:
(b1) ребуферизации,
(b2) замораживания,
(b3) размораживания, и
(b4) фасовки;
где указанный по меньшей мере один этап обработки на этой второй фазе проводят в присутствии композиции, содержащей
(i) по меньшей мере три аминокислоты, где комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию; а также
(ii) один или несколько дисахаридов;
при отношении аминокислота: сахар от 4:1 до 1:2 (по массе).
2. Способ по п.1, в котором биофармацевтический лекарственный продукт, полученный на фазе (b), дополнительно обрабатывают для хранения и/или применения в виде жидкой композиции.
3. Способ по п.2, в котором жидкая композиция предназначена для хранения и/или применения биофармацевтического лекарственного продукта в концентрации в диапазоне от 0,001 до <100 мг/мл.
4. Способ по п.2, в котором жидкая композиция предназначена для хранения и/или применения биофармацевтического лекарственного продукта в высокой концентрации в диапазоне от 100 до 500 мг/мл, и где композиция отличается тем, что содержит следующие вспомогательные вещества:
(i) по меньшей мере три аминокислоты, где комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию; и
(ii) один или несколько сахаров;
и где отношение между аминокислотами и сахаром корректируют от 4:1 до 1:1 (по массе).
5. Способ по п.3 или 4, в котором жидкую композицию дополнительно корректируют таким образом, чтобы отношение между представляющей интерес биомолекулой и по меньшей мере тремя аминокислотами из (i) составляло от 3,5:1 до 1:2 (по массе).
6. Способ по п.1, дополнительно включающий:
(c) третий этап сушки биофармацевтической лекарственной субстанции, полученной на этапе (b), для получения высушенного биофармацевтического лекарственного продукта, причем указанный этап сушки на этой третьей фазе проводят в присутствии композиции, содержащей:
(i) по меньшей мере три аминокислоты, где комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию; и
(ii) один или несколько сахаров;
и где отношение между интересующей биомолекулой и суммой вспомогательных веществ устанавливают в пределах от 1:1 до 1:10 (по массе).
7. Способ по п.6, в котором сушку биофармацевтического лекарственного продукта, полученного на стадии (b), осуществляют путем лиофилизации, распылительной сушки или распылительной сублимационной сушки.
8. Способ по п.6 или 7, в котором высушенный биофармацевтический лекарственный продукт, полученный на этапе (с), стерилизуют, предпочтительно стерилизуют на конечной стадии.
9. Способ по любому из п.6-8, дополнительно включающий этап восстановления высушенного биофармацевтического лекарственного продукта, полученного на этапе (с), для получения жидкой композиции,
отличающийся тем, что высушенный биофармацевтический лекарственный продукт восстанавливают для получения жидкого биофармацевтического лекарственного продукта в композиции, содержащей:
(i) по меньшей мере три аминокислоты, где комбинация указанных по меньшей мере трех аминокислот обеспечивает по меньшей мере одну положительно заряженную функциональную группу, по меньшей мере одну антиоксидантную функциональную группу, по меньшей мере одну осмолитическую функцию и по меньшей мере одну буферную функцию; и
(ii) один или несколько сахаров;
при отношении между аминокислотами и сахаром от 4:1 до 1:1 (по массе).
10. Способ по любому из пп.1-9, в котором композиция на этапе (а) содержит от 0,5 мг/мл до 10 мг/мл триптофана и от 0,5 мг/мл до 30 мг/мл гистидина.
11. Способ по любому из пп.1-10, в котором представляющую интерес биомолекулу выбирают из группы, состоящей из белков и пептидов, а также их смесей.
12. Способ по п.11, в котором представляющая интерес биомолекула является антигеном.
13. Способ по пп.3, 4, 5, 6 и 9, в котором отношение между аминокислотами и сахаром регулируют в диапазоне от 10:1 до 100:1.
14. Способ по пп.5 и 6, в котором отношение между представляющей интерес биомолекулой и суммой вспомогательных веществ устанавливают в пределах от 1:1 до 1:500.
| WO 2015140751 A1, 2015.09.24 | |||
| US 2014127227 A, 2014.05.08 | |||
| WO 2013001044 A1, 2013.01.03 | |||
| WO 2013055958 A1, 18.04.2013 | |||
| Электропневматический тормоз | 1924 |
|
SU23446A1 |
| WO 2015059284 A1, 2015.04.30. | |||

