Данная заявка претендует на приоритет предварительной заявки на патент США под номером 61/780621, зарегистрированной 13 марта 2013 года, и на приоритет предварительной заявки на патент США под номером 61/947850, зарегистрированной 4 марта 2014 года, каждая из которых во всей ее полноте включена в данный контекст путем ссылки.
Область техники, к которой относится изобретение
Данное изобретение относится к способам получения вызывающего апоптоз агента и химическим промежуточным продуктам для его получения. Также, данное изобретение относится к новым химическим промежуточным продуктам, связанным с предусматриваемыми в данном контексте способами.
Предпосылки создания изобретения
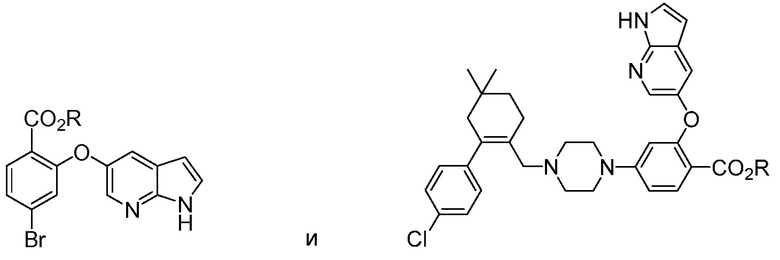
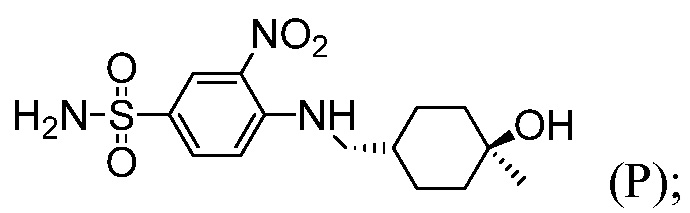
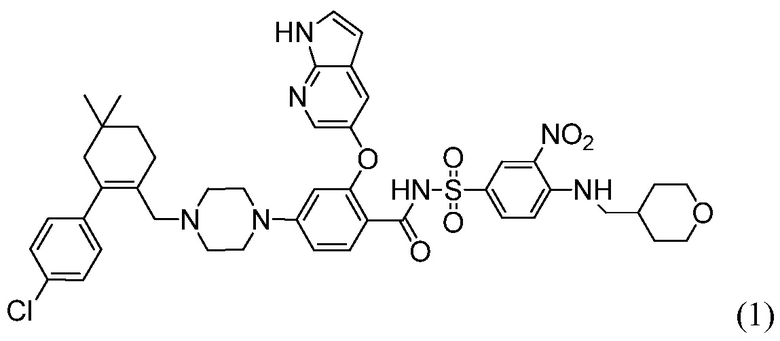
4-(4-{[2-(4-Хлорфенил)-4,4-диметилциклогекс-1-ен-1-ил]метил}пиперазин-1-ил)-N-({3-нитро-4-[(тетрагидро-2Н-пиран-4-илметил)амино]фенил}сульфонил)-2-(1Н-пирроло[2,3-b]пиридин-5-илокси)бензамид (в дальнейшем, «соединение 1») и 4-(4-{[2-(4-хлорфенил)-4,4-диметилциклогекс-1-ен-1-ил]метил}пиперазин-1-ил)-N-({3-нитро-4-[(1R,4R)-[4-гидрокси-4-метилциклогексил]метил)амино]фенил}сульфонил)-2-(1Н-пирроло[2,3-b]пиридин-5-илокси)бензамид (в дальнейшем, «соединение 2»), каждый, представляют собой эффективные и селективные ингибиторы Bcl-2, обладающие, между прочим, противоопухолевой активностью, в качестве вызывающих апоптоз агентов.
Соединение 1 имеет формулу
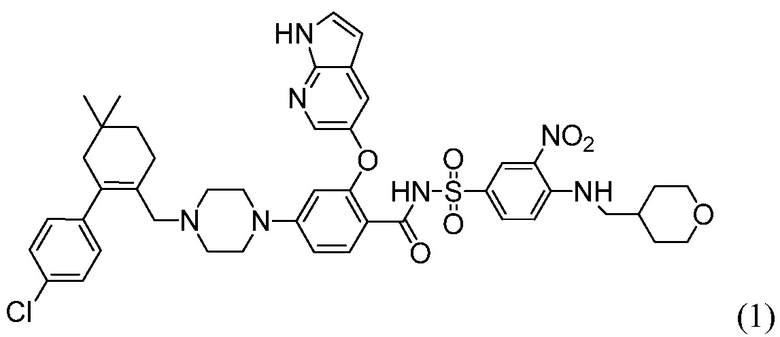
Соединение 2 имеет формулу:
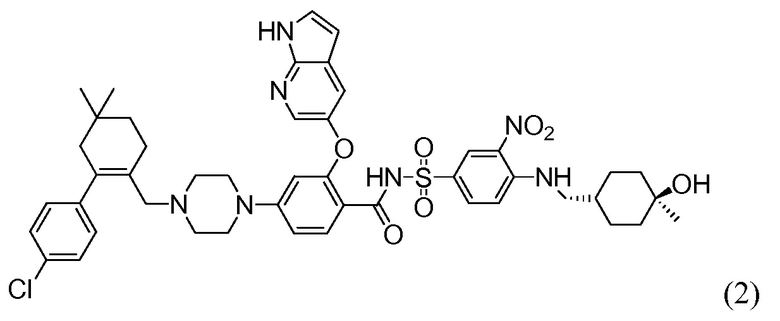
Соединение 1 в настоящее время является объектом продолжающихся клинических исследований в отношении лечения хронической лимфоцитарной лейкемии. В публикации патента США под номером 2010/0305122 раскрываются соединение 1, соединение 2 и другие соединения, которые обладают эффективным связыванием с белком семейства Bcl-2, и их фармацевтически приемлемые соли. В публикациях патентов США под номерами 2012/0108590 и 2012/0277210 раскрываются фармацевтические композиции, содержащие такие соединения, и способы лечения неопластических, иммунных или аутоиммунных заболеваний, включающие такие соединения. В публикации патента США под номером 2012/0129853 раскрываются способы лечения системной красной волчанки, волчаночного нефрита или синдрома Шегрена, включающие такие соединения. В публикации патента США под номером 2012/0157470 раскрываются фармацевтически приемлемые соли и кристаллические формы Соединения 1. Раскрытия патентов США 2010/0305122; 2012/0108590; 2012/0129853; 2012/0157470 и 2012/0277210 во всей их полноте включены в данный контекст путем ссылки.
Краткое изложение сущности изобретения
Данное изобретение относится к способам получения соединений формулы А1:
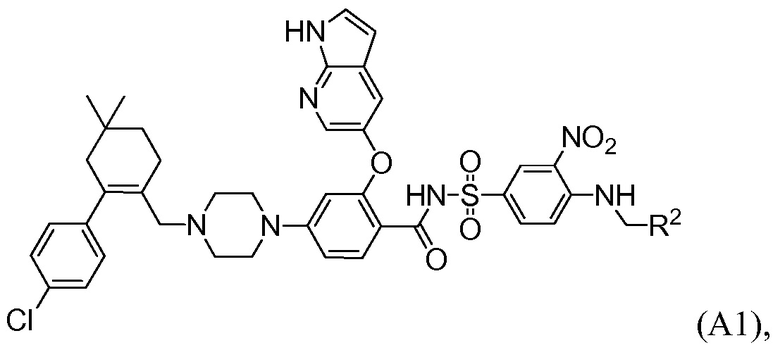
где R2 выбирают из: 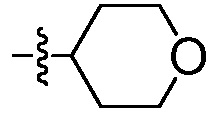 и
и 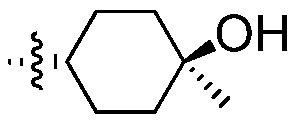 .
.
Также, данное изобретение относится к соединениям формул:
где R означает С1-С12-алкил; и к способам их получения.
Подробное описание изобретения
Данное изобретение относится к способу получения соединений формулы А1:
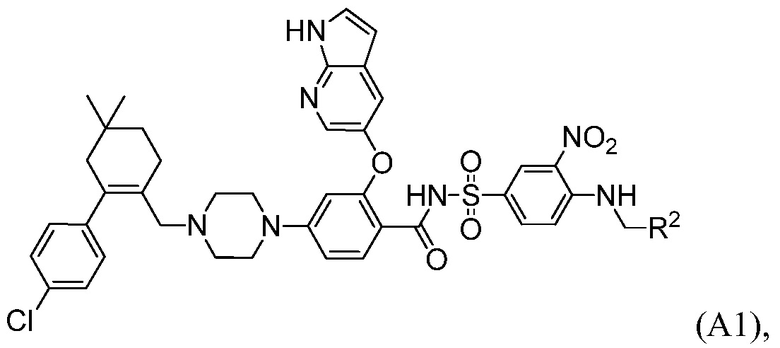
где R2 выбирают из: 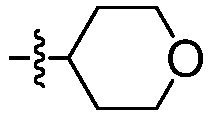 и
и  ,
,
который включает:
а) комбинирование соединения формулы (К):
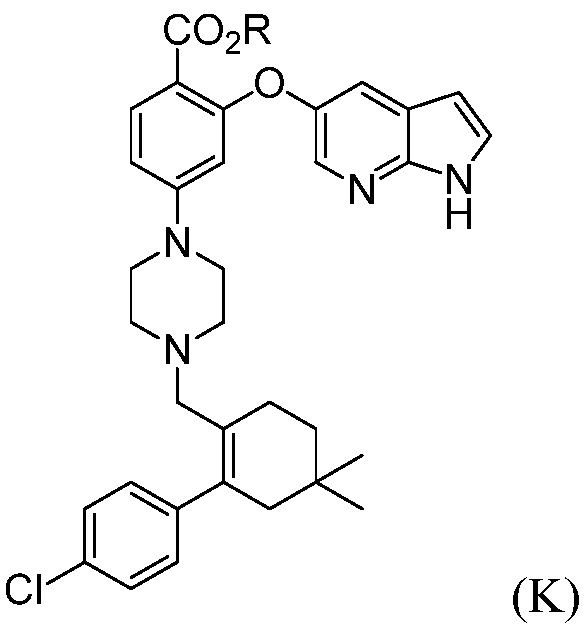
где R означает С1-С12-алкил,
с трет-бутоксидом, апротонным органическим растворителем и водой, до получения соединения формулы (L):
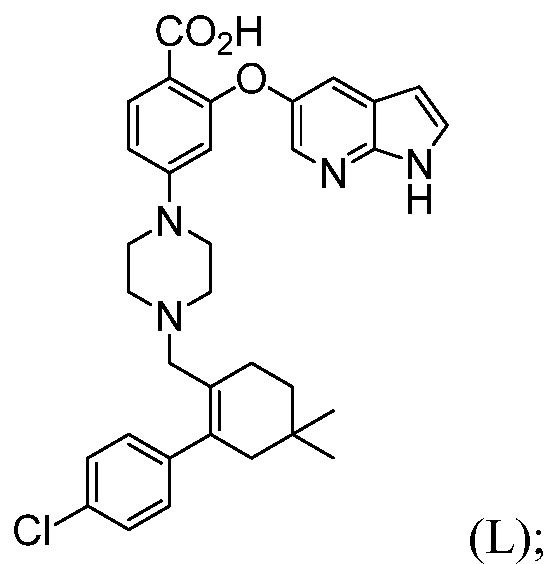
и
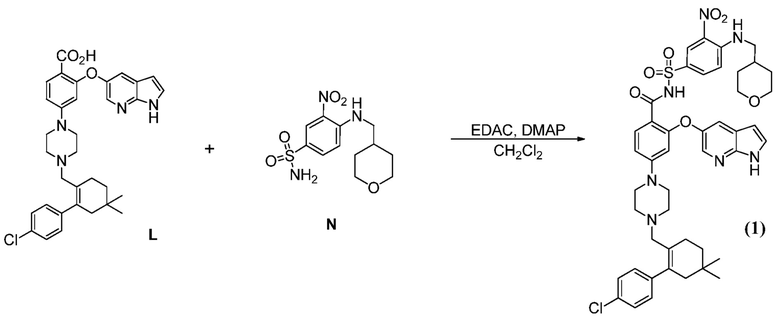
(b”) комбинирование соединения формулы (L) с 1-этил-3-(3-диметиламинопропил)карбодиимидгидрохлоридом (EDAC), 4-диметиламинопиридином (DMAP), органическим растворителем и или с соединением формулы (N), до получения соединения формулы (А1), где R2 означает: 
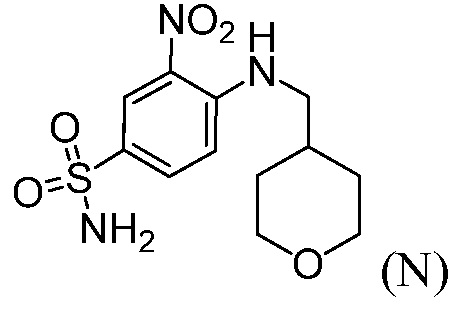
или с соединением формулы (Р), до получения соединения формулы (А1), где R2 означает: 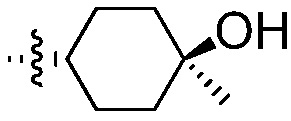
таким образом получая соединение формулы (А1).
В одном воплощении, R2 означает: 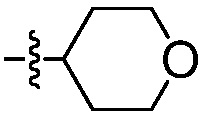 .
.
В другом воплощении, R2 означает: 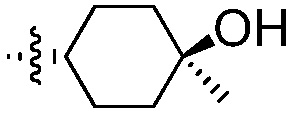 .
.
В некоторых воплощениях, R означает С1-С6-алкил. В некоторых воплощениях, R означает С1-С4-алкил. В некоторых воплощениях, R выбирают из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, трет-бутила, изобутила и необутила. В некоторых воплощениях, R означает трет-бутил.
В одном воплощении, способ, предусматриваемый в данном контексте, далее включает:
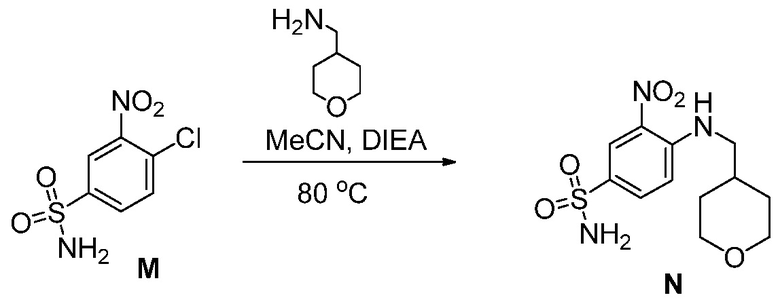
(с”) комбинирование соединения формулы (М):

с основанием в виде третичного амина, органическим растворителем и или (тетрагидро-2Н-пиран-4-ил)метанамином или его солью, до получения соединения формулы (N), или с (1R,4R)-4-(аминометил)-1-метилциклогексанолом или его солью, до получения соединения формулы (Р).
В одном воплощении, соль (1R,4R)-4-(аминометил)-1-метилциклогексанола, со стадии (с”), представляет собой соль п-толуолсульфоновой кислоты.
В другом воплощении, способ, предусматриваемый в данном контексте, далее включает:
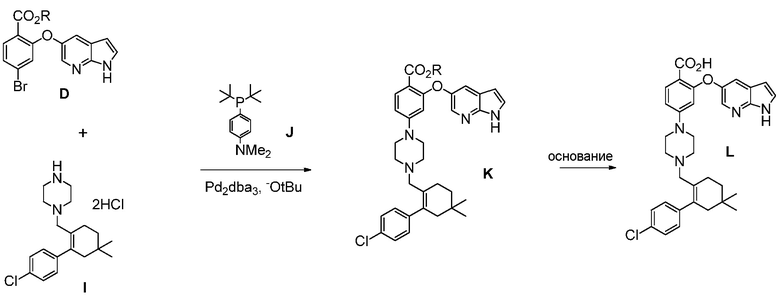
(d) комбинирование соединения формулы (D):
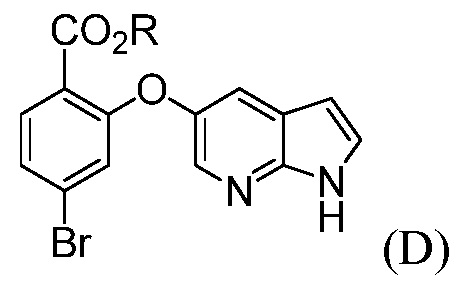
где R означает С1-С12-алкил,
с соединением формулы (I):

с источником палладия, трет-бутоксидом и с фосфиновым лигандом, в апротонном органическом растворителе, до получения соединения формулы (К).
В некоторых воплощениях, фосфиновый лиганд представляет собой соединение формулы (J):

В других воплощениях, фосфиновый лиганд выбирают из группы, состоящей из:
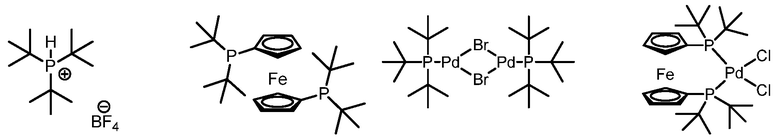
В другом воплощении, способ, предусматриваемый в данном контексте, далее включает:
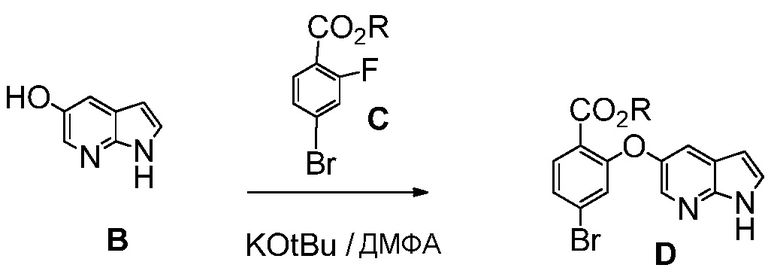
(е) комбинирование соединения формулы (В) с соединением формулы (С):

где R означает С1-С12-алкил,
и с трет-бутоксидом, в органическом растворителе, до получения соединения формулы (D).
В другом воплощении, способ, предусматриваемый в данном контексте, далее включает:
(f) комбинирование соединения формулы (А):

с R1MgX, в апротонном органическом растворителе;
где R1 означает С1-С6-алкил; и Х означает Cl, Br или I; и
(g) комбинирование С1-С12-алкилхлорформиата или ди(С1-С12-алкил)дикарбоната с продуктом со стадии (f), до получения соединения формулы (С).
В другом воплощении, способ, представленный в данном контексте, далее включает:
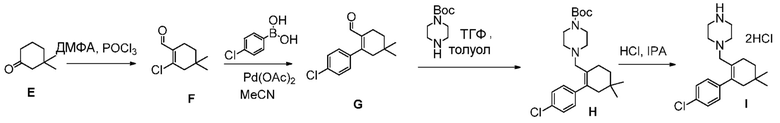
(h) комбинирование соединения формулы (Е):
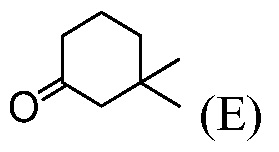
с ДМФА и POCl3, до получения соединения формулы (F):

(i) комбинирование соединения формулы (F) с источником палладия и 4-хлорфенилбороновой кислотой, в органическом растворителе, до получения соединения формулы (G):
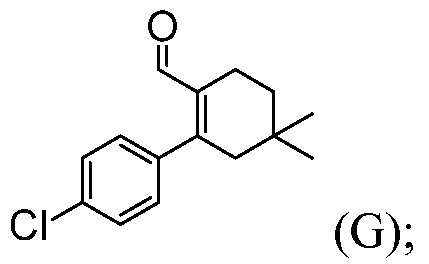
(j) комбинирование соединения формулы (G) с ВОС-пиперазином и триацетоксиборгидридом натрия, в органическом растворителе, до получения соединения формулы (Н):
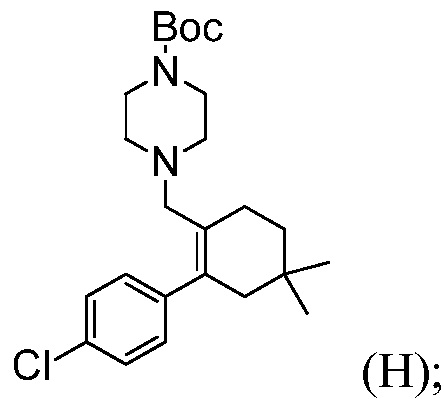
и
(k) комбинирование соединения формулы (Н) с соляной кислотой, до получения соединения формулы (I).
В одном воплощении, способ включает стадию (а), стадию (b”), стадию (c”) и стадию (d). В одном воплощении, способ включает стадию (а), стадию (b”), стадию (c”), стадию (d) и стадию (е). В одном воплощении, способ включает стадию (а), стадию (b”), стадию (c”), стадию (d), стадию (е), стадию (f) и стадию (g). В другом воплощении, способ включает стадию (а), стадию (b”), стадию (c”), стадию (d), стадию (е), стадию (f), стадию (g), стадию (h), стадию (i), стадию (j) и стадию (k).
В одном воплощении, способ включает стадии (а), (b”) и (d). В другом воплощении, способ включает стадии (а), (b”), (d) и (е). В другом воплощении, способ включает стадии (а), (b”), (d), (h), (i), (j) и (k). В другом воплощении, способ включает стадии (а), (b”), (c”), (d), (h), (i), (j) и (k). В другом воплощении, способ включает стадии (а), (b”), (d), (f), (g), (h), (i), (j) и (k). В другом воплощении, способ включает стадии (а), (b”), (d), (е), (f), (g), (h), (i), (j) и (k).
Также, данное изобретение относится к способу получения Соединения 1 формулы:
который включает:
(а) комбинирование соединения формулы (К):
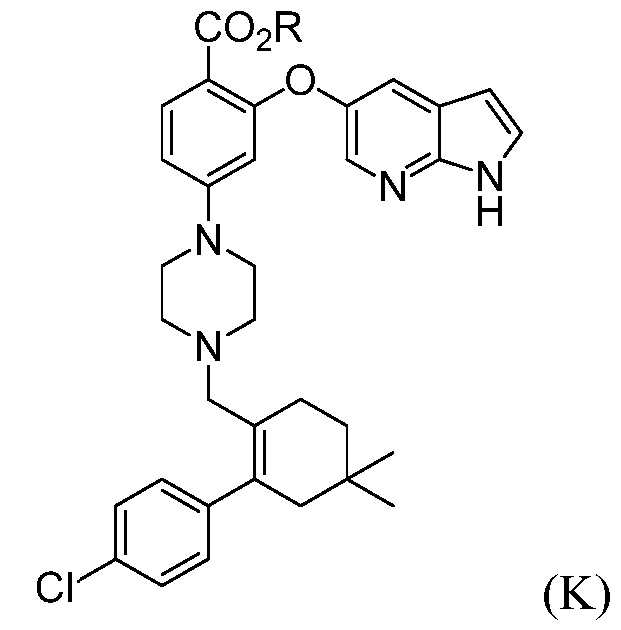
где R означает С1-С12-алкил,
с трет-бутоксидом, апротонным органическим растворителем и водой, до получения соединения формулы (L):
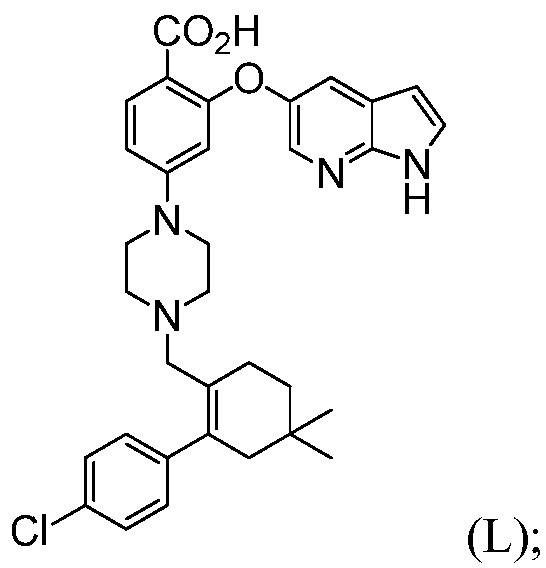
(b) комбинирование соединения формулы (L) с соединением формулы (N):
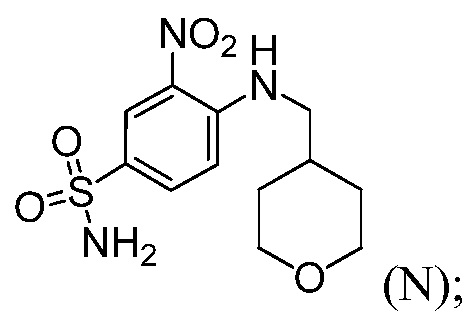
и с 1-этил-3-(3-диметиламинопропил)карбодиимидгидрохлоридом (EDAC), 4-диметиламинопиридином (DMAP) и органическим растворителем, до получения соединения формулы (1).
В некоторых воплощениях, R означает С1-С6-алкил. В некоторых воплощениях, R означает С1-С4-алкил. В некоторых воплощениях, R выбирают из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, трет-бутила, изобутила и необутила. В некоторых воплощениях, R означает трет-бутил.
В одном воплощении, способ получения Соединения 1 далее включает:
(с) комбинирование соединения формулы (М):
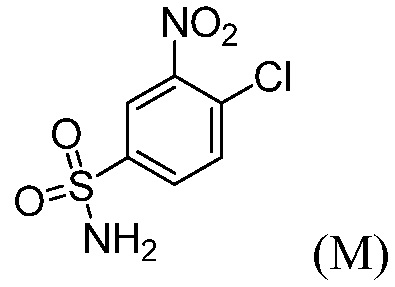
с (тетрагидро-2Н-пиран-4-ил)метанамином, основанием в виде третичного амина и органическим растворителем, до получения соединения формулы (N).
В другом воплощении, способ получения Соединения 1 далее включает:
(d) комбинирование соединения формулы (D):
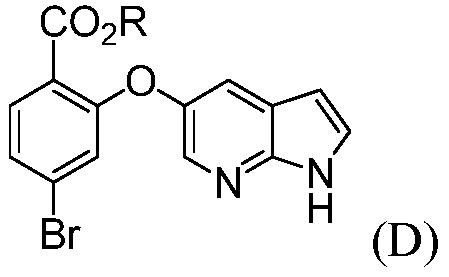
где R означает С1-С12-алкил,
с соединением формулы (I):
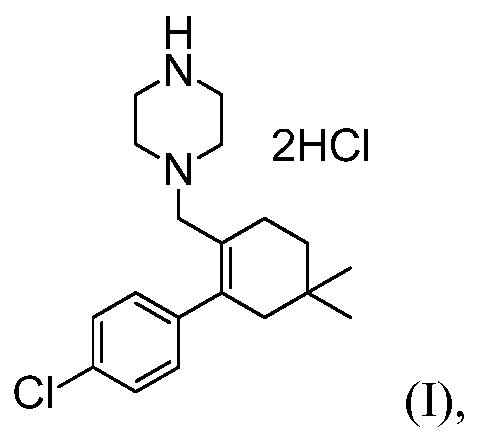
с источником палладия, трет-бутоксидом и фосфиновым лигандом, в апротонном органическом растворителе, до получения соединения формулы (К).
В некоторых воплощениях, фосфиновый лиганд представляет собой соединение формулы (J):
В других воплощениях, фосфиновый лиганд выбирают из группы, состоящей из:
В другом воплощении, способ получения Соединения 1 далее включает:
(е) комбинирование соединения формулы (В) с соединением формулы (С):
где R означает С1-С12-алкил,
и с трет-бутоксидом, в органическом растворителе, до получения соединения формулы (D).
В другом воплощении, способ получения Соединения 1 далее включает:
(f) комбинирование соединения формулы (А):
с R1MgX, в апротонном органическом растворителе;
где R1 означает С1-С6-алкил; и Х означает Cl, Br или I; и
(g) комбинирование С1-С12-алкилхлорформиата или ди(С1-С12-алкил)дикарбоната с продуктом со стадии (f), до получения соединения формулы (С).
В другом воплощении, способ получения Соединения 1 далее включает:
(h) комбинирование соединения формулы (Е):
с ДМФА и POCl3, до получения соединения формулы (F):
(i) комбинирование соединения формулы (F) с источником палладия и 4-хлорфенилбороновой кислотой, в органическом растворителе, до получения соединения формулы (G):
(j) комбинирование соединения формулы (G) с ВОС-пиперазином и триацетоксиборгидридом натрия, в органическом растворителе, до получения соединения формулы (Н):
и
(k) комбинирование соединения формулы (Н) с соляной кислотой, до получения соединения формулы (I).
В одном воплощении, способ получения Соединения 1 включает стадии (а)-(d). В одном воплощении, способ получения Соединения 1 включает стадии (а)-(е). В другом воплощении, способ получения Соединения 1 включает стадии (а)-(g). В другом воплощении, способ получения Соединения 1 включает стадии (а)-(k).
В одном воплощении, способ получения Соединения 1 включает стадии (а), (b) и (d). В другом воплощении, способ получения Соединения 1 включает стадии (а), (b), (d) и (е). В другом воплощении, способ получения Соединения 1 включает стадии (а), (b), (d), (h), (i), (j) и (k). В другом воплощении, способ получения Соединения 1 включает стадии (а), (b), (с), (d), (h), (i), (j) и (k). В другом воплощении, способ получения Соединения 1 включает стадии (а), (b), (d), (f), (g), (h), (i), (j) и (k). В другом воплощении, способ получения Соединения 1 включает стадии (а), (b), (d), (е), (f), (g), (h), (i), (j) и (k).
Также, данное изобретение относится к способу получения Соединения 2 формулы:
который включает:
(а) комбинирование соединения формулы (К):
где R означает С1-С12-алкил,
с трет-бутоксидом, апротонным органическим растворителем и водой, до получения соединения формулы (L):
(b’) комбинирование соединения формулы (L) с соединением формулы (Р):
и с 1-этил-3-(3-диметиламинопропил)карбодиимидгидрохлоридом (EDAC), 4-диметиламинопиридином (DMAP) и органическим растворителем, до получения соединения формулы (2).
В некоторых воплощениях, R означает С1-С6-алкил. В некоторых воплощениях, R означает С1-С4-алкил. В некоторых воплощениях, R выбирают из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, трет-бутила, изобутила и необутила. В некоторых воплощениях, R означает трет-бутил.
В одном воплощении, способ получения Соединения 2 далее включает:
(с’) комбинирование соединения формулы (М):
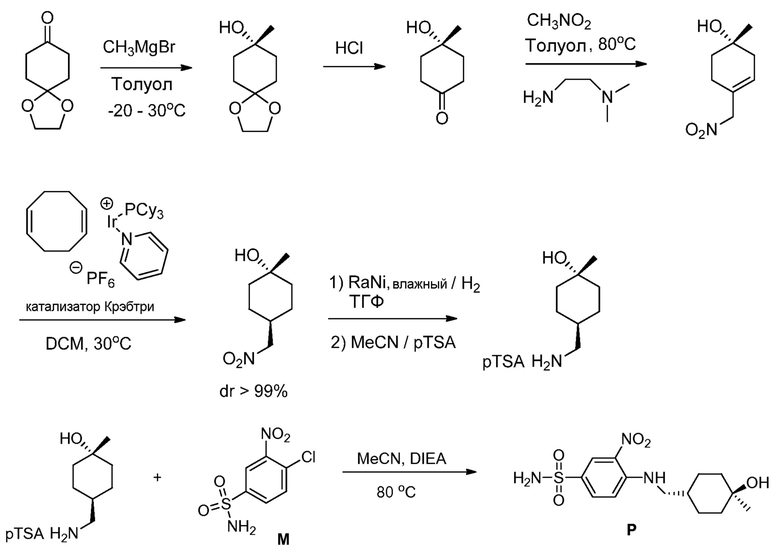
с (1R,4R)-4-(аминометил)-1-метилциклогексанолом или его солью, основанием в виде третичного амина и органическим растворителем, до получения соединения формулы (Р).
В одном воплощении, соль (1R,4R)-4-(аминометил)-1-метилциклогексанола со стадии (с’) представляет собой соль п-толуолсульфоновой кислоты.
В некоторых воплощениях, способ получения Соединения 2 далее включает стадию (d), как описано выше для получения Соединения 1.
В некоторых воплощениях, способ получения Соединения 2 далее включает стадию (е), как описано выше для получения Соединения 1.
В некоторых воплощениях, способ получения Соединения 2 далее включает стадию (f) и стадию (g), как описано выше для получения Соединения 1.
В некоторых воплощениях, способ получения Соединения 2 далее включает стадию (h), стадию (i), стадию (j) и стадию (k), как описано выше для получения Соединения 1.
В одном воплощении, способ получения Соединения 2 включает стадию (а), стадию (b’), стадию (c’) и стадию (d). В одном воплощении, способ получения Соединения 2 включает стадию (а), стадию (b’), стадию (c’), стадию (d) и стадию (е). В одном воплощении, способ получения Соединения 2 включает стадию (а), стадию (b’), стадию (c’), стадию (d), стадию (е), стадию (f) и стадию (g). В другом воплощении, способ получения Соединения 2 включает стадию (а), стадию (b’), стадию (c’), стадию (d), стадию (е), стадию (f), стадию (g), стадию (h), стадию (i), стадию (j) и стадию (k).
В одном воплощении, способ получения Соединения 2 включает стадии (а), (b’) и (d). В другом воплощении, способ получения Соединения 2 включает стадии (а), (b’), (d) и (е). В другом воплощении, способ получения Соединения 2 включает стадии (а), (b’), (d), (h), (i), (j) и (k). В другом воплощении, способ получения Соединения 2 включает стадии (а), (b’), (c’), (d), (h), (i), (j) и (k). В другом воплощении, способ получения Соединения 2 включает стадии (а), (b’), (d), (f), (g), (h), (i), (j) и (k). В другом воплощении, способ получения Соединения 2 включает стадии (а), (b’), (d), (е), (f), (g), (h), (i), (j) и (k).
В некоторых воплощениях, на стадии (а), трет-бутоксид выбирают из группы, состоящей из трет-бутоксида натрия и трет-бутоксида калия. В некоторых воплощениях, на стадии (а), трет-бутоксид представляет собой трет-бутоксид натрия. В некоторых воплощениях, на стадии (а), трет-бутоксид представляет собой трет-бутоксид калия.
В некоторых воплощениях, на стадии (а), апротонный органический растворитель выбирают из группы, состоящей из дихлорметана, хлороформа, ацетона, ацетонитрила, ТГФ, ДМФА, NMP, НМРА, диоксана, нитрометана, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, на стадии (а), апротонный органический растворитель представляет собой 2-метилтетрагидрофуран.
В некоторых воплощениях, на стадии (b), стадии (b’) и/или стадии (b”), органический растворитель выбирают из группы, состоящей из пентана, гексана, гептана, циклогексана, метанола, этанола, 1-пропанола, изопропанола, 1-бутанола, 2-бутанола, трет-бутанола, 2-бутанона, дихлорметана, хлороформа, тетрахлорида углерода, 1,2-дихлорэтана, ТГФ, ДМФА, НМРА, NMP, нитрометана, ацетона, уксусной кислоты, ацетонитрила, этилацетата, диэтилового эфира, диэтиленгликоля, глима, диглима, петролейного эфира, диоксана, МТВЕ, бензола, толуола, ксилола, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, на стадии (b), стадии (b’) и/или стадии (b”), органический растворитель выбирают из группы, состоящей из дихлорметана, хлороформа, ацетона, ацетонитрила, ТГФ, ДМФА, NMP, НМРА, диоксана, нитрометана, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, на стадии (b), стадии (b’) и/или стадии (b”), органический растворитель представляет собой дихлорметан.
В некоторых воплощениях, на стадии (с), стадии (с’) и/или стадии (с”), основание в виде третичного амина представляет собой N,N-диизопропилэтиламин.
В некоторых воплощениях, на стадии (с), стадии (с’) и/или стадии (с”), органический растворитель выбирают из группы, состоящей из пентана, гексана, гептана, циклогексана, метанола, этанола, 1-пропанола, изопропанола, 1-бутанола, 2-бутанола, трет-бутанола, 2-бутанона, дихлорметана, хлороформа, тетрахлорида углерода, 1,2-дихлорэтана, ТГФ, ДМФА, НМРА, NMP, нитрометана, ацетона, уксусной кислоты, ацетонитрила, этилацетата, диэтилового эфира, диэтиленгликоля, глима, диглима, петролейного эфира, диоксана, МТВЕ, бензола, толуола, ксилола, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, на стадии (с), стадии (с’) и/или стадии (с”), органический растворитель выбирают из группы, состоящей из дихлорметана, хлороформа, ацетона, ацетонитрила, ТГФ, ДМФА, NMP, НМРА, диоксана, нитрометана, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, на стадии (с), стадии (с’) и/или стадии (с”), органический растворитель представляет собой ацетонитрил.
В некоторых воплощениях, на стадии (d), соединение формулы (I) сначала комбинируют с основанием, до комбинирования на стадии (d). В некоторых воплощениях, основание представляет собой неорганическое основание. В некоторых воплощениях, основание представляет собой органическое основание. В некоторых воплощениях, основание выбирают из группы, состоящей из К3РО4, Na3PO4, NaOH, KOH, K2CO3 или Na2CO3. В некоторых воплощениях, основание представляет собой К3РО4. В некоторых воплощениях, на стадии (d), соединение формулы (I) сначала комбинируют с основанием, в одном или более растворителе(ях), до комбинирования на стадии (d).
В некоторых воплощениях, на стадии (d), источник палладия представляет собой Pd2dba3 или [(циннамил)PdCl]2. В некоторых воплощениях, на стадии (d), источник палладия представляет собой Pd2dba3.
В некоторых воплощениях, на стадии (d), трет-бутоксид выбирают из группы, состоящей из трет-бутоксида натрия и трет-бутоксида калия.
В некоторых воплощениях, на стадии (d), трет-бутоксид является безводным. В некоторых воплощениях, на стадии (d), трет-бутоксид представляет собой безводный трет-бутоксид натрия.
В некоторых воплощениях, на стадии (d), органический растворитель выбирают из группы, состоящей из пентана, гексана, гептана, циклогексана, метанола, этанола, 1-пропанола, изопропанола, 1-бутанола, 2-бутанола, трет-бутанола, 2-бутанона, дихлорметана, хлороформа, тетрахлорида углерода, 1,2-дихлорэтана, ТГФ, ДМФА, НМРА, NMP, нитрометана, ацетона, уксусной кислоты, ацетонитрила, этилацетата, диэтилового эфира, диэтиленгликоля, глима, диглима, петролейного эфира, диоксана, МТВЕ, бензола, толуола, ксилола, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, на стадии (d), органический растворитель выбирают из группы, состоящей из дихлорметана, хлороформа, ацетона, ацетонитрила, ТГФ, ДМФА, NMP, НМРА, диоксана, нитрометана, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, на стадии (d), апротонный органический растворитель представляет собой смесь из ТГФ и толуола.
В некоторых воплощениях, стадия (d) далее включает следующие стадии:
(1) комбинирование трет-бутоксида с соединением формулы (I), в апротонном органическом растворителе;
(2) комбинирование источника палладия, соединения формулы (J) и соединения формулы (D), в апротонном органическом растворителе; и
(3) добавление смеси со стадии (1) к смеси со стадии (2).
В некоторых воплощениях, на стадии (d), смесь, полученную на стадии (2), отфильтровывают до стадии (3).
В некоторых воплощениях, стадию (d) осуществляют в атмосфере азота или аргона.
В некоторых воплощениях, на стадии (d), каталитическое количество источника палладия используют соответственно количеству соединения (I). В некоторых воплощениях, источник палладия представляет собой Pd2dba3 и каталитическое количество Pd2dba3 составляет от примерно 0,5% мол. до примерно 2% мол. В одном воплощении, каталитическое количество Pd2dba3 составляет примерно 0,75% мол.
В некоторых воплощениях, на стадии (d), каталитическое количество соединения формулы (J) используют соответственно количеству соединения (I). В некоторых воплощениях, каталитическое количество соединения формулы (J) составляет от примерно 1% мол. до примерно 5% мол. В одном воплощении, каталитическое количество соединения формулы (J) составляет от примерно 1% мол. до примерно 4% мол. В одном воплощении, каталитическое количество соединения формулы (J) составляет от примерно 2% мол. до примерно 4% мол. В одном воплощении, каталитическое количество соединения формулы (J) составляет от примерно 1% мол. до примерно 2% мол. В одном воплощении, каталитическое количество соединения формулы (J) составляет от примерно 1% мол. до примерно 2% мол.
В некоторых воплощениях, на стадии (е), трет-бутоксид выбирают из группы, состоящей из трет-бутоксида натрия и трет-бутоксида калия. В некоторых воплощениях, на стадии (е), трет-бутоксид представляет собой трет-бутоксид натрия. В некоторых воплощениях, на стадии (е), трет-бутоксид представляет собой трет-бутоксид калия.
В некоторых воплощениях, на стадии (е), органический растворитель выбирают из группы, состоящей из пентана, гексана, гептана, циклогексана, метанола, этанола, 1-пропанола, изопропанола, 1-бутанола, 2-бутанола, трет-бутанола, 2-бутанона, дихлорметана, хлороформа, тетрахлорида углерода, 1,2-дихлорэтана, ТГФ, ДМФА, НМРА, NMP, нитрометана, ацетона, уксусной кислоты, ацетонитрила, этилацетата, диэтилового эфира, диэтиленгликоля, глима, диглима, петролейного эфира, диоксана, МТВЕ, бензола, толуола, ксилола, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, на стадии (е), органический растворитель выбирают из группы, состоящей из дихлорметана, хлороформа, ацетона, ацетонитрила, ТГФ, ДМФА, NMP, НМРА, диоксана, нитрометана, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, на стадии (е), органический растворитель представляет собой ДМФА.
В некоторых воплощениях, на стадии (f), R1 означает С1-С4-алкил. В некоторых воплощениях, R1 означает изопропил.
В некоторых воплощениях, на стадии (f), R означает метил и С1-С12-алкилхлорформиат представляет собой метилхлорформиат. В некоторых воплощениях, R означает этил и С1-С12-алкилхлорформиат представляет собой этилхлорформиат. В некоторых воплощениях, R означает трет-бутил и ди(С1-С12-алкил)дикарбонат представляет собой ди-трет-бутилдикарбонат.
В некоторых воплощениях, на стадии (f), органический растворитель выбирают из группы, состоящей из дихлорметана, хлороформа, ацетона, ацетонитрила, ТГФ, ДМФА, NMP, НМРА, диоксана, нитрометана, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, на стадии (f), апротонный органический растворитель представляет собой ТГФ.
В некоторых воплощениях, на стадии (i), источник палладия представляет собой Pd(OAc)2.
В некоторых воплощениях, на стадии (i), органический растворитель выбирают из группы, состоящей из пентана, гексана, гептана, циклогексана, метанола, этанола, 1-пропанола, изопропанола, 1-бутанола, 2-бутанола, трет-бутанола, 2-бутанона, дихлорметана, хлороформа, тетрахлорида углерода, 1,2-дихлорэтана, ТГФ, ДМФА, НМРА, NMP, нитрометана, ацетона, уксусной кислоты, ацетонитрила, этилацетата, диэтилового эфира, диэтиленгликоля, глима, диглима, петролейного эфира, диоксана, МТВЕ, бензола, толуола, ксилола, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, на стадии (i), органический растворитель выбирают из группы, состоящей из дихлорметана, хлороформа, ацетона, ацетонитрила, ТГФ, ДМФА, NMP, НМРА, диоксана, нитрометана, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, на стадии (i), органический растворитель представляет собой ацетонитрил.
В некоторых воплощениях, стадия (i) включает комбинирование тетрабутиламмонийбромида с соединением формулы (F), источником палладия и 4-хлорфенилбороновой кислотой, в органическом растворителе.
В некоторых воплощениях, на стадии (j), органический растворитель выбирают из группы, состоящей из пентана, гексана, гептана, циклогексана, метанола, этанола, 1-пропанола, изопропанола, 1-бутанола, 2-бутанола, трет-бутанола, 2-бутанона, дихлорметана, хлороформа, тетрахлорида углерода, 1,2-дихлорэтана, ТГФ, ДМФА, НМРА, NMP, нитрометана, ацетона, уксусной кислоты, ацетонитрила, этилацетата, диэтилового эфира, диэтиленгликоля, глима, диглима, петролейного эфира, диоксана, МТВЕ, бензола, толуола, ксилола, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, на стадии (j), органический растворитель выбирают из группы, состоящей из дихлорметана, хлороформа, ацетона, ацетонитрила, ТГФ, ДМФА, NMP, НМРА, диоксана, нитрометана, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, на стадии (j), органический растворитель представляет собой смесь из ТГФ и толуола. В некоторых воплощениях, смесь из ТГФ и толуола составляет примерно 1:1 по объему.
В некоторых воплощениях, стадия (j) далее включает получение соединения формулы (Н) в виде кристаллического твердого вещества. В некоторых воплощениях, стадия (j) далее включает:
(1) добавление водного раствора к смеси со стадии (j), до получения водной и органической фаз;
(2) отделение органической фазы от смеси со стадии (1);
(3) концентрирование органической фазы; и
(4) добавление органического растворителя к смеси со стадии (3), до получения соединения формулы (Н) в виде кристаллического твердого вещества.
В некоторых воплощениях стадии (4) стадии (j), органический растворитель представляет собой ацетонитрил. В некоторых воплощениях стадии (4) стадии (j), органический растворитель представляет собой ацетонитрил и смесь нагревают до температуры примерно 80°С.
В некоторых воплощениях, стадия (4) стадии (j) далее включает охлаждение смеси до температуры от примерно 10°С до примерно -10°С. В некоторых воплощениях, стадия (4) стадии (j) далее включает охлаждение смеси до температуры примерно -10°С и выделение соединения формулы (Н) в виде кристаллического твердого вещества путем фильтрования смеси.
В некоторых воплощениях, комбинирование на стадии (k) проводят в органическом растворителе. В некоторых воплощениях, органический растворитель выбирают из группы, состоящей из пентана, гексана, гептана, циклогексана, метанола, этанола, 1-пропанола, изопропанола, 1-бутанола, 2-бутанола, трет-бутанола, 2-бутанона, дихлорметана, хлороформа, тетрахлорида углерода, 1,2-дихлорэтана, ТГФ, ДМФА, НМРА, NMP, нитрометана, ацетона, уксусной кислоты, ацетонитрила, этилацетата, диэтилового эфира, диэтиленгликоля, глима, диглима, петролейного эфира, диоксана, МТВЕ, бензола, толуола, ксилола, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, органический растворитель представляет собой изопропанол.
В некоторых воплощениях, стадия (k) далее включает получение соединения формулы (I) в виде кристаллического твердого вещества. В некоторых воплощениях, комбинирование на стадии (k) проводят в органическом растворителе и стадия (k) далее включает выделение соединения формулы (I) в виде кристаллического твердого вещества путем фильтрования смеси.
В некоторых воплощениях, комбинирование на стадии (k) проводят в органическом растворителе и стадия (k) далее включает охлаждение смеси до температуры от примерно 10°С до примерно -10°С, до получения соединения формулы (I) в виде кристаллического твердого вещества.
В некоторых воплощениях, комбинирование на стадии (k) проводят в изопропаноле и стадия (k) далее включает охлаждение смеси до температуры от примерно 10°С до примерно -10°С, до получения соединения формулы (I) в виде кристаллического твердого вещества. В некоторых воплощениях, комбинирование на стадии (k) проводят в изопропаноле и стадия (k) далее включает охлаждение смеси до температуры примерно -5°С, до получения соединения формулы (I) в виде кристаллического твердого вещества, и выделение соединения формулы (I) в виде кристаллического твердого вещества путем фильтрования смеси.
Также, данное изобретение относится к способу получения соединения формулы (С):
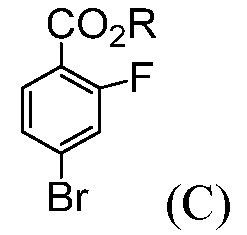
где R означает С1-С12-алкил,
который включает:
(а) комбинирование соединения формулы (А):
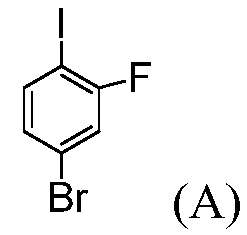
с R1MgX, в апротонном органическом растворителе; где R1 означает С1-С6-алкил; и Х означает Cl, Br или I; и
(b) комбинирование С1-С12-алкилхлорформиата или ди(С1-С12-алкил)дикарбоната с продуктом со стадии (а), до получения соединения формулы (С).
В некоторых воплощениях, R означает С1-С6-алкил. В некоторых воплощениях, R означает С1-С4-алкил. В некоторых воплощениях, R выбирают из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, трет-бутила, изобутила и необутила. В некоторых воплощениях, R означает трет-бутил.
В некоторых воплощениях, R1 означает С1-С4-алкил. В некоторых воплощениях, R1 означает изопропил.
В некоторых воплощениях способа получения соединения формулы (С), органический растворитель на стадии (а) выбирают из группы, состоящей из пентана, гексана, гептана, циклогексана, метанола, этанола, 1-пропанола, изопропанола, 1-бутанола, 2-бутанола, трет-бутанола, 2-бутанона, дихлорметана, хлороформа, тетрахлорида углерода, 1,2-дихлорэтана, ТГФ, ДМФА, НМРА, NMP, нитрометана, ацетона, уксусной кислоты, ацетонитрила, этилацетата, диэтилового эфира, диэтиленгликоля, глима, диглима, петролейного эфира, диоксана, МТВЕ, бензола, толуола, ксилола, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, органический растворитель на стадии (а) представляет собой ТГФ.
В одном воплощении, R означает С1-С6-алкил.
В одном воплощении, R выбирают из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, трет-бутила, изобутила и необутила.
В одном воплощении, R выбирают из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, трет-бутила, изобутила и необутила; и R1 означает изопропил.
В одном воплощении, R означает трет-бутил и R1 означает изопропил.
В некоторых воплощениях способа получения соединения формулы (С), на стадии (b), R означает метил и С1-С12-алкилхлорформиат представляет собой метилхлорформиат. В некоторых воплощениях, R означает этил и С1-С12-алкилхлорформиат представляет собой этилхлорформиат. В некоторых воплощениях, R означает трет-бутил и ди(С1-С12-алкил)дикарбонат представляет собой ди-трет-бутилдикарбонат.
Также, данное изобретение относится к способу получения соединения формулы (D):
где R означает С1-С12-алкил,
который включает:
(х) комбинирование соединения формулы (В):

с соединением формулы (С):
и трет-бутоксидом, в органическом растворителе, до получения соединения формулы (D).
В одном воплощении, R означает трет-бутил.
В некоторых воплощениях, способ получения соединения формулы (D) далее включает стадии (x’) и (x”):
(x’) комбинирование соединения формулы (А):
с R1MgX, в апротонном органическом растворителе; где R1 означает С1-С6-алкил; и Х означает Cl, Br или I;
(x”) комбинирование С1-С12-алкилхлорформиата или ди(С1-С12-алкил)дикарбоната с продуктом со стадии (x’), до получения соединения формулы (С).
В некоторых воплощениях, на стадии (х), трет-бутоксид выбирают из группы, состоящей из трет-бутоксида натрия и трет-бутоксида калия.
В некоторых воплощениях, органический растворитель, на стадии (х), выбирают из группы, состоящей из пентана, гексана, гептана, циклогексана, метанола, этанола, 1-пропанола, изопропанола, 1-бутанола, 2-бутанола, трет-бутанола, 2-бутанона, дихлорметана, хлороформа, тетрахлорида углерода, 1,2-дихлорэтана, ТГФ, ДМФА, НМРА, NMP, нитрометана, ацетона, уксусной кислоты, ацетонитрила, этилацетата, диэтилового эфира, диэтиленгликоля, глима, диглима, петролейного эфира, диоксана, МТВЕ, бензола, толуола, ксилола, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, органический растворитель, на стадии (х), представляет собой ДМФА.
В некоторых воплощениях, на стадии (x’), R1 означает С1-С4-алкил. В некоторых воплощениях, R1 означает изопропил.
В некоторых воплощениях, на стадии (x”), С1-С12-алкилхлорформиат представляет собой метилхлорформиат. В некоторых воплощениях, С1-С12-алкилхлорформиат представляет собой этилхлорформиат. В некоторых воплощениях, ди(С1-С12-алкил)дикарбонат представляет собой ди-трет-бутилдикарбонат.
В некоторых воплощениях, на стадии (x’), апротонный органический растворитель выбирают из группы, состоящей из дихлорметана, хлороформа, ацетона, ацетонитрила, ТГФ, ДМФА, NMP, НМРА, диоксана, нитрометана, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, на стадии (x’), апротонный органический растворитель представляет собой ТГФ.
Также, данное изобретение относится к соединению формулы (3):
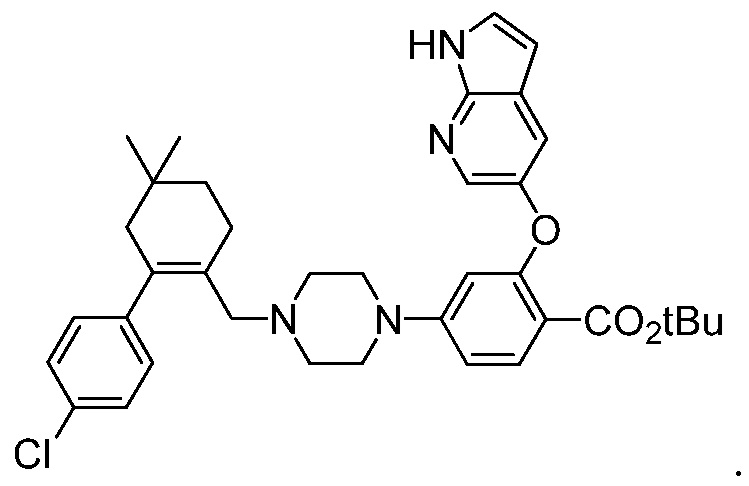
В одном воплощении, соединение формулы (3) получают посредством следующих стадий:
(y) комбинирование соединения формулы (В):
с соединением формулы (С):
где R означает трет-бутил,
и трет-бутоксидом, в органическом растворителе, до получения соединения формулы (D):
где R означает трет-бутил; и
(z) комбинирование соединения формулы (D), где R означает трет-бутил;
с соединением формулы (I):
источником палладия, трет-бутоксидом и фосфиновым лигандом, в апротонном органическом растворителе.
В одном воплощении, фосфиновый лиганд на стадии (z) представляет собой соединение формулы (J):
В других воплощениях, фосфиновый лиганд выбирают из группы, состоящей из:
В одном воплощении, на стадии (z), источником палладия является Pd2dba3.
В некоторых воплощениях, на стадии (z), апротонный органический растворитель выбирают из группы, состоящей из дихлорметана, хлороформа, ацетона, ацетонитрила, ТГФ, ДМФА, NMP, НМРА, диоксана, нитрометана, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, апротонный органический растворитель представляет собой смесь из ТГФ и толуола.
В некоторых воплощениях, на стадии (z), трет-бутоксид выбирают из группы, состоящей из трет-бутоксида натрия и трет-бутоксида калия.
В некоторых воплощениях, на стадии (z), трет-бутоксид представляет собой безводный трет-бутоксид натрия или безводный трет-бутоксид калия.
В некоторых воплощениях, стадия (z) далее включает следующие стадии:
(1) комбинирование трет-бутоксида с соединением формулы (I), в апротонном органическом растворителе;
(2) комбинирование источника палладия, соединения формулы (J) и соединения формулы (D), в апротонном органическом растворителе; и
(3) добавление смеси со стадии (1) к смеси со стадии (2).
В некоторых воплощениях, на стадии (z), смесь, полученную на стадии (2), отфильтровывают до стадии (3).
В некоторых воплощениях, стадию (z) осуществляют в атмосфере газа азота или аргона.
В некоторых воплощениях, на стадии (z), каталитическое количество источника палладия используют соответственно количеству соединения (I). В некоторых воплощениях, источник палладия представляет собой Pd2dba3 и каталитическое количество Pd2dba3 составляет от примерно 0,5% мол. до примерно 2% мол. В одном воплощении, каталитическое количество Pd2dba3 составляет примерно 0,75% мол.
В некоторых воплощениях, когда фосфиновый лиганд на стадии (z) представляет собой соединение формулы (J), каталитическое количество соединения формулы (J) используют соответственно количеству соединения (I). В некоторых воплощениях, каталитическое количество соединения формулы (J) составляет от примерно 1% мол. до примерно 5% мол. В одном воплощении, каталитическое количество соединения формулы (J) составляет от примерно 1% мол. до примерно 4% мол. В одном воплощении, каталитическое количество соединения формулы (J) составляет от примерно 2% мол. до примерно 4% мол. В одном воплощении, каталитическое количество соединения формулы (J) составляет от примерно 1% мол. до примерно 2% мол. В одном воплощении, каталитическое количество соединения формулы (J) составляет от примерно 1% мол. до примерно 2% мол.
В другом воплощении, данное изобретение относится к соединениям формул:
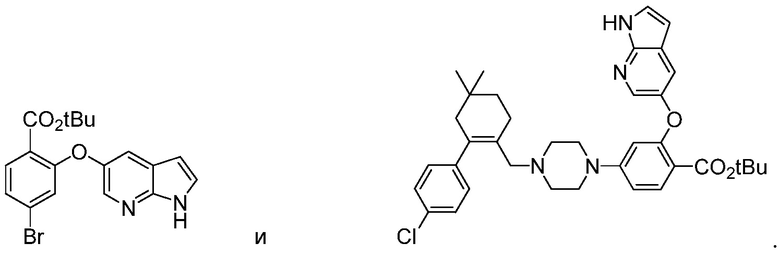
В некоторых воплощениях, способы, описанные в данном контексте, являются улучшенными способами для получения химическим путем в коммерческом масштабе Соединения 1 или Соединения 2. Без привязки к конкретной теории или механизму действия, способы, описанные в данном контексте, значительно улучшают общую эффективность и выход продукта, Соединения 1 или Соединения 2. Предыдущие способы (например, публикации патентов США под номерами 2010/0305122 и 2012/0157470 и публикации Международных заявок на патенты под номерами WO 2011/15096 и WO 2012/071336) найдены недостаточно пригодными для получения Соединения 1 в коммерческом масштабе. Таким образом, способы, предусмотренные в данном контексте, представляют собой улучшенные способы синтеза соединений в количествах, требуемых для клинического и/или коммерческого расширения. Улучшения относительно этих предыдущих способов включают, но не ограничиваясь этим, общий выход Соединения 1 или Соединения 2, общую эффективность способа и экономику, мягкие условия реакции, практичные способы выделения/очистки и «жизнеспособность» к коммерциализации.
Улучшенный способ, предусматриваемый в данном контексте, включает реакцию селективного нуклеофильного ароматического замещения («реакция SnAr») соединений (В) и (С), которую можно осуществлять в мягких условиях, в течение более короткого времени реакции, когда сравнивают с ранее описанными способами, как раскрытые, например, в публикациях патентов США под номерами 2010/0305122 и 2012/0157470 и в публикациях Международных заявок на патенты под номерами WO 2011/15096 и WO 2012/071336. Без ограничения теорией, улучшенная реакция SnAr соединений (В) и (С) не вызывает образования региоизомерных побочных продуктов, что делает необходимой дальнейшую очистку для удаления побочных продуктов, как это имеет место в случае ранее описанных способов. Реакция SnAr в предыдущих способах также требует более длительного времени реакции и жестких условий реакции, что, в результате, приводит к снижению общего выхода по сравнению со способами, описанными в данном контексте. Кроме того, предыдущие способы также требуют трудоемкой очистки от промежуточных продуктов, которая неосуществима в способе большого, коммерческого, масштаба. Способы, описанные в данном контексте, являются более конвергентными, чем предыдущие способы, в результате приводя к высокоэффективной реакции поперечного связывания соединения (D) и в виде свободного основания соединения (I) с высоким выходом. В некоторых воплощениях, в случае способов, описанных в данном контексте, используют кристаллические твердые промежуточные продукты (Н) и (I), которые позволяют проводить эффективную очистку путем кристаллизации для удаления примесей – преимущества, недоступные в ранее описанных способах.
Следующие схемы иллюстрируют одно или более воплощение(ий) способа, предусматриваемого в данном контексте. В некоторых воплощениях, соединение формулы (D) получают из соединения (В) и соединения (С), как показано на нижеприводимой Схеме 1. Соединение формулы (В) можно получать с помощью способов, известных в данной области, например, как показано в Международной заявке под номером WO 2000/047212 и в J. Am. Chem. Soc., 1959, 81: 743-747. Соединение формулы (С) можно получать с помощью способов, известных в данной области, например, как показано в Международной заявке под номером WO 2006/059801 и Tetrahedron Letters, 2008, 49(12), 2034-2037; или, как показано на Схеме 2.
Схема 1
Соединение формулы (С), указанное на Схеме 1, можно получать из коммерчески доступного соединения (А), как показано на нижеприводимой Схеме 2, где «R1MgX» представляет собой реактив Гриньяра, где R1 означает алкильную группу и Х означает Cl, Br или I. Электрофильный ацетилирующий реагент, на Схеме 2, может представлять собой, но не ограничиваясь этим, метилхлорформиат или этилхлорформиат или ВОС2О.
Схема 2

Типичная реакция, в соответствии со схемой 2, представлена ниже.

В другом воплощении, соединение формулы (I) получают из соединения (Е), как показано на нижеприводимой Схеме 3. Соединение (Е) является коммерчески доступным или его можно получать с помощью способов, известных в данной области, например, как показано в патенте США под номером 3813443 и в Proceedings of the Chemical Society, London, 1907, 22, 302.
Схема 3
В другом воплощении, соединение формулы (N) получают из соединения (М), как показано на нижеприводимой Схеме 4. Соединение (М) является коммерчески доступным или его можно получать с помощью способов, известных в данной области, например, как показано в GB 585940 и в J. Am. Chem. Soc., 1950, 72, 1215-1218.
Схема 4
В другом воплощении, соединение формулы (Р) получают из соединения (М), как показано на нижеприводимой Схеме 4’.
Схема 4’
В другом воплощении, соединение формулы (1) получают из соединения (D) и соединения (I), как показано на нижеприводимой Схеме 5. Соединение (J) можно получать с помощью способов, известных в данной области, например, как показано в Международной заявке на патент под номером WO 2009/117626 и в Organometallics, 2008, 27(21), 5605-5611.
Схема 5
В другом воплощении, соединение формулы (2) получают из соединения (L) и соединения (Р), как показано на нижеприводимой Схеме 6, где получение соединения (Р) осуществляют, как показано на Схеме 4’, а получение соединения (L) осуществляют, как показано на Схеме 5.
Схема 6

В некоторых воплощениях, получение соединения формулы (К) из соединения (D) и соединения (I) осуществляют на воздухе и/или в условиях чувствительности к влажности, и, кроме того, получение осуществляют в инертной атмосфере, например, используя газ азот или аргон.
Без привязки к конкретной теории, использование соединения (D) в качестве промежуточного продукта при получении соединения формулы (1) и соединения формулы (2), как показано на вышеприведенных Схемах 1-6, представляет собой улучшение по сравнению с ранее описанными способами для получения соединения формулы (1) и соединения формулы (2). В некоторых воплощениях, улучшения включают более высокие выхода продукта, более короткие времена реакции. В некоторых воплощениях, улучшения предусмотрены, когда R означает трет-бутил в соединении (D).
Схемы 1-6 представляют собой неограничивающие примеры способа, предусмотренного в данном контексте. Растворители и/или реагенты являются известными соединениями и могут быть взаимозаменимыми в соответствии с компетентностью квалифицированного специалиста в данной области.
Аббревиатуры, используемые на Схемах 1-6, означают следующее:
За исключением иначе указанного, температуры, при которых проводят реакции в соответствии со Схемами 1-6, не являются критическими. В некоторых воплощениях, когда указана температура при осуществлении реакции, эту температуру можно варьировать от примерно плюса до минуса, 0,1°С, 0,5°С, 1°С, 5°С или 10°С. В зависимости от того, какой растворитель используют в конкретной реакции, оптимальную температуру можно варьировать. В некоторых воплощениях, реакции проводят при наличии энергичного перемешивания, достаточного для поддержания по существу однородной диспергируемой смеси реагентов.
При проведении реакции, предусмотренной в данном контексте, ни скорость, ни порядок добавления реагентов не являются критическими, за исключением иначе указанного. За исключением иначе указанного, реакции проводят при атмосферном давлении. За исключением иначе указанного, точное количество реагентов не является критическим. В некоторых воплощениях, количество реагента можно варьировать на примерно 10% мол. или примерно 10% масс.
За исключением иначе указанного, органические растворители, используемые в случае способов, предусмотренных в данном контексте, можно выбирать из таковых, коммерчески доступных или иным образом известных квалифицированному специалисту в данной области. Подходящие растворители для данной реакции входят в рамки знаний квалифицированного специалиста и включают смеси растворителей. Примеры органических растворителей, предусмотренных в данном контексте для использования, включают, но не ограничиваясь этим: пентан, гексан, гептан, циклогексан, метанол, этанол, 1-пропанол, изопропанол, 1-бутанол, 2-бутанол, трет-бутанол, 2-бутанон, дихлорметан, хлороформ, тетрахлорид углерода, 1,2-дихлорэтан, тетрагидрофуран (ТГФ), диметилформамид (ДМФА), гексаметилфосфорамид (НМРА), N-метил-2-пирролидинон (NMP), нитрометан, ацетон, уксусную кислоту, ацетонитрил, этилацетат, диэтиловый эфир, диэтиленгликоль, глим, диглим, петролейный эфир, диоксан, метил-трет-бутиловый эфир (МТВЕ), бензол, толуол, ксилол, пиридин, 2-метилтетрагидрофуран и их смеси.
В некоторых воплощениях, органический растворитель, используемый в случае способов, предусмотренных в данном контексте, представляет собой апротонный органический растворитель. Как предусмотрено в данном контексте, апротонный растворитель является растворителем, который не содержит кислый атом водорода или атом водорода, который способен образовывать водородную связь (например, не связан с атомом кислорода или атомом азота). Апротонный органический растворитель можно выбирать из группы, состоящей из дихлорметана, хлороформа, ацетона, ацетонитрила, ТГФ, ДМФА, NMP, НМРА, диоксана, нитрометана, пиридина, 2-метилтетрагидрофурана и их смесей. В некоторых воплощениях, апротонный органический растворитель представляет собой ТГФ. В некоторых воплощениях, апротонный органический растворитель представляет собой ДМФА. В некоторых воплощениях, апротонный органический растворитель представляет собой ацетонитрил.
Как представлено в данном контексте, термин «основание в виде третичного амина» относится к амину, который замещен с помощью трех алкильных групп, например, триэтиламин или N,N-диизопропилэтиламин.
Как предусмотрено в данном контексте, термин «каталитическое количество» относится к менее, чем одному, молярному эквиваленту реагента или реагирующего вещества в данной реакции в виде установленного соответственно другому реагенту или реагирующему веществу в реакционной смеси. В некоторых воплощениях, каталитическое количество определяют в виде молярного процента соответственно другому реагенту или реагирующему веществу в реакционной смеси.
Как предусмотрено в данном контексте, термин «источник палладия» относится к источнику палладия в стабильном состоянии окисления, то есть, Pd(0), Pd(I), Pd(II) и/или Pd(IV). Палладий может быть свободным металлом, как например в форме порошка, или может быть связан с одним или более лигандом(ами), например, PdCl2, Pd2dba3, PdCl2(PPh3)2, Pd(PPh3)4, Pd(OAc)2 или [(циннамил)PdCl]2.
Как предусмотрено в данном контексте, термин «фосфиновый лиганд» относится к соединению формулы PR’3, где каждый R’ независимо выбирают из С1-С6-алкила или фенила, где арильная группа является необязательно замещенной с помощью С1-С6-алкила, фенила, триалкиламино, алкокси или галогена.
Как предусмотрено в данном контексте, за исключением иначе определенного, термин «примерно» означает, что значение или количество, к которому он относится, может варьироваться на ±5%, на ±2% или на ±1%.
Продукты, полученные с помощью любого способа, предусмотренного в данном контексте, можно выделять с помощью стандартных способов, таких как выпаривание или экстракция, и можно очищать с помощью стандартных способов, таких как дистилляция, перекристаллизация или хроматография.
ПРИМЕРЫ
Соединения следующих примеров представлены на вышеприведенных Схемах 1-6 и названы с использованием программного обеспечения Chemdraw® Ultra. В дополнение к аббревиатурам, описанным выше, принимая во внимание предусмотренные в данном контексте Схемы, в примерах используются следующие аббревиатуры:
«ВЭЖХ» = высокоэффективная жидкостная хроматография, «IP» = в процессе; «ML» = маточный раствор; «NLT» = не менее, чем; «NMT» = не более, чем; «RB» = круглодонная (колба); «RT» = комнатная температура; «sm» = исходное вещество; «DCM» = дихлорметан.
За исключением иначе указанного, соединения характеризуются с помощью ВЭЖХ и 1Н-ЯМР-анализа и используются в последующих введениях во взаимодействие, с очисткой или без очистки. 1Н-ЯМР-Анализ осуществляют при 400 МГц, за исключением иначе указанного. За исключением иначе определенного, выход/чистоту продукта определяют по массе, посредством кЯМР-анализа и/или посредством ВЭЖХ-анализа.
Пример 1: Синтез трет-бутил-4-бром-2-фторбензоата
(Соединение (С))
В реактор с рубашкой емкостью 100 мл, оснащенный механической мешалкой, вводят 4-бром-2-фтор-1-иодбензол, «Соединение (А)» (5 г, 1,0 экв.) и ТГФ (25 мл). Раствор охлаждают до температуры -5°С. Медленно добавляют 2 М раствор изопропилхлорида магния в ТГФ (10,8 мл, 1,3 экв.), поддерживая внутреннюю температуру ниже 0°С. Смесь перемешивают при температуре 0°С в течение 1 часа. Добавляют ди-трет-бутилдикарбонат (5,44 г, 1,5 экв.) в ТГФ (10 мл). Спустя 1 час, раствор гасят 10%-ным раствором лимонной кислоты (10 мл) и затем разбавляют 25%-ным раствором NaCl (10 мл). Слои разделяют и органический слой концентрируют почти досуха и обрабатывают с помощью ТГФ (3 раза по 10 мл). Сырое масло разбавляют с помощью ТГФ (5 мл), отфильтровывают для удаления неорганических веществ и концентрируют досуха. Сырое масло (6,1 г, эффективность = 67%, установленный по эффективности выход = 88%) используют на следующей стадии без дальнейшей очистки.
1H ЯМР (ДМСО-d6): δ 1,53 (с, 9H), 7,50-7,56 (м, 1H), 7,68 (дд, J=10,5, 1,9 Гц, 1H), 7,74 (т, J=8,2 Гц, 1H).
Пример 2: Синтез трет-бутил-2-((1Н-пирроло[2,3-b]пиридин-5-ил)окси)-4-бромбензоата (Соединение (D))
В трехгорлую колбу Мортона емкостью 3 л вводят 1Н-пирроло[2,3-b]пиридин-5-ол (80,0 г, 1,00 экв.), трет-бутил-4-бром-2-фторбензоат (193 г, 1,15 экв.) и безводный ДМФА (800 мл). Смесь перемешивают при температуре 20°С в течение 15 минут. Полученный раствор охлаждают до температуры от примерно нуля до 5°С. Медленно, в течение 30 минут, добавляют раствор трет-бутоксида натрия (62,0 г) в ДМФА (420 мл), все время поддерживая внутреннюю температуру не выше, чем 10°С, и промывают с помощью ДМФА (30 мл). Реакционную смесь перемешивают при температуре 10°С в течение 1 часа (не совсем белого цвета суспензия) и устанавливают внутреннюю температуру равной ~45°С в течение 30 минут. Реакционную смесь перемешивают при температуре 45-50°С в течение 7 часов и за протеканием реакции наблюдают с помощью ВЭЖХ (образцы IP: 92% конверсии, в % по ВЭЖХ). Раствор охлаждают до температуры ~20°С. Этот раствор перемешивают при температуре 20°С в течение ночи.
К реакционной смеси медленно добавляют воду (1200 мл), при температуре <30°С, в течение 1 часа (слегка экзотермическая реакция). Полученную суспензию доводят до температуры примерно 20°С и перемешивают не менее, чем в течение 2 часов. Сырой продукт собирают путем фильтрации и промывают водой (400 мл). Влажный фильтровальный осадок промывают гептаном (400 мл) и высушивают в вакууме при температуре 50°С в течение ночи, получая сырой продукт (236,7 г).
Перекристаллизация или повторное суспендирование: 230,7 г сырого продукта (по эффективности установлено: 200,7 г) вводят в трехгорлую колбу Мортона емкостью 3 л. Добавляют этилацетат (700 мл) и суспензию медленно нагревают до температуры кипения с обратным холодильником в течение 1 часа (остается небольшое количество твердых веществ). Медленно добавляют гептан (1400 мл) и смесь подвергают кипячению с обратным холодильником (78°С). Суспензию перемешивают, при температуре кипения с обратным холодильником, в течение 30 минут и медленно охлаждают до температуры примерно -10°С со скоростью, составляющей приблизительно 10°С/час, и перемешивают в течение 2 часов. Продукт собирают путем фильтрации и промывают гептаном (200 мл).
Твердое вещество высушивают в вакууме при температуре примерно 50°С в течение ночи, получая 194,8 г; выход выделенного продукта в виде не совсем белого цвета твердого вещества составляет 86%.
MS-ESI: 389,0 (М+1); температура плавления: 190-191°С (не корректировано).
1H ЯМР (ДМСО-d6): δ 1,40 (с, 9H), 6,41 (дд, J=3,4, 1,7 Гц, 1H), 7,06 (д, J=1,8 Гц, 1H), 7,40 (дд, J=8,3, 1,8 Гц, 1H), 7,51 (т, J=3,4 Гц, 1H), 7,58 (д, J=2,6 Гц, 1H), 7,66 (д, J=8,3 Гц, 1H), 8,03 (д, J=2,7 Гц, 1H), 11,72 (с, 1H, NH).
Пример 3: Синтез 2-хлор-4,4-диметилциклогексанкарбальдегида (Соединение (F))
В круглодонную колбу емкостью 500 мл вводят безводный ДМФА (33,4 г, 0,456 моль) и СН2Cl2 (80 мл). Раствор охлаждают до температуры <-5°С и медленно добавляют POCl3 (64,7 г, 0,422 моль), в течение 20 минут, при температуре <20°С (экзотермическая реакция), промывают с помощью СН2Cl2 (6 мл). Раствор слегка коричневого цвета доводят до температуры 20°С в течение 30 минут и перемешивают при температуре 20°С в течение 1 часа. Раствор охлаждают до температуры <5°С. Добавляют 3,3-диметилциклогексанон (41,0 г, чистота 90%, ~0,292 моль) и промывают с помощью СН2Cl2 (10 мл) (слегка экзотермическая реакция) при температуре <20°С. Раствор кипятят с обратным холодильником и перемешивают в течение ночи (21 час).
В трехгорлую круглодонную колбу емкостью 1000 мл, оснащенную механической мешалкой, вводят 130 г 13,6% масс. водного раствора тригидрата ацетата натрия, 130 г 12%-го насыщенного солевого раствора и 130 мл СН2Cl2. Смесь перемешивают и охлаждают до температуры <5°С. Вышеуказанную реакционную смесь (прозрачную и коричневого цвета) переносят, медленно ее гасят, все время поддерживая внутреннюю температуру <10°С. Реакционный сосуд ополаскивают с помощью СН2Cl2 (10 мл). Погашенную реакционную смесь перемешивают при температуре <10°С в течение 15 минут и повышают температуру до 20°С. Смесь перемешивают при температуре 20°С в течение 15 минут и оставляют отстаиваться в течение 30 минут (до некоторой степени эмульсия). Нижнюю органическую фазу отделяют. Верхнюю водную фазу снова экстрагируют с помощью СН2Cl2 (50 мл). Объединенные органические слои промывают смесью 12%-ный насыщенный солевой раствор (150 г) – 20%-ный водный раствор К3РО4 (40 г). Органический слой сушат над MgSO4, отфильтровывают и промывают с помощью СН2Cl2 (30 мл). Фильтрат концентрируют досуха в вакууме, получая масло коричневого цвета (57,0 г, эффективность = 90,9% масс., по кЯМР, ~100%). 1H ЯМР (CDCl3): δ 0,98 (с, 6H), 1,43 (т, J=6,4 Гц, 2H), 2,31 (тт, J=6,4, 2,2 Гц, 2H), 2,36 (т, J=2,2 Гц, 2H), 10,19 (с, 1H).
Пример 4: Синтез 2-(4-хлорфенил)-4,4-диметилциклогекс-1-енкарбальдегида (Соединение (G))
В толстостенную колбу емкостью 250 мл вводят 2-хлор-4,4-диметилциклогекс-1-енкарбальдегид (10,00 г), тетрабутиламмонийбромид (18,67 г) и ацетонитрил (10 мл). Смесь перемешивают при температуре 20°С в течение 5 минут. Добавляют 21,0% масс. водного раствора К2СО3 (76,0 г). Смесь перемешивают при комнатной температуре не менее, чем в течение 5 минут, затем за один раз полностью добавляют 4-хлорфенилбороновую кислоту (9,53 г). Смесь три раза вакуумируют и продувают азотом. За один раз полностью добавляют ацетат палладия (66 мг, 0,5% мол.), в атмосфере азота. Реакционную смесь три раза вакуумируют и продувают азотом (смесь окрашена в оранжевый цвет). Колбу заполняют азотом и нагревают до температуры ~35°С на масляной бане (температура бани составляет ~35°С). Смесь перемешивают при температуре 30°С в течение ночи (15 часов). Реакционную смесь охлаждают до комнатной температуры и извлекают IP образец из верхней органической фазы для определения завершения реакции, обычно исходное вещество составляет <2% (смесь окрашена в оранжевый цвет). Добавляют толуол (100 мл) и водный раствор 5% NaHCO3 - 2% L-цистеин (100 мл). Смесь перемешивают при температуре 20°С в течение 60 минут. Смесь отфильтровывают через слой целита для удаления твердого вещества черного цвета, промывают колбу и слой целита толуолом (10 мл). Верхнюю органическую фазу неоднократно промывают водным раствором 5% NaHCO3 - 2% L-цистеин (100 мл). Верхнюю органическую фазу промывают 25%-ным насыщенным солевым раствором (100 мл). Органический слой (105,0 г) анализируют (118,8 мг/г, 12,47 г продукта по анализу, выход по анализу составляет 87%) и концентрируют до ~1/3 объема (~35 мл). Раствор продукта непосредственно используют на следующей стадии, без выделения продукта. Однако, образец для анализа получают путем удаления растворителя, получая масло коричневого цвета.
1H ЯМР (CDC13): δ 1,00 (с, 6H), 1,49 (т, J=6,6 Гц, 2H), 2,28 (т, J=2,1 Гц, 2H), 2,38 (м, 2H), 7,13 (м, 2H), 7,34 (м, 2H), 9,47 (с, 1H).
Пример 5: Синтез трет-бутил-4-((4’-хлор-5,5-диметил-3,4,5,6-тетрагидро-[1,1’-бифенил]-2-ил)метил)пиперазин-1-карбоксилата (Соединение (Н))
В трехгорлую круглодонную колбу емкостью 2 л, оснащенную механической мешалкой, вводят раствор 4’-хлор-5,5-диметил-3,4,5,6-тетрагидро-[1,1’-бифенил]-2-карбальдегида (50,0 г) в толуоле (250 мл), ВОС-пиперазин (48,2 г) и безводный ТГФ (250 мл). Раствор желтого цвета перемешивают при температуре 20°С в течение 5 минут. В виде одной порции добавляют триацетоксиборгидрид натрия (52,7 г) (примечание: внутренняя температура повышается до ~29,5°С в течение 15 минут, может быть необходимым охлаждение). Смесь желтого цвета перемешивают при температуре ~25°С не менее, чем в течение 4 часов. Конверсию исходного вещества до продукта, составляющую 99,5%, наблюдают по ВЭЖХ, после 3 часов времени реакции.
Для гашения реакции медленно добавляют 12,5% масс. насыщенного солевого раствора (500 г). Смесь перемешивают при температуре 20°С не менее, чем в течение 30 минут, и оставляют отстаиваться не менее, чем в течение 15 минут. Нижнюю водную фазу (~560 мл) отделяют (примечание: остается какое-то количество эмульсии в верхней органической фазе). Органическую фазу промывают 10%-ным раствором лимонной кислоты (2 раза по 500 г). В колбу медленно вводят 500 г 5%-ного водного раствора NaHCO3. Смесь перемешивают при температуре 20°С не менее, чем в течение 30 минут, и оставляют отстаиваться не менее, чем в течение 15 минут. Верхнюю органическую фазу отделяют. Вводят 500 г 25%-ного водного насыщенного солевого раствора. Смесь перемешивают при температуре 20°С не менее, чем в течение 15 минут, и оставляют отстаиваться не менее, чем в течение 15 минут. Верхнюю органическую фазу концентрируют до объема ~200 мл в вакууме. Раствор доводят до температуры ~30°С и отфильтровывают неорганическую соль. Толуол (50 мл) используют в качестве средства для промывки. Объединенный фильтрат концентрируют до объема ~100 мл. Добавляют ацетонитрил (400 мл) и смесь нагревают до температуры ~80°С для достижения получения прозрачного раствора. Раствор медленно охлаждают до температуры 20°С со скоростью, составляющей 10°С/час, и перемешивают при температуре 20°С в течение ночи (продукт кристаллизуется при температуре ~45-50°С, если необходимо, затравочный материал можно добавлять при температуре 50°С). Суспензию продолжают медленно охлаждать до температуры ~10°С со скоростью, составляющей 10°С/час. Суспензию перемешивают при температуре ~ -10°С не менее, чем в течение 6 часов. Продукт собирают путем фильтрации и промывают предварительно охлажденным ацетонитрилом (100 мл). Твердое вещество высушивают в вакууме при температуре 50°С в течение ночи (72,0 г, выход 85%).
MS-ESI: 419 (М+1); температура плавления: 109-110°С (не корректировано).
1H ЯМР (CDC13): δ 1,00 (с, 6H), 1,46 (с, 9H), 1,48 (т, J=6,5 Гц, 2H), 2,07 (с, ушир, 2H), 2,18 (м, 4H), 2,24 (т, J=6,4 Гц, 2H), 2,80 (с, 2H), 3,38 (м, 4H), 6,98 (м, 2H), 7,29 (м, 2H).
Пример 6: Синтез 1-((4’-хлор-5,5-диметил-3,4,5,6-тетрагидро-[1,1’-бифенил]-2-ил)метил)пиперазиндигидрохлорида
(Соединение (I))
В трехгорлую круглодонную колбу емкостью 2,0 л, оснащенную механической мешалкой, вводят Вос-продукт восстановительного аминирования (Соединение (Н), 72,0 г) и IPA (720 мл). Смесь перемешивают при комнатной температуре в течение 5 минут и к суспензии добавляют 59,3 г концентрированного водного раствора хлороводорода. Внутреннюю температуру реакционной смеси доводят до ~65°С (получается прозрачный и бесцветный раствор). Реакционную смесь перемешивают при температуре ~65°С не менее, чем в течение 12 часов.
Суспензию продукта медленно охлаждают до температуры -5°С (10°С/час). Суспензию продукта перемешивают при температуре примерно -5°С не менее, чем в течение 2 часов, продукт собирают путем фильтрации. Влажный фильтровальный осадок промывают с помощью IPA (72 мл) и высушивают при температуре 50°С в вакууме в течение ночи, получая 73,8 г (выход 95%) желательного продукта в виде сольвата бис-гидрохлорида IPA (чистота >99,5%, площадь пика при 210 нм).
MS-ESI: 319 (М+1);
1H ЯМР (D2O): δ 1,00 (с, 6H), 1,19 (д, J=6,0 Гц, 6H, IPA), 1,65 (т, J=6,1 Гц, 2H), 2,14 (с, ушир, 2H), 2,26 (м, 2H), 3,36 (ушир, 4H), 3,55 (с, ушир, 4H), 3,82 (с, 2H), 4,02 (септет, J=6,0 Гц, 1H, IPA), 7,16 (д, J=8,1 Гц, 2H), 7,45 (д, J=8,1 Гц, 2H); 1H ЯМР (CDCl3): δ 0,86 (с, 6H), 1,05 (д, J=6,0 Гц, 6H, IPA), 1,42 (т, J=6,1 Гц, 2H), 2,02 (с, ушир, 2H), 2,12 (м, 2H), 3,23 (м, 4H), 3,4 (с, ушир, 4H), 3,68 (с, 2H), 3,89 (септет, J=6,0 Гц, 1H, IPA), 7,11 (д, J=8,1 Гц, 2H), 7,41 (д, J=8,1 Гц, 2H).
Пример 7: Синтез 3-нитро-4-(((тетрагидро-2Н-пиран-4-ил)метил)амино)бензолсульфонамида (Соединение (N))
В трехгорлую круглодонную колбу емкостью 500 мл, оснащенную механической мешалкой, вводят 4-хлор-3-нитробензолсульфонамид, Соединение М (10,0 г), диизопропилэтиламин (17,5 г), (тетрагидро-2Н-пиран-4-ил)метанамин (7,0 г) и ацетонитрил (150 мл). Внутреннюю температуру реакционной смеси устанавливают при 80°С и перемешивают не менее, чем в течение 12 часов.
Раствор продукта охлаждают до температуры 40°С и перемешивают не менее, чем в течение 1 часа, до тех пор, пока не наблюдают выпадения осадка. Суспензию продукта далее охлаждают до температуры 20°С. Медленно вводят воду (75 мл), в течение времени не менее, чем 1 час, и смесь охлаждают до температуры 10°С и перемешивают не менее, чем в течение 2 часов, перед сбором продукта путем фильтрации. Влажный фильтровальный осадок промывают смесью ацетонитрил:вода (40 мл, 1:1). Влажный фильтровальный осадок затем повторно суспендируют в воде (80 мл), при температуре 40°С, не менее, чем в течение 1 часа, перед сбором продукта путем фильтрации. Влажный фильтровальный осадок промывают водой (20 мл) и высушивают при температуре 75°С в вакууме, получая 12,7 г желательного продукта с чистотой 99,9% и установленным выходом по массе 91%.
1H ЯМР(ДМСО-d6): δ 1,25 (м, 2H), 1,60 (м, 2H), 1,89 (м, 1H), 3,25 (м, 2H), 3,33 (м, 2H), 3,83 (м, 2H), 7,27 (д, J=9,3 Гц, 1H), 7,32 (с, NH2, 2H), 7,81(дд, J=9,1, 2,3 Гц, 1H), 8,45 (д, J=2,2 Гц, 1H), 8,54 (т, J=5,9 Гц, 1H, NH).
Пример 8: Синтез трет-бутил-2-((1Н-пирроло[2,3-b]пиридин-5-ил)окси)-4-(4-((4’-хлор-5,5-диметил-3,4,5,6-тетрагидро-[1,1’-бифенил]-2-ил)метил)пиперазин-1-ил)бензоата (Соединение (К))
Общие рассмотрения: в случае этого химического процесса принимают во внимание воздух и чувствительность к влаге.
В то время как предшественников катализатора в их твердой, сухой форме можно обрабатывать и хранить на воздухе без особых предосторожностей, контакт с даже незначительными количествами растворителя может делать их поддающимися разложению. В результате, следы кислорода или других компетентных окислителей (например, содержащиеся в растворителе пероксиды) нужно удалять перед комбинированием предшественников катализатора с растворителем и нужно заботиться при использовании о предотвращении доступа кислорода во время реакции. Также, нужно заботиться об использовании сухого оборудования, растворителей и реагентов для предотвращения образования нежелательных побочных продуктов. трет-Бутоксид натрия, используемый в этой реакции, является гигроскопичным и его нужно должным образом обрабатывать и хранить перед использованием или во время использования.
В трехгорлую круглодонную колбу емкостью 2,0 л, оснащенную механической мешалкой, вводят бис-гидрохлорид (Соединение (I), 42,5 г) и толуол (285 мл). Добавляют 20%-ный раствор К3РО4 (285 мл) и двухфазную смесь перемешивают в течение 30 минут. Слои разделяют и органический слой промывают 25%-ным раствором NaCl (145 мл). Органический слой концентрируют до 120 г и используют в реакции связывания, без дальнейшей очистки.
NaOtBu (45,2 г) и Соединение (I) в виде раствора в толуоле (120 г раствора – 30 г установленная эффективность) комбинируют с ТГФ (180 мл) в подходящем реакторе и барботируют азотом в течение времени не менее, чем 45 минут. Pd2dba3 (0,646 г), Соединение (J) (0,399 г) и Соединение (D) (40,3 г) комбинируют во втором подходящем реакторе и продувают азотом, пока уровень кислорода достигнет не менее, чем 40 ч/млн. Используя давление азота, раствор, содержащий Соединение (I) и NaOtBu в смеси толуол/ТГФ, добавляют через толщиной 0,45 мкм проходной фильтр во второй реактор (катализатор, Соединение (J) и Соединение (D)) и ополаскивают с помощью барботируемого азотом ТГФ (30 мл).
Полученную смесь нагревают при температуре 55°С, при перемешивании, не менее, чем в течение 16 часов, затем охлаждают до температуры 22°С. Смесь разбавляют 12%-ным раствором NaCl (300 г), потом ТГФ (300 мл). Слои разделяют.
Органический слой перемешивают со свежеприготовленным раствором из L-цистеина (15 г), NaHCO3 (23 г) и воды (262 мл). Спустя 1 час слои разделяют.
Органический слой перемешивают со вторым свежеприготовленным раствором из L-цистеина (15 г), NaHCO3 (23 г) и воды (262 мл). Спустя 1 час слои разделяют. Органический слой промывают 12%-ным раствором NaCl (300 г), затем отфильтровывают через проходной фильтр толщиной 0,45 мкм. Отфильтрованный раствор концентрируют в вакууме до объема ~300 мл и три раза промывают гептаном (по 600 мл каждый раз) для удаления ТГФ.
Сырую смесь концентрируют до 6 объемов и разбавляют циклогексаном (720 мл). Смесь нагревают до температуры 75°С, выдерживают в течение 15 минут и потом охлаждают до температуры 65°С в течение времени не менее, чем 15 минут. Вводят затравочный материал и смесь выдерживают при температуре 65°С в течение 4 часов. Суспензию охлаждают до температуры 25°С в течение времени не менее, чем 8 часов, потом нагревают при температуре 25°С в течение 4 часов. Твердые вещества отфильтровывают и промывают циклогексаном (90 мл) и высушивают при температуре 50°С в вакууме.
Выделяют 52,5 г (выход 88,9%) твердого вещества белого цвета. Температура плавления (не корректировано): 154-155°С.
1H ЯМР (ДМСО-d6): δ 0,93 (с, 6H), 1,27 (с, 9H), 1,38 (т, J=6,4 Гц, 2H), 1,94 (с, 2H), 2,08-2,28 (м, 6H), 2,74 (с, 2H), 3,02-3,19 (м, 4H), 6,33 (дд, J=3,4, 1,9 Гц, 1H), 6,38 (д, J=2,4 Гц, 1H), 6,72 (дд, J=9,0, 2,4 Гц, 1H), 6,99-7,06 (м, 2H), 7,29 (д, J=2,7 Гц, 1H), 7,30-7,36 (м, 2H), 7,41-7,44 (м, 1H), 7,64 (т, J=6,7 Гц, 1H), 7,94 (д, J=2,7 Гц, 1H), 11,53 (с, 1H).
Пример 9: Синтез 2-((1Н-пирроло[2,3-b]пиридин-5-ил)окси)-4-(4-((4’-хлор-5,5-диметил-3,4,5,6-тетрагидро-[1,1’-бифенил]-2-ил)метил)пиперазин-1-ил)бензойной кислоты (Соединение (L))
Приготовление раствора: 10%-ный водный раствор КН2РО4: КН2РО4 (6 г) в воде (56 г); 2:1, гептан/2-МеТГФ:гептан (16 мл) в 2-МеТГФ (8 мл).
Соединение (К) (5,79 г), трет-бутоксид калия (4,89 г), 2-метилтетрагидрофуран (87 мл) и воду (0,45 мл) комбинируют в подходящем реакторе, в атмосфере азота, и нагревают при температуре 55°С до тех пор, пока не завершится реакция. Реакционную смесь охлаждают до температуры 22°С, дважды промывают 10%-ным раствором КН2РО4 (31 г). Органический слой затем промывают водой (30 г).
После удаления водного слоя, органический слой концентрируют до 4 объемов (~19 мл) и нагревают при температуре, не менее, чем 50°С. Медленно добавляют гептан (23 мл). Альтернативно, после удаления водного слоя, органический слой концентрируют до 5 объемов и нагревают до температуры, не менее, чем 70°С, и медленно добавляют 5 объемов гептана. Полученную суспензию охлаждают до температуры 10°С. Твердые вещества потом собирают путем вакуумной фильтрации, с рециркуляцией жидкостей, и фильтровальный осадок промывают смесью гептан/2-МеТГФ (24 мл, 2:1). Твердые вещества высушивают при температуре 80°С в вакууме, получая 4,0 г Соединения (L) с установленным по массе выходом приблизительно 85%.
1H ЯМР (ДМСО-d6): δ 0,91 (с, 6H), 1,37 (т, J=6A Гц, 2H), 1,94 (с, ушир, 2H), 2,15 (м, 6H), 2,71 (с, ушир, 2H), 3,09 (м, 4H), 6,31 (д, J=2,3 Гц, 1H), 6,34 (дд, J=3,4, 1,9 Гц, 1H), 6,7 (дд, J=9,0, 2,4 Гц, 1H), 7,02 (м, 2H), 7,32 (м, 2H), 7,37 (д, J=2,6 Гц, 1H), 7,44 (т, J=3,0 Гц, 1H), 7,72 (д, J=9,0 Гц, 1H), 7,96 (д, J=2J Гц, 1H) & 11,59 (м, 1H).
Пример 10: Синтез 4-(4-{[2-(4-хлорфенил)-4,4-диметилциклогекс-1-ен-1-ил]метил}пиперазин-1-ил)-N-({3-нитро-4-[(тетрагидро-2Н-пиран-4-илметил)амино]фенил}сульфонил)-2-(1Н-пирроло[2,3-b]пиридин-5-илокси)бензамида (Соединение (1))
Приготовление раствора перед введением во взаимодействие: 10%-ный раствор уксусной кислоты : уксусная кислота (37 мл) в воде (333 г); 5%-ный раствор NaHCO3 : NaHCO3 (9 г) в воде (176 г); 5%-ный раствор NaCl : NaCl (9 г) в воде (176 г).
Соединение (N) (13,5 г), DMAP (10,5 г), EDAC (10,7 г) и дихлорметан (300 мл) комбинируют в подходящем реакторе и перемешивают при температуре 25°С. Во второй подходящий реактор вводят кислоту (Соединение (L), 25 г), Et3N (8,7 г) и дихлорметан (120 мл). Полученный раствор кислоты (Соединение (L)) медленно вводят в исходную суспензию Соединения (N) и перемешивают до тех пор, пока не завершится реакция. Затем, в реакционную смесь, при непрерывном перемешивании, вводят N,N-диметилэтилендиамин (9,4 г). Реакционную смесь нагревают до температуры 35°С и дважды промывают 10%-ным раствором уксусной кислоты (185 мл). Нижний органический слой разбавляют еще дихлорметаном (75 мл) и метанолом (12,5 мл). Содержащий продукт органический слой потом промывают 5%-ным раствором NaHCO3 (185 мл) и после этого промывают 5%-ным раствором NaCl (185 мл), при температуре 35°С. Нижний органический слой отделяют и потом концентрируют до 8 объемов (~256 мл), разбавляют метанолом (26 мл) и нагревают до температуры 38°С. Медленно вводят этилацетат (230 мл). Полученную суспензию медленно охлаждают до температуры 10°С и затем отфильтровывают. Влажный фильтровальный осадок дважды промывают смесью дихлорметана и этилацетата (1:1, ~2 об., 64 мл). После высушивания фильтровального осадка при температуре 90°С, выделяют 32 г (выход 84%) Соединения (1).
1H ЯМР (ДМСО-d6): δ 0,90 (с, 6H), 1,24 (м, 2H), 1,36 (т, J=6,4 Гц, 2H), 1,60 (м, 2H), 1,87 (м, 1H), 1,93 (с, ушир, 2H), 2,12 (м, 2H), 2,19 (м, 4H), 2,74 (с, ушир, 2H), 3,06 (м, 4H), 3,26 (м, 4H), 3,83 (м, 2H), 6,17 (д, J=2,1 Гц, 1H), 6,37 (дд, J=3,4, 1,9 Гц, 1H), 6,66 (дд, J=9,1, 2,2 Гц, 1H), 7,01 (м, 2H), 7,31 (м, 2H), 7,48 (м, 3H), 7,78 (дд, J=9,3, 2,3 Гц, 1H), 8,02 (д, J=2, 61 Гц, 1H), 8,54 (д, J=2, 33 Гц, 1H), 8,58 (т, J=5,9 Гц, 1H, NH), 11,65 (м, 1H).
Пример 11: Синтез ((1R,4R)-4-гидрокси-4-метилциклогексил)метанаминий-4-метилбензолсульфоната
Стадия А: 1,49 г циклогександионмоноэтиленацеталя (1,0 экв.) и 15 мл толуола вводят в подходящий реактор. Смесь перемешивают в течение 30 минут при температуре 10°С. В другой реактор вводят 1,4 М раствор метилмагнийбромида (2,32 экв.) в смеси толуол-ТГФ (75-25) и перемешивают при температуре 15°С. Раствор исходного вещества, по каплям, добавляют к раствору реактива Гриньяра, при температуре приблизительно 10-20°С, в течение 4 часов (скорость добавления = 0,1 мл/мин). За протеканием реакции наблюдают посредством ТСХ. После завершения взаимодействия, реакционную смесь медленно вводят в 24%-ный раствор хлорида аммония (20 мл), при температуре 25°С. Реакционную смесь перемешивают и отстаивают, органический слой отделяют, а водный слой экстрагируют этилацетатом (3 раза по 20 мл). Объединенные органические слои отфильтровывают через слой сульфата натрия и фильтрат концентрируют путем дистилляции досуха. Выделяют 1,57 г сырых твердых веществ (выход 95%) и переносят на следующую стадию.
1H ЯМР (400 MГц, Хлороформ-d1) δ м.д. 3,88-4,01 (м, 4H), 1,85-1,96 (м, 2H), 1,08-1,64 (м, 7H). LCMS-(MS 310 и 292). Rf=0,074 по ТСХ (гексан-EtOАc=1-1).
Стадия В: 18 мл 0,005 н соляной кислоты (0,02 экв.) вводят к остатку после дистилляции со стадии А. Реакционную смесь перемешивают при температуре 70°С в течение 3 часов и осуществляют мониторинг посредством ТСХ. После завершения взаимодействия, реакционную смесь охлаждают до температуры 25°С и вводят в другой подходящий реактор, содержащий 22 мл 5%-ного раствора хлорида натрия. Реакционную смесь перемешивают до тех пор, пока не растворится вся соль, затем экстрагируют этилацетатом (8 раз по 200 мл). Объединенные органические слои отфильтровывают через слой сульфата натрия и фильтрат концентрируют путем дистилляции досуха. Продукт выделяют (выход 99,38%) и непосредственно используют на следующей стадии.
1H ЯМР (400 MГц, Хлороформ-d1) δ м.д. 2,68-2,80 (м, 2H), 2,16-2,39 (м, 3H), 1,77-2,04 (м, 4H), 1,41 (с, 3H), 1,33 (с, 1H).
Стадия С: Продукт со стадии В (0,25 г) растворяют в толуоле (5 мл) в трехгорлой колбе емкостью 25 мл, оснащенной ловушкой Дина-Старка. Через реактор барботируют азот для удаления воздуха. В реактор вводят 0,585 г нитрометана (5 экв.), затем 0,052 г N,N-диметилэтилендиамина (0,3 экв.). Реакционную смесь кипятят с обратным холодильником, воду удаляют с помощью ловушки Дина-Старка. Реакционную смесь перемешивают при кипячении с обратным холодильником в течение 1 часа и осуществляют мониторинг посредством ВЭЖХ-анализа. Реакционную смесь потом охлаждают до температуры 20°С, когда стабилизировался продукт согласно ВЭЖХ-анализу, затем обрабатывают с помощью EtOAc и гептана и концентрируют досуха. Остаток очищают на колонке CombiFlash (колонка, масса 12 г) при использовании смеси гексан/EtOAc = от 80-20 до 60-40. Фракции анализируют посредством ВЭЖХ и ТСХ, содержащие продукт фракции дистиллируют досуха. Получают 0,23 г концентрированного масла (выход 68,09%) и используют на стадии D.
1H ЯМР (400 MГц, Хлороформ-d1) δ 5,88-5,90 (ушир.с, 1H), 4,88-4,89 (ушир.с, 2H), 2,16-2,40 (м, 4H), 1,78-1,85 (м, 1H), 1,33 (с,3H).
Стадия D: Катализатор Крэбтри (0,471 г, 0,585 ммоль) вводят, в атмосфере азота, в емкостью 450 мл и с перемешиванием реактор Парра стандартной спецификации. Реактор продувают азотом и вводят раствор (S)-1-метил-4-(нитрометил)циклогекс-3-енола (34,88 г, 58,5 ммоль) в DCM (100 мл). Добавляют дополнительное количество подвергнутого барботажу азотом DCM (80 мл), реактор продувают аргоном, водородом и создают давление водорода 100 фунт/кв.дюйм. Смесь перемешивают в течение 4 часов при температуре 30°С. За протеканием реакции наблюдают посредством ЯМР, концентрируют до получения масла, обрабатывают 2 раза с помощью ТГФ (50 мл), затем разбавляют с помощью ТГФ (50 мл). Продукт далее переносят, для последующего восстановления при использовании никеля Ренея, на стадию Е.
1H ЯМР (Хлороформ-d1): δ 4,33 (д, J=7,3 Гц, 2H), 4,32 (J=6,5 Гц, 1H), 2,36-2,20 (м, 1H), 1,92-1,69 (м, 1H), 1,64-1,40 (м, 1H), 1,39-1,18 (м, 1H).
Стадия Е: Никель Ренея (*d/(d-1) или *7/6)=2,04 г (20% масс.), 3 раза промывают с последующей декантацией при использовании ТГФ. Никель Ренея, раствор из (1R,4R)-1-метил-4-(нитрометил)циклогексанола и ТГФ (50 мл), в атмосфере азота, вводят в емкостью 450 мл и с перемешиванием реактор Парра стандартной спецификации. Реактор продувают азотом, водородом и осуществляют гидрирование при давлении, составляющем 40 фунт/кв.дюйм, в течение 4 часов, при температуре 50°С. Реакцию контролируют посредством GC и, после завершения, реакционную смесь отфильтровывают через полипропиленовую фильтровальную воронку с диатомовой землей/полиэтиленовым фриттированным диском для удаления катализатора. ТГФ используют в качестве ополаскивающего средства для экстракции остаточного продукта из фильтровального осадка. Объединенный фильтрат представляет собой раствор янтарного цвета, который непосредственно переносят на следующую стадию.
1H ЯМР (400 MГц, Хлороформ-d1) δ 2,61 (д, J=6,5 Гц, 2H), 1,25-1,50 (м, 12H), 0,80-1,17 (м, 3H).
Стадия F: 9,86 г Раствора со стадии Е добавляют в круглодонную колбу емкостью 500 мл и дистиллируют досуха, дважды обрабатывают ацетонитрилом и затем растворяют в ацетонитриле (100 мл). К раствору добавляют гидрат 4-метилбензолсульфоновой кислоты (11,68 г), после чего твердое вещество осаждается и температуру повышают до 40°С. Суспензию перемешивают при температуре 50°С в течение 2 часов и охлаждают до температуры 20°С в течение 12 часов. Твердые вещества отфильтровывают и промывают с помощью 40 мл ацетонитрила. Влажный фильтровальный осадок высушивают в вакууме, получая 14,24 г продукта (выход 77%).
1H ЯМР (400 MГц, оксид дейтерия-d2) δ 2,79 (д, J=7,0 Гц, 2H), 1,48-1,68 (м, 5H), 1,31-1,46 (м, 2H), 0,90-1,29 (м,5H).
Пример 12: Синтез 4-({[(1R,4R)-4-гидрокси-4-метилциклогексил]метил}амино)-3-нитробензолсульфонамида (Соединение (Р))
4-Хлор-3-нитробензолсульфонамид (6,5 г, 27,5 ммоль) и ((1R,4R)-4-гидрокси-4-метилциклогексил)метанаминий-4-метилбензолсульфонат (11,26 г, 35,7 ммоль) комбинируют в 35 мл ацетонитрила и перемешивают. К суспензии добавляют N,N-диизопропилэтиламин (8,88 г, 68,7 ммоль), при температуре окружающей среды, приходя в результате к эндотермии (от 200°С до 17,5°С). Спустя 10 минут, реакционную смесь нагревают до температуры 80°С и отстаивают при этой же температуре в течение 24 часов. Реакцию подвергают мониторингу в отношении завершения посредством ВЭЖХ. После завершения реакции, реакционную смесь охлаждают до температуры 40°С. Спустя 15 минут добавляют воду (32,5 мл) и выдерживают в течение 30 минут. В течение 30 минут добавляют дополнительные 74,5 мл воды. Твердый продукт осаждается вскоре после добавления второй порции воды. После перемешивания в течение 1 часа, при температуре 40°С, содержащую продукт смесь оставляют охлаждаться до температуры 20°С, перемешивают в течение 12 часов и затем охлаждают до температуры 0°С, при перемешивании, в течение 2 дополнительных часов. Продукт отфильтровывают и высушивают в вакууме, получая 8,8 г продукта (выход 93%; чистота >99%, ч.д.а.).
1H ЯМР (400 MГц, ДМСО-d6) δ м.д. 8,52 (т, J=5,9 Гц, 1H), 8,45 (д, J=2,2 Гц, 1H), 7,80 (дд, J=9,1, 2,3 Гц, 1H), 7,24-7,30 (м, 3H), 4,23 (с, 1H), 1,60-1,74 (м, 3H), 1,52-1,57 (м, 2H), 1,26-1,40 (м, 2H), 1,06-1,25 (м, 5H).
Пример 13: Синтез 4-(4-{[2-(4-хлорфенил)-4,4-диметилциклогекс-1-ен-1-ил]метил}пиперазин-1-ил)-N-({3-нитро-4-[(1R,4R)-[4-гидрокси-4-метилциклогексил]метил)амино]фенил}сульфонил)-2-(1Н-пирроло[2,3-b]пиридин-5-илокси)бензамида (Соединение (2))
Сульфонамид, 4-((((1R,4R)-4-гидрокси-4-метилциклогексил)метил)амино)-3-нитробензолсульфонамид (8,00 г, 23,29 ммоль), EDAC-HCl (5,80 г, 30,3 ммоль) и DMAP (8,54 г, 69,9 ммоль) перемешивают в DCM (186 мл, 14 об.) до образования суспензии золотистого цвета. Добавляют раствор из кислоты, 2-((1Н-пирроло[2,3-b]пиридин-5-ил)окси)-4-(4-((4’-хлор-5,5-диметил-3,4,5,6-тетрагидро-[1,1’-бифенил]-2-ил)метил)пиперазин-1-ил)бензойной кислоты (13,3 г, 23,29 ммоль), и ТЕА (6,49 мл, 46,6 ммоль) в DCM (80 мл, 6 об.), в течение 2,5 часов, с помощью капельной воронки, затем ополаскивают с помощью 10 мл DCM. После перемешивания в течение 12 часов, добавляют N1,N1-диметилэтан-1,2-диамин (5,09 мл, 46,6 ммоль) и продолжают перемешивать при температуре 20°С в течение 5 часов. Реакционную смесь промывают 10%-ным раствором НОАс (3 раза по 130 мл). Органический слой промывают 5%-ным раствором NaHCO3 (140 мл) и 5%-ным раствором NaCl (140 мл). Органический слой сушат над Na2SO4 и концентрируют до 7 объемов DCM-раствора. В течение 2 часов, по каплям, добавляют метанол (10 об., 140 мл), и раствор охлаждают до температуры 15°С, после чего продукт осаждается. Содержащую продукт смесь охлаждают до температуры 5°С и перемешивают в течение 2 часов. После отфильтровывания твердого вещества и высушивания с помощью продувки азотом в течение 2 часов, получают 17,35 г продукта (выход 83%; чистота >99,5%, ч.д.а.).
1Н ЯМР (400 MГц, ДМСО-d6): δ м.д. 11,57-11,59 (ушир., 1H), 8,48-8,52 (м, 2H), 7,97 (д, J=2,6 Гц, 1H), 7,73 (дд, J=9,2, 2,3 Гц, 1H), 7,43-7,50 (м, 3H), 7,29-7,31 (м, 2H), 6,98-7,03 (м, 3H), 6,65 (дд, J=8,9, 2,3 Гц, 1H), 6,35 (дд,J=3,4, 1,8 Гц, 1H), 6,16 (д, J=2,2 Гц, 1H), 4,41-4,44 (м, 1H), 3,71-3,75 (м, 2H), 2,98-3,51 (м, 11H), 2,74-2,76 (м, 3H), 2,02-2,26 (м, 6H), 1,88-1,92 (м, 2H), 1,47-1,70 (м, 5H), 1,24-1,40 (м, 4H), 1,08 (с, 5H), 0,89 (с, 6H).
Все ссылки, цитированные в данном контексте, включены во всей их полноте путем ссылки. Тогда как способы, предусмотренные в данном контексте, описаны, принимая во внимание конкретные воплощения, квалифицированному специалисту в данной области должно быть очевидно, что можно осуществлять различные изменения и модификации без отступления от сущности и объема данного изобретения, как изложено в прилагаемой формуле изобретения.
Подразумевают, что воплощения, описанные выше, являются только примерными и квалифицированный специалист в данной области должен распознавать или должен быть способен установить, используя не более, чем обычное экспериментирование, многочисленные эквиваленты конкретных соединений, веществ и способов. Все такие эквиваленты входят в рамки данного изобретения и охвачены прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФОТОХРОМНЫЕ ОКСАЗИНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРОИЗВОДСТВА | 2002 |
|
RU2315042C2 |
| СПОСОБ СИНТЕЗА ПРОИЗВОДНЫХ АМИНО-МЕТИЛТЕТРАЛИНА | 2009 |
|
RU2512285C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ 3-(4-(2,4-ДИФТОРБЕНЗИЛОКСИ)-3-БРОМ-6-МЕТИЛ-2-ОКСОПИРИДИН-1(2Н)-ИЛ)-N,4-ДИМЕТИЛБЕНЗАМИДА | 2007 |
|
RU2411236C1 |
| МОДУЛЯТОРЫ ROR-ГАММА | 2012 |
|
RU2658013C2 |
| ПРОИЗВОДНЫЕ ТРИАЗОЛА КАК ИНГИБИТОРЫ 11-БЕТА-ГИДРОКСИСТЕРОИДДЕГИДРОГЕНАЗЫ-1 | 2003 |
|
RU2360910C2 |
| ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2528393C9 |
| ПРОТИВООПУХОЛЕВЫЕ СОЕДИНЕНИЯ | 2008 |
|
RU2489429C2 |
| ПОЗИТИВНЫЕ АЛЛОСТЕРИЧЕСКИЕ МОДУЛЯТОРЫ НИКОТИНОВОГО РЕЦЕПТОРА АЦЕТИЛХОЛИНА | 2012 |
|
RU2604737C2 |
| ПОГЛОЩАЮЩИЕ УФ-ИЗЛУЧЕНИЕ ТРИФУНКЦИОНАЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2009 |
|
RU2544540C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ 1, 3-БЕНЗОДИОКСОЛЬНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ | 2015 |
|
RU2697251C2 |
Изобретение относится к способу получения вызывающего апоптоз агента (А1)  и химическим промежуточным продуктам для его получения. Технический результат: разработан новый способ получения, позволяющий получать соединение A1 с высоким выходом, а кроме того, позволяющий использовать практичные способы выделения и очистки. 6 н. и 23 з.п. ф-лы, 1 табл., 13 пр.
и химическим промежуточным продуктам для его получения. Технический результат: разработан новый способ получения, позволяющий получать соединение A1 с высоким выходом, а кроме того, позволяющий использовать практичные способы выделения и очистки. 6 н. и 23 з.п. ф-лы, 1 табл., 13 пр.
1. Способ получения соединения формулы А1:
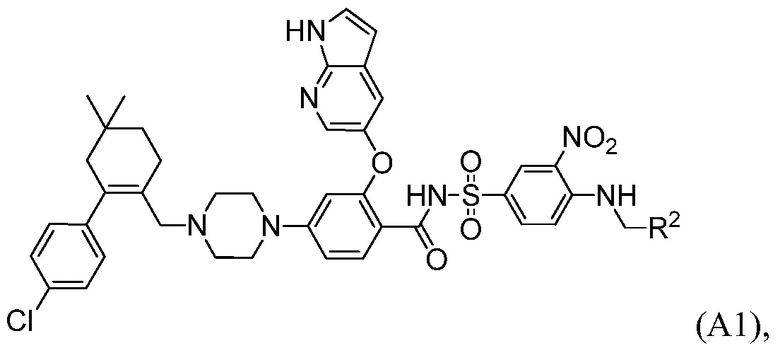
где R2 выбирают из: 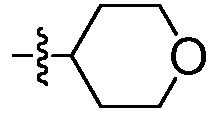 и
и 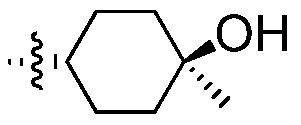 ,
,
включающий:
(а) комбинирование соединения формулы (К):
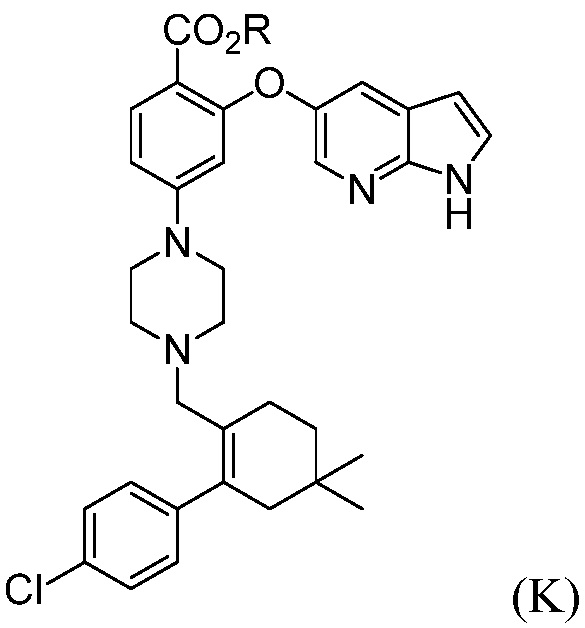 ,
,
где R означает С1-С12-алкил,
с трет-бутоксидом, апротонным органическим растворителем и водой, до получения соединения формулы (L):

и
(b”) комбинирование соединения формулы (L) с 1-этил-3-(3-диметиламинопропил)карбодиимидгидрохлоридом, 4-диметиламинопиридином, органическим растворителем и или с соединением формулы (N), до получения соединения формулы (А1), где R2 означает: 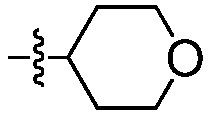 ,
,
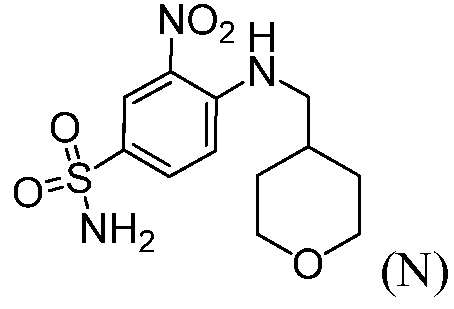
или с соединением формулы (Р), до получения соединения формулы (А1), где R2 означает:  ,
,
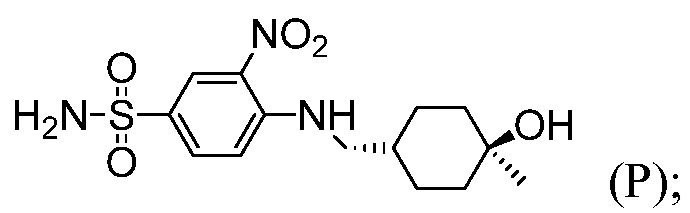
таким образом, получая соединение формулы (А1),
где соединение формулы (К) получают следующим образом:
(d) комбинирование соединения формулы (D):
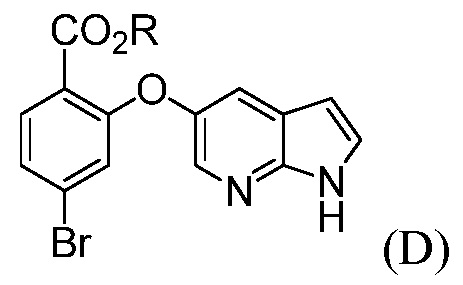
с соединением формулы (I):
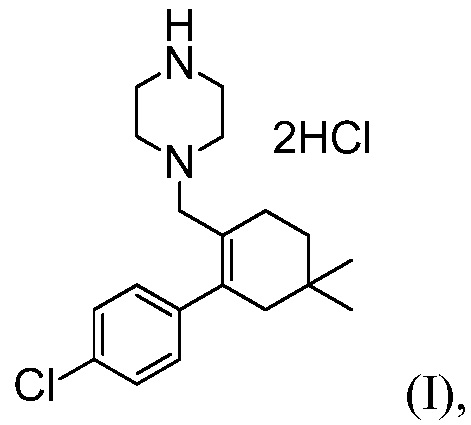
источником палладия, трет-бутоксидом и фосфиновым лигандом, в апротонном органическом растворителе, до получения соединения формулы (К).
2. Способ по п.1, где R2 означает:  , и стадия (b”) включает:
, и стадия (b”) включает:
(b”) комбинирование соединения формулы (L) с 1-этил-3-(3-диметиламинопропил)карбодиимидгидрохлоридом, 4-диметиламинопиридином, органическим растворителем и соединением формулы (N):
до получения соединения формулы (А1).
3. Способ по п.1, где R2 означает: , дополнительно включающий:
(с”) комбинирование соединения формулы (М):
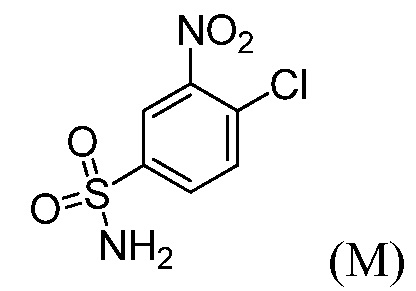
с основанием в виде третичного амина, органическим растворителем и (тетрагидро-2Н-пиран-4-ил)метанамином или его солью, до получения соединения формулы (N).
4. Способ по п.1, где R2 означает: , дополнительно включающий:
(с”) комбинирование соединения формулы (М):
с основанием в виде третичного амина, органическим растворителем и (1R,4R)-4-(аминометил)-1-метилциклогексанолом или его солью, до получения соединения формулы (Р).
5. Способ по любому одному из пп.1-4, где на стадии (а) трет-бутоксид выбирают из группы, состоящей из трет-бутоксида натрия и трет-бутоксида калия.
6. Способ по п.5, где на стадии (с”) основание в виде третичного амина представляет собой N,N-диизопропилэтиламин.
7. Способ по любому из пп.1-4, где соединение формулы (I) комбинируют с основанием перед комбинированием на стадии (d).
8. Способ по любому из пп.1-4, где на стадии (d) источник палладия представляет собой Pd2dba3 или [(циннамил)PdCl]2.
9. Способ по любому из пп.1-4, где фосфиновый лиганд на стадии (d) представляет собой соединение формулы (J):
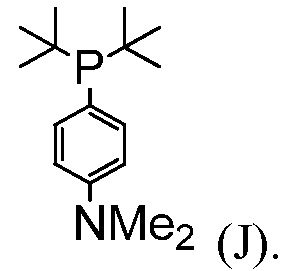
10. Способ по п.9, где источник палладия представляет собой Pd2dba3, каталитическое количество Pd2dba3 используют соответственно количеству соединения (I) и где каталитическое количество Pd2dba3 составляет от примерно 0,5% мол. до примерно 2% мол.
11. Способ по п.10, где каталитическое количество соединения формулы (J) используют соответственно количеству соединения (I) и где каталитическое количество соединения формулы (J) составляет от примерно 1% мол. до примерно 5% мол.
12. Способ по любому из пп.1-4, где на стадии (d) трет-бутоксид выбирают из группы, состоящей из трет-бутоксида натрия и трет-бутоксида калия.
13. Способ по любому из пп.1-4, где способ далее включает:
(е) комбинирование соединения формулы (В) с соединением формулы (С):
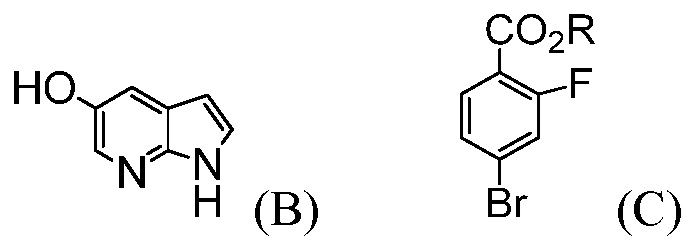
и с трет-бутоксидом, в органическом растворителе, до получения соединения формулы (D).
14. Способ по п.13, где на стадии (е) трет-бутоксид выбирают из группы, состоящей из трет-бутоксида натрия и трет-бутоксида калия.
15. Способ по п.13, дополнительно включающий:
(f) комбинирование соединения формулы (А):

с R1MgX, в апротонном органическом растворителе;
где R1 означает С1-С6-алкил и Х означает Cl, Br или I;
(g) комбинирование С1-С12-алкилхлорформиата или ди(С1-С12-алкил)дикарбоната с продуктом со стадии (f), до получения соединения формулы (С).
16. Способ по п.15, где на стадии (f) R1 означает изопропил.
17. Способ по п.15, где на стадии (f) R означает трет-бутил, а ди(С1-С12-алкил)дикарбонат представляет собой ди-трет-бутилдикарбонат.
18. Способ по любому из пп.1-4, дополнительно включающий:
(h) комбинирование соединения формулы (Е):
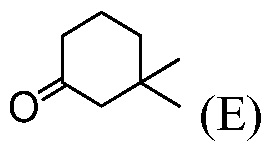
с диметилформамидом и POCl3, до получения соединения формулы (F):
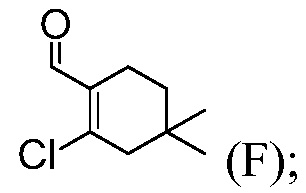
(i) комбинирование соединения формулы (F) с источником палладия и 4-хлорфенилбороновой кислотой, в органическом растворителе, до получения соединения формулы (G):
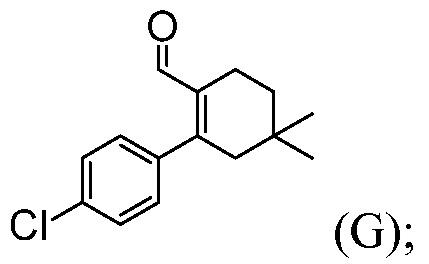
(j) комбинирование соединения формулы (G) с ВОС-пиперазином и триацетоксиборгидридом натрия, в органическом растворителе, до получения соединения формулы (Н):
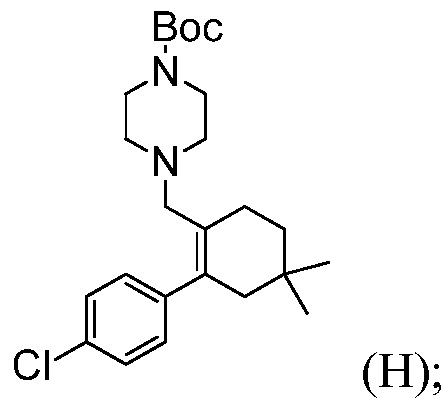 и
и
(k) комбинирование соединения формулы (Н) с соляной кислотой, до получения соединения формулы (I).
19. Способ по п.18, где на стадии (i) источник палладия представляет собой Pd(OAc)2.
20. Способ по п.18, где стадия (i) включает комбинирование тетрабутиламмонийбромида с соединением формулы (F), источником палладия и 4-хлорфенилбороновой кислотой, в органическом растворителе.
21. Способ по п.18, где стадия (j) дополнительно включает получение соединения формулы (Н) в виде кристаллического твердого вещества.
22. Способ по п.18, где стадия (k) дополнительно включает получение соединения формулы (I) в виде твердого кристаллического вещества.
23. Способ по любому одному из пп.1-4, где R выбирают из группы, состоящей из метила, этила, н-пропила, изопропила, н-бутила, трет-бутила, изобутила и необутила.
24. Способ по п.23, где R означает трет-бутил.
25. Способ получения соединения формулы (С):
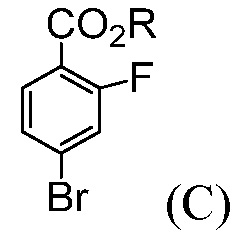 ,
,
где R означает С1-С12-алкил,
включающий:
(а) комбинирование соединения формулы (А):
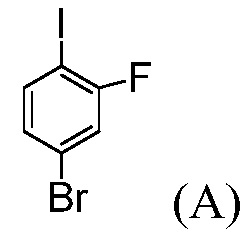
с R1MgX, в апротонном органическом растворителе; где R1 означает С1-С6-алкил и Х означает Cl, Br или I;
(b) комбинирование С1-С12-алкилхлорформиата или ди(С1-С12-алкил)дикарбоната с продуктом со стадии (а), до получения соединения формулы (С).
26. Соединение формулы:

27. Способ получения соединения по п.26, включающий:
(х) комбинирование соединения формулы (В):
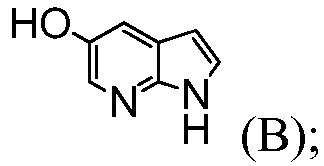
с соединением формулы (С):
,
где R означает трет-бутил,
и с трет-бутоксидом, в органическом растворителе, до получения соединения по п.27.
28. Соединение формулы:
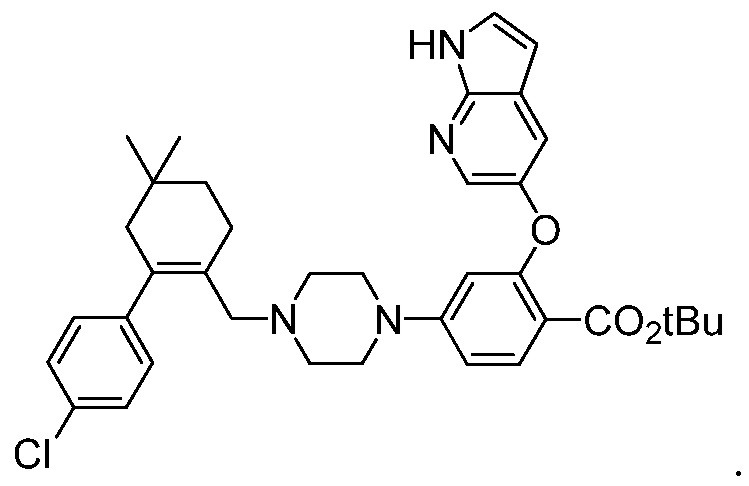
29. Способ получения соединения по п.28, включающий:
(y) комбинирование соединения формулы (В):
с соединением формулы (С):
,
где R означает трет-бутил,
и с трет-бутоксидом, в органическом растворителе, до получения соединения формулы (D):
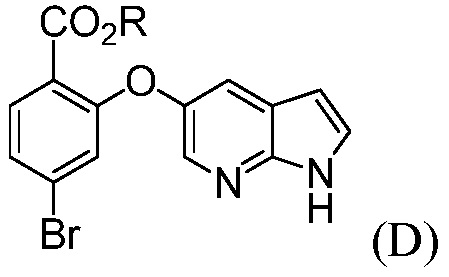 ,
,
где R означает трет-бутил;
(z) комбинирование соединения формулы (D), где R означает трет-бутил;
с соединением формулы (I):
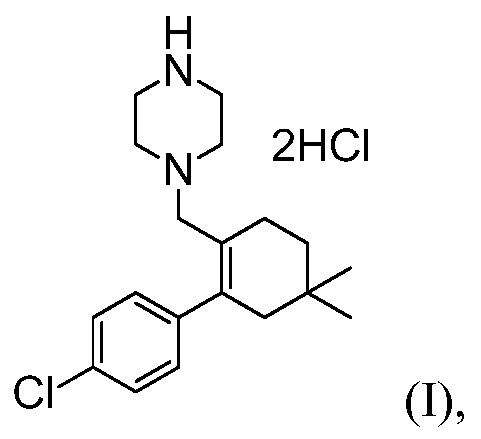
источником палладия, трет-бутоксидом и фосфиновым лигандом, в апротонном органическом растворителе, до получения соединения по п.28.
| WO 2011150016 A1, 01.12.2011 | |||
| WO 201207336 A1, 31.05.2012 | |||
| Gassman P G; Schenk; W N, A General Procedure for the Base-Promoted Hydrolysis of Hindered Esters at Ambient Temperatures J | |||
| Org | |||
| Chem, т.42, 5, стр | |||
| Весы | 1923 |
|
SU918A1 |
| Juan D | |||
| Arredondo, Hongmei Li and Jaume Balsells, Preparation of t-Butyl-3-Bromo-5-Formylbenzoate Through Selective Metal-Halogen Exchange Reactions, Org | |||
| Synth., 89, стр.460-470, 2012 | |||
| Rausis T., Schlosser M | |||
| The Basicity Gradient-Driven Migration of Iodine: Conferring Regioflexibility on the Substitution of Fluoroarenes, Eur | |||
| J | |||
| Org | |||
| Chem, стр.3351-3358, 2002 | |||
| RU 2011134634 A, 15.01.2010. | |||

