ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым соединениям и их применению при профилактическом и/или терапевтическом лечении сердечно-сосудистых заболеваний, и, в частности, лечении предгипертензии, гипертензии и/или фиброзных состояний.
Настоящее изобретение было разработано преимущественно для профилактического и/или терапевтического лечения сердечно-сосудистого заболевания и будет описано далее в данном документе со ссылкой на настоящую заявку. Однако следует принимать во внимание, что настоящее изобретение не ограничено данной конкретной областью применения.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Любое обсуждение уровня техники по всему настоящему описанию никоим образом не должно рассматриваться как допущение того, что такой уровень техники широко известен или образует часть общеизвестных знаний в данной области.
Гипертензия (высокое артериальное давление) поражает 26% взрослого населения по всему миру с частотой встречаемости 30-33% в западных странах. Ожидается, что частота встречаемости гипертензии в мире достигнет 29% к 2025 году вследствие вестернизации Индии и Китая. Современные исследования указывают на то, что менее чем 20% пациентов с гипертензией достигают рекомендованного целевого значения артериального давления (BP) и что для достижения этих целей >75% пациентов нуждаются в терапии с использованием нескольких антигипертензивных средств. Предгипертензия (немного повышенное артериальное давление) поражает 31% взрослых в США и без лечения может развиться в гипертензию.
Все доступные в настоящее время методы терапии имеют побочные эффекты:
• ингибиторы ангиотензинпревращающего фермента (ACEI) - кашель, ангионевротический отек, гиперкалиемия;
• блокаторы рецепторов ангиотензина (ARB) - ангионевротический отек, гиперкалиемия;
• блокаторы кальциевых каналов (ССВ) - гиперемия, отек ноги/лодыжки, запор;
• тиазидные диуретики - впервые выявленный диабет, подагра, гипонатриемия;
• бета (β)-блокаторы - впервые выявленный диабет, неспособность к физическим нагрузкам, брадикардия, скрытая гипогликемия у больных диабетом, и
• антагонисты альдостерона - гинекомастия, меноррагия, гиперкалиемия.
Необходимость применения комбинированной терапии повышает вероятность того, что пациенты будут испытывать побочные эффекты и, как следствие, не достигнут целевого значения BP.
Гипертензия и предгипертензия представляют собой основной фактор развития повреждения сердца, почек и кровеносных сосудов, приводя к замещению нормальной функциональной ткани рубцовой тканью или фиброзом. Некоторые из существующих антигипертензивных средств - ингибиторы АСЕ, ингибиторы ренина ARB и антагонисты альдостерона, способны замедлять прогрессирование замещения функциональной ткани фиброзом, однако ни для одного из них не было показано обращение существующего фиброза и восстановление нормального строения ткани. Таким образом, существует потребность в средствах, которые эффективны в существенном снижении значения BP и, таким образом, позволяют более значительной части пациентов достигнуть целевого значения BP при терапии одним средством, и/или обратить существующий фиброз, и/или восстановить нормальное строение ткани.
Целью настоящего изобретения является преодоление или устранение по меньшей мере одного из недостатков уровня техники или предоставление полезной альтернативы.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения неожиданно обнаружили, что некоторые новые терфенильные соединения обладают эффектами, снижающими артериальное давление, и/или противофиброзными эффектами. Эти эффекты можно наблюдать в исследованиях с внутривенным и/или пероральным введением дозы.
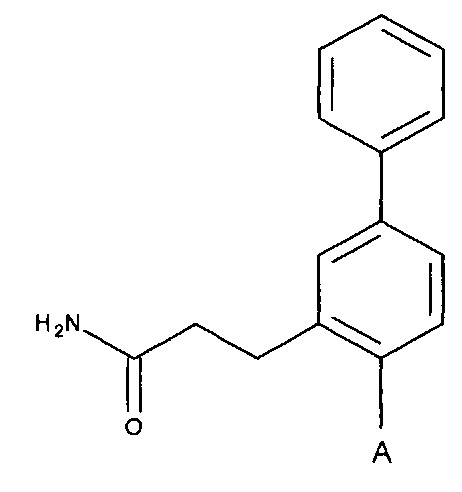
Согласно одному аспекту настоящее изобретение предусматривает соединение формулы
 ,
,
где
А выбран из группы, состоящей из
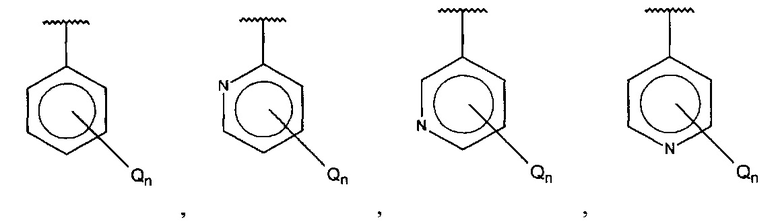
 и
и 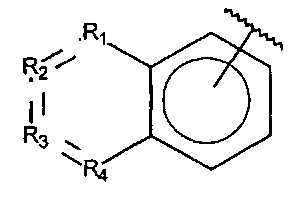 ,
,
Q независимо выбран из галогена, алкила, гидрокси, амино и замещенного амино;
n равняется 0, 1, 2, 3, 4 или 5;
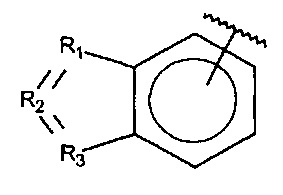
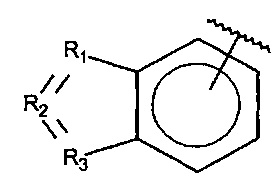
R1, R3 и R4 независимо представляют собой C, CH, CH2, О, N, NH или S, и
R2 представляет собой C, СН, CH2, N, NH, C-CF3, CH-CF3 или C=O,
или его стереоизомер или фармацевтически приемлемую соль,
где, если n равняется 1, Q не может представлять собой гидрокси.
В одном варианте осуществления Q представляет собой галоген, выбранный из группы, состоящей из F, Cl, Br и I.
В одном варианте осуществления Q представляет собой замещенный амино формулы -NHW, и где
W выбран из -CN, -SO2(X)aY и -CO(X)aY,
а равняется 0 или 1,
X выбран из -NH- и -O-, и
Y выбран из -H, -CH3, -CH2CH3, -CH2OH и -CH2CH2OH.
В одном варианте осуществления Q представляет собой замещенный амино, выбранный из группы, состоящей из -NHSO2CH3, -NHCOH, -NHCONHCH3, -NHCONHCH2CH3, -NHSO2NHCH3, -NHSO2NHCH2CH3, -NHCOCH3, -NHCOOCH3, -NHCOOCH2CH2OH, -NHCONH2 и -NHCN.
В одном варианте осуществления Q представляет собой алкил, выбранный из группы, состоящей из метила, этила, пропила, бутила и пентила.
В одном варианте осуществления A выбран из:
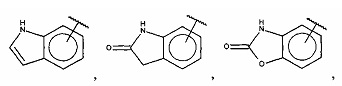
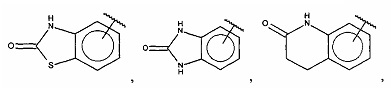
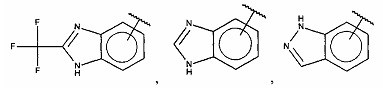
и 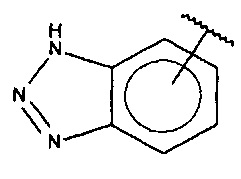 .
.
В одном варианте осуществления A представляет собой
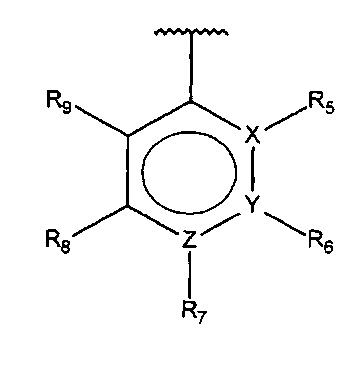 ,
,
где
X, Y или Z представляют собой C или N, где только один из X, Y или Z может представлять собой N,
R5-R9 независимо выбраны из водорода, галогена, алкила, гидрокси, амино и замещенного амино, при условии, что если один из R5-R9 представляет собой галоген, то по меньшей мере один из оставшихся R5-R9 не может представлять собой водород.
В одном варианте осуществления A представляет собой
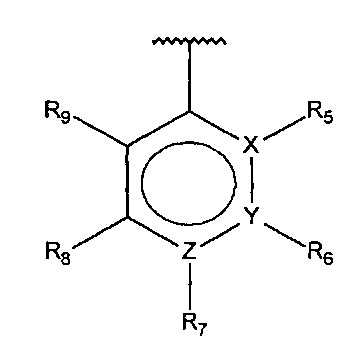 ,
,
где
X, Y или Z представляют собой C или N, где только один из X, Y или Z может представлять собой N,
R5-R9 независимо выбраны из водорода, галогена, алкила, гидрокси, амино и замещенного амино, при условии, что если один из R5-R9 представляет собой галоген, то по меньшей мере один из оставшихся R5-R9 должен представлять собой галоген, алкил, гидрокси, амино или замещенный амино.
В одном варианте осуществления A представляет собой
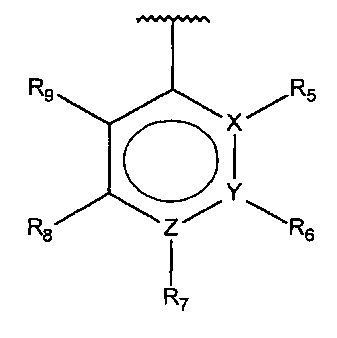 ,
,
где
X, Y или Z представляют собой C или N, где только один из X, Y или Z может представлять собой N,
R5-R9 независимо выбраны из водорода, галогена, алкила, гидрокси, амино и замещенного амино, при условии, что если один из R5-R9 представляет собой галоген, то по меньшей мере один из оставшихся R5-R9 должен представлять собой алкил, гидрокси, амино или замещенный амино.
В одном варианте осуществления A представляет собой
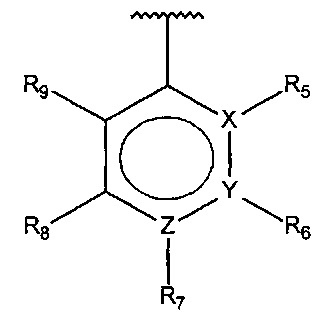 ,
,
где
X, Y или Z представляют собой C или N, где только один из X, Y или Z может представлять собой N,
R5-R9 независимо выбраны из водорода, галогена, алкила, гидрокси, амино и замещенного амино, при условии, что если один из R5-R9 представляет собой алкил, то по меньшей мере один из оставшихся R5-R9 не может представлять собой водород.
В одном варианте осуществления A представляет собой
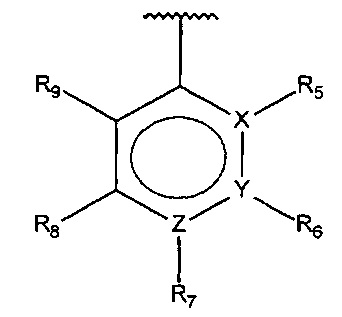 ,
,
где
X, Y или Z представляют собой C или N, где только один из X, Y или Z может представлять собой N,
R5-R9 независимо выбраны из водорода, галогена, алкила, гидрокси, амино и замещенного амино, при условии, что если один из R5-R9 представляет собой алкил, то по меньшей мере один из оставшихся R5-R9 должен представлять собой галоген, алкил, гидрокси, амино или замещенный амино.
В одном варианте осуществления A представляет собой
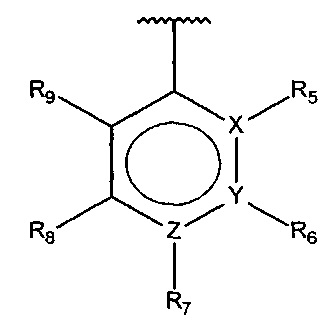 ,
,
где
X, Y или Z представляют собой C или N, где только один из X, Y или Z может представлять собой N,
R5-R9 независимо выбраны из водорода, галогена, алкила, гидрокси, амино и замещенного амино, при условии, что если один из R5-R9 представляет собой алкил, то по меньшей мере один из оставшихся R5-R9 должен представлять собой галоген, гидрокси, амино или замещенный амино.
В одном варианте осуществления A представляет собой
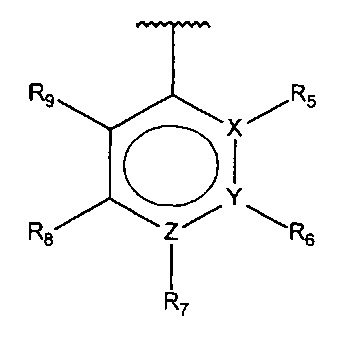 ,
,
где
X, Y или Z представляют собой C или N, где только один из X, Y или Z может представлять собой N,
R5-R9 независимо выбраны из водорода, галогена, алкила, гидрокси, амино и замещенного амино, при условии, что если один из R5-R9 представляет собой гидрокси, то по меньшей мере один из оставшихся R5-R9 не может представлять собой водород.
В одном варианте осуществления A представляет собой
 ,
,
где
X, Y или Z представляют собой C или N, где только один из X, Y или Z может представлять собой N,
R5-R9 независимо выбраны из водорода, галогена, алкила, гидрокси, амино и замещенного амино, при условии, что если один из R5-R9 представляет собой гидрокси, то по меньшей мере один из оставшихся R5-R9 должен представлять собой галоген, гидрокси, амино или замещенный амино.
В одном варианте осуществления A представляет собой
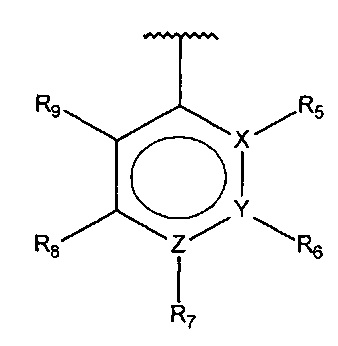 ,
,
где
X, Y или Z представляют собой C или N, где только один из X, Y или Z может представлять собой N,
R5-R9 независимо выбраны из водорода, галогена, алкила, гидрокси, амино и замещенного амино, при условии, что если один из R5-R9 представляет собой гидрокси, то по меньшей мере один из оставшихся R5-R9 должен представлять собой галоген, амино или замещенный амино.
В одном варианте осуществления A представляет собой  , Q представляет собой замещенный амино, и n равняется 1.
, Q представляет собой замещенный амино, и n равняется 1.
В одном варианте осуществления A представляет собой 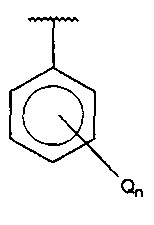 , Q представляет собой замещенный амино, и n равняется 2.
, Q представляет собой замещенный амино, и n равняется 2.
В одном варианте осуществления A представляет собой 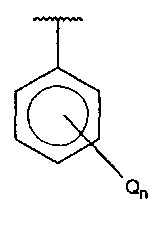 , Q представляет собой амино, и n равняется 1.
, Q представляет собой амино, и n равняется 1.
В одном варианте осуществления A представляет собой 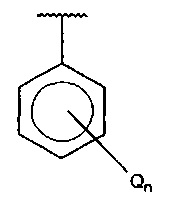 , Q представляет собой амино, и n равняется 2.
, Q представляет собой амино, и n равняется 2.
В одном варианте осуществления A представляет собой 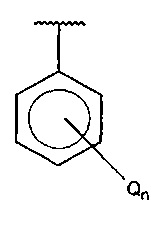 , Q представляет собой гидрокси, и n равняется 2.
, Q представляет собой гидрокси, и n равняется 2.
В одном варианте осуществления A представляет собой 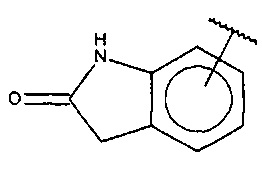 .
.
В одном варианте осуществления A представляет собой 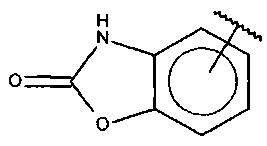 .
.
В одном варианте осуществления A представляет собой 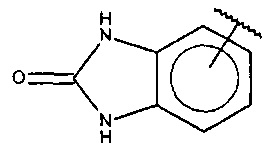 .
.
В одном варианте осуществления A представляет собой 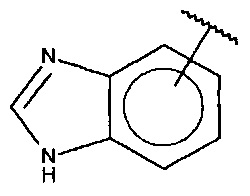 .
.
В одном варианте осуществления A представляет собой 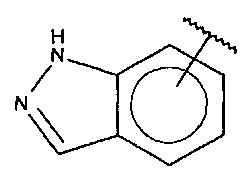 .
.
В одном варианте осуществления A представляет собой 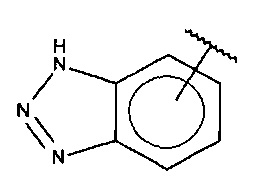 .
.
В одном варианте осуществления A представляет собой 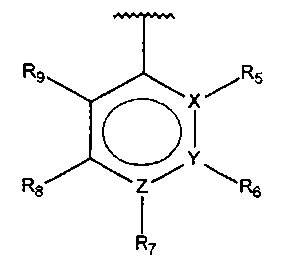 , X и Y представляют собой C, Z представляет собой N, и один из R5-R9 представляет собой амино.
, X и Y представляют собой C, Z представляет собой N, и один из R5-R9 представляет собой амино.
В одном варианте осуществления A представляет собой 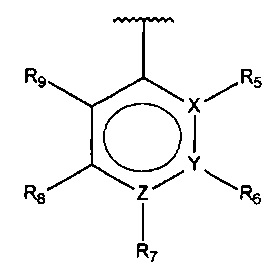 , все из X, Y и Z представляют собой C, один из R5-R9 представляет собой гидрокси, и по меньшей мере один из оставшихся R5-R9 представляет собой галоген.
, все из X, Y и Z представляют собой C, один из R5-R9 представляет собой гидрокси, и по меньшей мере один из оставшихся R5-R9 представляет собой галоген.
В одном варианте осуществления A представляет собой 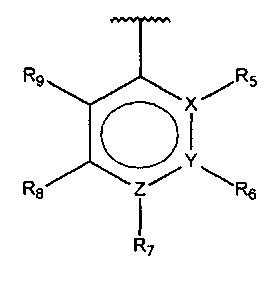 , все из Z, Y и Z представляют собой C, два из R5-R9 представляют собой гидрокси, и по меньшей мере один из оставшихся R5-R9 представляет собой галоген.
, все из Z, Y и Z представляют собой C, два из R5-R9 представляют собой гидрокси, и по меньшей мере один из оставшихся R5-R9 представляет собой галоген.
В одном варианте осуществления A представляет собой 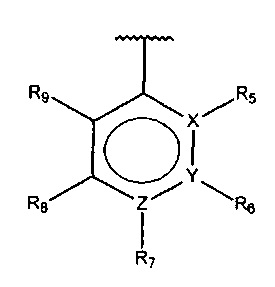 , все из Z, Y и Z представляют собой C, два из R5-R9 представляют собой гидрокси, и по меньшей мере один из оставшихся R5-R9 представляет собой алкил.
, все из Z, Y и Z представляют собой C, два из R5-R9 представляют собой гидрокси, и по меньшей мере один из оставшихся R5-R9 представляет собой алкил.
В одном варианте осуществления A представляет собой 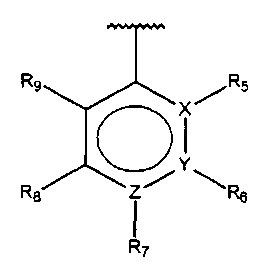 , все из Z, Y и Z представляют собой C, один из R5-R9 представляет собой алкил, и по меньшей мере один из оставшихся R5-R9 представляет собой гидрокси.
, все из Z, Y и Z представляют собой C, один из R5-R9 представляет собой алкил, и по меньшей мере один из оставшихся R5-R9 представляет собой гидрокси.
В одном варианте осуществления A представляет собой 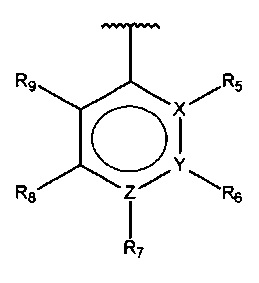 , все из Z, Y и Z представляют собой C, один из R5-R9 представляет собой галоген, и по меньшей мере один из оставшихся R5-R9 представляет собой гидрокси.
, все из Z, Y и Z представляют собой C, один из R5-R9 представляет собой галоген, и по меньшей мере один из оставшихся R5-R9 представляет собой гидрокси.
В одном варианте осуществления A представляет собой 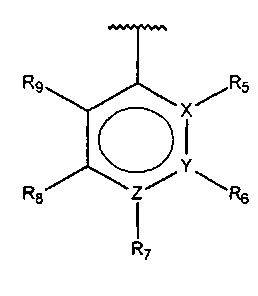 , все из Z, Y и Z представляют собой C, два из R5-R9 представляют собой галоген, и по меньшей мере один из оставшихся R5-R9 представляет собой гидрокси.
, все из Z, Y и Z представляют собой C, два из R5-R9 представляют собой галоген, и по меньшей мере один из оставшихся R5-R9 представляет собой гидрокси.
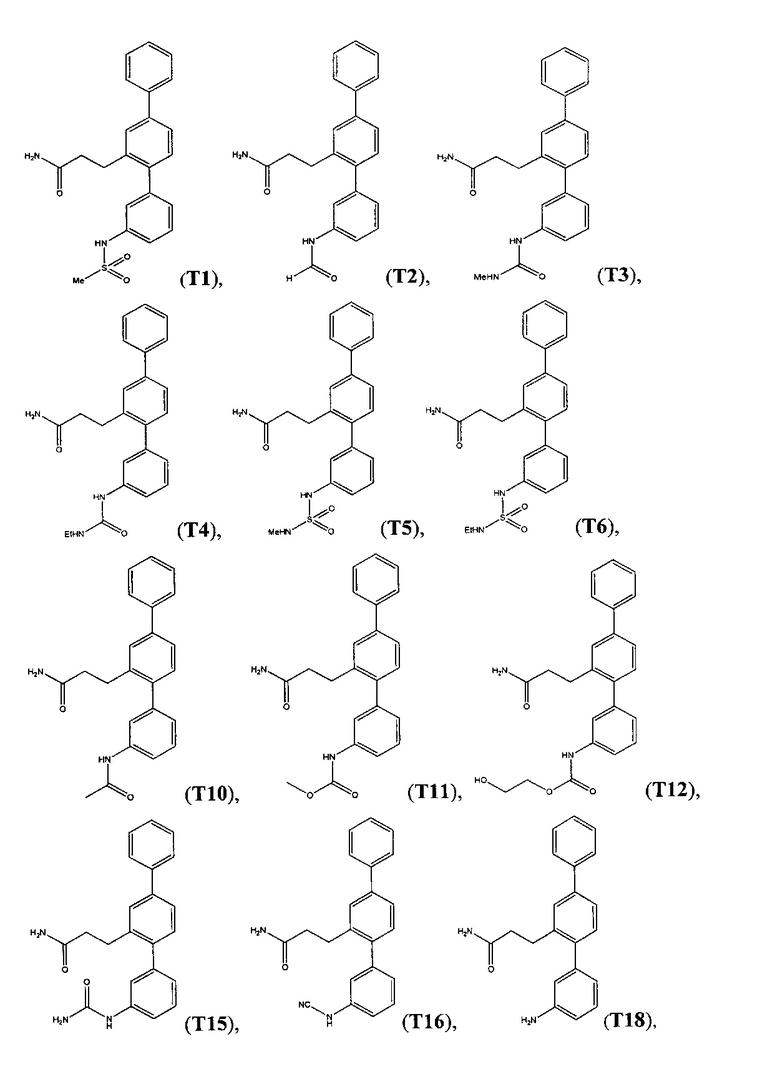
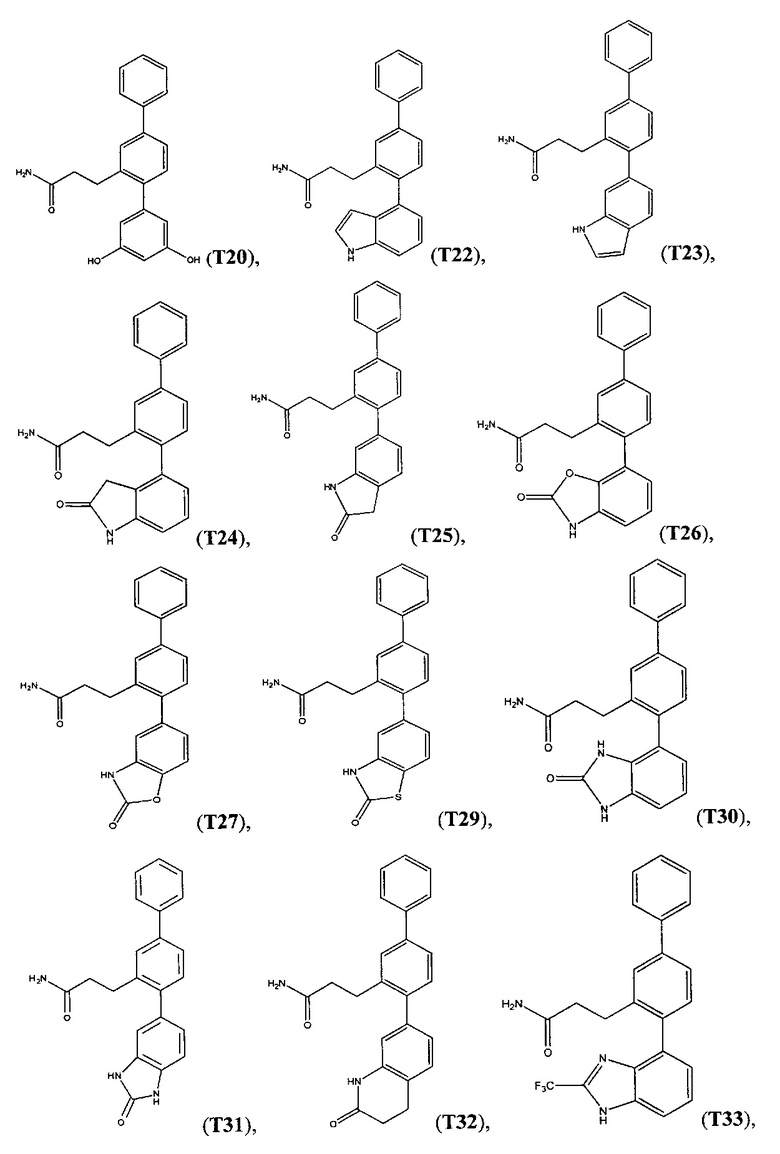
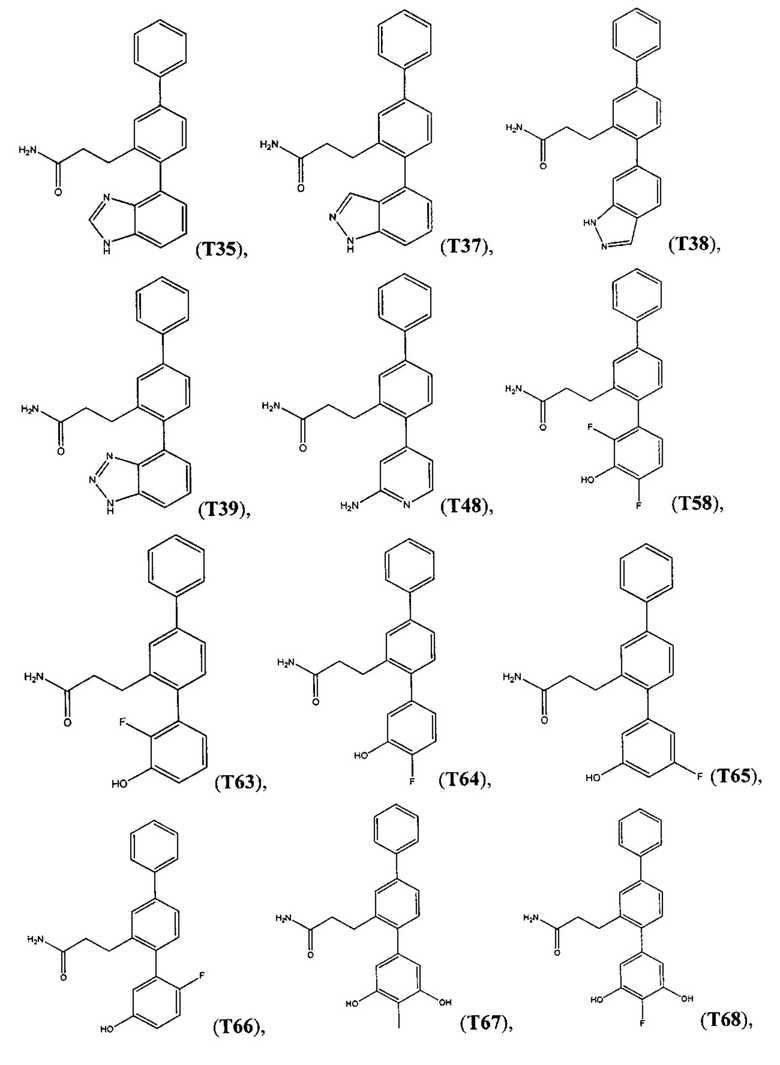
В одном варианте осуществления соединение выбрано из группы, состоящей из:
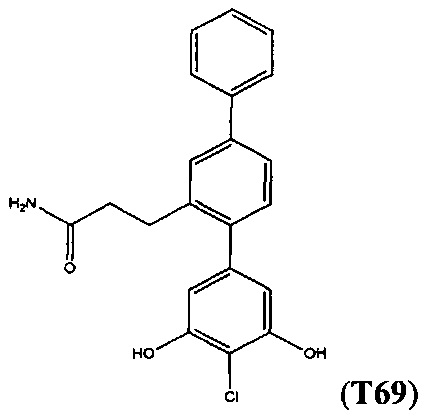 и
и 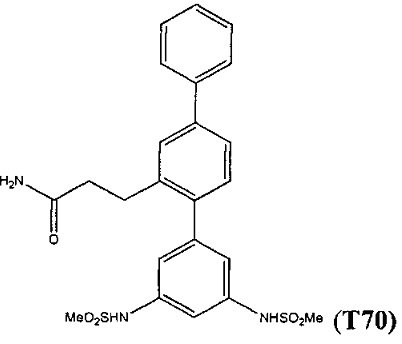 .
.
Согласно другому аспекту настоящее изобретение относится к фармацевтической композиции, содержащей соединение по настоящему изобретению и фармацевтически приемлемое вспомогательное вещество.
Согласно другому аспекту настоящее изобретение относится к способу терапевтического лечения гипертензии или предгипертензии у субъекта, включающему введение субъекту соединения согласно настоящему изобретению.
Согласно другому аспекту настоящее изобретение относится к способу терапевтического лечения фиброза у субъекта, включающему введение субъекту соединения согласно настоящему изобретению.
Согласно другому аспекту настоящее изобретение относится к способу профилактического лечения фиброза у субъекта, включающему введение субъекту соединения согласно настоящему изобретению.
Согласно другому аспекту настоящее изобретение относится к способу терапевтического лечения гипертензии и фиброза у субъекта, включающему введение субъекту соединения согласно настоящему изобретению.
Согласно другому аспекту настоящее изобретение относится к способу терапевтического лечения предгипертензии и фиброза у субъекта, включающему введение субъекту соединения согласно настоящему изобретению.
В одном варианте осуществления фиброз представляет собой фиброз миокарда или фиброз почки.
В другом варианте осуществления фиброз представляет собой фиброз миокарда и фиброз почки.
Согласно другому аспекту настоящее изобретение относится к соединению по настоящему изобретению для применения в терапевтическом лечении гипертензии или предгипертензии.
Согласно другому аспекту настоящее изобретение относится к соединению по настоящему изобретению для применения в терапевтическом лечении фиброза.
Согласно другому аспекту настоящее изобретение относится к соединению по настоящему изобретению для применения в профилактическом лечении фиброза.
Согласно другому аспекту настоящее изобретение относится к соединению по настоящему изобретению для применения в терапевтическом лечении гипертензии и фиброза.
Согласно другому аспекту настоящее изобретение относится к соединению по настоящему изобретению для применения в терапевтическом лечении предгипертензии и фиброза.
Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного препарата для терапевтического лечения гипертензии или предгипертензии.
Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного препарата для терапевтического лечения фиброза.
Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного препарата для профилактического лечения фиброза.
Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного препарата для терапевтического лечения гипертензии и фиброза.
Согласно другому аспекту настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного препарата для терапевтического лечения предгипертензии и фиброза.
Если контекст явно не требует иного, по всему описанию и формуле изобретения слова "содержит", "содержащий" и тому подобное должны быть истолкованы во включающем смысле в противоположность исключающему или исчерпывающему смыслу; то есть в смысле "включая без ограничения".
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фигура 1: Синтез 3-формилбифенил-4-ил-трифторметансульфоната.
Фигура 2: Синтез T1, Т2, Т10 и Т18.
Фигура 3: Синтез диэтил(карбоилметил)фосфоната.
Фигура 4: Синтез Т20.
Фигура 5: Синтез Т70.
Фигура 6: Синтез Т48.
Фигура 7: Синтез 3-(3-амино-3-оксопропил)бифенил-4-ил-трифторметансульфоната.
Фигура 8: Синтез Т25.
Фигура 9: Синтез индолонпинаколового эфира бороновой кислоты.
Фигура 10: Синтез Т31.
Фигура 11: Нормализованный относительно исходного уровня клеточный индекс для различных соединений в трех концентрациях, 62,5 мкМ (белые столбики), 125 мкМ (заштрихованные столбики) и 250 мкМ (столбики со сплошной заливкой) на гладкомышечных клетках сосудов крысы А10, определенный при помощи инструмента xCELLigence RTCA.
Фигура 12: Нормализованный относительно исходного уровня клеточный индекс для различных соединений в трех концентрациях, 62,5 мкМ (белые столбики), 125 мкМ (заштрихованные столбики) и 250 мкМ (столбики со сплошной заливкой) на эндотелиальных клетках бычьей аорты, определенный при помощи инструмента xCELLigence RTCA.
Фигура 13: Значения систолического (заштрихованные столбики) и диастолического (белые столбики) артериального давления в контрольных образцах и у обработанных крыс со спонтанной гипертензией (SHR) на 2,2% солевой диете через 4 недели после терапии. T1, Т2, Т20, Т31 и Т48 вводили при 500 пкмоль/кг/мин. в раствор для питья (5% этанола) в течение 4 недель, Т70 вводили при 100 пкмоль/кг/мин. в раствор для питья. * p<0,05, ** p<0,01, *** p<0,005 и **** p<0,0005 - систолическое артериальное давление при лечении по сравнению со систолическим артериальным давлением в контроле; # p<0,05, ## p<0,025 и ### p<0,005 - диастолическое артериальное давление при лечении по сравнению с диастолическим артериальным давлением в контроле.
Фигура 14: Связь между нормализованным относительно исходного уровня клеточным индексом для гладкомышечных клеток сосудов А10 и систолическим артериальным давлением для различных соединений.
Фигура 15: Связь между нормализованным относительно исходного уровня клеточным индексом для эндотелиальных клеток бычьей аорты и систолическим артериальным давлением для различных соединений.
Фигура 16: Фиброз миокарда, определенный количественно при помощи компьютеризированной гистоморфометрии на гистологических срезах, окрашенных с помощью трехцветной окраски по Массону, у SHR на 2,2% солевой диете через 14 недель и спустя 4 недели после обработки лекарственным средством, добавляемым в питьевой раствор, или для контроля, получавшего носитель. * p<0,005, ** p<0,001 и *** p<0,0005 по сравнению с 18-недельным контролем, обработанным носителем. # p<0,05, ## p<0,01, ### p<0,005 и #### p<0,0005 по сравнению с 14-недельным контролем. Последнее сравнение указывает на способность обращать существующую патологию.
Фигура 17: Интерстициальный фиброз в почке, определенный количественно при помощи компьютеризированной гистоморфометрии на гистологических срезах, окрашенных с помощью трехцветной окраски по Массону, у SHR на 2,2% солевой диете через 14 недель и спустя 4 недели после обработки лекарственным средством, добавляемым в питьевой раствор, или для контроля, получавшего носитель. * p<0,005, ** p<0,001 и *** p<0,0005 по сравнению с 18-недельным контролем, обработанным носителем. # p<0,05 по сравнению с 14-недельным контролем.
Последнее сравнение указывает на способность обращения существующей патологии.
Фигура 18: Связь между нормализованным относительно исходного уровня клеточным индексом для эндотелиальных клеток бычьей аорты и фиброзом миокарда для различных соединений.
Фигура 19: Связь между нормализованным относительно исходного уровня клеточным индексом для эндотелиальных клеток бычьей аорты и интерстициальным фиброзом почки для различных соединений.
Фигура 20: Микрофотографии сердца контрольных крыс (А) и крыс, обработанных в течение четырех недель при 500 пкмоль/кг/мин. Т1 (В), Т2 (С), Т20 (D) или Т31 (Е).
Фигура 21: Микрофотографии почки контрольных крыс (А) и крыс, обработанных в течение четырех недель при 500 пкмоль/кг/мин. Т1 (В), Т2 (С), Т20 (D) или Т31 (Е).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к некоторым новым терфенильным соединениям, которые характеризуются эффектами, снижающими артериальное давление, и/или противофиброзными эффектами при исследовании с пероральным введением дозы на экспериментальной животной модели. Что касается противофиброзной активности, соединения по настоящему изобретению являются эффективными в предотвращении фиброза, замедлении прогрессирования развившегося фиброза и/или снижении степени (обращении) развившегося фиброза. Это важное заключение в отношении диапазона и тяжести состояний, которые можно лечить с помощью соединений по настоящему изобретению.
Соединения по настоящему изобретению представлены формулой:
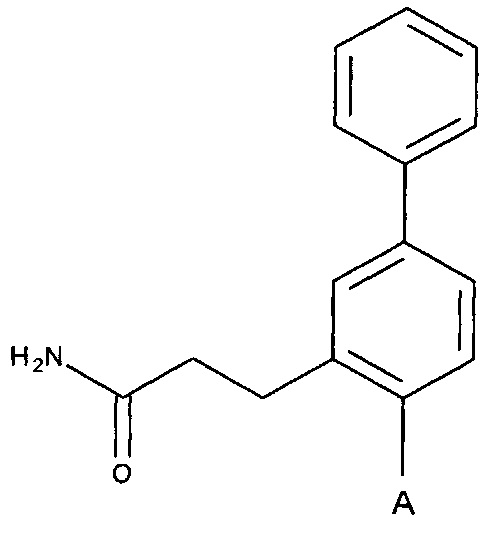 ,
,
где
А выбран из группы, состоящей из
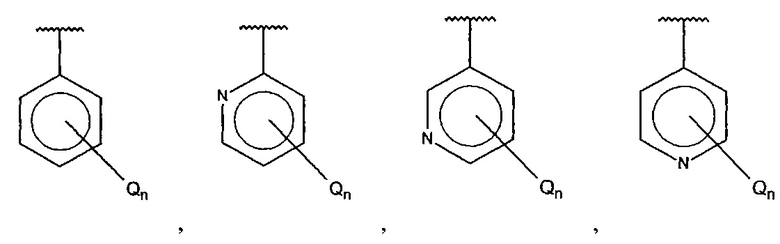
 и
и 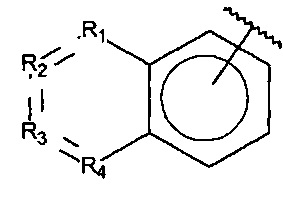 ,
,
Q независимо выбран из галогена, алкила, гидрокси, амино и замещенного амино;
n равняется 0, 1, 2, 3, 4 или 5;
R1, R3 и R4 независимо представляют собой C, СН, CH2, О, N, NH или S, и
R2 представляет собой C, СН, CH2, N, NH, C-CF3, CH-CF3 или C=O,
или их стереоизомером или фармацевтически приемлемой солью,
где, если n равняется 1, Q не может представлять собой гидрокси.
Следующие соединения являются конкретными, но не ограничивающими примерами соединений по настоящему изобретению:
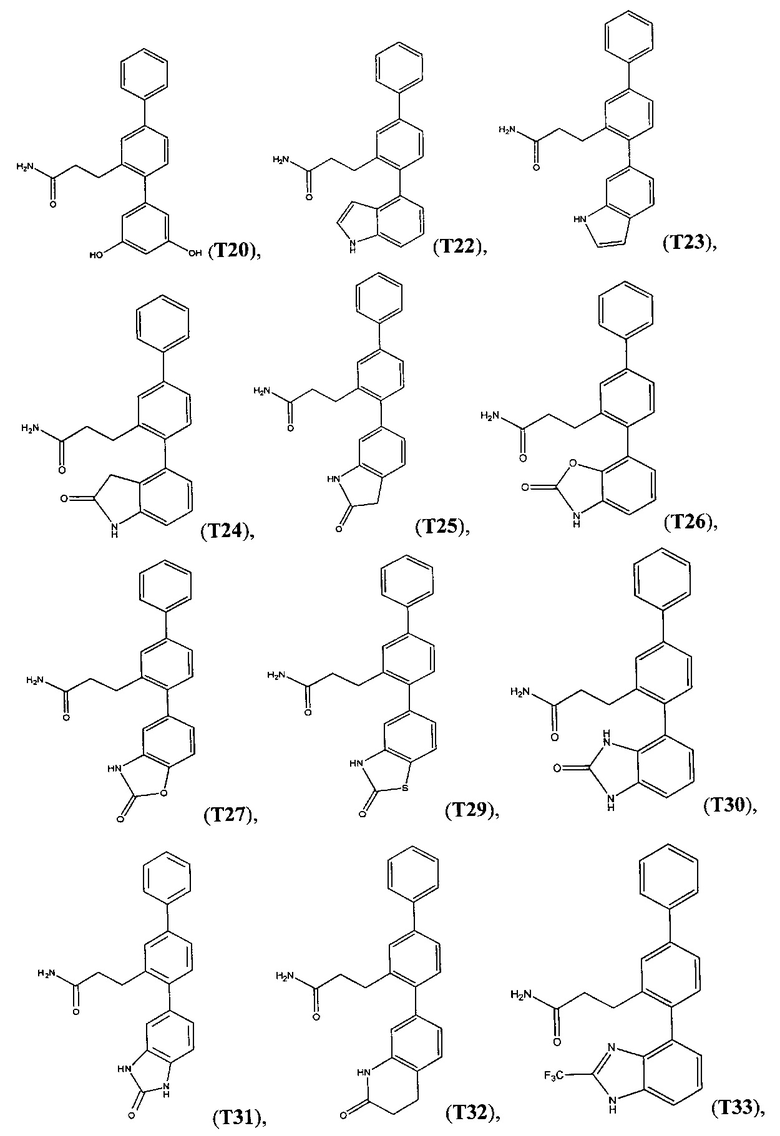
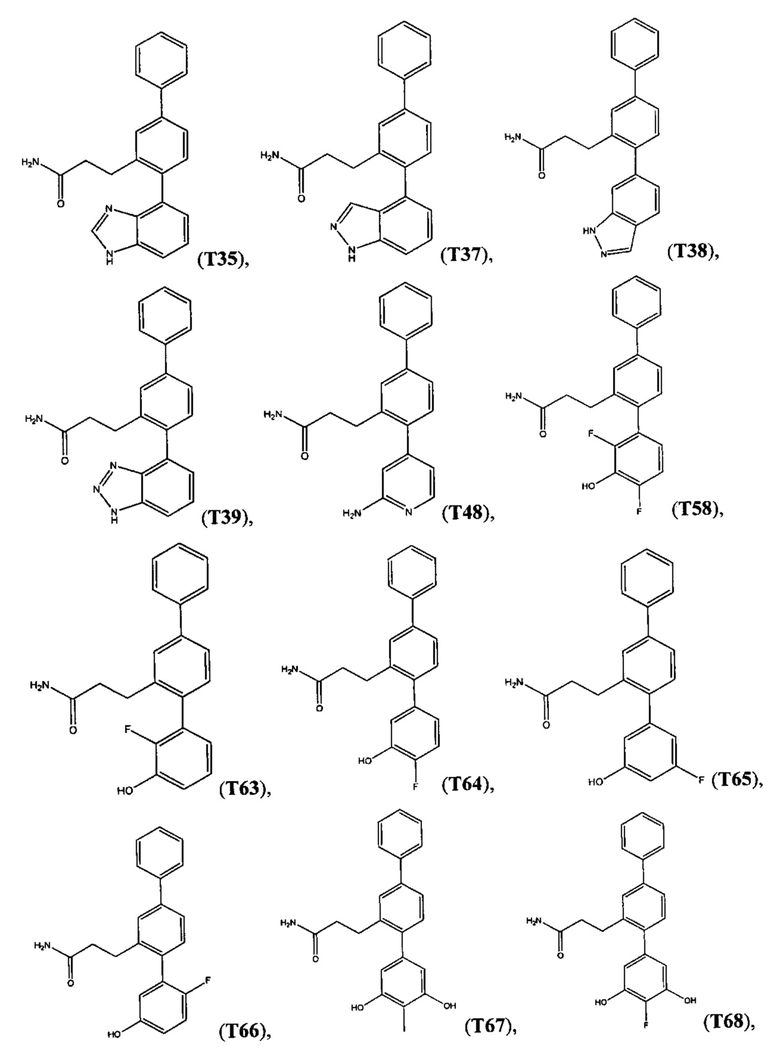
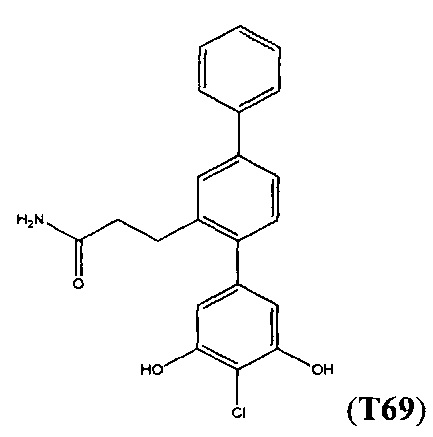 и .
и .
Применяемое в данном документе выражение "галоген" означает -F, -О, -Br или -I; выражение "гидрокси" означает -OH; выражение "амино" означает -NH2, и выражение "замещенный амино" включает -NHW, где W выбран из -CN, -SO2(X)aY и -CO(X)aY, а равняется 0 или 1, X выбран из -NH- и -O-, и Y выбран из -H, -CH3, -CH2CH3, -CH2OH и -CH2CH2OH.
Применяемые в данном документе аббревиатуры Me, Et, Ph, Ms представляют собой метил, этил, фенил и метансульфонил, соответственно. Более полный перечень аббревиатур, используемых специалистами в области органической химии в данной области техники, представлен в первом выпуске каждого тома Journal of Organic Chemistry; этот перечень обычно представлен в таблице под названием Стандартный перечень аббревиатур. Аббревиатуры, содержащиеся в упомянутом перечне, и все аббревиатуры, используемые специалистами в области органической химии в данной области техники, включены в данный документ с помощью ссылки.
Соединения по настоящему изобретению могут существовать в определенных геометрических или стереоизомерных формах. Настоящее изобретение предполагает все подобные соединения, включая цис- и транс-изомеры, (R)- и (S)-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры, их рацемические смеси и другие их смеси, как попадающие в объем настоящего изобретения. Все подобные изомеры, а также их смеси предназначены для включения в настоящее изобретение.
Если, например, необходим определенный энантиомер соединения по настоящему изобретению, его можно получить путем асимметричного синтеза или путем получения производного с хиральным вспомогательным веществом, где разделяют полученную диастереомерную смесь и отщепляют вспомогательную группу с получением необходимых чистых энантиомеров. В качестве альтернативы, диастереомерные соли могут быть образованы с помощью подходящей оптически активной кислоты или основания с последующим разделением образованных таким образом диастереомеров фракционной кристаллизацией или хроматографическими средствами, хорошо известными из уровня техники, и последующим восстановлением чистых энантиомеров.
В целом, соединения по настоящему изобретению могут быть получены с помощью способов, проиллюстрированных на общих схемах реакций, как, например, описано ниже, или путем их модификаций с использованием легко доступных исходных веществ, реагентов и общепринятых методик синтеза. В данных реакциях также можно воспользоваться вариантами, которые сами по себе известны, но не упомянуты в данном документе.
Настоящее изобретение также предусматривает фармацевтически приемлемые соли соединений. Выражение "фармацевтически приемлемая соль" включает соли присоединения как кислоты, так и основания, и относится к солям, которые сохраняют биологическую эффективность и свойства свободных оснований или кислот и которые не являются биологически или иным образом нежелательными. Фармацевтически приемлемые соли образуются с неорганическими или органическими кислотами или основаниям и могут быть получены in situ во время окончательного выделения и очистки соединений или путем отдельной реакции очищенного соединения в форме его свободного основания или кислоты с соответствующей органической или неорганической кислотой или основанием и выделения образованной таким образом соли.
Выражение "фиброз", применяемое в контексте настоящего изобретения, включает без ограничения фиброз миокарда и/или фиброз почки.
В дополнение к лечению развившегося фиброза соединения по настоящему изобретению могут использоваться профилактически у субъекта с риском развития фиброза. В качестве примера субъектов в категории риска развития фиброза выступают имеющие гипертензию, диабет, миокардит, ишемическую болезнь сердца, синдром Конна, феохромоцитому, генетическую предрасположенность и высокосолевой рацион, и/или получающие лекарственные средства, используемые при химиотерапии рака (такие как даунорубицин). Выражение "профилактический", применяемое в контексте настоящего изобретения, предназначено inter alia для охвата способов лечения, используемых для предупреждения или замедления развития фиброза у находящихся в группе риска. Субъекты, которым может быть предоставлено профилактическое лечение, уже могут иметь признаки ранних нарушений сердца на эхокардиографии.
Выражение "гипертензия", применяемое в контексте настоящего изобретения, указывает на значение артериального давления у взрослого выше приблизительно 139 мм рт.ст. для систолического и/или выше приблизительно 89 мм рт.ст. для диастолического.
Выражение "предгипертензия", применяемое в контексте настоящего изобретения, указывает на значение артериального давления у взрослого в диапазоне приблизительно 120-139 мм рт.ст. для систолического и/или приблизительно 80-89 мм рт.ст. для диастолического.
Настоящее изобретение также предусматривает фармацевтические композиции, которые включают соединения по настоящему изобретению в сочетании с приемлемыми фармацевтическими вспомогательными веществами. Выражение "фармацевтически приемлемое вспомогательное вещество", применяемое в контексте настоящего изобретения, означает любой фармацевтически приемлемый неактивный компонент композиции. Как хорошо известно из уровня техники, вспомогательные вещества включают разбавители, буферы, связующие вещества, смазывающие вещества, разрыхлители, красители, антиоксиданты/консерванты, регуляторы pH и т.д. Вспомогательные вещества выбирают, исходя из желаемых физических аспектов конечной формы: например, получение таблетки с желаемой твердостью и хрупкостью, являющейся быстро диспергируемой и легко проглатываемой и т.д. Требуемая скорость высвобождения активного вещества из композиции после ее приема также играет определенную роль в выборе вспомогательных веществ. Фармацевтические композиции могут включать в себя любой тип лекарственной формы, такой как таблетки, капсулы, порошки, жидкие составы, замедленного или длительного высвобождения, пластыри, средства для вдыхания через нос, назальные спреи и тому подобное. Физическая форма и содержание предусмотренных фармацевтических композиций представляют собой традиционные препараты, которые могут быть составлены специалистом в области фармацевтических составов и основаны на хорошо установленных принципах и композициях, описанных, например, в Remington: The Science and Practice of Pharmacy, 19th Edition, 1995; British Pharmacopoeia 2000, а также аналогичных текстах и руководствах по составам.
Например, если соединения или композиции подлежат введению перорально, их можно составить в виде таблеток, капсул, гранул, порошков или сиропов; или для парентерального введения их можно составить в виде инъекций (внутривенных, внутримышечных или подкожных), препаратов для капельного вливания или суппозиториев. Для применения через слизистую оболочку глаза их можно составить в виде глазных капель или глазных мазей. Эти составы можно получить с помощью обычных средств, и, если необходимо, активный ингредиент можно смешивать с любой традиционной добавкой, такой как вспомогательное вещество, связующее вещество, разрыхляющее средство, смазывающее вещество, модификатор лекарственных средств, солюбилизирующее средство, суспендирующее вспомогательное средство, эмульгирующее средство или покрывающее средство.
Когда соединение(-ия) по настоящему изобретению вводят в виде фармацевтических препаратов человеку и животным, их можно принимать per se или в виде фармацевтической композиции, содержащей, например, от 0,1 до 99,5% (более предпочтительно от 0,5 до 90%) активного ингредиента в комбинации с фармацевтически приемлемым носителем.
Дозировку соединения и частоту введения, которые следует использовать, также может легко определить практикующий врач для получения требуемого ответа.
Несмотря на то, что дозировка будет варьироваться в зависимости от симптомов, возраста и массы тела пациента, природы и тяжести расстройства, подлежащего лечению или профилактике, пути введения и формы лекарственного средства, в целом суточная доза от 0,0001 мг до 200 мг соединения по настоящему изобретению может быть подходящим эффективным количеством для взрослого пациента-человека, и ее можно вводить в виде одной дозы или в виде раздельных доз.
"Пациент" или "субъект", подлежащий лечению заявленным способом, может означать человека или субъекта, не относящегося к человеку.
"Эффективное количество" заявленного соединения в связи со способом лечения относится к количеству терапевтического средства в препарате, которое при применении в виде части требуемого режима дозирования обеспечивает пользу согласно клинически приемлемым стандартам лечения или профилактики определенного нарушения.
Настоящее изобретение далее будет описано более подробно со ссылкой на конкретные, но не ограничивающие примеры, описывающие конкретные композиции и способы применения. Однако следует понимать, что подробное описание конкретных процедур, композиций и способов включено исключительно для целей иллюстрации настоящего изобретения. В любом случае его не следует понимать в качестве ограничения широкого описания концепции изобретения, как изложено выше.
ПРИМЕРЫ
Пример 1 - Синтез 3-формилбифенил-4-ил-трифторметансульфоната
Синтетический путь, используемый для получения 3-формилбифенил-4-ил-трифторметансульфоната (14), показан на фигуре 1. Вкратце, использовали реакцию перекрестного сочетания Сузуки между 5-бром-2-гидроксибензальдегидом и фенилбороновой кислотой с получением 2-гидрокси-5-фенилбензальдегида (13), который затем вводили в реакцию с N-фенилтрифламидом с получением 3-формилбифенил-4-ил-трифторметасульфоната (14).
Получение 2-гидрокси-5-фенилбензальдегида (13)
5-Бромсалицилальдегид (2,49 г, 12,4 ммоль), фенилбороновую кислоту (1,51 г, 12,4 ммоль), палладия(II) ацетат (14 мг, 0,5 мол. %) и карбонат калия (5,14 г, 37,2 ммоль) перемешивали в дегазированной воде (75 мл) при температуре окружающей среды в течение 2 часов в атмосфере аргона. Реакцию контролировали при помощи TLC (1:1 дихлорметан/пентан). Добавляли воду (75 мл) и подкисляли реакционную смесь (pH 6) с помощью 10% HCl, затем экстрагировали этилацетатом (3х). Объединенные органические экстракты промывали солевым раствором, затем высушивали и концентрировали. Неочищенное вещество пропускали через короткую колонку силикагеля с элюированием смесью 1:1 дихлорметан/пентан, затем перекристаллизовывали из смеси этилацетат/пентан с получением 2-гидрокси-5-фенилбензальдегида (1,89 г, 77%) в виде темно-желтых кристаллов (при необходимости можно растирать в порошок с пентаном вместо перекристаллизации); т.пл. 100-101°C. 1Н ЯМР (400 МГц, CDCl3) δ 10,99 (s, 1Н); 9,97 (s, 1H); 7,78-7,73 (m, 2H); 7,56-7,52 (m, 2H); 7,47-7,41 (m, 2H); 7,37-7,32 (m, 1H); 7,09-7,04 (m, 1H). 13C ЯМР (100 МГц, CDCl3) δ 196,9, 161,2, 139,6, 136,0, 133,6, 132,1, 129,2, 127,6, 126,8, 121,0, 118,4. EIMS: масса/заряд 198 [M]+. HRMS рассчитанное для C13H10O2 198,0675, обнаруженное 198,0677.
Получение 3-формилбифенил-4-ил-трифторметансульфоната (14)
2-Гидрокси-5-фенилбензальдегид (13) (100 мг, 0,50 ммоль), N-фенилтрифлимид (180,0 мг, 0,51 ммоль) и карбонат калия (209 мг, 1,51 ммоль) перемешивали в сухом THF в герметично закрытой пробирке и нагревали при 120°C в течение 6 минут с помощью микроволнового излучения. Удаляли растворитель при пониженном давлении; добавляли воду и дихлорметан и разделяли слои. Водный слой дополнительно экстрагировали дихлорметаном (2х). Объединенные органические экстракты промывали солевым раствором (1х), затем высушивали и концентрировали. Очищали с помощью радиальной хроматографии с элюированием смесью 1:1 дихлорметан/пентан с получением 3-формилбифенил-4-ил-трифторметансульфоната (143 мг, 86%) в виде прозрачного бесцветного масла. 1Н ЯМР (200 МГц, CDCl3) δ 10,32 (s, 1Н); 8,17 (d, 1Н, J=2,4 Гц); 7,89 (dd, 1H, J=8,6, 2,5 Гц); 7,63-7,36 (m, 6Н). 13С ЯМР (125 МГц, CDCL3) δ 186,5, 149,1, 142,3, 138,0, 134,1, 129,2, 129,1, 128,8, 128,6, 127,2, 122,9, 118,7 (q, JCF-=320,9 Гц). 19F ЯМР(188 МГц, CDCl3) δ -73,2. EIMS: масса/заряд 330 [М]+. HRMS рассчитанное для C14H9F3O2S 330,0168, обнаруженное 330,0163.
Пример 2 - Синтез Т1, Т2, Т10 и Т18
Синтетический путь, используемый для получения T1, Т2, Т10 и Т18, показан на фигуре 2. Вкратце, проводили реакцию перекрестного сочетания 3-формилбифенил-4-ил-трифторметансульфоната (14) с 3-нитрофенилбороновой кислотой с получением нитротерфенила (17), который затем вводили в реакцию Хорнера-Уодсворта-Эммонса с диэтил(карбамоилметил)фосфонатом (18) с получением терфенилакриламида (19). С помощью гидрогенолиза соединения 19 одновременно восстанавливали олефиновые группы и нитрогруппы с получением 3-(3-амино-[1,1':4',1''-терфенил]-2'-ил)пропанамида (Т18), который затем использовали для получения 3-(3-(метилсульфонамидо)-[1,1':4',1''-терфенил]-2'-ил)пропанамида (Т1) посредством реакции с метансульфонилхлоридом, 3-(3-формамидо-[1,1':4',1''-терфенил]-2'-ил)пропанамида (Т2) посредством реакции с муравьиной кислотой и 3-(3-ацетамидо-[1,1':4',1''-терфенил]-2'-ил)пропанамида (Т10) посредством реакции с уксусным ангидридом.
Диэтил(карбамоилметил)фосфонат (18) получали в результате реакции Арбузова между 2-хлорацетамидом и триэтилфосфитом (полученным, как показано на фигуре 3).
Получение 3-нитро-[1,1':4',1''-терфенил]-2'-карбальдегида (17)
3-Формил-[1,1'-бифенил]-4-ил-трифторметансульфонат (14) (4,15 г, 12,60 ммоль), 3-нитрофенилбороновую кислоту (2,52 г, 15,10 ммоль), фосфат калия (4,01 г, 18,90 ммоль) и тетракис(трифенилфосфин)палладий(0) (0,33 г, 0,28 ммоль) в диоксане (50 мл) помещали в сосуд Шленка в атмосфере аргона. Добавляли дегазированный 1,4-диоксан (2 мл) и смесь продували аргоном. Реакционную смесь нагревали при 85°C до тех пор, пока не наблюдали полное превращение (контролируемое с помощью GCMS), обычно требующее время реакции в течение ночи. Неочищенное вещество очищали с помощью хроматографии (DCVC) с элюированием с применением градиента этилацетата в гептане (0-25% этилацетата) с получением 3-нитро-[1,1':4',1''-терфенил]-2-карбальдегида (17) в виде бледно-коричневатого твердого вещества (2,05 г, 67%) после восстановления не вступившего в реакцию трифлата (0,83 г); т.пл. 113,6-116,3°C (NB: Продукт был загрязнен ~25% 1Н ЯМР 3,3'-динитро-1,1'-бифенил). 1Н ЯМР (400 МГц, CDCl3) δ 10,02 (s, 1Н), 8,29 (m, 3Н), 7,92 (dd, 1H, J 8,0, 2,1 Гц), 7,72 (m, 1H), 7,66 (m, 3H), 7,50 (m, 3H), 7,42 (m, 1H). 13C ЯМР (100 МГц, DMSO-d6) δ 191,7, 147,8, 140,8, 140,5, 139,1, 138,4, 136,4, 133,8, 132,0, 131,9, 130,6, 129,9, 128,3, 127,0, 126,8, 124,2, 122,8. EIMS: масса/заряд обнаруженное: M+• 303,0880, для C19H13NO3 необходимо 303,0890. EIMS: масса/заряд 303 (М+•, 100%), 256 (52).
Получение (Е)-3-(3-нитро-[1,1':4',1''-терфенил]-2'-ил)акриламида (19)
3-нитро-[1,1':4',1''-терфенил]-2-карбальдегид (17) (2,35 г, 7,77 ммоль) и диэтил(карбамоилметил)фосфонат (18) (1,51 г, 7,75 ммоль) растворяли в сухом THF (100 мл) и медленно добавляли к интенсивно перемешиваемой суспензии порошкообразного гидроксида калия (0,86 г, 15,40 ммоль). После перемешивания в течение 1 часа при комнатной температуре вещество осаждали из реакционной смеси добавлением воды и диэтилового эфира с получением (E)-3-(3-нитро-[1,1':4',1''-терфенил]-2'-ил)акриламида (19) (1,8 г, 82%) в виде бледно-лимонного твердого вещества. Небольшую часть очищали с помощью хроматографии (DCVC) с элюированием с применением градиента этилацетата в DCM (0-20% этилацетата) для характеристики с получением (Е)-3-(3-нитро-[1,1':4',1''-терфенил]-2'-ил)акриламида (19) в виде бесцветного твердого вещества; т.пл. 206-210°C. 1Н ЯМР (400 МГц, DMSO-d6) δ 8,35-8,28 (m, 1Н), 8,18-8,15 (m, 1H), 8,02-7,98 (m, 1H), 7,85-7,76 (m, 5H), 7,56-7,41 (m, 4H), 7,49 (br s, 1H), 7,33 (d, 1H, J 15,7 Гц), 7,15 (br s, 1H), 6,78 (d, 1H, J 15,7 Гц). 13C ЯМР (100 МГц, DMSO-d6) δ 166,2, 147,8, 140,9, 140,6, 139,1, 138,3, 136,5, 136,2, 133,4, 131,1, 130,0, 129,0, 128,0, 127,8, 126,8, 125,0, 124,8, 123,8, 122,5. EIMS: масса/заряд обнаруженное: M+• 344,1153, для C21H16N2O3 необходимо 344,1155. EIMS: масса/заряд 344 (М+•, 37%), 326 (50), 252 (100).
Получение 3-(3-амино-[1,1':4',1''-терфенил]-2'-ил)пропанамида (Т18)
К раствору (E)-3-(3-нитро-[1,1':4',1''-терфенил]-2'-ил)акриламида (19) (1,70 г, 4,94 ммоль) в метаноле (50 мл) и этилацетат (25 мл) добавляли 10% палладий на углероде (50 вес. % воды). Реакционную смесь интенсивно перемешивали в автоклаве в атмосфере водорода при 140 фунтах на кв. дюйм в течение 2 часов. Реакционную смесь фильтровали через целит, хорошо промывая метанолом и этилацетатом. Фильтрат концентрировали, затем предварительно абсорбировали на целите и хроматографировали (DCVC) с элюированием с применением градиента метанола в DCM (0-3% метанола). Фракции, содержащие одно пятно на TLC, объединяли с получением 3-(3-амино-[1,1':4,1''-терфенил]-2'-ил)пропанамида (Т18) в виде бесцветного твердого вещества (0,92 г, 59%); т.пл. 157,3-157,9°C. 1Н ЯМР (400 МГц, DMSO-d6) δ 7,69 (d, 2Н, J 7,4 Гц), 7,59 (s, 1Н), 7,51-7,46 (m, 3Н), 7,37 (m, 1H), 7,23 (br s, 1H), 7,19 (d, 1H, J 7,9 Гц), 7,08 (m, 1H), 6,74 (br s, 1H), 6,57 (d, 1H, J 8,4 Гц), 6,52 (s, 1H), 6,46 (d, 1H, J 7,5 Гц), 5,13 (br s, 2H), 2,84 (m, 2H), 2,31 (m, 2H). 13С ЯМР (100 МГц, DMSO-d6) δ 173,5, 148,5, 141,5, 141,3, 140,1, 139,1, 128,8, 130,1, 128,9, 128,7, 127,3, 127,1, 126,6, 124,0, 116,5, 114,4, 112,6, 36,4, 28,3. EIMS: масса/заряд обнаруженное: M+• 316,1566, для C21H20N2O необходимо 316,1570. EIMS: масса/заряд 316 (М+•, 100%). ВЭЖХ чистота (40% ACN/H2O, 258 нм): 100,0%.
Получение 3-(3-(метилсульфонамидо)-[1,1':4',1''-терфенил]-2'-ил)пропанамида (Т1)
К суспензии 3-(3-амино-[1,1':4,1''-терфенил]-2'-ил)пропанамида (Т18) (0,50 г, 1,57 ммоль) в DCM (7 мл), охлажденной до -5°C, добавляли триэтиламин (0,33 мл, 2,36 ммоль), с последующим добавлением по каплям метансульфонилхлорида (0,21 г, 1,83 ммоль) при такой скорости, чтобы поддерживать температуру ниже 0°C (~20 минут). Реакционную смесь распределяли между 2М соляной кислотой и этилацетатом и слои разделяли. Органическую фазу еще раз промывали 2М соляной кислотой, насыщенным раствором бикарбоната и солевым раствором. Неочищенное вещество предварительно абсорбировали на целите и хроматографировали (DCVC) с элюированием с применением градиента метанола в DCM (0-3% метанола). Подобные фракции объединяли с получением 3-(3-(метилсульфонамидо)(1,1':4',1''-терфенил]-2'-ил)пропанамида (Т1) в виде бесцветных тонких игл (0,25 г, 41%); т.пл. 166,7-168,4°C. 1Н ЯМР (400 МГц, CDCl3) δ 8,12 (br s, 1Н), 7,58 (m, 2H), 7,53 (m, 1H), 7,47-7,31 (m, 5H), 7,27-7,24 (m, 2H), 7,19 (m, 1H), 7,12 (m, 1H), 5,87 (br s, 1H), 5,78 (br s, 1H), 2,99, (s, 3H), 2,94 (m, 2H), 2,43 (m, 2H). 13C ЯМР (100 МГц, DMSO-d6) δ 173,3, 141,8, 139,9 (два совпадающих сигнала), 139,5, 139,1, 138,4, 130,3, 129,3, 128,9, 127,5, 127,4, 126,7, 124,4, 124,3, 119,9, 118,2, 39,3, 36,2, 28,3. EIMS: масса/заряд обнаруженное: М+• 394,1341, для C22H22N2O3S необходимо 394,1346. EIMS: масса/заряд 394 (М+•, 12%), 376 (22), 256 (100). ВЭЖХ чистота (40% ACN/H2O, 256 нм): 99,84%.
Получение 3-(3-формамидо-[1,1':4',1''-терфенил]-2'-ил)пропанамида (Т2)
Раствор 3-(3-амино-[1,1':4,1''-терфенил]-2'-ил)пропанамида (Т18) (0,41 г, 1,30 ммоль) в муравьиной кислоте (5 мл) нагревали с обратным холодильником в течение 5 часов, затем концентрировали до сухого состояния. Неочищенное вещество предварительно абсорбировали на целите, затем хроматографировали (DCVC) с элюированием с применением градиента метанола в DCM (0-5% метанола). Подобные фракции объединяли с получением 3-(3-формамидо-[1,1':4,1''-терфенил]-2'-ил)пропанамида (Т2) в виде бесцветного твердого вещества (0,21 г, 47%); т.пл. 213°C. При этом он находился в виде смеси Е- и Z-амидных изомеров. 1Н ЯМР (400 МГц, DMSO-d6) δ 10,30 (s) и 10,22 (d, J 11,0 Гц; 1Н), 8,88 (d, J 11,0 Гц) и 8,31 (d, J 1,8 Гц; 1Н), 7,70 (m, 2Н), 7,63-7,19 (m, 10Н), 7,08 (m, 1Н), 6,76 (br s, 1Н), 2,83 (m, 2Н), 2,32 (m, 2Н). 13С ЯМР (50 МГц, DMSO-d6) δ 173,4, 162,7, 159,7, 142,0, 141,4, 140,2, 140,0, 139,9, 139,4, 139,3, 139,1, 138,3, 138,2, 130,2, 129,3, 128,9, 128,8, 127,5, 127,3, 126,7, 124,3, 124,2, 119,6, 117,8, 117,7, 116,0,36,2, 28,2 (ряд сигналов совпадал). EIMS: масса/заряд обнаруженное: М+• 344,1518, для C22H20N2O2 необходимо 344,1519. EIMS: масса/заряд 344 (М+•, 20%), 299 (34), 254 (100). ВЭЖХ чистота (50% ACN/H2O, 255 нм): 99,53%.
Получение 3-(3-ацетамидо-[1,1':4',1''-терфенил]-2'-ил)пропанамида (Т10)
Раствор 3-(3-амино-[1,1':4,1''-терфенил]-2'-ил)пропанамида (Т18) (0,42 г, 1,33 ммоль) и N,N-диметиламинопиридина (0,04 г, кат.) в уксусном ангидриде (7 мл) перемешивали при комнатной температуре в течение 20 часов. Реакционную смесь распределяли между водой и этилацетатом. Слои разделяли и промывали органический слой водой (2х) и солевым раствором и фильтровали через пробку из силикагеля 60, тщательно промывая этилацетатом. Фильтрат концентрировали до сухого состояния и перекристаллизовывали из метанола и 1,2-дихлорэтана с получением 3-(3-ацетамидо-[1,1':4,1''-терфенил]-2'-ил)пропанамида (T10) в виде бежевого твердого вещества (0,36 г, 75%); т.пл. 208-209°C. 1Н ЯМР (400 МГЦ, DMSO-d6) δ 10,03 (br s, 1Н), 7,70 (m, 2H), 7,62 (m, 2H), 7,58-7,47 (m, 4H), 7,40-7,35 (m, 2H), 7,23 (m, 2H), 7,02 (d, 1H, 7,7 Гц), 6,75 (br s, 1H), 2,83 (m, 2H), 2,31 (m, 2Н), 2,06 (s, 3Н). 13С ЯМР (100 МГц, DMSO-d6) δ 173,4, 168,4, 141,2, 140,4, 139,9, 139,3, 139,2, 139,1, 130,2, 128,9, 128,6, 127,4, 127,3, 126,7, 124,2, 123,6, 119,4, 117,5, 36,2, 28,2, 24,1. EIMS: масса/заряд обнаруженное: М+• 358,1666, для C23H22N2O2 необходимо 358,1676. EIMS: масса/заряд 358 (М+•, 8%), 299 (33), 254 (100). ВЭЖХ чистота (50% ACN/H2O, 255 нм): 99,53%.
Пример 3 - Синтез Т20
Синтетический путь, используемый для получения Т20, показан на фигуре 4. Вкратце, проводили реакцию перекрестного сочетания 3-формилбифенил-4-ил-трифторметансульфоната (14) с 3,5-диметоксифенилбороновой кислотой с получением диметокситерфенила (20), который затем вводили в реакцию Хорнера-Уодсворта-Эммонса с диэтил(карбамоилметил)фосфонатом (18) с получением терфенилакриламида (21). Гидрогенолиз соединения 21 приводил к образованию пропанамида (22), который затем деметилировали трибромидом с получением Т20.
Получение 3,5-диметокси-[1,1':4',1''-терфенил]-2'-карбальдегида (20)
К раствору 3,5-диметоксифенилбороновой кислоты (4,0 г, 22,0 ммоль),3-формилбифенил-4-ил-трифторметансульфоната (14) (6,6 г, 20,0 ммоль) и карбоната натрия (47,2 г, 40,0 ммоль) в дегазированной смеси диоксан/этанол/Н2О (5:1:1, 165 мл) добавляли тетракис(трифенилфосфин)палладий(0) (1,16 г, 1,0 ммоль). Реакционную смесь нагревали при 110°C в течение 2 часов в запаянной пробирке. Анализ при помощи TLC (1:2 DCM/PE) указывал на то, что трифлат был израсходован. Реакционную смесь концентрировали, затем переносили в воду и экстрагировали этилацетатом (3х). Объединенные органические экстракты промывали водой и солевым раствором, затем высушивали (MgSO4) и концентрировали. Неочищенное вещество фильтровали через короткую колонку силикагеля с элюированием смесью 1:1 DCM : PE с получением 3,5-диметокси-[1,1':4',1''-терфенил]-2'-карбальдегида (20) (6,1 г, 96%) в виде бледно-желтого твердого вещества. 1Н ЯМР (200 МГц, CDCl3) δ 10,09 (s, 1H), 8,26 (d, 1Н, J 1,8 Гц), 7,87 (dd, 1H, J 2,1, 8,0 Гц),7,68 (m, 2Н), 7,58-7,35 (m, 4Н),6,56 (s, совпадает, 3Н),3,84 (s, 6Н). 13С ЯМР (50 МГц, CDCl3) δ 192,4, 160,9, 144,8, 140,9, 139,6, 134,2, 132,0, 131,1, 129,1, 128,1, 127,2, 125,8, 108,6, 100,2, 55,6 (один сигнал не наблюдался). EIMS: масса/заряд обнаруженное: М+• 318,1255, для С21Н18О3 необходимо 318,1250. EIMS: масса/заряд 318 (М+•, 55%).
Получение (Е/Z)-3-(3,5-диметокси-[1,1':4',1''-терфенил]-2'-ил)акриламида (21)
3,5-Диметокси-[1,1':4',1''-терфенил]-2-карбальдегид (20) (6,1 г, 19,1 ммоль) и диэтил(карбамоилметил)фосфонат (18) (3,7 г, 19,1 ммоль) растворяли в сухом THF (180 мл) и медленно добавляли к интенсивно перемешиваемой суспензии порошкообразного КОН (2,1 г, 38,2 ммоль) в THF (70 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа в атмосфере аргона. Анализ при помощи TLC (1:2 DCM:PE) указывал на то, что был израсходован карбальдегид. THF удаляли при пониженном давлении и переносили остаток в воду и экстрагировали DCM (х3). Объединенные органические экстракты промывали солевым раствором (x1), затем высушивали (MgSO4) и концентрировали до ~50 мл. Раствор фильтровали через короткую колонку силикагеля с элюированием DCM с получением (E/Z)-3-(3,5-диметокси-[1,1':4',1''-терфенил]-2'-ил)акриламида (21) (2,5 г, 36%) в виде оранжевой пены. 1Н ЯМР (200 МГц, CDCl3) δ 7,85 (d, 1Н, J 1,7 Гц); 7,78-7,56 (m, 4H); 7,53-7,32 (m, 4H); 6,54-6,38 (m, 4H); 5,70 (brs, 2H); 3,81 (s, 6H). 13C ЯМР (50 МГц, CDCl3) δ 167,9, 160,8, 141,8, 141,7, 141,4, 140,9, 140,5, 133,4, 130,9, 129,1, 128,4, 127,9, 127,3, 125,7, 121,5, 108,3, 100,1, 55,7. EIMS: масса/заряд обнаруженное: М+• 359,1504, для C23H21O3N необходимо 359,1516. EIMS: масса/заряд 359 (М+•, 3%).
Получение 3-(3,5-диметокси-[1,1':4',1''-терфенил]-2'-ил)пропанамида (22)
(E/Z)-3-(3,5-Диметокси-[1,1':4',1''-терфенил]-2'-ил)акриламид (21) (2,5 г, 6,9 ммоль) и 10% палладий на углероде (50 вес. % В Н2О, 1,0 г) в метаноле (100 мл) перемешивали при комнатной температуре в атмосфере водорода при 50 фунтах на кв. дюйм в течение 2 часов. Реакционную смесь подвергали гравитационной фильтрации через бумагу GF, тщательно промывая метанолом, затем концентрировали. Остаток затем переносили в DCM и подвергали гравитационной фильтрации через бумагу GF, тщательно промывая DCM, затем концентрировали. Неочищенное вещество затем фильтровали через короткую колонку силикагеля, тщательно промывая DCM, затем элюируя необходимое соединение смесью 1:49 метанол:БСМ с получением 3-(3,5-диметокси-[1,1':4',1''-терфенил]-2'-ил)пропанамида (22) (2,2 г, 90%) в виде белого твердого вещества. 1Н ЯМР (200 МГц, CDCl3) δ 7,70-7,24 (m, 8Н); 6,50 (m, 3Н); 5,78 (br s, 1H); 5,34 (br s, 1H); 3,82 (s, 6H); 3,05 (m, 2H); 2,39 (m, 2H). 13C ЯМР (50 МГц, CDCl3) δ 174,8, 160,8, 143,3, 141,0, 140,8, 140,8, 138,6, 130,6, 129,0, 128,1, 127,6, 127,2, 125,1, 107,6, 99,2, 55,5, 37,2, 29,2. EIMS: масса/заряд обнаруженное: М+• 361,1672, для C23H23O3N необходимо 361,1672. EIMS: масса/заряд 361 (М+•, 100%).
Получение 3-(3,5-дигидрокси-[1,1':4',1''-терфенил]-2'-ил)пропанамида (Т20)
Раствор 3-(3,5-диметокси-[1,1':4',1''-терфенил]-2'-ил)пропанамида (22) (500 мг, 1,4 ммоль) растворяли в сухом DCM (5 мл) и охлаждали до -78°C в атмосфере аргона. Добавляли трибромид бора (2,9 мл, 2,9 ммоль, 1,0 М раствор в гексанах) и реакционную смесь нагревали до комнатной температуры в течение ночи. Раствор охлаждали (водяная баня со льдом) и медленно добавляли воду (5 мл) и метанол (2 мл). Слои разделяли и дополнительно экстрагировали водную фазу DCM (x2). Объединенные органические экстракты промывали 1,0 М тиосульфатом натрия (x1), водой (x1) и солевым раствором (x1), затем высушивали (MgSO4) и концентрировали. Очищали с помощью радиальной хроматографии с использованием градиентного элюирования (DCM→4:96 метанол:DCM→6:94 метанол:DCM→8:92 метанол:DCM) с получением 3-(3,5-дигидрокси-[1,1':4',1''-терфенил]-2'-ил)пропанамида (Т20) (122 мг, 26%) в виде белого твердого вещества; т.пл. 232-233°C. 1Н ЯМР (200 МГц, DMSO-d6) δ 9,32 (brs, 2Н); 7,72-7,64 (m, 2Н); 7,58 (d, 1Н, J 1,8 Гц); 7,53-7,33 (m, 4H); 7,24 (перекрывающийся, brs, 1H); 7,18 (перекрывающийся, d, 1Н, J 7,9 Гц); 6,75 (brs, 1Н); 6,23 (t, 1H, J 2,1); 6,15 (d, 2H, J 2,1 Гц); 2,84 (m, 2H); 2,31 (m, 2H). 13C ЯМР (100 МГц, DMSO-d6) δ 173,5, 158,1, 142,6, 140,9, 140,0, 139,0, 138,9, 129,9, 128,9, 127,3, 127,1, 126,6, 124,1, 107,1, 101,2, 36,3, 28,2. EIMS: масса/заряд обнаруженное: M+• 333,1344, для C21H19O3N необходимо 333,1359. EIMS: масса/заряд 333 (M+•, 94%). ВЭЖХ чистота (40% ACN/H2O, 264 нм): 95,97%.
Пример 4 - Синтез Т70
Синтетический путь, используемый для получения Т70, показан на фигуре 5. Вкратце, проводили реакцию перекрестного сочетания 3-формил-[1,1'-бифенил]-4-ил-трифторметансульфоната (14) с 3,5-динитрофенилпинаколовым эфиром бороновой кислоты (34) [полученным в результате реакции между 1-йодо-3,5-динитробензола и бис(пинаколато)диборана] с образованием 3,5-динитротерфенила (35). Последующая реакция Хорнера-Уодсворта-Эммонса с диэтил(карбамоилметил)фосфонатом (18) приводила к образованию 3,5-динитротерфенилакриламида (36). Соединение 36 затем гидрогенизировали с получением пропанамида (37), который вводили в реакцию с метансульфонилхлоридом с получением Т70.
Получение 2-(3,5-динитрофенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (34)
1-Йодо-3,5-динитробензол (5,00 г, 17,00 ммоль), биспинаколатодибор (4,75 г, 18,7 ммоль), ацетат калия (5,00 г, 51,00 ммоль) и аддукт дихлор[1,1'-бис(дифенилфосфино)ферроцен]палладий(II)-дихлорметан (0,35 г, 0,48 ммоль) в DMSO (80 мл) перемешивали при 70°C в течение 17 часов. Реакционную смесь охлаждали до температуры окружающей среды, разбавляли этилацетатом, затем промывали насыщенным раствором бикарбоната натрия и солевым раствором. Неочищенное вещество предварительно абсорбировали на целите, затем хроматографировали (DCVC) с элюированием с применением градиента этил ацетата в гептане (0-100% этилацетата). Подобные фракции объединяли с получением 2-(3,5-динитрофенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (34) в виде бледно-желтого твердого вещества (2,10 г, 40%); т.пл. 144,0-148,0°C. 1Н ЯМР (400 МГц, CDCl3) δ 9,08 (t, 1Н, J 2,2 Гц), 8,90 (d, 2Н, J 2,2 Гц), 1,37, (s, 12Н).
Получение 3,5-динитро-[1,1':4',1''-терфенил]-2'-карбальдегида (35)
Получали в соответствии со способом Р5 из 3-формил-[1,1'-бифенил]-4-ил-трифторметансульфоната (14) (1,77 г, 5,36 ммоль), 2-(3,5-динитрофенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (34) (1,81 г, 6,16 ммоль), тетракис(трифенилфосфин)палладия(0) (0,44 г, 0,38 ммоль) и водного раствора карбоната натрия (1 М) (11,0 мл, 11,0 ммоль) в толуоле (36 мл) и этаноле (7 мл). Твердое вещество отфильтровывали с поверхности раздела во время экстракции, которое, как оказалось, является требуемым продуктом (0,86 г, 46%). Экстракт этилацетата очищали с помощью хроматографии (DCVC) с элюированием с применением градиента дихлорметана в гептане (10-50% DCM) с получением дополнительного количества 3,5-динитро-[1,1':4',1''-терфенил]-2'-карбальдегида (35) в виде бледно-коричневатого твердого вещества (0,62 г, 33%) (общий выход: 79%); т.пл. 209-212°C. 1Н ЯМР (400 МГц, DMSO-d6) δ 10,04 (s, 1Н), 8,90 (s, 1Н), 8,74 (s, 2Н), 8,33 (s, 1Н), 8,14 (d, 1Н, J 8,0 Гц), 7,83 (d, 2Н, J 7,3 Гц), 7,72 (d, 1Н, J 8,0 Гц), 7,56 (m, 2Н), 7,47 (m, 1Н). 13С ЯМР (100 МГц, DMSO-d6) δ 192,0, 147,9, 141,2, 138,2, 138,1, 134,0, 132,3, 131,8, 130,0, 129,3, 128,5 (два совпадающих сигнала), 126,9, 117,7 (один сигнал не наблюдался). EIMS: масса/заряд обнаруженное: М+• 348,0731, для C19H12N2O5 необходимо 348,0741. EIMS: масса/заряд 348 (М+•, 100%).
Получение (Е)-3-(3,5-динитро-[1,1':4',1'-терфенил]-2'-ил)акриламида (36)
Получали в соответствии со способом, используемым для получения соединения 19, из 3,5-динитро-[1,1':4',1''-терфенил]-2'-карбальдегида (35) (1,75 г, 5,03 ммоль), диэтил(карбамоилметил)фосфоната (18) (1,09 г, 5,59 ммоль) и гидроксида натрия (0,50 г, 12,50 ммоль) в THF (70 мл). Неочищенное твердое вещество перекристаллизовывали из ацетона с получением (E)-3-(3,5-динитро-[1,1':4',1''-терфенил]-2'-ил)акриламида (36) в виде бледно-коричневатого твердого вещества (1,40 г, 72%); т.пл. 221-223°C. 1Н ЯМР (400 МГц, DMSO-d6) δ 8,90 (s, 1Н), 8,58 (s, 2H), 8,02 (s, 1H), 7,85 (d, 1H, J 8,0 Гц), 7,80 (d, 2H, J 7,6 Гц), 7,64 (d, 1H, J8,0 Гц), 7,56-7,43 (m, 4H), 7,32 (d, 1H, 3Jтран 15,7 Гц), 7,16 (br s, 1H), 6,76 (d, 1H, 3Jтранс 15,7 Гц). 13C ЯМР (100 МГц, DMSO-d6) δ 166,1, 148,0, 142,4, 141,3, 138,9, 136,2, 135,9, 133,8, 131,3, 129,7, 129,1, 128,2, 127,8, 126,9, 126,1, 125,3, 117,6. EIMS: масса/заряд обнаруженное: M+• 389,1000, для C21H15N3O5 необходимо 389,1006. EIMS: масса/заряд 389 (М+•, 42%), 252 (100).
Получение 3-(3,5-диамино-[1,1':4',1''-терфенил]-2'-ил)пропанамида (37)
Получали в соответствии со способом, используемым для получения Т18, из (Е)-3-(3,5-динитро-[1,1':4',1''-терфенил]-2'-ил)акриламида (36) (1,40 г, 3,60 ммоль) и 10% палладия на углероде (50 вес. % воды) (0,28 г) в метаноле (40 мл). Катализатор удаляли фильтрацией и фильтрат концентрировали до сухого состояния с получением 3-(3,5-диамино-[1,1':4',1''-терфенил]-2'-ил)пропанамида (37) в виде коричневатого твердого вещества (1,07 г, 90%); т.пл. 87,4-90,6°C. 1Н ЯМР (400 МГц, DMSO-d6) δ 7,68 (m, 2Н), 7,65 (m, 1H), 7,49-7,45 (m, 33Н), 7,36 (m, 1H), 7,22 (br s, 1H), 7,15 (d, 1H, J 7,9 Гц), 6,76 (br s, 1H), 5,83 (m, 1H), 5,75 (m, 2H), 4,79 (br s, 4H), 2,86 (m, 2H), 2,31 (m, 2H). 13C ЯМР (100 МГц, DMSO-d6) δ 173,7,148,9, 142,2, 141,9, 140,2, 139,0, 138,4, 129,9, 128,9, 127,2, 127,0, 126,6, 123,8, 104,0, 98,7, 36,6, 28,4. EIMS: масса/заряд обнаруженное: M+• 331,1678, для C21H21N3O необходимо 331,1679. EIMS: масса/заряд 331 (М+•, 67%), 287 (100), 273 (72).
Получение 3-(3,5-ди(метилсульфонамидо)-[1,1':4',1''-терфенил]-2'-ил)пропанамида (T70)
Получали в соответствии со способом, используемым для получения Т1, из 3-(3,5-диамино-[1,1':4',1''-терфенил]-2'-ил)пропанамида (37) (0,46 г, 1,38 ммоль), метансульфонилхлорида (2,56 мл, 3,30 ммоль) и триэтиламина (0,58 мл, 4,14 ммоль) в DCM (15 мл). Неочищенное вещество очищали с помощью хроматографии (DCVC) с элюированием с применением градиента метанола в DCM (0-5% метанола), и затем с помощью радиальной хроматографии с элюированием 3% метанолом в DCM с получением 3-(3,5-диметилсульфонамидо)-[1,1':4',1''-терфенил]пропанамида (Т70) в виде бежевого твердого вещества (0,15 г, 22%); т.пл. 227-230°C. 1Н ЯМР (400 МГц, DMSO-d6) δ 9,96 (s, 2Н), 7,73-7,66 (m, 2Н), 7,63 (d, 1H, J 1,9 Гц), 7,55 (dd, 1H, J 1,9, 7,9 Гц), 7,52-7,45 (m, 2H), 7,42-7,36 (m, 1H), 7,26 (d, 1H, J 7,9 Гц), 7,24 (br s, 1H), 7,19-7,15 (m, 1H), 6,91 (d, 2H, J 1,9 Гц), 6,77 (br s, 1H), 3,06 (s, 6H), 2,83 (t, 2H, J 8,0 Гц), 2,32 (t, 2H, J 8,0 Гц). 13C ЯМР (50 МГц, DMSO-d6) δ 173,3, 142,6, 139,8, 139,6, 139,5, 139,3, 139,1, 130,1, 128,9, 127,5 (два совпадающих сигнала), 126,7, 124,4, 114,8, 108,8, 39,3, 36,3, 28,3. EIMS: масса/заряд обнаруженное: М+• 487,1226, для C23H25N3O532S2 необходимо 487,1230. EIMS: масса/заряд 487 (М+•, 4%), 408 (75), 349 (100), 271 (78). ВЭЖХ чистота (40% ACN/H2O, 264 нм): 94,72%.
Пример 5 - Синтез Т48
Синтетический путь, используемый для получения Т48, показан на фигуре 6. Вкратце, проводили реакцию перекрестного сочетания 3-формил-[1,1'-бифенил]-4-ил-трифторметансульфонат (14) пиридилпинаколовым с эфиром бороновой кислоты (30-Ihle, N.С; Krause, А.Е. J. Org. Chem. 1996, 61, 4810) с получением терарила (31), который затем вводили в реакцию Хорнера-Уодсворта-Эммонса с диэтил(карбамоилметил)фосфонатом (18) с получением терарилакриламида (32). Гидрогенизация соединения 32 приводила к образованию пропанамида (33), из которого затем удаляли защитную группу с получением Т48.
Получение трет-бутил(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)карбамата (30)
2-Аминопиридин-4-пинаколовый эфир бороновой кислоты (2,0 г, 9,1 ммоль) перемешивали в виде суспензии в трет-бутаноле (30 мл) в атмосфере аргона. Медленно добавляли Вос-ангидрид (2,20 г, 10,0 ммоль) в трет-бутаноле (20 мл) и реакционную смесь перемешивали при 35°C в течение 18 часов. Анализ при помощи 1Н ЯМР показал, что было израсходовано исходное вещество, представляющее собой пинаколовый эфир. Реакционную смесь концентрировали при пониженном давлении и неочищенное вещество перемешивали в воде в течение 5 минут. Твердое вещество собирали фильтрацией и высушивали под вакуумом при 50°C с получением трет-бутил(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)карбамата (31) в виде белого твердого вещества (2,9 г, 98%); т.пл. 172-178,0°C. (Lit. 188-193°C). 1Н ЯМР (200 МГц, DMSO-d6) δ 9,75 (br s, 1H), 8,26 (dd, 1H, J 0,9,4,8 Гц), 8,08 (m, 1H), 7,18 (dd, 1H, J 0,7,4,8 Гц), 1,47 (s, 9H), 1,31 (s, 12H).
Получение трет-бутил(4-(3-формил-[1,1'-бифенил]-4-ил)пиридин-2-ил)карбамата (31)
К раствору трет-бутил(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)карбамата (30) (2,9 г, 8,9 ммоль), 3-формилбифенил-4-ил-трифторметансульфоната (14) (2,7 г, 8,1 ммоль) и карбоната натрия (1,7 г, 16,2 ммоль) в дегазированной смеси диоксан/этанол/H2O (5:1:1, 75 мл) добавляли тетраксис(трифенилфосфин)палладий(0) (467 мг, 0,40 ммоль). Реакционную смесь нагревали при 110°C в течение 2 часов в запаянной пробирке. Анализ при помощи 1Н ЯМР указывал на то, что был израсходован трифлат. Реакционную смесь концентрировали, затем переносили в DCM и выливали в воду. Слои разделяли и дополнительно экстрагировали водную фазу DCM (2х). Объединенные органические экстракты промывали водой (x1) и солевым раствором, затем высушивали и концентрировали до объема приблизительно 20-30 мл. Раствор фильтровали через короткую колонку силикагеля с элюированием DCM с получением трет-бутил(4-(3-формил-[1,1'-бифенил]-4-ил)пиридин-2-ил)карбамата (31) в виде желтого твердого вещества (1,5 г, 48%); т.пл. 168,8-171,5°C. 1Н ЯМР (200 МГц, DMSO-d6) δ 10,00 (перекрывающийся, s, 1Н), 9,98 (перекрывающийся, br s, 1H), 8,35 (dd, 1Н, J 0,7, 5,1 Гц), 8,20 (d, 1Н, J 1,9 Гц), 8,10 (dd, 1H, J 2,1,8,0 Гц), 7,88 (m, 1H), 7,79 (m, 2Н), 7,63 (d, 1Н, J 8,0 Гц), 7,59-7,40 (m, 3Н), 7,16 (dd, 1H, J 1,6, 5,1 Гц), 1,47 (s, 9Н). 13С ЯМР (50 МГц, DMSO-d6) δ 191,4, 152,8, 152,6, 147,8, 146,8, 141,2, 140,7, 138,4, 133,6, 132,0, 131,2, 129,2, 128,3, 126,8, 126,1, 119,4, 112,8, 79,7, 28,0. EIMS: масса/заряд обнаруженное: М+• 374,1611, для C23H22O3N2 необходимо 374,1625. EIMS: масса/заряд 374 (М+•, 7%), 57 (100).
Получение (Е)-трет-бутил(4-(3-(3-амино-3-оксопроп-1-ен-1-ил)-[1,1'-бифенил]-4-ил)пиридин-2-ил)карбамата (32)
Получали в соответствии со способом, используемым для получения соединения 19, из трет-бутил(4-(3-формил-[1,1'-бифенил]-4-ил)пиридин-2-ил)карбамата (31) (1,44 г, 3,85 ммоль), диэтил(карбамоилметил)фосфоната (18) (0,75 г, 3,85 ммоль) и гидроксида натрия (0,31 г, 7,70 ммоль) в THF (40 мл). (E)-Трет-бутил(4-(3-(3-амино-3-оксопроп-1-ен-1-ил)-[1,1'-бифенил]-4-ил)пиридин-2-ил)карбамат (3 2) осаждали из реакционной смеси при добавлении воды и диэтилового эфира в виде бесцветного твердого вещества (1,32 г, 83%); т.пл. 179,5-182,2°C. 1Н ЯМР (200 МГц, DMSO-d6) δ 9,95 (s, 1Н); 8,33 (m, 1H); 7,99 (d, 1H, J 1,6 Гц); 7,82-7,73 (m, 4H); 7,58-7,42 (m, 5H); 7,34 (d, 1H, J 15,8 Гц); 7,14 (br s, 1H); 6,99 (dd, 1H, J 1,5, 5,1 Гц); 6,77 (d, 1H, J 15,7 Гц); 1,46 (s, 9H). 13C ЯМР (50 МГц, DMSO-d6) δ 166,3, 152,7, 149,0, 147,7, 140,6, 139,2, 138,7, 136,4, 133,1, 130,4, 129,0, 128,0, 127,7, 126,8, 124,6, 124,6, 119,2, 112,5, 79,7, 28,0. EIMS: масса/заряд обнаруженное: M+• 415,1873, для C22H25O3N3 необходимо 415,1890. EIMS: масса/заряд 415 (М+•, 5%), 315 (58), 297 (64), 271 (100).
Получение (Е)-трет-бутил(4-(3-(3-амино-3-оксопропил)-[1,1'-бифенил]-4-ил)пиридин-2-ил)карбамата (33)
Получали в соответствии со способом, используемым для получения Т18, из (Е)-трет-бутил(4-(3-(3-амино-3-оксопроп-1-ен-1-ил)-[1,1'-бифенил]-4-ил)пиридин-2-ил)карбамата (32) (1,17 г, 2,80 ммоль) и 10% палладия на углероде (50 вес. % воды) (0,50 г)в метаноле (75 мл). Фильтрат концентрировали с получением трет-бутил(4-(3-(3-амино-3-оксопропил)-[1,1'-бифенил]-4-ил)пиридин-2-ил)карбамата (33) в виде бесцветного твердого вещества (1,05 г, 89%); т.пл. 161,5-164,5°C. 1Н ЯМР (400 МГц, DMSO-d6) δ 9,90 (s, 1Н), 8,32-8,29 (m, 1H), 7,78 (s, 1H), 7,73-7,69 (m, 2Н), 7,66 (s, 1H), 7,61-7,56 (m, 1Н), 7,53-7,47 (m, 2Н), 7,43-7,37 (m, 1H), 7,30-7,26 (m, 1H), 7,25 (br s, 1H), 7,09-7,05 (m, 1Н), 6,76 (br s, 1H), 2,88-2,81 (m, 2H), 2,36-2,29 (m, 2H), 1,47 (s, 9H). 13C ЯМР (50 МГц, DMSO-d6) δ 173,2, 152,8, 152,6, 150.3, 147,6, 140,1, 139,7, 138,9, 138,2, 129,8, 128,9, 127,6, 127,5, 126,7, 124,5, 118,8, 112.4, 79,6, 36,1, 28,04, 28,00. EIMS: масса/заряд обнаруженное: M+• 417,2028, для C25H27N3O3 необходимо 417,2047. EIMS: масса/заряд 417 (М+•, 5%), 317 (15), 284 (89), 258 (100).
Получение 3-(4-(2-аминопиридин-4-ил)-[1,1'-бифенил]-3-ил)пропанамида (Т48)
Смесь трет-бутил(4-(3-(3-амино-3-оксопропил)-[1,1'-бифенил]-4-ил)пиридин-2-ил)карбамата (33) (0,94 г, 2,26 ммоль) и TFA (7,0 мл) в DCM (10 мл) перемешивали при температуре окружающей среды в течение 3 часов. Реакционную смесь разделяли ледяной водой и этилацетатом, затем нейтрализовали гидроксидом натрия (~pH 6), затем подщелачивали до pH 10 1 М раствором карбоната натрия. Неочищенное вещество собирали фильтрацией, затем перекристаллизовывали из метанола с получением 3-(4-(2-аминопиридин-4-ил)-[1,1'-бифенил]-3-ил)пропанамида (Т48) в виде бесцветного твердого вещества (0,48 г, 67%); т.пл. 248-249°C. 1Н ЯМР (400 МГц, DMSO-d6) δ 7,95 (d, 1H, J 5,2 Гц), 7,73-7,66 (m, 2H), 7,62 (s, 1H), 7,57-7,52 (m, 1H), 7,52-7,45 (m, 2H), 7,42-7,35 (m, 1H), 7,27 (br s, 1H), 7,24-7,20 (m, 1H), 6,78 (br s, 1H), 6,48 (d, 1H, J 5,2 Гц), 6,39 (s, 1H), 5,99 (s, 2H), 2,84 (t, 2H, J 7,9 Гц), 2,33 (t, 2H, J 7,9 Гц). 13C ЯМР (100 МГц, DMSO-d6) δ 173,3, 159,9, 149,4, 147,7, 139,8, 139,7, 139,0, 138,9, 129,6, 128,9, 127,5, 127,4, 126,7, 124,4, 112,7, 107,8, 36,2, 28,1. EIMS: масса/заряд обнаруженное: М+• 317,1516, для C20H19N3O необходимо 317,1523. EIMS: масса/заряд 317 (М+•, 12%), 273 (53), 258 (100). ВЭЖХ чистота (35% ACN / 0,1% TFA, 291 нм): 98,76%.
Пример 6 - Синтез Т3, T11, Т12 и Т15 из Т18
Раствор 3-(3-амино-1,1':4',1''-терфенил-2'-ил)пропанамида (Т18, полученный в примере 2) (1 экв.) в дихлорметане (12,5 мл/ммоль) добавляли к раствору трифосгена (0,3 экв.) в дихлорметане (6,25 мл/ммоль). Добавляли триэтиламин (0,3 мл/ммоль) и смесь перемешивали при комнатной температуре в атмосфере азота в течение 30 минут. Добавляли амин или спирт (2-5 экв.) и смесь перемешивали при комнатной температуре в атмосфере азота. Беспримесную реакционную смесь очищали с помощью флэш-хроматографии (метанол/дихлорметан). Продукт суспендировали в смеси 1:1 дихлорметан/гексаны и выделяли фильтрацией.
С помощью этой процедуры получали следующие соединения.
3-{3-[(Метилкарбамоил)амино]-1,1':4',1'-терфенил-2'-ил}пропанамид (Т3)
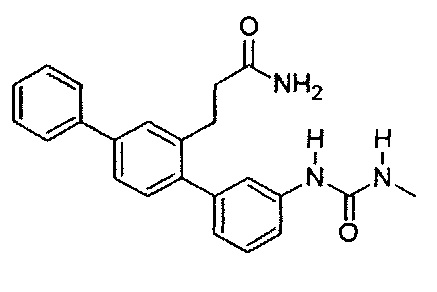
Белый порошок (82 мг, 27%). 1H ЯМР (400 МГц, DMSO-d6) 8,59 (s, 1Н), 7,70 (d, J=7,2 Гц, 2H), 7,61 (d, J=1,6 Гц, 1H), 7,43-7,56 (m, 4H), 7,26-7,42 (m, 3H), 7,23 (d, J=7,8 Гц, 2H), 6,87 (d, J=7,2 Гц, 1H), 6,72 (br. s., 1H), 6,06 (br. q, J=4,5 Гц, 1H), 2,79-2,87 (m, 2H), 2,64 (d, J=4,7 Гц, 3H), 2,27-2,35 (m, 2H); ЖХ-МС [M+H]+=374,2; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 100% при 220 нм.
Метил[2'-(3-амино-3-оксопропил)-1,1':4',1''-терфенил-3-ил]карбамат (T11)
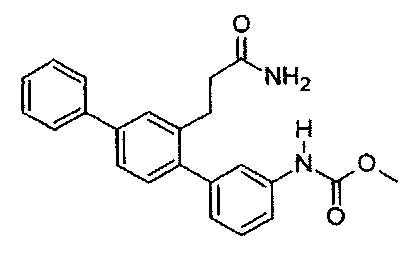
Белый порошок (131 мг, 55%). 1Н ЯМР (400 МГц, DMSO-d6) 9,74 (s, 1H), 7,70 (d, J=7,4 Гц, 2H), 7,62 (d, J=1,4 Гц, 1H), 7,42-7,58 (m, 5H), 7,32-7,41 (m, 2H), 7,23 (s, 2H), 6,99 (d, J=7,4 Гц, 1H), 6,73 (br. s., 1H), 3,68 (s, 3H), 2,78-2,87 (m, 2H), 2,26-2,35 (m, 2H); ЖХ-МС [M+H]+=375,3; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 99,4% при 220 нм.
2-Гидроксиэтил[2'-(3-амино-3-оксопропил)-1,1':4',1''-терфенил-2'-ил]карбамат (T12)
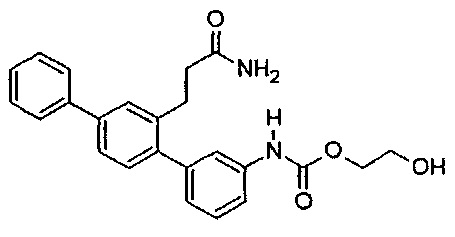
Белый порошок (129 мг, 50%). 1H ЯМР (400 МГц, DMSO-d6) 9,79 (s, 1H), 7,70 (d, J=7,2 Гц, 2H), 7,62 (d, J=1,6 Гц, 1Н), 7,42-7,58 (m, 5H), 7,31-7,41 (m, 2H), 7,15-7,27 (m, 2H), 6,98 (d, J=7,6 Гц, 1H), 6,73 (br. s., 1H), 4,81 (t, J=5,3 Гц, 1H), 4,11 (t, J=5,1 Гц, 2H), 3,63 (q, J=5,3 Гц, 2H), 2,76-2,88 (m, 2H), 2,25-2,37 (m, 2H); ЖХ-МС [M+H]+=405,1; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 99,4% при 220 нм.
3-[3-(Карбамоиламино)-1,1':4',1''-терфенил-2'-ил]пропанамид (Т15)
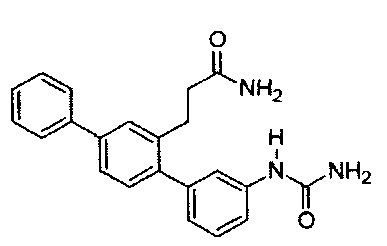
Белый порошок (74 мг, 32%). 1H ЯМР (400 МГц, DMSO-d6) 8,62 (s, 1Н), 7,70 (d, J=7,4 Гц, 2H), 7,61 (d, J=1,6 Гц, 1Н), 7,43-7,56 (m, 4H), 7,26-7,42 (m, 3Н), 7,18-7.26 (m, 2H), 6,88 (d, J=7,0 Гц, 1H), 6,72 (br. s., 1H), 5,87 (s, 2H), 2,78-2,87 (m, 2H), 2.27-2,36 (m, 2H); ЖХ-МС [M+H]+=360,3; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 97,2% при 220 нм.
Пример 7 - Синтез Т4 из Т18
Этилизоцианат (50 мкл, 0,63 ммоль) добавляли к раствору 3-(3-амино-1,1':4',1'-терфенил-2'-ил)пропанамида (155 мг, 0,49 ммоль) (Т18, полученный в примере 2) в дихлорметане (10 мл). Смесь перемешивали при комнатной температуре в атмосфере азота в течение 3 дней. Реакционную смесь выпаривали до сухого состояния. Остаток растворяли в смеси дихлорметана (10 мл) и метанола (2 мл), адсорбировали на силикагель 60 и очищали с помощью флэш-хроматографии (метанол/дихлорметан). Продукт суспендировали в смеси 1:1 дихлорметан/гексаны и выделяли фильтрацией с получением 3-{3-[(этилкарбамоил)амино]-1,1':4',1'-терфенил-2'-ил}пропанамида (Т4):
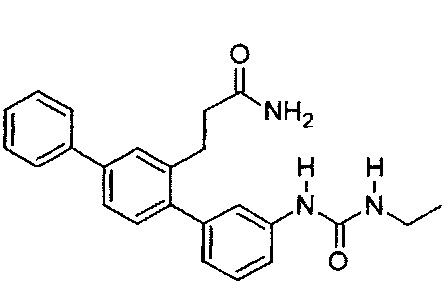
Белый порошок (115 мг, 60%). 1H ЯМР (400 МГц, DMSO-d6) 8,50 (s, 1H), 7,70 (d, J=7,4 Гц, 2H), 7,61 (d, J=1,6 Гц, 1H), 7,42-7,57 (m, 4H), 7,25-7,42 (m, 3H), 7,18-7,26 (m, 2H), 6,87 (d, J=7,2 Гц, 1H), 6,72 (br. s., 1H), 6,14 (t, J=5,5 Гц, 1H), 3,03-3,18 (m, 2H), 2,77-2,90 (m, 2H), 2,24-2,38 (m, 2H), 1,05 (t, J=7,1 Гц, 3Н); ЖХ-МС [M+H]+=388,3; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 98,5% при 220 нм.
Пример 8 - Синтез Т5 и Т6 из Т18
3-(3-Амино-1,1':4',1''-терфенил-2'-ил)пропанамид (Т18, полученный в примере 2) (1 экв.) и триэтиламин (1,3-2,0 экв.) растворяли в дихлорметане (18 мл/ммоль). По каплям добавляли раствор алкилсульфамоилхлорида (1,3-2,0 экв.) в дихлорметане (4 мл/ммоль). Смесь перемешивали при комнатной температуре в атмосфере азота в течение 1 часа. Беспримесную реакционную смесь очищали с помощью флэш-хроматографии (метанол/дихлорметан). Продукт суспендировали в смеси 1:1 дихлорметан/гексаны и выделяли фильтрацией.
С помощью этой процедуры получали следующие соединения.
3-{3-[(Метилсульфамоил)амино]-1,1':4',1''-терфенил-2'-ил}пропанамид (Т5)
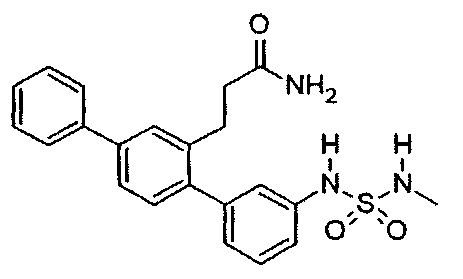
Белый порошок (60 мг, 30%). 1Н ЯМР (400 МГц, DMSO-d6) 9,77 (br. s., 1Н), 7,70 (d, J=7,2 Гц, 2H), 7,62 (d, J=1,6 Гц, 1H), 7,54 (dd, J=7,9, 1,7 Гц, 1H), 7,49 (t, J=7,6 Гц, 2H), 7,31-7,42 (m, 3H), 7,22-7,29 (m, 2H), 7,13-7,21 (m, 2H), 6,99 (d, J=7,6 Гц, 1H), 6,78 (br. s., 1H), 2,75-2,88 (m, 2H), 2,48 (неясный за счет DMSO-d6), 2,27-2,36 (m, 2Н); ЖХ-МС [М+Н]+=410,2; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 97,4% при 220 нм.
3-{3-[(Этилульфамоил)амино]-1,1':4',1''-терфенил-2'-ил}пропанамид (Т6)
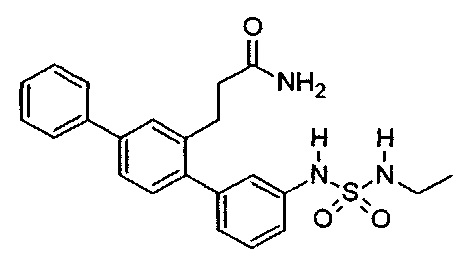
Белый порошок (69 мг, 34%). 1H ЯМР (400 МГц, DMSO-d6) 9,72 (br. s., 1H), 7,70 (d, J=7,2 Гц, 2H), 7,62 (d, J=1,6 Гц, 1H), 7,43-7,58 (m, 4H), 7,31-7,42 (m, 2H), 7,20-7,29 (m, 2H), 7,11-7,20 (m, 2H), 6,97 (d, J=7,6 Гц, 1H), 6,77 (br. s., 1H), 2,76-2,97 (m, 4H), 2,26-2,37 (m, 2H), 0,98 (t, J=7,2 Гц, 3Н); ЖХ-МС [M+H]+=424,3; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 99,6% при 220 нм.
Пример 9 - Синтез Т16 из Т18
3-(3-Амино-1,1':4',1''-терфенил-2'-ил)пропанамид (Т18, полученный в примере 2) (181 мг, 0,57 ммоль) растворяли в метаноле (3,8 мл) при легком нагревании. Добавляли ацетат калия (170 мг, 1,73 ммоль) и смесь охлаждали на водяной бане со льдом. По каплям добавляли раствор цианогена бромида (61 мг, 0,58 ммоль) в метаноле (1,1 мл). Смесь перемешивали на водяной бане со льдом в атмосфере азота в течение 1 часа, затем при комнатной температуре в атмосфере азота в течение ночи. Реакционную смесь выпаривали до сухого состояния. Остаток растворяли в 10% смеси метанол/дихлорметан (60 мл). Органическую фазу промывали водой (3×20 мл) и солевым раствором (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат выпаривали до сухого состояния. Остаток очищали с помощью флэш-хроматографии (метанол/дихлорметан). Продукт растворяли в этилацетате (20 мл) и органическую фазу промывали соляной кислотой (1 М, 3×20 мл) и солевым раствором (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат выпаривали до сухого состояния. Остаток суспендировали в смеси 1:1 дихлорметан/гексаны и выделяли фильтрацией. Полученный продукт очищали с помощью флэш-хроматографии (метанол/дихлорметан). Продукт суспендировали в смеси 1:1 дихлорметан/гексаны и выделяли фильтрацией с получением 3-[3-(цианоамино)-1,1':4',1''-терфенил-2'-ил]пропанамида (Т16):
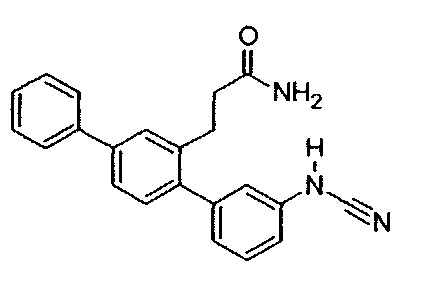
Белый порошок (68 мг, 35%). 1Н ЯМР (400 МГц, DMSO-d6) 10,30 (br. s., 1Н), 7,70 (d, J=7,2 Гц, 2H), 7,63 (d, J=1,6 Гц, 1H), 7,55 (dd, J=7,8, 1,8 Гц, 1H), 7,35-7,52 (m, 4H), 7,20-7,30 (m, 2H), 7,04 (d, J=7,6 Гц, 1H), 6,99 (dd, J=8,0, 1,8 Гц, 1H), 6,88 (s, 1H), 6,75 (br. s., 1H), 2,82 (t, J=7,8 Гц, 2H), 2,25-2,35 (m, 2H); ЖХ-МС [M+H]+=342,3; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 97,6% при 220 нм.
Пример 10 - Синтез 3-(3-амино-3-оксопропил)бифенил-4-ил-трифторметансульфоната
Синтез 3-(3-амино-3-оксопропил)бифенил-4-ил-трифторметансульфоната (7) показан на фигуре 7.
Получение 4-гидроксибифенил-3-карбальдегида (2)
5-Бромсалицилальдегид (1) (50,00 г, 0,249 моль), K2CO3 (103,13 г, 0,746 моль), фенилбороновую кислоту (30,33 г, 0,249 ммоль) и Pd(OAc)2 (0,28 г, 1,2 ммоль) добавляли в перемешиваемую магнитной мешалкой 2-л круглодонную колбу, содержащую свежедегазированную H2O (1,5 л, дегазирование продувкой N2 (4×2,5-л баллона)). Реакционную смесь перемешивали в атмосфере N2 в течение ночи и анализировали при помощи TLC (2 наблюдали, но 1 все еще присутствовал). Реакционную смесь перемешивали еще в течение 24 часов перед осторожным выливанием в HCl (водн., 0,2 М, 3 л) в течение нескольких часов, поддерживая pH смеси (~pH 2) добавлением небольших количеств HCl (водн., 33%). Смесь затем перемешивали в течение 1 часа с EtOAc (500 мл) и фильтровали через целит в отдельную воронку. Органический слой собирали и водный слой экстрагировали EtOAc (500 мл, промывая через фильтрат из целита) и два органических слоя объединяли, высушивали над MgSO4 и концентрировали с получением желтого твердого остатка (64 г). Остаток переносили в горячий EtOH (200 мл) и медленно добавляли H2O (200 мл) при интенсивном перемешивании и охлаждали до комнатной температуры в течение 48 часов. Полученный осадок собирали вакуумной фильтрацией и промывали смесью H2O/EtOH (1:1, 200 мл) и высушивали на воздухе с получением неочищенного бифенила 2 (41,97 г, содержащие 25 мол. % 1 в качестве примеси) в виде бледно-желтого твердого вещества. Неочищенный бифенил 2 (39,8 г, содержащий приблизительно 0,050 моль 1), фенилбороновую кислоту (6,02 г, 0,050 моль) и K2CO3 (40,76 г, 0,295 моль) добавляли в H2O (1,0 л) в 2-л круглодонную колбу с перемешиванием магнитной мешалкой. Реакционную смесь продували N2 (2×2,5-л баллона, в течение 15 минут) перед добавлением Pd(OAc)2 (223 мг, 1,0 ммоль) и медленным нагреванием до температуры флегмы в течение 3 часов в атмосфере N2. Добавляли дополнительное количество фенилбороновой кислоты (1,2 г, 9,84 ммоль) и продолжали перемешивание с обратным холодильником в течение 4 часов, затем охлаждали до комнатной температуры и отстаивали в течение выходных дней. Смесь выливали в HCl (водн., 3,3 М, 1,5 л) в течение 1 минуты и тщательно перемешивали в течение 10 минут перед сбором твердого вещества вакуумной фильтрацией и отсасыванием с высушиванием в течение 30 минут. Твердое вещество переносили в вакуумный эксикатор и высушивали в течение ночи с получением 39,7 г смеси 12:1 бифенила 2 (масс. экв. 36,6 г, 74%) и 1 (экв. 8 мол. % примеси). 1H ЯМР (400 МГц, CDCl3) 7,08 (d, J=8,61 Гц, 1Н), 7,32-7,39 (m, 1Н), 7,45 (t, J=7,43 Гц, 2Н), 7,55 (d, J=7,43 Гц, 2Н), 7,72-7,80 (m, 2Н), 9,93-10,00 (m, 1 Н), 11,01 (s, 1Н).
Получение 4-(бензилокси)бифенил-3-карбальдегида (3)
Перемешиваемую магнитной мешалкой смесь фенола 2 (38,10 г, 0,192 ммоль), K2CO3 (33,78 г, 0,250 моль) и бензилбромида (29,7 мл, 0,250 моль) в CH3CN (370 мл) в 500 мл круглодонной колбе медленно нагревали до 70°C в течение 3 часов и анализировали при помощи TLC (силикагель, смесь 10% EtOAc/гексан, визуализация при помощи УФ-лучей). TLC показала, что реакция протекала, однако некоторое количество фенола 2 оставалось. Реакционную смесь нагревали до температуры флегмы в течение 2 часов и затем анализировали при помощи TLC (реакция завершена, фенол 2 не наблюдался). Реакционную смесь охлаждали до комнатной температуры и переносили в 1-л коническую колбу и осторожно подкисляли HCl (водн., 2 М, 200 мл, наблюдалось некоторое выделение пузырьков газа, продолжавшееся до pH<2). Добавляли воду (200 мл) и экстрагировали EtOAc (3×500 мл). Экстракты высушивали над MgSO4 и концентрировали с получением светло-коричневого твердого вещества. Твердое вещество суспендировали в гексане (150 мл) и интенсивно перемешивали в течение 10 минут перед сбором продукта вакуумной фильтрацией и промыванием гексаном (2×60 мл) с получением соединения 3 в виде светло-коричневого порошка (44,50 г, 80%). 1Н ЯМР (400 МГц, CDCl3) 5,25 (s, 2 Н), 7,13 (d, J=9,00 Гц, 1Н), 7,29-7,39 (m, 2Н), 7,39-7,49 (m, 6Н), 7,57 (d, 7=7,43 Гц, 2Н), 7,77 (dd, J=8,61, 2,35 Гц, 1Н), 8,10 (d, J=2,35 Гц, 1Н), 10,60 (s, 1Н).
Получение (2Е)-3-[4-(бензилокси)бифенил-3-ил]проп-2-еновой кислоты (4)
Пиперидин (2,2 мл, 0,022 моль) добавляли к перемешиваемой магнитной мешалкой смеси альдегида (3) (44,5 г, 0,154 моль) и малоновой кислоты (19,25 г, 0,185 моль) в пиридине (250 мл) и медленно нагревали до слабого кипения в течение 5 часов. Выделение пузырьков газа отмечали, когда температура реакционной смеси приближалась к 90°C. TLC реакционной смеси (силикагель, смесь 10% EtOAc/гексан, визуализация с помощью УФ-лучей) показала лишь бледное пятно, соответствующее исходному альдегиду 3, и интенсивное пятно флуоресцирующего вещества на исходном уровне, соответствующее продукту (4). Реакционную смесь охлаждали до комнатной температуры и концентрировали на роторном вакуумном испарителе (60°C). Добавляли EtOAc (200 мл) и HCl (водн., 2 М, 200 мл) с получением густой жидкой массы из белой кашицы. Твердое вещество (соединение 4) собирали вакуумной фильтрацией и двухфазный фильтрат переносили в отдельную воронку. Органическую фазу собирали, промывали HCl (водн., 2 М, 1×100 мл), H2O (2×200 мл) и солевым раствором (1×75 мл), высушивали над MgSO4 и концентрировали с образованием дополнительного количества соединения 4 в виде бледно-желто-коричневого твердого вещества. Две собранные порции соединения 4 объединяли и высушивали в вакуумном эксикаторе с получением соединения 4 (48,5 г, 95%). 1Н ЯМР (400 МГц, CDCl3) 5,23 (s, 2Н), 6,64 (d, J=16,04 Гц, 1Н), 7,03 (d, J=8,61 Гц, 1Н), 7,34 (d, J=5,48 Гц, 2Н), 7,38-7,49 (m, 6Н), 7,55 (d, J=7,43 Гц, 3Н), 7,68-7,85 (m, 1Н), 8,22 (d, J=16,04 Гц, 1Н).
Получение (2Е)-3-[4-(бензилокси)бифенил-3-ил]проп-2-енамида (5)
Оксалилхлорид (25 мл, 0,29 моль) медленно добавляли с помощью капельной воронки в течение 30 минут к перемешиваемой магнитной мешалкой смеси карбоновой кислоты 4 (48,2 г, 0,146 моль) и DMF (0,8 мл) в CH2Cl2 (500 мл) в 1-л 3-горлую кругло донную колбу, оснащенную капельной воронкой, пробкой и масляным барботером. Температуру реакционной смеси поддерживали путем помещения сосуда на водяную баню во время добавления. При добавлении приблизительно 2/3 оксалилхлорида реакционная смесь становилась гомогенной с исчезновением суспендированного твердого вещества. Реакционную смесь перемешивали в течение еще 1 часа перед концентрированием реакционной смеси на роторном испарителе (60°C) с получением промежуточного хлорида кислоты в виде желтого твердого вещества. Промежуточной хлорид кислоты желтого цвета суспендировали в перемешиваемом магнитной мешалкой растворе 1,4-диоксана (200 мл) и в течение 15 минут добавляли раствор NH3 (31 мл, 28% в H2O, 0,438 моль) в 1,4-диоксане (200 мл). Температуру реакционной смеси поддерживали путем помещения сосуда на водяную баню во время добавления. Получали густую жидкую массу. Жидкую массу перемешивали при комнатной температуре еще в течение 30 минут перед выливанием смеси в 1-л коническую колбу и последующим добавлением H2O с получением конечного объема 1 л. Жидкую массу перемешивали в течение 5 минут и твердое вещество собирали вакуумной фильтрацией, промывая твердое вещество H2O (2×300 мл). Твердое вещество высушивали в вакуумном эксикаторе в течение ночи и затем на роторном испарителе (60°C, приблизительно 1-5 мм рт.ст.) с получением первой порции соединения 5 (41,6 г, 86%) в виде грязно-белого порошка. Водные фильтраты концентрировали до сухого состояния, добавляли H2O (200 мл) и твердое вещество собирали вакуумной фильтрацией с получением второй порции соединения 5 (8,12 г) в виде грязно-белого порошка. 1Н ЯМР (400 МГц, CDCl3) 5,23 (s, 2Н), 6,61-6,67 (m, 1H), 7,04 (d, J=8,61 Гц, 1H), 7,31-7,38 (m, 3Н), 7,44 (m, 7Н), 7,55 (m, 2Н), 7,78 (d, J=1,57 Гц, 1Н), 8,22 (d, J=16,04 Гц, 1Н).
Получение 3-(4-гидроксибифенил-3-ил)пропанамида (6)
Соединение 5 (41,55 г, 0,126 моль) суспендировали в перемешиваемой магнитной мешалкой смеси EtOAc (1 л) и NEt3 (1,5 мл). N2 (3×1-л баллона) барботировали через смесь перед добавлением Pd/C (10% вес/вес, 4,15 г) и помещением колбы под вакуум непосредственно перед обратным заполнением атмосферы Н2 из баллона. Баллон перезаряжали свежим H2 и открывали реакционную смесь и перемешивали в течение 6 часов, перезаряжая баллон H2 приблизительно каждые 1-2 часа (3 раза), и затем перемешивали в течение ночи. Баллон повторно перезаряжали H2 и реакционную смесь медленно охлаждали до температуры флегмы в течение 3 часов и затем охлаждали до комнатной температуры и перемешивали в атмосфере Н2 в течение 4 дней. Баллон повторно перезаряжали Н2 и реакционную смесь медленно охлаждали до температуры флегмы в течение 3 часов перед охлаждением и последующим барботированием N2 (2×1-л баллона) через реакционную смесь. Реакционную смесь фильтровали через целит, промывали подушку из целита EtOAc (2×150 мл) и фильтрат концентрировали на роторном испарителе (60°C) с получением желтого масла. Et2O добавляли к желтому маслу и затем удаляли на роторном испарителе с получением бледно-желтого порошка (неочищенный 6). TLC (силикагель, 70% EtOAc/гексан) бледно-желтого порошка показала несколько продуктов. Желтый порошок интенсивно перемешивали в гексане (150 мл) в течение 30 минут и твердое вещество собирали вакуумной фильтрацией, промывая гексаном (2×30 мл) с получением соединения 6 (30,22 г, содержащее приблизительно 15 мол. % неизвестной примеси) в виде бледно-желтого порошка. 1Н ЯМР (400 МГц, CDCl3) 2,69-2,75 (m, 2Н), 2,94-3,01 (m, 2Н), 5,57 (br. s., 2Н), 6,99 (d, J=8,22 Гц, 1Н), 7,29 (d, J=2,35 Гц, 2Н), 7,33-7,43 (m, 4Н), 7,53 (d, J=7,43 Гц, 2Н).
Получение 3-(3-амино-3-оксопропил)бифенил-4-ил-трифторметансульфоната (7)
1,1,1-Трифтор-N-фенил-N-[(трифторметил)сульфонил]метансульфонамид (PhNTf2) (42,5 г, 0,119 моль) добавляли к раствору 6 (24,0 г, 0,0995 моль) и NEt3 (15,3 мл, 0,109 моль) в CH3CN (480 мл) и реакционную смесь перемешивали в течение 1,5 часа. Добавляли дополнительное количество PhN(Tf)2 (2,25 г, 6,30 ммоль) и NEt3 (1,5 мл, 10,7 ммоль) и реакционную смесь перемешивали еще в течение 30 минут. В реакционной смеси были показаны лишь незначительные следовые количества оставшегося 6. Реакционную смесь концентрировали на роторном испарителе (60°C) с получением оранжевого масла. Небольшую аликвоту масла переносили в EtOAc (15 мл) и промывали Na2CO3 (водн., 2 М, 2×20 мл) и NaOH (водн., 0,5 М, 2×20 мл), высушивали над MgSO4 и концентрировали с получением порции неочищенного 7 (422 мг). Последующий анализ ВЭЖХ указал на то, что необходимый продукт находился в органической фазе. Первую порцию неочищенного 7 снова соединяли с оранжевым маслом и переносили в EtOAc (300 мл), промывали Na2CO3 (водн., 2 М, 2×250 мл), высушивали над MgSO4 и концентрировали на роторном испарителе (60°C) с получением оранжевого масла (65 г, 1Н ЯМР показал значительные примеси, в том числе NEt3). Масло повторно растворяли в EtOAc (300 мл) и промывали лимонной кислотой (водн., 10% вес/вес, 2×250 мл) и водой (2×350 мл), высушивали над MgSO4 и концентрировали на роторном испарителе (60°C) с получением оранжевого масла (59 г). Масло повторно переносили в EtOAc (300 мл) и промывали NaOH (водн., 0,5 М, 3×200 мл), HCl (2 М, 2×200 мл) и H2O (1×300 мл), высушивали над MgSO4 и концентрировали на роторном испарителе (60°C) с получением оранжевого масла, которое при отстаивании затвердевало. Данное твердое вещество суспендировали в Et2O (150 мл) и интенсивно перемешивали в течение 30 минут, собирали вакуумной фильтрацией и промывали Et2O (2×30 мл) с получением соединения 7 в виде белого порошка (12,3 г, 37%). 1Н ЯМР (400 МГц, CDCl3) 2,60 (m, J=7,63 Гц, 2Н), 3,13 (t, J=7,83 Гц, 2Н), 5,48 (br. s., 2Н), 7,32 (d, J=8,61 Гц, 1Н), 7,36-7,42 (m, 1Н), 7,45 (t, J=7,43 Гц, 2Н), 7,50 (dd, J=8,61, 1,96 Гц, 1Н), 7,54 (d, J=7,43 Гц, 2Н), 7,59 (d, J=1,96 Гц, 1Н).
Пример 11 - Синтез Т22 и Т23
Т22 и Т23 получали из 3-(3-амино-3-оксопропил)бифенил-4-ил-трифторметансульфоната (7, полученный в примере 10). Смесь 7 (1 экв.), гетероциклической бороновой кислоты (1,2 экв.) и карбоната калия (2 экв.) суспендировали в 1,4-диоксане (4 мл/ммоль) и воде (5 капель/ммоль). Азот барботировали через смесь в течение 15 минут. Добавляли тетракис(трифенилфосфин)палладий(0) (0,1 экв.) и смесь нагревали при 85°C в атмосфере азота в течение 20 часов. Смесь разбавляли этилацетатом и фильтровали. Остаток промывали этилацетатом (2×). Объединенные фильтраты выпаривали до сухого состояния и очищали с помощью флэш-хроматографии (метанол/дихлорметан). Продукт суспендировали в гексанах (4 мл) и выделяли фильтрацией.
С помощью этой процедуры получали следующие соединения.
3-[4-(1Н-Индол-4-ил)бифенил-3-ил]пропанамид (Т22)
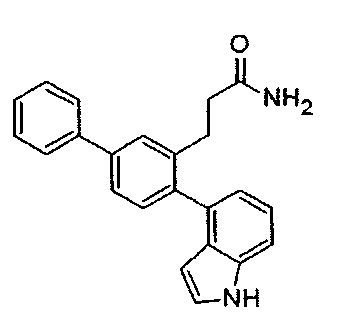
Бледно-бежевый порошок (58 мг, 32%). 1Н ЯМР (400 МГц, CDCl3) 5,27 (br. s., 1H), 7,64-7,71 (m, 2H), 7,58-7,63 (m, 1H), 7,54 (dd, J=7,8, 1,8 Гц, 1H), 7,40-7,50 (m, 4H), 7,36 (br. t, J=7,5 Гц, 1H), 7,28 (d, J=8,2 Гц, 1H), 7,22 (t, J=2,7 Гц, 1H), 7,03-7,09 (m, 1H), 6,27-6,33 (m, 1H), 4,96 (br. s., 1H), 4,88 (br. s., 1H), 2,99 (br. s., 2H), 2,23 (t, J=7,9 Гц, 2H); ЖХ-МС [M+H]+=341,2; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 97,1% при 220 нм.
3-[4-(1Н-Индол-6-ил)бифенил-3-ил]пропанамид (Т23)
Бледно-коричневый порошок (13 мг, 7%). 1Н ЯМР (400 МГц, CDCl3) 5,27 (br. s., 1H), 7,69 (d, J=8,0 Гц, 1H), 7,64 (d, J=7,2 Гц, 2H), 7,56 (d, J=1,6 Гц, 1H), 7,47-7,53 (m, 1H), 7,42-7,46 (m, 2H), 7,32-7,41 (m, 3H), 7,12 (dd, J=8,1, 1,3 Гц, 1H), 6,60 (br. s., 1H), 5,06 (br. s., 2H), 3,04-3,16 (m, 2H), 2,29-2,40 (m, 2H); ЖХ-МС [M+H]+=341,3; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 99,5% при 220 нм.
Пример 12 - Синтез Т29, Т38, Т63, Т64, Т65 и Т66
Т29, Т38, Т63, Т64, Т65 и Т66 получали из 3-(3-амино-3-оксопропил)бифенил-4-ил-трифторметансульфоната (7, полученный в примере 10). Смесь 7 (1 экв.), пинаколового эфира ароматической бороновой кислоты или гетероциклической бороновой кислоты (1,1 экв.) и карбоната калия (2-3 экв.) растворяли в смеси 1,4-диоксана (3,1 мл/ммоль), этанола (0,65 мл/ммоль) и воды (0,65 мл/ммоль). Азот барботировали через смесь в течение 10 минут. Добавляли тетракис(трифенилфосфин)палладий(0) (0,1 экв.) и смесь нагревали при 85°C в атмосфере азота в течение 20 часов. Смесь распределяли между этилацетатом и водой. Водную фазу экстрагировали этилацетатом. Объединенные экстракты этилацетата промывали водой и солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат выпаривали до сухого состояния и очищали с помощью флэш-хроматографии (метанол/дихлорметан). Продукт суспендировали в смеси 1:1 дихлорметан/гексаны и выделяли фильтрацией.
С помощью этой процедуры получали следующие соединения.
3-[4-(2-Оксо-2,3-дигидро-1,3-бензотиазол-5-ил)бифенил-3-ил]пропанамид (Т29)
Светло-коричневый порошок (39 мг, 11%). 1Н ЯМР (400 МГц, DMSO-d6) 11,96 (br. s., 1Н), 7,70 (d, J=7,2 Гц, 2H), 7,60-7,67 (m, 2H), 7,55 (dd, J=7,9, 1,7 Гц, 1H), 7,49 (t, J=7,6 Гц, 2H), 7,35-7,43 (m, 1H), 7,20-7,31 (m, 2H), 7,09-7,16 (m, 1H), 7,05 (d, J=l,2 Гц, 1H), 6,75 (br. s., 1H), 2,84 (t, J=7,8 Гц, 2H), 2,25-2,37 (m, 2H); ЖХ-МС [M+H]+=375,1; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 98,6% при 220 нм.
3-[4-(1Н-Индазол-6-ил)бифенил-3-ил]пропанамид (Т38)
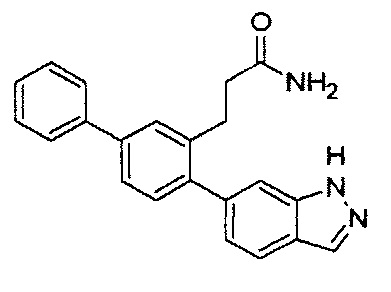
Бледно-желтый порошок (72 мг, 44%). 1Н ЯМР (400 МГц, DMSO-d6) 13,09 (s, 1H), 8,12 (s, 1H), 7,82 (d, J=8,2 Гц, 1H), 7,71 (d, J=7,4 Гц, 2H), 7,65 (d, J=1,4 Гц, 1H), 7,56 (dd, J=8,0, 1,8 Гц, 1H), 7,43-7,53 (m, 3H), 7,35-7,43 (m, 1H), 7,32 (d, J=7,8 Гц, 1H), 7,21 (br. s., 1H), 7,11 (dd, J=8,3, 0,9 Гц, 1H), 6,70 (br. s., 1H), 2,77-2,93 (m, 2H), 2,25-2,38 (m, 2H); ЖХ-МС [M+H]+=342,1; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 98,5% при 220 нм.
3-(2-Фтор-3-гидрокси-1,1':4',1''-терфенил-2'-ил)пропанамид (Т63)
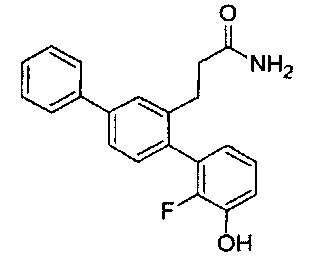
Белый порошок (87 мг, 53%). 1Н ЯМР (400МГц, DMSO-d6) 9,92 (br. s., 1Н), 7,70 (d, J=7,2 Гц, 2H), 7,64 (d, J=1,6 Гц, 1H), 7,54 (dd, J=7,9, 1,7 Гц, 1H), 7,49 (t, J=7,6 Гц, 2H), 7,35-7,42 (m, 1H), 7,16-7,29 (m, 2H), 7,03-7,10 (m, 1H), 6,94-7,02 (m, 1H), 6,65-6,78 (m, 2H), 2,72 (t, J=7,8 Гц, 2H), 2,28 (t, J=7,9 Гц, 2H); ЖХ-МС [M+H]+=336,2; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 99,3% при 220 нм.
3-(4-Фтор-3-гидрокси-1,1':4',1''-терфенил-2'-ил)пропанамид (Т64)
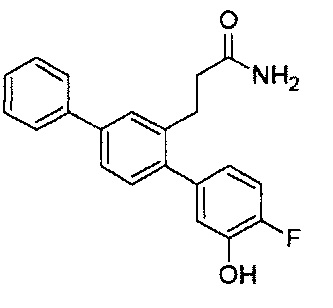
Белый порошок (98 мг, 61%). 1Н ЯМР (400 МГц, DMSO-d6) 9,95 (br. s., 1H), 7,69 (d, J=7,2 Гц, 2H), 7,60 (d, J=1,6 Гц, 1H), 7,43-7,55 (m, 3H), 7,33-7,42 (m, 1H), 7,12-7,28 (m, 3H), 6,90 (dd, J=8,5, 2,1 Гц, 1H), 6,65-6,80 (m, 2H), 2,76-2,87 (m, 2H), 2,25-2,36 (m, 2H); ЖХ-МС [M+H]+=336,2; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 99,6% при 220 нм.
3-(3-Фтор-5-гидрокси-1,1':4',1''-терфенил-2'-ил)пропанамид (Т65)
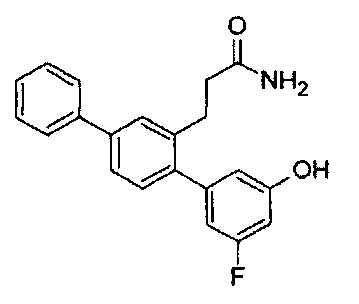
Белый порошок (89 мг, 55%). 1H ЯМР (400 МГц, DMSO-d6) 10,05 (br. s., 1Н), 7,69 (d, J=7,4 Гц, 2H), 7,61 (d, J=1,6 Гц, 1H), 7,44-7,56 (m, 3H), 7,34-7,42 (m, 1H), 7,20-7,31 (m, 2H), 6,76 (br. s., 1H), 6,54-6,64 (m, 3H), 2,83 (t, J=7,8 Гц, 2H), 2,32 (t, J=7,8 Гц, 2H); ЖХ-МС [M+H]+=336,2; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 99,6% при 220 нм.
3-(2-Фтор-5-гидрокси-1',1':4',1''-терфенил-2'-ил)пропанамид (Т66)
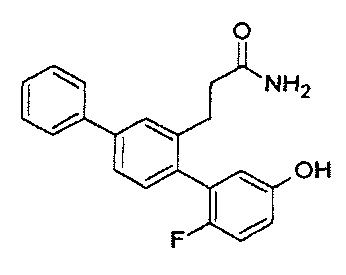
Белый порошок (75 мг, 45%). 1H ЯМР (400 МГц, DMSO-d6) 9,51 (s, 1Н), 7,70 (d, J=7,2 Гц, 2H), 7,63 (d, J=1,4 Гц, 1H), 7,54 (dd, J=7,9, 1,7 Гц, 1H), 7,49 (t, J=7,5 Гц, 2H), 7,35-7,42 (m, 1H), 7,19-7,29 (m, 2H), 7,10 (t, J=9,1 Гц, 1H), 6,79 (dt, J=8,6, 3,6 Гц, 1H), 6,74 (br. s., 1H), 6,66 (dd, J=6,3, 2,9 Гц, 1H), 2,68-2,78 (m, 2H), 2,24-2,34 (m, 2H); ЖХ-МС [M+H]+=336,2; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 97,6% при 220 нм.
Пример 13 - Синтез 3-[4-(4,4,5,5-тетраметил-1,3,2-диоксоборолан-2-ил)бифенил-3-ил]пропанамида
3-[4-(4,4,5,5-Тетраметил-1,3,2-диоксоборолан-2-ил)бифенил-3-ил]пропанамид (8) получали из 3-(3-амино-3-оксопропил)бифенил-4-ил-трифторметансульфоната (7, полученный в примере 10). Смесь 7 (1,81 г, 4,84 ммоль), бис(пинаколато)дибор (1,35 г, 5,31 ммоль), комплекс 1,1'-бис(дифенилфосфино)ферроценпалладия(II) дихлорида и дихлорметана (790 мг, 0,97 ммоль) и ацетат калия (1,43 г, 14,5 ммоль) суспендировали в безводном диметилсульфоксиде (31 мл) в атмосфере азота. Смесь нагревали при 85°C в атмосфере азота в течение 4 часов. Смесь разбавляли этилацетатом (90 мл) и элюировали через колонку из силикагеля с этилацетатом. Фракции, которые содержали основную полосу, промывали водой (2×200 мл) и солевым раствором (200 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат выпаривали до сухого состояния. Остаток очищали с помощью флэш-хроматографии (этилацетат/дихлорметан) с получением соединения 8:
Светло-коричневое масло, которое при отстаивании затвердевало (1,02 г, 59%). 1Н ЯМР (400 МГц, CDCl3) 7,91 (d, J=8,4 Гц, 1Н), 7,60 (d, J=7,2 Гц, 2Н), 7,40-7,50 (m, 4Н), 7,32-7,39 (m, 1Н), 5,82 (br. s., 1H), 5,33 (br. s., 1H), 3,22-3,31 (m, 2H), 2,51-2,59 (m, 2H), 1,38 (s, 12H).
Пример 14 - Синтез T24, T26, T27, T30, T32, Т33, Т35, Т37, Т39 и Т58
Т24, Т26, Т27, Т30, Т32, Т33, Т35, Т37, Т39 и Т58 получали из 3-[4-(4,4,5,5-тетраметил-1,3,2-диоксоборолан-2-ил)бифенил-3-ил]пропанамида (8, полученный в примере 13). Смесь 8 (1 экв.), ароматическую бороновую кислоту или гетероциклическую бороновую кислоту (1,1 экв.) и комплекс 1,1'-бис(дифенилфосфино)-ферроценпалладия(II) дихлорида и дихлорметана (0,1 экв.) растворяли в безводном N,N-диметилформамиде (10,6 мл/ммоль) в атмосфере азота. Добавляли дегазированный раствор карбоната натрия (2 М, 5,3 мл/ммоль). Смесь нагревали при 80°C в атмосфере азота. Смесь распределяли между этилацетатом и водой. Водную фазу экстрагировали этилацетатом. Объединенные экстракты этилацетата промывали водой (3×) и солевым раствором, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат выпаривали до сухого состояния и очищали с помощью флэш-хроматографии (метанол/дихлорметан). Продукт суспендировали в смеси 1:1 дихлорметан/гексаны и выделяли фильтрацией.
С помощью этой процедуры получали следующие соединения.
3-[4-(2-Оксо-2,3-дигидро-1Н-индол-4-ил)бифенил-3-ил]пропанамид (Т24)
Бледно-желтый порошок (72 мг, 58%). 1Н ЯМР (400 МГц, DMSO-d6) 10,46 (s, 1Н), 7,70 (d, J=7,2 Гц, 2H), 7,63 (d, J=1,4 Гц, 1H), 7,43-7,56 (m, 3H), 7,34-7,42 (m, 1H), 7,16-7,30 (m, 3H), 6,83 (t, J=7,3 Гц, 2H), 6,67 (br. s., 1H), 3,26 (br. s., 2H), 2,72 (br. t, J=7,0 Гц, 2H), 2,25 (t, J=7,7 Гц, 2H); ЖХ-МС [M+H]+=357,2; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 100,0% при 220 нм.
3-[4-(2-Оксо-2,3-дигидро-1,3-бензоксазол-7-ил)бифенил-3-ил]пропанамид (Т26)
Светло-оранжевый порошок (30 мг, 23%). 1Н ЯМР (400 МГц, DMSO-d6) 11,72 (br. s., 1H), 7,70 (d, J=7,4 Гц, 2H), 7,65 (s, 1H), 7,56 (dd, J=7,8, 1,2 Гц, 1H), 7,48 (t, J=7,6 Гц, 2H), 7,34-7,42 (m, 1H), 7,31 (d,.7=7,8 Гц, 1H), 7,15-7,26 (m, 2H), 7,10 (d, J=7,0 Гц, 1H), 7,02 (d,.7=7,4 Гц, 1H), 6,69 (br. s., 1H), 2,75 (t, J=8,0 Гц, 2H), 2,26 (t, J=8,0 Гц, 2H); ЖХ-МС [M+H]+=359,1; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 98,9% при 220 нм.
3-[4-(2-Оксо-2,3-дигидро-1,3-бензоксазол-5-ил)бифенил-3-ил]пропанамид (Т27)
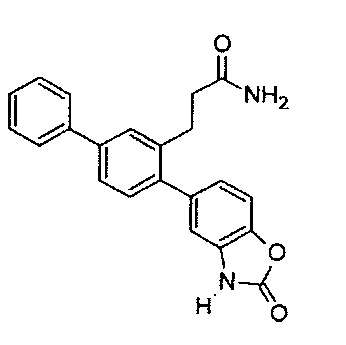
Бледно-оранжевый порошок (49 мг, 28%). 1Н ЯМР (400 МГц, DMSO-d6) 11,69 (br. s., 1H), 7,70 (d, J=7,4 Гц, 2Н), 7,62 (d, J=1,6 Гц, 1H), 7,45-7,57 (m, 3Н), 7,31-7,43 (m, 2Н), 7,17-7,30 (m, 2Н), 7,00-7,09 (m, 2Н), 6,72 (br. s., 1Н), 2,83 (t, J=7,8 Гц, 2H), 2,25-2,35 (m, 2H); ЖХ-МС [M+H]+=359,2; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 97,0% при 220 нм.
3-[4-(2-Оксо-2,3-дигидро-1Н-бензимидазол-4-ил)бифенил-3-ил]пропанамид (Т30)
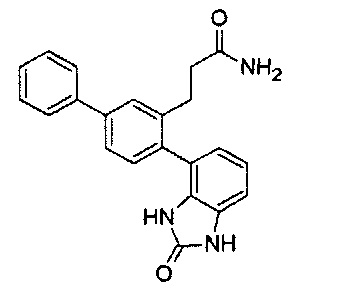
Светло-бежевый порошок (110 мг, 63%). 1Н ЯМР (400 МГц, DMSO-d6) 10,69 (s, 1H), 10,48 (s, 1H), 7,71 (d, J=7,4 Гц, 2H), 7,63 (d, J=1,6 Гц, 1H), 7,55 (dd, J=7,8, 1,8 Гц, 1H), 7,50 (t, J=7,6 Гц, 2H), 7,35-7,43 (m, 1H), 7,19-7,30 (m, 2H), 6,97-7,05 (m, 1H), 6,91-6,97 (m, 1H), 6,78-6,82 (m, 1H), 6,75 (br. s., 1H), 2,60-2,86 (m, 2H), 2,29 (br. s., 2H); ЖХ-МС [M+H]+=358,2; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 96,4% при 220 нм.
3-[4-(2-Оксо-1,2,3,4-Тетрагидрохинолин-7-ил)бифенил-3-ил]пропанамид (Т32)
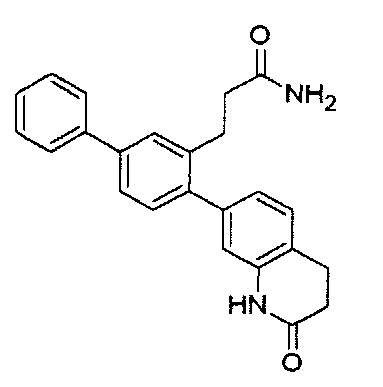
Бледно-бежевый порошок (118 мг, выход 66%). 1Н ЯМР (400 МГц, DMSO-d6) 10,13 (s, 1Н), 7,69 (d, J=7,4 Гц, 2H), 7,61 (d, J=1,6 Гц, 1H), 7,44-7,56 (m, 3H), 7,34-7,41 (m, 1H), 7,18-7,28 (m, 3H), 6,90 (dd, J=7,5, 1,5 Гц, 1H), 6,82 (d, J=1,2 Гц, 1H), 6,74 (br. s., 1H), 2,94 (t, J=7,5 Гц, 2H), 2,77-2,87 (m, 2H), 2,53 (неясный за счет DMSO-d6), 2,28-2,37 (m, 2H); ЖХ-МС [M+H]+=371,2; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 97,9% при 220 нм.
3-{4-[2-(Трифторметил)-1Н-бензимидазол-4-ил]бифенил-3-ил}пропанамид (Т33)
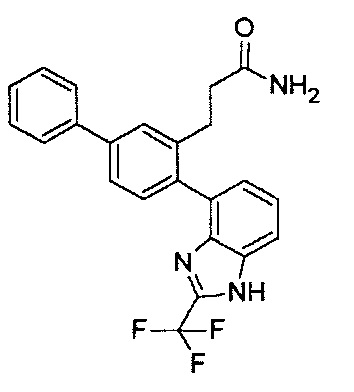
Белый порошок (107 мг, 53%). 1Н ЯМР (400 МГц, DMSO-d6) 13,81-14,10 (m, 1Н), 7,08-7,88 (m, 12H), 6,59-6,81 (m, 1H), 2,71 (br. t, J=7,3 Гц, 2H), 2,28 (m, 2H). Спектр расщеплялся на два соединения вследствие обмена водорода на бензимидазольном фрагменте; ЖХ-МС [М+Н]+=410,2; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 100% при 220 нм.
3-[4-(1Н-Бензимидазол-4-ил)бифенил-3-ил]пропанамид (Т35)
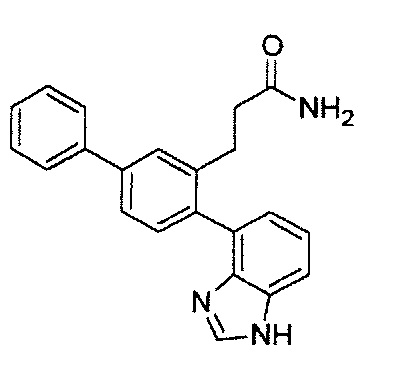
Светло-коричневый порошок (54 мг, 32%). 1Н ЯМР (400 МГц, DMSO-d6) 12,32-12,57 (m, 1Н), 8,16 (d, J=13,3 Гц, 1H), 7,03-7,79 (m, 12H), 6,54-6,75 (m, 1H), 2,67-2,81 (m, 2H), 2,20-2,33 (m, 2H). Спектр расщеплялся на два соединения вследствие обмена водорода на бензимидазольном фрагменте; ЖХ-МС [М+Н]+=342,2; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 96,9% при 220 нм.
3-[4-(1Н-Индазол-4-ил)бифенил-3-ил]пропанамид (Т37)
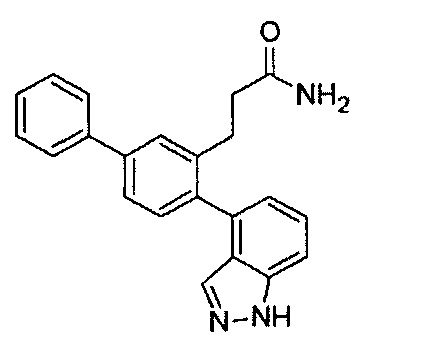
Бледно-коричневый порошок (69 мг, 42%). 1H ЯМР (400 МГц, DMSO-d6) 13,20 (s, 1Н), 7,68-7,79 (m, 4H), 7,54-7,63 (m, 2H), 7,50 (t, J=7,6 Гц, 2H), 7,32-7,47 (m, 3Н), 7,17 (br. s., 1H), 7,03 (d, J=6,8 Гц, 1H), 6,67 (br. s., 1H), 2,77 (t, J=7,8 Гц, 2H), 2,21-2,29 (m, 2H); ЖХ-МС [M+H]+=342,2; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 99,7% при 220 нм.
3-[4-(1Н-бензотриазол-4-ил)бифенил-3-ил]пропанамид (Т39)
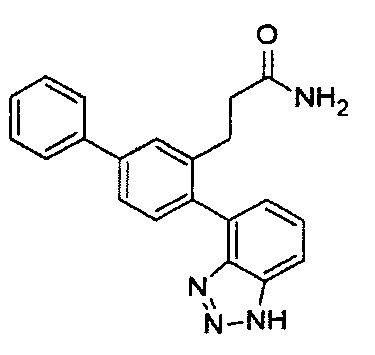
Бледно-желтый порошок (10 мг, 6%). 1Н ЯМР (400 МГц, DMSO-d6) 15,79 (br. s., 1Н), 7,81-8,05 (m, 1H), 7,68-7,79 (m, 3H), 7,62 (d, J=7,8 Гц, 1H), 7,52 (t, J=7,7 Гц, 3H), 7,29-7,45 (m, 3H), 7,21 (br. s., 1H), 6,71 (br. s., 1H), 2,68-2,80 (m, 2H), 2,27 (t, J=7,8 Гц, 2H); ЖХ-МС [M+H]+=343,2; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 98,2% при 220 нм.
3-(2,4-Дифтор-3-гидрокси-1,1':4',1''-терфенил-2'-ил)пропанамид (Т58).
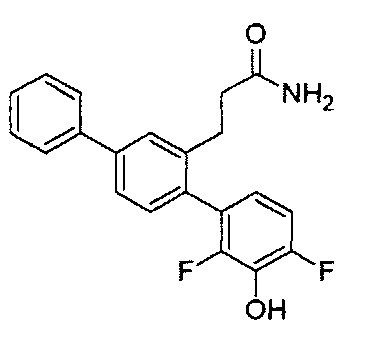
После первой экстракции этилацетатом/водой регулировали pH водного слоя до pH 6 добавлением 1 М соляной кислоты и продолжали исследование для общей процедуры. Получали в виде бледно-коричневого порошка (106 мг, 62%). 1Н ЯМР (400 МГц, DMSO-d6) 10,24 (br. s., 1Н), 7,70 (d, J=7,4 Гц, 2H), 7,64 (d, J=1,4 Гц, 1H), 7,55 (dd, J=7,9, 1,7 Гц, 1H), 7,49 (t, J=7,6 Гц, 2H), 7,35-7,43 (m, 1H), 7,17-7,27 (m, 2H), 7,11 (t, J=9,2 Гц, 1H), 6,65-6,81 (m, 2H), 2,71 (t, J=7,7 Гц, 2H), 2,28 (t, J=7,9 Гц, 2H); ЖХ-МС [M+H]+=354,3; ВЭЖХ (градиент вода/ACN + 0,1% TFA) 99,5% при 220 нм.
Пример 15 - Синтез 5-йодо-2-метилбензол-1,3-диола для применения при получении Т67
Смесь 4-хлор-3,5-диметоксианилина (3,0 г, 16,0 ммоль), палладия(II) ацетата (180 мг, 0,80 ммоль), 2-дициклогексилфосфино-2,4,6-триизопропилбифенила (XPhos) (381 мг, 0,80 ммоль), карбоната калия (6,73 г, 48,7 ммоль) и метилбороновой кислоты (1,15 г, 19,2 ммоль) в воде (100 мл) и диоксане (100 мл) нагревали до 100°C (температура масляной бани) в атмосфере азота в течение 18 часов. Реакцию не завершали и реакционную смесь нагревали до температуры флегмы еще в течение 3 часов, охлаждали до комнатной температуры, разбавляли водой (200 мл) и экстрагировали этилацетатом (3×150 мл), высушивали над сульфатом магния, концентрировали и очищали с помощью флэш-хроматографии (этилацетат/гексаны) с получением 3,5-диметокси-4-метиланилина (720 мг, 27%). 1Н ЯМР (400 МГц, CDCl3) 5,93 (s, 2Н), 3,77 (s, 6Н), 3,58 (bs, 2Н), 1,98 (s, 3Н). Нитрит натрия (340 мг, 4,93 ммоль) добавляли к смеси 3,5-диметокси-4-метиланилина (720 мг, 4,31 ммоль) в серной кислоте (1,1 мл) и воде (13 мл) при 0°C и перемешивали в течение 30 минут. Полученную смесь добавляли к предварительной нагретой смеси йодида натрия (2,58 г, 17,2 ммоль) и йода (555 мг, 2,19 ммоль) в серной кислоте (1,1 мл) и воде (13 мл) при 80°C, и смесь нагревали до температуры флегмы в течение 30 минут. Реакционную смесь охлаждали до комнатной температуры и разбавляли раствором сульфита натрия (20% вес/вес, 100 мл) и воды (100 мл) и экстрагировали этилацетатом (3×100 мл). Органические соединения объединяли, высушивали над сульфитом магния, концентрировали и очищали с помощью флэш-хроматографии (этилацетат/гексаны) с получением 5-йодо-1,3-диметокси-2-метилбензола в виде белого порошка (388 мг, 32%). 1Н ЯМР (400 МГц, CDCl3) 6,84 (s, 2Н), 3,79 (s, 6Н), 2,02 (s, 3Н). 5-Йодо-1,3-диметокси-2-метилбензол (388 мг, 1,39 ммоль) в дихлорметане (8 мл) охлаждали до 0°C перед медленным добавлением беспримесного трибромида бора (0,8 мл, 8 ммоль) в течение 1 минуты в атмосфере азота. Реакционную смесь медленно нагревали до комнатной температуры в течение 3 часов и перемешивали в течение 18 часов. Реакционную смесь медленно и осторожно выливали на ледяную воду (100 мл) и экстрагировали этилацетатом (3×60 мл), и промывали объединенные органические соединения солевым раствором (1×50 мл), высушивали над сульфатом магния, концентрировали и очищали с помощью флэш-хроматографии (этилацетат/гексаны) с получением 5-йодо-2-метилбензол-1,3-диола в виде белого порошка (260 мг, 74%). 1Н ЯМР (400 МГц, d6-DMSO) 9,44 (s, 2Н), 6,63 (s, 2Н), 1,87 (s, 3Н).
Пример 16 - Синтез 5-бром-2-фторбензол-1,3-диола для применения при получении Т68
Смесь оксона (1,44 г) в воде (2 мл) добавляли к раствору 5-бром-2-фтор-1,3-фенилендибороновой кислоты, пинаколового эфира (500 мг, 1,17 ммоль) в ацетоне (1,5 мл) в течение 1 минуты и перемешивали при комнатной температуре в течение 15 минут. Добавляли дополнительное количество оксона (0,512 г) и ацетона (1 мл) и перемешивали еще в течение 20 минут. Добавляли раствор бисульфита натрия (10% вес/вес, 10 мл) с последующим добавлением воды (10 мл) и экстрагировали дихлорметаном (3×20 мл), высушивали над сульфатом магния и очищали с помощью флэш-хроматографии (этилацетат/гексаны) с получением 5-бром-2-фторбензол-1,3-диола в виде белого порошка (159 мг, 66%). 1Н ЯМР (400 МГц, CDCl3) 6,73 (d, J=6,8 Гц, 2Н), 5,29 (bs, 2Н).
Пример 17 - Синтез 2-хлор-5-йодбензол-1,3-диола для применения при получении Т69
2-Хлор-5-йодбензол-1,3-диол синтезировали в 3 стадии из 3,5-диметоксианилина в соответствии с WO 2011/027106 А1 с модификацией для первой стадии получения 4-хлор-3,5-диметоксианилина, на которой реагент N-хлорсукцинимид добавляли по частям в виде суспензии в уксусной кислоте (50 мл) к раствору 3,5-диметоксианилина (10,01 г, 65,35 ммоль) в уксусной кислоте (50 мл) при 0°C и через 30 минут нагревали до комнатной температуры.
Пример 18 - Синтез Т67, Т68 и Т69
Т67, Т68 и Т69 получали из 3-[4-(4,4,5,5-тетраметил-1,3,2-диоксоборолан-2-ил)бифенил-3-ил]пропанамида (8, полученный в примере 13). Смесь 8 (1 экв.), замещенного бензол-1,3-диола (полученный в примерах 15-17) (1,1 экв.), Pd(dppf)C12.CH2Cl2 (0,1 экв.) и карбоната натрия (2 М, 5 мл/ммоль) в N,N-диметилформамиде (10 мл/ммоль) барботировали азотом в течение 5 минут перед нагреванием до 80°C в атмосфере азота в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры и распределяли между 1 М соляной кислотой и этилацетатом. Объединенные органические слои промывали солевым раствором, высушивали над сульфатом магния, концентрировали и очищали с помощью флэш-хроматографии (этилацетат/гексаны).
С помощью этой процедуры получали следующие соединения.
3-(3,5-Дигидрокси-4-метил-1,1':4',1''-терфенилш-2'-ил)пропанамид (Т67)
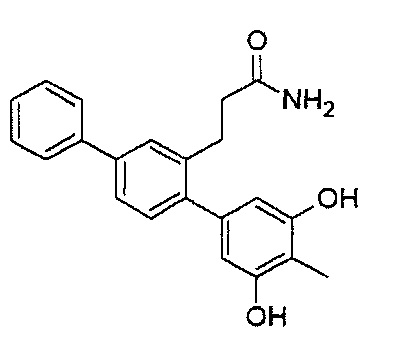
Белый порошок (121 мг, 59%). 1Н ЯМР (400 МГц, DMSO-d6) 9,17 (s, 2Н), 7,68 (d, J=7,43 Гц, 2Н), 7,58 (br. s, 1Н), 7,48 (t, J=7,80 Гц, 3Н), 7,37 (s, 1H), 7,23 (br. s., 1H), 7,17 (d, J=7,83 Гц, 1H), 6,76 (br. s, 1H), 6,25 (s, 2H), 3,33 (s, 3H), 2,85 (t, J=7,83 Гц, 2H), 2,30 (t, J=7,83 Гц, 2H). ЖХ-МС [M+H]+=348. ВЭЖХ (градиент вода/ACN + 0,1% TFA) 100% при 220 нм.
3-(4-Фтор-3,5-дигидрокси-1,1':4',1''-терфенш-2'-ил)пропанамид (Т68)
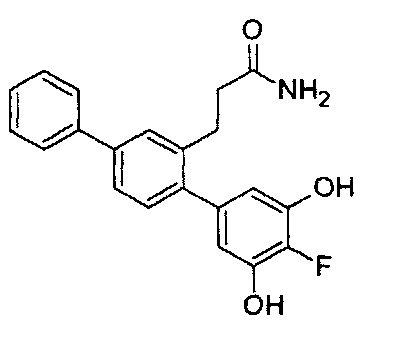
Бледно-желтое твердое вещество (82 мг, 41%). 1Н ЯМР (400 МГц, DMSO-d6): 9,70 (br. s, 2Н), 7,68 (d, J=7,43 Гц, 2H), 7,58 (s, 1Н), 7,43-7,53 (m, 3Н), 7,32-7,42 (m, 1H), 7,25 (br. s, 1H), 7,18 (d, J=7,83 Гц, 1H), 6,75 (br. s, 1H), 6,33 (d, J=7,43 Гц, 2H), 2,83 (t, J=7,80 Гц, 2H), 2,30 (t, J=8,20 Гц, 2H). ЖХ-МС [M+H]+=352. ВЭЖХ (градиент вода/ACN + 0,1% TFA) 96,1% при 220 нм.
3-(4-Хлор-3,5-дигидрокси-1,1':4',1''-терфенил-2'-ил)пропанамид (Т69)
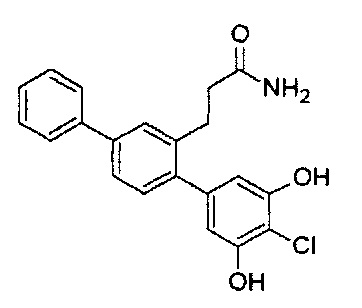
Белый порошок (26 мг, 12%). 1Н ЯМР (400 МГц, DMSO-d6) 9,93-10,12 (m, 2Н), 7,69 (d, J=7,43 Гц, 2Н), 7,59 (s, 1H), 7,43-7,54 (m, 3Н), 7,34-7,42 (m, 1Н), 7,25 (br. s., 1Н), 7,19 (d, J=7,83 Гц, 1H), 6,76 (br. s., 1Н), 6,38 (s, 2H), 2,84 (t, J=7,40 Гц, 2H), 2,30 (t, J=8,20 Гц, 2H). ЖХ-МС [М+Н]+=368. ВЭЖХ (градиент вода/ACN + 0,1% TFA) 100% при 254 нм.
Пример 19 - Синтез Т25
Синтетический путь, используемый для получения Т25, показан на фигуре 8. Вкратце, 3-формилбифенил-4-ил-трифторметансульфонат (14) вводили в реакцию Хорнера-Уодсворта-Эммонса с диэтил(карбамоилметил)фосфонатом (18) с получением бифенилакриламида (23), с которым проводили реакцию перекрестного сочетания с индолонпинаколовым эфиром бороновой кислоты (24) с получением акриламида (25). Последующая гидрогенизация соединения 25 приводила к образованию Т25.
Для синтеза Т25 требовался необходимый индолонпинаколовый эфир бороновой кислоты (24). С этой целью 1,4-дибром-2-нитробензол алкилировали диметилмалонатом с получением арилмалоната (26), который декарбоксилировали и циклизировали с получением броминдолона (27); его, в свою очередь, вводили в реакцию с бис(пинаколото)дибораном с получением индолонпинаколового эфира бороновой кислоты (24) (фигура 9).
Получение диметил-2-(4-бром-2-нитрофенил)малоната (26)
К смеси трет-бутоксида калия (21,6 г, 193,00 ммоль) в DMF (75 мл) добавляли диметилмалонат (22,40 мл, 196,00 ммоль). Реакция была экзотермической, и при этом осаждалось твердое вещество. Реакционную смесь нагревали до 90°C в течение 10 минут, затем охлаждали до температуры окружающей среды. 2,5-Дибромнитробензол (25,50 г, 91 ммоль) добавляли в виде твердого вещества. Реакционная смесь становилась пурпурной, и ее перемешивали при 90°C в течение 2 часов. После охлаждения до температуры окружающей среды ее выливали в 5% раствор ледяной соляной кислоты и переносили в отдельную воронку. Неочищенное вещество экстрагировали этилацетатом (2х). Объединенные экстракты этилацетата промывали водой и солевым раствором с получением ярко-желтого масла. Неочищенное масло предварительно абсорбировали на целите, затем хроматографировали (DCVC) с элюированием с применением градиента этилацетата в гептане (0-10% этилацетата). Подобные фракции объединяли и перекристаллизовывали из DCM и гептана с получением диметил-2-(4-бром-2-нитрофенил)малоната (26) в виде бледно-желтых игл (26,78 г, 87%); т.пл. 85,8-87,1°C. 1H ЯМР (400 МГц, CDCl3) δ 8,18 (d, 1H, J 2,1 Гц), 7,75 (dd, 1Н, J 2,1, 8,4 Гц), 7,40 (d, 1H, J 8,4 Гц), 5,26 (s, 1H), 3,78 (s, 6H).
Получение 6-броминдолин-2-она (27)
Хлорид лития (6,36 г, 156,0 ммоль) добавляли к раствору диметил-2-(4-бром-2-нифтрофенил)малоната (26) (26,0 г, 78,30 ммоль) в диметилсульфоксиде (100 мл) и нагревали при 100°C в течение 20 часов. Сразу после охлаждения до температуры окружающей среды реакционную смесь распределяли между этилацетатом и солевыми раствором. Слои разделяли, затем повторно промывали солевым раствором и концентрировали. Темно-коричневое масло растворяли в уксусной кислоте (100 мл) и добавляли железный порошок (17,50 г, 313,0 ммоль) (экзотермический эффект). Реакционную смесь затем нагревали при 110°C в течение 1 часа. Уксусную кислоту удаляли роторным испарением, а остаток растворяли в этилацетате и удаляли железный порошок фильтрацией через целит. Фильтрат промывали 1 М соляной кислотой и водой, затем фильтровали через бумагу для разделения фаз (1PS). Неочищенное вещество предварительно абсорбировали на целите, затем хроматографировали (DCVC) с элюированием хлороформом. Фракции, содержащие необходимое вещество, объединяли, предварительно абсорбировали на целите и повторно хроматографировали (DCVC) с элюированием с применением градиента этилацетата в гептане (20-80% этилацетата). Чистые фракции объединяли и перекристаллизовывали из DCM и метанола с получением 6-броминдолин-2-она (27) в виде желтых игл (4,32 г, 26%); т.пл. 208-214°C. 1Н ЯМР (400 МГц, CDCl3) δ 10,47 (br s, 1H), 7,14 (d, 1H, J 7,9 Гц), 7,09 (dd, 1H, J 1,8, 7,9 Гц), 6,94 (d, 1H, J 1,8 Гц), 3,44 (s, 2H).
Получение 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)индолин-2-она (24)
6-Броминдолин-2он (27) (2,00 г, 9,40 ммоль), биспинаколатодибор (6,00 г, 23,60 ммоль), ацетат калия (2,76 г, 28,2 ммоль) и аддукт дихлор[1,1'-бис(дифенилфосфино)ферроцен]палладий(II)-дихлорметан (0,40 г, 0,55 ммоль) в DMSO (30 мл) перемешивали при 90°C в течение 18 часов. Реакционную смесь охлаждали до температуры окружающей среды, затем распределяли между водой и этилацетатом. Слои разделяли и водную фазу снова экстрагировали этилацетатом (2х). Объединенные органические слои промывали водой и солевым раствором и концентрировали с получением пурпурного твердого вещества. Неочищенное вещество предварительно абсорбировали на целите и хроматографировали (DCVC) с элюированием с применением градиента этилацетата в гептане (0-50% этилацетата). Подобные фракции объединяли и перекристаллизовывали из DCM и РЕ с получением 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)индолин-2-она (24) в виде бесцветного твердого вещества в 2 порциях (1,33 г, 55%); т.пл. 178,5-181,4°C. 1Н ЯМР (200 МГц, CDCl3) δ 8,61 (br s, 1Н), 7,46 (d, 1H, J 7,4 Гц), 7,30 (s, 1H), 7,21 (d, 1H, J 7,4 Гц), 3,53 (s, 2H), 1,32 (s, 12H).
Получение (Е)-3-(3-амино-3-оксопроп-1-ен-1-ил)-[1,1'-бифенил]-4-ил-трифторметансульфоната (23)
3-Формилбифенил-4-ил-трифторметансульфонат (14) (3,80 г, 11,50 ммоль) и диэтил(2-амино-2-оксоэтил)фосфонат (18) (2,25 г, 11,50 ммоль) растворяли в сухом THF (100 мл) и медленно добавляли к интенсивно перемешиваемой суспензии порошкообразного гидроксида натрия (0,92 г, 23,00 ммоль). После перемешивания в течение 1 часа при комнатной температуре реакционную смесь распределяли между солевым раствором и этилацетатом. Желтый побочный продукт удаляли фильтрацией и слои разделяли. Органический слой концентрировали, и затем очищали с помощью хроматографии (DCVC) с элюированием с применением градиента этилацетата в гептане (0-20% этилацетата), и затем перекристаллизовывали из DCM и РЕ с получением (E)-3-(3-амино-3-оксопроп-1-ен-1-ил)-[1,1'-бифенил]-4-ил-трифторметансульфоната (23) в виде бежевого твердого вещества (0,82 г, 19%); т.пл. 130,6-132,3°C. 1Н ЯМР (400 МГц, DMSO-d6) δ 8,09-8,04 (m, 1H), 7,88-7,82 (m, 1H), 7,79-7,73 (m, 2Н), 7,65-7,41 (m, 6Н), 7,33 (br s, 1Н), 6,93 (d, 1H, 3Jтранс 16 Гц). 13C ЯМР (100 МГц, DMSO-d6) δ 165,6, 146,4, 141,1, 138,0, 130,0, 129,5, 129,1, 128,6, 128,4, 127,7, 127,1, 126,4, 122,8, 118,1 (q, J 321 Гц). EIMS: масса/заряд обнаруженное: M+• 371,0420, для C16H12F3NO432S необходимо 371,0434. EIMS: масса/заряд 371 (М+•, 62%), 195 (100), 167 (100).
Получение (Е)-3-(4-(2-оксиндолин-6-ил)-[1,1'-бифенил]-3-ил)акриламида (25)
Получали в соответствии со способом Р5 из (E)-3-(3-амино-3-оксопроп-1-ен-1-ил)-[1,1'-бифенил]-4-ил-трифторметансульфоната (23) (0,50 г, 1,35 ммоль), 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)индолин-2-она (24) (0,43 г, 1,68 ммоль), тетракис(трифенилфосфин)палладия(0) (0,100 г, 0,09 ммоль) и водного раствора карбоната натрия (1 М) (3,0 мл, 3,00 ммоль) в толуоле (10 мл) и этаноле (2 мл). Неочищенное вещество собирали фильтрацией из продукта водной обработки, затем очищали растиранием в DCM и метаноле с получением (E)-3-(4-(2-оксиндолин-6-ил)-[1,1'-бифенил]-3-ил)акриламида (25) в виде бледно-лимонных игл (0,36 г, 75%); т.пл. 263-267°C (Dec.). 1Н ЯМР (400 МГц, DMSO-d6) δ 10,47 (s, 1Н), 7,95 (s, 1H), 7,80-7,70 (m, 3H), 7,57-7,37 (m, 5H), 7,46 (br s, 1H), 7,32 (d, 1H, J 7,6 Гц), 7,12 (br s, 1H), 6,89 (d, 1H, J 7,6 Гц), 6,80-6,72 (m, 2H), 3,55 (s, 2H). 13C ЯМР (100 МГц, DMSO-d6) δ 177,4, 166,5, 143,9, 141,1, 139,6, 139,4, 138,8, 137,4, 133,1, 130,9, 129,0, 127,8, 127,6, 126,8, 125,3, 124,4, 124,3, 123,7, 122,6, 110,1, 35,6. EIMS: масса/заряд обнаруженное: M+• 354,1356, для C23H18N2O2 необходимо 354,1363. EIMS: масса/заряд 354 (М+• 13%), 310 (100), 309 (43).
Получение 3-(4-(2-оксоиндолин-6-ил)-[1,1'-бифенил]-3-ил)пропанамида (Т25)
Получали в соответствии со способом, используемым для получения Т18, из (Е)-3-(4-(2-оксиндолин-6-ил)-[1,1'-бифенил]-3-ил)акриламида (25) (0,11 г, 0,30 ммоль) и 10% палладия на углероде (50 вес. % воды) в метаноле (30 мл). Фильтрат концентрировали с получением 3-(4-(2-оксиндолин-6-ил)-[1,1'-бифенил]-3-ил)пропанамида (Т25) в виде бледно-желтого твердого вещества (0,96 г, 89%); т.пл. 219-222°C. 1Н ЯМР (400 МГц, DMSO-d6) δ 10,44 (s, 1Н), 7,74-7,65 (m, 2H), 7,65-7,58 (m, 1H), 7,56-7,43 (m, 3Н), 7,42-7,34 (m, 1H), 7,31-7,19 (m, 3H), 6,94-6,87 (m, 1H), 6,80-6,71 (m, 2H), 3,53 (s, 2H), 2,84 (t, 2H, J 7,9 Гц), 2,31 (t, 2H, J 7,9 Гц). 13C ЯМР (50 МГц, DMSO-d6) δ 176,5, 173,4, 143,8, 140,6, 140,2, 140,0, 139,2, 139,1, 130,3, 128,9, 127,4, 127,3, 126,7, 124,6, 124,2 (два совпадающих сигнала), 121,9, 109,7, 36,2, 35,6, 28,2. EIMS: масса/заряд обнаруженное: М+• 356,1531, для C23H20N2O2N необходимо 356,1531. EIMS: масса/заряд 356 (М+•, 100%), 297 (70). ВЭЖХ чистота (35% ACN / 0,1% TFA, 256 нм): 97,57%.
Пример 20 - Синтез Т31
Синтетический путь, используемый для получения Т31, показан на фигуре 10. Вкратце, проводили реакцию перекрестного сочетания 3-формилбифенил-4-ил-трифторметансульфонат (14) с бензимидазолонпинаколовым эфиром бороновой кислоты (24) с получением бензимидазолона (28), который затем вводили в реакцию Хорнера-Уодсворта-Эммонса с диэтил(карбамоилметил)фосфонатом (18) с получением бензимидазолонакриламида (29). Последующая гидрогенизация соединения 29 приводила к образованию Т31.
Получение 4-(2-оксо-2,3-дигидро-1Н-бензо[d]имидазол-5-ил)-[1,1'-бифенил]-3-карбальдегида (28)
К суспензии 2-оксо-2,3-дигидро-1Н-бензоимидазол-5-пинаколового эфира бороновой кислоты (24) (574 мг, 2,2 ммоль), 3-формилбифенил-4-ил-трифторметансульфоната (14) (663 мг, 2,0 ммоль) и карбоната натрия (426 мг, 4,0 ммоль) в дегазированной смеси диоксан/этанол/H2O (5:1:1, 20 мл) добавляли тетракис(трифенилфосфин)палладий(0) (116 мг, 0,1 ммоль). Реакцию нагревали при 110°C в течение 2 часов в герметично закрытой пробирке. Анализ при помощи TLC (1:2 DCM : PE) указывал на то, что был израсходован трифлат. Реакционную смесь концентрировали до сухого состояния, затем переносили в равных объемах DCM и воды и интенсивно перемешивали в течение 20 минут, обеспечивая разбиение всех комков и получение мелкодисперсного осадка. Твердое вещество собирали фильтрацией с помощью отвержденной обеззоленной бумаги (540) на воронке Бюхнера и затем тщательно промывали DCM и водой. Твердое вещество высушивали под вакуумом при 40°C с получением 4-(2-оксо-2,3-дигидро-1Н-бензо[d]имидазол-5-ил)-[1,1'-бифенил]-3-карбальдегида (28) (365 мг, 58%) в виде бледно-желтого твердого вещества. 1Н ЯМР (200 МГц, DMSO-d6) δ 10,82 (brs, 2H), 9,95 (s, 1Н), 8,15-7,96 (m, 2Н), 7,76 (m, 2Н), 7,68-7,36 (m, 4Н), 7,12-6,95 (m, 3Н). 13С ЯМР (50 МГц, DMSO-d6) δ 192,1, 155,4, 144,6, 139,1, 138,7, 133,7, 131,8, 131,7, 130,1, 130,0, 129,2, 128,0, 126,7, 125,1, 122,9, 109,7, 108,4 (один сигнал не наблюдался). EIMS: масса/заряд обнаруженное: М+• 314,1050, для C20H14O2N2 необходимо 314,1055. EIMS: масса/заряд 314 (М+•, 100%).
Получение (Е/Z)-3-(4-(2-оксо-2,3-дигидро-1Н-бензо[d]имидазол-5-ил)-[1,1'-бифенил]-3-ил)акриламида (29)
4-(2-Оксо-2,3-дигидро-1Н-бензо[d]имидазол-5-ил)-[1,1'-бифенил]-3-карбальдегид (28) (350 мг, 1,1 ммоль) и диэтил(карбамоилметил)фосфонат (18) (217 мг, 1,1 ммоль) растворяли в сухом THF (15 мл) и медленно добавляли к интенсивно перемешиваемой суспензии порошкообразного КОН (125 мг, 2,2 ммоль) в THF (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа в атмосфере аргона. Анализ при помощи TLC (1:99 метанол : DCM) указывал на то, что карбальдегид был израсходован. THF удаляли при пониженном давлении и остаток переносили в равных объемах DCM и воды и интенсивно перемешивали в течение 30 минут, обеспечивая разбиение всех комков и получение мелкодисперсного осадка. Твердое вещество собирали фильтрацией с помощью отвержденной обеззоленной бумаги (540) на воронке Бюхнера и затем тщательно промывали DCM и водой. Твердое вещество высушивали под вакуумом при 40°C с получением E/2)-3-(4-(2-оксо-2,3-дигидро-1Н-бензо[d]имидазол-5-ил)-[1,1'-бифенил]-3-ил)акриламида (29) (247 мг, 56%) в виде желтого/коричневого твердого вещества. 1Н ЯМР (200 МГц, DMSO-d6) δ 10,74 (brs, 1Н), 10,70 (brs, 1H), 7,93 (d, 1H, J 1,8), 7,80-7,67 (m, 3H), 7,58-7,34 (m, 6H), 7,14-6,98 (m, 2H), 6,94-6,83 (m, 2H), 6,74 (d, 1H, J 15,8 Гц). 13C ЯМР (100 МГц, DMSO-d6) δ 166,6, 155,4, 141,5, 139,5, 139,1, 137,8, 133,1, 131,7, 131,2, 129,8, 129,3, 129,0, 127,7, 127,5, 126,7, 124,4, 123,4, 122,4, 109,5, 108,3. EIMS: масса/заряд обнаруженное: M+• 355,1315, для C22H17O2F3 необходимо 355,1315. EIMS: масса/заряд 355 (М+•, 31%).
Получение 3-(4-(2-оксо-2,3-дигидро-1Н-бензо[d]имидазол-5-ил)-[1,1'-бифенил]-3-ил)пропанамида (Т31)
(E/Z)-3-(4-(2-Оксо-2,3-дигидро-1H-бензол[d]имидазол-5-ил)-[1,1'-бифенил]-3-ил)акриламид (29) (240 мг, 0,7 ммоль) и 10% палладий на углероде (50 вес. % в H2O, 100 мг) в метаноле (20 мл) перемешивали при комнатной температуре в атмосфере водорода при 50 фунтах на кв. дюйм в течение 2 часов. Реакционную смесь подвергали гравитационной фильтрации через бумагу GF, тщательно промывая метанолом, затем концентрировали. Очищали препаративной ВЭЖХ (смесь 55% метанол/H2O, 70 мл/мин., 280 нм, 300×40 мм колонка Deltaprep C18) с получением 3-(4-(2-оксо-2,3-дигидро-1Н-бензо[d]имидазол-5-ил)-[1,1'-бифенил]-3-ил)пропанамида (Т31) (177 мг, 73%) в виде розового твердого вещества; т.пл. 250-251°C. 1Н ЯМР (400 МГц, DMSO-d6) δ 10,67 (d, 2Н, J 7,8 Гц), 7,69 (m, 2Н), 7,60 (d, 1H, J 1,9 Гц), 7,53-7,45 (m, 3Н), 7,37 (m, 1Н), 7,28-7,21 (m, 2Н), 6,99 (d, 1H, J 7,8 Гц), 6,90 (dd, 1H, J 1,6, 7,9 Гц), 6,89-6,86 (m, 1Н), 6,74 (brs, 1H), 2,84 (m, 2Н), 2,30 (m, 2Н). 13С ЯМР (100 МГц, DMSO-d6) δ 173,4, 155,4, 140,9, 140,0, 139,3, 138,8, 133,1, 130,6, 129,7, 128,9, 128,8, 127,3, 127,2, 126,6, 124,1, 121,3, 109,0, 108,1, 36,1, 28,3. EIMS: масса/заряд обнаруженное: М+• 357,1469, для C22H19N3O2 необходимо 357,1472. EIMS: масса/заряд 357 (М+•, 30%). ВЭЖХ чистота (40% ACN/H2O, 282 нм): 94,51%.
Пример 21 - Скрининг in vitro
Систему xCELLigence SP (Roche) использовали для измерения изменений клеточного импеданса (клеточного индекса) после обработки эмбриональных гладкомышечных клеток сосудов А10 (АТСС, CRL-1476) исследуемым соединением. Этот in vitro анализ согласовывали с данными артериального давления, полученными на животной модели, описанной ниже в примере 22, так чтобы его можно было использовать для более быстрого скрининга большего количества соединений. В этой клеточной экспериментальной системе in vitro отрицательный профиль импеданса коррелирует со снижением артериального давления у крыс - снижение импеданса связано с вазодилатацией и увеличение импеданса связано с вазоконстрикцией (Stallaert W., Dorn J.F., van der Westhuizen E., Audet M. & Bouvier M. Impedance responses reveal β-adrenergic signaling pluridensitometry and allow classification of ligands with distinct signalling profiles, PLoS ONE 2012; 7(1):e29420, doi:10.1371/journal.pone.0029420).
Вкратце, 50 мкл среды для культивирования клеток (DMEM с низким уровнем глюкозы с добавлением 10% фетальной бычьей сыворотки при 37°C) добавляли в каждую лунку E-Plate 96 (Roche) и измеряли фоновый импеданс в каждой лунке. Затем 50 мкл суспензии клеток А-10 (10000 клеток/лунка) добавляли в соответствующие лунки E-Plate 96. Клеточный индекс контролировали для каждой лунки E-Plate 96 в RTCA SP Station внутри инкубатора для клеточных культур. После инкубирования в течение ночи на протяжении 16-20 часов при 5% CO2 и влажности 95% 100 мкл исследуемого соединения (исследуемые соединения готовили в DMSO и разбавляли средой для культивирования клеток до конечной концентрации DMSO 0,25%) добавляли в соответствующие лунки E-Plate 96 и значения клеточного индекса измеряли сразу же после обработки соединением через каждые 20 секунд в течение 3 часов. Значение клеточного индекса корректировали относительно исходного уровня путем вычитания клеточного индекса обработанных носителем клеток и осуществляли нормализацию путем деления на клеточный индекс в момент времени непосредственно перед добавлением соединения. Нормализованный относительно исходного уровня клеточный индекс в зависимости от времени наносили на график с помощью программного обеспечения Roche RTCA.
С помощью соединений может достигаться снижение артериального давления при взаимодействии с гладкомышечными клетками сосудов, вызывая расслабление этих клеток, приводящее к вазодилатации и снижению артериального давления. Они называются прямыми вазодилататорами. Ответ с отрицательным импедансом для гладкомышечных клеток сосудов А10 указывает на то, что исследуемое соединение является прямым вазодилататором (фигура 11).
Систему xCELLigence SP (Roche) также использовали для измерения изменений клеточного импеданса (клеточного индекса) после обработки эндотелиальных клеток бычьей аорты (Европейская коллекция клеточных культур) исследуемым соединением. Используемый способ является тем же, что и для эмбриональных гладкомышечных клеток сосудов А10, описанных выше, но со средой для культивирования клеток, дополненной 15% фетальной бычьей сывороткой вместо 10%.
Соединения могут взаимодействовать с эндотелиальными клетками сосудов, вызывая высвобождение веществ, таких как оксид азота и эндотелиальный фактор гиперполяризации, которые, в свою очередь, действуют на гладкомышечные клетки сосудов, вызывая вазодилатацию и снижение артериального давления. Такие соединения называются непрямыми вазодилататорами. Ответ с отрицательным импедансом для эндотелиальных клеток аорты указывает на то, что исследуемое соединение является непрямым вазодилататором (фигура 12).
Пример 22 - Скрининг in vivo
Пероральные исследования
Четырнадцатинедельных SHR (на 2,2% солевой диете; Glen Forrest Stockfeeders) случайным образом распределяли в группы контроля в начальный момент времени, обработки исследуемым соединением (100 или 500 пкмоль/кг/мин) в питьевом растворе или в контрольном питьевым растворе (5% этанол в деионизированной дистиллированной воде (n=5 в каждой группе). Крыс, распределенных в контрольную группу начального момента времени, анестезировали и собирали их сердца и почки, в то время как крыс, распределенных для обработки контролем и исследуемым соединением, взвешивали дважды в неделю и контролировали их потребление питьевого раствора, чтобы обеспечить регулирование концентрации исследуемого соединения в питьевом растворе для поддержания постоянной дозы в течение 4-недельного периода исследования. Значение артериального давления измеряли дважды в неделю при помощи плетизмографии с использованием хвостовой манжеты (PowerLab, ADInstruments, Касл Хилл, Новый Южный Уэльс, Австралия). Через 4 недели крыс анестезировали и собирали их сердца и почки для количественной оценки фиброза.
Количественная оценка фиброза
Для количественной оценки фиброзатканей тканевые срезы толщиной ≤3 мм фиксировали в 10% забуференном формалине в течение 24 часов, обрабатывали и заливали в парафин. Поперечные срезы толщиной три микрометра окрашивали с использованием трехцветной окраски по Массону. Минимум 20 случайных полей при х20 увеличении с поперечных срезов (5 на каждом из 2 уровней) оцифровывали и определяли степень фиброза как процент от площади поля каждого оцифрованного изображения с использованием Image-Pro Plus V. 7 (Media Cybernetics, Бетезда, Мэриленд, США), затем усредняли для определения уровня фиброза ткани для каждой крысы.
Результаты
Результаты среднего значения систолического артериального давления, наблюдаемые для животных на 2,2% солевой диете через 4 недели пероральной обработки 100 или 500 пкмоль/кг/мин T1, Т2, Т20, Т31, Т48 или Т70, показывали сниженное значение артериального давления по сравнению с контролем (фигура 13). Среднее диастолическое артериальное давление также снижалось по сравнению с контролем для T1, Т2, Т31 и Т70.
Результаты среднего систолического артериального давления для T1, Т2, Т20, Т31, Т48 и Т70 сравнивания с нормализованными относительно исходного уровня клеточными индексами соединений в отношении гладкомышечных клеток сосудов А10 (фигура 14) и эндотелиальных клеток бычьей аорты (фигура 15) и показали связь между результатами in vivo и in vitro.
Фиброз в сердце через 4 недели пероральной обработки 500 пкмоль/кг/мин. T1, Т2, Т20, Т31, Т48 или Т70 у 18-недельных SHR на 2,2% солевой диете снижался по сравнению с контролем (фигура 16).
Фиброз в почке через 4 недели пероральной обработки 500 пкмоль/кг/мин. T1, Т2, Т20, Т31, Т48 или Т70 у 18-недельных SHR на 2,2% солевой диете снижался по сравнению с контролем (фигура 17).
Нормализованные относительно исходного уровня клеточные индексы T1, Т2, Т20, Т31, Т48 и Т70 в отношении эндотелиальных клеток бычьей аорты сравнивали с результатами фиброза миокарда (фигура 18) и фиброза почки (фигура 19) для соединений и показали связь между результатами in vivo и in vitro.
Гистологические срезы из сердец (фигура 20) контрольных крыс (А) или крыс, обработанных в течение четырех недель 500 пкмоль/кг/мин Т1 (В), Т2 (С), Т20 (D) или Т31 (е) на 2,2% солевой диете, показали, что контроль характеризуется обширным фиброзом (см. стрелки), заметным в виде полосы светло-серого цвета в правой нижней четверти, продолжающейся по диагонали вверх и окружающей снаружи большой кровеносный сосуд, а также несколько мышечных волокон, при этом меньшие количества присутствуют на всей микрофотографии (мышечные волокна заметны в виде отдельных более темных серых областей). В срезах от обработанных T1, Т2, Т20 и Т31 крыс отчетливые области фиброза не присутствовали, мышечные волокна заметны на поперечном срезе в виде различных теней темно-серого цвета.
Гистологические срезы из почек (фигура 21) контрольных крыс (А) или крыс, обработанных в течение четырех недель 500 пкмоль/кг/мин Т1 (В), Т2 (С), Т20 (D) или Т31 (е) на 2,2% солевой диете, показали что, контроль характеризуется обширным фиброзом, заметным в виде толстых более светлых серых полос, полностью окружающих все канальцы, при этом в центре 2 канальца были облитерированы (стрелки). В срезах от крыс, обработанных T1, Т2, Т20 и Т31, фиброз был уменьшен до тонких полос, не полностью окружающих некоторые, но не все канальцы.
| название | год | авторы | номер документа |
|---|---|---|---|
| Композиции для лечения заболевания почек и/или печени | 2016 |
|
RU2712624C2 |
| Композиции для лечения фиброза и связанных с фиброзом состояний | 2016 |
|
RU2712140C2 |
| Композиции для лечения легочного фиброза | 2017 |
|
RU2747801C2 |
| КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ГИПЕРТЕНЗИИ И/ИЛИ ФИБРОЗА | 2014 |
|
RU2635564C2 |
| Композиции для лечения гипертензии и/или фиброза | 2017 |
|
RU2752088C1 |
| ХИМИЧЕСКИЕ СОЕДИНЕНИЯ 637: ПИРИДОПИРИМИДИНДИОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE4 | 2008 |
|
RU2479584C2 |
| 1-(2-ИЗОПРОПОКСИЭТИЛ)-2-ТИОКСО-1,2,3,5-ТЕТРАГИДРО-ПИРРОЛО[3,2-d]ПИРИМИДИН-4-ОН И ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 2005 |
|
RU2409578C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРРОЛО[3,2-d]ПИРИМИДИН-4-ОНА И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2005 |
|
RU2577858C2 |
| Новые 3-индол замещенные производные, фармацевтические композиции и способы применения | 2016 |
|
RU2672252C1 |
| ИНГИБИТОРЫ ФУРИНА | 2019 |
|
RU2799824C2 |
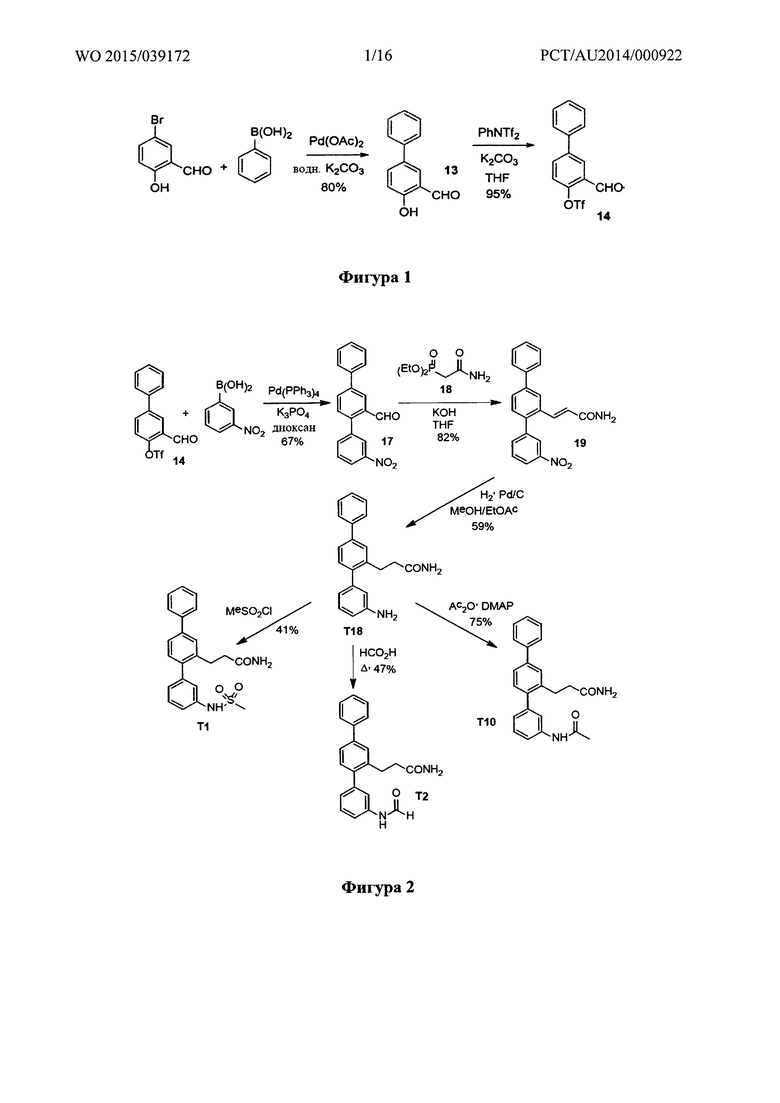
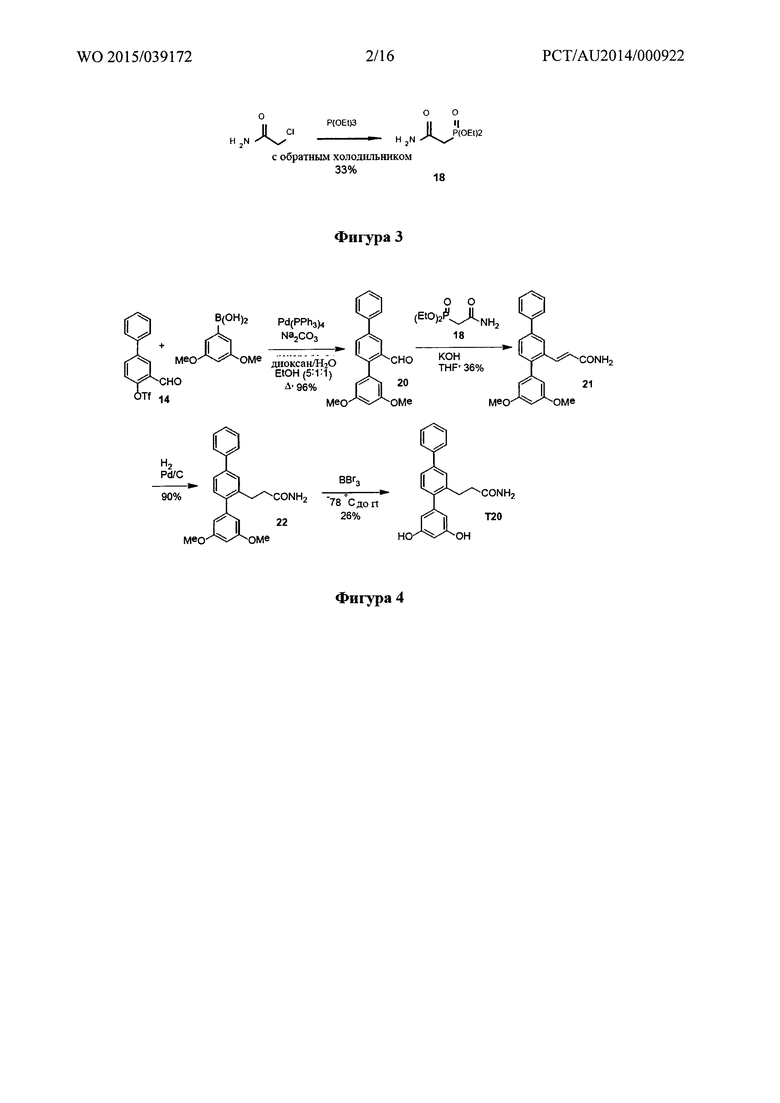
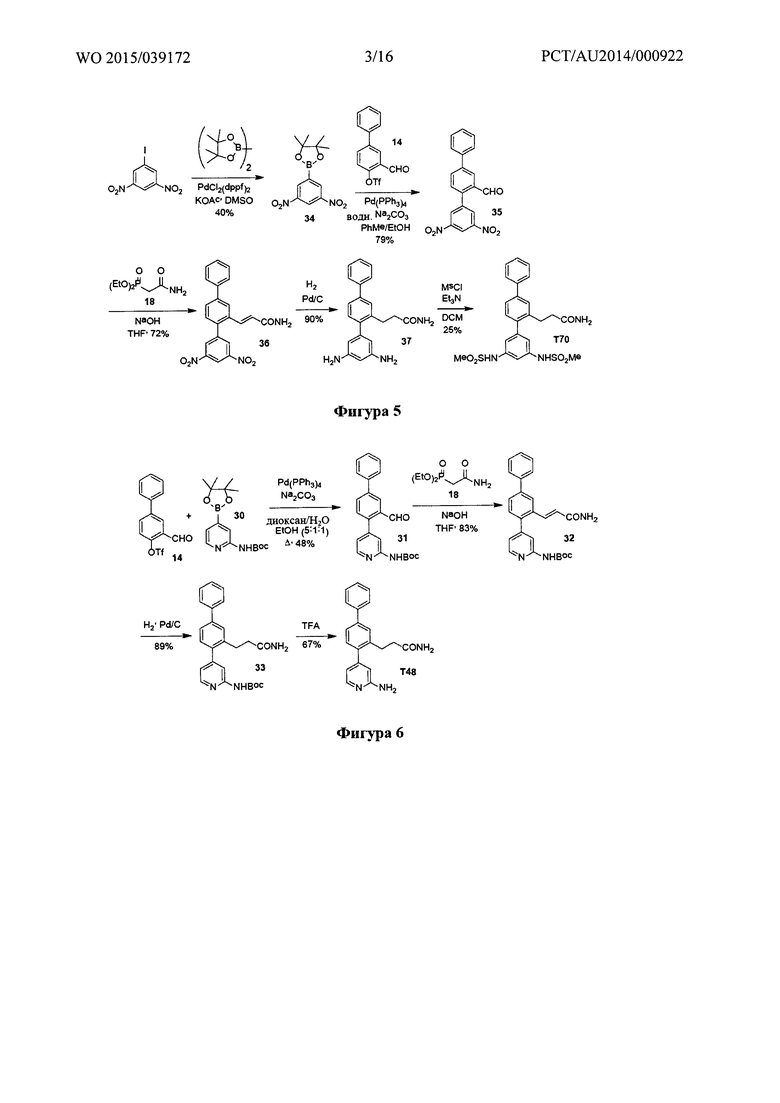
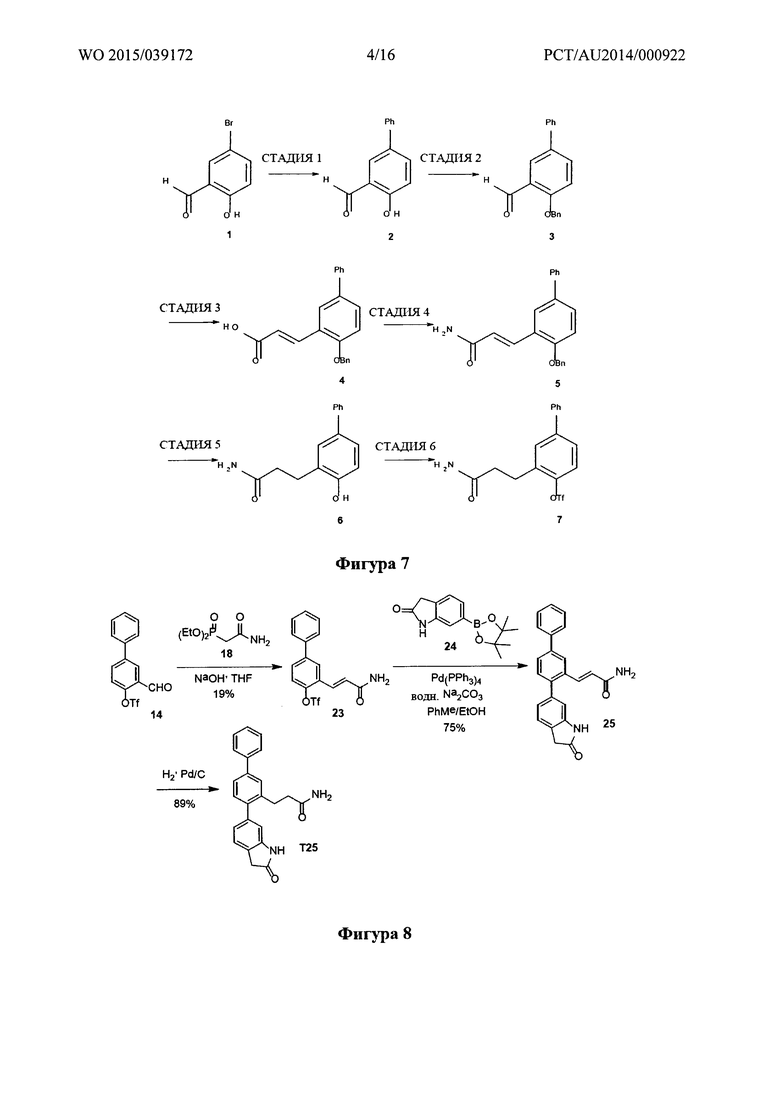

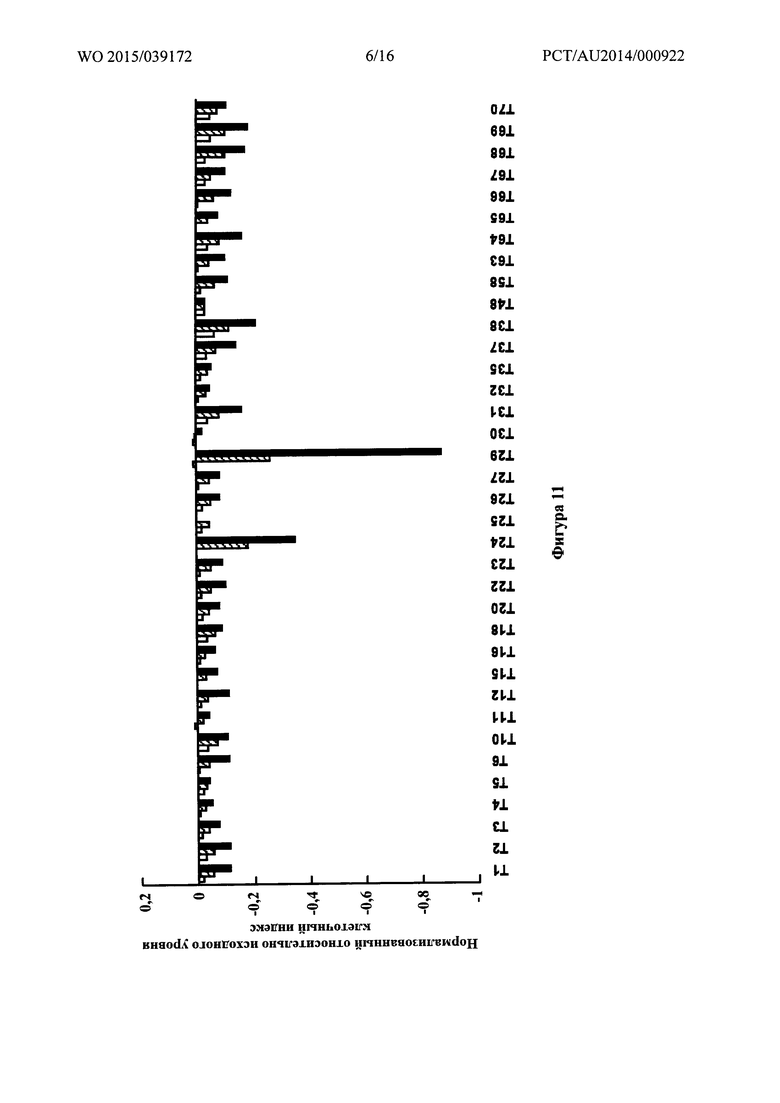
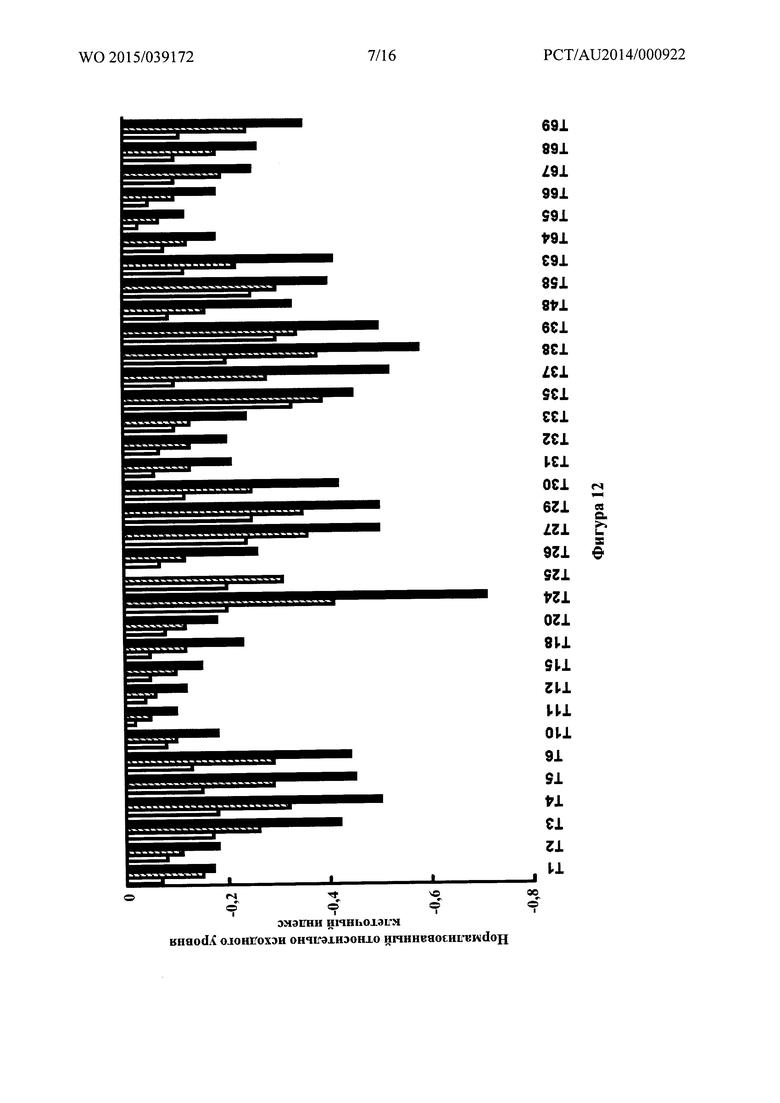
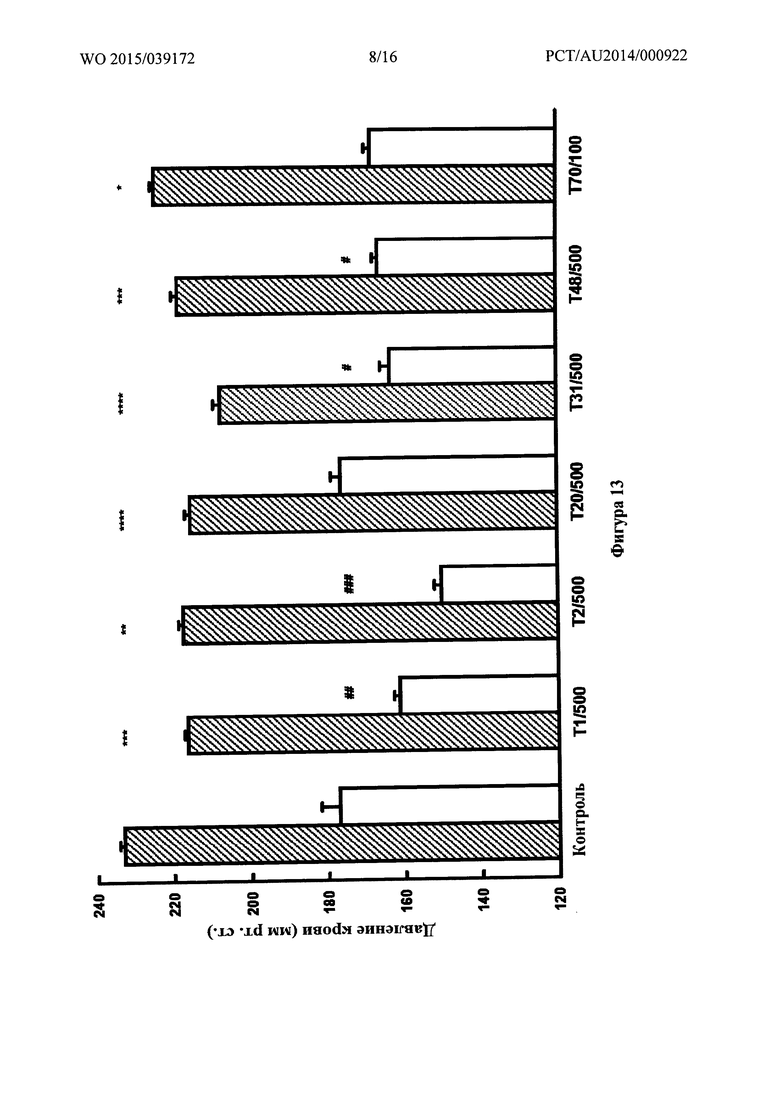
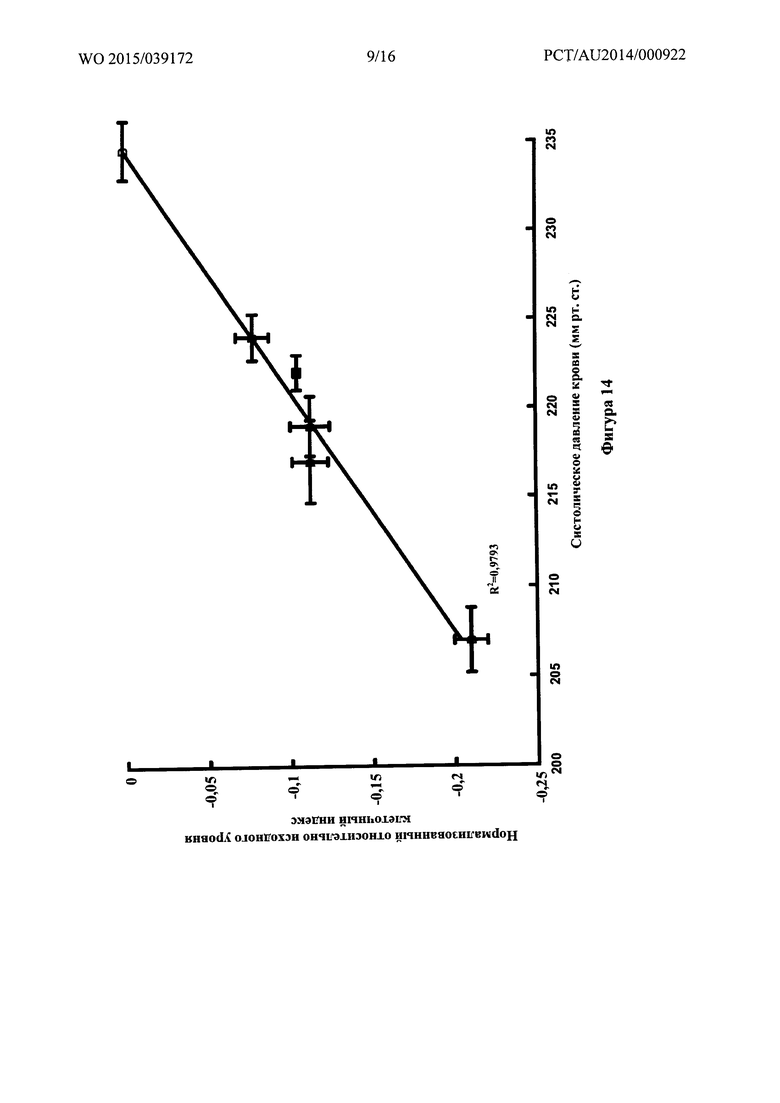
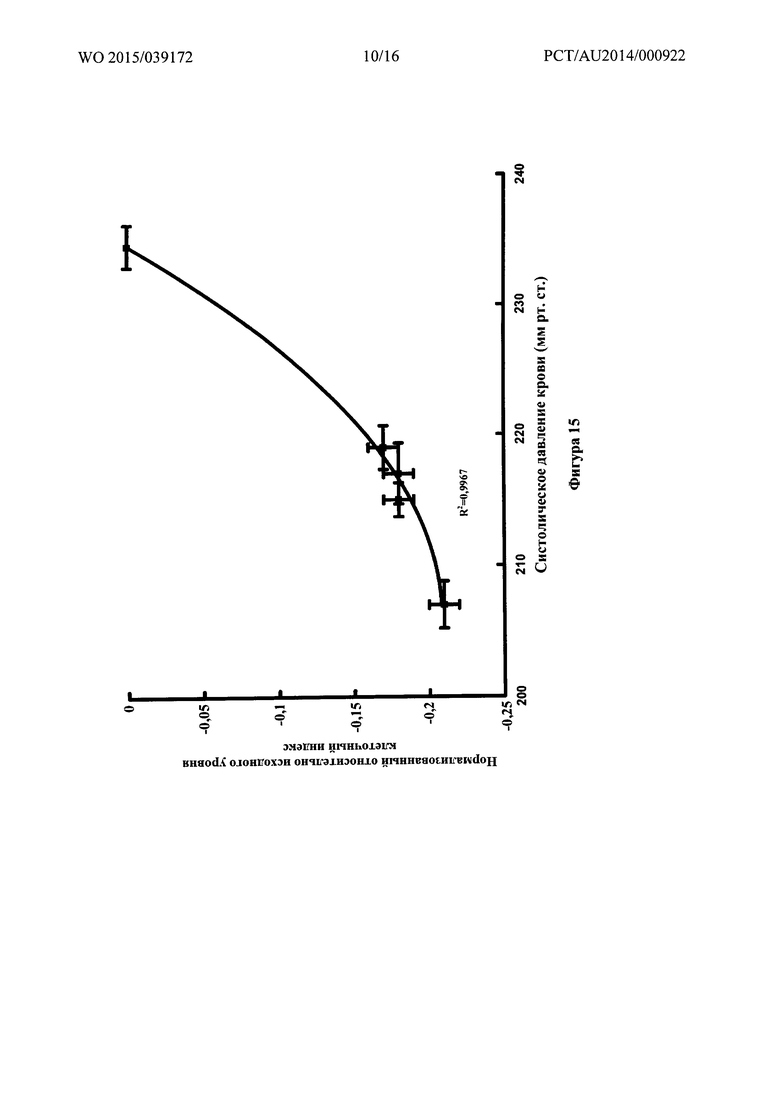
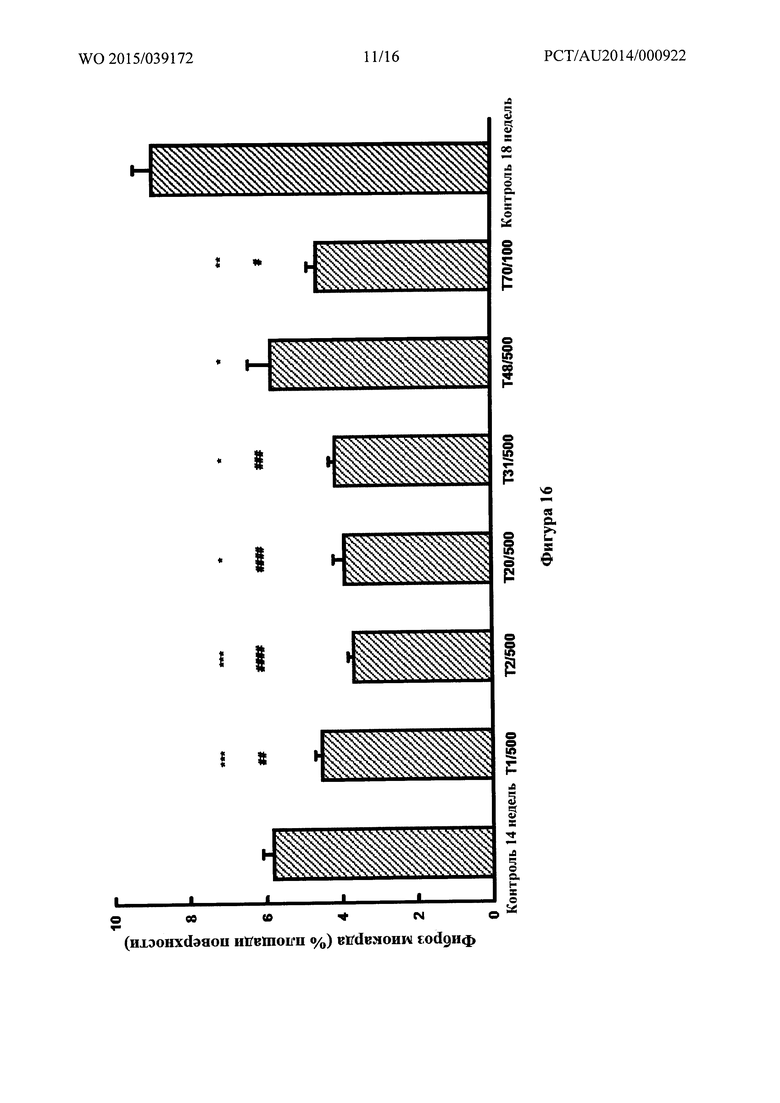
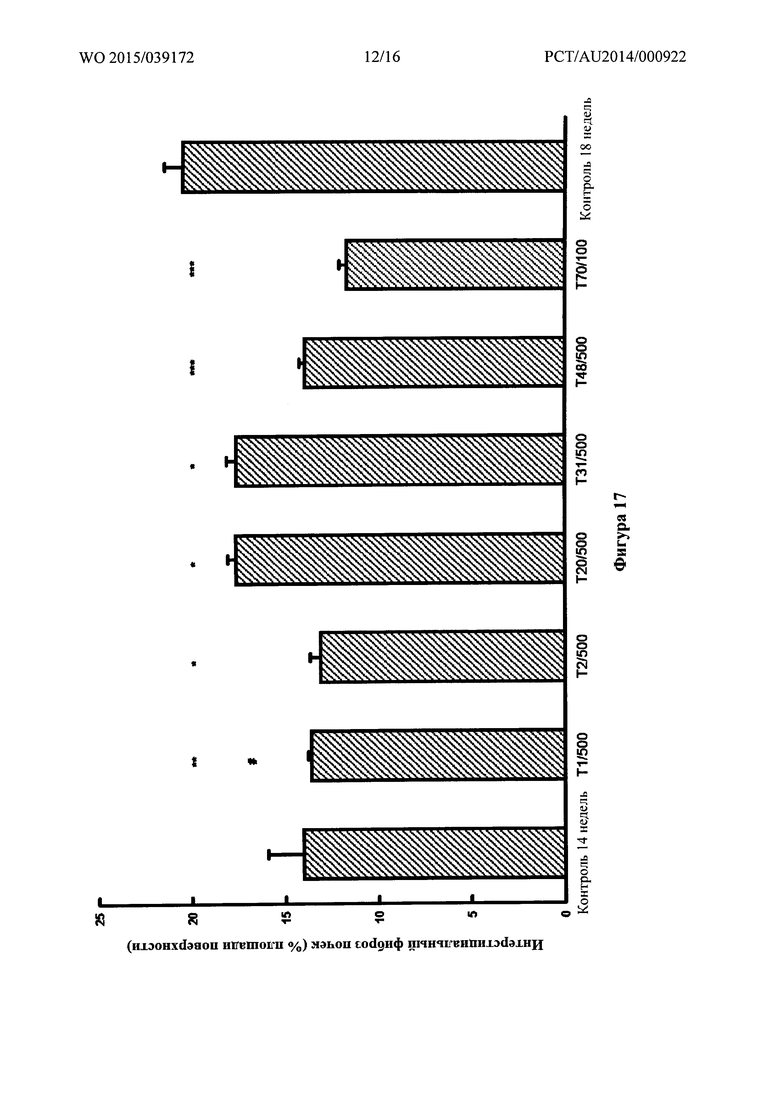
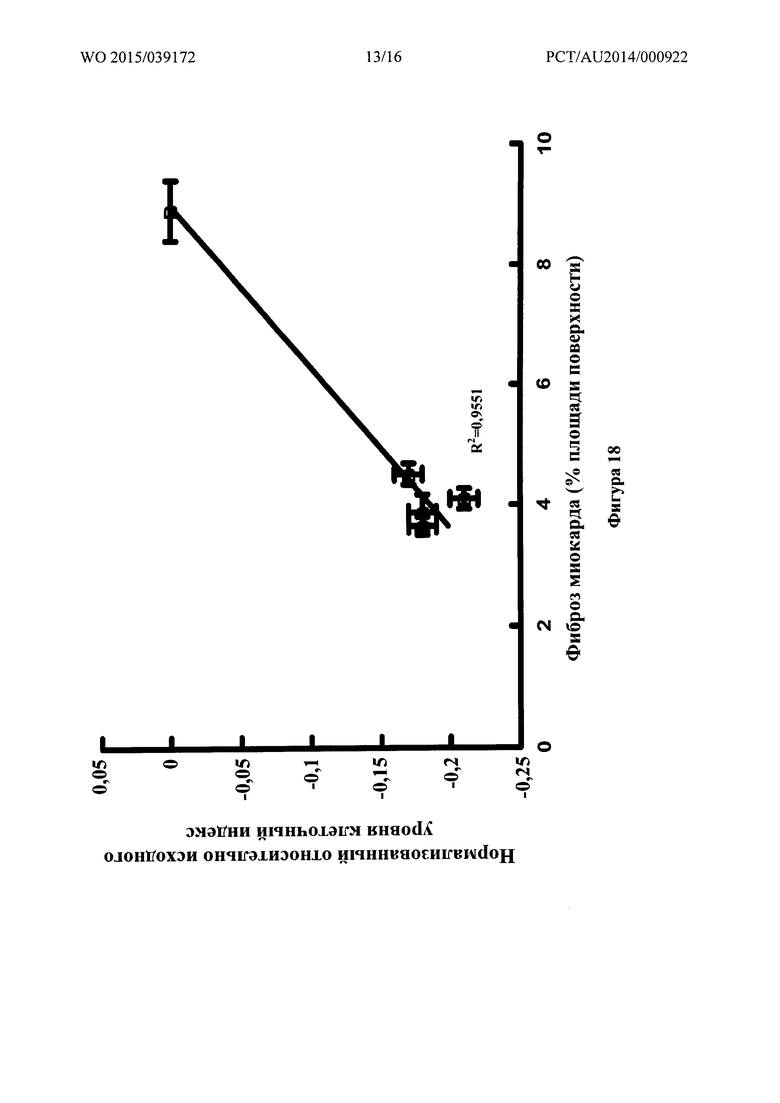
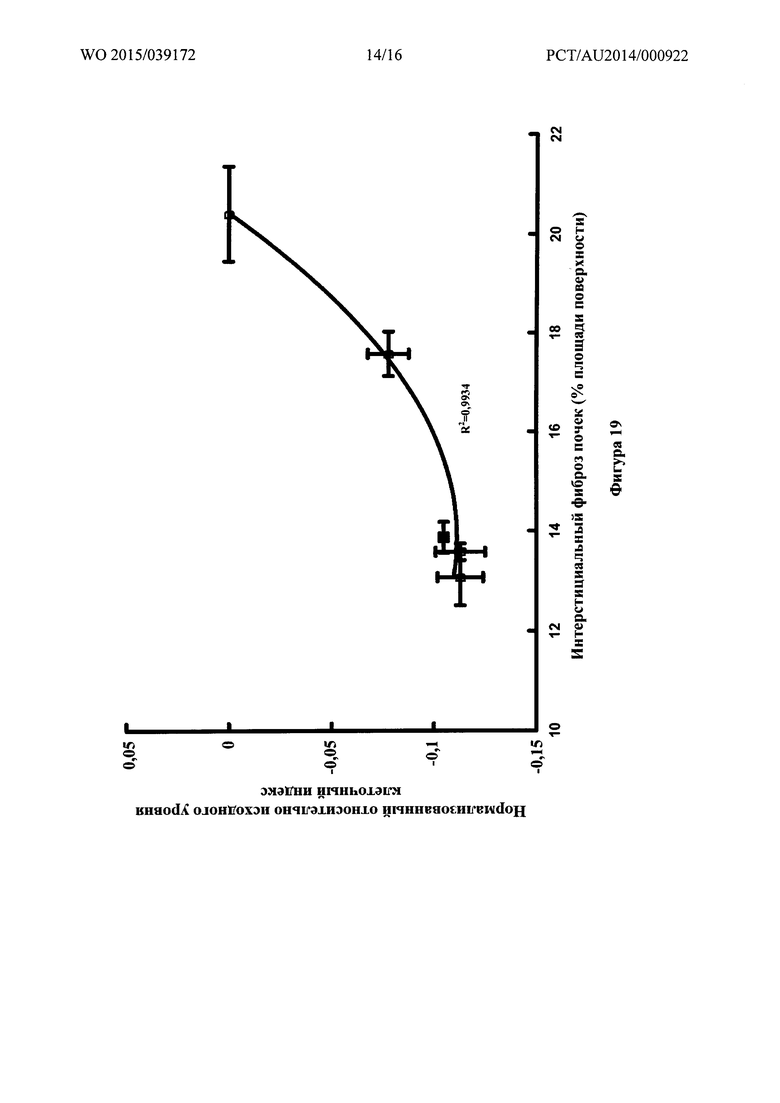
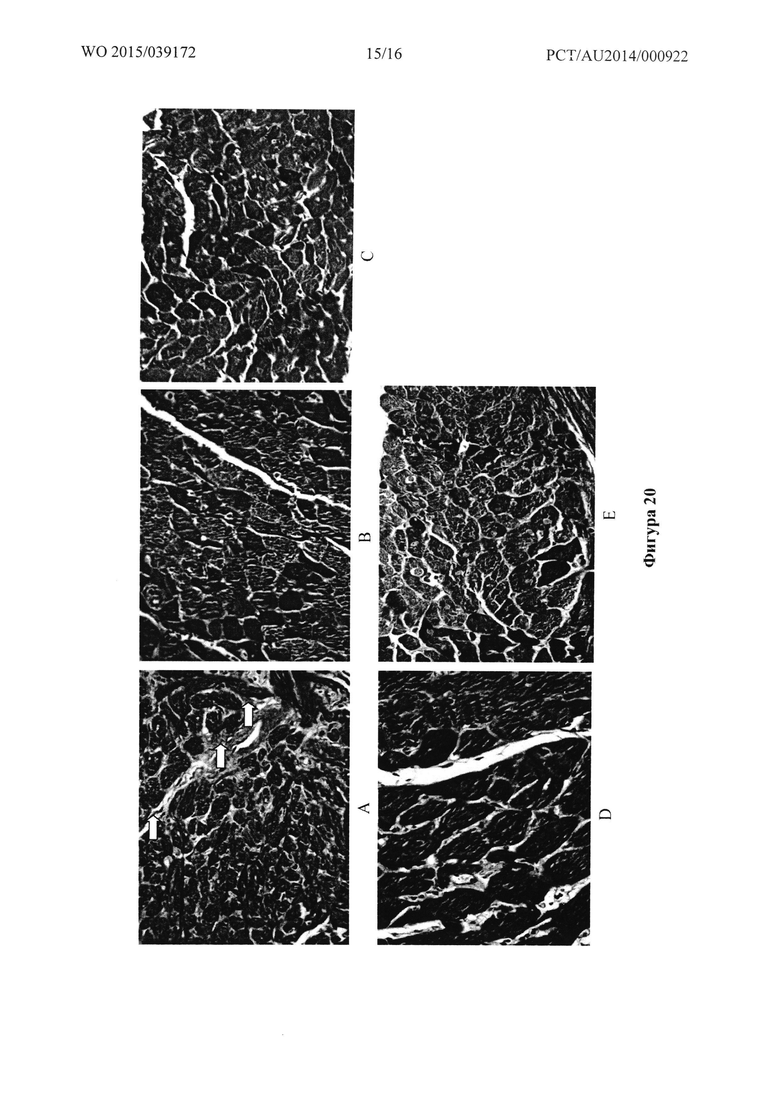
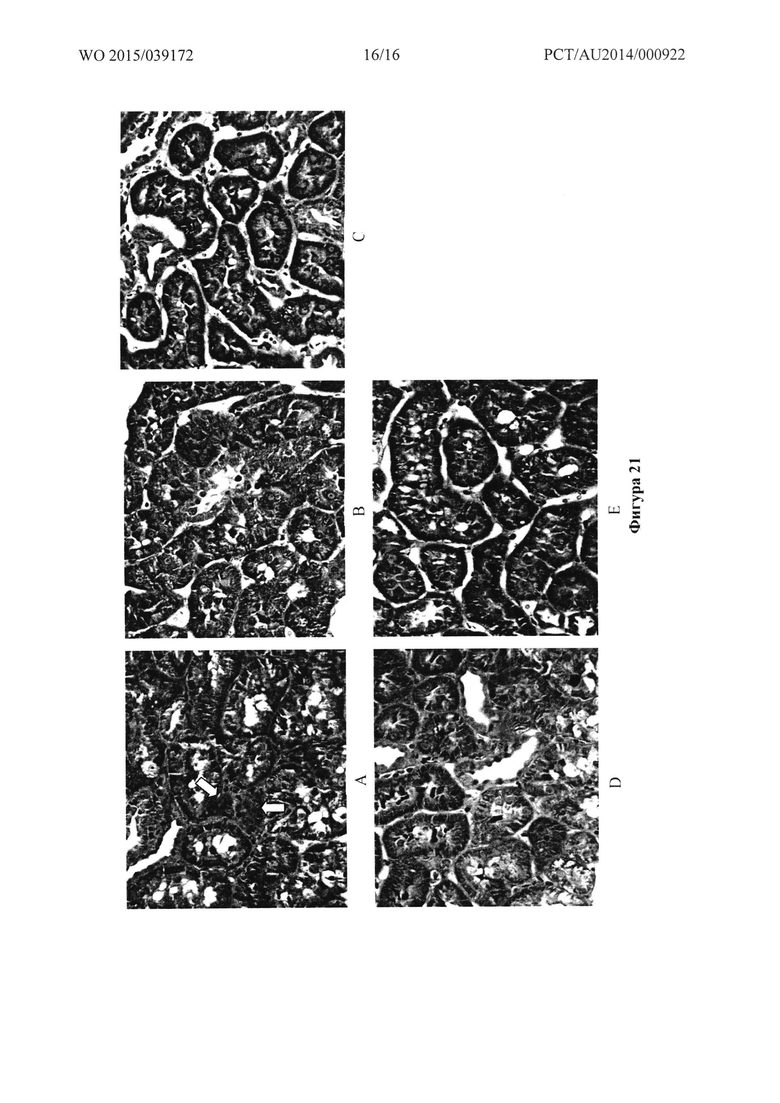
Изобретение относится к соединению формулы (1) или его фармацевтически приемлемой соли, где А выбран из группы, состоящей из фенила или пиридина, замещенных Qn, или конденсированных кольцевых структур, приведенных ниже; Q независимо выбран из галогена, алкила, гидрокси, амино и замещенного амино; n равняется 0, 1, 2, 3, 4 или 5; R1, R3 и R4 независимо представляют собой С, СН, CH2, О, N, NH или S, и R2 представляет собой С, СН, СН2, N, NH, C-CF3, CH-CF3 или С=O; причем, если n равняется 1, Q не может представлять собой гидрокси; замещенный амино имеет формулу -NHW, где W выбран из -CN, -SO2(X)aY и -CO(X)aY, а равняется 0 или 1, X выбран из -NH- и -О-, и Y выбран из -Н, -СН3, -СН2СН3, -СН2ОН и -СН2СН2ОН, и где связи между R1 и R2, R2 и R3, R3 и R4 независимо представляют собой двойные или одинарные связи. Также изобретение относится к фармацевтической композиции, обладающей эффектами, снижающими артериальное давление, и/или противофиброзными эффектами, содержащей эффективное количество соединения или его фармацевтически приемлемой соли формулы (1) и фармацевтически приемлемое вспомогательное средство. Соединения по изобретению предназначены для лечения гипертензии, предгипертензии и фиброза. 11 н. и 7 з.п. ф-лы, 22 пр., 21 ил.
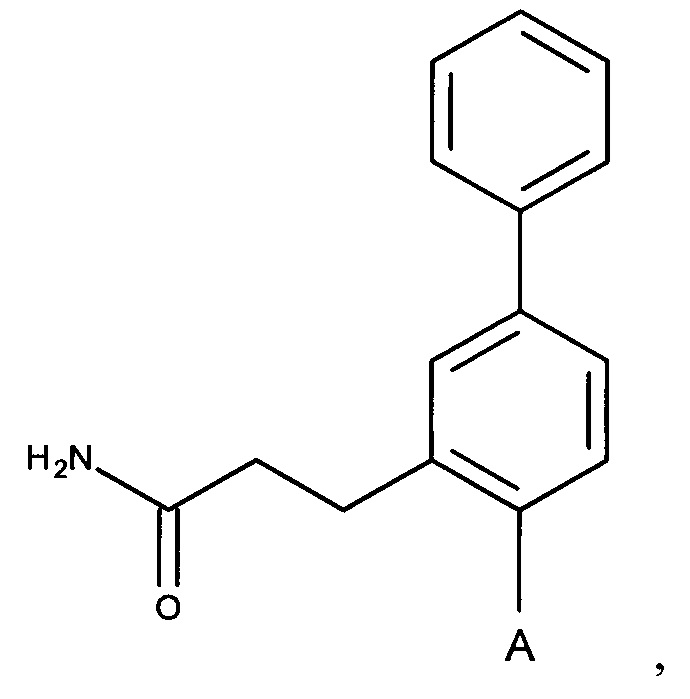
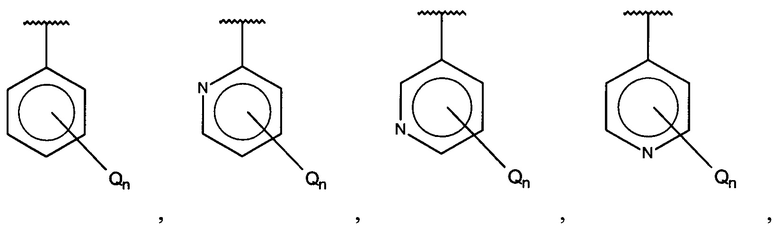
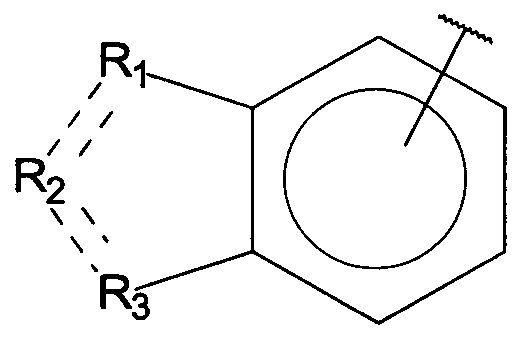 ,
, 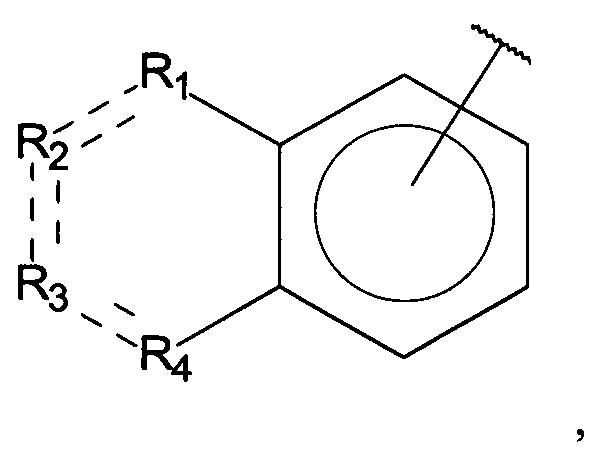
1. Соединение формулы:
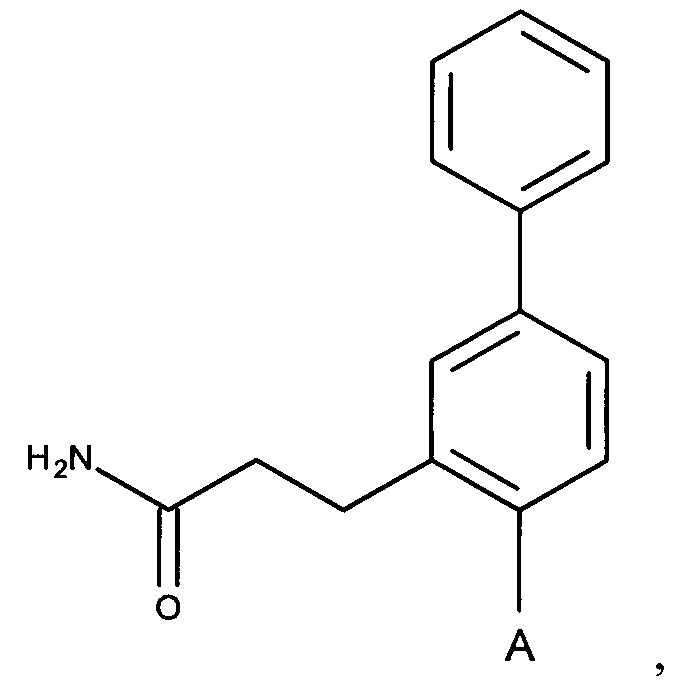
где
А выбран из группы, состоящей из
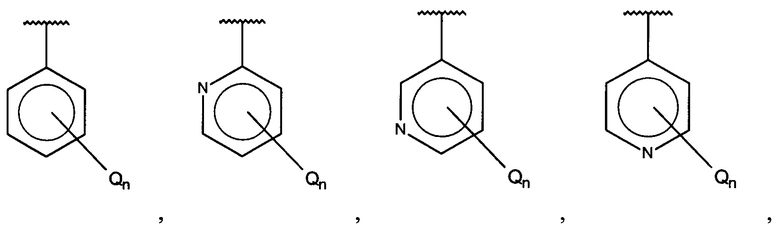
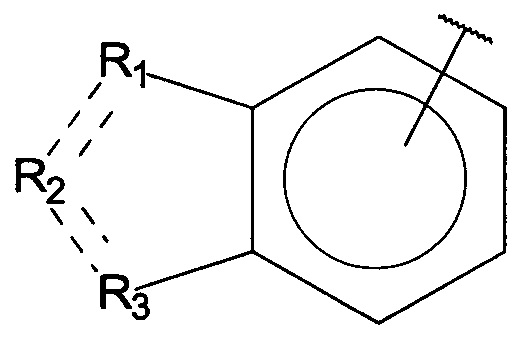 и
и 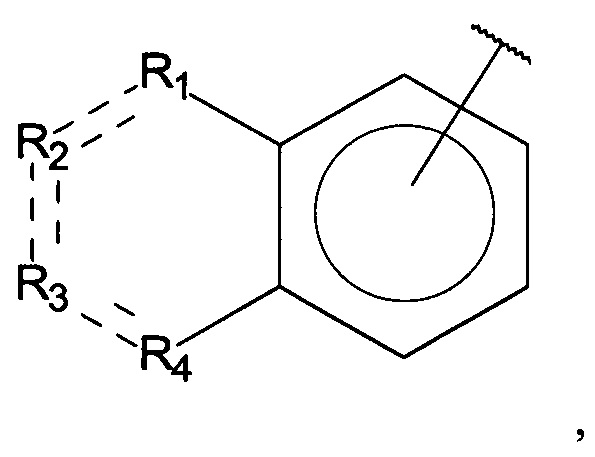
Q независимо выбран из галогена, алкила, гидрокси, амино и замещенного амино;
n равняется 0, 1, 2, 3, 4 или 5;
R1, R3 и R4 независимо представляют собой С, СН, CH2, О, N, NH или S, и R2 представляет собой С, СН, СН2, N, NH, C-CF3, CH-CF3 или С=O, или его фармацевтически приемлемая соль,
где, если n равняется 1, Q не может представлять собой гидрокси,
где замещенный амино имеет формулу -NHW, и где:
W выбран из -CN, -SO2(X)aY и -CO(X)aY,
а равняется 0 или 1,
X выбран из -NH- и -О-, и
Y выбран из -Н, -СН3, -СН2СН3, -СН2ОН и -СН2СН2ОН, и
где связи между R1 и R2, R2 и R3, R3 и R4 независимо представляют собой двойные или одинарные связи.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где Q представляет собой галоген, выбранный из группы, состоящей из F, Cl, Br и I.
3. Соединение или его фармацевтически приемлемая соль по п. 1, где Q представляет собой замещенный амино, выбранный из группы, состоящей из -NHSO2CH3, -NHCOH, -NHCONHCH3, -NHCONHCH2CH3, -NHSO2NHCH3, -NHSO2NHCH2CH3, -NHCOCH3, -NHCOOCH3, -NHCOOCH2CH2OH, -NHCONH2 и -NHCN.
4. Соединение или его фармацевтически приемлемая соль по п. 1, где Q представляет собой алкил, выбранный из группы, состоящей из метила, этила, пропила, бутила и пентила.
5. Соединение или его фармацевтически приемлемая соль по п. 1, где А выбран из:
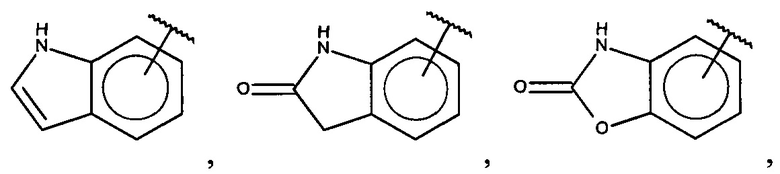
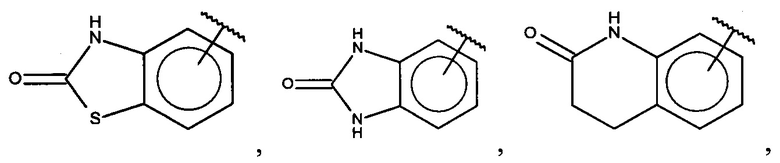
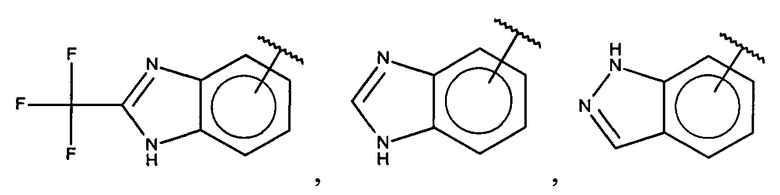
и 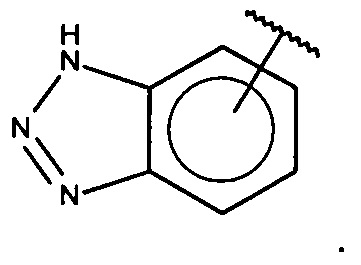
6. Соединение или его фармацевтически приемлемая соль по п. 1, где соединение выбрано из группы, состоящей из:
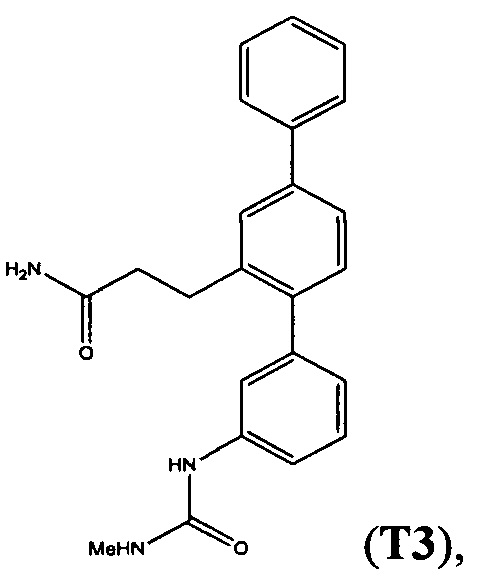
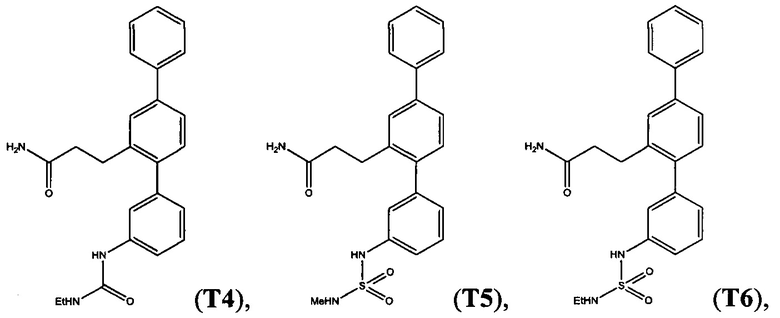
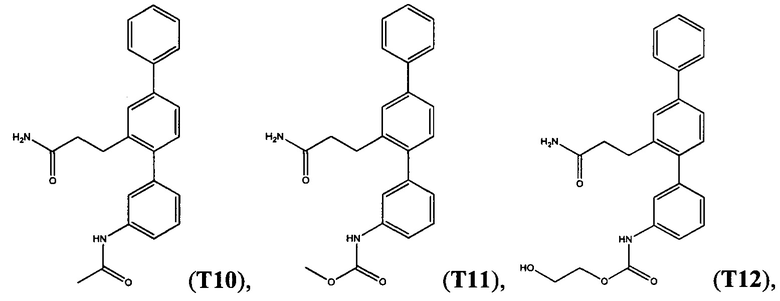
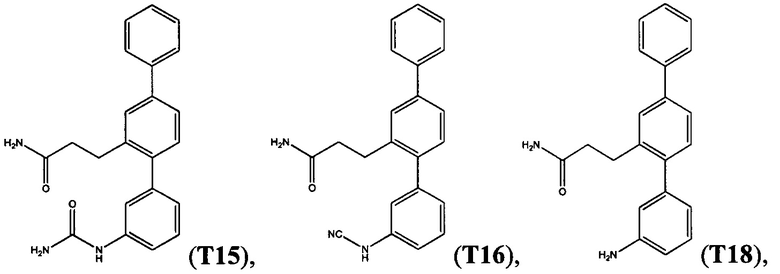
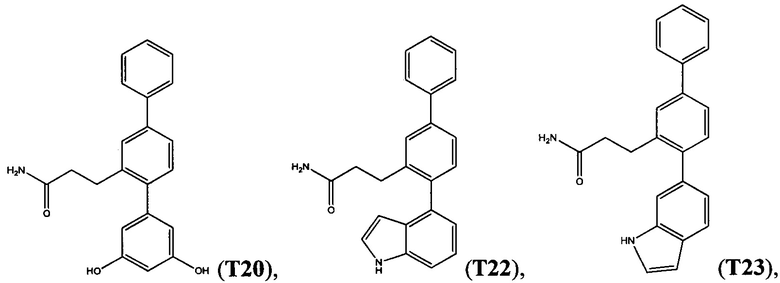
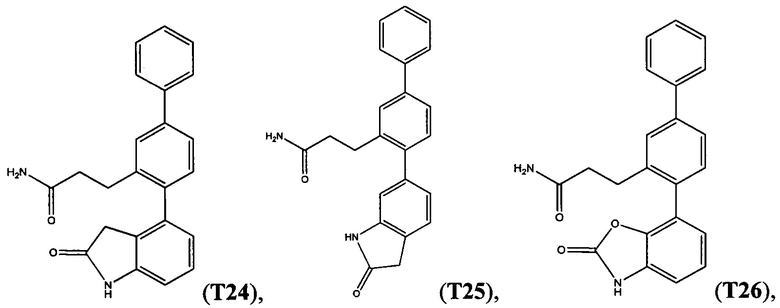
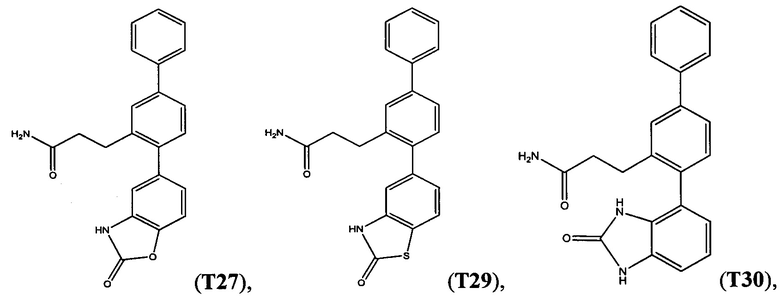
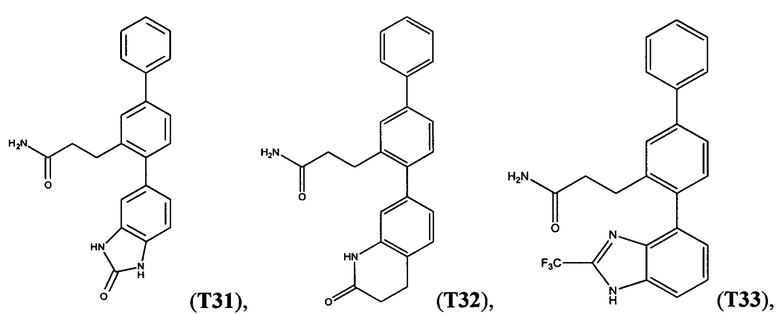
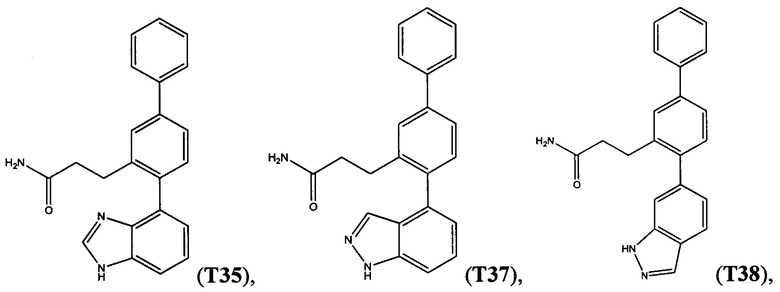
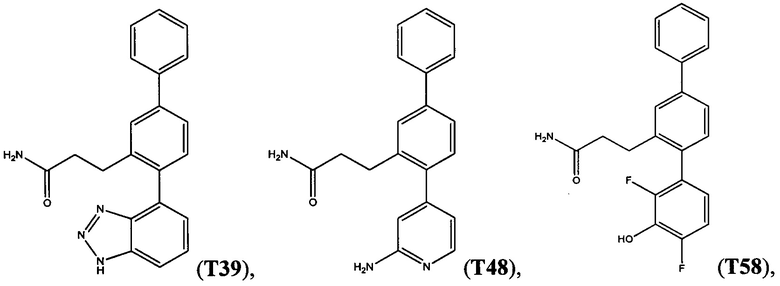
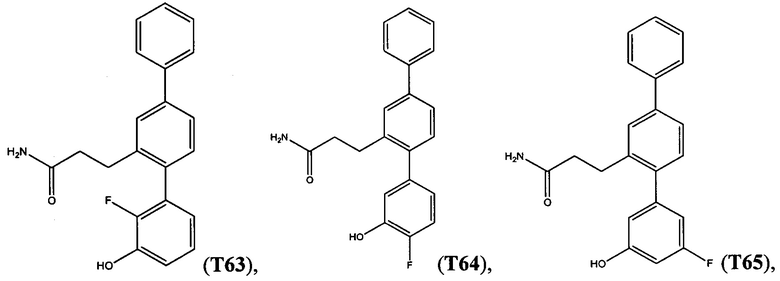
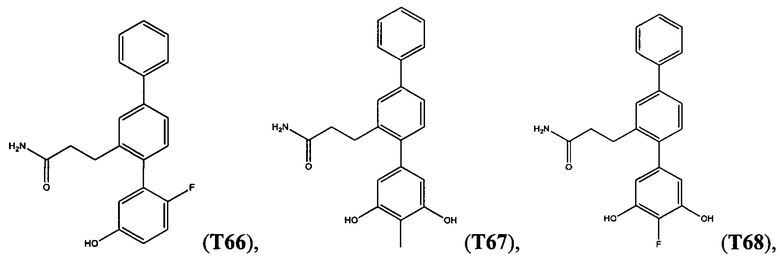
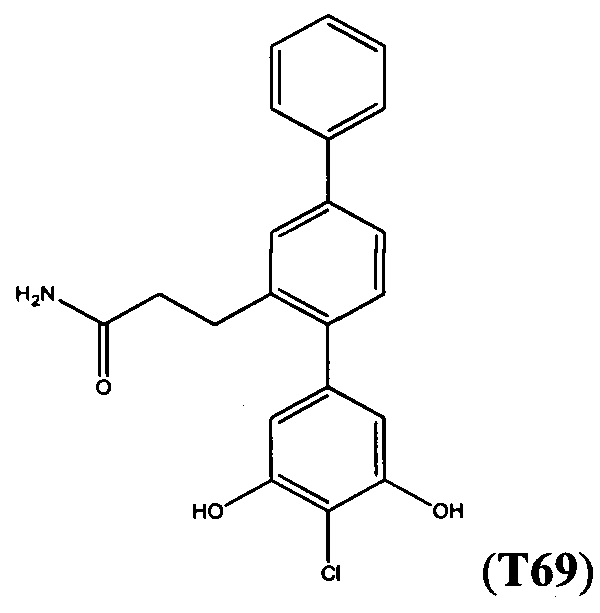 и
и 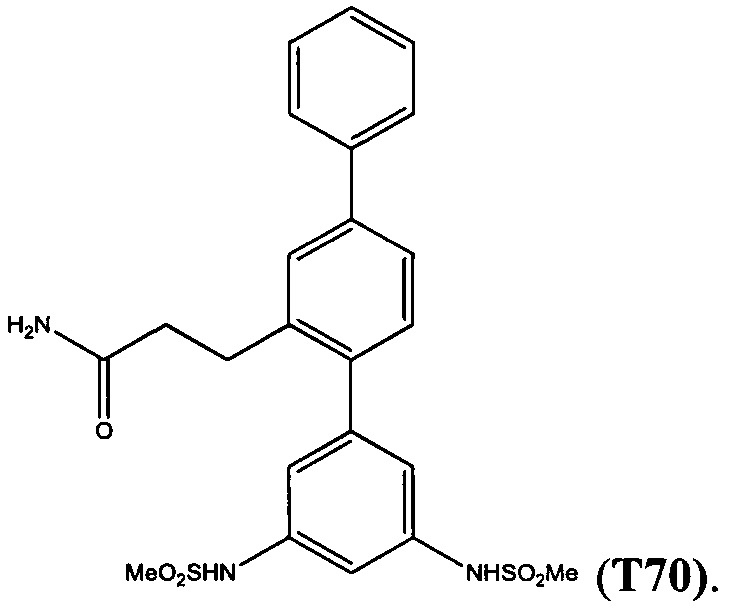
7. Фармацевтическая композиция, обладающая эффектами, снижающими артериальное давление, и/или противофиброзными эффектами, содержащая эффективное количество соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 и фармацевтически приемлемое вспомогательное средство.
8. Способ терапевтического лечения гипертензии или предгипертензии у субъекта, включающий введение субъекту соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 или композиции по п. 7.
9. Способ профилактического лечения фиброза у субъекта, включающий введение субъекту соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 или композиции по п. 7.
10. Способ терапевтического лечения фиброза у субъекта, включающий введение субъекту соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 или композиции по п. 7.
11. Способ терапевтического лечения гипертензии и фиброза у субъекта, включающий введение субъекту соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 или композиции по п. 7.
12. Способ лечения предгипертензии и фиброза у субъекта, включающий введение субъекту соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 или композиции по п. 7.
13. Способ по любому из пп. 9-12, где фиброз представляет собой фиброз миокарда или фиброз почки.
14. Способ по любому из пп. 9-12, где фиброз представляет собой фиброз миокарда и фиброз почки.
15. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 для изготовления лекарственного препарата для терапевтического лечения гипертензии или предгипертензии.
16. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 для изготовления лекарственного препарата для профилактического лечения фиброза.
17. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 для изготовления лекарственного препарата для терапевтического лечения фиброза.
18. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 для изготовления лекарственного препарата для терапевтического лечения гипертензии и фиброза или предгипертензии и фиброза.
| PETERS M | |||
| et al.: "A Modular synthesis of teraryl-based α-Helix mimetics, Part 1: Synthesis of core fragments with two electronically differentiated leaving groups", CHEMISTRY-A EUROPEAN JOURNAL, 2013, vol.19, pp.2442-2449 | |||
| HUANG, W.-J | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Приспособление для суммирования отрезков прямых линий | 1923 |
|
SU2010A1 |
| ЗАМЕЩЕННЫЕ ФЕНОКСИУКСУСНЫЕ КИСЛОТЫ, ОБЛАДАЮЩИЕ МОДУЛИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ РЕЦЕПТОРОВ CRTh2 | 2004 |
|
RU2372330C2 |

