ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым производным 3-(индол-3-ил)-пиридина, включая их фармацевтически приемлемые энантиомеры, соли и сольваты. Соединения по изобретению являются ингибиторами TDO2 (триптофан-2,3-диоксигеназы) и полезны в качестве терапевтических соединений, в частности в лечении и/или предупреждении рака.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Два десятилетия после того как была открыта важность катаболизма триптофана для поддержания иммунных привилегий плаценты (Munn, D.H. et al., Science, 1998, 281, 1191-1193), возрастающие свидетельства расширяют свою биологическую релевантность за пределы иммунной толерантности к чужому. Согласно общепринятой концепции, триптофан, являющийся незаменимой аминокислотой, подвергается катаболизму в местном микроокружении опухолей, иммунопривилегированных сайтах или сайтах воспаления (Mellor AL and Munn DH., Nat Rev Immunol, 2008, 8, 74-80). В этих тканях, раковые клетки, иммунные клетки или специализированные эпителиальные клетки (например синцитиотрофобласты в плаценте) создают иммуносупрессивное окружение в опухолях, которое блокирует противоопухолевые иммунные ответы в опухолях и дренирующих опухоль лимфоузлах путем индуцирования Т-клеточной анергии и апоптоза через истощение триптофана и накопление иммуносупрессивных катаболитов триптофана (Munn DH et al., J Exp Med., 1999, 189, 1363-1372; Fallarino F et al., Cell Death Differ., 2002, 9, 1069-1077).
Недавно обнаружено, что ключевой фермент в катаболизме триптофана, триптофан-2,3-диоксигеназа (TDO2), который считается ответственным за регулирование системных уровней триптофана в печени, конститутивно экспрессируется в широком ряде злокачественных новообразований, таких как, например, карцинома мочевого пузыря, гепатокарцинома, меланома, мезотелиома, нейробластома, саркома, карцинома молочной железы, лейкоз, почечно-клеточная карцинома, колоректальная карцинома, карцинома головы и шеи, карцинома легких, опухоль мозга, глиобластома, астроцитома, миелома и панкреатическая карцинома (Pilotte L et al., Proc NatI Acad Sci U S A, 2012, 109(7), 2497-502). Экспрессия TDO2 в опухолевых клетках предупреждает опухолевый надзор иммунной системой и тем самым предупреждает отторжение опухоли местно разлагающимся триптофаном (Opitz CA et al., Nature, 2011, 478(7368), 197-203). Первое доказательство этого было предложено посредством ингибирования TDO2 небольшой молекулой, которая ингибировала рост опухоли в опухолевой модели мастоцитомы Р815, с подходом профилактической вакцинации (Pilotte L et al., Proc NatI Acad Sci U S А, 2012, 109(7), 2497-502). Опухоли, экспрессирующие P815mTDO2, отторгались меньше по сравнению с опухолями Р815, трансфицированными пустым вектором, ясно демонстрируя преимущество роста для TDO2-экспрессирующих опухолей. Ингибирование TDO2-ингибитором сильно снижало рост опухоли в P815m TDO2 имплантированных опухолях. Противоопухолевая активность с ингибитором TDO2 в равной степени наблюдалась в Р815 контрольных имплантированных опухолях, негативных для TDO2, тем самым обеспечивая доказательство эффекта экспрессии TDO2 в иммунной системе животного. Эти эксперименты впервые дали ясное доказательство роли TDO2 в регулировании роста опухоли через экспрессию в раковой клетке, а также иммунном компартменте.
Согласуясь со своим профилем экспрессии в печени, TDO2 преимущественно обнаруживался в гепатоцеллюлярной карциноме (НСС) (Pilotte L et al., Proc NatI Acad Sci USA, 2012, 109(7), 2497-502). Ингибирование катаболизма триптофана и тем самым восстановление концентрации триптофана и снижение продукции последующих метаболитов может обеспечить пользу в лечении заболевания печени, прогрессирующего до стадии карциномы печени. Более конкретно: (1) несколько сообщений продемонстрировали доказательство того, что повышенная доступность триптофана через его восполнение преимущественна, например, при циррозе печени, обеспечивая непосредственное использование триптофана для синтеза белка (Ohta et al., Амино Acids, 1993, 10(4), 369-78); (2) имеется корреляция между повышенными сыворточными уровнями последующих метаболитов триптофана, таких как хинолиновая кислота, и печеночной дисфункцией у пациентов с циррозом печени (Lahdou et al., Hum Immunol, 2013, 74(1), 60-6), и (З) повышенная секреция другого метаболита триптофана, индол-3-молочной кислоты, ассоциирована с алкоголь-индуцированным заболеванием печени у мышей (Manna et al., J Proteome Res, 2011, 10(9), 4120-33). В контексте карциномы печени как таковой, очень высокая экспрессия RNA является хорошим индикатором терапевтической оценки ингибиторов TDO2 (Pilotte L et al., Proc NatI Acad Sci U S A, 2012, 109(7), 2497-502). Сказанное выше обеспечивает таким образом четкое логическое обоснование для модулирования активности TDO2 в контроле развития опухоли печени.
Помимо экспрессии в печени, TDO2 экпрессируется в нейронах, микроглии и астроцитах, и была показана потенциальная польза ингибирования TDO2 при глиоме в еще одной животной модели. Flatten и сотрудники продемонстрировали, что катаболит триптофана кинуренин, продуцируемый при экспрессии TDO в опухолевых клетках, подавляет противоопухолевые иммунные ответы и стимулирует выживание опухолевой клетки и подвижность через AHR аутокринным/паракринным образом (Opitz CA et al., Nature, 2011, 478(7368), 197-203). TDO-AHR путь активен в опухолях мозга человека и ассоциирован со злокачественным прогрессированием и плохим выживанием. Еще одно доказательство следует из накопления последующего метаболита, хинолиновой кислоты, которая накапливается в человеческих глиомах, и было ассоциировано со злокачественным фенотипом (Sahm et al., Cancer Res, 2013, 73(11), 3225-34). Здесь было показано, что катаболизм триптофана происходит также и в клетках микроглии. Таким образом, приведенные выше данные обеспечивают доказательство TDO2 нацеливания в глиоме с помощью проникающих в головной мозг малых молекул.
Другие типы опухолей, в которых была обнаружена TDO2 mRNA, представляют собой карциному молочной железы, мочевого пузыря, почечно-клеточную карциному, панкреатическую, колоректальную карциному, карциному головы и шеи и карциному легких, а также меланому, тем самым расширяя сферу нацеливания на TDO2 за пределы НСС и глиомы (Pilotte L et al., Proc NatI Acad Sci U S A, 2012, 109(7), 2497-502).
Усиленное разложение триптофана, наблюдаемое у пациентов с гинекологическими видами рака (карцинома яичников, рак шейки матки, рак эндометрия), дает дополнительное обоснование для нацеливания на TDO2 при этих видах рака (Sperner-Unterweger В et al, Immunology, 2011, 216 (3); 296-301).
Катаболизм триптофана при некоторых видах рака может также увеличиваться посредством экспрессии индоламин-2,3-диоксигеназы (IDO1) опухолевыми клетками (Uyttenhove, С. et al., Nat. Med., 2003, 9, 1269-1274).
Поскольку катаболизм триптофана индуцируется воспалительными медиаторами, а именно гамма-интерфероном (IFN-гамма), он, как полагают, представляет собой эндогенный механизм, который ограничивает избыточные иммунные ответы, тем самым предотвращая иммунопатологию. Однако в контексте рака, существует ясное свидетельство того, что супрессия противоопухолевых иммунных ответов при предраковых поражениях и установленных видах рака посредством катаболизма триптофана стимулирует рост опухоли, что делает такой катаболизм привлекательной мишенью для терапевтического вмешательства ( E and
E and  R., Expert Opin Ther Pat, 2013, 23(10), 1367-81). Поэтому, значительные усилия предпринимаются для идентификации селективных и эффективных ингибиторов катаболизма триптофана для усиления эффективности традиционной химиотерапии, иммунных контрольных точек (Holmgaard RB et al., J Ехр Med., 2013, 210(7), 1389-402) или терапевтических вакцин.
R., Expert Opin Ther Pat, 2013, 23(10), 1367-81). Поэтому, значительные усилия предпринимаются для идентификации селективных и эффективных ингибиторов катаболизма триптофана для усиления эффективности традиционной химиотерапии, иммунных контрольных точек (Holmgaard RB et al., J Ехр Med., 2013, 210(7), 1389-402) или терапевтических вакцин.
В контексте неврологических мозговых расстройств, экспрессия TDO2 продемонстрирована в нейронах, сосудистой сети мозга и, дополнительно в случае шизофрении, в астроглиальных клетках (Miller С et al., 2004, Neurobiology Dis, 15(3):618-29). Путь кинуренина считается в настоящее время терапевтической мишенью при когнитивных заболеваниях типа биполярного расстройства или синдрома Туретта и нейродегенеративных расстройств типа болезни Альцгеймера, заболевания двигательных нейронов типа бокового амиотрофического склероза, рассеянного склероза, болезни Гентингтона или болезни Паркинсона (Stone TW, 2013, Br J of Pharmacol, 169(6): 1211-27; Wu et al, 2013, Plos One, 8(4):e59749;  et al, 2012, J Neural Transm, 119(2):225-34; Widner et al, 2002, J Neural Transm, 109(2): 181-9; Comings et al, 1996, Pharmacogenetics, 6(4):307-18; Forrest2010, J Neurochem, 112(1):112-22).
et al, 2012, J Neural Transm, 119(2):225-34; Widner et al, 2002, J Neural Transm, 109(2): 181-9; Comings et al, 1996, Pharmacogenetics, 6(4):307-18; Forrest2010, J Neurochem, 112(1):112-22).
Когнитивные изменения, связанные с катаболизмом триптофана, были показаны у пациентов, инфицированных вирусом иммунодефицита типа-1 человека (HIV), называемые HIV-ассоциированное нейрокогнитивное расстройство (HAND) (Davies et al, 2010, Int J of Tryptophan Res, 3:121-40). В дополнение, Т-клеточную гипореактивность ассоциировали в последнее время с путем катаболизма триптофана у HIV-инфицированных пациентов, с возможным распространением на другие хронические инфекционные заболевания, такие как, например, гепатит С.
Некоторые ингибиторы TDO2 были предложены в WO 2010/008427 и в Dolusic, E. et al. (Dolusic et al., J. Med. Chem., 2011, 54, 5320-5334), однако либо их аффинность в отношении мишени ограничена, либо их фармакокинетические свойства не подходят для разработки в качестве лекарственного средства для использования у человека.
Таким образом, имеется необходимость в новых ингибиторах TDO2 с повышенной эффективностью для лечения и/или предупреждения рака.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению предложены новые ингибиторы TDO2, которые могут быть введены субъекту-млекопитающему, страдающему состоянием или заболеванием, при котором желательно модулирование, и в частности снижение, активности TDO2, включая, без ограничения, пациентов, у которых диагностирован рак, или любого субъекта, имеющего риск развития рака. Также предложены композиции, содержащие эти соединения, и их применение.
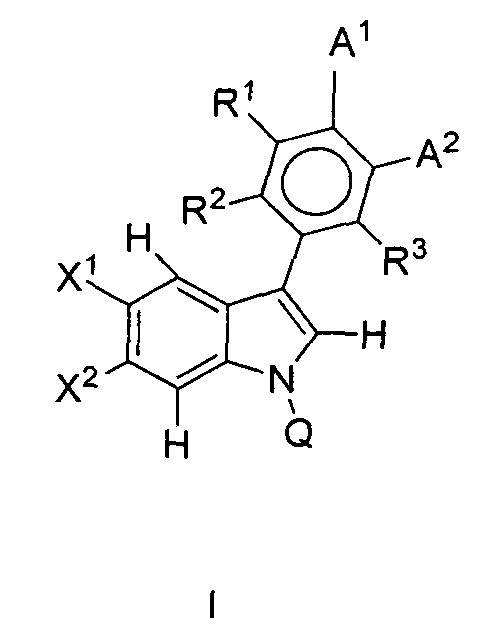
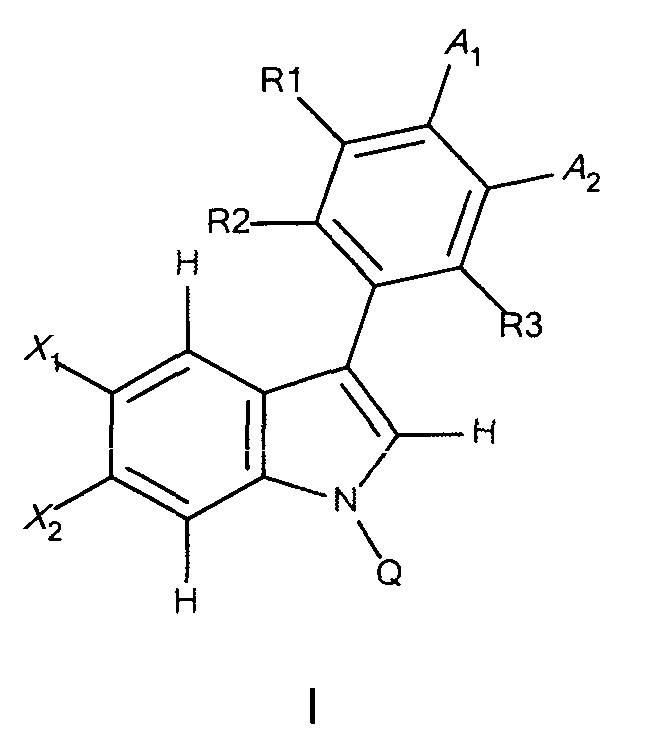
В одном аспекте предложено соединение формулы I или его фармацевтически приемлемые соль, растворитель или сольват, где A1, A2, Q, R1, R2, R3, X1 и X2 являются такими, как определено в данном описании.
В другом аспекте предложена фармацевтическая композиция, содержащая соединение формулы I или его фармацевтически приемлемые энантиомер, соль или сольват и по меньшей мере один фармацевтически приемлемый носитель, разбавитель, эксципиент и/или адъювант.
В еще одном аспекте предложено лекарственное средство, содержащее соединение формулы I или его фармацевтически приемлемые энантиомер, соль или сольват.
В еще одном аспекте предложено соединение формулы I или его фармацевтически приемлемые энантиомер, соль или сольват для применения в лечении и/или предупреждении рака, нейродегенеративных расстройств, таких как болезнь Паркинсона, болезнь Альцгеймера и болезнь Гентингтона, хронических вирусных инфекций, таких как HCV и HIV, депрессии и ожирения, или для применения в качестве ингибитора TDO2.
В еще одном аспекте предложен способ лечения и/или предупреждения рака, нейродегенеративных расстройств, таких как болезнь Паркинсона, болезнь Альцгеймера и болезнь Гентингтона, хронических вирусных инфекций, таких как HCV и HIV, депрессии и ожирения, или ингибирования TDO2. Способ включает введение соединения формулы I или его фармацевтически приемлемой соли.
В другом аспекте предложен способ получения соединения формулы I или его фармацевтически приемлемых энантиомера, соли или сольвата. Способ включает:
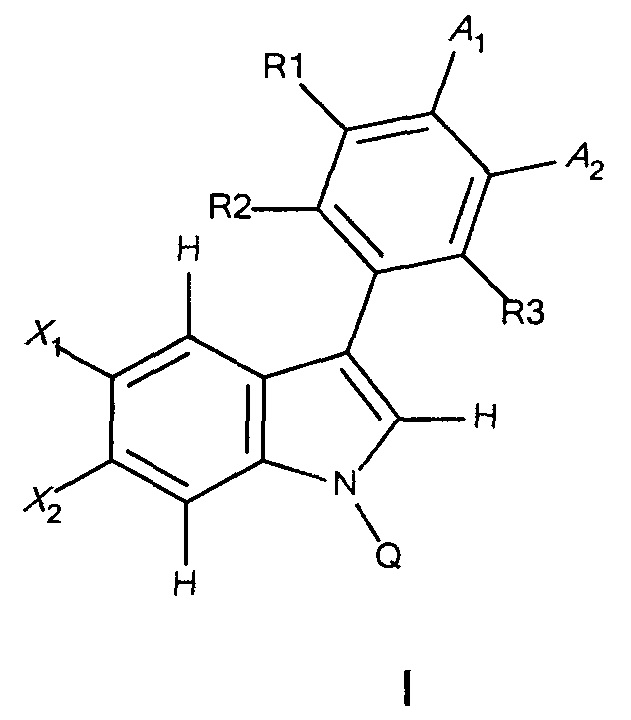
и его фармацевтически приемлемые энантиомер, соль или сольват, где X1, X2, R1, R2, R3, А1, А2 и Q являются такими, как определено в формуле I;
включающий:
(а1) взаимодействие соединения формулы (i)

где
X1 и X2 являются такими, как определено в формуле I;
Z1 представляет собой Q или амино-защитную группу, такую как, например, арилсульфонил, трет-бутоксикарбонил, метоксиметил, пара-метоксибензил, бензил или любая другая подходящая защитная группа, известная специалистам в данной области техники;
Y представляет собой галоген (предпочтительно йод, бром или хлор), группу алкилсульфонилокси, имеющую 1-6 атомов углерода (предпочтительно метилсульфонилокси или трифторметилсульфонилокси) или группу арилсульфонилокси, имеющую 6-10 атомов углерода (предпочтительно фенил- или пара-толилсульфонилокси), или любую уходящую группу, известную специалистам в данной области техники;
с соединением формулы (ii)
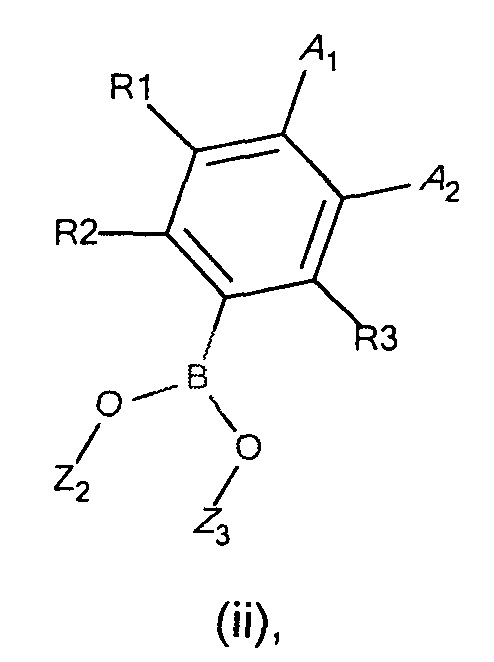
где
R1, R2, R3, А1, А2 и А3 являются такими, как определено в формуле I;
Z2 и Z3 представляют собой Н или алкильные группы, с возможностью образования кольца для Z2 и Z3;
с получением соединения формулы (iii),
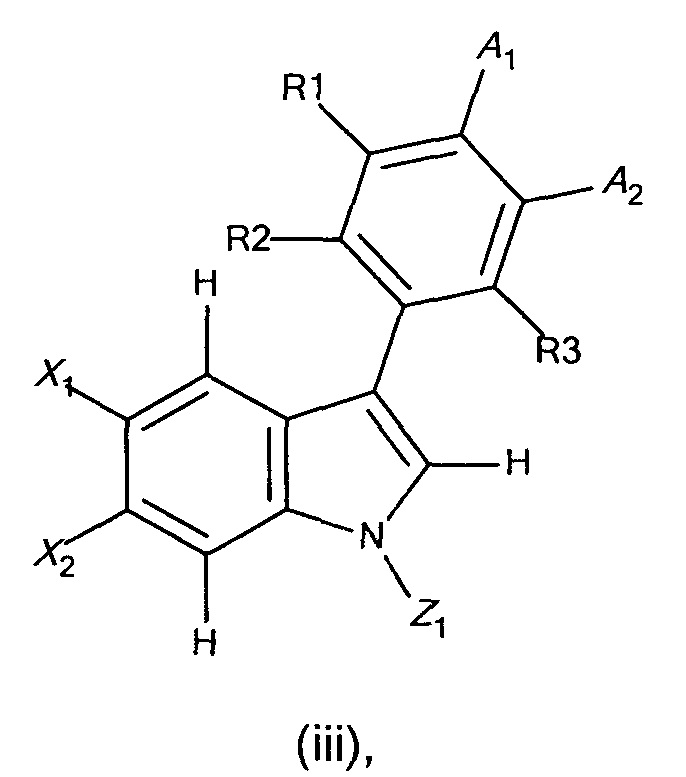
где X1, X2, R1, R2, R3, А1, А2 и Z1 являются такими, как определено выше;
и
(b1) в случае, когда Z1 не представляет собой Q, снятие защиты с индоламина соединения формулы (iii), с получением соединения формулы I.
Другие дополнительные аспекты и преимущества изобретения будут понятны из следующего подробного описания изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Соединения
В данном изобретении предложены соединения формулы I или его фармацевтически приемлемые энантиомер, соль или сольват. Если конкретно не указано иное, хотя ссылка для удобства сделана на формулу I и ее применения и способы получения, следует понимать, что ее подформула, т.е. формула II, охватывается этими описаниями. Формула I имеет структуру:
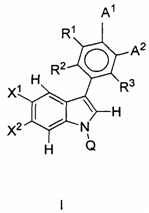 ,
,
или его фармацевтически приемлемые энантиомер, соль или сольват, где:
каждый из X1 и X2 независимо представляет собой Н, галоген, ОН, OR7 или С1-С4алкил;
R1, R2 и R3 независимо представляют собой: Н, галоген, циано, R7, ОН, OR7, NR7R8, CONR7, N(R7)COR8, SO2R7 или алкилNR7R8;
Q представляет собой Н, COR7 или CONR7R8;
R7 и R8 независимо представляют собой: (1) Н, (2) NH2, (3) прямой или разветвленный С1-С6алкил, возможно замещенный одним-тремя заместителями, выбранными из оксо, амино, ОН, галогена, С1-С4алкила, (4) C1-C3алкил-гетероцикл или гетероцикл, возможно замещенный пяти- или шестичленный гетероцикл, в котором заместитель представляет собой оксо, ОН, NH2 или С1-С3алкил, который является возможно замещенным;
А1 и А2 вместе образуют 5-членную конденсированную кольцевую структуру, содержащую SO2NR5R9, где R9 представляет собой атом водорода или возможно замещенную группу, выбранную из С1-С6алкила, арила, арилалкила, алкиларила, гетероарила, гетероарилалкила, алкилгетероарила или амино;
R5 и R6 независимо представляют собой: (1') Н, (2') оксо, (3') амино, (4') галоген или возможно замещенную группу, выбранную из:
(5') прямого или разветвленного С1-С6алкила, возможно замещенного заместителями в количестве до трех включительно, выбранными из галогена, гидроксила, OR9, COOR9, CONR9R10, NR9COR10, NR9R10, SO2R9, SO2NR9R10, NR9SO2R10, SOR9, арила или СО-алкила, где каждый из R9 и R10 независимо представляет собой атом водорода или возможно замещенную группу, выбранную из С1-С6алкила, гетероциклила, арила, арилалкила, алкиларила, гетероарила, гетероарилалкила, алкилгетероарила или амино;
(6') гетероциклила или группы С1-С2алкил-гетероциклил, где указанный гетероциклил возможно замещен заместителями в количестве до трех включительно, которые независимо представляют собой галоген, гидроксил, оксо, OR9, COOR9, CONR9R10, NR9OR10, NR9R10, SO2R9, SO2NR9R10, NR9SO2R10, SO2R9, арил, СО-алкил или алкил, где указанная алкильная группа возможно замещена одной или более чем одной группой, выбранной из галогена, гидроксила, амино или СООН; где каждый из R9 и R10 независимо представляет собой атом водорода или возможно замещенную группу, выбранную из С1-С6алкила, арила, арилалкила, алкиларила, гетероарила, гетероарилалкила, алкилгетероарила или амино;
(7') циклоалкила, возможно замещенного заместителями в количестве до трех включительно, выбранными из галогена, гидроксила, OR9, COOR9, CONR9R10, NR9OR10, NR9R10, SO2R9, SO2NR9R10, NR9SO2R10, SO2R9, арила, СО-алкила или С1-С6алкила, который возможно замещен одной или более чем одной группой, выбранной из галогена, гидроксила, амино или СООН; где каждый из R9 и R10 независимо представляет собой атом водорода или возможно замещенную группу, выбранную из С1-С6алкила, арила, арилалкила, алкиларила, гетероарила, гетероарилалкила, алкилгетероарила или амино. В некоторых воплощениях формулы I Q представляет собой Н. В некоторых воплощениях формулы I X1 и X2 независимо представляют собой Н, F или Cl, предпочтительно F. В некоторых воплощениях в соединении формулы I А2 представляет собой Н, галоген или ОН, предпочтительно Н.
В некоторых воплощениях формулы I X1 представляет собой Н, и X2 представляет собой F.
В некоторых воплощениях формулы I А1 и А2 вместе образуют 5-членную конденсированную кольцевую структуру, содержащую SO2NR5CR9, где R9 представляет собой атом водорода или возможно замещенную группу, выбранную из С1-С6алкила, арила, арилалкила, алкиларила, гетероарила, гетероарилалкила, алкилгетероарила или амино. В дополнительном воплощении А1 и А2 вместе образуют 5-членную конденсированную кольцевую структуру, содержащую SO2NR5CR9', где R9' представляет собой С1-С4алкил, ОН или галоген.
В другом воплощении каждый из X1 и X2 независимо представляет собой Н, галоген, ОН, OR7 или С1-С4алкил; R1, R2 и R3 независимо представляют собой: Н, галоген, циано, R7, OR7, NR7R8, CONR7, N(R7)COR8, SO2R7 или алкилNR7R8; Q представляет собой Н, COR7 или CONR7R8; R7 и R8 независимо представляют собой: (1) Н, (2) NH2, (3) прямой или разветвленный С1-С6алкил, возможно замещенный одним-тремя заместителями, выбранными из одного или более чем одного оксо, амино, ОН, галогена или С1-С4алкила, (4) С1-С3алкил-гетероцикл или (5) гетероцикл, где указанный гетероцикл из (4) или (5) представляет собой возможно замещенный пяти- или шестичленный гетероцикл, в котором заместитель представляет собой оксо, ОН, NH2 или С1-С3алкил, который возможно замещен одним-тремя заместителями, выбранными из одного или более чем одного галогена, алкила, ОН, оксо или амино.
В некоторых воплощениях А1 и А2 вместе образуют 5-членную конденсированную кольцевую структуру, содержащую SO2NR5CR9'R9, где R9' представляет собой Н, или каждый из R9' и R9 представляет собой метил, где когда R9' представляет собой Н, R9 представляет собой атом водорода, циклопропил или возможно замещенную группу, выбранную из С1-С6алкила, арила, арилалкила, алкиларила, гетероарила, гетероарилалкила или алкилгетероарила, где указанная возможно замещенная группа имеет один, два или три заместителя, выбранных из одного или более чем одного галогена, С1-С4алкила, ОН, оксо или амино.
В некоторых воплощениях R5 и R6 независимо представляют собой: (1') Н, (2') оксо, (3') амино, (4') галоген или возможно замещенную группу, выбранную из:
(5') прямого или разветвленного С1-С6алкила, возможно замещенного заместителями в количестве до трех включительно, выбранными из одного или более чем одного галогена, гидроксила, OR9, COOR9, CONR9R10, NR9COR10, NR9R10, SO2R9, SO2NR9R10, NR9SO2R10, SOR9, арила или СО-алкила,
(6') гетероцикпила или группы C1-С3алкил-гетероциклил, где указанный гетероциклил возможно замещен заместителями в количестве до трех включительно, которые выбраны из одного или более чем одного галогена, гидроксила, оксо, OR9, COOR9, CONR9R10, NR9COR10, NR9R10, SO2R9, SO2NR9R10, NR9SO2R10, SO2R9, арила, СО-алкила, пяти- или шестичленного гетероцикла, имеющего 2 атома N в своей структуре; пиперидина, замещенного F и тремя ОН, или алкилом, где указанная алкильная группа возможно замещена одной-тремя группами, выбранными из одного или более чем одного галогена, гидроксила, оксо, амино или СООН;
(7') циклоалкила, возможно замещенного заместителями в количестве до трех включительно, выбранными из галогена, гидроксила, OR9, COOR9, CONR9R10, NR9COR10, NR9R10, SO2R9, SO2NR9R10, NR9SO2R10, SO2R9, арила, СО-алкила или C1-С6алкила, который возможно замещен одной или более чем одной группой, выбранной из галогена, гидроксила, амино или СООН;
каждый из R9 и R10 независимо представляет собой атом водорода или возможно замещенную группу, выбранную из С1-С6алкила, где в случае замещения указанный С1-С6алкил имеет одну, две или три группы, выбранные из одного или более чем одного галогена, гидроксила, оксо, амино или СООН, гетероциклила, арила, арилалкила, алкиларила, гетероарила, гетероарилалкила или алкилгетероарила, где в случае замещения указанные арил, арилалкил, алкиларил, гетероарил, гетероарилалкил, алкилгетероарил имеют до трех заместителей включительно, которые представляют собой один или более чем один галоген, гидроксил, оксо, OR9, COOR9, CONR9R10, NR9COR10, NR9R10, SO2R9, SO2NR9R10, NR9SO2R10, SO2R9, СО-алкил или амино.
В других воплощениях А1 или А2 вместе образуют 5-членную конденсированную кольцевую структуру, содержащую SO2NR5CR9. В некоторых воплощениях R5 представляет собой С1-С3 алкил-гетероциклил, возможно замещенный заместителями в количестве до трех включительно, которые независимо представляют собой галоген, С1-С6 алкил, гидроксил, оксо, OR9, COOR9, CONR9R10, NR9COR10, NR9R10, SO2R9, SO2NR9R10, NR9SO2R10 или SO2R9.
В других воплощениях, в случае SO2NR5CR9, R9 представляет собой C1-С4алкил, который возможно замещен группой ОН или галогеном.
В некоторых воплощениях соединение формулы I находится в солевой форме. В другом воплощении предложена форма свободного основания (несолевая форма) соединения формулы I.
Кроме того, в данном изобретении предложено соединение формулы II:

или его фармацевтически приемлемые энантиомер, соль или сольват, где:
X2 представляет собой Н, галоген, ОН, OR7 или С1-С4алкил;
R7 представляет собой: (1) Н; (2) NH2; (3) прямой или разветвленный С1-С6 алкил, возможно замещенный одним-тремя заместителями, выбранными из оксо, амино, ОН, галогена или С1-С4алкила; (4) C1-C3алкил-гетероцикл, или (5) возможно замещенный пяти- или шестичленный гетероцикл, в котором заместитель представляет собой С1-С3алкил, который сам возможно замещен группой, выбранной из оксо, ОН или NH2;
R14 и R15 независимо представляют собой Н или возможно замещенную группу, выбранную из С1-С6алкила, арила, арилалкила, алкиларила, гетероарила, гетероарилалкила, алкилгетероарила или амино. В некоторых воплощениях R14 представляет собой Н, С1-С3алкил или ОН. В некоторых воплощениях R15 представляет собой Н, С1-С3алкил или ОН. В других воплощениях R15 определен как R5, и R14 определен как R9R9' формулы I в 5-членной конденсированной кольцевой структуре, содержащей SO2NR5CR9'R9 формулы I, где R9' представляет собой Н, или каждый из R9' и R9 представляет собой метил, где когда R9' представляет собой Н, R9 представляет собой атом водорода, циклопропил или возможно замещенную группу, выбранную из С1-С6алкила, арила, арилалкила, алкиларила, гетероарила, гетероарилалкила или алкилгетероарила, где указанная возможно замещенная группа имеет один, два или три заместителя, выбранных из одного или более чем одного галогена, С1-С4алкила, ОН, оксо или амино.
В некоторых воплощениях указанная возможно замещенная группа имеет один, два или три заместителя, выбранных из одного или более чем одного галогена, С1-С4алкила, ОН, оксо или амино.
В некоторых воплощениях формулы II X2 представляет собой галоген.
В некоторых воплощениях формулы II R14 представляет собой СН3.
В некоторых воплощениях соединение формулы II находится в солевой форме. В другом воплощении предложена форма свободного основания (несолевая форма) соединения формулы II.
Иллюстративные соединения формулы I перечислены в Таблице 1 ниже.
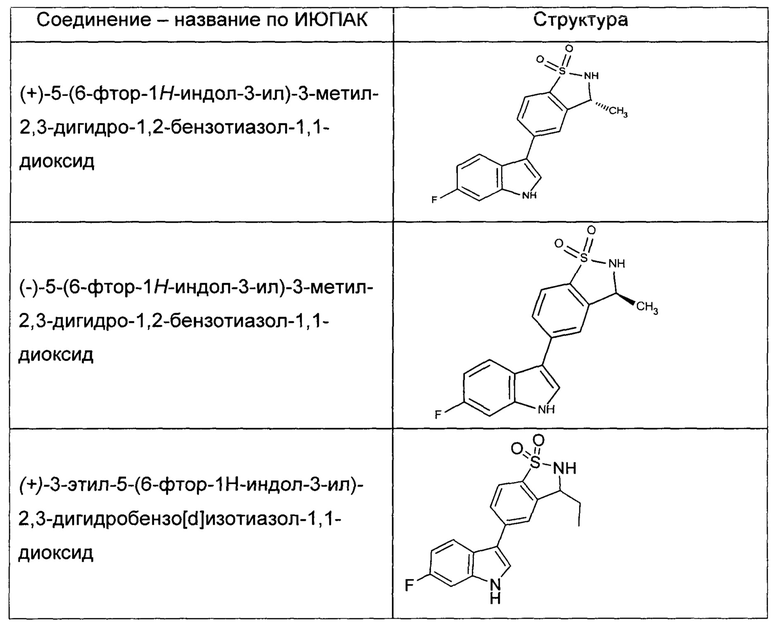
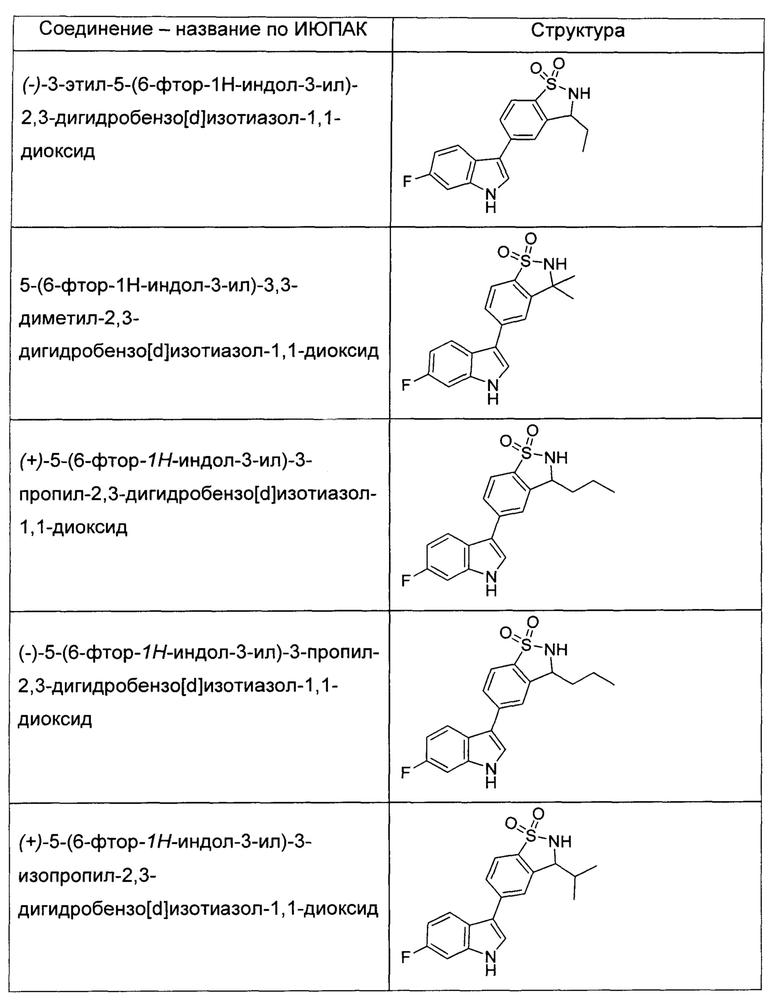
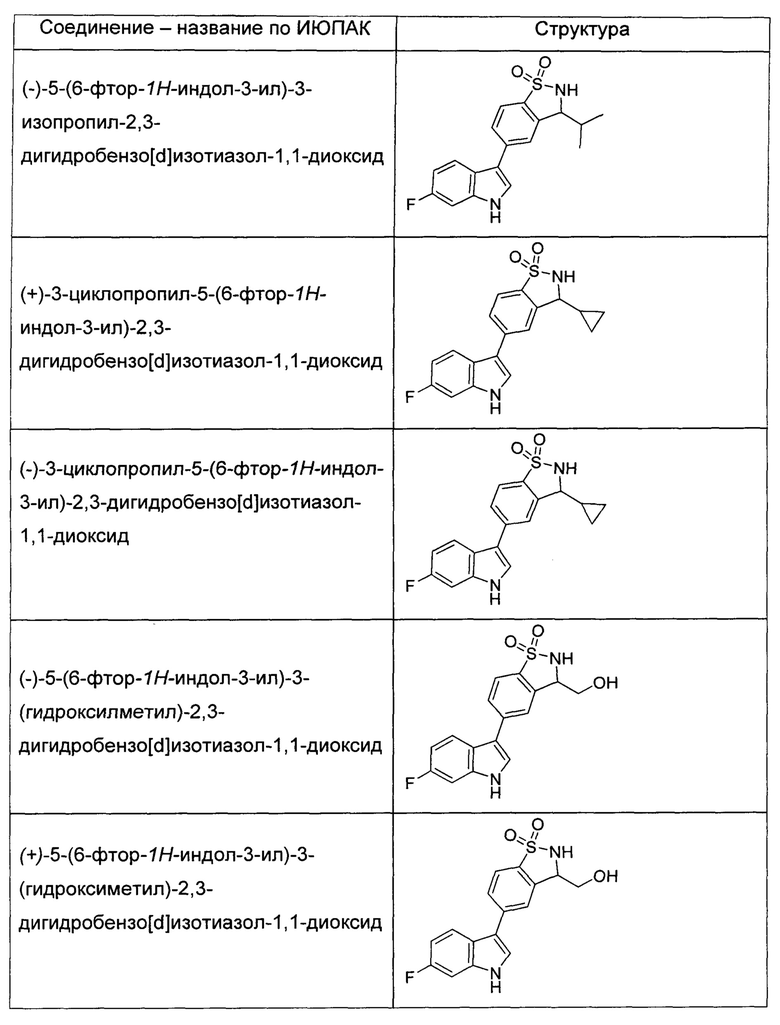
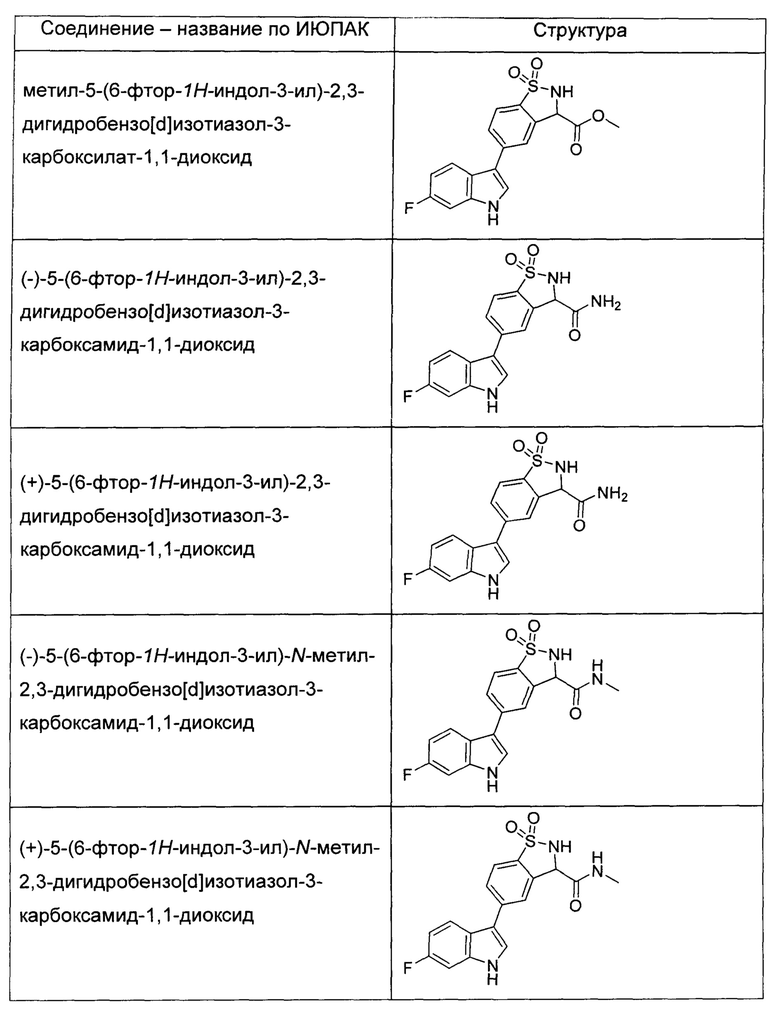
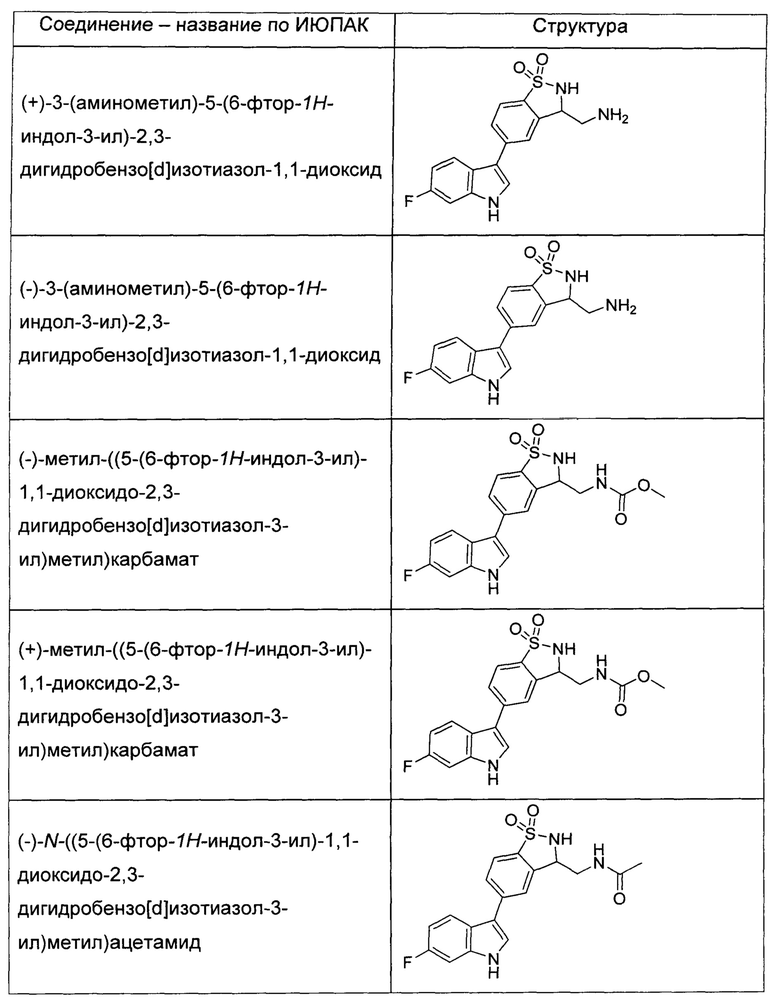

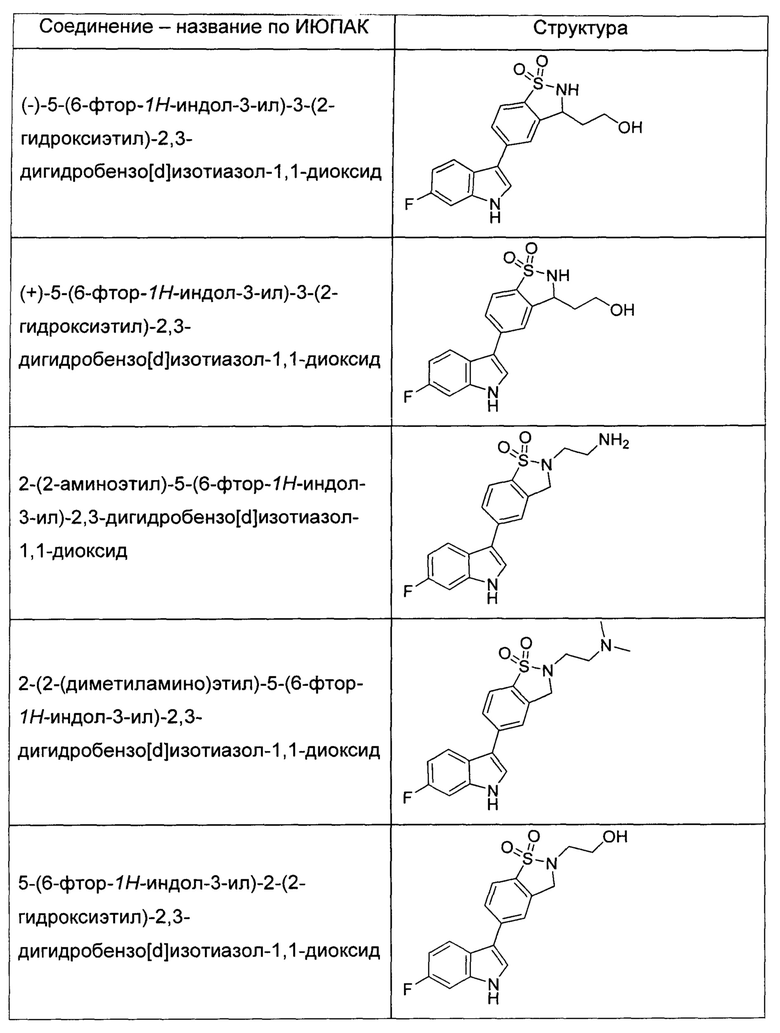
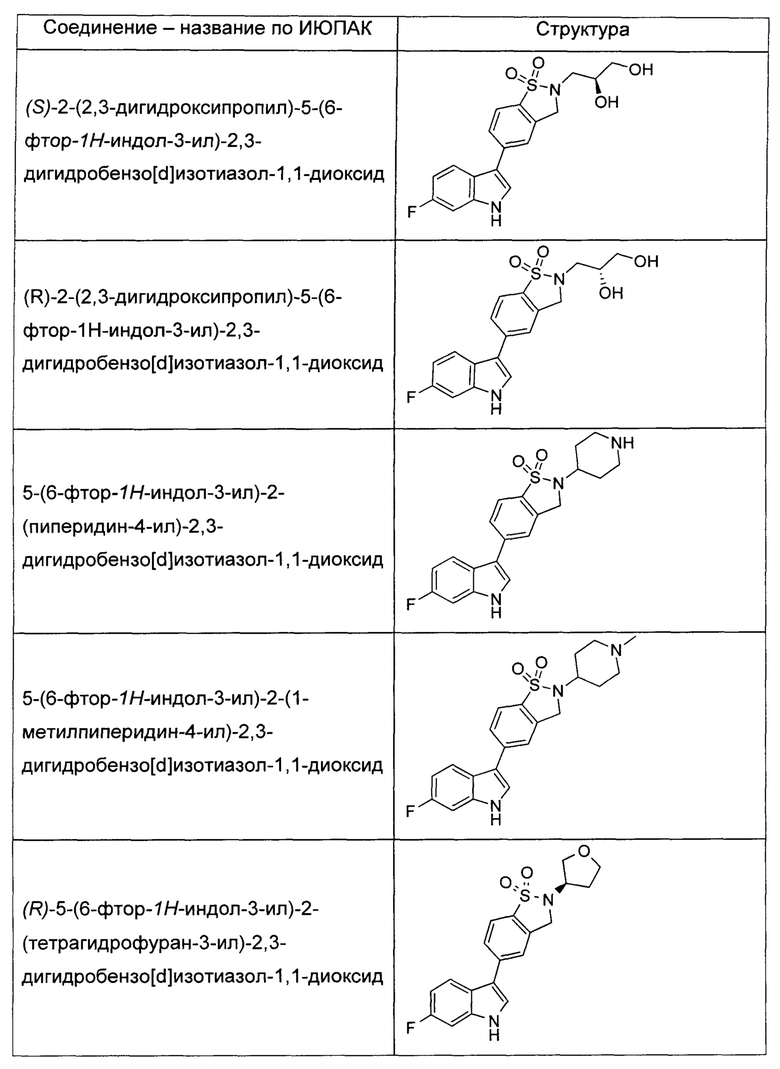
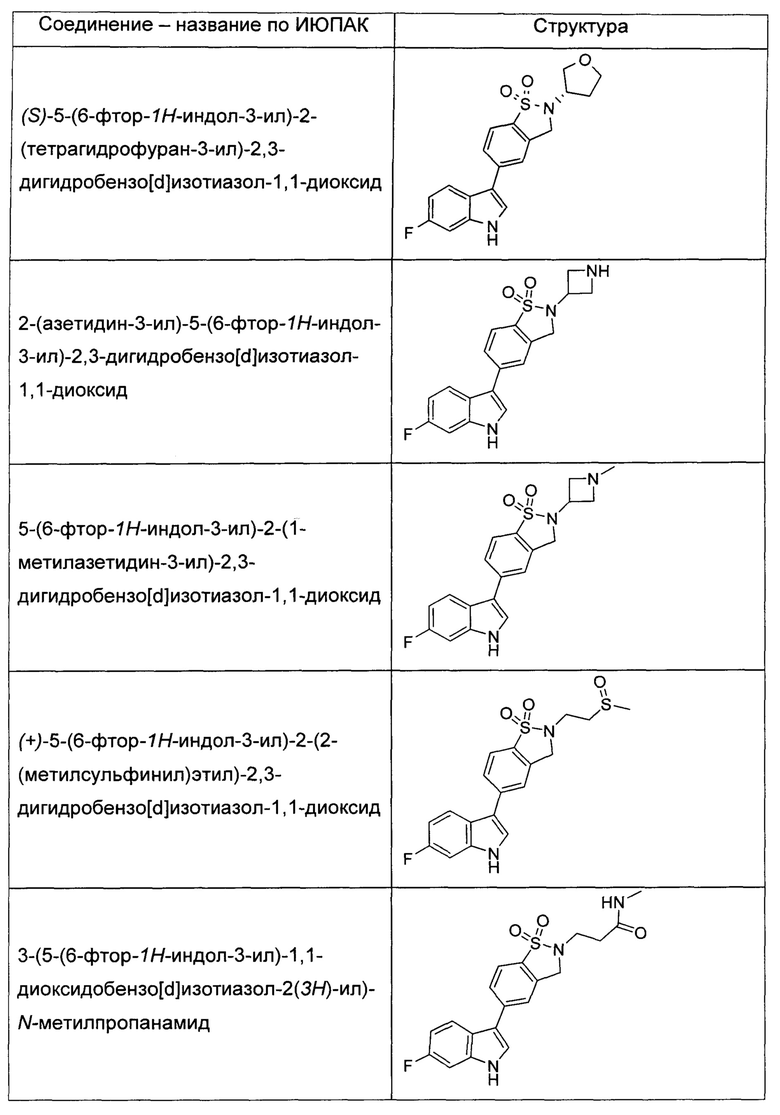
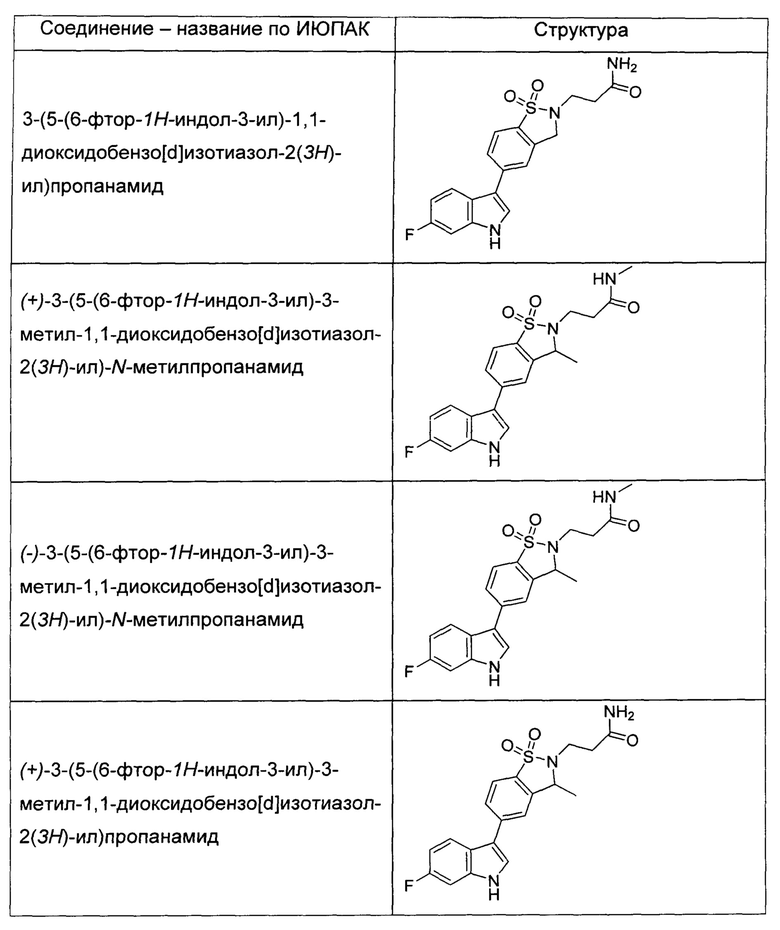
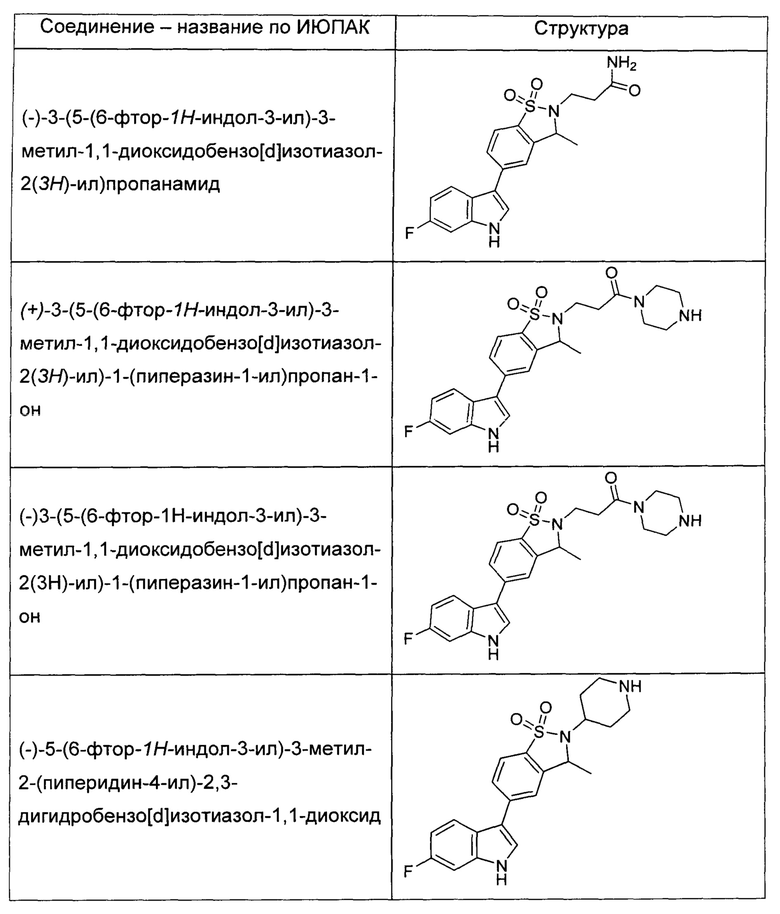
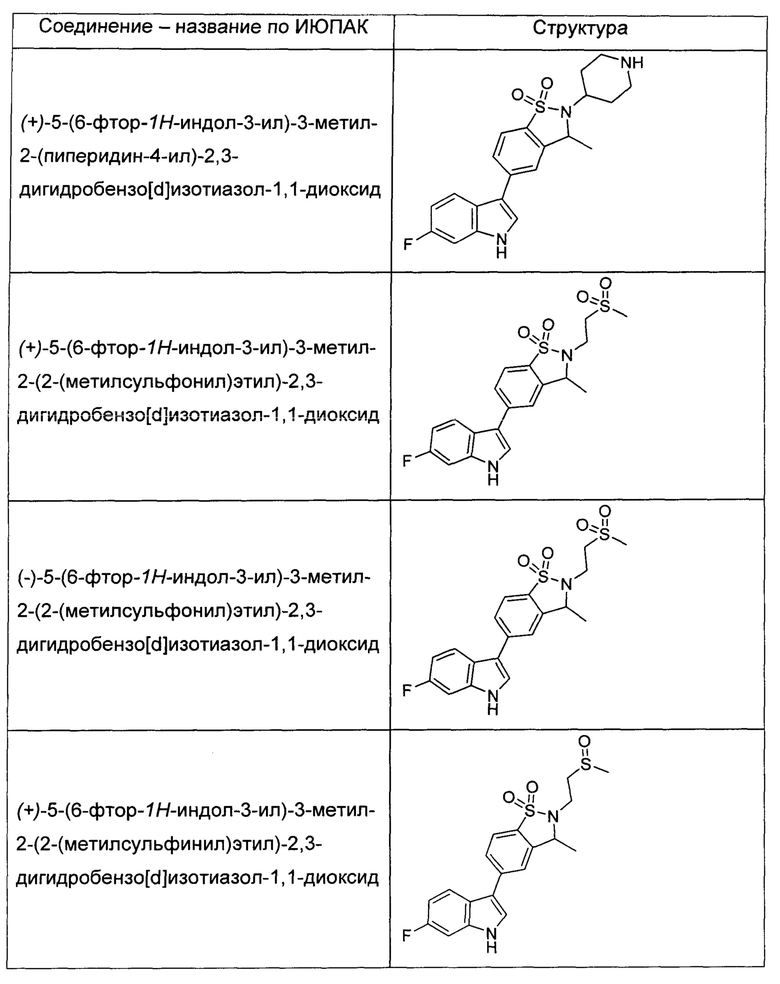
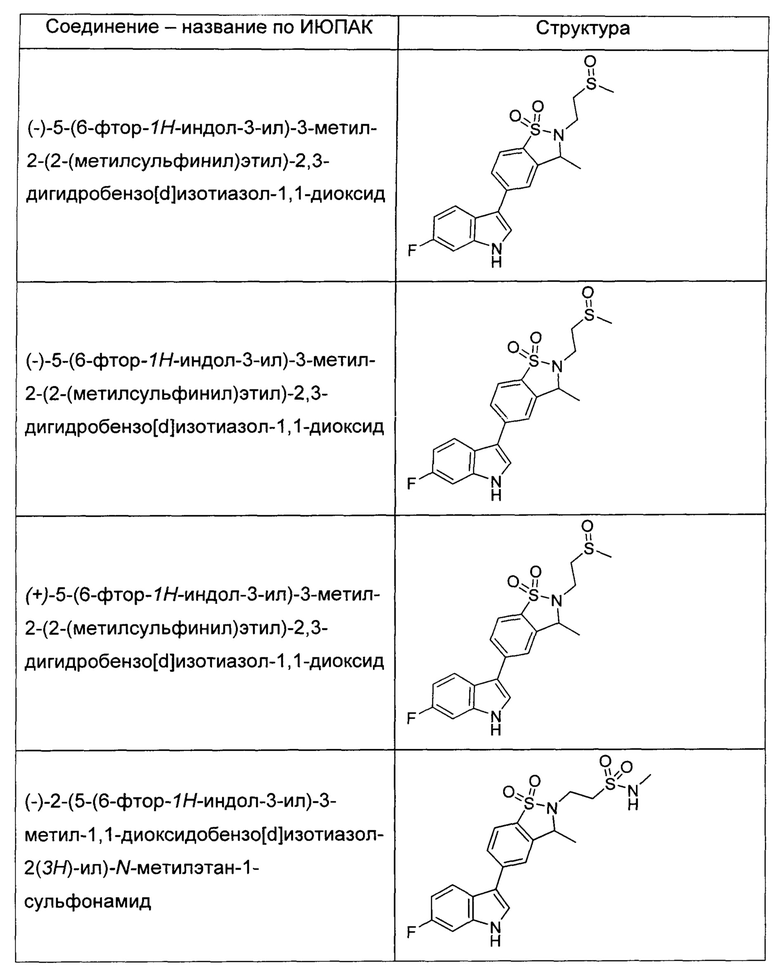
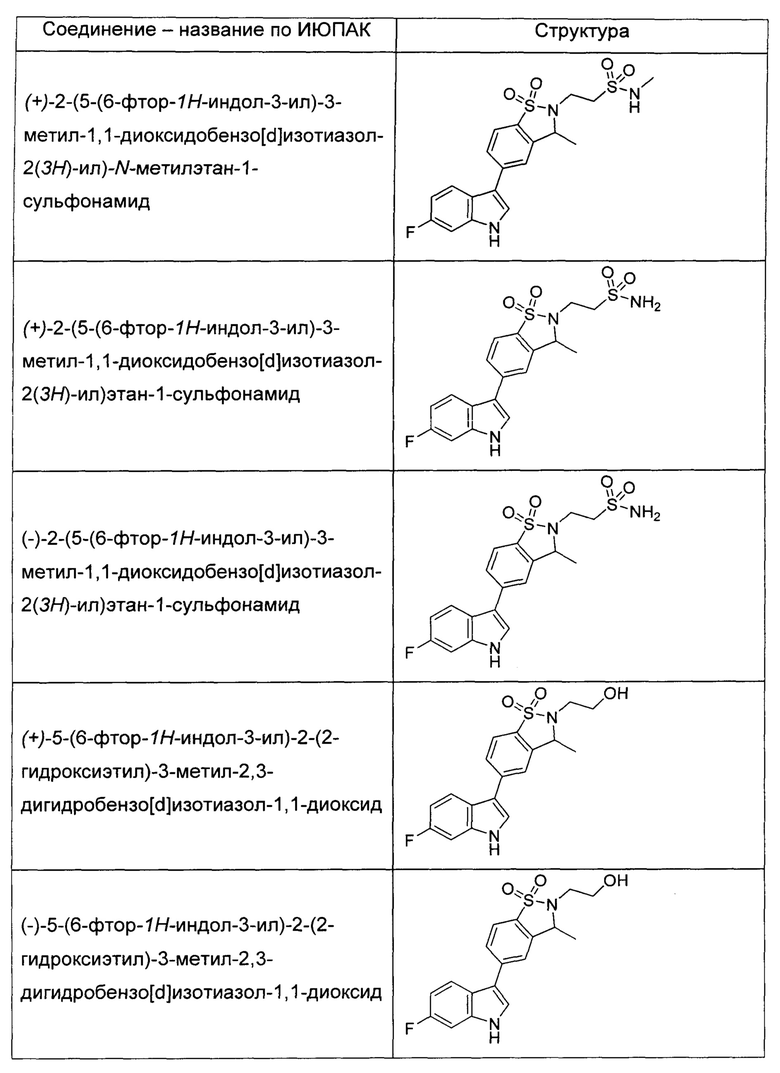
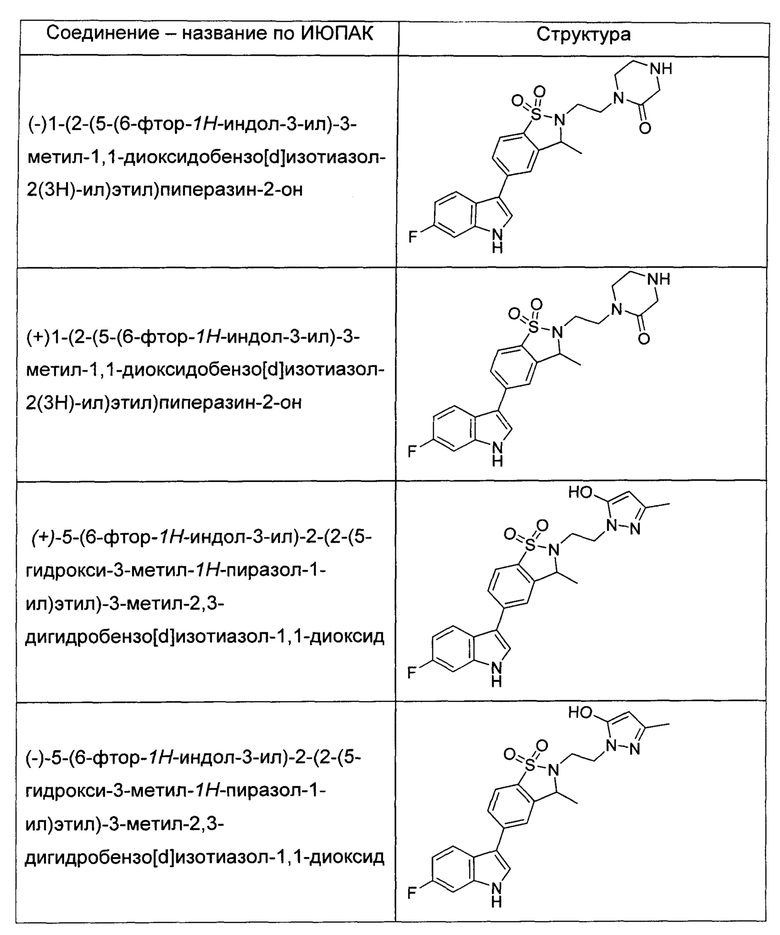
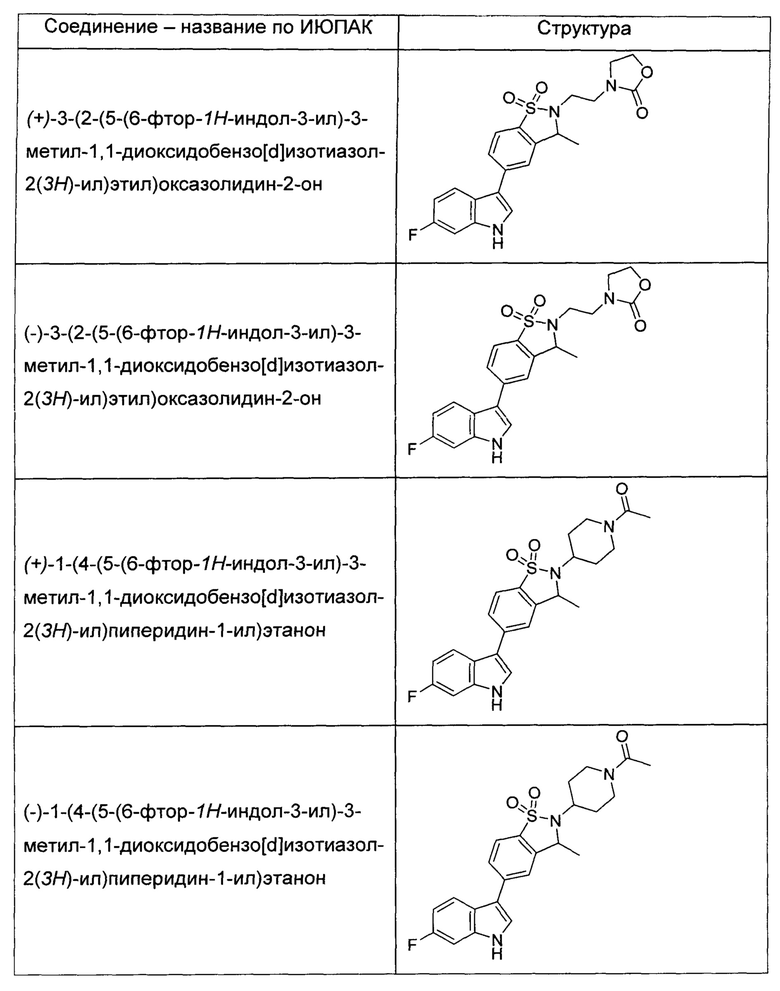
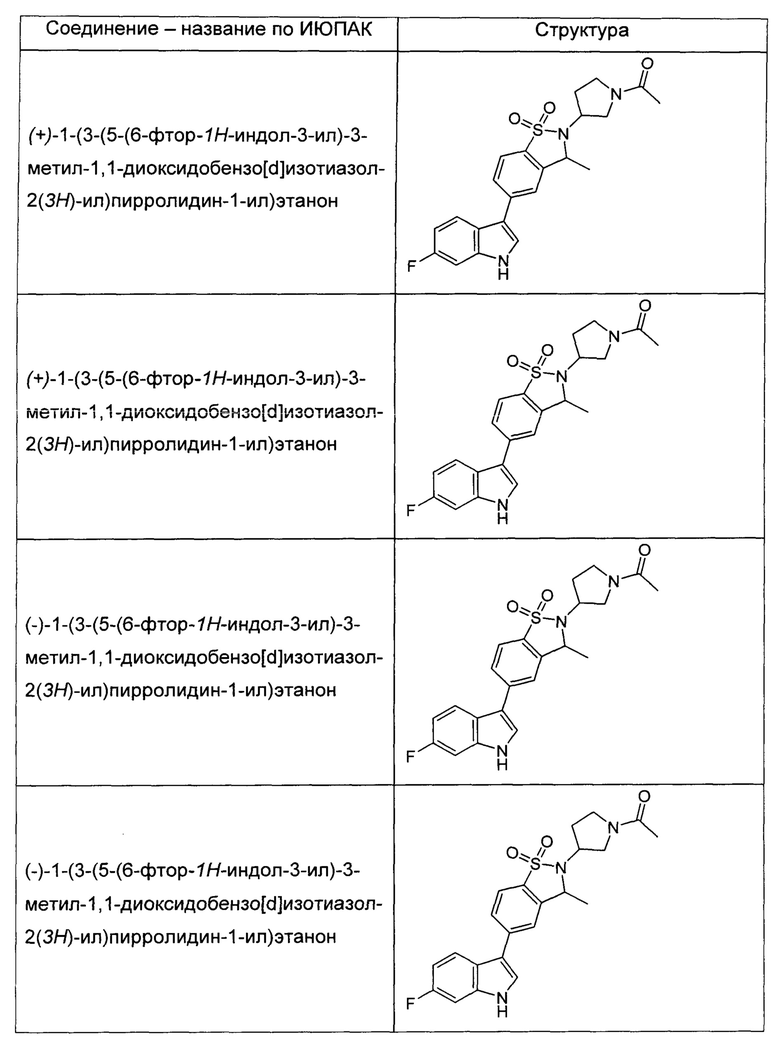
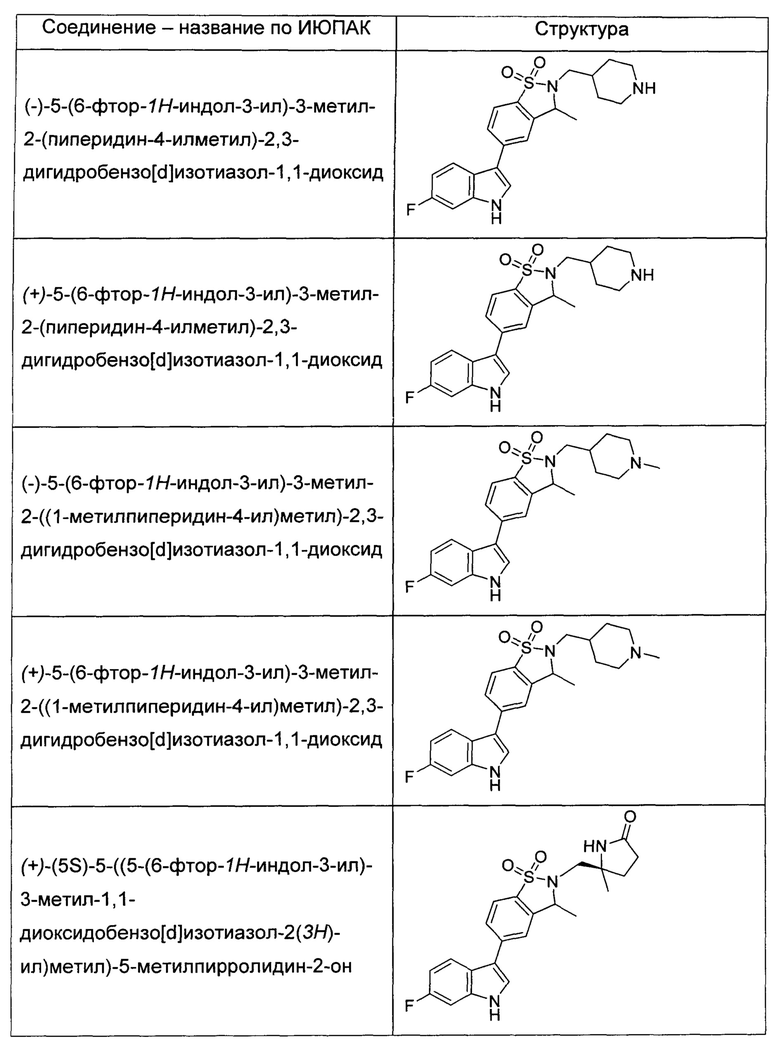
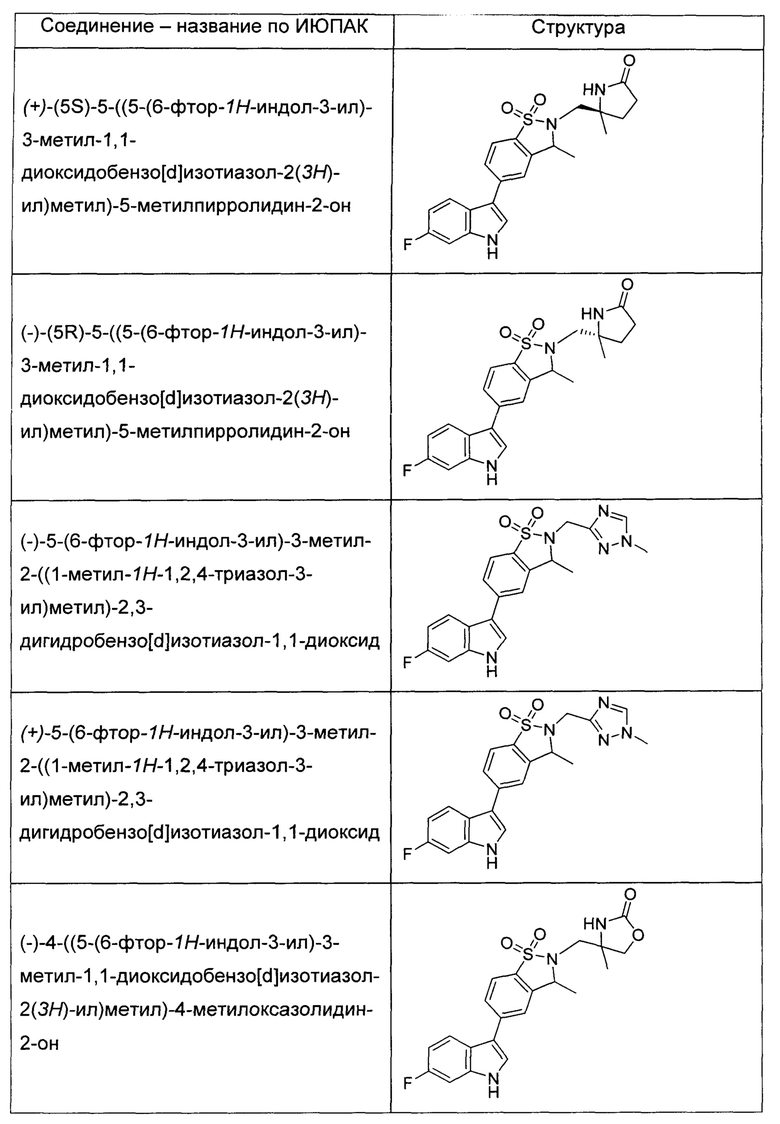
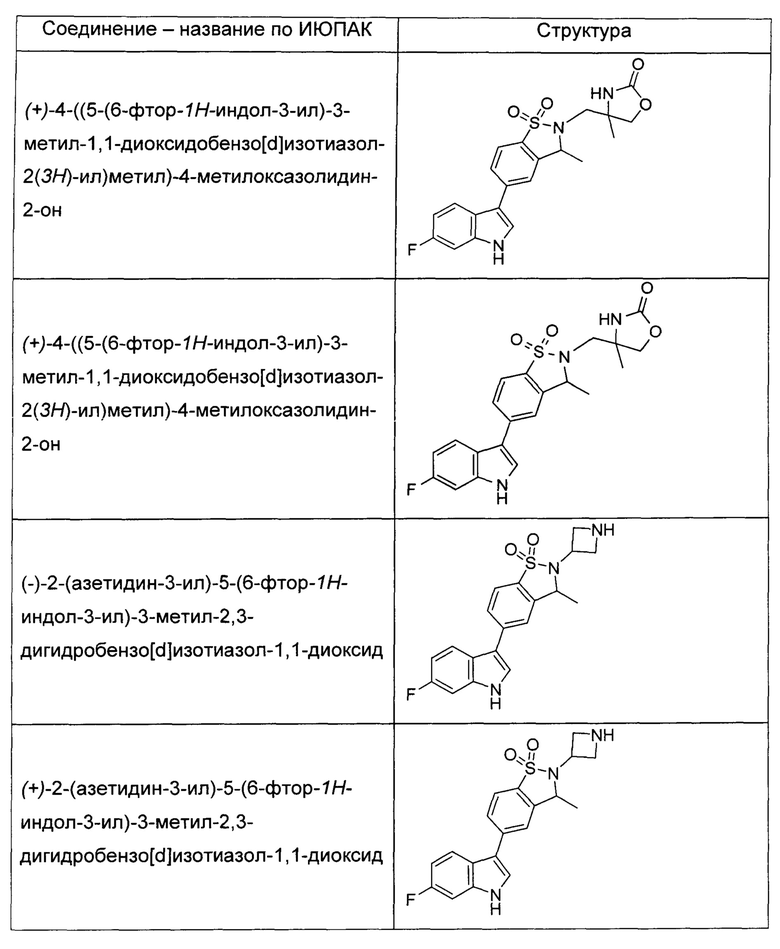
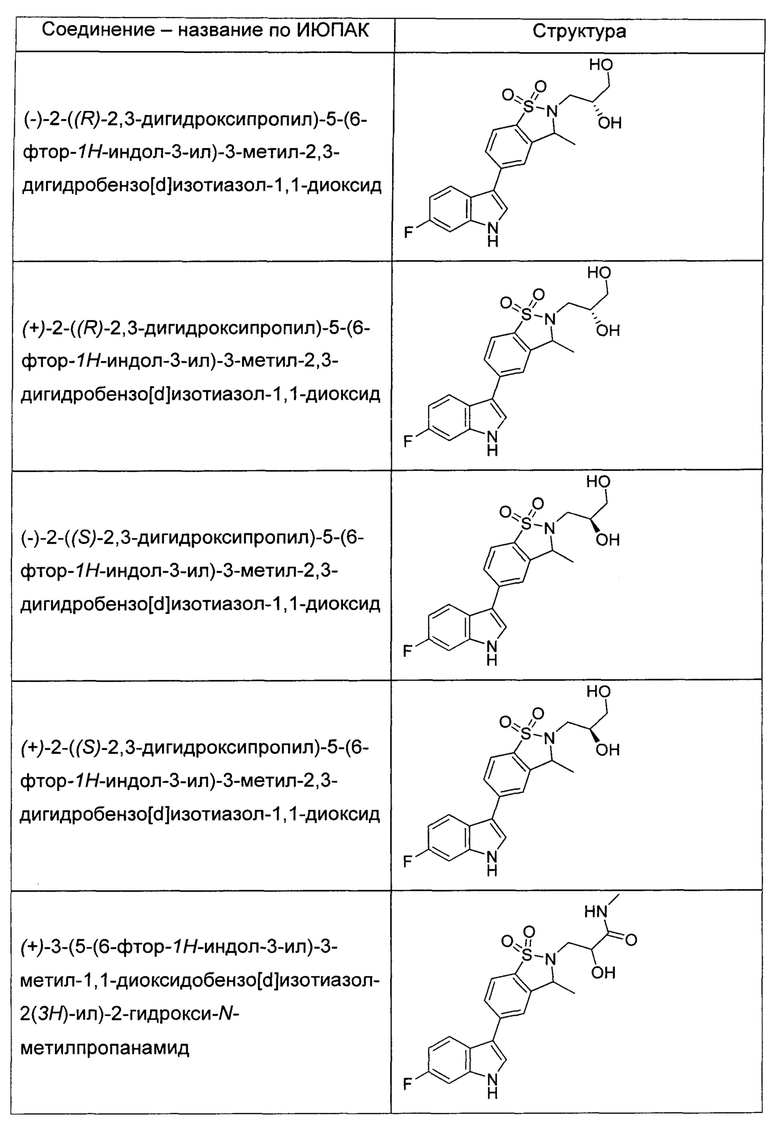
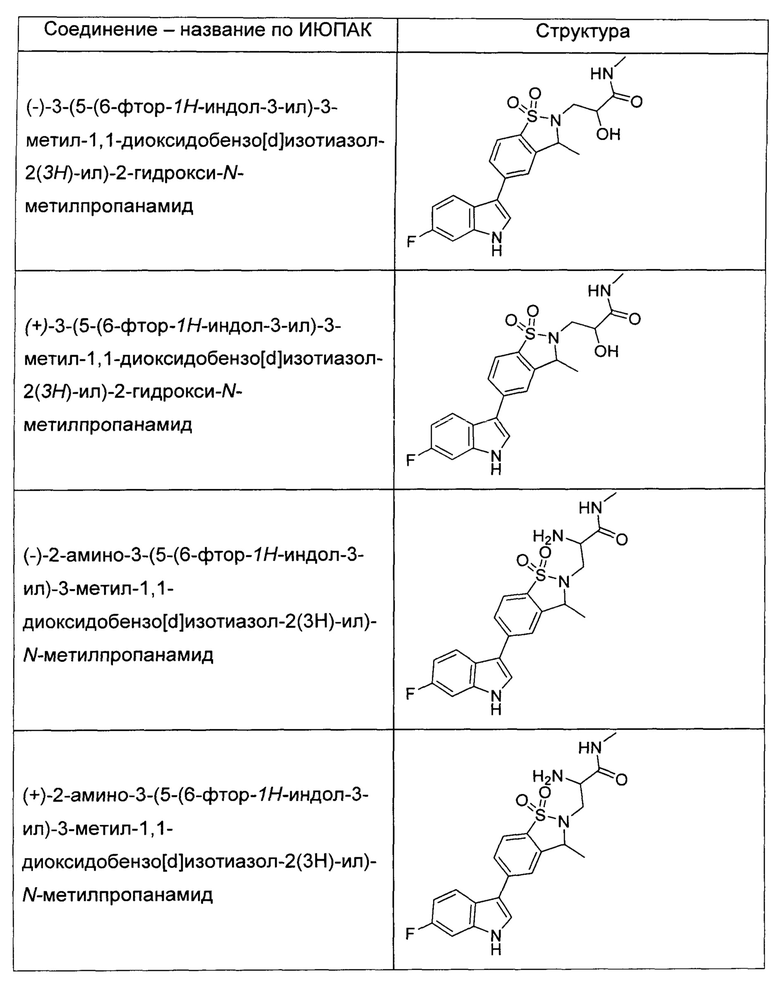
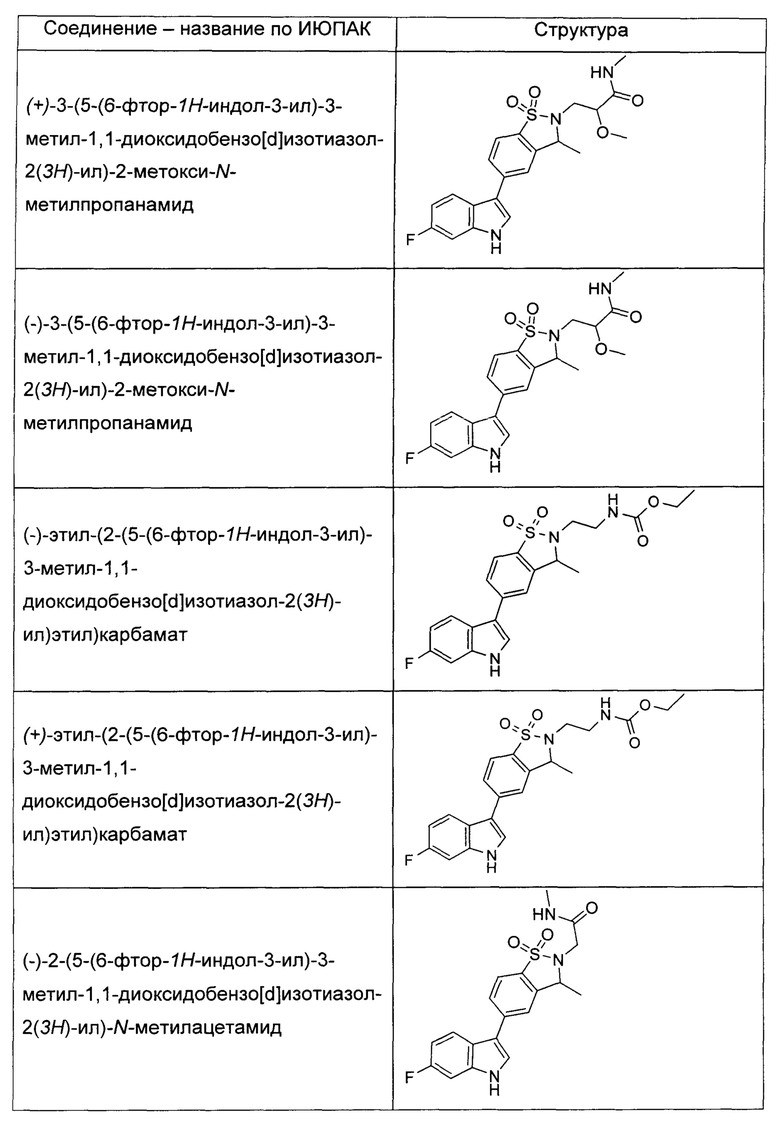
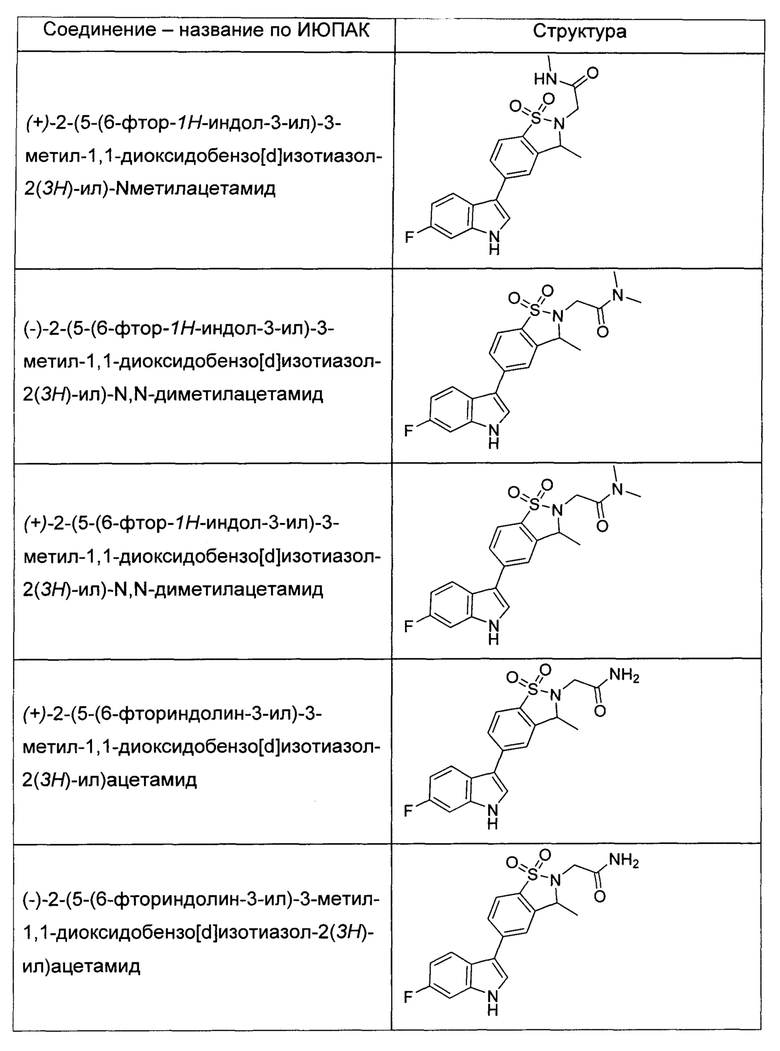
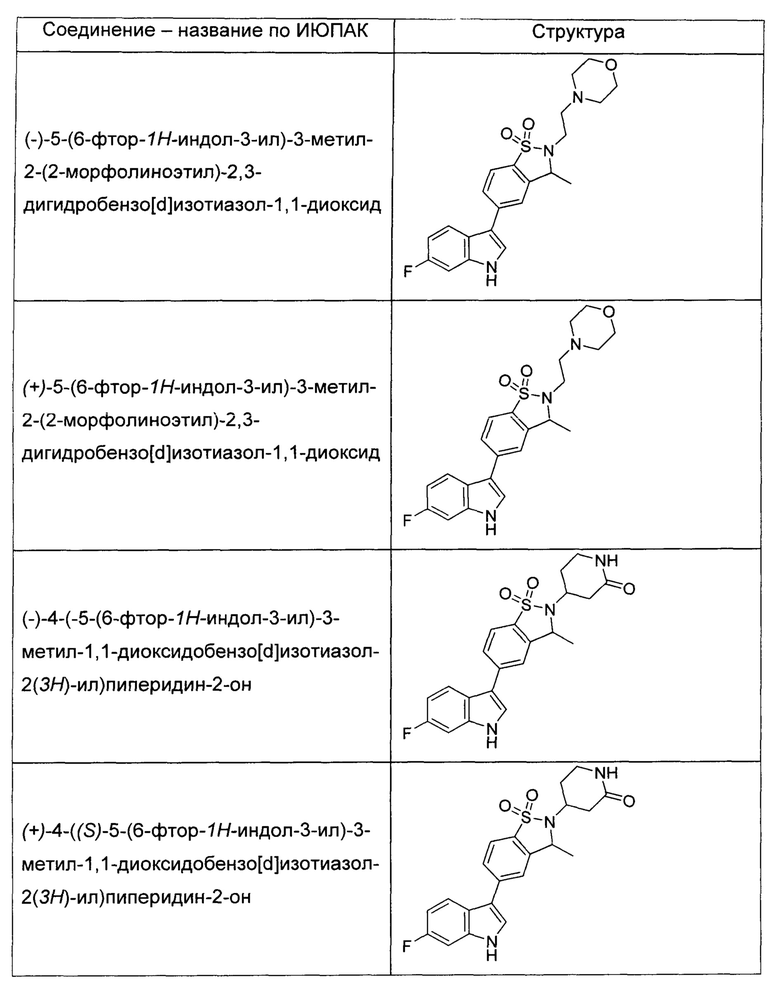
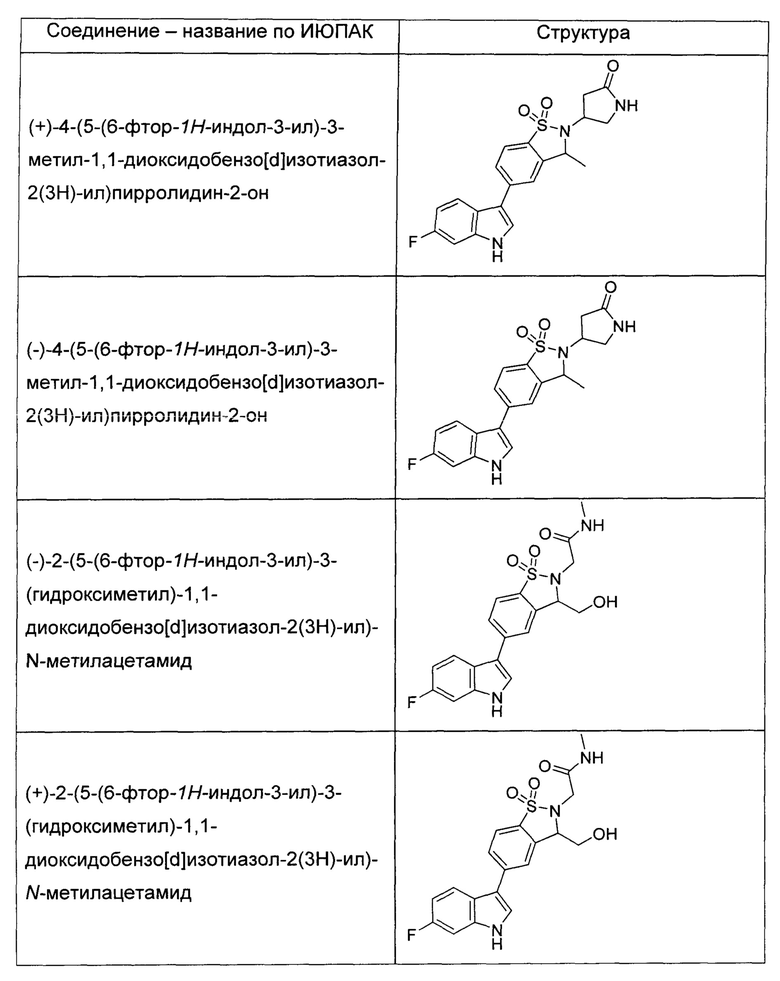
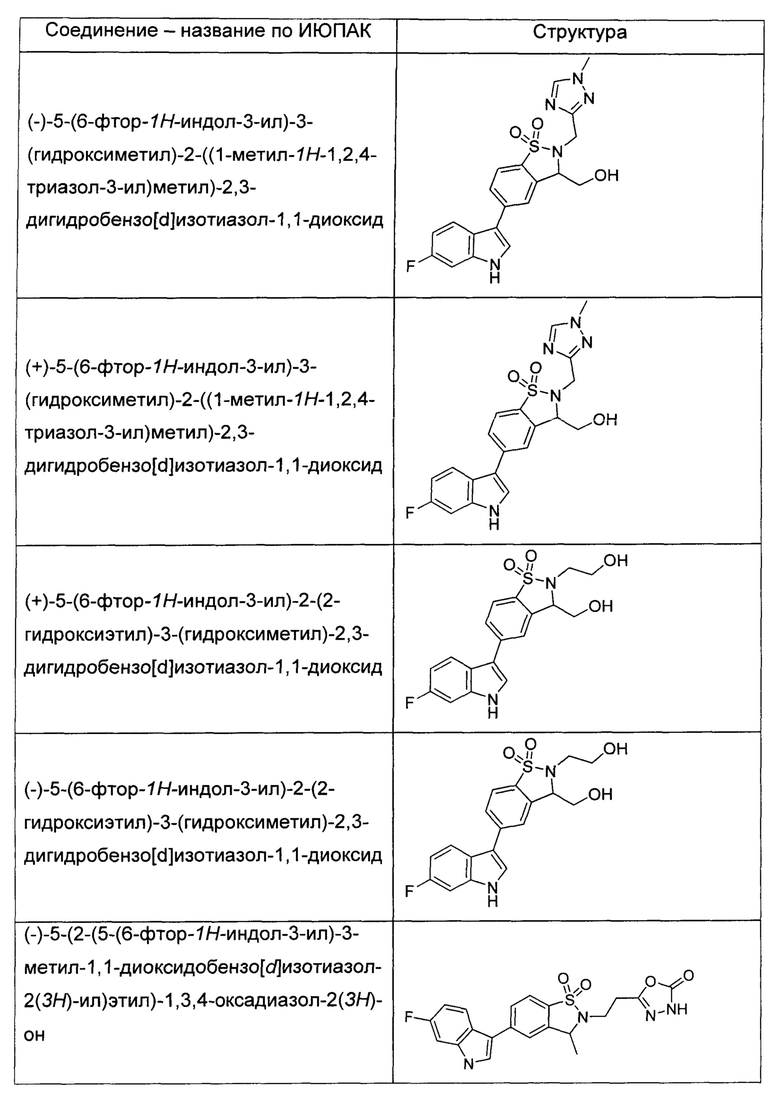
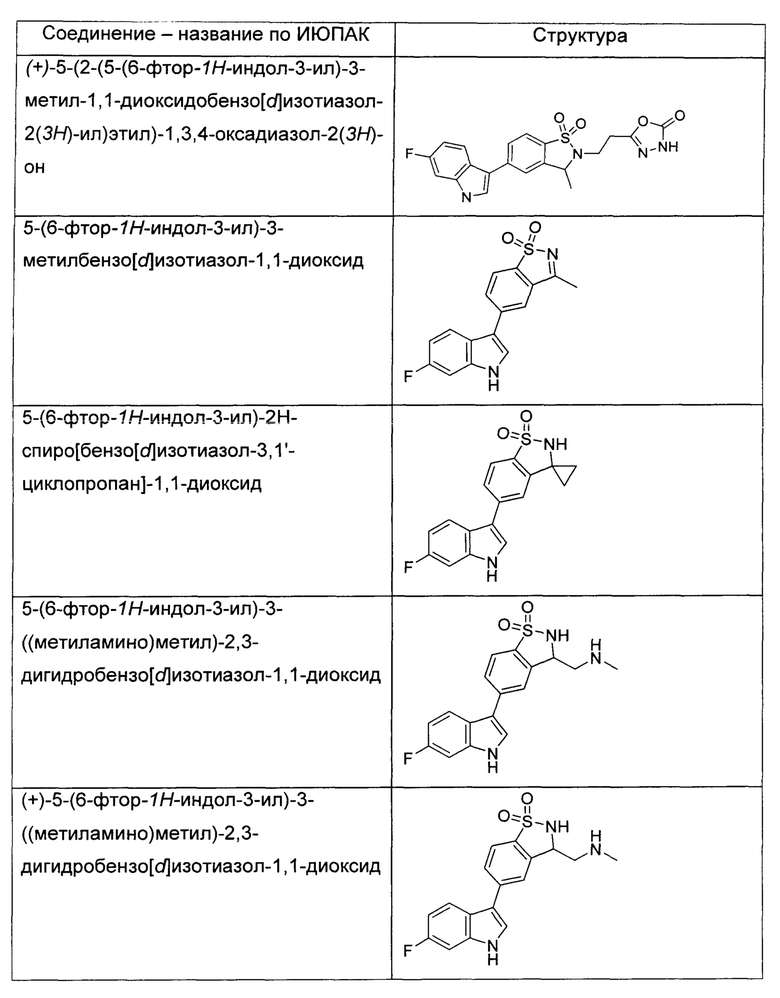
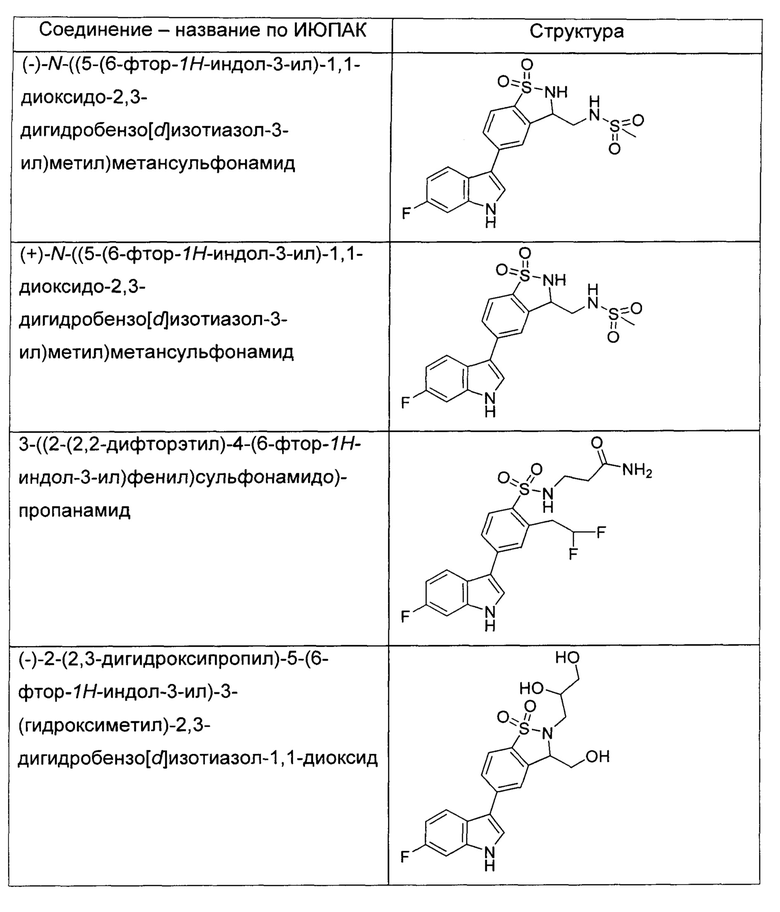
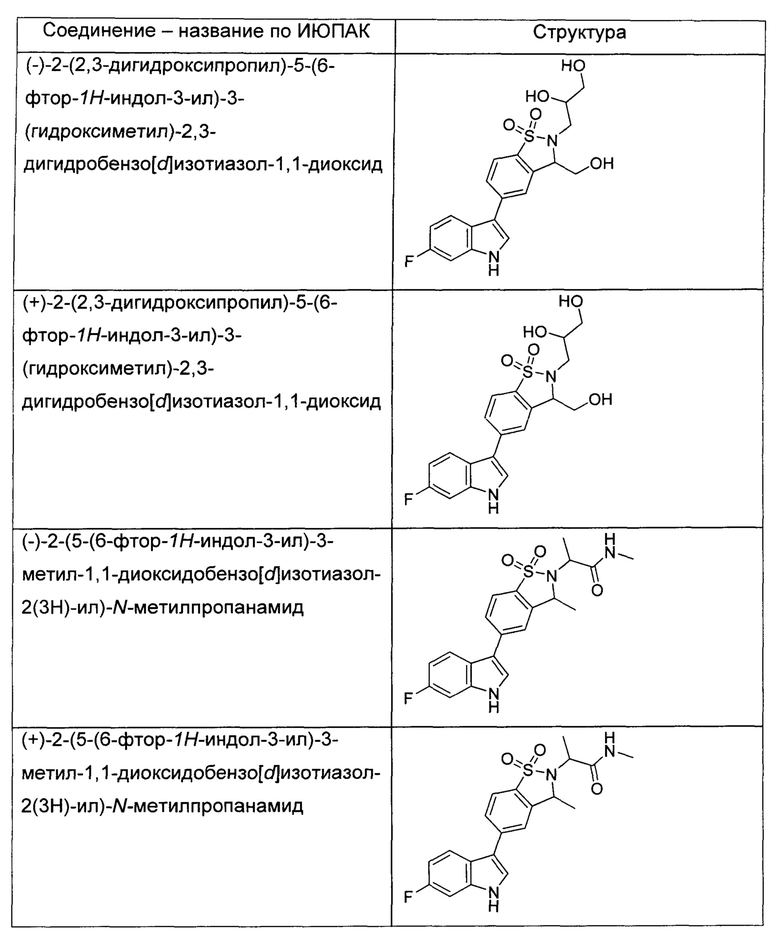
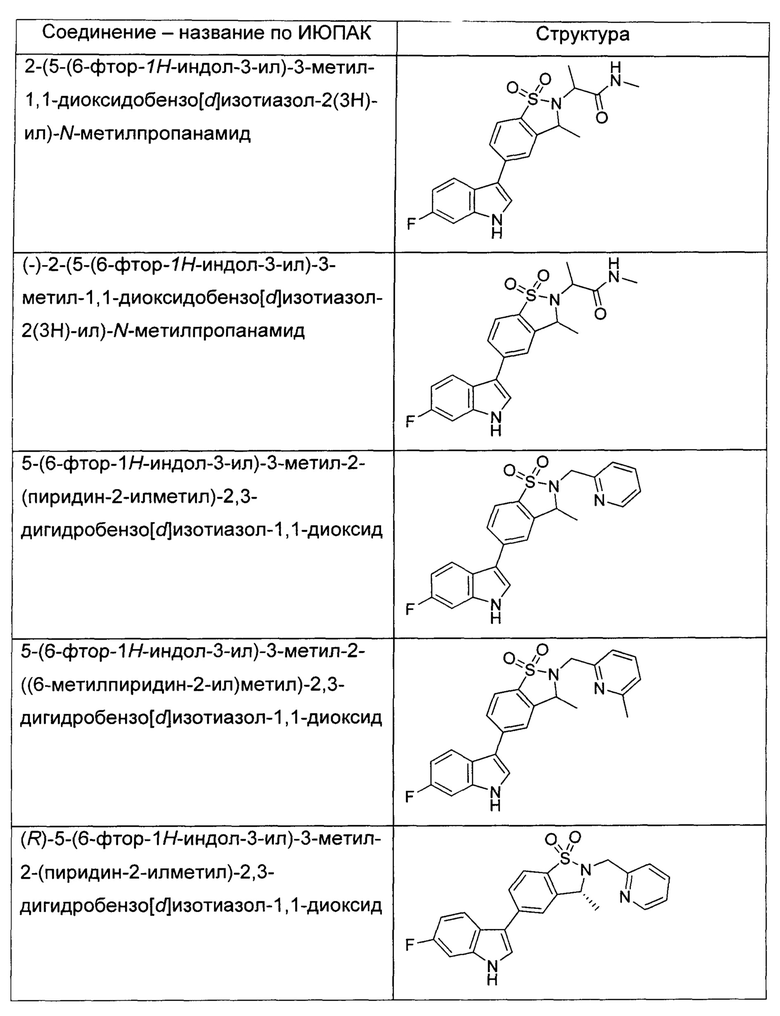
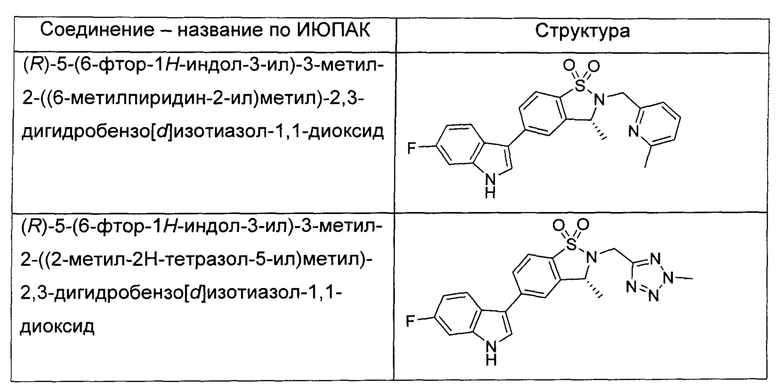
или их фармацевтически приемлемые энантиомеры, соли и сольваты. В одном воплощении выбрано соединение, которое является энантиомером. В другом воплощении выбрано соединение, которое представляет собой соль. В дополнительном воплощении выбрано соединение, которое представляет собой сольват. В еще одном воплощении выбрано соединение из Таблицы 1 формулы I (или ее подформул), которое является свободным основанием (не является солью). Также здесь охватываются соли данных формул, соли энантиомеров и сольваты таких солей.
Соединениям Таблицы 1 присвоены названия с использованием ChemBioDraw® Ultra версии 12.0 (PerkinElmer).
Соединения формулы I и ее подформул могут содержать асимметрический центр и поэтому могут существовать в виде различных стереоизомерных форм. Соответственно, настоящее изобретение включает все возможные стереоизомеры и включает не только рацемические соединения, но и индивидуальные энантиомеры, а также их нерацемические смеси. Когда требуется соединение в виде индивидуального стереоизомера, оно может быть получено стереоспецифическим синтезом, разделением конечного продукта или любого подходящего промежуточного соединения или хиральными хроматографическими способами, каждые из которых известны в данной области техники. Разделение конечного продукта, промежуточного соединения или исходного материала может быть осуществлено любым подходящим способом, известным в данной области техники.
Соединения по изобретению могут находиться в форме "фармацевтически приемлемых солей". Фармацевтически приемлемые соли соединений формулы 1 включают их соли присоединения кислоты и соли с основаниями. Подходящие соли присоединения кислоты получают из кислот, которые образуют нетоксичные соли. Примеры включают соли ацетат, лактобионат, бензолсульфонат, лаурат, адипат, аспартат, бензоат, бесилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камсилат, цитрат, цикламат, эдисилат, эсилат, формиат, фумарат, глюцептат, глюконат, глюкуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изэтионат, лактат, малат, малеат, малонат, манделат, битартрат, метилбромид, бромид, метилнитрат, эдетат кальция, мукат, напсилат, хлорид, клавуланат, бутил(N)-олеат, эдетат, эстолат, пантотенат, полигалактуронат, салицилат, глутамат, гликоллиларсанилат, сульфат, гексилрезорцинат, субацетат, гидрабамин, гидроксинафталоат, этолат, триэтиодид, валерат, мезилат, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, пироглутамат, сахарат, стеарат, сукцинат, таннат, тартрат, тозилат, трифторацетат и ксинафоат. Подходящие соли с основаниями получают из оснований, которые образуют нетоксичные соли. Примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина, лизина, магния, меглумина, оламина, орнитина, N,N-дибензилэтилендиамина, пиперазина, трис(гидроксиметил)аминометана, гидроксида тетраметиламмония, метилглюкамина, соли аммония, калия, натрия, трометамина, 2-(диэтиламино)этанола, этаноламина, морфолина, 4-(2-гидроксиэтил)-морфолина и цинка. Также могут быть образованы гемисоли кислот и оснований, например, гемисульфат и гемикальциевые соли. Предпочтительно, фармацевтически приемлемые соли включают гидрохлорид/хлорид, гидробромид/бромид, бисульфат/сульфат, нитрат, цитрат и ацетат.
Когда соединения по изобретению содержат кислотную группу, а также основную группу, соединения по изобретению могут также образовывать внутренние соли, и такие соединения входят в объем данного изобретения. Когда соединения по изобретению содержат водород-донорный гетероатом (например NH), изобретение также охватывает соли и/или изомеры, образуемые при переносе указанного атома водорода на основные группу или атом в молекуле.
Фармацевтически приемлемые соли соединений формулы 1 могут быть получены одним или более чем одним из следующих способов:
(1) путем взаимодействия соединения формулы I с требуемой кислотой;
(2) путем взаимодействия соединения формулы I с требуемым основанием;
(3) путем удаления чувствительной к кислоте или основанию защитной группы с подходящего предшественника соединения формулы I или путем раскрытия кольца подходящего циклического предшественника, например, лактона или лактама, с использованием требуемой кислоты; или
(4) путем превращения одной соли соединения формулы I в другую посредством взаимодействия с подходящей кислотой или посредством подходящей ионообменной колонки.
Все эти взаимодействия обычно осуществляют в растворе. Соль можно осадить из раствора и собрать посредством фильтрации или можно выделить путем выпаривания растворителя. Степень ионизации в соли может варьировать от полностью ионизированной до почти неионизированной.
Соединения по настоящему изобретению могут быть введены в форме фармацевтически приемлемых солей, которые являются такими как определено выше. Эти соли могут быть получены стандартными методами, например, путем взаимодействия свободной кислоты с подходящим органическим или неорганическим основанием. Когда присутствует основная группа, такая как амино, кислая соль, то есть гидрохлорид, гидробромид, ацетат, пальмоат и тому подобное, может быть использована в качестве дозированной формы.
Также, в случае присутствия спиртовой группы, могут быть использованы фармацевтически приемлемые сложные эфиры, например, ацетат, малеат, пивалоилоксиметил и тому подобное, и те сложные эфиры, которые известны в данной области техники для модификации характеристик растворимости или гидролиза для использования в качестве препаратов продолжительного высвобождения или пролекарственных препаратов.
Способ получения
Соединения формулы I могут быть получены различными путями с использованием реакций, известных специалистам в данной области техники.
Изобретение дополнительно относится к первому способу получения соединений формулы I
и их фармацевтически приемлемых энантиомеров, солей и сольватов, где X1, X2, R1, R2, R3, А1, А2 и Q являются такими, как определено в формуле I;
включающему:
(а1) взаимодействие соединения формулы (i)
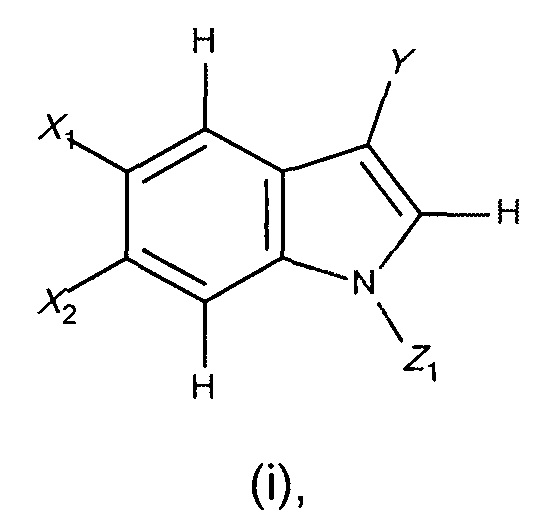
где
X1 и X2 являются такими, как определено в формуле I;
Z1 представляет собой Q или амино-защитную группу, такую как, например, арилсульфонил, трет-бутоксикарбонил, метоксиметил, пара-метоксибензил, бензил или любую другую подходящую защитную группу, известную специалистам в данной области техники;
Y представляет собой галоген (предпочтительно йод, бром или хлор), алкилсульфонилокси, имеющий 1-6 атомов углерода (предпочтительно метилсульфонилокси или трифторметилсульфонилокси), или арилсульфонилокси, имеющий 6-10 атомов углерода (предпочтительно фенил- или пара-толилсульфонилокси), или любую уходящую группу, известную специалистам в данной области техники;
с соединением формулы (ii)
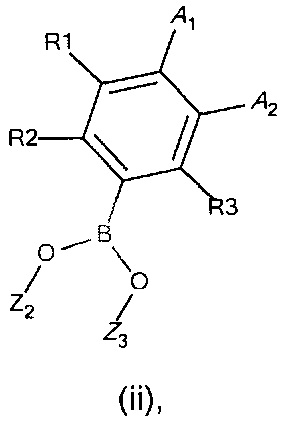
где
R1, R2, R3, А1, А2 и А3 являются такими, как определено в формуле I;
Z2 и Z3 представляют собой Н или алкильные группы, с возможностью для Z2 и Z3 образовывать кольцо;
с получением соединения формулы (iii),
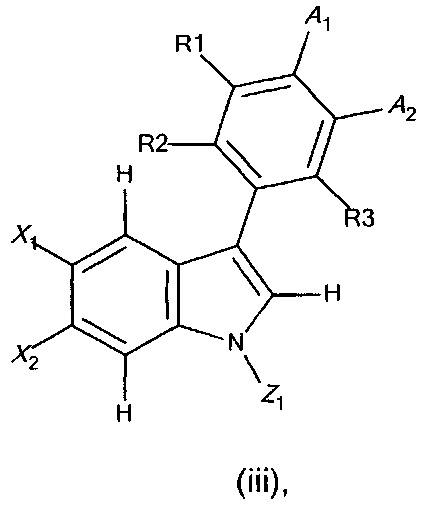
где X1, X2, R1, R2, R3, А1, А2 и Z1 являются такими, как определено выше;
и
(b1) в случае когда Z1 не представляет собой Q, снятие защиты с индол-амина соединения формулы (iii), с получением соединения формулы I.
Согласно одному воплощению, стадия (а1) может быть осуществлена с использованием или без использования катализатора, такого как, без ограничения, Pd2(dba)3, Pd(PPh3)4, дихлорбис(трифенилфосфин)палладий(II) или 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий(II), Pd(OAc)2 или Pd/C, в присутствии или в отсутствие дополнительного лиганда, такого как, без ограничения, X-Phos, S-Phos, P(oTol)3, PPh3, BINAP, P(tBu)3 или любой другой подходящий фосфиновый лиганд, известный специалистам в данной области техники.
Согласно одному воплощению, стадия (а1) может быть осуществлена в присутствии оснований, таких как, без ограничения, K3PO4, K2CO3, Na2CO3.
Согласно одному воплощению, стадия (а1) может быть осуществлена в присутствии подходящего растворителя, такого как, без ограничения, диоксан, THF, DMF, вода или их смеси, предпочтительно в смеси диоксана или THF и воды.
Согласно одному воплощению, стадия (а1) может быть осуществлена при температуре в диапазоне от 20°C до примерно 180°C, с использованием микроволнового облучения или без него, в течение периода времени в диапазоне от 10 минут до нескольких часов, например, от 10 минут до 24 часов.
Согласно одному воплощению, стадия снятия защиты (b1) может быть осуществлена, в зависимости от природы группы Z1, путем обработки основаниями, такими как, без ограничения, гидроксид натрия, гидроксид калия, карбонат калия. Согласно одному воплощению, снятие защиты может быть осуществлено в присутствии или в отсутствие подходящего растворителя, такого как, без ограничения, метанол, этанол, изопропанол, трет-бутанол, THF, DMF, диоксан, вода или их смесь. Согласно одному воплощению, снятие защиты может быть осуществлено при температуре в диапазоне от 20°C до 100°C, предпочтительно при примерно 85°C, в течение нескольких часов, например, от одного часа до 24 часов.
Согласно альтернативному воплощению, снятие защиты (b1) может быть осуществлено, в зависимости от природы группы Z1, в присутствии сильных кислот, таких как, без ограничения, HCl, TFA, HF, HBr. Согласно одному воплощению, снятие защиты может быть осуществлено в присутствии или в отсутствие подходящего растворителя, такого как метанол, этанол, изопропанол, трет-бутанол, THF, DMF, диоксан, вода или их смесь. Согласно одному воплощению, снятие защиты может быть осуществлено при температуре от примерно 20°C до примерно 100°C, в течение периода времени от 10 минут до нескольких часов, например, от 10 минут до 24 часов.
Также предложен второй способ получения соединений формулы I
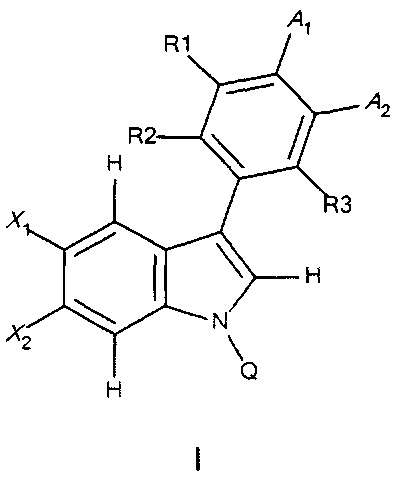
и их фармацевтически приемлемых энантиомеров, солей и сольватов, где X1, X2, R1, R2, R3, А1, А2 и Q являются такими, как определено в формуле I;
включающий:
(а2) взаимодействие соединения формулы (iv)

где
X1 и X2 являются такими, как определено в формуле I;
Z1 представляет собой Q или амино-защитную группу, такую как, например, арилсульфонил, трет-бутоксикарбонил, метоксиметил, пара-метоксибензил, бензил или любую другую подходящую защитную группу, известную специалистам в данной области техники;
Z2 и Z3 представляют собой Н или алкильные группы, с возможностью для Z2 и Z3 образовывать кольцо;
с соединением формулы (v)

где
R1, R2, R3, А1 и А2 являются такими, как определено в формуле I;
Y представляет собой галоген (предпочтительно йод, бром или хлор), алкилсульфонилокси, имеющий 1-6 атомов углерода (предпочтительно метилсульфонилокси или трифторметилсульфонилокси), или арилсульфонилокси, имеющий 6-10 атомов углерода (предпочтительно фенил- или пара-толилсульфонилокси), или любую уходящую группу, известную специалистам в данной области техники;
с получением соединения формулы (vi),
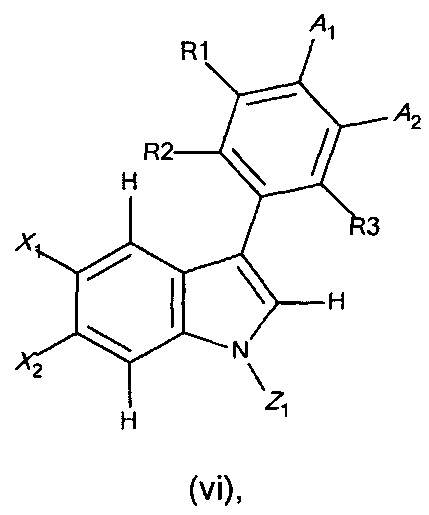
где X1, X2, R1, R2, R3, А1, А2 и Z1 являются такими, как определено выше;
и
(b2) в случае когда Z1 не представляет собой Q, снятие защиты с индол-амина соединения формулы (xii), с получением соединения формулы I (или ее подформул).
Согласно одному воплощению, стадия (а2) может быть осуществлена с использованием или без использования катализатора, такого как, без ограничения, Pd2(dba)3, Pd(PPh3)4, дихлорбис(трифенилфосфин)палладий(II) или 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий(II), Pd(OAc)2 или Pd/C, в присутствии или в отсутствие дополнительного лиганда, такого как, без ограничения, X-Phos, S-Phos, P(oTol)3, PPh3, BINAP, P(tBu)3 или любой другой подходящий фосфиновый лиганд, известный специалистам в данной области техники.
Согласно одному воплощению, стадия (а2) может быть осуществлена в присутствии оснований, таких как, без ограничения, K3PO4, K2CO3, Na2CO3.
Согласно одному воплощению, стадия (а2) может быть осуществлена в присутствии подходящего растворителя, такого как, без ограничения, диоксан, THF, DMF, вода или их смеси, предпочтительно в смеси диоксана или THF и воды.
Согласно одному воплощению, стадия (а2) может быть осуществлена при температуре в диапазоне от 20°C до примерно 180°C, с использованием микроволнового облучения или без него, в течение периода времени в диапазоне от 10 минут до нескольких часов, например, от 10 минут до 24 часов.
Согласно одному воплощению, стадия снятия защиты (b2) может быть осуществлена в условиях, описанных выше для снятия защиты (b1).
В общем, пути синтеза для любого индивидуального соединения формулы (I) будут зависеть от конкретных заместителей каждой молекулы и исходя из доступности необходимых готовых промежуточных соединений; опять же, таких факторов, которые учитываются специалистами в данной области техники.
Согласно дополнительному общему способу, соединения формулы I могут быть преобразованы в альтернативные соединения формулы I с использованием подходящих методик взаимопревращения, хорошо известных специалисту в данной области техники.
Соединения формулы I и связанных с ней формул могут дополнительно быть получены выделением соединений формулы I из одного из их функциональных производных путем обработки агентом сольволиза или гидрогенолиза.
Предпочтительные исходные материалы для сольволиза или гидрогенолиза представляют собой те, которые соответствуют формуле I и связанным с ней формулам, но содержат соответствующие защищенные аминогруппы и/или гидроксильные группы вместо одной или более свободных аминогрупп и/или гидроксильных групп, предпочтительно те, которые несут амино-защитную группу вместо атома Н, связанного с атомом N, в частности те, которые несут группу R*-N, в которой R* означает амино-защитную группу, вместо группы HN, и/или те, которые несут гидроксил-защитную группу вместо атома Н гидроксильной группы, например те, которые соответствуют формуле I, но несут группу -COOR**, в которой R** означает гидроксил-защитную группу, вместо группы -COOH.
Также возможно присутствие множества идентичных или разных защищенных аминогрупп и/или гидроксильных групп в молекуле исходного материала. Если присутствующие защитные группы отличаются одна от другой, они могут быть во многих случаях отщеплены по отдельности.
Термин "амино-защитная группа" в общем известен и относится к группам, которые подходят для защиты (блокирования) аминогруппы от химических реакций, но легко удаляются после осуществления требуемой химической реакции где-либо в молекуле. Обычно такие группы представляют собой, в частности, незамещенные или замещенные ацильные, арильные, аралкоксиметильные или аралкильные группы. Поскольку амино-защитные группы удаляют после осуществления требуемой реакции (или последовательности реакций), их тип и размер более не является важным; однако, предпочтение дается группам, имеющим 1-20, в частности 1-8, атомов углерода. Термин "ацильная группа" следует понимать в наиболее широком смысле в контексте способа по настоящему изобретению. Он включает ацильные группы, производные от алифатических, аралифатических, ароматических или гетероциклических карбоновых кислот или сульфоновых кислот и, в частности, группы алкоксикарбонил, арилоксикарбонил и особенно аралкоксикарбонил. Примерами таких ацильных групп являются алканоил, такой как ацетил, пропионил и бутирил; аралканоил, такой как фенилацетил; ароил, такой как бензоил и толил; арилоксиалканоил, такой как РОА; алкоксикарбонил, такой как метоксикарбонил, этоксикарбонил, 2,2,2-трихлорэтоксикарбонил, ВОС (трет-бутоксикарбонил) и 2-йодэтоксикарбонил; аралкоксикарбонил, такой как CBZ ("карбобензокси"), 4-метоксибензилоксикарбонил и FMOC; и арилсульфонил, такой как Mtr. Предпочтительные амино-защитные группы представляют собой ВОС и Mtr, CBZ, Fmoc, бензил и ацетил.
Термин "гидроксил-защитная группа" аналогичным образом известен в общем виде и относится к группам, которые подходят для защиты гидроксильной группы от химических реакций, но легко удаляются после осуществления требуемой химической реакции где-либо в молекуле. Обычно такие группы представляют собой вышеупомянутые незамещенные или замещенные арильные, аралкильные или ацильные группы, дополнительно также алкильные группы. Природа и размер гидроксил-защитных групп не важны, поскольку они опять же удаляются после осуществления требуемой химической реакции или последовательности реакций; предпочтение дается группам, имеющим 1-20, в частности 1-10, атомов углерода. Примеры гидроксил-защитных групп представляют собой, среди прочего, бензил, 4-метоксибензил, пара-нитробензоил, пара-толуолсульфонил, трет-бутил и ацетил, причем бензил и трет-бутил являются особенно предпочтительными.
Соединения формулы I и связанных с ней формул выделяют из их функциональных производных, в зависимости от используемой защитной группы, например, сильных неорганических кислот, таких как соляная кислота, перхлорная кислота или серная кислота, сильных органических карбоновых кислот, таких как трихлоруксусная кислота, трифторуксусная кислота (TFA), или сульфоновых кислот, таких как бензол- или пара-толуолсульфоновая кислота. Присутствие дополнительного инертного растворителя возможно, но не всегда обязательно. Подходящие инертные растворители предпочтительно представляют собой органические, например, карбоновые кислоты, такие как уксусная кислота, эфиры, такие как тетрагидрофуран или диоксан, амиды, такие как DMF, галогенированные углеводороды, такие как дихлорметан, и дополнительно также спирты, такие как метанол, этанол или изопропанол, и воду. Смеси вышеупомянутых растворителей также являются подходящими. TFA предпочтительно используют в избытке без добавления дополнительного растворителя, и перхлорную кислоту предпочтительно используют в смеси уксусной кислоты и 70%-ной перхлорной кислоты в соотношении 9:1. Температуры реакционных смесей для расщепления преимущественно составляют от примерно 0 до примерно 50°C, предпочтительно от 15 до 30°C (комнатная температура).
Группы ВОС, OtBu и Mtr можно, например, предпочтительно отщепить с использованием TFA в дихлорметане или используя приблизительно 3-5 н. HCl в диоксане при 15-30°C, а группу FMOC можно отщепить с использованием приблизительно 5-50% раствора диметиламина, диэтиламина или пиперидина в DMF при 15-30°C.
Защитные группы, которые могут быть удалены путем гидрогенолиза (например, CBZ, бензил, или высвобождение группы амидино из его оксадиазолового производного), можно отщепить, например, путем обработки водородом в присутствии катализатора (например катализатора на основе благородного металла, такого как палладий, преимущественно на подложке, такой как углерод). Подходящие растворители здесь представляют собой такие, как указаны выше, в частности, например, спирты, такие как метанол или этанол, или амиды, такие как DMF. Гидрогенолиз в общем осуществляют при температурах в диапазоне примерно от 0 до 100°C и давлениях в диапазоне от примерно 1 до 200 бар, предпочтительно при 20-30°C и 1-10 бар. Гидрогенолиз группы CBZ успешно осуществляют, например, на 5-10% Pd/C в метаноле или с использованием формиата аммония (вместо водорода) на Pd/C в смеси метанол/DMF при 20-30°C.
Примеры подходящих инертных растворителей представляют собой углеводороды, такие как гексан, петролейный эфир, бензол, толуол или ксилол; хлоруглеводороды, такие как трихлорэтилен, 1,2-дихлорэтан, тетрахлорметан, трифторметилбензол, хлороформ или дихлорметан; спирты, такие как метанол, этанол, изопропанол, н-пропанол, н-бутанол или трет-бутанол; эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран (THF) или диоксан; гликолевые эфиры, такие как монометиловый или моноэтиловый эфир этиленгликоля или диметиловый эфир этиленгликоля (диглим); кетоны, такие как ацетон или бутанон; амиды, такие как ацетамид, диметилацетамид, N-метилпирролидон (NMP) или диметилформамид (DMF); нитрилы, такие как ацетонитрил; сульфоксиды, такие как диметилсульфоксид (DMSO); дисульфид углерода; карбоновые кислоты, такие как муравьиная кислота или уксусная кислота; нитросоединения, такие как нитрометан или нитробензол; сложные эфиры, такие как этилацетат, или смеси указанных растворителей.
Сложные эфиры могут быть гидролизованы, например, с использованием HCl, H2SO4, или с использованием LiOH, NaOH или KOH в воде, смесях вода/THF, вода/THF/этанол или вода/диоксан, при температурах в диапазоне от 0 до 100°C.
Свободные аминогруппы могут быть дополнительно ацилированы традиционным способом с использованием хлорангидрида или ангидрида либо алкилированы с использованием незамещенного или замещенного алкилгалогенида, преимущественно в инертном растворителе, таком как дихлорметан или THF, и/или в присутствии основания, такого как триэтиламин или пиридин, при температурах в диапазоне от -60°C до +30°C.
Для всех способов введения и снятия защиты см. Philip J. Kocienski в "Protecting groups", Georg Thieme Verlag Stuttgart, New York, 1994 and, Theodora W. Greene and Peter G.M. Wuts in "Protective groups in Organic Synthesis", Wiley Interscience, 3rd Edition 1999.
Реакционные схемы, как описано в разделе Примеров, являются только иллюстративными и не должны интерпретироваться как ограничивающие объем изобретения каким-либо образом.
Применения
Соединение формулы I (включая ее подформулы, например формулы Ia, Ib и II) или фармацевтически приемлемые энантиомеры, соли и сольваты являются полезными в качестве активного ингредиента в фармацевтической композиции или препарате. В одном воплощении соединение используют в качестве ингибитора TDO2.
Соответственно, в конкретном предпочтительном воплощении соединения формулы I и подформул, включая, без ограничения, соединения из Таблицы 1 выше, или их фармацевтически приемлемые энантиомеры, соли и сольваты используют в качестве ингибиторов TDO2.
Соответственно, в другом аспекте эти соединения или их энантиомеры, соли и сольваты используют в синтезе фармацевтически активных ингредиентов, таких как ингибиторы TDO2.
В одном воплощении соединения формулы I и подформул, в частности соединения из Таблицы 1 выше, или их фармацевтически приемлемые энантиомеры, соли и сольваты используют для повышения иммунного распознавания и деструкции раковых клеток.
Соединения формулы I и подформул полезны в качестве лекарственных средств, в частности, в предупреждении и/или лечении рака.
В одном воплощении описанные здесь соединения или их фармацевтически приемлемые энантиомеры, соли и сольваты предназначены для использования в лечении и/или предупреждении рака, нейродегенеративных расстройств, таких как болезнь Паркинсона, болезнь Альцгеймера и болезнь Гентингтона, хронических вирусных инфекций, таких как HCV и HIV, депрессии и ожирения.
Также предложен способ лечения или предупреждения рака, нейродегенеративных расстройств, таких как болезнь Паркинсона, болезнь Альцгеймера и болезнь Гентингтона, хронических вирусных инфекций, таких как HCV и HIV, депрессии и ожирения, включающий введение млекопитающему, нуждающемуся в этом, терапевтически эффективного количества соединения по изобретению или его фармацевтически приемлемых энантиомеров, солей или сольватов.
Различные виды рака известны в данной области техники. Рак может быть метастатическим или неметастатическим. Рак может быть наследственным или спорадическим. В некоторых воплощениях рак выбран из группы, состоящей из: лейкоза и множественной миеломы. Дополнительные виды рака, которые можно лечить с использованием способов по изобретению, включают, например, доброкачественные и злокачественные солидные опухоли и доброкачественные и злокачественные несолидные опухоли.
Примеры солидных опухолей включают, без ограничения: рак желчевыводящих путей, рак мозга (включая глиобластомы и медуллобластомы), рак молочной железы, рак шейки матки, хориокарциному, рак толстой кишки, рак эндометрия, рак пищевода, рак желудка, интраэпителиальные неоплазмы (включая болезнь Боуэна и болезнь Педжета), рак печени, рак легких, нейробластомы, рак полости рта (включая плоскоклеточный рак), рак яичников (включая те виды, которые происходят из эпителиальных клеток, стромальных клеток, эмбриональных клеток и мезенхимальных клеток), рак поджелудочной железы, рак предстательной железы, рак прямой кишки, рак почки (включая аденокарциному и опухоль Вильмса), саркомы (включая лейомиосаркому, рабдомиосаркому, липосаркому, фибросаркому и остеосаркому), рак кожи (включая меланому, саркому Капоши, базальноклеточный рак и плоскоклеточный рак), рак яичек, включая эмбрионально-клеточные опухоли (семиномы и несеминомы, такие как тератомы и хориокарциномы), стромальные опухоли, эмбрионально-клеточные опухоли и рак щитовидной железы (включая аденокарциному щитовидной железы и медуллярный рак).
Примеры несолидных опухолей включают, без ограничения, гематологический рак. Как используют здесь, гематологический рак означает термин в данной области, который включает лимфоидные заболевания, миелоидные заболевания и СПИД-ассоциированные лейкозы.
Лимфоидные заболевания представляют собой, без ограничения, острый лимфоцитарный лейкоз и хронические лимфопролиферативные расстройства (например, лимфомы, миеломы и хронические лимфоидные лейкозы). Лимфомы включают, например, лимфому Ходжкина, неходжкинские лимфомы и лимфоцитарные лимфомы). Хронические лимфоидные лейкозы включают, например, Т-клеточные хронические лимфоидные лейкозы и В-клеточные хронические лимфоидные лейкозы.
В изобретении также предложен способ задержки у пациента начала возникновения рака, включающий введение пациенту, нуждающемуся в этом, фармацевтически эффективного количества соединения формулы I или фармацевтически приемлемого энантиомера, соли и сольвата.
Предпочтительно, пациент представляет собой теплокровного животного, более предпочтительно человека.
Соединения по изобретению особенно полезны в лечении и/или предупреждении рака.
В конкретном воплощениии соединения по изобретению особенно полезны в лечении и/или предупреждении рака.
В изобретении дополнительно предложено применение соединения формулы I или его фармацевтически приемлемых энантиомера, соли и сольвата для изготовления лекарственного средства для лечения и/или предупреждения рака.
Согласно дополнительному аспекту настоящего изобретения предложен способ модулирования активности TDO2 у пациента, предпочтительно теплокровного животного, предпочтительно млекопитающего и еще более предпочтительно человека, нуждающегося в таком лечении, включающий введение указанному пациенту эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемых энантиомера, соли и сольвата.
В дополнительном воплощении в изобретении предложено применение соединения формулы I (или ее подформул) или его фармацевтически приемлемых энантиомера, соли или сольвата для использования в лечении и/или предупреждении рака. В одном воплощении рак представляет собой карциному мочевого пузыря. В другом воплощении рак представляет собой гепатокарциному. В дополнительном воплощении рак представляет собой меланому. В еще одном воплощении рак представляет собой мезотелиому. В еще одном воплощении рак представляет собой нейробластому. В еще одном воплощении рак представляет собой саркому. В еще одном воплощении рак представляет собой карциному молочной железы. В еще одном воплощении рак представляет собой лейкоз. В еще одном воплощении рак представляет собой почечно-клеточную карциному. В еще одном воплощении рак представляет собой колоректальную карциному. В еще одном воплощении рак представляет собой карциному головы и шеи. В еще одном воплощении рак представляет собой карциному легких. В еще одном воплощении рак представляет собой опухоль мозга. В еще одном воплощении рак представляет собой глиобластому. В еще одном воплощении рак представляет собой астроцитому. В еще одном воплощении рак представляет собой миелому. В еще одном воплощении рак представляет собой панкреатическую карциному.
В другом воплощении в изобретении предложено применение соединения формулы I (или ее подформул) или его фармацевтически приемлемых энантиомера, соли или сольвата для использования в лечении нейродегенеративного расстройства. В одном воплощении расстройство представляет собой болезнь Паркинсона. В другом воплощении расстройство представляет собой болезнь Альцгеймера. В еще одном воплощении расстройство представляет собой болезнь Гентингтона.
В еще одном воплощении предложено применение соединения формулы I (или ее подформул) или его фармацевтически приемлемых энантиомера, соли или сольвата в лечении хронических вирусных инфекций, таких как HCV и HIV.
В еще одном воплощении предложено применение соединения формулы I (или ее подформул) или его фармацевтически приемлемых энантиомера, соли или сольвата в лечении депрессии.
В еще одном воплощении предложено применение соединения формулы I (или ее подформул) или его фармацевтически приемлемых энантиомера, соли или сольвата в лечении ожирения.
Для применения в таких лечениях предложенные здесь соединения могут быть изготовлены в виде композиций как следует ниже.
Композиции
В изобретении также предложены фармацевтические композиции, содержащие одно или более соединений формулы I и/или ее подформул или их фармацевтически приемлемых энантиомеров, солей и сольватов и по меньшей мере один фармацевтически приемлемый носитель, разбавитель, эксципиент и/или адъювант. Как указано выше, изобретение также охватывает фармацевтические композиции, содержащие, в дополнение к соединению по настоящему изобретению, его фармацевтически приемлемым энантиомеру, соли и сольвату в качестве активного ингредиента, дополнительные терапевтические агенты и/или активные ингредиенты.
Еще одним объектом данного изобретения является лекарственное средство, содержащее по меньшей мере одно соединение по изобретению или его фармацевтически приемлемые энантиомер, соль и сольват в качестве активного ингредиента.
Согласно дополнительному аспекту настоящего изобретения предложено применение соединения формулы I или его фармацевтически приемлемых энантиомера, соли и сольвата для изготовления лекарственного средства для модулирования активности TDO2 у пациента, нуждающегося в этом, включающее введение указанному пациенту эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемых энантиомера, соли и сольвата.
В целом, для фармацевтического применения соединения по изобретению могут быть изготовлены в виде фармацевтического препарата, содержащего по меньшей мере одно соединение по изобретению и по меньшей мере один фармацевтически приемлемый носитель, разбавитель, эксципиент и/или адъювант и возможно один или более дополнительных фармацевтически активных соединений.
В качестве неограничивающих примеров, такая композиция может быть представлена в форме, подходящей для перорального введения, для парентерального введения (такого как внутривенная, внутримышечная или подкожная инъекция или внутривенная инфузия), для местного введения (включая глазное), для введения путем ингаляции, посредством кожного пластыря, посредством имплантата, посредством суппозитория и так далее. Такие подходящие формы введения - которые могут быть твердыми, полутвердыми или жидкими, в зависимости от пути введения - а также способы и носители, разбавители и эксципиенты для использования в препарате, будут очевидны специалисту в данной области техники; ссылка сделана на последнюю редакцию Remington's Pharmaceutical Sciences.
Некоторые предпочтительные, но неограничивающие примеры таких препаратов включают таблетки, пилюли, порошки, лепешки, саше, облатки, эликсиры, суспензии, эмульсии, растворы, сиропы, аэрозоли, мази, кремы, лосьоны, мягкие и твердые желатиновые капсулы, суппозитории, капли, стерильные инъекционные растворы и стерильные упакованные порошки (которые обычно разводят перед использованием) для введения в виде болюса и/или для продолжительного введения, которые могут быть изготовлены с носителями, эксципиентами и разбавителями, подходящими per se для таких композиций, такими как лактоза, декстроза, сахароза, сорбит, маннит, крахмалы, аравийская камедь, фосфат кальция, альгинаты, трагакантовая камедь, желатин, силикат кальция, микрокристаллическая целлюлоза, поливинилпирролидон, полиэтиленгликоль, целлюлоза, (стерильная) вода, метилцеллюлоза, метил- и пропилгидроксибензоаты, тальк, стеарат магния, съедобные масла, растительные масла и минеральные масла или их подходящие смеси. Композиции возможно могут содержать другие вещества, которые общепринято используются в фармацевтических композициях, такие как смазывающие агенты, увлажняющие агенты, эмульгаторы и суспендирующие агенты, диспергирующие агенты, разрыхлители, объемообразующие агенты, наполнители, консерванты, подсластители, корригенты, регуляторы текучести, агенты высвобождения и так далее. Композиции также могут быть изготовлены с возможностью обеспечения быстрого, длительного или отсроченного высвобождения активного(ых) соединения(ий), содержащегося(ихся) в них.
В одном воплощении по меньшей мере одно соединение формулы I, ее подформул, или их энантиомер, соль или сольват, доставляют субъекту в количестве в диапазоне от примерно 0,01 мг/кг до примерно 600 мг/кг или в дозе от примерно 1 мг до примерно 500 мг. Однако могут быть выбраны более высокие или более низкие количества, например, принимая во внимание такие факторы, как показание, подлежащее лечению, и/или возраст и масса тела пациента.
Фармацевтические препараты по изобретению представлены предпочтительно в стандартной лекарственной форме и могут быть подходящим образом упакованы, например, в коробку, блистер, флакон, бутыль, саше, ампулу или в любой другой подходящий однодозовый или многодозовый сосуд или контейнер (который может быть надлежащим образом помечен); возможно с одним или более вкладышами, содержащими информацию о продукте и/или инструкции по использованию.
В зависимости от состояния, подлежащего предупреждению или лечению, а также пути введения, активное соединение по изобретению может быть введено в виде однократной суточной дозы, разделенной на одну или более доз в сутки, или по существу постоянно, например с использованием капельного вливания.
Определения
Как используют здесь, следующие термины имеют следующие ниже значения:
Когда группы могут быть замещенными, такие группы могут быть замещены одним или более заместителями и, предпочтительно, одним, двумя или тремя заместителями. Заместители могут быть выбраны, без ограничения, например, из группы, содержащей галоген, гидроксил, оксо, нитро, амидо, карбокси, амино, циано, галогеналкокси и галогеналкил. В некоторых воплощениях более одного заместителя может находиться по одному и тому же атому группы (например, диметил-замещение по N или С). В других воплощениях могут быть выбраны другие заместители, например как описаны и/или проиллюстрированы в примерах.
Термин "галоген" означает фтор (F), хлор (Cl), бром (Br) или йод (I).
Следующие определения используют в контексте описанных здесь соединений. В общем, количество атомов углерода, присутствующих в данной группе, обозначают "Сх-Cy", где х и y представляют собой нижний и верхний пределы, соответственно. Число атомов углерода, как используют в данных здесь определениях, относится к углеродному скелету и разветвлению углеродной цепи, но не включают атомы углерода заместителей, таких как алкоксильные заместители и им подобные. Если не указано иное, номенклатура заместителей, которые не определены здесь точно, определяется по названию слева направо от концевого участка функциональной группы, следующей за соседней функциональной группой в направлении к точке присоединения. Как используют здесь, "возможно замещенный" означает, что по меньшей мере 1 атом водорода возможно замещенной группы был заменен.
Термин "алкил" сам по себе или в составе другого заместителя относится к углеводородному радикалу формулы CnH2n+1, где n представляет собой число больше или равное 1. Алкильные группы могут содержать от 1 до 10 атомов углерода (включительно), то есть С1, С2, С3, С4, С5, С6, С7, С8, С9 или С10, то есть С1-С10 алкил. В некоторых воплощениях алкильные группы в данном изобретении содержат от 1 до 6 атомов углерода, предпочтительно от 1 до 4 атомов углерода, более предпочтительно от 1 до 3 атомов углерода. Алкильные группы могут быть линейными или разветвленными и могут быть замещены, как указано здесь. Подходящие алкильные группы включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил и трет-бутил, пентил и его изомеры (например, н-пентил, изопентил) и гексил и его изомеры (например, н-гексил, изогексил).
Термин "галогеналкил" один или в комбинации относится к алкильному радикалу, имеющему значение, как определено выше, где один или более водородов заменены галогеном, как он определен выше. Неограничивающие примеры таких галогеналкильных радикалов включают фторметил, дифторметил, трифторметил и тому подобное. В одном примере галогеналкил представляет собой С1-С6алкильную группу, замещенную по меньшей мере одним галогеном. В другом примере галогеналкил представляет собой С1-С4алкильную группу, замещенную по меньшей мере одним галогеном. Каждое замещение галогеном может быть выбрано независимо.
Термин "циклоалкил", как используют здесь, представляет собой циклическую алкильную группу, то есть моновалентную насыщенную или ненасыщенную углеводородную группу, имеющую 1 или 2 циклические структуры. Циклоалкил включает моноциклические или бициклические углеводородные группы. Циклоалкильные группы могут содержать 3 или более атомов углерода в кольце и в общем, согласно данному изобретению, содержат от 3 до 10, более предпочтительно от 3 до 8 атомов углерода, еще более предпочтительно от 3 до 6 атомов углерода. Примеры циклоалкильных групп включают, без ограничения, циклопропил, циклобутил, циклопентил, циклогексил, причем циклопропил является особенно предпочтительным.
Термин "гетероатом" относится к атому серы, азота или кислорода.
Когда по меньшей мере один атом углерода в циклоалкильной группе заменен гетероатомом, полученное кольцо называют здесь "гетероциклилом".
Термины "гетероциклил" или "гетероцикл", как используют здесь, сами по себе или в составе другой группы, относятся к неароматическим полностью насыщенным или частично ненасыщенным циклическим группам (например, 3-7-членным моноциклическим, 7-11-членным бициклическим или содержащим суммарно от 3 до 10 кольцевых атомов), имеющим по меньшей мере один гетероатом в по меньшей мере одном кольце, содержащем атомы углерода. Каждое кольцо гетероциклической группы, содержащей гетероатом, может иметь 1, 2, 3 или 4 гетероатома, выбранных из атомов азота, кислорода и/или серы, где гетероатомы азота и серы могут быть возможно окисленными и гетероатомы азота могут быть возможно кватернизированными. Гетероцикл может содержать от 3 до 7 атомов углерода (включительно) или целое число между ними. Любой из атомов углерода гетероциклической группы может быть замещен оксогруппой (например, пиперидон, пирролидинон). Гетероциклическая группа может быть присоединена по любому гетероатому или атому углерода данного кольца или кольцевой системы, где позволяет валентность. Кольца указанных поликольцевых гетероциклов могут быть конденсированными, связанными мостиковой связью и/или присоединены через один или более спироатомов. В одном воплощении гетероцикл представляет собой 4-, 5- или 6-членное кольцо с 1, 2 или 3 гетероатомами в своей структуре, выбранными из одного или более N или О. В одном воплощении гетероцикл представляет собой 5-членное кольцо, имеющее 3 N. Как используют здесь, в случае когда число гетероатомов конкретно указано, остальные члены гетероциклической структуры представляют собой атомы С. Неограничивающие примеры гетероциклических групп включают пиперидинил, азетидинил, тетрагидропиранил, пиперазинил, имидазолинил, морфолинил, оксетанил, пиразолидинил, имидазолидинил, изоксазолинил, оксазолидинил, изоксазолидинил, тиазолидинил, изотиазолидинил, индолил, индолинил, изоиндолинил, тетрагидрофуранил, тетрагидрохинолинил, тиоморфолинил, тиоморфолинилсульфоксид, тиоморфолинилсульфон, пирролизинил.
Термин "арил", как используют здесь, относится к полиненасыщенной ароматической углеводородной группе, имеющей одно кольцо (а именно фенил) или несколько ароматических колец, конденсированных вместе (например, нафтил) или ковалентно связанных, обычно содержащих 5-12 атомов; предпочтительно 6-10, где по меньшей мере одно кольцо является ароматическим. Ароматическое кольцо может возможно включать 1-2 дополнительных кольца (циклоалкил, гетероциклил либо гетероарил), конденсированных с ним. Арил также предназначен включать частично гидрированные производные карбоциклических (углерод-содержащих кольцевых) систем, перечисленных здесь. Неограничивающие примеры арила включают фенил, бифенилил, бифениленилнафталинил, инденил.
Термин "гетероарил", как используют здесь, сам по себе или как часть другой группы, относится, без ограничения, к ароматическим кольцам из 5-12 атомов углерода или кольцевым системам, содержащим 1-2 кольца, которые конденсированы вместе или ковалентно связаны, обычно содержащим 5-6 атомов, по меньшей мере одно из которых является ароматическим, где один или более атомов углерода в одном или более из этих колец заменен атомами кислорода, азота и/или серы, где гетероатомы азота и серы могут быть возможно окисленными и гетероатомы азота могут быть возможно кватернизированными. Такие кольца могут быть конденсированными с арильным, циклоалкильным, гетероарильным или гетероциклильным кольцом. Неограничивающие примеры такого гетероарила включают: пиридазинил, пиридинил, фуранил, тиофенил, пиразолил, имидазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, триазолил, оксадиазолил, тиадиазолил, тетразолил, оксатриазолил, тиатриазолил, пиримидинил, пиразинил, оксазинил, диоксинил, тиазинил, триазинил, индолил, индолизинил, изоиндолил, бензофуранил, изобензофуранил, бензотиофенил, изобензотиофенил, индазолил, бензимидазолил, хинолинил, изохинолинил, циннолинил, хиназолинил, хиноксалинил.
Термин "арилалкил" относится к любой группе -алкил-арил. Термин "алкиларил" относится к любой группе -арил-алкил.
Термин "гетероарилалкил" относится к любой группе -алкил-гетероарил. Термин "алкилгетероарил" относится к любой группе -гетероарил-алкил.
Термин "алкокси" относится к любой группе О-алкил. Термин "галогеналкокси" относится к любой группе О-галогеналкил.
Термин "оксо" относится к группировке =O.
Термин "амино" относится к группе -NH2 или любой группе, производной из нее путем замещения одного или двух атомов водорода органической алифатической или ароматической группой. Предпочтительно, производные группы -NH2 представляют собой "алкиламино" группы, то есть N-алкилгруппы, включающие моноалкиламино и диалкиламино. Неограничивающие примеры термина "амино" включают NH2, NHMe или NMe2, NHCOOH, NHCOOCH3, NHCOCH3 или N(CH3)COCH3.
Термин "амино-защитная группа" относится к группе, защищающей функциональную аминогруппу. Согласно предпочтительному воплощению, амино-защитная группа выбрана в группах, включающих: арилсульфонил, трет-бутоксикарбонил, метоксиметил, пара-метоксибензил или бензил.
Термин "уходящая группа" относится к молекулярному фрагменту, который уходит с парой электронов при гетеролитическом расщеплении связи. Согласно предпочтительному воплощению, уходящая группа выбрана в группах, включающих: галоген, предпочтительно йод, бром или хлор; алкилсульфонилокси, имеющий 1-6 атомов углерода, предпочтительно метилсульфонилокси или трифторметилсульфонилокси; или арилсульфонилокси, имеющий 6-10 атомов углерода, предпочтительно фенил- или пара-толилсульфонилокси.
Термин "сольват" используют здесь для описания соединения по данному изобретению, которое содержит стехиометрические или нестехиометрические количества одной или более чем одной молекулы фармацевтически приемлемого растворителя, например этанола. Как правило, сольват не изменяет значительно физиологическую активность или токсичность соединений и как таковой может функционировать в качестве фармакологических эквивалентов несольватированным соединениям формулы I и ее подформул, как определено здесь. Термин "сольват", как используют здесь, означает объединение, физическую ассоциацию и/или сольватирование соединения по настоящему изобретению с молекулой растворителя. Такая физическая ассоциация включает варьирование степеней ионного и ковалентного связывания, включая водородные связи. В некоторых случаях сольват может быть выделен, например когда одна или более молекул растворителя включены в кристаллическую решетку кристаллического твердого вещества. Таким образом, "сольват" охватывает как сольваты в растворе, так и поддающиеся выделению сольваты. "Сольват" может охватывать сольваты солей соединений формулы I.
Термин "гидрат" используют, когда молекула растворителя представляет собой воду, и может представлять собой неорганическую соль, содержащую nH2O, где n равен числу молекул воды на структурную единицу соли. N может быть равным 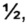
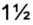 или целому числу от 1 до 10. Гидрат, который не имеет воды.
или целому числу от 1 до 10. Гидрат, который не имеет воды.
Соединения по изобретению включают соединения формулы I, как они здесь определены, включая все их полиморфы и кристаллические формы, пролекарства и их пролекарства, и меченые изотопами соединения формулы I.
Изобретение также в общем охватывает все фармацевтически приемлемые прелекарства и пролекарства соединений формулы I.
Термин "пролекарство", как используют здесь, означает фармакологически приемлемые производные соединений формулы I, такие как, например, сложные эфиры, чьи продукты биотрансформации in vivo генерирует биологически активное лекарственное средство. Пролекарства в общем характеризуются повышенной биодоступностью и легко метаболизируются в биологически активные соединения in vivo.
Термин "прелекарство", как используют здесь, означает любое соединение, которое будет модифицировано с образованием видов лекарственного средства, где модификация может иметь место либо внутри, либо вне организма, и либо до, либо после того как прелекарство достигнет области организма, для которой показано введение данного лекарственного средства.
Термин "пациент" относится к теплокровному животному, более предпочтительно человеку, который ожидает получения или получает медицинское лечение или который является или будет являться объектом медицинской процедуры.
Термин "человек" относится к субъекту обоих полов и любой стадии развития (то есть новорожденный, младенец, подросток, юноша, взрослый).
Термины "лечить", "проведение лечения" и "лечение", как используют здесь, означают облегчение, ослабление или подавление состояния или заболевания и/или его сопутствующих симптомов.
Термины "предупреждать", "предупреждение" и "предотвращение", как используют здесь, относится к способу задержки или препятствованию начала возникновения состояния или заболевания и/или его сопутствующих симптомов, не давая развиться состоянию или заболеванию у пациента, или снижения у пациента риска развития состояния или заболевания.
Термин "терапевтически эффективное количество" (или проще "эффективное количество"), как используют здесь, означает количество активного агента или активного ингредиента, которое является достаточным для достижения требуемого терапевтического или профилактического эффекта у пациента, которому его вводят.
Термин "введение" или его вариант (например "вводить"), означает предоставление активного агента и активного ингредиента, отдельно или в составе фармацевтически приемлемой композиции, пациенту, подлежащему лечению или предупреждению, или для воздействия на состояние, симптом или заболевание, подлежащее лечению или предупреждению.
Под "фармацевтически приемлемыми" понимают ингредиенты фармацевтической композиции, совместимые друг с другом и не вредные для пациента, принимающего их.
Термин "фармацевтический носитель", как используют здесь, означает носитель или инертную среду, используемые в качестве растворителя или разбавителя, в котором фармацевтически активный агент изготавливают и/или вводят. Неограничивающие примеры фармацевтических носителей включают кремы, гели, лосьоны, растворы и липосомы.
Термины "содержать", "содержит" и "содержащий" следует интерпретировать включительно, а не исключительно. Термины "состоит из", "состоящий из" и их варианты следует интерпретировать исключительно, а не включительно.
Как используют здесь, термин "примерно" означает вариабельность в пределах 10% от данного значения, если специально не указано иное.
ПРИМЕРЫ
Настоящее изобретение будет более понятным со ссылкой на следующие примеры. Эти примеры предназначены для иллюстративного описания конкретных воплощений изобретения и не предназначены ограничивать объем изобретения.
I. ХИМИЧЕСКИЕ ПРИМЕРЫ
Данные по масс-спектрометрии (МС), предложенные в описанных ниже Примерах, получали на следующем оборудовании: масс-спектр: LC/MS Agilent 6110 (электрораспылительная ионизация, ЭРИ) или a Waters Acquity SQD (ESI).
Данные ЯМР, предложенные в описанных ниже Примерах, получали на следующем оборудовании: Bruker Ultrashield™ 400 PLUS и Bruker Fourier 300 МГц, и TMS использовали в качестве внутреннего стандарта.
Микроволновую химию проводили на одномодовом микроволновом реакторе Initiator Microwave System EU от Biotage.
Очистку препаративной высокоэффективной жидкостной хроматографией (ВЭЖХ) осуществляли с управляемой по массе автоматизированной очисткой Fractionlynx от Waters, оборудованной колонкой Xbridge™ Prep С18 OBD 19×150 мм, 5 мкм, если не отмечено другое. Все ВЭЖХ-очистки осуществляли с использованием градиента CH3CN/H2O/NH4HCO3 (5 мМ), CH3CN/H2O/TFA (0,1%) или CH3CN/H2O/NH3H2O (0,1%).
Следующие сокращения использованы здесь и имеют указанные определения: ACN означает ацетонитрил; DMSO означает диметилсульфоксид; DCM означает дихлорметан; DIPEA означает диизопропилэтиламин; DMF означает N,N-диметилформамид; dppf означает 1,1'-бис(дифенилфосфино)-феррорцен; EtOH означает этанол; HATU означает 2-(1Н-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат метанаминий; Гц означает герц; KOAc означает ацетат калия; МеОН означает метанол; MeNH2 означает метиламид; BH3MeS означает диметилсульфид бора; BuOK означает трет-бутоксид калия; MeI означает метилйодид; МГц означает мегагерц; мМ означает микромолярный; мл означает милилитр; мин. означает минуты; mol означает моль; М+ означает молекулярный ион; [М+Н]+ означает протонированный молекулярный ион; н. означает нормальность; ЯМР означает ядерный магнитный резонанс; РЕ означает петролейный эфир; ЕА означает этилацетат; PPh3 означает трифенилфосфин; psi означает фунты на квадратный дюйм; м.д. означает частей на миллион; qd ро означает ежесуточно на прием; кт означает комнатную температуру; RT означает время удерживания; ТСХ означает тонкослойную хроматографию; TFA означает трифторуксусную кислоту; TEA означает триметиламин; СФХ означает сверхкритическую флюидную хроматографию; ЖХМС (также ЖХ-МС) означает жидкостную хроматографию/масс-спектрометрию; ВЭЖХ означает высокоэффективную жидкостную хроматографию; TBAF означает фторид тетра-н-бутиламмония; AIBN означает азобисизобутиронитрил; BNS означает бензолсульфоновую кислоту; TBDPSCI означает трет-бутилдифенилхлорсилан.
Промежуточное соединение 1: трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индол-1-карбоксилат
Стадия 1: трет-бутил-6-фтор-1Н-индол-1-карбоксилат
К раствору 6-фтор-1Н-индола (10,0 г, 74,0 ммоль) в DCM (200 мл) добавляли (Вос)2O (19,4 г, 88,9 ммоль), TEA (11,2 г, 15,4 ммоль) и DMAP (1,81 г, 14,8 ммоль). Реакционную смесь перемешивали при 18°C в течение 18 часов. Смесь промывали водн. HCl (1 М, 100 мл) и рассолом. Органический слой сушили, фильтровали и концентрировали с получением 17,4 г неочищенного продукта, который использовали на следующей стадии без очистки.
Стадия 2: трет-бутил-3-бром-6-фтор-1Н-индол-1-карбоксилат
К раствору трет-бутил-6-фтор-1Н-индол-1-карбоксилата (17,4 г, 74,0 ммоль) в DCM (200 мл) добавляли NBS (15,8 г, 88,8 ммоль). Реакционную смесь перемешивали при 40°C в течение 6 часов. Смесь промывали водой и рассолом. Органический слой сушили, фильтровали и концентрировали. Осадок очищали посредством хроматографии на силикагеле (РЕ/EtOAc 10:1) с получением указанного в заголовке соединения в виде белого твердого вещества.
Стадия 3: трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индол-1-карбоксилат
К раствору трет-бутил-3-бром-6-фтор-1Н-индол-1-карбоксилата (10,0 г, 32,0 ммоль) в диоксане (150 мл) добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (12,0 г, 47,0 ммоль), KOAc (9,30 г, 95,0 ммоль) и Pd(dppf)Cl2 (2,30 г, 3,10 ммоль). Реакционную смесь перемешивали при 90°C в течение 5 часов. Растворитель удаляли и добавляли DCM (300 мл). Смесь промывали рассолом, сушили и фильтровали. Фильтрат концентрировали и очищали посредством хроматографии на силикагеле (РЕ/EtOAc 10:1) с получением указанного в заголовке соединения (6,00 г, 50%) в виде белого твердого вещества.
Пример 1:
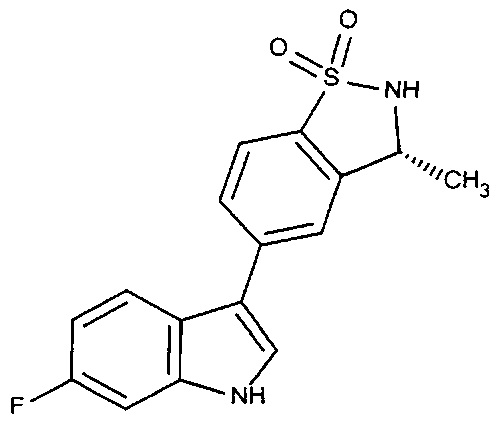
Пример 1: (+)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидро-1,2-бензотиазол-1,1-диоксид
Стадия 1: 4-бром-2-этилбензолсульфонилазид
Раствор 4-бром-2-этилбензол-1-сульфонилхлорида (2 г, 7,0 ммоль) в смеси вода/ацетон (1:1, 50 мл) перемешивали в круглодонной колбе и охлаждали в ледяной бане до 0°C в течение 15-20 минут. К этой сульфонилхлоридной смеси порциями добавляли азид натрия (0,92 г, 14,2 ммоль). Бледно-розовый раствор перемешивали при 20°C в течение 3 часов. Реакционный раствор концентрировали в вакууме при 25°C для удаления ацетона и неочищенный продукт экстрагировали с использованием этилацетата (3×15 мл). Органическую фазу затем промывали рассолом (10 мл/ммоль), сушили над сульфатом натрия и концентрировали в вакууме при 25°C с получением указанного в заголовке соединения (2,0 г, 98%) в виде масла.
Стадия 2: 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Раствор 4-бром-2-этилбензолсульфонилазида (500 мг, 1,72 ммоль) и Со(ТРР) (116 мг, 0,172 ммоль) в PhCl (5 мл) дегазировали с N2 трижды и затем перемешивали при 80-85°C в течение 24 часов. Добавляли дополнительное количество Со(ТРР) (116 мг, 0,172 ммоль) и раствор перемешивали при 80-85°C в течение еще 48 часов. Смесь концентрировали до сухого состояния и очищали посредством флэш-хроматографии (SiO2, петролейный эфир/EtOAc от 1/5 до 1/1) с получением неочищенного 5-бром-3-метил-2,3-дигидро-бензо[d]изотиазол-1,1-диоксида (380 мг, выход 84,1%) в виде красной смолы. 1Н ЯМР (400 МГц, CDCl3) d=7.72-7.62 (m, 2Н), 7.58-7.53 (m, 1Н), 4.76 (td, J=6,4, 12,3 Гц, 1Н), 4.63 (br. s., 1Н), 1.63 (d, J=6,5 Гц, 3Н).
Стадия 3: трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилат
К слегка белой суспензии трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индол-1-карбоксилата (524 мг, 1,45 ммоль), 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (380 мг, 1,45 ммоль) и K3PO4 (616 мг, 2,9 ммоль) в диоксане (15 мл) и H2O (5 мл) добавляли Pd(dppf)Cl2 (106 мг, 0,145 ммоль) в атмосфере азота, затем эту суспензию перемешивали при 100°C в течение 18 часов. Реакционную смесь разбавляли EtOAc (60 мл) и водой (20 мл). Слои разделяли, затем водный слой экстрагировали EtOAc (30 мл*2). Объединенные органические слои промывали рассолом (20 мл×2), сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилата (604 мг) в виде черного твердого вещества, которое использовали на следующей стадии без дополнительной очистки.
Стадия 4: 5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидро-1,2-бензотиазол-1,1-диоксид
К желтому раствору трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилата (0,604 г, 1,45 ммоль) в DCM (6 мл) добавляли TFA (3 мл) при 0°C. По окончании добавления черный раствор перемешивали при 20°C в течение 2 часов. Этот черный раствор концентрировали и обрабатывали водн. NaHCO3 (10 мл), экстрагировали DCM (10 мл Х5), сушили над NaSO4, фильтровали и концентрировали с получением неочищенного продукта (0,5 г), который очищали посредством флэш-хроматографии (SiO2, DCM/EtOAc от 10/1 до 3/1) с получением рацемического 5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидро-1,2-бензотиазол-1,1-диоксида (0,18 г, выход 45%) в виде черного твердого вещества. 150 мг рацемического продукта разделяли посредством препаративной хиральной СФХ с получением (+)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидро-1,2-бензотиазол-1,1-диоксида в качестве первого элюируемого пика (70 мг, 47%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) d=11.67 (br. s., 1H), 8.02-7.68 (m, 6H), 7.27 (dd, J=2,3, 10,0 Гц, 1H), 7.02 (dt, J=2,5, 9,3 Гц, 1H), 4.90-4.67 (m, 1H), 1.54 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 316,9 (М+Н)+. [α]20D +27,0° (с=2 мг/мл, EtOH).
Пример 2: (-)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидро-1,2-бензотиазол-1,1-диоксид
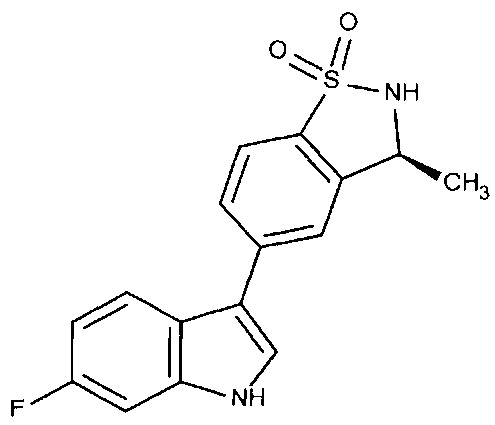
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 111 (70 мг, 47%, желтое твердое вещество). 1Н ЯМР (400 МГц, DMSO-d6) d=11.67 (br. s., 1H), 8.05-7.69 (m, 6H), 7.27 (dd, J=2,0, 10,0 Гц, 1H), 7.11-6.86 (m, 1H), 4.91-4.64 (m, 1H), 1.54 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 316,9 (М+Н)+. [α]20D -43,8° (с=1,3 мг/мл, EtOH).
Пример 3: (+)-3-этил-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Стадия 1: 1-бром-3-пропилбензол
К суспензии 1-(3-бромфенил)пропан-1-она (5000 мг, 23,5 ммоль) и KOH (3,95 г, 70,4 ммоль) в (СН2ОН)2 (28 мл) добавляли N2H4-H2O (4,15 г, 70,4 ммоль). Смесь перемешивали в атмосфере N2 при 200°C в течение 4 часов, затем при 140°C в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры и гасили 1 М HCl (500 мл) (рН 3-4), затем экстрагировали петролейным эфиром (150 мл). Органический слой промывали рассолом (100 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, петролейный эфир) с получением 1-бром-3-пропилбензола (4,0 г, 86%) в виде прозрачного масла. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 7.37-7.30 (m, 2Н), 7.19-7.09 (m, 2Н), 2.64-2.52 (m, 2Н), 1.69-1.61 (m, 2Н), 0.95 (t, J=7,3 Гц, 3Н).
Стадия 2: 4-бром-2-пропилбензол-1-сульфонилхлорид
Круглодонную колбу емкостью 100 мл продували N2 и загружали 1-бром-3-пропилбензолом (2 г, 10 ммоль) и хлороформом (25 мл), затем охлаждали в ледяной бане до 0°C.Добавляли по каплям в течение 10 мин хлорсульфоновую кислоту (7,02 г, 60,3 ммоль) и реакционную смесь перемешивали при 0°C в течение 1 часа, затем при 30°C в течение 16 часов. Неочищенную реакционную смесь осторожно вливали в ледяную воду (60 мл) и экстрагировали дихлорметаном (50 мл×2). Объединенные органические слои промывали рассолом (60 мл×3), затем сушили над Na2SO4, фильтровали и концентрировали. Осадок очищали посредством колоночной хроматографии (силикагель, петролейный эфир) с получением 4-бром-2-пропилбензол-1-сульфонилхлорида (1,6 г, 53%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 7.93 (d, J=8,5 Гц, 1Н), 7.63 (d, J=2,0 Гц, 1Н), 7.56 (dd, J=2,1, 8,7 Гц, 1H), 3.15-3.02 (m, 2Н), 1.85-1.70 (m, 2Н), 1.08 (t, J=7,3 Гц, 3Н).
Стадия 3: 4-бром-2-пропилбензолсульфонилазид
Раствор 4-бром-2-пропилбензол-1-сульфонилхлорида (1,6 г, 3,2 ммоль) в смеси вода/ацетон (16 мл/16 мл) охлаждали в ледяной бане, затем добавляли порциями азид натрия (523 мг, 8,04 ммоль). Реакционную смесь перемешивали при примерно 10°C в течение 1 часа, затем концентрировали и экстрагировали петролейным эфиром (30 мл). Органический слой промывали рассолом (20 мл), затем сушили над Na2SO4, фильтровали и концентрировали с получением 4-бром-2-пропилбензолсульфонилазида (1,6 г, 98%) в виде прозрачного масла. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 7.90 (d, J=8,5 Гц, 1Н), 7.61 (d, J=2,0 Гц, 1Н), 7.54 (dd, J=2,0, 8,5 Гц, 1Н), 3.00-2.88 (m, 2Н), 1.73 (dd, J=7,5, 15,6 Гц, 2Н), 1.05 (t, J=7,3 Гц, 3Н).
Стадия 4: 5-бром-3-этил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К желтому раствору 4-бром-2-пропилбензолсульфонилазида (1,63 г, 5,36 ммоль) в хлорбензоле (5 мл) добавляли 5,10,15,20-тетрафенил-21Н,23H-порфинкобальт(II) (180 мг, 0,268 ммоль) в атмосфере N2. Реакционную смесь барботировали N2 в течение 2 мин и нагревали до 80°C в течение 64 часов, затем концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 10-20% этилацетат/петролейный эфир) с получением смеси 5-бром-3-этил-2,3-дигидробензо[d]изотиазол-1,1-диоксида и 6-бром-3-метил-3,4-дигидро-2Н-бензо[е][1,2]тиазин-1,1-диоксида (1,0 г, 68% смесь) в виде черной смолы. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 7.69-7.59 (m, 3Н), 7.56-7.46 (m, 2Н), 7.39 (s, 1Н), 4.85 (br s, 1Н), 4.71-4.56 (m, 1Н), 4.45 (d, J=11,3 Гц, 1Н), 4.11-3.94 (m, 1Н), 3.00-2.88 (m, 1Н), 2.84-2.71 (m, 1Н), 2.10-1.95 (m, 1Н), 1.81 (td, J=7,5, 14,7 Гц, 1Н), 1.40 (d, J=6,5 Гц, 3Н), 1.08-0.99 (m, 3Н).
Стадия 5: трет-бутил-3-(3-этил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилат
К желтому раствору 5-бром-3-этил-2,3-дигидробензо[d]изотиазол-1,1-диоксида и 6-бром-3-метил-3,4-дигидро-2Н-бензо[е][1,2]тиазин-1,1-диоксида (600 мг, 2,17 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индол-1-карбоксилата (872 мг, 2,17 ммоль) и K3PO4 (922 мг, 4,35 ммоль) в смеси диоксан/H2O (8 мл/2 мл) добавляли Pd(dppf)Cl2 (153 мг, 0,217 ммоль) при 25°C в атмосфере N2. Красную суспензию перемешивали при 80°C в течение 2 часов, затем разбавляли водой (10 мл) и экстрагировали этилацетатом (15 мл×2). Объединенные органические слои промывали рассолом (10 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 20% этилацетат/петролейный эфир) с получением смеси трет-бутил-3-(3-этил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилата и трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-3,4-дигидро-2Н-бензо[е][1,2]тиазин-6-ил)-1Н-индол-1-карбоксилата (570 мг, 61% смесь) в виде не совсем белого твердого вещества.
Стадия 6: (+)-3-этил-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К прозрачному раствору трет-бутил-3-(3-этил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилата и трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-3,4-дигидро-2Н-бензо[е][1,2]тиазин-6-ил)-1Н-индол-1-карбоксилата (570 мг, 1,32 ммоль) в дихлорметане (12 мл) добавляли трифторуксусную кислоту (4 мл). Реакционную смесь перемешивали при 25°C в течение 1 часа, затем концентрировали. Осадок нейтрализовали до рН более 7 с использованием NaHCO3 (насыщ.) и экстрагировали этилацетатом (30 мл×2). Объединенные органические слои промывали рассолом (30 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Осадок очищали посредством колоночной хроматографии (силикагель, 30% этилацетат/петролейный эфир) с получением смеси четырех продуктов. Смесь разделяли посредством препаративной хиральной сверхкритической флюидной хроматографии (СФХ) с получением (+)-3-этил-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида в качестве первого элюируемого пика (38 мг, 9%) в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.66 (br s, 1Н), 7.98-7.82 (m, 5Н), 7.81-7.73 (m, 1Н), 7.26 (dd, J=2,3, 10,0 Гц, 1H), 7.01 (dt, J=2,4, 9,2 Гц, 1H), 4.70-4.61 (m, 1Н), 2.08-1.90 (m, 1Н), 1.75-1.60 (m, 1Н), 0.95 (t, J=7,3 Гц, 3Н); ЖХ-МС: m/z 331,2 (М+Н)+; [α]20D +43,8° (с=0,95 мг/мл, метанол).
Пример 4: (-)-3-этил-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Указанное в заголовке соединение выделили в качестве второго элюируемого пика из СФХ-очистки, описанной в Примере 3 (36 мг, 8%), в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.66 (br s, 1Н), 8.01-7.81 (m, 5Н), 7.80-7.75 (m, 1Н), 7.26 (dd, J=2,3, 9,8 Гц, 1Н), 7.05-6.95 (m, 1Н), 4.72-4.58 (m, 1Н), 2.10-1.95 (m, 1Н), 1.75-1.58 (m, 1Н), 0.95 (t, J=7,3 Гц, 3Н); ЖХ-МС: m/z 330,9 (М+Н)+; [α]20D -48° (с=1,0 мг/мл, метанол).
Пример 5: 5-(6-фтор-1Н-индол-3-ил)-3,3-диметил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
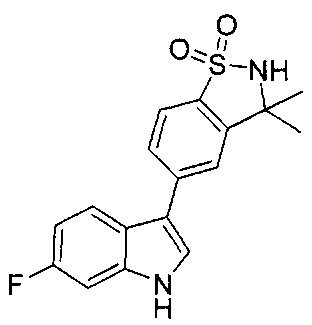
Следуя общей методике, изложенной в Примере 3, начиная с 1-бром-3-изопропилбензола, получили указанное в заголовке соединение в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1Н), 7.94-7.88 (m, 3Н), 7.88-7.81 (m, 2Н), 7.75 (d, J=8,0 Гц, 1Н), 7.27 (d, J=10,0 Гц, 1Н), 7.01 (t, J=9,3 Гц, 1Н), 1.60 (s, 6H); ЖХ-МС: m/z 352,9 (М+23)+.
Пример 6: (+)-5-(6-фтор-1H-индол-3-ил)-3-пропил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
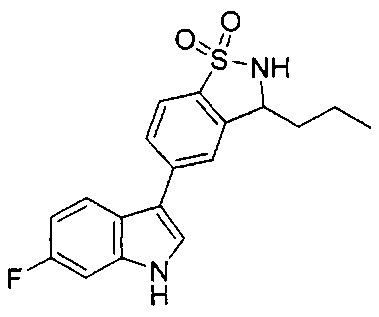
Следуя общей методике, изложенной в Примере 3, начиная с 1-бром-3-бутилбензола, получили указанное в заголовке соединение в качестве первого элюируемого пика в виде серого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.92-7.81 (m, 2Н), 7.80-7.72 (m, 2Н), 7.66 (s, 1Н), 7.16 (dd, J=2,3, 9,8 Гц, 1Н), 6.95 (dt, J=2,4, 9,2 Гц, 1Н), 4.74 (dd, J=3,5, 9,0 Гц, 1Н), 2.09-1.96 (m, 1Н), 1.82-1.69 (m, 1Н), 1.63-1.48 (m, 2Н), 1.01 (t, J=7,4 Гц, 3Н); ЖХ-МС: m/z 344,9 (М+Н)+; [α]20D +34,8° (с=2,3 мг/мл, метанол).
Пример 7: (-)-5-(6-фтор-1Н-индол-3-ил)-3-пропил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
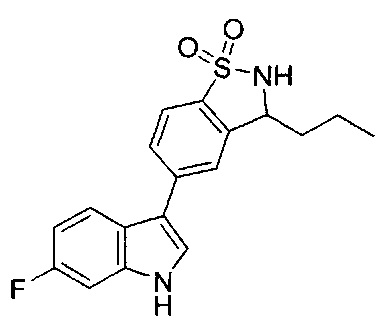
Следуя общей методике, изложенной в Примере 3, начиная с 1-бром-3-бутилбензола, получили указанное в заголовке соединение в качестве второго элюируемого пика в виде серого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.89-7.81 (m, 2Н), 7.78-7.71 (m, 2Н), 7.66 (s, 1Н), 7.15 (dd, J=2,4, 9,7 Гц, 1Н), 6.94 (dt, J=2,5, 9,2 Гц, 1Н), 4.73 (dd, J=3,4, 8,9 Гц, 1Н), 2.06-1.96 (m, 1Н), 1.81-1.69 (m, 1H), 1.60-1.48 (m, 2H), 1.01 (t, J=7,3 Гц, 3Н), ЖХ-МС: m/z 344,9 (М+Н)+; [α]20D -36,4° (с=2,5 мг/мл, метанол).
Пример 8: (+)-5-(6-фтор-1H-индол-3-ил)-3-изопропил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
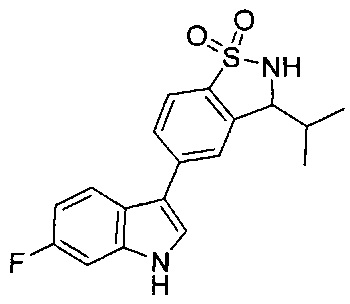
Стадия 1: 4-бром-2-изобутилбензолсульфонамид
Желтый раствор 4-бром-2-изобутилбензол-1-сульфонилхлорида (1000 мг, 3,21 ммоль) и NH4OH (5 мл) в безводном дихлорметане (50 мл) перемешивали при 25°C в течение 2 часов. Этот реакционный раствор разбавляли H2O (10 мл) и слои разделяли. Органический слой сушили над Na2SO4, фильтровали и концентрировали, затем очищали посредством колоночной хроматографии (петролейный эфир/этилацетат от 6/1 до 3/1) с получением 4-бром-2-изобутилбензолсульфонамида (900 мг, 96%) в виде коричневого твердого вещества.
Стадия 2: 4-бром-2-(1-бром-2-метилпропил)бензолсульфонамид
Желтый раствор 4-бром-2-изобутилбензолсульфонамида (200 мг, 0,684 ммоль), N-бромсукцинамида (NBS) (146 мг, 0,820 ммоль) и азобисизобутиронитрила (AIBN) (11 мг, 0,07 ммоль) в CCl4 (10 мл) перемешивали при 70°C в течение 12 часов. Этот реакционный раствор разбавляли H2O (10 мл) и слои разделяли. Органический слой сушили над Na2SO4, фильтровали и концентрировали, затем очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат от 6/1 до 3/1) с получением 4-бром-2-(1-бром-2-метилпропил)бензолсульфонамида (250 мг, 98) в виде коричневого твердого вещества.
Стадия 3: 5-бром-3-изопропил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Желтый раствор 4-бром-2-(1-бром-2-метилпропил)бензолсульфонамида (350 мг, 0,943 ммоль) и K2CO3 (261 мг, 1,89 ммоль) в смеси H2O/ацетон (5 мл/10 мл) перемешивали при 50°C в течение 12 часов, затем концентрировали и экстрагировали этилацетатом (20 мл×3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали, затем очищали посредством колоночной хроматографии (петролейный эфир/этилацетат 5/1) с получением 5-бром-3-изопропил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (200 мг, 85%) в виде желтой смолы.
Стадия 4: трет-бутил-3-(4-(2-(3-этокси-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)фенил)-6-фтор-1H-индол-1-карбоксилат
К раствору 5-бром-3-изопропил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (400 мг, 2,21 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (498 мг, 1,38 ммоль) и K3PO4 (586 мг, 2,76 ммоль) в смеси диоксан/H2O (12 мл/4 мл) добавляли Pd(dppf)Cl2 (101 мг, 0,138 ммоль) при 28°C в атмосфере N2. Красную суспензию перемешивали при 80°C в течение 16 часов, затем разбавляли водой (8 мл) и экстрагировали этилацетатом (15 мл×3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 5-30% этилацетат/петролейный эфир) с получением трет-бутил-3-(4-(2-(3-этокси-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)фенил)-6-фтор-1Н-индол-1-карбоксилата (300 мг, 24%) в виде желтой смолы.
Стадия 5: (+)-5-(6-фтор-1Н-индол-3-ил)-3-изопропил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Красный раствор трет-бутил-3-(4-(2-(3-этокси-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)фенил)-6-фтор-1Н-индол-1-карбоксилата (300 мг, 0,675 ммоль) в трифторуксусной кислоте и дихлорметане (5 мл/5 мл) перемешивали при 20°C в атмосфере N2 в течение 1 часа. Желтую суспензию концентрировали, затем разбавляли водным NaHCO3 (10 мл) и экстрагировали этилацетатом (20 мл×3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат 1/1, затем этилацетат/метанол 10/1) и препаративной ВЭЖХ с получением рацемического 5-(6-фтор-1Н-индол-3-ил)-3-изопропил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (30 мг, 13%) в виде белого твердого вещества. Этот рацемический продукт разделяли посредством препаративной хиральной СФХ с получением (+)-5-(6-фтор-1H-индол-3-ил)-3-изопропил-2,3-дигидробензо[d]изотиазол-1,1-диоксида в качестве первого элюируемого пика (10 мг, 4%) в виде бледно-желтого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.95-7.83 (m, 2Н), 7.80-7.75 (m, 2Н), 7.69 (s, 1Н), 7.18 (dd, J=2,4, 9.7 Гц, 1Н), 6.97 (dt, J=2,4, 9,2 Гц, 1Н), 4.77 (d, J=3,0 Гц, 1Н), 2.48-2.30 (m, 1Н), 1.18 (d, J=7,0 Гц, 3Н), 0.78 (d, J=6,8 Гц, 3Н); ЖХ-МС: m/z 344,9 (M+Na)+; [α]20D +35,3° (метанол, 0,001 г/мл).
Пример 9: (-)-5-(6-фтор-1H-индол-3-ил)-3-изопропил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
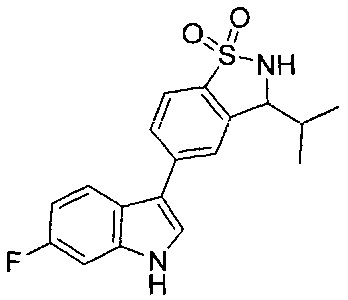
Указанное в заголовке соединение получили в качестве второго элюируемого пика из очистки, описанной в Примере 8, в виде бледно-желтого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.94-7.83 (m, 2Н), 7.80-7.75 (m, 2Н), 7.69 (s, 1Н), 7.18 (dd, J=2,4, 9,4 Гц, 1Н), 6.97 (dt, J=2,4, 9,2 Гц, 1Н), 4.77 (d, J=3,0 Гц, 1Н), 2.44-2.32 (m, 1Н), 1.18 (d, J=6,8 Гц, 3Н), 0.78 (d, J=6,8 Гц, 3Н); ЖХ-МС: m/z 344,9 (М+Н)+; [α]20D -25° (метанол, 0,001 г/мл).
Пример 10: (+)-3-циклопропил-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
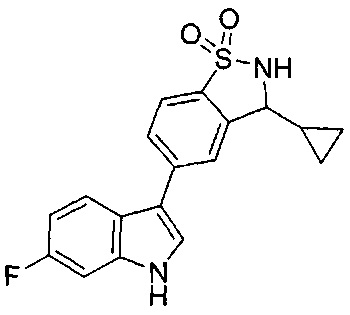
Стадия 1: 5-бром-3-циклопропилбензо[d]изотиазол-1,1-диоксид
Раствор 5-бромбензо[d]изотиазол-3(2Н)-он-1,1-диоксида (500 мг, 1,91 ммоль) в безводном THF (5,0 мл) выкачивали и заполняли с помощью азота за четыре цикла, затем медленно добавляли бромид циклопропилмагния (0,5 М в THF, 15,3 мл). Реакционную смесь нагревали до 40°C в течение 2 часов, затем охлаждали и гасили водным NH4Cl (насыщ.) (15 мл) и 2М HCl (0,5 мл). Реакционную смесь экстрагировали EtOAc (20 мл×3) и объединенные органические слои промывали водой (15 мл), затем концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 4-50% EtOAc/петролейный эфир) с получением 5-бром-3-циклопропилбензо[d]изотиазол-1,1-диоксида (270 мг, 50%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 8.62 (d, J=1,0 Гц, 1Н), 8.15-8.07 (m, 2Н), 2.91-2.79 (m, 1Н), 1.47 (qd, J=3,6, 7,7 Гц, 2Н), 1.34 (quin, J=3,9 Гц, 2Н).
Стадия 2: 5-бром-3-циклопропил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К прозрачному раствору 5-бром-3-циклопропилбензо[d]изотиазол-1,1-диоксида (344 мг, 0,35 ммоль) в МеОН (12 мл) добавляли NaBH4 (68 мг, 1,8 ммоль) при 15°C. Реакционную смесь перемешивали при 15°C в течение 1,5 часов, затем гасили водой (1,5 мл) и концентрировали. Неочищенный остаток очищали посредством колоночной хроматографии (силикагель, 4-50% EtOAc/петролейный эфир) с получением 5-бром-3-циклопропил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (222 мг, 64%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 8.16 (br s, 1Н), 7.93 (s, 1Н), 7.79 (d, J=4,5 Гц, 2Н), 4.22 (d, J=7,5 Гц, 1Н), 1.13 (d, J=16,1 Гц, 1Н), 0.71-0.60 (m, 2Н), 0.48-0.35 (m, 2Н).
Стадия 3: трет-бутил-3-(3-циклопропил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилат
Смесь 5-бром-3-циклопропил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (267 мг, 0,93 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индол-1-карбоксилата (351 мг, 0,97 ммоль), PdCl2(dppf) (69 мг, 0,09 ммоль) и K3PO4 (590 мг, 2,78 ммоль) в 1,4-диоксане (9 мл) барботировали азотом в течение 1 мин, затем перемешивали при 85°C в течение 2,5 часов. Реакционную смесь концентрировали и очищали посредством колоночной хроматографии (силикагель, 5-60% EtOAc/петролейный эфир) с получением трет-бутил-3-(3-циклопропил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилата (330 мг, 81%) в виде желтой смолы. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 8.12-8.05 (m, 2Н), 7.97 (s, 1Н), 7.94-7.85 (m, 3Н), 7.76-7.68 (m, 1Н), 7.26 (dt, J=2,5, 9,0 Гц, 1Н), 4.32 (dd, J=4,5, 7,5 Гц, 1Н), 1.75-1.59 (m, 8Н), 1.28-1.19 (m, 1Н), 0.76-0.57 (m, 2Н), 0.52-0.36 (m, 2Н); ЖХ-МС: m/z 443,1 (М+Н)+.
Стадия 4: (+)-3-циклопропил-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К желтому раствору трет-бутил-3-(3-циклопропил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (330 мг, 0,75 ммоль) в CH2Cl2 (8,0 мл) добавляли TFA (4,0 мл) при 18°C. Реакционную смесь перемешивали в течение 3 часов, затем концентрировали и снова растворяли в DCM (50 мл). Этот раствор нейтрализовали NaHCO3 (насыщ.) (15 мл) и слои разделяли. Водный слой подвергали обратной экстракции дихлорметаном (DCM) (20 мл×2) и объединенные органические слои концентрировали и очищали посредством колоночной хроматографии (силикагель, 10-65% EtOAc/петролейный эфир) с получением рацемического 3-циклопропил-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (190 мг, 74%) в виде желтой смолы. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (+)-3-циклопропил-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида в качестве первого элюируемого пика (55 мг, 29%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1Н), 8.01-7.83 (m, 5Н), 7.81-7.76 (m, 1Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.02 (dt, J=2,5, 9,3 Гц, 1Н), 4.28 (dd, J=4,0, 7,5 Гц, 1Н), 1.26-1.13 (m, 1Н), 0.78-0.59 (m, 2Н), 0.52-0.37 (m, 2Н); ЖХ-МС: m/z 343,1 (М+Н)+; [α]20D +39,55° (с=0,0015 г/мл, МеОН).
Пример 11: (-)-3-циклопропил-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
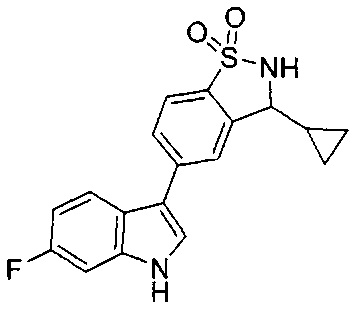
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 10 (60 мг, 32%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1Н), 8.00-7.85 (m, 5Н), 7.81-7.76 (m, 1Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.06-6.97 (m, 1Н), 4.31-4.24 (m, 1Н), 1.25-1.13 (m, 1Н), 0.77-0.59 (m, 2Н), 0.52-0.37 (m, 2Н); ЖХ-МС: m/z 343,1 (М+Н)+; [α]20D -33,46° (с=0,0017 г/мл, МеОН).
Пример 12: (-)-5-(6-фтор-1Н-индол-3-ил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
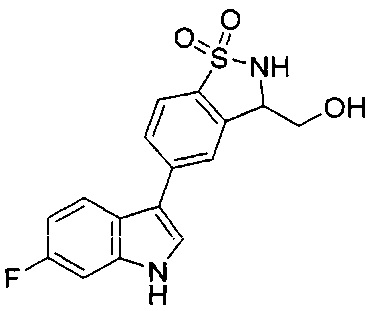
Стадия 1: метил-2-(2-(азидосульфонил)-5-бромфенил)ацетат
Желтый раствор метил-2-(5-бром-2-(хлорсульфонил)фенил)ацетата (10 г, 4,6 ммоль) в ацетоне (100 мл) и H2O (50 мл) охлаждали в ледяной бане, затем добавляли порциями азид натрия (3 г, 46 ммоль). Реакционную смесь перемешивали при 0°C в течение 1 часа, затем концентрировали и экстрагировали МТВЕ (3×50 мл). Объединенные органические слои промывали рассолом (10 мл), затем сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии (силикагель, 0-10% этилацетат/петролейный эфир) с получением метил-2-(2-(азидосульфонил)-5-бромфенил)ацетата (6 г, 59%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.89-8.01 (m, 1Н), 7.59-7.70 (m, 2Н), 4.05 (s, 2Н), 3.75 (s, 3Н).
Стадия 2: метил-5-бром-2,3-дигидробензо[d]изотиазол-3-карбоксилат-1,1-диоксид
К раствору метил-2-(2-(азидосульфонил)-5-бромфенил)ацетата (6 г, 19 ммоль) в хлорбензоле (15 мл) добавляли 5,10,15,20-тетрафенил-21Н,23H-порфинкобальт(II) (654 мг, 0,974 ммоль) в атмомфере N2. Коричневую суспензию барботировали N2 в течение 2 мин и нагревали до 80°C в течение 18 часов. Реакционную смесь охлаждали до температуры окружающей среды и концентрировали, затем очищали посредством колоночной хроматографии (силикагель, 0-30% этилацетат/петролейный эфир) с получением метил-5-бром-2,3-дигидробензо[d]изотиазол-3-карбоксилат-1,1-диоксида (3,5 г, 59%) в виде светло-коричневого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 7.86 (s, 1Н), 7.64-7.75 (m, 1Н), 7.58-7.63 (m, 1Н), 5.48-5.53 (br, 1Н), 5.26 (d, 1Н), 3.97 (s, 3Н).
Стадия 3: 1,1-диоксид 5-бром-2,3-дигидробензо[d]изотиазол-3-карбоновой кислоты
К раствору метил-5-бром-2,3-дигидробензо[d]изотиазол-3-карбоксилат-1,1-диоксида (2,5 г, 8,2 ммоль) в THF (25 мл) добавляли раствор LiOH-H2O (857 мг, 20,4 ммоль) в воде (25 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 10 мин, затем концентрировали для удаления тетрагидрофурана (THF) и промывали метил-трет-бутиловым эфиром (МТВЕ) (2×50 мл). Водную фазу подкисляли до рН 2 с использованием 2 н. HCl и экстрагировали МТВЕ (2×50 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением 1,1-диоксида 5-бром-2,3-дигидробензо[d]изотиазол-3-карбоновой кислоты (1,8 г, 76%) в виде светло-коричневого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 8.00 (s, 1Н), 7.84 (dd, J=8,28, 1,00 Гц, 1Н), 7.69-7.76 (m, 1Н), 5.38 (s, 1Н).
Стадия 4: 5-бром-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К раствору 1,1-диоксида 5-бром-2,3-дигидробензо[d]изотиазол-3-карбоновой кислоты (1,0 г, 3,4 ммоль) в безводном THF (20 мл) добавляли по каплям BH3-Me2S (2,1 г, 27 ммоль) при 0°C. Реакционную смесь перемешивали при температуре окружающей среды в течение 4 часов, затем осторожно вливали в ледяную воду (50 мл) (выделение газа) и экстрагировали МТВЕ (3×30 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Осадок очищали посредством колоночной хроматографии (силикагель, 0-10% метанол/дихлорметан) с получением 5-бром-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (650 мг, 68%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.88 (d, J=0,75 Гц, 1Н), 7.75-7.82 (m, 1Н), 7.64-7.73 (m, 1Н), 4.69 (t, J=5,77 Гц, 1Н), 3.73-3.87 (m, 2Н).
Стадия 5: трет-бутил-6-фтор-3-(3-(гидроксиметил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилат
К раствору 5-бром-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (550 мг, 1,98 ммоль) и трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (929 мг, 2,57 ммоль) в диоксане (10 мл) добавляли Cs2CO3 (1290 мг, 3,96 ммоль) и Pd(dppf)2Cl2 (145 мг, 0,198 ммоль). Светло-коричневый раствор барботировали N2 в течение 2 мин, герметично закрывали и перемешивали при 80°C в течение 2 часов. Реакционную смесь охлаждали до температуры окружающей среды, затем концентрировали и очищали посредством колоночной хроматографии (силикагель, 0-45% этилацетат/петролейный эфир) с получением трет-бутил-6-фтор-3-(3-(гидроксиметил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилата (500 мг, 59%) в виде светло-желтой смолы.
Стадия 6: (-)-5-(6-фтор-1Н-индол-3-ил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Рацемический трет-бутил-6-фтор-3-(3-(гидроксиметил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилат (200 мг) очищали посредством хиральной СФХ с получением двух энантиомеров. К раствору соединения пика 1 (100 мг, 0,231 ммоль) в метаноле (5 мл) добавляли HCl/метанол (10 мл, 4 М) при 0°C. Реакционную смесь нагревали до температуры окружающей среды и перемешивали в течение 2 часов, затем концентрировали. Неочищенный продукт очищали посредством препаративной ВЭЖХ с получением (-)-5-(6-фтор-1Н-индол-3-ил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (45 мг, 59%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.57-11.75 (m, 1Н), 7.85-8.00 (m, 5Н), 7.77-7.83 (m, 1Н), 7.27 (dd, J=9,91, 2,38 Гц, 1Н), 7.02 (td, J=9,29, 2,51 Гц, 1Н), 5.25 (t, J=5,52 Гц, 1Н), 4.67 (t, J=5,90 Гц, 1Н), 3.61-3.80 (m, 2Н); ЖХ-МС: m/z (M+Na)+ 354,9; [α]20D -40,3° (с=2,2 мг/мл, DMSO).
Пример 13: (+)-5-(6-фтор-1Н-индол-3-ил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид

К раствору соединения второго пика из хирального разделения, описанного в Примере 12 (100 мг, 0,231 ммоль), в метаноле (5 мл) добавляли HCl/метанол (10 мл, 4 М) при 0°C. Реакционную смесь нагревали до температуры окружающей среды и перемешивали в течение 2 часов, затем концентрировали. Неочищенный продукт очищали посредством препаративной ВЭЖХ с получением (+)-5-(6-фтор-1Н-индол-3-ил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (36 мг, 47%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.57-11.75 (m, 1Н), 7.85-8.00 (m, 5Н), 7.77-7.83 (m, 1Н), 7.27 (dd, J=9,91, 2,38 Гц, 1Н), 7.02 (td, J=9,29, 2,51 Гц, 1Н), 5.25 (t, J=5,52 Гц, 1Н), 4.67 (t, J=5,90 Гц, 1Н), 3.61-3.80 (m, 2Н); ЖХ-МС: m/z (M+Na)+ 354,9; [α]20D +37,3° (с=3 мг/мл, DMSO).
Пример 14: метил-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-3-карбоксилат-1,1-диоксид
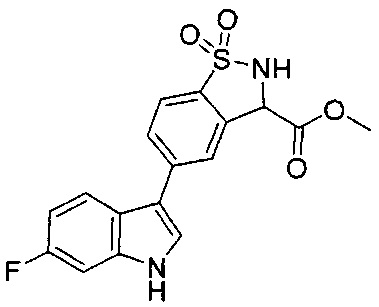
Стадия 1: метил-5-(1-(трет-бутоксикарбонил)-6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-3-карбоксилат-1,1-диоксид
В сосуд добавляли метил-5-бром-2,3-дигидробензо[d]изотиазол-3-карбоксилат-1,1-диоксид (Пример 12, Стадия 2, 200 мг, 0,653 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилат (236 мг, 0,653 ммоль) и Pd(Amphos)Cl2. Смесь закрывали крышечкой, затем выкачивали и снова заполняли с помощью N2 за два цикла. Добавляли толуол (13 мл) с последующим добавлением CsF (469 мг, 3,27 ммоль, 1 M в воде) и реакционную смесь нагревали до 70°C в течение 17 часов. Неочищенную реакционную смесь разбавляли EtOAc (20 мл) и водой (5 мл), затем слои разделяли. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали, затем очищали посредством колоночной хроматографии с получением метил-5-(1-(трет-бутоксикарбонил)-6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-3-карбоксилат-1,1-диоксида (180 мг, 60%) в виде светло-желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 7.93-8.01 (m, 1Н), 7.90 (d, J=8,53 Гц, 2Н), 7.82-7.86 (m, 1Н), 7.79 (s, 1Н), 7.65 (dd, J=8,78, 5,27 Гц, 1Н), 7.10 (d, J=2,51 Гц, 1Н), 5.48 (d, J=4,02 Гц, 1Н), 5.36 (d, J=4,27 Гц, 1Н), 3.96 (s, 3Н), 1.67-1.77 (m, 9Н).
Стадия 2: метил-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-3-карбоксилат-1,1-диоксид
К раствору метил-5-(1-(трет-бутоксикарбонил)-6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-3-карбоксилат-1,1-диоксида (180 мг, 0,391 ммоль) в безводном DCM (2 мл) добавляли HCl/МеОН (10 мл, 4 М). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем концентрировали и очищали посредством колоночной хроматографии с получением рацемического метил-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-3-карбоксилат-1,1-диоксида (120 мг, 85%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.71 (br s, 1Н), 8.36-8.62 (m, 1Н), 7.81-8.05 (m, 5Н), 7.29 (dd, J=9,79, 2,26 Гц, 1Н), 6.95-7.10 (m, 1Н), 5.59 (br s, 1Н), 3.78 (s, 3Н); ЖХ-МС: m/z 383,0 (M+Na)+.
Пример 15: (-)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-3-карбоксамид-1,1-диоксид
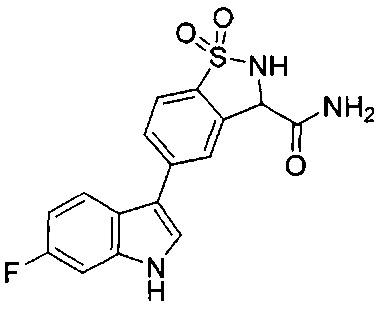
Стадия 1: трет-бутил-3-(3-карбамоил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилат
Раствор метил-5-(1-(трет-бутоксикарбонил)-6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-3-карбоксилат-1,1-диоксида (150 мг, 0,326 ммоль) в EtOH (15 мл) барботировали газообразным NH3 в течение 10 мин при -30°C. Реакционную смесь герметично закрывали и перемешивали при комнатной температуре в течение 18 часов, затем концентрировали с получением трет-бутил-3-(3-карбамоил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (150 мг, 100%) в виде светло-желтого твердого вещества.
Стадия 2: 5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-3-карбоксамид-1,1-диоксид
К раствору трет-бутил-3-(3-карбамоил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (150 мг, 0,337 ммоль) в EtOAc (2 мл) добавляли HCl/EtOAc (10 мл, 4М) при 0°C. Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем концентрировали и нейтрализовали NaHCO3 (насыщ.) (10 мл). Слои разделяли и органическую фазу концентрировали и очищали посредством колоночной хроматографии с получением указанного в заголовке соединения в виде рацемической смеси. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением 5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-3-карбоксамид-1,1-диоксида в качестве первого элюируемого пика (24 мг, 21%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.71 (br s, 1Н), 8.23 (d, J=5,02 Гц, 1Н), 8.03 (s, 1Н), 7.88-7.95 (m, 3Н), 7.79-7.86 (m, 1Н), 7.65 (s, 2Н), 7.29 (dd, J=9,79, 2,26 Гц, 1Н), 7.04 (td, J=9,22, 2,38 Гц, 1Н), 5.19 (d, J=5,02 Гц, 1Н); ЖХ-МС: m/z 367,9 (M+Na)+; [α]20D -31.83° (с=0,004 г/мл, DMSO).
Пример 16: (+)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-3-карбоксамид-1,1-диоксид
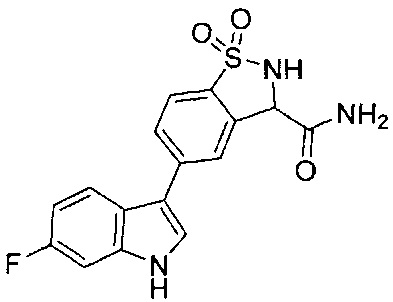
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 15 (25 мг, 21%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.71 (br s, 1Н), 8.23 (d, J=5,02 Гц, 1Н), 8.03 (s, 1Н), 7.88-7.95 (m, 3Н), 7.79-7.86 (m, 1H), 7.65 (s, 2H), 7.29 (dd, J=9,79, 2,26 Гц, 1H), 7.04 (td, J=9,22, 2,38 Гц, 1H), 5.19 (d, J=5,02 Гц, 1H); ЖХ-МС: m/z 368 (M+Na)+; [α]20D +33,66° (c=0,004 г/мл, DMSO).
Пример 17: (-)-5-(6-фтор-1H-индол-3-ил)-N-метил-2,3-дигидробензо[d]изотиазол-3-карбоксамид-1,1-диоксид
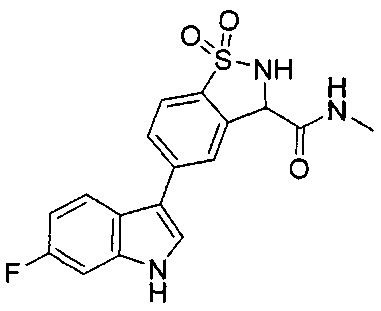
Раствор метил-5-(1-(трет-бутоксикарбонил)-6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-3-карбоксилат-1,1-диоксида (120 мг, 0,261 ммоль) в MeNH2/EtOH (5 мл, 30%) перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали с получением указанного в заголовке соединения в виде рацемической смеси. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением 5-(6-фтор-1Н-индол-3-ил)-N-метил-2,3-дигидробензо[d]изотиазол-3-карбоксамид-1,1-диоксида в качестве первого элюируемого пика (38 мг, 41%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.71 (br s, 1Н), 8.37 (d, J=5,52 Гц, 1Н), 8.16 (d, J=4,77 Гц, 1Н), 8.00-8.08 (m, 1Н), 7.80-7.96 (m, 4 Н), 7.29 (dd, J=9,79, 2,51 Гц, 1Н), 7.06 (td, J=9,22, 2,38 Гц, 1Н), 5.23 (d, J=5,02 Гц, 1Н), 2.67 (d, J=4,52 Гц, 3Н); ЖХ-МС m/z S 381,9 (M+Na)+; [α]20D -59,38° (с=0,0035 г/мл, DMSO).
Пример 18: (+)-5-(6-фтор-1H-индол-3-ил)-N-метил-2,3-дигидробензо[d]изотиазол-3-карбоксамид-1,1-диоксид
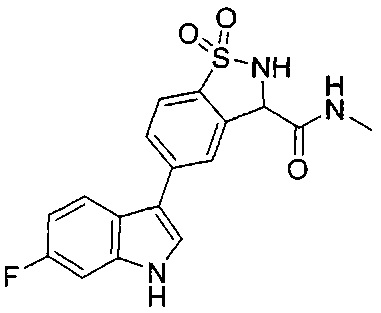
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 17 (35 мг, 37%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.71 (br s, 1Н), 8.37 (d, J=5,52 Гц, 1Н), 8.16 (d, J=4,77 Гц, 1Н), 8.00-8.08 (m, 1Н), 7.80-7.96 (m, 4Н), 7.29 (dd, J=9,79, 2,51 Гц, 1Н), 7.06 (td, J=9,22, 2,38 Гц, 1Н), 5.23 (d, J=5,02 Гц, 1Н), 2.67 (d, J=4,52 Гц, 3Н); ЖХ-МС: m/z 381,9 (M+Na)+; [α]20D +76,85° (с=0,0036 г/мл, DMSO).
Пример 19: (+)-3-(аминометил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
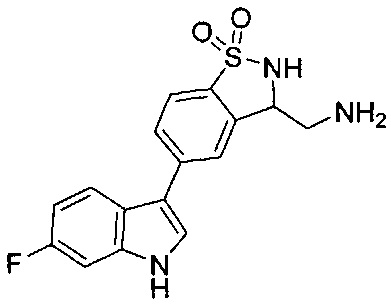
Стадия 1: N-(3-бромфенетил)-2,2,2-трифторацетамид
К охлажденному (ледяная баня) раствору 3-бромфенетиламина (21 г, 105 ммоль) и TEA (12,7 г, 125 ммоль) в DCM (150 мл) добавляли по каплям трифторуксусный ангидрид (24 г, 115 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем вливали в воду (200 мл) и экстрагировали DCM (2×200 мл). Объединенные органические слои концентрировали, затем разбавляли метил-трет-бутиловым эфиром (ТВМЕ) (250 мл) и H2O (50 мл). Слои разделяли и органическую фазу промывали рассолом (50 мл), затем сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного N-(3-бромфенетил)-O-(2,2,2-трифторацетил)гидроксиламина (33 г, более 100%) в виде коричневого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 9.49 (br s, 1Н), 7.47-7.35 (m, 2Н), 7.30-7.17 (m, 2Н), 3.42 (d, J=4,0 Гц, 2Н), 2.80 (t, J=7,0 Гц, 2Н).
Стадия 2: 4-бром-2-(2-(2,2,2-трифторацетамидо)этил)бензол-1-сульфонилхлорид
Раствор N-(3-бромфенетил)-2,2,2-трифторацетамида (33 г, 110 ммоль) в DCM (250 мл) охлаждали в ледяной бане, затем добавляли по каплям хлорсульфоновую кислоту (121 г). Реакционную смесь перемешивали в атмосфере N2 при 5°C в течение 4 часов, затем при комнатной температуре в течение 18 часов. Реакционную смесь осторожно вливали в ледяную воду (500 мл) и экстрагировали ТВМЕ (2×200 мл). Объединенные органические слои промывали рассолом (10 мл) и NaHCO3 (насыщ.) (50 мл), затем сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного 4-бром-2-(2-(2,2,2-трифторацетамидо)этил)бензол-1-сульфонилхлорида (35 г, 82%) в виде коричневого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 9.68 (br s, 1Н), 7.67 (s, 1Н), 7.52-7.25 (m, 2Н), 3.62-3.40 (m, 2Н), 3.27 (t, J=6,7 Гц, 2Н).
Стадия 3: 5-бром-2-(N-(трет-бутил)сульфамоил)фенетилацетат
В охлажденный (ледяная баня) раствор 4-бром-2-(2-(2,2,2-трифторацетамидо)этил)бензол-1-сульфонилхлорида (5 г, 13 ммоль) в DCM (50 мл) добавляли по каплям трет-бутиламин (4,0 мл, 38 ммоль). Реакционную смесь перемешивали в ледяной бане в течение 10 мин, затем концентрировали и очищали посредством колоночной хроматографии с получением 5-бром-2-(N-(трет-бутил)сульфамоил)фенетилацетата (5 г, 92%) в виде светло-желтого масла. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 9.45 (t, J=5,4 Гц, 1Н), 7.82 (d, J=8,5 Гц, 1Н), 7.71-7.60 (m, 2Н), 7.53 (d, J=2,0 Гц, 1Н), 3.54 (q, J=6,3 Гц, 2Н), 3.21 (t, J=6,7 Гц, 2Н), 1.13 (s, 9Н).
Стадия 4: N-(2-бром-2-(5-бром-2-(N-(трет-бутил)сульфамоил)-фенил)этил)-2,2,2-трифторацетамид
Раствор N-(5-бром-2-(N-(трет-бутил)сульфамоил)фенетил)-2,2,2-трифторацетамида (12 г, 28 ммоль), NBS (7,4 г, 42 ммоль) и AIBN (2,3 г, 14 ммоль) в CCl4 (350 мл) перемешивали при 80°C в течение 16 часов, затем концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением N-(2-бром-2-(5-бром-2-(N-(трет-бутил)-сульфамоил)фенил)этил)-2,2,2-трифторацетамида (3,8, 27%) в виде желтой смолы, и 6,9 г исходного материала было выделено.
Стадия 5: N-((5-бром-2-(трет-бутил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)-2,2,2-трифторацетамид
Смесь N-(2-бром-2-(5-бром-2-(N-(трет-бутил)сульфамоил)фенил)этил)-2,2,2-трифторацетамида (3,8 г, 7,4 ммоль) и K2CO3 (2,1 г, 15 ммоль) в DMF (60 мл) нагревали до 80°C в течение 2 часов, затем разбавляли водой (50 мл) и экстрагировали EtOAc (25 мл×2). Объединенные органические экстракты промывали рассолом (10 мл×3), затем сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением N-((5-бром-2-(трет-бутил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)-2,2,2-трифторацетамида (4,2 г, 100%) в виде желтой смолы.
Стадия 6: трет-бутил-3-(2-(трет-бутил)-1,1-диоксидо-3-((2,2,2-трифторацетамидо)метил)-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилат
К суспензии N-((5-бром-2-(трет-бутил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)-2,2,2-трифторацетамида (4,2 г, 9,8 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (3,5 г, 9,8 ммоль) и Cs2CO3 (6,4 мг, 20 ммоль) в диоксане (60 мл) и H2O (15 мл) добавляли PdCl2(dppf) (0,72 г, 0,98 ммоль). Реакционную смесь перемешивали при 80°C в атмосфере N2 в течение 1,5 часов, затем концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением трет-бутил-3-(2-(трет-бутил)-1,1-диоксидо-3-((2,2,2-трифторацетамидо)метил)-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилата (2,9 г, 51%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 9.37 (t, J=6,1 Гц, 1Н), 8.07-7.98 (m, 2Н), 7.94-7.88 (m, 2Н), 7.86-7.80 (m, 2Н), 7.25 (dt, J=2,5, 9,0 Гц, 1Н), 5.15 (dd, J=2,4, 6,4 Гц, 1Н), 3.88-3.79 (m, 1Н), 3.56-3.49 (m, 1Н), 1.66 (s, 9Н), 1.51 (s, 9Н).
Стадия 7: хиральный 3-(аминометил)-2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К раствору трет-бутил-3-(2-(трет-бутил)-1,1-диоксидо-3-((2,2,2-трифторацетамидо)метил)-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилата (500 мг, 0,857 ммоль) в МеОН (6 мл) и H2O (3 мл) добавляли NaOH (69 мг, 1,7 ммоль). Реакционную смесь перемешивали при 50°C в течение 9 часов, затем разбавляли EtOAc/МеОН (10/1: 30 мл). Слои разделяли и органическую фазу промывали рассолом (10 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Осадок очищали посредством колоночной хроматографии с получением рацемического 3-(аминометил)-2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]-изотиазол-1,1-диоксида (262 мг, 79%) в виде светло-желтой смолы. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением соединения пика 1 (112 мг, 33,1%) и пика 2 (105 мг, 31,6%), каждое в виде желтой смолы.
Стадия 8: (+)-3-(аминометил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Раствор хирального 3-(аминометил)-2-(трет-бутил)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (100 мг, 0,19 ммоль) (стадия 7, пик 1) в HCl/MeOH (10 мл) перемешивали при 30°C в течение 16 часов. Реакционную смесь концентрировали и очищали посредством препаративной ВЭЖХ с получением (+)-3-(аминометил)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (75 мг, 88%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.98 (dd, J=1,0, 8,3 Гц, 1Н), 7.95-7.89 (m, 2Н), 7.84 (d, J=8,0 Гц, 1Н), 7.73 (s, 1Н), 7.18 (dd, J=2,3, 9,8 Гц, 1Н), 6.96 (dt, J=2,5, 9,2 Гц, 1Н), 5.03 (dd, J=3,5, 8,8 Гц, 1Н), 3.56 (dd, J=3,6, 13,2 Гц, 1Н), 3.28-3.21 (m, 1Н); ЖХ-МС: m/z 353,9 (М+Na)+; [α]20D +84,38° (с=0,0035 г/мл, МеОН).
Пример 20: (-)-3-(аминометил)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
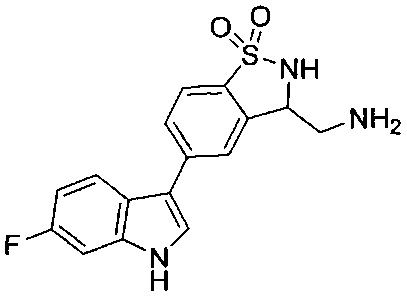
Раствор хирального 3-(аминометил)-2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (100 мг, 0,19 ммоль) (Пример 19, стадия 7, пик 2) в HCl (газ)/МеОН (10 мл) перемешивали при 30°C в течение 16 часов. Реакционную смесь концентрировали и очищали посредством препаративной ВЭЖХ с получением (-)-3-(амино-метил)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (72 мг, 84%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 8.01-7.96 (m, 1Н), 7.95-7.89 (m, 2Н), 7.84 (d, J=8,3 Гц, 1Н), 7.73 (s, 1Н), 7.18 (dd, J=2,3, 9,5 Гц, 1Н), 6.96 (dt, J=2,4, 9,2 Гц, 1Н), 5.03 (dd, J=3,5, 8,8 Гц, 1Н), 3.56 (dd, J=3,5, 13,1 Гц, 1Н), 3.24 (dd, J=8,8, 13,1 Гц, 1Н); ЖХ-МС: m/z 353,9 (М+Н)+; [α]20D -65,21° (с=0,0035 г/мл, МеОН).
Пример 21: (-)-метил-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)карбамат
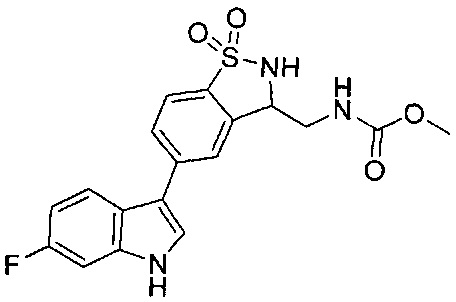
Стадия 1: метил-((2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)карбамат
К раствору 3-(аминометил)-2-(трет-бутил)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (300 мг, 0,774 ммоль) и N,N-диизопропилэтиламина (DIPEA) (200 мг, 1,55 ммоль) в DCM (5 мл) добавляли метилхлорформиат (81 мг, 0,852 ммоль) при 5°C. Реакционную смесь перемешивали в течение 30 минут, затем экстрагировали EtOAc (20 мл×2). Объединенные органические слои промывали рассолом (15 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного метил-((2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)карбамата (320 мг, выход 93%) в виде не совсем белого твердого вещества.
Стадия 2: (-)-метил-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)карбамат
Раствор метил-((2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)карбамата (320 мг, 0,718 ммоль) и TFA (8 мл) в DCM (5 мл) перемешивали при 50°C в течение 3 часов. Реакционную смесь концентрировали и нейтрализовали NaHCO3 (насыщ.), затем экстрагировали EtOAc (20 мл×2). Объединенные органические слои промывали рассолом (15 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством препаративной ВЭЖХ с получением 100 мг рацемического продукта. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (-)-метил-((5-(6-фтор-1Н-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)карбамата в качестве первого элюируемого пика (33 мг, 12%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.97-7.88 (m, 2H), 7.86 (s, 1H), 7.77 (d, J=8,0 Гц, 1H), 7.68 (s, 1H), 7.16 (dd, J=2,4, 9,7 Гц, 1H), 6.95 (dt, J=2,4, 9,2 Гц, 1H), 4.87-4.82 (m, 1H), 3.71-3.58 (m, 4H), 3.50 (dd, J=7,0, 14,1 Гц, 1H); ЖХ-МС: m/z, 411,9 (M+Na)+; [α]20D -85° (c=1 мг/мл, MeOH).
Пример 22: (+)-метил-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)карбамат
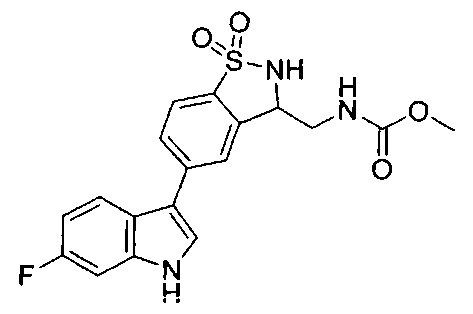
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 21 (37 мг, 13%), в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.95-7.88 (m, 2H), 7.86 (s, 1H), 7.77 (d, J=8,3 Гц, 1H), 7.68 (s, 1H), 7.16 (dd, J=2,4, 9,7 Гц, 1H), 6.95 (dt, J=2,5, 9,2 Гц, 1H), 4.85 (d, J=5,8 Гц, 1H), 3.69-3.59 (m, 4H), 3.50 (dd, J=6,9, 14,2 Гц, 1H); ЖХ-МС: m/z 411,8 (M+Na)+; [α]20D+89° (c=1 мг/мл, MeOH).
Пример 23: (-)-N-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)ацетамид
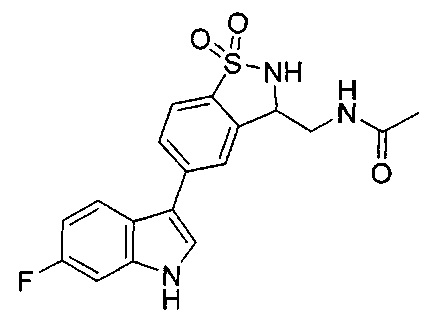
Стадия 1: N-((2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)ацетамид
К охлажденному (ледяная баня) раствору 3-(аминометил)-2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (100 мг, 0,258 ммоль) в DCM (5 мл) и DIPEA (0,09 мл, 0,52 ммоль) добавляли по каплям раствор ацетилхлорида (22 мг, 0,28 ммоль) в DCM (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, затем концентрировали и очищали посредством колоночной хроматографии с получением указанного в заголовке соединения в виде рацемической смеси. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением первого элюируемого пика указанного в заголовке соединения (пик 1, 37 мг, 23%) и второго элюируемого пика (пик 2, 25 мг, выход: 23%) в виде белой смолы.
Стадия 2: (-)-N-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)ацетамид
Раствор хирального N-((2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)ацетамида (стадия 1, пик 1, 25 мг, 0,07 ммоль) в HCl(газ)/MeOH (5 мл) перемешивали при 30°C в течение 18 часов. Реакционную смесь концентрировали и очищали посредством препаративной ТСХ с получением (-)-N-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)ацетамида (11 мг, 44%) в виде белого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.97-7.86 (m, 3H), 7.77 (d, J=8,0 Гц, 1H), 7.70-7.68 (m, 1H), 7.16 (dd, J=2,3, 9,5 Гц, 1H), 6.99-6.92 (m, 1H), 4.91-4.88 (m, 1H), 3.81 (dd, J=4,1, 13,9 Гц, 1H), 3.52 (dd, J=6,7, 13,9 Гц, 1H), 1.94 (s, 3H) ЖХ-МС: m/z 373,9 (M+H)+; [α]20D -67,65° (c=0,00136 г/мл, MeOH).
Пример 24: (+)-N-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)ацетамид
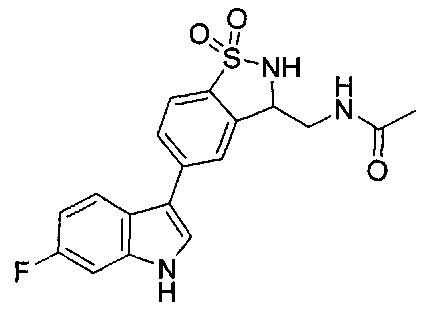
Раствор хирального N-((2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)ацетамида (Пример 23, стадия 1, пик 2, 37 мг, 0,09 ммоль) в HCl/MeOH (5 мл) перемешивали при 30°C в течение 7 часов. Реакционную смесь концентрировали и очищали посредством препаративной ТСХ с получением (+)-N-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)ацетамида (27 мг, 83%) в виде белого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.94-7.87 (m, 3H), 7.77 (d, J=8,0 Гц, 1H), 7.68 (s, 1H), 7.16 (dd, J=2,3, 9,8 Гц, 1H), 6.95 (dt, J=2,5, 9,2 Гц, 1H), 4.91-4.88 (m, 1H), 3.80 (dd, J=4,1, 13,9 Гц, 1H), 3.56-3.48 (m, 1H), 1.94 (s, 3H) ЖХ-МС: m/z 395,9 (M+Na)+; [α]20D +81,67° (c=0,0024 г/мл, MeOH).
Пример 25: (-)-3-((диметиламино)метил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
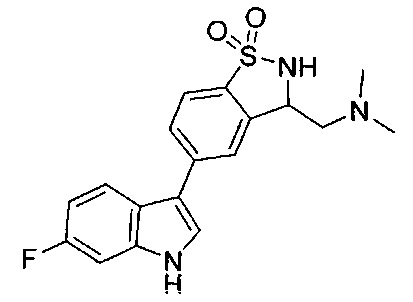
Стадия 1: 2-(трет-бутил)-3-((диметиламино)метил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К раствору трет-бутил-3-(2-(трет-бутил)-1,1-диоксидо-3-((2,2,2-трифторацетамидо)метил)-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (1,9 г, 3,3 ммоль) в MeOH (20 мл) и H2O (12 мл) добавляли NaOH (260 мг, 6,51 ммоль). Реакционную смесь перемешивали при 65°C в течение 30 часов, затем разбавляли водой (20 мл) и экстрагировали EtOAc (50 мл × 2). Объединенные органические слои промывали рассолом (50 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением 2-(трет-бутил)-3-((диметиламино)метил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (1,1 г, 87%) в виде не совсем белого твердого вещества.
Стадия 2: 2-(трет-бутил)-3-((диметиламино)метил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К раствору 3-(аминометил)-2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (200 мг, 0,516 ммоль) в MeOH (5 мл) добавляли формальдегид (37% в воде, 209 мг, 2,58 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 15 мин, затем добавляли NaBH3CN (49 мг, 0,77 ммоль) с последующим добавлением AcOH (31 мг, 0,52 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, затем экстрагировали EtOAc (20 мл × 2). Объединенные органические слои промывали рассолом (15 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного 2-(трет-бутил)-3-((диметиламино)метил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (200 мг, 93%) в виде желтого масла.
Стадия 3: (-)-3-((диметиламино)метил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Раствор 2-(трет-бутил)-3-((диметиламино)метил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (200 мг, 0,481 ммоль) и TFA (5 мл) в DCM (5 мл) перемешивали при 30°C в течение 2 часов, затем при 50°C в течение 2 часов. Реакционную смесь концентрировали, затем нейтрализовали NaHCO3 (насыщ.) и экстрагировали EtOAc (30 мл × 2). Объединенные органические слои промывали рассолом (15 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством препаративной ВЭЖХ с получением указанного в заголовке соединения в виде рацемической смеси. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (-)-3-((диметиламино)метил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида в качестве первого элюируемого пика (26 мг, 15%) в виде бесцветного масла. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.90-7.83 (m, 3H), 7.76 (d, J=8,0 Гц, 1H), 7.67 (s, 1H), 7.16 (dd, J=2,3, 9,5 Гц, 1H), 6.94 (dt, J=2,4, 9,2 Гц, 1H), 4.86 (dd, J=4,5, 9,3 Гц, 1H), 2.84 (dd, J=4,3, 12,8 Гц, 1H), 2.70 (dd, J=9,3, 12,8 Гц, 1H), 2.43 (s, 6H); ЖХ-МС: m/z 359,9 (M+H)+; [α]20D -270,66° (c=1 мг/мл, MeOH).
Пример 26: (+)-3-((диметиламино)метил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
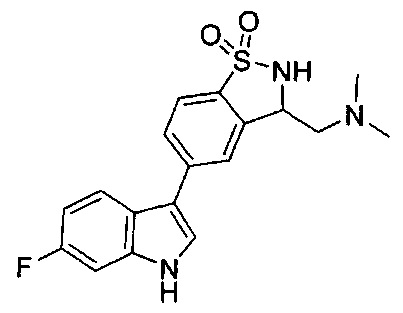
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 25 (25 мг, 14%), в виде белого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.92-7.81 (m, 3H), 7.75 (d, J=8,0 Гц, 1H), 7.65 (s, 1H), 7.15 (dd, J=2,3, 9,8 Гц, 1H), 6.94 (dt, J=2,3, 9,2 Гц, 1H), 4.83 (dd, J=4,5, 9,0 Гц, 1H), 2.84-2.75 (m, 1H), 2.66 (dd, J=9,0, 12,8 Гц, 1H), 2.40 (s, 6H); ЖХ-МС: m/z 359,7 (M+H)+; [α]20D+31° (c=1 мг/мл, MeOH).
Пример 27: (+)-N-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)-N-метилацетамид
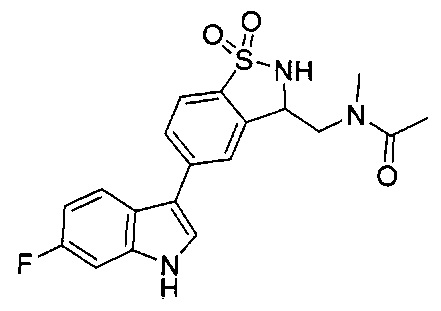
Стадия 1: трет-бутил-3-(2-(трет-бутил)-1,1-диоксидо-3-((2,2,2-трифтор-N-метилацетамидо)метил)-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилат
К раствору трет-бутил-3-(2-(трет-бутил)-1,1-диоксидо-3-((2,2,2-трифторацетамидо)метил)-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (430 мг, 0,737 ммоль) в DMF (5 мл) добавляли NaH (35 мг, 0,88 ммоль, 60% дисперсия в минеральном масле). Суспензию перемешивали при 10°C в течение 5 мин, затем добавляли метилйодид (MeI) (523 мг, 3,68 ммоль) и перемешивание продолжали при 10°C в течение 16 часов. Реакционную смесь гасили NH4Cl (насыщ.) (15 мл) и экстрагировали EtOAc (30 мл × 2). Объединенные органические слои промывали рассолом (15 мл × 3), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного трет-бутил-3-(2-(трет-бутил)-1,1-диоксидо-3-((2,2,2-трифторацетамидо)метил)-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (500 мг, более 100%) в виде желтого масла.
Стадия 2: 2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-3-((метиламино)-метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К раствору трет-бутил-3-(2-(трет-бутил)-1,1-диоксидо-3-((2,2,2-трифторацетамидо)метил)-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (500 мг, 0,837 ммоль) в MeOH (10 мл) и H2O (6 мл) добавляли NaOH (100 мг, 2,51 ммоль). Реакционную смесь перемешивали при 60°C в течение 16 часов, затем разбавляли водой (20 мл) и экстрагировали EtOAc (50 мл × 3). Объединенные органические слои промывали рассолом (50 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного 2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-3-((метиламино)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (330 мг, 98%) в виде не совсем белого твердого вещества.
Стадия 3: N-((2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)-N-метилацетамид
К раствору 2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-3-((метиламино)-метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (180 мг, 0,448 ммоль) и DIPEA (0,148 мл, 0,897 ммоль) в DCM (5 мл) добавляли по каплям раствор ацетилхлорида (38,7 мг/0,0351 мл, 0,493 ммоль) в DCM (1 мл). Реакционную смесь перемешивали при 10°C в течение 1 часа, затем концентрировали, разбавляли водой (10 мл) и экстрагировали DCM (20 мл × 3). Объединенные органические слои промывали рассолом (10 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением N-((2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)-метил)-N-метилацетамида (220 мг, более 100%) в виде не совсем белого твердого вещества.
Стадия 4: (+)-N-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)-N-метилацетамид
Раствор N-((2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)-N-метилацетамида (220 мг, 0,496 ммоль) в TFA (10 мл) перемешивали при 50°C в течение 32 часов. Реакционную смесь концентрировали, затем нейтрализовали NaHCO3 (насыщ.) (10 мл) и экстрагировали EtOAc (20 мл × 3). Объединенные органические слои промывали рассолом (20 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного продукта (200 мг). Этот неочищенный продукт очищали посредством препаративной ВЭЖХ с получением рацемического N-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)-N-метилацетамида (54 мг, выход 28%) в виде бесцветного масла. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (+)-N-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)-N-метилацетамида в качестве первого элюируемого пика (18 мг, 33%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1H), 8.20-7.76 (m, 6H), 7.34-7.21 (m, 1H), 7.02 (t, J=9,3 Гц, 1H), 4.96 (br s, 1H), 4.04-3.48 (m, 2H), 3.09-2.89 (m, 3H), 2.02 (d, J=3,8 Гц, 3H); ЖХ-МС: m/z 388,1 (M+H)+; [α]20D +35° (c=2 мг/мл, DMSO).
Пример 28: (-)-N-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)-N-метилацетамид
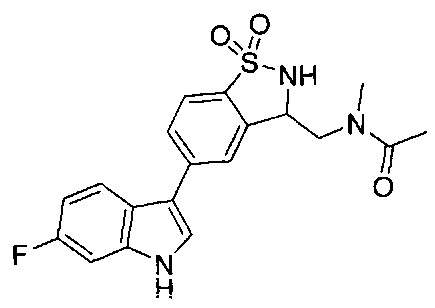
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 27 (18 мг, 33%), в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1H), 8.18-7.78 (m, 6H), 7.27 (d, J=9,8 Гц, 1H), 7.02 (t, J=9,2 Гц, 1H), 4.97 (d, J=9,5 Гц, 1H), 4.05-3.48 (m, 2H), 3.08-2.91 (m, 3H), 2.02 (d, J=4,8 Гц, 3H); ЖХ-МС: m/z 387,9 (M+H)+; [α]20D -30,5° (c=2 мг/мл, DMSO).
Пример 29: (-)-5-(6-фтор-1H-индол-3-ил)-3-(2-гидроксиэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
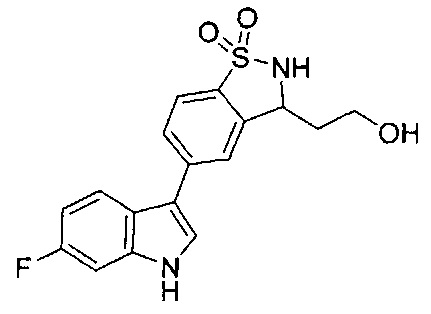
Стадия 1: 3-(3-бромфенил)пропан-1-ол
В охлажденный (ледяная баня) раствор 3-(3-бромфенил)пропановой кислоты (8,0 г, 35 ммоль) в безводном THF (100 мл) добавляли BH3-THF (100 мл, 1,0 М) через капельную воронку в атмосфере азота. Реакционную смесь перемешивали при 0°C в течение 1 часа, затем при дефлегмации в течение 16 часов. Реакционную смесь осторожно гасили HCl (37 мл, 2 М), затем концентрировали для удаления THF. Суспензию разбавляли DCM (300 мл), затем фильтровали для удаления твердых веществ. Слои разделяли и органическую фазу промывали водой (50 мл) и рассолом (50 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением 3-(3-бромфенил)пропан-1-ола (9,8 г, более 100%) в виде желтого масла, которое использовали на следующей стадии без дополнительной очистки.
Стадия 2: 3-(3-бромфенил)пропилацетат
К раствору 3-(3-бромфенил)пропан-1-ола (4,5 г, 16 ммоль) в DCM (50 мл) добавляли Ac2O (1,95 г, 19,1 ммоль), Et3N (4,83 г, 47,7 ммоль) и DMAP (97 мг, 0,79 ммоль). Реакционную смесь перемешивали при 16°C в течение 30 минут, затем гасили водой (10 мл) и слои разделяли. Органическую фазу промывали водой (20 мл) и рассолом (20 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенное масло очищали посредством колоночной хроматографии (силикагель, 0-50% EtOAc в петролейном эфире) с получением 3-(3-бромфенил)пропилацетата (4,0 г, 98%) в виде желтого масла. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 7.43 (s, 1H), 7.38 (td, J=1,9, 7,3 Гц, 1H), 7.27-7.21 (m, 2H), 3.98 (t, J=6,5 Гц, 2H), 2.65-2.61 (m, 2H), 1.99 (s, 3H), 1.90-1.80 (m, 2H).
Стадия 3: 3-(5-бром-2-(хлорсульфонил)фенил)пропилацетат
В охлажденный раствор 3-(3-бромфенил)пропилацетата (3,8 г, 15 ммоль) в CHCl3 (50 мл) добавляли по каплям хлорсульфоновую кислоту (17,2 г, 148 ммоль) через капельную воронку. Реакционную смесь перемешивали при 14°C в течение 3 часов, затем выливали на лед (200 г) и экстрагировали DCM (50 мл × 3). Объединенные органические слои промывали водой (50 мл), NaHCO3 (насыщ.) (50 мл), водой (50 мл) и рассолом (50 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением 3-(5-бром-2-(хлорсульфонил)фенил)пропилацетата (2,2 г, 42%) в виде желтого масла.
Стадия 4: 3-(5-бром-2-(N-(трет-бутил)сульфамоил)фенил)пропилацетат
К раствору 3-(5-бром-2-(хлорсульфонил)фенил)пропилацетата (1,7 г, 8,0 ммоль) в DCM (30 мл) добавляли t-BuNH2 (1,2 г, 16 ммоль). Реакционную смесь перемешивали при 14°C в течение 30 мин, затем гасили водой (15 мл) и концентрировали для удаления DCM. Осадок экстрагировали EtOAc (20 мл × 3) и объединенные органические слои промывали водой (20 мл) и рассолом (20 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением 3-(5-бром-2-(N-(трет-бутил)сульфамоил)фенил)пропилацетата (2,4 г, 81%) в виде желтого масла, которое использовали на следующей стадии без дополнительной очистки.
Стадия 5: 3-бром-3-(5-бром-2-(N-(трет-бутил)сульфамоил)фенил)-пропилацетат
К раствору 3-(5-бром-2-(N-(трет-бутил)сульфамоил)фенил)-пропилацетата (2,4 г, 6,118 ммоль) в CCl4 (40 мл) добавляли NBS (1,2 г, 6,73 ммоль) и AIBN (502 мг, 3,06 ммоль). Реакционную смесь перемешивали в атмосфере азота при 80°C в течение 6 часов, затем гасили водой (20 мл). Слои разделяли и органическую фазу промывали водой (20 мл × 2) и рассолом (20 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением 3-бром-3-(5-бром-2-(N-(трет-бутил)-сульфамоил)фенил)пропилацетата (3,0 г, более 100%) в виде коричневого масла.
Стадия 6: 2-(5-бром-2-(трет-бутил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)этилацетат
К раствору 3-бром-3-(5-бром-2-(N-(трет-бутил)сульфамоил)-фенил)пропилацетата (1,7 г, 3,5 ммоль) в MeCN (20 мл) добавляли K2CO3 (968 мг, 7,00 ммоль). Реакционную смесь перемешивали при 40°C в течение 16 часов, затем гасили водой (15 мл) и экстрагировали EtOAc (20 мл × 3). Объединенные органические слои промывали водой (20 мл) и рассолом (20 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением 2-(5-бром-2-(трет-бутил)-1,1-диоксидо-2,3-дигидробензо[d]-изотиазол-3-ил)этилацетата (580 мг, 42%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 7.66-7.59 (m, 2H), 7.56-7.54 (m, 1H), 4.82 (t, J=4,6 Гц, 1H), 4.28-4.23 (m, 1H), 4.06-3.99 (m, 1H), 2.34-2.30 (m, 2H), 1.88 (s, 3H), 1.55 (s, 9H).
Стадия 7: трет-бутил-3-(3-(2-ацетоксиэтил)-2-(трет-бутил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилат
Раствор 2-(5-бром-2-(трет-бутил)-1,1-диоксидо-2,3-дигидробензо[d]-изотиазол-3-ил)этилацетата (580 мг, 1,49 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (448 мг, 1,63 ммоль), PdCl2(dppf)CH2Cl2 (111 мг, 0,149 ммоль) и K3PO4 (946 мг, 4,46 ммоль) в 1,4-диоксане (10 мл) и воде (2,5 мл) барботировали азотом в течение 1 минуты, затем перемешивали при 80°C в течение 16 часов. Неочищенную реакционную смесь экстрагировали EtOAc (15 мл × 3) и объединенные органические слои промывали водой (15 мл) и рассолом (15 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии (силикагель, 0-40% EtOAc в петролейном эфире) с получением трет-бутил-3-(3-(2-ацетоксиэтил)-2-(трет-бутил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (460 мг, 57%) в виде желтой смолы.
Стадия 8: 2-(5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)этилацетат
Охлажденный (ледяная баня) раствор трет-бутил-3-(3-(2-ацетоксиэтил)-2-(трет-бутил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (660 мг, 1,21 ммоль) в TFA (6 мл) перемешивали при 6°C в течение 3 часов. Реакционную смесь концентрировали, затем разбавляли DCM (60 мл) и нейтрализовали NaHCO3 (насыщ.) (20 мл). Слои разделяли и органическую фазу промывали водой (20 мл) и рассолом (20 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного 2-(5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)этилацетата (280 мг, 60%) в виде белого твердого вещества.
Стадия 9: (-)-5-(6-фтор-1H-индол-3-ил)-3-(2-гидроксиэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К раствору 2-(5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)этилацетата (280 мг, 0,721 ммоль) в THF (20 мл) и воде (10 мл) добавляли LiOH-H2O (151 мг, 3,60 ммоль). Реакционную смесь перемешивали при 50°C в течение 2 часов, затем экстрагировали DCM (20 мл × 3). Водную фазу лиофилизировали, затем растворяли в DCM/MeOH (40 мл/4 мл) и фильтровали. Фильтрат концентрировали с получением рацемического 5-(6-фтор-1H-индол-3-ил)-3-(2-гидроксиэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (190 мг, 76%) в виде белого твердого вещества. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (-)-5-(6-фтор-1H-индол-3-ил)-3-(2-гидроксиэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида в качестве первого элюируемого пика (54 мг, 28%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1H), 7.95-7.90 (m, 2H), 7.88-7.77 (m, 4H), 7.26 (dd, J=2,4, 9,9 Гц, 1H), 7.01 (dt, J=2,4, 9,2 Гц, 1H), 4.81-4.74 (m, 1H), 4.70 (t, J=4,9 Гц, 1H), 3.67-3.57 (m, 2H), 2.22-2.13 (m, 1H), 1.83-1.74 (m, 1H); ЖХ-МС: m/z 347,1 (M+H)+; [α]20D -46,89° (c=1,045 мг/мл, MeOH).
Пример 30: (+)-5-(6-фтор-1H-индол-3-ил)-3-(2-гидроксиэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
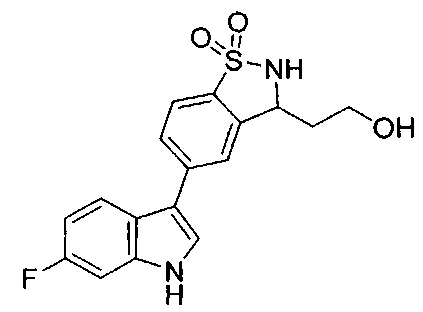
Стадия 1: (+)-5-(6-фтор-1H-индол-3-ил)-3-(2-гидроксиэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 29 (58 мг, 31%), в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1H), 7.95-7.90 (m, 2H), 7.89-7.76 (m, 4H), 7.26 (dd, J=2,4, 9,9 Гц, 1H), 7.01 (dt, J=2,4, 9,2 Гц, 1H), 4.77 (dd, J=3,0, 9,5 Гц, 1H), 4.70 (br s, 1H), 3.65-3.57 (m, 2H), 2.22-2.14 (m, 1H), 1.77 (dt, J=5,0, 9,2 Гц, 1H); ЖХ-МС: m/z 346,9 (M+H)+; [α]20D +44,1° (c=1,155 мг/мл, MeOH).
Пример 31: 2-(2-аминоэтил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
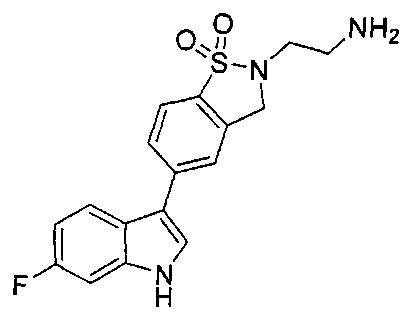
Стадия 1: 4-бром-2-метилбензол-1-сульфонилхлорид
В бесцветный раствор 1-бром-3-метилбензола (40 г, 230 ммоль) в CHCl3 (400 мл) добавляли хлорсульфоновую кислоту (164 г, 1400 ммоль) при 0°C через капельную воронку в течение 80 мин. Полученный коричневый раствор перемешивали при 0°C в течение 4 часов, затем выливали на лед (400 г) и экстрагировали дихлорметаном (150 мл × 3). Объединенные органические слои промывали водой (150 мл × 2) и рассолом (150 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением 4-бром-2-метилбензол-1-сульфонилхлорида (45 г, 71%) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 7.93 (dd, J=1,3, 8,5 Гц, 1H), 7.61 (s, 1H), 7.57 (d, J=8 Гц, 1H), 2.77 (s, 3H).
Стадия 2: 4-бром-2-метилбензолсульфонамид
К желтому раствору 4-бром-2-метилбензол-1-сульфонилхлорида (45 г, 170 ммоль) в 1,4-диоксане (350 мл) добавляли NH4OH (350 мл, 28% масс/об.) при 0°C через капельную воронку. Реакционную смесь перемешивали при 24°C в течение 10 часов, затем концентрировали. Полученное твердое вещество промывали водой (50 мл), затем растирали с эфиром. Суспензию фильтровали и перекристаллизовывали с ацетоном/петролейным эфиром (100 мл/250 мл) с получением 4-бром-2-метилбензолсульфонамида (39 г, 94%) в виде белого кристаллического вещества. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 7.88 (d, J=8,5 Гц, 1H), 7.51 (s, 1H), 7.48-4.46 (m, 1H), 4.83 (br s, 2H), 2.67 (s, 3H).
Стадия 3: 5-бромбензо[d]изотиазол-3(2H)-он-1,1-диоксид
В трехгорлую круглодонную колбу емкостью 500 мл, оборудованную магнитной мешалкой и встроенным термометром, загружали раствор H5IO6 (46,3 г, 203 ммоль) в MeCN (350 мл). Белую суспензию перемешивали при 23°C в течение 1 часа, затем добавляли CrO3 (254 мг, 2,54 ммоль) с последующим добавлением Ac2O (20,7 г, 203 ммоль). Реакционную смесь охлаждали до 0°C и добавляли 4-бром-2-метилбензолсульфонамид (6,35 г, 25,4 ммоль) при перемешивании при 0°C в течение 15 мин, затем при комнатной температуре в течение 16 часов. Полученную желтую суспензию фильтровали и промывали водой (300 мл). Белое твердое вещество сушили с получением 5-бромбензо[d]изотиазол-3(2H)-он-1,1-диоксида (5,6 г, 84%) в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 8.23-8.01 (m, 3H), 7.08 (br s, 1H).
Стадия 4: 5-бром-2,3-дигидробензо[d]изотиазол-1,1-диоксид
В охлажденную (ледяная баня) белую суспензию 5-бромбензо[d]изотиазол-3(2H)-он-1,1-диоксида (7,5 г, 29 ммоль) в безводном THF (160 мл) медленно добавляли BH3.MeS (10 М, 14,3 мл, 143 ммоль). Реакционную смесь перемешивали при 75°C в течение 2 часов, затем медленно гасили HCl (2 М, 150 мл) при выдерживании в ледяной бане. Смесь перемешивали в течение 1 часа, затем концентрировали и экстрагировали дихлорметаном (100 мл × 4). Объединенные органические слои промывали рассолом, затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением 5-бром-2,3-дигидробензо[d]изотиазол-1,1-диоксида (6,6 г, 92%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 7.91 (s, 1H), 7.84 (s, 1H), 7.80-7.72 (m, 2H), 4.39 (d, J=5,0 Гц, 2H). 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 7.67 (d, J=1,0 Гц, 2H), 7.57 (s, 1H), 4.89 (br s, 1H), 4.52 (s, 2H).
Стадия 5: трет-бутил-(2-гидроксиэтил)карбамат
К бесцветному раствору Boc2O (21,8 г, 0,1 моль) в дихлорметане (20 мл) добавляли этаноламин (6,71 г, 0,11 моль) порциями при 0°C в течение 15 мин. Реакционную смесь перемешивали при 28°C в течение 16 часов, затем гасили водой (20 мл) и экстрагировали дихлорметаном (30 мл × 3). Объединенные органические слои промывали водой (20 мл) и рассолом (20 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением трет-бутил-(2-гидроксиэтил)карбамата (17 г, 100%) в виде прозрачного масла. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 5.23 (br s, 1H), 3.63-3.61 (m, 2H), 3.23 (br s, 2H), 1.41 (s, 9H).
Стадия 6: 2-((трет-бутоксикарбонил)амино)этилметансульфонат
В охлажденный (ледяная баня) раствор трет-бутил-(2-гидроксиэтил)-карбамата (10,0 г, 62,0 ммоль) и Et3N (9,48 мл, 68,2 ммоль) в безводном дихлорметане (200 мл) добавляли раствор MsCl (8,53 г, 74,4 ммоль) в дихлорметане (100 мл) по каплям в течение 30 мин. Реакционную смесь перемешивали при 0°C в течение 1 часа, затем концентрировали. Неочищенный осадок разбавляли этилацетатом (100 мл) и водой (60 мл). Органический слой промывали водным 5% NaHCO3 (50 мл) и рассолом (50 мл), затем сушили над Na2SO4, фильтровали и концентрировали. Неочищенное масло очищали посредством колоночной хроматографии (силикагель, 0-50% этилацетат/петролейный эфир) с получением 2-((трет-бутоксикарбонил)-амино)этилметансульфоната (9,3 г, выход 62%) в виде бесцветной жидкости. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 5.03 (br s, 1H), 4.26 (t, J=5,1 Гц, 2H), 3.44 (d, J=4,8 Гц, 2H), 3.01 (s, 3H), 1.42 (s, 9H).
Стадия 7: трет-бутил-(2-(5-бром-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)карбамат
Белую суспензию 5-бром-2,3-дигидробензо[d]изотиазол-1,1-диоксида (500 мг, 2,02 ммоль) и K2CO3 (557 мг, 4,03 ммоль) в DMF (8 мл) перемешивали при 27°C в течение 1 часа, затем медленно добавляли раствор 2-((трет-бутоксикарбонил)амино)этилметансульфоната (723 мг, 3,02 ммоль) в DMF (2 мл). Реакционную смесь барботировали N2 в течение 1 мин и перемешивали при 80°C в течение 16 часов. Реакционную смесь охлаждали до температуры окружающей среды, разбавляли водой (30 мл) и экстрагировали этилацетатом (25 мл × 3). Объединенные органические слои промывали рассолом (20 мл × 2), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенное желтое твердое вещество очищали посредством колоночной хроматографии (силикагель, 0-50% этилацетат/петролейный эфир) с получением трет-бутил-(2-(5-бром-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-этил)карбамата (656 мг, 83%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 8.02 (s, 1H), 7.67 (s, 2H), 7.57 (s, 1H), 4.43 (s, 2H), 3.49-3.43 (m, 4H), 1.41 (s, 9H).
Стадия 8: трет-бутил-3-(2-(2-((трет-бутоксикарбонил)амино)этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилат
Желтый раствор трет-бутил-(2-(5-бром-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)карбамата (800 мг, 2,04 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (1,1 г, 2,5 ммоль), PdCl2(dppf)CH2Cl2(153 мг, 0,204 ммоль) и K3PO4 (1,3 г, 6,1 ммоль) в 1,4-диоксане (16 мл) и воде (4 мл) барботировали N2 в течение 1 мин и перемешивали при 80°C в течение 16 часов. Неочищенную реакционную смесь концентрировали, затем очищали посредством колоночной хроматографии (силикагель, 0-50% этилацетат/петролейный эфир) с получением трет-бутил-3-(2-(2-((трет-бутоксикарбонил)амино)этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (830 мг, 74%) в виде желтого масла. ЖХ-МС: m/z 568,1 (M+Na)+.
Стадия 9: 2-(2-аминоэтил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
В охлажденный (ледяная баня) желтый раствор трет-бутил-3-(2-(2-((трет-бутоксикарбонил)амино)этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (330 мг, 0,61 ммоль) в дихлорметане (4 мл) добавляли по каплям трифторуксусную кислоту (2 мл). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 часа, затем гасили водным NH3/H2O (5 мл) и экстрагировали дихлорметаном (10 мл × 3). Объединенные органические экстракты сушили над безводным Na2SO4, затем фильтровали и концентрировали. Неочищенное твердое вещество очищали посредством препаративной ВЭЖХ с получением 2-(2-аминоэтил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (24 мг, 20%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.74 (br s, 1H), 8.33 (s, 1H), 7.97-7.86 (m, 5H), 7.27 (dd, J=2,3, 9,8 Гц, 1H), 7.01 (dt, J=2,4, 9,2 Гц, 1H), 4.56 (s, 2H), 3.33-3.31 (m, 2H), 2.98 (t, J=5,8 Гц, 2H); ЖХ-МС: m/z 368,0 (M+Na)+.
Пример 32: 2-(2-(диметиламино)этил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
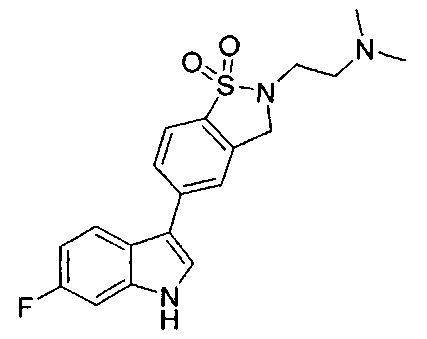
Стадия 1: 2-(2-(диметиламино)этил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К белой суспензии 2-(2-аминоэтил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (210 мг, 0,608 ммоль) в метаноле (10 мл) добавляли формальдегид (247 мг, 1,22 ммоль, 37% в воде) при 27°C. Реакционную смесь перемешивали при 27°C в течение 30 мин, затем охлаждали в ледяной бане и добавляли NaBH4 (46 мг, 1,22 ммоль). Реакционную смесь перемешивали в течение 16 часов, затем разбавляли водой (10 мл) и экстрагировали дихлорметаном (20 мл × 3). Объединенные органические слои промывали рассолом (10 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством препаративной ВЭЖХ с получением 2-(2-(диметиламино)этил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (11 мг, 5%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.67 (br s, 1H), 8.16 (s, 1H), 7.98-7.84 (m, 4H), 7.26 (dd, J=2,3, 9,8 Гц, 1H), 7.01 (dt, J=2,3, 9,3 Гц, 1H), 4.57 (s, 2H), 3.30 (t, J=6,5 Гц, 2H), 2.60 (t, J=6,5 Гц, 2H), 2.24 (s, 6H); ЖХ-МС: m/z 374,1 (M+H)+.
Пример 33: 5-(6-фтор-1H-индол-3-ил)-2-(2-гидроксиэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
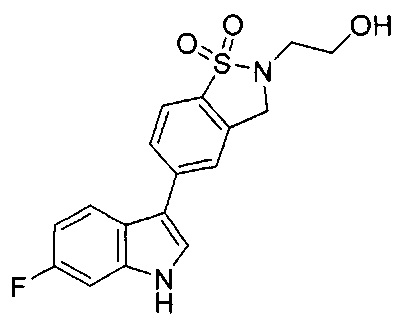
Стадия 1: 5-бром-2-(2-гидроксиэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Раствор 5-бром-2,3-дигидробензо[d]изотиазол-1,1-диоксида (300 мг, 1,21 ммоль), 2-бромэтанола (227 мг, 1,81 ммоль) и K2CO3 (334 мг, 0,242 ммоль) в DMF (5 мл) перемешивали в атмосфере N2 при 80°C в течение 16 часов. Реакционную смесь гасили водой (10 мл) и экстрагировали этилацетатом (15 мл × 3). Объединенные органические слои промывали водой (15 мл) и рассолом (15 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенное желтое масло очищали посредством колоночной хроматографии (силикагель, 0-50% этилацетат/петролейный эфир) с получением 5-бром-2-(2-гидроксиэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (249 мг, 70%) в виде белого твердого вещества.
Стадия 2: трет-бутил-6-фтор-3-(2-(2-гидроксиэтил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилат
Раствор 5-бром-2-(2-гидроксиэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (249 мг, 0,852 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (308 мг, 0,852 ммоль), PdCl2(dppf)CH2Cl2 (64 мг, 0,09 ммоль) и K3PO4 (543 мг, 2,56 ммоль) в 1,4-диоксане (8 мл) и воде (2 мл) барботировали N2 в течение 1 мин, затем перемешивали при 80°C в течение 16 часов. Неочищенную реакционную смесь экстрагировали этилацетатом (15 мл × 3). Объединенные органические слои промывали водой (15 мл) и рассолом (15 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии (силикагель, 0-100% этилацетат/петролейный эфир) с получением трет-бутил-6-фтор-3-(2-(2-гидроксиэтил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилата (290 мг, 76%) в виде желтого твердого вещества. ЖХ-МС: m/z 447,2 (M+H)+.
Стадия 3: 2-(5-(6-фтор-1H-индол-3-ил)-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этилацетат
В охлажденный (ледяная баня) раствор трет-бутил-6-фтор-3-(2-(2-гидроксиэтил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (290 мг, 0,55 ммоль) в этилацетате (3 мл) медленно добавляли HCl (4М в этилацетате, 6 мл). Желтый раствор перемешивали при 17°C в течение 12 часов. Реакционную смесь снова охлаждали в ледяной бане, затем добавляли трифторуксусную кислоту (3 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 2 часов. Реакционную смесь гасили водой (5 мл), концентрировали и значение pH полученного раствора доводили до 8 с использованием NaHCO3 (насыщ.) (15 мл). Водный раствор экстрагировали дихлорметаном (15 мл × 3). Объединенные органические слои промывали водой (15 мл) и рассолом (15 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. 2-(5-(6-Фтор-1H-индол-3-ил)-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-этилацетат (251 мг, более 100%) выделили в виде коричневого твердого вещества. ЖХ-МС: m/z 389,1 (M+H)+.
Стадия 4: 5-(6-фтор-1H-индол-3-ил)-2-(2-гидроксиэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
В охлажденный (ледяная баня) раствор 2-(5-(6-фтор-1H-индол-3-ил)-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этилацетата (251 мг, 0,52 ммоль) в THF (4 мл) медленно добавляли раствор LiOH.H2O (54 мг, 1,3 ммоль) в воде (2 мл). Реакционную смесь перемешивали при 50°C в течение 3 часов, затем концентрировали и экстрагировали дихлорметаном (15 мл). Органическую фазу сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством препаративной ВЭЖХ с получением 5-(6-фтор-1H-индол-3-ил)-2-(2-гидроксиэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (84 мг, 47%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.66 (br s, 1H), 7.96 (dd, J=5,3, 8,8 Гц, 1H), 7.91-7.84 (m, 4H), 7.26 (dd, J=2,4, 9,9 Гц, 1H), 7.03-6.98 (m, 1H), 4.94 (t, J=5,1 Гц, 1H), 4.59 (s, 2H), 3.71 (q, J=5,7 Гц, 2H), 3.26 (t, J=5,8 Гц, 2H); ЖХ-МС: m/z 347,1 (M+H)+.
Пример 34: (S)-2-(2,3-дигидроксипропил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
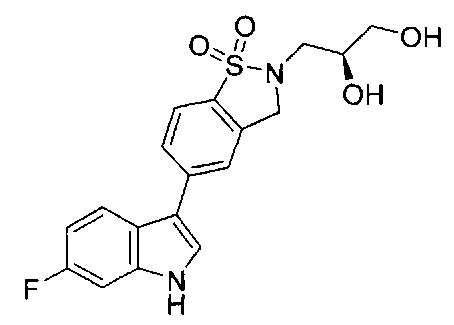
Стадия 1: (S)-5-бром-2-((2,2-диметил-1,3-диоксолан-4-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Раствор 5-бром-2,3-дигидробензо[d]изотиазол-1,1-диоксида (600 мг, 1,5 ммоль), (R)-4-(хлорметил)-2,2-диметил-1,3-диоксолана (328 мг, 2,2 ммоль) и K2CO3 (501 мг, 3,63 ммоль) в DMF (10 мл) перемешивали в атмосфере N2 при 80°C в течение 50 часов. Реакционную смесь гасили водой (10 мл) и экстрагировали этилацетатом (15 мл × 3). Объединенные органические слои промывали водой (10 мл) и рассолом (10 мл × 3), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенное коричневое масло очищали посредством колоночной хроматографии (силикагель, 0-30% этилацетат/петролейный эфир) с получением (S)-5-бром-2-((2,2-диметил-1,3-диоксолан-4-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (220 мг, 42%) в виде желтого масла.
Стадия 2: (S)-трет-бутил-3-(2-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилат
Раствор (S)-5-бром-2-((2,2-диметил-1,3-диоксолан-4-ил)-метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (220 мг, 0,607 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (219 мг, 0,61 ммоль), PdCl2(dppf)CH2Cl2 (45 мг, 0,06 ммоль) и K3PO4 (387 мг, 1,82 ммоль) в 1,4-диоксане (8 мл) и воде (2 мл) барботировали N2 в течение 1 минуты. Реакционную смесь перемешивали при 80°C в течение 16 часов, затем концентрировали и очищали посредством колоночной хроматографии (силикагель, 0-50% этилацетат/петролейный эфир) с получением (S)-трет-бутил-3-(2-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилата (416 мг, более 100%) в виде коричневого масла, которое использовали на следующей стадии без дополнительной очистки. ЖХ-МС: m/z 539,1 (M+H)+.
Стадия 3: (S)-2-(2,3-дигидроксипропил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К раствору (S)-трет-бутил-3-(2-((2,2-диметил-1,3-диоксолан-4-ил)метил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (416 мг, 0,81 ммоль) в дихлорметане (4 мл) добавляли трифторуксусную кислоту (2 мл). Реакционную смесь перемешивали при 16°C в течение 1 часа, затем концентрировали. Неочищенное твердое вещество нейтрализовали до pH 7 путем добавления NH3-H2O (0,5 мл), затем очищали посредством препаративной ВЭЖХ с получением (S)-2-(2,3-дигидроксипропил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (45 мг, 27%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.67 (br s, 1H), 7.97 (dd, J=5,3, 9,0 Гц, 1H), 7.92-7.85 (m, 4H), 7.26 (dd, J=2,3, 9,8 Гц, 1H), 7.01 (dt, J=2,4, 9,2 Гц, 1H), 5.05 (d, J=5,3 Гц, 1H), 4.73 (t, J=5,5 Гц, 1H), 4.68-4.53 (m, 2H), 3.79-3.76 (m, 1H), 3.45-3.36 (m, 3H), 3.04 (dd, J=7,5, 14,1 Гц, 1H); ЖХ-МС: m/z 377,0 (M+H)+; [α]20D -4,91° (c=1,0189 мг/мл, метанол).
Пример 35: (R)-2-(2,3-дигидроксипропил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
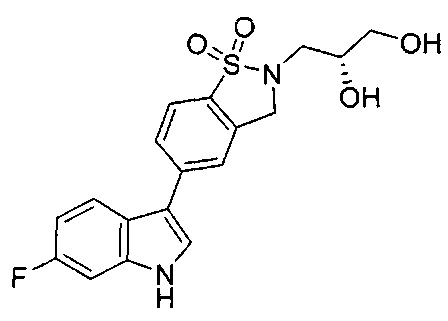
Следуя общим методикам, изложенным в Примере 34, используя (S)-4-(хлорметил)-2,2-диметил-1,3-диоксолан, получили указанное в заголовке соединение в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1H), 8.10-7.75 (m, 5H), 7.27 (dd, J=2,3, 9,8 Гц, 1H), 7.07-6.95 (m, 1H), 5.06 (d, J=5,3 Гц, 1H), 4.75 (t, J=5,5 Гц, 1H), 4.71-4.64 (m, 1H), 4.60-4.53 (m, 1H), 3.84-3.74 (m, 1H), 3.48-3.40 (m, 3H), 3.05 (dd, J=7,5, 14,1 Гц, 1H); LCMS: m/z 399,0 (M+Na)+; [α]20D +3,51° (c=1,14 мг/мл, метанол).
Пример 36: 5-(6-фтор-1H-индол-3-ил)-2-(пиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
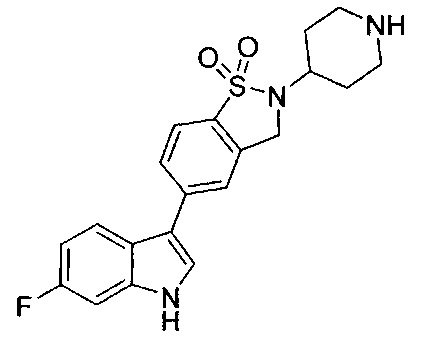
Стадия 1: трет-бутил-4-((метилсульфонил)окси)пиперидин-1-карбоксилат
В охлажденный (ледяная баня) раствор трет-бутил-4-гидроксипиперидин-1-карбоксилата (10 г, 50 ммоль) в дихлорметане (100 мл) добавляли триэтиламин (14 мл, 99 ммоль) и метансульфонилхлорид (6,8 г, 60 ммоль). Реакционную смесь нагревали до температуры окружающей среды и перемешивали в течение 1 часа, затем гасили водой (50 мл) и экстрагировали дихлорметаном (50 мл × 2). Объединенные органические слои промывали рассолом (50 мл), затем сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением трет-бутил-4-((метилсульфонил)окси)пиперидин-1-карбоксилата (15 г, выход более 100%) в виде бледно-желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 4.90-4.86 (m, 1H), 3.73-3.67 (m, 2H), 3.33-3.27 (m, 2H), 3.04 (s, 3H), 1.99-1.94 (m, 2H), 1.85-1.79 (m, 2H), 1.46 (s, 9H).
Стадия 2: трет-бутил-4-(5-бром-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-пиперидин-1-карбоксилат
Смесь 5-бром-2,3-дигидробензо[d]изотиазол-1,1-диоксида (1,0 г, 1,5 ммоль) и K2CO3 (411 мг, 2,97 ммоль) в DMF (20 мл) перемешивали при 27°C в течение 30 мин, затем добавляли трет-бутил-4-((метилсульфонил)окси)-пиперидин-1-карбоксилат (498 мг, 1,78 ммоль). Реакционную смесь выкачивали и заполняли N2 трижды, затем перемешивали при 80°C в течение 16 часов. Реакционную смесь гасили водой (30 мл) и экстрагировали этилацетатом (30 мл × 5). Объединенные органические слои промывали рассолом (20 мл × 3), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 0-40% этилацетат/петролейный эфир) с получением трет-бутил-4-(5-бром-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-пиперидин-1-карбоксилата (600 мг, выход 34,5%) в виде белого твердого вещества. ЖХ-МС: m/z 454,8 (M+Na)+.
Стадия 3: трет-бутил-3-(2-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилат
Желтый раствор трет-бутил-4-(5-бром-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пиперидин-1-карбоксилата (405 мг, 1,25 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (497 мг, 1,38 ммоль), PdCl2(dppf)CH2Cl2 (93,5 мг, 0,125 ммоль) и K3PO4 (797 мг, 3,76 ммоль) в 1,4-диоксане (8,0 мл) и воде (2,0 мл) барботировали N2 в течение 1 мин, затем перемешивали при 80°C в течение 4 часов. Реакционную смесь концентрировали и очищали посредством колоночной хроматографии (силикагель, 0-40% этилацетат/петролейный эфир) с получением трет-бутил-3-(2-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (498 мг, 68%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 7.96 (d, J=11.3 Гц, 1H), 7.88 (d, J=8,0 Гц, 1H), 7.78-7.78 (m, 2H), 7.67-7.62 (m, 2H), 7.11-7.06 (m, 1H), 4.46 (s, 2H), 4.24-4.18 (m, 2H), 3.89-3.83 (m, 1H), 2.94-2.88 (m, 2H), 2.08 (d, J=13,6 Гц, 2H), 1.87-1.77 (m, 2H), 1.71 (s, 9H), 1.48 (m, 9H); ЖХ-МС: m/z 608,0 (M+Na)+.
Стадия 4: 5-(6-фтор-1H-индол-3-ил)-2-(пиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
В бледно-желтый раствор трет-бутил-3-(2-(1-(трет-бутоксикарбонил)-пиперидин-4-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (400 мг, 0,683 ммоль) в этилацетате (6 мл) добавляли трифторуксусную кислоту (3 мл). Реакционную смесь перемешивали при 32°C в течение 2 часов, затем разбавляли метанолом (10 мл), охлаждали в ледяной бане и обрабатывали NH3-H2O (10 мл). Желтый раствор концентрировали и очищали посредством препаративной ВЭЖХ с получением 5-(6-фтор-1H-индол-3-ил)-2-(пиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (78 мг, 27%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1H), 8.32 (br s, 1H), 7.97-7.82 (m, 5H), 7.27 (dd, J=2,3, 9,8 Гц, 1H), 7.01 (dt, J=2,4, 9,2 Гц, 1H), 4.54 (s, 2H), 3.76-3.69 (m, 1H), 3.19 (d, J=11,5 Гц, 2H), 2.987-2.81 (m, 2H), 2.00-1.85 (m, 4H); ЖХ-МС: m/z 385,9 (M+H)+.
Пример 37: 5-(6-фтор-1H-индол-3-ил)-2-(1-метилпиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
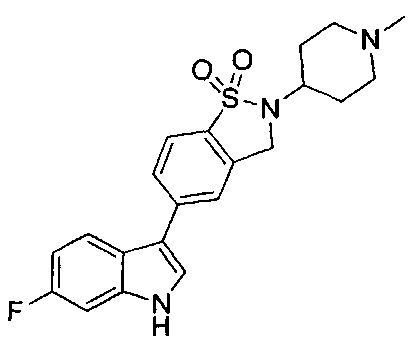
Желтый раствор 5-(6-фтор-1H-индол-3-ил)-2-(пиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (170 мг, 0,441 ммоль), формальдегида (40% в воде) (46 мг, 0,57 ммоль) и CH3COOH (2 капли) в дихлорметане (3 мл) перемешивали при 0°C в течение 15 мин, затем добавляли NaBH(OAc)3 (100 мг, 0,472 ммоль) в виде суспензии в дихлорметане (2 мл). Суспензию перемешивали при 0°C в течение 1 часа, затем гасили водой (5 мл) и экстрагировали дихлорметаном (10 мл × 3). Органические слои промывали рассолом (5 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенное твердое вещество очищали посредством препаративной ВЭЖХ с получением 5-(6-фтор-1H-индол-3-ил)-2-(1-метилпиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (40 мг, 23%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.67 (br s, 1H), 7.97-7.81 (m, 5H), 7.26 (dd, J=2,3, 9,8 Гц, 1H), 7.01 (dt, J=2,4, 9,2 Гц, 1H), 4.53 (s, 2H), 2.85 (d, J=12,0 Гц, 1H), 2.20 (s, 3H), 1.99-1.88 (m, 4H), 1.23 (br s, 4H); ЖХ-МС: m/z 399,9 (M+H)+.
Пример 38: (R)-5-(6-фтор-1H-индол-3-ил)-2-(тетрагидрофуран-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
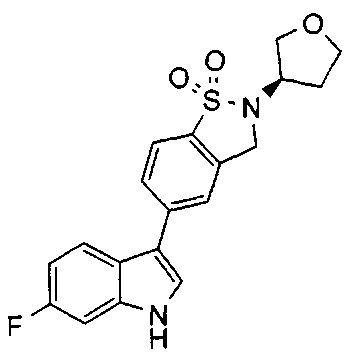
Следуя общей методике, изложенной в Примере 36, начиная с (S)-(+)-3-гидрокситетрагидрофурана, получили указанное в заголовке соединение в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.80-11.56 (m, 1H), 8.00-7.83 (m, 5H), 7.27 (dd, J=2,3, 9,8 Гц, 1H), 7.06-6.97 (m, 1H), 4.60-4.51 (m, 2H), 4.32-4.23 (m, 1H), 3.98-3.90 (m, 2H), 3.87-3.79 (m, 1H), 3.75-3.66 (m, 1H), 2.34-2.24 (m, 1H), 2.23-2.13 (m, 1H); ЖХ-МС: m/z 394,8 (M+Na)+; [α]20D +21,1° (c=0,95 мг/мл, DMSO).
Пример 39: (S)-5-(6-фтор-1H-индол-3-ил)-2-(тетрагидрофуран-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид

Следуя общей методике, изложенной в Примере 36, начиная с (R)-(+)-3-гидрокситетрагидрофурана, получили указанное в заголовке соединение в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.67 (br s, 1H), 7.99-7.82 (m, 5H), 7.26 (dd, J=2,5, 10,0 Гц, 1H), 7.01 (dt, J=2,3, 9,2 Гц, 1H), 4.60-4.51 (m, 2H), 4.31-4.23 (m, 1H), 3.98-3.91 (m, 2H), 3.82 (dd, J=6,0, 9,5 Гц, 1H), 3.74-3.66 (m, 1H), 2.33-2.23 (m, 1H), 2.22-2.13 (m, 1H); ЖХ-МС: m/z 394,8 (M+Na)+; [α]20D -27,36° (c=0,95 мг/мл, DMSO).
Пример 40: 2-(азетидин-3-ил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
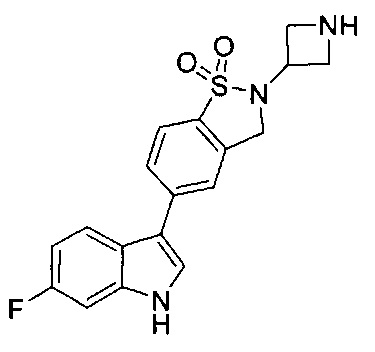
Следуя общим методикам, изложенным в Примере 36, используя N-Boc-3-ОН-азетидин, получили указанное в заголовке соединение в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.77 (br s, 1H), 8.35 (br s, 1H), 7.97-7.87 (m, 5H), 7.28 (dd, J=2,1, 9,9 Гц, 1H), 7.02 (dt, J=2,3, 9,3 Гц, 1H), 4.72 (s, 2H), 4.61 (d, J=8,0 Гц, 1H), 4.14 (br s, 4H); ЖХ-МС: m/z 357,9 (M+H)+.
Пример 41: 5-(6-фтор-1H-индол-3-ил)-2-(1-метилазетидин-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
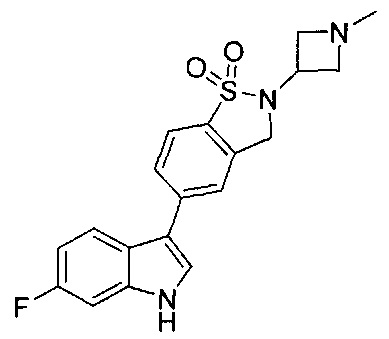
Стадия 1: 5-(6-фтор-1-(гидроксиметил)-1H-индол-3-ил)-2-(1-метилазетидин-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К раствору 2-(азетидин-3-ил)-5-(6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (150 мг, 0,318 ммоль) в метаноле (5 мл) добавляли формальдегид (37% в воде, 129 мг, 1,59 моль) при температуре окружающей среды. Реакционную смесь перемешивали при 27°C в течение 15 мин, затем охлаждали в ледяной бане и добавляли NaBH3CN (30 мг, 0,477 ммоль). Реакционную смесь медленно нагревали до 27°C и перемешивали в течение 12 часов, затем разбавляли водой (4 мл) и экстрагировали дихлорметаном (10 мл × 3). Объединенные органические слои промывали рассолом (10 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением 5-(6-фтор-1-(гидроксиметил)-1H-индол-3-ил)-2-(1-метилазетидин-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (134 мг, более 100%) в виде желтого твердого вещества.
Стадия 2: 5-(6-фтор-1H-индол-3-ил)-2-(1-метилазетидин-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К охлажденному (ледяная баня) желтому раствору 5-(6-фтор-1-(гидроксиметил)-1H-индол-3-ил)-2-(1-метилазетидин-3-ил)-2,3-дигидробензо[d]-изотиазол-1,1-диоксида (124 мг, 0,309 ммоль) в 1,4-диоксане (5 мл) медленно добавляли NH3/H2O (2,5 мл). Реакционную смесь перемешивали при 27°C в течение 48 часов, затем экстрагировали дихлорметаном (15 мл × 3). Объединенные органические слои промывали водой (10 мл) и рассолом (10 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенное желтое масло очищали посредством препаративной ТСХ (силикагель, 10% метанола в дихлорметане) с получением 5-(6-фтор-1H-индол-3-ил)-2-(1-метилазетидин-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (30 мг, 26%) в виде белого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.91-7.87 (m, 2H), 7.84 (s, 1H), 7.77 (d, J=8,3 Гц, 1H), 7.68 (s, 1H), 7.16 (dd, J=2,3, 9.5 Гц, 1H), 6.95 (dt, J=2,4, 9,2 Гц, 1H), 4.56 (s, 2H), 4.27-4.22 (m, 1H), 3.73-3.69 (m, 2H), 3.55-3.51 (m, 2H), 2.44 (s, 3H); ЖХ-МС: m/z 371,9 (M+H)+.
Пример 42: (+)-5-(6-фтор-1H-индол-3-ил)-2-(2-(метилсульфинил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
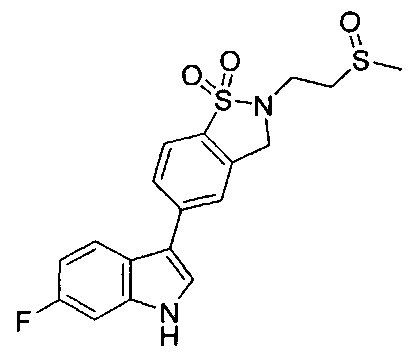
Стадия 1: 5-бром-2-(2-(метилтио)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Раствор 5-бром-2,3-дигидробензо[d]изотиазол-1,1-диоксида (1000 мг, 4,03 ммоль), (2-хлорэтил)(метил)сульфана (892 мг, 8,06 ммоль) и K2CO3 (1110 мг, 8.06 ммоль) в DMF (10 мл) выкачивали и заполняли N2 трижды, затем перемешивали при 85°C в течение 7 часов. Реакционную смесь разбавляли водой (30 мл) и экстрагировали этилацетатом (30 мл × 3). Объединенные органические слои промывали водой (30 мл × 2) и рассолом (30 мл × 3), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 0-50% этилацетат/петролейный эфир) с получением 5-бром-2-(2-(метилтио)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (1,3 г, 43%) в виде темно-желтого масла. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 7.90 (d, J=0,8 Гц, 1H), 7.86-7.84 (m, 1H), 7.81-7.79 (m, 1H), 4.54 (s, 2H), 3.41 (t, J=8,0 Гц, 2H), 2.79 (t, J=8,0 Гц, 2H), 2.12 (s, 3H).
Стадия 2: 5-бром-2-(2-(метилсульфинил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К желтому раствору 5-бром-2-(2-(метилтио)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (1000 мг, 3,103 ммоль) в безводном дихлорметане (50 мл) добавляли мета-хлорпероксибензойную кислоту (m-СРВА) (630 мг, 3,10 ммоль) при -25°C. Желтую суспензию перемешивали в течение 30 мин, затем промывали H2O (25 мл × 3). Слои разделяли и органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии (силикагель, 0-5% метанол/дихлорметан) с получением 5-бром-2-(2-(метилсульфинил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (652 мг, 62%) в виде желтого твердого вещества. ЖХ-МС: m/z 359,7 (M+Na)+.
Стадия 3: трет-бутил-6-фтор-3-(2-(2-(метилсульфинил)этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилат
Желтую смесь 5-бром-2-(2-(метилсульфинил)этил)-2,3-дигидробензо[d]-изотиазол-1,1-диоксида (857 мг, 2,53 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (1100 мг, 3,04 ммоль), PdCl2(dppf)CH2Cl2 (189 мг, 0,025 ммоль) и K3PO4 (1610 мг, 7,60 ммоль) в 1,4-диоксане (10 мл) и воде (3 мл) барботировали N2 в течение 1 минуты. Реакционную смесь перемешивали при 80°C в течение 3 часов, затем экстрагировали этилацетатом (25 мл × 3). Объединенные органические слои промывали водой (20 мл) и рассолом (25 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный материал очищали посредством колоночной хроматографии (силикагель, 0-5% метанол/дихлорметан) с получением трет-бутил-6-фтор-3-(2-(2-(метилсульфинил)этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (1,33 г, 79%) в виде черного масла. ЖХ-МС: m/z 493,0 (M+H)+.
Стадия 4: (+)-5-(6-фтор-1H-индол-3-ил)-2-(2-(метилсульфинил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К раствору трет-бутил-6-фтор-3-(2-(2-(метилсульфинил)этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (1,3 г, 2,0 ммоль) в дихлорметане (16 мл) добавляли трифторуксусную кислоту (8 мл) при 0°C. Черный раствор перемешивали при 26°C в течение 16 часов, затем концентрировали и разбавляли этилацетатом (10 мл) и NaHCO3 (насыщ.) (20 мл). Слои разделяли и водную фазу подвергали обратной экстракции этилацетатом (20 мл × 3). Объединенные органические слои промывали водой (20 мл) и рассолом (20 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 0-10% метанол/дихлорметан) с получением рацемического 5-(6-фтор-1H-индол-3-ил)-2-(2-(метилсульфинил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (450 мг, 57%) в виде желтого твердого вещества. ЖХ-МС: m/z 392,9 (M+H)+, 414,8 (M+Na)+. Этот рацемический продукт разделяли посредством препаративной хиральной СФХ с получением (+)-5-(6-фтор-1H-индол-3-ил)-2-(2-(метилсульфинил)этил)-2,3-дигидробензо[d]-изотиазол-1,1-диоксида в качестве первого элюируемого пика (68 мг, 26%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s, 1H), 7.98-7.87 (m, 5H), 7.27 (d, J=10,0 Гц, 1H), 7.01 (t, J=9,2 Гц, 1H), 4.59 (s, 2H), 3.62 (t, J=6,7 Гц, 2H), 3.26-3.19 (m, 1H), 3.07-3.04 (m, 1H), 2.65 (s, 3H); ЖХ-МС: m/z 393,1 (M+H)+; [α]20D +18.5° (c=1,515 мг/мл, DMSO).
Пример 43: 3-(5-(6-фтор-1H-индол-3-ил)-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилпропанамид
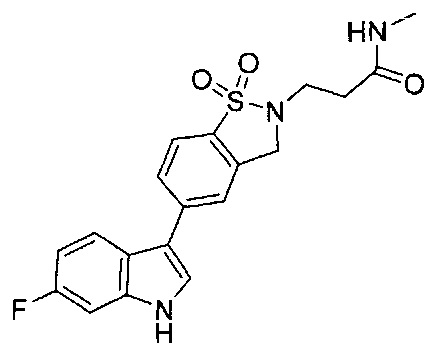
Стадия 1: этил-3-(5-бром-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-пропаноат
К раствору 5-бром-2,3-дигидробензо[d]изотиазол-1,1-диоксида (800 мг, 1,9 ммоль) в DMF (20 мл) добавляли этил-3-бромпропаноат (420 мг, 2,32 ммоль) и K2CO3 (535 мг, 3,87 ммоль), затем смесь барботировали N2 в течение 1 мин и перемешивали при 80°C в течение 16 часов. Неочищенную реакционную смесь разбавляли этилацетатом (20 мл) и водой (20 мл). Слои разделяли и водный слой подвергали обратной экстракции этилацетатом (30 мл × 2). Объединенные органические слои промывали рассолом (10 мл × 2), затем сушили над Na2SO4, фильтровали и концентрировали. Неочищенный материал очищали посредством колоночной хроматографии (силикагель, 2-20% этилацетат/петролейный эфир) с получением этил-3-(5-бром-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропаноата (530 мг, 79%) в виде серого твердого вещества.
Стадия 2: трет-бутил-3-(2-(3-этокси-3-оксопропил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилат
К желтому раствору этил-3-(5-бром-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропаноата (530 мг, 1,52 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (605 мг, 1,67 ммоль) и K3PO4 (646 мг, 3,04 ммоль) в смеси диоксан/H2O (12 мл/4 мл) добавляли Pd(dppf)Cl2 (111 мг, 0,152 ммоль) при 25°C в атмосфере N2. Реакционную смесь нагревали до 90°C и перемешивали в течение 14 часов, затем разбавляли этилацетатом (40 мл). Слои разделяли и органическую фазу промывали H2O (10 мл) и рассолом (15 мл × 2). Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 10-60% этилацетат/петролейный эфир) с получением трет-бутил-3-(2-(3-этокси-3-оксопропил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (220 мг, 29%) в виде желтой смолы и этил-3-(5-(6-фтор-1H-индол-3-ил)-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропаноата (360 мг, выход 60%) в виде желтой смолы.
Стадия 3: 3-(5-(6-фтор-1H-индол-3-ил)-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилпропанамид
Желтый раствор трет-бутил-3-(2-(3-этокси-3-оксопропил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (220 мг, 0,438 ммоль) в MeNH2/EtOH (30% масс./масс., 10 мл) перемешивали при 100°C в герметично закрытой пробирке в течение 14 часов. Реакционную смесь концентрировали и очищали посредством колоночной хроматографии (силикагель, 20-100% этилацетат/петролейный эфир), а затем посредством препаративной ВЭЖХ с получением 3-(5-(6-фтор-1H-индол-3-ил)-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилпропанамида (58 мг, 34%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1H), 8.01-7.81 (m, 6H), 7.26 (dd, J=2,5, 9,8 Гц, 1H), 7.06-6.96 (m, 1H), 4.50 (s, 2H), 3.45 (t, J=7,0 Гц, 2H), 2.60 (d, J=4,5 Гц, 3H), 2.56-2.51 (m, 2H); ЖХ-МС: m/z 410,0 (M+Na)+.
Пример 44: 3-(5-(6-фтор-1H-индол-3-ил)-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропанамид

Следуя общей методике, изложенной в Примере 43, используя 3-(5-бром-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропанамид, получили указанное в заголовке соединение в виде серого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1H), 8.06-7.77 (m, 5H), 7.48 (br s, 1H), 7.26 (d, J=9,5 Гц, 1H), 7.10-6.88 (m, 2H), 4.51 (s, 2H), 3.44 (t, J=6,8 Гц, 2H), 2.58-2.52 (m, 2H); ЖХ-МС: m/z для 396,0 (M+Na)+.
Пример 45: (+)-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилпропанамид
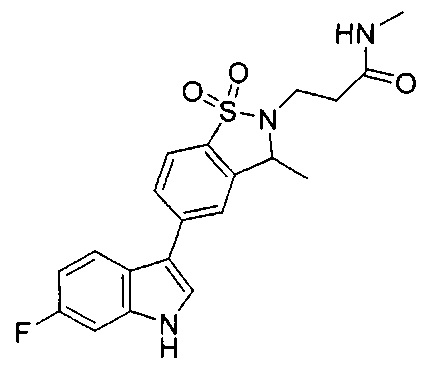
Стадия 1: Этил-3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропаноат:
Желтую смесь 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (1,5 г, 5,7 ммоль), этил-3-бромпропаноата (1,24 г, 6,87 ммоль) и K2CO3 (1,58 г, 11,4 ммоль) в DMF (15 мл) барботировали N2 в течение 1 мин и перемешивали при 80°C в течение 16 часов. Реакционную смесь разбавляли этилацетатом (50 мл) и водой (10 мл). Слои разделяли и водный слой подвергали обратной экстракции этилацетатом (20 мл × 2). Объединенные органические слои промывали рассолом (10 мл), затем сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 5-50% этилацетат/петролейный эфир) с получением этил-3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-пропаноата (1,6 г, 77%) в виде черного твердого вещества.
Стадия 2: трет-бутил-3-(4-(2-(3-этокси-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)фенил)-6-фтор-1H-индол-1-карбоксилат
К желтому раствору этил-3-(5-бром-3-метил-1,1-диоксидобензо[d]-изотиазол-2(3H)-ил)пропаноата (800 мг, 2,21 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (801 мг, 2,21 ммоль) и K3PO4 (938 мг, 4,42 ммоль) в смеси диоксан/H2O (12 мл/4 мл) добавляли Pd(dppf)Cl2 (162 мг, 0,221 ммоль) при 28°C в атмосфере N2. Полученную красную суспензию перемешивали при 80°C в течение 16 часов, затем разбавляли водой (8 мл) и экстрагировали этилацетатом (15 мл × 3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный материал очищали посредством колоночной хроматографии (силикагель, 5-30% этилацетат/петролейный эфир) с получением трет-бутил-3-(4-(2-(3-этокси-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)фенил)-6-фтор-1H-индол-1-карбоксилата (500 мг, 38%) в виде желтой смолы.
Стадия 3: (+)-3-(5-(4-(6-фтор-1H-индол-3-ил)фенил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилпропанамид
Желтый раствор трет-бутил-3-(4-(2-(3-этокси-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)фенил)-6-фтор-1H-индол-1-карбоксилата (500 мг, 0,84 ммоль) в MeNH2/EtOH (30% масс./масс., 40 мл) перемешивали при 50°C в герметично закрытой пробирке в течение 3 часов. Неочищенную реакционную смесь концентрировали и очищали посредством колоночной хроматографии (SiO2, петролейный эфир/эти л ацетат от 10/1 до 3/1) с получением рацемического 3-(5-(4-(6-фтор-1H-индол-3-ил)фенил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилпропанамида (300 мг, 58%) в виде красной смолы. Этот рацемический материал разделяли посредством препаративной хиральной СФХ с получением (+)-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилпропанамида в качестве первого элюируемого пика (90 мг, 27%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 8.63 (br s, 1H), 7.86-7.71 (m, 3H), 7.57 (s, 1H), 7.42 (d, J=2,3 Гц, 1H), 7.16 (dd, J=2,3, 9,3 Гц, 1H), 7.01 (dt, J=2,3, 9,2 Гц, 1H), 6.07 (br s, 1H), 4.50 (q, J=6,1 Гц, 1H), 3.80-3.61 (m, 2H), 2.84-2.61 (m, 5H), 1.59 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 401,9 (M+H)+; [α]20D +6,7° (c=0,0015 г/мл, метанол).
Пример 46: (-)-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилпропанамид
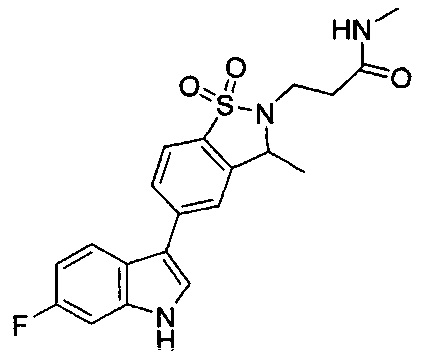
Указанное в заголовке соединение выделили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 45 (100 мг, 30%), в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 8.61 (br s, 1H), 7.89-7.70 (m, 3H), 7.57 (s, 1H), 7.42 (d, J=2,5 Гц, 1H), 7.16 (dd, J=2,3, 9,0 Гц, 1H), 7.01 (dt, J=2,4, 9,1 Гц, 1H), 6.07 (d, J=4,3 Гц, 1H), 4.50 (d, J=6,5 Гц, 1H), 3.84-3.59 (m, 2H), 2.91-2.59 (m, 5H), 1.59 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 401,9 (M+H)+; [α]20D -10,9° (c=0,0011 г/мл, метанол).
Пример 47: (+)-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропанамид
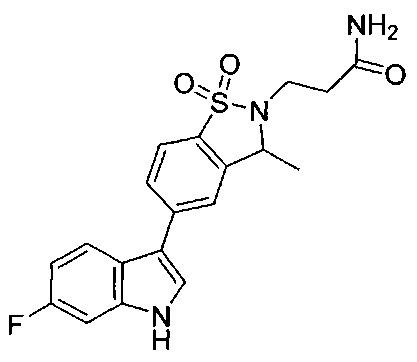
Стадия 1: 1-трет-бутил-2-метил-4-((4-бромфенил)сульфонил)-пиперазин-1,2-дикарбоксилат
К перемешиваемому раствору 1-трет-бутил-2-метилового эфира пиперазин-1,2-дикарбоновой кислоты (977 мг, 4,0 ммоль) в DCM (20 мл) при 0°C добавляли 4-бром-бензолсульфонилхлорид (1,02 мг, 4,0 ммоль). Затем добавляли по каплям TEA (404 мг, 4,0 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали и очищали посредством хроматографии на силикагеле (петролейный эфир/EtOAc от 20/1 до 5/1) с получением 1,66 г (90%) указанного в заголовке соединения в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 7.69 (d, J=8,8 Гц, 2H), 7.61 (d, J=8,8 Гц, 2H), 4.89-4.60 (m, 1H), 4.27-4.20 (m, 1H), 4.04-3.82 (m, 1H), 3.77 (s, 3H), 3.76-3.61 (m, 1H), 3.35-3.11 (m, 1H), 2.51 (dd, J=11,6, 4,0 Гц, 1H), 2.33 (td, J=11,6, 4,0 Гц, 1H), 1.44 (s, 9H).
Стадия 2: трет-бутил-4-((4-бромфенил)сульфонил)-2-(гидроксиметил)-пиперазин-1-карбоксилат
К перемешиваемому раствору 1-трет-бутил-2-метилового эфира 4-(4-бром-бензолсульфонил)-пиперазин-1,2-дикарбоновой кислоты (1,66 г, 3,59 ммоль) в безводном THF (20 мл) при 0°C добавляли LiAlH4 (137 мг, 3,59 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, а затем разбавляли EtOAc (100 мл) и водой (0,5 мл). Органическую смесь сушили над безводным Na2SO4 и фильтровали. Фильтрат концентрировали и очищали посредством хроматографии на силикагеле (петролейный эфир/EtOAc от 5/1 до 3/1) с получением 910 мг (58%) указанного в заголовке соединения в виде белого твердого вещества. 1H ЯМР (300 МГц, CDCl3) δ [м.д.] 7.69 (d, J=8,8 Гц, 2H), 7.61 (d, J=8,8 Гц, 2H), 4.28-4.20 (m, 1H), 4.04-3.93 (m, 1H), 3.91-3.71 (m, 3H), 3.70-3.64 (m, 1H), 3.19-3.09 (m, 1H), 2.44-2.27 (m, 2H), 1.99 (t, J=5,7 Гц, 1H), 1.42 (s, 9H).
Стадия 3: трет-бутил-3-(4-((4-(трет-бутоксикарбонил)-3-(гидроксиметил)-пиперазин-1-ил)сульфонил)фенил)-6-фтор-1H-индол-1-карбоксилат
Смесь трет-бутилового эфира 4-(4-бром-бензолсульфонил)-2-гидроксиметил-пиперазин-1-карбоновой кислоты (120 мг, 0,277 ммоль), трет-бутилового эфира 6-фтор-3-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-индол-1-карбоновой кислоты (Промежуточное соединение 1, 100 мг, 0,277 ммоль), K2CO3 (114 мг, 0,831 ммоль) и Pd(dppf)Cl2 (20 мг, 0,028 ммоль) в смеси диоксан/вода (10 мл/2 мл) перемешивали при 90°C в атмосфере N2 в течение 4 часов. Смесь охлаждали и разбавляли EtOAc (60 мл). Органический слой сушили над безводным Na2SO4 и фильтровали. Фильтрат концентрировали и очищали посредством хроматографии на силикагеле (петролейный эфир/EtOAc от 5/1 до 3/1) с получением 120 мг (74%) указанного в заголовке соединения в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 7.97 (d, J=9,2 Гц, 1H), 7.83 (d, J=8,8 Гц, 2H), 7.81-7.74 (m, 3H), 7.74-7.68 (m, 1H), 7.08 (td, J=8,8, 2,4 Гц, 1H), 4.29-4.21 (m, 1H), 4.02-3.95 (m, 1H), 3.95-3.79 (m, 2H), 3.79-3.71 (m, 2H), 3.17 (t, J=12,8 Гц, 1H), 2.49 (dd, J=12,0, 4,0 Гц, 1H), 2.41 (td, J=12,0, 4,0 Гц, 1H), 2.02 (brs, 1H), 1.70 (s, 9H), 1.42 (s, 9H).
Стадия 4: (4-((4-(6-фтор-1H-индол-3-ил)фенил)сульфонил)пиперазин-2-ил)метанол
К перемешиваемому раствору трет-бутилового эфира 3-[4-(4-трет-бутоксикарбонил-3-гидроксиметил-пиперазин-1-сульфонил)-фенил]-6-фтор-индол-1-карбоновой кислоты (120 мг, 0,204 ммоль) в безводном DCM (5 мл) добавляли TFA (3 мл) по каплям при 0°C. Смесь перемешивали при комнатной температуре в течение 3 часов, а затем добавляли EtOAc (60 мл) и TEA (5 мл). Смесь промывали водой (20 мл) и рассолом (20 мл × 2). Органический слой сушили над безводным Na2SO4 и фильтровали. Фильтрат концентрировали и очищали посредством препаративной ВЭЖХ (NH3H2O в качестве вспомогательного агента) с получением 36 мг (46%) указанного в заголовке соединения в виде белого твердого вещества. ЖХ-МС для C19H20FN3O3S+H+[M+H]+: рассчитано: 390,1; обнаружено: 390,1. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.67 (brs, 1H), 7.99-7.91 (m, 4H), 7.72 (d, J=8,4 Гц, 2H), 7.26 (dd, J=10,0, 2,0 Гц, 1H), 7.01 (td, J=8,8, 2,4 Гц, 1H), 4.67 (t, J=5,2 Гц, 1H), 3.58 (d, J=10,0 Гц, 1H), 3.45 (t, J=11,2 Гц, 1H), 3.27-3.16 (m, 1H), 2.91 (d, J=12,0 Гц, 1H), 2.70-2.60 (m, 2H), 2.21-2.13 (m, 1H), 1.89 (t, J=10,8 Гц, 1H).
Стадия 5: трет-бутил-3-(4-(2-(3-этокси-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)фенил)-6-фтор-1H-индол-1-карбоксилат
Следуя общей методике, описанной на стадиях 1-4 выше, начиная с 1-трет-бутил-3-метил-пиперазин-1,3-дикарбоксилата, получили указанное в заголовке соединение в виде белого твердого вещества. ЖХ-МС для C19H20FN3O3S-H-[M-H]-: рассчитано: 388,1; обнаружено: 388,1. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.66 (br s, 1H), 7.95-7.91 (m, 4H), 7.83 (d, J=8,4 Гц, 2H), 7.26 (dd, J=10,0, 2,4 Гц, 1H), 7.01 (td, J=8,8, 2,4 Гц, 1H), 4.76 (br s, 1H), 3.77-3.72 (m, 1H), 3.68-3.64 (m, 1H), 3.51-3.48 (m, 1H), 3.37-3.29 (m, 2H), 3.04-2.92 (m, 2H), 2.71-2.68 (m, 1H), 2.49-2.40 (m, 2H).
Стадия 6: Желтый раствор трет-бутил-3-(4-(2-(3-этокси-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)фенил)-6-фтор-1H-индол-1-карбоксилата (Стадия 5, 500 мг, 1,2 ммоль) в NH3/EtOH (30% масс./масс., 30 мл) перемешивали при 80°C в герметично закрытой пробирке в течение 14 часов. Реакционную смесь концентрировали и очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат от 2/1 до 1/4) с получением рацемического 3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропанамида (170 мг, 52%) в виде красного твердого вещества. Этот рацемический материал разделяли посредством препаративной хиральной СФХ с получением (+)-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропанамида в качестве первого элюируемого пика (90 мг, 27%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1H), 8.11-7.80 (m, 5H), 7.48 (br s, 1H), 7.31-7.24 (m, 1H), 7.02 (d, J=2,0 Гц, 1H), 6.95 (br s, 1H), 4.67 (d, J=6,5 Гц, 1H), 3.60-3.43 (m, 2H), 2.64-2.53 (m, 2H), 1.56 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 409,9 (M+Na)+; [α]20D +2° (c=0,002 г/мл, DMSO).
Пример 48: (-)-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропанамид
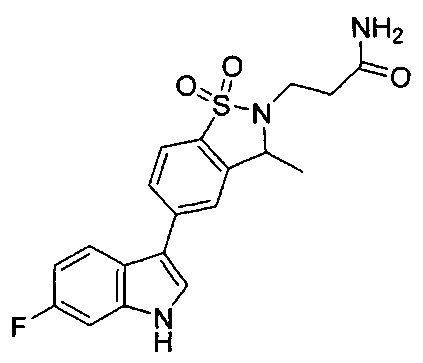
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 47 (80 мг, 24%), в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1H), 7.98-7.82 (m, 5H), 7.48 (br s, 1H), 7.28 (dd, J=2,4, 9,9 Гц, 1H), 7.02 (dt, J=2,4, 9,2 Гц, 1H), 6.95 (br s, 1H), 4.67 (q, J=6,7 Гц, 1H), 3.57-3.46 (m, 2H), 2.64-2.54 (m, 2H), 1.56 (d, J=6,3 Гц, 3H); ЖХ-МС: m/z 409,9 (M+Na)+; [α]20D -2,14° (c=0,0014 г/мл, DMSO).
Пример 49: (+)-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-1-(пиперазин-1-ил)пропан-1-он

Стадия 1: 3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропановая кислота
К желтому раствору трет-бутил-3-(2-(3-этокси-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (210 мг, 0,354 ммоль) в THF (5 мл) и H2O (5 мл) добавляли LiOH-H2O (47 мг, 1,1 ммоль). Реакционную смесь перемешивали при 28°C в течение 18 часов, затем концентрировали и разбавляли H2O (5 мл). Значение pH полученного раствора доводили до pH 6 с использованием 1 н. HCl, затем экстрагировали 5% метанолом в этилацетате (10 мл × 3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением неочищенной 3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропановой кислоты (202 мг, более 100%) в виде желтой смолы, которую использовали непосредственно на следующей стадии.
Стадия 2: трет-бутил-4-(3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропаноил)пиперазин-1-карбоксилат
Желтую суспензию 3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропановой кислоты (200 мг, 0,354 ммоль), трет-бутил-пиперазин-1-карбоксилата (132 мг, 0,708 ммоль) и DIPEA (0,123 мл, 0,708 ммоль) в безводном DMF (5 мл) перемешивали при 28°C в течение 10 мин, затем добавляли HATU (202 мг, 0,531 ммоль). Реакционную смесь перемешивали при 28°C в течение 6 часов, концентрировали, затем разбавляли H2O (10 мл) и экстрагировали этилацетатом (10 мл × 3). Объединенные органические слои промывали рассолом (5 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии (силикагель, 10-60% этилацетат/петролейный эфир) с получением 110 мг в виде рацемической смеси. Рацемический продукт очищали посредством хиральной СФХ с получением (+)-трет-бутил-4-(3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропаноил)пиперазин-1-карбоксилата (суммарно 46 мг, 17%) в виде желтого масла и (-)-трет-бутил-4-(3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропаноил)-пиперазин-1-карбоксилата (31 мг, выход 17%) в виде желтого масла.
Стадия 3: (+)-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-1-(пиперазин-1-ил)пропан-1-он
К раствору (+)-трет-бутил-4-(3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропаноил)-пиперазин-1-карбоксилата (46 мг, 0,08 ммоль) в дихлорметане (5 мл) добавляли HCl(газ)/этилацетат (4N) (5 мл). Реакционную смесь перемешивали при 28°C в течение 5 часов, затем концентрировали с получением твердого вещества, которое промывали этилацетатом (10 мл). Полученное твердое вещество сушили с получением (+)-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-1-(пиперазин-1-ил)пропан-1-она (42 мг, 100%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.75 (br s, 1H), 9,25 (br s, 1H), 7.97-7.87 (m, 4H), 7.86-7.82 (m, 1H), 7.27 (dd, J=2,4, 9,9 Гц, 1H), 7.06-6.96 (m, 1H), 4.76-4.68 (m, 1H), 3.69 (br s, 4H), 3.59-3.49 (m, 2H), 3.13-3.02 (m, 4H), 2.90-2.76 (m, 2H), 1.55 (d, J=6,3 Гц, 3H); LCMS: m/z 479,0 (M+Na)+; [α]20D +2,67° (c=1,5 мг/мл, метанол).
Пример 50: (-)3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-1-(пиперазин-1-ил)пропан-1-он
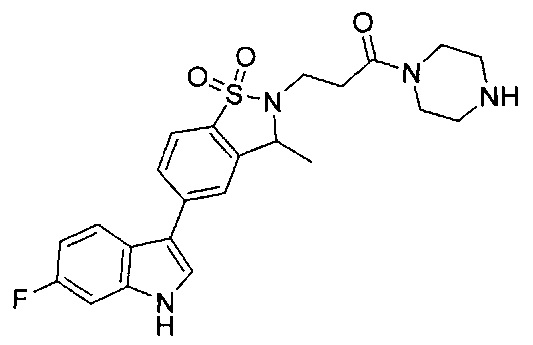
К раствору (-)-трет-бутил-4-(3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропаноил)пиперазин-1-карбоксилата (45 мг, 0,08 ммоль) в дихлорметане (5 мл) добавляли HCl (газ)/этилацетат(4N) (5 мл). Реакционную смесь перемешивали при 28°C в течение 5 часов, затем концентрировали с получением твердого вещества, которое промывали этилацетатом (10 мл). Твердое вещество сушили с получением (-)-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-1-(пиперазин-1-ил)пропан-1-она (40 мг, 100%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.76 (br s, 1H), 9.30 (br s, 1H), 7.98-7.88 (m, 4H), 7.87-7.81 (m, 1H), 7.27 (dd, J=2,4, 9,9 Гц, 1H), 7.01 (dt, J=2,4, 9,2 Гц, 1H), 4.72 (q, J=6,3 Гц, 1H), 3.70 (br s, 4H), 3.62-3.49 (m, 2H), 3.16-3.00 (m, 4H), 2.93-2.73 (m, 2H), 1.56 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 479,1 (M+Na)+; [α]20D -2,67° (c=1,3 мг/мл, метанол).
Пример 51: (-)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(пиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
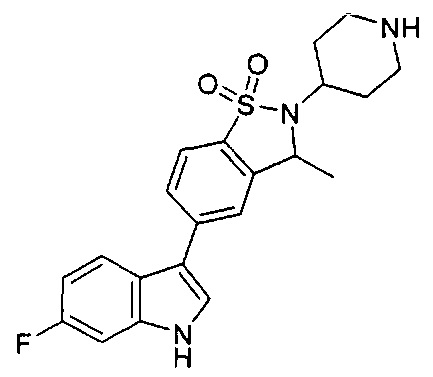
Стадия 1: трет-бутил-4-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пиперидин-1-карбоксилат
Коричневую суспензию 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (400 мг, 1,53 ммоль), трет-бутил-4-((метилсульфонил)окси)-пиперидин-1-карбоксилата (853 мг, 3,05 ммоль) и K2CO3 (211 мг, 1,53 ммоль) в DMF (5 мл) перемешивали при 80°C в течение 18 часов. Реакционную смесь охлаждали до температуры окружающей среды, затем вливали в воду (100 мл) и экстрагировали этилацетатом (3 × 20 мл). Объединенные органические слои промывали рассолом (2 × 10 мл), затем сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 0-20% этилацетат/петролейный эфир) с получением трет-бутил-4-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пиперидин-1-карбоксилата (250 мг, 37%) в виде светло-желтого твердого вещества.
Стадия 2: трет-бутил-4-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пиперидин-1-карбоксилат
В раствор трет-бутил-4-(5-бром-3-метил-1,1-диоксидобензо[d]-изотиазол-2(3H)-ил)пиперидин-1-карбоксилата (200 мг, 0,449 ммоль) и трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (211 мг, 0,584 ммоль) в диоксане (5 мл) добавляли водн. K3PO4 (2 М в воде, 191 мг, 0,898 ммоль) и Pd(dppf)2Cl2 (33 мг, 0,045 ммоль). Реакционный раствор барботировали N2 в течение 2 мин, герметично закрывали и перемешивали при 80°C в течение 3 часов. Неочищенную реакционную смесь охлаждали до температуры окружающей среды и концентрировали, затем очищали посредством колоночной хроматографии (силикагель, 0-20% этилацетат/петролейный эфир) с получением трет-бутил-3-(2-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (210 мг, 82%) в виде светло-желтой смолы. LCMS (M+Na, 622).
Стадия 3: (-)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(пиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Рацемический трет-бутил-3-(2-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилат (150 мг, 0,25 ммоль) разделяли посредством хиральной СФХ с получением двух пиков. Соединение первого пика (65 мг, 0,11 ммоль) растворяли в дихлорметане (5 мл) и HCl/этилацетате (4 М, 10 мл), затем перемешивали при температуре окружающей среды в течение 4 часов. Реакционную смесь концентрировали и очищали посредством препаративной ВЭЖХ с получением (-)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(пиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (10 мг, 21%). 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.66-11.87 (m, 1H), 8.63-9.08 (m, 1H), 7.88-8.02 (m, 4H), 7.75-7.86 (m, 1H), 7.29 (dd, J=9,79, 2,26 Гц, 1H), 6.98-7.07 (m, 1H), 4.90 (q, J=6,53 Гц, 1H), 3.91 (t, J=11,17 Гц, 1H), 3.29 (br s, 2H), 3.05 (br s, 2H), 2.06-2.33 (m, 4H), 1.61 (d, J=6,53 Гц, 3H); ЖХ-МС: m/z 399,9 (М+1)+; [α]20D -8,79° (c=1,1 мг/мл, DMSO).
Пример 52: (+)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(пиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид

Соединение второго пика из хирального разделения, описанного в Примере 51 (70 мг, 0,12 ммоль), растворяли в дихлорметане (5 мл), затем охлаждали до 0°C и добавляли по каплям HCl/этилацетат (10 мл, 4 М). Реакционную смесь перемешивали при температуре окружающей среды в течение 4 часов, концентрировали и очищали посредством препаративной ВЭЖХ с получением (+)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(пиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (10 мг, 24%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.79 (br s, 1H), 8.79-9,21 (m, 1H), 7.87-7.98 (m, 4H), 7.78-7.86 (m, 1H), 7.29 (dd, J=9,79, 2,26 Гц, 1H), 7.02 (m, 1H), 4.90 (q, J=6,53 Гц, 1H), 3.91 (t, J=11,17 Гц, 1H), 3.33 (br s, 2H), 3.05 (br s, 2H), 2.00-2.40 (m, 4H), 1.62 (d, J=6,53 Гц, 3H); ЖХ-МС: m/z 399,9 (М+1)+; [α]20D +8,1° (c=0,9 мг/мл, DMSO).
Пример 53: (+)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(2-(метилсульфонил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
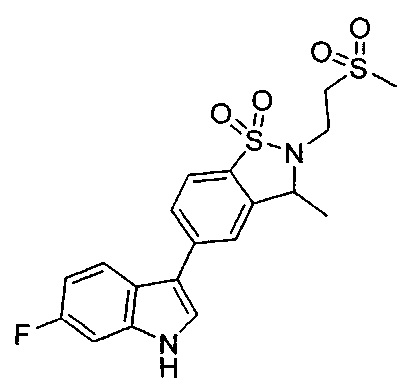
Стадия 1: 5-бром-3-метил-2-(2-(метилтио)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Красный раствор 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (400 мг, 1,53 ммоль), (2-хлорэтил)(метил)сульфана (169 мг, 1,534 ммоль) и K2CO3 (422 мг, 3,05 ммоль) в DMF (5 мл) выкачивали и заполняли N2 трижды, затем перемешивали при 85°C в течение 14 часов. Реакционную смесь разбавляли этилацетатом (50 мл) и промывали H2O (10 мл × 3). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного 5-бром-3-метил-2-(2-(метилтио)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (513 мг, 100%) в виде красной смолы.
Стадия 2: 5-бром-3-метил-2-(2-(метилсульфонил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Красный раствор 5-бром-3-метил-2-(2-(метилтио)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (513 мг, 1,53 ммоль) и оксона (938 мг, 1,53 ммоль) в THF/H2O (10 мл/2) перемешивали при 25°C в течение 1 часа. Реакционную смесь разбавляли этилацетатом (50 мл) и промывали H2O (10 мл × 3). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали. Неочищенный остаток очищали посредством колоночной хроматографии (SiO2, этилацетат/петролейный эфир от 1/5 до 1/1) с получением 5-бром-3-метил-2-(2-(метилсульфонил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (400 мг, 71%) в виде красного твердого вещества.
Стадия 3: трет-бутил-6-фтор-3-(3-метил-2-(2-(метилсульфонил)этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилат
Желтый раствор 5-бром-3-метил-2-(2-(метилсульфонил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (394 мг, 1,09 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (400 мг, 1,09 ммоль), PdCl2(dppf) (80 мг, 0,11 ммоль) и Cs2CO3 (710 мг, 2,18 ммоль) в диоксане (8 мл) и H2O (2 мл) перемешивали при 85°C в атмосфере N2 в течение 14 часов. Полученный черный раствор разбавляли этилацетатом (50 мл) и промывали рассолом (20 мл). Органическую фазу сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного трет-бутил-6-фтор-3-(3-метил-2-(2-(метилсульфонил)этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата в виде красной смолы.
Стадия 4: (+;-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(2-(метилсульфонил)-этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Красный раствор неочищенного трет-бутил-6-фтор-3-(3-метил-2-(2-(метилсульфонил)этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (570 мг, 1,09 ммоль) в смеси трифторуксусная кислота/дихлорметан (5 мл/5 мл) перемешивали при 20°C в атмосфере N2 в течение 2 часов. Полученную желтую суспензию концентрировали и нейтрализовали NaHCO3 (насыщ.) (10 мл), затем экстрагировали дихлорметаном (20 мл × 3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат от 1/1 до 1/4) с получением рацемического продукта (150 мг) в виде красного твердого вещества. Рацемическую смесь разделяли посредством препаративной хиральной СФХ с получением (+)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(2-(метилсульфонил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида в качестве первого элюируемого пика (65 мг, 14%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.72 (br s, 1H), 8.11-7.75 (m, 5H), 7.28 (dd, J=2,3, 9,8 Гц, 1H), 7.03 (dt, J=2,5, 9,3 Гц, 1H), 4.80 (q, J=6,5 Гц, 1H), 3.85-3.52 (m, 4H), 3.11 (s, 3H), 1.60 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 444,8 (M+Na)+; [α]20D +0,67° (c=0,0035 г/мл, DMSO).
Пример 54: (-)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(2-(метилсульфонил)-этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
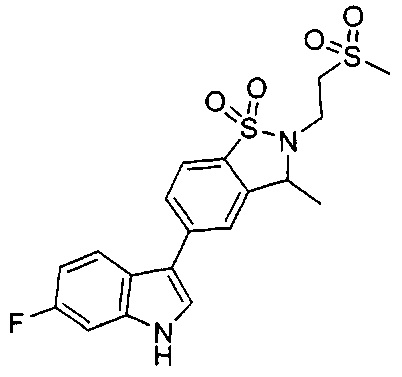
Указанное в заголовке соединение выделяли в качестве второго элюируемого пика из хирального разделения, описанного в Примере 53 (30 мг, 7%), в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.72 (br s, 1H), 8.09-7.83 (m, 5H), 7.28 (dd, J=2,3, 9,8 Гц, 1H), 7.03 (dt, J=2,4, 9,2 Гц, 1H), 4.80 (q, J=6,4 Гц, 1H), 3.84-3.50 (m, 4H), 3.16-3.05 (m, 3H), 1.60 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 444,8 (M+Na)+; [α]20D=-1,62° (c=0,0036 г/мл, DMSO).
Пример 55: (+)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(2-(метилсульфинил)-этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид

Стадия 1: 5-бром-3-метил-2-(2-(метилсульфинил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К желтому раствору 5-бром-3-метил-2-(2-(метилтио)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (1,05 г, 3,12 ммоль) в безводном дихлорметане (50 мл) добавляли m-СРВА (634 мг, 3,12 ммоль) при -25°C. Желтую суспензию перемешивали при -25°C в течение 1 часа, затем промывали H2O (10 мл × 2). Слои разделяли и органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Неочищенный материал очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат от 6/1 до 3/1) с получением 5-бром-3-метил-2-(2-(метилсульфинил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (1,0 г, 91%) в виде желтой смолы.
Стадия 2: трет-бутил-6-фтор-3-(3-метил-2-(2-(метилсульфинил)этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилат
Желтый раствор трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (513 мг, 1,42 ммоль), 5-бром-3-метил-2-(2-(метилсульфинил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (500 мг, 1,42 ммоль), PdCl2(dppf) (104 мг, 0,142 ммоль) и Cs2CO3 (925 мг, 2,84 ммоль) в диоксане (8 мл) и H2O (2 мл) перемешивали при 85°C в атмосфере N2 в течение 14 часов. Полученный черный раствор разбавляли этилацетатом (50 мл) и промывали рассолом (20 мл). Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного трет-бутил-6-фтор-3-(3-метил-2-(2-(метилсульфинил)этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата в виде красной смолы, которую использовали на следующей стадии без очистки.
Стадия 3: (+)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(2-(метилсульфинил)-этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Красный раствор трет-бутил-3-(4-(N-(3-амино-3-оксопропил)-сульфамоил)-3-изобутилфенил)-6-фтор-1H-индол-1-карбоксилата (719 мг, 1,42 ммоль) в смеси трифторуксусная кислота/дихлорметан (5 мл/5 мл) перемешивали при 20°C в атмосфере N2 в течение 1 часа. Желтую суспензию концентрировали, нейтрализовали NaHCO3 (насыщ.) (10 мл) и экстрагировали этилацетатом (20 мл × 3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (SiO2, от смеси петролейный эфир/этилацетат 1/1 до смеси этилацетат/метанол 10/1), а затем посредством препаративной ВЭЖХ с получением рацемического 5-(6-фтор-1H-индол-3-ил)-3-метил-2-(2-(метилсульфинил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида в виде красного твердого вещества. Смесь диастереомеров разделяли посредством препаративной хиральной СФХ с получением (+)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(2-(метилсульфинил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида в качестве первого элюируемого пика (40 мг, 24%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 8.63 (br s, 1H), 7.87-7.81 (m, 1H), 7.79-7.71 (m, 2H), 7.57 (s, 1H), 7.42 (d, J=2,8 Гц, 1H), 7.16 (dd, J=2,3, 9,0 Гц, 1H), 7.01 (dt, J=2,3, 9,2 Гц, 1H), 4.68 (q, J=6,5 Гц, 1H), 4.01-3.90 (m, 1H), 3.87-3.74 (m, 1H), 3.44-3.32 (m, 1H), 3.09 (td, J=5,8, 13,2 Гц, 1H), 2.76-2.66 (m, 3H), 1.65 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 428,8 (M+Na)+; [α]20D +25,1° (c=0,0016 г/мл, метанол).
Пример 56: (-)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(2-(метилсульфинил)-этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид

Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 55 (40 мг, 24%), в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 8.56 (br s, 1H), 7.87-7.73 (m, 3H), 7.59 (s, 1H), 7.43 (d, J=2,5 Гц, 1H), 7.16 (dd, J=2,3, 9,3 Гц, 1H), 7.02 (dt, J=2,4, 9,1 Гц, 1H), 4.70 (q, J=6,7 Гц, 1H), 4.02-3.90 (m, 1H), 3.82 (d, J=7,0 Гц, 1H), 3.43-3.32 (m, 1H), 3.14-3.05 (m, 1H), 2.71 (s, 3H), 1.66 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 428,8 (M+Na)+; [α]20D -20° (c=0,0016 г/мл, метанол).
Пример 57: (-)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(2-(метилсульфинил)-этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
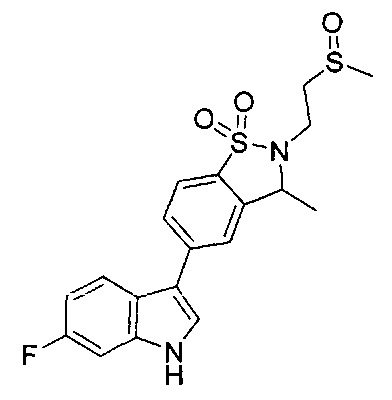
Указанное в заголовке соединение получили в качестве третьего элюируемого пика из хирального разделения, описанного в Примере 55 (40 мг, 24%), в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 8.56 (br s, 1H), 7.88-7.72 (m, 3H), 7.60 (s, 1H), 7.44 (d, J=2,8 Гц, 1H), 7.16 (dd, J=2,3, 9,3 Гц, 1H), 7.01 (dt, J=2,3, 9,2 Гц, 1H), 4.52 (q, J=6,4 Гц, 1H), 3.96-3.88 (m, 1H), 3.85-3.75 (m, 1H), 3.38 (ddd, J=6,0, 10,3, 13,3 Гц, 1H), 3.05-2.94 (m, 1H), 2.69 (s, 3H), 1.71 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 428,8 (M+Na)+; [α]20D -10° (с=0,0019 г/мл, метанол).
Пример 58: (+)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(2-(метилсульфинил)-этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
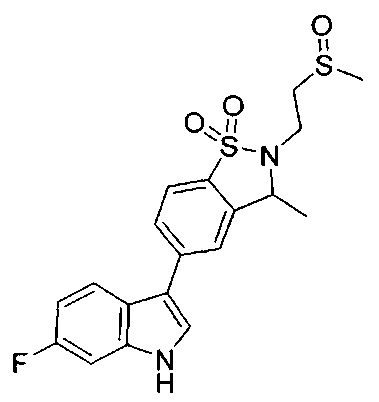
Указанное в заголовке соединение получили в качестве четвертого элюируемого пика из хирального разделения, описанного в Примере 55 (40 мг, 24%), в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 8.61-8.49 (m, 1H), 7.89-7.72 (m, 3H), 7.60 (s, 1H), 7.44 (d, J=2,5 Гц, 1H), 7.16 (dd, J=2,3, 9,3 Гц, 1H), 7.02 (dt, J=2,3, 9,2 Гц, 1H), 4.52 (q, J=6,3 Гц, 1H), 3.97-3.88 (m, 1H), 3.86-3.75 (m, 1H), 3.38 (ddd, J=6,0, 10,3, 13,6 Гц, 1H), 3.02-2.95 (m, 1H), 2.72-2.65 (m, 3H), 1.71 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 428,8 (M+Na)+; [α]20D +0,67° (с=0,0015 г/мл, метанол).
Пример 59: (-)-2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилэтан-1-сульфонамид
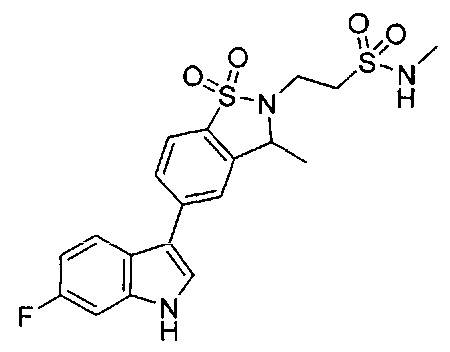
Стадия 1: 2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-этансульфонилхлорид
Красный раствор 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (500 мг, 1,9 ммоль), 2-хлорэтансульфонилхлорида (933 мг, 5,72 ммоль) и K2CO3 (1050 мг, 7,063 ммоль) в MeCN (15 мл) выкачивали и заполняли N2 трижды, затем перемешивали при 85°С в течение 48 часов. Желтую суспензию 2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)- этансульфонилхлорида использовали непосредственно на следующей стадии.
Стадия 2: 2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилэтансульфонамид
Суспензию 2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-этансульфонилхлорида (Стадия 1) охлаждали до 25°С и добавляли MeNH2 в H2O (10 мл). Реакционную смесь перемешивали при 25°С в течение 4 часов, затем разбавляли этилацетатом (50 мл) и промывали H2O (10 мл × 3). Органический слой сушили над Na2SO4, фильтровали и концентрировали, затем очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат от 3/1 до 1/1) с получением неочищенного 2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилэтансульфонамида (270 мг, 37%) в виде красной смолы.
Стадия 3: трет-бутил-6-фтор-3-(3-метил-2-(2-(N-метилсульфамоил)-этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилат
Желтый раствор трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (254 мг, 0,704 ммоль), 2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилэтан-сульфонамида (270 мг, 0,704 ммоль), PdCl2(dppf) (51,5 мг, 0,0704 ммоль) и Cs2CO3 (459 мг, 1,41 ммоль) в диоксане (8 мл) и H2O (2 мл) перемешивали при 85°С в атмосфере N2 в течение 14 часов. Полученный черный раствор разбавляли этилацетатом (50 мл) и промывали рассолом (20 мл). Органическую фазу сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного трет-бутил-6-фтор-3-(3-метил-2-(2-(N-метилсульфамоил)этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата в виде красной смолы, которую использовали на следующей стадии без очистки.
Стадия 4: (-)-2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилэтан-1-сульфонамид
Красный раствор трет-бутил-6-фтор-3-(3-метил-2-(2-(N-метилсульфамоил)этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (378 мг, 0,703 ммоль) в смеси трифторуксусная кислота/дихлорметан (5 мл / 5 мл) перемешивали при 20°С в атмосфере N2 в течение 1 часа. Желтую суспензию концентрировали и нейтрализовали NaHCO3 (насыщ.) (10 мл), затем экстрагировали этилацетатом (20 мл × 3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат от 1/1 до 1/4) с получением смеси энантиомеров. Рацемическую смесь разделяли посредством препаративной хиральной СФХ с получением (-)-2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилэтан-1-сульфонамида в качестве первого элюируемого пика (20 мг, 7%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.96-7.86 (m, 2Н), 7.85-7.79 (m, 2Н), 7.71 (s, 1H), 7.18 (dd, J=2,4, 9,7 Гц, 1H), 6.97 (dt, J=2,4, 9,2 Гц, 1H), 4.77 (d, J=6,5 Гц, 1H), 3.89-3.71 (m, 2Н), 3.63-3.47 (m, 2Н), 2.78 (s, 3H), 1.66 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 459,9 (M+Na)+; [α]20D -1,33° (с=0,003 г/мл, метанол).
Пример 60: (+)-2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилэтан-1-сульфонамид
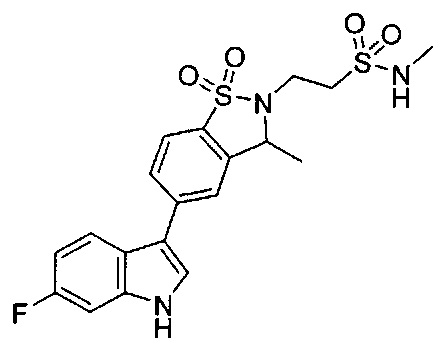
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 59 (20 мг, 7%), в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.95-7.86 (m, 2Н), 7.85-7.80 (m, 2Н), 7.71 (s, 1H), 7.18 (dd, J=2,4, 9,7 Гц, 1H), 6.97 (dt, J=2,4, 9,2 Гц, 1H), 4.78 (d, J=6,5 Гц, 1H), 3.90-3.71 (m, 2Н), 3.65-3.48 (m, 2Н), 2.78 (s, 3H), 1.67 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 459,9 (M+Na)+; [α]20D +0,42° (с=0,004 г/мл, метанол).
Пример 61: (+)-2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этан-1-сульфонамид
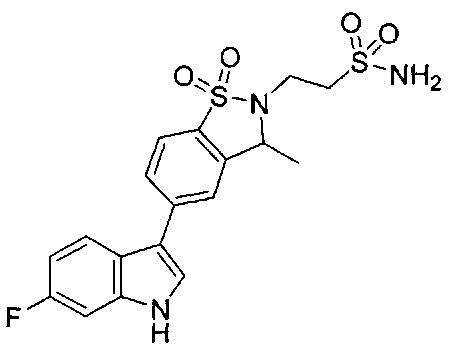
Стадия 1: 2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-этансульфонамид
Суспензию 2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-этансульфонилхлорида (737 мг, 1,9 ммоль) в MeCN (15 мл) охлаждали до 25°С и добавляли NH4OH (5 мл). Перемешивание продолжали при 25°С в течение 4 часов, затем реакционную смесь разбавляли этилацетатом (50 мл) и промывали H2O (10 мл × 3). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали, затем очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат от 3/1 до 1/1) с получением 2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-этансульфонамида (280 мг, 40%) в виде красной смолы.
Стадия 2: трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2-(2-сульфамоилэтил)-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилат
Желтый раствор трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (275 мг, 0,760 ммоль), 2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этансульфонамида (280 мг, 0,758 ммоль), PdCl2(dppf) (56 мг, 0,08 ммоль) и Cs2CO3 (495 мг, 1,52 ммоль) в диоксане (8 мл) и H2O (2 мл) перемешивали при 85°С в атмосфере N2 в течение 14 часов. Полученный черный раствор разбавляли этилацетатом (50 мл) и слои разделяли. Органический слой промывали рассолом (20 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2-(2-сульфамоилэтил)-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилата в виде красной смолы, которую использовали на следующей стадии без очистки.
Стадия 3: (+)-2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этан-1-сульфонамид
Красный раствор трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2-(2-сульфамоилэтил)-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилата (398 мг, 0,76 ммоль) в смеси трифторуксусная кислота/дихлорметан (5 мл/5 мл) перемешивали при 20°С в атмосфере N2 в течение 1 часа. Реакционную смесь концентрировали, затем нейтрализовали NaHCO3 (насыщ.) (10 мл) и экстрагировали этилацетатом (20 мл × 3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали, затем очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат от 1/1 до 1/4) с получением рацемического материала (100 мг) в виде красного твердого вещества. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (+)-2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этан-1-сульфонамида в качестве первого элюируемого пика (30 мг, 9%) в виде бледно-желтого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.96-7.86 (m, 2Н), 7.85-7.80 (m, 2Н), 7.71 (s, 1Н), 7.18 (dd, J=2,3, 9,5 Гц, 1Н), 7.01-6.94 (m, 1Н), 4.81-4.75 (m, 1Н), 3.94-3.78 (m, 2Н), 3.68-3.51 (m, 2H), 1.67 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z для 445,9 (M+Na)+; [α]20D+6° (c=0,002 г/мл, метанол).
Пример 62: (-)-2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этан-1-сульфонамид
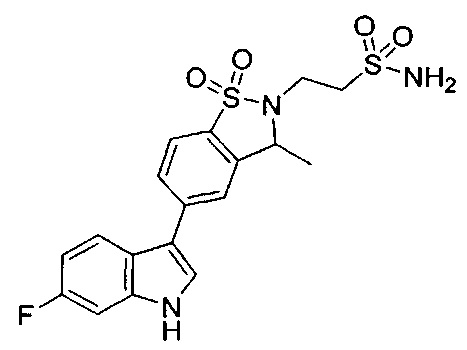
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 61 (30 мг, 9%), в виде бледно-желтого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.97-7.86 (m, 2Н), 7.86-7.80 (m, 2Н), 7.71 (s, 1Н), 7.19 (dd, J=2,4, 9,7 Гц, 1Н), 6.97 (dt, J=2,4, 9,2 Гц, 1Н), 4.78 (q, J=6,4 Гц, 1Н), 3.95-3.78 (m, 2Н), 3.68-3.51 (m, 2Н), 1.67 (d, J=6,3 Гц, 3Н); ЖХ-МС: m/z 445,9 (M+Na)+; [α]20D=-1,33° (с=0,003 г/мл, метанол).
Пример 63: (+)-5-(6-фтор-1Н-индол-3-ил)-2-(2-гидроксиэтил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
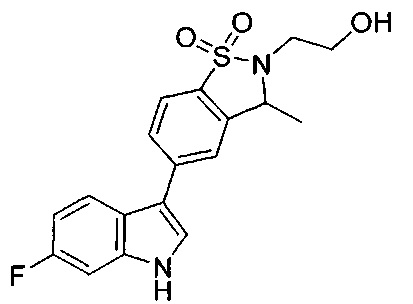
Стадия 1: 5-бром-2-(2-гидроксиэтил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Красный раствор 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (500 мг, 1,91 ммоль), 2-бромэтанола (477 мг, 3,81 ммоль) и K2CO3 (1050 мг, 7,63 ммоль) в MeCN (5 мл) выкачивали и снова заполняли с помощью N2 трижды, затем перемешивали при 85°С в течение 48 часов. Неочищенную реакционную смесь разбавляли этилацетатом (50 мл) и промывали Н2О (10 мл × 3). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали с получением 5-бром-2-(2-гидроксиэтил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (500 мг, 86%) в виде красной смолы.
Стадия 2: трет-бутил-6-фтор-3-(2-(2-гидроксиэтил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилат
Желтый раствор трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (500 мг, 1,38 ммоль), 5-бром-2-(2-гидроксиэтил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (500 мг, 1,63 ммоль), PdCl2(dppf) (101 мг, 0,138 ммоль) и Cs2CO3 (902 мг, 2,77 ммоль) в диоксане (8 мл) и H2O (2 мл) перемешивали при 85°С в атмосфере N2 в течение 14 часов. Полученный черный раствор разбавляли этилацетатом (50 мл) и слои разделяли. Органический слой промывали рассолом (20 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного трет-бутил-6-фтор-3-(2-(2-гидроксиэтил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата в виде красной смолы, которую использовали на следующей стадии без очистки.
Стадия 3: (+)-5-(6-фтор-1Н-индол-3-ил)-2-(2-гидроксиэтил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Красный раствор трет-бутил-6-фтор-3-(2-(2-гидроксиэтил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (637 мг, 1,38 ммоль) в смеси трифторуксусная кислота/дихлорметан (5 мл/5 мл) перемешивали при 20°С в атмосфере N2 в течение 1 часа. Желтую суспензию концентрировали, затем нейтрализовали NaHCO3 (насыщ.) (10 мл) и экстрагировали этилацетатом (20 мл × 3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (SiO2) петролейный эфир/этилацетат от 1/1 до 1/4) с получением рацемического продукта (150 мг) в виде красного твердого вещества. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (+)-5-(6-фтор-1Н-индол-3-ил)-2-(2-гидроксиэтил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида в качестве первого элюируемого пика (35 мг, 7%) в виде бледно-желтого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.95-7.86 (m, 2Н), 7.85-7.77 (m, 2Н), 7.70 (s, 1Н), 7.18 (dd, J=2,3, 9,5 Гц, 1Н), 6.97 (dt, J=2,1, 9,2 Гц, 1Н), 4.80 (q, J=6,6 Гц, 1Н), 3.94-3.83 (m, 2Н), 3.58-3.42 (m, 2Н), 1.66 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 360,9 (М+Н)+; [α]20D +15,7° (с=0,0021 г/мл, метанол).
Пример 64: (-)-5-(6-фтор-1H-индол-3-ил)-2-(2-гидроксиэтил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
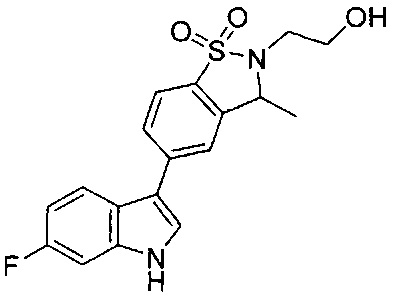
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 63 (40 мг, 8%), в виде бледно-желтого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.96-7.86 (m, 2Н), 7.84-7.78 (m, 2Н), 7.70 (s, 1Н), 7.18 (dd, J=2,5, 9,5 Гц, 1Н), 6.97 (dt, J=2,3, 9,2 Гц, 1Н), 4.84-4.77 (m, 1Н), 3.93-3.84 (m, 2Н), 3.59-3.42 (m, 2Н), 1.66 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 360,9 (М+Н)+; [α]20D -17° (с=0,002 г/мл, метанол).
Пример 65: (-)1-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этил)пиперазин-2-он
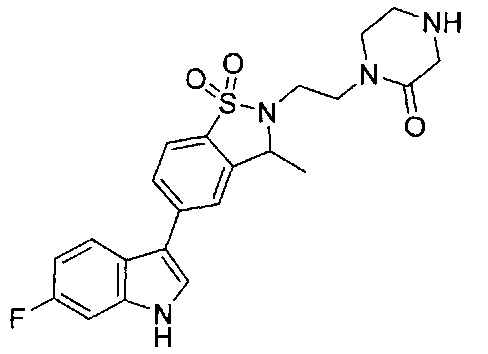
Стадия 1: 2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-этилметансульфонат
К раствору 5-бром-2-(2-гидроксиэтил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (900 мг, 2,94 ммоль) (Пример 173, стадия 1) и TEA (892 мг, 8,82 ммоль) в DCM (10 мл) добавляли метансульфонилхлорид (MsCl) (673 мг, 5,88 ммоль). Реакционную смесь перемешивали при 20°С в течение 16 часов, затем вливали в воду (100 мл) и экстрагировали DCM (100 мл × 2). Объединенные органические слои промывали рассолом (100 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного 2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этилметансульфоната (1,1 г) в виде желтого масла, которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2: трет-бутил-4-(2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этил)-3-оксопиперазин-1-карбоксилат
К суспензии 2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-этилметансульфоната (1,5 г, 3,9 ммоль) и t-BuOK (876 мг, 7,81 ммоль) в DMF (30 мл) добавляли трет-бутил-3-оксопиперазин-1-карбоксилат (938 мг, 4,68 ммоль). Реакционную смесь перемешивали при 80°С в течение 16 часов, затем вливали в воду (100 мл), экстрагировали EtOAc (100 мл × 2). Объединенные органические слои промывали рассолом (100 мл × 3), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением трет-бутил-4-(2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)-3-оксопиперазин-1-карбоксилата (1800 мг, неочищенный) в виде желтого масла, которое использовали на следующей стадии без дополнительной очистки.
Стадия 3: трет-бутил-3-(2-(2-(4-(трет-бутоксикарбонил)-2-оксопиперазин-1-ил)этил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилат
К суспензии трет-бутил-(3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-1-(метиламино)-1-оксопропан-2-ил)-карбамата (1,8 г, 3,7 ммоль) и трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индол-1-карбоксилата (1,5 г, 4,1 ммоль) в смеси диоксан/вода (20 мл/6 мл) добавляли Pd(dppf)Cl2 (270 мг, 0,369 ммоль) и K3PO4 (1,6 г, 7,4 ммоль). Реакционную смесь перемешивали при 80°С в течение 16 часов, затем концентрировали и очищали посредством колоночной хроматографии (силикагель, петролейный эфир/этилацетат (РЕ/ЕА) 4/1), а затем посредством препаративной ВЭЖХ с получением указанного в заголовке соединения в виде рацемической смеси (400 мг). Энантиомеры разделяли посредством препаративной хиральной СФХ с получением хирального трет-бутил-3-(2-(2-(4-(трет-бутоксикарбонил)-2-оксопиперазин-1-ил)этил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d1]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата в качестве первого элюируемого пика (200 мг) в виде белого твердого вещества и хирального трет-бутил-3-(2-(2-(4-(трет-бутоксикарбонил)-2-оксопиперазин-1-ил)-этил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата в качестве второго элюируемого пика (200 мг) в виде белого твердого вещества.
Стадия 4: (-)1-(2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этил)пиперазин-2-он
Раствор трет-бутил-3-(2-(2-(4-(трет-бутоксикарбонил)-2-оксопиперазин-1-ил)этил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (300 мг, 0,467 ммоль) (стадия 3, пик 1) в HCl/EA (10 мл) перемешивали при 20°С в течение 36 часов. Реакционную смесь концентрировали и сушили с получением (-)1-(2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этил)пиперазин-2-она (154 мг, 75%) в виде желтого твердого вещества. 1Н ЯМР (400МГц, DMSO-d6) δ [м.д.] 12.00 (br s, 1Н), 10.84-10.05 (m, 1Н), 8.43-7.80 (m, 5Н), 7.29 (br s, 1Н), 7.23-6.94 (m, 1Н), 4.81 (br s, 1Н), 3.83-3.46 (m, 10Н), 1.86-1.51 (m, 3Н); ЖХ-МС: m/z 443,1 (М+Н)+; [α]20D -15° (с=0,001 г/мл, DMSO).
Пример 66: (+)1-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[b]изотиазол-2(3Н)-ил)этил)пиперазин-2-он
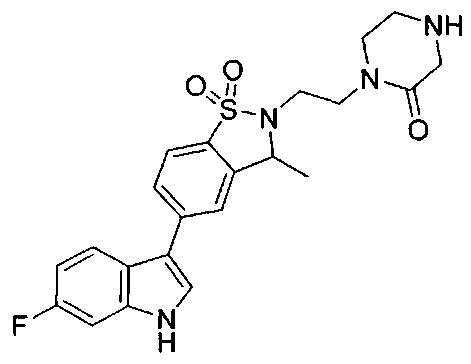
Раствор трет-бутил-3-(2-(2-(4-(трет-бутоксикарбонил)-2-оксопиперазин-1-ил)этил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилата (300 мг, 0,467 ммоль) (Пример 176, стадия 3, пик 2) в HCl/EA (10 мл) перемешивали при 20°С в течение 36 часов. Реакционную смесь концентрировали и сушили с получением (+)-1-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этил)-пиперазин-2-она (149 мг, 72%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.80 (br s, 1Н), 9,87 (br s, 1Н), 7.98-7.89 (m, 5Н), 7.28 (dd, J=2,3, 9,8 Гц, 1H), 7.01 (d, J=2,0 Гц, 1H), 4.80 (d, J=6,3 Гц, 1H), 3.70-3.37 (m, 10Н), 1.58 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 443,0 (М+Н)+; [α]20D 13° (с=0,001 г/мл, DMSO).
Пример 67: (+)-5-(6-фтор-1Н-индол-3-ил)-2-(2-(5-гидрокси-3-метил-1Н-пиразол-1-ил)этил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
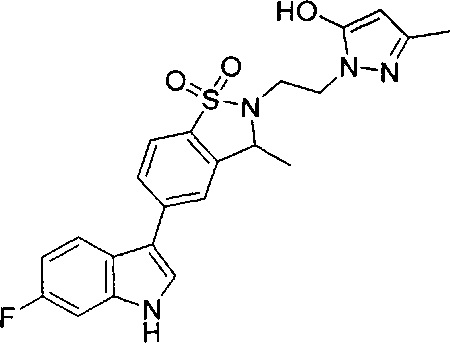
Стадия 1: хиральный трет-бутил-6-фтор-3-(3-метил-2-(2-((метилсульфонил)окси)этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилат
К желтому раствору хирального трет-бутил-6-фтор-3-(2-(2-гидроксиэтил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (200 мг, 0,434 ммоль) и DIPEA (112 мг, 0,869 ммоль) в дихлорметане (20 мл) добавляли MsCl (74,6 мг, 0,651 ммоль) при 0°С. Реакционную смесь перемешивали при 25°С в течение 2 часов, затем концентрировали с получением хирального трет-бутил-6-фтор-3-(3-метил-2-(2-((метилсульфонил)окси)этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилата (0,23 г, 100%) в виде желтой смолы, которую использовали непосредственно на следующей стадии.
Стадия 2: хиральный 5-(6-фтор-1Н-индол-3-ил)-2-(2-гидразинилэтил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Желтый раствор хирального трет-бутил-6-фтор-3-(3-метил-2-(2-((метилсульфонил)окси)этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (234 мг, 0,434 ммоль) в EtOH/NH2NH2H2O (10 мл/2 мл) перемешивали при 25°С в течение 18 часов. Реакционную смесь концентрировали с получением хирального 5-(6-фтор-1H-индол-3-ил)-2-(2-гидразинилэтил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (163 мг, 100%) в виде желтой смолы, которую использовали непосредственно на следующей стадии.
Стадия 3: (+)-5-(6-фтор-1Н-индол-3-ил)-2-(2-(5-гидрокси-3-метил-1Н-пиразол-1-ил)этил)-3-метил-2,3-дигидробензо[d]изотиазол-111-диоксид
Желтый раствор хирального 5-(6-фтор-1Н-индол-3-ил)-2-(2-гидразинилэтил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (163 мг, 0,435 ммоль) и этилацетоацетата (57 мг, 0,44 ммоль) в EtOH (10 мл) перемешивали при 25°С в течение 14 часов. Реакционную смесь концентрировали и очищали посредством препаративной ВЭЖХ с получением (+)-5-(6-фтор-1Н-индол-3-ил)-2-(2-(5-гидрокси-3-метил-1H-пиразол-1-ил)этил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (75 мг, 39%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1Н), 10.86 (br s, 1Н), 8.06-7.79 (m, 5Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.02 (dt, J=2,4, 9,2 Гц, 1Н), 5.18 (s, 1Н), 4.88-4.58 (m, 1Н), 4.21-3.74 (m, 2Н), 3.70-3.45 (m, 2Н), 2.15-1.94 (m, 3Н), 1.68-1.36 (m, 3Н); ЖХ-МС: m/z 440,9 (М+Н)+; [α]20D +2,2° (с=0,005 г/мл, DMSO).
Пример 68: (-)-5-(6-фтор-1Н-индол-3-ил)-2-(2-(5-гидрокси-3-метил-1Н-пиразол-1-ил)этил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
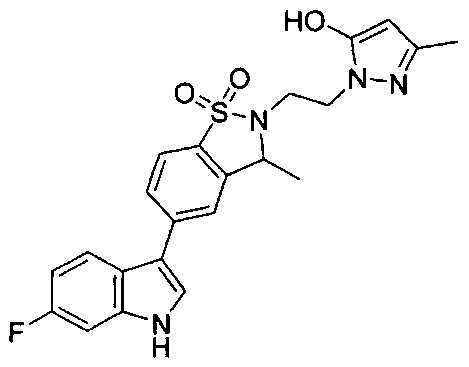
Следуя общей методике, изложенной в Примере 67, начиная с хирального трет-бутил-6-фтор-3-(3-метил-2-(2-((метилсульфонил)окси)-этил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата, получили указанное в заголовке соединение в виде желтого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1Н), 10.86 (br s, 1Н), 8.06-7.79 (m, 5Н), 7.27 (dd, J=2,3, 9,8 Гц, 1H), 7.02 (dt, J=2,4, 9,2 Гц, 1H), 5.18 (s, 1Н), 4.88-4.58 (m, 1Н), 4.21-3.74 (m, 2Н), 3.70-3.45 (m, 2Н), 2.15-1.94 (m, 3Н), 1.68-1.36 (m, 3Н); ЖХ-МС: m/z 440,9 (М+Н)+; [α]20D -2° (с=0,003 г/мл, DMSO).
Пример 69: (+)-3-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)оксазолидин-2-он
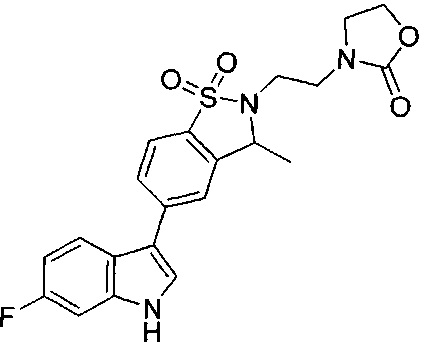
Стадия 1: 2-(2-оксооксазолидин-3-ил)этилметансульфонат
В охлажденный (ледяная баня) раствор 3-(2-гидроксиэтил)оксазолидин-2-она (3,0 г, 23 ммоль) и Et3N (2,55 г, 25,2 ммоль) в безводном дихлорметане (50 мл) добавляли по каплям раствор MsCl (3,14 г, 27,5 ммоль) в дихлорметане (10 мл) в течение 15 мин. Реакционную смесь перемешивали при 0°С в течение 2 часов, затем разбавляли водой (25 мл). Слои разделяли и органический слой промывали водным NaHCO3 (15 мл × 3), водой (15 мл) и рассолом (15 мл). Органический слой сушили над безводным Na2SO4, фильтровали и концентрировали с получением 2-(2-оксооксазолидин-3-ил)этилметан-сульфоната (3,0 г, 63%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 4.41-4.35 (m, 4Н), 3.74-3.61 (m, 4Н), 3.06 (s, 3Н).
Стадия 2: 3-(2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-этил)оксазолидин-2-он
К раствору 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (500 мг, 1,91 ммоль) в DMF (10 мл) добавляли K2CO3 (527 мг, 3,81 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 30 мин, затем добавляли 2-(2-оксооксазолидин-3-ил)этил-метансульфонат (649 мг, 2,48 ммоль) и перемешивание продолжали при 80°С в течение 16 часов. Черную смесь разбавляли водой (30 мл) и экстрагировали этилацетатом (25 мл × 3). Объединенные органические слои промывали водой (20 мл × 2) и рассолом (20 мл × 2), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенное масло очищали посредством колоночной хроматографии (силикагель, 50-100% этилацетат/петролейный эфир) с получением 3-(2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этил)оксазолидин-2-она (418 мг, 58%) в виде коричневого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 7.69-7.63 (m, 2Н), 7.57 (s, 1Н), 4.72- 4.69 (m, 1H), 4.36-4.31 (m, 2H), 3.83-3.73 (m, 2H), 3.68-3.64 (m, 2H), 3.48-3.41 (m, 2H), 1.57 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 398,7 (M+Na)+.
Стадия 3: трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2-(2-(2-оксооксазолидин-3-ил)этил)-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилат
Желтую смесь 3-(2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)оксазолидин-2-она (400 мг, 0,85 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (484 мг, 0,938 ммоль), PdCl2(dppf)CH2Cl2 (63,7 мг, 0,0853 ммоль) и K3PO4 (543 мг, 2,56 ммоль) в 1,4-диоксане (8 мл) и воде (2 мл) барботировали N2 в течение 1 минуты, затем перемешивали при 80°С в течение 16 часов. Реакционную смесь концентрировали и очищали посредством колоночной хроматографии (силикагель, 0-80% этилацетат/петролейный эфир) с получением трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2-(2-(2-оксооксазолидин-3-ил)этил)-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата 280 мг, 62%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 7.98-7.96 (m, 1Н), 7.88-7.86 (m, 1Н), 7.79-7.77 (m, 2Н), 7.66 (dd, J=5,4, 8,7 Гц, 1Н), 7.61 (s, 1Н), 7.09 (dt, J=2,4, 9,0 Гц, 1Н), 4.83-4.78 (m, 1Н), 4.39-4.33 (m, 2Н), 3.90-3.78 (m, 2Н), 3.74-3.68 (m, 2Н), 3.54-3.47 (m, 2Н), 1.72 (s, 9Н), 1.64 (d, J=6,5 Гц, 3Н). ЖХ-МС: m/z 530,0 (М+Н)+, 552,0 (M+Na)+.
Стадия 4: (+)-3-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)оксазолидин-2-он
В охлажденный (ледяная баня) желтый раствор трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2-(2-(2-оксооксазолидин-3-ил)этил)-2,3-дигидробензо[d]-изотиазол-5-ил)-1H-индол-1-карбоксилата (280 мг, 0,529 ммоль) в дихлорметане (5 мл) медленно добавляли трифторуксусную кислоту (2 мл). Реакционную смесь перемешивали при 27°С в течение 1 часа, затем концентрировали и нейтрализовали (рН=7-8) с использованием NH3/H2O (5 мл). Смесь экстрагировали дихлорметаном (15 мл × 3) и объединенные органические слои промывали рассолом (15 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенное желтое твердое вещество очищали посредством колоночной хроматографии (силикагель, 0-5% метанол/дихлорметан) с получением рацемического 3-(2-(5-(6-фтор-1Н-индол- 3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)оксазолидин-2-она (220 мг, 97%) в виде желтого твердого вещества. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (+)-3-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)-оксазолидин-2-она в качестве первого элюируемого пика (36 мг, 22%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s, 1Н), 7.96-7.84 (m, 5Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.04-6.99 (m, 1Н), 4.78 (q, J=6,4 Гц, 1Н), 4.26-4.21 (m, 2Н), 3.69-3.62 (m, 2Н), 3.53-3.50 (m, 2Н), 3.43-3.40 (m, 2Н), 1.57 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 430,0 (М+Н)+; [α]20D +16,0° (с=1 мг/мл, метанол).
Пример 70: (-)-3-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этил)оксазолидин-2-он
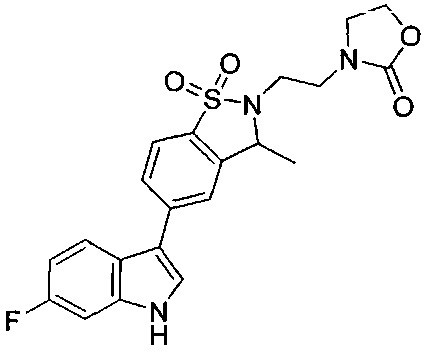
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 69 (33 мг, 20%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s, 1Н), 7.84-7.96 (m, 5Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.04-6.99 (m, 1Н), 4.78 (q, J=6,4 Гц, 1Н), 4.26-4.21 (m, 2Н), 3.72 -3.60 (m, 2Н), 3.53-3.50 (m, 2Н), 3.43-3.38 (m, 2Н), 1.57 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 429,9 (М+Н)+; [α]20D -46,0° (с=1 мг/мл, метанол).
Пример 71: (+)-1-(4-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пиперидин-1-ил)этанон

Стадия 1: трет-бутил-4-((метилсульфонил)окси)пиперидин-1-карбоксилат
В желтый раствор трет-бутил-4-гидроксипиперидин-1-карбоксилата (10 г, 50 ммоль) и триэтиламина (14 мл, 99 ммоль) в дихлорметане (100 мл) при 0°С добавляли по каплям метансульфонилхлорид (6,8 г, 60 ммоль). Реакционную смесь нагревали до температуры окружающей среды и перемешивали в течение 2 часов, затем гасили NaHCO3 (насыщ.) (50 мл). Слои разделяли и водную фазу экстрагировали дихлорметаном (50 мл × 2). Объединенные органические слои промывали водой (50 мл) и рассолом (50 мл), затем сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением трет-бутил-4-((метилсульфонил)окси)-пиперидин-1-карбоксилата (14 г, более 100%) в виде бледно-желтого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 4.91-4.85 (m, 1Н), 3.73-3.67 (m, 2Н), 3.33-3.27 (m, 2Н), 3.04 (s, 3Н), 1.99-1.94 (m, 2Н), 1.83-1.79 (m, 2Н), 1.46 (s, 9Н).
Стадия 2: трет-бутил-4-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пиперидин-1-карбоксилат
Раствор 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (500 мг, 1,91 ммоль), трет-бутил-4-((метилсульфонил)окси)пиперидин-1-карбоксилата (799 мг, 2,86 ммоль) и K2CO3 (527 мг, 3,81 ммоль) в DMF (10 мл) перемешивали в атмосфере N2 при 80°С в течение 19 часов. Реакционную смесь охлаждали до температуры окружающей среды и гасили водой (30 мл) и экстрагировали этилацетатом (30 мл × 3). Объединенные органические слои промывали водой (30 мл × 2) и рассолом (30 мл × 2), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 0-20% этилацетат/петролейный эфир) с получением трет-бутил-4-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пиперидин-1-карбоксилата (400 мг, 47%) в виде черного масла. ЖХ-МС: m/z 467,0 (M+Na)+.
Стадия 3: 5-бром-3-метил-2-(пиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
В охлажденный (ледяная баня) раствор трет-бутил-4-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пиперидин-1-карбоксилата (800 мг, 1,1 ммоль) в этилацетате (5 мл) медленно добавляли HCl (4 M в этилацетате, 10 мл). Реакционную смесь перемешивали при 21°С в течение 16 часов, затем концентрировали и разбавляли водой (8 мл) и этилацетатом (8 мл). Слои разделяли и водную фазу промывали этилацетатом (8 мл × 3), затем лиофилизировали с получением 5-бром-3-метил-2-(пиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (230 мг, 56%) в виде желтого твердого вещества. ЖХ-МС: m/z 364,9 (М+Н)+.
Стадия 4: 1-(4-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-пиперидин-1-ил)этанон
К желтому раствору 5-бром-3-метил-2-(пиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (230 мг, 0,54 ммоль) в дихлорметане (5 мл) добавляли триэтиламин (163 мг, 0,224 мл, 1,61 ммоль). Реакционную смесь перемешивали при 21°С в течение 15 мин, затем добавляли по каплям уксусный ангидрид (66 мг, 0,06 мл, 0,64 ммоль). Реакционную смесь перемешивали при 21°С в течение 2 часов, затем гасили NaHCO3 (насыщ.) (10 мл) и экстрагировали дихлорметаном (10 мл × 3). Объединенные органические слои промывали водой (10 мл) и рассолом (10 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением рацемического 1-(4-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-пиперидин-1-ил)этанона (480 мг, выход более 100%) в виде коричневого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. ЖХ-МС: m/z 408,9 (M+Na)+.
Стадия 5: трет-бутил-3-(2-(1-ацетилпиперидин-4-ил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилат
Раствор 1-(4-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-пиперидин-1-ил)этанона (280 мг, 0,723 ммоль), трет-бутил-6-фтор-3-(4,4,5,5 тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индол-1-карбоксилата (261 мг, 0,723 ммоль), PdCl2(dppf)CH2Cl2 (54 мг, 0,07 ммоль) и K3PO4 (460 мг, 2,17 ммоль) в 1,4-диоксане (6 мл) и воде (1,5 мл) барботировали N2 в течение 1 минуты. Реакционную смесь перемешивали при 80°С в течение 16 часов, затем экстрагировали этилацетатом (20 мл × 3). Объединенные органические слои промывали водой (20 мл) и рассолом (20 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного трет-бутил-3-(2-(1-ацетилпиперидин-4-ил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилата, который использовали на следующей стадии без дополнительной очистки. ЖХ-МС: m/z 542,2 (М+Н)+.
Стадия 6: (+)-1-(4-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пиперидин-1-ил)этанон
В охлажденный (ледяная баня) раствор трет-бутил-3-(2-(1-ацетил пиперидин-4-ил)-3-метил-1,1-диоксидо-2,3-дигидробензо(с1]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (410 мг, 0,53 ммоль) в этилацетате (8 мл) медленно добавляли HCl (4 M в этилацетате, 8 мл). Реакционную смесь перемешивали при 21°С в течение 16 часов, затем нейтрализовали (рН=7-8) с использованием NaHCO3 (насыщ.) (6 мл) и экстрагировали этилацетатом (10 мл × 3). Объединенные органические слои промывали водой (10 мл) и рассолом (10 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенное масло очищали посредством колоночной хроматографии (силикагель, 0-5% метанол/дихлорметан) с получением рацемического 1-(4-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пиперидин-1-ил)этанона (220 мг, 94%) в виде коричневого твердого вещества. ЖХ-МС: m/z 442,2 (М+Н)+; 464,2 (M+Na)+. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (+)-1-(4-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пиперидин-1-ил)этанона в качестве первого элюируемого пика (35 мг, 20%) в виде бледно-желтого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (brs, 1Н), 7.95-7.79 (m, 5Н), 7.28-7.26 (m, 1Н), 7.04-6.99 (m, 1Н), 4.93-4.90 (m, 1Н), 4.47 (t, J=11,9 Гц, 1Н), 3.93-3.87 (m, 1Н), 3.82-3.79 (m, 1Н), 3.17-3.13 (m, 1Н), 2.67-2.58 (m, 1Н), 2.02 (s, 3Н), 1.98-1.68 (m, 4Н), 1.56 (d, J=4,5 Гц, 3Н); ЖХ-МС: m/z 442,1 (М+Н)+; [α]20D +60,9° (с=1,05 мг/мл, метанол).
Пример 72: (-)-1-(4-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пиперидин-1-ил)этанон
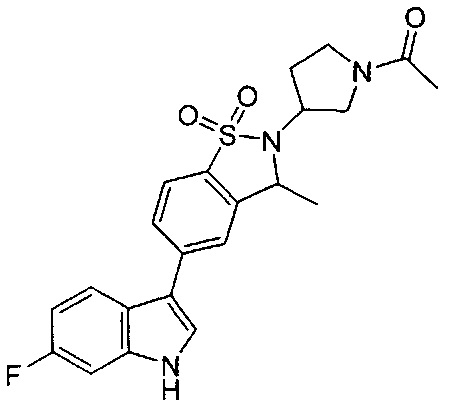
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 71, в виде бледно-желтого твердого вещества (50 мг, 28%). 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1Н), 7.95-7.79 (m, 5Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.04-6.99 (m, 1Н), 4.91 (q, J=6,1 Гц, 1Н), 4.47 (t, J=13,1 Гц, 1Н), 3.90-3.88 (m, 1Н), 3.80-3.76 (m, 1Н), 3.18-3.10 (m, 1Н), 2.67-2.59 (m, 1Н), 2.02 (d, J=2,5 Гц, 3Н),1.99-1.68 (m, 4Н), 1.56 (dd, J=2,4, 6,4 Гц, 3Н); ЖХ-МС: m/z 442,1 (М+Н)+; [α]20D -43,7° (с=1,03 мг/мл, метанол).
Пример 73: (+)-1-(3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пирролидин-1-ил)этанон (Пик 1)
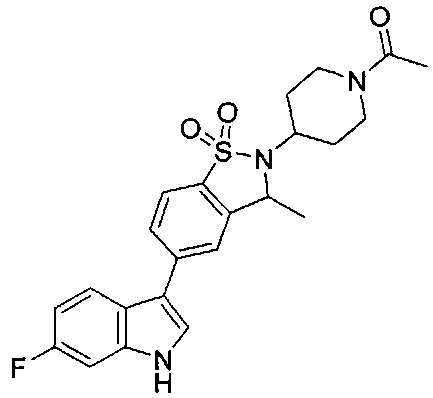
Следуя общей методике, изложенной в Примере 71, начиная с трет-бутил-3-гидроксипирролидин-1-карбоксилата, получили указанное в заголовке соединение в качестве первого элюируемого пика в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (s., 1Н), 7.96-7.89 (m, 4Н), 7.85-7.82 (m, 1H), 7.29-7.25 (m, 1H), 7.01 (dt, J=2,1, 9,1 Гц, 1H), 4.92-4.84 (m, 1H), 4.23-4.05 (m, 1H), 3.92-3.78 (m, 1H), 3.71-3.25 (m, 3Н), 2.47-2.27 (m, 2H), 1.97 (d, J=7,3 Гц, 3Н), 1.57 (dd, J=6,5, 9,8 Гц, 3Н); ЖХ-МС: m/z 428,1 (М+Н)+; [α]20D +1,9° (с=1,04 мг/мл, DMSO).
Пример 74: (+)-1-(3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пирролидин-1-ил)этанон
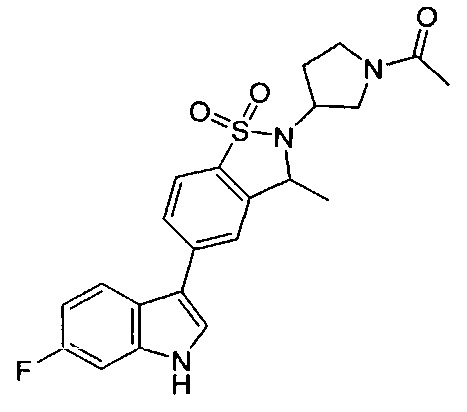
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 73 (48 мг, 6%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s, 1Н), 7.96-7.89 (m, 4Н), 7.84-7.83 (m, 1Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.02 (dt, J=2,3, 9,3 Гц, 1Н), 4.93-4.81 (m, 1Н), 4.22-4.06 (m, 1Н), 3.83-3.23 (m, 4Н), 2.39-2.17 (m, 2Н), 1.96 (d, J=1,3 Гц, 3Н), 1.61-1.50 (m, 3Н); ЖХ-МС: m/z 428,1 (М+Н)+; [α]20D +5,88° (с=1,02 мг/мл, DMSO).
Пример 75: (-)-1-(3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пирролидин-1-ил)этанон
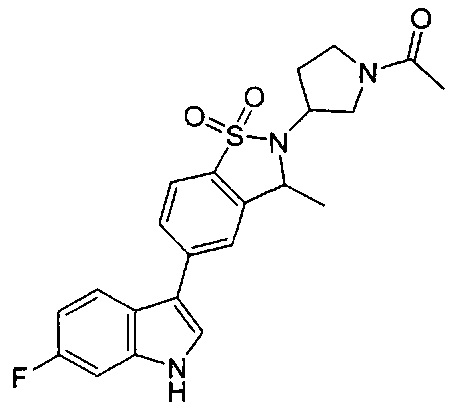
Указанное в заголовке соединение получили в качестве третьего элюируемого пика из хирального разделения, описанного в Примере 73 (52 мг, 6%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s, 1H), 7.96-7.89 (m, 4H), 7.85-7.82 (m, 1H), 7.28-7.26 (m, 1H), 7.01 (dt, J=2,3, 9,2 Гц, 1H), 4.92-4.84 (m, 1H), 4.23-4.05 (m, 1H), 3.92-3.78 (m, 1H), 3.72-3.25 (m, 3H), 2.39-2.22 (m, 2H), 1.97 (d, J=7,3 Гц, 3Н), 1.57 (dd, J=6,5, 9,5 Гц, 3Н); ЖХ-МС: m/z 428,1 (M+H)+; [α]20D -1,90° (c=1,05 мг/мл, DMSO).
Пример 76: (-)-1-(3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пирролидин-1-ил)этанон
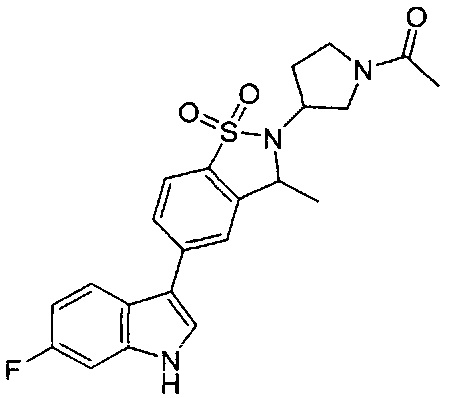
Указанное в заголовке соединение получили в качестве четвертого элюируемого пика из хирального разделения, описанного в Примере 73 (100 мг, 12%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1Н), 7.96-7.88 (m, 4Н), 7.85-7.83 (m, 1Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.02 (dt, J=2,3, 9,3 Гц, 1Н), 4.95-4.87 (m, 1Н), 4.22-4.04 (m, 1Н), 3.85-3.73 (m, 1Н), 3.67-3.45 (m, 2Н), 3.30-3.23 (m, 1Н), 2.44-2.17 (m, 2Н), 1.96 (d, J=1,3 Гц, 3Н), 1.56 (dd, J=6,7, 7,9 Гц, 3Н); ЖХ-МС: m/z 428,1 (М+Н)+; [α]20D -4,0° (с=1,00 мг/мл, DMSO).
Пример 77: (-)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(пиперидин-4-илметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
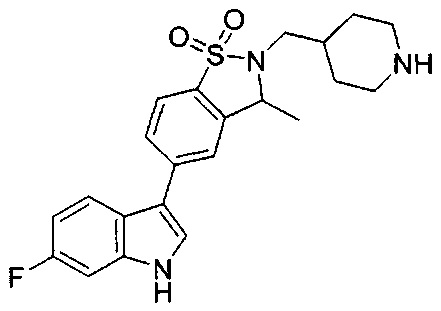
Следуя общей методике, изложенной в Примере 69, начиная с N-ВОС-4-пиперидинметанола, получили указанное в заголовке соединение в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.77 (br s, 1Н), 8.95 (d, J=10,0 Гц, 1Н), 8.70 (d, J=9,0 Гц, 1Н), 8.00-7.79 (m, 4Н), 7.28 (d, J=9,8 Гц, 1Н), 7.01 (t, J=9,2 Гц, 1H), 4.65 (q, J=6,3 Гц, 1H), 3.34-3.19 (m, 3Н), 3.08 (dd, J=8,8, 14,3 Гц, 1H), 2.96-2.78 (m, 2H), 2.09-1.89 (m, 3H),1.56 (d, J=6,5 Гц, 3Н), 1.48-1.30 (m, 2H); LCMS: m/z 414,0 (M+H)+, [α]20D -9,2° (c=0,4 мг/мл, DMSO).
Пример 78: (+)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(пиперидин-4-илметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
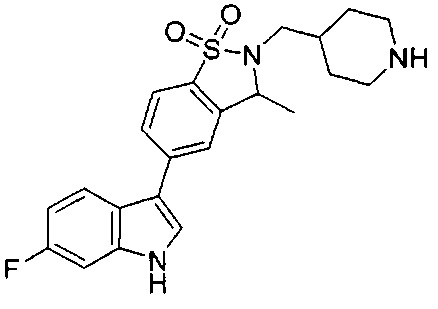
Указанное в заголовке соединение получили в качестве второго злюируемого пика из хирального разделения в Примере 78. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.76 (br s, 1Н), 8.89 (d, J=9,8 Гц, 1Н), 8.64 (d, J=8,5 Гц, 1Н), 7.97-7.88 (m, 4Н), 7.27 (dd, J=2,4, 9,9 Гц, 1Н), 7.01 (dt, J=2,3, 9,3 Гц, 1Н), 4.65 (q, J=6,1 Гц, 1Н), 3.34-3.19 (m, 3Н), 3.11-3.05 (m, 1Н), 2.95-2.79 (m, 2Н), 2.11-1.86 (m, 3Н), 1.56 (d, J=6,5 Гц, 3Н), 1.46-1.31 (m, 2Н); LCMS: m/z 414,0 (М+Н)+; [α]20D +33,3° (с=0,4 мг/мл, DMSO).
Пример 79: (-)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-((1-метилпиперидин-4-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
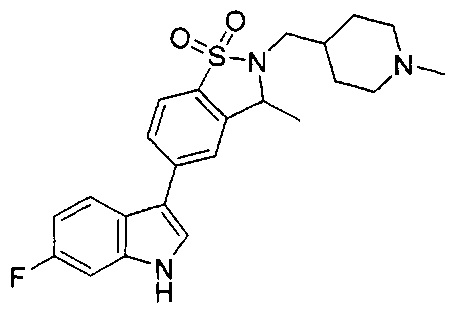
К белому раствору 5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(пиперидин-4-илметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (100 мг, 0,242 ммоль) в метаноле (5 мл) добавляли формальдегид (37% в воде, 98,1 мг, 1,21 ммоль) при 25°С. Реакционную смесь перемешивали при 25°С в течение 15 мин, затем охлаждали в ледяной бане и добавляли NaBH3CN (23 мг, 0,36 ммоль). Реакционную смесь нагревали до 25°С и перемешивали в течение 1 часа, затем концентрировали с получением рацемического 5-(6-фтор-1Н-индол-3-ил)-3-метил-2-((1-метил пиперидин-4-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением указанного в заголовке соединения в качестве первого элюируемого пика (46 мг, 44%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1Н), 7.96-7.81 (m, 5Н), 7.27 (dd, J=2,5, 9,8 Гц, 1Н), 7.01 (dt, J=2,5, 9,3 Гц, 1Н), 4.62 (q, J=6,4 Гц, 1Н), 3.20 (dd, J=5,9, 14,2 Гц, 1Н), 3.03 (dd, J=8,9, 13,9 Гц, 1Н), 2.86 (br s, 2Н), 2.24 (s, 3Н), 2.07-1.95 (m, 2Н), 1.86-1.69 (m, 3Н), 1.54 (d, J=6,5 Гц, 3Н), 1.32-1.17 (m, 2Н); LCMS: m/z 428,1 (M+H)+; [α]20D -8,3° (c=1,8 мг/мл, DMSO).
Пример 80: (+)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-((1-метилпиперидин-4-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
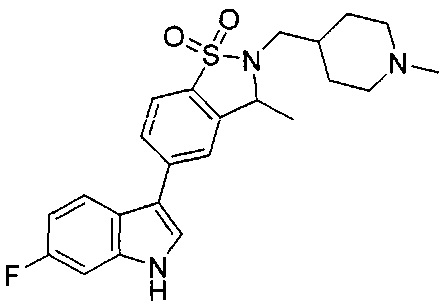
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 79 (42 мг, 41%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1Н), 7.98-7.79 (m, 5Н), 7.26 (d, J=9,5 Гц, 1Н), 7.01 (t, J=9,4 Гц, 1Н), 4.66-4.56 (m, 1Н), 3.20 (dd, J=5,4,14,4 Гц, 1Н), 3.09-2.98 (m, 1Н), 2.84 (br s, 2Н), 2.23 (s, 3Н), 1.99 (q, J=11,5 Гц, 2Н), 1.86-1.66 (m, 3Н), 1.54 (d, J=6,5 Гц, 3Н), 1.31-1.16 (m, 2Н); LCMS: m/z 428,1 (М+Н)+; [α]20D +7,1° (с=0,7 мг/мл, DMSO).
Пример 81: (+)-(5S)-5-((5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)метил)-5-метилпирролидин-2-он
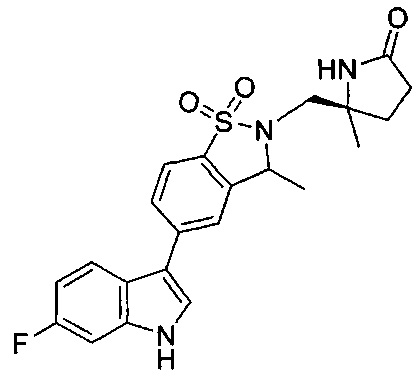
Следуя общей методике, изложенной в Примере 69, начиная с (S)-5-(гидроксиметил)-5-метилпирролидин-2-она, получили указанное в заголовке соединение в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) 6 [м.д.] 11.70 (br s, 1Н), 7.99-7.86 (m, 5Н), 7.76 (s, 1Н), 7.27 (dd, J=2,5, 9,8 Гц, 1Н), 7.01 (dt, J=2,1, 9,2 Гц, 1Н), 4.79 (q, J=6,4 Гц, 1Н), 3.29 (s, 1Н), 3.24-3.17 (m, 1Н), 2.30-2.14 (m, 3Н), 1.82-1.72 (m, 1Н), 1.56 (d, J=6,5 Гц, 3Н), 1.28 (s, 3Н); LCMS: m/z 427,9 [М+Н]+, LCMS: m/z 428,1 (М+Н)+, [α]20D +1,6° (с=4,2 мг/мл, DMSO).
Пример 82: (+)-(5S)-5-((5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)метил)-5-метилпирролидин-2-он
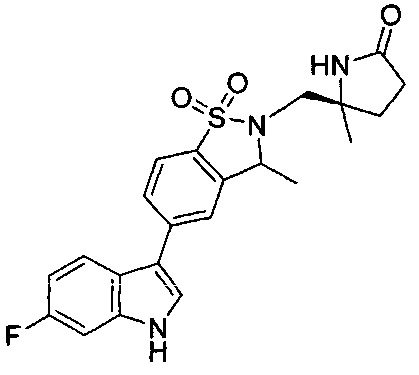
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения в Примере 81. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1Н), 7.97-7.85 (m, 5Н), 7.69 (s, 1Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.01 (dt, J=2,3, 9,3 Гц, 1Н), 4.83 (q, J=6,2 Гц, 1Н), 3.44 (d, J=15,3 Гц, 1Н), 3.15 (d, J=15,3 Гц, 1Н), 2.41-2.30 (m, 1Н), 2.23-2.11 (m, 2Н), 1.87-1.77 (m, 1Н), 1.55 (d, J=6,5 Гц, 3Н), 1.26 (s, 3Н); LCMS: m/z 427,9 (М+Н)+; [α]20D +24,7° (с=3,0 мг/мл, DMSO).
Пример 83: (-)-(5R)-5-((5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)метил)-5-метилпирролидин-2-он
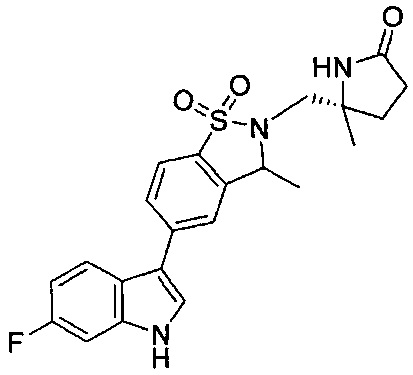
Следуя общей методике, изложенной в Примере 69, начиная с (R)-5-(гидроксиметил)-5-метилпирролидин-2-она, получили указанное в заголовке соединение в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 1.24-1.34 (s, 3Н), 1.57 (d, J=6,53 Гц, 3Н), 1.71-1.83 (m, 1Н), 2.14-2.31 (m, 3Н), 3.16-3.32 (m, 2Н), 4.79 (q, J=6,44 Гц, 1H), 7.02 (td, J=9,29, 2,51 Гц, 1H), 7.27 (dd, J=9,79, 2,26 Гц, 1H), 7.72-7.78 (m, 1Н), 7.84-8.03 (m, 5 Н), 11.70 (br s, 1H); ЖХ-МС: m/z 427,9 (М+Н)+; [α]20D -7,52° (с=0,62 мг/мл, DMSO).
Пример 84: (-)-(5R)-5-((5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)метил)-5-метилпирролидин-2-он
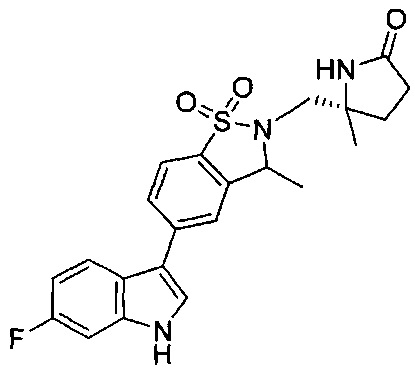
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 83. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 1.24-1.34 (s, 3Н), 1.57 (d, J=6,53 Гц, 3Н), 1.71-1.83 (m, 1Н), 2.14-2.31 (m, 3Н), 3.16-3.32 (m, 2Н), 4.79 (q, J=6,44 Гц, 1Н), 7.02 (td, J=9,29, 2,51 Гц, 1Н), 7.27 (dd, J=9,79, 2,26 Гц, 1Н), 7.72-7.78 (m, 1Н), 7.84-8.03 (m, 5Н), 11.70 (br s, 1Н); LCMS m/z 427,9 (М+1)+; [α]20D -97,3° (с=0,5 мг/мл, DMSO).
Пример 85: (-)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-((1-метил-1Н-1,2,4-триазол-3-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
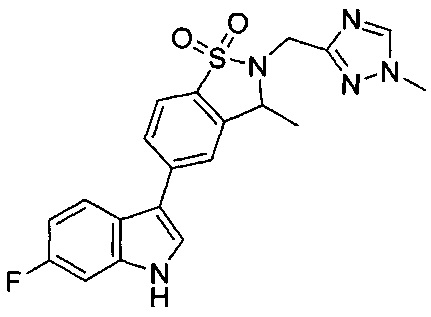
Следуя общей методике, изложенной в Примере 69, начиная с (1-метил-1Н-1,2,4-триазол-3-ил)метанола, получили указанное в заголовке соединение в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s, 1Н), 8.46 (s, 1Н), 7.98-7.83 (m, 5Н), 7.26 (dd, J=2,3, 9,8 Гц, 1Н), 7.00 (dt, J=2,5, 9,3 Гц, 1Н), 4.86 (q, J=6,4 Гц, 1Н), 4.51-4.39 (m, 2Н), 3.86 (s, 3Н), 1.54 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 411,8 (М+Н)+; [α]20D -15° (с=1,3 мг/мл, метанол).
Пример 86: (+)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-((1-метил-1H-1,2,4-триазол-3-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
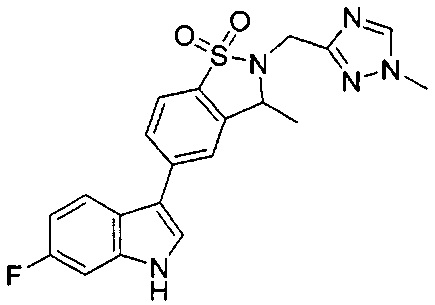
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 85. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s, 1Н), 8.46 (s, 1Н), 7.98-7.84 (m, 5Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.00 (dt, J=2,5, 9,3 Гц, 1Н), 4.86 (q, J=6,5 Гц, 1Н), 4.51-4.40 (m, 2Н), 3.86 (s, 3Н), 1.55 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 412,0 (М+Н)+; [α]20D +9° (с=1,3 мг/мл, метанол).
Пример 87: (-)-4-((5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)метил)-4-метилоксазолидин-2-он
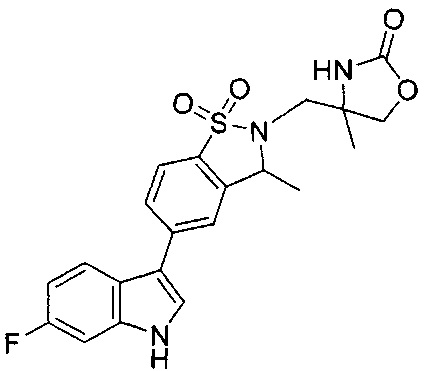
Следуя общей методике, изложенной в Примере 69, начиная с 4-(гидроксиметил)-4-метилоксазолидин-2-она, получили указанное в заголовке соединение в качестве первого элюируемого пика в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1Н), 7.97-7.85 (m, 5Н), 7.79 (s, 1Н), 7.27 (d, J=12,0 Гц, 1Н), 7.02 (t, J=9,5 Гц, 1Н), 4.89 (d, J=6,5 Гц, 1Н), 4.34 (d, J=9,0 Гц, 1Н), 4.05 (d, J=9,0 Гц, 1Н), 3.41 (s, 1Н), 3.28 (s, 1Н), 1.56 (d, J=6,5 Гц, 3Н), 1.33 (s, 3Н); ЖХ-МС: m/z 429,9 (М+Н)+; [α]20D -45° (с=1,3 мг/мл, метанол).
Пример 88: (-)-4-((5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)метил)-4-метилоксазолидин-2-он
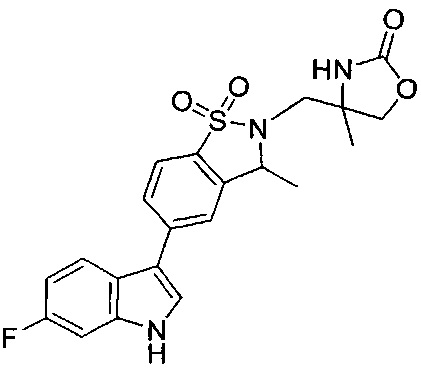
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 87 (50 мг, 25%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.71 (br s, 1Н), 7.99-7.85 (m, 6Н), 7.26 (d, J=10,0 Гц, 1Н), 7.01 (t. J=9,3 Гц, 1Н), 4.87-4.79 (m, 1Н), 4.40 (d, J=8,5 Гц, 1Н), 4.02 (d, J=8,5 Гц, 1Н), 3.39 (s, 1Н), 3.29 (s, 1Н), 1.57 (d, J=6,5 Гц, 3Н), 1.35 (s, 3Н); ЖХ-МС: m/z 429,9 (М+Н)+; [α]20D -2,38° (с=1,4 мг/мл, метанол).
Пример 89: (+)-4-((5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)метил)-4-метилоксазолидин-2-он
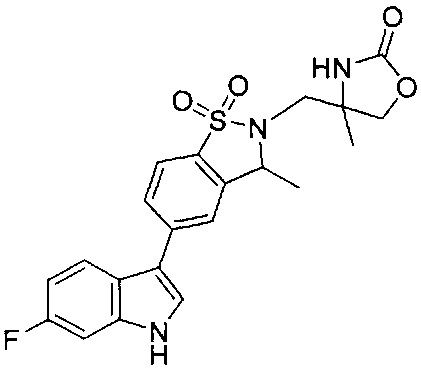
Указанное в заголовке соединение получили в качестве третьего элюируемого пика из хирального разделения, описанного в Примере 87 (23 мг, 12%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1Н), 7.97-7.86 (m, 5Н), 7.80 (s, 1Н), 7.27 (dd, J=2,4, 9,7 Гц, 1Н), 7.06-6.98 (m, 1Н), 4.93-4.85 (m, 1Н), 4.34 (d, J=8,8 Гц, 1Н), 4.05 (d, J=8,8 Гц, 1Н), 3.48-3.37 (m, 2Н), 1.56 (d, J=6,3 Гц, 3Н), 1.33 (s, 3Н); ЖХ-МС: m/z 430,0 (М+Н)+; [α]20D +50° (с=1,2 мг/мл, метанол).
Пример 90: (+)-4-((5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)метил)-4-метилоксазолидин-2-он
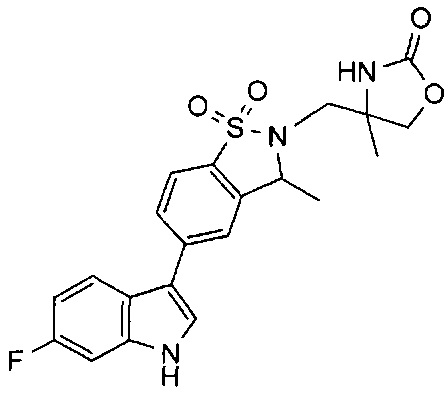
Указанное в заголовке соединение получили в качестве четвертого элюируемого пика из хирального разделения, описанного в Примере 87 (23 мг, выход 12%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1Н), 8.01-7.85 (m, 6Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.01 (dt, J=2,3, 9,2 Гц, 1Н), 4.83 (q, J=6,0 Гц, 1Н), 4.40 (d, J=9,0 Гц, 1Н), 4.02 (d, J=9,0 Гц, 1Н), 3.41-3.38 (m, 1Н), 3.30-3.24 (m, 1Н), 1.57 (d, J=6,5 Гц, 3Н), 1.36 (s, 3Н); ЖХ-МС: m/z 430,0 (М+Н)+; [α]20D +2,86° (с=1,4 мг/мл, метанол).
Пример 91: (-)-2-(азетидин-3-ил)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
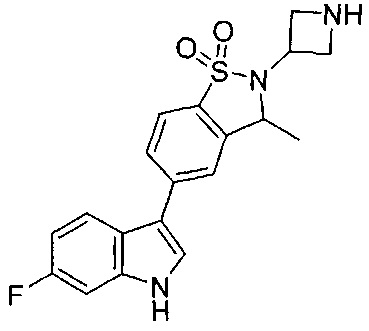
Следуя общей методике, изложенной в Примере 69, начиная с N-Вос-3-ОН-азетидина, получили указанное в заголовке соединение в качестве первого элюируемого пика в хиральном разделении в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.71 (br s, 1Н), 7.99-7.87 (m, 4Н), 7.86-7.80 (m, 1Н), 7.28 (dd, J=2,0, 10,0 Гц, 1Н), 7.07-6.98 (m, 1Н), 4.78 (d, J=6,5 Гц, 1Н), 4.32 (br s, 1Н), 4.22-3.69 (m, 3Н), 3.61 (br s, 2H), 1.54 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 393,9 (M+Na)+; [α]20D -35,3° (c=1,1 мг/мл, метанол).
Пример 92: (+)-2-(азетидин-3-ил)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
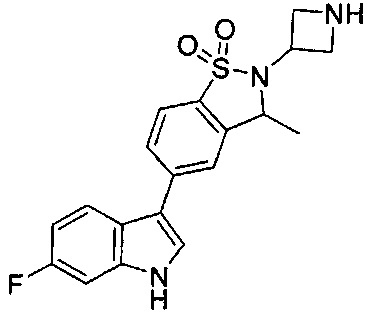
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 91, в виде белого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.94 (d, J=9,0 Гц, 1Н), 7.87 (dd, J=5,3, 8,8 Гц, 1Н), 7.83-7.78 (m, 2Н), 7.71 (s, 1Н), 7.17 (dd, J=2,3, 9,8 Гц, 1Н), 6.96 (dt, J=2,5, 9,3 Гц, 1Н), 4.80-4.74 (m, 1Н), 4.74-4.69 (m, 1Н), 4.68-4.62 (m, 2Н), 4.42-4.32 (m, 2Н), 1.62 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 394,0 (M+Na)+; [α]20D+7,5° (с=1,07 мг/мл, метанол).
Пример 93: (-)-2-((R)-2,3-дигидроксипропил)-5-(6-фтор-1H-индол-3-ил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
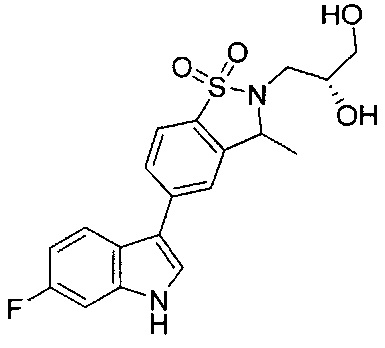
Стадия 1: 5-бром-2-(((R)-2,2-диметил-1,3-диоксолан-4-ил)метил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Суспензию 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (500 мг, 1,91 ммоль), (S)-4-(хлорметил)-2,2-диметил-1,3-диоксолана (287 мг, 1,91 ммоль) и K2CO3 (527 мг, 3,81 ммоль) в DMF (6 мл) перемешивали при 90°С в течение 48 часов. Реакционную смесь охлаждали до 25°С и вливали в воду (100 мл), затем экстрагировали этилацетатом (25 мл × 3). Объединенные органические слои промывали рассолом (15 мл × 2), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 30% петролейный эфир/этилацетат) с получением указанного в заголовке соединения (234 мг, 54%).
Стадия 2: трет-бутил-3-(2-((2,2-диметил-1,3-диоксолан-4-ил)метил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилат
К суспензии 5-бром-2-((2,2-диметил-1,3-диоксолан-4-ил)метил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (234 мг, 0,622 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (385 мг, 0,746 ммоль) и Cs2CO3 (405 мг, 1,24 ммоль) в диоксане (9 мл) и H2O (3 мл) добавляли PdCl2(dppf) (46 мг, 0,06 ммоль). Реакционную смесь перемешивали при 80°С в атмосфере N2 в течение 6 часов, затем твердые вещества отфильтровывали. Фильтрат концентрировали с получением неочищенного трет-бутил-3-(2-((2,2-диметил-1,3-диоксолан-4-ил)метил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилата, который использовали на следующей стадии без дополнительной очистки.
Стадия 3: (-)-2-((R)-2,3-дигидроксипропил)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К раствору трет-бутил-3-(2-((2,2-диметил-1,3-диоксолан-4-ил)метил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилата (320 мг, 0,602 ммоль) в дихлорметане (10 мл) добавляли трифторуксусную кислоту (5 мл). Реакционную смесь перемешивали при 30°С в течение 2 часов, затем концентрировали. Осадок разбавляли этилацетатом (20 мл) и H2O (15 мл). Смесь нейтрализовали NaHCO3 (насыщ.) до рН 7. Слои разделяли и органический слой сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, дихлорметан/метанол от 1/0 до 5/1) и препаративной ВЭЖХ с получением рацемического 2-((R)-2,3-дигидроксипропил)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидробензо[d]-изотиазол-1,1-диоксида. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением указанного в заголовке соединения в качестве первого элюируемого пика (20 мг, 9%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.92-7.85 (m, 2Н), 7.83-7.78 (m, 2Н), 7.69 (s, 1Н), 7.16 (dd, J=2,4, 9,7 Гц, 1Н), 6.95 (dt, J=2,5, 9,2 Гц, 1Н), 4.77-4.73 (m, 1Н), 4.60 (br s, 1H), 4.04-3.97 (m, 1H), 3.71-3.53 (m, 3H), 1.65 (d, J=6,3 Гц, 3Н); ЖХ-МС: m/z 390,9 (M+H)+; [α]20D -26,4° (c=0,43 мг/мл, DMSO).
Пример 94: (+)-2-((R)-2,3-дигидроксипропил)-5-(6-фтор-1Н- индол-3-ил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
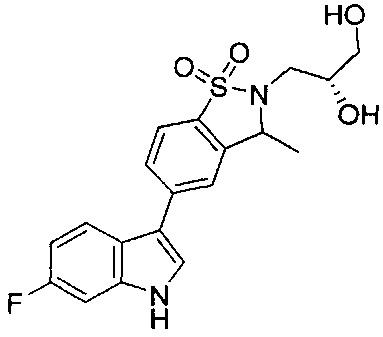
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 93 (40 мг, 18%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.92-7.84 (m, 2Н), 7.82-7.78 (m, 2Н), 7.68 (s, 1Н), 7.16 (dd, J=2,4, 9,7 Гц, 1Н), 6.95 (dt, J=2,4, 9,2 Гц, 1Н), 4.60 (br s, 1Н), 4.08-4.01 (m, 1Н), 3.68-3.59 (m, 2Н), 3.52 (dd, J=5,4, 14,7 Гц, 1Н), 3.39-3.33 (m, 1Н), 1.65 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 390,9 (М+Н)+, [α]20D =+36,1° (с=0,61 мг/мл, DMSO).
Пример 95: (-)-2-((S)-2,3-дигидроксипропил)-5-(6-фтор-1H-индол-3-ил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
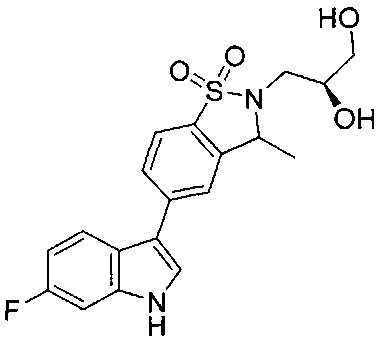
Следуя общей методике, изложенной в Примере 93, начиная с (R)-(+)-4-(хлорметил)-2,2-диметил-1,3-диоксолана, получили указанное в заголовке соединение в качестве первого элюируемого пика в хиральном разделении в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s, 1Н), 7.99-7.82 (m, 5Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.01 (dt, J=2,3, 9,2 Гц, 1Н), 5.02 (d, J=5,0 Гц, 1Н), 4.93 (q, J=6,2 Гц, 1Н), 4.73 (t, J=5,5 Гц, 1Н), 3.88-3.78 (m, 1Н), 3.50-3.41 (m, 3Н), 3.19-3.06 (m, 1Н), 1.56 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 390,9 (М+Н)+; [α]20D -16° (с=1,0 мг/мл, DMSO).
Пример 96: (+)-2-((S)-2,3-дигидроксипропил)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
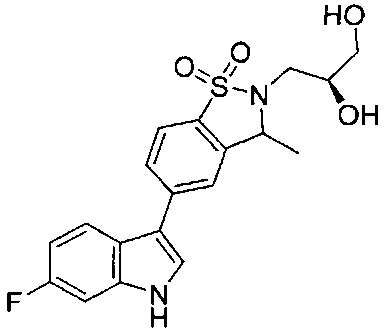
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 95 (65 мг, 32%), в виде желтого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s, 1Н), 7.99-7.81 (m, 5Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.01 (dt, J=2,5, 9,3 Гц, 1Н), 4.93 (d, J=5,0 Гц, 1Н), 4.75 (q, J=6,5 Гц, 1Н), 4.69 (t, J=5,5 Гц, 1Н), 3.83-3.74 (m, 1Н), 3.45-3.40 (m, 3Н), 3.19-3.06 (m, 1Н), 1.56 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 390,9; (М+Н)+; [α]20D +9,5° (с=1,05 мг/мл, DMSO).
Пример 97: (+)-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-гидрокси-N-метилпропанамид
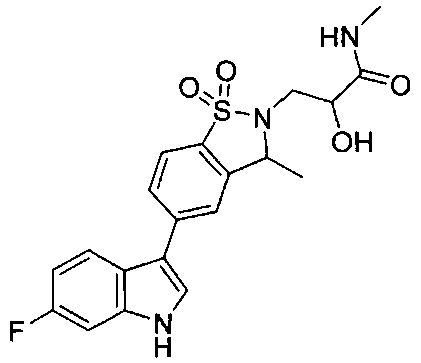
Стадия 1: метил-оксиран-2-карбоксилат
Метилакрилат (19 г, 220 ммоль) добавляли к 8% водному раствору NaOCl (300 мл) при 0°С. Бифазную смесь интенсивно перемешивали при 0°С в течение 30 мин, затем нагревали до 40°С в течение 2 часов. Реакционную смесь охлаждали до 25°С, затем экстрагировали дихлорметаном (200 мл × 2). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного метил-оксиран-2-карбоксилата (9 г, 39%) в виде прозрачного масла. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 3.79 (s, 3Н), 3.46 (dd, J=2,6, 4,1 Гц, 1Н), 3.02-2.90 (m, 2Н).
Стадия 2: метил-3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-гидроксипропаноат
Суспензию 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (3 г, 11 ммоль), метил-оксиран-2-карбоксилата (8,2 г, 80 ммоль) и Cs2CO3 (11 г, 34 ммоль) в DMF (30 мл) перемешивали при 80°С в течение 16 часов. Реакционную смесь охлаждали и разбавляли водой (100 мл) и экстрагировали этилацетатом (150 мл × 3). Объединенные органические слои промывали рассолом (200 мл × 3), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 30% этилацетат/петролейный эфир) с получением метил-3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-2-гидроксипропаноата (1,5 г, 36%) в виде черного масла.
Стадия 3: 3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-2-гидрокси-N-метилпропанамид
Желтый раствор метил-3-(5-бром-3-метил-1,1-диоксидобензо[d]-изотиазол-2(3Н)-ил)-2-гидроксипропаноата (1,5 г, 4,1 ммоль) в MeNH2 (30% в EtOH, 30 мл) перемешивали при 50°С в течение 3 часов. Реакционную смесь концентрировали с получением неочищенного 3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-2-гидрокси-N-метилпропанамида (1,8 г, более 100%) в виде черного масла, которое использовали непосредственно на следующей стадии.
Стадия 4: трет-бутил-6-фтор-3-(2-(2-гидрокси-3-(метиламино)-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата
Суспензию 3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-2-гидрокси-N-метилпропанамида (600 мг, 1,65 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индол-1-карбоксилата (895 мг, 2,48 ммоль), PdCl2(dppf) (121 мг, 0,165 ммоль) и K3PO4 (701 мг, 3,30 ммоль) в диоксане (12 мл) и H2O (4 мл) перемешивали при 80°С в атмосфере N2 в течение 2 часов. Реакционную смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (20 мл × 2). Объединенные органические слои промывали рассолом (20 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, этилацетат) с получением трет-бутил-6-фтор-3-(2-(2-гидрокси-3-(метиламино)-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (390 мг, 46%) в виде желтого твердого вещества.
Стадия 5: (+)-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-2-гидрокси-N-метилпропанамид
Раствор трет-бутил-6-фтор-3-(2-(2-гидрокси-3-(метиламино)-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (390 мг, 0,754 ммоль) и трифторуксусной кислоты (2 мл) в дихлорметане (10 мл) перемешивали при 20°С в атмосфере N2 в течение 2 часов. Реакционную смесь концентрировали, затем очищали посредством колоночной хроматографии (силикагель, этилацетат) с получением смеси диастереомеров. Диастереомеры разделяли посредством препаративной хиральной СФХ с получением (+)-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-гидрокси-N-метилпропанамида в качестве первого элюируемого пика (30 мг, 10%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1Н), 8.02-7.82 (m, 6Н), 7.27 (dd, J=2,5, 9,8 Гц, 1Н), 7.01 (dt, J=2,4, 9,2 Гц, 1Н), 6.04 (d, J=6,0 Гц, 1Н), 4.97 (q, J=6,4 Гц, 1Н), 4.30-4.17 (m, 1Н), 3.68 (dd, J=2,6, 15,4 Гц, 1Н), 3.28 (d, J=6,5 Гц, 1Н), 2.64 (d, J=4,5 Гц, 3Н), 1.53 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 440,0 (M+Na)+; [α]20D +20,3° (с=4 мг/мл, DMSO).
Пример 98: (-)-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-гидрокси-N-метилпропанамид
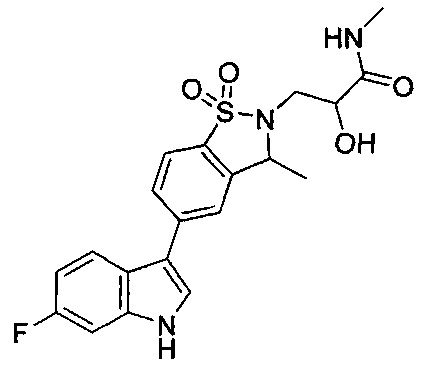
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 97 (30 мг, 10%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s, 1Н), 8.03-7.79 (m, 6Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.01 (dt, J=2,5, 9,3 Гц, 1Н), 6.05 (d, J=6,0 Гц, 1Н), 4.98 (q, J=6,4 Гц, 1Н), 4.29-4.18 (m, 1Н), 3.69 (dd, J=2,9, 15,2 Гц, 1Н), 3.28 (dd, J=8,5, 15,1 Гц, 1Н), 2.64 (d, J=4,8 Гц, 3Н), 1.53 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 440,2 (M+Na)+; [α]20D -8° (с=1 мг/мл, DMSO).
Пример 99: (+)-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-2-гидрокси-N-метилпропанамид
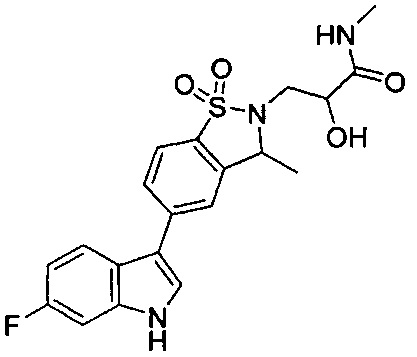
Указанное в заголовке соединение получили в качестве третьего элюируемого пика из хирального разделения, описанного в Примере 97 (33 мг, 10%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s, 1Н), 8.04-7.80 (m, 6Н), 7.27 (dd, J=2,5, 9,8 Гц, 1Н), 7.01 (dt, J=2,4, 9,2 Гц, 1Н), 5.94 (d, J=5,5 Гц, 1Н), 4.74 (q, J=6,3 Гц, 1Н), 4.18 (t, J=8,0 Гц, 1Н), 3.66 (dd, J=3,3, 14,6 Гц, 1Н), 3.27 (dd, J=7,5, 14,6 Гц, 1Н), 2.63 (d, J=4,5 Гц, 3Н), 1.56 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 440,0 (M+Na)+; [α]20D +19,25° (с=4 мг/мл, DMSO).
Пример 100: (-)-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-гидрокси-N-метилпропанамид
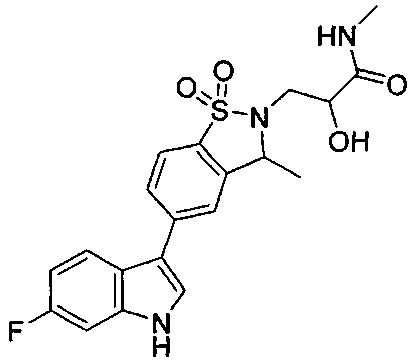
Указанное в заголовке соединение получили в качестве четвертого элюируемого пика из хирального разделения, описанного в Примере 97 (33 мг, 10%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1Н), 8.00-7.78 (m, 6Н), 7.27 (dd, J=2,4, 9,9 Гц, 1Н), 7.01 (dt, J=2,4, 9,3 Гц, 1Н), 5.95 (d, J=5,5 Гц, 1Н), 4.74 (q, J=6,7 Гц, 1Н), 4.17 (t, J=8,3 Гц, 1Н), 3.64 (dd, J=3,4, 14,7 Гц, 1H), 3.29-3.22 (m, 1H), 2.62 (d, J=4,8 Гц, 3Н), 1.55 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 440,0 (M+Na)+; [α]20D -7° (c=4 мг/мл, DMSO).
Пример 101: (-)-2-амино-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамид
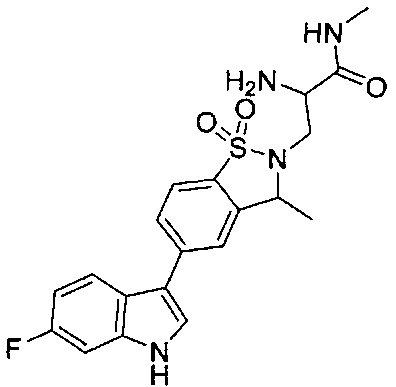
Стадия 1: 5-бром-2-(2-гидроксиэтил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К раствору метил-3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-гидроксипропаноата (Пример 207, 500 мг, 1,38 ммоль) и TEA (418 мг, 4,13 ммоль) в DCM (10 мл) добавляли MsCl (315 мг, 2,75 ммоль). Реакционную смесь перемешивали при 20°С в течение 16 часов, затем вливали в воду (10 мл) и экстрагировали DCM (10 × 2). Объединенные органические слои промывали рассолом (10 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного 5-бром-2-(2-гидроксиэтил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида в виде желтого масла, которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2: 2-азидо-3-(5-бром-3-метил-1,1-диоксидобензоЭДизотиазол-2(3Н)-ил)-N-метилпропанамид
К раствору метил-3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-гидроксипропаноата (600 мг, 1,36 ммоль) в DMF (10 мл) добавляли NaN3 (106 мг, 1,20 ммоль). Реакционную смесь перемешивали при 80°С в течение 16 часов, затем вливали в воду (20 мл) и экстрагировали DCM (20 мл × 2). Объединенные органические слои промывали рассолом (20 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного 2-азидо-3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н) ил)-N-метилпропанамида (450 мг) в виде желтого масла, которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 3: трет-бутил-(3-(5-бром-3-метил-1,1-диоксидобензо[d]-изотиазол-2(3Н)-ил)-1-(метиламино)-1-оксопропан-2-ил)-карбамат
Раствор метил-2-азидо-3-(5-бром-3-метил-1,1-диоксидобензо[d]-изотиазол-2(3Н)-ил)пропаноата (450 мг, 1,16 ммоль) и PPh3 (1210 мг, 4,62 ммоль) в THF (20 мл) и воде (0,5 мл) перемешивали при 60°С в течение 16 часов. К полученному желтому раствору добавляли TEA (468 мг, 4,63 ммоль) и (Вос)2O (757 мг, 3,47 ммоль). Реакционную смесь перемешивали при 20°С в течение 16 часов, затем вливали в воду (50 мл) и экстрагировали EtOAc (50 мл × 2). Объединенные органические слои промывали рассолом (50 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением трет-бутил-(3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-1-(метиламино)-1-оксопропанил)карбамата (250 мг, 47%) в виде желтого твердого вещества.
Стадия 4: хиральный трет-бутил-3-(2-(2-((трет-бутоксикарбонил)амино)-3-(метиламино)-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилат
К суспензии трет-бутил-(3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-1-(метиламино)-1-оксопропан-2-ил)-карбамата (200 мг, 0,433 ммоль) и трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индол-1-карбоксилата (172 мг, 0,476 ммоль) в смеси диоксан/вода (8 мл/3 мл) добавляли Pd(dppf)Cl2 (32 мг, 0,04 ммоль) и K3PO4 (184 мг, 0,865 ммоль). Реакционную смесь перемешивали при 80°С в течение 2 часов, затем концентрировали и очищали посредством колоночной хроматографии с получением трет-бутил-3-(2-(2-((трет-бутоксикарбонил)-амино)-3-(метиламино)-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилата (270 мг) в виде смеси диастереомеров. Смесь очищали посредством препаративной хиральной СФХ с получением четырех продуктов: (пик 1 (60 мг), пик 2 (60 мг), пик 3 (30 мг) и пик 4 (40 мг), каждый в виде желтого твердого вещества.
Стадия 5: (-)-2-амино-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамид
Раствор трет-бутил-3-(2-(2-((трет-бутоксикарбонил)амино)-3-(метиламино)-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]-изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (пик 1, 60 мг, 0,097 ммоль) в HCl/EtOAc (3 мл) перемешивали при 20°С в течение 20 часов. Реакционную смесь концентрировали, затем очищали посредством препаративной ВЭЖХ с получением (-)-2-амино-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамида (14 мг, 34%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.77 (br s, 1Н), 8.69 (br s, 1Н), 8.42 (br s, 2H), 7.99-7.88 (m, 4H), 7.33-7.26 (m, 1H), 7.06-6.99 (m, 1H), 4.76-4.68 (m, 1H), 4.06-3.98 (m, 1H), 3.70-3.62 (m, 1H), 3.60-3.53 (m, 1H), 2.69 (d, J=4,5 Гц, 3Н), 2.54 (s, 1H), 1.61 (d, J=6,3 Гц, 3Н); ЖХ-МС: m/z 439,1 (M+Na)+; [α]20D -4,09° (c=0,001 г/мл, DMSO).
Пример 102: (+)-2-амино-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамид
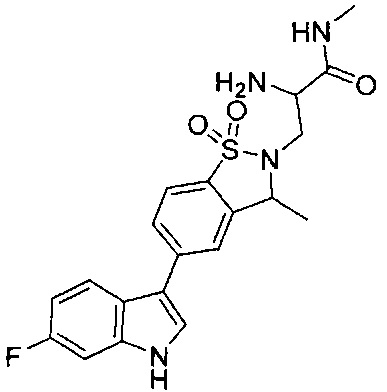
Раствор трет-бутил-3-(2-(2-((трет-бутоксикарбонил)амино)-3-(метиламино)-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]-изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилата (60 мг, 0,097 ммоль) (Пример 211, стадия 4, пик 2) в HCl/EA (3 мл) перемешивали при 20°С в течение 20 часов. Реакционную смесь концентрировали, затем очищали посредством препаративной ВЭЖХ с получением (+)-2-амино-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамида (9 мг, 23%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.77 (br s, 1Н), 8.69 (br s, 1Н), 8.42 (br s, 2Н), 7.99-7.88 (m, 4Н), 7.33-7.26 (m, 1Н), 7.06-6.99 (m, 1Н), 4.76-4.68 (m, 1Н), 4.06-3.98 (m, 1Н), 3.70-3.62 (m, 1Н), 3.60-3.53 (m, 1H), 2.69 (d, J=4,5 Гц, 3Н), 2.54 (s, 1H), 1.61 (d, J=6,3 Гц, 3Н); ЖХ-МС: m/z 439,0 (M+Na)+; [α]20D 0,8° (c=0,0075 г/мл, DMSO).
Пример 103: (-)-2-амино-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамид
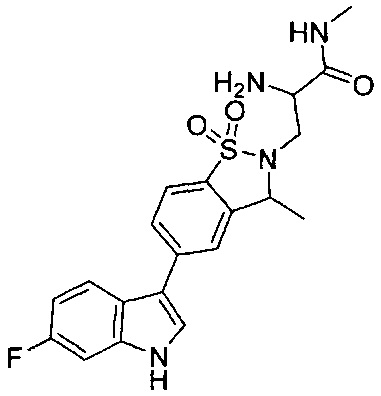
Раствор трет-бутил-3-(2-(2-((трет-бутоксикарбонил)амино)-3-(метиламино)-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]-изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилата (30 мг, 0,05 ммоль) (Пример 101, стадия 4, пик 3) в HCl/EA (3 мл) перемешивали при 20°С в течение 20 часов. Реакционную смесь перемешивали в течение еще 16 часов, затем концентрировали и очищали посредством препаративной ВЭЖХ с получением (-)-2-амино-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамида (7 мг, 36%) в виде бледно-желтого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.81 (br s, 1Н), 8.69 (d, J=4,5 Гц, 1Н), 8.61-8.45 (m, 2Н), 8.00-7.86 (m, 5Н), 7.27 (s, 1Н), 7.06-6.98 (m, 1Н), 4.94-4.86 (m, 1Н), 4.07-3.97 (m, 1Н), 3.73-3.57 (m, 2Н), 2.66 (d, J=4,5 Гц, 3Н), 1.54 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 438,9 (M+Na)+; [α]20D -21° (с=0,001 г/мл, DMSO).
Пример 104: (+)-2-амино-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамид
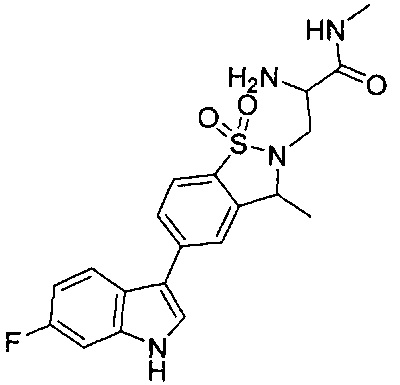
Раствор трет-бутил-3-(2-(2-((трет-бутоксикарбонил)амино)-3-(метиламино)-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]-изотиазол-5-ил)-6-фтор-1H-индол-карбоксилата (40 мг, 0,07 ммоль) (Пример 101, стадия 4, пик 4) в HCl/EA (3 мл) перемешивали при 20°С в течение 20 часов. Реакционную смесь перемешивали в течение еще 16 часов, затем концентрировали и очищали посредством препаративной ВЭЖХ с получением (+)-2-амино-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамида (12 мг, 43%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.80 (s, 1Н), 8.72-8.63 (m, 1Н), 8.50 (br s, 2Н), 7.99-7.87 (m, 5Н), 7.32-7.26 (m, 1Н), 7.06-6.99 (m, 1Н), 4.93-4.84 (m, 1Н), 4.06-3.97 (m, 1Н), 3.73-3.58 (m, 2Н), 2.67 (d, J=4,5 Гц, 3Н), 1.54 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 438,9 (M+Na)+; [α]20D +10,67° (с=0,001 г/мл, DMSO).
Пример 105: (+)-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-метокси-N-метилпропанамид
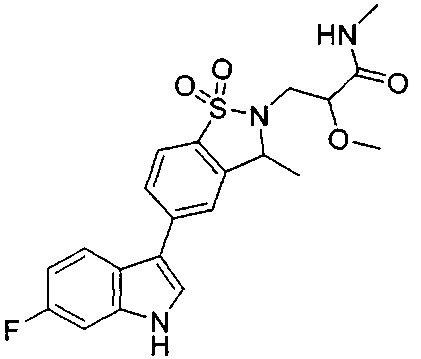
Стадия 1: N-2,2-триметил-1,3-диоксолан-4-карбоксамид
К раствору метил-2,2-диметил-1,3-диоксолан-4-карбоксилата (5,0 г, 31 ммоль) в EtOH (50 мл) добавляли MeNH2 (30% в EtOH, 9,7 г, 94 ммоль).
Реакционную смесь перемешивали при 25°С в течение 16 часов, затем концентрировали. Осадок растворяли в MeNH2 (30% в EtOH, 30 мл), затем переносили в герметично закрываемую пробирку и перемешивали при 50°С в течение 3 часов. Неочищенную реакционную смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (40 мл × 2). Объединенные органические слои промывали рассолом (20 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением N,2,2-триметил-1,3-диоксолан-4-карбоксамида (2,5 г, 50%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 6.60 (br s, 1Н), 4.49 (dd, J=5,5, 7,5 Гц, 1Н), 4.29 (t, J=8,3 Гц, 1Н), 4.09 (dd, J=5,3, 8,8 Гц, 1Н), 2.86 (d, J=5,0 Гц, 1Н), 1.49 (s, 3Н), 1.39 (s, 3Н). Стадия 2: 2,3-дигидрокси-N-метилпропанамид
К раствору N,2,2-триметил-1,3-диоксолан-4-карбоксамида (2,5 г, 16 ммоль) в дихлорметане (40 мл) добавляли трифторуксусную кислоту (20 мл). Реакционную смесь перемешивали при 25°С в течение 2 часов, затем концентрировали с получением 2,3-дигидрокси-N-метилпропанамида (2 г, более 100%) в виде черного масла, которое использовали непосредственно на следующей стадии. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 4.13-4.01 (m, 1Н), 3.82-3.63 (m, 2Н), 2.78 (s, 3Н).
Стадия 3: 3-((трет-бутилдифенилсили)окси)-2-гидрокси-N-метилпропанамид
К раствору 2,3-дигидрокси-N-метилпропанамида (2,0 г, 17 ммоль) в DMF (20 мл) добавляли имидазол (3,5 г, 52 ммоль) и трет-бутилдифенилсилилхлорид (TBDPSCI) (7,1 г, 26 ммоль). Реакционную смесь перемешивали при 25°С в течение 16 часов, затем разбавляли водой (60 мл) и экстрагировали этилацетатом (100 мл × 2). Объединенные органические слои промывали рассолом (150 мл × 3), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 40% этилацетат/петролейный эфир) с получением 3-((трет-бутилдифенилсилил)окси)-2-гидрокси-N-метилпропанамида (2,2 г, выход 36%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 7.64 (dd, J=1,4, 7,9 Гц, 4Н), 7.49-7.36 (m, 6Н), 6.77 (br s, 1Н), 4.22-4.14 (m, 1Н), 3.90 (dd, J=2,8, 5,8 Гц, 2Н), 3.25 (d, J=4,3 Гц, 1Н), 2.87 (d, J=5,0 Гц, 3Н), 1.08 (s, 9Н).
Стадия 4: 3-((трет-бутилдифенилсилил)окси)-2-метокси-N-метилпропанамид
К желтому раствору 3-((трет-бутилдифенилсилил)окси)-2-гидрокси-N-метилпропанамида (2,2 г, 6,2 ммоль) в безводном THF (20 мл) добавляли Mel (4,4 г, 31 ммоль) и Ag2O (11,4 г, 49,2 ммоль). Реакционную смесь перемешивали при 25°С в течение 16 часов, затем переносили в герметично закрываемую пробирку и добавляли Mel (20 г, 141 ммоль). Реакционную смесь герметично закрывали и перемешивали при 40°С в течение 16 часов. Полученную желтую суспензию фильтровали и фильтрат концентрировали, затем очищали посредством колоночной хроматографии (силикагель, 40-50% этилацетат/петролейный эфир) с получением 3-((трет-бутилдифенилсилил)-окси)-2-метокси-N-метилпропанамида (1,5 г, выход 66%) в виде желтого масла.
Стадия 5: 3-гидрокси-2-метокси-N-метилпропанамид
К желтому раствору 3-((трет-бутилдифенилсилил)окси)-2-метокси-N-метилпропанамида (2,2 г, 5,92 ммоль) в безводном THF (10 мл) добавляли фторид тетра-н-бутиламмония (TBAF) (1М в THF, 23,7 мл г, 23,7 ммоль). Реакционную смесь перемешивали при 25°С в течение 16 часов, затем разбавляли водой (20 мл) и экстрагировали этилацетатом (40 мл × 3). Объединенные органические слои промывали рассолом (30 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 40-80% этилацетат/петролейный эфир) с получением 3-гидрокси-2-метокси-N-метилпропанамида (300 мг, 38%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 6.71 (br s, 1Н), 3.87-3.82 (m, 2Н), 3.80-3.71 (m, 1Н), 3.33-3.20 (m, 1Н), 2.86 (d, J=5,0 Гц, 3Н).
Стадия 6: 2-метокси-3-(метиламино)-3-оксопропилметансульфонат
К желтому раствору 3-гидрокси-2-метокси-N-метилпропанамида (370 мг, 2,78 ммоль) в безводном дихлорметане (10 мл) добавляли DIPEA (718 мг, 5,56 ммоль) и MsCl (477 мг, 4,17 ммоль). Реакционную смесь перемешивали при 25°С в течение 1 часа, затем концентрировали, разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл × 3). Объединенные органические слои промывали водой (10 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением неочищенного 2-метокси-3-(метиламино)-3-оксопропилметансульфоната (350 мг, 60%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 6.73 (br s, 1Н), 4.60 (d, J=11,2 Гц, 1Н), 4.47 (dd, J=4,4, 11,2 Гц, 1Н), 3.94 (s, 1Н), 3.54 (s, 3Н), 3.04 (s, 3Н), 2.88 (d, J=5,2 Гц, 3Н).
Стадия 7: 3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-метокси-N-метилпропанамид
Раствор 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (434 мг, 1,7 ммоль), 2-метокси-3-(метиламино)-3-оксопропилметансульфоната (350 мг, 1,66 ммоль) и K2СО3 (458 мг, 3,31 ммоль) в DMSO (10 мл) перемешивали при 80°С в течение 2 часов. Неочищенную реакционную смесь разбавляли рассолом (20 мл) и экстрагировали этилацетатом (30 мл × 2). Объединенные органические слои промывали рассолом (20 мл × 3), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, этилацетат) с получением 3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-метокси-N-метилпропанамида (130 мг, выход 21%) в виде черного масла.
Стадия 8: трет-бутил-6-фтор-3-(2-(2-метокси-3-(метиламино)-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилат
Красную суспензию 3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-2-метокси-N-метилпропанамида (200 мг, 0,530 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (249 мг, 0,689 ммоль), PdCl2(dppf) (39 мг, 0,05 ммоль) и K3PO4 (225 мг, 1,06 ммоль) в диоксане (8 мл) и H2O (2 мл) перемешивали при 80°С в атмосфере N2 в течение 2 часов. Смесь разбавляли водой (15 мл) и экстрагировали этилацетатом (20 мл × 3). Объединенные органические слои промывали рассолом (15 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 80-100% этилацетат/петролейный эфир) с получением трет-бутил-6-фтор-3-(2-(2-метокси-3-(метиламино)-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (220 мг, выход 78%) в виде черного масла.
Стадия 9: (+)-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-метокси-N-метилпропанамид
К раствору трет-бутил-6-фтор-3-(2-(2-метокси-3-(метиламино)-3-оксопропил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (220 мг, 0,414 ммоль) в дихлорметане (10 мл) добавляли трифторуксусную кислоту (2 мл). Реакционную смесь перемешивали при 20°С в течение 16 часов, затем концентрировали и нейтрализовали до рН 7 с помощью NaHCO3 (насыщ.). Смесь экстрагировали этилацетатом (40 мл × 3) и объединенные органические слои промывали рассолом (15 мл), сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 80%-100% этилацетат/петролейный эфир) с получением смеси диастереомеров (150 мг). Диастереомеры разделяли посредством препаративной хиральной СФХ с получением (+)-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]-изотиазол-2(3H)-ил)-2-метокси-N-метилпропанамида в качестве первого элюируемого пика (28 мг, 16%) в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1Н), 8.10 (d, J=4,8 Гц, 1Н), 8.00-7.82 (m, 5Н), 7.27 (dd, J=2,4, 9,9 Гц, 1Н), 7.01 (dt, J=2,4, 9,1 Гц, 1Н), 4.81 (q, J=6,2 Гц, 1Н), 3.93 (dd, J=3,8, 7,8 Гц, 1Н), 3.59-3.51 (m, 1Н), 3.44 (d, J=5,5 Гц, 1Н), 3.33 (s, 3Н), 2.65 (d, J=4,5 Гц, 3Н), 1.54 (d, 7=6,5 Гц, 3Н); ЖХ-МС: m/z 432,1(М+Н)+; [α]20D +6,75° (с=4 мг/мл, DMSO).
Пример 106: (-)-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-метокси-N-метилпропанамид
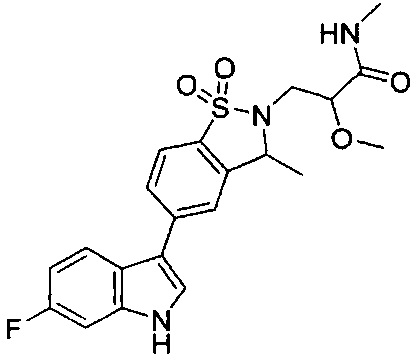
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 105 (21 мг, 12%), в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s, 1Н), 8.10 (d, J=4,8 Гц, 1Н), 8.05-7.76 (m, 5Н), 7.27 (dd, J=2,4, 9,9 Гц, 1Н), 7.01 (dt, J=2,4, 9,2 Гц, 1Н), 4.81 (q, J=6,5 Гц, 1Н), 3.94 (dd, J=3,5, 7,8 Гц, 1Н), 3.61-3.50 (m, 1Н), 3.43 (d, J=7,8 Гц, 1Н), 3.33 (s, 3Н), 2.65 (d, J=4,8 Гц, 3Н), 1.54 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 432,1 (М+Н)+; [α]20D -7° (с=4 мг/мл, DMSO).
Пример 107: (+)-3-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-2-метокси-N-метилпропанамид
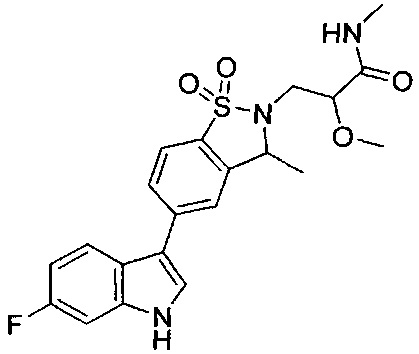
Указанное в заголовке соединение получили в качестве третьего элюируемого пика из хирального разделения, описанного в Примере 105 (21 мг, 12%), в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1Н), 8.06 (d, J=4,5 Гц, 1Н), 7.99-7.81 (m, 5Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.01 (dt, J=2,5, 9,3 Гц, 1Н), 4.71 (q, J=6,3 Гц, 1Н), 3.90 (dd, J=3,5, 7,3 Гц, 1Н), 3.62 (dd, J=3,5, 15,1 Гц, 1Н), 3.43 (d, J=2,5 Гц, 1Н), 3.36 (s, 3Н), 2.64 (d, J=4,8 Гц, 3Н), 1.54 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 432,2 (М+Н)+; [α]20D +3,25° (с=4 мг/мл, DMSO).
Пример 108: (-)-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-метокси-N-метилпропанамид
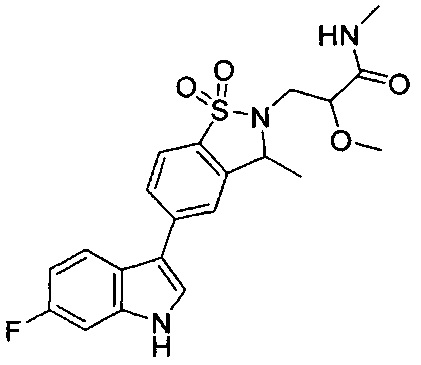
Указанное в заголовке соединение получили в качестве четвертого элюируемого пика из хирального разделения, описанного в Примере 105 (11 мг, 6%), в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1Н), 8.06 (d, J=4,8 Гц, 1Н), 7.98-7.81 (m, 5Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.01 (dt, J=2,4, 9,2 Гц, 1Н), 4.71 (q, J=6,3 Гц, 1Н), 3.90 (dd, J=3,4, 7,4 Гц, 1Н), 3.62 (dd, J=3,6, 14,9 Гц, 1Н), 3.48-3.47 (m, 1Н), 3.35 (s, 3Н), 2.64 (d, J=4,8 Гц, 3Н), 1.54 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 432,1 (М+Н)+; [α]20D -4,75° (с=4 мг/мл, DMSO).
Пример 109: (-)-этил-(2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)карбамат
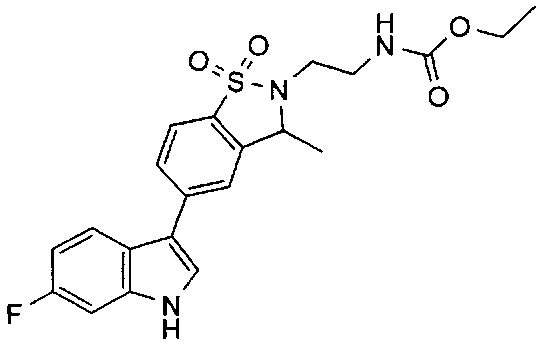
Стадия 1: трет-бутил-(2-(5-бром-3-метил-1,1-диоксидобензо-[d]изотиазол-2(3Н)-ил)этил)карбамат
Смесь 5-бром-3-метил-2,3-дигидро-1,2-бензотиазол-1,1-диоксида (300 мг, 1,14 ммоль), трет-бутил-н-(2-бромэтил)карбамата (282 мг, 1,26 ммоль) и K2CO3 (316 мг, 2,29 ммоль) в DMF (5,7 мл) барботировали N2 в течение 1 мин, затем перемешивали при 80°С в течение 16 часов. Реакционную смесь разбавляли этилацетатом (10 мл) и водой (10 мл). Слои разделяли и водный слой экстрагировали этилацетатом (10 мл × 2). Объединенные органические слои промывали NaHCO3 (насыщ.) (10 мл) и рассолом (10 мл), затем сушили (Na2SO4), фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 4-60% этилацетат/петролейный эфир) с получением трет-бутил-(2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)-карбамата (410 мг, 88%) в виде желтой смолы. ЖХ-МС: m/z 428,8 (M+Na)+.
Стадия 2: трет-бутил-3-(2-(2-((трет-бутоксикарбонил)амино)этил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилат
Коричневую смесь трет-бутил-(2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)карбамата (300 мг, 0,74 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (382 мг, 0,74 ммоль), PdCl2(dppf)-CH2Cl2 (55,3 мг, 0,074 ммоль) и K3PO4 (471 мг, 2,22 ммоль) в 1,4-диоксане (7,4 мл) барботировали N2 в течение 1 мин, затем перемешивали при 80°С в течение 5 часов. Реакционную смесь концентрировали и очищали посредством колоночной хроматографии (силикагель, 4-60% этилацетат/петролейный эфир) с получением трет-бутил-3-(2-(2-((трет-бутоксикарбонил)амино)этил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилата (367 мг, 87%) в виде желтой смолы. ЖХ-МС: m/z 582,0 (M+Na)+.
Стадия 3: 2-(2-аминоэтил)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К желтому раствору трет-бутил-3-(2-(2-((трет-бутокси карбон ил)-амино)этил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (245 мг, 0,44 ммоль) в CH2Cl2 (5 мл) добавляли трифторуксусную кислоту (3 мл) при 0°С.Реакционный раствор перемешивали при 25°С в течение 3 часов, разбавляли метанолом и концентрировали с получением неочищенного 2-(2-аминоэтил)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (288 мг, 100%) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии.
Стадия 4: этил-3-(2-(2-((этоксикарбонил)амино)этил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилат
В охлажденный (ледяная баня) раствор 2-(2-аминоэтил)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (157 мг, 0,44 ммоль) в CH2Cl2 (8 мл) добавляли Et3N (0,183 мл, 1,31 ммоль), а затем этилхлорформиат (110 мг, 1,01 ммоль). Реакционную смесь нагревали до 25°С и перемешивали в течение 3 часов. Реакционный раствор гасили водой (10 мл) и экстрагировали этилацетатом (10 мл × 2). Объединенные органические слои концентрировали и очищали посредством колоночной хроматографии (силикагель, 4-100% этилацетат/петролейный эфир) с получением этил-3-(2-(2-((этоксикарбонил)амино)-этил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]-изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилата (174 мг, 79%) в виде желтого твердого вещества. ЖХ-МС: m/z 526,0 (M+Na)+.
Стадия 5: (-)-этил-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этил)карбамат
К желтой смеси этил-3-(2-(2-((этоксикарбонил)амино)этил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилата (174 мг, 0,35 ммоль) в EtOH (8 мл) добавляли K2CO3 (95,5 мг, 0,69 ммоль). Реакционную смесь перемешивали при 25°С в течение 3 часов. Неочищенную реакционную смесь фильтровали и промывали метанолом, затем концентрировали. Неочищенный осадок очищали на колонке (силикагель, 5-100% этилацетат/петролейный эфир) с получением рацемического этил-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)-карбамата (74 мг, 50%) в виде желтого твердого вещества. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (-)-этил-(2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-этил)карбамата в качестве первого элюируемого пика (15 мг, 15%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.93-7.84 (m, 2Н), 7.83-7.77 (m, 2Н), 7.68 (s, 1Н), 7.16 (dd, J=2,0, 9,5 Гц, 1Н), 6.99-6.91 (m, 1Н), 4.73 (d, J=6,5 Гц, 1Н), 4.59 (s, 1Н), 4.08 (q, J=7,2 Гц, 2Н), 3.50-3.41 (m, 4Н), 1.62 (d, J=6,5 Гц, 3Н), 1.22 (t, J=7,0 Гц, 3Н); ЖХ-МС: m/z 432,0 (М+Н)+; [α]20D -5° (с=1,2 мг/мл, метанол).
Пример 110: (+)-этил-(2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо [d]изотиазол-2(3Н)-ил)этил)карбамат
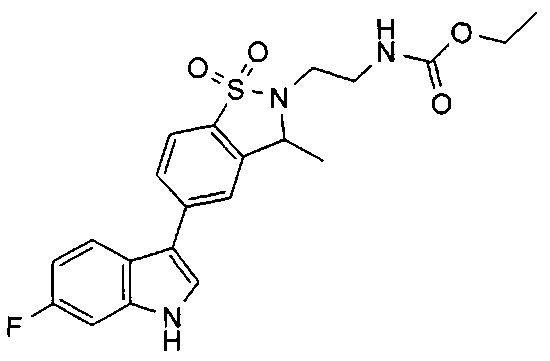
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 109 (25 мг, 24%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.92-7.82 (m, 2Н), 7.81-7.75 (m, 2Н), 7.67 (s, 1Н), 7.16 (dd, J=2,0, 10,0 Гц, 1Н), 6.94 (dt, J=2,0, 9,3 Гц, 1Н), 4.71 (d, J=6,5 Гц, 1Н), 4.60 (s, 1Н), 4.08 (q, J=7,0 Гц, 2Н), 3.48-3.41 (m, 4Н), 1.61 (d, J=6,5 Гц, 3Н), 1.22 (t, J=7,0 Гц, 3Н); ЖХ-МС: m/z 432,0 (М+Н)+; [α]20D +5,56° (с=3,6 мг/мл, метанол).
Пример 111: (-)-2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилацетамид
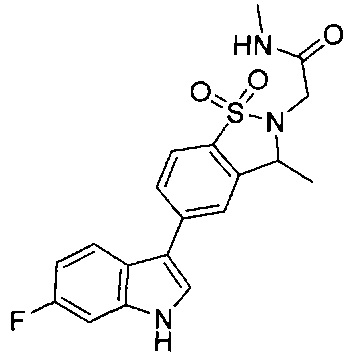
Стадия 1: трет-бутил-2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)ацетат
В коричневый раствор 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (1,2 г, 4,6 ммоль) в DMF (20 мл) добавляли трет-бутил-2-бромацетат (1,1 г, 5,5 ммоль) и K2СО3 (1,27 г, 9,16 ммоль), затем реакционную смесь барботировали N2 в течение 1 мин и перемешивали при 80°С в течение 16 часов. Реакционную смесь разбавляли этилацетатом (20 мл) и водой (20 мл). Слои разделяли и водный слой подвергали обратной экстракции этилацетатом (30 мл × 2). Объединенные органические слои промывали рассолом (10 мл × 2), затем сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 2-20% этилацетат/петролейный эфир) с получением трет-бутил-2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)ацетата (1,38 г, 80%) в виде черного твердого вещества.
Стадия 2: трет-бутил-2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)ацетат
К черному раствору трет-бутил-2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)ацетата (1,38 г, 3,67 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (1,57 г, 3,70 ммоль) и K3PO4 (1,43 г, 6,72 ммоль) в смеси диоксан/Н2О (30 мл/10 мл) добавляли Pd (dppf)Cl2 (246 мг, 0,336 ммоль) при 28°С в атмосфере N2. Реакционную смесь перемешивали при 90°С в течение примерно 6 часов, затем разбавляли этилацетатом (40 мл). Слои разделяли и органический слой промывали H2O (10 мл) и рассолом (15 мл × 2), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 10-40% этилацетат/петролейный эфир) с получением трет-бутил-2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-ацетата (597 мг, выход 38%) в виде желтой смолы.
Стадия 3: 2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)уксусная кислота
Желтый раствор трет-бутил-2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)ацетата (597 мг, 1,39 ммоль) в трифторуксусной кислоте (12 мл) перемешивали при 25°С в течение 71 часа. Реакционную смесь концентрировали с получением неочищенной 2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)уксусной кислоты (560 мг, более 100%) в виде зеленого твердого вещества, которое использовали непосредственно на следующей стадии.
Стадия 4: (-)-2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилацетамид
Раствор 2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]-изотиазол-2(3H)-ил)уксусной кислоты (175 мг, 0,467 ммоль), HCl соли метанамина (97 мг, 0,94 ммоль) и DIPEA (0,326 мл, 1,87 ммоль) в безводном DMF (5 мл) перемешивали при 25°С в течение 5 мин, затем добавляли HATU (267 мг, 0,701 ммоль). Реакционную смесь перемешивали при 25°С в течение 3 часов, затем разбавляли этилацетатом (30 мл) и промывали водой (10 мл) и рассолом (10 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали, затем очищали посредством колоночной хроматографии (силикагель, 30-100% этилацетат/петролейный эфир) с получением рацемического продукта (135 мг). Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (-)-2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилацетамида в качестве первого элюируемого пика (29 мг, выход 16%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1Н), 8.04-7.82 (m, 6Н), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.01 (dt, J=2,4, 9,2 Гц, 1Н), 4.92 (d, J=6,5 Гц, 1Н), 3.96-3.80 (m, 2Н), 2.63 (d, J=4,5 Гц, 3Н), 1.54 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 409,9 (M+Na)+; [α]20D -7,11° (с=1,97 мг/мл, DMSO).
Пример 112: (+)-2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилацетамид
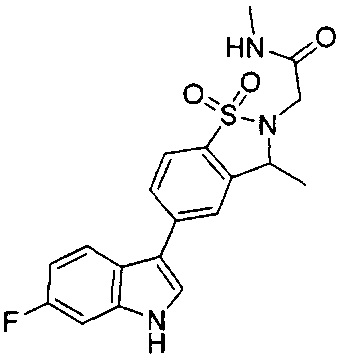
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 111 (16 мг, 9%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1Н), 8.09-7.80 (m, 6Н), 7.27 (dd, J=2,4, 9,9 Гц, 1Н), 7.01 (dt, J=2,5, 9,2 Гц, 1Н), 4.92 (d, J=6,5 Гц, 1Н), 3.89 (d, J=3,3 Гц, 2Н), 2.63 (d, J=4,8 Гц, 3Н), 1.54 (d, J=6,5 Гц, 3Н); LCMS: m/z 409,8 (M+Na)+; [α]20D +9,32° (c=2,11 мг/мл, DMSO).
Пример 113: (-)-2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N,N-диметилацетамид
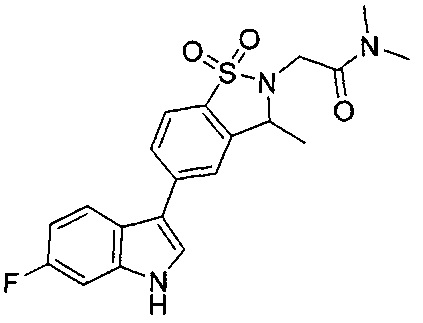
В охлажденный (ледяная баня) раствор 2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)уксусной кислоты (260 мг, 0,260 ммоль) и HCl соли диметиламина (42 мг, 0,51 ммоль) в безводном DMF (5 мл) добавляли DIPEA (133 мг, 1,03 ммоль). Реакционную смесь перемешивали на ледяной бане в течение 5 мин, затем добавляли HATU (147 мг, 0,385 ммоль) и перемешивание продолжали при 25°С в течение 2 часов. Реакционную смесь разбавляли этилацетатом (10 мл) и H2O (20 мл), затем слои разделяли. Водную фазу подвергали обратной экстракции этилацетатом (10 мл × 3) и объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 1-5% метанол/дихлорметан) и дополнительно очищали посредством препаративной ТСХ (этилацетат) с получением рацемического 2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]-изотиазол-2(3H)-ил)-N,N-диметилацетамида (76 мг, 74%) в виде желтой смолы. Энантиомеры разделяли посредством СФХ с получением (-)-2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N,N-диметилацетамида в качестве первого элюируемого пика (19 мг, 28%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s, 1H), 8.03-7.78 (m, 5H), 7.27 (dd, J=2,0, 9,8 Гц, 1H), 7.01 (dt, J=2,3, 9,3 Гц, 1H), 4.98 (q, J=6,4 Гц, 1H), 4.31-4.06 (m, 2H), 3.08-2.99 (m, 3H), 2.84 (s, 3H), 1.50 (d, J=6,8 Гц, 3H); ЖХ-МС: m/z 424,1 (M+Na)+; [α]20D -30,7° (c=2,9 мг/мл, DMSO).
Пример 114: (+)-2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N,N-диметилацетамид
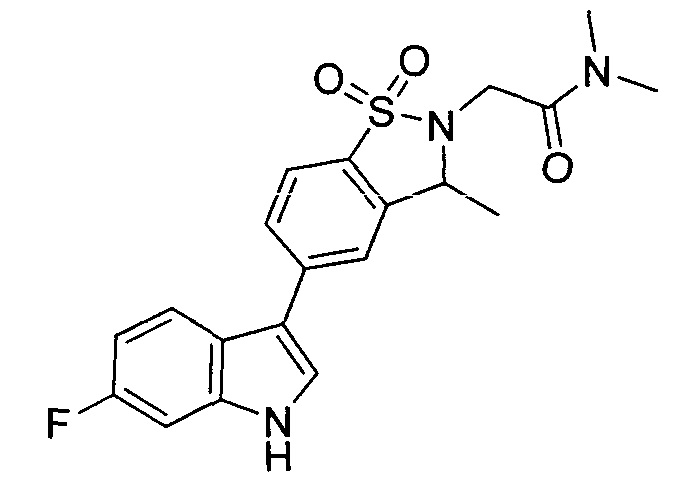
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 113 (10 мг, 15%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s, 1H), 8.00-7.82 (m, 5H), 7.27 (dd, J=2,0, 9,8 Гц, 1H), 7.01 (dt, J=2,3, 9,3 Гц, 1H), 4.96 (q, J=6,4 Гц, 1H), 4.31-4.06 (m, 2H), 3.08-2.99 (m, 3H), 2.84 (s, 3H), 1.54 (d, J=6,8 Гц, 3Н); LCMS: m/z 424,0 (M+Na)+; [α]20D +21,9° (c=2,55 мг/мл, DMSO).
Пример 115: (+)-2-(5-(6-фториндолин-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)ацетамид
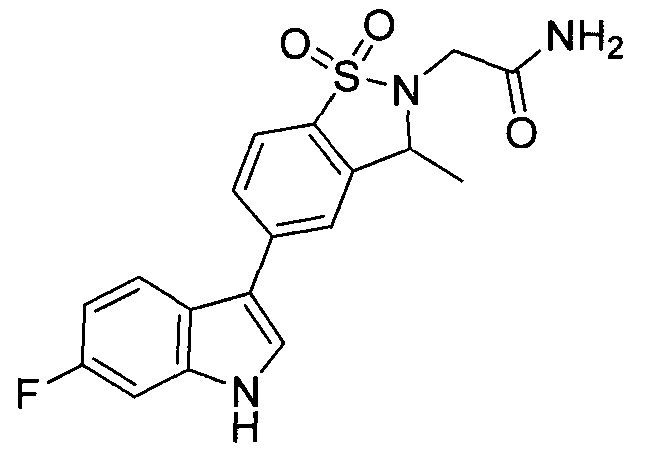
Светло-зеленый раствор 2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)уксусной кислоты (300 мг, 0,30 ммоль), твердого NH4Cl (32 мг, 0,59 ммоль) и DIPEA (153 мг, 1,19 ммоль) в безводном DMF (5 мл) перемешивали при 25°С в течение 3 мин, затем добавляли HATU (169 мг, 0,445 ммоль). Реакционную смесь перемешивали при 25°С в течение 3 часов, затем разбавляли этилацетатом (10 мл) и Н2О (20 мл). Слои разделяли и водную фазу подвергали обратной экстракции этилацетатом (10 мл × 3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 50-100% петролейный эфир/этилацетат) с получением рацемического продукта. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением 2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)ацетамида в качестве первого элюируемого пика (17 мг, 28%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1H), 7.99-7.83 (m, 5H), 7.45 (br s, 1H), 7.33-7.21 (m, 2H), 7.07-6.96 (m, 1H), 4.96 (q, J=6,3 Гц, 1H), 3.94-3.81 (m, 2H), 1.54 (d, J=6,3 Гц, 3H); LCMS: m/z 396,1 (M+Na)+; [α]20D +12,2° (c=2,45 мг/мл, DMSO).
Пример 116: (-)-2-(5-(6-фториндолин-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)ацетамид
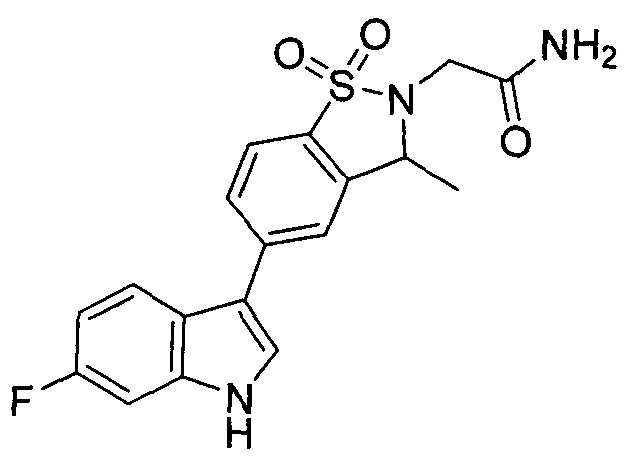
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 115 (18 мг, 30%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s, 1H), 8.00-7.82 (m, 5H), 7.44 (br s, 1H), 7.34-7.19 (m, 2H), 7.01 (t, J=8,9 Гц, 1H), 4.96 (q, J=6,0 Гц, 1H), 3.95-3.80 (m, 2H), 1.54 (d, J=6,3 Гц, 3H); LCMS: m/z 396,1 (M+Na)+; [α]20D -13,6° (c=2,55 мг/мл, DMSO).
Пример 117: (-)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(2-морфолиноэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
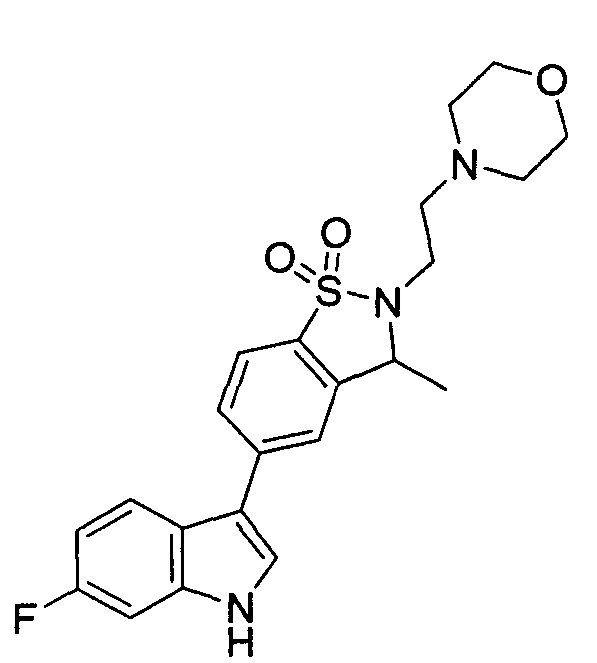
Стадия 1: 2-хлор-1-морфолиноэтанон
К раствору 2-хлорацетилхлорида (1,0 г, 8,8 ммоль) и морфолина (771 мг, 8,85 ммоль) в безводном дихлорметане (10 мл) добавляли триэтиламин (1,34 г, 13,3 ммоль). Реакционную смесь перемешивали при 25°С в течение 1 часа, затем концентрировали и очищали посредством колоночной хроматографии (силикагель, 30% этилацетат/петролейный эфир) с получением 2-хлор-1-морфолиноэтанона (1,1 г, 96%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 4.07 (s, 2H), 3.72 (td, J=4,9, 10,0 Гц, 4Н), 3.67-3.60 (m, 2H), 3.59-3.48 (m, 2H).
Стадия 2: 2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-1-морфолиноэтанон
Раствор 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (300 мг, 1,14 ммоль), 2-хлор-1-морфолиноэтанона (225 мг, 1,37 ммоль) и K2CO3 (316 мг, 2,29 ммоль) в DMSO (5 мл) перемешивали при 80°С в течение 18 часов. Полученную суспензию разбавляли рассолом (20 мл) и экстрагировали этилацетатом (20 мл × 2). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 50-100% этилацетат/петролейный эфир) с получением 2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-1-морфолиноэтанона (270 мг, выход 61%) в виде желтого масла.
Стадия 3: 5-бром-3-метил-2-(2-морфолиноэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К раствору 2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-1-морфолиноэтанона (270 мг, 0,694 ммоль) в безводном THF (10 мл) добавляли ВН3-THF (2,77 мл, 2,77 ммоль). Реакционную смесь перемешивали при 30°С в течение 48 часов, затем добавляли дополнительное количество ВН3-THF (2,77 мл, 2,77 ммоль) и перемешивание продолжали при 70°С в течение 16 часов. Реакционную смесь гасили 2 н. HCl и перемешивали при 40°С в течение 1 часа, затем разбавляли водой (10 мл) и нейтрализовали до рН 7 твердым NaHCO3. Смесь экстрагировали этилацетатом (20 мл × 2) и объединенные органические слои промывали рассолом (20 мл), сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, этилацетат) с получением 5-бром-3-метил-2-(2-морфолиноэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (250 мг, 96%) в виде желтого масла.
Стадия 4: трет-бутил-6-фтор-3-(3-метил-2-(2-морфолиноэтил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилат
Красную суспензию 5-бром-3-метил-2-(2-морфолиноэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (250 мг, 0,666 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (361 мг, 0,799 ммоль), PdCl2(dppf) (39 мг, 0,05 ммоль) и K3PO4 (226 мг, 1,7 ммоль) в диоксане (8 мл) и H2O (2 мл) перемешивали при 80°С в атмосфере N2 в течение 2 часов. Полученную смесь разбавляли водой (15 мл) и экстрагировали этилацетатом (20 мл × 3). Объединенные органические слои промывали рассолом (15 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 80% этилацетат/петролейный эфир) с получением трет-бутил-6-фтор-3-(3-метил-2-(2-морфолиноэтил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (180 мг, 64%) в виде желтого масла.
Стадия 5: (-)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(2-морфолиноэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К желтому раствору трет-бутил-6-фтор-3-(3-метил-2-(2-морфолиноэтил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (180 мг, 0,340 ммоль) в дихлорметане (8 мл) добавляли трифторуксусную кислоту (2 мл). Реакционную смесь перемешивали при 25°С в течение 1 часа, затем концентрировали и нейтрализовали до рН 7 с использованием NaHCO3 (насыщ.). Смесь экстрагировали этилацетатом (30 мл × 2) и объединенные органические слои промывали рассолом (20 мл), сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 3% метанол/дихлорметан) с получением рацемического продукта (130 мг) в виде желтого масла. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (-)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(2-морфолиноэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида в качестве первого элюируемого пика (40 мг, 27%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1H), 7.99-7.79 (m, 5H), 7.27 (dd, J=2,5, 9,8 Гц, 1H), 7.01 (dt, J=2,4, 9,2 Гц, 1H), 4.78 (q, J=6,4 Гц, 1H), 3.58 (t, J=4,6 Гц, 4H), 3.38-3.31 (m, 2H), 2.73-2.55 (m, 2H), 2.46 (d, J=5,8 Гц, 4Н), 1.55 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 430,1 (M+H)+; [α]20D -3,6° (с=1,1 мг/мл, метанол).
Пример 118: (+)-(6-фтор-1Н-индол-3-ил)-3-метил-2-морфолиноэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
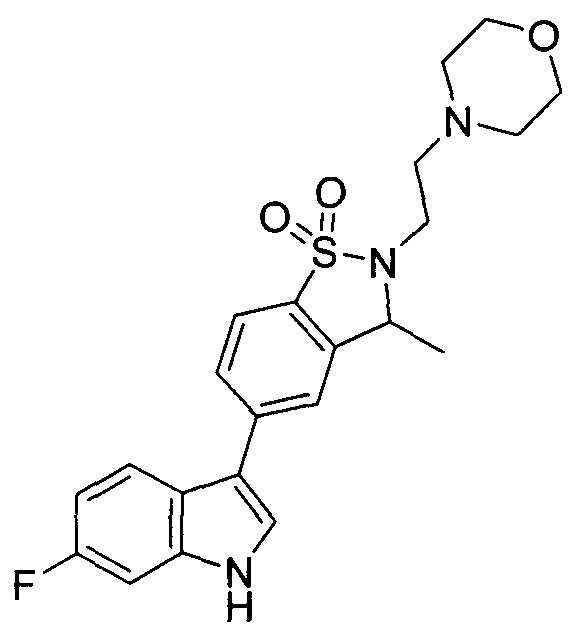
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 117 (45 мг, 31%), в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s, 1H), 8.02-7.79 (m, 5H), 7.27 (d, J=9,8 Гц, 1H), 7.01 (t, J=9,0 Гц, 1H), 4.78 (d, J=6,3 Гц, 1H), 3.58 (br s, 4H), 3.35-3.26 (m, 2H), 2.70-2.55 (m, 2H), 2.49-2.37 (m, 4H), 1.55 (d, J=6,0 Гц, 3Н); ЖХ-МС: m/z 430,0 (M+H)+; [α]20D +4,6° (c=1,3 мг/мл, метанол).
Пример 119: (-)-4-(-5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пиперидин-2-он
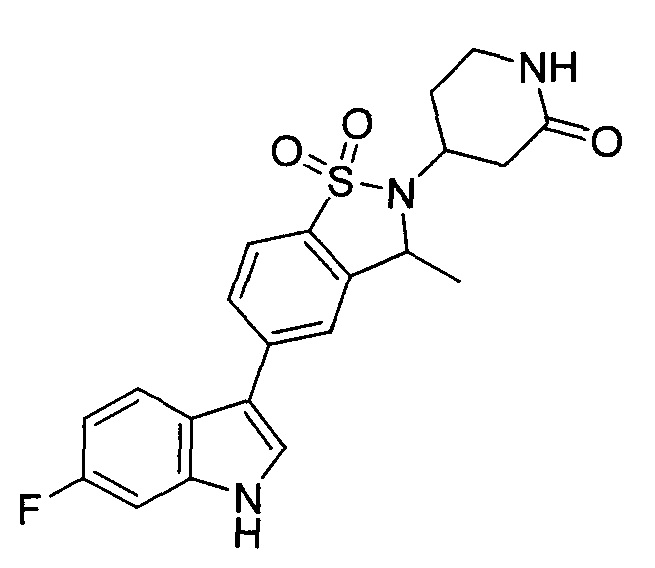
Стадия 1: 4-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-пиперидин-2-он
К раствору 5,6-дигидропиридин-2(1Н)-она (500 мг, 5,15 ммоль) в метаноле (15 мл) добавляли 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксид (2,7 г, 10 ммоль) и Cs2CO3 (1,68 г, 5,15 ммоль). Реакционную смесь перемешивали при 55°С в течение 2 суток, затем концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии (силикагель, этилацетат/метанол от 1/0 до 20/1) с получением указанного в заголовке соединения (475 мг, 13%) в виде желтого масла.
Стадия 2: трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2-(2-оксопиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилат
К суспензии 4-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-пиперидин-2-она (475 мг, 1,32 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (637 мг, 1,59 ммоль) и Cs2CO3 (862 мг, 0,334 ммоль) в диоксане (10 мл) и H2O (3 мл) добавляли PdCl2(dppf) (96,7 мг, 0,132 ммоль). Реакционную смесь перемешивали при 85°С в атмосфере N2 в течение 1,5 часов, затем концентрировали и очищали посредством колоночной хроматографии (силикагель, этилацетат/метанол от 1/0 до 10/1) с получением указанного в заголовке соединения (338 мг, 14%) в виде коричневого масла.
Стадия 3: (-)-4-(-5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пиперидин-2-он
Раствор трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2-(2-оксопиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (235 мг, 0458 ммоль) в HCl/метаноле (20 мл, 4М) перемешивали при 20°С в течение 6 часов. Реакционную смесь концентрировали и смесь диастереомеров разделяли посредством препаративной хиральной СФХ. Указанное в заголовке соединение получили в качестве первого элюируемого пика (25 мг, 13,2%) в виде белого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.91-7.85 (m, 2H), 7.80 (s, 1H), 7.75 (d, J=8,3 Гц, 1Н), 7.68 (s, 1H), 7.16 (dd, J=2,3, 9,5 Гц, 1H), 6.95 (dt, J=2,4, 9,2 Гц, 1H), 4.96-4.91 (m, 1H), 4.17-4.05 (m, 1H), 3.48-3.33 (m, 2H), 2.98-2.87 (m, 1H), 2.83-2.70 (m, 1H), 2.33-2.18 (m, 2H), 1.65 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 435,9 (M+Na)+; [α]20D -5,6° (c=3,3 мг/мл, метанол).
Пример 120: (+)-4-((S)-5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пиперидин-2-он

Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 119 (15 мг, 8%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.92-7.85 (m, 2H), 7.81 (s, 1H), 7.76 (d, J=8,3 Гц, 1H), 7.69 (s, 1H), 7.17 (dd, J=2,3, 9,8 Гц, 1H), 6.95 (dt, J=2,3, 9,2 Гц, 1H),4.94-4.90 (m, 1H), 4.15-4.04 (m, 1H), 3.46-3.34 (m, 2H), 2.93-2.77 (m, 2H), 2.38-2.30 (m, 2H), 1.65 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 436,0 (M+Na)+; [α]20D +4,8° (с=4,2 мг/мл, метанол).
Пример 121: (+)-4-(-5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пиперидин-2-он

Указанное в заголовке соединение получили в качестве третьего элюируемого пика из хирального разделения, описанного в Примере 119 (6 мг, 7%), в виде белого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.92-7.85 (m, 2H), 7.81 (s, 1H), 7.76 (d, J=8,0 Гц, 1Н), 7.69 (s, 1H), 7.17 (dd, J=2,1, 9,7 Гц, 1H), 6.95 (dt, J=2,4, 9,2 Гц, 1H), 4.91(d, J=6,3 Гц, 1H), 4.15-4.03 (m, 1H), 3.48-3.34 (m, 2H), 2.92-2.77 (m, 2H), 2.39-2.27 (m, 2H), 1.65 (d, J=6,5 Гц, 3Н); ЖХ-МС: m/z 435,9 (M+Na)+; [α]20D +3,7° (с=3,9 мг/мл, метанол).
Пример 122: (-)-4-(-5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пиперидин-2-он
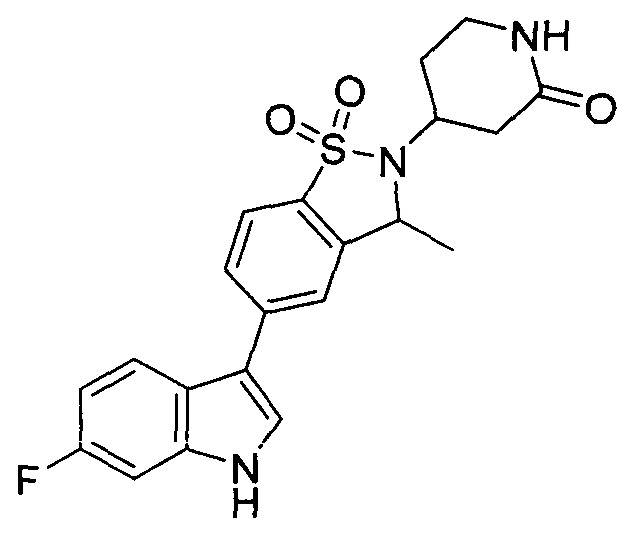
Указанное в заголовке соединение получили в качестве четвертого элюируемого пика из хирального разделения, описанного в Примере 119 (36 мг, 45%), в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.92-7.85 (m, 2H), 7.81 (s, 1H), 7.76 (d, J=8,0 Гц, 1H), 7.68 (s, 1H), 7.16 (dd, J=2,1, 9,7 Гц, 1H), 6.95 (dt, J=2,4, 9,2 Гц, 1H), 4.96- 4.91 (m, 1H), 4.16-4.07 (m, 1H), 3.46-3.34 (m, 2H), 2.96-2.88 (m, 1H), 2.81-2.72 (m, 1H), 2.30-2.23 (m, 2H), 1.65 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 435,9 (M+23)+; [α]20D -3,4° (с=3,6 мг/мл, метанол).
Пример 123: (+)4-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пирролидин-2-он
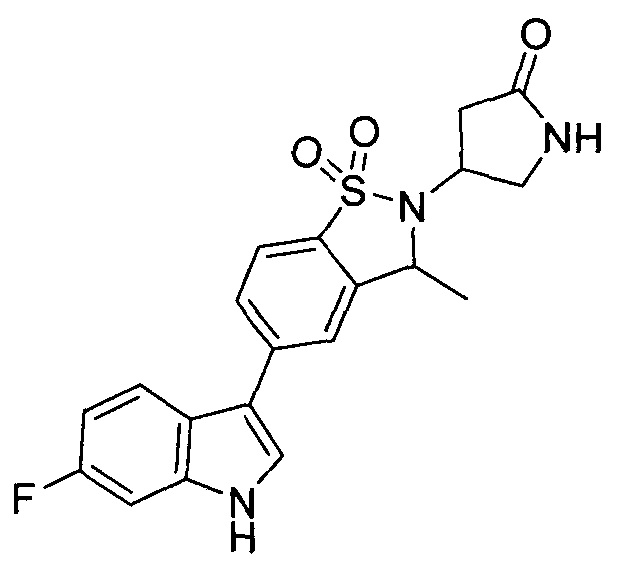
Стадия 1: 5-оксопирролидин-3-ил-метансульфонат
В охлажденную (ледяная баня) суспензию 4-гидрокси-2-пирролидона (1,0 г, 9,9 ммоль) в CH2Cl2 (33,0 мл) добавляли Et3N (1,52 мл, 10,9 ммоль) с последующим медленным добавлением MsCl (0,84 мл, 10,9 ммоль). Ледяную баню удаляли и реакционную смесь перемешивали в течение 1 часа. Неочищенную реакционную смесь концентрировали с получением 5-оксопирролидин-3-ил-метансульфоната (1,8 г, 100%) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2: 4-азидопирролидин-2-он
К раствору 5-оксопирролидин-3-ил-метансульфоната (6,0 г, 33 ммоль) в DMF (111 мл) добавляли NaN3 (6,5 г, 100 ммоль). Реакционную смесь нагревали при 60°С в течение 18 часов, затем охлаждали и разбавляли EtOAc (400 мл). Этот раствор промывали NaHCO3 (насыщ.) и дополнительно экстрагировали EtOAc (200 мл × 2). Объединенные органические слои промывали рассолом (200 мл × 2), затем сушили над безводным Na2SO4, фильтровали и концентрировали, причем оставалось некоторое количество EtOAc. Смесь разбавляли THF (150 мл) и добавляли при 25°С PPh3 (13,1 г, 50,1 ммоль, 1,5 экв.) и воду (50 мл). Реакционную смесь нагревали при 65°С в течение 18 часов, затем концентрировали и подкисляли до рН 1 с использованием 2М HCl. Водную фазу промывали CH2Cl2 (150 мл × 3), затем концентрировали с получением 4-аминопирролидин-2-она (959 мг, 21%) в виде коричневой смолы. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 8.43 (br s, 2H), 7.90-7.80 (m, 1H), 3.55 (dd, J=7,0, 11,0 Гц, 1H), 3.47-3.40 (m, 1H), 3.25 (dd, J=3,3, 10,8 Гц, 1H), 2.56 (dd, J=8,3, 17,3 Гц, 1H), 2.21 (dd, J=4,0, 17,1 Гц, 1H).
Стадия 3: 4-бром-2-этил-N-(5-оксопирролидин-3-ил)бензолсульфонамид
К желтому раствору 4-аминопирролидин-2-она (959 мг, 7,02 ммоль) в воде (1,0 мл) добавляли THF (34 мл), DIPEA (4,3 мл, 25 ммоль) и 4-бром-2-этилбензол-1-сульфонилхлорид (3,9 г, 14 ммоль) при 15°С. Реакционную смесь перемешивали при 15°С в течение 4,5 часов, затем разбавляли EtOAc (20 мл) и водой (10 мл). Слои разделяли и водный слой подвергали обратной экстракции EtOAc (40 мл × 2). Объединенные органические слои промывали рассолом (20 мл × 2) и NaHCO3 (насыщ.) (20 мл × 1), затем сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением 4-бром-2-этил-N-(5-оксопирролидин-3-ил)бензол-сульфонамида (685 мг, 28%) в виде желтого твердого вещества.
Стадия 4: 4-бром-2-(1-бромэтил)-N-(5-оксопирролидин-3-ил)-бензолсульфонамид
Желтую суспензию 4-бром-2-этил-N-(5-оксопирролидин-3-ил)бензол-сульфонамида (841 мг, 2,42 ммоль), NBS (560 мг, 3,15 ммоль, 1,3 экв.) и AIBN (199 мг, 1,21 ммоль, 0,5 экв.) в CCl4 (48,4 мл) нагревали при 70°С в течение 3 часов. Реакционную смесь концентрировали и разбавляли EtOAc (20 мл) и водой (15 мл). Слои разделяли и водный слой подвергали обратной экстракции EtOAc (15 мл × 2). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением 4-бром-2-(1-бромэтил)-N-(5-оксопирролидин-3-ил)бензолсульфонамида (814 мг, 79%) в виде желтого твердого вещества.
Стадия 5: 4-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-пирролидин-2-он
К смеси 4-бром-2-(1-бромэтил)-N-(5-оксопирролидин-3-ил)бензол-сульфонамида (814 мг, 1,91 ммоль) в ацетоне (35 мл) добавляли K2CO3 (528 мг, 3,82 ммоль) и воду (3,4 мл). Реакционную смесь нагревали до 50°С в течение 15 часов, затем концентрировали и очищали посредством колоночной хроматографии с получением 4-(5-бром-3-метил-1,1-диоксидобензо[d]-изотиазол-2(3H)-ил)пирролидин-2-она (470 мг, 71%) в виде желтого твердого вещества.
Стадия 6: трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2-(5-оксопирролидин-3-ил)-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилат
Смесь 4-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-пирролидин-2-она (457 мг, 1,32 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (478 мг, 1,32 ммоль), PdCl2(dppf) (99 мг, 0,13 ммоль) и K3PO4 (562 мг, 2,65 ммоль) в 1,4-диоксане (13 мл) и воде (0,5 мл) барботировали азотом в течение 60 секунд, затем перемешивали при 85°С в течение 2 часов. Реакционную смесь концентрировали и очищали посредством колоночной хроматографии с получением трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2-(5-оксопирролидин-3-ил)-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилата (476 мг, 72%) в виде желтой смолы. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 8.13 (s, 1H), 8.02-7.80 (m, 6Н), 7.25 (d, J=9,5 Гц, 1H), 4.88 (br s, 1H), 4.47 (br s, 1H), 3.17 (d, J=5,0 Гц, 2H), 2.71-2.56 (m, 2H), 1.66 (s, 9H), 1.58 (br s, 3H); ЖХ-МС: m/z 522,0 (M+Na)+.
Стадия 7: (+)-4-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]-изотиазол-2(3H)-ил)пирролидин-2-он
В охлажденный (ледяная баня) раствор трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2-(5-оксопирролидин-3-ил)-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилата (330 мг, 0,66 ммоль) в CH2Cl2 (10,0 мл) добавляли TFA (4,0 мл). Реакционную смесь перемешивали при 5°С в течение 4 часов, затем концентрировали. Неочищенный осадок нейтрализовали NaHCO3 (насыщ.) и разбавляли DCM. Слои разделяли и водный слой подвергали обратной экстракции CH2Cl2 (20 мл × 2). Объединенные органические слои концентрировали и очищали посредством колоночной хроматографии с получением 4-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]-изотиазол-2(3H)-ил)пирролидин-2-она (183 мг, 69%) в виде смеси диастереомеров. Смесь разделяли посредством препаративной хиральной СФХ с получением (+)-4-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидо-бензо[d]изотиазол-2(3H)-ил)пирролидин-2-она в качестве первого элюируемого пика (30 мг, 16,0%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.72 (br s, 1H), 7.97-7.89 (m, 4H), 7.86-7.80 (m, 2H), 7.27 (dd, J=2.5, 9,5 Гц, 1H), 7.01 (dt, J=2,5, 9,3 Гц, 1H), 4.86 (q, J=6,2 Гц, 1H), 4.46 (td, J=6,9, 14,3 Гц, 1H), 3.66-3.54 (m, 2H), 2.71-2.62 (m, 1H), 2.55 (d, J=6,5 Гц, 1Н), 1.57 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 421,9 (M+Na)+; [α]20D +4,62° (c=0,00087 г/мл, МеОН).
Пример 124: (-)-4-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидо-бензо[d]изотиазол-2(3H)-ил)пирролидин-2-он
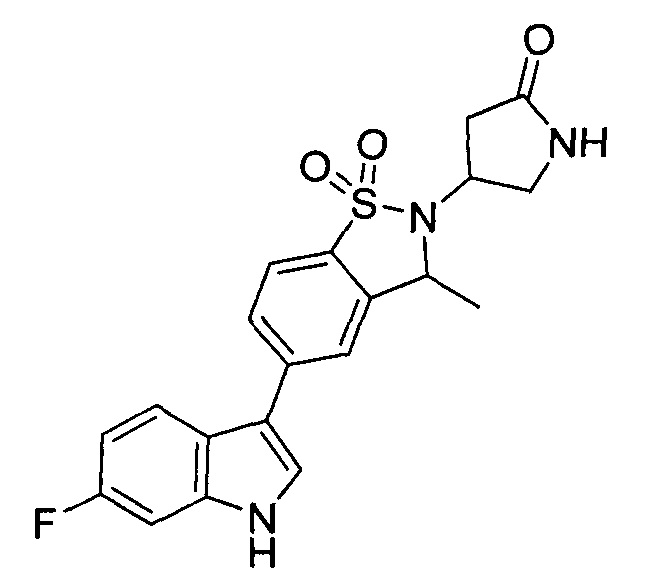
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 123 (25 мг, 14%), в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1H), 7.99-7.88 (m, 4H), 7.88-7.80 (m, 2H), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.01 (dt, J=2,5, 9,3 Гц, 1H), 4.86 (q, J=6,4 Гц, 1H), 4.46 (td, J=7,1, 14,4 Гц, 1H), 3.66-3.54 (m, 2H), 2.70-2.63 (m, 1H), 2.55 (d, J=6,5 Гц, 1H), 1.57 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 421,9 (M+Na); [α]20D -30,00° (c=0,00067 г/мл, МеОН).
Пример 125: (-)-4-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидо-бензо[d]изотиазол-2(3H)-ил)пирролидин-2-он
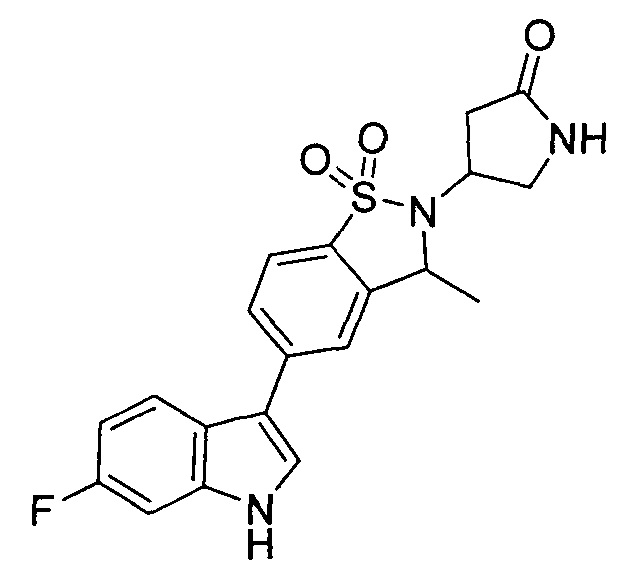
Указанное в заголовке соединение получили в качестве третьего элюируемого пика из хирального разделения, описанного в Примере 123 (25 мг, 14%), в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.72 (br s, 1H), 8.00-7.81 (m, 6H), 7.28 (d, J=8,0 Гц, 1H), 7.02 (dt, J=2,5, 9,3 Гц, 1H), 4.86 (q, J=6,5 Гц, 1H), 4.46 (td, J=7,1, 14,4 Гц, 1H), 3.72-3.65 (m, 1H), 3.48 (dd, J=5,5, 10,0 Гц, 1H), 2.65-2.53 (m, 2H), 1.59 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 421,9 (M+Na)+; [α]20D -10,00° (с=0,001 г/мл, МеОН).
Пример 126: (+)-4-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пирролидин-2-он
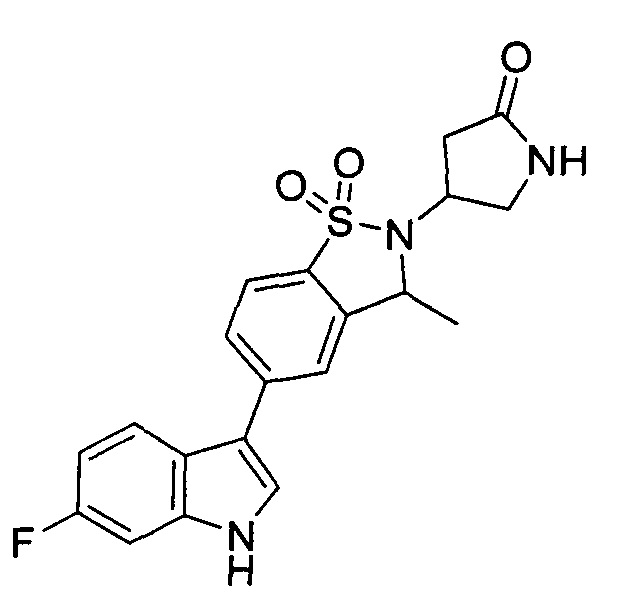
Указанное в заголовке соединение получили в качестве четвертого элюируемого пика из хирального разделения, описанного в Примере 123 (35 мг, 19%), в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1H), 8.00-7.80 (m, 6H), 7.27 (d, J=10,0 Гц, 1Н), 7.01 (t, J=9,0 Гц, 1Н), 4.93-4.81 (m, 1Н), 4.53-4.38 (m, 1Н), 3.67-3.53 (m, 2H), 2.70-2.62 (m, 1Н), 2.55 (d, J=7,0 Гц, 1Н), 1.57 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 421,9 (M+Na)+; [α]20D +12,50° (c=0,00080 г/мл, МеОН).
Пример 127: (-)-2-(5-(6-фтор-1H-индол-3-ил)-3-(гидроксиметил)-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилацетамид
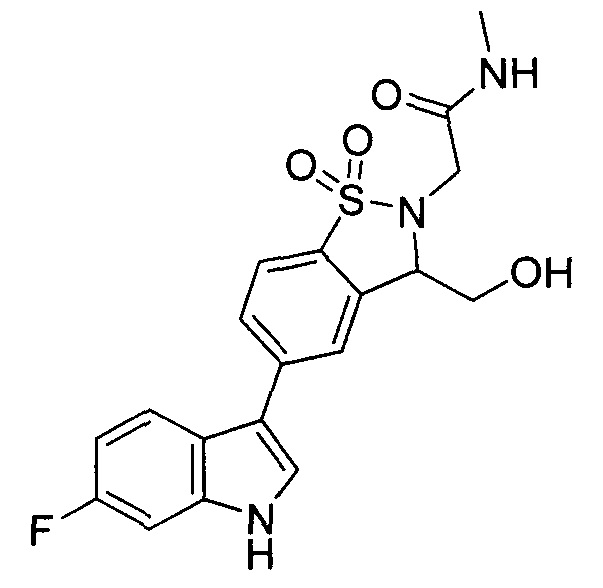
Стадия 1: трет-бутил-3-(2-(2-этокси-2-оксоэтил)-3-(гидроксиметил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилат
К суспензии трет-бутил-6-фтор-3-(3-(гидроксиметил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилата (Пример 112, 500 мг, 0,462 ммоль) и K2CO3 (192 мг, 1,39 ммоль) в DMF (5 мл) добавляли по каплям этил-2-бромацетат (232 мг, 1,39 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, затем вливали в воду (50 мл) и экстрагировали EtOAc (3×10 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением трет-бутил-3-(2-(2-этокси-2-оксоэтил)-3-(гидроксиметил)-1,1-диоксидо-2,3-дигидробензо[d]-изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (450 мг, 78%) в виде желтой смолы.
Стадия 2: Раствор трет-бутил-3-(2-(2-этокси-2-оксоэтил)-3-(гидроксиметил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (100 мг, 0,193 ммоль) (Стадия 1) в MeNH2/EtOH (30%, 5 мл) перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали и очищали посредством колоночной хроматографии с получением указанного в заголовке соединения в виде рацемической смеси. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (-)-2-(5-(6-фтор-1H-индол-3-ил)-3-(гидроксиметил)-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилацетамида в качестве первого элюируемого пика (28 мг, 18%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1H), 7.85-80.6 (m, 6H), 7.28 (dd, J=9,91, 2,38 Гц, 1H), 7.02 (td, J=9,22, 2,38 Гц, 1H), 5.25-5.40 (m, 1H), 4.85 (t, J=4,14 Гц, 1H), 3.77-4.14 (m, 4H), 2.59-2.74 (m, 3H); LCMS: m/z 404,0 (M+H)+; [α]20D -4,54° (c=0,0022 г/мл, DMSO).
Пример 128: (+)-2-(5-(6-фтор-1H-индол-3-ил)-3-(гидроксиметил)-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилацетамид
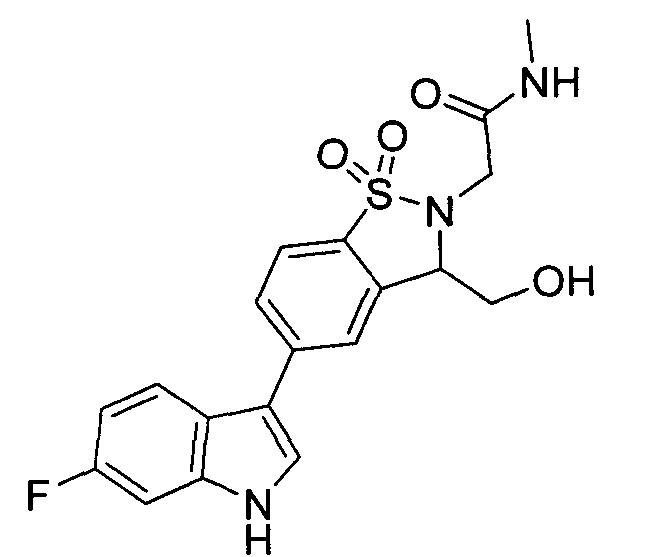
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 127 (33 мг, 21%), в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1H), 7.85-80.6 (m, 6H), 7.28 (dd, J=9,91, 2.38 Гц, 1H), 7.02 (td, J=9,22, 2,38 Гц, 1H), 5.25-5.40 (m, 1H), 4.85 (t, J=4,14 Гц, 1H), 3.77-4.14 (m, 4H), 2.59-2.74 (m, 3H); LCMS: m/z 404,1 (M+H)+; [α]20D +7,42° (c=0,0035 г/мл, DMSO).
Пример 129: (-)-5-(6-фтор-1Н-индол-3-ил)-3-(гидроксиметил)-2-((1-метил-1Н-1,2,4-триазол-3-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
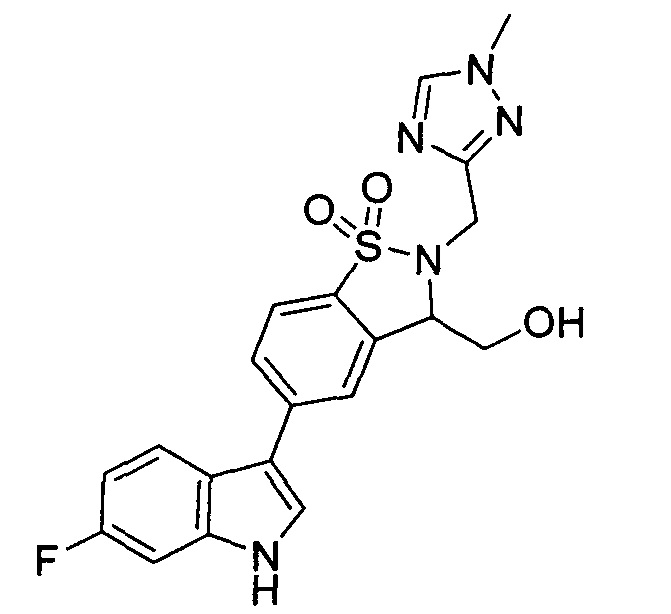
Стадия 1: (1-метил-1H-1,2,4-триазол-3-ил)метилметансульфонат
В охлажденный (ледяная баня) раствор (1-метил-1H-1,2,4-триазол-3-ил)-метанола (400 мг, 3,54 ммоль) и DIEA (0,98 мл, 7,1 ммоль) в CH2Cl2 (35 мл) добавляли MsCl (0,41 мл, 5,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем гасили NaHCO3 (насыщ.) (20 мл). Слои разделяли и водный слой подвергали обратной экстракции CH2Cl2 (15 мл × 2). Объединенные органические слои промывали рассолом (20 мл × 2), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением (1-метил-1Н-1,2,4-триазол-3-ил)метилметансульфоната (650 мг, 96%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 8.05 (s, 1H), 5.29 (s, 2H), 3.93 (s, 3H), 3.09 (s, 3Н).
Стадия 2: трет-бутил-6-фтор-3-(3-(гидроксиметил)-2-((1-метил-1H-1,2,4-триазол-3-ил)метил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилат
Смесь трет-бутил-6-фтор-3-(3-(гидроксиметил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилата (200 мг, 0,46 ммоль), (1-метил-1Н-1,2,4-триазол-3-ил)метилметансульфоната (133 мг, 0,69 ммоль) и K2CO3 (128 мг, 0,92 ммоль) в DMF (2,3 мл) барботировали азотом в течение 30 секунд, затем нагревали при 70°С в течение 12 часов. Реакционную смесь разбавляли EtOAc(10 мл) и водой (10 мл) и слои разделяли. Водный слой подвергали обратной экстракции EtOAc (10 мл × 2) и объединенные органические слои промывали рассолом (10 мл × 2), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением трет-бутил-6-фтор-3-(3-(гидроксиметил)-2-((1-метил-1Н-1,2,4-триазол-3-ил)метил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (196 мг, 80%) в виде желтой смолы.
Стадия 3: (-)-5-(6-фтор-1H-индол-3-ил)-3-(гидроксиметил)-2-((1-метил-1H-1,2,4-триазол-3-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К раствору трет-бутил-6-фтор-3-(3-(гидроксиметил)-2-((1-метил-1H-1,2,4-триазол-3-ил)метил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (196 мг, 0,37 ммоль) в EtOH (5 мл) и CH2Cl2 (2 мл) добавляли MeNH2/EtOH (25%, 5,0 мл, 32 ммоль). Реакционную смесь перемешивали при 15°С в течение 15 часов, затем концентрировали и очищали посредством колоночной хроматографии с получением 5-(6-фтор-1H-индол-3-ил)-3-(гидроксиметил)-2-((1-метил-1H-1,2,4-триазол-3-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (75 мг, 47%) в виде рацемической смеси. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (-)-5-(6-фтор-1H-индол-3-ил)-3-(гидроксиметил)-2-((1-метил-1H-1,2,4-триазол-3-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида в качестве первого элюируемого пика (15 мг, 20%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.72-11.65 (m, 1Н), 8.47 (s, 1H), 7.99-7.82 (m, 5H), 7.27 (dd, J=2,3, 9,8 Гц, 1H), 7.01 (dt, J=2,5, 9,3 Гц, 1H), 5.35 (t, J=6,0 Гц, 1H), 4.85 (t, J=4,0 Гц, 1H), 4.57 (q, J=16,2 Гц, 2Н), 4.00-3.86 (m, 2H), 3.85 (s, 3H); ЖХ-МС: m/z 427,9 (M+H)+; [α]20D -100,91° (с=0,00073 г/мл, МеОН).
Пример 130: (+)-5-(6-фтор-1H-индол-3-ил)-3-(гидроксиметил)-2-((1-метил-1H-1,2,4-триазол-3-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
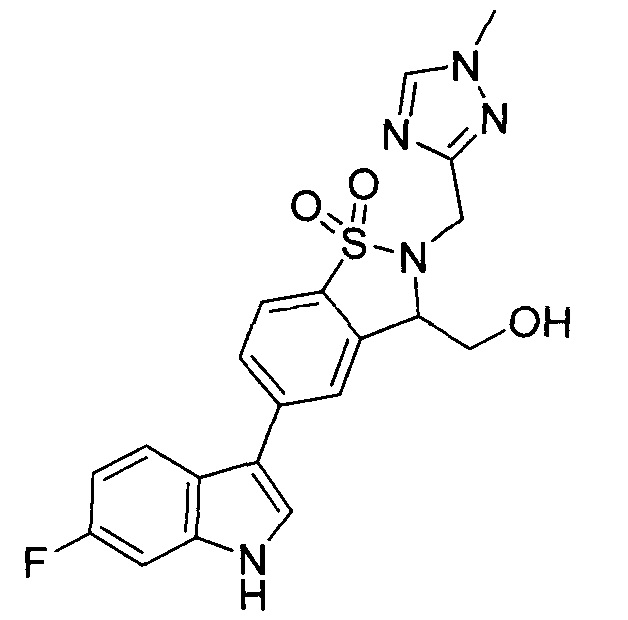
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 129 (20 мг, 27%), в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1H), 8.47 (s, 1H), 8.00-7.81 (m, 5H), 7.27 (d, J=9,5 Гц, 1Н), 7.01 (dt, J=2,5, 9,3 Гц, 1H), 5.36 (br s, 1H), 4.85 (t, J=4,0 Гц, 1H), 4.56 (q, J=16,6 Гц, 2Н), 4.01-3.86 (m, 2Н), 3.86-3.83 (m, 3H); ЖХ-МС: m/z 449,9 (M+Na)+; [α]20D +27,27° (c=0,00073 г/мл, МеОН).
Пример 131: (+)-5-(6-фтор-1H-индол-3-ил)-2-(2-гидроксиэтил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
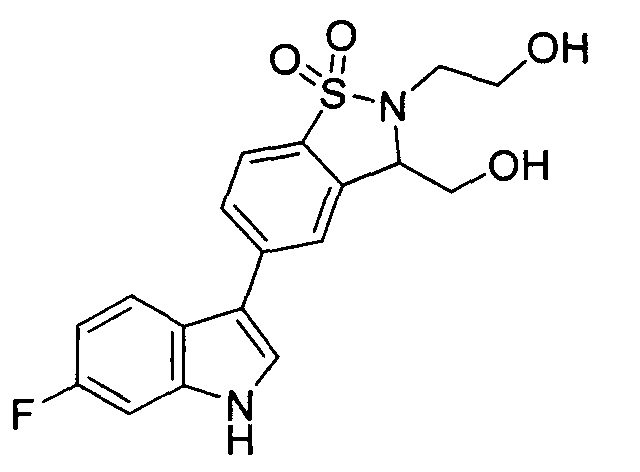
Стадия 1: трет-бутил-3-(2-(2-(трет-бутокси)-2-оксоэтил)-3-(гидроксиметил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилат
К желтой суспензии трет-бутил-6-фтор-3-(3-(гидроксиметил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (500 мг, 1,16 ммоль) (Пример 122) и K2CO3 (192 мг, 1,39 ммоль) в DMF (10 мл) добавляли трет-бутил-бромацетат (271 мг, 1,39 ммоль) при 20°С. Реакционную смесь перемешивали при 25°С в течение 2 часов, затем вливали в воду (20 мл) и экстрагировали EtOAc (3×10 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали, затем очищали посредством колоночной хроматографии с получением трет-бутил-3-(2-(2-(трет-бутокси)-2-оксоэтил)-3-(гидроксиметил)-1,1-диоксидо-2,3-дигидробензо-[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (540 мг, выход 85%) в виде желтой смолы.
Стадия 2: 2-(5-(6-фтор-1H-индол-3-ил)-3-(гидроксиметил)-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)уксусная кислота
Раствор трет-бутил-3-(2-(2-(трет-бутокси)-2-оксоэтил)-3-(гидроксиметил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (540 мг, 0,84 ммоль) в TFA (20 мл) перемешивали при 30°С в течение 18 часов, затем концентрировали с получением неочищенной 2-(5-(6-фтор-1H-индол-3-ил)-3-(гидроксиметил)-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)уксусной кислоты (360 мг), которую использовали непосредственно на следующей стадии.
Стадия 3: (+)-5-(2-((3-фторфенил)амино)винил)-2-(2-гидроксиэтил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Раствор неочищенной 2-(5-(6-фтор-1H-индол-3-ил)-3-(гидроксиметил)-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)уксусной кислоты (310 мг, 0,794 ммоль) в THF (3 мл) перемешивали в ледяной бане в течение 5 мин, затем медленно добавляли ВН3-DMS (0,5 мл, 10 М, 5 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 4 часов. Медленно добавляли дополнительное количество ВН3-DMS (1,0 мл, 1,0 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 14 часов. Неочищенную реакционную смесь охлаждали в ледяной бане, затем медленно гасили NH4Cl (насыщ.) до прекращения выделения газа. Смесь разбавляли EtOAc (20 мл) и NH4Cl (насыщ.) (10 мл), затем слои разделяли. Водный слой подвергали обратной экстракции EtOAc (10 мл × 3), затем объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали посредством препаративной ВЭЖХ с получением рацемического 5-(6-фтор-1H-индол-3-ил)-2-(2-гидроксиэтил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида в виде белого твердого вещества. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (+)5-(2-((3-фторфенил)амино)-винил)-2-(2-гидроксиэтил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида в качестве первого элюируемого пика (41 мг, 14%) в виде светло-желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.73-11.63 (m, 1Н), 7.91 (s, 5H), 7.31-7.24 (m, 1H), 7.06-6.97 (m, 1H), 5.20-5.13 (m, 1H), 5.03-4.96 (m, 1H), 4.76-4.69 (m, 1H),3.96-3.81 (m, 2H), 3.70 (d, J=5,8 Гц, 2H), 3.49 (s, 1H), 3.35-3.29 (m, 1H); LCMS: m/z 399,0 (M+Na)+; [α]20D +2,33° (c=0,0018 г/мл, МеОН).
Пример 132: (-)-5-(6-фтор-1H-индол-3-ил)-2-(2-гидроксиэтил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид

Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 131 (45 мг, 15%), в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1Н), 8.02-7.87 (m, 4H), 7.87-7.79 (m, 1H), 7.27 (dd, J=2,1, 9,9 Гц, 1Н), 7.01(dt, J=2,3, 9,2 Гц, 1Н), 5.16 (s, 1Н), 4.99 (t, J=5,3 Гц, 1Н), 4.72 (s, 1Н), 3.97-3.81 (m, 2H), 3.75-3.64 (m, 2H), 3.56-3.46 (m, 1Н), 3.35-3.28 (m, 1Н); ЖХ-МС: m/z для 399,0 (M+Na)+; [α]20D -1,72° (c=0,0018 г/мл, МеОН).
Пример 133: (-)-5-(2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)-1,3,4-оксадиазол-2(3H)-он
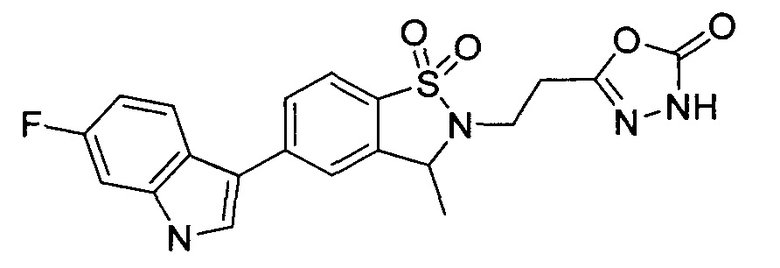
Стадия 1: 3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-пропангидразид
Желтый раствор этил-3-(5-бром-3-метил-1,1-диоксидобензо[d]-изотиазол-2(3H)-ил)пропаноата (600 мг, 1,66 ммоль) в EtOH/NH2NH2H2O (10 мл/2 мл) перемешивали при 25°С в течение 14 часов, затем концентрировали с получением 3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-пропангидразида (570 мг, 99%) в виде желтой смолы.
Стадия 2: 5-(2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-этил)-1,3,4-оксадиазол-2(3H)-он
К раствору 3-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-пропангидразида (500 мг, 1,44 ммоль) в DCE (30 мл) добавляли трифосген (213 мг, 0,718 ммоль) при 0°С. Реакционную смесь нагревали до 80°С и перемешивали в течение 2 часов, затем разбавляли дихлорметаном (50 мл) и промывали NaHCO3 (насыщ.) (10 мл). Органическую фазу сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (DCM/метанол от 120/1 до 10/1) с получением 5-(2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)-1,3,4-оксадиазол-2(3H)-она (484 мг, 90%) в виде желтого твердого вещества.
Стадия 3: (-)-5-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)-1,3,4-оксадиазол-2(3H)-он
К раствору 5-(2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)-1,3,4-оксадиазол-2(3H)-она (552 мг, 1,48 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (533 мг, 1,48 ммоль) и K3PO4 (626 мг, 2,95 ммоль) в смеси диоксан/Н2О (12 мл/4 мл) добавляли Pd(dppf)Cl2 (108 мг, 0,148 ммоль) при 28°С. Реакционную смесь перемешивали при 80°С в течение 16 часов, затем разбавляли водой (8 мл) и экстрагировали этилацетатом (15 мл × 3). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (силикагель, 50-100% этилацетат/петролейный эфир) с получением рацемического 5-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]-изотиазол-2(3H)-ил)этил)-1,3,4-оксадиазол-2(3H)-она (150 мг, 24%) в виде желтого твердого вещества. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (-)-5-(2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)-1,3,4-оксадиазол-2(3H)-она в качестве первого элюируемого пика (45 мг, 7%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.90 (d, J=1,3 Гц, 2Н), 7.84-7.77 (m, 2Н), 7.70 (s, 1H), 7.18 (dd, J=2,4, 9,7 Гц, 1Н), 6.97 (dt, J=2,4, 9,2 Гц, 1Н), 4.75 (d, J=6,3 Гц, 1Н), 3.75 (d, J=2,5 Гц, 2Н), 3.08 (t, J=6,9 Гц, 2Н), 1.64 (d, J=6,5 Гц, 3H); ЖХ-МС: m/z 428,9 (М+Н)+; [α]20D -4,67° (с=0,0015 г/мл, метанол).
Пример 134: (+)-5-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)этил)-1,3,4-оксадиазол-2(3H)-он

Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 133 (45 мг, 7%), в виде бледно-желтого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.94-7.85 (m, 2Н), 7.84-7.77 (m, 2H), 7.70 (s, 1H), 7.18 (dd, J=2,4, 9,7 Гц, 1Н), 6.97 (dt, J=2,5, 9,2 Гц, 1H), 4.75 (d, J=6,5 Гц, 1H), 3.75 (dt, J=2,6, 6,8 Гц, 2H), 3.07 (t, J=6,9 Гц, 2H), 1.64 (d, J=6,3 Гц, 3H); ЖХ-МС: m/z 428,9 (M+H)+; [α]20D +5,63° (с=0,0015 г/мл, метанол).
Пример 135: 5-(6-фтор-1Н-индол-3-ил)-3-метилбензо[d]изотиазол-1,1-диоксид
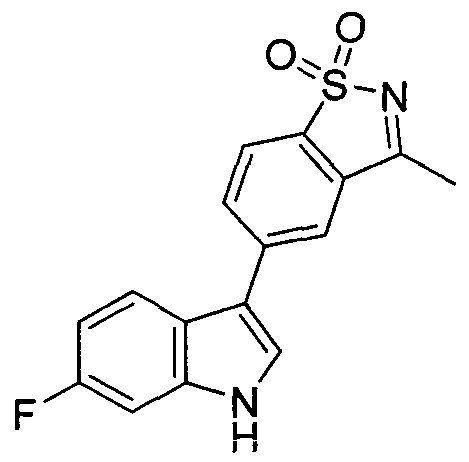
Стадия 1: 5-бром-3-метилбензо[d]изотиазол-1,1-диоксид
К раствору 5-бромбензо[d]изотиазол-3(2H)-он-1,1-диоксида (10 г, 38 ммоль) в безводном THF (191 мл) добавляли по каплям бромид метилмагния (3 М в Et2O, 38,2 мл) при 0°С. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 4 часов, затем охлаждали до 0°С. Медленно добавляли 6 M HCl (65 мл) и смесь перемешивали при комнатной температуре в течение 30 мин, затем концентрировали. Полученное твердое вещество разбавляли водой (200 мл) и отфильтровывали. Осадок на фильтре промывали водой (200 мл), затем сушили с получением 5-бром-3-метилбензо[d]изотиазол-1,1-диоксида (7,5 г, 76%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 8.32 (s, 1H), 8.14-8.07 (m, 2H), 2.71 (s, 3Н).
Стадия 2: 5-(6-фтор-1H-индол-3-ил)-3-метилбензо[d]изотиазол-1,1-диоксид
Смесь 5-бром-3-метилбензо[d]изотиазол-1,1-диоксида (200 мг, 0,54 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (194 мг, 0,538 ммоль), PdCl2(dppf)CH2Cl2 (20 мг, 0,03 ммоль) и K3PO4 (228 мг, 1,08 ммоль) в 1,4-диоксане (8 мл) и воде (2 мл) барботировали азотом в течение 1 минуты. Реакционную смесь перемешивали при 80°С в течение 32 часов, затем разбавляли водой (2 мл) и перемешивали в течение еще 16 часов при 80°С. Смесь концентрировали и очищали посредством колоночной хроматографии, а затем посредством очистки препаративной ВЭЖХ с получением трет-бутил-6-фтор-3-(3-метил-1,1-диоксидобензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (17 мг, выход 10%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, ацетонитрил-d3) δ [м.д.] 9,81 (br s, 1H), 8.12-8.05 (m, 2H), 8.00-7.90 (m, 2H), 7.74 (d, J=2,5 Гц, 1Н), 7.28 (dd, J=2,4, 9,9 Гц, 1Н), 7.03 (dt, J=2,3, 9,3 Гц, 1Н), 2.72 (s, 3H); ЖХ-МС: m/z 315,0 (М+Н)+.
Пример 136: 5-(6-фтор-1Н-индол-3-ил)-2Н-спиро[бензо[d]изотиазол-3,1'-циклопропан]-1,1-диоксид
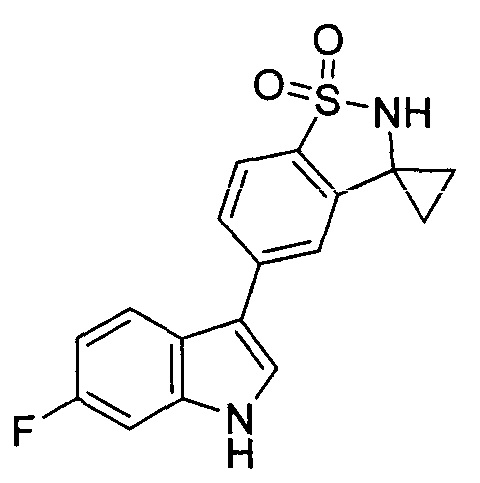
Стадия 1: 1-(3-хлорфенил)циклопропанамин
К раствору 3-бромбензонитрила (10 г, 55 ммоль) и Ti(O-iPr)4 (17,2 г, 18 мл, 60,4 ммоль) в THF (100 мл) добавляли по каплям EtMgBr (40,3 мл, 121 ммоль, 3М в THF) при -78°С. Реакционную смесь медленно нагревали до комнатной температуры в течение 1 часа, затем добавляли BF3⋅OEt2 (13,9 мл, 110 ммоль). Перемешивание продолжали в течение 16 часов, затем реакционную смесь гасили 1 н. HCl (150 мл). Смесь нейтрализовали 10% NaOH (200 мл) и экстрагировали EtOAc (300 мл). Этот органический раствор использовали непосредственно на следующей стадии.
Стадия 2: трет-бутил-(1-(3-хлорфенил)циклопропил)карбамат
К раствору 1-(3-бромфенил)циклопропанамина (12 г, 55 ммоль) в EtOAc (300 мл) добавляли Boc2O (18 г, 83 ммоль) и TEA (17 г, 165 ммоль). Реакционную смесь перемешивали при 10°С в течение 16 часов, затем концентрировали и очищали посредством колоночной хроматографии с получением трет-бутил-(1-(3-бромфенил)циклопропил)карбамата (1,8 г, 11%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 7.39-7.29 (m, 2H), 7.20-7.10 (m, 2H), 5.25 (br s, 1H), 1.52-1.37 (m, 9H), 1.33-1.18 (m, 4H).
Стадия 3: 2-(1-аминоциклопропил)-4-хлорбензол-1-сульфонилхлорид
В охлажденный (ледяная баня) раствор трет-бутил-(1-(3-бромфенил)-циклопропил)карбамата (1,8 г, 5,8 ммоль) в хлороформе (20 мл) добавляли хлорсульфоновую кислоту (4,0 г, 35 ммоль). Реакционную смесь перемешивали на ледяной бане в течение 16 часов, затем гасили ледяной водой (50 мл) и фильтровали. Осадок на фильтре промывали водой (15 мл) и DCM (20 мл) с получением побочного продукта 2-(1-аминоциклопропил)-4-бромбензол-сульфоновой кислоты (900 мг, 53%) в виде не совсем белого твердого вещества. Полученный фильтрат экстрагировали DCM (50 мл × 2) и объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением 2-(1-аминоциклопропил)-4-бромбензол-1-сульфонилхлорида (1 г, 56%) в виде черного масла, которое использовали непосредственно на следующей стадии без очистки. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 8.60 (br s, 3H), 7.88 (d, J=8,0 Гц, 1Н), 7.62 (d, J=1,5 Гц, 1Н), 7.36-7.21 (m, 1Н), 1.40-1.17 (m, 4H); LCMS, m/z 292,1 (M+H)+.
Стадия 4: 5-хлор-2Н-спиро[бензо[d]изотиазол-3,1'-циклопропан]-1,1-диоксид
Смесь 2-(1-аминоциклопропил)-4-хлорбензол-1-сульфонилхлорида (1,0 г, 3,2 ммоль) и Na2CO3 (1,0 г, 9,7 ммоль) в ацетоне (30 мл) и воде (30 мл) перемешивали при 10°С в течение 2 часов. Реакционную смесь концентрировали для удаления ацетона и водный раствор экстрагировали EtOAc (50 мл × 2). Объединенные органические слои промывали рассолом (10 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением 5-бром-2Н-спиро[бензо[d]изотиазол-3,1'-циклопропан]-1,1-диоксида (40 мг, 0,3%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 7.65-7.55 (m, 2H), 7.08 d, J=0,8 Гц, 1Н), 5.03 (br s, 1H), 1.64-1.59 (m,2H), 1.33 (d, J=2,0 Гц, 2H).
Стадия 5: трет-бутил-3-(1,1-диоксидо-2Н-спиро[бензо[d]изотиазол-3,1'-циклопропан]-5-ил)-6-фтор-1H-индол-1-карбоксилат
Смесь 5-бром-2Н-спиро[бензо[d]изотиазол-3,1'-циклопропан]-1,1-диоксида (40 мг, 0,15 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (79 мг, 0,22 ммоль), PdCl2(dppf)CH2Cl2 (5 мг, 0,01 ммоль) и K3PO4 (62 мг, 0,29 ммоль) в 1,4-диоксане (3 мл) и воде (1 мл) барботировали азотом в течение 1 минуты. Реакционную смесь перемешивали при 80°С в течение 2 часов, затем разбавляли водой (5 мл) и экстрагировали EtOAc (15 мл × 2). Объединенные органические слои промывали рассолом (15 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением трет-бутил-3-(1,1-диоксидо-2Н-спиро[бензо[d]изотиазол-3,1'-циклопропан]-5-ил)-6-фтор-1H-индол-1-карбоксилата (20 мг, 32%) в виде желтого масла.
Стадия 6: 5-(6-фтор-1H-индол-3-ил)-2Н-спиро[бензо[d]изотиазол-3,1'-циклопропан]-1,1-диоксид
Раствор трет-бутил-3-(1,1-диоксидо-2Н-спиро[бензо[d]изотиазол-3,1'-циклопропан]-5-ил)-6-фтор-1H-индол-1-карбоксилата (20 мг, 0,05 ммоль) и TFA (2 мл) в DCM (4 мл) перемешивали при 5°С в течение 1 часа. Реакционную смесь концентрировали и нейтрализовали NaHCO3 (насыщ.) (5 мл), затем экстрагировали EtOAc (15 мл × 2). Объединенные органические слои промывали рассолом (5 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением 5-(6-фтор-1H-индол-3-ил)-2Н-спиро[бензо[d]изотиазол-3,1'-циклопропан]-1,1-диоксида (10 мг, 65%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, ацетонитрил-d3) δ [м.д.] 9.70 (br s, 1H), 7.86 (dd, J=5,3, 8,8 Гц, 1Н), 7.81-7.74 (m, 2H), 7.63 (d, J=2,8 Гц, 1H), 7.31 (s, 1Н), 7.25 (dd, J=2,4, 9,9 Гц, 1Н), 6.98 (dt, J=2,5, 9,3 Гц, 1Н), 6.12 (s, 1Н), 1.50-1.46 (m, 2H), 1.44-1.40 (m, 2H); ЖХ-МС: m/z 329,0 (M+H)+.
Пример 137: 5-(6-фтор-1H-индол-3-ил)-3-((метиламино)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
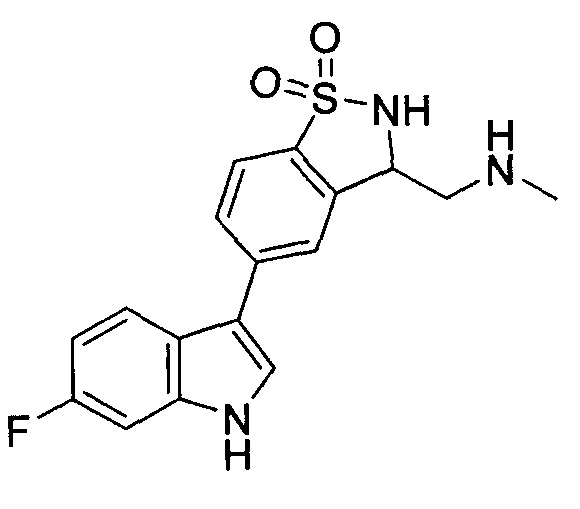
Стадия 1: 5-(6-фтор-1Н-индол-3-ил)-N-метил-2,3-дигидробензо[d]тиофен-3-карбоксамид-1,1-диоксид
Раствор метил-5-(1-(трет-бутоксикарбонил)-6-фтор-1H-индол-3-ил)-2,3-дигидробензо[d]изотиазол-3-карбоксилат-1,1-диоксида (200 мг, 0,43 ммоль) в MeNH2/EtOH (10 мл, 27% MeNH2 в EtOH) перемешивали при комнатной температуре в течение 2 часов, затем концентрировали и очищали посредством колоночной хроматографии с получением 5-(6-фтор-1Н-индол-3-ил)-N-метил-2,3-дигидробензо[d]изотиазол-3-карбоксамид-1,1-диоксида (140 мг, 89%) в виде не совсем белого твердого вещества.
Стадия 2: (-)-5-(6-фтор-1Н-индол-3-ил)-3-((метиламино)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К раствору 5-(6-фтор-1H-индол-3-ил)-N-метил-2,3-дигидробензо[d]-изотиазол-3-карбоксамид-1,1-диоксида (120 мг, 0,334 ммоль) в безводном THF (12 мл) добавляли ВН3-Me2S (0,334 мл, 3,34 ммоль) при 10°С. Реакционную смесь перемешивали при 40°С в течение 4 часов, затем добавляли по каплям 1 М HCl (15 мл) и перемешивание продолжали при 50°С в течение 2 часов. Неочищенную реакционную смесь нейтрализовали твердым NaHCO3 и экстрагировали EtOAc (30 мл × 2). Объединенные органические слои промывали рассолом (10 мл), затем сушили над безводным Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения в виде рацемической смеси. Энантиомеры разделяли посредством препаративной хиральной СФХ с получением (-)-2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-3-((метиламино)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида в качестве первого элюируемого пика (12 мг, 10%) в виде белого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 8.00-7.89 (m, 3H), 7.85 (d, J=8,0 Гц, 1H), 7.73 (s, 1H), 7.18 (dd, J=2,3, 9,5 Гц, 1Н), 6.96 (dt, J=2,5, 9,2 Гц, 1Н), 5.14-5.07 (m, 1H), 3.64 (d, J=12,5 Гц, 1H), 3.27 (br s, 1H), 2.81 (s, 3H); ЖХ-МС: m/z 346,0 (M+H)+; [α]20D -74° (c=0,002 г/мл, МеОН).
Пример 138: (+)-5-(6-фтор-1H-индол-3-ил)-3-((метиламино)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид

Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 137 (9 мг, выход 8%), в виде белого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 8.03-7.89 (m, 3H), 7.85 (d, J=8,0 Гц, 1H), 7.75 (s, 1H), 7.20 (dd, J=2,4, 9,7 Гц, 1Н), 6.98 (dt, J=2,4, 9,2 Гц, 1H), 5.14 (dd, J=3,5, 9,5 Гц, 1H), 3.67 (dd, J=3,5, 13,1 Гц, 1H), 3.32-3.26 (m, 1H), 2.83 (s, 3H); ЖХ-МС: m/z 346,0 (М+Н)+; [α]20D = +60,75° (с=0,004 г/мл, МеОН).
Пример 139: (-)-N-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)метансульфонамид
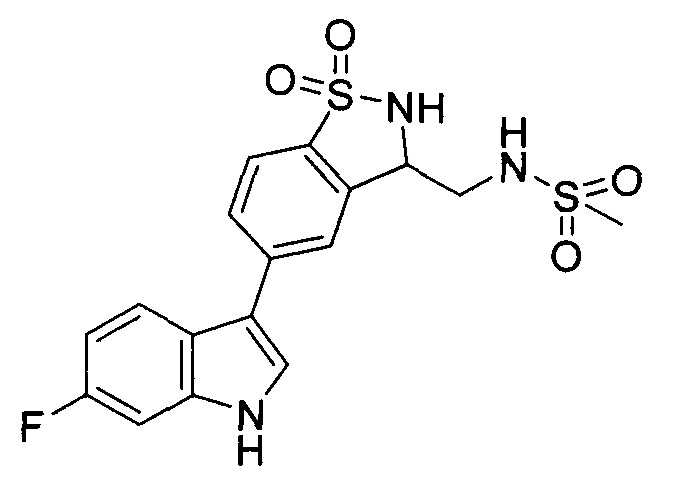
Стадия 1: хиральный N-((2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)метансульфонамид
В охлажденный (ледяная баня) раствор 3-(аминометил)-2-(трет-бутил)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (250 мг, 0,645 ммоль) и DIPEA (0,213 мл, 1,29 ммоль) в DCM (15 мл) добавляли по каплям MsCl (0,06 мл, 0,62 ммоль). Реакционную смесь перемешивали при 20°С в течение 8 часов, затем разбавляли водой (15 мл). Слои разделяли и органический слой сушили над безводным Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения в виде рацемической смеси (290 мг, 97%). Рацемический N-((2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)-метансульфонамид очищали посредством препаративной хиральной СФХ с получением двух энантиомеров: пик 1 (146 мг) и пик 2 (146 мг).
Стадия 2: (-)-N-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)метансульфонамид
К раствору хирального N-((2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)метансульфонамида (Стадия 1, Пик 1, 146 мг, 0,314 ммоль) в DCM (5 мл) добавляли HCl/MeOH (20 мл). Реакционную смесь перемешивали при 30°С в течение 16 часов, затем концентрировали и очищали посредством препаративной ВЭЖХ с получением (-)-N-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)метансульфонамида (70 мг, 55%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1H), 8.05 (d, J=5,3 Гц, 1Н), 7.98 (dd, J=5,3, 8,8 Гц, 1Н), 7.95-7.89 (m, 3H), 7.85-7.81 (m, 1Н), 7.39 (t, J=6,0 Гц, 1Н), 7.27 (dd, J=2,4, 9,9 Гц, 1Н), 6.99 (dt, J=2,5, 9,3 Гц, 1Н), 4.76 (q, J=5,6 Гц, 1Н), 3.46-3.40 (m, 1Н), 3.34-3.28 (m, 1Н), 2.96 (s, 3H); LCMS: m/z 409,8 [М+Н]+; [α]20D -72,3° (c=4,64 мг/мл, DMF).
Пример 140: (+)-N-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)метансульфонамид
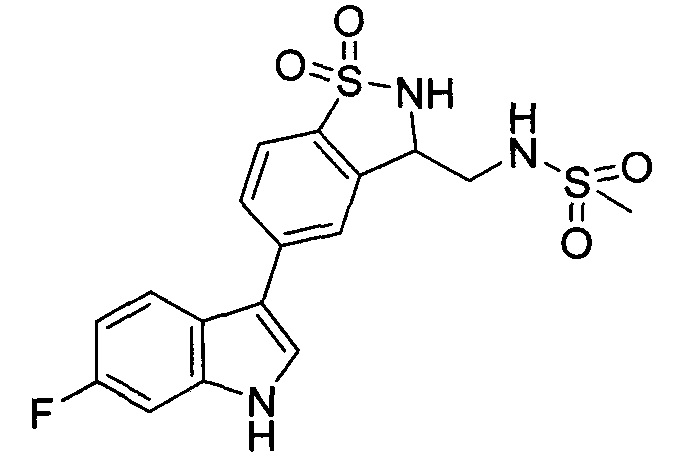
К раствору хирального N-((2-(трет-бутил)-5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)метансульфонамида (Пример 139, Стадия 1, Пик 2, 146 мг, 0,314 ммоль) в DCM (5 мл) добавляли HCl/MeOH (20 мл). Реакционную смесь перемешивали при 30°С в течение 16 часов, затем концентрировали и очищали посредством препаративной ВЭЖХ с получением (+)-N-((5-(6-фтор-1H-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]-изотиазол-3-ил)метил)метансульфонамида (70 мг, 55%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1H), 8.05 (d, J=5,0 Гц, 1Н), 7.99 (dd, J=5,3, 8,8 Гц, 1Н), 7.94 (s, 1Н), 7.93-7.89 (m, 2H), 7.86-7.82 (m, 1Н), 7.39 (t, J=6,1 Гц, 1Н), 7.28 (dd, J=2,3, 9,8 Гц, 1Н), 7.00 (dt, J=2,4, 9,2 Гц, 1Н), 4.77 (q, J=5,9 Гц, 1Н), 3.46-3.39 (m, 1H), 3.32 (d, J=7,0 Гц, 1Н), 2.99-2.94 (m, 3H); LCMS: m/z 409,8 [М+Н]+; [α]20D +37,3° (с=1,84 мг/мл, DMF).
Пример 141: (+)-2-(2,3-дигидроксипропил)-5-(6-фтор-1H-индол-3-ил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
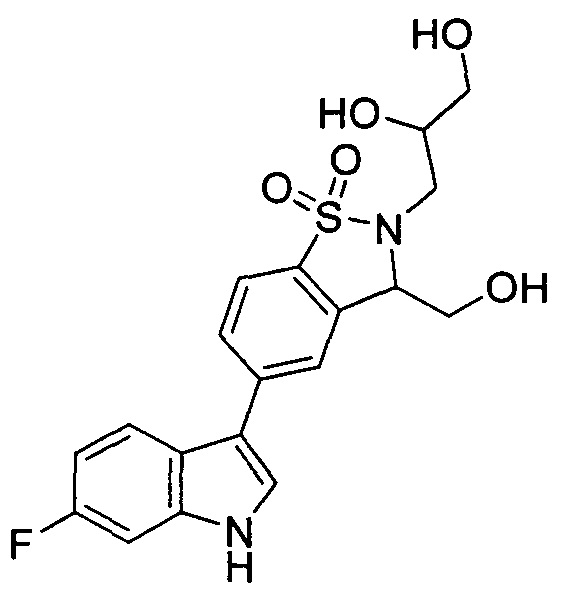
Стадия 1: (5-бром-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)-метилацетат
Раствор (5-бром-2-(трет-бутил)-1,1-диоксидо-2,3-дигидробензо[d]-изотиазол-3-ил)метилацетата (1,9 г, 5,1 ммоль) в TFA (25 мл) перемешивали при 30°С в течение 24 часов, затем концентрировали и разбавляли EtOAc (50 мл). Смесь промывали NaHCO3 (насыщ.) (10 мл) и рассолом (10 мл × 2), затем сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного 5-бром-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)-метилацетата (1,70 г, 100%) в виде желтой смолы. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 7.75-7.62 (m, 3H), 5.33 (d, J=4,5 Гц, 1Н), 4.98-4.86 (m, 1Н), 4.57 (dd, J=4,3, 11,5 Гц, 1Н), 4.27-4.21 (m, 1Н), 2.14-2.08 (m, 3H).
Стадия 2: (2-аллил-5-бром-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метилацетат
К суспензии (5-бром-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)-метилацетата (800 мг, 2,50 ммоль) и K2CO3 (1,0 г, 7,5 ммоль) в DMF (8 мл) добавляли 3-бромпроп-1-ен (363 мг, 3,0 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 25°С в течение 1 часа, затем вливали в воду (10 мл) и экстрагировали EtOAc (3×15 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением (2-аллил-5-бром-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метилацетата (620 мг, 69%) в виде желтой смолы. 1Н ЯМР (400 МГц, CDCl3) δ [м.д.] 7.75-7.62 (m, 3H), 6.00-5.85 (m, 1H), 5.42 (s, 1H), 5.37 (s, 1H), 5.32 (d, J=1,0 Гц, 1Н), 5.34 (s, 1H), 4.67-4.62 (m, 1H), 4.61-4.54 (m, 1H), 4.30 (d, J=4,5 Гц, 1H), 4.15-4.11 (m, 1H), 4.15-4.07 (m, 1H), 3.95 (dd, J=0,8, 8,0 Гц, 1H), 2.09-2.05 (m, 3Н).
Стадия 3: (5-бром-1,1-диоксидо-2-(оксиран-2-илметил)-2,3-дигидробензо-[d]изотиазол-3-ил)метилацетат
К раствору (2-аллил-5-бром-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метилацетата (420 мг, 1,17 ммоль) в DCM (15 мл) добавляли m-СРВА (947 мг, 4,66 ммоль) в двух порциях. Реакционную смесь перемешивали при 30°С в течение 18 часов, затем разбавляли DCM (10 мл) и промывали NaHCO3 (насыщ.) (5 мл), Na2SO3 (5 мл) и рассолом (5 мл). Органический слой сушили над Na2SO4, фильтровали и концентрировали, затем очищали посредством колоночной хроматографии (0-40% ЕА/РЕ) с получением (5-бром-1,1-диоксидо-2-(оксиран-2-илметил)-2,3-дигидробензо[d]изотиазол-3-ил)метилацетата (260 мг, 59%) в виде белого твердого вещества.
Стадия 4: (5-бром-2-(2,3-дигидроксипропил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метилацетат
К раствору (5-бром-1,1-диоксидо-2-(оксиран-2-илметил)-2,3-дигидробензо[d]изотиазол-3-ил)метилацетата (260 мг, 0,55 ммоль) в диоксане (4 мл) добавляли H2SO4 (4 мл) при 15°С. Реакционную смесь нагревали до 60°С и перемешивали в течение 6 часов, затем гасили NaHCO3 (насыщ.) (6 мл). Смесь экстрагировали ЕА (15 мл × 3) и объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного 5-бром-2-(2,3-дигидроксипропил)-1,1-диоксидо-2,3-дигидробензо-[d]изотиазол-3-ил)метилацетата (230 мг) в виде желтой смолы, которую использовали непосредственно на следующей стадии.
Стадия 5: трет-бутил-3-(3-(ацетоксиметил)-2-(2,3-дигидроксипропил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1Н-индол-1-карбоксилат
К раствору трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (211 мг, 0,583 ммоль), (5-бром-2-(2,3-дигидроксипропил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)-метилацетата (230 мг, 0,583 ммоль) и K3PO4 (248 мг, 1,17 ммоль) в смеси диоксан/H2O (8 мл/2 мл) добавляли Pd(dppf)Cl2 (43 мг, 0,06 ммоль) при 20°С. Реакционную смесь перемешивали при 90°С в течение 6 часов, затем разбавляли H2O (5 мл) и экстрагировали EtOAc (10 мл × 3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением трет-бутил-3-(3-(ацетоксиметил)-2-(2,3-дигидроксипропил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (376 мг) в виде желтого масла, которое использовали непосредственно на следующей стадии.
Стадия 6: 2-(2,3-дигидроксипропил)-5-(6-фтор-1H-индол-3-ил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
Раствор трет-бутил-3-(3-(ацетоксиметил)-2-(2,3-дигидроксипропил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (320 мг, 0,583 ммоль) в MeNH2/EtOH (30%, 10 мл) перемешивали в герметично закрытой пробирке при 60°С в течение 4 часов. Реакционную смесь концентрировали и очищали посредством колоночной хроматографии с получением указанного в заголовке соединения в виде смеси диастереомеров. Диастереомеры разделяли посредством препаративной хиральной СФХ с получением (+)-2-(2,3-дигидроксипропил)-5-(6-фтор-1H-индол-3-ил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида в качестве первого элюируемого пика (26 мг, 11%) в виде коричневого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.95-7.85 (m, 3H), 7.81 (s, 1H), 7.68 (s, 1H), 7.16 (dd, J=2,1, 9,7 Гц, 1H), 6.94 (dt, J=2,3, 9,2 Гц, 1H), 4.80 (s, 1H), 4.13-3.97 (m, 3H), 3.74-3.62 (m, 2Н), 3.60-3.46 (m, 2Н); LCMS: m/z 407,0 (M+H)+; [α]20D +12° (с=2,0 мг/мл, МеОН).
Пример 142: (-)-2-(2,3-дигидроксипропил)-5-(6-фтор-1H-индол-3-ил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
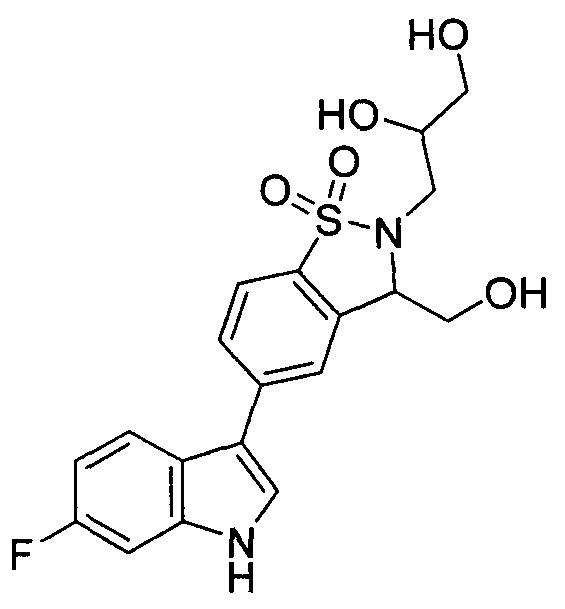
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 141 (18 мг, 8%), в виде желтого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.94-7.85 (m, 3H), 7.81 (s, 1H), 7.68 (s, 1H), 7.19-7.13 (m, 1H), 6.99-6.90 (m, 1H), 4.69-4.63 (m, 1H), 4.14-4.00 (m, 3H), 3.81 (dd, J=3,5, 14,8 Гц, 1H), 3.68-3.58 (m, 2H), 3.29-3.22 (m, 1H); LCMS: m/z 407,0 (М+Н)+; [α]20D -10° (с=2,2 мг/мл, МеОН).
Пример 143: (-)-2-(2,3-дигидроксипропил)-5-(6-фтор-1H-индол-3-ил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
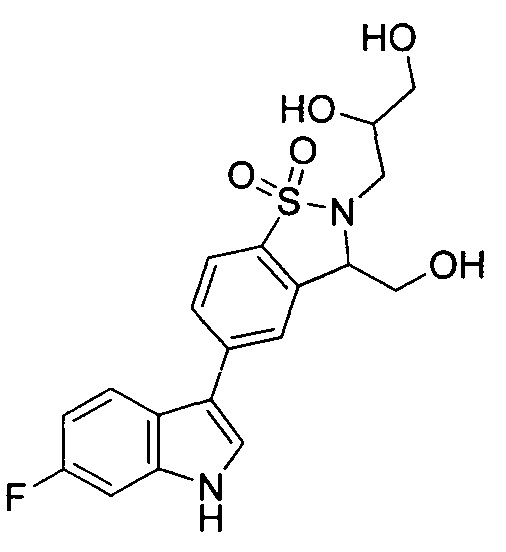
Указанное в заголовке соединение получили в качестве третьего элюируемого пика из хирального разделения, описанного в Примере 141 (15 мг, 6%), в виде серого твердого вещества. 1Н ЯМР (400 МГц, MeOD) δ [м.д.] 7.93-7.86 (m, 3H), 7.80 (d, J=8,0 Гц, 1H), 7.68 (s, 1H), 7.22-7.12 (m, 1H), 6.99-6.89 (m, 1H), 4.80 (t, J=4,1 Гц, 1H), 4.06 (d, J=4,0 Гц, 3H), 3.75-3.61 (m, 2H), 3.60-3.42 (m, 2H); LCMS: m/z 407,0 (M+H)+; [α]20D -15° (c=5,0 мг/мл, МеОН).
Пример 144: (+)-2-(2,3-дигидроксипропил)-5-(6-фтор-1H-индол-3-ил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
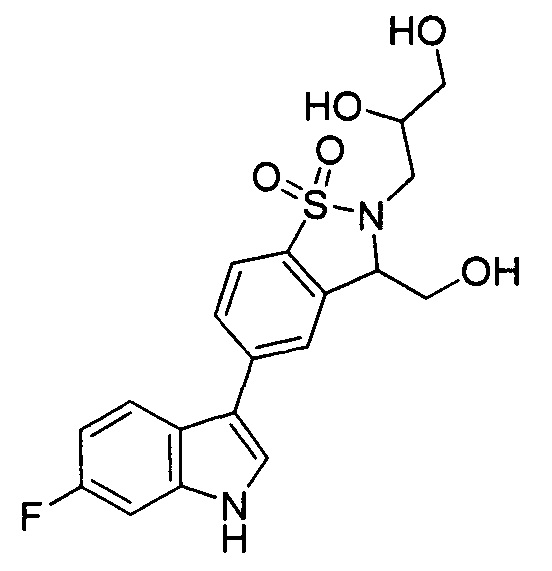
Указанное в заголовке соединение получили в качестве четвертого элюируемого пика из хирального разделения, описанного в Примере 141 (20 мг, 8%), в виде серого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.94-7.86 (m, 3H), 7.80 (d, J=8,3 Гц, 1H), 7.68 (s, 1H), 7.16 (dd, J=2,1, 9,7 Гц, 1H), 6.95 (dt, J=2,4, 9,2 Гц, 1H), 4.66 (t, J=4,3 Гц, 1H), 4.11-4.01 (m, 3H), 3.81 (dd, J=3,5, 15,1 Гц, 1H), 3.63 (dd, J=5,1, 11,2 Гц, 2Н), 3.29-3.23 (m, 1H); LCMS: m/z 407,0 (M+H)+; [α]20D +19,5° (c=2,0 мг/мл, МеОН).
Пример 145: (-)-2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилпропанамид
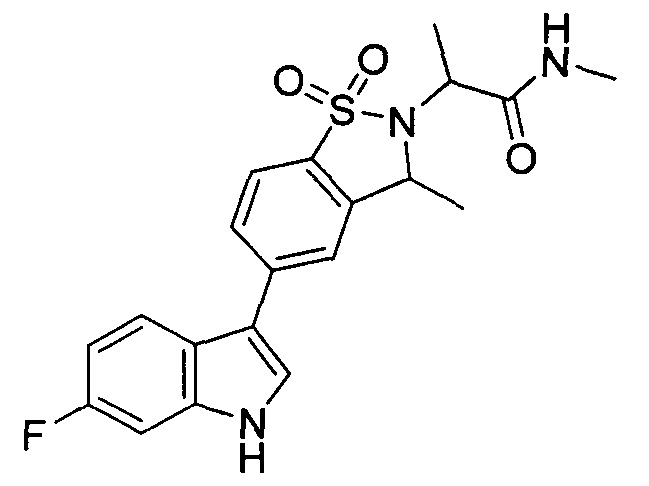
Стадия 1: этил-2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропаноат
К суспензии 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (1,2 г, 4,1 ммоль) и K2CO3 (1,7 г, 12 ммоль) в DMF (20 мл) добавляли этил-2-бромпропаноат (1,1 г, 6,2 ммоль). Реакционную смесь перемешивали в течение 1 часа, затем разбавляли EtOAc (60 мл) и промывали H2O (10 мл × 2) и рассолом (10 мл × 2). Органический слой сушили над Na2SO4, фильтровали и концентрировали, затем очищали посредством колоночной хроматографии с получением неочищенного этил-2-(5-бром-3-метил-1,1-диоксидобензо[d]-изотиазол-2(3H)-ил)пропаноата (1,2 г, 80%) в виде желтой смолы. 1H ЯМР (400 МГц, CDCl3) δ [м.д.] 7.69-7.61 (m, 2Н), 7.54 (s, 1H), 4.85 (d, J=6,8 Гц, 1H), 4.34 (q, J=7,3 Гц, 1H), 4.20 (q, J=7,1 Гц, 2Н), 1.74-1.68 (m, 3H), 1.57 (d, J=6,5 Гц, 3H), 1.23 (t, J=7,2 Гц, 3H); LCMS: m/z для 385,9 (M+Na)+.
Стадия 2: трет-бутил-3-(2-(1-этокси-1-оксопропан-2-ил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилат
К раствору трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-индол-1-карбоксилата (1,2 г, 3,3 ммоль), этил-2-(5-бром-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)пропаноата (1,2 г, 3,3 ммоль) и K3PO4 (1,4 г, 6,6 ммоль) в смеси диоксан/H2O (25 мл/5 мл) добавляли Pd(dppf)Cl2 (240 мг, 0,328 ммоль). Реакционную смесь перемешивали при 90°С в течение 16 часов, затем разбавляли H2O (20 мл) и экстрагировали EtOAc (20 мл × 3). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного трет-бутил-3-(2-(1-этокси-1-оксопропан-2-ил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (1,8 г, более 100%) в виде светло-желтого масла, которое использовали непосредственно на следующей стадии.
Стадия 3: 2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]-изотиазол-2(3H)-ил)-N-метилпропанамид
Раствор трет-бутил-3-(2-(1-этокси-1-оксопропан-2-ил)-3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-6-фтор-1H-индол-1-карбоксилата (1,7 г, 3,3 ммоль) в MeNH2/EtOH (30%, 30 мл) перемешивали при 60°С в течение 4 часов, затем концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии с получением указанного в заголовке соединения в виде смеси диастереомеров. Диастереомеры разделяли посредством препаративной хиральной СФХ с получением (-)-2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилпропанамида в качестве первого элюируемого пика (106 мг, 8%) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.71-11.64 (m, 1H), 7.97-7.84 (m, 5H), 7.82-7.76 (m, 1H), 7.30-7.24 (m, 1H), 7.04-6.97 (m, 1H), 5.41-5.33 (m, 1H), 4.38-4.29 (m, 1H), 2.54 (d, J=4,8 Гц, 3H), 1.62-1.48 (m, 6H); LCMS: m/z 402,0 (M+H)+; [α]20D -13,5° (c=2,0 мг/мл, МеОН).
Пример 146: (+)-2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилпропанамид
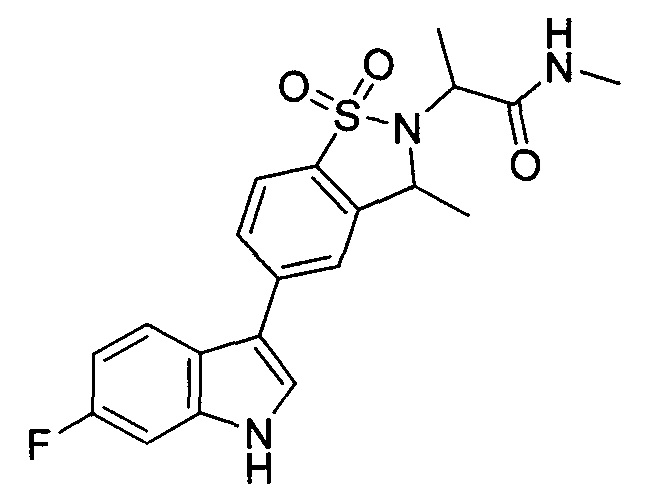
Указанное в заголовке соединение получили в качестве второго элюируемого пика из хирального разделения, описанного в Примере 145 (60 мг, 5%), в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.69 (br s,1H), 7.98-7.83 (m, 6H), 7.30-7.24 (m, 1H), 7.05-6.98 (m, 1H), 4.94-4.87 (m, 1H), 4.35-4.28 (m, 1H), 2.64 (d, J=4,5 Гц, 3H), 1.50 (d, J=6,5 Гц, 3H), 1.40 (d, J=7,0 Гц, 3H); LCMS: m/z 402,0 (M+H)+; [α]20D +13,5° (с=2,0 мг/мл, МеОН).
Пример 147: 2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилпропанамид
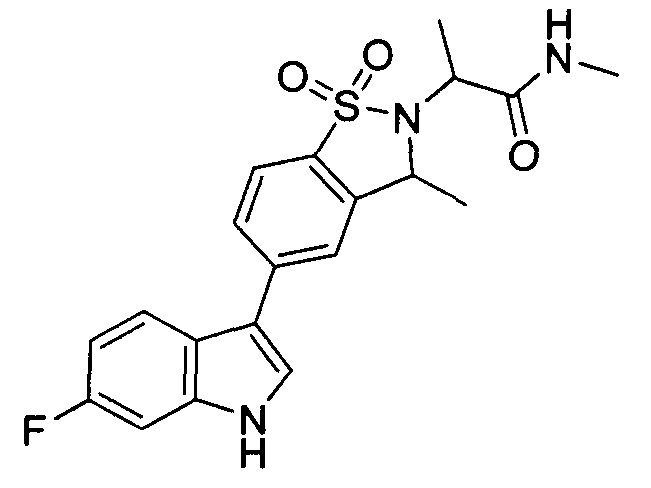
Указанное в заголовке соединение получили в качестве третьего элюируемого пика из хирального разделения, описанного в Примере 145 (93 мг, 7%), в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.68 (br s, 1Н), 7.97-7.85 (m, 5H), 7.82-7.76 (m, 1H), 7.27 (dd, J=2,3, 9,8 Гц, 1Н), 7.01 (d, J=2,0 Гц, 1H), 5.37 (d, J=6,8 Гц, 1H), 4.34 (d, J=7,3 Гц, 1H), 2.54 (d, J=4,5 Гц, 3H), 1.55 (dd, J=7,0, 12,3 Гц, 6H); LCMS: m/z для 402,0 (М+Н)+; [α]20D +12,17° (с=2,0 мг/мл, МеОН).
Пример 148: (-)-2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилпропанамид
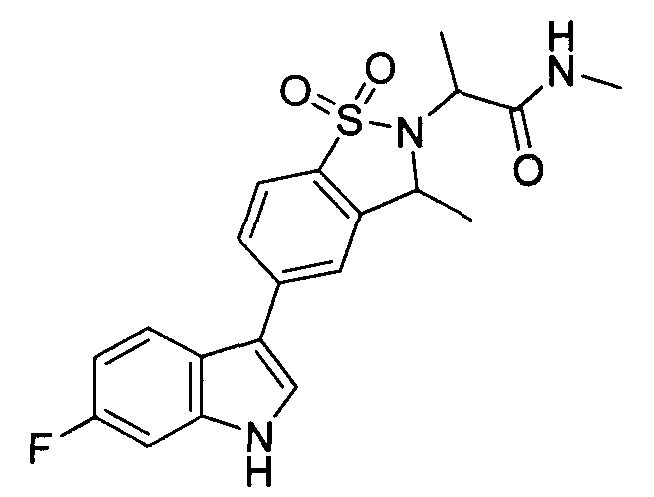
Указанное в заголовке соединение получили в качестве четвертого элюируемого пика из хирального разделения, описанного в Примере 145 (215 мг, 16%), в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.70 (br s, 1H), 8.00-7.83 (m, 6H), 7.27 (dd, J=1,9, 9,9 Гц, 1H), 7.01 (dt, J=2,1, 9,2 Гц, 1H), 4.91 (q, J=6,4 Гц, 1H), 4.32 (q, J=6,8 Гц, 1H), 2.64 (d, J=4,8 Гц, 3H), 1.50 (d, J=6,3 Гц, 3H), 1.40 (d, J=7,0 Гц, 3H); LCMS: m/z 402,0 (M+H)+; [α]20D -9,33° (с=2,0 мг/мл, МеОН).
Пример 149: 5-(6-фтор-1H-индол-3-ил)-3-метил-2-(пиридин-2-илметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
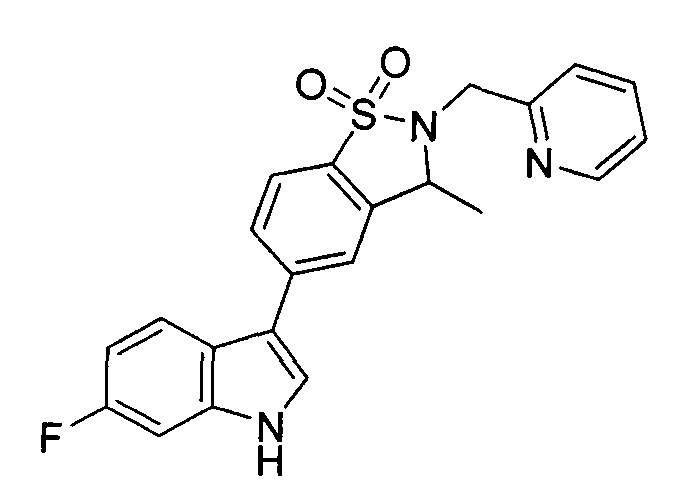
Стадия 1: трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилат
К смеси 5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (726 мг, 2,77 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индол-1-карбоксилата (1000 мг, 2,77 ммоль) и K2PO4 (1210 мг, 5,54 ммоль) в 1,4-диоксане (13,8 мл, с=0,2 М) и воде (4,6 мл, 0,6 М) добавляли Pd(dppf)Cl2 (158 мг, 0,19 ммоль) в атмосфере N2. По окончании добавления темно-красный раствор перемешивали при 70°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли EtOAc, промывали NH4Cl (насыщ.), сушили над Na2SO4, фильтровали и концентрировали. Осадок очищали посредством колоночной хроматографии (15-60% EtOAc/гептан) с получением трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилата (910 мг, 79%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 8.11 (s, 1Н), 7.83-7.98 (m, 6H), 7.18-7.32 (m, 1H), 4.79 (dd, J=6,42, 4,59 Гц, 1Н), 1.68 (s, 9H), 1.55 (d, J=6,72 Гц, 3H); ЖХ-МС: m/z 417 (M+H)+.
Стадия 2: трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2-(пиридин-2-илметил)-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилат
К смеси трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2,3-дигидробензо-[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (30,1 мг, 0,07 ммоль), и 2-(хлорметил)пиридин-HCl (18,5 мг, 0,11 ммоль) в диоксане (0,69 мл, 0,21 М) и DMF (0,69 мл, с=0,21 М) добавляли по каплям при комнатной температуре трет-бутоксид натрия. Смесь перемешивали при комнатной температуре в течение 5 мин, затем нагревали до 80°С и перемешивали в течение 1,5 часов. Реакционную смесь фильтровали и очищали посредством колоночной хроматографии с получением трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2-(пиридин-2-илметил)-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилата (18 мг, выход 55%).
Стадия 3: 5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(пиридин-2-илметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
К раствору трет-бутил-6-фтор-3-(3-метил-1,1-диоксидо-2-(пиридин-2-илметил)-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (18,0 мг, 0,036 ммоль) в DCM (0,0709 мл, с=0,5 М) добавляли HCl (0,177 мл, 4,0 М) в диоксане. Реакционную смесь перемешивали при 30°С в течение ночи, затем концентрировали с получением 5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(пиридин-2-илметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида (15,7 мг, выход 100%). 1H ЯМР (400 МГц, MeOD) δ [м.д.] 8.83 (d, J=5,01 Гц, 1Н), 8.60-8.70 (m, 1H), 8.26 (d, J=8,07 Гц, 1H), 8.05 (t, J=6,72 Гц, 1H), 7.94-7.99 (m, 1H), 7.84-7.91 (m, 3H), 7.73 (s, 1H), 7.18 (dd, J=9,66, 2,32 Гц, 1H), 6.96 (td, J=9,20, 2,38 Гц, 1H), 4.94-5.11 (m, 2H), 4.84 (d, J=6,60 Гц, 1H), 1.69 (d, J=6,48 Гц, 3H); ЖХ-МС: m/z 408 (M+H)+.
Пример 150: 5-(6-фтор-1H-индол-3-ил)-3-метил-2-((6-метилпиридин-2-ил)-метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
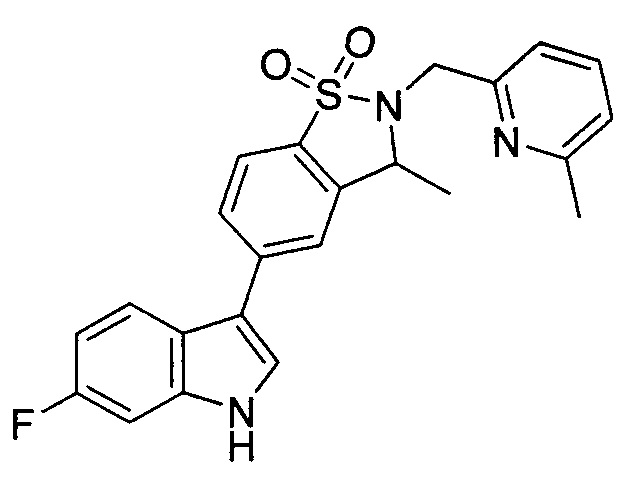
Следуя общей методике, изложенной в Примере 149, начиная с 2-(хлорметил)-6-метилпиридина, получили указанное в заголовке соединение в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 11.72 (br. s., 1H), 8.12 (br. s., 1H), 7.89-8.00 (m, 5H), 7.66 (br. s., 1H), 7.55 (br. s., 1H), 7.28 (dd, J=9,84, 2,38 Гц, 1H), 7.02 (td, J=9,26, 2.38 Гц, 1H), 4.67-4.89 (m, 3H), 2.66 (br. s., 3H), 1.54 (d, J=6,48 Гц, 3H); ЖХ-МС: m/z 422 (M+H)+.
Пример 151: (R)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-(пиридин-2-илметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
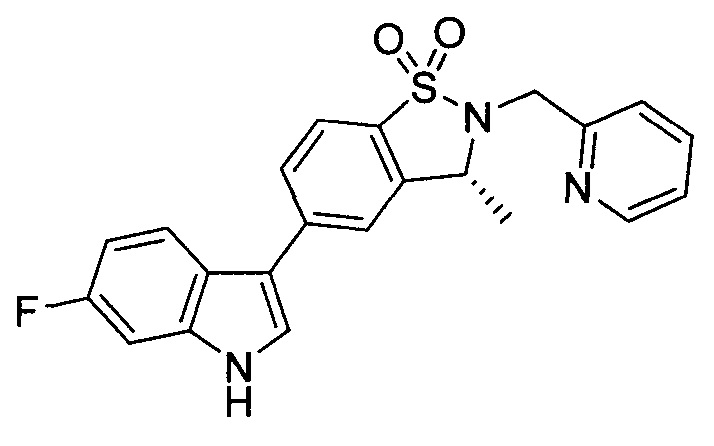
Стадия 1: (3R)-5-бром-3-метил-2,3-дигидро-1,2-бензотиазол-1,1-диоксид
Раствор муравьиной кислоты/триэтиламина (10,0 мл, 5:2) добавляли к раствору 5-бром-1,2-бензотиазол-3(2H)-он-1,1-диоксида (5,0 г, 19,2 ммоль) и RuCl(пара-цимен)[(R,R)-Ts-DPEN] (122 мг, 0,19 ммоль) при комнатной температуре с получением светло-янтарного раствора. Реакционную смесь перемешивали в течение ночи. Растворитель удаляли и это янтарное масло добавляли к 200 мл 50%-ного насыщенного NaHCO3. Водный слой экстрагировали EtOAc (3×150 мл) и объединенные органические экстракты промывали рассолом, сушили над MgSO4, фильтровали и концентрировали с получением рыжевато-коричневого твердого вещества. Это рыжевато-коричневое твердое вещество растирали с 50 мл МТВЕ с получением указанного в заголовке соединения (2 г, 40%, 96% ее) в виде белого твердого вещества. Фильтрат концентрировали и растирали снова с 2 мл МТВЕ с получением не совсем белого твердого вещества, которое дополнительно очищали посредством колоночной хроматографии (силикагель, 30% гептан-EtOAc) с получением указанного в заголовке соединения (1 г, 20%, 96% ее) в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 8.01 (s, 1Н), 7.93 (s, 1H), 7.68-7.81 (m, 2H), 4.70 (q, J=6,7 Гц, 1Н), 1.47 (d, J=6,7 Гц, 3H); ЖХ-МС: m/z 262/264 (M+H)+; [α]20D +42,9° (с=0,2, МеОН), [α]20D +30,3° (с=0,4, CHCl3).
Стадия 2: трет-бутил-(R)-6-фтор-3-(3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилат
К раствору (R)-5-бром-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида (890 мг, 3,4 ммоль), трет-бутил-6-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-индол-1-карбоксилата (1230 мг, 3,4 ммоль) и K3PO4 (1340 мг, 6,1 ммоль) в 1,4-диоксане (17,0 мл, 0,2 M) и деионизированной воде (5,7 мл, 0,6 M) добавляли Pd(dppf)Cl2 (194 мг, 0,24 ммоль) в атмосфере N2. По окончании добавления красный раствор перемешивали при 70°С в течение 2,2 часа. Реакционную смесь охлаждали до комнатной температуры и разбавляли EtOAc, промывали NH4Cl (насыщ.), сушили над Na2SO4, фильтровали и концентрировали. Неочищенный осадок очищали посредством колоночной хроматографии (10-55% EtOAc/гептан) с получением трет-бутил-(R)-6-фтор-3-(3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (1213 мг, 84%) в виде желтого твердого вещества. 1H ЯМР (400 МГц, DMSO-d6) δ [м.д.] 8.11 (s, 1Н), 7.84-7.98 (m, 6 H), 7.20-7.31 (m, 1H), 4.79 (dd, J=6.42, 4.46 Гц, 1H), 1.67 (s, 9H), 1.55 (d, J=6.72 Гц, 3H); ЖХ-МС: m/z 417 (М+Н)+.
Стадия 3: трет-бутил-(R)-6-фтор-3-(3-метил-1,1-диоксидо-2-(пиридин-2-ил-метил)-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилат
К смеси трет-бутил-(R)-6-фтор-3-(3-метил-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (60,0 мг, 0,144 ммоль) и 2-(хлорметил)пиридина HCl (42,5 мг, 0,26 ммоль)) в диоксане (0,69 мл, 0,21 M) и DMF (0,69 мл, с=0,21 M) добавляли по каплям при комнатной температуре трет-бутоксид натрия (1,0 M). Смесь перемешивали при комнатной температуре в течение 5 мин и затем нагревали до 80°С в течение 1,5 часов. Реакционную смесь фильтровали и очищали посредством колоночной хроматографии с получением трет-бутил-(R)-6-фтор-3-(3-метил-1,1-диоксидо-2-(пиридин-2-илметил)-2,3-дигидробензо[d]изотиазол-5-ил)-1H-индол-1-карбоксилата (30 мг, 40%).
Стадия 4: (R)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-((6-метилпиридин-2-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид-трет-бутил-(R)-6-фтор-3-(3-метил-1,1-диоксидо-2-(пиридин-2-илметил)-2,3-дигидробензо[d]изотиазол-5-ил)-1Н-индол-1-карбоксилата (29 мг, 0,06 ммоль) в EtOAc (0,2 мл) обрабатывали HCl (0,31 мл) в диоксане (4,0 M). Реакционную смесь перемешивали при 30°С в течение ночи. Растворитель удаляли и осадок очищали посредством препаративной ВЭЖХ с получением (R)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-((6-метилпиридин-2-ил)метил)-2,3-дигидробензо[d]-изотиазол-1,1-диоксида (19 мг, 77%) в виде белого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 8.54 (br. s., 1H), 7.76-7.96 (m, 5H), 7.63-7.74 (m, 2H), 7.33-7.42 (m, 1H), 7.15 (dd, J=9,60, 2,26 Гц, 1H), 6.94 (td, J=9,23, 2,32 Гц, 1H), 4.65-4.74 (m, 2H), 4.55-4.65 (m, 1H), 1.53 (d, J=6,48 Гц, 3H); ЖХ-МС: m/z 408 (M+H)+.
Пример 152: (R)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-((6-метилпиридин-2-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
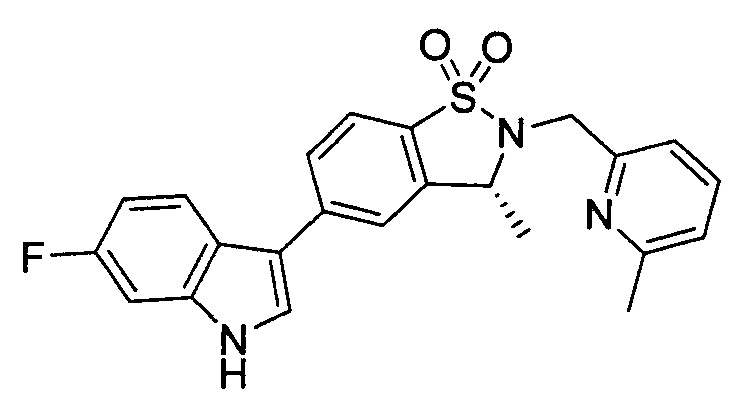
Следуя общей методике, изложенной в Примере 151, начиная с 2-(хлорметил)-6-метилпиридина, получили указанное в заголовке соединение в виде белого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.95-8.01 (m, 1Н), 7.87-7.94 (m, 2H), 7.85 (s, 1H), 7.71-7.81 (m, 2H), 7.54 (d, J=7,83 Гц, 1Н), 7.27 (d, J=7,70 Гц, 1Н), 7.21 (dd, J=9,72, 2,26 Гц, 1Н), 6.99 (td, J=9,17, 2,32 Гц, 1Н), 4.67-4.79 (m, 2H), 4.54-4.64 (m, 1Н), 2.61 (s, 3H), 1.58 (d, J=6,48 Гц, 3H); ЖХ-МС: m/z 422 (М+Н)+.
Пример 153: (R)-5-(6-фтор-1H-индол-3-ил)-3-метил-2-((2-метил-2Н-тетразол-5-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксид
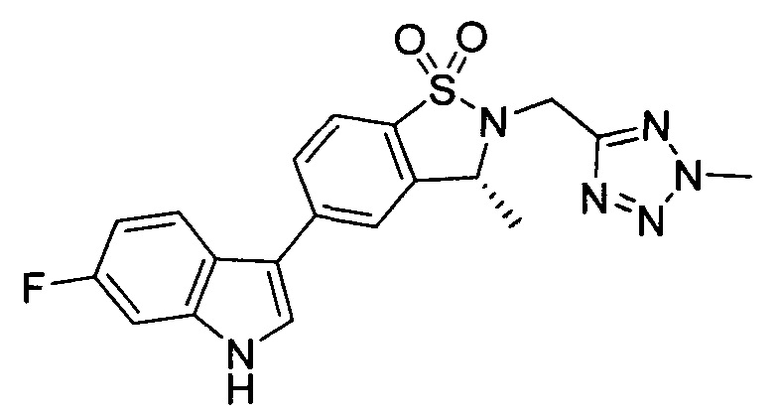
Следуя общей методике, изложенной в Примере 151, начиная с 5-(хлорметил)-2-метил-2Н-тетразола, получили указанное в заголовке соединение в виде белого твердого вещества. 1H ЯМР (400 МГц, MeOD) δ [м.д.] 7.76-7.95 (m, 4 Н), 7.68 (s, 1Н), 7.16 (dd, J=9,66, 2,32 Гц, 1Н), 6.95 (td, J=9,20, 2.38 Гц, 1Н), 4.84-4.91 (m, 1Н), 4.81 (s, 2H), 4.37 (s, 3H), 1.66 (d, J=6,48 Гц, 3H); ЖХ-МС: m/z 413 (M+H)+.
I. БИОЛОГИЧЕСКИЕ ПРИМЕРЫ
II.1. Анализ определения ферментативной активности TDO2
Соединения формулы I и ее подформул и их энантиомеры, соли и сольваты полезны в ингибировании ферментативной активности человеческой TDO2.
Для измерения активности TDO2, методику, описанную в Dolusic et al. J. Med. Chem.; 2011, 54, 5320-533, адаптируют следующим образом: реакционная смесь содержит (конечные концентрации) буфера фосфата калия (50 мМ, рН 7,5), аскорбиновую кислоту (0,25 М), метиленовый синий (0,125 мкМ), каталазу (40 единиц/мл, из бычьей печени, Sigma) и человеческий рекомбинантный фермент TDO2 (полученный как описано в Dolusic et al. J. Med. Chem.; 2011, 54, 5320-5334; 0,9 мкг) в отсутствие или в присутствии соединений по настоящему изобретению в тестируемых концентрациях (суммарный объем 112,5 мкл). Реакцию инициировали добавлением 37,5 мкл L-Trp (конечная концентрация 1 мМ) при комнатной температуре. Реакцию проводили при комнатной температуре в течение одного часа и останавливали добавлением 30 мкл 30% (масс./об.) трихлоруксусной кислоты.
Для превращения N-формилкинуренина в кинуренин реакционную смесь инкубировали при 65°С в течение 30 мин. Затем 150 мкл реакционной смеси смешивали с 120 мкл 2,5% (масс./об.) 4-(диметиламино)бензальдегида в уксусной кислоте и инкубировали в течение 5 мин при комнатной температуре. Концентрации кинуренина определяли путем измерения поглощения при 480 нм. Стандартную кривую строили с использованием чистого кинуренина. Активность TDO измеряли как описано выше с использованием десяти серийных концентраций соединений по настоящему изобретению. Данные подгоняли с использованием программного обеспечения Prism™ (GraphPad Software, Inc.), используя стандартные параметры.
В одном воплощении выбраны соединения, имеющие значение IC50 менее 2000 нМ, предпочтительно имеющие значение IC50 менее 1000 нМ.
II.2. Клеточный анализ определения активности TDO2
II.2. Клетки А172
Соединения формулы I ингибируют активность человеческой TDO2 в клетках, которые конститутивно экспрессируют TDO2, такие как клетки А172. А172 представляет собой клеточную линию, имеющую происхождение из клеток глиобластомы головного мозга человека. Клетки доступны в Американской коллекции типовых культур (АТСС®) как CRL-1620™.
Анализ (адаптированный от Pilotte L et al., Proc Natl Acad Sci USA, 2012, 109(7), 2497-502) осуществляли в 96-луночных плоскодонных планшетах с засеянными клетками человеческой глиобластомы А172, естественно экспрессирующих hTDO2 (получена как описано в Tilman et al., Mol Cancer, 2007, 17(6), 80), в концентрации 1,25×104 клеток/лунку в конечном объеме 200 мкл. Для определения TDO клетки инкубировали в течение ночи при 37°С при 5% CO2 в IMDM (Invitrogen), дополненной 2% FBS и 2% пенициллина/стрептомицина в присутствии соединений по настоящему изобретению в разных концентрациях.
Планшеты затем центрифугировали 5 мин при 1000 об/мин и 100 мкл супернатанта собирали в конический планшет, добавляли 30 мкл 30% ТСА и дополнительно центрифугировали при 3000 × g в течение 10 минут. 100 мкл супернатанта собирали в плоскодонный планшет, добавляли 100 мкл 2% (масс./об.) 4-(диметиламино)бензальдегида в уксусной кислоте и инкубировали в течение 5 мин при комнатной температуре. Концентрации кинуренина определяли путем измерения поглощения при 480 нм. Стандартную кривую строили с использованием чистого кинуренина. Активность TDO измеряли как описано выше с использованием десяти серийных концентраций соединений по настоящему изобретению. Данные подгоняли с использованием программного обеспечения Prism™ (GraphPad Software, Inc.), используя стандартные параметры.
Суммарные данные по биологической активности репрезентативных соединений в клетках глиобластомы головного мозга человека, как определено в вышеуказанном анализе, приведены в следующей таблице:
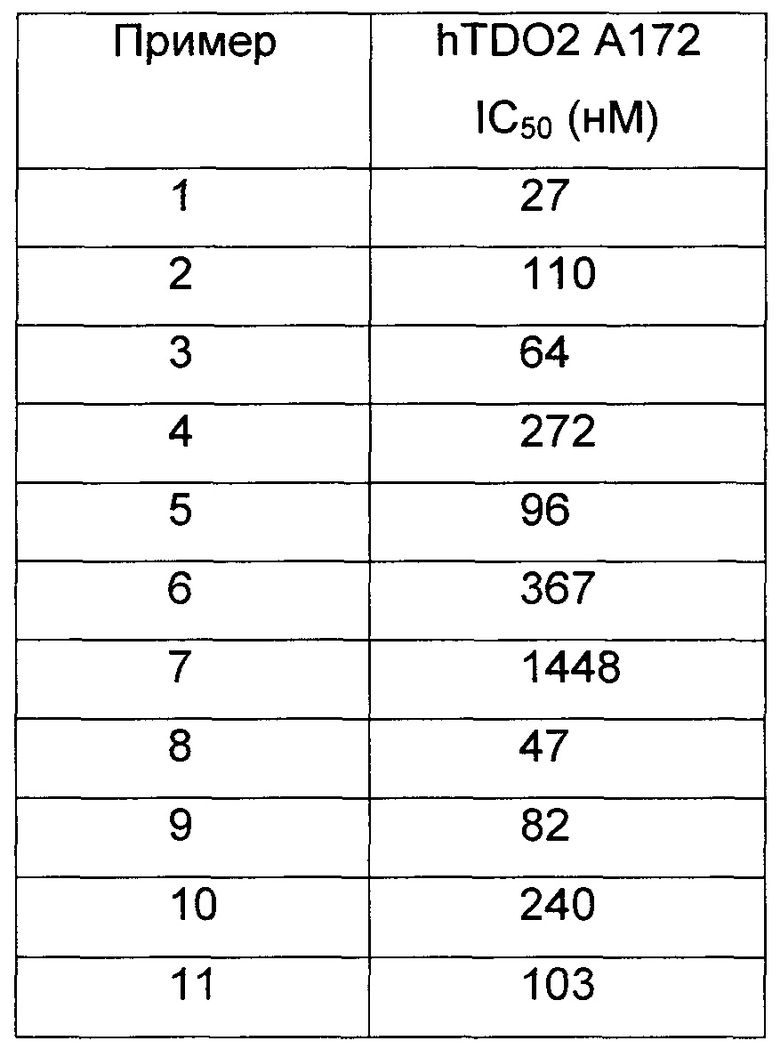
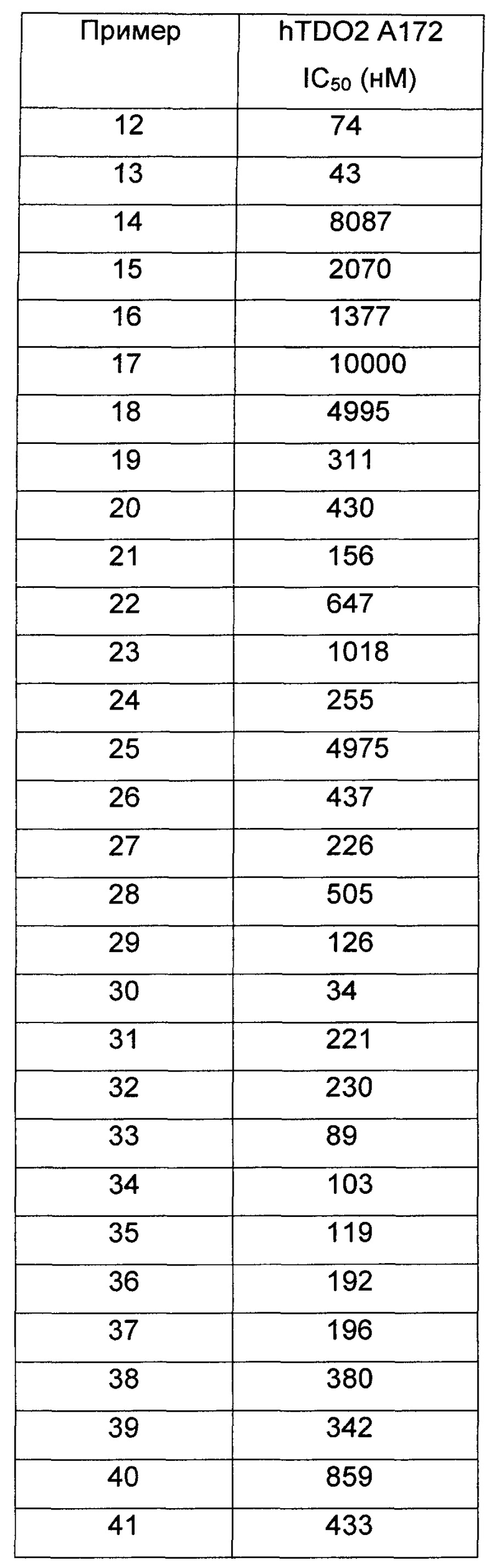
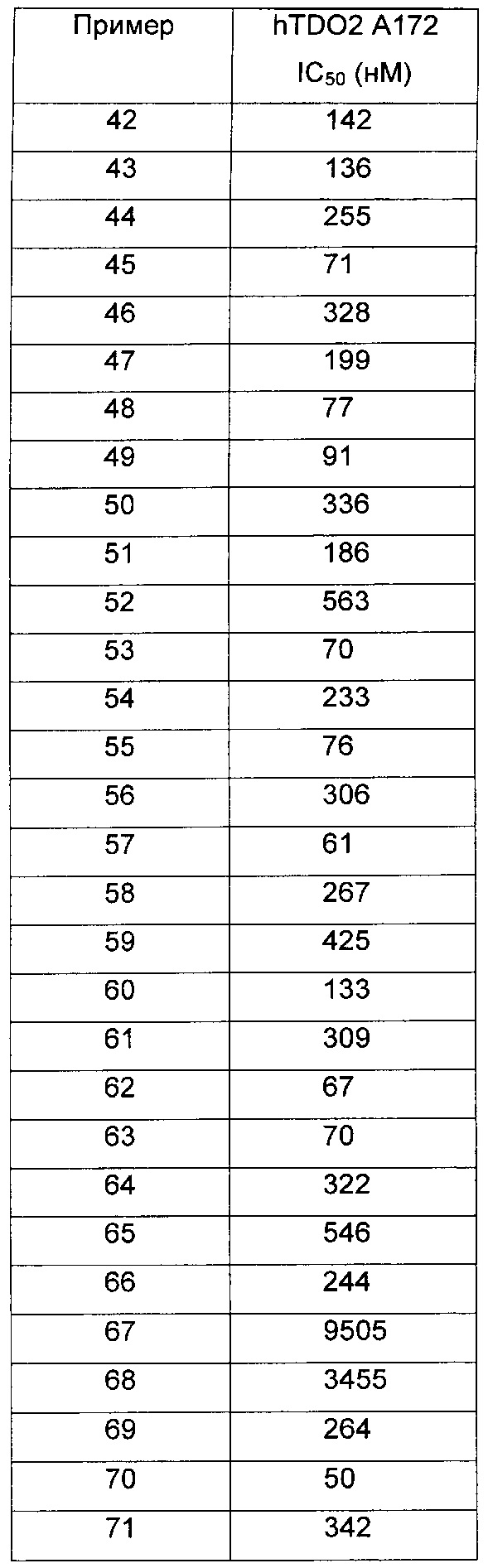
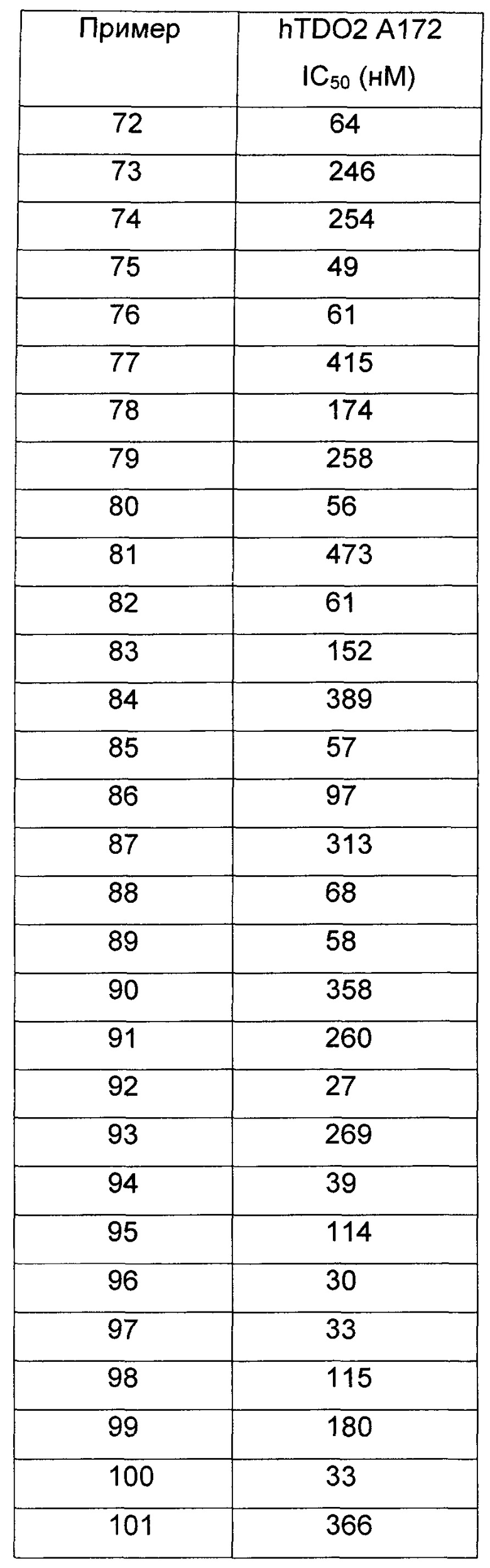

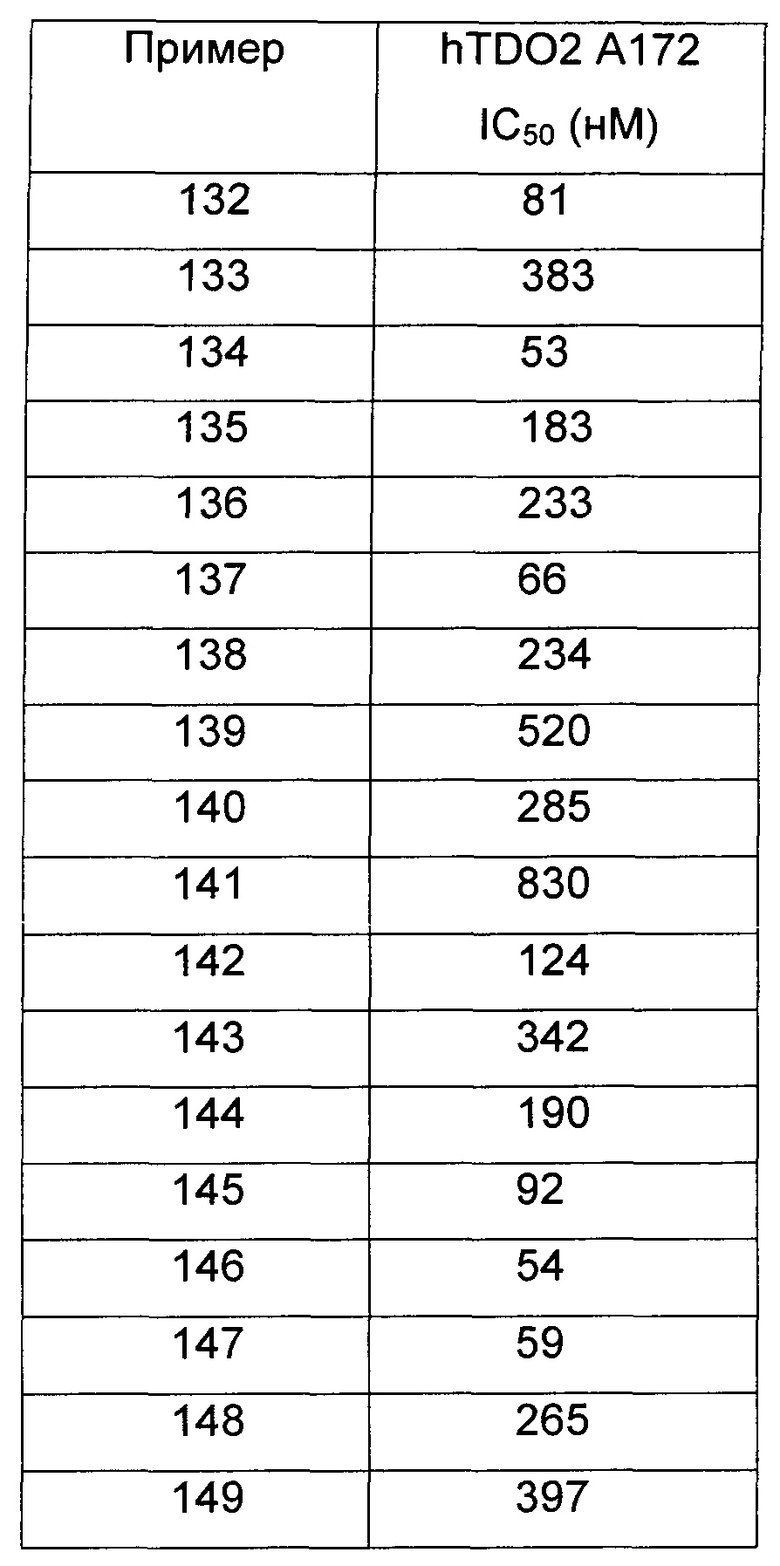
В одном воплощении выбраны соединения, имеющие значение IC50 менее 1000 нМ. В другом воплощении выбраны соединения, имеющие значение IC50 менее 300 нМ.
II.3. Фармакодинамический анализ определения активности TDO2 in vivo: повышение уровней триптофана в крови у мышей
Соединения по настоящему изобретению можно оценить на их способность повышать количество триптофана в крови в мышиной модели. Вкратце, самкам мышей BALB/c (возрастом 7-8 недель) давали либо суспензию одного из соединений по настоящему изобретению в 0,5% гидроксипропилметилцеллюлозе (НРМС) K4M (4000 мПа⋅с (сП), Methocell™, Dow chemical)/0,25% Tween® 20 (Sigma Aldrich) в различных дозах (30, 60 и 100 мг/кг), либо контрольный носитель (0,5% НРМС K4M/0,25% Tween 20), посредством принудительного перорального введения через зонд (объем дозирования 5 мл/кг, 10 мышей на группу). Через два часа отбирали кровь, получали плазму и определяли количество присутствующего триптофана посредством ЖХ-МС-МС (ВЭЖХ-колонка Unison UK-Фенил, 75×4,6, 3 мкм, скорость потока 0,8 мл/мин, 8-минутный градиент от 95% воды плюс 0,1% муравьиной кислоты/5% ацетонитрил плюс 0,1% муравьиной кислоты до 5% воды плюс 0,1% муравьиной кислоты/95% ацетонитрила плюс 0,1% муравьиной кислоты, время удерживания 2,4 мин; система API4000 MS-MS от АВ Sciex, ESI+ mode, основной ион 205,1, дочерний ион 146,1).
Все документы, цитированные в данном описании, а также приоритетная заявка PCT/IB2015/051957, дата подачи 17 марта 2015 года, и предварительная заявка на патент США No. 62/203032, дата подачи 10 августа 2015 года, включены в данное описание посредством ссылки. Хотя изобретение описано со ссылкой на конкретные воплощения, следует понимать, что модификации могут быть сделаны не отходя от идеи данного изобретения. Предусматривается, что такие модификации входят в объем прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЦИКЛОАЛКИЛНИТРИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2655380C2 |
| ГЕМИНАЛЬНО-ЗАМЕЩЕННЫЕ ЦИАНОЭТИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ JANUS КИНАЗ | 2014 |
|
RU2664533C2 |
| НОВОЕ СОЕДИНЕНИЕ БИФЕНИЛА ИЛИ ЕГО СОЛЬ | 2016 |
|
RU2726622C2 |
| ПРОИЗВОДНЫЕ АНИЛИНПИРИМИДИНА И ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2734849C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ОБЕСПЕЧИВАЮЩИЕ РАЗРУШЕНИЕ БЕЛКА IKAROS И БЕЛКА AIOLOS | 2020 |
|
RU2833608C2 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ ТЕРАПЕВТИЧЕСКОГО ЭФФЕКТА ИНГИБИТОРА LSD1 НА ОСНОВЕ ЭКСПРЕССИИ INSM1 | 2018 |
|
RU2789449C2 |
| СОЕДИНЕНИЯ, МОДУЛИРУЮЩИЕ ЭСТРОГЕНОВЫЕ РЕЦЕПТОРЫ | 2019 |
|
RU2826635C2 |
| N-(2-ЦИАНОГЕТЕРОЦИКЛИЛ)ПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2669922C2 |
| ПИРИДОНОВЫЕ И АЗАПИРИДОНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2011 |
|
RU2617405C2 |
| (АЗА)ИНДОЛ-, БЕНЗОТИОФЕН- И БЕНЗОФУРАН-3-СУЛЬФОНАМИДЫ | 2017 |
|
RU2767904C2 |
Изобретение относится к соединению формулы I:
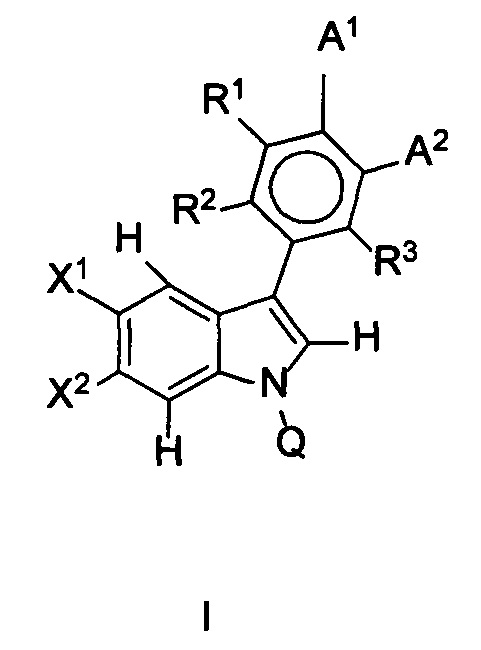
или его фармацевтически приемлемым энантиомеру или соли. В формуле I: каждый из X1 и X2 независимо представляет собой Н или галоген, R1, R2 и R3 независимо представляют собой Н, Q представляет собой Н, А1 и А2 вместе образуют 5-членную конденсированную кольцевую структуру, содержащую SO2NR5CR9'R9, где в указанной структуре R9' представляет собой Н, или каждый из R9' и R9 представляет собой метил, причем когда R9' представляет собой Н, R9 представляет собой атом водорода, циклопропил или C1-С6алкил, возможно замещенный одним, двумя или тремя заместителями, выбранными из галогена, С1-С4алкила, ОН, оксо, NH2, NHCH3, N(CH3)2, NHCOOCH3, NHCOCH3 и N(CH3)COCH3; либо группу -CH2NHSO2CH3 или -С(O)ОСН3. Группа R5 независимо представляет собой: (1') Н, (2') прямой или разветвленный C1-С6алкил, возможно замещенный заместителями в количестве до трех включительно, выбранными из одного или более чем одного гидроксила, OR9, CONR9R10, NR9COR10, NR9R10, SO2R9, SO2NR9R10, NR9SO2R10 и SOR9; или группу -CH2-CH2NHCOOCH2CH3, (3') гетероциклил или группу С1-С3алкил-гетероциклил, где указанный гетероциклил возможно замещен заместителями в количестве до трех включительно, которые выбраны из одного или более чем одного галогена, гидроксила, оксо, OR9, NR9R10, CO-алкила и C1-С6алкила, либо группу -(пиперазин-1-ил)пропан-1-он, (4') С3-С6циклоалкил, возможно замещенный заместителями в количестве до трех включительно, выбранными из галогена, гидроксила, OR9, CONR9R10, NR9COR10, NR9R10, SO2R9, SO2NR9R10, NR9SO2R10 и SO2R9, где каждый из R9 и R10 независимо представляет собой атом водорода или C1-С6алкил, (5') C1-С3алкил-гетероарил, где гетероарил возможно замещен группой -СН3 или группами -СН3 и -ОН, и где гетероарил представляет собой ароматическое кольцо, содержащее 5-6 атомов, в котором один или более атомов углерода заменены атомами азота. Также предложены фармацевтическая композиция, лекарственное средство и способ лечения. Соединения по изобретению могут применяться для лечения и/или предупреждения рака или ингибирования TDO2. 4 н. и 11 з.п. ф-лы, 2 табл., 153 пр.
1. Соединение формулы I:
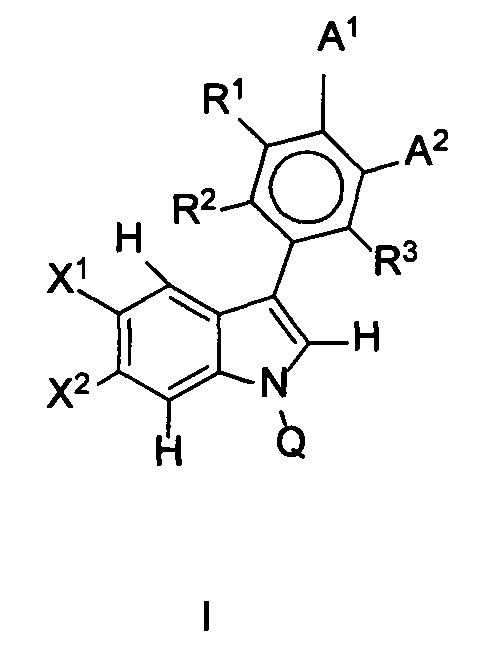
или его фармацевтически приемлемые энантиомер или соль, где:
каждый из X1 и X2 независимо представляет собой Н или галоген;
R1, R2 и R3 независимо представляют собой Н;
Q представляет собой Н;
А1 и А2 вместе образуют 5-членную конденсированную кольцевую структуру, содержащую SO2NR5CR9'R9, где в указанной структуре R9' представляет собой Н, или каждый из R9' и R9 представляет собой метил, причем когда R9' представляет собой Н, R9 представляет собой атом водорода, циклопропил или C1-С6алкил, возможно замещенный одним, двумя или тремя заместителями, выбранными из галогена, С1-С4алкила, ОН, оксо, NH2, NHCH3, N(CH3)2, NHCOOCH3, NHCOCH3 и N(CH3)COCH3; либо группу -CH2NHSO2CH3 или-С(O)ОСН3;
R5 независимо представляет собой:
(1') Н;
(2') прямой или разветвленный C1-С6алкил, возможно замещенный заместителями в количестве до трех включительно, выбранными из одного или более чем одного гидроксила, OR9, CONR9R10, NR9COR10, NR9R10, SO2R9, SO2NR9R10, NR9SO2R10 и SOR9; или группу -CH2-CH2NHCOOCH2CH3;
(3') гетероциклил или группу С1-С3алкил-гетероциклил, где указанный гетероциклил возможно замещен заместителями в количестве до трех включительно, которые выбраны из одного или более чем одного галогена, гидроксила, оксо, OR9, NR9R10, CO-алкила и C1-С6алкила; либо группу -(пиперазин-1-ил)пропан-1-он;
(4') С3-С6циклоалкил, возможно замещенный заместителями в количестве до трех включительно, выбранными из галогена, гидроксила, OR9, CONR9R10, NR9COR10, NR9R10, SO2R9, SO2NR9R10, NR9SO2R10 и SO2R9, где
каждый из R9 и R10 независимо представляет собой атом водорода или C1-С6алкил;
(5') C1-С3алкил-гетероарил, где гетероарил возможно замещен группой -СН3 или группами -СН3 и -ОН, и где гетероарил представляет собой ароматическое кольцо, содержащее 5-6 атомов, в котором один или более атомов углерода заменен атомами азота;
где гетероциклил, как используют здесь, сам по себе или в составе другой группы, относится к неароматическим полностью насыщенным или частично ненасыщенным 3-7-членным моноциклическим группам, имеющим 1, 2, 3 или 4 гетероатома, выбранных из атомов азота, кислорода и/или серы.
2. Соединение по п. 1, где X1 и X2 независимо представляют собой Н, F или Cl.
3. Соединение по п. 2, где X1 представляет собой Н, и X2 представляет собой F.
4. Соединение по п. 1, где R5 представляет собой C1-С3алкил-гетероциклил, возможно замещенный заместителями в количестве до трех включительно, которые независимо представляют собой галоген, C1-С6алкил, гидроксил, оксо, OR9, NR9R10.
5. Соединение по п. 1, где R9 представляет собой С1-С4алкил, который возможно замещен группой ОН или галогеном.
6. Соединение по п. 1, которое находится в форме соли.
7. Соединение по п. 1, которое не находится в форме соли.
8. Соединение по п. 1, выбранное из группы, состоящей из:
(+)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидро-1,2-бензотиазол-1,1-диоксида,
(-)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидро-1,2-бензотиазол-1,1-диоксида,
(+)-3-этил-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-3-этил-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
5-(6-фтор-1Н-индол-3-ил)-3,3-диметил-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-3-пропил-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-5-(6-фтор-1Н-индол-3-ил)-3-пропил-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-3-изопропил-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-5-(6-фтор-1Н-индол-3-ил)-3-изопропил-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-3-циклопропил-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-3-циклопропил-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-5-(6-фтор-1Н-индол-3-ил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
метил-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-3-карбоксилат-1,1-диоксида,
(-)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-3-карбоксамид-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-3-карбоксамид-1,1-диоксида,
(-)-5-(6-фтор-1Н-индол-3-ил)-N-метил-2,3-дигидробензо[d]изотиазол-3-карбоксамид-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-N-метил-2,3-дигидробензо[d]изотиазол-3-карбоксамид-1,1-диоксида,
(+)-3-(аминометил)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-3-(аминометил)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-метил((5-(6-фтор-1Н-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)карбамата,
(+)-метил((5-(6-фтор-1Н-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)карбамата,
(-)-N-((5-(6-фтор-1Н-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)ацетамида,
(+)-N-((5-(6-фтор-1Н-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)ацетамида,
(-)-3-((диметиламино)метил)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-3-((диметиламино)метил)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-N-((5-(6-фтор-1Н-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)-N-метилацетамида,
(-)-N-((5-(6-фтор-1Н-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)-N-метилацетамида,
(-)-5-(6-фтор-1Н-индол-3-ил)-3-(2-гидроксиэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-3-(2-гидроксиэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
2-(2-аминоэтил)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
2-(2-(диметиламино)этил)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
5-(6-фтор-1Н-индол-3-ил)-2-(2-гидроксиэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(S)-2-(2,3-дигидроксипропил)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(R)-2-(2,3-дигидроксипропил)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
5-(6-фтор-1Н-индол-3-ил)-2-(пиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
5-(6-фтор-1Н-индол-3-ил)-2-(1-метилпиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(R)-5-(6-фтор-1Н-индол-3-ил)-2-(тетрагидрофуран-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(S)-5-(6-фтор-1Н-индол-3-ил)-2-(тетрагидрофуран-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
2-(азетидин-3-ил)-5-(6-фтор-1Н-индол-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
5-(6-фтор-1Н-индол-3-ил)-2-(1-метилазетидин-3-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-2-(2-(метилсульфинил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
3-(5-(6-фтор-1Н-индол-3-ил)-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамида,
3-(5-(6-фтор-1Н-индол-3-ил)-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пропанамида,
(+)-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамида,
(-)-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамида,
(+)-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пропанамида,
(-)-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пропанамида,
(+)-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-1-(пиперазин-1-ил)пропан-1-она,
(-)3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-1-(пиперазин-1-ил)пропан-1-она,
(-)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(пиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(пиперидин-4-ил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(2-(метилсульфонил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(2-(метилсульфонил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(2-(метилсульфинил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(2-(метилсульфинил)этил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилэтан-1-сульфонамида,
(+)-2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилэтан-1-сульфонамида,
(-)-2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этан-1-сульфонамида,
(+)-5-(6-фтор-1Н-индол-3-ил)-2-(2-гидроксиэтил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-5-(6-фтор-1Н-индол-3-ил)-2-(2-гидроксиэтил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-1-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этил)пиперазин-2-она,
(+)-1-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этил)пиперазин-2-она,
(+)-5-(6-фтор-1Н-индол-3-ил)-2-(2-(5-гидрокси-3-метил-1Н-пиразол-1-ил)этил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-5-(6-фтор-1Н-индол-3-ил)-2-(2-(5-гидрокси-3-метил-1Н-пиразол-1-ил)этил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-3-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этил)оксазолидин-2-она,
(-)-3-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этил)оксазолидин-2-она,
(+)-1-(4-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пиперидин-1-ил)этанона,
(-)-1-(4-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пиперидин-1-ил)этанона,
(+)-1-(3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пирролидин-1-ил)этанона,
(-)-1-(3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пирролидин-1-ил)этанона,
5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(пиперидин-4-илметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(пиперидин-4-илметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-((1-метилпиперидин-4-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-((1-метилпиперидин-4-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-(5S)-5-((5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)метил)-5-метилпирролидин-2-она,
(-)-(5R)-5-((5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)метил)-5-метилпирролидин-2-она,
(-)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-((1-метил-1Н-1,2,4-триазол-3-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-((1-метил-1Н-1,2,4-триазол-3-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-4-((5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)метил)-4-метилоксазолидин-2-она,
(+)-4-((5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)метил)-4-метилоксазолидин-2-она,
(-)-2-(азетидин-3-ил)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-2-(азетидин-3-ил)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-2-((R)-2,3-дигидроксипропил)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-2-((R)-2,3-дигидроксипропил)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-2-((S)-2,3-дигидроксипропил)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-2-((S)-2,3-дигидроксипропил)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-гидрокси-N-метилпропанамида,
(-)-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-гидрокси-N-метилпропанамида,
(-)-2-амино-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамида,
(+)-2-амино-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамида,
2-амино-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамида,
(+)-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-метокси-N-метилпропанамида,
(-)-3-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-2-метокси-N-метилпропанамида,
(-)-этил-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этил)карбамата,
(+)-этил-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этил)карбамата,
(-)-2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N-метилацетамида,
(+)-2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилацетамида,
(-)-2-(5-(6-фтор-1H-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3H)-ил)-N,N-диметилацетамида,
(+)-2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N,N-диметилацетамида,
(+)2-(5-(6-фториндолин-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)ацетамида,
(-)-2-(5-(6-фториндолин-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)ацетамида,
(-)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(2-морфолиноэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(2-морфолиноэтил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-4-(-5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пиперидин-2-она,
(+)-4-((S)-5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пиперидин-2-она,
(+)-4-((R)-5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пиперидин-2-она,
(+)-4-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пирролидин-2-она,
(-)-4-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)пирролидин-2-она,
(-)-2-(5-(6-фтор-1Н-индол-3-ил)-3-(гидроксиметил)-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилацетамида,
(+)-2-(5-(6-фтор-1Н-индол-3-ил)-3-(гидроксиметил)-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилацетамида,
(-)-5-(6-фтор-1Н-индол-3-ил)-3-(гидроксиметил)-2-((1-метил-1Н-1,2,4-триазол-3-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-3-(гидроксиметил)-2-((1-метил-1Н-1,2,4-триазол-3-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-2-(2-гидроксиэтил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-5-(6-фтор-1Н-индол-3-ил)-2-(2-гидроксиэтил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-5-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этил)-1,3,4-оксадиазол-2(3Н)-она,
(+)-5-(2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)этил)-1,3,4-оксадиазол-2(3Н)-она,
5-(6-фтор-1Н-индол-3-ил)-3-((метиламино)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(+)-5-(6-фтор-1Н-индол-3-ил)-3-((метиламино)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-N-((5-(6-фтор-1Н-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)метансульфонамида,
(+)-N-((5-(6-фтор-1Н-индол-3-ил)-1,1-диоксидо-2,3-дигидробензо[d]изотиазол-3-ил)метил)метансульфонамида,
(+)-2-(2,3-дигидроксипропил)-5-(6-фтор-1Н-индол-3-ил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-2-(2,3-дигидроксипропил)-5-(6-фтор-1Н-индол-3-ил)-3-(гидроксиметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(-)-2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамида,
(+)-2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамида,
2-(5-(6-фтор-1Н-индол-3-ил)-3-метил-1,1-диоксидобензо[d]изотиазол-2(3Н)-ил)-N-метилпропанамида,
5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(пиридин-2-илметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
5-(6-фтор-1Н-индол-3-ил)-3-метил-2-((6-метилпиридин-2-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(R)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-(пиридин-2-илметил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида,
(R)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-((6-метилпиридин-2-ил)метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида или
(R)-5-(6-фтор-1Н-индол-3-ил)-3-метил-2-((2-метил-2Н-тетразол-5-ил)-метил)-2,3-дигидробензо[d]изотиазол-1,1-диоксида.
9. Фармацевтическая композиция для ингибирования активности TDO2 (триптофан-2,3-диоксигеназы), содержащая соединение по любому из пп. 1-8 или его фармацевтически приемлемые энантиомер или соль и по меньшей мере один фармацевтически приемлемый носитель, разбавитель, эксципиент и/или адъювант.
10. Лекарственное средство для ингибирования активности TDO2, содержащее соединение по любому из пп. 1-8 или его фармацевтически приемлемые энантиомер или соль.
11. Соединение по любому из пп. 1-8 или его фармацевтически приемлемые энантиомер или соль для применения в лечении и/или предупреждении рака.
12. Соединение по п. 11, где указанный рак выбран из группы, состоящей из: карциномы мочевого пузыря, гепатокарциномы, меланомы, мезотелиомы, нейробластомы, саркомы, карциномы молочной железы, лейкоза, почечно-клеточной карциномы, колоректальной карциномы, карциномы головы и шеи, карциномы легких, опухоли головного мозга, глиобластомы, астроцитомы, миеломы и панкреатической карциномы.
13. Соединение по любому из пп. 1-8 или 11 или его фармацевтически приемлемые энантиомер или соль для применения в качестве ингибитора TDO2 (триптофан-2,3-диоксигеназы).
14. Способ лечения и/или предупреждения рака или ингибирования TDO2, включающий введение субъекту, нуждающемуся в этом, соединения по любому из пп. 1-8 или 11.
15. Способ по п. 14, где указанный рак выбран из группы, состоящей из: карциномы мочевого пузыря, гепатокарциномы, меланомы, мезотелиомы, нейробластомы, саркомы, карциномы молочной железы, лейкоза, почечно-клеточной карциномы, колоректальной карциномы, карциномы головы и шеи, карциномы легких, опухоли головного мозга, глиобластомы, астроцитомы, миеломы и панкреатической карциномы.
| DOLUSIC E | |||
| et al, Tryptophan 2,3-Dioxygenase (TDO) Inhibitors 3-(2-Pyridyl)ethenyl)indoles as Potential Anticanser Immunomodulators, J | |||
| Med | |||
| Chem., 2011, v | |||
| Видоизменение прибора для получения стереоскопических впечатлений от двух изображений различного масштаба | 1919 |
|
SU54A1 |
| Прибор для нагревания перетягиваемых бандажей подвижного состава | 1917 |
|
SU15A1 |
| Приспособление для разводки рогов вил | 1925 |
|
SU5320A1 |
| DOLUSIC E | |||
| et al, Indoleamine 2,3-dioxygenase inhibitors: a patent review (2008-2012), Expert Opin | |||
| Ther | |||
| Patents, 2013, v | |||
| Прибор для равномерного смешения зерна и одновременного отбирания нескольких одинаковых по объему проб | 1921 |
|
SU23A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Сигнализационный аппарат для охраны касс | 1924 |
|
SU1367A1 |
| WO 2007050963 A1, 03.05.2007 | |||
| CN 0101265259 A, 17.09.2008 | |||
| WO 2008115804 A1, 25.09.2008 | |||
| EA 201170161 A1, 30.08.2011. | |||

