ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящая заявка испрашивает приоритет по предварительной заявке на патент Австралии №2016902978 (поданной 28 июля 2016 г.), содержание которой включено в данное описание во всей своей полноте.
Настоящее изобретение относится к соединениям и их применению для профилактического и/или терапевтического лечения легочного фиброза и связанных с ним состояний.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Любое обсуждение предшествующего уровня техники по всему описанию никоим образом не должно рассматриваться как признание того, что такой предшествующий уровень техники широко известен или образует часть известных общедоступных сведений в данной области.
Легочный фиброз представляет собой респираторное заболевание, при котором в легком накапливается избыток волокнистой соединительной ткани, что приводит к утолщению стенок легкого и уменьшению подачи кислорода в кровь. Вследствие этого субъекты с легочным фиброзом страдают от одышки.
Легочный фиброз может представлять собой вторичный эффект других заболеваний легких, таких как аутоиммунные расстройства, вирусные инфекции и бактериальные инфекции (такие как туберкулез) легкого, или быть результатом лучевой терапии при раке легкого или молочной железы. Легочный фиброз также может быть идиопатическим, при этом факторами риска считаются курение сигарет, факторы окружающей среды (например, связанное с родом занятий воздействие газов, дыма, химических веществ, асбестовых волокон или пыли) или генетическая предрасположенность.
Возможности лечения легочного фиброза весьма ограничены. Некоторые типы фиброза легких восприимчивы к кортикостероидам или другим иммуносупрессорам. Однако такие методы лечения приводят к различным результатам и не эффективны в случае субъектов с идиопатическим легочным фиброзом. Трансплантация легких является единственным терапевтическим вариантом, доступным в настоящее время в тяжелых случаях идиопатического легочного фиброза.
Легочный фиброз может приводить к развитию легочной гипертензии, правосторонней сердечной недостаточности, дыхательной недостаточности, гипоксии, кашлю, образованию тромбов, пневмонии и раку легкого.
Существует потребность в агентах, которые предотвращают или лечат легочный фиброз и связанные с ним состояния. В частности, существует потребность в агентах, которые предотвращают, ослабляют или замедляют прогрессирование легочного фиброза или ослабляют диагностированный легочный фиброз.
Задача настоящего изобретения заключается в преодолении или исправлении по меньшей мере одного из недостатков предшествующего уровня техники либо в предложении полезного альтернативного варианта.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Согласно одному из аспектов настоящего изобретения предложено соединение формулы:
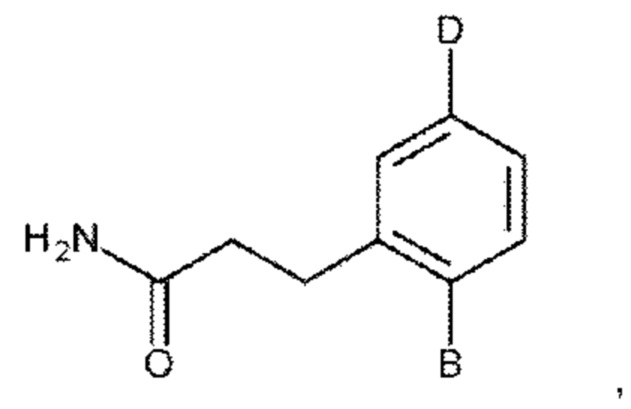
или его фармакологически приемлемые соль, стереоизомер, диастереомер, энантиомер, рацемат, гидрат и/или сольват,
где:
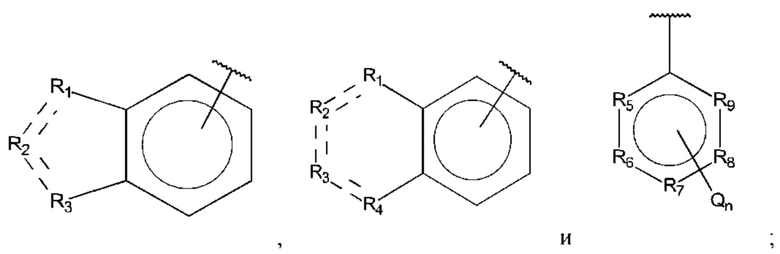
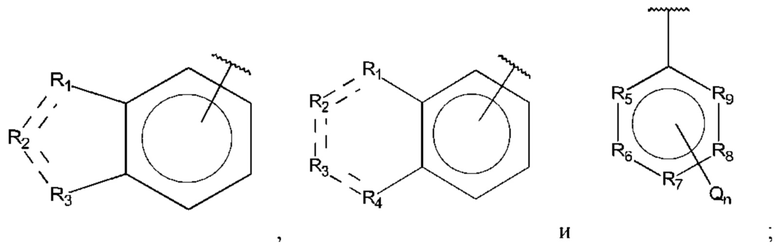
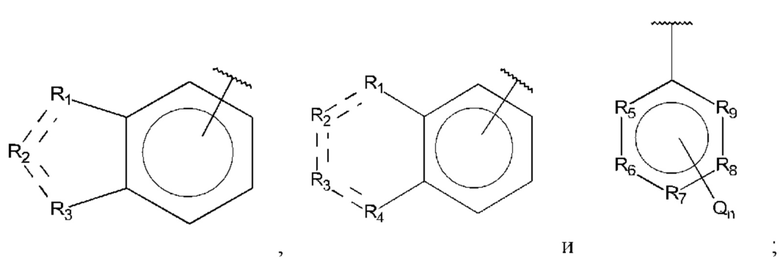
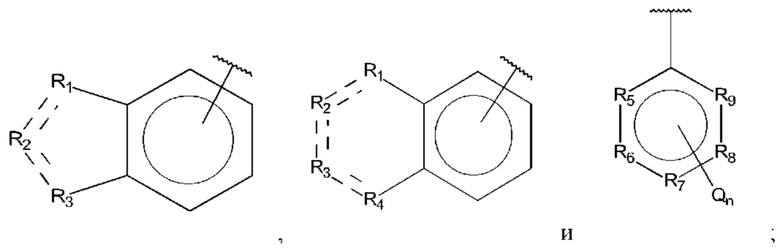
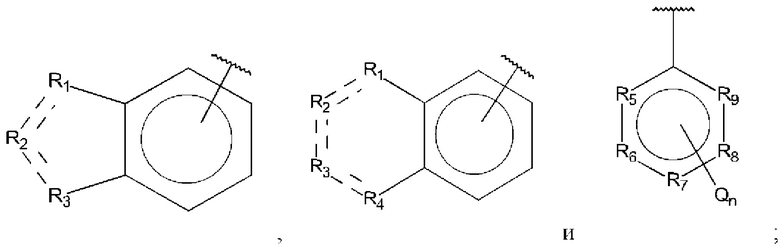
В выбран из группы, состоящей из:
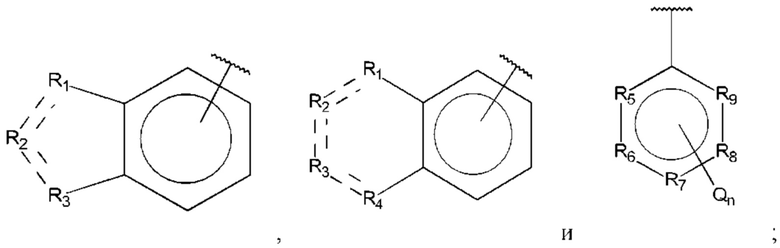
R1, R3 и R4 независимо представляют собой С, СН, CH2, О, N, NH или S;
R2 представляет собой С, СН, СН2, N, NH, C-CF3, CH-CF3 или С=O;
R5-R9 независимо представляют собой С или N;
Q независимо выбран из галогена, алкила, гидрокси, амино и замещенного амино;
n равно 0, 1, 2, 3, 4 или 5;
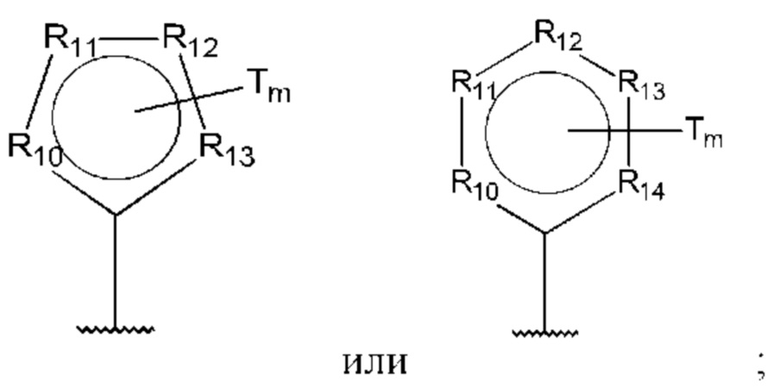
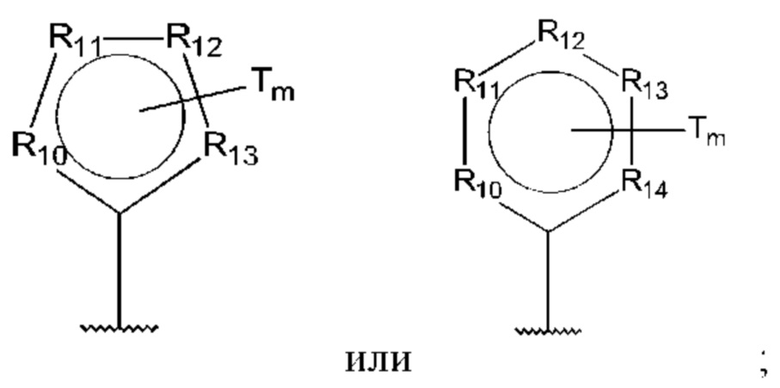
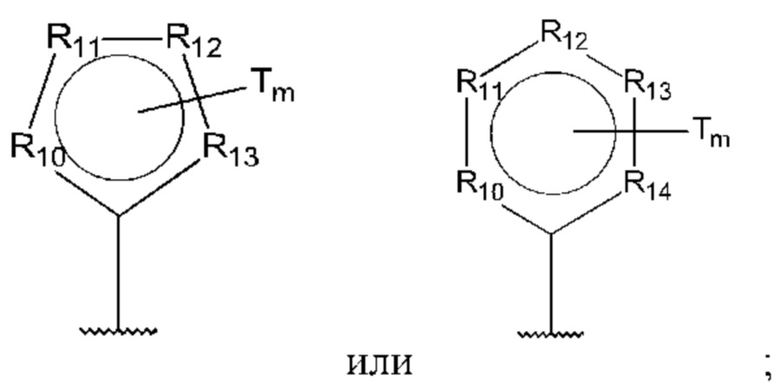
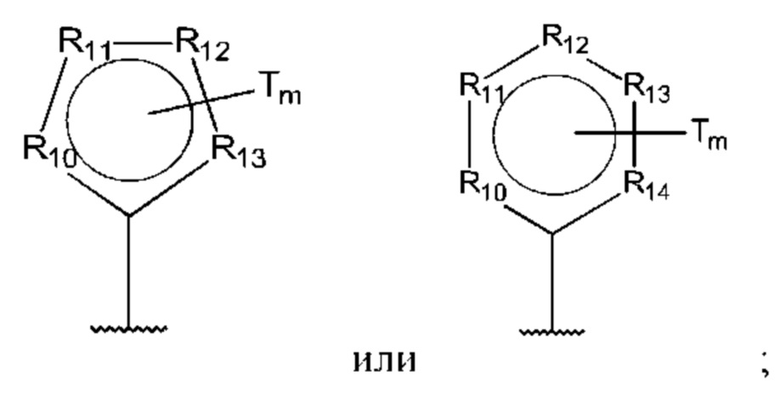
D представляет собой:

R10-R14 независимо представляют собой С, N, О или S;
Т независимо выбран из С1-6алкила, галогена, С0-6алкилкарбоновой кислоты, амино, гидрокси и C1-6алкокси; и
m равно 0, 1, 2, 3 или 4,
при этом D не может представлять собой незамещенный фенил, и Q не может представлять собой гидрокси, когда n равно 1, и все R5-R9 представляют собой С.
Согласно другому аспекту настоящего изобретения предложен способ профилактического или терапевтического лечения легочного фиброза или связанного с ним состояния у субъекта с легочным фиброзом или с риском развития легочного фиброза, включающий введение субъекту эффективного количества соединения формулы:
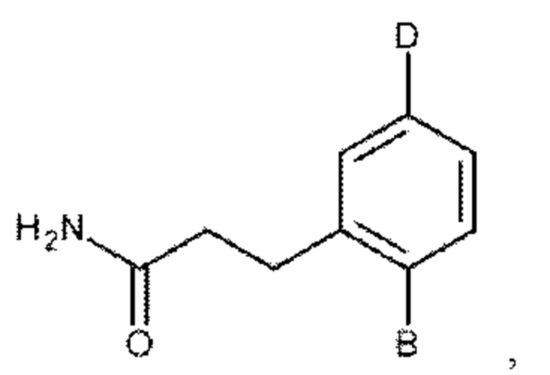
или его фармакологически приемлемых соли, стереоизомера, диастереомера, энантиомера, рацемата, гидрата и/или сольвата,
где:
В выбран из группы, состоящей из:
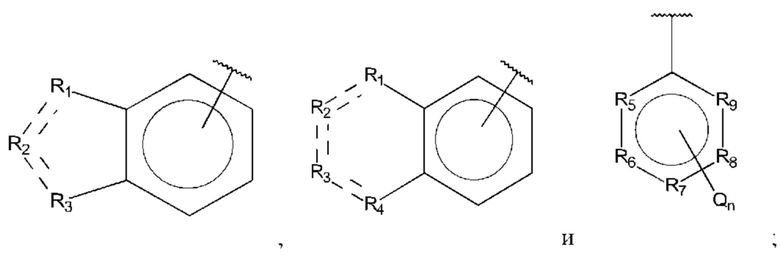
R1, R3 и R4 независимо представляют собой С, СН, СН2, О, N, NH или S;
R2 представляет собой С, СН, СН2, N, NH, C-CF3, CH-CF3 или С=O;
R5-R9 независимо представляют собой С или N;
Q независимо выбран из галогена, алкила, гидрокси, амино и замещенного амино;
n равно 0, 1, 2, 3, 4 или 5;
D представляет собой:
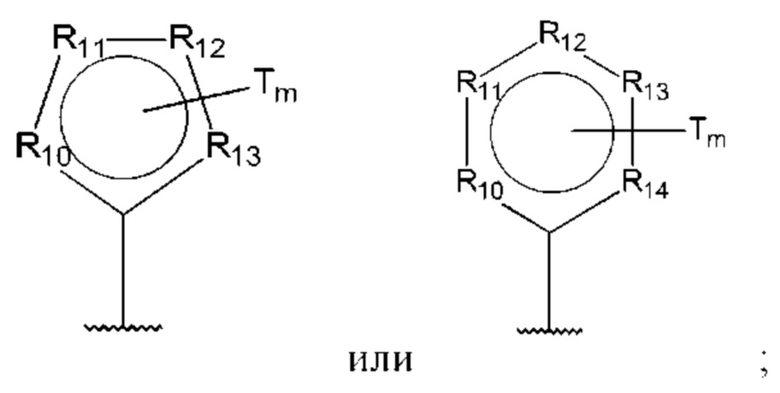
R10-R14 независимо представляют собой С, N, О или S;
Т независимо выбран из С1-6алкила, галогена, С0-6алкилкарбоновой кислоты, амино, гидрокси и C1-6алкокси; и
m равно 0, 1, 2, 3 или 4,
при этом D не может представлять собой незамещенный фенил, и Q не может представлять собой гидрокси, когда n равно 1, и все R5-R9 представляют собой С.
Согласно другому аспекту настоящего изобретения предложено применение соединения формулы:
или его фармакологически приемлемых соли, стереоизомера, диастереомера, энантиомера, рацемата, гидрата и/или сольвата,
для приготовления лекарственного средства для профилактического или терапевтического лечения легочного фиброза или связанного с ним состояния у субъекта с легочным фиброзом или с риском развития легочного фиброза,
где:
В выбран из группы, состоящей из:
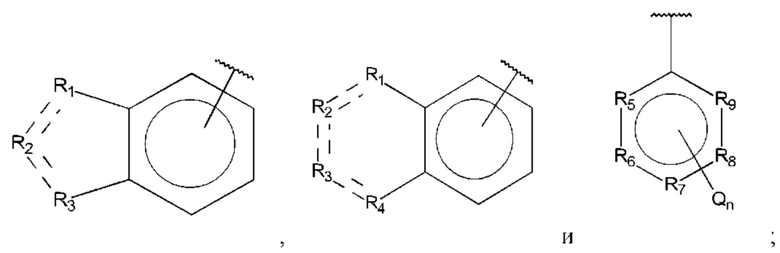
R1, R3 и R4 независимо представляют собой С, СН, СН2, О, N, NH или S;
R2 представляет собой С, СН, СН2, N, NH, C-CF3, CH-CF3 или С=O;
R5-R9 независимо представляют собой С или N;
Q независимо выбран из галогена, алкила, гидрокси, амино и замещенного амино;
n равно 0, 1, 2, 3, 4 или 5;
D представляет собой:
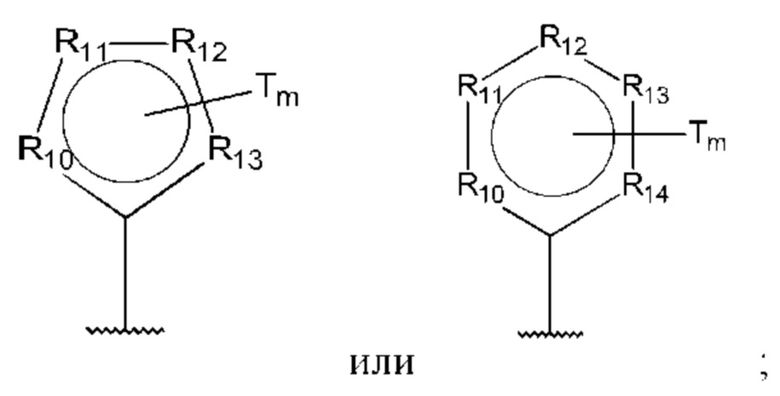
R10-R14 независимо представляют собой С, N, О или S;
Т независимо выбран из С1-6алкила, галогена, С0-6алкилкарбоновой кислоты, амино, гидрокси и С1-6алкокси; и
m равно 0, 1, 2, 3 или 4,
при этом D не может представлять собой незамещенный фенил, и Q не может представлять собой гидрокси, когда n равно 1, и все R5-R9 представляют собой С.
Согласно другому аспекту настоящего изобретения предложено соединение формулы:
где:
В выбран из группы, состоящей из:
Q независимо выбран из галогена, алкила, гидрокси, амино и замещенного амино;
n равно 0, 1, 2, 3, 4 или 5;
R1, R3 и R4 независимо представляют собой С, СН, СН2, О, N, NH или S;
R2 представляет собой С, СН, СН2, N, NH, C-CF3, CH-CF3 или С=O;
R5-R9 независимо представляют собой С или N;
D представляет собой:
R10-R14 независимо представляют собой С, N, О или S;
Т независимо выбран из С1-6алкила, галогена, С0-6алкилкарбоновой кислоты, амино, гидрокси и С1-6алкокси; и
m равно 0, 1, 2, 3 или 4,
при этом D не может представлять собой незамещенный фенил, и Q не может представлять собой гидрокси, когда n равно 1, и все R5-R9 представляют собой С,
для применения в способе профилактического или терапевтического лечения легочного фиброза или связанного с ним состояния у субъекта с легочным фиброзом или с риском развития легочного фиброза.
Согласно другому аспекту настоящего изобретения предложено соединение формулы:
или его фармакологически приемлемые соль, стереоизомер, диастереомер, энантиомер, рацемат, гидрат и/или сольват.
Согласно другому аспекту настоящего изобретения предложен способ профилактического или терапевтического лечения легочного фиброза или связанного с ним состояния у субъекта с легочным фиброзом или с риском развития легочного фиброза, включающий введение субъекту эффективного количества соединения формулы:
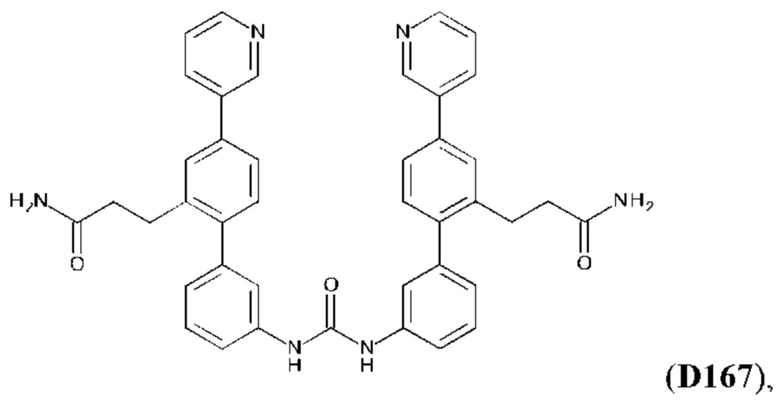
или его фармакологически приемлемых соли, стереоизомера, диастереомера, энантиомера, рацемата, гидрата и/или сольвата.
Согласно другому аспекту настоящего изобретения предложено применение соединения формулы:
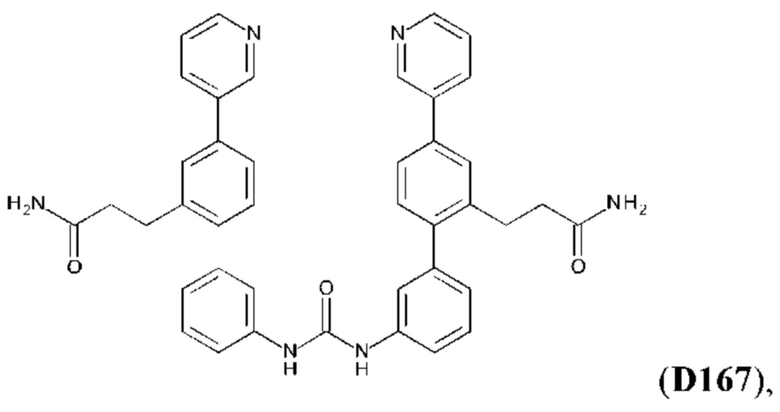
или его фармакологически приемлемых соли, стереоизомера, диастереомера, энантиомера, рацемата, гидрата и/или сольвата,
для изготовления лекарственного средства для профилактического или терапевтического лечения легочного фиброза или связанного с ним состояния у субъекта с легочным фиброзом или с риском развития легочного фиброза.
Согласно другому аспекту настоящего изобретения предложено соединение формулы:
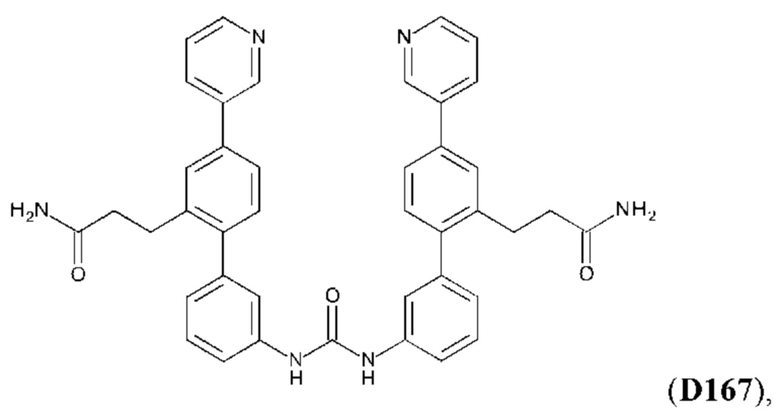
или его фармакологически приемлемых соли, стереоизомера, диастереомера, энантиомера, рацемата, гидрата и/или сольвата,
для применения в способе профилактического или терапевтического лечения легочного фиброза или связанного с ним состояния у субъекта с легочным фиброзом или с риском развития легочного фиброза.
Согласно другому аспекту настоящего изобретения предложен способ профилактического или терапевтического лечения легочного фиброза или связанного с ним состояния у субъекта с легочным фиброзом или с риском развития легочного фиброза, включающий введение субъекту эффективного количества соединения формул:
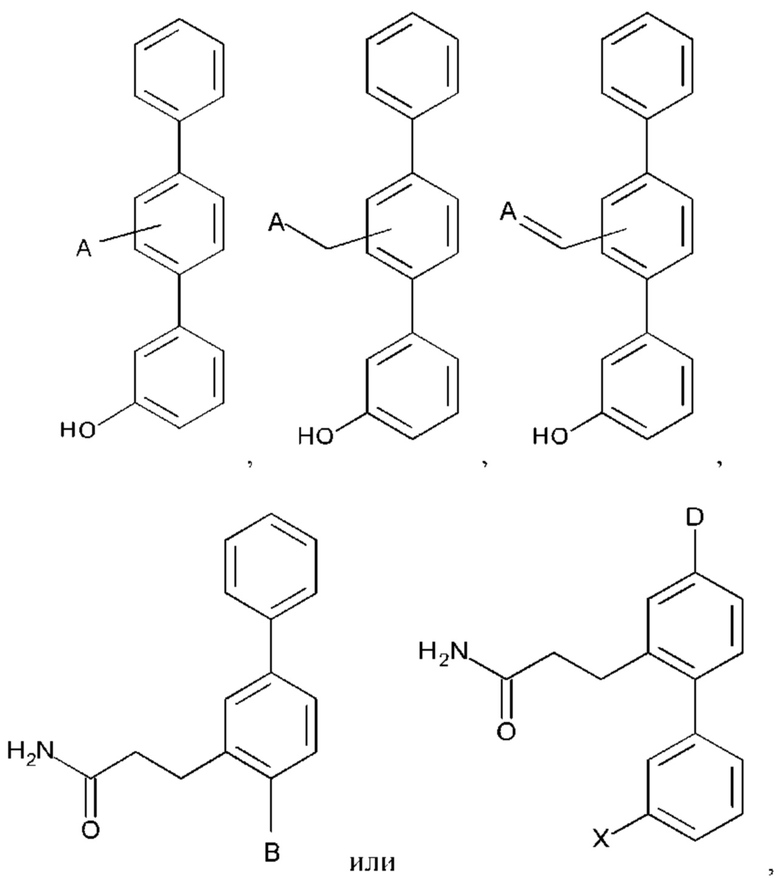
или его фармакологически приемлемых соли, стереоизомера, диастереомера, энантиомера, рацемата, гидрата и/или сольвата,
где:
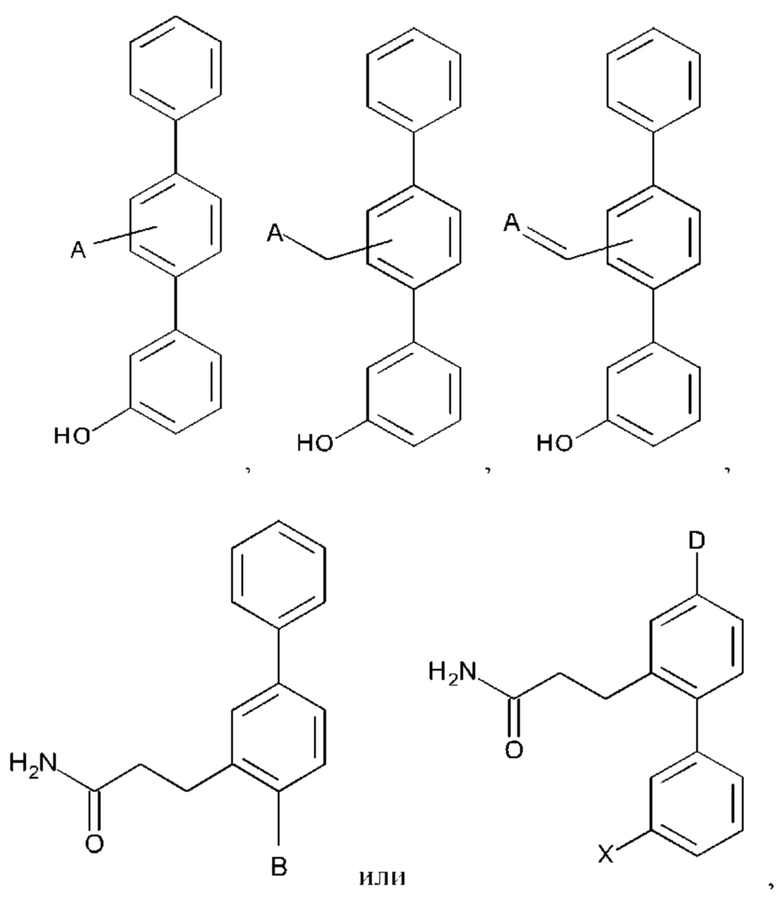
А выбран из возможно замещенного насыщенного, частично насыщенного или ненасыщенного 5- или 6-членного гетероциклила; возможно замещенного С1-6алкоксиламина; возможно замещенного С1-6алкиламина; возможно замещенной С0-6алкилкарбоновой кислоты; возможно замещенного С1-6алкилгидроксила; возможно замещенного насыщенного или ненасыщенного бициклического С0-6алкилгетероциклила; возможно замещенного насыщенного или ненасыщенного бициклического С1-6алкоксилгетероциклила;
В выбран из группы, состоящей из:
Q независимо выбран из галогена, алкила, гидрокси, амино и замещенного амино;
n равно 0, 1, 2, 3, 4 или 5;
R1, R3 и R4 независимо представляют собой С, СН, СН2, О, N, NH или S;
R2 представляет собой С, СН, СН2, N, NH, C-CF3, CH-CF3 или С=O;
R5-R9 независимо представляют собой С или N;
D представляет собой:
R10-R14 независимо представляют собой С, N, О или S;
Т независимо выбран из С1-6алкила, галогена, С0-6алкилкарбоновой кислоты, амино, гидрокси и С1-6алкокси;
m равно 0, 1, 2, 3 или 4; и
X представляет собой -ОН или  .
.
Согласно другому аспекту настоящего изобретения предложено применение соединения формул:
или его фармакологически приемлемых соли, стереоизомера, диастереомера, энантиомера, рацемата, гидрата и/или сольвата,
для изготовления лекарственного средства для профилактического или терапевтического лечения легочного фиброза или связанного с ним состояния у субъекта с легочным фиброзом или с риском развития легочного фиброза,
где:
А выбран из возможно замещенного насыщенного, частично насыщенного или ненасыщенного 5- или 6-членного гетероциклила; возможно замещенного С1-6алкоксиламина; возможно замещенного С1-6алкиламина; возможно замещенной С0-6алкилкарбоновой кислоты; возможно замещенного С1-6алкилгидроксила; возможно замещенного насыщенного или ненасыщенного бициклического С0-6алкилгетероциклила; и возможно замещенного насыщенного или ненасыщенного бициклического С1-6алкоксилгетероциклила;
В выбран из группы, состоящей из:
Q независимо выбран из галогена, алкила, гидрокси, амино и замещенного амино;
n равно 0, 1, 2, 3, 4 или 5;
R1, R3 и R4 независимо представляют собой С, СН, СН2, О, N, NH или S;
R2 представляет собой С, СН, СН2, N, NH, C-CF3, CH-CF3 или С=O;
R5-R9 независимо представляют собой С или N;
D представляет собой:
R10-R14 независимо представляют собой С, N, О или S;
Т независимо выбран из С1-6алкила, галогена, С0-6алкилкарбоновой кислоты, амино, гидрокси и С1-6алкокси;
m равно 0, 1, 2, 3 или 4; и
X представляет собой -ОН или  .
.
Согласно другому аспекту настоящего изобретения предложено соединение формул:
или его фармакологически приемлемые соль, стереоизомер, диастереомер, энантиомер, рацемат, гидрат и/или сольват,
где:
А выбран из возможно замещенного насыщенного, частично насыщенного или ненасыщенного 5- или 6-членного гетероциклила; возможно замещенного С1-6алкоксиламина; возможно замещенного С1-6алкиламина; возможно замещенной С0-6алкилкарбоновой кислоты; возможно замещенного С1-6алкилгидроксила; возможно замещенного насыщенного или ненасыщенного бициклического С0-6алкилгетероциклила; и возможно замещенного насыщенного или ненасыщенного бициклического С1-6алкоксилгетероциклила;
В выбран из группы, состоящей из:
Q независимо выбран из галогена, алкила, гидрокси, амино и замещенного амино;
n равно 0, 1, 2, 3, 4 или 5;
R1, R3 и R4 независимо представляют собой С, СН, СН2, О, N, NH или S;
R2 представляет собой С, СН, СН2, N, NH, C-CF3, CH-CF3 или С=O;
R5-R9 независимо представляют собой С или N;
D представляет собой:
R10-R14 независимо представляют собой С, N, О или S;
Т независимо выбран из С1-6алкила, галогена, С0-6алкилкарбоновой кислоты, амино, гидрокси и С1-6алкокси;
m равно 0, 1, 2, 3 или 4; и
X представляет собой -ОН или 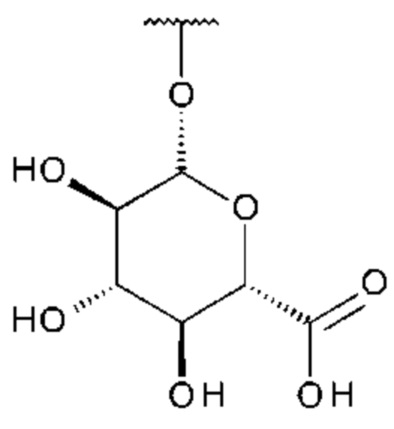 ,
,
для применения в способе профилактического или терапевтического лечения легочного фиброза или связанного с ним состояния у субъекта с легочным фиброзом или с риском развития легочного фиброза.
Согласно другому аспекту настоящего изобретения предложено соединение формулы
или его фармакологически приемлемые соль, стереоизомер, диастереомер, энантиомер, рацемат, гидрат и/или сольват.
В одном из воплощений насыщенный, частично насыщенный или ненасыщенный 5- или 6-членный гетероциклил содержит один атом или несколько атомов N, S или О, возможно замещен одним или более заместителями оксо, С1-6алкил, амино, гидроксил или галоген.
В одном из воплощений насыщенный, частично насыщенный или ненасыщенный 5- или 6-членный гетероциклил выбран из пирролила, пиразолила, имидазолила, триазолила, имидазолидинила, пирролидинила, пирролидинилидена, дигидропирролила, изоксазолила, дигидрооксазолила, изоксазолидинила, оксазолидинила и оксазолила, возможно замещенного одним заместителем или несколькими заместителями оксо, С1-6алкил, амино, гидроксил или галоген.
В одном из воплощений С1-6алкоксиламин представляет собой аминооксиметил.
В одном из воплощений С1-6алкиламин возможно замещен одним или более чем одним из С1-6алкила, С1-6галогеналкила, гидроксила или галогена, предпочтительно моно-, ди- или три-замещенного галогеналкила, наиболее предпочтительно трифторметана.
В одном из воплощений С0-6алкилкарбоновая кислота представляет собой карбоновую кислоту.
В одном из воплощений С1-6алкилгидроксил представляет собой метилгидроксил.
В одном из воплощений бициклический С0-6алкилгетероциклил выбран из индолила, изоиндолила, индолинила и изоиндолинила, возможно замещенного одним или более оксо, предпочтительно диоксо.
В одном из воплощений бициклический С1-6алкоксилгетероциклил выбран из индолила, изоиндолила, индолинила и изоиндолинила, возможно замещенного одним или более оксо, и где С1-6алкоксил представляет собой метокси или этокси.
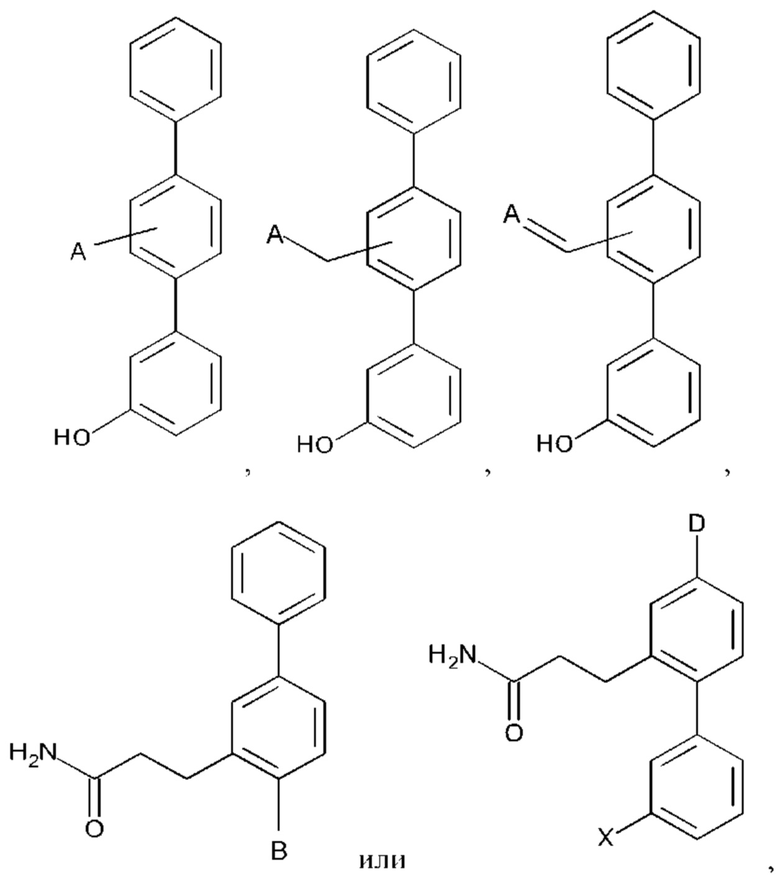
В одном из воплощений А выбран из:
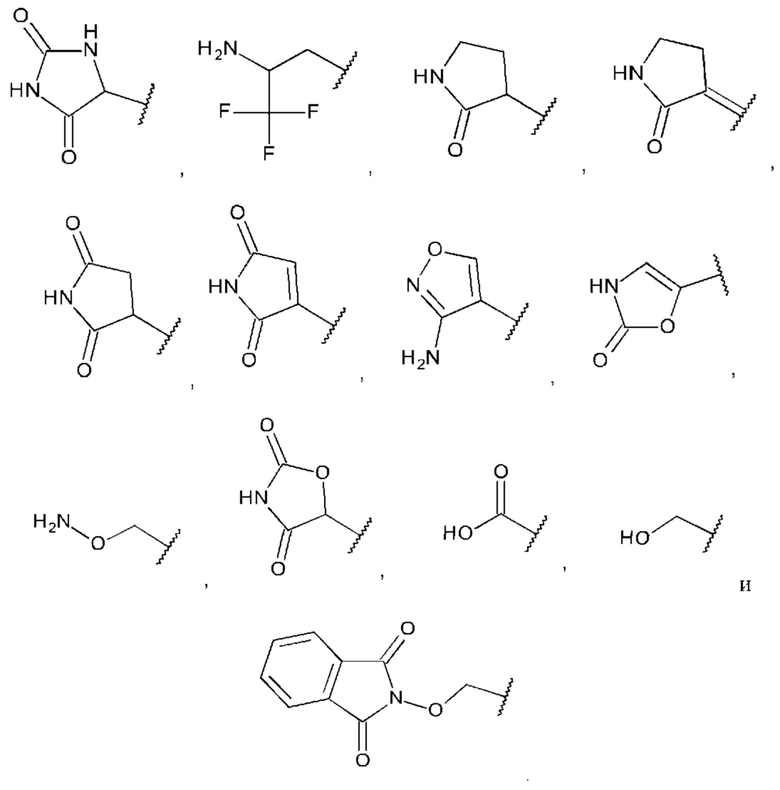
В одном из воплощений Q представляет собой галоген, выбранный из группы, состоящей из F, Cl, Br и I.
В одном из воплощений Q представляет собой замещенный амино формулы -NHW и где:
W выбран из -CN, -SO2(X1)aY и -CO(X1)aY,
а равно 0 или 1,
X1 выбран из -NH- и -О-, и
Y выбран из -Н, -СН3, -СН2СН3, -СН2ОН и -СН2СН2ОН.
В одном из воплощений Q представляет собой замещенный амино, выбранный из группы, состоящей из -NHSO2CH3, -NHCOH, -NHCONHCH3, -NHCONHCH2CH3, -NHSO2NHCH3, -NHSO2NHCH2CH3, -NHCOCH3, -NHCOOCH3, -NHCOOCH2CH2OH, -NHCONH2 и -NHCN.
В одном из воплощений Q представляет собой алкил, выбранный из группы, состоящей из метила, этила, пропила, бутила и пентила.
В одном из воплощений В выбран из:
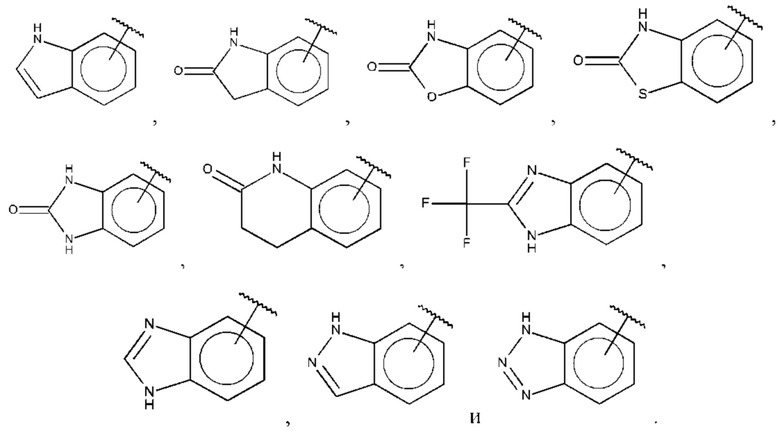
В одном из воплощений В выбран из:
где Q представляет собой замещенный амино, предпочтительно -NHSO2CH3, -NHCOH, -NHCONHCH3, -NHCONHCH2CH3, -NHSO2NHCH3, -NHSO2NHCH2CH3, -NHCOCH3, -NHCOOCH3, -NHCOOCH2CH2OH, -NHCONH2 или -NHCN, и n равно 1 или 2.
В одном из воплощений В выбран из:
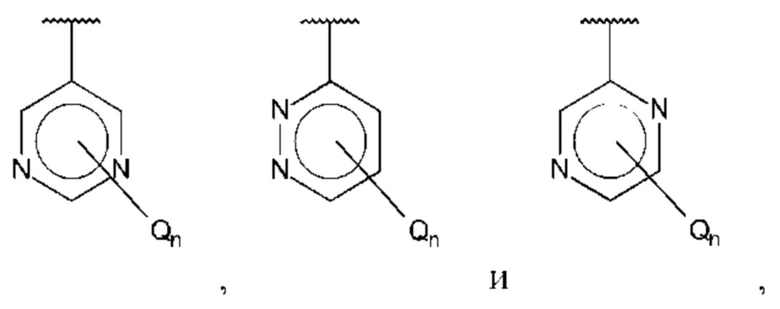
где Q представляет собой амино, и n равно 1 или 2.
В одном из воплощений В выбран из:
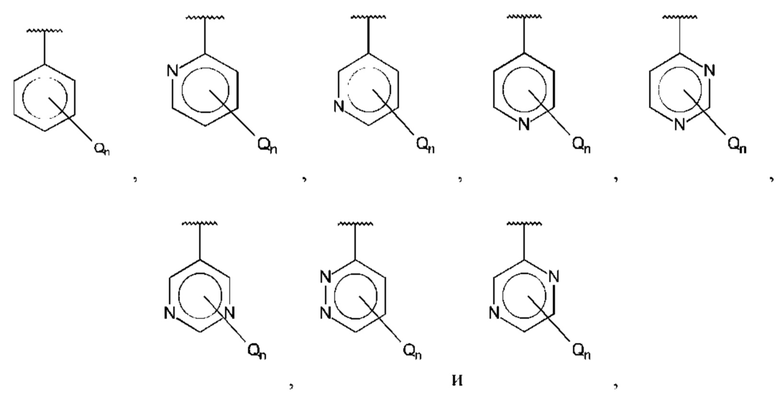
где Q представляет собой гидрокси, и n равно 1 или 2.
В одном из воплощений В выбран из:
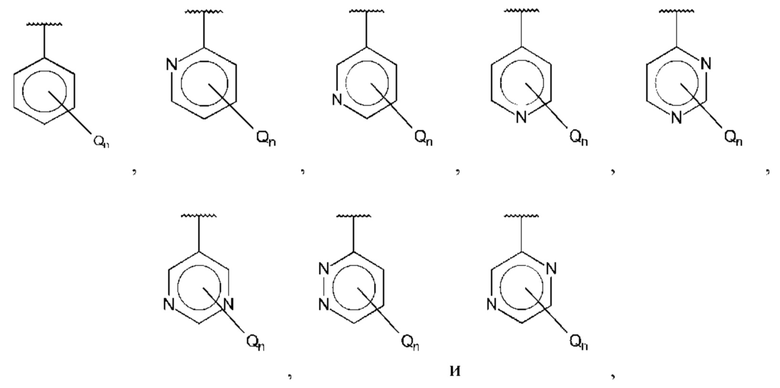
где Q представляет собой галоген, предпочтительно -F или -Cl, и n равно 1 или 2.
В одном из воплощений В выбран из:
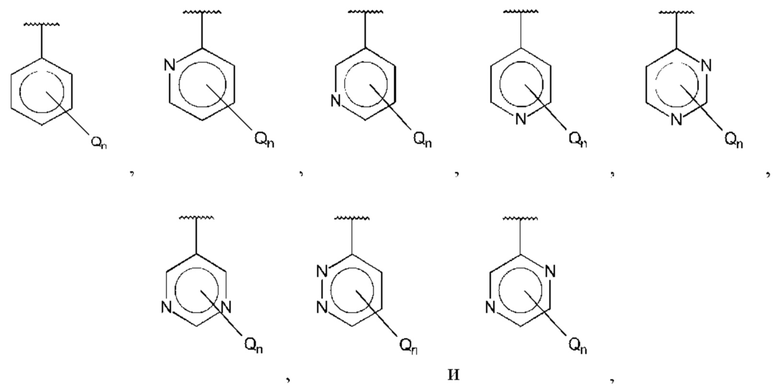
где Q представляет собой алкил, предпочтительно -СН3, и n равно 1 или 2.
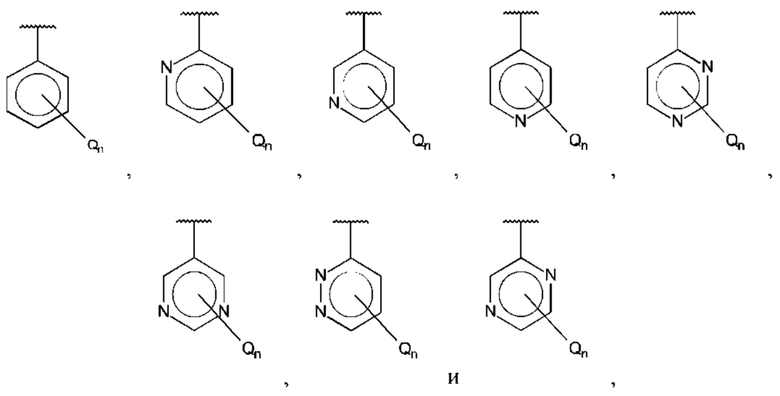
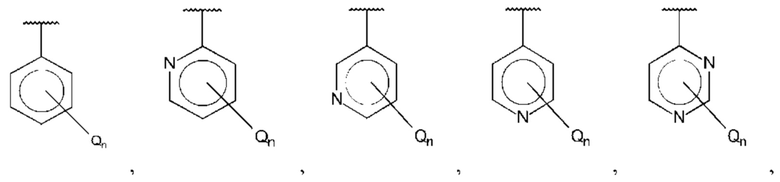
В одном из воплощений В выбран из:
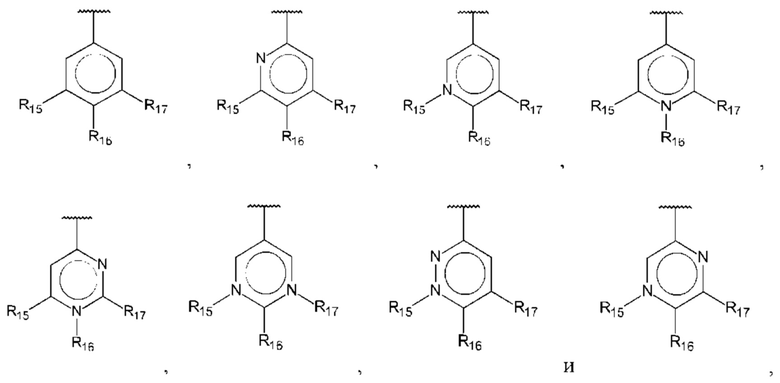
где R15-R17 независимо выбраны из галогена, алкила, гидрокси, амино и замещенного амино.
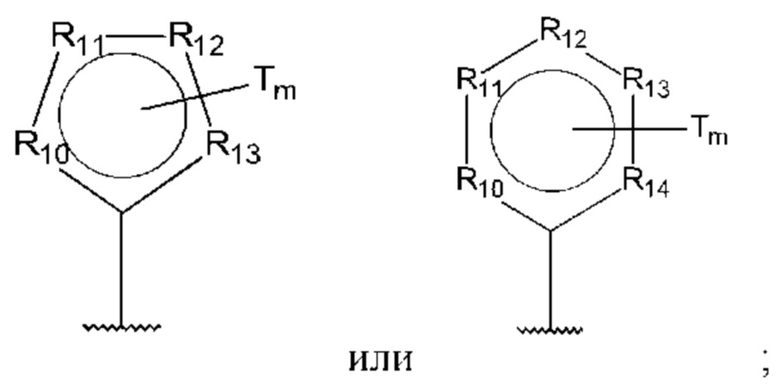
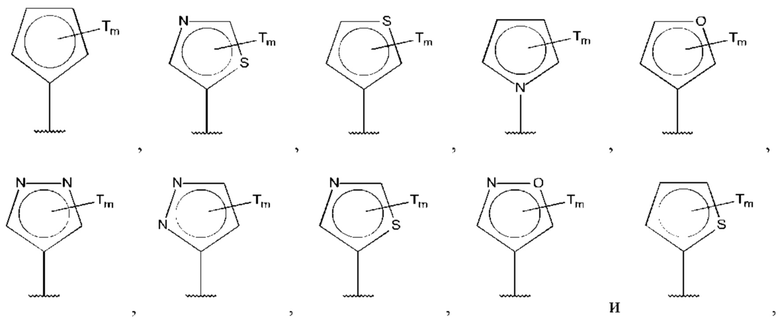
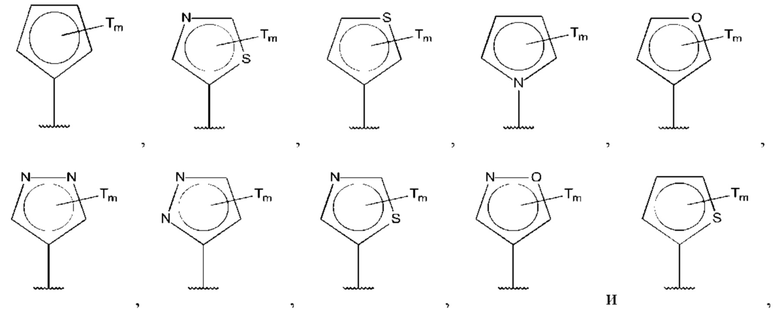
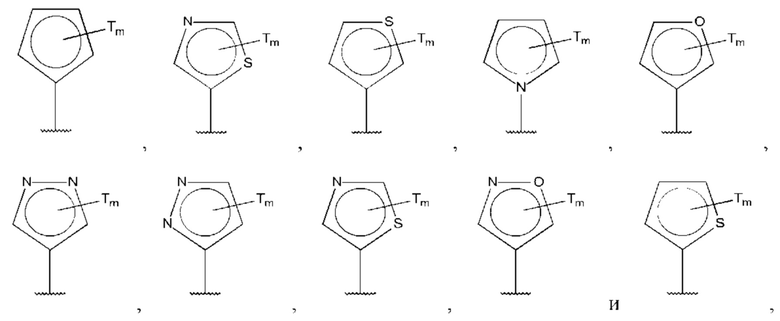
В одном из воплощений D выбран из:
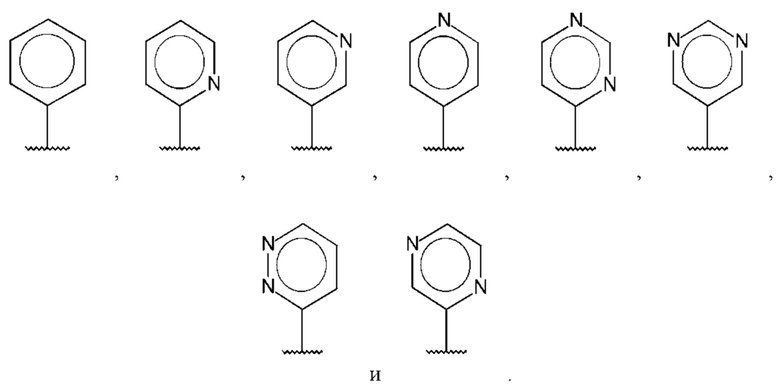
В одном из воплощений D выбран из:
где Т представляет собой алкил, предпочтительно -СН3, и m равно 1 или 2.
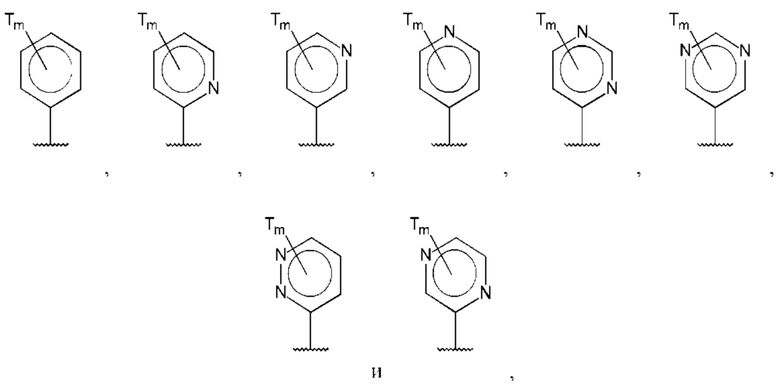
В одном из воплощений D выбран из:
где Т представляет собой С0-6алкилкарбоновую кислоту, предпочтительно -С(O)ОН, и m равно 1 или 2.
В одном из воплощений D выбран из:
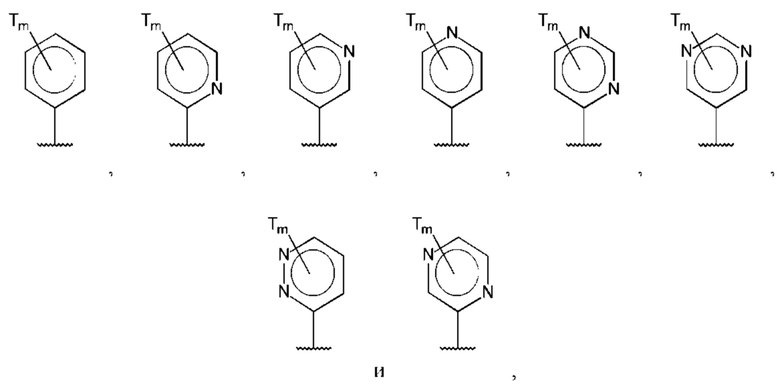
где Т представляет собой галоген, предпочтительно -F, и m равно 1 или 2.
В одном из воплощений D выбран из:
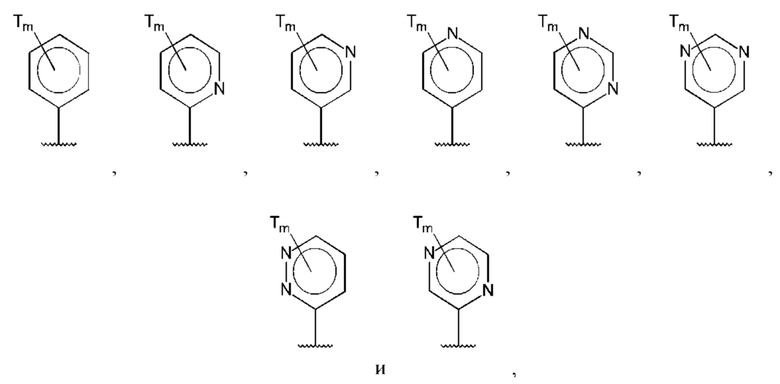
где Т представляет собой амино, и m равно 1 или 2.
В одном из воплощений D выбран из:
где Т представляет собой гидрокси, и m равно 1 или 2.
В одном из воплощений D выбран из:
где Т представляет собой С1-6алкокси, предпочтительно -ОСН3, и m равно 1 или 2.
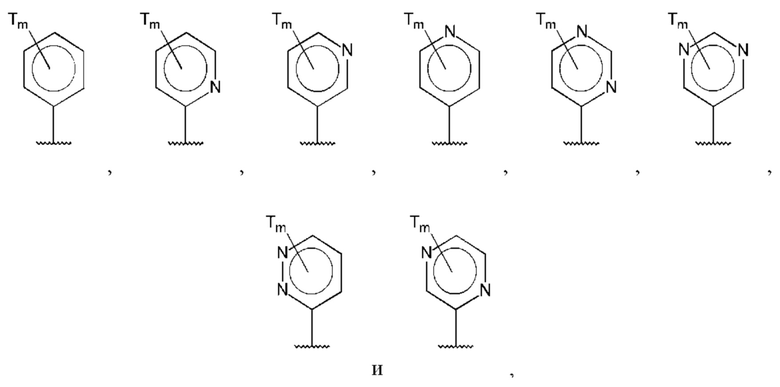
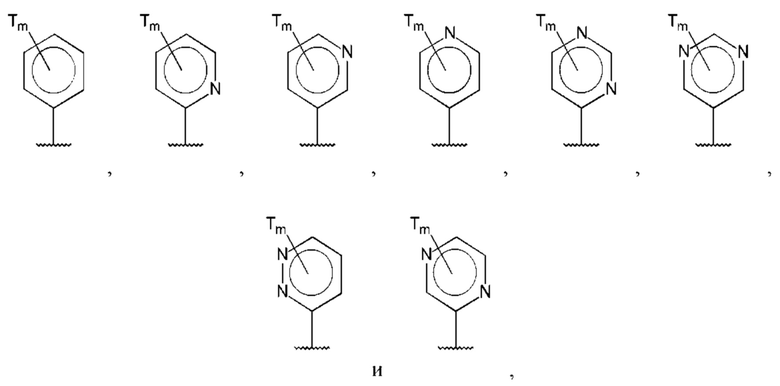
В одном из воплощений D выбран из:
В одном из воплощений D выбран из:
где Т представляет собой алкил, предпочтительно -СН3, и m равно 1 или 2.
В одном из воплощений D выбран из:
где Т представляет собой С0-6алкилкарбоновую кислоту, предпочтительно -С(O)ОН, и m равно 1 или 2.
В одном из воплощений D выбран из:
где Т представляет собой галоген, предпочтительно -F, и m равно 1 или 2.
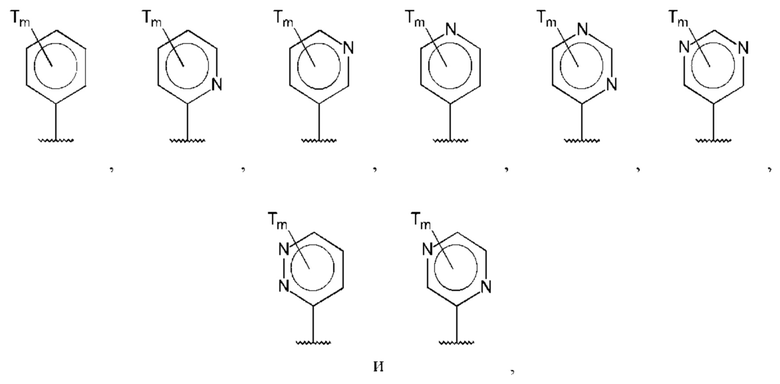
В одном из воплощений D выбран из:
где Т представляет собой амино, и m равно 1 или 2.
В одном из воплощений D выбран из:
где Т представляет собой гидрокси, и m равно 1 или 2.
В одном из воплощений D выбран из:
где Т представляет собой С1-6алкокси, предпочтительно -ОСН3, и m равно 1 или 2.
В одном из воплощений X представляет собой -ОН.
В одном из воплощений X представляет собой  .
.
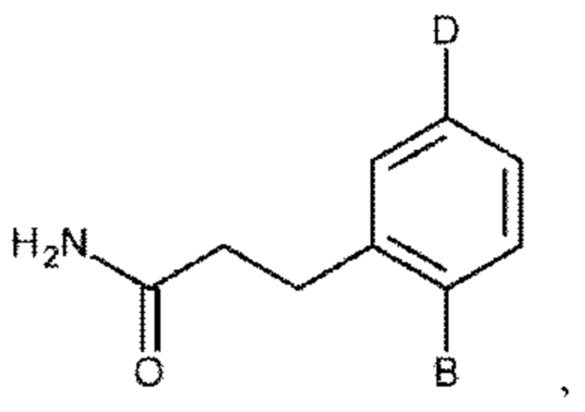
В одном из воплощений соединение выбрано из группы, состоящей из:
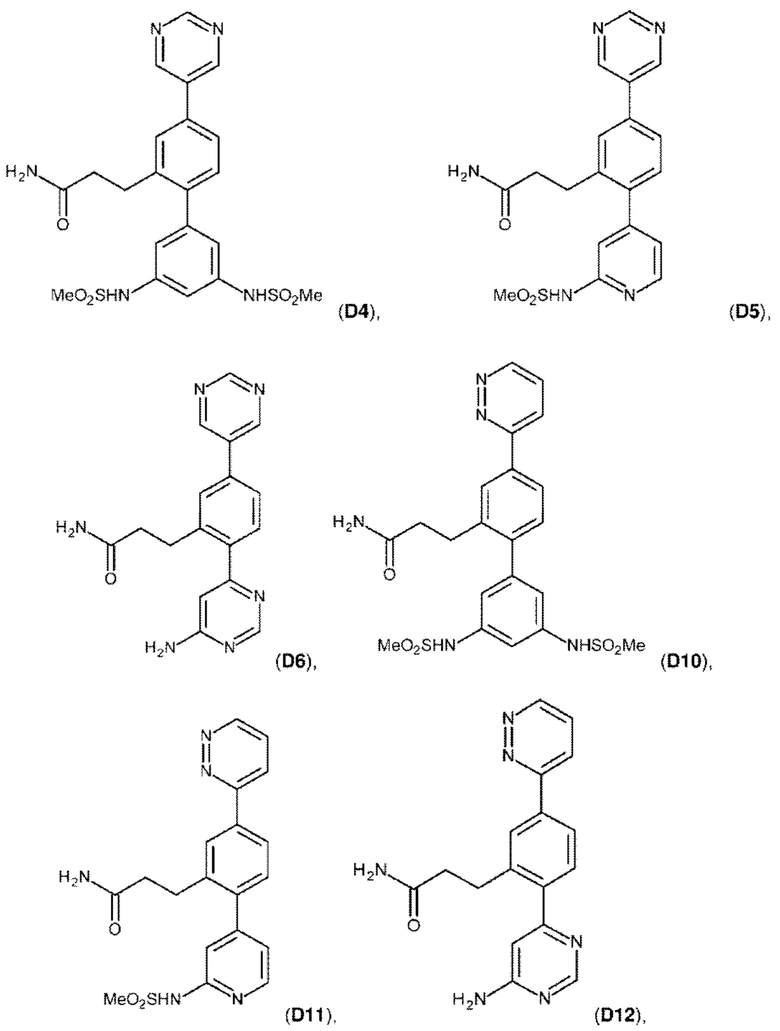
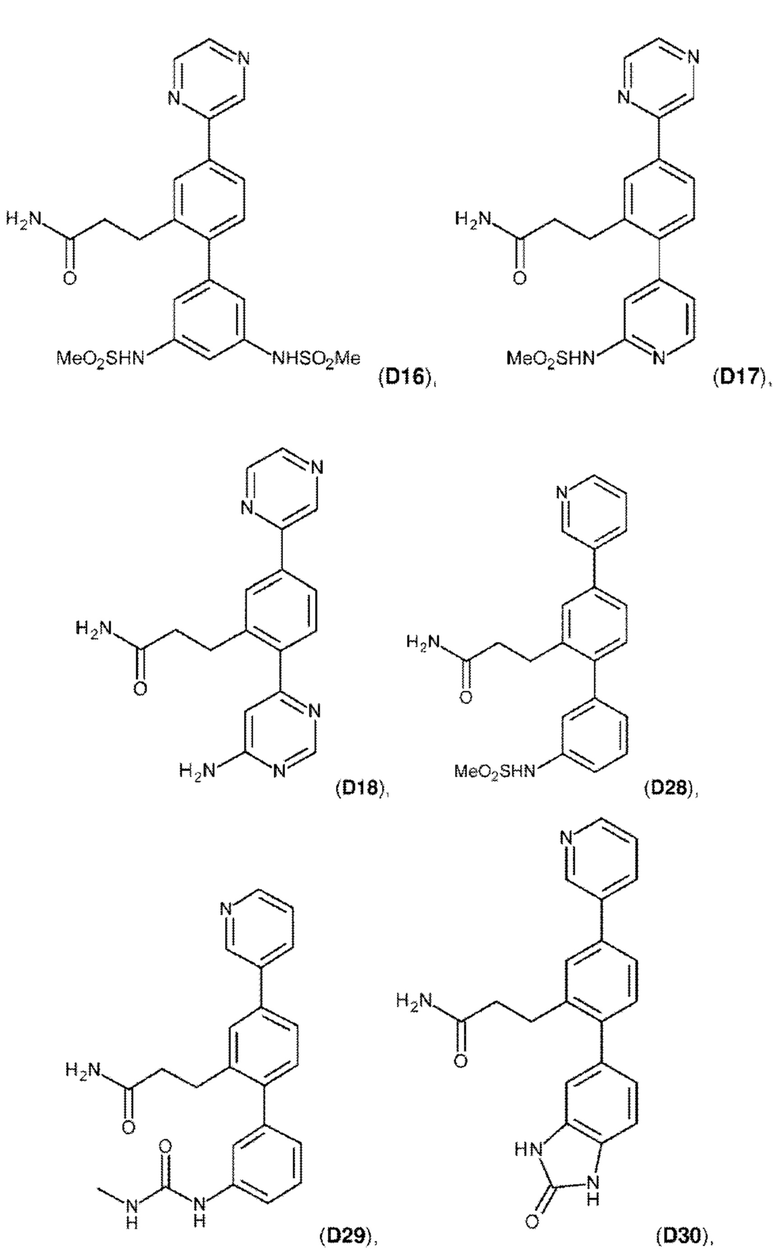
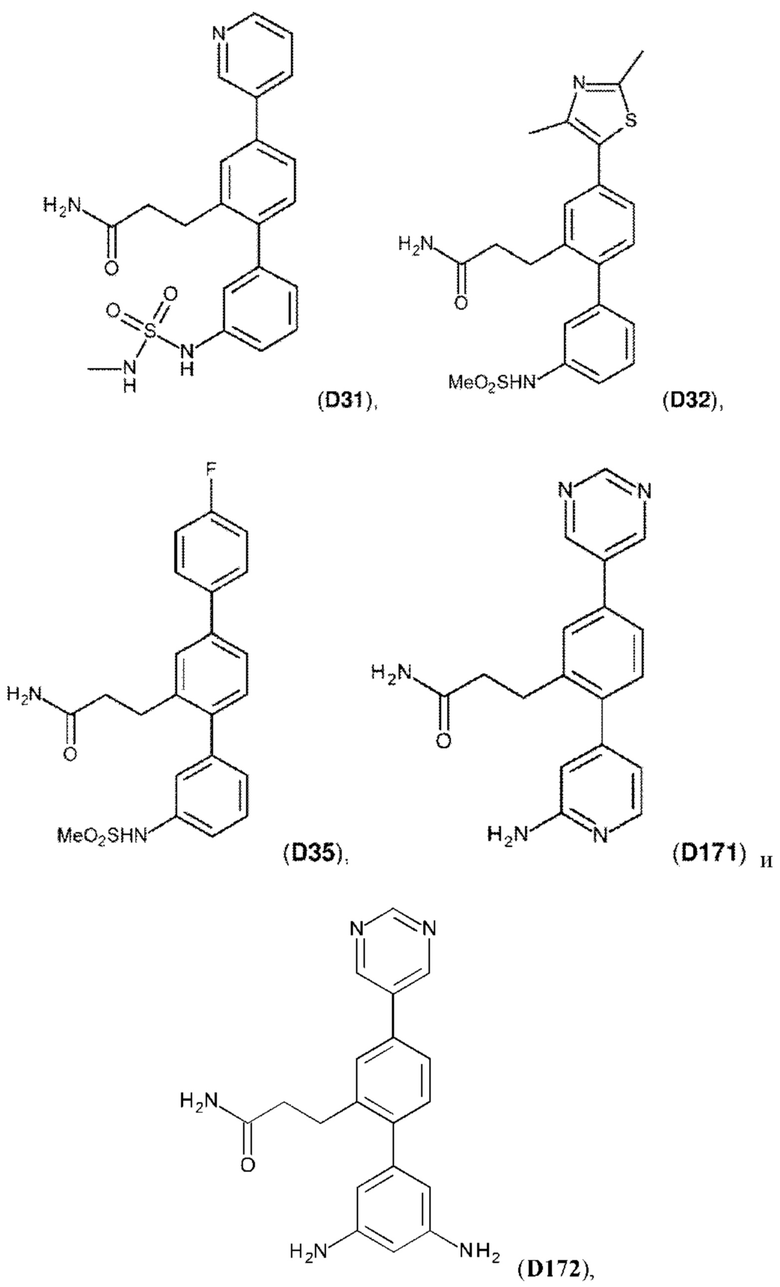
или их фармакологически приемлемых соли, стереоизомера, диастереомера, энантиомера, рацемата, гидрата и/или сольвата.
В одном из воплощений соединение выбрано из группы, состоящей из:
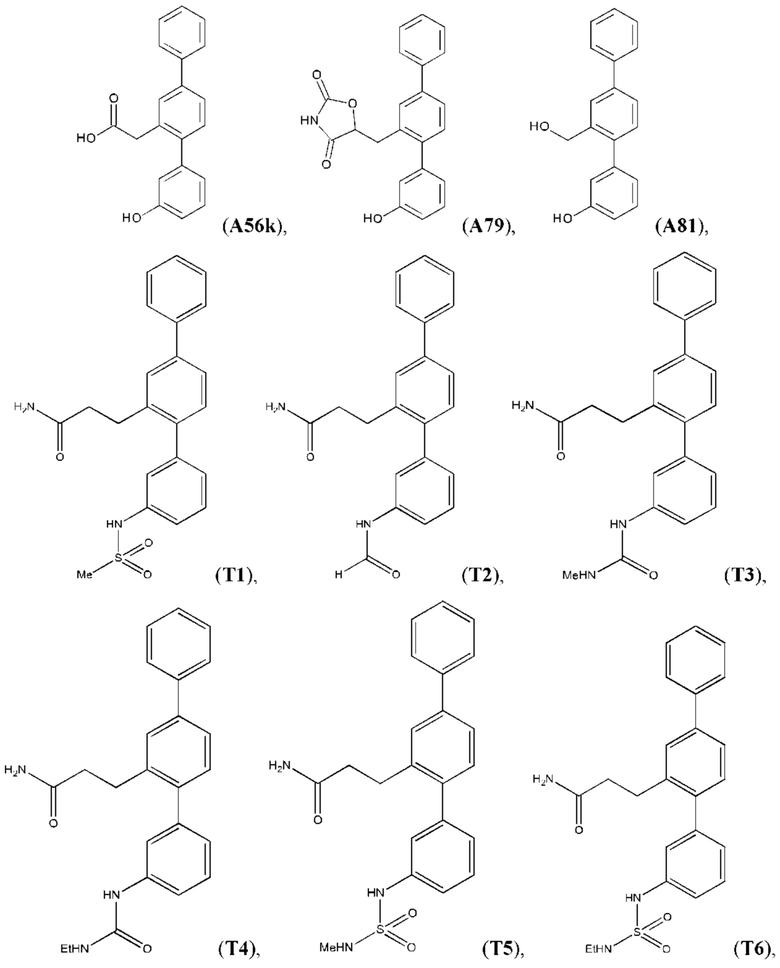
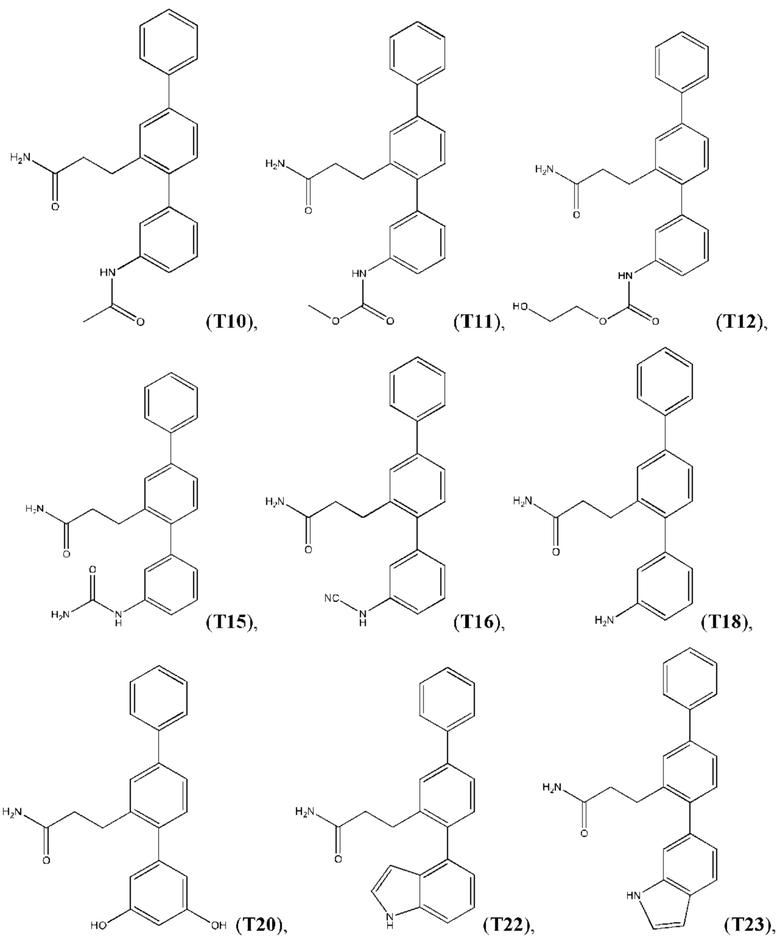
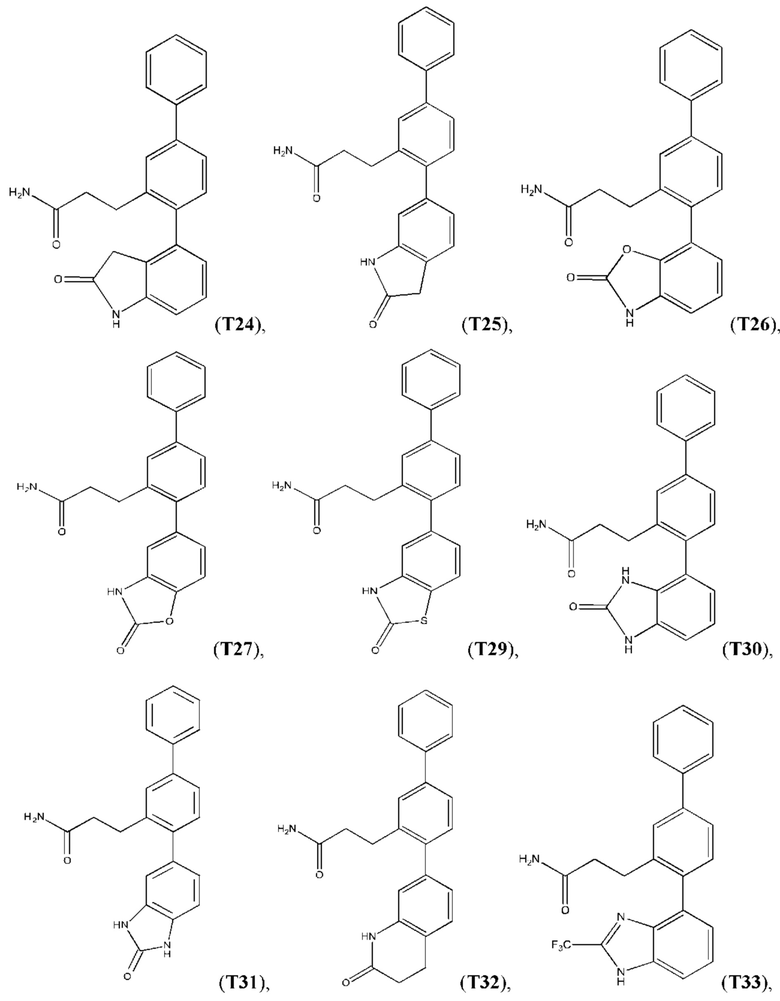
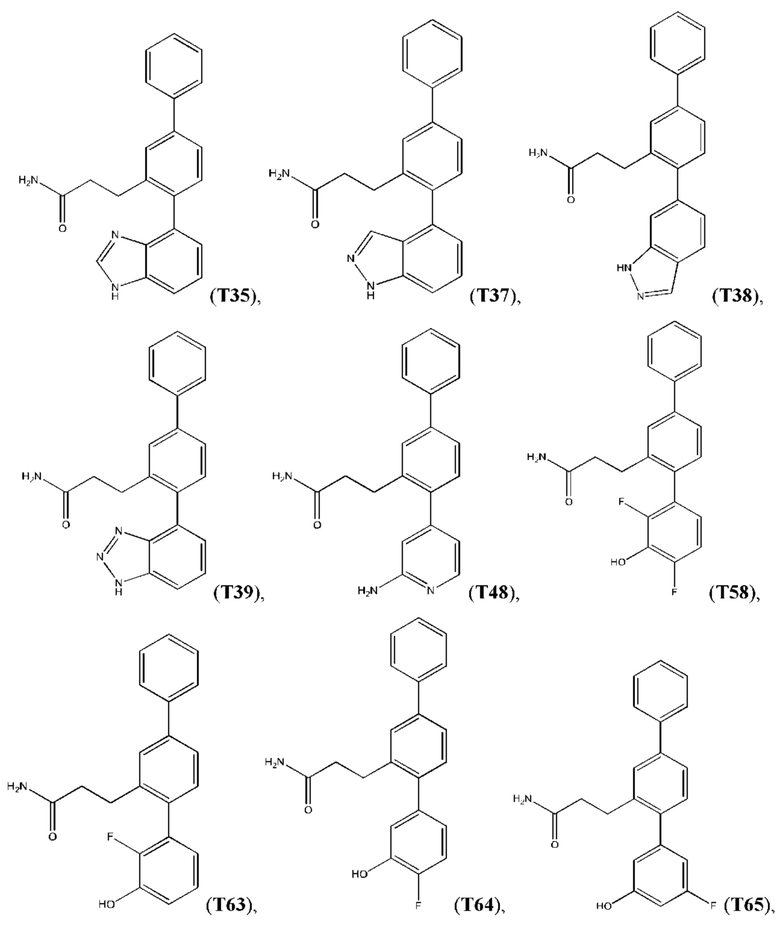
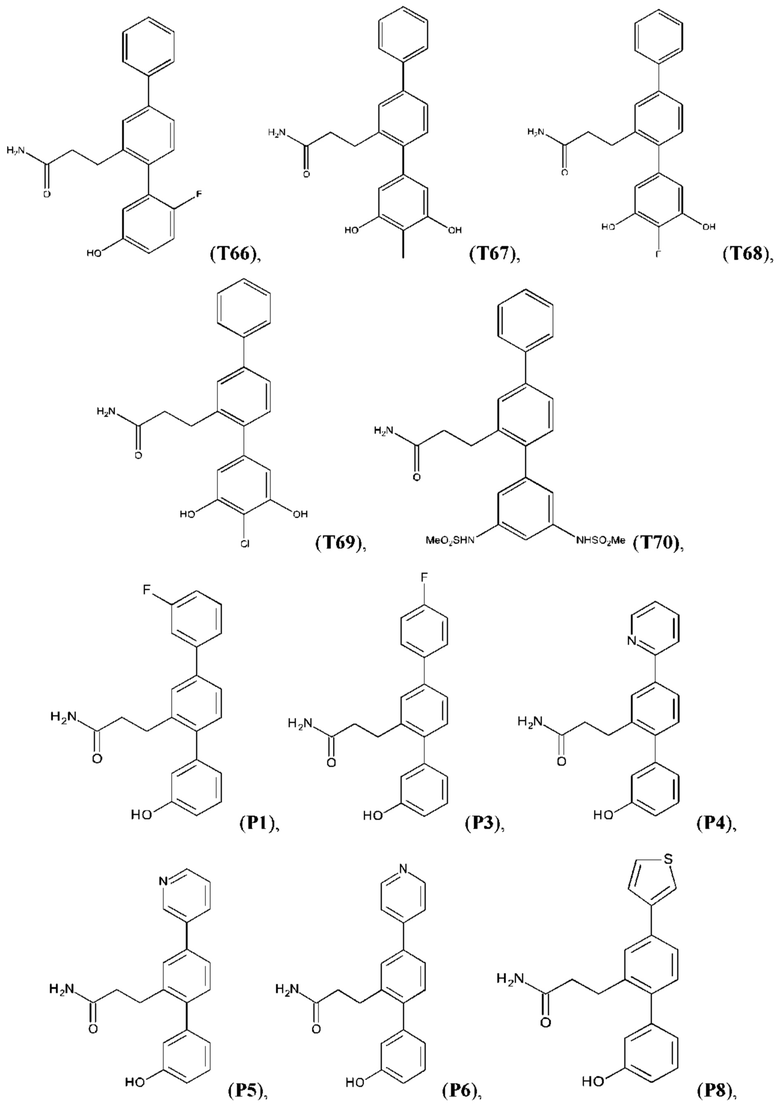
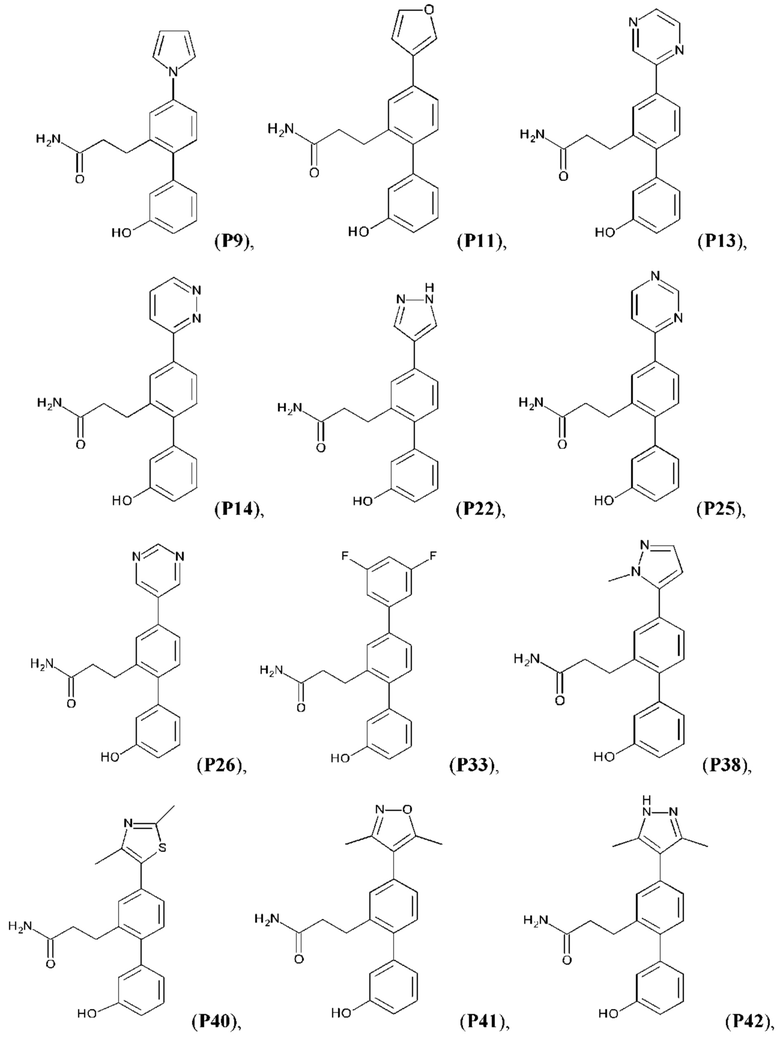
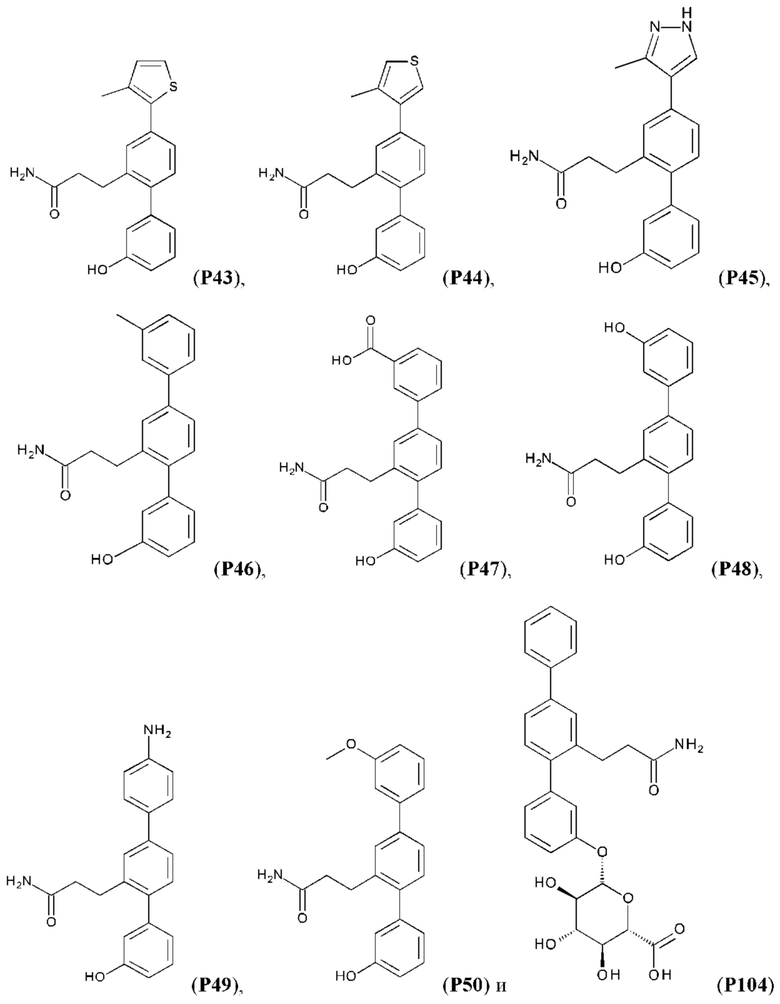
или их фармакологически приемлемых соли, стереоизомера, диастереомера, энантиомера, рацемата, гидрата и/или сольвата.
В одном из воплощений субъект с риском развития легочного фиброза подвергался воздействию газов, дыма, химических веществ, асбестовых волокон или пыли.
В одном из воплощений субъект с риском развития легочного фиброза имеет аутоиммунное расстройство, вирусную инфекцию или бактериальную инфекцию легкого.
В одном из воплощений субъект с риском развития легочного фиброза получал лучевую терапию по поводу рака легкого или молочной железы.
В одном из воплощений субъект с риском развития легочного фиброза имеет генетическую предрасположенность.
В одном из воплощений субъект с риском развития легочного фиброза является курильщиком.
В одном из воплощений связанное с легочным фиброзом состояние выбрано из легочной гипертензии, правосторонней сердечной недостаточности, дыхательной недостаточности, гипоксии, кашля, образования тромбов, пневмонии и рака легкого.
В одном из воплощений прогрессирование легочного фиброза предотвращают, ослабляют или замедляют.
В одном из воплощений ослабляют диагностированный легочный фиброз.
Если контекст явно не требует иного, то во всем описании и формуле изобретения слова «содержат», «содержащий» и тому подобные следует истолковывать во всеобъемлющем смысле, а не в исключительном или исчерпывающем смысле; то есть в смысле «включая, но не ограничиваясь этим».
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фиг. 1. Легочный фиброз у 16-недельных контрольных животных (через две недели после введения блеомицина) и на момент достижения возраста 20 недель после 4 недельной обработки соединениями VB0004, А6, А32, А79 или используемым в качестве контроля разбавителем. Все лекарственные средства вводили в дозе 500 пмоль/кг/мин в питьевом растворе (5%-ном этаноле). Используемым в качестве контроля разбавителем является сам питьевой раствор. * р<0,001 относительно 20-недельного контроля, # р<0,025 относительно 16-недельного контроля, ## р<0,001 относительно 16-недельного контроля.
Фиг. 2. Легочный фиброз (выраженный в виде процента от 20-недельного контроля) у 16-недельных контрольных животных (через две недели после введения блеомицина) и на момент достижения возраста 20 недель после 4 недельной обработки соединениями А32, А79, А6, VB0004, Р13, D30, D6, А81 и используемым в качестве контроля разбавителем (100%). Все лекарственные средства вводили в дозе 500 пмоль/кг/мин в питьевом растворе (5%-ном этаноле). Используемым в качестве контроля разбавителем является сам питьевой раствор.
Фиг. 3. Линейная зависимость между клеточным импедансом в эпителиальных клетках малых дыхательных путей человека и процентом уменьшения степени фиброза по сравнению с 20-недельным контролем (т.е. предотвращение развития фиброза легких) (R2=0,812).
Фиг. 4. Линейная зависимость между клеточным импедансом в эпителиальных клетках малых дыхательных путей человека и процентом уменьшения степени фиброза по сравнению с 16-недельным контролем (т.е. реверсирование диагностированного фиброза легких) (R2=0,802).
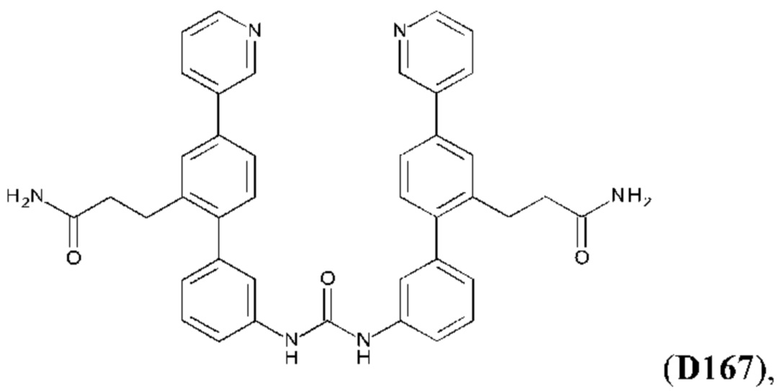
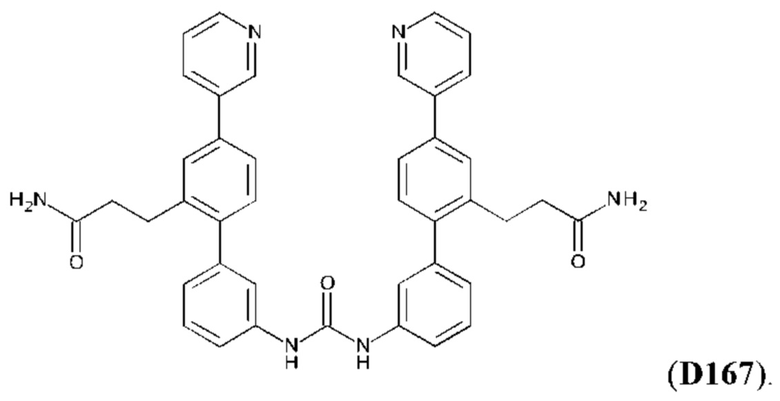
Фиг. 5. Клеточный импеданс, измеренный в эпителиальных клетках малых дыхательных путей человека, обработанных соединениями А6, А26, А27, А30, A32, A35, А56, А79, А81, D4, D5, D6, D10, D11, D12, D16, D17, D18, D28, D30, D31, D32, D35, D167, D171, D172, Р13, Р14, Р25, Р42, Р43, Р44 и VB0004. Обнаружено, что наблюдаемое отклонение в сторону отрицательных значений коррелировало с процентом уменьшения степени фиброза по сравнению с 20-недельным контролем, а также с процентом уменьшения по сравнению с 16-недельным контролем.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, которые эффективны для предупреждения, ослабления или замедления прогрессирования легочного фиброза или ослабления диагностированного легочного фиброза.
Соединения по настоящему изобретению представлены формулой:
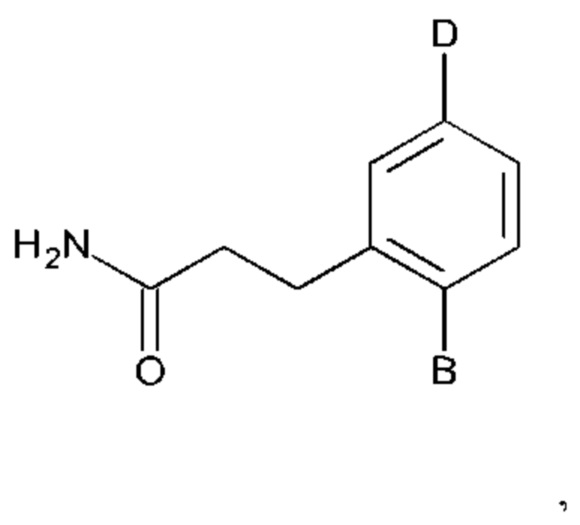
или их фармакологически приемлемые соль, стереоизомер, диастереомер, энантиомер, рацемат, гидрат и/или сольват,
где:
В выбран из группы, состоящей из:
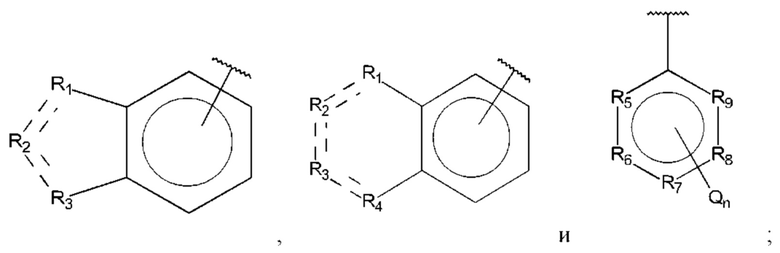
R1, R3 и R4 независимо представляют собой С, СН, СН2, О, N, NH или S;
R2 представляет собой С, СН, СН2, N, NH, C-CF3, CH-CF3 или С=O;
R5-R9 независимо представляют собой С или N;
Q независимо выбран из галогена, алкила, гидрокси, амино и замещенного амино;
n равно 0, 1, 2, 3, 4 или 5;
D представляет собой:
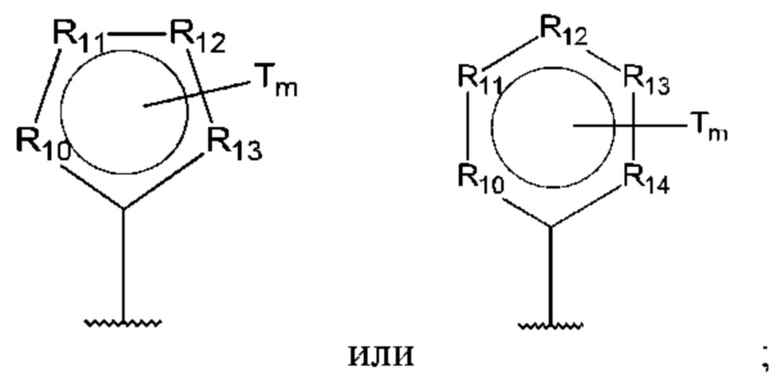
R10-R14 независимо представляют собой С, N, О или S;
Т независимо выбран из С1-6алкила, галогена, С0-6алкилкарбоновой кислоты, амино, гидрокси и С1-6алкокси; и
m равно 0, 1, 2, 3 или 4,
при этом D не может представлять собой незамещенный фенил, и Q не может представлять собой гидрокси, когда n равно 1, и все R5-R9 представляют собой С.
Дополнительные соединения по настоящему изобретению представлены формулой:
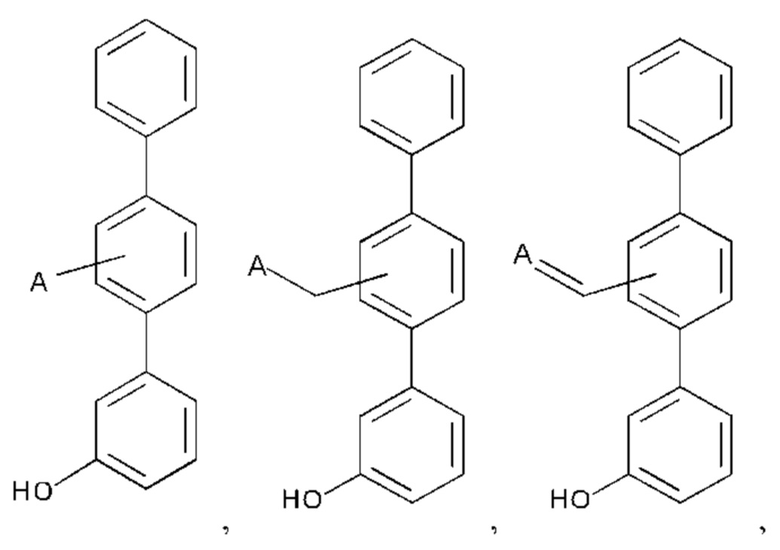
или их фармакологически приемлемые соль, стереоизомер, диастереомер, энантиомер, рацемат, гидрат и/или сольват,
где:
А выбран из возможно замещенного насыщенного, частично насыщенного или ненасыщенного 5- или 6-членного гетероциклила; возможно замещенного С1-6алкоксиламина; возможно замещенного С1-6алкиламина; возможно замещенной С0-6алкилкарбоновой кислоты; возможно замещенного С1-6алкилгидроксила; возможно замещенного насыщенного или ненасыщенного бициклического С0-6алкилгетероциклила; и возможно замещенного насыщенного или ненасыщенного бициклического С1-6алкоксилгетероциклила;
В выбран из группы, состоящей из:
Q независимо выбран из галогена, алкила, гидрокси, амино и замещенного амино;
n равно 0, 1, 2, 3, 4 или 5;
R1, R3 и R4 независимо представляют собой С, СН, СН2, О, N, NH или S;
R2 представляет собой С, СН, СН2, N, NH, C-CF3, CH-CF3 или С=O;
R5-R9 независимо представляют собой С или N;
D представляет собой:
R10-R14 независимо представляют собой С, N, О или S;
Т независимо выбран из C1-6алкила, галогена, С0-6алкилкарбоновой кислоты, амино, гидрокси и C1-6алкокси;
m равно 0, 1, 2, 3 или 4; и
X представляет собой -ОН или 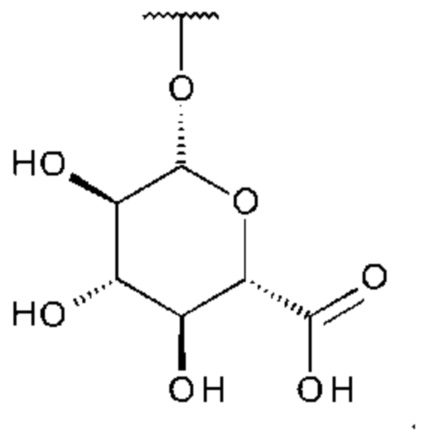
Дополнительное соединение по настоящему изобретению представляет собой:
Следующие далее соединения являются конкретными, но неограничивающими примерами соединений по настоящему изобретению:
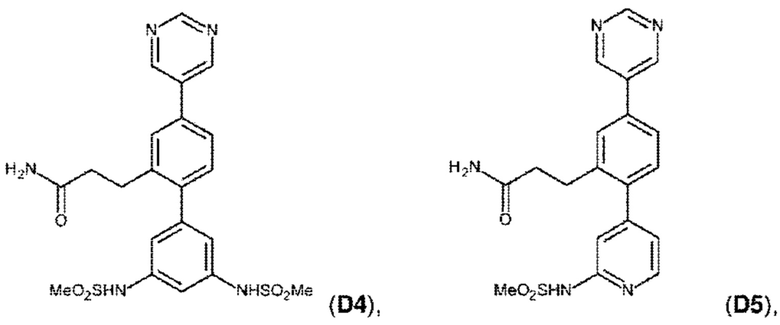
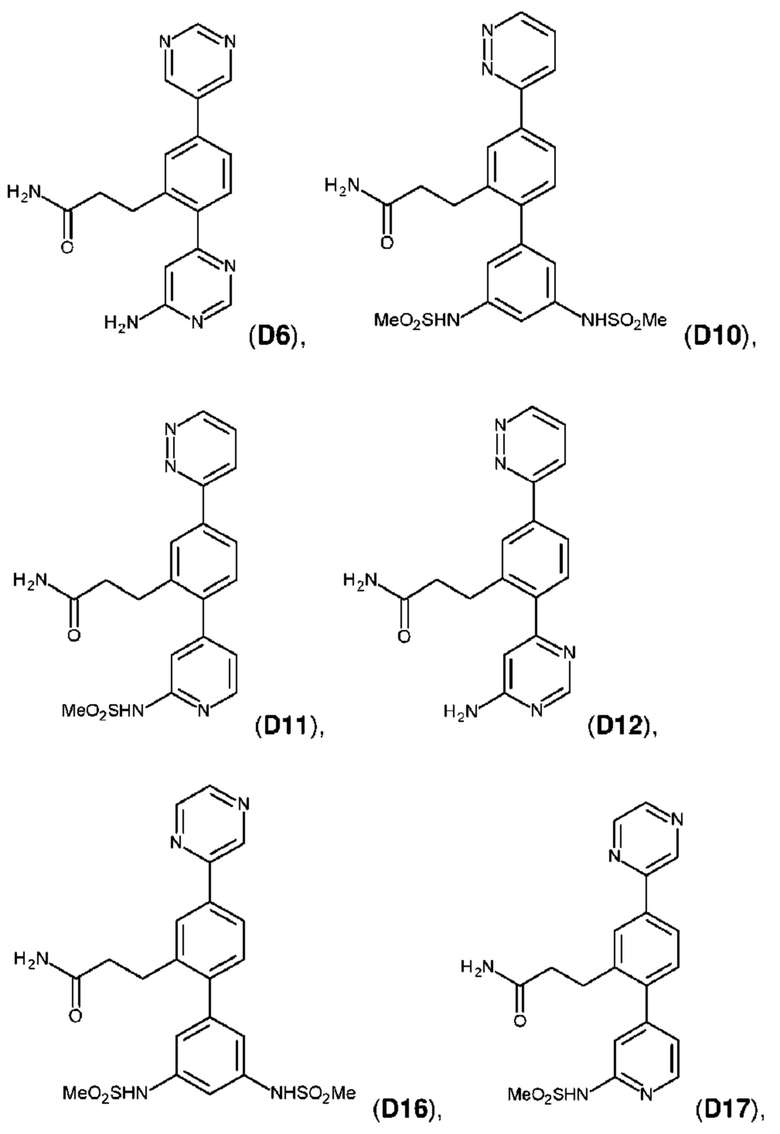
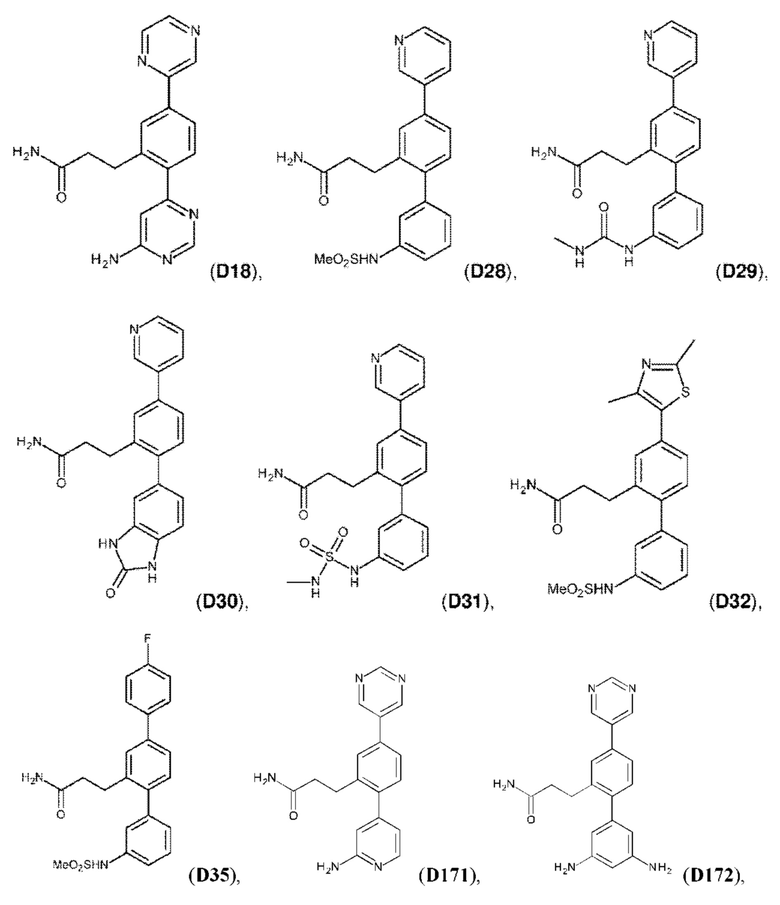
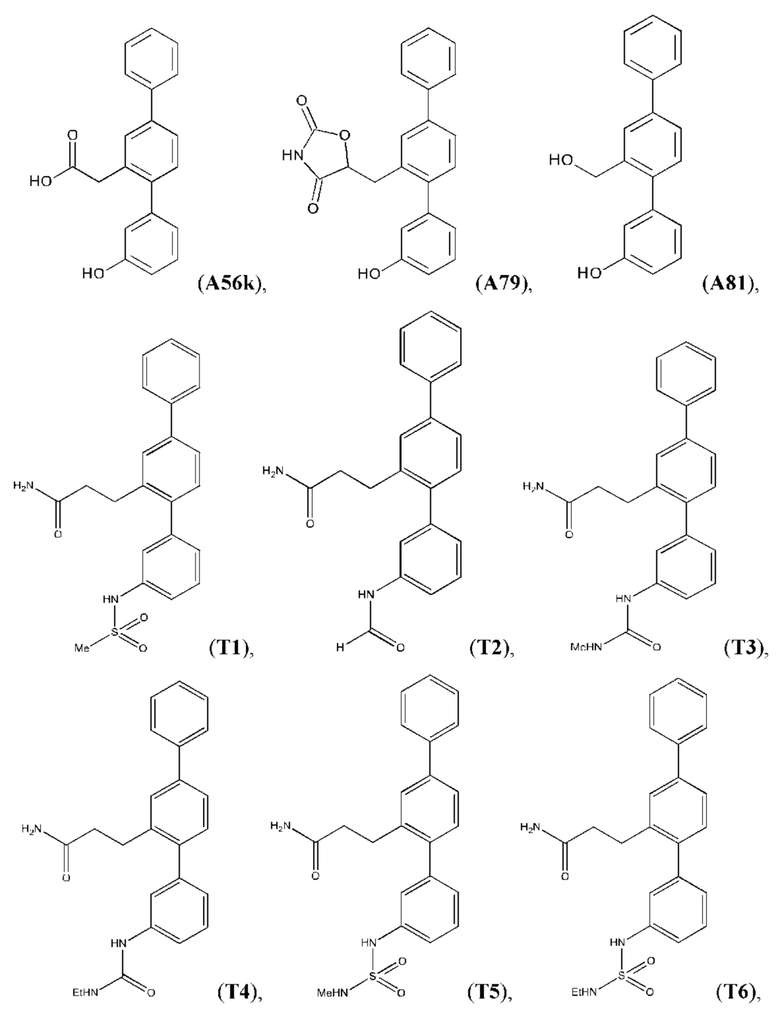
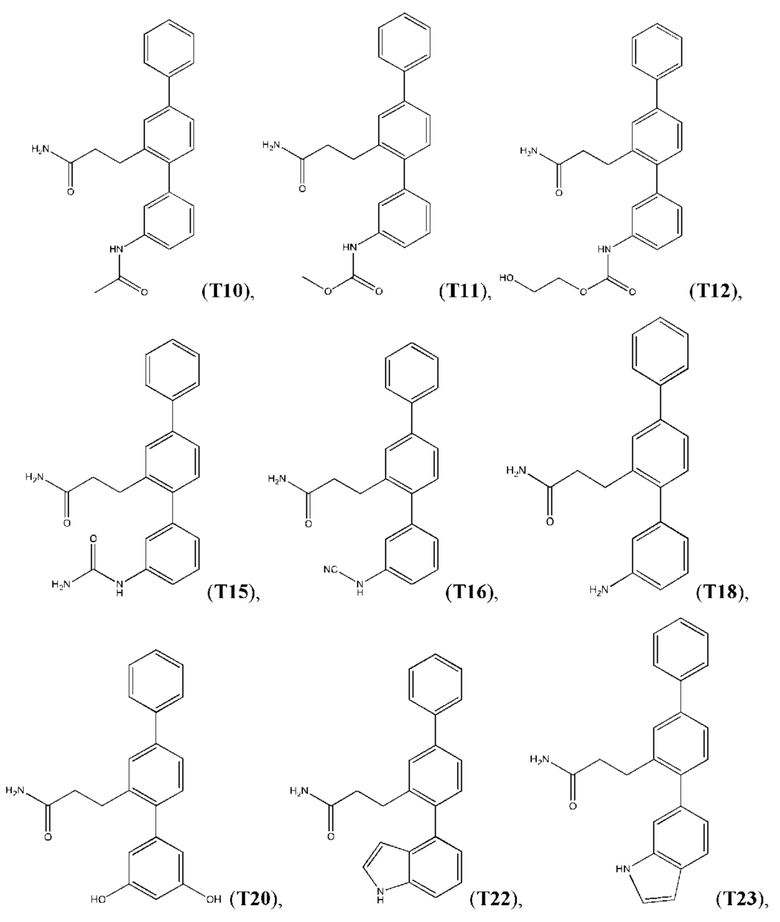
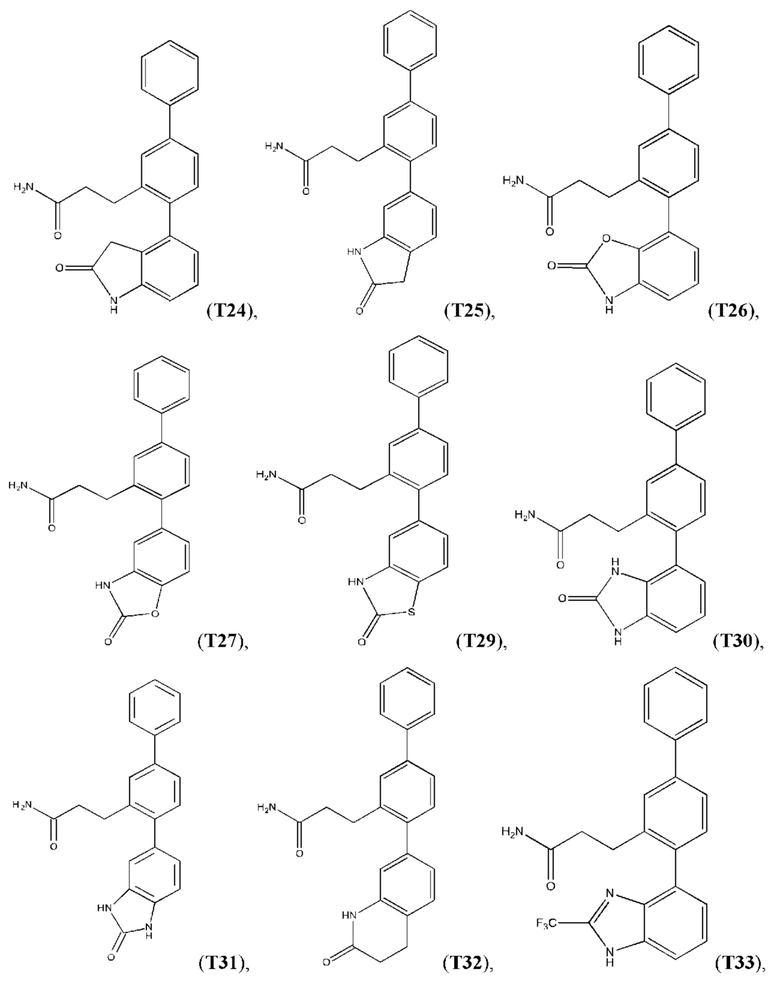
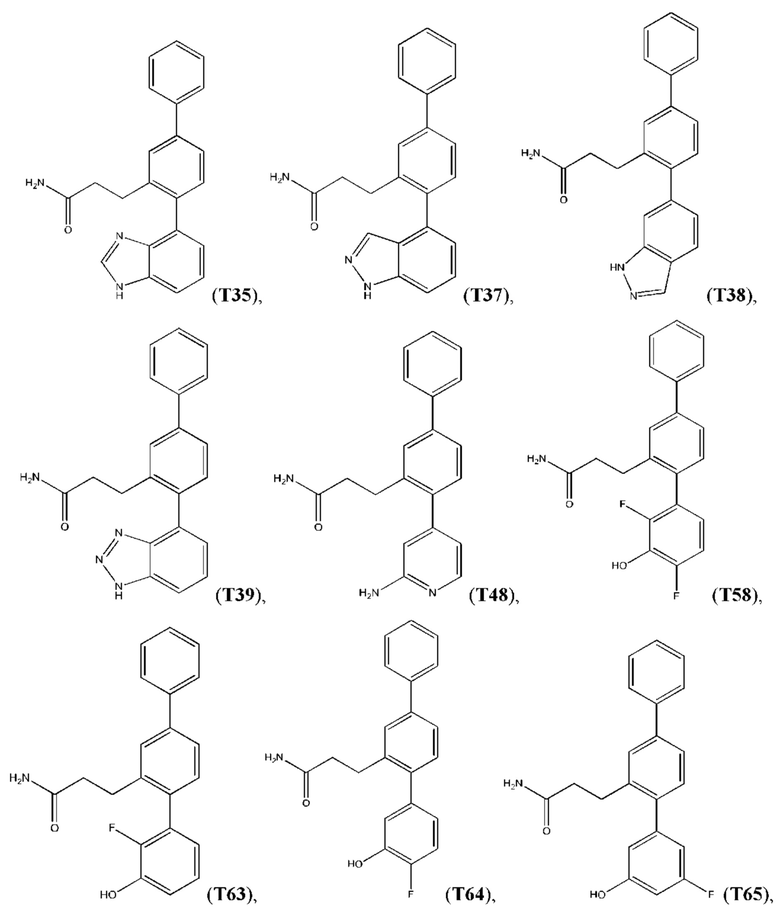
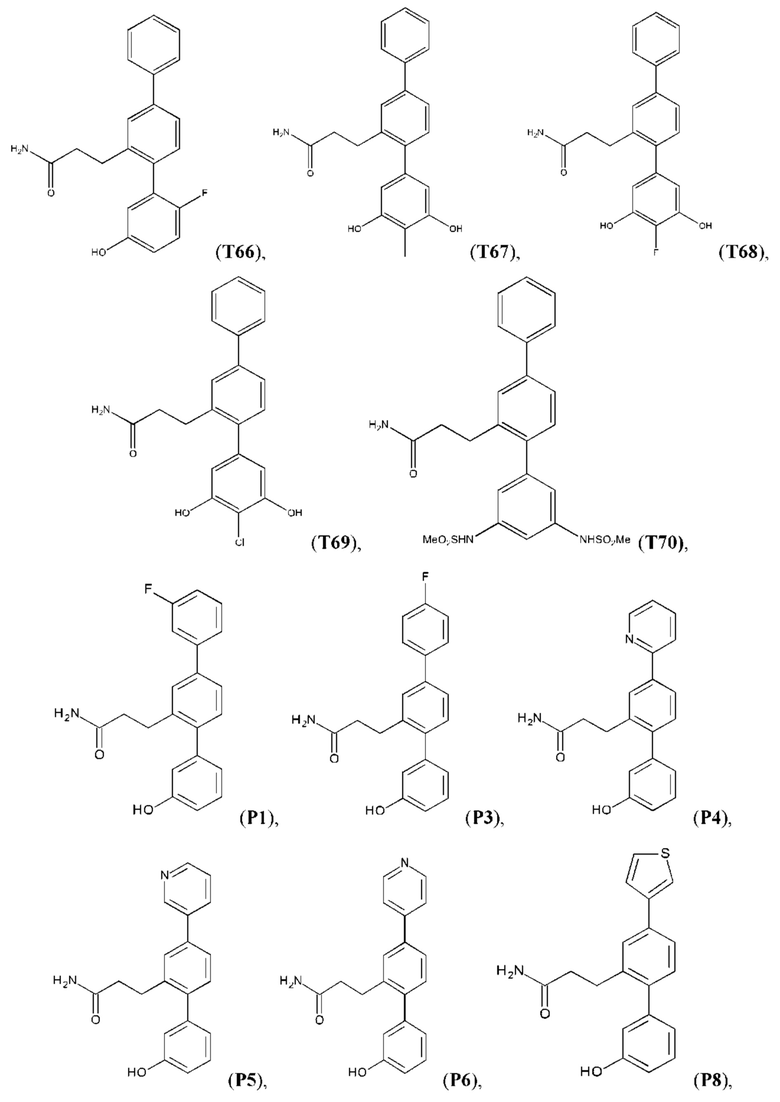
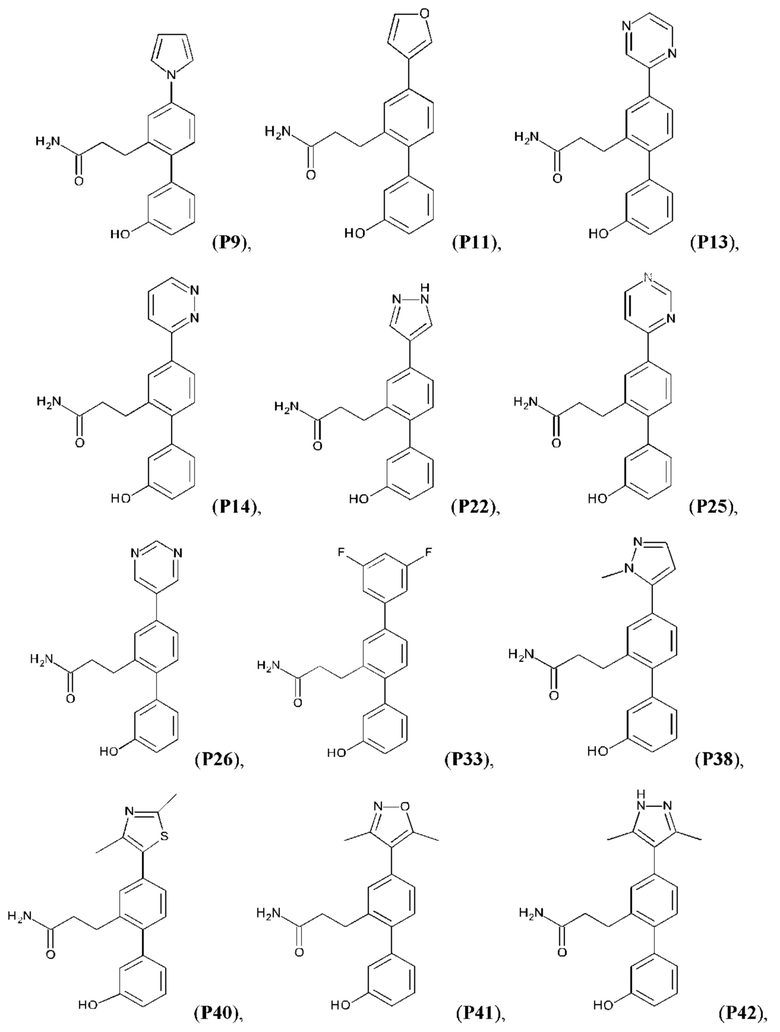
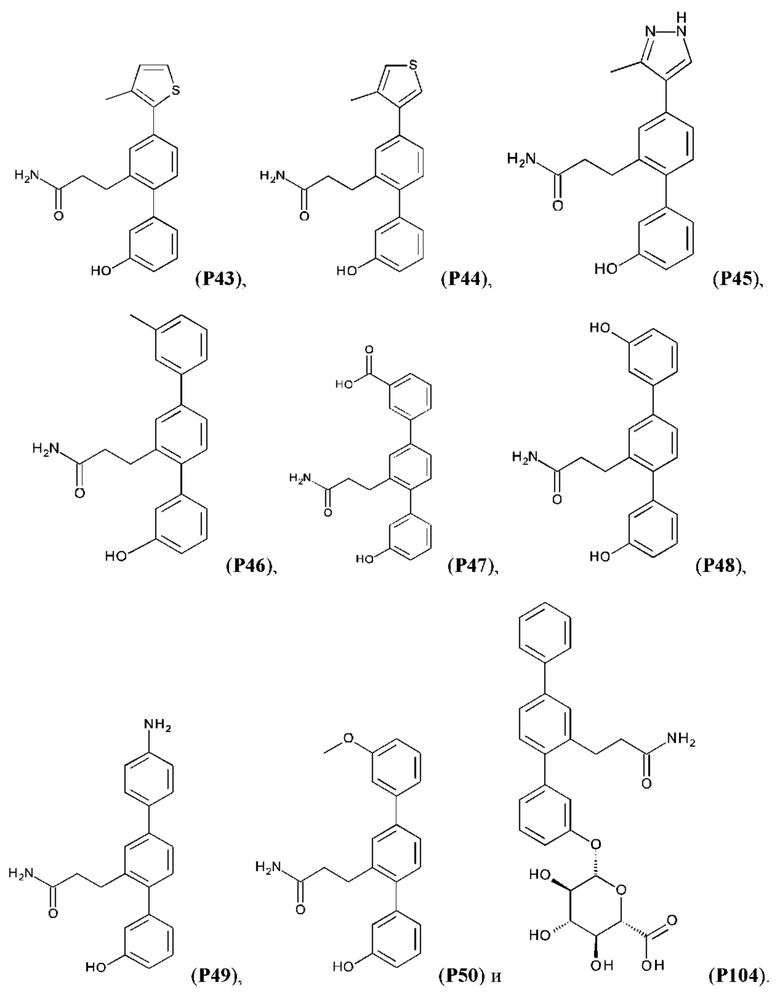
Использованный в данном описании термин «алкил», по отдельности или в комбинации, означает алкильный радикал с прямой или разветвленной цепью формулы -CnH(2n+1). Примеры алкилов включают метил, этил, н-пропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, изоамил, гексил, октил и тому подобное.
Использованный в данном описании термин «алкокси», по отдельности или в комбинации, означает алкил, соединенный с атомом кислорода, при этом термин «алкил» является таким, как определено выше. Примеры алкокси включают метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси и тому подобное.
Использованный в данном описании термин «галоген» обозначает -F, -Cl, -Br или -I.
Использованный в данном описании термин «гидрокси» обозначает -ОН.
Использованные в данном описании термины «амино» или «амин» обозначают -NH2.
Использованный в данном описании термин «замещенный амино» включает -NHW, где W выбран из -CN, -SO2(X)aY и -CO(X)aY, а равно 0 или 1, X выбран из -NH- и -О-, и Y выбран из -Н, -СН3, -СН2СН3, -СН2ОН и -СН2СН2ОН.
Использованный в данном описании термин «карбоновая кислота» обозначает -С(O)ОН.
Использованный в данном описании термин «окси» обозначает -О-.
Использованный в данном описании термин «оксо» обозначает =O.
Использованный в данном описании термин «глюкуронид» включает соединения, где глюкуроновая кислота связана с соединением посредством гликозидной связи.
Использованные в данном описании сокращения Me, Et, Ph, Ms представляют метил, этил, фенил и метансульфонил, соответственно. Более полный список сокращений, используемых специалистами обычной квалификации в области органической химии, содержится в первом номере каждого тома журнала по органической химии (Journal of Organic Chemistry); обычно этот список приводится в таблице, озаглавленной как «Стандартный список сокращений». Сокращения, содержащиеся в указанном списке, и все сокращения, используемые специалистами обычной квалификации в области органической химии, тем самым включены посредством ссылки.
Соединения по настоящему изобретению могут существовать в конкретных геометрических или стереоизомерных формах. Настоящее изобретение предусматривает все такие соединения, включая цис- и транс-изомеры, (R)- и (S)-энантиомеры, диастереомеры, (d)-изомеры, (l)-изомеры, их рацемические смеси и другие их смеси, которые попадают в объем данного изобретения. Подразумевается, что все такие изомеры, а также их смеси, включены в данное изобретение.
Если желателен, например, конкретный энантиомер соединения по настоящему изобретению, то он может быть получен путем асимметрического синтеза или посредством получения производного с использованием хирального вспомогательного вещества, при этом выполняют разделение полученной диастереомерной смеси и отщепляют группу вспомогательного вещества, получая чистые желаемые энантиомеры. Альтернативно, для образования диастереомерных солей могут быть использованы соответствующая(ее) оптически активная(ое) кислота или основание, после чего следует разделение образованных таким образом диастереомеров методами фракционной кристаллизации или хроматографическими методами, хорошо известными в данной области техники, и далее извлечение чистых энантиомеров.
В общем случае соединения по настоящему изобретению могут быть получены способами, проиллюстрированными на общих реакционных схемах, например, описанными ниже, или их модификациями, с использованием легко доступных исходных веществ, реагентов и традиционных методик синтеза. В этих реакциях также возможно использование вариантов, которые сами по себе известны, но здесь не упомянуты.
За исключением отмеченных случаев, способы синтеза соединений основаны на хорошо известных методах, описанных, например, в March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (2013) под редакцией Michael В. Smith; Advanced Organic Chemistry, Part A: Structure and Mechanisms (2008) и Advanced Organic Chemistry: Part B: Reaction and Synthesis (2010) под редакцией Francis A. Carey и Richard J. Sunberg; и Greene's Protective Groups in Organic Synthesis (2014) под редакцией Peter G.M. Wuts.
Настоящее изобретение также предусматривает фармацевтически приемлемые соли соединений. Термин «фармацевтически приемлемая соль» включает в себя соли присоединения как кислоты, так и основания, и относится к солям, которые сохраняют биологическую эффективность и свойства свободных оснований или кислот и которые не являются биологически или иным образом нежелательными. Фармацевтически приемлемые соли образуются с неорганическими или органическими кислотами или основаниями и могут быть получены in situ в ходе окончательного выделения и очистки соединений или в результате взаимодействия очищенного соединения в своей форме свободного основания или свободной кислоты по отдельности с подходящими органическими или неорганическими кислотой или основанием и выделения полученной таким образом соли.
Термин «легочный фиброз», как он использован в контексте настоящего изобретения, относится к образованию избытка волокнистой соединительной ткани в легком. Легочный фиброз может представлять собой вторичный эффект других заболеваний легких. Примеры таких заболеваний включают аутоиммунные расстройства, вирусные инфекции и бактериальные инфекции (такие как туберкулез). Легочный фиброз также может быть идиопатическим, при этом факторами риска считаются курение сигарет, факторы окружающей среды (например, связанное с родом занятий воздействие газов, дыма, химических веществ или пыли) или генетическая предрасположенность.
Помимо лечения диагностированного фиброза, соединения по настоящему изобретению могут быть использованы в профилактических целях субъектами с риском развития легочного фиброза. Примерами субъектов, попадающих в категорию риска развития легочного фиброза, являются субъекты, имеющие заболевания легких (такие как аутоиммунные расстройства, вирусные инфекции или бактериальные инфекции легких), субъекты, подвергающиеся воздействию газов, дыма, химических веществ, асбестовых волокон или пыли, либо имеющие генетическую предрасположенность, или субъекты, которые курят. Подразумевается, что термин «профилактический», как он использован в контексте настоящего изобретения, охватывает помимо прочего способы лечения, используемые для предупреждения или замедления развития фиброза в группе с таким риском.
В контексте настоящего изобретения термин «связанное с легочным фиброзом состояние» относится к любому состоянию, ассоциированному с легочным фиброзом или возникающее вследствие него. Примеры связанных с легочным фиброзом состояний включают легочную гипертензию, правостороннюю сердечную недостаточность, дыхательную недостаточность, гипоксию, кашель, образование тромбов, пневмонию и рак легкого.
Настоящее изобретение также предусматривает фармацевтические композиции, которые включают в себя соединения по настоящему изобретению вместе с фармацевтически приемлемыми эксципиентами. Термин «фармацевтически приемлемый эксципиент», как он использован в контексте настоящего изобретения, обозначает любой фармацевтически приемлемый неактивный компонент композиции. Как хорошо известно в данной области техники, эксципиенты включают разбавители, буферы, связующие вещества, смазывающие вещества, разрыхлители, красители, антиоксиданты/консерванты, средства для подведения рН и так далее. Эксципиенты выбирают с учетом желаемых физических аспектов конечной формы: например, для получения таблетки с желаемой твердостью и прочностью к истиранию, которая быстро диспергируется и легко проглатывается и так далее. В выборе эксципиентов также играет роль желаемая скорость высвобождения активного вещества из композиции после ее проглатывания. Фармацевтические композиции могут включать любой тип лекарственной формы, такой как таблетки, капсулы, порошки, жидкие композиции с модифицированным или длительным высвобождением, пластыри, лекарственные средства для вдыхания через нос, назальные спреи и тому подобное. Физическая форма и содержание рассматриваемых фармацевтических композиций относятся к традиционным препаратам, которые могут быть получены специалистами в области изготовления фармацевтических композиций, и основаны на ясно установленных принципах и технологиях изготовления, описанных, например, в Remington: The Science and Practice of Pharmacy, 19-oe издание, 1995; Британской фармакопее, 2000, и аналогичных относящихся к технологии приготовления текстов и руководств.
Например, если соединения или композиции подлежат пероральному введению, то они могут быть приготовлены в виде таблеток, капсул, гранул, порошков или сиропов; или в случае парентерального введения они могут быть приготовлены в виде инъекций (внутривенных, внутримышечных или подкожных), препаратов для капельной инфузии или суппозиториев. Для применения путем нанесения на слизистую оболочку глаз они могут быть приготовлены в виде глазных капель или глазных мазей. Эти композиции могут быть приготовлены традиционными способами, и при желании активный ингредиент может быть смешан с любым традиционным вспомогательным веществом, таким как эксципиент, связующее вещество, разрыхлитель, смазывающее вещество, корригент, солюбилизирующий агент, суспендирующая добавка, эмульгирующий агент или покрывающий агент.
Когда соединения по настоящему изобретению вводят в виде фармацевтических препаратов людям и животным, их можно принимать в чистом виде или в виде фармацевтической композиции, содержащей, например, 0,1-99,5% (более предпочтительно 0,5-90%) активного ингредиента в комбинации с фармацевтически приемлемым носителем.
Дозировка соединения и частота введения, которые должны быть использованы с целью получения желаемого ответа, также могут быть легко определены практикующим врачом.
Несмотря на то, что дозировка будет варьировать в зависимости от симптомов, возраста и массы тела пациента, характера и тяжести подлежащего лечению или предупреждению расстройства, пути введения и формы лекарственного средства, в общем случае суточная дозировка от 0,0001 мг до 200 мг соединения по настоящему изобретению может представлять собой подходящее эффективное количество для пациента, являющегося взрослым человеком, и ее можно вводить в виде разовой дозы или в виде разделенных доз.
«Пациент» или «субъект», подлежащий лечению способом по изобретению, может означать либо человека, либо не являющегося человеком субъекта.
«Эффективное количество» соединения по изобретению, в отношении способа лечения, относится к количеству терапевтического средства в препарате, которое при применении в качестве части желаемого режима введения обеспечивает благоприятное действие в соответствии с клинически приемлемыми стандартами для лечения или профилактики конкретного расстройства.
Теперь настоящее изобретение будет раскрыто более подробно со ссылкой на конкретные, но неограничивающие примеры, описывающие конкретные композиции и способы применения. Однако следует понимать, что подробное описание конкретных методик, композиций и способов включено исключительно с целью иллюстрации настоящего изобретения. Это никоим образом не должно рассматриваться как ограничение широкого описания концепции изобретения, изложенной выше.
ПРИМЕРЫ
Пример 1. Синтез соединений
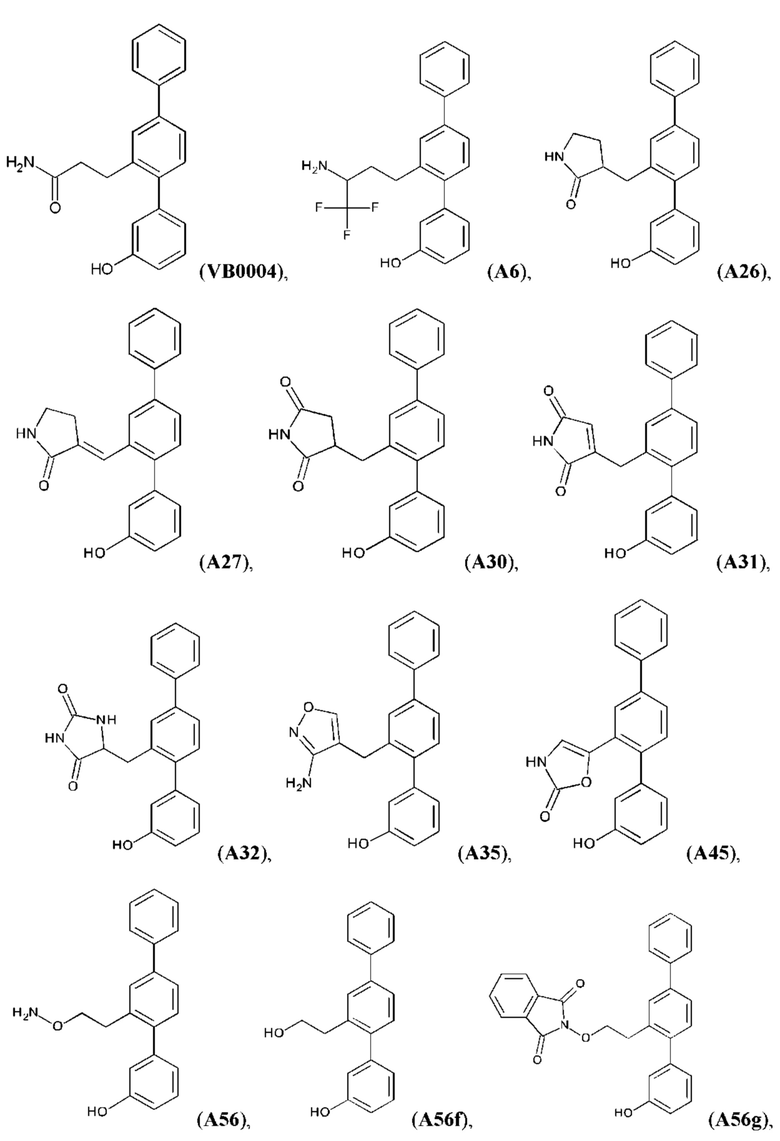
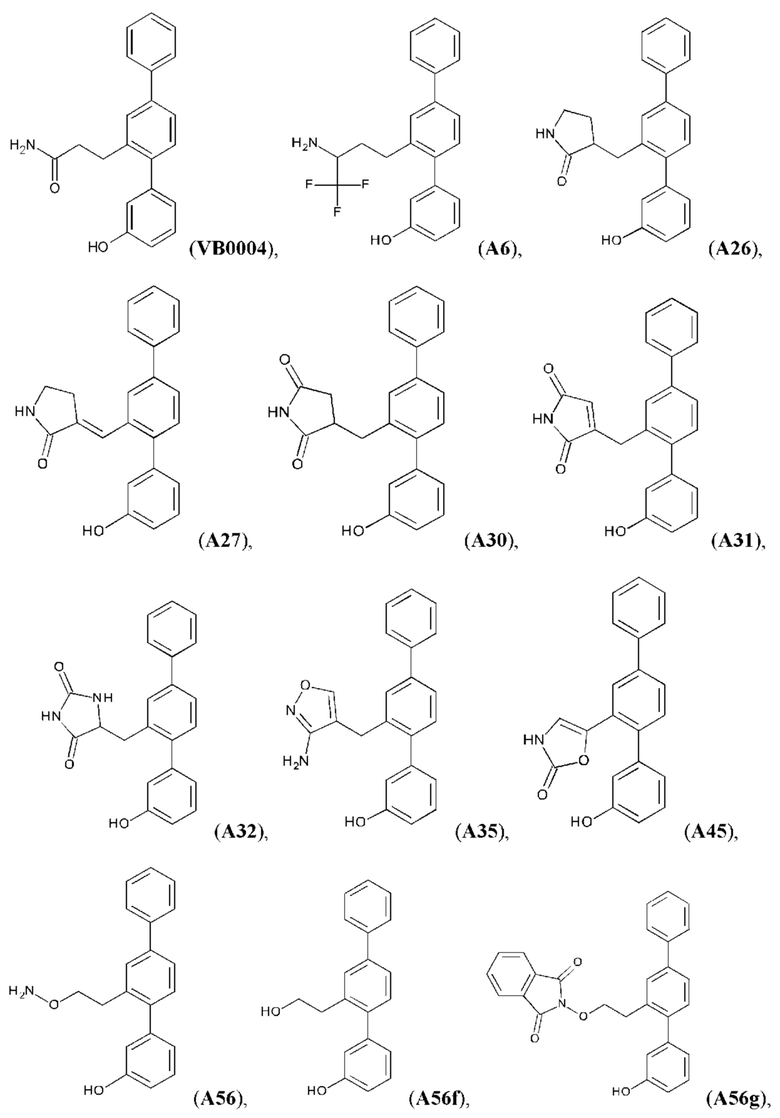
Синтез VB0004 описан в заявке PCT/AU 2014/000923 (WO 2015/039173), содержание которой тем самым включено в данное описание посредством ссылки во всей своей полноте.
Синтез А32, А6, А30, A56f, A56g, А56, A56k, А26, А27, А31, А35, А45, А79 и А81 описан в заявке PCT/AU 2016/000095 (WO 2016/145479), содержание которой тем самым включено в данное описание посредством ссылки во всей своей полноте.
Синтез T1, Т2, Т3, Т4, Т5, Т6, Т10, T11, Т12, Т15, Т16, Т18, Т20, Т22, Т23, Т24, Т25, Т26, Т27, Т29, Т30, Т31, Т32, Т33, Т35, Т37, Т38, Т39, Т48, Т58, Т63, Т64, Т65, Т66, Т67, Т68, Т69 и Т70 описан в заявке PCT/AU 2014/000922 (WO 2015/039172), содержание которой тем самым включено в данное описание посредством ссылки во всей своей полноте.
Синтез Р1, Р3, Р4, Р5, Р6, Р8, Р9, Р11, Р22, Р26, Р33, Р38, Р40, Р41, Р42, Р43, Р44, Р45, Р46, Р47, Р48, Р49, Р50 и Р104 описан в заявке PCT/AU 2016/000094 (WO 2016/145478), содержание которой тем самым включено в данное описание посредством ссылки во всей своей полноте.
Схема синтеза Р13 показана ниже.
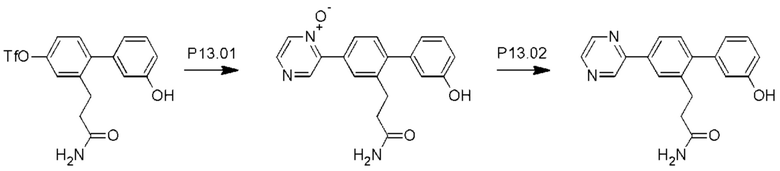
Стадия P13.01: синтез 3-[3'-гидрокси-4-(1-оксидопиразин-2-ил)бифенил-2-ил]пропанамида. Во флакон для микроволнового реактора емкостью 20 мл загружали пиразина N-оксид (Fagnou et al., JACS, 2005, 127: 18020-1) (0,346 г; 3,60 ммоль), карбонат калия (0,497 г; 3,60 ммоль), ацетат палладия (0,020 г; 0,089 ммоль), тетрафторборат три-пара-бутилфосфония (0,052 г; 0,179 ммоль) и пивалевую кислоту (0,055 г; 0,541 ммоль). Добавляли раствор 2-(3-амино-3-оксопропил)-3'-гидроксибифенил-4-ил-трифторметансульфоната (0,700 г; 1,798 ммоль) в толуоле (4 мл), флакон продували азотом в течение 10 мин, герметично закрывали и нагревали при температуре дефлегмации в течение 4 ч. После охлаждения добавляли хлороформ (10 мл), полученный осадок отфильтровывали и последовательно промывали дихлорметаном и смесью этилацетат/метанол. Органические экстракты объединяли, концентрировали и очищали флэш-хроматографией (ацетон/дихлорметан/метанол), получая указанное в заголовке соединение в виде бесцветного твердого вещества (0,460 г; 76%). 1Н ЯМР (400 МГц, диметилсульфоксид (DMSO)-d6) δ млн-1 9.56 (s, 1H), 8.81 (s, 1H), 8.52-8.49 (m, 1H), 8.45 (d, J=4,1 Гц, 1H), 7.78-7.73 (m, 2Н), 7.30-7.26 (m, 1H), 7.25 (d, J=8,0 Гц, 1H), 7.22 (br. s., 1H), 6.80 (ddd, J=0,8; 2,3; 8,2 Гц, 1H), 6.78-6.75 (m, 1H), 6.75-6.70 (m, 2Н), 2.82 (dd, J=6,9; 9,1 Гц, 2Н), 2.31-2.24 (m, 2Н). LCMS (жидкостная хроматография в сочетании с масс-спектрометрией) [М+Н]+=336,2.
Стадия Р13.02: синтез 3-[3'-гидрокси-4-(пиразин-2-ил)бифенил-2-ил]пропанамида (Р13). В круглодонную колбу в атмосфере азота загружали 3-[3'-гидрокси-4-(1-оксидопиразин-2-ил)бифенил-2-ил]пропанамид (0,460 г; 1,37 ммоль) и метанол (10 мл). Реакционную смесь продували, барботируя через нее азот в течение 5 мин, добавляли 10% палладий на угле (0,046 г) и реакционную смесь перемешивали в атмосфере водорода в течение 18 ч. Смесь фильтровали через нейлоновый фильтр, концентрировали и очищали флэш-хроматографией (метанол/дихлорметан), получая бледно-желтое твердое вещество. Это твердое вещество перекристаллизовывали из смеси хлороформ/метанол, получая указанное в заголовке соединение в виде бесцветного порошка (0,245 г; 56%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.54 (s, 1H), 9.28 (d, J=1,6 Гц, 1H), 8.74 (dd, J=1,6; 2,5 Гц, 1H), 8.62 (d, J=2,5 Гц, 1H), 8.10 (d, J=1,8 Гц, 1H), 8.00 (dd, J=2,0; 8,0 Гц, 1H), 7.30 (d, J=7,8 Гц, 1H), 7.26 (t, J=7,8 Гц, 2Н), 6.83-6.71 (m, 4Н), 2.86 (dd, J=6,9; 9,1 Гц, 2Н), 2.36-2.29 (m, 2Н). LCMS [М+Н]+=320,1, [M+Na]+=342,1.
Схема синтеза Р14 показана ниже.
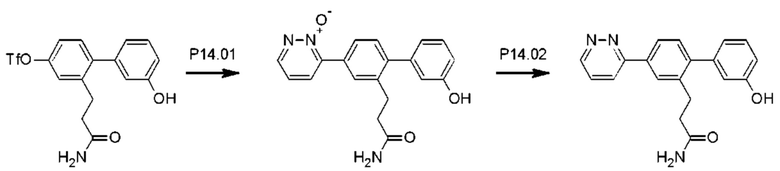
Стадия Р14.01: синтез 3-[3'-гидрокси-4-(2-оксидопиридазин-3-ил)бифенил-2-ил]пропанамида. Получали согласно стадии Р13.01. Реакционную смесь нагревали при температуре дефлегмации в течение 12 ч. Неочищенное вещество очищали флэш-хроматографией (дихлорметан/ацетон/метанол), получая указанное в заголовке соединение в виде желтой пены (0,380 г; 63%). 1Н ЯМР (400 МГц, МЕТАНОЛ-d4) δ млн-1 8.60 (dd, J=2,3; 5,3 Гц, 1H), 8.12 (dd, J=2,3; 8,0 Гц, 1H), 7.80 (d, J=1,6 Гц, 1H), 7.75 (dd, J=1,9; 7,9 Гц, 1H), 7.45 (dd, J=5,3; 8,0 Гц, 1H), 7.33 (d, J=8,0 Гц, 1H), 7.27 (t, J=7,8 Гц, 1H), 6.84-6.79 (m, 2Н), 6.78-6.76 (m, 1H), 2.99 (dd, J=7,1; 8,7 Гц, 2Н), 2.44-2.36 (m, 2Н). LCMS [М+Н]+=336,1; [M+Na]+=358,1.
Стадия Р14.02: синтез 3-[3'-гидрокси-4-(пиридазин-3-ил)бифенил-2-ил]пропанамида (Р14). Выполняли согласно стадии Р13.02 с добавлением раствора гидроксида аммония (2 мл) через 24 ч, получая указанное в заголовке соединение в виде бесцветного порошка (0,105 г; 55%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.54 (s, 1H), 9.22 (dd, J=1,6; 4,9 Гц, 1H), 8.24 (dd, J=1,6; 8,6 Гц, 1H), 8.13 (d, J=1,8 Гц, 1H), 8.00 (dd, J=2,0; 8,0 Гц, 1H), 7.80 (dd, J=4,9; 8,6 Гц, 1H), 7.32 (d, J=7,8 Гц, 1H), 7.29-7.22 (m, 2Н), 6.83-6.71 (m, 4Н), 2.91-2.83 (m, 2Н), 2.36-2.30 (m, 2Н). LCMS [М+Н]+=320,2; [M+Na]+=342,2.
Схема синтеза Р25 показана ниже.
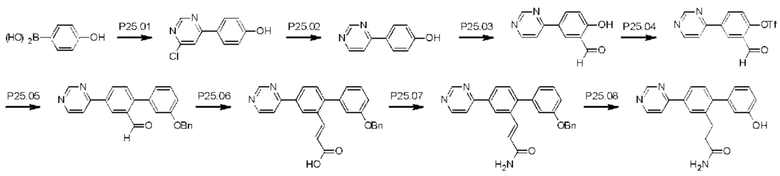
Стадия Р25.01: синтез 4-(6-хлорпиримидин-4-ил)фенола. В круглодонную колбу загружали 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенол (10,0 г; 0,045 моль), 4,6-дихлорпиримидин (8,12 г; 0,055 моль) и карбонат цезия (29,58 г; 0,092 моль) в растворе 1,4-диоксан/вода (9:1, 100 мл) и азот барботировали через эту смесь в течение 10 мин, после чего добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II)-дихлорметан (1,85 г; 2,27 ммоль) и реакционную смесь нагревали при температуре дефлегмации в течение 3 ч. Смесь охлаждали до комнатной температуры, фильтровали, переносили в делительную воронку и распределяли между этилацетатом (100 мл) и водой (100 мл). Водный слой отделяли и повторно экстрагировали этилацетатом (2×50 мл). Органические экстракты объединяли, сушили над сульфатом магния, фильтровали и концентрировали. Неочищенное вещество частично очищали флэш-хроматографией (этилацетат/гексаны), получая указанное в заголовке соединение (3,8 г) в виде смеси, которую непосредственно переносили на следующую стадию. LCMS [М+Н]+=207,1.
Стадия Р25.02: синтез 4-(пиримидин-4-ил)фенола. В круглодонную колбу в атмосфере азота загружали 4-(6-хлорпиримидин-4-ил)фенол (0,20 г; 0,968 ммоль), метанол (10 мл) и водный аммиак (1,5 мл 25%-ного раствора). Реакционную смесь продували, барботируя через нее азот в течение 5 мин, затем добавляли 10% палладий на угле (0,020 г) и реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 3 ч, затем фильтровали через фильтровальную бумагу. Фильтрат концентрировали досуха и остаток распределяли между этилацетатом (20 мл) и водой (10 мл). Водную фазу отделяли, подщелачивали 10%-ным водным раствором гидроксида натрия, далее экстрагировали этилацетатом (3×20 мл), значение рН доводили до 4 путем добавления 1 М раствора соляной кислоты и смесь экстрагировали раствором 9/1 дихлорметан/метанол. Органическую фазу отделяли, сушили над сульфатом магния, фильтровали и концентрировали, получая указанное в заголовке соединение в виде желтого твердого вещества (0,08 г; 80%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 10.08 (s, 1H), 9.13 (d, J=1,2 Гц, 1H), 8.74 (d, J=5,5 Гц, 1H), 8.12-8.06 (m, 2Н), 7.94 (dd, J=1,4; 5,5 Гц, 1H), 6.94-6.88 (m, 2Н). LCMS [М+Н]+=173,1.
Стадия Р25.03: синтез 2-гидрокси-5-(пиримидин-4-ил)бензальдегида. В круглодонную колбу загружали 4-(пиримидин-4-ил)фенол (1,9 г; 11,04 ммоль), трифторуксусную кислоту (22 мл) и гексамин (2,32 г; 16,5 ммоль) и реакционную смесь нагревали при температуре дефлегмации в течение 16 ч. После охлаждения до комнатной температуры добавляли воду (100 мл) и перемешивание продолжали в течение еще 30 мин, затем реакционную смесь переносили в делительную воронку и экстрагировали дихлорметаном (100 мл). Водный слой отделяли и далее экстрагировали дихлорметаном (3×100 мл). Органические экстракты объединяли, промывали водой (150 мл), рассолом (100 мл), сушили над сульфатом магния, фильтровали и концентрировали. Неочищенное вещество очищали флэш-хроматографией (этилацетат/дихлорметан/метанол), получая указанное в заголовке соединение в виде белого твердого вещества (0,750 г; 42%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 11.32 (br. s., 1H), 10.35 (s, 1H), 9.21 (d, J=1,3 Гц, 1H), 8.82 (d, J=5,5 Гц, 1H), 8.55 (d, J=2,3 Гц, 1H), 8.38 (dd, J=2,4; 8,7 Гц, 1H), 8.06 (dd, J=1,4; 5,5 Гц, 1H), 7.17 (d, J=8,7 Гц, 1H). LCMS [М+Н]+=201,1.
Стадия Р25.04: синтез 2-формил-4-(пиримидин-4-ил)фенил-трифторметансульфоната. В круглодонную колбу загружали 2-гидрокси-5-(пиримидин-4-ил)бензальдегид (0,75 г; 3,75 ммоль), ацетонитрил (30 мл) и карбонат калия (1,04 г; 17,5 ммоль). Добавляли N-фенил-бис(трифтор-метансульфонимид) (1,47 г; 4,12 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь концентрировали, распределяли между этилацетатом (50 мл) и водой (50 мл) и водную фазу далее экстрагировали этилацетатом (3×40 мл). Объединенные органические экстракты промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали досуха, получая указанное в заголовке соединение в виде желтого твердого вещества (1,4 г), которое использовали без дополнительной очистки. 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ млн-1 10.35 (s, 1H), 9.34 (d, J=1,3 Гц, 1H), 8.89 (d, J=5,3 Гц, 1H), 8.69 (d, J=2,3 Гц, 1H), 8.53 (dd, J=2,4; 8,7 Гц, 1H), 7.81 (dd, J=1,4; 5,3 Гц, 1H), 7.41 (d, J=7,9 Гц, 1H). LCMS [М+Н]+=333,0.
Стадия Р25.05: синтез 3'-(бензилокси)-4-(пиримидин-4-ил)бифенил-2-карбальдегида. Смесь 2-формил-4-(пиримидин-4-ил)фенил-трифторметансульфоната (1 экв.), гетероциклической бороновой кислоты (1,2 экв.) и карбоната калия (2 экв.) суспендировали в 1,4-диоксане (4 мл/ммоль) и воде (5 капель/ммоль). Через эту смесь в течение 15 мин барботировали азот. Добавляли тетракис(трифенилфосфин)палладий(0) (0,1 экв.) и смесь нагревали при 85°С в атмосфере азота в течение 20 ч. Смесь разбавляли этилацетатом и фильтровали. Остаток промывали этилацетатом (2×). Объединенные фильтраты упаривали досуха и очищали флэш-хроматографией (метанол/дихлорметан). Продукт суспендировали в гексанах (4 мл) и выделяли фильтрованием. Неочищенное вещество очищали флэш-хроматографией (этилацетат/дихлорметан), получая указанное в заголовке соединение в виде оранжевого масла (0,400 г; 91%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ млн-1 10.04 (s, 1H), 9.32 (d, J=1,3 Гц, 1H), 8.84 (d, J=5,4 Гц, 1H), 8.66 (d, J=1,8 Гц, 1H), 8.47 (dd, J=2,0; 8,1 Гц, 1H), 7.86 (dd, J=1,5; 5,4 Гц, 1H), 7.62 (d, J=8,5 Гц, 1H), 7.47-7.43 (m, 2Н), 7.43-7.38 (m, 3Н), 7.30 (m, 1H), 7.10 (ddd, J=0,9; 2,6; 8,4 Гц, 1H), 7.07-7.03 (m, 1H), 7.03-6.98 (m, 1H), 5.13 (s, 2H). LCMS [M+H]+=367,2; [M+Na]+=389,1.
Стадия P25.06: синтез (Е)-3-(2-(3-(бензилокси)фенил)-5-(пиримидин-4-ил)фенил)проп-2-еновой кислоты. В круглодонную колбу загружали 3'-(бензилокси)-4-(пиримидин-4-ил)бифенил-2-карбальдегид (0,350 г; 0,96 ммоль) и пиридин (11 мл). Добавляли малоновую кислоту (0,120 г; 1,15 ммоль), затем пиперидин (9,8 мг; 0,11 ммоль) и смесь нагревали при температуре дефлегмации в течение 48 ч. После охлаждения до комнатной температуры добавляли 1 М раствор соляной кислоты до тех пор, пока значение рН не достигало 1-2. Полученную суспензию охлаждали в ледяной бане, фильтровали и остаток экстрагировали этилацетатом (100 мл) и промывали рассолом (50 мл). Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали досуха, получая указанное в заголовке соединение в виде оранжевого твердого вещества (0,280 г; 71%), которое использовали без дополнительной очистки. 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.31 (d, J=1,1 Гц, 1H), 8.93 (d, J=5,4 Гц, 1H), 8.65 (d, J=1,8 Гц, 1H), 8.38-8.33 (m, 2Н), 7.60 (d, J=4,9 Гц, 1H), 7.57 (d, J=2,9 Гц, 1H), 7.49-7.38 (m, 5Н), 7.36-7.31 (m, 1H), 7.13 (ddd, J=0,8; 2,6; 8,3 Гц, 1H), 7.05-7.02 (m, 1H), 6.97-6.93 (m, 1H), 6.73 (d, J=15,9 Гц, 1H), 5.17 (s, 2Н). LCMS [М+Н]+=409,2; [M+Na]+=431,2.
Стадия Р25.07: синтез (Е)-3-(2-(3-(бензилокси)фенил)-5-(пиримидин-4-ил)фенил)проп-2-енамида. Оксалилхлорид (0,075 мл; 0,892 ммоль) по каплям добавляли в круглодонную колбу с загруженными в нее (E)-3-(2-(3'-(бензилокси)фенил)-5-(пиримидин-4-ил)фенил)проп-2-еновой кислотой (0,280 г; 0,686 ммоль) и дихлорметаном (2,3 мл). Добавляли каталитическое количество N,N-диметилформамида, смесь перемешивали при комнатной температуре в течение 3 ч и затем концентрировали. Остаток переносили в 1,4-диоксан (10 мл), охлаждали в ледяной бане и по каплям добавляли водный аммиак (0,15 мл; 25%-ный раствор). Через 30 мин выдерживания при комнатной температуре реакционную смесь концентрировали досуха, добавляли диэтиловый эфир и суспензию фильтровали. Твердый остаток переносили в горячий метанол, фильтровали и концентрировали досуха, получая указанное в заголовке соединение в виде бесцветного твердого вещества (0,267 г; 96%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.31 (d, J=1,2 Гц, 1H), 8.93 (d, J=5,4 Гц, 1H), 8.61 (d, J=1,8 Гц, 1H), 8.26 (dd, J=1,9; 8,1 Гц, 1H), 8.23 (dd, J=1,4; 5,5 Гц, 1H), 7.60 (br. s., 1H), 7.55 (d, J=8,1 Гц, 1H), 7.49-7.45 (m, 3Н), 7.45-7.37 (m, 3Н), 7.36-7.33 (m, 1H), 7.16 (br. s., 1H), 7.12 (ddd, J=0,8; 2,6; 8,3 Гц, 1H), 7.03-7.00 (m, 1H), 6.94 (td, J=1,0; 7,8 Гц, 1H), 6.81 (d, J=15,6 Гц, 1H), 5.16 (s, 2Н). LCMS [М+Н]+=408,1.
Стадия Р25.08: синтез 3-[3'-гидрокси-4-(пиримидин-4-ил)бифенил-2-ил]пропанамида (Р25). В круглодонную колбу в атмосфере азота загружали (E)-3-(2-(3-(бензилокси)фенил)-5-(пиримидин-4-ил)фенил)проп-2-енамид (0,250 г; 0,164 ммоль), метанол (10 мл) и водный аммиак (1,5 мл 25%-ного раствора). Реакционную смесь продували, барботируя через нее азот в течение 5 мин, затем добавляли 10% палладий на угле (0,025 г) и реакционную смесь перемешивали в атмосфере водорода при комнатной температуре в течение 18 ч, затем фильтровали через фильтровальную бумагу. Остаток промывали горячим метанолом и фильтрат концентрировали досуха, получая бесцветный остаток, который очищали обращенно-фазовой флэш-хроматографией (метанол/вода) и растиранием из метанола, получая указанное в заголовке соединение в виде беловатого твердого вещества (0,036 г; 18%). 1H ЯМР (400 МГц, МЕТАНОЛ-d4) δ млн-1 9.19 (d, J=1,2 Гц, 1H), 8.80 (d, J=5,5 Гц, 1H), 8.16 (d, J=1,8 Гц, 1H), 8.07-8.00 (m, 2Н), 7.35 (d, J=8,0 Гц, 1H), 7.27 (t, J=7,9 Гц, 1H), 6.84-6.76 (m, 3Н), 3.05-2.99 (m, 2Н), 2.46-2.39 (m, 2Н). LCMS [М+Н]+=320,1; [M+Na]+=342,1.
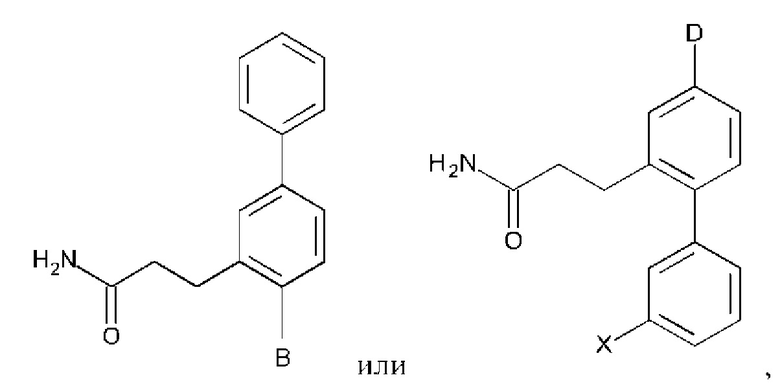
Схема синтеза реагентов для получения D4-D18 показана ниже:
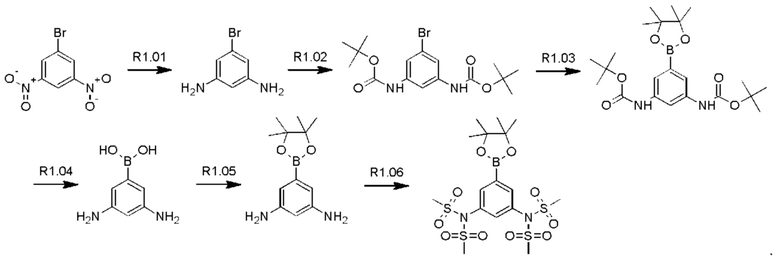
Стадия R1.01: синтез 5-бром-1,3-диаминобензола. В круглодонную колбу загружали 5-бром-1,3-динитробензол (5,0 г; 20,2 ммоль), этанол (240 мл) и воду (120 мл). Добавляли порошок железа (13,57 г; 24,3 ммоль), затем хлорид аммония (1,30 г; 24,3 ммоль) и полученную смесь перемешивали при 85°С в течение 16 ч. Реакционную смесь фильтровали и концентрировали досуха, получая указанное в заголовке соединение в виде желтого твердого вещества (4,0 г), которое использовали без очистки. 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ млн-1 6.26 (d, J=2,0 Гц, 2Н), 5.91 (t, J=2,0 Гц, 1H), 3.59 (ушир. s., 4Н). LCMS [М+Н]+=189,0.
Стадия R1.02: синтез ди-трет-бутил-(5-бромбензол-1,3-диил)бискарбамата. В круглодонную колбу загружали 5-бром-1,3-диаминобензол (3,7 г; 19,8 ммоль) и суспендировали в воде (20 мл). Добавляли ди-трет-бутил-дикарбонат (9,50 г; 43,56 ммоль), что приводило к нагреванию и выделению газа. Далее реакционную смесь разбавляли водой (80 мл), затем нагревали при 70°С. После охлаждения до комнатной температуры добавляли воду (100 мл), осадок собирали фильтрованием и тщательно промывали водой. Твердый остаток экстрагировали дихлорметаном (100 мл), сушили над сульфатом магния, фильтровали и концентрировали досуха, получая указанное в заголовке соединение в виде бледно-желтого твердого вещества (7,4 г; 96%). LCMS [M+Na]+=411,0.
Стадия R1.03: синтез ди-трет-бутил-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензол-1,3-диил)бискарбамата. В круглодонную колбу загружали ди-трет-бутил-(5-бромбензол-1,3-диил)бискарбамат (4,00 г; 10,3 ммоль), 1,4-диоксан (50 мл), бис(пинаколато)дибор (2,88 г; 11,36 ммоль) и ацетат калия (3,03 г; 30,9 ммоль). Через эту смесь в течение 5 мин барботировали азот, добавляли комплекс [1,1'-бис(дифенилфосфино)-ферроцен]дихлорпалладий(II)-дихлорметан (0,420 г; 0,515 ммоль) и реакционную смесь нагревали при 100°С в течение 20 ч. После охлаждения реакционную смесь распределяли между этилацетатом (150 мл) и водой (20 мл), органическую фазу промывали рассолом (20 мл), сушили над сульфатом магния, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (этилацетат/гексаны), получая указанное в заголовке соединение в виде бледно-желтого кристаллического порошка (0,55 г; 12%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ млн-1 7.75 (t, J=2,1 Гц, 1H), 7.35 (d, J=2,1 Гц, 2Н), 6.45 (s, 2Н), 1.50 (s, 18Н), 1.31 (s, 12Н).
Стадия R1.04: синтез 1,3-диамино-5-бензолбороновой кислоты. В круглодонную колбу загружали ди-трет-бутил-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензол-1,3-диил)бискарбамат (1,34 г; 3,08 ммоль) и хлороформ (10 мл), охлаждали до 0°С и добавляли трифторуксусную кислоту (2,4 мл; 30,8 ммоль). Реакционную смесь нагревали до комнатной температуры, перемешивали в течение 24 ч, затем концентрировали досуха. Неочищенный остаток распределяли между хлороформом (50 мл) и водой (50 мл), водную фазу промывали 10%-ным метанолом в хлороформе (50 мл), значение рН доводили до 7, добавляя гидрокарбонат натрия, и промывали дихлорметаном. Водную фазу концентрировали досуха, получая твердый остаток, который промывали дихлорметаном, переносили в метанол, фильтровали и концентрировали досуха, получая указанное в заголовке соединение в виде белого твердого вещества (0,50 г; колич.). 1Н ЯМР (400 МГц, МЕТАНОЛ-d4) δ млн-1 7.62 (ушир. s., 3Н), 7.40 (ушир. s., 1H). LCMS [М+Н]+=153,2.
Стадия R1.05: синтез 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-бензол-1,3-диамина. В круглодонную колбу загружали 1,3-диамино-5-бензолбороновую кислоту (0,50 г; 3,30 ммоль) и суспендировали в тетрагидрофуране (10 мл). Добавляли 2,3-диметил-2,3-бутандиол (0,39 г; 3,30 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 24 ч и затем разбавляли 1%-ным раствором метанола в дихлорметане (50 мл), сушили над сульфатом натрия, фильтровали и концентрировали досуха, получая указанное в заголовке соединение в виде светло-зеленого твердого вещества (0,850 г), которое использовали без очистки. LCMS [М+Н]+=235,2.
Стадия R1.06: синтез N,N'-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)(бензол-1,3-диил)диметансульфонамида. В круглодонную колбу загружали 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-бензол-1,3-диамин (0,10 г; 0,427 ммоль), хлороформ (2 мл) и охлаждали до 0°С. Добавляли триэтиламин (0,298 мл; 2,14 ммоль), затем по каплям добавляли метансульфонилхлорид (0,165 мл; 2,14 ммоль). Полученную смесь перемешивали в течение 2 ч, концентрировали досуха и остаток распределяли между хлороформом (50 мл) и водой (50 мл). Водную фазу промывали хлороформом (3×20 мл), органические фазы объединяли, сушили над сульфатом магния, фильтровали и концентрировали досуха. Твердый остаток растирали с эфиром, получая указанное в заголовке соединение в виде белого твердого вещества (0,10 г; 43%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ млн-1 7.90 (d, J=2,1 Гц, 2Н), 7.44 (t, J=2,1 Гц, 1H), 3.40 (s, 12Н), 1.35 (s, 12Н).
Схема синтеза дополнительных реагентов для получения D4-D18 показана ниже:
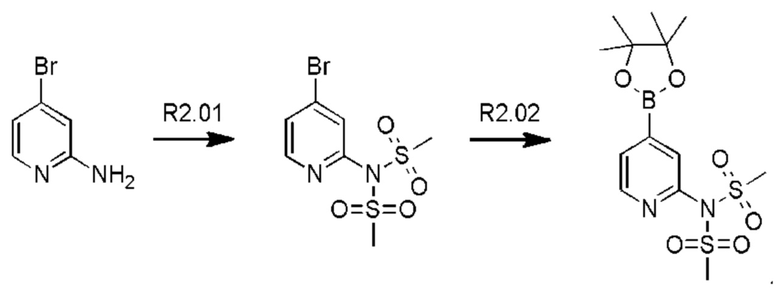
Стадия R2.01: синтез N-(4-бромпиридин-2-ил)-N-(метилсульфонил)метансульфонамида. В круглодонную колбу загружали 2-амино-4-бромпиридин (1,0 г; 5,78 ммоль) и хлороформ (10 мл), охлаждали до 0°С и добавляли триэтиламин (2,41 мл; 17,34 ммоль), затем по каплям добавляли метансульфонилхлорид (1,34 мл; 17,34 ммоль). Реакционную смесь перемешивали в течение 1 ч при 0°С, 1 ч при комнатной температуре и затем концентрировали досуха. Добавляли диэтиловый эфир (50 мл), суспензию перемешивали в течение 30 мин и фильтровали. Твердый остаток экстрагировали дихлорметаном (100 мл), промывали водой (50 мл), органическую фазу сушили над сульфатом магния, фильтровали и концентрировали досуха, получая указанное в заголовке соединение в виде белого порошка (1,3 г), содержащего приблизительно 15% моносульфонилированного продукта. LCMS [М+Н]+=330,9; [M+Na]+=352,9.
Стадия R2.02: синтез N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)-N-(метилсульфонил)метансульфонамида. Во флакон емкостью 20 мл для микроволнового реактора загружали N-(4-бромпиридин-2-ил)-N-(метилсульфонил)метансульфонамид (0,500 г; 1,52 ммоль), 1,4-диоксан (5 мл), бис(пинаколато)дибор (0,490 г; 1,67 ммоль) и ацетат калия (0,450 г; 4,56 ммоль) и через эту смесь в течение 5 мин барботировали азот. Добавляли комплекс [1,1'-бис(дифенилфосфино)-ферроцен]дихлорпалладий(II)-дихлорметан (0,062 г; 0,076 ммоль), флакон герметично закрывали и смесь нагревали при 90°С в течение 12 ч. После охлаждения добавляли этилацетат (100 мл), смесь переносили в делительную воронку и промывали водой (50 мл). Органическую фазу промывали 0,01 М раствором соляной кислоты, объединенные водные фазы повторно экстрагировали этилацетатом (2×20 мл), объединенные органические фазы промывали рассолом, сушили (сульфат магния), фильтровали и концентрировали досуха. Неочищенный остаток промывали смесью этилацетат/гексаны и фильтровали, получая указанное в заголовке соединение в виде белого твердого вещества (0,120 г; 23%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 8.62 (dd, J=0,9; 4,7 Гц, 1H), 7.79 (t, J=0,9 Гц, 1H), 7.71 (dd, J=0,9; 4,7 Гц, 1H), 3.65 (s, 6Н), 1.34 (s, 12Н). LCMS (бороновая кислота) [М+Н]+=295,0; [M+Na]+=317,0.
Схема синтеза D4 показана ниже:
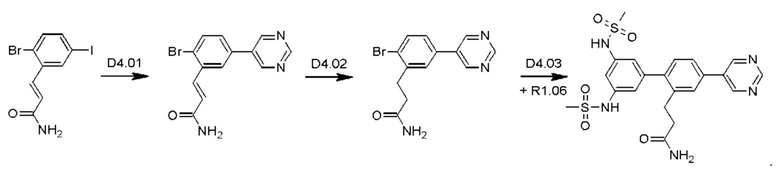
Стадия D4.01: синтез 3-(2-бром-5-(пиримидин-5-ил)фенил)проп-2-енамида. В круглодонную колбу загружали 3-(2-бром-5-иодфенил)проп-2-енамид (1,0 г; 1,70 ммоль), пиримидин-5-бороновую кислоту (0,211 г; 1,7 ммоль), карбонат цезия (1,11 г; 3,4 ммоль), 1,4-диоксан (20 мл) и воду (3,5 мл) и через эту смесь в течение 5 мин барботировали азот. Добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II)-дихлорметан (0,070 г; 0,08 ммоль) и смесь нагревали до температуры дефлегмации в течение 2 ч. После охлаждения добавляли этилацетат (50 мл), смесь переносили в делительную воронку и промывали водой (25 мл). Водную фазу повторно экстрагировали этилацетатом (25 мл), органические экстракты объединяли, сушили (сульфат магния), фильтровали и концентрировали досуха. Остаток перемешивали в дихлорметане (50 мл), твердое вещество фильтровали, промывали дихлорметаном и сушили, получая указанное в заголовке соединение в виде серого порошка (0,167 г; 32%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.23 (s, 1H), 9.22 (s, 2Н), 8.11 (d, J=2,15 Гц, 1H), 7.86 (d, J=8,40 Гц, 1H), 7.76 (dd, J=8,40; 2,34 Гц, 1H), 7.70 (d, J=15,63 Гц, 1H), 7.56 (bs, 1H), 6.83 (d, J=15,63 Гц, 1H), 7.27 (bs, 1H). LCMS [М+Н]+=304,1; 306,1; [M+Na]+=326,0; 328,0.
Стадия D4.02: синтез 3-(2-бром-5-(пиримидин-5-ил)фенил)пропанамида (заимствован из Monatshefte fur Chemie; vol. 147; nb. 3; (2016); p. 509-521). В круглодонную колбу с обратным холодильником загружали 3-(2-бром-5-(пиримидин-5-ил)фенил)проп-2-енамид (0,200 г; 0,658 ммоль), дихлорметан (10 мл), метанол (10 мл), азодикарбоксилат калия (0,639 г; 3,29 ммоль) и уксусную кислоту (0,188 мл; 3,29 ммоль). Реакционную смесь перемешивали при 45-50°С в течение 3 суток, затем охлаждали до комнатной температуры и разбавляли дихлорметаном (50 мл) и водой (50 мл). Смесь встряхивали, затем водный слой отделяли и далее экстрагировали дихлорметаном (50 мл). Органические экстракты объединяли, промывали рассолом (25 мл), сушили над сульфатом магния, фильтровали и концентрировали, получая указанное в заголовке соединение в виде коричневого порошка (0,147 г; 73%). 1H ЯМР (400 МГц, DMSO-d6) δ млн-1 9.20 (s, 1H), 9.14 (s, 2Н), 7.78 (d, J=2,34 Гц, 1H), 7.74 (d, J=8,40 Гц, 1H), 7.60 (dd, J=8,40; 2,34 Гц, 1H), 7.29-7.37 (m, 1H), 6.83 (bs, 1H), 2.94-3.02 (m, 2Н), 2.41-2.48 (m, 2Н). LCMS [М+Н]+=306,0; 308,0; [M+Na]+=328,0; 330,0; [М-Н+СН3ООН]-=350,0; 352,1.
Стадия D4.03: синтез 3-(3',5'-бис((метилсульфонил)амино)-4-(пиримидин-5-ил)бифенил-2-ил)пропанамида (D4). Во флакон для микроволнового реактора в атмосфере азота загружали 3-(2-бром-5-(пиримидин-5-ил)фенил)пропанамид (0,147 г; 0,480 ммоль), 3,5-бис((метилсульфонил)амино)фенилбороновую кислоту, пинаколовый эфир (0,262 г; 0,480 ммоль), 1,4-диоксан (5 мл), воду (0,5 мл) и карбонат цезия (0,626 г; 1,92 ммоль). Смесь перемешивали и продували, барботируя через нее азот в течение 5 мин, затем добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II)-дихлорметан (0,020 г; 0,024 ммоль). Реакционный сосуд закрывали и реакционную смесь перемешивали при температуре дефлегмации в течение 2,5 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом (50 мл) и водой (25 мл). рН водного слоя доводили до 5-6, используя раствор соляной кислоты, и эти два слоя тщательно смешивали. Водный слой отделяли и экстрагировали смесью дихлорметан/метанол (100 мл смеси 1:1). Органические экстракты объединяли, промывали рассолом (25 мл), сушили над сульфатом магния, фильтровали и концентрировали. Остаток растирали в горячем этилацетате (50 мл) в течение 10 мин, затем оставляли охлаждаться до комнатной температуры. Полученное твердое вещество собирали фильтрованием, промывали этилацетатом (3×15 мл) и сушили на воздухе. Твердое вещество растирали в горячей смеси дихлорметан/метанол (20 мл смеси 1:1), аналогичным образом получая указанное в заголовке соединение в виде бежевого порошка (0,058 г; 25%). Фильтраты, полученные в результате второго процесса растирания, содержали неочищенный продукт, который очищали флэш-хроматографией (дихлорметан/метанол), получая вторую порцию указанного в заголовке соединения в виде бежевого порошка (0,022 г; 9%). Его равномерно смешивали с первым образцом. 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.97 (bs, 2Н), 9.21 (s, 1H), 9.19 (s, 2Н), 7.78 (s, 1H), 7.71 (d, J=8,01 Гц, 1H), 7.34 (d, J=8,01 Гц, 1H), 7.24 (bs, 1H), 7.18 (s, 1H), 6.92 (d, J=1,56 Гц, 2Н), 6.76 (bs, 1H), 3.06 (s, 6Н), 2.84 (t, J=7,91 Гц, 2Н), 2.28-2.41 (m, 2Н). LCMS [М+Н]+=490,1; [M+Na]+=512,1; [М-Н]-=488,2.
Схема синтеза D5 показана ниже:
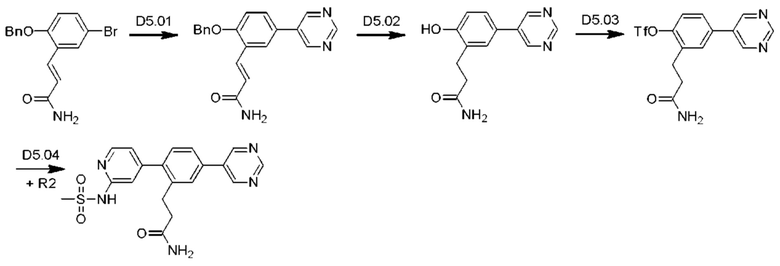
Стадия D5.01: синтез (2Е)-3-[2-(бензилокси)-5-(пиримидин-5-ил)фенил]проп-2-енамида. В круглодонную колбу загружали (2E)-3-[2-(бензилокси)-5-бромфенил]проп-2-енамид (1,5 г; 4,51 ммоль), пиримидин-5-бороновую кислоту (0,671 г; 5,41 ммоль), карбонат калия (1,24 г; 9,0 ммоль), 1,4-диоксан (25 мл), этанол (5 мл) и воду (2 мл) и через эту смесь в течение 5 мин барботировали азот. Добавляли тетракис(трифенилфосфин)палладий (0,26 г; 0,225 ммоль) и смесь нагревали до 80°С в течение 12 ч. После охлаждения добавляли этилацетат (20 мл), смесь фильтровали, органическую фазу отделяли, сушили (сульфат магния), фильтровали и концентрировали досуха. После растирания неочищенного вещества с этилацетатом получали указанное в заголовке соединение в виде беловатого твердого вещества (1,15 г; 77%). LCMS [М+Н]+=332,2.
Стадия D5.02: синтез 3-[2-гидрокси-5-(пиримидин-5-ил)фенил]пропанамида. В круглодонную колбу загружали (2E)-3-[2-(бензилокси)-5-(пиримидин-5-ил)фенил]проп-2-енамид (0,440 г; 1,33 ммоль), метанол (25 мл) и 25%-ный водный раствор аммиака (2,5 мл). Добавляли 10 масс. % палладий на угле (0,044 г) в атмосфере азота. Смесь заполняли водородом и перемешивали в атмосфере водорода при комнатной температуре в течение 48 ч. Реакционную смесь фильтровали через набивку целита и целит промывали кипящим метанолом (2×50 мл). Фильтраты объединяли, концентрировали и распределяли между дихлорметаном (50 мл) и водой (50 мл). рН водной фазы доводили до 4-5, используя разбавленную соляную кислоту (водн.), и повторно экстрагировали дихлорметаном (50 мл) и этилацетатом (50 мл). Органические экстракты объединяли, сушили (сульфат магния), фильтровали и концентрировали, получая указанное в заголовке соединение в виде беловатого твердого вещества (0,025 г; 8%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.78 (s, 1H), 9.08 (s, 1H), 9.00-9.06 (m, 2Н), 7.53 (d, J=2,34 Гц, 1H), 7.47 (dd, J=8,30; 2,44 Гц, 1H), 7.30 (ушир. s., 1H), 6.92 (d, J=8,40 Гц, 1H), 6.79 (ушир. s., 1H), 2.80 (t, J=7,72 Гц, 2Н), 2.40 (t, J=7,81 Гц, 2Н). Водную фазу концентрировали досуха, получая указанное в заголовке соединение в виде неочищенного беловатого твердого вещества (0,298 г).
Стадия D5.03: синтез 2-(3-амино-3-оксопропил)-4-(пиримидин-5-ил)фенил-трифторметансульфоната. В круглодонную колбу в атмосфере азота загружали 3-(2-гидрокси-5-(пиримидин-5-ил)фенил)пропанамид (0,323 г; 1,33 ммоль), карбонат калия (0,551 г; 3,98 ммоль) и ацетонитрил (20 мл). Реакционную смесь охлаждали до температуры ниже 10°С в ледяной бане и добавляли N-фенил-бис(трифторметансульфонимид) (0,498 г; 1,39 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 12 ч. Реакционную смесь концентрировали досуха, распределяли между этилацетатом (100 мл) и водой (100 мл). Водную фазу повторно экстрагировали этилацетатом (50 мл), органические фазы объединяли, промывали рассолом (25 мл), сушили (сульфат магния), фильтровали и концентрировали. Неочищенное вещество очищали флэш-хроматографией (дихлорметан/ацетон), получая указанное в заголовке соединение в виде беловатого твердого вещества (0,314 г; 63%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.23 (s, 1H), 9.18 (s, 2Н), 7.90-7.98 (m, 1H), 7.85 (dd, J=8,50; 2,25 Гц, 1H), 7.55 (d, J=8,40 Гц, 1H), 7.35 (bs, 1H), 6.84 (bs, 1H), 2.96 (t, J=7,62 Гц, 2Н), 2.51-2.57 (m, 2Н). LCMS [М+Н]+=376,0; [M+Na]+=398,0.
Стадия D5.04: синтез диэтил-(2-(6-аминопиримидин-4-ил)-5-(пиримидин-5-ил)бензил)пропандиоата (D5). В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали 2-(3-амино-3-оксопропил)-4-(пиримидин-5-ил)фенил-трифторметансульфонат (0,244 г; 0,649 ммоль), N-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)(пиридин-2-ил)метансульфонамид (0,513 г; 1,36 ммоль), карбонат калия (0,351 г; 2,54 ммоль), 1,4-диоксан (10,2 мл), этанол (3,9 мл) и воду (3,9 мл). Реакционную смесь продували, барботируя через нее азот в течение 2 мин, затем добавляли тетракис(трифенилфосфин)палладий (0,075 г; 0,652 ммоль) и реакционную смесь перемешивали при 85°С в течение 2,5 ч. Реакционную смесь охлаждали до комнатной температуры и распределяли между этилацетатом (75 мл) и водой (30 мл). Смесь переносили в делительную воронку, водный слой (рН 6-7) отделяли и оставляли стоять в течение пяти суток, за это время твердое вещество выпадало в осадок. Твердое вещество собирали фильтрованием и перемешивали в смеси дихлорметан/метанол (4:1, 50 мл). Твердые вещества собирали фильтрованием и промывали метанолом (10 мл), получая указанное в заголовке соединение. Органические фильтраты концентрировали, полученное твердое вещество перемешивали в кипящей смеси вода/метанол (5:1, 24 мл) и смесь фильтровали в горячем состоянии. Фильтрат охлаждали до комнатной температуры и оставляли стоять в течение 18 ч, за это время твердое вещество выпадало в осадок (указанное в заголовке соединение). Оставшиеся фильтраты концентрировали досуха, получая третью порцию указанного в заголовке соединения. Образцы равномерно смешивали, получая указанное в заголовке соединение в виде беловатого порошка (0,059 г; 23%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.22 (s, 1H), 9.19 (s, 2Н), 8.29 (s, 1H), 7.82 (d, J=1,56 Гц, 1H), 7.75 (dd, J=7,91; 1,86 Гц, 1H), 7.35 (d, J=8,01 Гц, 1H), 7.27 (bs, 1H), 7.01 (bs, 1H), 6.92 (s, 1H), 6.76 (bs, 1H), 3.29 (s, 3Н), 2.84 (t, J=7,81 Гц, 2Н), 2.34-2.42 (m, 2Н). LCMS [М+Н]+=398,1; [M+Na]+=420,1; [М-Н]-=396,1.
Схема синтеза D6 показана ниже:
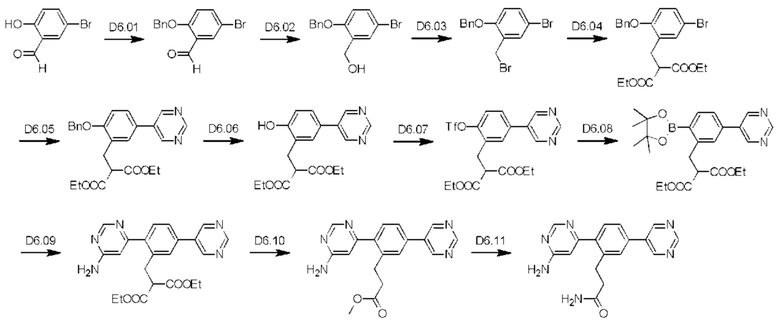
Стадия D6.01: синтез 2-(бензилокси)-5-бром-бензальдегида. В круглодонную колбу, оснащенную обратным холодильником, загружали 5-бром-2-гидроксибензальдегид (10,0 г; 0,050 моль), карбонат калия (8,94 г; 0,065 моль), ацетонитрил (100 мл) и бензилбромид (11,1 г; 0,065 моль). Реакционную смесь перемешивали при 70°С в течение 16 ч, затем охлаждали и концентрировали. Остаток распределяли между этилацетатом (100 мл) и водой (100 мл), водный слой отделяли и еще раз экстрагировали этилацетатом (100 мл). Органические экстракты объединяли, промывали рассолом (50 мл), сушили (сульфат магния), фильтровали и концентрировали. Масло перемешивали в гексанах (250 мл) с охлаждением в ледяной бане в течение 1 ч и полученные твердые вещества собирали вакуумной фильтрацией, промывали гексанами (3×25 мл) и сушили на воздухе, получая указанное в заголовке соединение в виде беловатого твердого вещества (10,5 г; 73%). 1H ЯМР (400 МГц, CDCl3) δ млн-1 10.46 (s, 1H), 7.95 (d, J=2,54 Гц, 1H), 7.60 (dd, J=8,89; 2,64 Гц, 1H), 7.32-7.46 (m, 5Н), 6.95 (d, J=8,99 Гц, 1H), 5.18 (s, 2Н). LCMS [M+Na]+=313,0; 315,0.
Стадия D6.02: синтез бензил-4-бром-2-(гидроксиметил)фенилового эфира. В оснащенную круглодонную колбу загружали 2-(бензилокси)-5-бром-бензальдегид (10,5 г; 0,036 моль) и метанол (200 мл). Реакционную смесь перемешивали и охлаждали в ледяной бане в течение 20 мин, затем добавляли порциями боргидрид натрия (1,51 г; 0,040 моль), наблюдая выделение газа. Реакционную смесь перемешивали и оставляли нагреваться до комнатной температуры в течение 1-2 ч. Реакционную смесь концентрировали почти досуха и остаток распределяли между этилацетатом (150 мл) и раствором бикарбоната натрия (150 мл). Водную фазу отделяли и далее экстрагировали этилацетатом (100 мл). Органические экстракты объединяли, промывали рассолом (50 мл), сушили (сульфат магния), фильтровали и концентрировали, получая указанное в заголовке соединение в виде оранжевого масла (10,6 г; 100%). 1Н ЯМР (400 МГц, CDCl3) δ млн-1 7.46 (d, J=2,34 Гц, 1H), 7.37-7.42 (m, 4Н), 7.31-7.37 (m, 2Н), 6.81 (d, J=8,79 Гц, 1H), 5.09 (s, 2Н), 4.70 (d, J=6,25 Гц, 2Н), 2.17 (t, J=6,45 Гц, 1H). LCMS [М-H2O+Н]+=275,0; 277,0; [M+Na]+=315,0; 317,0.
Стадия D6.03: синтез бензил-4-бром-2-(бромметил)фенилового эфира. В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали бензил-4-бром-2-(гидроксиметил)фениловый эфир (10,6 г; 0,036 моль) и безводный толуол (110 мл). Реакционную смесь перемешивали и охлаждали в ледяной бане в течение 20 мин, затем медленно добавляли трибромид фосфора (11,3 г; 0,042 моль). Реакционную смесь перемешивали и охлаждали в ледяной бане в течение 20 мин, затем оставляли нагреваться до комнатной температуры в течение 30 мин. После этого реакционную смесь перемешивали при температуре дефлегмации в течение 1,5 ч и охлаждали до комнатной температуры. К смеси добавляли воду (110 мл) и перемешивали в течение 15 мин, затем переносили в делительную воронку с этилацетатом (50 мл) и энергично встряхивали. Водный слой отделяли и далее экстрагировали этилацетатом (50 мл). Органические экстракты объединяли, промывали водой (50 мл), рассолом (50 мл), сушили (сульфат магния), фильтровали и концентрировали, получая указанное в заголовке соединение в виде темного масла (12,4 г; 96%). 1Н ЯМР (400 МГц, CDCl3) δ млн-1 7.43-7.49 (m, 3Н), 7.37-7.43 (m, 2Н), 7.32-7.37 (m, 2Н), 6.79 (d, J=8,79 Гц, 1H), 5.14 (s, 2Н), 4.52 (s, 2Н). GCMS (газовая хроматография в сочетании с масс-спектрометрией) m/z=354, 356, 358.
Стадия D6.04: синтез 1,3-диэтил-2-(5-бром-2-(фенилметокси)бензил)пропандиоата. В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали диэтилмалонат (5,52 г; 0,034 моль) и 1,2-диметоксиэтан (50 мл). К перемешиваемому раствору порциями добавляли гидрид натрия (1,26 г 60%-ной дисперсии в масле; 0,032 моль), при этом наблюдали бурное выделение газа. Реакционную смесь перемешивали в течение 20 мин, затем медленно добавляли раствор 4-бром-2-(бромметил)-1-(фенилметокси)бензола (10,2 г; 0,029 моль) в 1,2-диметоксиэтане (50 мл). Реакционную смесь перемешивали при температуре дефлегмации в течение 24 ч, затем охлаждали до комнатной температуры и гасили водой (50 мл). Смесь экстрагировали этилацетатом (2×100 мл) и органические экстракты объединяли, промывали рассолом (50 мл), сушили (сульфат магния), фильтровали и концентрировали. Масло очищали флэш-хроматографией (этилацетат/гексаны), получая указанное в заголовке соединение в виде бесцветного масла (11,3 г; 90%). 1H ЯМР (400 МГц, CDCl3) δ млн-1 7.35-7.43 (m, 4Н), 7.27-7.35 (m, 3Н), 6.75 (d, J=8,40 Гц, 1H), 5.09 (s, 2Н), 4.08-4.18 (m, 4Н), 3.83 (t, J=7,82 Гц, 1H), 3.21 (d, J=7,62 Гц, 2Н), 1.16-1.23 (m, 6Н). LCMS [М+Н]+=435,0; 437,0; [M+Na]+=457,0; 459,0.
Стадия D6.05: синтез 1,3-диэтил-2-(2-(фенилметокси)-5-(пиримидин-5-ил)бензил)пропандиоата. В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали 1,3-диэтил-2-(5-бром-2-(фенилметокси)бензил)пропандиоат (7,37 г; 16,9 ммоль), пиримидин-5-бороновую кислоту (2,73 г; 22,0 ммоль), карбонат калия (7,02 г; 50,7 ммоль), толуол (290 мл), этанол (185 мл) и воду (105 мл). Реакционную смесь продували, барботируя через нее азот в течение 10 мин, затем добавляли тетракис(трифенилфосфин)палладий (1,96 г; 1,69 ммоль). Реакционную смесь перемешивали при 85°С в течение 2,5 ч, затем охлаждали до комнатной температуры, разбавляли этилацетатом (100 мл) и переносили в делительную воронку. После энергичного встряхивания водный слой (рН примерно 10) отделяли и далее экстрагировали этилацетатом (100 мл). Органические экстракты объединяли, промывали рассолом (3×100 мл), сушили (сульфат магния), фильтровали и концентрировали. Неочищенное масло очищали флэш-хроматографией (этилацетат/гексаны), получая указанное в заголовке соединение в виде масла цвета темного янтаря (3,18 г; 43%). 1Н ЯМР (400 МГц, CDCl3) δ млн-1 9.15 (s, 1H), 8.88 (s, 2Н), 7.30-7.49 (m, 7Н), 7.02 (d, J=8,21 Гц, 1H), 5.19 (s, 2Н), 4.06-4.20 (m, 4Н), 3.92 (t, J=7,72 Гц, 1H), 3.34 (d, J=7,62 Гц, 2Н), 1.17 (t, J=7,13 Гц, 6Н). LCMS [М+Н]+=435,2; [M+Na]+=457,1.
Стадия D6.06: синтез 1,3-диэтил-2-(2-гидрокси-5-(пиримидин-5-ил)бензил)пропандиоата. В круглодонную колбу в атмосфере азота загружали 1,3-диэтил-2-(2-(фенилметокси)-5-(пиримидин-5-ил)бензил)пропандиоат (0,659 г; 1,52 ммоль) и этилацетат (10 мл). Реакционную смесь продували, барботируя через нее азот в течение 5 мин, затем добавляли 10% палладий на угле (0,066 г), после чего триэтиламин (0,211 мл). Реакционную смесь перемешивали в атмосфере водорода при 45°С в течение 3 суток, затем фильтровали через целит. Целит промывали этилацетатом (3×15 мл), фильтраты объединяли и концентрировали, получая указанное в заголовке соединение в виде кремового твердого вещества (0,471 г; 90%). 1Н ЯМР (400 МГц, CDCl3) δ млн-1 9.15 (s, 1H), 8.88 (s, 2Н), 7.37 (dd, J=8,21; 2,34 Гц, 1H), 7.34 (d, J=2,34 Гц, 1H), 7.03 (d, J=8,21 Гц, 1H), 4.21 (m, 4Н), 3.78 (t, J=7,03 Гц, 1H), 3.25 (d, J=7,03 Гц, 2Н), 1.24 (t, J=7,13 Гц, 6Н). LCMS [М+Н]+=345,2; [M+Na]+=367,1.
Стадия D6.07: синтез 1,3-диэтил-2-(((2-трифторметил)сульфонил)оксо)-5-(пиримидин-5-ил)-бензил)пропандиоата. В круглодонную колбу в атмосфере азота загружали 1,3-диэтил-2-(2-гидрокси-5-(пиримидин-5-ил)бензил)пропандиоат (0,471 г; 1,37 ммоль), карбонат калия (0,388 г; 2,80 ммоль) и ацетонитрил (10 мл). Реакционную смесь охлаждали до температуры ниже 10°С в ледяной бане и добавляли N-фенил-бис(трифторметансульфонимид) (0,513 г; 1,44 ммоль). Реакционную смесь перемешивали и оставляли нагреваться до комнатной температуры. Через 4,5 ч реакционную смесь наносили непосредственно на диоксид кремния и очищали флэш-хроматографией (этилацетат/гексаны), получая указанное в заголовке соединение в виде желтого масла (0,614 г; 94%). 1Н ЯМР (400 МГц, CDCl3) δ млн-1 9.26 (s, 1H), 8.92 (s, 2Н), 7.59 (d, J=2,1 Гц, 1H), 7.54 (dd, J=2,2; 8,5 Гц, 1H), 7.46 (d, J=8,6 Гц, 1H), 4.26-4.11 (m, 4Н), 3.76 (t, J=7,7 Гц, 1Н), 3.41 (d, J=7,8 Гц, 2Н), 1.22 (t, J=7,1 Гц, 6Н). LCMS [М+Н]+=477,1; [M+Na]+=499,0.
Стадия D6.08: синтез 1,3-диэтил-2-(5-(пиримидин-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пропандиоата. Во флакон для микроволнового реактора загружали 1,3-диэтил-2-(((2-трифторметил)сульфонил)оксо)-5-(пиримидин-5-ил)-бензил)пропандиоат (0,600 г; 1,26 ммоль), бис(пинаколато)дибор (0,800 г; 3,15 ммоль), ацетат калия (0,371 г; 3,78 ммоль) и 1,4-диоксан (10 мл). Реакционную смесь продували, барботируя через нее азот в течение 5 мин, затем добавляли комплекс [1,1'-бис(дифенилфосфино)-ферроцен]дихлорпалладий(II)-дихлорметан (0,103 г; 0,126 ммоль). Реакционный сосуд закрывали и смесь нагревали при 105°С в течение 3 ч в микроволновом реакторе. Реакционную смесь распределяли между этилацетатом (25 мл) и водой (25 мл) и рН водной фазы доводили до значения выше 10, используя раствор карбоната натрия. Органические фазы собирали, промывали рассолом (10 мл), сушили (сульфат магния), фильтровали и концентрировали. Неочищенное твердое вещество очищали флэш-хроматографией (этилацетат/гексаны), получая указанное в заголовке соединение в виде розового масла (0,457 г; 80%). 1Н ЯМР (400 МГц, CDCl3) δ млн-1 9.21 (s, 1H), 8.93 (s, 2Н), 7.95 (d, J=8,40 Гц, 1H), 7.42-7.46 (m, 2Н), 4.09-4.20 (m, 4Н), 3.75-3.81 (m, 1H), 3.54 (d, J=7,62 Гц, 2Н), 1.37 (s, 12Н), 1.16-1.22 (m, 6Н). LCMS [М+Н]+=455,2.
Стадия D6.09: синтез 1,3-диэтил-2-(2-(6-амино-пиримидин-4-ил)-5-(пиримидин-5-ил)бензил)пропандиоата. В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали 1,3-диэтил-2-(5-(пиримидин-5-ил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензил)пропандиоат (0,577 г; 1,27 ммоль), 4-амино-6-хлорпиримидин (0,181 г; 1,40 ммоль), карбонат калия (0,351 г; 2,54 ммоль), 1,4-диоксан (4 мл), этанол (1 мл) и воду (1 мл). Реакционную смесь продували, барботируя через нее азот в течение 2 мин, затем добавляли тетракис(трифенилфосфин)палладий (0,146 г; 0,127 ммоль) и реакционную смесь перемешивали при 85°С в течение 2,5 ч. Реакционную смесь охлаждали до комнатной температуры и распределяли между этилацетатом (100 мл) и водой (25 мл). Органические фазы собирали, промывали рассолом (25 мл), сушили (сульфат магния), фильтровали и концентрировали. Остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение (0,093 г; 17%) (LCMS [M+H]+=422,2) в виде сложной смеси с этил-3-(2-(6-аминопиримидин-4-ил)-5-(пиримидин-5-ил)фенил)пропаноатом (LCMS [М+Н]+=350,2). Водные промывки объединяли и подкисляли до рН примерно 3, используя 2 М раствор соляной кислоты, затем экстрагировали смесью дихлорметан/метанол (9:1, 3×25 мл). Органические экстракты объединяли, промывали рассолом (10 мл), сушили (сульфат магния), фильтровали и концентрировали, получая остаток (0,202 г), содержащий 2-(2-(6-аминопиримидин-4-ил)-5-(пиримидин-5-ил)бензил)-3-этокси-3-оксопропановую кислоту (LCMS [М+Н]+=394,1), указанное в заголовке соединение и другие примеси. Кислотные водные промывки концентрировали досуха и остаток экстрагировали смесью этилацетатом/метанол (9:1, 2×25 мл). Экстракты объединяли и концентрировали, получая остаток (0,228 г), содержащий (2-(6-аминопиримидин-4-ил)-5-(пиримидин-5-ил)бензил)пропандиовую кислоту (LCMS [М+Н]+=366,1), 2-(2-(6-аминопиримидин-4-ил)-5-(пиримидин-5-ил)бензил)-3-этокси-3-оксопропановую кислоту (LCMS [М+Н]+=394,1) и другие примеси. Остатки, содержащие указанное в заголовке соединение и другие гидролизованные аналоги, объединяли и использовали на следующей стадии.
Стадия D6.10: синтез метил-3-(2-(6-аминопиримидин-4-ил)-5-(пиримидин-5-ил)фенил)пропаноата. В круглодонную колбу, оснащенную обратным холодильником, загружали 1,3-диэтил-2-(2-(6-аминопиримидин-4-ил)-5-(пиримидин-5-ил)бензил)пропандиоат (0,523 г; 1,24 ммоль) и серную кислоту (5 мл 2 М водного раствора). Реакционную смесь перемешивали при температуре дефлегмации в течение 24 ч, затем охлаждали и трижды концентрировали из метанола (150 мл). Остаток перемешивали в метаноле при комнатной температуре в течение ночи, затем концентрировали. Остаток распределяли между этилацетатом (100 мл) и раствором бикарбоната натрия (50 мл), наблюдая выделение газа. Органические слои собирали и основный водный слой далее экстрагировали этилацетатом (50 мл). Органические слои объединяли, промывали рассолом (50 мл), сушили (сульфат магния), фильтровали и концентрировали. Остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде беловатого порошка (0,075 г; 18%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.17-9.23 (m, 3Н), 8.43 (d, J=0,98 Гц, 1H), 7.81 (d, J=1,76 Гц, 1H), 7.75 (dd, J=7,91; 1,86 Гц, 1H), 7.45 (d, J=8,01 Гц, 1H), 6.96 (s, 2Н), 6.52 (d, J=1,17 Гц, 1H), 3.55 (s, 3Н), 3.04 (t, J=7,91 Гц, 2Н), 2.67 (t, J=8,01 Гц, 2Н). LCMS [М+Н]+=336,2.
Стадия D6.11: синтез 3-(2-(6-аминопиримидин-4-ил)-5-(пиримидин-5-ил)фенил)пропанамида (D6). Во флакон для микроволнового реактора загружали метил-3-(2-(6-аминопиримидин-4-ил)-5-(пиримидин-5-ил)фенил)пропаноат (0,075 г; 0,224 ммоль) и раствор аммиака (5 мл 7 н. раствора в метаноле). Сосуд закрывали и реакционную смесь перемешивали при 60°С в течение 5 суток. Реакционную смесь охлаждали, наносили непосредственно на диоксид кремния и очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде розового порошка (0,051 г; 71%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.21 (s, 1H), 9.19 (s, 2Н), 8.40-8.46 (m, 1H), 7.76-7.80 (m, 1H), 7.73 (dd, J=8,01; 1,76 Гц, 1H), 7.45 (d, J=8,01 Гц, 1H), 7.26 (bs, 1H), 6.96 (s, 2Н), 6.74 (bs, 1H), 6.52 (d, J=1,17 Гц, 1H), 2.95-3.02 (m, 2Н), 2.36-2.43 (m, 2Н). LCMS [М+Н]+=321,1.
Схема синтеза D10 показана ниже:
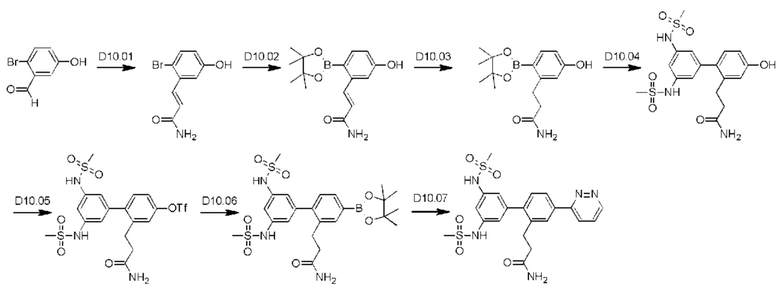
Стадия D10.01: синтез 3-(2-бром-5-гидроксифенил)проп-2-енамида (заимствован из Monatshefte fur Chemie; vol. 147; nb. 3; (2016); p. 509-521). В круглодонную колбу в атмосфере азота загружали (2-амино-2-оксоэтил)(трифенил)фосфонийхлорид (3,70 г; 10,4 ммоль) и метанол (40 мл) и раствор охлаждали в ледяной бане. Добавляли трет-бутилат калия (1,17 г; 10,4 ммоль) и смесь перемешивали в течение 10 мин. Добавляли 2-бром-5-гидроксибензальдегид (1,99 г; 9,90 ммоль) и реакционную смесь перемешивали в течение 15 мин с охлаждением в ледяной бане, затем оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разбавляли водой (125 мл) и рН доводили до значения ниже 4, используя раствор соляной кислоты. Водный слой экстрагировали дихлорметаном (100 мл) и органические экстракты собирали и концентрировали. Остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде темной вязкой смолы (2,30 г; 96%), состоящей из обоих Е- и Z-изомеров в соотношении приблизительно 1:0,3. 1H ЯМР (400 МГц, DMSO-d6) δ млн-1 9.88 (s, 1H), 9.64 (s, 0.3Н), 7.67-7.53 (m, 2Н), 7.51-7.42 (m, 1.3Н), 7.36 (d, J=8,8 Гц, 0,3Н), 7.22 (bs, 1,3Н), 7.11-7.03 (m, 1,3Н), 7.00 (d, J=2,9 Гц, 0,3Н), 6.76 (dd, J=2,9; 8,8 Гц, 1H), 6.68-6.61 (m, 0,6Н), 6.52 (d, J=15,6 Гц, 1H), 6.09 (d, J=15,6 Гц, 0,3H). LCMS [М+Н]+=242,1; 244,1; [M+Na]+=264,0; 266,0; [М-Н]-=240,0; 242,0.
Стадия D10.02: синтез 3-(5-гидрокси-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)проп-2-енамида. В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали 3-(2-бром-5-гидроксифенил)проп-2-енамид (8,83 г; 0,036 моль), бис(пинаколато)дибор (10,2 г; 0,040 моль), ацетат калия (14.3 г; 0,146 моль) и безводный диметилсульфоксид (250 мл). Реакционную смесь продували, барботируя через нее азот в течение 10 мин. Добавляли комплекс [1,1'-бис(дифенил-фосфино)ферроцен]дихлорпалладий(II)-дихлорметан (4,25 г; 0,005 моль) и реакционную смесь перемешивали при 85°С в течение ночи. Реакционную смесь охлаждали до комнатной температуры и добавляли воду (600 мл). Значение рН водного слоя доводили до рН ниже 2, используя раствор соляной кислоты, затем смесь экстрагировали этилацетатом (3×500 мл). Органические экстракты объединяли, промывали рассолом (250 мл), сушили над сульфатом магния, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (этилацетат/гексаны), получая указанное в заголовке соединение в виде оранжевого твердого вещества (5,57 г; 53%) как смесь Е- и Z-изомеров в соотношении приблизительно 2:1. 1H ЯМР (400 МГц, CDCl3) δ млн-1 8.36 (d, J=16,22 Гц, 1H), 7.76 (d, J=8,21 Гц, 1H), 7.59 (d, J=8,21 Гц, 0.5Н), 7.10 (d, J=2,15 Гц, 1,5Н), 7.00 (s, 0,5Н), 6.84 (dd, J=8,21; 2,34 Гц, 1.5Н), 6.41 (d, J=15,63 Гц, 0,5Н), 6.32 (d, J=16,02 Гц, 1H), 1.35 (s, 18Н). LCMS [М+Н]+=290,2; [M+Na]+=312,1; [М-Н]-=288,2.
Стадия D10.03: синтез 3-(5-гидрокси-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропенамида. В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали 3-(5-гидрокси-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)проп-2-енамид (5,57 г; 0,019 моль) и этанол (250 мл). Раствор продували, барботируя через него азот в течение 10 мин, затем добавляли 10% палладий на угле (0,56 г). Реакционную смесь перемешивали при температуре дефлегмации в атмосфере водорода в течение ночи, затем фильтровали в горячем состоянии через набивку целита и остаток промывали этанолом (3×50 мл). Фильтраты концентрировали, получая указанное в заголовке соединение в виде желтого твердого вещества (3,54 г; 63%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.60 (s, 1H), 7.47 (d, J=8,01 Гц, 1H), 7.11 (bs, 1H), 6.66 (bs, 1H), 6.59 (d, J=2,34 Гц, 1H) 6.56 (dd, J=8,11; 2,44 Гц, 1H), 2.94 (t, J=7,82 Гц, 2Н), 2.24 (t, J=7,82 Гц, 2Н), 1.27 (s, 12Н). LCMS [М+Н]+=292,2; [M+Na]+=314,1; [М-Н]-=290,1.
Стадия D10.04: синтез 3-(4-гидрокси-3',5'-бис((метилсульфонил)амино)бифенил-2-ил)пропенамида. Во флакон для микроволнового реактора загружали 3-(5-гидрокси-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропанамид (0,312 г; 1,07 ммоль), N,N'-(5-бромбензол-1,3-диил)бис(N-(метилсульфонил)метансульфонамид (0,562 г; 1,13 ммоль), карбонат калия (0,889 г; 6,43 ммоль), N,N-диметилформамид (12 мл) и воду (1,6 мл). Реакционную смесь продували, барботируя через нее азот в течение 5 мин, затем добавляли комплекс [1,1'-бис(дифенил-фосфино)ферроцен]-дихлорпалладий(II)-дихлорметан (0,088 г; 0,107 ммоль). Реакционную смесь нагревали при 105°С в течение 1 ч, затем охлаждали до комнатной температуры и разбавляли водой (10 мл). Значение рН доводили до рН ниже 4, используя раствор соляной кислоты, затем смесь концентрировали. Остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде янтарной смолы (0,334 г; 73%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.45 (bs, 1H), 7.20 (bs, 1H), 7.11 (t, J=2,05 Гц, 1H), 6.96 (d, J=8,21 Гц, 1H), 6.82 (d, J=1,95 Гц, 2Н), 6.71 (d, J=2,54 Гц, 2Н), 6.66 (dd, J=8,21; 2,54 Гц, 1H), 3.02 (s, 6Н), 2.61-2.70 (m, 2Н), 2.14-2.24 (m, 2Н). LCMS [М+Н]+=428,1; [M+Na]+=450,0; [М-Н]-=426,0.
Стадия D10.05: синтез 3-(3-амино-3-оксопропил)-3',5'-бис((метилсульфонил)амино)бифенил-трифторметансульфоната. В круглодонную колбу загружали 3-(4-гидрокси-3',5'-бис((метилсульфонил)-амино)бифенил-2-ил)пропанамид (0,796 г; 1,86 ммоль), N,N-диметилформамид (30 мл), ацетонитрил (50 мл) и карбонат калия (2,06 г; 14,9 ммоль). Реакционную смесь перемешивали в течение 15 мин, затем добавляли N-фенил-бис(трифторметансульфонимид) (0,698 г; 1,96 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, затем концентрировали. Остаток распределяли между смесью дихлорметан/метанол (5:1, 100 мл) и водой (50 мл). рН водного слоя доводили до значения примерно 4, используя раствор соляной кислоты, и слои тщательно перемешивали. Водный слой отделяли и промывали смесью дихлорметан/метанол (5:1, 100 мл). Органические слои объединяли, промывали рассолом (50 мл), сушили над сульфатом магния, фильтровали и концентрировали. Оставшийся N,N-диметилформамид удаляли упаривания смеси из ксилолов (150 мл) и остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде беловатого стеклообразного вещества (0,513 г; 49%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.96 (s, 2Н), 7.45 (d, J=2,34 Гц, 1H), 7.33-7.43 (m, 2Н), 7.23 (bs, 1H), 7.16 (s, 1H), 6.85 (d, J=1,95 Гц, 2Н), 6.77 (bs, 1H), 3.04 (s, 6Н), 2.73-2.82 (m, 2Н), 2.23-2.31 (m, 2Н). LCMS [М+Н]+=560,0; [М+Н]+=582,0; [М-Н]-=558,0.
Стадия D10.06: синтез 3-(3',5'-бис((метилсульфонил)амино)-1-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бифенил-3-ил)пропенамида. Во флакон для микроволнового реактора загружали 3-(3-амино-3-оксопропил)-3',5'-бис((метилсульфонил)амино)-бифенил-трифторметансульфонат (0,507 г; 0,906 ммоль), бис(пинаколато)дибор (0,230 г; 0,906 ммоль), безводный 1,4-диоксан (10 мл) и ацетат калия (0,445 г; 4,53 ммоль). Реакционную смесь продували, барботируя через нее азот в течение 2 мин, затем добавляли комплекс [1,1'-бис(дифенилфосфино)-ферроцен]дихлорпалладий(II)-дихлорметан (0,074 г; 0,091 ммоль). Флакон закрывали и реакционную смесь нагревали при 105°С в течение 1 ч в микроволновом реакторе, затем охлаждали до комнатной температуры и разбавляли смесью дихлорметан/метанол (5:1, 25 мл). рН смеси доводили до значения примерно 4, используя раствор соляной кислоты, затем фильтровали. Остаток промывали смесью дихлорметан/метанол (5:1, 75 мл) и фильтраты объединяли, сушили над сульфатом магния, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде беловатого стеклообразного вещества (0,324 г; 67%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.91 (bs, 2Н), 7.64 (s, 1H), 7.56 (d, J=7,42 Гц, 1H), 7.22 (bs, 1H), 7.13-7.20 (m, 2Н), 6.80-6.90 (m, 2Н), 6.72 (bs, 1H), 3.03 (s, 6Н), 2.70-2.80 (m, 2Н), 2.15-2.26 (m, 2Н), 1.31 (s, 12Н). LCMS [М+Н]+=538,2; [М+Н]+=560,2; [М-Н]-=536,2.
Стадия D10.07: синтез 3-(3',5'-бис((метилсульфонил)амино)-1-(пиридазин-3-ил)бифенил-3-ил)пропанамида (D10). Во флакон для микроволнового реактора загружали 3-(3',5'-бис((метилсульфонил)амино)-1-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бифенил-3-ил)пропанамид (0,100 г; 0,186 ммоль), 3-бромпиридазин (0,030 г; 0,186 ммоль), карбонат калия (0,129 г; 0,930 ммоль), N,N-диметилформамид (2 мл) и воду (0,26 мл). Реакционную смесь продували, барботируя через нее азот в течение 2 мин, затем добавляли комплекс [1,1'-бис(дифенил-фосфино)ферроцен]-дихлорпалладий(II)-дихлорметан (0,015 г; 0,019 ммоль) и флакон закрывали. Реакционную смесь перемешивали при 105°С в течение 1 ч в микроволновом реакторе, затем охлаждали до комнатной температуры и разбавляли смесью дихлорметан/метанол (5:1, 25 мл). рН смеси доводили до 5-6, используя раствор соляной кислоты, затем фильтровали. Остаток промывали смесью дихлорметан/метанол (5:1, 50 мл) и фильтраты объединяли, сушили над сульфатом магния, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде темного стеклообразного вещества (0,029 г; 32%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.95 (s, 2Н), 9.24 (dd, J=4,88; 1,56 Гц, 1H), 8.26 (dd, J=8,69; 1,47 Гц, 1H), 8.15 (d, J=1,76 Гц, 1H), 8.03 (dd, J=8,01; 1,95 Гц, 1H), 7.77-7.85 (m, 1H), 7.37 (d, J=7,82 Гц, 1H), 7.24 (bs, 1H), 7.19 (t, J=1,95 Гц, 1H), 6.93 (d, J=1,95 Гц, 2Н), 6.75 (bs, 1H), 3.06 (s, 6Н), 2.82-2.90 (m, 2Н), 2.30-2.38 (m, 2Н). LCMS [М+Н]+=490,1; [M+Na]+=512,0; [М-Н]-=488,1.
Схема синтеза D16 показана ниже:
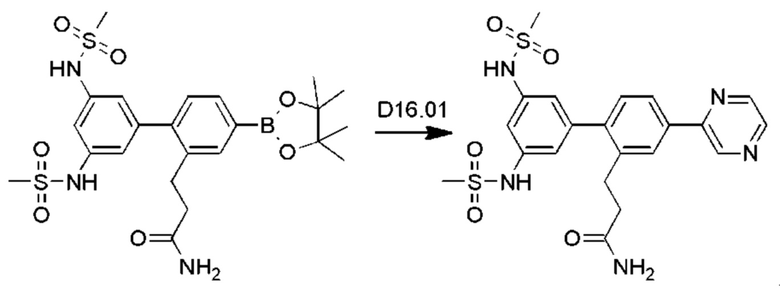
Стадия D16.01: синтез 3-(3',5'-бис((метилсульфонил)амино)-1-(пиразин-2-ил)бифенил-3-ил)пропанамида (D16). Выполняли согласно стадии D10.07, используя 2-хлорпиразин, с получением указанного в заголовке соединения в виде темного стеклообразного вещества (0,038 г; 39%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.96 (s, 2Н), 9.29 (s, 1H), 8.74 (s, 1H), 8.64 (d, J=2,34 Гц, 1H), 8.12 (s, 1H), 8.03 (d, J=8,01 Гц, 1H), 7.34 (d, J=8,01 Гц, 1H), 7.25 (bs, 1H), 7.19 (s, 1H), 6.93 (d, J=1,76 Гц, 2Н), 6.75 (bs, 1H), 3.06 (s, 6Н), 2.81-2.89 (m, 2Н), 2.33 (t, J=7,91 Гц, 2Н). LCMS [М+Н]+=490,1; [M+Na]+=512,0; [М-Н]-=488,1.
Схема синтеза D11 показана ниже:
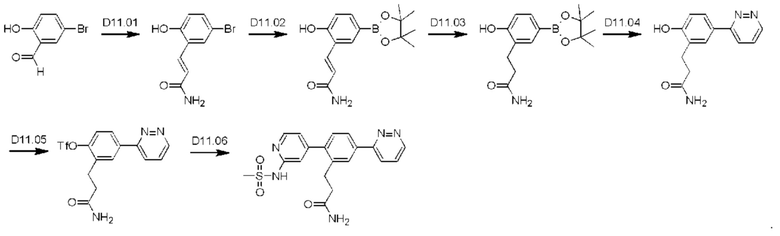
Стадия D11.01: синтез 3-(5-бром-2-гидроксифенил)проп-2-енамида (заимствован из Monatshefte fur Chemie; vol. 147; nb. 3; (2016); p. 509-521). В круглодонную колбу в атмосфере азота загружали (2-амино-2-оксоэтил)(трифенил)фосфонийхлорид (5,70 г; 16,0 ммоль) и метанол (60 мл) и раствор охлаждали в ледяной бане. Добавляли трет-бутилат калия (1,80 г; 16,0 ммоль) и смесь перемешивали в течение 20 мин. Добавляли 2-бром-5-гидроксибензальдегид (3,07 г; 15,3 ммоль) и реакционную смесь перемешивали в течение 20 мин с охлаждением в ледяной бане, затем оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разбавляли дихлорметаном (200 мл) и перемешивали в течение 20 мин, затем фильтровали. Остаток промывали смесью дихлорметан/метанол (примерно 5/1, 50 мл) и фильтраты объединяли и концентрировали. Остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде кремового твердого вещества (3,11 г; 84%). 1H ЯМР (400 МГц, DMSO-d6) δ млн-1 10.35 (s, 1Н), 7.49-7.59 (m, 2Н), 7.46 (bs, 1Н), 7.33 (dd, J=8,69; 2,44 Гц, 1H), 7.05 (bs, 1Н), 6.86 (d, J=8,79 Гц, 1H), 6.68 (d, J=16,02 Гц, 1H). LCMS [M+H]+=242,0; 244,0; [M+Na]+=263,9; 265,9; [M-H]-=240,0; 242,0.
Стадия D11.02: синтез 3-(2-гидрокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)проп-2-енамида. В каждый из трех флаконов для микроволнового реактора загружали 3-(5-бром-2-гидроксифенил)проп-2-енамид (1,04 г; 4,30 ммоль), бис(пинаколато)дибор (1,20 г; 4,73 ммоль), ацетат калия (1,69 г; 17,2 ммоль) и безводный 1,4-диоксан (15 мл). Реакционные смеси продували, барботируя через них азот в течение 5 мин, затем добавляли комплекс [1,1'-бис(дифенил-фосфино)ферроцен]-дихлорпалладий(II)-дихлорметан (0,351 г; 0,430 ммоль) и каждую реакционную смесь нагревали при 105°С в микроволновом реакторе в течение 1 ч. Реакционные смеси охлаждали до комнатной температуры, объединяли и фильтровали. Остаток промывали смесью дихлорметан/метанол (5:1, 250 мл) и фильтраты объединяли и концентрировали. Остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде коричневого твердого вещества (2,22 г; 60%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 7.79 (d, J=1,37 Гц, 1H), 7.66 (d, J=16,02 Гц, 1H), 7.48 (dd, J=8,01; 1,56 Гц, 2Н), 6.99 (bs, 1H), 6.89 (d, J=8,21 Гц, 1H), 6.65 (d, J=16,02 Гц, 1H), 1.28 (s, 12Н). LCMS [М+Н]+=290,1; [М-Н]-=288,1.
Стадия D11.03: синтез 3-(2-гидрокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропенамида (заимствован из Monatshefte fur Chemie; vol. 147; nb. 3; (2016); p. 509-521). В круглодонную колбу загружали 3-(2-гидрокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)проп-2-енамид (2,12 г; 7,33 ммоль), дихлорметан (50 мл), метанол (50 мл), азодикарбоксилат калия (8,55 г; 44.1 ммоль) и ледяную уксусную кислоту (5,28 г; 87.9 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь наносили непосредственно на диоксид кремния и очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде бледно-оранжевого кристаллического вещества (0,982 г; 46%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.75 (s, 1H), 7.39 (d, J=1,56 Гц, 1H), 7.34 (dd, J=8,01; 1,76 Гц, 1H), 7.27 (bs, 1H), 6.71-6.80 (m, 2Н), 2.71 (t, J=7,81 Гц, 2Н), 2.26-2.34 (m, 2Н), 1.26 (s, 12Н). LCMS [М+Н]+=292,2; [M+Na]+=314,1; [М-Н]-=290,2.
Стадия D11.04: синтез 3-(2-гидрокси-5-(пиридазин-3-ил)фенил)пропенамида. В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали 3-(2-гидрокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропанамид (0,982 г; 3,37 ммоль), 3-бромпиридазин (0,590 г; 3,71 ммоль), карбонат цезия (3,30 г; 10,1 ммоль), 1,4-диоксан (50 мл) и воду (9 мл). Реакционную смесь продували, барботируя через нее азот в течение 5 мин, затем добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II)-дихлорметан (0,275 г; 0,337 ммоль). Реакционную смесь перемешивали при 85°С в течение 3 ч, затем охлаждали до комнатной температуры. Реакционную смесь разбавляли смесью дихлорметан/метанол (5:1, 50 мл) и фильтровали. Остаток промывали смесью дихлорметан/метанол (5:1, 50 мл), фильтраты объединяли и концентрировали. Остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде темно-коричневого твердого вещества (0,124 г; 15%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.90 (s, 1H), 9.10 (dd, J=4,79; 1,47 Гц, 1H), 8.07 (dd, J=8,69; 1,47 Гц, 1H), 7.93 (d, J=2,34 Гц, 1H), 7.83 (dd, J=8,40; 2,34 Гц, 1H), 7.68 (dd, J=8,60; 4,88 Гц, 1H), 7.32 (bs, 1H), 6.93 (d, J=8,40 Гц, 1H), 6.77 (bs, 1H), 2.82 (t, J=7,72 Гц, 2Н), 2.36-2.44 (m, 2Н). LCMS [М+Н]+ = 244,2; [M+Na]+ = 266,1; [М-Н]- = 242,2.
Стадия D11.05: синтез 2-(3-амино-3-оксопропил)-4-(пиридазин-3-ил)фенил-трифторметансульфоната. В круглодонную колбу загружали 3-(2-гидрокси-5-(пиридазин-3-ил)фенил)пропанамид (0,124 г; 0,510 ммоль), ацетонитрил (25 мл) и карбонат калия (0,282 г; 2,04 ммоль). Добавляли N-фенил-бис(трифторметансульфонимид) (0,191 г; 0,535 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь концентрировали и остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде темно-коричневого твердого вещества (0,119 г; 62%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.27 (dd, J=4,88; 1,56 Гц, 1H), 8.25-8.31 (m, 2Н), 8.17 (dd, J=8,79; 2,34 Гц, 1H), 7.84 (dd, J=8,60; 4,88 Гц, 1H), 7.58 (d, J=8,60 Гц, 1H), 7.24 (d, J=8,21 Гц, 1H), 6.83 (bs, 1H), 2.99 (t, J=7,62 Гц, 2Н), 2.51-2.55 (m, 2Н). LCMS [М+Н]+ = 376,0; [M+Na]+ = 398,0; [М+HCO2H-Н]- = 420,0.
Стадия D11.06: синтез 3-(2-(2-((метилсульфонил)амино)пиридин-4-ил)-5-(пиридазин-3-ил)фенил)пропанамида (D11). Во флакон для микроволнового реактора загружали 2-(3-амино-3-оксопропил)-4-(пиридазин-3-ил)фенил-трифторметансульфонат (0,119 г; 0,317 ммоль), N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-пиридин-2-ил)-N-(метилсульфонил)метансульфонамид (0,131 г; 0,349 ммоль), карбонат калия (0,088 г; 0,634 ммоль), 1,4-диоксан (6,7 мл), этанол (1,7 мл) и воду (1,7 мл). Реакционную смесь продували, барботируя через нее азот в течение 5 мин, затем добавляли тетракис(трифенил-фосфин)палладий(0) (0,037 г; 0,032 ммоль) и флакон закрывали. Реакционную смесь перемешивали при 105°С в течение 3 ч в микроволновом реакторе, затем охлаждали до комнатной температуры и разбавляли смесью дихлорметан/метанол (5:1, 50 мл). Смесь (рН примерно 7) фильтровали и остаток сушили над сульфатом магния, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде коричневого твердого вещества (0,056 г; 44%). 1H ЯМР (400 МГц, DMSO-d6) δ млн-1 9.25 (dd, J=1,6; 4,9 Гц, 1H), 8.32 (bs, 1H), 8.27 (dd, J=1,6; 8,6 Гц, 1H), 8.18 (d, J=1,6 Гц, 1H), 8.07 (dd, J=1,8; 8,0 Гц, 1H), 7.82 (dd, J=4,9; 8,6 Гц, 1H), 7.38 (d, J=8,0 Гц, 1H), 7.28 (bs, 1H), 7.07 (d, J=4,7 Гц, 1H), 6.97 (s, 1H), 6.76 (bs, 1H), 3.33 (bs, 3Н), 2.91-2.82 (m, 2Н), 2.37 (dd, J=7,0; 9,0 Гц, 2Н). LCMS [М+Н]+ = 398,1; [M+Na]+ = 420,1; [М-Н]- = 396,1.
Схема синтеза D12 показана ниже:
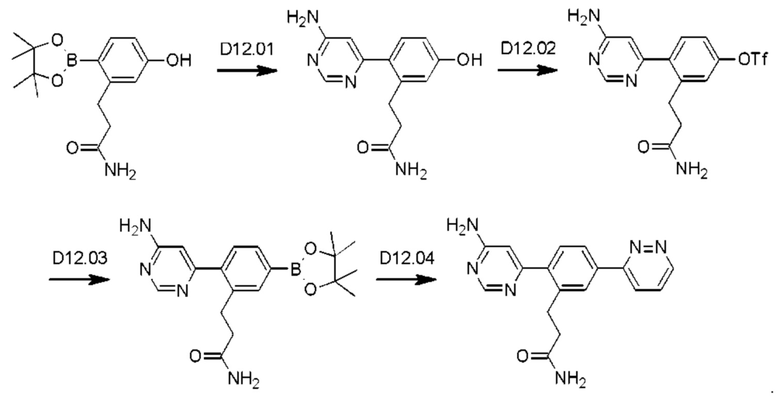
Стадия D12.01: синтез 3-(2-(6-аминопиримидин-4-ил)-5-гидроксифенил)пропанамида. В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали 3-(5-гидрокси-2-(пинаколборан)фенил)пропанамид (1,17 г; 4,02 ммоль), 4-амино-6-хлорпиримидин (0,573 г; 4,42 ммоль), карбонат цезия (3,93 г; 12,0 ммоль), 1,4-диоксан (50 мл) и воду (9 мл). Реакционную смесь продували, барботируя через нее азот в течение 5 мин, затем добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II)-дихлорметан (0,328 г; 0,402 ммоль). Реакционную смесь перемешивали при 85°С в течение 3 ч, затем охлаждали до комнатной температуры. Реакционную смесь разбавляли этилацетатом (100 мл) и водой (100 мл) и рН водной фазы доводили до 4, используя раствор соляной кислоты. Органические фазы собирали, сушили над сульфатом магния, фильтровали и концентрировали, получая неочищенный продукт. Водный слой концентрировали досуха и остаток экстрагировали смесью дихлорметан/метанол (1:1, 2×50 мл). Органические экстракты концентрировали, получая вторую порцию неочищенного продукта, которую объединяли с первой и очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде коричневого полутвердого вещества (0,743 г; 72%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.87 (bs, 1H), 8.55 (bs, 1H), 7.75 (bs, 2Н), 7.41 (bs, 1H), 7.18 (d, J=8,40 Гц, 1H), 6.63-6.90 (m, 3Н), 6.54 (s, 1H), 2.81 (t, J=7,62 Гц, 2Н), 2.35 (t, J=7,81 Гц, 2Н). LCMS [М+Н]+ = 259,1; [М-Н]- = 257,2.
Стадия D12.02: синтез 3-(3-амино-3-оксопропил)-4-(6-аминопиримидин-4-ил)фенил-трифторметансульфоната. В круглодонную колбу загружали 3-(2-(6-аминопиримидин-4-ил)-5-гидроксифенил)пропанамид (0,769 г; 2,98 ммоль), ацетонитрил (50 мл) и N,N-диметилформамид (25 мл). Добавляли карбонат калия (1,23 г; 8,93 ммоль) и реакционную смесь охлаждали в ледяной бане в течение 15 мин. Добавляли N-фенил-бис(трифторметансульфонимид) (1,12 г; 3,13 ммоль) и реакционную смесь оставляли медленно нагреваться до комнатной температуры в течение нескольких часов, затем нагревали при 50°С в течение 3 суток. Реакционную смесь охлаждали до комнатной температуры, концентрировали, добавляли этилацетат (250 мл) и перемешивали в течение 30 мин. Полученные твердые вещества удаляли фильтрованием и фильтраты концентрировали, затем снова упаривали из ксилолов (2×150 мл). Остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде коричневого воскообразного твердого вещества (0,689 г; 59%). 1H ЯМР (400 МГц, DMSO-d6) δ млн-1 8.42 (d, J=0,98 Гц, 1H), 7.38-7.50 (m, 3Н), 7.26 (bs, 1H), 7.00 (s, 2Н), 6.75 (bs, 1H), 6.48 (d, J=1,17 Гц, 1H), 2.92 (t, J=7,81 Гц, 2Н), 2.33 (t, J=7,72 Гц, 2Н). LCMS [М+Н]+ = 391,1; [М-Н]- = 389,0.
Стадия D12.03: синтез 3-(2-(6-аминопиримидин-4-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-пропанамида. Во флакон для микроволнового реактора загружали 3-(3-амино-3-оксопропил)-4-(6-аминопиримидин-4-ил)фенил-трифторметансульфонат (0,639 г; 1,64 ммоль), бис(пинаколато)дибор (1,04 г; 4,09 ммоль), ацетат калия (0,482 г; 4,91 ммоль) и безводный 1,4-диоксан (15 мл). Реакционную смесь продували, барботируя через нее азот в течение 5 мин, затем добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладий(II)-дихлорметан (0,134 г; 0,164 ммоль). Реакционную смесь нагревали при 105°С в течение 1 ч в микроволновом реакторе, затем охлаждали до комнатной температуры. Смесь фильтровали и остаток промывали смесью дихлорметан/метанол (4:1, 2×50 мл). Фильтраты объединяли и концентрировали и остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде коричневого твердого вещества (0,442 г; 73%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 8.40 (s, 1H), 7.62 (s, 1H), 7.54-7.59 (m, 1H), 7.28 (d, J=7,62 Гц, 1H), 7.24 (bs, 1H), 6.92 (s, 2H), 6.69 (bs, 1H), 6.45 (d, J=1,17 Гц, 1H), 2.84-2.93 (m, 2H), 2.24-2.33 (m, 2H), 1.31 (s, 12H). LCMS [M+H]+ = 369,3.
Стадия D12.04: синтез 3-(2-(6-аминопиримидин-4-ил)-5-(пиридазин-3-ил)фенил)пропанамида (D12). Во флакон для микроволнового реактора загружали 3-(2-(6-аминопиримидин-4-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пропанамид (0,224 г; 0,608 ммоль), 3-бромпиридазин (0,106 г; 0,669 ммоль), карбонат цезия (0,595 г; 1,82 ммоль), 1,4-диоксан (10 мл) и воду (1,8 мл). Реакционную смесь продували, барботируя через нее азот в течение 2 мин, затем добавляли комплекс [1,1'-бис(дифенил-фосфино)ферроцен]-дихлорпалладий(II)-дихлорметан (0,050 г; 0,061 ммоль) и флакон закрывали. Реакционную смесь перемешивали при 105°С в течение 80 мин в микроволновом реакторе, затем охлаждали до комнатной температуры и фильтровали. Остаток промывали этилацетатом (25 мл), затем смесью дихлорметан/метанол (5:1, 2×25 мл). Фильтраты объединяли, сушили над сульфатом магния, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде темно-красного стеклообразного вещества (0,088 г; 45%). 1H ЯМР (400 МГц, DMSO-d6) δ млн-1 9.24 (dd, J=1,5; 5,0 Гц, 1H), 8.44 (d, J=1,2 Гц, 1H), 8.27 (dd, J=1,6; 8,6 Гц, 1H), 8.14 (d, J=1,8 Гц, 1H), 8.05 (dd, J=2,0; 8,0 Гц, 1H), 7.81 (dd, J=4,9; 8,6 Гц, 1H), 7.47 (d, J=8,0 Гц, 1H), 7.28 (bs, 1H), 6.97 (s, 2Н), 6.72 (bs, 1H), 6.55 (d, J=1,2 Гц, 1H), 3.06-2.96 (m, 2Н), 2.39 (m, 2Н). LCMS [М+Н]+ = 321,2; [М-Н]- = 319,1.
Схема синтеза D18 показана ниже:
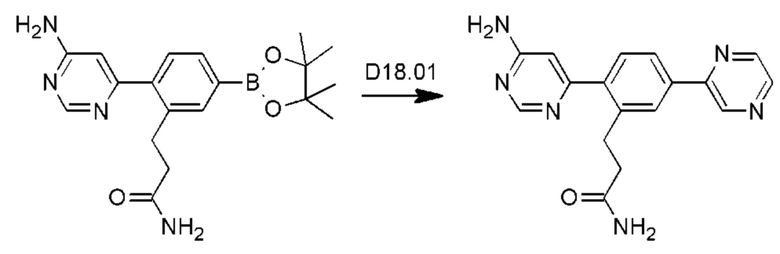
Стадия D18.01: синтез 3-(2-(6-аминопиримидин-4-ил)-5-(пиразин-2-ил)фенил)пропанамида (D18). Выполняли согласно стадии D12.04, используя 2-хлорпиразин, с получением указанного в заголовке соединения в виде темного стеклообразного вещества (0,066 г; 37%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.29 (s, 1H), 8.75 (s, 1H), 8.64 (d, J=2,3 Гц, 1H), 8.44 (s, 1H), 8.11 (s, 1H), 8.04 (d, J=8,0 Гц, 1H), 7.45 (d, J=8,0 Гц, 1H), 7.27 (bs, 1H), 6.95 (bs, 2Н), 6.73 (bs, 1H), 6.53 (s, 1H), 3.00 (t, J=7,7 Гц, 2Н), 2.38 (t, J=7,8 Гц, 2Н). LCMS [М+Н]+ = 321,2; [М-Н]- = 319,0.
Схема синтеза D17 показана ниже:
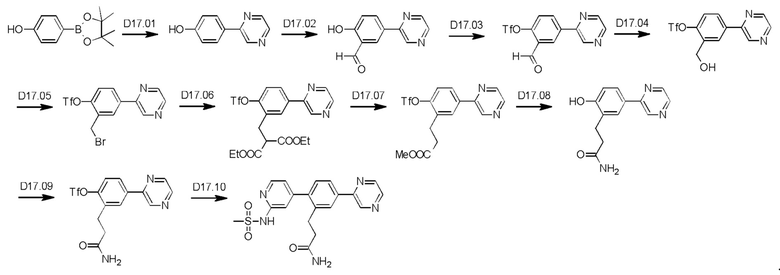
Стадия D17.01: синтез 4-(пиразин-2-ил)фенола. В круглодонную колбу в атмосфере азота загружали 4-гидроксифенилбороновую кислоту (10,0 г; 0,045 моль)), 2-хлорпиразин (6,25 г; 0,0545 моль) и карбонат цезия (29,58 г; 0,0908 моль), 1,4-диоксан (90 мл) и воду (10 мл). Добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]-дихлорпалладий(II)-дихлорметан (1,85 г; 2,27 ммоль) и реакционную смесь нагревали с обратным холодильником в течение 2 ч. Смесь охлаждали, разбавляли этилацетатом (100 мл), фильтровали и переносили в делительную воронку. Водную фазу отделяли, промывали этилацетатом (100 мл) и дихлорметаном (100 мл). Объединенные органические экстракты сушили (сульфат магния), фильтровали и концентрировали. Неочищенное вещество очищали флэш-хроматографией (этилацетат/дихлорметан), получая указанное в заголовке соединение в виде серого порошка (7,1 г; выход 91%). 1H ЯМР (400 МГц, DMSO-d6) δ млн-1 9.91 (s, 1H), 9.14 (d, J=1,5 Гц, 1H), 8.63 (dd, J=1,6; 2,4 Гц, 1H), 8.49 (d, J=2,4 Гц, 1H), 8.03-7.96 (m, 2Н), 6.94-6.87 (m, 2Н). LCMS [М+Н]+ = 173,1.
Стадия D17.02: синтез 2-гидрокси-5-(пиразин-2-ил)бензальдегида. В круглодонную колбу загружали 4-(пиразин-2-ил)фенол (4,3 г; 25,0 ммоль) и трифторуксусную кислоту (50 мл). Порциями добавляли гексамин (5,26 г; 37,5 ммоль) и раствор нагревали с обратным холодильником в течение 6 ч. Смесь охлаждали, разбавляли водой (200 мл), перемешивали в течение 30 мин и переносили в делительную воронку. Добавляли дихлорметан (200 мл), органическую фазу отделяли, промывали водой (100 мл) и рассолом (100 мл), сушили (сульфат магния), фильтровали и концентрировали досуха, получая указанное в заголовке соединение в виде серого порошка (2,11 г; 42%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ млн-1 11.20 (s, 1H), 10.04 (d, J=0,6 Гц, 1H), 9.06-9.01 (m, 1H), 8.65-8.60 (m, 1H), 8.52 (d, J=2,3 Гц, 1H), 8.33 (d, J=2,1 Гц, 1H), 8.22 (dd, J=2,1; 8,9 Гц, 1H), 7.15 (d, J=8,7 Гц, 1H). LCMS [М+Н]+ = 201,1.
Стадия D17.03: синтез 2-формил-4-(пиразин-2-ил)фенил-трифторметансульфоната. В круглодонную колбу загружали 2-гидрокси-5-(пиразин-2-ил)бензальдегид (3,5 г; 17,5 ммоль), карбонат калия (4,84 г; 35,0 ммоль) и ацетонитрил (100 мл). Добавляли N-фенил-бис(трифтор-метансульфонимид) (6,87 г; 19,2 ммоль) и смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли этилацетат (100 мл), смесь фильтровали и концентрировали. После очистки флэш-хроматографией (этилацетат/гексаны) получали указанное в заголовке соединение в виде прозрачного масла, которое отвердевало при стоянии (4,6 г; 92%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ млн-1 10.36 (s, 1H), 9.12 (d, J=1,4 Гц, 1H), 8.70 (dd, J=1,6; 2,5 Гц, 1H), 8.66 (d, J=2,2 Гц, 1H), 8.63 (d, J=2,4 Гц, 1H), 8.43 (dd, J=2,4; 8,7 Гц, 1H), 7.58 (d, J=8,6 Гц, 1H). LCMS [М+Н]+ = 333,0.
Стадия D17.04: синтез 2-(гидроксиметил)-4-(пиразин-2-ил)фенил-трифторметансульфоната. Выполняли согласно стадии D6.02, получая указанное в заголовке соединение в виде желтого твердого вещества (1,79 г; 51%). LCMS [М+Н]+ = 335,0.
Стадия D17.05: синтез 2-(бромметил)-4-(пиразин-2-ил)фенил-трифторметансульфоната. В круглодонную оснащенную колбу в атмосфере азота загружали трифенилфосфин (2,26 г; 8,62 ммоль) и дихлорметан (80 мл). Реакционную смесь перемешивали и охлаждали в ледяной бане в течение 20 мин, затем медленно добавляли 2-(гидроксиметил)-4-(пиразин-2-ил)фенил-трифторметансульфонат (1,44 г; 4,31 ммоль). Реакционную смесь перемешивали и охлаждали в ледяной бане в течение 20 мин, затем оставляли нагреваться до комнатной температуры и перемешивали в течение 2 ч. Смесь концентрировали и очищали флэш-хроматографией (этилацетат/гексаны), получая указанное в заголовке соединение в виде желтого масла (1,53 г; 89%). LCMS [М]+ = 397,9; 399,9.
Стадия D17.06: синтез 1,3-диэтил-2-[(((2-трифторметил)сульфонил)оксо)-5-(пиразин-2-ил)-бензил]пропандиоата. В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали диэтилмалонат (0,726 г; 4,54 ммоль) и тетрагидрофуран (80 мл). К перемешиваемому раствору порциями добавляли гидрид натрия (0,099 г 60%-ной дисперсии в масле; 4,16 ммоль), наблюдая бурное выделение газа. Реакционную смесь перемешивали в течение 10 мин, затем медленно добавляли раствор 2-(бромметил)-4-(пиразин-2-ил)фенил-трифторметансульфонат (0,75 г; 1,89 ммоль) в тетрагидрофуране (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем концентрировали. После очистки флэш-хроматографией (этилацетат/дихлорметан) получали указанное в заголовке соединение в виде бледно-желтого масла (1,05 г). LCMS [М+Н]+ = 477,1; [M+Na]+ = 499,0.
Стадия D17.07: синтез метил-3-[5-(пиразин-2-ил)-2-((трифторметансульфонил)-фенил]пропаноата. В круглодонную колбу загружали 1,3-диэтил-2-[(((2-трифторметил)-сульфонил)оксо)-5-(пиразин-2-ил)-бензил]пропандиоат (7,0 г; 14,7 ммоль) и водный раствор серной кислоты (2 М, 15 мл). Смесь нагревали с обратным холодильником в течение 24 ч, охлаждали и добавляли метанол (50 мл). Смесь концентрировали досуха, получая указанное в заголовке соединение в виде оранжевого масла (1,72 г; 45%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ млн-1 8.95 (d, J=1,5 Гц, 1H), 8.58 (dd, J=1,6; 2,4 Гц, 1H), 8.50 (d, J=2,4 Гц, 1H), 7.99 (d, J=2,2 Гц, 1H), 7.88 (dd, J=2,3; 8,6 Гц, 1H), 7.36 (d, J=8,6 Гц, 1H), 3.62 (s, 3Н), 3.08 (t, J=7,7 Гц, 2Н), 2.68 (t, J=7,7 Гц, 2Н). LCMS [М+Н]+ = 391,1; [M+Na]+ = 413,1.
Стадия D17.08: синтез 3-[2-гидрокси-5-(пиразин-2-ил)фенил]пропенамида. Во флакон для микроволнового реактора загружали метил-3-[5-(пиразин-2-ил)-2-((трифторметансульфонил)фенил]пропаноат (1,19 г; 3,07 ммоль) и метанольный раствор аммиака (7 н., 15 мл). Реакционный сосуд закрывали и смесь нагревали с обратным холодильником в течение 48 ч. После охлаждения смесь концентрировали и очищали флэш-хроматографией (метанол/дихлорметан), получая указанное в заголовке соединение в виде бледно-желтого масла (0,41 г; 55%). LCMS [М+Н]+ = 244,2; [M+Na]+ = 266,0.
Стадия D17.09: синтез 2-((3-амино-3-оксопропил)-4-(пиразин-2-ил)фенил)трифторметансульфоната. Выполняли согласно стадии D17.03, получая указанное в заголовке соединение в виде прозрачного масла, которое затвердевало при стоянии (0,360 г; 58%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ млн-1 8.96 (d, J=1,4 Гц, 1H), 8.58 (dd, J=1,6; 2,4 Гц, 1H), 8.50 (d, J=2,4 Гц, 1H), 8.03 (d, J=2,2 Гц, 1H), 7.89 (dd, J=2,3; 8,6 Гц, 1H), 7.35 (d, J=8,6 Гц, 1H), 5.37 (br. s., 1H), 5.24 (ушир. s., 1H), 3.14-3.08 (m, 2Н), 2.60-2.52 (m, 2Н). LCMS [М+Н]+ = 376,1; [M+Na]+ = 398,0.
Стадия D17.10: синтез 3-[2-(2-[(метилсульфонил)амино]пиридин-4-ил)-5-(пиразин-2-ил)фенил]пропанамида. Выполняли согласно стадии D 11.06, получая указанное в заголовке соединение в виде беловатого порошка (0,15 г; 70%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.30 (d, J=1,4 Гц, 1H), 8.75 (dd, J=1,5, 2,5 Гц, 1H), 8.65 (d, J=2,4 Гц, 1H), 8.31 (ушир. s., 1H), 8.15 (d, J=1,8 Гц, 1H), 8.06 (dd, J=1,9; 8,0 Гц, 1H), 7.36 (d, J=8,0 Гц, 1H), 7.28 (ушир. s., 1H), 7.05 (ушир. s., 1H), 6.95 (ушир. s., 1H), 6.76 (ушир. s., 1H), 2.85 (dd, J=6,6; 9,3 Гц, 2Н), 2.36 (dd, J=6,8; 8,9 Гц, 2Н), 2.30 (s, 4Н). LCMS [М+Н]+ = 398,1; [M+Na]+ = 420,0.
Схема синтеза D28 показана ниже:
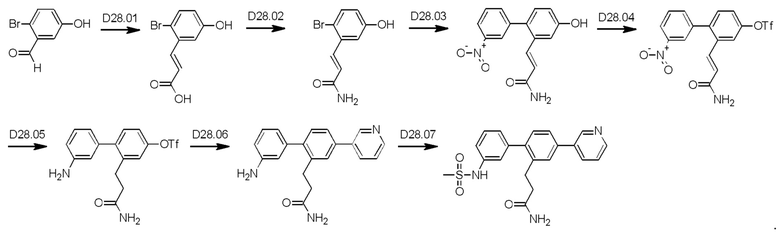
Стадия D28.01: синтез (2Е)-3-(2-бром-5-гидроксифенил)проп-2-еновой кислоты. В круглодонную колбу, оснащенную обратным холодильником, загружали 4-бром-3-формилфенол (25,3 г; 0,126 моль), пиридин (150 мл), малоновую кислоту (15,7 г; 0,151 моль) и пиперидин (1,50 мл). Реакционную смесь перемешивали при температуре дефлегмации в течение 6,5 ч, затем оставляли охлаждаться. К реакционной смеси добавляли 2 М раствор соляной кислоты (500 мл) и рН доводили до значения ниже 2, используя раствор концентрированной соляной кислоты. Смесь энергично перемешивали и охлаждали в ледяной бане до температуры ниже 10°С. Полученные твердые вещества собирали на воронке Бюхнера и промывали 2 М раствором соляной кислоты (200 мл) и водой (50 мл). Твердые вещества распределяли между этилацетатом (1,2 л) и 2 М раствором соляной кислоты (200 мл). Водный слой удаляли и органические слои промывали рассолом (200 мл), сушили над сульфатом магния, фильтровали и концентрировали, получая указанное в заголовке соединение в виде бледно-пурпурового порошка (24,8 г; 81%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 12.53 (bs, 1H), 9.90 (s, 1H), 7.75 (d, J=15,8 Гц, 1H), 7.47 (d, J=8,6 Гц, 1H), 7.19 (d, J=2,9 Гц, 1H), 6.80 (dd, J=8,7; 2,8 Гц, 1H), 6.40 (d, J=15,8; 2,8 Гц, 1H). LCMS [М]+ = 242,9; 244,9.
Стадия D28.02: синтез (2Е)-3-(2-бром-5-гидроксифенил)проп-2-енамида. В круглодонную колбу, оснащенную обратным холодильником, загружали (2E)-3-(2-бром-5-гидроксифенил)проп-2-еновую кислоту (10,0 г; 0,041 моль), дихлорметан (100 мл) и N,N-диметилформамид (0,20 мл). При энергичном перемешивании медленно добавляли оксалилхлорид (8,70 мл), наблюдая бурное выделение газа. По завершении добавления реакционную смесь перемешивали при температуре дефлегмации в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали почти досуха. Остаток снова упаривали из дихлорметана (50 мл) и переносили в 1,4-диоксан (100 мл). Этот раствор медленно выливали в перемешиваемую смесь 25%-ного водного раствора аммиака (30 мл) в 1,4-диоксане (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и концентрировали почти досуха. Остаток распределяли между этилацетатом (250 мл) и водой (150 мл), что приводило к образованию нерастворимого осадка. Смесь фильтровали и фильтраты объединяли в делительной воронке. Водный слой удаляли и органические слои промывали рассолом (25 мл), сушили над сульфатом магния, фильтровали и концентрировали, получая указанное в заголовке соединение в виде темно-красного твердого вещества (8,88 г; 89%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.89 (bs, 1H), 7.62 (bs, 1H), 7.59 (d, J=15,6, 1H), 7.45 (d, J=8,7 Гц, 1H), 7.21 (bs, 1H), 7.05 (d, J=2,7 Гц, 1H), 6.75 (dd, J=8,7; 2,8 Гц, 1H), 6.52 (d, J=15,6 Гц, 1Н). LCMS [M+H]+ = 241,9; 243,9; [M+Na]+ = 264,0; 266,0; [M-H]- = 240,0; 242,0.
Стадия D28.03: синтез (2Е)-3-(4-гидрокси-3'-нитробифенил-2-ил)проп-2-енамида. В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали (2E)-3-(2-бром-5-гидроксифенил)проп-2-енамид (2,00 г; 8,26 ммоль), 3-нитрофенилбороновую кислоту (2,76 г; 16,5 ммоль), карбонат калия (3,43 г; 24,8 ммоль), 1,4-диоксан (35 мл) и воду (1,5 мл). Реакционную смесь продували, барботируя через нее азот в течение 5 мин, затем добавляли тетракис(трифенилфосфин)палладий(0) (0,95 г; 0,83 ммоль). Реакционную смесь перемешивали при 85°С в течение 4 ч и охлаждали до комнатной температуры. Реакционную смесь концентрировали почти досуха и остаток разбавляли этилацетатом (100 мл) и водой (50 мл). Смесь фильтровали и твердые вещества промывали 2 М раствором соляной кислоты (10 мл). Фильтраты объединяли, рН водного слоя доводили до 4, используя 2 М раствор соляной кислоты, и смесь переносили в делительную воронку. Органические слои отделяли и водный слой далее экстрагировали этилацетатом (50 мл). Органические экстракты объединяли, промывали рассолом (25 мл), сушили над сульфатом магния, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (этилацетат/дихлорметан/метанол), получая указанное в заголовке соединение в виде желтого порошка (1,83 г; 78%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.89 (s, 1H), 8.20-8.27 (m, 1H), 8.04 (t, J=1,8 Гц, 1H), 7.67-7.78 (m, 2Н), 7.54 (bs, 1H), 7.17-7.30 (m, 2Н), 7.10 (d, J=2,5 Гц, 2Н), 6.92 (dd, J=8,4; 2,5 Гц, 1H), 6.52 (d, J=15,6 Гц, 1H). LCMS [М+Н]+ = 285,1; [M+Na]+ = 307,0; [М-Н]- = 283,1.
Стадия D28.04: синтез 2-[(1Е)(3-амино-3-оксопроп-1-ен-1-ил)-3'-нитробифенил-4-ил-трифторметансульфоната. В круглодонную колбу загружали (2E)-3-(4-гидрокси-3'-нитробифенил-2-ил)проп-2-енамид (1,83 г; 6,43 ммоль), ацетонитрил (30 мл) и карбонат калия (1,82 г; 13,2 ммоль). Желтую суспензию перемешивали энергично и охлаждали в ледяной бане в течение 15 мин. Порциями добавляли N-фенил-бис(трифтор-метансульфонимид) (2,53 г; 7,07 ммоль) и реакционную смесь оставляли нагреваться до комнатной температуры в течение 3 ч. Реакционную смесь концентрировали и остаток очищали флэш-хроматографией (этилацетат/гексаны), получая указанное в заголовке соединение в виде беловатого твердого вещества (2,77 г; колич.). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 8.34 (ddd, J=5,9; 3,5; 2,3 Гц, 1H), 8.16-8.20 (m, 1H), 7.85 (s, 1H), 7.80-7.84 (m, 2Н), 7.65 (d, J=1,4 Гц, 2Н), 7.57 (bs, 1H), 7.20 (d, J=15,6 Гц, 2Н), 6.70 (d, J=15,6 Гц, 1H). LCMS [М+Н]+ = 417,0; [M+Na]+ = 439,0; [М+HCO2]- = 461,1.
Стадия D28.05: синтез 3'-амино-2-(3-амино-3-оксопропил)бифенил-4-ил-трифторметансульфоната. В круглодонную колбу загружали 2-[(1E)-2-(3-амино-3-оксопроп-1-ен-1-ил)-3'-нитробифенил-4-ил-трифторметансульфонат (0,500 г; 1,20 ммоль) и этанол (25 мл). Смесь перемешивали и продували, барботируя через нее азот в течение 5 мин. К реакционной смеси добавляли 10% палладий на угле (0,050 г), промывали сильной струей водорода и перемешивали в атмосфере водорода в течение 20 ч. Реакционную смесь фильтровали через набивку целита, и целит промывали этанолом (3×15 мл). Этанольные фильтраты объединяли и концентрировали и остаток очищали флэш-хроматографией (этилацетат/гексаны), получая указанное в заголовке соединение в виде бледно-желтого кристаллического вещества (0,108 г; 23%). 1H ЯМР (400 МГц, CDCl3) δ млн-1 7.28 (m, 1H), 7.18-7.23 (m, 2Н), 7.11-7.17 (dd, 1H), 6.70 (ddd, J=8,0; 2,3; 1,0 Гц, 1H), 6.65 (dt, J=7,4; 1,3 Гц, 1H), 6.57-6.60 (m, 1H), 5.17 (brs, 2Н), 2.94-3.04 (m, 2Н), 2.27-2.35 (m, 2Н). LCMS [М+Н]+ = 389,1; [M+Na]+ = 411,1; [М+HCO2]- = 433,1.
Стадия D28.06: синтез 3-(3'-амино-4-(пиридин-3-ил)бифенил-2-ил)пропенамида. В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали 3'-амино-2-(3-амино-3-оксопропил)бифенил-4-ил-трифторметансульфонат (0,786 г; 2,02 ммоль), пиридин-3-бороновую кислоту (0,323 г; 2,63 ммоль), карбонат калия (0,839 г; 6,07 ммоль), воду (11,5 мл), этанол (20,5 мл) и 1,4-диоксан (32,0 мл). Смесь перемешивали и продували, барботируя через нее азот в течение 5 мин. Добавляли тетракис(трифенилфосфин)палладий(0) (0,234 г; 0,20 ммоль) и реакционную смесь перемешивали при 85°С в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой (75 мл) и этилацетатом (150 мл). Смесь фильтровали, переносили в делительную воронку и собирали органическую фазу. Водный слой далее экстрагировали этилацетатом (50 мл), органические экстракты объединяли, промывали рассолом (25 мл), сушили над сульфатом магния, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде бледно-желтой смолы (0,382 г; 60%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 8.92 (d, J=1,6 Гц, 1H), 8.58 (dd, J=4,8;, 1,7 Гц, 1H), 8.06-8.12 (m, 1H), 7.65 (d, J=1,7 Гц, 1H), 7.57 (dd, J=7,8; 2,0 Гц, 1H), 7.46-7.53 (m, 1H), 7.23 (d, J=8,0 Гц, 2H), 7.08 (t, J=7,8 Гц, 1H), 6.74 (s, 1H), 6.57 (dd, J=8,0; 1,4 Гц, 1H), 6.52 (t, J=1,9 Гц, 1H), 6.46 (d, J=7,4 Гц, 1H), 5.14 (s, 2Н), 2.80-2.88 (m, 2Н), 2.28-2.36 (m, 2Н). LCMS [М+Н]+ = 318,2; [M+Na]+ = 340,2; [М-Н]- = 316,.2.
Стадия D28.07: синтез 3-(3'-((метилсульфонил)амино)-4-(пиридин-3-ил)бифенил-2-ил)пропанамида (D28). В круглодонную колбу в атмосфере азота загружали 3-(3'-амино-4-(пиридин-3-ил)бифенил-2-ил)пропанамид (0,281 г; 0,885 ммоль), дихлорметан (15 мл) и триэтиламин (0,617 мл; 4,43 ммоль). Реакционную смесь охлаждали в ледяной бане в течение 15 мин, затем добавляли метансульфонилхлорид (0,069 мл; 0,885 ммоль). Реакционную смесь перемешивали в течение 3 ч в ледяной бане, затем оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь наносили непосредственно на диоксид кремния и очищали флэш-хроматографией (дихлорметан/метанол), получая частично очищенный продукт в виде беловатого твердого вещества. Неочищенный продукт перемешивали в смеси дихлорметан/гексаны (1:1) (25 мл) в течение нескольких часов, полученное твердое вещество собирали фильтрованием, промывали смесью дихлорметан/гексаны (1:1) (2×10 мл) и сушили под высоким вакуумом, получая указанное в заголовке соединение, содержащее 5 моль % примеси алкена с предыдущей стадии. Соединение подвергали процедуре гидрирования, используя 10%-ный палладий на угле в этаноле. После проведения обычной обработки указанное в заголовке соединение получали в виде беловатого порошка (0,029 г; 47%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 8.94 (ушир. s., 1H), 8.52-8.67 (m, 1H), 8.11 (d, J=7,23 Гц, 1H), 7.70 (ушир. s., 1H), 7.62 (d, J=7,42 Гц, 1H), 7.47-7.57 (m, 1H), 7.43 (t, J=7,52 Гц, 1H), 7.15-7.36 (m, 5Н), 7.10 (d, J=7,03 Гц, 1H), 6.75 (ушир. s., 1H), 3.04 (s, 3Н), 2.84 (t, J=7,23 Гц, 2Н), 2.33 (t, J=7,23 Гц, 2Н). LCMS [М+Н]+ = 396,1.
Схема синтеза D29 показана ниже:
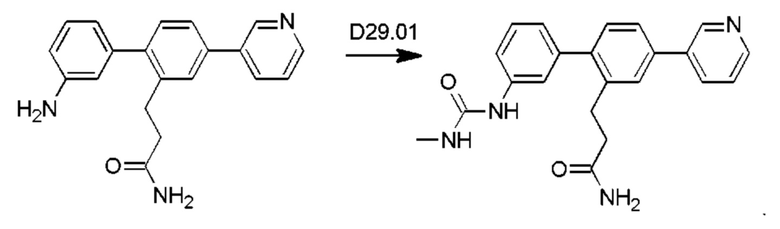
Стадия D29.01: синтез 3-(3'-((метилкарбамоил)амино)-4-(пиридин-3-ил)бифенил-2-ил)пропанамида (D29). В круглодонную колбу в атмосфере азота загружали карбонилдиимидазол (0,403 г; 2,48 ммоль) и ацетонитрил (30 мл) и перемешивали при 40°С. К реакционной смеси по каплям добавляли раствор 3-(3'-амино-4-(пиридин-3-ил)бифенил-2-ил)пропанамида (0,394 г; 1,24 ммоль) в ацетонитриле (30 мл) через капельную воронку с приспособлением для выравнивания давления, и реакционную смесь перемешивали при 40°С в течение 2 ч. Добавляли дополнительную аликвоту карбонилдиимидазола (0,403 г; 2,48 ммоль) и смесь перемешивали при 40°С в течение ночи. После охлаждения до комнатной температуры добавляли метиламин (4,5 мл 2 М раствора в тетрагидрофуране) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем наносили непосредственно на диоксид кремния и очищали флэш-хроматографией (дихлорметан/метанол), получая частично очищенный продукт, который повторно хроматографировали (дихлорметан/метанол), получая указанное в заголовке соединение в виде беловатого стеклообразного вещества (0,102 г; 22%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 8.93 (dd, J=2,4; 0,7 Гц, 1H), 8.61 (s, 1H), 8.58 (dd, J=4,7; 1,6 Гц, 1H), 8.07-8.14 (m, 1H), 7.68 (d, J=1,8 Гц, 1H), 7.60 (dd, J=7,8; 2,0 Гц, 1H), 7.51 (ddd, J=8,0; 4,8; 0,9 Гц, 1H), 7.47 (t, J=1,7 Гц, 1H), 7.25-7.38 (m, 3Н), 7.23 (ушир. s., 1H), 6.88 (dt, J=7,4; 1,4 Гц, 1H), 6.73 (ушир. s., 1H), 6.07 (q, J=4,4 Гц, 1H), 2.78-2.90 (m, 2Н), 2.60-2.69 (m, 3Н), 2.27-2.37 (m, 2Н). LCMS [М+Н]+ = 375,2; [М-Н]- = 373,2.
Схема синтеза D31 показана ниже:
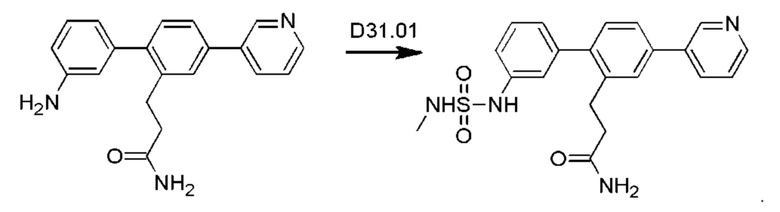
Стадия D31.01: синтез 3-(3'-((метилсульфамоил)амино)-4-(пиридин-3-ил)бифенил-2-ил)пропанамида (D31). В круглодонную колбу в атмосфере азота загружали 3-(3'-амино-4-(пиридин-3-ил)бифенил-2-ил)пропанамид (0,250 г; 0,788 ммоль), дихлорметан (10 мл) и триэтиламин (0,440 мл; 3,15 ммоль). Реакционную смесь перемешивали при комнатной температуре и добавляли раствор хлорангидрида метилсульфаминовой кислоты (0,256 г; 1,97 ммоль) в дихлорметане (10 мл) в виде порций по 2 мл. Реакционную смесь перемешивали при комнатной температуре в течение 48 ч, концентрировали и остаток перемешивали в воде (30 мл) и этилацетате (30 мл) вместе с небольшим количеством метанола, чтобы способствовать растворению. Органические слои отделяли, и водный слой (рН примерно 8) далее экстрагировали этилацетатом (20 мл). Органические экстракты объединяли, промывали водой (25 мл), рассолом (25 мл), сушили над сульфатом магния, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде беловатого порошка (0,063 г; 20%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.79 (s, 1H), 8.93 (dd, J=0,6; 2,3 Гц, 1H), 8.59 (dd, J=1,6; 4,7 Гц, 1H), 8.14-8.06 (m, 1H), 7.69 (d, J=1,8 Гц, 1H), 7.61 (dd, J=2,0; 7,8 Гц, 1H), 7.51 (ddd, J=0,8; 4,7; 8,0 Гц, 1H), 7.42-7.33 (m, 2Н), 7.29 (d, J=8,0 Гц, 1H), 7.25 (ушир. s., 1H), 7.21-7.14 (m, 2Н), 7.03-6.97 (m, 1H), 6.79 (ушир. s., 1H), 2.87-2.79 (m, 2Н), 2.48 (s, 3Н), 2.34 (dd, J=7,0; 9,0 Гц, 2Н). LCMS [М+Н]+ = 411,1; [М-Н]- = 409,2.
Схема синтеза D30 показана ниже:
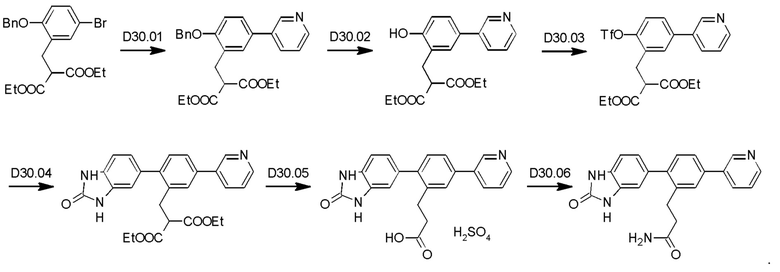
Стадия D30.01: синтез 1,3-Диэтил-2-(2-(фенилметокси)-5-(пирид-3-ил)бензил)пропандиоат. Выполняли согласно стадии D6.05, используя 3-пиридил-бороновую кислоту, с получением указанного в заголовке соединения в виде желтого масла (1,57 г; 79%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ млн-1 8.77 (dd, J=0,8; 2,3 Гц, 1H), 8.54 (dd, J=1,6; 4,7 Гц, 1H), 7.82-7.76 (m, 1H), 7.49-7.44 (m, 2Н), 7.44-7.37 (m, 4Н), 7.36-7.29 (m, 2Н), 7.01-6.96 (m, 1H), 5.18 (s, 2Н), 4.18-4.07 (m, 4Н), 3.93 (t, J=7,8 Гц, 1H), 3.34 (d, J=7,6 Гц, 2Н), 1.29-1.22 (m, 6Н). LCMS [М+Н]+ = 434,2.
Стадия D30.02: синтез 1,3-диэтил-2-(2-гидрокси-5-(пирид-3-ил)бензил)пропандиоата. Выполняли согласно стадии D6.06, получая указанное в заголовке соединение в виде бледно-желтого масла (0,847 г; 68%). 1Н ЯМР (400 МГц, ХЛОРОФОРМ-d) δ 8.75-8.81 (m, 1H), 8.54 (dd, J=4,79; 1,66 Гц, 1H), 7.80 (dt, J=7,86; 2,03 Гц, 1H), 7.30-7.40 (m, 3Н), 6.99 (d, J=8,21 Гц, 1H), 4.15-4.29 (m, 4Н), 3.79 (t, J=7,13 Гц, 1H), 3.24 (d, J=7,03 Гц, 2Н), 2.04 (s, 1H), 1.19-1.30 (m, 6Н). LCMS [М+Н]+ = 344,2.
Стадия D30.03: синтез 1,3-диэтил-2-[(((2-трифторметил)сульфонил)оксо)-5-(пирид-3-ил)-бензил]пропандиоата. Выполняли согласно стадии D6.07, получая указанное в заголовке соединение в виде неочищенного желтого масла (1,23 г). LCMS [М+Н]+ = 476,44.
Стадия D30.04: синтез 1,3-диэтил-2-(2-(8-оксо-7,9-диаза-бицикло[4.3.0]нона-1,3,5-триен-3-ил)-5-(пиридин-3-ил)-бензил)пропандиоата. Смесь 1,3-диэтил-2-[(((2-трифторметил)сульфонил)оксо)-5-(пирид-3-ил)-бензил]пропандиоата (1 экв.), пинаколового эфира ароматической бороновой кислоты или гетероциклической бороновой кислоты (1,1 экв.) и карбоната калия (2-3 экв.) растворяли в смеси 1,4-диоксана (3,1 мл/ммоль), этанола (0,65 мл/ммоль) и воды (0,65 мл/ммоль). Через эту смесь в течение 10 мин барботировали азот. Добавляли тетракис(трифенилфосфин)палладий(0) (0,1 экв.) и смесь нагревали при 85°С в атмосфере азота в течение 20 ч. Смесь распределяли между этилацетатом и водой. Водную фазу экстрагировали этилацетатом. Объединенные этилацетатные экстракты промывали водой и рассолом, сушили над безводным сульфатом натрия и фильтровали. Фильтрат упаривали досуха и очищали флэш-хроматографией (метанол/дихлорметан). Продукт суспендировали в смеси 1:1 дихлорметан/гексаны и выделяли фильтрованием. Беловатый порошок (0,422 г; 58%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 10.69 (s, 1H), 10.66 (s, 1H), 8.92 (s, 1H), 8.58 (d, J=3,5 Гц, 1H), 8.04-8.14 (m, 1H), 7.70 (d, J=1,6 Гц, 1H), 7.63 (dd, J=7,91; 1,7 Гц, 1H), 7.51 (dd, J=7,81; 4,9 Гц, 1H), 7.29 (d, J=8,0 Гц, 1H), 7.01 (d, J=8,0 Гц, 1H), 6.82-6.95 (m, 2Н), 3.89-4.03 (m, 4Н), 3.72 (t, J=7,9 Гц, 1H), 3.22 (d, J=8,0 Гц, 2Н), 0.99 (t, J=7,0 Гц, 6Н). LCMS [М+Н]+ = 460,1; [М-Н]- = 458,2.
Стадия D30.05: синтез 3-(2-(2-оксо-2,3-дигидро-1Н-бензимидазол-5-ил)-5-(пиридиний-3-ил)фенил)пропановой кислоты гидросульфата. В круглодонную колбу, оснащенную обратным холодильником, загружали 1,3-диэтил-2-(2-(8-оксо-7,9-диаза-бицикло [4.3.0]нона-1,3,5-триен-3-ил)-5-(пиридин-3-ил)-бензил)-пропандиоат (0,422 г; 0,918 ммоль) и серную кислоту (5 мл 2 М раствора в воде). Реакционную смесь перемешивали при температуре дефлегмации в течение 42 ч. Горячий раствор декантировали, отделяя от нерастворенного вещества, и оставляли медленно охлаждаться. Полученные твердые вещества собирали вакуумной фильтрацией, промывали водой (3×15 мл) и сушили на воздухе, получая указанное в заголовке соединение в виде беловатого порошка (0,305 г; 73%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 10.65 (s, 1H), 10.69 (s, 1H), 9.07 (s, 1H), 8.70 (d, J=3,9 Гц, 1H), 8.42 (d, J=7,4 Гц, 1H), 7.79-7.70 (m, 2Н), 7.66 (dd, J=1,9; 7,9 Гц, 1H), 7.32 (d, J=8,0 Гц, 1H), 7.01 (d, J=8,0 Гц, 1H), 6.94-6.83 (m, 2Н), 2.93-2.85 (m, 2Н), 2.48-2.43 (m, 2Н). LCMS [М+Н]+ = 360,1; [М-Н]- = 357,9.
Стадия D30.06: синтез 3-(2-(2-оксо-2,3-дигидро-1Н-бензимидазол-5-ил)-5-(пиридин-3-ил)фенил)пропанамида (D30). В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали 3-(2-(2-оксо-2,3-дигидро-1H-бензимидазол-5-ил)-5-(пиридиний-3-ил)фенил)пропановой кислоты гидросульфат (0,150 г; 0,328 ммоль) и безводный тетрагидрофуран (10 мл). Добавляли карбонилдиимидазол (0,106 г; 0,656 ммоль), триэтиламин (0,046 мл; 0,328 ммоль) и диметилацетамид (2 мл) и реакционную смесь перемешивали при комнатной температуре в течение ночи. По данным LCMS анализа реакция прошла не до конца, поэтому добавляли безводный ацетонитрил (20 мл) и реакционную смесь перемешивали при температуре дефлегмации в течение 1-2 ч. Реакционную смесь охлаждали до комнатной температуры и добавляли аммиак (4 мл 0,5 М раствора в диоксане; 2,00 ммоль). Реакционную смесь перемешивали при 40-45°С и выполняли еще 3 добавления аммиака (1 мл 0,5 М раствора в диоксане; 0,500 ммоль) в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Остаток разбавляли водой (25 мл) и значение рН доводили примерно до 10, используя раствор карбоната натрия. Смесь перемешивали и охлаждали в ледяной бане в течение 15 мин, полученное твердое вещество собирали фильтрованием и сушили на воздухе. Твердое вещество очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде беловатого порошка (0,036 г; 31%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 10.64 (s, 2Н), 8.90-8.95 (m, 1H), 8.58 (dd, J=4,7; 1,6 Гц, 1H), 8.05-8.13 (m, 1H), 7.67 (d, J=2,0 Гц, 1H), 7.58 (dd, J=7,8; 2,0 Гц, 1H), 7.50 (ddd, J=8,0; 4,7; 0,8 Гц, 1H), 7.28 (d, J=7,8 Гц, 1H), 7.22 (bs, 1H), 6.99 (d, J=7,8 Гц, 1H), 6.88-6.93 (m, 1H), 6.87 (s, 1H), 6.73 (bs, 1H), 2.81-2.89 (m, 2Н), 2.27-2.34 (m, 2Н). LCMS [М+Н]+ = 359,2; [М-Н]- = 357,1.
Синтез D32 показан ниже:
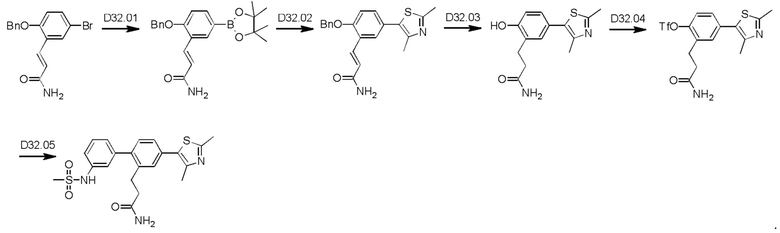
Стадия D32.01: синтез (2Е)-3-(2-(бензилокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)проп-2-енамида. В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали (2E)-3-[2-(бензилокси)-5-бромфенил]проп-2-енамид (2,0 г; 6,0 ммоль), бис(пинаколато)дибор (1,168 г; 6,62 ммоль), ацетат калия (2,36 г; 24,1 ммоль) и безводный 1,4-диоксан (60 мл). Смесь перемешивали и продували, барботируя через нее азот в течение 5 мин, затем добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II)-дихлорметан (0,492 г; 0,603 ммоль) и реакционную смесь перемешивали при температуре дефлегмации в течение 3 ч и затем при комнатной температуре в течение ночи. Смесь охлаждали до комнатной температуры, разбавляли этилацетатом (200 мл) и водой (125 мл), фильтровали и переносили в делительную воронку. Водный слой отделяли и далее экстрагировали этилацетатом (50 мл). Органические экстракты объединяли, промывали рассолом (25 мл), сушили над сульфатом магния, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (дихлорметан/ацетон), получая указанное в заголовке соединение в виде неочищенного темного масла (3,33 г). LCMS [M+H]+ = 380,26.
Стадия D32.02: синтез (2Е)-3-(2-(бензилокси)-5-(2,4-диметил-1,3-тиазол-5-ил)фенил)проп-2-енамида. В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали (2E)-3-(2-(бензилокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)проп-2-енамид (2,18 г; 5,73 ммоль), 5-бром-2,4-диметил-1,3-тиазол (1,10 г; 5,73 ммоль), карбонат цезия (3,74 г; 11,5 ммоль), 1,4-диоксан (25 мл) и воду (2,5 мл). Смесь перемешивали и продували, барботируя через нее азот в течение 5 мин, затем добавляли комплекс [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II)-дихлорметан (0,234 г; 0,287 ммоль) и реакционную смесь перемешивали при температуре дефлегмации в течение 2 ч. Смесь охлаждали до комнатной температуры и разбавляли этилацетатом (100 мл) и водой (50 мл). Смесь фильтровали, переносили в делительную воронку, водный слой отделяли и далее экстрагировали этилацетатом (50 мл). Органические экстракты объединяли, промывали рассолом (25 мл), сушили над сульфатом магния, фильтровали и концентрировали. Остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде коричневого порошка (0,890 г; 43%), содержащего неизвестные примеси. 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 7.72 (d, J=16,02 Гц, 1H), 7.56 (d, J=2,34 Гц, 1H), 7.45-7.52 (m, 3Н), 7.32-7.45 (m, 4Н), 7.23 (d, J=8,60 Гц, 1H), 7.10 (bs, 1H), 6.68 (d, J=16,02 Гц, 1H), 5.26 (s, 2Н), 2.61 (s, 3Н), 2.36 (s, 3Н). LCMS [М+Н]+ = 365,2; [M+Na]+ = 387,1.
Стадия D32.03: синтез 3-(5-(2,4-диметил-1,3-тиазол-5-ил)-2-гидроксифенил)пропанамида. В работающий под высоким давлением реактор загружали (2E)-3-(2-(бензилокси)-5-(2,4-диметил-1,3-тиазол-5-ил)фенил)проп-2-енамид (0,675 г; 2,44 ммоль), метанол (50 мл) и раствор аммиака (5 мл 25%-ного водного раствора). Смесь перемешивали и продували, барботируя через нее азот в течение 5 мин. К реакционной смеси добавляли 10%-ный палладий на угле (0,100 г), промывали сильной струей водорода и перемешивали в атмосфере водорода (20 бар (2 МПа)) при комнатной температуре в течение 4 суток. Смесь фильтровали через набивку целита и целит промывали горячим метанолом (3×50 мл). Фильтраты объединяли и концентрировали. Остаток очищали флэш-хроматографией (дихлорметан/метанол), получая указанное в заголовке соединение в виде бледно-коричневого твердого вещества (0,211 г; 31%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.68 (s, 1H), 7.29 (bs, 1H), 7.12 (d, J=2,34 Гц, 1H), 7.07 (dd, J=8,30; 2,44 Гц, 1H), 6.84 (d, J=8,21 Гц, 1H), 6.76 (bs, 1H), 2.75 (t, J=7,62 Гц, 2Н), 2.58 (s, 3Н), 2.35 (t, J=7,62 Гц, 2Н), 2.32 (s, 3Н). LCMS [М+Н]+ = 277,2; [M+Na]+ = 299,1; [М-Н]- = 275,1.
Стадия D32.04: синтез 2-(3-амино-3-оксопропил)-4-(2,4-диметил-1,3-тиазол-5-ил)фенил-трифторметансульфоната. В круглодонную колбу в атмосфере азота загружали 3-(5-(2,4-диметил-1,3-тиазол-5-ил)-2-гидроксифенил)пропанамид (0,211 г; 0,764 ммоль), ацетонитрил (10 мл) и карбонат калия (0,317 г; 2,29 ммоль). Реакционную смесь перемешивали в течение 15 мин, затем добавляли N-фенил-бис(трифторметансульфонимид) (0,286 г; 0,802 ммоль) и перемешивание продолжали в течение 4,5 ч при комнатной температуре. Реакционную смесь концентрировали и остаток распределяли между смесью дихлорметан/метанол (9:1, 50 мл) и промывали водой (20 мл). Водную фазу отделяли и экстрагировали смесью дихлорметан/метанол (9:1, 25 мл). Органические экстракты объединяли, промывали рассолом (15 мл), сушили над сульфатом магния, фильтровали и концентрировали, получая указанное в заголовке соединение в виде беловатого твердого вещества (0,338 г; колич.). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 7.53 (s, 1H), 7.44-7.48 (m, 2Н), 7.34 (bs, 1H), 6.84 (bs, 1H), 2.92 (t, J=7,42 Гц, 2Н), 2.63 (s, 3Н), 2.42-2.48 (m, 2Н), 2.39 (s, 3Н). LCMS [М+Н]+ = 409,0.
Стадия D32.05: синтез 4-(2,4-диметил-1,3-тиазол-5-ил)-3-(3'-((метилсульфонил)амино)бифенил-2-ил)пропанамида (D32). В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали 2-(3-амино-3-оксопропил)-4-(2,4-диметил-1,3-тиазол-5-ил)фенил-трифторметансульфонат (0,145 г; 0,355 ммоль), 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-N-фенилметансульфонамид (0,116 г; 0,390 ммоль), карбонат калия (0,147 г; 1,06 ммоль), 1,4-диоксан (1,75 мл), этанол (0,35 мл) и воду (0,35 мл). Реакционную смесь перемешивали и продували, барботируя через нее азот в течение 5 мин, затем добавляли тетракис(трифенилфосфин)палладий (0,021 г; 0,018 ммоль). Реакционную смесь перемешивали при 85°С в течение 2 ч, охлаждали до комнатной температуры и разбавляли этилацетатом (20 мл). Добавляли воду (5 мл) и рН водной фазы доводили до 5-6, используя 2 М раствор соляной кислоты. Эти два слоя тщательно перемешивали и водный слой отделяли. Органические слои промывали рассолом (10 мл), сушили над сульфатом магния, фильтровали и концентрировали. Остаток перемешивали в смеси этилацетат/гексаны (3:1, 40 мл) и осторожно нагревали. Через 15 мин суспензию охлаждали в ледяной бане и твердые вещества собирали фильтрованием, промывали смесью этилацетат/гексаны (1:1, 2×5 мл) и сушили, получая указанное в заголовке соединение в виде коричневого порошка (0,067 г; 32%). 1H ЯМР (400 МГц, DMSO-d6) δ млн-1 9.85 (bs, 1H), 7.36-7.45 (m, 2Н), 7.30-7.36 (m, 1H), 7.19-7.27 (m, 3Н), 7.17 (s, 1H), 7.08 (d, J=7,81 Гц, 1H), 6.73 (bs, 1H), 3.02 (s, 3Н), 2.79 (t, J=7,72 Гц, 2Н), 2.63 (s, 3Н), 2.42 (s, 3Н), 2.27 (t, J=7,72 Гц, 2Н). LCMS [М+Н]+ = 430,1; [М-Н]- = 428,2.
Схема синтеза D35 показана ниже:
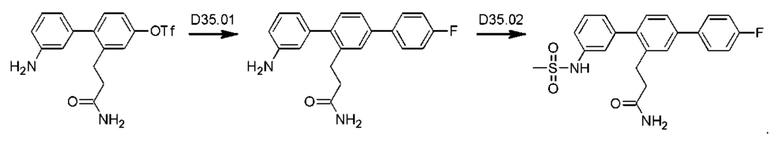
Стадия D35.01: синтез 3-(3-амино-4''-фтор-1,1':4':1''-терфенил-2'-ил)пропанамида. С применением способа, описанного на стадии D28.06, и 4-фторбензолбороновой кислоты получали указанное в заголовке соединение в виде беловатого порошка (0,064 г; 69%). 1Н ЯМР (400 МГц, CDCl3) δ млн-1 7.53-7.61 (m, 2Н), 7.48 (d, J=1,8 Гц, 1H), 7.43 (dd, J=8,0; 2,0 Гц, 1H), 7.28 (d, J=7,8 Гц, 1H), 7.21 (t, J=7,7 Гц, 1H), 7.09-7.17 (m, 2Н), 6.64-6.76 (m, 3Н), 5.15 (bs, 2Н), 3.00-3.10 (m, 2Н), 2.30-2.40 (m, 2Н). LCMS [М+Н]+ = 335,1; [M+Na]+ = 357,1; [М+HCO2]- = 379,1.
Стадия D35.02: синтез 3-(4''-фтор-3-((метилсульфонил)амино)-1,1':4',1''-терфенил-2'-ил)пропанамида (D35). В круглодонную колбу в атмосфере азота загружали 3-(3-амино-4''-фтор-1,1':4':1''-терфенил-2'-ил)пропанамид (0,064 г; 0,191 ммоль), дихлорметан (5 мл) и триэтиламин (0,106 мл; 0,766 ммоль). Реакционную смесь охлаждали в ледяной бане в течение 15 мин, затем добавляли метансульфонилхлорид (0,045 мл; 0,573 ммоль). Реакционную смесь перемешивали в течение 3 ч в ледяной бане, затем оставляли нагреваться до комнатной температуры и перемешивали в течение 48 ч. Реакционную смесь наносили непосредственно на диоксид кремния и очищали флэш-хроматографией (дихлорметан/метанол), получая частично очищенный продукт в виде смолы. Неочищенный продукт перемешивали в смеси дихлорметан/гексаны (1:1) (25 мл) в течение нескольких часов и полученное твердое вещество собирали фильтрованием, промывали смесью дихлорметан/гексаны (1:1) (2×10 мл) и сушили под высоким вакуумом, получая указанное в заголовке соединение в виде беловатого порошка (0,036 г; 46%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.84 (s, 1H), 7.70-7.78 (m, 2Н), 7.61 (d, J=1,8 Гц, 1H), 7.53 (dd, J=7,8; 2,0 Гц, 1H), 7.39-7.46 (m, 1H), 7.28-7.36 (m, 2Н), 7.15-7.28 (m, 4Н), 7.09 (d, J=7,6 Гц, 1H), 6.73 (bs, 1H), 3.03 (s, 3Н), 2.78-2.85 (m, 2H), 2.31 (dd, J=9,0; 7,0 Гц, 2H). LCMS [M+H]+ = 413,2; [M+Na]+ = 435,1; [M-H]- = 411,2.
Структура D167 показана ниже:
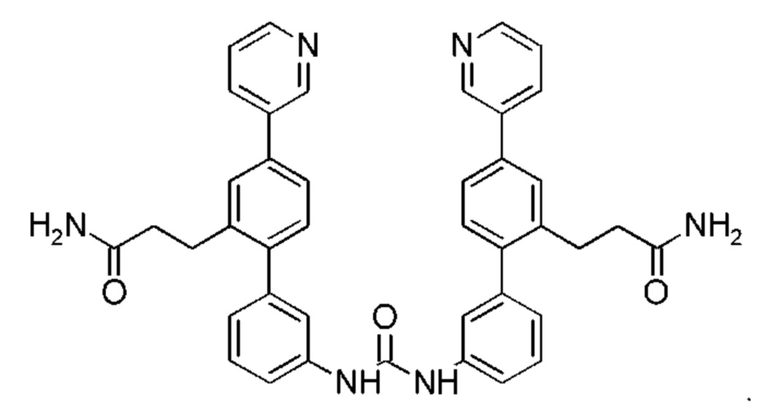
Синтез 3-(2-(3-((3-(2-(2-карбамоилэтил)-4-(пиридин-3-ил)фенилфениламино)формиламино)фенил)-5-(пиридин-3-ил)-фенил)пропенамида (D167). Выделяли в ходе синтезе соединения D29 после очистки флэш-хроматографией в виде медленнее элюирующегося соединения. Указанное в заголовке соединение получали в виде белого порошка (0,046 г; 11%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.00 (s, 2Н), 8.93 (dd, J=2,34; 0,78 Гц, 2Н), 8.59 (dd, J=4,79; 1,66 Гц, 2Н), 8.07-8.13 (m, 2Н), 7.69 (d, J=1,95 Гц, 2Н), 7.61 (dd, J=7,81; 1,95 Гц, 2Н), 7.48-7.55 (m, 4Н), 7.33-7.40 (m, 2Н) 7.45 (d, J=9,18 Гц, 2Н), 7.30 (d, J=7,82 Гц, 2Н), 7.24 (br. s., 2Н), 6.97 (d, J=7,62 Гц, 2Н), 6.74 (br. s., 2Н), 2.81-2.90 (m, 4Н), 2.29-2.38 (m, 4Н). LCMS [М+Н]+ = 661,3.
Схема синтеза D171 показана ниже:
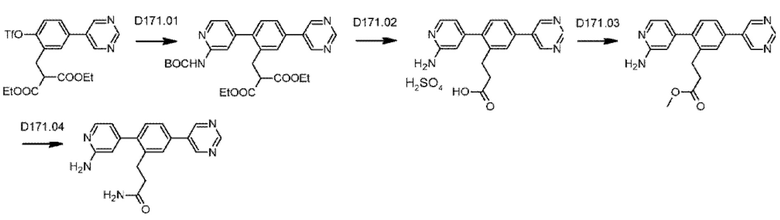
Стадия D171.01: синтез 1,3-диэтил-2-(2-(2-((1,1-диметилэтокси) формиламино)пиридин-4-ил)-5-(пиримидин-5-ил)бензил)пропандиоата. В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали 1,3-диэтил-2-(((2-трифторметил)сульфонил)оксо)-5-(пиримидин-5-ил)бензил)пропандиоат (0,400 г; 0,840 ммоль), (1,1-диметилэтокси)(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-иламино)метанон (0,296 г; 0,924 ммоль), карбонат калия (0,232 г; 1,68 ммоль), 1,4-диоксан (3,2 мл), этанол (0,8 мл) и воду (0,8 мл). Реакционную смесь продували, барботируя через нее азот в течение 2 мин, затем добавляли тетракис(трифенилфосфин)палладий (0,097 г; 0,084 ммоль) и реакционную смесь перемешивали при 85°С в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом (25 мл) и водой (15 мл). Смесь переносили в делительную воронку, водную фазу отделяли и далее экстрагировали этилацетатом (25 мл). Органические экстракты объединяли, промывали рассолом (10 мл), сушили (сульфат магния), фильтровали и концентрировали. После очистки флэш-хроматографией (этилацетат/гексаны) получали указанное в заголовке соединение в виде желтого твердого вещества (0,316 г; 72%). LCMS [М+Н]+ = 521,3.
Стадия D171.02: синтез 3-(2-(2-аминопиридин-4-ил)-5-(пиримидин-5-ил)фенил)пропановой кислоты гидросульфата. В круглодонную колбу, оснащенную обратным холодильником, загружали 1,3-диэтил-2-(2-(2-((1,1-диметилэтокси)формиламино)пиридин-4-ил)-5-(пиримидин-5-ил)бензил)пропандиоат (0,406 г; 0,780 ммоль) и серную кислоту (5 мл 2 М водного раствора). Реакционную смесь перемешивали при температуре дефлегмации в течение 24 ч, затем охлаждали и концентрировали. Значение рН доводили до 4-5 путем добавления раствора карбоната натрия и смесь перемешивали в течение 2 ч. Полученный желтый осадок собирали фильтрованием, промывали водой (3×10 мл) и сушили на воздухе, получая указанное в заголовке соединение в виде желтого твердого вещества (0,145 г; 44%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.21 (s, 1H), 9.20 (s, 2Н), 7.99 (d, J=5,67 Гц, 1H), 7.85 (d, J=1,76 Гц, 1H), 7.75 (dd, J=8,01; 1,95 Гц, 1H), 7.34 (d, J=8,01 Гц, 1H), 6.66 (d, J=4,88 Гц, 1H), 6.59 (s, 1H), 2.84-2.92 (m, 2Н), 2.52-2.58 (m, 2Н). LCMS [М]+ = 320,0.
Стадия D171.03: синтез метил-3-(2-(2-аминопиридин-4-ил)-5-(пиримидин-5-ил)фенил)пропаноата. В круглодонную колбу, оснащенную обратным холодильником, загружали 3-(2-(2-аминопиридин-4-ил)-5-(пиримидин-5-ил)фенил)пропановой кислоты гидросульфат (0,145 мг; 0,400 ммоль), метанол (75 мл) и соляную кислоту (1 мл 2 М водного раствора) и нагревали с обратным холодильником в течение 3 ч. Реакционную смесь фильтровали в горячем состоянии, остаток промывали метанолом (2×15 мл) и фильтраты объединяли и концентрировали. Остаток распределяли между этилацетатом (50 мл) и раствором карбоната натрия (50 мл). Водную фазу отделяли и далее экстрагировали этилацетатом (50 мл). Органические экстракты объединяли, промывали рассолом (20 мл), сушили над сульфатом магния и концентрировали досуха, получая указанное в заголовке соединение в виде желтого твердого вещества (0,048 г; 36%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.20 (s, 1H), 9.19 (s, 2Н), 7.97 (d, J=5,08 Гц, 1H), 7.82 (d, J=1,76 Гц, 1H), 7.72 (dd, J=7,81; 1,95 Гц, 1H), 7.30 (d, J=8,01 Гц, 1H), 6.47 (dd, J=5,18; 1,47 Гц, 1H), 6.38 (s, 1H), 6.02 (s, 2Н), 3.55 (s, 3Н), 2.87-2.94 (m, 2Н), 2.58-2.65 (m, 2Н). LCMS [М+Н]+ = 335,2.
Стадия D171.04: синтез 3-[2-(2-аминопиридин-4-ил)-5-(пиримидин-5-ил)фенил]пропенамида (D171). Выполняли согласно стадии D6.11, получая указанное в заголовке соединение в виде беловатого порошка (0,045 г; 41%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.21 (s, 1H), 9.18 (s, 2Н), 7.96 (d, J=5,08 Гц, 1H), 7.77 (d, J=1,76 Гц, 1H), 7.70 (dd, J=7,91; 1,86 Гц, 1H), 7.29 (d, J=7,82 Гц, 1H), 7.25 (ушир. s., 1H), 6.76 (ушир. s., 1H), 6.48 (dd, J=5,18; 1,47 Гц, 1H), 6.39 (s, 1H), 6.00 (s, 2Н), 2.81-2.89 (m, 2Н), 2.36 (dd, J=8,89; 6,94 Гц, 2Н). LCMS [М+Н]+ = 320,2.
Схема синтеза D172 показана ниже:
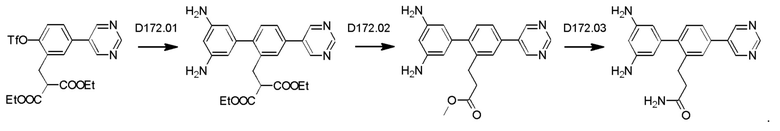
Стадия D172.01: синтез 1,3-диэтил-2-(2-(3,5-диаминофенил)-5-(пиримидин-5-ил)бензил)пропандиоата. В круглодонную колбу, оснащенную обратным холодильником, в атмосфере азота загружали 1,3-диэтил-2-(((2-трифторметил)сульфонил)оксо)-5-(пиримидин-5-ил)бензил)пропандиоат (0,157 г; 0,329 ммоль), 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензол-1,3-диамин (0,077 г; 0,329 ммоль), карбонат калия (0,091 г; 0,658 ммоль), 1,4-диоксан (2 мл), этанол (0,5 мл) и воду (0,5 мл). Реакционную смесь продували, барботируя через нее азот в течение 2 мин, затем добавляли тетракис(трифенилфосфин)палладий (0,038 г; 0,033 ммоль) и реакционную смесь перемешивали при 85°С в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом (50 мл) и водой (25 мл) и подщелачивали, используя карбонат натрия, до рН выше 10. Смесь переносили в делительную воронку, органическую фазу отделяли и промывали рассолом (10 мл), сушили (сульфат магния), фильтровали и концентрировали. После очистки флэш-хроматографией (этилацетат/гексаны) получали указанное в заголовке соединение в виде желтого твердого вещества (0,118 г; 82%). 1H ЯМР (400 МГц, ХЛОРОФОРМ-d) δ млн-1 9.21 (s, 0,5Н), 8.95 (s, 1,5Н), 7.63-7.71 (m, 0,5Н), 7.54 (d, J=4,88 Гц, 0,5Н), 7.42-7.50 (m, 2,5Н), 7.34 (d, J=8,40 Гц, 1H), 6.02-6.10 (m, 2,5Н), 4.09 (q, J=6,32 Гц, 4Н), 3.64 (ушир. s., 3Н), 3.54 (t, J=7,91 Гц, 1H), 3.36 (d, J=7,62 Гц, 2Н), 1.26 (t, J=7,03 Гц, 1H), 1.15 (t, J=7,13 Гц, 6Н). LCMS [М+Н]+ = 435,2.
Стадия D172.02: синтез метил-3-(2-(3,5-диамино-фенил)-5-(пиримидин-5-ил)фенил)пропаноата. Выполняли согласно стадии D6.10, получая указанное в заголовке соединение в виде янтарной смолы (0,105 г; 90%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.18 (s, 1H), 9.17 (s, 2Н), 7.72 (d, J=1,76 Гц, 1H), 7.51-7.58 (m, 1H), 7.22 (d, J=7,82 Гц, 1H), 5.83 (t, J=1,95 Гц, 1H), 5.73 (d, J=1,95 Гц, 2Н), 4.81 (s, 4Н), 3.56 (s, 3Н), 2.89-2.97 (m, 2Н), 2.54-2.61 (m, 2Н). LCMS [М+Н]+ = 349,2.
Стадия D172.03: синтез 3-(2-(3,5-диаминофенил)-5-(пиримидин-5-ил)фенил)пропенамида (D172). Выполняли согласно стадии D6.11, получая указанное в заголовке соединение в виде бежевого порошка (0,038 г; 38%). 1Н ЯМР (400 МГц, DMSO-d6) δ млн-1 9.18 (s, 1H), 9.15 (s, 2Н), 7.70 (d, J=1,76 Гц, 1H), 7.62 (dd, J=7,81; 1,95 Гц, 1H), 7.22 (d, J=8,01 Гц, 2Н), 6.75 (br. s., 1H), 5.84 (t, J=1,86 Гц, 1H), 5.74 (d, J=1,95 Гц, 2Н), 4.79 (s, 4Н), 2.82-2.92 (m, 2Н), 2.28-2.39 (m, 2Н). LCMS [М+Н]+ = 334,2.
Пример 2. Скрининг соединений in vivo
Спонтанно гипертензивных крыс (SHR) в возрасте четырнадцати недель подвергали анестезии, используя 3%-ный изофлуран, язык осторожно вытягивали вперед и с использованием микропипетки в дистальную зону ротоглотки наносили блеомицин в дозе 10 ед./100 г массы тела в 200 мкл обычного физиологического раствора, при этом нос остается слегка закрытым. Затем крыс оставляют оправиться от анестезии. SHR в возрасте 16 недель рандомизировали на контрольные (16 недель или 20 недель) или подвергаемые обработке группы. 16-недельных контрольных животных подвергали анестезии, проводили отбор образцов крови и сбор тканей. 20-недельные контрольные животные получали только питьевой раствор (5%-ный этанол), в то время как подвергаемые обработке группы получали тестируемое соединение, растворенное в питьевой воде, при этом концентрацию доводили два раза в неделю для поддержания дозы 500 пмоль/кг/мин в течение 4 недель. Крыс, достигших возраста 20 недель, подвергали анестезии, проводили отбор образцов крови и сбор тканей.
Для проведения гистологического исследования выполняли фиксацию легких в формалине и срезы для каждого их двух уровней окрашивали, используя трихром по Массону. Количественная оценка фиброза проводилась методом гистоморфометрии наблюдателем, не допущенным к рандомизационным кодам экспериментальной группы.
Степень фиброза легкого после 4 недельной обработки соединениями A32, А79, А6, VB0004, Р13, D30, D6 и А81 в дозе 500 пмоль/кг/мин уменьшалась по сравнению с 20-недельными контролями (Фиг. 1 и 2), и это демонстрирует, что данные соединения предотвращают развитие фиброза легких.
Степень фиброза легкого после 4 недельной обработки соединениями A32, А79, А6, VB0004, D30, D6 и А81 в дозе 500 пмоль/кг/мин уменьшалась по сравнению с 16-недельными контролями (Фиг. 1 и 2), и это демонстрирует, что данные соединения реверсируют диагностированный фиброз легких.
Пример 3. Скрининг соединений in vitro
Для измерения изменений в клеточном импедансе (клеточном индексе) после обработки эпителиальных клеток малых дыхательных путей человека (АТСС (Американская коллекция типовых культур)) тестируемым соединением использовали систему xCELLigence SP (Roche). В этой основанной на использовании клеток экспериментальной системе in vitro было обнаружено, что отрицательный наклон графика импеданса коррелирует с ослаблением фиброза легких (Фиг. 3 и 4).
Кратко, по 50 мкл среды для культивирования клеток (модифицированной Дульбекко среды Игла (DMEM) с низким содержанием глюкозы, дополненной 15% фетальной телячьей сыворотки, при 37°С) добавляли в каждую лунку 96-луночного Е-планшета (E-Plate 96; Roche) и измеряли базовый уровень импеданса в каждой лунке. Затем в соответствующие лунки E-Plate 96 добавляли по 50 мкл суспензии эпителиальных клеток малых дыхательных путей человека (10000 клеток/лунка). Мониторинг клеточного индекса для каждой лунки E-Plate 96 проводили с использованием прибора RTCA SP (работающий в режиме реального времени клеточный анализатор для одного планшета) Station в инкубаторе для клеточных культур. После инкубирования ночью в течение 16-20 часов при 5% СО2 и влажности 95% в соответствующие лунки E-Plate 96 добавляли по 100 мкл раствора тестируемого соединения (готовили растворы тестируемых соединений в DMSO и разбавляли средой для культивирования клеток до концентрации тестируемого соединения 10 мкМ, 20 мкМ или 30 мкМ при конечной концентрации DMSO 0,25%) и значения клеточного индекса измеряли сразу после обработки соединением через каждые 20 секунд в течение 3 часов. Значение клеточного индекса корректируют относительно базовой линии путем вычитания значения клеточного индекса для обработанных разбавителем клеток и нормируют путем деления на значение клеточного индекса для момента времени непосредственно перед добавлением соединения. График зависимости нормированного к базовой линии клеточного индекса как функцию от времени строят, используя программное обеспечение RTCA от Roche.
Характеризующиеся отрицательным импедансом ответы в эпителиальных клетках малых дыхательных путей человека наблюдали для А6, А26, А27, А30, A32, A35, А56, А79, А81, D4, D5, D6, D10, D11, D12, D16, D17, D18, D28, D30, D31, D32, D35, D167, D171, D172, Р13, Р14, Р25, Р42, Р43, Р44 и VB0004 (Фиг. 5), и это указывает на то, что данные соединения ослабляют фиброз легких.
| название | год | авторы | номер документа |
|---|---|---|---|
| Композиции для лечения заболевания почек и/или печени | 2016 |
|
RU2712624C2 |
| Композиции для лечения фиброза и связанных с фиброзом состояний | 2016 |
|
RU2712140C2 |
| ПОЛИГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МЕТАБОТРОПНОГО РЕЦЕПТОРА ГЛУТАМАТА | 2005 |
|
RU2381226C2 |
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2021 |
|
RU2822180C1 |
| ПРОИЗВОДНЫЕ 1,3,4-ОКСАДИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2021 |
|
RU2810081C1 |
| Новые производные имидазо[4,5-c] хинолинов и имидазо[4,5-c][1,5] нафтиридинов в качестве ингибиторов LRRK2 | 2016 |
|
RU2722149C1 |
| НОВЫЙ СПОСОБ СИНТЕЗА АГОМЕЛАТИНА | 2014 |
|
RU2683279C1 |
| Композиции для лечения гипертензии и/или фиброза | 2014 |
|
RU2661878C2 |
| ПРОИЗВОДНЫЕ ТИПА АЗАИНДАЗОЛА ИЛИ ДИАЗАИНДАЗОЛА ДЛЯ ЛЕЧЕНИЯ БОЛИ | 2013 |
|
RU2640046C2 |
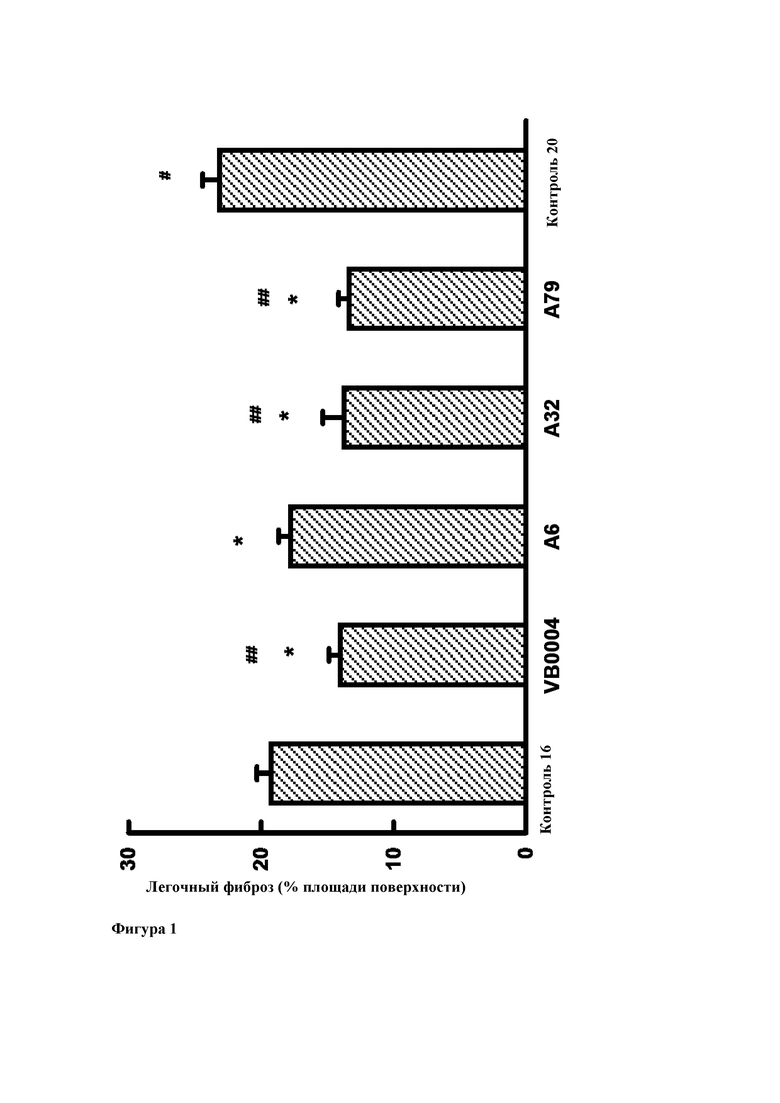
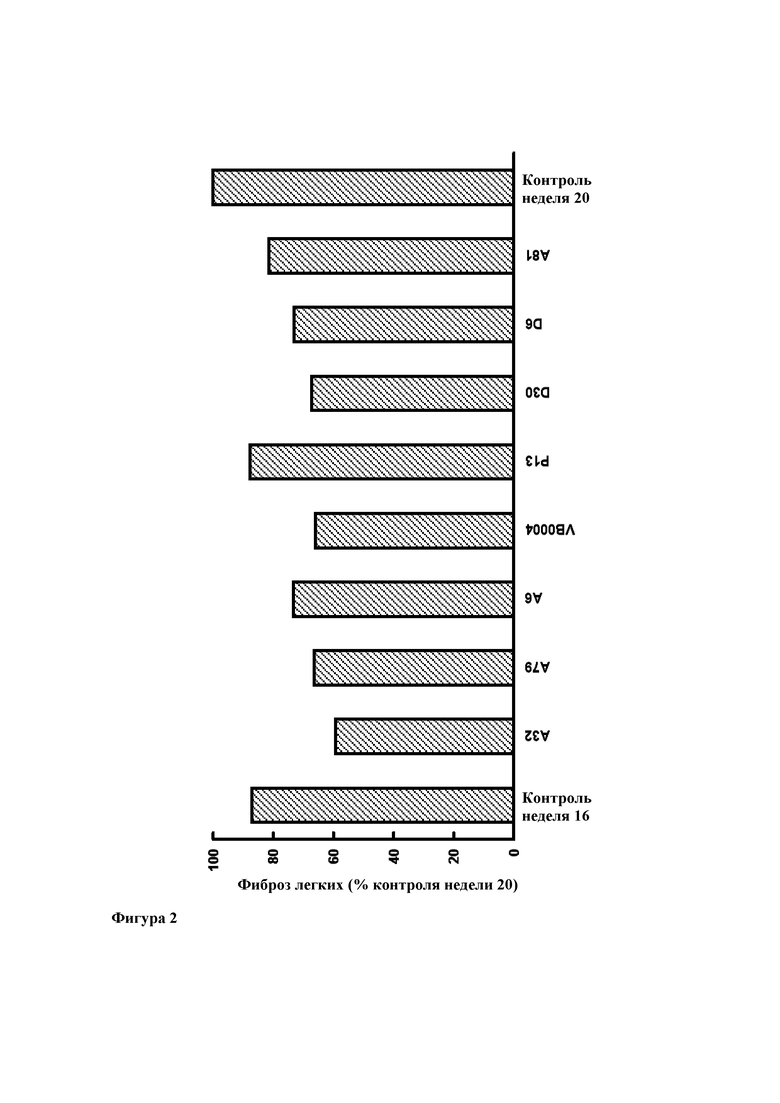
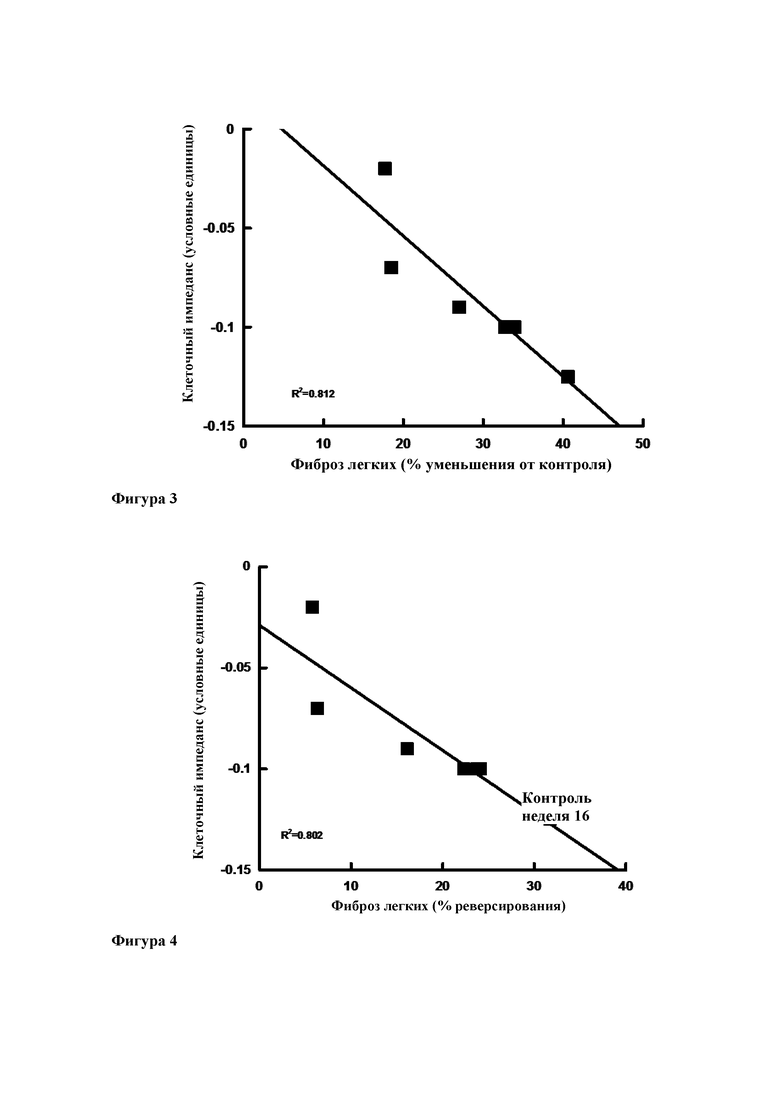
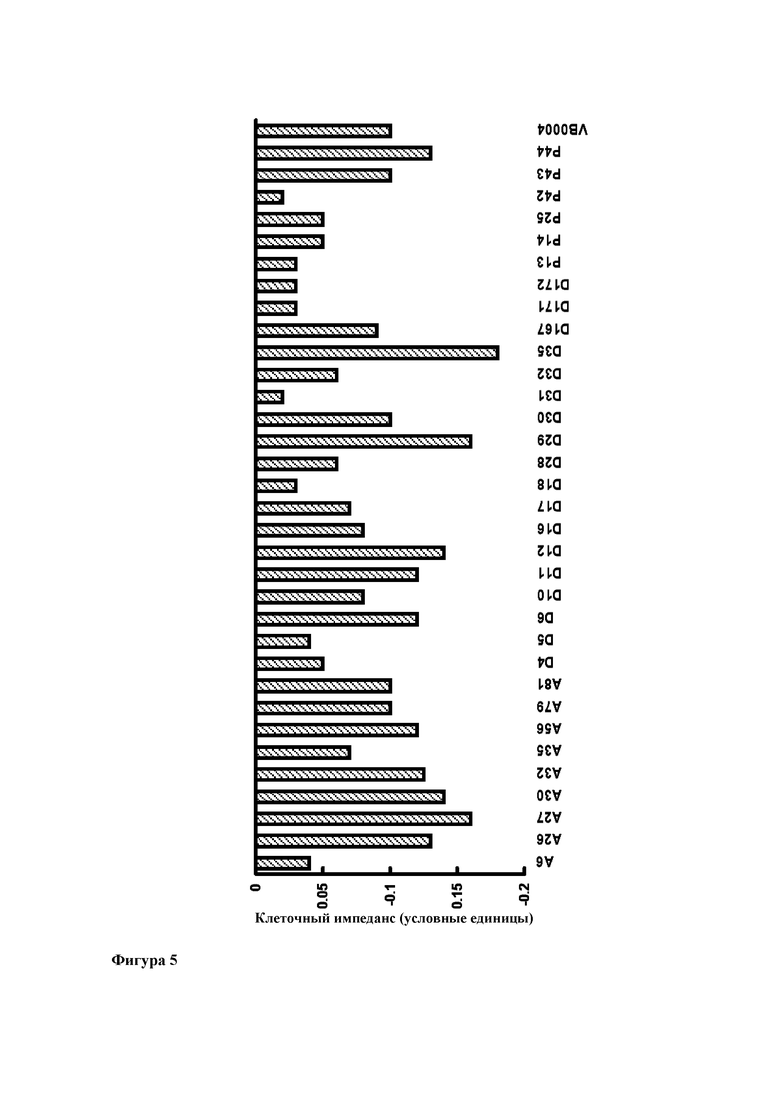
Настоящее изобретение относится к соединению формулы
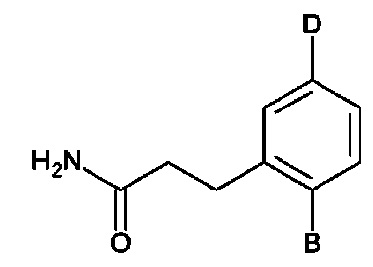 или его фармакологически приемлемой соли, где: В выбран из группы, состоящей из:
или его фармакологически приемлемой соли, где: В выбран из группы, состоящей из:
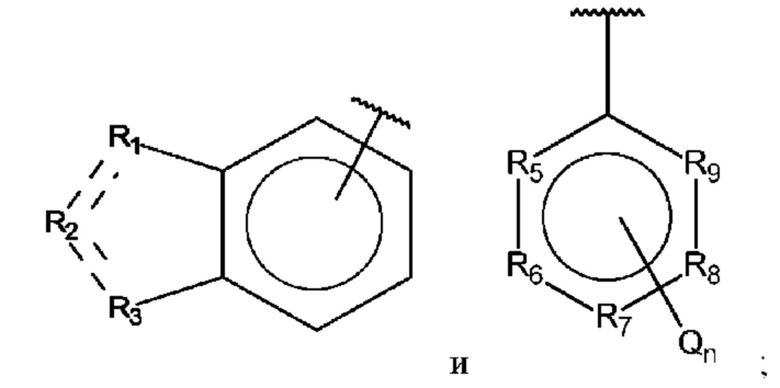 R1 и R3 независимо представляют собой СН или N; R2 представляет собой СН или С=O; R5-R9 независимо представляют собой С или N; Q независимо выбран из алкила, гидрокси, амино и замещенного амино, выбранного из группы, состоящей из -NHSO2CH3, -NHCOH, -NHCONHCH3, -NHCONHCH2CH3, -NHSO2NHCH3, -NHSO2NHCH2CH3 и -NHCOCH3; n равно 0, 1 или 2; D представляет собой:
R1 и R3 независимо представляют собой СН или N; R2 представляет собой СН или С=O; R5-R9 независимо представляют собой С или N; Q независимо выбран из алкила, гидрокси, амино и замещенного амино, выбранного из группы, состоящей из -NHSO2CH3, -NHCOH, -NHCONHCH3, -NHCONHCH2CH3, -NHSO2NHCH3, -NHSO2NHCH2CH3 и -NHCOCH3; n равно 0, 1 или 2; D представляет собой:
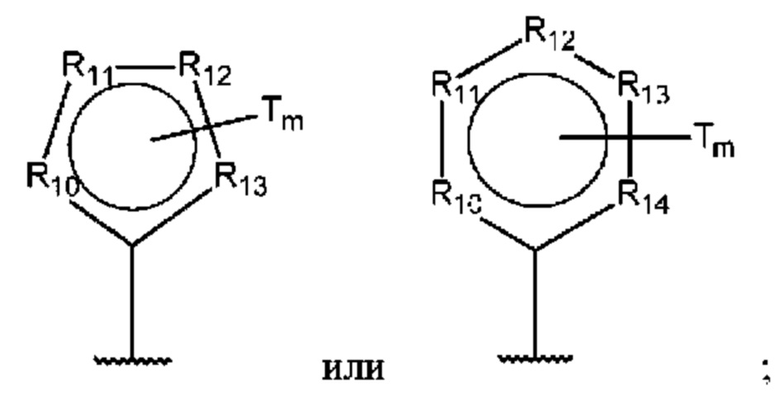 R10-R14 независимо представляют собой С, N или S; Т независимо выбран из С1-6алкила и галогена; и m равно 0, 1, 2, 3 или 4, при этом D не может представлять собой незамещенный фенил, и Q не может представлять собой гидрокси, когда n равно 1 и все R5-R9 представляют собой С. Кроме того, изобретение относится к соединению
R10-R14 независимо представляют собой С, N или S; Т независимо выбран из С1-6алкила и галогена; и m равно 0, 1, 2, 3 или 4, при этом D не может представлять собой незамещенный фенил, и Q не может представлять собой гидрокси, когда n равно 1 и все R5-R9 представляют собой С. Кроме того, изобретение относится к соединению 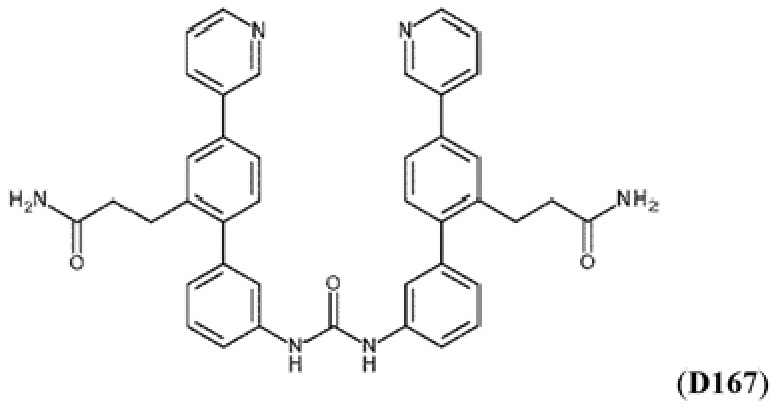 и его фармакологически приемлемой соли, к способу профилактического или терапевтического лечения легочного фиброза или связанного с ним состояния у субъекта с легочным фиброзом или с риском развития легочного фиброза, включающему введение вышеуказанных соединений, а также к применению данных соединений для изготовления лекарственного средства для профилактического или терапевтического лечения легочного фиброза. Технический результат: получены и описаны новые соединения, которые могут быть полезны в лечении легочного фиброза. 6 н. и 10 з.п. ф-лы, 5 ил., 3 пр.
и его фармакологически приемлемой соли, к способу профилактического или терапевтического лечения легочного фиброза или связанного с ним состояния у субъекта с легочным фиброзом или с риском развития легочного фиброза, включающему введение вышеуказанных соединений, а также к применению данных соединений для изготовления лекарственного средства для профилактического или терапевтического лечения легочного фиброза. Технический результат: получены и описаны новые соединения, которые могут быть полезны в лечении легочного фиброза. 6 н. и 10 з.п. ф-лы, 5 ил., 3 пр.
1. Соединение формулы:
или его фармакологически приемлемая соль,
где:
В выбран из группы, состоящей из:
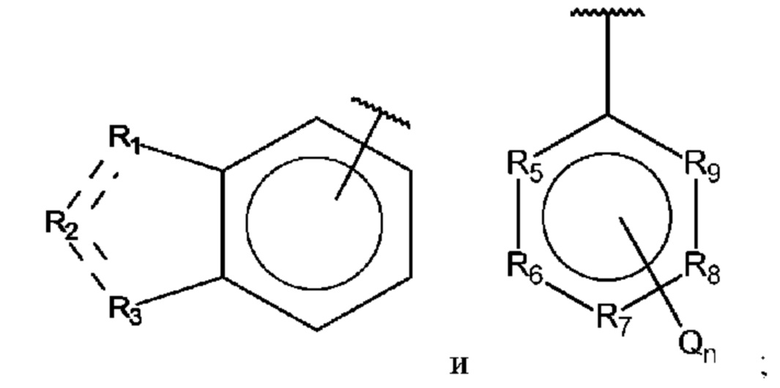
R1 и R3 независимо представляют собой СН или N;
R2 представляет собой СН или С=O;
R5-R9 независимо представляют собой С или N;
Q независимо выбран из алкила, гидрокси, амино и замещенного амино, выбранного из группы, состоящей из -NHSO2CH3, -NHCOH, -NHCONHCH3, -NHCONHCH2CH3, -NHSO2NHCH3, -NHSO2NHCH2CH3 и -NHCOCH3;
n равно 0, 1 или 2;
D представляет собой:
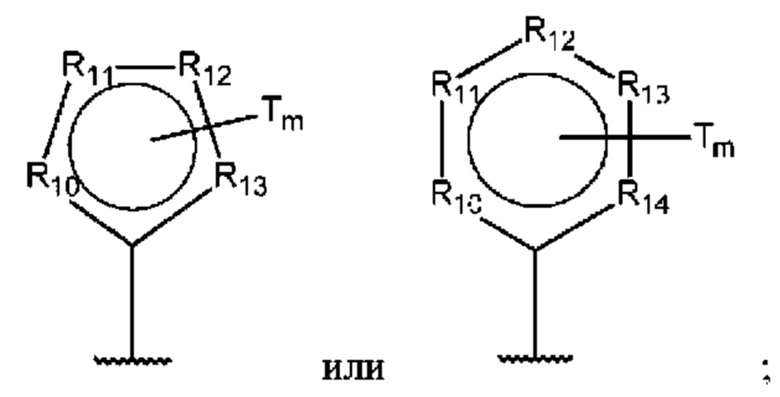
R10-R14 независимо представляют собой С, N или S;
Т независимо выбран из С1-6алкила и галогена; и
m равно 0, 1, 2, 3 или 4,
при этом D не может представлять собой незамещенный фенил, и Q не может представлять собой гидрокси, когда n равно 1 и все R5-R9 представляют собой С.
2. Соединение по п. 1, где соединение выбрано из группы, состоящей из:
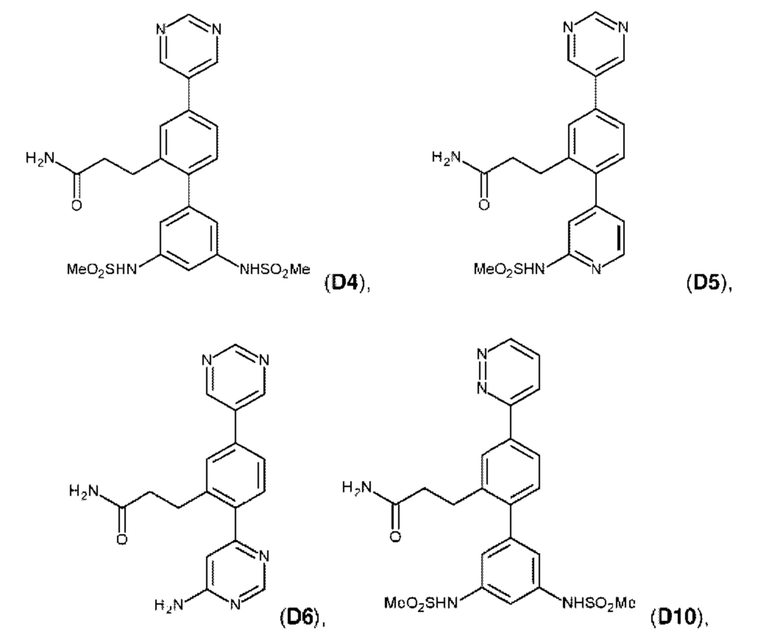
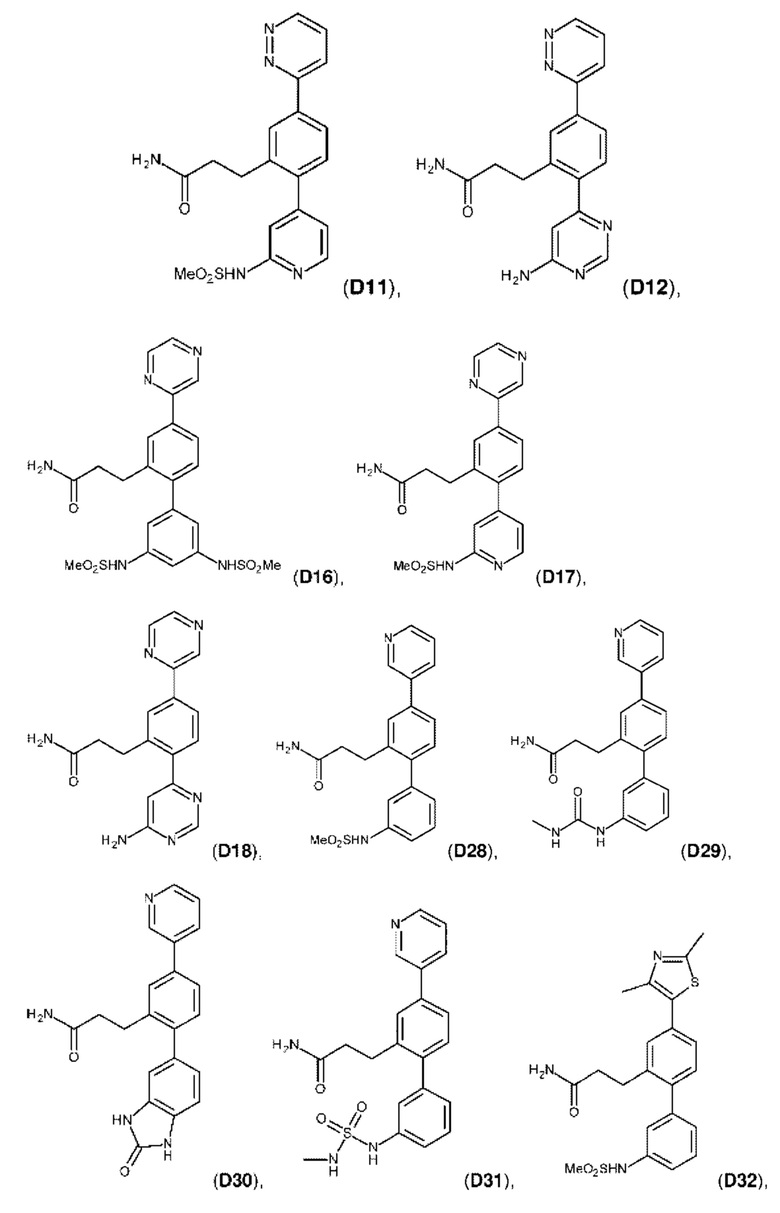
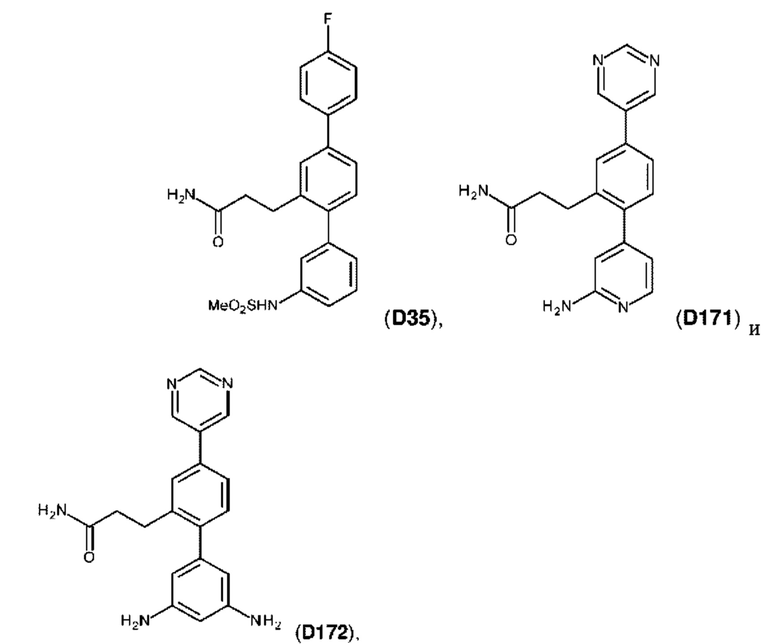
или их фармакологически приемлемой соли.
3. Соединение формулы:
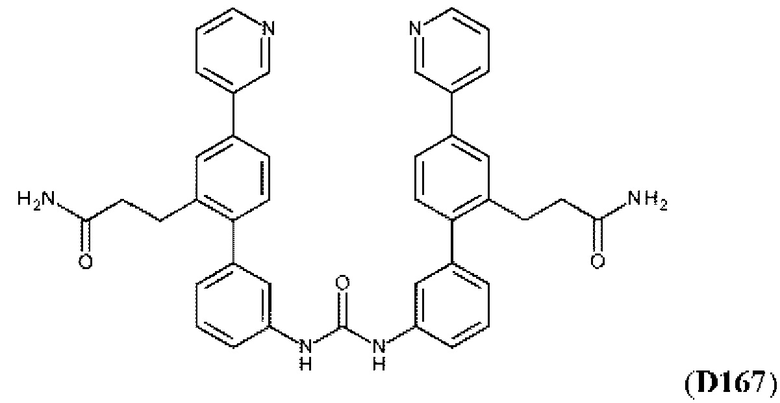
или его фармакологически приемлемая соль.
4. Способ профилактического или терапевтического лечения легочного фиброза или связанного с ним состояния у субъекта с легочным фиброзом или с риском развития легочного фиброза, включающий введение субъекту эффективного количества соединения по любому из пп. 1-3.
5. Способ профилактического или терапевтического лечения легочного фиброза или связанного с ним состояния у субъекта с легочным фиброзом или с риском развития легочного фиброза, включающий введение субъекту эффективного количества соединения формулы
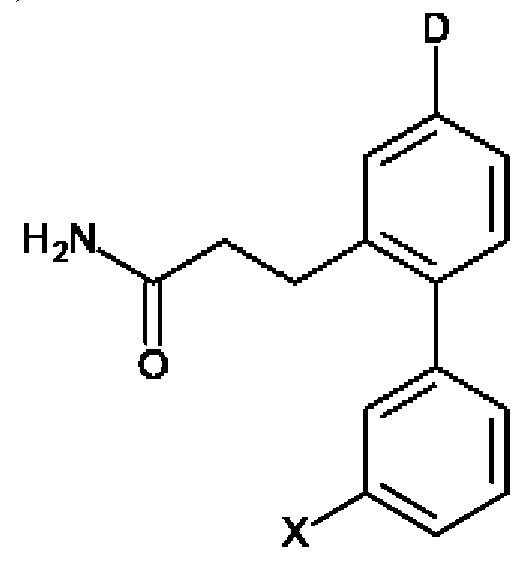
или его фармакологически приемлемой соли,
где:
D представляет собой:
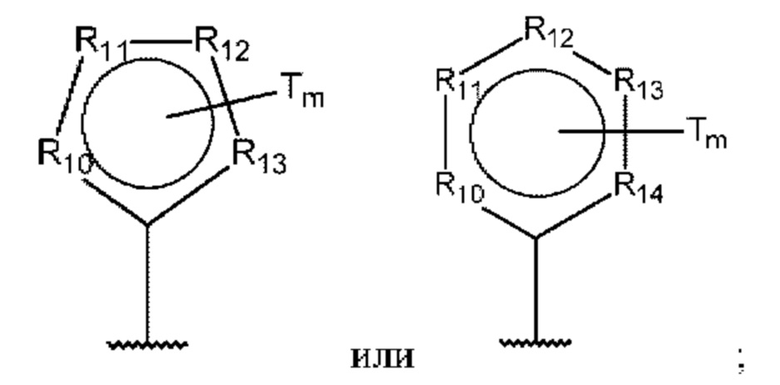
R10-R14 независимо представляют собой С, N или S;
Т независимо выбран из С1-6алкила и галогена;
m равно 0, 1, 2, 3 или 4; и
X представляет собой -ОН.
6. Способ по п. 5, где соединение выбрано из группы, состоящей из:
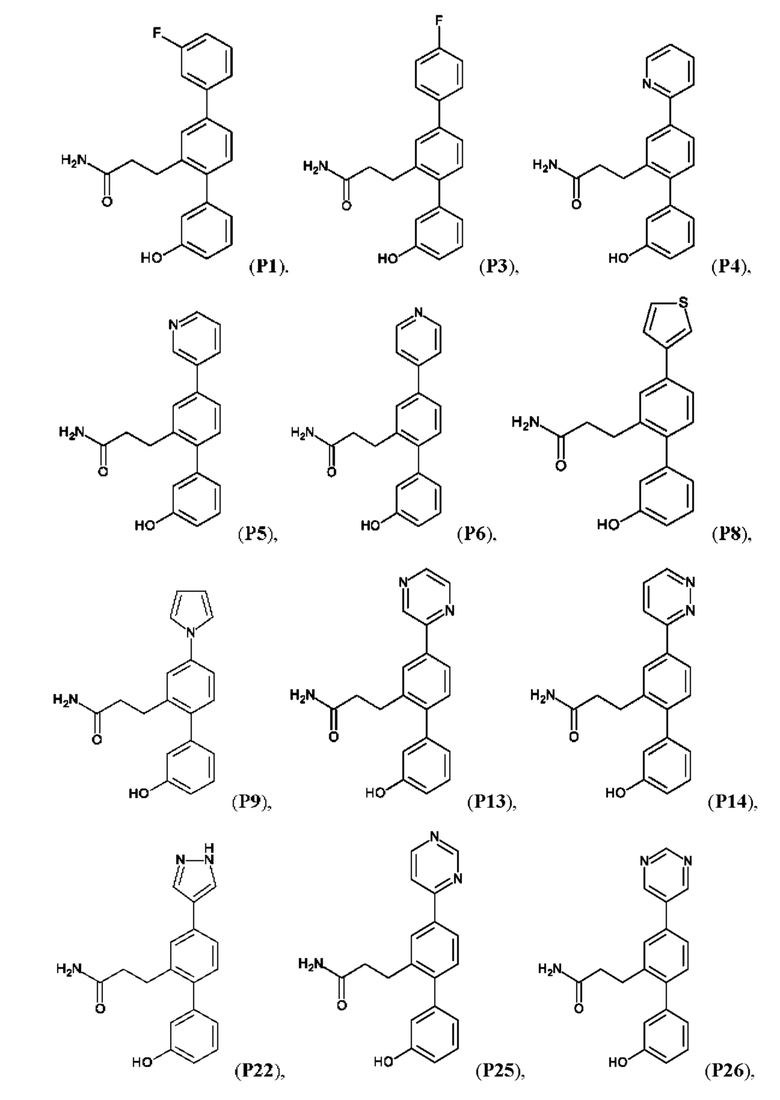

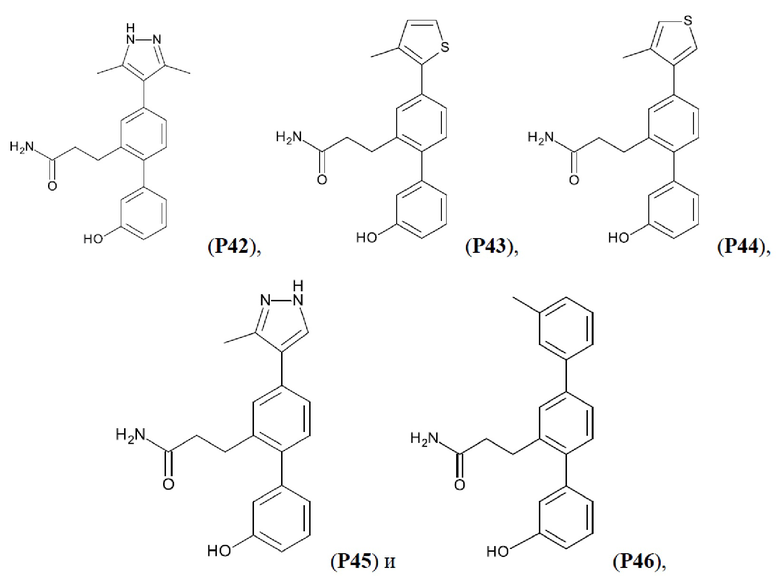
или их фармакологически приемлемой соли.
7. Способ по любому из пп. 4-6, где субъект с риском развития легочного фиброза подвергался воздействию газов, дыма, химических веществ, асбестовых волокон или пыли.
8. Способ по любому из пп. 4-6, где субъект с риском развития легочного фиброза имеет аутоиммунное расстройство, вирусную инфекцию или бактериальную инфекцию легкого.
9. Способ по любому из пп. 4-6, где субъект с риском развития легочного фиброза получал лучевую терапию по поводу рака легкого или молочной железы.
10. Способ по любому из пп. 4-6, где субъект с риском развития легочного фиброза имеет генетическую предрасположенность.
11. Способ по любому из пп. 4-6, где субъект с риском развития легочного фиброза является курильщиком.
12. Способ по любому из пп. 4-11, где связанное с легочным фиброзом состояние выбрано из легочной гипертензии, правосторонней сердечной недостаточности, дыхательной недостаточности, гипоксии, кашля, образования тромбов, пневмонии и рака легкого.
13. Способ по любому из пп. 4-12, где такое лечение предотвращает, ослабляет или замедляет прогрессирование легочного фиброза.
14. Способ по любому из пп. 4-13, где такое лечение ослабляет диагностированный легочный фиброз.
15. Применение соединения по любому из пп. 1-3 для изготовления лекарственного средства для профилактического или терапевтического лечения легочного фиброза или связанного с ним состояния у субъекта с легочным фиброзом или с риском развития легочного фиброза.
16. Применение соединения формулы:
или его фармакологически приемлемой соли,
где:
D представляет собой:
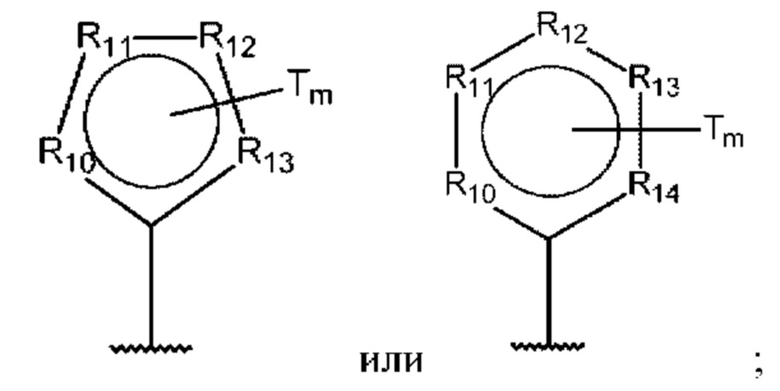
R10-R14 независимо представляют собой С, N или S;
Т независимо выбран из С1-6алкила и галогена;
m равно 0, 1, 2, 3 или 4; и
X представляет собой -ОН,
для изготовления лекарственного средства для профилактического или терапевтического лечения легочного фиброза или связанного с ним состояния у субъекта с легочным фиброзом или с риском развития легочного фиброза.
| WO 2015039172 А1, 26.03.2015 | |||
| WO 2016046782 A1, 31.03.2016 | |||
| Композиции для лечения фиброза и связанных с фиброзом состояний | 2016 |
|
RU2712140C2 |

