ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области медицины и в частности к способу лечения рака с применением комбинации плинабулина и таксана для лечения пациентов, страдающих от рака, и к ее неожиданному действию, которое состоит в значительном повышении безопасности таксана и значительном уменьшении побочных действий таксана, и к ее неожиданной эффективности при лечении больших опухолей в моделях на животных и у пациентов, страдающих от рака.
УРОВЕНЬ ТЕХНИКИ
Рак, в качестве основной причины вызванной заболеванием смертности в мире, превзошел сердечно-сосудистые заболевания. В США более 1 миллиона людей заболевают раком каждый год, тогда как в Китае каждый год раком заболевают более 3 миллионов людей. 45% всех новых случаев рака в мире возникают в Китае. Наиболее широко распространенное лечение рака представляет собой лечение химиотерапевтическими агентами, такими как таксаны. Тем не менее, вследствие непереносимых побочных действий таксана, включая высокие уровни нейтропении (степень 3-4 при 30-40%), его дозу нужно снижать в процессе применения или даже нужно завершить его применение, таким образом, уменьшая его эффективность и продолжительность лечения пациентов, страдающих от рака.
Рак легких является основной причиной смертности от рака в Соединенных Штатах, в Китае и в мире. Немелкоклеточная карцинома легких (НМККЛ) составляет приблизительно 80% из всех случаев рака легких. У большинства пациентов на момент диагностики НМККЛ данное заболевание уже переходит на позднюю стадию (становится распространенным). Стандартные одобренные методы лечения распространенной НМККЛ, как правило, включают последовательные линии химиотерапии агентами, включающими платины, таксаны, алкалоиды барвинка, пеметрексед и/или ингибиторы рецептора эпидермального фактора роста (EGFR).
Доцетаксел представляет собой соединение таксана, одобренное в качестве лекарственного средства второй линии для лечения НМККЛ в США, Европейском Союзе, Китае и множестве других стран. Доцетаксел действует путем разрушения сети микротрубочек в клетках. Его, как правило, вводят в виде 1-часовой внутривенной (в/в) инфузии раз в 3 недели в дозе, равной 75 мг/м2, после предварительного введения дексаметазона, чтобы минимизировать вероятность реакций гиперчувствительности и задержки жидкости. В 2 рандомизированных испытаниях доцетаксела у пациентов с НМККЛ, которых ранее лечили с помощью химиотерапевтического режима лечения на основе платины, средняя общая выживаемость (ОВ) находилась в диапазоне от 5,7 до 7,5 месяцев.
Самые распространенные нежелательные реакции при лечении доцетакселом включают инфекции, нейтропению, анемию, лихорадочную нейтропению, гиперчувствительность, тромбоцитопению и т.д. Несколько дополнительных химиотерапевтических агентов было одобрено в качестве лекарственных средств второй линии для лечения стадии IIIb/IV НМККЛ (пеметрексед, эрлотиниб и гефитиниб), но они приводят к клинически эквивалентной общей выживаемости. У различных типов пациентов с НМККЛ (стадия IIIb/IV, вторая линия) общая выживаемость для группы, получающей доцетаксел (75 мг/м2), составляла 8,2 месяца, что гораздо дольше, чем общая выживаемость для некоторых других лекарственных средств. Таким образом, доцетаксел все еще представляет собой лекарственное средство, выбираемое для лечения в рамках второй линии лечения НМККЛ.
Поскольку лечение раковых заболеваний все еще неудовлетворительно, существует явная нереализованная потребность медицины в дополнительных противораковых агентах для лечения пациентов, страдающих от рака, таких как пациенты с распространенной НМККЛ.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Цель настоящего изобретения состоит в том, чтобы предложить оптимальный способ лечения рака.
Другая цель настоящего изобретения состоит в том, чтобы предложить соответствующую комбинацию лекарственных средств, набор и их применение для лечения и/или предупреждения рака, в частности рака легких.
В первом аспекте настоящего изобретения предложена фармацевтическая комбинация, содержащая активный ингредиент (а) соединение таксана; и активный ингредиент (b) плинабулин.
В предпочтительном варианте реализации указанное соединение таксана включает паклитаксел, доцетаксел и абраксан.
В предпочтительном варианте реализации комбинация состоит из доцетаксела и плинабулина.
Во втором аспекте настоящего изобретения предложено применение фармацевтической комбинации, описанной в первом аспекте, для получения лекарственных средств для лечения и/или предотвращения рака.
В предпочтительном варианте реализации рак выбран из группы, состоящей из рака легких, рака толстого кишечника, рака печени, рака молочной железы, рака предстательной железы и множественной миеломы.
В третьем аспекте настоящего изобретения предложено применение композиции, набора или смеси, содержащей активный ингредиент (а) таксан и активный ингредиент (b) плинабулин, для лечения и/или предотвращения рака.
В одном предпочтительном варианте реализации при раковом заболевании размер опухоли составляет >3 см, >5 см или >7 см.
В 4-ом аспекте настоящего изобретения предложен набор, который включает:
(i) первый контейнер, содержащий первое лекарственное средство, которое содержит соединение таксана в качестве активного ингредиента (а) и необязательно фармацевтически приемлемый носитель; и
(ii) второй контейнер, содержащий второе лекарственное средство, которое содержит плинабулин в качестве активного ингредиента (b) и необязательно фармацевтически приемлемый носитель;
(iii) необязательную инструкцию, содержащую информацию о введении указанного активного ингредиента (а) в комбинации с указанным активным ингредиентом (b) для лечения и/или предотвращения рака.
Предпочтительно в инструкции указано, что плинабулин необходимо вводить путем инъекции через 1-24 часа после введения таксана.
Предпочтительно в одном наборе находится 8 флаконов первого лекарственного средства в количестве 20 мг на флакон и 2 флакона второго лекарственного средства в количестве 80 мг на флакон.
В 5-ом аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая:
активный ингредиент (а) таксан;
активный ингредиент (b) плинабулин; и
(с) фармацевтически приемлемый носитель.
В одном предпочтительном варианте реализации отношение (мг : мг) активного ингредиента (а) к активному ингредиенту (b) составляет от 1:100 до 50:1; предпочтительно от 1,5:1 до 4:1.
В одном предпочтительном варианте реализации совокупное количество активного ингредиента (а) и активного ингредиента (b) составляет 1-99% масс.; и более предпочтительно 5-90% масс. в указанной композиции.
В 6-ом аспекте настоящего изобретения предложено применение плинабулина для получения лекарственного средства, применяемого для уменьшения побочного эффекта таксана.
В одном предпочтительном варианте реализации указанный побочный эффект включает нейтропению, анемию, лихорадочную нейтропению, тромбоцитопению.
В 7-ом аспекте настоящего изобретения предложен способ уменьшения побочного эффекта таксана, отличающийся тем, что он включает этап введения плинабулина нуждающемуся в этом субъекту до, в то же время или после введения соединения таксана.
В 8-ом аспекте настоящего изобретения предложен способ лечения и/или предотвращения рака, включающий следующие этапы: введение нуждающемуся в этом млекопитающему активного ингредиента (а) таксана и активного ингредиента (b) плинабулина.
Предпочтительно сначала вводят активный ингредиент (а), а затем вводят активный ингредиент (b).
В 9-ом аспекте настоящего изобретения предложен способ применения комбинации плинабулина и доцетаксела для лечения рака у субъекта, в котором для повышения эффективности лечения рака плинабулин необходимо вводить путем инъекции через 1-24 часа после введения доцетаксела.
В одном предпочтительном варианте реализации субъект представляет собой пациента с большой опухолью различных типов рака.
В одном предпочтительном варианте реализации субъект представляет собой пациента на стадии IIIb/IV НМККЛ, имеющего по меньшей мере одну первичную опухоль легкого >3 см, предпочтительно >5 см и более предпочтительно >7 см.
В 10-ом аспекте настоящего изобретения предложен способ применения таксанов в комбинации с плинабулином для уменьшения токсичности таксанов, в частности нейтропении 3 и 4 степени.
Предложен способ применения плинабулина и доцетаксела для лечения пациента с НМККЛ для снижения вызванного доцетакселом повышения уровня нейтропении всех степеней и уменьшения количества используемого G-CSF.
Должно быть очевидно, что в настоящем изобретении технические признаки, конкретно описанные выше и ниже (такие как описанные в примерах), можно комбинировать друг с другом, посредством чего образуя новые или предпочтительные технические решения, которые не требуют конкретного упоминания в данной заявке.
ОПИСАНИЕ ЧЕРТЕЖЕЙ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
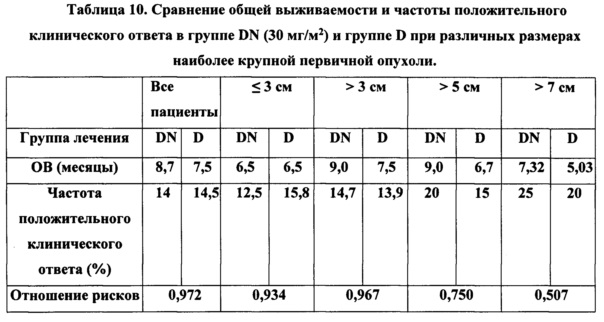
На фиг. 1 показано химическое строение плинабулина.
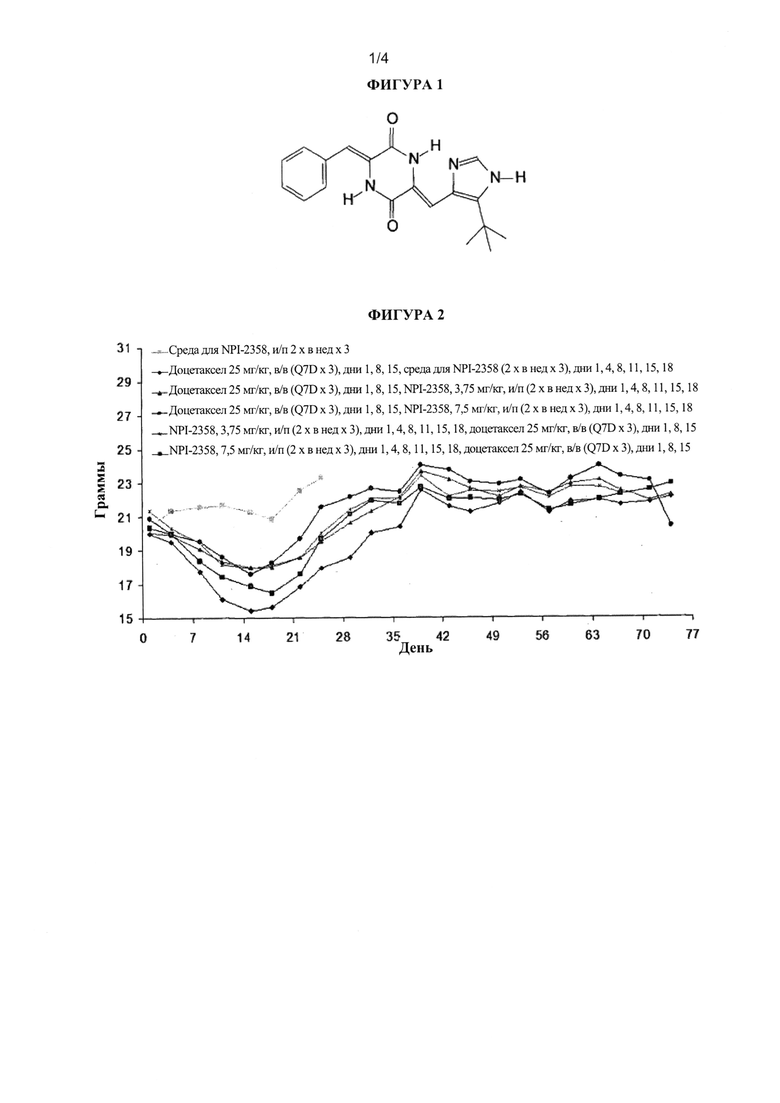
На фиг. 2 показан средний процент изменения массы в примере 1 (модель опухоли НМККЛ MV522). Добавление плинабулина (NPI-2358) к доцетакселу уменьшало связанную с доцетакселом потерю массы животного независимо от последовательности применения указанных лекарственных средств (с интервалом 2 часа друг от друга).
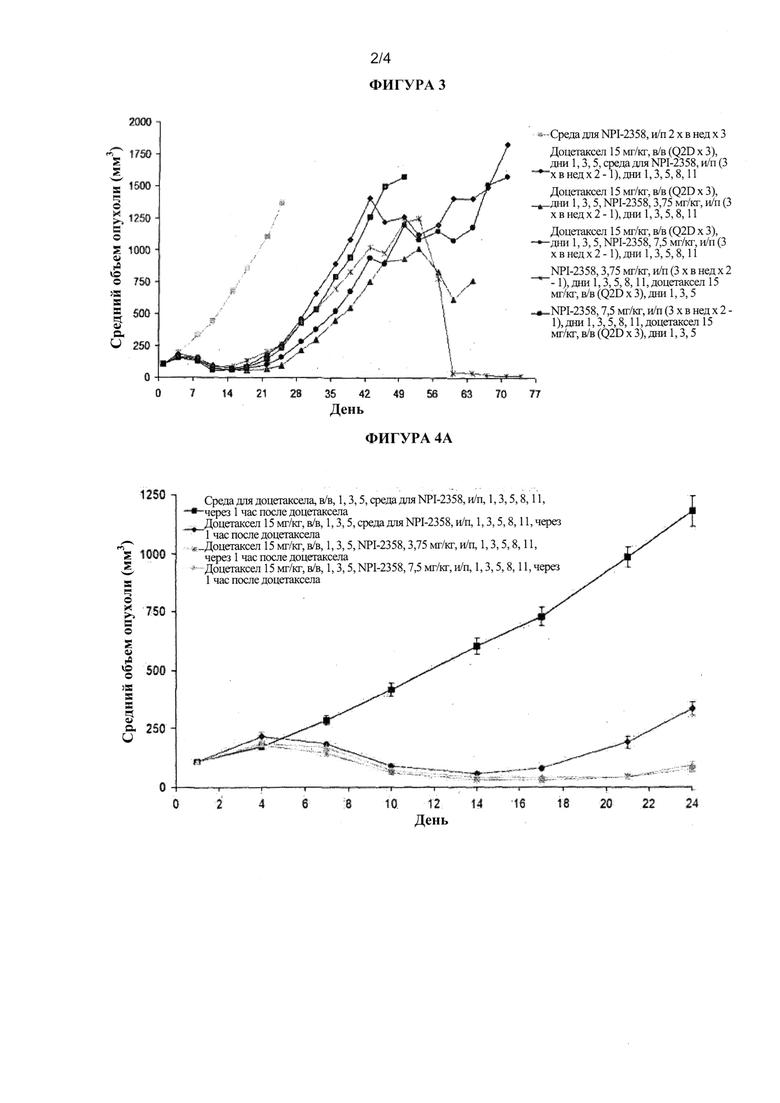
На фиг. 3 показан средний объем опухоли в примере 1 (модель опухоли НМККЛ MV522). Добавление плинабулина (NPI-2358) повышало противоопухолевую активность доцетаксела. Синергичное действие было более выраженным в группах, получавших сначала доцетаксел, а затем NPI-2358 2 часа спустя.
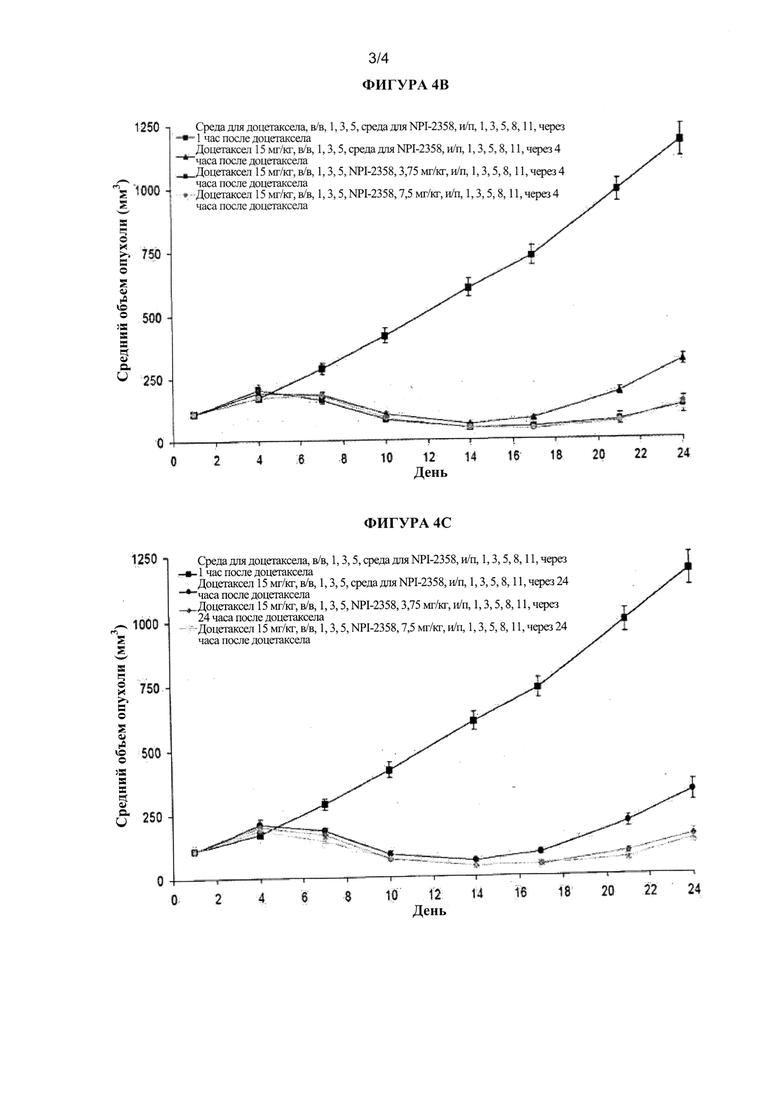
На фиг. 4А, фиг. 4B и фиг. 4С показан средний объем опухоли в примере 2 (модель опухоли НМККЛ MV522). Эффект усиления противоопухолевой активности плинабулина (NPI-2358), который добавляли к доцетакселу, был сходным в группах, получавших сначала доцетаксел, а затем плинабулин 1,4 или 24 часа спустя.
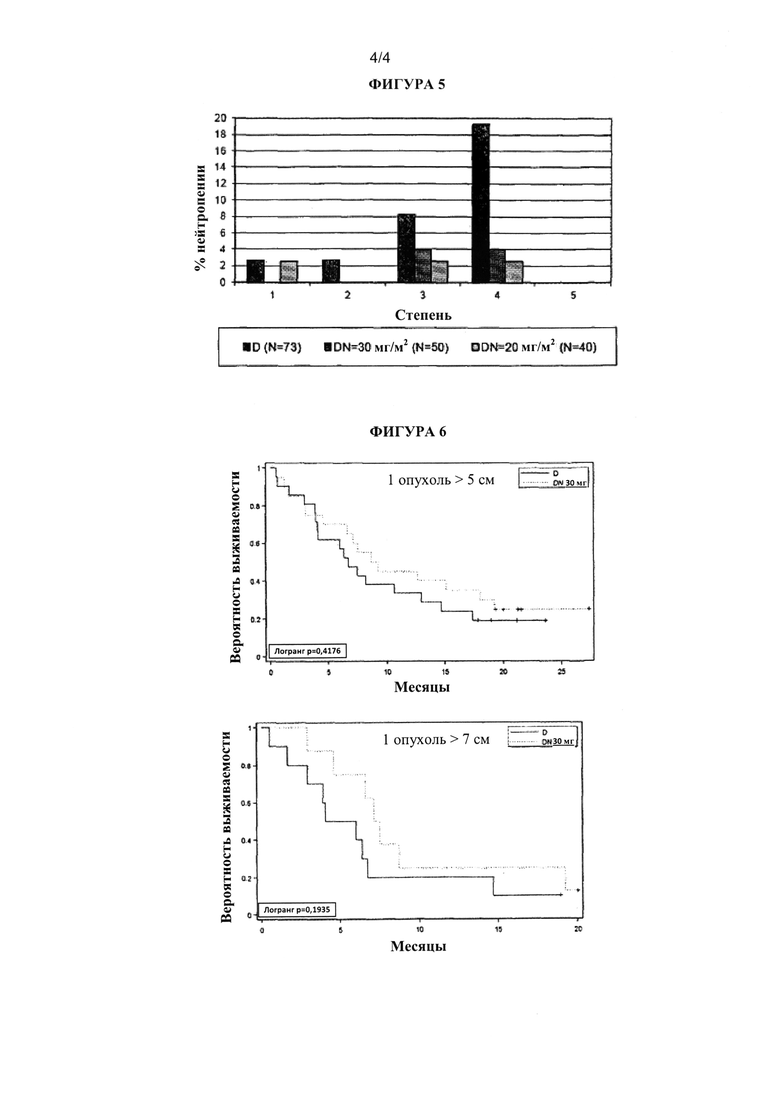
На фиг. 5 показано, что побочный эффект, такой как нейтропения различных степеней тяжести, уменьшался при добавлении плинабулина к лечению доцетакселом пациента с НМККЛ. Как 20 мг/м2, так и 30 мг/м2 плинабулина, который добавляли к доцетакселу, снижали уровень вызванной доцетакселом нейтропении (D - доцетаксел; DN - плинабулин и доцетаксел)
На фиг. 6 показана кривая общей выживаемости Каплана-Мейера у пациентов с НМККЛ (стадия IIIb/IV, по меньшей мере 1 предшествующая химиотерапия). Группа DN: 30 мг/м2 плинабулина и 75 мг/м2 доцетаксела; группа D: 75 мг/м2 доцетаксела. Эффект общей выживаемости возрастает в группе DN по сравнению с группой D с увеличением размера опухоли.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В ходе комплексного и интенсивного исследования и скрининга автор настоящего изобретения неожиданно разработал новый способ лечения рака путем применения плинабулина и доцетаксела в комбинации. В экспериментах показали преимущество их оптимальной эффективности, применяя модель НМККЛ у животных - MV522. Автор настоящего изобретения также открыл серию способов комбинированного применения плинабулина и доцетаксела для достижения оптимального преимущества эффективности и безопасности, проводя эксперименты с использованием модели НМККЛ у животных - MV522. На этом основании, настоящее изобретение завершено.
В одном варианте реализации настоящего изобретения оптимальный комбинированный способ может значительно уменьшить побочные действия таксана (такого как доцетаксел) как в модели НМККЛ у животных - MV522, так и у пациентов с НМККЛ, и снизить применение G-CSF у пациентов с НМККЛ, что является неожиданным открытием. Не было обнаружено другого повреждающего сосуды агента (VDA), оказывающего такое действие в комбинации с доцетакселом при лечении пациента, страдающего от рака.
В другом варианте реализации настоящего изобретения оптимальный комбинированный способ помог добиться наибольшей эффективности продления общей выживаемости пациентов с однозначно определяемой большой опухолью НМККЛ и животных в модели большой опухоли НМККЛ - MV522, что является новым и неожиданным открытием. Не было обнаружено другого VDA или антиангиогенного агента с лучшей эффективностью в популяции данных пациентов с однозначно определяемой большой опухолью НМККЛ.
СОЕДИНЕНИЕ ТАКСАНА ИЛИ ЕГО ПРОИЗВОДНЫЕ
В комбинации согласно настоящему изобретению один из важных активных ингредиентов представляет собой соединение таксана или его производные.
В настоящем изобретении термин "соединение таксана" или "таксан" означает представителя семейства таксана, который обладает противораковой активностью, близкой к таковой у паклитаксела, основанной на механизме, таком же или аналогичном механизму действия паклитаксела. Типичные соединения таксана включают, но не ограничены перечисленными: паклитаксел, доцетаксел, абраксан и т.д. В настоящем изобретении данный термин также включает производные и фармацевтически приемлемые соли указанных соединений.
В настоящем изобретении определенное количество соединения таксана предпочтительно вводят обычным способом и в обычной дозировке. Например, паклитаксел обычно вводят путем внутривенной инъекции 50-250 или 100-175 мг/м2.
ПЛИНАБУЛИН
В комбинации согласно настоящему изобретению другой важный активный ингредиент представляет собой плинабулин.
В данной заявке термины "плинабулин", "NPI-2358" и "трет-бутил-дегидрофенилагистин" взаимозаменяемы, и каждый из них обозначает синтетическое, низкомолекулярное химическое вещество с химическим наименованием 2,5-пиперазиндион-3-[[5-(1,1-диметилэтил)-1Н-имидазол-4-ил]метилен]-6-(фенилметилен)-(3Z,6Z)], как показано на фигуре 1. В настоящем изобретении указанные выше термины также включают фармацевтически приемлемые соли указанных соединений.
Открыли, что NPI-2358 является противораковым агентом. В WO 2004/054498 описана структура, синтез и применение NPI-2358.
Плинабулин ингибирует димеризацию мономеров тубулина. Механизм его действия заключается в воздействии на сосудистую сеть опухоли и, следовательно, в лишении кровоснабжения, необходимого для роста опухоли, и его классифицируют как повреждающий сосуды агент (VDA).
В настоящем изобретении определенное количество плинабулина предпочтительно вводят обычным способом и в обычной дозировке. Обычно плинабулин вводят путем внутривенной инъекции 10-50 или 20-30 мг/м2.
Комбинация, фармацевтическая композиция и набор.
В настоящем изобретении предложена комбинация, содержащая активный ингредиент (а) соединение таксана; и активный ингредиент (b) плинабулин.
Кроме того, предложена фармацевтическая композиция, содержащая активный ингредиент (а) соединение таксана; активный ингредиент (b) плинабулин; и (с) фармацевтически приемлемый носитель.
Лекарственные формы и способы получения фармацевтической композиции согласно настоящему изобретению особо не ограничены, и указанную композицию можно получить в различных лекарственных формах, таких как таблетки, капсулы, гранулы, агенты с пролонгированным высвобождением, инъекции и тому подобные лекарственные формы, с помощью процессов, обычных в данной области. Предпочтительная лекарственная форма представляет собой пероральную лекарственную форму.
В настоящем изобретении также предложен набор, включающий:
(i) первый контейнер, содержащий первое лекарственное средство, которое содержит соединение таксана в качестве активного ингредиента (а) и необязательно фармацевтически приемлемый носитель; и
(ii) второй контейнер, содержащий второе лекарственное средство, которое содержит плинабулин в качестве активного ингредиента (b) и возможно фармацевтически приемлемый носитель;
(iii) инструкцию, содержащую информацию о введении указанного активного ингредиента (а) в комбинации с указанным активным ингредиентом (b) для лечения и/или предотвращения рака.
Указанная комбинация, лекарственные формы и наборы согласно настоящему изобретению полезны для предотвращения и/или лечения рака.
Комбинацию согласно настоящему изобретению можно вводить совместно или последовательно. Предпочтительно, сначала вводят активный ингредиент (а) таксан, а затем вводят активный ингредиент (b) плинабулин в пределах 0,5-72 часов, предпочтительно в пределах 0,5-24 часов, более предпочтительно в пределах 1-24 часов, посредством этого значительно улучшая эффективность лечения и исполнительность пациента, и значительно уменьшая побочный эффект таксана, такой как нейтропения.
Разумеется, эффективную дозировку активных ингредиентов можно изменять в зависимости от способа введения и тяжести заболевания, от которого лечат.
ЛЕЧЕНИЕ КОМБИНАЦИЕЙ ПЛИНАБУЛИНА И ТАКСАНА
Согласно настоящему изобретению предложен оптимальный способ применения плинабулина и доцетаксела для лечения субъектов, страдающих от рака.
В настоящем изобретении предложен способ лечения и предотвращения рака путем применения двух активных ингредиентов, который включает введение субъекту-млекопитающему (такому как человек) эффективного количества активного ингредиента (а) соединения таксана и активного ингредиента (b) плинабулина, или введение первого лекарственного средства, содержащего активный ингредиент (а), и второго лекарственного средства - активного ингредиента (b).
Два указанных активных ингредиента или фармацевтические композиции согласно настоящему изобретению можно вводить обычными путями, включая (но не ограничиваясь перечисленными): внутримышечное, интраперитонеальное, внутривенное, подкожное, внутрикожное, пероральное или топическое введение. Предпочтительные пути введения включают пероральное введение.
Лекарственные средства согласно настоящему изобретению могут представлять собой твердые композиции для удобного введения, в частности таблетки и заполненные твердым веществом или заполненные жидкостью капсулы. Предпочтительно лекарственное средство или фармацевтическая композиция составлены в виде жидкой лекарственной формы, или лиофилизированной лекарственной формы, или другой подходящей для инъекции формы.
Более того, два указанных активных ингредиента или лекарственные средства согласно настоящему изобретению можно применять в комбинации с другими лекарственными средствами для лечения рака (такими как цисплатин, паклитаксел, противоопухолевые антитела).
В примерах настоящего изобретения автор настоящего изобретения оценил противоопухолевую активность плинабулина в ксенотрансплантатной модели рака легких MV522. Эффективность плинабулина in vivo определили в одельности и в комбинации с доцетакселом. Ингибирование роста опухоли (ИРО) плинабулином (3,75 и 7,5 мг/кг), доцетакселом (15 и 25 мг/кг) и их комбинацией определили в модели опухоли MV522. Также составили расписание введения комбинаций лекарственных средств, и вводили один из агентов через 2 часа после введения первого лекарственного средства. Важные конечные результаты данного эксперимента включали среднее ингибирование роста опухоли (ИРО) или регрессию опухоли, потерю массы животного, потенциальную токсичность и задержку роста опухоли (ЗРО). Важнейшим конечным результатом исследования ИРО был день, когда средний объем опухоли после введения среды для NPI-2358 достиг 1,2 см3. Конечным результатом исследования ЗРО был день, когда каждая опухоль достигла объема 1,5 см3. Кроме того, осуществляли корректировку для сравнения эффективности доцетаксела и комбинации NPI-2358 + доцетаксел в больших опухолях (1,5 см3).
Животным имплантировали раковые клетки, собранные из культуры ткани, и позволяли возникнуть опухолям в голых мышах. Лечение начинали, когда был достигнут средний объем опухоли, приблизительно равный 105 мм3. Не встречалась существенная потеря массы животного в любой из групп, в которой плинабулин был единственным агентом, демонстрируя, что данное лекарственное средство хорошо переносилось при данных дозах и графике. Как и ожидалось, наблюдали существенную потерю массы тела животного после лечения доцетакселом. Неожиданно, добавление NPI-2358 к доцетакселу в группах с комбинированным лечением уменьшало связанную с доцетакселом потерю массы тела (таблица 2).
Лечение плинабулином как единственным агентом вызывало незначительное уменьшение объема опухоли по сравнению с контролем средой, тогда как лечение доцетакселом приводило к сильному уменьшению массы опухоли. Важно отметить, что добавление плинабулина повышало противоопухолевую активность доцетаксела в данной модели.
Данный эффект был наиболее выражен в группах, получавших сначала доцетаксел, а затем плинабулин, например, 2 часа спустя (таблица 3). Более того, данная комбинация лекарственных средств более эффективно, чем отдельный агент доцетаксел, уменьшала массу больших опухолей (1,5 см3) (таблица 4).
Для того чтобы дополнительно исследовать схему введения комбинации плинабулина и доцетаксела, автор настоящего изобретения изучил эффективность in vivo плинабулина, который вводили через 1, 4 и 24 часа после лечения доцетакселом, в ксенотрансплантатной модели на мышах рака легких человека MV522. План эксперимента был аналогичен таковому для исследования на животных, которое обсуждалось выше. Добавление плинабулина к доцетакселу в группах с комбинированным лечением уменьшало связанную с доцетакселом потерю массы животного. Тенденция к потере массы была аналогичной среди различных групп, в которых плинабулин вводили через 1,4 либо 24 часа после лечения доцетакселом (таблица 6). Кроме того, добавление плинабулина повышало противоопухолевую активность доцетаксела.
Не наблюдалось существенного различия в массе опухоли, когда плинабулин вводили через 1, 4 либо 24 часа после лечения доцетакселом (таблица 7). В группах, в которых плинабулин вводили через 1 час после введения 15 мг/кг доцетаксела, наблюдали опухоли немного меньшего размера, чем в группах, которым плинабулин вводили через 4 и 24 часа (таблица 7).
После данного первоначального исследования животных со сходной массой опухоли лечили повторно, чтобы оценить влияние комбинации плинабулина и доцетаксела на большие опухоли. Комбинация плинабулина и доцетаксела вызывала более ярко выраженное уменьшение массы опухоли по сравнению с лечением доцетакселом отдельно. Неожиданно автор настоящего изобретения обнаружил, что комбинация плинабулина и доцетаксела эффективна против большой развившиеся опухоли (таблица 8).
Автор настоящего изобретения применил график лечения оптимальной комбинацией плинабулина и доцетаксела, полученный в ходе исследования на животных MV522, к пациентам с НМККЛ. Пациенты получали терапию в день 1 и день 8 в рамках 3-недельных циклов. Терапия в день 1 состояла из 75 мг/м2 доцетаксела, который вводили путем внутривенной инфузии (в/в) в течение 1 часа, после которой следовало введение 30 мг/м2 плинабулина 2 часа спустя (от момента начала инфузии доцетаксела) посредством внутривенной инфузии (в/в) в течение 30 минут. Терапия в день 8 состояла из 20 мг/м2 или 30 мг/м2 плинабулина, который вводили посредством внутривенной инфузии (в/в) в течение 30 минут.
Профиль переносимости плинабулина, который добавляли к доцетакселу, был лучше, чем у доцетаксела отдельно. Вследствие непереносимых побочных действий доцетаксела первоначальную дозу доцетаксела, равную 75 мг/м2, уменьшили у 10% пациентов (5 из 50) в группе лечения 30 мг/м2 плинабулина плюс доцетаксел, тогда как указанный процент в соответствующей группе лечения доцетакселом отдельно был гораздо выше у 18,2% пациентов (10 из 55).
Особенно интересно отметить, что у пациентов в группе лечения 30 мг/м2 плинабулина плюс доцетаксел наблюдали статистически значительно более низкую степень нейтропении (для всех событий и событий со степенью ≥3), чем у пациентов в соответствующей группе лечения доцетакселом отдельно.
Нейтропения представляет собой побочный эффект доцетаксела, который является наиболее тяжелым. Нейтропению наблюдали у 36,4% пациентов в данной группе лечения доцетакселом отдельно, что согласуется с накопленными данными. Наоборот, частота нейтропении в группе лечения 30 мг/м2 плинабулина плюс доцетаксел на уровне 8% была значимо ниже, чем в соответствующей группе лечения доцетакселом отдельно (р<0,01).
Результаты, аналогичные описанным выше, наблюдали в группе лечения 20 мг/м2 плинабулина плюс доцетаксел и в соответствующей группе лечения доцетакселом. В группе комбинированного лечения использовали меньше G-CSF по сравнению с группой, получающей доцетаксел.
Новое открытие, состоящее в том, что плинабулин может уменьшить уровень нейтропении, вызванной доцетакселом, было неожиданным.
Автор настоящего изобретения также осуществил всесторонние анализы и неожиданно обнаружил подгруппу, которая реагирует на комбинацию плинабулина и доцетаксела. Из всех проанализированных подгрупп та, в которой "диаметр по меньшей мере одной опухоли составлял больше, чем 3 см", представляла собой подгруппу, в которой комбинация 30 мг/м2 плинабулина плюс доцетаксел (общая выживаемость =11,5 М) приводила к наиболее значительному преимуществу общей выживаемости по сравнению с соответствующей группой, которую лечили доцетакселом (общая выживаемость =7,8 М) (фиг. 6). Указанное преимущество общей выживаемости в группе комбинированного лечения по сравнению с группой, получающей доцетаксел, сохранялось и в других группах с более крупными опухолями (1 опухоль >5 см или >7 см, фиг. 6). Автор настоящего изобретения обнаружил однозначно определяемую группу пациентов с большими опухолями, для лечения которых полезна оптимальная комбинация плинабулина и доцетаксела, о которой ранее не сообщалось ни в какой литературе. Ни для одного из одобренных нацеленных на сосудистую сеть опухоли агентов не выявили какое-либо предпочтение популяции пациентов с большими опухолями НМККЛ.
В общих словах, настоящее изобретение является первым, в котором обнаружили оптимальную комбинированную схему приема плинабулина и доцетаксела для достижения повышенной эффективности в однозначно определяемой популяции пациентов с большими опухолями и для существенного снижения уровня нейтропении, вызванной доцетакселом, у всех пациентов. Согласно настоящему изобретению предложено новое применение плинабулина в комбинации с другими химиотерапевтическими агентами для лечения больших опухолей во множестве раков. Согласно настоящему изобретению дополнительно предложено новое применение плинабулина, комбинированного с другими соединениями таксана, для уменьшения побочного действия соединений таксана - ослабляющей нейтропении.
Основные преимущества настоящего изобретения включают описанные далее преимущества.
(a) В настоящем изобретении раскрыто действие таксана в комбинации с плинабулином в отношении предотвращения рака (такого как рак легких) и предложен способ лечения путем применения оптимальной комбинации таксана и плинабулина.
(b) Комбинация таксана и плинабулина относительно безопасна.
(c) Последовательное введение таксана и плинабулина вызывает синергичное ингибирование опухолей, и обладает важной с медицинской точки зрения статистической значимостью, посредством чего переносимость таксана увеличивается и побочные действия уменьшаются.
Настоящее изобретение будет дополнительно проиллюстрировано ниже с сылкой на конкретные примеры. Должно быть очевидно, что данные примеры предназначены только для иллюстрированния настоящего изобретения, но не для ограничения объема настоящего изобретения. Экспериментальные способы без конкретных условий, описанных в следующих примерах, как правило, осуществляют при обычных условиях или следуя инструкциям производителя. Если не указано иное, доли и проценты рассчитывают по массе.
Пример 1.
Оценка плинабулина in vivo (NPI-2358) в качестве отдельного агента и в комбинации с доцетакселом в ксенотрансплантатной модели немелкоклеточной опухоли легкого человека MV522 у бестимусных мышей nu/nu.
Целью данного примера было определение потенциального аддитивного или синергичного действия плинабулина в комбинации с доцетакселом в модели MV522 путем исследования схемы применения комбинации лекарственных средств при введении одного из агентов через 2 часа после введения первого лекарственного средства. Значимые конечные результаты данного эксперимента включали среднее значение ингибирования роста опухоли (ИРО) или регрессию опухоли, задержку роста (ЗРО) большой опухоли, потерю массы и смертность.
План эксперимента.
Материалы и методы.
Информация о модели. Самок голых мышей (Hsd: бестимусные Nude-Foxnlnu) в возрасте 5-6 недель, весящих приблизительно 20 грамм, получали от Harlan, Inc. (Мэдисон, Висконсин). MV522 представляет собой обычную линию клеток метастатической немелкоклеточной опухоли легкого человека (US 7700615 или 7629380). Животным вводили путем подкожной (п/к) инъекции приблизительно 1×107 клеток MV522, собранных из культуры ткани. Когда опухоли доросли до размера приблизительно 105 кубических миллиметров (мм3) (через 3 дня после имплантации), животных в зависимости от размера опухоли попарно распределяли по группам лечения и контрольным группам; в каждой группе лечения находилось по восемь мышей. Животным делали отметки на ушах, и отслеживали их индивидуально на протяжении всего эксперимента.
План исследования и введение доз. Первоначальные дозы вводили в день 1 после попарного распределения. Эксперимент осуществляли в рамках исследования ингибирования роста опухоли (ИРО) и задержки роста опухоли (ЗРО). Конечный результат для группы 1 представлял собой достижение опухолью объема 1,2 см3 (ИРО) и конечный результат для группы лечения представлял собой достижение опухолью каждого отдельного животного объема 1,5 см3 (ЗРО). NPI-2358 в среде (8% солютол® HS15,12% PG, 80% D5W) вводили путем интраперитонеальной (и/п) инъекции три раза в неделю в течение двух недель - 1-дневная схема (3 × в нед × 2-1), или по схеме два раза в неделю в течение трех недель (2 × в нед × 3) в дозах, перечисленных ниже (таблица 1). В качестве отрицательных контролей вводили среду для NPI-2358 и среду для доцетаксела путем и/и инъекции по схеме 2 × в нед × 3 и путем в/в инъекции по схеме Q2D×3, соответственно. Доцетаксел вводили путем внутривенной (в/в) инъекции через хвостовую вену в дозе 15 мг/кг один раз в два дня, всего три введения (Q2D×3), или в дозе 25 мг/кг по схеме Q7D×3 (раз в 7 дней, всего три введения). Доцетаксел и среду для доцетаксела вводили за два часа до введения среды для NPI-2358 и NPI-2358, соответственно, в группах 7-9 и 12-14. В группах 10, 11, 15 и 16 NPI-2358 вводили за два часа до доцетаксела.
Сбор данных и статистический анализ.
Массы животных. Отдельные массы и средние массы в группе ± стандартное отклонение (СО) и процент изменения массы регистрировали два раза в неделю до завершения исследования, начиная в день 1. Регистрировали массы в группе в день 42 и самые низкие значения массы.
Состояние агонии/смертность. За животными наблюдали два раза в неделю для выявления общего состояния агонии и ежедневно для выявления смертности.
Объем опухоли. Индивидуальные и средние объемы опухоли в группах ± стандартная ошибка среднего значения регистрировали два раза в неделю через 24 часа после введения дозы до завершения исследования (средний объем опухоли в контроле =1,2 см3, день 25), начиная в день 1. Измерения опухоли преобразовывали в объем опухоли в кубических миллиметрах, применяя приведенную ниже формулу:
Объем опухоли (мм3)= ширина2 (мм) × длина (мм) ×0,52
Некроз опухоли. Степень некроза опухоли классифицировали при каждом измерении опухоли, используя следующий условный показатель:

Были описаны заметные различия в степени некроза опухоли между группами лечения и контрольной группой.
Ингибирование роста опухоли. Часть исследования, в которой анализировали ингибирование роста опухоли (ИРО), завершили в день 25, после того, как средний объем опухоли в указанной контрольной группе (группе 3, среда для NPI-2358) достиг размера, равного приблизительно 1,2 см3, которую проводили отдельно от исследования задержки роста опухоли (ЗРО). Мышей взвешивали и измеряли опухоли с помощью штангенциркуля. Значения ИРО рассчитывали для каждой группы, включающей подвергнутых лечению животных, применяя приведенную ниже формулу:

Животных, у которых наблюдался полный ответ опухоли на лечение, или животных, погибших по технической или связанной с лекарственным средством причине, исключали из конечных расчетов ИРО; тем не менее, животных, у которых наблюдался частичный ответ опухоли на лечение, включали в конечные расчеты ИРО. Критерии активности соединения, установленные Национальным институтом рака (NCI), представляют собой ИРО>58%. Значения ИРО для каждой группы лечения регистрировали по окончании исследования; данные расчеты проводили на основе данных в последний день исследования.
Задержка роста опухоли. Данную часть исследования завершили в день 74 по требованию заказчика. По окончании исследования ИРО (день 25) проверяли индивидуальные объемы опухоли в контрольной группе и группах лечения, и объемы опухоли, большие или равные указанному конечному результату для объема опухоли в исследовании ЗРО (1,5 см3), удаляли из исследования, и каждому животному присваивали значение дня умерщвления на основании дня, когда оно достигло конечного результата. По окончании исследования ЗРО рассчитывали срединный день умерщвления (СДУ) для контрольной (К) и каждой группы лечения (Л), и использовали полученные значения для определения задержки роста опухоли (Л-К), применяя следующие уравнения:

где ИДУ (индивидуальный день умерщвления) представляет собой день, в который каждое животное достигло конечного результата объема опухоли (1,5 см3); только животных, которые достигли данного конечного результата, включали в расчеты ЗРО. Общий или чистый log10 гибели клеток для каждой группы лечения определили, применяя следующие уравнения:
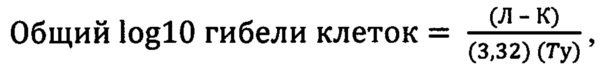
где Л - срединный день гибели для группы лечения, К - срединный день гибели для контрольной группы (группы, которой вводили среду для NPI-2358) и Ту представляет собой время удвоения объема опухоли, которое оценивают по логарифмически линейному графику логарифмической фазы роста опухоли (100-800 мм3) с течением времени в контрольной группе; 3,32 представляет собой количество удвоений, необходимое для того, чтобы популяция увеличилась на одну единицу log10. Данные, касающиеся массы и опухолей отдельных животных, погибших по технической или связанной с лекарственным средством причине, исключали из конечных расчетов для групп и статистической обработки. Долгожителей (ДГЖ, животных, не достигших конечного результата объема опухоли к заранее установленному периоду времени) не включили в данные расчеты. Для сравнения активности со стандартным агентом(ами) значения общего или чистого log10 гибели клеток преобразовывали в условную классификацию активности, описанную ниже:

Чистый log10 гибели клеток использовали для тестирования агентов, которые вводили в течение менее чем пяти дней совокупного лечения, тогда как активность агентов, которые вводили в течение пяти или более дней лечения, рассчитывали, применяя общий log гибели клеток; классификация активности от (+++) до (++++) необходима для обозначения частичных или полных ответов.
Частичный/полный ответ опухоли. Отдельных мышей, имеющих опухоли, размеры которых были меньше, чем в день 1, классифицировали как проявляющих частичный ответ (40), и значение регрессии опухоли в процентах (% РО) определяли, применяя приведенную ниже формулу:
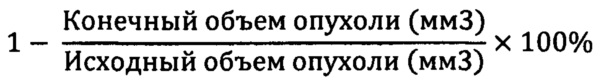
Если у многих животных в одной группе наблюдался частичный ответ опухоли на лечение, то определяли среднее значение ЧО. Отдельных мышей, у которых не обнаруживали пальпируемых опухолей, классифицировали как проявляющих полный ответ (ПО). Количество частичных и полных ответов и регрессию опухоли в процентах регистрировали для каждой группы лечения по окончании исследования; данные расчеты проводили на основе данных в последний день исследования.
Статистика ИРО. Проводили статистические анализы для сравнения конечного объема опухолей в группах лечения и контрольной группе. Для сравнения двух или более групп лечения использовали двусторонний однофакторный дисперсионный анализ (ANOVA), а затем критерий Даннета для множественных сравнений. Непарный двусторонний t-критерий Стьюдента использовали для сравнения одной группы лечения с контрольной группой. Данные, касающиеся массы и опухолей отдельных животных, погибших по технической или связанной с лекарственным средством причине, исключали из анализа. Тем не менее, данные, касающиеся массы и опухолей из животных, проявляющих частичные или полные ответы, включали в данные расчеты.
Статистика ЗРО. Логарифмический ранговый критерий использовали, чтобы определить статистически значимые различия в общей выживаемости, наблюдаемые между каждой группой лечения по сравнению с контрольной группой, и в используемом виде он эквивалентен критерию Мантеля-Хензеля. Если лекарственное средство оценивали при множестве концентраций при одинаковом пути и схеме введения, то также использовали логарифмический ранговый критерий для тренда. Данные, касающиеся массы и опухолей из отдельных животных, погибших по технической или связанной с лекарственным средством причине, исключали из анализа. Тем не менее, данные, касающиеся массы и опухолей из животных, проявляющих частичную или полную регрессию, или из долгожителей включали в данные расчеты. Все анализы осуществляли, применяя программное обеспечение GraphPad Prism® (версии 5.0). Результаты.
Ни в одной из групп, которым вводили плинабулин в качестве единственного агента, не обнаружили значимой потери массы животных, демонстрируя, что данное лекарственное средство хорошо переносилось при используемых дозах и схеме введения. Как и ожидали, наблюдали существенную потерю массы животных после лечения доцетакселом.
Было неожиданным, что добавление NPI-2358 к доцетакселу в группах с комбинированным лечением уменьшало связанную с доцетакселом потерю массы тела (таблица 2).
Лечение плинабулином в качестве отдельного агента вызывало незначительное уменьшение объема опухоли по сравнению с контролем средой, тогда как лечение доцетакселом приводило к сильному уменьшению массы опухоли. Важно отметить, что добавление NPI-2358 повышало противоопухолевую активность доцетаксела в данной модели.
Данный эффект был наиболее выражен в группах, получающих сначала доцетаксел, а затем NPI-2358 2 часа спустя (таблица 3). Более того, данная комбинация лекарственных средств наиболее эффективно уменьшала массу больших опухолей (1,5 см3), чем отдельный агент доцетаксел (таблица 4).
Пример 2.
Оценка плинабулина in vivo в комбинации с доцетакселом в ксенотрансплантатной модели немелкоклеточной опухоли легкого человека MV522 у бестимусных мышей nu/nu: схема введения лекарственного средства.
Целью данного примера было определение потенциального аддитивного или синергичного действия плинабулина (NPI-2358) в комбинации с доцетакселом путем введения NPI-2358 в различные моменты времени (через 1 час, 4 часа или 24 часа) после введения доцетаксела в модели MV522. Значимые конечные результаты для данного эксперимента включали среднее значение ингибирования роста опухоли (ИРО) или регрессию опухоли, задержку роста (ЗРО) большой опухоли, потерю массы и смертность.
План эксперимента.
Материалы и методы.
Информация о модели. Самок голых мышей (Hsd: бестимусные Nude-Foxnlnu) в возрасте 5-6 недель, весящих приблизительно 20 грамм, получали от Harlan, Inc. (Мэдисон, Висконсин). Животным вводили путем подкожной (п/к) инъекции приблизительно 1×107 клеток MV522, собранных из культуры ткани (1:1 матригель : среда). Когда опухоли доросли до размера приблизительно 100 кубических миллиметров (мм3), животных в зависимости от размера опухоли попарно распределяли по группам лечения и контрольным группам. Животным делали отметки на ушах, и отслеживали их индивидуально на протяжении всего эксперимента.
Исходный план исследования и введение доз. Первоначальные дозы вводили в день 1 после попарного распределения. Эксперимент осуществляли в рамках исследования ингибирования роста опухоли (ИРО) и задержки роста опухоли (ЗРО). Для ИРО конечный результат был достигнут, когда средний объем опухоли в группе 1 достиг 1,2 см3. Для ЗРО конечный результат был достигнут, когда опухоль каждого отдельного животного достигала объема 1,0 см3. Исходный раствор доцетаксела при концентрации 10 мг/мл разбавляли в 0,9% солевом растворе в каждый день введения доз и осуществляли внутривенное (в/в) введение. Схема и дозы каждого агента приведены в таблице 5. Среду для доцетаксела (0,9% солевой раствор; в/в; дни 1,3,5) и среду для NPI-2358 (8% солютол® HS15, 12% PG, 80% D5W; и/п; дни 1, 3, 5, 8, 11) вводили в качестве отрицательного контроля.
Сбор данных и статистический анализ.
Исследование в данном примере завершили в день 24, как только средний объем опухоли в указанной контрольной группе достиг размера, равного приблизительно 1,2 см3.
Массы животных. Индивидуальные массы и средние массы в группе ±СО, и процент изменения массы регистрировали два раза в неделю до завершения исследования, начиная в день 1. Регистрировали массы в группе до дня 24 или 53 и самые низкие значения массы.
Состояние агонии/смертность. Совпадает с примером 1.
Объем опухоли. Совпадает с примером 1.
Некроз опухоли. Совпадает с примером 1.
Частичный/полный ответ опухоли. Совпадает с примером 1.
Ингибирование роста опухоли. Совпадает с примером 1.
Статистика ИРО. Совпадает с примером 1.
Задержка роста опухоли. Совпадает с примером 1. Данную часть исследования завершили в день 80 или 53 по требованию заказчика. Долгожителям (ДГЖ, животным, не достигшим конечного результата объема опухоли к заранее установленному периоду времени) присваивали значение ИДУ в последний день исследования (день 80) и включали в данные расчеты. Для сравнения активности со стандартным агентом(ами) значения общего или чистого log10 гибели клеток преобразовывали в условную классификацию активности, описанную ниже:
Чистый log10 гибели клеток использовали для тестирования агентов, которые вводили в течение менее чем пяти дней совокупного лечения, тогда как активность агентов, которые вводили в течение пяти или более дней лечения, рассчитывали, применяя общий log гибели клеток; классификация активности от (+++) до (++++) необходима для обозначения частичных или полных ответов.
Статистика ЗРО. Совпадает с примером 1.
Результаты.
Для того, чтобы дополнительно изучить схему введения комбинации плинабулина и доцетаксела, автор настоящего изобретения исследовал эффективность плинабулина in vivo, который вводили через 1, 4 и 24 часа после лечения доцетакселом ксенотрансплантатной модели на мышах рака легких человека MV522. План эксперимента был аналогичен таковому в исследовании на животных, которое обсуждалось выше. Добавление плинабулина к доцетакселу в группах с комбинированным лечением уменьшало связанную с доцетакселом потерю массы тела животного. Тенденция к потере массы тела была сходной среди различных групп, в которых плинабулин вводили через 1, 4 или 24 часа после лечения доцетакселом (таблица 6).
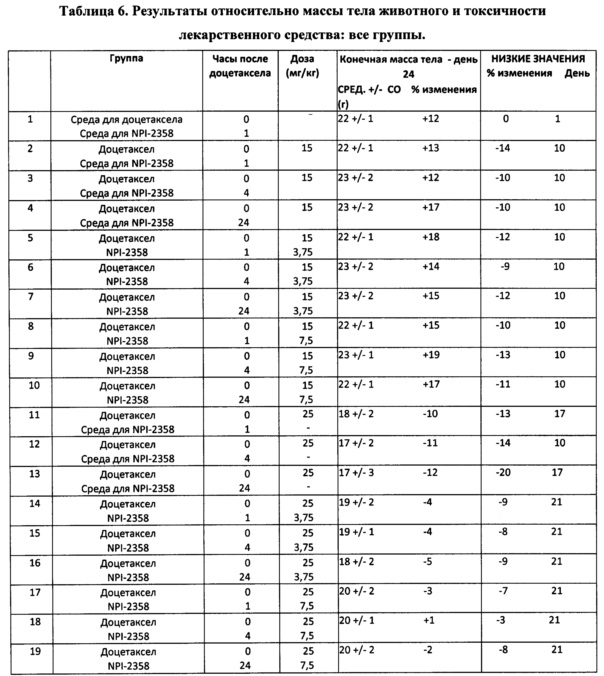
Кроме того, добавление плинабулина, похоже, увеличивало противоопухолевую активность доцетаксела. Не наблюдали значительного различия в массе опухоли в зависимости от введения плинабулина через 1, 4 или 24 часа после лечения доцетакселом (таблица 7). В группе, получившей плинабулин через 1 час после введения 15 мг/кг доцетаксела, возникали опухоли незначительно меньшего размера, чем в группах, которые получили плинабулин через 4 и 24 часа (таблица 7).
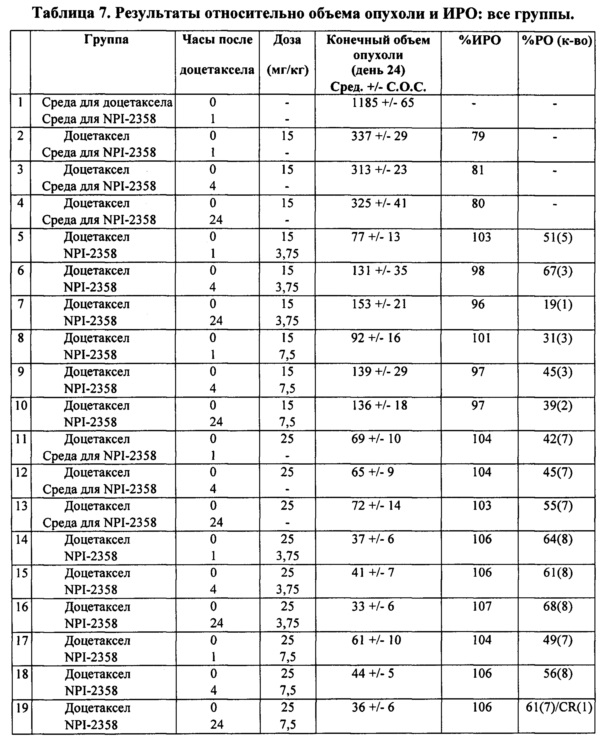
После данного первоначального исследования животных с аналогичными массами опухолей заново подвергали лечению, чтобы оценить влияние комбинации плинабулина и доцетаксела на большие опухоли. Комбинация плинабулина и доцетаксела приводила к более ярко выраженному уменьшению массы опухоли по сравнению с лечением доцетакселом отдельно. Неожиданно автор настоящего изобретения обнаружил, что комбинация плинабулина и доцетаксела эффективна против больших развившихся опухолей (таблица 8).
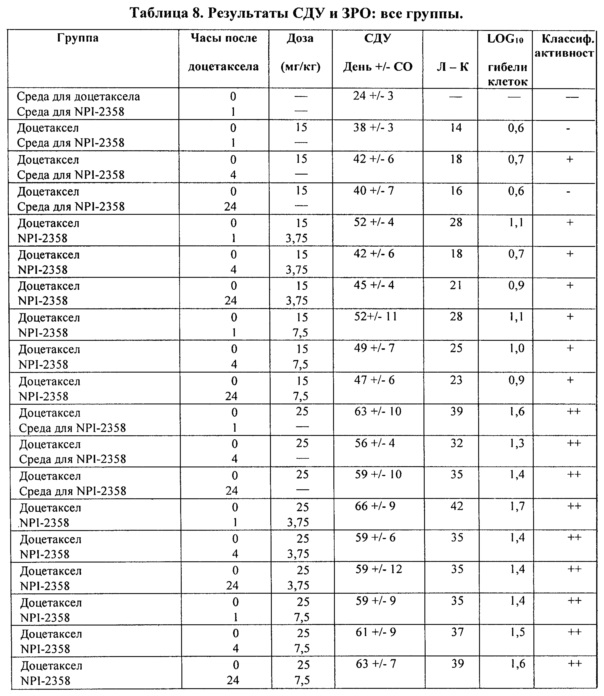
Пример 3.
Оценка оптимальной комбинации плинабулина и доцетаксела для лечения пациентов с распространенным немелкоклеточным раком легких.
ЦЕЛИ
Первичная.
Сравнить общую выживаемость пациентов с НМККЛ, которых лечили доцетакселом, с общей выживаемостью пациентов, которых лечили комбинацией доцетаксел + плинабулин.
Вторичная.
1. Сравнить частоту положительного клинического ответа, продолжительность ответа, 6-месячную выживаемость и выживаемость без прогрессирования заболевания у пациентов с НМККЛ, которых лечили доцетакселом, с таковыми у пациентов, которых лечили комбинацией доцетаксел + плинабулин;
2. Сравнить профиль безопасности и нежелательных явлений у пациентов, которых лечили доцетакселом, с таковыми у пациентов, которых лечили комбинацией доцетаксел + плинабулин.
ПЛАН ИССЛЕДОВАНИЯ
Это было открытое исследование у пациентов с распространенной НМККЛ, которая прогрессировала после лечения по меньшей мере 1 химиотерапевтическим режимом. Пациентов рандомизировали по группам, получающим либо доцетаксел плюс плинабулин (DN), либо доцетаксел отдельно (75 мг/м2) (D). Исследовали 2 группы дозирования:
1) Группа дозирования 30 мг/м2: приблизительно 110 пациентов нужно было рандомизировать (1:1) по группам, получающим либо доцетаксел плюс плинабулин при концентрации 30 мг/м2 (группа DN 30 мг/м2), либо доцетаксел отдельно (группа D);
2) Группа дозирования 20 мг/м2: приблизительно 57 пациентов нужно было рандомизировать (2:1) по группам, получающим либо доцетаксел плюс плинабулин при концентрации 20 мг/м2 (группа DN 20 мг/м2), либо доцетаксел (группа D) отдельно.
Режим дозирования
Пациенты получали терапию в день 1 и день 8 в рамках 3-недельных циклов.
Терапия в день 1 состояла из 75 мг/м2 доцетаксела, который вводили путем внутривенной инфузии (в/в) в течение 1 часа, а через 2 часа (от момента начала инфузии доцетаксела) следовало введение плацебо (группа D) или 30 мг/м2 либо 20 мг/м2 плинабулина (группа DN). DN вводили посредством внутривенной инфузии (в/в) в течение 30 минут. Пероральное введение дексаметазона (16 мг) осуществляли за день до, в тот же день и на следующий день после инфузии доцетаксела (день 1). Терапия в день 8 состояла из плацебо (группа D) или 30 мг/м2 либо 20 мг/м2 плинабулина (группа DN), который вводили посредством внутривенной инфузии (в/в) в течение 30 минут.
У пациентов, которые испытывали связанные с лекарственным средством возникшие после начала лечения нежелательные явления степени >2 (за исключением алопеции, анорексии и утомляемости) согласно классификации общих терминологических критериев нежелательных явлений (СТСАЕ, версии 3.0), лечение можно было отсрочить до момента, когда указанное нежелательное явление станет < степени 1. Лабораторные испытания безопасности должны удовлетворять следующим критериям перед лечением доцетакселом в начале каждого последующего цикла: ACT<2,5 × верхняя граница нормы, АЛТ<2,5 × верхняя граница нормы (<1,5 × верхняя граница нормы, если концентрация щелочной фосфатазы >2,5 × верхняя граница нормы); билирубин < верхняя граница нормы; гемоглобин >9 г/дл, абсолютное количество нейтрофилов >1,5×109/л и тромбоциты >100×109/л. Можно уменьшить дозу для пациентов, которые испытывают повторяющуюся или специфическую сильную токсичность.
ЦЕЛЕВАЯ ПОПУЛЯЦИЯ
Пациенты со стадией IIIb/IV немелкоклеточной карциномы легких, которая прогрессировала после лечения по меньшей мере одним химиотерапевтическим режимом.
КРИТЕРИИ ВКЛЮЧЕНИЯ
1. Мужчины и женщины в возрасте ≥18 лет.
2. Функциональный статус по шкале Восточной объединенной группы онкологов (ECOG)<1.
3. Патологически или гистологически подтвержденная распространенная немелкоклеточная карцинома легких (неоперабельная стадия IIIb или IV), которая прогрессировала после лечения по меньшей мере одним химиотерапевтическим режимом. Измеряемое проявление заболевания не требуется для включения в данное исследование.
4. Все нежелательные явления любой предшествующей химиотерапии, хирургического вмешательства или радиотерапии должны пройти до степени <2 по классификации СТСАЕ (версии 3.0).
5. Должны быть следующие результаты лабораторных исследований (не позднее 14 дней):
- Гемоглобин >9 г/дл
- Абсолютное количество нейтрофилов >1,5×109/л
- Количество тромбоцитов >100×109/л
- Билирубин в сыворотке < верхней границы нормы
- ACT и АЛТ <2,5 × верхняя граница нормы (<1,5 × верхняя граница нормы, если концентрация щелочной фосфатазы ≥2,5 × верхняя граница нормы).
6. Подписанное информированное согласие.
КРИТЕРИИ ИСКЛЮЧЕНИЯ
1. Проведение некоторой химиотерапии, введение биологического агента, иммунотерапии, лучевой терапии или экспериментального агента (терапевтического или диагностического) в течение 21 дня до получения исследуемого лекарственного средства. Обширное хирургическое вмешательство, отличное от диагностического хирургического вмешательства, в течение 6 недель до первого введения исследуемого лекарственного средства.
2. Значимый анамнез сердечно-сосудистых заболеваний:
- Инфаркт миокарда или ишемическая болезнь сердца в анамнезе;
- Клинически значимые аритмии; неконтролируемая аритмия или потребность в антиаритмических лекарственных средствах в анамнезе;
- Врожденный удлиненный интервал QT в анамнезе;
- Блокада левой ножки пучка Гиса;
- Результаты ЭКГ, указывающие на ишемическую болезнь сердца;
- Болезнь сердца класса III или IV по классификации Нью-Йоркской ассоциации кардиологов;
- Неконтролируемая гипертония: кровяное давление стабильно выше, чем 150 мм рт.ст. (систолическое) и 100 мм рт.ст. (диастолическое), несмотря на прием противогипертонического лекарственного средства.
3. Предшествующее лечение повреждающими сосуды опухоли агентами.
4. Предшествующая эпилепсия.
5. Метастазы в головном мозге. Пациентов, у которых обнаружены признаки или симптомы метастазов в головном мозге, следует подвергнуть визуализации с помощью КТ или МРТ. Пациентов, которые имеют метастазы в головном мозге, которых ранее лечили и снова подвергли визуализации после лечения, и поражения которых стабильны, без временного развития новых поражений, можно включить в исследование.
6. Значимый анамнез заболеваний желудочно-кишечного тракта, таких как илеус, кишечная непроходимость, геморрагическая диарея, воспалительное заболевание кишечника, активная неконтролируемая пептическая язва (сопутствующая терапия ранитидином или его эквивалентом и/или омепразолом или его эквивалентом приемлема).
7. В анамнезе была периоперационная лучевая терапия малого таза, лучевая терапия всей брюшной полости или остаточные симптомы ≥2 степени в желудочно-кишечном тракте после лучевой терапии.
8. Активная неконтролируемая бактериальная, вирусная или грибковая инфекция, требующая системной терапии.
9. Известное инфицирование вирусом иммунодефицита человека (ВИЧ) или активный гепатит А, В или С.
10. Пациенты, у которых ранее развивалась реакция гиперчувствительности на любой продукт, содержащий полисорбат 80, таксаны, солютол и/или пропиленгликоль.
11. Беременные или кормящие грудью женщины. Пациенты женского пола должны представлять собой женщин в постменопаузе, стерилизованных хирургическим путем, или они должны дать согласие на применение приемлемых способов контрацепции (т.е. гормональный контрацептив с барьерным методом, внутриматочное устройство, диафрагма со спермицидом или презерватив со спермицидом, воздержание) в течение всей продолжительности исследования и в течение одного месяца после окончания исследования. Пациенты-женщины с репродуктивным потенциалом должны иметь отрицательный результат тестирования на беременность по сыворотке в течение 10 дней перед первым введением исследуемого лекарственного средства. Пациенты мужского пола должны представлять собой мужчин, стерилизованных хирургическим путем, или должны дать согласие на применение приемлемых способов контрацепции.
12. Сопутствующее активное второе злокачественное новообразование, от которого указанный пациент получает лечение, исключая базально-клеточную карциному кожи или карциному in situ шейки матки.
13. Какие-либо медицинские состояния, которые, по мнению исследователя, будут приводить к избыточному риску для пациента. Примеры таких состояний включают инфекцию, требующую парентерального противоинфекционного лечения, гидронефроз, печеночную недостаточность, любое изменение психического состояния или любое психиатрическое состояние, которое будет препятствовать пониманию информированного согласия.
14. Нежелание или неспособность соблюдать процедуры, требуемые данным протоколом.
ПРОДОЛЖИТЕЛЬНОСТЬ ИССЛЕДОВАНИЯ
Стабильных и отвечающих на лечение пациентов лечили в данном исследовании до тех пор, пока он/она проявляли признаки клинической пользы (стабильного заболевания или ответа на лечение) в отсутствие неприемлемых нежелательных явлений. Завершить исследование планировали через 12 месяцев после того, как был привлечен последний пациент.
ИССЛЕДУЕМЫЕ ПРОДУКТЫ/ДОЗА/ПУТЬ/СХЕМА ПРИЕМА.
Плинабулин или плацебо.
Первоначальная доза плинабулина составляла 30 мг/м2 или 20 мг/м2. Корректировки дозы зависели от наблюдаемых нежелательных явлений. Вводимый объем изменялся в зависимости от прописанной дозы и площади поверхности тела пациента. Лекарственную форму поставляли в виде концентрированного раствора в 40% солютоле® HS-15/60% пропиленгликоле во флаконах янтарного цвета, содержащих 80 мг лекарственного средства в 20 мл (4 мг/мл), и хранили при комнатной температуре. Каждый флакон был предназначен для одноразового применения. Подходящий объем лекарственного средства (при концентрации 4 мг/мл во флаконе) разбавляли в 5% декстрозе в воде (D5W) при разведении 1:20 и вводили внутривенно (в/в) в периферические или центральные вены. Время инфузии можно увеличить в соответствии с клиническими показаниями по указанию спонсора. Плинабулин и плацебо должны быть защищены от света в течение всего времени, включая хранение, разбавление и введение. Плинабулин и плацебо следует вводить в течение 6 часов после разбавления.
Следует провести премедикацию с помощью противорвотных средств (включая схемы приема важных противорвотных средств, таких как ингибитор вещества Р, комбинации кортикостероида и/или антагониста дофамина), в соответствии с практикой лечебного учреждения перед каждым введением дозы плинабулина или плацебо. Антагонисты 5-НТ3 не следует в плановом порядке вводить после или между дозами плинабулина или плацебо, если только нет явной необходимости. Пациенты должны получать улучшающий перистальтику агент, такой как метоклопрамид, в рамках противорвотной терапии.
Перистальтику кишечника следует поддерживать в соответствии с практикой лечебного учреждения, которая применяется по отношению к лекарственным средствам, таким как винкристин, включая применение таких агентов, как размягчители стула, объемообразующие агенты, стимулирующие агенты и/или антагонисты дофамина, а также минимизируя применение ухудшающих перистальтику агентов, таких как опиаты, если только нет явных показаний, или контролируя вызванный опиатами запор с помощью таких агентов, как метилналтрексон, в соответствии с показаниями. Если развивается значительный запор, то его следует незамедлительно устранять, и введение плинабулина или плацебо следует отложить до устранения запора. Рекомендуется тщательное наблюдение для выявления признаков илеуса и ранняя диагностическая оценка с помощью рентгенографического и/или ультразвукового исследований.
Если наблюдается повышение систолического кровяного давления >20% после введения плинабулина или плацебо, то следует перорально ввести 10 мг амлодипина или эквивалентного блокатора кальциевых каналов за один час перед каждой последующей дозой. Повышение систолического кровяного давления выше 180 мм. рт.ст. следует контролировать с помощью нитропруссида или аналогичного лекарственного средства в соответствии с практикой лечебного учреждения.
Доцетаксел.
Первоначальная доза доцетаксела составляла 75 мг/м2. Корректировки дозы зависели от наблюдаемых нежелательных явлений.
Вводимый объем изменялся в зависимости от прописанной дозы и площади поверхности тела пациента.
Так как доцетаксел представляет собой стандартный одобренный и доступный для приобретения химиотерапевтический агент, исследователь и штат центра проведения клинического исследования должен иметь опыт применения доцетаксела и должен быть знаком с лекарственной формой и инструкцией по применению доцетаксела, предоставленной производителем.
Доцетаксел следует получить в аптеке медицинского учреждения и приготовить, в соответствии с протоколом медицинского учреждения. Введение следует осуществлять с помощью 1-часовой в/в инфузии в соответствии с протоколом медицинского учреждения в дозе, назначенной протоколом данного клинического испытания.
Дексаметазон (16 мг) вводили перорально за день до, в тот же день и на следующий день после инфузии доцетаксела (день 1). Можно применять аналогичную схему премедикации кортикостероидами в соответствии с местной практикой медицинского учреждения. Доза дексаметазона или другого кортикостероида должна быть соответствующим образом снижена для пациентов, уже получающих кортикостероиды.
ПРОЦЕДУРЫ
Скрининг (В течение 28 дней перед началом лечения (т.е. со дня - 28 по день 1)). Информированное согласие, медицинский анамнез и сопутствующие лекарственные средства; ЭКГ, рентгенографическая оценка опухоли и опухолевые маркеры, при необходимости.
Исходная оценка (в течение 14 дней до начала лечения, т.е. со дня - 14 по день 1). Медицинский осмотр, основные показатели жизнедеятельности, функциональный статус по шкале ECOG, применение сопутствующих лекарственных средств, лабораторные испытания безопасности. Женщины с репродуктивным потенциалом должны иметь отрицательный результат тестирования на беременность по сыворотке в течение 10 дней до начала лечения. Если в анамнезе были какие-либо значимые сердечно-сосудистые заболевания или показатели, предполагающие наличие значимых сердечно-сосудистых заболеваний, то следует получить консультацию кардиолога.
Фаза лечения. Перед инфузией исследуемого лекарственного средства проводили оценку безопасности (включая полный медицинский осмотр). Оценку безопасности (включая полный медицинский осмотр) проводили перед каждым последующим циклом (2+). Кроме того, проводили следующие оценки:
- Общий анализ крови с подсчетом лейкоцитарной формулы/тромбоцитов и клиническую биохимию осуществляли до 72 часов перед днем 1 каждого цикла; дополнительную оценку проводили в цикле 1/в день 15.
- Основные показатели жизнедеятельности (частоту сердечных сокращений, частоту дыхательных движений, кровяное давление и температуру) проверяли в дни инфузии непосредственно до и после каждой инфузии исследуемого лекарственного средства и через 30 и 60 минут после последней инфузии в первом цикле. Во время последующих циклов основные показатели жизнедеятельности проверяли до и после каждой инфузии в процессе медицинского осмотра.
Оценку ответа на лечение проводили во время остального периода второго цикла (и приблизительно каждые 2 цикла после этого).
Лечение продолжали до тех пор, пока не наблюдали признаки прогрессирования заболевания, неприемлемые связанные с лечением нежелательные явления, исследование не было закрыто или пациент был исключен из данного исследования (либо вследствие отзыва информированного согласия, либо по решению исследователя).
Визит по окончании исследования (вне исследования). Все пациенты, получившие по меньшей мере одну дозу исследуемого лекарственного средства и прекратившие лечение по любой причине, за исключением гибели, прошли данную оценку в течение 28 дней с момента последнего введения исследуемого лекарственного средства. Пациенты, которые прошли медицинский осмотр, оценку основных показателей жизнедеятельности, массы тела, документальное подтверждение функционального статуса по шкале ECOG и стандартные лабораторные анализы, включая тест на беременность, будут исключены из исследования.
Визиты последующего наблюдения. Визиты последующего наблюдения были необходимы для отслеживания продолжающихся связанных с лекарственным средством нежелательных явлений и выживаемости. За пациентами со связанными с лекарственным средством нежелательными явлениями ≥2 степени, за которыми наблюдали в рамках оценки по окончании исследования, следует наблюдать ежемесячно до тех пор, пока степень нежелательного явления не уменьшится до <1, или пока указанное нежелательное явление не сочтут хроническим, или пока пациент не получит другую противораковую терапию. Наблюдение за выживаемостью следует осуществлять с 3-месячными интервалами.
ОЦЕНКИ.
ЭФФЕКТИВНОСТЬ. Проводили сравнения конечных результатов эффективности между группой D и группой DN. Главным конечным результатом эффективности является общая выживаемость. Вторичные конечные результаты включают частоту положительного клинического ответа, продолжительность ответа, качество жизни, уровень нейтропении и применение G-CSF.
БЕЗОПАСНОСТЬ. О нежелательных явлениях самостоятельно сообщали сами пациенты, или их замечали во время медицинского осмотра, проверки основных показателей жизнедеятельности, функционального статуса по шкале ECOG и проведения лабораторных анализов.
СТАТИСТИЧЕСКАЯ ОБРАБОТКА.
Эффективность. Распределения общей выживаемости и любого другого времени до развития событий конечного результата суммировали, применяя способ Каплана-Мейера. Логарифмический ранговый критерий применяли для сравнения эффективности конечных результатов между группами лечения. Все статистические анализы осуществляли, применяя односторонние критерии с уровнем значимости 5%. Первичная цель данного исследования состояла в оценке влияния добавления плинабулина на общую выживаемость.
Безопасность. Всех пациентов оценивали в анализе безопасности, если они получали по меньшей мере одну дозу исследуемого лекарственного средства. Результаты анализа безопасности в данной части исследования были представлены в отдельных перечнях и сводных таблицах, включая таблицы частоты нежелательных явлений и таблицы частоты и изменения лабораторных переменных.
Результаты.
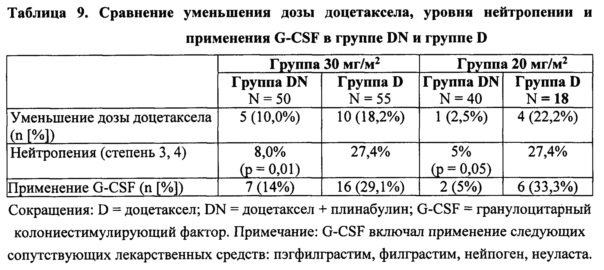
1) Нейтропении уменьшалась в группе, получающей комбинацию плинабулина и доцетаксела.
Профиль переносимости плинабулина, который добавляли к доцетакселу, был лучше, чем таковой для доцетаксела отдельно. Вследствие непереносимых побочных действий доцетаксела, первоначальную дозу доцетаксела, равную 75 мг/м2, уменьшили у 10% пациентов (у 5 из 50) в группе лечения комбинацией плинабулин 30 мг/м2 плюс доцетаксел, тогда как процент таких пациентов в соответствующей группе лечения доцетакселом отдельно был гораздо выше, на уровне 18,2% пациентов (10 из 55). Такой же результат получили для группы лечения 20 мг/м2 плинабулина, в которой меньшему проценту пациентов требовалось уменьшение дозы доцетаксела при лечении комбинацией (2,5%), чем в сопоставляемых группах D (22,2%).
Наблюдали более низкую частоту нейтропении у пациентов в группе DN 30 мг/м2 по сравнению с сопоставляемой группой D (8,0% по сравнению с 36,4%, р<0,001) и в группе DN 20 мг/м2 по сравнению с сопоставляемой группой D (7,5% по сравнению с 22,2%). В группе DN 30 мг/м2 (n=50) наблюдалась значительно более низкая частота нейтропении всех степеней, в частности нейтропении >3 степени, по сравнению с объединенной группой D (n=73), 8,0% по сравнению с 27,4% соответственно (р=0,010). Аналогичные результаты получили для группы 20 мг/м2 (5,0% по сравнению с 27,4% соответственно; р=0,050). Эффект уменьшения нейтропении показан на фиг. 5. Часть пациентов, которым требовался G-CSF, и уровень уменьшения дозы доцетаксела также были ниже в обоих группах DN по сравнению с группами D. Уменьшение процента пациентов, применяющих G-CSF, в группе комбинированного лечения DN по сравнению с группой комбинированного лечения D было статистически значимым на уровне 0,0013%.
2) Положительное влияние комбинации плинабулина и доцетаксела на общую выживаемость (ОВ) популяции пациентов с большими опухолями.
В описанном выше исследовании, в котором сравнивали комбинацию плинабулин (30 мг/м2, день 1 и день 8 каждого 21-дневного цикла) плюс доцетаксел (75 мг/м2, день 1 каждого цикла) с доцетакселом отдельно у пациентов с местнораспространенной или метастатической НМККЛ, которым не помогло по меньшей мере 1 предшествующее химиотерапевтическое лечение, в группе 30 мг/м2 не наблюдалось существенного различия в общей выживаемости группы лечения комбинацией плинабулин плюс доцетаксел (ОВ=8,7 М (месяца)) по сравнению с контрольной группой лечения доцетакселом (ОВ=7,5 М) (таблица 11, фиг. 6), выживаемости без прогрессирования заболевания (ВБП) и частоты положительного клинического ответа. Автор настоящего изобретения затем осуществил подробный анализ результатов, чтобы определить подгруппу, которая реагировала на комбинацию плинабулина и доцетаксела.
В результате анализов подгрупп выявили явное разделение по общей выживаемости при размере опухоли 3 см: в группе, в которой у пациентов была по меньшей мере 1 опухоль >3 см, >5 см, или >7 см, наблюдалось явное преимущество общей выживаемости в группе DN по сравнению с группой D, но во всех группах с опухолями <3 см не наблюдалось различий в общей выживаемости (6,45 М по сравнению с 6,47 М, таблица 11, фиг. 6). Чем больше был размер опухоли, тем более значительным было преимущество общей выживаемости, отношение рисков и частота положительного клинического ответа в группе комбинированного лечения по сравнению с группой, получающей доцетаксел отдельно (таблица 11, фиг. 6).
Таким образом, автор настоящего изобретения обнаружил однозначно определяемую группу пациентов с большими опухолями, которой приносит пользу оптимальная комбинация плинабулина и доцетаксела, которую никогда не описывали ранее в какой-либо литературе.
Все ссылочные материалы, упомянутые в настоящей заявке, включены в данную заявку посредством ссылки, как если бы они были отдельно включены посредством ссылки. Кроме того, должно быть очевидно, что после прочтения описанных выше идей специалист в данной области может осуществить множество их вариаций и модификаций, и данные эквиваленты также входят в объем, определенный прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ УМЕНЬШЕНИЯ НЕЙТРОПЕНИИ | 2018 |
|
RU2790989C2 |
| СПОСОБ ЛЕЧЕНИЯ РАКА, АССОЦИИРОВАННОГО С МУТАЦИЕЙ RAS | 2016 |
|
RU2736045C2 |
| КОМПОЗИЦИЯ И СПОСОБ УМЕНЬШЕНИЯ ВЫЗВАННОЙ ХИМИОТЕРАПИЕЙ НЕЙТРОПЕНИИ С ПОМОЩЬЮ ВВЕДЕНИЯ ПЛИНАБУЛИНА И АГЕНТА Г-КСФ | 2019 |
|
RU2798103C2 |
| СПОСОБ УМЕНЬШЕНИЯ НЕЙТРОПЕНИИ | 2017 |
|
RU2760348C2 |
| КОМПОЗИЦИЯ И СПОСОБ УМЕНЬШЕНИЯ ТРОМБОЦИТОПЕНИИ ПУТЕМ ВВЕДЕНИЯ ПЛИНАБУЛИНА | 2019 |
|
RU2808652C2 |
| КОМБИНИРОВАННАЯ ХИМИОТЕРАПИЯ | 2010 |
|
RU2587013C2 |
| КОМБИНИРОВАННАЯ ХИМИОТЕРАПИЯ | 2006 |
|
RU2429838C2 |
| СПОСОБ ЛЕЧЕНИЯ ОПУХОЛИ ГОЛОВНОГО МОЗГА | 2016 |
|
RU2728796C2 |
| СПОСОБЫ ЛЕЧЕНИЯ СОЛИДНЫХ ОПУХОЛЕЙ | 2017 |
|
RU2737934C2 |
| СПОСОБ И КОМПОЗИЦИИ ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ ТАКСАНОВ ПАЦИЕНТАМ | 1998 |
|
RU2205005C2 |
Настоящая группа изобретений относится к медицине, а именно к онкологии, и касается лечения рака. Для этого вводят плинабулин в сочетании с таксаном. Такое сочетанное введение обеспечивает возможность лечения опухолей большого размера - более 3 см, а также снижение нейтропении 3 или 4 степени, развившейся в результате монотерапии таксаном. 2 н. и 19 з.п. ф-лы, 6 ил., 10 табл., 3 пр.
1. Способ лечения вызванной таксаном нейтропении 3 или 4 степени у субъекта, включающий введение плинабулина указанному субъекту и введение таксана.
2. Способ по п. 1, включающий введение плинабулина перед введением таксана.
3. Способ по п. 1, включающий введение плинабулина после введения таксана.
4. Способ по любому из пп. 1-3, отличающийся тем, что указанное введение приводит к уровню нейтропении, более низкому, чем вызванный таксаном отдельно.
5. Способ по любому из пп. 1-3, отличающийся тем, что таксан представляет собой доцетаксел.
6. Способ по п. 5, включающий введение доцетаксела в дозе, приблизительно составляющей от 45 мг/м2 до 120 мг/м2.
7. Способ по п. 6, включающий введение доцетаксела в дозе, приблизительно составляющей от 50 мг/м2 до 75 мг/м2.
8. Способ по любому из пп. 1-3, отличающийся тем, что таксан представляет собой паклитаксел.
9. Способ по п. 8, включающий введение паклитаксела в дозе, приблизительно составляющей от 50 до 250 мг/м2.
10. Способ по любому из пп. 1-3, отличающийся тем, что таксан представляет собой абраксан.
11. Способ по любому из пп. 1-3, 6, 7 и 9, включающий введение плинабулина в дозе, приблизительно составляющей от 10 до 50 мг/м2.
12. Способ по п. 11, включающий введение плинабулина в дозе, приблизительно составляющей 20 мг/м2 или 30 мг/м2.
13. Способ по любому из пп. 1-3, 6, 7, 9 и 12, отличающийся тем, что массовое отношение дозировки таксана и дозировки плинабулина приблизительно составляет от 1,5:1 до 4:1.
14. Способ по п. 1 или 3, отличающийся тем, что введение плинабулина осуществляют приблизительно через 1-24 часа после введения таксана.
15. Способ по п. 14, отличающийся тем, что плинабулин вводят приблизительно через 2 часа после введения таксана.
16. Способ по любому из пп. 1-3, 6, 7, 9, 12 и 15, отличающийся тем, что степень вызванной таксаном нейтропении уменьшается на по меньшей мере 4% для нейтропении 3 степени.
17. Способ по любому из пп. 1-3, 6, 7, 9, 12 и 15, отличающийся тем, что степень вызванной таксаном нейтропении уменьшается на по меньшей мере 15% для нейтропении 4 степени.
18. Способ лечения рака у пациента, размер опухоли у которого больше, чем 3 см, включающий совместное введение таксана и плинабулина.
19. Способ по п. 18, отличающийся тем, что плинабулин вводят по меньшей мере через 1 час после введения таксана.
20. Способ по п. 18 или 19, отличающийся тем, что размер опухоли у пациента больше, чем 5 см.
21. Способ по любому из пп. 18-19, отличающийся тем, что размер опухоли у пациента больше, чем 7 см.
| RU 2011148945 A, 10.06.2013 | |||
| WO 2005077940 A1, 25.08.2005 | |||
| MILLWARD M | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| WILT JA et al | |||
| "Anal cancer in Alaska; a retrospective study of incidence, treatment, and outcomes" | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |

