РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет и преимущества на основании предварительной заявки США №61/714140, поданной 15 октября 2012 года, №61/714145, поданной 15 октября 2012 года, №61/780703, поданной 13 марта 2013 года, и №61/786277, поданной 14 марта 2013 года. Полное содержание каждой из данных предварительных заявок включено в настоящий документ посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
В настоящее время существует необходимость в разработке новых агентов в качестве ингибиторов активности EZH2, которые можно применять для лечения EZH2-опосредуемого нарушения (например, рака).
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Один из аспектов настоящего изобретения относится к содержащему заместители бензольному соединению, выбранному из
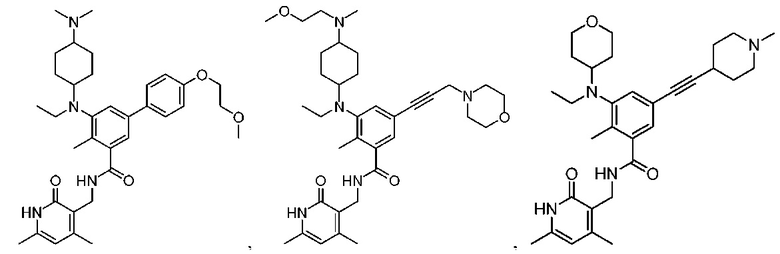 и их фармацевтически приемлемых солей.
и их фармацевтически приемлемых солей.
Например, указанное соединение представляет собой 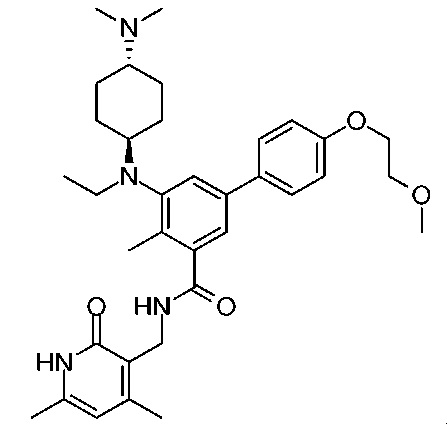 или его фармацевтически приемлемую соль.
или его фармацевтически приемлемую соль.
Например, соединение представляет собой 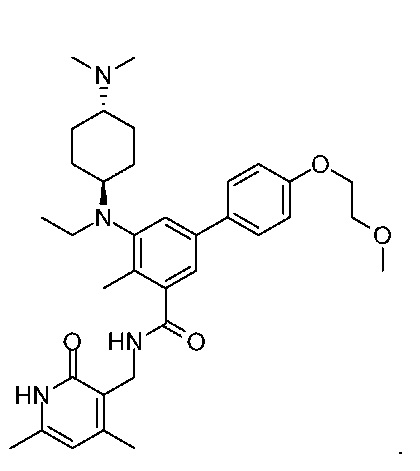
Например, соединение представляет собой 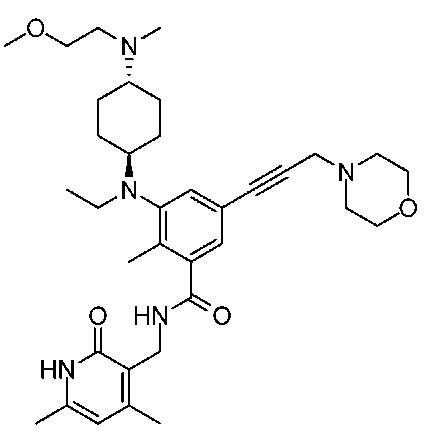 или его фармацевтически приемлемую соль.
или его фармацевтически приемлемую соль.
Например, соединение представляет собой 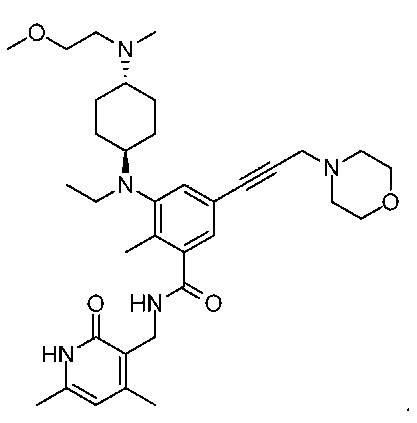
Например, соединение представляет собой 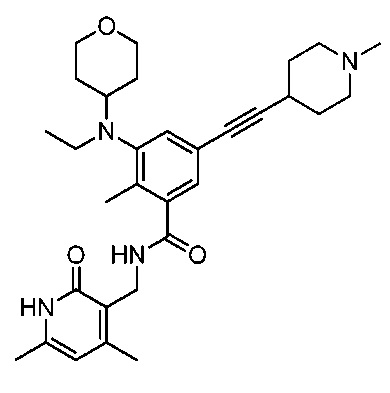 или его фармацевтически приемлемую соль.
или его фармацевтически приемлемую соль.
Например, соединение представляет собой 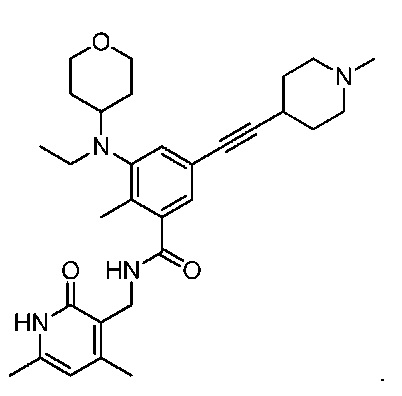
Согласно настоящему изобретению также предложены фармацевтические композиции, содержащие один или более фармацевтически приемлемых носителей и одно или более соединений, описанных в настоящем документе.
Другой аспект настоящего изобретения представляет собой способ лечения или предотвращения EZH2-опосредуемого нарушения. Указанный способ включает введение нуждающемуся в этом субъекту терапевтически эффективного количества одного или более соединений, описанных в настоящем документе. EZH2-опосредуемое нарушение представляет собой заболевание, нарушение или состояние, которое по меньшей мере отчасти опосредуется активностью EZH2. В одном из вариантов реализации EZH2-опосредуемое нарушение связано с повышенной активностью EZH2. В одном из вариантов реализации EZH2-опосредуемое нарушение представляет собой рак. EZH2-опосредуемый рак может представлять собой лимфому, лейкоз или меланому, например, диффузную крупноклеточную В-клеточную лимфому (ДКВЛ), неходжкинскую лимфому (НХЛ), фолликулярную лимфому, хронический миелогенный лейкоз (ХМЛ), острый миелоидный лейкоз, острый лимфоцитарный лейкоз, недифференцированный лейкоз или миелодиспластические синдромы (МДС). В одном из вариантов реализации EZH2-опосредуемый рак может представлять собой злокачественную рабдоидную опухоль или опухоль с дефицитом INI1. Гистологический диагноз злокачественной рабдоидной опухоли зависит от идентификации характерных рабдоидных клеток (крупные клетки с эксцентрично расположенными ядрами и обильной эозинофильной цитоплазмой) и иммуногистохимии с антителами к виментину, кератину и эпителиальному мембранному антигену. В большинстве злокачественных рабдоидных опухолей ген SMARCB1/INI1, расположенный в сегменте хромосомы 22q11.2, инактивирован, вследствие делеций и/или мутаций. В одном из вариантов реализации злокачественные рабдоидные опухоли могут представлять собой опухоль с дефицитом INI1.
Если не указано иное, любое описание способа лечения включает применение соединений для обеспечения такого лечения или профилактики, как указано в настоящем описании, а также применение соединений для получения лекарственного средства для лечения или предотвращения такого состояния. Указанное лечение включает лечение животных, являющихся человеком или не являющихся человеком, включая грызунов, и других моделей заболеваний.
Более того, соединения или способы, описанные в настоящем документе, можно применять для исследования (например, исследования эпигенетических ферментов) и для других нетерапевтических целей.
В некоторых вариантах реализации предпочтительные соединения, описанные в настоящем документе, обладают желаемыми фармакологическими и/или фармакокинетическими свойствами, например, имеют низкие скорости клиренса и/или ограниченный риск нежелательных взаимодействий лекарственных средств в комбинированной терапии, оцениваемый, например, по зависимому от времени и обратимому ингибированию ферментов системы цитохрома P-450.
Если не определено иное, все технические и научные термины, употребляемые в настоящем описании, имеют такое значение, которое обычно понятно специалисту в области техники, к которой относится настоящее изобретение. В настоящем описании формы единственного числа также включают множества объектов, если в контексте явным образом не указано иное. Несмотря на то, что при практическом применении или тестировании настоящего изобретения можно использовать способы и вещества, аналогичные или эквивалентные описанным в настоящем документе, ниже описаны подходящие способы и вещества. Содержание всех публикаций, заявок на патент, патентов и других источников, упомянутых в настоящем документе, включено посредством ссылки. Источники, указанные в настоящем описании, не признаются уровнем техники по отношению к заявленному изобретению. В случае противоречия настоящее описание, включая определения, будет иметь преимущественную силу. Кроме того, вещества, способы и примеры приведены только в качестве иллюстрации и не являются ограничивающими. В случае противоречия между химическими структурами и названиями соединений, описанных в настоящем документе, химические структуры будут иметь преимущественную силу.
Другие признаки и преимущества настоящего изобретения будут очевидны, исходя из следующего подробного описания и формулы изобретения.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг. 1 представляет собой график, демонстрирующий противоопухолевое действие перорально вводимого соединения 1 или соединения А в отношении ксенотрансплантата лимфомы KARPAS-422 у мышей. Данные представляют собой среднее значение ± SD (n = 10).
Фиг. 2 представляет собой график, демонстрирующий влияние соединения 1 или соединения А на массу тела мыши. Данные представляют собой среднее значение ± SD (n = 10).
Фиг. 3 представляет собой график, демонстрирующий концентрацию соединения 1 в опухоли в 7 день или 28 день после лечения или концентрацию соединения А в опухоли в 7 день после лечения. На данной фигуре «А»-«G» обозначают 7 дней после введения соединения 1 в дозах, составляющих 62,5, 83,3, 125, 166,7, 250, 333,3 и 500 мг/кг соответственно; «Н» и «I» обозначают 7 дней после введения соединения А в дозах, составляющих 125 и 250 мг/кг соответственно; и «J»-«L» обозначают 28 дней после введения соединения 1 в дозах, составляющих 62,5, 125 и 250 мг/кг соответственно.
Фиг. 4 представляет собой график, демонстрирующий концентрацию соединения 1 или соединения А в плазме крови в 7 день или 28 день после лечения. Верхняя пунктирная линия показывает наименьшую цитотоксическую концентрацию (LCC) соединения А с поправкой на связывание с белками плазмы крови (PPB), а нижняя пунктирная линия показывает LCC соединения 1 с поправкой на PPB.
Фиг. 5 представляет собой график, демонстрирующий глобальное метилирование H3K27me3 в опухолях KARPAS-422 у мышей, которых лечили соединением 1 или соединением А в течение 7 дней. На данной фигуре «А» обозначает лечение носителем; «В»-«Н» обозначают лечение соединением 1 в дозах, составляющих 62,5, 83,3, 125, 166,7, 250, 333,3 и 500 мг/кг соответственно; и «I» и «J» обозначают лечение соединением А в дозах, составляющих 125 и 250 мг/кг соответственно.
Фиг. 6 представляет собой график, демонстрирующий глобальное метилирование H3K27me3 в опухолях KARPAS-422 у мышей, которых лечили соединением 1 в течение 28 дней.
Фиг. 7 представляет собой график, демонстрирующий глобальное метилирование H3K27me3 в костном мозге мышей с ксенотрансплантатными опухолями KARPAS-422, которых лечили соединением 1 или соединением А в течение 7 дней. На данной фигуре «А» обозначает лечение носителем; «В»-«Н» обозначают лечение соединением 1 в дозах, составляющих 62,5, 83,3, 125, 166,7, 250, 333,3 и 500 мг/кг соответственно; и «I» и «J» обозначают лечение соединением А в дозах, составляющих 125 и 250 мг/кг соответственно.
Фиг. 8 представляет собой график, демонстрирующий глобальное метилирование H3K27me3 в костном мозге мышей с ксенотрансплантатными опухолями KARPAS-422, которых лечили соединением 1 в течение 28 дней. На данной фигуре «А» обозначает лечение носителем; «В»-«Е» обозначают лечение соединением 1 в дозах, составляющих 62,5, 125, 250 и 500 мг/кг соответственно; и «F» обозначает лечение соединением А в дозе, составляющей 250 мг/кг.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению предложены новые содержащие заместители бензольные соединения, способы синтеза для получения указанных соединений, содержащие их фармацевтические композиции и различные способы применения указанных соединений.
Типичные соединения согласно настоящему изобретению включают соединения, перечисленные в таблице 1.
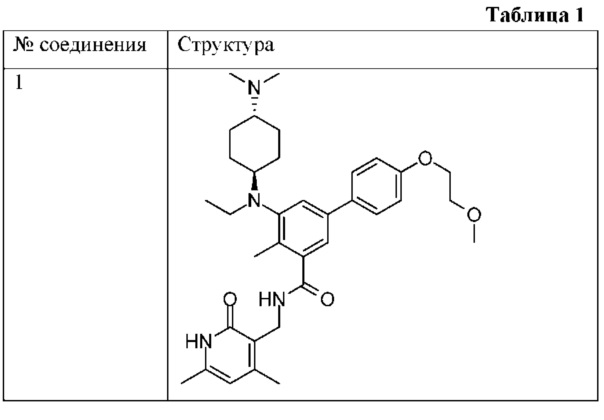
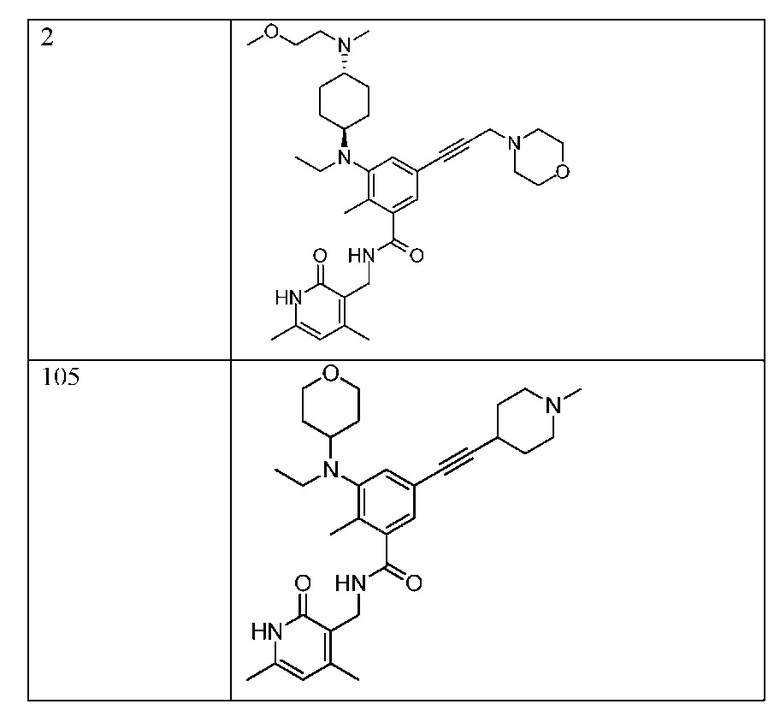
В настоящем описании структурная формула соединения в некоторых случаях для удобства представляет собой определенный изомер, но настоящее изобретение может включать все изомеры, такие как геометрические изомеры, оптические изомеры на основе асимметричного атома углерода, стереоизомеры, таутомеры, энантиомеры, ротамеры, диастереомеры, рацематы и т.п., при этом понятно, что не все изомеры могут обладать одинаковым уровнем активности. Кроме того, для соединений, представленных формулой, может быть характерен кристаллический полиморфизм. Следует отметить, что любая кристаллическая форма, смесь кристаллических форм или ее ангидрид или гидрат включены в объем настоящего изобретения.
Термин «изомерия» означает соединения, которые имеют одинаковые молекулярные формулы, но отличаются последовательностью связывания атомов или расположением атомов в пространстве. Изомеры, которые отличаются расположением атомов в пространстве, называются «стереоизомерами». Стереоизомеры, которые не являются зеркальными отображениями друг друга, называются «диастереоизомерами», и стереоизомеры, которые являются неналагающимися зеркальными отображениями друг друга, называются «энантиомерами» или иногда оптическими изомерами. Смесь, содержащая равные количества отдельных энантиомерных форм с противоположной хиральностью, называется «рацемической смесью».
Термин «геометрический изомер» означает диастереомеры, существование которых обусловлено затрудненным вращением вокруг двойных связей или циклоалкильного линкера (например, 1,3-циклобутил). Данные конфигурации различаются в названиях приставками «цис» и «транс», или Z и Е, которые указывают на то, что группы находятся по одну сторону или по разные стороны от двойной связи в молекуле в соответствии с правилами Кана-Ингольда-Прелога (Cahn-Ingold-Prelog).
Следует понимать, что соединения согласно настоящему изобретению могут быть изображены в виде стереоизомеров. Следует также понимать, что, когда соединения имеют стереоизомерные формы, все стереоизомерные формы включены в объем настоящего изобретения, при этом понятно, что не все стереоизомеры могут обладать одинаковым уровнем активности.
Кроме того, структуры и другие соединения, рассмотренные согласно настоящему изобретению, включают все их атропоизомеры, при этом понятно, что не все атропоизомеры могут обладать одинаковым уровнем активности. «Атропоизомеры» представляют собой тип стереоизомера, в котором атомы двух изомеров по-разному расположены в пространстве. Существование атропоизомеров обусловлено ограниченным вращением, вызванным затруднением вращения крупных групп вокруг центральной связи. Такие атропоизомеры, как правило, существуют в виде смеси, однако благодаря последним достижениям в области методов хроматографии в отдельных случаях стало возможным разделение смесей двух атропоизомеров.
«Таутомер» представляет собой один из двух или более структурных изомеров, которые существуют в равновесии, и легко превращается из одной изомерной формы в другую. Данное превращение приводит к формальному переносу атома водорода, сопровождающемуся переходом соседних сопряженных двойных связей. Таутомеры существуют в виде смеси набора таутомеров в растворе. В растворах, где возможна таутомеризация, будет достигаться химическое равновесие таутомеров. Точное соотношение таутомеров зависит от нескольких факторов, включая температуру, растворитель и рН. Понятие таутомеров, которые являются взаимопревращаемыми в результате таутомеризации, называется таутомерией.
Из различных возможных типов таутомерии обычно наблюдают два. В случае кето-енольной таутомерии происходит одновременный сдвиг электронов и атома водорода. Кольчато-цепная таутомерия является результатом реакции альдегидной группы (-CHO) в цепной молекуле сахара с одной из гидроксильных групп (-OH) в той же молекуле с образованием циклической (кольцевой) формы, демонстрируемой глюкозой.
В настоящем описании любой случай 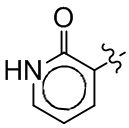 следует рассматривать как
следует рассматривать как 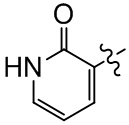 .
.
Распространенными таутомерными парами являются: кетон-енол, амид-нитрил, лактам-лактим, таутомерия амид-имидокислота в гетероциклических кольцах (например, в нуклеотидных основаниях, таких как гуанин, тимин и цитозин), имин-енамин и енамин-енамин. Примером кето-енольного равновесия является равновесие между пиридин-2(1Н)-онами и соответствующими пиридин-2-олами, как показано ниже.
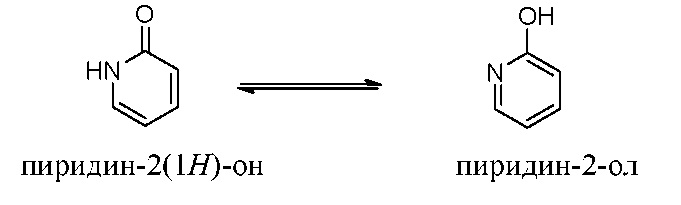
Следует понимать, что соединения согласно настоящему изобретению могут быть изображены в виде различных таутомеров. Следует также понимать, что, когда соединения имеют таутомерные формы, все таутомерные формы включены в объем настоящего изобретения, и название соединений не исключает какую-либо таутомерную форму. Очевидно, что некоторые таутомеры могут обладать более высоким уровнем активности, чем другие.
Термин «кристаллические полиморфы», «полиморфы» или «кристаллические формы» означает кристаллические структуры, в которые соединение (или его соль или сольват) может кристаллизоваться с различной кристаллической упаковкой при одном и том же элементном составе. Разные кристаллические формы обычно имеют разные рентгеновские дифрактограммы, инфракрасные спектры, температуры плавления, плотность, твердость, форму кристаллов, оптические и электрические свойства, стабильность и растворимость. Растворитель для перекристаллизации, скорость кристаллизации, температура хранения и другие факторы могут вызывать преобладание одной кристаллической формы. Кристаллические полиморфы соединений могут быть получены путем кристаллизации в различных условиях.
Соединения согласно настоящему изобретению включают сами соединения, такие как соединения любой из формул, описанных в настоящем документе. Соединения согласно настоящему изобретению могут также включать их соли и их сольваты при необходимости. Соль, например, может быть образована между анионом и положительно заряженной группой (например, амино) в содержащем заместители бензольном соединении. Подходящие анионы включают хлорид, бромид, йодид, сульфат, бисульфат, сульфамат, нитрат, фосфат, цитрат, метансульфонат, трифторацетат, глутамат, глюкуронат, глутарат, малат, малеат, сукцинат, фумарат, тартрат, тозилат, салицилат, лактат, нафталинсульфонат и ацетат (например, трифторацетат). Термин «фармацевтически приемлемый анион» относится к аниону, подходящему для образования фармацевтически приемлемой соли. Кроме того, соль также может быть образована между катионом и отрицательно заряженной группой (например, карбоксилатом) в содержащем заместители бензольном соединении. Подходящие катионы включают ион натрия, ион калия, ион магния, ион кальция и катион аммония, такой как ион тетраметиламмония. Содержащие заместители бензольные соединения также включают соли, содержащие четвертичные атомы азота.
Кроме того, соединения согласно настоящему изобретению, например, соли указанных соединений, могут существовать либо в гидратированной, либо в негидратированной (безводной) форме, либо в виде сольватов с молекулами другого растворителя. Неограничивающие примеры гидратов включают моногидраты, дигидраты и т.д. Неограничивающие примеры сольватов включают этанольные сольваты, ацетоновые сольваты и т.д.
Термин «сольват» означает формы присоединения растворителя, которые содержат либо стехиометрические, либо нестехиометрические количества растворителя. Некоторые соединения склонны удерживать фиксированное молярное соотношение молекул растворителя в твердом кристаллическом состоянии, таким образом образуя сольват. Если растворителем является вода, то образуемый сольват представляет собой гидрат; и если растворителем является спирт, то образуемый сольват представляет собой алкоголят. Гидраты образуются путем комбинации одной или более молекул воды с одной молекулой вещества, в котором вода сохраняет свое молекулярное состояние в виде Н2О.
В настоящем описании термин «аналог» относится к химическому соединению, которое является структурно схожим с другим соединением, но несколько отличается по составу (как в случае замены одного атома атомом другого элемента или в случае присутствия конкретной функциональной группы, или замены одной функциональной группы другой функциональной группой). Таким образом, аналог представляет собой соединение, схожее или сопоставимое по функции и внешнему виду, но не по структуре или происхождению, с исходным соединением.
В настоящем описании термин «производное» относится к соединениям, которые имеют общую базовую структуру и содержат в качестве заместителей различные группы, описанные в настоящем документе. Например, все соединения в таблице 1 представляют собой содержащие заместители бензольные соединения и содержат общую базовую основу.
Термин «биоизостер» относится к соединению, которое образуется в результате замены атома или группы атомов другим схожим в широком смысле атомом или группой атомов. Задачей биоизостерической замены является получение нового соединения, обладающего биологическими свойствами, схожими с исходным соединением. Биоизостерическая замена может быть на физико-химической или топологической основе. Примеры биоизостеров карбоновых кислот включают, но не ограничиваются ими, ацилсульфонимиды, тетразолы, сульфонаты и фосфонаты. См., например, Patani and LaVoie, Chem. Rev. 96, 3147-3176, 1996.
Настоящее изобретение включает все изотопы атомов, встречающихся в соединениях согласно настоящему изобретению. Изотопы включают такие атомы, которые имеют одинаковый атомный номер, но разные массовые числа. В качестве общего примера и без ограничения, изотопы водорода включают тритий и дейтерий, а изотопы углерода включают С-13 и С-14.
Согласно настоящему изобретению предложены способы синтеза соединений, описанных в настоящем документе. Согласно настоящему изобретению также предложены подробно рассмотренные способы синтеза различных описанных соединений согласно настоящему изобретению в соответствии со схемами, представленными в разделе «Примеры».
На всем протяжении настоящего описания в случае, когда композиции описаны как имеющие, включающие или содержащие конкретные компоненты, предполагается, что композиции также состоят или по существу состоят из перечисленных компонентов. Подобным образом, в случае, когда методы или способы описаны как имеющие, включающие или содержащие конкретные этапы способа, указанные способы также состоят или по существу состоят из перечисленных этапов способов. Кроме того, следует понимать, что порядок этапов или порядок выполнения определенных действий не имеет значения, если настоящее изобретение остается реализуемым. Более того, два или более этапов или действий можно осуществлять одновременно.
Способы синтеза согласно настоящему изобретению могут допускать широкий спектр функциональных групп, поэтому можно использовать различные содержащие заместители исходные вещества. Указанные способы, в целом, позволяют получить желаемое конечное соединение в конце или под конец общего способа, хотя в некоторых случаях может быть желательным дальнейшее превращение соединения в его фармацевтически приемлемую соль.
Соединения согласно настоящему изобретению могут быть получены различными способами с использованием коммерчески доступных исходных веществ, соединений, известных в литературе, или из легко получаемых промежуточных соединений путем использования стандартных способов и процедур синтеза, которые либо известны специалисту в данной области техники, либо будут очевидны специалисту в данной области техники в свете идей, описанных в настоящем документе. Стандартные способы и процедуры синтеза для получения органических молекул и трансформаций и манипуляций с функциональными группами можно найти в соответствующей научной литературе или в стандартных руководствах в данной области. Несмотря на отсутствие ограничений каким-либо одним или несколькими источниками, классические руководства, такие как Smith, M.B., March, J., March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th edition, John Wiley & Sons: New York, 2001; Greene, T.W., Wuts, P.G. M., Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons: New York, 1999; R. Larock, Comprehensive Organic Transformations, VCH Publishers (1989); L. Fieser and M. Fieser, Fieser and Fieser's Reagents for Organic Synthesis, John Wiley and Sons (1994); и L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995), содержание которых включено в настоящее описание посредством ссылки, являются полезными и общепризнанными эталонными руководствами по органическому синтезу, известными специалисту в данной области техники. Следующее описание способов синтеза предназначено для иллюстрации, а не для ограничения, общих процедур для получения соединений согласно настоящему изобретению.
Для специалиста в данной области техники очевидно, что для некоторых групп может быть необходима защита от условий реакций путем использования защитных групп. Защитные группы можно также использовать для различия схожих функциональных групп в молекулах. Перечень защитных групп и способы введения и удаления данных групп можно найти в Greene, T.W., Wuts, P.G. M., Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons: New York, 1999.
Предпочтительные защитные группы включают, но не ограничиваются ими:
Для гидроксильной группы: TBS, бензил, ТНР, Ac
Для карбоновых кислот: сложный бензиловый эфир, сложный метиловый эфир, сложный этиловый эфир, сложный аллиловый эфир
Для аминов: Cbz, BOC, DMB
Для диолов: Ac (×2) TBS (×2) или взятые вместе ацетониды
Для тиолов: Ac
Для бензимидазолов: SEM, бензил, PMB, DMB
Для альдегидов: диалкилацетали, такие как диметоксиацеталь или диэтилацетил.
В схемах реакций, описанных в настоящем документе, может быть получено несколько стереоизомеров. Когда не указан конкретный стереоизомер, следует понимать все возможные стереоизомеры, которые могут быть получены в результате реакции. Для специалиста в данной области техники очевидно, что реакции могут быть оптимизированы для получения предпочтительно одного изомера, или могут быть разработаны новые схемы для получения одного изомера. Если получают смеси, для разделения изомеров могут быть использованы такие методы, как препаративная тонкослойная хроматография, препаративная ВЭЖХ, препаративная хиральная ВЭЖХ или препаративная сверхкритическая жидкостная хроматография (SFC).
На всем протяжении настоящего описания используются следующие сокращения, которые определены ниже:
Соединения согласно настоящему изобретению можно удобным образом получать различными способами, известными специалисту в данной области техники. Соединения согласно настоящему изобретению c любой формулой, описанной в настоящем документе, могут быть получены в соответствии с процедурами, проиллюстрированными в приведенных ниже примерах, из коммерчески доступных исходных веществ или исходных веществ, которые могут быть получены с использованием процедур, описанных в литературе.
Специалисту в данной области техники следует отметить, что в случае последовательностей реакций и схем синтеза, описанных в настоящем документе, порядок некоторых этапов может быть изменен, например, введение и удаление защитных групп.
Соединения согласно настоящему изобретению ингибируют гистонметилтрансферазную активность EZH2 или его мутанта, и, соответственно, в соответствии с одним из аспектов настоящего изобретения некоторые соединения, описанные в настоящем документе, являются кандидатами для лечения или предотвращения некоторых состояний и заболеваний, при которых EZH2 играет роль. Согласно настоящему изобретению предложены способы лечения состояний и заболеваний, на течение которых можно влиять путем модуляции статуса метилирования гистонов или других белков, при этом указанный статус метилирования опосредуется по меньшей мере отчасти активностью EZH2. Модуляция статуса метилирования гистонов может в свою очередь влиять на уровень экспрессии целевых генов, активируемых путем метилирования, и/или целевых генов, подавляемых путем метилирования. Указанный способ включает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, полиморфа, сольвата или стереоизомера.
Если не указано иное, любое описание способа лечения включает применение соединений для обеспечения такого лечения или профилактики, как указано в настоящем описании, а также применение соединений для получения лекарственного средства для лечения или предотвращения такого состояния. Указанное лечение включает лечение животных, относящихся к человеку или не относящихся к человеку, включая грызунов, и других моделей заболеваний.
Другой аспект настоящего изобретения относится к способу модулирования активности EZH2, каталитической субъединицы комплекса PRC2, которая катализирует моно - триметилирование лизина 27 в гистоне Н3 (Н3-K27), у нуждающегося в этом субъекта. Например, указанный способ включает этап введения субъекту, страдающему раком, экспрессирующим мутантный EZH2, терапевтически эффективного количества соединения, описанного в настоящем документе, при этом указанное соединение (соединения) ингибирует гистонметилтрансферазную активность EZH2, осуществляя тем самым лечение рака.
Например, EZH2-опосредуемый рак выбран из группы, состоящей из фолликулярной лимфомы и диффузной крупноклеточной В-клеточной лимфомы (ДКВЛ) подтипа, представляющего собой лимфому из В-клеток герминативного центра (germinal center B cell-like subtype, GCB). Например, рак представляет собой лимфому, лейкоз или меланому. Предпочтительно, лимфома представляет собой неходжкинскую лимфому (НХЛ), фолликулярную лимфому или диффузную крупноклеточную В-клеточную лимфому. В качестве альтернативы, лейкоз представляет собой хронический миелогенный лейкоз (ХМЛ), острый миелоидный лейкоз, острый лимфоцитарный лейкоз или недифференцированный лейкоз.
Например, EZH2-опосредуемое предраковое состояние представляет собой миелодиспластические синдромы (МДС, ранее известные как предлейкоз).
Например, EZH2-опосредуемый рак представляет собой гематологическую злокачественную опухоль.
Соединение (соединения) согласно настоящему изобретению ингибирует гистонметилтрансферазную активность EZH2 или его мутанта, и, соответственно, согласно настоящему изобретению также предложены способы лечения состояний и заболеваний, на течение которых можно влиять путем модуляции статуса метилирования гистонов или других белков, при этом указанный статус метилирования опосредуется по меньшей мере отчасти активностью EZH2. В соответствии с одним из аспектов настоящего изобретения некоторые соединения, описанные в настоящем документе, являются кандидатами для лечения или предотвращения некоторых состояний и заболеваний. Модуляция статуса метилирования гистонов может в свою очередь влиять на уровень экспрессии целевых генов, активируемых путем метилирования, и/или целевых генов, подавляемых путем метилирования. Указанный способ включает введение субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения согласно настоящему изобретению.
В настоящем описании термины «субъект» и «нуждающийся в этом субъект» являются взаимозаменяемыми и оба из них относятся к субъекту, страдающему нарушением, при котором EZH2-опосредуемое метилирование белка играет роль, или субъекту, подвергающемуся повышенному риску развития такого нарушения относительно населения в целом. Термин «субъект» включает млекопитающее. Указанное млекопитающее может представлять собой, например, человека или соответствующее млекопитающее, не являющееся человеком, такое как примат, мышь, крыса, собака, кошка, корова, лошадь, коза, верблюд, овца или свинья. Субъект может также представлять собой птицу или домашнюю птицу. В одном из вариантов реализации млекопитающее представляет собой человека. Нуждающийся в этом субъект может представлять собой субъект, у которого ранее был диагностирован или установлен рак или предраковое состояние. Нуждающийся в этом субъект может также представлять собой субъект, имеющий рак или предраковое состояние (например, страдающий ими). В качестве альтернативы, нуждающийся в этом субъект может представлять собой субъект, подвергающийся повышенному риску развития такого нарушения относительно населения в целом (т.е. субъект, который предрасположен к развитию такого нарушения относительно населения в целом). Нуждающийся в этом субъект может страдать предраковым состоянием. Нуждающийся в этом субъект может страдать неподдающимся лечению или резистентным раком (т.е. раком, который не отвечает или еще не ответил на лечение). Субъект может быть невосприимчив в начале лечения или может стать невосприимчивым во время лечения. В некоторых вариантах реализации нуждающийся в этом субъект демонстрирует рецидив рака после ремиссии в процессе последней терапии. В некоторых вариантах реализации нуждающийся в этом субъект получил и на него не подействовали все известные эффективные виды терапии для лечения рака. В некоторых вариантах реализации, нуждающийся в этом субъект, получил по меньшей мере одну предшествующую терапию. В предпочтительном варианте реализации субъект страдает раком или раковым состоянием. Например, рак представляет собой лимфому, лейкоз, меланому или рабдомиосаркому. Предпочтительно, лимфома представляет собой неходжкинскую лимфому, фолликулярную лимфому или диффузную крупноклеточную В-клеточную лимфому. В качестве альтернативы, лейкоз представляет собой хронический миелогенный лейкоз (ХМЛ). Предраковое состояние представляет собой миелодиспластические синдромы (МДС, ранее известные, как предлейкоз).
В настоящем описании термин «лечение» или «лечить» описывает оказание помощи и уход за пациентом с целью борьбы с заболеванием, состоянием или нарушением, и включает введение соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, полиморфа или сольвата для облегчения симптомов или осложнений заболевания, состояния или нарушения, или для устранения указанного заболевания, состояния или нарушения. Термин «лечить» может также включать обработку клетки in vitro или лечение модели у животного.
Соединение согласно настоящему изобретению или его фармацевтически приемлемую соль, полиморф или сольват можно или также можно применять для предотвращения соответствующего заболевания, состояния или нарушения, или применять для идентификации подходящих кандидатов для данных целей. В настоящем описании термин «предотвращение», «предотвращать» или «защита от» описывает уменьшение или устранение появления симптомов или осложнений такого заболевания, состояния или нарушения.
Существуют данные о том, что точечные мутации гена EZH2 в одном аминокислотном остатке (например, Y641, A677 и A687) EZH2 связаны с лимфомой. Дополнительные примеры мутантов EZH2 и способов детектирования мутации, и способов лечения нарушений, связанных с мутациями, описаны, например, в публикации заявки на патент США № US 20130040906, полное содержание которой включено в настоящее описание посредством ссылки.
Специалист в данной области техники может обратиться к общим эталонным руководствам для подробного описания известных методов, рассмотренных в настоящем документе, или эквивалентных методов. Данные руководства включают Ausubel et al., Current Protocols in Molecular Biology, John Wiley and Sons, Inc. (2005); Sambrook et al., Molecular Cloning, A Laboratory Manual (3rd edition), Cold Spring Harbor Press, Cold Spring Harbor, New York (2000); Coligan et al., Current Protocols in Immunology, John Wiley & Sons, N.Y.; Enna et al., Current Protocols in Pharmacology, John Wiley & Sons, N.Y.; Fingl et al., The Pharmacological Basis of Therapeutics (1975), Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, PA, 18th edition (1990). К данным руководствам можно, конечно, также обращаться при получении или использовании аспекта настоящего изобретения.
В настоящем описании термин «комбинированная терапия» или «совместная терапия» включает введение соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, полиморфа или сольвата и по меньшей мере второго агента в рамках конкретной схемы лечения, направленной на обеспечение полезного эффекта в результате совместного действия данных терапевтических агентов. Полезный эффект такой комбинации включает, но не ограничивается ими, фармакокинетическое или фармакодинамическое совместное действие, обусловленное комбинацией терапевтических агентов.
Согласно настоящему изобретению также предложены фармацевтические композиции, содержащие соединение, описанное в настоящем документе, в комбинации по меньшей мере с одним фармацевтически приемлемым вспомогательным веществом или носителем.
«Фармацевтическая композиция» представляет собой состав, содержащий соединения согласно настоящему изобретению в форме, подходящей для введения субъекту. В одном из вариантов реализации фармацевтическая композиция представлена в нерасфасованной форме или в единичной лекарственной форме. Единичная лекарственная форма представляет любую из множества форм, включая, например, капсулу, пакет для внутривенного введения, таблетку, дозированный картридж в аэрозольном ингаляторе или флакон. Количество активного ингредиента (например, состава описанного соединения или его соли, гидрата, сольвата или изомера) в однократной дозе композиции является эффективным количеством и варьируется в соответствии с конкретным применяемым лечением. Для специалиста в данной области техники очевидно, что иногда необходимо проводить обычные изменения дозы в зависимости от возраста и состояния пациента. Доза будет также зависеть от пути введения. Предусмотрены различные пути, включая пероральный, через легкие, ректальный, парентеральный, трансдермальный, подкожный, внутривенный, внутримышечный, интраперитонеальный, ингаляционный, трансбуккальный, сублингвальный, внутриплевральный, интратекальный, интраназальный и т.п. Лекарственные формы для топического или трансдермального введения соединения согласно настоящему изобретению включают порошки, спреи, мази, пасты, кремы, лосьоны, гели, растворы, пластыри и препараты для ингаляции. В одном из вариантов реализации активное соединение смешано в стерильных условиях с фармацевтически приемлемым носителем и с любыми консервантами, буферами или пропеллентами, которые необходимы.
В настоящем описании термин «фармацевтически приемлемый» относится к соединениям, анионам, катионам, веществам, композициям, носителям и/или лекарственным формам, которые с медицинской точки зрения подходят для применения в контакте с тканями людей и животных, не вызывая при этом чрезмерной токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, в соответствии с разумным соотношением польза/риск.
Термин «фармацевтически приемлемое вспомогательное вещество» означает вспомогательное вещество, которое подходит для получения фармацевтической композиции, которое является в целом безопасным, нетоксичным и не является ни биологически, ни в другом отношении нежелательным, и включает вспомогательное вещество, приемлемое для применения в ветеринарии, а также применения в фармацевтике. В настоящем описании и формуле изобретения термин «фармацевтически приемлемое вспомогательное вещество» включает как одно, так и более одного такого вспомогательного вещества.
Фармацевтическую композицию согласно настоящему изобретению изготавливают таким образом, чтобы она была совместима с предполагаемым путем введения. Примеры путей введения включают парентеральное, например, внутривенное, внутрикожное, подкожное, пероральное (например, ингаляционное), трансдермальное (топическое) введение и введение через слизистую. Растворы или суспензии, используемые для парентерального, внутрикожного или подкожного применения, могут содержать следующие компоненты: стерильный разбавитель, такой как вода для инъекций, физиологический раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты, и агенты для регулирования тоничности, такие как хлорид натрия или декстроза. рН можно регулировать кислотами или основаниями, такими как хлористоводородная кислота или гидроксид натрия. Препарат для парентерального введения может быть заключен в ампулы, одноразовые шприцы или изготовленные из стекла или пластика флаконы, содержащие несколько доз.
Соединение или фармацевтическую композицию согласно настоящему изобретению можно вводить субъекту многими хорошо известными способами, используемыми в настоящее время для химиотерапевтического лечения. Например, для лечения раковых опухолей соединение согласно настоящему изобретению можно вводить путем инъекции непосредственно в опухоли, вводить путем инъекции в кровоток или полости тела, или принимать перорально, или применять через кожу с помощью пластырей. Выбранная доза должна быть достаточной для обеспечения эффективного лечения, но не настолько высокой, чтобы вызывать неприемлемые побочные эффекты. Предпочтительно, следует внимательно наблюдать за течением заболевания (например, рака, предракового состояния и т.п.) и состоянием здоровья пациента во время лечения и в течение разумного периода времени после лечения.
В настоящем описании термин «терапевтически эффективное количество» относится к количеству фармацевтического агента для лечения, облегчения или предотвращения установленного заболевания или состояния, или для оказания обнаруживаемого терапевтического или ингибирующего эффекта. Указанный эффект может быть обнаружен с помощью любого метода анализа, известного в данной области техники. Точное эффективное количество для субъекта будет зависеть от массы тела, объемов и состояния здоровья субъекта; характера и степени состояния; и лекарственного средства или комбинации лекарственных средств, выбранных для введения. Терапевтически эффективные количества для конкретной ситуации могут быть определены путем проведения рутинных экспериментов в рамках компетенции и по усмотрению врача-клинициста. В соответствии с предпочтительным аспектом заболевание или состояние, которое лечат, представляет собой рак. В соответствии с другим аспектом заболевание или состояние, которое лечат, представляет собой клеточное пролиферативное нарушение.
Для любого соединения терапевтически эффективное количество может быть изначально определено либо в анализах культур клеток, например, неопластических клеток, либо в моделях у животных, как правило, крыс, мышей, кроликов, собак или свиней. Модель у животного можно также использовать для определения соответствующего диапазона концентраций и пути введения. Такую информацию затем можно использовать для определения подходящих доз и путей введения у людей. Терапевтическая/профилактическая эффективность и токсичность могут быть определены с помощью стандартных фармацевтических процедур в культурах клеток или у экспериментальных животных, например, ED50 (доза, являющаяся терапевтически эффективной у 50% популяции) и LD50 (доза, являющаяся летальной для 50% популяции). Соотношение доз, соответствующих токсическому и терапевтическому эффектам, представляет собой терапевтический индекс, и он может быть выражен в виде соотношения LD50/ED50. Предпочтительными являются фармацевтические композиции, которые демонстрируют большие терапевтические индексы. Доза может варьироваться в пределах данного диапазона в зависимости от используемой лекарственной формы, чувствительности пациента и пути введения.
Дозу и введение корректируют для обеспечения достаточных уровней активного агента (агентов) или для поддержания желаемого эффекта. Факторы, которые можно учитывать, включают тяжесть болезненного состояния, общее состояние здоровья субъекта, возраст, массу тела и пол субъекта, питание, время и частоту введения, комбинацию (комбинации) лекарственных средств, чувствительность реакции и переносимость/ответ на терапию. Длительно действующие фармацевтические композиции можно вводить каждые 3-4 дня, каждую неделю или один раз в две недели в зависимости от периода полувыведения и скорости клиренса конкретного состава.
Фармацевтические композиции, содержащие активные соединения согласно настоящему изобретению, могут быть получены общеизвестным способом, например, с помощью традиционных способов смешивания, растворения, гранулирования, изготовления драже, растирания в порошок, эмульгирования, инкапсулирования, захвата или лиофилизации. Фармацевтические композиции могут быть получены традиционным способом с использованием одного или более фармацевтически приемлемых носителей, включающих вспомогательные вещества и/или дополнительные вещества, которые облегчают переработку активных соединений в препараты, подходящие для фармацевтического применения. Соответствующий состав, конечно, зависит от выбранного пути введения.
Фармацевтические композиции, подходящие для применения путем инъекций, включают стерильные водные растворы (в случае растворимости в воде) или дисперсии и стерильные порошки для немедленного приготовления стерильных растворов или дисперсий для инъекций. Для внутривенного введения подходящие носители включают физиологический раствор, бактериостатическую воду, Cremophor EL™ (BASF, Парсиппани, Нью-Джерси, США) или фосфатный буферный раствор (ФБР). Во всех случаях композиция должна быть стерильной и должна быть текучей в такой степени, чтобы существовала возможность легкого введения через шприц. Она должна быть стабильной в условиях изготовления и хранения, и должна быть защищена от загрязняющего действия микроорганизмов, таких как бактерии и грибы. Носитель может представлять собой растворитель или дисперсионную среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль, и т.п.) и подходящие смеси указанных веществ. Необходимая текучесть может быть сохранена, например, путем использования покрытия, такого как лецитин, путем поддержания требуемого размера частиц в случае дисперсии и путем использования поверхностно-активных веществ. Предотвращение действия микроорганизмов может быть обеспечено различными антибактериальными и противогрибковыми агентами, например, парабенами, хлорбутанолом, фенолом, аскорбиновой кислотой, тимеросалом и т.п. Во многих случаях будет предпочтительным включение в композицию изотонических агентов, например, сахаров, полиспиртов, таких как маннит и сорбит, и хлорида натрия. Пролонгированную абсорбцию композиций для инъекций можно обеспечить путем включения в композицию агента, который отсрочивает абсорбцию, например, моностеарата алюминия и желатина.
Стерильные растворы для инъекций могут быть приготовлены путем включения активного соединения в требуемом количестве в соответствующий растворитель совместно с одним ингредиентом или комбинацией ингредиентов, перечисленных выше, при необходимости, с последующей стерилизацией путем фильтрации. Как правило, дисперсии готовят путем включения активного соединения в стерильный носитель, содержащий основную дисперсионную среду и другие требуемые ингредиенты из перечисленных выше. В случае стерильных порошков для приготовления стерильных растворов для инъекций способы получения представляют собой вакуумную сушку и сушку методом сублимации, которая позволяет получить порошок активного ингредиента, а также любого желаемого дополнительного ингредиента из его предварительно стерилизованного путем фильтрации раствора.
Композиции для перорального введения, как правило, содержат инертный разбавитель или годный к употреблению фармацевтически приемлемый носитель. Данные композиции могут быть заключены в желатиновые капсулы или спрессованы в таблетки. Для целей перорального терапевтического введения активное соединение можно включать совместно со вспомогательными веществами и применять в форме таблеток, пастилок или капсул. Композиции для перорального введения также могут быть получены с использованием жидкого носителя для применения в виде жидкости для полоскания рта, в случае которой соединение в жидком носителе применяют перорально для полоскания рта, с последующим выплевыванием или проглатыванием. Фармацевтически совместимые связующие агенты и/или вещества, представляющие собой адъюванты, могут быть включены в композицию в качестве ее части. Таблетки, пилюли, капсулы, пастилки и т.п. могут содержать любой из следующих ингредиентов или соединений схожей природы: связующее вещество, такое как микрокристаллическая целлюлоза, трагакантовая камедь или желатин; вспомогательное вещество, такое как крахмал или лактоза, разрыхлитель, такой как альгиновая кислота, Примогель или кукурузный крахмал; смазывающее вещество, такое как стеарат магния или Sterotes; вещество, способствующее скольжению, такое как коллоидный диоксид кремния; подсластитель, такой как сахароза или сахарин; или ароматизатор, такой как перечная мята, метилсалицилат или апельсиновый ароматизатор.
Для введения путем ингаляции соединения доставляют в форме спрея-аэрозоля из находящейся под давлением емкости или дозирующего устройства, содержащего подходящий пропеллент, например, газ, такой как диоксид углерода, или распылителя.
Системное введение можно также осуществлять через слизистую или трансдермальным способом. Для введения через слизистую или трансдермального введения в составе используют проникающие вещества, необходимые для прохождения через соответствующий барьер. Такие проникающие вещества, в целом, известны в данной области техники и включают, например, для введения через слизистую, детергенты, соли желчных кислот и производные фусидовой кислоты. Введение через слизистую можно осуществлять путем применения назальных спреев или суппозиториев. Для трансдермального введения активные соединения включают в состав мазей, бальзамов, гелей или кремов, общеизвестных в данной области техники.
Активные соединения могут быть получены совместно с фармацевтически приемлемыми носителями, которые будут защищать соединение от быстрого выведения из организма, например, в виде состава с контролируемым высвобождением, включая имплантаты и микроинкапсулированные системы доставки. Могут быть использованы биоразлагаемые биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, сложные полиортоэфиры и полимолочная кислота. Способы получения таких составов очевидны специалисту в данной области техники. Указанные вещества также могут быть приобретены у Alza Corporation и Nova Pharmaceuticals, Inc. Липосомальные суспензии (включая липосомы, направленные на инфицированные клетки, с моноклональными антителами к вирусным антигенам) также можно применять в качестве фармацевтически приемлемых носителей. Они могут быть получены в соответствии со способами, известными специалисту в данной области техники, например, описанными в патенте США №4522811.
Особенно предпочтительным является получение композиций для перорального или парентерального введения в единичной лекарственной форме для удобства введения и однородности дозы. В настоящем описании термин «единичная лекарственная форма» относится к физически раздельным объектам, подходящим для использования в качестве однократных доз для введения субъекту, которого лечат; причем каждая доза содержит заранее определенное количество активного соединения, рассчитанное таким образом, чтобы обеспечить желаемый терапевтический эффект, совместно с необходимым фармацевтическим носителем. Описание единичных лекарственных форм согласно настоящему изобретению обусловлено и зависит непосредственно от уникальных характеристик активного соединения и конкретного достигаемого терапевтического эффекта.
При терапевтическом применении дозы фармацевтических композиций, применяемых в соответствии с настоящим изобретением, варьируются в зависимости от агента, возраста, массы тела и клинического состояния пациента-реципиента, и опыта и решения врача-клинициста или практикующего врача, осуществляющего терапию, наряду с другими факторами, влияющими на выбираемую дозу. В целом, доза должна быть достаточной для того, чтобы привести к замедлению и предпочтительно регрессии роста опухолей, а также предпочтительно к полной регрессии рака. Дозы могут находиться в диапазоне от примерно 0,01 мг/кг в сутки до примерно 5000 мг/кг в сутки. В соответствии с предпочтительными аспектами дозы могут находиться в диапазоне от примерно 1 мг/кг в сутки до примерно 1000 мг/кг в сутки. В соответствии с одним из аспектов доза будет находиться в диапазоне от примерно 0,1 мг/сутки до примерно 50 г/сутки; от примерно 0,1 мг/сутки до примерно 25 г/сутки; от примерно 0,1 мг/сутки до примерно 10 г/сутки; от примерно 0,1 мг до примерно 3 г/сутки; или от примерно 0,1 мг до примерно 1 г/сутки, в виде однократных, разделенных или непрерывных доз (при этом доза может быть скорректирована с учетом массы тела пациента в кг, площади поверхности тела в м2 и возраста в годах). Эффективное количество фармацевтического агента представляет собой такое количество, которое обеспечивает улучшение, которое можно объективно определить, отмечаемое врачом-клиницистом или другим квалифицированным наблюдателем. Например, регрессию опухоли у пациента можно измерить на основании диаметра опухоли. Уменьшение диаметра опухоли указывает на регрессию. На регрессию также указывает отсутствие повторного появления опухолей после прекращения лечения. В настоящем описании термин «способ эффективной дозы (dosage effective manner)» относится к количеству активного соединения для оказания желаемого биологического действия у субъекта или клетки.
Фармацевтические композиции могут быть заключены в емкость, упаковку или дозатор вместе с инструкциями по введению.
Соединения согласно настоящему изобретению способны также образовывать соли. Все данные формы также предусмотрены в пределах объема заявленного изобретения.
В настоящем описании термин «фармацевтически приемлемые соли» относится к производным соединений согласно настоящему изобретению, в которых исходное соединение модифицировано путем получения его кислых или основных солей. Примеры фармацевтически приемлемых солей включают, но не ограничиваются ими, соли, образованные неорганическими или органическими кислотами и основными остатками, такими как амины, щелочные или органические соли кислых остатков, таких как карбоновые кислоты, и т.п. Фармацевтически приемлемые соли включают обычные нетоксичные соли или четвертичные аммониевые соли исходного соединения, полученные, например, из нетоксичных неорганических или органических кислот. Например, такие обычные нетоксичные соли включают, но не ограничиваются ими, соли, полученные из неорганических и органических кислот, выбранных из 2-ацетоксибензойной, 2-гидроксиэтансульфоновой, уксусной, аскорбиновой, бензолсульфоновой, бензойной, бикарбоновой, угольной, лимонной, этилендиаминтетрауксусной, этандисульфоновой, 1,2-этансульфоновой, фумаровой, глюкогептоновой, глюконовой, глутаминовой, гликолевой, гликолиларсаниловой, гексилрезорциновой, гидрабаминовой, бромистоводородной, хлористоводородной, йодистоводородной, гидроксималеиновой, гидроксинафтойной, изэтионовой, молочной, лактобионовой, лаурилсульфоновой, малеиновой, яблочной, миндальной, метансульфоновой, напсиловой (napsylic), азотной, щавелевой, памовой, пантотеновой, фенилуксусной, фосфорной, полигалактуроновой, пропионовой, салициловой, стеариновой, подуксусной (subacetic), янтарной, сульфаминовой, сульфаниловой, серной, дубильной, винной, толуолсульфоновой, и часто встречающихся аминокислот, например, глицина, аланина, фенилаланина, аргинина и т.д.
Другие примеры фармацевтически приемлемых солей включают гексановую кислоту, циклопентанпропионовую кислоту, пировиноградную кислоту, малоновую кислоту, 3-(4-гидроксибензоил)бензойную кислоту, коричную кислоту, 4-хлорбензолсульфоновую кислоту, 2-нафталинсульфоновую кислоту, 4-толуолсульфоновую кислоту, камфорсульфоновую кислоту, 4-метилбицикло[2.2.2]окт-2-ен-1-карбоновую кислоту, 3-фенилпропионовую кислоту, триметилуксусную кислоту, трет-бутилуксусную кислоту, муконовую кислоту и т.п. Настоящее изобретение также включает соли, образующиеся при замещении присутствующего в исходном соединении кислого протона ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; или координировании с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и т.п. Очевидно, что в форме соли отношение соединения к катиону или аниону соли может составлять 1:1, или любое другое отношение, отличное от 1:1, например, 3:1, 2:1, 1:2 или 1:3.
Следует понимать, что все ссылки на фармацевтически приемлемые соли включают формы присоединения растворителя (сольваты) или кристаллические формы (полиморфы) этой соли, определенные в настоящем описании.
Соединения или их фармацевтически приемлемые соли вводят перорально, назально, трансдермально, через легкие, путем ингаляции, трансбуккально, сублингвально, интраперитонеально, подкожно, внутримышечно, внутривенно, ректально, внутриплеврально, интратекально и парентерально. В одном из вариантов реализации соединение вводят перорально. Для специалиста в данной области техники очевидны преимущества конкретных путей введения.
Режим дозирования с применением соединений выбирают в соответствии с различными факторами, включая тип, вид, возраст, массу тела, пол и состояние пациента; тяжесть состояния, которое лечат; путь введения; функцию почек и печени пациента; и конкретное используемое соединение или его соль. Квалифицированный врач или ветеринар может легко определить и назначить эффективное количество лекарственного средства, необходимое для предотвращения, противостояния или остановки прогрессирования состояния.
Способы включения в состав и введения описанных соединений согласно настоящему изобретению можно найти в Remington: the Science and Practice of Pharmacy, 19th edition, Mack Publishing Co., Easton, PA (1995). В одном из вариантов реализации соединения, описанные в настоящем документе, и их фармацевтически приемлемые соли применяют в фармацевтических препаратах в комбинации с фармацевтически приемлемым носителем или разбавителем. Подходящие фармацевтически приемлемые носители включают твердые инертные наполнители или разбавители и стерильные водные или органические растворы. Соединения будет присутствовать в таких фармацевтических композициях в количествах, достаточных для обеспечения желаемой величины дозы в диапазоне, приведенном в настоящем описании.
Все проценты и соотношения, используемые в настоящем описании, если не указано иное, приведены по массе. Другие признаки и преимущества настоящего изобретения очевидны из различных примеров. Приведенные примеры иллюстрируют различные компоненты и методику, подходящую для практического применения настоящего изобретения. Указанные примеры не ограничивают заявленное изобретение. Исходя из настоящего описания, специалист в данной области техники может определить и использовать другие компоненты и методику, подходящую для практического применения настоящего изобретения.
На схемах синтеза и в химических структурах, описанных в настоящем документе, соединения для простоты могут быть изображены в одной конкретной конфигурации (например, с или без конкретного указанного стереоизомера). Такие конкретные конфигурации или их отсутствие не ограничивают настоящее изобретение тем или иным изомером, таутомером, региоизомером или стереоизомером, а также не исключают смеси изомеров, таутомеров, региоизомеров или стереоизомеров; однако очевидно, что конкретный изомер, таутомер, региоизомер или стереоизомер может обладать более высоким уровнем активности, чем другой изомер, таутомер, региоизомер или стереоизомер.
Соединения, разработанные, выбранные и/или оптимизированные способами, описанными выше, после получения могут быть охарактеризованы с использованием различных анализов, известных специалисту в данной области техники, для определения, обладают ли указанные соединения биологической активностью. Например, молекулы могут быть охарактеризованы с помощью обычных анализов, включая, но не ограничиваясь ими, анализы, описанные ниже, для определения, обладают ли они ожидаемой активностью, активностью связывания и/или специфичностью связывания.
Кроме того, может быть использован высокопроизводительный скрининг для ускорения анализа с использованием таких методов анализа. Благодаря этому, становится возможным быстрый скрининг молекул, описанных в настоящем документе, на предмет активности с использованием методов, известных в данной области техники. Общие методики проведения высокопроизводительного скрининга описаны, например, в Devlin (1998) High Throughput Screening, Marcel Dekker; и в патенте США №5763263. В высокопроизводительных анализах можно использовать один или более различных методов анализа, включая, но не ограничиваясь ими, те, которые описаны ниже.
Содержание всех публикаций и патентных документов, приведенных в настоящем описании, включено в настоящее описание посредством ссылки так же, как если бы содержание каждой такой публикации или документа было специально включено в настоящее описание отдельно. Ссылка на публикации и патентные документы не является признанием того, что любой из указанных документов относится к соответствующему уровню техники, а также не является признанием этого в отношении содержания или даты. После описания настоящего изобретения в письменной форме, для специалиста в данной области техники очевидно, что настоящее изобретение можно практически применять в различных вариантах и что вышеприведенное описание и примеры ниже приведены для целей иллюстрации и не ограничивают следующую формулу изобретения.
ПРИМЕРЫ
Пример 1. Синтезы соединений согласно настоящему изобретению
Общие эксперименты
ЯМР
1H-ЯМР-спектры получали с использованием CDCl3, если не указано иное, и регистрировали на 400 или 500 МГц с использованием магнитных инструментов (500 МГц) Varian или Oxford instruments. Указанная мультиплетность представляет собой s = синглет, d = дублет, t = триплет, q = квартет, quint = квинтет, sxt = секстет, m = мультиплет, dd = дублет дублетов, dt = дублет триплетов; br означает широкий сигнал.
ЖХ-МС и ВЭЖХ
Масса: сверхэффективная ЖХ Waters Acquity. ВЭЖХ: Продукты анализировали с помощью Shimadzu SPD-20A с колонкой YMC ODS-M80 150×4,5 мм или колонкой YMC-Pack Pro C18 150×4,6 мм при 1,0 мл/мин. Подвижная фаза представляла собой MeCN:H2O=3:2 (содержащая 0,3% SDS и 0,05% H3PO4). Продукты очищали путем ВЭЖХ/МС (MeOH-H2O, содержащая 0,1% гидроксид аммония) с использованием Waters AutoPurification System с масс-детектором 3100.
HCl соль 3-(аминометил)-4,6-диметил-1,2-дигидропиридин-2-она
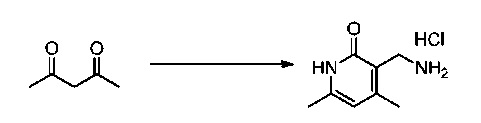
К раствору 2-цианоацетамида (8,40 г, 100 ммоль) и ацетилацетона (10,0 г, 100 ммоль) в H2O (200 мл) добавляли K2CO3 (4,00 г, 28,9 ммоль). Смесь перемешивали при комнатной температуре в течение 22 часов. Затем выпавшее в осадок твердое вещество фильтровали с использованием воронки Бюхнера, промывали ледяной H2O и сушили под вакуумметрическим давлением с получением 4,6-диметил-2-оксо-1,2-дигидропиридин-3-карбонитрила (13,5 г, выход 91%).
К раствору 4,6-диметил-2-оксо-1,2-дигидропиридин-3-карбонитрила (10,0 г, 67,5 ммоль) в МеОН (1,50 л) и конц. HCl (30 мл) добавляли 10% Pd(OH)2 (19 г) в атмосфере N2. Газообразный N2 вытесняли газообразным H2, и смесь перемешивали в течение 26 часов при комнатной температуре в атмосфере водорода. Газообразный H2 вытесняли газообразным N2. Смесь фильтровали через Целит, промывали MeOH и концентрировали. Остаток растирали с EtOH, собирали с помощью воронки Бюхнера и сушили под вакуумметрическим давлением с получением соединения, указанного в названии, в виде белого твердого вещества (11,5 г, 90%). 1H ЯМР (400 МГц, ДМСО-d6): δ м.д. 11,86 (шир.с, 1H), 5,98 (с, 1H), 3,78 (м, 2H), 2,20 (с, 3H), 2,16 (с, 3H).
5-Бром-2-метил-3-нитробензойная кислота
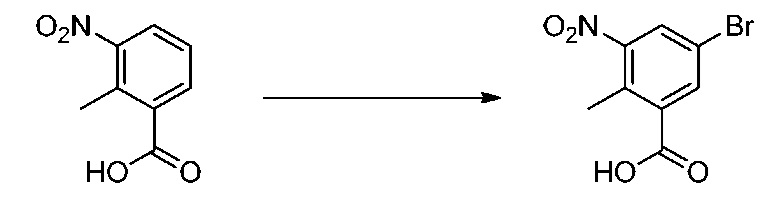
К перемешиваемому раствору 2-метил-3-нитробензойной кислоты (5,00 г, 27,6 ммоль) в H2SO4 (20 мл) добавляли 1,3-дибром-5,5-диметилгидантоин (4,34 г, 15,20 ммоль) при 0°С. Реакционную смесь перемешивали при 0°С в течение 5 ч. Реакционную смесь вливали в ледяную воду, полученное выпавшее в осадок твердое вещество собирали, промывали водой и сушили в вакууме с получением соединения, указанного в названии, в виде белого твердого вещества (7,28 г, количественный выход). 1H-ЯМР (400 МГц, ДМСО-d6) δ м.д.; 8,31 (с, 1H), 8,17 (с, 1H), 2,43 (с, 3H).
Метил-5-бром-2-метил-3-нитробензоат
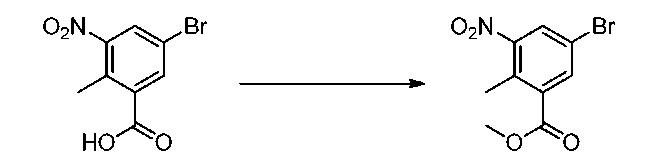
К перемешиваемому раствору 5-бром-2-метил-3-нитробензойной кислоты (7,28 г, 28,0 ммоль) в ДМФА (100 мл) добавляли натрия карбонат (11,9 г, 112 ммоль) и метилиодид (15,9 г, 112 ммоль). Реакционную смесь перемешивали при 60°С в течение 8 часов. После завершения реакции реакционную смесь фильтровали и промывали этилацетатом. Объединенный фильтрат промывали водой и водную фазу повторно экстрагировали этилацетатом. Объединенные органические фазы сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения, указанного в названии, в виде твердого вещества. (7,74 г, количественный выход). 1H-ЯМР (400 МГц, CDCl3) δ (м.д.); 8,17 (с, 1H), 7,91 (с, 1H), 3,96 (с, 3H), 2,59 (с, 3H).
Метил-3-амино-5-бром-3-метилбензоат
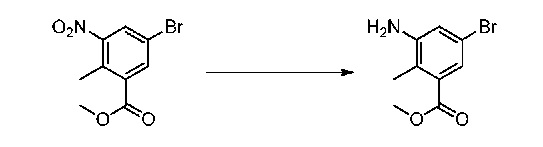
К перемешиваемому раствору метил-5-бром-2-метил-3-нитробензоата (7,60 г, 27,7 ммоль) в водн. EtOH (100 мл EtOH и 20 мл H2O) добавляли аммония хлорид (4,45 г, 83,1 ммоль) и железо (4,64 г, 83,1 ммоль). Реакционную смесь перемешивали при 80°С в течение 5 часов. Затем смесь фильтровали через Целит и слой Целита промывали этилацетатом. Объединенный фильтрат концентрировали в вакууме. Полученный остаток растворяли в этилацетате и воде. Водную фазу экстрагировали этилацетатом (два раза). Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения, указанного в названии, в виде масла коричневого цвета (6,67 г, 99%). 1H-ЯМР (400 МГц, CDCl3) δ м.д.; 7,37 (с, 1H), 6,92 (с, 1H), 3,94 (с, 3H), 3,80 (шир.с, 2H), 2,31 (с, 3H).
Соединение 1:

1 этап: Синтез метил-5-(((транс)-4-((трет-бутоксикарбонил)амино)циклогексил)(этил)амино)-4'-гидрокси-4-метил-[1,1'-бифенил]-3-карбоксилата
К перемешиваемому раствору метил-5-бром-3-(((транс)-4-((трет-бутоксикарбонил)амино)циклогексил)(этил)амино)-2-метилбензоата (10 г, 21,3 ммоль, см., например, WO 2012142504 (номер патентного реестра 41478-507001WO)) и (4-гидроксифенил)бороновой кислоты (3,5 г, 25,3 ммоль) в смеси диоксана (225 мл) и воды (75 мл) добавляли Na2CO3 (8,01 г, 75,5 ммоль), и раствор продували аргоном в течение 30 минут. Затем добавляли Pd(PPh3)4 (2,4 г, 2,07 ммоль) и снова продували аргоном в течение еще 15 минут. Реакционную массу нагревали при 100°С в течение 4 часов. По завершении реакционную смесь разбавляли водой и экстрагировали этилацетатом. Объединенную органическую фазу сушили над сульфатом натрия. В результате удаления растворителя при пониженном давлении с последующей очисткой путем колоночной хроматографии получали соединение, указанное в названии (8,9 г, выход 87%).
2 этап: Синтез метил-5-(((транс)-4-((трет-бутоксикарбонил)амино)циклогексил)(этил)амино)-4'-(2-метоксиэтокси)-4-метил-[1,1'-бифенил]-3-карбоксилата
К перемешиваемому раствору метил-5-(((транс)-4-((трет-бутоксикарбонил)амино)циклогексил)(этил)амино)-4'-гидрокси-4-метил-[1,1'-бифенил]-3-карбоксилата (0,6 г, 1,24 ммоль) и 1-бром-2-метоксиэтана (0,519 г, 3,73 ммоль) в ацетонитриле (6 мл) добавляли Cs2CO3 (0,485 г, 1,49 ммоль), и реакционную смесь перемешивали при 80°С в течение 12 часов. По завершении к ней добавляли воду и экстрагировали этилацетатом. Объединенные органические фазы сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенное соединение очищали путем колоночной хроматографии с получением соединения, указанного в названии (0,6 г, выход 76,5%).
3 этап: Синтез трет-бутил-((транс)-4-((5-(((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)карбамоил)-4'-(2-метоксиэтокси)-4-метил-[1,1'-бифенил]-3-ил(этил)-амино)-циклогексил)карбамата
Водный NaOH (0,066 г, 1,66 ммоль в 5 мл H2O) добавляли к раствору метил-5-(((транс)-4-((трет-бутоксикарбонил)амино)циклогексил)(этил)амино)-4'-(2-метоксиэтокси)-4-метил-[1,1'-бифенил]-3-карбоксилата (0,6 г, 1,11 ммоль) в EtOH (10 мл) и перемешивали при 60°С в течение 1 часа. После завершения реакции удаляли этанол при пониженном давлении, и остаток подкисляли с использованием лимонной кислоты и доводили до рН 4 с использованием лимонной кислоты. Осуществляли экстракцию с использованием 10% метанола в ДХМ. Объединенные органические фазы сушили, концентрировали с получением соответствующей кислоты (0,5 г, выход 85,6%).
Затем вышеуказанную кислоту (0,5 г, 0,95 ммоль) растворяли в ДМСО (5 мл) и к ней добавляли 3-(аминометил)-4,6-диметилпиридин-2(1Н)-он (0,288 г, 1,90 ммоль) и триэтиламин (0,096 г, 0,950 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 минут перед добавлением к ней PyBop (0,741 г, 1,42 ммоль) и продолжали перемешивать в течение ночи при комнатной температуре. После завершения реакции реакционную массу выливали на лед и осуществляли экстракцию с использованием 10% MeOH/ДХМ. Объединенные органические фазы сушили над сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного вещества, которое затем очищали путем колоночной хроматографии с получением соединения, указанного в названии (0,45 г, выход 71,8%).
4 этап: Синтез N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-(((транс)-4-(диметиламино)-циклогексил)(этил)-амино)-4'-(2-метоксиэтокси)-4-метил-[1,1'-бифенил]-3-карбоксамида
К перемешиваемому раствору трет-бутил-((транс)-4-((5-(((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)карбамоил)-4'-(2-метоксиэтокси)-4-метил-[1,1'-бифенил]-3-ил(этил)амино)циклогексил)карбамата (0,45 г, 0,681 ммоль) в ДХМ (5 мл) при 0°С добавляли ТФУ (1 мл) и реакционную смесь перемешивали в течение 2 часов при комнатной температуре. После завершения реакционную смесь концентрировали досуха. Затем остаток подщелачивали Na2CO3 (водн.) до рН 8 и водную фазу экстрагировали 20% метанолом в ДХМ. Объединенные органические фазы сушили над Na2SO4, и растворитель удаляли при пониженном давлении с получением Вос-незащищенного соединения (0,3 г, выход 78,7%).
К перемешиваемому раствору Вос-незащищенного соединения (0,3 г, 0,535 ммоль) в дихлорметане (3 мл) добавляли раствор формальдегида (35-41% водн.) (0,056 г, 1,87 ммоль) при 0°С и перемешивали в течение 20 минут. Затем добавляли NaBH(OAc)3 (0,28 г, 1,33 ммоль) и перемешивали в течение 2 часов при 0°С. По завершении реакции добавляли воду и экстрагировали 20% метанолом в ДХМ. Объединенные органические фазы сушили над Na2SO4 и удаляли растворитель при пониженном давлении. Неочищенное соединение очищали путем преп. ВЭЖХ с получением соединения, указанного в названии (0,1 г, выход 31,7%).
ЖХ-МС: 589,75 (M+1)+; ТФУ-соль: 1H ЯМР (ДМСО-d6, 400 МГц) δ 11,47 (шир.с, 1H), 9,48 (шмр.с, 1H), 8,21 (шир.с, 1H), 7,57 (д, 2H, J=8,0 Гц), 7,40 (с, 1H), 7,23 (с, 1H), 7,03 (д, 2H, J=8,8 Гц), 5,87 (с, 1H), 4,29 (д, 2H, J=4,4 Гц), 4,14-4,12 (м, 2H), 3,69-3,66 (м, 2H), 3,32 (с, 3H), 3,13 (м, 4H), 2,69-2,68 (м, 6H), 2,24 (с, 3H), 2,21 (с, 3H), 2,11 (с, 3H), 1,96 (м, 4H), 1,44 (м, 4H), 0,85 (т, 3H, J=6,8 Гц).
Соединение 2:
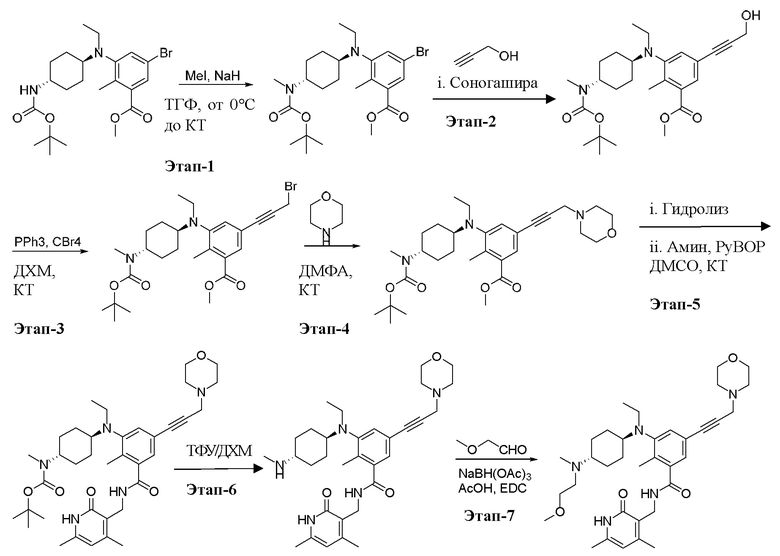
1 этап: Синтез метил-5-бром-3-(((транс)-4-((трет-бутоксикарбонил)-(метил)-амино)циклогексил)(этил)амино)-2-метилбензоата
К перемешиваемому раствору метил-5-бром-3-(((транс)-4-((трет-бутоксикарбонил)амино)циклогексил)(этил)амино)-2-метилбензоата (3 г, 6,41 ммоль, см., например, WO2012142504) в ТГФ (30 мл) добавляли NaH (0,184 г, 7,69 ммоль) при 0°С и перемешивали при этой же температуре в течение 20 минут. Затем добавляли метилиодид (9,10 г, 64,10 ммоль) при 0°С и реакционную смесь перемешивали в течение ночи при комнатной температуре. По завершении реакционную смесь гасили ледяной водой и экстрагировали дихлорметаном. Объединенные органические фазы промывали водой, сушили, концентрировали при пониженном давлении. Неочищенное соединение очищали путем колоночной хроматографии с получением неочищенного соединения, указанного в названии, которое использовали без дополнительной очистки (3 г, выход 97,4%).
2 этап: Синтез метил-3-(((транс)-4-((трет-бутоксикарбонил)-(метил)амино)циклогексил)(этил)амино)-5-(3-гидроксипроп-1-ин-1-ил)-2-метилбензоата
К перемешиваемому раствору метил-5-бром-3-(((транс)-4-((трет-бутоксикарбонил)(метил)амино)циклогексил)(этил)амино)-2-метилбензоата (2 г, 4,14 ммоль) в сухом толуоле добавляли CuI (0,015 г, 0,079 ммоль), PPh3 (0,043 г, 0,165 ммоль), PdCl2(PPh3)2 (0,058 г, 0,082 ммоль), N,N-диизопропиламин (1,08 г, 10,78 ммоль) и реакционную смесь продували аргоном в течение 15 минут. К ней добавляли проп-2-ин-1-ол (0,46 г, 8,29 ммоль), реакционную смесь нагревали при 80°С в герметичных условиях в течение 5 часов. По завершении ее гасили водой и экстрагировали этилацетатом. Органическую фазу сушили над Na2SO4. Неочищенное соединение очищали путем колоночной хроматографии с получением соединения, указанного в названии (1,2 г, выход 63,2%).
3 этап: Синтез метил-5-(3-бромпроп-1-ин-1-ил)-3-(((транс)-4-((трет-бутоксикарбонил)(метил)амино)циклогексил)(этил)амино)-2-метилбензоата:
К перемешиваемому раствору метил-3-(((транс)-4-((трет-бутоксикарбонил)(метил)амино)циклогексил)(этил)амино)-5-(3-гидроксипроп-1-ин-1-ил)-2-метилбензоата (1,2 г, 2,62 ммоль) в ДХМ (15 мл) добавляли PPh3 (1,37 г, 5,22 ммоль) и CBr4 (1,7 г, 5,10 ммоль) при 0°С, и реакционную смесь перемешивали в течение 4 часов при комнатной температуре. По завершении реакцию гасили ледяной водой и экстрагировали дихлорметаном. Объединенные органические фазы промывали водой, сушили, концентрировали при пониженном давлении. Неочищенное вещество очищали путем колоночной хроматографии с получением соединения, указанного в названии (0,5 г, выход 38,5%).
4 этап: Синтез метил-3-(((транс)-4-((трет-бутоксикарбонил)(метил)амино)циклогексил)(этил)амино)-2-метил-5-(3-морфолинопроп-1-ин-1-ил)бензоата
К перемешиваемому раствору метил-5-(3-бромпроп-1-ин-1-ил)-3-(((транс)-4-((трет-бутоксикарбонил)-(метил)амино)циклогексил)(этил)амино)-2-метилбензоата (1 экв.) в ДМФА добавляли морфолин (5 экв.) и реакционную смесь перемешивали в течение 12 часов при комнатной температуре. По завершении реакцию гасили ледяной водой и экстрагировали дихлорметаном. Объединенные органические фазы промывали водой, сушили, концентрировали при пониженном давлении с получением желаемого неочищенного соединения, указанного в названии, которое использовали на следующем этапе без дополнительной очистки (выход 98,7%)
5 этап: Синтез трет-бутил-((транс)-4-((3-(((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)карбамоил)-2-метил-5-(3-морфолинопроп-1-ин-1-ил)фенил)(этил)амино)циклогексил)(метил)карбамата
NaOH (1,5 экв.) добавляли к раствору метил-3-(((транс)-4-((трет-бутоксикарбонил)(метил)амино)циклогексил)(этил)амино)-2-метил-5-(3-морфолинопроп-1-ин-1-ил)бензоата (1 экв.) в EtOH:H2O (9:1) и перемешивали при 60°С в течение 1 часа. После завершения реакции удаляли этанол при пониженном давлении и подкисляли с использованием разбавленной HCl до рН 6, и доводили до рН 4 с использованием лимонной кислоты. Осуществляли экстракцию с использованием 10% метанола в ДХМ. Объединенные органические фазы сушили, концентрировали с получением соответствующей кислоты.
Затем вышеуказанную кислоту (1 экв.) растворяли в ДМСО и к ней добавляли 3-(аминометил)-4,6-диметилпиридин-2(1Н)-он (2 экв.) и триэтиламин (1 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 15 минут перед добавлением к ней PyBop (1,5 экв.) и продолжали перемешивать в течение ночи при комнатной температуре. После завершения реакции реакционную массу выливали на лед и осуществляли экстракцию с использованием 10% MeOH/ДХМ. Объединенные органические фазы сушили над Na2SO4 и концентрировали при пониженном давлении с получением неочищенного вещества, которое затем очищали сначала водой, а затем промывали ацетонитрилом с получением желаемого соединения, указанного в названии (выход 69,4%).
6 этап: Синтез N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-3-(этил((транс)-4-(метиламино)циклогексил)амино)-2-метил-5-(3-морфолинопроп-1-ин-1-ил)бензамида
К перемешиваемому раствору трет-бутил-((транс)-4-((3-(((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)карбамоил)-2-метил-5-(3-морфолинопроп-1-ин-1-ил)фенил)(этил)амино)циклогексил)(метил)карбамата (1 экв.) в ДХМ при 0°С добавляли ТФУ (3 экв.) и реакционную смесь перемешивали в течение 2 часов при комнатной температуре. После завершения реакционную смесь концентрировали досуха. Затем остаток подщелачивали Na2CO3 (водн.) до рН 8 и водную фазу экстрагировали 20% метанолом в ДХМ. Объединенные органические фазы сушили над Na2SO4 и растворитель удаляли при пониженном давлении с получением соединения, указанного в названии (выход 99%), которое использовали в следующей реакции без дополнительной очистки.
7 этап: Синтез N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-3-(этил((транс)-4-((2-метоксиэтил)(метил)амино)циклогексил)амино)-2-метил-5-(3-морфолинопроп-1-ин-1-ил)бензамида
К перемешиваемому раствору N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-3-(этил((транс)-4-(метиламино)циклогексил)амино)-2-метил-5-(3-морфолинопроп-1-ин-1-ил)бензамида (1 экв.) в дихлорэтане добавляли 2-метоксиацетальдегид (10 экв.) и уксусную кислоту (6 экв.) при 0°С, и перемешивали в течение 20 минут. Затем добавляли NaBH(OAc)3 (3 экв.) и перемешивали в течение 2 часов при 0°С. По завершении реакции добавляли воду и экстрагировали 20% метанолом в ДХМ. Объединенные органические фазы сушили над Na2SO4 и удаляли растворитель при пониженном давлении. Неочищенное соединение очищали путем преп. ВЭЖХ с получением целевой молекулы (0,1 г, выход 33,6%).
ЖХ-МС: 606,65 (M+1)+; ТФУ-соль: 1H ЯМР (ДМСО-d6, 400 МГц) δ 11,50 (шир.с, 1H), 9,22 (шир.с, 1H), 8,18 (т, 1H), 7,24 (с, 1H), 7,09 (с, 1H), 5,86 (с, 1H), 4,26-4,25 (м, 4H), 3,66-3,59 (м, 4H), 3,48-3,36 (м, 3H), 3,29-3,17 (м, 7H), 3,04-3,01 (м, 3H), 2,69-2,68 (м, 4H), 2,20 (с, 3H), 2,19 (с, 3H), 2,11 (с, 3H), 2,00-1,92 (м, 2H), 1,82-1,73 (м, 3H), 1,46 (м, 4H), 0,78 (т, 3H, J=6,4 Гц).
Альтернативная схема синтеза соединения 2:
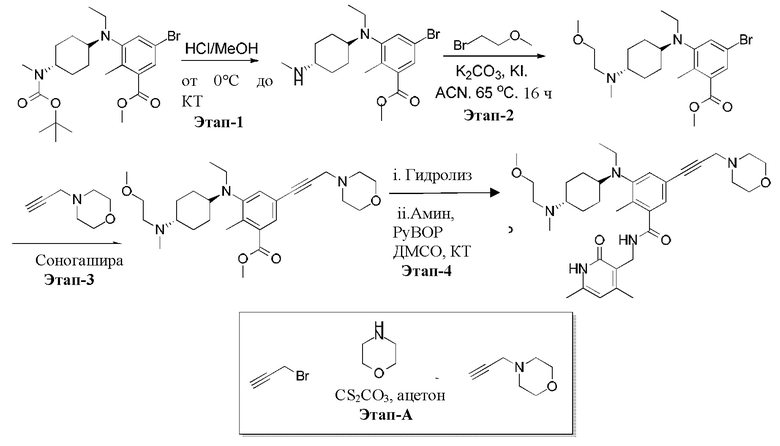
Этап А: Синтез 4-(проп-2-ин-1-ил)морфолина:
К перемешиваемому раствору пропаргилбромида (50 г, 420 ммоль) в ацетоне (300 мл) добавляли Cs2CO3 (136,5 г, 420 ммоль) при 0°С. Затем по каплям добавляли морфолин (36,60 г, 420 ммоль) в ацетоне (200 мл) и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. По завершении фильтровали реакционную массу, и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в названии (50 г, неочищенное). Выделенное соединение использовали непосредственно на следующем этапе сочетания без дополнительной очистки.
1 этап: Синтез метил-5-бром-3-(этил((транс)-4-(метиламино)циклогексил)амино)-2-метилбензоата:
К перемешиваемому раствору метил-5-бром-3-(((транс)-4-((трет-бутоксикарбонил)(метил)амино)циклогексил)(этил)амино)-2-метилбензоата (30 г, 62,24 ммоль) в метаноле (100 мл) при 0°С добавляли раствор HCl в метаноле (500 мл) и реакционную смесь перемешивали в течение 2 часов при комнатной температуре. После завершения реакционную смесь концентрировали досуха. Остаток подщелачивали Na2CO3 (водн.) до рН 8 и водную фазу экстрагировали 10% метанолом в ДХМ (200 мл × 3). Объединенные органические фазы сушили над Na2SO4 и удаляли растворитель при пониженном давлении с получением соединения, указанного в названии, в виде бесцветного масла (25 г, неочищенное). Выделенное соединение использовали на следующем этапе без дополнительной очистки.
2 этап: синтез метил-5-бром-3-(этил((транс)-4-((2-метоксиэтил)(метил)амино)циклогексил)амино)-2-метилбензоата:
К перемешиваемому раствору неочищенного метил-5-бром-3-(этил((транс)-4-(метиламино)циклогексил)амино)-2-метилбензоата (25 г, 65,44 ммоль), 1-бром-2-метоксиэтана (18,19 г, 130,8 ммоль) в ацетонитриле (250 мл) добавляли K2CO3 (18,06 г, 130,8 ммоль) и KI (6,51 г, 39,21 ммоль). Полученную реакционную массу перемешивали при 65°С в течение 16 часов. По завершении реакционную смесь разбавляли водой (300 мл) и экстрагировали ДХМ (500 мл × 3). Объединенные органические фазы промывали водой, сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенное соединение очищали путем колоночной хроматографии на силикагеле с получением соединения, указанного в названии (20 г, выход 69,3%).
1H ЯМР (ДМСО-d6, 400 МГц) δ 7,55 (с, 1H), 7,45 (с, 1H), 3,82 (с, 3H), 3,32 (м, 4H), 3,20 (с, 3H), 3,05 (кв., 2H), 2,61 (м, 1H), 2,32 (с, 3H), 2,30 (м, 1H), 2,15 (с, 3H), 1,77-1,67 (м, 4H), 1,37-1,31(м, 2H), 1,24-1,18 (м, 2H), 0,78 (т, 3H, J=6,8 Гц).
3 этап: Синтез метил-3-(этил((транс)-4-((2-метоксиэтил)(метил)амино)циклогексил)амино)-2-метил-5-(3-морфоилинопроп-1-ин-1-ил)бензоата:
Раствор метил-5-бром-3-(этил((транс)-4-((2-метоксиэтил)(метил)амино)циклогексил)амино)-2-метилбензоата (30 г, 68,02 ммоль), 4-(проп-2-ин-1-ил)морфолина (25,51 г, 204 ммоль) и триэтиламина (20,61 г, 204 ммоль) в ДМФА (300 мл) барботировали аргоном в течение 20 минут. Затем добавляли CuI (3,87 г, 20,36 ммоль) и Pd (PPh3)4 (7,85 г, 6,79 ммоль), и барботировали аргоном в течение еще 20 минут. Реакционную смесь нагревали при 105°С в течение 4 часов, а затем охлаждали до комнатной температуры. Реакцию гасили водой (100 мл) и водную фазу экстрагировали 10% МеОН/ДХМ (400 мл × 3). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали путем колоночной хроматографии на силикагеле с получением соединения, указанного в названии (21 г, выход 63,7%)
1H ЯМР (ДМСО-d6, 400 МГц) δ 7,46 (с, 1H), 7,32 (с, 1H), 3,82 (с, 3H), 3,62-3,57 (м, 6H), 3,50 (с, 2H), 3,35-3,32 (м, 2H), 3,21 (с, 3H), 3,17 (м, 1H), 3,05 (кв., 2H), 2,61-2,58 (м, 2H), 2,38 (с, 3H), 2,33 (м, 1H), 2,18 (м, 2H), 1,77-1,70 (м, 4H), 1,36-1,20 (м, 4H), 0,77 (т, 3H, J=6,8 Гц), 3H слился с пиком растворителя.
4 этап: Синтез N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-3-(этил((транс)-4-((2-метоксиэтил)(метил)амино)циклогексил)амино)-2-метил-5-(3-морфолинопроп-1-ин-1-ил)бензамида:
Водный NaOH (2,59 г, 64,91 ммоль в 10 мл H2O) добавляли к раствору метил-3-(этил((транс)-4-((2-метоксиэтил)(метил)амино)циклогексил)амино)-2-метил-5-(3-морфолинопроп-1-ин-1-ил)бензоата (21 г, 43,29 ммоль) в EtOH (100 мл) и перемешивали при 60°С в течение 1 часа. После завершения реакции удаляли этанол при пониженном давлении, и остаток подкисляли с использованием разбавленной HCl до рН 4 с использованием лимонной кислоты. Осуществляли экстракцию с использованием 10% MeOH/ДХМ (200 мл × 3). Объединенные органические фазы сушили, концентрировали с получением соответствующей кислоты (15,5 г, выход 76%).
К раствору вышеуказанной кислоты (15,5 г, 32,90 ммоль) в ДМСО (50 мл) добавляли 3-(аминометил)-4,6-диметилпиридин-2(1Н)-он (10 г, 65,80 ммоль) и триэтиламин (23 мл, 164,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 минут перед добавлением к ней PyBop (25,66 г, 49,34 ммоль) при 0°С и дополнительно перемешивали в течение ночи при комнатной температуре. После завершения реакционную массу вливали в ледяную воду (100 мл) и осуществляли экстракцию с использованием 10% MeOH/ДХМ (200 мл × 3). Объединенные органические фазы сушили над Na2SO4 и концентрировали при пониженном давлении. Неочищенное соединение очищали путем колоночной хроматографии на щелочном оксиде алюминия, элюируя MeOH:ДХМ, с получением соединения, указанного в названии (11 г, выход 55,3%).
ЖХ-МС: 606,50 (M+1)+; 1H ЯМР (MeOD, 400 МГц) δ 7,23 (с, 1H), 7,09 (с, 1H), 6,11 (с, 1H), 4,46 (с, 2H), 3,74-3,72 (м, 4H), 3,51 (с, 2H), 3,47 (т, 2H, J=5,6 Гц,), 3,32 (с, 3H), 3,07 (кв., 2H, J=7,2 Гц), 2,64-2,63 (м, 7H), 2,38 (м, 1H), 2,37 (с, 3H), 2,27 (с, 3H), 2,26 (с, 3H), 2,25 (с, 3H), 1,89-1,86 (м, 4H), 1,50-1,30 (м, 4H), 0,83 (т, 3H, J=7,2 Гц).
Соединение 105:
Метил-5-бром-2-метил-3-[(оксан-4-ил)амино]бензоат
К перемешиваемому раствору метил-3-амино-5-бром-2-метилбензоата (40,2 г, 165 ммоль) в CH2Cl2 (500 мл) и АсОН (60 мл) добавляли дигидро-2H-пиран-4-он (17,3 г, 173 ммоль) и триацетоксиборгидрид натрия (73,6 г, 330 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Затем добавляли насыщенный водный раствор NaHCO3 и смесь разделяли. Водную фазу экстрагировали CH2Cl2 и объединенную органическую фазу концентрировали в вакууме. Остаток растирали с этиловым эфиром и собирали полученный осадок с получением соединения, указанного в названии, в виде белого твердого вещества (39,1 г, 72%). 1H-ЯМР (400 МГц, ДМСО-d6) δ м.д.; 7,01 (с, 1H), 6,98 (с, 1H), 5,00 (д, J=7,6 Гц, 1H), 3,84-3,87 (м, 2H), 3,79 (с, 3H), 3,54-3,56 (м, 1H), 3,43 (м, 2H), 2,14 (с, 3H), 1,81-1,84 (м, 2H), 1,47-1,55 (м, 2H).
Метил-5-бром-3-[этил(оксан-4-ил)амино]-2-метилбензоат
К перемешиваемому раствору метил-5-бром-2-метил-3-[(оксан-4-ил)амино]бензоата (39,1 г, 119 ммоль) в CH2Cl2 (400 мл) и АсОН (40 мл) добавляли ацетальдегид (24,7 г, 476 ммоль) и триацетоксиборгидрид натрия (79,6 г, 357 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Затем добавляли насыщенный водный раствор NaHCO3 и смесь разделяли. Водную фазу экстрагировали CH2Cl2 и объединенную органическую фазу концентрировали в вакууме. Остаток очищали путем колоночной хроматографии на силикагеле (SiO2 гептан/EtOAc = 3/1) с получением соединения, указанного в названии, в виде вязкого масла (44,1 г, количественный выход). 1H-ЯМР (400 МГц, ДМСО-d6) δ ppm; 7,62 (s, 1H), 7,52 (s, 1H), 3,80 (m, 5H), 3,31 (m, 2H), 2,97-3,05 (m, 2H), 2,87-2,96 (m, 1H), 2,38 (s, 3H), 1,52-1,61 (m, 2H), 1,37-1,50 (m, 2H), 0,87 (t, J=6,8 Гц, 3H).
трет-Бутил-4-((3-(этил(тетрагидро-2H-пиран-4-ил)амино)-5-(метоксикарбонил)-4-метилфенил)этинил)пиперидин-1-карбоксилат
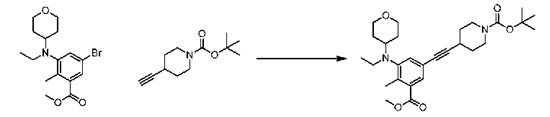
К раствору метил-5-бром-3-(этил(тетрагидро-2Н-пиран-4-ил)амино)-2-метилбензоата (1,80 г, 5,05 ммоль) и трет-бутил-4-этинилпиперидин-1-карбоксилата (1,80 г, 8,59 ммоль) в ДМФА (40 мл) добавляли триэтиламин (2,82 мл, 20,2 ммоль) и йодид меди (I) (0,096 г, 0,505 ммоль). Реакционную смесь дегазировали путем барботирования азотом в течение 15 минут. Затем вводили тетракис(трифенилфосфин)палладий (0) (0,292 г, 0,253 ммоль) и дегазировали в течение еще 10 минут путем барботирования азотом. Реакционную смесь нагревали при 80°С в течение 6 часов. Реакцию гасили насыщенным раствором NaHCO3, экстрагировали МТБЭ (3×40 мл), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали путем хроматографии (0%-40% AcOEt/гептан) с получением соединения, указанного в названии (2,40 г, выход 98%). 1H-ЯМР (500 МГц) δ м.д.; 7,65 (с, 1H), 7,28 (с, 1H), 3,97 (шир.д, J=11,3 Гц, 2H), 3,90 (с, 3H), 3,76 (м, 2H), 3,34 (дт, J=2,0, 11,7 Гц, 2H), 3,24 (ддд, J=3,4, 8,8, 12,2 Гц, 2H), 3,08 (шир.с, 2H), 2,98 (шир.с, 1H), 2,80 (дддд, J=3,9, 3,9, 3,9, 3,9 Гц, 1H), 2,52 (с, 3H), 1,87 (м, 2H), 1,60-1,74 (м, 6H), 1,48 (с, 9H), 0,89 (т, J=6,8 Гц, 3H) ); МС (ESI) [M+H]+ 485,4.
5-((1-(трет-Бутоксикарбонил)пиперидин-4-ил)этинил)-3-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-метилбензойная кислота
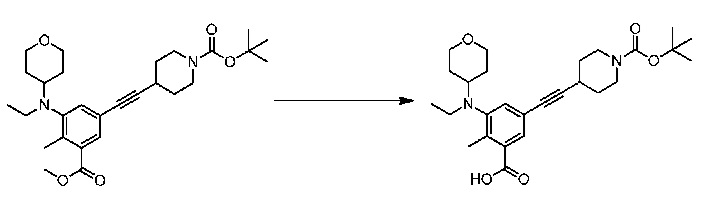
К раствору трет-бутил-4-((3-(этил(тетрагидро-2H-пиран-4-ил)амино)-5-(метоксикарбонил)-4-метилфенил)этинил)пиперидин-1-карбоксилата (2,4 г, 4,95 ммоль) в этаноле (20,0 мл) добавляли раствор гидроксида натрия (0,565 г, 14,1 ммоль) в воде (3,0 мл) при комнатной температуре. Реакционную смесь нагревали при 60°С в течение 6 часов. Реакцию смесь гасили 1 М HCl (5 мл), а затем избытком раствора лимонной кислоты с доведением рН до 5. Смесь концентрировали с удалением EtOH и оставшуюся водную фазу экстрагировали AcOEt (2×40 мл). Органические фазы объединяли, сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали путем хроматографии (10-100% AcOEt/гептан) с получением соединения, указанного в названии (2,30 г, выход 99%). 1H-ЯМР (500 МГц) δ м.д.; 7,82 (с, 1H), 7,35 (с, 1H), 3,98 (шир.д, J=11,3 Гц, 2H), 3,77 (м, 2H), 3,35 (дт, J=1,5, 11,3 Гц, 2H), 3,25 (ддд, J=3,4, 8,3, 12,2 Гц, 2H), 3,11 (шир.с, 2H), 3,00 (шир.с, 1H), 2,81 (дддд, J=3,9, 3,9, 3,9, 3,9 Гц, 1H), 2,60 (с, 3H), 1,88 (м, 2H), 1,60-1,78 (м, 6H), 1,48 (с, 9H), 0,90 (т, J=6,8 Гц, 3H); МС (ESI) [M+H]+ 471,4.
трет-Бутил-4-((3-(((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)карбамоил)-5-(этил(тетрагидро-2H-пиран-4-ил)амино)-4-метилфенил)этинил)пиперидин-1-карбоксилат
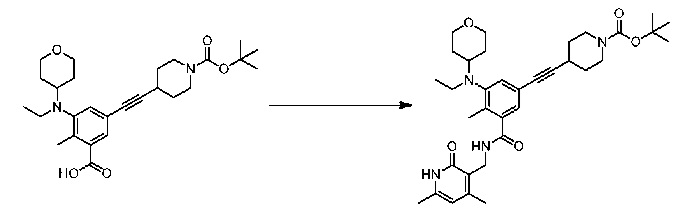
К раствору 5-((1-(трет-бутоксикарбонил)пиперидин-4-ил)этинил)-3-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-метилбензойной кислоты (1,06 г, 2,25 ммоль) в ДМСО (5,8 мл) при комнатной температуре добавляли триэтиламин (0,90 мл, 6,44 ммоль) и (4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метанаминий хлорид (0,405 г, 2,15 ммоль). Прозрачный раствор становился гетерогенным. Затем добавляли HOBT (0,493 г, 3,22 ммоль) и EDC (0,617 г, 3,22 ммоль), и полученную реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили водой (80 мл) и суспензию перемешивали в течение 1 часа при комнатной температуре. Фильтровали суспензию, и осадок на фильтре промывали водой (2×20 мл). Сушили собранное твердое вещество под вакуумом с получением соединения, указанного в названии (1,27 г, выход 98%). 1H-ЯМР (500 МГц, CD3OD) δ м.д.; 7,22 (с, 1H), 7,08 (д, J=1,0 Гц 1H), 6,11 (с, 1H), 4,45 (с, 2H), 3,92 (шир.д, J=10,8 Гц, 2H), 3,78 (дд, J=4,4, 5,4 Гц, 1H), 3,75 (дд, J=4,4, 5,4 Гц, 1H), 3,36 (т, J=11,7 Гц, 2H), 3,21 (шир.т, J=8,3 Гц, 2H), 3,07 (кв., J=7,3 Гц, 2H), 3,01 (дддд, J=3,9, 3,9, 11,3, 11,3 Гц, 1H), 2,84 (дддд, J=3,4, 3,4, 3,9, 3,9 Гц, 1H), 2,38 (с, 3H), 2,28 (с, 3H), 2,25 (с, 3H), 1,88 (м, 2H), 1,70 ((шир.д, J=12,2 Гц, 2H), 1,60 (м, 4H), 1,47 (с, 9H), 0,87 (т, J=7,3 Гц, 3H); МС (ESI) [M+H]+ 605,6.
N-((4,6-Диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-3-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-метил-5-(пиперидин-4-ил-этинил)бензамид
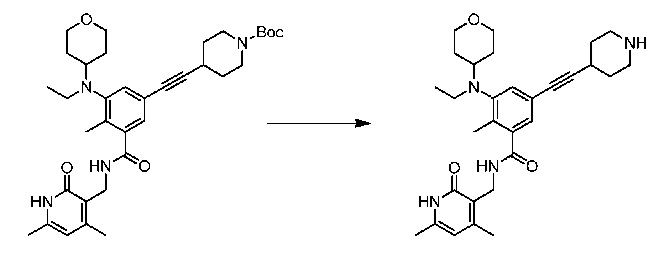
К раствору трет-бутил-4-((3-(((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)карбамоил)-5-(этил(тетрагидро-2H-пиран-4-ил)амино)-4-метилфенил)этинил)пиперидин-1-карбоксилата (250 мг, 0,413 ммоль) в ДХМ (3 мл) добавляли 4 М HCl в 1,4-диоксане (3 мл, 12,0 ммоль) при 20°С. Смесь перемешивали при 20°С в течение 1 часа. ЖХ-МС показывала завершение реакции. Реакционную смесь напрямую концентрировали, и остаток растворяли в ДХМ, а затем нейтрализовали насыщ. NaHCO3/солевым раствором. Органическую фазу сушили (Na2SO4) и фильтровали. И фильтрат концентрировали. Остаток использовали для алкилирования без дополнительной очистки (209 мг, 100%). 1H-ЯМР (500 МГц, CD3OD) δ м.д. 7,21 (шир.с, 1H), 7,07 (шир.с, 1H), 6,11 (с, 1H), 4,46 (с, 2H), 3,95-3,89 (м, 2H), 3,39-3,34 (м, 2H), 3,08 (кв., J=7,0 Гц, 2H), 3,06-2,98 (м, 3H), 2,79-2,72 (м, 1H), 2,72-2,65 (м, 2H), 2,38 (с, 3H), 2,28 (с, 3H), 2,25 (с, 3H), 1,94-1,88 (м, 2H), 1,73-1,68 (м, 2H), 1,68-1,56 (м, 4H), 0,85 (т, J=7,0 Гц, 3H); МС (ESI) [M+H]+ 505,5.
N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-3-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-метил-5-((1-метилпиперидин-4-ил)этинил)бензамид
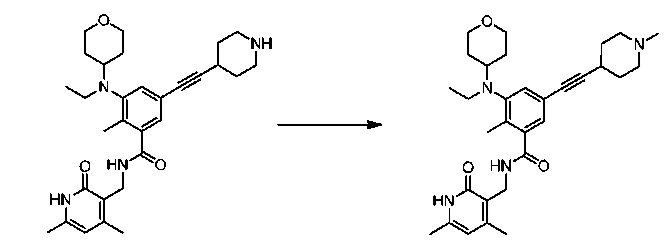
К раствору N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-3-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-метил-5-(пиперидин-4-ил-этинил)бензамида (100 мг, 0,198 ммоль) в метаноле (5 мл) добавляли 35% формальдегид в Н2О (0,155 мл, 1,98 ммоль) при 0°С. После перемешивания при 0°С в течение 10 минут добавляли цианоборогидрид натрия (24,9 мг, 0,396 ммоль). Полученную смесь перемешивали при 0°С в течение 1 часа. ЖХ-МС показывала завершение реакции. Реакцию гасили насыщ. NaHCO3/солевым раствором и экстрагировали EtAOc/гептаном. Органическую фазу сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали путем хроматографии (10 г колонка, МеОН/ДХМ=1:9, а затем МеОН/7 М NH3 в MeOH/ДХМ=1:1:8) с получением соединения, указанного в названии (96,0 мг, 93%). 1H-ЯМР (500 МГц, CD3OD) δ м.д. 7,22 (шир.с, 1H), 7,08 (шир.с, 1H), 6,10 (с, 1H), 4,46 (с, 2H), 3,94-3,87 (м, 2H), 3,35-3,30 (м, 2H), 3,07 (кв, J=7,0 Гц, 2H), 3,04-2,97 (м, 1H), 2,79-2,71 (м, 2H), 2,67-2,58 (м, 1H), 2,38 (с, 3H), 2,28 (с, 3H), 2,28 (с, 3H), 2,25 (с, 3H), 2,28-2,21 (м, 2H), 1,97-1,91 (м, 2H), 1,78-1,67 (м, 4H), 1,64-1,54 (м, 2H), 0,85 (т, J=7,0 Гц, 3H); МС (ESI) [M+H]+ 519,4.
Пример 2: протокол и общие методы биологического анализа
Протокол анализов ферментов дикого типа и мутантных ферментов PRC2
Общие материалы. S-аденозилметионин (SAM), S-аденозилгомоцистеин (SAH), бицин, KCl, Твин20, диметилсульфоксид (ДМСО) и желатин кожи крупного рогатого скота (BSG) приобретали у Sigma-Aldrich с максимально высоким уровнем чистоты. Дитиотреитол (DTT) приобретали у EMD. 3H-SAM приобретали у American Radiolabeled Chemicals с удельной активностью 80 Ки/ммоль. 384-луночные покрытые стрептавидином планшеты Flashplates приобретали у PerkinElmer.
Субстраты. Пептиды, характерные для остатков 21-44 гистона H3 человека, содержащие либо немодифицированный лизин 27 (H3K27me0), либо диметилированный лизин 27 (H3K27me2), были синтезированы с С-концевой аффинной меткой, состоящей из глицинового (G) линкера и биотинилированного лизина (K), и С-концевым амидным кэпом компанией 21st Century Biochemicals. Указанные пептиды очищали путем высокоэффективной жидкостной хроматографии (ВЭЖХ) до степени чистоты, составляющей более 95%, и подтверждали путем жидкостной хроматографии-масс-спектрометрии (ЖХ-МС). Последовательности приведены ниже.
H3K27me0: ATKAARKSAPATGGVKKPHRYRPGGK(биотин)-амид (SEQ ID NO: 101)
H3K27me2: ATKAARK(me2)SAPATGGVKKPHRYRPGGK(биотин)-амид (SEQ ID NO: 102)
Олигонуклеосомы эритроцитов цыпленка выделяли из крови цыпленка в соответствии с установленными процедурами.
Рекомбинантные комплексы PRC2. Комплексы PRC2 человека очищали в виде 4-компонентных ферментных комплексов, совместно экспрессируемых в клетках Spodoptera frugiperda (sf9) с использованием бакуловирусной системы экспрессии. Экспрессируемые субъединицы представляли собой EZH2 (NM_004456) дикого типа или Y641F, N, H, S или C-мутанты EZH2, полученные из конструкции EZH2 дикого типа, EED (NM_003797), Suz12 (NM_015355) и RbAp48 (NM_005610). Субъединица EED содержала N-концевую FLAG-метку, которую использовали для очистки всего 4-компонентного комплекса от клеточных лизатов sf9. Чистота комплексов соответствовала или превышала 95% по данным электрофореза в полиакриламидном геле в присутствии додецилсульфата натрия (SDS-ПААГ) и анализа на Agilent Bioanalyzer. Исходные концентрации ферментов (как правило, 0,3-1,0 мг/мл) определяли с использованием анализа методом Бредфорда относительно стандарта - бычьего сывороточного альбумина (BSA).
Общая процедура анализов ферментов PRC2 с использованием пептидных субстратов. Все анализы выполняли в буфере, состоящем из 20 мМ бицина (рН=7,6), 0,5 мМ DTT, 0,005% BSG и 0,002% Твин20, приготовленном в день использования. Соединения в 100% ДМСО (1 мкл) вносили в полипропиленовые 384-луночные планшеты с V-образным дном (Greiner) с использованием Platemate 2×3, оснащенного 384-канальной пипетирующей головкой (Thermo). ДМСО (1 мкл) добавляли в колонки 11, 12, 23, 24, ряды А-H для контроля максимального сигнала, и SAH, известный продукт и ингибитор PRC2 (1 мкл), добавляли в колонки 11, 12, 23, 24, ряды I-P для контроля минимального сигнала. Добавляли коктейль (40 мкл), содержащий фермент PRC2 дикого типа и пептид H3K27me0 или любой из Y641-мутантных ферментов и пептид H3K27me2, с помощью Multidrop Combi (Thermo). Соединения оставляли инкубироваться совместно с PRC2 в течение 30 минут при 25°С, затем добавляли коктейль (10 мкл), содержащий смесь нерадиоактивного и 3H-SAM, для инициации реакции (конечный объем = 51 мкл). Во всех случаях конечные концентрации были следующими: концентрация фермента дикого типа или мутантного фермента PRC2 составляла 4 нМ, концентрация SAH в лунках контроля минимального сигнала составляла 1 мМ, и концентрация ДМСО составляла 1%. Конечные концентрации остальных компонентов указаны в таблице 2, ниже. Анализы прекращали путем добавления нерадиоактивного SAM (10 мкл) до конечной концентрации 600 мкМ, который разводил 3H-SAM до уровня, при котором его включение в пептидный субстрат больше не было детектируемым. Затем 50 мкл реакционной смеси из 384-луночного полипропиленового планшета переносили в 384-луночный планшет Flashplate и биотинилированные пептиды оставляли связываться с покрытой стрептавидином поверхностью в течение по меньшей мере 1 часа перед промывкой три раза 0,1% Твин20 в устройстве для промывки планшетов Biotek ELx405. Затем планшеты прочитывали на планшет-ридере TopCount PerkinElmer с измерением количества 3H-меченого пептида, связанного с поверхностью Flashplate, измеряемого в виде числа распадов в минуту (dpm) или, в качестве альтернативы, рассматриваемого как число импульсов в минуту (cpm).
Конечные концентрации компонентов для каждого варианта анализа на основе идентичности EZH2 (EZH2 дикого типа или Y641-мутантный EZH2)
Общая процедура анализа фермента PRC2 дикого типа с использованием субстрата, представляющего собой олигонуклеосому. Анализы выполняли в буфере, состоящем из 20 мМ бицина (рН=7,6), 0,5 мМ DTT, 0,005% BSG, 100 мМ KCl и 0,002% Твин20, приготовленном в день использования. Соединения в 100% ДМСО (1 мкл) вносили в полипропиленовые 384-луночные планшеты с V-образным дном (Greiner) с использованием Platemate 2×3, оснащенного 384-канальной пипетирующей головкой (Thermo). ДМСО (1 мкл) добавляли в колонки 11, 12, 23, 24, ряды А-H для контроля максимального сигнала, и SAH, известный продукт и ингибитор PRC2 (1 мкл), добавляли в колонки 11, 12, 23, 24, ряды I-P для контроля минимального сигнала. Добавляли коктейль (40 мкл), содержащий фермент PRC2 дикого типа и олигонуклеосому эритроцитов цыпленка, с помощью Multidrop Combi (Thermo). Соединения оставляли инкубироваться совместно с PRC2 в течение 30 минут при 25°С, затем добавляли коктейль (10 мкл), содержащий смесь нерадиоактивного и 3H-SAM, для инициации реакции (конечный объем = 51 мкл). Конечные концентрации были следующими: концентрация фермента PRC2 дикого типа составляла 4 нМ, концентрация нерадиоактивного SAM составляла 430 нМ, концентрация 3H-SAM составляла 120 мМ, концентрация олигонуклеосомы эритроцитов цыпленка составляла 120 нМ, концентрация SAH в лунках контроля минимального сигнала составляла 1 мМ, и концентрация ДМСО составляла 1%. Анализ прекращали путем добавления нерадиоактивного SAM (10 мкл) до конечной концентрации 600 мкМ, который разводил 3H-SAM до уровня, при котором его включение в субстрат, представляющий собой олигонуклеосому эритроцитов цыпленка, больше не было детектируемым. Затем 50 мкл реакционной смеси из 384-луночного полипропиленового планшета переносили в 384-луночный планшет Flashplate и нуклеосомы эритроцитов цыпленка иммобилизовали на поверхности планшета, который затем три раза промывали 0,1% Твин20 в устройстве для промывки планшетов Biotek ELx405. Затем планшеты прочитывали на планшет-ридере TopCount PerkinElmer с измерением количества 3H-меченой олигонуклеосомы эритроцитов цыпленка, связанной с поверхностью Flashplate, измеряемого в виде числа распадов в минуту (dpm) или, в качестве альтернативы, рассматриваемого как число импульсов в минуту (cpm).
Расчет % ингибирования
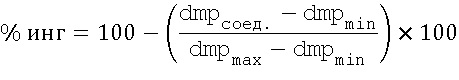 ,
,
где dpm = число распадов в минуту, соед. = сигнал в аналитической лунке, и min и max представляют собой соответствующие контроли минимального и максимального сигнала.
Четырехпараметрическая подгонка IC50
 ,
,
где обычно допускаются колебания максимального значения и минимального значения, но они могут быть зафиксированы на уровне 100 или 0 соответственно при 3-параметрической подгонке. Обычно допускается колебание коэффициента Хилла, но он также может быть зафиксирован на уровне 1 при 3-параметрической подгонке. Y представляет собой % ингибирования, а Х представляет собой концентрацию соединения.
Значения IC50 для анализов ферментов PRC2 с использованием пептидных субстратов (например, EZH2 дикого типа и Y641F) представлены в таблице 3 ниже.
Анализ метилирования WSU-DLCL2
Суспензию клеток WSU-DLCL2 приобретали у Немецкой коллекции микроорганизмов и клеточных культур (DSMZ, Брауншвейг, Германия). Среду RPMI/Glutamax, пенициллин-стрептомицин, термоинактивированную фетальную бычью сыворотку и Д-ФБР приобретали у Life Technologies, Гранд-Айленд, Нью-Йорк, США. Буфер для экстракции и буфер для нейтрализации (5×) приобретали у Active Motif, Карлсбад, Калифорния, США. Антитела кролика к гистону H3 приобретали у Abcam, Кембридж, Массачусетс, США. Антитела кролика к H3K27me3 и HRP-конъюгированные антитела к IgG кролика приобретали у Cell Signaling Technology, Данверс, Массачусетс, США. «Суперчувствительный» субстрат TMB получали от BioFX Laboratories, Оуингс Милс, Мэриленд, США. Бычий сывороточный альбумин, не содержащий IgG, приобретали у Jackson ImmunoResearch, Вест Грув, Пенсильвания, США. ФБР с Твин (10X ФБР-T) приобретали у KPL, Гейтерсберг, Мэриленд, США. Серную кислоту приобретали у Ricca Chemical, Арлингтон, Техас, США. Планшеты Immulon для твердофазного иммуноферментного анализа (ELISA) приобретали у Thermo, Рочестер, Нью-Йорк, США. Планшеты для культур клеток с V-образным дном приобретали у Corning Inc., Корнинг, Нью-Йорк, США. Полипропиленовые планшеты с V-образным дном приобретали у Greiner Bio-One, Монро, Северная Каролина, США.
Суспензию клеток WSU-DLCL2 выдерживали в питательной среде (RPMI 1640 с добавлением 10% об./об. термоинактивированной фетальной бычьей сыворотки и 100 единиц/мл пенициллина-стрептомицина) и культивировали при 37°С в атмосфере 5% СО2. В условиях анализа клетки инкубировали в среде для анализа (RPMI 1640 с добавлением 20% об./об. термоинактивированной фетальной бычьей сыворотки и 100 единиц/мл пенициллина-стрептомицина) при 37°С в атмосфере 5% СО2 на шейкере для планшетов.
Клетки WSU-DLCL2 высевали в среде для анализа в концентрации 50000 клеток на мл в 96-луночные планшеты для культур клеток с V-образным дном по 200 мкл на лунку. Соединение (1 мкл) из 96-луночных исходных планшетов добавляли непосредственно в клеточный планшет с V-образным дном. Планшеты инкубировали на шейкере для титровальных планшетов при 37°С, 5% СО2 в течение 96 часов. После четырех дней инкубации планшеты центрифугировали при 241 × g в течение пяти минут и среду осторожно аспирировали из каждой лунки клеточного планшета, не будоража осадок клеток. Осадок ресуспендировали в 200 мкл Д-ФБР и планшеты снова центрифугировали при 241 × g в течение пяти минут. Аспирировали надосадочную жидкость и в каждую лунку добавляли холодный (4°С) буфер для экстракции (100 мкл). Планшеты инкубировали при 4°С на орбитальном шейкере в течение двух часов. Планшеты центрифугировали при 3427 × g × 10 минут. Надосадочную жидкость (80 мкл на лунку) переносили в соответствующую лунку в 96-луночный полипропиленовый планшет с V-образным дном. В полипропиленовый планшет с V-образным дном, содержащий надосадочную жидкость, добавляли буфер для нейтрализации 5X (20 мкл на лунку). Полипропиленовые планшеты с V-образным дном, содержащие неочищенный препарат гистонов (CHP), инкубировали на орбитальном шейкере × пять минут. Неочищенные препараты гистонов добавляли (2 мкл на лунку) в каждую соответствующую лунку в параллельные 96-луночные планшеты (дубликаты) для ELISA, содержащие 100 мкл покрывающего буфера (1X ФБР + BSA 0,05% масс./об.). Планшеты герметизировали и инкубировали в течение ночи при 4°С. На следующий день планшеты три раза промывали 1X ФБР-T 300 мкл на лунку. Лунки блокировали в течение двух часов 300 мкл на лунку разбавителя для ELISA ((ФБР (1x) BSA (2% масс./об.) и Твин20 (0,05% об./об.)). Планшеты три раза промывали 1X ФБР-T. Для планшета для детектирования гистона Н3 добавляли 100 мкл на лунку антител к гистону Н3 (Abcam, ab1791), разведенных 1:10000 в разбавителе для ELISA. Для планшета для детектирования триметилирования H3K27 добавляли 100 мкл на лунку анти-H3K27me3, разведенных 1:2000 в разбавителе для ELISA. Планшеты инкубировали в течение 90 минут при комнатной температуре. Планшеты три раза промывали 300 мкл 1X ФБР-T на лунку. Для детектирования гистона H3 добавляли 100 мкл HRP-конъюгированных антител к IgG кролика, разведенных 1:6000 в разбавителе для ELISA, на лунку. Для детектирования H3K27me3 добавляли 100 мкл HRP-конъюгированных антител к IgG кролика, разведенных 1:4000 в разбавителе для ELISA, на лунку. Планшеты инкубировали при комнатной температуре в течение 90 минут. Планшеты четыре раза промывали 300 мкл 1X ФБР-T на лунку. Добавляли 100 мкл субстрата TMB на лунку. Планшеты с гистоном H3 инкубировали в течение пяти минут при комнатной температуре. Планшеты H3K27me3 инкубировали в течение 10 минут при комнатной температуре. Реакцию останавливали 1Н серной кислотой (100 мкл на лунку). Поглощение для каждого планшета считывали при 450 нм.
Сначала определяли соотношение для каждой лунки: 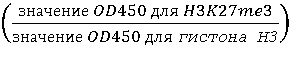 .
.
Каждый планшет содержал восемь контрольных лунок для обработки только ДМСО (минимальное ингибирование), а также восемь контрольных лунок для максимального ингибирования (фоновые лунки).
Рассчитывали средние значения соотношения для каждого контрольного типа и их использовали для определения процента ингибирования для каждой тестируемой лунки в планшете. Тестируемое соединение подвергали серийному трехкратному разведению в ДМСО для получения всего десяти тестируемых концентраций, начиная с 25 мкМ. Определяли процент ингибирования и получали кривые IC50 с использованием дубликатов лунок для каждой концентрации соединения. Значения IC50 для данного анализа представлены в таблице 3 ниже.
Процент ингибирования = 100-

Анализ пролиферации клеток
Суспензию клеток WSU-DLCL2 приобретали у DSMZ (Немецкая коллекция микроорганизмов и клеточных культур, Брауншвейг, Германия). Среду RPMI/Glutamax, пенициллин-стрептомицин, термоинактивированную фетальную бычью сыворотку приобретали у Life Technologies, Гранд-Айленд, Нью-Йорк, США. Полипропиленовые 384-луночные планшеты с V-образным дном приобретали у Greiner Bio-One, Монро, Северная Каролина, США. 384-луночные белые непрозрачные планшеты для культур клеток приобретали у Perkin Elmer, Уолтем, Массачусетс, США. Cell-Titer Glo® приобретали у Promega Corporation, Мэдисон, Висконсин, США. Планшет-ридер SpectraMax M5 приобретали у Molecular Devices LLC, Саннивейл, Калифорния, США.
Суспензию клеток WSU-DLCL2 выдерживали в питательной среде (RPMI 1640 с добавлением 10% об./об. термоинактивированной фетальной бычьей сыворотки) и культивировали при 37°С в атмосфере 5% СО2. В условиях анализа клетки инкубировали в среде для анализа (RPMI 1640 с добавлением 20% об./об. термоинактивированной фетальной бычьей сыворотки и 100 единиц/мл пенициллина-стрептомицина) при 37°С в атмосфере 5% СО2.
Для оценки влияния соединений на пролиферацию линии клеток WSU-DLCL2 экспоненциально растущие клетки высевали в 384-луночные белые непрозрачные планшеты с плотностью 1250 клеток/мл в конечном объеме 50 мкл среды для анализа. Подготавливали исходный планшет с соединением путем осуществления 3-кратных серийных разведений с получением растворов 9 концентраций, полученных в трех повторностях, в ДМСО, начиная с 10 мМ (конечная максимальная концентрация соединения в анализе составляла 20 мкМ, а для ДМСО составляла 0,2%). 100 нл аликвоту из исходного планшета с соединением добавляли в соответствующую лунку в клеточном планшете. Контроль, демонстрирующий 100% ингибирование, состоял из клеток, обрабатываемых 200 нМ конечной концентрацией стауроспорина, и контроль, демонстрирующий 0% ингибирование, состоял из клеток, обрабатываемых ДМСО. После добавления соединений аналитические планшеты инкубировали в течение 6 дней при 37°С, 5% СО2, относительной влажности >90% в течение 6 дней. Жизнеспособность клеток измеряли по квантованию АТФ, присутствующего в культурах клеток, добавляя 35 мкл реагента Cell Titer Glo® в клеточные планшеты. Люминесценцию считывали на SpectraMax M5. Концентрацию, ингибирующую жизнеспособность клеток на 50%, определяли с использованием 4-параметрической подгонки нормализованных кривых доза-ответ. Значения IC50 для данного анализа также представлены в таблице 3 ниже. Масс-спектральные данные для данных соединений также приведены в таблице 3 ниже.
Пример 3: Выведение наименьшей цитотоксической концентрации (LCC)
Точно установлено, что пролиферация клеток происходит путем деления клетки, которое приводит к удвоению количества клеток после деления относительно количества клеток до деления. При фиксированном наборе условий окружающей среды (например, рН, ионная сила, температура, плотность клеток, содержание белков и факторов роста в среде и т.п.) клетки будут пролиферировать путем последовательного удвоения (т.е. деления) в соответствии со следующим уравнением, при условии, что присутствует достаточное количество питательных веществ и других необходимых факторов.

где Nt представляет собой количество клеток в момент времени (t) после начала периода наблюдения, N0 представляет собой количество клеток в начале периода наблюдения, t представляет собой время после начала периода наблюдения, и tD представляет собой временной интервал, необходимый для удвоения клеток, также называемый временем удвоения. Уравнение A.1 может быть преобразовано в более удобную форму экспоненциального уравнения по основанию е с использованием равенства 0,693 = ln(2).
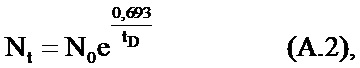
Константа скорости пролиферации клеток (kp) обратно пропорциональна времени удвоения, как показано ниже.

Объединение уравнения А.2 и А.3 приводит к

Таким образом, в соответствии с уравнением А.4 ожидается, что количество клеток будет экспоненциально увеличиваться со временем во время раннего периода роста клеток, называемого логарифмической фазой роста. Экспоненциальные уравнения, подобные уравнению А.4, можно линеаризовать, взяв натуральный логарифм каждой части.
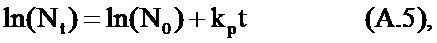
Таким образом, ожидается, что график зависимости ln(Nt) от времени даст восходящую прямую линию с наклоном, равным kp, и отрезком, отсекаемым на оси Y, равным ln(N0).
Изменения условий окружающей среды могут привести к изменению скорости пролиферации клеток, которая поддается количественному определению, как изменения константы скорости пролиферации kp. Среди условий, которые могут привести к изменению скорости пролиферации, находится введение в систему антипролиферативного соединения в начале периода наблюдения (т.е. при t=0). Когда антипролиферативное соединение оказывает немедленное влияние на пролиферацию клеток, ожидают, что графики зависимости ln(Nt) от времени по-прежнему, будут линейными при всех концентрациях соединения с уменьшением значений kp при увеличении концентраций соединения.
В зависимости от механизма антипролиферативного действия некоторые соединения могут не сразу вызывать изменение скорости пролиферации. Вместо этого, может наблюдаться период задержки, прежде чем соединение начнет действовать. В таких случаях график зависимости ln(Nt) от времени окажется двухфазным и можно будет определить момент времени, в который начинается действие соединения, в виде точки излома между фазами. Независимо от того, является ли действие соединения на пролиферацию немедленным, или оно начинается после периода задержки, константу скорости пролиферации при каждой концентрации соединения лучше всего определять по наклону кривой зависимости ln(Nt) от времени от момента времени, при котором начинается действие соединения, до конца периода наблюдения в эксперименте.
Соединение, наносимое на растущие клетки, может влиять на наблюдаемую пролиферацию одним из двух общих способов: путем ингибирования дальнейшего деления клеток (цитостаз) или путем уничтожения клеток (цитотоксичность). Если соединение является цитостатическим, повышение концентрации соединения будет уменьшать значение kp до тех пор, пока не прекратится дальнейшее деление клеток. В этот момент скорость роста клеток и, следовательно, значение kp будет равно нулю. С другой стороны, если соединение является цитотоксическим, то значение kp будет состоять из двух констант скорости: константы скорости для продолжающегося роста клеток в присутствии соединения (kg) и константы скорости для уничтожения клеток соединением (kd). Таким образом, общая константа скорости пролиферации в случае фиксированной концентрации соединения будет представлять собой разницу между абсолютными значениями данных противоположных констант скоростей.

В случае концентраций соединения, при которых скорость роста клеток превышает скорость уничтожения клеток, величина kp будет иметь положительное значение (т.е. kp > 0). В случае концентраций соединения, при которых скорость роста клеток ниже скорости уничтожения клеток, величина kp будет иметь отрицательное значение (т.е. kp < 0), и количество клеток будет уменьшаться со временем, что указывает на сильную цитотоксичность. Когда kg точно соответствует kd, общая константа скорости пролиферации, kp, будет иметь значение, равное нулю. Таким образом, можно определить наименьшую цитотоксическую концентрацию (LCC) как такую концентрацию соединения, которая приводит к значению kp, равному нулю, поскольку любая концентрация больше данной будет приводить к отчетливо наблюдаемой цитотоксичности. Внимание: в случае концентраций ниже LCC есть вероятность того, что будет происходить уничтожение клеток, но со скоростью, которая меньше скорости пролиферации остаточных клеток. В данном случае обработка не направлена на определение биологических особенностей действия соединения. Наоборот, в данном случае целью является просто определение практического параметра, с помощью которого можно объективно количественно определить концентрацию соединения, при которой скорость уничтожения клеток превышает рост новых клеток. Действительно, LCC представляет собой контрольную точку или критическую концентрацию, выше которой наблюдается выраженная цитотоксичность, а не цитотоксическую концентрацию как таковую. В этом отношении LCC можно рассматривать подобно другим физическим контрольным показателям, таким как критическая концентрация мицеллообразования (CMC), используемая для определения концентрации липида, детергента или других поверхностно-активных веществ, выше которой все молекулы включаются в мицеллярные структуры.
Традиционно, влияние антипролиферативных соединений на рост клеток чаще всего количественно определяли по значению IC50, которое определяется как концентрация соединения, которая снижает скорость пролиферации клеток до половины, наблюдаемой в отсутствии соединения (т.е. для контрольного образца, представляющего собой носитель или растворитель). Однако IC50 не позволяет исследователю различать цитостатические и цитотоксические соединения. LCC, напротив, легко позволяет провести такое различие, а также количественно определить концентрацию, при которой происходит переход к устойчивому цитотоксическому поведению.
Если временное окно наблюдения ограничивают окном между началом действия и концом эксперимента, то данные будут, в целом, хорошо подходить для линейного уравнения в случае отражения на графике в виде зависимости ln(Nt) от времени (см. выше). Из соответствий данного типа может быть определено значение kp в случае каждой концентрации тестируемого соединения. Перестроенный график зависимости значения kp от концентрации соединения ([I]) будет иметь форму нисходящей изотермы с максимальным значением kmax при [I]=0 (определенным по контрольному образцу, представляющему собой носитель или растворитель) и минимальным значением kmin при бесконечной концентрации соединения.
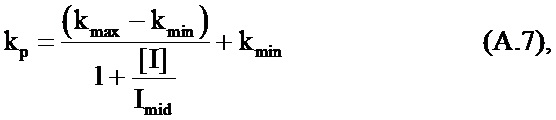
где Imid представляет собой концентрацию соединения, дающую значение kp посередине между значениями kmax и kmin (следует отметить, что значение Imid не то же самое, что IC50, за исключением случая совершенно и полностью цитостатического соединения). Таким образом, подгонка данных перестроенного графика к уравнению A.7 позволяет оценить kmax, kmin и Imid. Если соединение является цитостатическим (как определено в настоящем описании), значение kmin не может быть меньше нуля. Для цитотоксических соединений kmin будет меньше нуля, и абсолютное значение kmin будет напрямую связано с эффективностью соединения в отношении уничтожения клеток.
Подобранные значения, полученные из уравнения А.7, можно также использовать для определения значения LCC. По определению, когда [I]=LCC, kp=0. Таким образом, в данных условиях уравнение А.7 становится
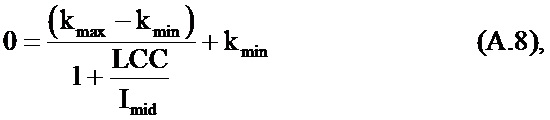
Алгебраическое преобразование уравнения А.8 позволяет получить уравнение для LCC.
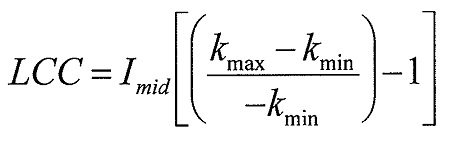
Данный анализ легко выполнять с помощью программного обеспечения для подгонки нелинейных кривых и его можно использовать во время анализов активности соединений на основе клеток на протяжении всего процесса поиска и разработки лекарственных средств. Таким образом, LCC может стать ценным показателем для оценки зависимости активности соединения от его структуры (SAR).
Пример 4: анализы in vivo
Мыши
Возраст самок мышей линии SCID® Fox Chase (CB17/Icr-Prkdcscid/IcrIcoCrl, Charles River Laboratories) или бестимусных мышей линии nude (Crl:NU(Ncr)-Foxn1nu, Charles River Laboratories) составлял 8 недель, а масса тела (МТ) находилась в диапазоне 16,0-21,1 г в 1 день исследования. Животных обеспечивали водой без ограничения (обратный осмос 1 ч./млн. Cl) и питанием NIH 31 Modified and Irradiated Lab Diet®, состоящим из 18,0% сырого белка, 5,0% сырого жира и 5,0% сырой клетчатки. Животных содержали на облученной подстилке Enrich-o'cobs™M в стационарных микроизоляторах в условиях 12-часового светового цикла при 21-22°C (68-72°F) и влажности 40-60%. Все процедуры выполняли в соответствии с рекомендациями Руководства по содержанию и использованию лабораторных животных (Guide for Care and Use of Laboratory Animals) относительно обездвиживания, условий содержания, оперативных вмешательств, регулирования корма и жидкости, и ветеринарной помощи.
Культура клеток опухоли
Линии клеток лимфомы человека получали из различных источников (ATCC, DSMZ), например, Karpas-422 получали от DSMZ. Линии клеток хранили в виде суспензионных культур в среде RPMI-1640, содержащей 100 единиц/мл натриевой соли пенициллина G, 100 г/мл стрептомицина, 1% HEPES и 1% L-глутамин. В указанную среду добавляли 20% фетальную бычью сыворотку. Клетки культивировали в колбах для тканевых культур в увлажненном инкубаторе при 37°C, в атмосфере 5% CO2 и 95% воздуха.
Имплантация опухоли in vivo
Линии клеток лимфомы человека, например, клетки Karpas-422, собирали во время середины логарифмической фазы роста и ресуспендировали в базовых средах RPMI-1640 и 50% Матригеле™ (BD Biosciences) (RPMI : Матригель = 1:1). Каждая мышь получала 1×107 клеток (0,2 мл суспензии клеток) подкожно в правый бок. Опухоли измеряли штангенциркулем в двух измерениях для контроля роста, когда средний объем приближался к желаемому диапазону 80-120 мм3. Размер опухоли в мм3 рассчитывали следующим образом:
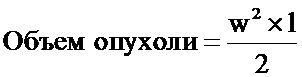
где w = ширина и l = длина опухоли в мм. Массу опухоли можно оценить путем предположения, что 1 мг эквивалентен 1 мм3 объема опухоли. Через 10-30 дней мышей с опухолями 145-150 мм3 распределяли по группам лечения со средним объемом опухоли 147 мм3.
Тестируемые препараты
Тестируемые соединения хранили при комнатной температуре в защищенном от света месте. В каждый день лечения получали свежие составы соединений путем суспендирования порошков в 0,5% натрийкарбоксиметилцеллюлозе (NaCMC) и 0,1% Твин® 80 в деионизированной воде. Носитель, 0,5% NaCMC и 0,1% Твин® 80 в деионизированной воде, использовали для лечения контрольных групп в соответствии с теми же схемами. Составы хранили в защищенном от света месте при 4°С перед введением. Если не указано иное, указанные соединения, которые тестировали в данном эксперименте, были в форме конкретных солей, указанных в данном абзаце.
План лечения
Мышей лечили дозами соединений в диапазоне 62,5-500 мг/кг два раза в сутки (2 раза в сутки каждые 12 часов) через желудочный зонд на протяжении различного количества дней. Каждую дозу доставляли в объеме 0,2 мл/20 г мыши (10 мл/кг) и корректировали в соответствии с последней зарегистрированной массой отдельных животных. Максимальная продолжительность лечения составляла 28 дней.
Средний объем опухоли (MTV) и анализ ингибирования роста опухоли (TGI)
Эффективность лечения определяли в последний день лечения. Для каждой группы определяли MTV(n), средний объем опухоли для определенного количества животных, n, оцениваемый в последний день. Процент ингибирования роста опухоли (% TGI) может быть определен несколькими способами. В соответствии с первым способом разницу между MTV(n) указанной контрольной группы и MTV(n) группы, получавшей лечение лекарственным средством, выражают в виде процента от MTV(n) контрольной группы:
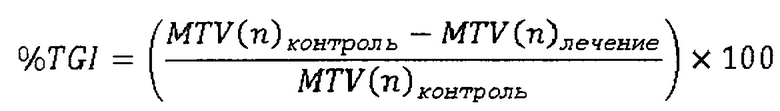
Другой способ расчета % TGI заключается в том, что учитывают изменение размера опухоли с 1 дня по день n, при этом n представляет собой последний день лечения.

Токсичность
Массу тела животных определяли каждый день в дни 1-5, а затем два раза в неделю до завершения исследования. Мышей часто осматривали на предмет явных признаков каких-либо нежелательных побочных эффектов, связанных с лечением, которые документировали. Приемлемую токсичность для максимальной переносимой дозы (MTD) определяли как среднюю потерю массы тела в группе, составляющую менее 20%, во время тестирования и не более 10% летальности в результате смерти, связанной с лечением (TR). Смерть классифицируют как TR, если она связана с побочными эффектами лечения, о чем свидетельствуют клинические признаки и/или аутопсия, или вызвана неизвестными причинами во время периода введения доз. Смерть классифицируют как не связанную с лечением (NTR), если есть подтверждение тому, что смерть не связана с побочными эффектами лечения. NTR-смерть во время интервала между введением доз, как правило, будет отнесена к категории NTRa (вследствие несчастного случая или ошибки со стороны человека) или NTRm (вследствие подтвержденного аутопсией распространения опухоли путем инвазии и/или метастазирования). Животные, получающие лечение перорально, которые умирают по неизвестным причинам во время периода введения доз, могут быть классифицированы как NTRu, когда показатели группы не подтверждают отнесение к классу TR, и аутопсия для исключения ошибки, связанной с введением доз, не представляется возможной.
Получение образцов
В 7 или 28 день во время исследований у мышей забирали образцы предварительно установленным способом для оценки целевого ингибирования опухолей. Опухоли собирали у указанных мышей в условиях с отсутствием РНКаз и разрезали. Замороженную опухолевую ткань каждого животного подвергали мгновенному замораживанию в жидком N2 и измельчали с использованием ступки и пестика.
Статистический и графический анализ
Все статистические и графические анализы выполняли с помощью Prism 3.03 (GraphPad) для Windows. Для проверки статистической значимости между контрольными группами и группами, получавшими лечение, на протяжении всего времени лечения использовали дисперсионный анализ (ANOVA) с повторными измерениями с последующим апостериорным критерием множественного сравнения Даннета или двухфакторный ANOVA. Prism сообщает результаты в виде незначимых (ns) при P>0,05, значимых (обозначены «*») при 0,01<P<0,05, очень значимых («**») при 0,001<P<0,01 и в высшей степени значимых («***») при P <0,001.
Экстракция гистонов
Для выделения гистонов 60-90 мг опухолевой ткани гомогенизировали в 1,5 мл буфера для ядерной экстракции (10 мМ Трис-HCl, 10 мМ MgCl2, 25 мМ KCl, 1% Тритон Х-100, 8,6% сахароза, а также таблетка ингибитора протеаз 1836145 Roche) и инкубировали на льду в течение 5 минут. Ядра собирали путем центрифугирования при 600 g в течение 5 минут при 4°С и один раз промывали ФБР. Удаляли надосадочную жидкость и экстрагировали гистоны 0,4 Н холодной серной кислотой в течение одного часа при перемешивании на вортексе каждые 15 минут. Экстракты осветляли путем центрифугирования при 10000 g в течение 10 минут при 4°С и переносили в чистую микроцентрифужную пробирку, содержащую 10x объем ледяного ацетона. Гистоны осаждали при -20°С в течение 2 часов - в течение ночи, осаждали путем центрифугирования при 10000 g в течение 10 минут и ресуспендировали в воде.
ELISA
Гистоны получали в эквивалентных концентрациях в покрывающем буфере (ФБР + 0,05% BSA) с получением 0,5 нг/мкл образца, и 100 мкл образца или стандарта добавляли в двух повторностях в два 96-луночных планшета для ELISA (Thermo Labsystems, Immulon 4HBX #3885). Планшеты герметизировали и инкубировали в течение ночи при 4°С. На следующий день планшеты промывали 3x 300 мкл/лунка ФБР-T (ФБР + 0,05% Твин 20; 10Х ФБР-T, KPL #51-14-02) в устройстве для промывки планшетов Bio Tek. Планшеты блокировали 300 мкл/лунка разбавителя (ФБР + 2% BSA + 0,05% Твин 20), инкубировали при комнатной температуре в течение 2 часов и промывали 3x ФБР-T. Все антитела разводили в разбавителе. В каждый планшет добавляли 100 мкл/лунка анти-H3K27me3 (CST #9733, 50% исходный раствор глицерина 1:1000) или антитела к общему H3 (Abcam ab1791, 50% глицерин 1:10000). Планшеты инкубировали в течение 90 минут при комнатной температуре и промывали 3x ФБР-T. 100 мкл/лунка анти-Rb-IgG-HRP (Cell Signaling Technology, 7074) добавляли в разведении 1:2000 в планшет H3K27Me3 и в разведении 1:6000 в планшет Н3, и инкубировали в течение 90 минут при комнатной температуре. Планшеты промывали 4x ФБР-T. Для детектирования добавляли 100 мкл/лунка субстрата ТМВ (BioFx Laboratories, #TMBS) и планшеты инкубировали в темноте при комнатной температуре в течение 5 минут. Реакцию останавливали 100 мкл/лунка 1Н H2SO4. Поглощение при 450 нм считывали на ридере для микропланшетов SpectraMax M5.
7-дневное ФД исследование
Для тестирования того, может ли соединение модулировать гистоновую метку H3K27me3 в опухолях in vivo, мышей с ксенотрансплантатными опухолями Karpas-422 лечили соединением в дозах, составляющих 62,5, 83,3, 125, 166,7, 250, 333,3 или 500 мг/кг, два раза в сутки или носителем (в соответствии со схемой введения два раза в сутки) в течение 7 дней. В каждой группе было 5 животных. Животных подвергали эвтаназии через 3 часа после введения последней дозы, и опухоль сохраняли в замороженном состоянии, как описано выше. После экстракции гистонов образцы включали в анализы ELISA с использованием антител к триметилированному состоянию гистона H3K27 (H3K27me3) или общему гистону Н3. На основе этих данных рассчитывали отношение глобально метилированного к общему H3K27. Средние соотношения глобального метилирования для всех групп, измеренные путем ELISA, показывают целевой диапазон ингибирования по сравнению с носителем. План данного эксперимента представлен в таблице 4А.
Схема введения доз
Пример 5: исследование эффективности с увеличением доз в ксенотрансплантатной модели Karpas-422
Для тестирования того, может ли соединение индуцировать противоопухолевый эффект in vivo, мышей с ксенотрансплантатными опухолями Karpas-422 лечили соединением в дозах, составляющих, например, 62,5, 125, 250 или 500 мг/кг, два раза в сутки в течение 28 дней. В каждой группе было 10 мышей для обеспечения эффективности эксперимента. Измеряли рост опухоли на протяжении курса лечения, составляющего 28 дней, для групп, получающих лечение носителем и тестируемым соединением.
Гистоны экстрагировали из опухолей, собранных в конце исследования в 28 день для когорты оценки эффективности (через 3 часа после введения последней дозы для обеих когорт). Метильную метку H3K27me3 оценивали на предмет модуляции при лечении в зависимости от дозы.
План данного эксперимента представлен в таблице 4В.
Схема введения доз
Объем опухоли рассчитывали на основе измерений штангенциркулем по формуле для объема вытянутого эллипсоида (L×W2)/2, где L и W представляют собой соответствующие ортогональные измерения длины и ширины (мм).
Данные выражали в виде среднего значения ± стандартное отклонение (SD). Различия в объеме опухоли между группами, получавшими лечение носителем и получавшими лечение соединением, анализировали путем дисперсионного анализа (ANOVA) с повторными измерениями с последующим критерием множественного сравнения типа Даннетта. Значение P<0,05 (двусторонний) считается статистически значимым. Статистические анализы выполняли с использованием пакета программного обеспечения Prism 5, версия 5.04 (GraphPad Software, Inc., Калифорния, США).
Указанные выше исследования показали, что как соединение 1, так и соединение А демонстрировали остановку роста и регрессию опухоли в ксенотрансплантатной модели Karpas-422, и хорошо переносились. См., например, Фиг. 1 и 2. Кроме того, фармакокинетические и фармакодинамические свойства соединения 1 или соединения А из вышеуказанных исследований проиллюстрированы на Фиг. 3-8. Фиг. 3 представляет собой график, демонстрирующий концентрацию соединения 1 в опухоли в 7 день или 28 день после лечения или концентрацию соединения А в опухоли в 7 день после лечения. На данной фигуре «А»-«G» обозначают 7 дней после введения соединения 1 в дозах, составляющих 62,5, 83,3, 125, 166,7, 250, 333,3 и 500 мг/кг соответственно; «Н» и «I» обозначают 7 дней после введения соединения А в дозах, составляющих 125 и 250 мг/кг соответственно; и «J»-«L» обозначают 28 дней после введения соединения 1 в дозах, составляющих 62,5, 125 и 250 мг/кг соответственно. Примечание: для образцов в 28 день опухоли были слишком малы для анализа в группе, получавшей лечение 250 мг/кг соединения А, и группе, получавшей лечение 500 мг/кг соединения 1; только 1 из 10 и 5 из 10 были достаточно большими для анализа в группах, получавших лечение 250 мг/кг соединения 1 и 125 мг/кг соединения 1 соответственно. Фиг. 4 представляет собой график, демонстрирующий концентрацию соединения 1 или соединения А в плазме крови в 7 день или 28 день после лечения. Верхняя пунктирная линия показывает LCC соединения А с поправкой на PPB, а нижняя пунктирная линия показывает LCC соединения 1 с поправкой на PPB. Фиг. 5 представляет собой график, демонстрирующий глобальное метилирование H3K27me3 в опухолях KARPAS-422 у мышей, которых лечили соединением 1 или соединением А в течение 7 дней. На данной фигуре «А» обозначает лечение носителем; «В»-«Н» обозначают лечение соединением 1 в дозах, составляющих 62,5, 83,3, 125, 166,7, 250, 333,3 и 500 мг/кг соответственно; и «I» и «J» обозначают лечение соединением А в дозах, составляющих 125 и 250 мг/кг соответственно. Фиг. 6 представляет собой график, демонстрирующий глобальное метилирование H3K27me3 в опухолях KARPAS-422 у мышей, которых лечили соединением 1 в течение 28 дней. Фиг. 7 представляет собой график, демонстрирующий глобальное метилирование H3K27me3 в костном мозге мышей с ксенотрансплантатными опухолями KARPAS-422, которых лечили соединением 1 или соединением А в течение 7 дней. На данной фигуре «А» обозначает лечение носителем; «В»-«Н» обозначают лечение соединением 1 в дозах, составляющих 62,5, 83,3, 125, 166,7, 250, 333,3 и 500 мг/кг соответственно; и «I» и «J» обозначают лечение соединением А в дозах, составляющих 125 и 250 мг/кг соответственно. Фиг. 8 представляет собой график, демонстрирующий глобальное метилирование H3K27me3 в костном мозге мышей с ксенотрансплантатными опухолями KARPAS-422, которых лечили соединением 1 в течение 28 дней. На данной фигуре «А» обозначает лечение носителем; «В»-«Е» обозначают лечение соединением 1 в дозах, составляющих 62,5, 125, 250 и 500 мг/кг соответственно; и «F» обозначает лечение соединением А в дозе, составляющей 250 мг/кг.
В примерах 6-8 ниже эксперименты и анализы проводили с использованием способов и методов, аналогичных описанным, например, в J. Lin, Pharmaceutical Research, 2006, 23(6):1089-1116; L. Di et al., Comb Chem High Throughput Screen. 2008, 11(6):469-76; M. Fonsi et al., Journal of Biomolecular Screening, 2008, 13:862; E. Sjögren et al., Drug Metab. Dispos. 2012, 40:2273-2279; T.D. Bjornsson, et al., Drug Metab Dispos, 2003, 31(7):815-832; J.B. Houston and K.E. Kenworthy, Drug Metab Dispos, 2000, 28(3):246-254; и S.W. Grimm et al., Drug Metab Dispos, 2009, 37(7):1355-1370, полное содержание каждого из которых включено в настоящее описание посредством ссылки.
Пример 6
Оценка метаболической стабильности в микросомах печени
Метаболическую стабильность соединений 1, 2 и 105 оценивали в микросомах печени пяти видов, включая мышей, крыс, собак, обезьян и людей.
Методы: осуществляли инкубацию в 96-луночных планшетах, содержащих 250 мкл общего объема, состоящего из 100 ммоль/л калийфосфатного буфера (рН 7,4), 1 мг/мл микросом печени, тестируемого соединения (т.е. соединений 1, 2 или 105) в 8 концентрациях и 2 мг/мл НАДФН. Концентрации соединений, используемые для инкубации, находились в диапазоне от 45,7 нМ до 100 мкМ. Добавление НАДФН использовали для начала реакции, и инкубацию осуществляли на встряхивающей водяной бане при 37°С в течение до 60 минут. Реакции прекращали путем добавления равного объема стоп-раствора, содержащего внутренний стандарт (IS). Затем образцы центрифугировали на центрифуге с охлаждением при 3000 об/мин. в течение не менее 5 минут перед анализом. Степень истощения в инкубационных смесях для тестируемого соединения, определенную путем использования ЖХ/времяпролетной масс-спектрометрии (TOFMS), использовали для определения значений собственного клиренса с помощью микросом печени. Системы ЖХ/TOFMS состояли из автоматического пробоотборника Shimadzu SIL-HTC (Киото, Япония), двух насосов LC-20AD (Shimadzu Corp.) и термостата колонки (CTO-20AC; Shimadzu Corp.) с времяпролетным масс-спектрометром (AB SCIEX Qstar Elite, AB Sciex, Фостер-Сити, Калифорния). Площади пиков тестируемого соединения и IS интегрировали с помощью Analyst QS (версия 2.0, Applied Biosystems, Фостер-Сити, Калифорния, США). Активированную столкновениями диссоциацию (CAD) с использованием азота использовали для генерации ионов продукта. Оптимизированные параметры оборудования были в режиме положительной ионизации.
Количественное определение путем ЖХ/МС/МС было основано на соотношениях площадей пиков тестируемого соединения и IS. Для расчета и интеграции площадей пиков для соединений 1, 2 или 105 и IS использовали Analyst QS 2.0 (AB Sciex, Фостер-Сити, Калифорния, США). Расчеты проводили с использованием Excel (Office 2010, Microsoft Corp., Редмонд, Вашингтон, США) и GraphPad Prism v. 5.02 (GraphPad Software Inc., Ла-Хойя, Калифорния, США). Данные анализировали и сообщали на основе соответствующих SOP, таких как описаны в J. Lin, Pharmaceutical Research, 2006, 23(6): 1089-1116; и Di L et al., Comb Chem High Throughput Screen. 2008 11(6): 469-76.
Истощение тестируемого соединения, используемого для значений Km и Vmax в случае микросом печени, рассчитывали путем построения графика зависимости от времени для определения скорости истощения. Затем скорости истощения наносили на график с использованием соответствующей кинетической модели. Рассчитывали данные на основе следующих уравнений:
Михаэлиса-Ментен: Clint (мкл/мин/мг микросом печени) = Vmax/Km
Хилла: Clint (мкл/мин/мг микросом печени) = Clmax = Vmax/Ks·(n-1)/(n(n-1)1/n)
Clint (мкл/мин/г печени) = Clint (мкл/мин/мг микросом)⋅масштабный коэффициент или Clint (мкл/мин/106 клеток)⋅масштабный коэффициент.
Результаты приведены в таблице 5 ниже.
Все три тестируемых соединения, т.е. соединения 1, 2 и 105, демонстрировали низкий метаболический клиренс в микросомах печени человека (HLM). Также наблюдали видовое различие метаболического клиренса. Яванские макаки, в целом, демонстрировали более высокий клиренс, чем другие виды.
Пример 7
Оценка индукции CYP
Оценивали индукцию для каждого из соединений 1, 2 и 105 в криоконсервированных гепатоцитах человека от одного донора.
Методы: гепатоциты получали от BD Biosciences (Уоборн, Массачусетс, США), а соответствующие среды и ядерный краситель DAPI приобретали у Life Technologies (Дарем, Северная Каролина, США). Модифицированную по способу Дульбекко среду Игла (DMEM), фосфатный буферный раствор Дульбекко (Д-ФБР), заменимые аминокислоты 100x MEM, 100x раствор пенициллина/стрептомицина/глутамина, 2x трипановый синий приобретали у Mediatech (Манассас, Вирджиния, США). Фетальную бычью сыворотку (FBS) приобретали у Tissue Culture Biologicals (Тулар, Калифорния, США). 24-луночные планшеты, покрытые коллагеном, для анализа мРНК приобретали у BD Biosciences (Сан-Хосе, Калифорния, США). Предварительно разработанные зонды и праймеры использовали в двух тройных анализах для оценки изменения мРНК. Положительные контроли представляли собой β-нафтофлавон для CYP1A (1 и 10 мкмоль/л), фенобарбитал для CYP2B6 (100 мкмоль/л и 1 ммоль/л) и рифампицин для CYP2C9 и CYP3A (1 и 10 мкмоль/л). ДМСО использовали в качестве контроля носителем (отрицательного контроля). Анализы, специфичные в отношении формы CYP, выполняли после периода лечения, и клетки подсчитывали для определения жизнеспособности.
Криоконсервированные гепатоциты подвергали оттаиванию на водяной бане 37°C и высевали в соответствии с инструкциями производителя. Одну пробирку с гепатоцитами добавляли в 50 мл коническую пробирку, содержащую среду для извлечения криоконсервированных гепатоцитов (CHRM) от Life Technologies. Клетки центрифугировали на центрифуге Beckman с ротором GH 3.8 при 800 об/мин. в течение 10 минут. Надосадочную жидкость удаляли и клетки ресуспендировали в средах для планшетов для подсчета. После подсчета клетки ресуспендировали в количестве 0,75 миллиона жизнеспособных клеток/мл. Суспензию (0,5 мл/лунка) добавляли в 24-луночный планшет, покрытый коллагеном, или после добавления 60 мкл DMEM, содержащей 10% FBS, пенициллин/стрептомицин/глутамин и заменимые аминокислоты MEM, 80 мкл суспензии добавляли в каждую лунку 96-луночного планшета, покрытого коллагеном. После перемешивания и встряхивания для обеспечения лучшего покрытия планшета клетки помещали в инкубатор тканевой культуры при 5% CO2 и 37°C, и оставляли прилипать в течение ночи. Затем клетки обрабатывали с использованием поддерживающих сред для гепатоцитов CellzDirect. На клетки воздействовали соединением 1, 2 или 105 (1 или 10 мкмоль/л), или носителем в течение 48 часов. Во время обработки поддерживающие среды и тестируемые соединения пополняли каждые 24 часа.
После завершения инкубации клетки промывали Д-ФБР. После промывки Д-ФБР клетки фиксировали с использованием 3,7% п-формальдегида в Д-ФБР в течение одного часа. Удаляли формальдегид и добавляли 0,6 мкмоль/л DAPI в Д-ФБР. Клетки окрашивали DAPI в течение 20 минут, а затем три раза промывали Д-ФБР. Клетки подсчитывали с использованием ArrayScan II (Cellomics, Питтсбург, Пенсильвания, США) с 5X линзой объектива. Оставшуюся фракцию гепатоцитов рассчитывали с использованием количества клеток, обнаруженных в условиях обработки, деленного на клетки, обнаруженные в случае контроля носителем.
После обработки клетки лизировали с использованием буфера RLT из набора Qiagen RNeasy. мРНК выделяли с использованием набора RNeasy с обработкой ДНКазой с использованием протоколов производителя. Концентрацию мРНК измеряли с помощью Nanodrop ND-1000 (Уилмингтон, Делавэр, США). Концентрацию мРНК нормализовали для каждого образца в пределах донора. Обратную транскрипцию осуществляли с использованием набора для синтеза кДНК Superscript VILO от Life Technologies, следуя инструкции производителя. После синтеза кДНК проводили количественную ПЦР в реальном времени с использованием системы 7500 Fast Real Time PCR от Applied Biosystems, дочерней компании Life Technologies, находящейся в полной собственности. Компоненты реакции для ПЦР в реальном времени состояли из: 10 мкл Taqman Fast Advanced Master Mix (Life Technologies), 2 мкл кДНК, 5 мкл воды, не содержащей нуклеазу (Life Technologies), 1 мкл FAM-меченого HS00984230_m1 для анализа с ограниченным праймером для β-2 микроглобулина, 1 мкл VIC-меченого HS00167927_m1 для CYP1A2 или HS04183483_g1 для CYP2B6 для анализа с ограниченным праймером и 1 мкл NED-меченого HS04260376_m1 для CYP2C9 или HS00604506_m1 для CYP3A4 для анализа с ограниченным праймером. Расчеты кратного различия по сравнению с контролем носителем выполняли с помощью программного обеспечения 7500, версия 2.0.5 (Life Technologies) с использованием метода ΔΔCt. Расчеты Emax и EC50 выполняли с использованием GraphPad Prism.
Как показано в таблице 6 выше, соединение 1 не демонстрировало значительную индукцию (за исключением возможно CYP2C9); соединение 2 не демонстрировало индукцию (с несколько более низкой активностью); и соединение 105 демонстрировало индукцию CYP2B6, CYP2C9 и CYP3A. Не наблюдали значительной потери жизнеспособности клеток.
Пример 8
Оценка ингибирования CYP
Возможное ингибирование ферментативной активности цитохромов P450 (CYP) человека соединением 1, 2 или 105 оценивали с использованием объединенных в пул микросом печени человека.
Методы: потенциал соединений 1, 2 и 105 в отношении конкурентного ингибирования определяли путем оценки в нескольких концентрациях в реакциях с маркером CYP вблизи их соответствующих значений Km для построения кривых IC50 в микросомах печени человека (HLM). Потенциал в отношении зависимой от времени инактивации (TDI) также оценивали для CYP3A4/5 путем оценки значений KI и kinact в соответствующих случаях.
Суспензию, содержащую PB, HLM, CYP-селективный маркерный субстрат и тестируемый ингибитор, добавляли в 96-луночный планшет. Планшеты предварительно инкубировали на водяной бане 37°C в течение приблизительно 2 минут. Реакцию инициировали путем добавления НАДФН в каждую лунку 96-луночного планшета. Конечные концентрации для PB, HLM и НАДФН составляли 100 ммоль/л (рН 7,4), 0,1 мг/мл и 2,3 ммоль/л соответственно. Маркерные субстраты CYP и ингибиторы CYP, используемые в качестве положительных контролей, и их соответствующие концентрации приведены ниже. Конечная концентрация MeOH, используемая при каждой инкубации, не превышала 0,8%.
По окончании соответствующего времени инкубации в соответствующие лунки добавляли равный объем гасящего раствора, содержащего IS. Стандартные образцы и образцы контроля качества (QC) готовили с использованием аналогичных компонентов, что и тестируемые образцы. Холостую пробу получали с использованием аналогичного гасящего раствора, что и для других образцов, но не содержащего IS. Планшеты центрифугировали в течение 5 минут на настольной центрифуге при 3000 об/мин. Образцы анализировали путем ЖХ/МС/МС. Условия инкубации и положительные контроли приведены в таблице 7 ниже.
Для оценки TDI, первичной инкубации, суспензию, содержащую PB, HLM и исходные растворы ингибиторов, добавляли в 96-луночный планшет. Планшеты предварительно инкубировали на водяной бане 37°C в течение приблизительно 2 минут. Реакцию инициировали путем добавления НАДФН в каждую лунку 96-луночного планшета и проводили в течение 0, 5, 10, 15, 20 и 30 минут. Для контролей с отсутствием НАДФН, раствор PB заменяли на исходный раствор НАДФН. Конечные концентрации для PB, HLM и НАДФН составляли 100 ммоль/л (рН 7,4), 0,2 мг/мл и 2,3 ммоль/л соответственно. В соответствующие моменты времени 12,5 мкл суспензии для первичной инкубации 20-кратно разводили с получением предварительно инкубированной смеси для вторичной инкубации, содержащей CYP-селективные маркерные субстраты, для оценки остаточной активности. Используемые маркерные субстраты представляли собой тестостерон (250 мкмоль/л), а положительный контроль представлял собой тролеандомицин.
Используемая система ВЭЖХ представляла собой систему ВЭЖХ Shimadzu (Киото, Япония), состоящую из автоматического пробоотборника SIL-HTC, дегазатора DGU-14A, трех насосов LC-10ADvp и термостата колонки CTO-10ACvp. Образцы анализировали на масс-спектрометре с тремя квадрупольными линзами API4000QTrap (АВ Sciex, Фостер-Сити, Калифорния) с использованием ионизации распылением в режиме определения положительных ионов.
Расчет и интеграцию площадей пиков для всех контролируемых метаболитов и IS производили с помощью Analyst 1.6 (AB Sciex, Фостер-Сити, Калифорния). Количественное определение осуществляли с использованием калибровочных кривых, построенных путем построения графика зависимости соотношения площадей пиков метаболита и IS от концентраций калибровочных стандартов, и производили с помощью квадратичной регрессии с 1/x2 заданием массы (y = а х2 + bx + c). В расчетах процента ингибирования использовали Excel (Office 2010, Microsoft Corp., Редмонд, Вашингтон) и данные наносили на график с использованием GraphPad Prism (версия 5.02, GraphPad Software, Inc., Ла-Хойя, Калифорния), исходя из одного сайта связывания. Номинальные концентрации тестируемых соединений и положительных контролей подвергали логарифмическому преобразованию в GraphPad Prism. С использованием графиков зависимости остаточной активности CYP3A4/5 от времени предварительной инкубации оценивали скорость потери активности CYP (λ) путем нелинейной регрессии. Затем оценивали параметры kinact и KI путем подгонки к следующему уравнению:
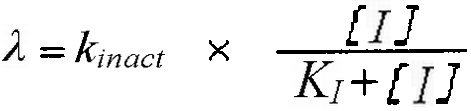
где [I] представляет собой начальную концентрацию соединения 1, 2 или 105.
Обратимое ингибирование
Оценка зависимой от времени инактивации (TDI)
Как показано в таблице 8 выше, соединения 1 и 2 не демонстрировали значительное обратимое ингибирование; а соединение 105 демонстрировало потенциально значимое ингибирование CYP2D6 и CYP3A4 (мидазолам). Кроме того, как показано в таблице 9 выше, все соединения 1, 2 и 105 демонстрировали время-, концентрацию- и НАДФН-зависимую инактивацию CYP3A4/5 (с использованием мидазолама в качестве маркера).
Полное содержание каждого из патентных документов и научных статей, указанных в настоящем описании, включено посредством ссылки для всех целей.
Настоящее изобретение может быть реализовано в других конкретных формах в пределах его сущности или существенных характеристик. Соответственно, приведенные выше варианты реализации являются во всех отношениях иллюстративными, а не ограничивающими настоящее изобретение, описанное в настоящем документе. Таким образом, объем настоящего изобретения определяется прилагаемой формулой изобретения, а не приведенным выше описанием, и все изменения, которые подпадают под значение и диапазон эквивалентности формулы изобретения, включены в него.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ЛЕЧЕНИЯ РАКА | 2013 |
|
RU2677276C2 |
| СОЛЕВАЯ ФОРМА ИНГИБИТОРА ГИСТОН-МЕТИЛТРАНСФЕРАЗЫ EZH2 ЧЕЛОВЕКА | 2013 |
|
RU2658911C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ИНГИБИТОРОМ EZH2 | 2018 |
|
RU2754131C1 |
| АРИЛ-ИЛИ ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2632193C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2013 |
|
RU2699546C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ СОЕДИНЕНИЯ ТРИАЗОЛОПИРИМИДИНА | 2017 |
|
RU2754856C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2629118C2 |
| ПРИМЕНЕНИЕ ИНГИБИТОРА EZH2 В КОМБИНАЦИИ С ИНГИБИТОРОМ BTK В ПОЛУЧЕНИИ ЛЕКАРСТВЕННОГО СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ ОПУХОЛИ | 2018 |
|
RU2762893C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2013 |
|
RU2827936C2 |
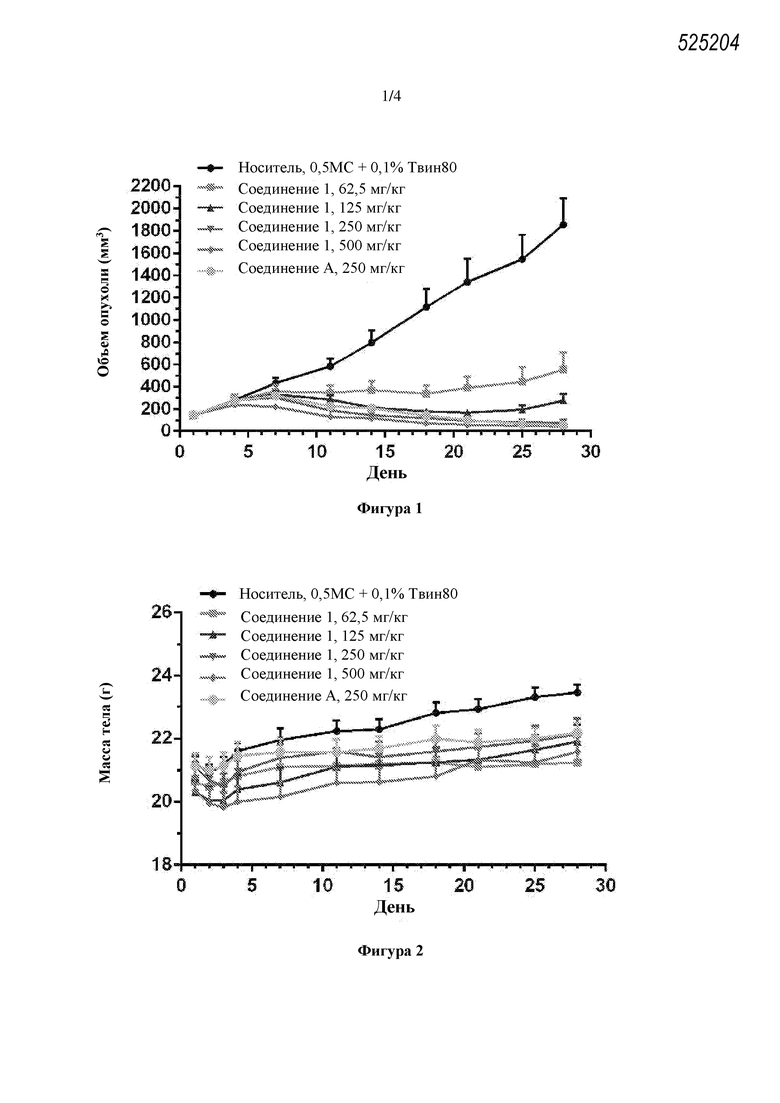
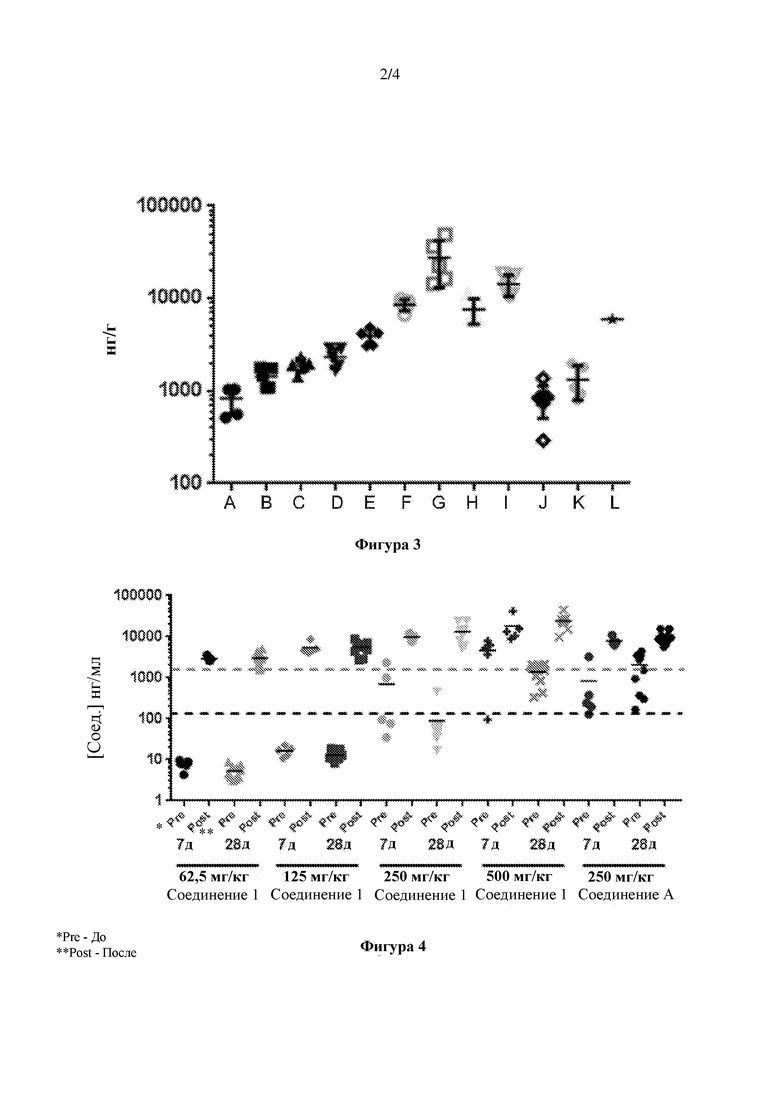
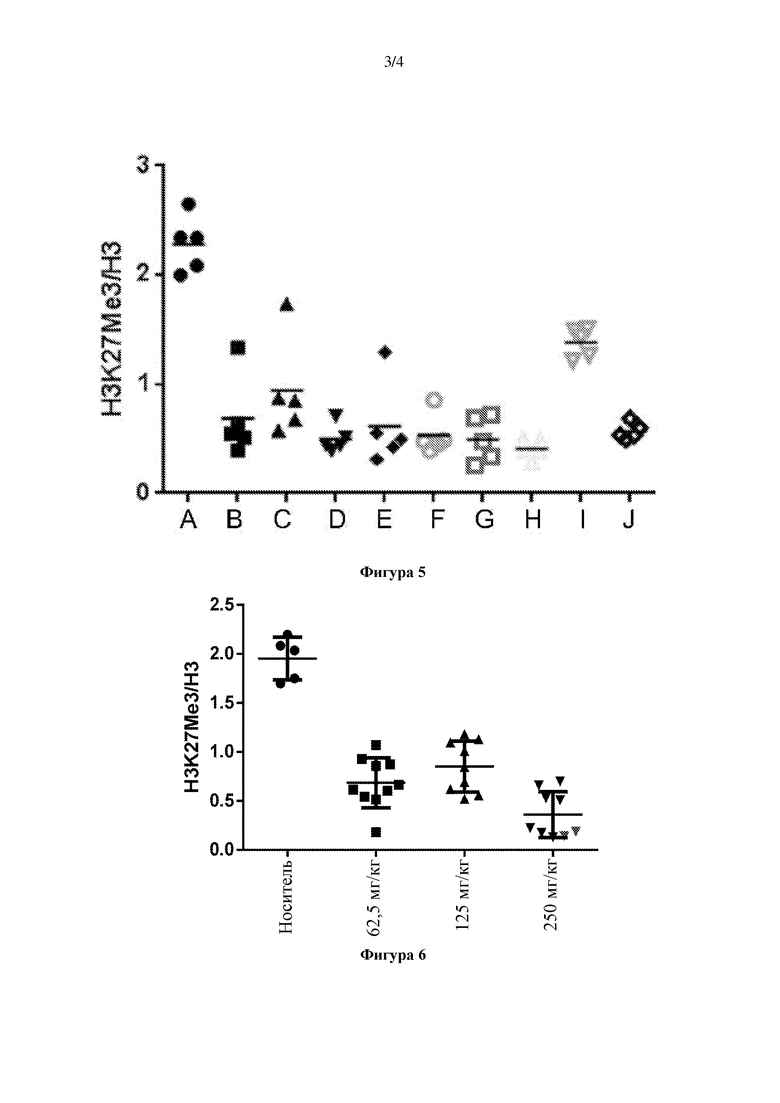
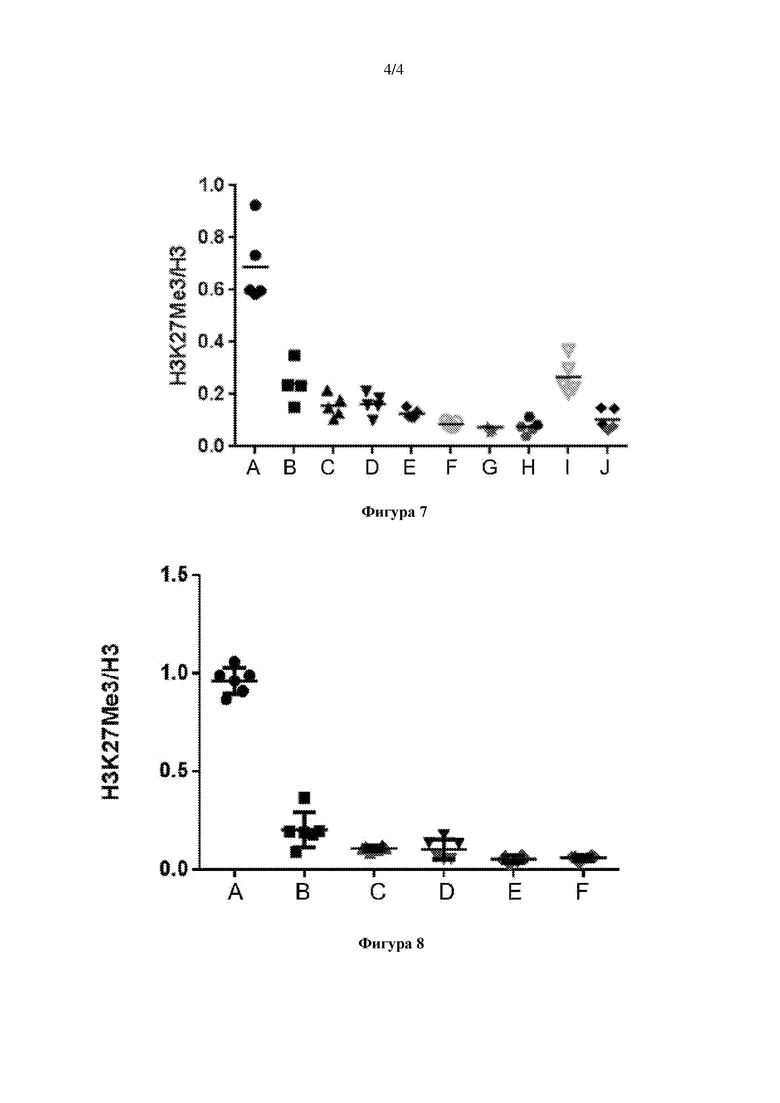
Изобретение относится к соединению, выбранному из группы, состоящей из
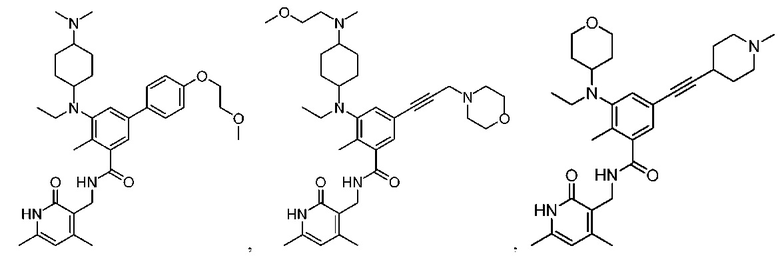
и его фармацевтически приемлемым солям. Изобретение также относится к фармацевтическим композициям, к способу лечения EZH2-опосредуемого нарушения, к применению соединения для изготовления лекарственного средства. Технический результат: получены новые соединения, обладающие свойствами ингибиторов активности EZH2. 5 н. и 11 з.п. ф-лы, 8 ил., 9 табл., 8 пр.
1. Соединение, выбранное из группы, состоящей из
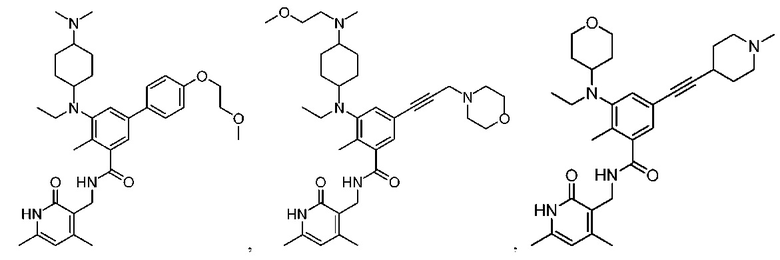
и их фармацевтически приемлемых солей.
2. Соединение по п. 1, отличающееся тем, что указанное соединение представляет собой 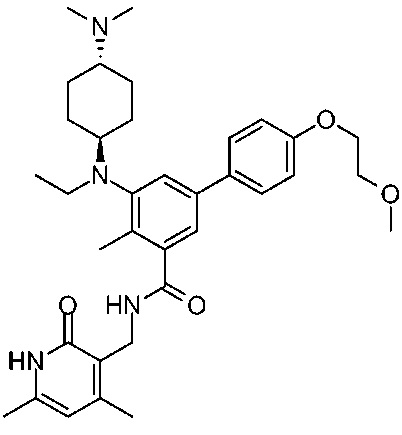 или его фармацевтически приемлемую соль.
или его фармацевтически приемлемую соль.
3. Соединение по п. 1, отличающееся тем, что указанное соединение представляет собой 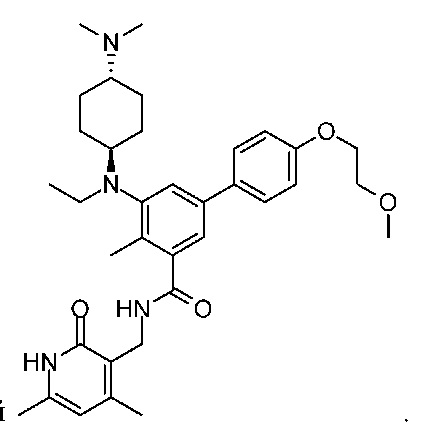 .
.
4. Соединение по п. 1, отличающееся тем, что указанное соединение представляет собой 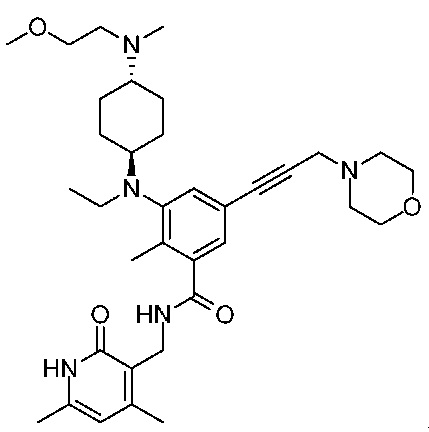 или его фармацевтически приемлемую соль.
или его фармацевтически приемлемую соль.
5. Соединение по п. 1, отличающееся тем, что указанное соединение представляет собой 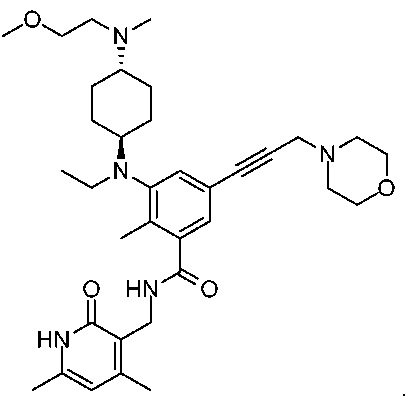 .
.
6. Соединение по п. 1, отличающееся тем, что указанное соединение представляет собой 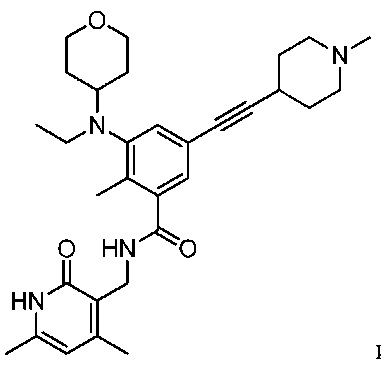 или его фармацевтически приемлемую соль.
или его фармацевтически приемлемую соль.
7. Соединение по п. 1, отличающееся тем, что указанное соединение представляет собой 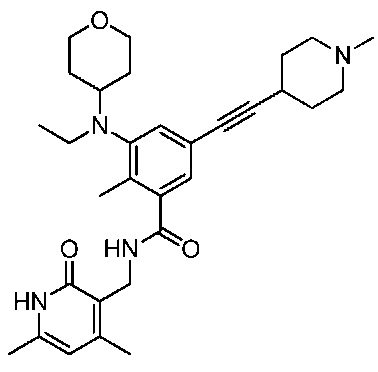 .
.
8. Фармацевтическая композиция, обладающая способностью ингибировать активность EZH2, содержащая терапевтически эффективное количество соединения по любому из пп. 1-7 и фармацевтически приемлемый носитель.
9. Способ лечения EZH2-опосредуемого нарушения, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения по любому из пп. 1-7.
10. Способ по п. 9, отличающийся тем, что EZH2-опосредуемое нарушение представляет собой рак.
11. Способ по п. 10, отличающийся тем, что рак представляет собой лимфому, лейкоз или меланому.
12. Способ по п. 10, отличающийся тем, что рак представляет собой диффузную крупноклеточную В-клеточную лимфому (ДКВЛ), неходжкинскую лимфому (НХЛ), фолликулярную лимфому, хронический миелогенный лейкоз (ХМЛ), острый миелоидный лейкоз, острый лимфоцитарный лейкоз, недифференцированный лейкоз или миелодиспластические синдромы (МДС).
13. Способ по п. 10, отличающийся тем, что рак представляет собой злокачественную рабдоидную опухоль или опухоль с дефицитом INI1.
14. Соединение по любому из пп. 1-7 для применения в способе лечения EZH2-опосредуемого нарушения.
15. Применение соединения по любому из пп. 1-7 для изготовления лекарственного средства для лечения EZH2-опосредуемого нарушения.
16. Фармацевтическая композиция, обладающая способностью ингибировать гистонметилтрансферазную активность EZH2 или его мутанта, содержащая терапевтически эффективное количество соединения по любому из пп. 1-7 в качестве основного ингредиента и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество.
| WO 2011140324 A1, 10.11.2011 | |||
| WO 2012118812 A2, 07.09.2012 | |||
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ МОДУЛЯТОРОВ ПУТИ HEDGEHOG | 2007 |
|
RU2413718C2 |

