ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к комбинации ингибитора EZH2 и ингибитора BTK, а также к ее применению в получении лекарственного средства для лечения опухоли.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Лимфома представляет собой лимфоидное злокачественное новообразование, образующееся в лимфатических узлах и/или внеузловых лимфоидных тканях. В зависимости от наличия клеток Рид - Штернберга (R-S клеток) по данным патологического анализа лимфому можно классифицировать как лимфому Ходжкина (ЛХ) и неходжкинскую лимфому (НХЛ). В конце 2015 г. заболеваемость злокачественной лимфомой в Китае составляла 8,82/100000, что составляет одиннадцатую часть от общей заболеваемости для всех видов опухолей. Заболеваемость злокачественной лимфомой у мужчин выше, чем у женщин, и составляет 5,30/100000 и 3,52/100000 соответственно. В 2015 г. смертность от злокачественной лимфомы в Китае составляла 5,21/100000, занимая 10 место среди всех случаев смерти от опухолевых заболеваний.
В Азии 90% пациентов с лимфомой составляют пациенты с НХЛ, у которых в патологическом анализе имеются лимфоциты, гистиоциты или ретикулоциты в различной степени дифференцировки. В соответствии с естественным течением НХЛ можно разделить на три основных клинических типа, а именно высокоинвазивную, инвазивную и индолентную лимфому. В зависимости от происхождения от различных лимфоцитов ее можно классифицировать как B-клеточную лимфому, T-клеточную лимфому и лимфому из естественных киллеров (NK). Основная функция B-клеток состоит в секреции различных антител для защиты организма от различных внешних инвазий.
Метилтрансфераза гистонов, кодируемая геном EZH2, представляет собой каталитический компонент репрессивного комплекса 2 группы поликомб(Polycomb repressive complex 2; PRC2). Концентрации EZH2 в раковых тканях аномально повышены по сравнению со здоровыми тканями, а наиболее высокая экспрессия EZH2 отмечается в распространенных опухолях или при плохом прогнозе. При некоторых типах опухолей гиперэкспрессия EZH2 происходит одновременно с амплификацией гена EZH2. В ряде экспериментальных исследований, посвященных малой интерферирующей РНК (siРНК)/малой РНК, образующей шпильки (shРНК), показано, что снижение экспрессии EZH2 в линиях опухолевых клеток может ингибировать пролиферацию, миграцию и инвазию, или ангиогенез, и приводить к апоптозу. В WO 2017084494 (PCT/CN 2016/104318, дата подачи 2 ноября 2016 г.) описан ингибитор EZH2, имеющий следующую структуру:
Тирозинкиназа Брутона (BTK) входит в состав подсемейства тирозинкиназ и относится к семейству Tecкиназ. Она преимущественно экспрессируется в B-клетках и распределена в лимфатической системе, системе гемопоэза и клетках крови. B-клеточный рецептор (BCR) играет принципиально важную роль в регуляции пролиферации и выживания различных лимфом, включая подтипы хронического лимфоцитарного лейкоза (ХЛЛ) и неходжкинской лимфомы (НХЛ), мантийноклеточной лимфомы (МКЛ) и диффузной B-крупноклеточной лимфомы (ДВКЛ). Кроме того, в клинической практике доказано влияние B-клеток на патогенез ревматоидного артрита, системной красной волчанки, рассеянного склероза и других заболеваний иммунной системы. Тирозинкиназа Брутона (BTK) является ключевой протеинкиназой в сигнальном пути BCR. Она может регулировать созревание и дифференцировку нормальных B-клеток, а также тесно связана с различными заболеваниями, связанными с нарушениями B-клеточной лимфоидной ткани. Таким образом, нацеленный на BTK низкомолекулярный ингибитор может быть полезен в лечении B-клеточных злокачественных новообразований и аутоиммунных заболеваний. В WO 2016007185 A1 (дата публикации 14 января 2014 г.) описан ингибитор BTK, имеющий следующую структуру:
B-клетки, расположенные в зародышевом центре, называют B-клетками зародышевого центра (GCB-клетками). GCB-клетки делятся очень быстро и усиленно вырабатывают антитела с высокой аффинностью, которые помогают бороться с инвазивными инфекциями, а остальные GCB-клетки являются апоптическими. В связи с быстрым делением GCB-клеток и одновременной перегруппировкой VDJ репарация ДНК ослабляется. Таким образом, зародышевый центр является движущей силой образования лимфомы. К сожалению, в этом случае также происходят мутации во многих других генах, что, в конечном счете, приводит к образованию лимфомы, такой как диффузная B-крупноклеточная лимфома и фолликулярная лимфома, из клеток, подобных B-клеткам зародышевого центра.
Показано, что B-клеточная лимфома, имеющая происхождение из зародышевого центра, характеризуется стойкой активацией мутаций BTK и EZH2 (Y641, Y646, A682, A692 и т.п.) или их гиперэкспрессией. Комбинация ингибитора BTK и ингибитора EZH2 способна одновременно ингибировать пролиферацию опухолевых клеток, вызванную аномальной (или избыточной) активацией мутаций BTK и EZH2 (или их гиперэкспрессией), что приводит к синергическому противоопухолевому действию.
В заявках на патенты WO 2014168975 A1 (дата публикации 16 октября 2014 г.), WO 2014166820 A1 (дата публикации 16 октября 2014 г.) и WO 2015146159 A1 (дата публикации 1 октября 2015 г.) описаны комбинации ингибитора EZH2 и ингибитора BTK в лечении B-клеточных пролиферативных заболеваний. В настоящем изобретении предложено применение комбинации ингибитора EZH2 и ингибитора BTK в получении лекарственного средства для лечения опухоли, в которой ингибитор EZH2 и ингибитор BTK имеют новые структуры, а их комбинация обладает синергическим действием.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Техническая задача, решаемая настоящим изобретением, состоит в обеспечении применения комбинации ингибитора EZH2 и ингибитора BTK в получении лекарственного средства для лечения опухоли, где комбинация обладает синергическим действием.
Технические решения настоящего изобретения состоят в следующем.
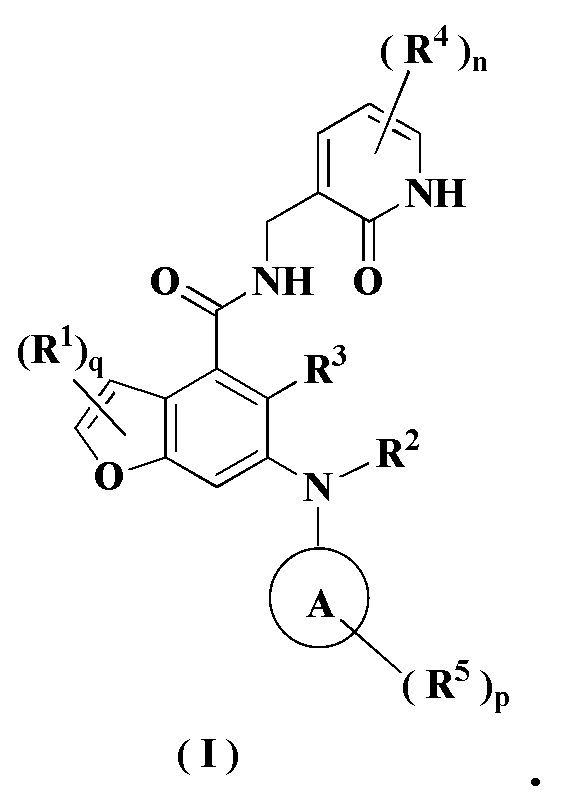
В настоящем изобретении предложено применение комбинации ингибитора EZH2 и ингибитора BTK в получении лекарственного средства для лечения опухоли, отличающейся тем, что ингибитор EZH2 представляет собой соединение формулы (I)
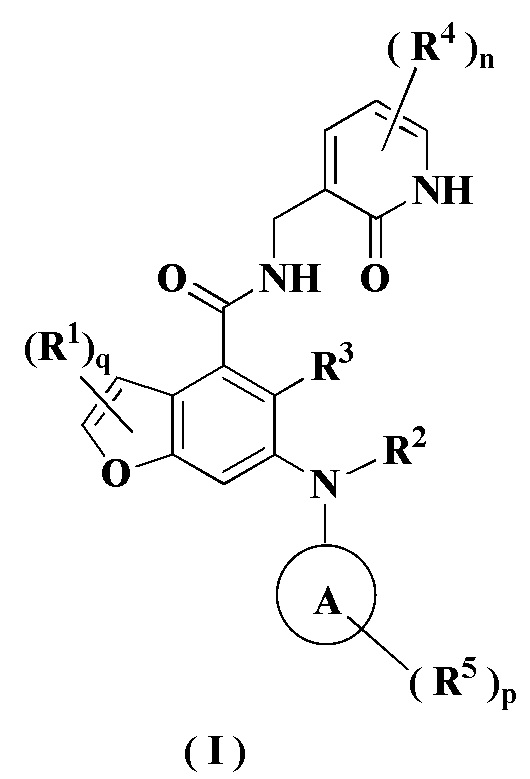
или его фармацевтически приемлемую соль, или его стереоизомер,
где
кольцо A выбрано из группы, состоящей из гетероциклила и циклоалкила;
каждый из R1 является одинаковым или разным, и каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, амино, нитро, гидрокси, циано, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6, -S(O)mR6, -S(O)mNR7R8 и -(CH2)xRa, где каждый из алкила, галогеналкила, гетероциклила, арила и гетероарила независимо и необязательно замещен одним или более заместителями, выбранными из группы, состоящей из алкила, галогеналкила, галогена, амино, нитро, циано, гидрокси, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
Ra выбран из группы, состоящей из галогена, циклоалкила, гетероциклила и -NR7R8, где каждый из циклоалкила и гетероциклила независимо и необязательно замещен одним или более заместителями, выбранными из группы, состоящей из алкила, галогеналкила, галогена, амино, нитро, циано, гидрокси, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R2 представляет собой водород или алкил, где алкил необязательно замещен одной или более заместителями, выбранными из группы, состоящей из галогена, гидрокси, циано, циклоалкила и гетероциклила;
R3 выбран из группы, состоящей из водорода, алкила, галогена, циано, алкокси и галогеналкила;
каждый из R4 является одинаковым или разным, и каждый независимо выбран из группы, состоящей из водорода, алкила, галогеналкила, гидрокси, амино, алкокси, галогеналкокси, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6, -S(O)mR6, -S(O)mNR7R8 и -NR7R8;
каждый из R5 является одинаковым или разным, и каждый независимо выбран из группы, состоящей из водорода, алкила, оксо, галогена, галогеналкила, гидрокси, амино, алкокси, галогеналкокси, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6, -S(O)mR6, -S(O)mNR7R8 и -NR7R8;
R6 выбран из группы, состоящей из водорода, алкила, галогеналкила, алкокси, гидроксиалкила, гидрокси, амино, циклоалкила, гетероциклила, арила и гетероарила;
R7 и R8 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из водорода, алкила, алкокси, гидроксиалкила, гидрокси, амино, алкоксикарбонила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из алкила, амино, циклоалкила, гетероциклила, арила и гетероарила независимо и необязательно замещен одним или более заместителями, выбранными из группы, состоящей из алкила, галогена, гидрокси, амино, алкоксикарбонила, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
m равно 0, 1 или 2;
n равно 0, 1, 2 или 3;
p равно 0, 1, 2, 3, 4 или 5;
q равно 0, 1 или 2; и
x равно 0, 1, 2 или 3.
Предпочтительно ингибитор EZH2 представляет собой соединение формулы (IA)
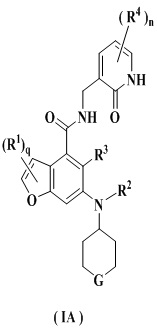
или его фармацевтически приемлемую соль, или его стереоизомер,
где
G выбран из группы, состоящей из CRbRc, C=O, NRd, S(O)m и кислорода;
каждый из Rb и Rc независимо выбран из группы, состоящей из водорода, алкила, алкокси, галогена, амино, нитро, гидрокси, циано, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6, -S(O)mR6 и -NR7R8;
Rd выбран из группы, состоящей из водорода, алкила, циклоалкила, галогеналкила, гидроксиалкила, гетероциклила, арила, гетероарила, -C(O)R6, -C(O)OR6 и -S(O)mR6; и
R1-R4, R6-R8, n, m и q являются такими, как определено в п. 1.
Еще более предпочтительно ингибитор EZH2 представляет собой соединение формулы (IB)
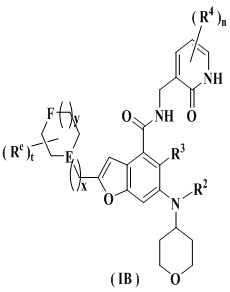
или его фармацевтически приемлемую соль,
где
E представляет собой CH или азот;
F выбран из группы, состоящей из CRbRc, C=O, NRd и кислорода;
каждый из Rb и Rc независимо выбран из группы, состоящей из водорода, алкила, алкокси, галогена, амино, нитро, гидрокси, циано, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6, -S(O)mR6 и -NR7R8;
Rd выбран из группы, состоящей из водорода, алкила, циклоалкила, галогеналкила, гидроксиалкила, гетероциклила, арила, гетероарила, -C(O)R6, -C(O)OR6 и -S(O)mR6;
каждый из Re является одинаковым или разным, и каждый независимо выбран из группы, состоящей из водорода, алкила, галогеналкила, галогена, амино, нитро, циано, гидрокси, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
t равно 0, 1, 2, 3, 4 или 5;
x равно 0, 1, 2 или 3;
y равно 0, 1, 2 или 3; и
R2-R4, R6-R8, m и n являются такими, как определено в п. 1.
Еще более предпочтительно ингибитор EZH2 представляет собой соединение формулы (IC)
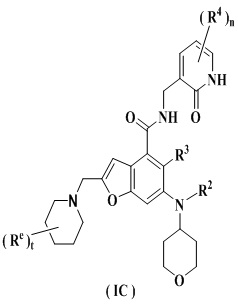
или его фармацевтически приемлемую соль,
где
каждый из Re является одинаковым или разным, и каждый независимо выбран из группы, состоящей из водорода, алкила и галогена;
t равно 0, 1, 2, 3, 4 или 5; и
R2-R4 и n являются такими, как определено в п. 1.
Еще более предпочтительно ингибитор EZH2 представляет собой соединение формулы (ID)
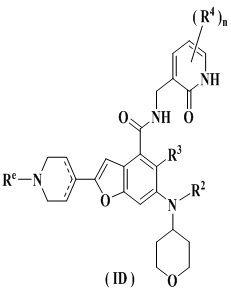
или его фармацевтически приемлемую соль,
где
Re выбран из группы, состоящей из водорода, алкила, галогеналкила, галогена, амино, нитро, циано, гидрокси, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила; и
R2-R4 и n являются такими, как определено в п. 1.
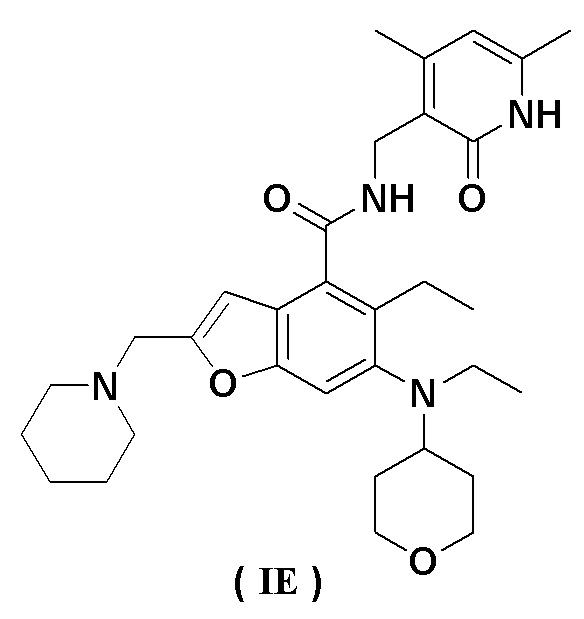
Еще более предпочтительно ингибитор EZH2 представляет собой соединение формулы (IE)
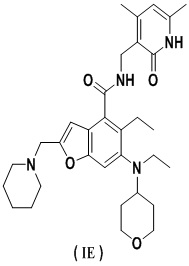
или его фармацевтически приемлемую соль.
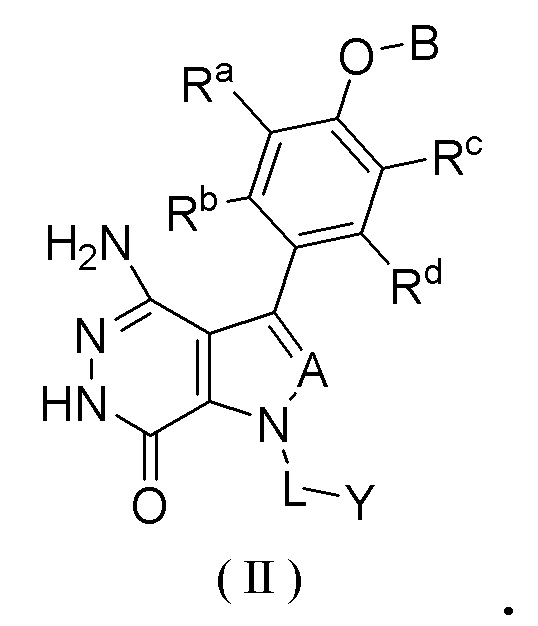
В описанных выше воплощениях изобретения ингибитор BTK представляет собой соединение формулы (II)
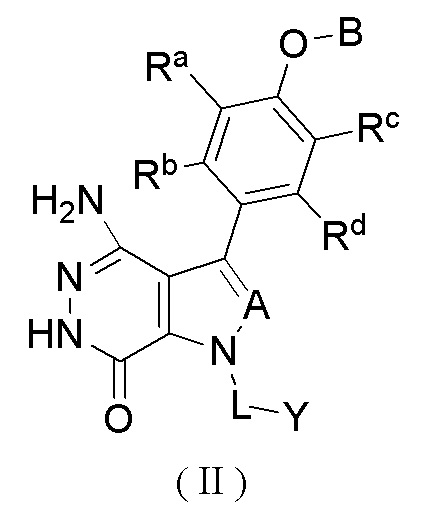
или его фармацевтически приемлемую соль, или его стереоизомер,
где
A выбран из группы, состоящей из CR1 и N;
CR1 выбран из группы, состоящей из водорода, галогена и необязательно замещенного алкила, где заместитель выбран из группы, состоящей из галогена, гидрокси, циано, нитро, карбокси, амино, алкила, алкокси и галогеналкила;
каждый из Ra, Rb, Rc и Rd независимо выбран из группы, состоящей из водорода, галогена, гидрокси, циано, нитро, необязательно замещенного алкила и необязательно замещенного алкокси, где заместитель выбран из группы, состоящей из галогена, гидрокси, циано, нитро, карбокси, амино, алкила, алкокси и галогеналкила;
B выбран из группы, состоящей из водорода, необязательно замещенного циклоалкила, необязательно замещенного гетероциклила, необязательно замещенного арила и необязательно замещенного гетероарила, где заместитель выбран из группы, состоящей из галогена, гидрокси, циано, нитро, карбокси, амино, алкила, алкокси и галогеналкила;
L выбран из группы, состоящей из связи и необязательно замещенного алкила; и
Y выбран из группы, состоящей из необязательно замещенного циклоалкила, необязательно замещенного гетероциклила, необязательно замещенного арила и необязательно замещенного гетероарила, где заместитель выбран из группы, состоящей из галогена, гидрокси, циано, нитро, карбокси, амино, алкила, алкилкарбонила, алкинилкарбонила и галогеналкила;
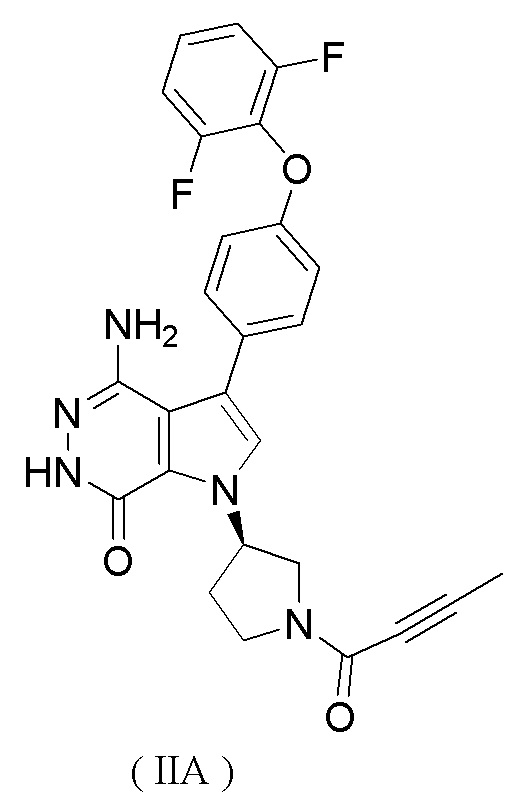
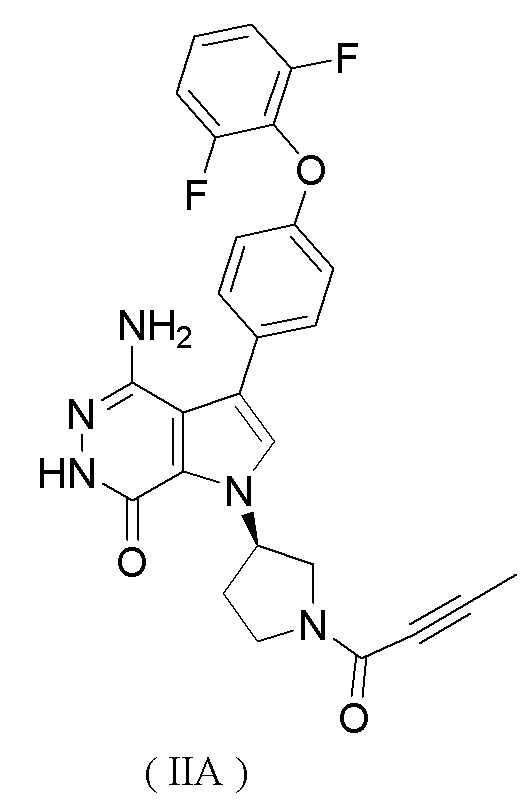
Предпочтительно ингибитор BTK представляет собой соединение формулы (IIA)

или его фармацевтически приемлемую соль.
В одном воплощении настоящего изобретения фармацевтически приемлемая соль выбрана из группы, состоящей из фосфата, гидрохлорида, метансульфоната, малеата, малата, пара-толуолсульфоната и безилата.
В другом воплощении настоящего изобретения ингибитор BTK выбран из группы, состоящей из ибрутиниба, акалабрутиниба, MSC-2364447, спебрутиниба, HM-71224, плевитрекседа, GS-4059, GDC-0853, SNS-062, CGP-53716, идоксифена, BTG-511, баноксантрона, глюкарпидазы, поликлонального антитела к дигоксину, поливалентного иммунного Fab ямкоголовых (овечий, BTG) и отеликсизумаба.
В описанных выше воплощениях изобретения комбинация необязательно содержит третий компонент, выбранный из группы, состоящей из ингибитора HDAC, ингибитора CDK4/6, ингибитора ALK, ингибитора JAK2, ингибитора Bcl-2, ингибитора Hsp90, глюкокортикоида, винка-алкалоида, антиметаболита, повреждающего ДНК агента, леналидомида, ритуксимаба, PKC пертурбагена, ингибитора Lyn/Fyn, ингибитора Syk, ингибитора PI3K, ингибитора PKCβ, ингибитора IKK, 20s протеасомы, IRF-4, антитела к IRAK4, антитела к CXCR4, антитела к CXCR5, антитела к GLS, антитела к PLK, антитела к CD20, ингибитора Topo II, ингибитора ДНК метилтрансферазы, ингибитора Ras/MAPK и ингибитора рецептора 1 фактора роста фибробластов (FGFR1); где ингибитор HDAC предпочтительно выбран из группы, состоящей из панобиностата лактата, белиностата, хиндамида, ромидепсина, вориностата, бексаностата и энтиностата; ингибитор CDK4/6 предпочтительно выбран из группы, состоящей из палбоциклиба, блинатумомаба, тиагабина гидрохлорида и итолизумаба; ингибитор Bcl-2 предпочтительно выбран из группы, состоящей из венетоклакса, облимерсена натрия, ABT-737 и HA14-1; ингибитор Hsp90 предпочтительно выбран из группы, состоящей из себелипазы альфа и ретаспимицина гидрохлорида; ингибитор JAK2 предпочтительно выбран из группы, состоящей из тофацитиниба цитрата, руксолитиниба фосфата, лестауртиниба, момелотиниба дигидрохлорида, пефицитиниба и филготиниба; PKC пертурбаген предпочтительно выбран из группы, состоящей из тепренона, препарата TruHeal, HO/03/03, сотрастаурина, энзастаурина и GF109203X; ингибитор ALK предпочтительно выбран из группы, состоящей из алектиниба гидрохлорида, церитиниба, кризотиниба, бендамустина, кармустина, лумостина, хлорметина гидрохлорида и NVP-TAE684; ингибитор PI3K предпочтительно выбран из группы, состоящей из GS-1101, IPI-145, BKM120, BEZ235, GDC-0941, Amg319, CAL-101 и A66; ингибитор IKK предпочтительно выбран из группы, состоящей из ауранофина, BAY 86-9766 и RDEA-119.
В описанных выше воплощениях изобретения комбинация обладает синергическим действием.
В настоящем изобретении предложен способ лечения опухоли, включающий введение пациенту описанных выше ингибитора EZH2 и ингибитора BTK.
В соответствии с применением по настоящему изобретению опухоль представляет собой лимфому, предпочтительно неходжкинскую лимфому и более предпочтительно B-клеточное пролиферативное заболевание; где B-клеточное пролиферативное заболевание выбрано из группы, состоящей из диффузной B-крупноклеточной лимфомы (ДВКЛ), хронического лимфолейкоза (ХЛЛ), лимфомы из малых лимфоцитов (МЛЛ), МЛЛ с высоким риском ХЛЛ или без риска ХЛЛ, фолликулярной лимфомы (ФЛ), мантийноклеточной лимфомы (МКЛ), опухоли из предшественников B-клеток, лимфобластного лейкоза (или лимфомы) из предшественников B-клеток, опухоли из зрелых (периферических) B-клеток, лимфоплазмацитарной лимфомы (или иммунобластомы), слизисто-ассоциированной внеузловой лимфомы, волосатоклеточного лейкоза, плазмацитомы (или миеломы из плазматических клеток), макроглобулинемии Вальденстрема, множественной миеломы, лимфомы из клеток маргинальной зоны, лимфомы Беркитта (ЛБ), неберкитовской B-клеточной лимфомы высокой стадии или внеузловой B-клеточной лимфомы из клеток маргинальной зоны, острого или хронического миелогенного (или миелоидного) лейкоза, миелодиспластического синдрома и острого лимфобластного лейкоза.
В настоящем изобретении предложена комбинация описанных выше ингибитора EZH2 и ингибитора BTK для применения в качестве лекарственного средства для лечения опухоли.
В соответствии с применением по настоящему изобретению отношение ингибитора EZH2 к ингибитору BTK составляет 0,001-1000, предпочтительно 0,01-100, еще более предпочтительно 0,1-10 и более предпочтительно 1:1, 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, 1:10, 1:11, 1:12, 1:13, 1:14, 1:15, 1:16, 1:17, 1:18, 1:19, 1:20, 1:21, 1:22, 1:23, 1:24, 1:25, 1:26, 2:1, 2:3, 2:5, 2:7, 2:9, 2:11, 2:13, 2:15, 2:17, 2:19, 2:21, 3:1, 3:2, 3:4, 3:5, 3:7, 3:8, 3:10, 3:11, 3:13, 3:14, 3:16, 3:17, 3:19, 3:20, 4:1, 4:3, 4:5, 4:7, 4:9, 4:11, 4:13, 4:15, 4:17, 4:19, 4:21, 5:1, 5:2, 5:3, 5:4, 5:6, 5:7, 5:8, 5:9, 5:11, 5:12, 5:13, 5:14, 5:16, 5:17, 5:18, 5:19, 5:21, 6:1, 6:5, 6:7, 6:11, 6:13, 6:17, 6:19, 7:1, 7:2, 7:3, 7:5, 7:6, 7:8, 7:9, 7:10, 7:11, 7:12, 7:13, 7:15, 7:16, 7:17, 7:18, 7:19, 7:20, 8:1, 8:3, 8:5, 8:7, 8:9, 8:11, 8:13, 8:15, 8:17, 8:19, 9:1, 9:2, 9:4, 9:5, 9:7, 9:8, 9:10, 9:11, 9:13, 9:14, 9:16, 9:17, 9:19, 9:20, 10:1, 10:3, 10:7, 10:9, 10:11, 10:13, 10:17 или 10:19.
В соответствии с применением по настоящему изобретению содержание ингибитора EZH2 составляет 0,1-5000 мг и предпочтительно 1-2000 мг.
В соответствии с применением по настоящему изобретению содержание ингибитора BTK составляет 0,1-2000 мг и предпочтительно 1-1000 мг.
В настоящем изобретении вводимая доза ингибитора EZH2 составляет 0,1-5000 мг и предпочтительно 10 мг, 50 мг, 100 мг, 150 мг, 200 мг, 250 мг, 300 мг, 350 мг, 400 мг, 400 мг, 500 мг, 550 мг, 600 мг, 650 мг, 700 мг, 750 мг, 800 мг, 8500 мг, 900 мг, 950 мг, 1000 мг, 1200 мг, 1250 мг, 1300 мг, 1400 мг, 1500 мг, 1600 мг, 1700 мг, 1800 мг, 1900 мг, 2000 мг, 2100 мг, 2200 мг, 2300 мг, 2400 мг, 2500 мг, 2600 мг, 2700 мг, 2800 мг, 2900 мг, 3000 мг, 3500 мг, 4000 мг, 4500 мг или 5000 мг; вводимая доза ингибитора BTK составляет 0,1-2000 мг, и предпочтительно 10 мг, 20 мг, 30 мг, 50 мг, 80 мг, 90 мг, 100 мг, 150 мг, 160 мг, 200 мг, 250 мг, 300 мг, 350 мг, 500 мг, 650 мг, 700 мг, 750 мг, 800 мг, 850 мг, 900 мг, 950 мг, 1000 мг, 1200 мг, 1300 мг, 1400 мг, 1500 мг, 1600 мг, 1800 мг, 1900 мг или 2000 мг.
Способ введения комбинации по настоящему изобретению выбран из группы, состоящей из одновременного введения, совместного введения после приготовления отдельной лекарственной формы и последовательного введения после приготовления отдельной лекарственной формы.
Настоящее изобретение дополнительно относится к применению комбинации ингибитора EZH2 и ингибитора BTK в получении лекарственного средства для лечения опухоли, где рекомендуемая периодичность введения ингибитора EZH2 составляет один раз в сутки или два раза в сутки, а рекомендуемая периодичность введения ингибитора BTK составляет один раз в сутки.
Важным является то, что комбинация ингибитора EZH2 и ингибитора BTK по настоящему изобретению обладает синергическим действием.
Настоящее изобретение также относится к фармацевтической композиции ингибитора EZH2 и ингибитора BTK, содержащей один или более фармацевтически приемлемых носителей, эксципиентов и/или разбавителей. Фармацевтическая композиция может быть включена в любую из фармацевтически приемлемых дозированных форм. Например, фармацевтический состав ингибитора EZH2 и ингибитора BTK можно готовить в виде таблетки, капсулы, пилюли, гранулы, раствора, суспензии, сиропа, инъекционной формы (включая раствор для инъекций, стерильный порошок для инъекций и концентрированный раствор для инъекций), суппозитория, ингаляционного препарата или спрея.
Кроме того, фармацевтическую композицию по настоящему изобретению можно также вводить нуждающемуся в таком лечении пациенту или субъекту любым подходящим способом введения, например пероральным, парентеральным, ректальным, внутрилегочным или местным способом введения. Для перорального введения фармацевтическую композицию можно готовить в лекарственной форме для перорального введения, например в твердой лекарственной форме для приема внутрь, такой как таблетка, капсула, пилюля, гранула и т.п.; или в пероральной жидкой лекарственной форме, такой как пероральный раствор, пероральная суспензия, сироп и т.п. При включении в пероральную лекарственную форму фармацевтическая композиции может дополнительно содержать подходящий наполнитель, связующее вещество, разрыхлитель, смазывающее вещество и т.п.
Фармацевтическую композицию ингибитора EZH2 и ингибитора BTK по настоящему изобретению можно вводить отдельно или в комбинации с одним или более терапевтических агентов. Соответственно, в некоторых предпочтительных воплощениях изобретения фармацевтическая композиция дополнительно содержит один или более терапевтических агентов.
Объединяемые компоненты (например, ингибитор EZH2, ингибитор BTK и второй терапевтический агент) можно вводить одновременно или последовательно по отдельности. Например, второй терапевтический агент можно вводить до, во время или после совместного введения ингибитора EZH2 и ингибитора BTK по настоящему изобретению. Кроме того, объединяемые компоненты можно также вводить совместно в одной и той же лекарственной форме или в отдельных и различных лекарственных формах.
В настоящем изобретении термин "комбинированное введение" или "совместное введение" относится к способу введения, включающему различные ситуации, в которых два лекарственных средства вводят последовательно или одновременно. Термин "одновременно" в настоящем документе означает, что ингибитор EZH2 и ингибитор BTK вводят в ходе одного и того же цикла введения, например, два лекарственных средства вводят в течение двух дней или одного дня. Термин "последовательное или поочередное" введение включает ситуации, в которых ингибитор EZH2 и ингибитор BTK вводят соответственно в разных циклах введения. Все эти способы введения относятся к комбинированному введению по настоящему изобретению.
Термин "эффективное количество" по настоящему изобретению охватывает достаточное количество для ослабления или предотвращения симптома или признака медицинского состояния. Термин "эффективное количество" также относится к достаточному количеству, чтобы обеспечить диагностику или способствовать ей. Эффективное количество для конкретного пациента в медицине или ветеринарии может изменяться в зависимости от таких факторов, как подлежащее лечению состояние, общее состояние здоровья пациента, пути введения и вводимой дозы и степени тяжести побочных эффектов. Эффективное количество может представлять собой максимальную дозу или режим введения, позволяющий избежать значимых побочных или токсических эффектов.
ОПРЕДЕЛЕНИЯ
В описании и в формуле настоящей заявки, если не указано иное, используемые в настоящем документе научные и технические термины имеют значения, обычно понятные специалисту в данной области техники. Однако для лучшего понимания настоящего изобретения приведены определения и пояснения некоторых относящихся к изобретению терминов. Кроме того, если определения и пояснения терминов, приведенные в настоящей заявке, не совпадают с их значениями, обычно понимаемыми специалистами в данной области техники, преимущество имеют определения и пояснения терминов, приведенные в настоящей заявке.
Используемый в настоящем изобретении термин "галоген" или "атом галогена" относится к атому фтора, атому хлора, атому брома или атому йода.
Используемый в настоящем изобретении термин "циано" относится к группе -CN.
Используемый в настоящем изобретении термин "гидрокси" относится к группе -OH.
Используемый в настоящем изобретении термин "амино" относится к группе -NH.
Используемый в настоящем изобретении термин "карбокси" относится к группе -COOH.
Используемый в настоящем изобретении термин "карбонил" относится к группе -CO-.
Используемый в настоящем изобретении термин "нитро" относится к группе -NO2.
Используемый в настоящем изобретении термин "алкил" относится к нормальному или разветвленному алкилу, имеющему от 1 до 20 атомов углерода, включая, например, "C1-6 алкил", "C1-4 алкил" и т.п. Конкретные примеры алкила включают без ограничений: метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, 2-метилбутил, нео-пентил, 1-этилпропил, н-гексил, изогексил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 2-этилбутил, 1,2-диметилпропил и т.п.
Используемый в настоящем изобретении термин "алкинил" относится к нормальному или разветвленному алкинилу, имеющему от 2 до 20 атомов углерода и по меньшей мере одну углерод-углеродную тройную связь, включая, например, "C2-6 алкинил", "C2-4 алкинил" и т.п. Примеры алкинила включают без ограничений: этинил, пропинил, 2-бутинил, 2-пентинил, 3-пентинил, 4-метил-2-пентинил, 2-гексинил, 3-гексинил, 5-метил-2-гексинил и т.п.
Используемый в настоящем изобретении термин "циклоалкил" относится к насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей от 3 до 14 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно от 3 до 8 атомов углерода, наиболее предпочтительно от 5 до 6 атомов углерода, и наиболее предпочтительно циклоалкил представляет собой циклопропил. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т.п., и предпочтительно циклопропил или циклогексенил. Полициклический циклоалкил включает циклоалкил, имеющий спиро-кольцо, конденсированное кольцо или кольцо с внутренним мостиком.
Используемый в настоящем изобретении термин "конденсированный циклоалкил" относится к циклической структуре, имеющей от 4 до 15 атомов углерода, образованной из двух или более циклических структур, присоединенных друг к другу посредством двух смежных атомов. Конденсированный циклоалкил включает, например, "6-11-членный конденсированный циклоалкил", "5-9-членный конденсированный циклоалкил", "7-10-членный конденсированный циклоалкил", "9-10-членный конденсированный циклоалкил" и т.п. Атомы углерода циклической структуры могут быть необязательно окисленными. Примеры конденсированного циклоалкила включают без ограничений:  ,
,  ,
,  ,
, 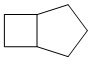 ,
, 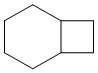 ,
, 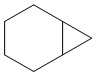 ,
, 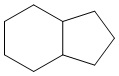 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  и т.п.
и т.п.
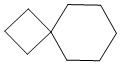
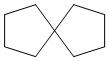
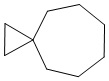
Используемый в настоящем изобретении термин "спиро-циклоалкил" относится к циклической структуре, имеющей от 5 до 15 кольцевых атомов углерода, образованной из двух или более циклических структур, присоединенных друг к другу посредством одного атома углерода. Атомы углерода циклической структуры могут быть необязательно окисленными. Спиро-циклоалкил включает, например, "6-11-членный спиро-циклоалкил", "5-10-членный спиро-циклоалкил", "7-8-членный спиро-циклоалкил", "9-10-членный спиро-циклоалкил" и т.п. Конкретные примеры спиро-циклоалкила включают без ограничений:  ,
,  ,
, 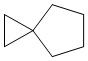 ,
, 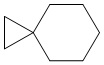 ,
, 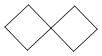 ,
, 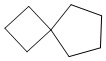 ,
,  ,
,  ,
,  ,
,  ,
, 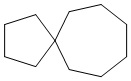 ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 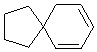 ,
, 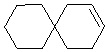 ,
, 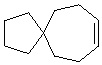 ,
, 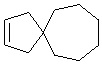 и т. п.
и т. п.
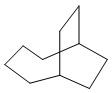
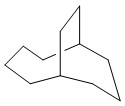
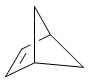
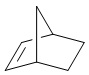
Используемый в настоящем изобретении термин "мостиковый циклоалкил" относится к циклической структуре, имеющей от 5 до 15 кольцевых атомов углерода, образованной из двух или более циклических структур, присоединенных друг к другу посредством двух несмежных атомов углерода. Атомы углерода циклической структуры могут быть необязательно окисленными. Мостиковый циклоалкил включает, например, "мостиковый 6-11-членный циклоалкил", "мостиковый 7-10-членный циклоалкил", "мостиковый 9-10-членный циклоалкил" и т.п. Конкретные примеры мостикового циклоалкила включают без ограничений:  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  и т.п.
и т.п.
Используемый в настоящем изобретении термин "гетероциклил" относится к 3-14-членной насыщенной или частично ненасыщщенной моноциклической или полициклической углеводородной группе, где один или более кольцевых гетероатомов представляют собой гетероатомы, выбранные из группы, состоящей из N, O и S(O)m (где m представляет собой целое число от 0 до 2), но за исключением -O-O-, -O-S- или -S-S- в кольце, при этом остальные кольцевые атомы представляет собой атомы углерода. Предпочтительно гетероциклил имеет от 3 до 12 кольцевых атомов, из которых от 1 до 4 представляют собой гетероатомы, более предпочтительно от 3 до 8 кольцевых атомов и более предпочтительно от 5 до 6 кольцевых атомов. Неограничивающие примеры моноциклического гетероциклила включают пирролидинил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил, пиранил, тетрагидрофуранил и т.п. Полициклический гетероциклил включает гетероциклил, имеющий спирокольцо, конденсированное кольцо или кольцо с внутренним мостиком.
Используемый в настоящем изобретении термин "конденсированный гетероциклил" относится к циклической структуре, имеющей от 4 до 15 кольцевых атомов (из которых по меньшей мере один кольцевой атом представляет собой гетероатом, например атом азота, атом кислорода или атом серы), образованной из двух или более циклических структур, присоединенных друг к другу посредством двух смежных атомов. Кольцевые атомы (например, атомы углерода, атомы азота или атомы серы) циклической структуры необязательно могут быть окисленными. Конденсированный гетероциклил включает, например, "4-12-членный конденсированный гетероциклил", "5-9-членный конденсированный гетероциклил", "6-11-членный конденсированный гетероциклил", "7-9-членный конденсированный гетероциклил", "9-10-членный конденсированный гетероциклил" и т.п. Конкретные примеры конденсированного гетероциклила включают без ограничений: пирролидиноциклопропил, циклопентаноазациклопропил, пирролидиноциклобутил, пирролидинопирролидинил, пирролидинопиперидил, пирролидинопиперазинил, пирролидиноморфолинил, пиперидиноморфолинил, бензопирролидинил, тетрагидроимидазо[4,5-c]пиридил, 3,4-дигидрохиназолинил, 1,2-дигидрохиноксалинил, бензо[d][1,3]диоксациклопентенил, 1,3-дигидроизобензофурил, 2H-хроменил, 2-оксо-2H-хроменил, 4H-хроменил, 4-оксо-4H-хроменил, хроманил, 4H-1,3-бензоксазинил, 4,6-дигидро-1H-фуро[3,4-d]имидазолил, 3a,4,6,6a-тетрагидро-1H-фуро[3,4-d]имидазолил, 4,6-дигидро-1H-тиено[3,4-d]имидазолил, 4,6-дигидро-1H-пирроло[3,4-d]имидазолил, бензоимидазолидинил, октагидро-бензо[d]имидазолил, декагидрохинолинил, гексагидротиеноимидазолил, гексагидрофуроимидазолил, 4,5,6,7-тетрагидро-1H-бензо[d]имидазолил, октагидроциклопентено[c]пирролил, дигидроиндолил, дигидроизоиндолил, бензоксазолидинил, бензотиазолидинил, 1,2,3,4-тетрагидроизохинолил, 1,2,3,4-тетрагидрохинолил, 4H-1,3-бензооксазинил и т.п.
Используемый в настоящем изобретении термин "спиро-гетероциклил" относится к циклической структуре, имеющей от 5 до 15 кольцевых атомов (из которых по меньшей мере один кольцевой атом представляет собой гетероатом, например атом азота, атом кислорода или атом серы), образованной из двух или более циклических структур, присоединенных друг к другу посредством одного кольцевого атома. Кольцевые атомы (например, атомы углерода, атомы азота или атомы серы) циклической структуры необязательно могут быть окисленными. Спиро-гетероциклил включает, например, "5-11-членный спиро-гетероциклил", "6-11-членный спиро-гетероциклил", "6-9-членный спиро-гетероциклил", "9-10-членный спиро-гетероциклил" и т.п. Конкретные примеры спиро-гетероциклила включают без ограничений:  ,
,  ,
, 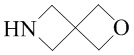 ,
, 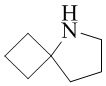 ,
, 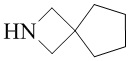 ,
, 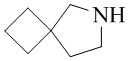 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  и т. п.
и т. п.
Используемый в настоящем изобретении термин "мостиковый гетероциклил" относится к циклической структуре, имеющей от 5 до 15 кольцевых атомов (из которых по меньшей мере один кольцевой атом представляет собой гетероатом, например атом азота, атом кислорода или атом серы), образованной из двух или более циклических структур, присоединенных друг к другу посредством двух несмежных кольцевых атомов. Кольцевые атомы (например, атомы углерода, атомы азота или атомы серы) циклической структуры могут быть необязательно окисленными. Мостиковый гетероциклил включает, например, "мостиковый 5-10-членный гетероциклил", "мостиковый 6-11-членный гетероциклил", "мостиковый 6-9-членный гетероциклил", "мостиковый 7-9-членный гетероциклил" и т.п. Конкретные примеры мостикового гетероциклила включают без ограничений:  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 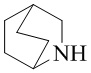 ,
, 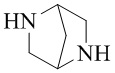 ,
, 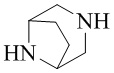 ,
, 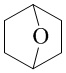 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 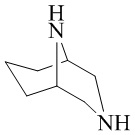 ,
,  и т.п.
и т.п.
Используемый в настоящем изобретении термин "галогеналкил" относится к производной группе "алкила", в которой один или более атомов водорода замещены одним или более "атомов галогена", а термины "атом галогена" и "алкил" являются такими, как определено выше.
Используемый в настоящем изобретении термин "гидроксиалкил" относится к производной группе "алкила", в которой один или более атомов водорода замещены одной или более групп "гидрокси", а термин "алкил" является таким, как определено выше.
Используемые в настоящем изобретении термины "алкокси, галогеналкокси, алкилкарбонил, алкоксикарбонил, алкилкарбониламино, алкиламинокарбонил, диалкиламинокарбонил, алкиламинокарбокси, галогеналкилкарбонил, циклоалкилалкил, циклоалкилкарбонил, гетероциклилкарбонил, алкиламино, алкиламиноалкил или диалкиламино" относятся к группе со связью алкил-O-, галогеналкил-O-, алкил-C(O)-, алкил-O-C(O)-, алкил-C(O)-NH-, алкил-NH-C(O)-, (алкил)2-NH-C(O)-, алкил-C(O)-O-, галогеналкил-C(O)-, циклоалкил-алкил-, циклоалкил-C(O)-, гетероциклил-C(O)-, алкил-NH-, алкил-NH-алкил- или (алкил)2-N-, где термины "алкил, галогеналкил, циклоалкил и гетероциклил" являются такими, как определено выше.
Используемый в настоящем изобретении термин "арил" относится к 6-14-членному полностью углеродному моноциклическому кольцу или полициклическому конденсированному кольцу (т.е. каждое кольцо в системе имеет общую пару смежных атомов углерода с другим кольцом в системе), имеющему конъюгированную π-электронную систему, предпочтительно к 6-8-членному арилу, более предпочтительно к фенилу, антрилу и фенантрилу и наиболее предпочтительно к фенилу. Кольцо арила может быть конденсировано с кольцом гетероарила, гетероциклила или циклоалкила, где кольцом, связанным с исходной структурой, является арильное кольцо. Его неограничивающие примеры включают:
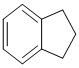 ,
, 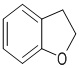 ,
, 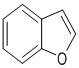 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  и
и 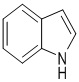 .
.
Используемый в настоящем изобретении термин "гетероарил" относится к 5-15-членному полностью углеродному моноциклическому кольцу или к полициклической группе из конденсированных колец, имеющей конъюгированную π-электронную систему, которые дополнительно имеют от 1 до 4 гетероатомов, выбранных из группы, состоящей из O, S и N. Гетероарил предпочтительно представляет собой 5-8-членный гетероарил и более предпочтительно 5- или 6-членный гетероарил. Конкретные примеры гетероарила включают без ограничений фурил, тиенил, пирролил, тиазолил, изотиазолил, тиадиазолил, оксазолил, изоксазолил, оксадиазолил, имидазолил, пиразолил, 1,2,3-триазолил, 1,2,4-триазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, пиридил, 2-пиридонил, 4-пиридонил, пиримидил, пиридазинил, пиразинил, 1,2,3-триазинил, 1,3,5-триазинил, 1,2,4,5-тетразинил, азациклогептатриенил, 1,3-диазациклогептатриенил, азациклооктатетраенил и т.п. Кольцо гетероарила может быть конденсировано с кольцом арила, гетероциклила или циклоалкила, где кольцом, связанным с исходной структурой, является гетероарильное кольцо. Его неограничивающие примеры включают:
 ,
, 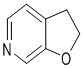 ,
, 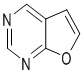 ,
,  ,
, 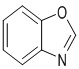 ,
,  ,
,  ,
,  и
и  .
.
Используемое в настоящем изобретении выражение "атомы углерода, атомы азота или атомы серы окислены" относится к образованию структуры C=O, N=O, S=O или SO2.
"Замещенный" относится к одному или более атомам водорода в группе, предпочтительно атомам водорода в количестве вплоть до 5, более предпочтительно от 1 до 3, независимо замещенным соответствующим количеством заместителей. Само собой разумеется, что заместители существуют только в возможном химическом положении. Специалист в данной области техники может, не прилагая чрезмерных усилий, экспериментальным или теоретическим путем определить, где возможно, а где невозможно замещение. Например, комбинация амино- или гидроксигрупп, имеющих свободные атомы водорода и углерода, с ненасыщенными связями (такими как олефинные связи) может быть неустойчивой.
Преимущества настоящего изобретения
По сравнению с предшествующим уровнем техники техническое решение настоящего изобретения обладает следующими преимуществами:
Комбинированное введение ингибитора EZH2 и ингибитора BTK по настоящему изобретению обладает значительным ингибирующим действием, а также значительным синергическим действием на пролиферацию клеток SU-DHL-4 и SU-DHL-6; комбинированное введение также обладает значительным ингибирующим действием, а также значительным синергическим действием на пролиферацию клеток B-клеточной лимфомы DOHH-2.
ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
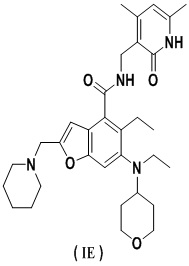
На Фиг. 1 показано ингибирующее действие комбинированного введения ингибитора EZH2 и ингибитора BTK по настоящему изобретению (молярное отношение соединения A к соединению B равно 1:2) и введения отдельного компонента (соединения B, соединения A) на пролиферацию клеток SU-DHL-4.
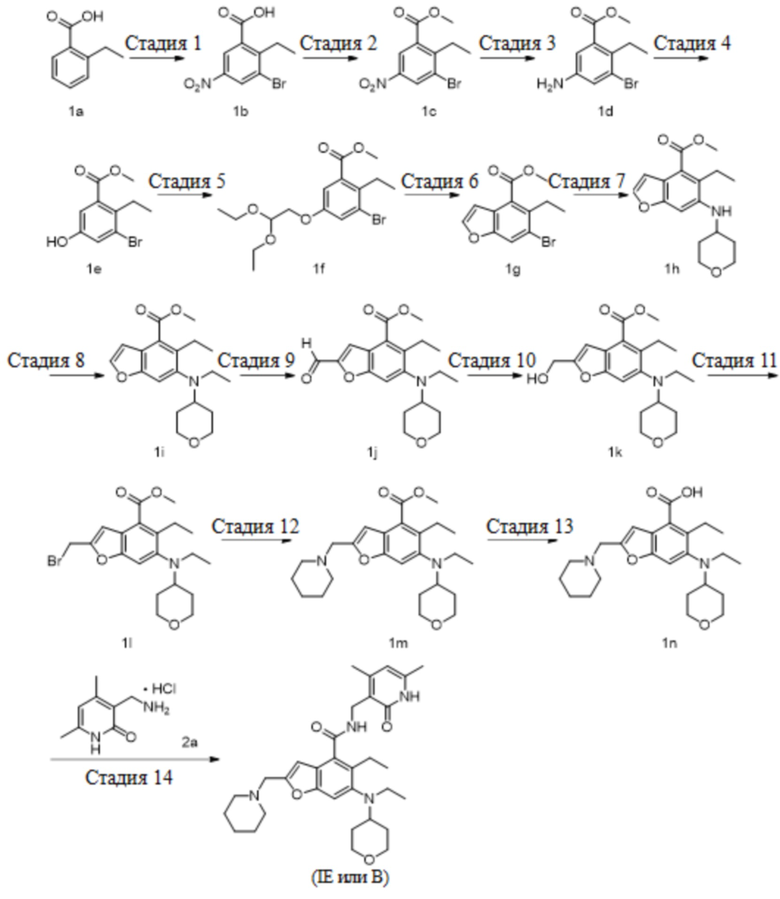
На Фиг. 2 показано ингибирующее действие комбинированного введения ингибитора EZH2 и ингибитора BTK по настоящему изобретению (молярное отношение соединения A к соединению B равно 1:4) и введения отдельного компонента (соединения B, соединения A) на пролиферацию клеток SU-DHL-6.
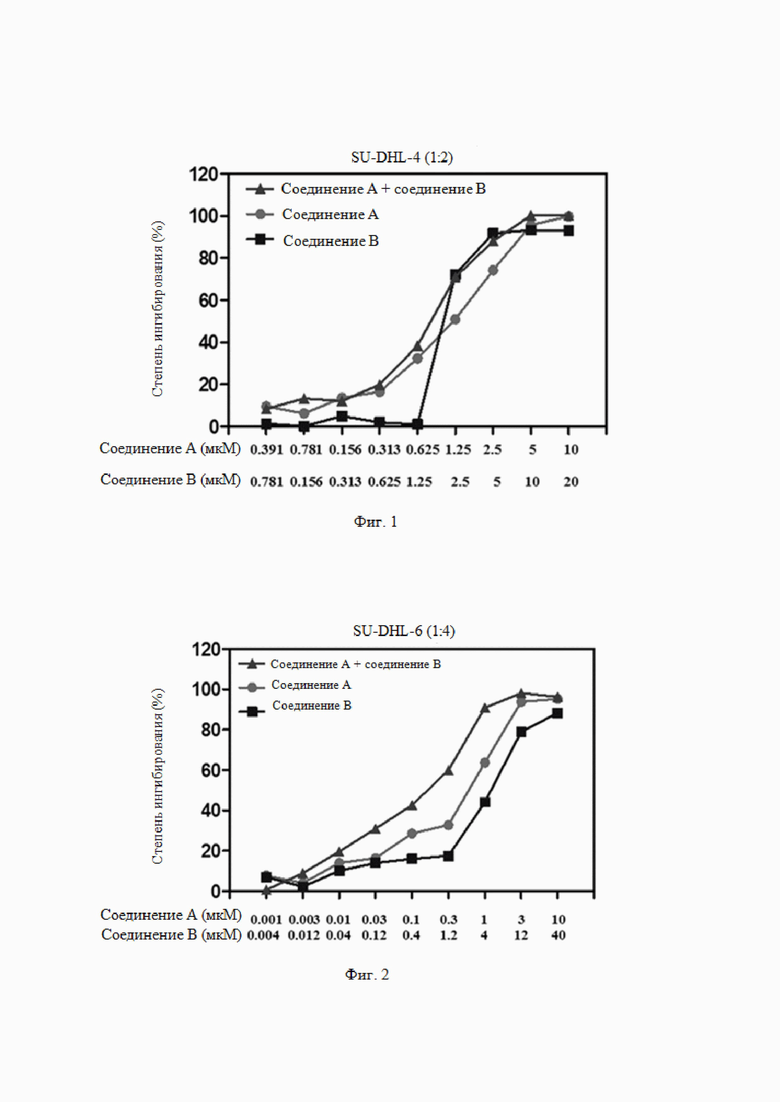
На Фиг. 3 показана эффективность комбинированного введения ингибитора EZH2 и ингибитора BTK по настоящему изобретению (комбинации соединения B и соединения A) и введения отдельного компонента (соединения B, соединения A) в модели подкожной трансплантации опухоли у бестимусных мышей, инокулированных клетками лимфомы DOHH-2.
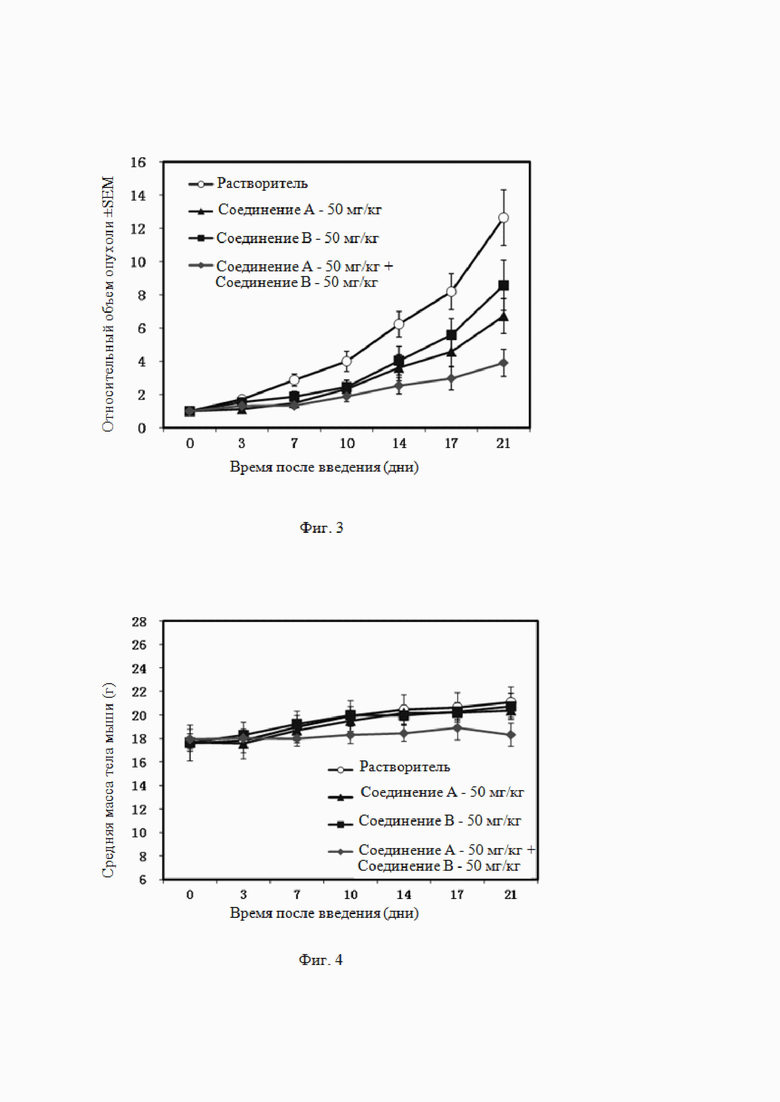
На Фиг. 4 показано действие комбинированного введения ингибитора EZH2 и ингибитора BTK по настоящему изобретению (комбинации соединения B и соединения A) и введения отдельного компонента (соединения B, соединения A) на массу тела бестимусных мышей, подкожно инокулированных клетками лимфомы DOHH-2.
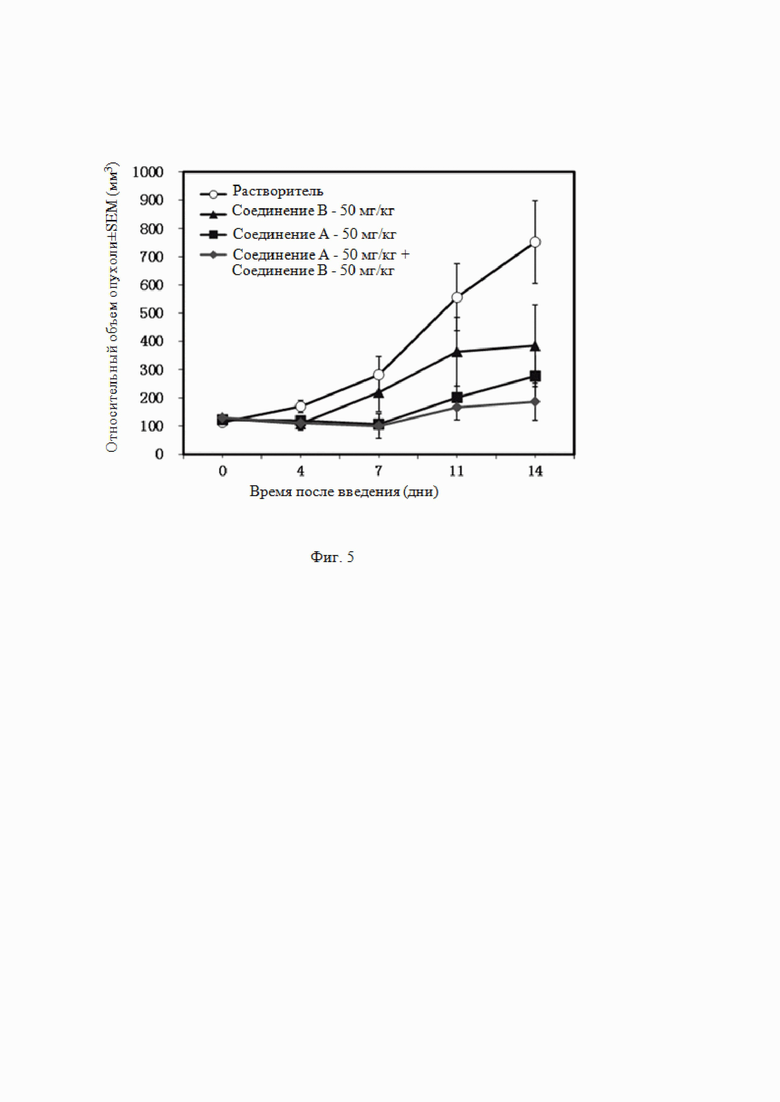
На Фиг. 5 показана эффективность комбинированного введения ингибитора EZH2 и ингибитора BTK по настоящему изобретению (комбинации соединения B и соединения A) и введения отдельного компонента (соединения B, соединения A) в модели подкожной трансплантации опухоли у бестимусных мышей, инокулированных клетками лимфомы SU-DHL-4.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Ниже приведены примеры экспериментальных решений по медицинскому применению композиции по настоящему изобретению в лечении диабета для демонстрации благоприятной активности и полезных технических эффектов композиции по настоящему изобретению. Однако должно быть понятно, что приведенные ниже экспериментальные решения представляют собой исключительно примеры настоящего изобретения и не предназначены для ограничения объема настоящего изобретения. На основании идей настоящего описания специалист в данной области техники может производить подходящие модификации или изменения технических решений настоящего изобретения без отклонения от сущности и объема настоящего изобретения.
Сравнительный пример 1. Получение N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксамида, представленного формулой (IE) (соединение B)
Стадия 1
3-Бром-2-этил-5-нитробензойная кислота
2-Этилбензойную кислоту 1a (20,0 г, 133 ммоль, получена способом, описанным в Journal of the American Chemical Society, 1991, 113 (13), 4931-6) добавляли к 150 мл серной кислоты, после чего добавляли порциями нитрат натрия (11,3 г, 133 ммоль) в ледяной бане. Реакционный раствор перемешивали в течение 3 часов, затем добавляли порциями N-бромсукцинимид (2,6 г, 14,5 ммоль). Реакционную систему перемешивали в течение 1 часа при 60°C. После завершения реакции реакционный раствор наливали в ледяную воду, тщательно перемешивали и фильтровали. Фильтрат промывали водой и концентрировали под пониженным давлением с получением указанного в заголовке неочищенного соединения 3-бром-2-этил-5-нитробензойной кислоты 1b (35 г) в виде белого твердого вещества, которое использовали непосредственно в следующей стадии без очистки.
Стадия 2
Метил 3-бром-2-этил-5-нитробензоат
Неочищенную 3-бром-2-этил-5-нитробензойную кислоту 1b (35 г, 128 ммоль) растворяли в 200 мл N,N-диметилформамида, затем добавляли йодметан (21,8 г, 153 ммоль) и карбонат калия (35,3 г, 255 ммоль). Реакционную систему перемешивали в течение 2 часов при комнатной температуре. После завершения реакции реакционный раствор концентрировали под пониженным давлением. К реакционному раствору добавляли избыток воды и экстрагировали этилацетатом. Органические фазы объединяли, промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением указанного в заголовке неочищенного продукта метил-3-бром-2-этил-5-нитробензоата 1c (36 г) в виде желтого масла, которое использовали непосредственно в следующей стадии без очистки.
Стадия 3
Метил-5-амино-3-бром-2-этилбензоат
Неочищенный метил-3-бром-2-этил-5-нитробензоат 1c (35,0 г, 121 ммоль) добавляли к смеси 250 мл этанола и 150 мл воды. Реакционный раствор нагревали до 70°C, добавляли хлорид аммония (52,8 г, 969 ммоль), затем добавляли порциями порошок железа (34 г, 606 ммоль). Реакционную систему перемешивали в течение 2 часов при 70°C. После завершения реакции реакционный раствор фильтровали через целит в горячем виде. Фильтрационный кек промывали горячим этанолом, затем фильтрат объединяли и концентрировали под пониженным давлением. Добавляли этилацетат и насыщенный раствор бикарбоната натрия. Две фазы разделяли, и водную фазу экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с н-гексаном и этилацетатом в качестве элюента с получением указанного в заголовке продукта метил-5-амино-3-бром-2-этилбензоата 1d (22,0 г, выход 70%) в виде желтого твердого вещества.
Стадия 4
Метил-3-бром-2-этил-5-гидроксибензоат
Метил-5-амино-3-бром-2-этилбензоат 1d (15,0 г, 58 ммоль) растворяли в 10 мл ацетонитрила, затем добавляли 200 мл 10% серной кислоты. Реакционный раствор тщательно перемешивали и охлаждали до 3°C в бане с ледяной солью, впоследствии добавляли по каплям 10 мл предварительно приготовленного раствора нитрита натрия (4,4 г, 64 ммоль). Реакционный раствор перемешивали в течение 4 часов при указанной выше температуре, добавляли по каплям 200 мл 50% серной кислоты, затем перемешивали в течение 1 часа при 90°C. После завершения реакции реакционный раствор экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с н-гексаном и этилацетатом в качестве элюента с получением указанного в заголовке продукта метил-3-бром-2-этил-5-гидроксибензоата 1e (5,5 г, выход 37%) в виде коричневого твердого вещества.
Стадия 5
Метил-3-бром-5-(2,2-диэтоксиэтокси)-2-этилбензоат
Метил-3-бром-2-этил-5-гидроксибензоат 1e (35 г, 135 ммоль) растворяли в 200 мл N,N-диметилформамида, затем добавляли 2-бром-1,1-диэтоксиэтан (40 г, 202 ммоль) и карбонат калия (37 г, 269 ммоль). Реакционную систему перемешивали при 120°C в течение 12 часов. После завершения реакции реакционный раствор концентрировали под пониженным давлением для удаления N,N-диметилформамида. К реакционному раствору добавляли воду и экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с н-гексаном и этилацетатом в качестве элюента с получением указанного в заголовке продукта метил-3-бром-5-(2,2-диэтоксиэтокси)-2-этилбензоата 1f (40 г, выход 80%) в виде светло-желтого масла.
Стадия 6
Метил-6-бром-5-этилбензофуран-4-карбоксилат
Полифосфорную кислоту (30 г) добавляли к 400 мл толуола. Реакционный раствор нагревали до 100°C, добавляли 50 мл предварительно приготовленного раствора метил-3-бром-5-(2,2-диэтоксиэтокси)-2-этилбензоата 1f (40 г, 107 ммоль) в толуоле при перемешивании. Реакционную систему перемешивали в течение 16 часов при 100°C. После завершения реакции надосадочную жидкость декантировали. К остатку добавляли воду и этилацетат. Две фазы разделяли, и водную фазу экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором карбоната натрия и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с н-гексаном и этилацетатом в качестве элюента с получением указанного в заголовке продукта метил-6-бром-5-этилбензофуран-4-карбоксилата 1g (11,8 г, выход 39%) в виде желтого твердого вещества.
Стадия 7
Метил-5-этил-6-((тетрагидро-2H-пиран-4-ил)амино)бензофуран-4-карбоксилат
Метил-6-бром-5-этилбензофуран-4-карбоксилат 1g (11,0 г, 39 ммоль), тетрагидро-2H-пиран-4-амин (5,89 г, 58 ммоль), трис(дибензилиденацетон)дипалладий (3,6 г, 3,9 ммоль), (0,9 ммоль) бис(дифенилфосфино)-1,1’-динафталин (4,86 г, 7,8 ммоль) и карбонат цезия (38 г, 117 ммоль) растворяли в 100 мл толуола. Реакционную систему перемешивали в течение 12 часов при 100°C. После завершения реакции реакционный раствор фильтровали через целит, и фильтрационный кек промывали этилацетатом. Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с н-гексаном и этилацетатом в качестве элюента с получением указанного в заголовке продукта метил-5-этил-6-((тетрагидро-2H-пиран-4-ил)амино)бензофуран-4-карбоксилата 1h (10,0 г, выход 85%) в виде желтого твердого вещества.
Стадия 8
Метил-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)бензофуран-4-карбоксилат
Метил-5-этил-6-((тетрагидро-2H-пиран-4-ил)амино)бензофуран-4-карбоксилат 1h (10,0 г, 0,033 ммоль) растворяли в 150 мл 1,2-дихлорэтана, затем добавляли ацетальдегид (7,2 г, 0,165 ммоль) и уксусную кислоту (9,9 г, 0,165 ммоль). Реакционный раствор перемешивали в течение 1 часа и добавляли триацетоксиборгидрид натрия (20,8 г, 0,1 ммоль). Реакционный раствор перемешивали в течение 12 часов при комнатной температуре. После завершения реакции реакционный раствор концентрировали под пониженным давлением, нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с н-гексаном и этилацетатом в качестве элюента с получением указанного в заголовке продукта метил-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)бензофуран-4-карбоксилата 1i (7,8 г, выход 71%) в виде белого твердого вещества.
МС m/z (ЖХ-МС): 332,4 [M+1]
Стадия 9
Метил-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-формилбензофуран-4-карбоксилат
Метил-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)бензофуран-4-карбоксилат 1i (1,6 г, 4,8 ммоль) растворяли в 25 мл тетрагидрофурана. Реакционный раствор охлаждали до -70°C и добавляли по каплям 2,0 M диизопропиламида лития (3,6 мл, 7,3 ммоль) в атмосфере аргона. Реакционный раствор перемешивали в течение 90 минут и добавляли N,N-диметилформамид (536 мг, 7,3 ммоль). Реакционный раствор перемешивали в течение 2 часов, затем медленно подогревали до комнатной температуры. К реакционному раствору добавляли избыток хлорида аммония, тщательно перемешивали и экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с н-гексаном и этилацетатом в качестве элюента с получением указанного в заголовке продукта метил-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-формилбензофуран-4-карбоксилата 1j (1,3 г, выход 75%) в виде желтого масла.
МС m/z (ионизация электрораспылением; ИЭР): 360,2 [M+1]
Стадия 10
Метил-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-(гидроксиметил)бензофуран-4-карбоксилат
Метил-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-формилбензофуран-4-карбоксилат 1j (1,4 г, 3,9 ммоль) растворяли в 5 мл тетрагидрофурана и 10 мл метанола, затем добавляли боргидрид натрия (222 мг, 5,8 ммоль). Реакционный раствор перемешивали в течение 30 минут при комнатной температуре. После завершения реакции реакционный раствор концентрировали под пониженным давлением, добавляли воду и насыщенный раствор бикарбоната натрия и экстрагировали три раза этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с н-гексаном и этилацетатом в качестве элюента с получением указанного в заголовке продукта метил-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-(гидроксиметил)бензофуран-4-карбоксилата 1k (1,4 г, выход 99%) в виде желтого масла.
Стадия 11
Метил-2-(бромметил)-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)бензофуран-4-карбоксилат
Метил-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-(гидроксиметил) бензофуран-4-карбоксилат 1k (1,0 г, 2,8 ммоль) растворяли в 30 мл тетрагидрофурана, затем добавляли по каплям трибромид фосфора (1,12 г, 4,2 ммоль). Реакционный раствор перемешивали в течение 12 часов при комнатной температуре. После завершения реакции реакционный раствор нейтрализовали насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением с получением указанного в заголовке неочищенного продукта метил-2-(бромметил)-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)бензофуран-4-карбоксилата 11 (1,15 г) в виде желтого масла, которое использовали непосредственно в следующей стадии без очистки.
Стадия 12
Метил-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-(пиперидин-1-илметил) бензофуран-4-карбоксилат
Неочищенный метил 2-(бромметил)-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)бензофуран-4-карбоксилат 1l (1,15 г, 2,7 ммоль) растворяли в 15 мл ацетонитрила, затем добавляли по каплям 10 мл предварительно приготовленного раствора пиперидина (362 мг, 4,3 ммоль) в ацетонитриле. Реакционный раствор перемешивали в течение 30 минут при комнатной температуре. После завершения реакции реакционный раствор концентрировали под пониженным давлением и добавляли этилацетат и насыщенный раствор бикарбоната натрия. Две фазы разделяли, и водную фазу экстрагировали этилацетатом. Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с дихлорметаном и метанолом в качестве элюента с получением указанного в заголовке продукта метил-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-(пиперидин-1-илметил) бензофуран-4-карбоксилата 1m (1,2 г, выход 99%) в виде желтого масла.
МС m/z (ЖХ-МС): 429,2 [M+1]
Стадия 13
5-Этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоновая кислота
Метил-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-(пиперидин-1-илметил) бензофуран-4-карбоксилат 1m (1,2 г, 2,7 ммоль) растворяли в смеси 5 мл тетрагидрофурана и 20 мл метанола, затем добавляли 5 мл 4 М раствора гидроксида натрия. Реакционный раствор перемешивали в течение 12 часов при 60°C. После завершения реакции добавляли концентрированную хлористоводородную кислоту для доведения pH реакционного раствора до 4. Смесь концентрировали под пониженным давлением, и остаток растворяли в смешанном растворителе из дихлорметана и метанола (об. : об. = 5 : 1) и фильтровали. Фильтрационный кек промывали смешанным растворителем из дихлорметана и метанола (об.:об.=5:1). Фильтрат концентрировали под пониженным давлением с получением указанного в заголовке неочищенного продукта 5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоновой кислоты 1n (1,1 г) в виде желтого твердого вещества, которое использовали непосредственно в следующей стадии без очистки.
МС m/z (ЖХ-МС): 415,2 [M+1]
Стадия 14
Получение соединения формулы (IE) (определенного как соединение B)
5-Этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-(пиперидин-1-илметил) бензофуран-4-карбоновую кислоту 1n (1,0 г, 2,4 ммоль) растворяли в 30 мл N,N-диметилформамида, затем добавляли 1-этил-3-(3-диметиламинопропил)карбодиимид (696 мг, 3,6 ммоль), 1-гидроксибензотриазол (490 мг, 3,6 ммоль) и N,N-диизопропилэтиламин (1,56 г, 12,1 ммоль). Реакционный раствор перемешивали в течение 1 часа, затем добавляли 3-(аминометил)-4,6-диметилпиперидин-2(1H)-она гидрохлорид 2a (593 мг, 3,0 ммоль, получен способом, описанным в заявке на патент WO 2014097041). Реакционный раствор перемешивали в течение 12 часов при комнатной температуре. После завершения реакции к реакционному раствору добавляли избыток воды и экстрагировали смещанным растворителем из дихлорметана и метанола (об. : об. = 8 : 1). Органические фазы объединяли, промывали водой и насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с дихлорметаном и метанолом в качестве элюента с получением указанного в заголовке продукта N-((4,6-диметил-2-оксо-1,2-дигидропиридин-3-ил)метил)-5-этил-6-(этил(тетрагидро-2H-пиран-4-ил)амино)-2-(пиперидин-1-илметил)бензофуран-4-карбоксамида 2 (750 мг, выход 57%) в виде белого твердого вещества.
МС m/z (ИЭР): 549,7 [M+1]
1H ЯМР (400 МГц, ДМСО-d6): δ 11.48 (s, 1H), 8.15 (t, 1H), 7.39 (s, 1H), 6.46 (s, 1H), 5.86 (s, 1H), 4.32 (d, 2H), 3.83 (d, 2H), 3.54 (s, 2H), 3.21 (t, 2H), 3.01-3.07 (m, 2H), 2.92-2.97 (m, 1H), 2.77-2.82 (m, 2H), 2.39 (brs, 4H), 2.23 (s, 3H), 2.11 (s, 3H), 1.64-1.67 (brd, 2H), 1.47-1.55 (m, 6H), 1.36-1.37 (brd, 2H), 1.02 (t, 3H), 0.82 (t, 3H).
Пример 1. Действие композиции по настоящему изобретению на пролиферацию клеток DOHH-2 in vitro
Исследуемые соединения: соединение формулы (IE) (определено как соединение B, полученное способом, описанным в WO 2017084494 (заявка на патент PCT/CN 2016/104318), см. сравнительный пример 1), и соединение формулы (IIA) (определено как соединение A, полученное способом, описанным в заявке на патент WO 2016007185 A1).
Линия клеток: клетки B-клеточной лимфомы человека DOHH-2 (приобретены в Немецкой коллекции микроорганизмов и клеточных культур (DSMZ)), выращенные in vitro в среде RPMI 1640, содержащей 10% фетальную телячью сыворотку (ФТС).
Приготовление раствора исследуемого соединения
Все исследуемые соединения готовили в виде исходного раствора 10 ммоль/л в диметилсульфоксиде (ДМСО) и при использовании готовили в желаемой концентрации со средой, не содержащей сыворотку.
Экспериментальная методика
Определенное количество клеток в логарифмической фазе роста высевали в 96-луночный культуральный планшет. Через 24 часа к клеткам добавляли исследуемые соединения в различных концентрациях (1-100000 нмоль/л) и инкубировали в течение 72 часов. В каждую лунку добавляли рабочий раствор 3-4,5-диметилтиазол-2-ил-2,5-дифенилтетразолия бромида (MTT). Через 4 часа клетки подвергали лизису с помощью трехкомпонентного раствора и измеряли значение оптической плотности (OD) при длине волны 570 нм с помощью ридера для микропланшетов.
Анализ данных
Ингибирование роста клеток рассчитывали по следующей формуле:
Степень ингибирования = (значение OD контрольной лунки - значение OD лунки с добавлением лекарственного средства) / значение OD контрольной лунки ×100%;
Половину эффективной концентрации IC50 рассчитывали, используя метод нелинейной регрессии, в соответствии со степенью ингибирования при каждой концентрации.
При комбинированном введении отношение концентраций соединения A и соединения B составляло 1:10. Показатель аддитивности (CI) рассчитывали с помощью программы Calcu-Syn, используя метод медианы эффекта для оценки взаимосвязи между двумя соединениями при комбинированном введении (CI<1 относится к синергическому эффекту, CI=1 относится к аддитивному эффекту, CI>1 относится к антагонистическому эффекту).
Экспериментальные результаты
Таблица 1. Действие отдельного соединения на пролиферацию клеток DOHH-2
Таблица 2. Ингибирующее действие комбинированного введения на пролиферацию клеток DOHH-2
Вывод из эксперимента
Из приведенных выше табличных данных видно, что комбинированное введение соединения A и соединения B обладает синергическим ингибирующим действием на пролиферацию клеток DOHH-2 in vitro.
Пример 2. Действие композиции по настоящему изобретению на пролиферацию клеток SU-DHL-4 и SU-DHL-6 in vitro
Исследуемые соединения: соединение формулы (IE) (определено как соединение B, полученное способом, описанным в WO 2017084494 (заявка на патент PCT/CN 2016/104318), см. сравнительный пример 1), и соединение формулы (IIA) (определено как соединение A, полученное способом, описанным в заявке на патент WO 2016007185 A1).
Линия клеток: клетки B-клеточной лимфомы человека SU-DHL-4 и SU-DHL-6 (приобретены в Американской коллекции типовых культур (ATCC)), выращенные in vitro в среде RPMI 1640, содержащей 10% фетальную телячью сыворотку (ФТС).
Приготовление раствора исследуемого соединения
Все исследуемые соединения готовили в виде исходного раствора 10 ммоль/л в ДМСО и при использовании готовили в желаемой концентрации со средой, не содержащей сыворотку.
Экспериментальная методика
Определенное количество клеток в логарифмической фазе роста высевали в 96-луночный культуральный планшет. Через 24 часа к клеткам добавляли исследуемые соединения в различных концентрациях (1-40000 нмоль/л) и инкубировали в течение 72 часов. В каждую лунку добавляли рабочий раствор MTT. Через 4 часа клетки подвергали лизису с помощью трехкомпонентного раствора (10% додецилсульфата натрия (SDS)), 5% изобутанола, 0,012 моль/л HCl) при 37°C и измеряли значение OD при длине волны 570 нм с помощью ридера для микропланшетов.
Анализ данных
Ингибирование роста клеток рассчитывали по следующей формуле:
Степень ингибирования = (значение OD контрольной лунки - значение OD лунки с добавлением лекарственного средства) / значение OD контрольной лунки ×100%;
Половину эффективной концентрации IC50 рассчитывали, используя метод нелинейной регрессии, в соответствии со степенью ингибирования при каждой концентрации.
При комбинированном введении отношение концентраций соединения A и соединения B составляло 1:2 (для SU-DHL-4) и 1:4 (для SU-DHL-6). Показатель аддитивности (CI) рассчитывали с помощью программы Calcu-Syn, используя метод медианы эффекта для оценки взаимосвязи между двумя соединениями при комбинированном введении (CI<1 относится к синергическому эффекту, CI=1 относится к аддитивному эффекту, CI>1 относится к антагонистическому эффекту).
Экспериментальные результаты
Таблица 3. Действие отдельного соединения на пролиферацию клеток SU-DHL-4 и SU-DHL-6
Таблица 4. Ингибирующее действие комбинированного введения на пролиферацию клеток SU-DHL-4 и SU-DHL-6
Вывод из эксперимента
Из приведенных выше табличных данных видно, что комбинированное введение соединения A и соединения B обладает синергическим ингибирующим действием на пролиферацию клеток SU-DHL-4 и SU-DHL-6 in vitro.
Пример 3: Эффективность композиции по настоящему изобретению при подкожной трансплантации опухоли бестимусным мышам, инокулированным клетками фолликулярной лимфомы человека DOHH-2
Исследуемые соединения: соединение формулы (IE) (определено как соединение B, полученное способом, описанным в WO 2017084494 (заявка на патент PCT/CN 2016/104318), см. сравнительный пример 1), и соединение формулы (IIA) (определено как соединение A, полученное способом, описанным в заявке на патент WO 2016007185 A1).
Экспериментальные животные: Бестимусных мышей линии BALB/cA-nude, возраст 5-6 недель, самок, приобретали у компании Shanghai Lingchang Biotechnology Co., Ltd., лицензия на использование лабораторных животных №: SCXK (Шанхай) 2013-0018 и сертификат животных №: 2013001818958, условия поставки: класс "свободны от специфических патогенных микроорганизмов" (SPF).
Приготовление раствора исследуемого соединения
Все исследуемые соединения готовили с 0,2% Твин 80 и 0,5% карбоксиметилцеллюлозы (КМЦ) и разводили до соответствующей концентрации.
Экспериментальная методика
(1) Бестимусным мышам подкожно инокулировали клетки лимфомы DOHH-2. Когда опухоли вырастали до 100-200 мм3, животных группировали случайным образом (день 0). Доза и режим введения представлены в таблице 5.
(2) Наблюдение и регистрация данных: объем опухоли измеряли 2-3 раза в неделю, мышей взвешивали и регистрировали данные.
(3) Измерение опухоли и конечная точка
Конечная точка, в основном, зависит от того, происходит ли задержка роста опухоли или излечение мыши. Объем опухоли (в мм3) измеряли два раза в неделю с помощью штангенциркуля в двух измерениях.
Объем опухоли (V) рассчитывают как:
V=0.5×a×b2, где a и b представляют собой длину и ширину соответственно;
T/C (%)=(T-T0)/(C-C0)×100, где T и C представляют собой объем опухоли в конце эксперимента; T0 и C0 представляют собой объем опухоли в начале эксперимента. Значение T/C (в процентах) является показателем противоопухолевой эффективности.
(4) Анализ данных: получали сводную статистику, включая среднее и стандартную ошибку среднего (SEM), статистический анализ различий объема опухоли между группами и анализ данных, полученных в результате взаимодействия между лекарственными средствами, который проводили в оптимальный момент времени обработки после последнего введения препарата (день 21 после группировки). Для сравнения объема опухоли и массы опухоли между группами проводили односторонний дисперсионный анализ. При отсутствии статистической значимости полученной F-статистики (p<0,001, дисперсия обработки по сравнению с дисперсией ошибки) сравнение между группами проводили с помощью критерия Геймса-Ховелла. Для анализа всех данных использовали программу SPSS17.0, и P<0,05 считали статистически значимой.
Экспериментальные результаты:
Таблица 5. Действие комбинированного введения на пролиферацию клеток DOHH-2
(мм3)
День 0
(мм3)
День 21
День 21
День 21
День 21
День 0: время первого введения; п/о: пероральное введение; 2 р/сут: два раза в сутки; величина P относится к сравнению с растворителем; *P<0,05, сравнение с комбинацией соединение A 50 мг/кг + соединение B 50 мг/кг, с помощью t-критерия Стьюдента; количество мышей в начале исследования, группа, получавшая растворитель, n=10, группа, получавшая препарат, n=6.
Вывод из эксперимента
Из данных, приведенных в таблице 5, видно, что соединение A (50 мг/кг, п/о, QD×21) ингибировало рост подкожно трансплантированной опухоли у бестимусных мышей, инокулированных клетками DOHH-2, и степень ингибирования роста опухоли составляла 49% (P<0,05 по сравнению с растворителем). Соединение B (50 мг/кг, п/о, QD×21) обладает определенным ингибирующим действием на клетки DOHH-2, и степень ингибирования роста опухоли составляла 35% (P>0,05 по сравнению с растворителем). При введении двух соединений в комбинации степень ингибирования роста опухоли увеличивалась до 75%, и эффективность была статистически значимо сильнее, чем соединения A или соединения B по отдельности (P<0,05 по сравнению с отдельным соединением, см. Фиг. 3). На Фиг. 4 показано, что комбинация соединения A и соединения B не вызывает статистически значимой потери массы и других симптомов.
Таким образом, комбинированное действие ингибитора BTK соединения A и ингибитора EZH2 соединения B по настоящему изобретению превышает действие отдельного соединения, и такая комбинация обладает синергическим действием.
Пример 4: Эффективность композиции по настоящему изобретению при подкожной трансплантации опухоли мышам, инокулированным клетками B-клеточной лимфомы человека SU-DHL-4
Исследуемые соединения: соединение формулы (IE) (определено как соединение B, полученное способом, описанным в WO 2017084494 (заявка на патент PCT/CN 2016/104318), см. сравнительный пример 1), и соединение формулы (IIA) (определено как соединение A, полученное способом, описанным в заявке на патент WO 2016007185 A1).
Экспериментальные животные: Мышей линии SCID.BG, возраст 5-6 недель, самок, приобретали у компании Shanghai Lingchang Biotechnology Co., Ltd., лицензия на использование лабораторных животных №: SCXK (Шанхай) 2013-0018 и сертификат животных №: 2013001820833, условия поставки: класс "свободны от специфических патогенных микроорганизмов" (SPF).
Приготовление раствора исследуемого соединения
Все исследуемые соединения готовили с 0,2% Твином 80 с добавлением 0,5% КМЦ и разводили до соответствующей концентрации.
Экспериментальная методика:
(1) Мышам инокулировали подкожно клетки SU-DHL-4 (клетки B-клеточной лимфомы SU-DHL-4 приобретали в ATCC). Когда опухоли вырастали до 100-150 мм3, животных группировали в соответствии с объемом опухоли (день 0). Доза и режим введения представлены в таблице 6.
(2) Наблюдение и регистрация данных: объем опухоли измеряли 2-3 раза в неделю, мышей взвешивали и регистрировали данные.
(3) Измерение опухоли и конечная точка
Конечная точка, в основном, зависит от того, происходит ли задержка роста опухоли или излечение мыши. Объем опухоли (в мм3) измеряли два раза в неделю с помощью штангенциркуля в двух измерениях.
Объем опухоли (V) рассчитывают как:
V=0.5×a×b2, где a и b представляют собой длину и ширину соответственно;
T/C (%)=(T−T0)/(C−C0)×100, где T и C представляют собой объем опухоли в конце эксперимента; T0 и C0 представляют собой объем опухоли в начале эксперимента. Значение T/C (в процентах) является показателем противоопухолевой эффективности.
Степень ингибирования опухоли (TGI)(%)=100-T/C(%);
В случае регрессии опухоли степень ингибирования опухоли (TGI)(%)=100-(T-T0)/T0×100
Если объем опухоли меньше первоначального объема, т.е. T<T0 или C<C0, это определяют как частичную регрессию (partial regression, PR); если опухоль исчезает полностью, это определяют как полную регрессию (complete regression, CR).
(4) Анализ данных: получали сводную статистику, включая среднее и стандартную ошибку среднего (SEM), статистический анализ различий объема опухоли между группами и анализ данных, полученных в результате взаимодействия между лекарственными средствами, который проводили в оптимальный момент времени обработки после последнего введения препарата (день 14 после группировки). Для сравнения объема опухоли и массы опухоли между группами проводили односторонний дисперсионный анализ. При получении статистически не значимого результата F-статистики (p<0,001, дисперсия обработки по сравнению с дисперсией ошибки), для сравнения объема опухоли в двух группах проводили односторонний статистический анализ методом Манна - Уитни, и статистически значимым считали P<0,05.
Экспериментальные результаты:
Таблица 6. Действие комбинированного введения при подкожной трансплантации опухоли мышам, инокулированным клетками B-клеточной лимфомы человека SU-DHL-4
(мм3)
День 0
(мм3)
День 14
День 14
День 14
День 0: время первого введения; п/о: пероральное введение; 2 р/сут: два раза в сутки; *P<0,05, **P<0,01, сравнение с растворителем.
Вывод из эксперимента
Из данных, приведенных в таблице 6, видно, что соединение A (50 мг/кг, п/о, QD×14) ингибировало рост подкожно трансплантированной опухоли у мышей, инокулированных клетками SU-DHL-4, и степень ингибирования роста опухоли составляла 76%; у 1/8 мышей опухоль регрессировала частично, и у 1/8 мышей полностью. Соединение B (50 мг/кг, п/о, QD×14) приводило к степени ингибирования роста опухоли 60% для клеток SU-DHL-4; опухоль частично регрессировала у 2/8 мышей и полностью регрессировала у 1/8 мышей. При введении соединения A и соединения B в комбинации степень ингибирования опухолевого роста увеличивалась до 91%; опухоль частично регрессировала у 2/8 мышей и полностью регрессировала у 1/8 мышей; и эффективность была статистически значимо сильнее по сравнению с соединением A или соединением B по отдельности (см. Фиг. 5). Комбинированное введение двух соединений приводило к статистически значимому ингибированию роста опухоли после подкожной трансплантации опухоли мышам, инокулированным клетками B-клеточной лимфомы человека SU-DHL-4 и Вызывало частичную или полную регрессию опухоли. При введении двух соединений в комбинации эффективность улучшалась, и у мышей, несущих опухоль, масса тела снижалась, но была устойчивой к соединениям.
Таким образом, комбинированное действие ингибитора BTK соединения A и ингибитора EZH2 соединения B по настоящему изобретению превышает действие отдельного соединения, и такая комбинация обладает синергическим действием.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО БЕНЗОФУРАНА | 2018 |
|
RU2777624C2 |
| ПРОИЗВОДНОЕ БЕНЗОФУРАНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2016 |
|
RU2727198C2 |
| ИНГИБИТОРЫ EZH2 ЧЕЛОВЕКА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2013 |
|
RU2704445C2 |
| ИМИДАЗОПИРИМИДИНЫ КАК ИНГИБИТОРЫ EED И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2836176C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2013 |
|
RU2699546C2 |
| БЕНЗАМИДНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2801647C2 |
| Ингибиторы BTK | 2020 |
|
RU2830171C1 |
| СПОСОБЫ ЛЕЧЕНИЯ РАКА | 2013 |
|
RU2677276C2 |
| N-(ФЕНИЛСУЛЬФОНИЛ) БЕНЗАМИДЫ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ BCL-2 | 2020 |
|
RU2831159C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
Изобретение относится к применению комбинации ингибитора EZH2 и ингибитора BTK в получении лекарственного средства для лечения опухоли, отличается тем, что ингибитор EZH2 представляет собой соединение формулы (IE) или его фармацевтически приемлемую соль, ингибитор BTK представляет собой соединение формулы (IIA) или его фармацевтически приемлемую соль, где опухоль выбрана из группы, состоящей из диффузной B-крупноклеточной лимфомы и фолликулярной лимфомы. 6 з.п. ф-лы, 5 ил., 6 табл., 4 пр.
1. Применение комбинации ингибитора EZH2 и ингибитора BTK в получении лекарственного средства для лечения опухоли, отличающееся тем, что ингибитор EZH2 представляет собой соединение формулы (IE)

или его фармацевтически приемлемую соль,
ингибитор BTK представляет собой соединение формулы (IIA)
или его фармацевтически приемлемую соль,
где опухоль выбрана из группы, состоящей из диффузной B-крупноклеточной лимфомы и фолликулярной лимфомы.
2. Применение по п. 1, характеризующееся тем, что фармацевтически приемлемая соль выбрана из группы, состоящей из фосфата, гидрохлорида, метансульфоната, малеата, малата, пара-толуолсульфоната и безилата.
3. Применение по п. 1, характеризующееся тем, что отношение количества ингибитора EZH2 к количеству ингибитора BTK составляет 0,001–1000.
4. Применение по п. 3, характеризующееся тем, что отношение количества ингибитора EZH2 к количеству ингибитора BTK составляет 0,01–100.
5. Применение по п. 3, характеризующееся тем, что отношение количества ингибитора EZH2 к количеству ингибитора BTK составляет 0,1–10.
6. Применение по п. 1, характеризующееся тем, что вводимая доза ингибитора EZH2 составляет 1–2000 мг; а вводимая доза ингибитора BTK составляет 1–1000 мг.
7. Применение по п. 1, характеризующееся тем, что вводимая доза ингибитора EZH2 составляет 10 мг, 50 мг, 100 мг, 150 мг, 200 мг, 300 мг, 400 мг, 800 мг или 1600 мг, а вводимая доза ингибитора BTK составляет 10 мг, 20 мг, 50 мг, 80 мг, 100 мг, 150 мг, 160 мг, 200 мг, 250 мг, 300 мг, 350 мг, 500 мг или 650 мг.
| WO 2014168975 A1, 16.10.2014 | |||
| WO 2013067300 A1, 10.05.2013 | |||
| WO 2016007185 A1, 14.01.2016 | |||
| US 20150065483 A1, 05.03.2015 | |||
| WO 2013049770 A2, 04.04.2013 | |||
| RU 2014117632 A, 10.11.2015. |

