Область техники
Изобретение относится к способам получения биоразлагаемых фосфатсодержащих полимерных материалов, аналогов костной ткани.
Получаемый композит может быть использован для приготовления различных изделий медицинского назначения:
- крепежных винтов, шпилек, пинов для сращивания поврежденных костей,
- заполнителя костных пустот, генерирующих кальций по мере биоразложения имплантата,
- биоразлагаемых пленок и нитей повышенной прочности по сравнению с ненаполненными материалами,
- основы для выращивания новых тканей,
- лекарств пролонгированного действия,
а также в быту для изделий одноразового использования (бутылок, стаканчиков, тарелок, ножей, ложек, вилок и т.д.).
Создание полностью биоразлагаемых композитных материалов на основе алифатических сложных полиэфиров и фосфатов кальция имеет многоцелевое медицинское назначение, Главнейшее из них - получение достаточно прочных к механическим нагрузкам материалов, пригодных для изготовления крепежных изделий с внутренней, непосредственной фиксацией сложных переломов костей, в том числе плохо поддающихся внешней фиксации с помощью гипса или различных металлических конструкций.
Непосредственное введение фосфата кальция, главного компонента костной ткани, в то место в организме, где необходима срочная ее регенерация, преследует также цель снижения (или даже ликвидации) кальциевого дефицита в месте повреждения. Очень важным при этом становится механизм, по которому происходит разборка поставляемого кальция и последующая сборка его во вновь возникающей (регенерируемой) костной ткани. Оптимизация разборки снижает уровень кислотности, возникающей от резорбируемого композита и отрицательную реакцию окружающих тканей соответственно, оптимизация последующей сборки во многом определяется структурными особенностями конструируемого композита.
Уровень техники
Наиболее интенсивно изучаемыми и, следовательно, особенно перспективными исходными компонентами для создания полностью биоразлагаемых композитов следует принять следующие объекты:
- в качестве биоразлагаемой полимерной матрицы наиболее целесообразной следует считать гомополимеры полилактидного (полигликолидного) или полиоксикислотного типа (алифатические сложные полиэфиры). На втором месте по значимости могут стать сополимеры полилактида с полигликолидом блочного типа с фазово-разделенной микроструктурой, в которой полигликолидные участки являются местами локализации возникающей решетки, обеспечивающей повышенную прочность матрицы и, наконец, индивидуальный полигликолид, обеспечивающий наиболее высокий модуль на изгиб, вследствие высокой кристалличности и до недавнего времени считавшийся наиболее приемлемым для создания крепежных изделий, но проигрывающий полилактиду в сроках биоразложения. Остальные полилактоны могут иметь вспомогательное назначение и применяться только в виде добавок.
- достаточно важными компонентами биоразлагаемой полимерной матрицы следует считать полимеры оксикислот, индивидуального или смешанного типа, получаемые гомо- или совместной поликонденсацией. Целесообразность такого варианта продиктована не только значимостью этих продуктов в качестве возможной замены относительно дорогостоящего лактида, но и в качестве перспективных модификаторов фосфатов кальция, создающих необходимое сродство между органической частью композита и его неорганической составляющей.
- основные фосфатные наполнители - гидроксиапатит Ca10(PO4)6(OH)2 и трикальцийфосфат Са3(РO4)2. Считается, что первый более приемлем, т.к., попадая в живой организм, сразу начинает работать, в отличие от второго, который должен пройти предварительную стадию гидратации. Не исключаются и комбинации с другими фосфатами кальция, в частности с меньшей степенью замещения фосфора, а также соединения кальция вообще не содержащие фосфора типа Са(ОН)2, СаСO3 и т.д. Основное назначение указанных комбинаций - изменение характеристического соотношения Са/Р в диапазоне расширенном с естественного для костной ткани (1,67) до 1,0-1,9.
- состав композита по содержанию наполнителя определяется в основном выбираемым балансом между прочностными характеристиками получаемого композита и сроком его службы. Моделирование живой костной ткани по высокому содержанию наполнителя (70-77%) к настоящему времени имеет смысл только для изделий, не несущих достаточно высоких (необходимых для скрепления) механических нагрузок, например для заполнения костных пустот, скаффолдов и т.д. Преимущественное содержание наполнителя, на которое следует ориентироваться и которое создает необходимый баланс свойств при нынешнем уровне техники, характеризуется на уровне 20%.
- типы совмещения (комбинирования) фосфатных наполнителей и полимерной матрицы. Самый простой из них - экструдерное совмещение через расплав или с помощью растворителя. Однако экструдерный вариант не приводит к достаточно удовлетворительным результатам, т.к. не обеспечивает необходимую дезагрегацию наполнителя и степень его прививки к полимерной матрице. Наиболее часто применяемым в настоящее время следует считать растворный вариант совмещения.
- дисперсность исходного наполнителя и характер его распределения в получаемом композите также весьма важная характеристика, которая в значительной мере определяет прочностные характеристики материала в целом.
Следует отметить, что разработанные к настоящему времени методы синтеза фосфатов (преимущественно мокрые, Баринов С.М., Комлев B.C. Биокерамика на основе фосфатов кальция. Москва. Наука 2005) позволяют реализовать наноуровневый их размер, однако удержать высокую дисперсность при последующем получении композита представляется достаточно сложной задачей, т.к. получаемые нанокристаллы весьма склонны к агрегации. Решение указанной проблемы возможно путем создания максимально возможного сродства наполнителя к матрице, которое достигается химической прививкой фрагментов полимерной матрицы к поверхности наполнителя.
Учитывая вышесказанное, основное внимание при обзоре существующего уровня техники в данной области было направлено на выявление способов получения органоминеральных (фосфатсодержащих) биоразлагаемых композитов с улучшенной совместимостью полимер-наполнитель, которая в свою очередь достигается с помощью обязательной предварительной модификации поверхности наполнителя органической составляющей композита полимеризационными, поликонденсационными или полимераналогичными превращениями, проводимыми в жидкой органической среде, после удаления которой конструируется конечная полимерная матрица с дисперсно расределенным в ней модификатом.
Уровень техники, существующий на данный момент в данной области, может быть представлен следующей подборкой выявленных патентов-аналогов и научных публикаций.
Известен способ модификации гидроксиапатита полилилактидом, осуществляемый полимеризацией лактида в присутствии дисперсии гидроксиапатита. Полимеризацию проводили в растворе ксилола под действием октоата олова (содержание 500-5000 ppm), при температуре 90-120°С, в течение 3-48 часов. Дисперсию гидроксиапатита медленно вводили в раствор лактида при постоянном перемешивании. Полученную дисперсию подвергали далее центрифугированию для отделения ее от не прореагировавшего мономера, промывали несколько раз хлороформом, сушили и приготавливали пластинки для механических испытаний при температуре 180°С и давлении 15 МПа. Реализованная степень прививки на поверхность гидроксиапатита, привела к заметному повышению механических свойств композита даже при относительно низкой молекулярной массе полимерной матрицы (Mw=11645).
Недостатком данного способа полимеризационной модификации наполнителя является многостадийность, длительность проведения процесса, необходимость применения катализатора и растворителей.
(Полимеризационная прививка L-лактида на поверхность нано-кристаллов гидроксиапатита. Z. Hong, X. Qiu, J. Sun, M. Deng, X. Chen, X. Jing. Polymer 45, (2004), 6699-6706).
Известен метод поверхностной 2-х стадийной модификации наночастиц гидроксиапатита с помощью прививки на них L-молочной кислоты и L-лактида. Суть метода заключается в том что молочная кислота на первой стадии прививается на гидроксиапатит в растворе тетрагидрофурана (заменяемым далее на толуол) при 60°С с образованием кальций карбоксилатных и гидроксильных связей на поверхности, а лактид, на второй стадии, только гидроксильных. Расплавная полимеризация лактида в присутствии полученных привитых фосфатов, проводимая под действием октоата олова, приводит к композитам, обладающим при степени модификации 15% наибольшим повышением прочностных характеристик композита (прочность на разрыв повышается с 45 Мпа до 67 МПа и модуль с 1,7 ГПа до 2,1 ГПа).
Недостатком способа является применение двух промежуточных растворителей и связанной с этим многостадийностью процесса, применением катализатора на стадии полимеризации, необходимостью удаления каталитических остатков из композита для применения его в медицинских целях.
(Поверхностно модифицированный L-молочной кислотой гидроксиапатит и его последующая полимеризационная прививка L-лактидом. X. Qiu, Z. Hong, J. Hu, L. Chen, X. Chen, X. Jing. Biomacromolecules, 2005, 6, 1193-1199).
В более поздней аналогичной работе методом предварительной модификации поверхности дисперсного гидроксиапатита с помощью молочной кислоты и последующей полимеризации L-лактида в присутствии полученного модификата синтезированы образцы композитов с содержанием гидроксиапатита 5, 10 и 20 вес %. Модификацию проводили в растворе тетрагидрофурана с последующей добавкой толуола и выдерживанием системы 6-8 часов при температуре 85°С, после чего наполнитель промывали этилацетатом. Полимеризацию осуществляли при температуре 130°С в течение 24 часов. Готовый композит промывали хлороформом и сушили в вакууме. Оптимальная степень наполнения, повышающая прочность на разрыв до 55 МПа, составила 10%. Констатировано и повышение термостабильности композита по сравнению с ненаполненными образцами.
Предложенный способ модификации ГАП характеризуется теми же недостатками, что и предыдущая работа, а именно, применением растворителей (тетрагидрофурана и толуола), выдержкой системы достаточно длительное время при повышенной температуре (85°С), применением этилацетата для очистки композита.
(J. Li, X.L. Lu, Y.F. Zheng. Effect of surface modified hydroxyapatite on the tensile property improvement of HA/PLA composite. Applied Surface Science, 255, (2008), 494-497).
В последующей работе приведены результаты медицинских (in vivo) испытаний, которые тем не менее показали улучшение эксплуатационных свойств предложенного композита.
(Cell responses and hemocompatibility of g-HA/PLA composites.
LI Jia, ZHENG Wei. ZHENG YuFeng, LOU Xia. Sci China Life Sci April (2011) Vol. 54 No. 4.).
Известен способ получения органоминерального биодеградируемого полиэфирного композита с широкой дисперсностью наполнителя от нанометрового до микрометрового уровня, в котором минеральная составляющая - гидроксиапатит, а органическая - полимолочная кислота, получаемая конденсацией молочной кислоты в присутствии толуола или в отсутствие растворителя.
Получение композита начинали с модификации и увеличения поверхности ГАП путем повышения содержания на его поверхности гидроксильных групп, через которые, по мнению авторов, образуется далее ковалентная связь между наполнителем и полимерной матрицей. Увеличение поверхности ГАП и одновременно прививку к нему биоразлагаемого полимера проводили в тетрагидрофуране или толуоле постепенным введением молочной кислоты, лактата кальция или олигомера молочной кислоты. Режим введения 0-80°С, в течение 0,5-5 часов, при постоянном перемешивании. Далее твердый осадок отделяли фильтрованием и проводили дегидратацию с помощью азеотропной отгонки воды с толуолом, в течение 1-48 часов.
Допускалась и непосредственная дегидратация молочной кислоты без участия толуола при температуре 80-120°С и постоянном перемешивании в течение 0,5-5 часов с последующей промывкой привитого полимера тетрагидрофураном или этанолом с целью отделения его от не привитого. Однако количество вводимой кислоты по отношению к ГАП было достаточно большим, что приводило к растворению ГАП в кислоте, отсутствию модификации, вызывало неизбежное слипание и комкование его частиц и не позволяло квалифицировать данный вариант как «сухой».
Получение собственно композита (на второй стадии) с содержанием 0,1-70% ГАП осуществляли растворной или расплавной (solvent free) полимеризацией лактонов биоразлагаемого типа в присутствии модифицированного на первой стадии ГАП, катализатор полимеризации - октоат олова, время полимеризации 12-L-полилактидом нано-гидроксиапатита под действием микроволнового излучения, который осуществлялся без применения каких-либо органических растворителей. Прививку производили в расплаве лактида при температуре 140°С, соотношении гидроксиапатит/лактид, выбранном в интервале 1:50-1:400, при мощности микроволнового излучения 50W и наличии октоата олова в качестве катализатора полимеризации. При этом, в зависимости от выбранных условий, достигаемая степень прививки составляла 14,9-35,9%, а частицы гидроксиапатита уменьшались в размерах по сравнению с первоначальными. Наличие прививки доказано методом ИК-спектроскопии, ТГА- и рентгеновским анализом, а размер частиц исследован методом седиментации в органическом растворителе.
Предложенный процесс прививки гидроксиапатита основан на полимеризационном методе модификации, осуществляемым с помощью микроволнового нагрева системы, характеризуется достаточно высокой температурой (140°С) и продолжительностью (45 мин), достигаемая при этом конверсия мономера не сообщается.
(B. Luo, С. Hsu, J. Yang, C. Zhou. Синтез нано-гидроксиапатита, поверхностно привитого поли-L-лактидом под действием микроволнового излучения. Advanced Materials Reserch Vols. 204-210, 2011, 1929-1933).
Известен способ получения биоразлагаемого высокомодульного композита, состоящего, по крайней мере, из 3-х компонент, фибриллярного наполнителя, привитого на него биоразлагаемого полимера и одноименного, предварительно полученного полимера (полилактида) большой молекулярной массы. Образование привитого полимера катализируется специально вводимым катализатором или реакционными функциональными группами, имеющимися на поверхности фибрилл наполнителя. В качестве катализаторов предложен широкий перечень кислых и основных реагентов, включающий соединения 4-х валентного титана и олова, сульфоновую и серную кислоты, амины, алкоголяты калия или натрия, а также органические основания, цеолиты, индивидуальные или смешанные оксиды металлов (магния и алюминия). Полимерная составляющая - 35% лактида и 65% полилактида, фибриллярный наполнитель - преимущественно очищенная целлюлоза (характеристическое отношение L/D=45), предпочтительная каталитическая группа - октоат олова, трифенилфосфин, изопропоксид титана.
Подчеркивается, что чрезвычайно важным фактором для образования высокомодульного композита является наличие прочной и надежной связи между фибриллами наполнителя и высокомолекулярной полимерной матрицей. Посредником между этими составляющими является привитой, относительно низкомолекулярный полимер, одноименного состава с матрицей. Полезную роль в создании этой связи выполняют также катализаторы, способные к трансэтерификации. Смешение осуществлялось в обычных реакторах промышленного типа или экструдерах, полимеризация при температурах 25-400°С.
Недостатком указанного способа получения биоразлагаемого композита является многокомпонентность, обязательное наличие фибриллярного наполнителя, трудно поддающегося биоразложению и подвергаемого для этого модификации (целлюлоза), гомополимера лактида, приготавливаемого заранее и катализаторов полимеризации.
(Пат. США №8569428. 29.10.2013).
Известны многочисленные примеры модификации поверхности гидроксиапатита агентами, повышающими сродство гидрофобного эфира к гидрофильному наполнителю. Указанные агенты не являются химическими аналогами полиэфира, а лишь выполняют роль посредников между наполнителем и матрицей, улучшающих их совместимость. Посредники не только увеличивают число поверхностно-активных групп наполнителя, но в некоторых случаях активируют их и придают им каталитическую функцию, что позволяет проводить последующее полимеризационное наполнение композита без участия традиционных оловоорганических катализаторов. Следующие примеры иллюстрируют данную тенденцию.
Поверхностная модификация гидроксиапатита додециловым спиртом, проводимая простым гидротермальным нагревом системы, резко увеличивает стабильность его дисперсий в этиловом спирте. Указанный прием достаточно нагляден, но не гарантирует свою полезность при создании биоразлагаемого композита.
(L. Borum-Nicolas, O.C. Wilson Jr. Biomaterials, 24, 2003, 3671-3679).
Способ модификации цинксодержащего гидроксиапатита винной кислотой, проводимый в растворе тетрагидрофурана. Наличие прививки доказано методами ИК- и ЯМР-спектроскопии, которая составляла, в зависимости от соотношения кислота/ГАП 4,8-14,7%.
Недостатком указанной модификации является использование тетрагидрофурана, необходимость применения каталитически активного (цинксодержащего) гидроксиапатита и возможность появления сшитых структур вследствие 4-х функциональности винной кислоты.
(Thouraya Turki, Masseoud Othmani, Christophe Goze Вас, Ferid Rachdi, Khaled Bouzouita. Surface modification of zinc-containing hydroxyapatite by tartaric acid. Applied Surface Science, 284, (2013). 66-71).
Способ модификации гидроксиапатита лимонной кислотой. Модификацию проводили в растворе диметилформамида при 150°С, 11 час, путем медленного введения раствора кислоты в суспендированную дисперсию гидроксиапатита, подвергаемому перемешиванию и ультразвуковой обработке. Последующее компаундирование модификата с полилактидом (или сополимером с гликолидом), осуществляемое через раствор, показало 20%-ое улучшение механических свойств композита при содержании наполнителя в количестве 3%.
Недостатком способа является применение диметилформамида, высокая температура процесса, длительность и применение ультразвуковой обработки композита.
(Effect of new surface-grafting method for nano-hidroxiapatite on the dispersion and mechanical enhancement for poly(lactide-co-glycolide).
L.X Jiang, L.Y. Jiang, L.J, Xu, C.T. Han, C.D. Xiong.
eXPRESS Poymer Letters Vol. 8, №2 (2014) 133-141).
В последнее время все чаще в патентной и научной литературе появляются сообщения, в которых для создания биоразлагаемых кальцийфосфатсодержащих композитов используются азотсодержащие соединения, которые не только выполняют роль посредников между наполнителем и полимерной матрицей, но и являются достаточно активными безметальными катализаторами полимеризации лактонов и поликонденсации оксикислот. Среди таких соединений можно выделить алифатические амины, имины, аминокислоты, производные гуанидина, хитозан и т.д. Следующие примеры иллюстрируют данную тенденцию.
Предложен нано-гидроксиапатит, модифицированный L-лизином и полилактидный композит его содержащий. Модификацию проводили в диметилформамиде при температуре 150°С в течение 9 часов. Полимеризационное наполнение осуществляли лактидом при 140°С в течение 20 часов. Композит, содержащий 10 и 20% отмытого модификата, показал существенное возрастание прочности на изгиб и растяжение по сравнению с ненаполненным материалом (около 20%). Испытания in vitro продемонстрировали улучшенную биосовместимость.
Недостатком указанного предложения, как и в цитированных выше, является применение высококипящего растворителя, высокая температура модификации и длительность процесса.
(Effect of L-lysine-assisted surface grafting for nano-hydroxyapatite on mechanical properties and in vitro bioactivity of poly(lactic acide-co-glycolic acid.
Jiang Liuyun, Jiang Lixin, Xiong Chengdong, Xu Lijuan, Li Ye. Journal of Biomaterials Appications 0(0) 1-9 2015).
В работах китайских авторов активно разрабатывается креатинин (производное гуанидина) в качестве катализатора конденсации молочной кислоты и полимеризации лактида. Показано, что при относительно небольшом содержании указанного соединения (0,1% к мономеру) с помощью двухстадийного процесса (расплавная конденсация и твердофазная деполимеризация) возможно получение высокомолекулярного неокрашенного полимера (Mw=120000), сохраняющего высокую изотактичность (на уровне 98%), что в свою очередь ведет к повышению его термостойкости. Первая, поликонденсационная стадия, несомненно повышает сродство получаемого полимера к гидроксиапатиту с помощью добавки креатинина, однако о последующем создании фосфатсодержащего композита в данном варианте не сообщается.
(Isotactic polycondensation of L-lactic acid with biogenic creatinine. Wei Jiang, Wei Huang, Na Cheng, Yundiao, Xupeng Zong, Hong Li, Quanxing Zhang. Polymer 53 (2012) 5476-5479.
(Syntesis of high molecular weight poly(L-lactic acid) and poly(D-lactic acid) with improved thermal stability via melt/solid polycondensation catalyzed biogenic creatinine. Polymer 55 (2014) 1491-1496).
Среди посредников полимерного типа, улучшающих совместимость, выделяется хитозан, которым модифицируют и гидроксипатит и полиэфир. На основе получаемых модификатов создаются 3-х компонентные аминосодержащие биоразлагаемые композиты. Помимо посреднической функции хитозан может сообщать и ряд других полезных свойств композиту, а именно, ранозаживления, катализатора полимеризации лактонов, дополнительного (совместно с гидроксиапатитом) усиливающего активного наполнителя, пластификатора и т.д. Следующие примеры иллюстрируют сказанное.
Наиболее показательна в этом плане работа китайских авторов, в которой предложен 3-х компонентный биоразлагаемый композит, где в качестве промежуточного слоя между гидроксиапатитом и полилактидом внедрен хитозан. Способ состоит из нескольких стадий:
- первоначальное in situ соосаждение гидроксиапатита совместно с хитозаном путем медленного введения фосфорной кислоты в дисперсию гидроокиси кальция, суспендированную в растворе хитозана,
- очистка и осушка системы,
- прививка полученного порошка молочной кислотой,
- повторная очистка и осушка системы,
- растворное наполнение модифицированного порошка готовым полилактидом.
При высокой степени наполнения (60% и 67%) авторам удалось продемонстрировать реальное повышение физико-механических свойств композита и существенно улучшить его поведение in vitro.
Предложенный способ, так же, как и ранее рассмотренные, характеризуется 2-х стадийной поверхностной модификацией гидроксиапатита через хитозан и неизбежной при этом многостадийностью процесса.
(Fabrication and in Vitro Investigation of Nanohidroxyapatite, Chitosan, Poly(L-lactic acid) Ternary Biocomposite. C.Y. Zhang. C.L. Zhang. J.F. Wang, C.H. Lu, Z. Zhuang, X.P. Wang, Q.F. Fang. J. Appl. Polymer. Sci. 2013, DOI: 10.1002/APP. 37795.).
В работе японских авторов предложен способ получения композита, содержащего пористый гидроксиапатит и сополимер лактида с гликолидом. Исходные пористые таблетки гидроксиапатита, диаметром 2 мм, размером пор 20 ммк и степенью пористости около 40% заполнялись смесью лактида и гликолида, содержащей 2% энзима липазы ММ в качестве катализатора полимеризации. Полимеризация проводилась при температуре 100°С в течение 9 дней, по истечении которых поры гидроксиапатита оказывались полностью заполненными полимером. Прочностные характеристики при этом составили 90-54 МПа сопротивления изгибу и 1,3-0,57 МПа и1/2 сопротивления разлому, что близко к значениям кортикальной кости человека. Способ позволяет получать монолитный композит с высоким содержанием непрерывной фазы наполнителя, но также характеризуется многостадийностью и длительностью процесса получения (9 дней).
(In situ preparation of poly(L-lactic acid-co-glycolic acid)|hydroxyapatite composites as artificial bone materials. Y. Takeoka, M. Hayasyi, N. Sugiyama, M. Yoshizawa-Fujta, M. Aizawa and M. Rikukawa. Polymer Journal, (2015), 47, 164-170).
Известен метод, предложенный Российскими авторами твердофазной «сухой» сополимеризации хитозана и лактида, который осуществили пропуском смеси твердых компонент через 2-х шнековый экструдер при температуре ниже точки плавления лактида 90-120°С. Под действием интенсивного механического воздействия и возникающих при этом механохимических реакций получен амфифильный сополимер хитозана и лактида, который привит на боковые аминогруппы хитозана в количестве от 3 до 10 звеньев. Амфифильность полученного сополимера использована для стабилизации микросфер в системе вода-масло.
Достоинством метода является демонстрация возможности использования механохимических реакций в отсутствии каких либо растворителей для связывания компонент, широко используемых в создании биоразлагаемых композитов, однако о реализации указанной возможности с использованием гидроксиапатита не сообщается.
(Polylactide-based microspheres prepared using-state copolymerized chitosan and d,l-lactide. T.S. Demina, T.A. Akopova, L.V. Vladimirov, A.N. Zelenetskii, E.A. Markvicheva, Ch. Grandfils. Materials Science Engineering С 59, (2016), 333-338).
Французскими авторами аналогичный сополимер получен 2-х вариантным «мокрым» способом. Первый - путем непосредственной поликонденсации молочной кислоты в водно-кислотном растворе хитозана. Реакция катализирована р-толуолсульфокислотой и осуществлена при температуре 80°С в течение 24 часов. Второй вариант - методом раскрытия кольца лактида также в предварительно полученном водно-кислотном растворе хитозана. Реакция катализирована триэтаноламином и проведена при температуре 80°С в течение 24 часов. О выходе обоих типов сополимеров не сообщается, однако ориентировочная степень прививки в обоих вариантах оценена в 46-47%. Оба варианта свидетельствуют в пользу того, что прививка лактида на хитозан возможна и в водной среде.
Недостаток метода состоит в том, что он отражает только возможность получения посредника - сополимера хитозана и полимолочной кислоты (или полилактида) и не затрагивает возможность дальнейшего синтеза фосфатсодержащего композита.
(Different Pla Grafting Techniques on Chitosan. N.E. Suyatma, A. Copinet, E. Legin-Copinet, F. Fricoteax, V. Coma. J. Polym Environ. (2011), 19:166-171).
Известен метод получения хитозана, модифицированного полимолочной кислотой. Модификацию осуществляли через промежуточную вставку 4,4-диизоцианатдифенилметана, вставляемую в полимолочную кислоту и сшивающую ее гидроксильные концевые группы, после чего проводили сшивку концевых карбоксильных групп с помощью хитозана. Степень прививки предлагаемым методом может быть очень высокой, достигающей 30-кратного избытка хитозана по отношению к молочной кислоте.
Недостаток метода аналогичен варианту, цитированному выше.
(Method for preparing chitosan С grafting polylactic acid. CN 101003632 (A) - 2007-07-25).
Наиболее близким техническим решением по отношению к заявляемому является китайский патент CN №102940908 (А) (Absorbable interference screw for repairing interior and posterior cruciate ligaments and preparation method of screw) от 2013-02-27, который принят в качестве прототипа. В указанном прототипе предложен двухстадийный способ получения биоразлагаемого композита и крепежных винтов на его основе, представляющего собой смесь полилактида и предварительно модифицированного олигомером молочной кислоты гидроксиапатита. Предложенный способ состоял из следующих операций:
- на первой стадии получали смешанный раствор олигомера молочной кислоты и ГАП путем смешения ацетонового раствора олигомера и водного золя ГАП. Массовое соотношение олигомера молочной кислоты и ацетона в интервале 1:3(10), олигомера молочной кислоты и ГАП - 1(10):1.
- полученный раствор далее разбавляли водой при перемешивании до соотношения вода : ацетон в пределах 1,5(3):1, фильтровали выпавший осадок и сушили, получая сухой смешанный материал. Для улучшения фильтруемости предпочтительно вызревание смеси в течение 4-24 час.
- после осушки смешанный материал обрабатывался смесью ксилола и ацетона (объемное соотношение 1:1(5), в интервале температур 130-140°С, в течение 24-36 часов. Цель обработки - повышение степени поверхностной прививки ГАП и реализация зернистой структуры модифицируемого материала.
- удаление ксилола центрифугированием с последующей обработкой несколько раз хлороформом и сушкой приводила к искомому продукту, модифицированному ГАП, поверхностно-привитому олигомером молочной кислоты.
Дальнейшие операции включали смешение полученного материала с различными биоразлагаемыми полимерами с варьируемой молекулярной массой в пределах 40000-200000 и характеристической вязкостью в пределах 1-2,5 дл/г. Смешение производили в экструдере с получением прутка при температуре 120-230°. Предпочтительный диаметр прутка 9-14 мм, получение из него винтов 7-12 мм (метрическая резьба М7, М8, М10, M12). Нарезание резьбы производилось с применением охлаждающей жидкости.
Улучшенный вариант модификации ГАП включал применение ультразвука на стадии смешения ацетонового раствора полимолочной кислоты и водного золя ГАП с целью улучшения полноты смешения и однородности раствора. Время облучения 15-30 мин.
Как видно из приведенного описания, в данном патенте реализован метод формирования композита, использующий золь-гель переход системы, который позволяет гибко регулировать размер возникающих частиц. Однако достигается это только применением комбинированных растворителей определенного состава (в данном случае это смесь вода-ацетон), причем возникающая при этом степень прививки не достаточна и вводится дополнительная операция со сменой комбинированного растворителя (ксилол-ацетон) и длительным временем выдерживания системы (24-36 часов) при высокой температуре 130-140°С.
Таким образом, предложенный способ получения композита характеризуется применением смеси органических растворителей, по крайней мере, на стадии модификации, необходимостью их последующего удаления и разделения, длительностью процесса и требует относительно высоких температур для реализации.
Из приведенного обзора уровня техники, отражающего суть заявляемого объекта, можно выделить следующую основную тенденцию, а именно, сближение сродства практически несовместимых компонент композита, минерального фосфатного наполнителя и органической полиэфирной матрицы. Наиболее действенным способом указанного сближения признается создание ковалентных связей между ними. Предложено достаточно большое разнообразие вариантов создания композитов, с помощью которых удается получить не только более тонкое распределение частиц наполнителя в полимерной матрице, но и химическое связывание их, что в свою очередь улучшает физико-механические характеристики композита в целом. Суммируя основные приемы, применяемые объекты и выбираемые условия, можно констатировать что:
- создание химических связей осуществляется преимущественно с помощью функциональных групп наполнителя, имеющихся изначально или создающихся специально путем различных химических превращений,
- полимерная матрица поставляется в виде готового полимера или создается по ходу синтеза композита высокотемпературной катализированной полимеризацией лактонов или поликонденсацией оксикислот,
- совмещение компонент композита осуществляется в большинстве случаев через раствор, реже через расплав полимерной матрицы или комбинированием обоих приемов.
Таким образом, большинство выявленных способов получения композитов, аналогов заявляемого, характеризуются применением органических растворителей и необходимостью их последующего тщательного удаления, достаточно высокой температурой катализированного синтеза полимерной матрицы, многостадийностью и длительностью проведения отдельных операций.
Раскрытие изобретения
Техническим результатом заявленного изобретения является исключение использования органического растворителя при сниженной (по сравнению с однотипными реакциями)температуре проведения способа, уменьшении времени проведения синтеза с использованием однотипного простого оборудования для любых составов компонент.
Технический результат достигается тем, что предложен способ получения биоразлагаемого композита для использования в качестве аналога костной ткани, характеризующийся тем, что гидроксиапатит и полилактид подвергают совместной механохимической обработке путем диспергирования при температуре 20-40°C, в интервале 3000-7000 об/мин диспергатора, при следующем исходном соотношении гидроксиапатит/полилактид % вес: 20-80/90-10.
Изобретение поясняется чертежами:
Фиг. 1 Фотографии исходного промышленного полилактида в гранулах (левая верхняя чашка) и продуктов совместного его диспергирования с гидроксиапатитом, разделенных с помощью сита (0,25 мм) где: Первая мелкая фракция (нижняя правая чашка), вторая, более крупная (нижняя левая чашка), изношенные гранулы исходного полимера (верхняя правая чашка).
Фиг. 2 ТГА-кривые фракций композита, полученного «сухим» методом из гидроксиапатита и промышленного полилактида при их совместном диспергировании, (кривые обозначены в тексте).
Фиг. 3 ИК-спектры образцов, полученных при «сухой» модификации гидроксиапатита высокомолекулярным полил актидом: фракция диспергированного продукта, прошедшего через сито 0.25 мм (красный); после последующей пятикратной отмывки в хлористом метилене, центрифугирования и сушки (зеленый); индивидуальный гидроксиапатит (синий).
Фиг. 4 ИК-спектр не осаждаемой в центрифуге фракции, выделенной после обработки гидроксиапатита, модифицированного высокомолекулярным полилактидом, хлористым метиленом (фиолетовая кривая). На этом же фигуре приведен ИК-спектр исходного высокомолекулярного полилактида (красная кривая).
Фиг. 5 ТГА-кривая образца модификата гидроксиапатита, полученного «сухим» методом с помощью молочной кислоты (первая часть).
Фиг. 6 ИК-спектры конденсатов молочной кислоты, полученных «сухой» конденсацией на гидроксиапатите в одну и две стадии: порционное введение молочной кислоты, чередуемое с диспергированием (первая стадия, синяя кривая 2), порционная доконденсация образца, полученного на первой стадии, с последующей промывкой его в хлористом метилене, центрифугированием и сушкой (вторая стадия, красная кривая 3); индивидуальный гидроксиапатит (коричневая кривая 1).
Фиг. 7 ИК-спектр конденсата молочной кислоты, полученного при порционном введении гидроксиапатита в кислоту (синяя кривая 1); конденсат, полученный термически в отсутствие катализатора (красная кривая 2).
Фиг. 8 ИК-спектр образца, полученного «сухой» механически активированной полимеризацией лактида в присутствии гидроксиапатита с последующей термообработкой при 150°С и отмывкой хлористым метиленом где:
гидроксиапатит, модифицированный полилактидом (бирюзовая кривая 2);
индивидуальный гидроксиапатит (красная кривая 1).
Фиг. 9 Кривые ТГА продукта где: черная кривая 2 полученного «сухой» модификацией гидроксиапатита винной мезо-кислотой; красная кривая 1 полученного «мокрой» модификацией гидроксиапатита винной мезо-кислотой.
Фиг. 10 ТГА-кривая продукта, полученного «мокрой» модификацией гидроксиапатита лимонной кислотой.
Фиг. 11 ИК-спектр поликонденсата лимонной кислоты, полученного «мокрым» способом на гидроксиапатите (синяя кривая 2); индивидуальный гидроксиапатит (красная кривая 1).
Фиг. 12 ИК-спектр поликонденсата лимонной кислоты в диапазоне 520-950 см-1, полученного «мокрым» способом на гидроксиапатите (синяя кривая 2); индивидуальный гидроксиапатит (красная кривая 1).
В таблице приведена кинетика оседания в воде гидроксиапатита, модифицированного лимонной кислотой «сухим» методом.
Осуществление изобретения
Заявляемый способ получения биоразлагаемого композита основан на двух принципах, не выявленных в патентной и научной литературе применительно к заявляемому объекту. Первый основан на полном отказе от каких либо органических растворителей при синтезе композита и который квалифицируется нами как «сухой». Второй основан на использовании интенсивного механического воздействия на совмещаемые компоненты, провоцируя механохимические реакции между ними. Оба принципа объединены и могут работать одновременно.
Отличительными признаками «сухого» метода являются:
- обязательное использование сухих фосфатов кальция в порошкообразном состоянии различной степени дисперсности, задаваемой его предшествующим синтезом,
- модифицирующий органический биоразлагаемый компонент может быть твердым (полилактид, полигликолид или их сополимеры высокой молекулярной массы 100000-200000), хрупким (относительно низкой молекулярной массы, предпочтительно 5000-10000), жидким олигомером (ММ=1000-3000), кристаллическим (гликолид, лактид) или жидким лактоном ((ε-капролактон). Таким образом, охватывается практически весь диапазон молекулярных масс полимерной составляющей композита от мономера до конечного полимера,
- при применении полимерной составляющей в жидком состоянии, например при использовании жидкого полиэфирного олигомера молочной кислоты, расплава лактона при температуре выше его Тпл. и т.д. обязательным является условие, при котором в процессе отверждения жидкого модифицирующего агента (в результате полимеризации или поликонденсации) смесь остается сухой, сыпучей, практически не изменяющей своей степени дисперсности. В этом случае частицы не слипаются, не агрегируют, смесь не комкуется и не прилипает к стенкам сосуда, что позволяет сохранять развитую поверхность системы в целом. Указанное условие соблюдается при непрерывной или порционной медленной подаче полимерной составляющей мелкими порциями на модифицируемый наполнитель и достаточно интенсивном его перемешивании,
- применение интенсивного механического воздействия на отдельно взятые компоненты или их смесь преследует цель не только дезагрегации частиц наполнителя, но и их активации, которая способствует протеканию механохимических процессов на их поверхности,
- образующиеся в результате «сухой» модификации гидроксиапатита порошки имеют достаточно низкий насыпной вес, хорошо поддаются перемешиванию, в том числе потоком инертного газа, что создает возможность проведения процесса в так называемом «кипящем слое».
Сущность предлагаемого способа создания биоразлагаемого композита состоит в следующем.
В качестве наполнителя используют порошок гидроксиапатита, полученный методом «мокрого» синтеза, имеющий размер частиц в достаточно широком диапазоне (от нанометрового до субмикронного), остаточную влажность 3-5%, степень кристалличности 30%, твердость по Моосу 5.
В качестве модификатора гидроксиапатита, по одному из вариантов, берут твердый полилактид (полигликолид), который может иметь различные молекулярно-массовые и механические характеристики, в частности:
- полилактид промышленного типа; в предлагаемом способе используется в виде цилиндрических гранул диаметром 3 мм, высотой 5 мм;
- полигликолид, более хрупкий, обладающий повышенной, по сравнению с полилактидом, прочностью;
- полимолочная кислота (полилактид поликонденсационного происхождения), с молекулярной массой 10-100 тыс., предпочтительно 5-10 тыс. Полилактид такого типа достаточно хрупок, легко разрушается под действием интенсивных ударных нагрузок, но обладает некоторой слипаемостью получаемых частиц, особенно в нижней части диапазона молекулярных масс. Применяется в виде частиц произвольной формы, линейный размер которых не превышает 10-15 мм или в виде порошка. Полигликолид (или полигликолевая кислота) применяется с аналогичными характеристиками.
В качестве модификаторов возможно также применение кристаллических мономеров (лактид, гликолид), структура которых легко разрушается под механическим воздействием с образованием вакансий, на которых может осуществляться химическое взаимодействие смешиваемых компонент.
По другому варианту, примененному в предлагаемом способе, в качестве модификатора используют также жидкие компоненты, такие как молочная кислота и ее концентрированные водные растворы (содержание кислоты 80-90%), жидкие олигомеры молочной кислоты, расплавы мономеров (лактонов) при температуре выше их точки плавления, оксикислоты с повышенным или несбалансированным содержанием функциональных групп. Основное условие, которое при этом должно неукоснительно соблюдаться, заключается в том, чтобы количество добавляемого реагента не превышало некоторое критическое (текущее) значение, при котором начинается слипание частиц наполнителя, их агломерация и смесь в результате этого не может квалифицироваться как «сухая».
Указанный режим в значительной мере определяется типом и количеством добавляемого в единицу времени реагента, его смачивающей способностью по отношению к наполнителю, интенсивностью перемешивания, температурой, скоростью превращения (прививки) модификатора, его степенью конверсии и т.д. Данные условия подбираются экспериментально для каждой пары компонент и регламентируются оптимальным количеством добавляемого модификатора, отнесенного к массе модифицируемого наполнителя в единицу времени. В большинстве случаев при обычном перемешивании (число оборотов мешалки 1-5 об/сек.) соотношение масса модификатора/масса фосфата не должно превышать значений, установленных в пределах 0,005-0,05 за 10 мин. Подача модификатора может производиться порционно или непрерывно в пределах указанного диапазона. В начале процесса модификации выбирается минимальное соотношение, по мере развития процесса оно может приближаться к максимальным значениям. В случае высокооборотного перемешивания (3000-5000 об/мин) смесь приобретает свойства аэрозоля и скорость введения модификатора может быть повышена в 3-5 раз (при условии интенсивного отвода паров воды в случае поликонденсационного типа модификации).
В качестве диспергирующих (перемешивающих) устройств при получении заявляемого композита возможно применение диспергаторов (мешалок) различного типа, как низко - (30-120 об/мин), так и высокооборотных (3000-5000 об/мин). Последние позволяют более эффективно активировать наполнитель, перерабатывать твердые полимеры с высокой долей механохимических взаимодействий и результативных (прививочных) реакций между смешиваемыми компонентами соответственно. Низкооборотные вполне приемлемы для пар гидроксиапатит/жидкие олигомеры (оксикислоты), предпочтительно с высокой смачивающей способностью жидкого компонента к порошку наполнителя. «Сухой» режим проведения процесса в этом случае гарантирует сохранение высокоразвитой поверхности наполнителя и быстрое и эффективное удаление паров воды, выделяющихся в результате поликонденсации (аналог пленочных испарителей).
В случае 2-х стадийного проведения процесса получения композита возможно сочетание низко - и высокооборотных перемешивающих устройств. Например, получение модификатора на первой стадии поликонденсацией молочной кислоты на гидроксиапатите с применением низкооборотных устройств. На второй стадии получения конечного композита - с. применением высокооборотных диспергаторов. Соответствующее конструктивное оформление аппарата позволит вести процесс одностадийно, последовательно разделяя его на необходимые режимы.
Диспергирующие устройства различного типа хорошо известны, разнообразны по техническому оформлению и широко применяются в различных отраслях промышленности (в том числе и химической), например лакокрасочной для смешения твердых дисперсных пигментных красителей, как друг с другом, так и с отверждаемой жидкой основой (краскотерки).
Твердую полимерную основу композита, взятую в виде предварительно отформованных гранул произвольной формы, можно далее превращать в композит частично (предпочтительно не менее 20%) или полностью, отделяя, при необходимости, непрореагировавшую часть с помощью сит с различным размером ячеек (преимущественно 0,25 мм). Состав композита в этом случае определяется исходной дозировкой компонент или с учетом доли непрореагировавшей фракции.
Исходный состав композита выбирается в диапазоне гидроксиапатит/полимер, % вес 10-90/90-10, предпочтительно 20-80/80-20. При применении поликонденсационного метода получения композита с помощью оксикислот или их функциональных олигомеров предельное количество взятого модификатора может быть уменьшено до соотношения гидроксиапатита/полимер =95/5, однако получаемый продукт может быть далее использован преимущественно в качестве модификатора и нуждается в дополнительном полимерном наполнении.
Температура, время необходимое для получения композита, а также применяемое устройство, зависят от природы модификатора. При использовании твердых полимеров предпочтительны высокооборотные устройства (до 7000 об/мин), тогда процесс может осуществляться при комнатной температуре и время, необходимое для достижения высокой конверсии полимера (50-100%) может составлять 5-10 мин. Поликонденсационная модификация предпочтительна при повышенных температурах (50-100°С), хотя установлено, что она в принципе возможна (при соответствующем интенсивном перемешивании) и при комнатной температуре, но с применением вакуума. В зависимости от выбранной температуры продолжительность получения композита методом поликонденсации может варьироваться в пределах 2-5 часов.
Пример 1
Пример иллюстрирует возможность получения композита «сухим» методом. Отличительной особенностью предлагаемого примера является использование в качестве и модификатора и полимерной основы одного и того же промышленного полилактида, имеющего достаточно высокую молекулярную массу (ММ=180000), что позволяет получить композит в одну стадию без каких либо дополнительных операций.
Смешение и модификация производилось в диспергаторе лопастного типа с заточенными краями лопастей при числе оборотов 4000-5000 об/мин. В диспергатор загружается 3,117 г порошка гидроксиапатита и 2,0 г высокомолекулярного полимера в виде цилиндрических гранул диаметром и высотой 3 мм (средний вес одной гранулы 0,3 г). Состав полученной исходной смеси 60,9% гидроксиапатита и 39,1% полилактида. Диспергирование производится в один прием, с небольшим промежуточным интервалом, необходимым для охлаждения смеси (превышение по отношению к комнатной на 10-15°С). При общем времени диспергирования 8 мин (за вычетом времени охлаждения) полученная смесь содержала порошок и гранулы, отшлифованные до идеально сферической формы (средний вес одной гранулы 0,07 г). Общая потеря массы гранул (конверсия) введенного полимера составила 76,7%.
Ситовое фракционирование полученной сухой смеси производилось с помощью сита с размером ячейки 0,25 мм. Получено 3 фракции, самая мелкая (первая), прошедшая сквозь сито, составила 66,4% от исходной смеси. Средняя (вторая) - 23,02% и вышеупомянутые круглые (изношенные) гранулы - третья фракция. Ее содержание составило 10,6%. Третья фракция легко отделяется от первых двух простым встряхиванием смеси на ровной, слегка наклоненной поверхности (откатом). Поскольку она не содержала прилипшего порошка первых двух фракций ее можно идентифицировать как чистый (остаточный) полимер. Все три полученные фракции и исходный полимер представлены на фотографии (фиг. 1).
Определение насыпного веса первой и второй фракций (0,576 г/см3 и 0,685 г/см3 соответственно) показали, что модификация и полимерное наполнение гидроксиапатита «сухим» методом в данном варианте приводит к более плотной упаковке получаемого порошка по сравнению с чистым гидроксиапатитом (0,397 г/см3).
Экстракцию 1-ой и 2-ой фракций модификата производили с целью определения их различия в соотношении минерального наполнителя и органического полимера.
Отделение самой мелкой (первой) фракции осуществлялось простой (естественной) декантацией взвеси, полученной распределением ее в хлористом метилене (концентрация 8,9 г на 100 мл растворителя). Верхней границы в течение 3-х часов не зафиксировано, а кинетика декантации нижнего (видимого) слоя осадка составила: 60% за 8 мин, 80% за 40 мин, 90% за 90 мин, 95% за 170 мин.
Сравнение времен полного оседания индивидуального (не модифицированного) гидроксиапатита, взятого без каких либо добавок (3-4 мин, хлористый метилен), и подвергнутого модификации полимером (см. выше) показывает существенное их различие, что свидетельствует в пользу достаточно эффективного образования связей между гидроксиапатитом и полилактидом при их совместном «сухом» диспергировании.
Содержание не осаждаемой путем естественной декантации фракции составило 65,5%, что заметно выше исходного содержания введенного полимера (39,1%).
Отделение осаждаемой части первой фракции с помощью центрифуги с 2-х кратной промежуточной ее промывкой растворителем показали ее содержание 48,5%, что существенно ниже содержания первоначально введенного гидроксиапатита (60,9%).
Приведенные различия в содержании осаждаемой и не осаждаемой фракций, определяемых до и после модификации, доказывают возникновение достаточно больших количеств привитого (органоминерального) (со)полимера под действием интенсивного механического воздействия на исходные компоненты получаемого композита.
Содержание полимерной составляющей во второй фракции ситового разделения образца оказалось существенно более высоким и трудно определяемым из-за очень высокой вязкости раствора, получаемого в хлорметилене. Ориентировочное содержание осаждаемой части составило 32,3%.
Приводится сравнительный пример, доказывающий эффективность выбранных условий для совместного диспергирования компонент. С целью получения равномерно диспергируемого по полимеру исходного материала произвольной формы (гранул, кусочков, пленок и т.д.) в ходе его диспергирования предложенным «сухим» методом произведено предварительное усреднение (смешение) компонент через растворитель. Показано, что в этом случае характер диспергирования существенно отличается от того, когда компоненты берутся порознь. Получаемые дисперсии не имеют порошкообразного вида, бесформенны, а изнашивание таких предварительно подготовленных композитов происходит с большим трудом. Приведенный сравнительный пример четко указывает на то, что в выбранном варианте важен эффект абразивного (шлифующего) воздействия порошка фосфата на изнашиваемый полимер (твердость апатитов по минералогической шкале равна 5). При этом механохимические реакции между ними становятся более результативными.
Можно констатировать, что прививка полилактида на порошок гидроксиапатита, также как и прививка гидроксиапатита на полилактид, несомненно имеет место при их совместном «сухом» диспергировании. Соотношение указанных реакций, выражающееся в появлении осаждаемых и не осаждаемых в растворителе фракций при экстрактивном их разделении, может быть очень широким и, в конечном итоге, определять размер вовлекаемых в композит частиц.
Сделанный вывод подтверждается методами ТГА и ИК-спектроскопии.
На фиг. 2 представлены термогравиметрические кривые фракций модификата, полученного «сухим» совместным диспергированием гидроксиапатита и промышленно производимого полилактида. Модификат с помощью сита разделен на различающиеся по размерам частиц фракции, из которых анализированы только первая (образец 107.1 на фиг. 2) и вторая (образец 107.2 на фиг. 2), т.к. третья представляла собой остаток чистого исходного полимера.
Из фиг. 2 видно, что содержание минеральной составляющей в первой, самой мелкой, фракции, отделенной с помощью сита, равно 69,5% (черная кривая 1, 107.1), что удовлетворительно совпадает с исходным содержанием, заданным при составлении смеси и поправленным с учетом не полного расхода полимера при диспергировании (67%). Содержание минеральной (фосфатной) составляющей во второй фракции (красная кривая 2, образец 107.2, фиг. 2) оказалось равным 20%, что согласуется с экстрактивным разделением, показавшим 32,3%, но которое, в отличие от данного метода, дополняется еще и привитым полимерным фрагментом (формальное его содержание 11,3%).
Более детальное определение фрагментарного состава полученного модификата производилось комбинированием предварительного экстрактивного разделения с последующим методом ТГА. Первая фракция, подвергнутая 5-ти кратному чередованию промывок и центрифугирования, показала резкое увеличение содержания фосфата до 89% (фиолетовая кривая 3, образец 107.1.1), в то время как содержание не осаждаемой фракции увеличилась всего до 25% (серая кривая 4, образец 107.1.2). Для сравнения приведена кривая образца, подвергнутого однократной промывке (голубая кривая 5, образец 107.1. otm). Заниженное содержание нелетучей составляющей в не осаждаемой экстракцией части можно отнести за счет повышенной летучести привитых частиц совместно с разлагаемыми органическими продуктами в ходе термического анализа вследствие достижения ими наноразмерного уровня дисперсности, которое может в значительной мере искажать наблюдаемый результат.
ИК-спектральный анализ исследованных фракций, полученных ситовым и экстракционным разделением модификата, представлен на фиг. 3 и фиг. 4.
На фиг. 3 представлены ИК-спектры первой фракции, полученной после просеивания модификата на сите 0,25 мм (красная кривая 1), той же фракции, подвергнутой пятикратной промывке хлористым метиленом (зеленая кривая 2) и исходного гидроксиапатита (синяя кривая 3).
В сравнении со спектром исходного гидроксиапатита в ИК-спектрах просеянного модифицированного и отмытого продуктов наблюдаются новые полосы поглощения с максимумами в области 1744 см-1 и 1593 см-1. Полоса в области 1744 см-1 может быть отнесена к колебаниям карбонильной группы полилактида или его фрагмента, привитого на гидроксиапатит. Полоса при 1593 см-1 может быть отнесена к колебаниям небольшого количества воды, адсорбированной на поверхности гидроксиапатита [Hong Z., Qiu X., Sun J., Deng M., Chen X., Jing X. // Grafting polymerization of L-lactide on the surface of hydroxyapatite nano-cristals // Polymer 2004; v. 45, p. 6699-6706]. Важно подчеркнуть, что ИК-спектр полученного отмытого продукта (зеленая кривая 2) практически совпадает со спектром продукта, полученного после просеивания на первой стадии, что свидетельствует о том, что в просеянном продукте практически присутствует только исходный и модифицированный гидроксиапатит с привитыми фрагментами полилактида. В случае присутствия не привитого полилактида, интенсивность полосы поглощения карбонильной группы полилактида должна была бы существенно снизиться из-за его растворения и удаления в процессе отмывки.
На фиг. 4 представлен ИК-спектр не осаждаемой в центрифуге фракции, выделенной после обработки гидроксиапатита, модифицированного высокомолекулярным полилактидом, хлористым метиленом (фиолетовая кривая 1). На этом же рисунке приведен ИК-спектр исходного высокомолекулярного полилактида (красная кривая 2). На присутствие в спектре не осаждаемой в центрифуге фракции гидроксиапатита однозначно указывает наличие в ИК-спектре полос поглощения при 563, 602 и 1032 см-1, обязанных различным колебаниям фосфатных групп гидроксиапатита. Полученный результат является доказательством прививки полилактида к гидроксиапатиту, что и приводит к вовлечению гидроксиапатита в среду растворителя полимера. Следует подчеркнуть, что индивидуальный гидроксиапатит в хлористом метилене не растворяется, а при центрифугировании его дисперсии в метилендихлориде быстро и количественно осаждается.
По результатам данного примера можно заключить, что возможность совместного диспергирования порошкообразного гидроксиапатита и гранулированного промышленного полилактида, несмотря на его высокие исходные физико-механические характеристики, вполне осуществима и сопровождается эффективным взаимодействием наполнителя и полимерной матрицы. Процесс осуществляется «сухим» методом и приводит к достаточно высокой степени связанности компонент. Предложенный метод не имеет аналогов в литературе и является патентночистым.
Пример 2
Следующий пример «сухого» совместного диспергирования полилактида и гидроксиапатита демонстрирует возможность получения модифицированного наполнителя при малом исходном содержании модификатора (полимера).
В лопастной высокооборотный диспергатор загружают 0,4305 г гранулированного высокомолекулярного полилактида. Форма гранул круглая, диаметр 1,2-1,4 мм. Туда же загружают 4,755 г гидроксиапатита. Весовой состав исходной смеси 8,3% полимера и 91,7% апатита. Диспергирование проводят в один прием 4 раза продолжительностью по 2 мин каждое с перерывами на охлаждение смеси. Полученный порошок просеивают на сите с размером ячейки 0,25 мм. Получают три фракции: первая, прошедшая сквозь сито, составила 93,2%, вторая, не прошедшая, 4,8% и третья, остатки гранул, 1,9%. Конверсия полимера в композит составила 55,6%. Насыпной вес полученного порошка 0,457 г/см3 (насыпной вес исходного гидроксиапатита 0,397 г/см3). Содержание оседающей в хлорметилене (отмытой) фракции 79,6%, Содержание не оседающей фракции, увеличилось по сравнению с первоначальным содержанием полимера (8,3%) до 20,4%, демонстрируя тем самым наличие привитой фракции, несмотря на низкое исходное содержание полимера.
Пример 3
Пример иллюстрирует возможность «сухой» конденсации молочной кислоты на гидроксиапатите с образованием привитого органоминерального (блок)-сополимера. Отличительной особенностью данного примера является тщательно дозированная подача кислоты, предотвращающая слипание (агломерацию) частиц наполнителя.
В стакан емкостью 200 мл, помещенный на электроплитку, засыпают 18,737 г порошкообразного гидроксиапатита. Стакан снабжен стеклянной мешалкой, вращающейся со скоростью 120 об/мин и термометром, определяющим температуру порошка. Включают электрообогрев, мешалку и, по достижении температуры 120-130°С, постепенно, небольшими каплями, порционно, вводят d,l-молочную кислоту (МК, концентрация 85%). Вес каждой вводимой порции кислоты в начале процесса конденсации не превышает соотношение МК/ГА=0,06-0,08, которое можно увеличивать к концу процесса до 0,1-0,2. Время между порциями ввода кислоты составляет не менее 15 мин. В промежутках фиксируют уменьшение веса модификата за счет свободной убыли имеющейся первоначально и образующейся при конденсации воды. В процессе конденсации отмечается хорошее распределение жидкой фазы кислоты по порошку, отсутствие его слипаемости или комкования и достаточно быстрое уменьшение веса содержимого за счет большой поверхности испарения. По окончании ввода кислоты (общее ее содержание составило 10,949 г) смесь продолжают нагревать 1 час при температуре 100-120°С и по охлаждении фиксируют количество связанной кислоты, которое составило 25,2% (в расчете на полученный продукт), что соответствует 74,8%-ой степени наполнения гидроксиапатитом полученного модификата.
Ориентировочное определение степени прививки производилось на основании результатов следующих операций:
- растворение навески модификата в хлористом метилене (концентрация 10 г на 100 мл),
- естественная седиментация полученной дисперсии при комнатной температуре в течение 1 часа,
- разделение оседающей и не оседающей фракций путем отбора полученного раствора,
- сушка полученных фракций до постоянного веса и определение их содержания во взятой пробе.
В данном примере содержание не оседающей фракции составило 50,5% от веса взятой навески. Для сравнения гидроксиапатит, не подвергнутый модификации в соответствии с приведенным примером, полностью оседает в хлористом метилене в аналогичных условиях за 3 мин, не оставляя никаких следов растворенного вещества в растворителе.
Следует отметить, что принятый в данном техническом решении метод разделения привитой (не оседающей в естественных условиях за 1 час) фракции от формально не привитой (оседающей за то же время) является ориентировочным, т.к. полимер, фиксированный на гидроксиапатите, распределяется как на мелких (субмикронных или наноразмерных) его частицах (не оседающая фракция), так и на их агрегатах (оседающая фракция с размером агрегированных частиц от микрона и более), что создает широкое распределение весовых соотношений частица/полимер, которое и определяет их способность к осаждению в тех или иных условиях. Точное определение степени прививки полимера на гидроксиапатит представляет известные трудности, т.к. предполагает отдельное определение неорганической составляющей в каждой фракции (например, методом ТГА). Естественная седиментация и принудительное осаждение с помощью центрифуги производят лишь некоторое разграничение в размерах разделяемых частиц, сохраняя наличие привитого полимера в обеих указанных фракциях.
Пример 4
Пример иллюстрирует модификацию гидроксиапатита конденсацией на нем молочной кислоты «сухим» методом с промежуточной его активацией в диспергаторе.
Условия эксперимента (первая часть):
- реакция проводилась в стеклянном стакане емкостью 150 мл.
- D,L-молочная кислота использована в виде 85%-ого водного раствора.
- гидроксиапатит взят в виде порошка, который предварительно никак не обрабатывался и не подвергался предварительной сушке.
- способ введения «молочная кислота в гидроксиапатит» осуществлялся порционно, весовое отношение молочная кислота/гидроксиапатит в каждой порции, с целью предотвращения слипания и комкования частиц, не превышал величины 0,05, причем первые порции были в 2-3 раза меньше последующих. После каждого ввода кислоты производилось немедленное и осторожное ручное перемешивание смеси с целью равномерного распределения кислоты.
- промежуток времени между каждым введением молочной кислоты не менее 30 мин.
- температурный режим конденсации 120-130°С.
- произведено 4 ввода молочной кислоты, после второго и четвертого ввода осуществлен размол порошкообразного конденсата в диспергаторе, время каждого диспегирования 3-4 мин (число оборотов лопастей диспергатора 5000 об/мин).
- отвод продуктов реакции (преимущественно воды) осуществлялся естественным испарением, без вакуумирования или принудительной продувки газом.
Полученный материальный баланс:
- взято 28,4195 г гидроксиапатита
- введено 6,976 г молочной кислоты (суммарно)
- получено 32,6 порошка модифицированной смеси с формальным содержанием наполнителя 87,16% (12,84% органической части)
Анализ модификата на наличие привитой фракции экстракцией растворителем:
- проба порошка модификата дважды обработана хлористым метиленом, без промедления центрифугирована (число оборотов ротора 3000-4000 об/мин) и быстро высушена до постоянного веса. Растворимая часть отделена, суммирована и также высушена. Зафиксировано, что из введенной органической части (12,84%) связано наполнителем 4,09%, что составляет 31,85% от введенного модификатора.
Через несколько дней проведена 2-х кратная повторная промывка исходного порошка в хлористом метилене и отделение с помощью центрифуги растворимой фракции, причем выдержка его в растворителе увеличена до 2-х часов. Повторное определение не осаждаемой центрифугой фракции показало 8,75%-е ее содержание. Отнесенное к введенной органической (полимерной) фракции (12,84%), это составило уже 68%-ое связывание полимера наполнителем. Обнаруженный эффект свидетельствует или о продолжении реакции прививки в твердой фазе при хранении смеси при комнатной температуре или о влиянии растворителя на возможность взаимодействия наполнителя с полимером.
О высокой степени прививки в этом случае свидетельствует и седиментационная кривая выдержанного образца в хлористом метилене. За 10 мин оседает 45% взвеси (определено по верхней границе раздела), за 25 мин 60%, за 60 мин 74%, за 100 мин 77%.
Анализ методом ТГА (фиг. 5).
Из полученной зависимости следует, что определенное этим методом содержание неорганической составляющей (черная кривая 1, 81,5%) меньше, чем это определено методом экстракции (зеленая кривая 2, 87,16%). С поправкой на содержание летучих (воды) в исходном гидроксиапатите (1,5%, красная кривая 3) это не ликвидирует получаемое расхождение, которое по всей вероятности связано с частичным уносом нанофракции фосфата потоком испаряемой при высокой температуре органической составляющей в ходе термогравиметрического анализа.
ИК-спектральный анализ (фиг. 6)
В сравнении со спектром исходного гидроксиапатита (коричневая кривая 1) в ИК-спектрах конденсатов молочной кислоты, полученных «сухой» конденсацией на гидроксиапатите, появляются новые полосы поглощения при 1582 см-1 и 1313 см-1 (синяя кривая 2). Указанные полосы подобны полосам поглощения при 1583 см-1 и 1316 см-1 в спектре DL-лактата кальция, которые приписываются колебаниям группы -СОО- лактата. Наличие данных полос свидетельствует об образовании лактата кальция на поверхности гидроксиапатита. Кроме того, наблюдается возникновение новой достаточно широкой полосы поглощения с максимумом при 1737 см-1, предполагающей наличие сложноэфирных связей, которые могут возникать в результате реакции поликонденсации молочной кислоты на поверхности гидроксиапатита, приводящей на первой стадии к образованию олигомеров молочной кислоты, привитых к поверхности гидроксиапатита.
Условия эксперимента (вторая часть):
- в качестве объекта для проведения дальнейшей модификации образца взят порошок, полученный в первой части примера.
- дальнейшее порционное введение молочной кислоты осуществлено семью порциями с интервалом 15-20 мин.
- отношение веса порционно вводимой кислоты к гидроксиапатиту, также как и в первой части примера, не превышало 0,05.
- температурный режим обогрева порошкообразной смеси аналогичен первой части, 120-130°С.
- дополнительное диспергирование в процессе доконденсации молочной кислоты не производилось.
Полученный материальный баланс:
- взято 30,09 г порошка, полученного в первой части,
- суммарно введено 6,59 г молочной кислоты,
- получено 32,84 г модификата.
Анализ модификата экстрактивным методом.
- двойная отмывка полученного модификата хлористым метиленом, центрифугирование его, отделение и сушка неосаждаемой фракции показали увеличение содержания в ней органической части с 12,84% до 17,07%, демонстрируя тем самым, что не все вакантные места на гидроксиапатите, несмотря на интенсивное промежуточное диспергирование, были израсходованы для прививки в ходе первой части конденсации.
ИК-спектральный анализ (фиг.6).
Доконденсация молочной кислоты в присутствии модифицированного на первой стадии гидроксиапатита, приводит к сужению полосы и смещению ее максимума с 1737 см-1 до 1751 см-1 (вторая стадия, красная кривая 3). Указанное смещение полосы поглощения карбонильной группы может быть связано с увеличением молекулярной массы продукта поликонденсации. Отметим, что полоса поглощения карбонильной группы в высокомолекулярной полимолочной кислоте (или полилактиде) находится при 1754 см-1. Следует подчеркнуть, что полосы поглощения полимера молочной кислоты наблюдаются после пятикратной отмывки образца в метиленхлориде с последующим центрифугированием и окончательной сушки, в процессе которых не привитый полимер растворяется и удаляется. Таким образом, наличие полосы 1751 см-1 свидетельствует о прививке полимера молочной кислоты к поверхности гидроксиапатита. Об этом же свидетельствует наличие в ИК-спектре полос при 2889 см-1, 2939 см-1 и 2989 см-1. Эти полосы обусловлены колебаниями связи С-Н в метальных и метановых группах полимера молочной кислоты, привитого на гидроксиапатит.
Приводим некоторые характерные (качественные) явления, наблюдаемые по ходу проводимой реакции:
- в процессе порционного введения молочной кислоты в указанных условиях какого либо слипания частиц, комкования смеси, прилипания ее к стенкам стеклянного стакана не отмечалось. Ориентировочная толщина слоя кислоты при условии равномерного ее распределения по поверхности порошка, удельной поверхности фосфата не менее 20 м2/г и выбранном режиме подачи кислоты не может превышать микронного уровня.
- даже при осторожном перемешивании горячего порошка наблюдалось образование тонкого пылевидного (аэрозольного типа) облачка, качественно свидетельствующего о малом размере частиц. Указанное явление особенно выражено при диспергировании порошка, в связи с чем, выгрузка его из диспергатора производилась после полного остывания смеси.
- ориентировочный подсчет скорости конденсации молочной кислоты (по убыли веса) показал, что она существенно выше по сравнению с обычными условиями (в массе конденсата). Обнаруженное явление несомненно связано с большой поверхностью испарения продуктов реакции (преимущественно воды), каталитической активностью гидроксиапатита, а также промежуточной его активацией. Зафиксировано также, что конденсация молочной кислоты в «сухом» варианте в принципе может протекать при температуре, сниженной до комнатной.
- отмечалось, что первые порции кислоты, вводимые в гидроксиапатит, несколько замедленнее смачивали порошок по сравнению с последующими. После 2-ой-3-ей порции новая порция кислоты распределялась в порошке (смачивала его) практически в течение нескольких секунд.
Пример 5
Пример иллюстрирует возможность получения композита с помощью «обращенного» варианта, в соответствии с которым гидроксиапатит вводится небольшими порциями в расплав полимолочной кислоты.
Целесообразность такого, сравнительного с «сухим», расплавного варианта обуславливается потенциальной возможностью уменьшения отрицательного влияния высокой склонности частиц гидроксиапатита к агрегации друг с другом, повышению их каталитической активности и, как следствие, увеличению степени прививки с полимолочной кислотой. В этом случае наполнитель, введенный в кислоту малыми порциями будет успевать дезагрегироваться прежде, чем будет окклюдирован конденсатом, затрудняющим более дисперсное его распределение.
Условия получения композита
В стеклянный стакан емкостью 50 мл загружается 19,895 г d,1-молочной кислоты (85%-ый раствор). Кислота нагревается до температуры 120°С и выдерживается при данной температуры до 15%-ой потери массы. Гидроксиапатит вводится далее малыми порциями, не превышающими 10% от взятой первоначально кислоты, при постоянном перемешивании (20-30 об/мин) с промежутками времени между вводами гидроксиапатита 20-30 мин, общее число вводов - 11. Зафиксировано, что первые порции (1-5) гидроксиапатита распределяются до визуально гомогенного состояния, далее появляется небольшая мелкодисперсная взвесь, которая не исчезает до конца процесса. По окончании ввода гидроксиапатита проведена доводочная конденсация кислоты при температуре 120°С в течение 4 часов. Итоговое содержание гидроксиапатита в конденсате 4,7%.
Экстрактивное разделение фракций с помощью хлористого метилена (естественное отстаивание взвеси в течение нескольких дней) показало практически полный переход наполнителя в не оседающую фракцию, что свидетельствует о высокой степени прививки полученного конденсата на наполнитель.
ИК-спектр композита также подтверждает результат экстрактивного разделения фракций.
На фиг. 7 представлены ИК-спектры конденсата молочной кислоты, полученного при порционном введении гидроксиапатита в кислоту (синяя кривая 1), и конденсата, полученного термически в отсутствие наполнителя (красная кривая 2). Фиксируется появление новых полос с максимумами при 1600 см-1 и 1310 см-1, которые были отнесены к колебаниям группы -СОО- лактата. Наличие данных полос свидетельствует об образовании лактата кальция на поверхности гидроксиапатита и о прививке полимера молочной кислоты через него к гидроксиапатиту.
Пример 6
Пример иллюстрирует возможность «сухой» модификации гидроксиапатита L-лактидом с помощью высокоскоростного диспергатора.
Данный вариант модификации предпочтителен тем, что предоставляет возможность одновременного (низкотемпературного) осуществления и активации гидроксиапатита и инициирования полимеризации с участием образующихся на гидроксиапатите активных центров. Инициирующая активность не активированного гидроксиапатита в полимеризации лактида известна, однако для этого требуется высокая температура (160°С) и достаточно продолжительное время (2-3 часа).
Последовательность модификационных операций осуществлялась следующим образом. В диспергатор загружали 4,5485 г порошкообразного гидроксиапатита и 1,5845 г лактида, содержащего примерно 10-3 моль карбоксильных групп на моль мономера, исходное содержание наполнителя составляло 74,16%. Диспергирование проводилось в один прием с небольшими перерывами на охлаждение смеси. Общее время активного воздействия - 6 мин. Отмечается, что диспергированная смесь становится очень сухой, тонкой и сильно пылящей при малейшем движении воздуха. Насыпной вес полученного порошка снижен до 0,184 г/см3.
Испытание полученной смеси в хлорметилене (концентрация дисперсии 4,25 г/100 мл) показало, что при естественной седиментации за 2 часа оседает не более 60% от веса введенного (исходного) порошка, а остальная часть подвешена в объеме без дополнительных признаков последующего оседания. Применение центрифуги для однократного разделения увеличивает содержание осадка до 70%, а чередование нескольких отмывок осаждаемого порошка с центрифугированием доводит его содержание до 85% от исходного. Это, однако, не означает, что содержание привитой фракции при естественном выстаивании определяется завышенным. В зависимости от типа осаждения (естественного или принудительного) происходит перераспределение фракций с различным соотношением минеральной и органической составляющих между осаждаемой и неосаждаемой частями композита.
Часть подвергнутой диспергированию и модификации смеси лактида и гидроксиапатита прогревается до температуры 160°С в течение получаса, констатируется некоторое спекание частиц и увеличение насыпного веса до 0,3236 г/см3.
ИК-спектр модификата (фиг. 8)
Для образца, полученного «сухой» механически активированной полимеризацией лактида под действием индивидуального гидроксиапатита (красная кривая 1) с последующей его термообработкой при 150°С, пятикратной отмывкой хлористым метиленом и сушкой, зафиксированы новые полосы при 1743 см-1 и 1596 см-1, а также в области 2890-2990 см-1 (бирюзовая кривая 2). Полоса при 1743 см-1 и полосы в области 2890-2990 см-1 могут быть отнесены, соответственно, к колебаниям С=0 группы полилактида и колебаниям связи С-Н в метальных и метановых группах полилактида. Наличие этих полос свидетельствует о прививке полилактида к гидроксиапатиту. Широкая полоса с максимумом при 1596 см-1 может представлять собой суперпозицию полос, обязанных следовым количествам воды в гидроксиапатите и колебаниям группы -СОО-, возникающей в результате присутствия карбоксильных групп, в форме молочной кислоты или ее димера в исходном лактиде.
На основании полученных данных можно сделать заключение, что прививочные реакции полилактида на гидроксиапатит имеют место не только в случае конденсационных превращений (см. пример 3 и 4), но и полимеризационных. Обращает на себя внимание, что указанные превращения происходят достаточно быстро благодаря активирующему механическому воздействию, оказываемому при совместном «сухом» твердофазном диспергировании гидроксиапатита и лактида.
Пример 7
Пример иллюстрирует возможность получения биоразлагаемого композита на основе гидроксиапатита и оксикислот с повышенным сбалансированным или несбалансировнным содержанием гидроксильных и карбоксильных функциональных групп (винная и лимонная кислоты соответственно). Для сравнения приведены примеры с применением «сухого» и «мокрого» методов синтеза.
«Мокрая» модификация гидроксиапатита с применением винной кислоты.
Гидроксиапатит в количестве 0,6733 г диспергируют в 50 мл воды и к полученному молоку малыми порциями, не превышающими 10% от взятого гидроксиапатита, при постоянном перемешивании добавляют винную кислоту. По ходу процесса фиксируют рН раствора, который колеблется от 6,0 до 5,0 между вводами кислоты. Промежутки времени между очередными порциями составляют 30 мин, которые необходимы для того, чтобы рН не выходила за указанный выше интервал. По мере добавления винной кислоты меняется вид дисперсии гидроксиапатита, она становится более «тяжелой» и быстрее оседающей. Подвешенная фракция отсутствует. Общее количество введенной кислоты 0,684 г. По окончании ввода дисперсию отстаивают, полученный осадок отделяют от раствора и сушат до постоянного веса.
Материальный баланс:
Взято 0,6733 г гидроксиапатита (49,6% в исходной смеси)
Введено 0,684 г винной кислоты (мезо-форма) (50,4%)
Получено 1,127 г высушенного осадка (90,4% от суммы введенных компонент)
Выход полимера винной кислоты 0,4387 г
Степень превращения винной кислоты 64,13%
Содержание гидроксиапатита в осадке, определенное экстракцией, 60,0% (при условии отсутствия его растворимой в воде формы).
По данным ТГА содержание гидроксиапатита (точнее не удаляемого при высоких температурах остатка) составило 52% (см. фиг. 9, красная кривая 1, чистый гидроксиапатит черная кривая 2)). Расхождение по содержанию минеральной составляющей, определенное методом экстракции и ТГА, несомненно связано со спецификой образующегося полимерного покрытия, которая обусловлена высокой склонностью винной кислоты к образованию полимерных сеток.
Возможно, что этой же спецификой объясняется и весьма неожиданный вид полученной кривой ТГА, который характеризуется не плавной потерей веса с увеличением температуры, а по крайней мере тремя, ярко выраженными изломами в интервалах температур 150-225°С, 300-350°С, и 450-525°С, причем последний из них выходит за пределы обычно наблюдаемых температур окончательного распада органической составляющей, характерных для полиэфиров. Указанный феномен может быть объяснен тем, что в данном варианте распад проходит через стадию образования элементарного углерода, дальнейшее окисление которого, образование и удаление газообразных продуктов (СО, СO2), происходит при более высоких температурах, чем это характерно для обычного (несшитого) полимерного покрытия.
Испытание полученного высушенного осадка в хлористом метилене показало его полную оседаемость и отсутствие каких либо растворимых фракций.
Полученные результаты позволяют с большой долей вероятности считать, что в принятых условиях наблюдается количественное образование сшитого полимера винной кислоты и отложение его на частицах гидроксиапатита.
«Сухая» модификация гидроксиапатита с применением винной кислоты.
В отличие от «мокрого» (водного) способа модификации, который требует порционного введения винной кислоты, «сухой» способ вполне допускает одноразовый ввод модификатора, причем в достаточно больших количествах. Указанная особенность обусловлена твердым (кристаллическим) состоянием винной кислоты при комнатной температуре, что предпочтительно для совместного диспергирования 2-х порошков. Следует также отметить, что удвоенное количество функциональных групп по сравнению с молочной кислотой создает большее многообразие возможных структур при ее конденсации.
Типовой пример «сухой» модификации гидроксиапатита винной кислотой состоял в следующем:
- взято 1,70 г мезо-винной кислоты (24,3% в смеси) и 5,30 г сухого гидроксиапатита (75,7%),
- диспергирование проведено постадийно, 4 раза по одной минуте, с интервалом 10 мин,
- определен насыпной вес полученного порошка, который составил 0,5543 г/см3,
- естественная оседаемость в воде (концентрация раствора 10 г на 100 мл), характеризовалась следующими данными: за 2 мин осело 26,6%, 4 мин - 46,7%, 8 мин - 66,7%, 18 мин - 73,7%, 40 мин - 91,1%, сутки - 100%,
- центрифугирование быстро и полностью осаждает модификат в воде, содержание растворимой части в растворе - незначительно,
- рН жидкой части - нейтрально и показывает полное отсутствие кислых продуктов в ней, это позволяет считать, что винная кислота практически полностью связалась гидроксиапатитом и содержание его в полученном модификате близко к исходной смеси (75,7%).
Определение нелетучего остатка методом ТГА (фиг. 9, черная кривая 2), как и в предыдущем варианте (мокром), показала завышенное его содержание (88%) по сравнению с исходной дозировкой (75,7%). Объяснение этого расхождения несомненно связано со спецификой полимерного (сшитого) покрытия на гидроксиапатите, создаваемого на основе винной кислоты.
Общий вывод по данному «сухому» варианту модификации состоит в том, что по сравнению с другими вышеприведенными он отличается легкостью, наличием сшитой полимерной сетки и перспективен для комбинирования с другими оксикислотами, прежде всего с молочной, наличие которой позволит регулировать степень сшиваемости возникающего полимерного покрытия.
«Мокрая» модификация гидроксиапатита лимонной кислотой.
Предыдущими примерами, проведенными с винной кислотой, было показано, что образование полимера на поверхности гидроксиапатита вполне возможно и в водной среде. Наиболее вероятная реакция полимерообразования - поликонденсационного типа, за счет гидроксильных и карбоксильных групп оксикислоты. Полное отсутствие такого взаимодействия в гомогенных водных растворах винной кислоты указывает на то, что необходимым условием протекания этой реакции является наличие сорбционно активной поверхности, которую собственно и предоставляет гидроксиапатит. Первые сложноэфирные звенья, образовавшиеся на поверхности, выводятся из равновесия и по существу превращают равновесную реакцию этерификации в гомогенных условиях, в неравновесную - в гетерогенных. С целью доказательства того, что указанное явление не является характерным только для винной кислоты были поставлены эксперименты с другой оксикислотой - лимонной.
Типовое испытание проводилось в следующих условиях:
- 1,200 г гидроксиапатита диспергировали в 200 мл воды,
- 2,400 г кристаллической лимонной кислоты вводили мелкими порциями в полученное молоко в течение 10 дней. Вводимые порции не превышали 8-10% от взятого гидроксиапатита. Конечное содержание введенных компонент составляло 33% по гидроксиапатиту и 6% по лимонной кислоте. «Вызревание» осадка, увеличение его объема, происходило при комнатной температуре и периодическом перемешивании. По окончании обозначенного срока, осадок отфильтровывали и сушили до постоянного веса. Выход осадка составил ориентировочно 55-60% от суммы введенных компонент.
Образец модификата, исследованный методом ТГА, показал конечное содержание неорганической части около 60% и наличие 2-х изломов на кривой, первый - низкотемпературный, с температурным интервалом 100-150°С и второй, очень широкий 300-575°С (см. фиг. 10).
Его необычность, также как и в случае с винной кислотой, состоит в том, что потеря веса продолжается при температурах, существенно завышенных по сравнению с обычными полимерами, полученными на основе полилактида или полимолочной кислоты, также в присутствии гидроксиапатита. Однако, возможность к сеткообразованию в случае лимонной кислоты, по сравнению с винной, несомненно ниже и, по-видимому, с более активным вовлечением гидроксиапатитного кальция и карбоксильных групп лимонной кислоты.
ИК-спектр представлен на фиг. 11, из которого видно наличие большого числа новых полос, в области 667-1616 см-1, которые несомненно ответственны за лимонную кислоту, ее олигомеры и более высокомолекулярные продукты поликонденсации, а также интенсивная широкая полоса в области 3000-3600 см-1, обязанная молекулам воды, связанным водородными связями (синяя кривая 2, исходный гидроксиапатит - красная кривая 1).
Однако на приведенном фрагменте данных спектров в диапазоне 500-950 см-1 (Фиг. 12 ИК-спектр поликонденсата лимонной кислоты в диапазоне 520-950 см- 1, полученного «мокрым» способом на гидроксиапатите (синяя кривая 2); индивидуальный гидроксиапатит (красная кривая 1)), четко видно, что в результате поликонденсации наблюдается значительное уменьшение концентрации гидроксильных групп гидроксиапатита, которое выражается в резком снижении интенсивности полосы при 627 см-1, обязанной колебаниям ОН-групп гидроксиапатита (фиг. 12, красная кривая 1). Наблюдаемое уменьшение концентрации гидроксильных групп гидроксиапатита может быть объяснено протеканием реакции поликонденсации с участием карбоксильных групп лимонной кислоты, приводящей к образованию привитого продукта.
«Сухая» модификация гидроксиапатита лимонной кислотой
В лопастной диспергатор загружают 5,4234 г гидроксиапатита и 2,5015 г лимонной кислоты (1-водной). Весовое соотношение компонент 68,44% гидроксиапатита и 31,56% лимонной кислоты. Совместное диспергирование компонент производят в течение 5 мин. Получают мелкодисперсный порошок с насыпным весом 0,425 г/см. Пробу порошка обрабатывают хлористым метиленом и отстаивают в течение 10 мин. Отделяют растворную часть от осадка, и обе фракции высушивают. Содержание растворимой фракции составило 1,95%. Другую пробу диспергируют в воде и фиксируют оседание порошка во времени (по верхней границе). Определение повторяют после выдержки исходного порошка в течение 24 часов. Соответствующие кинетические данные приведены в таблице.
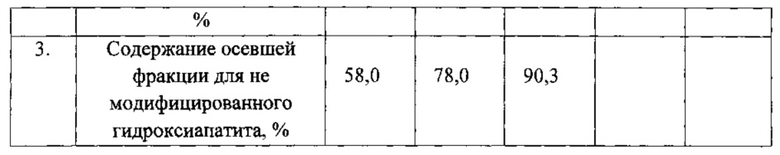
Из приведенных данных следует, что взаимодействие лимонной кислоты с поверхностью гидроксиапатита продолжается после их совместного диспергирования, приводит к связыванию кислоты и повышению веса частиц соответственно.
В предлагаемом способе получения композита для охарактеризования получаемых продуктов использованы следующие методики и измерительные приборы.
Степень прививки полимера на гидроксиапатит определялась фракционированием композита, осуществляемым в растворе хлористого метилена. Разделение на фракции проводили или естественной седиментацией получаемых дисперсий или с применением центрифуги. Определение минеральной составляющей в каждой фракции производили также методом ТГА.
ТГА измерения проводились на приборе фирмы Perkin Elmer модель Pyris 1 TGA. Измерение проводилось в потоке азота 100 мл/мин. Выдержка (выход на режим) при 50°С - 5 мин. Нагрев в диапазоне 50-700°С. Скорость нагрева 10°С/мин.
Доказательство наличия прививки проводили по соответствующим характеристическим полосам поглощения в ИК-спектрах, которые снимали методом нарушенного полного внутреннего отражения (НПВО) на ИК-Фурье спектрометре The Thermo Scientific Nicolet iS5 с приставкой iD5 с кристаллом алмаза. ИК-спектры записывали в диапазоне 4000-500 см-1 с разрешением 4 см-1 при числе сканов 32 при комнатной температуре. Все спектры были подвергнуты ATR коррекции с использованием программного средства OMNIC.
Определение насыпного веса получаемых порошков производили в соответствии с общими правилами, принятыми в ситовом анализе порошков, с точностью в третьем знаке после запятой, путем усреднения не менее трех измерений.
Лабораторный диспергатор. Рабочий объем 260 мл, имел 2-х лопастной, измельчающий, остро заточенный, винт. Корпус выполнен из нержавеющей стали, крышка - из полиметилметакрилата. Число оборотов лопастей 3000-5000 об/мин (зависит от нагрузки при диспергировании). Корпус диспергатора имеет специальные внутренние выступы, которые поворачивают часть потока измельчаемых компонент на определенный угол с целью увеличения числа соударений частиц друг с другом, их взаимного трения и усиления абразивного износа полимера под действием фосфатного наполнителя.
По сравнению с анализированным уровнем техники и выбранным прототипом предлагаемый способ получения биоразлагаемого композита на основе гидроксиапатита и алифатических полиэфиров обеспечивает:
- полное отсутствие каких либо органических растворителей в синтезе и связанной с этим необходимостью их тщательного удаления из композита и последующей регенерации для повторного использования,
- возможность получения при комнатной или сниженной (по сравнению с однотипными реакциями) температуре,
- сниженное время синтеза,
- применение одного и того же полимера и в качестве модификатора и наполнителя полимерной матрицы композита одновременно,
- широкий диапазон степени наполнения,
- малое число стадий.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения органомодифицированного гидроксиапатита | 2019 |
|
RU2703645C1 |
| Способ получения органомодифицированного монтмориллонита (ММТ) | 2018 |
|
RU2704190C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОРИСТЫХ ПОЛИМЕРНЫХ БИОДЕГРАДИРУЕМЫХ ИЗДЕЛИЙ ДЛЯ РЕГЕНЕРАЦИИ КОСТНОЙ ТКАНИ | 2006 |
|
RU2327709C2 |
| Полимерный композит с эффектом памяти формы для 3D-печати медицинских изделий | 2016 |
|
RU2631890C1 |
| БИОСОВМЕСТИМЫЙ БИОРАЗЛАГАЕМЫЙ СКАФФОЛД НА ОСНОВЕ ПОЛИМЕРНОГО КОМПОЗИТА, СОДЕРЖАЩЕГО НАНОЧАСТИЦЫ ГИДРОКСИАПАТИТА | 2019 |
|
RU2756551C2 |
| Биоактивная полимерная нить для осуществления послойной 3D-печати | 2015 |
|
RU2637841C2 |
| СПОСОБ НАНЕСЕНИЯ БИОАКТИВНОГО ПОКРЫТИЯ НА ОСНОВЕ ХИТОЗАНА НА ПОЛИМЕРНЫЕ ПОРИСТЫЕ КОНСТРУКЦИИ | 2015 |
|
RU2600652C1 |
| Биоактивный полимерный пористый каркас | 2016 |
|
RU2665175C2 |
| КОМПОЗИТ ДЛЯ 3D-ПЕЧАТИ МЕДИЦИНСКИХ ИЗДЕЛИЙ | 2018 |
|
RU2679632C1 |
| КОМПОЗИТ ДЛЯ 3D-ПЕЧАТИ МЕДИЦИНСКИХ ИЗДЕЛИЙ | 2018 |
|
RU2679127C1 |
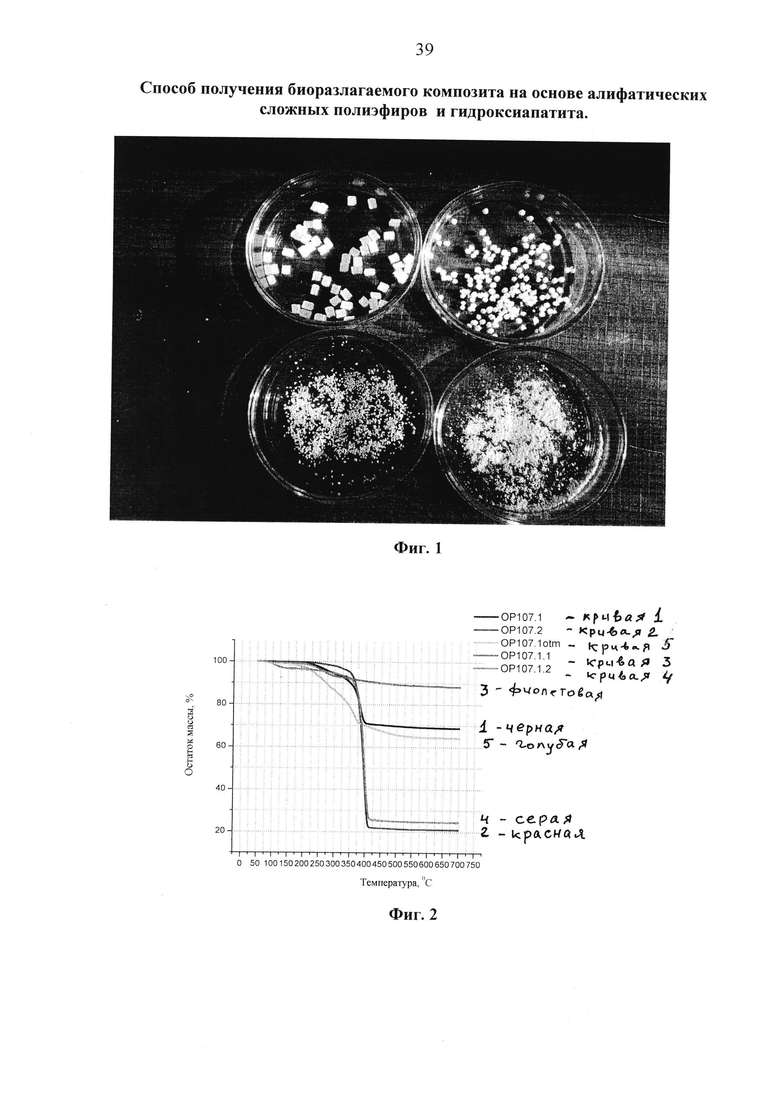
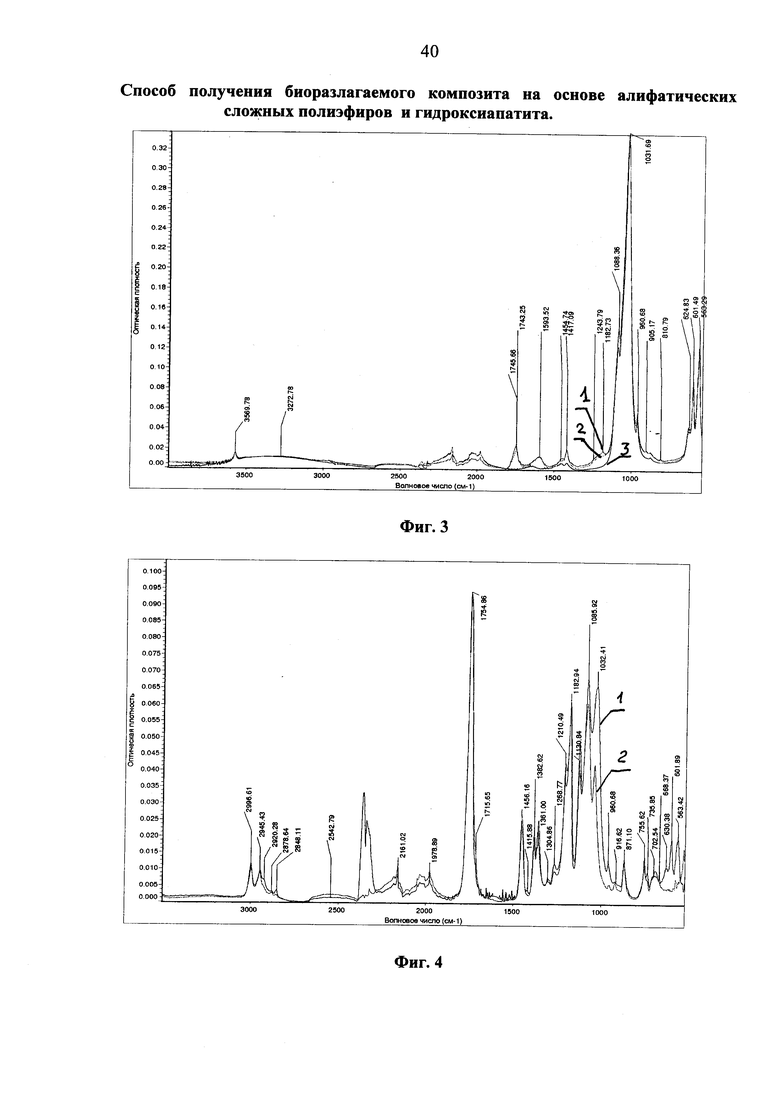
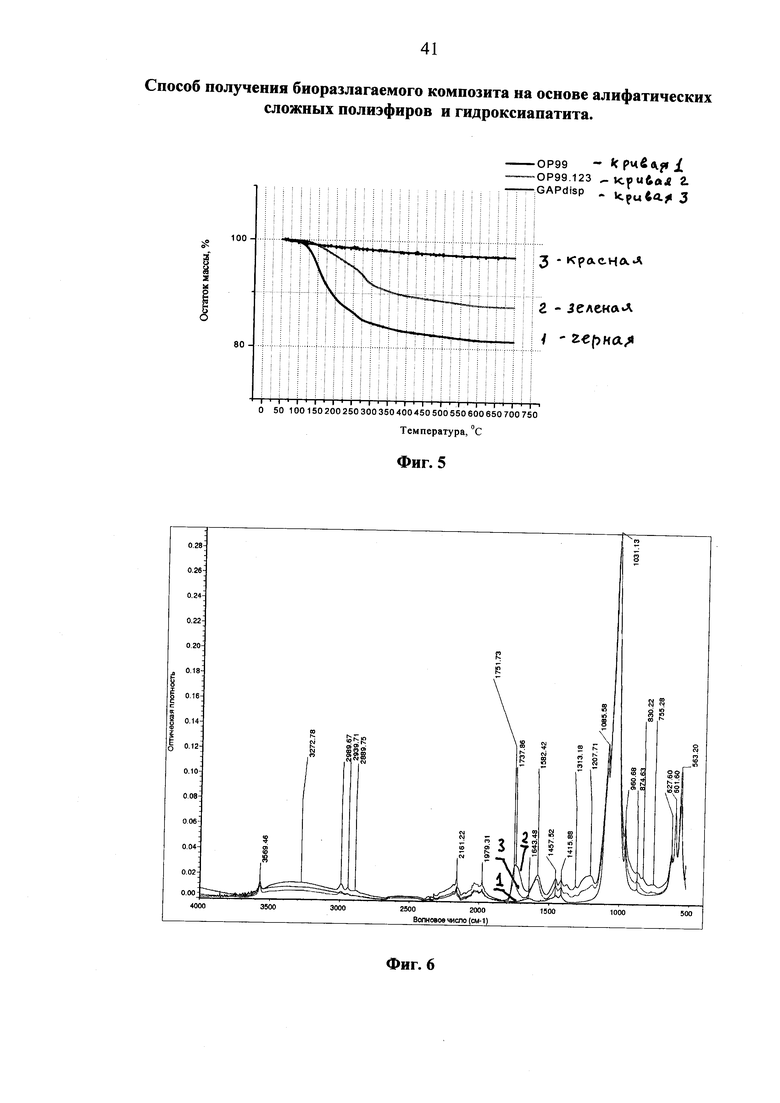
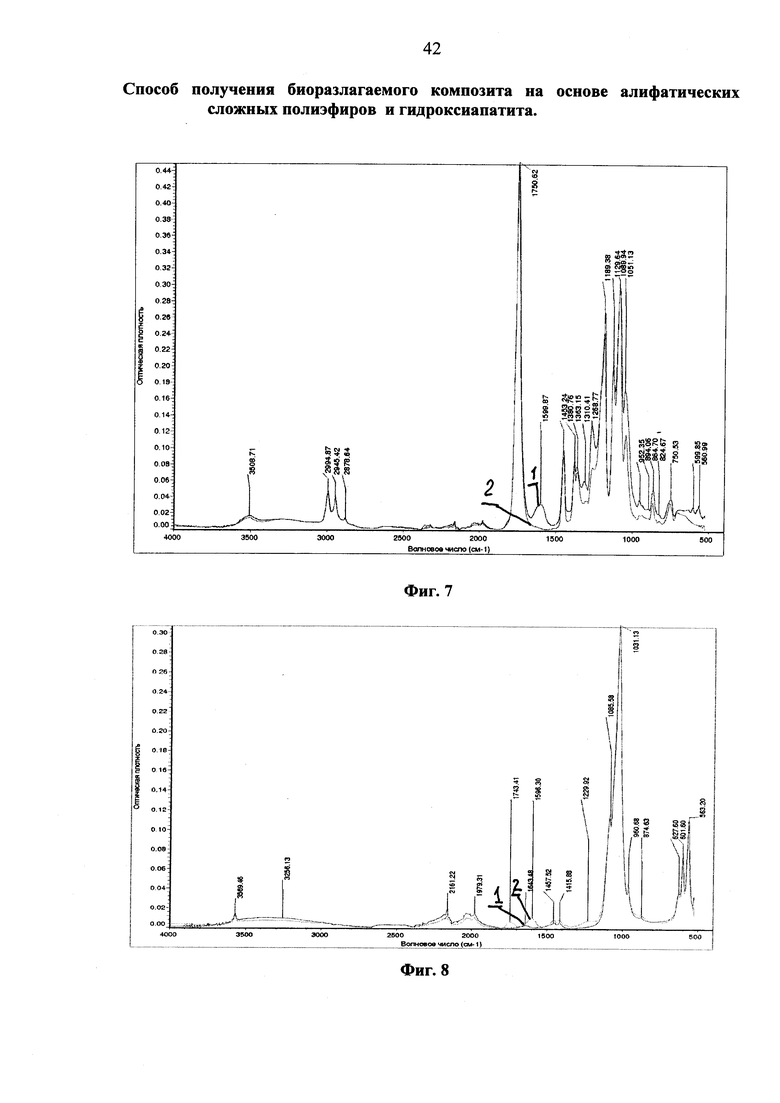
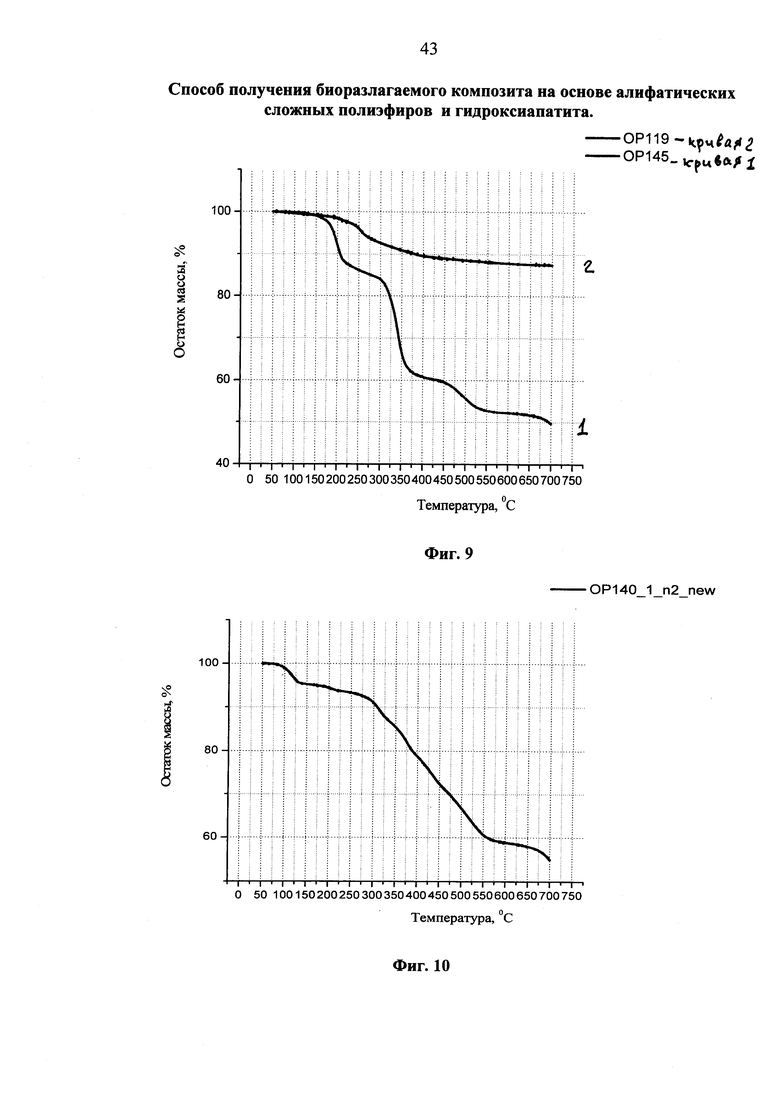
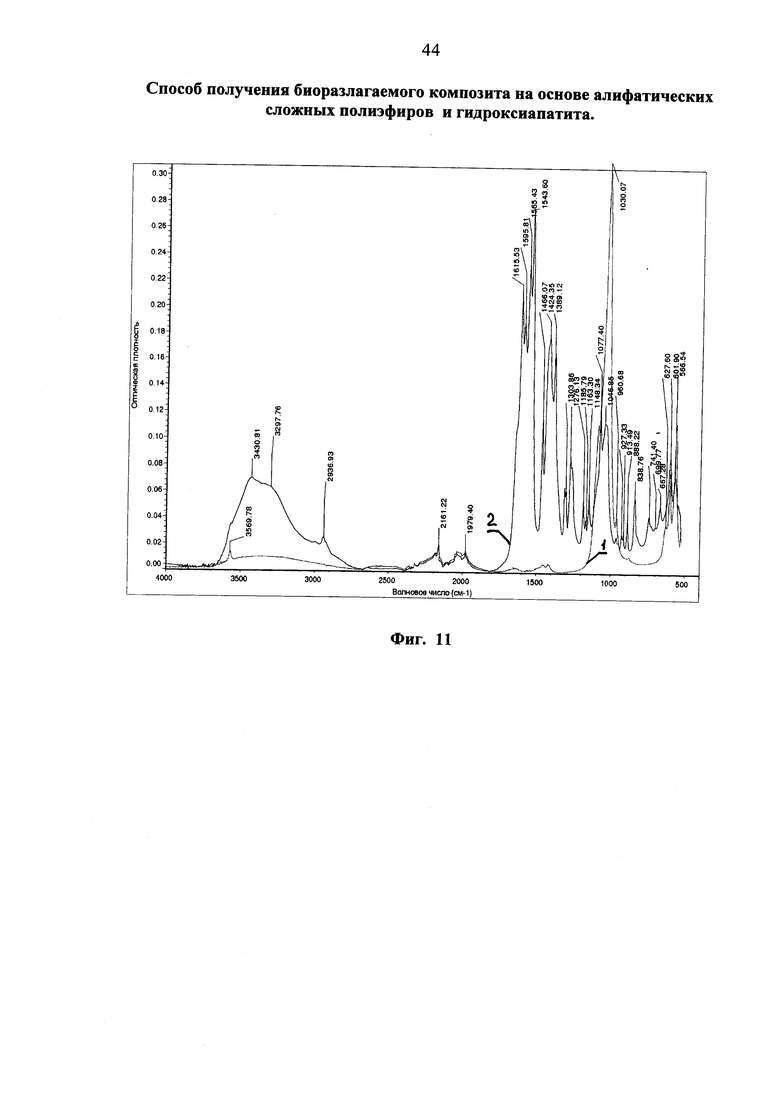
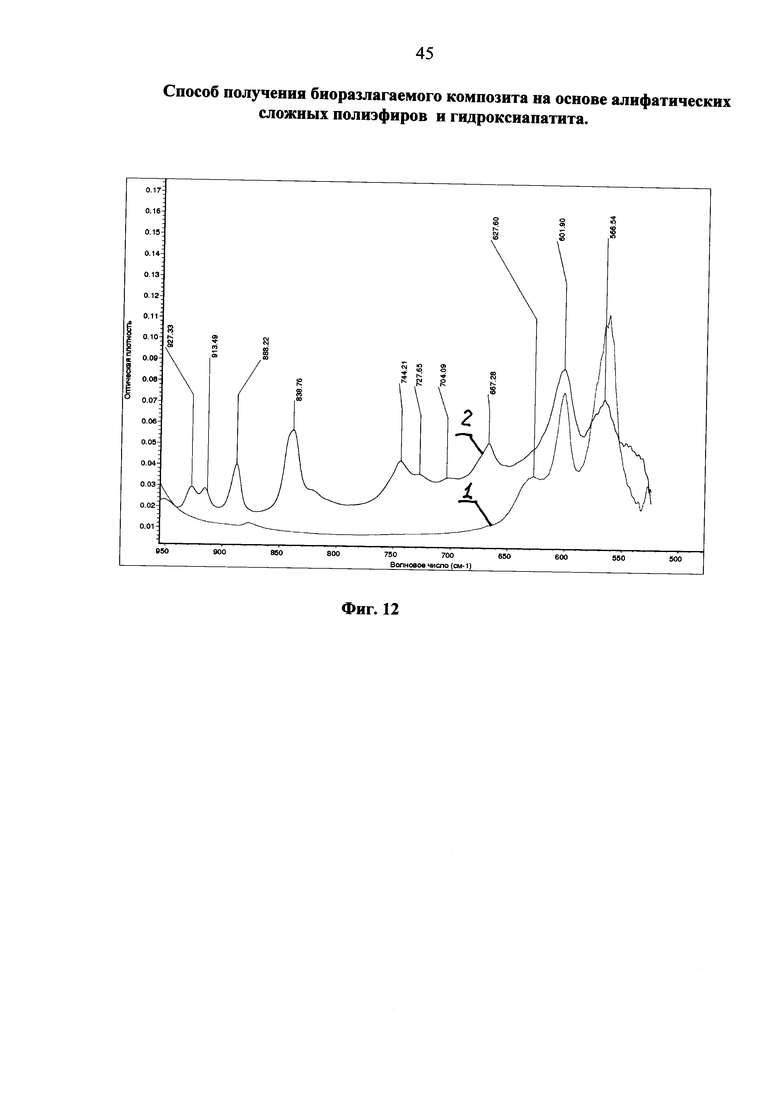
Изобретение относится к медицинской химии, а именно к биоразлагаемым фосфатсодержащим полимерным материалам, использующимся в качестве аналогов костной ткани, и раскрывает способ получения биоразлагаемого композита. Способ характеризуется тем, что синтез композита, который включает в себя взаимную прививку полимера и наполнителя, осуществляют в отсутствие органического растворителя одно- или двухстадийным способом, методом совместной механохимической обработки компонентов, проводящейся при комнатной температуре в интервале 3000-7000 об/мин диспергатором при исходном соотношении гидроксиапатит / полилактид 10-90/90-10%. Способ может использоваться для получения композита при комнатной температуре при сниженном времени синтеза. Изобретение позволяет получать биоразлагаемый композит более быстрым, чем в аналогах, синтезом при отсутствии органического растворителя. 12 ил., 7 пр., 1 табл.
Способ получения биоразлагаемого композита для использования в качестве аналога костной ткани, характеризующийся тем, что гидроксиапатит и полилактид подвергают совместной механохимической обработке путем диспергирования при температуре 20-40°C, в интервале 3000-7000 об/мин диспергатора, при следующем исходном соотношении гидроксиапатит/полилактид, мас.%: 20-80/90-10.
| N.L | |||
| IGNJATOVICH et al | |||
| Synthesis and application of hydroxyapatite/polylactide composite biomaterial | |||
| Applied Surface Science November, 2004, 238, 1-4, PP | |||
| Мяльно-трепальный станок | 1921 |
|
SU314A1 |
| P | |||
| TORMALA et.al., Surgical biocomposite material and a method for producing the material | |||
| Evaluation of hydroxyapatite/poly (L-lactide) composites: physicochemical properties J | |||
| Mater | |||
| Sci., Mater, 1993, N 4, PP | |||
| Способ окисления боковых цепей ароматических углеводородов и их производных в кислоты и альдегиды | 1921 |
|
SU58A1 |
| Y | |||
| SHIKINAMI, et al | |||
| Bioresorbable devices made of forged composites of hydroxyapatite (HA) particles and poly-L-lactide (PLLA): part I | |||
| Basic characteristics, Biomaterials, 1999, N 20, PP | |||
| Аппарат для перегонки углеводородных масел | 1925 |
|
SU859A1 |
| T | |||
| FURUKAWA et al | |||
| Biodegradation behavior of ultra-high-strength hydroxyapatite/poly (L-lactide) composite rods for internal fixation of bone fractures | |||
| Biomaterials, 2000, N 21, PP | |||
| Коромысла весов | 1921 |
|
SU889A1 |
| ЖАРКОВ А.В | |||
| Повышение эффективности оcтеопластики челюстей с помощью полимера полилактида, наполненного гидроксиаппатитом | |||
| Диссертация на соискание ученой степени кандидата медицинских наук | |||
| Москва, 2006, C.1-24. | |||

