ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к области терапевтического лечения ревматоидного артрита. Точнее, изобретение относится к применению антагонистов рецептора интерлейкина-6 (IL-6R), таких как анти-IL-6R антитела, в комбинации с модифицирующими заболевание антиревматическими лекарственными средствами для лечения ревматоидного артрита.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Оценивают, что приблизительно 0,5-1% взрослой популяции Северной Америки и Европы страдают от ревматоидного артрита (РА (RA)). РА в два раза чаще поражает женщин, чем мужчин, и наиболее часто женщин в возрасте старше 40 лет.
РА характеризуется персистирующим синовитом и прогрессирующей деструкцией хряща и кости во множестве суставов. Принципиальной особенностью заболевания является симметричный полиартрит, характерно вовлекающий мелкие суставы кистей и стоп. Воспалительный процесс также может поражать другие органы, особенно костный мозг (анемия), глаза (склерит, эписклерит), легкие (интерстициальный пневмонит, плеврит), сердце (перикардит) и кожу (узлы, лейкоцитокластический васкулит). Системное воспаление характеризуется лабораторными нарушениями, такими как анемия, повышенная скорость оседания эритроцитов, фибриноген и C-реактивный белок (CRP), и клиническими симптомами слабости, потери массы тела, мышечной атрофии в области пораженных суставов. Наличие высоких титров поликлональных ревматоидных факторов и антител к антициклическому цитруллинированному пептиду (анти-CCP) обеспечивает свидетельства иммунной дисрегуляции. По оценке от 65% до 70% пациентов с РА имеют прогрессирующее заболевание, которое приводит к деструкции суставов, инвалидизации и преждевременной смерти.
В области техники существует необходимость в улучшенных схемах лечения для облегчения симптомов, ассоциированных с РА.
СУЩНОСТЬ
Настоящее описание представляет способ лечения ревматоидного артрита у пациента, нуждающегося в этом. Способ включает введение пациенту эффективного количества сарилумаба (SAR153191) и члена группы, состоящей из лефлуномида, сульфасалазина и гидроксихлорохина. В определенных вариантах осуществления изобретения пациента ранее неэффективно лечили от ревматоидного артрита путем введения антагониста TNF-α. В частности, пациент мог получать лечение антагонистом TNF-α в течение, по меньшей мере, трех месяцев или мог не переносить антагонист TNF-α. Антагонистом TNF-α может быть этанерцепт, инфликсимаб, адалимумаб, голимумаб или цертолизумаб. В других вариантах осуществления изобретения пациента ранее неэффективно лечили от ревматоидного артрита путем введения метотрексата.
Сарилумаб можно вводить в дозе от 50 до 150 мг в неделю или от 100 до 200 мг раз в две недели.
В определенных специфических вариантах осуществления изобретения пациенту вводят сарилумаб и лефлуномид. Лефлуномид можно вводить перорально. Лефлуномид также можно вводить пациенту в дозе от 10 до 20 мг в сутки.
В других специфических вариантах осуществления изобретения пациенту вводят сарилумаб и сульфасалазин. Сульфасалазин можно вводить перорально. Сульфасалазин также можно вводить пациенту в дозе от 1000 до 3000 мг в сутки.
В других специфических вариантах осуществления изобретения пациенту вводят сарилумаб и гидроксихлорохин. Гидроксихлорохин можно вводить перорально. Гидроксихлорохин также можно вводить пациенту в дозе между 200 и 400 мг в сутки.
В некоторых вариантах осуществления изобретения в результате лечения пациент достигает 20% или 50% улучшения по шкале основного заболевания Американской Коллегии Ревматологов после 12 недель лечения. В других вариантах осуществления изобретения в результате лечения пациент достигает 20%, 50% или 70% улучшения по шкале основного заболевания Американской Коллегии Ревматологов после 24 недель лечения.
В некоторых вариантах осуществления изобретения в результате лечения пациент достигает более низкого балла активности заболевания после 12 недель лечения, чем пациент имел до лечения. Балл активности заболевания может быть меньше или равным 2,6 через 12 недель. Балл активности заболевания может снижаться более чем на 1,2 от начала лечения к 12 неделям. Балл активности заболевания может быть меньше или равным 3,2 через 12 недель. Балл активности заболевания может снижаться более чем на 0,6 от начала лечения к 12 неделям. Балл активности заболевания может быть меньше или равным 5,1 через 12 недель.
В некоторых вариантах осуществления изобретения в результате лечения после около 24 недель лечения пациент достигает более низкого балла активности заболевания, чем имел до лечения. Балл активности заболевания может быть меньше или равным 2,6 через 24 недели. Балл активности заболевания может снижаться более чем на 1,2 от начала лечения к 24 неделям. Балл активности заболевания может быть меньше или равен 3,2 через 24 недели.
Балл активности заболевания может снижаться более чем на 0,6 от начала лечения к 24 неделям. Балл активности заболевания может быть меньше или равным 5,1 через 24 недели.
Настоящее описание также представляет способ лечения ревматоидного артрита у пациента, нуждающегося в этом, включающий введение пациенту эффективного количества сарилумаба и метотрексата, где пациента ранее неэффективно лечили от ревматоидного артрита путем введения анти-TNF-α препарата. В определенных вариантах осуществления изобретения пациента ранее неэффективно лечили от ревматоидного артрита путем введения метотрексата. Метотрексат можно вводить пациенту в дозе от 10 до 25 мг в неделю.
В определенных вариантах осуществления изобретения пациентом является млекопитающее. Млекопитающим может быть человек. В определенных вариантах осуществления изобретения человек происходит из Азии или Тихоокеанского региона. Людям, происходящим из Азии или Тихоокеанского региона, можно вводить от 6 до 25 мг метотрексата в неделю.
В определенных вариантах осуществления изобретения пациента ранее неэффективно лечили от ревматоидного артрита путем введения антагониста TNF-α. В частности, пациента могли лечить в течение, по меньшей мере, трех месяцев с помощью антагониста TNF-α или он мог не переносить антагонист TNF-α. Антагонистом TNF-α может быть этанерцепт, инфликсимаб, адалимумаб, голимумаб или цертолизумаб. В других вариантах осуществления изобретения пациента ранее неэффективно лечили от ревматоидного артрита путем введения метотрексата.
Сарилумаб можно вводить в дозе от 50 до 150 мг в неделю или от 100 до 200 мг раз в две недели.
В некоторых вариантах осуществления изобретения в результате лечения пациент достигает 20% или 50% улучшения по основной шкале заболевания Американской Коллегии Ревматологов после 12 недель лечения. В других вариантах осуществления изобретения в результате лечения пациент достигает 20%, 50% или 70% улучшения по основной шкале заболевания Американской Коллегии Ревматологов после 24 недель лечения.
В некоторых вариантах осуществления изобретения в результате лечения в течение 12 недель пациент достигает более низкого балла активности заболевания, чем имел до лечения. Балл активности заболевания может быть меньше или равным 2,6 через 12 недель. Балл активности заболевания может снижаться более чем на 1,2 между началом лечения и 12 неделями. Балл активности заболевания может быть меньше или равным 3,2 через 12 недель. Балл активности заболевания может снижаться на более чем 0,6 между началом лечения и 12 неделями. Балл активности заболевания может составлять менее чем или равняться 5,1 через 12 недель.
В некоторых вариантах осуществления изобретения в результате лечения пациент достигает более низкого балла активности заболевания после 24 недель лечения, чем пациент имел до начала лечения. Балл активности заболевания может быть меньше или равным 2,6 через 24 недели. Балл активности заболевания может снижаться на более чем 1,2 между началом лечения и 24 неделями. Балл активности заболевания может быть меньше или равным 3,2 через 24 недели. Балл активности заболевания может снижаться более чем на 0,6 между началом лечения и 24 неделями. Балл активности заболевания может быть меньше или равным 5,1 через 24 недели.
Описание также представляет фармацевтическую композицию, включающую эффективное количество сарилумаба и члена группы, состоящей из лефлуномида, сульфасалазина и гидроксихлорохина. Сарилумаб может присутствовать в количестве от 50 до 150 мг на дозу или от 100 до 200 мг на дозу.
В определенных специфических вариантах осуществления изобретения композиция включает сарилумаб и лефлуномид. Лефлуномид может присутствовать в пероральной лекарственной форме. Лефлуномид может присутствовать в композиции в количестве от 10 до 20 мг на дозу.
В других специфических вариантах осуществления изобретения композиция включает сарилумаб и сульфасалазин. Сульфасалазин может присутствовать в пероральной лекарственной форме. Сульфасалазин может присутствовать в композиции для пациента в количестве от 1000 до 3000 мг в сутки.
В других специфических вариантах осуществления изобретения композиция включает сарилумаб и гидроксихлорохин.
Гидроксихлорохин может присутствовать в пероральной лекарственной форме. Гидроксихлорохин может присутствовать в композиции для пациента в дозе от 200 до 400 мг в сутки.
Примеры вариантов осуществления изобретения перечислены ниже.
Вариант осуществления изобретения 1: Способ лечения ревматоидного артрита у пациента, нуждающегося в этом, включающий введение пациенту эффективного количества сарилумаба (SAR153191) и члена группы, состоящей из лефлуномида, сульфасалазина и гидроксихлорохина.
Вариант осуществления изобретения 2: Способ по варианту осуществления изобретения 1, где пациента ранее неэффективно лечили от ревматоидного артрита путем введения антагониста TNF-α.
Вариант осуществления изобретения 3: Способ по варианту осуществления изобретения 2, где антагонист TNF-α является биологическим антагонистом анти-TNF-α.
Вариант осуществления изобретения 4: Способ по варианту осуществления изобретения 2 или 3, где пациента лечили антагонистом TNF-α в течение, по меньшей мере, трех месяцев.
Вариант осуществления изобретения 5: Способ по варианту осуществления изобретения 2 или 3, где пациент не переносит антагонист TNF-α.
Вариант осуществления изобретения 6: Способ по варианту осуществления изобретения 2 или 3, где антагонист TNF-α выбирают из группы, состоящей из этанерцепта, инфликсимаба, адалимумаба, голимумаба и сертолизумаба.
Вариант осуществления изобретения 7: Способ по варианту осуществления изобретения 2 или 3, где пациента ранее лечили от ревматоидного артрита путем введения метотрексата.
Вариант осуществления изобретения 8: Способ по варианту осуществления изобретения 1, где сарилумаб вводят в дозе от 50 до 150 мг в неделю.
Вариант осуществления изобретения 9: Способ по варианту осуществления изобретения 1, где сарилумаб вводят в дозе от 100 до 200 мг один раз в две недели.
Вариант осуществления изобретения 10: Способ по варианту осуществления изобретения 1, где пациенту вводят сарилумаб и лефлуномид.
Вариант осуществления изобретения 11: Способ по варианту осуществления изобретения 10, где лефлуномид вводят перорально.
Вариант осуществления изобретения 12: Способ по варианту осуществления изобретения 10, где лефлуномид вводят пациенту в дозе от 10 до 20 мг в сутки.
Вариант осуществления изобретения 13: Способ по варианту осуществления изобретения 1, где пациенту вводят сарилумаб и сульфасалазин.
Вариант осуществления изобретения 14: Способ по варианту осуществления изобретения 13, где сульфасалазин вводят перорально.
Вариант осуществления изобретения 15: Способ по варианту осуществления изобретения 13, где сульфасалазин вводят пациенту в дозе от 1000 до 3000 мг в сутки.
Вариант осуществления изобретения 16: Способ по варианту осуществления изобретения 1, где пациенту вводят сарилумаб и гидроксихлорохин.
Вариант осуществления изобретения 17: Способ по варианту осуществления изобретения 16, где гидроксихлорохин вводят перорально.
Вариант осуществления изобретения 18: Способ по варианту осуществления изобретения 16, где гидроксихлорохин вводят пациенту в дозе от 200 до 400 мг в сутки.
Вариант осуществления изобретения 19: Способ по любому из вариантов осуществления изобретения 1-18, где пациент достигает 20% улучшения по основной шкале заболевания Американской Коллегии Ревматологов после 12 недель лечения.
Вариант осуществления изобретения 20: Способ по любому из вариантов осуществления изобретения 1-18, где пациент достигает 50% улучшения по основной шкале заболевания Американской Коллегии Ревматологов после 12 недель лечения.
Вариант осуществления изобретения 21: Способ по любому из вариантов осуществления изобретения 1-18, где пациент достигает 20% улучшения по основной шкале заболевания Американской Коллегии Ревматологов после 24 недель лечения.
Вариант осуществления изобретения 22: Способ по любому из вариантов осуществления изобретения 1-18, где пациент достигает 50% улучшения по основной шкале заболевания Американской Коллегии Ревматологов после 24 недель лечения.
Вариант осуществления изобретения 23: Способ по любому из вариантов осуществления изобретения 1-18, где пациент достигает 70% улучшения по основной шкале заболевания Американской Коллегии Ревматологов после 24 недель лечения.
Вариант осуществления изобретения 24: Способ по любому из вариантов осуществления изобретения 1-18, где пациент достигает более низкого балла активности заболевания после 12 недель лечения, чем пациент имел до лечения.
Вариант осуществления изобретения 25: Способ по любому из вариантов осуществления изобретения 1-18, где балл активности заболевания меньше или равен 2,6 через 12 недель.
Вариант осуществления изобретения 26: Способ по любому из вариантов осуществления изобретения 1-18, где балл активности заболевания снижается на более чем 1,2 от начала лечения до 12 недель.
Вариант осуществления изобретения 27: Способ по любому из вариантов осуществления изобретения 1-18, где балл активности заболевания меньше или равен 3,2 через 12 недель.
Вариант осуществления изобретения 28: Способ по любому из вариантов осуществления изобретения 1-18, где балл активности заболевания снижается более чем на 0,6 от начала лечения до 12 недель.
Вариант осуществления изобретения 29: Способ по любому из вариантов осуществления изобретения 1-18, где балл активности заболевания меньше или равен 5,1 через 12 недель.
Вариант осуществления изобретения 30: Способ по любому из вариантов осуществления изобретения 1-18, где пациент достигает более низкого балла активности заболевания через 24 недели лечения, чем пациент имел до лечения.
Вариант осуществления изобретения 31: Способ по любому из вариантов осуществления изобретения 1-18, где балл активности заболевания меньше или равен 2,6 через 24 недели.
Вариант осуществления изобретения 32: Способ по любому из вариантов осуществления изобретения 1-18, где балл активности заболевания снижается на более чем 1,2 от начала лечения до 24 недель.
Вариант осуществления изобретения 33: Способ по любому из вариантов осуществления изобретения 1-18, где балл активности заболевания меньше или равен 3,2 через 24 недели.
Вариант осуществления изобретения 34: Способ по любому из вариантов осуществления изобретения 1-18, где балл активности заболевания снижается на более чем 0,6 от начала лечения до 24 недель.
Вариант осуществления изобретения 35: Способ по любому из вариантов осуществления изобретения 1-18, где балл активности заболевания меньше или равен 5,1 через 24 недели.
Вариант осуществления изобретения 36: Способ лечения ревматоидного артрита у пациента, нуждающегося в этом, включающий введение пациенту эффективного количества сарилумаба и метотрексата, где пациента ранее неэффективно лечили от ревматоидного артрита путем введения антагониста анти-TNF-α.
Вариант осуществления изобретения 37: Способ по варианту осуществления изобретения 36, где пациента ранее неэффективно лечили от ревматоидного артрита путем введения метотрексата.
Вариант осуществления изобретения 38: Способ по варианту осуществления изобретения 36, где метотрексат вводят пациенту в дозе от 10 до 25 мг в неделю.
Вариант осуществления изобретения 39: Способ по варианту осуществления изобретения 36, где пациентом является млекопитающее.
Вариант осуществления изобретения 40: Способ по варианту осуществления изобретения 37, где млекопитающим является человек.
Вариант осуществления изобретения 41: Способ по варианту осуществления изобретения 38, где человек происходит от уроженцев Азии или Тихоокеанского региона.
Вариант осуществления изобретения 42: Способ по варианту осуществления изобретения 39, где людям, происходящим от уроженцев Азии или Тихоокеанского региона, вводят от 6 до 25 мг метотрексата в неделю.
Вариант осуществления изобретения 43: Способ по варианту осуществления изобретения 36, где пациента лечили антагонистом TNF-α в течение, по меньшей мере, трех месяцев.
Вариант осуществления изобретения 44: Способ по варианту осуществления изобретения 36, где пациент не переносит антагонист TNF-α.
Вариант осуществления изобретения 45: Способ по варианту осуществления изобретения любого из вариантов осуществления изобретения 36-44, где антагонистом TNF-α является биологический антагонист анти-TNF-α.
Вариант осуществления изобретения 46: Способ по варианту осуществления изобретения 44, где антагонист TNF-α выбирают из группы, состоящей из этанерцепта, инфликсимаба, адалимумаба, голимумаба и сертолизумаба.
Вариант осуществления изобретения 47: Способ по варианту осуществления изобретения 36, где сарилумаб вводят в дозе от 50 до 150 мг в неделю.
Вариант осуществления изобретения 48: Способ по варианту осуществления изобретения 36, где сарилумаб вводят в дозе от 100 до 200 мг раз в две недели.
Вариант осуществления изобретения 49: Способ по любому из вариантов осуществления изобретения 36-48, где пациент достигает 20% улучшения по основной шкале заболевания Американской Коллегии Ревматологов после 12 недель лечения.
Вариант осуществления изобретения 50: Способ по любому из вариантов осуществления изобретения 36-48, где пациент достигает 50% улучшения по основной шкале заболевания Американской Коллегии Ревматологов после 12 недель лечения.
Вариант осуществления изобретения 51: Способ по любому из вариантов осуществления изобретения 36-48, где пациент достигает 20% улучшения по основной шкале заболевания Американской Коллегии Ревматологов после 24 недель лечения.
Вариант осуществления изобретения 52: Способ по любому из вариантов осуществления изобретения 36-48, где пациент достигает 50% улучшения по основной шкале заболевания Американской Коллегии Ревматологов после 24 недель лечения.
Вариант осуществления изобретения 53: Способ по любому из вариантов осуществления изобретения 36-48, где пациент достигает 70% улучшения по основной шкале заболевания Американской Коллегии Ревматологов после 24 недель лечения.
Вариант осуществления изобретения 54: Способ по любому из вариантов осуществления изобретения 36-48, где пациент достигает более низкого балла активности заболевания после 12 недель лечения, чем пациент имел до лечения.
Вариант осуществления изобретения 55: Способ по любому из вариантов осуществления изобретения 36-48, где балл активности заболевания меньше или равен 2,6 через 12 недель.
Вариант осуществления изобретения 56: Способ по любому из вариантов осуществления изобретения 36-48, где балл активности заболевания снижается более чем на 1,2 между началом лечения и 12 неделями.
Вариант осуществления изобретения 57: Способ по любому из вариантов осуществления изобретения 36-48, где балл активности заболевания меньше или равен 3,2 через 12 недель.
Вариант осуществления изобретения 58: Способ по любому из вариантов осуществления изобретения 36-48, где балл активности заболевания снижается более чем на 0,6 от начала лечения до 12 недель.
Вариант осуществления изобретения 59: Способ по любому из вариантов осуществления изобретения 36-48, где балл активности заболевания меньше или равен 5,1 через 12 недель.
Вариант осуществления изобретения 60: способ по любому из вариантов осуществления изобретения 36-48, где пациент достигает более низкого балла активности заболевания после 24 недель лечения, чем пациент имел до лечения.
Вариант осуществления изобретения 61: Способ по любому из вариантов осуществления изобретения 36-48, где балл активности заболевания меньше или равен 2,6 через 24 недели.
Вариант осуществления изобретения 62: Способ по любому из вариантов осуществления изобретения 36-48, где балл активности заболевания снижается более чем на 1,2 между началом лечения и 24 неделями.
Вариант осуществления изобретения 63: Способ по любому из вариантов осуществления изобретения 34-45, где балл активности заболевания меньше или равен 3,2 через 24 недели.
Вариант осуществления изобретения 64: Способ по любому из вариантов осуществления изобретения 34-45, где балл активности заболевания снижается более чем на 0,6 между началом лечения и 24 неделями.
Вариант осуществления изобретения 65: Способ по любому из вариантов осуществления изобретения 34-4 5, где балл активности заболевания меньше или равен 5,1 через 24 недели.
Вариант осуществления изобретения 66: Фармацевтическая композиция, включающая эффективное количество сарилумаба и члена группы, состоящей из лефлуномида, сульфасалазина и гидроксихлорохина.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фигуре 1 приведена схема многоцентрового, двойного слепого, плацебоконтролируемого 12-недельного исследования, включавшего введение в параллельных шести группах SAR153191 или плацебо, которые применяли еженедельно подкожно с сопутствующей терапией МТХ.
На фигуре 2 приведена диаграмма, показывающая ACR20 ответ (%) на проводимое лечение на 12 неделе.
На фигуре 3 приведена диаграмма, показывающая ACR50 и ACR70 ответы (%) на проводимое лечение на 12 неделе.
ПОДРОБНОЕ ОПИСАНИЕ
Описание обеспечивает фармацевтические композиции и способы применения таких композиций для лечения ревматоидного артрита (РА) и облегчения, по меньшей мере, одного симптома РА. Такие композиции включают, по меньшей мере, одно антитело, которое специфически связывается с человеческим рецептором интерлейкина-6 (hlL-6R) и, по меньшей мере, одно антиревматическое лекарственное средство, модифицирующее заболевание (DMARD).
AHTM-hlL-6R антитела
Настоящее описание включает способы, которые включают введение пациенту человеческого антитела или его антиген-связывающего фрагмента, которые специфически связываются с hlL-6R. Как используется в настоящем описании, термин "hlL-6R" обозначает рецептор человеческого цитокина, который специфически связывается с человеческим интерлейкином-6 (IL-6). В определенных вариантах осуществления изобретения антитело, которое вводят пациенту, специфически связывается с внеклеточным доменом hlL-6R. Внеклеточный домен hlL-6R показан в последовательности аминокислот SEQ ID NO: 1.
Если специфически не указано иначе, термин «антитело», как используется в настоящем описании, необходимо понимать как охватывающий молекулы антител, включающие две тяжелые цепи иммуноглобулина и две легкие цепи иммуноглобулина (т.е. "полные молекулы антител"), а также их антиген-связывающие фрагменты. Термины "антиген-связывающая часть" антитела, "антиген-связывающий фрагмент" антитела и подобные, как используются в настоящем описании, включают любые естественные, ферментативно получаемые, синтетические или генетически модифицированные полипептиды или гликопротеины, которые специфически связывают антиген с образованием комплекса. Антиген-связывающие фрагменты антител могут быть получены, например, из целых молекул антител с использованием любых подходящих стандартных методик, таких как протеолитическое расщепление или рекомбинантные методики генной инженерии, включающие манипуляции и экспрессию ДНК, кодирующей вариабельный и (необязательно) постоянный домен антитела. Такие ДНК известны и/или легко доступны из, например, коммерческих источников, библиотек ДНК (включая, например, библиотеки фагов-антител), или могут быть синтезированы. ДНК может быть секвенирована и обработана химически или с использованием методик молекулярной биологии, например, для расположения одного или более вариабельных и/или постоянных доменов в подходящей конфигурации, или для внесения кодонов, создания цистеиновых остатков, модификации, добавления или удаления аминокислот и др.
Неограничивающие примеры антиген-связывающих фрагментов включают: (i) Fab фрагменты; (ii) F(abʹ)2 фрагменты; (iii) Fd фрагменты; (iv) Fv фрагменты; (v) одноцепочечные молекулы Fv (scFv); (vi) dAb фрагменты; и (vii) минимальные распознаваемые единицы, состоящие из остатков аминокислот, которые имитируют гипервариабельный участок антитела (например, выделенный участок, определяющий комплементарность (CDR)). Другие инженерные молекулы, такие как диатела, триотела, тетратела и минитела, также охвачены выражением "антиген-связывающий фрагмент", как используется в настоящем описании.
Антиген-связывающий фрагмент антитела обычно включает, по меньшей мере, один вариабельный домен. Вариабельный домен может быть любого размера или состава аминокислот и обычно включает, по меньшей мере, один CDR, который соседствует или находится внутри рамки считывания с одной или более каркасными последовательностями. В антиген-связывающих фрагментах, имеющих VH домен, ассоциированный с VL доменом, VH и VL домены могут быть расположены относительно друг друга в любом подходящем порядке. Например, вариабельный участок может быть димерным и содержать димеры VH-VH, VH-VL или VL-VL. Альтернативно, антиген-связывающий фрагмент антитела может содержать мономерный VH или VL домен.
В определенных вариантах осуществления изобретения антиген-связывающий фрагмент антитела может содержать, по меньшей мере, один вариабельный домен, ковалентно связанный с одним постоянным доменом. Неограничивающие, примерные конфигурации вариабельных и постоянных доменов, которые могут быть обнаружены в рамках антиген-связывающего фрагмента антитела по настоящему изобретению, включают: (i) VH-CH1; (ii) VH-CH2; (iii) VH-H3; (iv) VH-CH1-CH2; (V) VH-CH1-CH2-CH3; (vi) VH-CH2-CH3; (vii) VH-CL; (viii) VL-CH1; (ix) VL-CH2; (x) VL-CH3; (xi) VL-CH1-CH2; (xii) VL-CH1-CH2-CH3; (xiii) VL-CH2-CH3; и (xiv) VL-CL. В любой конфигурации вариабельного и постоянного доменов, включая любую примерную конфигурацию, перечисленную выше, вариабельные и постоянные домены могут быть непосредственно связаны друг с другом или могут быть связаны посредством полного или частичного петлевого или линкерного участка. Петлевой участок может состоять из, по меньшей мере, 2 (например, 5, 10, 15, 20, 40, 60 или более) аминокислот, что дает гибкое или полугибкое соединение между соседними вариабельными и/или постоянными доменами в одиночной молекуле полипептида. Более того, антиген-связывающий фрагмент антитела по настоящему изобретению может включать гомодимер или гетеродимер (или другой мультимер) любого из вариабельных и постоянных доменов конфигураций, перечисленных выше, в нековалентной ассоциации друг с другом и/или с одним или более мономерными VH или VL доменами (например, посредством дисульфидной связи(ей)).
Термин "специфически связывается" обозначает, что антитело или его антиген-связывающий фрагмент образуют комплекс с антигеном, который является относительно стабильным в физиологических условиях. Специфическое связывание может быть охарактеризовано константой диссоциации, по меньшей мере, около 1×10-6 M или меньше. В других вариантах осуществления изобретения, константа диссоциации составляет, по меньшей мере, около 1×10-7 M, 1×10-8 M или 1×10-9 M. Методы для определения, будут ли две молекулы специфически связываться, хорошо известны в области техники и включают, например, равновесный диализ, поверхностный плазмонный резонанс и подобные.
Как целые молекулы антител, антиген-связывающие фрагменты могут быть моноспецифическими или мультиспецифическими (например, биспецифическими). Мультиспецифический антиген-связывающий фрагмент антитела обычно включает, по меньшей мере, два различных вариабельных домена, где каждый вариабельный домен способен специфически связываться с отдельным антигеном или с различными эпитопами того же антигена. Любые мультиспецифические форматы антител, включая примерные форматы биспецифических антител, описанные в настоящем описании, могут быть адаптированы для применения в контексте антиген-связывающего фрагмента антитела по настоящему изобретению с использованием рутинных методик, доступных в области техники.
В специфическом варианте осуществления изобретения антитело или фрагмент антитела для использования в способе по изобретению, может быть мультиспецифическим антителом, которое может быть специфичным к различным эпитопам одного целевого полипептида или может содержать антиген-связывающие домены, специфические к эпитопам более чем одного целевого полипептида. Примерный формат биспецифического антитела, который может быть использован в контексте настоящего изобретения, включает применение первого домена иммуноглобулина (Ig) CH3 и второго домена Ig CH3, где первый и второй домены Ig CH3 отличаются друг от друга на, по меньшей мере, одну аминокислоту и где, по меньшей мере, отличие одной аминокислоты уменьшает связывание биспецифического антитела с белком A по сравнению с биспецифическим антителом, не имеющим различий аминокислот. В одном варианте осуществления изобретения первый домен Ig CH3 связывает белок A и второй домен Ig CH3 содержит мутацию, которая снижает или прекращает связывание белка A, такую как модификация H95R (по нумерации экзонов IMGT; H435R по EU нумерации). Второй CH3 может дополнительно включать модификацию Y96F (по IMGT; Y436F по EU). Дополнительные модификации, которые могут быть обнаружены во втором CH3, включают: D16E, L18M, N44S, K52N, V57M и V82I (по IMGT; D356E, L358M, N384S, K392N, V397M и V422I по EU) в случае антител IgG1; N44S, K52N и V82I (IMGT; N384S, K392N и V422I по EU) в случае антител IgG2; и Q15R, N44S, K52N, V57M, R69K, E79Q и V82I (по IMGT; Q355R, N384S, K392N, V397M, R409K, E419Q и V422I по EU) в случае антител IgG4. Вариации формата биспецифических антител, описанных выше, предусматриваются в рамках настоящего изобретения.
В других специфических вариантах осуществления изобретения антителом является сарилумаб (SAR153191). Вариабельный участок тяжелой цепи сарилумаба показан ниже как SEQ ID NO: 2.
Вариабельный участок легкой цепи сарилумаба показан ниже как SEQ ID NO: 3.
"Нейтрализующее" или "блокирующее" "антитело, как используется в настоящем описании, относится к антителу, чье связывание с hlL-6R приводит к ингибированию биологической активности hlL-6. Такое ингибирование биологической активности hlL-6 может быть оценено путем измерения одного или более индикаторов биологической активности hlL-6, известных в области техники, таких как клеточная активация, индуцированная hlL-б, и hlL-6 связывание с hlL-6R (см. примеры ниже).
Полностью человеческие анти-IL-GR антитела, описанные в настоящем описании, могут включать одну или более замен, вставок и/или делеций аминокислот в каркасе и/или CDR участках вариабельных доменов тяжелой и легкой цепи по сравнению с соответствующими исходными последовательностями. Такие мутации могут быть легко определены путем сравнения последовательностей аминокислот, описанных в настоящем описании, с исходными последовательностями, доступными из, например, общественных баз данных последовательностей антител. Настоящее изобретение включает антитела и их антиген-связывающие фрагменты, которые получают из любой последовательности аминокислот, описанных в настоящем описании, где одна или более аминокислот в одном или более каркасе и/или участков мутирована к первоначальному виду соответствующего исходного остатка(ов) или к замене консервативной аминокислоты (натуральной или ненатуральной) соответствующего исходного остатка(ов) (такие изменения последовательности называют в настоящем описании как "исходные мутации к первоначальному виду"). Обычный специалист в области техники, начиная с последовательностей вариабельного участка тяжелых и легких цепей, описанных в настоящем описании, может легко получить множество антител и антител-связывающих фрагментов, которые включают одну или более отдельных исходных мутаций к первоначальному виду или их комбинаций. В определенных вариантах осуществления изобретения, все каркасы и/или остатки CDR в доменах VH и/или VL мутированы обратно в исходную последовательность. В другом варианте осуществления изобретения, только определенные остатки мутированы обратно в исходную последовательность, например, только мутированные остатки, обнаруженные среди первых 8 аминокислот FR1 или среди последних 8 аминокислот FR4, или мутированные остатки обнаруживаются только в рамках CDR1, CDR2 или CDR3. Более того, антитела по настоящему изобретению могут содержать любую комбинацию двух или более исходных мутаций к первоначальному состоянию в рамках каркаса и/или участков CDR, т.е., где определенные отдельные остатки мутированы обратно в исходную последовательность, тогда как другие определенные остатки, которые отличаются от исходной последовательности, сохраняются. При получении антитела и антиген-связывающие фрагменты, которые содержат одну или более исходных мутаций к первоначальному состоянию, могут быть легко исследованы в отношении одного или более желаемых свойств, таких как улучшенная специфичность связывания, улучшенная афинность связывания, улучшенные или усиленные антагонистические или агонистические биологические свойства (какие могут быть), сниженная иммуногенность и др. Антитела и антиген-связывающие фрагменты, полученные таким обычным способом, охватываются настоящим изобретением.
Термин "эпитоп" относится к антигенной детерминанте, которая взаимодействует со специфическим участком связывания антигена в вариабельном участке молекулы антитела, известном как паратоп. Один антиген может иметь более чем один эпитоп. Эпитопы могут быть или конформационными или линейными. Конформационный эпитоп получают путем частичной юкстапозиции аминокислот из различных сегментов линейной полипептидной цепи. Линейный эпитоп представляет собой таковой, полученный из соседних остатков аминокислот в полипептидной цепи. В определенных обстоятельствах эпитоп может содержать на антигене фрагменты сахаридов, фосфорильные группы или сульфонильные группы.
Анти-hlL-6R может быть сарилумаб (SAR153191). В одном варианте осуществления изобретения сарилумаб определяют как антитело, включающее вариабельный участок тяжелой цепи SEQ ID NO: 2 и вариабельный участок легкой цепи SEQ ID NO: 3.
DMARD
Антиревматические лекарственные средства, модифицирующие заболевание (DMARD), включают метотрексат, сульфасалазин, гидроксихлорохин и лефлуномид. В соответствии с композициями и методами по описанию, DMARD можно вводить, как указано далее. Метотрексат можно вводить от 10 до 25 мг в неделю перорально или внутримышечно. В другом варианте осуществления изобретения, метотрексат вводят в дозе от 6 до 25 мг/неделю перорально или внутримышечно пациентам Азиатско-Тихоокеанского региона или таким, которые имеют происхождение от лиц Азиатско-Тихоокеанского региона. Азиатско-Тихоокеанский регион включает Тайвань, Южную Корею, Малайзию, Филиппины, Таиланд и Индию. В определенном варианте осуществления изобретения метотрексат вводят в дозе между 6 и 12, 10 и 15, 15 и 20 и 20 и 25 мг в неделю. В другом варианте осуществления изобретения, метотрексат вводят в дозе 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 или 25 мг в неделю. Лефлуномид можно вводить в дозе от 10 до 20 мг перорально в сутки. В определенном варианте осуществления изобретения, лефлуномид можно вводить в дозе между 10 и 12, 12 и 15, 15 и 17 и 18 и 20 мг в сутки. В другом варианте осуществления изобретения, лефлуномид вводят в дозе 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 мг в сутки. Сульфасалазин можно вводить в дозе от 1000 до 3000 мг перорально в сутки. В определенном варианте осуществления изобретения, сульфасалазин можно вводить в дозе между 1000 и 1400, 1400 и 1800, 1800 и 2200, 2200 и 2600, и 2600 и 3000 мг в сутки. В другом варианте осуществления изобретения сульфасалазин вводят в дозе 1000, 1100, 1200, 1300, 1400, 1500, 1600, 1700, 1800, 1900, 2000, 2100, 2200, 2300, 2400, 2500, 2600, 2700, 2800, 2900 или 3000 мг в сутки. Гидроксихлорохин можно вводить в дозе от 200 до 400 мг перорально в сутки. В определенном варианте осуществления изобретения гидроксихлорохин можно вводить в дозе между 200 и 240, 240 и 280, 280 и 320, 320 и 360 и 360 и 400 в сутки. В другом варианте осуществления изобретения, гидроксихлорохин можно вводить в дозе 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390 или 400 мг в сутки.
Терапевтическое применение и композиции
Способы, описанные в настоящем описании, включают введение пациенту терапевтически эффективного количества анти-hlL-6R антитела и DMARD. Как используется в настоящем описании, фраза "терапевтически эффективное количество" обозначает дозу анти-hlL-6R антитела и DMARD, которая дает определяемое улучшение одного или более симптомов, ассоциированных с ревматоидным артритом, или которая вызывает биологический эффект (например, снижение уровня определенного биомаркера), который коррелирует с лежащим в основе патологическим механизмом(ами), дающим развитие состояния или симптома(ов) ревматоидного артрита. Например, доза анти-hlL-6R антитела с одним или более DMARD, которая вызывает улучшение любого из следующих симптомов или состояний, называется "терапевтически эффективным количеством": хроническая анемия, лихорадка, депрессия, слабость, ревматоидные узелки, васкулит, нейропатия, склерит, перикардит, синдром Фелти и/или деструкция суставов.
Определяемое улучшение также может быть определено с использованием критериев классификации ревматоидного артрита Американской Коллегии ревматологов (ACR). Например, 20% (ACR20), 50% (ACR50) или 70% (ACR70) улучшение от исходного может быть использовано для демонстрации определяемого улучшения.
Шкала активности заболевания (DAS28) может быть использована для демонстрации определяемого улучшения. DAS28 представляет собой комплексную шкалу числа болезненных суставов на основании 28 суставов, числа отечных суставов на основании 28 суставов, оценки общего состояния здоровья и маркера воспаления, который может быть оценен путем измерения уровня C-реактивного белка (CRP). Ответ заболевания может быть представлен с использованием критериев ответа Европейской лиги против ревматизма (EULAR). Хороший ответ по такому критерию является улучшением более чем на 1,2 балла DAS28 с настоящим баллом более чем или равным 3,2. Умеренный ответ представляет собой улучшение более чем на 0,6, но менее чем на 1,2 балла DAS28 и представляет собой балл более чем 3,2. Отсутствие ответа представляет собой улучшение на менее чем 0,6 балла DAS28 и представляет собой балл более чем 5,1. Ремиссия DAS28 представляет собой балл DAS28 менее чем 2,6.
В соответствии с методами настоящего изобретения терапевтически эффективное количество анти-hlL-6R антитела, которое вводят пациенту, варьируется в зависимости от возраста и размера (например, массы тела или площади поверхности тела) пациента, а также пути введения и других факторов, хорошо известных обычному специалисту в области техники. В определенных вариантах осуществления изобретения доза вводимого пациенту анти-hlL-6R антитела составляет от около 10 мг до около 500 мг. Например, настоящее изобретение включает способы, где около 10 мг, около 15 мг, около 20 мг, около 25 мг, около 30 мг, около 35 мг, около 40 мг, около 45 мг, около 50 мг, около 55 мг, около 60 мг, около 65 мг, около 70 мг, около 75 мг, около 80 мг, около 85 мг, около 90 мг, около 95 мг, около 100 мг, около 105 мг, около 1 10 мг, около 115 мг, около 120 мг, около 125 мг, около 130 мг, около 135 мг, около 140 мг, около 145 мг, около 150 мг, около 155 мг, около 160 мг, около 165 мг, около 170 мг, около 175 мг, около 180 мг, около 185 мг, около 190 мг, около 195 мг, около 200, около 205 мг, около 210 мг, около 215 мг, около 220 мг, около 225 мг, около 230 мг, около 235 мг, около 240 мг, около 245 мг, около 250 мг, около 255 мг, около 260 мг, около 265 мг, около 270 мг, около 275 мг, около 280 мг, около 285 мг, около 290 мг, около 295 мг, около 300, около 325 мг, около 350 мг, около 375 мг, около 400 мг, около 425 мг, около 450 мг, около 475 мг, около 500 мг или более анти-hlL-6R антитела вводят пациенту в неделю.
В одном варианте осуществления изобретения hlL-6R антитело вводят в дозе 100-150 мг в неделю. В другом варианте осуществления изобретения, hlL-6R антитело вводят в дозе 100-200 мг один раз в две недели. В других вариантах осуществления изобретения, hlL-6R антитело вводят в дозе от около 100 до около 150 мг в неделю. В другом варианте осуществления изобретения hlL-6R антитело вводят в дозе около 100, 150 или 200 мг один раз в две недели.
Количество анти-hlL-6R антитела, которое вводят пациенту, может быть выражено в миллиграммах антитела на килограмм массы тела пациента (т.е. мг/кг). Например, методы по настоящему изобретению включают введение анти-hlL-6R антитела пациенту в суточной дозе от около 0,01 до около 100 мг/кг, от около 0,1 до около 50 мг/кг или от около 1 до около 10 мг/кг массы тела пациента.
Методы по настоящему изобретению включают введение пациенту нескольких доз анти-hlL-6R антитела в течение установленных периодов времени. Например, анти-hlL-6R антитела можно вводить около 1-5 раз в сутки, около 1-5 раз в неделю, около 1-5 раз в месяц или около 1-5 раз в год. В определенном варианте осуществления изобретения, методы по изобретению включают введение пациенту первой дозы анти-hlL-6R антитела в первую временную точку, с последующим введением, по меньшей мере, второй дозы анти-hlL-6R антитела пациенту во вторую временную точку. Первая и вторая дозы в определенных вариантах осуществления изобретения, могут содержать одно и то же количество анти-hlL-6R антитела. Например, первая и вторая дозы могут каждая содержать от около 10 мг до около 500 мг, от около 20 мг до около 300 мг, от около 100 мг до около 200 мг или от около 100 мг до около 150 мг антитела. Период времени между первой и второй дозами может составлять от около нескольких часов до нескольких недель. Например, вторая временная точка (т.е. время, когда вводят вторую дозу) может составлять от около 1 часа до около 7 недель после первой временной точки (т.е. времени, когда вводят первую дозу). В соответствии с определенным примерным вариантом осуществления изобретения по настоящему изобретению вторая временная точка может составлять около 1 часа, около 4 часов, около 6 часов, около 8 часов, около 10 часов, около 12 часов, около 24 часов, около 2 дней, около 3 дней, около 4 дней, около 5 дней, около 6 дней, около 7 дней, около 2 недель, около 4 недель, около 6 недель, около 8 недель, около 10 недель, около 12 недель, около 14 недель или дольше после первой временной точки. В определенных вариантах осуществления изобретения вторая временная точка составляет около 1 недели или около 2 недель. Третью и последующие дозы можно подобным образом вводить на всем протяжении схемы лечения пациента.
Изобретение обеспечивает способы с использованием терапевтических композиций, включающих анти-IL-6R антитела и их антиген-связывающие фрагменты и одно или более DMARD. Терапевтические композиции по изобретению вводят с подходящими носителями, вспомогательными веществами и другими средствами, которые включают в композиции для обеспечения улучшенного переноса, доставки, переносимости и подобного. Множество соответствующих композиций может быть обнаружено в формуляре, известном всем фармацевтам: Remingtonʹs Pharmaceutical Sciences, Mack Publishing Company, Easton, PA. Такие композиции включают, например, порошки, пасты, мази, желе, воска, масла, липиды, пузырьки, содержащие липиды (катионные или анионные) (такие как LIPOFECTIN™), конъюгаты ДНК, безводные поглощающие пасты, эмульсии типа масло-в-воде и вода-в-масле, эмульсии карбовоска (полиэтиленгликоли различной молекулярной массы), полутвердые гели и полутвердые смеси, содержащие карбовоск. См. также Powell et al. "Compendium of excipients for parenteral formulations" PDA (1998) J Pharm Sci Technol 52: 238-311.
Доза может варьироваться в зависимости от возраста и массы тела пациента, целевого органа мишени, состояний, пути введения и подобного. Известны различные системы доставки, и они могут быть использованы для введения фармацевтической композиции по изобретению, например, инкапсуляция в липосомы, микрочастицы, микрокапсулы, рецептор-опосредованный эндоцитоз (см., например, Wu et al. (1987) J. Biol. Chem. 262: 4429-4432). Способы введения включают, но не ограничиваются, внутрикожный, внутримышечный, интраперитонеальный, внутривенный, подкожный, интраназальный, эпидуральный и пероральный путь. Композицию можно вводить любым обычным путем, например путем инфузии или болюсной инъекции, путем абсорбции через эпителиальные или слизисто-кожные оболочки (например, слизистую оболочку полости рта, слизистую оболочку прямой кишки и кишечника и др.) и можно вводить вместе с другими биологически активными агентами. Введение может быть системным или местным. hlL-6R антитела можно вводить подкожно. DMARD можно вводить перорально или внутримышечно.
Фармацевтическая композиция также может быть доставлена в пузырьках, в частности липосомах (см. Langer (1990) Science 24 9: 1527-1533). В определенных ситуациях фармацевтическая композиция может быть доставлена в системе с контролируемым высвобождением, например с использованием помпы или полимерных материалов. В другом варианте осуществления изобретения система с контролируемым высвобождением может быть помещена вблизи мишени композиции, таким образом требуя только часть системной дозы.
Инъекционные препараты могут включать лекарственные формы для внутривенных, подкожных, внутрикожных и внутримышечных инъекций, местных инъекций, капельных инфузий и др. Такие инъекционные препараты могут быть получены широко известными методами. Например, инъекционные препараты могут быть получены, например, путем растворения, суспендирования и эмульгирования антитела или его соли, описанных выше, в стерильной водной среде или масляной среде, обычно используемых для инъекций. В качестве водной среды для инъекций, существуют, например физиологический солевой раствор, изотонический раствор, содержащий глюкозу и другие вспомогательные средства и др., которые могут быть использованы в комбинации с соответствующим растворяющим средством, таким как спирт (например, этанол), многоатомный спирт (например, пропиленгликоль, полиэтиленгликоль), неионное поверхностно-активное вещество [например, полисорбат 80, HCO-50 (полиоксиэтиленовый (50 моль) аддукт гидрогенизированного касторового масла)], и др. В качестве масляной среды используют, например, кунжутное масло, соевое масло и др., которые могут быть использованы в комбинации с растворителем, таким как бензилбензоат, бензиловый спирт и др. Инъекционный раствор, полученный таким образом, может быть заполнен в соответствующую ампулу.
Преимущественно фармацевтические композиции для перорального или парентерального применения, описанные выше, получают в лекарственных формах в стандартной дозе, содержащей дозы активных ингредиентов. Такие лекарственные формы в стандартной дозе включают, например, таблетки, пилюли, капсулы, инъекции (ампулы), суппозитории и др. Количество содержащегося DMARD обычно составляет от около 5 до 3000 мг на лекарственную форму в стандартной пероральной дозе в зависимости от специфического используемого DMARD. Количество содержащегося hlL-6R антитела обычно составляет от около 100 до 200 мг на подкожную лекарственную форму.
В соответствии с методами, описанными в настоящем описании, анти-hlL-6R антитела (или фармацевтическую композицию, включающую антитела) можно вводить пациенту с использованием любого приемлемого устройства или механизма. Например, введение может быть осуществлено с использованием шприца и иглы или многоразовой ручки и/или шприц-ручки. Методы по настоящему изобретению включают применение множества многоразовых ручек и/или шприц-ручек для введения анти-hlL-6R антитела (или фармацевтической композиции, включающей антитела). Примеры таких устройств включают, но не ограничиваются AUTOPEN™ (Owen Mumford, Inc., Woodstock, UK), ручку DISETRONIC™ (Disetronic Medical Systems, Bergdorf, Switzerland), ручку HUMALOG MIX 75/25™, ручку HUMALOG™, ручку HUMALIN 70/30™ (Eli Lilly и Co., Indianapolis, IN), NOVOPEN™ I, II и III (Novo Nordisk, Copenhagen, Denmark), NOVOPEN JUNIOR™ (Novo Nordisk, Copenhagen, Denmark), ручку BD™ (Becton Dickinson, Franklin Lakes, NJ), OPTIPEN™, OPTIPEN PRO™, OPTIPEN STARLET™, и OPTICLIK™ (sanofi-aventis, Frankfurt, Germany), называя только несколько. Примеры многоразовых ручек и/или шприц-ручек, применяющихся для подкожной доставки фармацевтической композиции по настоящему изобретению, включают, но не ограничиваются ручку SOLOSTAR™ (sanofi-aventis), FLEXPEN™ (Novo Nordisk), и KWIKPEN™ (Eli Lilly), аутоинжектор SURECLICK™ (Amgen, Thousand Oaks, CA), PENLET™ (Haselmeier, Stuttgart, Germany), EPIPEN (Dey, L.P.), и ручку HUMIRA™ (Abbott Labs, Abbott Park, IL), называя только несколько.
Применение микроинфузора для введения пациенту анти-hlL-6R антител (или фармацевтически приемлемой композиции, включающей антитела) также предусматривается в настоящем описании. Как используется в настоящем описании, термин "микроинфузор" обозначает устройство для подкожной доставки, созданное для медленного введения больших объемов (например, до около 2,5 мл или более) терапевтической композиции в течение длительного периода времени (например, около 10, 15, 20, 25, 30 или более минут). См., например, US 6629949; US 6659982; и Meehan et al., J. Controlled Release 46: 107-116 (1996). Микроинфузора являются особенно применимыми для доставки больших доз терапевтических белков, содержащихся в высокой концентрации (например, около 100, 125, 150, 175, 200 или более мг/мл) и/или вязких растворов.
Комбинированная терапия
Настоящее изобретение включает способы лечения ревматоидного артрита, которые включают введение пациенту, нуждающемуся в таком лечении, анти-hlL-6R антител в комбинации с, по меньшей мере, одним дополнительным терапевтическим средством. Примеры дополнительных терапевтических средств, которые могут быть введены в комбинации с анти-hlL-6R антителом в осуществлении методов по настоящему изобретению, включают, но не ограничиваются DMARD, и любые другие соединения, известные для лечения, профилактики или облегчения ревматоидного артрита у человека. Специфические неограничивающие примеры дополнительных терапевтических средств, которые можно вводить в комбинации с анти-hlL-6R антителами в контексте метода по настоящему изобретению, включают, но не ограничиваются метотрексат, сульфасалазин, гидроксихлорохин и лефлуномид. В настоящих способах дополнительные терапевтические средства(о) можно вводить одновременно или последовательно с анти-hlL-6R антителами. Например, для одновременного введения может быть получена фармацевтическая композиция, которая содержит и анти-hlL-6R антитела и, по меньшей мере, одно дополнительное терапевтическое средство. Количество дополнительного терапевтического средства, которое вводят в комбинации с анти-hlL-6R антителами в осуществлении методов настоящего изобретения, может быть легко определено с использованием обычных методов, известных и легкодоступных в области техники.
Описание изобретения обеспечивает фармацевтические композиции, включающие любое из следующего:
Композиция, включающая от 100 до 150 мг сарилумаба (SAR153191) и 10-25 мг метотрексата.
Композиция, включающая от 100 до 200 мг сарилумаба (SAR153191) и 10-25 мг метотрексата.
Композиция, включающая от 100 до 150 мг сарилумаба (SAR153191) и 6-25 мг метотрексата.
Композиция, включающая от 100 до 200 мг сарилумаба (SAR153191) и 6-25 мг метотрексата.
Композиция, включающая от 100 до 150 мг сарилумаба (SAR153191) и 10-20 мг лефлуномида.
Композиция, включающая от 100 до 200 мг сарилумаба (SAR153191) и 10-20 мг лефлуномида.
Композиция, включающая от 100 до 150 мг сарилумаба (SAR153191) и 1000-3000 мг сульфасалазина.
Композиция, включающая от 100 до 200 мг сарилумаба (SAR153191) и 1000-3000 мг сульфасалазина.
Композиция, включающая от 100 до 150 мг сарилумаба (SAR153191) и 200-400 мг гидроксихлорохина.
Композиция, включающая от 100 до 200 мг сарилумаба (SAR153191) и 200-400 мг гидроксихлорохина.
Описание изобретения обеспечивает способы облегчения симптомов, ассоциированных с ревматоидным артритом, включающие любое из следующего:
Способ, включающий введение пациенту, нуждающемуся в этом, от 100 до 150 мг сарилумаба (SAR153191) и 10-25 мг метотрексата в неделю.
Способ, включающий введение пациенту, нуждающемуся в этом, от 100 до 200 мг сарилумаба (SAR153191) один раз в две недели и 10-25 мг метотрексата в неделю.
Способ, включающий введение пациенту, нуждающемуся в этом, от 100 до 150 мг сарилумаба (SAR153191) и 6-25 мг метотрексата в неделю.
Способ, включающий введение пациенту, нуждающемуся в этом, от 100 до 200 мг сарилумаба (SAR153191) один раз в две недели и 6-25 мг метотрексата в неделю.
Способ, включающий введение пациенту, нуждающемуся в этом, от 100 до 150 мг сарилумаба (SAR153191) в неделю и 10-20 мг лефлуномида в сутки.
Способ, включающий введение пациенту, нуждающемуся в этом, от 100 до 200 мг сарилумаба (SAR153191) один раз в две недели и 10-20 мг лефлуномида в сутки.
Способ, включающий введение пациенту, нуждающемуся в этом, от 100 и 150 мг сарилумаба (SAR153191) в неделю и 1000-3000 мг сульфасалазина в сутки.
Способ, включающий введение пациенту, нуждающемуся в этом, от 100 до 200 мг сарилумаба (SAR153191) один раз в две недели и 1000-3000 мг сульфасалазина в сутки.
Способ, включающий введение пациенту, нуждающемуся в этом, от 100 до 150 мг сарилумаба (SAR153191) в неделю и 200-400 мг гидроксихлорохина в сутки.
Способ, включающий введение пациенту, нуждающемуся в этом, от 100 до 200 мг сарилумаба (SAR153191) один раз в две недели и 200-400 мг гидроксихлорохина в сутки.
Биомаркеры
Настоящее описание включает методы лечения ревматоидного артрита путем введения пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества человеческого антитела или его антиген-связывающего фрагмента, которое специфически связывается с hlL-6R, и терапевтически эффективного количества одного или более DMARD, где уровень одного или более PA-ассоциированных биомаркеров у пациента модифицируется (например, увеличивается, снижается и др., как может быть) после введения. В связанном аспекте настоящее изобретение включает методы для снижения PA-ассоциированного биомаркера у пациента путем введения пациенту терапевтически эффективного количества человеческого антитела или его антиген-связывающего фрагмента, которые специфически связываются с hlL-6R, и терапевтически эффективного количества одного или более DMARD.
Примеры PA-ассоциированных биомаркеров включают, но не ограничиваются, например, высокочувствительный C-реактивный белок (hsCRP), амилоид сыворотки A (SAA), скорость оседания эритроцитов (СОЭ), гепсидин сыворотки, интерлейкин-6 (IL-6), и гемоглобин (Hb). Как понимает обычный специалист в области техники, повышение или снижение PA-ассоциированного биомаркера может быть определено путем сравнения уровня биомаркера, измеренного у пациента в определенную временную точку после введения анти-IL-6R антитела, с уровнем биомаркера, измеренным у пациента перед введением (т.е. "исходное измерение"). Определенная временная точка, в которой может быть измерен биомаркер, может составлять, например через около 4 часа, 8 часов, 12 часов, 1 день, 2 дня, 3 дня, 4 дня, 5 дней, 6 дней, 7 дней, 8 дней, 9 дней, 10 дней, 15 дней, 20 дней, 35 дней, 40 дней или более после введения анти-hlL-6R антитела.
В соответствии с определенным вариантом осуществления изобретения по настоящему изобретению у пациента может проявляться снижение уровня одного или более из hsCRP, SAA, ESR и/или гепсидина после введения пациенту анти-hlL-6R антитела. Например, через приблизительно 12 недель после еженедельного введения анти-hlL-6R антител и одного или более DMARD пациент может проявлять одно или более из следующего: (i) снижение hsCRP на около 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% или более; (ii) снижение SAA на около 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% или более; (iii) снижение СОЭ на около 15%, 20%, 25%, 30%, 35%, 40%, 45%, 50%, 55% или более; и/или (iv) снижение гепсидина на около 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75% или более.
В соответствии с определенным другим вариантом осуществления изобретения по настоящему изобретению пациент может иметь повышение уровня одного или более из НЬ или IL-6 после введения пациенту анти-hlL-6R антител и одного или более DMARD. Например, через приблизительно 12 недель еженедельного введения анти-hlL-6R антител и одного или более DMARD у пациента может развиться одно или более из следующего: (v) повышение Hb на около 0,5%, 1,0%, 1,5%, 2,0%, 2,5%, 3,0%, 3,5%, 4,0%, 4,5%, 5,0%, 5,5%, 6,0% или более; и/или (vi) повышение IL-6 на около 100%, 150%, 200%, 250%, 300%, 350%, 400%, 450%, 500%, 550%, 600%, 650%, 700%, 750%, 800% или более.
Настоящее изобретение включает методы для определения того, является ли субъект подходящим пациентом, которому будет полезным введение анти-hlL-6R антител. Например, если у пациента перед получением анти-hlL-6R антител и/или одного или более DMARD, имелся уровень PA-ассоциированного биомаркера, который подтверждает патологическое состояние, следовательно, субъекта определяют как подходящего пациента, для которого введение анти-hlL-6R антитела будет полезным. В соответствии с определенным примерным вариантом осуществления изобретения пациент может быть идентифицирован как хороший кандидат для терапии анти-hlL-6R/DMARD, если у пациента имеется одно или более из следующего: (i) уровень hsCRP более чем около 4 мг/л (например, около 4,5 мг/л, около 5,0 мг/л, около 5,5 мг/л, около 6,0 мг/л, около 7,0 мг/л, около 10,0 мг/л, около 15,0 мг/л, около 20,0 мг/л, или более); (ii) уровень SAA более чем около 3800 нг/мл (например, около 4000 нг/мл, 4500 нг/мл, около 5000 нг/мл, около 5500 нг/мл, около 6000 нг/мл, около 10000 нг/мл, около 20000 нг/мл, около 25000 нг/мл, около 30000 нг/мл, около 35000 нг/мл, около 40000 нг/мл, около 45000 нг/мл, или более); (iii) СОЭ более чем около 15 мм/ч (например, около 16 мм/ч, около 17 мм/ч, около 18 мм/ч, около 19 мм/ч, около 20 мм/ч, около 21 мм/ч, около 22 мм/ч, около 25 мм/ч, около 30 мм/ч, около 35 мм/ч, около 40 мм/ч, около 45 мм/ч, около 50 мм/ч, или более); и/или (iv) уровень гепсидина более чем около 60 нг/мл (например, около 62 нг/мл, около 64 нг/мл, около 68 нг/мл, около 70 нг/мл, около 72 нг/мл, около 74 нг/мл, около 76 нг/мл, около 78 нг/мл, около 80 нг/мл, около 82 нг/мл, около 84 нг/мл, около 85 нг/мл, около 90 нг/мл, около 95 нг/мл, около 100 нг/мл, около 105 нг/мл, или более). Дополнительные критерии, такие как другие клинические показатели РА, могут быть использованы в комбинации с любым из вышеперечисленных PA-ассоциированных биомаркеров для определения пациента, как подходящего кандидата для анти-hlL-6R терапии.
Популяция пациентов
В определенных вариантах осуществления изобретения методы и композиции, описанные в настоящем описании, применяют у специфических популяций пациентов. Такие популяции включают пациентов, которых ранее лечили от ревматоидного артрита с помощью схем лечения, иных, чем комбинация анти-hlL-6R антител и одного или более DMARD. Такие схемы лечения включают анти-TNF-α терапию, например схему лечения биологическими анти-TNF-α. Биологические анти-TNF-α антагонисты включают этанерцепт, инфликсимаб, адалимумаб, голимумаб и сертолизумаб пегол. Такие схемы лечения также включают терапию DMARD в отсутствие анти-hlL-6R антител.
DMARD, используемые в таком лечении, включают метотрексат, сульфасалазин, гидроксихлорохин и лефлуномид. DMARD можно вводить отдельно или в комбинации с другой терапией, которая не является анти-hlL-GR антителами. В специфическом варианте осуществления изобретения предшествующей схемой лечения был метотрексат. В другом варианте осуществления изобретения лечение метотрексатом сохраняли у пациента, получающего лечение анти-hlL-6R антителами. В определенных вариантах осуществления изобретения пациенту ранее вводили и анти-TNF-α и DMARD. Лечение можно проводить последовательно в любом порядке или одновременно. В определенных вариантах осуществления изобретения такое лечение получали пациенты в течение 2 лет перед получением комбинации анти-hlL-6R антитела и одного или более DMARD. В других вариантах осуществления изобретения такое лечение получали в течение 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 лет перед получением комбинации анти-hlL-6R антитела и одного или более DMARD.
В определенных вариантах осуществления изобретения методы и композиции, описанные в настоящем описании, применяют у специфической популяции пациентов, которые получали одну или более схем лечения, описанных выше, где проводимое лечение не было эффективным. Как используется в настоящем описании, лечение не считается эффективным, когда доза анти-TNF-α и DMARD не приводит к определяемому улучшению одного или более симптомов, ассоциированных с ревматоидным артритом, или не вызывает биологического эффекта (например, снижения уровня биологического биомаркера), который коррелирует с лежащим в основе патологическим механизмом(ами), дающим состояние или симптом(ы) ревматоидного артрита.
В другом примере лечение не считается эффективным, когда доза анти-TNF-α не приводит к определяемому улучшению одного или более симптомов, ассоциированных с ревматоидным артритом, и не вызывает биологического эффекта (например, снижения уровня определенного биомаркера), который коррелирует с лежащим в основе патологическим механизмом(ами), дающими развитие состояния или симптома(ов).
В другом примере лечение не считается эффективным, когда доза анти-hlL-6R антитела и DMARD не приводит к определяемому улучшению одного или более симптомов, ассоциированных с ревматоидным артритом, и не вызывает биологического эффекта, который коррелирует с лежащим в основе патологическим механизмом(ами), дающим состояние или симптом(ы) ревматоидного артрита.
В определенном варианте осуществления изобретения сарилумаб вводят пациенту, которого ранее неэффективно лечили DMARD. Как используется в настоящем описании, лечение DMARD не было эффективным, когда после лечения у пациента все еще присутствовало "активное заболевание". Пациенты имеют активное заболевание, когда у них имеется, по меньшей мере, 8 из 68 болезненных суставов и 6 из 66 отечных суставов, и высокочувствительный C-реактивный белок (hs-CRP) >10 мг/л (>1,0 мг/дл). В специфическом варианте осуществления изобретения пациентов ранее неэффективно лечили МТХ. В таком примере пациенты получали непрерывное лечение МТХ 10-25 мг/неделю (или согласно местным рекомендациям, если диапазон доз отличается) в течение, по меньшей мере, 12 недель и стабильную дозу МТХ в течение минимум 8 недель и все еще имеют PA от умеренной до тяжелой активности, определяемой как: (i) по меньшей мере, 8 из 68 болезненных суставов и 6 из 66 отечных суставов, и (ii) высокочувствительный C-реактивный белок (hs-CRP) >10 мг/л (>1,0 мг/дл).
Например, лечение, которое не дало улучшения любого из следующих симптомов или состояний, расценивается неэффективным: хроническая анемия, лихорадка, депрессия, слабость, ревматоидные узелки, васкулит, нейропатия, склерит, перикардит, синдром Фелти и/или деструкция суставов.
Определяемое улучшение также может быть выявлено с использованием критериев классификации ревматоидного артрита Американской Коллегии ревматологов (ACR). Например, 20% (ACR20), 50% (ACR50) или 70% (ACR70) улучшение от исходного может быть использовано для демонстрации определяемого улучшения.
Шкала активности заболевания (DAS28) может быть использована для демонстрации определяемого улучшения. DAS28 представляет собой комплексную шкалу количества болезненных суставов на основании 28 суставов, количества отечных суставов на основании 28 суставов, оценку общего состояния здоровья и маркеры воспаления, которые могут быть оценены путем измерения уровня C-реактивного белка (CRP). Ответ заболевания может быть представлен с использованием критериев ответа Европейской Лиги ревматизма (EULAR). Хороший ответ по таким критериям представляет собой улучшение более чем на 1,2 балла DAS28 с настоящим баллом больше или равным 3,2. Умеренный ответ представляет собой улучшение более чем на 0,6, но менее чем или равное 1,2 балла DAS28 и настоящий балл более чем 3,2. Отсутствие ответа представляет собой улучшение менее чем на 0,6 балла DAS28 и настоящий балл более чем 5,1. Ремиссия DAS28 представляет собой балл DAS28 менее чем 2,6. Определяемое улучшение также может быть показано путем измерения улучшения любого из компонентов DAS28 шкалы.
ПРИМЕРЫ
Пример 1. Комбинация Сарилумаба и Метотрексата является эффективной в лечении ревматоидного артрита у пациентов, у которых лечение Метотрексатом оказалось неэффективным.
Международное, двойное слепое, плацебоконтролируемое, рандомизированное исследование проводили среди пациентов с ревматоидным артритом с неадекватным ответом на метотрексат (МТХ). Пациенты, которых включали в исследование, соответствовали следующим критериям. Пациенты должны были иметь активное заболевание, определяемое как: по меньшей мере, 6 из 66 отечных суставов и 8 из 68 болезненных суставов и; hs-CRP >6 мг/л. Пациенты также должны были проходить непрерывное лечение метотрексатом (МТХ) - 10-25 мг/нед. (или 6-25 мг/нед. для пациентов из Азиатско-Тихоокеанского региона) в течение 12 недель.
Исследование включает две части. Первой частью (Часть A) исследования было 12-недельное, дозозависимое исследование в 6 группах, предназначенное для выбора двух наилучших схем лечения, основанного на эффективности (уменьшение признаков и симптомов) и безопасности. Вторая часть (Часть B) исследования представляет собой 52-недельное исследование для подтверждения эффективности и безопасности таких двух выбранных схем лечения в отношении облегчения признаков и симптомов, ингибирования прогрессирования структурного повреждения, улучшения физической функции, и индукции основного клинического ответа.
Дизайн настоящего исследования с непрерывным переходом между фазами обусловлен тем фактом, что Часть B исследования у пациентов начинают сразу после того, как последний пациент был рандомизирован в Части A, не ожидая выбора дозы, основанного на его результатах. Следовательно, пациенты части B принадлежат к 2 отдельным когортам в соответствии со временем их включения.
Когорту 1 пациентов рандомизировали до выбора дозы: этих пациентов рандомизировали в шесть групп (как в Части А). После выбора дозы пациенты, рандомизированные на схемы двух выбранных доз и плацебо, продолжали участие в течение 52 недель, а рандомизированные в три другие группы прекращали участие в настоящем исследовании, но им предлагали вступить в открытое продолжение (см. LTS11210).
Когорту 2 пациентов рандомизировали после выбора дозы: таких пациентов рандомизировали в три группы, две выбранные дозы и плацебо.
Часть A
Пациентов оценивали на визите скрининга для подтверждения диагноза, активности заболевания, соответствия исследованию и верификации сопутствующей терапии. Проводили полное обследование и лабораторные анализы, включая гематологию, биохимический профиль, липидный профиль, печеночные ферменты и реагенты острой фазы, HbA1c, гепатит B и C и тест на беременность по сыворотке крови для женщин детородного возраста. Также проводили исследование ЭКГ. Для исключения туберкулеза проводили Тест PPD и QuantiFERON, а также рентгенограмму грудной клетки (при отсутствии документированной отрицательной рентгенограммы за последние 3 месяца).
После подтверждения соответствия пациентов рандомизировали сбалансированным образом в указанном международном многоцентровом, двойном слепом, плацебоконтролируемом 12-недельном исследовании лечения в параллельных шести группах SAR153191 или плацебо, вводимых еженедельно подкожно с сопутствующей терапией МТХ. Дозы показаны на фиг. 1.
Метотрексат вводили каждому пациенту, как это было до исследования. Доза составляла 10-25 мг/нед. или 6-25 мг/нед. для пациентов Азиатско-Тихоокеанского региона; Тайвань, Южная Корея, Малайзия, Филиппины, Таиланд и Индия.
В течение первого визита пациентам напоминали список запрещенных лекарственных препаратов, и то, что они должны продолжать принимать МТХ в их обычной стабильной дозе до конца исследования с фолиевой кислотой для предотвращения токсичности МТХ согласно местным рекомендациям. Пациентов учили готовить и самостоятельно вводить IMP и напоминали делать инъекции точно через 7 дней. В моменты введения вне визитов в центр, SAR153191 вводил самостоятельно пациент, обученный социальный работник или обученный профессионал.
Пациентам проводили шесть дополнительных визитов на 2, 4, 6, 8, 10 и 12 неделе. Оценку эффективности и лабораторные исследования, включая гематологию, биохимический профиль, липидный профиль, ферменты печени и белки острой фазы проводили на всем протяжении исследования для возможности расчета основных показателей эффективности и отслеживания аспектов безопасности. На визите рандомизации и в Недели 2, 4, 8 и 12, эксперт, независимый от Исследователя и данных пациента, проводил полное обследование суставов в отношении числа болезненных суставов и числа отечных суставов с целью расчета балла ACR (основная конечная точка). С целью сохранения заслепления Исследователь, Спонсор и пациент были заслеплены относительно уровня CRP и IL6 сыворотки в течение исследования.
Четкий мониторинг нежелательных явлений, включая потенциальные инфекции, оцениваемые частично путем наблюдения за температурой тела, проводили на каждом визите. Наличие туберкулеза проверяли посредством специфического обследования пациента (проверка любых признаков или симптомов или контакт с активным ТВ). Неврологические нарушения (анамнез и физикальное обследование) или аутоиммунные диатезы (ANA, антитела ds-DNA) исследовали исходно и в конце лечения.
Специфические образцы крови и мочи получали в течение исследования для анализа потенциальных биомаркеров, которые могут быть предвестниками ответа заболевания или нежелательных явлений. Они включают один образец для ДНК (после подписания пациентом специфической формы информированного согласия) и несколько образцов, полученных последовательно на всем протяжении исследования, для анализа профиля экспрессии РНК и белков биомаркеров. Образцы также получали в соответствующие временные точки для оценки фармакокинетических параметров и антител к SAR153191.
Пациентов, преждевременно прекративших участие, оценивали на визите окончания лечения с проведением полного клинического и лабораторного обследования. Их расценивали как не давших ответа в отношении балла ACR.
На визите окончания лечения всем пациентам назначали визит наблюдения после лечения. Пациентам, которые завершили период лечения, было предложено вступить в открытое длительное продолжение исследования безопасности с SAR153191.
Результаты
Пациенты, получавшие лечение сарилумабом (REGN88/SAR153191) в комбинации со стандартным лечением PA, метотрексатом (МТХ), достигли достоверного или клинически значимого улучшения признаков и симптомов средней тяжести - тяжелого ревматоидного артрита (РА) по сравнению с пациентами, получавшими только МТХ. Среди 306 пациентов проводили дозозависимое, международное, рандомизированное, в нескольких группах, двойное слепое, плацебоконтролируемое исследование, в котором сравнивали пять различных схем лечения сарилумабом в комбинации с МТХ, с плацебо плюс МТХ. Основной конечной точкой была доля пациентов, достигших, по меньшей мере, 20% улучшения симптомов PA (ACR20) через 12 недель.
Ответ на дозу наблюдали у пациентов, получавших сарилумаб в комбинации с МТХ. ACR20 ответ через 12 недель наблюдали у 49,0% пациентов, получавших наименьшую дозу сарилумаба, и 72,0% пациентов, получавших наивысшую дозу, по сравнению с 46,2% пациентов, получавших плацебо и МТХ (p=0,02, скоррелированный для множественности, для наивысшей дозы сарилумаба) (фиг. 2). Наиболее частые нежелательные явления (>5%), наиболее часто выявляемые в группах с активным лечением, включают инфекции (несерьезные), нейтропению и изменения показателей функции печени. Типы и частоты нежелательных явлений согласовывались с таковыми, ранее сообщаемыми для ингибирования IL-б. Частота серьезных нежелательных явлений среди пяти групп лечения сарилумабом и группой плацебо были сопоставимыми.
Сарилумаб также продемонстрировал достоверное преимущество по сравнению с плацебо во вторичных конечных точках, включая баллы ACR50, ACR70, и DAS28, дополнительные показатели клинической активности, используемые в исследованиях PA. Точнее:
- ответ ACR50 через 12 недель наблюдали у 22% пациентов, получавших наименьшую дозу сарилумаба, и 30% пациентов, получавших наивысшую дозу, по сравнению с 15% пациентов, получавших плацебо и МТХ (фиг. 3);
- ACR70 также был достоверно выше в группе 200 мг q2w по сравнению с плацебо. ACR70 ответ через 12 недель наблюдали у 16% пациентов, получавших наивысшую дозу, по сравнению с 2% пациентов, получавших плацебо и МТХ (фиг. 3).
Полученные результаты обеспечивают доказательства того, что блокада IL-6R сарилумабом представляет собой многообещающую новую противовоспалительную исследуемую терапию для уменьшения патологических симптомов PA.
Часть B
Пациентов оценивали на визите скрининга для подтверждения диагноза, активности заболевания, приемлемости для участия в исследовании и уточнения сопутствующей терапии. Исследователь проверяет, является ли пациент положительным или по антициклическим цитруллинированным пептидным антителам (CCP) или положительному ревматоидному фактору (RF) или он/она имеет подтвержденную эрозию кости по рентгенографии. Если необходимо, для пациентов, которые являются и CCP и RF отрицательными и не имеют рентгенографии, проводят скрининговую рентгенографию оцениваемую централизованно и ее расценивают также как исходное рентгеновское исследование.
Группа 1: Пациенты, рандомизированные до выбора дозы
Набор для длительного продолженного исследования безопасности начинают сразу после того, как последний пациент был рандомизирован в часть А. После подтверждения приемлемости пациентов рандомизируют сбалансированном образом, стратифицированным по первичному биологическому применению и по регионам, в международном, многоцентровом, двойном слепом, плацебоконтролируемом исследовании лечения в параллельных 6 группах SAR153191 (5 активных схем введения) или плацебо, вводимых подкожно еженедельно с сопутствующей терапией МТХ.
В начале каждого визита для пациентов группы 1, Исследователь проверяет по перечню IVRS, что пациент все еще "подходит" для исследования, т.е. что пациент не должен прекращать участие из-за рандомизации в невыбранную группу. Действительно, когда из Части A выбирают основные схемы лечения, только пациенты, рандомизированные в соответствующие группы или плацебо, все еще рассматриваются приемлемыми для исследования и продолжают участие в исследовании в течение 52 недель. Другие пациенты (рандомизированные в невыбранные схемы лечения) расценивались как более не подходящие по IVRS. Исследователь предлагает таким пациентам участие в открытом продолженном исследовании с SARI53191 в наивысшей дозе, доступной в момент включения пациента.
Исходная рандомизация остается заслепленной для всех пациентов.
Группа 2: Пациенты, рандомизированные после выбора дозы. Основная часть
В день 1 после подтверждения приемлемости к участию пациентов рандомизируют сбалансированным образом, стратифицированными по предшествующему биологическому применению и по регионам, в международном, многоцентровом, двойном слепом, плацебоконтролируемом исследовании в 3 параллельных группах, SAR153191 (2 основные схемы лечения) или плацебо, вводимых подкожно с сопутствующей терапией МТХ.
Обе группы
В каждой группе пациентов оценивали на неделе 2, неделе 4 и каждые 4 недели до недели 28 и затем каждые 8 недель до недели 52 в отношении показателей эффективности и безопасности и лабораторных исследований.
Такие же процедуры, как описано в Части A, применяют в Части B. Кроме того, рентгеновское исследование суставов кисти и стопы проводят исходно, на неделе 24 и неделе 52. Рентгенограммы, лишенные какой-либо информации о пациенте, отправляют исследователям в центр для расчета балла Sharp (специфическая система оценки деструкции суставов). Также добавляют фармакоэкономические оценки, такие как SF-36.
С недели 16 пациентам с отсутствием эффективности, определяемым как менее чем 20% улучшение от исходного или в количестве отечных суставов (SJC) или количестве болезненных суставов (TJC) на 2 последовательных визитах, или любое другое четкое отсутствие эффективности по мнению Исследователя, предлагают перейти на открытое лечение SAR153191 наивысшей доступной дозой в момент перехода в группу спасательного лечения и продолжить участие в исследовании в соответствии с планируемым расписанием визитов. Образцы крови для лабораторных анализов получают через две недели после перехода по причинам безопасности. Их расценивают как не-ответчиков в отношении первичной конечной точки (ACR20). Такие пациенты остаются в исследовании и продолжают все визиты.
В выбранных странах пациенты, которые не соответствовали критериям эффективности лечения части B визит 7/неделя 16 или после этого, преждевременно прекращают лечение и не могут принимать участие в открытой группе спасательного лечения. Вместо этого пациентам проводят визит наблюдения для оценки безопасности через 6 недель после визита завершения лечения.
Для любого пациента, который преждевременно прекращает участие в исследовании или которого преждевременно переводят на открытый SAR153191, в момент отмены или перехода проводят дополнительное рентгеновское исследование, если рентгеновское исследование не проводили в течение предшествующих 3 месяцев (окно 3 месяца между 2 рентгеновскими обследованиями используют во избежание избыточного воздействия рентгеновского излучения).
Пациентам, завершившим Часть B (включая таковых в открытой переходной группе), предлагают войти в открытое продолженное исследование в максимальной дозе в момент включения. Всем пациентам назначают визит наблюдения после лечения. Если пациент соглашается вступить в открытое длительное продолженное исследование SAR153191 и подходит для этого, визит наблюдения после завершения лечения не проводят.
Пример 2. Комбинация Сарилумаба и DMARD является эффективной в лечении ревматоидного артрита у пациентов, у которых неэффективно лечение антагонистом TNF-α и Метотрексатом
Международное двойное слепое, плацебоконтролируемое, рандомизированное исследование проводили у пациентов с ревматоидным артритом с неадекватным ответом на метотрексат (МТХ) и, по меньшей мере, один антагонист TNF-α. Пациенты, которых включали в исследование, соответствовали следующим критериям. Пациенты имели, по мнению исследователя, неадекватный ответ на, по меньшей мере, один антагонист TNF-α после лечения в течение, по меньшей мере, 3 месяцев за последние 2 года или пациенты не переносили, по меньшей мере, 1 антагонист TNF-α, что приводило к прекращению лечения. Антагонисты TNF-α включали этанерцепт, инфликсимаб, адалимумаб, голимумаб и/или сертолизумаб пегол. Пациенты должны были иметь активное заболевание, определяемое как: по меньшей мере, 6 из 66 отечных суставов и 8 из 68 болезненных суставов и hs-CRP ≥8 мг/л. Пациенты также должны иметь непрерывное лечение одним или комбинацией DMARD в течение, по меньшей мере, 12 недель перед началом и стабильную дозу(ы) в течение, по меньшей мере, 6 недель перед скринингом. Такие DMARD включают метотрексат (МТХ) - 10-25 мг/нед. (или 6-25 мг/нед. для пациентов Азиатско-Тихоокеанского региона; лефлуномид (LEF) - 10-20 мг в сутки; сульфасалазин (SSZ) - 1000-3000 мг в сутки; или гидроксихлорохин (HCQ) - 200-400 мг в сутки.
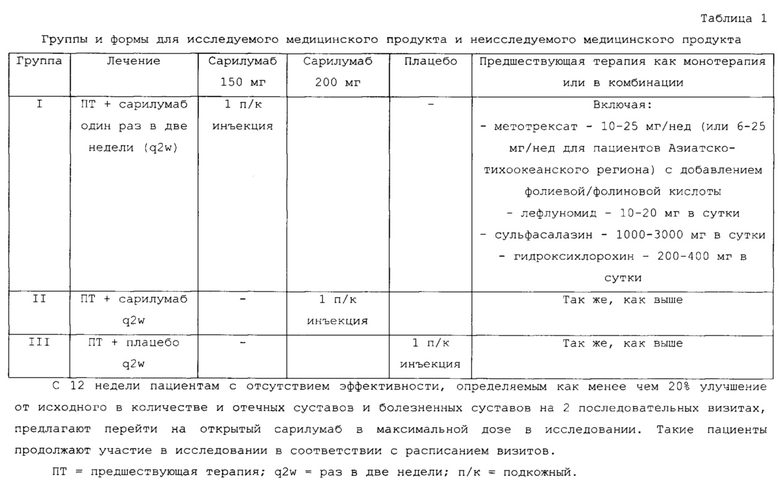
Подкожное введение проводят в живот или бедро. Каждую дозу вводят самостоятельно (если возможно), в одной инъекции. Участки п/к инъекций можно чередовать между 4 квадрантами живота (за исключением пупка или области талии) или бедра (передняя и боковая поверхность).
Пациентов и/или их непрофессиональных помощников обучают приготовлению и введению IMP в начале периода двойного слепого лечения. Такой тренинг должен быть задокументирован в карте пациента. Координатор исследования или уполномоченное лицо могут вводить первую инъекцию, включающую исходную дозу, как часть обучающей процедуры в День 1 (Визит 2). В дни, когда пациент приходит на визит в центр, IMP вводят после клинических процедур и сбора крови. Для доз, вводимых не в исследовательском центре, выдают дневники для записи информации, касающейся таких инъекций; такие дневники сохраняют как первичные данные в карте пациента. Если пациент неспособен или не желает вводить IMP, должны быть сделаны назначения для квалифицированного персонала центра и/или помощника для введения IMP в дозах, которые не вводят в исследовательском центре.
Если визит по исследованию не проведен в центре, как назначено, дозу вводят пациенты и/или их помощник(и), как назначено.
Лечение продолжается в течение 24 недель. С недели 12 пациентам при отсутствии эффективности, определяемой как менее чем 20% улучшение от исходного в SJC и TJC на 2 последовательных визитах, предлагают перейти на открытый сарилумаб в максимальной дозе в исследовании. Такие пациенты продолжают участие в исследовании в соответствии с расписанием визитов.
В проводимом исследовании сарилумаб вводят на фоне DMARD терапии, расцениваемой как предшествующая терапия. Все пациенты должны продолжать получать непрерывное лечение одним или комбинацией небиологических DMARD в качестве предшествующей терапии в течение, по меньшей мере, 12 недель перед началом и в стабильной дозе(ах) в течение, по меньшей мере, 6 недель перед скринингом:
- метотрексат (МТХ) - 10-25 мг/нед. (или 6-25 мг/нед. для пациентов Азиатско-Тихоокеанского региона) с дополнением фолиевой/фолиновой кислотой,
- лефлуномид (LEF) - 10-20 мг в сутки,
- сульфасалазин (SSZ) - 1000-3000 мг в сутки,
- гидроксихлорохин (HCQ) - 200-400 мг в сутки.
Каждую дозу DMARD записывают на всем протяжении исследования в индивидуальную регистрационную карту. В любой момент времени доза DMARD может быть снижена по причине безопасности или переносимости. Любое изменение дозы и дата начала новой дозы должны быть зафиксированы в ИРК на каждом визите. DMARD не выдаются и не поставляются Спонсором как IMP.
Все пациенты, принимающие МТХ, получают фолиевую/фолиновую кислоту в соответствии с местными рекомендациями в стране, где проводится исследование. Доза, путь и расписание введения фолиевой/фолиновой кислоты записывают с сопутствующим лечением.
Сарилумаб и соответствующее плацебо обеспечивают в идентично маркированных стеклянных заранее заполненных шприцах. Каждый заранее заполненный шприц содержит 1,14 мл раствора сарилумаба (SAR153191) или соответствующего плацебо.
Создается перечень номеров наборов. Рандомизированный список создается посредством интерактивной системы голосового ответа (IVRS). Оба списка рандомизации и наборов лечения загружены в IVRS.
Номера наборов лечения получает Исследователь в момент рандомизации пациента и при последующих назначенных визитах пациента посредством IVRS, которая доступна 24 часа в сутки.
В соответствии с двойным слепым дизайном Исследователи остаются заслепленными к исследуемому лечению и не имеют доступа к рандомизации (кодам лечения) за исключением обстоятельств, описанных в разделе 8.7.
Пациентов рандомизируют в одну из групп лечения посредством централизованной системы рандомизации с использованием IVRS. Пациент расценивается рандомизированным, когда системой IVRS выдается номер лечения.
На визите скрининга, Визите 1, координатор в центре связывается с IVRS для получения номера пациента для каждого пациента, который дал информированное согласие. Каждому пациенту присваивается номер пациента, ассоциированный с центром и назначенный в хронологическом порядке в каждом центре.
Назначение лечения проводится пациенту в соответствии с центральным списком рандомизации посредством IVRS, стратифицированным по региону и количеству предшествующих анти-TNF (1 против >1). На визите 2 (День 1) после подтверждения того, что пациент подходит для участия в лечении, координатор центра связывается с IVRS с целью получения первого назначенного лечения (номера наборов). Пациентов рандомизируют для получения или одной из 2 групп лечения сарилумабом или соответствующего плацебо. Соотношение рандомизации составляет 1:1:1 (сарилумаб 150 мг:сарилумаб 200 мг:соответствующее плацебо). На последующих визитах выдачи в течение периода лечения координатор центра звонит в IVRS для получения назначений последующих наборов лечения. Подтверждающий факс/e-mail отправляются в центр после каждого назначения.
Рандомизированного пациента определяют как пациента, который зарегистрирован и которому назначен рандомизационный номер от IVRS, как задокументировано в протоколе IVRS. IMP также записывают и отслеживают в исследовательских формах IMP центра.
Композиции и методы по настоящему описанию не ограничиваются рамками специфического варианта осуществления изобретения, описанного в настоящем описании. Действительно, различные модификации изобретения в добавление к таковым, описанным в настоящем описании, очевидны специалисту в области техники из выше представленного описания. Такие модификации попадают в рамки приложенной формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ДИАГНОСТИКИ И ЛЕЧЕНИЯ РЕВМАТОИДНОГО АРТРИТА | 2020 |
|
RU2828027C2 |
| КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ | 2009 |
|
RU2531548C2 |
| КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ АРТРИТА | 2009 |
|
RU2563360C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ АНТИТЕЛА К IL6R, ДЛЯ ЛЕЧЕНИЯ УВЕИТА И МАКУЛЯРНОГО ОТЕКА, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2747193C2 |
| АНТИТЕЛА, НЕЙТРАЛИЗУЮЩИЕ GM-CSF, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ РЕВМАТОИДНОГО АРТРИТА ИЛИ В КАЧЕСТВЕ АНАЛЬГЕТИКОВ | 2014 |
|
RU2714919C2 |
| АНТИТЕЛО К РЕЦЕПТОРУ IL-6 ДЛЯ ЛЕЧЕНИЯ ЮВЕНИЛЬНОГО ИДИОПАТИЧЕСКОГО АРТРИТА | 2020 |
|
RU2822089C2 |
| СПОСОБЫ ЛЕЧЕНИЯ РЕВМАТОИДНОГО АРТРИТА С ПРИМЕНЕНИЕМ АНТАГОНИСТОВ IL-17 | 2011 |
|
RU2582937C2 |
| СПОСОБ ЛЕЧЕНИЯ ПОВРЕЖДЕНИЯ СУСТАВОВ | 2006 |
|
RU2457860C2 |
| ЛЕЧЕНИЕ РЕВМАТОИДНОГО АРТРИТА | 2003 |
|
RU2322238C2 |
| ПРИМЕНЕНИЕ АНТАГОНИСТОВ IL-17 ДЛЯ ТОРМОЖЕНИЯ ПРОГРЕССИРОВАНИЯ СТРУКТУРНЫХ ПОВРЕЖДЕНИЙ У ПАЦИЕНТОВ С ПСОРИАТИЧЕСКИМ АРТРИТОМ | 2015 |
|
RU2697383C2 |





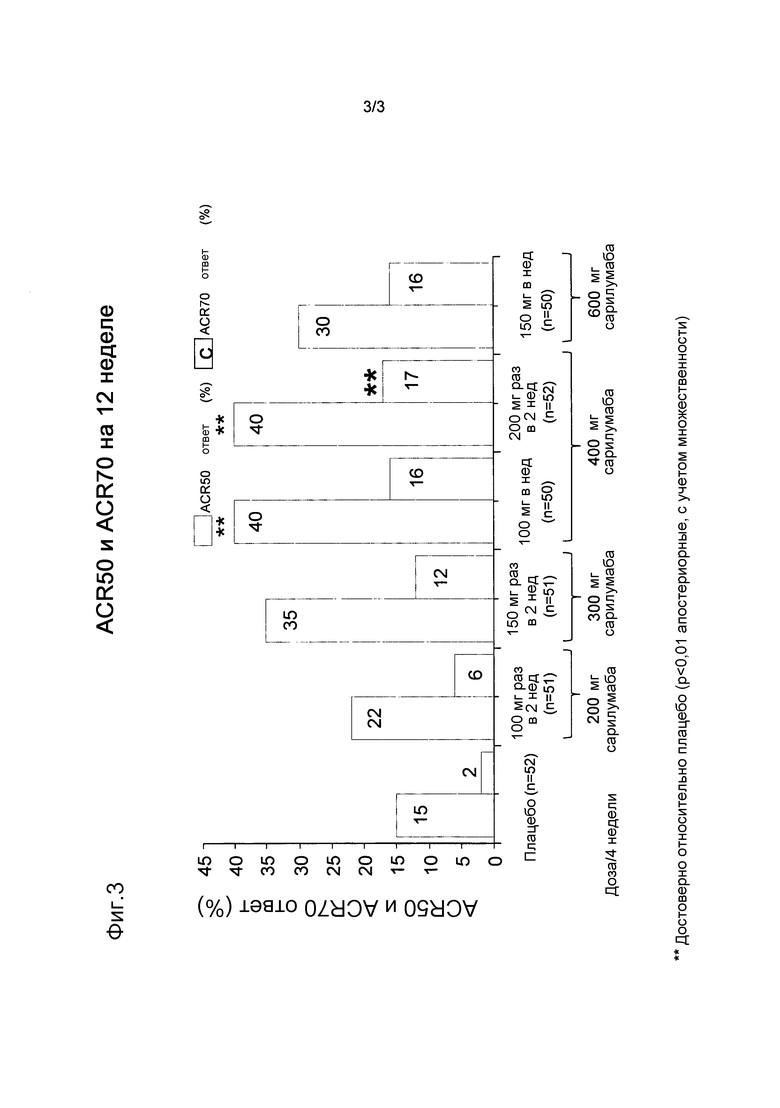
Изобретение относится к медицине, а именно к ревматологии, и может быть использовано для лечения ревматоидного артрита (РА). Для этого пациенту вводят эффективное количество антитела и метотрексата, где указанное антитело включает вариабельный участок тяжелой цепи последовательности SEQ ID NO: 2 и вариабельный участок легкой цепи последовательности SEQ ID NO: 3 и указанное антитело вводят в дозе от 100 мг до 200 мг раз в две недели и где указанного пациента ранее лечили от ревматоидного артрита путем введения метотрексата и ранее неэффективно лечили от РА путем введения антагониста TNF-α, выбранного из группы, состоящей из этанерцепта, инфликсимаба, адалимумаба, голимумаба и/или сертолизумаба пегол. Способ позволяет повысить эффективность лечения РА и облегчить его симптомы за счет специфического связывания антитела или его антиген-связывающего фрагмента с рецептором человеческого интрлейкина-6 (hIL-6R). 9 з.п. ф-лы,3 ил., 1 табл., 2 пр.
1. Способ лечения ревматоидного артрита у пациента, включающий введение указанному пациенту эффективного количества антитела и метотрексата, где указанное антитело включает вариабельный участок тяжелой цепи последовательности SEQ ID NO: 2 и вариабельный участок легкой цепи последовательности SEQ ID NO: 3 и указанное антитело вводят в дозе от 100 мг до 200 мг раз в две недели и где указанного пациента (1) ранее лечили от ревматоидного артрита путем введения метотрексата и (2) ранее неэффективно лечили от ревматоидного артрита путем введения антагониста TNF-α, выбранного из группы, состоящей из этанерцепта, инфликсимаба, адалимумаба, голимумаба и/или сертолизумаба пегол.
2. Способ по п. 1, где указанного пациента ранее неэффективно лечили от ревматоидного артрита путем введения метотрексата.
3. Способ по п. 2, где метотрексат вводят в дозе от 6 до 25 мг в неделю.
4. Способ по п. 1, где антитело вводят в дозе 150 мг раз в каждые две недели или 200 мг раз в каждые две недели.
5. Способ по любому из пп. 1-4, где пациент достигает по меньшей мере 20% улучшения по критериям основного заболевания Американской Коллегии ревматологов после 12 недель лечения.
6. Способ по любому из пп. 1-4, где пациент достигает по меньшей мере 50% улучшения по критериям основного заболевания Американской Коллегии ревматологов после 12 недель лечения.
7. Способ по любому из пп. 1-4, где пациент достигает по меньшей мере 70% улучшения по критериям основного заболевания Американской Коллегии ревматологов после 12 недель лечения.
8. Способ по любому из пп. 1-3, где пациента лечили анти-TNF-α антагонистом в течение, по меньшей мере, 3 месяцев за последние 2 года или пациент не переносит, по меньшей мере, один антагонист TNF-α.
9. Способ по любому из пп. 1-4, где антитело представляет собой сарилумаб.
10. Способ по любому из пп. 1-4, где указанное антитело вводят пациенту подкожно.
| NISHIMOTO N | |||
| et al | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |
| столбец Discussion, абзацы 1 и 3 | |||
| REICHERT J.M | |||
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Двигатель внутреннего горения | 1929 |
|
SU14226A1 |
| ЛЕЧЕНИЕ АУТОИММУННЫХ ЗАБОЛЕВАНИЙ У ПАЦИЕНТА С НЕАДЕКВАТНЫМ ОТВЕТОМ НА ИНГИБИТОР TNF-АЛЬФА | 2004 |
|
RU2358762C9 |
| МАЗУРОВ В.И | |||
| под ред | |||
| Клиническая ревматология | |||
| Руководство для практических врачей // СПб, "Фолиант", 2001, с | |||
| Прибор для массовой выработки лекал | 1921 |
|
SU118A1 |

