РОДСТВЕННЫЕ ЗАЯВКИ
Настоящая заявка испрашивает приоритет предварительной заявки на патент США № 62/799698, поданной 31 января 2019 г., предварительной заявки на патент США № 62/851474, поданной 22 мая 2019 г., предварительной заявки на патент США № 62/935395, поданной 14 ноября 2019 г., и заявки на европейский патент № 19306553.9, поданной 3 декабря 2019 г.; каждая из которых включена в данный документ посредством ссылки во всей своей полноте.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к области терапевтического лечения ювенильного идиопатического артрита, такого как системный ювенильный идиопатический артрит и ювенильный идиопатический артрит с полиартикулярным течением (который включает полиартикулярный и распространившийся олигоартикулярный ювенильный идиопатический артрит). Определенные аспекты настоящего изобретения относятся к применению антагонистов рецептора интерлейкина-6 (IL-6R), таких как антитела к IL-6R, для лечения системного ювенильного идиопатического артрита и ювенильного идиопатического артрита с полиартикулярным течением.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Ювенильный идиопатический артрит (JIA) представляет собой наиболее распространенное ревматическое заболевание детского возраста. JIA определяется Международной лигой ассоциаций ревматологов (ILAR) как артрит неизвестной этиологии с началом в возрасте до 16 лет, который сохраняется в течение по меньшей мере 6 недель, когда другие известные состояния были исключены (Petty, R.E. et al., 2001. J Rheumatol. 2004;31(2):390-2; Giannini, E.H. et al., 1997 Arthritis Rheum. 40(7):1202-9; и Macaubas, C. et al., 2009 Nat Rev Rheumatol. 5(11):616-26). Состояние включает семь подтипов согласно определению ILAR, включая JIA с полиартикулярным течением и системный JIA. См. Petty, R.E. et al., 2001. J Rheumatol. 2004;31(2):390-2, который включен в данный документ посредством ссылки во всей своей полноте.
Известного лекарства от JIA не существует. В то время как традиционные способы лечения могут использоваться для лечения пораженных индивидов, необходимы более эффективные способы лечения персистирующих типов JIA.
КРАТКОЕ ОПИСАНИЕ
В настоящем изобретении предусмотрены inter alia способы лечения JIA у субъекта, нуждающегося в этом, включающие введение эффективного количества антитела, которое специфически связывает IL-6R.
В варианте осуществления JIA представляет собой системный JIA (sJIA).
В варианте осуществления антитело, которое специфически связывается с IL-6R, содержит последовательность вариабельного участка тяжелой цепи под SEQ ID NO: 1 и последовательность вариабельного участка легкой цепи под SEQ ID NO: 2.
В различных вариантах осуществления антитело содержит вариабельный участок тяжелой цепи (VH) и вариабельный участок легкой цепи (VL), где VH содержит три определяющие комплементарность участка (CDR), расположенные в последовательности под SEQ ID NO: 1, и где VL содержит три CDR, расположенные в последовательности под SEQ ID NO: 2. В различных вариантах осуществления антитело к IL-6R или его антигенсвязывающий фрагмент содержат три определяющие комплементарность участка тяжелой цепи (HCDR; т. е. HCDR1, HCDR2 и HCDR3) и три определяющие комплементарность участка легкой цепи (LCDR; т. е. LCDR1, LCDR2 и LCDR3), где HCDR1 содержит аминокислотную последовательность под SEQ ID NO: 3; HCDR2 содержит аминокислотную последовательность под SEQ ID NO: 4; HCDR3 содержит аминокислотную последовательность под SEQ ID NO: 5; LCDR1 содержит аминокислотную последовательность под SEQ ID NO: 6; LCDR2 содержит аминокислотную последовательность под SEQ ID NO: 7 и LCDR3 содержит аминокислотную последовательность под SEQ ID NO: 8.
В варианте осуществления антитело представляет собой сарилумаб.
В различных вариантах осуществления с антителом не вводят никакое другое модифицирующее заболевание противоревматическое лекарственное средство (DMARD). В некоторых вариантах осуществления субъекту вводят по меньшей мере одно другое DMARD. В варианте осуществления по меньшей мере одно другое DMARD вводят субъекту параллельно с антителом или одновременно с ним.
В некоторых вариантах осуществления субъект страдает от по меньшей мере одного симптома sJIA, такого как артрит в по меньшей мере 1 суставе в течение по меньшей мере 6 недель с сопутствующей или предшествующей лихорадкой, продолжающейся по меньшей мере 2 недели; кратковременная эритематозная сыпь преимущественно на туловище и конечностях; генерализованная лимфаденопатия; гепатомегалия и/или спленомегалия; полисерозит; потеря веса; усталость; недомогание; лихорадка; повышенное количество лейкоцитов в периферической крови (WBC) (от 25000 до 50000/мл3); повышенное количество тромбоцитов (например, >1×106), заметно повышенная скорость оседания эритроцитов (ESR) >100 мм/ч; анемия и/или высокий уровень ферритина по сравнению со здоровым субъектом. В варианте осуществления лечение субъекта включает уменьшение, замедление, остановку прогрессирования или иное ослабление любого из этих симптомов (или любой их комбинации).
Как используется в данном документе "симптом", связанный с JIA, включает любое клиническое или лабораторное (например, диагностическое) проявление, связанное с JIA, и не ограничивается тем, что субъект может чувствовать или наблюдать.
В различных вариантах осуществления субъект страдает по меньшей мере одним симптомом sJIA, таким как хромота; скованность при пробуждении; нежелание субъекта пользоваться рукой или ногой; пониженный уровень активности; ежедневная лихорадка с высокими пиками (например, достигающая приблизительно 40°C или по меньшей мере 40°C); отек суставов; и/или трудности с мелкой моторикой. В варианте осуществления способа наблюдается улучшение в отношении по меньшей мере одного симптома sJIA у субъекта после введения антитела.
В различных вариантах осуществления способа антитело улучшает по меньшей мере одну оценку или показатель, такие как ACR JIA (например, ACR30 JIA, ACR50 JIA, ACR70 JIA, ACR90 JIA и ACR100 JIA), компонент ACR JIA (например, количество суставов с активным артритом, количество суставов с ограниченным диапазоном движений, общая оценка активности заболевания врачом, реагенты острой фазы, такие как скорость оседания эритроцитов или С-реактивный белок (CRP), опросник оценки здоровья детей, общая оценка самочувствия пациентом (или родителем)) и/или уменьшение лихорадки (например, у субъектов с лихорадкой при первом введении антитела), снижение по сравнению с исходным уровнем количества применяемых кортикостероидов (например, глюкокортикоидов) и/или индекс активности заболевания, представляющего собой ювенильный артрит (такой какиндекс-27 активности заболевания, представляющего собой ювенильный артрит). В различных вариантах осуществления способа улучшение характеризуется по меньшей мере одной оценкой или показателем, такими как ACR30 JIA, ACR50 JIA, ACR70 JIA, ACR90 JIA и/или ACR100 JIA. В различных вариантах осуществления способа улучшение характеризуется по меньшей мере одной оценкой или показателем, такими как общая оценка индекса активности заболевания врачом, оценка общего самочувствия пациентом или родителем, опросник оценки здоровья детей, количество суставов с активным артритом, количество суставов с ограниченным движением, высокочувствительный С-реактивный белок и/или уменьшение лихорадки (например, у субъектов с лихорадкой при первом введении антитела). В различных вариантах осуществления улучшение характеризуется по меньшей мере одним биомаркером. В различных вариантах осуществления способа антитело приводит к клинически неактивному заболеванию, как определено критериями Уоллиса. Критерии Уоллиса определены как общая оценка врача по VAS <1/10, отсутствие активного артрита, отсутствие активного увеита и CRP <10 мг/л. В различных вариантах осуществления способа антитело приводит к клинически неактивному заболеванию, как определено индексом-27 активности заболевания, представляющего собой ювенильный артрит, CRP ≤1. В различных вариантах осуществления способа антитело приводит к клинически неактивному заболеванию или низкой активности заболевания, как определено индексом-27 активности заболевания, представляющего собой ювенильный артрит, CRP ≤3,8.
В варианте осуществления способа у субъекта наблюдается недостаточный ответ на текущее лечение, и субъект рассматривается как кандидат на лечение биологическим модифицирующим заболевание противоревматическим лекарственным средством.
В варианте осуществления способа возраст субъекта составляет от приблизительно 1 года до приблизительно 17 лет. В других вариантах осуществления возраст субъекта составляет от приблизительно 4 до приблизительно 6 лет или от приблизительно 12 до приблизительно 18 лет.
В варианте осуществления способа у субъекта имеются либо 5 или более активных суставов, либо 2 активных сустава и системные симптомы (например, лихорадка). В варианте осуществления способа у субъекта имеются либо 5 или более активных суставов, либо 2 или более активных сустава и лихорадка sJIA выше приблизительно 37,5 °C в течение по меньшей мере 3 из любых 7 последовательных дней, несмотря на глюкокортикоиды в дозе, стабильной в течение по меньшей мере 3 дней.
"Активный сустав" представляет собой сустав с (i) отеком внутри сустава, не вызванным деформацией, и/или (ii) ограничением движений либо с болью, либо с чувствительностью.
В варианте осуществления способа JIA представляет собой JIA с полиартикулярным течением (pcJIA). В некоторых вариантах осуществления pcJIA представляет собой распространенный олигоартикулярный JIA. В различных вариантах осуществления pcJIA представляет собой полиартикулярный JIA, положительный в отношении ревматоидного фактора (RF). В некоторых вариантах осуществления pcJIA представляет собой RF-отрицательный полиартикулярный JIA. В варианте осуществления способа возраст субъекта составляет от приблизительно 12 до приблизительно 14 лет или от приблизительно 7 до приблизительно 9 лет.
В варианте осуществления антитело, которое специфически связывается с IL-6R, содержит последовательность вариабельного участка тяжелой цепи под SEQ ID NO: 1 и последовательность вариабельного участка легкой цепи под SEQ ID NO: 2.
В различных вариантах осуществления антитело содержит VH и VL, где VH содержит три CDR, расположенные в последовательности под SEQ ID NO: 1, и где VL содержит три CDR, расположенные в последовательности под SEQ ID NO: 2. В различных вариантах осуществления антитело к IL-6R или его антигенсвязывающий фрагмент содержат три HCDR (т. е. HCDR1, HCDR2 и HCDR3) и три LCDR (т. е. LCDR1, LCDR2 и LCDR3), где HCDR1 содержит аминокислотную последовательность под SEQ ID NO: 3; HCDR2 содержит аминокислотную последовательность под SEQ ID NO: 4; HCDR3 содержит аминокислотную последовательность под SEQ ID NO: 5; LCDR1 содержит аминокислотную последовательность под SEQ ID NO: 6; LCDR2 содержит аминокислотную последовательность под SEQ ID NO: 7 и LCDR3 содержит аминокислотную последовательность под SEQ ID NO: 8.
В варианте осуществления антитело представляет собой сарилумаб.
В различных вариантах осуществления с антителом не вводят никакое другое DMARD. В некоторых вариантах осуществления субъекту вводят по меньшей мере одно другое DMARD. В варианте осуществления по меньшей мере одно другое DMARD вводят субъекту параллельно с антителом или одновременно с ним.
В некоторых вариантах осуществления субъект страдает от по меньшей мере одного симптома RF-положительного полиартикулярного JIA. В некоторых вариантах осуществления субъект страдает по меньшей мере одним симптомом RF-положительного полиартикулярного JIA, таким как деформирующий симметричный полиартрит, который может развиться в подвывих сустава (например, запястья и/или большого пальца); контрактура сустава (например, проксимальных и дистальных межфаланговых суставов, разрастание костей проксимальных межфаланговых суставов и деформации пальцев, такие как деформации по типу "лебединой шеи" или "бутоньерки"); хроническое синовиальное воспаление; потеря суставного хряща и эрозия околосуставной кости; нормоцитарная хроническая анемия; повышенные ESR и С-реактивный белок; количество лейкоцитов; бессимптомный артрит шейного отдела позвоночника и/или микрогнатия. В различных вариантах осуществления у субъекта имеется подвывих сустава (например, запястья и/или большого пальца) и/или контрактура сустава (например, проксимальных и дистальных межфаланговых суставов, разрастание костей проксимальных межфаланговых суставов и деформации пальцев, такие как деформации по типу "лебединой шеи" или "бутоньерки"). В варианте осуществления лечение субъекта включает уменьшение, замедление, остановку прогрессирования или иное ослабление любого из этих симптомов (или любой их комбинации). В варианте осуществления способа возраст субъекта составляет от приблизительно 12 года до приблизительно 14 лет.
В различных вариантах осуществления субъект страдает от по меньшей мере одного симптома RF-отрицательного полиартикулярного JIA. В различных вариантах осуществления субъект страдает от по меньшей мере одного симптома RF-отрицательного полиартикулярного JIA, такого как симметричный полиартрит с пониженной подвижностью, мышечная слабость и/или сниженное физическое функционирование. В варианте осуществления лечение субъекта включает уменьшение, замедление, остановку прогрессирования или иное ослабление любого из этих симптомов (или любой их комбинации). В варианте осуществления способа возраст субъекта составляет от приблизительно 7 года до приблизительно 9 лет.
В варианте осуществления субъект страдает от по меньшей мере одного симптома распространенного олигоартикулярного JIA. В различных вариантах осуществления субъект страдает по меньшей мере от одного симптома распространенного олигоартикулярного JIA, такого как асептический воспалительный синовит, который поражает более 4 суставов (например, крупные суставы, такие как колени, лодыжки, запястья) через первые 6 месяцев заболевания; хромота при ходьбе; хронический передний увеит; хронический артрит колена или лодыжки, приводящий к разрастанию кости этой конечности с последующим несоответствием длины ног; атрофия мышц (например, мышц-разгибателей, таких как латеральная широкая мышца бедра, четырехглавая мышца при поражении колена) и/или сгибательные контрактуры в коленях или запястьях. В варианте осуществления лечение субъекта включает уменьшение, замедление, остановку прогрессирования или иное ослабление любого из этих симптомов (или любой их комбинации).
В различных вариантах осуществления субъект страдает от по меньшей мере одного симптома pcJIA, такого как хромота; скованность при пробуждении; нежелание субъекта пользоваться рукой или ногой; пониженный уровень активности; отек суставов; и/или трудности с мелкой моторикой.
В варианте осуществления способа наблюдается улучшение в отношении по меньшей мере одного симптома pcJIA у субъекта после введения антитела. В различных вариантах осуществления способа улучшение характеризуется по меньшей мере одной оценкой или показателем, такими как ACR30 JIA, ACR50 JIA, ACR70 JIA, ACR90 JIA и ACR100 JIA, и определяется как улучшение ≥3/6 из следующих переменных базового набора при ухудшении не более 1/6 из них (i) общая оценка активности заболевания врачом (например, с помощью визуальной аналоговой шкалы (VAS)), (ii) оценка общего самочувствия пациентом или родителем, (iii) опросник оценки здоровья детей (например, индекс инвалидизации по опроснику оценки здоровья детей (CHAQ-DI)), (iv) количество суставов с активным артритом, (v) количество суставов с ограниченным движением, и (vi) индекс воспаления (например, высокочувствительный С-реактивный белок). В варианте осуществления ответ ACR30 JIA представляет собой улучшение на ≥30% от исходного уровня ≥3/6 из переменных базового набора при ухудшении на ≥30% не более 1/6 из них.
В различных вариантах осуществления способа улучшение активности заболевания характеризуется по меньшей мере одним индексом активности заболевания, представляющего собой ювенильный артрит (таким как индекс-27 активности заболевания, представляющего собой ювенильный артрит, который включает 4 показателя: общую оценку активности заболевания врачом, общую оценку самочувствия родителем/пациентом, количество суставов с активным заболеванием и индекс воспаления (уровень hs-CRP или ESR)).
В различных вариантах осуществления способа улучшение характеризуется по меньшей мере одной оценкой или показателем, такими как общая оценка активности заболевания врачом, оценка общего самочувствия пациентом или родителем, опросник оценки здоровья детей, количество суставов с активным артритом, количество суставов с ограниченным движением, высокочувствительный С-реактивный белок и/или индекс активности заболевания, представляющего собой ювенильный артрит (такой как индекс-27 активности заболевания, представляющего собой ювенильный артрит).
В различных вариантах осуществления способа антитело приводит к клинически неактивному заболеванию, как определено критериями Уоллиса. Критерии Уоллиса определены как общая оценка врача по VAS <1/10, отсутствие активного артрита, отсутствие активного увеита и CRP <10 мг/л. В различных вариантах осуществления способа антитело приводит к клинически неактивному заболеванию, как определено индексом-27 активности заболевания, представляющего собой ювенильный артрит, CRP ≤1. В различных вариантах осуществления способа антитело приводит к клинически неактивному заболеванию или низкой активности заболевания, как определено индексом-27 активности заболевания, представляющего собой ювенильный артрит, CRP ≤3,8.
В варианте осуществления способа возраст субъекта составляет от приблизительно 2 года до приблизительно 17 лет.
В различных вариантах осуществления субъект страдает от JIA в течение по меньшей мере 6 месяцев. В варианте осуществления субъект страдал артритом, который поражал до 4 суставов в течение по меньшей мере первых 6 месяцев заболевания, а затем развился до 5 или более суставов, пораженных через по меньшей мере первые 6 месяцев. В некоторых вариантах осуществления субъект страдал артритом, который поражал до 4 суставов в течение первых 6 месяцев заболевания, а затем развился до 5 или более суставов, пораженных через первые 6 месяцев.
В варианте осуществления способа субъект имеет по меньшей мере 5 суставов с активным артритом. В варианте осуществления способа субъект страдает артритом, который поражает 5 или более суставов в течение первых 6 месяцев заболевания. В варианте осуществления способа артрит является отрицательным по ревматоидному фактору. В качестве альтернативы в варианте осуществления артрит является положительным по ревматоидному фактору.
В варианте осуществления способа антитело вводят подкожно. В различных вариантах осуществления способа антитело вводят каждую неделю или каждые две недели.
В варианте осуществления способа антитело вводят в дозе, составляющей от приблизительно 2 мг/кг до приблизительно 4 мг/кг. В варианте осуществления дозу вводят один раз в неделю или один раз в 2 недели. В различных вариантах осуществления способа антитело применяют в дозе, составляющей приблизительно 2 мг/кг, каждую неделю, приблизительно 2,5 мг/кг, каждую неделю, приблизительно 2 мг/кг, каждые две недели, приблизительно 2,5 мг/кг, каждые две недели, приблизительно 3 мг/кг, каждые две недели, и приблизительно 4 мг/кг, каждые две недели. В различных вариантах осуществления способа антитело применяют в дозах, перечисленных в таблицах 1-5.
В некоторых вариантах осуществления способа вес тела субъекта составляет менее 30 кг.
В некоторых вариантах осуществления способа вес тела субъекта составляет по меньшей мере приблизительно 30 кг. В некоторых вариантах осуществления способа вес тела субъекта составляет менее 60 кг.
В некоторых вариантах осуществления способа возраст субъекта составляет от 1 года до 17 лет.
В некоторых вариантах осуществления способа субъект представляет собой человека, а антитело представляет собой антитело человека.
В настоящем изобретении дополнительно предусмотрен способ лечения pcJIA у нуждающегося в этом субъекта, включающий введение эффективного количества антитела, которое специфически связывает IL-6R, где антитело, которое специфически связывается с IL-6R, содержит последовательность вариабельного участка тяжелой цепи под SEQ ID NO: 1 и последовательность вариабельного участка легкой цепи под SEQ ID NO: 2, где антитело вводят в дозе, составляющей от приблизительно 2 мг/кг до приблизительно 4 мг/кг в неделю или в две недели, где вес тела субъекта больше или равен 10 кг и меньше или равен 60 кг.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей от приблизительно 2 мг/кг до приблизительно 3 мг/кг, каждые две недели или в дозе, составляющей приблизительно 2 мг/кг, каждую неделю.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей от приблизительно 2 мг/кг до приблизительно 3 мг/кг, один раз в две недели или в дозе, составляющей приблизительно 2 мг/кг, один раз в неделю.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей приблизительно 2 мг/кг или приблизительно 3 мг/кг, каждые две недели.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей приблизительно 2 мг/кг или приблизительно 3 мг/кг, один раз в две недели.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей 2 мг/кг или 3 мг/кг, каждые две недели или 2 мг/кг каждую неделю.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей 2 мг/кг или 3 мг/кг, один раз в две недели или 2 мг/кг один раз в неделю.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 10 кг и меньше 30 кг, и антитело вводят в дозе, составляющей от приблизительно 2,5 мг/кг до приблизительно 4 мг/кг, каждые две недели или в дозе, составляющей приблизительно 2,5 мг/кг, каждую неделю.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 10 кг и меньше 30 кг, и антитело вводят в дозе, составляющей от приблизительно 2,5 мг/кг до приблизительно 4 мг/кг, один раз в две недели или в дозе, составляющей приблизительно 2,5 мг/кг, один раз в неделю.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 10 кг и меньше 30 кг, и антитело вводят в дозе, составляющей приблизительно 2,5 мг/кг или приблизительно 4 мг/кг, каждые две недели.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 10 кг и меньше 30 кг, и антитело вводят в дозе, составляющей приблизительно 2,5 мг/кг или приблизительно 4 мг/кг, один раз в две недели.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 10 кг и меньше 30 кг, и антитело вводят в дозе, составляющей 2,5 мг/кг или 4 мг/кг, каждые две недели или 2,5 мг/кг каждую неделю.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 10 кг и меньше 30 кг, и антитело вводят в дозе, составляющей 2,5 мг/кг или 4 мг/кг, один раз в две недели или 2,5 мг/кг один раз в неделю.
В настоящем изобретении дополнительно предусмотрен способ лечения pcJIA у нуждающегося в этом субъекта, включающий введение эффективного количества антитела, которое специфически связывает IL-6R, где антитело, которое специфически связывается с IL-6R, содержит последовательность вариабельного участка тяжелой цепи под SEQ ID NO: 1 и последовательность вариабельного участка легкой цепи под SEQ ID NO: 2, где вес тела субъекта больше или равен 30 кг и меньше 33 кг, и антитело вводят в дозе, составляющей 61,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 33 кг и меньше 37,5 кг, и антитело вводят в дозе, составляющей 70 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 37,5 кг и меньше 42 кг, и антитело вводят в дозе, составляющей 78,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 42 кг и меньше 46,5 кг, и антитело вводят в дозе, составляющей 87,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 46,5 кг и меньше 50,5 кг, и антитело вводят в дозе, составляющей 96,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 50,5 кг и меньше 55 кг, и антитело вводят в дозе, составляющей 105 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 55 кг и меньше 59,5 кг, и антитело вводят в дозе, составляющей 113,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 59,5 кг и меньше 64 кг, и антитело вводят в дозе, составляющей 122,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 64 кг и меньше 68 кг, и антитело вводят в дозе, составляющей 131,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 68 кг и меньше 72,5 кг, и антитело вводят в дозе, составляющей 140 мг, один раз в две недели или один раз в неделю; или где вес тела субъекта больше или равен 72,5 кг, и антитело вводят в дозе, составляющей 148,75 мг, один раз в две недели или один раз в неделю.
В настоящем изобретении дополнительно предусмотрен способ лечения pcJIA у нуждающегося в этом субъекта, включающий введение эффективного количества антитела, которое специфически связывает IL-6R, где антитело, которое специфически связывается с IL-6R, содержит последовательность вариабельного участка тяжелой цепи под SEQ ID NO: 1 и последовательность вариабельного участка легкой цепи под SEQ ID NO: 2, где вес тела субъекта больше или равен 30 кг и меньше 31 кг, и антитело вводят в дозе, составляющей 87,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 31 кг и меньше 34 кг, и антитело вводят в дозе, составляющей 96,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 34 кг и меньше 37 кг, и антитело вводят в дозе, составляющей 105 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 37 кг и меньше 39,5 кг, и антитело вводят в дозе, составляющей 113,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 39,5 кг и меньше 42,5 кг, и антитело вводят в дозе, составляющей 122,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 42,5 кг и меньше 45 кг, и антитело вводят в дозе, составляющей 131,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 45 кг и меньше 48,5 кг, и антитело вводят в дозе, составляющей 140 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 48,5 кг и меньше 51,5 кг, и антитело вводят в дозе, составляющей 148,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 51,5 кг и меньше 54,5 кг, и антитело вводят в дозе, составляющей 157,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 54,5 кг и меньше 57 кг, и антитело вводят в дозе, составляющей 166,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 57 кг и меньше 63 кг, и антитело вводят в дозе, составляющей 175 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 63 кг, и антитело вводят в дозе, составляющей 192,5 мг, один раз в две недели или один раз в неделю.
В настоящем изобретении дополнительно предусмотрен способ лечения pcJIA у нуждающегося в этом субъекта, включающий введение эффективного количества антитела, которое специфически связывает IL-6R, где антитело, которое специфически связывается с IL-6R, содержит последовательность вариабельного участка тяжелой цепи под SEQ ID NO: 1 и последовательность вариабельного участка легкой цепи под SEQ ID NO: 2, где вес тела субъекта больше или равен 10 кг и меньше 12,5 кг, и антитело вводят в дозе, составляющей 26,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 12,5 кг и меньше 16 кг, и антитело вводят в дозе, составляющей 35 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 16 кг и меньше 19,5 кг, и антитело вводят в дозе, составляющей 43,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 19,5 кг и меньше 23 кг, и антитело вводят в дозе, составляющей 52,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 23 кг и меньше 26,5 кг, и антитело вводят в дозе, составляющей 61,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 26,5 кг и меньше 30 кг, и антитело вводят в дозе, составляющей 70 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 30 кг и меньше 37,5 кг, и антитело вводят в дозе, составляющей 70 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 37,5 кг и меньше 42 кг, и антитело вводят в дозе, составляющей 78,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 42 кг и меньше 46,5 кг, и антитело вводят в дозе, составляющей 87,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 46,5 кг и меньше 50,5 кг, и антитело вводят в дозе, составляющей 96,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 50,5 кг и меньше 55 кг, и антитело вводят в дозе, составляющей 105 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 55 кг и меньше 59,5 кг, и антитело вводят в дозе, составляющей 113,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 59,5 кг и меньше 64 кг, и антитело вводят в дозе, составляющей 122,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 64 кг и меньше 68 кг, и антитело вводят в дозе, составляющей 131,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 68 кг и меньше 72,5 кг, и антитело вводят в дозе, составляющей 140 мг, один раз в две недели или один раз в неделю; или где вес тела субъекта больше или равен 72,5 кг, и антитело вводят в дозе, составляющей 148,75 мг, один раз в две недели или один раз в неделю.
В настоящем изобретении дополнительно предусмотрен способ лечения pcJIA у нуждающегося в этом субъекта, включающий введение эффективного количества антитела, которое специфически связывает IL-6R, где антитело, которое специфически связывается с IL-6R, содержит последовательность вариабельного участка тяжелой цепи под SEQ ID NO: 1 и последовательность вариабельного участка легкой цепи под SEQ ID NO: 2, где вес тела субъекта больше или равен 10 кг и меньше 12,5 кг, и антитело вводят в дозе, составляющей 43,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 12,5 кг и меньше 14,5 кг, и антитело вводят в дозе, составляющей 52,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 14,5 кг и меньше 16,5 кг, и антитело вводят в дозе, составляющей 61,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 16,5 кг и меньше 19 кг, и антитело вводят в дозе, составляющей 70 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 19 кг и меньше 21 кг, и антитело вводят в дозе, составляющей 78,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 21 кг и меньше 23,5 кг, и антитело вводят в дозе, составляющей 87,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 23,5 кг и меньше 25,5 кг, и антитело вводят в дозе, составляющей 96,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 25,5 кг и меньше 27,5 кг, и антитело вводят в дозе, составляющей 105 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 27,5 кг и меньше 30 кг, и антитело вводят в дозе, составляющей 113,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 30 кг и меньше 39,5 кг, и антитело вводят в дозе, составляющей 113,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 39,5 кг и меньше 42,5 кг, и антитело вводят в дозе, составляющей 122,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 42,5 кг и меньше 45 кг, и антитело вводят в дозе, составляющей 131,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 45 кг и меньше 48,5 кг, и антитело вводят в дозе, составляющей 140 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 48,5 кг и меньше 51,5 кг, и антитело вводят в дозе, составляющей 148,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 51,5 кг и меньше 54,5 кг, и антитело вводят в дозе, составляющей 157,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 54,5 кг и меньше 57 кг, и антитело вводят в дозе, составляющей 166,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 57 кг и меньше 60,5 кг, и антитело вводят в дозе, составляющей 175 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 60,5 кг и меньше 63 кг, и антитело вводят в дозе, составляющей 175 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 63 кг, и антитело вводят в дозе, составляющей 192,5 мг, один раз в две недели или один раз в неделю.
В настоящем изобретении дополнительно предусмотрен способ лечения sJIA у нуждающегося в этом субъекта, включающий введение эффективного количества антитела, которое специфически связывает IL-6R, где антитело, которое специфически связывается с IL-6R, содержит последовательность вариабельного участка тяжелой цепи под SEQ ID NO: 1 и последовательность вариабельного участка легкой цепи под SEQ ID NO: 2, где антитело вводят в дозе, составляющей от приблизительно 2 мг/кг до приблизительно 2,5 мг/кг в неделю или от приблизительно 3 мг/кг до приблизительно 4 мг/кг в две недели, где вес тела субъекта больше или равен 30 кг и меньше или равен 210 кг.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей приблизительно 2 мг/кг, каждую неделю или в дозе, составляющей приблизительно 3 мг/кг, каждые две недели.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей приблизительно 2 мг/кг, один раз в неделю или в дозе, составляющей приблизительно 3 мг/кг, один раз в две недели.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей 2 мг/кг, каждую неделю или в дозе, составляющей 3 мг/кг, каждые две недели.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей 2 мг/кг, один раз в неделю или в дозе, составляющей 3 мг/кг, один раз в две недели.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 30 кг и меньше или равен 210 кг, и антитело вводят в дозе, составляющей приблизительно 2,5 мг/кг, каждую неделю или в дозе, составляющей приблизительно 4 мг/кг, каждые две недели.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 30 кг и меньше или равен 210 кг, и антитело вводят в дозе, составляющей приблизительно 2,5 мг/кг, один раз в неделю или в дозе, составляющей приблизительно 4 мг/кг, один раз в две недели.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 30 кг и меньше или равен 210 кг, и антитело вводят в дозе, составляющей 2,5 мг/кг, каждую неделю или в дозе, составляющей 4 мг/кг, каждые две недели.
В некоторых вариантах осуществления способа вес тела субъекта больше или равен 30 кг и меньше или равен 210 кг, и антитело вводят в дозе, составляющей 2,5 мг/кг, один раз в неделю или в дозе, составляющей 4 мг/кг, один раз в две недели.
В настоящем изобретении показано антитело для применения при лечении JIA у нуждающегося в этом субъекта, где антитело специфически связывает IL-6R.
В варианте осуществления JIA представляет собой sJIA.
В варианте осуществления антитело, которое специфически связывается с IL-6R, содержит последовательность вариабельного участка тяжелой цепи под SEQ ID NO: 1 и последовательность вариабельного участка легкой цепи под SEQ ID NO: 2.
В различных вариантах осуществления антитело содержит VH и VL, где VH содержит три CDR, расположенные в последовательности под SEQ ID NO: 1, и где VL содержит три CDR, расположенные в последовательности под SEQ ID NO: 2. В различных вариантах осуществления антитело к IL-6R или его антигенсвязывающий фрагмент содержат три HCDR (т. е. HCDR1, HCDR2 и HCDR3) и три LCDR (т. е. LCDR1, LCDR2 и LCDR3), где HCDR1 содержит аминокислотную последовательность под SEQ ID NO: 3; HCDR2 содержит аминокислотную последовательность под SEQ ID NO: 4; HCDR3 содержит аминокислотную последовательность под SEQ ID NO: 5; LCDR1 содержит аминокислотную последовательность под SEQ ID NO: 6; LCDR2 содержит аминокислотную последовательность под SEQ ID NO: 7 и LCDR3 содержит аминокислотную последовательность под SEQ ID NO: 8.
В варианте осуществления антитело представляет собой сарилумаб.
В различных вариантах осуществления с антителом не вводят никакое другое DMARD. В некоторых вариантах осуществления субъекту вводят по меньшей мере одно другое DMARD. В варианте осуществления по меньшей мере одно другое DMARD вводят субъекту параллельно с антителом или одновременно с ним.
В некоторых вариантах осуществления субъект страдает по меньшей мере одним симптомом sJIA, таким как артрит в по меньшей мере 1 суставе в течение по меньшей мере 6 недель с сопутствующей или предшествующей лихорадкой, продолжающейся по меньшей мере 2 недели; кратковременная эритематозная сыпь преимущественно на туловище и конечностях; генерализованная лимфаденопатия; гепатомегалия и/или спленомегалия; полисерозит; потеря веса; усталость; недомогание; лихорадка; повышенное количество WBC (от 25000 до 50000/мл3); повышенное количество тромбоцитов (например, >1×106), заметно повышенная ESR >100 мм/ч; анемия и/или высокий уровень ферритина по сравнению со здоровым субъектом. В варианте осуществления лечение субъекта включает уменьшение, замедление, остановку прогрессирования или иное ослабление любого из этих симптомов (или любой их комбинации).
В различных вариантах осуществления субъект страдает по меньшей мере одним симптомом sJIA, таким как хромота; скованность при пробуждении; нежелание субъекта пользоваться рукой или ногой; пониженный уровень активности; ежедневная лихорадка с высокими пиками (например, достигающая приблизительно 40°C или по меньшей мере 40°C); отек суставов; и/или трудности с мелкой моторикой. В варианте осуществления антитела наблюдается улучшение в отношении по меньшей мере одного симптома sJIA у субъекта после введения антитела.
В различных вариантах осуществления антитело улучшает по меньшей мере одну оценку или показатель, такие как JIA ACR (, например, JIA ACR30, JIA ACR50, JIA ACR70, JIA ACR90, и JIA ACR100), компонент ACR JIA (например, количество суставов с активным артритом, количество суставов с ограниченным диапазоном движений, общая оценка активности заболевания врачом, реагенты острой фазы, такие как скорость оседания эритроцитов или С-реактивный белок, опросник оценки здоровья детей, общая оценка самочувствия пациентом (или родителем)) и/или уменьшение лихорадки (например, у субъектов с лихорадкой при первом введении антитела), снижение по сравнению с исходным уровнем количества применяемых кортикостероидов (например, глюкокортикоидов) и/или индекс активности заболевания, представляющего собой ювенильный артрит (такой какиндекс-27 активности заболевания, представляющего собой ювенильный артрит). В различных вариантах осуществления антитела улучшение характеризуется по меньшей мере одной оценкой или показателем, такими как ACR30 JIA, ACR50 JIA, ACR70 JIA, ACR90 JIA и/или ACR100 JIA. В различных вариантах осуществления антитела улучшение характеризуется по меньшей мере одной оценкой или показателем, такими как общая оценка индекса активности заболевания врачом, оценка общего самочувствия пациентом или родителем, опросник оценки здоровья детей, количество суставов с активным артритом, количество суставов с ограниченным движением, высокочувствительный С-реактивный белок и/или уменьшение лихорадки (например, у субъектов с лихорадкой при первом введении антитела). В различных вариантах осуществления улучшение характеризуется по меньшей мере одним биомаркером.
В различных вариантах осуществления способа антитело приводит к клинически неактивному заболеванию, как определено критериями Уоллиса. Критерии Уоллиса определены как общая оценка врача по VAS <1/10, отсутствие активного артрита, отсутствие активного увеита и CRP <10 мг/л. В различных вариантах осуществления способа антитело приводит к клинически неактивному заболеванию, как определено индексом-27 активности заболевания, представляющего собой ювенильный артрит, CRP ≤1. В различных вариантах осуществления способа антитело приводит к клинически неактивному заболеванию или низкой активности заболевания, как определено индексом-27 активности заболевания, представляющего собой ювенильный артрит, CRP ≤3,8.
В варианте осуществления применения у субъекта наблюдается недостаточный ответ на текущее лечение, и субъект рассматривается как кандидат на лечение биологическим модифицирующим заболевание противоревматическим лекарственным средством.
В варианте осуществления применения возраст субъекта составляет от приблизительно 1 года до приблизительно 17 лет. В других вариантах осуществления возраст субъекта составляет от приблизительно 4 до приблизительно 6 лет или от приблизительно 12 до приблизительно 18 лет.
В варианте осуществления применения у субъекта имеются либо 5 или более активных суставов, либо 2 активных сустава и системные симптомы (например, лихорадка). В варианте осуществления антитела у субъекта имеются либо 5 или более активных суставов, либо 2 или более активных сустава и лихорадка sJIA выше приблизительно 37,5 °C в течение по меньшей мере 3 из любых 7 последовательных дней, несмотря на глюкокортикоиды в дозе, стабильной в течение по меньшей мере 3 дней.
В варианте осуществления применения JIA представляет собой pcJIA. В некоторых вариантах осуществления pcJIA представляет собой распространенный олигоартикулярный JIA. В различных вариантах осуществления pcJIA представляет собой RF-положительный полиартикулярный JIA. В некоторых вариантах осуществления pcJIA представляет собой RF-отрицательный полиартикулярный JIA.
В варианте осуществления антитело, которое специфически связывается с IL-6R, содержит последовательность вариабельного участка тяжелой цепи под SEQ ID NO: 1 и последовательность вариабельного участка легкой цепи под SEQ ID NO: 2.
В различных вариантах осуществления антитело содержит VH и VL, где VH содержит три CDR, расположенные в последовательности под SEQ ID NO: 1, и где VL содержит три CDR, расположенные в последовательности под SEQ ID NO: 2. В различных вариантах осуществления антитело к IL-6R или его антигенсвязывающий фрагмент содержат три HCDR (т. е. HCDR1, HCDR2 и HCDR3) и три LCDR (т. е. LCDR1, LCDR2 и LCDR3), где HCDR1 содержит аминокислотную последовательность под SEQ ID NO: 3; HCDR2 содержит аминокислотную последовательность под SEQ ID NO: 4; HCDR3 содержит аминокислотную последовательность под SEQ ID NO: 5; LCDR1 содержит аминокислотную последовательность под SEQ ID NO: 6; LCDR2 содержит аминокислотную последовательность под SEQ ID NO: 7 и LCDR3 содержит аминокислотную последовательность под SEQ ID NO: 8.
В варианте осуществления антитело представляет собой сарилумаб.
В различных вариантах осуществления с антителом не вводят никакое другое DMARD. В некоторых вариантах осуществления субъекту вводят по меньшей мере одно другое DMARD. В варианте осуществления по меньшей мере одно другое DMARD вводят субъекту параллельно с антителом или одновременно с ним.
В некоторых вариантах осуществления субъект страдает от по меньшей мере одного симптома RF-положительного полиартикулярного JIA. В некоторых вариантах осуществления субъект страдает по меньшей мере одним симптомом RF-положительного полиартикулярного JIA, таким как деформирующий симметричный полиартрит, который может развиться в подвывих сустава (например, запястья и/или большого пальца); контрактура сустава (например, проксимальных и дистальных межфаланговых суставов, разрастание костей проксимальных межфаланговых суставов и деформации пальцев, такие как деформации по типу "лебединой шеи" или "бутоньерки"); хроническое синовиальное воспаление; потеря суставного хряща и эрозия околосуставной кости; нормоцитарная хроническая анемия; повышенные ESR и С-реактивный белок; количество лейкоцитов; бессимптомный артрит шейного отдела позвоночника и/или микрогнатия. В различных вариантах осуществления у субъекта имеется подвывих сустава (например, запястья и/или большого пальца) и/или контрактура сустава (например, проксимальных и дистальных межфаланговых суставов, разрастание костей проксимальных межфаланговых суставов и деформации пальцев, такие как деформации по типу "лебединой шеи" или "бутоньерки"). В варианте осуществления лечение субъекта включает уменьшение, замедление, остановку прогрессирования или иное ослабление любого из этих симптомов (или любой их комбинации).
В различных вариантах осуществления субъект страдает от по меньшей мере одного симптома RF-отрицательного полиартикулярного JIA. В различных вариантах осуществления субъект страдает от по меньшей мере одного симптома RF-отрицательного полиартикулярного JIA, такого как симметричный полиартрит с пониженной подвижностью, мышечная слабость и/или сниженное физическое функционирование. В варианте осуществления лечение субъекта включает уменьшение, замедление, остановку прогрессирования или иное ослабление любого из этих симптомов (или любой их комбинации).
В варианте осуществления субъект страдает по меньшей мере одним симптомом распространенного олигоартикулярного JIA, таким как асептический воспалительный синовит, который поражает более 4 суставов (например, крупные суставы, такие как колени, лодыжки, запястья) через первые 6 месяцев заболевания; хромота при ходьбе; хронический передний увеит; хронический артрит колена или лодыжки, приводящий к разрастанию кости этой конечности с последующим несоответствием длины ног; атрофия мышц (например, мышц-разгибателей, таких как латеральная широкая мышца бедра, четырехглавая мышца при поражении колена); и/или сгибательные контрактуры в коленях или запястьях. В варианте осуществления лечение субъекта включает уменьшение, замедление, остановку прогрессирования или иное ослабление любого из этих симптомов (или любой их комбинации).
В различных вариантах осуществления субъект страдает от по меньшей мере одного симптома pcJIA, такого как хромота; скованность при пробуждении; нежелание субъекта пользоваться рукой или ногой; пониженный уровень активности; отек суставов; и/или трудности с мелкой моторикой.
В варианте осуществления антитела наблюдается улучшение в отношении по меньшей мере одного симптома pcJIA у субъекта после введения антитела. В различных вариантах осуществления антитела улучшение характеризуется по меньшей мере одной оценкой или показателем, такими как ACR30 JIA, ACR50 JIA, ACR70 JIA, ACR90 JIA и ACR100 JIA, и определяется как улучшение ≥3/6 из следующих переменных базового набора при ухудшении не более 1/6 из них (i) общая оценка активности заболевания врачом (например, с помощью визуальной аналоговой шкалы (VAS)), (ii) оценка общего самочувствия пациентом или родителем, (iii) опросник оценки здоровья детей (например, CHAQ-DI), (iv) количество суставов с активным артритом, (v) количество суставов с ограниченным движением, и (vi) индекс воспаления (например, высокочувствительный С-реактивный белок). В варианте осуществления ответ ACR30 JIA представляет собой улучшение на ≥30% от исходного уровня ≥3/6 из переменных базового набора при ухудшении на ≥30% не более 1/6 из них.
В различных вариантах осуществления антитела улучшение активности заболевания характеризуется по меньшей мере одним индексом активности заболевания, представляющего собой ювенильный артрит (таким как индекс-27 активности заболевания, представляющего собой ювенильный артрит, который включает 4 показателя: общую оценку активности заболевания врачом, общую оценку самочувствия родителем/пациентом, количество суставов с активным заболеванием и индекс воспаления (уровень hs-CRP или ESR)).
В различных вариантах осуществления антитела улучшение характеризуется по меньшей мере одной оценкой или показателем, такими как общая оценка активности заболевания врачом, оценка общего самочувствия пациентом или родителем, опросник оценки здоровья детей, количество суставов с активным артритом, количество суставов с ограниченным движением, высокочувствительный С-реактивный белок и/или индекс активности заболевания, представляющего собой ювенильный артрит (такой как индекс-27 активности заболевания, представляющего собой ювенильный артрит).
В варианте осуществления антитела возраст субъекта составляет от приблизительно 2 лет до приблизительно 17 лет.
В различных вариантах осуществления субъект страдает от JIA в течение по меньшей мере 6 месяцев. В варианте осуществления субъект страдал артритом, который поражал до 4 суставов в течение по меньшей мере первых 6 месяцев заболевания, а затем развился до 5 или более суставов, пораженных через по меньшей мере первые 6 месяцев. В некоторых вариантах осуществления субъект страдал артритом, который поражал до 4 суставов в течение первых 6 месяцев заболевания, а затем развился до 5 или более суставов, пораженных через первые 6 месяцев.
В варианте осуществления антитела субъект имеет по меньшей мере 5 суставов с активным артритом. В варианте осуществления антитела субъект имеет артрит, который поражает 5 или более суставов в течение первых 6 месяцев заболевания. В варианте осуществления антитела артрит является отрицательным по ревматоидному фактору. В качестве альтернативы в варианте осуществления артрит является положительным по ревматоидному фактору.
В варианте осуществления антитело вводят подкожно. В различных вариантах осуществления антитело вводят каждую неделю или каждые две недели.
В варианте осуществления антитело вводят в дозе, составляющей от приблизительно 2 мг/кг до приблизительно 4 мг/кг. В варианте осуществления дозу вводят один раз в неделю или один раз в 2 недели. В различных вариантах осуществления антитело применяют в дозе, составляющей приблизительно 2 мг/кг, каждую неделю, приблизительно 2,5 мг/кг, каждую неделю, приблизительно 2 мг/кг, каждые две недели, приблизительно 2,5 мг/кг, каждые две недели, приблизительно 3 мг/кг, каждые две недели, и приблизительно 4 мг/кг, каждые две недели. В различных вариантах осуществления антитело применяют в дозах, перечисленных в таблицах 1-5.
В некоторых вариантах осуществления вес тела субъекта составляет по меньшей мере приблизительно 10 кг. В некоторых вариантах осуществления вес тела субъекта составляет менее 30 кг.
В некоторых вариантах осуществления вес тела субъекта составляет по меньшей мере приблизительно 30 кг.
В некоторых вариантах осуществления вес тела субъекта составляет менее 60 кг.
В различных вариантах осуществления возраст субъекта составляет от 1 года до 17 лет.
В определенных вариантах осуществления субъект представляет собой человека, а антитело представляет собой антитело человека.
В настоящем изобретении дополнительно предусмотрено антитело для применения при лечении pcJIA у нуждающегося в этом субъекта, где антитело специфически связывает IL-6R, где антитело, которое специфически связывается с IL-6R, содержит последовательность вариабельного участка тяжелой цепи под SEQ ID NO: 1 и последовательность вариабельного участка легкой цепи под SEQ ID NO: 2, где антитело вводят в дозе, составляющей от приблизительно 2 мг/кг до приблизительно 4 мг/кг в неделю или в две недели, где вес тела субъекта больше или равен 10 кг и меньше или равен 60 кг.
В некоторых вариантах осуществления вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей от приблизительно 2 мг/кг до приблизительно 3 мг/кг, каждые две недели или в дозе, составляющей приблизительно 2 мг/кг, каждую неделю.
В определенных вариантах осуществления вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей от приблизительно 2 мг/кг до приблизительно 3 мг/кг, один раз в две недели или в дозе, составляющей приблизительно 2 мг/кг, один раз в неделю.
В различных вариантах осуществления вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей приблизительно 2 мг/кг или приблизительно 3 мг/кг, каждые две недели.
В некоторых вариантах осуществления вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей приблизительно 2 мг/кг или приблизительно 3 мг/кг, один раз в две недели.
В различных вариантах осуществления вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей 2 мг/кг или 3 мг/кг, каждые две недели или 2 мг/кг каждую неделю.
В определенных вариантах осуществления вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей 2 мг/кг или 3 мг/кг, один раз в две недели или 2 мг/кг один раз в неделю.
В некоторых вариантах осуществления вес тела субъекта больше или равен 10 кг и меньше 30 кг, и антитело вводят в дозе, составляющей от приблизительно 2,5 мг/кг до приблизительно 4 мг/кг, каждые две недели или в дозе, составляющей приблизительно 2,5 мг/кг, каждую неделю.
В различных вариантах осуществления вес тела субъекта больше или равен 10 кг и меньше 30 кг, и антитело вводят в дозе, составляющей от приблизительно 2,5 мг/кг до приблизительно 4 мг/кг, один раз в две недели или в дозе, составляющей приблизительно 2,5 мг/кг, один раз в неделю.
В определенных вариантах осуществления вес тела субъекта больше или равен 10 кг и меньше 30 кг, и антитело вводят в дозе, составляющей приблизительно 2,5 мг/кг или приблизительно 4 мг/кг, каждые две недели.
В некоторых вариантах осуществления вес тела субъекта больше или равен 10 кг и меньше 30 кг, и антитело вводят в дозе, составляющей приблизительно 2,5 мг/кг или приблизительно 4 мг/кг, один раз в две недели.
В различных вариантах осуществления вес тела субъекта больше или равен 10 кг и меньше 30 кг, и антитело вводят в дозе, составляющей 2,5 мг/кг или 4 мг/кг, каждые две недели или 2,5 мг/кг каждую неделю.
В определенных вариантах осуществления вес тела субъекта больше или равен 10 кг и меньше 30 кг, и антитело вводят в дозе, составляющей 2,5 мг/кг или 4 мг/кг, один раз в две недели или 2,5 мг/кг один раз в неделю.
В настоящем изобретении дополнительно предусмотрено антитело для применения при лечении pcJIA у нуждающегося в этом субъекта, где антитело специфически связывает IL-6R, где антитело, которое специфически связывается с IL-6R, содержит последовательность вариабельного участка тяжелой цепи под SEQ ID NO: 1 и последовательность вариабельного участка легкой цепи под SEQ ID NO: 2, где вес тела субъекта больше или равен 30 кг и меньше 33 кг, и антитело вводят в дозе, составляющей 61,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 33 кг и меньше 37,5 кг, и антитело вводят в дозе, составляющей 70 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 37,5 кг и меньше 42 кг, и антитело вводят в дозе, составляющей 78,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 42 кг и меньше 46,5 кг, и антитело вводят в дозе, составляющей 87,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 46,5 кг и меньше 50,5 кг, и антитело вводят в дозе, составляющей 96,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 50,5 кг и меньше 55 кг, и антитело вводят в дозе, составляющей 105 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 55 кг и меньше 59,5 кг, и антитело вводят в дозе, составляющей 113,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 59,5 кг и меньше 64 кг, и антитело вводят в дозе, составляющей 122,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 64 кг и меньше 68 кг, и антитело вводят в дозе, составляющей 131,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 68 кг и меньше 72,5 кг, и антитело вводят в дозе, составляющей 140 мг, один раз в две недели или один раз в неделю; или где вес тела субъекта больше или равен 72,5 кг, и антитело вводят в дозе, составляющей 148,75 мг, один раз в две недели или один раз в неделю.
В настоящем изобретении дополнительно предусмотрено антитело для применения при лечении pcJIA у нуждающегося в этом субъекта, где антитело специфически связывает IL-6R, где антитело, которое специфически связывается с IL-6R, содержит последовательность вариабельного участка тяжелой цепи под SEQ ID NO: 1 и последовательность вариабельного участка легкой цепи под SEQ ID NO: 2, где вес тела субъекта больше или равен 30 кг и меньше 31 кг, и антитело вводят в дозе, составляющей 87,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 31 кг и меньше 34 кг, и антитело вводят в дозе, составляющей 96,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 34 кг и меньше 37 кг, и антитело вводят в дозе, составляющей 105 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 37 кг и меньше 39,5 кг, и антитело вводят в дозе, составляющей 113,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 39,5 кг и меньше 42,5 кг, и антитело вводят в дозе, составляющей 122,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 42,5 кг и меньше 45 кг, и антитело вводят в дозе, составляющей 131,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 45 кг и меньше 48,5 кг, и антитело вводят в дозе, составляющей 140 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 48,5 кг и меньше 51,5 кг, и антитело вводят в дозе, составляющей 148,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 51,5 кг и меньше 54,5 кг, и антитело вводят в дозе, составляющей 157,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 54,5 кг и меньше 57 кг, и антитело вводят в дозе, составляющей 166,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 57 кг и меньше 63 кг, и антитело вводят в дозе, составляющей 175 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 63 кг, и антитело вводят в дозе, составляющей 192,5 мг, один раз в две недели или один раз в неделю.
В настоящем изобретении дополнительно предусмотрено антитело для применения при лечении pcJIA у нуждающегося в этом субъекта, где антитело специфически связывает IL-6R, где антитело, которое специфически связывается с IL-6R, содержит последовательность вариабельного участка тяжелой цепи под SEQ ID NO: 1 и последовательность вариабельного участка легкой цепи под SEQ ID NO: 2, где вес тела субъекта больше или равен 10 кг и меньше 12,5 кг, и антитело вводят в дозе, составляющей 26,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 12,5 кг и меньше 16 кг, и антитело вводят в дозе, составляющей 35 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 16 кг и меньше 19,5 кг, и антитело вводят в дозе, составляющей 43,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 19,5 кг и меньше 23 кг, и антитело вводят в дозе, составляющей 52,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 23 кг и меньше 26,5 кг, и антитело вводят в дозе, составляющей 61,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 26,5 кг и меньше 30 кг, и антитело вводят в дозе, составляющей 70 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 30 кг и меньше 37,5 кг, и антитело вводят в дозе, составляющей 70 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 37,5 кг и меньше 42 кг, и антитело вводят в дозе, составляющей 78,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 42 кг и меньше 46,5 кг, и антитело вводят в дозе, составляющей 87,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 46,5 кг и меньше 50,5 кг, и антитело вводят в дозе, составляющей 96,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 50,5 кг и меньше 55 кг, и антитело вводят в дозе, составляющей 105 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 55 кг и меньше 59,5 кг, и антитело вводят в дозе, составляющей 113,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 59,5 кг и меньше 64 кг, и антитело вводят в дозе, составляющей 122,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 64 кг и меньше 68 кг, и антитело вводят в дозе, составляющей 131,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 68 кг и меньше 72,5 кг, и антитело вводят в дозе, составляющей 140 мг, один раз в две недели или один раз в неделю; или где вес тела субъекта больше или равен 72,5 кг, и антитело вводят в дозе, составляющей 148,75 мг, один раз в две недели или один раз в неделю.
В настоящем изобретении дополнительно предусмотрено антитело для применения при лечении pcJIA у нуждающегося в этом субъекта, где антитело специфически связывает IL-6R, где антитело, которое специфически связывается с IL-6R, содержит последовательность вариабельного участка тяжелой цепи под SEQ ID NO: 1 и последовательность вариабельного участка легкой цепи под SEQ ID NO: 2, где вес тела субъекта больше или равен 10 кг и меньше 12,5 кг, и антитело вводят в дозе, составляющей 43,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 12,5 кг и меньше 14,5 кг, и антитело вводят в дозе, составляющей 52,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 14,5 кг и меньше 16,5 кг, и антитело вводят в дозе, составляющей 61,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 16,5 кг и меньше 19 кг, и антитело вводят в дозе, составляющей 70 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 19 кг и меньше 21 кг, и антитело вводят в дозе, составляющей 78,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 21 кг и меньше 23,5 кг, и антитело вводят в дозе, составляющей 87,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 23,5 кг и меньше 25,5 кг, и антитело вводят в дозе, составляющей 96,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 25,5 кг и меньше 27,5 кг, и антитело вводят в дозе, составляющей 105 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 27,5 кг и меньше 30 кг, и антитело вводят в дозе, составляющей 113,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 30 кг и меньше 39,5 кг, и антитело вводят в дозе, составляющей 113,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 39,5 кг и меньше 42,5 кг, и антитело вводят в дозе, составляющей 122,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 42,5 кг и меньше 45 кг, и антитело вводят в дозе, составляющей 131,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 45 кг и меньше 48,5 кг, и антитело вводят в дозе, составляющей 140 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 48,5 кг и меньше 51,5 кг, и антитело вводят в дозе, составляющей 148,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 51,5 кг и меньше 54,5 кг, и антитело вводят в дозе, составляющей 157,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 54,5 кг и меньше 57 кг, и антитело вводят в дозе, составляющей 166,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 57 кг и меньше 60,5 кг, и антитело вводят в дозе, составляющей 175 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 60,5 кг и меньше 63 кг, и антитело вводят в дозе, составляющей 175 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 63 кг, и антитело вводят в дозе, составляющей 192,5 мг, один раз в две недели или один раз в неделю.
В настоящем изобретении дополнительно предусмотрено антитело для применения при лечении sJIA у нуждающегося в этом субъекта, где антитело специфически связывает IL-6R, где антитело, которое специфически связывается с IL-6R, содержит последовательность вариабельного участка тяжелой цепи под SEQ ID NO: 1 и последовательность вариабельного участка легкой цепи под SEQ ID NO: 2, где антитело вводят в дозе, составляющей от приблизительно 2 мг/кг до приблизительно 2,5 мг/кг в неделю или от приблизительно 3 мг/кг до приблизительно 4 мг/кг в две недели, где вес тела субъекта больше или равен 30 кг и меньше или равен 210 кг.
В некоторых вариантах осуществления вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей приблизительно 2 мг/кг, каждую неделю или в дозе, составляющей приблизительно 3 мг/кг, каждые две недели.
В различных вариантах осуществления вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей приблизительно 2 мг/кг, один раз в неделю или в дозе, составляющей приблизительно 3 мг/кг, один раз в две недели.
В определенных вариантах осуществления вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей 2 мг/кг, каждую неделю или в дозе, составляющей 3 мг/кг, каждые две недели.
В некоторых вариантах осуществления вес тела субъекта больше или равен 30 кг и меньше или равен 60 кг, и антитело вводят в дозе, составляющей 2 мг/кг, один раз в неделю или в дозе, составляющей 3 мг/кг, один раз в две недели.
В различных вариантах осуществления вес тела субъекта больше или равен 30 кг и меньше или равен 210 кг, и антитело вводят в дозе, составляющей приблизительно 2,5 мг/кг, каждую неделю или в дозе, составляющей приблизительно 4 мг/кг, каждые две недели.
В определенных вариантах осуществления вес тела субъекта больше или равен 30 кг и меньше или равен 210 кг, и антитело вводят в дозе, составляющей приблизительно 2,5 мг/кг, один раз в неделю или в дозе, составляющей приблизительно 4 мг/кг, один раз в две недели.
В некоторых вариантах осуществления вес тела субъекта больше или равен 30 кг и меньше или равен 210 кг, и антитело вводят в дозе, составляющей 2,5 мг/кг, каждую неделю или в дозе, составляющей 4 мг/кг, каждые две недели.
В различных вариантах осуществления вес тела субъекта больше или равен 30 кг и меньше или равен 210 кг, и антитело вводят в дозе, составляющей 2,5 мг/кг, один раз в неделю или в дозе, составляющей 4 мг/кг, один раз в две недели.
В определенных вариантах осуществления антитело содержится в фармацевтической композиции. В некоторых вариантах осуществления композиция представляет собой водный раствор объемом менее приблизительно 1 мл. В различных вариантах осуществления композиция представляет собой водный раствор объемом приблизительно от 0,5 до 1 мл. В определенных вариантах осуществления композиция представляет собой водный раствор объемом приблизительно от 0,75 до 1 мл. В некоторых вариантах осуществления композиция представляет собой водный раствор объемом приблизительно от 0,5 до 0,75 мл. В различных вариантах осуществления композиция представляет собой водный раствор объемом приблизительно от 0,15 до 0,25 мл. В определенных вариантах осуществления композиция представляет собой водный раствор объемом приблизительно от 0,25 до 0,5 мл.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фиг. 1 представлено изображение схемы исследования для двух когорт по дозам для многонационального многоцентрового открытого 2-фазного и 2-частного исследования среди детей и подростков в возрасте от 2 до 17 лет с pcJIA, которые показали недостаточный ответ на текущую терапию или у которых наблюдалась ее непереносимость, или которые рассматривались в качестве кандидатов на биологическое DMARD. 2 фазы исследования представляли собой начальную 12-недельную основную фазу лечения, за которой следовала 144-недельная фаза дополнительного лечения. Сокращения. EOS=конец исследования, EOT=конец лечения, FDA=Управление по контролю качества пищевых продуктов и лекарственных средств, f/u=последующее наблюдение, IMP=исследуемый лекарственный препарат, PK=фармакокинетика, sIL-6Rα=α-субъединица растворимого рецептора интерлейкина 6, V=визит, Wk=неделя.
Примечания. Все пациенты должны пройти визит в конце лечения (EOT) (V27, неделя 156) по завершении лечения (последняя инъекция IMP на неделе 154 для когорты дозы 1 и когорты дозы 2 и на неделе 155 для когорты дозы 3) или во время досрочного окончательного прекращения лечения (независимо от фазы лечения).
Для пациента, который преждевременно прекратил исследуемый вид лечения в течение 12-недельной основной фазы лечения, требовался дополнительный PK-визит через 2 недели после визита EOT для взятия образца крови (V88), и во время визита EOT измеряли IL-6 и общий sIL-6Rα.
Все пациенты должны совершить визит последующего наблюдения после лечения (V28) через 6 недель после визита EOT, т. е. на неделе 162 для пациентов, которые завершили лечение, и EOT+6 недель для пациентов, которые преждевременно прекратили лечение. Однако пациенты, которые преждевременно прекратили лечение во время основной фазы лечения, должны были продолжать совершать визиты исследования в соответствии с графиком протокола без введения лечения до недели 12 (в соответствии с руководствами FDA в отношении недостающих данных).
На фиг. 2 представлено графическое изображение плана исследования 12-недельной основной фазы лечения, описанной на фиг. 1.
На фиг. 3 представлен график, показывающий средние (со стандартным отклонением; SD) функциональные концентрации сарилумаба в сыворотке крови в течение периода времени после первого подкожного введения сарилумаба пациентам с pcJIA. Сокращения на графике включают q2w=один раз в две недели, qw=один раз в неделю, SC=подкожно, SD=стандартное отклонение. Концентрации после лечения ниже LLOQ были заменены на LLOQ/2.
На фиг. 4 представлен график, показывающий средние (со стандартным отклонением; SD) наблюдаемые остаточные концентрации сарилумаба в сыворотке крови в течение периода времени после первого подкожного введения сарилумаба пациентам с pcJIA.
На фиг. 5 представлен график, показывающий частоту ответа ACR30 JIA (hs-CRP) (наблюдаемую во время лечения) в течение 12-недельной основной фазы лечения.
На фиг. 6 представлен график, показывающий частоту ответа ACR30 JIA (hs-CRP) (подход с подстановкой данных субъектов, не ответивших на лечение) в течение 12-недельной основной фазы лечения.
На фиг. 7 представлено изображение схемы исследования для двух когорт по дозам для многонационального многоцентрового открытого последовательного 2-фазного исследования среди детей и подростков в возрасте от 1 до 17 лет (или применяются возрастные требования страны) с sJIA с недостаточным ответом на стандартную терапию или у которых наблюдается ее непереносимость, и которые будут получать SC инъекции сарилумаба q2w или qw.
На фиг. 8 представлено изображение схемы исследования, показывающее распределение пациентов. ALT=аланинаминотрансфераза; QW=каждую неделю; Q2W=каждые 2 недели.
На фиг. 9A-9D представлены графики, показывающие доли пациентов, достигших пороговых значений ответа ACR JIA, и среднее изменение JADAS-27-CRP по сравнению с исходным уровнем на неделе 12, которые наблюдали во время лечения. CRP=C-реактивный белок; JADAS-27-CRP=индекс активности заболевания, представляющего собой ювенильный артрит с подсчетом 27 суставов и CRP; ACR30 JIA/70/90=ответ ювенильного идиопатического артрита, составляющий 30/70/90% согласно Американской коллегии ревматологов; SE=стандартная ошибка.
На фиг. 10A-10B представлены графики средней концентрации CRP и доли пациентов с неопределяемым CRP (<0,2 мг/л) во время основной фазы лечения, которые наблюдали во время лечения. BL=исходный уровень; CRP=C-реактивный белок; SE=стандартная ошибка.
На фиг. 11A-11C представлены графики доли пациентов, у которых наблюдали ответы ACR JIA, рассчитанные путем подстановки данных субъектов, не ответивших на лечение. CRP=C-реактивный белок. ACR30 JIA/70/90=ответ ювенильного идиопатического артрита, составляющий 30/70/90% согласно Американской коллегии ревматологов
ПОДРОБНОЕ ОПИСАНИЕ
В настоящем изобретении предусмотрены фармацевтические композиции и способы применения этих композиций для лечения JIA (например, sJIA и pcJIA) и улучшения в отношении по меньшей мере одного симптома нарушения. Эти композиции включают по меньшей мере одно антитело, которое специфически связывает рецептор интерлейкина-6 человека (hIL-6R).
Эффективность антитела для лечения JIA обычно измеряется с использованием стандартных в области техники способов, обычно используемых клиницистами и ревматологами, например с использованием ответа ювенильного идиопатического артрита ACR30/50/70/90/100, компонента ACR, изменения в применении глюкокортикоидов (для sJIA) и индекса-27 активности заболевания, представляющего собой ювенильный артрит (JADAS). См. Consolaro et al. Development and Validation of a Composite Disease Activity Score for Juvenile Idiopathic Arthritis. Arthritis & Rheumatism. 2009 May;61(5):658-666), который включен в данный документ посредством ссылки во всей своей полноте. Оценка ACR JIA хорошо известна клиницистам и может быть легко определена специалистами в области диагностики и лечения JIA. Оценка ACR JIA также известна как педиатрическая ACR (и является ее синонимом). Неограничивающие описания, относящиеся к ACR JIA, представлены в Giannini et al. (1994) "Preliminary core of set of outcome variables for use in JRA clinical trials" Arthritis Rheum. 37 Suppl 9:S428, который включен в данный документ посредством ссылки для всех целей.
Применяемый в формуле изобретения в разделе "Краткое описание" и разделе "Подробное описание" в данном документе термин "приблизительно" в количественных терминах относится к величине, составляющей плюс или минус 10% от значения, которое он изменяет (с округлением к ближайшему целому числу, если значение не является делимым, таким как число молекул или нуклеотидов). Например, фраза "приблизительно 100 мг" будет охватывать от 90 мг до 110 мг включительно, фраза "приблизительно 2500 мг" будет охватывать от 2250 мг до 2750 мг. При применении процента термин "приблизительно" относится к величине, составляющей плюс или минус 10% относительно этого процента. Например, фраза "приблизительно 20%" будет охватывать 18-22%, а фраза "приблизительно 80%" будет охватывать 72-88% включительно. Кроме того, в тех местах, где выражение "приблизительно" применяется в данном документе в сочетании с количественным термином, следует понимать, что в дополнение к значению плюс или минус 10% также предполагается и описывается точное значение количественного термина. Например, термин "приблизительно 23%" явно предполагает, описывает и включает точно 23%.
Следует отметить, что форма единственного числа относится к одному или нескольким из таких объектов; например, под термином "симптом" понимают один или несколько симптомов. В связи с этим формы единственного числа, термины "один или несколько" и "по меньшей мере один" могут применяться в данном документе взаимозаменяемо.
Кроме того, выражение "и/или", применяемое в данном документе, следует рассматривать в качестве конкретного раскрытия каждого из двух указанных признаков или компонентов с другим или без другого. Таким образом, термин "и/или", применяемый во фразе, такой как "А и/или В", в данном документе, предназначен для включения "А и В", "А или В", "А" (отдельно) и "В" (отдельно). Подобным образом термин "и/или", применяемый во фразе, такой как "А, В и/или С", предназначен для охвата каждого из следующих аспектов: А, В и С; A, B или C; A или C; A или B; B или C; A и C; A и B; B и C; A (отдельно); B (отдельно) и C (отдельно).
Следует понимать, что во всех случаях, где аспекты описываются в данном документе словами "содержащий", также предусмотрены иные аналогичные аспекты, описываемые с использованием терминов "состоящий из" и/или "состоящий по сути из".
JIA
Артрит представляет собой отек в суставе или ограничение диапазона движений в суставе с болью или чувствительностью в суставах, которые сохраняются в течение по меньшей мере 6 недель, наблюдаются врачом и не возникают вследствие первичных механических нарушений. (Petty RE, et al. International League of Associations for Rheumatology Classification of Juvenile Idiopathic Arthritis: Second Revision, Edmonton, 2001. J Rheumatol. 2004 Feb;31(2):390-2), который включен в данный документ посредством ссылки во всей своей полноте).
JIA представляет собой аутоиммунное нарушение, включающее воспаление суставов. Нарушение обычно проявляется у пациентов до исполнения 16 лет и может продолжаться в зрелом возрасте. JIA поражает 1 ребенка из 1000. Типичные симптомы включают хромоту, скованность при пробуждении, нежелание ребенка использовать руку или ногу, снижение уровня активности, постоянная лихорадка, отек суставов и трудности с мелкой моторикой. JIA включает 7 подтипов (например, системный JIA, RF-положительный полиартикулярный JIA и RF-отрицательный полиартикулярный JIA), классифицированных по возрасту начала, диапазону и характеристикам заболевания в первые 6 месяцев после начала, как определено ILAR в 2001 году.
При sJIA воспаление суставов может не проявляться в начале заболевания, когда заметные признаки носят внесуставной характер, что отсрочивает постановку диагноза, но в конечном итоге у большинства пациентов воспаление развивается по полиартикулярному типу. Клинические признаки (определенные по классификации ILAR) включают артрит с сопутствующей/предшествующей ежедневной лихорадкой в течение по меньшей мере 2 недель и одно или более из кратковременной эритематозной сыпи лососевого цвета, генерализованной лимфаденопатии, гепато/спленомегалии и серозита. В некоторых вариантах осуществления критерии исключения для sJIA включают псориаз или псориаз у родственника первой степени родства; наличие лейкоцитарного антигена человека B27 (HLA-B27); анкилозирующий спондилит, энтезит-ассоциированный артрит, сакроилеит с сопутствующим хроническим воспалительным заболеванием кишечника, болезнь Рейтера у родственника первой степени родства; и обнаружение ревматоидного фактора (RF) по меньшей мере 2 раза с интервалом по меньшей мере 3 месяца. sJIA составляет 10% случаев JIA и, как известно, возникает в любом возрасте в детском и подростковом периодах. С точки зрения исходов известно, что пациенты с sJIA характеризуются 50% рецидивов в 1 год; в 25% случаев тяжелого деструктивного заболевания суставов; общими аномалиями роста и синдромом активации макрофагов.
Лихорадка sJIA классически представляет собой ежедневную лихорадку с пиками, которая может сопровождаться одним или несколькими из кратковременной (нефиксированной) эритематозной сыпи, генерализованного увеличения лимфатических узлов, гепатомегалии и/или спленомегалии и серозита. (Petty RE, et al. International League of Associations for Rheumatology Classification of Juvenile Idiopathic Arthritis: Second Revision, Edmonton, 2001. J Rheumatol. 2004 Feb;31(2):390-2). Пик начала sJIA наблюдается у пациентов в возрасте от 4 до 6 лет и иногда может возникать в подростковом периоде у пациентов в возрасте от 12 до 18 лет.
Используемый в данном документе термин "ювенильный идиопатический артрит с полиартикулярным течением" или "pcJIA" включает RF-положительный полиартикулярный JIA, RF-отрицательный полиартикулярный JIA и распространенный олигоартикулярный JIA.
Полиартикулярный JIA, подтип pcJIA, поражает по меньшей мере 5 суставов. Могут быть поражены как крупные, так и мелкие суставы, часто с симметричным двусторонним распределением, часто с поражением суставов с весовой нагрузкой и мелких суставов рук. Артрит может сопровождать субфебрильная лихорадка. Наличие ревматоидного фактора (RF) различает 2 формы полиартикулярного JIA: RF-положительный и RF-отрицательный полиартикулярный JIA.
Полиартикулярный JIA, положительный по ревматоидному фактору (RF), составляет относительно небольшую долю всех детей и подростков с JIA (3-5%), но это единственная подгруппа JIA, напоминающая ревматоидный артрит (RA) взрослых, деформирующий симметричный полиартрит, вызванный хроническим синовиальным воспалением, потерей суставного хряща и эрозией околосуставной кости. Признаками RF-положительного pcJIA являются средний возраст начала от 12 до 14 лет, выраженное преобладание женского пола (соотношение женщины/мужчины 13:1), симметричное поражение мелких и крупных суставов, выработка поликлональных IgM RF или антител к CCP, генетическая предрасположенность (ассоциация с аллелем HLA-DR4), клиническое течение, характеризующееся нормоцитарной хронической анемией (ретикулоэндотелиальная блокада), и белки острой фазы (повышенные ESR и С-реактивный белок), которые редко спонтанно нормализуются, а также повышенный уровень лейкоцитов. Связь с образованием твердых подвижных и безболезненных ревматоидных узелков возможна, но является редкой. Ожидается, что результаты лабораторных исследований будут более серьезными, чем результаты, связанные с олигоартритом. Долгосрочные последствия включают подвывих суставов (запястья и больших пальцев), контрактуры суставов (проксимальных и дистальных межфаланговых суставов, разрастание костей проксимальных межфаланговых суставов и деформации пальцев (например, по типу "лебединой шеи" или "бутоньерки"). Бессимптомный артрит шейного отдела позвоночника, связанный с уменьшением разгибания, может привести к подвывиху, как правило, позвонков C2-C3 и сращению задних позвоночных элементов. Артрит височно-нижнечелюстного сустава также может протекать бессимптомно и приводить к микрогнатии (Petty, R.E. et al., 2004. J Rheumatol. 31(2):390-2).
RF-отрицательный полиартикулярный JIA проявляется в более молодом возрасте (в позднем детстве, от 7 до 9 лет), чем RF-положительный полиартикулярный JIA, и составляет от 11 до 28% всех детей и подростков с JIA. Он может быть не таким деструктивным и персистирующим, как RF-положительное заболевание, но, по определению, поражает 5 или более суставов. Радиологические изменения при RF-отрицательном заболевании происходят позже, чем при RF-положительном заболевании. Серьезные ограничения движений обычно сопровождаются мышечной слабостью и снижением физического функционирования.
Олигоартикулярный JIA (oJIA), другой подтип pcJIA, является наиболее распространенным подтипом ювенильного артрита, составляющим приблизительно 50% всех пациентов с JIA в США и Западной Европе. Начало приходится на возраст от 1 до 5 лет с пиком в возрасте от 2 до 3 лет. Он определяется как асептический воспалительный синовит, который обычно поражает до 4 суставов (обычно более крупные суставы, такие как колени, лодыжки, запястья) и не связан с такими конституциональными симптомами, как лихорадка, потеря веса, усталость или системные признаки воспаления. Тем не менее, если в течение первых 6 месяцев заболевания поражается более 4 суставов, это обозначается как распространенный олигоартрит в отличие от персистирующего oJIA, который проявляется поражением до 4 суставов на протяжении всего периода заболевания. Дети часто выглядят хорошо, несмотря на хромоту при ходьбе. oJIA сопряжен с риском развития хронического переднего увеита, особенно при наличии антинуклеарных антител (ANA) и при начале заболевания в раннем детстве. Как правило, вначале он протекает бессимптомно и требует скринингового исследования с помощью офтальмологической щелевой лампы. Хронический артрит колена или лодыжки может привести к разрастанию кости этой конечности с последующим несоответствием длины ног. Обнаруживаются атрофия мышц, часто мышц-разгибателей (например, большой широкой мышцы бедра, четырехглавой мышцы при поражении колена) и/или сгибательные контрактуры в коленях и, реже, запястьях.
Современные методы лечения JIA включают стероиды, например традиционные синтетические DMARD и биологические DMARD. Разработка новых лекарственных средств, которые способны селективно ингибировать отдельные молекулы и сигнальные пути, дает надежду на улучшение показателей ремиссии при минимизации связанных с заболеванием повреждений и связанных с лечением побочных эффектов (Giancane, et al. Juvenile Idiopathic Arthritis: Diagnosis and Treatment. Rheumatology and Therapy. 2016;3(2):187-207).
Одним из кандидатов для лечения JIA является ингибирование интерлейкина-6 (IL-6). IL-6 является ключевым цитокином с широким диапазоном биологической активности, включая регуляцию иммунной реактивности, ответ острой фазы, воспаление, онкогенез и гемопоэз (Kishimoto, et al. The Cytokine Handbook (London: Academic Press). 2003, pp. 281-304). Было обнаружено, что сверхсинтез IL-6 играет патологическую роль при хронических воспалительных заболеваниях.
IL-6 взаимодействует непосредственно с субъединицей IL-6Rα, а пара IL-6/IL-6Rα образует высокоаффинный комплекс с субъединицей гликопротеина 130 (gp130). IL6Rα также существует в растворимой форме, которая участвует в транссигнализации и способствует тому, что IL6 влияет на клетки, которые не экспрессируют IL6Rα, в том числе синовиальные клетки в суставе (Rose-John et al., J Leukoc Biol. 2006; 80(2), 227-36).
Сарилумаб, также обозначаемый как SAR153191 или REGN88, представляет собой рекомбинантное моноклональное антитело к IgG1-каппа с полностью человеческой последовательностью, направленной против альфа-субъединицы рецепторного комплекса IL-6 (IL-6Rα). Сарилумаб является эффективным и специфическим ингибитором передачи сигнала IL-6. В результате связывания IL-6Rα с высокой аффинностью сарилумаб блокирует связывание IL-6 и прерывает опосредованный цитокинами сигнальный каскад. В определенных вариантах осуществления интерлейкин-6 является основным элементом в этиологии ревматических состояний, и ингибирование передачи сигнала посредством него является важной частью механизма действия сарилумаба. В анализах ex vivo сарилумаб не демонстрировал антителозависимую клеточную цитотоксичность (ADCC) или комплементзависимую цитотоксичность (CDC) для соответствующих типов клеток, где связывание сарилумаба было подтверждено анализом на клеточном сортере с активацией флуоресценции (FACS) (Committee for Medicinal Products for Human Use, Assessment Report, April 27, 2017 EMA/292840/2017, доступно по адресу https://www.ema.europa.eu/documents/assessment-report/kevzara-epar-public-assessment-report_en.pdf).
Антитела
В настоящем изобретении предусмотрены способы, которые включают введение субъекту антитела или его антигенсвязывающего фрагмента, которые специфически связываются с hIL-6R. Используемый в данном документе термин "hIL-6R" означает цитокиновый рецептор человека, который специфически связывает IL-6 человека. В определенных вариантах осуществления антитело, которое вводят пациенту, специфически связывается с внеклеточным доменом hIL-6R.
Термин "антитело", применяемый в данном документе, относится к молекулам иммуноглобулина, содержащим четыре полипептидные цепи, две тяжелые (Н) цепи и две легкие (L) цепи, соединенные между собой дисульфидными связями, а также их мультимерам (например, IgM). Каждая тяжелая цепь содержит вариабельную область тяжелой цепи (в данном документе обозначенную аббревиатурой HCVR или VH) и константную область тяжелой цепи. Константная область тяжелой цепи содержит три домена: CH1, CH2 и CH3. Каждая легкая цепь содержит вариабельную область легкой цепи (в данном документе обозначенную аббревиатурой LCVR или VL) и константную область легкой цепи. Константная область легкой цепи содержит один домен (CL1). Области VH и VL могут быть дополнительно подразделены на гипервариабельные участки, называемые CDR, которые чередуются с более консервативными участками, называемыми каркасными участками(FR). Каждая VH и VL состоит из трех CDR и четырех FR, расположенных от амино-конца к карбокси-концу в следующем порядке: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. В некоторых вариантах осуществления FR антитела (или его антигенсвязывающей части) могут быть идентичны последовательностям зародышевой линии человека или могут быть изменены естественным или искусственным путем. Аминокислотная консенсусная последовательность может быть определена на основании анализа "бок-о-бок" двух или более CDR.
Термин "антитело", применяемый в данном документе, также включает антигенсвязывающие фрагменты целых молекул антител. Термины "антигенсвязывающая часть" антитела, "антигенсвязывающий фрагмент" антитела и т. п., применяемые в данном документе, включают любой встречающийся в природе, получаемый ферментативным путем, синтетический или полученный с помощью генной инженерии полипептид или гликопротеин, который специфически связывает антиген с образованием комплекса. Антигенсвязывающие фрагменты антитела могут быть получены, например, из целых молекул антител с помощью любых подходящих стандартных методик, таких как протеолитическое расщепление или рекомбинантные технологии генной инженерии, включающие манипуляцию с ДНК, кодирующей вариабельные и необязательно константные домены антител, и ее экспрессию. Такая ДНК известна и/или легкодоступна, например, из коммерческих источников, библиотек ДНК (в том числе, например, библиотек "фаг-антитело"), или ее можно синтезировать. ДНК можно секвенировать и с ней можно проводить химические манипуляции или манипуляции с помощью методик молекулярной биологии, например, для расположения одного или нескольких вариабельных и/или константных доменов в подходящей конфигурации или для введения кодонов, создания цистеиновых остатков, модификации, добавления или удаления аминокислот и т. д.
Неограничивающие примеры антигенсвязывающих фрагментов включают: (i) Fab-фрагменты; (ii) F(ab')2-фрагменты; (iii) Fd-фрагменты; (iv) Fv-фрагменты; (v) одноцепочечные молекулы Fv (scFv); (vi) dAb-фрагменты и (vii) минимальные распознающие единицы, состоящие из аминокислотных остатков, имитирующих гипервариабельный участок антитела (например, выделенный CDR, такой как пептид CDR3), или пептид c ограниченной конформационной свободой FR3-CDR3-FR4. Другие сконструированные молекулы, такие как домен-специфические антитела, однодоменные антитела, антитела с удаленным доменом, химерные антитела, CDR-привитые антитела, диатела, триатела, тетратела, минитела, нанотела (например, моновалентные нанотела, бивалентные нанотела), иммунофармацевтические препараты на основе модульного белка малого размера (SMIP) и вариабельные домены IgNAR акулы, также включены в выражение "антигенсвязывающий фрагмент", применяемое в данном документе.
Антигенсвязывающий фрагмент антитела, как правило, будет содержать по меньшей мере один вариабельный домен. Вариабельный домен может быть любого размера или аминокислотного состава и будет, как правило, содержать по меньшей мере одну CDR, которая прилегает к рамке считывания с одной или несколькими каркасными последовательностями или находится в ней. В антигенсвязывающих фрагментах, содержащих домен VH, связанный с доменом VL, домены VH и VL могут располагаться относительно другу друга в любом подходящем порядке. Например, вариабельный участок может быть димерным и содержать димеры VH-VH, VH-VL или VL-VL. В качестве альтернативы антигенсвязывающий фрагмент антитела может содержать мономерный домен VH или VL.
В определенных вариантах осуществления антигенсвязывающий фрагмент антитела может содержать по меньшей мере один вариабельный домен, ковалентно связанный с по меньшей мере одним константным доменом. Неограничивающие иллюстративные конфигурации вариабельных и константных доменов, которые можно выявить в антигенсвязывающем фрагменте антитела, включают: (i) VH-CH1; (ii) VH-CH2; (iii) VH-CH3; (iv) VH-CH1-CH2; (v) VH-CH1-CH2-CH3; (vi) VH-CH2-CH3; (vii) VH-CL; (viii) VL-CH1; (ix) VL-CH2; (x) VL-CH3; (xi) VL-CH1-CH2; (xii) VL-CH1-CH2-CH3; (xiii) VL-CH2-CH3; и (xiv) VL-CL. В любой конфигурации вариабельных и константных доменов, в том числе любых иллюстративных конфигурациях, перечисленных выше, вариабельные и константные домены могут быть либо непосредственно связаны друг с другом, либо могут быть связаны с помощью целой или неполной шарнирной или линкерной области. Шарнирная область в различных вариантах осуществления может состоять из по меньшей мере 2 (например, 5, 10, 15, 20, 40, 60 или больше) аминокислот, что приводит к образованию гибкой или полугибкой связи между смежными вариабельными и/или константными доменами в одной молекуле полипептида. Кроме того, антигенсвязывающий фрагмент антитела в различных вариантах осуществления может содержать гомодимер или гетеродимер (или другой мультимер) из любых конфигураций вариабельных и константных доменов, перечисленных выше, в нековалентной ассоциации друг с другом и/или с одним или более мономерными доменами VH или VL (например, с помощью дисульфидной(дисульфидных) связи(связей)).
В конкретных вариантах осуществления антитело или фрагмент антитела для применения в способе по настоящему изобретению могут представлять собой мультиспецифическое антитело, которое может быть специфическим по отношению к различным эпитопам одного целевого полипептида или может содержать антигенсвязывающие домены, специфические по отношению к эпитопам более одного целевого полипептида. Иллюстративное антитело в биспецифическом формате, которое можно применять в контексте настоящего изобретения, включает применение CH3-домена первого иммуноглобулина (Ig) и CH3-домена второго иммуноглобулина Ig, где CH3-домены первого и второго Ig отличаются друг от друга по меньшей мере одной аминокислотой и где различие по меньшей мере в одну аминокислоту уменьшает связывание биспецифического антитела с белком A по сравнению с биспецифическим антителом, в котором отсутствует различие по аминокислотам. В одном варианте осуществления первый CH3-домен Ig связывает белок А и второй CH3-домен Ig содержит мутацию, которая уменьшает или устраняет связывание с белком А, такую как модификация H95R (по нумерации экзонов по IMGT; H435R по нумерации по EU). Второй CH3 может дополнительно содержать модификацию Y96F (по IMGT; Y436F по EU). Дополнительные модификации, которые можно выявить во втором CH3, включают: D16E, L18M, N44S, K52N, V57M и V82I (по IMGT; D356E, L358M, N384S, K392N, V397M и V422I по EU) в случае с антителами IgG1; N44S, K52N и V82I (IMGT; N384S, K392N и V422I по EU) в случае с антителами IgG2 и Q15R, N44S, K52N, V57M, R69K, E79Q и V82I (по IMGT; Q355R, N384S, K392N, V397M, R409K, E419Q и V422I по EU) в случае с антителами IgG4. Варианты формата биспецифических антител, описанные выше, рассматриваются в объеме настоящего изобретения. Любой формат мультиспецифических антител, в том числе форматы иллюстративных биспецифических антител, раскрытых в данном документе, в различных вариантах осуществления могут быть адаптированы для применения в контексте антигенсвязывающего фрагмента антитела к IL-6R с помощью стандартных методик, доступных в уровне техники.
Полностью человеческие моноклональные антитела к IL-6R могут содержать одну или несколько аминокислотных замен, вставок и/или делеций в каркасных участках и/или участках CDR вариабельных доменов тяжелой и легкой цепей по сравнению с соответствующими последовательностями зародышевой линии. Такие мутации можно легко определить посредством сравнения аминокислотных последовательностей, раскрытых в данном документе, с последовательностями зародышевой линии, доступными, например, из публичных баз данных последовательностей антител. Настоящее изобретение включает антитела и их антигенсвязывающие фрагменты, полученные из любых аминокислотных последовательностей, раскрытых в данном документе, где одна или несколько аминокислот в пределах одной или нескольких каркасных областей и/или областей CDR подвергнуты обратной мутации в соответствующий(соответствующие) остаток(остатки) зародышевой линии или в консервативную аминокислотную замену (природную или неприродную) соответствующего(соответствующих) остатка(остатков) зародышевой линии (такие изменения последовательностей упоминаются в данном документе как "обратные мутации зародышевой линии"). Специалист в данной области техники, начиная с последовательностей вариабельных областей тяжелой и легкой цепей, раскрытых в данном документе, может легко получить множество антител и антигенсвязывающих фрагментов, которые содержат одну или несколько отдельных обратных мутаций зародышевой линии или их комбинации. В определенных вариантах осуществления все остатки каркасных областей и/или остатки CDR в доменах VH и/или VL подвергнуты обратной мутации в последовательность зародышевой линии. В других вариантах осуществления только определенные остатки подвергнуты обратной мутации в последовательность зародышевой линии, например, только подвергнутые мутации остатки, обнаруженные в первых 8 аминокислотах FR1 или в последних 8 аминокислотах FR4, или только подвергнутые мутации остатки, обнаруженные в CDR1, CDR2 или CDR3. Кроме того, антитела по настоящему изобретению могут содержать любую комбинацию из двух или более обратных мутаций зародышевой линии в пределах каркасных областей и/или областей CDR, т. е. где определенные отдельные остатки подвергнуты обратной мутации до последовательности зародышевой линии, в то время как определенные другие остатки, которые отличаются от последовательности зародышевой линии, поддерживаются. Сразу после получения антитела и антигенсвязывающие фрагменты, которые содержат одну или несколько обратных мутаций зародышевой линии, могут быть легко протестированы в отношении одного или нескольких требуемых свойств, таких как улучшенная специфичность связывания, повышенная аффинность связывания, улучшенные или усиленные биологические антагонистические или агонистические свойства (в случае необходимости), сниженная иммуногенность и т. д. Антитела и антигенсвязывающие фрагменты, полученные при помощи этого общего способа, охвачены настоящим изобретением.
Константная область антитела важна с точки зрения способности антитела связывать комплемент и опосредовать клеточнозависимую цитотоксичность. Таким образом, изотип антитела может быть выбран на основании того, требуется ли антителу опосредовать цитотоксичность.
Термин "антитело человека", применяемый в данном документе, предназначен для включения антител, содержащих вариабельные и константные области, полученные из последовательностей иммуноглобулинов зародышевой линии человека. Антитела человека, представленные в настоящем изобретении, в различных вариантах осуществления при этом могут включать аминокислотные остатки, не кодируемые последовательностями иммуноглобулинов зародышевой линии человека (например, мутации, вводимые случайным или сайт-специфическим мутагенезом in vitro или соматической мутацией in vivo), например, в CDR и, в некоторых вариантах осуществления, в CDR3. Однако термин "антитело человека", применяемый в данном документе, не предназначен для включения антител, в которых последовательности CDR, полученные из зародышевой линии другого вида млекопитающего, такого как мышь, привиты на каркасные последовательности человека.
Термин "рекомбинантное антитело человека", применяемый в данном документе, предназначен для включения всех антител человека, которые получены, экспрессированы, созданы или выделены посредством рекомбинантных способов, таких как антитела, экспрессируемые с помощью рекомбинантного вектора экспрессии, трансфицированного в клетку-хозяина (описанные дополнительно ниже), антитела, выделенные из комбинаторной библиотеки рекомбинантных антител человека (описанные дополнительно ниже), антитела, выделенные из животного (например, мыши), которое является трансгенным по генам иммуноглобулинов человека (см., например, Taylor et al. (1992) Nucl. Acids Res. 20:6287-6295, включен в данный документ посредством ссылки во всей своей полноте), или антитела, полученные, экспрессированные, созданные или выделенные посредством любых других способов, которые предусматривают сплайсинг последовательностей генов иммуноглобулинов человека с другими последовательностями ДНК. Такие рекомбинантные антитела человека имеют вариабельные и константные области, полученные из последовательностей иммуноглобулинов зародышевой линии человека. В определенных вариантах осуществления, однако, такие рекомбинантные антитела человека подвергают мутагенезу in vitro (или, если применяют животное, трансгенное по последовательностям Ig человека, то соматическому мутагенезу in vivo) и, таким образом, аминокислотные последовательности областей VH и VL рекомбинантных антител представляют собой последовательности, которые хотя и получены из последовательностей VH и VL зародышевой линии человека и родственны им, могут не существовать в природе в пределах репертуара антител зародышевой линии человека in vivo.
Антитела человека могут встречаться в двух формах, что связано с гетерогенностью шарнирных участков. В варианте осуществления молекула иммуноглобулина содержит стабильную четырехцепочечную конструкцию массой примерно 150-160 кДа, в которой димеры удерживаются вместе посредством межцепочечной дисульфидной связи, которая связывает тяжелые цепи. В другом варианте осуществления димеры не соединены межцепочечными дисульфидными связями, и образуется молекула массой приблизительно 75-80 кДа, состоящая из ковалентно связанных легкой и тяжелой цепей (полуантитело). Эти варианты осуществления/формы крайне сложно разделить даже после аффинной очистки.
Частота появления второй формы в различных изотипах интактных IgG обусловлена без ограничения структурными различиями, связанными с изотипом шарнирной области антитела. Единственная аминокислотная замена в шарнирной области шарнира IgG4 человека может значительно снизить появление второй формы (Angal et al., (1993) Molecular Immunology 30:105, включен посредством ссылки во всей своей полноте) до уровней, обычно наблюдаемых при применении шарнира IgG1 человека. Настоящее изобретение охватывает в различных вариантах осуществления антитела с одной или несколькими мутациями в шарнирной области, области CH2 или CH3, что может быть необходимым, например, в получении, для повышения выхода необходимой формы антитела.
Термин "выделенное антитело", применяемый в данном документе, означает антитело, которое было идентифицировано и отделено и/или извлечено из по меньшей мере одного компонента своего естественного окружения. Например, антитело, которое было отделено или удалено из по меньшей мере одного компонента организма, или из ткани или клетки, в которой антитело изначально присутствует или продуцируется естественным путем, представляет собой "выделенное антитело". В различных вариантах осуществления выделенное антитело также включает антитело in situ в рекомбинантной клетке. В других вариантах осуществления выделенные антитела представляют собой антитела, которые были подвергнуты по меньшей мере одной стадии очистки или выделения. В различных вариантах осуществления выделенное антитело может практически не содержать другого клеточного материала и/или других химических соединений.
Термин "специфически связывает" и т. п. означает, что антитело или его антигенсвязывающий фрагмент образуют комплекс с антигеном, который является сравнительно устойчивым в физиологических условиях. Способы определения наличия специфического связывания антитела с антигеном хорошо известны из уровня техники и включают, например, равновесный диализ, поверхностный плазмонный резонанс и т. п. Например, антитело, которое "специфически связывает" IL-6R, применяемое в данном документе, включает антитела, которые связывают IL-6R или его часть с KD менее приблизительно 1000 нМ, менее приблизительно 500 нМ, менее приблизительно 300 нМ, менее приблизительно 200 нМ, менее приблизительно 100 нМ, менее приблизительно 90 нМ, менее приблизительно 80 нМ, менее приблизительно 70 нМ, менее приблизительно 60 нМ, менее приблизительно 50 нМ, менее приблизительно 40 нМ, менее приблизительно 30 нМ, менее приблизительно 20 нМ, менее приблизительно 10 нМ, менее приблизительно 5 нМ, менее приблизительно 4 нМ, менее приблизительно 3 нМ, менее приблизительно 2 нМ, менее приблизительно 1 нМ или менее приблизительно 0,5 нМ, измеренной в анализе поверхностного плазмонного резонанса. Специфическое связывание можно также характеризовать с помощью константы диссоциации, составляющей по меньшей мере приблизительно 1×10-6 M или меньше. В других вариантах осуществления константа диссоциации составляет по меньшей мере приблизительно 1×10-7 M, 1×10-8 M или 1×10-9 M. Выделенное антитело, которое специфически связывает IL-6R человека, может, однако, характеризоваться перекрестной реактивностью в отношении других антигенов, таких как молекулы IL-6R от других (отличных от человека) видов.
Термин "поверхностный плазмонный резонанс", применяемый в данном документе, относится к оптическому феномену, который обеспечивает возможность анализа взаимодействий в режиме реального времени посредством выявления изменений концентраций белков в биосенсорной матрице, например, с помощью системы BIACORE (отдел Biacore Life Sciences в GE Healthcare, Пискатауэй, Нью-Джерси).
Предполагается, что термин "KD", применяемый в данном документе, относится к равновесной константе диссоциации взаимодействия антитела и антигена.
Термин "эпитоп" относится к антигенной детерминанте, которая взаимодействует со специфическим антигенсвязывающим сайтом в вариабельной области молекулы антитела, известной как паратоп. Один антиген может иметь более одного эпитопа. Таким образом, различные антитела могут связываться с различными областями на антигене и могут оказывать различные биологические эффекты. Эпитопы могут быть конформационными или линейными. Конформационный эпитоп образован пространственно сближенными аминокислотами из различных сегментов линейной полипептидной цепи. Линейный эпитоп образован смежными аминокислотными остатками в полипептидной цепи. При определенных условиях эпитоп может включать фрагменты сахаридов, фосфорильных групп или сульфонильных групп антигена.
Антитела к IL-6R, применимые для способов, описанных в данном документе, в различных вариантах осуществления могут включать одну или несколько аминокислотных замен, вставок и/или делеций в каркасных участках и/или участках CDR вариабельных доменов тяжелой и легкой цепей по сравнению с соответствующими последовательностями зародышевой линии, из которых были получены антитела. Такие мутации можно легко определить посредством сравнения аминокислотных последовательностей, раскрытых в данном документе, с последовательностями зародышевой линии, доступными, например, из публичных баз данных последовательностей антител. В различных вариантах осуществления настоящее изобретение включает способы, предусматривающие применение антител и их антигенсвязывающих фрагментов, полученных из любых аминокислотных последовательностей, раскрытых в данном документе, где одна или несколько аминокислот в одной или нескольких каркасных областях и/или областях CDR подвергаются мутации по отношению к соответствующему(соответствующим) остатку(остаткам) последовательности зародышевой линии, из которой антитело получено, или в соответствующий(соответствующие) остаток(остатки) другой последовательности зародышевой линии человека, или по отношению к консервативной аминокислотной замене соответствующего(соответствующих) остатка(остатков) зародышевой линии (такие изменения последовательностей упоминаются в данном документе собирательно как "мутации зародышевой линии"). Можно конструировать многочисленные антитела и антигенсвязывающие фрагменты, которые содержат одну или несколько отдельных мутаций зародышевой линии или их комбинации. В определенных вариантах осуществления все остатки каркасных областей и/или CDR в доменах VH и/или VL подвергнуты обратной мутации в остатки, встречающиеся в исходной последовательности зародышевой линии, из которой было получено антитело. В других вариантах осуществления только определенные остатки подвергнуты обратной мутации в исходную последовательность зародышевой линии, например, подвергнуты мутации только остатки, расположенные в первых 8 аминокислотах FR1 или в последних 8 аминокислотах FR4, или подвергнуты мутации только остатки, расположенные в CDR1, CDR2 или CDR3. В других вариантах осуществления один или несколько остатков каркасных областей и/или CDR подвергнуты мутации в соответствующий(соответствующие) остаток(остатки) последовательности другой зародышевой линии (т. е. последовательности зародышевой линии, которая отличается от последовательности зародышевой линии, из которой изначально получено антитело). Кроме того, антитела могут содержать любую комбинацию из двух или более мутаций зародышевой линии в каркасных областях и/или областях CDR, например, где определенные отдельные остатки подвергнуты мутации в соответствующий остаток последовательности определенной зародышевой линии, в то время как определенные другие остатки, которые отличаются от последовательности исходной зародышевой линии, сохраняются или подвергнуты мутации в соответствующий остаток последовательности другой зародышевой линии. Сразу после получения антитела и антигенсвязывающие фрагменты, которые содержат одну или несколько мутаций зародышевой линии, могут быть легко протестированы в отношении одного или нескольких требуемых свойств, таких как улучшенная специфичность связывания, повышенная аффинность связывания, улучшенные или усиленные биологические антагонистические или агонистические свойства (в случае необходимости), сниженная иммуногенность и т. д. Настоящее изобретение охватывает применение антител и антигенсвязывающих фрагментов, полученных с помощью этого общего способа.
Настоящее изобретение также включает способы, предусматривающие применение антител к IL-6R, содержащие варианты любых из аминокислотных последовательностей HCVR, LCVR и/или CDR, раскрытых в данном документе, с одной или несколькими консервативными заменами. Например, настоящее изобретение включает применение антител к IL-6R с аминокислотными последовательностями HCVR, LCVR и/или CDR с, например, 10 или менее, 8 или менее, 6 или менее, 4 или менее и т. д. консервативными аминокислотными заменами относительно любой из аминокислотных последовательностей HCVR, LCVR и/или CDR, раскрытых в данном документе.
Согласно настоящему изобретению, антитело к IL-6R или его антигенсвязывающий фрагмент в различных вариантах осуществления содержат HCVR, LCVR и/или CDR, содержащие любую из аминокислотных последовательностей антител к IL-6R, описанных в патенте США № 7582298, включенном в данный документ посредством ссылки во всей своей полноте. В определенных вариантах осуществления антитело к IL-6R или его антигенсвязывающий фрагмент содержат HCDR в виде HCVR, содержащего аминокислотную последовательность под SEQ ID NO: 1, и LCDR в виде LCVR, содержащего аминокислотную последовательность под SEQ ID NO: 2. Согласно определенным вариантам осуществления антитело к IL-6R или его антигенсвязывающий фрагмент содержит три HCDR (т. е. HCDR1, HCDR2 и HCDR3) и три LCDR (т. е. LCDR1, LCDR2 и LCDR3), где HCDR1 содержит аминокислотную последовательность под SEQ ID NO: 3; HCDR2 содержит аминокислотную последовательность под SEQ ID NO: 4; HCDR3 содержит аминокислотную последовательность под SEQ ID NO: 5; LCDR1 содержит аминокислотную последовательность под SEQ ID NO: 6; LCDR2 содержит аминокислотную последовательность под SEQ ID NO: 7 и LCDR3 содержит аминокислотную последовательность под SEQ ID NO: 8. В других конкретных вариантах осуществления антитело к IL-6R или его антигенсвязывающий фрагмент содержат HCVR, содержащий аминокислотную последовательность под SEQ ID NO: 1, и LCVR, содержащий аминокислотную последовательность под SEQ ID NO: 2.
В другом варианте осуществления антитело к IL-6R или его антигенсвязывающий фрагмент содержат тяжелую цепь, содержащую аминокислотную последовательность под SEQ ID NO: 9, и легкую цепь, содержащую аминокислотную последовательность под SEQ ID NO: 10. В некоторых вариантах осуществления внеклеточный домен hIL-6R содержит аминокислотную последовательность под SEQ ID NO: 11. Согласно определенным иллюстративным вариантам осуществления способы по настоящему изобретению предусматривают применение антитела к IL-6R, обозначаемого и известного из уровня техники как сарилумаб, или его биологического эквивалента.
Аминокислотная последовательность под SEQ ID NO: 1 представляет собой EVQLVESGGGLVQPGRSLRLSCAASRFTFDDYAMHWVRQAPGKGLEWVSGISWNSGRIGYADSVKGRFTISRDNAENSLFLQMNGLRAEDTALYYCAKGRDSFDIWGQGTMVTVSS.
Аминокислотная последовательность под SEQ ID NO: 2 представляет собой DIQMTQSPSSVSASVGDRVTITCRASQGISSWLAWYQQKPGKAPKLLIYGASSLESGVPSRFSGSGSGTDFTLTISSLQPEDFASYYCQQANSFPYTFGQGTKLEIK.
Аминокислотная последовательность под SEQ ID NO: 3 представляет собой RFTFDDYA.
Аминокислотная последовательность под SEQ ID NO: 4 представляет собой ISWNSGRI.
Аминокислотная последовательность под SEQ ID NO: 5 представляет собой AKGRDSFDI.
Аминокислотная последовательность под SEQ ID NO: 6 представляет собой QGISSW.
Аминокислотная последовательность под SEQ ID NO: 7 представляет собой GAS.
Аминокислотная последовательность под SEQ ID NO: 8 представляет собой QQANSFPYT.
Аминокислотная последовательность под SEQ ID NO: 9 представляет собой
EVQLVESGGGLVQPGRSLRLSCAASRFTFDDYAMHWVRQAPGKGLEWVSGISWNSGRIGYADSVKGRFTISRDNAENSLFLQMNGLRAEDTALYYCAKGRDSFDIWGQGTMVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK.
Аминокислотная последовательность под SEQ ID NO: 10 представляет собой
DIQMTQSPSSVSASVGDRVTITCRASQGISSWLAWYQQKPGKAPKLLIYGASSLESGVPSRFSGSGSGTDFTLTISSLQPEDFASYYCQQANSFPYTFGQGTKLEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC.
Последовательность под SEQ ID NO: 11 представляет собой MVAVGCALLAALLAAPGAALAPRRCPAQEVARGVLTSLPGDSVTLTCPGVEPEDNATVHWVLRKPAAGSHPSRWAGMGRRLLLRSVQLHDSGNYSCYRAGRPAGTVHLLVDVPPEEPQLSCFRKSPLSNVVCEWGPRSTPSLTTKAVLLVRKFQNSPAEDFQEPCQYSQESQKFSCQLAVPEGDSSFYIVSMCVASSVGSKFSKTQTFQGCGILQPDPPANITVTAVARNPRWLSVTWQDPHSWNSSFYRLRFELRYRAERSKTFTTWMVKDLQHHCVIHDAWSGLRHVVQLRAQEEFGQGEWSEWSPEAMGTPWTESRSPPAENEVSTPMQALTTNKDDDNILFRDSANATSLPVQD.
Термин "биологический эквивалент", применяемый в данном документе, относится к молекуле, характеризующейся аналогичной биологической доступностью (коэффициентом и степенью доступности) после введения в той же молярной дозе и в аналогичных условиях (например, одинаковый путь введения), c тем, чтобы с учетом как эффективности, так и безопасности ожидаемый эффект был, по сути, таким же, как и у сопоставляемой молекулы. Две фармацевтические композиции, содержащие антитело к IL-6R, являются биологически эквивалентными, если они являются фармацевтически эквивалентными, то есть это означает, что они содержат одинаковое количество активного ингредиента (например, антитела к IL-6R) в одной и той же лекарственной форме, при одном и том же пути введения, и соответствуют одинаковым или сопоставимым стандартам. Биологическую эквивалентность можно определить, например, посредством исследования in vivo, сравнивая фармакокинетические параметры двух композиций. Обычно используемые в исследованиях биологической эквивалентности параметры включают пиковую концентрацию в плазме крови (Cmax) и площадь под кривой концентрации лекарственного средства в плазме крови в зависимости от времени (AUC).
Настоящее изобретение в определенных вариантах осуществления относится к способам, включающим введение субъекту антитела, содержащего вариабельную область тяжелой цепи, содержащую последовательность под SEQ ID NO: 1 и вариабельную область легкой цепи, содержащую последовательность под SEQ ID NO: 2.
В настоящем изобретении предусмотрены фармацевтические композиции, содержащие такое антитело, и способы применения этих композиций.
В различных вариантах осуществления антитело, содержащее вариабельную область тяжелой цепи SEQ ID NO: 1 и вариабельную область легкой цепи, содержащую последовательность под SEQ ID NO: 2, представляет собой антитело, которое специфически связывает hIL-6R. См. международную публикацию № WO2007/143168, включенную в данный документ посредством ссылки во всей своей полноте. В варианте осуществления антитело содержит вариабельную область тяжелой цепи, содержащую последовательность под SEQ ID NO: 9, и вариабельную область легкой цепи, содержащую последовательность под SEQ ID NO: 10. В различных вариантах осуществления антитело представляет собой сарилумаб.
DMARD
DMARD представляют собой лекарственные средства, которые используются при ревматоидном артрите и JIA для замедления прогрессирования заболевания.
DMARD подразделяются на синтетические (sDMARD) и биологические (bDMARD). sDMARD неисчерпывающе включают метотрексат, сульфасалазин, лефлуномид и гидроксихлорохин. bDMARD неисчерпывающе включают адалимумаб, голимумаб, этанерцепт, абатацепт, инфликсимаб, ритуксимаб и тоцилизумаб.
В некоторых вариантах осуществления с антителом не вводят никакое другое DMARD. В некоторых вариантах осуществления субъекту вводят по меньшей мере одно другое DMARD. В варианте осуществления по меньшей мере одно другое DMARD вводят субъекту параллельно с антителом или одновременно с ним.
В некоторых вариантах осуществления у субъекта наблюдали недостаточный ответ на текущее лечение JIA. В некоторых вариантах осуществления у субъекта наблюдали недостаточный ответ на текущее лечение JIA, и его рассматривают в качестве кандидата для лечения с помощью bDMARD. В некоторых вариантах осуществления текущее лечение субъекта представляет собой лечение на основе кортикостероида, sDMARD и/или bDMARD. В качестве примера, но не в качестве ограничения, в некоторых вариантах осуществления кортикостероид выбран из преднизона, преднизолона и метилпреднизолона.
Способы введения и составы
Способы, описанные в данном документе, предусматривают введение терапевтически эффективного количества антитела к IL-6R субъекту. Применяемый в данном документе термин "эффективное количество" или "терапевтически эффективное количество" представляет собой терапевтичекую дозу, которая приводит к лечению sJIA и/или pcJIA. Используемый в данном документе термин "лечение" означает приведение к нормализации одного или нескольких симптомов, ассоциированных с sJIA и/или pcJIA, или приведение к биологическому эффекту (например, снижению уровня определенного биомаркера), который связан с лежащим(ежащими) в основе патологическим(патологическими) механизмом(механизмами), приводящим(приводящими) к состоянию или симптому(симптомам). Например, доза антитела к IL-6R, которая вызывает улучшение любого из следующих симптомов или состояний, связанных с ювенильным идиопатическим артритом, считается "терапевтически эффективным количеством": хромота, скованность при пробуждении, нежелание субъекта пользоваться рукой или ногой, пониженный уровень активности, постоянная лихорадка, отек сустава и трудности с мелкой моторикой.
"Улучшение" ассоциированного с JIA симптома в различных вариантах осуществления относится к снижению частоты появления симптома JIA, которое может коррелировать с улучшением в одном или нескольких ассоциированных с JIA тестах, оценках или показателях (как описано в данном документе). Например, улучшение может коррелировать с увеличением по сравнению с исходным уровнем одного или нескольких критериев ACR JIA и/или снижением по сравнению с исходным уровнем одного или нескольких из следующего: использования кортикостероидов, индекса активности заболевания, представляющего собой ювенильный артрит или лихорадки. В одном варианте осуществления улучшение может включать уменьшение исходного уровня скованности (например, сустава с ограниченным движением). Используемое в данном документе выражение "исходный уровень" по отношению к ассоциированному с JIA параметру означает числовое значение ассоциированного с JIA параметра для пациента до или в течение всего периода введения антитела по настоящему изобретению. Обнаруживаемое "улучшение" также может быть обнаружено с использованием по меньшей мере одного теста, оценки или показателя, описанных в данном документе. В различных вариантах осуществления улучшение обнаруживается с использованием по меньшей мере одного, выбранного из группы, состоящей из: ответа ювенильного идиопатического артрита согласно Американской коллегии ревматологов (ACR) (например, ACR30 JIA, ACR50 JIA, ACR70 JIA, ACR90 JIA и ACR100 JIA). В различных вариантах осуществления улучшение характеризуется по меньшей мере одной оценкой или показателем, такими как общая оценка индекса активности заболевания врачом, оценка общего самочувствия пациентом или родителем, опросник оценки здоровья детей, количество суставов с активным артритом, количество суставов с ограниченным движением, высокочувствительный С-реактивный белок. В некоторых вариантах осуществления улучшение симптома sJIA представляет собой снижение лихорадки (например, у субъектов с лихорадкой при первом введении антитела). В различных вариантах осуществления улучшение характеризуется по меньшей мере одним биомаркером. В некоторых вариантах осуществления улучшение характеризуется повышением уровня по меньшей мере одного биомаркера. В некоторых вариантах осуществления улучшение характеризуется снижением уровня по меньшей мере одного биомаркера.
В другом примере лечение не являлось эффективным, когда доза антитела к IL-6R не приводит к выявляемому улучшению одного или нескольких параметров или симптомов, ассоциированных с JIA, или которая не вызывает биологического эффекта, который коррелирует с лежащим(лежащими) в основе патологическим(патологическими) механизмом(механизмами), вызывающим(вызывающими) состояние или симптом(симптомы) JIA.
В соответствии с некоторыми из этих вариантов осуществления антитело к IL-6R вводят подкожно. В соответствии с некоторыми из этих вариантов осуществления антителом к IL-6R является сарилумаб.
В соответствии со способами настоящего изобретения терапевтически эффективное количество антитела к IL-6R, которое вводят субъекту, будет варьировать в зависимости от возраста и размера (например, веса тела или площади поверхности тела) субъекта, а также пути введения и других факторов, хорошо известных специалистам в данной области.
В определенных вариантах осуществления доза антитела варьирует в зависимости от веса тела субъекта. В различных вариантах осуществления, если субъект имеет вес тела, составляющий по меньшей мере 30 кг (например, до 60 кг, 70 кг, 80 кг, 90 кг или 100 кг), то субъекту вводят дозу, составляющую 2,0 мг/кг один раз в 2 недели. В других вариантах осуществления, если субъект имеет вес тела, составляющий по меньшей мере 30 кг и менее чем 40, 50, 60, 70, 80, 90 или 100 кг, то субъекту вводят дозу, составляющую 2,0 мг/кг один раз в 2 недели. В некоторых вариантах осуществления, если субъект имеет вес тела, составляющий по меньшей мере 30 кг, то субъекту вводят дозу, составляющую 3,0 мг/кг один раз в 2 недели. В некоторых вариантах осуществления, если субъект имеет вес тела, составляющий по меньшей мере 30 кг, то субъекту вводят дозу, составляющую 4-6 мг/кг один раз в 2 недели. В других вариантах осуществления, если субъект имеет вес тела, составляющий по меньшей мере 30 кг и менее чем 40, 50, 60, 70, 80, 90 или 100 кг, то субъекту вводят дозу, составляющую 3,0 мг/кг один раз в 2 недели. В определенных вариантах осуществления, если субъект имеет вес тела, составляющий по меньшей мере 30 кг, то субъекту вводят дозу, составляющую 2,0 мг/кг, один раз в неделю. В других вариантах осуществления, если субъект имеет вес тела, составляющий по меньшей мере 30 кг и менее чем 40, 50, 60, 70, 80, 90 или 100 кг, то субъекту вводят дозу, составляющую 2,0 мг/кг, один раз в неделю. В различных вариантах осуществления, если субъект имеет вес тела, составляющий 10-<30 кг, то субъекту вводят дозу, составляющую 2,5 мг/кг каждые 2 недели. В определенных вариантах осуществления, если субъект имеет вес тела, составляющий 10-<30 кг, то субъекту вводят дозу, составляющую 4 мг/кг каждые 2 недели. В некоторых вариантах осуществления, если субъект имеет вес тела, составляющий 10-<30 кг, то субъекту вводят дозу, составляющую 2,5 мг/кг в неделю. В различных вариантах осуществления, если субъект имеет вес тела, составляющий 10-<30 кг, то субъекту вводят дозу, составляющую 5-7 мг/кг каждые 2 недели. В различных вариантах осуществления, если субъект имеет вес тела, составляющий 10-<30 кг, то субъекту вводят дозу, составляющую 4 мг/кг каждые 2 недели.
В определенных вариантах осуществления доза антитела к IL-6R, вводимая субъекту, составляет от приблизительно 10 мг до приблизительно 600 мг. В некоторых вариантах осуществления доза антитела, вводимая субъекту, составляет от приблизительно 25 мг до приблизительно 200 мг. В различных вариантах осуществления доза антитела, вводимая субъекту, составляет от приблизительно 60 мг до приблизительно 200 мг. Например, настоящее изобретение включает (без ограничения) способы, где приблизительно 10 мг, приблизительно 15 мг, приблизительно 20 мг, приблизительно 25 мг, приблизительно 30 мг, приблизительно 35 мг, приблизительно 40 мг, приблизительно 45 мг, приблизительно 50 мг, приблизительно 55 мг, приблизительно 60 мг, приблизительно 65 мг, приблизительно 70 мг, приблизительно 75 мг, приблизительно 80 мг, приблизительно 85 мг, приблизительно 90 мг, приблизительно 95 мг, приблизительно 100 мг, приблизительно 105 мг, приблизительно 110 мг, приблизительно 115 мг, приблизительно 120 мг, приблизительно 125 мг, приблизительно 130 мг, приблизительно 135 мг, приблизительно 140 мг, приблизительно 145 мг, приблизительно 150 мг, приблизительно 155 мг, приблизительно 160 мг, приблизительно 165 мг, приблизительно 170 мг, приблизительно 175 мг, приблизительно 180 мг, приблизительно 185 мг, приблизительно 190 мг, приблизительно 195 мг, приблизительно 200 мг, приблизительно 205 мг, приблизительно 210 мг, приблизительно 215 мг, приблизительно 220 мг, приблизительно 225 мг, приблизительно 230 мг, приблизительно 235 мг, приблизительно 240 мг, приблизительно 245 мг, приблизительно 250 мг, приблизительно 255 мг, приблизительно 260 мг, приблизительно 265 мг, приблизительно 270 мг, приблизительно 275 мг, приблизительно 280 мг, приблизительно 285 мг, приблизительно 290 мг, приблизительно 295 мг, приблизительно 300 мг, приблизительно 325 мг, приблизительно 350 мг, приблизительно 375 мг, приблизительно 400 мг, приблизительно 425 мг, приблизительно 450 мг, приблизительно 475 мг, приблизительно 500 мг, приблизительно 525 мг, приблизительно 550 мг, приблизительно 575 мг, приблизительно 600 мг или более антитела к IL-6R вводят пациенту один раз в неделю или один раз в две недели.
Используемое в данном документе выражение "вводимый от приблизительно 2,0 до приблизительно 4,0 мг/кг" означает, что указанное вещество вводят при любом значении в пределах установленного диапазона, включая конечные точки диапазона. Например, "доза антитела к IL-6R, вводимая пациенту, составляет от 2,0 мг/кг до 4,0 мг/кг" предусматривает введение 2,0 мг/кг антитела к IL-6R, 4,0 мг/кг антитела к IL-6R и всех доз между ними.
В различных вариантах осуществления антитело к IL-6R вводят в дозе, составляющей приблизительно от 25 до 150 мг один раз в неделю или от 40 мг до 200 мг каждые две недели. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей от 25 до 50 мг один раз в неделю или один раз в две недели. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей от 50 до 75 мг один раз в неделю или один раз в две недели. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей от 75 до 100 мг один раз в неделю или один раз в две недели. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей от 100 до 125 мг один раз в неделю или один раз в две недели. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей от 125 до 150 мг один раз в неделю или один раз в две недели. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей от 150 до 175 мг один раз в неделю или один раз в две недели. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей от 175 до 200 мг один раз в неделю или один раз в две недели. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей 100 мг один раз в неделю. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей 150 мг один раз в неделю. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей 200 мг один раз в неделю. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей от 100 до 150 мг один раз в неделю. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей от 100 до 200 мг, один раз в две недели. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей от 150 до 200 мг, один раз в две недели. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей приблизительно 100 или приблизительно 150 мг, один раз в две недели. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей приблизительно 100, 150 или 200 мг, один раз в две недели. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей 100 мг, один раз в две недели. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей 150 мг, один раз в две недели. В одном варианте осуществления антитело к IL-6R вводят в дозе, составляющей 200 мг, один раз в две недели.
В различных вариантах осуществления способа антитело применяют в дозах, перечисленных в таблицах 2-5.
Количество антитела к IL-6R, которое вводят пациенту, можно выражать в миллиграммах антитела на килограмм веса тела пациента (т. е. мг/кг). Например, способы по настоящему изобретению предусматривают введение антитела к IL-6R пациенту в суточной дозе, составляющей от приблизительно 0,01 до приблизительно 100 мг/кг, от приблизительно 0,1 до приблизительно 50 мг/кг или от приблизительно 1 до приблизительно 10 мг/кг веса тела пациента. В определенных вариантах осуществления антителом к hIL6R является сарилумаб, и его вводят в дозе, составляющей от приблизительно 2 мг/кг до приблизительно 4 мг/кг. В различных вариантах осуществления антителом к hIL6R является сарилумаб и его вводят в дозе, составляющей от приблизительно 2 мг/кг до приблизительно 3 мг/кг. В определенных вариантах осуществления антителом к hIL6R является сарилумаб и его вводят в дозе, составляющей от приблизительно 2,5 мг/кг до приблизительно 4 мг/кг. В различных вариантах осуществления антитело к hIL6R представляет собой сарилумаб и его вводят в дозе, составляющей от приблизительно 2 мг/кг до приблизительно 3 мг/кг с частотой один раз в неделю (qw) или один раз в две недели (q2w). В некоторых вариантах осуществления антитело к hIL6R вводят в дозе, составляющей приблизительно от 2 мг/кг до 4 мг/кг, с частотой один раз в неделю (qw) или один раз в две недели (q2w). В некоторых вариантах осуществления антитело к hIL6R вводят в дозе, составляющей приблизительно от 2,5 мг/кг до 4 мг/кг, с частотой один раз в неделю (qw) или один раз в две недели (q2w). В различных вариантах осуществления антитело к hIL6R вводят в дозе, составляющей приблизительно 1,0, 1,5, 2,0, 2,5, 3,0, 3,5, 4,0, 4,5 или 5,0 мг/кг. В некоторых вариантах осуществления антитело к hIL6R содержит VH и VL, где VH содержит три CDR, расположенные в последовательности SEQ ID NO: 1, и где VL содержит три CDR, расположенные в последовательности SEQ ID NO: 2. В других вариантах осуществления антитело к hIL6R представляет собой сарилумаб.
Способы по настоящему изобретению предусматривают введение многократных доз антитела к IL-6R пациенту в течение определенного промежутка времени. Например, антитело к IL-6R можно вводить приблизительно 1-5 раз в день, приблизительно 1-5 раз в неделю, приблизительно 1-5 раз в месяц или приблизительно 1-5 раз в год. В определенных вариантах осуществления в способах по настоящему изобретению предусмотрено введение первой дозы антитела к IL-6R пациенту в первой временной точке с последующим введением по меньшей мере второй дозы антитела к IL-6R пациенту во второй временной точке. В определенных вариантах осуществления первая и вторая дозы могут содержать одинаковое количество антитела к IL-6R. Например, каждая из первой и второй доз может содержать от приблизительно 10 мг до приблизительно 500 мг, от приблизительно 20 мг до приблизительно 300 мг, от приблизительно 100 мг до приблизительно 200 мг или от приблизительно 100 мг до приблизительно 150 мг антитела. Период времени между первой и второй дозами может составлять от приблизительно нескольких часов до нескольких недель. Например, вторая временная точка (т. е. время, когда вводят вторую дозу) может располагаться через от приблизительно 1 часа до приблизительно 7 недель после первой временной точки (т. е. времени, когда вводят первую дозу). В соответствии с определенными иллюстративными вариантами осуществления настоящего изобретения вторая временная точка может располагаться через приблизительно 1 час, приблизительно 4 часа, приблизительно 6 часов, приблизительно 8 часов, приблизительно 10 часов, приблизительно 12 часов, приблизительно 24 часа, приблизительно 2 дня, приблизительно 3 дня, приблизительно 4 дня, приблизительно 5 дней, приблизительно 6 дней, приблизительно 7 дней, приблизительно 2 недели, приблизительно 4 недели, приблизительно 6 недель, приблизительно 8 недель, приблизительно 10 недель, приблизительно 12 недель, приблизительно 14 недель или более после первой временной точки. В определенных вариантах осуществления вторая временная точка располагается через приблизительно 1 неделю или приблизительно 2 недели. Третью и последующие дозы можно вводить аналогичным образом в течение курса лечения пациента. В настоящем изобретении предусмотрены способы применения терапевтических композиций, содержащих антитела к IL-6R или их антигенсвязывающие фрагменты и необязательно одно или несколько дополнительных терапевтических средств. Терапевтические композиции по настоящему изобретению будут вводить с подходящими носителями, вспомогательными веществами и другими средствами, которые включены в составы, для обеспечения улучшенных переноса, доставки, переносимости и т. п. Большое количество соответствующих составов можно найти в справочнике, известном всем химикам-фармацевтам: Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, включенном в данный документ посредством ссылки во всей своей полноте. Эти составы включают, например, порошки, пасты, мази, желе, воски, масла, липиды, везикулы, содержащие липиды (катионные или анионные) (такие как LIPOFECTIN), конъюгаты ДНК, безводные абсорбционные пасты, эмульсии типа "масло в воде" и "вода в масле", эмульсии карбовакс (полиэтиленгликоли с различной молекулярной массой), полутвердые гели и полутвердые смеси, содержащие карбовакс. См. также Powell et al. "Compendium of excipients for parenteral formulations" PDA (1998) J Pharm Sci Technol 52:238-311, включенный в данный документ посредством ссылки во всей своей полноте.
Известны различные системы доставки, и их можно применять для введения фармацевтической композиции по настоящему изобретению, например, инкапсуляция в липосомы, микрочастицы, микрокапсулы, опосредованный рецепторами эндоцитоз (см., например, Wu et al. (1987) J. Biol. Chem. 262:4429-4432, включенный в данный документ посредством ссылки во всей своей полноте). Способы введения включают без ограничения внутрикожные, внутримышечные, внутрибрюшинные, внутривенные, подкожные, интраназальные, эпидуральные и пероральные пути. Композицию можно вводить любым удобным путем, например, путем инфузии или болюсной инъекции, путем абсорбции через эпителиальные или слизистые оболочки (например, слизистую оболочку полости рта, слизистую оболочку прямой кишки и кишечника и т. д.) и можно вводить вместе с другими биологически активными средствами. Введение может быть системным или местным. Антитело к IL-6R можно вводить подкожно.
Фармацевтическую композицию также можно доставлять в везикуле, такой как липосома (см. Langer (1990) Science 249:1527-1533, включенный в данный документ посредством ссылки во всей своей полноте). В определенных ситуациях фармацевтическую композицию можно доставлять в системе с контролируемым высвобождением, например, с применением насоса или полимерных материалов. В другом варианте осуществления систему с контролируемым высвобождением можно поместить вблизи от мишени композиции, при этом требуется лишь часть системной дозы.
Инъекционные препараты могут включать лекарственные формы для внутривенных, подкожных, внутрикожных и внутримышечных инъекций, для местной инъекции, капельных инфузий и т. д. Эти инъекционные препараты можно получать посредством известных способов. Например, инъекционные препараты можно получать, например, растворением, суспендированием или эмульгированием антитела или его соли, описанных выше, в стерильной водной среде или масляной среде, обычно применяемых для инъекций. В качестве водной среды для инъекций существуют, например, физиологический раствор, изотонический раствор, содержащий глюкозу и другие вспомогательные средства, и т. д., которые можно применять в комбинации с соответствующим солюбилизирующим средством, таким как спирт (например, этанол), многоатомный спирт (например, пропиленгликоль, полиэтиленгликоль), неионогенное поверхностно-активное вещество [например, полисорбат 80, HCO-50 (полиоксиэтиленовый (50 моль) аддукт гидрогенизированного касторового масла)] и т. д.). В качестве масляной среды применяют, например, кунжутное масло, соевое масло и т. д., которые могут применять в комбинации с солюбилизирующим средством, таким как бензилбензоат, бензиловый спирт и т. д. Раствор для инъекции, полученный таким образом, можно помещать в соответствующую ампулу.
Антитело обычно составляют как описано в данном документе и в международной публикации № WO2011/085158, которая включена в данный документ посредством ссылки во всей своей полноте.
В различных вариантах осуществления антитело вводят в виде водного буферного раствора с pH приблизительно 6,0, содержащего
- приблизительно 21 мМ гистидина,
- приблизительно 45 мМ аргинина,
- приблизительно 0,2% (вес/об.) полисорбата 20,
- приблизительно 5% (мас./об.) сахарозы и
- от приблизительно 100 мг/мл до приблизительно 200 мг/мл антитела.
В другом варианте осуществления антитело вводят в виде водного буферного раствора с pH приблизительно 6,0, содержащего
- приблизительно 21 мМ гистидина,
- приблизительно 45 мМ аргинина,
- приблизительно 0,2% (вес/об.) полисорбата 20,
- приблизительно 5% (мас./об.) сахарозы и
- по меньшей мере приблизительно 130 мг/мл антитела.
В другом варианте осуществления антитело вводят в виде водного буферного раствора с pH приблизительно 6,0, содержащего
- приблизительно 21 мМ гистидина,
- приблизительно 45 мМ аргинина,
- приблизительно 0,2% (вес/об.) полисорбата 20,
- приблизительно 5% (мас./об.) сахарозы и
- приблизительно 131,6 мг/мл антитела.
В другом варианте осуществления антитело вводят в виде водного буферного раствора с pH приблизительно 6,0, содержащего
- приблизительно 21 мМ гистидина,
- приблизительно 45 мМ аргинина,
- приблизительно 0,2% (вес/об.) полисорбата 20,
- приблизительно 5% (мас./об.) сахарозы и
- приблизительно 175 мг/мл антитела.
В других вариантах осуществления антитело вводят в виде водного буферного раствора с pH 6,0, содержащего
- 21 мМ гистидина,
- 45 мМ аргинина,
- 0,2% (вес/об.) полисорбата 20,
- 5% (мас./об.) сахарозы и
- от 100 мг/мл до 200 мг/мл антитела.
В другом варианте осуществления антитело вводят в виде водного буферного раствора с pH 6,0, содержащего
- 21 мМ гистидина,
- 45 мМ аргинина,
- 0,2% (вес/об.) полисорбата 20,
- 5% (мас./об.) сахарозы и
- по меньшей мере 130 мг/мл антитела.
В другом варианте осуществления антитело вводят в виде водного буферного раствора с pH 6,0, содержащего
- 21 мМ гистидина,
- 45 мМ аргинина,
- 0,2% (вес/об.) полисорбата 20,
- 5% (мас./об.) сахарозы и
- 131,6 мг/мл антитела.
В другом варианте осуществления антитело вводят в виде водного буферного раствора с pH 6,0, содержащего
- 21 мМ гистидина,
- 45 мМ аргинина,
- 0,2% (вес/об.) полисорбата 20,
- 5% (мас./об.) сахарозы и
- 175 мг/мл антитела.
Преимущественно фармацевтические композиции для перорального или парентерального применения, описанные выше, получают в лекарственных формах в стандартной дозе, подходящей для подбора дозы активных ингредиентов. Такие лекарственные формы в стандартной дозе включают, например, таблетки, пилюли, капсулы, инъекционные формы (ампулы), суппозитории и т. д.
В соответствии со способами, раскрытыми в данном документе, антитело к IL-6R (или фармацевтический состав, содержащий антитело) можно вводить пациенту с помощью любого приемлемого устройства или механизма. Например, введение можно выполнять с применением шприца и иглы или с помощью шприца-ручки многократного использования и/или автоинъекторного устройства доставки. Способы по настоящему изобретению предусматривают применение многочисленных шприцев-ручек многоразового использования и/или автоинъекторных устройств доставки для введения антитела к IL-6R (или фармацевтического состава, содержащего антитело). Примеры таких устройств включают без ограничения AUTOPEN (Owen Mumford, Inc., Вудсток, Великобритания), шприц-ручку DISETRONIC (Disetronic Medical Systems, Бургдорф, Швейцария), шприц-ручку HUMALOG MIX 75/25, шприц-ручку HUMALOG, шприц-ручку HUMALIN 70/30 (Eli Lilly and Co., Индианаполис, Индиана), NOVOPEN I, II и III (Novo Nordisk, Копенгаген, Дания), NOVOPEN JUNIOR (Novo Nordisk, Копенгаген, Дания), шприц-ручку BD (Becton Dickinson, Франклин-Лэйкс, Нью-Джерси), OPTIPEN, OPTIPEN PRO, OPTIPEN STARLET и OPTICLIK (Sanofi-Aventis, Франкфурт, Германия). Примеры шприцов-ручек однократного применения и/или автоинъекторных устройств доставки, применяемых при подкожной доставке фармацевтической композиции по настоящему изобретению, включают без ограничения шприц-ручку SOLOSTAR (Sanofi-Aventis), FLEXPEN (Novo Nordisk) и KWIKPEN (Eli Lilly), автоинъектор SURECLICK (Amgen, Таузанд-Окс, Калифорния), PENLET (Haselmeier, Штутгарт, Германия), EPIPEN (Dey, L.P.) и шприц-ручку HUMIRA (Abbott Labs, Норт-Чикаго, Иллинойс), при этом упомянуты только несколько.
В одном варианте осуществления антитело вводят с помощью предварительно заполненного шприца. В другом варианте осуществления антитело вводят с помощью предварительно заполненного шприца с защитным устройством. Например, защитное устройство предупреждает случайное ранение, полученное медработником. В различных вариантах осуществления антитело вводят с помощью предварительно заполненного шприца с защитным устройством ÈRIS (West Pharmaceutical Services Inc.). См. также патенты США №№ 5215534 и 9248242, включенные в данный документ посредством ссылки во всей их полноте.
В другом варианте осуществления антитело вводят с помощью автоинъектора. В различных вариантах осуществления антитело вводят с помощью автоинъектора с технологией PUSHCLICK (SHL Group). В различных вариантах осуществления автоинъектор представляет собой устройство, состоящее из шприца, которое обеспечивает введение субъекту дозы композиции и/или антитела. См. также патенты США №№ 9427531 и 9566395, включенные в данный документ посредством ссылки во всей их полноте.
Применение микроинфузора для доставки антитела к IL-6R (или фармацевтической композиции, содержащей антитело) пациенту также предусмотрено в данном документе. Применяемый в данном документе термин "микроинфузор" означает устройство для подкожной доставки, разработанное для медленного введения больших объемов (например, до приблизительно 2,5 мл или больше) терапевтического состава в течение продолжительного периода времени (например, приблизительно 10, 15, 20, 25, 30 или более минут). См., например, U.S. 6629949; US 6659982; и Meehan et al., J. Controlled Release 46:107-116 (1996), включенные в данный документ посредством ссылки во всей их полноте. Микроинфузоры являются особенно применимыми для доставки больших доз терапевтических белков, содержащихся в растворах с высокой концентрацией (например, приблизительно 100, 125, 150, 175, 200 мг/мл или более) и/или вязких растворах.
Антитело по настоящему изобретению в различных вариантах осуществления вводят субъектам, которые страдают от типа JIA, например от системного ювенильного идиопатического артрита. В определенных вариантах осуществления способы, раскрытые в данном документе, включают стадию выбора субъекта, страдающего от типа JIA, например от системного ювенильного идиопатического артрита. Диагностику системного ювенильного идиопатического артрита проводят в соответствии с критериями классификации JIA ILAR 2001 (Petty RE, et al. International League of Associations for Rheumatology Classification of Juvenile Idiopathic Arthritis Second Revision, Edmonton, 2001. J Rheumatol. 2004 Feb;31(2):390-2). В некоторых вариантах осуществления возраст пациентов составляет от приблизительно 1 года до приблизительно 17 лет. В различных вариантах осуществления возраст пациентов составляет от приблизительно 4 до приблизительно 6 лет, от приблизительно 12 до приблизительно 18 лет, от приблизительно 12 до приблизительно 14 лет или от приблизительно 7 до приблизительно 9 лет. В различных вариантах осуществления у субъекта проявляется артрит в 5 активных суставах. В некоторых вариантах осуществления у субъекта проявляется артрит в 2 активных суставах с лихорадкой системного ювенильного идиопатического артрита (при температуре выше 37,5 °C) в течение по меньшей мере 3 из любых 7 последовательных дней, несмотря на прием глюкокортикоидов в стабильной дозе в течение по меньшей мере 3 дней. В различных вариантах осуществления у субъекта наблюдали недостаточный ответ на текущее лечение, и его рассматривают в качестве кандидата для лечения с помощью bDMARD.
Антитело по настоящему изобретению в различных вариантах осуществления вводят субъектам, страдающим от pcJIA. В определенных вариантах осуществления способы, раскрытые в данном документе, включают стадию выбора субъекта, страдающего от типа JIA, например от pcJIA. Диагностику RF-отрицательного или RF-положительного подтипа полиартикулярного ювенильного идиопатического артрита или подтипа распространенного олигоартикулярного ювенильного идиопатического артрита проводят в соответствии с критериями классификации JIA ILAR 2001 (Petty RE, et al. International League of Associations for Rheumatology Classification of Juvenile Idiopathic Arthritis: Second Revision, Edmonton, 2001. J Rheumatol. 2004 Feb;31(2):390-2). В некоторых вариантах осуществления возраст пациентов составляет от приблизительно 2 лет до приблизительно 17 лет. В различных вариантах осуществления возраст пациентов составляет от приблизительно 4 до приблизительно 6 лет, от приблизительно 12 до приблизительно 18 лет, от приблизительно 12 до приблизительно 14 лет или от приблизительно 7 до приблизительно 9 лет. В различных вариантах осуществления у субъекта проявляется артрит в по меньшей мере 5 активных суставах в соответствии с определением активного артрита Американской коллегией ревматологов. В различных вариантах осуществления у субъекта наблюдали недостаточный ответ на текущее лечение, и его рассматривают в качестве кандидата для лечения с помощью bDMARD.
В определенных вариантах осуществления недостаточный ответ на предыдущее лечение относится к субъектам, у которых JIA является плохо контролированным после получения предыдущего лечения (например, инъекции глюкокортикоидов в сустав и/или метотрексата) в максимальной переносимой типичной дозе. В одном варианте осуществления недостаточный ответ на предыдущее лечение относится к субъектам, у которых наблюдается умеренная или высокая активность заболевания и признаки плохого прогноза, несмотря на предыдущее лечение. В различных вариантах осуществления недостаточный ответ на предыдущее лечение относится к субъектам с симптомом JIA (например, любым симптомом, перечисленным в данном документе), в отношении которого не наблюдали улучшения или ухудшения, несмотря на предыдущее лечение.
Эффективность различных способов лечения sJIA и pcJIA можно оценить с помощью любого из способов, описанных в данном документе, например, по индексу-27 активности заболевания, представляющего собой ювенильный артрит (JADAS). (Consolaro et al. Development and Validation of a Composite Disease Activity Score for Juvenile Idiopathic Arthritis. Arthritis & Rheumatism. 2009 May;61(5):658-666).
Все публикации, упомянутые в данном документе, включены в данный документ посредством ссылки во всей их полноте для всех предназначений.
СПИСОК СОКРАЩЕНИЙ
ACR: Американская коллегия ревматологов
ADA: антитело к лекарственному средству
AE: нежелательное явление
AESI: нежелательное(нежелательные) явление(явления), представляющее(представляющие) особый интерес
ALP: щелочная фосфатаза
ALT: аланинаминотрансфераза
ANA: антинуклеарные антитела
ANC: абсолютное количество нейтрофилов
антитела к dsDNA: антитела к двухцепочечной ДНК
AST: аспартатаминотрансфераза
BP: кровяное давление
BUN: азот мочевины крови
CHAQ: опросник оценки здоровья детей
COX-2: ингибиторы циклооксигеназы 2
CRF: индивидуальная регистрационная карта
CRP: C-реактивный белок
CSR: отчет о клиническом исследовании
CYP: цитохром
DEC: комитет по повышению дозы
DMARD: модифицирующее заболевание противоревматическое лекарственное средство
DMC: комитет по мониторингу данных
ДНК: дезоксирибонуклеиновая кислота
EBV: вирус Эпштейна-Барр
e-CRF: электронная индивидуальная регистрационная карта
EOS: конец исследования
EOT: конец лечения
ESR: скорость оседания эритроцитов
GCP: надлежащая клиническая практика
HAQ-DI: индекс инвалидизации по опроснику оценки здоровья
HBc-Ab: антитело к коровому антигену вируса гепатита B
ВИЧ: вирус иммунодефицита человека
HLGT: групповой термин высокого уровня
HR: частота сердечных сокращений
hs-CRP: высокочувствительный C-реактивный белок
ICF: форма информированного согласия
ICH: Международный совет по гармонизации
IEC: независимый комитет по вопросам этики
IgG: иммуноглобулин G
IL-6: интерлейкин 6
IL-6R: рецептор IL-6
ILAR: Международная лига ассоциаций ревматологов
IMP: исследуемый лекарственный препарат
IRB: Институциональный наблюдательный совет
IVRS: интерактивная система, обеспечивающая доступ путем голосового ответа
IWRS: интерактивная система, обеспечивающая доступ через интернет
JADAS: индекс активности заболевания, представляющего собой ювенильный артрит
JAK: Янус-киназа
JIA: ювенильный идиопатический артрит
ACR JIA: ответ ювенильного идиопатического артрита согласно Американской коллегии ревматологов
LDH: лактатдегидрогеназа
LDL: липопротеин низкой плотности
LFT: функциональные пробы печени, функциональная проба печени
LLOQ: нижний предел количественного определения
mAB: моноклональное антитело
MAS: синдром активации макрофагов
mTSS: модифицированный общий счет Шарпа
MTX: метотрексат
NIMP: неисследуемый лекарственный препарат
NSAID: нестероидные противовоспалительные лекарственные средства
oJIA: олигоартикулярный ювенильный идиопатический артрит
pcJIA: полиартикулярное течение ювенильного идиопатического артрита
PCSA: потенциально клинически значимое отклонение от нормы
PD: фармакодинамика
PK: фармакокинетика
PopPK: популяционная фармакокинетика
PPD: очищенное производное белка
PT: предпочтительный термин
q2w: один раз в две недели
qw: один раз в неделю
RA: ревматоидный артрит
RF: ревматоидный фактор
SAE: серьезное нежелательное явление.
SC: подкожно
SD: стандартное отклонение.
sJIA: системный ювенильный идиопатический артрит
SOC: системно-органный класс
SUSAR: прогнозированная непредвиденная серьезная нежелательная реакция
TB: туберкулез
TEAE: нежелательное явление, возникшее в ходе лечения
ULN: верхняя граница нормы
VAS визуальная аналоговая шкала
WBC: лейкоциты
ПРИМЕРЫ
Следующие примеры изложены для того, чтобы обеспечить специалистов в данной области полным раскрытием и описанием получения и применения способов и композиций, описанных в настоящем изобретении, и они не предполагают ограничения объема того, что авторы настоящего изобретения рассматривают в качестве своего изобретения. Были приложены усилия для обеспечения точности в отношении используемых чисел (например, количеств, температуры и т. п.), однако, необходимо учитывать некоторые экспериментальные ошибки и отклонения.
Пример 1. Открытое исследование с подбором дозы сарилумаба с последовательными нарастающими повторными дозами, вводимыми путем подкожной (SC) инъекции, у детей и подростков в возрасте от 2 до 17 лет с pcJIA с последующей фазой дополнительного лечения. (Исследование № DRI13925, название исследования: открытое исследование с подбором дозы сарилумаба с нарастающими повторными дозами у детей и подростков с pcJIA)
Контролируемое испытание фазы 2 (№ NCT02776735) начинали для тестирования моноклонального антитела IgG1 человека к IL-6Rα, сарилумаба, вводимого подкожно при лечении pcJIA. Это исследование было многонациональным многоцентровым открытым 2-фазным и 2-частным исследованием с участием детей и подростков в возрасте от 2 до 17 лет (или применяются возрастные требования страны) с pcJIA, у которых наблюдали недостаточный ответ на текущую терапию или у которых наблюдали ее непереносимость, или которых рассматривали в качестве кандидатов на биологическое модифицирующее заболевание противоревматическое лекарственное средство (DMARD). 2 фазы исследования представляли собой начальную 12-недельную основную фазу лечения, за которой следовала 144-недельная фаза дополнительного лечения (см.фиг. 1 и фиг. 2).
Общий дизайн и план исследования.
12-недельная основная фаза лечения
В это многонациональное многоцентровое открытое исследование включали пациентов в возрасте от 2 до 17 лет с диагнозом RF-положительный или RF-отрицательный pJIA или eoJIA в соответствии с критериями классификации JIA ILAR 2001 (1). Пациенты имели ≥5 активных суставов и должны были рассматриваться по мнению исследователей в качестве кандидатов на bDMARD.
Пациенты получали сарилумаб путем подкожной (SC) инъекции, которую проводило профессиональное лицо, осуществляющее уход, в исследовательском центре. Схемы введения доз тестировали в 2 группах по весу тела (группа A, ≥30-60 кг и группа B, 10-<30 кг) поэтапно, начиная со схемы с наименьшей дозой сарилумаба в группе с более высоким весом тела, группе A (фиг. 2). Схемы введения доз в группах A/B представляли собой 2,0/2,5 мг/кг Q2W, 3,0/4,0 мг/кг Q2W и 2,0/2,5 мг/кг QW для схем введения доз 1, 2 и 3 соответственно. Начало схемы введения доз 1 в группе B разрешали после обзора данных от первых 3 пациентов, включенных в ту же схему введения доз в группе A, через по меньшей мере 4 недели лечения. Переход к схемам с более высокими дозами в каждой группе по весу тела разрешали после обзора данных по безопасности, эффективности, фармакокинетике (PK) и фармакодинамике (PD) после того, как первые 3 пациента, включенные в текущую группу по весу тела и дозе, завершали 6 недель лечения. Включение в группу дозы 3 группы B разрешали после анализа данных по всем другим группам по весу тела и дозе. Решения о повышении дозы принимал Комитет по повышению дозы (DEC). DEC мог объявить дозировку неэффективной, если первые 3 пациента, включенные в эту группу по дозе и весу тела, не достигали ответа ACR30 JIA к неделе 6, при этом прекращали включение в данную группу по дозе и весу тела и переводили пациентов с данной схемы на стандарт лечения, утвержденный исследователем. Безопасность сарилумаба официально контролировал на протяжении всего исследования независимый комитет по мониторингу данных, в который входили 3 детских ревматолога.
Основная цель исследования заключалась в изучении PK профиля 3 схем введения доз сарилумаба, применяемых в течение 12 недель, с целью выбора одной схемы для дальнейшего исследования. Схемы введения доз выбирали на основе моделирования и симуляции, чтобы обеспечить воздействия, аналогичные схемам введения доз, оцененным при RA у взрослых. Схема введения доз 1 соответствовала 150 мг каждые 2 недели (Q2W), самой низкой одобренной дозе для взрослых; схема введения доз 2 соответствовала 200 мг Q2W, рекомендуемой одобренной дозе для взрослых; и схема введения доз 3 соответствовала 150 мг каждую неделю (QW), самой высокой дозе, испытанной у взрослых, которая продемонстрировала большую эффективность, чем плацебо с приемлемым профилем безопасности (20).
12-недельная основная фаза лечения (проиллюстрированная на фиг. 2) была разделена на 3 части. Первую часть с подбором дозы с последовательными нарастающими дозами, в которой исследовали 3 схемы введения доз (таблица 1) в каждой из 2 групп по весу тела: пациенты ≥30 кг и ≤60 кг (группа A) и пациенты <30 кг и ≥10 кг (группа B), у 6 поддающихся оценке пациентов на дозу и группу по весу тела (всего приблизительно 36 пациентов), начиная с группы A и когорты дозы 1. Вторую последующую часть, где приблизительно 24 дополнительных пациентов (по 12 в каждой группе по весу тела, ≥30 кг (группа A) и <30 кг и ≥10 кг (группа B)) включают непосредственно в выбранную схему введения доз, чтобы получить в общей сложности 18 пациентов, подлежащих оценке, на каждую группу по весу тела при выбранной схеме введения доз. Третью часть, где приблизительно 28 дополнительных пациентов (ограничение 70% в каждой группе по весу: пациенты ≥30 кг [группа A] и пациенты <30 кг и ≥10 кг [группа B]) включат непосредственно в выбранную схему введения доз, чтобы получить в общей сложности приблизительно 100 пролеченных пациентов за все исследование. Эти пациенты будут совершать те же визиты в исследовательский центр в течение 12-недельной основной фазы лечения, что и пациенты, набранные для подбора дозы и второй части; однако пациенты в третьей части не будут совершать визиты для отбора образцов PK сарилумаба между исходным уровнем и неделей 2.
12-недельная основная фаза лечения продолжалась от визита 2 (исходный уровень - неделя 0) до времени введения исследуемого лекарственного препарата (IMP) во время визита 12. В течение 12-недельной основной фазы лечения пациентов в группе с более низким весом тела (группа B: <30 кг и ≥10 кг) случайным образом распределили на график 1 или график 2 отбора образцов PK сарилумаба, чтобы минимизировать количество забираемой крови и количество визитов при сохранении оценки первичной конечной точки.
- График 1. Исходный уровень, день 3, день 8, неделя 2, неделя 4, неделя 8 и неделя 12
- График 2. Исходный уровень, день 5, день 12, неделя 2, неделя 4, неделя 8 и неделя 12.
Оценки Данные представлены для 3 схем введения доз в обеих группах по весу тела и представлены от исходного уровня исследования до недели 12.
Первичной конечной точкой было PK воздействие, включая максимальную концентрацию в сыворотке крови, площадь под кривой концентрации в сыворотке крови в зависимости от времени, рассчитанную с использованием метода трапеций в течение интервала между дозами, и концентрацию в сыворотке, наблюдаемую перед введением лечения во время повторного введения дозы. Вторичными конечными точками были PD, включая концентрацию высокочувствительного С-реактивного белка (CRP), безопасность и клиническую эффективность. Эффективность определялась долей пациентов, достигших ответа ACR30 JIA/70/90, изменением по сравнению с исходным уровнем компонентов ответа ACR JIA и средним изменением индекса активности ювенильного артрита с подсчетом 27 суставов и оценкой CRP (JADAS-27-CRP), все на 12 неделе.
Кроме того, после завершения включения субъектов для подбора дозы и второй части, в третью часть включат приблизительно 28 дополнительных пациентов непосредственно в выбранную схему введения доз, чтобы получить в общей сложности приблизительно 100 пролеченных пациентов для всего исследования в соответствии с рекомендацией органа здравоохранения. Эти пациенты будут совершать те же визиты в исследовательский центр в течение 12-недельной основной фазы лечения, что и пациенты, набранные для подбора дозы и второй части; однако пациенты в третьей части не будут совершать визиты для отбора образцов PK сарилумаба между исходным уровнем и неделей 2.
Дополнительный период лечения в фазе дополнительного лечения позволит собрать данные о долгосрочной безопасности и клиническом ответе на сарилумаб у пациентов с pcJIA. Эта фаза дополнительного лечения длится до 144 недель для пациентов, включенных в часть подбора дозы и во вторую часть, и до 84 недель для пациентов, включенных в третью часть. Пациентам, включенным в часть подбора дозы, которые уже находятся в фазе дополнительного лечения на момент выбора схемы введения доз и которые не получали выбранную схему введения доз, будут корректировать схему введения доз в соответствии с выбранной дозой.
Чтобы свести к минимуму количество забираемой крови, пациентов группы B (<30 кг и ≥10 кг) при подборе дозы и во второй части будут рандомизировать на 1 из 2 различных графиков отбора образцов PK сарилумаба. Анализы PK объединят эти графики отбора образцов для описания PK профиля сарилумаба. Сбор образцов PK не планируется между исходным уровнем и неделей 2 для пациентов, включенных в третью часть. Общий объем забираемой крови для исследования не будет превышать 3% от общего объема крови в течение 4 недель и не будет превышать 1% от общего объема крови за один раз (39).Кроме того, запланированные частые оценки контролируют риск для участников исследования.
Наконец, период последующего наблюдения после лечения в течение 6 недель после последнего визита лечения для оценки TEAE, PK и PD сарилумаба является подходящим, принимая во внимание наблюдаемые профили PK в программе RA у взрослых.
Статистический анализ.
Популяционную модель PK объединенных данных разработали с использованием нелинейного моделирования со смешанными эффектами для описания PK профиля сарилумаба. Данные PD, безопасности и эффективности анализировали только с использованием описательной статистики. Конечные точки эффективности анализировали на основе данных, наблюдаемых во время лечения (отсутствие подстановки недостающих данных). Кроме того, результаты ACR30 JIA/70/90 также представляли с использованием метода подстановки данных субъектов, не ответивших на лечение (статус не ответивших на лечение автоматически присваивается пациентам с недостаточными данными или после прекращения лечения).
144-недельная/84-недельная фаза дополнительного лечения.
IMP на визите 12 рассматривали как первый IMP для фазы дополнительного лечения. Только пациентам, достигшим ответа ACR30 JIA на визите 12 (неделя 12), разрешили продолжить исследование в фазе дополнительного лечения. Пациенты продолжали придерживаться той же схемы введения доз сарилумаба, которую им назначили на 12-недельную основную фазу лечения исследования, до тех пор, пока не определяли выбранную схему введения доз. После выбора схемы введения доз пациентам, которые еще не придерживались этой схемы введения доз, корректировали схему введения доз в соответствии с выбранной схемой введения доз, и они придерживались нового графика визитов с более частым мониторингом (для PK, безопасности и эффективности) в течение первых 12 недель, по сравнению с пациентами, у которых схему введения доз не изменяли.
Пациентов, преждевременно прекративших прием исследуемого средства для лечения, оценивали с использованием процедуры EOT на визите 27. Этих пациентов просили вернуться для оценки в конце исследования (EOS) через 6 недель после визита EOT (EOT+6 недель). Для пациентов, которые прекратили исследуемый вид лечения в течение 12-недельной основной фазы лечения, проводили дополнительную оценку PK сарилумаба через 2 недели после визита EOT (EOT+2 недели), а во время визита EOT измеряли IL-6 и sIL6R. Этим пациентам предлагали совершать все визиты и оценки в соответствии с графиком протокола, за исключением введения сарилумаба, до визита 12.
Для пациентов, включенных в третью часть, запланирована 84-недельная фаза дополнительного лечения. IMP на визите 12 рассматривают как первый IMP для фазы дополнительного лечения. За пациентами, включенными в третью часть с выбранной схемой введения доз, будут совершать последующее наблюдение, как указано в таблице 32.
Протестированные схемы введения доз
Для каждой группы по весу тела определяли 3 схемы введения последовательных нарастающих доз, которые должны быть протестированы, на основе PK моделирования со следующим обоснованием.
- Когорта дозы 1. Доза, нацеленная на PK воздействие, аналогичное таковому при дозе сарилумаба 150 мг q2w, самая низкая эффективная схема введения доз для взрослых пациентов с RA.
- Когорта дозы 2. Доза, нацеленная на PK воздействие, аналогичное таковому при дозе сарилумаба 200 мг q2w, рекомендуемая схема введения доз для взрослых пациентов с RA.
- Когорта дозы 3. Доза, нацеленная на PK воздействие, аналогичное таковому при дозе сарилумаба 150 мг qw, которая приводила к самым высоким уровням воздействия в исследованиях хронического введения доз у взрослых пациентов с RA.
≥30 кг и ≤60 кг
(6 пациентов)
(6 пациентов)
(6 пациентов)
<30 кг и ≥10 кг
(6 пациентов)
(6 пациентов)
(6 пациентов)
Сокращения. Qw=один раз в неделю, q2w=один раз в две недели.
Дозу (мг), вводимую пациентам, рассчитывали на исходном уровне. Доза и соответствующий объем лекарственного препарата оставались неизменными в течение 12-недельной основной фазы лечения исследования, независимо от изменения веса тела пациента. В фазе дополнительного лечения вес тела пациента измеряли при каждом визите, и дозу корректировали с учетом увеличения веса тела только в том случае, если расчет показывал необходимость увеличения дозы. Доза была ограничена 150 мг для когорты дозы 1 и когорты дозы 3 и 200 мг для когорты дозы 2 соответственно. Объемы сарилумаба, подлежащие инъекции, для когорт доз 1-3 представлены в таблицах 2-5 ниже.
Таблицы 2 и 3 показывают информацию о дозах для пациента из группы с высоким весом тела (группа A). Пациенты с высоким весом тела представляют собой пациентов, которые весят ≥30 кг и ≤60 кг для части подбора дозы и ≥30 кг для дополнительных пациентов во второй части. Таблицы 4 и 5 показывают информацию о дозах для пациента из группы с низким весом тела (группа B). <30 кг и ≥10 кг.
Таблицы 3 и 4 показывают информацию о дозах для пациента из группы с низким весом тела (группа
B): <30 кг и ≥10 кг.
следующий объем
следующий объем
Объем, подлежащий введению при каждой инъекции в течение 12-недельной основной фазы лечения, рассчитывали на основе веса тела на исходном уровне (визит 2) и поддерживали в течение всей 12-недельной основной фазы лечения. Объем, подлежащий введению в течение фазы дополнительного лечения, регулировали в зависимости от веса тела, измеряемого при каждом визите, в случае увеличения веса тела, и если расчет показывал необходимость увеличения дозы. Если выбранная схема введения доз соответствует одной из 3 заранее определенных когорт доз, пациенты, впоследствии включенные в или перешедшие на выбранную схему введения доз, использовали соответствующую дозу, указанную в таблицах 2-5.
Продолжительность участия в исследовании для каждого пациента
Общая продолжительность исследования (на одного пациента) составляла до 166 недель для пациентов, включенных в часть подбора дозы и во вторую часть, и до 106 недель для пациентов, включенных в третью часть.
До 4 недель+3 дня скрининга (до 31 дня).
12-недельная основная фаза лечения.
До 144-недельной фазы дополнительного лечения для пациентов, включенных в часть подбора дозы и во вторую часть, и до 84 недель фазы дополнительного лечения для пациентов, включенных в третью часть.
6-недельное последующее наблюдение после лечения.
Для всех визитов приемлемы временные рамки ± 3 дня для когорт доз 1 и 2 и ± 1 день для когорты дозы 3 с использованием дня 1 в качестве эталона, за исключением следующего.
Для визита 3 (день 3), визита 4 (день 5) и визита 5 (день 8) не допускается окно визита, когда происходит отбор образцов PK. Для пациентов, включенных в третью часть, визиты для отбора образцов PK, т. е. визиты 3, 4, 5 и 6, не применимы в течение основной фазы лечения.
Визит 6 (день 12) ±1 день для всех 3 когорт доз.
Визит 7 (неделя 2) ±1 день для всех 3 когорт доз.
Визит 8 (неделя 4) ±2 дня для когорт доз 1 и 2 и ±1 день для когорты дозы 3.
Критерии включения
Критерии включения для данного исследования приведены ниже.
1. Пациенты мужского и женского пола в возрасте ≥2 и ≤17 лет (или применяются возрастные требования страны) на момент скринингового визита.
2. Диагноз RF-отрицательного или RF-положительного полиартикулярного подтипа JIA или подтипа oJIA в соответствии с критериями классификации JIA ILAR 2001 (2) с по меньшей мере 5 активными суставами в соответствии с определением ACR для "активного артрита" при скрининге (таблица 6).
3. Пациент с недостаточным ответом на текущее лечение, которого рассматривают в качестве кандидата на bDMARD согласно заключению исследователя.
4. Пациент, достигший установленного законом возраста согласия, или родитель(родители), или законный(законные) опекун(опекуны) подписывают с указанием даты письменное информированное согласие, одобренное комитетом по вопросам этики. Согласие несовершеннолетнего пациента должно быть получено на основе местных руководств, зрелости пациента и его интеллектуальных способностей для понимания информации, связанной с исследованием. В случаях, связанных с эмансипированными или зрелыми несовершеннолетними, обладающими достаточной способностью принимать решения, или когда это иным образом разрешено законом, подписанное информированное согласие будут получать непосредственно от родителя(родителей) или законного(законных) опекуна(опекунов).
Критерии исключения
Пациентов исключали из исследования по следующим причинам: вес тела <10 кг или >60 кг; подтип JIA, отличный от RF-положительного, RF-отрицательного или eoJIA; предшествующее лечение антагонистом IL-6 или IL-6R; лечение любым другим биологическим средством в течение 5 периодов полужизни первой дозы сарилумаба; использование парентеральных или внутрисуставных инъекций глюкокортикоидов в течение 4 недель до исходного уровня исследования; активная или перенесенная оппортунистическая инфекция, включая туберкулез; введение живой аттенуированной вакцины в течение 4 недель до исходного уровня; или любое состояние, которое может отрицательно повлиять на участие пациента в исследовании. Пациентов, получавших нестероидные противовоспалительные лекарственные средства (NSAID), пероральные глюкокортикоиды или традиционные синтетические модифицирующие заболевание противоревматические лекарственные средства (csDMARD), исключали, если доза NSAID или пероральных глюкокортикоидов была нестабильной в течение 2 недель до исходного уровня или если дозировка пероральных глюкокортикоидов превышала эквивалентную дозу преднизона, составляющую 0,5 мг/кг/день (или 30 мг/день), если доза csDMARD была нестабильной в течение 6 недель до исходного уровня или если дозировка csDMARD превышала рекомендуемую дозировку, указанную на местной этикетке. Письменное информированное согласие требовали и получали от всех пациентов/опекунов в соответствии с местными нормативными руководствами.
признаки,
определенные
классификацией
ILAR
исключения (сноска a)
(% от общего JIA)*
Средний диапазон
-Тендосиновит, увеит, васкулит
Два пика
-ранний: 1-3 года
-поздний: 6-12 лет
Деструктивное заболевание суставов
5 или
более суставов
в первые 6
месяцев
заболевания.
-Тесты на RF
положительные дважды
с интервалом по меньшей мере
2 месяца
-Теносиновит,
ревматоидные
узелки,
васкулит,
синдром
Шегрена
10-18 лет
- Встречается в позднем детстве
- Тяжелое деструктивное заболевание суставов
°Персистирующий
°Распространенный
4 суставов
в течение
заболевания
-Поражает более
4 суставов
в течение
заболевания
1-5 лет
- Хронический увеит, особенно ANA+
- Несоответствие длины ног
- Деструктивное
заболевание суставов
- Терапия как при
pcJIA
а. Псориаз или псориаз в анамнезе у пациентов или родственников первой степени родства;
b. артрит у HLA B27-положительных мужчин, начинающийся в возрасте старше 6 лет;
c. анкилозирующий спондилит, энтезит-ассоциированный артрит, сакроилеит с сопутствующим воспалительным заболеванием кишечника, синдром Рейтера, острый передний увеит или 1 из этих заболеваний в анамнезе у родственников первой степени родства;
d. наличие IgM ревматоидного фактора в по меньшей мере 2 случаях с интервалом по меньшей мере 3 месяца;
е. наличие у пациентов системного JIA.
Исследуемые виды лечения
Исследуемый лекарственный препарат (IMP)
Сарилумаб, mAb к IL-6R (моноклональное антитело к альфа-субъединице рецептора интерлейкина 6).
Состав
Лекарственный препарат сарилумаб предоставляли в концентрации 175 мг/мл в водном буферном носителе, pH 6,0. Его поставляли во флаконе объемом 5 мл, заполненном 2,7 мл сарилумаба с экстрагируемым объемом 2,0 мл.
Путь(пути) введения
Сарилумаб вводили подкожно в живот или бедро при самостоятельной инъекции или также в предплечье (в латеральную поверхность) профессиональным или непрофессиональным лицом, осуществляющим уход. Предпочтительно, чтобы места SC инъекции чередовались между 4 квадрантами живота (кроме области пупка или талии) или бедра (спереди и сбоку).
Пациентам, которые придерживались схемы введения доз 1 или 2 (q2w), инъекции выполняло профессиональное лицо, осуществляющее уход, в течение 12-недельной основной фазы лечения исследования. В отношении пациентов, которые придерживались схемы введения доз 3 (еженедельные инъекции), приняли меры, обеспечивающие введение IMP квалифицированным персоналом центра или домашней медсестрой для доз, введение которых не запланировано в исследовательском центре. На фазе дополнительного лечения исследования, если пациенты или их родитель(родители), законный(законные) опекун(опекуны) или лицо(лица), осуществляющее(осуществляющие) уход, желали и имели возможность выполнять инъекции, домашние инъекции разрешали. В этих случаях требовалось и проводилось обучение для подготовки и введения IMP, начиная с визита 10 (неделя 8) на основной фазе лечения. Это обучение задокументировали в файле исследования пациентов. Пациентам, или родителю(родителям), или законному(законным) опекуну(опекунам) разрешали вводить инъекции под наблюдением/контролем исследователя(исследователей) или представителя(представителей) при визите 10 (неделя 8), визите 11 (неделя 10) и визите 12 (неделя 12) до разрешения домашних инъекций на фазе дополнительного лечения.
В дни, когда пациент совершает визит исследования, IMP вводили после клинических процедур и сбора крови. Предоставляли дневники для записи информации, относящейся к этим инъекциям; эти дневники хранили в качестве исходных данных в файлах исследования пациентов. Если лицо, осуществляющее уход, не могло или не желало вводить IMP, принимали меры, обеспечивающие введение IMP квалифицированным персоналом центра для доз, введение которых не запланировано в исследовательском центре.
Схема введения доз
Сарилумаб вводили q2w или qw. Однако разрешалось окно введения сарилумаба, которое составляло ±3 дня для когорт доз 1 и 2 и ±1 день для когорты дозы 3 в соответствии с протоколом с учетом исключительных обстоятельств, например ожидаемых результатов лабораторных исследований, продолжающихся нежелательных явлений (AE), сложностей планирования пациентов, кроме визита 5 (день 8). Для визита 5 (день 8) не было окна введения. Окно введения составляло только ± 1 день для когорт доз 1, 2 и 3 пациентов при визите 7 (неделя 2) и ±1 день для когорты дозы 3 или ±2 для когорт доз 1 и 2 при визите 8 (неделя 4).
Обратите внимание, что передозировку (случайная или преднамеренная) IMP определяли как по меньшей мере двойную дозу в течение интервала менее 11 дней для введения q2w и менее 6 дней для еженедельного введения.
Следующую дозу сарилумаба вводили q2w или qw.
Когорта дозы 1.
- Группа A (≥30 кг и ≤60 кг): 2 мг/кг q2w
- Группа B (<30 кг и ≥10 кг): 2,5 мг/кг q2w.
Когорта дозы 2.
- Группа A (≥30 кг и ≤60 кг): 3 мг/кг q2w
- Группа B (<30 кг и ≥10 кг): 4 мг/кг q2w.
Когорта дозы 3.
- Группа A (≥30 кг и ≤60 кг): 2 мг/кг qw
- Группа B (<30 кг и ≥10 кг): 2,5 мг/кг qw.
Изменение дозы
Дозу (мг), вводимую пациентам, рассчитывали на исходном уровне. Доза и соответствующий объем лекарственного препарата оставались неизменными в течение 12-недельной основной фазы лечения исследования, независимо от изменения веса тела пациента. В фазе дополнительного лечения вес тела пациента измеряли при каждом визите, и дозу корректировали с учетом увеличения веса тела только в том случае, если расчет показывал необходимость увеличения дозы. Доза была ограничена 150 мг и 200 мг для когорт доз 1 и 3 и когорты дозы 2 соответственно. Объемы инъекции в зависимости от веса тела пациента подробно описаны в таблицах 2-5.
Способ распределения пациентов в группу лечения
Список рандомизированных номеров лечебных наборов формировали централизованно. IMP упаковывали в соответствии с этим списком. Пациентам назначали лечебный набор с помощью интерактивной системы, обеспечивающей доступ путем голосового ответа (IVRS) или интерактивной системы, обеспечивающей доступ через интернет (IWRS).
Пациентов, которые соответствуют критериям включения, будут включать и им будут выдавать лечебные наборы в 2 группах по весу тела (группа A: ≥30 кг и ≤60 кг для пациентов, включенных во время части подбора дозы, и ≥30 кг для пациентов, включенных во время второй и третьей частей; группа B: <30 кг и ≥10 кг для всех 3 частей).
- Для части подбора дозы и второй части запланированное соотношение включения для 2 групп по весу тела составляет 1:1.
Для третьей части нет заранее определенного коэффициента включения для 2 групп по весу тела, но каждая группа по весу тела будет ограничена 70% (т. е. 20 пациентами). В результате наиболее несбалансированное включение в группу по весу тела будет 20 против 8 пациентов в третьей части.
Упаковка и маркировка
IMP предоставляли в наборах для лечения пациентов, содержащих маркированные флаконы.
Условия хранения
Исследуемый препарат хранили в холодильнике при температуре от 2oC до 8oC в исследовательском центре или дома.
ОЦЕНКА ФАРМАКОКИНЕТИКИ, ЭФФЕКТИВНОСТИ И БЕЗОПАСНОСТИ
Планировалось, что пациенты пройдут оценку 12-недельной основной фазы лечения в соответствии с приведенным ниже графиком (таблица 7).
Фармакокинетические измерения и сроки
График отбора образцов крови в 12-недельной основной фазе лечения можно найти в таблице 7. Если у пациента наблюдали SAE, для определения концентрации сарилумаба отбирали образцы крови в момент начала и завершения события, если это возможно, или в течение короткого времени после начала и завершения. Для пациентов, которые преждевременно прекратили исследуемый вид лечения во время основной фазы лечения (во время или до визита 12), проводили дополнительную оценку PK сарилумаба через 2 недели после запланированной оценки EOT (через 3 недели после последней инъекции IMP для пациентов когорты доз 3 или через 4 недели после последней инъекции IMP [для пациентов когорт доз 1 и 2]). Этих пациентов просили вернуться для оценки в конце исследования (EOS) через 6 недель после визита EOT. Должны регистрировать дату сбора образца. У пациентов с преждевременным прекращением вида лечения образцы крови должны собирать при визите EOS, если это возможно. Образцы сыворотки крови анализировали в отношении концентрации сарилумаба с использованием утвержденного метода твердофазного иммуноферментного анализа (ELISA).
(12 недель)
PK
EOT+2 недели (для пациентов, которые прекращают исследуемый вид лечения во время основной фазы лечения или не собираются переходить в фазу дополнительного лечения)
D1 (+3)
(максимум
31 день)
(±1)
(±1)
(±1 или 3)
(±1 или 3)
согласия пациентаd
анамнез
прием лекарственных препаратов/вакцинация в анамнезе
дневник соблюдения требований леченияe
(12 недель)
PK
EOT+2 недели (для пациентов, которые прекращают исследуемый вид лечения во время основной фазы лечения или не собираются переходить в фазу дополнительного лечения)
D1 (+3)
(максимум
31 день)
(±1)
(±1)
(±1 или 3)
(±1 или 3)
ВИЧg
тест QuantiFERON-TB для пациентов >5 летh
(12 недель)
PK
EOT+2 недели (для пациентов, которые прекращают исследуемый вид лечения во время основной фазы лечения или не собираются переходить в фазу дополнительного лечения)
D1 (+3)
(максимум
31 день)
(±1)
(±1)
(±1 или 3)
(±1 или 3)
(12 недель)
PK
EOT+2 недели (для пациентов, которые прекращают исследуемый вид лечения во время основной фазы лечения или не собираются переходить в фазу дополнительного лечения)
D1 (+3)
(максимум
31 день)
(±1)
(±1)
(±1 или 3)
(±1 или 3)
Группа A (≥30 кг и ≤60 кг)
Группа B (<30 кг и ≥10 кг)
График 1
(12 недель)
PK
EOT+2 недели (для пациентов, которые прекращают исследуемый вид лечения во время основной фазы лечения или не собираются переходить в фазу дополнительного лечения)
D1 (+3)
(максимум
31 день)
(±1)
(±1)
(±1 или 3)
(±1 или 3)
а. Для пациентов, которые прекращают лечение в течение 12-недельной основной фазы лечения (во время или до визита 12), будут проводить дополнительную оценку PK сарилумаба через 2 недели после визита EOT (EOT+2 недели). Для пациентов, включенных в третью часть, визит 88 не применим.
b. Отбор образцов для определения PK сарилумаба между визитом 2 и визитом 7 варьирует между пациентами: визит 3 (день 3), визит 4 (день 5), визит 5 (день 8), визит 6 (день 12) для группы A (≥30 кг); во время следующих визитов и дней: визит 3 (день 3), визит 5 (день 8), для графика 1 группы B (<30 кг); во время следующих визитов и дней визитов: визит 4 (день 5), визит 6 (день 12) для графика 2 группы B (<30 кг). Отбор образцов для определения PK сарилумаба можно проводить дома. Будут предоставлять дневник PK, чтобы фиксировать дату и время сбора образца. Для визита 3 (день 3), визита 4 (день 5) и визита 5 (день 8) окно для сбора образцов не допускается. Для визита 6 (день 12) допускается окно ±1 день. Для пациентов, включенных в третью часть, визиты 3, 4, 5 и 6 не применимы. Однако перед вторым введением сарилумаба необходимо провести дополнительное гематологическое исследование (подробности см. в сноске "o").
c. По усмотрению исследователей лабораторные исследования, упомянутые в критерии исключения E 24, можно повторять посредством повторного тестирования в центральной лаборатории между скрининговым визитом и первым введением IMP, чтобы гарантировать пригодность пациента с учетом критерия исключения E 24. Специальная форма согласия, одобренная на местном уровне, будет подписана пациентами, которым требуется генетическое тестирование в отношении синдрома Жильбера (согласие/согласие пациента должно быть получено до проведения данного исследования, и должны соблюдаться местные нормативные требования).
d. Перед проведением всех скрининговых оценок пациент (если он/она достигли установленного законом возраста согласия в соответствии с местными нормативными требованиями), родитель(родители) или законный(законные) опекун(опекуны) должны подписать с указанием даты форму письменного информированного согласия, одобренную EC. Пациент, родитель(родители) и законный(законные) опекун(опекуны) получат информацию о цели(целях) исследования и процедурах от исследователя. Отдельные формы письменного согласия должны быть получены от родителя(родителей) или законного опекуна(опекунов), которые разрешают его/ее ребенку участвовать в дополнительном сборе образцов слюны для фармакогеномного исследования и дают разрешение спонсору оставить их оставшиеся/неиспользованные образцы крови для будущих исследований
e. . Отдельная (одобренная на местном уровне) форма информированного согласия заполняется всеми пациентами, которым требуется генетическое тестирование в отношении болезни Жильбера в соответствии с местными нормативными требованиями. Подписанные формы согласия пациентов должны быть получены от пациентов в соответствии с местными нормативными требованиями и его/ее зрелостью в понимании информации об исследовании.
f. Домашний дневник для введения IMP, который необходимо заполнить для IMP, вводимого дома.
g. Полное физикальное обследование будут осуществлять во время визита 1 (от дня -28 до дня -1, вплоть до 31 дня), визита 2 (день 1, неделя 0), визита 8 (неделя 4), визита 9 (неделя 6) и визита 12 (неделя 12), где обследование включает обследование кожи, носовых полостей, глаз, ушей, дыхательной, сердечно-сосудистой, желудочно-кишечной, нервной, лимфатической и опорно-двигательной систем.
h. Необязательно. На основании заключения исследователя во время скрининга могут определять титр вируса Эпштейна-Барр (EBV), включая IgG и IgM; на основании семейного и медицинского анамнеза пациента, а также заключения исследователя, во время скрининга могут определять поверхностный антиген вируса гепатита B (HBs-Ag), антитело к поверхностному антигену вируса гепатита B (HBs-Ab), общие антитела к коровому антигену вируса гепатита B (HBc-Ab) и антитело к вирусу гепатиту C (HCV-Ab). Серологическое исследование в отношении ВИЧ будут проводить только на основании заключения исследователя для пациентов с подозрением на ВИЧ. Только для Аргентины и Германии: серологические исследования в отношении вирусов гепатита B и C и HIV должны проводить во время скринингового визита для всех пациентов, чтобы проверить соответствующий критерий исключения E 14.
i. Кожную пробу очищенного белкового продукта (PPD) должны проводить у пациентов в возрасте ≤5 лет до визита 2 исходного уровня (день 1, неделя 0). Пациентов следует обследовать в течение 48-72 часов после проведения кожной пробы PPD. Пациентам, которых не обследовали в течение 72 часов, кожную пробу следует повторить. При определении результатов кожных проб следует учитывать оценку уровня риска (контакт с TB и/или недавняя иммиграция из страны с высокой распространенностью TB) и анамнез вакцинаций. См. все подробности в критерии исключения E 13. Анализ высвобождения гамма-интерферона (IFN-r), тест QuantiFERON-TB, будут проводить для пациентов в возрасте >5 лет. (1) После первоначального скрининга в отношении TB, если результат PPD или QuantiFERON отрицательный, но клиническое подозрение на TB выше среднего, пациента должны повторно обследовать на наличие TB в любое время в ходе исследования на основе оценки исследователя. Тест QuantiFERON-TB будут рассматривать в более молодой группе пациентов (≤5 лет) на основании местного наличия PPD, местных нормативных требований проведения скрининга в отношении TB и заключения исследователя.
j. Рентгенографию грудной клетки могут проводить пациентам только в том случае, если это сочтут необходимым на основании заключения исследователя или в соответствии с местными руководствами по скринингу в отношении TB до начала биологической терапии для пациентов с JIA, у которых рентгенографию грудной клетки не проводили в течение 3 месяцев до визита 2 исходного уровня (день 1, неделя 0).
k. Исследуемый лекарственный препарат следует вводить один раз в две недели пациентам в когортах со схемами введения доз 1 и 2 и один раз в неделю для пациентов в когорте со схемой введения доз 3. Будут принимать меры для того, чтобы домашние медсестры вводили IMP для пациентов в когорте со схемой введения доз 3 для промежуточных визитов (введение на дому, если возможно). За пациентами следует наблюдать в течение по меньшей мере 30 минут после введения IMP в отношении любых признаков или симптомов реакции гиперчувствительности. Только для Германии: после первого введения IMP за пациентом необходимо наблюдать в течение по меньшей мере 1 часа для оценки местной переносимости. В части подбора дозы и второй части пациенты, которые не достигли ответа JIA Американской коллегии ревматологов (ACR) 30 к концу 12-недельного основного периода лечения, будут отстранены от исследуемого вида лечения для получения стандартного лечения в соответствии с клиническим заключением исследователя. Ответ ювенильного идиопатического артрита ACR, включая ответ ACR30 JIA, будет предоставлен спонсором исследователю для оценки этого критерия только после визита 12. Для пациентов, завершивших основную фазу лечения на визите 12 (неделя 12), но не продолжающих исследование в фазе дополнительного лечения в расширенной популяции по причинам, отличным от отсутствия ответа ACR30 JIA, инъекцию IMP на визите 12 проводить не будут.
l. Исследуемый лекарственный препарат будут выдавать пациентам, которые находятся в когорте со схемой введения доз 3, в течение основной фазы лечения.
m. Рост будут измерять с помощью ростомера в исследовательских центрах во время исследования.
n. Базовый набор ювенильного идиопатического артрита ACR включает общую оценку тяжести заболевания врачом, общую оценку самочувствия пациента или родителя(родителей)/опекуна(опекунов), количество суставов с активным артритом (определяется как отек, не вызванный деформацией ИЛИ ограничение движений либо с болью, либо с чувствительностью, либо с обоими признаками), количество суставов с ограничением движений, опросник оценки здоровья детей (CHAQ), hs-CRP (2,3,4). Во время скрининга будут оценивать только количество суставов с активным артритом и количество суставов с ограничением движений.
о. Принцип подсчета индекса активности заболевания, представляющего собой ювенильный артрит объяснялся в данном документе.
p. Гематологическое исследование (кровь следует забирать ДО введения лекарственного средства): гемоглобин, гематокрит, количество эритроцитов (RBC) и морфология эритроцитов (если количество эритроцитов аномальное), лейкоцитарная формула (WBC) (нейтрофилы, лимфоциты, моноциты, эозинофилы, базофилы), количество тромбоцитов, абсолютное количество нейтрофилов (ANC). Дополнительное гематологическое исследование должны проводить на визите 6 (день 12±1 день) для когорт со схемами введения доз 1 и 2, чтобы получить результаты до введения второй дозы сарилумаба (визит 7: неделя 2, день 15) или до или во время визита 5 (день 8) для когорты со схемой введения доз 3 (не перед визитом 4 [день 5]), чтобы получить результаты до введения второй дозы сарилумаба. Для пациентов, включенных в часть подбора дозы и во вторую часть, дополнительное гематологическое исследование могут проводить в центральной лаборатории или в местной лаборатории, чтобы подтвердить количество нейтрофилов и количество тромбоцитов перед введением второй дозы сарилумаба. Для пациентов, включенных в третью часть без визита 6 в исследовательский центр, дополнительное гематологическое исследование будут проводить в местной лаборатории перед введением второй дозы сарилумаба, но не ранее, чем в день 12. Если используется местная лаборатория, образец центральной лаборатории для гематологического исследования все равно должен быть собран перед введением IMP на визите 7 (день 15, неделя 2), как запланировано для когорт со схемами введения доз 1 и 2, и на визите 5 (день 8) для когорты со схемой введения доз 3. Для всех пациентов дополнительная гематологическая лабораторная оценка во время визита 2 (день 1) должна быть рассмотрена перед введением второй дозы сарилумаба во время визита 5 (день 8) для пациентов в когорте со схемой введения доз 3 или на визите 7 (неделя 2, день 15) для когорт со схемами введения доз 1 и 2.
q. Биохимическое исследование (кровь следует забирать ДО введения лекарственного средства). Полное биохимическое исследование будут проводить во время скринингового визита, визита 2 исходного уровня (день 1, неделя 0) и визита 12 (неделя 12) или только на EOT: натрий, калий, хлор, бикарбонат, азот мочевины крови, креатинин и клиренс креатинина, скорость клубочковой фильтрации (с использованием модифицированной формулы Шварца), кальций, фосфат, общий белок, альбумин, аланинаминотрансфераза (ALT), аспартатаминотрансфераза (AST), щелочная фосфатаза (ALP), общий билирубин, конъюгированный билирубин и неконъюгированный билирубин. На всех остальных визитах будут тестировать только ALT, AST, ALP, общий билирубин, конъюгированный билирубин, неконъюгированный билирубин и альбумин.
r. Липидограмма натощак (кровь следует забирать ДО введения лекарственного средства). Триглицериды (TG), общий холестерин, холестерин липопротеинов высокой плотности (HDL), холестерин липопротеинов низкой плотности (LDL). Требуется, чтобы пациенты не принимали пищу по меньшей мере в течение 8 часов до исследования.
s. Необязательно. Уровни HbA1c будут измерять только на основании медицинского анамнеза пациента и заключения исследователя.
t. Анализ мочи с помощью индикаторной полоски: удельный вес, pH, глюкоза, кровь, белок, нитраты, лейкоцитарная эстераза, билирубин. Если какой-либо параметр по анализу с помощью индикаторной полоски отклоняется от нормы, образец мочи следует отправить в центральную лабораторию для анализа. В случае положительного результата в отношении белков центральная лаборатория проводит микроскопический анализ.
u. Для женщин, у которых начались менструации, во время скринингового визита в обязательном порядке проводят исследование сыворотки на наличие беременности.
v. Для женщин, у которых начались менструации, местное исследование мочи на наличие беременности следует проводить во время визита 2 (неделя 0, день 1), визита 8 (неделя 4), визита 10 (неделя 8) и визита 12 (неделя 12). Исследование мочи на наличие беременности можно проводить местно. Наличие беременности следует проверять с помощью исследования мочи на наличие беременности до воздействия IMP и EOT.
w. Образцы крови будут собирать ДО введения IMP в дни введения доз в течение периода лечения. Если у пациента наблюдают SAE, для определения концентрации сарилумаба и оценки антител к лекарственному средству (ADA) следует собирать образцы крови в момент начала и завершения события, если это возможно, или в течение короткого времени после начала и завершения. В части подбора дозы и во второй части, если пациент преждевременно прекращает исследуемый вид лечения в течение 12-недельной основной фазы лечения, требуется дополнительный визит PK через 2 недели после визита EOT для отбора образцов крови (визит 88).
x. Для пациентов, которые преждевременно прекращают исследуемый вид лечения во время основной фазы лечения, при оценке EOT будут измерять IL-6 и общий sIL-6R.
y. Родитель(Родители) или законный(законные) опекун(опекуны) должны подписать отдельную форму информированного согласия перед необязательным забором образца слюны для фармакогеномного исследования. Образцы предпочтительнее собирать во время визита 2 исходного уровня (день 1, неделя 0), но их можно собирать во время любого визита. Пациент также может подписать форму информированного согласия в зависимости от его/ее возраста, местных нормативных требований и степени зрелости в отношении понимания информации об исследовании. Пациент по-прежнему пригоден для участия в исследовании, если он/она или его/ее родитель(родители) или его/ее законный(законные) опекун(опекуны) не желают, чтобы он/она участвовали в заборе образцов слюны.
Фармакокинетические переменные
PK переменные сарилумаба включали максимальные наблюдаемые концентрации в сыворотке (Cmax) и площадь под кривой зависимости концентрации в сыворотке от времени (AUC), рассчитанную с использованием метода трапеций в течение интервала между введениями доз (AUC0-τ), после первой дозы, концентрацию, наблюдаемую перед введением средства лечения при повторном введении доз (Ctrough) от исходного уровня до недели 12.
Популяционную модель PK (PopPK) объединенных данных разработали с использованием нелинейного моделирования со смешанными эффектами для описания PK профиля сарилумаба. Следующие PK параметры рассчитывали с использованием модели PopPK для функционального сарилумаба в сыворотке крови (анализ PopPK должен быть представлен в отдельном отчете). РК параметры включали без ограничения параметры, перечисленные в таблице 8.
Таблица 8. Перечень фармакокинетических параметров и определений
Фармакокинетический параметр
Определение_____________________________________________
Cmax Максимальная наблюдаемая концентрация в сыворотке крови
Ctrough Концентрация, наблюдаемая перед введением средства лечения при повторном введении доз
tmax Время до достижения Cmax
AUC0-τ Площадь под кривой концентрации в сыворотке в зависимости от времени, рассчитанная с помощью метода трапеций в течение интервала между введениями доз (τ)
Оценки эффективности
Ответ ACR JIA
В этом исследовании использовали уровень ответа ACR JIA для оценки признаков и симптомов (см. таблицу 7). ACR JIA 30/50/70/90/100 (ответ ACR30 JIA/50/70/90/100 определяли как пациента, у которого 3 из 6 переменных базового набора улучшились по меньшей мере на 30%/50%/70%/90%/100% по сравнению с исходным уровнем, при этом не более 1 из оставшихся переменных ухудшились более чем на 30%.
Базовый набор для ACR JIA включал 6 переменных.
1. Общая оценка активности заболевания врачом
2. Оценка общего самочувствия пациентом/родителем
3. Функциональные способности, определяемые опросником оценки здоровья детей (CHAQ)
4. Количество суставов с активным артритом (от 0 до 71 сустава)
5. Количество суставов с ограничением движений (от 0 до 67 суставов)
6. Высокочувствительный C-реактивный белок.
В течение 12-недельной основной фазы лечения базовый набор заболевания ACR JIA будут оценивать при каждом визите в исследовательский центр, за исключением визита 3 (день 3), визита 4 (день 5), визита 5 (день 8) и визита 6 (день 12). В течение фазы дополнительного лечения (для пациентов, которые продолжают принимать текущую дозу (придерживаются выбранной схемы введения доз), базовый набор ACR JIA будут периодически оценивать на неделях 24, 48, 72, 96 (EOT для третьей части), 120 (не применимо для третьей части), 144 (неприменимо для третьей части) и EOT (неделя 156 для части подбора дозы и второй части/неделя 96 для третьей части). Для пациентов, которые меняют выбранную схему введения доз во время фазы дополнительного лечения.
Общая оценка активности заболевания врачом
Исследователя просили оценивать активность заболевания пациента по горизонтальной визуально-аналоговой шкале (VAS) длиной 100 миллиметров (мм) с ориентирами, где 0 считалось наилучшей активностью заболевания, а 100 считалось наихудшей активностью заболевания.
Оценка общего самочувствия пациентом/родителем
Оценку общего самочувствия пациентом/родителем измеряли с помощью горизонтальной VAS длиной 100 мм. Форму просили заполнять пациента или одного и того же родителя или опекуна, чтобы обеспечить постоянство.
Опросник оценки здоровья детей (CHAQ)
CHAQ представлял собой опрос или инструмент для самостоятельного заполнения для детей ≥8 лет и для заполнения родителем/доверенным лицом для детей младше 8 лет. CHAQ представлял собой общий показатель состояния здоровья детей в возрасте от 1 до 19 лет. Всего оценка состояла из 43 пунктов, и ее выполнение занимало приблизительно 10 минут. Опросник CHAQ заполняли перед введением доз IMP и в последующие моменты времени (в соответствии с руководством, предоставленным отделом исследований экономики здравоохранения и исходов). Период оценки составлял 1 неделю (Singh, G. et al. 1994 Arthritis Rheum 1994 Dec;37(12):1761-9).
Медианные баллы по шкале CHAQ, соответствующие инвалидизации легкой степени, от легкой до умеренной степени и умеренной степени, о которых сообщали при создании CHAQ, составляли 0,13, 0,63 и 1,75 соответственно (Dempster, H. et al., 2001 Arthritis Rheum. 44(8):1768-74).
Чтобы устранить несоответствия, которые могли быть внесены в результате роста и
развития, родителей просили отмечать только те трудности, которые возникли из-за болезни (например, если ребенок не мог выполнять какое-либо действие, потому что его/ее возраст был слишком маленьким, ответ отмечали как "не применимо"). Варианты ответа, оценивающие трудности, основывались на 5-балльной шкале Лайкерта
(0=без каких-либо трудностей, 1=с некоторыми трудностями, 2=с большими трудностями, 3=невозможно выполнить и 4=неприменимо).
CHAQ был разделен на 3 части.
1. Индекс инвалидизации. 41 пункт для оценки способности функционировать в повседневной жизни, далее разделенный на 8 подшкал/сфер: одевание и уход за внешностью; подъем; прием пищи; ходьба; гигиена; дотягивание; хват и активная деятельность. Для каждой сферы оценивали степень сложности выполнения повседневных функций, необходимость использования специальных вспомогательных средств или устройств и необходимость помощи со стороны другого человека.
Для расчета CHAQ-DI сначала рассчитывали балл по каждой сфере.
- Вопрос с наивысшей оценкой определял балл для этой функциональной области
- Если использовались вспомогательные средства или устройства или требовалась помощь для выполнения задач в определенной области, для соответствующей функциональной области регистрировали минимальный балл 2
- 8 подшкал/сфер усреднили для расчета среднего балла, который представлял собой индекс инвалидизации (с диапазоном от 0 до 3).
Более низкие значения индекса инвалидизации указывают на лучшее состояние здоровья/лучшее качество жизни, связанное со здоровьем/меньшее количество признаков и симптомов, в то время как более высокие значения индекса инвалидизации указывают на худшее состояние здоровья/худшее HRQL/большее количество признаков и симптомов (Singh, G. et al. 1994 Arthritis Rheum. 1994 Dec;37(12):1761-90; Klepper S. 2003 Arthritis Rheum. 49(3):435-43). Минимальным клинически значимым улучшением индекса инвалидизации по CHAQ являлось снижение балла на 0,13. Минимальным клинически значимым ухудшением индекса инвалидизации по CHAQ являлся изменение медианного значения на 0,75 (Dempster, H. et al., 2001 Arthritis Rheum. 44(8):1768-74).
2. Индекс дискомфорта. Определяется степенью боли за последнюю неделю, оцененной по VAS (с ориентирами "0 - нет боли" и "100 - очень сильная боль"). Для расчета индекса дискомфорта по CHAQ расстояние от левого края VAS в пункте 67 до отметки респондента измеряли и умножали на 0,2. Диапазон составлял от 0 до 3.
3. Состояние здоровья. Показатель общей оценки заболевания пациентом или родителем.
Для расчета балла состояния здоровья по CHAQ расстояние от левого края VAS в пункте 69 до отметки респондента измеряли и умножали на 0,2. Диапазон составлял от 0 до 3.
Количество суставов с активным артритом и количество суставов с ограниченными движениями
"Активный сустав" определяли как сустав с отеком внутри сустава, не вызванным деформацией ИЛИ с ограничением движений с болью или чувствительностью. Семьдесят один сустав оценивали в отношении активного заболевания путем подсчета количества активных суставов (Bazso, A. et al., 2009 J Rheumatol. 36(1):183-90; Beukelman, T. et al., 2011 Arthritis Care Res (Hoboken). 63(4):465-82).
71 сустав включал шейный отдел позвоночника (считается как 1 сустав), височно-нижнечелюстной (2 сустава, справа и слева), грудино-ключичный (2 сустава), акромиально-ключичный (2 сустава), плечевой (2 сустава), локтевой (2 сустава), запястный (2 сустава), пястно-фаланговый (всего 10 суставов, по 5 с каждой стороны), проксимальный межфаланговый (всего 10 суставов, по 5 с каждой стороны), дистальный межфаланговый (всего 8 суставов, 4 с каждой стороны), тазобедренный (2 сустава), коленный (2 сустава), голеностопный (2 сустава), подтаранный (2 сустава), предплюсне-плюсневый (2 сустава), плюснефаланговый (10 суставов, 5 с каждой стороны) суставы и межфаланговый сустав стопы (10 суставов, по 5 с каждой стороны).
67 суставов, исследованных в отношении ограничения движений, представляли собой те же суставы, что и те, которые исследовали в отношении активного заболевания, за исключением грудино-ключичного (n=2) и акромиально-ключичного (n=2). Формальный подсчет суставов проводил обученный эксперт по оценке. Чувствительность суставов определяли как боль, вызванную давлением в суставах большим и указательным пальцами эксперта по оценке.
Высокочувствительный C-реактивный белок (hs-CRP)
При каждом визите оценивали hs-CRP. См. таблицу 7. Уровни высокочувствительного С-реактивного белка прямо коррелировали с активностью IL-6R. Ожидалось, что активные схемы введения доз будут оказывать эффект резкого снижения уровня С-реактивного белка (CRP).
Индекс активности заболевания, представляющего собой ювенильный артрит
В этом исследовании использовали индекс JADAS для оценки активности заболевания (см. таблицу 7). JADAS включал 4 показателя.
1. Общая оценка активности заболевания врачом, измеренная с помощью VAS длиной 10 сантиметров (см), где 0=отсутствие активности и 10=максимальная активность.
2. Общая оценка самочувствия родителем/пациентом, измеренная с помощью VAS длиной 10 см, где 0=очень хорошо, а 10=очень плохо.
3. Количество суставов с активным заболеванием.
4. Индекс воспаления. Уровень hs-CRP или ESR.
Скорость оседания эритроцитов нормализована по шкале от 0 до 10 по следующей формуле: (ESR[мм/час]-20)/10, где перед расчетом значение ESR <20 мм/час преобразовано в 0, а значения ESR >120 мм/час преобразовано в 120.
Или hs-CRP нормализован по шкале от 0 до 10 по следующей формуле: (CRP[мг/л]-10)/10, где перед расчетом значения CRP <10 мг/л преобразованы в 0, а значения CRP >110 мг/л преобразованы в 110.
JADAS рассчитывали как простую линейную сумму баллов по его 4 компонентам. См. Consolaro, A. et al. 2009 Arthritis Rheum. 61(5):658-66. JADAS-27 включал следующие суставы: шейный отдел позвоночника, локтевые, запястные суставы, пястно-фаланговые суставы (от первого до третьего), проксимальные межфаланговые суставы, тазобедренные, коленные и голеностопные суставы. JADAS оказался действенным инструментом для оценки активности заболевания при JIA и был потенциально применим в стандартном клиническом лечении, обследовании путем наблюдения и клинических испытаниях (там же).
Фармакодинамические измерения и сроки
Фармакодинамические эффекты сарилумаба оценивали путем измерения следующих биомаркеров: hs-CRP, IL-6 и общего sIL-6Rα.
Концентрации высокочувствительного CRP измеряли с помощью анализа методом нефелометрии (HS) с LLOQ 0,20 мг/л в Covance CLS.
Концентрации IL-6 в сыворотке крови определяли с помощью утвержденного количественного иммуноферментного сэндвич-анализа с LLOQ 3,12 пг/мл в Covance CLS
Концентрации общего sIL-6Rα в сыворотке измеряли с помощью утвержденного метода ELISA с LLOQ 15 нг/мл (DOH1163) в Covance.
График забора образов крови можно найти на схеме исследования в таблице 7.
Клинико-лабораторные оценки
Сводную статистику (включая число, среднее значение, медианное значение, SD, минимум и максимум) всех лабораторных переменных (значения центральной лаборатории и изменения по сравнению с исходным уровнем) рассчитывали для каждого визита или оценки исследования (исходный уровень, каждая временная точка после исходного уровня, конечная точка) по когорте дозы и организовывали по биологической функции. Частоту PCSA и аномальные лабораторные показатели в любое время в течение периода TEAE обобщали по биологической функции и когорте дозы. Для липидов на исходном уровне использовали классификацию NCEP APTIII. Обобщали частоту нейтропении, тромбоцитопении и лимфопении по максимальной степени тяжести.
Анализ фармакокинетических данных
Концентрации функционального сарилумаба в сыворотке обобщали с использованием описательной статистики. Все PK анализы проводили при использовании PK популяции. Она состояла из всех пациентов из группы безопасности, у которых было по меньшей мере 1 непропущенное значение концентрации сарилумаба после введения дозы. Остаточные концентрации функционального сарилумаба в сыворотке крови были обобщены с использованием описательной статистики (включая число, арифметические и геометрические средние значения, SD, SE среднего значения (SEM), CV, минимум, медиану и максимум) по дозе, в общем и по группе по весу тела для каждого визита. Образцы считались непригодными для этих анализов, если время предыдущего введения дозы составляло <11 дней или >17 дней до времени отбора образцов для схем с введением каждые две недели; <5 дней или >9 дней до времени отбора образцов для схем с введением каждую неделю. Концентрации ниже LLOQ были установлены на ноль для образцов перед введением дозы (неделя 0). Другие концентрации ниже LLOQ были заменены на LLOQ/2. Модель PopPK разработали с использованием нелинейного моделирования со смешанными эффектами для описания PK профиля сарилумаба. Модель PopPK представляли в отдельном от CSR документе.
Анализ фармакодинамических данных
Концентрации IL-6, общего sIL-6Rα и hs-CRP в сыворотке крови от исходного уровня до недели 12 обобщали с использованием описательной статистики (включая число, арифметические и геометрические средние значения, SD, SEM, CV, минимум, медиану и максимум) по дозе и по группе по весу тела для каждого визита.
Анализ фармакокинетико-фармакодинамических данных
Исследовательские анализы выполняли для выяснения взаимосвязи "воздействие-ответ" с ключевыми конечными точками эффективности и/или биомаркерами. Результаты взаимосвязи "воздействие-ответ" с ключевыми конечными точками эффективности представляли в отдельном от CSR документе.
Демографические данные и другие характеристики на исходном уровне
Приведенные в данном документе результаты относятся к части подбора дозы 12-недельной основной фазы данного исследования.
Демографические данные
Демографические данные и характеристики пациентов описывали для популяции, получавшей лечение, в таблице 9 по группе лечения. В целом, пациенты были в основном женского пола (64,3%) и европеоидной расы (78,6%) с одинаковым распределением по группам по весу тела и дозе. Средний вес и возраст составляли 45,0 кг и 13,0 лет в группе A и 19,7 кг и 5,2 года в группе B соответственно. Девять (21,4%) пациентов набрали в странах Южной Америки и 33 (78,6%) пациентов включили в исследование в западных регионах и в остальной части мира. Эти демографические характеристики были сопоставимы по трем когортам доз в каждой группе по весу тела. Подробные характеристики на исходном уровне представлены в таблице 9.
Медицинский анамнез
Всего было 9 [69,2%], 11 [78,6%] и 9 [60,0%] пациентов в когортах доз 1, 2 и 3 соответственно, с каким-либо медицинским анамнезом. Наиболее частым первичным SOC являлись инфекции и инвазии, наблюдаемые в общей сложности у 11 пациентов, более половины из которых наблюдали в когорте дозы 2 (6 [42,9%] пациентов). У одного пациента из группы A, относящегося к когорте дозы 2, в анамнезе были проблемы с кишечником из-за хирургической процедуры за 3,5 года до включения в исследование, что соответствовало критерию исключения 21 и впоследствии привело к окончательному прекращению исследования.
На исходном уровне пациенты характеризовались высокой активностью заболевания, со средним значением JADAS-27-CRP, составляющим 20,6, средним количеством активных суставов 13,9 и средним значением CRP, составляющим 9,5 мг/л. Сопутствующее и предшествующее использование лекарственных препаратов было сбалансировано между группами по весу тела (таблица 21).
(BMI) (кг/м2)
Сокращения. BMI=индекс массы тела
Регион 1 (западные страны). Чехия, Германия, Испания, Нидерланды, Великобритания, Финляндия, Франция и Италия.
Регион 2 (Южная Америка). Аргентина, Чили и Мексика
Регион 3 (Остальная часть мира). Польша и Россия
Процентные доли рассчитаны с применением количества оцененных пациентов в качестве знаменателя.
Характеристики заболевания в начальный момент времени
Характеристики заболевания на исходном уровне описывали для популяции, получавшей лечение, в таблице 10 по группе лечения. Эти характеристики заболевания на исходном уровне были сопоставимы по трем когортам доз в каждой группе по весу тела. Распределение пациентов с RF-отрицательным pcJIA, RF-положительным pcJIA и e-oJIA составляло 64,3%, 21,4% и 14,3% соответственно. Распределение RF-отрицательного pcJIA было одинаковым в 2 группах по весу тела, в то время как в группе A было приблизительно в два раза больше пациентов с RF-положительным pcJIA и в два раза меньше пациентов с e-oJIA по сравнению с группой B.
Как и ожидалось, исходя из возраста когорт доз, средняя продолжительность JIA с момента постановки диагноза была выше в группе А (2,6 года [диапазон: от 0,3 до 15,1 года]), чем в группе B (1,1 года [диапазон: от 0,1 до 7,2 года]) в когортах доз. Медианное количество активных суставов и суставов с ограниченными движениями составляло 10,0 (диапазон: от 5,0 до 45,0) и 8,0 (диапазон: от 1,0 до 46,0) соответственно, с распределением 15,5 (диапазон: от 5,0 до 45,0) и 8,5 (диапазон: от 1,0 до 46,0) в группе A и 8,5 (от 5,0 до 32,0) и 7,0 (диапазон: от 1,0 до 34,0) в группе B. Медианное значение JADAS-27-CRP на исходном уровне составляло 19,3 (диапазон: от 8,5 до 35,5) (23,2 [диапазон: от 9,2 до 33,8] и 16,8 [диапазон: от 8,5 до 35,5] в группах A и B соответственно), что отражает популяцию с высокоактивным заболеванием. Медианное значение hs-CRP на исходном уровне составляло 1,1 мг/л (диапазон: от 0,1 до 86,5) (1,0 мг/л [диапазон: от 0,1 до 45,9] и 1,6 мг/л [диапазон: от 0,1 до 86,5] в группах A и B соответственно). Распределение ANC было одинаковым по группам по весу тела и по дозе.
В целом 76,2% пациентов, включенных в исследование, получали сопутствующее лечение csDMARD и 31,0% получали сопутствующие пероральные глюкокортикоиды; 28,6% пациентов получали ≥1 курса лечения bDMARD, которое было прекращено до начала исследования. Другие характеристики пациентов и заболевания были сопоставимы по группам по весу тела.
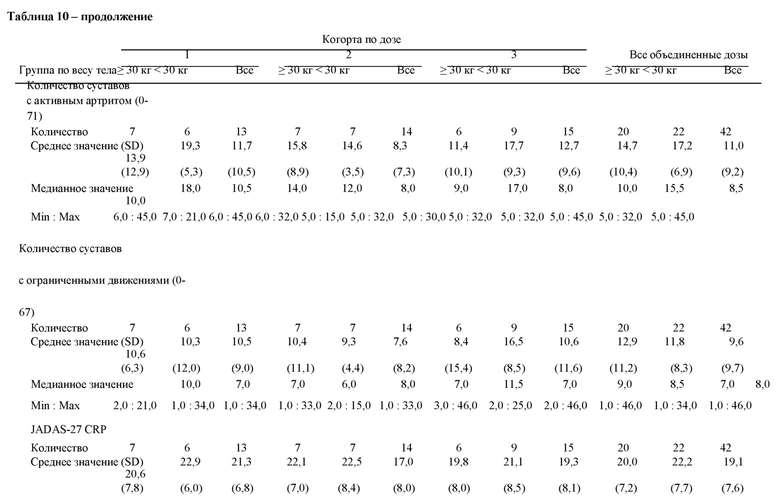


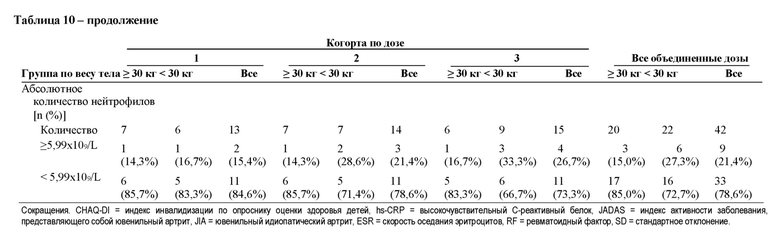
Определение соблюдения режима лечения
Определенное введение считали не соответствующим режиму лечения, если пациент не принимал запланированную дозу IMP, как того требует протокол. Процент соблюдения режима лечения для пациента определяли как количество введений, которые соответствовали режиму лечения, деленное на общее количество введений, которые были запланированы для приема пациентом к дате последней дозы или до этого.
Общий показатель соблюдения режима лечения составил 97,6%. Показатель соблюдения режима приема IMP составил 96,4%. Показатели соблюдения режима лечения были одинаковыми во всех группах по весу тела и дозе. У одного пациента в когорте дозы 2 группы B показатель соблюдения режима лечения составлял <80% из-за прерывания приема доз, вызванного нейтропенией. Ни один из пациентов не получил неправильную дозу или передозировку.
Из 42 включенных пациентов (группа A=20, группа B=22) 34 завершили 12-недельную основную фазу лечения (группа A=17; группа B=17) и 8 пациентов прекратили лечение: 5 вследствие нежелательных явлений, возникших в ходе лечения (AE), 2 вследствие отсутствия эффективности по мнению исследователя и 1 вследствие плохого соблюдения режима лечения (фиг. 8).
Фармакокинетическая и фармакодинамическая оценка
Концентрация функционального сарилумаба в сыворотке крови
Все концентрации функционального сарилумаба в сыворотке перед введением дозы были ниже LLOQ (0,3125 мг/л). Индивидуальные функциональные концентрации в сыворотке крови в течение 12-недельной основной фазы лечения с описательной статистикой анализировали для каждой группы лечения. Наблюдаемые средние значения (SD) концентрации сарилумаба после первого SC введения показаны на фиг. 3. Профили наблюдаемого среднего значения (SD) остаточной концентрации сарилумаба с течением времени показаны на фиг. 4.
Фармакокинетические параметры
Описательные статистические данные для PK параметров сарилумаба, оцененные на основе популяционной модели PK (POH0516), показаны в таблице 11A после первого введения дозы и в таблице 11B после повторного введения доз. Сарилумаб демонстрировал нелинейную PK с опосредованным мишенью распределением лекарственного средства у пациентов, страдающих pcJIA (фиг. 3). AUC0-τ представляет собой AUC с интервалом между дозами 2 недели для схемы q2w или одна неделя для схемы qw. После повторных SC доз сарилумаба в течение недель 10-12 "AUC0-14 дней" использовали для сравнения воздействия различных схем q2w и qw (AUC0-14 дней=2 x AUC0-τ для схемы qw и AUC0-14 дней=AUC0- τ для схемы q2w).
по дозе
по весу тела
мг/кг
мг/л
мг/л
≥ 30 кг
[8,63]
[57,7]
[0,620]
< 30 кг
[9,68]
[66,2]
[0,470]
≥ 30 кг
[15,2]
[133]
[1,69]
< 30 кг
[17,5]
[158]
[4,34]
≥ 30 кг
[10,8]
[60,8]
[7,87]
< 30 кг
[8,63]
[48,2]
[5,96]
по дозе
по весу тела
мг/кг
мг/л
мг/л
мг/
л
≥ 30 кг
[12,6]
[116]
[1,40]
< 30 кг
[13,7]
[101]
[1,27]
≥ 30 кг
[24,4]
[250]
[8,25]
< 30 кг
[31,1]
[331]
[13,1]
≥ 30 кг
[39,3]
[254]
[31,4]
< 30 кг
[29,9]
[197]
[27,3]
Сокращения. AUC0 τ: площадь под кривой концентрации в сыворотке в зависимости от времени в течение интервала между дозами τ, равного 2 неделям (схема q2w) или одной неделе (схема qw), Cmax=максимальная наблюдаемая концентрация в сыворотке, Ctrough=концентрации, наблюдаемые перед введением средства лечения во время повторного введения доз, CV=коэффициент вариации, pcJIA=ювенильный идиопатический артрит с полиартикулярным течением, q2w=один раз в две недели, qw=один раз в неделю, SC=подкожно.
Полученное из популяционной модели PK байесовское воздействие
После повторного SC введения q2w и qw в течение недель 10-12 воздействие сарилумаба увеличивалось более чем пропорционально дозе, при этом значение AUC0-14 дней увеличивалось в 2,4-2,6 раза при увеличении дозы сарилумаба в 1,5-1,6 раза в когорте дозы 2 по сравнению с когортой дозы 1, и увеличивалось в 3,4-4,4 раза при увеличении дозы сарилумаба в 2 раза в когорте дозы 3 по сравнению с когортой дозы 1.
Наблюдали накопление до значений в 1,9-2,2 раза больше для когорт доз 1 и 2 и накопление до значений в 4,1-4,5 раза больше для когорты дозы 3 на неделе 12 по сравнению с первой дозой. (Таблица 22) Схемы введения доз, испытанные в 2 группах по весу тела, достигли аналогичного воздействия для каждой когорты дозы. Вариабельность Cmax и AUC0-τ после повторных SC доз у пациентов, страдающих pcJIA, находилась в диапазоне от 13,6 до 36,6%.
Проводили анализ данных 12-недельного исследования по подбору дозы. Данные анализировали для пациентов, разделенных по весу тела на 2 группы: группу A (30-60 кг) и группу B (от 10 до <30 кг). Как описано выше, пациенты получали последовательные нарастающие дозы сарилумаба. Доза 1 (группа A/B): 2,0/2,5 мг/кг q2w; доза 2 (группа A/B): 3/4 мг/кг q2w; доза 3 (группа A/B): 2,0/2,5 мг/кг qw. Дозы для pcJIA были нацелены на достижение воздействия, аналогичного 150 мг q2w, 200 мг q2w и 150 мг qw для взрослых. Как описано выше, первичным исходом являлась фармакокинетика (PK), а вторичными исходами являлись безопасность, PD и эффективность сарилумаба.
Данные показывают, что в исследование включили 42 пациента (20/22 в группах A/B), средний возраст которых составлял 13,0/5,2 года. На исходном уровне средняя продолжительность pcJIA, количество активных суставов и значение JADAS27-CRP составляли 4,6/1,7 года, 17,2/11,0 и 22,2/19,1 в группах A/B соответственно. Как и у взрослых пациентов, сарилумаб демонстрировал нелинейную PK с опосредованным мишенью распределением лекарственного средства. После повторных SC введений воздействие увеличивалось более чем пропорционально дозе, и накапливалось до значений в 1,9-4,5 раза больше за 12 недель. Воздействие сарилумаба было одинаковым в обеих группах по весу тела для каждой дозы (таблица 12) и сопоставимо с соответствующими дозами для взрослых. Нежелательные явления, возникшие в ходе лечения, зарегистрировали у 36/42 (85,7%) пациентов (сопоставимо по группам по дозе и весу тела); инфекции (28/42, 66,7%) являлись наиболее частыми AE. Выявили 12 случаев нейтропении степени 3/4, в основном в когорте дозы 3 (n=6) и в группе B (n=8). Ни один из них не был связан с инфекцией; все разрешились за несколько дней. В целом, 4 пациента прекратили лечение из-за нейтропении и 1 пациент прекратил лечение из-за повышения уровня аланинаминотрансферазы. Не наблюдали серьезных AE, перфорации желудочно-кишечного тракта и летальных исходов. К неделе 12, как наблюдали в течение приема средства лечения все пациенты достигли ACR30 JIA; 50%, 62% и 100% пациентов достигли ACR70 JIA при дозах 1, 2 и 3 соответственно; среднее изменение JADAS27-CRP по сравнению с исходным уровнем для доз 1, 2 и 3 составило -74,6%, -73,1% и -87,9% соответственно
.

Фармакокинетические выводы
Сарилумаб демонстрировал нелинейную PK с опосредованным мишенью распределением лекарственного средства у пациентов, страдающих pcJIA. После повторного SC введения воздействие сарилумаба увеличивалось более чем пропорционально дозе, и накапливалось до значений в 1,9-4,5 раза больше за 12 недель. Для каждой когорты дозы схемы введения доз, испытанные в 2 группах по весу тела, показали одинаковое воздействие. Для дополнительного анализа данных группы пациентов A (30-60 кг) и пациентов группы B (от 10 до <30 кг) наблюдали, что сарилумаб проявлял нелинейную PK с опосредованным мишенью распределением лекарственного средства. Кроме того, все схемы введения доз оказались эффективными в отношении снижения активности заболевания. Профиль безопасности соответствовал классовым эффектам; более высокую частоту случаев нейтропении наблюдали при дозе 3 и у пациентов с весом тела от 10 до <30 кг.
Концентрации CRP, ESR, IL-6 и sIL-6R
Описательную статистику концентраций hs-CRP, ESR, IL-6 и общего sIL-6Rα от исходного уровня до недели 12 анализировали для каждой группы лечения. Профили средней (SE) концентрации CRP, ESR, IL-6 и sIL-6Rα с течением времени получали для каждой когорты. После SC введения сарилумаба среднее подавление CRP и увеличение sIL-6Rα было больше в когортах доз 2 и 3, чем в когорте дозы 1. Средние концентрации CRP были снижены в течение 2-4 недель в когортах доз 2 и 3, причем большее и более быстрое снижение наблюдали в когортах доз 2 и 3, чем в когорте дозы 1 (фиг. 10A). Изменение средней концентрации CRP от исходного уровня до 12 недели составило 7,4-5,8 мг/л, 14,6-0,5 мг/л и 6,5-0,1 мг/л при дозах 1, 2 и 3 соответственно. Все пациенты достигли нормальной концентрации CRP при дозах 2 и 3 в течение недель 2-6. На неделе 4 большинство пациентов достигли уровня CRP ниже предела обнаружения (<0,2 мг/л) при дозах 2 и 3 (71,4% и 83,3% соответственно, как наблюдали в течение приема средства лечения) по сравнению с 25,0% при дозе 1. Этот эффект сохранялся с течением времени 66,7% и 90,9% пациентов достигли уровня CRP <0,2 мг/л на неделе 12 при дозах 2 и 3 соответственно, по сравнению с 30,0% при дозе 1 (фиг. 10B).
Высокое среднее значение CRP в когорте дозы 1 группы А было в первую очередь обусловлено одним пациентом, у которого уровень CRP оставался повышенным (40-110 мг/л) в течение 12-недельной основной фазы лечения.
Скорость оседания эритроцитов на неделе 12 соответствует тому, что наблюдали для CRP. В целом, средняя скорость оседания эритроцитов (ESR) снизилась, а средняя концентрация IL-6 и sIL-6Rα увеличилась на неделе 12. Как и в случае уровней CRP, большее снижение наблюдали в когортах доз 2 и 3 со средним изменением от исходного уровня до недели 12 с 22,2 до 18,2 мм/ч, с 21,4 до 6,9 мм/ч и с 21,9 до 4,4 мм/ч в когортах доз 1, 2 и 3 соответственно.
Фармакодинамико-фармакокинетическая взаимосвязь биомаркеров
Исследовательские анализы выполняли для выяснения взаимосвязи "воздействие-ответ" с биомаркерами PD. Отдельные концентрации CRP, ESR, IL-6 и sIL-6Rα наносили на график в зависимости от минимальной концентрации в сыворотке на неделе 12. Не было очевидной взаимосвязи между отдельными уровнями IL-6 и остаточными концентрациями сарилумаба. Данные показывают, что уровень общего sIL-6Rα увеличивался с увеличением остаточных концентраций сарилумаба после повторных доз у отдельных пациентов, страдающих pcJIA. Уровни С-реактивного белка были ниже (≤1 мг/л) у отдельных пациентов с остаточными концентрациями сарилумаба >1 мг/л, по сравнению с пациентами с остаточными концентрациями сарилумаба ≤1 мг/л. Подавление CRP было больше у пациентов в когортах доз 2 и 3, чем у пациентов в когорте дозы 1. Скорость оседания эритроцитов была ниже (≤10 мм/час) у отдельных пациентов с остаточными концентрациями сарилумаба >1 мг/л, по сравнению с пациентами с остаточными концентрациями сарилумаба ≤1 мг/л. Подавление ESR было больше у пациентов в когортах доз 2 и 3, чем у пациентов в когорте дозы 1.
После SC введения сарилумаба подавление CRP и увеличение sIL-6Rα было больше в когортах доз 2 и 3, чем в когорте дозы 1. В целом, средние концентрации ESR снизились, а средние концентрации IL-6 и sIL-6Rα увеличились по сравнению с исходным уровнем на неделе 12.
Безопасность 2-недельной основной фазы лечения . Общее воздействие (пациенто-годы лечения) было одинаковым во всех группах по дозе и весу тела: 4,6 и 5,0 пациенто-годы в группах A и B соответственно.
Серьезных AE и летальных исходов не наблюдали (таблица 23). Наиболее часто наблюдаемыми AE являлись инфекции и нейтропения (n=28 [66,7%] и n=11 [26,2%] соответственно). Частота инфекций была одинаковой во всех группах по дозе и весу тела и в основном была следствием инфекций верхних дыхательных путей (n=9 [21,4%]; таблица 25). О серьезных инфекциях не сообщали. Случаев пневмонии или туберкулеза не выявляли. Произошел один случай кандидоза ротовой полости (когорта дозы 3, группа А), который разрешился спонтанно без корректирующего лечения и без прерывания лечения сарилумабом. Ни один из пациентов не прекратил лечение из-за какой-либо инфекции.
Предварительно установленный лабораторный мониторинг выявил 12 пациентов с преходящей нейтропенией 3/4 степени (28,6%). Из этих пациентов 3 получали дозу 1 (2 в группе A, 1 в группе B), 3 получали дозу 2 (1 в группе A, 2 в группе B) и 6 получали дозу 3 (1 в группе A, 5 в группе B; таблица 24). У всех пациентов отсутствовали симптомы, но для 3 требовалось прерывание лечения в соответствии с протоколом (по 1 в когорте дозы 2, группе B; когорте дозы 3, группах A и B). Ни один из случаев нейтропении не был связан с инфекцией и все они разрешились в течение нескольких дней. Лабораторный мониторинг, включающий количество эритроцитов, количество тромбоцитов, уровень липидов и оценку функции печени, не выявил каких-либо дополнительных потенциально клинически значимых отклонений. В частности, не наблюдали случаев вызванного лекарственным средством поражения печени, как это определено законом Хая (21).
Окончательное прекращение лечения в соответствии с протоколом вследствие AE имело место у 5 пациентов (11,9%). Из этих случаев 1 представлял собой нейтропению 3 степени на неделе 2 приема в когорте дозы 1, группе А; 3 представляли собой нейтропению 4 степени в когорте дозы 3, группе B, и 1 представлял собой повышение аланинаминотрансферазы (ALT) в когорте дозы 1, группе A (>5 × верхний предел нормы [ULN] на неделе 4).
Семь пациентов сообщили о местных реакциях после инъекции (4 в группе A и 3 в группе B). В основном эти реакции представляли собой эритему и боль; все были от легкой до умеренной по интенсивности; ни одна не привела к прекращению лечения. Два пациента дали положительный результат на антитела к лекарственному средству на неделе 12: один в когорте дозы 1, группе A (положительный результат на нейтрализующие антитела; лечение прекращено из-за ALT >5 × ULN на неделе 4); один в когорте дозы 3, группе B (отсутствие положительного результата на нейтрализующие антитела; лечение прекращено из-за нейтропении 4 степени после первой инъекции).
ОЦЕНКА ЭФФЕКТИВНОСТИ
Частота ответов ACR JIA
В данном исследовании для оценки признаков и симптомов использовали уровень ответа ACR JIA. Ответ ACR JIA 30/50/70/90/100 определяли как пациента, у которого 3 из 6 переменных базового набора улучшились по меньшей мере на 30%/50%/70%/90%/100% по сравнению с исходным уровнем, при этом не более одной из оставшихся переменных ухудшились более чем на 30%.
Частоту ответов ACR JIA рассчитывали методом непосредственного наблюдения для тех, кто принимал средство лечения ("как наблюдали в течение приема средства лечения"), и путем подстановки для пациентов, которые прекратили лечение ("подход с подстановкой данных субъектов, не ответивших на лечение", когда пациенты, прекратившие лечение, автоматически считались не ответившими на лечение на визитах после прекращения лечения). Частота ответов ACR JIA во всех группах по дозе и весу тела на неделе 12 обобщены в таблице 13. Оценки точности частоты ответов ACR JIA показаны в таблице 33.
Как наблюдали в течение приема средства лечения, на неделе 12 частота ответов ACR30 JIA составляла 100% во всех группах по дозе и весу тела. Частота ответов ACR70 JIA составляла 50,0%, 61,5% и 100% в когорте доз 1, 2 и 3 соответственно. Согласно расчетам подхода с подстановкой данных субъектов, не ответивших на лечение, на неделе 12 частота ответов ACR30 JIA составила 76,9%, 92,9% и 73,3% в когортах доз 1, 2 и 3 соответственно. Частота ответов ACR70 JIA составляла 38,5%, 57,1% и 73,3% в когорте доз 1, 2 и 3
соответственно.
В целом, при подходе с наблюдением в течение приема средства лечения возникновение ответа ACR30 JIA наблюдали с первой оценки на неделе 2 во всех группах по дозе и весу тела. Частота достигла плато 100% на неделях 4 и 8 для когорты дозы 2, групп A и B соответственно, и на неделях 6 и 8 для когорты дозы 3, групп A и B соответственно, в то время как частота ответов в когорте дозы 1 была неустойчивой до недели 12 (фиг. 5). Результаты подхода с подстановкой данных субъектов, не ответивших на лечение, показаны на фиг. 6.
Компоненты ACR JIA
Собирали данные в отношении компонентов ACR JIA в течение 12-недельной основной фазы лечения. Обобщенная информация о компонентах ACR JIA на неделе 12 представлена в таблице 14. На неделе 12 все когорты доз и группы по весу тела показали снижение по сравнению с исходным уровнем по всем компонентам ACR JIA, которое в целом оказалось более выраженным в когортах доз 2 и 3 по сравнению с когортой дозы 1. Количество суставов с активным артритом уменьшилось у пациентов во всех когортах доз в обеих группах по весу тела в течение 12-недельной основной фазы лечения. Наибольшее уменьшение среднего значения на неделе 12 наблюдали у пациентов в когорте дозы 3, группе А (-16,5). Количество суставов с ограниченными движениями уменьшилось у пациентов во всех когортах доз в обеих группах по весу тела в течение 12-недельной основной фазы лечения. Наибольшее уменьшение среднего значения на неделе 12 наблюдали у пациентов в когорте дозы 3, группе B (-11,3). Общая оценка врачом по VAS уменьшилась у пациентов во всех когортах доз в обеих группах по весу тела в течение 12-недельной основной фазы лечения. Наибольшее уменьшение среднего значения на неделе 12 наблюдали у пациентов в когорте дозы 3, группе B (-5,0). Общая оценка пациентом по VAS уменьшилась у пациентов во всех когортах доз в обеих группах по весу тела в течение 12-недельной основной фазы лечения. Наибольшее уменьшение среднего значения на неделе 12 наблюдали у пациентов в когорте дозы 3, группе B (-5,7). В целом оценка по CHAQ-DI уменьшилась у пациентов во всех когортах доз в обеих группах по весу тела в течение 12-недельной основной фазы лечения. Наибольшее уменьшение среднего значения на неделе 12 наблюдали у пациентов в группе B, когорте дозы 2 (-1,0). Средние концентрации hs-CRP снизились во всех когортах доз для обеих групп по весу тела на неделе 12. Средние концентрации CRP снизились в течение 2-4 недель в когортах доз 2 и 3. Высокое среднее значение CRP в когорте дозы 1 группы А было в первую очередь обусловлено одним пациентом, у которого уровень CRP оставался повышенным (40-110 мг/л) в течение 12-недельной основной фазы лечения. Наибольшее уменьшение среднего значения на неделе 12 наблюдали у пациентов в группе B, когорте дозы 2 (-15,2).
Индекс активности заболевания, представляющего собой ювенильный артрит
Активность заболевания оценивали по оценке JADAS-27 CRP. Анализировали оценку JADAS-27 CRP с течением времени в каждой когорте дозы и группе по весу тела. Обобщенные данные по JADAS-27 (ESR) в течение 12-недельной основной фазы лечения представлены в таблице 15. Сообщенные исследователем TEAE показаны в таблице 30. Данные показывают, что по сравнению с исходным уровнем все когорты доз и группы по весу тела показали снижение оценки JADAS-27 CRP с недели 2. На неделе 12 изменение среднего процентного значения по сравнению с исходным уровнем составило -74,6%, -73,1% и -87,9% в когортах доз 1, 2 и 3 соответственно.
ACR100
ACR100
a Пациенты, прекратившие лечение, автоматически считаются не ответившими на лечение на визитах после прекращения лечения.
(≥30 кг)
(<30 кг)
B
Доза 1 Доза 2 Доза 3 Доза 1 Доза 2 Доза 3 Доза 1 Доза 2 Доза 3
Количество активных суставов (0-71)
Исходный уровень
Общая оценка врачом по VAS (0-10)
Исходный уровень
Общая оценка пациентом по VAS (0-10)
Исходный уровень
Количество
(≥30 кг)
(<30 кг)
B
Доза 1 Доза 2 Доза 3 Доза 1 Доза 2 Доза 3 Доза 1 Доза 2 Доза 3
CHAQ-DI (0-3)
Исходный уровень
hs-CRP (мг/л)
Исходный уровень
Количество
Сокращения. CHAQ-D1=индекс инвалидизации по опроснику оценки здоровья ребенка, hs-CRP=высокочувствительный C-реактивный белок, VAS=визуальная аналоговая шкала.
значения
значения
значения
значения
значения
значения
V12/Неделя 12
значения
Выводы в отношении эффективности
Улучшение признаков и симптомов, оцениваемое с помощью ответа ACR JIA, наблюдали во всех когортах доз и группах по весу тела с недели 2 с использованием либо подхода с наблюдением в течение приема средства лечения, либо подхода с подстановкой данных субъектов, не ответивших на лечение. Ни одна когорта дозы не прекратила лечение преждевременно из-за недостаточной эффективности. Два пациента прекратили лечение из-за недостаточной эффективности по заключению исследователя, 1 в когорте дозы 1, группе B на неделе 10, а другой в когорте дозы 3, группе B на неделе 4. При использовании подхода с наблюдением в течение приема средства лечения на неделе 12 частота ответа ACR30 JIA составила 100% (34/34) во всех когортах доз и группах по весу тела, при этом когорты доз 2 и 3 приводили к более быстрому и устойчивому ответу ACR30 JIA по сравнению с когортой дозы 1 (фиг. 9A). На неделе 12, согласно расчетам с помощью подхода с подстановкой данных субъектов, не ответивших на лечение, 77%, 93% и 73% пациентов достигли ACR30 JIA (когорты доз 1, 2 и 3 соответственно; фиг. 11). Большая часть пациентов также достигла более высоких пороговых значений ответа: 50%, 62% и 100% пациентов достигли ответов ACR70 JIA и 40%, 31% и 64% пациентов достигли ответов ACR90 JIA в когортах доз 1, 2 и 3 соответственно на неделе 12, как наблюдали в течение приема средства лечения (фиг. 9В, 9С; результаты в соответствии с подходом с подстановкой данных субъектов, не ответивших на лечение, показаны на фиг. 11В, 11С). Все компоненты ACR JIA уменьшались с помощью каждой из 3 схем введения доз сарилумаба (таблица 26).
Наблюдали тенденцию к более быстрому ответу ACR JIA в когортах доз 2 и 3 по сравнению с когортой дозы 1. При подходе с наблюдением в течение приема средства лечения частота ответов ACR30 JIA достигала плато 100% в течение первых 2 месяцев лечения сарилумабом для когорт доз 2 и 3, групп A и B, в то время как частота ответов в когорте дозы 1 была неустойчивой до недели 12.
Снижение активности заболевания, оцененное с помощью JADAS-27 CRP, наблюдали у пациентов во всех когортах доз и группах по весу тела на неделе 12 с изменением среднего процентного значения по сравнению с исходным уровнем -74,6%, -73,1% и -87,9% в когортах доз 1, 2, и 3 соответственно, и наблюдали также с недели 2 и далее (фиг. 9D). В ретроспективном анализе на 12-й неделе 20 пациентов достигли низкой активности заболевания (LDA; JADAS-27-CRP ≤3,8): 6, 5 и 9 пациентов в когортах доз 1, 2 и 3 соответственно. Клинически неактивное заболевание (CID), определенное как JADAS-27-CRP ≤1, было достигнуто у 3 пациентов (1 в когорте дозы 2 и 2 в когорте дозы 3). CID, как определено критериями Уоллиса, было достигнуто у 9 пациентов (3, 2, и 4 пациентов в когортах доз 1, 2 и 3 соответственно) (22). У тринадцати пациентов не имелось активных суставов на неделе 12, у 4 в каждой из когорт доз 1 и 2 и у 5 в когорте дозы 3 (таблица 27).
ОБСУЖДЕНИЕ
Экспериментальные схемы введения доз сарилумаба, протестированные в исследовании, выбирали на основе моделирования и симуляции данных PK сарилумаба у взрослых пациентов с RA. Диапазон веса тела, состав тела и метаболизм лекарственных средств у пациентов детского возраста отличаются от таковых у взрослых, а ответ на лечение и побочные эффекты могут варьировать в зависимости от возраста. В результате прямая экстраполяция доз для детей из доз для взрослых без тестирования в педиатрических клинических испытаниях не может быть рекомендована (23, 24). Клиренс моноклональных антител часто увеличивается нелинейным образом относительно увеличения веса тела (25), поэтому, учитывая диапазон веса тела и возраста в популяции pcJIA, скорректированные по весу тела дозы были сочтены подходящими для данного исследования. Схемы введения доз, протестированные на пациентах, страдающих pcJIA, выбирали на основе популяционной симуляции PK для достижения аналогичного воздействия в обеих группах по весу тела, которое было бы сопоставимо с воздействием при схемах введения доз, изученным у взрослых пациентов с RA. Результаты исследования подтвердили точность первоначальной схемы модели на основе веса тела. Сарилумаб демонстрировал нелинейную PK с опосредованным мишенью распределением лекарственного средства. Скорректированные по весу тела схемы введения доз дали сопоставимое воздействие в двух группах по весу тела, при этом достигнутые воздействия были сопоставимы со схемами с фиксированными дозами, протестированными у взрослых с RA.
В данном исследовании лечение сарилумабом было связано со снижением средних значений CRP и снижением ESR, демонстрируя функциональное ингибирование IL-6 у пациентов, страдающих pcJIA (20). В частности, известно, что снижение уровня CRP является прямым следствием нейтрализации IL-6 и указывает на эффективность нейтрализации IL-6 у пациентов с RA (26-28). Снижение средних значений CRP было более выражено в когортах доз 2 и 3, при этом большее количество этих пациентов достигло и поддерживало уровни CRP ниже предела обнаружения (<0,2 мг/л) высокочувствительного метода, использованного в данном исследовании.
Ответ ACR JIA наблюдали с недели 2 при всех схемах введения доз сарилумаба. На основании ACR70 JIA уменьшение признаков и симптомов заболевания было более выраженным и устойчивым в когортах доз 2 и 3 по сравнению с когортой дозы 1. По оценке JADAS-27-CRP больше пациентов достигли CID или LDA в когорте дозы 3, чем в когортах доз 1 или 2. Эти результаты эффективности совпадают с четкими тенденциями, наблюдаемыми в результатах PD (т. е. концентрациях CRP), что дополнительно подтверждает взаимосвязь между нейтрализацией IL-6 и терапевтическим ответом (20, 29). Вместе с менее устойчивым эффектом в отношении CRP при дозе 1, эти результаты подтверждают исключение когорты дозы 1 для дальнейшего исследования.
Частота ответов на лечение, наблюдаемая в данном исследовании, выигрывала при сравнении с данными, опубликованными для большей когорты пациентов, страдающих pcJIA, получавших лечение блокатором IL-6R тоцилизумабом (18). Текущие результаты предполагают, что лечение сарилумабом может соответствовать целям недавних руководств, опубликованных международной целевой группой, которые установили конечной целью лечения ремиссию заболевания и рекомендовали, чтобы улучшение активности заболевания ≥50% было достигнуто в течение 3 месяцев, а также чтобы цели лечения поддерживались после достижения (11). В описанное в данном документе исследование включали пациентов с установленным заболеванием, у трети из которых уже было неудачным ≥1 лечение с помощью биологического средства лечения. Тем не менее, из 24 пациентов в когортах доз 2 или 3, через 3 месяца 23 и 19 достигли ACR50 JIA и ACR70 JIA соответственно, 14 достигли LDA по JADAS-27-CRP, а 9 не имели активных суставов. Долгосрочное последующее наблюдение во время фазы дополнительного лечения позволит лучше понять способность сарилумаба соответствовать долгосрочным целям лечения и поддерживать стойкую клиническую ремиссию после ее достижения.
Типы AE, наблюдаемые при приеме сарилумаба в исследовании, соответствовали тем, которые наблюдали при лечении сарилумабом у взрослых пациентов и классовым эффектам антагонистов IL-6 у пациентов детского возраста (18, 30-32). Сообщали о нескольких местных реакциях в месте инъекции, и все они были от легкой до умеренной по интенсивности, что свидетельствует о хорошей местной переносимости; это важно для пациентов детского возраста, поскольку страх боли от инъекции шприцем является частым явлением. Наиболее частыми AE являлись инфекции и нейтропения. Частота инфекции в основном была обусловлена инфекциями верхних дыхательных путей, которые распространены в детской популяции, и была сбалансирована по группам по весу тела и дозе. О серьезных инфекциях, инфекциях, приводящих к прекращению лечения, и оппортунистических инфекциях не сообщали.
В соответствии с протоколом исследования абсолютное количество нейтрофилов (ANC) периодически оценивали в течение периода приема средства лечения, в том числе после первой дозы сарилумаба и до введения второй. Этот тщательный гематологический мониторинг с первой оценкой после введения дозы в день 12 для схем Q2W и в дни 5-8 для схемы QW привел к выявлению 12 случаев нейтропении 3/4 степени. У взрослых пациентов, получавших сарилумаб или тоцилизумаб, самое низкое значение ANC регистрировали через несколько дней (до дня 7) после первого введения средства лечения, после чего значения ANC восстанавливались, что согласуется с классовыми эффектами (33). Основываясь на этих данных исследований у взрослых, сроки проведения оценок в данном исследовании помогли объяснить численный дисбаланс событий в когорте дозы 3 (n=6, включая 4 случая, выявленных в день 5) по сравнению с 2 другими схемами введения доз (n=3 при обеих дозах 1 и 2).
Из 12 случаев нейтропении 3/4 степени 8 произошли в группе B. Очевидную тенденцию к увеличению частоты нейтропении у более молодых пациентов с меньшим весом тела наблюдали также у пациентов, страдающих pcJIA, которых лечили тоцилизумабом (34, 35). Нет окончательного механизма, объясняющего, почему более молодые пациенты более склонны к нейтропении, чем более взрослые пациенты детского возраста или взрослые, но в целом сообщали о большей распространенности нейтропении у детей (36). Нейтропения была причиной большинства случаев прекращения терапии, вызванных AE (n=4/5): 3 случая представляли собой нейтропению 4 степени, которая привела к прекращению лечения в соответствии с критериями, определенными протоколом, и 1 случай представлял собой нейтропению 3 степени у пациента, у которого исходное значение ANC было ниже 2000/мм3. Важно отметить, что ни один из случаев нейтропении 3/4 степени, выявленных в ходе данного исследования, не был связан с инфекцией, и большинство случаев разрешилось в течение нескольких дней. Это наблюдение согласуется с данными, полученными у детей и взрослых, которых лечили тоцилизумабом или сарилумабом, с добавлением дополнительных данных, подтверждающих вывод о том, что нейтропения, вызванная ингибированием IL-6, не приводит к увеличению риска инфекции (18, 32-34, 37). Хотя все доступные данные подтверждают вывод о том, что нейтропения, вызванная ингибированием IL-6, не связана с повышенным риском инфекций, данные о нейтропении, полученные в данном исследовании, вместе с динамикой ANC у взрослых после введения тоцилизумаба или сарилумаба, выступают против выбора когорты дозы 3 (32, 33, 37). Этот осторожный выбор снижает риск нейтропении, особенно у детей младшего возраста, которые относятся к группе повышенного риска. Кроме того, использование схемы введения доз с введениями один раз в 2 недели в когорте дозы 2 обеспечивает более длительное время для восстановления ANC.
После обзора всех данных из 12-недельной части исследования по подбору дозы, когорту дозы 2 выбрали в качестве оптимальной схемы введения сарилумаба для включения дополнительных пациентов во вторую часть основной фазы лечения исследования, а также для продолжения во второй части для пациентов, которые уже были включены в когорты доз 1 или 3. Схема когорты дозы 2 продемонстрировала благоприятный баланс между эффективностью и безопасностью по сравнению с когортами 1 и 3. Эта схема введения доз обеспечивала аналогичное воздействие и отражала профиль эффективности эквивалентной схемы введения доз для взрослых 200 мг Q2W, рекомендованной одобренной дозировки для взрослых с RA (20, 30, 31). Когорта дозы 2 также придерживалась графика введения доз один раз в 2 недели (по сравнению со схемой когорты дозы 3 с введениями один раз в неделю), который снижает стресс от инъекций, что особенно актуально у детей, и, как упоминалось выше, увеличивает время восстановления ANC.
Дополнительные данные относительно безопасности и эффективности сарилумаба у пациентов, страдающих pcJIA, с течением времени в более крупной когорте пациентов будут предоставлены путем включения дополнительных пациентов, которые получат выбранную схему введения доз и за которыми будут проводить последующее наблюдение в фазе дополнительного лечения. Как уже отмечалось, данное исследование не было слепым и не включало плацебо или активный препарат сравнения. Это было связано с этическими проблемами, связанными с подверганием детей неэффективному лечению, с учетом потенциального хронического и тяжелого характера заболевания и наличия доступных данных об ингибировании IL-6 у взрослых и детей с артритом (18, 38). Эти проблемы были отражены в схеме исследования, с последовательным подходом к тестируемым дозам и частым мониторингом, в использовании независимого комитета по мониторингу данных для формальной оценки безопасности сарилумаба на протяжении всего исследования, в использовании DEC для мониторинга ответа пациента на лечение, а также в способности DEC прекратить лечение пациентов любой дозой, считающейся неэффективной, и вернуть их к стандартному лечению, одобренному исследователем.
В заключение, у пациентов, страдающих pcJIA, сарилумаб демонстрировал нелинейную PK с опосредованным мишенью распределением лекарственного средства, демонстрировал профиль безопасности, соответствующий классовым эффектам антагонистов IL-6, и снижал активность заболевания. Данная 12-недельная часть подбора дозы основной фазы исследования показала, что когорта дозы 2 (3,0 мг/кг Q2W для пациентов весом 30-60 кг и 4,0 мг/кг Q2W для пациентов весом от 10 до <30 кг) достигла воздействия, аналогичного таковому при одобренной дозировке 200 мг Q2W для взрослых с RA. Когорта дозы 2 обеспечивает хороший баланс между безопасностью/переносимостью и эффективностью у пациентов, страдающих pcJIA, и будет изучаться в дальнейшем как оптимальная схема введения доз сарилумаба при pcJIA.
Пример 2. Открытое исследование с подбором дозы сарилумаба с последовательными нарастающими повторными дозами, вводимыми путем подкожной (SC) инъекции, у детей и подростков в возрасте от 1 до 17 лет с системным ювенильным идиопатическим артритом (sJIA) с последующей фазой дополнительного лечения. (Исследование № DRI13926, название исследования: исследование с подбором дозы сарилумаба с повторными дозами у детей и подростков с системным ювенильным идиопатическим артритом)
Планируется контролируемое испытание фазы 2 [NCT02991469] для тестирования моноклонального антитела IgG1 человека к IL-6Rα, сарилумаба, вводимого подкожно для лечения sJIA. Данное исследование нацелено на детей и подростков в возрасте от 1 до 17 лет с sJIA, подтипом JIA, который напоминает болезнь Стилла у взрослых, относительно редкое воспалительное заболевание (оценочная заболеваемость во всем мире составляет 0,22 и 0,34 на 100000 для мужчин и женщин соответственно; Bennett, A.N. et al., Adult onset Still's disease and collapsing glomerulopathy: successful treatment with intravenous immunoglobulins and mycophenolate mofetil. Rheumatology (Oxford). 2004;43(6):795-9). В данной детской популяции планируется протестировать 3 схемы введения доз сарилумаба.
План исследования
Описание протокола
Исследование DRI13926 представляет собой многонациональное многоцентровое открытое последовательное 2-фазное исследование среди детей и подростков в возрасте от 1 до 17 лет (или применяются возрастные требования страны) с sJIA с недостаточным ответом на стандартную терапию или у которых наблюдается ее непереносимость, и которые будут получать SC инъекции сарилумаба q2w или qw. 2 фазы представляют собой следующие.
1. 12-недельная основная фаза лечения, разделенная на 2 части.
- Первую часть с подбором дозы с последовательными нарастающими дозами, в которой будут исследовать по меньшей мере 2 схемы введения доз в 2 группах по весу тела: пациенты весом ≥30 кг и ≤60 кг (группа A) и пациенты <30 кг и ≥10 кг (группа B), у 6 поддающихся оценке пациентов на схему введения доз и группу по весу тела (всего по меньшей мере 24 пациента). Включение пациентов в исследование будет разделено по группе по весу тела и схеме введения доз, начиная с группы A (≥30 кг и ≤60 кг) и когорты дозы 2.
- Вторую последующую часть, где приблизительно 24 дополнительных пациентов (по 12 в каждой группе по весу тела ≥30 кг [группа A] и <30 кг и ≥10 кг [группа B]) включат непосредственно в выбранную схему введения доз (определенную на основе совокупных данных от пациентов, включенных в первую часть), чтобы получить в общей сложности 18 пациентов, подлежащих оценке, на каждую группу по весу тела при выбранной схеме введения доз.
12-недельная основная фаза лечения продолжается от визита 2 (исходный уровень - неделя 0) до времени введения исследуемого лекарственного препарата (IMP) во время визита 12 (неделя 12). В течение 12-недельной основной фазы лечения, чтобы свести к минимуму количество забираемой крови и количество визитов при сохранении оценки первичной конечной точки, пациентов группы B (<30 кг и 210 кг) случайным образом распределят в следующие графики отбора образцов PK сарилумаба 1 или 2.
- График 1. Исходный уровень, день 3, день 8, неделя 2, неделя 4, неделя 8 и неделя 12
- График 2. Исходный уровень, день 5, день 12, неделя 2, неделя 4, неделя 8 и неделя 12
2. 144-недельная фаза дополнительного лечения.
IMP на визите 12 рассматривают как первый IMP для фазы дополнительного лечения.
Только пациентам, достигшим ответа ACR JIA 30 (при отсутствии лихорадки) на визите 12 (неделя 12), разрешат продолжить исследование в фазе дополнительного лечения. Пациенты продолжат придерживаться той же схемы введения доз сарилумаба, которую им назначили на 12-недельную основную фазу лечения исследования, до тех пор, пока не определят выбранную схему введения доз. После выбора схемы введения доз пациентам, которые еще не придерживались этой схемы введения доз, будут корректировать схему введения доз в соответствии с выбранной схемой введения доз, и они будут придерживаться нового графика визитов с более частым мониторингом (для PK, безопасности и эффективности) в течение первых 12 недель, по сравнению с пациентами, у которых схему введения доз не изменяли.
Пациентов, преждевременно прекративших прием исследуемого средства для лечения, будут оценивать с использованием процедуры EOT на визите 27. Этих пациентов попросят вернуться для оценки в конце исследования (EOS) через 6 недель после визита EOT (EOT+6 недель). Для пациентов, которые прекратят исследуемый вид лечения в течение 12-недельной основной фазы лечения, будут проводить дополнительную оценку PK сарилумаба через 2 недели после визита EOT (EOT+2 недели), а во время визита EOT будут измерять IL-6 и sIL-6R. Этим пациентам предложат совершать все визиты и оценки в соответствии с графиком протокола, за исключением введения сарилумаба, до визита 12.
Протестированные схемы введения доз
Для каждой группы по весу тела определяли 3 схемы введения последовательных нарастающих доз, которые изначально были запланированы для тестирования, на основе PK моделирования со следующим обоснованием.
- Когорта дозы 1: доза, нацеленная на PK воздействие, аналогичное таковому при дозе сарилумаба 150 мг q2w, самой низкой
эффективной дозе для взрослых пациентов с RA
- Когорта дозы 2: доза, нацеленная на PK воздействие, аналогичное таковому при дозе сарилумаба 200 мг q2w, рекомендуемой схеме введения доз для взрослых пациентов с RA
- Когорта дозы 3: доза, нацеленная на PK воздействие, аналогичное таковому при дозе сарилумаба 150 мг qw, которая
приводила к самым высоким уровням воздействия в исследованиях хронического введения доз у взрослых пациентов с RA. См. таблицу 1 для получения информации о дозах по группам по весу тела и по когортам доз, которые будут использоваться в данном исследовании. См. также фиг. 7.
Дозу (мг), вводимую пациентам, будут рассчитывать на исходном уровне. Доза и соответствующий объем лекарственного препарата будут оставаться неизменными в течение 12-недельной основной фазы лечения исследования, независимо от изменения веса тела пациента. В фазе дополнительного лечения вес тела пациента будут измерять при каждом визите, и дозу будут корректировать с учетом увеличения веса тела только в том случае, если расчет показывает необходимость увеличения дозы. Доза будет ограничена 200 мг для когорты дозы 2 и 150 мг для когорты дозы 3 соответственно. Объемы сарилумаба, подлежащие инъекции, для когорт доз представлены в таблицах 2-5 ниже.
Несмотря на то, что 12-недельная основная фаза лечения начинается с визита 2 (исходный уровень - неделя 0) и продолжается до визита 12 (неделя 12) включительно, IMP на визите 12 будут вводить только для пациентов, которые собираются переходить на фазу дополнительного лечения, и после визита 12 завершаются оценки, включая оценки эффективности, безопасности и PK/PD.
Продолжительность участия в исследовании для каждого пациента
Ожидается, что общая продолжительность исследования (на одного пациента) составит до 166 недель
- До 4 недель+3 дня скрининга (до 31 дня)
- 12-недельный основной период лечения
- До 144-недельной фазы дополнительного лечения
- 6-недельное последующее наблюдение после лечения
Для всех визитов приемлемы временные рамки ± 3 дня для когорты дозы 2 (включая любую потенциальную промежуточную дозу q2w) и ± 1 день для когорты дозы 3 (включая любую потенциальную промежуточную дозу qw) с использованием дня 1 в качестве эталона, за исключением того, что
- для визита 3 (день 3), визита 4 (день 5) и визита 5 (день 8) не допускается окно визита, когда происходит отбор образцов PK.
- Визит 6 (день 12) ±1 день для всех когорт доз.
- Визит 7 (неделя 2) ±1 день для всех когорт доз.
- Визит 8 (неделя 4) ±2 дня для когорты дозы 2 и ±1 день для когорты дозы 3.
Правила прекращения лечения при нейтропении 3/4 степени при sJIA
При нейтропении 4 степени без признаков инфекции было рекомендовано временное прекращение лечения сарилумабом, которое могло быть возобновлено на основании тщательной оценки исследователем соотношения пользы и риска после того, как значение ANC стало больше 1000/мм3. Снижение ANC является фармакодинамическим классовым эффектом антагонистов интерлейкина 6 (IL-6), который наблюдается как у взрослых, так и у пациентов детского возраста, которых лечат антителами к IL-6. После повторного введения доз сарилумаба в долгосрочных исследованиях взрослых с ревматоидным артритом (RA) снижение ANC было, как правило, преходящим и не было связано с повышенным риском инфекций. У пациентов детского возраста, несмотря на то, что в настоящее время доступно ограниченное количество опубликованных данных, использование антагонистов IL-6 не было связано с повышенным риском инфекций у пациентов с нейтропенией 3/4 степени. Таблица 31 показывает возникновение нейтропении 3 степени и 4 степени по наименьшему значению количества нейтрофилов, зарегистрированному в исследовании.
Отбор пациентов
Критерии включения
1. Пациенты мужского и женского пола в возрасте ≥1 и ≤17 лет на момент скринингового визита.
2. Диагноз подтипа sJIA в соответствии с критериями классификации JIA ILAR 2001 JIA Petty, R.E. et al. 2001. J Rheumatol. 2004;31(2):390-2) со следующими признаками на скрининге:
- ≥5 активных суставов или
- ≥2 активных сустава при скрининге с лихорадкой системного JIA >37,5°C в течение 3 дней, предшествующей исходному уровню, или в течение по меньшей мере 3 из любых 7 последовательных дней во время скрининга, несмотря на глюкокортикоиды в стабильной дозе в течение по меньшей мере 3 дней.
3. Пациент с недостаточным ответом на текущее лечение, которого рассматривают в качестве кандидата на bDMARD согласно заключению исследователя.
4. Пациент, достигший установленного законом возраста согласия, или родитель(родители), или законный(законные) опекун(опекуны) подписывают с указанием даты письменное информированное согласие, одобренное комитетом по вопросам этики (EC). Согласие несовершеннолетнего пациента должно быть получено на основе местных руководств, зрелости пациента и его интеллектуальных способностей для понимания информации, связанной с исследованием. В случаях, связанных с эмансипированными или зрелыми несовершеннолетними, обладающими достаточной способностью принимать решения, или когда это иным образом разрешено законом, подписанное информированное согласие будут получать непосредственно от родителя(родителей) или законного(законных) опекуна(опекунов).
Исследуемые виды лечения
Исследуемый лекарственный препарат
Сарилумаб, mAb к IL-6R.
Состав
Лекарственный препарат сарилумаб будут предоставлять в концентрации 175 мг/мл в водном буферном носителе, pH 6,0. Лекарственный препарат будут поставлять во флаконе объемом 5 мл, заполненном 2,7 мл сарилумаба с экстрагируемым объемом 2,0 мл.
Путь(пути) введения
Сарилумаб будут вводить подкожно в живот или бедро при самостоятельной инъекции или также в предплечье (в латеральную поверхность) профессиональным или непрофессиональным лицом, осуществляющим уход. Предпочтительно, чтобы места SC инъекции чередовались между 4 квадрантами живота (кроме области пупка или талии) или бедра (спереди и сбоку).
Пациентам, которые придерживаются схемы введения доз 2 (q2w), инъекции будет выполнять профессиональное лицо, осуществляющее уход, в течение 12-недельной основной фазы лечения исследования. См. фиг. 1. В отношении пациентов, которые придерживаются схемы введения доз 3 (еженедельные инъекции), необходимо принять меры, обеспечивающие введение IMP квалифицированным персоналом центра или домашней медсестрой для доз, введение которых не запланировано в исследовательском центре. На фазе дополнительного лечения исследования, если пациенты или их родитель(родители), законный(законные) опекун(опекуны) или лицо(лица), осуществляющее(осуществляющие) уход, желали и имели возможность выполнять инъекции, домашние инъекции будут разрешаться. В этих случаях будет требоваться и проводиться обучение для подготовки и введения IMP, начиная с визита 10 (неделя 8) на основной фазе лечения. Это обучение необходимо документировать в файле исследования пациентов. Пациентам, или родителю(родителям), или законному(законным) опекуну(опекунам) разрешают вводить инъекции под наблюдением/контролем исследователя(исследователей) или представителя(представителей) на визите 10 (неделя 8), визите 11 (неделя 10) и визите 12 (неделя 12) до разрешения на домашние инъекции на фазе дополнительного лечения.
В дни, когда пациент совершает визит исследования, IMP будут вводить после клинических процедур и сбора крови. Будут предоставлять дневники для записи информации, относящейся к этим инъекциям; эти дневники будут хранить в качестве исходных данных в файлах исследования пациентов. Если лицо, осуществляющее уход, не может или не желает вводить IMP, необходимо принимать меры, обеспечивающие введение IMP квалифицированным персоналом центра для доз, введение которых не запланировано в исследовательском центре.
Схема введения доз
Сарилумаб следует вводить q2w или qw. Однако разрешается окно введения сарилумаба, которое составляет ±3 дня для когорты дозы 2 (включая любую потенциальную промежуточную дозу q2w) ±1 день для когорты дозы 3 (включая любую потенциальную промежуточную дозу qw) в соответствии с протоколом с учетом исключительных обстоятельств, например ожидаемых результатов лабораторных исследований, продолжающихся нежелательных явлений (AE), сложностей планирования пациентов, кроме визита 5 (день 8). Для визита 5 (день 8) нет окна введения. Окно введения будет составлять только ± 1 день для когорт доз 2 и 3 пациентов на визите 7 (неделя 2) и ±1 день для когорты дозы 3 или ±2 для когорты дозы 2 на визите 8 (неделя 4). Обратите внимание, что передозировку (случайная или преднамеренная) IMP определяют как по меньшей мере двойную дозу в течение интервала менее 11 дней для введения q2w и менее 6 дней для еженедельного введения.
Следующая доза сарилумаба подлежит введению q2w или qw.
Когорта дозы 2.
- Группа A (>30 кг и ≤60 кг): 3 мг/кг q2w
- Группа B (<30 кг и 210 кг): 4 мг/кг q2w.
Когорта дозы 3.
- Группа A (>30 кг и ≤60 кг): 2 мг/кг qw
- Группа B (<30 кг и 210 кг): 2,5 мг/кг qw.
Когорту дозы 3 также тестируют как схему с введением дозы один раз в две недели, но с сохранением такого же PK воздействия. Следовательно диапазон следующий:
- пациенты с весом тела менее 30 кг: от 5 до 7 мг/кг q2w (доза 2 представляет собой 4 мг/кг q2w),
- пациенты с весом тела 30 кг и более: от 4 до 6 мг/кг q2w (доза 2 представляет собой 3 мг/кг q2w)
Изменение дозы
Дозу (мг), вводимую пациентам, будут рассчитывать на исходном уровне. Доза и соответствующий объем лекарственного препарата будут оставаться неизменными в течение 12-недельной основной фазы лечения исследования, независимо от изменения веса тела пациента. В фазе дополнительного лечения вес тела пациента будут измерять при каждом визите, и дозу будут корректировать с учетом увеличения веса тела только в том случае, если расчет показывает необходимость увеличения дозы. Доза будет ограничена 200 мг для когорты дозы 2 и 150 мг для когорты дозы 3 соответственно. Подлежащие введению объемы для когорт доз 2-3 в зависимости от веса тела пациента описаны в данном документе. См. таблицы 2-5.
Упаковка и маркировка
IMP будут предоставлять в наборах для лечения пациентов, содержащих маркированные флаконы. Содержание инструкции по применению соответствует региональным нормативной спецификации и требованиям. Один набор содержит один флакон.
Условия хранения и срок хранения
Исследуемый препарат следует хранить в холодильнике при температуре от 2oC до 8oC в исследовательском центре или дома.
(12 недель)
PK
EOT+2 недели
(для пациентов,
которые
прекращают
исследуемый вид лечения
во время основной
фазы
лечения или не
собираются переходить
в фазу
дополнительного лечения
D-1 (+ 3)
(Максимум
31 день)
(±1)
(±1)
3)
3)
3)
(12 недель)
PK
EOT+2 недели
(для пациентов,
которые
прекращают
исследуемый вид лечения
во время основной
фазы
лечения или не
собираются переходить
в фазу
дополнительного лечения
D-1 (+ 3)
(максимум
31 день)
(±1)
(±1)
3)
3)
3)
(2 измерения BP в каждый запланированный момент времени)
(12 недель)
PK
EOT+2 недели
(для пациентов,
которые
прекращают
исследуемый вид лечения
во время основной
фазы
лечения или не
собираются переходить
в фазу
дополнительного лечения
D-1 (+ 3)
(максимум
31 день)
(±1)
(±1)
3)
3)
3)
(12 недель)
PK
EOT+2 недели
(для пациентов,
которые
прекращают
исследуемый вид лечения
во время основной
фазы
лечения или не
собираются переходить
в фазу
дополнительного лечения
D-1 (+ 3)
(максимум
31 день)
(±1)
(±1)
3)
3)
3)
Группа A (≥30 кг и ≤60 кг)
Группа B (<30 кг и ≥10 кг)
График 1
Группа B (<30 кг и ≥10 кг)
График 2
(12 недель)
PK
EOT+2 недели
(для пациентов,
которые
прекращают
исследуемый вид лечения
во время основной
фазы
лечения или не
собираются переходить
в фазу
дополнительного лечения
D-1 (+ 3)
(максимум
31 день)
(±1)
(±1)
3)
3)
3)
Сокращения. ALP=щелочная фосфатаза, ANA=антинуклеарные антитела, ANC=абсолютное количество нейтрофилов, BP=кровяное давление, ДНК=дезоксирибонуклеиновая кислота, D=день, EBV=вирус Эпштейна-Барр, EOT=конец лечения, ESR=скорость оседания эритроцитов, HIV=вирус иммунодефицита человека, hs-CRP=высокочувствительный C-реактивный белок, Ig=иммуноглобулин, IVRS=интерактивная система, обеспечивающая доступ путем голосового ответа, IL=интерлейкин, IMP=исследуемый лекарственный препарат, JADAS=индекс активности заболевания, представляющего собой ювенильный артрит, ACR JIA=ответ ювенильного идиопатического артрита согласно Американской коллегии ревматологов, PK=фармакокинетика, PPD=очищенное производное белка, q2w=один раз в две недели, SAE=серьезное нежелательное явление, sIL-6R=растворимый рецептор интерлейкина-6, sJIA=системный ювенильный идиопатический артрит, TB=туберкулез, V=визит, Wk=неделя.
a Для пациентов, которые прекращают исследуемый вид лечения в течение 12-недельной основной фазы лечения (во время или до визита 12), будут проводить дополнительную оценку PK через 2 недели после визита EOT (EOT+2 недели).
b Отбор образцов для определения фармакокинетики во время следующих визитов и дней: визит 3 (D3), визит 4 (D5), визит 5 (D8), визит 6 (D12) для группы A (≥30 кг); во время следующих визитов и дней: визит 3 (D3), визит 5 (D8), для графика 1 группы B (<30 кг); во время следующих визитов и дней визитов: визит 4 (D5), визит 6 (D12) для графика 2 группы B (<30 кг). Отбор образцов для определения PK можно проводить дома. Для визита 3 (D3), визита 4 (D5) и визита 5 (D8) окно для сбора образцов не допускается. Для визита 6 (D12) допускается окно ±1 день. Пожалуйста, обратитесь к дополнительному гематологическому исследованию в течение периода отбора образцов для определения PK.
c По усмотрению исследователей лабораторные исследования, упомянутые в критерии исключения 27, можно повторять посредством повторного тестирования в центральной лаборатории между скрининговым визитом и первым введением IMP, чтобы гарантировать пригодность пациента с учетом критерия исключения 27. Специальная форма согласия, одобренная на местном уровне, будет подписана пациентами, которым требуется генетическое тестирование в отношении синдрома Жильбера (согласие/согласие пациента должно быть получено до проведения данного исследования, и должны соблюдаться местные нормативные требования).
d Перед проведением всех скрининговых оценок пациент (если он/она достигли установленного законом возраста согласия в соответствии с местными нормативными требованиями), родитель(родители) или законный(законные) опекун(опекуны) должны подписать с указанием даты форму письменного информированного согласия, одобренную EC. Пациент, родитель(родители) и законный(законные) опекун(опекуны) получат информацию о цели(целях) исследования и процедурах от исследователя. Отдельные формы письменного согласия должны быть получены от родителя(родителей) или законного опекуна(опекунов), которые разрешают его/ее ребенку участвовать в дополнительном сборе образцов слюны для фармакогеномного исследования и дают разрешение спонсору оставить их оставшиеся/неиспользованные образцы крови для будущих исследований Отдельная (одобренная на местном уровне) форма информированного согласия заполняется всеми пациентами, которым требуется генетическое тестирование в отношении болезни Жильбера в соответствии с местными нормативными требованиями. Подписанные формы согласия пациентов должны быть получены от пациентов в соответствии с местными нормативными требованиями и его/ее зрелостью в понимании информации об исследовании.
e Дневник пациента для введения IMP, который необходимо заполнить для IMP, вводимого дома.
f Полное физикальное обследование будут осуществлять во время визита 1 (от D -28 до D -1, вплоть до 31 дня), визита 2 (D1, неделя 0), визита 8 (неделя 4), визита 9 (неделя 6) и визита 12 (неделя 12), где обследование включает обследование кожи, носовых полостей, глаз, ушей, дыхательной, сердечно-сосудистой, желудочно-кишечной, нервной, лимфатической и опорно-двигательной систем.
g Необязательно. На основании заключения исследователя во время скрининга могут определять титр вируса Эпштейна-Барр (EBV), включая IgG и IgM; на основании семейного и медицинского анамнеза пациента, а также заключения исследователя, во время скрининга могут определять поверхностный антиген вируса гепатита B (HBs-Ag), антитело к поверхностному антигену вируса гепатита B (HBs-Ab), общие антитела к коровому антигену вируса гепатита B (HBc-Ab) и антитело к вирусу гепатиту C (HCV-Ab). Серологическое исследование в отношении ВИЧ будут проводить только на основании заключения исследователя для пациентов с подозрением на ВИЧ.
Только для Германии: серологические исследования в отношении вирусов гепатита B и C и HIV должны проводить во время скринингового визита для всех пациентов, чтобы проверить соответствующий критерий исключения 18.
i. Кожную пробу очищенного белкового продукта (PPD) должны проводить у пациентов в возрасте ≤5 лет до визита 2 исходного уровня (D1, неделя 0). Пациентов следует обследовать в течение 48-72 часов после проведения кожной пробы PPD. Пациентам, которых не обследовали в течение 72 часов, кожную пробу следует повторить. При определении результатов кожных проб следует учитывать оценку уровня риска (контакт с TB и/или недавняя иммиграция из страны с высокой распространенностью TB). См. все подробности в критерии исключения 17. Анализ высвобождения гамма-интерферона (IFN-r), тест QuantiFERON-TB, будут проводить для пациентов в возрасте >5 лет (1). После первоначального скрининга в отношении TB, если результат PPD или QuantiFERON отрицательный, но клиническое подозрение на TB выше среднего, пациента должны повторно провести скрининг на наличие TB в любое время в ходе исследования на основе оценки исследователя. Тест Quantiferon TB будут рассматривать в более молодой группе пациентов (≤5 лет) на основании местного наличия PPD, местных нормативных требований проведения скрининга в отношении TB и заключения исследователя.
Рентгенографию грудной клетки могут проводить пациентам только в том случае, если это сочтут необходимым на основании заключения исследователя или в соответствии с местными руководствами по скринингу в отношении TB до начала биологической терапии для пациентов с JIA, у которых рентгенографию грудной клетки не проводили в течение 3 месяцев до визита 2 исходного уровня (D1, неделя 0).
j Исследуемый лекарственный препарат следует вводить q2w пациентам в когорте дозы 2 и один раз в неделю для пациентов в когорте дозы 3. Будут принимать меры для того, чтобы домашние медсестры вводили IMP для пациентов в когорте дозы 3 для промежуточных визитов (введение на дому, если возможно). За пациентами следует наблюдать в течение по меньшей мере 30 минут после введения IMP в отношении любых признаков или симптомов реакции гиперчувствительности. Пациенты, которые не достигли ответа ювенильного идиопатического артрита по Американской коллегии ревматологов (ACR JIA) 30 к концу 12-недельного основного периода лечения, будут отстранены от исследуемого вида лечения для получения стандартного лечения в соответствии с клиническим заключением исследователя. Ответ ACR JIA, включая ответ ACR30 JIA, будет предоставлен спонсором исследователю для оценки этого критерия только после V12. Для пациентов, завершивших основную фазу лечения на V12, но не продолжающих исследование в фазе дополнительного лечения по причинам, отличным от отсутствия ответа ACR30 JIA, инъекцию IMP на визите 12 проводить не будут.
Только для Германии: после первого введения IMP за пациентом необходимо наблюдать в течение по меньшей мере 1 часа для оценки местной переносимости.
k Исследуемый лекарственный препарат будут выдавать пациентам, которые находятся в когорте дозы 3, в течение основной фазы лечения.
l Дневник температуры/сыпи пациента будет включать регистрацию тимпанической температуры и сообщение о сыпи (при ее наличии) дома каждый день между скринингом и визитами исходного уровня и в течение по меньшей мере 7 дней до каждого последующего запланированного визита с оценкой базового набора эффективности ACR JIA. Температуру следует измерять минимум два раза в день в фиксированные моменты, при пробуждении и перед сном, а также в любое время при подозрении на лихорадку.
m Следует собрать дополнительные данные о росте, измеренном максимально близко к моменту времени за 1 год до исходного уровня. Рост будут измерять с помощью ростомера в исследовательских центрах во время исследования.
n Базовый набор ювенильного идиопатического артрита ACR включает общую оценку тяжести заболевания врачом, общую оценку самочувствия пациента или родителя(родителей)/опекуна(опекунов), количество суставов с активным артритом (определяется как отек, не вызванный деформацией ИЛИ ограничение движений либо с болью, либо с чувствительностью, либо с обоими признаками), количество суставов с ограничением движений, опросник оценки здоровья детей (CHAQ), hs-CRP и лихорадку (Petty, R.E. et al., International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31(2):390-2; Giannini, E.H. et al. Preliminary definition of improvement in juvenile arthritis. Arthritis Rheum. 1997;40(7):1202-9; 4. Beukelman, T. et al.,, 2011 American College of Rheumatology recommendations for the treatment of juvenile idiopathic arthritis: initiation and safety monitoring of therapeutic agents for the treatment of arthritis and systemic features. Arthritis Care Res (Hoboken). 2011;63(4):465-82. Во время скрининга будут оценивать только лихорадку, количество суставов с активным артритом и количество суставов с ограничением движений. Принцип подсчета индекса JADAS объясняется в данном документе.
o Системные признаки sJIA включают лихорадку, кратковременную эритематозную сыпь лососевого цвета, генерализованное увеличение лимфатических узлов, гепатомегалию спленомегалию, серозит и другие осложнения при скрининге, исходном уровне и визите 12. На неделе 2, неделе 4, неделе 6, неделе 8 и неделе 10 будут оценивать только лихорадку и кратковременную эритематозную сыпь лососевого цвета.
p Гематологическое исследование (кровь следует забирать ДО введения лекарственного средства): гемоглобин, гематокрит, количество эритроцитов (RBC) и морфология эритроцитов (если количество эритроцитов аномальное), количество лейкоцитов (WBC), лейкоцитарная формула (нейтрофилы, лимфоциты, моноциты, эозинофилы, базофилы), количество тромбоцитов, абсолютное количество нейтрофилов (ANC). Дополнительное гематологическое исследование должны проводить на визите 6 (день 12±1 день) для пациентов когорты дозы 2, чтобы получить результаты до визита 7 (день 15, неделя 2), до введения второй дозы сарилумаба. Для пациентов когорты дозы 3 следует провести дополнительное гематологическое исследование до или во время визита 5 (день 8), но не ранее, чем визит 4 (день 5), и результаты необходимо проанализировать перед второй инъекцией сарилумаба во время визита 5 (день 8). Дополнительное гематологическое исследование могут проводить в центральной лаборатории или в местной лаборатории, чтобы подтвердить, что ANC и количество тромбоцитов не находятся в установленных протоколом пределах для временного или окончательного прекращения приема исследуемого лекарственного средства. Если используется местная лаборатория, образец центральной лаборатории для гематологического исследования все равно должен быть собран перед введением IMP на визите 7 (день 15, неделя 2), как запланировано для когорты 2, и на визите 5 (день 8) для когорты 3. Для всех пациентов гематологическая лабораторная оценка визита 2 (день 1) должна быть рассмотрена перед введением второй дозы сарилумаба на визите 5 (день 8) для пациентов в когорте дозы 3 или на визите 7 (неделя 2, день 15) для когорты дозы 2.
q Биохимическое исследование (кровь следует забирать ДО введения лекарственного средства). Полное биохимическое исследование будут проводить во время скринингового визита, визита 2 исходного уровня (день 1, неделя 0) и визита 12 (неделя 12) или только на EOT: натрий, калий, хлор, бикарбонат, азот мочевины крови, креатинин и клиренс креатинина, скорость клубочковой фильтрации, кальций, фосфат, общий белок, альбумин, аланинаминотрансфераза (ALT), аспартатаминотрансфераза (AST), щелочная фосфатаза (ALP), лактатдегидрогеназа (LDH), общий билирубин, конъюгированный билирубин и неконъюгированный билирубин. На всех остальных визитах будут тестировать только ALT, AST, ALP, LDH, общий билирубин, конъюгированный билирубин, неконъюгированный билирубин и альбумин.
r Липидограмма натощак (кровь следует забирать ДО введения лекарственного средства): Триглицериды (TG), общий холестерин, холестерин липопротеинов высокой плотности (HDL), холестерин липопротеинов низкой плотности (LDL). Требуется, чтобы пациенты не принимали пищу по меньшей мере в течение 8 часов до исследования.
s Необязательно. Уровни HbA1c будут измерять только на основании медицинского анамнеза пациента и заключения исследователя.
t hs-CRP будут измерять в дни 3 и 8 у пациентов с весом тела ≥30 кг (группа A). Для пациентов с весом тела <30 кг (группа B) в группе графика PK 1, hs-CRP будут измерять в дни 3 и 8. Для пациентов с весом тела <30 кг (группа B) в группе графика PK 2, hs-CRP будут измерять в дни 5 и 12.
u При подозрении на синдром активации макрофагов (MAS) необходимо назначить анализ на ферритин, анализ крови (эритроциты, лейкоциты, тромбоциты и гемоглобин), анализы на AST/ALT, триглицериды и фибриноген, если это необходимо по заключению исследователя. В данном документе описаны клинические и лабораторные особенности диагностики MAS.
v Анализ мочи с помощью индикаторной полоски: удельный вес, pH, глюкоза, кровь, белок, нитриты, лейкоцитарная эстераза, билирубин. Если какой-либо параметр по анализу с помощью индикаторной полоски отклоняется от нормы, образец мочи следует отправить в центральную лабораторию для анализа. В случае положительного результата в отношении белков центральная лаборатория проводит микроскопический анализ.
w Для женщин, у которых начались менструации, во время скринингового визита в обязательном порядке проводят исследование сыворотки на наличие беременности.
x Для женщин, у которых начались менструации, исследование сыворотки на наличие беременности следует проводить на скрининге и местное исследование мочи на наличие беременности следует проводить во время визита 2 (неделя 0, день 1), визита 8 (неделя 4), визита 10 (неделя 8) и визита 12 (неделя 12). Исследование мочи на наличие беременности можно проводить местно. Наличие беременности следует проверять с помощью исследования мочи на наличие беременности до воздействия IMP и EOT.
y Образцы крови будут собирать ДО введения IMP в дни введения доз в течение периода лечения. Если у пациента наблюдают SAE, для определения концентрации сарилумаба и оценки антител к лекарственному средству (ADA) следует собирать образцы крови в момент начала и завершения события, если это возможно, или в течение короткого времени после начала и завершения/*98+-.
z Для пациентов, которые преждевременно прекращают исследуемый вид лечения во время основной фазы лечения, при оценке EOT будут измерять IL-6 и общий sIL-6R.
aa Родитель(родители) или законный(законные) опекун(опекуны) должны подписать форму письменной информации для субъекта перед забором образца слюны для фармакогеномного исследования. Образцы предпочтительнее собирать во время визита 2 исходного уровня (D1, неделя 0), но их можно собирать во время любого визита. Пациент также может подписать WSI в зависимости от его/ее возраста, местных нормативных требований и степени зрелости в отношении понимания информации об исследовании. Пациент по-прежнему пригоден для участия в исследовании, если он/она или его/ее родитель(родители) или его/ее законный(законные) опекун(опекуны) не желают, чтобы он/она участвовали в заборе образцов слюны.
(до 144 недель воздействия сарилумаба после V12)
после лечения
(6 недель)
EOT
обследованиеd
Введение (IMP)e
температуры/сыпи
пациентаf
(до 144 недель воздействия сарилумаба после V12)
после лечения
(6 недель)
EOT
sJIAi
(до 144 недель воздействия сарилумаба после V12)
после лечения
(6 недель)
EOT
менструирующих женщинn
в сыворотке крови (PK)o
Сокращения. AE=нежелательное явление, ALP=щелочная фосфатаза, ALT=аланинаминотрансфераза, ANC=абсолютное количество нейтрофилов, AST=аспартатаминотрансфераза, ANA=антинуклеарные антитела, BP=кровяное давление, ДНК=дезоксирибонуклеиновая кислота, D=день, EOS=конец исследования, EOT=конец лечения, ESR=скорость оседания эритроцитов, EBV=вирус Эпштейна-Барр, HIV=вирус иммунодефицита человека, hs-CRP=высокочувствительный C-реактивный белок, IMP=исследуемый лекарственный препарат, IVRS=интерактивная система, обеспечивающая доступ путем голосового ответа, IL=интерлейкин, JADAS=индекс активности заболевания, представляющего собой ювенильный артрит, ACR JIA=ответ ювенильного идиопатического артрита согласно Американской коллегии ревматологов, PK=фармакокинетика, PPD=очищенное производное белка, q2w=один раз в две недели, SAE=серьезное нежелательное явление, sJIA=системный ювенильный идиопатический артрит, TB=туберкулез, V=визит, Wk=неделя.
a Визит 27 является запланированным визитом конца лечения (EOT) в рамках исследования. Если пациенты преждевременно прекращают исследуемое лечение, EOT произойдет через 1 неделю после последней инъекции IMP для когорты дозы 3 и через 2 недели после последней инъекции IMP для когорты дозы 2.
b Визит 28 является запланированным визитом конца исследования (EOS) в рамках исследования. Если пациенты преждевременно прекращают исследуемое лечение, этих пациентов попросят вернуться для оценки EOS через 6 недель после визита EOT (EOT+6 недель). Этот визит EOS (EOT+6 недель) будет применим ко всем пациентам как в основной фазе лечения, так и в фазе дополнительного лечения.
c Дневник пациента для введения IMP, который необходимо заполнить для IMP, вводимого дома.
d Полное физикальное обследование будут осуществлять во время визита 15 (неделя 24), визита 18 (неделя 48), визита 20 (неделя 72), визита 22 (неделя 96), визита 24 (неделя 120), визита 26 (неделя 144), визита 27 EOT и визита 28 EOS, где обследование включает обследование кожи, носовых полостей, глаз, ушей, дыхательной, сердечно-сосудистой, желудочно-кишечной, нервной, лимфатической и опорно-двигательной систем.
e IMP следует вводить q2w пациентам в когорте дозы 2 и один раз в неделю для пациентов в когорте дозы 3. Для пациентов, которые завершили фазу дополнительного лечения, если при выбранной схеме введения доз введение производят один раз в две недели (q2w), то последнюю инъекцию IMP будут производить на неделе 154; если при выбранной схеме введения доз введение производят один раз в неделю (qw), то последнюю инъекцию IMP будут производить на неделе 155. Пациенты будут проходить оценку EOT на визите 27 (неделя 156). Для пациентов, которые преждевременно прекратили исследуемый вид лечения во время фазы дополнительного лечения, будет предпочтительнее пройти оценку EOT через 2 недели после последней инъекции IMP для пациентов когорты дозы 2 и через 1 неделю после последней инъекции IMP для пациентов когорты дозы 3. В случае, если пациент не может придерживаться запланированного графика, визит EOT будет использован для оценки при следующем визите, определенном протоколом, после последней инъекции IMP. Пациентов просят вернуться в центр для оценки EOS через 6 недель после визита EOT. За пациентами следует наблюдать в течение по меньшей мере 30 минут после введения IMP в отношении любых признаков или симптомов реакции гиперчувствительности. Во время визита 27 (неделя 156) инъекции IMP производить не будут.
f Дневник температуры/сыпи пациента будет включать регистрацию тимпанической температуры и сообщение о сыпи (при ее наличии) дома в течение по меньшей мере 7 дней до каждого последующего запланированного визита с оценкой базового набора эффективности ACR JIA. Температуру следует измерять минимум два раза в день в фиксированные моменты, при пробуждении и перед сном, а также в любое время при подозрении на лихорадку.
g Следует собрать дополнительные данные о росте, измеренном максимально близко к моменту времени за 1 год до исходного уровня. Рост будут измерять с помощью ростомера в исследовательских центрах во время исследования.
h Базовый набор для ACR JIA включает общую оценку тяжести заболевания врачом, общую оценку самочувствия пациентом или родителем/законным опекуном, количество суставов с активным артритом, который определяется как отек в суставе, не вызванный деформацией ИЛИ ограничение движений либо с болью, либо с чувствительностью, либо с обоими признаками), количество суставов с ограничением движений, опросник оценки здоровья детей (CHAQ), hs-CRP и лихорадку. Принцип подсчета индекса JADAS объясняется в данном документе.
o Системные признаки sJIA включают лихорадку, кратковременную эритематозную сыпь лососевого цвета, генерализованное увеличение лимфатических узлов, гепатомегалию спленомегалию, серозит и другие осложнения на неделе 156 (визит EOT). На неделе 24, неделе 48, неделе 72, неделе 96, неделе 120, неделе 144 и неделе 162 (визит EOS) будут оценивать только лихорадку и кратковременную эритематозную сыпь лососевого цвета.
j Гематологическое исследование (кровь следует забирать ДО введения лекарственного средства): гемоглобин, гематокрит, количество эритроцитов (RBC) и морфология эритроцитов (если количество эритроцитов аномальное), лейкоцитарная формула (WBC) (нейтрофилы, лимфоциты, моноциты, эозинофилы, базофилы), количество тромбоцитов, абсолютное количество нейтрофилов (ANC).
k Биохимическое исследование (кровь следует забирать ДО введения лекарственного средства). Будут тестировать ALT, AST, ALP, лактатдегидрогеназу (LDH), общий билирубин, конъюгированный билирубин, неконъюгированный билирубин и альбумин. Полное биохимическое исследование должны провести на визите 27 EOT.
l Липидограмма (кровь следует забирать ДО введения лекарственного средства). Триглицериды (TG), общий холестерин, холестерин липопротеинов высокой плотности (HDL), холестерин липопротеинов низкой плотности (LDL). Требуется, чтобы пациенты не принимали пищу по меньшей мере в течение 8 часов до исследования.
m При подозрении на MAS необходимо назначить анализ на ферритин, анализ крови (эритроциты, лейкоциты, тромбоциты и гемоглобин), анализы на AST/ALT, триглицериды и фибриноген, если это необходимо по заключению исследователя. Обратитесь к клиническим и лабораторным особенностям диагностики MAS, описанным в данном документе.
n Для женщин, у которых начались менструации, исследование мочи на наличие беременности следует проводить до введения инъекции IMP при каждом запланированном визите и на EOT. Исследование мочи на наличие беременности можно проводить местно.
o Образцы крови будут собирать ДО введения IMP в дни введения доз в течение периода лечения. Если у пациента наблюдают SAE, для определения концентрации сарилумаба и оценки антител к лекарственному средству (ADA) следует собирать образцы крови в момент начала и завершения события, если это возможно, или в течение короткого времени после начала и завершения.
(в общем до 144 недель воздействия сарилумаба после V12)
наблюдение
после лечения
(6 недель)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
225
(±1
или 3)
281
(±1
или 3)
337
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(EOT
+ 6 недель)
0
2
4
6
8
12
16
20
24
32
40
48
60
72
84
96
108
120
132
между последним визитом пациента в течение периода приема лечения и EOT
+ 6 недель)
(в общем до 144 недель воздействия сарилумаба после V12)
наблюдение
после лечения
(6 недель)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
225
(±1
или 3)
281
(±1
или 3)
337
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(EOT
+ 6 недель)
0
2
4
6
8
12
16
20
24
32
40
48
60
72
84
96
108
120
132
между последним визитом пациента в течение периода приема лечения и EOT
+ 6 недель)
(в общем до 144 недель воздействия сарилумаба после V12)
наблюдение
после лечения
(6 недель)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
225
(±1
или 3)
281
(±1
или 3)
337
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(EOT
+ 6 недель)
0
2
4
6
8
12
16
20
24
32
40
48
60
72
84
96
108
120
132
между последним визитом пациента в течение периода приема лечения и EOT
+ 6 недель)
(в общем до 144 недель воздействия сарилумаба после V12)
наблюдение
после лечения
(6 недель)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
225
(±1
или 3)
281
(±1
или 3)
337
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(±1
или 3)
(EOT
+ 6 недель)
0
2
4
6
8
12
16
20
24
32
40
48
60
72
84
96
108
120
132
между последним визитом пациента в течение периода приема лечения и EOT
+ 6 недель)
Сокращения. AE=нежелательное явление, ALP=щелочная фосфатаза, ANA=антинуклеарные антитела, ANC=абсолютное количество нейтрофилов, BP=кровяное давление, ДНК=дезоксирибонуклеиновая кислота, D=день, EOT=конец лечения, EOS=конец исследования, ESR=скорость оседания эритроцитов, hs-CRP=высокочувствительный C-реактивный белок, IMP=исследуемый лекарственный препарат, IVRS=интерактивная система, обеспечивающая доступ путем голосового ответа,
JADAS=индекс активности заболевания, представляющего собой ювенильный артрит, ACR JIA=ответ ювенильного идиопатического артрита согласно Американской коллегии ревматологов, PK=фармакокинетика, PPD=очищенное производное белка, SAE=серьезное нежелательное явление, sJIA=системный ювенильный идиопатический артрит, TB=туберкулез, V=визит, Wk=неделя.
a Визит 27 является запланированным визитом конца лечения (EOT) в рамках исследования. Если пациенты преждевременно прекращают исследуемое лечение, EOT произойдет через 1 неделю после последней инъекции IMP для когорты дозы 3 и через 2 недели после последней инъекции IMP для когорты дозы 2.
b Визит 28 является запланированным визитом конца исследования (EOS) в рамках исследования. Если пациенты преждевременно прекращают исследуемое лечение, этих пациентов попросят вернуться для оценки EOS через 6 недель после визита EOT (EOT+6 недель). Этот визит EOS (EOT+6 недель) будет применим ко всем пациентам как в основной фазе лечения, так и в фазе дополнительного лечения.
c Дневник пациента для введения IMP, который необходимо заполнить для IMP, вводимого дома.
d Полное физикальное обследование включает обследование кожи, носовых полостей, глаз, ушей, дыхательной, сердечно-сосудистой, желудочно-кишечной, нервной, лимфатической и опорно-двигательной систем
e Исследуемый лекарственный препарат следует вводить q2w пациентам в когорте дозы 2 и один раз в неделю для пациентов в когорте дозы 3. Для пациентов, которые завершили фазу дополнительного лечения, если при выбранной схеме введения доз введение производят один раз в две недели (q2w), то последнюю инъекцию IMP будут производить за 2 недели до EOT; если при выбранной схеме введения доз введение производят один раз в неделю (qw), то последнюю инъекцию IMP будут производить за 1 неделю до EOT. Пациенты будут проходить оценку EOT на визите 27 (неделя 156). Для пациентов, которые преждевременно исследование во время фазы дополнительного лечения, будет предпочтительнее пройти оценку EOT через 2 недели после последней инъекции IMP для пациентов когорты дозы 2 и через 1 неделю после последней инъекции IMP для пациентов когорты дозы 3. В случае, если пациент не может придерживаться запланированного графика, визит EOT будет использован для оценки при следующем визите, определенном протоколом, после последней инъекции IMP. Пациентов просят вернуться в центр для оценки EOS через 6 недель после визита EOT. За пациентами следует наблюдать в течение по меньшей мере 30 минут после введения IMP в отношении любых признаков или симптомов реакции гиперчувствительности. Во время визита 27 (неделя 156) инъекции IMP производить не будут.
f Дневник температуры/сыпи пациента будет включать регистрацию тимпанической температуры и сообщение о сыпи (при ее наличии) дома в течение по меньшей мере 7 дней до каждого последующего запланированного визита с оценкой базового набора эффективности ACR JIA. Температуру следует измерять минимум два раза в день в фиксированные моменты, при пробуждении и перед сном, а также в любое время при подозрении на лихорадку.
g Следует собрать дополнительные данные о росте, измеренном максимально близко к моменту времени за 1 год до исходного уровня. Рост будут измерять с помощью ростомера в исследовательских центрах во время исследования.
h Базовый набор ювенильного идиопатического артрита ACR включает общую оценку тяжести заболевания врачом, общую оценку самочувствия пациентом или родителем/законным опекуном, количество суставов с активным артритом, который определяется как отек в суставе, не вызванный деформацией ИЛИ ограничение движений либо с болью, либо с чувствительностью, либо обоими признаками, количество суставов с ограничением движений, опросник оценки здоровья детей (CHAQ), hs-CRP и лихорадку. Принцип подсчета индекса JADAS объясняется в данном документе.
i Будут оценивать только лихорадку и кратковременную эритематозную сыпь лососевого цвета.
j Гематологическое исследование (кровь следует забирать ДО введения лекарственного средства): гемоглобин, гематокрит, количество эритроцитов (RBC) и морфология эритроцитов (если количество эритроцитов аномальное), лейкоцитарная формула (WBC) (нейтрофилы, лимфоциты, моноциты, эозинофилы, базофилы), количество тромбоцитов, абсолютное количество нейтрофилов (ANC).
k Биохимическое исследование (кровь следует забирать ДО введения лекарственного средства). Будут тестировать ALT, AST, ALP, лактатдегидрогеназу (LDH), общий билирубин, конъюгированный билирубин, неконъюгированный билирубин и альбумин. Полное биохимическое исследование должны провести на визите 27 EOT.
l Липидограмма (кровь следует забирать ДО введения лекарственного средства). Триглицериды (TG), общий холестерин, холестерин липопротеинов высокой плотности (HDL), холестерин липопротеинов низкой плотности (LDL). Требуется, чтобы пациенты не принимали пищу по меньшей мере в течение 8 часов до исследования.
m При подозрении на MAS необходимо назначить анализ на ферритин, анализ крови (эритроциты, лейкоциты, тромбоциты и гемоглобин), анализы на AST/ALT, триглицериды и фибриноген, если это необходимо по заключению исследователя.
n Для женщин, у которых начались менструации, исследование мочи на наличие беременности следует проводить до введения инъекции IMP при запланированных визитах, упомянутых в схеме исследования выше. Исследование мочи на наличие беременности можно проводить местно.
o Образцы крови будут собирать ДО введения IMP в дни введения доз в течение периода лечения. Если у пациента наблюдают SAE, для определения концентрации сарилумаба и оценки антител к лекарственному средству (ADA) следует собирать образцы крови в момент начала и завершения события, если это возможно, или в течение короткого времени после начала и завершения.
Процедура обработки образцов для определения фармакокинетики
Чрезвычайно важно собрать все образцы крови как можно ближе к дням, указанным в протоколе (см. график отбора образцов в схеме исследования в таблице 17). Причины пропущенных или утерянных образцов крови необходимо документировать. Могут использоваться специальные процедуры для сбора, хранения и транспортировки сыворотки крови. См. таблицу 20.
Условия транспортировки
сыворотки крови
80°C (предпочтительно)
В сухом льду
В сухом льду
a Для визита исследования с забором крови для образцов для определения как PK, так и антител к сарилумабу (например, визит 2 исходного уровня) рекомендуется взять 1 мл крови. Сыворотку крови разделят поровну на 2 аликвоты для получения одной аликвоты сыворотки для определения PK и одной аликвоты сыворотки для определения антител к сарилумабу.
Биоаналитический способ
Уровни функционального сарилумаба и антител к сарилумабу в сыворотке крови будут определять с использованием утвержденных биоаналитических способов.
Фармакокинетические параметры
Модель PopPK будут разрабатывать с использованием нелинейного моделирования со смешанными эффектами для описания PK профиля сарилумаба. Следующие PK параметры будут рассчитывать с использованием модели PopPK для функционального сарилумаба в сыворотке крови. PK параметры будут включать без ограничений следующее: Cmax, Ctrough, tmax и AUC0-τ.
среднее значение (SD)
(2,000)
*ANC=абсолютное количество нейтрофилов; bDMARD=биологическое DMARD; CHAQ-DI=индекс инвалидизации опросника оценки здоровья детей; CRP=C-реактивный белок; csDMARD=традиционное синтетическое DMARD; DMARD=модифицирующее заболевание противоревматическое лекарственное средство; ESR=скорость оседания эритроцитов; GC=глюкокортикоид; JADAS-27 CRP=индекс активности заболевания, представляющего собой ювенильный артрит, оценка 27 суставов с С-реактивным белком; JIA=ювенильный идиопатический артрит; n=общее количество пациентов, которые соответствовали критерию хотя бы один раз во время лечения; QW=каждую неделю; Q2W=каждые 2 недели; RF=ревматоидный фактор; SD=стандартное отклонение; VAS=визуальная аналоговая шкала; yrs=годы.
†Доза 1, 2,0 мг/кг Q2W для пациентов весом 30-60 кг и 2,5 мг/кг Q2W для пациентов весом 10-<30 кг.
‡Доза 2, 3,0 мг/кг Q2W для пациентов весом 30-60 кг и 4,0 мг/кг Q2W для пациентов весом 10-<30 кг.
§Доза 3, 2,0 мг/кг QW для пациентов весом 30-60 кг и 2,5 мг/кг QW для пациентов весом 10-<30 кг.
*AUC0-τ=площадь под кривой концентрации в сыворотке крови в зависимости от времени в течение интервала между дозами τ, равного 2 неделям (схема Q2W) или 1 неделе (схема QW); Cmax=максимальная наблюдаемая концентрация в сыворотке; Ctrough=концентрация, наблюдаемая перед введением средства лечения во время повторного введения доз; CV% = коэффициент вариации; n=общее количество пациентов, которые соответствовали критерию хотя бы один раз во время лечения; QW=один раз в неделю; Q2W=один раз в 2 недели; SC=подкожно.
†Схемы введения доз Q2W тестировали в течение недель 10-12; схемы введения доз QW тестировали в течение недель 11-12.
(30-60 кг)
(10-<30 кг)
*AE=Нежелательные явления, возникшие в ходе лечения; ALT=аланинаминотрансфераза; n=общее количество пациентов, которые соответствовали критерию хотя бы один раз во время лечения; nE=количество явлений; PY=пациенто-годы; QW=каждую неделю; Q2W=каждые 2 недели; SAE=серьезное AE. Все термины в соответствии с MedDRA 21.0.
†Доза 1, 2,0 мг/кг Q2W для пациентов весом 30-60 кг и 2,5 мг/кг Q2W для пациентов весом 10-<30 кг.
‡Доза 2, 3,0 мг/кг Q2W для пациентов весом 30-60 кг и 4,0 мг/кг Q2W для пациентов весом 10-<30 кг.
§Доза 3, 2,0 мг/кг QW для пациентов весом 30-60 кг и 2,5 мг/кг QW для пациентов весом 10-<30 кг.
≥500-1000 клеток/мм3
<500 клеток/мм3
*n=общее количество пациентов, которые соответствовали критерию хотя бы один раз во время лечения; QW=каждую неделю; Q2W=каждые 2 недели.
†Доза 1, 2,0 мг/кг Q2W для пациентов весом 30-60 кг и 2,5 мг/кг Q2W для пациентов весом 10-<30 кг.
‡Доза 2, 3,0 мг/кг Q2W для пациентов весом 30-60 кг и 4,0 мг/кг Q2W для пациентов весом 10-<30 кг.
§Доза 3, 2,0 мг/кг QW для пациентов весом 30-60 кг и 2,5 мг/кг QW для пациентов весом 10-<30 кг.
*nE=количество явлений; PY=пациенто-годы; QW=каждую неделю; Q2W=каждые 2 недели. Все термины в соответствии с MedDRA 21.0.
†Доза 1, 2,0 мг/кг Q2W для пациентов весом 30-60 кг и 2,5 мг/кг Q2W для пациентов весом 10-<30 кг.
‡Доза 2, 3,0 мг/кг Q2W для пациентов весом 30-60 кг и 4,0 мг/кг Q2W для пациентов весом 10-<30 кг.
§Доза 3, 2,0 мг/кг QW для пациентов весом 30-60 кг и 2,5 мг/кг QW для пациентов весом 10-<30 кг.
*ACR=Американская коллегия ревматологов; BL=исходный уровень; CHAQ-DI=индекс инвалидизации опросника оценки здоровья детей; CRP=C-реактивный белок; ACR30 JIA/70/90=ответ ювенильного идиопатического артрита, составляющий 30/70/90% согласно Американской коллегии ревматологов; QW=каждую неделю; Q2W=каждые 2 недели; SE=стандартная ошибка; VAS=визуальная аналоговая шкала.
†Доза 1, 2,0 мг/кг Q2W для пациентов весом 30-60 кг и 2,5 мг/кг Q2W для пациентов весом 10-<30 кг.
‡Доза 2, 3,0 мг/кг Q2W для пациентов весом 30-60 кг и 4,0 мг/кг Q2W для пациентов весом 10-<30 кг.
§Доза 3, 2,0 мг/кг QW для пациентов весом 30-60 кг и 2,5 мг/кг QW для пациентов весом 10-<30 кг.
-Группа по весу тела A: 30-60 кг, группа по весу тела B: 10-<30 кг.
*CRP=C-реактивный белок; JADAS-27-CRP=индекс активности заболевания, представляющего собой ювенильный артрит с подсчетом 27 суставов и CRP; LDA=низкая активность заболевания; n=общее количество пациентов, которые соответствовали критерию хотя бы один раз во время лечения; QW=каждую неделю; Q2W=каждые 2 недели; VAS=визуальная аналоговая шкала.
†Доза 1, 2,0 мг/кг Q2W для пациентов весом 30-60 кг и 2,5 мг/кг Q2W для пациентов весом 10-<30 кг.
‡Доза 2, 3,0 мг/кг Q2W для пациентов весом 30-60 кг и 4,0 мг/кг Q2W для пациентов весом 10-<30 кг.
§Доза 3, 2,0 мг/кг QW для пациентов весом 30-60 кг и 2,5 мг/кг QW для пациентов весом 10-<30 кг.
Группа по весу тела A: 30-60 кг, группа по весу тела B: 10-<30 кг.
**Один пациент не выполнил все компоненты оценки JADAS-27-CRP на неделе 12 и был удален.
††Критерии Уоллиса определены как общая оценка врача по VAS <1/10, отсутствие активного артрита, отсутствие активного увеита и CRP <10 мг/л.
Вторичная(вторичные) конечная(конечные) точка(точки)
12-недельная основная фаза лечения
Следующие параметры будут анализировать на 12-недельной открытой основной фазе лечения.
- Безопасность
o Неблагоприятные события, показатели жизненно важных функций, физикальное обследование, лабораторные показатели
o Оценка приемлемости (местная переносимость)
- Эффективность
O Частота ответов на терапию ACR30 JIA/50/70/90/100 (при отсутствии лихорадки) на неделе 12
o Изменение по сравнению с исходным уровнем в отдельных компонентах ACR JIA на неделе 12
o Изменение по сравнению с исходным уровнем индекса-27 активности заболевания, представляющего собой ювенильный артрит (JADAS), на неделе 12
O Изменения в использовании глюкокортикоидов, которые будут оценивать описательно с использованием линейного графика эквивалентной преднизону дозы глюкокортикоидов от исходного уровня до конца 12-недельного основного периода лечения для каждого отдельного пациента по когортам доз и по группам по весу тела
- Фармакодинамика
O Изменения в биомаркерах, связанных с IL-6 (например, изменение уровней высокочувствительного С-реактивного белка [hs-CRP], IL-6, sIL-6R)
Фаза дополнительного лечения
- Безопасность
o Неблагоприятные события, показатели жизненно важных функций, физикальное обследование, лабораторные показатели
o Оценка приемлемости (местная переносимость)
- Эффективность
o Частота ответов ACR JIA 30/50/70/90/100 (при отсутствии лихорадки) на неделях 24, 48 и каждые 24 недели до конца исследования. Изменение по сравнению с исходным уровнем индекса-27 активности заболевания, представляющего собой ювенильный артрит, на неделях 24, 48 и каждые 24 недели до последнего визита
o - Изменение по сравнению с исходным уровнем в отдельных компонентах ACR JIA на неделях 24 и 48 и каждые 24 недели до конца исследования
o - Доля пациентов, получающих глюкокортикоиды, по категориям доз (эквивалентная преднизону доза глюкокортикоидов 20,5 мг/кг, 20,2 мг/кг и <0,5 мг/кг, <0,2 мг/кг) на неделях 24, 48 и каждые 24 недели до конца исследования по сравнению с исходным уровнем
o - Доля пациентов, не принимающих глюкокортикоиды и у которых не наблюдают обострений JIA на неделях 24, 48 и каждые последующие 24 недели до конца исследования
O Изменения в использовании глюкокортикоидов, которые будут оценивать описательно с использованием линейного графика эквивалентной преднизону дозы глюкокортикоидов от исходного уровня до конца периода лечения для каждого отдельного пациента по когортам доз и по группам по весу тела
Исследовательская конечная точка
12-недельный основной период лечения
- Доля пациентов с лихорадкой всвязи с системным JIA на исходном уровне, у которых лихорадку не наблюдали на неделе 12
- Доля пациентов с сыпью всвязи с системным JIA на исходном уровне, у которых сыпь не наблюдали на неделе 12
Фаза дополнительного лечения
· Оценка роста в годы 1, 2 и 3, включая показатели скорости роста и стандартного отклонения роста (SD), для пациентов, которые никогда не получали гормон роста и не достигли стадии 5 по Таннеру к концу первого года лечения.
· Доля пациентов с лихорадкой на исходном уровне, у которых лихорадку не наблюдали на неделях 24, 48 и каждые 24 недели до конца исследования.
Доля пациентов с сыпью всвязи с системным JIA на исходном уровне, у которых сыпь не наблюдали на неделях 24, 48 и каждые 24 недели до конца исследования.
Конечные точки оценки эффективности
Ответ ACR JIA
В данном исследовании будет использоваться рейтинговая шкала ACR JIA для оценки признаков и симптомов. ACR JIA 30/50/70/90/100 будут оценивать на неделях 12, 24, 48, 72, 96, 120, 144 и 156 (EOT). Ответ ACR JIA 30/50/70/90/100 (при отсутствии лихорадки) определяют как пациента, у которого 3 из 6 переменных базового набора улучшились по меньшей мере на 30%/50%/70%/90%/100% по сравнению с исходным уровнем, при этом не более 1 из оставшихся переменных ухудшились более чем на 30%.
Базовый набор для ACR JIA включает 6 переменных плюс лихорадку для sJIA.
- Общая оценка активности заболевания врачом
- Оценка общего самочувствия пациентом/родителем
- Функциональные способности, определяемые опросником оценки здоровья детей (CHAQ)
- Количество суставов с активным артритом (от 0 до 71 сустава)
- Количество суставов с ограничением движений (от 0 до 67 суставов)
- Высокочувствительный C-реактивный белок
- Лихорадка (в течение предшествующих 7 дней)
- Базовый набор для ACR JIA будут оценивать при каждом визите в исследовательский центр, за исключением визита 3 (день 3), визита 4 (день 5), визита 5 (день 8), визита 6 (день 12) и визита EOS. Температуру следует измерять за 7 дней до оценки ACR.
Оценки переменных базового набора для ACR30 JIA и изменения по сравнению с исходным уровнем для каждой когорты доз и группы по весу тела будут обобщены по визитам с использованием средних значений, стандартных ошибок и соответствующих 95% CI. Ответы ACR30 JIA/50/70/90/100 с использованием ESR вместо hs-CRP также будет обобщены с использованием тех же методов.
Общая оценка активности заболевания врачом
Общую оценку активности заболевания врачом будут проводить во время скрининга, на исходном уровне перед введением IMP и при каждом визите в центр до визита EOT. Исследователя попросят оценивать активность заболевания пациента по горизонтальной визуально-аналоговой шкале (VAS) длиной 100 мм с ориентирами, где 0 считается наилучшей активностью заболевания, а 100 считается наихудшей активностью заболевания.
Оценка общего самочувствия пациентом/родителем
Оценку общего самочувствия пациентом/родителем будут измерять с помощью горизонтальной VAS длиной 100 мм во время скрининга, на исходном уровне перед введением дозы и при каждом визите в центр до визита EOT. Форму необходимо просить заполнять пациента или одного и того же родителя или того же опекуна, чтобы обеспечить постоянство.
Опросник оценки здоровья детей (CHAQ)
CHAQ представляет собой опрос или инструмент для самостоятельного заполнения для детей ≥8 лет и для заполнения родителем/доверенным лицом для детей младше 8 лет. CHAQ представляет собой общий показатель состояния здоровья детей в возрасте от 1 до 19 лет. Коэффициент корреляции Спирмена между значениями индекса инвалидизации, полученными из опросников, заполняемых родителями, и значениями индекса инвалидизации, полученными из опросников, заполняемых другими детьми (>8 лет), составил 0,84 (n=29; P <0,001), что свидетельствует о том, что родители могут давать точные ответы за своих детей. Очевидную валидность инструмента оценивали группой из 20 медицинских работников и родителей 22 здоровых детей. Всего оценка состоит из 43 пунктов, и ее выполнение занимает приблизительно 10 минут. CHAQ будут заполнять перед введением доз IMP и в последующие моменты времени. Период оценки составляет 1 неделю (Klepper, S.E. et al.. 2003 Arthritis Rheum. 49(3):435-43; Singh, G. et al., 1994 Arthritis Rheum. 37(12):1761-9). В этом случае фактическую дату и данные CHAQ будут регистрировать в eCRF.
Медианные баллы по шкале CHAQ, соответствующие инвалидизации легкой степени, от легкой до умеренной степени и умеренной степени, о которых сообщали при создании CHAQ, составляли 0,13, 0,63 и 1,75 соответственно (Dempster, H. et al., 2001 Arthritis Rheum. 44(8):1768-7).
Чтобы устранить несоответствия, которые могли быть внесены в результате роста и развития, родителей просят отмечать только те трудности, которые возникли из-за болезни (например, если ребенок не может выполнять какое-либо действие, потому что его/ее возраст слишком мал, ответ отмечают как "не применимо"). Варианты ответа, оценивающие трудности, основываются на 5-балльной шкале Лайкерта (0=без каких-либо трудностей, 1=с некоторыми трудностями, 2=с БОЛЬШИМИ трудностями, 3=невозможно выполнить и 4=неприменимо).
Часть 1 CHAQ представляет собой индекс инвалидизации, который содержит 41 пункт, оценивающий способность функционировать в повседневной жизни. Они далее разделены на 8 следующих подшкал/сфер. 1. Одевание и уход за внешностью; 2. подъем; 3. прием пищи; 4. ходьба; 5. гигиена; 6. дотягивание; 7. хват и 8. активная деятельность. Для каждой сферы оценивали (a) степень сложности выполнения повседневных функций (пункты 6-9, 12-13, 16-18, 21-22, 34-38, 41-44, 47-51 и 54-58); (b) необходимость использования специальных вспомогательных средств или устройств (пункты 24-27, 60-62) и (c) необходимость помощи со стороны другого человека (пункты 29-30, 64-65).
- Чтобы рассчитать индекс инвалидизации опросника оценки здоровья детей (CHAQ-DI), сначала рассчитывается балл по каждой сфере.
o Вопрос с наивысшей оценкой определяет балл для этой функциональной области
o Если используются вспомогательные средства или устройства или требуется помощь для выполнения задач в определенной области, для соответствующей функциональной области регистрируют минимальный балл 2
o 8 подшкал/сфер усредняют для расчета среднего балла, который представляет собой индекс инвалидизации (с диапазоном от 0 до -3).
Более низкие значения индекса инвалидизации указывают на лучшее состояние здоровья/лучшее качество жизни, связанное со здоровьем (HRQL)/меньшее количество признаков и симптомов, в то время как более высокие значения индекса инвалидизации указывают на худшее состояние здоровья/худшее качество жизни, связанное со здоровьем (HRQL)/большее количество признаков и симптомов. Минимальным клинически значимым улучшением индекса инвалидизации по CHAQ является снижение балла на 0,13. Минимальным клинически значимым ухудшением индекса инвалидизации по CHAQ является изменение медианного значения на 0,75 (20, 19).
Часть 2 CHAQ представляет собой индекс дискомфорта, а часть 3 CHAQ представляет собой показатель состояния здоровья; оба измеряются на отдельных 15-сантиметровых шкалах.
Индекс дискомфорта CHAQ определяется степенью боли за последнюю неделю, оцененной по VAS (с ориентирами "0 - нет боли" и "100 - очень сильная боль").
· Для расчета индекса дискомфорта по CHAQ (пункт 67)
- необходимо измерить расстояние от левого края VAS в пункте 67 до отметки респондента и умножить на 0,2. Диапазон составляет от 0 до 3. Значение индекса дискомфорта можно перемасштабировать по шкале от 0 до 100.
Часть 3 CHAQ, показатель состояния здоровья, измеряет общую оценку заболевания пациентом или родителем.
· Для расчета состояния здоровья по CHAQ (пункт 69)
- необходимо измерить расстояние от левого края VAS в пункте 69 до отметки респондента и умножить на 0,2. Диапазон составляет от 0 до 3. Значение состояния здоровья можно перемасштабировать по шкале от 0 до 100.
Количество суставов с активным артритом и количество суставов с ограниченными движениями
Активный сустав определяют как сустав с:
- отеком в суставе, не вызванным деформацией ИЛИ
- ограничением движений, либо с болью, либо с чувствительностью
Семьдесят один (71) сустав будут оценивать в отношении активного заболевания путем подсчета количества активных суставов с отеком, не вызванным деформацией ИЛИ ограничением движений, либо с болью, либо с болезненностью, либо с обоими признаками (Bazso, A. et al., 2009 J Rheumatol. 36(1):183-90; National Cholesterol Education Program (NCEP): highlights of the report of the expert panel on blood cholesterol levels in children and adolescents. Pediatrics. 1992;89(3);495-501).
Шейный отдел позвоночника (считается как 1 сустав), височно-нижнечелюстной (2 сустава, справа и слева), грудино-ключичный (2 сустава), акромиально-ключичный (2 сустава), плечевой (2 сустава), локтевой (2 сустава), запястный (2 сустава), пястно-фаланговый (всего 10 суставов, по 5 с каждой стороны), проксимальный межфаланговый (всего 10 суставов, по 5 с каждой стороны), дистальный межфаланговый (всего 8 суставов, 4 с каждой стороны), тазобедренный (2 сустава), коленный (2 сустава), голеностопный (2 сустава), подтаранный (2 сустава), предплюсне-плюсневый (2 сустава), плюснефаланговый (10 суставов, 5 с каждой стороны) суставы и межфаланговый сустав стопы (10 суставов, по 5 с каждой стороны), всего 71 сустав.
Шестьдесят семь (67) суставов, которые будут оценивать в отношении ограничения движений, представляют собой те же суставы, что и те, которые исследовали в отношении активного заболевания, за исключением грудино-ключичного (n=2) и акромиально-ключичного (n=2). Формальный подсчет суставов будет проводить обученный эксперт по оценке. Чувствительность суставов определяют как боль, вызванную давлением в суставах большим и указательным пальцами эксперта по оценке.
Высокочувствительный C-реактивный белок
Высокочувствительный С-реактивный белок будут оценивать на визите 1 (от дня -28 до дня -1, вплоть до 31 дня), визите 2 исходного уровня (неделя 0, день 1), визите 7 (неделя 2), визите 8 (неделя 4), визите 9 (неделя 6), визите 10 (неделя 8), визите 11 (неделя 10), визите 12 (неделя 12) и на каждом визите в фазе дополнительного лечения с визита 13 (неделя 16) до визита 27 EOT (неделя 156) для пациентов, которые продолжают принимать выбранную дозу; с визита 101 (неделя 0) до визита 108 (неделя 12), на визите 109 (неделя 24), визите 112 (неделя 48), визите 114 (неделя 72), визите 116 (неделя 96), визите 118 (неделя 120) и визите 27 EOT. Для пациентов с весом тела <30 кг (группа B) в группе графика PK 1, hs-CRP будут измерять в дни 3 и 8. Для пациентов с весом тела <30 кг (группа B) в группе графика PK 2, hs-CRP будут измерять в дни 5 и 12. Уровни высокочувствительного С-реактивного белка прямо коррелируют с активностью IL-6R. Ожидается, что активные схемы введения доз будут оказывать эффект резкого снижения уровня CRP.
индекс активности заболевания, представляющего собой ювенильный артрит
JADAS включает 4 показателя.
1. Общая оценка активности заболевания врачом, измеренная с помощью VAS длиной 10 см, где 0=отсутствие активности и 10=максимальная активность.
2. Общая оценка самочувствия родителем/пациентом, измеренная с помощью VAS длиной 10 см, где 0=очень хорошо, а 10=очень плохо.
3. Количество суставов с активным заболеванием.
4. Скорость оседания эритроцитов нормализована по шкале от 0 до 10 по следующей формуле:
[ESR(мм/час)-20]/10
Перед расчетом значение ESR <20 мм/час преобразовано в 0, а значения ESR >120 мм/час преобразовано в 120.
JADAS будут рассчитывать как простую линейную сумму баллов по его 4 компонентам (Consolaro, A. et al., 2009 Arthritis Rheum. 61(5):658-623). Общую оценка и изменение по сравнению с исходным уровнем JADAS-27 будут обобщать по визитам (включая количество, среднее значение, стандартную ошибку, SD, медианное значение, минимум и максимум) для каждой когорты дозы и группы по весу тела (если это возможно).
JADAS-27 включает следующие суставы: шейный отдел позвоночника, локтевые, запястные суставы, пястно-фаланговые суставы (от первого до третьего), проксимальные межфаланговые суставы, тазобедренные, коленные и голеностопные суставы. JADAS оказался действенным инструментом для оценки активности заболевания при JIA и был потенциально применим в стандартном клиническом лечении, обследовании путем наблюдения и клинических испытаниях (23). JADAS-27 будут рассчитывать на визите 12 (неделя 12), визите 15 (неделя 24), визите 18 (неделя 48),
визите 20 (неделя 72), визите 22 (неделя 96), визите 24 (неделя 120), визите 26 (неделя 144) и визите 27 EOT (неделя 156) для пациентов, которые продолжают принимать выбранную дозу; с визита 101 (неделя 0) до визита 106 (неделя 12), на визите 109 (неделя 24), визите 112 (неделя 48), визите 114 (неделя 72), визите 116 (неделя 96), визите 118 (неделя 120) и визите 27 EOT для пациентов, которые перешли на прием выбранной дозы.
Системные признаки sJIA
Информацию о системных признаках sJIA, включая лихорадку, кратковременную эритематозную сыпь лососевого цвета, генерализованное увеличение лимфатических узлов, гепатомегалию спленомегалию, серозит и другие осложнения, будут собирать на скрининговом визите 1 (от дня -28 до дня -1, вплоть до 31 дня) для подтверждения диагноза, на визите 2 (неделя 0), визите 12 (неделя 12) и визите 27 (неделя 156); на визите 15 (неделя 24), визите 18 (неделя 48) и каждые 24 недели до конца исследования будут собирать только информацию о лихорадке и кратковременной эритематозной сыпи лососевого цвета.
Общие системные признаки, включая лихорадку и сыпь, связанные с sJIA, будут собирать на всех визитах при оценке базовых компонентов ACR JIA.
Сыпь, связанная с sJIA, представляет собой кратковременную эритематозную сыпь лососевого цвета.
Отсутствие сыпи определяется как отсутствие сыпи, связанной с sJIA, зарегистрированной в дневнике пациента за 7 дней до дня оценки.
Отсутствие лихорадки определяют как отсутствие показателей температуры ≥37,5oC в течение 7 дней до визита, когда оценивают базовые компоненты ACR JIA.
Эффект лечения в отношении лихорадки и сыпи будут оценивать на неделе 12 по сравнению с исходным уровнем.
Долю пациентов с лихорадкой из-за системного JIA, у которых на исходном уровне не наблюдали лихорадку, по каждой когорте дозы, в целом и по группам по весу тела (при наличии достаточных данных) будут обобщать по визитам (т. е. на неделях 12, 24, 48 и каждые 24 недели до конца исследования).
Долю пациентов с сыпью из-за системного JIA, у которых на исходном уровне не наблюдали сыпь, по каждой когорте дозы, в целом и по группам по весу тела (при наличии достаточных данных) будут обобщать по визитам (т. е. на неделях 12, 24, 48 и каждые 24 недели до конца исследования).
Оценка использования глюкокортикоидов
Долю пациентов, получающих глюкокортикоиды, по категориям доз (20,5 мг/кг, 20,2 мг/кг и <0,5 мг/кг, <0,2 мг/кг), а также долю пациентов, не получающих глюкокортикоиды и у которых не наблюдают обострений JIA на неделях 24, 48, 72 и 104 будут обобщать по каждой когорте дозы и группе по весу тела для сравнения с долей на исходном уровне. Эквивалентную преднизону дозу глюкокортикоидов в мг/кг/день в определенный момент времени (т. е., недели 0, 24, 48, 72 или 104) будут рассчитывать на основе глюкокортикоидов, принятых в течение 2 недель до данного момента времени. Обострение JIA в определенный момент времени (т. е. недели 24, 48, 72 или 104) определяют как отсутствие сохранения ответа ACR50 JIA, лихорадку (любой показатель температуры ≥37,5°C) или ESR 220 мм/час между предыдущим визитом и данным моментом времени.
Эквивалентную преднизону дозу глюкокортикоидов (первичное значение и изменение по сравнению с исходным уровнем) будут обобщать по визитам (среднее значение, стандартное отклонение, медианное значение и т. д.) по когортам доз и группам по весу тела. Кроме того, эквивалентную преднизону дозу глюкокортикоидов при каждом визите в течение периода лечения (как основного, так и периода применения ранее установленной дозы в расширенной популяции) будут наносить на график для каждого отдельного пациента по когортам доз и по группам по весу тела.
Оценки роста
Влияние на рост будут оценивать с использованием показателей скорости роста и SD роста для пациентов, которые никогда не получали гормон роста и не достигли стадии 5 по Таннеру к концу первого года лечения.
Скорость роста (сантиметры в год) будут рассчитывать на основе изменения роста и временного интервала между измерениями роста. Скорости роста перед лечением будут оценивать с использованием роста, измеренного максимально близко к моменту времени за 1 год до исходного уровня, при условии, что измерения получали в период от 9 до 24 месяцев до исходного уровня. Впоследствии будут рассчитывать скорости роста в годы 1 и 2 лечения. Будут использовать измерения роста, максимально близкие к пороговым моментам времени в 1 год и 2 года.
Рост будут измерять в положении стоя с помощью настенного ростомера. При измерении роста детей будут измерять без обуви, головных уборов или украшений для волос с фиксированным прямым углом над головой, при этом голова, плечи, ягодицы и пятки прижаты к стене и ступни прижаты друг к другу. Из-за незначительных изменений позы ребенка при каждом измерении будут производить три последовательных измерения для каждого ребенка. Среднее значение трех измерений будут считать истинным ростом ребенка, если измерения находятся в пределах 0,3 см. Если измерения отличаются друг от друга >0,3 см, их будут повторять и усреднять, когда они будут находиться в пределах допустимого диапазона. Измерения, используемые для расчета скорости роста, будут производить как указано в графике оценок.
Рост будут измерять с помощью утвержденных методов в исследовательском центре. Ожидаемую нормальную скорость роста в течение определенного интервала для данного пациента будут рассчитывать как разницу среднего значения роста по Всемирной организации здравоохранения (ВОЗ; Всемирная организация здравоохранения. Нормы роста детей WHO Anthro (версия 2011) and macros. URL: http://www.who.int/childgrowth/software/en/) для возраста и пола пациента, деленную на изменение возраста. Скорости роста будут сравнивать с ожидаемыми нормальными скоростями роста до лечения и в течение 1, 2 и 3 лет лечения.
Коэффициенты SD роста будут рассчитывать (см. ниже) с использованием норм ВОЗ (там же). Эталонные популяции характеризуются средними значениями коэффициентов SD, равными 0, и значения от -2 до +2 обычно считаются нормальным диапазоном. Коэффициент SD = (наблюдаемое значение - медианное значение эталонной популяции)/значение SD эталонной популяции.
Чтобы оценить влияние в отношении роста, изменение по сравнению с исходным уровнем коэффициента SD роста в годы 1, 2 и 3 лечения будут описательно обобщать с использованием среднего значения, SD и 95% CI. Доли пациентов со скоростями роста, превышающими ожидания ВОЗ, будут обобщать до лечения и в течение 1, 2 и 3 лет лечения. Средние значения скоростей роста будут сравнивать с ожидаемыми нормальными скоростями роста по ВОЗ до лечения и в течение 1, 2 и 3 лет.
Физикальное обследование
Полное физикальное обследование будут проводить на скрининговом визите 1 (от дня -28 до дня -1, вплоть до 31 дня), визите 2 (день 1, неделя 0), визите 8 (неделя 4), визите 9 (неделя 6), визите 12 (неделя 12), визите 15 (неделя 24), визите 18 (неделя 48), визите 20 (неделя 72), визите 22 (неделя 96), визите 24 (неделя 120), визите 26 (неделя 144) и визите 27 EOT (неделя 156) для пациентов, которые продолжают принимать выбранную дозу; на визите 101 (неделя 0), визите 104 (неделя 6), визите 106 (неделя 12), визите 109 (неделя 24), визите 112 (неделя 48), визите 114 (неделя 72), визите 116 (неделя 96), визите 118 (неделя 120), визите 27 EOT и визите 28 EOS для пациентов, которые перешли на прием выбранной дозы. О любых клинически значимых отклонениях необходимо сообщать в eCRF пациента как о медицинском анамнезе, если их наблюдают и про них было уже известно на визите 1 (от дня -28 до дня -1, вплоть до 31 дня), и о них необходимо сообщать как о AE, если их наблюдают на визите 2 (день 1, неделя 0) и на последующих визитах. О любом клинически значимом отклонении при физикальном обследовании, о котором известно на скрининговом визите, необходимо сообщать как о медицинском анамнезе, а не как о AE. О клинически значимых отклонениях или ухудшении по сравнению с исходным уровнем, о которых сообщают после скринингового визита, следует сообщать как о AE.
Вес тела
Вес тела следует определять у пациента в нижнем белье или в очень легкой одежде (без верхней одежды и аксессуаров) и без обуви, а также с пустым мочевым пузырем. На протяжении всего исследования рекомендуется использовать одни и те же весы. Вес тела определяют с точностью до 0,1 кг.
На протяжении всего исследования рекомендуется использовать одни и те же весы. Вес тела будут определять на скрининговом визите 1 (от дня -28 до дня -1, вплоть до 31 дня), визите 2 исходного уровня (день 1, неделя 0), визите 12 (неделя 12) и на всех визитах в фазе дополнительного лечения для пациентов, которые продолжают принимать выбранную дозу; с визита 101 (неделя 0) до визита 119 (неделя 132), на визите 27 EOT и визите 28 EOS, кроме визита 102 (неделя 2) и визита 104 (неделя 6), для пациентов, которые перешли на прием выбранной дозы.
Фармакодинамические параметры
Фармакодинамические эффекты сарилумаба будут оценивать путем измерения следующих биомаркеров: hs-CRP, IL-6, sIL-6R.
График проведения оценок
График забора образов крови можно найти на схеме исследования (см. таблицы 17-19). IL-6 и общий sIL-6R будут измерять на визите 2 исходного уровня (неделя 0) и визите 12 (неделя 12). Для пациентов, которые преждевременно прекращают исследуемый вид лечения во время основной фазы лечения, при оценке EOT будут измерять IL-6 и общий sIL-6R.
Фармакогеномика
Родитель(родители) или законный(законные) опекун(опекуны) должны будут подписать отдельную форму ICF для забора образцов слюны. Образцы дезоксирибонуклеиновой кислоты для геномных исследований будут иметь двойной код в соответствии с руководством 15 Международного совета по гармонизации (ICH). Образцы дезоксирибонуклеиновой кислоты могут храниться до 15 лет после последней даты отчета о клиническом исследовании (CSR) и могут использоваться в исследовательских целях.
Целью геномного анализа является выявление геномных ассоциаций с клиническим ответом или ответом биомаркеров на целевую модуляцию, с прогнозом и прогрессированием заболевания или с другими показателями клинического исхода. Эти данные могут быть использованы или объединены с данными, собранными из других исследований, для идентификации геномных маркеров, которые могут предсказать ответ и выяснить механизмы заболевания. Анализы могут включать определение последовательности или исследования однонуклеотидного полиморфизма генов-кандидатов и окружающих геномных областей. Также могут проводить полногеномные исследования, включая (без ограничения) анализ однонуклеотидного полиморфизма, геномное секвенирование и секвенирование транскриптома. Если показано, можно также провести геномный анализ для выявления маркеров, связанных с токсичностью. Пациенты являются пригодными для исследования, даже если они и их родители или законные опекуны не желают участвовать в сборе образцов для фармакогеномных исследований.
Любые неиспользованные или оставшиеся образцы сыворотки крови, собранные для измерения концентрации лекарственного средства или ADA, могут быть сохранены и использованы для поисковых исследований биомаркеров, связанных с sJIA, ингибированием пути IL-6Rα антителами, ответом на лечение (PD и/или прогностическим), для исследования неожиданных AE или для выявления маркеров, связанных с токсичностью. Образцы могут храниться до 15 лет после даты CSR или в соответствии с местными нормативными требованиями.

Сокращения. HLA-B27=человеческий лейкоцитарный антиген-B27, ILAR=Международная лига ассоциаций ревматологов, JIA=ювенильный идиопатический артрит, RF=ревматоидный фактор.
a Специфические критерии исключения: a=псориаз или псориаз у родственника первой степени родства; b=наличие лейкоцитарного антигена человека B27, мужской пол и возраст старше 6 лет;
c=анкилозирующий спондилит, энтезит-ассоциированный артрит, сакроилеит с сопутствующим хроническим воспалительным заболеванием кишечника, болезнь Рейтера у
родственника первой степени родства; d=обнаружение ревматоидного фактора (RF) по меньшей мере 2 раза с интервалом по меньшей мере 3 месяца.
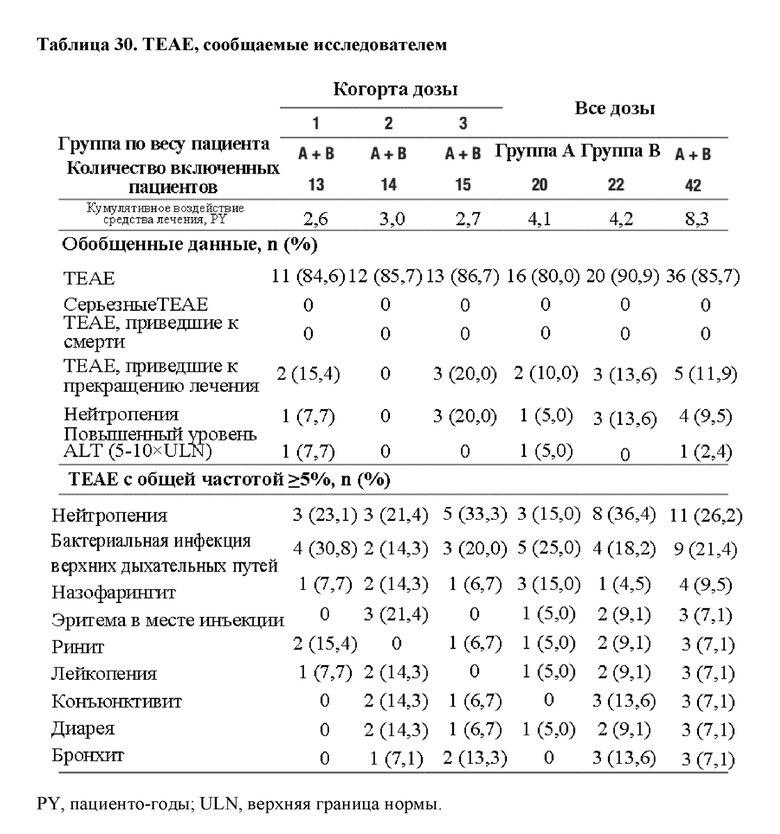
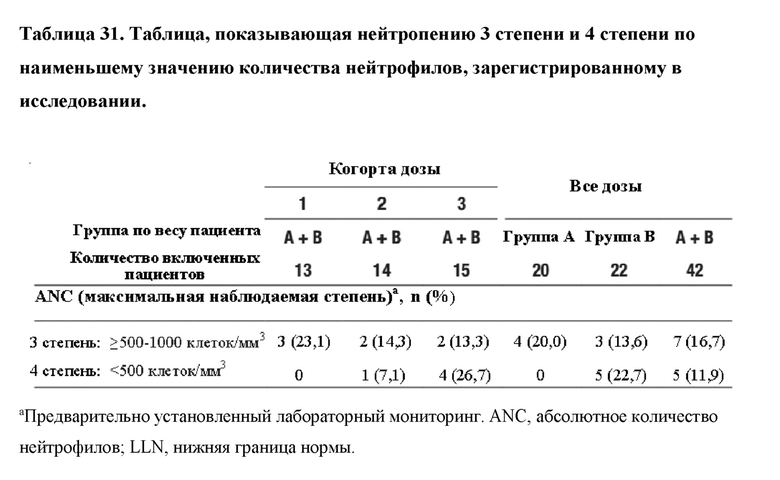
(до 84 недель воздействия сарилумаба после V12 для третьей части [часть 3])
после лечения
(6 недель)
(±1 или 3)
(±1 или 3)
(±1 или 3)
(±1 или 3)
(±1 или 3)
(±1 или 3)
(±1 или 3)
(±1 или 3)
(±1 или 3)
(±1 или 3)
части 3
Wk 102 для части 3
a Визиты 13, 14, 16 и 17 не применимы для пациентов в третьей части.
b Визит 16.1 применим только к пациентам третьей части.
c Визит 27 (неделя 96 для третьей части) является запланированным визитом конца лечения (EOT) в рамках исследования. Если пациенты преждевременно прекращают исследуемое лечение, EOT произойдет через 2 недели после последней инъекции IMP.
d Визит 28 (неделя 102 для третьей части) является запланированным визитом конца исследования (EOS) в рамках исследования. Если пациенты преждевременно прекращают исследуемое лечение, этих пациентов попросят вернуться для оценки EOT через 2 недели после последней инъекции IMP и для оценки EOS через 6 недель после визита EOT (EOT +6 недель). Визиты EOT и EOS будут применимы ко всем пациентам, преждевременно прекратившим лечение как в основной фазе лечения, так и в фазе дополнительного лечения.
e Домашний дневник для введения IMP, который необходимо заполнить для IMP, вводимого дома.
f Полное физикальное обследование будут осуществлять во время визита 15 (неделя 24), визита 18 (неделя 48), визита 20 (неделя 72) и визита 27 (EOT), где обследование включает обследование кожи, носовых полостей, глаз, ушей, дыхательной, сердечно-сосудистой, желудочно-кишечной, нервной, лимфатической и опорно-двигательной систем.
g Исследуемый лекарственный препарат (IMP) следует вводить один раз в две недели (q2w). За пациентами следует наблюдать в течение по меньшей мере 30 минут после введения IMP в отношении любых признаков или симптомов реакции гиперчувствительности. Последнюю инъекцию IMP произведут на неделе 94; затем пациенты будут проходить оценку EOT на визите 27 (неделя 96 для третьей части). Во время визита 27 инъекции IMP производить не будут.
h Рост будут измерять с помощью ростомера в исследовательских центрах во время исследования.
i Базовый набор ювенильного идиопатического артрита по Американской коллегии ревматологов (ACR JIA) включает общую оценку тяжести заболевания врачом, общую оценку самочувствия пациентом или родителем, количество суставов с активным артритом (определяется как отек в суставе, не вызванный деформацией ИЛИ ограничение движений либо с болью, либо с чувствительностью, либо с обоими признаками), количество суставов с ограничением движений, опросник оценки здоровья детей (CHAQ) и hs-CRP. Если пациент преждевременно прекращает лечение до недели 48, исследование hs-CRP необходимо провести в центральной лаборатории на EOT.
j Подсчет индекса активности заболевания, представляющего собой ювенильный артрит
k Гематологическое исследование (кровь следует забирать ДО введения лекарственного средства): гемоглобин, гематокрит, количество эритроцитов (RBC) и морфология эритроцитов (если количество эритроцитов аномальное), лейкоцитарная формула (WBC) (нейтрофилы, лимфоциты, моноциты, эозинофилы, базофилы), количество тромбоцитов, абсолютное количество нейтрофилов (ANC). Если пациент преждевременно прекращает лечение до недели 48, исследование необходимо провести в центральной лаборатории на EOT.
l Биохимическое исследование (кровь следует забирать ДО введения лекарственного средства). Будут тестировать ALT, AST, ALP, общий билирубин, конъюгированный билирубин, неконъюгированный билирубин и альбумин. Полное биохимическое исследование следует провести на визите 27 EOT (неделя 96 для третьей части). Если пациент преждевременно прекращает лечение до недели 48, исследование необходимо провести в центральной лаборатории на EOT.
m Липидограмма (кровь следует забирать ДО введения лекарственного средства). Триглицериды (TG), общий холестерин, холестерин липопротеинов высокой плотности (HDL), холестерин липопротеинов низкой плотности (LDL). Требуется, чтобы пациенты не принимали пищу по меньшей мере в течение 8 часов до исследования. На EOT исследование будут проводить только для пациентов, преждевременно прекративших лечение до недели 48, и его должна проводить центральная лаборатория.
n На EOT исследование ANA/антитела к dsDNA будут проводить только для пациентов, преждевременно прекративших лечение до недели 48, и его должна проводить центральная лаборатория.
o Для женщин, у которых начались менструации, исследования мочи на наличие беременности следует проводить до введения IMP при каждом запланированном визите и на EOT. Исследование мочи на наличие беременности можно проводить местно.
p Образцы крови будут собирать ДО введения IMP в дни введения доз в течение периода лечения. Если у пациента наблюдают SAE, для определения концентрации сарилумаба и оценки антител к лекарственному средству (ADA) следует собирать образцы крови в момент начала и завершения события, если это возможно, или в течение короткого времени после начала и завершения.
Примечание. "Размер выборки" представляет собой примерное количество пациентов, которых включат в выбранную дозу и которые завершат 12-недельный основной период лечения; предполагали процент выбывания 15%.
Ссылки
1. Petty RE, Southwood TR, Manners P, Baum J, Glass DN, Goldenberg J, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol 2004;31:390-2.
2. Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet (London) 2007;369:767-78.
3. Gabriel SE, Michaud K. Epidemiological studies in incidence, prevalence, mortality, and comorbidity of the rheumatic diseases. Arthritis Res Ther 2009;11:229-29.
4. Oberle EJ, Harris JG, Verbsky JW. Polyarticular juvenile idiopathic arthritis - epidemiology and management approaches. Clin Epidemiol 2014;6:379-93.
5. Shepherd J, Cooper K, Harris P, Picot J, Rose M. The clinical effectiveness and cost-effectiveness of abatacept, adalimumab, etanercept and tocilizumab for treating juvenile idiopathic arthritis: a systematic review and economic evaluation. Health Technol Assess (Winchester) 2016;20:1-222.
6. Hissink Muller PCE, Brinkman DMC, Schonenberg D, Koopman-Keemink Y, Brederije ICJ, Bekkering WP, et al. A comparison of three treatment strategies in recent onset non-systemic Juvenile Idiopathic Arthritis: initial 3-months results of the BeSt for Kids-study. Pediatr Rheumatol Online J 2017;15.
7. Lovell DJ, Johnson AL, Huang B, Gottlieb BS, Morris PW, Kimura Y, et al. Risk, timing, and predictors of disease flare after discontinuation of anti-tumor necrosis factor therapy in children with polyarticular forms of juvenile idiopathic arthritis with clinically inactive disease. Arthritis Rheumatol (Hoboken) 2018;70:1508-18.
8. Wallace CA, Giannini EH, Spalding SJ, Hashkes PJ, O'Neil KM, Zeft AS, et al. Trial of early aggressive therapy in polyarticular juvenile idiopathic arthritis. Arthritis Rheum 2012;64:2012-21.
9. Guzman J, Oen K, Tucker LB, Huber AM, Shiff N, Boire G, et al. The outcomes of juvenile idiopathic arthritis in children managed with contemporary treatments: results from the ReACCh-Out cohort. Ann Rheum Dis 2015;74:1854-60.
10. Simonini G, Ferrara G, Pontikaki I, Scoccimarro E, Giani T, Taddio A, et al. Flares after withdrawal of biologic therapies in juvenile idiopathic arthritis: clinical and laboratory correlates of remission duration. Arthritis Care Res 2018;70:1046-51.
11. Ravelli A, Consolaro A, Horneff G, Laxer RM, Lovell DJ, Wulffraat NM, et al. Treating juvenile idiopathic arthritis to target: recommendations of an international task force. Ann Rheum Dis 2018;77:819-28.
12. Swart J, Giancane G, Horneff G, Magnusson B, Hofer M, Alexeeva Е, et al. Pharmacovigilance in juvenile idiopathic arthritis patients treated with biologic or synthetic drugs: combined data of more than 15,000 patients from Pharmachild and national registries. Arthritis Res Ther 2018;20:285.
13. De Benedetti F, Robbioni P, Massa M, Viola S, Albani S, Martini A. Serum interleukin-6 levels and joint involvement in polyarticular and pauciarticular juvenile chronic arthritis. Clin Exp Rheumatol 1992;10:493-8.
14. Spirchez M, Samasca G, Iancu M, Bolba C, Miu N. Relation of interleukin-6, TNF-alpha and interleukin-1alpha with disease activity and severity in juvenile idiopathic arthritis patients. Clin Lab 2012;58:253-60.
15. De Benedetti F. Targeting interleukin-6 in pediatric rheumatic diseases. Curr Opin Rheumatol 2009;21:533-7.
16. De Benedetti F, Pignatti P, Gerloni V, Massa M, Sartirana P, Caporali R, et al. Differences in synovial fluid cytokine levels between juvenile and adult rheumatoid arthritis. J Rheumatol 1997;24:1403-9.
17. KEVZARA. Инструкция по применению лекарственного средства в США. Доступно по ссылке: https://www-accessdata-fda-gov/drugsatfda_docs/label/2017/761037s000lbl.pdf [доступно по состоянию на 14 марта 2018 г.].
18. Brunner HI, Ruperto N, Zuber Z, Keane C, Harari O, Kenwright A, et al. Efficacy and safety of tocilizumab in patients with polyarticular-course juvenile idiopathic arthritis: results from a phase 3, randomised, double-blind withdrawal trial. Ann Rheum Dis 2015;74:1110-17.
19. Маркировка продукта ACTEMRA (тоцилизумаб). [доступно по состоянию на 24 мая 2019 г.].
20. Huizinga TW, Fleischmann RM, Jasson M, Radin AR, van Adelsberg J, Fiore S, et al. Sarilumab, a fully human monoclonal antibody against IL-6Ralpha in patients with rheumatoid arthritis and an inadequate response to methotrexate: efficacy and safety results from the randomised SARIL-RA-MOBILITY Part A trial. Ann Rheum Dis 2014;73:1626-34.
21. Руководство по лечению травмы печени, вызванной лекарственными средствами: Дорегистрационная клиническая оценка. Доступно по ссылке: https://www-fda-gov/media/116737/download [доступно по состоянию на 09 июля 2019 г.].
22. Wallace CA, Giannini EH, Huang B, Itert L, Ruperto N. American College of Rheumatology provisional criteria for defining clinical inactive disease in select categories of juvenile idiopathic arthritis. Arthrit Care Res 2011;63:929-36.
23. Germovsek E, Barker CIS, Sharland M, Standing JF. Pharmacokinetic-pharmacodynamic modeling in pediatric drug development, and the importance of standardized scaling of clearance. Clin Pharmacokinet 2019;58:39-52.
24. Barker CIS, Standing JF, Kelly LE, Hanly Faught L, Needham AC, Rieder MJ, et al. Pharmacokinetic studies in children: recommendations for practice and research. Arch Dis Child 2018;103:695.
25. Gill KL, Machavaram KK, Rose RH, Chetty M. Potential sources of inter-subject variability in monoclonal antibody pharmacokinetics. Clin Pharmacokinet 2016;55:789-805.
26. Kojima T, Yabe Y, Kaneko A, Hirano Y, Ishikawa H, Hayashi M, et al. Monitoring C-reactive protein levels to predict favourable clinical outcomes from tocilizumab treatment in patients with rheumatoid arthritis. Modern rheumatology 2013;23:977-85.
27. Madhok R, Crilly A, Watson J, Capell HA. Serum interleukin 6 levels in rheumatoid arthritis: correlations with clinical and laboratory indices of disease activity. Ann Rheum Dis 1993;52:232-4.
28. Nishimoto N, Terao K, Mima T, Nakahara H, Takagi N, Kakehi T. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood 2008;112:3959-64.
29. Emery P, Keystone E, Tony HP, Cantagrel A, van Vollenhoven R, Sanchez A, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis 2008;67:1516-23.
30. Fleischmann R, van Adelsberg J, Lin Y, Castelar-Pinheiro GD, Brzezicki J, Hrycaj P, et al. Sarilumab and nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis and inadequate response or intolerance to tumor necrosis factor inhibitors. Arthritis Rheumatol (Hoboken) 2017;69:277-90.
31. Genovese MC, Fleischmann R, Kivitz AJ, Rell-Bakalarska M, Martincova R, Fiore S, et al. Sarilumab plus methotrexate in patients with active rheumatoid arthritis and inadequate response to methotrexate: results of a Phase III study. Arthritis Rheumatol (Hoboken) 2015;67:1424-37.
32. Burmester GR, Lin Y, Patel R, van Adelsberg J, Mangan EK, Graham NM, et al. Efficacy and safety of sarilumab monotherapy versus adalimumab monotherapy for the treatment of patients with active rheumatoid arthritis (MONARCH): a randomised, double-blind, parallel-group phase III trial. Ann Rheum Dis 2017;76:840-47.
33. Emery P, Rondon J, Parrino J, Lin Y, Pena-Rossi C, van Hoogstraten H, et al. Safety and tolerability of subcutaneous sarilumab and intravenous tocilizumab in patients with rheumatoid arthritis. Rheumatology (Oxford) 2018;58:849-58.
34. Pardeo M, Wang J, Ruperto N, Alexeeva E, Chasnyk V, Schneider R, et al. Neutropenia during tocilizumab treatment is not associated with infection risk in systemic or polyarticular-course juvenile idiopathic arthritis. J Rheumatol 2019. Доступно по ссылке: https://www-ncbi-nlm-nih-gov/pubmed/30824645 [доступно по состоянию на 29 августа 2019 г.].
35. Отчет об оценке Европейского агентства по лекарственным средствам в отношении ACTEMRA (тоцилизумаб). Доступно по ссылке: https://www-ema-europa-eu/en/documents/variation-report/roactemra-h-c-955-ii-0072-epar-assessment-report-variation_en.pdf [доступно по состоянию на 28 мая 2019 г.].
36. Hsieh MM, Everhart JE, Byrd-Holt DD, Tisdale JF, Rodgers GP. Prevalence of neutropenia in the U.S. population: age, sex, smoking status, and ethnic differences. Ann Intern Med 2007;146:486-92.
37. Moots RJ, Sebba A, Rigby W, Ostor A, Porter-Brown B, Donaldson F, et al. Effect of tocilizumab on neutrophils in adult patients with rheumatoid arthritis: pooled analysis of data from phase 3 and 4 clinical trials. Rheumatology (Oxford) 2017;56:541-49.
38. Genovese MC, van Adelsberg J, Fan C, Graham NMH, van Hoogstraten H, Parrino J, et al. Two years of sarilumab in patients with rheumatoid arthritis and an inadequate response to MTX: safety, efficacy and radiographic outcomes. Rheumatology 2018;57:1423-31.
39. European Medicines Agency. Ethical considerations for clinical trials on medicinal products conducted with the paediatric population. Recommendations of the ad hoc group for the development of implementing guidelines for Directive 2001/20/EC relating to good clinical practice in the conduct of clinical trials on medicinal products for human use London: European Medicines Agency. 2008.
--->
ПЕРЕЧЕНЬ ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> SANOFI BIOTECHNOLOGY
<120> КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ЛЕЧЕНИЯ ЮВЕНИЛЬНОГО ИДИОПАТИЧЕСКОГО АРТРИТА
<130> 619859-SA9-244PC
<160> 11
<170> PatentIn версия 3.5
<210> 1
<211> 116
<212> БЕЛОК
<213> Homo sapiens
<220>
<221> MISC_FEATURE
<222> (1)..(116)
<223> вариабельный участок тяжелой цепи сарилумаба
<400> 1
Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Arg
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Arg Phe Thr Phe Asp Asp Tyr
20 25 30
Ala Met His Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ser Gly Ile Ser Trp Asn Ser Gly Arg Ile Gly Tyr Ala Asp Ser Val
50 55 60
Lys Gly Arg Phe Thr Ile Ser Arg Asp Asn Ala Glu Asn Ser Leu Phe
65 70 75 80
Leu Gln Met Asn Gly Leu Arg Ala Glu Asp Thr Ala Leu Tyr Tyr Cys
85 90 95
Ala Lys Gly Arg Asp Ser Phe Asp Ile Trp Gly Gln Gly Thr Met Val
100 105 110
Thr Val Ser Ser
115
<210> 2
<211> 107
<212> БЕЛОК
<213> Homo sapiens
<220>
<221> MISC_FEATURE
<222> (1)..(107)
<223> вариабельный участок легкой цепи сарилумаба
<400> 2
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Val Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Arg Ala Ser Gln Gly Ile Ser Ser Trp
20 25 30
Leu Ala Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Gly Ala Ser Ser Leu Glu Ser Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Ser Tyr Tyr Cys Gln Gln Ala Asn Ser Phe Pro Tyr
85 90 95
Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys
100 105
<210> 3
<211> 8
<212> БЕЛОК
<213> Homo sapiens
<220>
<221> MISC_FEATURE
<222> (1)..(8)
<223> определяющая комплементарность область 1 тяжелой цепи
<400> 3
Arg Phe Thr Phe Asp Asp Tyr Ala
1 5
<210> 4
<211> 8
<212> БЕЛОК
<213> Homo sapiens
<220>
<221> MISC_FEATURE
<222> (1)..(8)
<223> определяющая комплементарность область 2 тяжелой цепи
<400> 4
Ile Ser Trp Asn Ser Gly Arg Ile
1 5
<210> 5
<211> 9
<212> БЕЛОК
<213> Homo sapiens
<220>
<221> MISC_FEATURE
<222> (1)..(9)
<223> определяющая комплементарность область 3 тяжелой цепи
<400> 5
Ala Lys Gly Arg Asp Ser Phe Asp Ile
1 5
<210> 6
<211> 6
<212> БЕЛОК
<213> Homo sapiens
<220>
<221> MISC_FEATURE
<222> (1)..(6)
<223> определяющая комплементарность область 1 легкой цепи
<400> 6
Gln Gly Ile Ser Ser Trp
1 5
<210> 7
<211> 3
<212> БЕЛОК
<213> Homo sapiens
<220>
<221> MISC_FEATURE
<222> (1)..(3)
<223> определяющая комплементарность область 2 легкой цепи
<400> 7
Gly Ala Ser
1
<210> 8
<211> 9
<212> БЕЛОК
<213> Homo sapiens
<220>
<221> MISC_FEATURE
<222> (1)..(9)
<223> определяющая комплементарность область 3 легкой цепи
<400> 8
Gln Gln Ala Asn Ser Phe Pro Tyr Thr
1 5
<210> 9
<211> 446
<212> БЕЛОК
<213> Homo sapiens
<220>
<221> MISC_FEATURE
<222> (1)..(446)
<223> Тяжелая цепь
<400> 9
Glu Val Gln Leu Val Glu Ser Gly Gly Gly Leu Val Gln Pro Gly Arg
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Arg Phe Thr Phe Asp Asp Tyr
20 25 30
Ala Met His Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ser Gly Ile Ser Trp Asn Ser Gly Arg Ile Gly Tyr Ala Asp Ser Val
50 55 60
Lys Gly Arg Phe Thr Ile Ser Arg Asp Asn Ala Glu Asn Ser Leu Phe
65 70 75 80
Leu Gln Met Asn Gly Leu Arg Ala Glu Asp Thr Ala Leu Tyr Tyr Cys
85 90 95
Ala Lys Gly Arg Asp Ser Phe Asp Ile Trp Gly Gln Gly Thr Met Val
100 105 110
Thr Val Ser Ser Ala Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala
115 120 125
Pro Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala Leu Gly Cys Leu
130 135 140
Val Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly
145 150 155 160
Ala Leu Thr Ser Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Ser
165 170 175
Gly Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro Ser Ser Ser Leu
180 185 190
Gly Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys Pro Ser Asn Thr
195 200 205
Lys Val Asp Lys Lys Val Glu Pro Lys Ser Cys Asp Lys Thr His Thr
210 215 220
Cys Pro Pro Cys Pro Ala Pro Glu Leu Leu Gly Gly Pro Ser Val Phe
225 230 235 240
Leu Phe Pro Pro Lys Pro Lys Asp Thr Leu Met Ile Ser Arg Thr Pro
245 250 255
Glu Val Thr Cys Val Val Val Asp Val Ser His Glu Asp Pro Glu Val
260 265 270
Lys Phe Asn Trp Tyr Val Asp Gly Val Glu Val His Asn Ala Lys Thr
275 280 285
Lys Pro Arg Glu Glu Gln Tyr Asn Ser Thr Tyr Arg Val Val Ser Val
290 295 300
Leu Thr Val Leu His Gln Asp Trp Leu Asn Gly Lys Glu Tyr Lys Cys
305 310 315 320
Lys Val Ser Asn Lys Ala Leu Pro Ala Pro Ile Glu Lys Thr Ile Ser
325 330 335
Lys Ala Lys Gly Gln Pro Arg Glu Pro Gln Val Tyr Thr Leu Pro Pro
340 345 350
Ser Arg Asp Glu Leu Thr Lys Asn Gln Val Ser Leu Thr Cys Leu Val
355 360 365
Lys Gly Phe Tyr Pro Ser Asp Ile Ala Val Glu Trp Glu Ser Asn Gly
370 375 380
Gln Pro Glu Asn Asn Tyr Lys Thr Thr Pro Pro Val Leu Asp Ser Asp
385 390 395 400
Gly Ser Phe Phe Leu Tyr Ser Lys Leu Thr Val Asp Lys Ser Arg Trp
405 410 415
Gln Gln Gly Asn Val Phe Ser Cys Ser Val Met His Glu Ala Leu His
420 425 430
Asn His Tyr Thr Gln Lys Ser Leu Ser Leu Ser Pro Gly Lys
435 440 445
<210> 10
<211> 214
<212> БЕЛОК
<213> Homo sapiens
<220>
<221> MISC_FEATURE
<222> (1)..(214)
<223> Легкая цепь
<400> 10
Asp Ile Gln Met Thr Gln Ser Pro Ser Ser Val Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Arg Ala Ser Gln Gly Ile Ser Ser Trp
20 25 30
Leu Ala Trp Tyr Gln Gln Lys Pro Gly Lys Ala Pro Lys Leu Leu Ile
35 40 45
Tyr Gly Ala Ser Ser Leu Glu Ser Gly Val Pro Ser Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Phe Ala Ser Tyr Tyr Cys Gln Gln Ala Asn Ser Phe Pro Tyr
85 90 95
Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<210> 11
<211> 358
<212> БЕЛОК
<213> Homo sapiens
<220>
<221> MISC_FEATURE
<222> (1)..(358)
<223> внеклеточный домен рецептора интерлейкина-6 человека
<400> 11
Met Val Ala Val Gly Cys Ala Leu Leu Ala Ala Leu Leu Ala Ala Pro
1 5 10 15
Gly Ala Ala Leu Ala Pro Arg Arg Cys Pro Ala Gln Glu Val Ala Arg
20 25 30
Gly Val Leu Thr Ser Leu Pro Gly Asp Ser Val Thr Leu Thr Cys Pro
35 40 45
Gly Val Glu Pro Glu Asp Asn Ala Thr Val His Trp Val Leu Arg Lys
50 55 60
Pro Ala Ala Gly Ser His Pro Ser Arg Trp Ala Gly Met Gly Arg Arg
65 70 75 80
Leu Leu Leu Arg Ser Val Gln Leu His Asp Ser Gly Asn Tyr Ser Cys
85 90 95
Tyr Arg Ala Gly Arg Pro Ala Gly Thr Val His Leu Leu Val Asp Val
100 105 110
Pro Pro Glu Glu Pro Gln Leu Ser Cys Phe Arg Lys Ser Pro Leu Ser
115 120 125
Asn Val Val Cys Glu Trp Gly Pro Arg Ser Thr Pro Ser Leu Thr Thr
130 135 140
Lys Ala Val Leu Leu Val Arg Lys Phe Gln Asn Ser Pro Ala Glu Asp
145 150 155 160
Phe Gln Glu Pro Cys Gln Tyr Ser Gln Glu Ser Gln Lys Phe Ser Cys
165 170 175
Gln Leu Ala Val Pro Glu Gly Asp Ser Ser Phe Tyr Ile Val Ser Met
180 185 190
Cys Val Ala Ser Ser Val Gly Ser Lys Phe Ser Lys Thr Gln Thr Phe
195 200 205
Gln Gly Cys Gly Ile Leu Gln Pro Asp Pro Pro Ala Asn Ile Thr Val
210 215 220
Thr Ala Val Ala Arg Asn Pro Arg Trp Leu Ser Val Thr Trp Gln Asp
225 230 235 240
Pro His Ser Trp Asn Ser Ser Phe Tyr Arg Leu Arg Phe Glu Leu Arg
245 250 255
Tyr Arg Ala Glu Arg Ser Lys Thr Phe Thr Thr Trp Met Val Lys Asp
260 265 270
Leu Gln His His Cys Val Ile His Asp Ala Trp Ser Gly Leu Arg His
275 280 285
Val Val Gln Leu Arg Ala Gln Glu Glu Phe Gly Gln Gly Glu Trp Ser
290 295 300
Glu Trp Ser Pro Glu Ala Met Gly Thr Pro Trp Thr Glu Ser Arg Ser
305 310 315 320
Pro Pro Ala Glu Asn Glu Val Ser Thr Pro Met Gln Ala Leu Thr Thr
325 330 335
Asn Lys Asp Asp Asp Asn Ile Leu Phe Arg Asp Ser Ala Asn Ala Thr
340 345 350
Ser Leu Pro Val Gln Asp
355
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ДИАГНОСТИКИ И ЛЕЧЕНИЯ РЕВМАТОИДНОГО АРТРИТА | 2020 |
|
RU2828027C2 |
| КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ РЕВМАТОИДНОГО АРТРИТА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2012 |
|
RU2664697C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ АСТМЫ ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-33 | 2020 |
|
RU2838119C2 |
| КОМПОЗИЦИИ, СОДЕРЖАЩИЕ АНТИТЕЛА К IL6R, ДЛЯ ЛЕЧЕНИЯ УВЕИТА И МАКУЛЯРНОГО ОТЕКА, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2747193C2 |
| СПОСОБ ЛЕЧЕНИЯ АТОПИЧЕСКОГО ДЕРМАТИТА ПОСРЕДСТВОМ ВВЕДЕНИЯ ИНГИБИТОРА ИЛ-4R | 2019 |
|
RU2801204C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ПАЦИЕНТОВ С ГЕТЕРОЗИГОТНОЙ СЕМЕЙНОЙ ГИПЕРХОЛЕСТЕРИНЕМИЕЙ (HEFH) | 2015 |
|
RU2735521C2 |
| СПОСОБЫ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ АСТМЫ ПОСРЕДСТВОМ ВВЕДЕНИЯ АНТАГОНИСТА IL-4R | 2015 |
|
RU2713406C2 |
| АНТИТЕЛА, НЕЙТРАЛИЗУЮЩИЕ GM-CSF, ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ РЕВМАТОИДНОГО АРТРИТА ИЛИ В КАЧЕСТВЕ АНАЛЬГЕТИКОВ | 2014 |
|
RU2714919C2 |
| АНТИТЕЛА К FGFR2 В КОМБИНАЦИИ С ХИМИОТЕРАПЕВТИЧЕСКИМИ СРЕДСТВАМИ ПРИ ЛЕЧЕНИИ РАКА | 2018 |
|
RU2797560C2 |
| АНТИТЕЛА К B7-H1 И К CTLA-4 ДЛЯ ЛЕЧЕНИЯ НЕМЕЛКОКЛЕТОЧНОГО РАКА ЛЕГКОГО | 2015 |
|
RU2806210C2 |
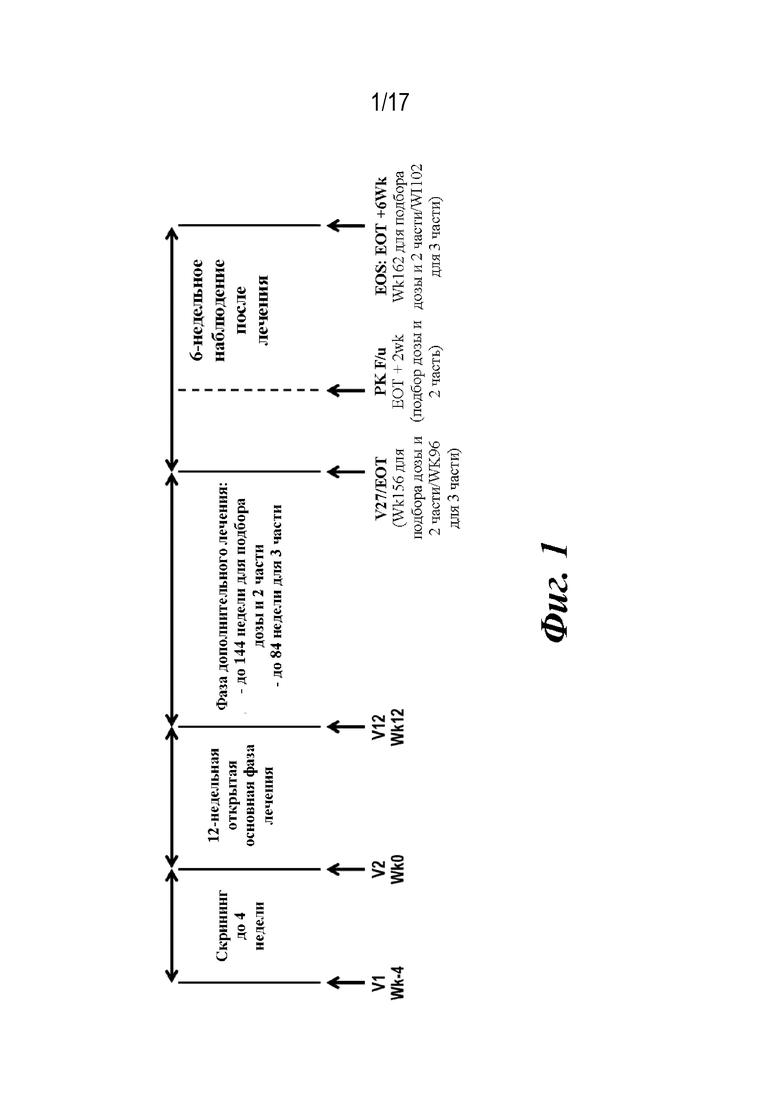
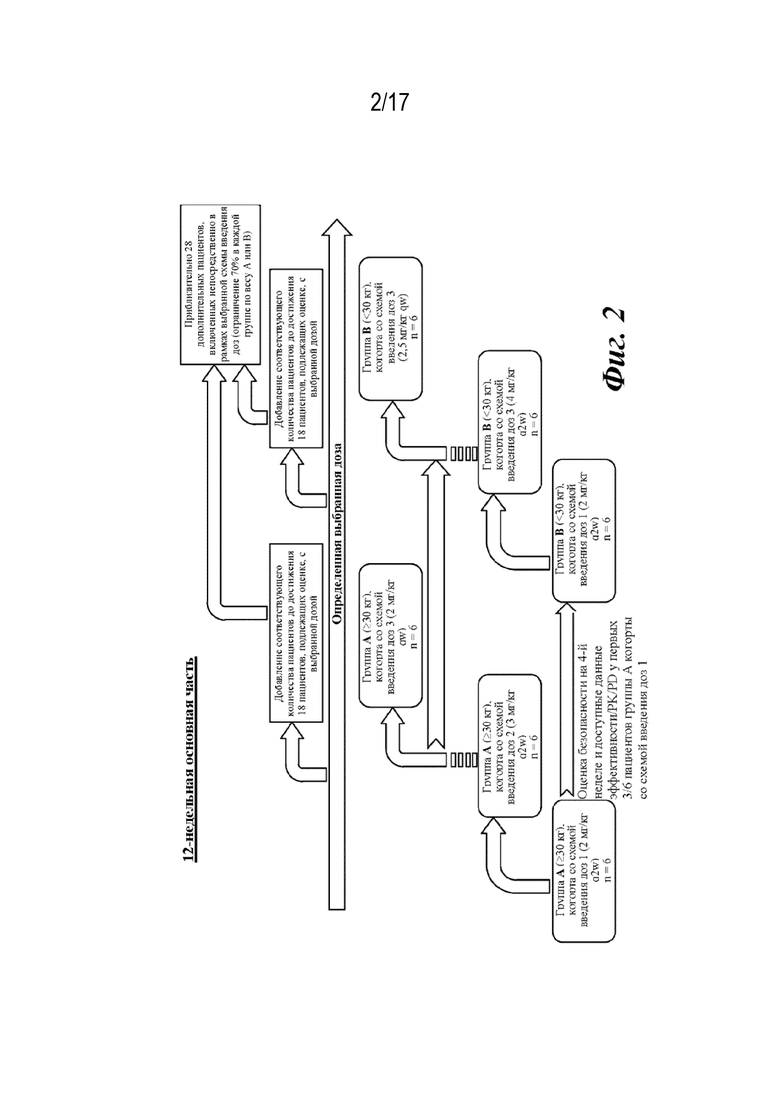
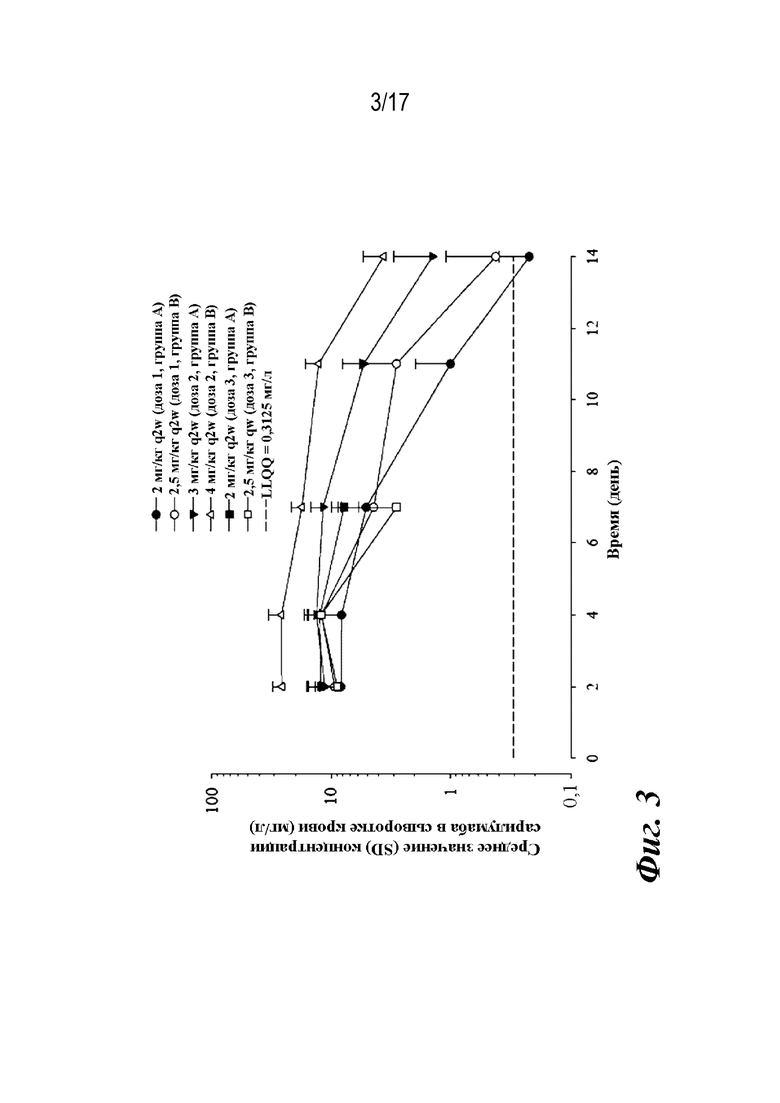
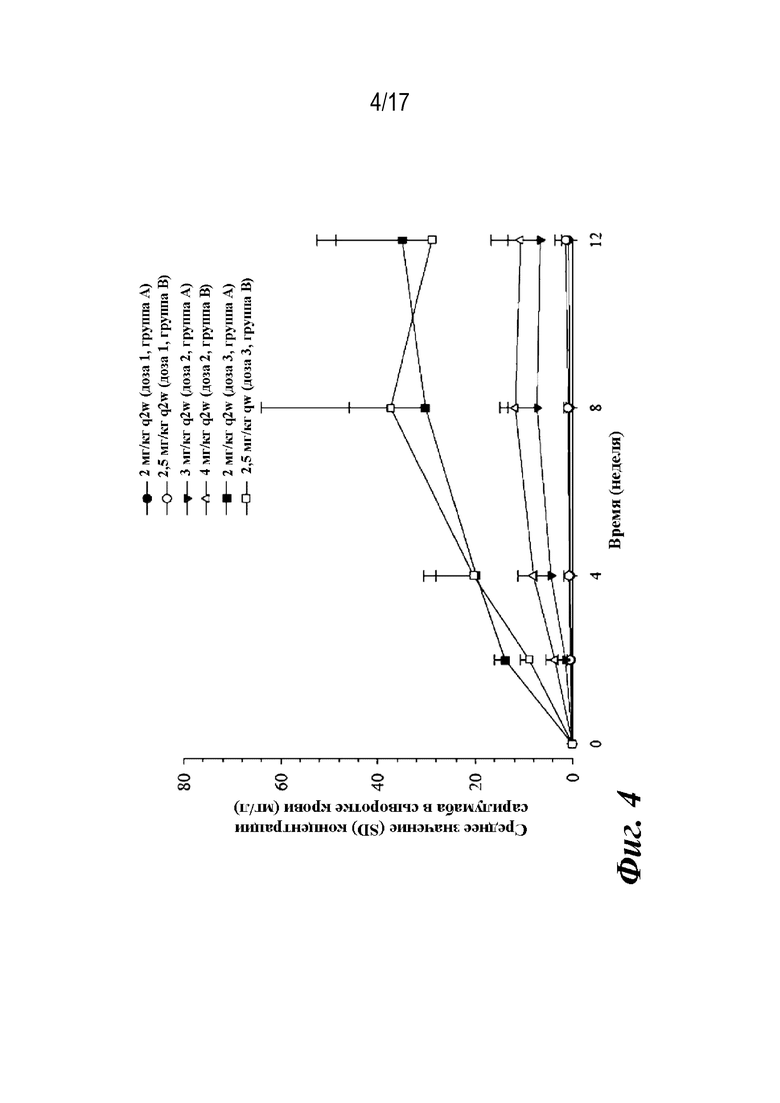
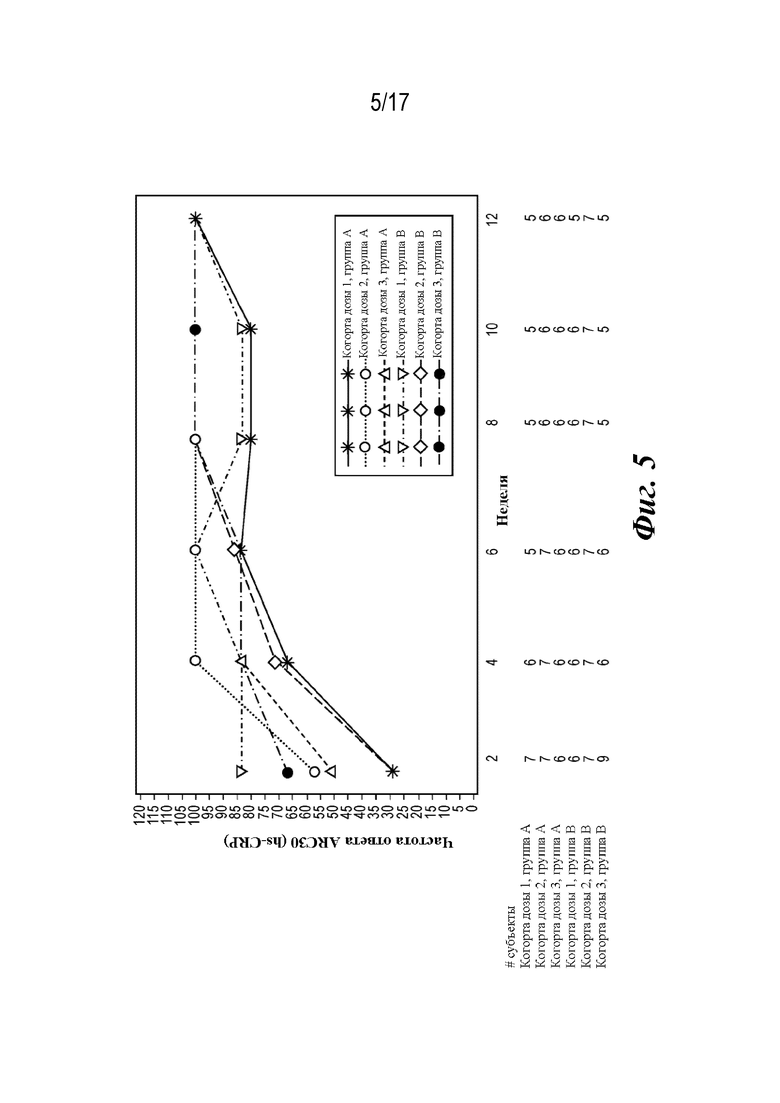

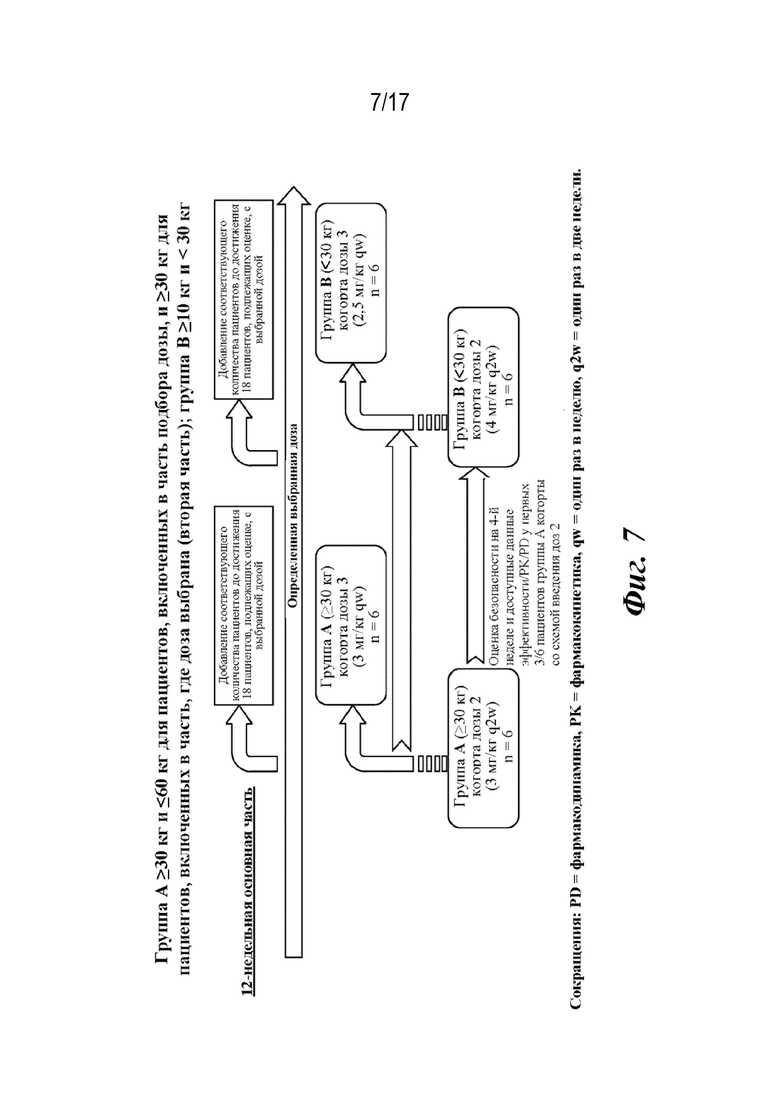
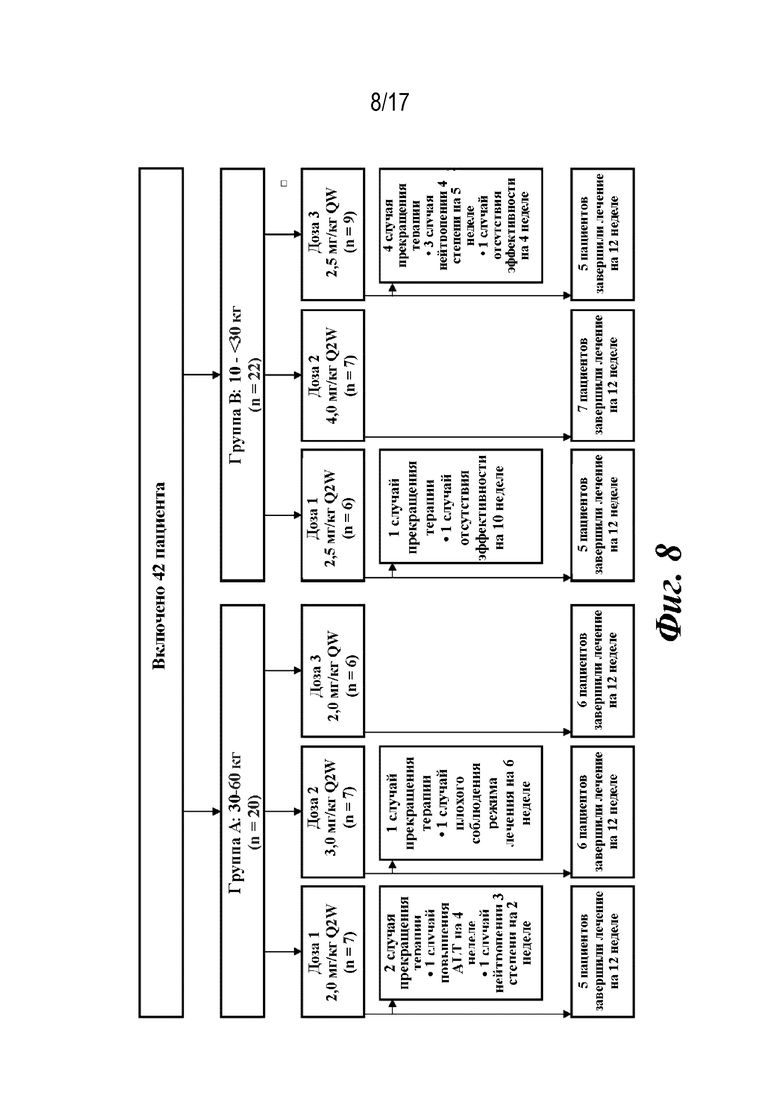
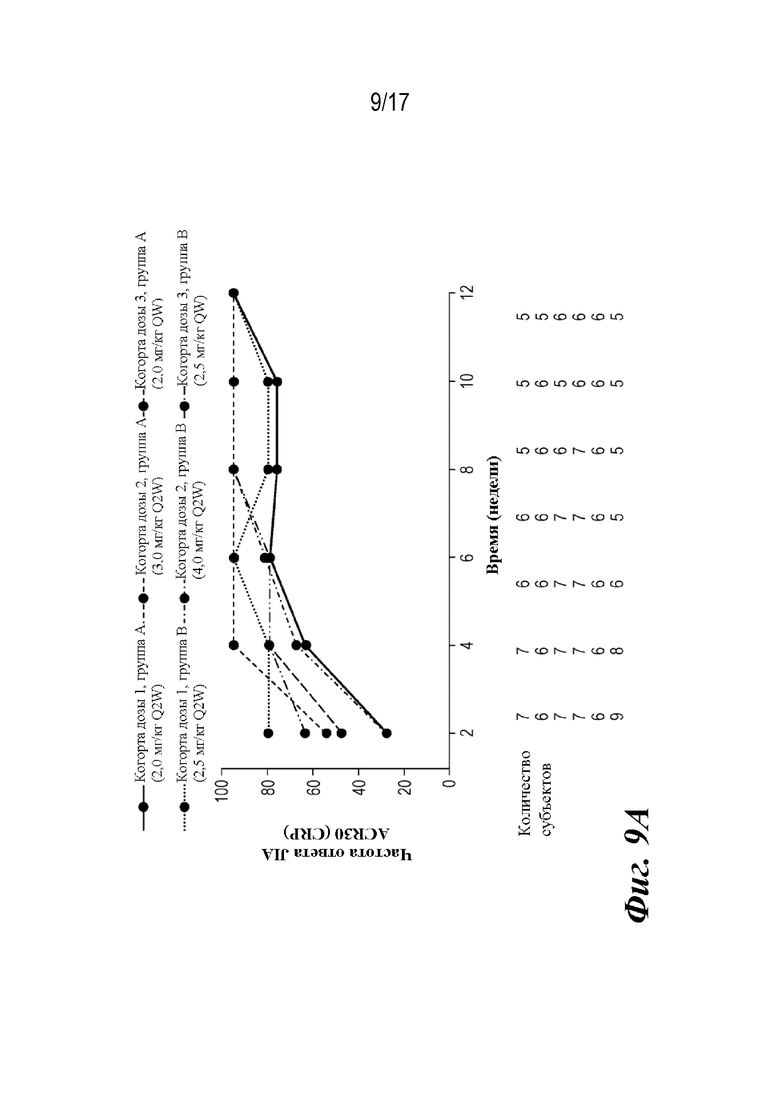
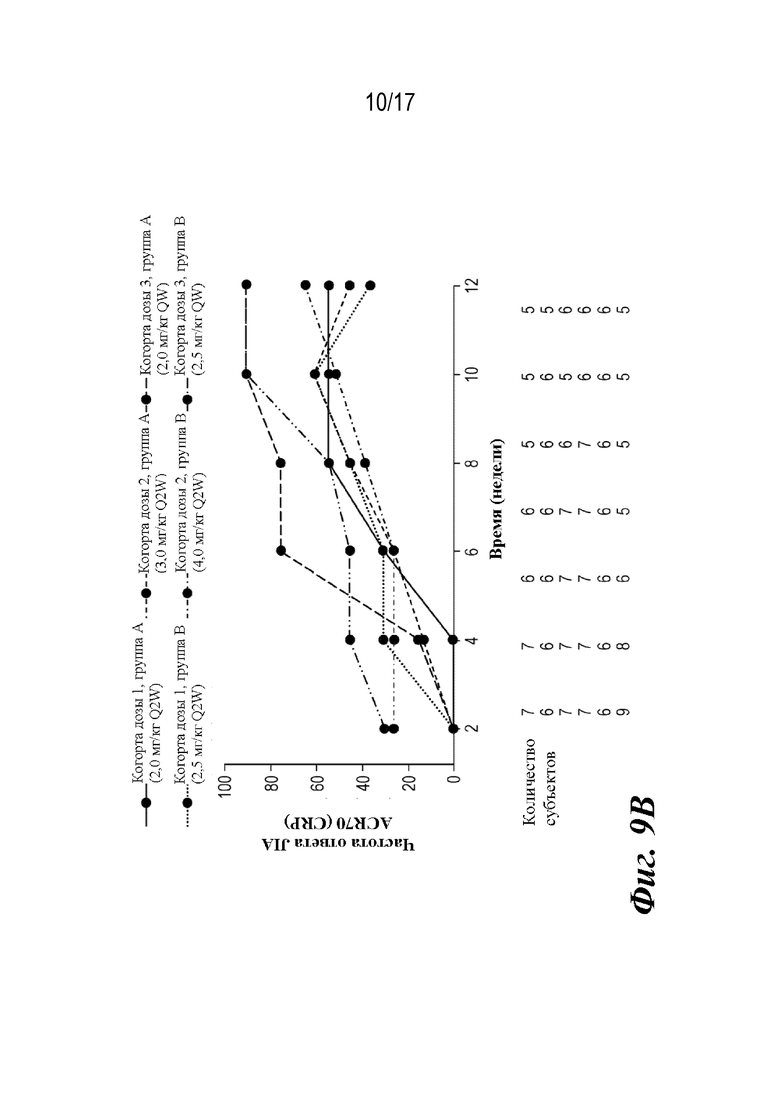
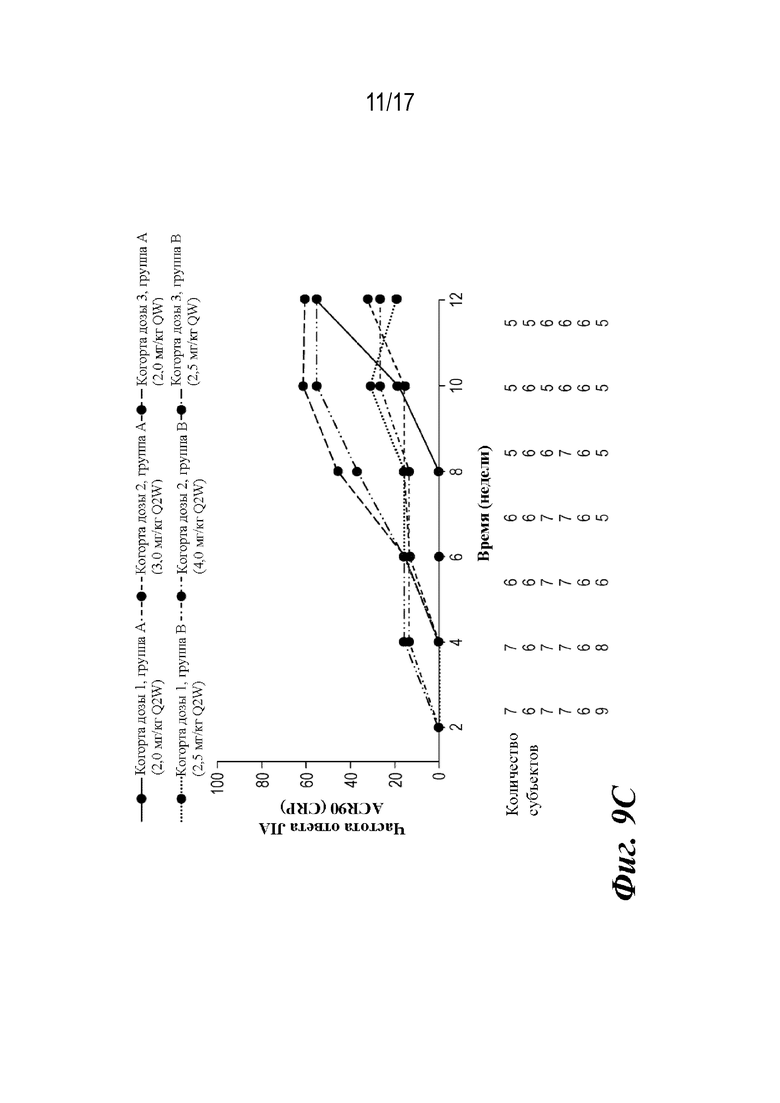
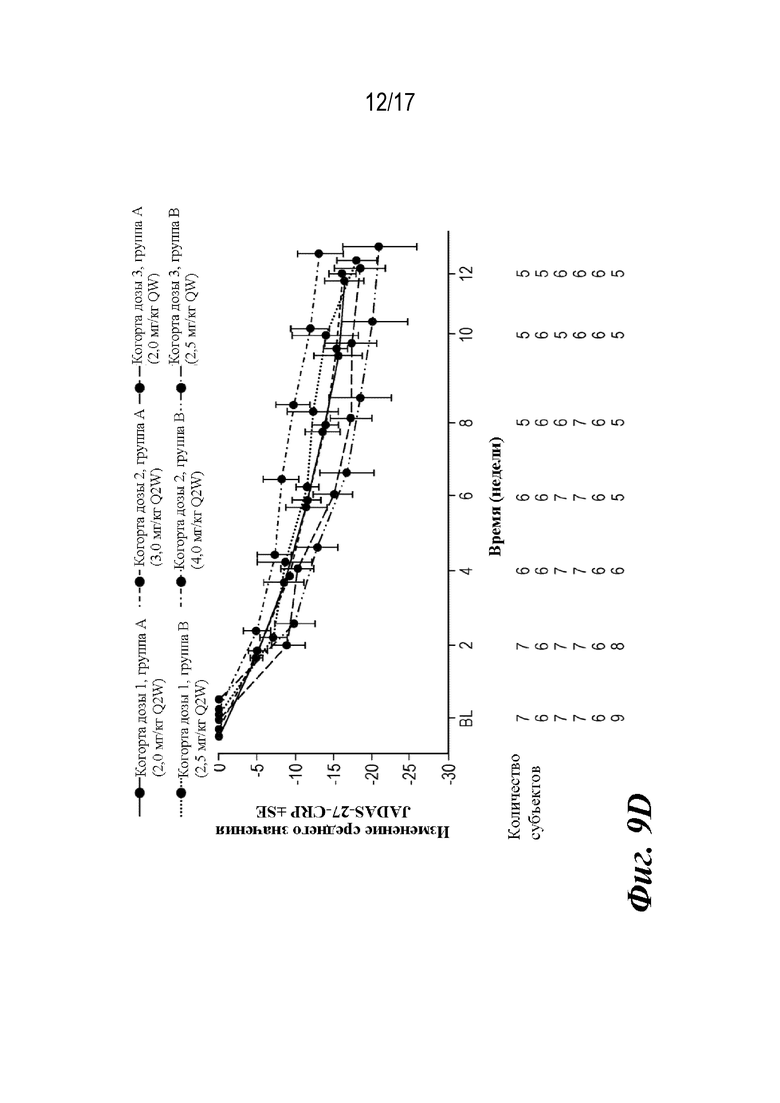
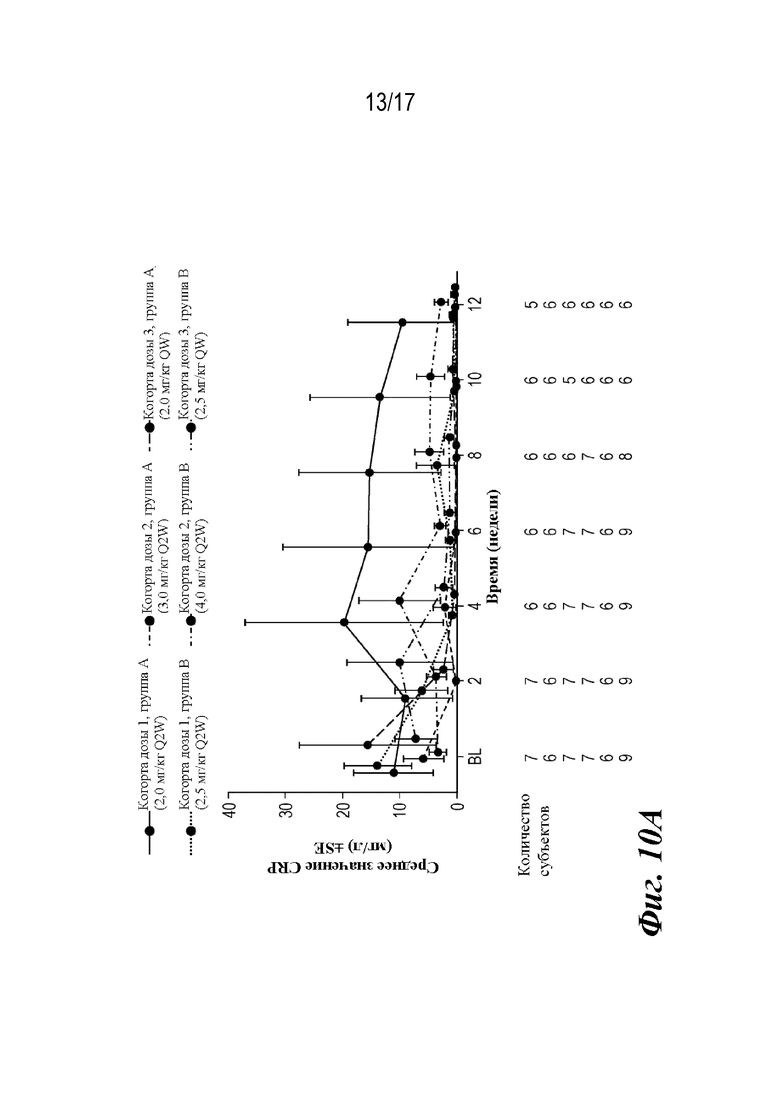
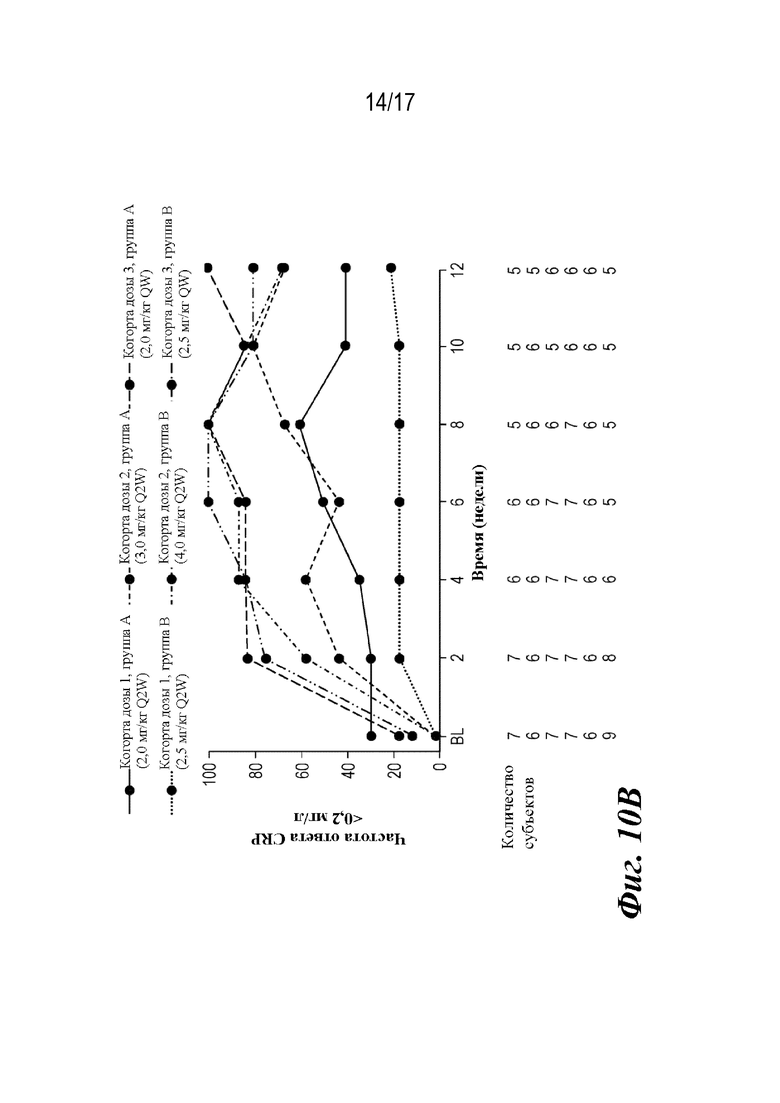
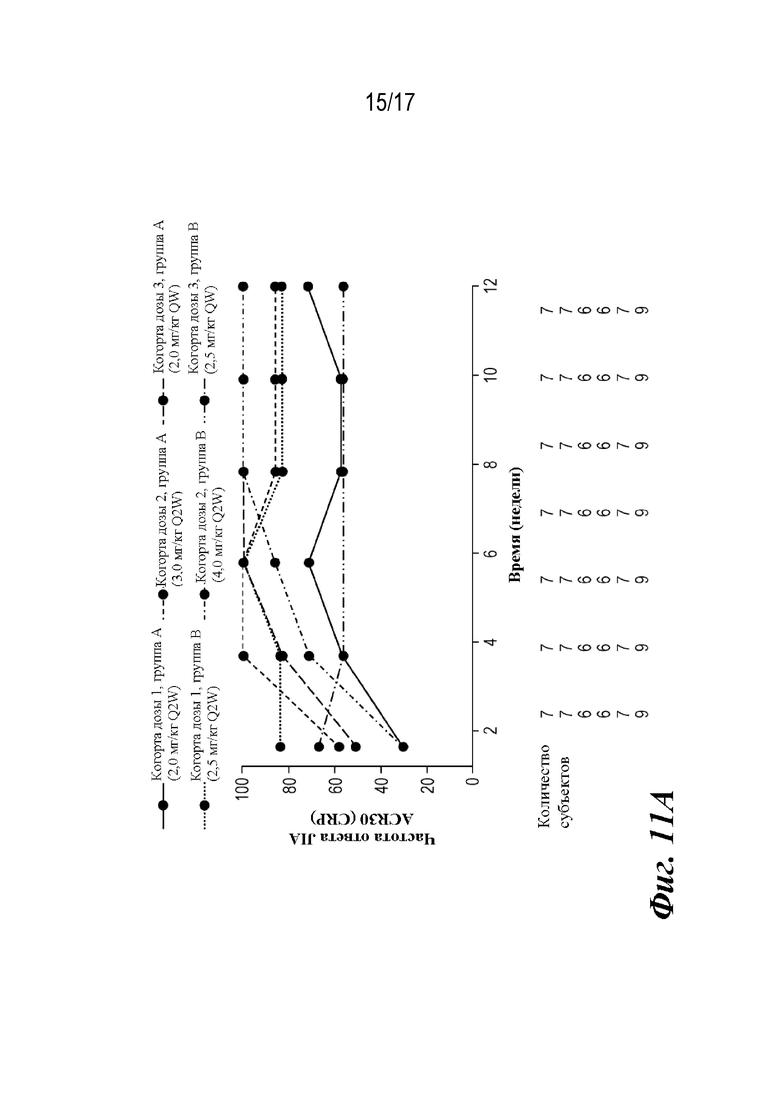
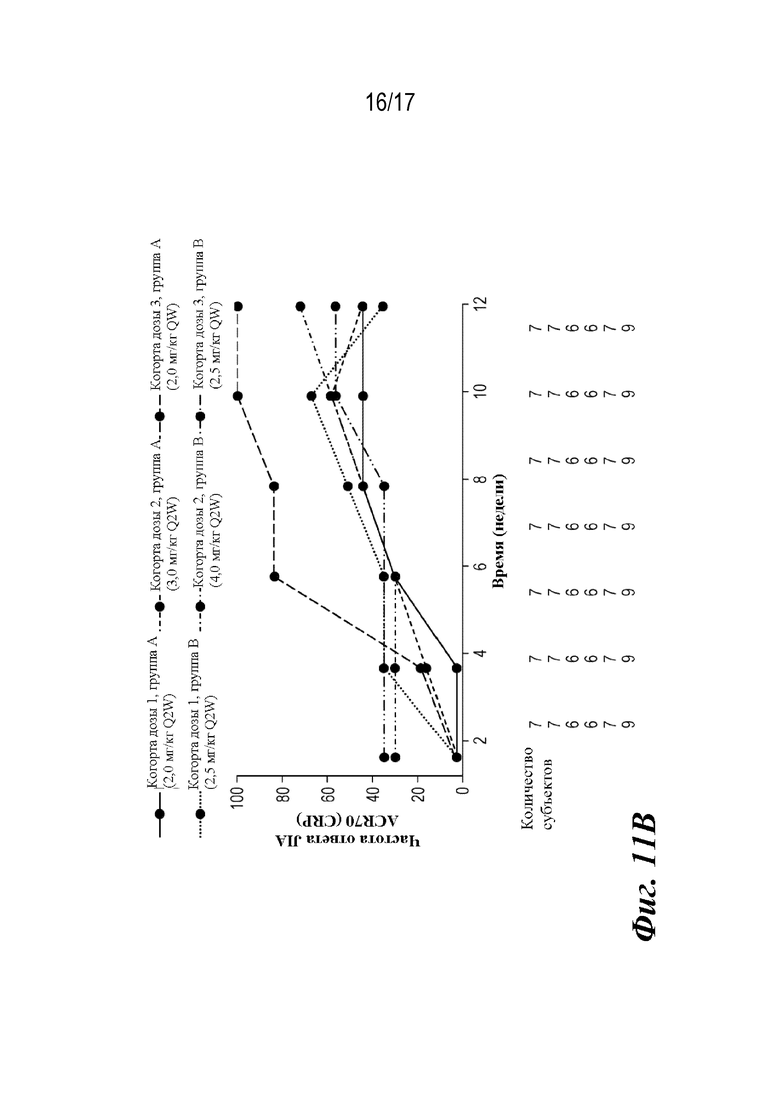

Группа изобретений относится к способам лечения системного ювенильного идиопатического артрита с полиартикулярным течением с помощью антитела, которое специфически связывает рецептор интерлейкина-6 человека (IL-6R). Антитело вводится подкожно в дозе от 2 до 4 мг/кг один раз в неделю или один раз в две недели. Возраст субъекта может составлять от 2 до 17 лет. Изобретение обеспечивает эффективные и безопасные способы лечения персистирующих типов JIA. 3 н. и 13 з.п. ф-лы, 11 ил., 33 табл., 2 пр.
1. Способ лечения ювенильного идиопатического артрита (JIA) у нуждающегося в этом субъекта, включающий введение эффективного количества антитела, которое специфически связывает рецептор интерлейкина 6 (IL-6) и содержит последовательность тяжелой цепи под SEQ ID NO: 9 и последовательность легкой цепи под SEQ ID NO: 10, где антитело вводится подкожно в дозе, составляющей от 2 до 4 мг/кг один раз в неделю или один раз в две недели.
2. Способ по п. 1, где JIA представляет собой JIA с полиартикулярным течением (pcJIA).
3. Способ по п. 1 или 2, где у субъекта наблюдался недостаточный ответ на лечение, включающее кортикостероид, синтетическое модифицирующее заболевание противоревматическое лекарственное средство (sDMARD) и/или биологическое DMARD, и субъект рассматривается как кандидат на лечение биологическим модифицирующим заболевание противоревматическим лекарственным средством (DMARD).
4. Способ по любому из пп. 1-3, где лечение субъекта включает улучшение показателя согласно Американской коллегии ревматологов (ACR) ювенильного идиопатического артрита у субъекта, улучшение в отношении компонента ACR JIA у субъекта, снижение уровня кортикостероидов, вводимых субъекту, по сравнению с уровнем до введения антитела, снижение индекса активности заболевания, представляющего собой ювенильный артрит, у субъекта, достижение клинически неактивного заболевания по критериям Уоллиса или любую их комбинацию.
5. Способ по любому из пп. 1-4, где субъект:
(a) имеет пять или более активных суставов; или
(b) страдает артритом в двух или более суставах с сопутствующей или предшествующей ежедневной лихорадкой с пиками в течение по меньшей мере 3 дней, которая сопровождается одним или несколькими из кратковременной эритематозной сыпи; генерализованного увеличения лимфатических узлов; гепатомегалии, спленомегалии, серозита или их комбинации.
6. Способ по любому из пп. 1-5, где доза антитела вводится в фармацевтической композиции.
7. Способ по любому из пп. 1-6, где возраст субъекта составляет от 1 года до 17 лет, субъект имеет вес тела, составляющий по меньшей мере 10 кг, менее чем 30 кг или менее чем 60 кг.
8. Способ лечения JIA с полиартикулярным течением (pcJIA) у нуждающегося в этом субъекта, включающий введение эффективного количества антитела, которое специфически связывается с рецептором IL-6 и содержит последовательность тяжелой цепи под SEQ ID NO: 9 и последовательность легкой цепи под SEQ ID NO: 10, где антитело вводится в дозе, составляющей от 2 до 4 мг/кг один раз в неделю или один раз в две недели, где возраст субъекта составляет от 2 до 17 лет.
9. Способ по п. 8, где субъект имеет вес тела больше или равный 10 кг и меньше или равный 60 кг.
10. Способ лечения JIA с полиартикулярным течением (pcJIA) у нуждающегося в этом субъекта, включающий введение эффективного количества антитела, которое специфически связывает рецептор IL-6 и содержит последовательность тяжелой цепи под SEQ ID NO: 9 и последовательность легкой цепи под SEQ ID NO: 10, где антитело вводится подкожно и
(а) где вес тела субъекта больше или равен 30 кг и меньше 33 кг, и антитело вводят в дозе, составляющей 61,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 33 кг и меньше 37,5 кг, и антитело вводят в дозе, составляющей 70 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 37,5 кг и меньше 42 кг, и антитело вводят в дозе, составляющей 78,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 42 кг и меньше 46,5 кг, и антитело вводят в дозе, составляющей 87,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 46,5 кг и меньше 50,5 кг, и антитело вводят в дозе, составляющей 96,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 50,5 кг и меньше 55 кг, и антитело вводят в дозе, составляющей 105 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 55 кг и меньше 59,5 кг, и антитело вводят в дозе, составляющей 113,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 59,5 кг и меньше 64 кг, и антитело вводят в дозе, составляющей 122,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 64 кг и меньше 68 кг, и антитело вводят в дозе, составляющей 131,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 68 кг и меньше 72,5 кг, и антитело вводят в дозе, составляющей 140 мг, один раз в две недели или один раз в неделю; или где вес тела субъекта больше или равен 72,5 кг, и антитело вводят в дозе, составляющей 148,75 мг, один раз в две недели или один раз в неделю;
(b) где вес тела субъекта больше или равен 30 кг и меньше 31 кг, и антитело вводят в дозе, составляющей 87,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 31 кг и меньше 34 кг, и антитело вводят в дозе, составляющей 96,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 34 кг и меньше 37 кг, и антитело вводят в дозе, составляющей 105 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 37 кг и меньше 39,5 кг, и антитело вводят в дозе, составляющей 113,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 39,5 кг и меньше 42,5 кг, и антитело вводят в дозе, составляющей 122,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 42,5 кг и меньше 45 кг, и антитело вводят в дозе, составляющей 131,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 45 кг и меньше 48,5 кг, и антитело вводят в дозе, составляющей 140 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 48,5 кг и меньше 51,5 кг, и антитело вводят в дозе, составляющей 148,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 51,5 кг и меньше 54,5 кг, и антитело вводят в дозе, составляющей 157,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 54,5 кг и меньше 57 кг, и антитело вводят в дозе, составляющей 166,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 57 кг и меньше 63 кг, и антитело вводят в дозе, составляющей 175 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 63 кг, и антитело вводят в дозе, составляющей 192,5 мг, один раз в две недели или один раз в неделю;
(c) где вес тела субъекта больше или равен 10 кг и меньше 12,5 кг, и антитело вводят в дозе, составляющей 26,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 12,5 кг и меньше 16 кг, и антитело вводят в дозе, составляющей 35 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 16 кг и меньше 19,5 кг, и антитело вводят в дозе, составляющей 43,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 19,5 кг и меньше 23 кг, и антитело вводят в дозе, составляющей 52,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 23 кг и меньше 26,5 кг, и антитело вводят в дозе, составляющей 61,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 26,5 кг и меньше 30 кг, и антитело вводят в дозе, составляющей 70 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 30 кг и меньше 37,5 кг, и антитело вводят в дозе, составляющей 70 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 37,5 кг и меньше 42 кг, и антитело вводят в дозе, составляющей 78,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 42 кг и меньше 46,5 кг, и антитело вводят в дозе, составляющей 87,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 46,5 кг и меньше 50,5 кг, и антитело вводят в дозе, составляющей 96,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 50,5 кг и меньше 55 кг, и антитело вводят в дозе, составляющей 105 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 55 кг и меньше 59,5 кг, и антитело вводят в дозе, составляющей 113,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 59,5 кг и меньше 64 кг, и антитело вводят в дозе, составляющей 122,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 64 кг и меньше 68 кг, и антитело вводят в дозе, составляющей 131,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 68 кг и меньше 72,5 кг, и антитело вводят в дозе, составляющей 140 мг, один раз в две недели или один раз в неделю; или где вес тела субъекта больше или равен 72,5 кг, и антитело вводят в дозе, составляющей 148,75 мг, один раз в две недели или один раз в неделю; или
(d) где вес тела субъекта больше или равен 10 кг и меньше 12,5 кг, и антитело вводят в дозе, составляющей 43,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 12,5 кг и меньше 14,5 кг, и антитело вводят в дозе, составляющей 52,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 14,5 кг и меньше 16,5 кг, и антитело вводят в дозе, составляющей 61,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 16,5 кг и меньше 19 кг, и антитело вводят в дозе, составляющей 70 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 19 кг и меньше 21 кг, и антитело вводят в дозе, составляющей 78,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 21 кг и меньше 23,5 кг и антитело вводят в дозе, составляющей 87,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 23,5 кг и меньше 25,5 кг, и антитело вводят в дозе, составляющей 96,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 25,5 кг и меньше 27,5 кг, и антитело вводят в дозе, составляющей 105 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 27,5 кг и меньше 30 кг, и антитело вводят в дозе, составляющей 113,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 30 кг и меньше 39,5 кг, и антитело вводят в дозе, составляющей 113,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 39,5 кг и меньше 42,5 кг, и антитело вводят в дозе, составляющей 122,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 42,5 кг и меньше 45 кг, и антитело вводят в дозе, составляющей 131,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 45 кг и меньше 48,5 кг, и антитело вводят в дозе, составляющей 140 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 48,5 кг и меньше 51,5 кг, и антитело вводят в дозе, составляющей 148,75 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 51,5 кг и меньше 54,5 кг, и антитело вводят в дозе, составляющей 157,5 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 54,5 кг и меньше 57 кг, и антитело вводят в дозе, составляющей 166,25 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 57 кг и меньше 60,5 кг, и антитело вводят в дозе, составляющей 175 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 60,5 кг и меньше 63 кг, и антитело вводят в дозе, составляющей 175 мг, один раз в две недели или один раз в неделю; где вес тела субъекта больше или равен 63 кг, и антитело вводят в дозе, составляющей 192,5 мг, один раз в две недели или один раз в неделю.
11. Способ по п. 9, где:
если вес тела субъекта больше чем или равен 10 кг и меньше чем 30 кг, антитело вводится в дозе от 2,5 до 4 мг/кг один раз в две недели или в дозе 2,5 мг/кг один раз в неделю или,
если вес тела субъекта больше чем или равен 30 кг и меньше чем или равен 60 кг, антитело вводится в дозе от 2 до 3 мг/кг один раз в две недели или в дозе 2 мг/кг один раз в неделю.
12. Способ по п. 11, где:
если вес тела субъекта больше чем или равен 30 кг и меньше чем или равен 60 кг, антитело вводится в дозе 3 мг/кг один раз в две недели или,
если вес тела субъекта больше чем или равен 10 кг и меньше чем 30 кг, антитело вводится в дозе 4 мг/кг один раз в две недели.
13. Способ по любому из пп. 1-12, где субъект страдает от по меньшей мере одного симптома pcJIA, где по меньшей мере один симптом представляет собой хромоту, скованность при пробуждении; нежелание пользоваться рукой или ногой; пониженный уровень активности; отек суставов; и/или трудности с мелкой моторикой.
14. Способ по любому из пп. 1-13, где возраст субъекта составляет 12-17 лет.
15. Способ по любому из пп. 1-14, где способ дополнительно содержит введение по меньшей мере одного другого модифицирующего заболевание противоревматического лекарственного средства (sDMARD).
16. Способ по любому из пп. 1-15, где антитело представляет собой сарилумаб.
| НЕСУЩИЙ БЛОК ДЛЯ ПЕРЕМЕЩЕНИЯ ПО МЕНЬШЕЙ МЕРЕ ОДНОГО ПОТОКА ОБЪЕКТОВ И СПОСОБ ОБРАБОТКИ ЗОН ВНЕШНИХ ПОВЕРХНОСТЕЙ НЕСКОЛЬКИХ ОБЪЕКТОВ | 2013 |
|
RU2776735C2 |
| Токарный резец | 1924 |
|
SU2016A1 |
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| TURNIER, J.L | |||
| et.al | |||
| Tocilizumab for treating juvenile idiopathic | |||

