Область изобретения
Изобретение относится к фармацевтике, а именно к системам доставки лекарственных веществ белковой природы.
В настоящем изобретении используется уникальная методика двойного ининкапсулирования для защиты, контролируемого высвобождения активного вещества и улучшения биодоступности биомолекул в желудочно-кишечном тракте.
Уровень техники
В наши дни в результате развития сферы биотехнологий на коммерческом уровне производится огромное количество пептидных и белковых препаратов. Клиническая разработка препаратов таких типов невозможна без специального технического оснащения, в отличие от традиционно используемых препаратов на основе малых молекул. Пероральный прием препаратов является самым распространенным способом приема, но он, как правило, невозможен в случае пептидных и белковых препаратов. Основной причиной низкой биодоступности биопрепаратов при пероральном приеме является досистемное ферментативное расщепление и затрудненное проникновение в кишечную мембрану. В последние десятилетия был досконально изучен механизм всасывания макромолекулярных препаратов из желудочно-кишечного тракта (ЖКТ), а также барьеры, препятствующие всасыванию в ЖКТ. Были испытаны различные методы преодоления этих барьеров и стратегии разработки безопасных и эффективных систем для перорального приема белков. Пероральный прием пептидов и белков все еще вызывает значительные трудности и является объектом многих фармакологических исследований.
Для увеличения биодоступности биологических препаратов при пероральном приеме может быть эффективна стратегия, включающая в себя повышение уровня проникновения или использование ингибиторов протеазы в качестве добавок. Эта стратегия также может повысить воспроизводимую биодоступность. Несмотря на то что эти методы могут успешно применяться в лаборатории, они все еще не признаются большинством практикующих врачей и регулирующих органов. Использование ингибиторов ферментов в ходе долгосрочной терапии остается спорным вопросом из-за возможного всасывания нежелательных белков, нарушения усвоения питательных белков и стимуляции секреции протеазы в результате ответной регуляции. Была изучена стратегия модулирования проницаемости плотных контактов для стимулирования параклеточного переноса молекул препарата. Токсин ZOT (Zonula Occludens toxin), хитозан, тиолированные полимеры и Pz-пептид демонстрируют выраженную способность к повышению всасывания макромолекулярных препаратов. Однако вероятно, данная стратегия также может вызвать вопросы, связанные с безопасностью. После открытия плотного контакта стимулируется перенос не только препаратов, но также потенциально токсичных или нежелательных молекул, присутствующих в ЖКТ.
Поэтому в данном случае подходящим вариантом может быть использование систем-носителей для доставки частиц, таких как гидрогели, наночастицы, микросферы, и систем доставки лекарственных средств на основе липидов (например, наноэмульсий, липосом и твердых липидных наночастиц), которые направлены на защиту пептидов и белков от ферментативного распада и которые способны модулировать перенос препарата.
Одна из наиболее важных задач исследований доставки лекарственных веществ в нужную точку организма заключается в разработке нано- и микросистем, способных доставлять лекарственные вещества в нужную точку в заданных участках тела с требуемым высвобождением при каждом конкретном случае лечения и в правильной лекарственной форме.
Обычно системы доставки лекарственных веществ в виде микрочастиц, предназначенные для орального приема, включают в себя технологии приготовления лекарственных веществ в форме гранул, микрокапсул, липосом, микрочастиц и наночастиц, которые обеспечивают модулированное высвобождение и всасывание данного лекарственного средства [1].
Очень быстро растет использование полимерных материалов для медицинских целей. Полимеры нашли применение в различных областях биомедицины, таких как: имплантация (вживление) медицинских устройств и искусственных органов, инженерия тканей, протезирование, офтальмология, стоматология, репарация кости, системы доставки лекарственных веществ в нужную точку организма. Среди них использование природных биополимеров для самого широкого круга применений в науке о жизни обладает такими преимуществами как биосовместимость и биоразлагаемость, обеспечивающими таким образом экологическую безопасность и возможность получения широкого ряда модифицируемых химическим и ферментативным путем производных веществ для конкретных случаев применения.
Метод ионотропного гелеобразования, при котором для формирования гранул используются природные полисахариды, очень прост и осуществляется в обычных условиях. Обратимое физическое сшивание путем электростатического взаимодействия вместо химического сшивания предотвращает возможную токсичность реагентов и другие нежелательные эффекты.
Использование этой технологии для разработки системы доставки лекарственного препарата с контролируемым высвобождением обладает несколькими преимуществами:
- Маскировка вкуса,
- Улучшение стабильности формуляции при длительном хранении,
- Улучшение стабильности формуляции в желудочно-кишечном тракте,
- Улучшение адгезионных свойств этих формуляций со слизистой оболочкой,
- Селективная доставка лекарственного препарата в конкретном месте организма,
- Устранение опасности интоксикации в связи с их способностью равномерно распределяться по всему желудочно-кишечному тракту,
- Улучшение биодоступности,
- Улучшение терапевтической эффективности.
Полисахариды [2, 3] как класс природных полимеров привлекли очень большое внимание в области разработок систем доставки лекарственных средств в организме частично из-за их биосовместимости и биоразлагаемости. Альгинат, представляющий собой природный биополимер, находит все возрастающее применение в различных областях. Он успешно использовался в течение многих лет в пищевой промышленности и в промышленности производства напитков как загуститель, желирующее вещество и коллоидный стабилизатор. Он также обладает несколькими уникальными свойствами, которые открыли для него область применения в качестве матрицы для ининкапсулирования и/или доставки к тканям целого ряда белков, лекарственных веществ и клеток. Эти свойства включают в себя: (i) создание относительно инертной водной среды внутри этой матрицы; (ii) процесс упаковки в капсулу при умеренной комнатной температуре без использования органических растворителей; (iii) высокую проницаемость геля, которая обеспечивает высокие скорости диффузии макромолекул; (iv) возможность регулирования этой проницаемости с помощью простых операций покрытия оболочкой; и (v) растворение и биоразложение этой системы при нормальных физиологических условиях [4].
Альгинат - это водорастворимый линейный полианионный полисахарид, экстрагируемый из крупной бурой водоросли, состоящий из чередующихся звеньев остатков связанной в положении 1-4 α-L-гулуроновой и β-D-маннуроновой кислоты [5]. Гелевые гранулы получают путем преобразования альгината из золя в гель, осуществляемого перекрестным сшиванием альгината, в основном, двухвалентными катионами. Гулуроновая кислота ответственна за образование геля из альгината с помощью этих катионов в растворе. Матрица из альгината, состоящая из открытой решетки, образует пористые гранулы.
Другим примером природных полисахаридов является хитозан - биосовместимый, биоразлагаемый, нетоксичный линейный сополимер, состоящий из звеньев β (1-4)-связанных 2-амино-2-деокси-D-глюкозы (D-глюкозамин) и 2-ацетамидо-2-деокси-D-глюкозы (N-ацетил-D-глюкозамин), и обладающий структурным сходством с целлюлозой (состоит из звеньев β (1-4)-связанной D-глюкозы). Хитозан - это N-деацетилированное производное хитина, хотя это N-деацетилирование никогда не бывает законченным, в котором ряд аминогрупп является доступным, что превращает его в поликатионный полисахарид. В зависимости от объема/степени деацетилирования, встречаются различные марки хитозана. Из-за своего свойства гелеобразования он используется при разработке системы доставки лекарственных веществ к тканям [6].
Пектины представляют собой семейство растительных полисахаридов, которые также обладают гелеобразующей способностью. D-галактуроновая кислота, связанная вместе с помощью α 1→4 гликозидных связей образует макромолекулярные звенья. Полигалактуроновые звенья выстроены в тройную винтовую спираль и прерываются связями с β, L-рамнозой. Полигалактуроновая кислота частично этерифицирована метальными группами, а свободные кислотные группы могут быть частично или полностью нейтрализованы катионными ионами. Соотношение этерифицированных групп галактуроновой кислоты к суммарному количеству групп галактуроновой кислоты - называемое степенью этерификации СЭ - имеет жизненно важное влияние на свойства пектина, такие как растворимость, гелеобразование и тенденция к связыванию двухвалентными ионами. СЭ в 50% делит коммерческие пектины на пектин с высоким содержанием сложного эфира (ВМ) и на пектин с низким содержанием сложного эфира (НМ). Эти две группы пектина желатинируются разными механизмами. Катионные ионы могут реагировать со свободными карбоксильными группами в цепях пектина с низким содержанием метоксильных групп, приводя к образованию структуры геля с поперечными связями, которая не растворима в воде. Нерастворимость поперечно-связанного пектината в водных средах вместе со склонностью пектина к ферментативному разложению делает этот поперечно-связанный пектинат кандидатом для различных систем доставки лекарственных средств к тканям, в особенности для доставки лекарственных средств в область толстой кишки.
Гранулы на основе полисахаридов можно производить различными способами. Один из способов их приготовления - это способ ионотропного гелеобразования. При способе ионотропного гелеобразования полисахариды вводят в реакцию с противоионами. Из-за образования комплексных соединений между противоположно заряженными продуктами реакции полисахариды подвергаются ионному гелеобразованию и выпадают в осадок, образуя сферические частицы. Эти гранулы обычно отделяют фильтрованием, промывают дистиллированной водой и сушат.
При остальных способах используются чувствительные к температуре полимеры, которые подвергаются спонтанному гелеобразованию в зависимости от чувствительного к температуре фазового перехода.
Свойства гранул из катион-связанных альгинатов, например, зависят от структуры, состава и молекулярного веса полимерных гранул [7]. Гибкость полимерного раствора зависит от концентрации звеньев a-D-маннуроновой кислоты и a-L-глуроновой кислоты в ряду MG>MM>GG. Гранулы с самой низкой сжимаемостью, с большей пористостью, механической прочностью и наивысшей стабильностью в отношении одновалентных катионов состоят главным образом из a-L-глуроновой кислоты, со средним содержанием не менее 70% и со средней длиной звена a-L-глуроновой кислоты 15. В то время как гелевые элементы, приготовленные с низким содержанием a-L-глуроновой кислоты, будут более эластичными, то те, которые приготовляются с высоким содержанием а-глуроновой кислоты, будут более хрупкими. В дополнение к этому, альгинат будет образовывать стабильные гелевые элементы в температурном диапазоне 0-100°С, однако чем выше температура в ходе формования, тем менее жесткими будут получаемые гелевые элементы. Полимер из альгината обладает очень сильной биоадгезионной способностью, что вновь делает его жизнеспособным кандидатом для доставки лекарственных средств к слизистым оболочкам желудочно-кишечного тракта.
Хитозан - это слабое основание, и он нерастворим в воде и в органических растворителях, однако, он растворим в разбавленном водном растворе кислоты (рН<6,5), что может преобразовывать звенья глюкозамина в растворимую форму 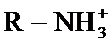
Гранулы диаметром более 1,0 мм обычно приготавливают капельным методом, путем переноса в виде капель раствора полимера и какого-то лекарственного вещества в перемешиваемый раствор для образования поперечных связей [9]. Размер иглы и вязкость раствора полимера будут определять диаметр получаемой гранулы, при этом при большей игле и при более вязких растворах будут производиться гранулы большего диаметра. Наряду с этим вязкость полимера также часто влияет на форму вырабатываемых гранул. При возрастании концентрации полимера вырабатываемые гранулы становятся более сферическими.
Когда требуются гранулы меньше 0,2 мм в диаметре, можно использовать три методики для получения микрогранул: технология вибрирующего сопла, коацервация и эмульгирование.
Обычно при технологии вибрирующего сопла микрогранулы получают из хорошо перемешиваемых растворов соответствующего раствора полимера и вещества, которые загружаются в шприц, установленный на насосе или через сосуд под давлением. Технология вибрирующего сопла основана на принципе, основанном на том, что струи ламинарного потока жидкости разбиваются на капельки одинаковых размеров с помощью наложенной вибрации. Выбираемая частота вибрации определяет качество получаемых капелек. Размер шариков задают предварительно в диапазоне от 0,15 мм до 2 мм со сферической формой, при узком разбросе размеров (стандартное отклонение <5%) и с производительностью до 6.000 гранул в секунду. Цепочка из капелек заряжается электростатически, что заставляет капельки отталкиваться друг от друга, устраняя таким образом возможность слипания. Капельки падают в отверждающий раствор, в котором гранулы быстро полимеризуются, или в охлаждающую камеру, в которой гранулы затвердевают. В случае приготовления микрогранул размер гранул может регулироваться типом полимера, диаметром сопла, производительностью шприцевого насоса и расстоянием между соплом и поверхностью раствора, обеспечивающего образование поперечных связей.
Другая методика приготовления микрогранул - это комплексная коацервация противоположно заряженных полиэлектролитов. С помощью этой методики при специфических условиях концентрации полиионов, рН и ионной силы эта смесь будет разделяться на плотную фазу, способную к слиянию, и на разбавленную равновесную фазу. Например, комплексная коацервация между альгиновой кислотой и хитозаном была достигнута распылением раствора альгината натрия в раствор хитозана, проводя к получению прочных микрогранул, которые оставались стабильными в широком диапазоне рН [10].
Заключение в капсулы активных компонентов, таких как ферменты, лекарственные препараты, витамины, масла, клетки, наночастицы, нанолипосомы, можно выполнять путем ионотропного и зависящего от температуры гелеобразования. В зависимости от заключаемого в капсулу объекта, например, водо- и маслорастворимого, гранула может делиться на матричные гранулы и капсулы с содержимым на масляной основе и с водорастворимой полимерной оболочкой, соответственно. Поведение молекул в отношении диффузии внутри и вне гранулы может модифицироваться путем добавления дополнительной мембраны.
Существует три механизма выделения активного элемента, связанные с заключением лекарственного вещества в капсулы в виде матриц на основе полисахаридов: диффузия заключаемого в капсулу лекарственного вещества через поры полимерной структуры и его высвобождение в результате разложения полимера.
В ходе исследования с помощью электронного микроскопа были выявлены размеры пор гранул альгината кальция в диапазоне диаметров от 5 до 300 нм. Диффузия небольших молекул не зависит от матрицы, в то время как диффузия более крупных молекул, таких как белки, также как и заключенных в капсулу объектов, например, липосом и наночастиц (НЧ), будет зависеть от молекулярного веса альгинатной матрицы и от размера пор. Увеличение концентрации альгината внутри гранул может снизить скорость диффузии крупных молекул и из альгинатных гранул. Кроме того, гели с более крупными звеньями α-L-глуроновой кислоты дают структуры с более крупными порами и обеспечивают наиболее высокие скорости диффузии белков. В дополнение к этому, на скорость диффузии может влиять заряд заключенного в матрицу белка. Белок с четким положительным зарядом будет создавать различные виды взаимодействия с отрицательно заряженным полимером и будет ингибировать диффузию из этой матрицы [11].
Заключенные в капсулу объекты могут высвобождаться вследствие разложения геля альгината с поперечными связями. Агент, способствующий образованию хелатных соединений, таких как лактат, цитрат, фосфат, или высокая концентрация ионов могут быть использованы для удаления создающих поперечные связи двухвалентных ионов, что приводит к разрушению матричного геля [12].
Как было описано в предыдущем разделе, высвобождение в ткани заключенных в гранулы объектов регулируется диффузией и/или разложением матрицы на полимерной основе, и эти объекты можно охарактеризовать иначе как зависимые от разбухания матрицы. Поведение в отношении разбухания зависит от рН.
Гранулы, например, на основе альгинатов слегка разбухают в желудке, что дает незначительное выделение лекарственного вещества при рН 1-4, однако разбухание постепенно усиливается при уровне рН в кишечнике, что приводит к глубокому высвобождению лекарственного вещества [13, 14]. Другая система, то есть с гелевыми гранулами из смеси альгината и хитозана, приготовленными на основе двойного перекрестного сшивания, когда гомогенный раствор альгината и хитозана в разных пропорциях вводили в виде капель вначале в раствор хлористого кальция (агент поперечного сшивания альгината), а затем во второй агент поперечного сшивания - сульфат натрия, - давала сравнимый рисунок выделения лекарственного вещества. Результаты этого исследования продемонстрировали, что незначительное высвобождение (1-3%) получали в моделируемых жидких средах желудка (SGF, 4 часа), в то время как 50-80% этого лекарственного вещества выделялось в моделируемых жидких средах кишечника (SIF, 3 часа), а 87-97% - в моделируемых жидких средах толстой кишки (SCF, 3 часа), что показало, что гранулы с двойными поперечными связями имеют потенциально низкую способность выделения лекарственного вещества, специфичного для кишечника или толстой кишки [15].
Высвобождение активных беков и пептидов в полностью работоспособном состоянии в целевую точку организма часто ограничивается технологией и физиологическими барьерами желудочно-кишечного тракта (ЖКТ). Один путь решения этих затруднений заключается в капсулировании, которое может стабилизировать биологически активные вещества во время технологических процессов и при хранении, обеспечивать защиту от низкого рН и ферментативного разложения после применения и регулировать высвобождение заключенного в капсулу биологически активного вещества в требуемой целевой точке организма. Липосомы (фосфолипидные пузырьки) являются одной такой платформой, которая используется для инкапсулирования и управляемого высвобождении лекарственного вещества и которая успешно и в течение длительного времени используется в производстве фармацевтических препаратов [16, 17], косметических средств [18] и пищевых продуктов [19, 20]. Показано, что в этих целях используют в основном большие однослойные однослойные липосомы (LUV) в диапазоне размеров 50-200 нм, так как этот диапазон размеров представляет собой компромисс между эффективностью дозировки липосом (которая возрастает с возрастанием размера), стабильностью липосом (которая снижается при увеличении размера) и способностью проникновения в клетки (которая снижается с увеличением размера).
В частности, наноразмерные биоразлагаемые системы для доставки лекарственных веществ (СДЛВСДЛВ, <1 μM) обладают следующими преимуществами:
- Они могут обеспечивать наличие как олеофильной, так и водной среды в одной системе, и поэтому они пригодны для ининкапсулирования гидрофобных, амфипатических и гидрофильных лекарственных веществ.
- Они биосовместимы из-за их биоразлагаемости и низкой токсичности.
- Они могут служить в качестве механизма для управляемого высвобождения лекарственных препаратов в жидких средах организма.
- Они могут вводиться в организм многими путями, включая внутривенный, оральный, внутримышечный, подкожный, глазной и легочный.
Что касается высвобождения белков в нужной точке организма, то затруднения по стабильности в ЖКТ представляют собой главный недостаток, связанный с оральным использованием липосом [21]. Эта нестабильность возникает вследствие значительных колебания уровней рН и присутствия липаз и солей желчных кислот в желудочной и кишечной жидкости, при этом все вышеперечисленное может дестабилизировать липосомы [22]. Однако полностью функциональные пептиды и белки, которые денатурируются физиологическими условиями в ЖКТ, как сообщалось, захватываются пейеровыми бляшками в кишечнике если они доставляются с помощью липосом, хотя только в очень малых концентрациях [23].
В течение длительного времени предпринимаются усилия по преодолению сложностей в отношении нестабильности липосом, предназначенных для орального приема, путем нанесения на липосомы полимерных покрытий [24-27] или сахаросодержащими цепочками муцина или полиэтиленгликолем или путем получения липосом с гелевым заполнителем [28-31], в то время как гелеобразующий полимер заполняет липосомы и затем подвергается поперечному сшиванию на месте. Когда альгинат включали в состав заполнителя многослойных липосом (MLV), это приводило к повышению стабильности при кислотной рН; однако, при нейтральной рН этот альгинат вызывал разбухание и агрегацию липосом [32].
СОКРАЩЕНИЯ
СДЛВ - система доставки лекарственных веществ
БСА - бычий сывороточный альбумин
ФИТС - флуоресцеинизотиоцианат
СЭМ - сканирующий электронный микроскоп
MTS - 3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2Н-тетразолий
Рарр - коэффициент видимой проницаемости.
ДПФХ - 1,2-дипальмитоил-sn-глицеро-3-фосфохолин
ДМФХ - 1,2-димиристоил-sn-глицеро-3-фосфохолин
ДОФЭ - 1,2-диолеоил-sn-глицеро-3-фосфоэтаноламин
ДМФА - Fluorenylmethyloxycarbonyl (9-флуоренилметилоксикарбонил)
ТФК - трифторуксусная кислота
ДДВ - бидистиллят воды
ЭИ эффективность ининкапсулирования
КЗ - коэффициент загрузки
СП - содержание препарата
ИПД - индекс полидисперсности
ИКС - искусственная кишечная среда
ВП - высвобождение препарата
ФЛ - фосфолипиды
ПЭГ - полиэтиленгликоль
ДСФЭ дистеарилфосфатидилэтаноламин
DE частично деэтерифицированный пектин.
Краткое описание фигур
Схема 1 Меченый пептид.
Рис.1 ВЭЖХ хроматограмма неочищенного пептида-ФИТЦ 10-мера, сделанная хроматографом Alliance Waters с использованием многоволнового детектора флуоресценции 2475 на длине волны: 485/528 нм и анализ МС время-пролетной ионизацией лазерной десорбции с использованием матрицы.
Рис. 2 ВЭЖХ хроматограмма очищенного продукта. Чистота пептидов и Mw были определена прибором Voyager DE-Pro MALDI-TOF (ABI) (В) с использованием α-циано 4-гидроксикоричной кислоты в 0,1% ТХК: АЦН (1:1) в качестве матрицы в линейно-положительном режиме.
Рис. 3 - Хроматограмма высокоэффективной жидкостной хроматографии 10-звенного полимерного пептида-ФИТС, инкапсулированного в большие однослойные липосомы, полученная на хроматографе Alliance Waters с использованием многоволнового флуоресцентного детектора модели 2475 на длине волны 485/528 нм.
На Рис. 4А отображены пустые однослойные липосомы LUV на основе ДМФХ, меченных оптической меткой LR-PE (х 88К); на В и С - анализ ПЭМПЭМ однослойных липосом на основе ДМФХ и ДПФХ, соответственно, меченных оптической меткой LR-PE, с инкапсулированным пептидом - ФИТСФИТС (х 88К); на D и Е отображены однослойные липосомы однослойные LUV на основе DМРС:ДОФЕ и DРРС:ДОФЕ, соответственно, меченные оптической меткой LR-PE, с инкапсулированным пептидом -ФИТС (х 88К).
Рис. 5 отображает анализ ПЭМ однослойных липосом LUV: А отображает однослойные липосомы LUV на основе ДМФХ:РF 127, меченных оптической меткой LR-PE, с инкапсулированным пептидом - ФИТС (х 25К); В отображает однослойные липосомы LUV на основе ДПФХ:РF 127, меченных оптической меткой LR-PE, с инкапсулированным пептидом - ФИТС (х 25К).
Рис. 6 отображает накопительную кинетику высвобождения лекарственного вещества, бычего сывороточного альбумина БСА-ФИТС (А) и пептида-ФИТС (В) из однослойных липосом LUV в SIF, согласно описанию в разделе Методики.
Рис. 7 отображает влияние составов однослойных липосом LUV, в который загружен пептид - ФИТС, на жизнеспособность клеток Сасо-2, с оценкой согласно методике MTS.
Рис. 8 отображает распределение в клетках Сасо-2 х пептида - ФИТС (зеленый) через 4 часа инкубирования с различными составами: (L27) LUV на основе ДМФХ; (L28) LUV на основе ДПФХ; (L28) LUV на основе способствующих слиянию ДМФХ/ДОФЕ; (L29) LUV на основе способствующих слиянию ДПФХ/ДОФЕ; (L35) LUV на основе ДМФХ. Обработанные клетки Сасо-2 анализировали с помощью Софокусного лазерного сканирующего микроскопа Fluoview 300 (CLSM, компания Olympus, Пенсильвания, США). Примечание: Красное окрашивание свидетельствует о распределении LUV, меченного с помощью LR-PE.
Рис. 9 отображает лиофилизованные гранулы хитозана с заключенными в него липосомами, наблюдаемые с помощью Сканирующего электронного микроскопа для окружающей среды (ESEM).
Рис. 10 отображает влажные гранулы из хитозана, наблюдаемые с помощью оптического микроскопа с увеличением 40.
Рис. 11 отображает накопительную кинетику высвобождения LUV, LR-PE, микрогранул на основе пектина (пектин - LUV) и микрогранул на основе хитозана (хитозан - LUV) в FaSSGF.
Рис. 12 отображает накопительную кинетику высвобождения LUV (А) и пептида - ФИТС (В) из гранулна основе хитозана в SIF, согласно описанию в разделе Методики.
Рис. 13 отображает влажные гранулы из пектина, наблюдаемые с помощью оптического микроскопа с увеличением 40.
Рис. 14 отображает влияние рН на плотность на кросс-линкера в пектинате кальция (СаР) (А) и концентрации CaCl2 на плотность на кросс-линкера в приготовленных матрицах СаР, (В) - оценка с помощью беспламенной Земановской атомной абсорбционной спектроскопии.
Рис. 15 отображает влияние времени инкубирования (А-30 секунд, В-60 секунд и С-30 минут) и увеличения концентраций CaCl2 (кружки 0,1%, треугольники 1,0% и квадратики 10,0% вес/объем) на стабильность in situ приготовленных матриц СаР, что выражается выделением свободного пектина в среду разбавителя, что определяется с помощью GPC. Отображаются средние значения по 3 измерениям.
Рис. 16 отображает суммарную фракцию пектина (A-DE 30%, B-DE 16%), растворяемого (% от исходного количества) при 1-20 минутах в среде растворителя, как функцию концентрации фосфатов, что определяется с помощью GPC. Отображаются средние значения по 3 измерениям.
Рис. 17 отображает накопительную кинетику высвобождения БСА-ФИТС из гранул на основе пектина в SIF, согласно описанию в разделе Методики.
Краткое изложение существа изобретения
В настоящем изобретении используется уникальная методика двойного ининкапсулирования для защиты, контролируемого высвобождения лекарственного вещества и улучшения биодоступности биомолекул, которые способны подвергаться ферментативному разложению в желудочно-кишечном тракте. Данные системы содержат липосомы, задача которых состоит в защите и улучшении биодоступности заключенной в капсулу биомолекулы, которые в свою очередь заключаются во второй объект, состоящий из гранул, состоящих из природных полисахаридов, что позволяет, с одной стороны, защитить липосомы от суровых условий окружающей среды и позволяет управлять высвобождением липосом, с другой стороны. Гранулы важны для создания сцепляющихся со слизистой оболочкой систем, которые представляют собой мощный инструмент для увеличения времени нахождения заключенных в капсулу веществ в организме, позволяя таким образом перенос заключенного в капсулу лекарственного вещества сквозь кишечные клетки и улучшение биодоступности биомолекул. Предлагаемая технология является развитием липосомальных лекарственных форм и предназначена для создания возможности пероральной доставки липосом в неизменном виде. Преимуществом является - стабильности липосом и заключенного в них лекарственного вещества в верхних отделах ЖКТ, т.к. в чистом виде липосомы разрушаются в верхних отделах ЖКТ в чрезвычайно короткий промежуток времени. Липосомы являются средством доставки лекарственного вещества, предназначенным для переноса действующего вещества в клетку или за какой-либо барьер, в данном изобретении описывается частный способ доставки липосом в кишечник, кроме того, позволяющий использовать липосомы в твердой лекарственной форме.
В одном аспекте изобретение относится к пероральной системе доставки лекарственного вещества белковой природы, из липосом, включенных в частицы поперечносшитого природного полисахарида.
Пероральная система доставки обеспечивает уменьшение вдвое степень разрушения действующего вещества белковой природы в желудочном соке в течение 30 минут, относительно степени разрушения вещества белковой природы, инкапсулированного только в липосомы.
Пероральная система доставки обеспечивает уменьшение на 70% степень разрушения действующего вещества белковой природы в желудочном соке в течение 30 минут, относительно степени разрушения вещества белковой природы, инкапсулированного только в липосомы.
Для получения частиц предпочтительно используют пектин с низкой степенью этерификации, наиболее предпочтительно со степенью этерификации 10-40%.
Для поперечного сшивания могут быть использованы соли поливалентных металлов, предпочтительно соли Са+2 или Al+3, наиболее предпочтительно соли Са+2.
Для получения частиц может быть использован хитозан, а для поперечного сшивания - полианионы, например - триполифосфат.
В другом аспекте пероральная система доставки лекарственного вещества белковой природы обеспечивает высвобождение веществ белковой природы в кишечной среде, предпочтительно в течение 1-48 ч.
Еще изобретение относится к защитной оболочке пероральной системы доставки состоящей из пектина поперечносшитого поливалентным металлом при следующем содержании в системе доставки в масс. % (за 100% принята сухая масса системы доставки):
Предпочтительно защитная оболочка состоит из пектина поперечносшитого поливалентным металлом при следующем содержании в масс. %:
Предпочтительно в качестве поливалентного металла использован Са+2 или Al+3, наиболее предпочтительно - Са+2.
Другой вариант защитной оболочки пероральной системы доставки состоит из хитозана поперечносшитого полианионом при следующем содержании в системе доставки (за 100% принята сухая масса системы доставки) в масс. %:
Предпочтительно защитная оболочка состоит из хитозана поперечносшитого полианионом при следующем содержании в масс. %:
Предпочтительно в качестве полианиона использован триполифосфат.
В противоположность обычному капельному методу [9], который страдает от нескольких недостатков, которые заключаются: (i) в относительно большом размере получаемых гранул, в диапазоне от 1 до 10 мм, что зависит главным образом от размера иглы сопла, вязкости используемого полимера матрицы и прочности перекрестно-сшивающего агента; (ii) в высокой вариабельности размеров, вызванной ручным процессом изготовления и разным расстоянием между иглой сопла и раствором перекрестно-сшивающего агента, который задается в различных вариантах приготовления; и (iii) в высокой вариабельности кинетики высвобождения лекарственного вещества в ткани, вызванной вариабельностью размеров приготавливаемых гранул Более успешно может быть использована наложенная вибрация, что способствует разделению капелек на объекты равного размера. Наложенная вибрация» или «технология вибрирующего сопла» - представляет собой техническое решение, состоящее из мембраны расположенной над выпускным соплом, причем мембрана вибрирует с регулируемой частотой и постоянной амплитудой, тем самым дозирует объем капли раствора выбрасываемой из сопла. Использование наложенной вибрации позволяет преодолеть недостатки и делает возможной:
- Перевод данной технологии на промышленные масштабы производства,
- Воспроизводимое формирование гранул с размером гранул от 0,08 до 2 мм при равномерном и узком по диапазону разбросе размеров (разброс размеров <5%),
- Гибкость - Формирование гранулы и капсулы с помощью одного и того же инструмента одноэтапно,
- Одношаговое нанесение покрытия на гранулу с дополнительной оболочкой для регулирования высвобождением лекарственного вещества в ткань,
- Приготовление стерильных составов и манипулирование ими в стерильных условиях,
- Уменьшение вариабельности в высвобождении лекарственных веществ.
В настоящем изобретении использован метод наложенной вибрации для формирования составов гранул с размерами от 0,08 до 2 мм на основе природных полисахаридов с помощью ионотропного гелеобразования, имеющего целью инкапсулирование составов на базе липосом, содержащих долю терапевтических веществ, таких как белки и пептиды.
Данное изобретение также относится к фармацевтическим соединениям, образующим микрогранулы, состоящие из природных полисахаридов, некоторого количества липидного соединения согласно данному изобретению, причем это количество достаточно для достижения определенного биологического эффекта на целевом участке организма. Фармацевтический состав согласно изобретению обычно содержит активную субстанцию, пептид, белок, небольшую молекулу, которая отвечает за биологическую активность орально вводимого наполнителя. Этот фармацевтический состав согласно изобретению обычно содержит, в дополнение к указанному липидному соединению, физиологически приемлемый носитель. Этот физиологически приемлемый носитель, используемый согласно данному изобретению, в общем относится к инертным, нетоксичным твердым или жидким веществам, предпочтительно не реагирующим с этим биологически активным липидом согласно данному изобретению.
Для ионотропного гелеобразования могут использоваться природные полисахариды, такие как пектин, альгинат, хитозан, триметилхитозан, каррагенан, карбоксиметилцеллюлоза, сульфат целлюлозы, гиалуроновая кислота, декстрансульфат, относящиеся к типу разбухающих. Противоионы, используемые для ионотропного гелеобразования, могут быть разделены на две основных категории: противоионы с низким молекулярным весом (например,. CaCl2, BaCl2, MgCl2, CuCl2, ZnCl2, CoCl2, AlCl3, FeCl3, сульфат натрия, пирофосфат, триполифосфат, тетраполифосфат, октаполифосфат, гексаметасульфат и [Fe(CN)6]-4/[Fe(CN)6]-3; ионы с высоким молекулярным весом (например, октилсульфат, лаурилсульфат, гексадецилсульфат, цетилстеарилсульфат).
Величина рН имеет влияние на формирование перекрестно-сшитых альгинатных гранул. Гранулы из перекрестно-связанного альгината, хитозана или пектина успешно формируются при рН 2,5-5,0. При избытке кросс-линкера гранулы формируются также при рН 6,0. В нашем изобретении использовали рН в диапазоне от 2,5 до 8,0.
Системы для формирования гелевых гранул, состоящие из пектина, альгината и хитозана, представляют собой главным образом системы, которые исследуются в настоящее время [5-8]. В данном изобретении системы, состоящие из пектина/ CaCl2 и хитозана/ТРР, использовались в основном для приготовления гелевых микрогранул.
Для разработки микрогранул с двойной перекрестной сшивкой могут использоваться нижеследующие сочетания, такие как альгинат/хитозан, относящиеся к типу разбухающих, при этом могут использоваться двухвалентные или трехвалентные ионы в качестве сшивающих агентов для альгината, а сульфат натрия, пирофосфат, триполифосфат, тетраполифосфат, октаполифосфат могут использоваться в качестве сшивающих агентов для хитозана.
Для индуцируемого температурой гелеобразования могут использоваться температурно-чувствительные полимеры, которые подвергаются спонтанному гелеобразованию в зависимости от их температурно-зависимого фазового перехода. Желатин и воски, относящиеся к типам разбухающих, будут формировать гель при понижении температуры. С другой стороны, микрокристаллическая целлюлоза и оксипропилцеллюлоза, относящиеся к типам разбухающих, растворимы в холодной воде, но нерастворимы в горячей. После разогрева такого раствора могут формироваться гелевые структуры при температурах гелеобразования в пределах от 50 до 90°С.
Газообразующие агенты, такие как карбонат кальция, бикарбонат натрия, при их добавлении в указанный состав для получения пористых гелевых шариков при изготовлении гранул будут оказывать влияние на размер и форму и пористость гелевых шариков, позволяя регулировать кинетику высвобождения лекарственного вещества.
Ниженазванные полимеры добавляются в качестве добавок к основному формирующему гранулы компоненту в целях обеспечения регулирования формирование гранул, стабильность и кинетику высвобождения лекарственного средства: оксипропилцеллюлоза, микрокристаллическая целлюлоза, оксиэтил метакрилат, N-(2-оксипропил) метакрилат, N-винил-2-пирролидон, N-изопропилакриламид, винилацетат, акриловая кислота, метакриловая кислота, полиэтиленгликольакрилат/метакрилат, полиэтиленгликольдиакрилат/диметакрилат, относящиеся к типам разбухающих.
На свойства гранул, приготовленных путем ионотропного гелеобразования, влияют состав и параметры технологического процесса. Это утверждение подтверждается несколькими статьями, в которых сообщается, что эффективность высвобождения лекарственных веществ и/или инкапсулирования зависит от типа лекарственного вещества и его характеристик, от типа и концентрации полимера [33, 34], от включения различных добавок [35-37], от весового соотношения полимера лекарственного вещества [38], также как и от переменных процесса, таких как время отверждения [40], тип и концентрация агента сшивания [33, 34, 39], условия сушки [40].
Диапазон концентрации полимера, который мы используем в нашем изобретении для приготовления гранул, лежит в пределах 0,5-5% и предпочтительно в пределах 0,7-3%.
В литературе сообщалось, что при увеличении диапазона концентрации используемого хлористого кальция получали гранулы меньших размеров [6, 18]. В нашем изобретении диапазон концентрации используемого хлористого кальция лежал в пределах 0,1М-1М, и предпочтительно в пределах 0,1М-0,48М. Для приготовления гранул на основе хитозана в нашем изобретении используемый диапазон концентрации ТРР находился в пределах 1-50 мг/мл и предпочтительно в пределах от 10 до 30 мг/мл.
Было выявлено, что на размер и форму гранул в значительной степени влияло время отверждения. Увеличение времени отверждения с 1 до 30 минут привело к уменьшению средних значений торцевого диаметра гранул [40]. В нашем изобретении применяемое время отверждения находилось в пределах от 1 до 45 минут и предпочтительно от 3 до 30 минут.
Сушка может оказывать влияние на размер и форму гранул. Существует три общих способа производства сухих гранул: сушка из замороженного состояния, сушка на воздухе в температурном диапазоне в пределах от 25 до 40°С и сушка в вакуумных условиях при диапазоне температур 25-40°С [40].
Обычно гранулы после лиофильной сушки остаются с тем же размером и сферичностью, что и до сушки, и их внутренняя структура очень пористая [40]. Такие характеристики высушенных из замороженного состояния гранул вызваны быстрой сублимацией замерзшей воды из альгинатной матрицы, что приводит к образованию пор на местах прежних кристаллов льда из-за отсутствия времени для усадки.
С другой стороны, сообщалось, что высушенные на воздухе гранулы обычно существенно сжимались и уменьшались в размерах при сушке. Однако поверхность высушенных на воздухе гранул была намного более гладкой, чем поверхность высушенных лиофильным путем. Кроме того, они не были такими пористыми, как гранулы, высушенные из замороженного состояния [40].
Все три способа были протестированы в нашем изобретении при получении составов с сухими гранулами.
Температура растворов как полимера, так и/или кросс-линкера имеет огромное влияние на параметры гранул. Было выявлено, что повышение (температуры) раствора кросс-линкера облегчает поперечное сшивание гранул. В нашем изобретении использованный температурный диапазон находился в пределах от 20 до 70°С.
Липидная матрица согласно данному изобретению предпочтительно содержит физиологически переносимый формирующий липосому липид или сочетание физиологически переносимых формирующих липосому липидов. Формирующие липосому липиды обычно бывают такими, которые имеют глицериновую основу, в которых по крайней мере одна из гидроксильных групп замещается ацильной цепочкой, фосфатной группой, комбинацией или производными последней и может содержать химически реакционноспособную группу (такую, как амин, кислота, альдегид или спирт) в головной группе. Обычно ацильные цепочки имеют в длину от 14 до примерно 24 атомов углерода и имеют варьирующиеся степени насыщения, являясь полностью, частично или негидрированными липидами. В дополнение к этому липидная матрица может быть из природного источника, полусинтетическим или полностью синтетическим липидом, и нейтральной, заряженной положительно или отрицательно.
Липидная матрица обычно содержит фосфолипиды., обычно глицерофосфолипид. Примеры глицерофосфофлипидов включают три типа липидов: (i) цвиттерионные фосфолипиды, которые включают, например, фосфатидилхолин (PC), фосфатидилхолин из яичного желтка (ЕРС), фосфатидилхолин PC из гидрогенизированных соевых бобов (HSPC), димиристил фосфатидилхолин (ДМФХ), дипальмитил фосфатидилхолин (ДПФХ) сфингомиелин (SM); (ii) отрицательно заряженные фосфолипиды: которые включают в себя, например, фосфатидилсерин (PS), фосфатидилинозитол (PI), фосфатидиновая кислота (РА), фосфатидилглицерин (PG), димиристил фосфатидилглицерин (DMPG); и (iii) катионные фосфолипиды, которые включают, например, фосфатидилхолин и сфингомиелин, в которых сложный эфир фосфокислоты является О-метилированным.
Одним параметром, использованным для выбора подходящих фосфолипидов, применяемых в наших препаратах, являлась их температура фазового перехода из упорядоченного твердого состояния (SO) в жидкое разупорядоченное состояние (LD), которая во всех случаях ниже 60°С, что важно для сохранения целостности лечебных препаратов на бежовой основе во время приготовления СДЛВ.
Липидный комплекс также может включать в себя другие компоненты, обычно применяемые в приготовлении липидных комплексов (например, для стабилизации). Примеры таких прочих компонентов включают в себя, не будучи ограниченными приведенным перечнем, спирты жирного ряда, жирные кислоты и/или холестериновые эфиры или любые другие фармацевтически переносимые наполнители, которые могут влиять на поверхностный заряд, на подвижность мембран и содействовать включению биологически активного липида в липидный комплекс. Примеры стеринов включают в себя холестерин, холестерилгемисукцинат, холестерилсульфат или любые другие производные холестерина.
Компоненты этого липидного комплекса могут выбираться для достижения специфической степени текучести или жесткости, для управления стабильностью комплекса при хранении, также как и при доставке, например, в ЖКТ, и для регулирования скорости высвобождения этого терапевтического компонента. Жидкие комплексы, имеющие более жесткую структуру, например, липосомы в гелевой фазе (упорядоченное твердое состояние) или в жидкокристаллическое текучее состояние (разупорядоченное жидкое состояние), получают включением в их состав относительно жесткого липида, например, липида, имеющего относительно высокую температуру фазового перехода из упорядоченного твердого состояния в разупорядоченное жидкое состояние, такую, как температура выше комнатной. Жесткие, то есть насыщенные липиды, имеющие длинные ацильные цепочки, содействуют более высокой жесткости мембран в этом комплексе. Известно, что липидные компоненты, такие как холестерин, способствуют приобретению жесткости в липидных структурах, в особенности, они обеспечивают уменьшение свободного объема, уменьшая таким образом проницаемость.
Похожим образом высокая текучесть липидов достигается путем добавления относительно текучего липида, обычно такого, у которого относительно низкая температура фазового перехода из жидкой в жидкокристаллическую фазу, например, при комнатной температуре или ниже ее, более предпочтительно при температуре целевого тела или несколько ниже ее. В нашем изобретении мы использовали способствующий слиянию компонент, такой как даолеилфосфатидилэтаноламин (ДОФЕ). ДОФЕ приобретает недвухслойную структуру (шестиугольную, НII) в водной среде при нейтральной рН. Увеличение пропорции ДОФЕ в липосомах управляет переходом в инвертированную шестиугольную фазу из пластинчатой структуры и, следовательно, дестабилизирует мембрану липосом, имея предрасположенность к слиянию с клеточной мембраной. Эти липосомы также считаются дестабилизирующимися в кислотной среде ядрышек, быстро теряя свое содержимое. Можно использовать в качестве варианта молекулы добавления.
Существуют многочисленные полимеры, которые могут быть включены в состав липосом для повышения стабильности. Обычно используемые в качестве липидных модификаторов полимеры включают в себя, не будучи при этом ограниченными нижеприведенными: полиэтиленгликоль (ПЭГ), полисиаловую кислоту, полилактическое соединение (также называемое полилактид), полигликолевую кислоту (также называемую полигликолид), полимолочную-полигликолевую кислоту, поливиниловый спирт, поливинилпирролидон, полимеметоксазолин, полиэтоксазолин, полиоксиэтилоксазолин, полиоксипропилоксазолин, полиаспартамид, полиоксипропил, метакриламид, полиметакриламид, полидиметилакриламид, поливинилметилэфир, полиоксиэтилакрилат, производные целлюлозы, такие как оксиметилцеллюлоза или оксиэтилцеллюлоза. Эти полимеры могут использоваться в виде гомополимеров или в виде блочных или статистических сополимеров.
Наиболее часто используемые и коммерчески доступные липиды, преобразованные путем дериватизации в липополимеры, - это такие, которые основаны на фосфатидилэтаноламине, обычно на дистеарилфосфатидилэтаноламине (ДСФЭ).
Когда этот липидный комплекс представлен в форме липосомы, то эта липосома может быть в форме многослойных пузырьков (MLV), больших однослойных пузырьков (LUV), малых однослойных пузырьков (SUV) или многопузырьковых пузырьков (MW), также как и в других двухслойных структурах, известных в данной области техники. Размер и число слоев липосомы будет зависеть от порядка приготовления, и от выбора их пути введения в организм. При оральном введении предпочтительными липосомами являются липосомы с диапазоном размеров от 50 до 300 нм в диаметре (LUV).
Физиологически усваиваемый носитель, используемый согласно данному изобретению, включает обычные носители, используемые для орального введения, такие как желатиновые капсулы, относящиеся к типу разбухающих.
Нами был выбран видимый спектральный зонд - флуоресцеинизотиоцианат-меченый БСА (БСА-ФИТЦ) и ФИТЦ-меченый 10-мер пептид в качестве модели лекарственного вещества белковой природы в целях оценки эффективности их включения в липосомы и их количества в липосомах, эффективности включения липосом с конкретным лекарственным веществом в изготавливаемые гранулы, морфологической оценки приготовленных липосом и содержимого, включенного в их капсулу, в виде микрогранул, состоящих из природных полисахаридов, кинетику высвобождения лекарственного вещества из этих гранул в свободной форме и/или в инкапсулированной форме в моделируемых жидких средах желудка, усвоения клетками кишечника лекарственного вещества и переноса через эпителий кишечных клеток. Возможность осуществления изобретения подтверждается, но не ограничивается примерами:
Пример 1. Получение меченных модельных белков
Материалы
БСА-ФИТЦ, молибдат аммония, 4-амино-2-нафтил-4-сульфореагент, алгинат, хитозан, PF-127, были приобретены у компании «Сигма-Элдрич» (Сент-Луис, Миссури, США).
1,2-дипальмитоил-sn-глицеро-3-фосфохолин (ДПФХ), 1,2-димиристоил-sn-глицеро-3-фосфохолин (ДМФХ), 1,2-диолеоил-sn-глицеро-3-фосфоэтаноламин-N-(лиссамин родамин В сульфонил) (аммониевая соль), 1,2-диолеоил-sn-глицеро-3-фосфоэтаноламин (ДОФЭ), и холестерин были приобретены у компании «Аванти Полар Липидс» («Алабастер», Алабама, США).
Свойства пептидов
Модель пептида 10-мера, со свойствами, сходными с химико-физическими характеристиками Дегареликса, была синтезирована твердофазным синтезом пептидов (ТФСП) компанией «Сигма-Элдрич» (WYPA10, Реховот, Израиль).
Последовательность пептидов
FMOC-Trp-Tyr-Phe-Ser-Tyr-Tyr-Leu-Lys-Prol-AIa-MBHA-смола
где боковые цепи Trp, Tyr, Ser и Lys защищены.
Мечение пептидов
Для этого связанный со смолой пептид был вначале увеличен в ДМФА в течение 60 мин, и 9-флуоренилметоксикарбонил был удален путем инкубации в 20% пиперидине в ДМФ в течение 20 минут и после этого еще 10 минут с последующей промывкой в ДМФ (х4) и ДХМ (х4). После этого, 4 экв. связующего вещества Fmoc-6-Ahx-COOH (HSX-Fmoc), было соединено с пептидом (30 мин инкубации) после предварительной активации 1Н-7-азабензотриазол-1-ил)-1,1,3,3-тетраметил урониевым гексафторфосфатом (4 экв., HATU 2), гидроксибензотриазолом (4 экв., НОВТ) и N,N-диизопропилэтиламином (4 экв., диизопропилэтиламин) в течение 2 мин. Затем 9-флуоренилметоксикарбонил связывающего вещества был удален, как описано ранее.
Затем 4 экв. флуоресцеинизотиоцианата (ФИТЦ) были связаны с пептидом с использованием связывающего вещества в присутствии диизопропилэтиламина путем его инкубации в течение 24 часов с последующим промыванием ДМФ (х4) и ДХМ (х4).
Пептид и его боковые защитные группы были удалены из смолы путем инкубации в течение 2 часов с использованием трифторуксусной кислоты/дистиллированной деионизированной воды/фенола/триэтилсилана (88/5/5/2 об/об). Меченый пептид (схема 1) был осажден холодным диэтиловым эфиром и его избыток был удален аргоном. Процедуру повторили (Рис. 1).
Структурная формула представлена на схеме 1
C97H111N15O20S
Точный вес: 1837.79
Молекулярная масса: 1839.07
Мг/экв.: 1838.79 (100.0%), 1837.79 (93.5%), 1839.79 (63.1%), 1840.80 (18.5%), 1840.79 (7.7%), 1841.80 (7.0%), 1838.78 (5.9%), 1840.78 (4.9%), 1839.78 (4.4%), 1841.79 (4.1%), 1842.80 (2.1%), 1842.79 (1.2%)
С, 63.35; Н, 6.08; N, 11.42; 0,17.40; S, 1.74
Очистка пептида ВЭЖХ (рис. 2):
- Колонка: Agilent Luna С18-2 250×4,6
- Температура колонки: 40°С
- Объем введения: 100 мкл
- Раствор А: 0,1% трифторуксусная кислота (ТФК); Раствор В: ацетонитрил
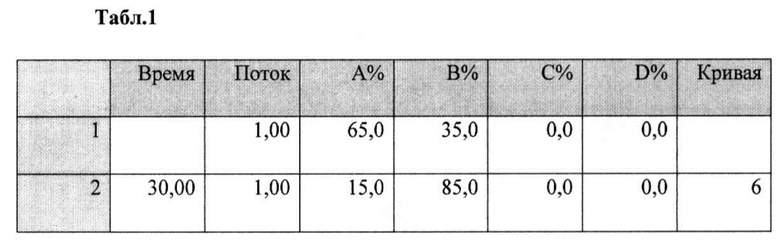
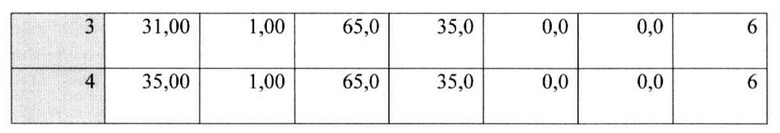
- Градиент (табл. 1)
Пример 2. Приготовление больших однопленочных пузырьков (LUV), заключающих в себе образец белка, и оценка физико-химических характеристик однослойных пузырьков LUV, в которые введен образец лекарственного вещества.
Методы
Получение больших однослойных пузырьков (LUV), содержащих белкок:
В качестве липосомобразующих липидов использовали ДМФХ и ДПФХ во всех липосомных составах. В качестве пространственного липосомного стабилизатора использовали 1,2-дистеарил-sn-глицеро-3-фосфоэтаноламин-N-[метокси(полиэтиленгликоль)-2000]-аммониевую соль (ПЭГ-ДСФЭ). В качестве обеспечивающего слияние липида использовали диолеоилфосфатидилэтаноламин (ДОФЕ), а холестерин использовали в качестве модулятора свойств мембран и высвобождения лекарственных веществ, в то время как 2-диолеил-sn-глицеро-3-фосфоэтаноламин-N-(лиссамин родамин В сульфонил)-аммониевая соль (LR-PE) использовалась в качестве флуоресцентного маркера для обеспечения отслеживания липосомы.
В случае получения липосом методом гидрирования, смешивали соответствующие количества маточного(ых) раствора(ов) липидов в трет-бутаноле с образцом лекарственного вещества, такого как ФИТС-БСА или десятичленного пептидного полимера-ФИТС, и оставляли в лиофильной сушилке на ночь, обычно в соотношении липид: лекарственное вещество в 100/1-10/1. Затем добавляли карбонатный буфер 3,6 мМ для гидратации лиофилизованных липидов.
В случае приготовления липосом методом впрыскивания этанола, когда соответствующее количество маточного(ых) раствора(ов) липидов в этаноле постепенно добавляли к ДДВ, предварительно смешанного с образцом лекарственного вещества ФИТС-БСА или десятичленного пептидного полимера-ФИТС, в целях получения соотношения EtOH/ДДВ в 1:10 (обычно при соотношении липид/лекарственное вещество в 100/1-10/1).
Затем, в обоих случаях, эти дисперсии вращали со скоростью 2.000 об/мин и перемешивали с помощью магнита на 500 об/мин для получения MLV. Затем MLV обрабатывали ультразвуком в ванне ультразвукового дезинтегратора при 37°С в случае с ДМФХ и при 50°С в случае с ДПФХ.
Большие однослойные пузырьки (LUV~100 нм) приготавливали экструзией MLV 11 раз через карбонатный фильтр с размером пор 0,8-мкм, с последующей 11-кратной экструзией через фильтр с размером пор 0,4 и 0,2 мкм (Компания Poretics, Ливермор, Калифорния) при использовании экструзионной системы компании Avanti Polar Lipids (Липоид, США). Затем составы подвергали двукратному диализу против 200 объемов 0,9% NaCl в течение 1 часа каждый, с последующим однократным диализом в течение 24 часов против 400 объемов 10%-й сахарозы.
Концентрацию ФЛ (PC плюс 2kПЭГ-ДСФЭ в некоторых случаях) определяли путем определения содержания фосфора в липиде с помощью модифицированного метода Бартлетта.
Определение размера и электрокинетического потенциала:
Разброс размеров и электрокинетический потенциал липосом с загруженным белком замеряли при 25°С с использованием прибора Zetasizer Nano-Z, Компании Malvern Instruments Ltd, Малвем, Соединенное Королевство. Для этого 10 мкл суспензий LUV разбавляли в 1 мл фильтрованного ДДВ и анализировали.
Анализ ПЭМ:
Составы 10 Мм LUV наносили на углеродную сетку. После этого на эту углеродную сетку добавляли уранилацетат (2% вес/вес) в виде отрицательной краски и инкубировали 20 секунд. Для визуализации LUV использовали микроскоп Phillips ПЭМ СМ 12.
Оценка эффективности инкапсулирования лекарственного вещества в липосомы:
Для этого 25 мкл LUV растворяли в 0,1М NaOH/1% Triton X при вращении и обработке ультразвуком и инкубировали при 37°С в течение 2 часов. Затем образцы центрифугировали 5 минут при 14000 об/мин для получения светлого всплывшего вещества. Отбирали 250 мкл от этого всплывшего вещества и анализировали на флуоресценцию с помощью многоканального спектрофотометра для прочтения планшетов (Компания BioTek, Вермонт, США) при ех485/em525 с чувствительностью 50.
Эффективность ининкапсулирования лекарственных веществ (ЭИ) определяли по стандартной кривой лекарственного вещества в 1%-ном Triton X после 2-х часового инкубирования при 37°С согласно нижеследующему уравнению:
ЭИ (%)=100×([лекарственное вещество]/[ФЛ]) после загрузки/([лекарственное вещество]/[ФЛ]) до загрузки.
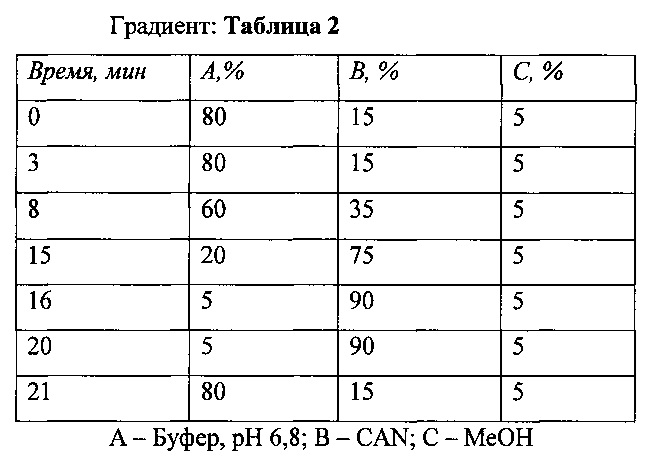
В виде альтернативы с помощью высокоэффективной жидкостной хроматографии замеряли концентрацию пептида-ФИТС в LUV (Рис. 3).
- Температура колонки - 40°С
- Скорость протока - 1,5 ml/min
- Детектор: флуориметрический с длиной волны: 485/528 нм
- Инъецируемый объем: 10 мкл.
Результаты
Для инкапсулирования в различных LUV выбирали два зонда-образца, белок, ФИТС-БСА или десятичленный пептидный полимер - ФИТС. В нашем изобретении большие однослойные пузырьки LUV получали с помощью технологий как тонкопленочной гидратации, так и инъекции растворителя, как описано в Методике. В обоих случаях инкапсулирование образца лекарственного вещества осуществляли путем пассивного включения, которое происходило при гидратации липида водным раствором, содержащим это лекарственное вещество, или при формировании пузырька в водном растворе, содержащем это лекарственное вещество.
Наши результаты показали, что с помощью обоих методов мы сумели приготовить большие однослойные пузырьки LUV (Таблицы 3А и В). Были получены и описаны различные составы с липосомами, варьирующиеся по типу липосом и по вводимым добавкам (ПЭГ-ДСФЭ и LR-PE) с исходным соотношением белок/ФЛ в 1/100 или 5/100 (Таблицы 3А и В). Очистку неинкапсулированного лекарственного вещества проводили диализом.
Соотношения лекарственного вещества на выходе всегда были выше, чем соотношения на входе, что согласуется с более высокой потерей ФЛ (определяется с помощью модифицированного метода Бартлетта), чем в образцах лекарственного вещества (Таблицы 3А и В). Обычно состав липосом не влиял на загрузку лекарственного вещества (Таблицы 3А и В). Все приготовленные LUV демонстрировали высокую ЭИ, находящуюся в пределах от 70 до 100%, независимо от типа липида (Таблицы 3А и В). Размер LUV, содержащего БСА-ФИТС, во всех случаях был в нанодиапазоне (102-406 нм, Таблицы 3А и В) с индексом полидисперсности (PDI), который почти во всех случаях был ниже 0,3, что соответствует формированию гомогенной популяции. Повышение содержания пептида-ФИТС в 3.4-раза (LUV №35 и 36 в Таблице 3В) позволило увеличить содержание лекарственного вещества до 4% (вес/вес). Увеличение объема БСА-ФИТС в 5 раз (LUV №13 и 14 в Таблице 3А) позволило повысить содержание лекарственного вещества до 6,3 и 10,6% соответственно (вес/вес).
Изображения ПЭМ, отображенные на Рис. 4, свидетельствовали о двухслойном LUV (А, В, С) с заключенным в капсулу лекарственным препаратом (окрашенные в темный цвет группы карбоновой кислоты при пептиде-ФИТС В и С) по сравнению с неокрашенным пустым пузырьком LUV, представленным на Рис. 4А. Составы на основе ДПФХ (Рис. 4С) свидетельствуют о формировании как малых (100 нм), так и относительно больших LUV (~400 нм) по сравнению с однородной популяцией в случае LUV на основе ДМФХ (Рис. 4В), что отразилось на их размере, замеренном с помощью DLS (Таблица 3В). Добавление обеспечивающего слияние липида выявило образование типичных шестиугольных структур с заключенным в капсулу лекарственным веществом (темные точки внутри шестиугольников, Рис. 4Е). Использование PF-127 в качестве компонента состава оказалось неблагоприятным по всем измеренным параметрам: размер (Рис 5: Было выявлено увеличение 25К по сравнению с 88К на Рис. 4) и ЭИ (LUV №31 и 32 в Таблице 3В).
Пример 3. Оценка высвобождения лекарственного вещества из липосом.
Методы
Для этого 100 мкл LUV диспергировали вначале в 250 мкл моделированных жидкостей желудка (SIF, USP, рН 6.8) и проверяли на флуоресценцию с помощью многоканального считывателя планшетов (Компания BioTek, Вермонт, США) при ex485/em525 с чувствительностью 70 сразу же после того, как на препаратах определяли общую величину FL. Отдельно пузырьки LUV в указанных количествах диспергировали в 250 мкл SIF и инкубировали в 2250 мкл SIF в течение 2, 4 и 24 час. при 37°С со встряхиванием при 50 об/мин. По окончании инкубационного периода 500 мкл инкубируемой среды загружали в микроконусы аппарата Nanosep Pal, имеющие поры в 100 кДа, и центрифугировали при 14000 в течение 12 мин. После этого эту среду анализировали на флуоресценцию с помощью многоканального считывателя планшетов (Компания BioTek, Вермонт, США) при ех485/em525 с чувствительностью 70. Высвобождение лекарственного вещества (ВВ) определяли с помощью стандартной кривой лекарственного вещества в SIF сразу же после приготовления и после инкубирования в течение 2, 4 и 24 час при 37°С со встряхиванием при 50 об/мин при 37°С согласно нижеследующему уравнению:
BB(%)=100×W/Z,
Где:
W - количество модельного пептида, перешедшего в инкубационную среду, мг;
Z - количественное содержание пептида в образце взятом для анализа, мг.
Результаты:
Высвобождение лекарственного вещества, стабильность in vivo и биораспределение определяются размером пузырьков, их поверхностным зарядом, гидрофобностью поверхности и текучестью мембран. Проницаемость мембран может подстраиваться путем выбора состава фосфолипидов и с помощью добавок, таких как молекулы холестерина. В нашем исследовании на проницаемость мембран влияли тип ФЛ и используемый холестерин.
Как можно видеть на Рис. 6А, первоначальное взрывное высвобождение БСА-ФИТС наблюдали в течение первых 4 час, за которыми следовало более медленное высвобождение в течение последующих 20 часов при этом 1/2 срока действия лекарственного вещества превышает по времени 24-часовой инкубационный период, за исключением LUV №7 и 29 (Т 1/2 составляет 4 часа). В соответствии с переходным состоянием липидов значения кинетики и высвобождения были намного выше в случае LUV на основе ДМФХ, чем на основе ДПФХ, при этом пониженное БСА холестерина также снижало кинетику высвобождения. С другой стороны, начальное взрывное высвобождение устранялось, когда использовали пептид-ФИТС в качестве образца лекарственного вещества с дополнительным высвобождением в диапазоне от 5 до 30% в течение 36 часов (Рис. 6В).
Пример 4. Оценка токсичности LUV в отношении клеток Сасо-2
Методы
Клеточную линию Сасо-2 карциномы толстой кишки человека получали из Американской коллекции типовых культур (АТСС, Манассас, Виргиния). Клетки Сасо-2 выращивали в полной среде DMEM. Клеточные линии выдерживали при t 37°С в инкубаторе с 5% СО2 с водяной рубашкой.
Оценивали цитотоксичность приготовленных составов. Отсутствие цитотоксичности определяли с помощью MTS - прибора CellTiter 96® AQueous Анализ проводился с использованием растворов нового соединения тетразолия [3-(4,5-диметилтиазол-2-ил)-5-(3-карбоксиметоксифенил)-2-(4-сульфофенил)-2Н-тетразолий - внутренняя соль; MTS] и электрон-связывающего реагента (феназинметосульфат) PMS, где MTS восстанавливается биологически клетками в продукт формазан, который растворим в среде для культивирования тканей, и абсорбцию продукта формазана можно измерять на 490 нм. Для этого клетки Сасо-2 высевали в 96-ячеечный планшет при 4×104 клеток/мл (8×103/на ячейку) и давали прикрепиться и расти в течение 48 часов. Затем 50 мкл этих исследуемых соединений вводили в исследуемые ячейки и инкубировали в течение 4 или 24 часов. По окончании инкубационного периода инкубационную среду удаляли, затем клетки промывали с помощью PBS и инкубировали в свежей среде до 24 часов, и определяли жизнеспособность клеток с помощью анализа MTS.
Результаты
Клеточная линия Сасо-2, производная от прямокишечной карциномы человека, стала установленной in vitro моделью для прогноза абсорбции лекарственного вещества в кишечнике человека. При выращивании на полупроницаемых мембранах клетки Сасо-2 дифференцируются в высоко функционализированный эпителиальный барьер с замечательным морфологическим и биохимическим сходством с цилиндрическим эпителием тонкой кишки.
С помощью метода MTS была определена токсичность LUV с инкапсулированным белком (Рис. 7) в отношении клеток Сасо-2 через 24 часа инкубирования. На Рис. 7 показано, что все испытанные составы были нетоксичны при любой испытанной концентрации.
Пример 5. Оценка проникновения LUV
В клетки кишечника
Методы
Использовали софокусный лазерный сканирующий микроскоп (CLSM) для оценки эффективности внутриклеточного проникновения LUV. Для анализа на CLSM клетки Сасо-2 высеивали на покровные стекла в 8-ячеечных планшетах с плотностью 5×104 клеток/см2. После культивирования в течение 48 часов культуральную среду сливали, и клетки дважды промывали с использованием культуральной среды без красного красителя Fenol. После этого добавляли 0,4 мл LUV, загруженных лекарственным веществом, разбавленных в культуральной среде без красного красителя Fenol при нетоксичных концентрациях, и клетки инкубировали в течение 4 часов при 37°С. По окончании инкубационного периода растворы с образцами сливали, клетки промывали трижды культуральной средой без красного красителя Fenol и анализировали с помощью Софокусного лазерного сканирующего микроскопа Fluoview 300 (CLSM, Компания Olympus, Пенсильвания, США).
Результаты
Проникновение в клетки Сасо-2 липосомных носителей визуализировали с использованием микроскопа CLSM (Рис. 8). Анализ с помощью CLSM показал, что клетки Сасо-2 (в основном цитоплазма) эффективно поглощали LUV (состав, главным образом способствующий слиянию, L29 и L30) лекарственное вещество - пептид, меченный ФИТС (зеленый). Это отразилось и в увеличении их эффективного переноса сквозь монослой Сасо-2. Увеличение содержания пептида в этом составе (LUV №35 в Таблице 3В) обеспечивало возросшее высвобождение пептида в эти клетки (L35 на Рис. 8).
Пример 6. Инкапсулирование загруженного лекарственным веществом LUV в микрогранулы хитозана
Методы
Гранулы готовили растворением хитозана (85% деацетилирование, 20-300 сР) в уксусной кислоте (3%) при 80°С до растворения при концентрации 0,7% (вес/объем). LUV добавляли к хитозану так, чтобы весовое соотношение всех сухих веществ было соответственно 1:4. После перемешивания на магнитной мешалке в течение получаса гомогенную и не содержащую пузырей дисперсию распыляли с помощью инкапсулирующего устройства Buchi 390 через сопло 0,2 мм в 100 мл 2%-ного раствора кросс-линкера - триполифосфата (ТРР). В процессе получения гранул использовали следующие параметры оборудования:
- Насос - 30 мл/мин
- Вращение - 50 об/мин
- Частота - 308
- Поток - 2500.
Образцы промывали 3 раза с помощью ДДВ. Часть влажных заполненных гранул с инкапсулированными липосомами визуализировали с помощью оптического микроскопа с увеличением 40. Гранулы собирали вместе, погружали в жидкий азот и затем высушивали из замороженного состояния (Компания Labconco, Миссури, США) в течение ночи. Выход гранул всегда находился в диапазоне от 90 до 100%. Сухие гранулы анализировали с помощью (ESEM, модель Quanta 200).
Результаты
Для приготовления частиц из хитозана использовали метод ионотропного гелеобразования. Наши результаты, представленные на Рис 9, 10 и в Таблице 4, показали, что сферические гранулы успешно формируются с помощью метода наложенной вибрации с использованием некапсулирующего устройства.
Гранулы на основе хитозана визуализированные с помощью SEM продемонстрировали наличие гладкой поверхности со средним размером примерно 190 мкм (Рис. 9 и Таблица 4). Влажные гранулы были все сферическими, независимо от параметра исследуемого процесса обработки (Рис. 10).
Пример 7. Оценка эффективности инкапсулирования (ЭИ) в гранулы на основе хитозана LUV с загруженным лекарственным веществом
Методы
Для оценки эффективности загрузки липосом в гранулы использовали маркер липосом LR-PE. Для оценки эффективности инкапсулирования лекарственного вещества, пептид, меченный ФИТС, инкапсулировали в LUV. Для этого 0,5 мг гранул (См. примеры 6 и 7) диспергировали в 200 мкл 0.1 М NaOH/1% Triton и сразу же проверяли на флуоресценцию LR-PE с помощью Считывателя микропланшетов Synergy НТ (Компания BioTek, Висконсин, США) при ех.525/em.590. Затем образцы инкубировали при 37°С в течение 4 час с целью дезинтеграции гранул и липосом и оценки заключенного в капсулу пептида-ФИТС. После инкубирования образцы центрифугировали на 14000 об/мин в течение 5 минут и затем проверяли на флуоресценцию пептида - ФИТС с помощью Считывателя микропланшетов Synergy НТ (Компания BioTek, Висконсин, США) при ex.485/em.525.%-ное значение эффективности ЭИ оценивали по стандартным кривым пептида- ФИТС и LR-PE в 0,1 M NaOH/1% Triton X.
Результаты
Микрогранулы из хитозана показали среднюю эффективность инкапсулирования (ЭИ) LUV в размере 51% для LUV (Таблица 4). Сходным образом, сравнимые значения ЭИ были обнаружены в отношении пептида- ФИТС, инкапсулированного в LUV.
Пример 8. Оценка высвобождения лекарственного вещества в свободном виде и инкапсулированного в LUV, из гранул на основе хитозана и пектина в моделированных желудочных жидкостях (FaSSGF)
LUV были приготовлены как описано в примере 2. 0,01 мол.% 2-диолеил-sn-глицеро-3-фосфоэтаноламин-N-(лиссамин родамин В сульфонил)-аммониевая соль (LR-PE) использовалась в качестве флуоресцентного маркера липосомы для обеспечения отслеживания целостности липосомы.
Гранулы на основе хитозана готовили растворением хитозана (85% деацетилирование, 20-300 сР) в уксусной кислоте (3%) при 80°С до растворения при концентрации 0,7% (вес/объем). LUV добавляли к хитозану так, чтобы весовое соотношение всех сухих веществ было соответственно 1:19. После перемешивания на магнитной мешалке в течение получаса гомогенную и не содержащую пузырей дисперсию распыляли с помощью инкапсулирующего устройства Buchi 390 через сопло 0,45 мм в 200 мл 2%-ного раствора кросс-линкера - триполифосфата и инкубировали 30 мин.
Гранулы на основе пектина готовили на основе 1%-го раствора пектина (35%-е метилирование; GENU® pectin AG-1-Z, CP Kelco) путем его растворения в воде при 60°С и последовательной деэстерифицикации в течение 12 часов, путем изменения рН до 8,0 с помощью 0,1М NaOH. LUV добавляли к пектину так, чтобы весовое соотношение всех сухих веществ было соответственно 1:19. После перемешивания на магнитной мешалке в течение получаса гомогенную и не содержащую пузырей дисперсию распыляли с помощью инкапсулирующего устройства Buchi 390 через сопло 0,45 мм в 200 мл 250 мМ раствора кросс-линкера - CaCl2 и инкубировали 30 мин.
Гранулы собирали вместе, добавляли маннитол, погружали в жидкий азот и затем высушивали из замороженного состояния (Компания Labconco, Миссури, США) в течение ночи.
Раствор моделирующий среду желудка (Fast-State Simulated Gastric Fluid (FaSSGF) был приготовлен согласно публикации [41] с некоторыми изменениями:
Для сравнения стабильности липосом с липосомами инкапсулированными в матрицу гранул на основе хитозана и пектина в растворе моделирующий среду желудка (FaSSGF),
неинкапсулированные или инкапсулированные в матрицу гранул липосомы, помещали в диализные кассеты с порами в 10 кДа (Slide-A-Lyzer Dialysis Cassettes, Pierce) и инкубировали в течение часа при скорости вращения в 75 rpm на приборе растворения Dissolution USP apparatus II в защищенных от света стаканах.
Пробы отбирали через 15, 30 и 60 мин инкубации и анализировали на наличие флюоресцентного маркера - LR-PE в среде растворения и в образце с помощью ВЖХ (Alliance, Waters) используя флюоресцентный детектор на длине волны 570 нм.
Результаты:
Приготовленные микрогранулы продемонстрировали способность препятствовать высвобождению липосомального маркера, косвенно демонстрируя способность предохранять липосомы в растворе моделирующем среду желудка в отличие от свободных липосом, которые показали нарастающую распадаемость, 20-68%, после 15-60 мин нахождения в растворе моделирующем среду желудка (Рис. 11), кинетику похожую на описанную в сноске 42. Подобная кинетика высвобождения свободного маркера (LR-PE) в сравнении с меченными LUV продемонстрировала приемлемость экспериментальной системы (Рис. 11). С другой стороны, полученные экспериментальные данные описывающие кинетику высвобождения маркера из липосом инкапсулированных в матрицу гранул на основе хитозана и пектина подтвердили способность микрогранул предохранять целостность липосом в течение 30 мин, время типичное для нахождения в желудке. Микрогранулы на основе пектина показали лучшую способность предохранять липосомы в растворе моделирующем среду желудка (12-24% высвобождения после 30-60 мин нахождения в растворе моделирующем среду желудка) по сравнению с микрогранулами на основе хитозана (16-43% высвобождения после 30-60 мин нахождения в растворе моделирующем среду желудка), находящей объяснение в частичной протонации аминогрупп хитозана в кислой среде с их последующим разбуханием и разложением (Рис. 11),
Пример 9. Оценка высвобождения лекарственного вещества в свободном виде и инкапсулированного в LUV, из гранул на основе хитозана в моделированных кишечных жидкостях (SIF)
Методы
Для этого 2 мг гранул (Пример 6) диспергировали в 800 мкл SIF (USP) и инкубировали в течение 48 часов при 37°С при перемешивании во встряхивателе при 50 об/мин. По окончании инкубационного периода образцы центрифугировали при 1.000 об/мин в течение 2 минут, и 200 мкл всплывшего вещества анализировали на флуоресценцию следующим образом:
- Для высвобождения LUV образцы анализировали с помощью Считывателя микропланшетов Synergy НТ (Компания BioTek, Висконсин, США) при ех.525/em.590.
- Для высвобождения пептида - ФИТС образцы центрифугировали в течение 12 минут при 14.000 об/мин на центробежных фильтрующих устройствах Microcon (с фильтром с ячейками 100 кДа) для отделения высвобожденного пептида-ФИТС, и этот фильтрат анализировали на флуоресценцию с помощью Считывателя микропланшетов Synergy НТ при ex.485/em.525.
Результаты
Приготовленные микрогранулы продемонстрировали длительное высвобождение как LUV так и меченого пептида в SIF. 20% пузырьков LUV было высвобождено из микрогранул на основе хитозана в течение 48 час инкубирования (Рис. 12А). Высвобождение пептида было несколько выше, так как 40% пептида-ФИТС высвобождалось из микрогранул на основе хитозана в течение 48 часов инкубирования, что согласуется с высвобождением этого пептида из LUV, который далее высвобождаются в инкубационную среду (Рис. 12В).
Пример 10. Инкапсулирование загруженных лекарственным веществом LUV в пектиновые микрогранулы
Методы
Готовили 1%-й раствор пектина с низким молекулярным весом (261 кДа, 34%-е метилирование; Unipectine ОВ 700, Cargill, Франция) и деэтерифицировали его в течение 12 часов путем изменения рН до 8,0 с помощью 0,1М NaOH. Затем рН доводили до 4,0.
В раствор пектина добавляли LUV с обеспечением весового соотношения всех сухих веществ 1:4,75, соответственно. После перемешивания на магнитной мешалке в течение получаса гомогенную и не содержащую пузырей дисперсию распыляли в 100 мл 50, 100 или 200 мМ раствора кросс-линкера, CaCl2. Затем образцы промывали 3 раза в ДДВ.
Результаты
Для получения пектиновых гранул использовали метод ионотропного гелеобразования. Наши результаты, представленные на Рис. 13 и в Таблице 4 показали, что успешно сформированы сферические гранулы.
Влажные гранулы были все сферическими, независимо от параметра исследуемого процесса обработки, с однородным распределением по размерам (Рис. 13).
Пример 11. Влияние рН и концентрации кросс-линкера на содержание Са в пектиновых гранулах
Методы
Готовили 4%-й раствор пектина с низким молекулярным весом (261 кДа, 34%-е метилирование; Unipectine ОВ 700, Cargill, Франция) и деэтерифицировали его, 12 часов, путем изменения рН до 8,0 с помощью 0,1М NaOH. Раствор пектина разбавляли до 1,5% и доводили рН до 4,0. После нагрева до 60°С пектин сшивали с помощью кросс-линкера, CaCl2 в течение 30 мин, используя следующие концентрации CaCl2 - 30 мМ (0,33%) 60 мМ (0,66%), 120 мМ (1,32%), 240 мМ (2,64%) или 480 мМ (5,28%). Также оценивали влияние рН (4, 5, 6, 7, 8) на плотность кросс-линкера. Избыток Са удаляли диализом с помощью трубки для диализа 14 кДа, в течение 12 часов. Количество кросс-линкера определяли с помощью беспламенной Земановской атомной абсорбционной спектроскопии (AAS) при длине волны 212 нм. Для этого образцы высушивали из замороженного состояния с помощью лиофилизатора Labconco (Миссури, США) в течение ночи, затем разводили в 5 мл 70%-ной HNO3 и минерализовали нагреванием в течение 10 минут при 60°С. Затем образцы разводили с помощью ДДВ содержащей 0,1% HNO3. Калибровочные кривые включали 5 стандартов раствора CaCl2.
Результаты
Было выявлено, что увеличение в пектине связанных ионов Са зависело от концентрации раствора CaCl2 (Рис. 14В), но не зависело от рН (Рис. 14А). Наивысшие уровни содержания Са - 68 мкг/мг - для всех концентраций были достигнуты после 30-минутного связывания кросс-линкером (данные не приводятся).
Пример 12. Кинетика поперечного сшивания пектина
Методы
Genu х-2947 (DE=15,0%, молекулярный вес MW=122000) использовался для исследования кинетики поперечного сшивания. Матрицы готовили путем сшивания кросс-линкером 1,5%-ного раствора пектина CaCl2 - 0,1, 1 и 10% вес/объем в течение 30 секунд, 1 минуты или 30 минут. Избыток Са удаляли диализом с помощью трубки для диализа 14 кДа, в течение 12 часов. Образцы высушивали из замороженного состояния с помощью лиофилизатора Labconco (Миссури, США) в течение 12 часов. Кинетику реакции поперечного сшивания определяли оценкой концентрации пектина в среде разбавителя и анализировали с помощью гель-фильтрационной хроматографии (GFC), используя пятиточечную калибровочную кривую. Образцы фильтровали через 0,45 мкм фильтр-шприц перед инекцией.
Результаты
Высвобождение свободного пектина из матриц, в которых образованы поперечные связи, указывает на степень сшивания пектина. Следовательно, самое низкое значение растворенного пектина должно согласовываться с более высокой степенью образования поперечных связей.
Результаты испытания на высвобождение пектина в процессе инкубирования матриц СаР в SIF (USP), при различных длительностях инкубирования и концентрациях раствора CaCl2, представлены на Рис. 15А, В, С. На Рис. 15А, В и С представлено, что растворение свободного пектина находится в прямой зависимости как от концентрации раствора CaCl2, так и от времени инкубирования матриц в раствор CaCl2. Повышение концентрации раствора CaCl2 может привести к снижению степени растворения свободного пектина. Это было более очевидным для более короткого инкубирования. Таким же образом, продолжительность погружения пектина в раствор CaCl2 может повлиять на растворимость свободного пектина, в частности, для нижнего диапазона концентраций солевого раствора Са. Можно отметить, что чем больше было время инкубирования, тем меньше растворенного (свободного) пектина находилось в растворе. Этот факт показывает, что поперечное сшивание пектина in-situ - это кинетический процесс, при котором за пределами химического сродства ионов Са к галактуроновой кислоте также может быть задействован процесс диффузии CaCl2 в матрицы пектин/СаР. Было выявлено, что очень короткое время погружения (примерно 30 секунд) пригодно только для более высоких концентраций кросс-линкера (1% или 10%), при которых отмечается постоянная величина растворенного пектина в SIF (Рис. 15). 30 минут было достаточно для соответствующего сшивания кросс-линкером при всех испытываемых концентрациях (Рис. 15С).
Пример 13. Влияние DE пектина на его кинетику растворения
Методы
Genu х-2947 (DE=15,0% и молекулярный вес MW=122000) и Genu х-4952 (DE=30% и молекулярный вес MW=114700) использовались для исследования влияния DE на кинетику растворения пектина. Матрицы готовили путем сшивания кросс-линкером 1,5%-ного раствора пектина с помощью 4% CaCl2 в течение 30 минут. Избыток Са удаляли диализом с помощью трубки для диализа 14 кДа, в течение 12 часов. Образцы высушивали из замороженного состояния с помощью лиофилизатора Labconco (Миссури, США) в течение ночи. Кинетику растворения пектина в растворяющей среде анализировали с помощью GPC. Система GPC включала в себя насос для высокоэффективной жидкостной хроматографии Waters 510 HPLC, нагретый до 40°С дифференциальный рефрактометр Waters 410 и автосэмплер (Waters 717). Использовали колонку Shodex ОН рак КВ-806, разогретую до 35°С. Использовали дегазированную, предварительно отфильтрованную подвижную фазу USPXXII SIF с протоком 1 мл/мин. Образцы фильтровали через фильтр шприцевого типа на 0,45 мкм перед инъекцией.
Средой-растворителем был раствор фосфатного буфера (жидкость кишечника TS, ЖКТ, фосфат 50 мМ), который готовили согласно Фармакопее США, XXII (Стр. 1789). Растворы фосфатного буфера (PBS) с разными концентрациями фосфата (5, 15, 25 мМ) готовили разведением IFT.
Результаты
Корреляция между процентом связанного Са и растворением свободного пектина из матриц СаР приведена на Рис. 16, где сравниваются пектины с разными DE, 30 и 16% (Рис. 16А и В, соответственно). Как можно видеть, содержание связанного Са сопоставимо с растворением пектина в SIF. Чем ниже содержание связанных ионов Са, имеющихся в СаР с более высоким DE (Рис. 16А), тем выше значение растворенного пектина из СаР.
Пример 14. Оценка эффективности инкапсулирования (ЕЕ) LUV с загруженным лекарственным веществом в гранулы на основе пектина
Методы
Для оценки эффективности загрузки БСА, меченного с помощью ФИТС, 0,5 мг гранул (См. пример 10) диспергировали в 200 мкл 0,1 М NaOH/1% Triton. Затем образцы инкубировали при 37°С в течение 4 час для дезинтеграции гранул и липосом и для оценки количества заключенных в гранулы ФИТС-БСА. После инкубирования образцы центрифугировали 5 минут при 14.000 об/мин для получения надосадочной жидкости, которую анализировали на флуоресценцию. БСА-ФИТС с помощью Считывателя микропланшетов Synergy НТ при ex.485/em.525.% Эффективность ЭИ оценивали согласно стандартным кривым для БСА-ФИТС в 0,1 М NaOH/1% Triton X.
Результаты
Микрогранулы из пектина показали высокую эффективность инкапсулирования (ЭИ) БСА-ФИТС, заключенного в LUV, равную 70% (Таблица 4).
Пример 15. Оценка высвобождения БСА-ФИТС из гранул на основе пектина в моделированных жидкостях кишечника (SIF)
Методы
Для этого 2 мг гранул (Пример 10) диспергировали в 800 мкл SIF (USP) и инкубировали в течение 1,5 и 48 час при 37°С с перемешиванием во встряхивателе при 50 об/мин. По окончании инкубационного периода образцы центрифугировали при 1000 об/мин в течение 2 минут, и 200 мкл надосадочной жидкости анализировали на флуоресценцию следующим образом: для высвобождения БСА-ФИТС, образцы центрифугировали 12 минут при 14.000 об/мин в центробежных фильтрующих устройствах Microcon (с фильтром на 100 кДа) для отделения высвободившихся БСА-ФИТС, и этот фильтрат анализировали на флуоресценцию с помощью Считывателя микропланшетов Synergy НТ при ex.485/em.525.
Результаты
Приготовленные микрогранулы продемонстрировали длительное высвобождение меченного БСА в SIF. Высвобождение БСА возросло от 5 до 80% в течение 1,5-48 часов инкубирования для всех приготовленных составов (Рис. 17).
Литература
1. Gaikwad A, Pathade Р, Bidkar S, Pawar A. Journal of Pharmacy Research 2011; 4:300-303.
2. A.J. Domb, A. Benitolila and D. Teomin, Acta Polym., 1998, 49, 526-533.
3. Gombotz WR, Wee SF.. Adv Drug Deliv Rev 1998; 31:267-85.
4. George M, Abraham ТЕ. J Control Release. 2006; 114:1-14.
5. Wong TW, Chan LW, Kho SB, Heng PWS. J Control Release 2002; 84:99-114.
6. Martinsen, A., Skjak-Braek, G., Smidsrod, O., Zanetti, F., Paoletti, S., Carbohydr. Polym. 1991, 15: 171-193.
7. Elquin, Y.M., Biomaterials 1995, 18: 1157-1161.
8. V.R. Sinha, A.K. Singla, S. Wadhawan, R. Kaushik, R. Kumria, K. Bansal, S. Dhawan. 2004. International Journal of Pharmaceutics, 214: 1-33.
9. Daly, M.M., Knorr, D., Biotech. Prog. 1998, 4: 76-81.
10. Murnper, R.J., Hoffman, A.S., Puolakkainen, P., Bouchard, L.S., Gombotz, W.R.,. J. Control. Release, 1994, 30: 241-251.
11. Sutherland, I.W., Alginates, in T. Hayashi, Prog Polym Sci., 1994, 19, 663-702.
12. D. Byrom (Ed.), Biomaterials; Novel Materials from BiologicalSources, Stockton, New York, 1991, pp.309-331.
13. Takka S, Ocak OH, Acarturk F. Eur J Pharm Sci, 1998; 6:241-6.
14. Pillay V, Fassihi R. J Control Release. 1999; 20; 59(2):229-42.
15. Xu Y, Zhan C, Fan L, Wang L, Zheng H. Int J Pharm. 2007, 24; 336(2):329-37.
16. Choi, M. J.; Maibach, H. I. Skin Pharmacol. Physiol. 2005, 18 (5), 209-219.
17. Tanaka, Т.; Taneda, K.; Kobayashi, H.; Okumura, K.; Muranishi, S.; Sezaki, H. Chem. Pharm. Bull. 1975, 23 (12), 3069-3074.
18. Vanlerberghe, G. Liposomes in cosmetics how and why? In Handbook of Nonmedical Applications of Liposomes; Lasic, D., Barenholz, Y., Eds.; CRC Press: Boca Raton, FL, 1996; p 77.
19. Dufour, P.; Laloy, E.; Vuillemard, J. C; Simard, R. Liposomes in cheesemaking. In Handbook of Nonmedical Applications of Liposomes; Lasic, D., Barenholz, Y., Eds.; CRC Press: Boca Raton, FL, 1996; pp 158-164.
20. Picon, A.; Gaya, P.; Medina, M.; Nunez, M. Biotechnol. Lett. 1997, 19 (4), 345-348.
21. Singh, R.; Singh, S.; Lillard, J. W., Jr. J. Pharm. Sci. 2008, 97 (7), 2497-2523.
22. Keller, В. C. Trends Food. Sci. Technol. 2001, 12(1), 25-31.
23. Yoshie, M.; Megumi, H.; Yasuko, Т.; Naoto, S.; Tsuneji, N. J. Pharm. Sci. 1996, 85, 440-445.
24. Cohen, S.; Bano, M. C; Chow, M.; Langer, R. Biochim. Biophys. Acta 1991, 1063 (1), 95-102.
25. Dai, C; Wang, В.; Zhao, H.; Li, B. Colloids Surf B, 2005, 42 (3-4), 253-258.
26. Dai, C; Wang, В.; Zhao, H.; Li, В.; Wang, J. Colloids Surf B, 2006, 47 (2), 205-210.
27. Dhoot, N.О.; Wheatley, M.A. J. Pharm. Sci. 2003, 92 (3), 679-689.
28. Hong, J.S.; Vreeland, W.N.; Lacerda, S.H.; Locascio, L.E.; Gaitan, M.; Raghavan, S.R. Langmuir, 2008, 24 (8), 4092-4096.
29. Huang, L. Preparation of solid core liposomes. U.S. Patent 4, 839, 111, 1989.
30. Monshipouri, M.; Rudolph, A.S. J. Microencapsulation, 1995, 12 (2), 117-127.
31. Tiwari, S.; Goyal, A.K.; Khatri, K.; Mishra, N.; Vyas, S.P. J. Microencapsulation 2009, 26 (1), 75-82.
32. Smith, AM., Jaime-Fonseka, M.R,,. Grover, L.M., Bakalis, S. J. Agric. Food Chem. 2010, 58, 4719-4724.
33. Acarturk F, Takka S. J Microencapsul. 1999; 16: 291-301.
34. Yan Pan, Ying-jian Li, Hui-ying Zhao, Jun-min Zheng, Hui Xu, Gang Wei, Jin-song Hao, Fu-de Cui. International Journal of Pharmaceutics, 2002: 249, 139-147.
35. Bodmeier, R., Wang, J. J Pharm Sci. 1993; 82: 191-194.
36. Takka S, Acarturk F. Pharmazie. 1999; 54: 137-139.
37. George M, Abraham T E. Int J Pharm. 2007; 335: 123-129.
38. Tang Y D, Venkatraman S S, Boey Y C, Wang L W. Int J Pharm. 2007; 336: 159-165.
39. Østberg T, Vesterhus L, Graffher C. Int J Pharm. 1993; 97:183-193.
40. Smrdel, P., Bogatag, M., Mrhar, A.. Sci Pharm. 2008; 76: 77-89.
41. Dressman, J. Journal of Pharmacy and Pharmacology. 2006; 58 (8): 1079-1089.
42. Shunwen, H. Integrity and stability of oral liposomes containing bile salts studied in simulated and ex vivo gastrointestinal media. International Journal of Pharmaceutics 2013; 441: 693-700.
| название | год | авторы | номер документа |
|---|---|---|---|
| Применение соли альгината в качестве сорбента липидов, фармацевтическая композиция и лекарственный препарат для лечения ожирения | 2014 |
|
RU2646788C2 |
| ЛИПОСОМНЫЕ КОМПОЗИЦИИ, ИСПОЛЬЗУЕМЫЕ ДЛЯ ДОСТАВКИ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2015 |
|
RU2757110C2 |
| ТРАНСЛЕГОЧНАЯ ЛИПОСОМА ДЛЯ РЕГУЛИРОВАНИЯ ДОСТАВКИ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2008 |
|
RU2493874C2 |
| ЛИПОСОМНЫЕ КОМПОЗИЦИИ, ИСПОЛЬЗУЕМЫЕ ДЛЯ ДОСТАВКИ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2005 |
|
RU2574926C2 |
| СИСТЕМА ДОСТАВКИ ЛЕКАРСТВА | 1999 |
|
RU2202340C2 |
| СРЕДСТВО ДЛЯ ДОСТАВКИ АКТИВНОГО АГЕНТА | 2017 |
|
RU2669354C1 |
| ЛИПИДНО-ПОЛИМЕРНАЯ СИСТЕМА ОДНОВРЕМЕННОЙ ДОСТАВКИ ДВУХ АНТИБАКТЕРИАЛЬНЫХ СОЕДИНЕНИЙ | 2023 |
|
RU2838145C1 |
| СИСТЕМА ДОСТАВКИ ВЕЩЕСТВА БЕЛКОВОЙ ПРИРОДЫ В ВИДЕ НАНОЧАСТИЦ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2013 |
|
RU2566069C2 |
| Композиция для пероральной доставки биоактивных агентов | 2014 |
|
RU2688136C1 |
| КОМПОЗИЦИЯ ДЛЯ ЭНТЕРОСОЛЮБИЛЬНОГО ПОКРЫТИЯ ПРИРОДНОГО ПРОДУКТА, СОДЕРЖАЩЕГО ЛЕКТИН | 2004 |
|
RU2315595C2 |
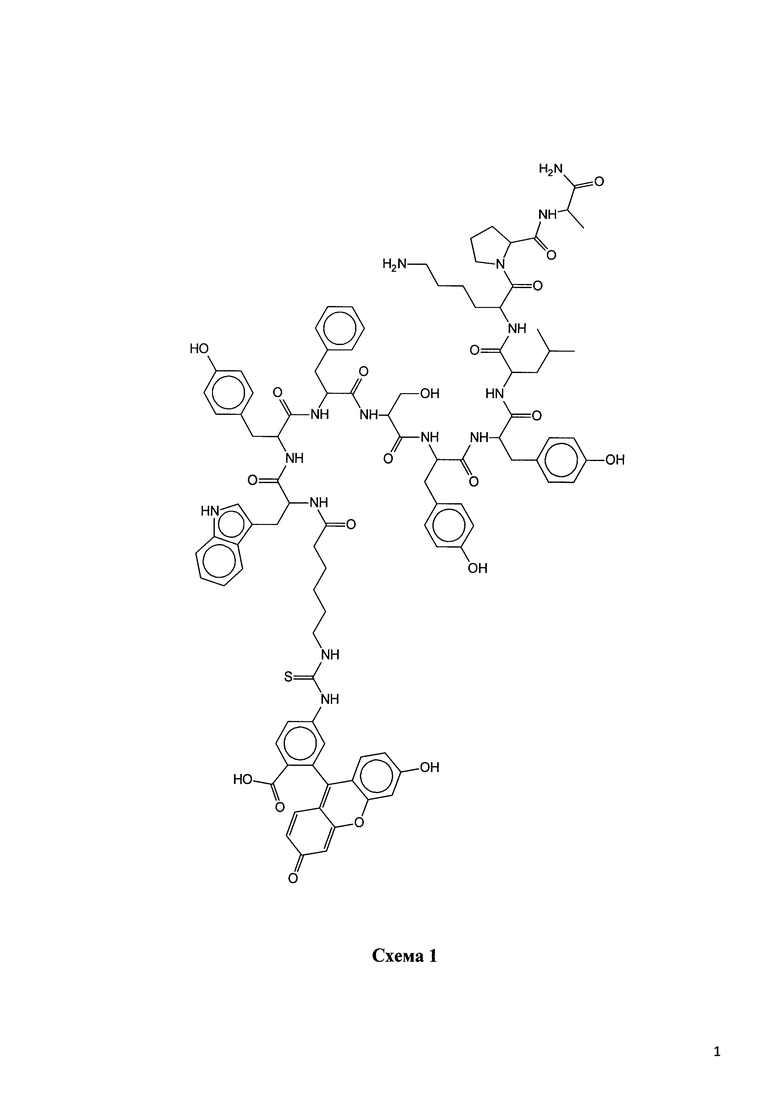
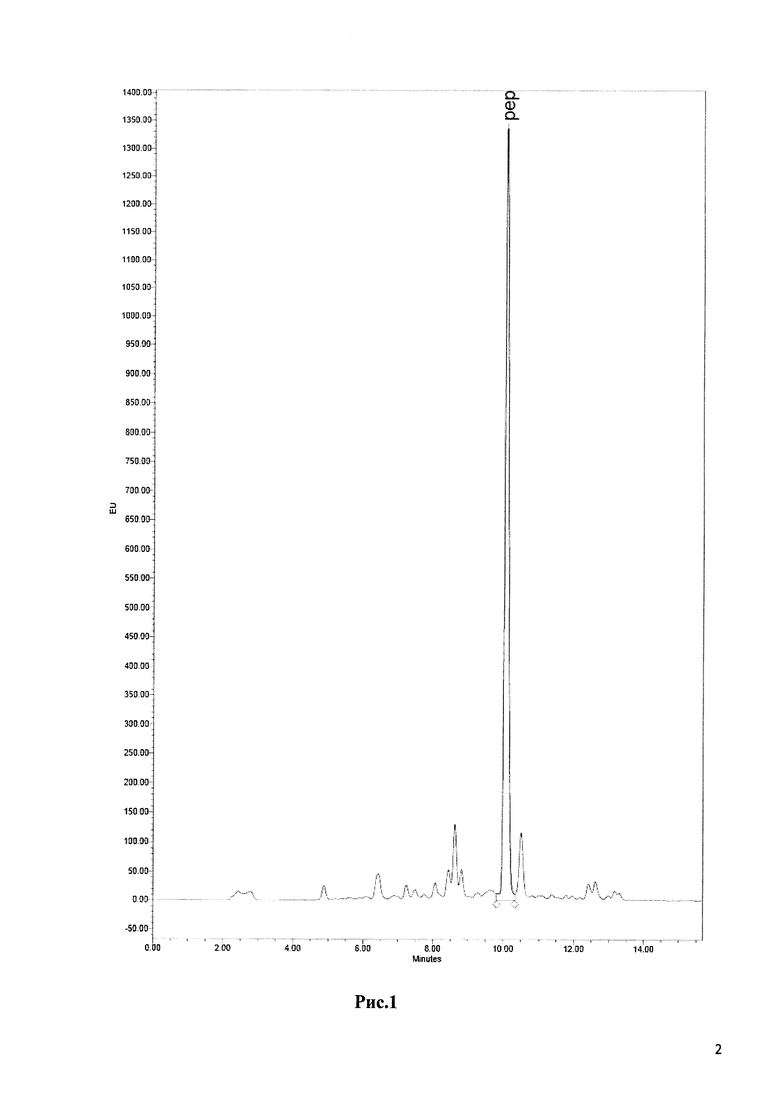
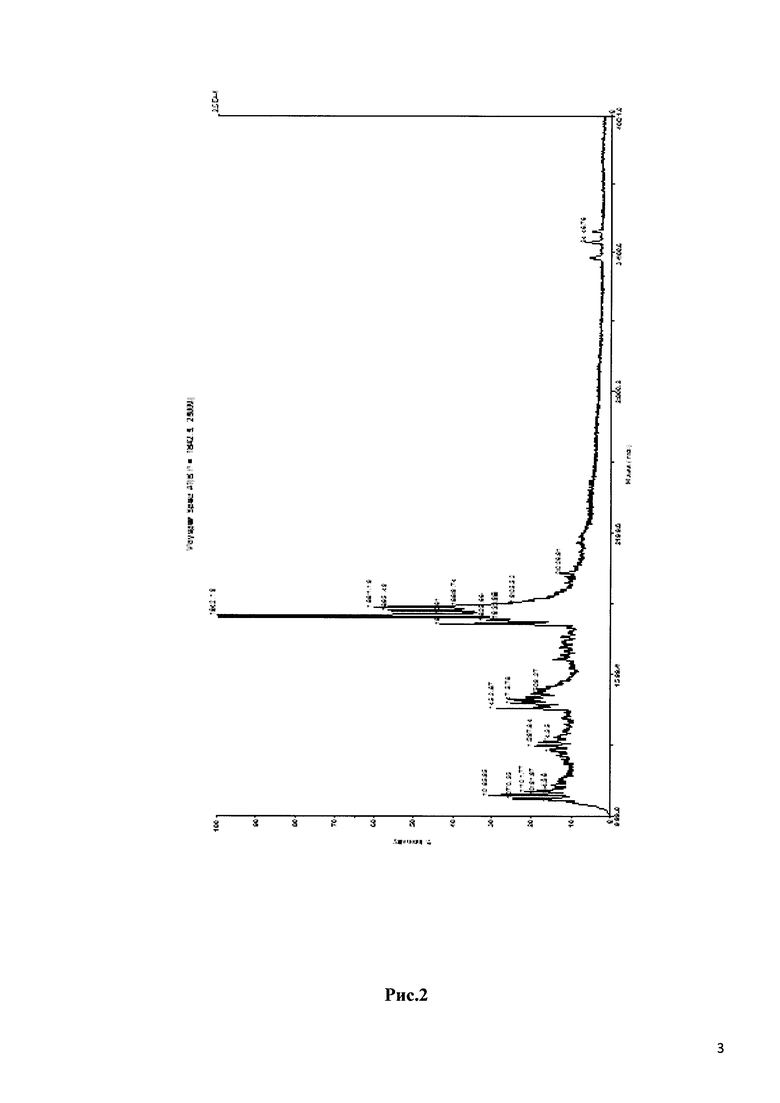

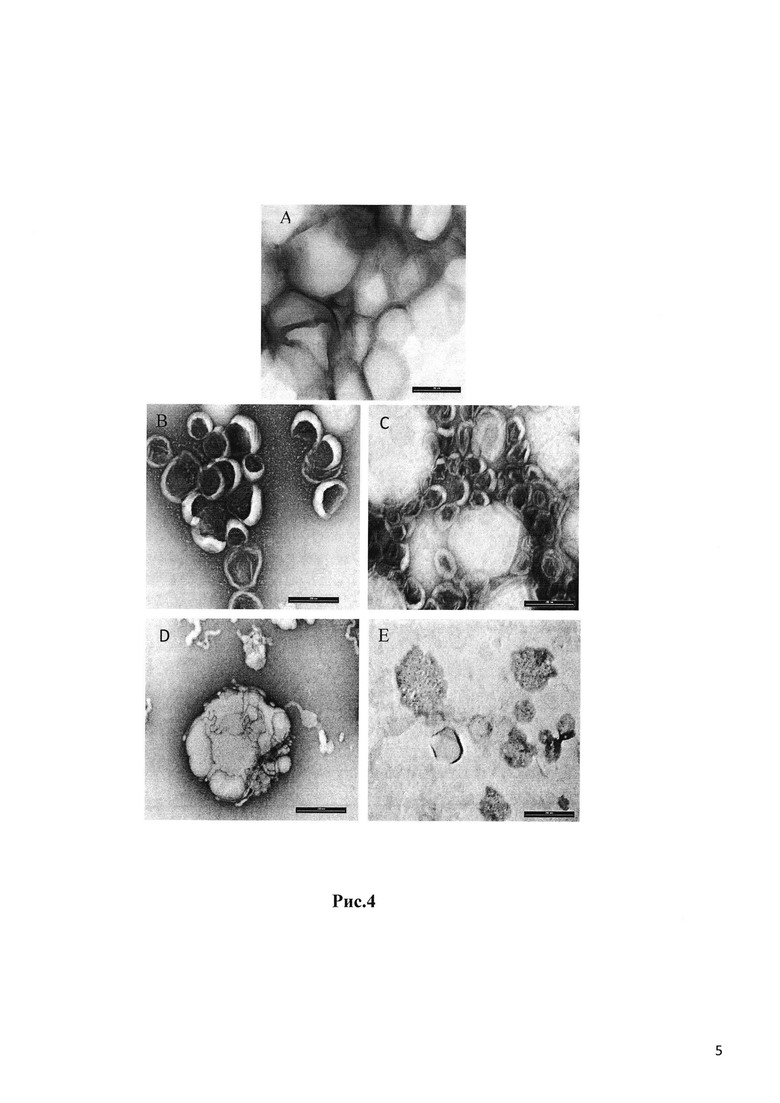
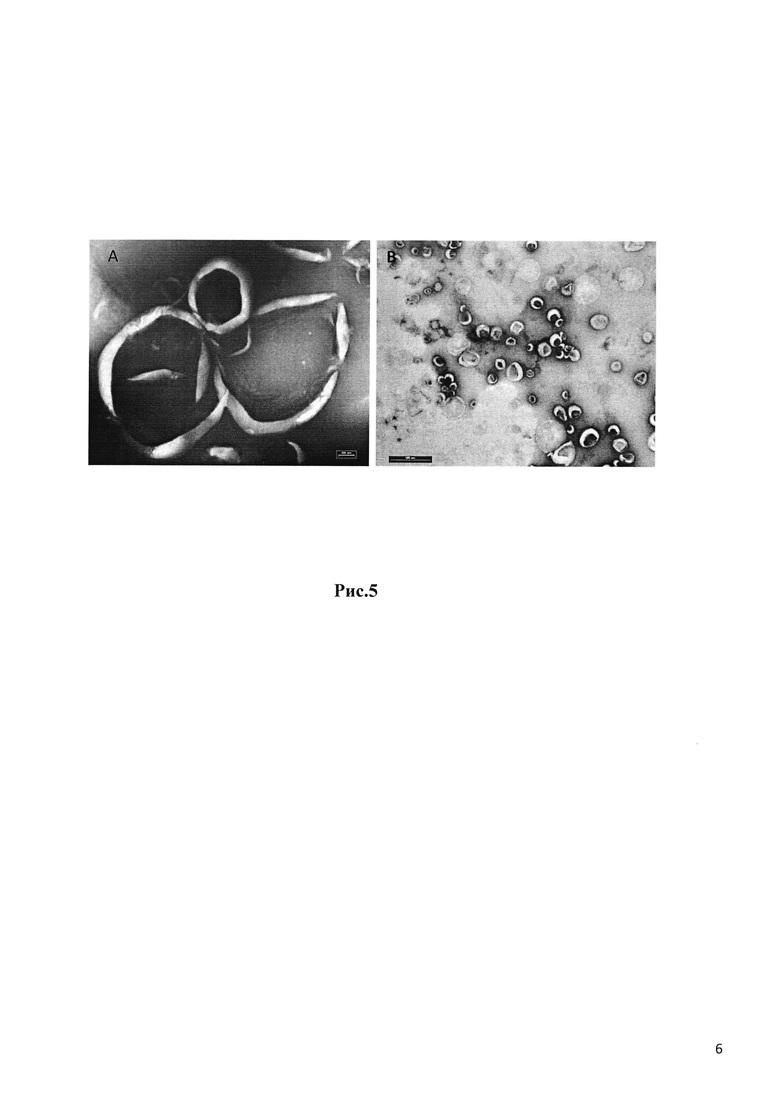
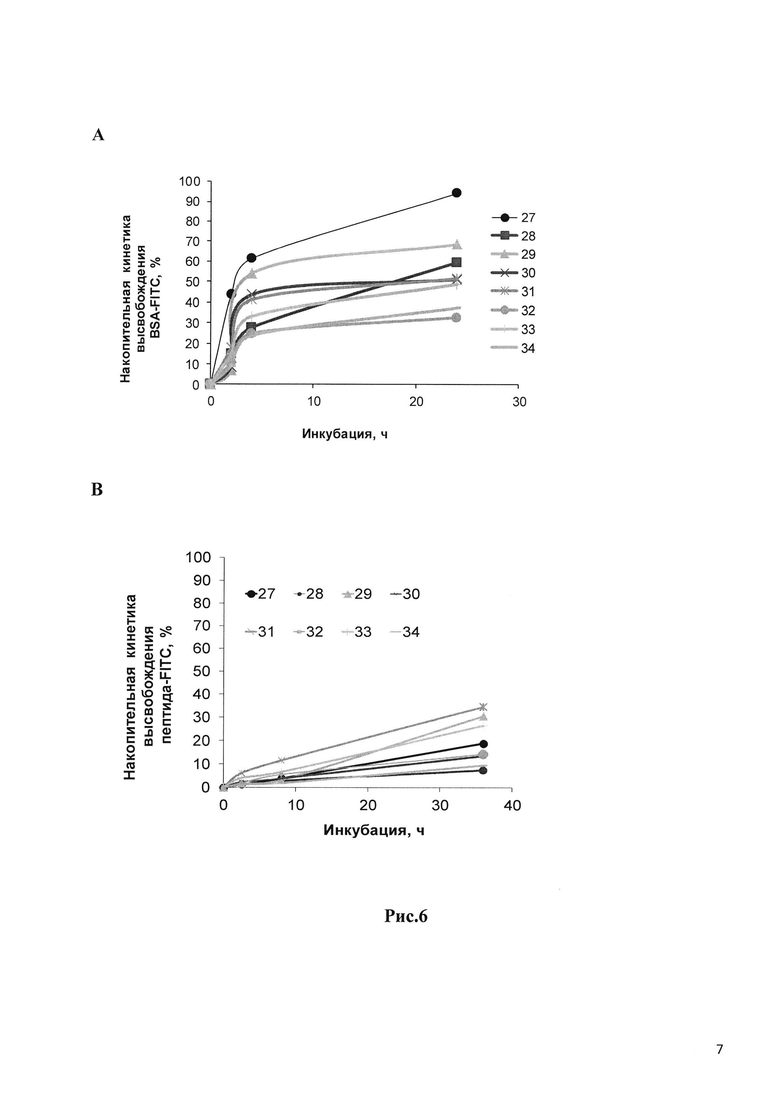
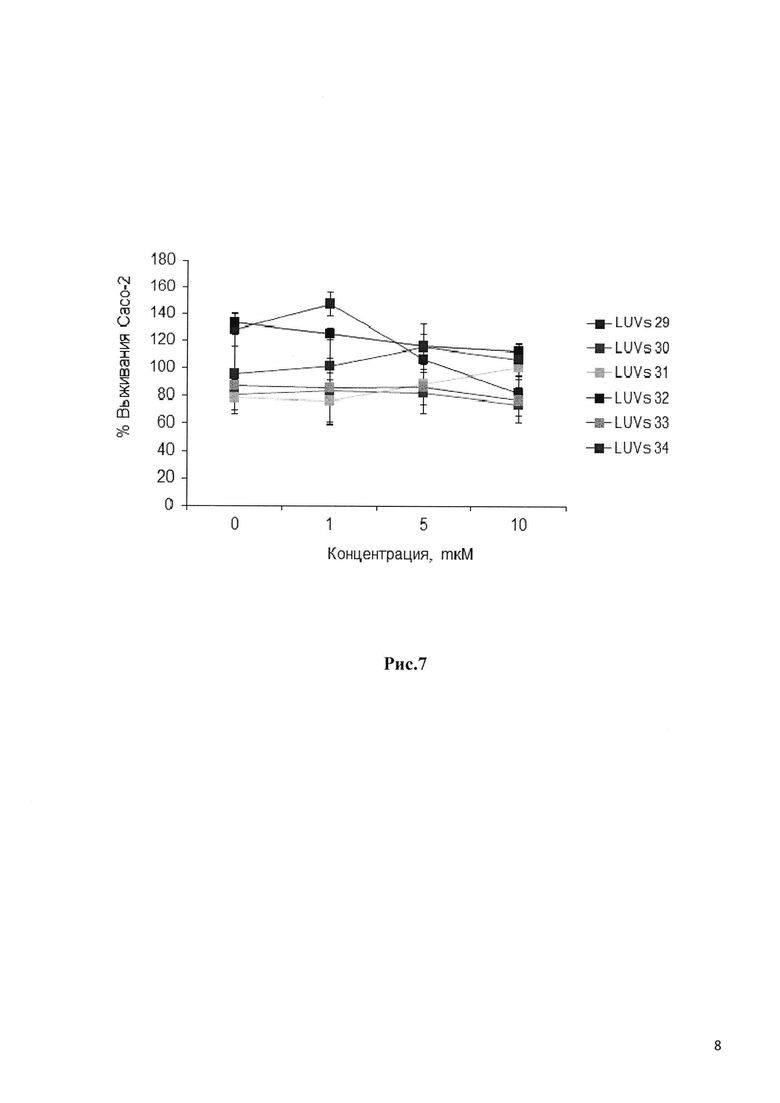
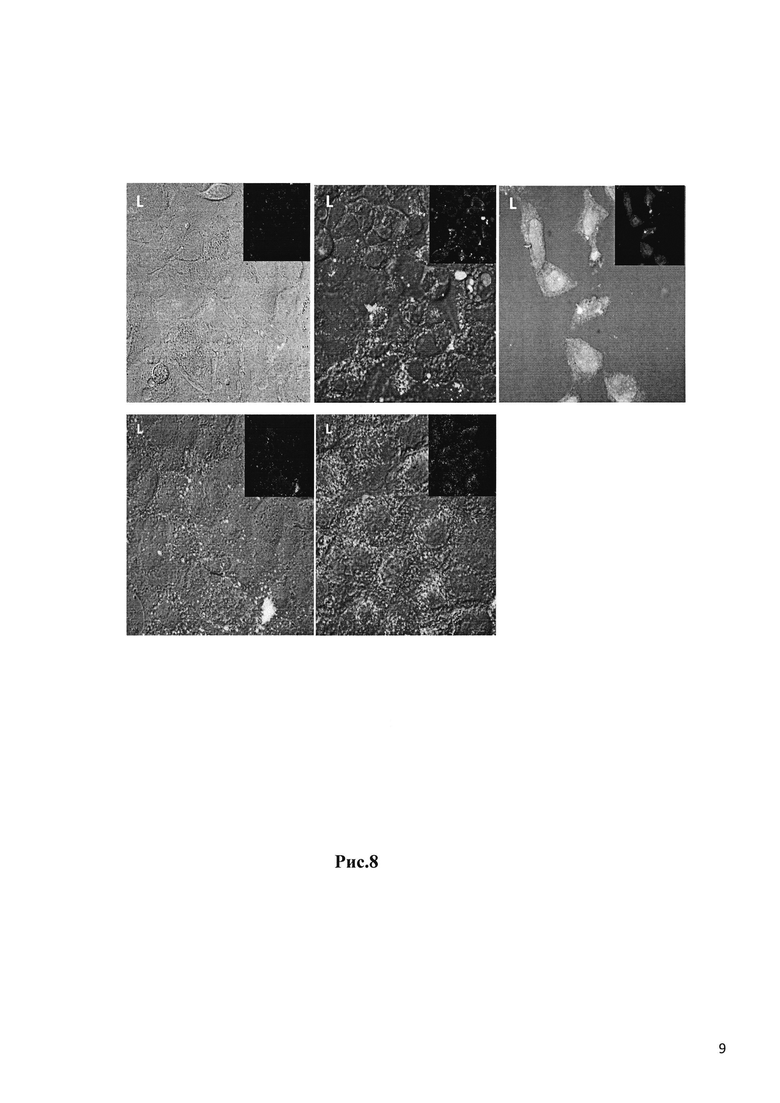


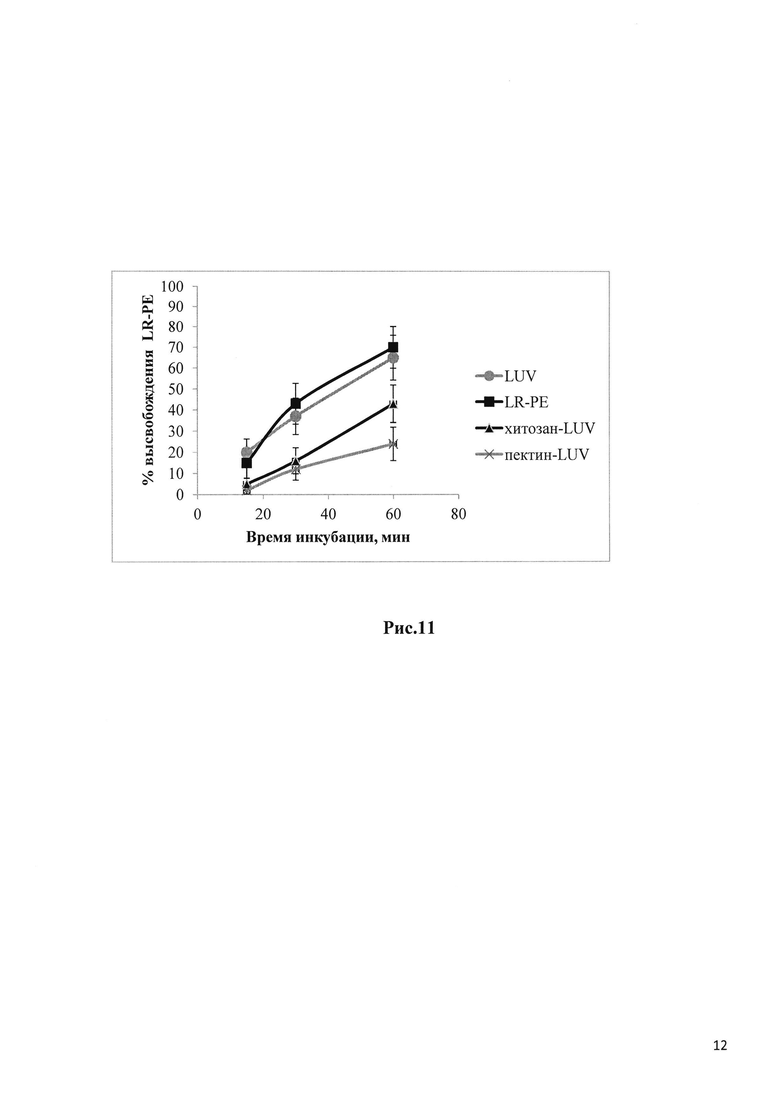
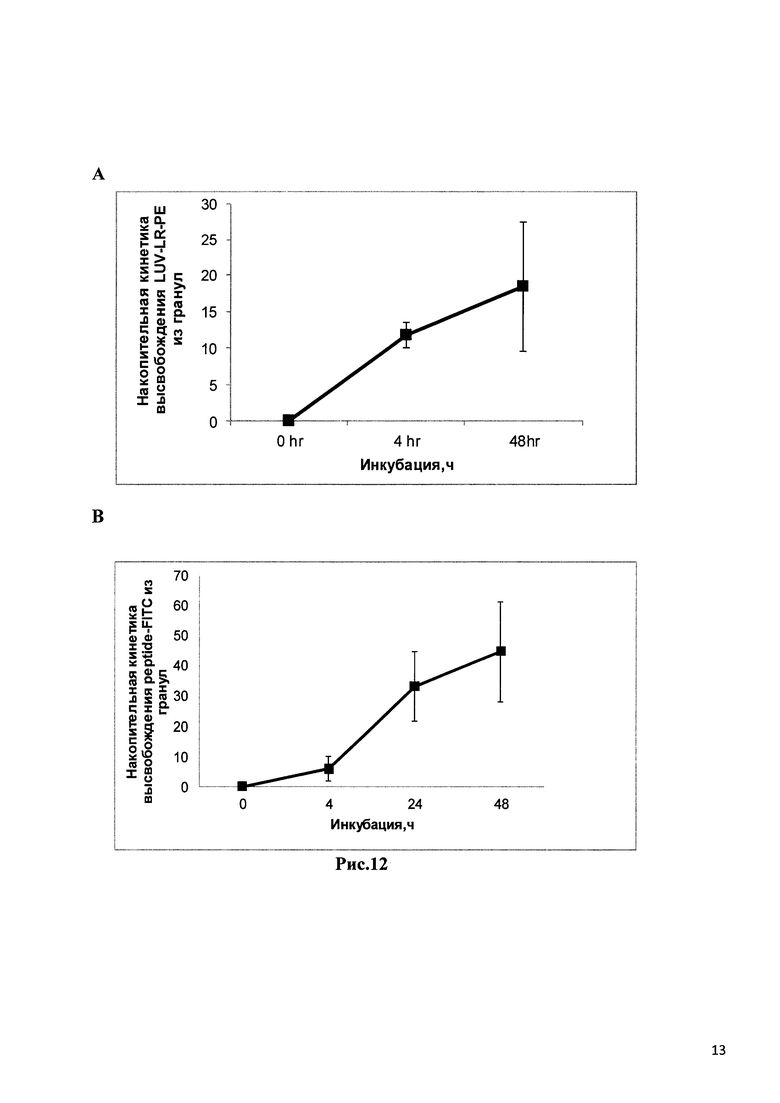


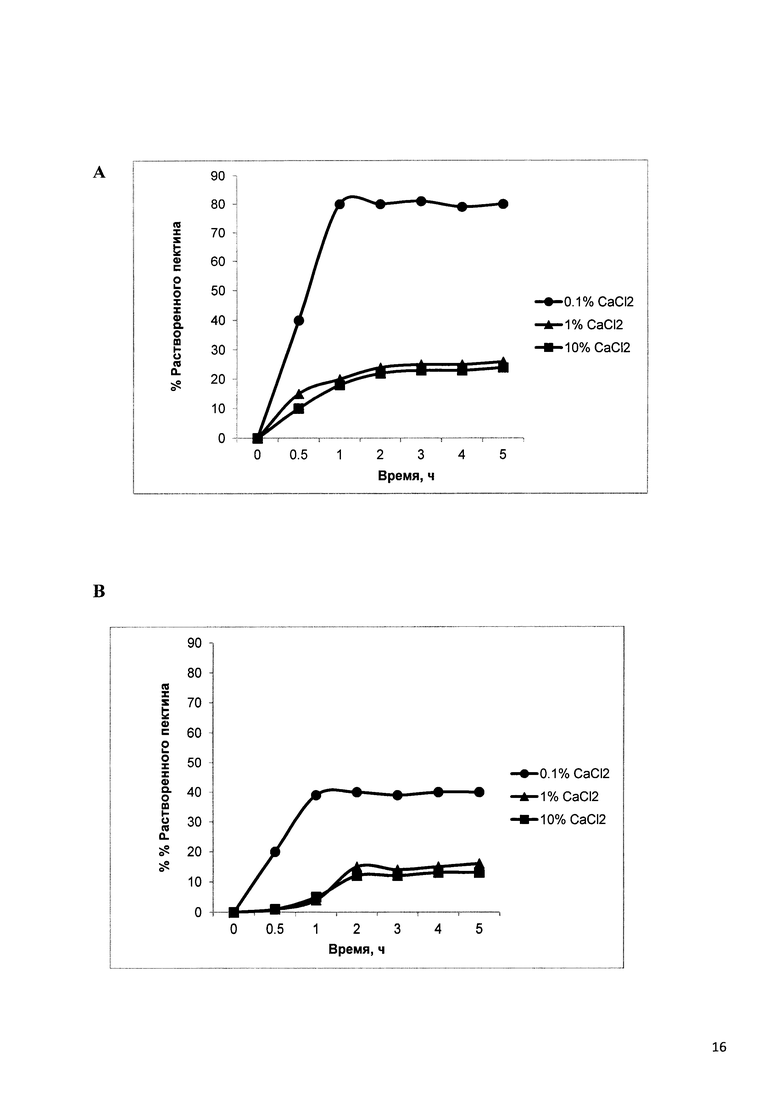

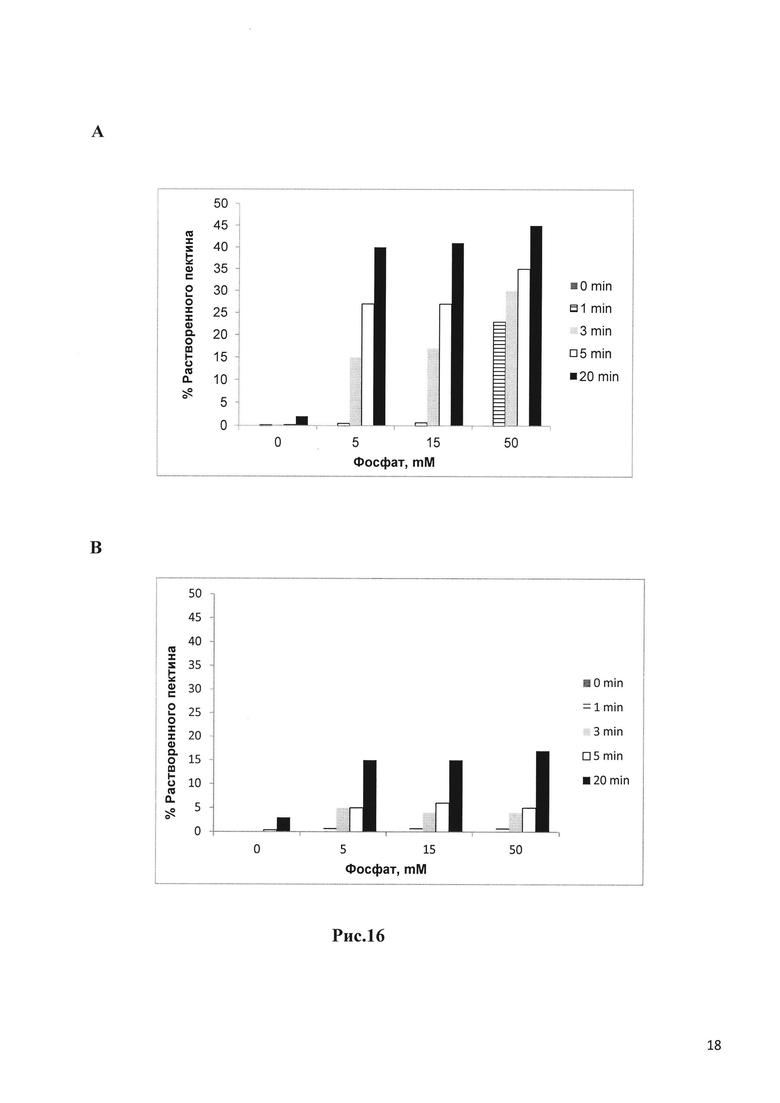
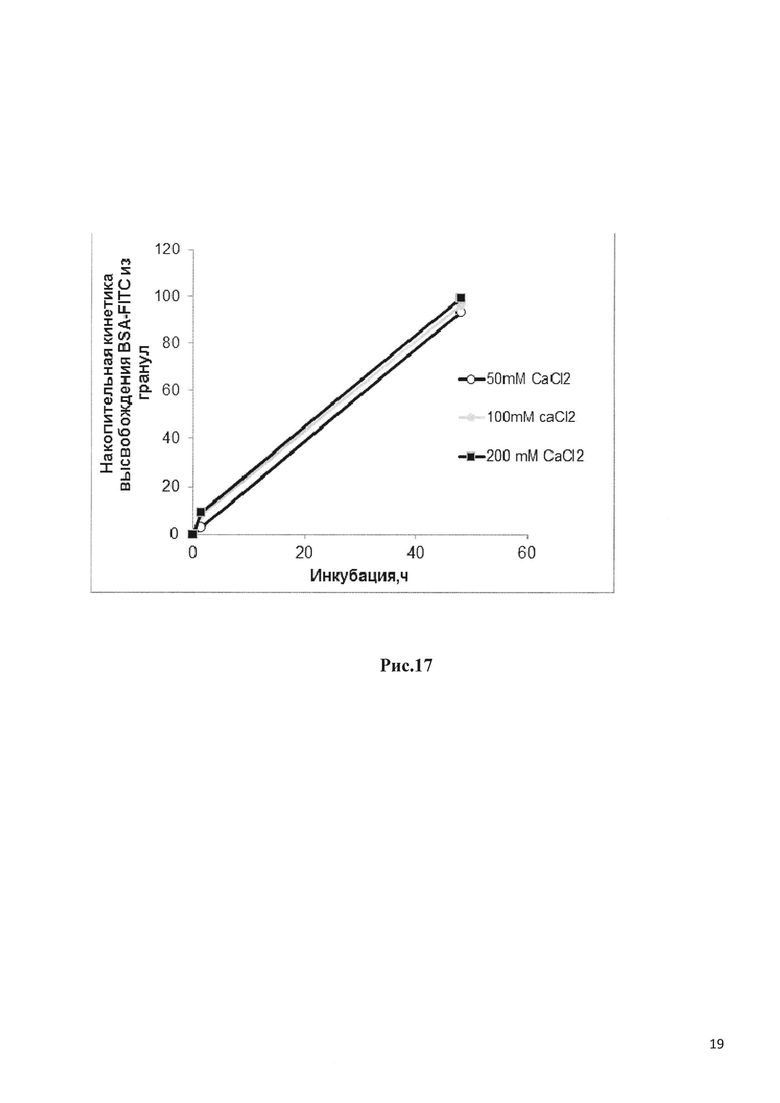
Группа изобретений относится к медицине и касается пероральной системы доставки лекарственного вещества белковой природы из липосом, включенных в частицы поперечносшитого природного полисахарида, выбранного из поперечносшитого поливалентным металлом пектина и поперечносшитого полианионом хитозана. Группа изобретений также касается защитной оболочки указанной пероральной системы доставки. Группа изобретений обеспечивает увеличение биодоступности лекарственного вещества белковой природы. 3 н. и 13 з.п. ф-лы, 17 ил., 2 табл., 15 пр.
1. Пероральная система доставки лекарственного вещества белковой природы из липосом, включенных в частицы поперечносшитого природного полисахарида, выбранного из поперечносшитого поливалентным металлом пектина и поперечносшитого полианионом хитозана.
2. Пероральная система доставки по п. 1 для доставки лекарственного вещества белковой природы в кишечник.
3. Пероральная система доставки по п. 2 для доставки лекарственного вещества белковой природы в течение 1-48 ч.
4. Пероральная система доставки по п. 1, где для получения частиц используют пектин с низкой степенью этерификации.
5. Пероральная система доставки по п. 4, где для получения частиц используют пектин со степенью этерификации 10-40%.
6. Пероральная система доставки по п. 1, где для поперечного сшивания используют соли Са+2 или Al+3.
7. Пероральная система доставки по п. 6, где для поперечного сшивания используют соли Са+2
8. Пероральная система доставки по п. 1, где для поперечного сшивания используют триполифосфат.
9. Пероральная система доставки по п. 1 в виде лиофилизата.
10. Защитная оболочка пероральной системы доставки по п. 1, состоящая из пектина, поперечносшитого поливалентным металлом, при следующем содержании в системе доставки в пересчете на сухую массу, масс. %:
11. Защитная оболочка по п. 10, состоящая из пектина, поперечносшитого поливалентным металлом, при следующем содержании в системе доставки в пересчете на сухую массу, масс. %:
12. Защитная оболочка по п. 10, в которой в качестве поливалентного металла использован Са+2 или Al+3.
13. Защитная оболочка по п. 12, в которой в качестве поливалентного металла использован Са+2.
14. Защитная оболочка пероральной системы доставки по п. 1, состоящая из хитозана, поперечносшитого полианионом, при следующем содержании в системе доставки в пересчете на сухую массу, масс. %:
15. Защитная оболочка по п. 14, состоящая из хитозана, поперечносшитого полианионом, при следующем содержании в системе доставки в пересчете на сухую массу, масс. %:
16. Защитная оболочка по п. 1, в которой в качестве полианиона использован триполифосфат.
| WO 2005030173 A1, 07.04.2005 | |||
| WO 2006103657 A2, 05.10.2006 | |||
| DHAWAN S., et al., Evaluation of mucoadhesive properties of chitosan microspheres prepared by different methods.AAPS PharmSciTech | |||
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| МУХАМЕДЖАНОВА М.Ю | |||
| и др., Процессы гелеобразования и реологические свойства умеренно-концентрированных водных растворов цитрусового пектина в присутствии ионов поливалентных металлов | |||
| Химия растительного сырья | |||
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |

