ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение в целом относится к липосомам, и, в частности, к липосомным композициям, используемым для доставки лекарственных и диагностических соединений.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Липосомы, или липидные двухслойные везикулы, используют или предлагают использовать для различных целей: исследовательских, промышленных и медицинских, в частности, в качестве носителей диагностических и лекарственных соединений in vivo. См, например, Lasic, D. Liposomes: from physic to applications. Elsevier, Amsterdam, 1993. Lasic, D, и Papahadjopoulos, D., eds. Medical Applications of Liposomes. Elsevier, Amsterdam, 1998. Липосомы обычно характеризуются наличием внутреннего пространства, отделенного от окружающей среды мембраной из одного или более бислоев, таким образом, формируется микроскопический мешочек или везикула. Двухслойные мембраны липосом обычно формируются липидами, то есть синтетическими или природными амфифильными молекулами, которые имеют пространственно разделенные гидрофильные и гидрофобные домены. См. Lasic D., 1993, supra. Двухслойные мембраны липосом могут быть также сформированы амфифильными полимерами и сурфактантами (полимеросомами, ниосомами). Обычно липосома выполняет функцию носителя множества объектов, включая (без ограничений указанными) химическое соединение, комбинации соединений, высокомолекулярный комплекс синтетического или природного происхождения, генетический материал, живой организм, а также части и производные перечисленных выше объектов, которые обладают полезными свойствами или проявляют полезную активность. Для этих целей липосомы получают таким образом, чтобы они содержали необходимый объект. Процесс включения желаемого объекта в липосому обычно обозначают термином "нагрузка". Включенный в липосому объект может полностью или частично располагаться во внутреннем пространстве липосомы, находиться в пределах двухслойной мембраны липосомы или быть связанным с наружной поверхностью липосомной мембраны. Включение объектов в липосомы также обозначают терминами инкапсулирование или захват, и все три указанных термина используются в настоящем описании взаимозаменяемо и имеют одинаковое значение.
При инкапсулировании вещества в липосому обычно преследуется цель защитить указанное вещество от деструктивного влияния окружающей среды и обеспечить возможность проявления активности указанным веществом в том месте, где данная активность наиболее предпочтительна, и избежать ее реализации там, где это бесполезно или нежелательно. Указанное явление обеспечивается доставкой. Например, лекарственное средство в составе липосомы может быть защищено от ферментного расщепления в организме, при этом указанное средство будет высвобождаться из липосомы и оказывать нужный лечебный эффект в том месте, где локализуется патологический процесс.
В идеале, такие липосомы могут быть приготовлены таким образом, чтобы включать желаемое соединение (i) с высокой нагрузочной эффективностью, которая выражается как процентное соотношение количества инкапсулировавшегося вещества и общего количества вещества, задействованного в процессе; (ii) большое количество инкапсулированного вещества на одну единицу липосомного бислойного материала; (iii) высокую концентрацию инкапсулированного вещества, и (iv) в стабильной форме, что подразумевает незначительное высвобождение инкапсулированного вещества из липосомы во время хранения или непосредственно перед тем, как липосома достигнет места или среды, где вещество, заключенное в липосому должно проявлять свою внутреннюю активность.
Таким образом, существует необходимость создания различных липосомных композиций, которые можно использовать для доставки множества соединений, в частности, лекарственных, диагностических и контрастирующих веществ.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение основано на открытии, что замещенный аммоний и полианион могут использоваться для нагрузки и сохранения веществ внутри липосом. Соответственно, настоящее изобретение относится к способам и липосомным композициям, используемым для доставки различных соединений, в частности, терапевтических агентов, а именно, веществ, применяемых для диагностики, прогнозирования, тестирования, скринирования, лечения и профилактики нежелательных состояний, например, заболеваний у живых организмов, таких, как люди, растения или животные.
Согласно одному варианту своего осуществления, настоящее изобретение относится к композиции, включающей липосому в среде, при этом внутри указанной липосомы находится замещенный аммоний
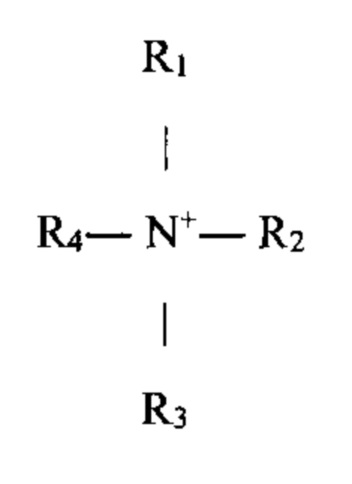
где каждый R1, R2, R3 и R4 независимо друг от друга представляют собой водород или органическую группу, имеющую, включительно, вплоть до 18 атомов углерода, при этом, по меньшей мере один из R1, R2, R3 или R4 представляет собой органическую группу, при этом органическая группа представляет собой углеводородную группу, имеющую вплоть до 8 атомов углерода, и является алкил, алкилиден, гетероциклический алкил, циклоалкил, арил, алкенил или циклоалкенил группой или их гидрокси-замещенным производным, и необязательно имеет в составе своей углеводородной цепи атомы S, О или N, формируя эфир, сложный эфир, тиоэфир, амин или амидную связь, при этом по меньшей мере три из R1, R2, R3 или R4 представляют собой органические группы, или же замещенный аммоний представляет собой стерически затрудненный аммоний, такой, как, например, в котором по меньшей мере одна из органических групп имеет вторичный или третичный атом углерода, непосредственно связанный с атомом азота аммония. Предпочтительно, соединение замещенного аммония, инкапсулированное в липосому, имеет отрицательный логарифм константы кислотной диссоциации (депротонирования) (pKa), равный, по меньшей мере 8.0, по меньшей мере, 8.5, по меньшей мере, 9.0, по меньшей мере, 9.5 или, по меньшей мере, 10.0, что определяется в водном растворе при комнатной температуре.
Согласно другому варианту своего осуществления, настоящее изобретение относится к композиции, включающей липосому в среде, при этом во внутреннем пространстве липосомы находится полианион, при этом указанный полианион представляет собой полианионизированный полиол или полианионизированный сахар. Липосома, предпочтительно, имеет трансмембранный градиент, который способствует эффективной инкапсуляции вещества в липосому. Согласно одному варианту осуществления настоящего изобретения, трансмембранный градиент представляет собой градиент аммония, четвертичного аммония, или первичного, вторичного или третичного замещенного аммониевого соединения, который в водном растворе при комнатной температуре имеет константу кислотной диссоциации (депротонирования) (pKa), равную, по меньшей мере, 8.0, по меньшей мере, 8.5, по меньшей мере, 9.0, по меньшей мере, 9.5 или, по меньшей мере, 10.0. Липосома может также содержать инкапсулированное соединение, например, лекарственное или выявляемое соединение, или полностью катионную органическую молекулу.
Согласно еще одному варианту осуществления настоящего изобретения, композиция, в соответствии с настоящим изобретением, содержит вещество, инкапсулированное в липосомы в соответствии с настоящим изобретением. Предпочтительно, вещество заключено во внутреннее пространство липосомы. Например, во внутреннем пространстве липосомы может содержаться противоопухолевый лекарственный агент, при этом уровень токсичности данной композиции для организма остается таким же или даже меньшим, чем в том случае, когда противовопухолевый агент вводится вне указанной композиции.
Согласно еще одному варианту осуществления настоящего изобретения, композиция в соответствии с настоящим изобретением представляет собой липосомную композицию, содержащую соединение камптотецина. Указанная композиция обладает противоопухолевой активностью, которая, по меньшей мере, в два раза, по меньшей мере, в четыре раза или, по меньшей мере, в 10 раз превышает противоопухолевую активность камптотецина при его введении вне указанной композиции, при этом токсичность композиции не увеличивается, и, по меньшей мере, в два раза, или, по меньшей мере, в четыре раза ниже, чем токсичность камптотецина при его введении вне указанной композиции. Согласно одному варианту осуществления настоящего изобретения, соединение камптотецина представляет собой пролекарственное средство и находится в составе липосомы в дозе по меньшей мере 0,1 мг, по меньшей мере, 0,2, по меньшей мере, 0,3 мг, по меньшей мере, 0,5 мг или по меньшей мере 1 мг на 1 мг вещества липосомной мембраны, то есть липидов. Соединение камптотецина, предпочтительно, полностью инкапсулировано во внутреннее пространство липосомы. Согласно одному варианту, соединение камптотецина представляет собой иринотекан (СРТ-11).
Согласно еще одному варианту осуществления настоящего изобретения, композиция в соответствии с настоящим изобретением представляет собой липосомную композицию алкалоидов барвинка (винкаалкалоидов) или их производных. Указанная композиция сохраняется внутри липосомы в течение 24 часов после 24 часов экспозиции в крови млекопитающего in vivo, что составляет, по меньшей мере, 50%, по меньшей мере, 60%, по меньшей мере, 70% исходной лекарственной нагрузки. Винкаалкалоид или его производное, предпочтительно, инкапсулировано во внутреннее пространство липосомы. Одним примером млекопитающего является крыса. Примерами винкаалкалоидов и их производных являются винкристин, винбластин и винорелбин.
Согласно еще одному варианту своего осуществления, настоящее изобретение относится к способу инкапсулирования объекта в липосому. Указанный способ включает обеспечение контакта липосом в соответствии с настоящим изобретением с веществом, например, лекарственным или контрастным веществом. Предпочтительно, указанный контакт осуществляют при таких условиях, когда концентрация замещенного аммония или полианиона, заявленного в соответствии с настоящим изобретением, в среде, меньше, чем во внутреннем пространстве липосом. Согласно одному варианту осуществления настоящего изобретения, контакт липосомной композиции и вещества происходит в водной среде.
Согласно еще одному варианту своего осуществления, настоящее изобретение относится к способу инкапсулирования объекта в липосому. Указанный способ включает обеспечение контакта липосомных композиций в соответствии с настоящим изобретением с предшественником вещества, при этом указанный предшественник может превращаться в активное вещество при определенных условиях, а также обеспечение таких условий внутри липосом, которые будут способствовать образованию активного вещества из его предшественника. Согласно одному варианту, вещество представляет собой органическое соединение, и предшественник является основным его производным.
Согласно еще одному варианту своего осуществления, настоящее изобретение относится к набору для приготовления веществ, заключенных в липосомы. Указанный набор включает контейнер с липосомой в соответствии с настоящим изобретением и, возможно, контейнер с веществом и/или инструкции по применению, то есть инкапсулированию вещества в липосому.
КРАТКОЕ ОПИСАНИЕ ФИГУР
На Фигуре 1 представлена фармакокинетика липидов липосом (окружности) и лекарственного средства (треугольники) в крови после внутривенного введения СРТ-11 нагруженных липосом крысам. Липосомы нагружают с помощью ТЕА-Pn способа (См. Пример 9).
На Фигуре 2 представлена динамика соотношения лекарственное средство/липосома в крови крыс in vivo после внутривенного болюсного введения СРТ-11-нагруженных липосом, полученных с помощью ТЕА-Pn способа (См. Пример 9).
На Фигуре 3 представлена противоопухолевая активность свободного СРТ-11 и липосомального СРТ-11 в отношении ксенотрансплантатов ВТ-474 рака легкого человека у голых мышей. "Контроль" включает мышей, получавших лечение только лекарственным средством или носителем без липосом (См. Пример 10).
На Фигуре 4 представлена динамика массы тела животных (голых мышей, имеющих ВТ-474 опухоль) во время лечения свободным СРТ-11 или липосомальным СРТ-11. "Контроль" включает мышей, получавших лечение только лекарственным средством или носителем без липосом (См. Пример 10).
На Фигуре 5 представлена динамика соотношения лекарственное средство/липосома в крови крыс in vivo после внутривенного болюсного введения липосом, нагруженных СРТ-11, с помощью TEA-SOS способа (См. Пример 14).
На Фигуре 6 представлена противоопухолевая активность свободного и липосомального СРТ-11 в отношении ксенотрансплантатов НТ-29 рака кишечника человека у голых мышей. На панели захвата представлен способ инкапсулирования лекарственного средства и доза на одну инъекцию. "Контроль" включает мышей, получавших лечение только лекарственным средством или носителем без липосом (См. Пример 15).
На Фигуре 7 представлена динамика массы тела животных (голых мышей с НТ-29 опухолью) во время лечения свободным СРТ-11 и липосомным СРТ-11. В прямых рамках выделены стандартные отклонения данных. "Контроль" включает мышей, получавших лечение только лекарственным средством или носителем без липосом (См. Пример 15).
На Фигуре 8А представлена фармакокинетика липидов липосом в крови после внутривенного болюсного введения липосом, нагруженных топотеканом, крысам. On-panel caption представлен способ инкапсуляции лекарственного средства и содержание лекарственного средства в липосомах (См. Пример 24).
На Фигуре 8В представлена динамика соотношения лекарственное средство/липосома в крови крыс in vivo после внутривенного болюсного введения липосом, нагруженных Топотеканом. На вставке представлен способ инкапсуляции лекарственного средства и содержание лекарственного средства в липосомах (См. Пример 24).
На Фигуре 9 представлена цитотоксичность свободного, липосомального или HER-2-направленного иммунолипосомального Топотекана (способ TEA-Pn) in vitro в отношении SKBr-З клеток карциномы легкого (См. Пример 27).
На Фигуре 10 представлена цитотоксичность свободного, липосомального или HER-2-направленного иммунолипосомального Топотекана (способ TEA-SOS) in vitro в отношении SKBr-3 клеток карциномы легкого (См. Пример 32).
На Фигуре 11 представлена противоопухолевая активность различных композиций Топотекана (ТРТ) в отношении ВТ-474 ксенотрансплантатов карциномы легкого человека у голых мышей. "Контроль" включает мышей, получавших лечение только лекарственным средством и носителем без липосом (См. Пример 29).
На Фигуре 12 представлена динамика массы тела животных во время лечения голых мышей, имеющих ВТ-474 опухоль, свободным Топотеканом (ТРТ), липосомальным Топотеканом (Ls-TPT) или анти-НЕРч.-2 иммунолипосомальным Топотеканом (F5 ILs-TPT). "Контроль" включает мышей, получавших лечение только лекарственным средством или носителем без липосом. (См. Пример 29).
На Фигуре 13А представлена противоопухолевая активность различных композиций Топотекана в отношении ВТ-474 ксенотрансплантатов карциномы легкого человека у голых мышей. Свободный Топотекан (свободный ТРТ) или липосомальный Топотекан (Ls-TPT) вводили в дозе, составляющей одну восьмую их максимальной толерантной дозы. Величина ошибки показывает стандартное отклонение данных. "Контроль" включает мышей, получавших лечение только лекарственным средством или носителем без липосом (См. Пример 31).
На Фигуре 13 В представлена противоопухолевая активность различных композиций Топотекана в отношении ВТ-474 ксенотрансплантатов карциномы легкого человека у голых мышей. Свободный Топотекан (свободный ТРТ) или липосомальный Топотекан (Ls-TPT) вводили в дозе, составляющей одну четвертую их максимальной толерантной дозы. Величина ошибки показывает стандартное отклонение данных. "Контроль" включает мышей, получавших лечение только лекарственным средством или носителем без липосом (См. Пример 31).
На Фигуре 13С представлена противоопухолевая активность различных композиций Топотекана в отношении ВТ-474 ксенотрансплантатов карциномы легкого человека у голых мышей. Свободный Топотекан (свободный ТРТ) или липосомальный Топотекан (Ls-ТРТ) вводили в дозе, составляющей одну вторую их максимальной толерантной дозы. Величина ошибки показывает стандартное отклонение данных. "Контроль" включает мышей, получавших лечение только лекарственным средством или носителем без липосом (См. Пример 31).
На Фигуре 13D представлена противоопухолевая активность различных композиций Топотекана в отношении ВТ-474 ксенотрансплантатов карциномы легкого человека у голых мышей. Свободный Топотекан (свободный ТРТ) или липосомальный Топотекан (Ls-ТРТ) вводили в их максимальной толерантной дозе. Величина ошибки показывает стандартное отклонение данных. "Контроль" включает мышей, получавших лечение только лекарственным средством или носителем без липосом (См. Пример 31).
На Фигуре 14 представлена динамика массы тела животных во время лечения голых мышей, имеющих ВТ-474 опухоль, свободным Топотеканом (ТРТ) или липосомальным Топотеканом (Ls-TPT), которые вводят в их максимальной толерантной дозе. "Контроль" включает мышей, получавших лечение только лекарственным средством или носителем без липосом. (См. Пример 31).
На Фигуре 15 представлена цитотоксичность свободного 6-(3-аминопропил)-эллиптицина (свободный АЕ), липосомального 6-(3-аминопропил)-эллиптицина (Ls-AE) или HER-2-направленного иммунолипосомального 6-(3-аминопропил)-эллиптицина (F5 Ils-АЕ) в отношении ВТ-474 клеток карциномы легкого in vitro (См. Пример 35).
На Фигуре 16 представлена in vitro цитотоксичность свободного 6-(3-аминопропил)-эллиптицина (свободный АРЕ), липосомального 6-(3-аминопропил)-эллиптицина (Ls-APE) или EGFR-направленного иммунолипосомального 6-(3-аминопропил)-эллиптицина (С225-Ils-APE) в отношении клеток карциномы легкого с низкой (MCF-7) или высокой (MDA-МВ468) экспрессией EGF-рецептора. (См. Пример 36).
На Фигуре 17 представлены характеристики фармакокинетики липосомных композиций 6-(3-аминопропил)эллиптицина (АРЕ) в крови: фармакокинетика липосомных липидов (рамка А, незакрашенные кружки), лекарственного средства (рамка В, закрашенные кружки) в крови и динамика соотношения лекарственное средство/липосомы (рамка С) после внутривенного болюсного введения липосом, содержащих винорелбин, крысам (См. Пример 43).
На Фигуре 19 представлена фармакокинетика липосомных липидов в крови после внутривенного болюсного введения липосом, нагруженных винорелбином, крысам.
Липосомы нагружают, используя предварительно захваченный декстрансульфат триэтиламмония (DS-TEA), декстрансульфат аммония (DS-A) или сульфат аммония (S-A). (См. Пример 44).
На Фигуре 20 представлена динамика соотношения лекарственное средство/липосома в крови крыс in vivo после внутривенного болюсного введения липосом, нагруженных винорелбином с использованием предварительно захваченного декстрансульфата триэтиламмония (DS-TEA), декстрансульфат аммония (DS-A) или сульфата аммония (S-A). (См. Пример 44).
На Фигуре 21 представлена фармакокинетка липосомных липидов в крови после внутривенного болюсного введения липосом, нагруженных винорелбином, крысам. Липосомы нагружают, используя предварительно захваченный сукросеоктасульфат триэтиламмония (TEA-SOS); липосомы имеют размер, как показано во вставке на рисунке. (См. Пример 45).
На Фигуре 22 представлена динамика соотношения лекарственное средство/липосома в крови крыс in vivo после внутривенного болюсного введения липосом, нагруженных винорелбином. Липосомы нагружают, используя предварительно захваченный сукрооктасульфат триэтиламмония (TEA-SOS); липосомы имеют размер, как показано на вставке на рисунке. (См. Пример 45).
На Фигуре 23 представлена фармакокинетика липосомных липидов в крови крыс после внутривенного болюсного введения винорелбина в составе липосом (Ls-VRB) или анти-HER2-иммунолипосом (F5-Ils-VRB), полученных с помощью TEA-SOS способа. (См. Пример 46).
На Фигуре 24 представлена динамика соотношения лекарственное средство/липосома в крови крыс in vivo после внутривенного болюсного введения винорелбина в составе липосом (Ls-VRB) или анти-НЕ112-иммунолипосом (F5-Ils-VRB), полученных с помощью TEA-SOS способа. (См. Пример 46).
На Фигуре 25 представлена in vitro цитотоксичность свободного винорелбина (свободный VRB), липосомального винорелбина (Ls-VRB) или HER-2-направленного иммунолипосомального винорелбина (F5-Ils-VRB) в отношении MDA-MB-453 клеток карциномы легкого человека, избыточно экспрессирующих HER-2. (См. Пример 48).
На Фигуре 26 представлена in vitro цитотоксичность свободного винорелбина (свободный VRB), липосомального винорелбина (Ls-VRB) или HER-2-направленного иммунолипосомального винорелбина (F5-Ils-VRB) в отношении CaLu-3 клеток немелкоклеточного рака легкого человека, избыточно экспрессирующих HER-2 (См. Пример 49).
На Фигуре 27 представлена in vitro цитотоксичность свободного винорелбина (свободный VRB), липосомального винорелбина (Ls-VRB) или HER-2-направленного иммунолипосомального винорелбина (F5-Ils-VRB/SOS-TEA) в отношении SKBr-3 клеток рака легкого человека, избыточно экспрессирующих HER-2. (См. Пример 50).
На Фигуре 28 представлена противоопухолевая активность свободного винорелбина (свободный VRB) или липосомального винорелбина (Ls VRB) в отношении НТ-29 ксенотрансплантатов рака толстого кишечника человека у голых мышей. "Контроль" включает мышей, получавших лечение только лекарственным средством или носителем без липосом. Величина ошибки показывает стандартное отклонение данных. (См. Пример 51).
На Фигуре 29 представлена динамика средней массы тела животных во время лечения голых мышей, имеющих НТ-29 опухоль, свободным винорелбином (свободный VRB), липосомальным винорелбином (Ls VRB) или только носителем (физиологический раствор). Величина ошибки показывает стандартное отклонение данных. (См. Пример 51).
На Фигуре 30 представлена противоопухолевая активность свободного винорелбина (свободный VRB) или липосомального винорелбина (Ls VRB) на сингенной мышиной модели С-26 карциномы толстого кишечника. Дозы лекарственного средства на одну инъекцию приведены на рисунке. Величина ошибки показывает стандартное отклонение данных. "Контроль" включает мышей, получавших лечение только лекарственным средством или носителем без липосом (См. Пример 52).
На Фигуре 31 представлена динамика средней массы тела мышей, имеющих сингенную С-26 опухоль толстого кишечника, во время лечения различными дозами свободного винорелбина (свободный VRB), липосомального винорелбина (Ls VRB) или только носителем (физиологический раствор). Дозы лекарственного средства на одну инъекцию приведены на рисунке. (См. Пример 52).
На Фигуре 32 представлена противоопухолевая активность свободного винорелбина (свободный VRB) или sc-Fv-F5-конъюгированного, анти-HER-2 иммунолипосомального винорелбина, полученного с помощью TEA-SOS способа (F5-Ils-VRB TEA-SOS), анти-HER2 иммунолипосомального винорелбина, полученного с помощью ТЕА-Pn способа (F5-Ils-VRB ТЕА-Pn), в отношении ВТ-474 ксенотрансплантатов карциномы легкого человека, избыточно экспрессирующих HER-2, у голых мышей. "Контроль" включает мышей, получавших лечение только лекарственным средством или носителем без липосом. (См. Пример 53).
На Фигуре 33 представлена динамика средней массы тела мышей, имеющих ксенотрансплантаты карциномы легких (ВТ-474), избыточно экспрессирующие HER2, во время лечения свободным винорелбином, scFvF-5-конъюгированным, анти-HER2 иммунолипосомальным винорелбином, полученным с помощью TEA-SOS способа, анти-HER2 иммунолипосомальным винорелбином, полученным с помощью ТЕА-Pn способа, или только носителем. Для расшифровки символов, см. вставку на Рисунке 32. (См. Пример 53).
На Фигуре 34 представлена противоопухолевая активность свободного винорелбина (свободное лекарственное средство) или scFv-F5-конъюгированного, анти-HER2 иммунолипосомального винорелбина, полученного при использовании различных количеств PEG-липида, в отношении ксенотрансплантатов карциномы легких человека (ВТ-474), избыточно экспрессирующих HER2, у голых мышей. Величина ошибки показывает стандартное отклонение данных. "Контроль" включает мышей, получавших лечение только лекарственным средством или носителем без липосом (См. Пример 54).
На Фигуре 35 представлена противоопухолевая активность свободного винорелбина (свободный NAV), липосомального винорелбина (NAV Lip) или FC225Fab'-конъюгированного, анти-EGFR-иммунолипосомального винорелбина (C225-NAV Lip) в отношении ксенотрансплантатов глиобластомы (U87) человека, избыточно экспрессирующей EGFR), у голых мышей. "Контроль" включает мышей, получавших лечение только лекарственным средством или носителем без липосом (См. Пример 55).
На Фигуре 36 представлена фармакокинетика липосомальных липидов и динамика соотношения лекарственное средство/липосомные липиды в крови крыс после внутривенного болюсного введения доксорубицина в составе липосом, полученных с помощью сульфата триэтиламмония (См. Пример 56).
На Фигуре 37 представлена противоопухолевая активность липосомального доксорубицина (Ls-Dox) или scFv Р5-конъюгированного, анти-HER2 иммунолипосомального доксорубицина (F5 ILs-Dox), полученного с помощью различных количеств PEG-липида, в отношении ксенотрансплантатов карциномы легких человека (ВТ-474), избыточно экспрессирующих HER2, у голых мышей. На вставке показаны количество PEG-липида, выраженное в мол. % от липосомных фосфолипидов. "Контроль" включает мышей, получавших лечение только лекарственным средством и носителем без липосом. (См. Пример 57).
На Фигуре 38 представлена фармакокинетика липосомального винбластина в крови у крыс.(См. Пример 58).
На Фигуре 39 представлена динамика соотношения лекарственное средство/липосомальные липиды в крови крыс после внутривенного болюсного введения липосомального винбластина (См. Пример 58).
На Фигуре 40 представлена in vitro цитотоксичность свободного винкристина (свободный VCR), липосомального винкристина (Ls-VCR), или HER2-направленного иммунолипосомального винкристина (F5-ILs-VCR) в отношении SKBr-3 клеток карциномы легкого человека, избыточно экспрессирующих HER2 (См. Пример 61).
На Фигуре 41 представлена фармакокинетика липосомальных липидов в крови крыс после внутривенного введения винкристина в составе липосом различного размера (размеры представлены на вставке). (См. Пример 62).
На Фигуре 42 представлена динамика соотношения лекарственное средство/липосомальные липиды в крови у крыс после внутривенного болюсного введения винкристина в составе липосом различного размера (размеры представлены на вставке). (См. Пример 62).
На Фигуре 43 представлена противоопухолевая активность свободного винкристина (свободный VCR), липосомального винкристина, полученного с помощью способа с использованием цитрата триэтиламмония (Ls-VCR Цитрат), липосомального винкристина, полученного с помощью способа с использованием сукрооктасульфата триэтиламмония (Ls-VCR SOS) или scFv Р5-конъюгированного, aHTH-HER2 иммунолипосомального винкристина, полученного с помощью способа с использованием сукрооктасульфата триэтиламмония (F5 ILs-VCR SOS), в отношении ксенотрансплантатов карциномы легких человека (ВТ-474), избыточно экспрессирующих HER2, у голых мышей. "Контроль" включает мышей, получавших только лекарственное средство и носитель без липосом. (См. Пример 64).
На Фигуре 44 представлена динамика средней массы тела мышей, имеющих ксенотрансплантаты карциномы легких человека (ВТ-474), избыточно экспрессирующие HER2, во время лечения свободным винкристином (свободный VCR), липосомальным винкристином, полученным с помощью способа с использованием цитрата триэтиламмония (Ls-VCR Цитрат), липосомального винкристина, полученного с помощью способа с использованием сукрооктасульфата триэтиламмония (Ls-VCR SOS), scFv F5-конъюгированного, анти-HER2 иммунолипосомального винкристина, полученного с помощью способа с использованием сукрооктасульфата триэтиламмония (F5 ILs-VCR SOS), или только носителем (физиологический раствор, контроль). (См. Пример 64).
На Фигуре 45 представлена противоопухолевая активность свободного винкристина (винкристин), липосомального винкристина (nt-vcr) или С225 Fab'-конъюгированного, анти-EGFR иммунолипосомального винкристина (C225-vcr) в отношении ксенотрансплантатов опухоли головного мозга людей (U87), избыточно экспрессирующих EGFRvIII, у голых мышей. "Контроль" включает мышей, получавших только лекарственное средство и носитель без липосом. (См. Пример 65).
На Фигуре 46 представлена фармакокинетика СРТ-11 и динамика присутствия СРТ-11 в активной (лактонной) в крови крыс, выраженная в процентах, после внутривенного болюсного введения липосомального СРТ-11. (См. Пример 69).
На Фигуре 47 представлена фармакокинетика СРТ-11 и динамика присутствия СРТ-11 в активной (лактонной) форме в крови крыс, выраженная в процентах, после внутривенного болюсного введения раствора СРТ-11 (свободный СРТ-11). (См. Пример 69).
ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
ИЗОБРЕТЕНИЯ
Настоящее изобретение, в целом, относится к способам и липосомным композициям, используемым для доставки различных объектов, в особенности лекарственных и контрастных соединений. Авторами изобретения было обнаружено, что замещенный аммоний и полианион могут быть использованы для инкапсулирования объектов (то есть соединений) и их сохранения внутри липосом. Соответственно, настоящее изобретение относится к липосомным композициям и наборам, содержащим замещенный аммоний и/или полианион, а также к способам создания указанных липосомных композиций.
Согласно одному варианту своего осуществления, настоящее изобретение относится к липосомной композиции, содержащей в своем внутреннем пространстве одно или более соединений замещенного аммония, имеющее формулу

где каждый R1, R2, R3 и R4 независимо друг от друга представляет собой атом водорода или органическую группу, при этом по меньшей мере один из R1, R2, R3 и R4 представляет собой органическую группу, такую, как алкил, алкилиден, гетероциклический алкил, циклоалкил, арил, алкенил или циклоалкенил, их гидрокси-замещенное производное, факультативно имеющее в своей углеводородной цепи атомы S, О или N, с формированием, например, эфира (включая ацеталь и кеталь), сложного эфира, сульфида (тиоэфира), амина или амидной связи. В том случае, если менее, чем три радикала из R1, R2, R3 и R4 представляют собой органические группы, тогда, согласно настоящему изобретению, по меньшей мере одна, и, предпочтительно, две из органических групп имеют вторичные или третичные углеродные атомы (то есть углеродные атомы, имеющие 2 или 3 углерод-углеродные связи, соответственно), непосредственно связанные с азотом аммония, то есть замещенный аммоний представляет собой стерически затрудненный аммоний. В целом известно, что наличие третичного аммония, такого, как ион незамещенного аммония (NH4+), а также ионов первичного и вторичного алкиламмония с прямой цепью во внутреннем пространстве липосомы, заявленной в соответствии с настоящим изобретением, способствует усиленной инкапсуляции слабых амфифильных оснований, например, с помощью механизма "активной", "дистанционной" или "управляемой трансмембранным градиентом" нагрузки (Haran, et al., Biochim. Biophys. Acta, 1993, v. 1152. p. 253-258; Maurer-Spurej, et al., Biochim. Biophys. Acya, 1999, v. 14161, p. 1-10). Поскольку указанные соединения аммония имеют в своем составе атомы водорода, которые легко вступают в реакции нуклеофильного замещения, химически реагируют с соединениями, заключенными во внутреннее пространство липосом и, таким образом, обладают способностью нарушать химическую целостность соединений во время или после процесса их инкапсуляции в липосомы. Следовательно, желательно, чтобы захваченное соединение замещенного аммония было более химически инертным и не имело химических свойств, которые являются нестабильными или способствуют тому, что аммоний легко вступает в реакции с компонентами липосомы, к которым могут относиться вещества, заключенные в ее внутреннее пространство. Авторами изобретения неожиданно было обнаружено, что липосомные композиции, содержащие в своем внутреннем пространстве замещенный третичный или четвертичный аммоний, не имеющий в своем составе замещенного атома водорода, или стерически затрудненный первичный или вторичный аммоний, в котором дополнительный атом водорода пространственно защищен соседней объемной органической группой, например, такой аммоний, который в своем составе имеет один или два вторичных или третичных атомов углерода, связанных с азотом аммония, не только обладают исключительной способностью к инкапсуляции вещества, но также обеспечивают улучшенную стабильность указанного инкапсулированного вещества, например, лекарственного средства, и препятствуют его преждевременному высвобождению из липосомы в живом организме.
Согласно одному варианту осуществления настоящего изобретения, заключенное в липосомы соединение замещенного аммония является фармацевтически инертным, то есть оно не вызывает нежелательный физиологический ответ при введении в живой организм, например, в организм животного или человека, при этом количество вещества мембраны липосом является достаточным для доставки эффективной дозы заключенного в липосому соединения. Согласно другому варианту осуществления настоящего изобретения, замещенный аммоний, заявленный в соответствии с настоящим изобретением, имеет приемлемый уровень токсичности по отношению к живому организму. Обычно приемлемый уровень токсичности подразумевает, что токсичная доза, то есть максимальная толерантная доза (MTD, от англ. Maximum Tolerated Dose), или доза, вызывающая летальность в 50% случаев (LD50) замещенного аммония, заявленного в соответствии с настоящим изобретением, по меньшей мере в два раза, по меньшей мере в четыре раза, по меньшей мере в восемь раз или по меньшей мере в десять раз выше, чем токсичная доза соединения, заключенного в липосому, то есть лекарственного средства, инкапсулированного в липосому, заявленную в соответствии с настоящим изобретением. Например, сульфат триэтиламмония имеет приемлемый уровень токсичности в соответствии с настоящим изобретением, пока его LD50 приблизительно в 40 раз выше, чем LD50 доксорубицина, противоопухолевого средства. Уровни токсичности или те физиологические ответы, которые вызывают соединения замещенного аммония в организме, равно как и аналогичные показатели соединений, представляющих интерес, в том случае, если они еще не определены, могут быть легко установлены с помощью стандартных методик, хорошо известных специалистам в области биомедицины. См., например, S.C. Gad. Drug Safety Evaluation, Wiley, New York, 2002. Один из способов определения токсичности свободного и/или заключенного в липосому лекарственного средства описан в Примере 16 настоящего изобретения.
Согласно одному предпочтительному варианту осуществления настоящего изобретения, замещенные органические группы среди R1, R2, R3 или R4 имеют размер и физико-химические свойства, которые обеспечивают формирование соединениями замещенного аммония в водной среде истинного (молекулярного) раствора, а не мицелия, бислоя или аналогичных самоформирующихся систем. Таким образом, замещенный аммоний, заявленный в соответствии с настоящим изобретением, предпочтительно, незначительно или вовсе не проникает в бислойный участок липосомы, что сводит к минимуму риск дестабилизации, растворения или рассасывания липосом, содержащих замещенный аммоний.
Органическая группа замещенного аммония обычно имеет в своем составе углеводород, включающий вплоть до 8 атомов углерода, вплоть до 6 атомов углерода или вплоть до 4 атомов углерода, в целом вся замещенная органическая группа имеет в своем составе вплоть до 18, вплоть до 16, вплоть до 12 или вплоть до 9 атомов углерода. Указанные замещенные углеводородные группы могут включать любые комбинации взаимосвязанных первичных, вторичных или третичных атомов углерода, а также циклоалкильные группы, связанные на своих концах непосредственно с атомом азота аммония с формированием гетероциклической структуры, либо с атомом углерода водород-замещенной группы аммония. Указанные замещенные алкильные группы также могут иметь в своем составе гетероатомы, например, атомы кислорода, азота или серы, расположенные в углеродных цепях с формированием функциональных групп, например, эфирных, ацетальных, аминовых или сульфидных групп, а также функциональных групп, таких, как гидроксильные группы, связанные с углеродной цепью алкильной группы. Примерами органических групп, заявленных в соответствии с настоящим изобретением, являются, без ограничений указанными, алкилы, алкилидены, гетероциклические алкилы, циклоалкилы, арилы, алкенилы, циклоалкенилы или гидрокси-замещенные их производные, например, гидрокси-замещенный алкилиден, формирующий кольцо с включением атома N замещенного аммония.
Согласно другому варианту осуществления настоящего изобретения, замещенный аммоний представляет собой гетероциклический аммоний, то есть аммоний, в котором по меньшей мере два радикала R1, R2, R3 или R4 формируют кольцо: стерически затрудненный первичный аммоний; или стерически затрудненный вторичный аммоний. В целом, первичный или вторичный аммоний представляет собой любой замещенный аммоний, в котором один или два радикала R1, R2, R3 или R4 замещены алкильными группами, которые стерически уплотняют молекулу, например, любой замещенный аммоний с одним или двумя радикалами R1, R2, R3 или R4, замещенными одной или двумя циклоалкильными группами или алкильными группами, имеющими, по меньшей мере, один вторичный или третичный алкильный атом углерода, связанный с атомом азота замещенного аммония. Примерами таких гетероциклов, стерически затрудненных первичных аммониевых соединений и стерически затрудненных вторичных аммониевых соединений, являются, без ограничений указанными, изопропилэтиламмоний, изопропилметиламмоний, диизопропиламмоний, терт-бутилэтиламмоний, дициклогексиламмоний, протонизированные формы морфолина, пиридина, пиперидина, пирролидина, пиперазина, tert-бутиламин, 2-амино-2-метилпропанол-1,2-амино-2-метил-пропандиол-1,3, и tris-(гидроксиэтил)-аминометан. Указанные соединения замещенного аммония являются коммерчески доступными в форме различных солей, или могут быть легко поучены на основе соответствующих аминов путем нейтрализации кислотами.
Согласно еще одному варианту осуществления настоящего изобретения, замещенный аммоний представляет собой третичный или четвертичный аммоний, включая, без ограничений указанными, триметиламмоний, триэтиламмоний, трибутиламмоний, диэтилметиламмоний, диизопропилэтиламмоний, триизопропиламмоний, N-метилморфолиний, N-гидроксиэтилпиперидиний, N-метилпирролидиний и N,N'-диметилпиперазоний, тетраметиламмоний и тетрабутиламмоний. Указанные соединения замещенного аммония являются коммерчески доступными в форме различных солей, или могут быть легко получены на основе соответствующих аминов путем нейтрализации кислотами.
Согласно еще одному варианту осуществления настоящего изобретения, соединение замещенного аммония, заявленного в соответствии с настоящим изобретением, является полностью катионным соединением, которое при условии полной инкапсуляции обычно в водном растворе при рН от 2 до 8, имеет полный положительный заряд, например, в результате ионизации (протонирования) атома азота.
Согласно еще одному варианту осуществления настоящего изобретения, соединение первичного, вторичного или третичного замещенного аммония, инкапсулированное в липосому, имеет отрицательный логарифм константы кислотной диссоциации (депротонирования) (pKa), равный, по меньшей мере, 8.0, по меньшей мере, 8.5, по меньшей мере 9.0, по меньшей мере, 9.5, или по меньшей мере 10.0, что определяется в водном растворе при комнатной температуре (обычно при 25°С). Показатель pKa является известной характеристикой аммониевых соединений, который в целом характеризует их основные свойства, а способы определения pKa являются рутинными. Величины pKa для многих аминов и их протонированных форм (аммониевых соединений) известны и приведены в специальных таблицах в руководствах по химии и фармакологии. См., например, IUPAC Handbook of Pharmaceutical Salts, ed. By P.H. Stahl and C.G. Wermuth, Wiley-VCH, 2002; CRC Handbook of Chemistry and Physics, 82nd Edition, ed. by D.R. Lide, CRC Press, Florida, 2001, p. 8-44 и 8-56. В общем, чем больше величина pKa, тем более выражены основные свойства соединения. Примерные соединения замещенного аммония, а также незамещенного аммония (представленные как их конъюгированные аминовые основания), имеют следующие значения pKa: пирролидин, 11,31; пиперидин, 11.12; диизопропиламин, 11,05; диэтиламин, 10,93; триэтиламин, 10.75; диметиламин, 10.73; терт-бутиламин, 10,68; циклогексиламин, 10,66; метиламин, 10,66; этиламин, 10,65; пропиламин, 10,54; изопропиламин, 10,53; N-этилпиперидин, 10,45; дициклогексиламин, 10,4; N-метилпиперидин, 10,38; диэтилметиламин, 10,35; диметилпропиламин, 10,15; триметиламин, 9,8; пиперазин, 9.73 (I), 5,53 (II); 2-амино-2-метилпропанол, 9,69; N,N'-диметилпиперазин, 9,66 (I), 5.2 (II); диэтил-(2-гидроксиэтил)амин, 9,58; этаноламин, 9,5; N-гидроксиэтилпирролидин, 9,44; диэтаноламин, 9,28; аммоний, 9,27; диметил-(2-гидроксиэтил)амин, 8.83; 2-амино-2-метилпропандиол-1,3, 8,8; морфолин, 8,5; трис-(гидроксиметил)-аминометан, 8,3; N-метилгликамин, 8,03; триэтаноламин, 7,76; N-этилморфолин, 7,67; N-гидроксиэтилморфолин, 7,39; имидазол, 7,03; пиридин, 5,23. Как правило, замещение алкильной или циклоалкильной группы аммониевого соединения водородом приводит к уменьшению pKa. Необходимо отметить, что гидроксильные или эфирные свойства замещенных алкильных групп или наличие ароматических свойств азотсодержащих гетероциклических групп уменьшают величину pKa, по сравнению с аналогичными замещенными аммониевыми соединениями, не имеющими групп с гидроксильными или эфирными свойствами. Соединения, имеющие в своем составе более чем одну аммониевую группу, обычно имеют величину pKa второй и замещенной аммониевой группы значительно меньшую, чем таковая величина первой группы. Авторы настоящего изобретения неожиданно обнаружили, что соединения замещенного аммония с большими величинами pKa, сформированные более сильными основными аминами, являются более эффективными для стабилизации лекарственного средства внутри липосом, чем соединения, сформированные слабыми аминами. Например, IHP и SOS соли триэтиламмония (pKa=10.75) являются значительно более эффективными, чем соответствующие соли триэтиламмония (pKa=7.76), для стабилизации иринотекана внутри липосом in vivo (Пример 73).
Замещенный аммоний в составе липосомной композиции, заявленной в соответствии с настоящим изобретением, может находиться в любой подходящей форме, например, в форме соли. К подходящим солям относятся фармацевтически доступные соли. См., например, Р.Н. Wermuth (eds), Handbook of Pharmaceutical Salts, Wiley-VCH, Weinheim, 2002. Согласно одному варианту осуществления настоящего изобретения, замещенный аммоний представляет собой соль, имеющую в своем составе один или более полианионов, заявленных в соответствии с настоящим изобретением. Оптимально, противоион (анион) в соли замещенного аммония, заявленной в соответствии с настоящим изобретением, обеспечивает растворимость соли в воде и является фармацевтически инертным, способным формировать преципитаты или гели при контактировании с терапевтическим или контрастным веществом, и/или в меньшей степени проникает через мембрану липосом, чем замещенный аммоний или его недиссоциирующая аминная форма. В целом, соль замещенного аммония, заявленного в соответствии с настоящим изобретением, формирует истинный раствор в интралипосомальном, например, водном пространстве, и не образует значительное количество конденсированных фаз, таких, как мицелий, бислой, гель или кристаллическая фаза. Соответствующее количество замещенного аммония и солеобразующего аниона, например, полианиона, находится в точке стехиометрического равновесия или около нее, и, обычно, имеет рН в пределах от 3 до 9, чаще, от 4 до 8 в зависимости, например, от константы диссоциации конъюгированного основания иона замещенного аммония.
В целом, замещенный аммоний находится во внутреннем пространстве липосом, заявленных в соответствии с настоящим изобретением. Согласно одному варианту осуществления настоящего изобретения, замещенный аммоний частично или полностью удален из среды, окружающей липосому. Это может быть достигнуто с помощью подходящих способов, хорошо известных специалистам в данной области, например, растворения, ионнобменной хроматографии, хроматографии с исключением по размеру, диализа, ультрафильтрации, преципитации т.д.
Согласно еще одному варианту своего осуществления, настоящее изобретение относится к липосомной композиции, содержащей полианион. Полианион, заявленный в соответствии с настоящим изобретением, может представлять собой любое химическое соединение, имеющее в своем составе более чем одну отрицательно заряженную группу, что обеспечивает полный отрицательный ионный заряд более чем двух единиц во внутреннем пространстве липосом, например, водном пространстве. Полианион, заявленный в соответствии с настоящим изобретением, может представлять собой двухвалентный анион, трехвалентный анион, поливалентный анион, полимерный поливалентный анион, полианиозированный полиол или полианиозированный сахар. Примерами указанных двух- и трехвалентных анионов являются сульфат, фосфат, пирофосфат, тартрат, сукцинат, малеат, борат и цитрат (без ограничений указанными). Согласно предпочтительному варианту осуществления настоящего изобретения, полианион, заявленный в соответствии с настоящим изобретением, представляет собой полианионный полимер, имеющий органический (углеродный) или неорганический скелет и множество анионных функциональных групп, то есть функциональных групп, ионизирующихся с отрицательным зарядом в водном растворе, которые интегрированы или прикреплены к скелету. Полимер представляет собой природное или синтетическое соединение, обычно с высокой молекулярной массой, состоящее из повторяющихся связанных между собой единиц, каждая из которых представляет собой относительно легкую и простую молекулу. Примерами полианионных полимеров являются полифосфат, поливинилсульфат, поливинилсульфонат, анионизированные полиакриловые полимеры, анионизированные, например, полисульфонированные полиамины, такие, как полисульфонированный поли(этиленимин); полисульфатированные, поликарбоксилированные или полифосфорилированные полисахариды; кислые полиаминокислоты; полинуклеотиды; другие полифосфорилированные, полисульфатированные, полисульфонированные, полиборированные или поликарбоксилированные полимеры. Указанные поливалентные анионы и полимеры хорошо известны специалистам в данной области, многие из них являются коммерчески доступными. Полимерный анион, заявленный в соответствии с настоящим изобретением, предпочтительно, является биологически разрушаемым, то есть в живом организме распадается на нетоксичные единицы. Примером такого биологически разрушаемого полимерного аниона является полифосфат.
Согласно другому предпочтительному варианту осуществления настоящего изобретения, полианион представляет собой полианиозированный полиол или полианиозированный сахар. Полиол представляет собой органическую молекулу, имеющую множество гидроксильных групп, связанных, например, с линейным, разветвленным или циклическим углеродным скелетом. Таким образом, полиол может быть охарактеризован термином полигидроксилированное соединение. Предпочтительно, большинство атомов углерода в полиоле являются гидроксилированными. Полиолы (полиатомарные спирты) представляют собой молекулы, которые хорошо известны специалистам в данной области. Могут использоваться полиолы с прямыми цепями (линейными или разветвленными) и циклические полиолы. Примерами полиолов, заявленных в соответствии с настоящим изобретением, являются (без ограничений указанными): этиленгликоль, глицерол, треитол, эритритол, пентаэритритол, маннитол, глицитол, сорбитол, сорбитан, ксилитол, лактитол, малтитол, фруктитол и иноситол. Сахар обычно имеет в своем составе циклический ацеталь, циклический кеталь, кетон или альдегидную группу, или их аддукты, в пределах группы взаимосвязанных предварительно гидроксилированных атомов углерода. Сахара обычно представляют собой существующие в природе соединения. Гидролиз Сахаров в водной среде приводит к образованию единиц, называемых моносахаридами. Обычно в водном растворе молекулы моносахаридов, состоящие из пяти или шести атомов углерода, формируют циклический гемиацеталь, кольцевую структуру. Предпочтительно, сахара, заявленные в соответствии с настоящим изобретением, представляют собой моносахариды или дисахариды, последние состоят из одной или двух моносахаридных единиц, каждая из которых содержит от трех до семи, предпочтительно, от трех до шести атомов углерода. Примерами Сахаров, заявленных в соответствии с настоящим изобретением, без ограничений указанными, являются: моносахаридные гексозы, такие, как глюкоза (декстроза), галактоза, манноза, фруктоза; моносахаридные пентозы, такие, как ксилоза, рибоза, арабиноза, и дисахариды, такие, как лактоза, трехалоза, сукроза, мальтоза и целлобиоза. Также могут быть использованы соединения, состоящие из нескольких взаимосвязанных единиц Сахаров с формированием кольца (циклодекстрины), и их производные. Восстановление Сахаров является одним из способов получения полиолов. Более стабильные "невосстановленные" и "неметаболизируемые" дисахариды, такие, как сахароза и трегалоза, являются предпочтительными. Различные полиолы, моносахариды и дисахариды являются коммерчески доступными.
Полианионизированный полиол или сахар представляет собой полиол или сахар, в котором гидроксильные группы полностью или частично модифицированы или замещены анионными группами (анионизированы). Следовательно, полианионизированный полиол или полианионизированный сахар имеет в своем составе молекулу полиола или сахара вместе со связанными анионными группами. Примерами анионных групп являются, без ограничений указанными, карбоксилат, карбонат, тиокарбонат, дитиокарбонат, фосфат, фосфонат, сульфат, сульфонат, нитрат и борат. Предпочтительно, чтобы по меньшей мере одна анионная группа полианионизированного сахара или полиола представляла собой сильную анионную группу, то есть такую группу, которая более чем на 50% ионизируется при различных значениях рН, например, при рН от 3 до 12, предпочтительно, от 2 до 12, в водной среде, или, альтернативно, имеет логарифм константы диссоциации (pKa) равный 3 или менее, предпочтительно, 2 или менее. Полианионизация полиола или сахара может быть получена с помощью различных химических процессов, которые хорошо известны специалистам в данной области. Например, взаимодействие полиолов и/или Сахаров с триоксидом серы или хлорсульфоновой кислотой в пиридине или 2-пиколине приводит к этерификации всех или некоторых гидроксильных групп серной кислотой (сульфатированию), что обеспечивает полисульфатирование сахара или полиола.
Примером сульфатированного сахара, заявленного в соответствии с настоящим изобретением, является сульфатированная сахароза, включая, без ограничений указанными, сахарозы гексасульфат, сахарозы гептасульфат и сахарозы октасульфат (См. Ochi. K., et al., 1980, Chem. Pharm. Bull. v. 28, p. 638-641). Аналогично, взаимодействие с фосфорированным оксихлоридом или диэтилхлорфосфатом в присутствии основного катализатора приводит к образованию полифосфорилированных полиолов и Сахаров. Полифосфорилированные полиолы также можно выделить из природных источников. Например, инозитол полифосфаты, такие, как инозитол гексафосфат (phytic acid) может быть выделен из злаков. Примеры различных сульфатированных, сульфонированных и фосфорилированных Сахаров и полиолов, подходящих для использования в соответствии с настоящим изобретением, приведены, например, в патенте США 5,783,568 и патенте США 5,281,237, которые приведены здесь в качестве ссылки. Авторами настоящего изобретения было неожиданно обнаружено, что полианионизированные полигидроксилированные соединения только с сильными кислыми группами, например, группами, имеющими pKa менее 3.0, предпочтительно, менее 2.0, такие, как, например, сульфат моноэфиры (pKa 1.0 и менее), обеспечивают лучшее сохранение инкапсулированного вещества в липосоме, чем полианионизированные полигидроксилированные соединения, имеющие более слабые кислые группы, такие, как фосфат моноэфиры (шаг 1, pKa 1.5; шаг 2, pKa около 6; см. Stahl и Wermuth, Op. cit., 2002). Пример 73, приведенный ниже, иллюстрирует это открытие. Синтез полиолов и/или Сахаров, имеющих в своем составе более чем одну молекулу борной кислоты также приводит к образованию полианионизированного (полиборированного) продукта. Взаимодействие полиолов и/или Сахаров с дисульфидом углерода в присутствии щелочей приводит к образованию полианионизированных (полидитиокарбонированных, поликсантогенированных) производных. Полианионизированное производное сахара или полиола может быть получено на основании свободной кислоты и нейтрализовано подходящим основанием, например, гидроксидом щелочного металла, гидроксидом аммония или, предпочтительно, замещенным амином, например, амином, соответствующим замещенному аммонию, заявленному в соответствии с настоящим изобретением, в neat форме или в форме гидроксида замещенного аммония, что приводит к образованию полианионной соли замещенного аммония, заявленного в соответствии с настоящим изобретением. Альтернативно, натриевая, калиевая, кальциевая, бариевая или магниевая соль полианионизированного полиола/сахара может быть выделена и превращена в подходящую форму, например, в форму соли замещенного аммония, с помощью любого известного способа, например, с помощью обмена ионов.
Полианион, заявленный в соответствии с настоящим изобретением, обычно имеет плотность заряда, составляющую, по меньшей мере, две, три или четыре отрицательно заряженные группы на одну единицу, например, на один атом углерода или кольцо в углеродной цепи или на одну моносахаридную единицу в сахаре. Полианионизированный сахар или циклический полиол, заявленный в соответствии с настоящим изобретением, имеет, по меньшей мере, 75% доступных полианионизированных гидроксильных групп, и, более предпочтительно, 100% доступных полианионизированных гидроксильных групп. Кроме того, полианионизированное соединение внутри липосом, заявленных в соответствии с настоящим изобретением, находится в количестве, достаточном для улучшения доставки и высвобождения вещества, инкапсулированного в липосомы, в то место, где оно проявляет свою активность, но при этом препятствует преждевременному высвобождению инкапсулированного вещества, то есть до того, как липосома достигнет места своего действия.
В соответствии с настоящим изобретением, уровень полианионизации внутри липосомы может использоваться для регулирования характеристик высвобождения липосом, например, скорости высвобождения и кинетики вещества, инкапсулированного в липосомы. В целом уровень полианионизации может регулироваться на основании количества полианионизированного сахара или полиола по отношению к общему количеству анионов или, в случае если полианион является единственным типом аниона, на основании процентного соотношения полианионизации по отношению к общей способности к полианионизации полианиона, например, полианионизированного сахара или полиола или их смеси внутри липосом, заявленных в соответствии с настоящим изобретением. Согласно одному варианту осуществления настоящего изобретения, полианионизированный сахар или полиол находится в смеси с одним или более другими анионами и, чем меньше количество полианионизированного сахара или полиола по сравнению с количеством других анионов, тем быстрее действующее вещество высвобождается из липосом.
В том случае, если инкапсулированное вещество высвобождается из липосом в месте своего действия слишком медленно, желаемая скорость высвобождения может быть достигнута с помощью использования смеси полианионизированного сахара или полиола с одним или более моновалентным или поливалентным анионом, например, хлоридом, сульфатом, фосфатом и т.д. Альтернативно, можно использовать смеси полианионизированных Сахаров и полиолов с различной степенью полианионизации. Согласно одному варианту осуществления настоящего изобретения, степень полианионизации внутри липосом, заявленных в соответствии с настоящим изобретением, находится в пределах от 0.1% до 99%, от 10% до 90% или от 20% до 80% от общего количества анионов внутри липосом, например, с инкапсулированным соединением.
В целом, липосомная композиция, заявленная в соответствии с настоящим изобретением, может содержать один или более полианион, заявленный в соответствии с настоящим изобретением, в любой подходящей форме, например, в форме кислоты или соли, включающей полианион и катион. Количество полианиона, например, полианионизированного сахара или полиола может быть стехиометрически эквивалентно количеству катиона или отличаться от последнего. Согласно одному варианту осуществления настоящего изобретения, липосомная композиция, заявленная в соответствии с настоящим изобретением, содержит одну или более полианионизированную соль катиона, при этом существует градиент концентрации или рН градиент через липосомные мембраны. Согласно другому варианту осуществления настоящего изобретения, липосомная композиция, заявленная в соответствии с настоящим изобретением, содержит одну или более полианионизированную соль замещенного аммония, заявленного в соответствии с настоящим изобретением. Согласно еще одному варианту осуществления настоящего изобретения, липосомная композиция, заявленная в соответствии с настоящим изобретением, содержит полианион во внутреннем пространстве липосом, при этом полианион частично или полностью уделен из среды, содержащей липосомы, с помощью способов, хорошо известных специалистам в данной области, например, растворения, ионообменной хроматографии, хроматографии с исключением по размеру, диализа, ультрафильтрации, абсорбции, преципитации и т.д. Согласно еще одному варианту осуществления настоящего изобретения, липосомы с инкапсулированным полианионом, например, полианионизированным полиолом или полианионизированным сахаром, также имеют трансмембранный градиент, способствующий сохранению активного вещества внутри липосом. Примеры такого трансмембранного градиента включают рН градиент, градиент электрохимических потенциалов, градиент аммониевых ионов, градиент замещенных аммониевых ионов, или градиент растворимости. Градиент замещенных аммониевых ионов обычно включает замещенную форму аммониевого иона, имеющую, по меньшей мере, одну C-N связь, такую, как первичный, вторичный, третичный или четвертичный аммоний. Способы создания трансмембранного градиента являются рутинным и широко используются при создании липосом.
Согласно еще одному варианту осуществления настоящего изобретения, липосомная композиция, заявленная в соответствии с настоящим изобретением, содержит одно или более соединение замещенного аммония и/или полианиона, заявленного в соответствии с настоящим изобретением, а также химическое или биологическое вещество, например, лекарственное средство или контрастное соединение. Например, вещество, содержащееся в липосомной композиции, заявленной в соответствии с настоящим изобретением, может представлять собой лекарственное средство, чернила, магнитное вещество, удобрение, краситель, биокатализатор, усилитель вкуса или запаха, отбеливатель, или любое вещество, которое можно определить с помощью любого способа, известного специалистам, например, магнитно-резонансной визуализации (MRI), оптической визуализации, флуоресцентной/люминесцентной визуализации, или ядерных визуализирующих методик. Обычно вещество, включенное в состав липосомной композиции, заявленной в соответствии с настоящим изобретением, представляет собой слабое основание и проникает через мембрану, (то есть является липофильным), например, таким веществом может быть вещество, имеющее в своем составе амин или азот.
Согласно одному варианту осуществления настоящего изобретения, вещество, содержащееся в липосомной композиции, заявленной в соответствии с настоящим изобретением, представляет собой лекарственное средство.
Согласно другому варианту осуществления настоящего изобретения, вещество, содержащееся в липосомной композиции, представляет собой противоопухолевое лекарственное средство. Частичный список некоторых общеизвестных коммерчески доступных (или находящихся на стадии разработки) противоопухолевых агентов с классификацией представлен ниже.
Классы соединений по структуре: Фторпиримидины-5-FU, Фтордеоксиуридины, Фторафур, 5'-деоксифторуридин, UFT, S-1 Капецитабин; пиримидиновые Нуклеозиды - Деоксицитидин, Цитозина Арабинозид, 5-Азацитозин, Гемцитабин, 5-Азацитозин-Арабинозид; Пурины - 6-Меркаптопурин, Тиогуанин, Азатиоприн, Аллопуринол, Кладрибин, Флударабин, Пентостатин, 2-Хлораденозин; Аналоги платины - Цисплатин, Карбоплатин, Оксалиплатин, Тетраплатин, Платина-DACH, Ормаплатин, CI-973, JM-216; Антрациклины/Антраценедионы - Доксорубицин, Даунорубицин, Епирубицин, Идарубицин, Митоксантрон; Эпиподофиллотоксины - Этопозид, Тенипозид; Камптотецины - Иринотекан, Топотекан, Луртотекан, Силатекан, 9-Амино Камптотецин, 10,11-Метилдиоксикамптотецин, 9-Нитрокамптотецин, TAS 103, 7-(4-метил-пиперазино-метилен)-10,11-этилендиокси-20(S)-камптотецин, 7-(2-N-изопропиламно)этил)-20(S)-камптотецин; Гормоны и их аналоги: Диэтилстилбестрол, Тамоксифен, Торемефин, Дролоксифен, Медроксипрогестерона ацетат, Магестрола ацетат, Аминоглутетимид, Тестолактон и др.; Ферменты, Протеины и Антитела - Аспарагиназа, Интерлейкины, Интерфероны, Леупролид, Пегаспарагаза и др.; Алкалоиды винки - Винкристин, Винбластин, Винорелбин, Виндезин; Таксаны - Паклитаксель, Доцетаксель.
Классы соединений по механизму действия: Антигормональные соединения - см. классификацию гормонов и их аналогов, Анастрозол; Антифолаты - Метотрексат, Аминоптерин, Триметрексат, Триметоприм, Пиритрексим, Пириметамин, Эдатрексат, MDAM; Антимикротубулярные агенты - Таксаны и Алкалоиды винки; Алкилирующие агенты (классические и неклассические) - азотистые mustard (Мехлорэтамин, Хлорамбуцил, Мелфалан, Урацила mustard), Оксафосфорины (Изофамид, Циклофосфамид, Перфосфамид, Трофосфамид), Алкилсульфонаты (Бусульфан), препараты Нитрозомочевины (Кармустин, Ломустин, Стрептозоцин), Тиотепа, Дакарбазин и другие; Антиметаболиты - пурины, пиримидины и нуклеозиды, перечисленные выше; Антибиотики - Антрациклины/Антраценедионы, Блеомицин, Дактиномицин, Митомицин, Пликамицин, Пентостатин, Стрептозоцин; Ингибиторы топоизомеразы - Камптотецины (Торо I), Эпиподофиллотоксины, m-AMSA, Эллиптицины (Торо II); Антивирусные средства - AZT, Зальцитабин, Гемцитабин, Диданозин и др; Мисцеллярные Цитотоксические Агенты - Гидроксимочевина, Митотан, Токсины слияния, PZA, Бриостатин, Ретиноиды, Масляная кислота и ее производные; Пентосан, Фумагиллин и др.
В дополнение к указанному, противораковое средство включает (без ограничения указанными) ингибитор топоизомеразы, алкалоид барвинка, например, винкристин, винбластин, винорелбин, винфлунин и винпоцетин, деполимеризующий или дестабилизирующий микротрубочки агент, стабилизирующий микротрубочки агент, например, таксан, аминоалкильный или аминоацильный аналог паклитакселя или доцетакселя, например, 2'-[3-(N,N-Диэтиламино)пропионил]паклитаксель, 7-(N,N-Диметилглицил)паклитаксель и 7-L-аланилпаклитаксель, алкилирующий агент, рецептор-связывающий агент, ингибитор тирозинкиназы, ингибитор фосфатазы, зависимый от циклина ингибитор киназы, энзимный ингибитор, ингибитор ауроракиназы, нуклеотид, полинуклеотид и ингибитор фарнезилтрансферазы.
В другом случае средство, содержащееся в липосомах, заявленных согласно настоящему изобретению, представляет собой терапевтический агент антрациклинового ряда или его производные, терапевтический агент камптотецинового ряда или его производные, терапевтический агент эллиптицинового ряда или его производные, алкалоиды или их производные барвинка, вортманнин, его аналоги и производные, или пиразолопиримидиновые соединения со свойствами ингибитора ауроракиназы.
В другом случае средство, содержащееся в липосомах, заявленных согласно настоящему изобретению, представляет собой антрациклиновое соединение, например, доксорубицин, даунорубицин, митомицин С, эпирубицин, пирарубицин, рубидомицин, карциномицин, N-ацетиладриамицин, рубидазон, 5-имидодауномицин, N-ацетилдауномицин, даунорулин, митоксантрон; камптотециновое соединение, например, камптотецин, 9-аминокамптотецин, 7-этилкамптотецин, 10-гидроксикамптотецин, 9-нитро камптотецин, 10 11-металл ендиоксикамптотецин, 9-амино-10,11-метилендиоксикамптотецин, 9-хлор-10,11-метил ендиоксикамптотецин, иринотекан, топотекан, луртотекан, силатекан, (7-(4-метилпиперазинометилен)-10,11-этилендиокси-20(S)-камптотецин, (7-(4-метилпиперазинометилен)-10,11-метил ендиокси-20(S)-камптотецин, (7-(2-N-изопропиламино)этил)-20(S)-камптотецин; эллиптициновое соединение, например, эллиптицин, 6-3-аминопропил-эллиптицин, 2-диэтиламиноэтил-эллиптицин и их соли, дателлиптиум, ретеллиптин.
В другом случае средство, содержащееся в липосомах, заявленных согласно настоящему изобретению, представляет собой фармацевтическое соединение, выбранное из группы, включающей (без ограничений указанными): антигистаминовые производные этилендиамина (бромфенифамин, дифенигидрамин); антипротозойные средства: хинолоны (иодохинол); амидины (пентамидин); антигельминтные средства (пирантел); антишистосомальные средства (оксаминихин); противогрибковые производные триазола (фликоназол, итраконазол, кетоконазол, миконазол); антимикробные цефалоспорины (цефазолин, цефонисид, цефотаксим, цефтазимид, цефуоксим); антимикробные бата-лактамовые производные (азтреопам, цефметазол, цефокситин); антимикробные средства эритромициновой группы (эритромицин, азитромицин, кларитромицин, олеандомицин); пенициллины (бензилпенициллин, феноксиметилпенициллин, хлоксациллин, метициллин, нафциллин, оксациллин, карбенициллин); тетрациклины; другие антимикробные средства, новобиоцин, спектиномицин, ванкомицин; антимикобактериальные средства: аминосалициловая кислота, капреомицин, этамбутол, изониазид, пиразинамид, рифабутин, рифампин, хлофазим; антивирусные адамантаны: амантадин, римантадин; хинидиновые производные: хлороквин, гидроксихлороквин, промаквин, квионон; антимикробные квионолоны: ципрофлоксацин, эноксацин, ломефлоксацин, налидиксиновая кислота, офлоксацин; сульфонамиды; анимикробные средства для лечения мочевыводящих путей: метенамин, нитрофурантоин, триметоприм; нитроимидазолы: метронидазол; холинергические четвертичные соединения аммония (амбетиниум, неостигмин, физостигмин); аминоакридины против болезни Альцгеймера (такрин); лекарственные средства против болезни Паркинсона (бензтропин, бипериден, проциклидин, триэксилэнидил); антимускариновые агенты (атропин, хиосциамин, скополамин, пропантелин); адренергические дофамины (албутерол, добутамин, эфедрин, эпинефрин, норепинефрин, изопротеренол, метапроперенол, салметрол, тербуталин); эрготаминовые производные; миорелаксанты или курановые соединения; миорелаксанты центрального действия; баклофен, циклобензепин, дентролен; никотин; бета-адреноблокаторы (ацебутил, амиодарон); бензодиазепины (дитиазем); антиаритмические препараты (диизопирамид, энкаидин, анестетики местного действия - прокаин, прокаинамид, лидокаин, флекаимид), квинидин; ингибиторы АСЕ: каптоприл, энелаприлат, фосинопрол, квинаприл, рамиприл; антилипидемические средства: флувастатин, гемфиброзил, ингибиторы HMG-coA (правастатин); гипотензивные препараты: хлонидин, квинабенз, празоцин, гуанетидин, гранадрил, гидралазин; и некоронарные сосудорасширяющие средства: дипиридамол.
В соответствии с настоящим изобретением, средство, содержащееся в заявленных липосомах, представляет собой пролекарственное средство, например, пролекарственное средство или агент, способный превращаться в нужную форму посредством одной или более стадий конверсии в условиях, связанных, например, с изменением рН или энзиматическим расщеплением лабильной цепи. Такая конверсия может осуществляться после высвобождения пролекарственного средства из липосомы в определенном участке действия лекарственного средства с липосомой. Однако, пролекарственное средство может быть превращено в нужное активное вещество внутри липосомы до использования липосомы в качестве вектора доставки, например, при введении пациенту. Лекарственное средство может быть модифицировано в пролекарственное средство с тем, чтобы его было легче поместить в липосомы, и затем оно может быть конвертировано обратно в нужное лекарственное средство при помещении его в заявленные липосомы. Таким образом, лекарственного средства, которые обычно не могут быть эффективным образом помещены в липосомы с помощью методов активации, дистанцирования или других основанных на использовании градиента методов, в соответствии с заявленным изобретением могут быть помещены, например, во внутреннее пространство липосом в нативной, немодифицированной форме.
Полностью катионные соединения, представляющие собой соединения, способные поддерживать полный ионный заряд в условиях нагружения липосом, в основном, соединения, содержащие титруемые амины, как известно, эффективно помещаются в липосомы, обладающие трансмембранным ионным градиентом. Если нужное средство представляет собой органическое соединение и не является полностью катионным соединением, содержащим титруемый амин, может быть получено его производное, имеющее необходимые ионные свойства, посредством подходящей модификации, например, методом, описанным в WO 96/25147 (Woodle et al.). Например, аминогруппа может быть введена этерификацией гидроксильной группы соединения с аминокислотой. Альтернативно, гидрофобная группа может быть ведена в растворимое в воде соединение, чтобы оно могло проникнуть в липосомную мембрану и затем переместиться из мембраны в интралипосомальный компартмент, т.е. внутрь липосом. Другая полезная модификация способного нагружаться в липосомы пролекарственного средства - это формирование карбонильной аддуктивной группы, например, гидразоновой, оксимной, ацетальной или кетальной. Модифицированная амино-содержащая группа может быть гидролизована или химически выщеплена из модифицированного соединения после его помещения в липосомы в соответствии с настоящим изобретением. Обычные способы внутрилипосомальной регенерации соединения из пролекарственного средства включают гидролиз, фотолиз, радиолиз, тиолиз, аммонолиз, восстановление, замещение, оксидацию или элиминацию. Эти способы могут быть осуществлены (без ограничений указанным) изменением рН или действием ферментов. Например, паклитаксель или доцетаксель, неионные вещества, конвертируются в 2'-(диэтиламинопропионил)- или 7'-(диэтиламинопропионил)-сложные эфиры, которые являются слабыми основаниями (пролекарственные средства). После помещения в липосомы любым известным методом, включающим (без ограничений указанными) методы активации, дистанцирования, основанные на трансмембранном градиента методы или основанные на градиенте растворимости методы, а также (или) методы, заявленные в соответствии с настоящим изобретением, внутрилипосомальный 2'-(диэтиламинопропионил)-паклитаксель превращается в исходный паклитаксель за счет стимуляции гидролиза при увеличении рН до около 7. Таким образом, липосомное инкапсулирование молекулы нейтрального таксана в пределах ее внутреннего пространства обеспечивается, когда отношение лекарственное средство/липид составляет около 0.05 молей на моль липосомного липида, без помощи гидрофильных ковалентных модификаций молекулы таксана (например, прикреплением ПЭГа), использования циклодекстринтаксановых комплексов или же таксан-солюбилизирующих формирующих мицеллы сурфактантов.
В соответствии с настоящим изобретением, липосомы, содержащиеся в липосомной композиции, заявленной согласно настоящему изобретению, могут быть любыми известными липосомами или же липосомами, которые будут созданы в дальнейшем. В целом, липосомы, согласно настоящему изобретению, могут иметь любую липосомную структуру, например, структуру, имеющую внутреннее пространство, отделенное от внешней среды одним или более липидным бислоем, или же любую микрокапсулярную структуру, имеющую полупроницаемую мембрану с липофильной центральной частью, отделенной мембраной от внутреннего пространства. Липидный бислой может иметь любую структуру из амфифильных молекул, имеющих гидрофильную часть и гидрофобную часть. Обычно амфифильные молекулы в бислое структурированы в два двухмерных слоя, в которых гидрофобные части ориентированы внутрь слоя, а гидрофильные - наружу. Амфифильные молекулы, образующие липосомы в соответствии с настоящим изобретением, могут быть любыми известными или позднее обнаруженными амфифильными молекулами, например, липидами синтетического или природного происхождения или биосовместимыми липидами. Липосомы согласно настоящему изобретению могут быть также сформированы амфифильными полимерами и сурфактантами, например, полимеросомами и ниосомами. Для целей настоящего описания (без ограничений) указанные липосомоформирующие материалы также называются липидами.
В соответствии с настоящим изобретением, липосомы, содержащиеся в липосомной композиции, заявленной согласно настоящему изобретению, могут также быть нацеливающими липосомами, например, липосомами, содержащими одно или более нацеливающих соединений или модификаторов биораспределения на своей поверхности. Нацеливающим соединением может быть любой агент, способный специфически связываться или взаимодействовать с нужной мишенью. В одном случае нацеливающим соединением является лиганд. Лиганд, заявленный согласно настоящему изобретению, предпочтительно связывается с клеткой или интернализуется в клетку, в которой заключенное в липосому вещество проявляет желаемый эффект (клетка-мишень). Примерами лигандов, подходящих для осуществления настоящего изобретения, являются фолиевая кислота, белки, скажем, трансферрин, ростовой фактор, энзим, пептид, рецептор, антитело или фрагмент антитела, такой, как Fab', Fv, одноцепочечный Fv, антитело с одним доменом или любой другой полипептид, содержащий антиген-связывающие участки (CDRs) молекулы антитела. Направляемая лигандом липосома, когда нацеливающим веществом является антитело или его антиген-связывающий фрагмент, называется иммунолипосомой. В предпочтительном случае липосома, несущая нацеливающую молекулу, например, лиганд, интернализуется клеткой-мишенью. В другом случае нацеливающее вещество представляет собой лиганд, который специфически взаимодействует с тирозинкиназным рецептором, например, EGFR, HER2, HER3, HER4, PD-GFR, VEGFR, bFGFR или IGFR. В еще одном случае нацеливающее вещество специфически взаимодействует с рецептором ростового фактора, рецептором ангиогенного фактора, рецептором трансферрина, молекулой клеточной адгезии или рецептором витамина.
В соответствии с настоящим изобретением, липосомы, содержащиеся в липосомной композиции, заявленной согласно настоящему изобретению, проявляют трансмембранный концентрационный градиент замещенного аммония и/или полианиона. Предпочтительно, более высокая концентрация указанных веществ свойственна внутреннему пространству липосом. Кроме того, липосомная композиция согласно данному изобретению может обладать одним или более трансмембранным градиентом помимо градиента, созданного замещенным аммонием и/или полианионом. Например, липосомы, содержащиеся в липосомной композиции, заявленной согласно настоящему изобретению, могут дополнительно проявлять градиент рН, ионный градиент, электрохимический градиент и/или градиент растворимости.
В соответствии с настоящим изобретением, липосомы, содержащиеся в липосомной композиции, заявленной согласно настоящему изобретению, могут быть включены в набор, содержащий контейнер с липосомами, и, необязательно, контейнер с лекарственным средством и инструкциями, например, процедурными или информационными, имеющими отношение к использованию липосомной композиции при одном или более применениях. Такая инструкция может быть представлена по-разному, например, на плотной бумаге, в электронном виде или в виде доступа к базе данных или веб-сайту, содержащему инструкцию.
Липосомная мембранная композиция в соответствии с настоящим изобретением может быть получена любым подходящим методом, известным из уровня техники, или тем, который будет открыт позднее специалистом в данной области. В целом, для получения заявленных липосом могут быть использованы различные липидные компоненты. Обычно такие компоненты включают (без ограничений указанными) (1) незаряженные липидные компоненты, например, холестерол, церамид, диацилглицерол, ацил(простые полиэфиры) или алкилполи(простые эфиры); (2) нейтральные фосфолипиды, например, диацилфосфатидилхолины, сфингомиелины и диацилфосфатидилэтанол амины; (3) анионные липиды, например, диацилфосфатидилсерин, диацилфосфатидилглицерол, диацилфосфатидат, кардиолипин, диацилфосфатидилинозитол, диацилглицеролхемисукцинат, диацилглицеролхемиглутарат, холестерилхемисукцинат, холестерилхемиглутарат и подобные указанным соединения; (4) конъюгированные с полимером липиды, например, N-метокси(поли(этиленгликоль)диацилфосфатидилэтаноламин, поли(этиленгликоль)-диацилглицерол, поли(этиленгликль)-церамид; и (5) катионные липиды, например, 1,2-диацил-3-триметиламмоний-пропан (DOTAP), диметилдиоктадециламмоний бромид (DDAB) и 1,2-диацил-зп-глицеро-3-этилфосфохолин. Могут быть также использованы моноацилзамещенные производные указанных липидов, а также их ди- и моноалкил-аналоги.
Для выполнения или модификации каких-либо функций могут быть использованы различные компоненты липидов. Например, фосфолипид может быть использован как основной липид, формирующий везикулы. Включение холестерола полезно для поддержания ригидности мембран и уменьшения вытекания лекарственного препарата из липосом. Конъюгированные с полимерами липиды могут быть применены для получения липосом с целью увеличения времени их циркуляции в крови за счет снижения клиренса липосом в печени и селезенке или же для улучшения стабильности липосом и предотвращения их агрегации во время хранения, но при этом эффект увеличения времени их циркуляции отсутствует. В то время как включение ПЭГ-липидов в количестве 1 моль % или выше в липиды липосом по предположениям увеличивает время их циркуляции в крови в несколько раз (см., например, патент США 5013556), авторы изобретения неожиданно обнаружили, что полученные в соответствии с изобретением липосомы циркулируют в крови достаточно долго, а добавление ПЭГ-липида к липосомам увеличивает время их существования в циркуляции менее, чем в два раза, если вообще увеличивает. Кроме того, модулирующие заряд липиды (титруемые липиды) могут быть использованы для обеспечения доставки инкапсулированных в липосомы веществ к цитозольным или ядерным мишеням за счет облегчения высвобождения некоторых классов таких веществ в ограниченное пространство эндосомальных путей.
В одном случае заявленные липосомы включают лецитин, холестерол и амфипатический полимер. Лецитин, включаемый в состав липосом согласно настоящему изобретению, может быть природным лецитином, гидрогенизированным природным лецитином, синтетическим лецитином, 1,2-дистеароил-лецитином, дипальмитоил-лецитином, димиристоил-лецитином, диолеоил-лецитином, 1-стеароил-2-олеоил-лецитином или 1-пальмитоил-2-олеоил-лецитином, а амфипатический полимер может быть может быть липидным производным полиэтиленгликоля, например, фосфатидилэтаноламином полиэтиленгликоля, полиэтиленгликоль-диацилглицеролом или церамидным производным полиэтиленгликоля, причем мол. масса полиэтиленгликоля составляет от около 250 до около 20000, предпочтительно, от около 500 до около 5000. В другом случае амфипатический полимер находится в количестве по меньшей мере 0.1 моль % от липосом-формирующего липида липосом, согласно заявленному изобретению. Еще в одном случае амфипатический полимер находится в количестве по меньшей мере от 0. 1 моль % до 1 моль % от липосом-формирующего липида липосом, согласно заявленному изобретению. Предпочтительно, амфипатический полимер является нейтральным полимером, т.е. условиях нагружения липосом лекарственным средством его полный ионный заряд, равен нулю; таким полимером может быть, например, ПЭГ-диацилглицерол, ПЭГ-диалкилглицерол или ПЭГ-церамид. Неожиданно было обнаружено, что включение ионно-нейтральных амфипатических липидов до содержания ПЭГ-липида около 5,7 моль % от общей липидной фракции обеспечивает высокую эффективность нагружения липосом, например, алколоидами барвинка, такими, как винорелбин, в то время как при использовании анионно-заряженных PEG-DSPE в состав липосом эффективность их нагружения существенно снижается при содержании ПЭГ-липида 1,6 моль % или более (Пример 72).
В еще одном варианте осуществления изобретения заявленные липосомы содержит производное камптотецина, например, камптотециновое пролекарственное средство, такое, как иринотекан, и содержит лецитин и холестерол, например, при молярном соотношении 3:2, и амфипатический полимер, например, в количестве по меньшей мере 0.1 моль % или менее от липосом-формирующего липида.
Заявленные в соответствии с настоящим изобретением липосомы могут быть получены любым способом, который известен, или который станет известным в будущем. См., например, G. Gregoriadis (под редакцией), Liposome Technology, vol. 1-3, первое издание. 1983; второе издание, 1993, CRC Press, Boca Ration, FL. Примеры способов, которыми могут быть получены липосомы, заявленные согласно настоящему изобретению, включают экструзию, обратно-фазовое выпаривание, озвучивание, инъекцию растворителя (например, этанола), микрофлюидизацию, детергентный диализ, инъекцию простого эфира и дегидратацию/регидратацию. Размер липосом может быть проконтролирован определением размера пор мембран, используемых для экструзии при низком давлении, и числа проходов при микрофлюидизации или любым другим подходящим способом. В одном случае нужные липиды сначала гидратируют тонкопленочной гидратацией или инъекцией этанола и далее формируют их размер путем экструзии через мембраны с заданным размером пор, наиболее часто составляющим 0,05 мкм, 0,08 мкм или 0,1 мкм.
Липосомные композиции, содержащие замещенный аммоний и/или полианион в соответствии с настоящим изобретением включают липосомы, которые могут получены любым подходящим способом, например, формированием липосом в присутствии замещенного аммония и/или полианиона, например, находящихся в виде соли. Замещенный аммоний и/или полианион за пределами липосом может быть удален или разбавлен после формирования липосом или до их нагружения или заключения в них активного вещества. Альтернативно, липосомные композиции, содержащие замещенный аммоний и/или полианион в соответствии с настоящим изобретением, могут быть получены ионообменным методом непосредственно или через промежуточную стадию с использованием свободной кислоты в градиенте замещенного аммония, например, соли замещенного аммония с полианионизированным сахаром или полиолом. Такие липосомы могут быть нейтрализованы при использовании амина или его соли с летучей кислотой, например, карбоната. Полученный раствор липосом может быть использован непосредственно или, альтернативно, содержащая его соль может быть удалена, если это требуется, путем выпаривания и кристаллизации после растворения в водной среде.
Предпочтительно, липосомные композиции, согласно настоящему изобретению, имеют трансмембранный концентрационный градиент замещенного аммония и/или полианиона, например, концентрация соли замещенного аммония и/или полианиона внутри липосомы выше, обычно, по меньшей мере, в 100 раз по сравнению с концентрацией соли замещенного аммония и/или полианиона во внешней среде липосом.
В одном случае концентрация соли замещенного аммония и/или полианиона внутри липосомы выше, обычно, по меньшей мере, в 100 раз по сравнению с концентрацией соли замещенного аммония и/или полианиона во внешней среде липосом и составляет, по меньшей мере, около 10 мМ, 50 мМ, 0,1 М, 0,2 М, 0,5 М, 0,6 М, 0,7 М или 1,0 М, причем молярность вычисляют по замещенному аммонию. В другом случае концентрация соли замещенного аммония и/или полианиона внутри липосомы выше, обычно, по меньшей мере, в 100 раз по сравнению с концентрацией соли замещенного аммония и/или полианиона во внешней среде липосом и составляет около 0, 65 М или около 1,0 М.
Кроме того, липосомная композиция согласно изобретению обычно имеет внешний рН, совместимый со стабильностью нужного вещества в процессе нагружения липосом или полезный для поддержания такой стабильности, при сохранении эффективности нагружения, например, выше 90%. Например, рН в диапазоне 4-7 или 4,5-6,5 предпочтителен. В частности, согласно настоящему изобретению нагружение липосом камптотециновым соединением, например, топотеканом или иринотеканом, наилучшим образом осуществляется при рН внешней среды в диапазоне от около 4,0 до около 7,0, более предпочтительно, в диапазоне от около 5,0 до 6,5. Нагружение липосом винкаалкалоидами (барвинка), например, винкристином, винорелбином или винбластином наилучшим образом осуществляется при рН внешней среды в диапазоне от около 5,0 до около 7,0, более предпочтительно при рН 6,5.
В соответствии с заявленным изобретением, нужное вещество может быть нагружено или заключено в липосомы путем его инкубирования с липосомами, заявленными согласно настоящему изобретению, в водной среде при подходящей температуре; например, температура выше температуры фазового перехода липидного компонента липосом в процессе их нагружения нужным веществом снижается до значения ниже температуры фазового перехода липидного компонента липосом после их нагружения. Время инкубации обычно зависит от природы липидного компонента, природы вещества, заключаемого в липосомы, и температуры инкубации. Обычно, время инкубации составляет от нескольких минут до нескольких часов. Поскольку обеспечивается высокая эффективность нагружения, а именно более 85%, обычно более 90%, как правило не возникает необходимости удаления невключившегося в липосомы вещества. Если такая необходимость существует, невключившееся вещество может быть удалено различными способами, например, хроматографией с исключением по размеру, диализом, ультрафильтрацией, адсорбцией или преципитацией. Неожиданно было обнаружено, что поддержание низкой ионной силы в процессе инкубирования заявленных липосом с нагружаемым веществом, в частности, таким, как производное камптотецина или алкалоид барвинка, с последующим увеличением ионной силы в конце инкубирования приводит к повышению эффективности нагружения, лучшему удалению невключившегося вещества и лучшей устойчивости липосом к агрегации. Обычно, инкубирование проводят, например, в водном растворе при ионной силе менее, чем эквивалентной 50 мМ NaCl, или, более предпочтительно, менее, чем эквивалентной 30 мМ. После инкубирования раствор концентрированной соли, например, NaCl может быть добавлен для увеличения ионной силы более, чем 50 мМ NaCl или, более предпочтительно, более 100 мМ NaCl. Не желая ограничиваться какой-либо теорией, авторы изобретения предполагают, что увеличение ионной силы кислотной диссоциации нужного вещества через мембрану липосом высвобождает практически все вещество, заключенное во внутреннее пространство липосом.
В целом, отношение нагружаемого вещества к липиду липосом, например, отношение нагружаемого лекарственного средства к липиду, полученное в результате его инкапсулирования, зависит от количества включившегося вещества во внутрь липосом, концентрации включившегося замещенного амина и/или полианиона, например, соли, физико-химических свойств инкапсулированного вещества и типа используемого противоиона (аниона), например, полианиона. Поскольку применительно к заявленным в соответствии с настоящим изобретением композициям и/или способам обеспечивается высокая эффективность нагружения липосом, отношение нагружаемого вещества к липиду липосом для вещества, инкапсулированного в липосомы, составляет более 80%, более 90% и, обычно, более 95%, что определяется на основании количества инкапсулируемого вещества и липида липосом, вовлеченных в процесс нагружения (отношение "захвата"). Действительно, в самом общем случае наблюдается 100% инкапсулирования (количественный показатель). Отношение нагружаемого вещества к липиду липосом может быть выражено в весовых показателях (весовое количество вещества в расчете на вес или молярную единицу липосомного липида) или в молярных показателях (число молей вещества в расчете на вес или молярную единицу липсомного липида). Одна единица отношения нагружаемого вещества к липиду липосом может быть преобразована в другие единицы обычными способами, как проиллюстрировано ниже. Весовое отношение вещества в заявленных липсомах обычно составляет, по меньшей мере, 0,05, 0,1, 0,2, 0,35, 0,5 или, по меньшей мере, 0,65 мг вещества в расчете на мг липида. При молярном отношении согласно заявленному изобретению отношение нагружаемого вещества к липиду липосом, обычно составляет, по меньшей мере, от около 0,02 до около 5, предпочтительно, по меньшей мере, от 0,1 до около 2 и, более предпочтительно, от около 0,15 до около 1,5 молей лекарственного средства в расчете на моль липосомного липида. В одном случае отношение нагружаемого вещества к липиду липосом, например, отношение при нагружении лекарственным средством, а именно производными камптотецина, составляет, по меньшей мере, 0,1, например, 0,1 моля производного камптотецина в расчете на один моль липосомного липида и, предпочтительно, по меньшей мере, 0,2. В другом случае отношение нагружаемого вещества к липиду липосом, например, отношение при нагружении лекарственным средством, составляет, по меньшей мере, 300 мг вещества (винкаалкалоиды или их производные) в расчете на мг формирующего липосомы липида. Еще в одном случае отношение нагружаемого вещества к липиду липосом, например, отношение при нагружении лекарственным средством, составляет, по меньшей мере, 500 мг вещества (например, производное камптотецина или камптотециновое пролекарственное средство) в расчете на мг формирующего липосомы липида. Неожиданно было обнаружено, что согласно изобретению обеспечивается стабильное и прочное количество липосомного инкапсулирования производными камптотецина, например, иринотеканом при отношении нагружаемого вещества к липиду липосом более 0,8 мМ вещества в расчете на 1 г липосомного липида, более 1,3 мМ вещества в расчете 1 г липосомного липида и даже 1,7 мМ вещества в расчете на 1 г липосомного липида (см. Пример 74).
Если липосомы содержат фосфолипид, целесообразно выражать содержащееся в них лекарственное средство в весовых (массовых) единицах, т.е. весовое количество вещества рассчитывается на молярную единицу фосфолипида липосом, например, мг лекарственного средства/мМ фосфолипида. Однако, специалист в данной области знаний должен понимать, что содержание лекарственного средства может быть эквивалентным образом выражено независимо от присутствия фосфолипидов в липосоме и, более того, выражено, например, через молярное количество вещества в расчете на единицу (весовую или молярную) липосомного липида. Скажем, содержание лекарственного средства в липосоме, состоящей из 3 молярных частей дистеароилфосфатидилхолина (DSPC, мол. масса 790), 2 молярных частей холестерола (мол. масса 387) и 0,015 молярных частей полиэтиленгликоль-дистеароилфосфатидилэтаноламина (PEG-DSPE, мол. масса 2750), в которую инкапсулирован доксорубицин (мол. масса 543,5) при отношении лекарственное средство/липид 150 мг/мМ фосфолипида, может быть эквивалентным образом выражено в единицах мг лекарственного средства/мг общего липида следующим образом.
(a) Вычисляют молярное количество компонентов липосомного липида, нормализованное к молярной единице липосомных фосфолипидов (DSPC и PEG-DSPE в этом примере) делением молярного количества компонента на общее молярное количество липосомных фосфолипидов:
DSPC 3/(3+0,015)=0,99502;
холестерол 2/(3+0,015)=0,66335
PG-DSPE 0,015/(3+0,015)=0,00498.
(b) Вычисляют весовое количество общего липосомного липида по отношению к единице молярного количества липосомного фосфолипида и мол. массе липосомных компонентов:
общий липид, мг/мМ фосфолипида = 0,99502×790+0,66335×387+0,00498×2750=1056, 48.
(c) Вычисляют массовое количество лекарственного средства по отношению к единице массы общего липида делением содержания лекарственного средства в массовых единицах на молярную единицу фосфолипида на значение, полученное на стадии (b):
доксорубицин, мг/мг общего липида = 150/1056,48=0,14198.
(d) Вычисляют ммолярное количество лекарственного средства по отношению к единице массы общего липида делением значения, полученного на стадии (с) на мол. массу лекарственного средства (в данном случае 543,5):
доксорубицин, мМ/г общего липида = 0,14198/543,5×1000=0,261.
(e) Вычисляют молярную долю фосфолипидов в липосомном липидном матриксе: молярная доля фосфолипидов = (общее число молей фосфолипидов)/(общее
количество молей липидов)=(3+0,015)/(3+2+0,015)=0,6012.
(f) Вычисляют молярное отношение доксорубицина к общему липиду.
Доксорубицин, моль/моль общего липида = (молярная доля фосфолипидов) × (доксорубицин, г/моль фосфолипидов) / (мол. масса доксорубицина) = 0,6012×150/543,5=0,166.
Таким образом, существует взаимосвязь между отношением лекарственное средство/липид и лекарственное средство/фосфолипид, выраженное в различных единицах. Используемый здесь термин "липид" включает (без ограничений указанным) любые формирующие мембрану липосомы компоненты липосомной мембраны, такие как, например, полимеры и/или детергенты.
Липосомы, содержащие замещенный аммоний и/или соль полианиона, согласно настоящему изобретению, обычно имеют осмотическую силу (осмолярность), которая обеспечивает стабильность липосом по отношению к осмотическому повреждению (набухание и/или разрыв) без нарушения нагружающей способности липосом. В одном случае осмолярность липосом, согласно настоящему изобретению, находится в интервале от 0,1 до 1,5 моль/кг или, предпочтительно, от 0,2 до 1,0 моль/кг. Неожиданно было обнаружено, что заявленные согласно изобретению липосомы стабильны по отношению к неблагоприятному эффекту высокой внутрилипосомальной осмотической силы на нагружающую способность липосом. К внутрилипосомальной осмолярности 0,727 моль/кг липосомы хорошо толерабельны, что приводит к почти количественному нагружению их лекарственным препаратом до теоретически максимального стехиометрического обмена внутрилипосомального замещенного аммония на молекулы лекарственного средства (в случае иринотекана одна молекула лекарственного средства на одну молекулу замещенного аммония), даже при том, что осмолярность внелипосомальной водной среды в процессе совместного инкубирования лекарственного средства и липосом близка к физиологическому значению около 0,3 моль/кг (Пример 74).
В целом, липосомы, заявленные согласно настоящему изобретению, достаточно стабильны во время хранения, например, это определяют по процентному содержанию включившегося вещества, которое высвобождается из липосом, или все еще находящегося внутри липосом после некоторого периода времени, прошедшего от момента первоначального включения вещества в липосомы. Например, заявленные липосомы стабильны при 4°C по меньшей мере в течение 6 месяцев, т.е. менее 10% включившегося вещества высвобождается из липосом в течение 6 месяцев, прошедших с момента первоначального включения вещества в липосомы. В одном случае заявленные липосомы стабильны при 4°C по меньшей мере в течение 2 лет, т.е. менее 20% включившегося вещества высвобождается из липосом в течение 2 лет, прошедших с момента первоначального включения вещества в липосомы.
Важно, что включившееся в липосомы вещество остается внутри липосом до того момента, когда липосомы достигнут нужного участка действия, например, в случае липосомальной доставки противоракового лекарственного средства к опухоли в организме больного. Заявленные липосомы обнаруживают неожиданную стабильность по отношению к "утечке" (высвобождению) включившегося вещества в условиях in vivo, например, в крови млекопитающего. Время, необходимое для 50%-ного высвобождения включившегося вещества, например, лекарственного средства из липосом (время полувысвобождения) в кровь крысы in vivo составляет более 24 ч. В частности, липосомы, нагруженные винкаалкалоидами, например, винбластином, винкристином и винорелбином, обладают значительной стабильностью по отношению к высвобождению из них лекарственного средства in vivo (время полу-высвобождения составляет, по меньшей мере, 24 ч или количество вещества, оставшегося в липосомах через 24 ч, находящихся в крови in vivo, составляет, по меньшей мере, 50% от значения, измеренного перед введением липосом). Обычно время полу-высвобождения составляет более 33 ч или же количество вещества, оставшегося в липосомах через 24 ч, находящихся в крови in vivo, составляет, по меньшей мере, 60% от значения, измеренного перед введением липосом; даже более часто время полу-высвобождения составляет более 46 ч или количество вещества, оставшегося в липосомах через 24 ч, находящихся в крови in vivo, составляет, по меньшей мере, 70% от значения, измеренного перед введением липосом. Иногда время полу-высвобождения включившегося вещества в крови составляет более 93 ч и даже более 120 ч. Липосомы, нагруженные производными камптотецина, такими, как топотекан и иринотекан, также обнаруживают исключительную стабильность в крови in vivo, а именно 79-85% первоначально включенного в липосомы лекарственного средства остается в них чрез 24 ч. Немаловажно, что заявленные липосомы, имеющие такую низкую скорость высвобождения лекарственного средства в крови in vivo, обнаруживают значительную противоопухолевую активность in vivo, превышающую активность свободного лекарственного препарата (при введении в форме раствора).
Заявленные в соответствии с изобретением липосомы проявляют неожиданное сочетание высокой эффективности включившегося в них терапевтического агента и низкой токсичности. В целом, активность включившегося в липосомы терапевтического агента, согласно изобретению, например, антинеопластическая активность производного камптотецина в организме млекопитающего, составляет, по меньшей мере, от эквивалентной до, по меньшей мере, в два раза превышающей или, по меньшей мере, в четыре раза превышающей активность терапевтического агента, если он вводится в том же самом количестве посредством обычной нелипосомальной процедуры, например, без использования заявленных липосом. При этом токсичность включившегося в липосомы вещества не повышена и, по меньшей мере, в два раза, или, по меньшей мере, в три раза, или, по меньшей мере, в четыре раза ниже, чем токсичность вещества терапевтического агента, который вводится в том же самом количестве и по той же самой схеме, но в свободной, нелипосомальной форме. Например, хорошо известно, что включение в липосомы противораковых производных камптотецина обычными методами среди прочего приводит к увеличению его токсичности (более низкая максимальная толерантная доза, менее 50% летальной дозы) по сравнению с неинкапсулированным препаратом. См. патент США 6355268, патент США 6465008, Colbern, et al. Clinical Cancer Res. 1998, v. 4, p. 3077-3082, Tardi, et al. Cancer Res., 2000, v. 60, p. 3389-3393, Emerson, et al. Clinical Cancer Res. 2000, v. 6, p. 2903-2912. Инкапсулированный в липосомы камптотециновые пролекарственные средства, такие, как иринотекан (СРТ-11), который растворим в воде и представляет собой катионное камптотециновое пролекарственное средство, имеет значительно более высокую (по меньшей мере, в 4 раза и даже, по меньшей мере, в 10 раз) противоопухолевую активность, измеренную на модели опухоли in vivo, по сравнению с лекарственным среством, не заключенным в липосомы, т.е. находящимся в свободной (растворимой) форме. Это даже более важно, поскольку терапевтическое соединение, например, камптотециновое пролекарственное средство, требует энзиматической активации, например, путем активации эндогенной неспецифической карбоксилэстеразы, но в соответствии с настоящим изобретением надежно заключено во внутреннее пространство липосом. С другой стороны, неожиданно, токсичность камптотецинового пролекарственного средства, такого, как СРТ-11 в липосомальной форме (весовое отношение лекарственное средство/липид более 0,1, например, 0,2-0,6 или более), согласно настоящему изобретению, более, чем в 2 раза, более, чем в 3 раза и даже более, чем в 4 раза ниже, чем пролекарственное средство СРТ-11, находящееся в свободной, неинкапсулированной форме. Более того, достигается пролонгированное высвобождение лекарственного средства СРТ-11 из липосом in vivo, так что более, чем с 50% и даже более, чем 70% (79-86%) исходного препарата все еще остается в липосомах через 24 ч после введения их в кровяное русло, а время высвобождения превышает 24 ч, обычно 48 ч. Пролонгированное сохранение лекарственного средства in vivo ассоциируется с высоким противоопухолевым эффектом. Неожиданно оказалось, что наиболее медленное высвобождение СРТ-11 и наиболее высокая противоопухолевая активность наблюдаются в липосомах, содержащих полианионизированное сахарное производное низкой мол. массы (октасульфат сахарозы) по сравнению с полимерным анионом (полифосфатом) (Пример 15).
В соответствии с другим вариантом осуществления изобретения, заявленная липосомная композиция может быть представлена фармацевтической композицией, содержащей заявленные липосомы и носитель, например, фармацевтически приемлемый носитель. Примерами фармацевтически приемлемых носителей являются физиологический раствор, изотоническая декстроза, изотоническая сахароза, раствор Рингера и раствор Хэнкса. Можно добавлять буфер для поддержания оптимального рН для стабильности при хранении. Например, рН между 6,0 и 7,5, более предпочтительно, около 6,5 является оптимальным для стабильности липидов липосомных мембран и обеспечивает сохранение в липосомах включенного в них вещества. Гистидин, гидроксиэтилпиперазин-этилсульфонат (HEPES), морфолино-этилсульфонат (MES), сукцинат, тартрат и цитрат, обычно при концентрации 2-20 мМ, являются примерами буферных систем. Другие подходящие носители включают, например, воду, забуференный водный раствор, 0,4%-ный NaCl, 0,3% - глицин и т.д. Белок, углевод или полимерные стабилизаторы и агенты, повышающие ригидность, например, желатин, декстран или поливинилпирролидон, также могут быть добавлены. Ригидность композиции может поддерживаться на физиологическом уровне 0,25-0,35 моль/кг с помощью глюкозы или более инертного соединения, такого, как лактоза, сахароза, маннитол или декстрин. Указанные композиции могут быть стерилизованы при использовании удобных, хорошо известных методик, например, фильтрованием. Полученные водные растворы могут быть упакованы для использования или отфильтрованы в асептических условиях и лиофилизированы; затем лиофилизированные препараты разводят стерильной водной средой перед применением.
Фармацевтическая липосомная композиция может также содержать другие фармацевтически приемлемые добавки, необходимые в определенных физиологических условиях, такие, как соединения, регулирующие рН и ригидность, буферные агенты и т.д., например, ацетат натрия, лактат натрия, хлорид натрия, хлорид калия, хлорид кальция и т.д. Кроме того, суспензия липосом может включать защищающие липиды агенты, которые предохраняют липиды от свободно-радикального или пероксидного повреждения при хранении. Могут быть использованы липофильные свободно-радикальные вещества, например, токоферол и растворимые в воде железо-специфичные хелаторы, такие, как ферриоксамин.
Концентрация липосом в жидкой фармацевтической композиции, согласно настоящему изобретению, может широко варьировать, т.е. составлять (обычно) от менее чем около 0,05% или от, по меньшей мере, около 2-10% до 30-50% по весу и может быть непосредственным образом выбрана в зависимости от объема жидкости, вязкости и т.д. в соответствии с используемым методом применения. Например, концентрация может быть повышена при необходимом снижении объема вводимой жидкости при лечении конкретных заболеваний. Это особенно желательно в отношении больных, страдающих связанной с атеросклерозом сердечной недостаточностью или серьезной гипертензией. Альтернативно, липосомные фармацевтические композиции, содержащие раздражающие липиды, могут быть разбавлены до более низких концентраций с целью уменьшения воспаления при области введения препарата.
Количество вводимой липосомной фармацевтической композиции зависит от терапевтически активного вещества, инкапсулированного в липосомы, природы заболевания, типа используемых липосом и решения лечащего врача. В целом, количество вводимой липосомной фармацевтической композиции должно быть достаточно для доставки терапевтически эффективной дозы препарата в область действия.
Количество вводимой липосомной фармацевтической композиции для доставки терапевтически эффективной дозы препарата в область действия может быть определено рутинными методами in vivo и in vitro, обычно используемыми для тестирования лекарственных средств. См., например, D.B. Budman, А.Н. Calvert, E.K. Rowinsky (под редакцией). Handbook of Anticancer Drug Development, LWW, 2003. Терапевтически эффективные дозы различных препаратов хорошо известны специалистам в данной области знаний; согласно настоящему изобретению терапевтический агент, доставляемый с помощью заявленной липосомной композиции, обеспечивает, по меньшей мере, ту же самую или двукратную, четырехкратную или десятикратную активность по отношению к активности при введении этого же препарата обычными методами без использования липосом. Обычно дозы терапевтически активных препаратов для липосомной фармацевтической композиции в соответствии с настоящим изобретением находятся в пределах от около 0, 005 до около 500 мг в расчете на кг веса тела, более часто, между примерно 0,1 мг и примерно 100 мг терапевтически активного препарата на кг веса тела.
Как правило, липосомную фармацевтическую композиции в соответствии с настоящим изобретением приготавливают для местного или инъецируемого введения как в жидкой форме, так и в виде суспензии. Однако если приготавливают твердые формы, подходящие для введения в виде раствора или суспензии, то необходимо приготовить также и жидкие носители. Композиция может быть получена и в форме таблеток с энетерическим покрытием или гелевых капсул в соответствии с методами, известными из уровня техники.
Липосомная фармацевтическая композиция в соответствии с настоящим изобретением может быть введена любым способом, приемлемым с медицинской точки зрения, что зависит от заболевания и степени его тяжести. Возможные пути введения включают инъекции, в том числе парентеральные (внутримышечные, подкожные, внутривенные, внутриартериальные, интраперитонеальные, внутрисуставные, внутриэпидуральные, интратекальные и другие, а также пероральный, назальный, офтальмический, ректальный, вагинальный, местный, пульмонарный (например, путем вдыхания) способы. Для доставки заключенных в липосомы лекарственных средств согласно настоящему изобретению к опухолям центральной нервной системы используют медленную поддерживаемую внутричерепную инъекцию липосом непосредственно в опухоль (конвекционно-усиленная доставка, CED). См. Saito, et al., Cancer Research, vol. 64, p. 2572-2579, 2004; Mamot, et al., J. Neuro-Oncology, vol. 68, p. 1-9, 2004. Композиции также могут быть доставлены к поверхности тканей. Поддерживаемое высвобождение, зависимое от рН высвобождение или другие специфические химические условия или условия, обеспечиваемые окружением, также включены в объем изобретения, например, инъекции в виде депо или распадаемых имплантов.
ПРИМЕРЫ
Нижеследующие примеры предназначаются для того, чтобы иллюстрировать, но не ограничивать каким-либо образом данное изобретение в части его объема или формы, прямо или косвенно. Поскольку они носят общий характер, исходя из того, что может быть использовано, иные процедуры, приемы и способы или методики, известные специалистам в данной области, также альтернативно могут использоваться.
Пример 1. Приготовление растворов солей замещенного аммония.
Растворы сульфатов триалкиламмония и диалкиламмония, пригодные для загрузки лекарственных средств (например, доксорубицина) в липосомы, готовили разбавлением серной кислоты водой до концентрации 0,25 М с последующим титрованием раствора серной кислоты одним из ряда аминов. Замещенными аминами, использованными в данном примере, были триэтиламин, триметиламин, диметиламин, диэтиламин или диэтаноламин. После прибавления аминов полученный в результате раствор разбавили до конечной концентрации 0,2 М замещенного аммония. Осмолярность определяли с помощью осмометра, используя точку росы. Свойства полученных растворов замещенных алкиламмонийных сульфатных солей показаны ниже в таблице 1.
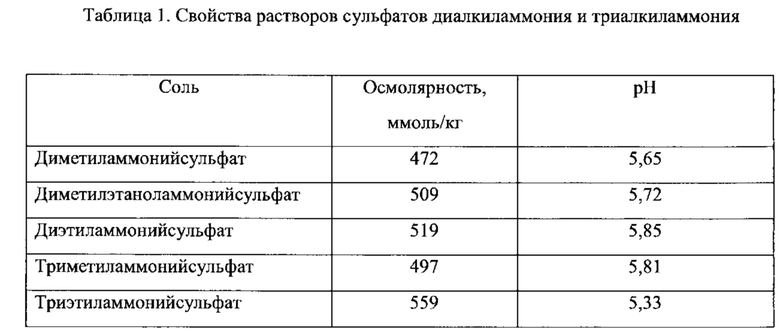
Пример 2. Получение липосом с захваченными диалкиламмониевыми и триалкиламмониевыми солями и загрузка вещества в эти липосомы.
Дистеароилфосфатидилхолин (DSPC), холестерин (Chol) и N-(метоксиполи(этиленгликоль)-оксикарбонил)-дистеароилфосфатидилэтано-ламин (PEG-DSPE) (приготовленный из поли(этиленгликоль) с мол. массой 2000) были сорастворены в хлороформе в мольном соотношении 3:2:0,015, и хлороформ удаляли при 55-60°C в ротационном испарителе. Высушенная липидная пленка была затем гидратирована раствором одного (каждого) из приведенных в примере 1 диалкил- или триалкиламмониевых сульфатов при 60°C в течение 30 минут. Липидную суспензию экструдировали под давлением через два объединенных в набор поликарбонатных мембранных фильтра с протравленными желобами и с размером пор 0,1 μм (Corning Nuclepore). Размер липосом, определяемый методом квазиупругого светорассеяния составлял приблизительно 110-120 нм. Неинкапсулированные соли триалкиламмония или диалкиламмония удаляли из внешней среды липосом гель-фильтрацией с помощью колонки с поперечносшитым декстрановым гелем (Сефадекс G-75, Amersham Pharmacia Biotechnology) при элюировании HEPES-буферным солевым раствором, рН 7,2-7,4, и собирали липосомы в виде фракции свободного объема колонки. Доксорубицин в форме гидрохлорида USP (лиофилизированный порошок, содержащий 5 вес. частей лактозы на 1 часть доксорубицина) добавили к липосомам в концентрации 150 μг лекарственного средства на 1 μмоль фосфолипида липосомы. Смесь инкубировали при 55°C в течение 45 мин., охлаждали на льду в течение 10 минут и неинкапсулированное лекарственное средство удалили гель-фильтрационной хроматографией, используя колонку с Сефадексом G-75, элюируя HEPES-солевым буферным раствором, рН 7,4. Наличие свободного доксорубицина (характеризующееся появлением более медленно движущейся полосы, окрашенной в красный цвет) визуально не детектировано. Нагруженные доксорубицином липосомы исследовали на фосфолипиды и доксорубицин в соответствии с примером 70 и 71 (спектрофотометрический метод), соответственно. Получившиеся в результате уровни нагруженности лекарственным средством приведены ниже в таблице 2.
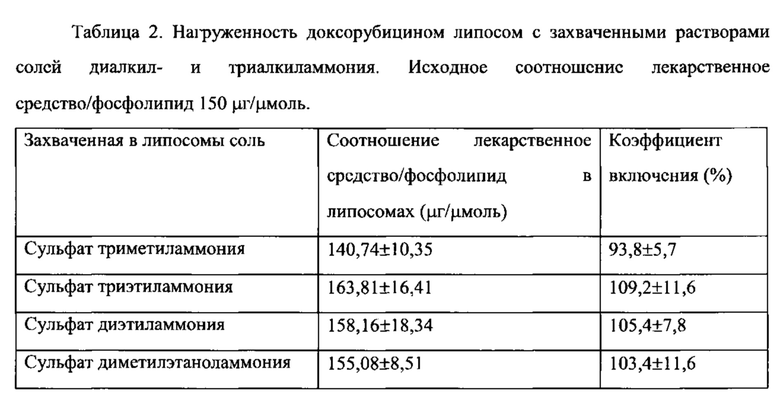
Пример 3. Получение липосом, содержащих различные диалкил-, триалкил- и гетероцикл-замещенные аммониевые соли серной кислоты, и нагружение этих липосом доксорубицином
Растворы сульфатов замещенного аммония готовили как описано в примере 1, используя коммерчески доступные алкилзамещенные, гидроксиалкилзамещенные и гетероциклические амины. Липосомы сформировали как в примере 1, за исключением того, что вместо стадии гидратации липидной пленки чистые липиды растворяли в этаноле (приблизительно 100 μл этанола на каждые 50 μл фосфолипидов) и смешивали с раствором соли замещенного аммония при 60-65°C так, что получившаяся липидная дисперсия содержала около 10% об. этанола.
Нагружение доксорубицином осуществляли путем добавления раствора доксорубицина (2 мг/мл в HEPES-буферном солевом растворе рН 6,5) к липосомам в соотношении 155 μг лекарственного средства/μмоль фосфолипида липосомы (PL) и нагревания до 58°C в течение 45 минут в горячей водяной бане. Полученные липосомы отделяли от оставшегося неинкапсулированного доксорубицина и анализировали на содержание лекарственного средства и липида как описано в примере 1. Результаты показаны в таблице 2.
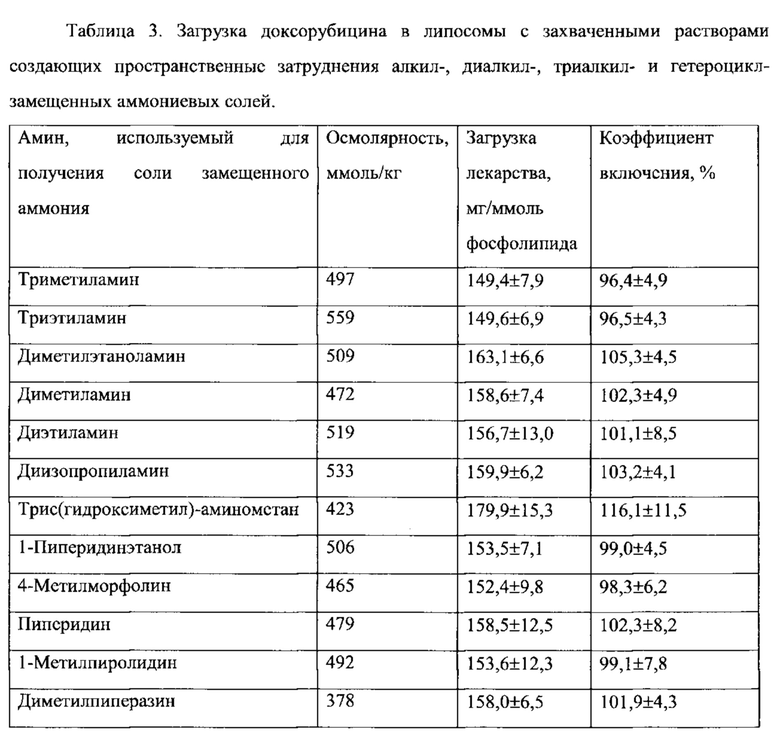
Пример 4. Получение раствора триэтил-аммоний-полифосфата (TEA-Pn)
Линейный поли(фосфат) натрия, имеющий 13-18 фосфатных групп на молекулу (Phosphate Glass, CALGON®, полученный от Sigma Chemical Company) был растворен в воде до концентрации примерно 1,3 М фосфата. Раствор пропустили через колонку, набитую 120 мл зернистой катионообменной смолы на основе сульфонированного полистирол-дивинилбензольного сополимера (Dowex 50W × 8-200, Dow Chemical G.) в водородной форме. Колонку предварительно уравновесили 3-3,6 М водным раствором HCl для того, чтобы привести смолу в водородную форму, и промыли деионизированной водой до нейтрального значения рН. Пятнадцать мл раствора полифосфата натрия нанесли на колонку и элюировали деионизированной водой. Элюент с колонки подвергали мониторингу, используя детектор проводимости. Вытекающее из колонки количество жидкости, которое соответствовало пику проводимости, оттитровывали чистым триэтиламином до рН 5,5-6,0. Раствор проверяли на остаточный натрий потенциометрическим методом, используя натрий-чувствительный стеклянный электрод, и на содержание фосфата, используя анализ неорганического фосфата, как описано в примере 1. Раствор с содержанием остаточного натрия менее, чем 1%, был разбавлен до конечной концентрации фосфата 0,55 М. Этот раствор обычно имеет TEA концентрацию 0,52-0,55 М, рН 5,5-6,0 и осмолярность 430-480 ммоль/кг.
Пример 5. Удаление незахваченных полифосфатных солей из препаратов липосом
Согласно методике Kirpotin et al., Biochemistry 36:66-75, 1997, получали липосомы (120 нм по размеру) с включенным флуоресцентным маркером трисульфонатом 8-гидроксипирена и смешивали их с раствором полифосфоната натрия. Эту смесь загружали на колонки, работающие на исключение по размеру, заполненные гранулами поперечносшитого декстрана (Сефадекс G-75), гранулами 6% агарозы (Сефароза 6B-Cl) или гранулами 4% агарозы (Сефароза 4B-Cl), все - из Amersham Pharmacia, и элюировали MES-Dextrose буфером (рН 5,5). Элюенты проверяли на содержание фосфата с помощью метода определения фосфата Bartlett (1959) и на содержание липосом спектрофотометрией. При проведении гель-хроматографических исследований Сефароза Cl-6B обеспечила полное отделение полифосфата от липосом при соотношении объема пробы и объема слоя в колонке, равном 13.
Пример 6. Приготовление раствора сахарозооктасульфата триэтиламмония (TEA-SOS)
Сахарозооктасульфат натрия (эквивалентный вес 144.8) - это натриевая соль производного сахарозы, в котором все гидроксильные группы образовали эфиры с серной кислотой. Сахарозооктасульфат натрия был закуплен в Toronto Research Chemicals, Toronto, Canada, p/n S 699020. Шесть граммов сахарозооктасульфата натрия растворили в 16,57 мл деионизированной воды до конечной концентрации около 2,5 Н сульфатных групп. Раствор подвергли ионному обмену как описано в примере 4. Раствор сахарозооктасерной кислоты, полученный как элюент с ионообменной колонки, был оттирован чистым триэтиламином до рН 5,7 (точка нейтрализации) и определены рН и осмолярность раствора. В полученном растворе была определена концентрация триэтиламмония, равная 0,643 М, рН 5,7 и осмолярность 530 ммоль/кг. Наличие остаточного натрия не могло быть определено потенциометрическим методом (менее, чем 0,1%).
Пример 7. Липосомы, нагруженные Иринотеканом (СРТ-11) с использованием солей замещенного аммония: получение и in vitro выделение лекарственного средства в присутствии плазмы крови.
В этом примере сульфат, цитрат, пирофосфат, трифосфат и линейный полифосфат (13-18 mer) изучались как анионы в растворах захваченных в липосомы солей замещенного аммония. Были выбраны фосфатные полимеры благодаря их биодеградируемости и из-за того, что полифосфаты естественным образом содержатся в клетках, в противоположность другим синтетическим полимерным анионам (полиакрилат, декстрансульфат и т.д.). Кроме того, вязкость растворов полифосфатов с низкой молекулярной массой была ниже, чем у других полимеров, что делает полифосфаты более подходящими и пригодными для процесса.
Для приготовления растворов солей были использованы следующие материалы.
1. Полифосфат натрия, NaO-[PO3Na]n-Na, n=13-18, закуплен у Sigma (Product № Р-8510, «Phosphate Glass, Practical Grade», известный также как гексаметафосфат натрия или под брендовым наименованием CALGON);
2. Триполифосфат пентанатрия; Na5P3O10, закупленный у Sigma (Product № Т-5883); 3. Декагидрат тетранатрия пирофосфата, Na4P2O7⋅10H2O, закупленный у Sigma (Product № Р-9146).
3. Ионообменные смолы Dowex 50Wx4 (4% поперечносшитая сульфонированная полистироловая смола, 100-200 меш), закупленная у Sigma (Product №50x4-200) или Dowex HCR-W2 (8% поперечносшитая сульфонированная полистироловая смола 50-100 меш), закупленная у Sigma (Product №1-8880), использовались взаимозаменительно. Смолы были промыты при декантации следующим образом: три раза деионизированной водой, два раза 1 н HCl (трехкратным избытком по сравнению с объемом смолы), три раза водой, два раза 1 н NaOH, три раза водой, три раза 1 н HCl, три раза водой. После декантации смолы были в H+-форме.
4. Триметиламин (ТМА), 40% водный раствор, от Aldrich Chemical Co. (Product №43, 326-8). Концентрации устанавливались кислотным титрованием до около 5,9 N.
5. Триэтиламин (TEA), 99%, ВЭЖХ-степень, от Fisher (Product №04884). Концентрация, согласно кислотному титрованию, составляла 6,9-7,1 N.
Вода была очищена обратным осмосом, ионообменом и удалением органики до достижения свободного от органических примесей качества «16-18 MOhm».
Водные растворы пирофосфатных, трифосфатных и полифосфатных солей готовили методом ионообмена. Растворы полифосфата натрия (3 г в 25 мл воды), пирофосфата (4 г в 27 мл воды) или полифосфата (6,7 г в 30 мл воды) были загружены в колонку, содержащую 100 мл (объем слоя) ионообменной смолы, приготовленной как указано выше. Колонку элюировали водой, фракции собирали. Фракции, показывающие кислый рН (рН<3) объединяли. Тройные 0,5-мл аликвоты объединенной фракции, содержащей фосфаты (фосфорную кислоту), разбавляли 20 мл воды и титровали 0,100 н NaOH до конечной точки рН 4,5-5,0 (Fisher раствор для анализа) для определения нормальности. Объединенные фракции после ионообмена были оттитрованы триметиламином (чтобы получить соли триметиламмония) до рН 5,4-5,5. После титрования растворы разбавляли, если необходимо, чтобы получить конечную концентрацию триметиламмония, близкую к 0,5 Н.
Сульфаты триметиламмония и триэтиламмония готовили разбавлением 1,39 мл концентрированной (17,9 М) серной кислоты 80 мл воды к титрованию разбавленного раствора чистым триэтиламином или водным триметиламином под контролем pH-метра до точки эквивалентности (рН 5,1-5,5). Объем доводили водой до 100 мл.
Раствор цитрата триметиламмония готовили растворением 1,572 г моногидрата лимонной кислоты ACS от Sigma (Product № С-1909) в 20 мл воды и титрованием раствора водным триметиламином до точки эквивалентности. Объем доводили водой до 25 мл.
Растворы фильтровали через ацетатцеллюлозный фильтр с размером пор 0,2 им с применением давления. Осмолярность и рН растворов измеряли с помощью осмометра (работающего с давлением пара) и pH-метра со стеклянным и каломельным электродами, соответственно. Нормальность аниона в фосфатных растворах определяли спектрофотометрическим методом с помощью голубого фосфомолибдата (см. пример 70) после гидролиза кислотой (5 мин, 100°C, 3 Н H2SO4). Нормальность по аниону принимали в расчет только по кислотным функциональным группам, которые существенно ионизированы при рН 5,5. Нормальность катионная определялась на основе добавленного основания триалкиламмония. Полученные растворы имели следующие свойства (таблица 4):
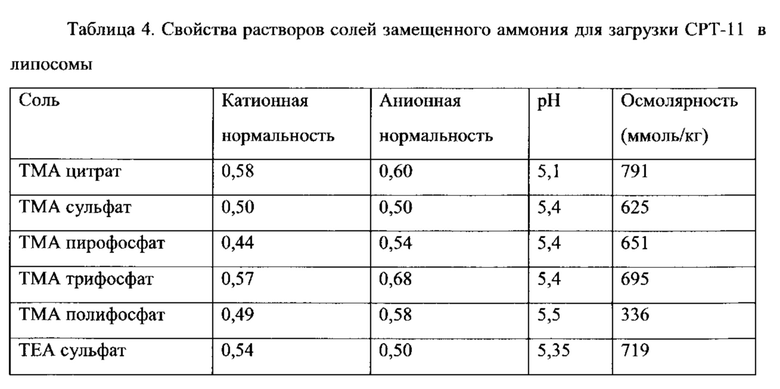
Холестерол и DSPC были закуплены у Avanti Polar Lipids, Alabaster, Alabema, USA. PEG-DSPE (PEG мол. масса 2000) был от Shearwater Polymers, Huntsville, AL, USA. DSPC, холестерол и PEG-DSPE в весовом соотношении 3:1:0,1 (мольное соотношение приблизительно 3:2:0,03) были растворены в хлороформе (Fisher; Optima-grade, стабилизирован амиленом) при 60 мг/мл DSPC. Раствор разливали в Pyrex пробирки по 30 мг DSPC (0,5 мл) на пробирку и медленно выпаривали при пониженном давлении, используя роторный испаритель при 60°C. Липидные пленки высушивали под вакуумом (100 микрон ртутного столба, масляный насос) в течение 30-60 минут при комнатной температуре.
Сухие липидные пленки гидратировали при осторожном встряхивании в вышеупомянутых водных растворах солей при 60°C в течение 15-20 минут. Липиды образовали суспензию, напоминающую молоко (мультиламеллярные пузырьки). Эту молочную суспензию подвергали пяти циклам замораживания в смеси сухого льда и изопропанола (-80°C, 3 минуты) и оттаивания в водяной бане при 60°C в течение 3 минут. Затем суспензию липидов экструдировали 10 раз (двойным ударом) через два объединенных поликарбонатных мембранных фильтра (Nucleopore, Whatman, размер пор 0,1 дм), используя управляемый вручную совершающий возвратно-поступательное движение экструдер (Avanti Polar Lipids), нагретый до 60°C.
Экструдированные липосомы выдерживали при 60°C в течение пяти минут и резко охлаждали в ледяной воде (0-4°C) в течение пяти минут. Затем липосомы выделяли из раствора, создающего градиент соли, в загружающий буфер (буфер загрузки), содержащий MES-Dextrose (50 г/л Dextrose ACS, 0,975 г/л 2-(N-морфолино)-этансульфокислоты (MES), и значительное количество 5 М NaOH, чтобы довести рН до 6.4) гель-хроматографией на Сефадоксе G-75. Липосомы выявлялись во фракции свободного объема (приблизительно 30% объема слоя в колонке).
Препарат СРТ-11 (Иринотекан гидрохлорид), содержащий 0,860 мг СРТ-11 основания на 1 мг твердого вещества, был растворен в 0,001 н HCl для того, чтобы получить основной (исходный) раствор 16,5 мг/мл СРТ-11 основания. Этот раствор смешали с липосомами в MES-Dextrose буфере для достижения соотношения 150 μг СРТ-11 на 1 μмоль фосфолипидов липосомы. Смесь инкубировали при 55°C в водяной бане, осторожно встряхивая (приблизительно каждые пять минут) в течение 30 минут, а затем быстро вылили в ледяную воду (0-4°C). Липосомы отделяли от неинкапсулированного лекарственного средства гель-хроматографией на Сефадексе G-75, используя MES-Dextrose в качестве элюента. Инкапсулированное лекарственное средство определяли спектрофотометрическим способом (пример 71), а фосфолипиды определяли, используя экстракционный анализ (пример 70).
Выделение in vitro лекарственного средства в полученных таким образом СРТ-11-нагруженных липосомах в присутствии 50% плазмы человека изучали следующим образом. Замороженную донорскую плазму человека размораживали 37°C, центрифугировали при 14 500 g в течение 10 минут и фильтровали через 0,45-μм ацетатцеллюлозный фильтр с орошением. Препарат липосом, нагруженных СРТ-11, был стерилизован пропусканием через 0,2-μм свободный от сурфактантов ацетатцеллюлозный (SFCA) стерильный фильтр с орошением. 0,5 мл липосом смешивали с 0,5 мл плазмы в стерильных 1,5 мл Eppendorf пробирках, их закрывали и инкубировали при 37°C, поместив на вращающуюся подставку, в течение 24 часов. Образец слепого опыта содержал 0,5 мл стерильного MES-Dextrose вместо липосом. Липосомы выделяли гель-хроматографией на гранулированном 2% геле поперечно-сшитой агарозы (Сефароза Cl-2B, Pharmacia; 10 мл объема слоя), используя 144 мМ NaCl, 5 мМ HEPES-Na, рН 7,4 буфер (HBS-5). Липосомы появлялись во фракции свободного объема, в то время как белки плазмы и выделившееся лекарственное средство (если таковое было) удерживались гелем. Липосомные фракции исследовали на содержание СРТ-11 и фосфолипидов и определяли соотношение (окончательное значение) лекарственное средство/фосфолипиды. Значения для образцов слепого опыта (только плазма) вычитали из значений для образцов, содержащих липосомы. Долю лекарственного средства, остающегося в липосомах после инкубации, определяли делением окончательного значения соотношения лекарственное средство/липиды к исходному соотношению лекарственное средство/липиды (соотношение лекарственное средство/липиды до инкубации с плазмой). Данные по загрузке и выделению объединены и приведены в таблице 5.
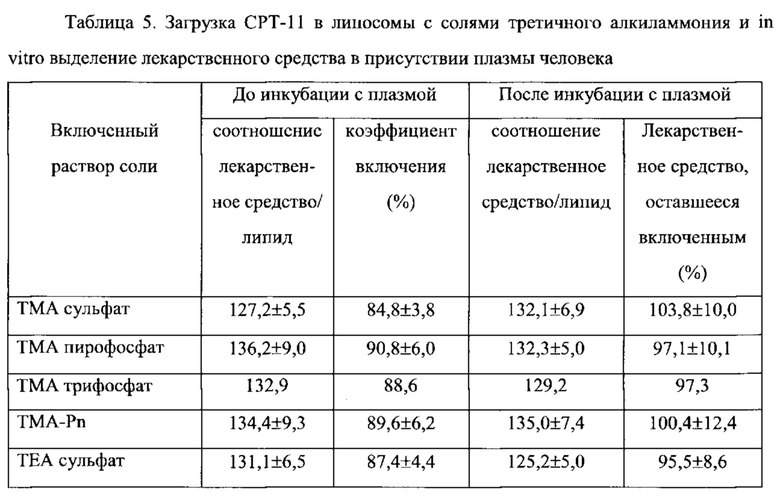
Пример 8. In vivo стабильность липосом, нагруженных СРТ-11 с использованием пирофосфатов, трифосфатов, полифосфатов, цитратов и сульфатов триалкиламмония
Хотя липосомы с кампотецином могут демонстрировать приемлемую просачиваемость лекарственного средства в плазму крови in vitro, это лекарственное средство может более быстро просачиваться в кровоток in vivo. Поэтому ряд составов СРТ-11 липосом проверили на стабильность лекарственного средства в них при помещении в кровоток in vivo с помощью исследования на мышах методом намеченных временных точек.
Получали липосомы и нагружали их СРТ-11, как описано в примере 6 со следующими модификациями. Вместо использования PEG-DSPE от Shearwater Polymers, был использован аналогичный PEG-DSPE от Avanti Polar Lipids. Чтобы произвести количественное определение липидной основы липосом в пробах крови/ткани, использовали неизменную радиоактивную метку [3Н]-холестерил-гексадециловый эфир ([3HJ-CHE; Amersham, USA), ее добавили к раствору липидов в хлороформе в количестве 0,25 мКи/ммоль фосфолипидов. Липидный раствор распределяли в Pyrex пробирки по 12 мг DSPC/пробирку и создавали липидные пленки с помощью роторного испарителя и сушки в вакууме. Липидные пленки гидратировали в 0,7 мл в градиент-образующих растворах солей замещенного аммония. Концентрацию липидов в липосомах с включенными фосфатсодержащими солями определяли подсчитывая сцинтилляцию радиоактивности. Препараты без включенных фосфатсодержащих солей также исследовались на фосфолипиды без экстракции, описанной в примере 70, и использовались как стандарты радиоактивности липидов. Часть из смесей липосом и лекарств, приготовленных для загрузки, сохраняли и исследовали для того, чтобы подтвердить изначальное соотношение до загрузки добавляемого СРТ-11 к липидам липосомы, т.е. в момент перед загрузкой (исходное соотношение). Усредненное значение объема и распределение стандартного отклонения размера липосом определяли методом квазиупругого светорассеяния (QELS), используя модель Гаусса. Свойства этих липосом объединены и показаны в таблице 6.
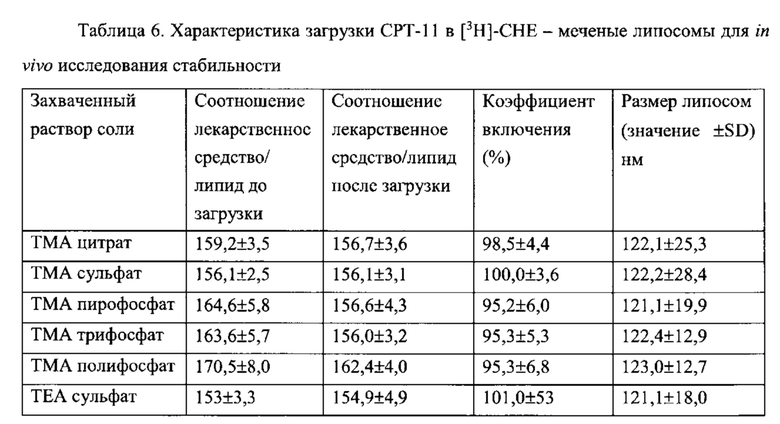
Самкам CD-мышей (Charles River) 6-недельного возраста в хвостовую вену инъецировали эти липосомные СРТ-11 составы в дозе 10 мг/кг (0,2 мг СРТ-11 на мышь) в двух повторностях. Через восемь часов мышей анестезировали и обескровливали путем прямого прокола сердца. Кровь собирали в гепаринизированные шприцы (10-20 ил 1000 Ед/мл гепарина USP) и переносили в две взвешенные пробирки, содержащие 0,4 мл забуференного фосфатом физиологического раствора (PBS), содержащего 0,04% ЭДТА (Gibco BRL), хранящегося на льду. Эти пробирки взвешивали, чтобы определить массу образцов крови, кровяные клетки отделяли центрифугированием при 3000 g в течение 5 минут и супернатанты, содержащие PBS-разбавленную плазму, хранили для использования в анализах липидов и лекарственного средства в липосомах. СРТ-11 количественно определяли путем флуорометрического анализа (пример 71). Липосомные липиды количественно определяли, подсчитывая корректируемую гашением сцинтилляцию радиоактивности. Стандартную радиоактивность липосом и фосфолипидов обеспечивали параллельно с образцами плазмы. Долю лекарственного средства, которая осталась невключенной, подсчитывали делением соотношения лекарственного средства/радиоактивности в образцах плазмы к соотношению лекарственного средства/радиоактивности в инъецируемых липосомах. Вследствие быстрого удаления свободного СРТ-11 из крови (см. пример 69) и известной стабильности [3Н]-СНЕ в зависимости от замены липидов, результаты анализов считаются показательными в отношении содержания в крови липосомального СРТ-11 и липидов. Долю вводимой дозы липидов (% I.D.), остающейся в кровотоке, вычисляли, принимая за 100% инъецируемого болюса, вводимого в кровоток; объем крови принимали за 6,3% массы тела мыши, и гематокрит - 45%. Результаты собраны и показаны в таблице 7.
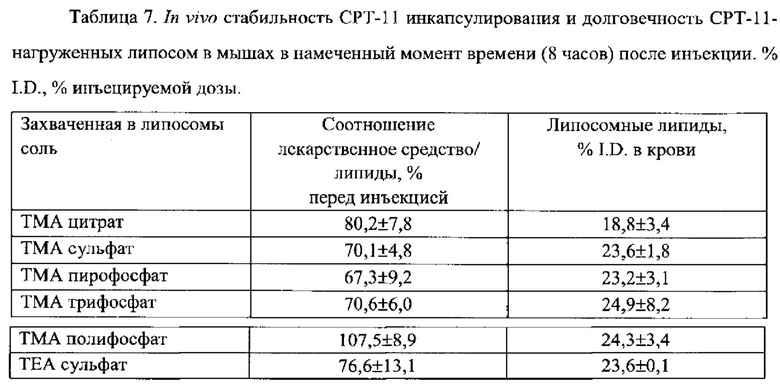
Все препараты показали уровень инкапсулирования лекарственного средства, составляющий 70-80% после 8 часов в крови in vivo, по отношению к уровню перед инъекцией, в то время как липосомы, содержащие полифосфат, оказались самыми стабильными (степень инкапсулирования оставалась на уровне почти 100%).
Пример 9. Фармакокинетические характеристики в крови СРТ-11 липосом, полученных при использовании полифосфата триэтиламмония.
Состав липосомального СРТ-11 с использованием полифосфата триэтиламмония готовили как выделено в примере 3. Липиды - DSPC, холестерин и N-(метокси-поли(этиленгликоль) (М.м. 2000)-оксикарбонил) - DSPE (PEG-DSPE) (все от Avanti Polar Lipids, Inc.) - были объединены в виде сухих порошков при мольном соотношении 3:2:0,015 и растворены в 100% этаноле (USP качества, приблизительно 0,15 мл/100 мг липидов) при 62-65°C. Для изучения фармакокинетики 3Н-холестерил-гексадециловый эфир (3Н-СНЕ, получен от Amersham Pharmacia) был добавлен к липидам в количестве 0,5 мКи фосфолипидов как неизмененного радиоактивной метки липидов. Водный раствор TEA-Pn (0,5 М триэтиламмоний, рН 5,7-6,2) готовили как в примере 4. Раствор ТЕА-Pn (10-кратный по объему от добавленного этанола) смешали с липидным раствором при 60-65°C и перемешивали при этой температуре до тех пор, пока не образовалась гомогенная молочная суспензия мультиламеллярных пузырьков. Эту суспензию экструдировали 15 раз через 2 объединенных поликарбонатных фильтра с протравленными желобами (Corning Nuclepore) с размером пор 100 нм с использованием экструдера, работающего при давлении аргона (Lipex Biomembranes), при 60-65°C и получившиеся в результате одноламеллярные липосомы быстро охлаждали на льду и затем давали липосом достичь уровня комнатной температуры. Этанол и невключенные полифосфатные соли удаляли гель-хроматографией на колонке с Sepharose CL-4B с элюцией MES-Dextrose буфером (5 мМ MES, 50 г/л декстрозы, рН доводили NaOH до 6,5).
Основной раствор СРТ-11 (Иринотекан гидрохлорид), содержащий 20 мг/мл Иринотекана основания в воде, добавили к липосомам в концентрации лекарственное средство/липиды 150-200 мг/мл фосфолипидов, и эту смесь инкубировали при осторожном встряхивании в течение 45-60 минут при 60-62°C. Инкубированную смесь быстро охлаждали и инкубировали в течение 10 минут при 0°C, затем давали достичь комнатной температуры. 1/20 объема 2,88 М NaCl добавили для достижения физиологического значения ионной силы, чтобы улучшить перемещение связанного с мембранами СРТ-11 (в противоположность лекарству, инкапсулированному внутрь липосомного внутреннего пространства). Неинкапсулированное лекарственное средство удаляли гель-хроматографией на колонке с Сефадексом G-25 или G-75 (Amersham Pharmacia) с элюцией HBS-6,5 буфером (5 мМ 2-(4-2-гидроксиэтил)-пиперазино)-этилсульфокислота (HEPES), 144 мМ NaCl, рН 6,5). Липосомные фракции, элюированные в свободном объеме, объединяли, стерилизовали фильтрацией при размере пор 0,2 мк и хранили при 4-6°C до использования. Липосомы были исследованы в отношении липидной концентрации, концентрации лекарственного средства и размера частиц как в примере 7. Эти липосомы имели средний размер 108 нм и содержание СРТ-11, соответствующее 139±18 мг СРТ-11 основания на ммоль фосфолипидов.
Долговечность липосомных липидов и лекарственное средство в липосомах в крови и характеристики выделения лекарственного средства из липосом in vivo изучали на самках Spraque-Dawley крыс (190-210 г) с введенными в центральную вену катетерами. Крысам инъецировали 0,2-0,3 мл болюсных липосом с 3Н-СНЕ-меченным Иринотеканом (0,05 ммоль фосфолипидов или 7-8 мг СРТ-11 на кг массы тела). Отбирали пробы крови в различные моменты времени после инъекции с помощью обработанного гепарином шприца. Отобранный объем крови восполняли фосфатным физиологическим солевым раствором. Пробы крови разбавляли 0,3 мл ледяным PBS, содержащим 0,04% ЭДТА, взвешивали, отделяли клетки крови центрифугированием. Надосадочные супернатанты собирали и исследовали на СРТ-11, используя флуорометрический метод примера 71, а для липосом измеряли радиоактивную сцинтилляцию липидной метки, используя общеизвестные методы. Препараты липосом с известной концентрацией лекарственного средства и 3Н-СНЕ-липид использовали как стандарты. Стандарты радиоактивности содержат равные количества разбавленной плазмы крыс для учета гашения. Количество СРТ-11 и липосомных липидов в крови вычисляли, принимая объем крови в мл как 6,5% массы тела в граммах и гематокрит 40%. Общее количество липидов и лекарственного средства в крови выражали в % инъецируемой дозы (% ID., % ID) и наносили против постинъекционного временного периода. Долю лекарственного средства, оставшегося в липосомах, вычисляли делением соотношения лекарственное средство/липид в пробах крови на соотношение лекарственное средство/липид в инъецируемых липосомах (принято за 100%). Поскольку графики в целом показали хорошее соответствие с моноэкспоненциальными кинетическими характеристиками (линейность на полулогарифмической шкале), время полужизни в крови для лекарственного средства, липида и просачивания лекарственного средства из липосом вычисляли, исходя из наилучшего соответствия данных уравнению моноэкспоненциального убывания, используя TREND-опцию программного обеспечения Microcoft EXEL (Microsoft Corp., USA). Результаты показаны на Фиг. 1. Исходя из наилучшего соответствия параметров, время полужизни в крови для липида и для лекарственного средства было 16,4 часа и 6,61 часа соответственно. При этих условиях свободный СРТ-11 удаляется из кровотока очень быстро (см. пример 69).
Соотношение лекарственное средство/липид в крови обнаруживает двухфазный характер выделения СРТ-11 из липосом (Фиг. 2). Спустя первые 24 часа характер выделения лекарственного средства был линейным во времени (R=0,992), показывая очевидность нулевого порядка кинетики выделения. Только после того, как выделилось примерно 75% лекарственного средства на 24-ом часу, последующее выделение носило нелинейный характер. В течение 24 часов липосомы выделяли лекарственное средство с постоянной скоростью примерно 3,6% от начальной загрузки в час. Так что 50% лекарственного средства просочилось в период приблизительно 14 часов. Нулевой порядок характера выделения лекарственного средства является привлекательным свойством составов для непрерывного выделения, так как скорость выделения лекарственного средства остается постоянной в течение периода времени.
Пример 10. Антиопухолевый эффект СРТ-11 липосом, полученных с исопльзованием полифосфата триэтиламмония, в отношении рака молочной железы, привитого бесшерстным мышам.
Антиопухолевое действие СРТ-11 липосом изучали на модели карциномы молочной железы ВТ-474 человека, эстрогензависимой дуктальной аденокарциномы, которая обуславливает сверхэкспрессию C-ErbB2 (HER2) рецептора. ВТ-474 клетки получали из American Type Culture Collection (Rockville, MD). ВТ-474 подлиния с более высокой скоростью роста была получена из быстрорастущего опухолевого узелка ксенотрансплантата, выращенного как описано ниже. Клетки культивировали in vitro в RPMI-1460 среде с 10% фетальной сыворотки теленка, 0,1 мг/мл сульфата стрептомицина и 100 Ед/мл пенициллина G в Т-150 колбах, и разводили (1:3) каждую неделю. Самкам мышей NCR nu/nu (возраст 4-6 недель; 4-6 недель; Taconic Farms) подкожно имплантировали (в основание хвоста) гранулы, непрерывно в течение 60 дней выделяющие 0,72 мг 17β-эстрадиол (Innovative Research of America, Inc.) и через два дня инокулировали подкожно в зону верхней части спины 0,1 мл суспензии, содержащей 2×107 ВТ-474 клеток в среде для роста клеток. Рост опухоли проверяли пальпированием и с помощью измерений опухоли циркулем по длине (больший размер) и ширине (меньший размер) дважды в неделю. Размеры опухоли определяли дважды в неделю по измерениям циркулем, используя формулу (Geran, R.I., et al, 1972 Cancer Chemother. Per. 3:1-88): Объем опухоли = [(длина) × (ширина)2]/2
Липосомальный СРТ-11 получали как описано в примере 8 (соотношение лекарственное средство/фосфолипид 192 мг/ммоль; средний размер липосомы 86,8 нм). Свободный и липосомальный СРТ-11 разбавили MES-декстрозной средой до 5 мг/мл СРТ-11 основания. Животным инъецировали в хвостовую вену свободный СРТ-11, липосомальный СРТ-11 или среду только на 14, 18, 21 и 25-ый дни после инокуляции опухоли. Составы, содержащие лекарственное средство, давали в дозе 50 мг СРТ-11/кг на инъекцию, что есть среднее из доз СРТ-11, указываемых в литературе по исследованию опухолей на моделях грызунов.
Для того, чтобы оценить токсичность в зависимости от лечения, животных взвешивали дважды в неделю. Наблюдения делали до 60-ого дня после инокуляции, в течение этого времени гранула, добавляющая эстроген, исчерпывалась. Средние объемы опухолей в группах отмечали на графиках и через некоторое время сравнивали. Как показано на Фиг. З, пока свободный СРТ-11 уменьшал скорость роста опухоли, в группе, которая получала лечение липосомами, опухоли резко регрессировали. В то время как на 36-ой день в контрольной группе опухоли достигли максимально возможного размера, в среднем соответствующего 3500 мм3, а на 46,-ой день в группе, леченной свободным лекарственным средством, опухоли были около 1000 мм3 в среднем, а в тот же самый момент времени ни у одного из животных в группе, которая получала лечение липосомами, не было пальпируемых опухолей.
Токсичность в зависимости от лечения оценивалась по динамике веса тела животного (Фиг. 4). Ни в одной группе не обнаруживалось никакой значительной токсичности. Вес животных в контрольной группе непрерывно рос. Небольшое снижение среднего веса животных, получавших липосомальный СРТ-11, было около 3,3% - на последний лень лечения. Эта потеря веса была обратимой, и вес животных вернулся к ожидаемой величине. Это снижение значения веса тела не было статистически значимым по Student's t-тесту по сравнению с весом до лечения (р=0,274). Таким образом, все виды лечения допустимы без значительной токсичности.
Так, липосомы с СРТ-11, полученные загрузкой лекарственного средства через предварительно захваченную имеющую стерические затруднения замещенную соль захваченного аммония (триэтиламмоний) полианионного, биодеградируемого полимера (полифосфата), показали повышенную долговечность в крови, свойство непрерывного выделения и повышенную антиопухолевую активность в изученных моделях опухолей без предполагаемого увеличения токсичности.
Пример 11. Сравнительные оценки содержащих СРТ-11 липосом, полученных с использованием предварительно захваченных солей триэтиламмония: влияние размера липосом, соотношения лекарственное средство/липид и природы ранее включенного аниона.
Готовили два прототипа составов СРТ-11-нагруженных липосом, в одном использовались липосомы с предварительно захваченным ТЕА-Pn, а в другом - с предварительно захваченным TEA-SOS. Получение этих липосом включало следующие технологические стадии.
1) Сочетание липидов сорастворением в этаноле
Матричная липидная композиция состояла из 1,2-дистеароил-SN-фосфатидилхолина (DSPC) (М.м. 790) 3 мольные части (59,8% моль.); холестерина (Chol) (М.м. 387) 2 мольные части (39,9% моль.) и М-(омега-метокси-поли(этиленгликоль)-оксикарбонил)-1,2-дистеароилфосфатидил-этаноламина (М.м. 2787) (PEG-DSPE) 0,015 мольных частей (приблизит.0,3% моль.). DSPC и PEG-DSPE были закуплены у Avanti Polar Lipids, Birmingham, Alabama. Холестерин (высокой степени чистоты) был закуплен у Calbiochem. Сухие липиды взвешивали с точностью ±0,1 мг в контейнере из боросиликатного стекла и соединяли с абсолютным этанолом в соотношении, подходящем для стадии диспергирования липидов, приведенной ниже. Из-за высокой температуры перехода DSPC (55°C) растворение обычно проводили при 55-60°C до тех пор, пока не получали прозрачного раствора.
2) Получение ТЕА-Pn и TEA-SOS растворов. Полифосфат натрия (n=13-18) был получен из Sigma Chemical Co., p/n P 8510. Сахарозооктасульфат натрия был закуплен у Toronto Research Chemicals, Toronto, Canada, p/n S699020. Соли взвешивали и растворяли в воде, чтобы получить 1,2-2,5 н растворы. Анионообменные смолы Dowex 50Wx8-200 или Dowex HCR-W2 в H+-форме (поставляются Sigma) использовалось для того, чтобы перевести натриевые соли в свободные кислоты. Перед первым использованием смолы промывали, перемешивая их с 3 объемами следующих растворов с последующим декантированием: (1) 1,0-1,2 М водный раствор HCl - 2 раза; (2) Вода - 2 раза; (3) 1,0-1,2 М водный раствор NaOH - 2 раза; (4) Вода - 2 раза; (5) 1,0-1,2 М водный раствор HCl - 2 раза. Суспензию промытой смолы в воде загружали в хроматографическую колонку подходящего размера, чтобы получить, по меньшей мере, 8 мл установленной смолы на каждый мл растворов натриевых солей. Затем смолу уравновешивали, пропустив 2 объема колонки 3,0-3,6 М водной HCl, затем - 5 объемов колонки воды, пока электропроводность элюата не опускалась ниже 1 микро-S. После использования колонки регенерировали, последовательно пропуская: 1-1,2 М HCl - 3 колоночных объема; 3,0-3,6 М HCl - 2 колоночных объема; вода - по меньшей мере, 5 колоночных объемов, или пока электропроводность элюата не станет ниже 1 μS, и хранили под водой, отфильтрованной на 0,2-мкм фильтре, при комнатной температуре. Растворы Pn и SOS-натриевых солей наносили на окрашенную поверхность колонки (приблизительно 1 мл на каждые 4 мл объема загруженной смолы) и обеспечивали протекание под действием силы тяжести со скоростью около 1-2 мл/мин при объеме слоя смолы 75-150 мл. Элюцию с колонки осуществляли с помощью воды. В полученном элюате исследовали электропроводность. Фракции с электропроводностью 10 mS и выше собирали. Если требовались более концентрированные растворы поликислот, сбор фракции начинали при 20-50 mS, но за счет отчасти большей потери градиент-образующей соли. В случае полифосфорной кислоты собранный раствор хранят охлажденным (0-4°C) до стадии титрования амина из-за гидролитической нестабильности фосфодиэфирных связей при низких значениях рН. Собранные элюаты должны иметь рН менее 0,7 (обычно около 0,4) и электропроводность около 120-200 mS. Обычно стадию титрования амина проводят сразу из-за стабильности полифосфата при низких значениях рН. ВЭЖХ-очищенный триэтиламин (99,5 + % чистоты) от Fisher, p/n 04884, был использован для титрования кислых растворов, полученных после ионного обмена. Нормальность чистого TEA определялась потенциометрическим титрованием. 0,100-мл аликвоты TEA (0,100 мл) отбирали в 20 мл воды в трех повторностях. Эти аликвоты титровали 0,1 н стандартным раствором HCl до конечной точки рН 5,5-6,0 (стеклянный электрод). Вычисленная нормальность (7,07 н) была близка к теоретической величине 7,17 н. Отмеренный объем раствора полифосфорной кислоты (Pn) или сахарозооктасерной кислоты (SOS) оттитровывали чистым TEA под контролем рН (стеклянный электрод). Для того, чтобы амин диспергировался, требовалось тщательное перемешивание. Титрование до конечной точки достигало значения рН 5,6-6,2. Объем добавляемого TEA тщательно регистрировали. Объем оттитрованного раствора измеряли, а концентрацию TEA вычислили, исходя из объема добавленного TEA и нормальности. Воду добавляли по мере необходимости, чтобы довести концентрацию TEA до требуемого значения 0,55±0,05 н или 0,65±0,03 н, как показано ниже. Количество остаточного натрия в полученных растворах ТЕА-Pn или TEA-SOS определяли потенциометрически, используя натрий-селективный стеклянный электрод (Corning). Один мл раствора разбавляли 19 мл воды, и концентрацию натрия определяли, используя инкрементный метод согласно инструкции изготовителя электродов. Количество остаточного натрия было меньше, чем 1 мМ, обычно менее, чем 0,5 мМ. Полученные ТЕА-Pn или TEA-SOS растворы пропускали под давлением через стерильный ацетатцеллюлозный фильтр. Измеряли конечные значения рН и осмолярности растворов и регистрировали эти значения. Использовались рН-каломель в микрокомбинации с полностью стеклянным электродом для измерений рН и осмометр, учитывающий давление пара и точку росы - для измерения осмолярности. До использования растворы хранили в охлажденном виде.
3) Приготовление дисперсии липидов в градиент-образующем буфере путем смешения этанолсодержащего раствора липидов с градиент-образующим буфером.
Липиды были диспергированы в градиент-образующем растворе соли с использованием метода этанольного смешения. Все стадии выполнялись при 60-65°С. Липиды растворяли в 100% этаноле USP при концентрации около 0,5-0,6 М DSPC в химически инертной стеклянной колбе грушевидной формы или пробирке. Градиент-образующий раствор соли (ТЕА-Pn или TEA-SOS) был предварительно подогрет до 60-65°С и немедленно добавлен к этанолсодержащему липидному раствору, компоненты таза тщательно только перемешивались при вращении и/или при вихревом движении. Конечное количество этанола было около 10% об. Для препаратов с содержанием фосфолипидов более 0,1 ммоль, получившуюся в результате суспензию помещали в роторный испаритель при 60-65°С и создавали вакуум при вращении до тех пор, пока отгонка этанола не прекращалась, что показывало прекращение пенообразования. Если содержание фосфолипидов соответствовало 0,1 ммоль или менее, этанол не удалялся из липидной дисперсии на этой стадии. Полученные липидные суспензии хранили при 60-65°С и использовали точно перед стадией экструзии.
4) Последовательная экструзия липидной дисперсии через мембраны с определенным размером пор
Для липидных суспензий объемом до 1 мл использовали экструдер возвратнопоступательного действия, работающий при ручном управлении, поставляемый Avanti Polar Lipids. Экструдер снабжен 19-мм фильтровальными мембранами с протравленными желобами и термостатирован в металлическом нагревающем блоке. Для объемов от 1 до 10 мл использовался термостатированный, управляемый сжатым газом, однонаправленный проточный экструдер от Lipex Biomembranes. Экструдер загружен 25 мм фильтровальными мембранами. Суспензию липидов быстро экструдировали при 60-65°С, используя ручную загрузку или давление газа, соответственно, через серию объединенных по 2 поликарбонатных мембранных фильтров (фильтры от Corning-Nuclepore and Osmonics Corp. были одинаково пригодны), имеющих номинальный размер пор 100 нм, 80 нм или 50 нм. В тех случаях, если влияние размера липосом представляло интерес, экструзию прекращали в точке 100 нм, 80 нм или 50 нм. Точный тип используемых фильтров и количество стадий экструзии показано ниже для каждого опыта. Экструдированные липосомы сохраняли при 60-65°С около 15 минут и быстро охлаждали до 2-4° на ледяной бане. После примерно 15 минутного выдерживания на ледяной бане липосомам давали нагреться до комнатной температуры.
5) Удаление экстралипосомального градиент-образующего буфера и перенос липосом в буфер загрузки лекарственного средства. Неинкапсулированные градиент-образующие соли удаляли и липосомы перенесли в буфер для загрузки лекарственного средства с помощью хроматографии исключения по размеру (sige-exclusion chromatography - SEC). Фильтрация при тангенциальном направлении потока, диализ через полые волокна, другие приемы очистки могут использоваться в расширенном масштабировании. Эти процедуры выгодно использовать для того, чтобы обеспечить полное удаление электролипосомальных полианионов обработкой липосом анионообменной смолой (например, Dowex-1 или Dowex-2 поперечносшитый четвертичным аммонием полистирол). Буфер для загрузки лекарственного средства содержал 50 г/л безводной декстрозы USP и 5 мМ проверенного на тканевых культурах HEPES в воде с доведенным с помощью NaOH рН 6,5. Буфер был профильтрован под вакуумом через 0,2-микронный нейлоновый фильтр (Whatman). Экструдированные липосомы были подвергнуты хроматографии на колонке с Сефарозой CL-4B (Pharmacia) и элюированы буфером для загрузки лекарственного средства. Липосомы появлялись во фракции свободного пространства, были собраны с учетом мутности элюата, в объеме, примерно вдвое превышающем нанесенный объем. В элюированных липосомах исследовали концентрации фосфолипидов согласно примеру 70, размер частиц - по QELS и хранили их при 4-6°С.
6) Инкубирование липосом с лекарственным средством. Исходный раствор РСТ-11 (Иринотекан гидрохлорид) готовили непосредственно перед смешиванием с липосомами растворением гидрохлорида Иринотекана в виде с достижением концентрации 20 мг/мл Иринотекана основания. рН раствора был между 4,0 и 5,0. Раствор лекарственного средства фильтровали через 0,2-микронный полиэфирсульфоновый (PES) стерильный фильтр под давлением. Аликвоты липосом в буфере для загрузки лекарственного средства, полученных на стадии 5, смешивали при комнатной температуре с исходным раствором Иринотекана с достижением исходного соотношения лекарственное средство/липид в диапазоне 0,15-0,55 г лекарственного средства на ммоль фосфолипида липосомы. Конкретные значения исходных соотношений лекарственное средство/липид показаны ниже, где это уместно. рН смесей был доведен 1 М NaOH до 6,5, смеси в стеклянных емкостях инкубировали при термостатировании в водяной бане при 58-62°С при легком перемешивании в течение 30-45 минут, быстро охлаждали в ледяной бане (0-2°С) и оставляли при этой температуре на 15 минут. Затем липосомам давали нагреться до комнатной температуры для следующей стадии (удаление неинкапсулированного лекарственного средства и перенос в буфер для хранения). Эта стадия привела к достижению значения коэффициента инкапсулирования более чем 95%, как правило 98-100%, во всем диапазоне исследованных соотношений лекарственное средство/липид.
7) Удаление неинкапсулированного СРТ-11, перенос липосом в буфер для хранения, финальная фильтрация и хранение. Неинкапсулированное лекарственное средство было удалено, а липосомы перенесли в буфер для хранения, используя хроматографию с исключением по размеру. Буфер для хранения содержал 20 мМ HEPES, 135 мМ NaCl, рН 6,5 (доведено NaOH) в виде, перед использованием он был отфильтрован через 0,2-микронный вакуум-фильтр. Сразу после этого проводили гель-хроматографию на Сефадексе G-75 (Amersham Pharmacia Biotech) согласно описанному выше - стадия 2: СРТ-11 липосомы, элюированные с колонки (фракция свободного объема), исследовали на фосфолипиды в липосомах и СРТ-11 (путем спектрофотометрии) см. примеры 70 и 71) и значения размера частиц с замеренным объемом - по QELS. Если это было необходимо, концентрацию лекарственного средства доводили до значения, входящего в интервал 2,0-4,0 мг/мл. Липосомы фильтровали через 0,2-микронные полиэфирсульфонные стерильные фильтры и асептически распределяли в стерильные полипропиленовые емкости (Corning Cryo-Vials) или 4-мл стеклянные пробирки с боросиликатными прикручивающимися крышечками примерно на 70% по объему пробирки. Пробирки асептически закрывали (в воздухе), маркировали и хранили при 4-6°С.
Пример 12. Влияние соотношения лекарственное средство/липид на коэффициент включения лекарственного средства и in vivo удерживание лекарственного средства TEA-Pn-содержащими липосомами
Липосомы с захваченным водным 0,65 н раствором ТЕА-Pn, рН 6,1, осмолярностью 531 ммоль/кг, готовили после процедур примера 11. Липидную дисперсию экструдировали десять раз через два объединенных поликарбонатных фильтра с размером пор 100 нм. Липосомная липидная матрица включала также [3Н]-СНЕ при 0,5 мКи/ммоль фосфолипида. Размер липосом перед загрузкой лекарственного средства был 98,5±34,3 нм. Липосомы загружали при исходных соотношениях лекарственное средство-фосфолипид, равных 200, 300, 400 и 500 мг СРТ-11/ммоль фосфолипида. Количества лекарственного средства и фосфолипидов в липосомах определяли спектрофотометрическим способом в соответствии с примером 71 и анализом с помощью фосфомолибдата голубого фосфолипидной экстракции - расщепления по примеру 72, соответственно.
Чтобы оценить скорость выделения лекарственного средства in vivo, способ примера 8 был следующим.
Липосомы вводили через хвостовую вену 6-недельным самкам Swiss Webster мышей (масса тела животного 18-22 г) при дозе инъекции 5 мг СРТ-11/кг. Спустя 8 и 24 часа после инъекции, мыши были подвергнуты анестезии (в каждой группе по 3 животных) и обескровлены прямым проколом сердца. Кровь смешивали с 0,4 мл ледяного 0,04% EDTA в PBS, кровяные клетки отделяли центрифугированием, концентрацию СРТ-11 в плазме измеряли спектрофлуорометрическим способом как описано в примере 71. Липиды определяли измерением количества [3Н]-СНЕ, учитывая корректируемую гашением сцинтилляцию жидкости, а количество лекарственного средства, оставшегося в липосомах, вычисляли, деля определенное соотношение лекарственное средство/липид на соотношение лекарственное средство/липид в инъецируемых липосомах. Вследствие быстрого удаления свободного СРТ-11 из крови, приводящего к снижению его уровня в крови, принимается, что все исследуемое лекарственное средство было в липосомальной форме.
Результаты показаны в таблице 8. Разница между величинами, характеризующими удерживание лекарственного средства в разных группах, статистически незначительна. В результате проведенных исследований был сделан вывод, что повышение дозы загрузки лекарственного средства до 500 мг/ммоль не будет отрицательно влиять на включаемость лекарственного средства или in vivo стабильность. Это соотношение принимали при дальнейших анализах.
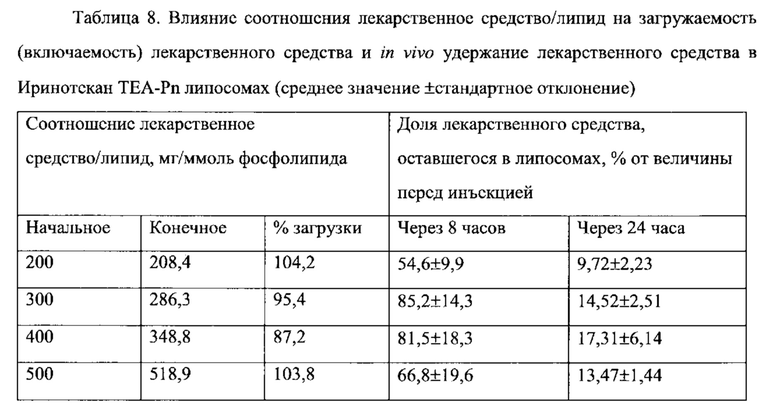
Пример 13. Коэффициент включения лекарственного средства при загружении СРТ-11 в TEA-SOS-содержащие липосомы: влияние размера липосом и in vivo удержание лекарственного средства в исследовании на мышах.
Липосомы, содержащие захваченные растворы, готовили как описано в примере 11, используя градиент-образующий раствор, имеющий 0,643 н TEA-SOS, рН 5,7, осмолярность 530 ммоль/кг. Липидную дисперсию десять раз экструдировали через два поликарбонатных фильтра с размером пор 50 нм, 80 нм или 100 нм. Липосомная липидная матрица содержала также [3Н]-СНЕ при 1,5 мКи/ммоль фосфолипидов липосомы. Размер липосом определяли динамическим светорассеянием. Липосомы нагружали СРТ-11 при начальных соотношениях лекарственное средство/фосфолипид приблизительно 550 Иринотекан/ммоль фосфолипида. Нагруженные лекарственным средством липосомы измеряли с помощью QELS и исследовали как описано в примерах 70 и 71.
Самкам Swiss Webster мышей (8-10 недельного возраста, вес в среднем 27-30 г) инъецировали в хвостовую вену эти СРТ-11 липосомные составы при дозе лекарственного средства 10 мг/кг. Мышей умерщвляли через 24 часа, собирали кровь и исследовали на СРТ-11 и липосомные липиды как в примере 11. Результаты показаны в таблице 9.
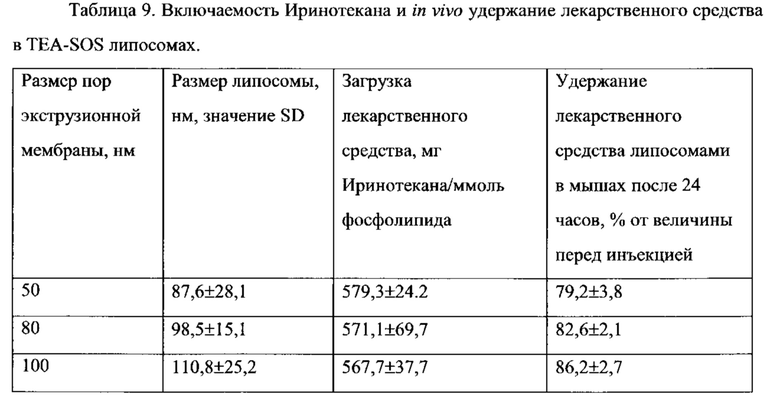
Удивительно, что липосомы с сахарозооктасульфатом триэтиламмония, неполимерным полианионным органическим гидроксилированным органическим соединением (сахаром), обеспечивали существенно лучшее (в 4-5 раз) in vivo удерживание лекарственного средства в липосомах по сравнению с аналогичными липосомами с полианионным полимером (полифосфатом).
Пример 14. Фармакокинетические характеристики в крови крыс СРТ-11-нагруженных SOS-TEA липосом
Липосомы (экструзионная мембрана с размером пор 100 нм) готовили как описано в примере 12. Эти липосомы вводили внутривенно в дозе 10 мг СРТ-11/кг двум самкам Spraque Dawley крыс (Harlan) 9-недельного возраста (массой около 200 г) с установленным в центральной вене катетером с дозой 10 мг СРТ-11/кг (17,6 μмоль фосфолипида/кг). Пробы крови отбирали в намеченные моменты времени и анализировали на содержание лекарственного средства и липосомных липидов как в примере 9. Эти данные выражали в % инъецируемой дозы липидов/мл плазмы и в % лекарственного средства, удержанного в липосоме в каждый момент времени, нанесенных на графике против постинъекционного времени, и времени полужизни для липосомного липида, а также времени полужизни для выделения лекарственного средства из липосом и вычисляли при наилучшем соответствии на основе моноэкспоненциальной кинетической модели (Фиг. 5). Время полужизни выделения лекарственного средства из СРТ-11 нагруженных TEA-SOS липосом было 56,8 часов, гораздо более продолжительное, чем у аналогичных ТЕА-Pn липосом.
Пример 15. Антиопухолевая активность свободного СРТ-11 и СРТ-11, включенного в ТЕА-Pn и TEA-SOS-содержащие липосомы, в бестимусных бесшерстных мышах с подкожными ксенотрансплантатами карциномы кишечника человека (НТ-29).
Липосомы получали как в примере 11, используя ТЕА-Pn раствор с 0,65 М TEA, рН 6,1 и осмолярностью 531 ммоль/кг или TEA-SOS раствор с 0,643 М TEA, рН 5,7, и осмолярностью 530 ммоль/кг. Экструзия включала 10 пассажей через две объединенные поликарбонатные мембраны размером пор 100 нм. Получившиеся в результате ТЕА-Pn и TEA-SOS липосомы имели размер 112,3±15,5 нм и 120,5±42,5 нм, соответственно (значение ± SD распределения размеров).
Липосомы нагружали CPT-11 при начальном соотношении лекарственное средство/фосфолипиды 500 мг/ммоль. Полученные липосомы содержали лекарственное средство в концентрации 465,6±26,5 (93% коэффициент загрузки) и 499,9±22,5 мг (100% коэффициент загрузки) СРТ-11/ммоль фосфолипида для ТЕА-Pn и TEA-SOS составов, соответственно.
НТ-29 клетки получали из American Type Culture Collection, Rockville, MD и культивировали в DMEM среде, содержащей 10% фетальной сыворотки теленка, 50 Ед/мл пенициллина G и 50 μг/мл сульфата стрептомицина, при 37°С в атмосфере 5% СО2, как рекомендовано поставщиком. NCR пи/пи гомозиготные бестимусные бесшерстные мыши-самцы (возраст - 6 недель, вес по меньшей мере - 16 г) были получены от Charles River. Мышей подкожно заражали в правый бок 0,1 мл суспензии, содержащей 5×106 клеток, суспендированных в ростовой среде без антибиотиков. Одиннадцать дней спустя животных с размером опухоли между 150 мм3 и 350 мм3 собирали в группы для лечения следующим образом. Животных классифицировали согласно размеру опухоли и поделили на 6 категорий по убывающему размеру опузоли. Было сформировано шесть групп для лечения (по 11 животных в группе) путем произвольного выбора животного, соответствующего каждой размерной категории, так что в каждой группе для лечения было равно представлены опухоли всех размеров.
Начиная с 13-го дня животные получали 4 инъекции в хвостовую вену с интервалом 4 дня следующих препаратов: 1) Контроль (HEPES-забуференный солевой раствор, рН 6,5); 2) Свободный СРТ-11 50 мг/кг, введенный в виде свежеприготовленного 5 мг/мл раствора в небуферированном физиологическом растворе; 3) ТЕА-Pn липосомальный СРТ-11 при 25 мг/кг на инъекцию; 4) ТЕА-Pn липосомальный СРТ-11 при 50 мг/кг на инъекцию; 5) TEA-SOS липосомальный СРТ-11 при 25 мг/кг на инъекцию; 6) TEA-SOS липосомальный СРТ-11 при 50 мг/кг на инъекцию. Вес животного и размер опухоли регистрировали дважды в неделю как описано в примере 10. Вес опухоли вычитали из результатов взвешивания животного, чтобы получить вес тела животного. Животных наблюдали в течение 60 дней после инокуляции опухоли. Когда вес опухоли в группе достигал 20% веса тела животного, животных группы умерщвляли. В некоторых группах наблюдали полную регрессию опухолей без признаков возобновления роста опухоли по завершении исследования. Ткани из района инокуляции опухоли от этих животных собирали и консервировали для патологического анализа на остаточные опухолевые клетки.
Результаты этого исследования показаны на Фиг. 6 и 7. Свободный СРТ-11 обладал только минорным эффектом на рост опухоли. Все липосомы обладали явно выраженным эффектом, приводящим к регрессии опухоли, за которым следует возобновление ее роста у большинства животных. Доза 50 мг/кг была более эффективной, чем 25 мг/кг и в ТЕА-Pn, и в TEA-SOS СРТ-11 липосомах. Среднее время удвоения опухоли, вычисленное по данным размеров опухоли (Фиг. 7) было: контроль - 4,2 дня; свободное лекарственное средство, 50 мг/кг - 4,8 дня; ТЕА-Pn липосомальное лекарственное средство, 25 мг/кг - 43,6 дня; ТЕА-Pn липосомальное лекарственное средство, 50 мг/кг - 47,5 дня; TEA-SOS липосомальное лекарственное средство при 25 мг/кг - 48,2 дня; TEA-SOS липосомальное лекарственное средство при 50 мг/кг - более 56 дней (время удвоения не было достигнуто). Таким образом, липосомальный СРТ-11, приготовленный согласно настоящему изобретению, был, по меньшей мере, почти в 10 раз более активным, чем свободное лекарственное средство, данное в той же дозе и по той же схеме. Неожиданно TEA-SOS СРТ-11 липосомы оказались заметно более Эффективными в уменьшении роста опухоли, чем ТЕА-Pn СРТ-11 липосомы, введенные в той же самой дозе. Так как в группах, получавших лечение свободным лекарственным средством и TES-Pn липосомальным лекарственным средством при 50 мг/кг на инъекцию, не было животных без повторного роста опухоли, в группах, получавших 25 мг/кг каждого липосомального состава, у одного животного (9,1%) не было опухоли в конце исследования, а в группе, получавшей 50 мг/кг TEA-SOS липосомального СРТ-11 состава, в конце исследования у 4 животных (36,4%) не было опухоли без признаков повторного роста опухоли.
Лекарственное средство обладало некоторой токсичностью. Животные, получившие свободный СРТ-11, но не липосомальный СРТ-11, испытывали временное недомогание (потеря резвости, живости, поза нахохленности, собранный в складки мех, меньшая подвижность) в течение около одного часа после инъекции лекарственного средства. Животные, получившие свободный СРТ-11, страдали от долговременной потери примерно 6% веса во время лечения и так и не поправлялись. Животные, получившие оба липосомалные СРТ-11 составы, испытывали кратковременную потерю веса во время между второй и третьей инъекциями, составляющую в среднем около 5% (при 25 мг/кг) или около 9% (при 50 мг/кг) того веса, который был до лечения и в конце концов возвращались к нормальному весу. Следовательно, токсичность липосомального лекарственного средства была не более токсичности свободного (нелипосомального) лекарственного средства, в то время как эффективность липосомального лекарственного средства была существенно выше. Потеря веса была обратимой, когда лечение лекарственным средством заканчивалось, и все животные возвращались к своему весу без неизлечимо-болезненного состояния или гибели из-за токсичности лекарственного средства. В конце животные достигали веса, сопутствующего регрессии опухоли. В контрольной группе (с физраствором) у животных с развившимися большими по размеру опухолями наблюдалась потеря веса, которая с очевидностью была следствием болезни, связанной с опухолью. В целом было доказано, что липосомальный состав лекарственного средства, которое было загружено в липосомы с предварительно включенным полианионизированным сахаром (сахарозо-октасульфатом), является самым эффективным, при этом менее токсичным, чем нелипосомальное лекарственное средство.
Пример 16. Токсичность свободного и липосомального СРТ-11, исследованная на мышах.
Сравнивали острую токсичность свободного СРТ-11 и СРТ-11, включенного в липосомы согласно способу настоящего изобретения, определяя максимально допустимую дозу (МТД) при осуществлении единичной внутривенной инъекции обычным (иммунокомпетентным) мышам.
Использовались следующие материалы:
1) Препарат СРТ-11 (гидрохлорид Иринотекана), содержащий гидрохлорид Иринотекана - 98,9% (ВЭЖХ) и воду 7,6%. В этом исследовании лекарственные составы готовили по принципу «как есть», без поправки на содержание влаги или содержание Иринотекана основания.
2) Липосомальный СРТ-11 (Ls-CPT-11) готовили по примеру 11 с использованием липидной матрицы DSPC 200 мольных частей, холестерола 133 мольные части, PEG-DSPE 1 мольная часть; захваченный раствор TEA-SOS, имеющий 0,65 М TEA, рН 6,4; лекарственное средство включали в липосомы в 5 мМ HEPES буфера, 5% декстрозы, рН 6,5, при 60°С в течение 30 минут при начальном соотношении лекарственное средство/липид 500 мг лекарственного средства на ммоль фосфолипида. Коэффициент включения был более 99%. Размер липосом (среднее значение размера ± стандартное отклонение по QELS): 101137 нм. Липосомы получали в среде, 20 мМ HEPES-Na, 135 мМ NaCl; рН 6,5. Концентрации лекарственного средства в инъецируемых составах были такими, которые приведены ниже в таблицах.
3) Раствор свобоного СРТ-11. Готовили исходный раствор свободного лекарственного средства, растворяя гидрохлорид Иринотекана в 5% водном растворе декстрозы с концентрацией 22 мг/мл, стерилизовали фильтрацией (размер пор 0,2 μм). Этот исходный раствор разбавили стерильным 5% раствором декстрозы перед инъекцией.
4) Животные. Самки Swiss Webster мышей, возраст 6-8 недель, от Harlan, USA. Определение МТД в целом осуществляли по протоколу, принятому согласно Исследовательской Терапевтической Программе Национального Института Рака в США. Этот протокол имеет три следующих этапа:
1 этап: стадия поиска диапазонов при коэффициенте повышения дозы 1,8. Группам, состоящим из двух животных, инъецировали в хвостовую вену повышающиеся дозы свободного или липосомального Иринотекана, начиная с дозы 60 мг/кг и продолжая это повышение с коэффициентом увеличения дозы 1,8, пока у каких-либо животных не наблюдалась летальность или предсмертное состояние (в пределах >1 дня после инъекции). При этом отмечали дозу, которая на ступень ниже той, которая вызывала гибель или предсмертное состояние.
2 этап: стадия поиска диапазонов при коэффициенте повышения дозы 1,15. Группам, состоящим из двух животных, инъецировали в хвостовую вену увеличивающиеся дозы свободного или липосомального Иринотекана, начиная с дозы, зарегистрированной в примере 1, и повышали дозу с коэффициентом повышения дозы 1,15 до тех пор, пока у каких либо животных не наступала гибель или предсмертное состояние. Дозу, предшествующую той дозе, при которой наблюдалась гибель или предсмертное состояние, регистрировали как экспериментальную МТД.
3 этап: Стадия подтверждения. Группе из 5 животных инъецировали внутривенно (хвостовая вена) свободный или липосомальный Иринотекан в экспериментальной МТД, определенной на 2-ом этапе. Животных наблюдали в течение 7 дней, дважды в неделю регистрировали массу тела и сравнивали с массой перед инъекцией. Следили в целом за состоянием здоровья животных (потеря резвости, вылизывание и приглаживание шерсти, интерес к кормлению, выделение экскрементов, состояние меха, шкурки, состояние слизистых оболочек, способность передвигаться, дыхание, положение тела, позы). Если в течение периода наблюдений не происходило гибели животных, не наблюдалось прогрессирующей потери жизненных сил или потери веса более чем на 15% от значения веса перед инъекций, делали заключение о том, что эта доза считается подтвержденной как МТД (максимально допустимая доза) при неотложной единичной инъекции. Если любой из вышеперечисленных эффектов имеет место, опыт повторяют при следующей более низкой дозе с коэффициентом 1,15.
Для того, чтобы получить дополнительные статистические данные по стадии подтверждения, отслеживали динамику изменения веса тела животных в период выживания, который длится вплоть до 11 дней после инъекции. Доза липосомального Иринотекана, превышающая 324 мг/кг, была невозможна для введения из-за ограничений по концентрации и объема введения. Результаты показаны в таблице 10.
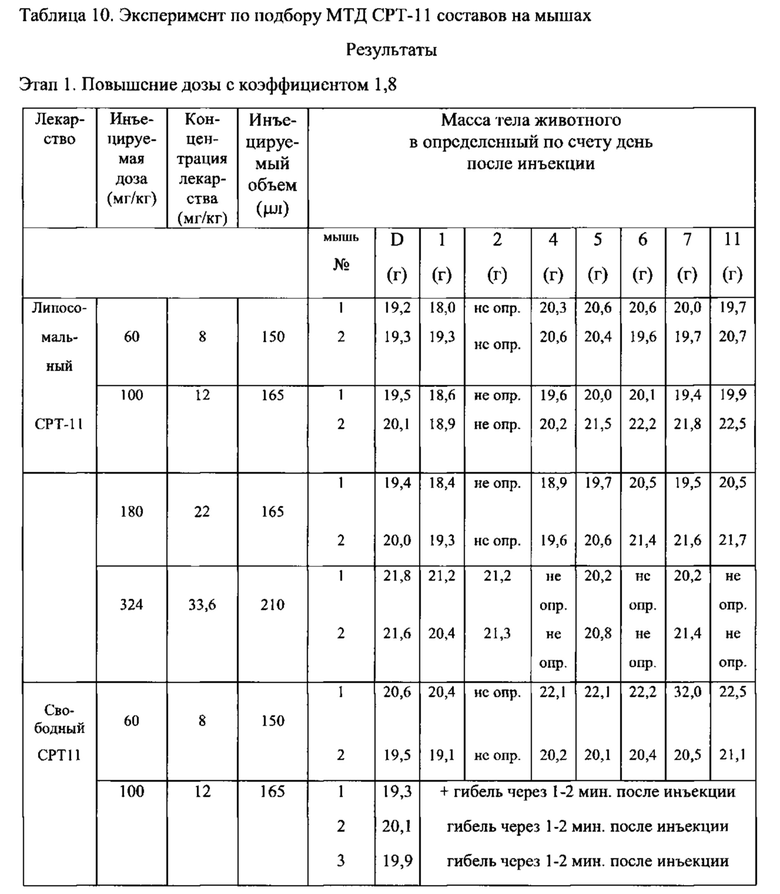
После инъекции все мыши, леченные СРТ-11, имели болезненные симптомы, дыхание с одышкой наблюдалось около 1 часа, потом - состояние снова стало нормальным.
После инъекции все мыши с липосомальным СРТ-11 выглядели нормально.
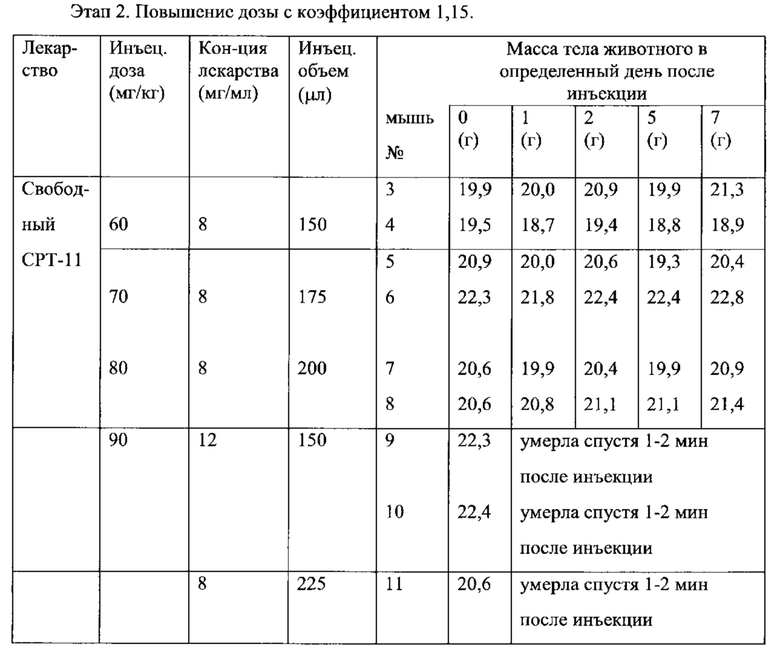
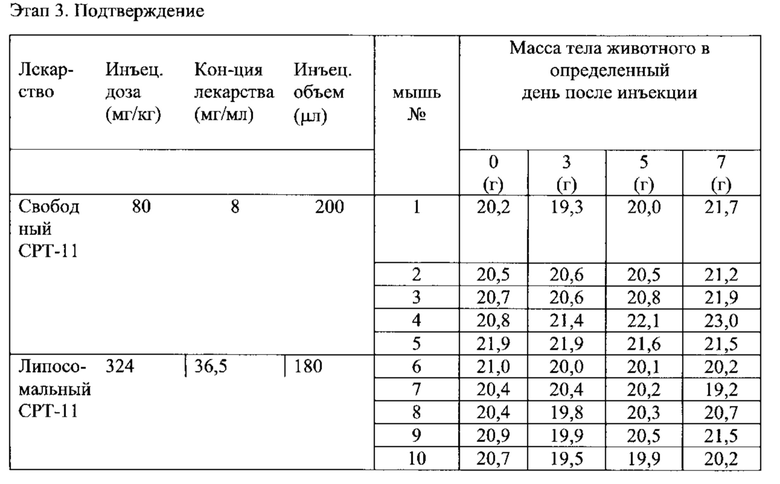
Таким образом, тогда как MTD свободного СРТ-11 была 80 мг/кг, МТД липосомального СРТ-11 не достигалась даже при более высокой введенной дозе 324 мг/кг. Следовательно, включение СРТ-11 в липосомы, согласно изобретению, понизило токсичность лекарственного средства, по меньшей мере, в 4 раза.
Пример 17. Стабильность при хранении СРТ-11-нагруженных TEA-SOS-липосомы в зависимости от просачивания (утечки) лекарственного средства.
Приготовили пять серий липосомального СРТ-11 с использованием TEA-SOS метода (пример 11) и при начальном соотношении лекарственное средство/липид 500-550 мг/ммоль фосфолипида. Липосомы готовили, используя мембранную экструзию с помощью поликарбонатной мембраны с размером пор 80 нм или 100 нм, как показано ниже в таблице. Липосомы стерильно отфильтровали через фильтр с 0,2 μм размером пор и хранили при 3,4-14,5 мг/мл СРТ-11 в 135 мМ NaCl, 20 мМ HEPES-Na, рН 6,5 (буфер хранения), при 4-8°С. После указанного срока хранения просочившееся лекарственное средство удаляли гель-хроматографией на Сефадексе G-75, используя буфер хранения в качестве элюента. Концентрации лекарственного средства и фосфолипида в липосомах до и после гель-хроматографии исследовали спектрофотометрическим методом и методом голубого фосфомолибдата и кислотного расщепления, соответственно, как описано в примерах 70 и 71. СРТ-11 липосомы, полученные в соответствии с настоящим изобретением, были очень стабильными. Утечка СРТ-11 из этих липосом во время хранения была меньше 5% через 6 месяцев (Таблица 10).
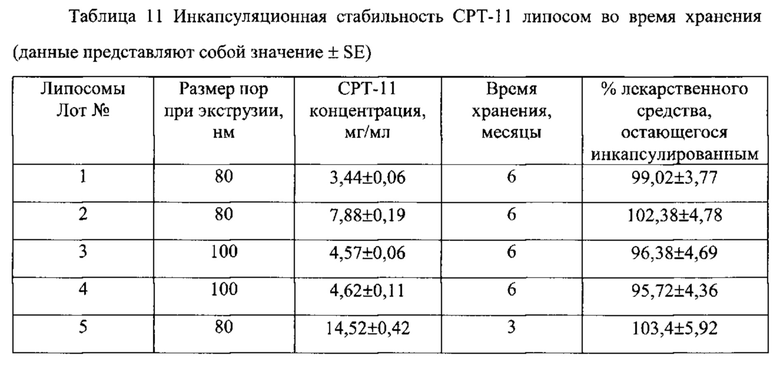
Пример 18. Липосомы, нагруженные Топотеканом
Липосомы с захваченным ТЕА-Pn раствором и TEA-SOS раствором получали как описано в примере 11. Исходный раствор солянокислого топотекана (Glaxo Smith-Kline, PA, USA) готовили непосредственно перед смешиванием с липосомами путем растворения солянокислого Топотекана в воде при 15-20 мг/мл, подсчитывая подлинное текущее содержание Топотекана ⋅ HCl. рН доводили до 3,0 с помощью 1 н HCl. Раствор лекарственного средства фильтровали под давлением через 0,2-микронный полиэфирсульфонный (PES) стерильный фильтр. Аликвоты ТЕА-Pn или TEA-SOS-содержащих липосом в буфере для загрузки лекарственного средства смешивали при комнатной температуре с исходным раствором Топотекана ⋅ HCl с достижением начального соотношения лекарственное средство/липид в диапазоне 0,15-0,45 г/ммоль липосомного фосфолипида. Предпочтительное соотношение было 0,35 г Топотекана ⋅ HCl на ммоль липосомного фосфолипида. Эти смеси в стеклянных контейнерах инкубировали в термостатированной водяной бане при 55-62°С со слабым встряхиванием в течение 30-60 минут, быстро охлаждали в ледяной бане (0-2°С) и оставили при этой температуре в течение 5-15 минут. Эта стадия обеспечила коэффициент инкапсулирования 89-90% (ТЕА-Pn градиент) или 97-100% (TEA-SOS градиент). Неинкапсулированный Топотекан удаляли и переносили липосомы в буфер для хранения, используя колоночную хроматографию исключения по размеру. Перед нанесением на колонку ионную силу препарата липосом повышали смешиванием с 1/20 объема 2,88 М водного раствора хлорида натрия, и смесь инкубировали в течение около 15 минут. Неожиданно было обнаружено, что регулирование ионной силы липосомной среды от низкого значения во время нагружения (обычно эквивалентного менее чем 20 мМ NaCl) до более высокого значения выше 20 мМ NaCl, прдпочтительно до 50 м NaCl и выше, улучшало удаление нинкапсулированного лекарственного средства и повышало стабильность Топотекан-нагруженных липосом против агрегации, возможно, за счет облегчения удаления связанного с мембранами Топотекана, в противоположность лекарству, инкапсулированному внутри липосом. Окончание процедуры следует в примере 11, стадия 7. Результаты - см. ниже в таблице 12.
Пример 19. Получение специфичных к HER2 иммунолипосомальных составов Топотекана
Иммунолипосомы с Топотеканом могут специфически интернализоваться раковыми клетками, сверхэкспрессирующими HER2 (С-ErbB-2) поверхностный рецептор онкопротеина тирозинкиназы, были получены конъюгированием липосом, нагруженных топотеканом, со специфичным к HER2 Fv фрагментом одноцепочечного антитела человека, F5, отобранного из библиотеки фагового дисплея по его высокой интернационализации в HER2-сверхэкспресирующие клетки (Poul et al. 2000, J. Molecular Biology, v. 301, p. 1149-1161). F5 представляет собой 27-KDДа белок, который связывается с экстрацеллюлярным доменом HER2 рецептора с аффинностью около 150 нм, вызывающей быструю интернационализацию (Neve, et al., Bioplys. Biochim. Res. Commun., v.280, p. 274-279). Для липосомной конъюгации можно воспользоваться следующими методами (US патент №6210707 и Nielsen et al. (2002), Biochim. Biophys. Acta, v. 1591, p. 109-118). Сначала готовили гидрофильный липополимерный конъюгат F5. С-конец аминокислотной цепочки F5 имел дополнительную терминальную цистеиновую группу (F5 Cys). F5 Cys-конструкт экспрессировали в E. coli и выделяли из бактериального лизата хроматографией на колонке с носителем, содержащим Белок А. Элюированные фракции с белком А адсорбировали на анионообменной смоле, чтобы удалить пирогены и хозяйскую ДНК, и обрабатывали тиол-восстанавливающим агентом, чтобы высвободить тиольную группу терминального цистеина. Восстановленный F5Cys был затем очищен ионообменной хроматографией с использованием SP Сефарозы быстропроточной (Amersham Pharmacia). Очищенный белок конъюгировали со взаимодействующим с тиолом липид-поли(этиленгликоль) линкером, N-(3-(N-малеимидо)пропиониламидо)-поли(оксиэтилен)-оксикарбонил)-1,2-дистеароилфосфатидил этаноламином (Mal-PEG-DSPE), производным PEG с м.м. 2000 (Фигура 4.1), поставляемый Avanti Polar Lipids, Inc., Alabama, USA. Белок и линкер инкубировали в водном буферном растворе при молярном соотношении 1:4, непрорегировавший линкер был подавлен 1 мМ цистином. Во время реакции терминальный цистеин F5 Cys ковалентно присоединяют к малеимидогруппе линкера. Полученный F5-PEG-DSPE конъюгат был водорастворимым в форме мицелл, имея явно высокий молекулярный вес (500-850 кДа) и его отделяли от непрореагировавшего протеина (около 25%) хроматографией исключения по размеру. Количество белка в очищенном конъюгате определяли УФ спектрофотометрией при 280 нм, а количество линкера исследовали спектрофотометрическим методом, идентичным тому, что использовался для обсчета фосфолипидов (см. пример 70). Очищенный F5-PEG-DSPE конъюгат был стабилен в воде, вполне иммунореактивен, и был устойчив к денатурации и потере реактивности в течение, по меньшей мере, 1 часа при 65°С, и по меньшей мере, 3-х месяцев при 37°С.
Для того, чтобы приготовить специфичный к HER2 иммунолипосомальный Топотекан, нагруженные Топотеканом липосомы примера 18 смешивали с F5-PEG-DSPE в водном солевом буфере при соотношении 15 микрограмм белка на 1 микромоль фосфолипида (около 45 F5 копий на липосому). Смесь инкубировали при 60°С в течение 40 минут, охлаждали на льду и хроматографировали на колонке с Сефарозой CL-4B (гранулы поперечносшитого 4% агарозного геля, Amersham Pharmacia), чтобы удалить остаточный мицеллярный конъюгат, неконъюгированный белок и следы экстралипосомного лекарственного средства, которые могли выделиться во время инкубирования. Липосомы с мембрано-включенным F5-PEG-DSPE элюировали 5 мМ HEPES-144 мМ NaCl-буфером, рН 7,4, собирали в свободном пространстве колонки, стерильно отфильтровали и расфасовали для хранения (4-6°С). Содержание F5, включенного в липосомы, было, как правило, более 80% добавленного конъюгата. Это определяли по SDS-PAGE липосом при количественном определении денситометрией окрашенной Coomassie F5 полосы. Концентрации лекарственного средства и липида в иммунолипосомных препаратах определяли аналогично ненацеленным липосомам. Свойства Топотекан-липосом и F5-иммунолипосом (Примеры 18 19) объединены и показаны в таблице 12.
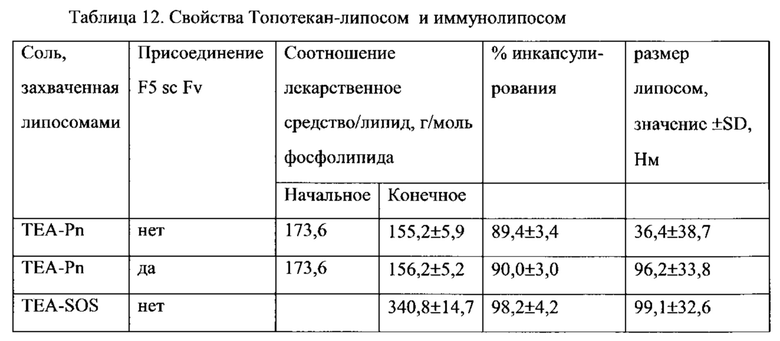
Пример 20. Влияние рН буфера загрузки и соотношения лекарственное средство/липид на характер загружения топотекана в липосомы.
Липосомы (DSPC /Chol/ PEG-DSPE, 3:2:0,015 - молярное соотношение) с захваченными 0,5 н ТЕА-Pn, рН 6,2, осмолярностью 413 ммоль/кг, готовили, используя метод этанольной инъекции (пример 18), экструдировали через два объединенных поликарбонатных фильтра с размером пор 100 нм - 5 раз и с размером пор 50 нм - 10 раз. В качестве буфера загрузки использовали 5 мМ MES, с 50 г/л декстрозы, с поддержанием различных значений рН в диапазоне 5,0-6,5. Размер липосом был 73,1±21,3 нм, определенный по QELS. Липосомы нагружали, смешивая исходный раствор Топотекана (20 мг/мл) с липосомами в буфере загрузки при начальном соотношении лекарственного средства и фосфолипида 100 мг/ммоль, инкубируя смесь при 60°С в течение 45 мин., охлаждая ее на льду и удаляя неинкапсулированное лекарственное средство на колонке с Сефадексом G-75 с элюцией 200 мМ HEPES, 135 мМ NaCl, рН 6,5. Топотекан и фосфолипид были учтены методом спектрофотометрии (примеры 70 и 71). Результаты (Таблица 13) показали, что загрузка Топотекана была почти количественной в диапазоне рН 5,5-6,5.

Влияние соотношения лекарственного средства и липида (0,15-0,45 мг/ммоль фосфолипида) на коэффициент включения также изучалось. Липосомы с включенным ТЕА-Pn (0,5 М TEA, рН 5,8, осмолярность 480 ммоль/кг) готовили как описано выше, за исключением того, что конечную стадию экструзии проводили 10 раз через два объединенных 0,08 μм поликарбонатных фильтра. Загрузку осуществляли при рН 6,5. Размер липосом по QELS был 93,1±15,1 нм. Результаты (Таблица 14) показали, что коэффициент включения (инкапсулирования) был свыше 85% во всем анализируемом диапазоне соотношений лекарственное средство/липид.
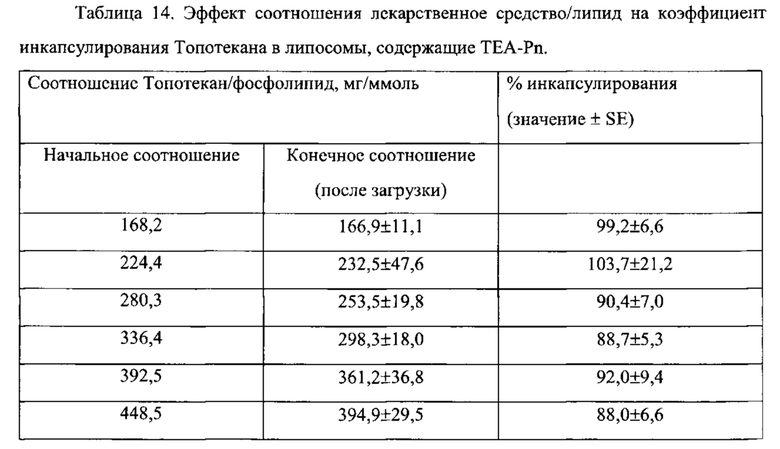
Пример 21. Стабильность Топотекан-липосом in vitro в присутствии плазмы
Липосомы (DSPC/Chol/PEG-DSPE, мольное соотношение 3:2:0,015) с захваченным 0,5 н ТЕА-Pn, рН 6,2, осмолярностью 413 ммоль/кг, готовили как описано в примере 18. Липосомы с размером 96,4±29,3 нм получали экструзией 10 раз через два объединенных поликарбонатных фильтра с размером пор 100 нм. Для подсчета липосомных липидов в плазме [3Н]-СНЕ включали в липидный раствор при 0,5 μКи/μмоль DSPC. Топотекан загружали при рН 6,0, 58°С в течение 45 минут при соотношении лекарственное средство/фосфолипид 150 мг/ммоль. Эффективность включения составляла 148,48±10,26 μг Топотекана/μмоль фосфолипида (99,0±6,8%).
Липосомы инкубировали с 50% человеческой плазмы в мультилуночном приспособлении для микродиализа (Spectra-Рог MicroDialyzer, 10 лунок, Spectrum, USA). Донорскую плазму человека разбавляли водным раствором HEPES-солевым буфером (20 мМ HEPES, 135 мМ NaCl), рН 6,5, содержащим 0,02% азида натрия, и заливали ее в нижний резервуар диализера (32 мл). Ячейки (0,4 мл) были отделены от резервуара поликарбонатной мембраной с размером пор 30 нм, чтобы избежать свободного прохождения белков плазмы и маленьких молекул, но не липосом. Липосомы смешивали с заданными количествами плазмы и HEPES-буферного солевого раствора, чтобы достигнуть концентрации 2,5 мМ фосфолипида и 50% об. плазмы. Устройство инкубировали при 37°С, и содержимое резервуара медленно перемешивалось. После 8 часов инкубации содержимое нижнего резервуара было заменено свежей 50% плазмой. В указанные моменты времени (см. ниже) 50-μм аликвоты были отобраны из лунок и подвергнуты хроматографии на колонках, содержащих 2,2-2,4 мл Сефарозы CL-2B, с элюентом - HEPES - буферным солевым раствором для отделения липосом от белков плазмы и свободного лекарственного средства. Липосомы собирали в фракциях свободного объема. Топотекан учитывали флуорометрическим способом при возбуждении 384 нм и эмиссии при 524 нм после солюбилизации образцов плазмы в 90% водном растворе изопропанола - 0,1 н HCl, липиды количественно учитывали с использованием сцинтилляции, подсчитывая [3Н]-СНЕ (с коррекцией гашения). Определенное в каждом конкретном случае соотношение лекарственного средства и липида сравнивали с начальным соотношением до инкубации, чтобы получить % Топотекана, который оставался инкапсулированным в каждый момент времени. После 8 часов инкубации количество лекарственного средства, остающегося в липосоме, было около 55% от его первоначальной величины (Таблица 15).
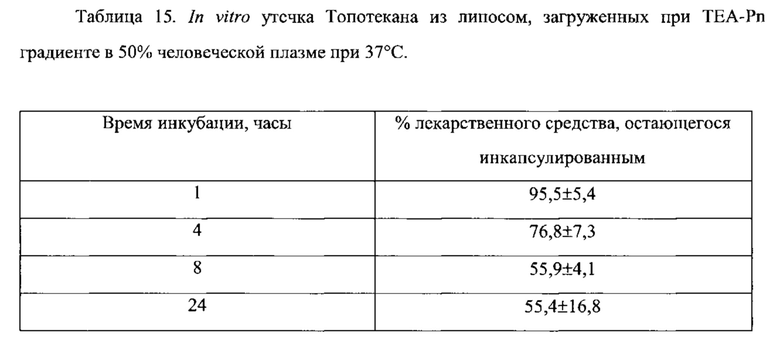
Пример 22. Топотекан-липосомы с захваченным ТЕА-Pn градиентом при различных соотношениях лекарственное средство/липид: in vivo утечка лекарственного средства и долговечность циркулирования в кровотоке мышей.
Липосомы (DSPC/Chol/PEG-DSPE при молярном соотношении 3:2:0,15, содержание [3Н]-СНЕ при 0,5 мКи/ммоль DSPC) с инкапсулированным градиент-образующим солевым раствором (0,5 н ТЕА-Pn, рН 6,2, осмолярность 413 ммоль/кг) готовили как в примере 18, используя 12-кратную экструзию через два объединенных поликарбонатных фильтра с размером пор 100 нм. Размер липосом составлял 107,7±19,1 нм с помощью QELS. Липосомы в 5 мМ HEPES, 50 г/л декстрозы, рН 6,5, смешивали с исходным водным раствором Топотекана (20 мг/мл) при соотношениях лекарственное средство/фосфолипид в диапазоне 130-360 μг/μмоль с последующей инкубацией смеси при 58°С в течение 45 минут, помещением на лед на 15 минут и удалением неинкапсулированного лекарственного средства путем хроматографии на Сефадексе G-75. Самкам FvB мышей (возраст 12 недель) инъецировали липосомы в хвостовую вену в дозе 5 мг Топотекана на кг массы тела (приблизительно 0,2 мг Топотекана/животное) в трех повторностях. В указанные моменты времени, обычно через 8 часов или спустя 24 часа после инъекций, мышам давали анастезию, обескровливали и пробы крови исследовали на содержание лекарственного средства и липиды липосом как в примере 8. Результаты показаны в таблице 16. Через 24 часа около 6-32% первоначально введенного (загруженного) лекарственного средства оставалось инкапсулированным. Более высокая степень загрузки лекарственного средства (более 200 мг/ммоль фосфолипида) приводила к более длительному периоду просачивания.
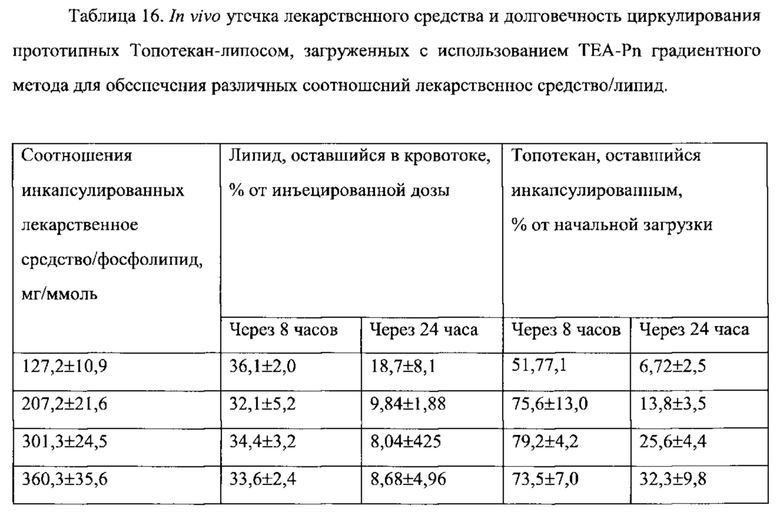
Пример 23. In vivo утечка лекарственного средства и долговечность циркулирования Топотекан-липосом, загруженных с использованием различных захваченных солей аммония и триэтиламмония.
Липосомы на основе DSPE, холестерола, PEG-DSPE (3:1:0,1 по весу), содержащие также [3Н]-СНЕ при 0,22 мКи/ммоль DSPE, получали как примере 18, за исключением 10-кратной экструзии через 2 объединенных фильтра с размером пор 200 нм, 10-кратный экструзии через 2 объединенных фильтра с размером пор 100 нм и 20-кратной экструзии через 2 объединенных фильтра с размером пор 50 нм. Липосомы содержали следующие солевые растворы.
0,5 н Раствор декстран-сульфата аммония (A-DS) готовили из декстран-сульфата натрия (М.м. 5000), поставляемого Sigma, и переводили в аммониевую соль ионообменной процедурой, аналогичной той, что представлена в примере 4. Раствор декстран-серной кислоты сразу же титровали 12,4 М водным раствором аммиака. A-DS раствор имел рН 5,66 и осмолярность 208 ммоль/кг.
0,48 н сахарозо-октасульфат аммония (A-SOS) готовили аналогично описанному в примере 6, только для титрования использовали гидроксид аммония. Раствор имел рН 6,27, осмолярность 258 ммоль/кг.
0,47 М сахарозо-октасульфат триэтиламмония (TEA-SOS) готовили как в примере 6. Раствор имел рН 6,6 осмолярность 297 ммоль/кг.
Топотекан загружали в липосомы в водном растворе 10 мМ MES-Na, 50 г/л декстрозы, рН 6,5, путем инкубирования липосом с лекарственным средством при 61-62°С и начальном соотношении лекарственное средство/фосфолипид 346±1 мг/ммоль в течение 40 минут с последующим инкубированием на льду в течение 10 минут. Липосомы очищали от неинкапсулированного лекарственного средства хроматографией на Сефадексе G-75, элюент-водный 2 мМ гистидин, 144 мМ NaCl, рН 6,6 (HCl).
Самцам Swiss Webster мышей 7-9 недельного возраста инъецировали в хвостовую вену эти липосомные составы с Топотеканом в дозе 5 мг Топотекана на кг веса тела (приблизительно 0,2 мг Топотекана/животное) в трех повторностях. Через 8 часов или 24 часа после инъекции кровь собирали и исследовали на Топотекан и липосомные липиды как в примере 22.
Результаты представлены ниже в таблице 17.
В то время как все три липосомные составы продемонстрировали очень близкую долговечность циркуляции, характеризующуюся примерно 23-28% остающихся в крови через 24 часа после инъекции (в расчете на инъецируемую дозу), неожиданным оказалось, что утечка лекарственного средства из TEA-SOS липосом и A-SOS липосом была лучше, чем в A-DS липосомах, как в плане величины (примерно 2-кратное увеличение просачиваемости лекарственного средства), так и в плане статистической значимости (статистическая значимость на 95% достоверном уровне согласно 2-полосному неспаренному t-тексту Стьюдента р=0,0257 и р=0,00995 соответственно; и по Mann's U-тесту расхождение было осуществлено при α=0,01). Просачиваемость лекарственного средства в TEA-SOS содержащих Топотекан липосомах была лучше, чем в A-SOS содержащих Топотекан липосомах.
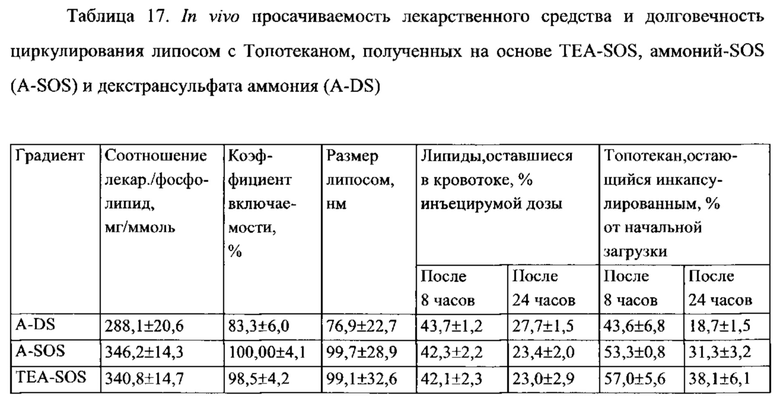
Пример 24. Фармакокинетические характеристики в плазме лекарственного средства и липидов липосомального Топотекана, исследованные на крысах
Параметры долговечности циркулирования в кровотоке и просачивания Топотекана оценивали с помощью анализа на крысах. Липосомы (DSPC/Холестерол/PEG-DSPE молярное соотношение 3:2:0,15) готовили методом смешивания с этанолом и экструзии и загружали Топотекан, используя TEA Pn градиент или ТЕА-сахарозооктасульфатный (TEA-SOS) градиент как описано в примере 18 и загружали при различных соотношениях лекарственное средство/липид (15-450 мг/ммоль фосфолипида). Для обсчитывания липидной матрицы липосомный липид содержал [3Н]-СНЕ при 6,5-1,5 мКи/ммоль DSPC.
Самкам Spraque Dawley крыс (возраст 6-8 недель, массой около 200 г) через введенный в центральную вену катетер внутривенно вводили (через катетер) липосомы с Топотеканом при дозе 4-5 мг/кг веса тела. Катетер промыли физраствром. В выбранные моменты времени (в пределах 48 часов после инъекции) пробы крови (0,2-0,3 мл) отбирали через катетер в гепаринизированные шприцы, смешивали с 0,4 мл холодного забуференного солевого раствора с 0,04% ЭДТА, клетки крови отделяли центрифугированием, а супернатанты (PBS-разбавленная плазма) исследовали на содержание липидов учетом 3Н-СНЕ радиоактивности (с коррекцией гашения) и Топотекана методом флуорометрии (Пример 71). Результаты анализа были скорректированы с учетом разбавления плазмы, обсчитаны с учетом веса взятой пробы крови и приняты как гематокрит 40%. Общее содержание лекарственного средства и липидов в крови было вычислено, исходя из объема крови, принятого как 6,5% от веса тела. Долю оставшегося в липосомах Топотекана определяли сравнением соотношения лекарственное средство/липид в инъецируемых липосомах. В таблице 18, приведенной ниже, собраны значения «полужизни» липида, лекарственного средства и «полужизни» для выделения лекарственного средства, а также другие свойства липосом. Фармакокинетические (РК) кривые показаны на фигурах 8А (липид) и 8В (соотношение лекарственное средство/липид). В целом кривые РК в крови хорошо соответствуют простой экспонентной модели (R2 0,984-0,999). Несмотря на размер 90-100 нм и очень малое количество PEG-ированных липидов (0,3 моль. %), липосомы неожиданно показали хорошую долговечность циркулирования (полужизнь в плазме для липидного компонента была в пределах 11-16 часов). Более медленное выделение Топотекана (время полужизни составляет 22,9 часа) наблюдалось у липосом, нагруженных с использованием TEA-SOS.
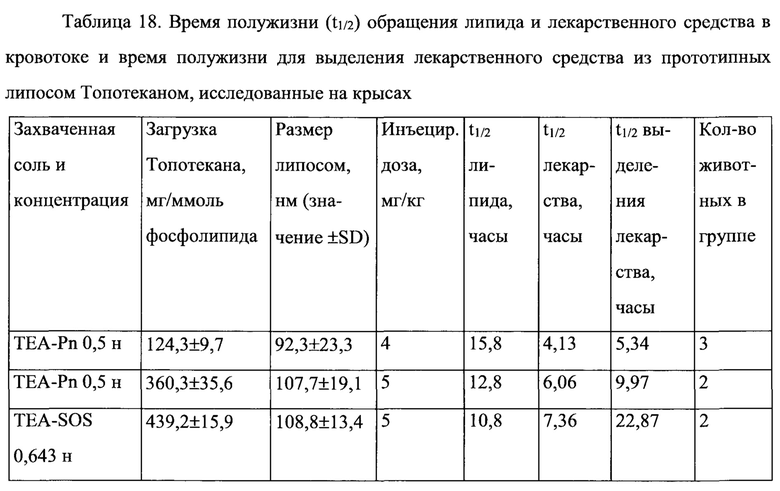
Пример 25. Устойчивость лекарственного средства к просачиванию во время хранения липосом с Топотеканом.
Образцы нескольких ранее описанных составов, которые готовили для вышеописанных исследований, хранили при 4-6°С в течение разных по продолжительности периодов времени, чтобы оценить стабильность инкапсулированного Топотекана в отношении утечки или просачивания из липосом. Образцы липосом пропускали через колонки с Сефадексом G-75, элюировали 20 мМ HEPES, 135 мМ NaCl, pH 6,5, чтобы удалить лекарственное средство, которое вне липосом, и анализировали на содержание лекарственного средства спектрофотометрий, а на содержание липида, подсчитывая [3Н]-СНЕ радиоактивность. Результаты (Таблица 19) показали хорошую удерживаемость Топотекана в липосомах в процессе хранения.
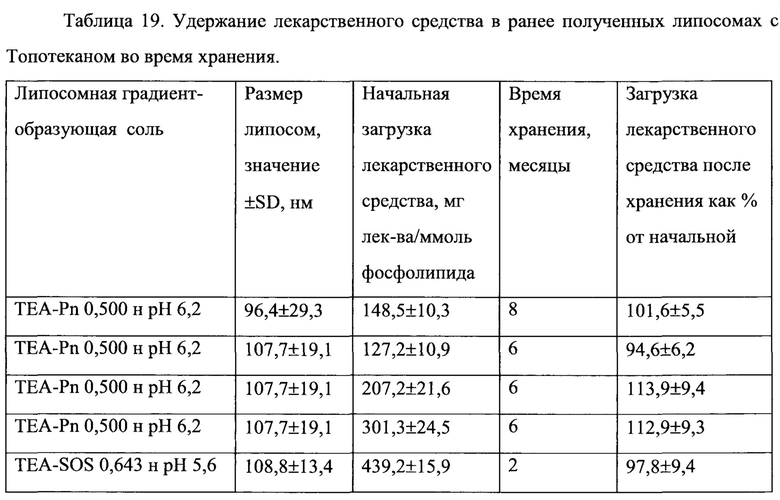
Пример 26. In vitro поглощение липосомального и иммунолипосомального Топотекана HER2-сверхэкспрессирующими раковыми клетками.
Это исследование касалось способности нагруженных Топотеканом иммуннолипосом, специфичных к HER2, полученных в соответствии с данным изобретением, доставлять Топотекан специально в клетки, сверхэкспрессирующие HER2 при культивировании клеточной культуры. Эти (иммуно) липосомы готовили и нагружали Топотеканом, используя ТЕА-Pn метод примера 19. Клетки карциномы молочной железы человека (SKBr-3, ATCC), сверхэкспрессирующие HER-2, выращивали в модифицированной McCoy 5A среде (без трицина), содержащей дополнительно 10% фетальной сыворотки теленка, 50 μг/мл сульфата стрептомицина и 50 Ед/мл пенициллина G (полная ростовая среда), в Т-75 колбах при 37°С, 5% CO2. Клетки собирали при трипсинизации, инокулировали в 24-луночные планшеты для клеточных культур при концентрации 150000 клеток/на лунку в 0,5 мл полной ростовой среды и давали адаптироваться в течение ночи. Среду заменяли 0,5 мл полной ростовой среды, содержащей состав Топотекана в выбранных концентрациях в пределах диапазона 0,01-0,1 мМ фосфолипида. Для каждого условия использовали лунки в трех повторностях. Контрольные лунки инкубировали в отсутствии лекарственного средства и/или липосом (чтобы получить фоновую интерпретацию для исследования). Планшеты инкубировали при медленном встряхивании при 37°С, 5% CO2 в течение 4-8 часов. Среду удаляли отсасыванием, а клетки промывали 4 раза порциями по 1 мл холодным сбалансированным солевым раствором Hanks', содержащим соли Са и Mg. Клетки солиболизировали добавлением 0,1 мл 1% Тритон Х-100 в воде и количество лекарственного средства в клеточных лизатах определяли флуорометрическим способом (пример 71). Стандартную кривую получали при значении 10-2500 нг Топотекана/на лунку, и она соответствовала многочлену второго порядка (чем объясняется убывание при более высоких концентрациях лекарственного средства) после вычитания фоновой (собственной) автофлуоресценции клеток. При использовании микроплатированного флуорометра выбирали фильтр 400/30 нм для возбуждения, 530/25 нм - для эмиссии. И кюветный, и микроплатированный флуорометры давали одни и те же результаты.
Результаты обоих экспериментов объединены в таблице 20, приведенной ниже. Имеет место заметное поглощение клетками HER2-нацеленного липосомального лекарственного средства (в 50-300 раз выше, чем ненацеленного липосомального Топотекана). Интересно, что поглощение свободного Топотекана было также значительно ниже, чем HER2-нацеленного иммунолипосомального Топотекана. Это может быть объяснено быстрым гидролизом лактонного кольца камптотецина в молекуле Топотекана в среде для роста клеток в присутствии сыворотки, что приводит к образованию карбоксильной формы лекарственного средства, которая может обладать более низкой клеточной проницаемостью и более низкой цитотоксичностью. В итоге была подтверждена способность нацеленных в клетки, интернализуемых, конъюгированных с лигандом иммунолипосом к выделению Топотекана внутри клеток.
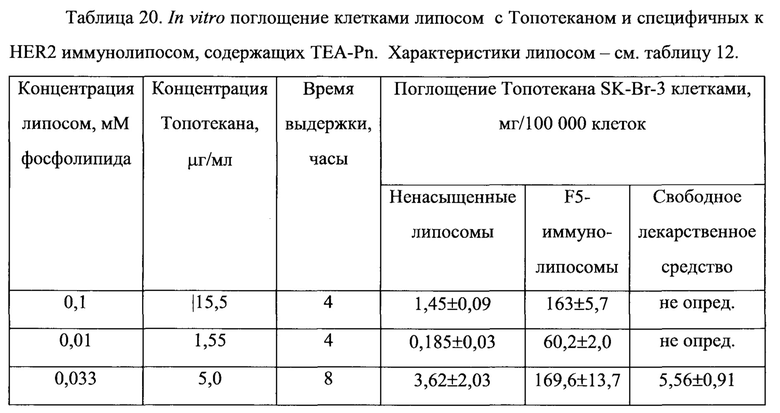
Пример 27. Цитотоксичность липосомального и иммунолипосомального Топотекана в отношении раковых клеток, сверхэкспрессирующих HER-2, in vitro.
Как только способность специфичных к HER-2 Топотекан-иммунолипосом к внутриклеточному выделению лекарственного средства в НЕК2-сверхэкспрессирующие раковые клетки была установлена (пример 26), стало важно обеспечить, чтобы интернализованные липосомы могли выделять лекарственное средство в его активной форме. С этой целью изучалась in vitro цитотоксичность свободного Топотекана (т.е. Топотекана в растворе), липосомального Топотекана и анти-HER2-иммунолипосомального Топотекана. Готовили препараты липосомального Топотекана и выращивали и собирали SKBr-3 клетки как описано в примере 26. Клетки засевали в 96-луночные планшеты для клеточных культур в концентрации 5000 клеток в 0,1 мл полной ростовой среды, в трех повторностях, и оставляли адаптироваться на ночь. Самые крайние продольные и поперечные ряды (лунок) в планшете оставляли свободными. Стерильные препараты Топотекансодержащих липосом, иммунолипосом или свободного лекарственного средства (свежеприготовленного - путем разбавления исходного раствора Топотекана с концентрацией 20 мг/мл, рН 3, небуферированным физраствором до концентрации 2 мг/мл) были разбавлены полной средой с лекарственным средством для достижения концентраций, начиная с 90,30 или 10 μг/мл и далее серийно - с разбавлением в среде при коэффициенте 3. Среду в лунках заменяли 0,2 мл разведенным раствором лекарственного средства относительно липосом (лекарственное средство/липосомы) и инкубировали при 37°С, 5% CO2, в течение определенного времени (4-6 часов). Одну ячейку в каждом ряду инкубировали со средой, не содержащей лекарственного средства, в качестве контроля (без обработки). Среду, содержащую лекарственное средство, удаляли из лунок, клетки промывали 0,2 мл среды, не содержащей лекарственное средство, и 0,2 мл свежей среды без лекарственного средства добавляли во все лунки. Планшеты инкубировали в течение 4 дней при 37°С, 5% CO2. Без замены среды в каждую лунку добавили 0,03 мл раствора тетразолиевого красителя с концентрацией 2 мг/мл (Thiazolyl Blue, MTT) (Sigma Chemical Со.) в бессыворотчной среде. Планшеты инкубировали еще 2-3 часа при 37°С, 5% CO2. Среду удаляли, в лунки вносили по 0,2 мл 70% об. водного изопропанола, 0,075 н HCl и осторожно встряхивали до тех пор, пока формазановый краситель не растворится (15-30 минут). Оптическую плотность растворов формазона определяли, используя микроплатный фотометр при 540 нм. Жизнеспособность клеток как %-ную долю необработанного контроля вычисляли в виде отношения оптической плотности содержимого в экспериментальных лунках к оптической плотности содержимого в лунках с необработанными клетками, с поправкой на фоновое значение. Строя график, эти данные наносили против концентрации лекарственного средства и дозу IC50 определяли графически по пересечению кривой с линией 50% жизнеспособности в координатах «жизнеспособность-концентрация».
Эти результаты представлены на фигуре 9. Доза лекарственного средства, приводящая к 50% ингибированию роста (IC50) для свободного Топотекана или ненацеленного липосомального Топотекана была свыше 30 μг/мл, для F5-иммунолипосомального Топотекана - 0,15 μг/мл. Эти результаты соответствовали данным по поглощению нацеливаемого лекарственного средства.
Пример 28. Сравнение стабильности и фармакокинетических характеристик в плазме для липосомального и F5-иммунолипосомального Топотекана, осуществленное на мышах.
Липосомы с Топотеканом, содержащие радиоактивную липидную метку [3H]-CHE при 1,5 мКи/ммоль фосфолипида, готовили согласно примерам 11 и 19, используя процедуры смешения этанола с липидным раствором и последующей экструзии при следующих условиях: 0,643 н сахарозооктасульфат триэтиламмония, экструзия через поликарбонатные мембраны: 15 пассажей через 2 объединенных РСТЕ фильтра с размером пор 80 нм; загрузка Топотекана: начальное соотношение лекарственное средство/фосфолипид, равное 350 мг/ммоль (вычислено из расчета на Топотекан в форме свободного основания); F5scFv конъюгацию выполняли согласно описанному в Примере 19. Липосомы имели следующие характеристики:
Размер по QELS: среднее значение согласно оценке: 101,2 нм; стандартное отклонение 20,1 нм.
Инкапсулирование лекарственного средства: липосомы с Топотеканом (Topo-Ls) 359,3±27,4 мг/ммоль фосфолипида; Топотекан F5scFv-иммунолипосомы (торо-F5-ILs) 326±15,9 мг/ммоль фосфолипида.
Исследование проводили, в основном, как в примере 22. Группы по 9 мышей-самцов Swiss Webster (возраст 8-10 недель, масса 24-27 г) получали инъекции в хвостовую вену Topo-Ls, Topo-F5Ls или свежеприготовленного раствора Топотекана с концентрацией 1 мг/мл в незабуференном физрастворе и в дозе 5 мг Топотекана основания на кг массы тела (эквивалентно дозе липида 14-16 μмоль фосфолипида на кг массы тела). Через 1 час, 8 часов или 24 часа после инъекции трех животных обескровливали посредством прямого укола в сердце над анестезией с применением Кетамина/Ксилазина, кровь собрали в пробирки, содержащие PBS-ЭДТА, и анализировали на содержание Топотекана (флуорометрией) и липосомные липиды (подсчитывая радиоактивную сцинтилляцию). Количественное содержание лекарственного средства и липида, остающихся в крови в заданные моменты времени, вычисляли, принимая введенную дозу за 100% и принимая, что количество крови в животном составляет 6,3% веса тела животного, а фракция отделенных клеток крови - 45%. Доля лекарственного средства, остающегося инкапсулированным в липосомах в каждый из указанных определенных моментов времени, вычислялась отдельно для каждого животного путем сравнения соотношения радиоактивности лекарственного средства и липида в образцах плазмы с аналогичным же соотношением инъецируемых липосом. Количество свободного Топотекана в образцах плазмы, отобранной через 1 час после инъекции, было меньше, чем 1% инъецируемой дозы (в действительности, они были ниже предела определения применяемого аналитического метода); поэтому группу, получавшую свободный Топотекан, с более поздним сроком не исследовали. Из-за быстрого клиренса крови и низких уровней свободного Топотекана в крови, было сделано предположение, что практически весь Топотекан, обнаруженный в крови во все указанные точки времени, представля собой включенный в липосомы (инкапсулированый) Топотекан.
Эти результаты приведены ниже в таблице 21.
Примечательно, что липосомы, полученные согласно изобретению, сохраняли 79-85% от исходного уровня загрузки даже через 24 часа после инъекции в кровоток животных. Различия между средними значениями содержания в плазме липида или лекарственного средства между группами, получавшими липосомы и иммунолипосомы, были в пределах 1,8-13,6% и приближались или были в пределах диапазона ошибки опыта. Вероятности недействительности предположения в отношении липосомной и иммунолипосомной группы в части количеств лекарственного средства или липида, вычисленные в каждый момент времени с использованием t-тест Стьюдента, были в пределах 0,543±0,938. Было сделано заключение о том, что различия в остаточных значениях содержания лекарственного средства или липида в крови между двумя препаратами были ничтожно малы и статистически неразличимы.
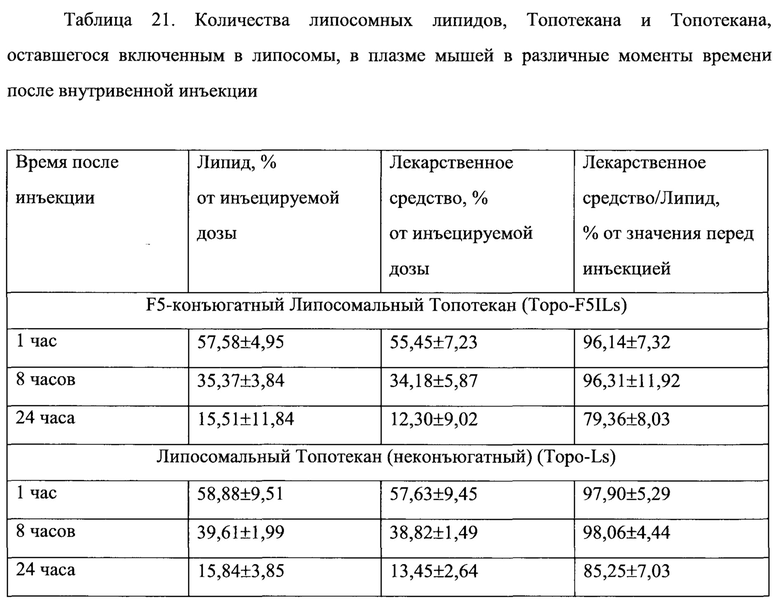
Пример 29. Антиопухолевая эффективность липосомального и анти-HER2-иммунолипосомальиого Топотекана в ВТ-474 ксенотрансплантатах.
В этом исследовании использовались первые упомянутые иммунолипосомы с Топотеканом, в которых использовали триэтиламмоний - полифосфатный градиент для включения лекарственного средства. Эти липосомы получали, в основном, по способам, описанным в примерах 11 и 19. Компоненты липидной матрицы DSPC (Avanti Polar Lipids; 3 моль. части), Холестерол (Calbiochem, 98,3%, 2 моль. части) и метокси-PEG (2000)-DSPE Avanti Polar Lipids, 0,015 моль. частей) - соединяли с 100% этанолом USP с получением раствора, содержащего 0,5 мМ фосфолипида при 60°С. Этанольно-липидный раствор разбавляли при 60°С водным раствором полифосфата триэтиламмония (0,608 М триэтиламина, 0,65 Н фосфата, рН 6,1, осмолярность 531 ммоль/кг), тщательно перемешивали и экструдировали 10 раз через 2 объединенных поликарбонатных фильтра с размером пор 100 нм (Nucleopore, Coming), используя термостатируемый экструдер, работающий под давлением газа (Lipex Biomembranes) при 60°С. Экструдированные липосомы охлаждали на льду и неинкапсулированный полифосфат триэтиламмония удаляли гель-хроматографией на Сефарозе CL-4B, используя содержащий 5% декстрозы 5 мМ HEPES-Na буфер, рН 6,5, в качестве элюента. Размер липосом был 103,8±35,1 по QELS. Липосомы в этом буфере инкубировали с гидрохлоридом Топотекана при 60°С в течение 30 минут при соотношении 0,35 мг Топотекана основания на μмоль фосфолипида. В конце инкубации липосомы охлаждали на льду и хроматографировали на Сефадексе G-75 с элюентом 20 мМ HEPES-Na, 135 мМ NaCl, рН 6,5, чтобы удалить неинкапсулированное лекарственное средство. Содержание лекарственного средства определяли флуорометрией, а содержание липида анализом фосфата, как сообщалось ранее. Полученный таким образом липосомальный Топотекан имел 365,4±23,1 мг Топотекана основания на ммоль фосфолипида. Для того, чтобы получить HER2-нацеленные иммунолипосомы с Топотеканом, порцию этого препарата липосом с Топотеканом инкубировали с очищенным конъюгатом анти-HER2 scFv F5 и малеимидо-PEG-DSPE линкера в целом так, как описано в примере 19. Кратко говоря, F5-PEG-DSPE конъюгат в водном 10 мМ растворе цитрата натрия, содердащем 10% сахарозы, соединяли с липосомами с Топотеканом в соотношении 0,15 мг белка на ммоль липосомного фосфолипида и инкубировали при 60°С в течение 30 минут. Инкубационную смесь охлаждали на льду и хроматографировали на Сефарозе CL-4B с элюцией элюентом 20 мМ HEPES-Na, 135 мМ NaCl, рН 6,5, чтобы удалить невключенный scFv конъгат. Соотношение количеств лекарственного средства и липида уменьшилось до 14% после этой дополнительной инкубации.
Составы липосом с Топотеканом и иммунолипосом, содержащих 1-2 мг/мл Топотекана, пропускали через 0,2 микронный стерильный шприцевой фильтр, расфасовывали асептически в полипропиленовые сосуды и хранили при 4-6°С в течение до 1 месяца перед использованием.
Свободный Топотекан был свежеприготовленным при растворении порошка гидрохлорида Топотекана (2 мг/мл) в 5% растворе декстрозы, его стерилизовали, пропуская через 0,2-микронный шприцевой фильтр.
Была использована модель ксенотрансплантата аденокарциномы молочной железы человеа ВТ-477, сверхэкспресирующая HER-2, как описано в примере 10.
На 13-ый день после пересадки опухоли животных с опухолями в пределах 120-350 кубических миллиметров отобрали и произвольно разделили на группы по 12 животных: 1 контрольную группу и 3 группы для проведения лечения. На 14-ый, 18-ый и 21-ый день после пересадки опухоли мышам вводили внутривенно инъекции составов с Топотеканом с дозой 5 мг/кг веса тела, или такого же объема физраствора. Состояние здоровья животных ежедневно отслеживали. Размеры опухоли и массу тела регистрировали дважды в неделю вплоть до 53-го дня после пересадки опухоли. Животных, у которых опухоли достигали 20% от массы тела, или те, у которых прогрессирующая потеря массы тела достигла 20% или более, подвергали эвтаназии.
На фигурах 11 и 12 показаны данные, характеризующие рост опухоли и массу тела животного, соответственно. Препараты липосомального Топотекана были более активными в подавлении роста опухолей, чем свободное лекарственное средство, a F5-нацеленный липосомальный препарат был более активным, чем такой же, но ненацеленный. Средние размеры опухолей в конце периода наблюдения значительно различались в группах, подвергающихся лечению (Р величины по неспаренному 2-полосному Student's t-тесту были 1,2×10-6 для свободного v. иммунолипосомального лекарственного средства, 0,000114 для свободного v. липосомального лекарственного средства и 0,00718 для липосомального v. иммунолипосомального лекарственного средства). Таким образом, липосомально инкапсулированный Топотекан был более активным, чем свободное лекарственное средство, а анти-HER2-иммунолипосомальный Топотекан был более активным, чем ненацеленное липосомальное лекарственное средство. В группе с лечением липосомами и иммунолипосомами, после первоначальной регрессии, повторный рост опухоли происходил в течение 10 дней, считая от последнего проведенного лечения. В группе, где лечили свободным лекарственным средством, не наблюдалось регрессии опухоли. Отмечалось, что липосомальные препараты Топотекана при данной дозе были более токсичными, чем свободное лекарственное средство. Имела место гастроинтестинальная токсичность. У животных, получавших липосомальный Топотекан, развивалась диарея и потеря веса, достигающая в среднем 14% от исходного. В то время как в группе, леченной ненацеленными липосомами, животные выздоравливали, за исключением одного (12,5%), которое имело устойчивую 15% потерю веса в конце исследования, 5 животных (41,6%) в группе леченных F5-нацеленными липосомами окончательно заболели и умерли; а еще два животных (16,7%) имели устойчивую потерю веса около 15%. В контрольной группе и группе, получавшей свободное лекарственное средство, не наблюдалось потери веса или вызванной лечением заболеваемости.
Пример 30. Максимально допустимая доза (MTD) свободного и линосомального Топотекана, установленная на мышах, получавших внутривенные инъекции в течение 3 недель.
В этом исследовании использовали препарат липосомального Топотекана, полученного по примеру 29, за исключением того, что раствор полифосфата триэтиламмония был заменен раствором сахарозооктасульфата триэтиламмония с 0,65 М триэтиламмония, рН 6,2; и для экструзии использовали 80-нм поликарбонатные мембранные фильтры вместо 100-нм. Размер липосом по объему и весу, определенный методом квазиупругого светорассеяния в Gaussian приближении (QELS) был 95,1±19,6 нм (среднее значение ± SD); соотношение лекарственное средство-липид было 369,1±18,3 мг/ммоль фосфолипида. Пяти-шестинедельным мышам-самкам Swiss-Webster (18-20 г), в группах по два животных, делали три внутривенные (хвостовая вена) инъекции свободного или липосомального Топотекана по графику «раз-в-неделю», начиная с дозы 2 мг/кг Топотекана основания за инъекцию с увеличением ее для каждой последующей группы в 1,8 раза до достижения дозы 37,8 мг/кг. Иммунолипосомальный Топотекан не включали в это исследование. Вес тела животных и общее состояние здоровья регистрировали ежедневно. Прогрессирующая потеря веса более чем в 20% случаев или естественная смерть в любое время любого из двух животных в группе в течение периода десяти дней с начала эксперимента позволяли судить о токсичности дозы. В соответствии со смертностью животных или весом данные по MTD определяли как падение в пределах диапазона 11,7-21 мг/кг для свободного Топотекана к 2,0-3,6 мг/кг для липосомального (Прототип 2) Топотекана. Во втором опыте мыши получали инъекции свбодного, липосомального или F5-иммунолипосомального Топотекана (полученного из липосомального Топотекана этого примера, который описан в примере 29) с дозами от 2,0 мг/кг (липосомальный/иммунолипосомальный Топотекан) или 12 мг/кг (свободный Топотекан) с увеличением дозы для каждой последующей группы в 1,15 раза до тех пор, пока не будет достигнута доза, превосходящая нижний предел установленного интервала MTD. Самую высокую дозу, которая не приводила к смерти или окончательному заболеванию любого из животных, считали MTD, и было обнаружено, что она составляет 18,4 мг/кг для свободного Топотекана, 3,0 мг/кг для липосомального Топотекана, 3,0 мг/кг для иммунолипосомального Топотекана. Таким образом, липосомальный Топотекан показал большую токсичность, чем свободные лекарственного средства.
Пример 31. Антиоухолевая эффективность липосомального Топотекана в ВТ-474 модельных ксенотрансплантатах в диапазоне 0,125-1,0 × MTD
В этом исследовании использовались липосомы с Топотеканом и F5-иммунолипосомы из примера 30. ВТ-474 подкожные трансплантаты были пересажены бесшерстным мышам как в примере 29. На 18-ый день заражения клетками опухоли животных с опухолями (105-345 кубич. мм в среднем - 200 кубич. мм) произвольно разделили группы для лечения по 6 животных в каждой группе и контрольную группу из 8 животных. Животные получали свободный или липосомальный Топотекан в дозе 1 × MTD, 0,5 × MTD, 0,25 × MTD или 0,125 × MTD в три i. v. (хвостовая вена) инъекции на 19, 23 и 27 день после пересадки опухоли. Контрольная группа получала инъекции физиологического раствора. Размеры опухоли и вес тела животного регистрировали как в примере 29. Для того, чтобы получить вес тела животного, вес опухоли (вычисленный по ее размеру и при принятии условий ее плотности, равной 1,0) вычитали из общего измеренного веса животного. Все составы (препараты) при MTD показали антиопухолевую активность (фигуры 13A-13D). Существенной разницы в эффективности между свободным или липосомальным лекарственным средством, заданными в соответствующей их MTD или в их идентичных долях (1/2, 1/4 или 1/8). Так что инкапсулирование лекарственного средства в липосомы с использованием TEA-SOS градиента приводило к почти 6-кратному увеличению антиопухолевой активности, но не только при таком же увеличении токсичности лекарственного средства. Динамика веса тела животных показала, что все введения лекарственного средства были нетоксичными, за исключением лечения свободным Топотеканом при MTD, которое показало кратковременное снижение веса тела (около 15% от начальной величины), которое позже восстановилось (фигура 14).
Пример 32. Получение и нацеленная in vitro цитотоксичность липосом с Топотеканом методом захвата сахарозооктасульфата триэтиламмония.
Липосомальный Топотекан получали, в основном, следуя процедуре примера 18, используя захваченный раствор TEA-SOS, имеющий 643 мМ TEA, pH 5,7, осмолярность 530 ммоль/кг и соотношение лекарственное средство/фосфолипид 170 мг/ммоль. Липосомы имели соотношение 155 мг лекарственного средства на ммоль фосфолипида; 90%-коэффициент включения и размер частиц 105 нм. Эти липосомы инкубировали с мицеллярным раствором F5-PEG-DSPE конъюгата при приблизительно 30 scFv на липосомы (15 мг антитела/ммоль фосфолипида) при 60°С в течение 1 часа, в целом так, как описано в примере 19. Антитело-конъюгированные липосомы отделяли с помощью SEC, используя Сефарозу CL-4B и соединяли с HBS-6,5 HEPES-буферным солевым раствором. В процессе присоединения анти-HER2 scFv (F5) заметных изменений соотношения лекарственное средство/липид не было.
Поглощение препаратов Топотекана раковыми клетками определяли следующим образом. HER2-сверхэкспрессирующие клетки карциномы молочной железы человека (SK-Br-3, АТСС НТВ-30) помещали в 24-луночные планшеты для культивирования клеток при дозе 150000 клеток на лунку и оставляли на ночь. Клетки (в трех повторностях) инкубировали с F5-нацеленным и ненацеленным липосомальным Топотеканом в полной ростовой среде при концентрации липосом 0,1 мМ и 0,01 мМ в течение 4 часов при 57°С.Клетки промывали 4 раза сбалансированным солевым раствором HanK's, солюбилизировали в смеси 0,1% Тритона-Х-100 - 70% подкисленного изопропанола (1:10) и количество связанного с клетками Топотекана на лунку определяли флуорометрией. Результаты (значение ± стандартное отклонение) объединены и приведены в таблице 22. Нацеленные липосомы доставили в 100-300 раз больше лекарственного средства в клетки-мишени, чем ненацеленные липосомы.
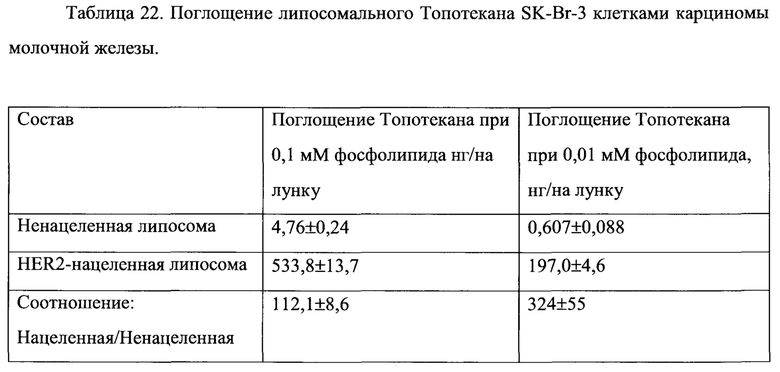
Цитотоксичность этих препаратов Топотекана в отношении SKBr-3 клеток рака молочной железы определялась как описано в примере 27. SKBr-3 клетки засевали в 96-луночноые планшеты в дозе 5000 клеток/на лунку, оставляли на ночь и инкубировали при повышающихся концентрациях (0,004-30 μг/мл) свободного, липосомального или F5-иммунолиполисомального Топотекана в среде для роста клеток в течение 4 часов при 37°С.Среду, содержащую лекарственное средство, удаляли, а клеткам давали расти в среде без лекарственного средства в течение 72 часов. Количество жизнеспособных клеток на лунку определяли, используя Thiazolyl Blue (MTT) тетразолиевый анализ, и выражали в % по сравнению с контролем (необработанные клетки). Результаты представлены на фигуре 10. Иммунолипосомы с Тепотеканом были более цитотоксичными (IC50 0,15-0,5 μг/мл), чем ненацеленные липосомы с Топотеканом (IC50≥3,1 μг/мл) и свободный Топотекан (IC50≥2,3 μг/мл).
Пример 33. In vivo стабильность липосом с Топотеканом различных размеров.
Липосомы, содержащие ТЕА-Pn, получали как в примере 22, используя 12-кратную экструзию через поликарбонатные мембраны с размером пор 100 нм или дополнительно - 12 раз через поликарбонатные мембраны с размером пор 50 нм. Топотекан (ТРТ) был добавлен при соотношении 150 μг на μмоль фосфолипида. Загрузку завершали при 58°С в течение 45 минут в бане с горячей водой с последующим охлаждением на льду. Эффективность включения для экструдированных при 50 нм и 100 нм липосом составила 126,80±19,24 μг ТРТ/μмоль PL (84,5±12,8%) и 148,48±10,26 μг ТРТ/μмоль PL (99,0±6,8%), соответственно. Мышам-самкам Swiss-Webster (по 3 в группах) инъецировали внутривенно один или два состава Ls-TPT в дозе 5 мг ТРТ/кг. Мышей умерщвляли через 6 часов и кровь собирали. Плазму анализировали на ТРТ и липосомный липид как описано в примере 22. Результаты показаны в таблице 23.
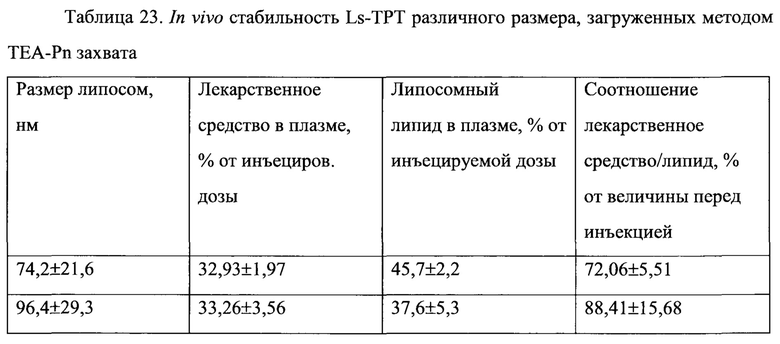
Пример 34. Синтез и включение в липосомы 6-(3-аминопропил)эллиптицина (6-АРЕ)
6-(3-аминопропил)эллиптицин получили из эллиптицина двухстадийным способом, основанным на процедуре Werbel et al., J. Med. Chem. 1986, v. 29, p.1321-1322.
501,4 мг эллиптицина основания (NSC 71795 (Aldrich Chemical Co.) перемешивали с приблизительно 100 мг гидрида натрия (Sigma, промыт безводным петролейным эфиром) в 5 мл сухого диметилформамида (DMF) при комнатной температуре в течение 30 минут. К этой смеси по каплям добавили раствор 678 мг N-бромпропилфталимида (Aldrich) в 2 мл сухого DMF. Реакционную смесь, окрашенную в лиловый цвет, перемешивали под аргоном в течение ночи, обрабатывали 1 мл воды и вливали в 60 мл воды. Смесь дважды экстрагировали 25 мл метиленхлорида, экстракт высушили над безводным сульфатом натрия и пропустили через слой нейтрального оксида алюминия. Слой оксида алюминия дважды прополаскивали в 10 мл метиленхлорида и объединенные фильтрат и промывочная жидкость были высушены под вакуумом. Продукт вместе с 20 мл абсолютного этанола и 2 мл безводного гидразина перемешивали в течение ночи при комнатной температуре. Полученный осадок был отфильтрован под вакуумом, фильтрат желтого цвета был разбавлен 50 мл 0,2 н NaOH и проэкстрагирован двумя порциями (75 мл и 50 мл) хлороформа. Хлороформный экстракт был высушен над Na2SO4 и доведен до сухости в вакууме. Неочищенный продукт (выход 408 мг) хроматографировали на колонке с диоксидом кремния 60 и элюировали изократно смесью хлороформа-метанола (7:3 по объему), насыщали сухим триметиламином. Было показано, что фракции, элюированные во второй окрашенной в желтый цвет полосе, после непрореагировавшего эллиптицина, содержали целевое соединение примерно с 30% выходом. Структура была подтверждена 1Н-ЯМР ТСХ: Rf 0,29-0,31 (Диоксид кремния 60; CHCl3-NaOH 7:3 по объему, насыщ. триметиламином). Эллиптицин, R1 0,81-0,83. Полученное соединение превращали в дигидрохлоридную соль растворением в безводном этаноле и титрованием 6 н раствором HCl в сухом изопропаноле. Оранжевые кристаллы дигидрохлорида 6-АРЕ (NSC 176328) отфильтровывали, промывали эфиром и сушили в вакууме. Выход дигидрохлорида 86%.
Липосомы получали гидратированием липидной пленки DSPC, холестерина и PEG (М.м. 2000) - DSPE (3:2:0,015 мольное соотношение) в растворе полифосфата триметиламмония (ТМА-Pn) при 0,5 М ТМА, рН 5,6, температуре 60°С, с последующими шестью циклами быстрого замораживания (-78°С) и таяния (60°С) и 10-кратной экструзии через два объединенных поликарбонатных фильтра с размером пор 50 нм. Неинкапсулированный ТМА-Pn удаляли, используя колонку с Сефарозой CL-4B с элюцией HEPES-Декстроза (5 мМ HEPES, 5% Декстрозы, рН 5,5). Размер липосом был 85,7±32,1 нм.
Концентрированный 6-АРЕ раствор (10 мг/мл) добавляли к ТМА-Pn-содержащим липосомам при соотношении лекарственное средство/фосфолипид 100 μг АРЕ/μмоль фосфолипида, смесь инкубировали при 58°С в течение 45 минут и быстро охлаждали на льду в течение 15 минут. Неинкапсулированное лекарственное средство удаляли гель-хроматографией на колонке с Сефадексом G-75 с элюцией HEPES-Декстрозным буфером (5 мМ HEPES-Na, 5% декстрозы, рН 6,5). Липосомы с включенным АРЕ затем оценивали спектрофотометрическим методом как описано в примере 71, а липосомный фосфолипид определяли экстракционным анализом по примеру 70. Инкапсулирование лекарственного средства было практически количественным.
Пример 35. Изготовление HER2-нацеленного иммунолипосомального 6-АРЕ и in vitro цитотоксичность 6-АРЕ препаратов, специфичных к Вт-474 клеткам рака молочной железы, сверхэкспрессирующим HER2.
Липосомы с инкапсулированным 6-АРЕ (Ls-APE) готовили как описано в примере 34. Анти-HER2-иммунолипосомы с инкапсулированным 6-АРЕ (F5-ILs-APE) были получены из Ls-APE способом примера 19. Основанный на МТТ анализ жизнеспособности клеток по примеру 27 использовали для определения цитотоксичности 6-АРЕ, доставленного (к клеткам) в виде раствора, Ls-APE или в виде HER2-нацеленных F5-ILs-АРЕ, специфичных к сверхэкспрессирующим HER2 клеткам карциномы молочной железы человека (Вт-474). Эти клетки выдерживали в среде, содержащей лекарственное средство, в течение 6 часов, а затем - в среде без лекарственного средства в течение 3 дней. Результаты показаны на фигуре 15. IC50 для свободного АРЕ составило 0,26 μг АРЕ/мл, для F5-ILs-APE было 0,756 μг АРЕ/мл, а для ненацеленных Ls-APE было 51,0 μг АРЕ/мл. Разница активности для нацеленного и ненацеленного липосомального 6-АРЕ составила 67,5 раза, что показало значительный эффект нацеленного средства доставки лекарственного средства.
Пример 36. EGFR-нацеленные иммунолипосомальиые составы 6-АРЕ и цитотоксичность в отношении раковых клеток in vitro.
6-АРЕ-нагруженные липосомы получали как описано в примере 34. EGFR-нацеленные иммунолипосомами получали присоединением EGFR-специфичных Fab'-фрагментов антитела следующим образом. EGFR-специфичное IgG MAb C225 (cetuximab, ERBITUX™, Imclone Systems) было расщеплено пепсином с получением (Fab')2 фрагментов. Очищенные (Fab')2 фрагменты обрабатывали 10-20 мМ 2-меркаптоэтиламина в течение 15 минут при 37°С и полученные Fab' фрагменты очищали гель-фильтрацией, используя Сефадекс G-25. Содержание реакционноспособных тиольных групп было обычно около 0,9 тиольных групп на молекулу белка (вычислено с использованием реагента Ellmann'a). C22Fab' ковалентно конъюгировали с амфифильным линкером Mal-PEG-DSPE (Avanti Polar Lipids, AL) в водном растворе при рН 6,2-6,5 и мольном соотношении 1:4 в течение 2-4 часов при комнатной температуре или в течение ночи при 4-6°C с получением C225Fab'-PEG-DSPE конъюгата с выходом 30-50% по белку. Этот мицеллообразующий конъюгат отделили от непрореагировавшего белка колоночной хроматографией исключения по размеру на гранулированном геле из 3% агарозы - 4% полиакриламида (Ultrogel АсА 34, получен из Sigma Chem Co.) с элюцией HBS-6,5 буфером. Конъюгат выделился во фракциях свободного объема. Иммунолипосомальный 6-АРЕ готовили инкубированием этих липосом с C225 Fab'-PEG-DSPE с получением липосом, нагруженных лекарственным средством, при соотношении 30 мг C225 белка/ммоль липосомного фосфолипида в течение 30 минут при 60°С, охлажденном на льду в течение 15 минут и очисткой иммунолипосом гель-хроматографией на колонке с Сефарозой CL-4B и элюцией HBS-6,5 буфером (липосомы появлялись в свободном объеме колонки или вблизи него).
MDA-MB-468 EGFR-сверхэкспрессирующие клетки рака молочной железы человека и MCF-7 клетки рака молочной железы человека с низкой степенью экспрессии EGFR (АТСС, Rockville, MD) культивировали в их среде, рекомендуемой для роста клеток, и изучали цитотоксичность свободного, липосомального и анти-EGFR-иммунолипосомального 6-АРЕ по отношению к этим клеткам согласно способу примера 27. Эти клетки инкубировали со средой, содержащей лекарственное средство, в течение 6 часов, а затем - со средой, не содержащей лекарственного средства, в течение 3 дней. Результаты показаны на фигуре 16. В MDA-MB-468 клетках IC50 для свободного 6-АРЕ составил около 0,1 μг/мл, а для C225-ILs-APE около 0,9 μг/мл. В MCF-7 клетках IC50 составил (около 0,1) для свободного 6-АРЕ около 0,5 μг/мл, а для C225-ILs-APE около 14 μг/мл. IC50 для Ls-APE в обеих линиях клеток была более 30 μг/мл. Так что EGFR-нацеленные 6-АРЕ-нагруженные иммунолипосомы продемонстрировали антигенспецифичную цитотоксическую активность в сверхэкспрессирующих EGFR клетках рака молочной железы MDA-MB-468, но не в MCF-7 клетках рака молочной железы, для которых не характерна сверхэкспрессия EGFR. В MCF-7 клетках нацеленные и ненацеленные 6-АРЕ липосомы были одинаково активны.
Пример 37. Фармакокинетика липосомального 6-АРЕ, изученного на крысах.
Липососы с захваченными ТЕА-Pn раствором (557 мМ фосфатных групп, 500 мМ TEA, pH 5,8, осмолярность 480 ммоль/кг) и липидной композицией DSPC, холестерола и PEG-DSPE (мольное соотношение 3:2:0,015) готовили как в примере 11. Этанольный раствор липидов соединили при 60°C с 10 объемами водного ТЕА-Pn раствора и экструдировали 10 раз через две объединенные поликарбонатные мембраны с размером пор 80 нм. Неинкапсулированный ТЕА-Pn удаляли с использованием колонки с Сефарозой CL-4B и элюцией MES-Декстрозой (5 мМ MES-Na, 5% Декстрозы, pH 5,5). Размер липосом по QELS был 92,3±23,3 нм. Неизменяемая радиоактивная липидная метка [3H]-СНЕ была включена в липидную матрицу при 0,5 мКи/ммоль фосфолипида. Липосомы были загружены 6-АРЕ, как описано в примере 34.
Исследования фармакокинетики проводили по протоколу примера 9. Крысам-самкам Sim Albino (9 недель, 200 г) инъецировали i.v. 6-АРЕ в дозе 10 мг/мл. Отбирали кровь в предназначенные моменты времени и плазму анализировали на 6-АРЕ флуорометрией. Аликвоты плазмы (0,05-0,2 мл) смешивали с 1-2 мм 90% водного изопропанола - 0,1 н HCl и 6-АРЕ количественно определяли по флуоресценции как в примере 71. Содержание липида количественно оценивали по [3H]-CHE радиоактивной сцинтилляции.
Результаты показаны на фигуре 17. Время полужизни (t1/2) лекарственного средства в крови составляло 13,7 часов, а липида липосомы - 16,6 часов (группа А). Время полужизни выделения лекарственного средства из липосомы была 77,9 часов, показывая совершенно замечательную стабильность инкапсулирования (группа В).
Пример 38. Синтез и инкапсулирование в липосомы 2-(2-(N,N-диэтиламино)этил)эллиптициния (2-DAE).
2-(2-(N,N-диэтиламино)этил-эллиптициний-хлорид (NSC 359449) является противораковым производным эллиптицина, который получают алкилированием эллиптицина 2-(N,N-диэтиламино)этилхлоридом в метаноле в присутствии триэтиламина (см. Werbel L.M, Angelo M, Fry D.M, Worth D.F. J. Med. Chem. 1986, 29:1321-1322).
Липосомы, содержащие захваченные ТЕА-Pn, готовили как описано в примере 37. 2-DAE-2HCl инкубировали с ТЕА-Pn липосомами в 5 мМ HEPES-Na, 5% Декстрозы, рН 7,4, при соотношении 2-DAE/фосфолипид 100 μг/μмоль. Количество загруженного лекарственного средства было 88,2 μг АРЕ/μмоль PL (эффективность 88,2%).
Пример 39. Фармакокинетика липосомального 2-DAE, исследованная на крысах.
Фармакокинетику липосомального 2-DAE в крови (пример 38) изучали на крысах как в примере 37. t1/2 для 2-DAE был 17,8 часов, а для липосомной липидной матрицы -18,2 часа (А). Время полужизни выделения лекарственного средства из липосомы в крови t1/2 = 677 час.(В). Таким образом, эти липосомы были чрезвычайно стабильными в плане просачиваемости лекарственного средства в кровоток.
Пример 40. Включение винорелбина в липосомы с помощью ТЕА-Pn метода. Влияние рН.
Липосомы получали методом впрыскивания этанола как в примере 11, используя ТЕА-Pn раствор 0,608 M TEA, 0,65 M фосфатных групп, рН 6,1 и осмолярность 531 ммоль/кг, и 15-кратную экструзию липидной суспензии через две объединенные поликарбонатные мембраны с размером пор 100 нм. Размер пор полученных липосом по QELS был 108,3±17,1 нм. Винорелбин (VRB) в виде исходного раствора битартрата винорелбина 10 мг/мл USP добавили к липосомам в водном 5 мМ HEPES-Na, 5% декстрозы, рН 6,5, при соотношении лекарственное средство/фосфолипид 350 μг/μмоль, рН доводили до нужной величины, используя 1-5 н NaOH. Смесь инкубировали при 58±2°С в течение 30 минут. Смесь затем охлаждали на льду в течение 15 минут, а неинкапсулированное лекарственное средство удаляли гель-фильтрационной хроматографией на Сефадаксе G-75 с элюцией HBS-6,5 буфером (20 мМ HEPES-Na, 135 мМ NaCl, рН 6,5). Аликвоты очищенных липосом были затем солюбилизированы в кислом изопропаноле и проанализированы на содержание винорелбина, используя спектрофотометрию при 270 нм. Фосфолипиды липосом количественно определяли после экстракции, используя анализ фосфатов Bartlett'a (1959) смесью метанола и хлороформа.
Подсчитанные соотношения лекарственное средство/липид после загрузки были такими, что указаны в таблице 24. Включение винорелбина было количественным (т.е. практически 100%) и не зависело от рН в изученном диапазоне значений.
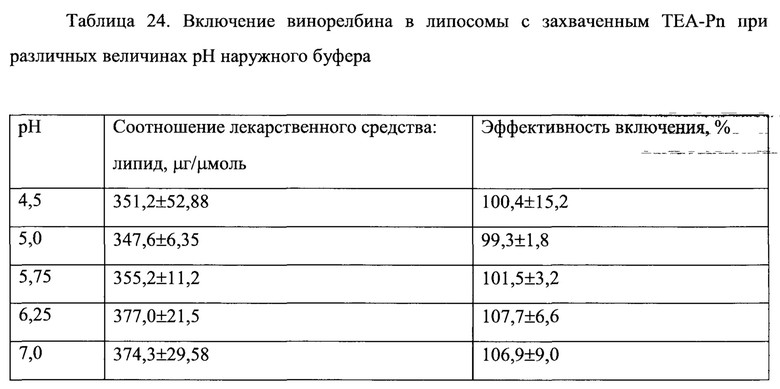
Пример 41. Липосомальный винорелбин, полученный ТЕА-Pn методом при различных соотношениях лекарственное средство/липид: эффективность включения и in vivo стабильность, исследованные на мышах.
Липосомы с захваченным ТЕА-Pn раствором получали согласно примеру 40, за исключением того, что [3H]-СНЕ включали в липидную матрицу при 1,5 мКи/ммоль фосфолипида. Размер липосом по QELS был 98,5±34,3 нм. Липосомы смешивали с битартратом винорелбина USP в водном буфере 5 мМ HEPES-Na, 5% декстрозы, рН 6,5, при соотношении лекарственное средство/фосфолипид 150-450 мг VRB/ммоль, и инкубировали при 58±2°С в течение 30 минут. Доведения рН не проводили, затем добавляли лекарственное средство. Нагруженные винорелбином липосомы (Ls-VRB) отделяли и анализировали на содержание лекарственного средства и фосфолипида как в примере 40.
Самкам Swiss Webster мышей возрастом 5-6 недель, по 3 животных в группе, внутривенно инъецировали Ls-VRB-Pn в дозе 5 мг VRB/кг. Липидная доза варьировалась согласно степени загрузки и могла быть определена по вышеуказанному вычисленному соотношению лекарственное средство/липид. Через 8 часов или 24 часа после инъекции животным давали анестезию, обескровливали, кровь собирали на льду во взвешенные пробирки, содержащие известные количества PBS с 0,04% ЭДТА. Клетки крови отделяли центрифугированием, а супернатанты исследовали на липосомные липиды подсчетом [3Н]-СНЕ радиоактивной сцинтилляции винорелбина с помощью ВЭЖХ следующим образом. В пробы впрыскивали винбластин (внутривенный стандарт), экстрагировали диэтиловым эфиром, выпаривали и остатки растворяли в подвижной фазе, состоящей из водного 50 мМ ацетата триэтиламмония (рН5,5) и ацетонитрила (58:42 по объему). Пробы загружали на колонку с диоксидом кремния с C18-обращенной фазой (Supeico С-18 колонка, 250 мм × 4 мм i.d. размер частиц 5 μм), которой предшествовала С-18 защитная колонка. Элюцию с колонки проводили изократически вышеуказанной подвижной фазой при скорости 1,0 мл/мин. VRB детектировали с помощью детектора поглощения при 280 нм. Типичное время удерживания для VRB и винбластина (внутренний стандарт) составило 9,1 мин. и 7,8 мин. соответственно.
Эти результаты показаны в таблице 25. Эффективность включения уменьшалась с повышением соотношения лекарственное средство/липид от практически 100% при 150 мг/ммоль до примерно 66% при 450 мг/ммоль. При этом отмечалось, что добавление батартрата винорелбина при соотношениях свыше 250 мг винорелбина на ммоль фосфолипида вызывало значительное подкисление суспензии липосом (рН < 4,0), что приводило к снижению эффективности включения. Таким образом, была установлена необходимость контроля рН на стадии загрузки лекарственного средства. Количества липосомной матрицы, определенные в крови через 8 часов, были в пределах от 30,4±6,6% от инъецируемой дозы (% id) до 38,6±5,2% независимо от абсолютного значения инъецируемого липида. Через 24 часа они были еще от 6,4% ID до 14,8% ID липидной матрицы, которую можно определить в крови. Количество лекарственного средства, которое оставалось инкапсулированным через 8 часов, колебалось от 37% до 65%. Однако, через 24 часа после инъекции уровни лекарственного средства падали ниже предела определения используемого аналитического метода.
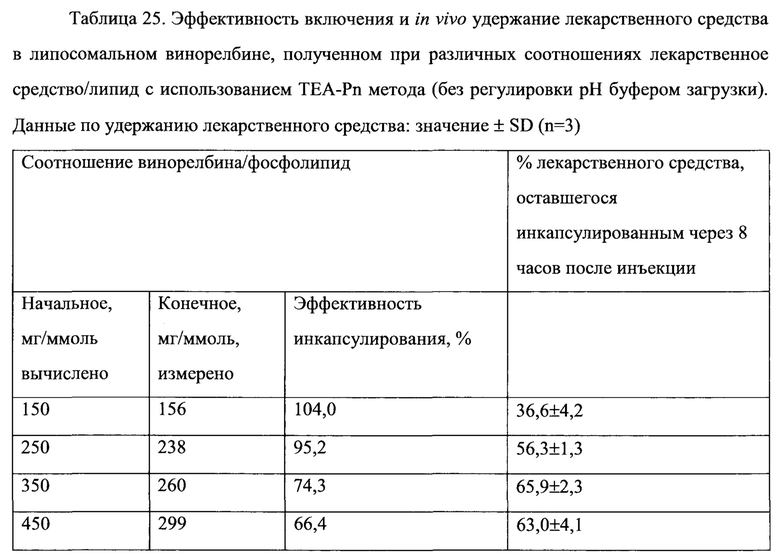
Пример 42. Включение винорелбина в липосомы с использованием TEA-SOS метода при различных соотношениях лекарственное средство/липид.
TEA-SOS липосомы для включения лекарственного средства готовили по примеру 40, за исключением того, что TEA-SOS раствор с 0,65 М TEA, pH 5,4, осмолярностью 521 ммоль/кг использовали вместо ТЕА-Pn раствора, а липосомы экструдировали через поликарбонатные мембраны с размером пор 80 нм. Размер липосом по QELS был 86,6±12,9 нм. VRB добавляли к липосомам в водном 5 мМ HEPES-Na, 5% Декстрозы, pH 6,5, при различных соотношениях лекарственное средство/фосфолипид и смесь затем инкубировали при 60°С в течение 30 минут. VRB-нагруженные липосомы были затем выделены и подвергнуты анализу как в примере 40.
Вычисленные значения соотношения лекарственное средство/липид для VRB липосом показана в таблице 26. Примечательно, что в отличие от полимерного аниона, способствующего загрузке, включение винорелбина в липосомы с полианионизированным сахаром (сахарозооктасульфат) было практически количественным независимо от соотношения лекарственное средство/липид, достигающего почти 450 мг VRB/ммоль фосфолипида, и только немного меньшим (88%) при 550 мг VRB/ммоль фосфолипида.
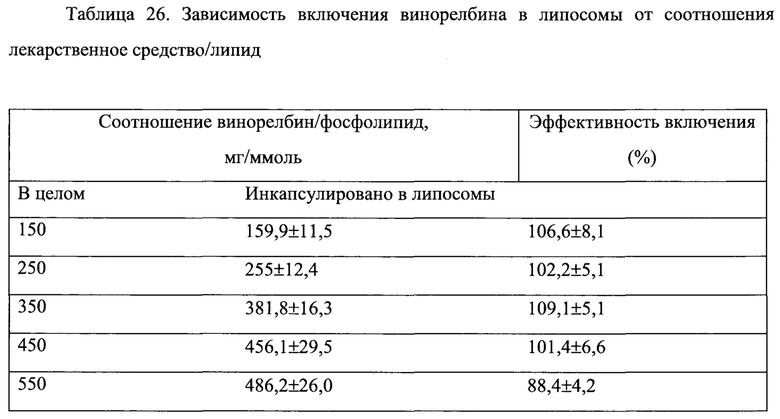
Пример 43. Получение HER2-нацеленных иммунолипосом с включенным винорелбином ТЕА-Pn методом и сравнительные фармакокинетические данные HER2-нацеленных и ненацеленных липосом с винорелбином, полученные на крысах.
Анти-HER2 scFv F5-PEG-DSPE конъюгат получали как в примере 19. HER2-нацеленные иммунолипосомы с винорелбином получали инкубацией ненацеленных липосом и винорелбином (пример 41, нагруженные при соотношении лекарственное средство/фосфолипид 350 мг/ммоль) с F5-PEG-DSPE конъюгатом (пример 19) в водном 20 мМ HEPES-Na, 135 мМ NaCl, pH 6,5, буфере при соотношении белок/фосфолипид 15 мг/ммоль при 60°С в течение 30 минут. Невключенный F5 конъюгат удаляли гель-хроматографией на колонке с Сефарозой 4 В и с элюцией тем же самым буфером. Ненацеленные липосомы (Ls-Pn-VRB) и HER2-нацеленные липосомы (F5-ILs-Pn-VRB) вводили i.v. самкам Albino крыс (возраст 8-9 недель, 200 г) в дозе 5 мг VRB/кг. В различные моменты времени кровь собирали как описано в примере 9 и анализировали на содержание VRB и липида как в примере 41. Время полужизни в крови для липосомных липидов и время выделения 50% лекарственного средства были вычислены на основе графиков «концентрация липида - время» или графиков «соотношение лекарственное средство/липид - время», соответственно, находя наилучшее соответствие моноэкспоненциальной кинетике, исползуя MICROSOFT EXCEL (Microsoft Corp.) при развертывании TREND функции. Результаты (фигура 18) показали, что оба типа липосом, как нацеленные, так и ненацеленные липосомы с винорелбином, имели идентичные фармакокинетические характеристики для лекарственного средства и для липида при времени полужизни липида около 12,1 часа и времени выделения 50% лекарственного средства, равном примерно 4,3 часа.
Пример 44. Получение и сравнительная in vivo стабильность липосом с винорелбином, получаемых с использованием солей аммония и замещенного аммония.
Раствор декстрансульфата аммония (DS-A) с рН 5,8, 0,65 М NH4+, осмолярностью 390 ммоль/кг и раствор декстрансульфата триэтиламмония (DS-TEA) с рН 6,0, 0,65 М NH4+, осмолярностью 465 ммоль/кг готовили из декстрансульфата с мол, массой 1000 (Sigma Chemical Co.) в соответствии со способом примера 4, используя титрование 12,4 М водным аммиаком или чистым триэтиламином, соответственно. Водный раствор сульфата аммония (S-A), 325 мМ, рН 5,1, осмолярностью 703 ммоль/кг, готовили из сульфата аммония аналитической степени чистоты. Все три раствора содержали менее чем 1% Na+ от общего содержания катионов. Липосомы, включающие эти растворы, были получены смешиванием с этанолом с последующей экструзией - методом примера 41 (DSPC/Холестерол/PEG-DSPE 3:2:0,015 мольное соотношение). Радиоактивную липидную метку [3H]-СНЕ включили в липидную матрицу при 1,5 мКи/ммоль фосфолипида. Стадия экструзии состояла из 10 пассажей через две объединенных 0,1 μм поликарбонатные мембраны. VRB добавили к липосомам в 5 мМ HEPES-Na, 5% Декстрозы, рН 6,5 при соотношении лекарственное средство/фосфолипид 350 мг/ммоль, рН доводили до 6,5, используя 1 н NaOH, и смесь инкубировали при 58-60°С в течение 30 минут. Затем реакционную смесь охладили на льду в течение 15 минут и неинкапсулированное лекарственное средство удаляли гельфильтрационной хроматографией на Сефадексе G-75 и элюцией водным 20 мМ HEPES-Na, 135 мМ NaCl, pH 6,5. Очищенные нагруженные винорелбином липосомы анализировали на VRB спектрофотометрически и на фосфолипид - анализом на фосфаты по Bartlett (1959) (См. примеры 70, 71). Фармакокинетические характеристики в крови для липосомного липида и лекарственного средства изучали на крысах как в примере 43.
Результаты показаны на фигурах 19-20 и в таблице 27. Липосомы, нагруженные декстрансульфатом триэтиламмония, сравнивали с липосомами, нагруженными с использованием аммониевой соли декстрансульфата. Неожиданно оказалось, что те липосомы, что были нагружены с помощью соли триэтиламмония, были значительно более стабильными, чем тем, что были нагружены с помощью соли аммония. Фармакокинетика самого липосомального носителя была близка аналогичным данным трех различных составов и была поэтому прежде всего зависима от применяемой липидной композиции. Утечка винорелбина из Ls-VRB, нагруженных с использованием декстрансульфата триэтиламмония, была почти в три раза медленнее, чем из липосом, нагруженных с помощью декстрансульфата аммония. Липосомы, нагруженные с помощью аммонийсульфата, имели самую высокую скорость утечки лекарственного средства из них.
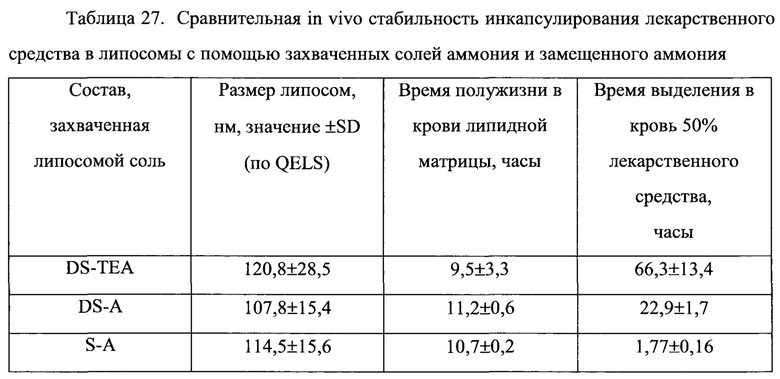
Пример 45. Получение и in vivo стабильность нагруженных винорелбином липосом с различным размером.
[3H]-СНЕ-меченные липосомы (1,5 мКи/ммоль фосфолипида) с захваченным раствором сахарозооктасульфата триэтиламмония (0,65 М TEA, pH 6,4, осмолярность 502 ммоль/кг) получали смешением с этанолом и экструзией по способу примера 11. Стадия экструзии включала 15 пассажей через две объединенные поликарбонатные мембраны с размером пор 0,05, 0,08 или 0,1 μм. Загрузка винорелбина, выделение липосом с винорелбином и оценку свойств этих липосом проводилась по способу, описанному в примере 40. Крысы-самки Albino (возраст 8-9 недель, 200 г) использовались для изучения липосомной in vivo стабильности. Липосомные липиды и фармакокинетика лекарственного средства изучались на крысах как в примере 43.
Результаты показаны на фигурах 21, 22 и в таблице 28, приведенной ниже. Липосомы, экструдированные через 0,05, 0,08 и 0,1 μм поликарбонатные фильтры, сравнивали, и было показано, что они обладают близкими фармакокинетическими свойствами в плане как лекарственного средства, так и липосомного носителя, а также аналогичной степенью просачиваемости (утечки) содержимого. Выделение лекарственного средства из липосом в кровь характеризовалось временем (продолжительностью) выделения 50% лекарственного средства в пределах примерно 40-80 часов, значительно выше 24 часов.

Пример 46. Получение HER2-нацеленных липосом с винорелбином с использованием метода захвата TEA-SOS и фармакокинетические характеристики HER2 scFv-нацеленного и ненацеленного иммуиолипосомального винорелбина, исследованные на крысах.
Липосомы получали, нагружали винорелбином при соотношении лекарственное средство/фосфолипид 350 мг/ммоль и исследовали как описано в примере 43, за исключением того, что TEA-SOS раствор примера 45 был заменен ТЕА-Pn раствором. Стадия экструзии включала 15 пассажей через поликарбонатные фильтры с размером пор 0,08 μм. Размер липосом по QELS составлял 95,0±26 нм. F5 scFv - сшитые анти-HER2-иммунолипосомы с винорелбином получали из липосом с винорелбином, а фармакокинетику липосомальных липидов и лекарственного средства HER2-нацеленного и ненацеленного липосомального винорелбина изучали на крысах как описано в примере 43. Время полужизни циркулирующего в крови липосомального липида было 11,4 часа и 10,3 часа, а время выделения 50% лекарственного средства составило 30,9 часа и 30,3 часа для F5-ILs-VRB и Ls-VRB, соответственно. Таким образом, фармакокинетические характеристики для липида и для лекарственного средства Ls-VRB и F5-ILs-VRB были очень близкими, показывая, что введение scFv-PEG-DSPE конъюгата и не влияет на клиренс самого носителя, и не приводит к повышенной просачиваемости (утечке) лекарственного средства через носитель в процессе циркулирования (фигуры 23, 24).
Пример 47. Получение и фармакокинетические свойства липосом с винорелбином, включающих неионные липидные производные поли-(этиленгликоля).
Метокси-PEG (мол. вес 2000)-производное синтетического С20-керамида (PEG-керамид) было получено от Northern Lipids, Inc., Canada. Метокси-PEG (Мол. вес. 2000)-дистеароилглицерин (PEG-DSG)(SUNBRIGHT G-S20) был получен от NOF Corp., Japan. Липосомы, имеющие липидную композицию DSPC, холестерин и PEG-липид (PEG-керамид или PEG-DSG) при мольном соотношении 3:2:0,3 и захваченном TEA-SOS растворе (М TEA, рН 6,4, осмолярность 502 ммоль/кг) готовили методом смешивания с этанолом и экструзией по примеру 11. Стадия экструзии включала два пассажа через два объединенных поликарбонатных мембранных фильтра с размером пор 0,2 μм и 10 пассажей с использованием фильтров с размером пор 0,08 μм. Липосомы нагружали винорелбином при соотношении лекарственное средство/фосфолипид 350 мг/ммоль, характеризуемым размером, концентрацией липида и лекарственного средства, и их фармакокинетика изучалась на крысах как описано в примере 46. Оба состава показали пролонгированное время циркуляции липидной матрицы и медленное выделение лекарственного средства in vivo при почти 50% доле лекарственного средства, остающегося инкапсулированным после 24 часов в крови in vivo, как показано в таблице 29 ниже.
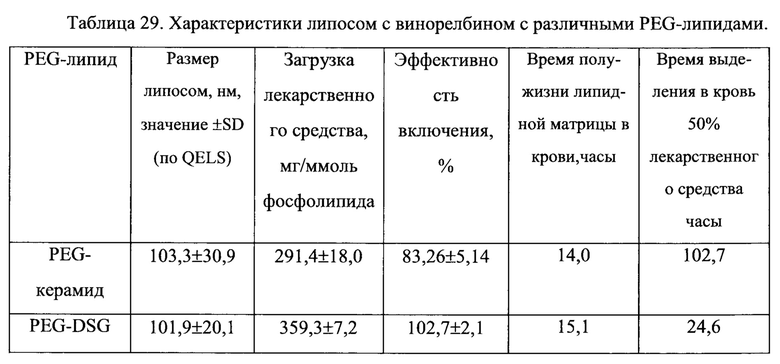
Примечательно, что повышенное PEG-илирование этих липосом (содержание PEG липида около 5,7 моль. % от общего количества липидов) практически не оказывало влияния на долговечность циркуляции липосом в крови, сопоставимую с подобными, подобранными по размеру липосомами, имеющими низкую степень PEG-илирования около 0,3 моль. % от общего количества липида) (Пример 45, 109,6 нм, t1/2=14,3 часа; 98,5 нм, t1/2=13,0 часов).
Пример 48. Получение HER2- нацеленного липосомального винорелбина и цитотоксичность свободного, HER2-нацеленного и ненацеленного липосомального винорелбина в отношении MDA-MB-453 клеток in vitro.
Винорелбин-нагруженные липосомы (Ls-VRB) получали как в примере 42 (без [3Н]-СНЕ), используя загрузку лекарственного средства при рН 6,0 и 350 μг винорелбина/цмоль фосфолипида. Анти-HER2 иммунолипосомальный винорелбин (F5-ILs-VRB) был получен инкубированием этих липосом с F5-PEG-DSPE конъюгатом, как описано в примерах 19 и 42, выше, за исключением того, что [3Н]-СНЕ не добавляли. «Свободный» винорелбин получали разбавлением раствора битартрата винорелбина (10 мг/мл) USP в среде для культуры клеток.
MDA-MB-453 представляет собой клетки аденокарциномы молочной железы человека (American Type Culture Collection, Rockville, MD), которые умеренно сверхэкспрессируют HER2 рецептор (около от 3×104 до 1×105 копий на клетки). Цитотоксичность VRB, выделившегося как свободное лекарственное средство, как ненацеленный липосомальный винорелбин или как HER2-нацеленный (F5)-иммунолипосомальный винорелбин, специфичный к MDA-MB-453 клеткам, определяли как указано в примере 27, за исключением того, что клетки помещали в 96-луночные микротитровальные планшеты в рекомендуемых поставщиком условиях роста (Leibowitz L-15 с 10% фетальной сыворотки теленка, без подачи СО2) при концентрации 10000 клеток/на лунку, и композиции лекарственного средства добавляли в серии 1:3 ступенчатых разбавлений, начиная с 0,03-0,1 мг/мл. Данные по жизнеспособности клеток наносят, строя график, против концентрации лекарственного средства (фигура 25) и концентрации лекарственного средства, требовавшиеся для уменьшения жизнеспособности клеток до 50% (IC50), вычисляли по графику, IC50 Р5-нацеленных липосом и винорелбином (0,06 μг/мл) близка к IC50 свободного лекарственного средства (0,07 μг/мм) и существенно ниже IC50 для ненацеленных липосом (2,2 μг/мл). Это показывает 37-кратное увеличение активности как результат выделения лекарственного средства, специфично нацеленного на раковые клетки.
Пример 49. Цитотоксичность свободного, HER2-нацеленного и ненацеленного липосомального винорелбина в отношении CaLu-3 клеток in vitro.
Липосомы и методы предыдущего примера (пример 48) использовались для изучения цитотоксичности свободного винорелбина, Ls-VRB и F5-ILs-VRB в HER2-суперэкспрессирующих CaLu-3 клетках крупноклеточной карциномы легкого человека (American Type Culture Collection, Rockville, MD). Клетки выращивали в RPMI среде с 10% фетальной сыворотки теленка в присутствии 5% СО2.
Результаты показаны на фигуре 26. IC50 для свободного VRB было 1,2 μг/мл, 10 μг/мл для F5-ILS-VRB и 50 μт/мл до ненацеленного Ls-VRB. Это показывает 5-кратное увеличение активности инкапсулированного в липосоме лекарственного средства как функции направленной доставки в клетки.
Пример 50. Цитотоксичность свободного, HER2-нацеленного и ненацеленного лпосомального винорелбина в отношении SKBr-3 клеток in vitro.
Липосомы и методы примера 18 использовались для изучения цитотоксичности свободного винорелбина, Ls-VRB и F5-ILs-VRB в HER2-суперэкспрессирующих SKBк-3 клетках карциномы молочной железы человека (American Type Culture Collection, Rockville, MD), за исключением того, что клетки выращивали на модифицированной Мс Coy 5А среде с 10% фетальной сыворотки теленка в присутствии 5% СО2, помещали в планшеты с концентрацией 5000 клеток/на лунку и инкубировали лекарственное средство с клетками в течение 6 часов.
Результаты показаны на фигуре 27. IC50 для свободного VRB было 0,28 μг/мл, 0,17 μг/мл - для F5-ILs-VRB и 0,8 μг/мл - для ненацеленного Ls-VRB. Это показывает 4,7-кратное увеличение активности лекарственного средства как функции нацеленной доставки.
Пример 51. In vivo противоопухолевая активность липосомального винорелбина в НТ29 ксенотрансплантатах рака кишечника человека, изученная на мышах.
Маленькие униламеллярные (однослойные) липосомы (93,2±26,4 по QELS) готовили из дистеароилфосфатидилхолина, холестерола и PEG-DSPW (3:2:0,045 мольное соотношение) гидратацией из концентрированного этанольного раствора в водный раствор сахарозооктасульфата триэтиламмония (0,6 М триэтиламмоний, рН 5,7-6,2) с последующей неоднократной экструзией через поликарбонатные мембраны (размер пор 100 нм), удалением экстралипосомальной полианионной соли и нагружением винорелбином путем инкубации липосом в изоосмотическом буфере рН 6,5, соотношение лекарственное средство/липид равно 325 мг VRB) ммоль фосфолипида при 60°С, как описано в примере 42.
Самкам BALB/c гомозиготных бесшерстных мышей (6-8 недель, вес 17-20 г) инъецировали подкожно в боковую область 1×106 НТ-29 клеток карциномы кишечника человека (American Type Culture Collection, Rockville, MD). Начиная с 16-го дня после инокулирования опухоли, когда опухоль достигала в диаметре 5-8 мм, мышей произвольно делили на группы по шесть животных в каждой группе и обрабатывали свободным или липосомальным винорелбином в дозе 5 мг/кг через хвостовую вену каждые три дня, введя в общей сложности четыре инъекции. В контрольной группе мышей обрабатывали равным объемом физраствора. Размер опухоли каждой мыши измеряли с помощью циркуля, а объем опухоли вычисляли по формуле: (длина опухоли) × (ширина опухоли) 2/2. Чтобы оценить токсичность, связанную с лечением, животных взвешивали также дважды в неделю. Было показано, что липосомальный винорелбин, значительно более эффективен в подавлении роста НТ-29 опухолей, чем свободный винорелбин, вызывая регрессию опухолей, в то время как в группах, где применялось свободное лекарственное средство, опухоли по-прежнему продолжали расти (фигура 28). Было небольшое изменение веса животных во время курса лечения, показывающего, что лечение было успешным, хорошо воспринималось и что включение в липосомы не увеличивало токсичности лекарственного средства (фигура 29).
Пример 52. In vivo противоопухолевая активность липосомального винорелбина по отношению к С-26 сингенным опухолям рака кишечника мыши.
Липосомальный винорелбин и свободный винорелбин готовили по примеру 48. Мышам-самцам BALB/c (6-8 недель, вес 17-20 г) вводили подкожно 2×105 С-26 клеток карциномы кишечника мыши. На 17-ый день после инокуляции, когда диаметр опухоли достигал 5-8 мм, мышей произвольно разделили на шесть групп для лечения по 5 животных в группе. Мышам с опухолями инъецировали через хвостовую вену свободный винорелбин в концентрации 6 мг/кг, 8 мг/кг, или 12 мг/кг и липосомальный винорелбин в концентрации 4 мг/кг или 6 мг/кг каждые три дня в виде четырех инъекций. В контрольной группе мышам вводили равный объем обычного физраствора. Размеры опухоли и вес тела животных определяли как в примере 51. Липосомальный винорелбин, даже при 4 мг/кг, был значительно более эффективен в уменьшении роста опухоли, чем свободное лекарственное средство при 12 мг/кг (фигура 30.). Вес тела животных в курсе лечения показал небольшое изменение (< 10% уменьшение), показывающее, что токсичность липосомального винорелбина не увеличивалась по сравнению со свободным лекарственным средством (фигура 31).
Пример 53. In vivo противоопухолевая активность HER2 - нацеленного липосомального винорелбина в отношении ВТ-474 трансплантированной опухоли рака молочной железы человека, изученная на мышах: влияние загрузки противоиона.
VRB - нагруженные липосомы размером 99,5±10,2 нм получали ТЕА-Pn методом примера 41 и TEA-SOS методом примера 42 соответственно, при этом не добавлялся [3Н]-СНЕ. VRB загружали при соотношении лекарственное средство/фосфолипид, равном 350 мг/ммоль. HER2-нацеленный липосомальный винорелбин получали инкубированием этих липосом с F5-PEG-DSPE конъюгатом (см. пример 19) как описано в примере 43. Трансплантаты ВТ 474 HER2- сверхэкспрессирующей карциномы молочной железы человека выращивали на гомозиготных бесшерстных мышах как в примере 10. На 25-ый день после заражения клетками опухоли, когда опухоли достигали размера почти 200 мм3 (порядка 144-309 мм3), мышей произвольно разделили на четыре группы, по восемь животных в группе и обрабатывали i.v. в дозе 5 мг/кг VRB, F5-ILS-VRB с Pn в качестве противоиона или F5-ILs-VRB с SOS в качестве противоиона при еженедельной дозе 5 мг/кг за все три инъекции. Контрольная группа получала равный объем обычного физраствора. Размер опухоли и вес тела животных регистрировали как в примере 10. HER2-нацеленной липосомальный винорелбин, загруженный с использованием сахарозооктасульфата, был заметно более эффективным в подавлении роста опухоли, чем точно такой же нацеленный конструкт, загруженный с использованием поли(фосфата), и оба иммунолипосомальных препарата оказались значительно более эффективными, чем свободный винорелбин при введении в дозе 5 мг VRB на кг (фигура 32). Получившие лекарственное средство мыши характеризовались небольшим изменением веса, что показано хорошую переносимость лекарственного средства (фигура 33).
Пример 54. In vivo противоопухолевая активность HER2-нацеленного липосомального винорелбина в отношении ВТ-474 трансплантированной опухоли рака молочной железы человека, изученная на мышах: влияние PEG-илирования.
Липосомы на основе DSPC и холестерола при молярном соотношении 3:2 получали согласно примеру 48 гидратацией липидной матрицы DSPC, холестерола и PEG-дистероилглицерина с PEG мол. массы 2000 (GS-20, NOF Corp., Japan) при молярном соотношении 3:2:0,015 («0,5% PEG») или 3:2:0,3 («10% PEG») посредством этанольного метода в водном сахарозооктасульфате триэтиламмония с последующей мембранной экструзией соответственно примеру 48. VRB загружали в липосомы при соотношении лекарственное средство/фосфолипид 350 мг/ммоль. F5 иммунолипосомальный винорелбин получали инкубированием этих липосом с F5-PEG-DSPE конъюгатом (пример 19) как описано в примере 43.
Бесшерстных мышей с введенными ВТ-474 ксенотрансплантатами обрабатывали i.v. свободным VRB, F5-ILs-VRB-«0,5% PEG» или F5-ILs-VRB- «10%PEG» в дозе 5 мг/кг как в примере 53. Как показано на фигуре 34, F5-ILs-VRB с большей степенью PEG-илирования, обеспеченной неионным PEG производным липида PEG-DSG, был значительно более эффективным в подавлении роста опухоли, чем F5-ILs-VRB с более низким количеством PEG-DSG, хотя оба препарата были более активными, чем свободное лекарственное средство.
Пример 55. In vivo противоопухолевая активность EGFR-нацеленного липосомального винорелбина в отношении U87 ксенотрансплантированной опухоли рака мозга человека, изученная на мышах.
Липосомы (размером 86,6±12,9 нм по QELS) с инкапсулированным 0,65 М TEA-SOS раствором получали и нагружали VRB согласно примеру 42. Анти-EGFR-иммунолипосомальный VRB (С225 Fab'-ILs-VRB) получали инкубированием VRB липосом с PEG-DSPE конъюгатом Fab'-фрагментов анти-EGFR антитела, как описано в примере 36.
Самцам NCR nu/nu мышей (5-6 недель, 17-20 г) подкожно в область бока инъецировали 1×107 U87 клеток глиобластомы человека (АТСС), суспендированных в ростовой среде при общем объеме 150 μл. Когда опухоль достигла среднего размера 250 мм3, мышей произвольно разделили на четыре группы по 10-12 животных. Трижды в неделю мышам делали i.v. инъекции «свободного» VRB, ненацеленного Ls-VRB или С225 Fab'-ILs-VRB в дозе 5 мг VRB/кг.Контрольная группа получала такой же объем физраствора. Размер опухолей и вес тела животных регистрировались как в примере 10. C225-Fab'-ILs-VRB был значительно более эффективным в подавлении роста трансплантатов EGFR-сверхэкспрессирующей опухоли рака головного мозга человека, чем ненацеленный липосомальный винорелбин и свободный винорелбин в той же дозе (фигура 35).
Пример 56. Получение и фармакокинетика доксорубицина, инкапсулированного в липосомы методом триэтиламмонийсульфата.
Липосомы с различным составом липидной матрицы (как показано в нижеприведенной таблице) получали как описано в примере 2. N-Глутарил-DSPE (Glu-DSPE) был от Avanti Polar Lipids, AL, USA. Чистая липидная пленка образовывалась из липидного раствора в хлороформе при роторном испарении, следы летучих веществ удалялись под вакуумом (90 μм Hg, 2 часа), липидную пленку гидратировали в растворе триэтиламмоний-сульфата (TEA-SO4) /0,65 н TEA), подвергали шести циклам быстрого замораживания и оттаивания и экструдировали через два объединенных поликарбонатных фильтра с размером пор 0,1 μм десять раз и через два объединенных поликарбонатных фильтра с размером пор 0,05 μм десять раз. Для обсчета липидной матрицы в пробах крови в липидную матрицу включали [3Н]-СНЕ при 0,5-1,5 мКи/ммоль фосфолипида. Липосомы с захваченным раствором TEA-SO4 нагружали доксорубицином согласно примеру 2. Липосомы в HEPES-буферном солевом растворе (20 мМ HEPES-Na, 135 мМ NaCl, рН 6,5) инкубировали с гидрохлоридом доксорубицина (при соотношениях лекарственное средство/фосфолипид 140-170 мг/ммоль) при 60°С в течение 45 минут с последующим охлаждением на льду и удалением неинкапсулированного доксорубицина гель-хроматографией. Доксорубицин анализировали спектрофотометрией (пример 71), а анализ фосфолипидов проводили методом Bartlett'a (пример 70).
Свойства полученных в результате липосом объединены в таблице 30, ниже.
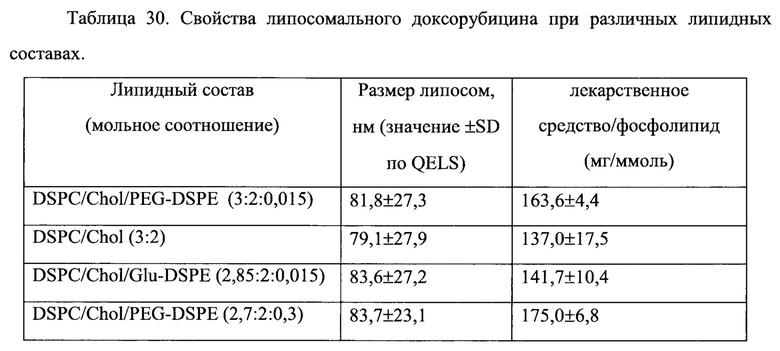
Пример 57. Доксорубицин-нагруженные липосомы и анти-HER2 иммунолипосомы, полученные ТЕА-сульфатным методом: получение и in vivo противоопухолевая активность в отношении HER2-сверхэкспрессирующих ксенотрансплантатов рака молочной железы человека.
Доксорубицин-нагруженные липосомы с различным липидным составом и свойствами (перечисленными в таблице, ниже) получали как описано в примере 56. Доксорубицин-нагруженные анти-HER2 иммунолипосомы готовили из доксорубицин-нагруженных липосом совместной инкубацией с анти-HER2 scFv F5-PEG-DSPE конъюгатом (приблизительно 30 scFv/липосома) как описано в примере 19. На NCR пи/пи мышах выращивались подкожные ксенотрансплантаты (ВТ-474) опухоли молочной железы человека, животных (в группах по 10-12 мышей) обрабатывали липосомальным или анти-HER2 иммунолипосомальным доксорубицином в дозе 5 мг/кг еженедельно один раз в течение трех недель, пока опухоли не достигали среднего размера 200 мм3, а рост опухоли и вес тела животных подвергали мониторингу как описано в примере 29. Для составов ненацеленного липосомального доксорубицина изучали липидные композиции, которые не содержали PEG-DSPE, содержали 0,5% моль. PEG-DSPE или 10% моль. PEG-DSPE (здесь количества PEG-DSPE выражены в мольных % к липосомальному фосфолипиду). Результаты (фигура 37, таблица 31) показали, что все процедуры по введению доксорубицина были эффективными в плане торможения роста опухоли. На основе размеров опухоли на 53-ий день после заражения различия в ингибировании роста опухоли среди трех ненацеленных типов липосом не увеличились до статистической значимости (ANOVA ρ=0,08), а иммунолипосомальный доксорубицин был значительно более эффективным, чем ненацеленный липосомальный доксорубицин (ANOVA ρ=5,5×10-10), притом что «10% PEG-DSPE» состав был более эффективным, чем «0,5% PEG-DSPE» (t - тест Стьюдента, ρ=0,027). В «10% PEG-DSPE» F5-ILs группе опухоли регрессировали до 1 мм3 или меньше у 67% животных, тогда как в «0,5% PEG-DSPE» F5-ILs группе только у 9%. В контрольной группе (введение физраствора) опухоли превышали приемлемую предельную степень веса тела на 15% на 38-43 день.
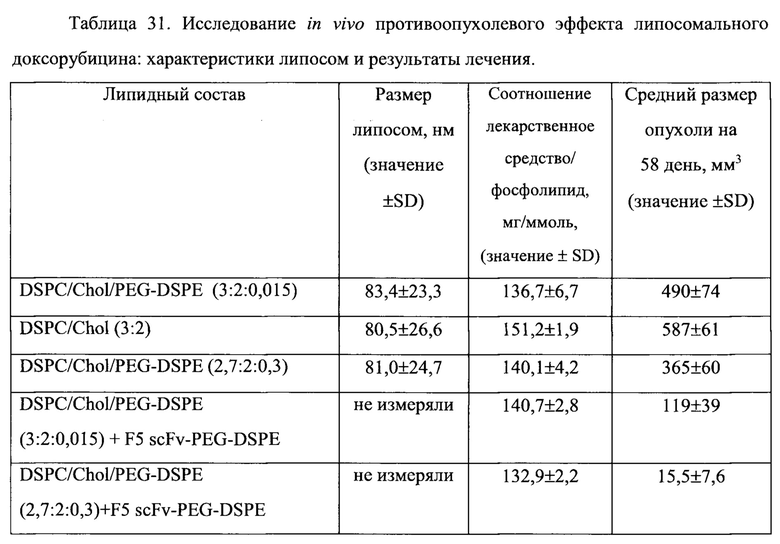
Пример 58. Получение липосомального винбластина и фармакокинетика в крови липосомального винбластина у крыс.
Липосомы с захваченным водным раствором TEA-SOS (0,65 М TEA, рН 6,4, осмолярность 502 ммоль/кг) и с размером 99,5±10,2 нм (значение ±SD по QELS) получали способом по примеру 11, используя двукратную экструзию через две объединенные поликарбонатные мембраны с размером пор 0,2 μм и десятикратную через две объединенные поликарбонатные мембраны с размером 0,08 μм. Винбластин (VBL) в форме винбластин-сульфата USP добавляли при соотношении лекарственное средство/фосфолипид 150 мг/ммоль. рН смеси лекарственное средство/липосомы доводили до 6,5, используя 1 н NaOH и смесь затем инкубировали при 60°С в течение 30 минут.Реакцию потом охлаждали на льду в течение 15 минут и неинкапсулированное лекарственное средство удаляли гельфильтрационной хроматографией с Сефадексом G-75, проводя элюцию 5 мМ HEPES-Na, 135 мМ NaCl, рН 6,5. Очищенные липосомы затем подвергали анализу на VBL спектрофотометрией и на фосфолипиды методом Bartlett'a как в примерах 70 и 71. [3Н]-СНЕ включали в состав при соотношении 1,5 мКи/ммоль фосфолипида. Липосомальный винбластин содержал 152,4±12,0 мг VBL/ммоль фосфолипида (количественное инкапсулирование).
Фармакокинетику в крови липосомального винбластина изучали на самках крыс Albino (возраст 8-9 недель, 200 г) при дозе 5 мг VBL/кг как описано в примере 9. Винбластин количественно определяли в пробах плазмы крови как описано в примере 41 (используя винорелбин как внутренний стандарт). Липосомы с винбластином показали хорошую долговечность циркуляции (время полужизни липидного компонента в плазме составило 12,8±0,04 часа) (фигура 38) и очень хорошую стабильность в плане просачиваемости лекарственного средства через липосомы, обеспечивающую после 24 часов сохранение инкапсулированным более чем 70% винбластина по сравнению с первоначальной загрузкой (Фигура 39). Было установлено, что пост-инъекционное время достижения выделения 50% инкапсулированного лекарственного средства составляло 40,6±1,2 часа.
Пример 59. Получение липосом, нагруженных винкристином с помощью TEA-SOS методам и влияние рН на эффективность включения.
Липосомы с размером 86,6±12,9 нм (по QELS), липидным составом DSPC/Chol/PEG-DSPE в молярном соотношении 3:2:0,015 и захваченным водным раствором TEA-SOS (0,65 М TEA, рН 5,4, осмолярность 521 ммоль/кг) готовили способом примера 11, используя стадию 16-кратной экструзии через две объединенные поликарбонатные мембраны с размером пор 0,08 μм. Винокристин (VCR) добавили к липосомам в 5 мМ HEPES-Na водном буфере с 5% декстрозы, рН 6,5 в виде сульфата винкристина при соотношении лекарственное средство/фосфолипид 350 μг винкристина/ммоль фосфолипида, рН доводили до указанного значения, используя 1 н NaOH, смесь инкубировали при 60°С в течение 30 минут, охлаждали на льду 15 минут и липосомы отделяли от неинкапсулированного лекарственного средства, используя гель-фильтрационную хроматографию на Сефадексе G-75 с элюцией HBS-6,5 (20 мМ HEPES, 135 мМ NaCl, рН 6,5). Очищенные липосомы анализировали затем на винкристин спектрофотометрическим методом при поглощении при 265 нм после солюбилизации в кислом изопропаноле, а на содержание фосфолипидов - методом фосфатов Bartlett'a (1959).
Результаты показаны ниже в таблице 32. Загрузка лекарственного средства была более 90% в интервале рН 4,5-7,5 и практически количественной при рН 5,0-7,5. При рН 3,5, т.е. при рН, который наблюдается в смеси липосом после добавления лекарственного средства, но без доведения рН, степень включения была значительно ниже.
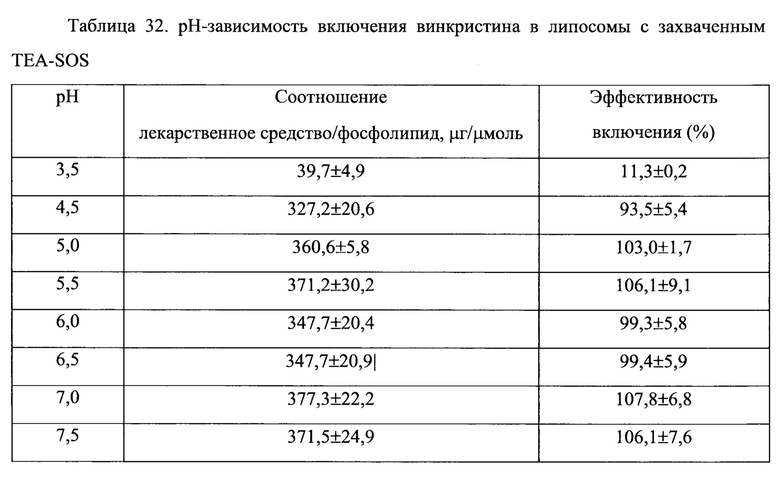
Пример 60. Получение липосом, нагруженных винкристином с помощью TEA-SOS метода: влияние соотношения лекарственное средство/липид на эффективность включения.
SOS-TEA - содержащие липосомы получали как в примере 59 и загружали винкристин-сульфатом при соотношении лекарственное средство/фосфолипид 150-550 μг винкристина/пмоль фосфолипида при рН 6,5 в соответствии с процедурой примера 59. Липосомы, отделенные от неинкапсулированного лекарственного средства, затем подвергали анализу на VCR спектрофотометрией и на липосомные фосфолипиды с помощью метода Bartlett'a (1959). Эффективность включения лекарственного средства была выше 90% во всем исследованном диапазоне соотношений лекарственное средство/липид и была практически количественной при 150-450 μг винкристина/μмоль фосфолипида (таблица 33).
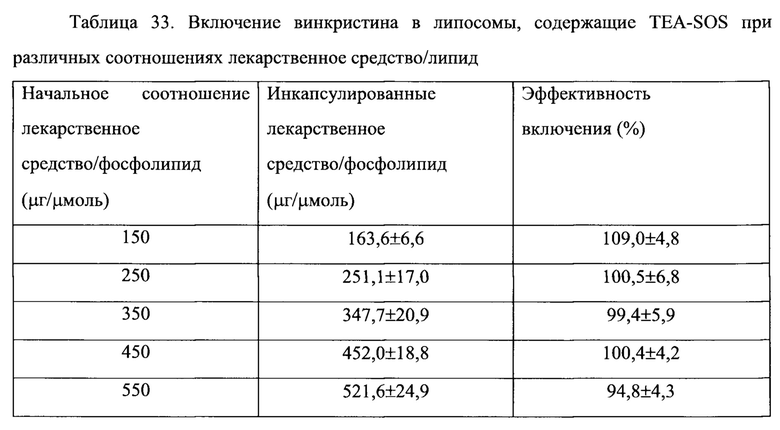
Пример 61. Получение иммунолипосомального винкристина и цитотоксичность липосомального и иммунолипосомального винкристина в отношении раковых клеток in vitro.
Липосомальный винкристин (Ls-VCR) получали как описано в примере 59 при соотношении лекарственное средство/фосфолипид 350 мг/ммоль. HER2-специфичный F5-иммунолипосомалный винкристин (F5-ILs-VCR) готовили из липосомального винкристина соинкубацией с анти-HER2 scFv F5-PEG-DSPE конъюгатом как описано в примере 19. Раствор «свободного» винкристина (VCR) готовили растворением сульфата винкристина USP в воде с последующей стерилизующей фильтрацией. Цитотоксичность VCR, Ls-VCR и F5-ILs-VCR в отношении HER2-сверхэкспрессирующих клеток SKBr-3 (АТСС) карциномы молочной железы человека определяли анализом основанной на МТТ жизнеспособности клеток по процедуре 27, согласно которой клетки инокулировали в 96-луночные микротитровальные планшеты в концентрации 5000 клеток/на лунку, оставляли на ночь, инкубировали со средой, содержащей лекарственное средство, в течение 4 часов, с последующей инкубацией в среде, не содержащей лекарственное средство, в течение 3 дней. Результаты показаны на фигуре 40. IC50 составляла 75 нг/мл для свободного VCR, 11 нг/мл для F5-ILs-VCR и 3 μг/мл для Ls-VCR. Нацеленный липосомальный винкристин, полученный согласно изобретению, был в 6,8 раз более активным, чем свободное лекарственное средство, и в 273 раза более активный, чем ненацеленное липосомальное лекарственное средство, показывая значительное увеличение противораковой активности как проявление специфичной в отношении клеток доставки лекарственного средства.
Пример 62. Фармакокинетика Ls-VCR в крови крыс.
Липосомы с захваченным SOS-TEA раствором (0,65 М TEA, рН 5,8, осмолярность 530 ммоль/кг) и липидным составом DSPC/Chol/PEG-DSPE (молярное соотношение 3:2:0,015), содержащим также [3Н]-СНЕ при 1,5 мКи/ммоль фосфолипида, готовили способом примера 11, используя стадию 10-кратной экструзии через две объединенные поликарбонатные мембраны с размером пор от 80 до 100 нм. Липосомы загружали VCR при рН 6,5, соотношении лекарственное средство/фосфолипид 350 мг/ммоль, как описано в примере 59. VCR-нагруженные липосомы вводили i.v. самкам Albino крыс (180-220 г) при дозе 5 мг VCR/кг, и фармакокинетику лекарственного средства и липосомного липида в крови изучали как описано в примере 9. Количество VCR в пробах крови определяли ВЭЖХ как описано в примере 41, за исключением того, объемное соотношение водного ацетата триэтиламмония (рН 5,5) и ацетонитрила в подвижной фазе было 65:35. Типичное время удерживания составляло для VCR 8,8 мин. Результаты показаны на фигуре 41 и в таблице 34. Оба препарата имели большую долговечность циркуляции (время полужизни в крови составляло 12-17 часов). Липосомальный винкристин был замечательно стабилен в отношении утечки лекарственного средства в обоих препаратах (время полужизни более 120 часов) (фигура 42).
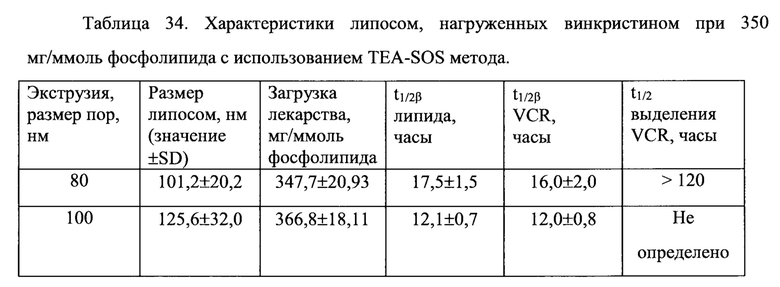
Пример 63. Фармакокинетика Ls-VCR в крови крыс при различных соотношениях лекарственное средство/липид.
Липосомы с захваченным SOS-TEA раствором (0,65 М TEA, рН 6,4, осмолярность 485 ммоль/кг) и липидным составом DSPC/Chol/PEG-DSPE (молярное соотношение 3:2:0,015), содержащим также [3Н]-СНЕ при 1,5 мКи/ммоль фосфолипида, получали по способу примера 11 с использованием 10-кратной экструзии через две объединенные поликарбонатные мембраны с размером пор 50 нм или 80 нм. Липосомы загружали винкристином при рН 6,5 как описано в примере 59 добавлением исходного 20 мг/мл водного раствора сульфата винкристина при вычисленном соотношении лекарственное средство/липид 100,200 или 350 мг/ммоль фосфолипида. Эффективность загрузки лекарственного средства была выше 96% для всех препаратов. VCR-нагруженные липосомы вводили i.v. самкам Albino крыс (возраст 8-9 недель, 190-220 г) в дозе 5 мг VCR/кг, и фармакокинетику лекарственного средства и липосомного липида в крови изучали как описано в примере 62. Результаты показаны в таблице 35. Липосомальный винкристин имел хорошую долговечность циркуляции (время полужизни лекарственного средства составило около 20-30 часов) и был исключительно стабильным при всех исследованных размерах (липосом) и соотношениях лекарственное средство/липид (время выделения 1/2 лекарственного средства было свыше 93 часов).
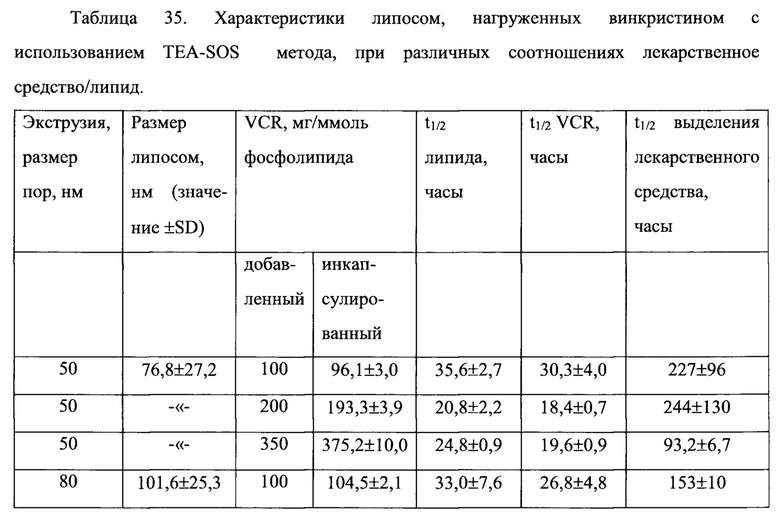
Пример 64. Получение HER2-нацеленного липосомального винкристина и противоопухолевая активность ненацеленного и HER2-нацеленного липосомального винкристина в отношении HER2-сверхэкспрессирующих ксенотрансплантатов опухоли рака молочной железы человека у мышей.
Винкристин-нагруженные липосомы (Ls-VCR-SOS) были получены с использованием TEA-SOS метода по примеру 63 (с исключением [3Н]-СНЕ компонента) с помощью экструдирования через мембрану с размером пор 50 нм и загрузки лекарственного средства при соотношении лекарственное средство/фосфолипид 100 мг/ммоль. F5 иммунолипосомальный винкристин (F5-ILs-VCR) получали инкубированием Ls-VCR-SOS с aHTH-HER2 scFv F5-PEG-DSPE конъюгатом (пример 19) как описано в примере 43. Винкристин-нагруженные липосомы с использованием ТЕА-цитрата (Ls-VCR-Cit) были получены подобно Ls-VCR-SOS липосомам, за исключением того, что раствором цитрата триэтиламмония (полученным титрованием водной лимонной кислоты чистым триэтиламином до рН 5,1; и доведением концентрации до 0,65 М триэтиламина) был заменен TEA-SOS раствор. Схема исследования соответствовала способу примера 10. Подкожные опухоли ксенотрансплантатов ВТ-474 клеток карциномы молочной железы человека выращивали в бесшерстных мышах и, когда размер опухоли достигал 250 мм3 (порядка 144-309 мм3), мышей в группах по 8-9 животных обрабатывали свободным VCR, Ls-VCR или F5-ILs-VCR раз в неделю i.v. при дозе 2 мг VCR/кг в течение трех недель, начиная с 19-го дня после заражения клетками опухоли. Размеры опухоли и вес тела животных регистрировали как описано в примере 10. В контрольной группе мыши получали физраствор в том же объеме. Различия в размерах опухоли между группами, получавшими лечение, статистически оценивали на 63-ий день после инокуляции опухоли, используя Mann-Whitney тест.Динамика среднего размера опухоли в группах показана на фигуре 43. F5-ILs-VCR продемонстрировал максимальную эффективность по сравнению и с Ls-VCR, и со свободным VCR, вызывая к 63-ему дню полную регрессию опухоли у шести из восьми животных (75%). Ls-VCR-Cit также был эффективным, вызывая полную регрессию опухоли у еще наблюдавшихся на 63-ий день у двух из девяти животных (22%), однако, он был менее эффективным, чем F5-ILs-VCR (ρ<0,005). Ls-VCR-SOS и свободный VCR были одинаково эффективны (ρ<0,2) и менее эффективны, чем F5-ILs-VCR или Ls-VCR-Cit. Таким образом, удивительно, что при клеточной направленности доставки лекарственного средства липосомальное лекарственное средство, инкапсулированное с использованием поливалентного аниона настоящего изобретения, показало большую эффективность, чем лекарственное средство, включенное в липосому с помощью несвязывающего аниона. Динамика веса животных показала, что все липосомальные препараты VCR были менее токсичны, чем свободный VCR, вызывая меньшую потерю веса в процессе лечения (фигура 44).
Пример 65. Получение EGFR-нацеленного липосомального винкристина и противоопухолевая активность ненацеленного и EGFR-нацеленного липосомального винкристина в отношении EGFR-сверхэкспрессирующих ксенотрансплантатов рака мозга человека, изученная на мышах.
Винкристин-нагруженные липосомы (Ls-VCR) получали, используя TEA-SOS метод, как в примере 64, EGFR-нацеленный иммунолипосомальный винкристин получали соинкубацией липосом с анти-HER2 Fab' С225 Fab-PEG-DSPE конъюгатом как описано в примере 36.
Самцам NCR пи/пи мышей (5-6 недель, весом 17-20 г) подкожно инъецировали в область бока 0,15 мл культуральной среды для роста клеток, содержащей 1×107 U87 клеток глиобластомы человека, стабильно экспрессирующих мутантный рецептор EGFR vIII эпидермального фактора роста (HER1). Но 11-ый день, когда значение размера опухоли достигло 300-400 мм3, мышей произвольно разделили на четыре группы по 10-12 животных в группе. Лечение свободным VCR (сульфат винкристина в физрастворе - 1 мг/мл), Ls - VCR или C225Fab-ILs-VCR при i.v. дозе 1,5 мг/кг проводили на 11,18 и 25 дни после инокуляции опухоли. Мышам в контрольной группе аналогично вводили равный объем физраствора. Размеры опухоли и вес тела животных регистрировали как в примере 10. Результаты показаны на фигуре 45. Все животные, получившие препараты VCR, показали замедление роста опухолей, сравнимое с контрольными животными. Значительной разницы между группами, леченными свободным винкристином и Ls-VCR, не наблюдалось. EGFR-нацеленный C225Fab-ILs-VCR был более эффективен, чем свободный или ненасыщенный липосомальный VCR.
Пример 66. Получение липосом с захваченным раствором инозитолгексафосфата триэтиламмония.
Полианионизированный полиол, додеканатриевая соль инозитолгексафосфата (ТЕА-IHP), был получен от Sigma (St. Louis, МО). Водный раствор, содержащий 0,65 М триэтиламмония и 0,681 М фосфатных групп, рН 6,5, осмолярность 718 ммоль/кг, готовили ионным обменом используя поперечносшитую сульфополистирольную смолу Dowex 50Wx8-200 с последующим титрованием чистым TEA и разбавлением водой согласно способу примера 4. Остаточное содержание натрия было меньше, чем 1% от суммы катионов. Сухие липиды (150 дмоль DSPC, 100 μмоль Choi, 0,75 дмоль PEG-DSPE) растворяли в 0,5 мл 100% этанола USP при 60°С и смешивали с 4,5 мл раствора инозитолгексафосфата триэтиламмония, предварительно нагретого до той же температуры. Этанол частично удаляли роторным испарением при 30-40 мм Hg и 40-45°С, пока смесь не прекращала кипеть. Липидную суспензию затем экструдировали при 60-65°С 15 раз через две объединенные поликарбонатные мембраны с размером пор 0,1 μм. Полученные липосомы имели размер 104,3±39,0 нм по QELS. Неинкапсулированный IHP триэтиламмония удаляли гельхроматографией на колонке с Сефарозой 4В с элюцией 5 мМ HEPES-Na буфером 5% декстрозы, рН 6,5, и липосомы анализировали на количественное содержание фосфолипидов методом Bartlett'a с экстракцией согласно примеру 70.
Пример 67. Загрузка лекарств в липосомы с захваченным раствором TEA-IHP.
Липосомы примера 67 нагружали СТР11 или винорелбином. Винорелбин загружали при соотношении лекарственное средство/фосфолипид 175 или 350 г/моль, а СРТ-11 при соотношении 250 или 500 г/моль. Лекарственные средства добавляли к липосомам в HEPES-декстрозном буфере (пример 67) при начальных соотношениях лекарственное средство/фосфолипид, показанных ниже (см. таблицу 36). Если необходимо, рН доводили до 6,5-6,8 с помощью 1 н NaOH. Смеси инкубировали при 60°С в течение 30 минут, охлаждали на льду 15 минут и хроматографировали на гельфильтрационной колонке с Сефадексом G-25 с элюцией 5 мМ HEPES-Na, 145 мМ NaCl, рН 6,5. Аликвоты очищенных липосом солюбилизировали в подкисленном метаноле и анализировали спектрофотометрией (пример 71). Фосфолипиды количественно оценивали методом Bartlett'a (1959) с экстракцией (пример 70). Оба лекарственных средства включались в липосомы количественно (т.е. практически 100%), как показано ниже, в таблице 36.
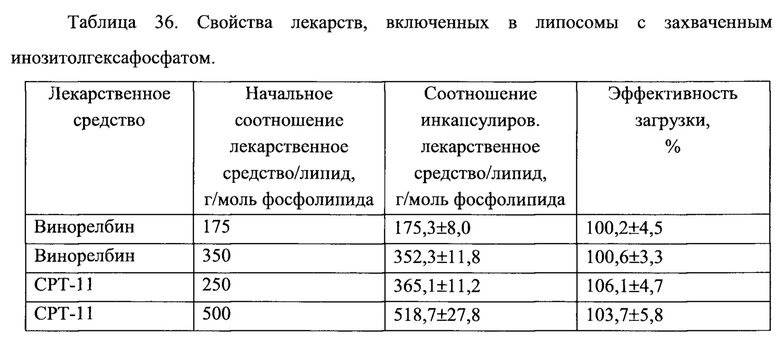
Пример 68. Химическая стабильность свободного или липосомального СРТ-11 в присутствии мышиной плазмы in vitro.
СРТ-11, который является пролекарственным средством, в организме подвергается химической трансформации и превращается в активное лекарственное средство, известное как SN-38. И SN-38, и СРТ-11 превращаются также из их активных лактонных форм в неактивные продукты, известные как SN-38 - или СРТ-11-карбоксилаты. В этом примере изучалось влияние включения в липосомы СРТ-11 в соответствии с настоящим изобретением на химическое превращение СРТ-11 в эти продукты в присутствии плазмы крови. Липосомы с захваченным сахарозооктасульфатом триэтиламмония (0,65 М TEA, рН 6,4, осмолярность 485 ммоль/кг) и липидным составом, включающим DSPC, холестерол и PEG-DSPE в молярном соотношении 3:2:0,015, готовили согласно примеру 11, используя 10-кратную экструзию через два объединенных поликарбонатных фильтра с размером пор 0,08 μм. Получали липосомы с размером 87,4±19,2 нм по QELS. СРТ-11 загружали при соотношении примерно 500 мг СРТ-11 основания на ммоль липосомного фосфолипида инкубацией в водном 5 мМ HEPES-Na, 5% декстрозы, рН 6,5, при 60°С в течение 30 минут с последующим охлаждением на льду в течение 15 минут.СРТ-11-нагруженные липосомы затем очищали на колонке с Сефадексом G-75 с элюцией HEPES-солевым буфером (5 мМ HEPES, 145 мМ NaCl, рН 6,5). Полученные липосомы содержали 536,5±20,1 мг СРТ-11 на ммоль фосфолипида. Раствор свободного СРТ-11 готовили непосредственно растворением гидрохлорида Иринотекана USP в концентрации 1 мг/мл в 144 мМ водного NaCl, подкисленного разбавленной HCl до рН 3. Десяти - цл аликвоты свободного или липосомального СРТ-11 или свободного СРТ-11 смешивали с 90 μм стабилизированной гепарином плазмы мышей (Harlan Bioproducts, USA) и инкубировали при 37°С в водяной бане со встряхиванием. В определенные моменты времени образцы липосом, в трех повторностях, хроматографировали на колонках с Сефарозой CL-4B, с исключением по размеру (2 мл объема слоя) с элюцией HBS-6,5 и фракции, содержащие лекарственное средство, определяли флуоресценцией. Первый (свободный объем) и второй (замыкающий) пики, содержащие лекарственное средство, собирали и считали их включенными в липосомы фракциями, которые выделяют лекарственное средство. Эти образцы экстрагировали 400 μл ледяного метанола при перемешивании (Vortex) в течение 10 с с последующим центрифугированием при 14000 g в течение 5 минут. Супернатанты исследовали на СРТ-11 и продукты его превращения методом ВЭЖХ, используя модификацию Warner и Burke, J. Chromatogr. Ser. В Biomed. Sci. Appl. 1997, vol. 691, p. 161-71. Подвижная фаза состояла из 3% ацетата триэтиламмония рН 5,5 (раствор А) и ацетонитрила (раствор В), выходящих со скоростью 1,0 мл/мин при линейном градиенте от 20% об. В до 50% об. В за 14 минут. Элюированные продукты определяли флуоресценцией с возбуждением при 375 нм и эмиссии при 500 нм. Время удерживания составило 5,3 мин. (СРТ-11 карбоксилат), 6,8 мин. (SN-38 карбоксилат), 9,3 мин. (СРТ-11) и 11,0 мин. (SN-38). Результаты (таблица 37) показали, что, несмотря на то, что свободный СРТ-11 и СРТ-11, выделенный из липосом, подверглись превращению, внутрилипосомальный СРТ-11 оставался абсолютно стабильным.
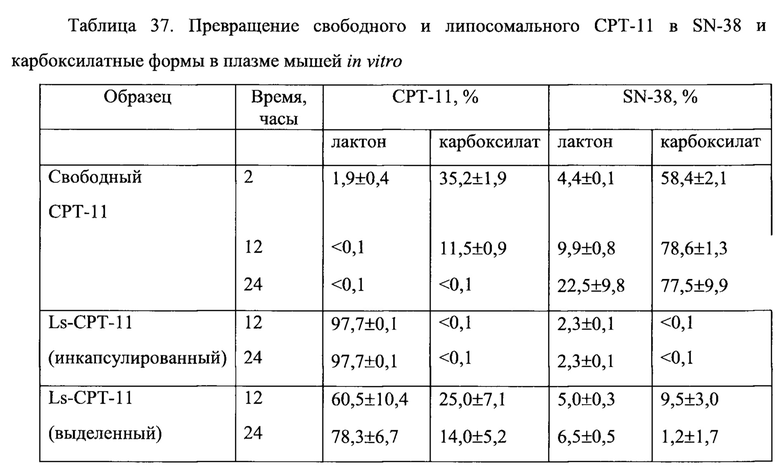
Пример 69. In vivo химическая стабильность свободного или липосомального СРТ-11, изученная на крысах.
Липосомальный СРТ-11 получали как в примере 68, используя сахарозооктасульфат триэтиламмония с 0,65 М TEA, рН 6,4 и осмолярностью 502 ммоль/кг. Размер липосом был 98,5±18,4 нм. и инкапсулирование СРТ-11 осуществлялось при 510,1±16,5 мг СРТ-11/ммоль фосфолипида. Липосомальный и свободный СРТ-11 вводили внутривенно в дозе 25 мг/кг самкам крыс Albino (180-220 г) с установленным в центральную вену катетером и пробы крови отбирали через определенные интервалы времени в течение периода 48 часов. Пробы крови смешивали с ледяным PBS, содержащим 0,04% ЭДТА, и быстро отцентрифугировали, чтобы удалить клетки крови. Аликвоты супернатанта исследовали на СРТ-11, SN-38 и их карбоксилатные производные с помощью ВЭЖХ как в примере 68. Результаты показаны на фигурах 46 и 47. В то время как СРТ-11 очищался очень быстро при отсутствии детектируемости спустя 30 минут, липосомальный СРТ-11 был непрерывно циркулирующим (t1/2 15,2 часа) с 37,8% лекарственного средства в крови на 24-ом часу и спустя 18 часов приблизительно 10% лекарственного средства еще было в кровотоке. Недетектируемой оказалась конверсия липосомальной формы СРТ-11, SN-38, а также карбоксилатная форма СРТ-11. Свободный СРТ-11, например вводимый в виде раствора, исчезал из кровотока совсем быстро (время полужизни составляло около 16 минут) и конверсия лекарственного средства до карбоксилатной формы была заметной.
Пример 70. Количественный учет фосфолипидов в липосомах.
Модифицированный метод I кислотного расщепления голубым фосфомолибдатом. Этот метод был модифицирован Bartlett (1959).
10-20 мл аликвот липосом помещают в пробирки из термоустойчивого стекла, подвергают расщеплению, нагреванием с 0,5 мл 10 N серной кислоты в течение 2 часов при 110-130°С, минерализуют добавлением 50 мл 9% пероксида водорода и нагревают еще 30 минут, пока не прекращается выделение пероксида водорода, детектируемое с помощью бумажной полоски. Образцы после расщепления разбавляют при комнатной температуре 1 мл 0,2% водного молибдата аммония, смешивают с 0,1 мл 5% водной аскорбиновой кислоты и инкубируют в кипящей водяной бане в течение 10 минут. Поглощение восстановленного фосфомолибдатного комплекса измеряют при 800 нм и сравнивали с калибровочной кривой, конкурентно полученной с использованием стандартных растворов неорганического фосфата.
Модифицированный метод II кислотного расщепления голубым фосфомолибдатиом.
Этот метод является модификацией метода Morrison (1964). 5 μл аликвоты липосом, содержащих 1-10 мМ фосфолипида, смешивают с 60 μл концентрированной серной кислоты и 10 μл 30% пероксида водорода в пробирках из термоустойчивого стекла. Смеси нагревают при 200-220°С в течение 10 минут, разбавляют 0,7 μл деионизированной воды, смешивают с 10 μл 10% водного сульфита натрия, инкубируют в кипящей водяной бане в течение 5 минут и охлаждают при комнатной температуре. Добавляют 260 μл 2% водного молибдата аммония и 10 μл 10% водной аскорбиновой кислоты, пробы инкубируют в кипящей водяной бане 10 минут. Пробы быстро охлаждают до комнатной температуры и поглощение восстановленного фосфомолибдатного комплекса определяют при 825 нм по сравнению со слепым опытом. Количество фосфолипида определяют по калибровочной кривой, полученной в тех же условиях при использовании стандартных растворов с 2, 4, 6, 8 и 10 мМ дигидрофосфата натрия.
Экстракционный метод. 25-100 μл аликвоты липосом экстрагируют 3 раза 200 μл порциями смеси метанол-хлороформ (1:2 по объему). Органические фазы собирают в пробирку из термоустойчивого стекла, растворители удаляют в вакууме. Остатки обрабатывают Юн серной кислотой и затем определяют фосфор по вышеприведенному методу I.
Если иное не оговорено, результаты анализов представлены как значение + стандартное отклонение (полученное на основе трех повторностей).
Пример 71. Количественное определение лекарственного средства в липосомах.
Спектрофотометрическое количественное определение.
Аликвоты липосом (10-50 μл) смешивают с 1 мл 70% об. водного изопропанола, содержащего 0,075-0,1 н HCl и измеряют поглощение против слепого опыта при следующих длинах волн: доксорубицин - 485 нм; СРТ-11 и топотекан - 372 нм, эллиптицины - 306 нм, винорелбин - при 270 нм, винкристин и винбластин - 265 нм. Количество лекарственного средства определяют в сравнении с построенной конкурентно калибровочной кривой.
Флуорометрическое количественное определение.
Аликвоты липосомсодержащих проб (например, плазмы крови) разбавляют подкисленным изопропанолом (0,02-0,1 нм аликвоты: 1 мл 70% изопропанол - 0,075 н HCl; >0,1 мл аликвоты: 90% изопропанол - 0,1 н HCl до 1 мл). Если происходит осаждение белков, пробы инкубируют на льду в течение 1 -2 часов и очищают центрифугированием в течение 10 минут при 12100 xg. Флуоресценцию супернатантов измеряют при следующих длинах волн: СРТ-11, возбуждение 370 нм, излучение 423-425 нм; Топотекан, возбуждение 380-385 нм, возбуждение (эмиссия ?) 520-525 нм; эллиптицины, возбуждение 306 нм, излучение 520 нм. Содержание лекарственного средства вычисляют по конкурентно построенным калибровочным кривым после вычитания значений холостого опыта.
Пример 72. Влияние липополимеров на эффективность включения винорелбина в липосомы.
Липосомы, в состав которых входили DSPC - 200 мольных частей, холестерол - 133 мольные части и липиды на основе поли(этиленгликоля) (мол. масса 2000) PEG-DSPE (1-20 мольных частей или PEG-DSG (20 мольных частей) и содержащие инкапсулированный 0,65 М раствор TEA-SOS, получали в соответствии со способом примера 11, используя при экструзии мембрану с размером пор 80 нм. Липосомы загружали винорелбином при соотношении лекарственное средство/фосфолипид. 350 мг/ммоль и очищали от невключенного лекарственного средства согласно способу по примеру 40. В липосомах определяли содержание лекарственного средства и липида, как описано в примерах 70, 71, и размер липосом по QELS с использованием Gaussian принципа тождественности объема весу. Результаты (таблица 38) показали, что хотя анионное PEG-производное, PEG-DSPE в количестве менее 1% моль липосомного фосфолипида (0,3% моль от общего липидного содержания) оказывало отрицательное воздействие на эффективность включения лекарственного средства в липосомы, нейтральное производное, PEG-DSG не оказывало влияния на эффективность включения даже при концентрации 9,1% моль, от общего содержания фосфолипида в липосомах (5,7% моль, от общего липидного количества).
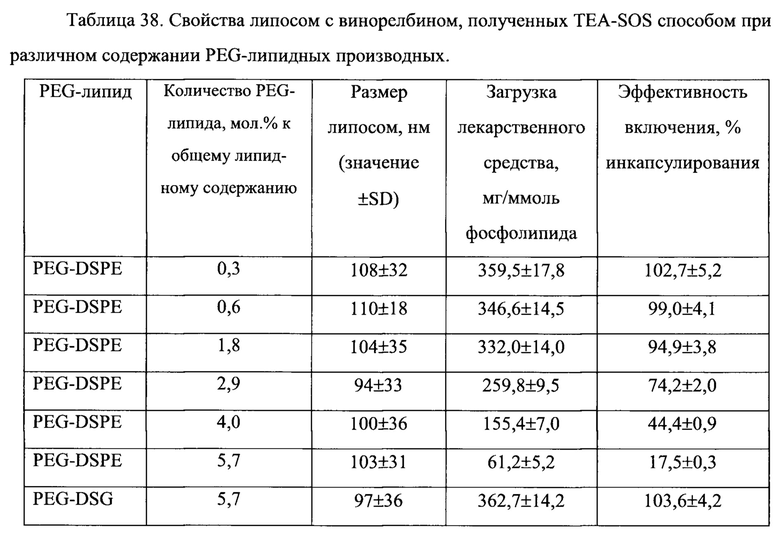
Пример 73. Влияние внутрилипосомального захватывающего лекарственное средство агента на долговечность сохранения СРТ-11 в кровотоке, исследованное на мышах.
Липосомы с захваченным 0,65 н растворами инозитолгексафосфата (IHP, фитиновая кислота) триэтиламмония (TEA) или триэтаноламмония (ТЕОА) или сахарозооктасульфата TEA или ТЕОА получали и нагружали СРТ-11 при 500 г/моль фосфолипида с последующими общими приемами примера 66. Эти липосомы внутривенно вводили Swiss-Webster мышам в дозе 5 мг СРТ-11/кг веса. Через 24 часа мышей подвергали анестезированию и обескровливали пункцией сердца. Кровь собирали, анализировали на содержание СРТ-11 в плазме крови путем ВЭЖХ, как описано в примере 68, и количество лекарственного средства, оставшегося в крови, выражали в % от инъецируемой дозы (% ID). TEAO-IHP был менее эффективен в повышении долговечности сохранения лекарственного средства в крови, чем ТЕА-IHP, TEOA-SOAS и TEA-SOS (Таблица 39).

Пример 74. Включение лекарственного средства в липосомы, содержащие 1,05 н сахарозооктасульфат диэтиламмония.
Водный раствор 1,05 н сахарозооктасульфата диэтиламмония (DEA-SOS), рН 6,0, осмолярнось 727 ммоль/кг, получали путем ионного обмена/титрования по примеру 6, используя чистый диэтиламин (99,5% чистоты). Липидную матрицу составляли из 3 мольных частей DSPC, 2 мольных частей холестерола и 0,015 мольных частей PEG-2000-DSPE для липосом (средний размер 92,4 нм по методу «объем-вес») в присутствии раствора DEA-SOS и загружали СРТ-11 в липосомы при различных исходных соотношениях лекарственное средство/липид, используя способ примера 11. Неинкапсулированное лекарственное средство удаляли гель-хроматографией, а количество инкапсулированного лекарственного средства на единицу липида (конечное соотношение лекарственное средство/липид) определяли. Эффективность инкапсулирования вычисляли как % конечного соотношения лекарственное средство/липид к начальному соотношению. Результаты показаны в таблице 40. Уровень загрузки достигал своего максимума приблизительно 1,76 моль лекарственного средства на моль фосфолипида (1,67-1,70 моль лекарственного средства/г общего липида), что хорошо согласуется с содержанием (1,78 моль диэтиламмония/моль фосфолипида), основанным на содержании диэтиламмония в липосомах, допуская стехиометрический обмен интралипосомальных ионов диэтиламмония на молекулы лекарственного средства и вычисленным объемом захвата внутри липосом, представляющим приблизительно 1,7 л/моль фосфолипида.
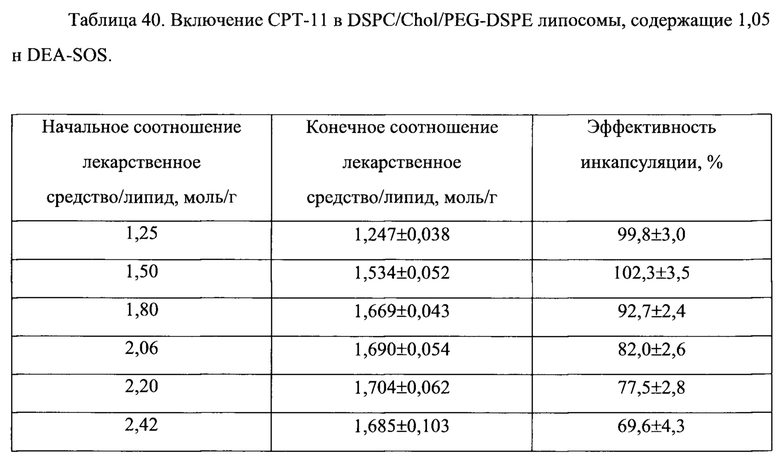
Если не указано иное, результаты анализов представлены как значение + стандартное отклонение трех повторностей. Фармакокинетические данные в плазме крыс представлены как значение ± стандартное отклонение двух повторностей.
Хотя изобретение описано с отсылкой к предпочтительному в настоящий момент варианту осуществления, следует понимать, что могут быть сделаны различные модификации без искажения идеи изобретения. Соответственно объем изобретения следует определять согласно прилагаемой формуле изобретения вместе с полнообъемными эквивалентами, к которым относятся пункты формулы изобретения. Ссылки на полное содержание всех цитируемых статей и ссылок, включая описания к патентам и патентным заявкам, включены здесь для использования в любых целях.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛИПОСОМНЫЕ КОМПОЗИЦИИ, ИСПОЛЬЗУЕМЫЕ ДЛЯ ДОСТАВКИ ЛЕКАРСТВЕННЫХ СРЕДСТВ | 2005 |
|
RU2574926C2 |
| ЛИПОСОМА, ИМЕЮЩАЯ ВНУТРЕННЮЮ ВОДНУЮ ФАЗУ, СОДЕРЖАЩУЮ СОЛЬ СУЛЬФОБУТИЛОВОГО ЭФИРА ЦИКЛОДЕКСТРИНА | 2010 |
|
RU2575793C2 |
| ЛИПОСОМАЛЬНАЯ КОМПОЗИЦИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2734900C1 |
| СТАБИЛИЗИРУЮЩИЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КАМПТОТЕЦИНА | 2016 |
|
RU2833053C2 |
| СТАБИЛИЗИРОВАННЫЕ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ КАМПТОТЕЦИНА | 2016 |
|
RU2732567C2 |
| ЛИПОСОМАЛЬНЫЙ ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ЕГО ИЗГОТОВЛЕНИЯ | 2007 |
|
RU2494729C2 |
| Способ доставки гранзима Б в клетки млекопитающих | 2021 |
|
RU2796685C2 |
| ПРИМЕНЕНИЕ ЛИПОСОМЫ МИТОКСАНТРОНА ГИДРОХЛОРИДА ДЛЯ ЛЕЧЕНИЯ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ | 2021 |
|
RU2806277C1 |
| СОЗДАНИЕ ПРОФИЛЯ КОНТРОЛИРУЕМОГО ВЫСВОБОЖДЕНИЯ ЛЕКАРСТВЕННОГО ВЕЩЕСТВА ПРИ ИСПОЛЬЗОВАНИИ ЛИПОСОМНОЙ КОМПОЗИЦИИ В ВОДНЫХ И БЕЗВОДНЫХ РАСТВОРАХ | 2014 |
|
RU2712157C2 |
| ПРОТИВООПУХОЛЕВОЕ СРЕДСТВО НА ОСНОВЕ ИММУНОЛИПОСОМАЛЬНОЙ БИОЛОГИЧЕСКОЙ КОНСТРУКЦИИ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ВЕКТОРНОЙ ДОСТАВКИ В ЦЕНТРАЛЬНУЮ НЕРВНУЮ СИСТЕМУ ПРИ ОПУХОЛЕВОМ ПРОЦЕССЕ | 2007 |
|
RU2336901C1 |
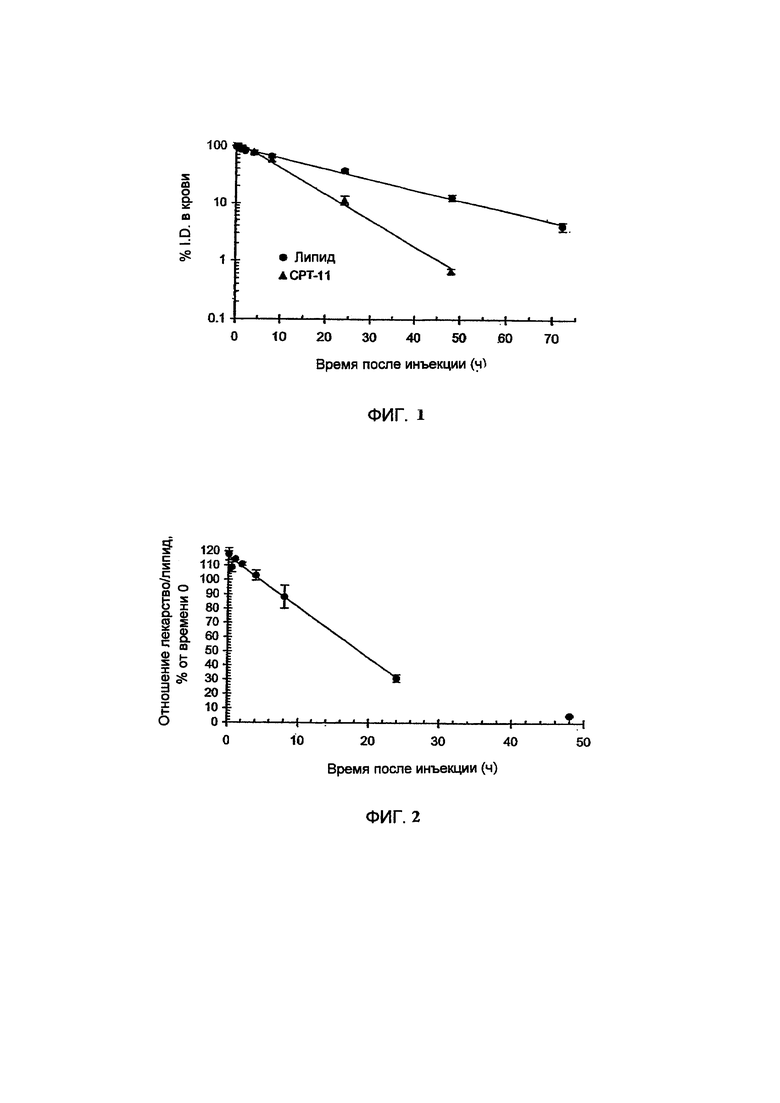
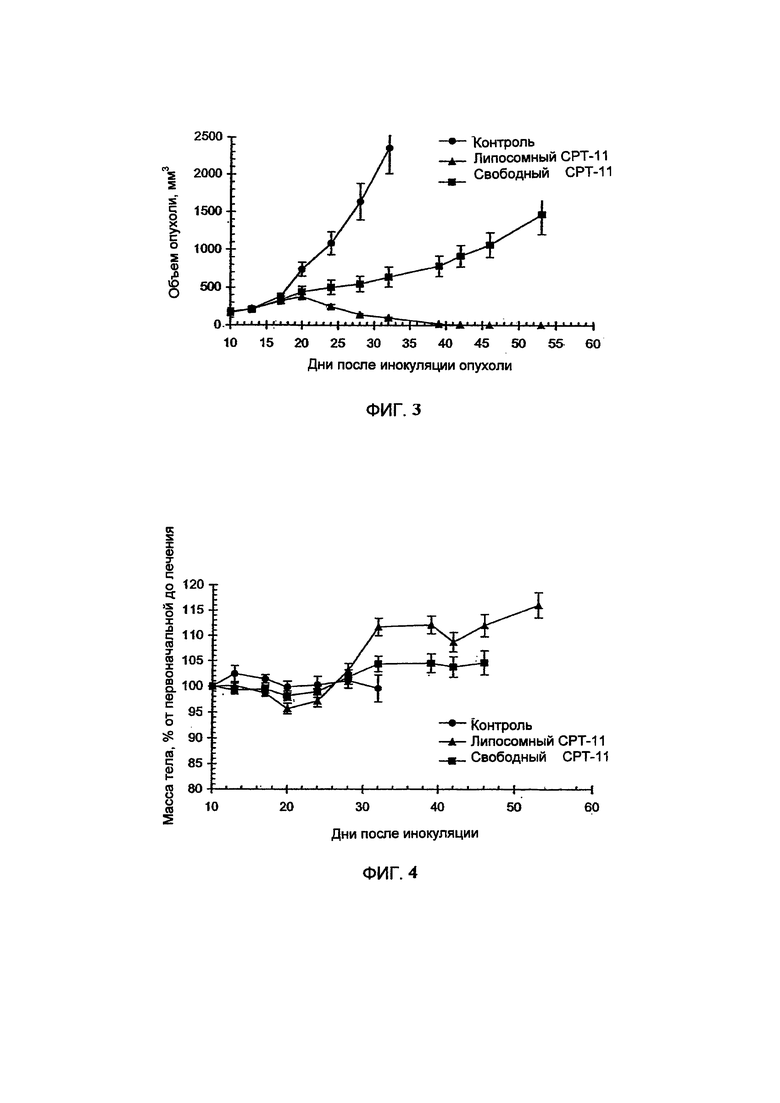
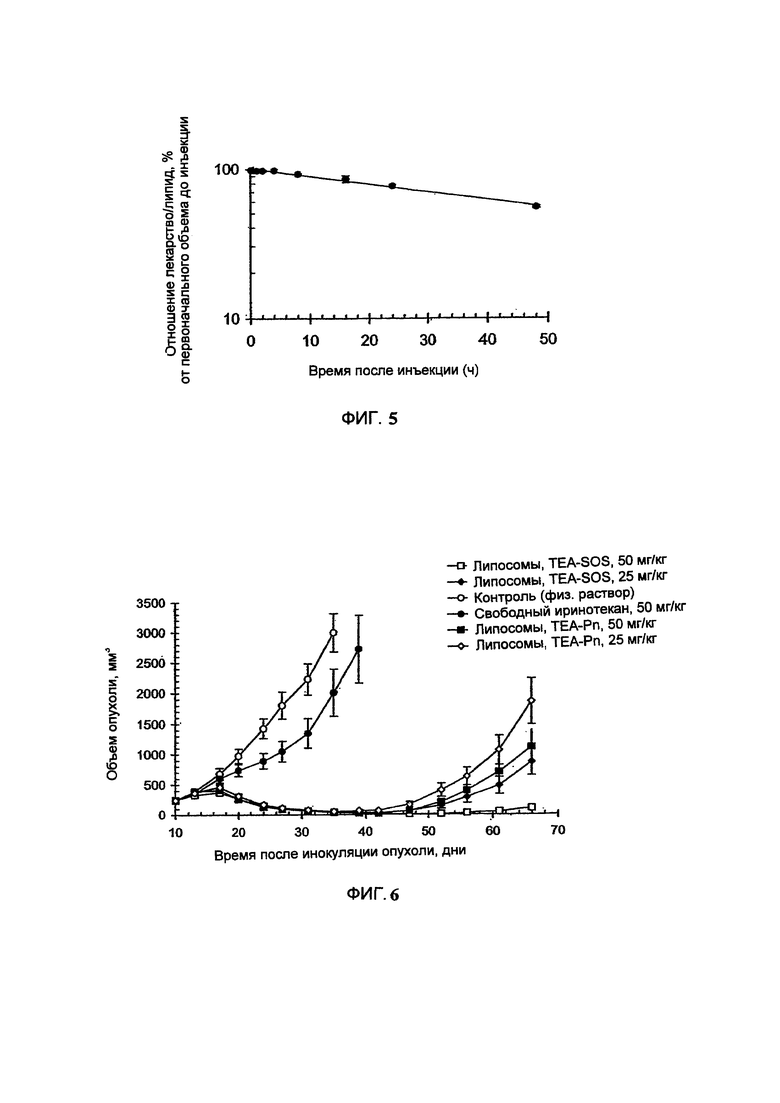
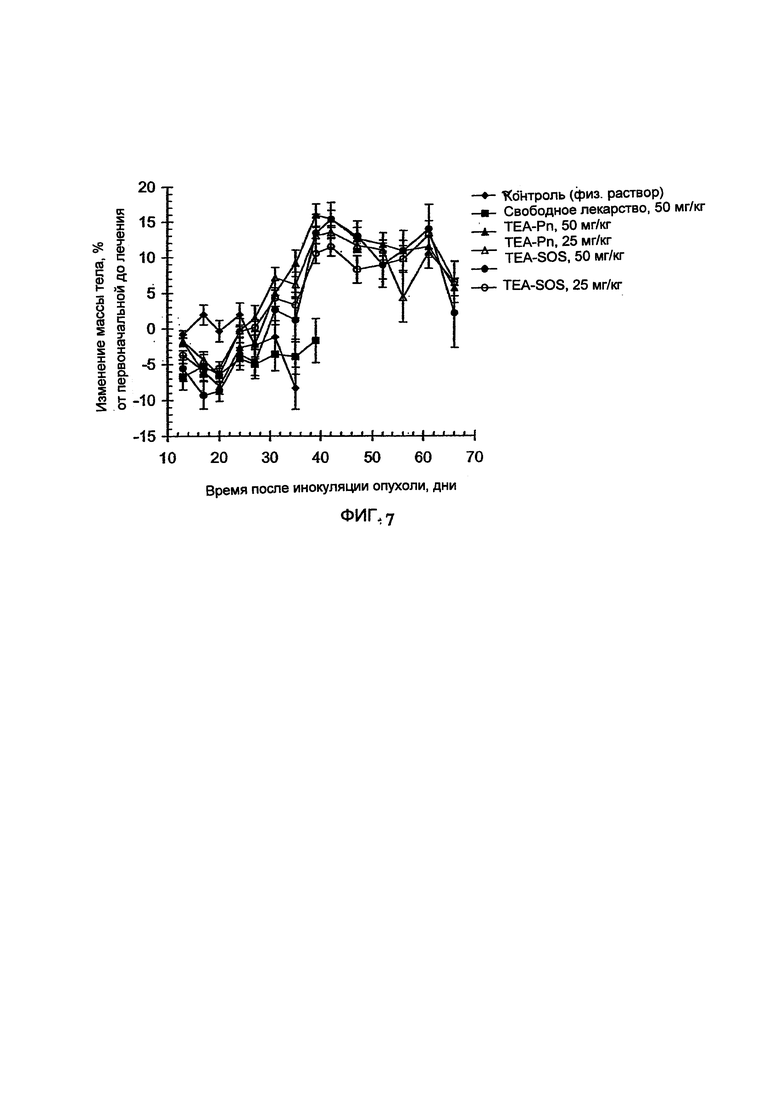
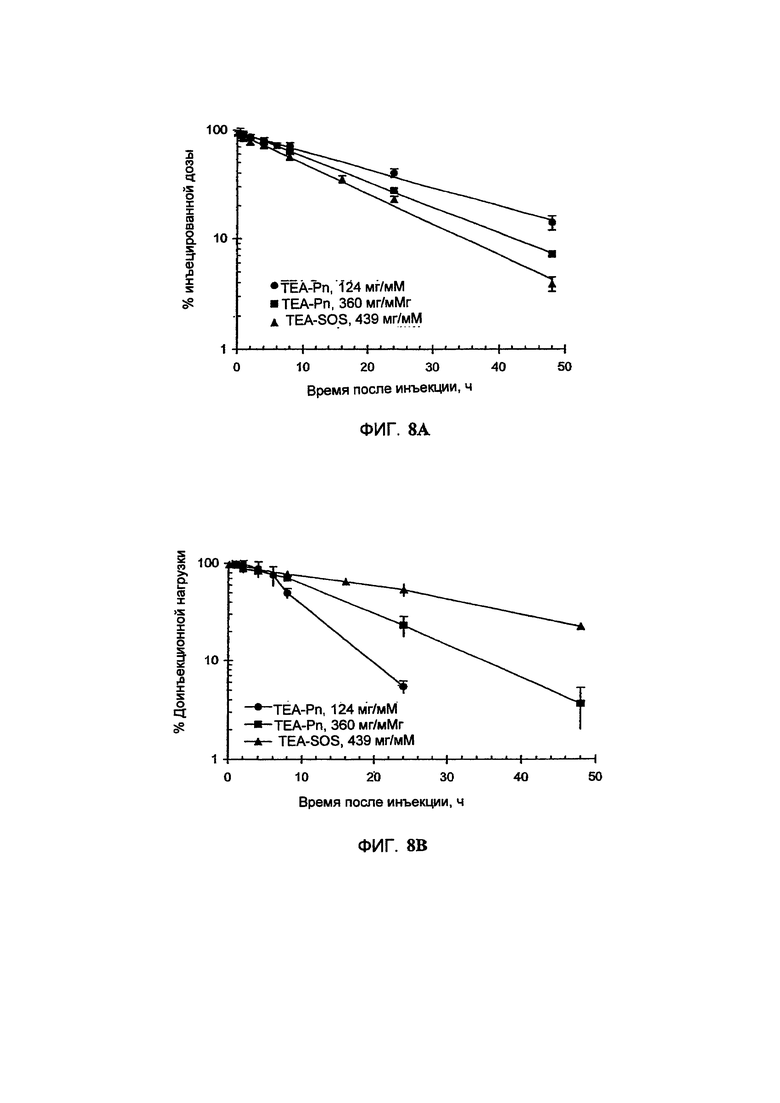
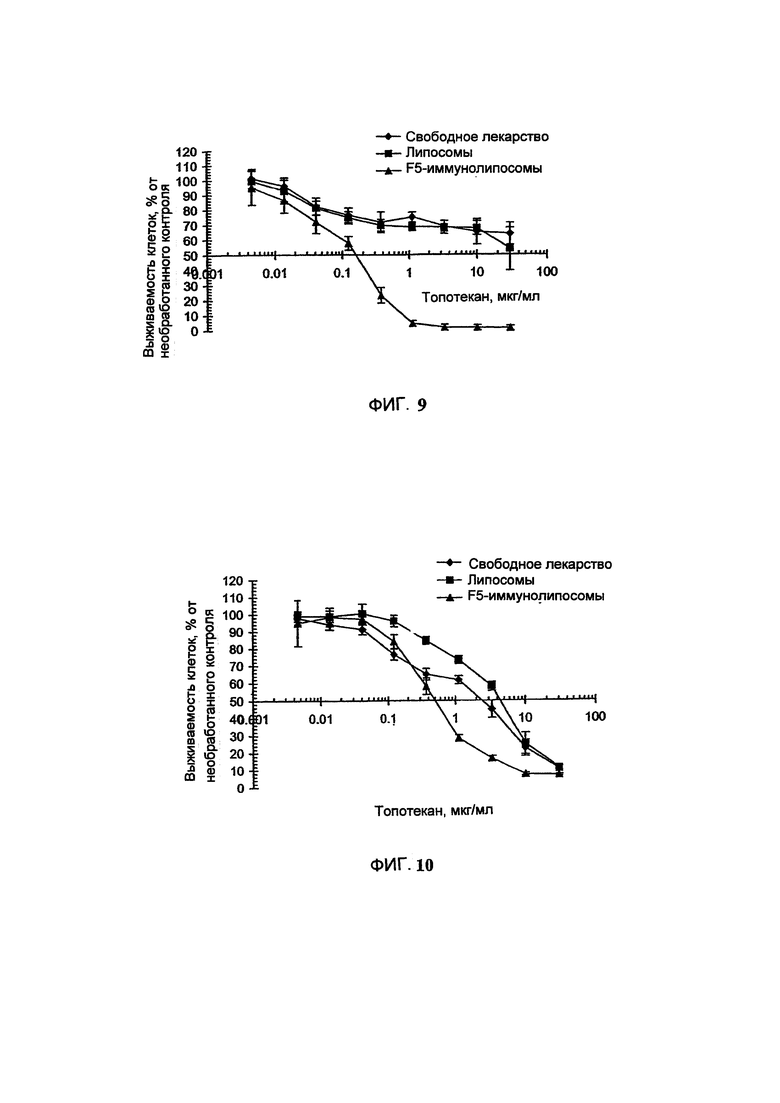
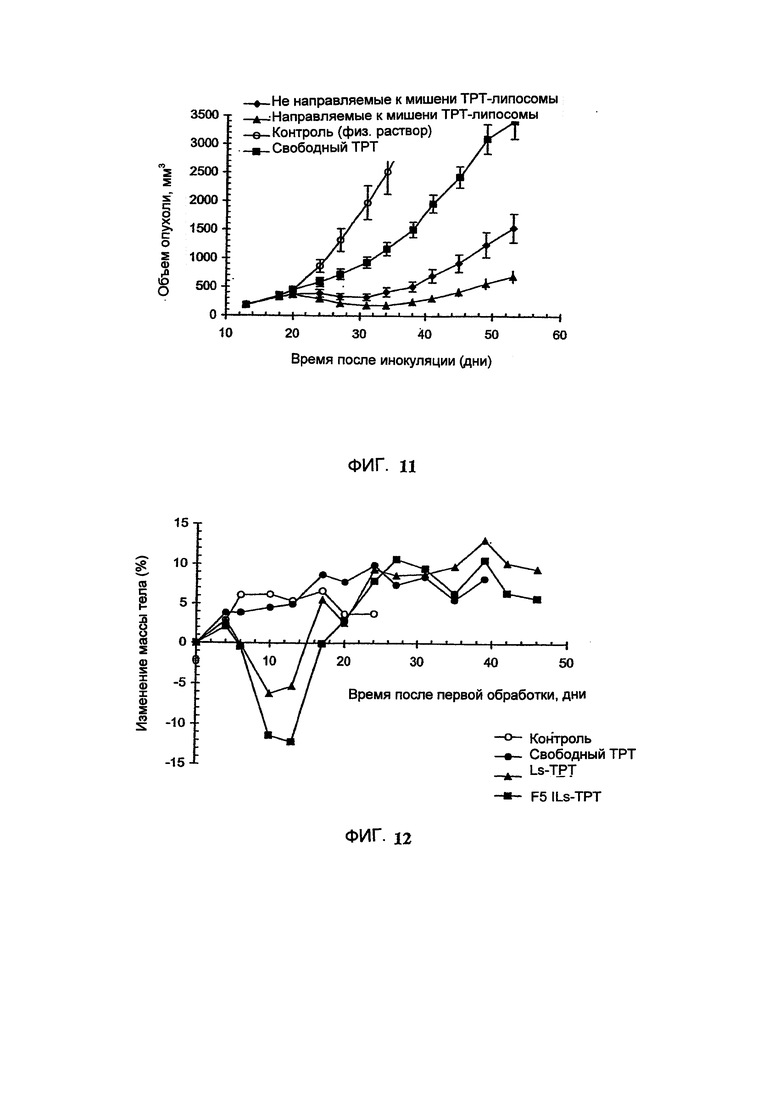
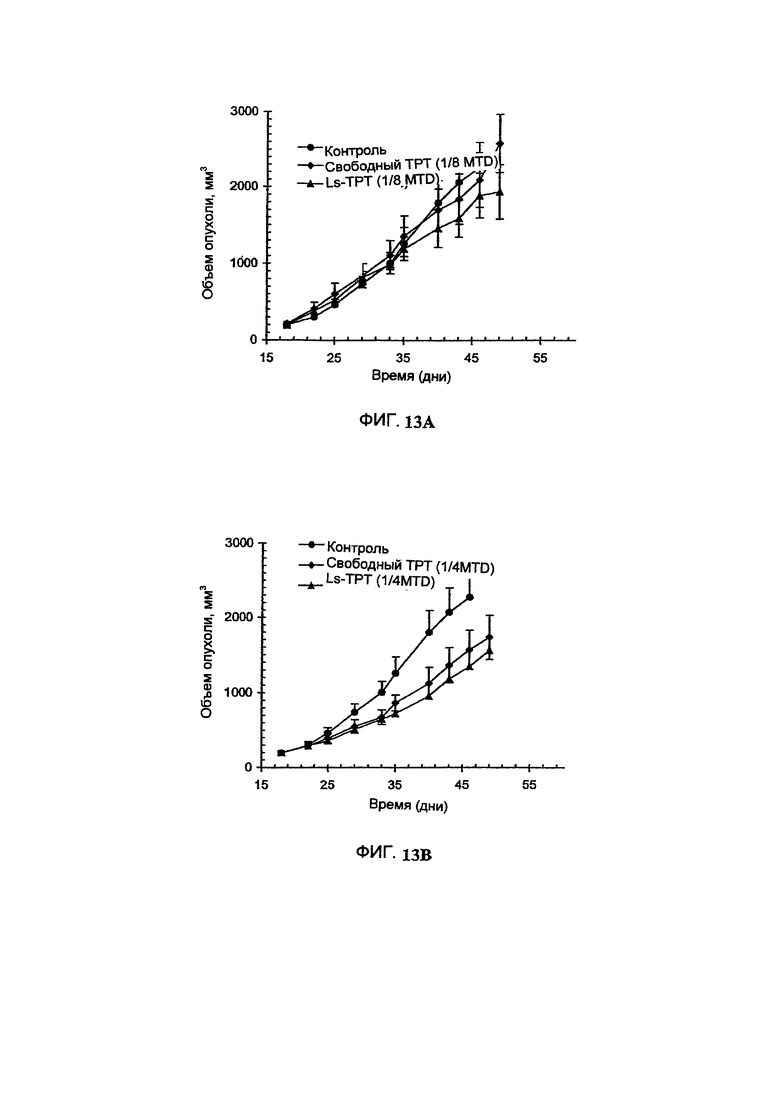
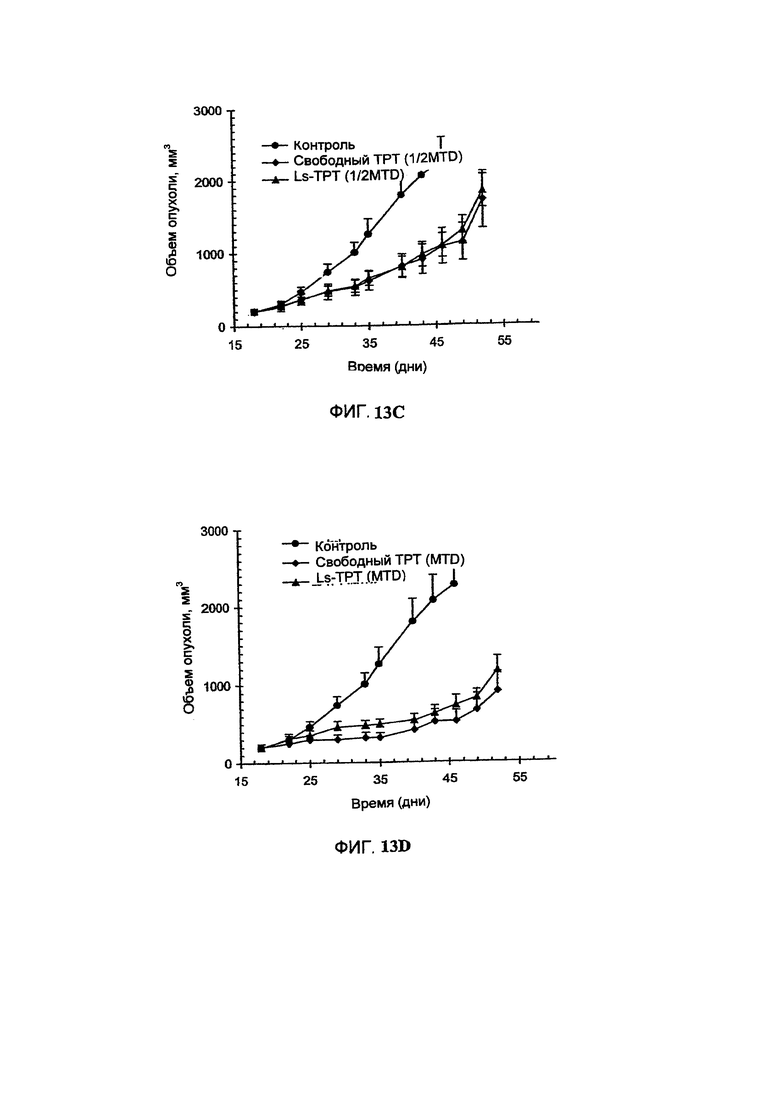
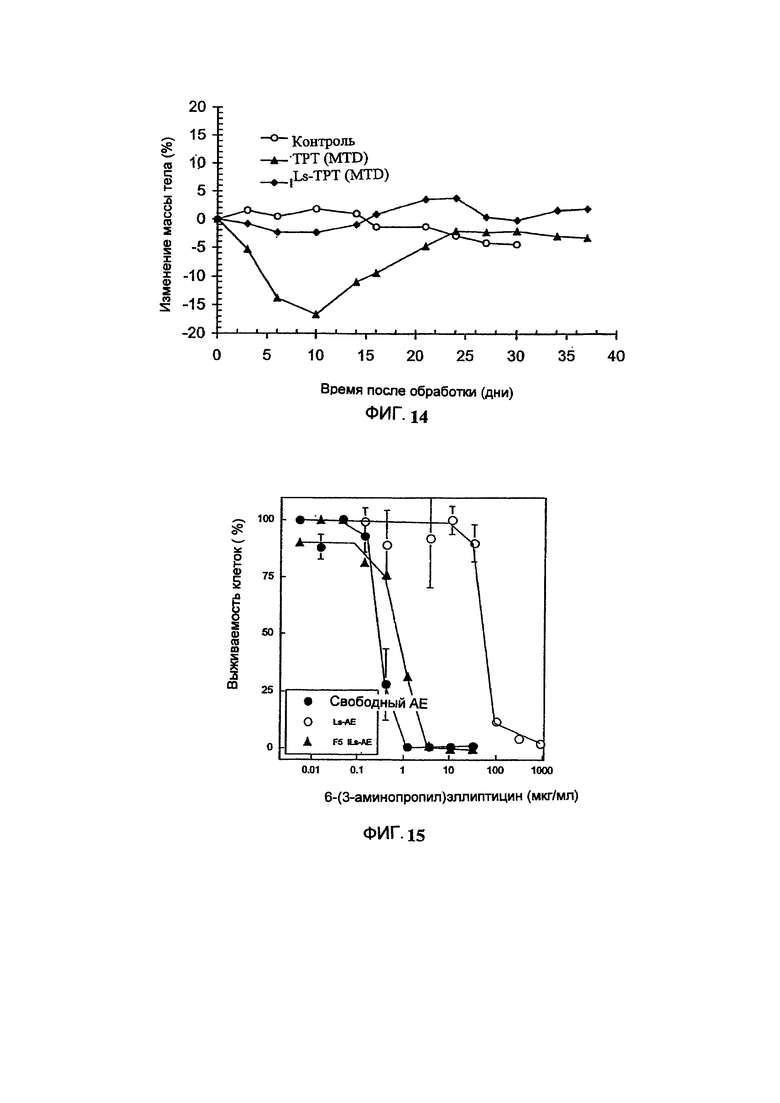
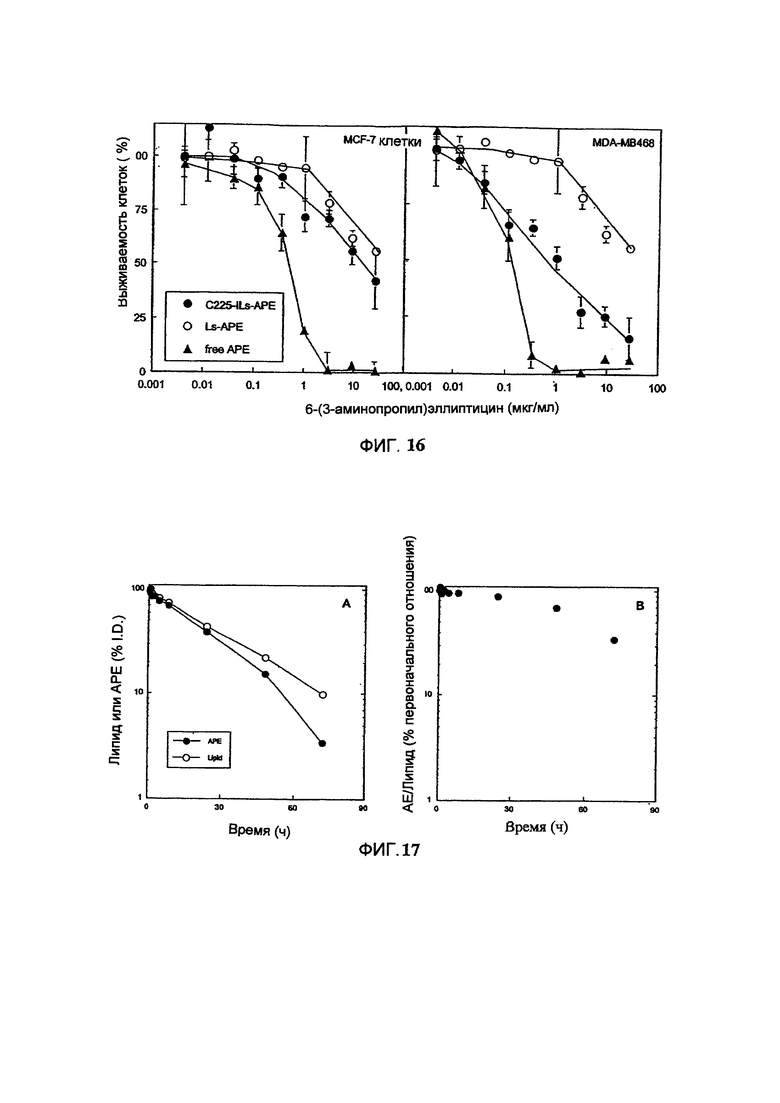
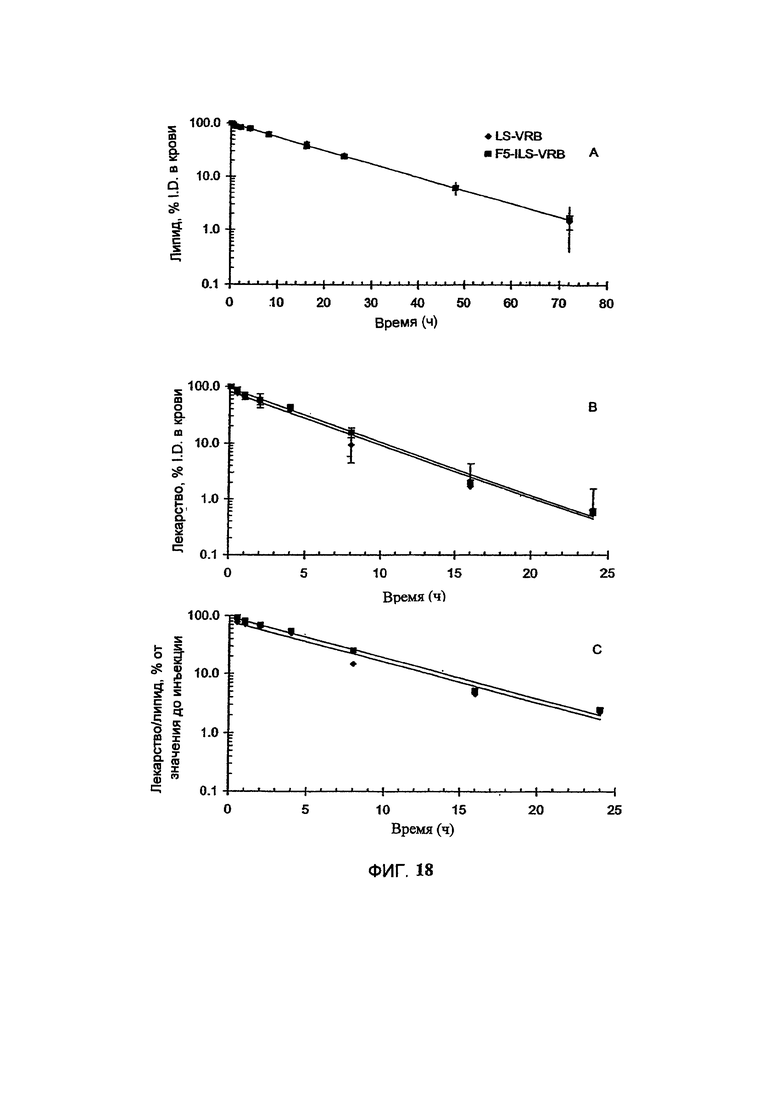
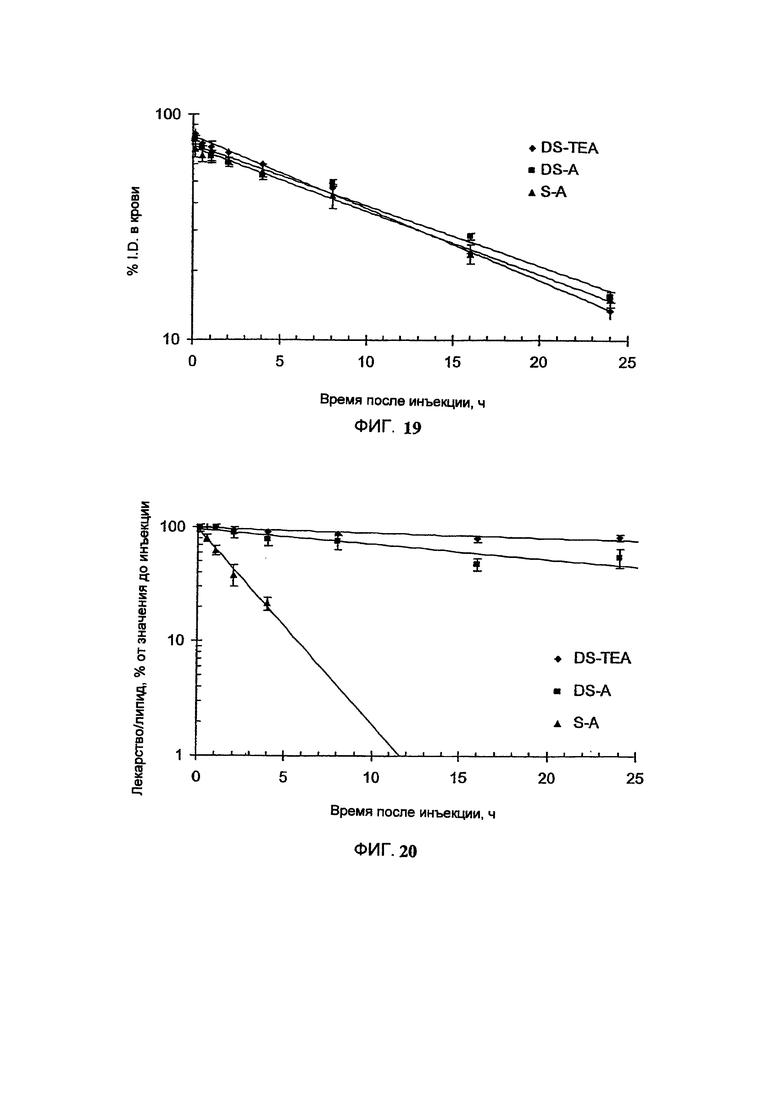
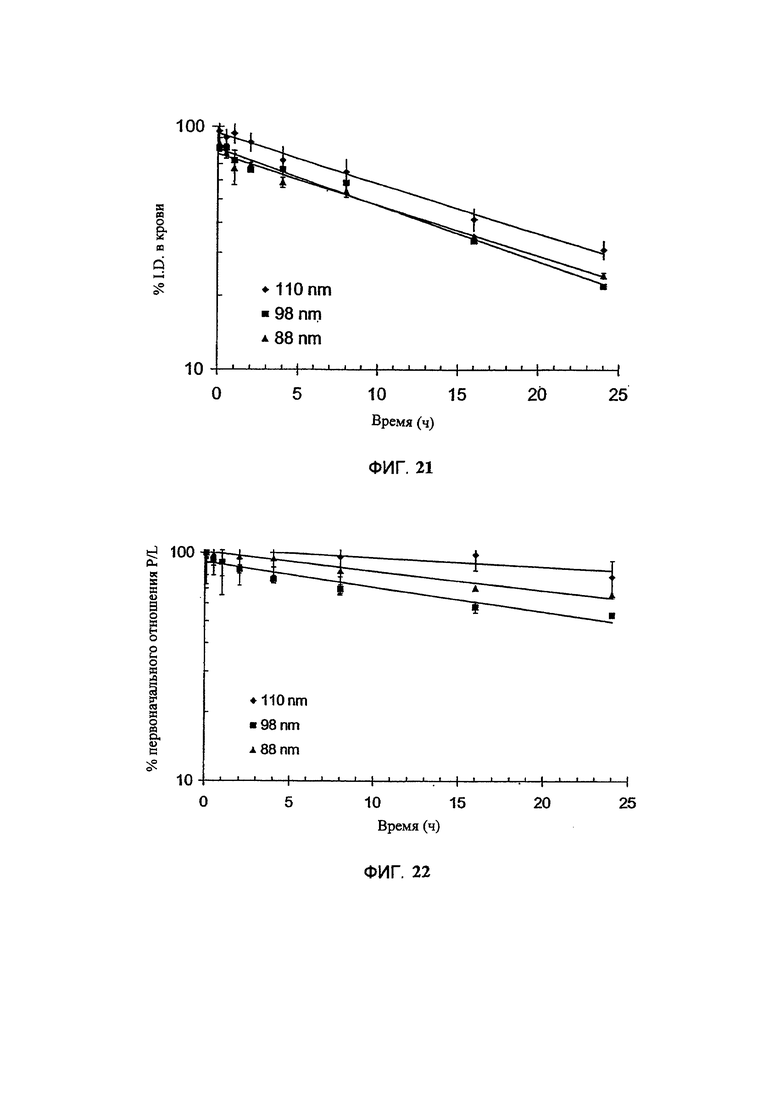
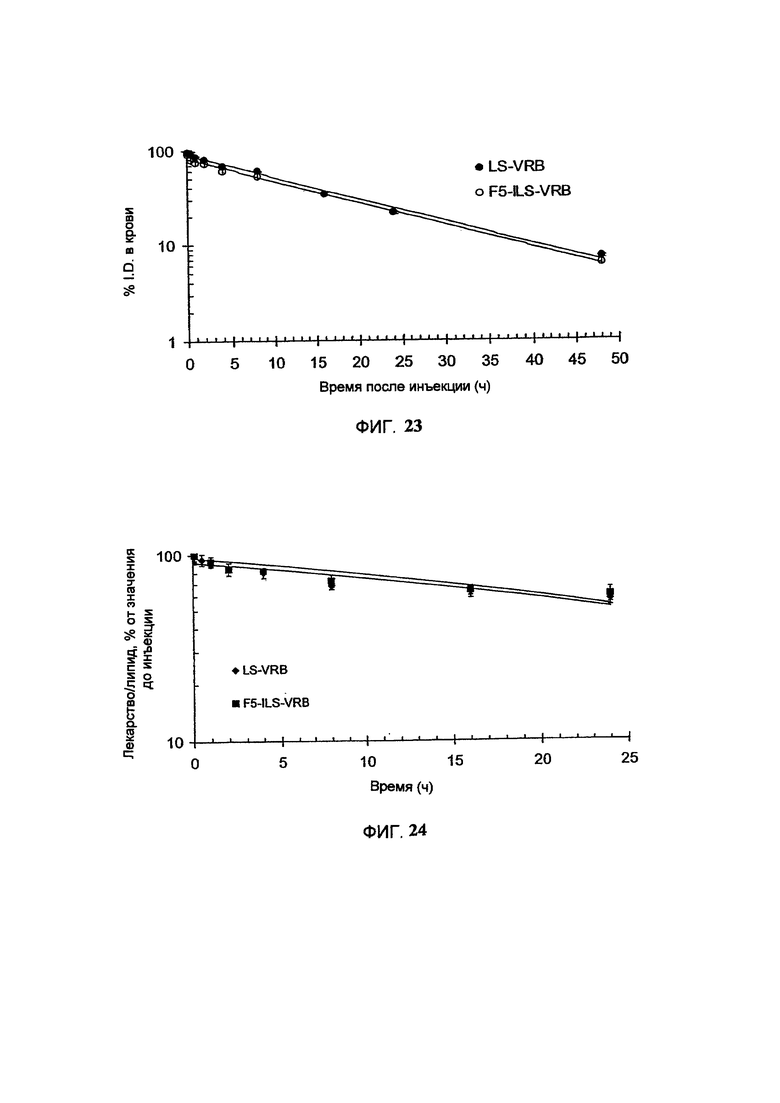
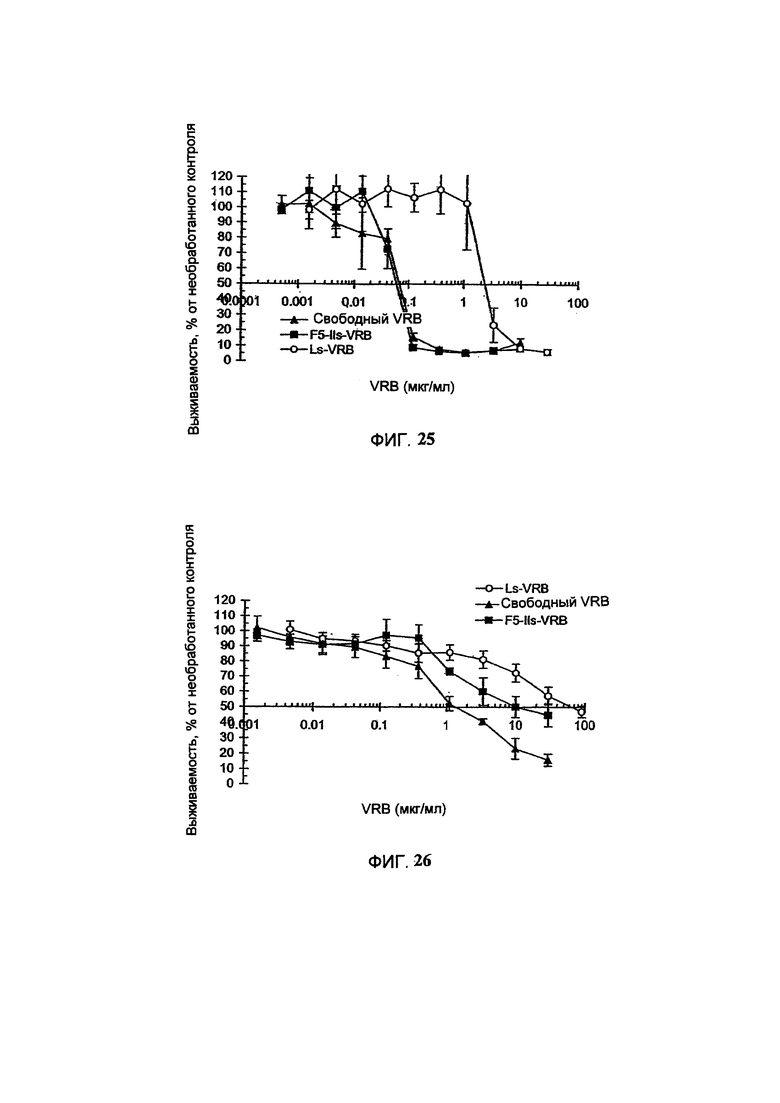
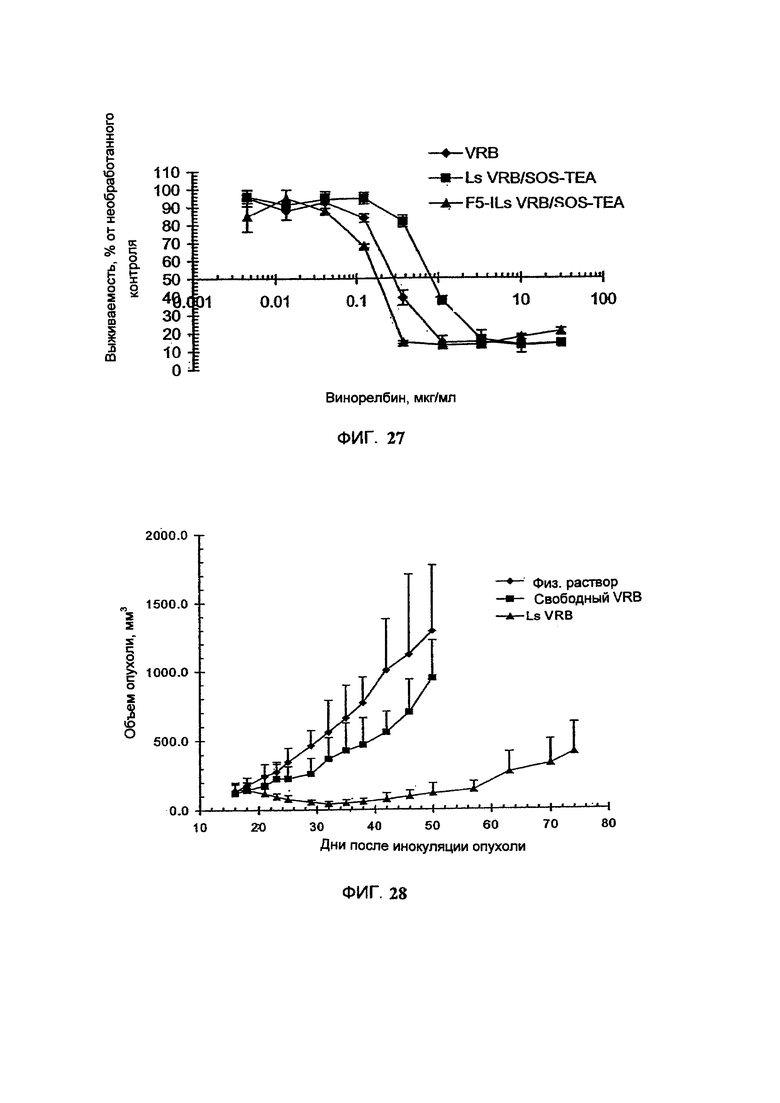
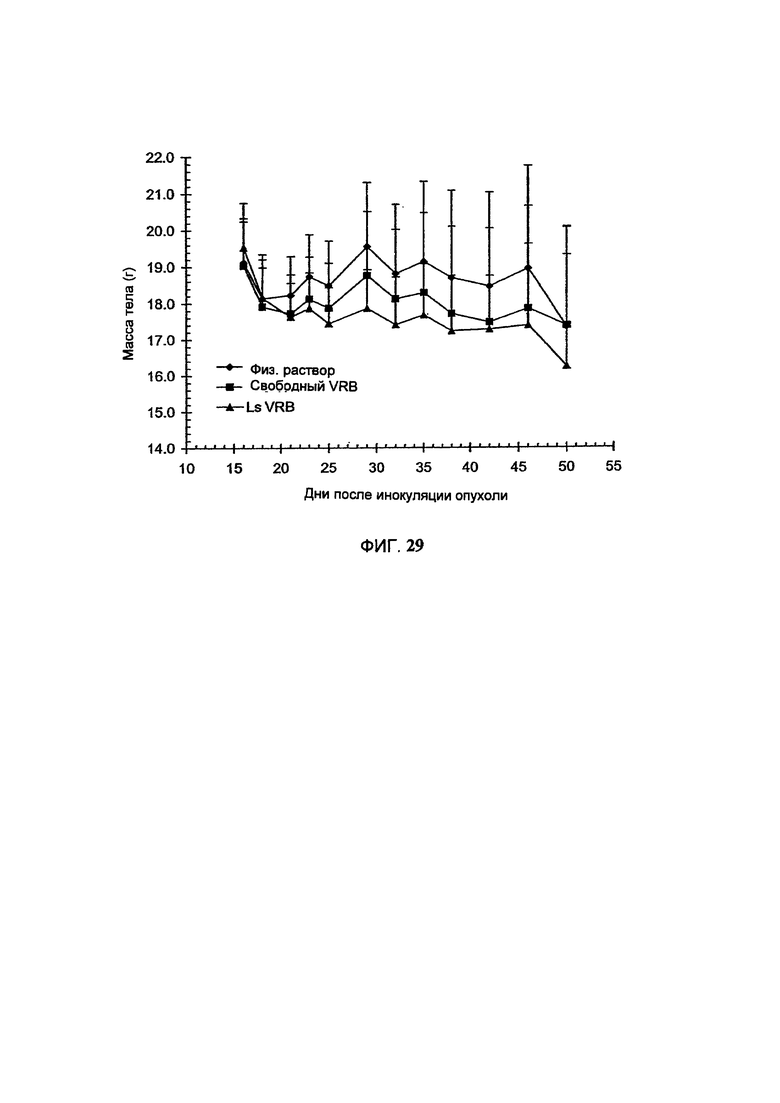
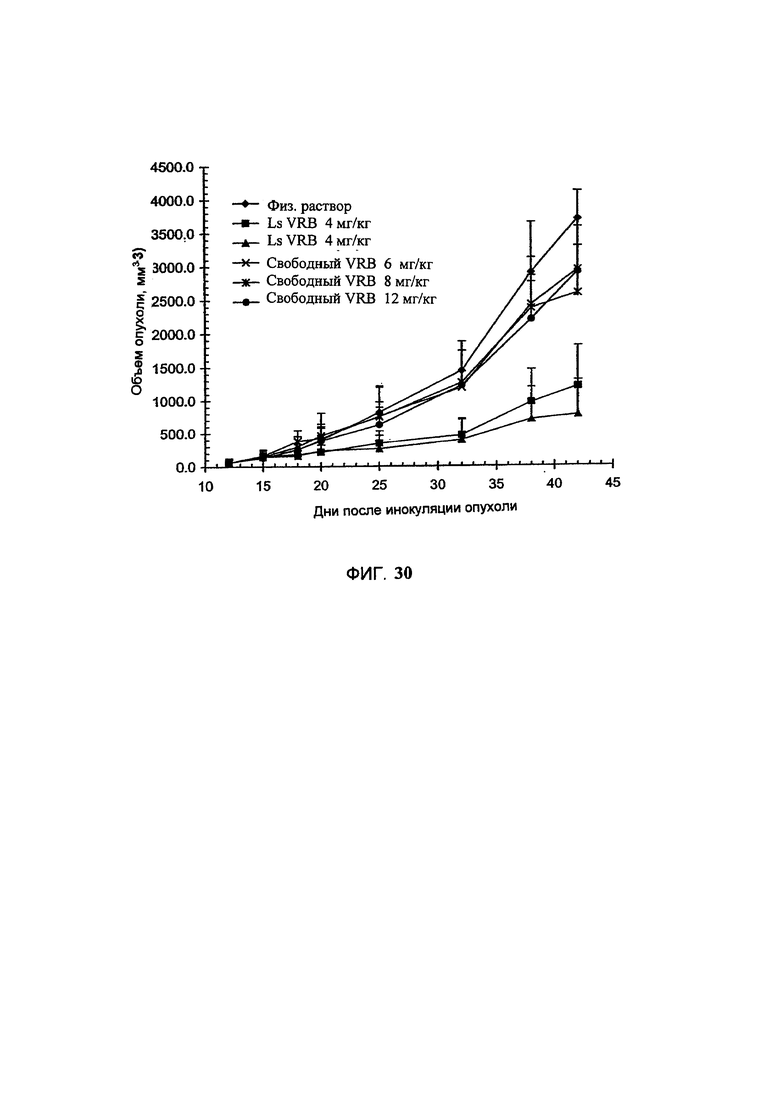
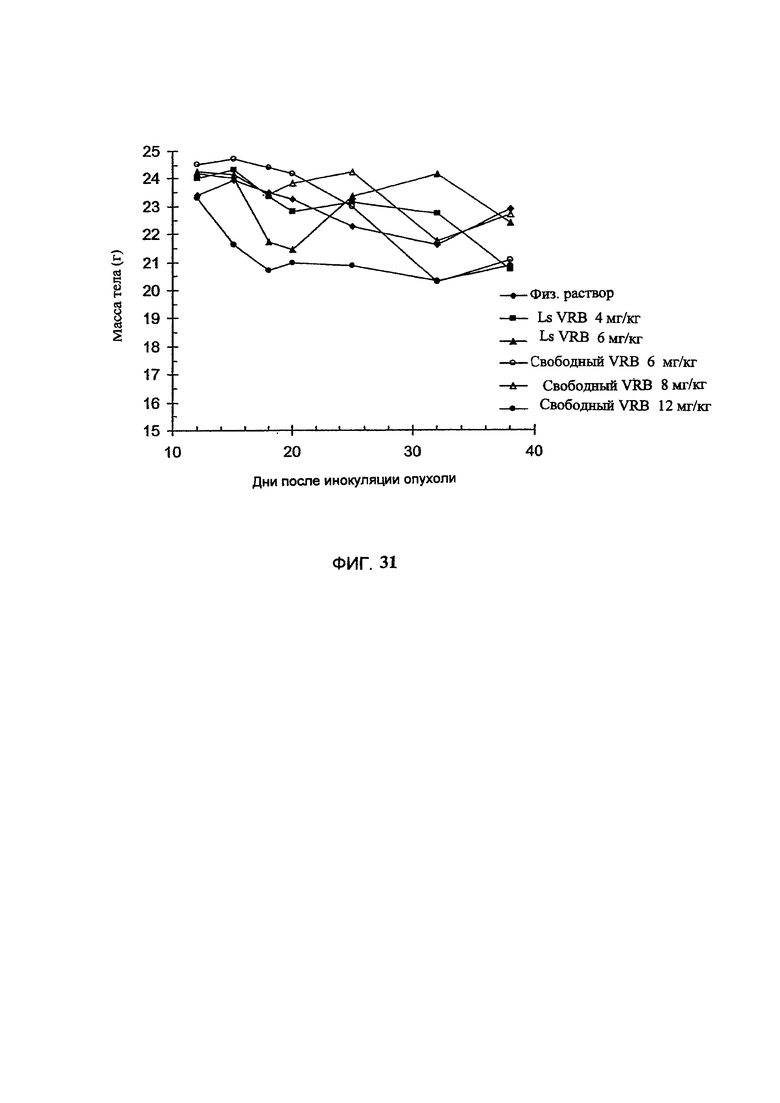
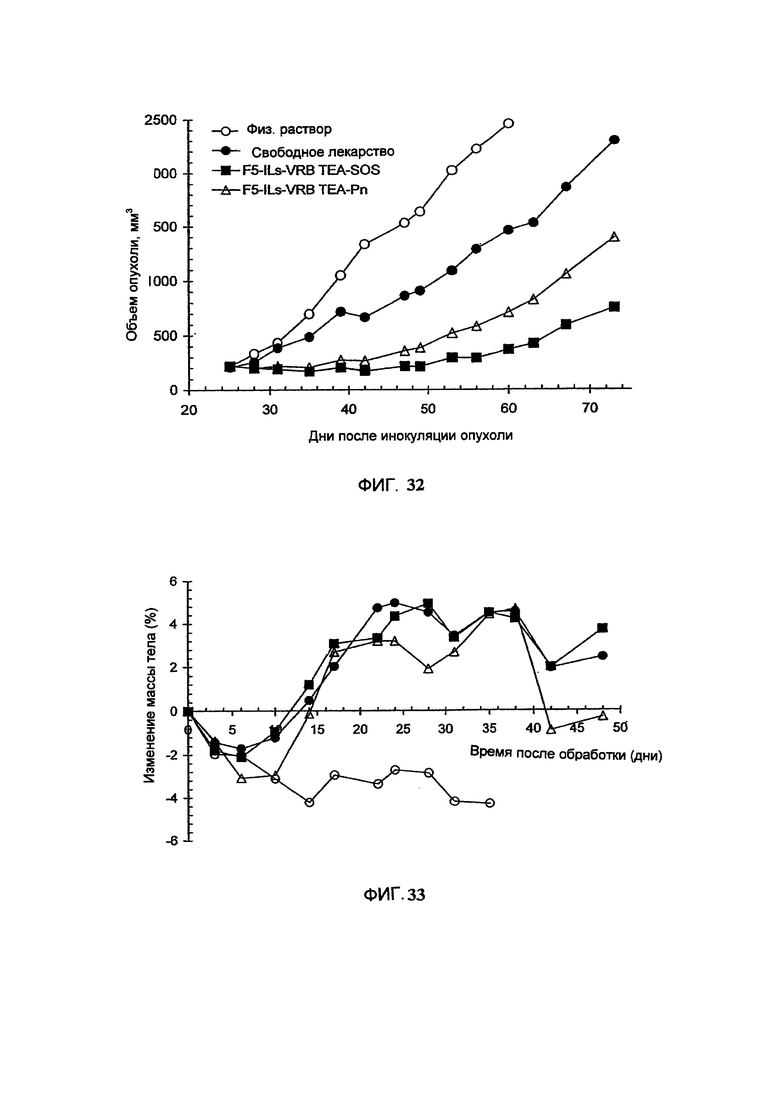
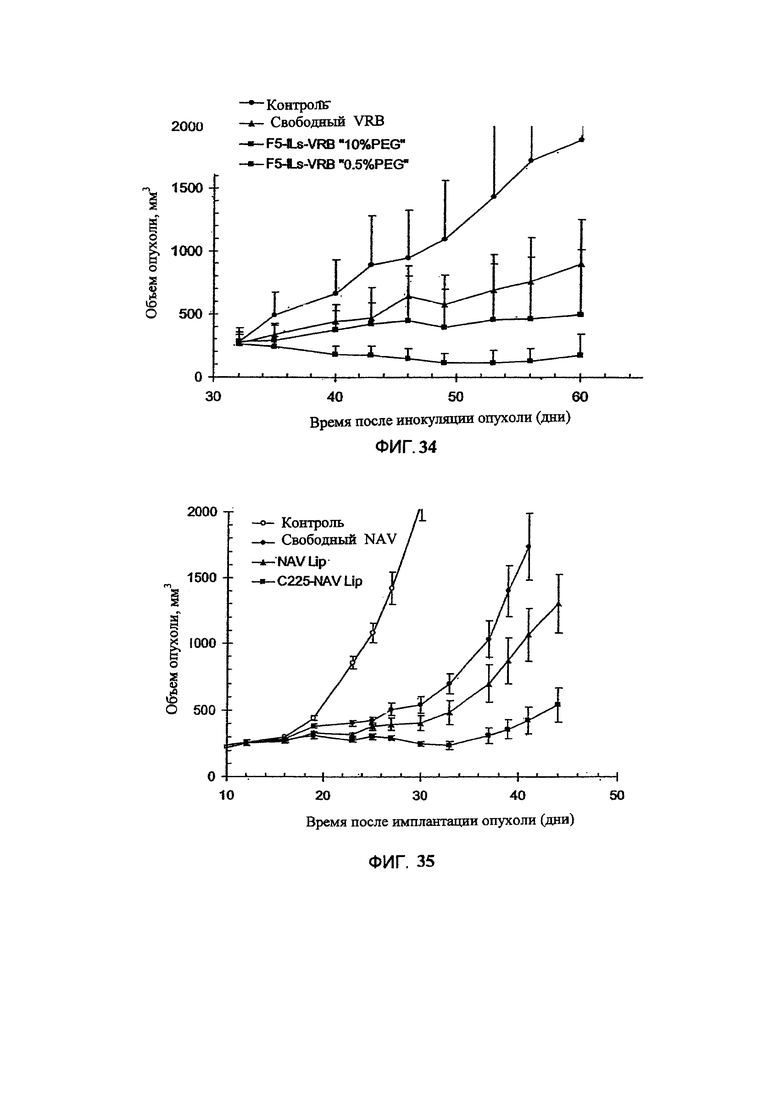
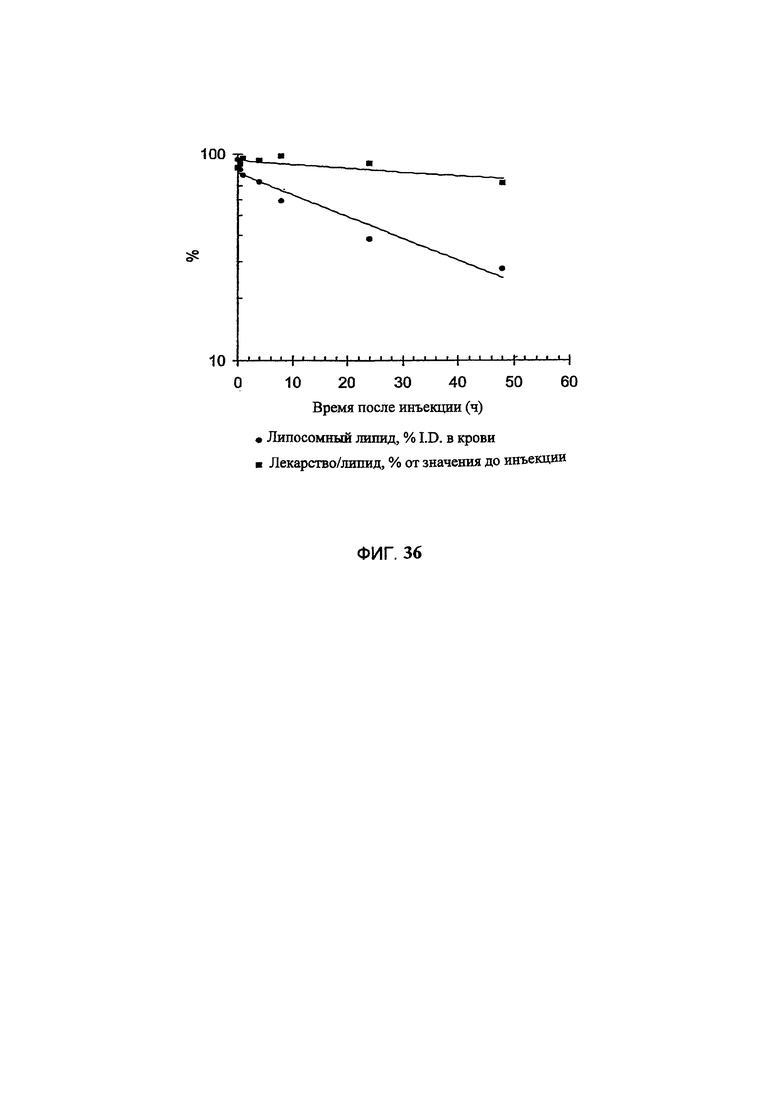
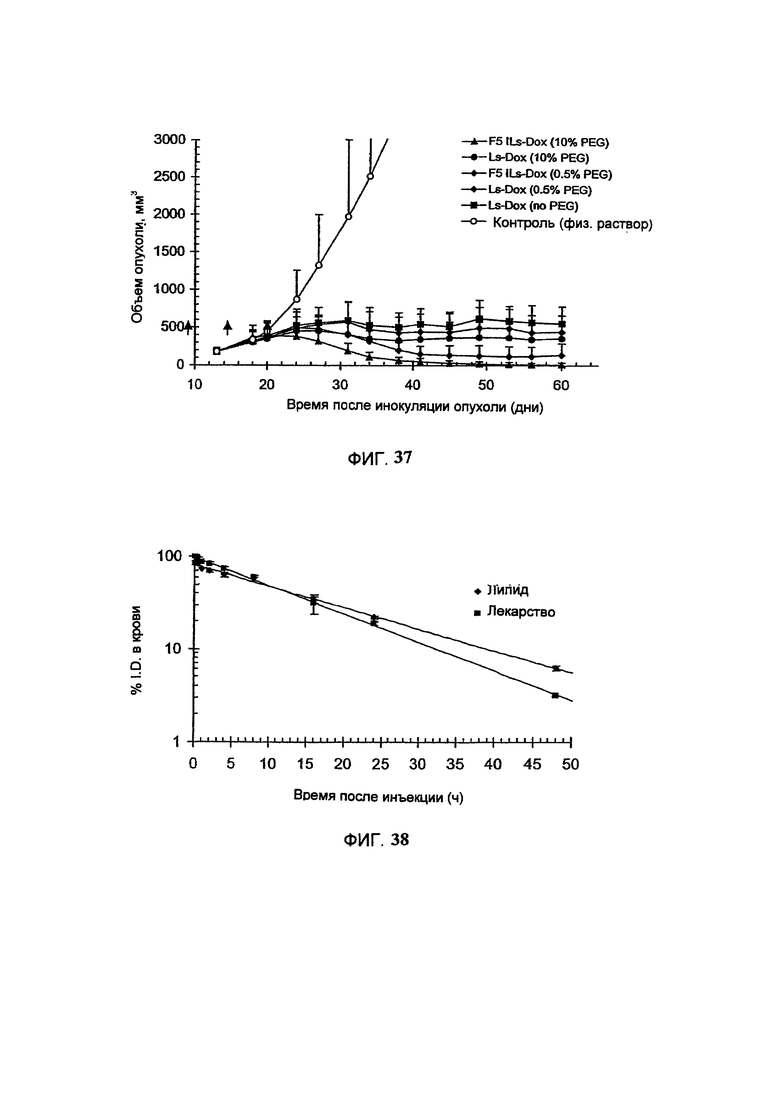
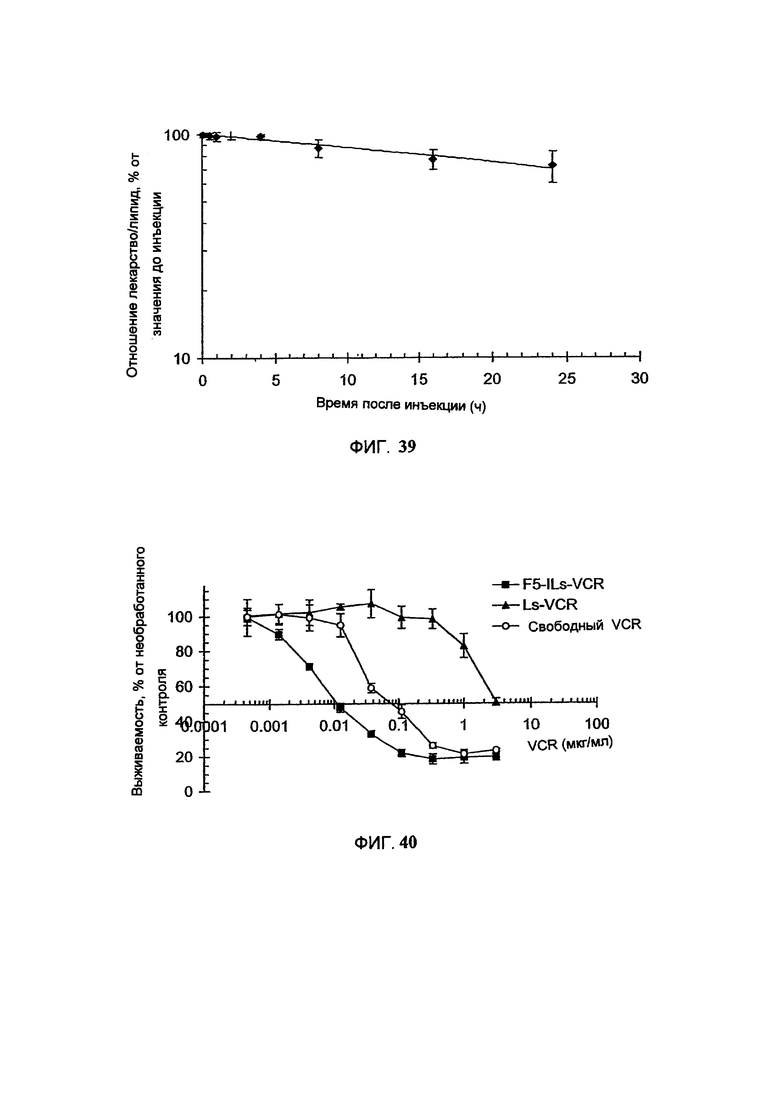
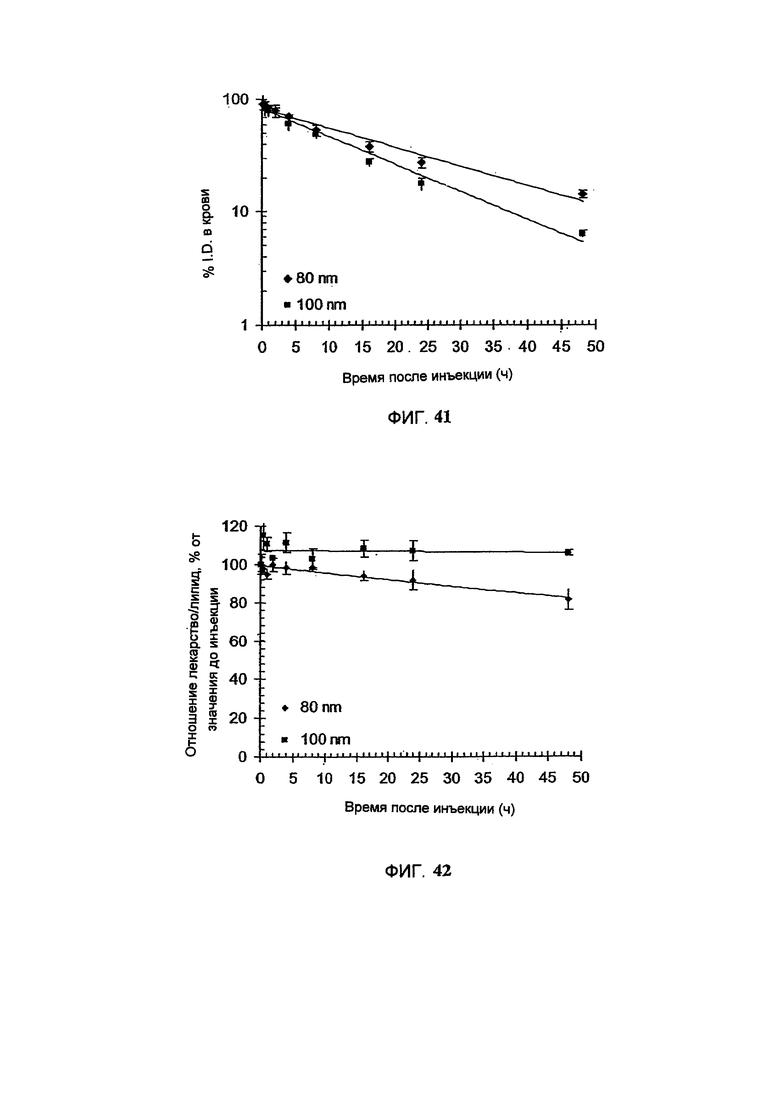
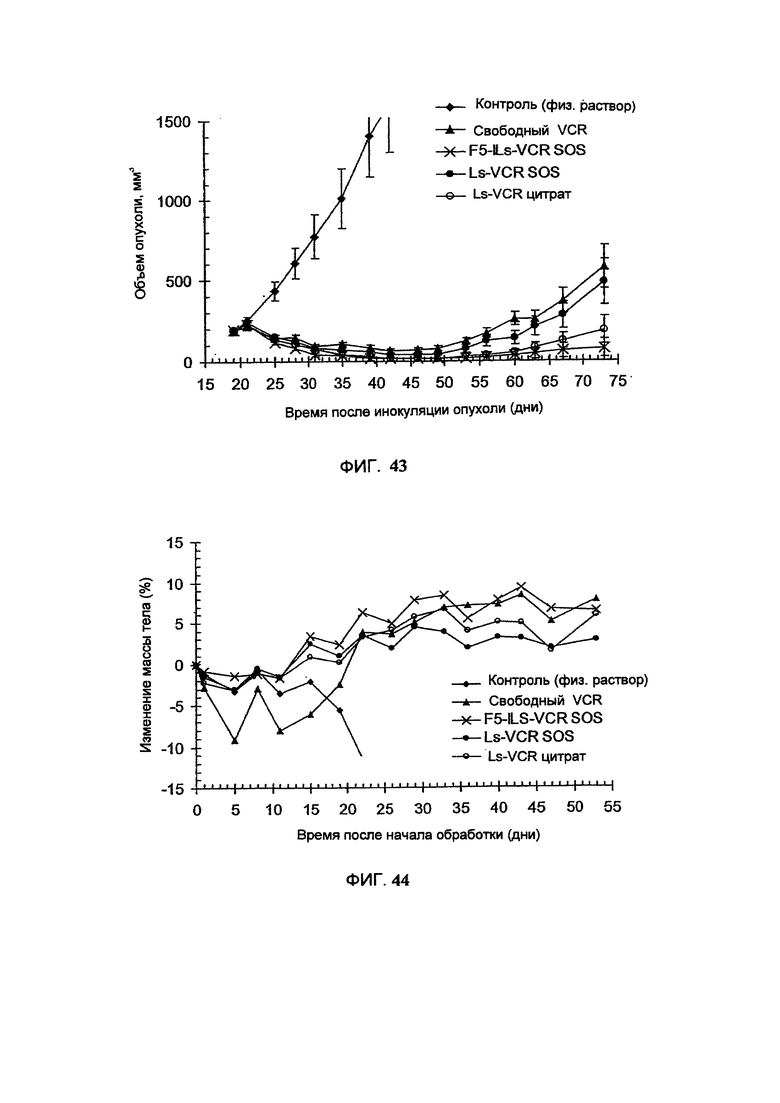
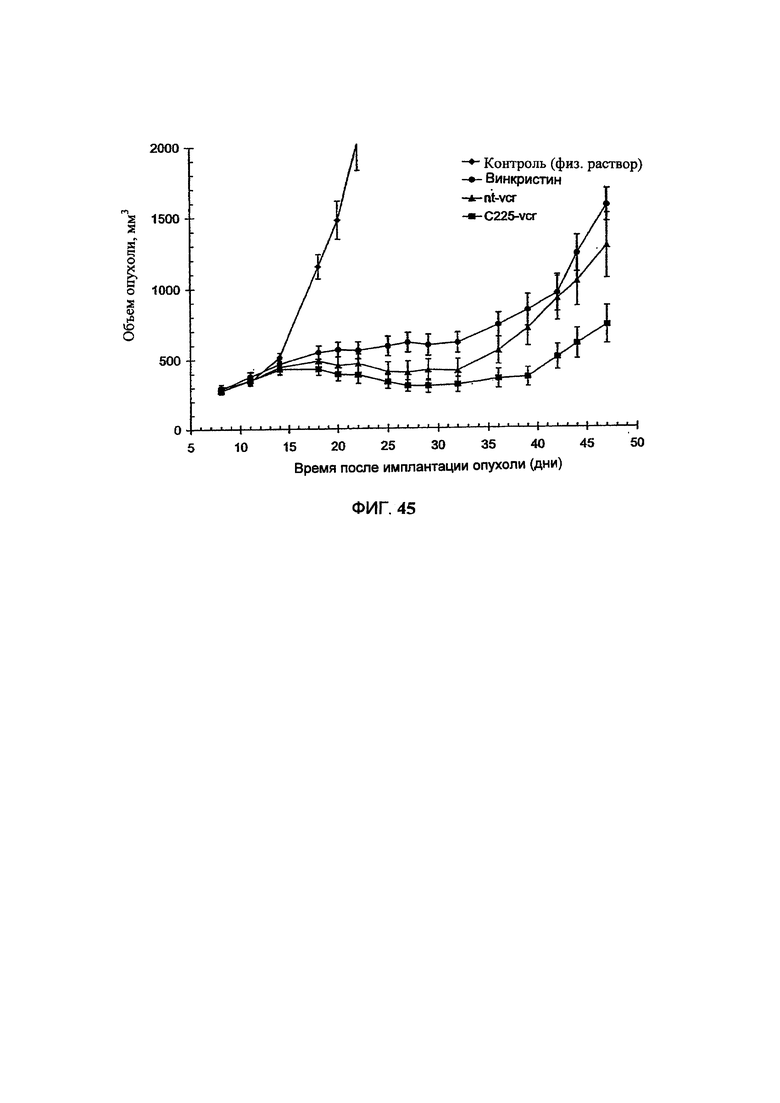
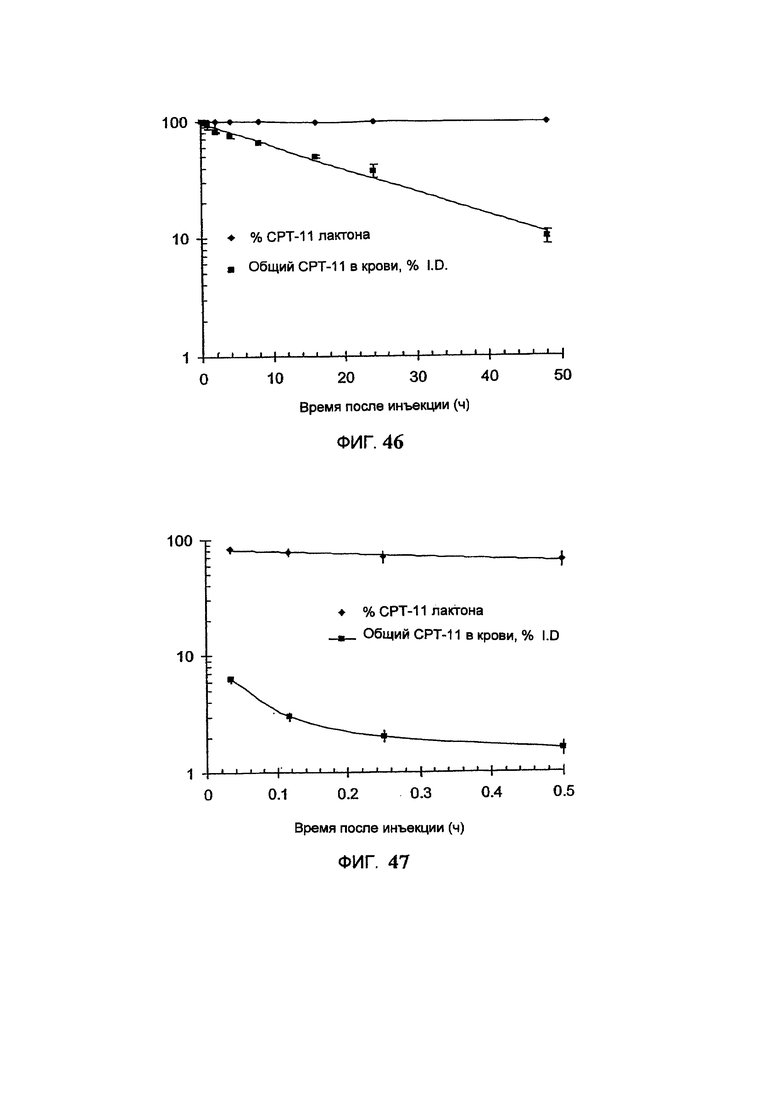
Группа изобретений относится к области медицины, а именно к противоопухолевой иринотекановой липосомной композиции, содержащей иринотекан сахарозооктасульфат, инкапсулированный в липидные везикулы, содержащие один или более фосфолипидов, причем липосомная композиция содержит суммарное количество в диапазоне от 150 до 550 мг основания иринотекана на ммоль суммарных фосфолипидов, а также относится к противоопухолевой иринотекановой липосомной композиции, содержащей липосомы, в которых инкапсулирован иринотекан в форме соли, выбранной из пирофосфата, трифосфата и инозитгексафосфата, причем указанная композиция содержит лецитин, холестерин и амфипатический полимер, и указанная композиция имеет от около 0,15 до около 1,5 моль иринотекана на моль суммарного липида, также относится к противоопухолевой иринотекановой липосомной композиции, содержащей липидные везикулы, в виде липосомной дисперсии, где в липидных везикулах инкапсулирован иринотекан и сахарозооктасульфат, так что иринотекан и сахарозооктасульфат образуют гель или осадок в виде соли, причем указанная композиция содержит лецитин, холестерин и амфипатический полимер, и указанная композиция имеет от около 0,15 до около 1,5 моль иринотекана на моль суммарного липида, также относится к противоопухолевой иринотекановой липосомной композиции, содержащей липосому в водной среде, причем липосома содержит 1,2-дистеароил-SN-фосфатидилхолин, холестерол и N-(омега-метоксиполи(этиленгликоль)оксикарбонил)-1,2-дистеароилфосфатидилэтаноламин в мольном соотношении 3:2:0,015, и внутри липосомы захвачены иринотекан и сахарозооктасульфат. Группа изобретений обеспечивает создание липосомных композиций, которые не только обладают исключительной способностью к инкапсуляции вещества, но также обеспечивают улучшенную стабильность указанного инкапсулированного вещества, например лекарственного средства, и препятствуют его преждевременному высвобождению из липосомы в живом организме. 5 н. и 17 з.п. ф-лы, 47 ил., 40 табл., 74 пр.
1. Противоопухолевая иринотекановая липосомная композиция, содержащая иринотекан сахарозооктасульфат, инкапсулированный в липидные везикулы, содержащие один или более фосфолипидов, причем липосомная композиция содержит суммарное количество в диапазоне от 150 до 550 мг основания иринотекана на ммоль суммарных фосфолипидов.
2. Противоопухолевая иринотекановая липосомная композиция по п.1, содержащая всего около 500 мг основания иринотекана на ммоль суммарных фосфолипидов.
3. Противоопухолевая иринотекановая липосомная композиция по п.1, причем липидные везикулы дополнительно содержат лецитин, холестерин и амфипатический полимер.
4. Противоопухолевая иринотекановая липосомная композиция по п.3, причем лецитин представляет собой диацилфосфатидилхолин.
5. Противоопухолевая иринотекановая липосомная композиция по п.3, причем амфипатический полимер представляет собой полиэтиленгликоль-липидное производное.
6. Противоопухолевая иринотекановая липосомная композиция по п.3, причем амфипатический полимер содержит полиэтиленгликоль-липидное производное, имеющее поли(этиленгликолевую) часть с молекулярной массой в диапазоне от около 250 до около 20000.
7. Противоопухолевая иринотекановая липосомная композиция по п.3, где амфипатический полимер выбран из полиэтиленгликоль-фосфатидилэтаноламина, полиэтиленгликоль-диацилглицерина и полиэтиленгликоль-церамидного производного.
8. Противоопухолевая иринотекановая липосомная композиция по п.3, причем лецитин представляет собой 1,2-дистеароил-sn-глицеро-3-фосфохолин (DSPC) и амфипатический полимер представляет собой полиэтиленгликоль-липидное производное, имеющее поли(этиленгликолевую) часть с молекулярной массой в диапазоне от около 250 до около 20000.
9. Противоопухолевая иринотекановая липосомная композиция по п.1, содержащая незаряженный липидный компонент и нейтральный фосфолипид.
10. Противоопухолевая иринотекановая липосомная композиция по п.9, содержащая один или более незаряженный(х) липидный(х) компонет(ов), выбранных из: холестерина, церамида, диацилглицерина, ацил(простого полиэфира) и алкилполи(простого эфира).
11. Противоопухолевая иринотекановая липосомная композиция по п.9 или 10, содержащая нейтральный фосфолипид, выбранный из диацилфосфатидилхолина, сфингомиелина и диацилфосфатидилэтаноламина.
12. Противоопухолевая иринотекановая липосомная композиция по п.2, имеющая суммарную концентрацию свободного основания иринотекана от около 3,44 до 4,62 мг/мл иринотекановой липосомной композиции.
13. Противоопухолевая иринотекановая липосомная композиция, содержащая липосомы, в которых инкапсулирован иринотекан в форме соли, выбранной из пирофосфата, трифосфата и инозитгексафосфата, указанная композиция содержит лецитин, холестерин и амфипатический полимер, и указанная композиция имеет от около 0,15 до около 1,5 моль иринотекана на моль суммарного липида.
14. Противоопухолевая иринотекановая липосомная композиция, содержащая липидные везикулы, в виде липосомной дисперсии, где в липидных везикулах инкапсулирован иринотекан и сахарозооктасульфат, так что иринотекан и сахарозооктасульфат образуют гель или осадок в виде соли, причем указанная композиция содержит лецитин, холестерин и амфипатический полимер, и указанная композиция имеет от около 0,15 до около 1,5 моль иринотекана на моль суммарного липида.
15. Противоопухолевая иринотекановая липосомная композиция, содержащая липосому в водной среде, причем липосома содержит 1,2-дистеароил-SN-фосфатидилхолин, холестерол и N-(омега-метоксиполи(этиленгликоль)оксикарбонил)-1,2-дистеароилфосфатидилэтаноламин в мольном соотношении 3:2:0,015, и внутри липосомы захвачены иринотекан и сахарозооктасульфат.
16. Противоопухолевая иринотекановая липосомная композиция по п.15, содержащая от около 0,15 до около 1,5 моль иринотекана на моль суммарного липида.
17. Противоопухолевая иринотекановая липосомная композиция по п.15, при милимоли иринотекана на грамм суммарных липосомальных липидов составляют от 0,8 до 1,3.
18. Противоопухолевая иринотекановая липосомная композиция по п.16 или 17, где внутри липосомы также захвачен триэтиламмоний.
19. Противоопухолевая иринотекановая липосомная композиция по п.16 или 17, отличающаяся тем, что N-(омега-метокси-поли(этиленгликоль)оксикарбонил)-1,2- дистеароилфосфатидилэтаноламин представляет собой N-(омега-метокси-поли(этиленгликоль) (с молекулярной массой 2000)-оксикарбонил)-1,2-дистеароилфосфатидилэтаноламин.
20. Фармацевтическая композиция, содержащая противоопухолевую иринотекановую липосомную композицию по п.16 или 17, фармацевтически приемлемый носитель и буферное вещество.
21. Фармацевтическая композиция по п.20, где фармацевтическая композиция приготовлена как инъекционная.
22. Фармацевтическая композиция по п.20 или 21, отличающаяся тем, что N-(омега-метокси-поли(этиленгликоль)оксикарбонил)-1,2-дистеароилфосфатидилэтаноламин представляет собой N-(омега-метокси-поли(этиленгликоль) (с молекулярной массой 2000)-оксикарбонил)-1,2-дистеароилфосфатидилэтаноламин.
| US 5785987 A, 28.07.1998 | |||
| US 6110491 A, 29.08.2000 | |||
| RU 98116122 A, 27.06.2000. |

