Настоящее изобретение относится к непрерывному способу получения пропиленоксида, который отличается тем, что на последующей стадии регенерации растворителя ацетонитрила, поток S1, содержащий растворитель ацетонитрил и, по меньшей мере, один компонент, который имеет нормальную точку кипения, выше, чем нормальная точка кипения ацетонитрила, и который отличается тем, что десятичный логарифм коэффициента разделения октанола-воды (log KOW), по меньшей мере, одного компонента B, измеренный при 25°C, составляет больше нуля, разделяется на два потока S2 и S3, и который отличается тем, что общая масса S3 по отношению к общей массе S1 находится в диапазоне от 0,01 до 25%. Поток S3 подвергают фракционированию паровой и жидкой фаз, а поток S4, полученный в результате фракционирования паровой и жидкой фаз и обедненный, по меньшей мере, одним компонентом B, необязательно после дополнительной обработки, подвергают рециркуляции в виде потока растворителя в реакцию эпоксидирования.
В частности, в непрерывных способах промышленного масштаба эпоксидирования пропена в ацетонитриле в качестве растворителя, одной из ключевых особенностей способа в целом является рециркуляция растворителя обратно в стадию эпоксидирования. Предпочтительный способ, который позволяет эффективно рециркулировать ацетонитрил описан в патенте WO 2011/006690 A1. Данный документ описывает способ отделения ацетонитрила из воды, причем данный способ может быть преимущественно включен в непрерывный способ получения пропиленоксида в ацетонитриле в качестве растворителя. При осуществлении данного способа эпоксидирования, было обнаружено, что, хотя способ и позволяет добиться отличных результатов, в частности в отношении рециркуляции ацетонитрила, определенные примеси, содержащиеся в, по меньшей мере, одном из исходных материалов в ацетонитриле или в перекиси водорода, используемых для реакции эпоксидирования, или полученные в ходе реакции эпоксидирования в качестве вторичных продуктов или побочных продуктов или образовавшиеся во время, по меньшей мере, одной из стадий обработки, которые предпочтительно проводят после реакции эпоксидирования, могут, как правило, накапливаться в рециркулирующем потоке ацетонитрила. Эти примеси дополнительно могут оказывать негативное влияние на производительность гетерогенного катализатора, который предпочтительно используется в способе эпоксидирования, в частности катализатор на основе цеолита, имеющий каркасную структуру MWW и содержащий титан (Ti). Такое снижение производительности может выражаться или в снижении селективности, и/или активности катализатора.
Таким образом, целью настоящего изобретения является создание экономически выгодного непрерывного способа получения пропиленоксида в ацетонитриле в качестве растворителя, который позволяет, по сути, избежать накопления таких примесей в рециркулирующем потоке растворителя ацетонитрила.
Обычно, если такие примеси накапливаются в определенном потоке, поток подвергают одной или более подходящим стадиям разделения, например, стадиям перегонки, которые, если осуществляются в подходящих условиях перегонки, могут привести к получению потока, обедненного примесями. Тем не менее, в частности, в способах промышленных масштабов, использование таких стадий разделения для воздействия на рециркулирующий поток растворителя обязательно влечет за собой значительные инвестиции и высокое энергопотребление, в связи с, как правило, высокими величинами расхода, которые, таким образом, требуют использования аппаратов с большой пропускной способностью.
Тем не менее, неожиданно было обнаружено, что для отделения примесей из рециркулирующего потока ацетонитрила при непрерывном способе получения пропиленоксида, этот недостаток может быть устранен при условии, что лишь часть определенного рециркулирующего потока будет подвергаться отделению примесей, при этом не подвергая обработке основную часть данного определенного потока. В отношении примесей, которые считаются критически важными, было неожиданно обнаружено, что производительность катализатора может быть обеспечена в течение очень длительного периода времени, несмотря на то, что только указанная меньшая часть рециркулирующего потока подвергается отделению примесей.
Таким образом, настоящее изобретение относится к непрерывному способу получения пропиленоксида, который включает в себя:
(a) реагирование пропена, необязательно смешанного с пропаном, с перекисью водорода в реакционном аппарате в присутствии ацетонитрила в качестве растворителя, с получением потока S0 на выходе реакционного аппарата, при этом S0 содержит пропиленоксид, ацетонитрил, воду, по меньшей мере, один дополнительный компонент B, необязательно пропен и необязательно пропан, и отличается тем, что нормальная точка кипения, по меньшей мере, одного компонента B выше, чем нормальная точка кипения ацетонитрила, и который отличается тем, что десятичный логарифм коэффициента разделения октанола-воды (log KOW), по меньшей мере, одного компонента B составляет больше нуля;
(b) отделение пропиленоксида из S0, необязательно после отделения пропена и необязательно пропана, с получением потока S1, который содержит ацетонитрил, воду и, по меньшей мере, один дополнительный компонент B;
(c) разделение S1 на два потока S2 и S3, при котором общая масса S3 по отношению к общей массе S1 находится в диапазоне от 0,01 до 25%;
(d) воздействие на поток S3 посредством фракционирования паровой и жидкой фаз в установке фракционирования, с получением потока паровой фракции S4, обедненного, по меньшей мере, одним компонентом B, и получением жидкого кубового потока S4b, обедненного ацетонитрилом;
(e) рециркуляцию, по меньшей мере, части потока S4, необязательно после обработки, в стадию (а).
Стадия (a)
В соответствии со стадией (а) согласно настоящему изобретению, пропен, необязательно смешанный с пропаном, вступает в реакцию с перекисью водорода в реакционном аппарате в присутствии ацетонитрила в качестве растворителя.
Как правило, отсутствуют какие-либо определенные ограничения в отношении реакции пропена, необязательно смешанного с пропаном, с перекисью водорода, при условии, что поток S0 будет получен на выходе реакционного аппарата, при этом поток S0 содержит пропиленоксид, ацетонитрил, воду, по меньшей мере, один дополнительный компонент B, и необязательно пропен, и необязательно пропан.
Как правило, можно использовать чистый или практически чистый пропен в качестве исходного материала и в виде потока, который подвергают эпоксидированию на стадии (а). Предпочтительно, используют смесь пропена и пропана. Если смесь пропена и пропана используется в качестве потока, который подвергают эпоксидированию на стадии (а), весовое соотношение пропена: пропана предпочтительно составляет, по меньшей мере, 7:3. Например, может использоваться коммерчески доступный пропен, который может представлять собой либо пропен полимерного сорта, либо пропен химического сорта. Как правило, пропен полимерного сорта имеет содержание пропена в диапазоне от 99 до 99,8 вес. % и содержание пропана в диапазоне от 0,2 до 1 вес. %. Пропен химического сорта, как правило, имеет содержание пропена в диапазоне от 92 до 98 вес. % и содержание пропана в диапазоне от 2 до 8 вес. %. Согласно предпочтительному варианту осуществления настоящего изобретения, смесь пропена и пропана, которую подвергают эпоксидированию, имеет содержание пропена в диапазоне от 99 до 99,8 вес. % и содержание пропана в диапазоне от 0,2 до 1 вес. %. Таким образом, способ согласно настоящему изобретению предпочтительно включает в себя:
(a) реагирование пропена, смешанного с пропаном, с перекисью водорода в реакционном аппарате в присутствии ацетонитрила в качестве растворителя, с получением потока S0 на выходе реакционного аппарата, причем S0 содержит пропиленоксид, ацетонитрил, воду, по меньшей мере, один дополнительный компонент B, пропан и необязательно пропен, и который отличается тем, что нормальная точка кипения, по меньшей мере, одного компонента B выше, чем нормальная точка кипения ацетонитрила, и который отличается тем, что десятичный логарифм коэффициента разделения октанола-воды (log KOW), по меньшей мере, одного компонента B составляет больше нуля.
Предпочтительно, реакцию эпоксидирования на стадии (а) проводят в присутствии, по меньшей мере, одного подходящего катализатора, предпочтительно в присутствии, по меньшей мере, одного подходящего гетерогенного катализатора. Еще более предпочтительно, по меньшей мере, один подходящий катализатор содержит, по меньшей мере, один цеолит, который, в частности, содержит титан (Ti). Предпочтительно, по меньшей мере, один цеолит, содержащий Ti, имеет каркасную структуру MWW. Еще более предпочтительно, такой цеолит, содержащий Ti и имеющий каркасную структуру MWW, который называют здесь TiMWW, содержит, по меньшей мере, один дополнительный гетероатом, кроме Ti. Среди таких дополнительных гетероатомов, цинк (Zn) является наиболее предпочтительным. Такой цеолит, содержащий Zn и Ti, и имеющий каркасную структуру MWW, называют здесь ZnTiMWW.
Катализаторы, особенно предпочтительно титановые цеолитные катализаторы, и еще более предпочтительно TiMWW или ZnTiMWW, в частности ZnTiMWW, могут быть использованы в виде порошка, гранул, микросфер, формованных изделий, имеющих, например, форму шариков, цилиндров, колес, звездочек, сфер и т.д., или в виде экструдатов, таких как, экструдаты, имеющие, например, длину от 1 до 10, более предпочтительно от 1 до 7, еще более предпочтительно от 1 до 5 мм, и диаметр от 0,1 до 5 мм, более предпочтительно от 0,2 до 4 мм и особенно предпочтительно от 0,5 до 2 мм.
Получение таких предпочтительных катализаторов TiMWW описано, например, в патенте US 2007043226 A1, в частности, в Примерах 3 и 5 патента US 2007043226 A1.
Что касается предпочтительного катализатора ZnTiMWW, более предпочтительно использовать данный катализатор в виде микропорошка или в виде формованного изделия, отличающегося тем, что формованное изделие предпочтительно содержит указанный микропорошок.
Указанный катализатор ZnTiMWW в виде микропорошка предпочтительно отличается следующими особенностями и вариантами, включая комбинации вариантов в соответствии с указанными зависимостями:
1. Микропорошок, частицы которого имеют значение DV10, по меньшей мере, 2 мкм, причем указанный микропорошок содержит мезопоры, имеющие средний диаметр пор (4V/A) в диапазоне от 2 до 50 нм, как определено методом ртутной порометрии в соответствии с DIN 66133, и содержит, в расчете на вес микропорошка, по меньшей мере, 95 вес. % микропористого цеолитного материала без содержания алюминия структурного типа MWW, содержащего титан и цинк (ZnTiMWW). Значение DV10 следует понимать согласно определению в соответствии со Справочным примером 2 настоящего изобретения.
2. Микропорошок согласно варианту 1, имеющий значение Dv10 в диапазоне от 2 до 5,5 мкм, предпочтительно от 3 до 5,5 мкм.
3. Микропорошок согласно варианту 1 или 2, имеющий значение Dv50 в диапазоне от 7 до 25 мкм и значение Dv90 в диапазоне от 26 до 85 мкм. Значения Dv50 и Dv90 следует понимать согласно определению в соответствии со Справочным примером 2 настоящего изобретения.
4. Микропорошок согласно любому из вариантов 1-3, в котором мезопоры имеют средний диаметр пор (4V/A) в диапазоне от 10 до 50 нм, предпочтительно от 15 до 40 нм, более предпочтительно от 20 до 30 нм, как определено методом ртутной порометрии в соответствии с DIN 66133.
5. Микропорошок согласно любому из вариантов 1-4, который дополнительно содержит макропоры, имеющие средний диаметр пор (4V/A) в диапазоне от более чем 50 нм, причем указанные макропоры предпочтительно имеют средний диаметр пор в диапазоне от 0,05 до 3 мкм, как определено методом ртутной порометрии в соответствии с DIN 66133.
6. Микропорошок согласно любому из вариантов 1-5, в котором микропоры ZnTiMWW имеют средний диаметр пор в диапазоне от 1,10 до 1,16 нм, как определено методом адсорбции азота в соответствии с DIN 66135.
7. Микропорошок согласно любому из вариантов 1-6, содержащий, в расчете на вес микропорошка, по меньшей мере, 99 вес. %, предпочтительно, по меньшей мере, 99,7 вес. % ZnTiMWW.
8. Микропорошок согласно любому из вариантов 1-7, в котором ZnTiMWW содержит цинк в количестве от 1,0 до 2,0 вес. %, предпочтительно от 1,2 до 1,9 вес. %, более предпочтительно от 1,4 до 1,8 вес. %, в пересчете на Zn и в расчете на вес ZnTiMWW.
9. Микропорошок согласно любому из вариантов 1-8, в котором ZnTiMWW содержит титан в количестве от 1,0 до 2,0 вес. %, предпочтительно от 1,2 до 1,8 вес. %, более предпочтительно от 1,4 до 1,6 вес. %, в пересчете на Ti и в расчете на вес ZnTiMWW.
10. Микропорошок согласно любому из вариантов 1-9, имеющий степень кристалличности, как определено рентгенодифракционным (XRD) анализом, по меньшей мере, 80%, предпочтительно, по меньшей мере, 85%.
11. Микропорошок согласно любому из вариантов 1-10, содержащий в расчете на общий вес микропорошка и в пересчете на элемент, менее 0,001 вес. %, предпочтительно менее 0,0001 вес. % благородного металла, предпочтительно выбранного из группы, состоящей из золота, серебра, платины, палладия, иридия, рутения, осмия и смеси двух или более из них, более предпочтительно выбранного из группы, состоящей из золота, платины, золота и смеси двух или более из них.
12. Микропорошок согласно любому из вариантов 1-11, содержащий в расчете на общий вес микропорошка и в пересчете на элемент, менее 0,1 вес. %, предпочтительно менее 0,01 вес. % бора.
13. Микропорошок согласно любому из вариантов 1-12, имеющий объемную плотность в диапазоне от 80 до 100 г/мл.
14. Микропорошок согласно любому из вариантов 1-13, который представляет собой распылительный порошок, предпочтительно получаемый или полученный посредством распылительной сушки.
Кроме того, указанный катализатор ZnTiMWW в виде формованного изделия предпочтительно отличается следующими особенностями и вариантами, включая комбинации вариантов в соответствии с указанными зависимостями:
1. Формованное изделие, содержащее микропористый цеолитный материал без содержания алюминия структурного типа MWW, содержащее титан и цинк (ZnTiMWW), причем указанное формованное изделие предпочтительно содержит микропорошок, содержащий, в расчете на вес микропорошка, по меньшей мере, 95 вес. % микропористого цеолитного материала без содержания алюминия структурного типа MWW, содержащего титан и цинк (ZnTiMWW), причем указанное формованное изделие более предпочтительно содержит микропорошок согласно любому из вариантов микропорошка 1-14, как описано выше, формованное изделие предпочтительно дополнительно содержит, по меньшей мере, одно связующее вещество, предпочтительно связующее вещество на основе диоксида кремния.
2. Формованное изделие согласно варианту 1, содержащее мезопоры, имеющие средний диаметр пор в диапазоне от 4 до 40 нм, предпочтительно от 20 до 30 нм, как определено методом ртутной порометрии в соответствии с DIN 66133.
3. Формованное изделие согласно варианту 1 или 2, имеющее степень кристалличности, как определено рентгенодифракционным (XRD) анализом, по меньшей мере, 55%, предпочтительно в диапазоне от 55 до 75%.
4. Формованное изделие согласно любому из вариантов 1-3, содержащее микропорошок в количестве в диапазоне от 70 до 80 вес. % и связующее вещество на основе диоксида кремния в количестве от 30 до 20 вес. %, причем микропорошок вместе со связующим веществом на основе диоксида кремния составляют, по меньшей мере, 99 вес. % формованного изделия, и которое отличается тем, что формованное изделие имеет концентрацию силанольных групп по отношению к общему числу атомов Si не более 6%, предпочтительно не более 3%, как определено методом ядерного магнитного резонанса при вращении образца под магическим углом 29Si MAS ЯМР. Концентрацию силанольных групп следует понимать согласно определению в соответствии со Справочным примером 3 настоящего изобретения.
5. Формованное изделие согласно любому из вариантов 1-4, представляющее собой заготовку, имеющую круглое поперечное сечение и диаметр в диапазоне от 1,5 до 1,7 мм, и имеющую прочность на раздавливание, по меньшей мере, 5 H, предпочтительно в диапазоне от 5 до 20 H, более предпочтительно в диапазоне от 12 до 20 H, причем прочность на раздавливание определяется с помощью испытательной установки для определения прочности на раздавливание Z2.5/TS1S в соответствии с методом, как описано в Справочном примере 4 настоящего изобретения.
6. Формованное изделие согласно любому из вариантов 1-5, причем 29Si-ЯМР-спектр указанного формованного изделия содержит шесть пиков в следующих положениях
пик 1 при -98 +/- x частей на миллион (ч.н.м.),
пик 2 при -104 +/- x ч.н.м.,
пик 3 при -110 +/- x ч.н.м.,
пик 4 при -113 +/- x ч.н.м.,
пик 5 при -115 +/- x ч.н.м.,
пик 6 при -118 +/- x ч.н.м.,
где x в любом из пиков составляет 1,5, предпочтительно 1,0, более предпочтительно 0,5,
где Q, которое определено как
Q=100*{[a1+a2]/[a4+a5+a6]}/a3
составляет не более 1,6, предпочтительно не более 1,4 и более предпочтительно не более 1,3, где [a1+a2] представляет собой сумму площадей пиков 1 и 2, и [a4+a5+a6] представляет собой сумму площадей пиков 4, 5 и 6, и a3 представляет собой площадь пика 3. Данные характеристики 29Si-ЯМР следует понимать согласно определению в соответствии со Справочным примером 5 настоящего изобретения.
7. Формованное изделие согласно любому из вариантов 1-6, имеющее водопоглощение в диапазоне от 3 до 8 вес. %, предпочтительно от 4 до 7 вес. %, более предпочтительно от 4,5 до 6,5 вес. %. Водопоглощение следует понимать согласно определению в соответствии со Справочным примером 6 настоящего изобретения.
8. Формованное изделие согласно любому из вариантов 1-7, причем инфракрасный спектр указанного формованного изделия содержит полосу в области 3746 см-1 +/- 20 см-1 и полосу в области 3678 см-1 +/- 20 см-1, где соотношение интенсивности полосы в области 3746 см-1 +/- 20 см-1 по отношению к полосе в области 3678 см-1 +/- 20 см-1, составляет не более 1,5, предпочтительно не более 1,4, более предпочтительно не более чем 1,3, более предпочтительно меньше, не более 1,2. Данные ИК характеристики следует понимать согласно определению в соответствии со Справочным примером 7 настоящего изобретения.
Предпочтительный способ получения предпочтительного катализатора ZnTiMWW и соответствующее определение характеристик данного катализатора ZnTiMWW описаны в Справочном примере 1 настоящего изобретения.
Таким образом, настоящее изобретение также относится к вышеописанному способу, который отличается тем, что на стадии (a), пропен подвергают реакции с перекисью водорода в присутствии гетерогенного катализатора, причем указанный гетерогенный катализатор предпочтительно содержит цеолит, предпочтительно титановый цеолит, более предпочтительно титановый цеолит структурного типа MWW (TiMWW), более предпочтительно цинксодержащий титановый цеолит структурного типа MWW (ZnTiMWW).
Таким образом, способ согласно настоящему изобретению предпочтительно включает в себя:
(a) реагирование пропена, смешанного с пропаном, с перекисью водорода в присутствии гетерогенного катализатора, причем указанный гетерогенный катализатор предпочтительно содержит цеолит, предпочтительно титановый цеолит, более предпочтительно титановый цеолит структурного типа MWW (TiMWW), более предпочтительно цинксодержащий титановый цеолит структурного типа MWW (ZnTiMWW), в реакционном аппарате в присутствии ацетонитрила в качестве растворителя, с получением потока S0 на выходе реакционного аппарата, причем S0 содержит пропиленоксид, ацетонитрил, воду, по меньшей мере, один дополнительный компонент B, пропан и необязательно пропен, и отличается тем, что нормальная точка кипения, по меньшей мере, одного компонента B выше, чем нормальная точка кипения ацетонитрила, и который отличается тем, что десятичный логарифм коэффициента разделения октанола-воды (log KOW) по меньшей мере, одного компонента B составляет больше нуля.
Обычно реакция на стадии (a) может проводиться любым подходящим способом. Так, например, она может быть проведена в реакторе периодического действия или, по меньшей мере, в одном реакторе полунепрерывного действия или, по меньшей мере, в одном реакторе непрерывного действия. Предпочтительным является непрерывный режим работы, при котором реакцию предпочтительно проводят при температуре в диапазоне от -10 до 120°C, более предпочтительно от 30 до 90°C, более предпочтительно от 30 до 65°C. Предпочтительно, температура, при которой проводится реакция не поддерживается постоянной во время реакции, а регулируется непрерывно или ступенчато, чтобы обеспечить постоянную конверсию перекиси водорода, как определено в потоке S0 на выходе из реактора, в котором осуществляется реакция эпоксидирования на стадии (а). Предпочтительно, реакцию на стадии (а) проводят, по меньшей мере, в одном реакторе непрерывного действия, таком как трубчатый реактор или кожухотрубный реактор, который предпочтительно содержит, по меньшей мере, одну охлаждающую рубашку, окружающую, по меньшей мере, одну трубку. Если реакцию на стадии (а) проводят в таком реакторе, содержащем, по меньшей мере, одну охлаждающую рубашку, термин "температура реакции", используемый здесь, относится к температуре охлаждающей среды на входе в охлаждающую рубашку. Как правило, вследствие деактивации катализатора, температура реакции непрерывно или ступенчато повышается. Предпочтительно, температура реакции непрерывно или ступенчато повышается не более, чем на 1°C/сутки, более предпочтительно менее чем на 1°C/сутки. Предпочтительно, конверсия перекиси водорода, которую предпочтительно поддерживают постоянной, составляет, по меньшей мере, 80%, более предпочтительно, по меньшей мере, 85%, более предпочтительно, по меньшей мере, 90%, более предпочтительно находится в диапазоне от 90 до 95%. Принцип определения предпочтительной конверсии перекиси водорода описан в Примере 1, раздел 1.1) ниже. Давление, по меньшей мере, в одном реакторе, как правило, находится в диапазоне от 3 до 100 бар, предпочтительно от 15 до 45 бар. В особенно предпочтительных вариантах осуществления способа согласно настоящему изобретению, реакцию проводят при температурах и давлении, при которых реакционная смесь является жидкой и отсутствует газовая фаза в, по меньшей мере, одном реакторе, в котором могут находиться две или более жидких фазы. Молярное соотношение пропена и перекиси водорода в отношении исходных материалов, подаваемых в, по меньшей мере, один реактор, в котором осуществляют эпоксидирование на стадии (а), предпочтительно находится в диапазоне от 0,9:1 до 3,0:1, более предпочтительно от 0,98:1 до 1,6:1, более предпочтительно от 1,0:1 до 1,5:1. Количество ацетонитрила, подаваемого в, по меньшей мере, один реактор, регулируют таким образом, что концентрация перекиси водорода в общем потоке, подаваемом в, по меньшей мере, один реактор, в котором осуществляют эпоксидирование на стадии (а), предпочтительно находилась в диапазоне от 2 до 20 вес. %, более предпочтительно от 5 до 12 вес. %, в расчете на полную массу общего потока.
Предпочтительно, общий поток, подаваемый в, по меньшей мере, один реактор эпоксидирования, то есть загрузка реактора, содержит от 50 до 80 вес. %, более предпочтительно от 60 до 70 вес. % ацетонитрила, от 7 до 14 вес. %, более предпочтительно от 8 до 11 вес. % пропена, от 5 до 12 вес. %, более предпочтительно от 6 до 10 вес. % перекиси водорода и от 10 до 25 вес. %, предпочтительно от 12 до 20 вес. % воды.
Предпочтительно, реакцию на стадии (а) проводят в две или более стадии, предпочтительно в две или три стадии, наиболее предпочтительно в две стадии. Предпочтительно, двухстадийная реакция включает в себя:
(a1) реагирование пропена, необязательно смешанного с пропаном, с перекисью водорода, предпочтительно в присутствии гетерогенного катализатора, причем указанный гетерогенный катализатор предпочтительно содержит цеолит, предпочтительно титановый цеолит, более предпочтительно титановый цеолит структурного типа MWW (TiMWW), более предпочтительно цинксодержащий титановый цеолит структурного типа MWW (ZnTiMWW), в реакционном аппарате в присутствии ацетонитрила в качестве растворителя, с получением потока S0-a1 на выходе реакционного аппарата, причем S0-a1 содержит пропиленоксид, ацетонитрил, воду, необязательно, по меньшей мере, один дополнительный компонент B, необязательно пропан, необязательно пропен, и непрореагировавшую перекись водорода;
(a2) отделение пропиленоксида из S0-a1, с получением потока S0-a2-1, обогащенного пропиленоксидом и обедненного перекисью водорода, и потока S0-а2-2, обедненного пропиленоксидом и содержащего непрореагировавшую перекись водорода, ацетонитрил и воду;
(a3) воздействие на поток S0-a2-2, предпочтительно после смешивания с пропеном, необязательно смешанным с пропаном, посредством условий реакции эпоксидирования, предпочтительно в присутствии гетерогенного катализатора, причем указанный гетерогенный катализатор предпочтительно содержит цеолит, предпочтительно титановый цеолит, более предпочтительно титановый цеолит структурного типа MWW (TiMWW), более предпочтительно цинксодержащий титановый цеолит структурного типа MWW (ZnTiMWW), в реакционном аппарате с получением потока S0-a3 на выходе реакционного аппарата, причем S0-a3 содержит пропиленоксид, ацетонитрил, воду, необязательно, по меньшей мере, один дополнительный компонент B, необязательно пропан и необязательно пропен;
отличается тем, что либо S0-a1 и/или S0-a3 содержит, по меньшей мере, один компонент B, и отличается тем, что нормальная точка кипения, по меньшей мере, одного компонента B выше, чем нормальная точка кипения ацетонитрила, и отличается тем, что десятичный логарифм коэффициента разделения октанола-воды (log KOW) по меньшей мере, одного компонента B составляет больше нуля.
На предпочтительной схеме способа согласно настоящему изобретению, как показано на Фигуре 1 ниже, поток (5) представляет собой предпочтительный поток S0-a1, поток (6) представляет собой предпочтительный поток S0-a2-1, поток (7) представляет собой предпочтительный поток S0-a2-2, и поток (9) представляет собой предпочтительный поток S0-a3. Поток (8) на Фигуре 1 представляет собой предпочтительный поток пропена, необязательно смешанного с пропаном, который предпочтительно смешивают на стадии (a3).
Предпочтительно, потоки S0-a2-1 и S0-a3 вместе составляют поток S0 согласно настоящему изобретению.
Что касается предпочтительных условий реакции эпоксидирования на стадии (a1), делается ссылка на предпочтительную реакцию эпоксидирования, как описано выше. Перекись водорода может быть отделена в соответствии с (a2) с помощью любых подходящих способов. Перекись водорода предпочтительно отделяют перегонкой с использованием одной или более перегонных колонн, предпочтительно одной перегонной колонны. Данная перегонная колонна предпочтительно работает при условиях, позволяющих получать головной (верхний) поток, который содержит перекись водорода в количестве не более 100 весовых частей на миллион, в расчете на общую массу головного потока, предпочтительно, не содержащего по существу перекиси водорода. Кроме данная перегонная колонна предпочтительно работает при условиях, позволяющих получать головной поток, который содержит, по меньшей мере, 80%, более предпочтительно, по меньшей мере, 90%, более предпочтительно, по меньшей мере, 95% пропиленоксида, содержащегося в подаваемом потоке S0-a1. Предпочтительно, данная перегонная колонна имеет от 15 до 45, предпочтительно от 20 до 40 теоретических тарелок и работает при давлении в верхней части колонны в диапазоне от 0,5 до 1,2 бар, предпочтительно от 0,7 до 1,1 бар. Флегмовое число данной перегонной колонны предпочтительно находится в диапазоне от 0,05:1 до 0,5:1, более предпочтительно от 0,1:1 до 0,2:1. Кубовый поток, получаемый из перегонной колонны на стадии (a2), содержащий по существу всю непрореагировавшую перекись водорода стадии (a1) и дополнительно содержащий ацетонитрил, воду, предпочтительно подают на стадию (a3). Что касается стадии (a3), предпочтительно использовать адиабатический реактор, предпочтительно адиабатический шахтный реактор. Условия эпоксидирования на стадии (a3) предпочтительно выбирают таким образом, чтобы обеспечить конверсию перекиси водорода на выходе (a3), по меньшей мере, 99%, предпочтительно, по меньшей мере, 99,5%, более предпочтительно, по меньшей мере, 99,9%, в расчете на перекись водорода, подаваемого на стадии (a1). На стадии (a3) предпочтительно использовать тот же катализатор, что и на стадии (a1). Что касается пропена, который предпочтительно вводят в реактор, используемый на стадии (a3), делается ссылка на пропен, который уже рассматривался выше в контексте стадии (а). Таким образом, например, можно использовать пропен химического сорта или пропен полимерного сорта, при этом предпочтение отдается пропену полимерного сорта. Если стадии (a1) и (a3) выполняются, реакторы предпочтительно работают таким образом, чтобы общая конверсия пропена, с учетом конверсии на стадии (a1) и конверсии на стадии (a3) составляла, по меньшей мере, 65%, более предпочтительно, по меньшей мере, 70%, более предпочтительно, по меньшей мере, 75%.
В зависимости от конкретных условий эпоксидирования на стадии (а), поток S0 может содержать любые возможные количества пропиленоксида, ацетонитрила, воды, по меньшей мере, одного дополнительного компонента B, необязательно пропена и необязательно пропана. Предпочтительно, от 90 до 97 вес. %, более предпочтительно от 92 до 97 вес. %, более предпочтительно от 95 до 97 вес. % потока S0 состоит из ацетонитрила, воды и пропиленоксида, и от 0,01 до 3 вес. %, более предпочтительно от 0,015 до 2 вес. %, более предпочтительно от 0,02 до 0,1 вес. ч.н.м. потока S0 состоит из, по меньшей мере, одного компонента B. Термин "… вес. % потока S0 состоит из, по меньшей мере, одного компонента B" относится к общему количеству всех компонентов B, содержащихся в потоке S0. Более предпочтительно, от 90 до 97 вес. %, более предпочтительно от 92 до 97 вес. %, более предпочтительно от 95 до 97 вес. % потока S0 состоит из ацетонитрила, воды и пропиленоксида, от 0,05 до 7 вес. %, более предпочтительно от 0,1 до 6 вес. %, более предпочтительно от 0,15 до 4 вес. % состоит из пропена и необязательно пропана, и от 0,01 до 3 вес. %, предпочтительно от 0,015 до 2 вес. %, более предпочтительно от 0,02 до 1 вес. ч.н.м. потока S0 состоит из, по меньшей мере, одного компонента B.
Согласно настоящему изобретению десятичный логарифм коэффициента разделения октанола-воды (log KOW) по меньшей мере, одного компонента B составляет больше нуля. Коэффициент разделения октанола-воды (log KOW) является параметром хорошо известным специалистам в данной области. Для полноты, его определение и расчет описаны в Справочном примере 8 ниже.
Как правило, по меньшей мере, один компонент B, содержащийся в потоке S0, либо представляет собой вторичный продукт и/или побочный продукт, полученный во время реакции эпоксидирования на стадии (a), и/или соединение, которое образуется во время, по меньшей мере, одной из стадий обработки, которую предпочтительно проводят после стадии (а), и которые накапливаются, если определенные технологические потоки предпочтительного интегрированного способа рециркулируются в стадию (а), и/или содержатся в качестве примесей в, по меньшей мере, одном из исходных материалов, используемых на стадии (а), такие, как примеси в ацетонитриле или примеси в перекиси водорода.
Предпочтительно, по меньшей мере, один компонент B представляет собой пропионитрил, 1-нитропропан, 2-нитропропан, 3-метилбутаннитрил, н-пентаннитрил, 1-пентанол, 2-пентанол, 2-бутанон, 2-пентанон, 2-гексанон, 4-метил -2-гептанон, 2,6-диметил-4-гептанол, 4,6-диметил-2-гептанол, 2,6-диметил-4-гептанон, 4,6-диметил-2-гептанон, 2,6-диметил-4,6-гептандиол, 2,4-диметил-оксазолин, 2,5-диметилоксазолин, цис-2,4-диметил-1,3-диоксолан, транс-2,4-диметил-1,3-диоксолан, по меньшей мере, одну примесь, содержащуюся в перекиси водорода, используемой на стадии (a), или комбинацию двух или более из этих соединений.
Предпочтительно, по меньшей мере, один компонент B включает в себя пропионитрил, 1-нитропропан, 2-нитропропан, 2,6-диметил-4-гептанол, 4,6-диметил-2-гептанол, 2,6-диметил-4-гептанон, или комбинацию двух или более из этих соединений. Более предпочтительно, по меньшей мере, один компонент B включает в себя комбинацию из трех или более таких соединений, более предпочтительно комбинацию из четырех или более таких соединений, более предпочтительно комбинацию из пяти или более из таких соединений. Более предпочтительно, по меньшей мере, один компонент B включает в себя комбинацию пропионитрила, 1-нитропропана, 2-нитропропана, 2,6-диметил-4-гептанола, 4,6-диметил-2-гептанола, 2,6-диметил-4-гептанона.
Что касается, по меньшей мере, одной примеси, содержащейся в перекиси водорода, используемой на стадии (а), данная, по меньшей мере, одна примесь представляет собой алкилфосфат, такой как трис-(2-этилгексил) фосфат, нониловый спирт, такой как диизобутилкарбинол, алкилциклогексиловый эфир, такой как 2 метил-циклогексилацетат, N,N-диалкилкарбонамиды, такие как N,N-дибутилпропионамид, N-алкил-N-арилкарбонамиды, такие как N-этил-N-фенилбензамид, N,N-диалкилкарбамат, такой как 2-этилгексил-N-бутилкарбамат, тетраалкилмочевину, такую как тетра-н-бутилмочевину, циклоалкилмочевину, такую как дигексилпропенмочевину, фенилалкилмочевину, такую как N,N-дибутил-N'-метил-N'-фенилмочевину, N-алкил-2-пирролидон, такой как октил пирролидон, N-алкил капролактам, такой как н-октил капролактам, C8-C12 алкильные ароматические соединения, дибутиламин, дибутилформамид, 1-бутанол, масляный альдегид, 2-этилгексанол, 2-этилантрахинон, 2-этил-5,6,7,8-тетрагидроантрахинон, или комбинацию двух или более из этих соединений.
Возможно, чтобы реагирование пропена, смешанного с пропаном, с перекисью водорода, в присутствии гетерогенного катализатора, причем указанный гетерогенный катализатор предпочтительно содержит цеолит, предпочтительно титановый цеолит, более предпочтительно титановый цеолит структурного типа MWW (TiMWW), более предпочтительно цинксодержащий титановый цеолит структурного типа MWW (ZnTiMWW), в реакционном аппарате в присутствии ацетонитрила в качестве растворителя, например, реагирование пропена на стадии (a1) и/или (a3) осуществлялось в присутствии, по меньшей мере, одной калиевой соли, которую растворяют в соответствующих смесях, которые подвергают условиям эпоксидирования на стадии (а), например, на стадиях (a1) и/или (a3). Предпочтительно, по меньшей мере, одну калиевую соль выбирают из группы, состоящей из, по меньшей мере, одной неорганической калиевой соли, по меньшей мере, одной органической калиевой соли и комбинаций, по меньшей мере, одной неорганической калиевой соли и, по меньшей мере, одной органической калиевой соли, в которых, по меньшей мере, одна из, по меньшей мере, одной калиевой соли представляет собой органическую калиевую соль. Более предпочтительно, по меньшей мере, одну калиевую соль выбирают из группы, состоящей из, по меньшей мере, одной неорганической калиевой соли, выбранной из группы, состоящей из гидроксида калия, галогенидов калия, нитрата калия, сульфата калия, гидросульфата калия, перхлората калия, по меньшей мере, одной органической калиевой соли, выбранной из группы, состоящей из калиевых солей алифатических насыщенных монокарбоновых кислот, предпочтительно содержащих 1, 2, 3, 4, 5 или 6 атомов углерода, карбоната калия и гидрокарбоната калия, и комбинации, по меньшей мере, одной из, по меньшей мере, одних неорганических калиевых солей и, по меньшей мере, одной из, по меньшей мере, одних органических калиевых солей. Более предпочтительно, по меньшей мере, одну калиевую соль выбирают из группы, состоящей из, по меньшей мере, одной неорганической калиевой соли, выбранной из группы, состоящей из гидроксида калия, хлорида калия, нитрата калия, по меньшей мере, одной органической калиевой соли, выбранной из группы, состоящей из формиата калия, ацетата калия, карбоната калия и гидрокарбоната калия, и комбинации, по меньшей мере, одной из, по меньшей мере, одних неорганических калиевых солей и, по меньшей мере, одной из, по меньшей мере, одних органических калиевых солей.
Таким образом, настоящее изобретение также относится к способу, который отличается тем, что стадия (a) включает в себя реагирование пропена, необязательно смешанного с пропаном, с перекисью водорода в реакционном аппарате в присутствии ацетонитрила в качестве растворителя и в присутствии, по меньшей мере, одной растворенной калиевой соли, с получением потока S0 на выходе реакционного аппарата, при этом S0 содержит пропиленоксид, ацетонитрил, воду, по меньшей мере, один дополнительный компонент B, необязательно пропен и необязательно пропан, и отличается тем, что нормальная точка кипения, по меньшей мере, одного компонента B выше, чем нормальная точка кипения ацетонитрила, и который отличается тем, что десятичный логарифм коэффициента разделения октанола-воды (log KOW), по меньшей мере, одного компонента B составляет больше нуля.
Стадия (b)
В соответствии со стадией (b) способа согласно настоящему изобретению, пропиленоксид отделяют из потока S0, с получением потока S1, который по сравнению с S0, обеднен пропиленоксидом и содержит ацетонитрил, воду и, по меньшей мере, один дополнительный компонент B. Если S0 дополнительно содержит пропен и/или пропан, предпочтительно, чтобы пропен и/или пропан также были отделены из S0 с получением потока S1, который по сравнению с S0, обеднен пропиленоксидом, пропеном и/или пропаном, и который содержит ацетонитрил, воду и, по меньшей мере, один дополнительный компонент B. Кроме того, если S0 дополнительно содержит кислород, предпочтительно, чтобы кислород также был отделен из S0 с получением потока S1, который по сравнению с S0, обеднен пропиленоксидом и кислородом, и который содержит ацетонитрил, воду и, по меньшей мере, один дополнительный компонент B. Предпочтительно, 50, полученный в соответствии со способом согласно настоящему изобретению, содержит пропен, пропан и, необязательно кислород, и, наряду с пропиленоксидом, пропен, пропан и, необязательно, кислород отделяют из S0 с получением потока 51, который по сравнению с S0, обеднен пропиленоксидом, пропеном и пропаном и, необязательно кислородом, и который содержит ацетонитрил, воду и, по меньшей мере, один дополнительный компонент B.
Что касается отделения пропена и/или пропана, и/или кислорода из S0, не существует никаких особых ограничений. В частности, все возможные последовательности разделения отдельных компонентов и все возможные способы отделения, такие как перегонка, являются возможными. Таким образом, можно отделить пропен и/или пропан и, необязательно кислород вместе с пропиленоксидом из S0 с получением потока S1. Отделенный поток, обогащенный пропеном и/или пропаном и необязательно кислородом, затем предпочтительно подвергают соответствующим стадиям отделения и/или стадиям обработки, в результате которых получают поток, который по существу состоит из пропиленоксида в качестве полноценного продукта. Предпочтительно поток S0 подвергают первой стадии отделения, в ходе которой отделяют пропен и необязательно пропан. Если S0 дополнительно содержит кислород, предпочтительно, чтобы кислород отделяли вместе с пропеном и/или пропаном.
Таким образом, настоящее изобретение относится к способу, как описано выше, включающему в себя (b) отделение пропиленоксида из S0, после отделения пропена и необязательно пропана, с получением потока S1, содержащего ацетонитрил, воду и, по меньшей мере, один дополнительный компонент B. Кроме того, настоящее изобретение относится к способу, как описано выше, включающему в себя (b) отделение пропиленоксида из S0, после отделения пропена и пропана, с получением потока S1, содержащего ацетонитрил, воду и, по меньшей мере, один дополнительный компонент B. Кроме того, настоящее изобретение относится к способу, как описано выше, включающему в себя (b) отделение пропиленоксида из S0, после отделения пропена, пропана и необязательно кислорода, с получением потока S1, содержащего ацетонитрил, воду и, по меньшей мере, один дополнительный компонент B. Кроме того, настоящее изобретение относится к способу, как описано выше, включающему в себя (b) отделение пропиленоксида из S0, после отделения пропена, пропана и кислорода, с получением потока S1, содержащего ацетонитрил, воду и, по меньшей мере, один дополнительный компонент B.
Таким образом, предпочтительно, чтобы стадия (b) способа согласно настоящему изобретению включала в себя стадию (I), которая отличается тем, что пропен, необязательно вместе с пропаном и кислородом, который необязательно дополнительно содержится в S0, отделяются из S0 с получением потока S01, обогащенного пропиленоксидом, ацетонитрилом, водой и, по меньшей мере, одним компонентом B, причем данный поток S01 обеднен пропеном, необязательно пропаном и кислородом; и дополнительно включала стадию (II), которая отличается тем, что пропиленоксид отделяют из S01, с получением потока S02, обогащенного ацетонитрилом, водой и, по меньшей мере, одним компонентом B, причем поток S02 обеднен пропиленоксидом.
Что касается отделения на стадии (I), не существует никаких особых ограничений. Предпочтительно, отделение проводят таким образом, чтобы, по меньшей мере, 90 вес. %, более предпочтительно, по меньшей мере, 95 вес. %, более предпочтительно, по меньшей мере, 98 вес. %, более предпочтительно, по меньшей мере, 99 вес. % потока S01 состояло из ацетонитрила, воды, по меньшей мере, одного компонента B и пропиленоксида. Предпочтительно, для отделения на стадии (I) используют установку фракционирования. Кроме того, предпочтительно, отделение на стадии (I) осуществляется, по меньшей мере, в одной перегонной колонне, более предпочтительно в одной перегонной колонне. В данной перегонной колонне, S01 предпочтительно получают в виде кубового потока. Предпочтительно, данная перегонная колонна имеет от 10 до 30, более предпочтительно от 15 до 25 теоретических тарелок. Перегонная колонна предпочтительно работает при верхнем давлении от 0,5 до 1,2 бар, более предпочтительно от 0,7 до 1,1 бар. Чтобы облегчить указанную задачу отделения, было обнаружено, что полезно добавлять либо жидкий ацетонитрил, либо жидкую смесь ацетонитрила с водой в верхнюю часть колонны. Считается, что данное наружное орошение служит захватывающим агентом, который, среди прочего, предотвращает отделение пропиленоксида через верхнюю часть перегонной колонны. В соответствии с предпочтительным вариантом осуществления настоящего изобретения, используют часть кубового потока перегонной колонны, предпочтительно используемого на стадии (II). Возможно также, чтобы поток TL2, описанный ниже или его часть, использовались в качестве захватывающего агента. Количество TL2 будет недостаточным, и другой поток должен быть добавлен. Предпочтительно, весовое соотношение количества ацетонитрила, подаваемого в качестве наружного орошения в верхнюю часть перегонной колонны, и массы потока S0, подаваемого в перегонную колонну, и подвергаемого разделению в перегонной колонне, находится в диапазоне от 1:1 до 4:1, предпочтительно от 1,5:1 до 3:1. Температура наружного орошения, как правило, находится в диапазоне от 2 до 20°C, предпочтительно в диапазоне от 5 до 15°C. Согласно настоящему изобретению, предпочтительно, по меньшей мере, 85 об. %, более предпочтительно, по меньшей мере, 90 об. %, более предпочтительно, по меньшей мере, 93 об. % головного потока перегонной колонны согласно (I) состоит из пропена, кислорода и необязательно пропана. В зависимости от содержания кислорода, данный головной поток может подвергаться дальнейшей подходящей стадии обработки, которая отличается тем, что содержание кислорода соответственно уменьшается с тем, чтобы обеспечить, например, возможность рециркуляции потока, обедненного кислородом, в одну или более стадий настоящего изобретения, например, в качестве исходного материала для стадии (а) способа согласно изобретению, например, стадии (a1) или стадии (a3), или в качестве части потока P, описанного ниже. Если содержание кислорода в указанном головном потоке уменьшается, предпочтительно снижать кислород посредством реакции с водородом в присутствии подходящего катализатора. Такими катализаторами, например, являются катализаторы, содержащие олово и, по меньшей мере, один благородный металл, как описано в патенте WO 2007/000396 A1, в частности, в Примере 1 патента WO 2007/000396 A1. Возможно также использовать катализаторы, содержащие медь в элементарной и/или оксидной форме на носителе, которые отличаются тем, что медь присутствует на носителе в количестве от 30 до 80 вес. %, в расчете на весь катализатор и в пересчете на CuO. Такие катализаторы могут быть получены, например, согласно примеру в патенте EP 0427062 A2, катализатор 2, стр. 4, строки с 41 по 50 (соответствующий патенту US 5,194,675). Для уменьшения содержания кислорода также возможными являются другие подходящие способы. Необязательно, указанный головной поток, перед тем, как подвергнутся гидрогенизации, может сжиматься и частично конденсироваться, в результате чего получают поток жидкости, который по существу состоит из пропена и необязательно пропана и ацетонитрила и, который содержит незначительные количества воды. Неконденсированная часть состоит в основном из пропена и необязательно пропана и кислорода, и содержит незначительное количество воды, и отличается тем, что, по сравнению с основным потоком, содержание кислорода увеличивается, при этом все же оставаясь в таком диапазоне, чтобы смесь была невоспламеняемой. Такой поток, обогащенный кислородом, затем подвергают гидрогенизации.
Как упоминалось выше, перед использованием потока S01 в качестве потока S1 в соответствии с настоящим изобретением, особенно предпочтительно отделить пропиленоксид от S01 на стадии (II) для получения потока S02, который по существу не содержит пропиленоксида. Что касается отделения на стадии (II), не существует никаких особых ограничений. Предпочтительно, отделение проводят таким образом, чтобы, по меньшей мере, 90 вес. %, более предпочтительно, по меньшей мере, 95 вес. %, более предпочтительно, по меньшей мере, 99 вес. % потока S02 состояло из ацетонитрила, воды и, по меньшей мере, одного компонента B. Более предпочтительно, весовое соотношение ацетонитрила и воды в S02 составляет больше 1:1, предпочтительно в диапазоне от 2:1 до 10:1, более предпочтительно от 2,5:1 до 5:1. Предпочтительно, для отделения на стадии (II) используют установку фракционирования. Кроме того, предпочтительно, отделение на стадии (II) осуществляется, по меньшей мере, в одной перегонной колонне, более предпочтительно в одной перегонной колонне. Предпочтительно, данная перегонная колонна имеет от 50 до 80, более предпочтительно от 60 до 70 теоретических тарелок. Перегонная колонна предпочтительно работает при верхнем давлении от 0,2 до 2 бар, более предпочтительно от 0,4 до 1 бара. Необязательно, по меньшей мере, один подходящий полярный растворитель или смесь двух или более полярных растворителей, предпочтительно воды, могут добавляться в верхнюю часть колонны в качестве экстракционного агента. В соответствии с вариантом осуществления способа согласно настоящему изобретению, отделение в соответствии со стадией (III) может осуществляться посредством:
- введения S01 в экстрактивную перегонную колонну;
- дополнительного введения полярного экстракционного растворителя или смеси двух или более из них, предпочтительно воды, в указанную экстрактивную перегонную колонну;
- отгонки пропиленоксида из верхней части указанной экстрактивной перегонной колонны в виде головного потока, которая отличается тем, что головной поток содержит только незначительные количества ацетонитрила, например, 500 ч.н.м. или менее;
- сжатия указанного головного потока, полученного в верхней части колонны на предыдущей стадии, посредством, по меньшей мере, одного компрессора с получением сжатого пара;
- конденсации сжатого пара, полученного на предыдущей стадии, и возврата, по меньшей мере, части теплоты конденсации, по меньшей мере, в один ребойлер, используемый в экстрактивной перегонной колонне.
Из данной перегонной колонны в соответствии со стадией (II), получают головной поток, который содержит, по меньшей мере, 90 вес. %, предпочтительно, по меньшей мере, 95 вес. %, более предпочтительно, по меньшей мере, 99 вес. % пропиленоксида. Кроме того, из данной перегонной колонны, предпочтительно получают поток S02 в виде кубового потока, который предпочтительно содержит не более 500 вес. ч.н.м., предпочтительно не более 100 вес. ч.н.м., более предпочтительно не более 60 вес. ч.н.м. пропиленоксида, в расчете на массу потока S02.
В зависимости от требований к качеству пропиленоксида, возможно использовать данную фракцию пропиленоксида без дополнительной очистки. Тем не менее, также возможна дальнейшая очистка указанной фракции пропиленоксида, например, посредством, по меньшей мере, одной дополнительной стадии перегонки.
Из перегонной колонны в соответствии со стадией (II) или необязательно после дополнительной стадии перегонки получают поток пропиленоксида, который отличается тем, что, по меньшей мере, 99,5 вес. %, более предпочтительно, по меньшей мере, 99,9 вес. %, более предпочтительно, по меньшей мере, 99,999 вес. % указанного потока состоит из пропиленоксида. Таким образом, настоящее изобретение также относится к композиции, содержащей, по меньшей мере, 99,999 вес. % пропиленоксида, получаемого или полученного согласно способу, описанному выше, и включающему стадию отделения (II).
Таким образом, настоящее изобретение предпочтительно относится к способу, описанному выше, который отличается тем, что стадия (b) включает в себя:
(I) отделение пропена, необязательно вместе с пропаном и кислородом, который необязательно дополнительно содержится в S0, из потока S0, с получением потока S01, обогащенного пропиленоксидом, ацетонитрилом, водой и, по меньшей мере, одним компонентом B, который отличается тем, что предпочтительно, по меньшей мере, 99 вес. % потока S01 состоит из ацетонитрила, воды, по меньшей мере, одного компонента B и пропиленоксида; и которое отличается тем, что для отделения предпочтительно используют установку фракционирования, и которое отличается тем, что предпочтительно в верхней части установки фракционирования, жидкий ацетонитрил, необязательно смешанный с жидкой водой, добавляют в качестве захватывающего агента;
(II) отделение пропиленоксида из потока S01, с получением потока S02, обогащенного ацетонитрилом, водой и, по меньшей мере, одним компонентом B, который отличается тем, что, по меньшей мере, 95 вес. % потока S02 состоит из ацетонитрила, воды и, по меньшей мере, одного компонента B, и который отличается тем, что весовое соотношение ацетонитрила и воды составляет больше 1:1.
Предпочтительно, поток S02, полученный на стадии (b), предпочтительно на стадии (II), включенной в стадию (а), подвергают стадии (с) в качестве потока S1.
Предпочтительно, от 90 до 99,9 вес. %, более предпочтительно от 95 до 99,8 вес. %, более предпочтительно от 99 до 99,7 вес. % потока S1 состоит из ацетонитрила и воды, и, предпочтительно, от 0,01 до 5 вес. %, более предпочтительно от 0,015 до 3 вес. %, более предпочтительно от 0,02 до 2 вес. % потока S1 состоит из, по меньшей мере, одного компонента B.
Необязательно, по меньшей мере, часть потока S02 отводится и используется в качестве захватывающего агента в установке фракционирования в соответствии со стадией (I), как описано выше. Предпочтительно, при использовании в качестве захватывающего агента, от 15 до 35%, более предпочтительно от 20 до 35% потока S02 отводят и предпочтительно добавляют в верхнюю часть установки фракционирования, используемой на стадии (I).
Необязательная дополнительная стадия (стадии), включенная в стадию (b)
В зависимости от конкретных условий во время предшествующих стадий способа согласно настоящему изобретению, в частности стадий (а), (I) и (II), кубовый поток, полученный из перегонной колонны в соответствии со стадией (II), может также содержать определенное количество гидроперекисей, например, определенные количества перекиси водорода и/или определенные количества органических гидроперекисей, например, 1-гидропероксипропанол-2 и/или 2-гидропероксипропанол-1. Предпочтительно, кубовый поток, полученный из перегонной колонны в соответствии со стадией (III) может содержать не более 2 вес. %, более предпочтительно не более 1 вес. % таких гидроперекисей в целом, в расчете на массу кубового потока. Для того, чтобы снизить содержание гидроперекиси и, таким образом, не допустить накопления гидроперекисей, которые, как полагают, возможно оказывают вредное влияние, связанное с образованием нежелательных побочных продуктов и аспектами безопасности, касающимися разложения гидроперекисей, возможно подвергнуть указанный кубовый поток, полученный из перегонной колонны согласно стадии (II), по меньшей мере, одной стадии дальнейшей обработки. Указанное накопление, в частности, возникает при реализации высокоинтегрированного способа согласно изобретению. Хотя любой подходящий способ, по меньшей мере, частичного удаления таких гидроперекисей является возможным, особенно предпочтительным является каталитическое восстановление, предпочтительно каталитическая гидрогенизация гидроперекисей. В качестве подходящего катализатора, следует упомянуть катализатор, который описан в патенте US 2004068128 A1, в частности, в параграфах [0053] - [0076]. Предпочтительные катализаторы выбирают из группы, состоящей из гетерогенных катализаторов, содержащих Ru, Ni, Pd, Pt, либо отдельно, либо в виде смеси двух или более из них, в качестве активного металла на подходящем носителе. Особенно подходящий катализатор, а именно катализатор на носителе, содержащий 5 вес. % Pd на активированном угле, описан в примере E2 патента US 2004068128 A1. Давление во время гидрогенизации, как правило, находится в диапазоне от 1 до 100 бар (абс), предпочтительно от 1 до 10 бар (абс), и температура во время гидрогенизации, как правило, находится в диапазоне от 0 до 180°C, предпочтительно от 25 до 120°C, более предпочтительно от 65 до 85°C. Парциальное давление водорода вовремя гидрогенизации предпочтительно находится в диапазоне от более чем 1 до 20 бар, более предпочтительно от 2 до 15 бар, более предпочтительно от 3 до 13 бар. Если гидрогенизацию проводят в неподвижном слое, который является предпочтительным, время пребывания жидкости, которую пропускают через реактор гидрогенизации, как правило, находится в диапазоне от 1 секунды (сек.) до 1 часа (ч), предпочтительно от 10 сек. до 20 минут (мин.), в частности, от 30 сек. до 5 мин. В зависимости от условий реакции, используемых для восстановления, предпочтительно гидрогенизации кубового потока, полученного из перегонной колонны согласно стадии (II), возможно потребуется отделение полученного потока от катализатора, предпочтительно катализатора гидрогенизации и/или непрореагировавшего восстанавливающего агента, предпочтительно водорода и/или побочных продуктов гидрогенизации, предпочтительно CO и/или метана. В частности, поток, полученный в результате восстановления, предпочтительно гидрогенизации, содержит, по меньшей мере, 95 вес. % ацетонитрила и воды, в расчете на общую массу кубового потока, отличающегося тем, что весовое соотношение ацетонитрила и воды предпочтительно составляет больше, чем 1:1. Как правило, можно использовать данный поток, полученный в результате гидрогенизации и предпочтительно отделения катализатора, в качестве потока S1 согласно настоящему изобретению.
В зависимости от конкретных условий во время последующих стадий настоящего изобретения, например, стадий (а), (I) и (II), а также стадии восстановления, предпочтительно стадии гидрогенизации, поток, полученный в результате восстановления, предпочтительно гидрогенизации, может содержать определенные количества ацетальдегида и необязательно дополнительные низкокипящие соединения, такие как, например, пропионовый альдегид и ацетон. Как правило, этот поток может содержать до 2000 весовых частей на миллион, предпочтительно до 1000 вес. ч.н.м., более предпочтительно до 300 вес. ч.н.м. ацетальдегида и других низкокипящих соединений в целом, в расчете на общую массу данного потока. Для того, чтобы снизить содержание ацетальдегида и необязательно содержание других низкокипящих соединений и, таким образом, не допустить накопления этих соединений, которое, в частности, возникает при реализации высокоинтегрированного способа согласно изобретению, предпочтительно подвергать данный поток, по меньшей мере, одной стадии дальнейшей обработки. Хотя любой подходящий способ, по меньшей мере, частичного удаления ацетальдегида является возможным, особенно предпочтительно отделять ацетальдегид от потока посредством перегонки. Отделение в соответствии с этой стадией предпочтительно проводят в, по меньшей мере, одной перегонной колонне, более предпочтительно в одной перегонной колонне. Предпочтительно, данная колонна имеет от 15 до 40, более предпочтительно от 20 до 35 теоретических тарелок. Перегонная колонна предпочтительно работает при верхнем давлении в диапазоне от 0,7 до 2 бар, более предпочтительно от 1,1 до 2 бар.
Из данной перегонной колонны получают кубовый поток, который предпочтительно содержит не более 200 вес. ч.н.м., предпочтительно не более 100 вес. ч.н.м., более предпочтительно не более 50 вес. ч.н.м. ацетальдегида и других низкокипящих соединений в целом, в расчете на общую массу потока. Предпочтительно, по меньшей мере, 98 вес. %, более предпочтительно, по меньшей мере, 98,5 вес. %, более предпочтительно, по меньшей мере, 98,7 вес. % кубового потока состоит из ацетонитрила, воды и, по меньшей мере, одного компонента B. Предпочтительно, по меньшей мере, 98 вес. %, более предпочтительно, по меньшей мере, 98,5 вес. %, более предпочтительно, по меньшей мере, 98,7 вес. % кубового потока состоит из ацетонитрила, воды и, по меньшей мере, одного компонента B, причем весовое соотношение ацетонитрила и воды составляет больше, чем 1:1. Как правило, можно использовать данный кубовый поток в качестве потока S1 в способе согласно настоящему изобретению. Согласно возможному варианту осуществления настоящего изобретения, такая стадия перегонки не выполняется.
Таким образом, настоящее изобретение также относится к способу, как описано выше, который отличается тем, что стадия (b) дополнительно включает в себя:
(IIIa) воздействие на поток S02, полученный на стадии (II), посредством гидрогенизации; и/или
(IIIb) воздействие на поток, полученный на стадии (II) или (IIIa), посредством перегонки для получения кубового потока,
и отличающийся тем, что гидрогенизированный поток, полученный на стадии (IIIa), или кубовый поток, полученный на стадии (IIIb), подвергают стадии (с) в качестве потока S1.
Таким образом, настоящее изобретение также относится к способу, как описано выше, который отличается тем, что стадия (b) дополнительно включает в себя:
(IIIa) воздействие на поток, полученный на стадии (II), посредством гидрогенизации с получением потока S1 и воздействие на поток S1 посредством стадии (c).
Таким образом, настоящее изобретение также относится к способу, как описано выше, который отличается тем, что стадия (b) дополнительно включает в себя:
(IIIb) воздействие на поток, полученный на стадии (II), посредством стадии перегонки, предпочтительно осуществляемой в перегонной колонне, работающей при верхнем давлении от 0,7 до 2 бар, более предпочтительно от 1,1 до 2 бар, с получением потока S1 и воздействие на поток S1 посредством стадии (с).
Кроме того, настоящее изобретение относится к способу, как описано выше, который отличается тем, что стадия (b) дополнительно включает в себя:
(IIIa) воздействие на поток S02, полученный на стадии (II), посредством стадии гидрогенизации, предпочтительно стадии каталитической гидрогенизации, причем катализатор предпочтительно представляет собой гетерогенные катализаторы, содержащие Ru, Ni, Pd, Pt, либо отдельно, либо в виде смеси двух или более из них, в качестве активного металла на подходящем носителе, в частности Pd на активированном угле; причем указанную гидрогенизацию предпочтительно проводят при давлении во время гидрогенизации в диапазоне от 1 до 100 бар(абс), предпочтительно от 1 до 10 бар(абс), и при температуре во время гидрогенизации в диапазоне от 0 до 180°C, предпочтительно от 25 до 120°C, более предпочтительно от 65 до 85°C;
(IIIb) воздействие на поток, полученный на стадии (IIIa), посредством стадии перегонки, предпочтительно осуществляемой в перегонной колонне, работающей при верхнем давлении от 0,7 до 2 бар, более предпочтительно от 1,1 до 2 бар, с получением потока S1 и воздействие на поток S1 посредством стадии (с).
Как упоминалось выше, предпочтительно, чтобы стадия (b) способа согласно настоящему изобретению не включала в себя ни (IIIa), ни (IIIb).
Стадия (c)
В соответствии со стадией (с) способа согласно настоящему изобретению, поток S1 разделяют на два потока S2 и S3, причем поток S3 подвергают, в качестве частичного потока согласно настоящему изобретению, воздействию посредством стадии (d), как описано здесь ниже. Термин "разделяют на два потока", как используется в данном контексте настоящего изобретения, как правило, включает в себя варианты осуществления, согласно которым поток S1 разделяют на более чем два потока при условии, что будут получены потоки S2 и S3, как определено здесь. Не существует никаких особых ограничений касательно того, какая часть S1 отделена в качестве потока S3. Предпочтительно, общая масса S3 по отношению к общей массе S1 составляет менее 50%, более предпочтительно менее 40%, более предпочтительно менее 30%. Более предпочтительно, общая масса S3 по отношению к общей массе S1 составляет, по меньшей мере, 0,01%. Более предпочтительно, общая масса S3 по отношению к общей массе S1 находится в диапазоне от 0,01 до 25%. Более предпочтительно, общая масса S3 по отношению к общей массе S1 находится в диапазоне от 0,05 до 20%, предпочтительно от 0,1 до 15%, более предпочтительно от 0,2 до 10%, более предпочтительно от 0,5 до 5%. Предпочтительные возможные диапазоны составляют от 0,5 до 1,5% или от 1,0 до 2,0%, или от 1,5 до 2,5%, или от 2,0 до 3,0%, или от 2,5 до 3,5%, или от 3,0 до 4,0%, или от 3,5 до 4,5%, или от 4,0 до 5,0%.
Стадия (d)
В соответствии со стадией (d) способа согласно настоящему изобретению, поток S3 подвергают фракционированию паровой и жидкой фаз в установке фракционирования, с получением потока паровой фракции S4, обедненного, по меньшей мере, одним компонентом B, и с получением жидкого кубового потока S4b, обедненного ацетонитрилом.
Как правило, не существует никаких особых ограничений в отношении стадии (d) при условии, что будет получен поток пара S4, который является обедненным, по меньшей мере, одним компонентом B, и который может быть возвращен в способ согласно настоящему изобретению. Неожиданно, тем не менее, было обнаружено, что особенно предпочтительно, если концентрация ацетонитрила в потоке S4b находится в определенном диапазоне. Было обнаружено, что такой определенный диапазон позволяет поддерживать концентрацию ацетонитрила в жидком кубовом потоке S4b на максимально низком уровне, таким образом, не допуская слишком высокой потери ацетонитрила, и одновременно, позволяет отделять очень большое количество, по меньшей мере, одного компонента B посредством жидкого кубового потока S4b. Данный определенный диапазон концентрации ацетонитрила в жидком кубовом потоке S4b, полученном на стадии (d) может составлять от 1 до 50 вес. %, от 2 до 45 вес. % или от 5 до 40 вес. %. Предпочтительно, на стадии (d) фракционирование паровой и жидкой фаз осуществляют в установке фракционирования, таким образом, что от 10 до 30 вес. %, предпочтительно от 10 до 25 вес. % жидкого кубового потока S4b состоит из ацетонитрила. Более предпочтительно, на стадии (d) фракционирование паровой и жидкой фаз осуществляют в установке фракционирования, таким образом, что от 10 до 30 вес. %, предпочтительно от 10 до 25 вес. % жидкого кубового потока S4b состоит из ацетонитрила и от 0,1 до 10 вес. %, предпочтительно от 0,25 до 5 вес. % жидкого кубового потока S4b состоит из, по меньшей мере, одного дополнительного компонента B.
Таким образом, настоящее изобретение предпочтительно относится к способу, описанному выше, отличающемуся тем, что на стадии (с), общая масса S3 по отношению к общей массе S1 находится в диапазоне от 0,5 до 5%, и отличающемуся тем, что на стадии (d) фракционирование паровой и жидкой фаз осуществляют в установке фракционирования, таким образом, что от 10 до 25 вес. % жидкого кубового потока S4b состоит из ацетонитрила. Более предпочтительно, настоящее изобретение относится к способу, описанному выше, отличающемуся тем, что на стадии (с), общая масса S3 по отношению к общей массе S1 находится в диапазоне от 0,5 до 5%, и отличающемуся тем, что на стадии (d) фракционирование паровой и жидкой фаз осуществляют в установке фракционирования, таким образом, что от 10 до 25 вес. % жидкого кубового потока S4b состоит из ацетонитрила и от 0,1 до 10 вес. % жидкого кубового потока S4b состоит из, по меньшей мере, одного дополнительного компонента B.
Как правило, не существует никаких особых ограничений в отношении того, каким образом осуществляют фракционирование паровой и жидкой фаз в установке фракционирования при условии, что будет достигнута вышеупомянутая концентрация ацетонитрила в жидком кубовом потоке S4b. В частности, давление и/или температура, и/или количество теоретических тарелок установки фракционирования, и/или флегмовое число будут соответствующим образом регулироваться специалистом в данной области техники.
Предпочтительно на стадии (d), фракционирование паровой и жидкой фаз осуществляют в установке фракционирования при абсолютном давлении в диапазоне от 0,1 до 10 бар, более предпочтительно от 0,5 до 5 бар, более предпочтительно от 1 до 2 бар.
Предпочтительно, на стадии (d), количество теоретических тарелок в установке фракционирования находится в диапазоне от 1 до 100, более предпочтительно от 2 до 25, более предпочтительно от 3 до 10.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения, установка фракционирования на стадии (d) работает с орошением (флегмой). Хотя, как правило, можно использовать любой подходящий поток в качестве флегмы, предпочтительно использовать часть потока S4, предпочтительно после конденсации, в качестве флегмы. Предпочтительно, флегмовое число находится в диапазоне от 0,01:1 до 10:1, более предпочтительно от 0,1:1 до 5:1, более предпочтительно от 0,5:1 до 2:1. Термин "флегмовое число", как используется в данном контексте, определяется как соотношение потока флегмы и потока S4, и является измерением того, какая часть материала, поднимающегося к вверху установки фракционирования возвращается обратно в установку фракционирования в качестве флегмы. Согласно данному варианту, предпочтительно подавать поток S3 в установку фракционирования между отпарной секцией и ректификационной секцией установки фракционирования.
В соответствии с другим предпочтительным вариантом осуществления настоящего изобретения, установка фракционирования на стадии (d) работает без орошения (флегмы). Согласно данному варианту, предпочтительно подавать поток S3 в верхнюю часть установки фракционирования. В этом случае, как правило, можно оперировать установкой фракционирования как отпарной колонной с ребойлером или как отпарной колонной без ребойлера. Если установка фракционирования спроектирована как отпарная колонна с ребойлером, предпочтительно, чтобы, по меньшей мере, один теплообменник был соответственно расположен в кубовой (нижней) части установки фракционирования, чтобы обеспечить испарение кубового потока установки фракционирования, в которой пар для отпаривания генерируется внутри. Если установка фракционирования спроектирована как отпарная колонна без ребойлера, предпочтительно, чтобы, по меньшей мере, один внешний питающий поток пара использовался в качестве пара для отпаривания, и не использовался, по меньшей мере, один теплообменник, расположенный в кубовой части установки фракционирования. В целом, можно комбинировать, по меньшей мере, один теплообменник, расположенный в кубовой части установки фракционирования и, по меньшей мере, один внешний питающий поток пара. Предпочтительно, в случае, если установка фракционирования работает без флегмы, чтобы установка фракционирования работала как отпарная колонна с ребойлером.
Предпочтительно, на стадии (d) от 95 до 99,99 вес. %, предпочтительно от 97 до 99,9 вес. %, более предпочтительно от 98 до 99,9 вес. % потока S4 состоит из ацетонитрила и воды, причем от 0,0001 до 0,2 вес. %, предпочтительно от 0,001 до 0,15 вес. %, более предпочтительно от 0,005 до 0,1 вес. % потока S4 состоит из, по меньшей мере, одного компонента B.
Жидкий кубовый поток S4b, полученный из установки фракционирования в соответствии со стадией (d), в целом может подвергаться дальнейшим стадиям обработки. Предпочтительно, жидкий кубовый поток S4b исключают, и поскольку поток S3, отведенный от S2, предпочтительно составляет лишь незначительную часть S2, причем данная незначительная часть неожиданно эффективно предотвращает накопление концентрации, по меньшей мере, одного компонента B в высокоинтегрированном способе согласно настоящему изобретению, простое исключение S4b без дальнейшей обработки является экономически выгодным.
Как правило, можно использовать более одной установки фракционирования на стадии (d), например, две, три, четыре или более установок фракционирования на стадии (d). Независимо друг от друга, каждая установка фракционирования может работать с флегмой или без флегмы, как описано выше. В случае, когда используется более одной установки фракционирования, можно отделить поток S3 от S4 и разделить таким образом, отделенный поток S3, на два или более потока S31, S32, и так далее, и пустить каждый субпоток в установку фракционирования с получением головных потоков пара S41, S42, и так далее, которые могут быть соответствующим образом объединены, чтобы получить поток S4. Кроме того, в случае, когда используется более одной установки фракционирования, можно отделить поток S31 от S4, отделить поток S32 от оставшейся части потока S4, и так далее, и пустить каждый субпоток в установку фракционирования с получением головных потоков пара S41, S42, и так далее, которые могут быть соответствующим образом объединены, чтобы получить поток S4. Кроме того, также возможно использование более одной установки фракционирования на стадии (d), при котором, по меньшей мере, две установки фракционирования соединены последовательно и, при котором головной поток или кубовый поток первой установки фракционирования подается как сырьевой поток во вторую установку фракционирования, соединенную последовательно с первой установкой фракционирования. Также можно использовать более двух установок фракционирования, при котором, по меньшей мере, две установки работают параллельно и, по меньшей мере, две установки соединены последовательно. В соответствии со способом согласно настоящему изобретению, предпочтительно, чтобы на стадии (d) использовалась всего одна установка фракционирования, которая предпочтительно работает, как описано выше.
Стадия (e)
Согласно стадии (е) способа согласно настоящему изобретению, по меньшей мере, часть потока S4 рециркулируют в стадию (а) способа согласно настоящему изобретению. Как правило, можно рециркулировать поток S4 или его часть без каких-либо дополнительных стадий обработки в стадию (а). Предпочтительно, S4 или его часть подвергают последующей стадии обработки перед рециркуляцией в стадию (а).
Предпочтительно, в соответствии со стадией (е) способа согласно настоящему изобретению, по меньшей мере, часть потока S2, рециркулируют в стадию (а) способа согласно настоящему изобретению. Как правило, можно рециркулировать поток S2 или его часть без каких-либо дополнительных стадий обработки в стадию (а). Предпочтительно, S2 или его часть подвергают последующей стадии обработки перед рециркуляцией в стадию (а). В случае, когда S2 или его часть подвергают последующей стадии обработки перед рециркуляцией в стадию (а), и в случае, когда во время такой стадии обработки, весовое соотношение ацетонитрила и, по меньшей мере, одного компонента B возрастает по сравнению с весовым соотношением потока S2, указанное весовое соотношение после стадии обработки является ниже, чем соответствующее весовое соотношение потока S4.
Таким образом, настоящее изобретение относится к способу, описанному выше, отличающемуся тем, что стадия (е) включает в себя рециркуляцию, по меньшей мере, части потока S4, необязательно после обработки, в стадию (а), и рециркуляцию, по меньшей мере, части потока S2, необязательно после обработки, в стадию (а).
Предпочтительно, во время стадии обработки S4, поток S4 или его часть объединяют с, по меньшей мере, частью потока S2. Предпочтительно, во время стадии обработки S2, поток S2 объединяют с, по меньшей мере, частью потока S4. Полученный соответственно объединенный поток рециркулируют, необязательно после обработки, в стадию (а). Более предпочтительно, полный поток S4, необязательно после отделения его части, используемой в качестве флегмы в установке фракционирования, используемой на стадии (d), и полный поток S2 соответствующим образом объединены, и объединенный поток рециркулируют, необязательно после обработки, в стадию (а). Более предпочтительно, поток S4 или его часть конденсируют и объединяют с потоком S2, с получением жидкого потока. Предпочтительно, полный поток S4, необязательно после отделения его части, используемой в качестве флегмы в установке фракционирования, используемой на стадии (d), конденсируют и объединяют с потоком S2, с получением жидкого потока. Предпочтительно, данный жидкий поток подвергают последующей стадии обработки перед рециркуляцией в стадию (а).
Таким образом, настоящее изобретение относится к способу, описанному выше, отличающемуся тем, что стадия (е) включает в себя объединение, по меньшей мере, части потока S4 и, по меньшей мере, части потока S2, и рециркуляцию объединенного потока, необязательно после обработки, в стадию (а).
В соответствии с настоящим изобретением, указанная последующая стадия обработки объединенного потока предпочтительно включает в себя отделение ацетонитрила-воды, в результате которого получают поток, обогащенный ацетонитрилом, который, необязательно после дальнейшей обработки, предпочтительно рециркулируют в стадию (а).
Таким образом, настоящее изобретение относится к способу, описанному выше, отличающемуся тем, что стадия (е) включает в себя объединение, по меньшей мере, части потока S4 и, по меньшей мере, части потока S2, воздействие на объединенный поток посредством отделения ацетонитрила-воды с получением потока, обогащенного ацетонитрилом, и рециркуляцию потока, обогащенного ацетонитрилом, необязательно после дополнительной обработки, в стадию (а).
Таким образом, настоящее изобретение также относится к способу, описанному выше, отличающемуся тем, что стадия (е) включает в себя обработку потока S4, причем указанная обработка включает в себя объединение, по меньшей мере, части потока S4, предпочтительно после конденсации, с потоком S2, с получением предпочтительно жидкого потока, воздействие на указанный предпочтительно жидкий поток посредством отделения ацетонитрила-воды с получением потока, обогащенного ацетонитрилом и рециркуляцию указанного потока, обогащенного ацетонитрилом, необязательно после дополнительной обработки, в стадию (а).
Что касается указанного отделения ацетонитрила-воды, не существует никаких особых ограничений. Предпочтительно, отделение ацетонитрила-воды включает в себя добавление потока P, предпочтительно содержащего, по меньшей мере, 95 вес. %, в расчете на общую массу потока P, C3, где C3 представляет собой пропен, необязательно смешанный с пропаном, предпочтительно добавление жидкого потока P,
- либо в поток S2, в котором полученный поток объединяют с, по меньшей мере, частью потока S4 с получением предпочтительно жидкого потока S5;
- либо в, по меньшей мере, часть потока S4, в котором полученный поток объединяют с потоком S2 с получением предпочтительно жидкого потока S5;
- либо, предпочтительно, в жидкий поток, полученный в результате объединения, по меньшей мере, части потока S2 и, по меньшей мере, части потока S4.
Также можно добавить первую часть потока P в, по меньшей мере, часть потока S4 и добавить вторую часть потока P в поток S2, и объединить два полученных потока с получением предпочтительно жидкого потока S5. Предпочтительно жидкий поток P предпочтительно содержит, по меньшей мере, 95 вес. %, в расчете на общую массу потока P, C3, где C3 представляет собой пропен, необязательно смешанный с пропаном. Что касается C3, предпочтительно, чтобы минимальное весовое соотношение пропена и пропана составляло 7:3. В контексте настоящего изобретения, все варианты, касающиеся добавления потока P, описанные выше, охватываются термином "воздействие на объединенный поток посредством отделения ацетонитрила-воды с получением потока, обогащенного ацетонитрилом", как используется в контексте стадии (е) выше.
Предпочтительно, по меньшей мере, 95 вес. % потока P состоит из пропена или смеси пропена с пропаном. Если P содержит смесь пропена и пропана, весовое соотношение пропена и пропана будет составлять, по меньшей мере, 7:3. Таким образом, потоки пропена могут использоваться в качестве потока P или C3, которые имеют различное содержание пропана. Например, в качестве P или C3 можно использовать коммерчески доступный пропен, который может представлять собой либо пропен полимерного сорта, либо пропен химического сорта. Как правило, пропен полимерного сорта будет иметь содержание пропена от 99 до 99,8 вес. % и содержание пропана от 0,2 до 1 вес. %. Пропен химического сорта, как правило, будет иметь содержание пропена от 92 до 98 вес. % и содержание пропана от 2 до 8 вес. %. В соответствии с предпочтительным вариантом осуществления настоящего изобретения, используется поток P, по меньшей мере, 95 вес. % которого, состоит из C3, в котором C3 представляет собой смесь пропена и пропана, и содержание пропена в C3 находится в диапазоне от 92 до 98 вес. %, предпочтительно от 94 до 97 вес. %, а содержание пропана в C3 находится в диапазоне от 2 до 8 вес. %, предпочтительно от 3 до 6 вес. %.
Что касается количества P, предпочтительно, чтобы поток P добавлялся таким образом, чтобы в потоке S5, весовое соотношение C3 и ацетонитрила находилось в диапазоне от 0,2:1 до 5:1, предпочтительно от 0,5:1 до 2:1. Таким образом, предпочтительно, от 90 до 99,9 вес. %, более предпочтительно от 95 до 99,8 вес. %, более предпочтительно от 98 до 99,5 вес. % потока S5 состоит из ацетонитрила, воды и C3, и предпочтительно от 0,01 до 3 вес. %, предпочтительно от 0,015 до 2,5 вес. %, более предпочтительно от 0,02 до 1,5 вес. % потока S5 состоит из, по меньшей мере, одного компонента B, в котором весовое соотношение ацетонитрила и воды составляет предпочтительно больше, чем 1:1 и, в котором весовое отношение C3 и ацетонитрила находится предпочтительно в диапазоне от 0,2:1 до 5:1, более предпочтительно от 0,5:1 до 2:1, в котором весовое соотношение пропена и пропана в C3 составляет, по меньшей мере, 7:3.
Предпочтительно поток S5 подвергают воздействию соответствующей температуры и соответствующего давления, причем в результате такой обработки посредством воздействия температуры и давления образуются две жидкие фазы L1 и L2. Было обнаружено, что для разделения на данные фазы L1 и L2 целесообразно подвергать поток S5 воздействию максимально возможной низкой температуры, при условии, что температура остается все еще подходящей; например, температура не должна быть настолько низкой, что образуется твердая фаза, такая как лед. Предпочтительно, жидкую фазу L1, обогащенную ацетонитрилом, соответствующим образом отделяют от L2 и рециркулируют в стадию (а), необязательно после дополнительной обработки. Что касается обработки посредством воздействия температуры и давления, не существует никаких особых ограничений, при условии, что будут образованы две фазы L1 и L2, где L1 является обогащенной ацетонитрилом. Предпочтительно, S5 доводят до температуры не более 92°C. В соответствии с настоящим изобретением, предпочтительно довести поток S5 до температуры в диапазоне от 5 до 90°C, предпочтительно от 10 до 80°C, более предпочтительно от 15 до 70°C, более предпочтительно от 20 до 60°C и более предпочтительно от 25 до 45°C. Предпочтительно, поток S5 подвергается воздействию давления, по меньшей мере, 10 бар, таким образом, чтобы S5 находился по существу или полностью в жидкой форме. Термин "по существу в жидкой форме", используемый в контексте настоящего изобретения, относится к варианту осуществления, согласно которому, по меньшей мере, 95 вес. %, более предпочтительно, по меньшей мере, 99 вес. % и более предпочтительно, по меньшей мере, 99,9 вес. % потока S5 находится в жидкой форме после воздействия на него вышеуказанных температур и давления. В соответствии с настоящим изобретением, предпочтительно подвергать поток S5 воздействию давления, по меньшей мере, 15 бар, более предпочтительно давления в диапазоне от 15 до 50 бар, более предпочтительно от 15 до 40 бар, более предпочтительно от 15 до 30 бар, и более предпочтительно от 15 до 25 бар.
Доведение потока S5 до вышеуказанной температуры может быть достигнуто любым подходящим способом. В соответствии с настоящим изобретением, предпочтительно использовать один или более подходящих теплоносителей, например воду, в подходящем аппарате, например, кожухотрубном теплообменнике. Воздействие на поток S5 вышеупомянутым давлением может быть достигнуто любым подходящим способом. Согласно настоящему изобретению, предпочтительно использовать подходящий насос, такой как центробежный насос или радиальный насос.
Предпочтительно, по меньшей мере, 95 вес. %, предпочтительно, по меньшей мере, 98 вес. % L1 состоит из C3, ацетонитрила, воды и, по меньшей мере, одного компонента B, причем содержание воды в L1 составляет менее 10 вес. %, предпочтительно находится в диапазоне от 1 до 5 вес. %, в расчете на общую массу L1.
Предпочтительно, по меньшей мере, 98 вес. % L2 состоит из C3, ацетонитрила, воды и, по меньшей мере, одного компонента B, причем содержание C3 в L2 составляет не более 5 вес. %, в расчете на общую массу L2, и содержание ацетонитрила в L2 составляет менее 45 вес. %, предпочтительно находится в диапазоне от 10 до 35 вес. %, в расчете на общую массу L2.
Согласно настоящему изобретению температуры и давление, как описано выше, обеспечивают существование двух различных жидких фаз L1 и L2. Предпочтительно, две различных жидких фазы L1 и L2 соответственно отделены друг от друга. Как правило, для такого разделения двух жидких фаз, могут применятся все возможные способы. Подходящими аппаратами, используемыми для разделения L1 и L2, например, являются гравитационные отстойники, отстойники с коалесцирующими вспомогательными устройствами, такими как переливные камеры, наклонный тарельчатый сепаратор, коалесцирующие фильтры, такие как, например, подушки, пласты, слои пористых или волокнистых твердых частиц, или мембраны, постадийные смесители-отстойники, гидроциклоны, центрифуги, подходящие колонны с подводом энергии или без него. Как правило, возможным является периодический режим или непрерывный режим. Предпочтительно использовать гравитационный отстойник, например, вертикальный или горизонтальный гравитационный отстойник. Еще более предпочтительно использовать горизонтальный гравитационный отстойник. Было обнаружено, что из-за значительной разницы плотности и низкой вязкости, достигаемых для жидких фаз L1 и L2 в соответствии со способом согласно изобретению, может использоваться гравитационный отстойник, один из простейших аппаратов. Согласно настоящему изобретению, можно добавлять, по меньшей мере, один агент, улучшающий разделение жидких фаз, например, по меньшей мере, один подходящий антиэмульгирующий, деэмульгирующий или разрушающий эмульсии агент. Как правило, можно добавлять указанный агент, улучшающий разделение жидких фаз, в поток S4 или S5, или S4 и S5. Количество добавляемого агента, улучшающего разделение жидких фаз, предпочтительно составляет не более 1 вес. %, в расчете на общую массу S4 и/или S5. Как правило, количество будет меньше, чем 1 вес. %, например, меньше 0,5 вес. % или меньше 0,1 вес. %. Подходящие агенты известны специалисту в данной области. Можно сделать ссылку, например, на K.J. Lissant, Making and Breaking Emulsions, Res. Lab., Petrolite Corp., St. Louis, Missouri, USA, in: K.J. Lissant (ed.), Emulsion Technology (1974), chapter 2, pp 111-124, Dekker, New York; and to S.E. Taylor, Chem. Ind. (1992), pp 770-773.
Таким образом, стадия (e) способа согласно настоящему изобретению предпочтительно включает в себя:
(i) получение предпочтительно жидкого потока S5, посредством добавления предпочтительно жидкого потока P в поток S2, или, по меньшей мере, в часть потока S4, или в жидкий поток, полученный в результате объединения S2 и, по меньшей мере, части S4,
в котором P содержит, по меньшей мере, 95 вес. % C3, в расчете на общую массу потока P,
в котором C3 представляет собой пропен, необязательно смешанный с пропаном, с минимальным весовым соотношением пропена и пропана 7:3, и
в котором P предпочтительно добавляют в таком количестве, чтобы в потоке S5, весовое соотношение C3 и ацетонитрила находилось в диапазоне от 0,2:1 до 5:1, предпочтительно от 0,5:1 до 2:1;
(ii) воздействие на поток S5 посредством температуры не более 92°C и давления, по меньшей мере, 10 бар, предпочтительно температуры в диапазоне от 5 до 90°C и давления в диапазоне от 15 до 50 бар, более предпочтительно температуры в диапазоне от 25 до 45°C и давления в диапазоне от 15 до 25 бар, с получением первой жидкой фазы L1 и второй жидкой фазы L2,
в котором, по меньшей мере, 95 вес. %, предпочтительно, по меньшей мере, 98 вес. % L1 состоит из C3, ацетонитрила, воды и, по меньшей мере, одного компонента B, причем содержание воды в L1 составляет менее 10 вес. %, предпочтительно находится в диапазоне от 1 до 5 вес. %, в расчете на общую массу L1, и
в котором, по меньшей мере, 95 вес. %, предпочтительно, по меньшей мере, 98 вес. % L2 состоит из C3, ацетонитрила, воды и, по меньшей мере, одного компонента B, причем содержание C3 в L2 составляет не более 5 вес. %, в расчете на общую массу L2, и содержание ацетонитрила в L2 составляет менее 45 вес. %, предпочтительно находится в диапазоне от 10 до 35 вес. %, в расчете на общую массу L2;
(iii) отделение L1 от L2, преимущественно в гравитационном отстойнике;
(iv) рециркуляцию L1 в качестве потока, обогащенного ацетонитрилом, необязательно после дополнительной обработки, в стадию (а).
Поток L2
Предпочтительно, способом согласно настоящему изобретению, получают жидкую фазу L2, которая по существу состоит из воды и ацетонитрила, причем весовое соотношение ацетонитрила: воды в L2 составляет менее 1. Термин "по существу состоит из ацетонитрила и воды", используемый в контексте настоящего изобретения относится к жидкой фазе L2, отличающейся тем, что, по меньшей мере, 90 вес. % L2 состоит из ацетонитрила и воды. Предпочтительно, по меньшей мере, 95 вес. %, более предпочтительно, по меньшей мере, 97 вес. % и еще более предпочтительно, по меньшей мере, 98 вес. % L2 состоит из C3, ацетонитрила и воды, причем содержание C3 в L2 составляет не более 5 вес. %, предпочтительно не более 3 вес. % и более предпочтительно не более 2 вес. %, в расчете на общую массу L2. Что касается ацетонитрила, соответствующее содержание в L2 предпочтительно составляет менее 45 вес. %, более предпочтительно находится в диапазоне от 10 до 40 вес. %, более предпочтительно от 10 до 35 вес. %, в расчете на общую массу L2.
Как правило, жидкая фаза L2 может быть использована в любом подходящем способе. Например, можно использовать жидкую фазу L2 в качестве потока, который подается в реакцию окисления, или стадию обработки, следующую за указанной реакцией окисления, в которой ацетонитрил используют в качестве растворителя и, в которой окисляется пропен, например, реакцию эпоксидирования, в которой ацетонитрил используют в качестве растворителя и, в которой пропен окисляется перекисью водорода, с получением пропиленоксида.
Предпочтительно жидкую фазу L2, перед тем, как использовать в подходящем способе, подвергают, по меньшей мере, одной дополнительной стадии разделения. Предпочтительный способ указанного разделения включает в себя воздействие на жидкую фазу L2 посредством стадии перегонки. Предпочтительно, перегонку проводят соответствующим образом так, чтобы получить поток TL2, который содержит от 75 до 95 вес. %, предпочтительно от 80 до 85 вес. % ацетонитрила, в расчете на общую массу TL2. Как правило, перегонка L2 может осуществляться в одной, двух или более перегонных колоннах. Если перегонка проводится в одной перегонной колонне, точка росы в верхней части указанной перегонной колонны составляет, как правило, по меньшей мере, 40°C, предпочтительно находится в диапазоне от 40 до 80°C, более предпочтительно в диапазоне от 40 до 65°C. Как правило, количество теоретических тарелок находится в диапазоне от 10 до 25. Обычные флегмовые числа находятся в диапазоне от 0,5 до 3. При таком способе поток TL2 получают в виде головного потока из перегонной колонны. Соответствующий кубовый поток, BL2, предпочтительно по существу не содержит ацетонитрила. В данном контексте термин "по существу не содержит ацетонитрила" относится к варианту, согласно которому содержание ацетонитрила в BL2 составляет не более 500 вес. ч.н.м., предпочтительно не более 300 вес. ч.н.м., более предпочтительно не более 100 вес. ч.н.м., в расчете на общую массу BL2.
Неожиданно было обнаружено, что можно подвергнуть жидкую фазу L2 специально предназначенной стадии перегонки, которая позволяет осуществлять высокоинтегрированный способ термической перегонки. Таким образом, было установлено, что разделение L2 предпочтительно проводят с использованием способа перегонки с двумя уровнями давления, отличающегося тем, что в первой перегонной колонне, перегонку проводят при верхнем давлении, которое выше, чем верхнее давление второй перегонной колонны, которая соединена с указанной первой перегонной колонной, и отличающегося тем, что конденсатор, используемый для конденсации головного потока первой перегонной колонны, одновременно используется в качестве испарителя второй перегонной колонны. В соответствии с данным предпочтительным вариантом, жидкий поток L2 предпочтительно вводят в указанную первую перегонную колонну, из которой получают первый кубовый поток и первый головной поток. Предпочтительно, указанная первая перегонная колонна работает при условиях, которые позволяют получать головной поток пара VTL2, который содержит от 50 до 70 вес. %, предпочтительно от 55 до 65 вес. % ацетонитрила, в расчете на общую массу VTL2. Как правило, указанная первая перегонная колонна работает при давлении в верхней части колонны в диапазоне от 10 до 20 бар, предпочтительно от 10 до 15 бар. Как правило, первая перегонная колонна имеет от 10 до 25, предпочтительно от 15 до 20 теоретических тарелок. Как правило, флегмовое число указанной первой перегонной колонны находится в диапазоне от 0,25:1 до 2:1, предпочтительно от 0,25:1 до 1:1. Соответствующий кубовый поток, полученный из первой перегонной колонны, предпочтительно по существу не содержит ацетонитрила. В данном контексте термин "по существу не содержит ацетонитрила" относится к варианту, согласно которому содержание ацетонитрила в кубовом потоке первой перегонной колонны составляет не более 500 вес. ч.н.м., предпочтительно не более 300 вес. ч.н.м., более предпочтительно не более 100 вес. ч.н.м., в расчете на общую массу кубового потока первой перегонной колонны. В дальнейшем, указанный кубовый поток, полученный из указанной первой перегонной колонны, необязательно смешанный с кубовым потоком, полученным из второй перегонной колонны, как описано здесь ниже, называют потоком BL2. В способе перегонки с двумя уровнями давления, по меньшей мере, часть, предпочтительно весь VTL2 соответственно конденсируют, и данный конденсированный поток подают во вторую перегонную колонну, из которой получают второй кубовый поток и второй головной поток. Предпочтительно, указанная вторая перегонная колонна работает при условиях, которые позволяют получать головной поток TL2, который содержит от 75 до 95 вес. %, предпочтительно от 80 до 85 вес. % ацетонитрила, в расчете на общую массу TL2. Как правило, указанная вторая перегонная колонна работает при давлении в верхней части колонны в диапазоне от 1 до 5 бар, предпочтительно от 1 до 2 бар. Как правило, вторая перегонная колонна имеет от 8 до 20, предпочтительно от 10 до 15 теоретических тарелок. Как правило, флегмовое число указанной второй перегонной колонны находится в диапазоне от 0,5 до 5, предпочтительно от 1 до 3. Соответствующий кубовый поток, полученный из второй перегонной колонны, предпочтительно по существу не содержит ацетонитрила. В данном контексте термин "по существу не содержит ацетонитрила" относится к варианту, согласно которому содержание ацетонитрила в кубовом потоке второй перегонной колонны составляет не более 500 вес. ч.н.м., предпочтительно не более 300 вес. ч.н.м., более предпочтительно не более 100 вес. ч.н.м., в расчете на общую массу кубового потока второй перегонной колонны.
Предпочтительно, TL2 полученный из соответствующей перегонной колонны, по меньшей мере, частично, предпочтительно полностью рециркулируют в способ согласно изобретению. Более предпочтительно, TL2 объединяют либо с S4 и/или S5, и/или с P, и необязательно также объединяют с TL1, как описано здесь ниже.
Если поток S5 содержит, по меньшей мере, один пропиленгликоль, поток BL2, полученный из указанной перегонной колонны, предпочтительно содержит, по меньшей мере, один пропиленгликоль в количестве от 1 до 5 вес. %, более предпочтительно в количестве от 2 до 5 вес. % в расчете на общую массу BL2, тогда как поток TL2 по существу не содержит, по меньшей мере, одного пропиленгликоля. В данном контексте настоящего изобретения, термин "TL2 по существу не содержит, по меньшей мере, одного пропиленгликоля" относится к варианту, согласно которому содержание, по меньшей мере, одного пропиленгликоля в TL2 составляет не более 500 вес. ч.н.м. TL2 по существу не содержит, по меньшей мере, одного пропиленгликоля, предпочтительно составляет не более 200 вес. ч.н.м. Если BL2 не содержит или по существу не содержит пропиленгликоля, предпочтительно подавать BL2 непосредственно на соответствующую установку очистки сточных вод, например, установку биологической очистки сточных вод. Было установлено, что не требуется никакой специальной очистки сточных вод, полученных способом согласно изобретению, что делает данный способ еще более экономичным и безопасным для окружающей среды. Если BL2 содержит, по меньшей мере, один пропиленгликоль в значительных количествах, например, в количестве от 1 до 5 вес. %, более предпочтительно в количестве от 2 до 5 вес. %, в расчете на общую массу BL2, может быть предпочтительно подавать BL2 на соответствующую стадию отделения пропиленгликоля, на которой, по меньшей мере, один пропиленгликоль соответствующим образом отделяют от воды и/или, на которой два или более различных пропиленгликолей отделяют друг от друга. Данный способ отделения, по меньшей мере, одного пропиленгликоля от BL2 может осуществляться, например, посредством выпаривания смеси, по меньшей мере, на двух, предпочтительно трех стадиях испарения и/или перегонки, предпочтительно трех стадиях испарения, при снижающемся рабочем давлении, предпочтительно в диапазоне от 1,5 до 5,5 бар при температуре от 111 до 155°C, с последующим давлением от 1,3 до 5,0 бар при температуре от 107 до 152°C, и последующим давлением от 0,7 до 4,0 бар при температуре от 90 до 144°C, получая таким образом смесь BL2-a и смесь BL2'-b; и отделяя смесь В12-а, по меньшей мере, посредством одной дополнительной стадии перегонки, с получением смеси BL2-I, содержащей, по меньшей мере, 70 вес. % воды, и смеси BL2-II, содержащей менее 30 вес. % воды. Особенно предпочтительно дополнительно разделять смесь BL2-b на смесь BL2-Ia, содержащую, по меньшей мере, 90 вес. % воды, и смесь BL2-Ib, содержащую менее 95 вес. % воды с помощью обратного осмоса. Предпочтительно, по меньшей мере, один пропиленгликоль отделяют от смеси BL2-II, предпочтительно смешанной со смесью BL2-Ib, посредством, по меньшей мере, одной дополнительной стадии перегонки. Более предпочтительно, смесь BL2'-b и BL2-I объединяют и дополнительно разделяют на смесь BL2-Ia, содержащую, по меньшей мере, 90 вес. % воды, и смесь BL2-Ib, содержащую менее 95 вес. % воды с помощью обратного осмоса.
Таким образом, настоящее изобретение также относится к способу, описанному выше, отличающемуся тем, что
(aa) L2 вводят в первую перегонную колонну, из которой получают головной поток пара VTL2, содержащий от 50 до 70 вес. % ацетонитрила, в расчете на общую массу головного потока VTL2, причем перегонку предпочтительно проводят при верхнем давлении от 10 до 20 бар; и
(bb) по меньшей мере, посредством частичной конденсации VTL2, полученного на стадии (aa), и введения конденсированного потока во вторую перегонную колонну, из нее получают TL2 в виде головного потока, причем перегонку предпочтительно проводят при верхнем давлении от 1 до 5 бар,
и отличающемуся тем, что предпочтительно, конденсатор, используемый для конденсации VTL2, одновременно используется в качестве испарителя второй перегонной колонны.
Поток L1
Согласно настоящему изобретению, предпочтительно, чтобы поток L1, отделенный в соответствии со стадией (iii), рециркулировали в стадию (а) после дополнительной обработки.
Предпочтительно, данная последующая обработка служит для отделения C3, предпочтительно части C3, от ацетонитрила. Возможным способом, например, является выпаривание жидкой фазы L1 посредством декомпрессии при подходящем давлении. Предпочтительно, чтобы температуру жидкой фазы поддерживали по существу постоянной во время декомпрессии. Посредством такой декомпрессии, C3 получают в газообразной форме. После этого можно рециркулировать, по меньшей мере, часть такого газообразного потока C3, после соответствующего сжатия, с получением жидкого потока, например, по меньшей мере, части потока P.
Предпочтительно, L1, подвергают фракционированию, более предпочтительно, перегонке, в результате которой получают поток, обогащенный ацетонитрилом, и который предпочтительно рециркулируют в стадию (а), необязательно после обработки. Предпочтительно, указанный поток, обогащенный ацетонитрилом, рециркулируют в стадию (а) без дополнительной проработки. Более предпочтительно, такую перегонку проводят соответствующим образом так, чтобы получить поток TL1, который содержит, по меньшей мере, 90 вес. %, предпочтительно, по меньшей мере, 95 вес. % C3, в расчете на общую массу TL1. Предпочтительно, в соответствии с такой перегонкой получают поток BL1, который предпочтительно, по меньшей мере, на 95 вес. %, более предпочтительно, по меньшей мере, на 98 вес. % состоит из C3, ацетонитрила, воды и, по меньшей мере, одного компонента B. Более предпочтительно, содержание C3 в BL1 находится в диапазоне от 7 до 18 вес. %, предпочтительно от 10 до 15% вес, в каждом случае в расчете на общую массу BL1.
Как правило, такая перегонка L1 может проводиться любым подходящим способом. Например, могут использоваться одна, две или более перегонных колонн при условии, что будут получены вышеупомянутые потоки TL1 и BL1. Предпочтительно, на указанной стадии перегонки, используется одна перегонная колонна. Более предпочтительно, указанную перегонку осуществляют при точке росы в верхней части указанной перегонной колонны, по меньшей мере, 40°C, предпочтительно в диапазоне от 40 до 80°C, более предпочтительно в диапазоне от 40 до 70°C. Предпочтительно, количество теоретических тарелок находится в диапазоне от 10 до 20. Предпочтительные флегмовые числа находятся в диапазоне от 0,01:1 до 0,2:1, например, от 0,05:1 до 0,15:1.
Таким образом, настоящее изобретение также относится к способу, описанному выше, который дополнительно включает в себя обработку L1, причем указанная обработка включает в себя воздействие на поток L1 посредством стадии перегонки, в результате которой получают кубовый поток BL 1, отличающийся тем, что, по меньшей мере, 95 вес. %, предпочтительно, по меньшей мере, 98 вес. % потока BL1 состоит из C3, ацетонитрила, воды и, по меньшей мере, одного компонента B, и отличающийся тем, что содержание C3 в BL1 находится в диапазоне от 7 до 18 вес. %, предпочтительно от 10 до 15 вес. %, и рециркуляцию BL1 в качестве потока, обогащенного ацетонитрилом, необязательно без дополнительной обработки, в стадию (а). Предпочтительно, от 0,01 до 5 вес. %, более предпочтительно от 0,015 до 3 вес. %, более предпочтительно от 0,02 до 2 вес. % потока BL1 состоит из, по меньшей мере, одного компонента B. В частности, перегонная колонна работает подходящим способом, например, посредством регулирования подвода энергии в отстойнике, что ведет к получению потока BL1, имеющего содержание пропена, которое, при подаче обратно в реакцию эпоксидирования в виде рециркулирующего потока, дает в результате молярное соотношение пропена и перекиси водорода в потоке (1) в диапазоне от 0,9:1 до 3,0:1, более предпочтительно от 0,98:1 до 1,6:1, более предпочтительно от 1,0:1 до 1,5:1, например от 1,2:1 до 1,4:1.
Таким образом, настоящее изобретение относится к способу, описанному выше, который включает в себя обработку L1, которая включает в себя воздействие на поток L1 посредством стадии перегонки, в результате которой получают кубовый поток BL1, отличающийся тем, что, по меньшей мере, 95 вес. %, предпочтительно, по меньшей мере, 98 вес. % потока BL1 состоит из C3, ацетонитрила, воды и, по меньшей мере, одного компонента B, и отличающийся тем, что содержание C3 в BL1 находится в диапазоне от 7 до 18 вес. %, предпочтительно от 10 до 15 вес. %, и рециркуляцию BL1 в качестве потока, обогащенного ацетонитрилом, предпочтительно без какой-либо дополнительной обработки, в стадию (а). Предпочтительно, от 0,01 до 5 вес. %, более предпочтительно от 0,015 до 3 вес. %, более предпочтительно от 0,02 до 2 вес. % потока BL1 состоит из, по меньшей мере, одного компонента B.
Далее было установлено, что комбинирование отделения L1 от L2 согласно изобретению и последующего отделения TL1 от BL1 позволяет обеспечить высокоинтегрированную схему способа согласно настоящему изобретению. С одной стороны, поток TL1 особенно подходит для рециркуляции в способ согласно изобретению в качестве, по меньшей мере, части потока P. Если, кроме, по меньшей мере, части TL1, в поток S1 дополнительно добавляют C3, такой еще один источник C3 может быть соответствующим образом выбран. Например, дополнительный C3 может быть добавлен в виде свежего пропена, например, в виде пропена химического сорта, содержащего около 95 вес. % пропена и 5 вес. % пропана. Все другие подходящие источники дополнительного C3 являются возможными, например, поток C3, полученный с центральных технологических площадок (Verbund site) или т.п.. Кроме того, было установлено, что чем больше C3 рециркулируют через TL1, тем более эффективным является разделение фаз в соответствии со стадиями (i)-(iii) способа согласно изобретению, в том, что касается полного разделения S1, насколько это возможно. Таким образом, предпочтительно, чтобы, по меньшей мере, часть TL1, предпочтительно весь поток TL1, рециркулировали в стадию (ii).
Как правило, возможно, что перегонка с частичным потоком может быть предусмотрена в другом месте в последующем способе эпоксидирования, предпочтительно в месте с доступом к потоку растворителя ацетонитрила. Предпочтительно таким возможным местом расположения будет место, в котором поток растворителя ацетонитрила не содержит или по существу не содержит пропена и необязательно пропана, и/или в котором поток растворителя ацетонитрила не содержит или по существу не содержит перекиси водорода. Более предпочтительно, таким возможным местом расположения будет местоположение после стадии реакции эпоксидирования (а) и перед местом, в котором смешивают поток P, перед жидкостно-жидкостным разделением на стадии (ii). Более предпочтительно, таким возможным местом расположения будет местоположение после места, в котором пропиленоксид удаляется из потока ацетонитрила на стадии (b) и перед местом, в котором смешивают поток P, перед жидкостно-жидкостным разделением на стадии (ii). Наиболее предпочтительно, месторасположением перегонки с частичным потоком является расположение, как описано выше, где S3 в качестве части потока S1 подвергают перегонке. Кроме того, как правило, возможно, что в более чем одном месторасположении в последующем способе эпоксидирования, размещают перегонку с частичным потоком, согласно которой часть, предпочтительно незначительную часть потока ацетонитрила подвергают перегонке.
Настоящее изобретение иллюстрируется следующими примерами и сравнительными примерами.
ПРИМЕРЫ
Пример 1: Предпочтительный способ согласно изобретению - общая компоновка
Что касается сокращений, делается ссылка на схему согласно Фиг. 1, в целом описанную в разделе "Описание Фигуры" ниже. Все указанные значения давления представляют собой значения абсолютного давления.
1.1 Получение потока S0 (стадия (а))
а) Эпоксидирование в главном реакторе эпоксидирования (установка эпоксидирования А)
Главный реактор A представляет собой вертикально установленный кожухотрубный реактор с 5 трубками (длина трубок: 12 м, внутренний диаметр трубки: 38 мм), каждая трубка оснащается аксиально размещенной многоточечной термопарой с 10 равномерно расположенными точками измерения, заключенными в подходящем термокармане с диаметром 18 мм. Каждую трубку заполнили 17,5 кг формованными изделиями, содержащими катализатор ZnTiMWW, полученными согласно Справочному примеру 1, раздел 1.8 (формованные изделия, подвергнутые последующей обработке). Свободное пространство, оставшееся в конечном итоге, было заполнено стеатитовыми сферическими гранулами (диаметром 3 мм). Теплоту реакции отводили посредством циркуляции термостатированного теплоносителя (водно-гликолевая смесь) на стороне межтрубного пространства в потоке, параллельном загрузке. Скорость потока теплоносителя регулировали таким образом, чтобы разность температур между входом и выходом не превышала 1°C. Температуру реакции, упоминаемую здесь ниже, определяли как температуру теплоносителя, поступающего в кожух реактора. На выходе реактора, давление регулировали посредством регулятора давления и поддерживали постоянным на уровне 20 бар.
Реактор загружали снизу жидким однофазным потоком (1). Поток 1 получали посредством смешивания трех потоков (2), (3) и (4). Температуру потока (1) активно не регулировали, но, как правило, она находилась в диапазоне от 20 до 40°C:
- Поток (2), имеющий скорость потока 85 кг/ч, по меньшей мере, 99,5 вес. % потока (2) состоит из ацетонитрила, воды и пропена. Данный поток (2) поступает из кубовой части установки рециркуляции ацетонитрила (J).
- Поток (3), имеющий скорость потока 15 кг/ч, представляет собой водный раствор перекиси водорода, имеющей концентрацию перекиси водорода 40 вес. % ("сырой/промытый" сорт от Solvay с общим содержанием органического углерода (ТОС) в диапазоне от 100 до 400 мг/кг. Водный раствор перекиси водорода подают из резервуара для хранения, что позволяет осуществлять непрерывную подачу, и подается с помощью подходящего дозирующего насоса.
- Поток (4) представляет собой подпиточный поток чистого ацетонитрила (химический сорт, от Ineos, чистота 99,9%, содержащий 70-180 вес. частей на миллион пропионитрила, 5-20 вес. ч.н.м. ацетамида и менее 100 вес. ч.н.м. воды в качестве примесей). Достаточно свежий ацетонитрил добавляют, чтобы компенсировать потери в процессе. В обычных условиях, добавляют в среднем от 100 до 150 г/ч подпиточного ацетонитрила.
- Необязательно, дополнительный поток может быть использован, и поток 1 получают посредством смешивания четырех потоков (2), (3), (4) и указанного дополнительного потока. Дополнительный поток представляет собой водный поток, который содержит, по меньшей мере, одну растворенную калиевую соль, например, дигидрофосфат калия. Дополнительный поток может подаваться из резервуара для хранения, что позволяет осуществлять непрерывную подачу, и может подаваться с помощью подходящего дозирующего насоса. Возможная концентрация калиевой соли составляет, например, 2,5 вес. %, возможная скорость подачи дополнительного потока составляет, например, 370 г/ч.
Из выходного потока, на выходе установки эпоксидирования A, каждые 20 минут отбирали пробы, чтобы определить концентрацию перекиси водорода с помощью метода на основе сульфата титанила, и рассчитать конверсию перекиси водорода. Конверсия перекиси водорода была определена как 100 x (l-mout/min), где min представляет собой молярный расход H2O2 в питающей смеси реактора и mout представляет собой молярный расход H2O2 на выходе реактора. На основе соответственно полученных значений конверсии перекиси водорода, регулируют температуру теплоносителя на входе, чтобы поддерживать конверсию перекиси водорода по существу постоянной в диапазоне от 90 до 92%. Температуру теплоносителя на входе установили на уровне 30°C в начале данного прогона свежей партии катализатора эпоксидирования и увеличивали, при необходимости, для поддержания конверсии перекиси водорода в указанном диапазоне. Необходимое повышение температуры, как правило, составляло менее 1°C/сутки.
b) Промежуточное удаление пропиленоксида (перегонная установка B)
После сброса давления, поток, выходящий из установки эпоксидирования A (поток (5)), направляли в колонну промежуточного удаления пропиленоксида (перегонная установка В), работающую при давлении около 1,1 бара. Колонна имела высоту 6 м, диаметр 200 мм была оборудована 30 барботажными тарелками, испарителем и конденсатором. Питающую смесь подавали в колонну над барботажной тарелкой 25 (считая сверху). Головной поток, выходящий из колонны при 50°C, в основном содержал пропиленоксид, непрореагировавший пропен и небольшое количество кислорода, образовавшегося как побочный продукт. Этот поток частично конденсировали (T=15-25°C), и конденсированную жидкость использовали в качестве внутреннего потока флегмы, тогда как газообразную часть (поток (6)) направляли в разделительную колонну легких фракций (перегонная установка D).
Температура в кубовой части колонны промежуточного удаления пропиленоксида составляла около 80°C. Кубовый поток (поток (7)) почти не содержал пропиленоксида (<300 вес. частей на миллион) и представлял собой смесь ацетонитрила (около 78-80 вес. %), воды (около 18-20 вес. %), непрореагировавшего эпоксида-водорода и тяжелокипящих соединений, имеющих нормальную точку кипения выше 100°C, причем основным высококипящим соединением является пропиленгликоль. Данный кубовый поток (7) затем охладили до 35°C и перекачивали насосом в реактор финишной обработки (установка эпоксидирования C, см. раздел c) ниже) с использованием подходящего дозирующего насоса.
с) Эпоксидирование в реакторе финишной обработки (установка эпоксидирования C)
Общий питающий поток для реактора финишной обработки C получали посредством смешивания потока (7), полученного в соответствии с разделом b) выше, с потоком (8) жидкого пропена полимерного сорта, содержащего пропан (чистота ≥99,5%, скорость подачи: 0.9 кг/ч, при температуре окружающей среды). Оба потока (7) и (8) смешивали с помощью статического смесителя и подавали в кубовую часть реактора финишной обработки C.
Реактор финишной обработки C представляет собой реактор с неподвижным слоем, работающий адиабатически. В данном контексте, термин "адиабатический" относится к режиму работы, в соответствии с которым активное охлаждение не осуществляется, и в соответствии с которым реактор финишной обработки соответствующим образом изолирован, чтобы минимизировать тепловые потери. Реактор финишной обработки C имел длину 4 м и диаметр 100 мм. Реактор был заполнен 9 кг такого же катализатора эпоксидирования, который был использован в главном реакторе эпоксидирования A. Свободное пространство было заполнено стеатитовыми сферическими гранулами (диаметром 3 мм). Рабочее давление в реакторе финишной обработки С составляло 10 бар и поддерживалась постоянным с помощью соответствующего регулятора давления на выходе реактора. На выходе реактора финишной обработки C каждые 20 минут отбирали пробы, чтобы определить концентрацию перекиси водорода с помощью метода на основе сульфата титанила.
Поток, выходящий из реактора финишной обработки С, поток (9), подвергали снижению давления в испарительном барабане, и оба потока - жидкость и газ, из данного барабана подавали в разделительную колонну легкокипящих соединений (перегонная установка D).
Поток (6), полученный из верхней части колонны промежуточного удаления пропиленоксида (перегонная установка В), и поток (9), полученный в качестве отходящего потока из реактора финишной обработки C (установка эпоксидирования С), в совокупности образуют поток S0 согласно настоящему изобретению.
Данный поток S0 имеет в среднем содержание ацетонитрила от 69 до 70 вес. %, содержание пропиленоксида 9,8 вес. %, содержание воды 17 вес. %, содержание пропена около 3 вес. %, содержание пропана около 0,05 вес. %, содержание перекиси водорода около 250 вес. ч.н.м., содержание пропиленгликоля около 0,1 вес. % и содержанием кислорода около 150 вес. частей на миллион.
1.2 Отделение пропиленоксида из потока S0 с получением потока S1 (стадия (b))
a) Отделение легкокипящих соединений из потоков (6) и (9) (поток S0) с получением потока (11) (поток S01 в соответствии со стадией (I) согласно настоящему изобретению)
Головной поток из колонны промежуточного удаления пропиленоксида (перегонная установка В) (поток (6), см. раздел 1.1b) выше) и выходящий поток со сниженным давлением из реактора финишной обработки C (поток (9), см. раздел 1.1c) выше) направили в разделительную колонну легкокипящих соединений (перегонная установка D), работающую при давлении 1,1 бара. Перегонная колонна имела длину 8,5 м, диаметр 170 мм и была оснащен 40 барботажными тарелками, испарителем в кубовой части и конденсатором в верхней части. Колонна работала в смешанном режиме как промывная / перегонная колонна. В качестве промывочного агента отбирали часть кубового потока перегонной установки E (поток 14, около 20-30 кг/ч), охлажденного до 10°C, и подавали в верхнюю часть колонны. Жидкий и газовый входящие потоки вводили в колонну в разных точках. Точка подачи жидкого потока (поток (6), а также жидкая часть потока (9)) размещалась выше барботажной тарелки 37; газовый поток вводили в колонну выше барботажной тарелки 28 (считая сверху).
Газовый поток (10) на выходе из охлаждающих средств в верхней части колонны содержал в основном пропен, пропан (который содержится в качестве примеси в используемом пропене полимерного сорта), кислород, образовавшийся в качестве побочного продукта и небольшие количества других легкокипящих соединений (ацетонитрил (около 4,7 об. %), пропиональдегид (около 200 об. ч.н.м.), ацетон (около 100 об. ч.н.м., H2 (около 400 об. ч.н.м.), CO2 (около 400 об. ч.н.м.) и ацетальдегид (около 100 об. ч.н.м.)), и по существу не содержал пропиленоксида (менее 300 об. ч.н.м.). Данный головной поток был направлен на факел для утилизации.
Кубовый поток из разделительной колонны легкокипящих соединений (поток (11), то есть поток S01 согласно настоящему изобретению), имеющий температуру 70°C, имел содержание пропена от 100 до 200 вес. частей на миллион.
b) Отделение пропиленоксида из потока (11) (поток S01) с получением потока S02 в соответствии со стадией (II) согласно настоящему изобретению
Поток S01, полученный в соответствии с разделом 1.2а) выше, ввели в перегонную колонну (перегонная установка Е), чтобы отделить пропиленоксид от потока S01. Колонна имела высоту 50 м и диаметр 220 мм и была оснащена насадкой (Sulzer ВХ64) с общей длиной насадки 27,5 м, разделенной на 8 слоев длиной 3060 мм каждый, и двумя слоями длиной 1530 мм каждый. Между каждым слоем были установлены промежуточные распределители потока. Колонна работала при верхнем давлении 750 мбар. Точка подачи потока S01 была расположена ниже четвертого слоя насадки, считая от верха.
Головной поток из колонны конденсировали и частично возвращали в колонну в виде флегмы (флегмовое число составляло приблизительно 5:1). Остаток (поток (12)), имеющий скорость потока 10,1 кг/ч, отбирали в качестве головного погона и он по существу состоял из пропиленоксида, имеющего чистоту более 99,9 вес. %.
Кубовый испаритель работал таким образом, чтобы концентрация пропиленоксида в кубовом потоке была ниже 100 вес. частей на миллион. Полученная температура кубового потока составляла около 69°C. Поток S02 затем разделяли на две части. Основную его часть (поток (13), с расходом примерно 85 кг/ч) направляли на следующую перегонную колонну (перегонная установка F). Остаток (поток (14), расход 20-30 кг/ч) охлаждали и рециркулировали в верхнюю часть разделительной колонны легкокипящих соединений (перегонная установка D) в качестве промывочного агента, как описано выше в разделе 1.2 а).
Данный поток S02 имел содержание ацетонитрила около 80 вес. %, содержание пропиленоксида менее 100 вес. ч.н.м., содержание воды около 20 вес. %, содержание пропиленгликоля около 0,1 вес. % и содержание гидроксипропанола около 0,1 вес. %.
с) Отделение легкокипящих соединений из потока (13) (поток S02) с получением потока (16) (поток S1 в соответствии со стадией (IIIb) согласно настоящему изобретению)
Поток S02, полученный в соответствии с разделом 1.2b) выше, ввели в разделительную колонну легкокипящих соединений (перегонная установка F). Данная разделительная колонна легкокипящих соединений имела высоту 8 м и номинальный диаметр 150 мм, и была оснащена 35 барботажными тарелками. Колонна работала при верхнем давлении 2 бара, и поток S02 вводили выше барботажной тарелки 7 (считая от нижней).
Полученный головной поток (поток (15), скорость потока около 1 кг/ч) отводили из колонны с температурой от 40 до 45°C и не конденсировали, поскольку колонна работала без внутреннего потока флегмы. Кроме ацетонитрила (6500 об. ч.н.м.), данный головной поток содержал в основном азот, который использовался для поддержания рабочего давления колонны на значении 2 бара, и небольшие количества легкокипящих соединений (ацетальдегид (900 об. ч.н.м.), кислород (300 об. ч.н.м.) и пропиональдегид (320 об. ч.н.м.). Данный головной поток был направлен на факел для утилизации.
Испаритель в отстойнике работал посредством подачи в него постоянного количества (5 кг/ч) насыщенного пара при давлении 16 бар. Температура кубовой части колонны составляла 100°C. Кубовый поток, поток S1 согласно настоящему изобретению, в основном, состоял из ацетонитрила и воды, остаток составляли высококипящие соединения. Данный поток S1 имел содержание ацетонитрила около 80 вес. % и с содержание воды около 20 вес. %.
1.3 Разделение потока S1 на потоки S2 и S3 (стадия (c))
Согласно настоящему изобретению, на стадии (c), поток S1, со скоростью потока 86 кг/ч, полученный в соответствии с разделом 1.2c) выше, был разделен на два потока, потоки S2 (поток (16а согласно Фиг. 1) и S3 (Поток 17 согласно Фиг. 1). Поток S2 имел скорость потока 84 кг/ч, а поток S3 имел скорость потока 2 кг/ч. Поток S3, 2,3% потока S1, подавали для обработки в перегонную установку частичного потока G (перегонная колонна частичного потока).
1.4 Перегонка частичного потока, полученного из потока S1 (стадия (d))
Перегонная колонна частичного потока имела высоту 9,5 м и диаметр 85 мм и была оснащена 6,5 м металлической структурированной насадкой Rombopak 9М, установленной с тремя идентичными слоями. Ниже первого слоя структурированной насадки, считая сверху, поток S3 вводили в перегонную колонну частичного потока. Температура подаваемого потока находилась в диапазоне от 89°C±5°C. Колонна работала при верхнем давлении 1,5 бара и имела перепад давления менее 10 мбар. Флегма не применялась.
Количество пара, подаваемого в кубовый испаритель, регулировали таким образом, чтобы концентрация ацетонитрила в кубовой части находилась в диапазоне от 10 до 25 вес. %. При используемом давлении, была получена температура в кубовой части колонны в диапазоне от 94 до 98°C. В зависимости от соответствующих количеств и химической природы тяжелокипящих соединений, содержащихся в потоке S3, кубовая часть состояла либо из одной, либо из двух жидких фаз. Если присутствует, верхняя органическая фаза составляла менее 10 вес. % от общего количества кубового потока. Из кубовой части, удаляли непрерывный поток 50 г/ч и после анализа, выбраковывали. Данный поток в основном состоял из воды (72-85 вес. %) и ацетонитрила (10-24 вес. %). Сумма всех анализируемых высококипящих компонентов (27 компонентов) варьировалась в диапазоне 4-10 вес. %.
Внешний головной конденсатор применяли для полной конденсации головного потока пара, выходящего из перегонной колонны частичного потока (поток S4 согласно настоящему изобретению).
Конденсированный поток S4 имел содержание ацетонитрила около 80 вес. % и содержание воды около 20 вес. %.
1.5 Рециркуляция потока S4 (стадия (4))
а) Получение жидкого потока S5 согласно стадии (I)
Конденсированный поток S4 (поток 18 согласно Фиг. 1) смешали с потоком S2 (поток (16а) согласно Фиг. 1). Таким образом, конденсированный поток S2 был закачан обратно в основной поток растворителя ацетонитрила согласно способу. Смешивание происходило в точке, расположенной ниже точки, в которой поток S3 отделяли от потока S1.
Данный объединенный поток, имеющий расход 86 кг/ч, смешали с жидким потоком P (далее обозначенным как поток (23) на Фиг. 1) с получением потока S5. Поток P представлял собой поток свежего пропена, содержащий пропан (полимерного сорта, с чистотой >96 вес. %, сжиженный под давлением, со скоростью подачи: 10 кг/ч).
В соответствии с данным конкретным вариантом осуществления настоящего изобретения, для того, чтобы получить поток S5, объединенный поток S2 и S4 затем смешивали с двумя другими потоками: первым из этих потоков является поток (19) согласно Фиг. 1, причем указанный поток был получен из верхней части перегонной установки I. Вторым из этих потоков является поток (22) согласно Фиг. 1, причем указанный поток был получен из установки рециркуляции ацетонитрила J. Оба потока (19) и (22) описаны подробно ниже.
b) Регулирование температуры потока S5 и разделение жидких фаз L1 и L2 (стадии (ii) и (iii))
Поток S5, имеющий скорость потока 150 кг/ч ± 10 кг/ч, затем подавали в установку смесителя-отстойника, работающую при 18 барах и температуре в диапазоне от 30±5°C. Резервуар отстойника имел объем 5,3 литра. Были получены две жидкие фазы L1 и L2, водная фаза L2 и органическая фаза L1. Верхнюю органическую фазу L1 удалили из резервуара отстойника в виде потока (20), нижнюю водную фазу L2 удалили из резервуара отстойника в виде потока (21). Поток (20) имел скорость потока в диапазоне 130 кг/ч ± 13 кг/ч.
Поток (20) затем подавали в установку рециркуляции ацетонитрила J, поток (21) подавали в установку регенерации ацетонитрила I, из которой был получен поток (19), упомянутый выше.
Поток (20), полученный таким образом, имел содержание ацетонитрила около 46 вес. %, содержание пропена около 51 вес. % и содержание воды около от 3 до 4 вес. %.
Поток (21), полученный таким образом, имел содержание ацетонитрила около 21 вес. %, содержание воды около 79 вес. % и содержание пропена менее 0,5 вес. %.
c) Регенерация ацетонитрила (установка регенерации ацетонитрила I)
Чтобы рециркулировать максимально возможное количество растворителя и
свести к минимуму потери ацетонитрила, поток (21) вводили в перегонную колонну, из которой поток (19), также называемый как поток TL2, был получен в виде головного потока, который в свою очередь рециркулировали в поток растворителя, как описано выше.
Для этой цели использовали перегонную колонну высотой 9,5 м и диаметром 100 мм, оборудованную 50 барботажными тарелками. Колонна работала при верхнем давлении 1,5 бар с флегмовым числом 1:4. Поток (21) подавали в колонну выше барботажной тарелки 26 (считая снизу).
Температура в кубовой части составляла около 113°C, и кубовый продукт состоял в основном из воды, содержащей высоко кипящие побочные продукты. Типичный состав кубового потока был следующим (вес. % в скобках): вода (>99,0), пропиленгликоль (0,5), ацетонитрил (не более 0,001), дипропиленгликоль (0,06), ацетамид (0,01), уксусная кислота (0,03), ТОС (2,4)). После необязательного измерения и анализа, данный поток был выбракован.
Головной продукт (поток (19) = поток TL2) имел следующие типичные диапазоны состава (вес. % в скобках): ацетонитрил (75-80), вода (15-20), низкокипящие соединения (например, пропен, 1). Как описано выше поток (19) рециркулировали в питающий поток, который подавали в установку смесителя-отстойника.
d) Рециркуляция ацетонитрила (установка рециркуляции ацетонитрила J), стадия (iv)
Для рециркуляции ацетонитрила, поток (20), полученный из установки смесителя-отстойника Н, ввели в перегонную колонну высотой 10 м и номинальным диаметром 200 мм, оборудованную 40 барботажными тарелками. Колонна работала при верхнем давлении 18 бар с флегмовым числом 1:4. Поток (20) подавали в колонну выше барботажной тарелки 26 (считая сверху). Головной продукт (поток (22)), также называемый как поток TL1, содержащий в основном пропен (приблизительно 97 об. %) с небольшими количествами пропана (приблизительно 1-3 об. %), возвращали в питающую смесь установки смесителя-отстойника Н, как описано выше. Таким образом, избыточный пропен был удален из потока (20) и рециркулирован.
Кубовый поток (поток (2), также называемый как поток BL1) имел температуру в диапазоне от 106 до 110°C. Точные рабочие параметры колонны, например, подвод энергии в отстойнике, регулируют таким образом, чтобы количество пропена, возвращенного в реактор с потоком (2), находилось в таком диапазоне, чтобы молярное соотношение пропена и перекиси водорода в потоке (1) составляло около 1:1,3. Для получения вышеупомянутой скорости подачи 15 кг/ч водного раствора перекиси водорода, это означает, что условия должны быть отрегулированы таким образом, чтобы скорость потока пропена в потоке (2) составляла около 9,7 кг/ч.
Перед подачей потока (2) в главный реактор эпоксидирования А, ацетонитрил (поток (4), химического сорта от Ineos, с чистотой 99,9%, содержащий 70-180 вес. ч.н.м. пропионитрила, 5-20 вес. ч.н.м. ацетамида и <100 вес. ч.н.м. воды в качестве примесей) необязательно добавляли, чтобы компенсировать возможные потери растворителя. Точное количество необходимого дополнительно добавляемого ацетонитрила зависит от потерь в выходящих потоках и побочных продуктах, но также от количества образцов, взятых для аналитики. Типичное количество дополнительно добавляемого ацетонитрила для описанной выше схемы способа может быть в диапазоне от 100 до 150 г/ч.
Пример 2а: Сравнительный (без перегонки с частичным потоком, без гидрогенизации)
Способ, описанный выше в Примере 1, был вначале принят в эксплуатацию с использованием свежей загрузки катализатора эпоксидирования и свежего ацетонитрила (такого же качества, как и для подпиточного потока (4), см. раздел 1.5 d) выше), но без использования перегонки частичного потока согласно изобретению. Таким образом, из потока (16) (поток S1), не отделяли поток (17) (поток S3) и не подвергали перегонке в установке G. Поток 1 смешивали как таковой с потоками (19), (22) и (23).
Начальную температуру для контура охлаждающей среды главного реактора эпоксидирования установили на уровне 30°C. В начале, конверсия перекиси водорода в главном реакторе эпоксидирования A была почти полной. В течение 24 часов, конверсия перекиси водорода начала снижаться, и когда она достигла требуемого значения приблизительно 90% (примерно через 100-200 часов), температуру охлаждающей среды медленно повышали, чтобы поддерживать конверсию перекиси водорода в главном реакторе эпоксидирования A на постоянном уровне. Скорость повышения температуры охлаждающей среды всегда составляла меньше, чем 1°C/сутки.
Затем установку эксплуатировали, как описано выше в Примере 1, на протяжении 441 часов. В конце данного периода температура охлаждающей среды главного реактора эпоксидирования составляла 35°C. В этот момент, несколько компонентов (либо побочных продуктов реакции эпоксидирования и/или примесей в подаваемых потоках, которые не присутствовали в начале прогона) накопилось в контуре растворителя. Накопление возрастало линейно без признаков достижения устойчивого состояния. Концентрация компонентов, которые накопились в потоке (2), рециркулирующем потоке ацетонитрила, полученном из установки J, спустя 441 часов на потоке представлена в Таблице А.
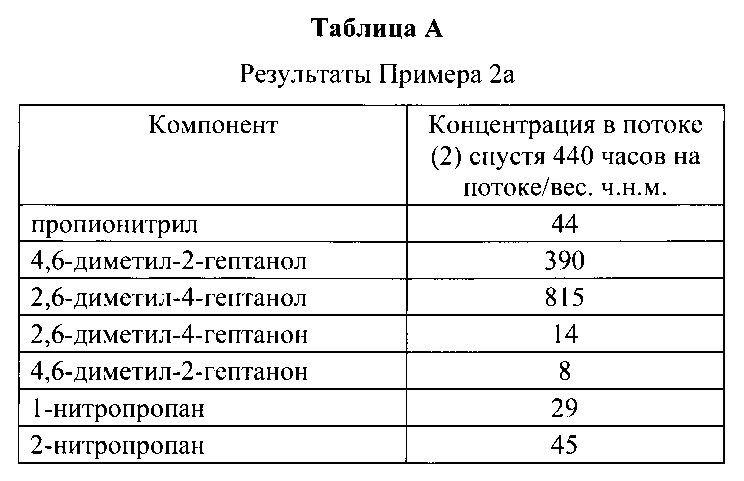
Данный опыт показывает, что при отсутствии перегонки с частичным потоком согласно изобретению, общий способ, включая рециркуляцию растворителя, страдает от накопления нескольких соединений в контуре растворителя. В отношении концентрации этих соединений не было достигнуто устойчивого состояния.
Пример 2b: Согласно изобретению (с перегонкой с частичным потоком, без гидрогенизации)
Прогон, как описано в Примере 2а, продолжали, и при t=441 часов на потоке, перегонка с частичным потоком (установка G) была введена в эксплуатацию. Затем прогон продолжали до тех пор, пока не было достигнуто время на потоке 1800 часов. В этот период поток S3 с постоянной скоростью потока 2 кг/ч (±0,1 кг/ч) отбирали из потока S1 и подавали в перегонную колонну (установка G), что соответствует примерно 2,3% от общего объема потока S1. Кубовый поток с постоянной скоростью потока 40 г/ч (±10 г/ч) удаляли из кубовой части перегонной колонны (установка G) и выбраковывали. Состав данного кубового потока спустя 1800 часов на потоке был следующим (вес. % в скобках): вода (77,5), пропиленгликоль (6,1), ацетонитрил (14,1), дипропиленгликоль (0,20), трипропиленгликоль (0,12), ацетамид (0,16), 2,6-диметил-4-гептанол (0,16), 4,6-диметил-2-гептанол (0,08), 1-нитропропан (0,004), 2-нитропропан (0,004), гидроксиацетон (0,4), уксусная кислота (0,6), аммиак (0,02), ТОС (0,02), кислотное число = 1,4 мг/г (определено в соответствии с DIN EN IS0 2114). Концентрация примесей в контуре растворителя (в потоке (2) непосредственно перед началом перегонки с частичным потоком (при 441 часов на потоке) и в конце прогона с перегонкой с частичным потоком (спустя 1329 часов на потоке) приведена в Таблице B.

В конце прогона, все соответствующие концентрации в потоке (2) достигли устойчивого состояния, и никакого накопления больше не наблюдалось. Данный пример согласно настоящему изобретению четко показывает, что используя перегонку с частичным потоком согласно изобретению, в соответствии с которой лишь незначительную фракцию потока S1 отделяют и подвергают перегонке, можно остановить накопление побочных продуктов и примесей во время регенерации растворителя и можно достичь устойчивого состояния при очень низких уровнях концентрации. Кром того, пример показывает, что способ перегонки с частичным потоком согласно изобретению позволяет даже значительно уменьшить концентрацию побочных продуктов и примесей, накопленных в контуре растворителя ацетонитрила.
Пример также показывает, что достаточно обработать небольшой боковой погон, чтобы получить желаемый результат, таким образом обеспечивая значительную экономию энергии и инвестиций.
Пример 3a: Сравнительный (без перегонки с частичным потоком, с гидрогенизацией)
В новом прогоне, способ, описанный выше в Примере 1, был вначале принят в эксплуатацию с использованием свежей загрузки катализатора эпоксидирования и свежего ацетонитрила (такого же качества, как и для подпиточного потока (4), см. раздел 1.5d) выше), но без использования перегонки частичного потока согласно изобретению. Таким образом, из потока (16) (поток S1), не отделяли поток (17) (поток S3) и не подвергали перегонке в установке G. Поток 1 смешивали как таковой с потоками (19), (22) и (23).
В этом примере, поток (13) (поток S02)) пропускали через реактор гидрогенизации (не показан на Фиг. 1), расположенный после установки E и перед установкой F. Реактор гидрогенизации представляет собой трубчатый реактор диаметром 53 мм и высотой 3,25 м, заполненный неподвижным слоем катализатора (0,3 вес. % Pd на Al2O3, заготовки диаметром 4 мм, H0-13 S4 от BASF SE, работал адиабатически. Реактор функционировал как насадочная барботажная колонна, в которой газ и жидкость протекали параллельно из кубовой части в верхнюю часть реактора при давлении около 15 бар. Водород подавали с постоянной скоростью 100 г/ч. Температуру жидкого питающего потока (13) в реактор гидрогенизации доводили до 70°C и поддерживали постоянной в течение всего прогона. На выходе реактора гидрогенизации, давление снижали до 1 бар, и разделяли жидкую фазу и газовую фазу, выходящие из реактора гидрогенизации. Газовую фазу отбраковывали, а жидкую фазу подавали в установку F, как описано выше.
Начальную температуру контура охлаждающей среды главного реактора эпоксидирования A установили на уровне 30°C. В начале, конверсия перекиси водорода в главном реакторе эпоксидирования A была почти полной. В течение 24 часов, конверсия перекиси водорода начала снижаться, и когда она достигла требуемого значения приблизительно 90% (примерно через 100-200 часов), температуру охлаждающей среды медленно повышали, чтобы поддерживать конверсию перекиси водорода в главном реакторе эпоксидирования A на постоянном уровне. Скорость повышения температуры охлаждающей среды всегда составляла меньше, чем 1°C/сутки.
Затем установку эксплуатировали, как описано выше в Примере 1, на протяжении 864 часов. В конце данного периода температура охлаждающей среды главного реактора эпоксидирования составляла 39,2°C. В этот момент, несколько компонентов (либо побочных продуктов реакции эпоксидирования и/или примесей в подаваемых потоках, которые не присутствовали в начале прогона) накопилось в контуре растворителя. Накопление возрастало линейно без признаков достижения устойчивого состояния. Концентрация компонентов, которые накопились в потоке (2), рециркулирующем потоке ацетонитрила, полученном из установки J, спустя 864 часов на потоке представлена в Таблице С.

Данный опыт показывает, что при отсутствии перегонки с частичным потоком согласно изобретению, общий способ, включая рециркуляцию растворителя, страдает от накопления нескольких соединений в контуре растворителя. В отношении концентрации этих соединений не было достигнуто устойчивого состояния.
Пример 3b: Согласно изобретению (с перегонкой с частичным потоком, с гидрогенизацией)
Прогон, как описано в Примере 3a, продолжали, и при t=864 часов на потоке, перегонка с частичным потоком (установка G) была введена в эксплуатацию. Затем прогон продолжали до тех пор, пока не было достигнуто время на потоке 1600 часов.
В этот период поток S2 с постоянной скоростью потока 2 кг/ч (±0,1 кг/ч) отбирали из потока S1 и подавали в перегонную колонну (установка G), что соответствует примерно 2,3% от общего объема потока S1. Кубовый поток с постоянной скоростью потока 50 г/ч удаляли из кубовой части перегонной колонны (установка G) и после анализа выбраковывали.
Состав потока S2 после достижения устойчивого состояния был следующим: вода (76,1), пропиленгликоль (0,43), пропионитрил (0,11), ацетонитрил (14,1), дипропиленгликоль (0,20), трипропиленгликоль (0,13), ацетамид (0,17), 2,6-диметил-4-гептанол (0,14), 4,6-диметил-2-гептанол (0,12), 1-нитропропан (0,10), 2-нитропропан (0,11), гидроксиацетон (0,34), уксусная кислота (0,46), аммиак (0,03), TOC (0,02), кислотное число = 1,4 мг/г.
Концентрация примесей в контуре растворителя (в потоке (2) непосредственно перед началом перегонки с частичным потоком (при 864 часов на потоке) и в конце опыта (спустя 1600 часов на потоке) приведена в Таблице D.
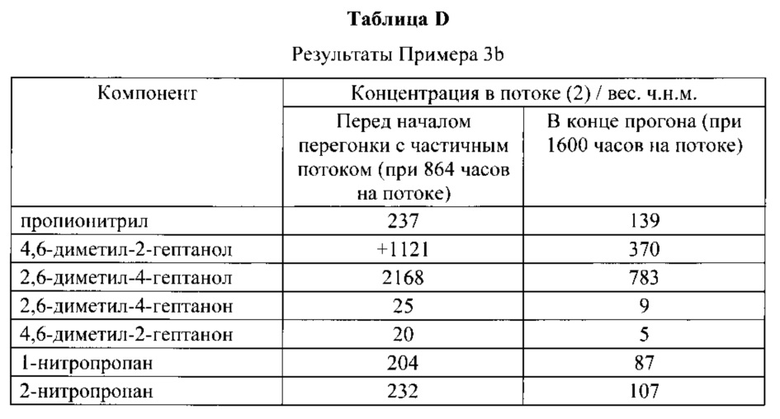
В период между 1370-1580 часов на потоке, все концентрации в потоке (2) достигли устойчивого состояния, и никакого накопления больше не наблюдалось. До завершения прогона никакого накопления больше не наблюдалось.
Данный пример согласно настоящему изобретению четко показывает, что используя перегонку с частичным потоком согласно изобретению, в соответствии с которой лишь незначительную фракцию потока S1 отделяют и подвергают перегонке, можно остановить накопление побочных продуктов и примесей во время регенерации растворителя и можно достичь устойчивого состояния при очень низких уровнях концентрации. Кром того, пример показывает, что способ перегонки с частичным потоком согласно изобретению позволяет даже значительно уменьшить концентрацию побочных продуктов и примесей, накопленных в контуре растворителя ацетонитрила.
Пример также показывает, что достаточно обработать небольшой боковой погон, чтобы получить желаемый результат, таким образом обеспечивая значительную экономию энергии и инвестиций.
Пример 4а: Согласно изобретению (с автономной перегонкой с частичным потоком, ректификацией с флегмой)
Для дальнейшей иллюстрации изобретения провели периодические перегонки. Для первой перегонки используемый технологический растворитель ацетонитрил из общей компоновки, как описано выше, был обработан посредством добавления выбранных компонентов с целью представления худшего случая в отношении уровней примесей. Начальную загрузку 3912 г данного раствора известного состава (см. таблицу E) поместили в лабораторный аппарат для перегонки, который состоит из колонны диаметром 43 мм, оснащенной 4 м насадкой Montz A3-1000.
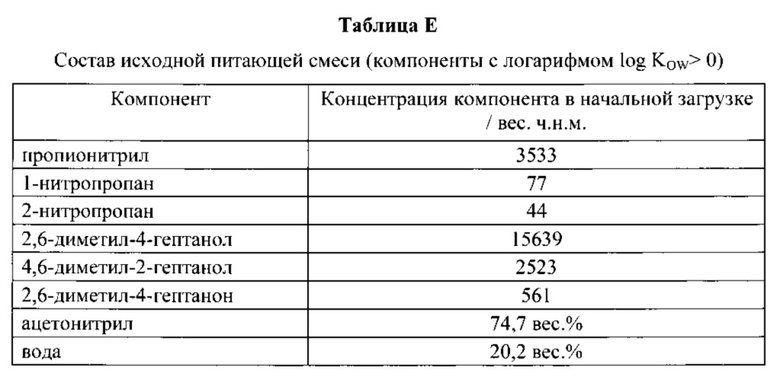
Эту смесь перегоняли с флегмой при давлении 950 мбар, используя флегмовое число 1.
В ходе перегонки количество и состав дистиллята регистрировали, состав определяли с помощью откалиброванной газовой хроматографии для всех органических компонентов и с помощью титрования по методу Карла Фишера для воды. Через равные промежутки времени из оставшегося отстойника также отбирали пробы и определяли концентрацию воды и ацетонитрила. С помощью этих данных, может быть получено процентное содержание каждого компонента, который был собран в дистилляте, а также концентрация ацетонитрила в отстойнике во время перегонки. Для иллюстрации изобретения лучше всего рассмотреть количество примесей, собранных в головной части, в зависимости от концентрации ацетонитрила в отстойнике. Таблица F показывает процентное содержание нежелательных компонентов, которые отгоняются в верхней части колонны, в момент, когда концентрация ацетонитрила в отстойнике составляла 35,2 вес. %. В этот момент в общей сложности количество 3211 г было отогнано в верхней части колонны, содержащее 92% ацетонитрила, первоначально присутствующего в загрузке. Это означает, что в таких условиях 8% ацетонитрила в загрузке будут потеряны. Примеси, которые отгоняются в верхней части колонны, будут возвращены в систему, что является нежелательным, поскольку необходимо низкое процентное содержание примесей в дистилляте.
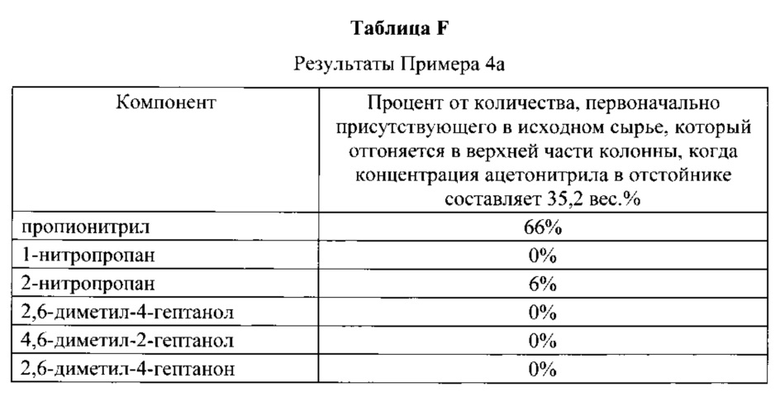
Пример показывает, что используя перегонную колонну с флегмой можно очень эффективно удерживать большую часть нежелательных побочных продуктов в отстойнике, и даже две трети относительно легкокипящего пропионитрила могут удерживаться в отстойнике. Тем не менее, это может быть достигнуто только посредством принятия потери 8% первоначально загружаемого ацетонитрила.
Пример 4b: Согласно изобретению (с автономной перегонкой с частичным потоком, продолжение примера 4а)
Перегонку предыдущего Примера 4а продолжали до тех пор, пока концентрация ацетонитрила в отстойнике не составляла 16,1 вес. %. В этот момент в общей сложности количество 3411 г было отогнано в верхней части колонны, содержащее 97% ацетонитрила, первоначально присутствующего в загрузке. Это означает, что в таких условиях 3% ацетонитрила в загрузке будут потеряны. Таблица G показывает процентное содержание нежелательных компонентов, которые отгоняются в верхней части колонны, на этой стадии.
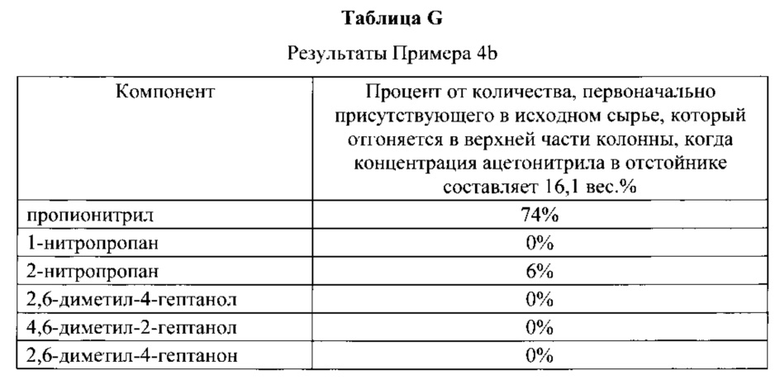
Пример согласно изобретению, показывает, что используя перегонную колонну с флегмой можно очень эффективно удерживать большую часть нежелательных побочных продуктов в отстойнике, и даже четверть относительно легкокипящего пропионитрила может удерживаться в отстойнике, при этом теряя только 3% первоначально загружаемого ацетонитрила.
Пример 4c: Продолжение Примера 4b
Перегонку предыдущего Примера 4b продолжали до тех пор, пока концентрация ацетонитрила в отстойнике не составляла 4 вес. %. В этот момент в общей сложности количество 3554 г было отогнано в верхней части колонны, содержащее 99% ацетонитрила, первоначально присутствующего в загрузке. Это означает, что в таких условиях только 1% ацетонитрила в загрузке будет потерян. Таблица Н показывает процентное содержание нежелательных компонентов, которые отгоняются в верхней части колонны, на этой стадии.
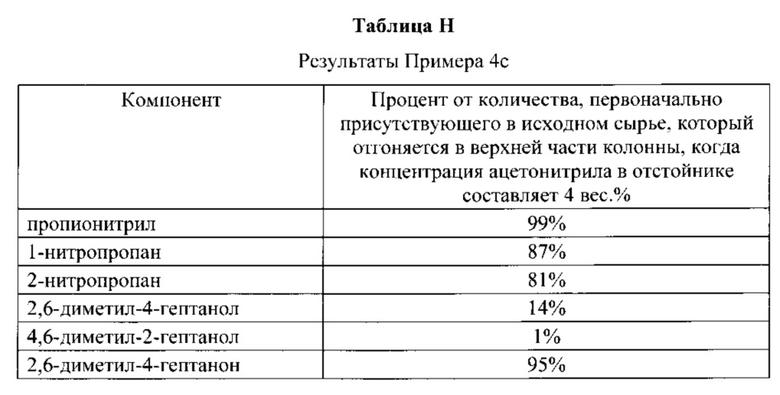
Данный пример показывает, что если концентрация ацетонитрила в отстойнике снижается слишком сильно, в попытке свести к минимуму потери ацетонитрила, эффективность отделения побочных продуктов, хотя и достигается до некоторой степени, значительно снижается.
Пример 5: Согласно изобретению (с автономной перегонкой с частичным потоком, с ректификацией без флегмы)
Для дальнейшей иллюстрации изобретения провели вторую периодическую перегонку в таком же перегонном аппарате, как и в Примерах 3a-c, но с флегмовым числом 0. Для данной перегонки другую партию используемого технологического растворителя ацетонитрила из пилотной установки, описанной выше, обработали посредством добавления выбранных компонентов с целью представления худшего случая в отношении уровней примесей.
Начальную загрузку 3934 г данного раствора известного состава (см. таблицу J) поместили в куб перегонного аппарата и перегоняли при давлении 950 мбар.
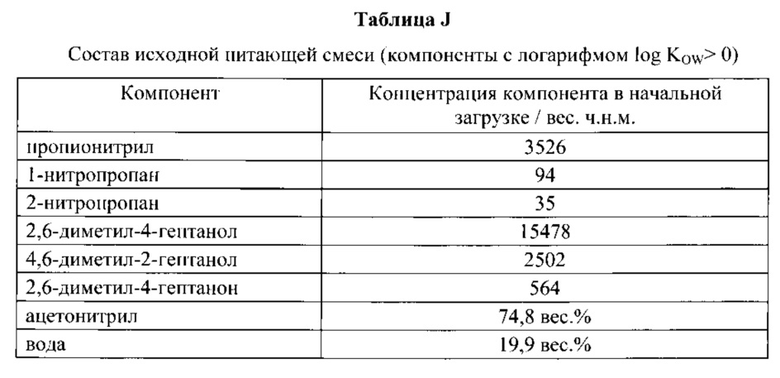
Как и в Примере 4а, во время перегонки количество и состав дистиллята регистрировали. Через равные промежутки времени из отстойника также отбирали пробы и определяли концентрацию воды и ацетонитрила. С помощью этих данных, может быть получено процентное содержание каждого компонента, который был собран в дистилляте, а также концентрация ацетонитрила в отстойнике во время перегонки. Таблица K показывает процентное содержание нежелательных компонентов, которые отгоняются в верхней части колонны, в момент, когда концентрация ацетонитрила в отстойнике составляла 37,1 вес. %. В этот момент в общей сложности количество 3255,7 г было отогнано в верхней части колонны, содержащее 90% ацетонитрила, первоначально присутствующего в загрузке. Это означает, что в таких условиях 10% ацетонитрила в загрузке будут потеряны. Примеси, которые отгоняются в верхней части колонны, будут возвращены в систему, что является нежелательным, поскольку необходимо низкое процентное содержание примесей в дистилляте.

Пример показывает, что используя перегонную колонну без флегмы можно эффективно удерживать большую часть нежелательных побочных продуктов (2,6-диметил-4-гептанол) в отстойнике. Тем не менее, это может быть достигнуто только посредством принятия потери 10% первоначально загружаемого ацетонитрила.
Пример 5b: Согласно изобретению (с автономной перегонкой с частичным потоком, продолжение примера 5a)
Перегонку предыдущего Примера 5a продолжали до тех пор, пока концентрация ацетонитрила в отстойнике не составляла только 18,2 вес. %. В этот момент в общей сложности количество 3459,4 г было отогнано в верхней части колонны, содержащее 96% ацетонитрила, первоначально присутствующего в загрузке. Это означает, что в таких условиях только 4% ацетонитрила в загрузке будут потеряны. Таблица L показывает процентное содержание нежелательных компонентов, которые отгоняются в верхней части колонны, на этой стадии.

Пример показывает, что используя перегонную колонну даже с флегмовым числом, равным нулю, можно очень эффективно удерживать диметилгептанолы в отстойнике. Для всех других компонентов степень удержания ниже, но все же значительно меньше, чем 100% утечки обратно в систему с дистиллята, при этом теряется только 4% первоначально загружаемого ацетонитрила. Хотя данный пример показывает, что даже можно работать при флегмовом числе, равным нулю, тем не менее предпочтительно работать с флегмовым числом, большим нуля.
Пример 5 с: Продолжение Примера 5b
Перегонку предыдущего Примера 5b продолжали до тех пор, пока концентрация ацетонитрила в отстойнике не составляла только 3,9 вес. %. В этот момент в общей сложности количество 3724,4 г было отогнано в верхней части колонны, содержащее 98% ацетонитрила, первоначально присутствующего в загрузке. Это означает, что в таких условиях только 2% ацетонитрила в загрузке будет потеряно. Таблица M показывает процентное содержание нежелательных компонентов, которые отгоняются в верхней части колонны, на этой стадии.

Данный пример показывает, что если концентрация ацетонитрила в отстойнике снижается слишком сильно, в попытке свести к минимуму потери ацетонитрила, эффективность отделения побочных продуктов, хотя и достигается до некоторой степени, значительно снижается. Некоторые компоненты, такие как 2-нитропропан, не могут быть удержаны в отстойнике.
Справочный пример 1: Получение катализатора эпоксидирования (ZnTiMWW)
1.1 Получение борсодержащего MWW
470,4 кг деионизированной воды загрузили в сосуд. При перемешивании при 70 оборотах в минуту (об./мин), 162,5 кг борной кислоты суспендировали в воде. Суспензию перемешивают в течение еще 3 часов. Затем добавили 272,5 кг пиперидина и смесь перемешивали в течение еще одного часа. В полученный раствор добавили 392,0 кг Ludox® AS-40 и полученную смесь перемешивали при 70 об./мин в течение еще одного часа. Полученную в результате смесь перенесли в кристаллизационный сосуд и нагревали до 170°C в течение 5 часов при собственном давлении и при перемешивании (50 об./мин). Температуру 170°C поддерживали по существу постоянной в течение 120 часов; в данный промежуток 120 часов смесь перемешивали при 50 об./мин. Затем смесь охлаждали до температуры 50-60°C в течение 5 часов. Водная суспензия, содержащая B-MWW, имела pH 11,3, как определено с помощью измерения pH-электродом. Из указанной суспензии, B-MWW отделили фильтрованием. Осадок на фильтре промывали деионизированной водой до тех пор, пока промывочная вода не имела проводимость меньше, чем 700 мкСм/см. Полученный таким образом осадок на фильтре подвергли распылительной сушке в распылительной башне при следующих условиях сушки распылением:
Распылительная башня состояла из вертикально расположенного цилиндра, имеющего длину 2650 мм, диаметр 1200 мм, причем данный цилиндр был конически зауженным в нижней части. Длина конуса составляла 600 мм. В головной части цилиндра размещались распылительные средства (двухкомпонентное сопло). Материал, высушенный распылением, отделяли от сушильного газа в фильтре, установленном на выходе распылительной башни, а затем сушильный газ пропускали через скруббер. Суспензию пропускали через внутреннее отверстие сопла, и распылительный газ пропускали через кольцевую щель, окружающую отверстие. Материал, высушенный распылением, затем подвергали обжигу при 650°C в течение 2 часов. Обожженный материал имел содержание бора (В) 1,9 вес. %, содержание кремния (Si) 41 вес. %, и общее содержание органического углерода (TOC) 0,18 вес. %.
1.2 Получение не содержащего бора MWW
На основе высушенного распылением материала, полученного в соответствии с разделом 1.1 выше, подготовили 4 партии не содержащего бора цеолита MWW. В каждой из первых 3 партий, использовали 35 кг материала, высушенного распылением, полученного в соответствии с разделом 1.1, и 525 кг воды. В четвертой партии, использовали 32 кг материала, высушенного распылением, полученного в соответствии с разделом 1.1, и 480 кг воды. В общей сложности были использованы 137 кг высушенного распылением материала, полученного в соответствии с разделом 1.1, и 2025 кг воды. Для каждой партии, соответствующий объем воды подавали в сосуд, оснащенный конденсатором флегмы. При перемешивании при 40 об./мин, данное количество материала, высушенного распылением, суспендировали в воде. Затем сосуд закрыли и запустили в работу конденсатор флегмы. Скорость перемешивания увеличили до 70 об./мин. При перемешивании при 70 об./мин, содержимое сосуда нагревали до 100°C в течение 10 часов и выдерживали при этой температуре в течение 10 часов. Затем содержимое сосуда охладили до температуры ниже 50°C. Полученный не содержащий бора цеолитный материал структурного типа MWW отделили от суспензии посредством фильтрации под давлением азота 2,5 бар и промыли четыре раза деионизованной водой. После фильтрации осадок на фильтре сушили в потоке азота в течение 6 часов. Не содержащий бора цеолитный материал, полученный в 4 партиях (625,1 кг осадка на фильтре, высушенного азотом, в общей сложности), имел остаточную влажность 79%, как было определено с помощью инфракрасной (ИК) шкалы при 160°C. Используя осадок на фильтре, высушенный азотом, имеющий остаточную влажность 79%, полученный в соответствии с разделом а) выше, получили водную суспензию с деионизированной водой, с содержанием твердых веществ 15 вес. %. Данную суспензию подвергли распылительной сушке в распылительной башне при следующих условиях сушки распылением:
Распылительная башня состояла из вертикально расположенного цилиндра, имеющего длину 2650 мм, диаметр 1200 мм, причем данный цилиндр был конически зауженным в нижней части. Длина конуса составляла 600 мм. В головной части цилиндра размещались распылительные средства (двухкомпонентное сопло). Материал, высушенный распылением, отделяли от сушильного газа в фильтре, установленном на выходе распылительной башни, а затем сушильный газ пропускали через скруббер. Суспензию пропускали через внутреннее отверстие сопла, и распылительный газ пропускали через кольцевую щель, окружающую отверстие. Материал MWW, высушенный распылением, имел содержание бора (В) 0,08 вес. %, содержание кремния (Si) 42 вес. %, и общее содержание органического углерода (ТОС) 0,23 вес. %.
1.3 Получение TiMWW
На основе не содержащего бора материала MWW, полученного в соответствии с разделом 1.2 выше, был получен цеолитный материал структурного типа MWW, содержащий титан (Ti), называемый далее как TiMWW. Синтез проводили посредством двух опытов, описаны ниже как a) и b):
54,16 кг не содержащего бора цеолитного материала структурного типа MWW загрузили в первый сосуд А. Во второй сосуд В загрузили 200,00 кг деионизированной воды и перемешивали при 80 об./мин. При перемешивании добавили 118,00 кг пиперидина и во время добавления температуру смеси увеличили приблизительно на 15°C. Затем добавили 10,90 кг тетрабутилортотитаната и 20,00 кг деионизированной воды. Перемешивание продолжали в течение 60 минут. Затем смесь сосуда В перенесли в сосуд А, и начали перемешивание в сосуде А (70 об./мин). В сосуд А поместили 24,00 кг деионизированной воды и перенесли ее в сосуд В. Затем смесь в сосуде В перемешивали в течение 60 минут при 70 об./мин. В начале перемешивания, pH смеси в сосуде В составлял 12,6, как определено с помощью pH-электрода. После указанного перемешиванием при 70 об./мин, частоту снизили до 50 об./мин. и смесь в сосуде B нагревали до температуры 170°C в течение 5 часов. При постоянной скорости перемешивания 50 об./мин температуру смеси в сосуде B выдерживали, по существу, при постоянной температуре 170°C в течение 120 часов при собственном давлении. Во время данной кристаллизации TiMWW наблюдалось повышение давления до 10,6 бар. Затем полученную суспензию, содержащую TiMWW, имеющую pH 12,6 охлаждали в течение 5 часов. Охлажденную суспензию подвергли фильтрации и отделенный маточный раствор направили на сброс сточных вод. Осадок на фильтре промыли четыре раза деионизированной водой под давлением азота 2,5 бар. После последней стадии промывки, осадок на фильтре сушили в потоке азота в течение 6 часов. Используя 246 кг указанного фильтровального осадка, получили водную суспензию с деионизированной водой с содержанием твердых веществ 15 вес. %. Данную суспензию подвергли распылительной сушке в распылительной башне при следующих условиях сушки распылением:
Распылительная башня состояла из вертикально расположенного цилиндра, имеющего длину 2650 мм, диаметр 1200 мм, причем данный цилиндр был конически зауженным в нижней части. Длина конуса составляла 600 мм. В головной части цилиндра размещались распылительные средства (двухкомпонентное сопло). Материал, высушенный распылением, отделяли от сушильного газа в фильтре, установленном на выходе распылительной башни, а затем сушильный газ пропускали через скруббер. Суспензию пропускали через внутреннее отверстие сопла, и распылительный газ пропускали через кольцевую щель, окружающую отверстие. Материал TiMWW, высушенный распылением, полученный в первом опыте, имел содержание кремния (Si) 37 вес. %, содержание титана (Ti) 2,4 вес. % и общее содержание органического углерода (TOC) 7,5 вес. %.
b) Второй опыт
Второй опыт был проведен таким же способом, как и первый опыт, описанный в разделе а) выше. Высушенный распылением материал TiMWW, полученный во втором опыте, имел содержание кремния (Si) 36 вес. %, содержание титана (Ti) 2,4 вес. % и общее содержание органического углерода (TOC) 8 вес. %.
1.4 Кислотная обработка TiMWW
Каждый из двух высушенных распылением материалов TiMWW, полученных в первом и втором опыте, описанных в разделах 1.3а) и 1.3b) выше, подвергли кислотной обработке, как описано ниже в разделах a) и b). В разделе c) ниже, описано, каким образом смесь материалов, полученных в разделах a) и b), высушили распылением. В разделе d) ниже, описано, каким образом обжигали материал, высушенный распылением.
a) Кислотная обработка материала, высушенного распылением, полученного в соответствии с разделом 1.3а).
TiMWW согласно 1.3.а):
670,0 кг деионизированной воды загрузили в сосуд. Добавили 900 кг азотной кислоты и 53,0 кг высушенного распылением TiMWW добавили при перемешивании при 50 об./мин. Полученную смесь перемешивали в течение еще 15 минут. Затем скорость перемешивания увеличили до 70 об./мин. В течение 1 часа смесь в сосуде нагревали до 100°C и выдерживали при этой температуре и под собственным давлением в течение 20 часов при перемешивании. Полученную таким образом смесь затем охлаждали в течение 2 часов до температуры менее 50°C. Охлажденную смесь фильтровали и осадок на фильтре промывали шесть раз деионизированной водой под давлением азота 2,5 бар. После последней стадии промывки, осадок на фильтре сушили в потоке азота в течение 10 часов. Промывочная вода после шести стадий промывки имела pH около 2,7. Было получено 225,8 кг высушенного отфильтрованного осадка.
b) Кислотная обработка материала, высушенного распылением, полученного в соответствии с разделом 1.3b)
Кислотную обработку высушенного распылением материала, полученного в соответствии с разделом 1.3b), проводили таким же способом, как и кислотную обработку высушенного распылением материала, полученного в соответствии с разделом 1.3a), как описано в разделе 1.4a). Промывочная вода после шести стадий промывки имела pH около 2,7. Было получено 206,3 кг высушенного отфильтрованного осадка.
c) Распылительная сушка смеси материалов, полученных в разделах 1.4a) и 1.4b)
Используя 462,1 кг смеси отфильтрованных осадков, полученных в разделах 1.4a) и 1.4b), получили водную суспензию с деионизированной водой с содержанием твердых веществ 15 вес. %. Данную суспензию подвергли распылительной сушке в распылительной башне при следующих условиях сушки распылением:
диаметр 4 мм
Распылительная башня состояла из вертикально расположенного цилиндра, имеющего длину 2650 мм, диаметр 1200 мм, причем данный цилиндр был конически зауженным в нижней части. Длина конуса составляла 600 мм. В головной части цилиндра размещались распылительные средства (двухкомпонентное сопло). Материал, высушенный распылением, отделяли от сушильного газа в фильтре, установленном на выходе распылительной башни, а затем сушильный газ пропускали через скруббер. Суспензию пропускали через внутреннее отверстие сопла, и распылительный газ пропускали через кольцевую щель, окружающую отверстие. Обработанный кислотой материал TiMWW, высушенный распылением, имел содержание кремния (Si) 42 вес. %, содержание титана (Ti) 1,6 вес. % и общее содержание органического углерода (ТОС) 1,7 вес. %.
d) Обжиг высушенного распылением материала, полученного в соответствии с разделом 1.4.с)
Материал, высушенный распылением, затем подвергали обжигу при температуре 650°C во вращающейся печи в течение 2 часов. Обожженный материал имел содержание Si 42,5 вес. %, содержание Ti 1,6 вес. % и содержание ТОС 0,15 вес. %.
1.5 Пропитка TiMWW цинком (Zn)
Обработанный кислотой, высушенный распылением и обожженный материал, полученный согласно разделу 1.4d) затем подвергли стадии пропитки.
Пропитку проводили в 3 партиях a)-c) следующим образом:
a) В сосуде, оснащенном конденсатором флегмы, в течение 30 минут был подготовлен раствор 840 кг деионизированной воды и 5,13 кг дигидрата ацетата цинка. При перемешивании (40 об./мин), 28 кг обожженного материала TiMWW, полученного в соответствии с 1.4d) суспендировали. Затем сосуд закрыли, и конденсатор флегмы запустили в работу. Скорость перемешивания была увеличена до 70 об./мин.
b) В сосуде, оснащенном конденсатором флегмы, в течение 30 минут был подготовлен раствор 840 кг деионизированной воды и 5,13 кг дигидрата ацетата цинка. При перемешивании (40 об./мин), 28 кг обожженного материала TiMWW, полученного в соответствии с 1.4d) суспендировали. Затем сосуд закрыли, и конденсатор флегмы запустили в работу. Скорость перемешивания была увеличена до 70 об./мин.
c) В сосуде, оснащенном конденсатором флегмы, в течение 30 минут был подготовлен раствор 930 кг деионизированной воды и 5,67 кг дигидрата ацетата цинка. При перемешивании (40 об./мин), 31 кг обожженного материала TiMWW, полученного в соответствии с 1.4 d) суспендировали. Затем сосуд закрыли, и конденсатор флегмы запустили в работу. Скорость перемешивания была увеличена до 70 об./мин.
Во всех партиях a)-c), полученную смесь в сосуде нагревали до 100°C в течение 1 часа и выдерживали с обратным холодильником в течение 4 часов при скорости перемешивания 70 об./мин. Затем смесь охлаждали в течение 2 часов до температуры менее 50°C. В каждой партии a)-c), охлажденную суспензию подвергли фильтрации, а маточный раствор направили на сброс сточных вод.
Осадок на фильтре промыли пять раз деионизированной водой под давлением азота 2,5 бар. После последней стадии промывки, осадок на фильтре сушили в потоке азота в течение 10 часов. В партии а) в конечном итоге получили 106,5 кг отфильтрованного осадка, высушенного азотом. В партии b) в конечном итоге получили 107,0 кг отфильтрованного осадка, высушенного азотом. В партии с) в конечном итоге получили 133,6 кг отфильтрованного осадка, высушенного азотом.
Полученный таким образом высушенный материал TiMWW, пропитанный Zn (ZnTiMWW), для каждой партии, имел содержание Si 42 вес. %, содержание Ti 1,6 вес. %, содержание Zn 1,4 вес. % и ТОС 1,4 вес. %.
1.6 Получение микропорошка
Используя 347,1 кг смеси отфильтрованных осадков, полученных согласно разделу 1.5 выше, получили водную суспензию с деионизированной водой с содержанием твердых веществ 15 вес. %. Данную суспензию подвергли распылительной сушке в распылительной башне при следующих условиях сушки распылением:
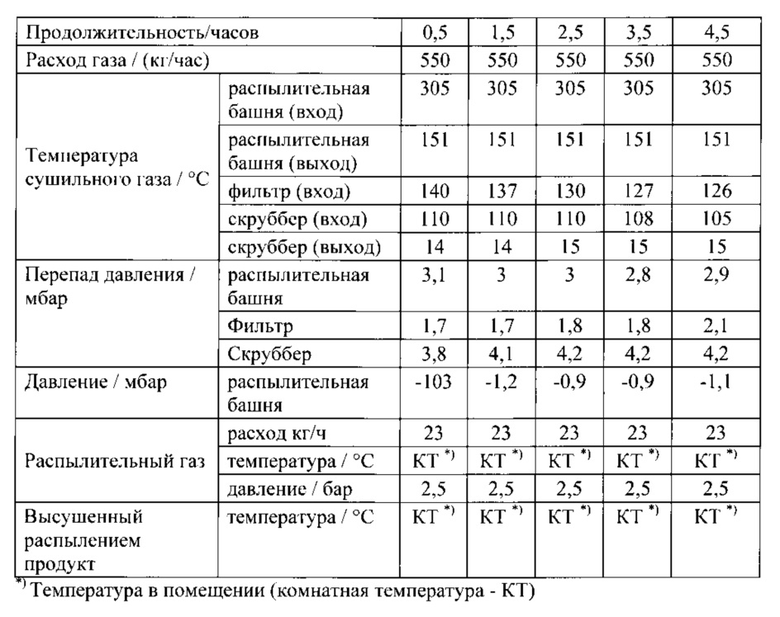
Распылительная башня состояла из вертикально расположенного цилиндра, имеющего длину 2650 мм, диаметр 1200 мм, причем данный цилиндр был конически зауженным в нижней части. Длина конуса составляла 600 мм. В головной части цилиндра размещались распылительные средства (двухкомпонентное сопло). Материал, высушенный распылением, отделяли от сушильного газа в фильтре, установленном на выходе распылительной башни, а затем сушильный газ пропускали через скруббер. Суспензию пропускали через внутреннее отверстие сопла, и распылительный газ пропускали через кольцевую щель, окружающую отверстие. Высушенный распылением материал, полученный таким способом, имел содержание цинка (Zn) 1,4 вес. %, содержание титана (Ti) 1,7 вес. %, содержание кремния (Si) 40 вес. % и общее содержание органического углерода (TOC) 0,27 вес. %. Затем высушенный распылением продукт подвергали обжигу в течение 2 часов при 650°C в атмосфере воздуха во вращающейся печи с выходом 76,3 кг обожженного высушенного распылением ZnTiMWW. Полученный таким образом обожженный материал, высушенный распылением, имел содержание цинка (Zn) 1,4 вес. %, содержание титана (Ti) 1,7 вес. %, содержание кремния (Si) 42 вес. % и содержание углерода (С) 0,14 вес. %. Объемная плотность обожженного высушенного распылением ZnTiMWW составляла 90 г/л (грамм/литр).
1.7 Получение формованного изделия
Используя обожженный высушенный распылением материал ZnTiMWW, полученный в соответствии с разделом 1.6 выше, было получено, высушено и подвергнуто обжигу формованное изделие. Для этой цели 22 партии, каждая с использованием 3,4 кг обожженного высушенного распылением материала ZnTiMWW, полученного в Примере 1, 0,220 кг Walocel™ (Walocel MW 15000 GB, Wolff Cellulosics GmbH & Co. KG, Germany), 2,125 кг Ludox® AC-40 и 6,6 л деионизированной воды, подготовили следующим образом:
3,4 кг ZnTiMWW и 0,220 кг Walocel подвергали разминанию в бегунковой мельнице в течение 5 минут. Затем во время дальнейшего разминания непрерывно добавляли 2,125 кг Ludox. Спустя еще 10 минут начали добавлять 6 л деионизированной воды. Еще через 30 минут, добавили дополнительно 0,6 л деионизированной воды. После общего времени подготовки 50 минут, замешанная масса стала пригодной для экструзии. Затем замешанную массу подвергли экструзии при 65-80 бар, при этом экструдер охлаждали водой в процессе экструзии. Для каждой партии, время экструзии находилось в диапазоне от 15 до 20 минут. Потребляемая электрическая мощность на каждую партию во время экструзии составляла 2,4 А. Экструзионную головку использовали для получения цилиндрических заготовок диаметром 1,7 мм. На выпускном отверстии экструзионной головки, заготовки не подвергали резке по длине. Заготовки, полученные таким образом, сушили в течение 16 часов при 120°C в сушильной камере на воздухе. В целом (сумма 22 партий) получили 97,1 кг белых заготовок диаметром 1,7 мм. 65,5 кг высушенных заготовок подвергли обжигу во вращающейся печи при 550°C в течение 1 часа на воздухе с выходом 62,2 кг обожженных заготовок. После этого заготовки пропустили через сито (размер ячейки 1,5 мм) и выход после просеивания составил 57,7 кг. Полученные таким образом формованные изделия имели объемную плотность 322 г/л (грамм на литр) и имели содержание Zn 1,2 вес. %, содержание Ti 1,4 вес. %, содержание Si 43 вес. % и содержание С 0,13 вес. %. Содержание натрия (Na) составляло 0,07 вес. %.
1.8 Последующая обработка формованного изделия
Используя обожженные заготовки, полученных в соответствии с разделом 1.7 выше, стадию последующей обработки выполнили следующим образом:
590 кг деионизированной воды загрузили в сосуд. Затем добавили 29,5 кг обожженных формованных изделий, полученных в соответствии с разделом 1.7 выше. Сосуд закрыли (герметично) и полученную смесь нагревали до температуры 145°C в течение 1,5 часов и выдерживали при этой температуре при собственном давлении (около 3 бар) в течение 8 часов. Затем смесь охлаждали в течение 2 часов. Обработанные водой заготовки подвергли фильтрации и промыли деионизованной водой. Полученные заготовки нагревали в сушильной камере на воздухе в течение 1 часа до температуры 120°C и выдерживали при данной температуре в течение 16 часов. Затем, высушенный материал нагревали на воздухе до температуры 450°C в течение 5,5 часов и выдерживали при данной температуре в течение 2 часов. После этого заготовки просеивали (размер ячейки 1,5 мм) и выход после просеивания составил 27,5 кг. Полученные таким образом обработанные водой формованные изделия имели объемную плотность 340 г/л (грамм на литр) и имели содержание Zn 1,3 вес. %, содержание Ti 1,4 вес. %, содержание Si 43 вес. % и содержание C 0,10 вес. %.
Справочный пример 2: Определение значений Dv10, Dv50 и Dv90
1. Получение образца: 1,0 г микропорошка суспендировали в 100 г деионизированной воды и перемешивали в течение 1 минуты.
2. Используемый аппарат и соответствующие параметры:
- Mastersizer S, исполнение с удлиненной станиной 2.15, серийный №33544-325; поставщик: Malvern Instruments GmbH, Herrenberg, Germany (Германия)
Справочный пример 3: Определение концентрации силанольных групп
Для определения концентрации силанольных групп, опыты согласно методу ядерного магнитного резонанса при вращении образца под магическим углом 29Si MAS ЯМР проводили при комнатной температуре на спектрометре VARIAN Infinityplus-400 с использованием 5,0 мм ZrO2 роторов. Спектры 29Si MAS ЯМР регистрировали при 79,5 МГц с использованием 1,9 мкс пи/4 импульса с 10 сек. задержкой повторного цикла и 4000 сканов. Все 29Si-спектры регистрировали на образцах, с вращением при 6 кГц, а химические сдвиги были определены относительно 4,4-диметил-4-силапентан-1-сульфоната натрия (DSS). Для определения концентрации силанольных групп, данный спектр 29Si MAS ЯМР восстанавливали из свертки соответствующих Гауссовых-Лоренцевых форм линий. Концентрацию силанольных групп по отношению к общему числу атомов Si получили посредством интегрирования восстановленных из свертки спектров 29Si MAS ЯМР.
Справочный пример 4: Определение прочности формованного изделия на раздавливание
Предел прочности на раздавливание, как указано в контексте настоящего изобретения, следует понимать как параметр, определенный с помощью испытательной установки для определения прочности на раздавливание Z2.5/TS1S, поставщик Zwick GmbH & Co., D-89079 Ulm, Germany (Германия). Что касается основных характеристик и принципов работы данной установки, ссылка делается на соответствующие руководство по эксплуатации "Register 1: Betriebsanleitung / Sicherheitshandbuch fur die  Z2.5/TS1S", version 1.5, December 2001 by Zwick GmbH & Co. Technische Dokumentation, August-Nagel-Strasse 11, D-89079 Ulm, Germany. С помощью указанной установки, данную заготовку, как описано в Справочном примере 1, подвергали воздействию возрастающего усилия с помощью плунжера, имеющего диаметр 3 мм, до тех пор, пока заготовка не была раздавлена. Усилие, при котором заготовка была раздавлена, называют прочностью заготовки на раздавливание. Установка оснащена неподвижным горизонтальным столом, на котором размещается заготовка. Плунжер, который может свободно перемещаться в вертикальном направлении, создает усилие, воздействующее на заготовку по отношению к неподвижному столу. Аппарат работал с предварительным усилием 0,5 H и скоростью сдвига при предварительном усилии 10 мм/мин и последующей скоростью испытания 1,6 мм/мин. Вертикально перемещаемый плунжер был соединен с датчиком нагрузки для считывания показателей усилия и во время измерения перемещался в направлении неподвижного поворотного стола, на котором было размещено формованное испытуемое изделие (заготовка), таким образом, воздействуя на заготовку посредством ее прижимания к столу. Плунжер воздействовал на заготовки перпендикулярно их продольной оси. Управление опытом осуществлялось с помощью компьютера, который регистрировал и оценивал результаты измерений. Полученные значения представляют собой средние значения измерений для 10 заготовок в каждом конкретном случае.
Z2.5/TS1S", version 1.5, December 2001 by Zwick GmbH & Co. Technische Dokumentation, August-Nagel-Strasse 11, D-89079 Ulm, Germany. С помощью указанной установки, данную заготовку, как описано в Справочном примере 1, подвергали воздействию возрастающего усилия с помощью плунжера, имеющего диаметр 3 мм, до тех пор, пока заготовка не была раздавлена. Усилие, при котором заготовка была раздавлена, называют прочностью заготовки на раздавливание. Установка оснащена неподвижным горизонтальным столом, на котором размещается заготовка. Плунжер, который может свободно перемещаться в вертикальном направлении, создает усилие, воздействующее на заготовку по отношению к неподвижному столу. Аппарат работал с предварительным усилием 0,5 H и скоростью сдвига при предварительном усилии 10 мм/мин и последующей скоростью испытания 1,6 мм/мин. Вертикально перемещаемый плунжер был соединен с датчиком нагрузки для считывания показателей усилия и во время измерения перемещался в направлении неподвижного поворотного стола, на котором было размещено формованное испытуемое изделие (заготовка), таким образом, воздействуя на заготовку посредством ее прижимания к столу. Плунжер воздействовал на заготовки перпендикулярно их продольной оси. Управление опытом осуществлялось с помощью компьютера, который регистрировал и оценивал результаты измерений. Полученные значения представляют собой средние значения измерений для 10 заготовок в каждом конкретном случае.
Справочный пример 5: Спектры 29Si-ЯМР в твердом состоянии в отношении структур Q3 и Q4
Все опыты 29Si ЯМР твердого тела проводили с использованием спектрометра Bruker Avance при ларморовой частоте 300 МГц 1H (Bruker BioSpin, Germany). Образцы были упакованы в 7 мм ZrO2 роторы и измерялись при вращении под магическим углом при 5 кГц при комнатной температуре. Прямые спектры поляризации 29Si были получены с использованием (пи/2)-импульсного возбуждения с 5 мкс длительностью импульса, 29Si несущей частотой, соответствующей -65 ч.н.м. в спектре, и задержкой повторного цикла сканирования 120 сек. Сигнал получали за 25 мс при 45 кГц протонной развязки высокой мощности и накапливали в течении 10-17 часов. Спектры обрабатывали с использованием Bruker TopSpin с 30 Гц экспоненциальным уширением линии, ручным фазированием и ручной коррекцией базовой линии по всей ширине спектра. Спектры определяли относительно полимера Q8M8 в качестве внешнего вторичного стандарта, задав резонанс M группы триметилсилила как 12,5 частей на миллион. Спектры затем согласовывали с набором гауссовых форм линий, в соответствии с числом различаемых резонансов. Относительно текущих анализируемых спектров, были использованы 6 линий в общей сложности, что составляет пять различных максимумов пиков (приблизительно при -118, -115, -113, -110 и -104 ч.н.м.) плюс четко видимое плечо при -98 ч.н.м. Корректировку проводили с использованием DMFit (Massiot et al., Magnetic Resonance in Chemistry, 40 (2002) pp 70-76). Пики вручную установили на видимых максимумах пиков или плече. Затем положение пика и ширину линии оставили незафиксированными, т.е. откорректированные пики не были зафиксированы в определенном положении. Результат корректировки был численно устойчивым, то есть, искажения в начальной настройке корректировки, как описано выше, приводили к аналогичным результатам. Площади откорректированных пиков в дальнейшем использовали как нормализованные согласно корректировки посредством DMFit. Для количественной оценки изменений спектра, было рассчитано соотношение, которое отражает изменения площадей пиков "по левую руку" и "по правую руку", следующим образом. Шесть пиков, как описано выше, были маркированы как 1, 2, 3, 4, 5 и 6, а соотношение Q было рассчитан по формуле 100*{[a1+a2]/[а4+а5+а6]}/a3. В данной формуле ai, i=1…6 представляет собой площадь откорректированного пика, которому было присвоено данный номер.
Справочный пример 6: Адсорбция / десорбция воды
Измерения изотерм адсорбции/десорбции воды проводились с помощью прибора VTI SA от TA Instruments согласно программы ступенчатых изотерм. Опыт состоял из прогона или серии прогонов, выполненных на образце материала, который поместили в чашу микровесов внутри прибора. Перед началом измерений, из образца удалили остаточную влагу посредством нагревания образца до 100°C (скорость нагрева 5°C/мин) и выдерживали образец в течение 6 часов в потоке N2. После программы сушки, температуру в камере снизили до 25°C и выдерживали изотермический в течение измерений. Микровесы были откалиброваны и вес высушенного образца измерили (максимальное отклонение веса 0,01 вес. %). Поглощение воды образцом измеряли как увеличение веса по сравнению с весом сухого образца. Во-первых, кривую адсорбции измеряли посредством увеличения относительной влажности (RH) (выраженной в вес. % воды в атмосфере внутри камеры), воздействию которой подвергали образцы, и посредством измерения поглощения воды образцом при равновесии. Относительную влажность увеличивали с шагом 10 вес. % от 5 до 85% и на каждой стадии система контролировала значение RH и осуществляла мониторинг веса образца до достижения условий равновесия, регистрируя поглощение веса. Общий объем адсорбированной образцом воды был поглощен после того, как образец подвергли воздействию 85 вес. % RH. Во время измерения десорбции, RH снижали с 85 вес. % до 5 вес. % с шагом 10% и изменение веса образца (поглощение воды) контролировали и регистрировали.
Справочный пример 7: Измерения инфракрасной спектроскопии на основе преобразования Фурье
Измерения инфракрасной спектроскопии на основе преобразования Фурье (Фурье-ИК-спектроскопия) были выполнены с помощью спектрометра Nicolet 6700. Формованное изделие было измельчено в порошок, который затем прессовали в самоподдерживающийся осадок без использования каких-либо добавок. Осадок ввели в ячейку высокого вакуума (HV), размещенную в приборе Фурье-ИК-спектроскопии. Перед измерением образец предварительно обработали в условиях высокого вакуума (10-5 мбар) в течение 3 часов при 300°C. Спектры были получены после охлаждения ячейки до 50°C. Спектры регистрировали в диапазоне от 4000 до 800 см-1 с разрешением 2 см-1. Полученные спектры представлены на графике, имеющем на оси x волновые числа (см-1) и на оси y поглощающую способность (в условных единицах, у.е.). Для количественного определения высот пиков и соотношения между этими пиками проводилась коррекция базовой линии. Изменения в области от 3000 до 3900 см-1 были проанализированы и для сравнения нескольких образцов, в качестве эталона была взята полоса при 1880±5 см-1.
Справочный пример 8: Определение и расчет коэффициента разделения октанола-воды KOW
Коэффициент разделения октанола-воды KOW данного соединения определяется как соотношение химической концентрации указанного соединения в октаноловой фазе и химической концентрации указанного соединения в водной фазе в двухфазной системе 1-октанола и воды при температуре 25°C.
Коэффициент разделения октанола-воды KOW данного соединения определяют при помощи метода встряхивания колбы, который состоит из растворения соединения в объеме высокой чистоты 1-октанола и деионизированной воды (предварительно смешанного и калибруемого не менее 24 часов) и измерения концентрации соединения в каждой фазе 1-октанола и водной фазе достаточно точным методом, предпочтительно с помощью UV/VIS спектроскопии (в видимой и инфракрасной области). Данный метод описан в Руководстве по испытаниям химических веществ ОЭСР: OECD Guideline for the testing of chemicals, number 107, adopted on July 27, 1995.
ОПИСАНИЕ ФИГУР
Фиг. 1 изображает блок-схему предпочтительного способа согласно настоящему изобретению. На Фиг. 1, буквы и цифры имеют следующие значения:
(1)-(23) потоки согласно особенно предпочтительному способу, описанному в примерах.
S0, S01, S02, S1, S2, S3, S4, S4B, S5, L1, L2, TL1, TL2, TL2, BL2 - потоки согласно предпочтительному способу, описанному в общем описании и примерах.
ССЫЛКИ НА ИЗВЕСТНЫЙ УРОВЕНЬ ТЕХНИКИ
- WO 2011/006990 A1
- US 2007043226 A1
- WO 2007/000396 А1
- EP 0427062 A2
- US 5,194,675
- US 2004068128 A1
- K.J. Lissant, Making and Breaking Emulsions, Res. Lab., Petrolite Corp., St. Louis, Missouri, USA, in: K.J. Lissant (ed.), Emulsion Technology (1974), chapter 2, pp 111-124, Dekker, New York
- S.E. Taylor, Chem. Ind. (1992), pp 770-773
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕРЕГОНКА С ЧАСТИЧНЫМ ПОТОКОМ | 2015 |
|
RU2705993C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОПИЛЕНОКСИДА | 2014 |
|
RU2673676C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОПИЛЕНОКСИДА | 2014 |
|
RU2671638C2 |
| Способ получения пропиленоксида | 2014 |
|
RU2678844C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОПИЛЕНОКСИДА | 2017 |
|
RU2734823C2 |
| ИЗВЛЕЧЕНИЕ ПРОПЕНА ПОСРЕДСТВОМ ОЧИСТКИ В СКРУББЕРЕ СО СМЕСЬЮ РАСТВОРИТЕЛЬ/ВОДА | 2018 |
|
RU2766954C2 |
| РЕГЕНЕРАЦИЯ ТИТАНСОДЕРЖАЩЕГО ЦЕОЛИТА | 2014 |
|
RU2673798C9 |
| СПОСОБ РЕГЕНЕРАЦИИ КАТАЛИЗАТОРА НА ОСНОВЕ ТИТАН-СОДЕРЖАЩЕГО ЦЕОЛИТА ДЛЯ ЭПОКСИДИРОВАНИЯ ПРОПИЛЕНА | 2016 |
|
RU2702349C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОПИЛЕНОКСИДА | 2017 |
|
RU2740395C2 |
| СПОСОБ ОЧИСТКИ ПРОПИЛЕНОКСИДА | 2017 |
|
RU2741991C2 |
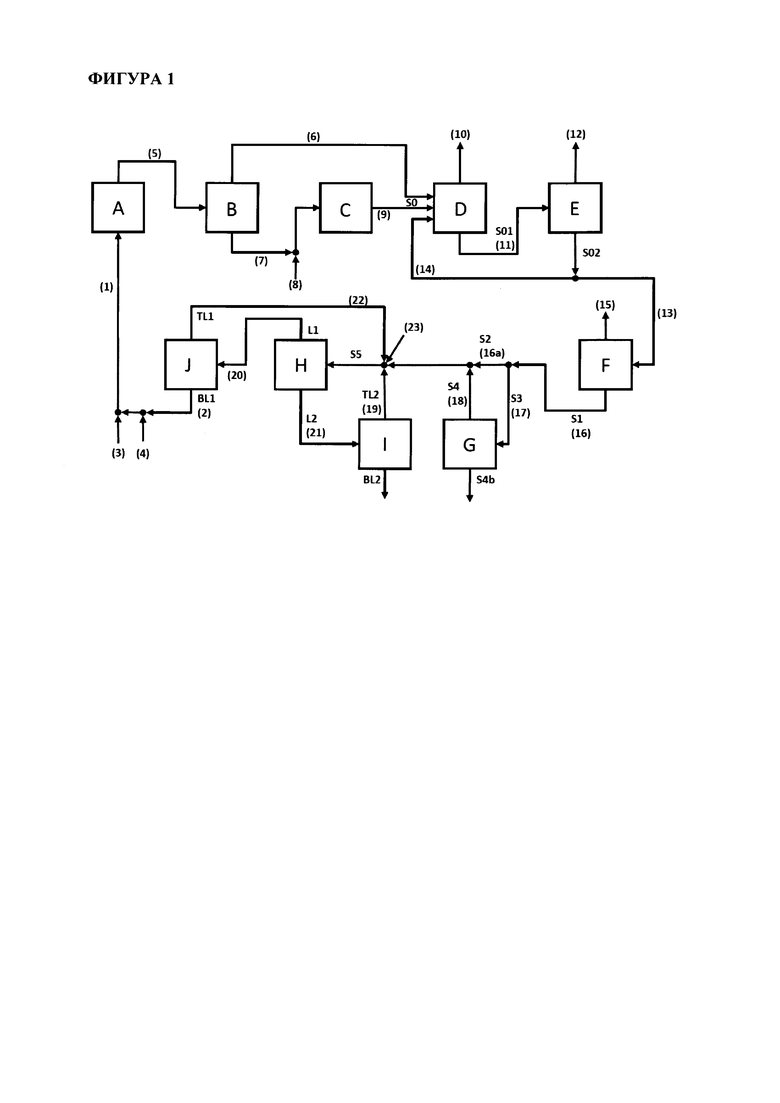
Настоящее изобретение относится к непрерывному способу получения пропиленоксида, который включает в себя: (a) реагирование пропена, необязательно смешанного с пропаном, с перекисью водорода в реакционном аппарате в присутствии ацетонитрила в качестве растворителя с получением потока S0 на выходе реакционного аппарата, при этом S0 содержит пропиленоксид, ацетонитрил, воду, по меньшей мере один дополнительный компонент В, необязательно пропен и необязательно пропан, и отличается тем, что нормальная точка кипения по меньшей мере одного компонента В выше, чем нормальная точка кипения ацетонитрила, и отличается тем, что десятичный логарифм коэффициента разделения октанола-воды (log Kow) по меньшей мере, одного компонента В составляет больше нуля; (b) отделение пропиленоксида из S0, необязательно после отделения пропена и необязательно пропана, с получением потока S1, который содержит ацетонитрил, воду и по меньшей мере один дополнительный компонент В; (c) разделение S1 на два потока S2 и S3, при котором общая масса S3 по отношению к общей массе S1 находится в диапазоне от 0,01 до 25%; (d) воздействие на поток S3 посредством фракционирования паровой и жидкой фаз в установке фракционирования с получением потока паровой фракции S4, обедненного по меньшей мере одним компонентом В, и с получением жидкого кубового потока S4b, обедненного ацетонитрилом; (e) рециркуляцию по меньшей мере части потока S4, необязательно после обработки, в стадию (а) и рециркуляцию по меньшей мере части потока S2, необязательно после обработки, в стадию (а). Технический результат – создание экономически выгодного непрерывного способа получения пропиленоксида в ацетонитриле в качестве растворителя, который позволяет избежать накопления примесей в рециркулирующем потоке растворителя ацетонитрила. 18 з.п. ф-лы, 1 ил., 13 табл., 19 пр.
1. Непрерывный способ получения пропиленоксида, который включает в себя:
(a) реагирование пропена, необязательно смешанного с пропаном, с перекисью водорода в реакционном аппарате в присутствии ацетонитрила в качестве растворителя с получением потока S0 на выходе реакционного аппарата, при этом S0 содержит пропиленоксид, ацетонитрил, воду, по меньшей мере один дополнительный компонент В, необязательно пропен и необязательно пропан, и отличающийся тем, что нормальная точка кипения по меньшей мере одного компонента В выше, чем нормальная точка кипения ацетонитрила, и отличающийся тем, что десятичный логарифм коэффициента разделения октанола-воды (log Kow) по меньшей мере одного компонента В составляет больше нуля;
(b) отделение пропиленоксида из S0, необязательно после отделения пропена и необязательно пропана, с получением потока S1, который содержит ацетонитрил, воду и по меньшей мере один дополнительный компонент В;
(c) разделение S1 на два потока S2 и S3, при котором общая масса S3 по отношению к общей массе S1 находится в диапазоне от 0,01 до 25%;
(d) воздействие на поток S3 посредством фракционирования паровой и жидкой фаз в установке фракционирования с получением потока паровой фракции S4, обедненного по меньшей мере одним компонентом В, и с получением жидкого кубового потока S4b, обедненного ацетонитрилом;
(e) рециркуляцию по меньшей мере части потока S4, необязательно после обработки, в стадию (а) и рециркуляцию по меньшей мере части потока S2, необязательно после обработки, в стадию (а).
2. Способ по п. 1, отличающийся тем, что на стадии (с) общая масса S3 по отношению к общей массе S1 находится в диапазоне от 0,05 до 20%, предпочтительно от 0,1 до 15%, более предпочтительно от 0,2 до 10%, более предпочтительно от 0,5 до 5%.
3. Способ по п. 1 или 2, отличающийся тем, что от 90 до 99,9 вес.%, предпочтительно от 95 до 99,8 вес.%, более предпочтительно от 99 до 99,7 вес.% потока S1 состоит из ацетонитрила и воды, и отличающийся тем, что от 0,01 до 5 вес.%, предпочтительно от 0,015 до 3 вес.%, более предпочтительно от 0,02 до 2 вес.% потока S1 состоит из по меньшей мере одного компонента В.
4. Способ по п.1 или 2, отличающийся тем, что на стадии (d) фракционирование паровой и жидкой фаз осуществляют в установке фракционирования таким образом, что от 10 до 30 вес.%, предпочтительно от 10 до 25 вес.% жидкого кубового потока S4b состоит из ацетонитрила и от 0,1 до 10 вес.%, предпочтительно от 0,25 до 5 вес.% жидкого кубового потока S4b состоит из по меньшей мере одного дополнительного компонента В.
5. Способ по п. 1 или 2, отличающийся тем, что на стадии (d) фракционирование паровой и жидкой фаз осуществляют в установке фракционирования при абсолютном давлении в диапазоне от 0,1 до 10 бар, предпочтительно от 0,5 до 5 бар, более предпочтительно от 1 до 2 бар.
6. Способ по п.1 или 2, отличающийся тем, что на стадии (d) количество теоретических тарелок в установке фракционирования находится в диапазоне от 1 до 100, предпочтительно от 2 до 25, более предпочтительно от 3 до 10.
7. Способ по п. 1 или 2, отличающийся тем, что часть потока S4 используется после конденсации в виде флегмы, причем флегмовое число предпочтительно находится в диапазоне от 0,01:1 до 10:1, более предпочтительно от 0,1:1 до 5:1, более предпочтительно от 0,5:1 до 2:1.
8. Способ по п. 1 или 2, отличающийся тем, что установка фракционирования работает без флегмы и S3 подается в верхнюю часть установки фракционирования.
9. Способ по п. 1 или 2, отличающийся тем, что от 95 до 99,99 вес.%, предпочтительно от 97 до 99,9 вес.%, более предпочтительно от 98 до 99,9 вес.% потока S4 состоит из ацетонитрила и воды, и отличающийся тем, что от 0,0001 до 0,2 вес.%, предпочтительно от 0,001 до 0,15 вес.%, более предпочтительно от 0,005 до 0,1 вес.% потока S4 состоит из по меньшей мере одного компонента В.
10. Способ по п. 1, отличающийся тем, что стадия (е) включает в себя обработку потока S4, причем указанная обработка включает в себя объединение по меньшей мере части потока S4, предпочтительно после конденсации, с потоком S2 и получение предпочтительно жидкого потока, воздействие на указанный предпочтительно жидкий поток посредством отделения ацетонитрила-воды с получением потока, обогащенного ацетонитрилом, и рециркуляцию указанного потока, обогащенного ацетонитрилом, необязательно после дополнительной обработки, в стадию (а).
11. Способ по п. 10, отличающийся тем, что стадия (е) включает в себя:
(i) получение предпочтительно жидкого потока S5 посредством добавления предпочтительно жидкого потока Р в поток S2, или по меньшей мере в часть потока S4, или в жидкий поток, полученный в результате объединения S2 и по меньшей мере части S4, в котором Р содержит по меньшей мере 95 вес.% С3 в расчете на общую массу потока Р,
в котором С3 представляет собой пропен, необязательно смешанный с пропаном, с минимальным весовым соотношением пропена и пропана 7:3, и
в котором Р предпочтительно добавляют в таком количестве, чтобы в потоке S5 весовое соотношение С3 и ацетонитрила находилось в диапазоне от 0,2:1 до 5:1, предпочтительно от 0,5:1 до 2:1;
(ii) воздействие на поток S5 посредством температуры не более 92°С и давления по меньшей мере 10 бар, предпочтительно температуры в диапазоне от 5 до 90°С и давления в диапазоне от 15 до 50 бар, более предпочтительно температуры в диапазоне от 25 до 45°С и давления в диапазоне от 15 до 25 бар с получением первой жидкой фазы L1 и второй жидкой фазы L2,
в котором по меньшей мере 95 вес.%, предпочтительно по меньшей мере 98 вес.% L1 состоит из С3, ацетонитрила, воды и по меньшей мере одного компонента В, причем содержание воды в L1 составляет менее 10 вес.%, предпочтительно находится в диапазоне от 1 до 5 вес.%, в расчете на общую массу L1, и
в котором по меньшей мере 95 вес.%, предпочтительно по меньшей мере 98 вес.% L2 состоит из С3, ацетонитрила, воды и по меньшей мере одного компонента В, причем содержание С3 в L2 составляет не более 5 вес.%, в расчете на общую массу L2, и содержание ацетонитрила в L2 составляет менее 45 вес.%, предпочтительно находится в диапазоне от 10 до 35 вес.%, в расчете на общую массу L2;
(iii) отделение L1 от L2, преимущественно в гравитационном отстойнике;
(iv) рециркуляцию L1 в качестве потока, обогащенного ацетонитрилом, необязательно после дополнительной обработки, в стадию (а).
12. Способ по п. 11, дополнительно включающий в себя обработку L1, причем указанная обработка включает в себя воздействие на L1 посредством стадии перегонки, в результате которой получают кубовый поток BL1, отличающийся тем, что по меньшей мере 95 вес.%, предпочтительно по меньшей мере 98 вес.% BL1 состоит из С3, ацетонитрила, воды и по меньшей мере одного компонента В, и отличающий тем, что содержание СЗ в BL1 находится в диапазоне от 7 до 18 вес.%, предпочтительно от 10 до 15%вес, и рециркуляцию BL1 в качестве потока, обогащенного ацетонитрилом, необязательно без дополнительной обработки, в стадию (а).
13. Способ по п.12, отличающийся тем, что от 0,01 до 5 вес.%, предпочтительно от 0,015 до 3 вес.%, более предпочтительно от 0,02 до 2 вес.% потока BL1 состоит из по меньшей мере одного компонента В.
14. Способ по п.1, отличающийся тем, что стадия (b) включает в себя:
(I) отделение пропена, необязательно вместе с пропаном и кислородом, который необязательно дополнительно содержится в S0, из потока S0 с получением потока S01, обогащенного пропиленоксидом, ацетонитрилом, водой и по меньшей мере одним компонентом В, отличающегося тем, что предпочтительно по меньшей мере 99 вес.% потока S01 состоит из ацетонитрила, воды, по меньшей мере одного компонента В и пропиленоксида; и отличающегося тем, что для отделения предпочтительно используют установку фракционирования, и отличающегося тем, что предпочтительно в верхней части установки фракционирования жидкий ацетонитрил, необязательно смешанный с жидкой водой, добавляют в качестве захватывающего агента;
(II) отделение пропиленоксида из потока S01 с получением потока S02, обогащенного ацетонитрилом, водой и по меньшей мере одним компонентом В, отличающегося тем, что по меньшей мере 95 вес.% потока S02 состоит из ацетонитрила, воды и по меньшей мере одного компонента В, отличающегося тем, что весовое соотношение ацетонитрила и воды составляет больше 1:1, и отличающегося тем, что поток S02 предпочтительно подвергают стадии (с) в качестве потока S1.
15. Способ по п. 14, отличающийся тем, что стадия (b) дополнительно включает в себя:
(IIIа) воздействие на поток S02, полученный на стадии (II), посредством гидрогенизации и/или
(IIIb) воздействие на поток, полученный на стадии (II) или (IIIа), посредством перегонки для получения кубового потока, и отличающийся тем, что гидрогенизированный поток, полученный на стадии (IIIа), или кубовый поток, полученный на стадии (IIIb), подвергают стадии (с) в качестве потока S1.
16. Способ по п. 1 или 2, отличающийся тем, что на стадии (а) пропен подвергают реакции с перекисью водорода в присутствии гетерогенного катализатора, причем указанный гетерогенный катализатор предпочтительно содержит цеолит, предпочтительно титановый цеолит, более предпочтительно титановый цеолит структурного типа MWW (TiMWW), более предпочтительно цинксодержащий титановый цеолит структурного типа MWW (ZnTiMWW).
17. Способ по п. 1 или 2, отличающийся тем, что от 90 до 97 вес.%, предпочтительно от 92 до 97 вес.%, более предпочтительно от 95 до 97 вес.% потока S0 состоит из ацетонитрила, воды и пропиленоксида, и отличающийся тем, что от 0,01 до 3 вес.%, предпочтительно от 0,015 до 2 вес.%, более предпочтительно от 0,02 до 1 вес.% потока S0 состоит из по меньшей мере одного компонента В.
18. Способ по п. 1 или 2, отличающийся тем, что по меньшей мере один компонент В представляет собой пропионитрил, 1-нитропропан, 2-нитропропан, 3-метилбутаннитрил, н-пентаннитрил, 1-пентанол, 2-пентанол, 2-бутанон, 2-пентанон, 2-гексанон, 4-метил-2-гептанон, 2,6-диметил-4-гептанол, 4,6-диметил-2-гептанол, 2,6-диметил-4-гептанон, 4,6-диметил-2-гептанон, 2,6-диметил-4,6-гептандиол, 2,4-диметил-оксазолин, 2,5-диметилоксазолин, цис-2,4-диметил-1,3-диоксолан, транс-2,4-диметил-1,3-диоксолан, по меньшей мере одну примесь, содержащуюся в перекиси водорода, используемой на стадии (а), или комбинацию двух или более из этих соединений.
19. Способ по п. 18, отличающийся тем, что по меньшей мере одна примесь, содержащаяся в перекиси водорода, используемой на стадии (а), представляет собой алкилфосфат, такой как трис-(2-этилгексил)-фосфат, нониловый спирт, такой как диизобутилкарбинол, алкилциклогексиловый эфир, такой как 2-метил-циклогексилацетат, N,N-диалкилкарбонамиды, такие как N,N-дибутилпропионамид, N-алкил-N-арилкарбонамид, такой как N-этил-N-фенилбензамид, N,N-диалкилкарбамат, такой как 2-этилгексил-N-бутилкарбамат, тетраалкилмочевину, такую как тетра-н-бутилмочевину, циклоалкилмочевину, такую как дигексилпропенмочевину, фенилалкилмочевину, такую как N,N-дибутил-N'-метил-N'-фенилмочевину, N-алкил-2-пирролидон, такой как октил-пирролидон, N-алкил-капролактам, такой как н-октил-капролактам, или комбинацию двух или более из этих соединений.
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОПИЛЕНОКСИДА | 2004 |
|
RU2324689C2 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| Колосоуборка | 1923 |
|
SU2009A1 |

