Настоящее изобретение относится к способу получения пропиленоксида, где пропен отделяют посредством дистилляции от вытекающего потока из зоны эпоксидирования, причем указанный вытекающий поток содержит пропиленоксид, воду, органический растворитель, и пропен, где на вытекающий поток воздействуют условиями дистилляции в узле дистилляции, и конденсированную часть газообразного верхнего потока, которая обогащена пропеном, возвращают в верхнюю часть узла дистилляции.
Пропиленоксид является важным промежуточным соединением в химической промышленности. Подходящий способ получения пропиленоксида начинается с пропена и использует применение пероксида водорода в качестве окисляющего агента, ацетонитрила в качестве растворителя и катализатора эпоксидирования, содержащий цеолит на основе титана. Из-за его важности для промышленных процессов желательно осуществлять реакцию эпоксидирования настолько эффективно насколько возможно и очищать пропиленоксид эффективно и до высокой степени. Реакция эпоксидирования приводит к смеси, содержащей растворитель, воду и пропиленоксид. Поскольку эпоксидирование обычно проводят с избытком пропена, полученная смесь содержит также различные количества пропена. Особенно в промышленных непрерывных процессах эпоксидирования пропена в растворителе, одной особенностью всего процесса является рециркуляция пропена обратно на стадию эпоксидирования. Поскольку пропиленоксид является летучим, отделение непрореагировавшего пропена является сложной задачей, если необходимо избежать уноса пропиленоксида. Унос пропиленоксида потребовал бы дальнейших стадий обработки, потому что возврат пропиленоксида в реактор эпоксидирования приведет к усиленному образованию нежелательных побочных продуктов.
В WO 2008/118265 A раскрывается экстрактивная дистилляция с метанолом и/или водой для отделения пропена от пропиленоксида. Разделяемая смесь образуется в результате эпоксидирования пропена с пероксидом водорода или H2/O2-смесями и содержит метанол в качестве растворителя. Таким образом, смеси метанола и воды являются предпочтительными растворителями для экстрактивной дистилляции, которые поступают с последующей стадии процесса. Недостатком способа является то, что в процессе создается дополнительная внутренняя петля, что приводит к увеличению потоков, а также к риску накопления побочных продуктов в петле. В частности увеличение гидравлической нагрузки приводит к более высокому потреблению энергии. В WO 2004/037802 A раскрывается аналогичный способ, основанный на процессе, где пропиленоксид получают из пропена и пероксида водорода в ацетонитриле. Недостатки сопоставимы с описанными выше в отношении WO 2008/118265 A.
Хотя точки кипения пропиленоксида и пропена отличаются друг от друга, разделение обоих соединений является сложной задачей. Их разделение при обычной дистилляции, когда верхний конденсатор работает с охлаждающей водой, температура конденсации на верху должна быть выше 40°C, поэтому требуется верхнее давление по меньшей мере 16,5 бар. Однако давление 16,5 бар приведет к температуре отстойника по меньшей мере 140°C, что приведет к термическому разложению пропиленоксида. Даже если для работы верхнего конденсатора потребуется холодная вода (температура 5°C), это также приведет к тому, что верхнее давление будет составлять по меньшей мере 6,5 бар. Это давление привело бы к температуре отстойника по меньшей мере 100°C, при которой пропиленоксид также термически разлагается в значительной степени.
Следовательно, задачей настоящего изобретения было обеспечение способа разделения пропиленоксида и пропена, который является эффективным и позволяет существенно избежать как разложения пропиленоксида в отстойнике, так и потерь отводимого сверху пропиленоксида. Способ должен быть экономически выгодным и особенно должен позволять снизить энергозатраты на разделение пропиленоксида и пропена.
Неожиданно было обнаружено, что, если при дистиллятивном отделении пропена от вытекающего потока, причем указанный вытекающий поток содержит пропиленоксид, воду, органический растворитель, пропен, и пропан; конденсированную часть газообразного верхнего потока S0, которая обогащена пропеном, возвращают в верхнюю часть узла дистилляции, на отделение пропена от пропиленоксидаможет оказываться положительное влияние, при этом можно избежать разложения пропиленоксида и снизить потребление энергии.
Поэтому настоящее изобретение относится к способу получения пропиленоксида, включающему
(i) обеспечение потока, содержащего пропен, пероксид водорода или источник пероксида водорода, воду и органический растворитель;
(ii) пропускание жидкого потока поступающего материала, обеспеченного на стадии (i), в зону эпоксидирования, содержащую катализатор эпоксидирования, содержащий цеолит на основе титана, и воздействие на жидкий поток поступающего материала условиями реакции эпоксидирования в зоне эпоксидирования, с получением реакционной смеси, содержащей пропен, пропиленоксид, воду и органический растворитель;
(iii) удаление вытекающего потока из зоны эпоксидирования, причем вытекающий поток содержит пропиленоксид, воду, органический растворитель и пропен;
(iv) отделение пропена от вытекающего потока посредством дистилляции, включающее
(iv.1) воздействие на вытекающий поток условиями дистилляции в узле дистилляции, с получением газообразного верхнего потока S0, обогащенного пропеном, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции, и жидкого нижнего потока S01, обогащенного пропиленоксидом, водой и органическим растворителем, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции;
(iv.2) возврат конденсированной части потока S0 в верхнюю часть узла дистилляции.
Предпочтительно, способ представляет собой непрерывный способ.
В общем, возможно применять чистый или по существу чистый пропен в качестве исходного вещества и в качестве части потока, подвергаемого эпоксидированию на стадии (ii). Предпочтительно, смесь пропена и пропена применяют. Наиболее предпочтительно применяется техническая марка пропилена в соответствии с международными нормами, например, ASTM D5273 или DIN 51622.
Поэтому, настоящее изобретение также относится к способу получения пропиленоксида, включающему
(i) обеспечение потока, содержащего пропен, пропан, пероксид водорода или источник пероксида водорода, воду и органический растворитель;
(ii) пропускание жидкого потока поступающего материала, обеспеченного на стадии (i), в зону эпоксидирования, содержащую катализатор эпоксидирования, содержащий цеолит на основе титана, и воздействие на жидкий поток поступающего материала условиями реакции эпоксидирования в зоне эпоксидирования, с получением реакционной смеси, содержащей пропен, пропан, пропиленоксид, воду и органический растворитель;
(iii) удаление вытекающего потока из зоны эпоксидирования, причем вытекающий поток содержит пропиленоксид, воду, органический растворитель, пропен, и пропан;
(iv) отделение пропена и пропана от вытекающего потока посредством дистилляции, включающее
(iv.1) воздействие на вытекающий поток условиями дистилляции в узле дистилляции, с получением газообразного верхнего потока S0, обогащенного пропеном и пропаном по сравнению с вытекающим потоком, подвергаемым условиям дистилляции, и жидкого нижнего потока S01, обогащенного пропиленоксидом, водой и органическим растворителем, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции;
(iv.2) возврат конденсированной части потока S0 в верхнюю часть узла дистилляции.
Если смесь пропена и пропана применяют как часть потока, обеспеченного на стадии (i), и подвергают эпоксидированию на стадии (ii), массовое соотношение пропена : пропана предпочтительно составляет по меньшей мере 7:3. Например, коммерчески доступный пропен может применяться, который может представлять собой либо пропен полимерной марки, либо пропен химической марки. Как правило, пропен полимерной марки имеет содержание пропена в интервале от 99 до 99.8 мас. % и содержание пропана в интервале от 0.2 до 1 мас. %. Пропен химической марки, как правило, имеет содержание пропена в интервале от 92 до 98 мас. % и содержание пропана в интервале от 2 до 8 мас. %. Согласно предпочтительному варианту выполнения настоящего изобретения смесь пропена и пропан подвергают эпоксидированию, которая имеет содержание пропена в интервале от 99 до 99.8 мас. % и содержание пропана в интервале от 0.2 до 1 мас. %.
Вытекающий поток
В общем, нет никаких конкретных ограничений в отношении состава вытекающего потока, при условии что он содержит пропиленоксид, воду, органический растворитель, пропен, и необязательно пропан. Предпочтительно, по меньшей мере 95 мас. %, более предпочтительно от 95 до 100 мас. %, более предпочтительно от 98 до 100 мас. %, более предпочтительно от 99 до 100 мас. %, вытекающего потока, удаленного на стадии (iii), состоит из пропиленоксида, органического растворителя, воды, пропена, кислорода и необязательно пропана.
Предпочтительно, вытекающий поток, удаленный на стадии (iii), содержит пропиленоксид в количестве от 5 до 20 мас. %, более предпочтительно от 8 до 18 мас. %, более предпочтительно от 10 до 14 мас. %, на основе общей массы вытекающего потока; органический растворитель в количестве от 60 до 75 мас. %, более предпочтительно от 65 до 70 мас. %, на основе общей массы вытекающего потока; воду в количестве от 10 до 25 мас. %, более предпочтительно от 15 до 20 мас. %, на основе общей массы вытекающего потока; пропен в количестве от 1 до 5 мас. %, более предпочтительно от 3 до 4.5 мас. %, на основе общей массы вытекающего потока; кислород в количестве от 0.05 до 1 мас. %, более предпочтительно от 0.1 до 0.5 мас. %, на основе общей массы вытекающего потока; и необязательно пропан в количестве от 0.1 до 2 мас. %, более предпочтительно от 0.2 до 1 мас. %, на основе общей массы вытекающего потока.
Условия дистилляции
Согласно стадии (iv), вытекающий поток подвергают на стадии (iv.1) условиям дистилляции в узле дистилляции. В общем, нет никаких конкретных ограничений в отношении конструкции узла дистилляции, при условии что она является подходящей для осуществления отделения пропена. Предпочтительно, узел дистилляции, применяемый на стадии (iv), представляет собой по меньшей мере одну дистилляционную колонну, более предпочтительно одну дистилляционную колонну, где дистилляционная колонна имеет от 3 до 50, предпочтительно от 5 до 15, более предпочтительно от 6 до 10, более предпочтительно от 7 до 9, теоретических тарелок. Предпочтительно, ректификационная секция узла дистилляции состоит из от 50 до 75 %, предпочтительно от 60 до 65 %, теоретических тарелок, и стриппинг-секция узла дистилляции состоит из от 25 до 50 %, предпочтительно от 35 до 40 %, теоретических тарелок. Предпочтительно, узел дистилляции, применяемый на стадии (iv), эксплуатируют при верхнем давлении от 0.5 до 2.8 бар, предпочтительно от 0.6 до 2.5 бар, более предпочтительно от 0.8 до 1.5 бар. Предпочтительно, узел дистилляции, применяемый на стадии (iv), эксплуатируют при верхней температуре в интервале от -70 до -30°C, предпочтительно от -60 до -40°C, более предпочтительно от -55 до -45°C.
Зона эпоксидирования
Согласно стадии (ii), жидкий поток поступающего материала, обеспеченный на стадии (i), подвергают условиям реакции эпоксидирования в зоне эпоксидирования, где получают реакционную смесь, содержащую пропен, пропиленоксид, воду и органический растворитель.
В общем, нет никаких конкретных ограничений в отношении конструкции зоны эпоксидирования, при условии, что она является подходящей для осуществления, предпочтительно непрерывной, реакции эпоксидирования. Предпочтительно, зона эпоксидирования согласно стадии (ii) содержит одну или более подзон эпоксидирования, где данная подзона эпоксидирования предпочтительно состоит из одного или более реакторов эпоксидирования, где, в отношении конструкции одного или более реакторов эпоксидирования, нет никаких конкретных ограничений при условии, что реакторы являются подходящими для осуществления непрерывной реакции эпоксидирования.
Предпочтительно, зона эпоксидирования согласно стадии (ii) содержит первую подзону эпоксидирования, состоящую из одного или более реакторов эпоксидирования A. Термин "первая подзона эпоксидирования", как применяется в контексте настоящего изобретения, относится к подзоне эпоксидирования, в которую жидкий поток поступающего материала, обеспеченный на стадии (i), пропускают, где зона эпоксидирования согласно (ii) может содержать дополнительные подзоны эпоксидирования, которые расположены ниже по ходу потока от первой подзоны эпоксидирования. Если первая подзона эпоксидирования состоит из двух или более реакторов эпоксидирования A, предпочтительно два или более реактора эпоксидирования A расположены параллельным образом. В этом случае предпочтительно, что на стадии (ii), жидкий поток поступающего материала, обеспеченный на стадии (i), пропускают в по меньшей мере один из реакторов эпоксидирования A. Возможно, например, что, в то время как жидкий поток поступающего материала, обеспеченный на стадии (i), пропускают в по меньшей мере один из реакторов эпоксидирования A, по меньшей мере один из реакторов A выводят из работы, например в целях поддержания и/или для регенерации катализатора, содержащегося в по меньшей мере одном из реакторов A. Если первая подзона эпоксидирования содержит два или более реакторов эпоксидирования A, действующие реакторы работают по существу одинаково, так что в каждом работающем реакторе эпоксидирования А заданные условия эпоксидирования находятся в одинаковом диапазоне в каждом реакторе.
Условия эпоксидирования согласно стадии (ii) содержат температуру эпоксидирования TN, где TN представляет собой температуру теплопередающей среды, применяемой для установления температуры реакционной смеси в первой подзоне реакции эпоксидирования согласно стадии (ii), где предпочтительно указанную температуру регулируют путем пропускания теплопередающей среды через рубашку одного или более реакторов эпоксидирования A, где TN предпочтительно представляет собой температуру теплопередающей среды перед установлением температуры реакционной среды, предпочтительно температуру теплопередающей среды на входе в рубашку одного или более реакторов эпоксидирования A. Если первая подзона эпоксидирования содержит два или более реакторов эпоксидирования A, температура эпоксидирования TN относится к температуре эпоксидирования TN данного работающего реактора A первой подзоны эпоксидирования.
Предпочтительно условия эпоксидирования согласно стадии (ii) содержат первое давление реакции эпоксидирования в интервале от 14 до 100 бар, более предпочтительно в интервале от 15 до 32 бар, более предпочтительно в интервале от 15 до 25 бар. Первое давление реакции эпоксидирования определяется как абсолютное давление на выходе из первой подзоны эпоксидирования. Если первая подзона эпоксидирования содержит два или более реактора эпоксидирования A, первое давление реакции эпоксидирования относится к абсолютному давлению на выходе из данного работающего реактора эпоксидирования A первой подзоны эпоксидирования.
Согласно первому предпочтительному варианту выполнения настоящего изобретения, зона эпоксидирования согласно стадии (ii) состоит из первой подзоны эпоксидирования.
Согласно второму предпочтительному варианту выполнения настоящего изобретения, зона эпоксидирования согласно стадии (ii) дополнительно содержит вторую подзону эпоксидирования, состоящую из одного или более реакторов эпоксидирования B, где, если вторая подзона эпоксидирования содержит два или более реакторов эпоксидирования B, два или более реактора эпоксидирования B расположены параллельно, где вторая подзона эпоксидирования расположена ниже по ходу потока от первой подзоны эпоксидирования. В этом случае, предпочтительно, что на стадии (ii), вытекающий поток, полученный из первой подзоны эпоксидирования, необязательно после подходящей промежуточной обработки, пропускают в по меньшей мере один из реакторов эпоксидирования B. Возможно, например, что, в то время как вытекающий поток, полученный из первой подзоны эпоксидирования, необязательно после подходящей промежуточной обработки, пропускают в по меньшей мере один из реакторов эпоксидирования B, по меньшей мере один из реакторов B выводят из работы, например в целях поддержания и/или для регенерации катализатора, содержащегося в по меньшей мере одном из реакторов B. Если втора подзона эпоксидирования содержит два или более реактора эпоксидирования B, действующие реакторы работают по существу одинаково, так что в каждом работающем реакторе эпоксидирования B заданные условия эпоксидирования находятся в одинаковом диапазоне в каждом реакторе. В общем, возможно, что в дополнение к первой подзоне эпоксидирования и второй подзоне эпоксидирования, зона эпоксидирования согласно стадии (ii) содержит по меньшей мере одну дополнительную подзону эпоксидирования, расположенную ниже по ходу потока от второй подзоны эпоксидирования. Предпочтительно, согласно второму предпочтительному варианту выполнения настоящего изобретения, зона эпоксидирования согласно стадии (ii) состоит из первой подзоны эпоксидирования и второй подзоны эпоксидирования.
Предпочтительно, условия эпоксидирования согласно стадии (ii) содержат второе давление реакции эпоксидирования в интервале от 14 до 100 бар, предпочтительно в интервале от 14.5 до 32 бар, более предпочтительно в интервале от 15 до 25 бар. Второе давление реакции эпоксидирования определяется как абсолютное давление на выходе из второй подзоны эпоксидирования. Если вторая подзона эпоксидирования содержит два или более реакторов эпоксидирования B, второе давление реакции эпоксидирования относится к абсолютному давлению на выходе из данного работающего реактора B второй подзоны эпоксидирования.
Предпочтительно, условия эпоксидирования согласно стадии (ii) содержат загрузку катализатора эпоксидирования во второй подзоне эпоксидирования в интервале от 0.001 до 0.5 ч-1, более предпочтительно в интервале от 0.005 до 0.3 ч-1, более предпочтительно в интервале от 0.01 до 0.2 ч-1, где загрузка катализатора эпоксидирования определяется как соотношение массового расхода в кг/ч пероксида водорода, содержащегося в потоке поступающего материала, пропускаемом во вторую подзону эпоксидирования, относительно количества в кг катализатора эпоксидирования, содержащего цеолит на основе титана, содержащегося во второй подзоне эпоксидирования согласно стадии (ii).
Предпочтительно, температура реакционной смеси во второй подзоне реакции эпоксидирования не регулируется посредством пропускания теплопередающей среды через рубашку одного или более реакторов эпоксидирования B. Более предпочтительно, вторая подзона эпоксидирования представляет собой по существу адиабатическую подзону эпоксидирования. Более предпочтительно, втора подзона эпоксидирования представляет собой адиабатическую подзону эпоксидирования.
Поток, обеспеченный на стадии (i), содержит органический растворитель, который предпочтительно представляет собой органический растворитель эпоксидирования, более предпочтительно один или более из метанола, ацетонитрила, трет.-бутанола, пропионитрила, более предпочтительно один или более из метанола, ацетонитрила.
Добавления к вытекающему потоку и катализатор эпоксидирования
Предпочтительно, поток, обеспеченный на стадии (i), необязательно реакционная смесь, полученная на стадии (ii), и необязательно вытекающий поток, удаленный на стадии (iii), дополнительно содержат по меньшей мере одну соль калия, где по меньшей мере одну соль калия выбирают из группы, состоящей из по меньшей мере одной неорганической соли калия, по меньшей мере одной органической соли калия, и комбинаций по меньшей мере одной неорганической соли калия и по меньшей мере одной органической соли калия.
Предпочтительно, по меньшей мере одну соль калия выбирают из группы, состоящей из по меньшей мере одной неорганической соли калия, выбранной из группы, состоящей из гидроксида калия, галогенидов калия, нитрата калия, сульфата калия, гидросульфата калия, перхлората калия, калиевых солей фосфорной оксикислоты, по меньшей мере одной органической соли калия, выбранной из группы, состоящей из калиевых солей алифатических насыщенных монокарбоновых кислот, предпочтительно имеющих 1, 2, 3, 4, 5 или 6 атомов углерода, карбоната калия и гидрокарбоната калия, и комбинации по меньшей мере о одной неорганической калиевой соли и по меньшей мере одной органической калиевой соли.
Более предпочтительно, по меньшей мере одну калиевую соль выбирают из группы, состоящей из по меньшей мере одной неорганической калиевой соли, выбранной из группы, состоящей из гидроксида калия, хлорида калия, нитрата калия, гидрофосфата калия, дигидрофосфата калия, по меньшей мере одной органической калиевой соли, выбранной из группы, состоящей из формиата калия, ацетата калия, карбоната калия, и гидрокарбоната калия, и комбинации по меньшей мере одной неорганической калиевой соли и по меньшей мере одной органической калиевой соли. Более предпочтительно, по меньшей мере одна калиевая соль содержит по меньшей мере одно из дигидрофосфата калия, дикалия гидрофосфата или формиатп калия.
Цеолит на основе титана, содержащийся в катализаторе эпоксидирования, предпочтительно представляет собой цеолит на основе титана, имеющий каркасную структуру типа ABW, ACO, AEI, AEL, AEN, AET, AFG, AFI, AFN, AFO, AFR, AFS, AFT, AFX, AFY, AHT, ANA, APC, APD, AST, ASV, ATN, ATO, ATS, ATT, ATV, AWO, AWW, BCT, BEA, BEC, BIK, BOG, BPH, BRE, CAN, CAS, CDO, CFI, CGF, CGS, CHA, CHI, CLO, CON, CZP, DAC, DDR, DFO, DFT, DOH, DON, EAB, EDI, EMT, EPI, ERI, ESV, ETR, EUO, FAU, FER, FRA, GIS, GIU, GME, GON, GOO, HEU, IFR, ISV, ITE, ITH, ITW, IWR, IWW, JBW, KFI, LAU, LEV, LIO, LOS, LOV, LTA, LTL, LTN, MAR, MAZ, MEI, MEL, MEP, MER, MMFI, MFS, MON, MOR, MSO, MTF, MTN, MTT, MTW, MWW, NAB, NAT, NEES, NON, NPO, OBW, OFF, OSI, OSO, PAR, PAU, PHI, PON, RHO, RON, RRO, RSN, RTE, RTH, RUT, RWR, RWY, SAO, SAS, SAT, SAV, SBE, SBS, SBT, SFE, SFF, SFG, SFH, SFN SFO, SGT, SOD, SSY, STF, STI, STT, TER, THO, TON, TSC, UEI, UFI, UOZ, USI, UTL, VET, VFI, VNI, VSV, WEI, WEN, YUG, ZON или смешанную структуру двух или более из этих каркасных структур, более предпочтительно цеолит на основе титана, имеющий каркасную структуру MFI, каркасную структуру MEL, каркасную структуру MWW, каркасную структуру ITQ, каркасную структуру BEA, каркасную структуру MOR, или смешанную структуру двух или более из этих каркасных структур, более предпочтительно каркасную структуру MFI или каркасную структуру MWW.
Катализатор эпоксидирования, содержащий цеолит на основе титана, может использоваться во всех возможных формах, включая порошок, микропорошок, предпочтительно распылительный порошок, или в виде формованного изделия, содержащего порошок, или в виде формованного изделия, содержащего микропорошок, предпочтительно распылительный порошок. Предпочтительно, катализатор, содержащий цеолит на основе титана, используется в виде формованного изделия, содержащего порошок или микропорошок, предпочтительно распылительный порошок, более предпочтительно в виде формованного изделия, содержащего микропорошок, предпочтительно распылительный порошок. Более предпочтительно, катализатор, содержащий цеолит на основе титана, присутствует в зоне эпоксидирования в виде формованного слоя, предпочтительно в виде катализатора с псевдоожиженным слоем или катализатора с фиксированным слоем, более предпочтительно в виде катализатора с фиксированным слоем.
В первом предпочтительном варианте выполнения настоящего изобретения, цеолит на основе титана, содержащийся в катализаторе эпоксидирования, представляет собой цеолит на основе титана, имеющий каркасную структуру MFI, предпочтительно TS-1. Предпочтительно, цеолит на основе титана, содержащийся в катализаторе эпоксидирования, представляет собой цеолит на основе титана, имеющий каркасную структуру типа MFI, предпочтительно TS-1, органический растворитель содержит метанол, и поток, обеспеченный на стадии (i), необязательно реакционная смесь, полученная на стадии (ii), и необязательно вытекающий поток, удаленный на стадии (iii), предпочтительно содержат по меньшей мере одну калиевую соль, более предпочтительно по меньшей мере одну неограническую калиевую соль, которая предпочтительно содержит по меньшей мере одно из дигидрофосфата калия или дикалия гидрофосфат.
Поэтому, настоящее изобретение также относится к способу получения пропиленоксида, включающему
(i) обеспечение потока, содержащего пропен, пероксид водорода или источник пероксида водорода, воду и органический растворитель, который содержит метанол;
(ii) пропускание жидкого потока поступающего материала, обеспеченного на стадии (i), в зону эпоксидирования, содержащую катализатор эпоксидирования, содержащий цеолит на основе титана, который имеет каркасную структуру MFI, предпочтительно TS-1, и воздействие на жидкий поток поступающего материала условиями реакции эпоксидирования в зоне эпоксидирования, с получением реакционной смеси, содержащей пропен, пропиленоксид, воду и органический растворитель;
(iii) удаление вытекающего потока из зоны эпоксидирования, причем вытекающий поток содержит пропиленоксид, воду, органический растворитель, и пропен;
(iv) отделение пропена от вытекающего потока посредством дистилляции, включающее
(iv.1) воздействие на вытекающий поток условиями дистилляции в узле дистилляции, с получением газообразного верхнего потока S0, обогащенного пропеном, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции, и жидкого нижнего потока S01, обогащенного пропиленоксидом, водой и органическим растворителем, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции;
(iv.2) возврат конденсированной части потока S0 в верхнюю часть узла дистилляции,
где поток, обеспеченный на стадии (i), необязательно реакционная смесь, полученную на стадии (ii), и необязательно вытекающий поток, удаленный на стадии (iii), предпочтительно содержат по меньшей мере одну калиевую соль, предпочтительно по меньшей мере одну неорганическую калиевую соль, которая предпочтительно содержит по меньшей мере одно или дигидрофосфата калия или дикалия гидрофосфата.
во втором предпочтительном варианте выполнения настоящего изобретения, цеолит на основе титана, предпочтительно цеолит на основе титана, имеющий каркасную структуру MWW, содержит по меньшей мере один из Al, B, Zr, V, Nb, Ta, Cr, Mo, W, Mn, Fe, Co, Ni, Zn, Ga, Ge, In, Sn, Pb, Pd, Pt, Au, предпочтительно по меньшей мере один из B, Zr, V, Nb, Ta, Cr, Mo, W, Mn, Fe, Co, Ni, Zn, Ga, Ge, In, Sn, Pb, Pd, Pt, Au, более предпочтительно Zn.
Предпочтительно, цеолит на основе титана представляет собой свободный от алюминия цеолитный материал с каркасной структурой MWW, содержащий титан, предпочтительно в количестве от 0.5 до 5 мас. %, более предпочтительно от 1 до 2 мас. %, как вычислено на основе элементарного титана и на основе содержащего титан цеолита, и содержащий цинк, предпочтительно в количестве от 0.5 до 5 мас. %, предпочтительно от 1 до 2 мас. %, как вычислено на основе элементарного цинка и на основе общей массы свежего, то есть неиспользованного содержащего титан цеолита. Термин “свободный от алюминия” в контексте настоящего изобретения относится к варианту выполнения настоящего изобретения, согласно которому содержание алюминия в цеолитном материале составляет 0.05 мас. частей на миллион самое большее, предпочтительно 0.03 мас. частей на миллион самое большее, более предпочтительно 0.02 мас. частей на миллион самое большее, на основе общей массы цеолитного материала. Значения мас. % относится к варианту выполнения настоящего изобретения, согласно которому цеолитный материал находится в сухом состоянии, предпочтительно после сушки в течение по меньшей мере десяти часов при 80°C при давлении менее 1013.25 гПа.
Более предпочтительно, цеолит на основе титана, содержащийся в катализаторе эпоксидирования, представляет собой цеолит на основе титана с каркасной структурой MWW, который предпочтительно свободен от алюминия и содержит цинк, органический растворитель представляет собой ацетонитрил, и поток, обеспеченный на стадии (i), необязательно реакционная смесь, полученная на стадии (ii), и необязательно вытекающий поток, удаленный на стадии (iii), предпочтительно содержат по меньшей мере одну калиевую соль, предпочтительно по меньшей мере одну органическую калиевую соль, которая предпочтительно содержит по меньшей мере формиат калия.
Поэтому, настоящее изобретение также относится к способу получения пропиленоксида, включающему
(i) обеспечение потока, содержащего пропен, пероксид водорода или источник пероксида водорода, воду и органический растворитель, который содержит ацетонитрил;
(ii) пропускание жидкого потока поступающего материала, обеспеченного на стадии (i), в зону эпоксидирования, содержащую катализатор эпоксидирования, содержащий цеолит на основе титана, который представляет собой цеолит на основе титана с каркасной структурой MWW, который предпочтительно свободен от алюминия и содержит цинк, и воздействие на жидкий поток поступающего материала условиями реакции эпоксидирования в зоне эпоксидирования, с получением реакционной смеси, содержащей пропен, пропиленоксид, воду и органический растворитель;
(iii) удаление вытекающего потока из зоны эпоксидирования, причем вытекающий поток содержит пропиленоксид, воду, органический растворитель, и пропен;
(iv) отделение пропена от вытекающего потока посредством дистилляции, включающее
(iv.1) воздействие на вытекающий поток условиями дистилляции в узле дистилляции, с получением газообразного верхнего потока S0, обогащенного пропеном, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции, и жидкого нижнего потока S01, обогащенного пропиленоксидом, водой и органическим растворителем, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции;
(iv.2) возврат конденсированной части потока S0 в верхнюю часть узла дистилляции,
где поток, обеспеченный на стадии (i), необязательно реакционная смесь, полученную на стадии (ii), и необязательно вытекающий поток, удаленный на стадии (iii), предпочтительно содержат по меньшей мере одну калиевую соль, предпочтительно по меньшей мере одну органическую калиевую соль, которая предпочтительно содержит по меньшей мере формиат калия.
Поток, содержащий пропен, необязательно пропан, пероксид водорода или источник пероксида водорода, воду, органический растворитель и необязательно по меньшей мере одну калиевую соль, обеспеченный на стадии (i), предпочтительно является жидким.
Поток, обеспеченный на стадии (i)
В общем, поток, содержащий пропен, пероксид водорода или источник пероксида водорода, воду и органический растворитель; может быть обеспечен на стадии (i) обычным способом. Предпочтительно, поток, содержащий пропен, пероксид водорода или источник пероксида водорода, воду и органический растворитель, обеспеченный на стадии (i),получают из двух или более потоков. Более предпочтительно, поток обеспечивают на стадии (i) посредством объединения по меньшей мере трех отдельных потоков, где первый поток содержит пероксид водорода или источник пероксида водорода, второй поток содержит пропен и необязательно пропан, и третий поток содержит органический растворитель и необязательно воду.
Предпочтительно, как уже описано выше, поток, содержащий пропен, дополнительно содержит пропан, где предпочтительно по меньшей мере 98 мас. %, более предпочтительно по меньшей мере 99 мас. %, более предпочтительно по меньшей мере 99.5 мас. %, более предпочтительно по меньшей мере 99.9 мас. % потока состоит из пропена и пропана. Предпочтительно, массовое соотношение пропена и пропана в потоке составляет по меньшей мере 7:3. Например, коммерчески доступный пропен, который может представлять собой либо пропен полимерной марки, либо пропен химической марки. Как правило, пропен полимерной марки имеет содержание пропена в интервале от 99 до 99.8 мас. % и содержание пропана в интервале от 0.2 до 1 мас. %. Пропен химической марки, как правило, имеет содержание пропена в интервале от 92 до 98 мас. % и содержание пропана в интервале от 2 до 8 мас. %. Предпочтительно, применяют поток, имеющий содержание пропена в интервале от 99 до 99.8 мас. % и содержание пропана в интервале от 0.2 до 1 мас. %.
Поток, содержащий пероксид водорода, может быть получен в соответствии любым возможным способом. Можно получить поток, содержащий пероксид водорода, посредством преобразования серной кислоты в пероксопиросерную кислоту посредством анодного окисления с одновременным выделением водорода на катоде. Гидролиз пероксопиросерной кислоты затем приводит к получению с помощью пероксомоносерной кислоты пероксида водорода и серной кислоты, которую, таким образом, получают обратно. Получение пероксида водорода из элементов также возможно. В зависимости от конкретного способа получения, поток, содержащий пероксид водорода, может представлять собой, например, водный или водный/метанольный поток пероксида водорода, предпочтительно водный поток пероксида водорода. В случае использования водного питающего потока пероксида водорода, содержимое потока по отношению к пероксида водорода, как правило, находится в интервале от 3 до 85 мас. %, предпочтительно от 25 до 75 мас. %, более предпочтительно от 30 до 50 мас. %, как например от 30 до 40 мас. % или от 35 до 45 мас. % от 40 до 50 мас. %. Предпочтительно, по меньшей мере 25 мас. %, более предпочтительно по меньшей мере 30 мас. %, более предпочтительно по меньшей мере 35 мас. % потока, содержащего пероксид водорода, состоит из воды и пероксида водорода. Предпочтительными диапазонами являются от 30 до 80 мас. % или от 35 до 75 мас. % или от 40 до 70 мас. %.
Согласно настоящему изобретению, предпочтительно использовать поток, содержащий пероксид водорода, который был получен в виде неочищенного раствора пероксида водорода посредством экстракции смеси, полученной согласно способу, известному как способ с использованием антрахинона, с помощью которого производится практически вся продукция перекиси водорода в мире (см, например, Ullmann's Encyclopedia of Industrial Chemistry, 5th edition, volume A 13 (1989) pages 443-466), который отличается тем, что используется раствор антрахинона, содержащий алкильную группу, предпочтительно, имеющую от 2 до 10 атомов углерода, более предпочтительно, по меньшей мере, 5 атомов углерода, например, 5 атомов углерода или 6 атомов углерода, и в котором используемый растворитель обычно состоит из смеси двух различных растворителей, где предпочтительно ни один из растворителей не представляет собой вещество, содержащее азот. Данный раствор антрахинона обычно называют рабочим раствором. В данном способе, пероксид водорода, который образуется в ходе способа с использованием антрахинона, обычно отделяют посредством экстракции из соответствующего рабочего раствора после цикла гидрогенизации/повторного окисления. Указанная экстракции может быть выполнена предпочтительно, по существу, с чистой водой, при этом получают неочищенный водный раствор пероксида водорода. Хотя, как правило, можно дополнительно очистить полученный таким образом неочищенный водный раствор пероксида водорода посредством перегонки, предпочтительно, согласно настоящему изобретению, использовать такой неочищенный водный раствор пероксида водорода, который не был подвергнут очистке с помощью перегонки. Кроме того, как правило, можно подвергнуть неочищенный водный раствор пероксида водорода дальнейшей стадии экстракции, которая отличается тем, что используется подходящий экстракционный агент, предпочтительно органический растворитель. Более предпочтительно, органический растворитель, используемый для данной дополнительной стадии экстракции, является тем же самым растворителем, который используют в способе на основе антрахинона. Предпочтительно, экстракцию проводили с использованием только одного из растворителей в рабочем растворе и наиболее предпочтительно с использованием только наиболее неполярного растворителя рабочего раствора. В случае, если неочищенный водный раствор пероксида водорода подвергают такой дополнительной стадии экстракции, получают так называемый неочищенный промытый раствор пероксида водорода. В соответствии с предпочтительным вариантом осуществления настоящего изобретения, неочищенный промытый раствор пероксида водорода используется в качестве питающего потока пероксида водорода. Получение неочищенного раствора описывается, например, в европейской патентной заявке ЕР 1.122.249 А1. Что касается термина "по существу чистой воды", сделана ссылка на параграф 10, стр. 3 заявки ЕР 1.122.249 А1, которая включена в виде ссылки. Пероксид водорода также может быть обработан для удаления следов металла, например, как описано в WO 2015/049327 A1, перед применением.
Возможно, что пероксид водорода получают in situ в зоне эпоксидирования из водорода и кислорода, предпочтительно в присутствии подходящего катализатора на основе благородного металла, содержащегося в зоне эпоксидирования согласно стадии (b). Подходящий катализатор на основе благородного металла предпочтительно содержит один или более из палладия, платины, серебра, золота, родия, иридия, рутения и осмия. Предпочтительно, катализатор на основе благородного металла содержит палладий. Катализатор на основе благородного металла предпочтительно нанесен на носитель, где носитель предпочтительно содержит один или более из SiO2, Al2O3, B2O3, GeO2, Ga2O3, ZrO2, TiO2, MgO, углерода и один или более из цеолитов, предпочтительно один или более из цеолитов на основе титанов. Более предпочтительно, носитель содержит катализатор эпоксидирования, содержащий цеолит на основе титана. Если пероксид водорода получают в зоне эпоксидирования согласно стадии (b) in situ из водорода и кислорода, поток, обеспеченный на стадии (a), содержит пропен и предпочтительно пропан, водород, кислород, воду и ацетонитрил.
Сброс давления
Согласно стадии (iv), пропен отделают от вытекающего потока посредством дистилляции. Предпочтительно, перед стадией (iv), вытекающий поток, удаленный согласно стадии (iii), подвергают сбросу давления, предпочтительно до давления от 0.5 до 2.8 бар, более предпочтительно от 0.6 до 2.5 бар, более предпочтительно от 0.8 до 1.5 бар. В общем, нет никаких конкретных ограничений в отношении того как подвергают сбросу давления вытекающий поток. Предпочтительно, вытекающий поток подвергают сбросу давления в испарительный барабан. Предпочтительно, в результате сброса давления вытекающего потока получают газообразный поток и жидкий поток, где газообразный и жидкий потоки предпочтительно пропускают по отдельности в узел дистилляции, применяемый согласно стадии (iv), предпочтительно в одной и той же теоретической тарелке узла дистилляции, применяемой согласно стадии (iv).
Поток S0
Согласно стадии (iv.1), газообразный верхний поток S0 получают, который обогащен пропеном по сравнению с вытекающим потоком, подвергаемым условиям дистилляции. В общем, нет никаких конкретных ограничений в отношении условий, при которых S0 отводят. Предпочтительно, S0, удаленный из узла дистилляции, применяемого на стадии (iv), имеет давление от 0.5 до 2.8 бар, более предпочтительно от 0.6 до 2.5 бар, более предпочтительно от 0.8 до 1.5 бар, и температуру в интервале от -70 до -30°C, более предпочтительно от -60 до -40°C, более предпочтительно от -55 до -45°C.
В общем, состав газообразного верхнего потока S0, полученного на стадии (i), не подлежит каким-либо ограничениям, при условии что он обогащен пропеном, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции. Предпочтительно, по меньшей мере 80 об. %, более предпочтительно по меньшей мере 85 об. %, более предпочтительно по меньшей мере 89 об. % потока S0 состоит из пропена. Предпочтительно, S0 содержит самое крайнее 0.1 мас. %, более предпочтительно в интервале от 1 до 250 мас. частей на миллион пропиленоксида.
Конденсированная часть S0
В общем, нет никаких конкретных ограничений в отношении того, где конденсированную часть потока S0 возвращают согласно стадии (iv.2), при условии что это происходит в верхнюю часть узла дистилляции. Предпочтительно, конденсированную часть потока S0 возвращают в верхнюю часть узла дистилляции на стадии (iv.2) вверху узла дистилляции или внутри ректификационной секции узла дистилляции, более предпочтительно вверху узла дистилляции.
В общем, нет никаких конкретных ограничений в отношении того, как много потока S0 конденсируется и образует конденсированную часть потока S0, которую возвращают в верхнюю часть узла дистилляции согласно стадии (iv.2). В общем обычно поток S0 конденсируют полностью или конденсируют только часть потока S0. Предпочтительно, только часть S0 конденсируется. Более предпочтительно, часть потока S0, которую конденсируют, с получением конденсированной части потока S0, регулируют так, что концентрация кислорода неконденсированной части потока S0 составляет менее 10 об. %, предпочтительно менее 7 об. %, наиболее предпочтительно менее 5 об. %.
Предпочтительно, от 50 до 90 мас. %, более предпочтительно от 60 до 85 мас. %, более предпочтительно от 65 до 80 мас. %, потока S0, которые образую конденсированную часть потока S0, возвращают в верхнюю часть узла дистилляции на стадии (iv.2).
В общем, нет никаких конкретных ограничений в отношении того как получают конденсированную часть потока S0, которую возвращают в верхнюю часть узла дистилляции согласно стадии (iv.2). Предпочтительно, конденсация достигается посредством сжатия до давления в интервале от 5 до 20 бар, более предпочтительно в интервале от 10 до 19 бар, более предпочтительно в интервале от 12 до 18 бар и регулирования температуры в интервале от 20 до 50°C, более предпочтительно от 25 до 40°C, более предпочтительно от 32 до 38°C.
В общем, не требуется никакая дополнительная регулировка в отношении давления или температуры конденсированной части потока S0 перед его возвратом в узел дистилляции согласно стадии (iv.2) Таким образом, возможно, что конденсированная часть потока S0, которую возвращают в верхнюю часть узла дистилляции на стадии (iv.2), имеет температуру в интервале от 20 до 50°C, предпочтительно в интервале от 30 до 40°C, более предпочтительно в интервале от 32 до 38°C. “Конденсированная часть потока S0” относится к все еще сжатому состоянию, означающему состояние потока, подлежащего возврату, перед входом в узел дистилляции, т.e. имеющему давление в интервале от 5 до 20 бар, более предпочтительно в интервале от 10 до 19 бар, более предпочтительно в интервале от 12 до 18 бар. На входе в узел дистилляции, поток (конденсированная часть потока S0) пропускается из-за разложения, таким образом также приводя к уменьшению температуры.
Согласно предпочтительному варианту выполнения настоящего изобретения, конденсированная часть потока S0 подвергают теплообмену с потоком S0 перед возвратом в верхнюю часть узла дистилляции на стадии (iv.2).
Предпочтительно, температуру конденсированной части потока S0 уменьшают после сжатия и перед возвратом в верхнюю часть узла дистилляции на стадии (iv.2) посредством теплообмена с потоком S0 на от 35 до 80 K, предпочтительно на от 45 до 65 K, более предпочтительно на от 55 до 65 K. “Теплообмен с потоком S0” относится к потоку S0, имеющему температуру, при которой S0 удаляют из узла дистилляции, применяемого на стадии (iv). Предпочтительно, температуру конденсированной части потока S0 самое максимальное доводят до температуры потока S0 при его удалении из узла дистилляции, применяемого на стадии (iv). Более предпочтительно, температура конденсированной части потока S0 доводят самое максимальное до температуры в интервале от -70 до -30°C, более предпочтительно от -60 до -40°C, более предпочтительно от -55 до -45°C.
Согласно стадии (iv.1), вытекающий поток, удаленный на стадии (iii), воздействуют условиями дистилляции в узле дистилляции, с получением газообразного верхнего потока S0, обогащенного пропеном, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции, и жидкого нижнего потока S01, обогащенного пропиленоксидом, водой и органическим растворителем, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции.
Предпочтительно, способ дополнительно содержит в дополнение к стадиям (i), (ii), (iii) и (iv)
(v) отделение пропиленоксида от S01, с получением потока S02, предпочтительно в качестве нижнего потока, который обогащен органическим растворителем и водой по сравнению с S01.
Предпочтительно, узел дистилляции применяют для разделения на стадии (v), который предпочтительно представляет собой по меньшей мере одну дистилляционную колонну, более предпочтительно одну дистилляционную колонну, который имеет предпочтительно от 30 до 80, более предпочтительно от 40 до 60 теоретических тарелок и предпочтительно эксплуатируют при верхнем давлении от 0.2 до 2 бар, более предпочтительно от 0.4 до 1 бар и предпочтительно при нижней температуре в интервале от 40 до 80°C, предпочтительно от 60 до 70°C.
В отношении стадии (v) нет никаких конкретных ограничений. Предпочтительно, разделение осуществляют так, что по меньшей мере 95 мас. % S02 состоит из органического растворителя и воды, где предпочтительно, массовое соотношение органического растворителя и воды в потоке S02 составляет более 1:1. Предпочтительно, S02, полученный в качестве нижнего потока, содержит 100 мас. частей на миллион, предпочтительно 50 мас. частей на миллион, самое большее пропиленоксида, на основе массы S02.
Предпочтительно, на стадии (v) дополнительный поток S03 получают, предпочтительно в качестве верхнего потока, содержащий пропиленоксид и обедненный в отношении органического растворителя и воды по сравнению с S01. Более предпочтительно, поток S03, полученный на стадии (v), предпочтительно в качестве верхнего потока, содержит по меньшей мере 90 мас. %, более предпочтительно по меньшей мере 95 мас. %, более предпочтительно по меньшей мере 98 мас. % пропиленоксида.
Предпочтительно, S03 расщепляется на по меньшей мере два потока S03a и S03b, и S03a, который содержит от 70 до 90 мас. %, более предпочтительно от 80 до 85 мас. % S03, возвращают в узел дистилляции, применяемый на стадии (v), предпочтительно вверху узла дистилляции.
Возможно, что поток S03, более предпочтительно поток S03b, подвергают дальнейшим стадиям обработки, описанным в настоящей заявке далее.
Возможно, что способ дополнительно содержит, в дополнение к стадиям (i), (ii), (iii), (iv) и (v)
(vi) отделение пропиленоксида от потока S03 или потока S03b, предпочтительно от потока S03b, с получением потока пропиленоксида S04 обогащенного пропиленоксидом по сравнению с потоком S03, полученным на стадии (v).
Предпочтительно, узел дистилляции применяют для разделения на стадии (vi), который предпочтительно представляет собой по меньшей мере одну дистилляционную колонну, более предпочтительно одну дистилляционную колонну, которая имеет предпочтительно от 30 до 80, более предпочтительно от 50 до 60 теоретических тарелок и предпочтительно эксплуатируют при верхнем давлении от 0.5 до 5 бар, более предпочтительно от 2 до 4 бар и предпочтительно при нижней температуре в интервале от 50 до 90°C, предпочтительно от 65 до 75°C.
Возможно, что поток пропиленоксида S04 удаляют из узла дистилляции, применяемого на стадии (vi), в верхней части узла дистилляции, предпочтительно в качестве верхнего потока. Предпочтительно, поток пропиленоксида S04, полученный на стадии (vi), содержит по меньшей мере 99.800 мас. %, более предпочтительно по меньшей мере 99.990 мас. %, более предпочтительно по меньшей мере 99.995 мас. %, более предпочтительно по меньшей мере 99.998 мас. %, пропиленоксида.
Возможно, что на стадии (vi) дополнительный поток S05 получают, предпочтительно в качестве нижнего потока, который обогащен органическим растворителем и водой по сравнению с S02 и который предпочтительно содержит 50 мас. частей на миллион самое большее пропиленоксида, на основе массы S05.
Способ получения пропиленоксида, в частности обеспечивая поток, содержащий пропен, пероксид водорода или источник пероксида водорода, воду и органический растворитель на стадии (i), пропуская жидкий поток поступающего материала, обеспеченный на стадии (i), в зону эпоксидирования, содержащую катализатор эпоксидирования, содержащий цеолит на основе титана, и воздействия на жидкий поток поступающего материала условиями реакции эпоксидирования в зоне эпоксидирования, с получением реакционной смеси, содержащей пропен, пропиленоксид, воду и органический растворитель, является непрерывным.
Настоящее изобретение далее проиллюстрировано следующими вариантами выполнения настоящего изобретения и комбинациями вариантов выполнения настоящего изобретения посредством приведенных зависимостей и обратных ссылок.
1. Способ получения пропиленоксида, включающий
(i) обеспечение потока, содержащего пропен, пероксид водорода или источник пероксида водорода, воду и органический растворитель;
(ii) пропускание жидкого потока поступающего материала, обеспеченного на стадии (i), в зону эпоксидирования, содержащую катализатор эпоксидирования, содержащий цеолит на основе титана, и воздействие на жидкий поток поступающего материала условиями реакции эпоксидирования в зоне эпоксидирования, с получением реакционной смеси, содержащей пропен, пропиленоксид, воду и органический растворитель;
(iii) удаление вытекающего потока из зоны эпоксидирования, причем вытекающий поток содержит пропиленоксид, воду, органический растворитель и пропен;
(iv) отделение пропена от вытекающего потока посредством дистилляции, включающее
(iv.1) воздействие на вытекающий поток условиями дистилляции в узле дистилляции, с получением газообразного верхнего потока S0, обогащенного пропеном, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции, и жидкого нижнего потока S01, обогащенного пропиленоксидом, водой и органическим растворителем, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции;
(iv.2) возврат конденсированной части потока S0 в верхнюю часть узла дистилляции.
2. Способ согласно варианту выполнения настоящего изобретения 1, включающий
(i) обеспечение потока, содержащего пропен, пропан, пероксид водорода или источник пероксида водорода, воду и органический растворитель;
(ii) пропускание жидкого потока поступающего материала, обеспеченного на стадии (i), в зону эпоксидирования, содержащую катализатор эпоксидирования, содержащий цеолит на основе титана, и воздействие на жидкий поток поступающего материала условиями реакции эпоксидирования в зоне эпоксидирования, с получением реакционной смеси, содержащей пропен, пропан, пропиленоксид, воду и органический растворитель;
(iii) удаление вытекающего потока из зоны эпоксидирования, причем вытекающий поток содержит пропиленоксид, воду, органический растворитель, пропен, и пропан;
(iv) отделение пропена и пропана от вытекающего потока посредством дистилляции, включающее
(iv.1) воздействие на вытекающий поток условиями дистилляции в узле дистилляции, с получением газообразного верхнего потока S0, обогащенного пропеном и пропаном по сравнению с вытекающим потоком, подвергаемым условиям дистилляции, и жидкого нижнего потока S01, обогащенного пропиленоксидом, водой и органическим растворителем по сравнению с вытекающим потоком, подвергаемым условиям дистилляции;
(iv.2) возврат конденсированной части потока S0 в верхнюю часть узла дистилляции.
3. Способ согласно варианту выполнения настоящего изобретения 1 или 2, где по меньшей мере 95 мас. %, предпочтительно от 95 до 100 мас. %, более предпочтительно от 98 до 100 мас. %, более предпочтительно от 99 до 100 мас. %, вытекающего потока, удаленного на стадии (iii), состоит из пропиленоксида, органического растворителя, воды, пропена, кислорода и необязательно пропана.
4. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 3, где вытекающий поток, удаленный на стадии (iii), содержит пропиленоксид в количестве от 5 до 20 мас. %, предпочтительно от 8 до 18 мас. %, более предпочтительно от 10 до 14 мас. %, на основе общей массы вытекающего потока; органический растворитель в количестве от 60 до 75 мас. %, предпочтительно от 65 до 70 мас. %, на основе общей массы вытекающего потока; воду в количестве от 10 до 25 мас. %, предпочтительно от 15 до 20 мас. %, на основе общей массы вытекающего потока; пропен в количестве от 1 до 5 мас. %, предпочтительно от 3 до 4.5 мас. %, на основе общей массы вытекающего потока; кислород в количестве от 0.05 до 1 мас. %, предпочтительно от 0.1 до 0.5 мас. %, на основе общей массы вытекающего потока; и необязательно пропан в количестве от 0.1 до 2 мас. %, предпочтительно от 0.2 до 1 мас. %, на основе общей массы вытекающего потока.
4. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 3, где узел дистилляции, применяемый на стадии (iv), представляет собой по меньшей мере одну дистилляционную колонну, предпочтительно одну дистилляционную колонну, где дистилляционная колонна имеет от 3 до 50, предпочтительно от 5 до 15, более предпочтительно от 6 до 10, более предпочтительно от 7 до 9, теоретических тарелок.
5. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 4, где ректификационная секция узла дистилляции состоит из от 50 до 75 %, предпочтительно от 60 до 65 %, теоретических тарелок, и стриппинг-секция узла дистилляции состоит из от 25 до 50 %, предпочтительно от 35 до 40 %, теоретических тарелок.
6. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 5, где узел дистилляции, применяемый на стадии (iv), эксплуатируют при верхнем давлении от 0.5 до 2.8 бар, предпочтительно от 0.6 до 2.5 бар, более предпочтительно от 0.8 до 1.5 бар.
7. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 6, где узел дистилляции, применяемый на стадии (iv), эксплуатируют при верхней температуре в интервале от -70 до -30°C, предпочтительно от -60 до -40°C, более предпочтительно от -55 до -45°C.
8. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 7, где перед стадией (iv), вытекающий поток, удаленный согласно стадии (iii), подвергают сбросу давления.
9. Способ согласно варианту выполнения настоящего изобретения 8, где в результате сброса давления вытекающего потока получают газообразный поток и жидкий поток.
10. Способ согласно варианту выполнения настоящего изобретения 9, где газообразный и жидкий потоки пропускают по отдельности узел дистилляции, применяемый согласно стадии (iv), предпочтительно в одной и той же теоретической тарелке узла дистилляции, применяемого согласно стадии (iv).
11. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 10, где поток, содержащий пропен, пероксид водорода или источник пероксида водорода, воду и органический растворитель, обеспеченный на стадии (i), получают из двух или более потоков.
12. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 11, где S0, удаленный из узла дистилляции, применяемого на стадии (iv), имеет давление от 0.5 до 2.8 бар, предпочтительно от 0.6 до 2.5 бар, более предпочтительно от 0.8 до 1.5 бар и температуру в интервале от -70 до -30°C, предпочтительно от -60 до -40°C, более предпочтительно от -55 до -45°C.
13. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 12, где по меньшей мере 80 об. %, более предпочтительно по меньшей мере 85 об. %, более предпочтительно по меньшей мере 89 об. % потока S0 состоит из пропена.
14. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 13, где часть потока S0, которую конденсируют, с получением конденсированной части потока S0, регулируют так, что концентрация кислорода неконденсированной части потока S0 составляет менее 10 об. %, предпочтительно менее 7 об. %, наиболее предпочтительно менее 5 об. %.
15. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 14, где конденсация достигается посредством сжатия до давления в интервале от 5 до 20 бар, предпочтительно в интервале от 10 до 19 бар, более предпочтительно в интервале от 12 до 18 бар и регулирования температуры в интервале от 20 до 50°C, предпочтительно от 25 до 40°C, более предпочтительно от 32 до 38°C.
16. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 15, где от 50 до 90 мас. %, предпочтительно от 60 до 85 мас. %, более предпочтительно от 65 до 80 мас. %, потока S0, которые образую конденсированную часть потока S0, возвращают в верхнюю часть узла дистилляции на стадии (iv.2).
17. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 16, где конденсированную часть потока S0 возвращают в верхнюю часть узла дистилляции на стадии (iv.2) вверху узла дистилляции или внутри ректификационной секции узла дистилляции, предпочтительно вверху узла дистилляции.
18. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 17, где конденсированная часть потока S0, которую возвращают в верхнюю часть узла дистилляции на стадии (iv.2), имеет температуру в интервале от 20 до 50°C, предпочтительно в интервале от 30 до 40°C, более предпочтительно в интервале от 32 до 38°C.
19. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 18, где конденсированная часть потока S0 подвергают теплообмену с потоком S0 перед возвратом в верхнюю часть узла дистилляции на стадии (iv.2).
20. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 19, где температуру конденсированной части потока S0 уменьшают после сжатия и перед возвратом в верхнюю часть узла дистилляции на стадии (iv.2) на от 35 до 80 K, предпочтительно на от 45 до 65 K, более предпочтительно на от 55 до 65 K.
21. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 20, где поток, обеспеченный на стадии (i), необязательно реакционная смесь, полученная на стадии (ii), и необязательно вытекающий поток, удаленный на стадии (iii), дополнительно содержат по меньшей мере одну калиевую соль, где по меньшей мере одну калиевую соль выбирают из группы, состоящей из по меньшей мере одной неорганической калиевой соли, по меньшей мере одной органической калиевой соли и комбинаций по меньшей мере одной неорганической калиевой соли и по меньшей мере одной органической калиевой соли.
22. Способ согласно варианту выполнения настоящего изобретения 21, где по меньшей мере одну калиевую соль выбирают из группы, состоящей из по меньшей мере одной неорганической калиевой соли, выбранной из группы, состоящей из гидроксида калия, галогенидов калия, нитрата калия, сульфата калия, гидросульфата калия, перхлората калия, калиевых солей фосфорной оксикислоты, по меньшей мере одной органической соли калия, выбранной из группы, состоящей из калиевых солей алифатических насыщенных монокарбоновых кислот, предпочтительно имеющих 1, 2, 3, 4, 5 или 6 атомов углерода, карбоната калия и гидрокарбоната калия, и комбинации по меньшей мере одной неорганической калиевой соли и по меньшей мере одной органической калиевой соли.
23. Способ согласно варианту выполнения настоящего изобретения 21 или 22, где по меньшей мере одну калиевую соль выбирают из группы, состоящей из по меньшей мере одной неорганической калиевой соли, выбранной из группы, состоящей из гидроксида калия, хлорида калия, нитрата калия, гидрофосфата калия, дигидрофосфата калия, по меньшей мере одной органической калиевой соли, выбранной из группы, состоящей из формиата калия, ацетата калия, карбоната калия и гидрокарбоната калия, и комбинации по меньшей мере одной неорганической калиевой соли и по меньшей мере одной органической калиевой соли.
24. Способ согласно любому из вариантов выполнения настоящего изобретения 21 - 23, где по меньшей мере одна калиевая соль содержит по меньшей мере одно из дигидрофосфата калия, дикалия гидрофосфат или формиата калия.
25. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 24, где органический растворитель представляет собой органический растворитель эпоксидирования, и представляет собой предпочтительно одно или более из метанола, ацетонитрила, трет-бутанола, пропионитрила, более предпочтительно одно или более из метанола, ацетонитрила.
26. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 25, где цеолит на основе титана, содержащийся в катализаторе эпоксидирования представляет собой цеолит на основе титана, имеющий каркасную структуру типа ABW, ACO, AEI, AEL, AEN, AET, AFG, AFI, AFN, AFO, AFR, AFS, AFT, AFX, AFY, AHT, ANA, APC, APD, AST, ASV, ATN, ATO, ATS, ATT, ATV, AWO, AWW, BCT, BEA, BEC, BIK, BOG, BPH, BRE, CAN, CAS, CDO, CFI, CGF, CGS, CHA, CHI, CLO, CON, CZP, DAC, DDR, DFO, DFT, DOH, DON, EAB, EDI, EMT, EPI, ERI, ESV, ETR, EUO, FAU, FER, FRA, GIS, GIU, GME, GON, GOO, HEU, IFR, ISV, ITE, ITH, ITW, IWR, IWW, JBW, KFI, LAU, LEV, LIO, LOS, LOV, LTA, LTL, LTN, MAR, MAZ, MEI, MEL, MEP, MER, MMFI, MFS, MON, MOR, MSO, MTF, MTN, MTT, MTW, MWW, NAB, NAT, NEES, NON, NPO, OBW, OFF, OSI, OSO, PAR, PAU, PHI, PON, RHO, RON, RRO, RSN, RTE, RTH, RUT, RWR, RWY, SAO, SAS, SAT, SAV, SBE, SBS, SBT, SFE, SFF, SFG, SFH, SFN SFO, SGT, SOD, SSY, STF, STI, STT, TER, THO, TON, TSC, UEI, UFI, UOZ, USI, UTL, VET, VFI, VNI, VSV, WEI, WEN, YUG, ZON или смешанную структуру двух или более из этих каркасных структур, более предпочтительно цеолит на основе титана, имеющий каркасную структуру MFI, каркасную структуру MEL, каркасную структуру MWW, каркасную структуру ITQ, каркасную структуру BEA, каркасную структуру MOR, или смешанную структуру двух или более из этих каркасных структур, более предпочтительно каркасную структуру MFI или каркасную структуру MWW.
27. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 26, где цеолит на основе титана, содержащийся в катализаторе эпоксидирования представляет собой цеолит на основе титана с каркасной структурой MFI, предпочтительно TS-1.
28. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 27, где цеолит на основе титана, содержащийся в катализаторе эпоксидирования, представляет собой цеолит на основе титана, имеющий каркасную структуру типа MFI, предпочтительно TS-1, растворитель эпоксидирования содержит метанол, и поток, обеспеченный на стадии (i), необязательно реакционная смесь, полученная на стадии (ii), и необязательно вытекающий поток, удаленный на стадии (iii), предпочтительно содержат по меньшей мере одну калиевую соль, предпочтительно по меньшей мере одну неорганическую калиевую соль, которая предпочтительно содержит по меньшей мере один из дигидрофосфата калия или дикалия гидрофосфата.
29. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 26, где цеолит на основе титана, предпочтительно цеолит на основе титана, имеющий каркасную структуру MWW, содержит по меньшей мере один из Al, B, Zr, V, Nb, Ta, Cr, Mo, W, Mn, Fe, Co, Ni, Zn, Ga, Ge, In, Sn, Pb, Pd, Pt, Au, предпочтительно по меньшей мере один из B, Zr, V, Nb, Ta, Cr, Mo, W, Mn, Fe, Co, Ni, Zn, Ga, Ge, In, Sn, Pb, Pd, Pt, Au, более предпочтительно Zn.
30. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 26 или 29, где цеолит на основе титана представляет собой свободный от алюминия цеолитный материал с каркасной структурой MWW, содержащий титан, предпочтительно в количестве от 0.5 до 5 мас. %, более предпочтительно от 1 до 2 мас. %, как вычислено на основании элементарного титана и на основе общей массы цеолита, содержащего титан, и содержащий цинк, предпочтительно в количестве от 0.5 до 5 мас. %, предпочтительно от 1 до 2 мас. %, как вычислено на основании элементарного цинка и на основе общей массы цеолита, содержащего титан.
31. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 26 или 29 - 30, где цеолит на основе титана, содержащийся в катализаторе эпоксидирования, представляет собой цеолит на основе титана с каркасной структурой MWW, предпочтительно свободный от алюминия и содержащий цинк, органический растворитель содержит ацетонитрил, и поток, обеспеченный на стадии (i), необязательно реакционная смесь, полученная на стадии (ii), и необязательно вытекающий поток, удаленный на стадии (iii), предпочтительно содержат по меньшей мере одну калиевую соль, предпочтительно по меньшей мере одну органическую калиевую соль, которая предпочтительно содержит по меньшей мере формиат калия.
32. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 31, где поток, содержащий пропен, необязательно пропан, пероксид водорода или источник пероксида водорода, воду и органический растворитель эпоксидирования и необязательно по меньшей мере одну калиевую соль, обеспеченный на стадии (i), является жидким.
33. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 32, в дополнение к стадиям (i), (ii), (iii) и (iv), дополнительно содержит
(v) отделение пропиленоксида от S01, с получением потока S02, предпочтительно в качестве нижнего потока, который обогащен органическим растворителем и водой по сравнению с S01.
34. Способ согласно варианту выполнения настоящего изобретения 33, где узел дистилляции применяют для разделения на стадии (v), который предпочтительно представляет собой по меньшей мере одну дистилляционную колонну, более предпочтительно одну дистилляционную колонну, которая имеет предпочтительно от 30 до 80, более предпочтительно от 40 до 60 теоретических тарелок и предпочтительно эксплуатируют при верхнем давлении от 0.2 до 2 бар, более предпочтительно от 0.4 до 1 бар и предпочтительно при нижней температуре в интервале от 40 до 80°C, предпочтительно от 60 до 70°C.
35. Способ согласно любому из вариантов выполнения настоящего изобретения 33 - 34, где по меньшей мере 95 мас. % потока S02 состоит из органического растворителя и воды, где предпочтительно, массовое соотношение органического растворителя и воды в потоке S02 составляет более 1:1.
36. Способ согласно любому из вариантов выполнения настоящего изобретения 33 - 36, где S02, полученный в качестве нижнего потока, содержит 100 мас. частей на миллион, предпочтительно 50 мас. частей на миллион, самое большее пропиленоксида, на основе массы S02.
37. Способ согласно любому из вариантов выполнения настоящего изобретения 33 - 36, где на стадии (v) дополнительный поток S03 получают, предпочтительно в качестве верхнего потока, содержащий пропиленоксид и обедненный в отношении органического растворителя и воды по сравнению с S01.
38. Способ согласно варианту выполнения настоящего изобретения 37, где поток S03, полученный на стадии (v), предпочтительно в качестве верхнего потока, содержит по меньшей мере 90 мас. %, более предпочтительно по меньшей мере 95 мас. %, более предпочтительно по меньшей мере 98 мас. % пропиленоксида.
39. Способ согласно варианту выполнения настоящего изобретения 37 или 38, где S03 расщепляется на по меньшей мере два потока S03a и S03b, и S03a, который содержит от 70 до 90 мас. %, более предпочтительно от 80 до 85 мас. % of S03, возвращают в узел дистилляции, применяемый на стадии (v), предпочтительно вверху узла дистилляции.
40. Способ согласно любому из вариантов выполнения настоящего изобретения 37 - 39, в дополнение к стадиям (i), (ii), (iii), (iv) и (v), дополнительно содержит
(vi) отделение пропиленоксида от потока S03 или потока S03b, предпочтительно от потока S03b, с получением потока пропиленоксида S04, обогащенного пропиленоксидом по сравнению с потоком S03, полученным на стадии (v).
41. Способ согласно варианту выполнения настоящего изобретения 40, где узел дистилляции применяют для разделения на стадии (vi), который предпочтительно представляет собой по меньшей мере одну дистилляционную колонну, более предпочтительно одну дистилляционную колонну, которая имеет предпочтительно от 30 до 80, более предпочтительно от 50 до 60 теоретических тарелок и предпочтительно эксплуатируют при верхнем давлении от 0.5 до 5 бар, более предпочтительно от 2 до 4 бар и предпочтительно при нижней температуре в интервале от 50 до 90°C, предпочтительно от 65 до 75°C.
42. Способ согласно варианту выполнения настоящего изобретения 40 или 41, где поток пропиленоксида S04 удаляют из узла дистилляции, применяемого на стадии (vi), в верхней части узла дистилляции, предпочтительно в качестве верхнего потока.
43. Способ согласно любому из вариантов выполнения настоящего изобретения 40 - 42, где поток пропиленоксида S04, полученный на стадии (vi), состоит из по меньшей мере 99.800 мас. %, более предпочтительно по меньшей мере 99.990 мас. %, более предпочтительно по меньшей мере 99.995 мас. %, более предпочтительно по меньшей мере 99.998 мас. %, пропиленоксида.
44. Способ согласно любому из вариантов выполнения настоящего изобретения 40 - 43, где на стадии (vi) дополнительный поток S05 получают, предпочтительно в качестве нижнего потока, который обогащен органическим растворителем и водой по сравнению с S02 и который предпочтительно содержит 50 мас. частей на миллион самое большее пропиленоксида, на основе массы S05.
45. Способ согласно любому из вариантов выполнения настоящего изобретения 1 - 44, который представляет собой непрерывный способ.
Настоящее изобретение дополнительно иллюстрируется следующими ссылочными примерами, сравнительными примерами и примерами.
Примеры
Ссылочный пример 1: Получение катализатора, содержащего цеолит, содержащий титан, имеющий тип каркасной структуры MWW
1.1 Получение борсодержащего цеолита со структурой MWW (BMWW)
В 2 м3 реактор с мешалкой сначала загрузили 470,4 кг деионизированной воды. После запуска мешалки при 70 оборотах в минуту, добавили борную кислоту (162,5 кг) и суспензию перемешивали в течение 3 часов. Затем добавили пиперидин (272,5 кг) за один раз, что вызвало повышение температуры от 28°C до 46°C. В данный раствор добавили коллоидный диоксид кремния (Ludox AS040, 392,0 кг). Затем реактор медленно нагревали до 170°C в течение 5 часов и затем выдерживали при этой температуре при перемешивании в течение 120 часов. Максимальное давление в ходе реакции составляло 9,3 бар. Затем реактор охладили до 50°C. Полученный гель имел значение рН 11,3 и вязкость 15 мПа⋅с при 20°C. Затем гель фильтровали и фильтровальный осадок промывали деионизированной водой до тех пор, пока электрическая проводимость фильтрата не составляла менее 500 мкСм/см. Фильтровальный осадок затем суспендировали в деионизированной воде и суспензию высушили распылением при температуре 235°C с использованием азота в качестве газа-носителя. Полученный белый порошок (174,3 кг) содержал 3,5 мас. % воды. Затем данный белый порошок подвергли обжигу при 650°C во вращающейся печи с выходом 138,2 кг борсодержащего цеолита структурного типа MWW (BMWW) в виде белого порошка.
1.2 Деборирование BMWW водой
В 5 м3 реактор с мешалкой загрузили 125 кг BMWW, полученного в соответствии с предыдущей стадией 1.1, и 3750 кг деионизированной воды. Реактор затем медленно нагревали до 100°C в течение 1 часа при перемешивании при 70 оборотах в минуту, а затем выдерживали при этой температуре в течение 20 часов и, наконец, охладили до температуры ниже 50°C перед тем, как подвергнуть его фильтрации. Фильтровальный осадок затем промывали деионизированной водой до тех пор, пока электрическая проводимость фильтрата не составляла менее 15 мкСм/см. Фильтровальный осадок затем сушили в течение 6 часов в потоке азота. Затем фильтровальный осадок удалили и суспендировали в 850 кг деионизированной воды. Данную суспензию затем высушили распылением при температуре 235°C с использованием азота в качестве газа-носителя. Высушенный распылением материал весил 118,5 кг и содержал 42,5 мас. % Si, 0,06 мас. % B и 0,23 мас. % С (общего органического углерода, (ТОС)).
1.3 Получение титансодержащего цеолита структурного типа MWW (TiMWW)
В 2 м3 реактор с мешалкой сначала загрузили 111,2 кг высушенного распылением материала, полученного в соответствии с предыдущей стадией 1.2. В отдельный 2 м3 реактор с мешалкой поместили 400 кг деионизированной воды. После запуска мешалки при 80 оборотах в минуту, добавили пиперидин (244,0 кг). После того, как добавление пиперидина было завершено, смесь перемешивали в течение 5 минут перед тем, как был добавлен тетрабутилортотитанат (22,4 кг). Трубку, через которую добавляли титанат, затем промыли 40 кг деионизированной воды. Затем смесь перемешивали в течение 1 часа перед добавлением в первый реактор с мешалкой, содержащий высушенный распылением порошок, при перемешивании (50 оборотов в минуту). Затем реактор нагревали до 170°C и выдерживали при этой температуре в течение 120 часов, затем охлаждали до 50°C. Максимальное давление в ходе реакции составляло 10,6 бар. Охлажденную суспензию затем фильтровали и фильтровальный осадок промывали деионизированной водой до тех пор, пока электрическая проводимость фильтрата не составляла менее 1300 мкСм/см и было достигнуто приблизительно нейтральное значение рН. Фильтровальный осадок затем сушили в потоке азота в течение 6 часов. Фильтровальный осадок, содержащий около 80 мас. % воды, непосредственно использовали для следующей стадия. Фильтровальный осадок, полученный на предыдущей стадии, и 1000 кг деионизированной воды загрузили в 2 м3 реактор с мешалкой. Затем добавили 1900 кг азотной кислоты (53 мас. % в воде) при перемешивании при 70 оборотах в минуту. Затем реактор нагревали до 100°C и выдерживали при этой температуре в течение 20 часов перед охлаждением до 50°C. Полученную суспензию затем фильтровали и фильтровальный осадок промывали деионизированной водой до тех пор, пока электрическая проводимость не составляла менее 10 мкСм/см и фильтрат не был приблизительно нейтральным. Затем фильтровальный осадок сушили в потоке азота в течение 6 часов. Данный фильтровальный осадок затем суспендировали в воде и высушили распылением при температуре 235°C с использованием азота в качестве газа-носителя. Получили 96 кг высушенного распылением порошка. Этот материал затем подвергли обжигу во вращающейся печи при температуре 650°C. Получили 84 кг титанового цеолита структурного типа MWW (TiMWW) в виде порошка, содержащего 43 мас. % Si, 2,0 мас. % Ti и 0,2 мас. % C (ТОС). Объем пор, определяемый методом ртутной порометрии в соответствии с DIN 66133, составил 7,3 мл/г, а удельная площадь поверхности по методу Брюнера - Эммета - Теллера (BET), определяемая в соответствии с DIN 66131, составила 467 м2/г.
1.4 Получение содержащего цинк TiMWW (ZnTiMWW) посредством импрегнирования
a) В сосуде, оборудованном обратным холодильником, в течение 30 минут готовили раствор 981 кг деионизированной воды и 6,0 кг дигидрата ацетата цинка. При перемешивании (40 оборотов в минуту) суспендировали 32,7 кг кальцинированного Ti-MWW материала, полученного согласно пункту 1.3 выше. Затем сосуд закрывали и включали обратный холодильник. Скорость перемешивания увеличивали до 70 оборотов в минуту.
b) В сосуде, оснащенном обратным холодильником, в течение 30 мин готовят раствор 585 кг деионизированной воды и 3,58 кг дигидрата ацетата цинка. При перемешивании (40 оборотов в минуту) суспендировали 19,5 кг кальцинированного материала Ti-MWW, полученного согласно пункту 1.3 выше. Затем сосуд закрывали и включали обратный холодильник. Скорость перемешивания увеличили до 70 оборотов в минуту.
Во всех партиях а) и b) смесь в сосуде нагревали до 100°С в течение 1 ч и выдерживали при кипячении с обратным холодильником в течение 2-х часов при перемешивании 70 оборотов в минуту. Затем смесь охлаждали в течение 2 ч до температуры ниже 50°С. Для каждой партии а) и b) охлажденную суспензию подвергали фильтрованию, а маточный раствор переносили с удалением отработавшей воды. Остаток на фильтре промывали пять раз деионизированной водой под давлением азота 2,5 бар. После последней стадии промывки остаток на фильтре высушили в потоке азота в течение 10 часов. Всего было получено 297 кг высушенного на воздухе остатка на фильтре. Полученный таким образом Zn-импрегнированный TiMWW материал (ZnTiMWW) имел содержание Si 42 мас. %, содержание Ti 1,8 мас. %, содержание Zn 1,3 мас. %.
Из 297 кг смеси остатка на фильтре, полученной выше, была приготовлена водная суспензия с деионизированной водой, причем суспензия имела содержание твердого вещества 15 мас. %. Эту суспензию подвергали распылительной сушке в башне с распылительным орошением со следующими условиями распылительной сушки:
- применяемое устройство: башня с распылительным орошением с одним соплом
- способ работы: азот напрямую
- конфигурация: осушитель - фильтр - скруббер
- доза: шланговый насос VF 10 (поставщик: Verder)
- сопло с диаметром 4 мм (поставщик: Niro)
- материал фильтра: Nomex® иглопробивной материал 10 м2
*) комнатная температура
Башня с распылительным орошением состояла из вертикально расположенного цилиндра, имеющего длину 2650 мм, диаметром 1200 мм, цилиндр которой был конически сужен у дна. Длина конуса составляла 600 мм. Во главе цилиндра располагались распыляющие средства (двухкомпонентное сопло). Высушенный распылением материал отделяли от сушильного газа в фильтре ниже по ходу потока от башня с распылительным орошением, а затем сушильный газ пропускали через скруббер. Суспензию пропускали через внутреннее отверстие сопла, и сопло для газа проходило через кольцевую щель, окружающую отверстие. Полученный таким образом высушенный распылением материал имел содержание Zn 1,4 мас. %, содержание Ti 1,7 мас. %, содержание Si 41 мас. % и содержание ТОС <0,5 мас. %. Высушенный распылением продукт затем подвергали кальцинированию в течение 2 ч при 650°C на воздухе во вращающейся печи, получая 43.8 кальцинированного высушенного распылением ZnTiMWW. Полученный таким образом кальцинированный высушенный распылением материал имел содержание Zn 1,3 мас. %, содержание Ti 1,8 мас. %, содержание Si 42,5 мас. % и содержание С <0,1 мас. %. Объемная плотность кальцинированного высушенного распылением ZnTiMWW составляла 90 г/л (грамм/литр). Мезопоры микропорошка имели средний диаметр пор (4 V/А) 20,2 нм, как определено посредством ртутной порометрии согласно DIN 66133. Макропоры микропорошка имели средний диаметр пор (4 V/A) 67,6 нм, как определено посредством ртутной порометрии согласно DIN 66133. Микропоры ZnTiMWW, содержащиеся в микропорошке, имели средний диаметр пор 1,06 нм, как определено адсорбцией азота в соответствии с DIN 66134 (метод Horward-Kawazoe). Значение Dv10 частиц микропорошка составляло 4,10 мкм. Значение Dv50 частиц микропорошка составляло 8,19 мкм. Значение Dv90 частиц микропорошка составляло 14,05 мкм. Степень кристаллизации, определяемая с помощью XRD, составляла (77 ± 10)%, средний размер кристаллов составлял 35,0 нм +/- 10%. Было обнаружено, что кристаллическая фаза имеет чистую структуру MWW. Никаких других кристаллических фаз титана, таких как анатаз, рутил или брукит, или кристаллический цинковый силикат (Zn2SiO4), такой как виллемит, не обнаружено.
1.5 Получение формованных изделий, содержащих ZnTiMWW и связующее на основе диоксида кремния
Исходя из кальцинированного высушенного распылением материала ZnTiMWW, полученного в соответствии с пунктом 1.4 выше, формованное изделие получили, высушили и кальцинировали. Для этого готовили 12 партий, каждую из 3,5 кг кальцинированного высушенного распылением материала ZnTiMWW, полученного выше, 0,226 кг Walocel™ (Walocel MW 15000 GB, Wolff Cellulosics GmbH & Co. KG, Германия), 2,188 кг Ludox® AS- 40 и 6,6 л деионизированной воды, следующим образом:
3,5 кг ZnTiMWW и 0,226 кг Walocel подвергали замешиванию на бегуне в течение 5 минут. Затем во время дальнейшего замешивания непрерывно добавляли 2,188 кг Ludox. Через 10 минут добавили 6 л деионизированной воды. Спустя еще 30 минут добавляли еще 0,6 л деионизированной воды. После общего времени 50 минут замешанная масса стала экструдируемой. После этого замешанную массу подвергали экструзии под давлением 65-80 бар, где экструдер охлаждали водой во время процесса экструзии. На партию время экструзии составляло от 15 до 20 минут. Потребляемая мощность для каждой партии в ходе экструзии составляла 2,4 А. Использовалась головка червячного пресса, позволяющая производить цилиндрические нити диаметром 1,7 мм. На выходе из головки червячного пресса нити не подвергались разрезанию по длине. Полученные таким образом нити сушили в течение 16 ч при 120°С в сушильной камере под воздухом. Всего (сумма для 12 партий) было получено 56 кг белых нитей диаметром 1,7 мм. 56 кг высушенных прядей подвергали прокаливанию во вращающейся печи при 550°С в течение 1 ч на воздухе, получая 52 кг прокаленных нитей. После этого нити просеивали (размер ячейки 1,5 мм), и выход после просеивания составлял 50,0 кг. Полученные таким образом формованные изделия имели объемную плотность 322 г/л (грамм на литр) и имели содержание Zn 1,1 мас. %, содержание Ti 1,4 мас. %, содержание Si 43 мас. % и содержание C <0,1 мас. %. Мезопоры микропорошка имели средний диаметр пор (4V/А) 20,9 нм, как определено посредством ртутной порометрии согласно DIN 66133. Макропоры микропорошка имели средний диаметр пор (4 V/A) 50,0 нм, как определено посредством ртутной порометрии согласно DIN 66133. Степень кристаллизации, определяемая с помощью XRD, составляла (70 ± 10)%, средний размер кристалла составлял 32,5 нм +/- 10%. Прочность на раздавливание формованных изделий, как определено согласно способу с использованием испытательной машины для испытаний на раздавливание Z2.5/TS01S, составляла 4,4 Н (стандартное отклонение: 0,5 Н). Минимальное значение, обнаруженное при тестировании 10 образцов, составляло 3,5 Н, максимальное значение 5,1 Н. Согласно 229Si MAS ЯМР после того, как кривая была деконволюционирована надлежащими гауссово-лоренцевыми линиями, четко наблюдалось шесть пиков. Соотношение Q3 / Q4, как было обнаружено, составляло 2.2. Общее количество адсорбированной воды, как определено согласно Ссылочному примеру 6 формованного изделия, составляло 6,9 мас. %. Поверхность по Ленгмюру определяют с помощью поглощения азота при 77 К согласно DIN 66133, и она составляла 518 м²/г, удельная площадь поверхности по БЭТ, определяемая поглощением азота при 77 K согласно DIN 66133, составляет 373 м²/г. Общий объем проникновения, определенный согласно ртутной порометрии согласно DIN 66133, составлял 1,3 мл / г (миллилитр / грамм), соответствующая общая площадь пор 100,2 м²/г. Было обнаружено, что кристаллическая фаза формованных изделий имеет чистую структуру MWW. Никакие другие фазы кристаллического титана, такие как анатаз, рутил или брукит, или кристаллический цинковый силикат (Zn2SiO4), такой как виллемит, не обнаружены с помощью XRD.
Исходя из кальцинированных нитей, стадию последующей обработки выполняли следующим образом: 1000 кг деионизированной воды заполняли в сосуд. Затем добавляли 50 кг кальцинированных формованных изделий. Судно закрывали (герметично), и полученную смесь нагревали до температуры 145°С в течение 1,5 ч и выдерживали при этой температуре при автогенном давлении (около 3 бар) в течение 8 часов. Затем смесь охлаждали в течение 2 часов. Обработанные водой нити подвергали фильтрации и промывали деионизированной водой. Полученные нити нагревали в сушильной камере под воздухом в течение 1 ч до температуры 120°C и выдерживали при этой температуре в течение 16 часов. Затем высушенный материал нагревали под воздухом до температуры 450°С в течение 5,5 ч и выдерживали при этой температуре в течение 2 часов. После этого нити просеивали (размер ячейки 1,5 мм), а выход после просеивания составлял 49,1 кг. Полученные таким образом обработанные водой формованные изделия имели объемную плотность 332 г/л (грамм на литр) и имели содержание Zn 1,1 мас. %, содержание Ti 1,4 мас. %, содержание Si 42 мас. %, и содержание С <0,10 мас. %. Мезопоры микропорошка имели средний диаметр пор (4 V/A) 22,1 нм, как определено посредством ртутной порометрии согласно DIN 66133. Макропоры микропорошка имели средний диаметр пор (4 V/A) 52,0 нм, как определено посредством ртутной порометрии согласно DIN 66133. Степень кристаллизации, определяемая с помощью XRD, составляла (69 ± 10)%, средний размер кристалла составлял 30,5 нм +/- 10%. Прочность на раздавливание формованных изделий, как определено согласно способу с использованием испытательной машины для испытаний на раздавливание Z2.5/TS01S, составляла 13,6 Н (стандартное отклонение: 2,5 Н). Минимальное значение, обнаруженное при тестировании 10 образцов, составляло 10,2 Н, максимальное значение 17,6 Н. Согласно 229Si MAS ЯМР после того, как кривая была деконволюционирована надлежащими гауссово-лоренцевыми линиями, четко наблюдалось шесть пиков. Соотношение Q3 / Q4, как было обнаружено, составляло 1.39. Общее количество адсорбированной воды формованного изделия составляло 6,9 мас. %. Соотношение интенсивности полосы инфракрасного диапазона в области (3746 +/- 20) см-1, приписываемой свободным силанольным группам, относительно полосы инфракрасного диапазона в области 3688 ± 20 см-1, приписываемой вицинальным силанольным группам, было меньше 1,4. Поверхность по Ленгмюру определяют с помощью поглощения азота при 77 К согласно DIN 66133, и она составляла 421 м²/г, удельная площадь поверхности по БЭТ, определяемая поглощением азота при 77 K согласно DIN 66133, составляет 303 м²/г. Общий объем проникновения, определенный согласно ртутной порометрии согласно DIN 66133, составлял 1,3 мл/г (миллилитр / грамм), соответствующая общая площадь пор 98.7 м²/г. Было обнаружено, что кристаллическая фаза формованных изделий имеет чистую структуру MWW. Никакие другие фазы кристаллического титана, такие как анатаз, рутил или брукит, или кристаллический цинковый силикат (Zn2SiO4), такой как виллемит, не обнаружены с помощью XRD.
Ссылочный пример 2: Характеризация катализатора
Ссылочный пример 2.1: Определение значений Dv10, Dv50 и Dv90
1,0 г микропорошка суспендировали в 100 г деионизированной воды и перемешивали в течение 1 минуты. Образец подвергли измерению в аппарате, используя следующие параметры: Mastersizer S, исполнение с удлиненной станиной 2.15, серийный № 33544-325; поставщик: Malvern Instruments GmbH, Herrenberg, Germany (Германия); фокусное расстояние - 300RF мм; длина луча - 10,00 мм; модуль - MS017; затенение - 16,9%; дисперсионная модель - 3$$D; аналитическая модель - полидисперсная, коррекция - отсутствует.
Ссылочный пример 2.2: Определение концентрации силанольных групп в формованных изделиях согласно настоящему изобретению
Для определения концентрации силанольных групп, опыты согласно методу ядерного магнитного резонанса при вращении образца под магическим углом 29Si MAS ЯМР проводили при комнатной температуре на спектрометре VARIAN Infinityplus-400 с использованием 5,0 мм ZrO2 роторов. Спектры 29Si MAS ЯМР регистрировали при 79,5 МГц с использованием 1,9 мкс π/4 (микросекунды пи/4) импульса с 10 сек. задержкой повторного цикла и 4000 сканов. Все 29Si-спектры регистрировали на образцах, с вращением при 6 кГц, а химические сдвиги были определены относительно 4,4-диметил-4-силапентан сульфоната натрия (DSS). Для определения концентрации силанольных групп, данный спектр 29Si MAS ЯМР восстанавливали из свертки соответствующих Гауссовых-Лоренцевых форм линий. Концентрацию силанольных групп по отношению к общему числу атомов Si получили посредством интегрирования восстановленных из свертки спектров 29Si MAS ЯМР.
Ссылочный пример 2.3: Определение прочности формованных изделий на раздавливание
Предел прочности на раздавливание, как указано в контексте настоящего изобретения, следует понимать как параметр, определенный с помощью испытательной установки для определения прочности на раздавливание Z2.5/TS01S, поставщик Zwick GmbH & Co., D-89079 Ulm, Germany (Германия). Что касается основных характеристик и принципов работы данной установки, ссылка делается на соответствующие руководство по эксплуатации "Register 1: Betriebsanleitung / Sicherheitshandbuch für die Material-Prüfmaschine Z2.5/TS01S ", version 1.5, December 2001 by Zwick GmbH & Co. Technische Dokumentation, August-Nagel-Strasse 11, D-89079 Ulm, Germany. С помощью указанной установки, данную заготовку подвергали воздействию возрастающего усилия с помощью плунжера, имеющего диаметр 3 мм, до тех пор, пока заготовка не была раздавлена. Усилие, при котором заготовка была раздавлена, называют прочностью заготовки на раздавливание. Установка оснащена неподвижным горизонтальным столом, на котором размещается заготовка. Плунжер, который может свободно перемещаться в вертикальном направлении, создает усилие, воздействующее на заготовку по отношению к неподвижному столу. Установка работала с предварительным усилием 0,5 Н и скоростью сдвига при предварительном усилии 10 мм/мин и последующей скоростью испытания 1,6 мм/мин. Вертикально перемещаемый плунжер был соединен с датчиком нагрузки для считывания показателей усилия и во время измерения перемещался в направлении неподвижного поворотного стола, на котором было размещено испытуемое формованное изделие (заготовка), таким образом, воздействуя на заготовку посредством ее прижимания к столу. Плунжер воздействовал на заготовки перпендикулярно их продольной оси. Управление опытом осуществлялось с помощью компьютера, который регистрировал и оценивал результаты измерений. Полученные значения представляют собой среднее значение измерений для 10 полосок в каждом случае.
Ссылочный пример 2.4: Спектры 29Si-ЯМР в твердом состоянии в отношении структур Q3 и Q4
Эффект от обработки водой согласно изобретению формованного изделия в отношении структур Q3 и Q4 в материале определяли посредством сравнения изменений в спектрах 29Si-ЯМР в твердом состоянии в сопоставимых условиях. Все опыты 29Si ЯМР твердого тела проводили с использованием спектрометра Bruker Avance при ларморовой частоте 300 МГц 1Н (Bruker BioSpin, Germany). Образцы были упакованы в 7 мм ZrO2 роторы и измерялись при вращении под магическим углом при 5 кГц при комнатной температуре. Прямые спектры поляризации 29Si были получены с использованием (пи/2)-импульсного возбуждения с 5 мкс длительностью импульса, 29Si несущей частотой, соответствующей -65 частей на миллион в спектре, и задержкой повторного цикла сканирования 120 сек. Сигнал получали за 25 мс при 45 кГц протонной развязки высокой мощности и накапливали в течении 10-17 часов. Спектры обрабатывали с использованием Bruker TopSpin с 30 Гц экспоненциальным уширением линии, ручным фазированием и ручной коррекцией базовой линии по всей ширине спектра. Спектры определяли относительно полимера Q8M8 в качестве внешнего вторичного стандарта, задав резонанс M группы триметилсилила как 12,5 частей на миллион. Спектры затем согласовывали с набором гауссовых форм линий, в соответствии с числом различаемых резонансов. Относительно текущих анализируемых спектров, были использованы 6 линий в общей сложности, что составляет пять различных максимумов пиков (приблизительно при -118, -115, -113, -110 и -104 частей на миллион) плюс четко видимое плечо при -98 частей на миллион Корректировку проводили с использованием DMFit (Massiot et al., Magnetic Resonance in Chemistry, 40 (2002) pp 70-76). Пики вручную установили на видимых максимумах пиков или плече. Затем положение пика и ширину линии оставили незафиксированными, т.е. откорректированные пики не были зафиксированы в определенном положении. Результат корректировки был численно устойчивым, то есть, искажения в начальной настройке корректировки, как описано выше, приводили к аналогичным результатам. Площади откорректированных пиков в дальнейшем использовали как нормализованные согласно корректировки посредством DMFit. После обработки водой согласно изобретению, наблюдалось уменьшение интенсивности сигнала "по левую руку" спектра, область, которая включает силанольные структуры Q3 (здесь особенно: около и выше -104 частей на миллион, т.е. "слева" от -104 частей на миллион). Кроме того, наблюдалось увеличение интенсивности сигнала "по правую руку" спектра (здесь ниже -110 частей на миллион, т.е. "справа" от -110 частей на миллион), область которого включает в себя исключительно структуры Q4. Для количественной оценки изменений спектра, было рассчитано соотношение, которое отражает изменения площадей пиков "по левую руку" и "по правую руку", следующим образом. Шесть пиков, как описано выше, были маркированы как 1, 2, 3, 4, 5 и 6, а соотношение Q было рассчитан по формуле 100 * { [a1+a2] / [a4+a5+a6] } / a3. В данной формуле ai, i=1..6 представляет собой площадь откорректированного пика, которому было присвоено данный номер.
Ссылочный пример 2.5: Адсорбция / десорбция воды - Водопоглощение
Измерения изотерм адсорбции/десорбции воды проводились с помощью прибора VTI SA от TA Instruments согласно программы ступенчатых изотерм. Опыт состоял из прогона или серии прогонов, выполненных на образце материала, который поместили в чашу микровесов внутри прибора. Перед началом измерений, из образца удалили остаточную влагу посредством нагревания образца до 100°C (скорость нагрева 5°C/минута) и выдерживали образец в течение 6 часов в потоке N2. После программы сушки, температуру в камере снизили до 25°C и выдерживали изотермический в течение измерений. Микровесы были откалиброваны и вес высушенного образца измерили (максимальное отклонение веса 0,01 мас. %). Поглощение воды образцом измеряли как увеличение веса по сравнению с весом сухого образца. Во-первых, кривую адсорбции измеряли посредством увеличения относительной влажности (RH) (выраженной в мас. % воды в атмосфере внутри камеры), воздействию которой подвергали образцы, и посредством измерения поглощения воды образцом при равновесии. Относительную влажность увеличивали с шагом 10 мас. % от 5 до 85% и на каждой стадии система контролировала значение RH и осуществляла мониторинг веса образца до достижения условий равновесия, регистрируя поглощение веса. Общий объем адсорбированной образцом воды был поглощен после того, как образец подвергли воздействию 85 мас. % RH. Во время измерения десорбции, RH снижали с 85 мас. % до 5 мас. % с шагом 10% и изменение веса образца (водопоглощения) контролировали и регистрировали.
Ссылочный пример 2.6: Измерения инфракрасной спектроскопии на основе преобразования Фурье
Измерения инфракрасной спектроскопии на основе преобразования Фурье (Фурье-ИК-спектроскопия) были выполнены с помощью спектрометра Nicolet 6700. Формованное изделие было измельчено в порошок, который затем прессовали в самоподдерживающийся осадок без использования каких-либо добавок. Осадок ввели в ячейку высокого вакуума (HV), размещенную в приборе Фурье-ИК-спектроскопии. Перед измерением образец предварительно обрабатывали в условиях высокого вакуума (10-5 мбар) в течение 3 часов при 300°C. Спектры были получены после охлаждения ячейки до 50°C. Спектры регистрировали в диапазоне от 4000 до 800 см-1 с разрешением 2 см-1. Полученные спектры представлены на графике, имеющем на оси х волновые числа (см-1) и на оси y поглощающую способность (в условных единицах, у.е.). Для количественного определения высот пиков и соотношения между этими пиками проводилась коррекция базовой линии. Изменения в области от 3000 до 3900 см-1 были проанализированы и для сравнения нескольких образцов в качестве эталона была взята полоса при 1880 ±5 см-1.
Ссылочный пример 2.7: Определение степени кристалличности с помощью рентгенодифракционного (XRD) анализа
Степень кристалличности цеолитных материалов в соответствии с настоящим изобретением определяли с помощью рентгенодифракционного (XRD) анализа. Данные собирали с помощью стандартного дифрактометра Брэгга-Брентано (Bragg-Brentano) с рентгеновской трубкой с медным анодом в качестве источника рентгеновских лучей и энергодисперсионным точечным детектором. Угловой диапазон от 2° до 70° (2 тета) сканировали с размером шага 0,02°, при этом переменная щель расходимости была задана для постоянной длины выборки 20 мм с подсветкой. Затем данные анализировали с использованием программного обеспечения TOPAS V4, с помощью которого были смоделированы острые дифракционные пики с использованием метода Паули (Pawley), содержащие элементарную ячейку со следующими исходными параметрами: а = 14,4 ангстрем (1 ангстрем = 10-10 м) и с = 25,2 ангстрем в пространственной группе P6/mmm. Они были доработаны, чтобы соответствовать данным. Независимые пики были вставлены в следующих положениях. 8.4°, 22,4°, 28,2° и 43°. Они были использованы для описания аморфного содержания. Содержание кристаллической структуры описывает интенсивность кристаллического сигнала по отношению к общей интенсивности рассеянного излучения. Модель также включала в себя линейный фон, коррекции по фактору Лоренца (Lorentz) и поляризации, параметры кристаллической решетки, пространственную группу и размер кристаллитов.
Ссылочный пример 3: Процесс эпоксидирования
Основным реактором был вертикально установленный трубчатый реактор с 3 трубами (длина труб: 12 м, внутренний диаметр трубы: 38 мм), причем каждая труба была оборудована аксиально размещенной многоточечной термопарой с 10 равноотстоящими точками измерения, заключенными в подходящую защитную гильзу диаметром 18 мм. В каждую трубу было загружено 3,2 кг формованных изделий ZnTiMWW катализатора, полученных согласно Ссылочному примеру 1 (пост-обработанные формованные изделия). Свободное пространство, в результате оставшееся, было заполненными сферами оксида алюминия высокой чистоты (Denstone ® 99, диаметр 5-6 мм). Теплота реакции удалялась путем циркуляции термостатируемой теплопередающей среды (смесь вода/гликоль) во межтрубном пространстве прямотоком по отношению к подаче. Скорость потока теплопередающей среды регулировали таким образом, чтобы разница температур между входом и выходом охлаждающей среды не превышала 1°C. Температура реакции, указанная ниже, также упоминаемая как TR, определялась как температура теплопередающей среды, поступающей в оболочку реактора. На выходе из реактора давление контролировали регулятором давления и поддерживали постоянным при 20 бар (abs). Выходящий поток, покидающий основной реактор, отбирали каждые 20 минут для определения концентрации пероксида водорода с использованием метода на основе титанилсульфата и для расчета превращения пероксида водорода. Превращение пероксида водорода определялось как 100 x (1-mout/min), где min представляет собой молярную скорость потока H2O2 при подпитке реактора, и mout представляет собой молярную скорость потока H2O2 на выходе из реактора. На основе полученных значений превращения пероксида водорода температура на входе теплопередающей среды была отрегулирована таким образом, чтобы конверсия пероксида водорода была по существу постоянной в интервале от 95 до 97%. Температура на входе теплопередающей среды была установлена при 30°C в начале данного цикла со свежей порцией катализатора эпоксидирования и при необходимости увеличивалась для поддержания превращения пероксида водорода в указанном диапазоне. Требуемое повышение температуры обычно составляло менее 0,1 К/д. Выходящий поток, выходящий из основного реактора эпоксидирования, пропускался через узел теплообмена, чтобы регулировать температуру до 40°C. Поток, выходящий из узла теплообмена, подавался в конечный реактор.
Конечный реактор представлял собой реактор с вертикальным неподвижным слоем, работавший адиабатически и при подаче снизу. В этом контексте термин «адиабатический» относится к режиму работы, согласно которому никакое активное охлаждение не проводится, и согласно которому конечный реактор соответствующим образом изолирован для минимизации потерь тепла. Конечный реактор имел длину 4 м и диаметр 100 мм. Реактор заполняли 4,5 кг того же катализатора эпоксидирования, который использовался в основном реакторе эпоксидирования. Оставшееся пространство заполнялось сферами оксида алюминия высокой чистоты (Denstone ® 99, диаметром 3 мм). Рабочее давление конечного реактора составляло 10 бар, которое поддерживалось постоянным подходящим регулятором давления на выходе из реактора. Выходящий поток из конечного реактора отбирали каждые 20 мин для определения концентрации пероксида водорода с использованием метода на основе титанилсульфата. Выходящий поток конечного реактора затем применяли в качестве потока поступающего материала для дистилляции, как описано в примерах ниже.
В основной реактор снизу подавали 101 кг / час жидкого монофазного потока следующего состава: 63,7 мас. % ацетонитрила, 12,7 мас. % пропена, 7,1 мас. % H2O2, 14,8 мас. % воды и 150 мас. частей на миллион формиата калия. Баланс был по пропану и высококипящим органическим примесям. Температуру потока поступающего материала в основной реактор доводили до около 30°С перед подачей в основной реактор.
Эпоксидирование проводилось непрерывно.
Вытекающий поток ниже по ходу потока от клапана регулирования давления конечного реактора дозировался с использованием подходящего массового расходомера, и пробы отбирались через равные промежутки времени для анализа. Органические компоненты и O2 анализировали в двух отдельных газовых хроматографах. Содержание пероксида водорода определяли колориметрическим методом с использованием титанилсульфата. Вытекающий поток содержал 63,7 мас. % ацетонитрила, 18,6 мас. % воды, 11,8 мас. % пропиленоксида, 4,0 мас. % пропена, 0,14 мас. % пропиленгликоля, 0,5 мас. % пропана, 0,03 мас. % кислорода и 0,018 мас. % H2O2. Баланс по смеси высококипящих органических побочных продуктов и примесей.
Сравнительные примеры и примеры согласно настоящему изобретению
1.1 Моделирования
Данные сравнительных примеров и примеров согласно настоящему изобретению были получены при моделировании с программным обеспечением AspenONE V8.6 (компания Aspentech). Термодинамические данные, использованные при моделировании, были взяты из Dortmunder Datenbank (http://www.ddbst.com/). Потребителями энергии считаются только компрессоры с электроприводом и паровые котлы. Затраты на электроэнергию, связанные с охлаждением воды (энергия для насосов охлаждающей воды), не учитывались, поскольку они были незначительными по сравнению с другими затратами энергии. Сравнение результатов было выполнено на основе рассчитанных значений тепловой и электрической энергии в МВт соответственно.
Сравнительный пример 1
Вытекающий поток конечного реактора, который представлял поток поступающего материала (F) для всех примеров, был постоянным в отношении массового расхода, а также состава. Для расчета взяли высококипящее соединение, состоящее исключительно из пропиленгликоля, который так или иначе является основным компонентом. Массовый расход потока F был установлен на 100 т/ч. Поток F был жидким, имел давление 1,1 бар и температуру 46.7°C.
Установка показана на Фиг. 1.
Для всех расчетов были установлены следующие граничные условия: 1) концентрация пропиленоксида в дистилляте (поток T1, S0) была установлена равной 70 мас. частей на миллион; 2) концентрация пропена в отстойнике (поток B, S01) была установлена на уровне 100 мас. частей на миллион. Поток R, используемый в качестве промывочной жидкости, содержал 76,3 мас. % ацетонитрила, 22,3 мас. % воды, остальное - пропиленгликоль. Этот поток подавали в верхнюю часть колонны в виде жидкости с температурой 10°С и скоростью потока 32,4 т/ч.
Дистилляционная колонна работали при верхнем давлении 1.0 бар и была вычислена с 8 теоретическими тарелками (включая ребойлер). Точка подачи потока F находилась на тарелке 5, считая сверху.
При этих граничных условиях, поток T1 (S01), который в основном состоял из пропена, пропана и O2, имел массовый расход 4.65 т/ч и температуру 11.3°C (= температура вверху колонны). Этот поток затем сжали до 16.7 бар и охладили до 36.6°C. Нижний поток B (S01) отбирали при температуре 69.1°C.
В этих условиях совокупная нагрузка ребойлера и электрическая мощность для сжатия увеличилась до 3.26 МВт.
Сравнительный пример 2
Граничные условия для этого примера были такими же, как для сравнительного примера 1, но в качестве промывочного потока R использовался водный поток. Этот поток содержал 98,7 мас. % воды, остальное - пропиленгликоль. Поток подавали вверху колонны при 10°C и массовом расходе 49,6 т/ч (поскольку вода является более плохим абсорбентом, для выполнения требований по разделению пришлось использовать большее количество). Верхний поток T1 (S0) имел температуру 9,7°С (= температура в верхней части колонны) и скорость потока 4,8 т/ч. Этот поток затем сжимали до 16,7 бар и охлаждали до 36,6°С. Температура потока B (S01) составляла 68,9°C. В этих условиях совокупная нагрузка ребойлера и электрическая мощность для сжатия увеличилась до 5.03 МВт.
Пример 1
Граничные условия примера 1 были такими же, как в сравнительном примере 1, за исключением того, что не использовался внешний промывочный поток. Вместо этого конденсированная часть S0, в данном случае часть сжиженного верхнего потока Т4, полученного после сжатия, охлаждения и разделения газа/жидкости, подавалась обратно в верхнюю часть колонны в виде обратного потока.
Установка схематически показана на Фиг. 2.
Температура в верхней части колонны теперь составляла -47,8°C (= температура T1, S0). Массовый расход потока T1 (S0) составил 19,0 т/ч. Он был сжат до 16,7 бар и охлажден до 35,1°С, тогда как большая часть потока конденсировалась. Полученный поток T2 был разделен на газообразный поток T3 и жидкий поток T4. Из жидкого потока T4 14,6 т/ч было возвращено в виде потока R на вершину дистилляционной колонны, где он отгонялся. Остаток потока Т4, поток Т5 был отведен.
Температура в нижней части колонны составляла теперь 67,8°C (= температура B, S01). В этих условиях совокупная нагрузка ребойлера и электрическая мощность для сжатия увеличились до 3,13 МВт и, таким образом, на 4% ниже, чем энергия, требуемая в лучшем сравнительном примере (сравнительный пример 1).
Пример 2
Пример 2 идентичен примеру 1, за исключением того, что обратный поток R1, который представляет собой часть потока T4, подлежащую возврату в колонну, был предварительно охлажден посредством теплообмена с потоком T1 (S0) на вершине колонны с получением предварительно охлажденного обратного потока R2, который образовывал конденсированную часть потока S0.
Установка схематически показана на Фиг. 3.
Температура в верхней части башни осталась такой же, как в примере 1 (= температура T1, S0), но массовый расход потока T1 (S01) теперь составлял только 13,0 т / ч. Этот поток сжимали до 16,7 бар и охлаждали до 35,1°С, тогда как большая часть потока конденсировалась (поток Т2). Из жидкого потока T4 отбирали 8,7 т/ч и предварительно охлаждали путем теплообмена с потоком T1 (S0). После теплообмена температура предварительно охлажденного обратного потока R2, который образовывал конденсированную часть потока S0, составляла -28,7°С. Этот поток был затем возвращен на вершину дистилляционной колонны, где он мелькал.
Температура на дне колонны осталась такой же, как в примере 1 (= температура B, S0). В этих условиях совокупная нагрузка ребойлера и электрическая мощность для сжатия составляли до 2,7 МВт и, таким образом, были на 16% ниже, чем энергия, требуемая в лучшем сравнительном примере (сравнительный пример 1).
Краткое описание чертежей
Фиг. 1 показывает блок-схему способа согласно сравнительному примеру 1. На Фиг. 1, буквы и цифры имеют следующие значения:
F, T1, T2, B потоки согласно особенно предпочтительному способу, как описано в примерах
S0, S1a потоки согласно особенно предпочтительному способу, как описано в общем описании и в примерах
Фиг. 2 показывает блок-схему способа согласно примеру 1. На Фиг. 2, буквы и цифры имеют следующие значения:
F, T1, T2, T3,
T4, T5, R, B потоки согласно особенно предпочтительному способу, как описано в примерах
S0, S1a потоки согласно особенно предпочтительному способу, как описано в общем описании и в примерах
Фиг. 3 показывает блок-схему способа согласно примеру 2. На Фиг. 3, буквы и цифры имеют следующие значения:
F, T1, T2, T3,
T4, T5, R1, R2, B потоки согласно особенно предпочтительному способу, как описано в примерах
S0, S1a потоки согласно особенно предпочтительному способу, как описано в общем описании и в примерах
Процитированная литература
- WO 2008/118265 A
- WO 2004/037802 A
- Ullmann's Encyclopedia of Industrial Chemistry, 5th edition, volume A 13 (1989) pages 443-466
- EP 1 122 249 A1
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОЧИСТКИ ПРОПИЛЕНОКСИДА | 2017 |
|
RU2741991C2 |
| ИЗВЛЕЧЕНИЕ ПРОПЕНА ПОСРЕДСТВОМ ОЧИСТКИ В СКРУББЕРЕ СО СМЕСЬЮ РАСТВОРИТЕЛЬ/ВОДА | 2018 |
|
RU2766954C2 |
| СПОСОБ ОЧИСТКИ ПРОПИЛЕНОКСИДА | 2017 |
|
RU2740188C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОПИЛЕНОКСИДА | 2017 |
|
RU2734823C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭТИЛЕНОКСИДА | 2017 |
|
RU2756603C2 |
| СПОСОБ РЕГЕНЕРАЦИИ КАТАЛИЗАТОРА НА ОСНОВЕ ТИТАН-СОДЕРЖАЩЕГО ЦЕОЛИТА ДЛЯ ЭПОКСИДИРОВАНИЯ ПРОПИЛЕНА | 2016 |
|
RU2702349C2 |
| ПЕРЕГОНКА С ЧАСТИЧНЫМ ПОТОКОМ | 2015 |
|
RU2705993C2 |
| ПЕРЕГОНКА С ЧАСТИЧНЫМ ПОТОКОМ | 2014 |
|
RU2665473C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОПИЛЕНОКСИДА | 2014 |
|
RU2671638C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФОРМОВАННОГО ИЗДЕЛИЯ, СОДЕРЖАЩЕГО ЦИНК И ТИТАНСОДЕРЖАЩИЙ ЦЕОЛИТ | 2018 |
|
RU2762171C2 |
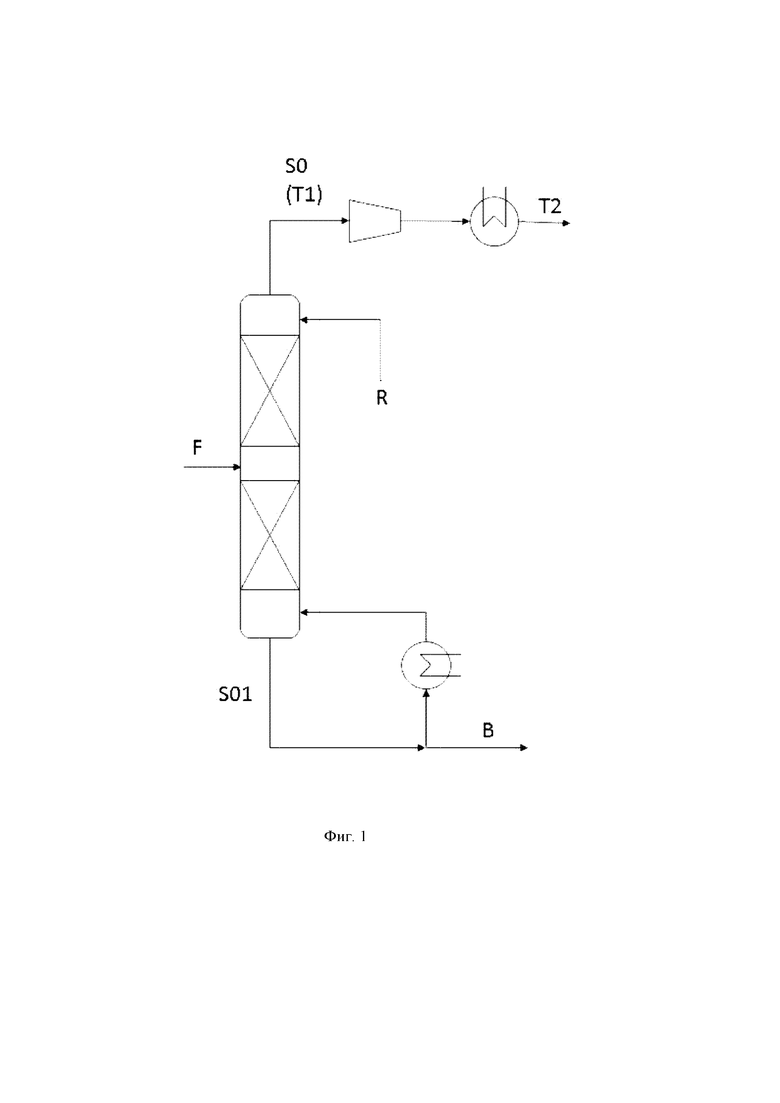

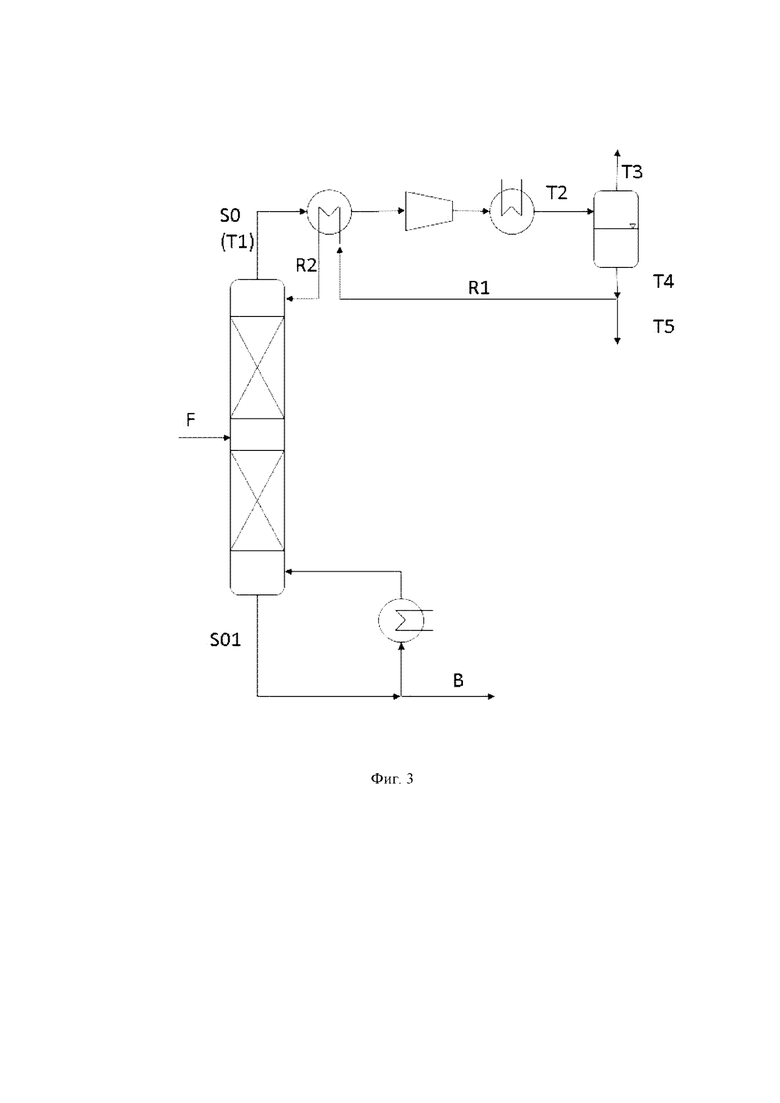
Настоящее изобретение относится к способу получения пропиленоксида, включающему: (i) обеспечение потока, содержащего пропен, пероксид водорода или источник пероксида водорода, воду и органический растворитель; (ii) пропускание жидкого потока поступающего материала, обеспеченного на стадии (i), в зону эпоксидирования, содержащую катализатор эпоксидирования, содержащий цеолит на основе титана, и воздействие на жидкий поток поступающего материала условиями реакции эпоксидирования в зоне эпоксидирования, с получением реакционной смеси, содержащей пропен, пропиленоксид, воду и органический растворитель; (iii) удаление вытекающего потока из зоны эпоксидирования, причем вытекающий поток содержит пропиленоксид, воду, органический растворитель, и пропен; (iv) отделение пропена от вытекающего потока посредством дистилляции, включающее (iv.1) воздействие на вытекающий поток условиями дистилляции в узле дистилляции, с получением газообразного верхнего потока S0, обогащенного пропеном, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции, и жидкого нижнего потока S01, обогащенного пропиленоксидом, водой и органическим растворителем, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции; (iv.2) возврат конденсированной части потока S0 в верхнюю часть узла дистилляции. Предлагаемый способ позволяет существенно избежать как разложения пропиленоксида в отстойнике, так и потерь отводимого сверху пропиленоксида, а также позволяет снизить энергозатраты на разделение пропиленоксида и пропена. 14 з.п. ф-лы, 7 пр., 3 ил.
1. Способ получения пропиленоксида, включающий
(i) обеспечение потока, содержащего пропен, пероксид водорода или источник пероксида водорода, воду и органический растворитель;
(ii) пропускание жидкого потока поступающего материала, обеспеченного на стадии (i), в зону эпоксидирования, содержащую катализатор эпоксидирования, содержащий цеолит на основе титана, и воздействие на жидкий поток поступающего материала условиями реакции эпоксидирования в зоне эпоксидирования, с получением реакционной смеси, содержащей пропен, пропиленоксид, воду и органический растворитель;
(iii) удаление вытекающего потока из зоны эпоксидирования, причем вытекающий поток содержит пропиленоксид, воду, органический растворитель и пропен;
(iv) отделение пропена от вытекающего потока посредством дистилляции, включающее
(iv.1) воздействие на вытекающий поток условиями дистилляции в узле дистилляции, с получением газообразного верхнего потока S0, обогащенного пропеном, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции, и жидкого нижнего потока S01, обогащенного пропиленоксидом, водой и органическим растворителем, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции;
(iv.2) возврат конденсированной части потока S0 в верхнюю часть узла дистилляции.
2. Способ по п. 1, включающий
(i) обеспечение потока, содержащего пропен, пропан, пероксид водорода или источник пероксида водорода, воду и органический растворитель;
(ii) пропускание жидкого потока поступающего материала, обеспеченного на стадии (i), в зону эпоксидирования, содержащую катализатор эпоксидирования, содержащий цеолит на основе титана, и воздействие на жидкий поток поступающего материала условиями реакции эпоксидирования в зоне эпоксидирования, с получением реакционной смеси, содержащей пропен, пропан, пропиленоксид, воду и органический растворитель;
(iii) удаление вытекающего потока из зоны эпоксидирования, причем вытекающий поток содержит пропиленоксид, воду, органический растворитель, пропен и пропан;
(iv) отделение пропена и пропана от вытекающего потока посредством дистилляции, включающее
(iv.1) воздействие на вытекающий поток условиями дистилляции в узле дистилляции, с получением газообразного верхнего потока S0, обогащенного пропеном и пропаном по сравнению с вытекающим потоком, подвергаемым условиям дистилляции, и жидкого нижнего потока S01, обогащенного пропиленоксидом, водой и органическим растворителем, по сравнению с вытекающим потоком, подвергаемым условиям дистилляции;
(iv.2) возврат конденсированной части потока S0 в верхнюю часть узла дистилляции.
3. Способ по п. 1 или 2, где узел дистилляции, применяемый на стадии (iv), представляет собой по меньшей мере одну дистилляционную колонну, предпочтительно одну дистилляционную колонну, где дистилляционная колонна имеет от 3 до 50, предпочтительно от 5 до 15, более предпочтительно от 6 до 10, более предпочтительно от 7 до 9, теоретических тарелок.
4. Способ по п. 1 или 2, где ректификационная секция узла дистилляции состоит из от 50 до 75%, предпочтительно от 60 до 65%, теоретических тарелок, и стриппинг-секция узла дистилляции состоит из от 25 до 50%, предпочтительно от 35 до 40%, теоретических тарелок.
5. Способ по п. 1 или 2, где узел дистилляции, применяемый на стадии (iv), эксплуатируют при верхнем давлении от 0,5 до 2,8 бар, предпочтительно от 0,6 до 2,5 бар, более предпочтительно от 0,8 до 1,5 бар.
6. Способ по п. 1 или 2, где узел дистилляции, применяемый на стадии (iv), эксплуатируют при верхней температуре в интервале от -70 до -30°C, предпочтительно от -60 до -40°C, более предпочтительно от -55 до -45°C.
7. Способ по п. 1 или 2, где поток S0, удаленный из узла дистилляции, применяемого на стадии (iv), имеет давление от 0,5 до 2,8 бар, предпочтительно от 0,6 до 2,5 бар, более предпочтительно от 0,8 до 1,5 бар, и температуру в интервале от -70 до -30°C, предпочтительно от -60 до -40°C, более предпочтительно от -55 до -45°C.
8. Способ по п. 1 или 2, где часть потока S0, которую конденсируют, с получением конденсированной части потока S0, регулируют так, что концентрация кислорода неконденсированной части потока S0 составляет менее 10 об.%, предпочтительно менее 7 об.%, наиболее предпочтительно менее 5 об.%.
9. Способ по п. 1 или 2, где конденсация достигается посредством сжатия до давления в интервале от 5 до 20 бар, предпочтительно в интервале от 10 до 19 бар, более предпочтительно в интервале от 12 до 18 бар, и регулирования температуры в интервале от 20 до 50°C, предпочтительно от 25 до 40°C, более предпочтительно от 32 до 38°C.
10. Способ по п. 1 или 2, где от 50 до 90 мас.%, предпочтительно от 60 до 85 мас.%, более предпочтительно от 65 до 80 мас.%, потока S0, которые образуют конденсированную часть потока S0, возвращают в верхнюю часть узла дистилляции на стадии (iv.2).
11. Способ по п. 1 или 2, где конденсированную часть потока S0 возвращают в верхнюю часть узла дистилляции на стадии (iv.2) вверху узла дистилляции или внутри ректификационной секции узла дистилляции, предпочтительно вверху узла дистилляции.
12. Способ по п. 1 или 2, где конденсированная часть потока S0, которую возвращают в верхнюю часть узла дистилляции на стадии (iv.2), имеет температуру в интервале от 20 до 50°C, предпочтительно в интервале от 30 до 40°C, более предпочтительно в интервале от 32 до 38°C.
13. Способ по п. 1 или 2, где конденсированную часть потока S0 подвергают теплообмену с потоком S0 перед возвратом в верхнюю часть узла дистилляции на стадии (iv.2).
14. Способ по п. 1 или 2, где температуру конденсированной части потока S0 уменьшают после сжатия и перед возвратом в верхнюю часть узла дистилляции на стадии (iv.2) на от 35 до 80 K, предпочтительно на от 45 до 65 K, более предпочтительно на от 55 до 65 K.
15. Способ по п. 1 или 2, который представляет собой непрерывный способ.
| WO 2015010990 A1, 29.01.2015 | |||
| WO 2008118265 A1, 02.10.2008 | |||
| WO 2009008493 A2, 15.01.2009 | |||
| WO 2004037802 A1, 06.05.2004 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПРОПИЛЕНОКСИДА | 2004 |
|
RU2324689C2 |

